User login
New diabetes guidelines put patient front and center
The 2016 updates of the American Diabetes Association’s Standards of Medical Care in Diabetes bring an enhanced focus on patient-centered care, evidence-based updates for atherosclerotic cardiovascular disease, and a dedicated section on obesity (Diabetes Care 2016;39[Suppl. 1]:S4-S5. doi: 10.2337/dc16-S003).
According to Dr. Robert E. Ratner, chief scientific and medical officer for the American Diabetes Association in Alexandria, Va., the focus of care for individuals with diabetes is to achieve a holistic approach coordinating patient-centered disease management to maximize the benefits of an integrated team approach. The 2016 guideline puts that mission front and center.

When formulating its yearly updates, the ADA looks for new information “that makes a significant change in the practice of medicine” and that will affect patient care, Dr. Ratner said in an interview. “We are trying to make the recommendations both thorough and evidence based.”
The guidelines were formulated by a professional practice committee chaired by Dr. William H. Herman, professor of epidemiology and internal medicine at the University of Michigan, Ann Arbor. According to the guidelines, the appointed committee “adheres to the Institute of Medicine Standards for Developing Trustworthy Clinical Practice Guidelines,” and the draft guidelines were made publicly available for comment before publication.
What’s different for 2016? Foremost is a change in approach, to more patient-centered care. Guidelines always address best practices at the population health level, Dr. Ratner said. However, “for each individual patient, interventions must be tailored, and the intervention must be adjusted to meet individual needs.”

One example is a relaxation in hemoglobin A1c (HbA1c) targets for the infirm and the elderly, based on data from large Veterans Affairs studies and from the National Health and Nutrition Examination Survey that show the harms of inappropriately low HbA1c levels for fragile populations. The updated guidelines encourage those caring for these patients to “back off and be more patient centered,” said Dr. Ratner.
The practice guidelines also acknowledge new information regarding the risk of euglycemic diabetic ketoacidosis with the use of sodium-glucose cotransporter 2 (SGLT2) inhibitors.
These changes strive to take into account new research, while acknowledging that in some areas there’s still a paucity of data, said Dr. Ratner. For example, recommendations regarding use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors for certain individuals with dyslipidemia and diabetes are included, though study of this new class of medications is ongoing; the guideline includes an evidence rating system to allow practitioners to rate the strength of recommendations when making individualized treatment decisions.
The guideline also gives the option of adding ezetimibe to a moderate statin dose for some individuals with diabetes and dyslipidemia, acknowledging the findings of the IMPROVE-IT trial. Other atherosclerotic cardiovascular disease updates include considering aspirin therapy for women 50 years and older who have diabetes with at least one additional major risk factor.
A new section devoted to obesity management acknowledges its critical importance in type 2 diabetes mellitus (T2DM). While continuing to emphasize the importance of lifestyle changes, such as healthful eating and physical activity, the guideline acknowledges that there’s a role for pharmacotherapy and surgical treatment. In addition to providing information on approved anti-obesity medication, the guideline addresses the benefit of bariatric surgery for a select group of patients. “We can’t ignore the data” that point toward a role for these interventions for some patients, said Dr. Ratner. The guideline weighs the advantages and disadvantages of each approach.
Other updates include a recommendation for testing all adults for diabetes beginning at age 45 years, without regard to weight; clarifying testing guidelines to emphasize that no one testing strategy is preferred over another; slightly relaxing HbA1c targets for pregnant women to 6%-6.5%; and enhancing guidelines for hospitalized patients with diabetes.
The complete practice guideline, which runs to 119 pages, is accompanied by an abridged version that provides a basic framework for diabetes management. Dr. Ratner said that the complete standards of care are appropriate for endocrinologists and others who focus on diabetes management, while the abridged version is a resource for primary care providers who may see some patients with diabetes in the mix of their caseload. Both documents are available to view or download free of charge. A summary of key updates precedes the full text of the guidelines, so that those using last year’s guidelines can familiarize themselves quickly with changes in the new version.
Dr. Herman reported chairing data and safety monitoring boards for Merck Sharp & Dohme, and Lexicon Pharmaceuticals. He has served as the editor for the Americas of Diabetic Medicine, and as ad hoc editor in chief of Diabetes Care.
On Twitter @karioakes
The 2016 updates of the American Diabetes Association’s Standards of Medical Care in Diabetes bring an enhanced focus on patient-centered care, evidence-based updates for atherosclerotic cardiovascular disease, and a dedicated section on obesity (Diabetes Care 2016;39[Suppl. 1]:S4-S5. doi: 10.2337/dc16-S003).
According to Dr. Robert E. Ratner, chief scientific and medical officer for the American Diabetes Association in Alexandria, Va., the focus of care for individuals with diabetes is to achieve a holistic approach coordinating patient-centered disease management to maximize the benefits of an integrated team approach. The 2016 guideline puts that mission front and center.
When formulating its yearly updates, the ADA looks for new information “that makes a significant change in the practice of medicine” and that will affect patient care, Dr. Ratner said in an interview. “We are trying to make the recommendations both thorough and evidence based.”
The guidelines were formulated by a professional practice committee chaired by Dr. William H. Herman, professor of epidemiology and internal medicine at the University of Michigan, Ann Arbor. According to the guidelines, the appointed committee “adheres to the Institute of Medicine Standards for Developing Trustworthy Clinical Practice Guidelines,” and the draft guidelines were made publicly available for comment before publication.
What’s different for 2016? Foremost is a change in approach, to more patient-centered care. Guidelines always address best practices at the population health level, Dr. Ratner said. However, “for each individual patient, interventions must be tailored, and the intervention must be adjusted to meet individual needs.”
One example is a relaxation in hemoglobin A1c (HbA1c) targets for the infirm and the elderly, based on data from large Veterans Affairs studies and from the National Health and Nutrition Examination Survey that show the harms of inappropriately low HbA1c levels for fragile populations. The updated guidelines encourage those caring for these patients to “back off and be more patient centered,” said Dr. Ratner.
The practice guidelines also acknowledge new information regarding the risk of euglycemic diabetic ketoacidosis with the use of sodium-glucose cotransporter 2 (SGLT2) inhibitors.
These changes strive to take into account new research, while acknowledging that in some areas there’s still a paucity of data, said Dr. Ratner. For example, recommendations regarding use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors for certain individuals with dyslipidemia and diabetes are included, though study of this new class of medications is ongoing; the guideline includes an evidence rating system to allow practitioners to rate the strength of recommendations when making individualized treatment decisions.
The guideline also gives the option of adding ezetimibe to a moderate statin dose for some individuals with diabetes and dyslipidemia, acknowledging the findings of the IMPROVE-IT trial. Other atherosclerotic cardiovascular disease updates include considering aspirin therapy for women 50 years and older who have diabetes with at least one additional major risk factor.
A new section devoted to obesity management acknowledges its critical importance in type 2 diabetes mellitus (T2DM). While continuing to emphasize the importance of lifestyle changes, such as healthful eating and physical activity, the guideline acknowledges that there’s a role for pharmacotherapy and surgical treatment. In addition to providing information on approved anti-obesity medication, the guideline addresses the benefit of bariatric surgery for a select group of patients. “We can’t ignore the data” that point toward a role for these interventions for some patients, said Dr. Ratner. The guideline weighs the advantages and disadvantages of each approach.
Other updates include a recommendation for testing all adults for diabetes beginning at age 45 years, without regard to weight; clarifying testing guidelines to emphasize that no one testing strategy is preferred over another; slightly relaxing HbA1c targets for pregnant women to 6%-6.5%; and enhancing guidelines for hospitalized patients with diabetes.
The complete practice guideline, which runs to 119 pages, is accompanied by an abridged version that provides a basic framework for diabetes management. Dr. Ratner said that the complete standards of care are appropriate for endocrinologists and others who focus on diabetes management, while the abridged version is a resource for primary care providers who may see some patients with diabetes in the mix of their caseload. Both documents are available to view or download free of charge. A summary of key updates precedes the full text of the guidelines, so that those using last year’s guidelines can familiarize themselves quickly with changes in the new version.
Dr. Herman reported chairing data and safety monitoring boards for Merck Sharp & Dohme, and Lexicon Pharmaceuticals. He has served as the editor for the Americas of Diabetic Medicine, and as ad hoc editor in chief of Diabetes Care.
On Twitter @karioakes
The 2016 updates of the American Diabetes Association’s Standards of Medical Care in Diabetes bring an enhanced focus on patient-centered care, evidence-based updates for atherosclerotic cardiovascular disease, and a dedicated section on obesity (Diabetes Care 2016;39[Suppl. 1]:S4-S5. doi: 10.2337/dc16-S003).
According to Dr. Robert E. Ratner, chief scientific and medical officer for the American Diabetes Association in Alexandria, Va., the focus of care for individuals with diabetes is to achieve a holistic approach coordinating patient-centered disease management to maximize the benefits of an integrated team approach. The 2016 guideline puts that mission front and center.
When formulating its yearly updates, the ADA looks for new information “that makes a significant change in the practice of medicine” and that will affect patient care, Dr. Ratner said in an interview. “We are trying to make the recommendations both thorough and evidence based.”
The guidelines were formulated by a professional practice committee chaired by Dr. William H. Herman, professor of epidemiology and internal medicine at the University of Michigan, Ann Arbor. According to the guidelines, the appointed committee “adheres to the Institute of Medicine Standards for Developing Trustworthy Clinical Practice Guidelines,” and the draft guidelines were made publicly available for comment before publication.
What’s different for 2016? Foremost is a change in approach, to more patient-centered care. Guidelines always address best practices at the population health level, Dr. Ratner said. However, “for each individual patient, interventions must be tailored, and the intervention must be adjusted to meet individual needs.”
One example is a relaxation in hemoglobin A1c (HbA1c) targets for the infirm and the elderly, based on data from large Veterans Affairs studies and from the National Health and Nutrition Examination Survey that show the harms of inappropriately low HbA1c levels for fragile populations. The updated guidelines encourage those caring for these patients to “back off and be more patient centered,” said Dr. Ratner.
The practice guidelines also acknowledge new information regarding the risk of euglycemic diabetic ketoacidosis with the use of sodium-glucose cotransporter 2 (SGLT2) inhibitors.
These changes strive to take into account new research, while acknowledging that in some areas there’s still a paucity of data, said Dr. Ratner. For example, recommendations regarding use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors for certain individuals with dyslipidemia and diabetes are included, though study of this new class of medications is ongoing; the guideline includes an evidence rating system to allow practitioners to rate the strength of recommendations when making individualized treatment decisions.
The guideline also gives the option of adding ezetimibe to a moderate statin dose for some individuals with diabetes and dyslipidemia, acknowledging the findings of the IMPROVE-IT trial. Other atherosclerotic cardiovascular disease updates include considering aspirin therapy for women 50 years and older who have diabetes with at least one additional major risk factor.
A new section devoted to obesity management acknowledges its critical importance in type 2 diabetes mellitus (T2DM). While continuing to emphasize the importance of lifestyle changes, such as healthful eating and physical activity, the guideline acknowledges that there’s a role for pharmacotherapy and surgical treatment. In addition to providing information on approved anti-obesity medication, the guideline addresses the benefit of bariatric surgery for a select group of patients. “We can’t ignore the data” that point toward a role for these interventions for some patients, said Dr. Ratner. The guideline weighs the advantages and disadvantages of each approach.
Other updates include a recommendation for testing all adults for diabetes beginning at age 45 years, without regard to weight; clarifying testing guidelines to emphasize that no one testing strategy is preferred over another; slightly relaxing HbA1c targets for pregnant women to 6%-6.5%; and enhancing guidelines for hospitalized patients with diabetes.
The complete practice guideline, which runs to 119 pages, is accompanied by an abridged version that provides a basic framework for diabetes management. Dr. Ratner said that the complete standards of care are appropriate for endocrinologists and others who focus on diabetes management, while the abridged version is a resource for primary care providers who may see some patients with diabetes in the mix of their caseload. Both documents are available to view or download free of charge. A summary of key updates precedes the full text of the guidelines, so that those using last year’s guidelines can familiarize themselves quickly with changes in the new version.
Dr. Herman reported chairing data and safety monitoring boards for Merck Sharp & Dohme, and Lexicon Pharmaceuticals. He has served as the editor for the Americas of Diabetic Medicine, and as ad hoc editor in chief of Diabetes Care.
On Twitter @karioakes
FROM DIABETES CARE

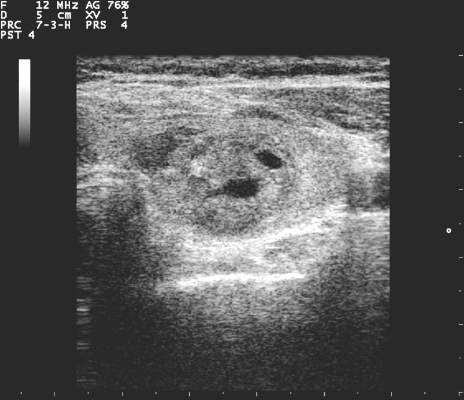
Wide variation in clinical management of thyroid nodules seen in first-ever survey
LAKE BUENA VISTA, FLA. – When making diagnostic and treatment decisions about thyroid nodules, many endocrinologists are not following current clinical practice guidelines, according to a recent survey. Further, according to a recent international survey of endocrinologists, there is wide regional variation in the use of molecular testing and calcitonin levels.
Dr. Nicole Vietor of the department of endocrinology at Walter Reed National Military Medical Center, Bethesda, Md., and her collaborators contacted members of the American Thyroid Association (ATA), the Endocrine Society (TES), and the American Association of Clinical Endocrinologists (AACE). Members of these societies were contacted directly by investigators and asked to complete a web-based survey regarding their practices for diagnosing and managing thyroid nodules.

The survey consisted of 36 questions, with an index case with variations presented to respondents, who then answered questions about diagnostic and management decisionmaking practices. The hypothetical index patient was a 52 year old woman with an incidental finding of a 1.5 cm right thyroid nodule. The patient was healthy and without risk factors; the patient’s nodule was not palpable on physical exam and she had no cervical lymphadenopathy.
Almost all respondents (99.4%) would order a thyroid-stimulating hormone for initial lab testing. Other commonly ordered exams included free T4 levels and thyroid peroxidase antibody requested by 41.5% and 24.3% of respondents, respectively. Fewer than 15% of respondents would have ordered any further lab exams.
All but 1.5% of respondents would order an anatomic or functional test for this index patient, with 57.2% ordering a thyroid ultrasound to be performed in radiology and 52.1% ordering an ultrasound in clinic (multiple responses were permitted). Cervical lymph nodes were included in the initial ultrasound assessment by 68.5% of respondents. Overall, more than half (56.6%) of thyroid ultrasounds were performed by endocrinologists, and about a third (31.9%) done by radiologists.
Practice variation from guidelines became apparent when respondents were asked how various nodule characteristics affected the decision to perform a fine needle aspiration (FNA). For a 1.5-cm solid hypoechoic nodule, 93.8% of respondents would perform FNA, while two thirds (67.0%) would perform an FNA for a 0.7-cm hypoechoic nodule with microcalcifications.
When performing FNAs, 83.3% of respondents use ultrasound to guide the biopsy, with most operators performing two (19.2%), three (28.5%), or four (23.0%) passes per nodule.
Of the 897 respondents, 80.5% were TES members, 56.5% were AACE members, and 44.5% were ATA members; most respondents belonged to more than one society. Almost two thirds of respondents (63.0%) were from North America, while 12.2% were from Europe, 10.8% were from Latin America, 6.5% were from Asia, 5.6% were from the Middle East or Africa, and just 1.9% were from Oceania. More men (60.2%) than women responded.
The AACE issued clinical practice guidelines for the management of thyroid nodules in 2010, as did the ATA in 2009 and 2015.
“In summary, management of a thyroid nodule is highly variable and differs from societal guidelines in multiple areas,” wrote Dr. Vietor and her colleagues in the presentation abstract.
On Twitter @karioakes
LAKE BUENA VISTA, FLA. – When making diagnostic and treatment decisions about thyroid nodules, many endocrinologists are not following current clinical practice guidelines, according to a recent survey. Further, according to a recent international survey of endocrinologists, there is wide regional variation in the use of molecular testing and calcitonin levels.
Dr. Nicole Vietor of the department of endocrinology at Walter Reed National Military Medical Center, Bethesda, Md., and her collaborators contacted members of the American Thyroid Association (ATA), the Endocrine Society (TES), and the American Association of Clinical Endocrinologists (AACE). Members of these societies were contacted directly by investigators and asked to complete a web-based survey regarding their practices for diagnosing and managing thyroid nodules.
The survey consisted of 36 questions, with an index case with variations presented to respondents, who then answered questions about diagnostic and management decisionmaking practices. The hypothetical index patient was a 52 year old woman with an incidental finding of a 1.5 cm right thyroid nodule. The patient was healthy and without risk factors; the patient’s nodule was not palpable on physical exam and she had no cervical lymphadenopathy.
Almost all respondents (99.4%) would order a thyroid-stimulating hormone for initial lab testing. Other commonly ordered exams included free T4 levels and thyroid peroxidase antibody requested by 41.5% and 24.3% of respondents, respectively. Fewer than 15% of respondents would have ordered any further lab exams.
All but 1.5% of respondents would order an anatomic or functional test for this index patient, with 57.2% ordering a thyroid ultrasound to be performed in radiology and 52.1% ordering an ultrasound in clinic (multiple responses were permitted). Cervical lymph nodes were included in the initial ultrasound assessment by 68.5% of respondents. Overall, more than half (56.6%) of thyroid ultrasounds were performed by endocrinologists, and about a third (31.9%) done by radiologists.
Practice variation from guidelines became apparent when respondents were asked how various nodule characteristics affected the decision to perform a fine needle aspiration (FNA). For a 1.5-cm solid hypoechoic nodule, 93.8% of respondents would perform FNA, while two thirds (67.0%) would perform an FNA for a 0.7-cm hypoechoic nodule with microcalcifications.
When performing FNAs, 83.3% of respondents use ultrasound to guide the biopsy, with most operators performing two (19.2%), three (28.5%), or four (23.0%) passes per nodule.
Of the 897 respondents, 80.5% were TES members, 56.5% were AACE members, and 44.5% were ATA members; most respondents belonged to more than one society. Almost two thirds of respondents (63.0%) were from North America, while 12.2% were from Europe, 10.8% were from Latin America, 6.5% were from Asia, 5.6% were from the Middle East or Africa, and just 1.9% were from Oceania. More men (60.2%) than women responded.
The AACE issued clinical practice guidelines for the management of thyroid nodules in 2010, as did the ATA in 2009 and 2015.
“In summary, management of a thyroid nodule is highly variable and differs from societal guidelines in multiple areas,” wrote Dr. Vietor and her colleagues in the presentation abstract.
On Twitter @karioakes
LAKE BUENA VISTA, FLA. – When making diagnostic and treatment decisions about thyroid nodules, many endocrinologists are not following current clinical practice guidelines, according to a recent survey. Further, according to a recent international survey of endocrinologists, there is wide regional variation in the use of molecular testing and calcitonin levels.
Dr. Nicole Vietor of the department of endocrinology at Walter Reed National Military Medical Center, Bethesda, Md., and her collaborators contacted members of the American Thyroid Association (ATA), the Endocrine Society (TES), and the American Association of Clinical Endocrinologists (AACE). Members of these societies were contacted directly by investigators and asked to complete a web-based survey regarding their practices for diagnosing and managing thyroid nodules.
The survey consisted of 36 questions, with an index case with variations presented to respondents, who then answered questions about diagnostic and management decisionmaking practices. The hypothetical index patient was a 52 year old woman with an incidental finding of a 1.5 cm right thyroid nodule. The patient was healthy and without risk factors; the patient’s nodule was not palpable on physical exam and she had no cervical lymphadenopathy.
Almost all respondents (99.4%) would order a thyroid-stimulating hormone for initial lab testing. Other commonly ordered exams included free T4 levels and thyroid peroxidase antibody requested by 41.5% and 24.3% of respondents, respectively. Fewer than 15% of respondents would have ordered any further lab exams.
All but 1.5% of respondents would order an anatomic or functional test for this index patient, with 57.2% ordering a thyroid ultrasound to be performed in radiology and 52.1% ordering an ultrasound in clinic (multiple responses were permitted). Cervical lymph nodes were included in the initial ultrasound assessment by 68.5% of respondents. Overall, more than half (56.6%) of thyroid ultrasounds were performed by endocrinologists, and about a third (31.9%) done by radiologists.
Practice variation from guidelines became apparent when respondents were asked how various nodule characteristics affected the decision to perform a fine needle aspiration (FNA). For a 1.5-cm solid hypoechoic nodule, 93.8% of respondents would perform FNA, while two thirds (67.0%) would perform an FNA for a 0.7-cm hypoechoic nodule with microcalcifications.
When performing FNAs, 83.3% of respondents use ultrasound to guide the biopsy, with most operators performing two (19.2%), three (28.5%), or four (23.0%) passes per nodule.
Of the 897 respondents, 80.5% were TES members, 56.5% were AACE members, and 44.5% were ATA members; most respondents belonged to more than one society. Almost two thirds of respondents (63.0%) were from North America, while 12.2% were from Europe, 10.8% were from Latin America, 6.5% were from Asia, 5.6% were from the Middle East or Africa, and just 1.9% were from Oceania. More men (60.2%) than women responded.
The AACE issued clinical practice guidelines for the management of thyroid nodules in 2010, as did the ATA in 2009 and 2015.
“In summary, management of a thyroid nodule is highly variable and differs from societal guidelines in multiple areas,” wrote Dr. Vietor and her colleagues in the presentation abstract.
On Twitter @karioakes
AT THE 15TH INTERNATIONAL THYROID CONGRESS
Key clinical point: A first-ever international survey of endocrinologists showed wide variation in clinical management of thyroid nodules.
Major finding: Respondents reported performing more fine needle aspiration (FNA) biopsies of thyroid nodules than recommended by practice guidelines.
Data source: Web-based survey of 897 members of three different professional organizations.
Disclosures: No disclosures were identified.
Have the conversation early: research proxies in Alzheimer’s disease
Many patients with Alzheimer’s disease can still identify a trusted individual to make decisions about participation in research, even when their cognitive abilities have declined to the point that they may not be able to give informed consent for participation in a particular research protocol.
This preserved ability can leave a window of time during which physicians and others caring for individuals with Alzheimer’s disease (AD) can offer patients the opportunity to identify a “research proxy” who can help the patient make future decisions about research participation.
Patients’ core values and “authenticity of character” are often relatively preserved even after cognitive abilities and memory have begun to decline, according to research conducted by Dr. Scott Kim, a senior investigator in the department of bioethics at the National Institutes of Health.

Speaking in Minneapolis at the University of Minnesota’s ethics forum entitled “Research with Human Participants: the National Debates,” Dr. Kim shared findings from a study examining decisional capability in patients with AD.
Dr. Kim and his colleagues interviewed 188 people with AD to determine their capacity to appoint a proxy and to consent to a lower-risk and a higher-risk clinical trial. They used a widely used competence assessment tool, along with an instrument that they designed to assess capacity to appoint a proxy. Capacity determinations were made by five psychiatrists who each independently viewed all interviews (Arch Gen Psychiatry. 2011 Feb; 68[2]:214-20).
Overall, fewer than half of patients (82 of 188) were deemed capable of consenting to the low-risk trial and fewer than 1 in 5 (31 of 188) were found capable of consenting to the high-risk trial. However, 38% (40 of 106) of patients who couldn’t consent to the low risk trial were still found capable of identifying a research proxy, as were 54% (86 of 157) of those incapable of consenting to the high risk trial.
Dr. Kim and his colleagues concluded that “providing for an appointed surrogate even after the onset of AD, which might best be done in the very-early stages of the illness, may help address key ethical challenges to AD research.”
Clarifying a proxy for research decision making is an important and separate process than designating a power of attorney for health care. Though clauses about research decision making can be written into power-of-attorney documents, participation in research is a separate – and slightly thornier – question than health care decision making. Not only do many research studies offer no direct benefit to participants, participating in research may carry some risk for the patient.
Still, many individuals with Alzheimer’s disease wish to participate in research, Dr. Kim said. Some may hope for some direct benefit from a particular clinical trial; others hope knowledge learned will help family members who may be at risk for dementia; for some, their desire to participate is purely altruistic.
Using a legally appointed representative as a research proxy can lend flexibility and authenticity to these choices, without diminishing the legal and ethical clarity these decisions require, Dr. Kim said. If the proxy is appointed early and functions as a “concurrent proxy” – meaning that decisions are made jointly early in the course of AD – then that proxy can learn more about the patient’s core values and wishes through the shared decisions they make. Though this joint process doesn’t fit well into our “relentlessly individualistic” view of competence, it can work very well, Dr. Kim said.
Whether an individual has decision-making capacity, said Dr. Kim, is a functional decision that should be based on the task-specific abilities that are relevant to the decision at hand. Most adults are presumed to have capacity to give consent for research participation.
In current thinking about who is or is not competent to give consent for participation in research, decisions about capacity should not be based on a diagnosis, such as “senility,” or a label, such as having an “unsound mind.” Rather, the threshold for capacity is fluid, and is affected by context, especially the balance of risk and benefit.
Still, when it comes to research participation, “ideological concerns often trump careful review of clinical data and realities,” said Dr. Kim.
Addressing a real-world concern, Dr. Kim said in an interview, “Health disparities often translate into the stage at which people seek help.” This means that for those with barriers to health care access, the window of capacity to appoint a research proxy may have closed by the time dementia is first diagnosed, shutting the patient out of research participation.
The Alzheimer’s Association also addresses the question of capacity and proxy appointment, noting that “the proxy’s consent can be either a research-specific advance directive or the proxy’s judgment of the individual’s “best interests.”
Physicians interested in drawing up a research proxy, Dr. Kim said, should work with their institutional review boards to devise a format that includes information about the types of research and the degree of risk the patient is willing to undergo. Since not every circumstance can be foreseen, Dr. Kim recommended leaving some flexibility in these documents to be used “only as guidance, not as a hard and fast rule.”
As genetic underpinnings are being found for more and more neurodegenerative diseases, a greater number of patients will be offered the opportunity for research participation. For Dr. Kim, the research proxy offers a path to participation in research for many of these patients. “Even after the diagnosis of Alzheimer’s disease, it is usually possible to obtain a valid proxy directive. As much as possible, involve the patient with dementia in the decision-making process; there are many, retained, preserved abilities that are ethically relevant.”
Dr. Kim’s study was supported by a grant from the National Institute of Mental Health.
On Twitter @karioakes
Many patients with Alzheimer’s disease can still identify a trusted individual to make decisions about participation in research, even when their cognitive abilities have declined to the point that they may not be able to give informed consent for participation in a particular research protocol.
This preserved ability can leave a window of time during which physicians and others caring for individuals with Alzheimer’s disease (AD) can offer patients the opportunity to identify a “research proxy” who can help the patient make future decisions about research participation.
Patients’ core values and “authenticity of character” are often relatively preserved even after cognitive abilities and memory have begun to decline, according to research conducted by Dr. Scott Kim, a senior investigator in the department of bioethics at the National Institutes of Health.
Speaking in Minneapolis at the University of Minnesota’s ethics forum entitled “Research with Human Participants: the National Debates,” Dr. Kim shared findings from a study examining decisional capability in patients with AD.
Dr. Kim and his colleagues interviewed 188 people with AD to determine their capacity to appoint a proxy and to consent to a lower-risk and a higher-risk clinical trial. They used a widely used competence assessment tool, along with an instrument that they designed to assess capacity to appoint a proxy. Capacity determinations were made by five psychiatrists who each independently viewed all interviews (Arch Gen Psychiatry. 2011 Feb; 68[2]:214-20).
Overall, fewer than half of patients (82 of 188) were deemed capable of consenting to the low-risk trial and fewer than 1 in 5 (31 of 188) were found capable of consenting to the high-risk trial. However, 38% (40 of 106) of patients who couldn’t consent to the low risk trial were still found capable of identifying a research proxy, as were 54% (86 of 157) of those incapable of consenting to the high risk trial.
Dr. Kim and his colleagues concluded that “providing for an appointed surrogate even after the onset of AD, which might best be done in the very-early stages of the illness, may help address key ethical challenges to AD research.”
Clarifying a proxy for research decision making is an important and separate process than designating a power of attorney for health care. Though clauses about research decision making can be written into power-of-attorney documents, participation in research is a separate – and slightly thornier – question than health care decision making. Not only do many research studies offer no direct benefit to participants, participating in research may carry some risk for the patient.
Still, many individuals with Alzheimer’s disease wish to participate in research, Dr. Kim said. Some may hope for some direct benefit from a particular clinical trial; others hope knowledge learned will help family members who may be at risk for dementia; for some, their desire to participate is purely altruistic.
Using a legally appointed representative as a research proxy can lend flexibility and authenticity to these choices, without diminishing the legal and ethical clarity these decisions require, Dr. Kim said. If the proxy is appointed early and functions as a “concurrent proxy” – meaning that decisions are made jointly early in the course of AD – then that proxy can learn more about the patient’s core values and wishes through the shared decisions they make. Though this joint process doesn’t fit well into our “relentlessly individualistic” view of competence, it can work very well, Dr. Kim said.
Whether an individual has decision-making capacity, said Dr. Kim, is a functional decision that should be based on the task-specific abilities that are relevant to the decision at hand. Most adults are presumed to have capacity to give consent for research participation.
In current thinking about who is or is not competent to give consent for participation in research, decisions about capacity should not be based on a diagnosis, such as “senility,” or a label, such as having an “unsound mind.” Rather, the threshold for capacity is fluid, and is affected by context, especially the balance of risk and benefit.
Still, when it comes to research participation, “ideological concerns often trump careful review of clinical data and realities,” said Dr. Kim.
Addressing a real-world concern, Dr. Kim said in an interview, “Health disparities often translate into the stage at which people seek help.” This means that for those with barriers to health care access, the window of capacity to appoint a research proxy may have closed by the time dementia is first diagnosed, shutting the patient out of research participation.
The Alzheimer’s Association also addresses the question of capacity and proxy appointment, noting that “the proxy’s consent can be either a research-specific advance directive or the proxy’s judgment of the individual’s “best interests.”
Physicians interested in drawing up a research proxy, Dr. Kim said, should work with their institutional review boards to devise a format that includes information about the types of research and the degree of risk the patient is willing to undergo. Since not every circumstance can be foreseen, Dr. Kim recommended leaving some flexibility in these documents to be used “only as guidance, not as a hard and fast rule.”
As genetic underpinnings are being found for more and more neurodegenerative diseases, a greater number of patients will be offered the opportunity for research participation. For Dr. Kim, the research proxy offers a path to participation in research for many of these patients. “Even after the diagnosis of Alzheimer’s disease, it is usually possible to obtain a valid proxy directive. As much as possible, involve the patient with dementia in the decision-making process; there are many, retained, preserved abilities that are ethically relevant.”
Dr. Kim’s study was supported by a grant from the National Institute of Mental Health.
On Twitter @karioakes
Many patients with Alzheimer’s disease can still identify a trusted individual to make decisions about participation in research, even when their cognitive abilities have declined to the point that they may not be able to give informed consent for participation in a particular research protocol.
This preserved ability can leave a window of time during which physicians and others caring for individuals with Alzheimer’s disease (AD) can offer patients the opportunity to identify a “research proxy” who can help the patient make future decisions about research participation.
Patients’ core values and “authenticity of character” are often relatively preserved even after cognitive abilities and memory have begun to decline, according to research conducted by Dr. Scott Kim, a senior investigator in the department of bioethics at the National Institutes of Health.
Speaking in Minneapolis at the University of Minnesota’s ethics forum entitled “Research with Human Participants: the National Debates,” Dr. Kim shared findings from a study examining decisional capability in patients with AD.
Dr. Kim and his colleagues interviewed 188 people with AD to determine their capacity to appoint a proxy and to consent to a lower-risk and a higher-risk clinical trial. They used a widely used competence assessment tool, along with an instrument that they designed to assess capacity to appoint a proxy. Capacity determinations were made by five psychiatrists who each independently viewed all interviews (Arch Gen Psychiatry. 2011 Feb; 68[2]:214-20).
Overall, fewer than half of patients (82 of 188) were deemed capable of consenting to the low-risk trial and fewer than 1 in 5 (31 of 188) were found capable of consenting to the high-risk trial. However, 38% (40 of 106) of patients who couldn’t consent to the low risk trial were still found capable of identifying a research proxy, as were 54% (86 of 157) of those incapable of consenting to the high risk trial.
Dr. Kim and his colleagues concluded that “providing for an appointed surrogate even after the onset of AD, which might best be done in the very-early stages of the illness, may help address key ethical challenges to AD research.”
Clarifying a proxy for research decision making is an important and separate process than designating a power of attorney for health care. Though clauses about research decision making can be written into power-of-attorney documents, participation in research is a separate – and slightly thornier – question than health care decision making. Not only do many research studies offer no direct benefit to participants, participating in research may carry some risk for the patient.
Still, many individuals with Alzheimer’s disease wish to participate in research, Dr. Kim said. Some may hope for some direct benefit from a particular clinical trial; others hope knowledge learned will help family members who may be at risk for dementia; for some, their desire to participate is purely altruistic.
Using a legally appointed representative as a research proxy can lend flexibility and authenticity to these choices, without diminishing the legal and ethical clarity these decisions require, Dr. Kim said. If the proxy is appointed early and functions as a “concurrent proxy” – meaning that decisions are made jointly early in the course of AD – then that proxy can learn more about the patient’s core values and wishes through the shared decisions they make. Though this joint process doesn’t fit well into our “relentlessly individualistic” view of competence, it can work very well, Dr. Kim said.
Whether an individual has decision-making capacity, said Dr. Kim, is a functional decision that should be based on the task-specific abilities that are relevant to the decision at hand. Most adults are presumed to have capacity to give consent for research participation.
In current thinking about who is or is not competent to give consent for participation in research, decisions about capacity should not be based on a diagnosis, such as “senility,” or a label, such as having an “unsound mind.” Rather, the threshold for capacity is fluid, and is affected by context, especially the balance of risk and benefit.
Still, when it comes to research participation, “ideological concerns often trump careful review of clinical data and realities,” said Dr. Kim.
Addressing a real-world concern, Dr. Kim said in an interview, “Health disparities often translate into the stage at which people seek help.” This means that for those with barriers to health care access, the window of capacity to appoint a research proxy may have closed by the time dementia is first diagnosed, shutting the patient out of research participation.
The Alzheimer’s Association also addresses the question of capacity and proxy appointment, noting that “the proxy’s consent can be either a research-specific advance directive or the proxy’s judgment of the individual’s “best interests.”
Physicians interested in drawing up a research proxy, Dr. Kim said, should work with their institutional review boards to devise a format that includes information about the types of research and the degree of risk the patient is willing to undergo. Since not every circumstance can be foreseen, Dr. Kim recommended leaving some flexibility in these documents to be used “only as guidance, not as a hard and fast rule.”
As genetic underpinnings are being found for more and more neurodegenerative diseases, a greater number of patients will be offered the opportunity for research participation. For Dr. Kim, the research proxy offers a path to participation in research for many of these patients. “Even after the diagnosis of Alzheimer’s disease, it is usually possible to obtain a valid proxy directive. As much as possible, involve the patient with dementia in the decision-making process; there are many, retained, preserved abilities that are ethically relevant.”
Dr. Kim’s study was supported by a grant from the National Institute of Mental Health.
On Twitter @karioakes
EXPERT ANALYSIS FROM RESEARCH WITH PARTICIPANTS: THE NATIONAL DEBATES

FDA proposes ban on indoor tanning for minors
The Food and Drug Administration wants to prohibit indoor tanning for minors and is seeking stricter regulations to enhance the safety of indoor tanning and use of sunlamp products for all users.
“Today’s action is intended to help protect young people from a known and preventable cause of skin cancer and other harms. Individuals under 18 years are at greatest risk of the adverse health consequences of indoor tanning,” said acting FDA Commissioner Stephen Ostroff in a news release outlining the two proposed rules.

Currently, 1.6 million minors tan indoors yearly, according to data from the 2013 National Youth Risk Behavior Survey, cited by the FDA. Although many states require parental consent before minors can use tanning facilities, a national standard is lacking.
An additional measure in the first proposed rule requires adults who use commercial tanning beds to sign a “risk acknowledgment certification” statement before the first tanning session and every 6 months thereafter. These restrictions are appropriate, the FDA statement said, because of the increased risk of skin cancer, notably melanoma, and other risks associated with indoor tanning. According to the proposed rule, some research has found that “doses of UVA radiation emitted by high-power sunlamp products may be up to 10-15 times higher than that of the midday sun, resulting in an intense amount of exposure that does not exist in nature.”
The second proposed ruleis aimed at tanning facilities and sunlamp manufacturers, and contains measures to bring about safer use of tanning devices. These rules would set limits on the amount of light allowed through protective eyewear when tanning, make an emergency “off” switch mandatory on tanning beds, improve warning signs and tanning bulb labeling, and prohibit device modifications without FDA recertification.
According to the FDA, these regulations would affect more than 30,000 tanning salons, health clubs, and spas that offer indoor tanning. “These proposed rules are meant to help adults make their decisions based on truthful information and to ensure manufacturers and tanning facilities take additional steps to improve the safety of these devices,” Dr. Ostroff said.
The rules and opportunities to comment are available starting Monday, Dec. 21 at www.regulations.gov. Public comments will be open for 90 days.
On Twitter @karioakes
The Food and Drug Administration wants to prohibit indoor tanning for minors and is seeking stricter regulations to enhance the safety of indoor tanning and use of sunlamp products for all users.
“Today’s action is intended to help protect young people from a known and preventable cause of skin cancer and other harms. Individuals under 18 years are at greatest risk of the adverse health consequences of indoor tanning,” said acting FDA Commissioner Stephen Ostroff in a news release outlining the two proposed rules.
Currently, 1.6 million minors tan indoors yearly, according to data from the 2013 National Youth Risk Behavior Survey, cited by the FDA. Although many states require parental consent before minors can use tanning facilities, a national standard is lacking.
An additional measure in the first proposed rule requires adults who use commercial tanning beds to sign a “risk acknowledgment certification” statement before the first tanning session and every 6 months thereafter. These restrictions are appropriate, the FDA statement said, because of the increased risk of skin cancer, notably melanoma, and other risks associated with indoor tanning. According to the proposed rule, some research has found that “doses of UVA radiation emitted by high-power sunlamp products may be up to 10-15 times higher than that of the midday sun, resulting in an intense amount of exposure that does not exist in nature.”
The second proposed ruleis aimed at tanning facilities and sunlamp manufacturers, and contains measures to bring about safer use of tanning devices. These rules would set limits on the amount of light allowed through protective eyewear when tanning, make an emergency “off” switch mandatory on tanning beds, improve warning signs and tanning bulb labeling, and prohibit device modifications without FDA recertification.
According to the FDA, these regulations would affect more than 30,000 tanning salons, health clubs, and spas that offer indoor tanning. “These proposed rules are meant to help adults make their decisions based on truthful information and to ensure manufacturers and tanning facilities take additional steps to improve the safety of these devices,” Dr. Ostroff said.
The rules and opportunities to comment are available starting Monday, Dec. 21 at www.regulations.gov. Public comments will be open for 90 days.
On Twitter @karioakes
The Food and Drug Administration wants to prohibit indoor tanning for minors and is seeking stricter regulations to enhance the safety of indoor tanning and use of sunlamp products for all users.
“Today’s action is intended to help protect young people from a known and preventable cause of skin cancer and other harms. Individuals under 18 years are at greatest risk of the adverse health consequences of indoor tanning,” said acting FDA Commissioner Stephen Ostroff in a news release outlining the two proposed rules.
Currently, 1.6 million minors tan indoors yearly, according to data from the 2013 National Youth Risk Behavior Survey, cited by the FDA. Although many states require parental consent before minors can use tanning facilities, a national standard is lacking.
An additional measure in the first proposed rule requires adults who use commercial tanning beds to sign a “risk acknowledgment certification” statement before the first tanning session and every 6 months thereafter. These restrictions are appropriate, the FDA statement said, because of the increased risk of skin cancer, notably melanoma, and other risks associated with indoor tanning. According to the proposed rule, some research has found that “doses of UVA radiation emitted by high-power sunlamp products may be up to 10-15 times higher than that of the midday sun, resulting in an intense amount of exposure that does not exist in nature.”
The second proposed ruleis aimed at tanning facilities and sunlamp manufacturers, and contains measures to bring about safer use of tanning devices. These rules would set limits on the amount of light allowed through protective eyewear when tanning, make an emergency “off” switch mandatory on tanning beds, improve warning signs and tanning bulb labeling, and prohibit device modifications without FDA recertification.
According to the FDA, these regulations would affect more than 30,000 tanning salons, health clubs, and spas that offer indoor tanning. “These proposed rules are meant to help adults make their decisions based on truthful information and to ensure manufacturers and tanning facilities take additional steps to improve the safety of these devices,” Dr. Ostroff said.
The rules and opportunities to comment are available starting Monday, Dec. 21 at www.regulations.gov. Public comments will be open for 90 days.
On Twitter @karioakes

FDA panel votes down expanded ezetimibe indications
An advisory committee of the Food and Drug Administration, unconvinced of a clinically meaningful effect based on the IMPROVE-IT trial, voted against approving an expanded indication for ezetimibe (Zetia) in combination with simvastatin (Zocor).
Merck, which markets the combination as Vytorin in the United States, had come before the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) seeking an indication to reduce the risk of cardiovascular events in patients with coronary heart disease.
The committee’s 10-5 “no” vote came after a day of testimony and discussion that revolved around the results of the 18,000 patient, 7-year Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT). The trial enrolled patients soon after they had experienced an acute coronary syndrome (ACS) event. The sponsor’s presentation of the primary endpoint of the trial showed a modest treatment effect. For the composite primary efficacy endpoint of cardiovascular death, nonfatal MI, unstable angina requiring hospitalization, coronary revascularization, or nonfatal stroke, for the patients randomized to receive simvastatin 40 mg plus ezetimibe 10 mg (Vytorin), compared with those on simvastatin 40 mg alone, the hazard ratio was 0.936 (95% confidence interval, 0.887-0.988; P = .016) (N Engl J Med. 2015 Jun 18;372[25]:2387-97).

“From what I’ve heard today, I was not persuaded that the treatment effect was clinically meaningful,” said Dr. Sanjay Kaul, professor of medicine at the University of California, Los Angeles, and attending cardiologist at Cedars-Sinai Medical Center, also in Los Angeles. Other committee members were more blunt: Dr. Milton Packer, a distinguished scholar in cardiovascular science at Dallas’ Baylor University, said of the treatment effect, “You blink and you miss it.”
The effect size was more notable for two prespecified subgroups of the IMPROVE-IT population, those with diabetes and those aged 75 years and older. The meaning of the stronger effect in these two populations, who may have represented a higher risk subset of the larger study, was debated at length by the committee.
“Subgroup analyses are tempting but they can be treacherous,” said Dr. Kaul. “At this point, it is hard to ignore that 27% of the cohort is where most of the benefit is found. This is difficult to operationalize.”
The committee also expressed concern that follow-up for the primary endpoint was 91%, meaning that data were missing for thousands of patient-years of follow-up. “Missingness matters,” said Thomas R. Fleming, Ph.D., a professor in the department of biostatistics at the University of Washington, Seattle. “The goal of clinical research should be to obtain statistically reliable evidence regarding clinically meaningful effects,” and the missing data could endanger that reliability, he said.
Echoing the comments of several of the committee members who voted in favor of the expanded recommendations, Dr. Michael Blaha said “I think there’s a modest unmet need for add-on lipid-lowering therapy” for individuals with coronary disease. “The trial is positive, though the effect is modest. ... I voted ‘yes’ consistent with the way I would use it in clinical practice,” said Dr. Blaha, director of clinical research at the Ciccarone Center for the Prevention of Heart Disease at Johns Hopkins Hospital, Baltimore.

EMDAC chair Dr. Robert J. Smith, professor of medicine at Brown University, Providence, R.I., voiced the consensus of the committee in praising IMPROVE-IT as a “remarkable trial in terms of scope, duration, and quality.” Dr. Blaha pointed out that this was the trial that really showed that for cardiovascular risk and LDL levels, “Lower is better.”
The duration of the trial was noted by several committee members as giving reassuring information about the overall safety profile of ezetimibe/simvastatin. Some committee members, notably the patient representative Debra McCall of Murrieta, Calif., expressed concern about the slightly increased numbers of patients who suffered hemorrhagic stroke in the ezetimibe/simvastatin arm.
The FDA panelists were cleared of any potential conflicts of interest.
On Twitter @karioakes
An advisory committee of the Food and Drug Administration, unconvinced of a clinically meaningful effect based on the IMPROVE-IT trial, voted against approving an expanded indication for ezetimibe (Zetia) in combination with simvastatin (Zocor).
Merck, which markets the combination as Vytorin in the United States, had come before the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) seeking an indication to reduce the risk of cardiovascular events in patients with coronary heart disease.
The committee’s 10-5 “no” vote came after a day of testimony and discussion that revolved around the results of the 18,000 patient, 7-year Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT). The trial enrolled patients soon after they had experienced an acute coronary syndrome (ACS) event. The sponsor’s presentation of the primary endpoint of the trial showed a modest treatment effect. For the composite primary efficacy endpoint of cardiovascular death, nonfatal MI, unstable angina requiring hospitalization, coronary revascularization, or nonfatal stroke, for the patients randomized to receive simvastatin 40 mg plus ezetimibe 10 mg (Vytorin), compared with those on simvastatin 40 mg alone, the hazard ratio was 0.936 (95% confidence interval, 0.887-0.988; P = .016) (N Engl J Med. 2015 Jun 18;372[25]:2387-97).
“From what I’ve heard today, I was not persuaded that the treatment effect was clinically meaningful,” said Dr. Sanjay Kaul, professor of medicine at the University of California, Los Angeles, and attending cardiologist at Cedars-Sinai Medical Center, also in Los Angeles. Other committee members were more blunt: Dr. Milton Packer, a distinguished scholar in cardiovascular science at Dallas’ Baylor University, said of the treatment effect, “You blink and you miss it.”
The effect size was more notable for two prespecified subgroups of the IMPROVE-IT population, those with diabetes and those aged 75 years and older. The meaning of the stronger effect in these two populations, who may have represented a higher risk subset of the larger study, was debated at length by the committee.
“Subgroup analyses are tempting but they can be treacherous,” said Dr. Kaul. “At this point, it is hard to ignore that 27% of the cohort is where most of the benefit is found. This is difficult to operationalize.”
The committee also expressed concern that follow-up for the primary endpoint was 91%, meaning that data were missing for thousands of patient-years of follow-up. “Missingness matters,” said Thomas R. Fleming, Ph.D., a professor in the department of biostatistics at the University of Washington, Seattle. “The goal of clinical research should be to obtain statistically reliable evidence regarding clinically meaningful effects,” and the missing data could endanger that reliability, he said.
Echoing the comments of several of the committee members who voted in favor of the expanded recommendations, Dr. Michael Blaha said “I think there’s a modest unmet need for add-on lipid-lowering therapy” for individuals with coronary disease. “The trial is positive, though the effect is modest. ... I voted ‘yes’ consistent with the way I would use it in clinical practice,” said Dr. Blaha, director of clinical research at the Ciccarone Center for the Prevention of Heart Disease at Johns Hopkins Hospital, Baltimore.
EMDAC chair Dr. Robert J. Smith, professor of medicine at Brown University, Providence, R.I., voiced the consensus of the committee in praising IMPROVE-IT as a “remarkable trial in terms of scope, duration, and quality.” Dr. Blaha pointed out that this was the trial that really showed that for cardiovascular risk and LDL levels, “Lower is better.”
The duration of the trial was noted by several committee members as giving reassuring information about the overall safety profile of ezetimibe/simvastatin. Some committee members, notably the patient representative Debra McCall of Murrieta, Calif., expressed concern about the slightly increased numbers of patients who suffered hemorrhagic stroke in the ezetimibe/simvastatin arm.
The FDA panelists were cleared of any potential conflicts of interest.
On Twitter @karioakes
An advisory committee of the Food and Drug Administration, unconvinced of a clinically meaningful effect based on the IMPROVE-IT trial, voted against approving an expanded indication for ezetimibe (Zetia) in combination with simvastatin (Zocor).
Merck, which markets the combination as Vytorin in the United States, had come before the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) seeking an indication to reduce the risk of cardiovascular events in patients with coronary heart disease.
The committee’s 10-5 “no” vote came after a day of testimony and discussion that revolved around the results of the 18,000 patient, 7-year Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT). The trial enrolled patients soon after they had experienced an acute coronary syndrome (ACS) event. The sponsor’s presentation of the primary endpoint of the trial showed a modest treatment effect. For the composite primary efficacy endpoint of cardiovascular death, nonfatal MI, unstable angina requiring hospitalization, coronary revascularization, or nonfatal stroke, for the patients randomized to receive simvastatin 40 mg plus ezetimibe 10 mg (Vytorin), compared with those on simvastatin 40 mg alone, the hazard ratio was 0.936 (95% confidence interval, 0.887-0.988; P = .016) (N Engl J Med. 2015 Jun 18;372[25]:2387-97).
“From what I’ve heard today, I was not persuaded that the treatment effect was clinically meaningful,” said Dr. Sanjay Kaul, professor of medicine at the University of California, Los Angeles, and attending cardiologist at Cedars-Sinai Medical Center, also in Los Angeles. Other committee members were more blunt: Dr. Milton Packer, a distinguished scholar in cardiovascular science at Dallas’ Baylor University, said of the treatment effect, “You blink and you miss it.”
The effect size was more notable for two prespecified subgroups of the IMPROVE-IT population, those with diabetes and those aged 75 years and older. The meaning of the stronger effect in these two populations, who may have represented a higher risk subset of the larger study, was debated at length by the committee.
“Subgroup analyses are tempting but they can be treacherous,” said Dr. Kaul. “At this point, it is hard to ignore that 27% of the cohort is where most of the benefit is found. This is difficult to operationalize.”
The committee also expressed concern that follow-up for the primary endpoint was 91%, meaning that data were missing for thousands of patient-years of follow-up. “Missingness matters,” said Thomas R. Fleming, Ph.D., a professor in the department of biostatistics at the University of Washington, Seattle. “The goal of clinical research should be to obtain statistically reliable evidence regarding clinically meaningful effects,” and the missing data could endanger that reliability, he said.
Echoing the comments of several of the committee members who voted in favor of the expanded recommendations, Dr. Michael Blaha said “I think there’s a modest unmet need for add-on lipid-lowering therapy” for individuals with coronary disease. “The trial is positive, though the effect is modest. ... I voted ‘yes’ consistent with the way I would use it in clinical practice,” said Dr. Blaha, director of clinical research at the Ciccarone Center for the Prevention of Heart Disease at Johns Hopkins Hospital, Baltimore.
EMDAC chair Dr. Robert J. Smith, professor of medicine at Brown University, Providence, R.I., voiced the consensus of the committee in praising IMPROVE-IT as a “remarkable trial in terms of scope, duration, and quality.” Dr. Blaha pointed out that this was the trial that really showed that for cardiovascular risk and LDL levels, “Lower is better.”
The duration of the trial was noted by several committee members as giving reassuring information about the overall safety profile of ezetimibe/simvastatin. Some committee members, notably the patient representative Debra McCall of Murrieta, Calif., expressed concern about the slightly increased numbers of patients who suffered hemorrhagic stroke in the ezetimibe/simvastatin arm.
The FDA panelists were cleared of any potential conflicts of interest.
On Twitter @karioakes
FROM AN FDA ADVISORY PANEL MEETING

New antivirals, more screening could slash hepatitis C cases
Hepatitis C could be reduced by 90% or more in the United States by the year 2040 with the use of direct-acting antivirals, near-universal screening, and enhanced treatment capacity, according to a study in Clinical Infectious Diseases.
A research team led by Jeffrey Townsend, Ph.D., of Yale School of Public Health, New Haven, Conn., said that new direct-acting antivirals (DAAs) alone could reduce the prevalence of hepatitis C virus (HCV) by 80% by the year 2040. When near-universal screening and enhanced treatment capacity are added to the equation, HCV could be eliminated in the United States, though cost and reimbursement issues may impede implementation.
“The key finding is that a fourfold increase to the number of patients treated each year could virtually eliminate HCV from the noninjecting population within a decade,” said Dr. Townsend, senior author of the study, in a statement accompanying the study [Clin Infect Dis. 2015. doi: 10.1093/cid/civ894].
First author David Durham, Ph.D., also of the Yale School of Public Health, and his collaborators analyzed currently available data and constructed a sophisticated model to generate projections of how future DAA treatment will change HCV prevalence. Dr. Durham and his colleagues also built models to account for varying levels of screening in the general population and for people who inject drugs (PWIDs).
More than 5 million people with chronic hepatitis C infection are thought to live in the United States, and just about 100,000 of those currently receive treatment each year. The treatment burden and relatively low cure rate of interferon-based therapies have historically been significant impediments for many patients. New DAAs promise a sustained virologic response (SVR) in up to 95% of patients with a well-tolerated, once-daily, one-pill several-week regimen.
Without enhanced screening or treatment rates, the model predicted an 80% decline in U.S. HCV prevalence by the year 2040. The 80% benchmark would be reached by the year 2025 if the annual treatment rate increased to 400,000, with 256,315 fewer HCV-related deaths through 2040. Just doubling the treatment rate to 200,000 patients per year would result in an 80% decrease in HCV prevalence by 2031, with 143,055 fewer deaths by 2040.
However, “more than half of those with chronic HCV are unaware of their status, including up to two-thirds of people who inject drugs,” wrote Dr. Durham and his coauthors. Therefore, enhanced HCV screening, especially among PWIDs, could provide a significant further reduction in morbidity and mortality from HCV.
Targeted screening of PWIDs to achieve a 20% screening rate (compared with the current 4.1%), combined with a treatment rate of at least 30%, would yield a 90% reduction of prevalence in HCV by 2040. Universal screening, combined with a treatment rate of at least 20%, would achieve a 90% reduction in the incidence of new infections by 2040, according to the model.
The drop in prevalence in the models took into account not only an increased SVR rate, but also the reduced transmission rate when more persons with HCV achieve an SVR. This dynamic transmission analysis “captures the impact of treatment on both chronic disease and transmission dynamics, distinguishes screening and treatment as separate but related public health objectives, and accounts for underreporting … of national HCV prevalence,” according to Dr. Durham and his coauthors.
Morbidity and mortality were accounted for by projecting the number of HCV patients in each treatment and screening condition who would spontaneously clear disease, become chronically infected, develop increasing levels of liver disease and cirrhosis, require transplant, become reinfected, or die.
As always, real world considerations temper what’s achievable. For DAAs, a chief factor is the cost of the regimens, which currently hovers around $90,000. “The improvement of health outcomes expected from greater acceptance might be tempered by the high costs of treatment and by limited insurance coverage and willingness to treat PWIDs,” Dr. Durham and his coauthors wrote.
Study limitations included some “simplifying assumptions” in the modeling, which did not account for the varying incidence of fibrosis and transmissibility among different HCV genotypes. Still, wrote Dr. Durham and his collaborators, “our analysis provides a forecast for the potential impact of DAAs in reducing HCV-associated liver disease, demonstrating that achievable expansion of HCV treatment at current screening rates can substantially reduce morbidity and mortality.”
The study was funded by the Notsew Orm Sands Foundation and Merck. Dr. Durham, Dr. Galvani, and Dr. Townsend have consulted for and received research funding from Sanofi Pasteur and Merck. Dr. Elbasha is employed by Merck and holds stock and stock options in the company. The other authors reported no conflicts of interest.
On Twitter @karioakes
Hepatitis C could be reduced by 90% or more in the United States by the year 2040 with the use of direct-acting antivirals, near-universal screening, and enhanced treatment capacity, according to a study in Clinical Infectious Diseases.
A research team led by Jeffrey Townsend, Ph.D., of Yale School of Public Health, New Haven, Conn., said that new direct-acting antivirals (DAAs) alone could reduce the prevalence of hepatitis C virus (HCV) by 80% by the year 2040. When near-universal screening and enhanced treatment capacity are added to the equation, HCV could be eliminated in the United States, though cost and reimbursement issues may impede implementation.
“The key finding is that a fourfold increase to the number of patients treated each year could virtually eliminate HCV from the noninjecting population within a decade,” said Dr. Townsend, senior author of the study, in a statement accompanying the study [Clin Infect Dis. 2015. doi: 10.1093/cid/civ894].
First author David Durham, Ph.D., also of the Yale School of Public Health, and his collaborators analyzed currently available data and constructed a sophisticated model to generate projections of how future DAA treatment will change HCV prevalence. Dr. Durham and his colleagues also built models to account for varying levels of screening in the general population and for people who inject drugs (PWIDs).
More than 5 million people with chronic hepatitis C infection are thought to live in the United States, and just about 100,000 of those currently receive treatment each year. The treatment burden and relatively low cure rate of interferon-based therapies have historically been significant impediments for many patients. New DAAs promise a sustained virologic response (SVR) in up to 95% of patients with a well-tolerated, once-daily, one-pill several-week regimen.
Without enhanced screening or treatment rates, the model predicted an 80% decline in U.S. HCV prevalence by the year 2040. The 80% benchmark would be reached by the year 2025 if the annual treatment rate increased to 400,000, with 256,315 fewer HCV-related deaths through 2040. Just doubling the treatment rate to 200,000 patients per year would result in an 80% decrease in HCV prevalence by 2031, with 143,055 fewer deaths by 2040.
However, “more than half of those with chronic HCV are unaware of their status, including up to two-thirds of people who inject drugs,” wrote Dr. Durham and his coauthors. Therefore, enhanced HCV screening, especially among PWIDs, could provide a significant further reduction in morbidity and mortality from HCV.
Targeted screening of PWIDs to achieve a 20% screening rate (compared with the current 4.1%), combined with a treatment rate of at least 30%, would yield a 90% reduction of prevalence in HCV by 2040. Universal screening, combined with a treatment rate of at least 20%, would achieve a 90% reduction in the incidence of new infections by 2040, according to the model.
The drop in prevalence in the models took into account not only an increased SVR rate, but also the reduced transmission rate when more persons with HCV achieve an SVR. This dynamic transmission analysis “captures the impact of treatment on both chronic disease and transmission dynamics, distinguishes screening and treatment as separate but related public health objectives, and accounts for underreporting … of national HCV prevalence,” according to Dr. Durham and his coauthors.
Morbidity and mortality were accounted for by projecting the number of HCV patients in each treatment and screening condition who would spontaneously clear disease, become chronically infected, develop increasing levels of liver disease and cirrhosis, require transplant, become reinfected, or die.
As always, real world considerations temper what’s achievable. For DAAs, a chief factor is the cost of the regimens, which currently hovers around $90,000. “The improvement of health outcomes expected from greater acceptance might be tempered by the high costs of treatment and by limited insurance coverage and willingness to treat PWIDs,” Dr. Durham and his coauthors wrote.
Study limitations included some “simplifying assumptions” in the modeling, which did not account for the varying incidence of fibrosis and transmissibility among different HCV genotypes. Still, wrote Dr. Durham and his collaborators, “our analysis provides a forecast for the potential impact of DAAs in reducing HCV-associated liver disease, demonstrating that achievable expansion of HCV treatment at current screening rates can substantially reduce morbidity and mortality.”
The study was funded by the Notsew Orm Sands Foundation and Merck. Dr. Durham, Dr. Galvani, and Dr. Townsend have consulted for and received research funding from Sanofi Pasteur and Merck. Dr. Elbasha is employed by Merck and holds stock and stock options in the company. The other authors reported no conflicts of interest.
On Twitter @karioakes
Hepatitis C could be reduced by 90% or more in the United States by the year 2040 with the use of direct-acting antivirals, near-universal screening, and enhanced treatment capacity, according to a study in Clinical Infectious Diseases.
A research team led by Jeffrey Townsend, Ph.D., of Yale School of Public Health, New Haven, Conn., said that new direct-acting antivirals (DAAs) alone could reduce the prevalence of hepatitis C virus (HCV) by 80% by the year 2040. When near-universal screening and enhanced treatment capacity are added to the equation, HCV could be eliminated in the United States, though cost and reimbursement issues may impede implementation.
“The key finding is that a fourfold increase to the number of patients treated each year could virtually eliminate HCV from the noninjecting population within a decade,” said Dr. Townsend, senior author of the study, in a statement accompanying the study [Clin Infect Dis. 2015. doi: 10.1093/cid/civ894].
First author David Durham, Ph.D., also of the Yale School of Public Health, and his collaborators analyzed currently available data and constructed a sophisticated model to generate projections of how future DAA treatment will change HCV prevalence. Dr. Durham and his colleagues also built models to account for varying levels of screening in the general population and for people who inject drugs (PWIDs).
More than 5 million people with chronic hepatitis C infection are thought to live in the United States, and just about 100,000 of those currently receive treatment each year. The treatment burden and relatively low cure rate of interferon-based therapies have historically been significant impediments for many patients. New DAAs promise a sustained virologic response (SVR) in up to 95% of patients with a well-tolerated, once-daily, one-pill several-week regimen.
Without enhanced screening or treatment rates, the model predicted an 80% decline in U.S. HCV prevalence by the year 2040. The 80% benchmark would be reached by the year 2025 if the annual treatment rate increased to 400,000, with 256,315 fewer HCV-related deaths through 2040. Just doubling the treatment rate to 200,000 patients per year would result in an 80% decrease in HCV prevalence by 2031, with 143,055 fewer deaths by 2040.
However, “more than half of those with chronic HCV are unaware of their status, including up to two-thirds of people who inject drugs,” wrote Dr. Durham and his coauthors. Therefore, enhanced HCV screening, especially among PWIDs, could provide a significant further reduction in morbidity and mortality from HCV.
Targeted screening of PWIDs to achieve a 20% screening rate (compared with the current 4.1%), combined with a treatment rate of at least 30%, would yield a 90% reduction of prevalence in HCV by 2040. Universal screening, combined with a treatment rate of at least 20%, would achieve a 90% reduction in the incidence of new infections by 2040, according to the model.
The drop in prevalence in the models took into account not only an increased SVR rate, but also the reduced transmission rate when more persons with HCV achieve an SVR. This dynamic transmission analysis “captures the impact of treatment on both chronic disease and transmission dynamics, distinguishes screening and treatment as separate but related public health objectives, and accounts for underreporting … of national HCV prevalence,” according to Dr. Durham and his coauthors.
Morbidity and mortality were accounted for by projecting the number of HCV patients in each treatment and screening condition who would spontaneously clear disease, become chronically infected, develop increasing levels of liver disease and cirrhosis, require transplant, become reinfected, or die.
As always, real world considerations temper what’s achievable. For DAAs, a chief factor is the cost of the regimens, which currently hovers around $90,000. “The improvement of health outcomes expected from greater acceptance might be tempered by the high costs of treatment and by limited insurance coverage and willingness to treat PWIDs,” Dr. Durham and his coauthors wrote.
Study limitations included some “simplifying assumptions” in the modeling, which did not account for the varying incidence of fibrosis and transmissibility among different HCV genotypes. Still, wrote Dr. Durham and his collaborators, “our analysis provides a forecast for the potential impact of DAAs in reducing HCV-associated liver disease, demonstrating that achievable expansion of HCV treatment at current screening rates can substantially reduce morbidity and mortality.”
The study was funded by the Notsew Orm Sands Foundation and Merck. Dr. Durham, Dr. Galvani, and Dr. Townsend have consulted for and received research funding from Sanofi Pasteur and Merck. Dr. Elbasha is employed by Merck and holds stock and stock options in the company. The other authors reported no conflicts of interest.
On Twitter @karioakes
FROM CLINICAL INFECTIOUS DISEASES
Key clinical point: Hepatitis C could be reduced by 90% or more in the United States by the year 2040 with the use of direct-acting antivirals, near-universal screening, and enhanced treatment capacity.
Major finding: Targeted screening of 20% of persons who inject drugs, combined with a 30% treatment rate, would reduce HCV prevalence by 90% by the year 2040.
Data source: Dynamic modeling of HCV prevalence, screening, and treatment, using currently available data to model public health scenarios to the year 2040.
Disclosures: The study was funded by the Notsew Orm Sands Foundation and Merck. Dr. Durham, Dr. Galvani, and Dr. Townsend have consulted for and received research funding from Sanofi Pasteur and Merck. Dr. Elbasha is employed by Merck and holds stock and stock options in the company. The other authors reported no conflicts of interest.
FDA sheds light on flibanserin approval
The Food and Drug Administration is providing a rare look into the agency’s decision-making process surrounding the approval of flibanserin (Addyi) for female hypoactive sexual desire disorder (HSDD) earlier this year.
Officials in the FDA’s Division of Bone, Reproductive, and Urologic Products shared their perspective on the factors behind the drug’s approval in a commentary published online in the New England Journal of Medicine on Dec. 9 (doi: 10.1056/NEJMp1513686). “In the face of divergent views, we at the FDA think it’s important to clarify why flibanserin was approved after being rejected twice,” Dr. Hylton Joffe, the division’s director, and his colleagues wrote.
Reviewing the data underpinning flibanserin’s three bids for approval, Dr. Joffe and his coauthors emphasized concerns that the initial data presented an unfavorable benefit-risk profile, given the drug’s known risks for hypotension and syncope, especially when taken in combination with alcohol or drugs that are moderate or strong cytochrome P-450 3A4 inhibitors.
Flibanserin won the day eventually because there had been convincing evidence presented regarding efficacy (though with modest effect size), there was no previous pharmacologic treatment for female HSDD, and phase III clinical trial data regarding flibanserin’s real-world use were reassuring, according to the FDA.
The agency’s review team required black box warnings regarding flibanserin’s risks, a Risk Evaluation and Mitigation Strategy (REMS), and ongoing post-marketing studies for safety.
Dr. Joffe and his colleagues acknowledged the divisive public discussions about flibanserin and in particular, the accusations of gender bias in the FDA’s scrutiny of the drug. Some of this criticism, Dr. Joffe and his coauthors wrote, was leveled by Even the Score, “an advocacy campaign partly funded by Sprout Pharmaceuticals, flibanserin’s sponsor after Boehringer Ingelheim sold the rights to the drug.”
Read the full Perspectives article in the New England Journal of Medicine.
On Twitter @karioakes
The Food and Drug Administration is providing a rare look into the agency’s decision-making process surrounding the approval of flibanserin (Addyi) for female hypoactive sexual desire disorder (HSDD) earlier this year.
Officials in the FDA’s Division of Bone, Reproductive, and Urologic Products shared their perspective on the factors behind the drug’s approval in a commentary published online in the New England Journal of Medicine on Dec. 9 (doi: 10.1056/NEJMp1513686). “In the face of divergent views, we at the FDA think it’s important to clarify why flibanserin was approved after being rejected twice,” Dr. Hylton Joffe, the division’s director, and his colleagues wrote.
Reviewing the data underpinning flibanserin’s three bids for approval, Dr. Joffe and his coauthors emphasized concerns that the initial data presented an unfavorable benefit-risk profile, given the drug’s known risks for hypotension and syncope, especially when taken in combination with alcohol or drugs that are moderate or strong cytochrome P-450 3A4 inhibitors.
Flibanserin won the day eventually because there had been convincing evidence presented regarding efficacy (though with modest effect size), there was no previous pharmacologic treatment for female HSDD, and phase III clinical trial data regarding flibanserin’s real-world use were reassuring, according to the FDA.
The agency’s review team required black box warnings regarding flibanserin’s risks, a Risk Evaluation and Mitigation Strategy (REMS), and ongoing post-marketing studies for safety.
Dr. Joffe and his colleagues acknowledged the divisive public discussions about flibanserin and in particular, the accusations of gender bias in the FDA’s scrutiny of the drug. Some of this criticism, Dr. Joffe and his coauthors wrote, was leveled by Even the Score, “an advocacy campaign partly funded by Sprout Pharmaceuticals, flibanserin’s sponsor after Boehringer Ingelheim sold the rights to the drug.”
Read the full Perspectives article in the New England Journal of Medicine.
On Twitter @karioakes
The Food and Drug Administration is providing a rare look into the agency’s decision-making process surrounding the approval of flibanserin (Addyi) for female hypoactive sexual desire disorder (HSDD) earlier this year.
Officials in the FDA’s Division of Bone, Reproductive, and Urologic Products shared their perspective on the factors behind the drug’s approval in a commentary published online in the New England Journal of Medicine on Dec. 9 (doi: 10.1056/NEJMp1513686). “In the face of divergent views, we at the FDA think it’s important to clarify why flibanserin was approved after being rejected twice,” Dr. Hylton Joffe, the division’s director, and his colleagues wrote.
Reviewing the data underpinning flibanserin’s three bids for approval, Dr. Joffe and his coauthors emphasized concerns that the initial data presented an unfavorable benefit-risk profile, given the drug’s known risks for hypotension and syncope, especially when taken in combination with alcohol or drugs that are moderate or strong cytochrome P-450 3A4 inhibitors.
Flibanserin won the day eventually because there had been convincing evidence presented regarding efficacy (though with modest effect size), there was no previous pharmacologic treatment for female HSDD, and phase III clinical trial data regarding flibanserin’s real-world use were reassuring, according to the FDA.
The agency’s review team required black box warnings regarding flibanserin’s risks, a Risk Evaluation and Mitigation Strategy (REMS), and ongoing post-marketing studies for safety.
Dr. Joffe and his colleagues acknowledged the divisive public discussions about flibanserin and in particular, the accusations of gender bias in the FDA’s scrutiny of the drug. Some of this criticism, Dr. Joffe and his coauthors wrote, was leveled by Even the Score, “an advocacy campaign partly funded by Sprout Pharmaceuticals, flibanserin’s sponsor after Boehringer Ingelheim sold the rights to the drug.”
Read the full Perspectives article in the New England Journal of Medicine.
On Twitter @karioakes
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
ASH: Carfilzomib doubles progression free survival in head-to-head trial with bortezomib
In a head-to-head clinical trial, carfilzomib (Kyprolis) plus dexamethasone doubled the length of progression-free survival (PFS) when compared with bortezomib (Velcade) plus dexamethasone for individuals with relapsed or refractory multiple myeloma. Response rates were also significantly higher for those receiving carfilzomib, though neither medication could overcome high-risk cytogenetic factors.
The international multi-center phase 3 open-label ENDEAVOR study (clinicaltrials.gov ID: NCT01568866) compared outcomes for a total of 929 patients who were randomized 1:1 to receive one of the treatments. Randomization was stratified to account for prior treatments, including prior proteasome treatment, as well as disease severity by International Staging System stage.
Reporting for the ENDEAVOR investigators, Dr. Meletios Dimopoulos and his co-authors published their results in The Lancet simultaneously with a presentation at the annual meeting of the American Society of Hematology [Lancet Oncol, http://dx.doi.org/10.1016/S1470-2045(15)00464-7] . Dr. Dimopoulos is a professor in the department of clinical therapeutics at the school of medicine of the National and Kapodistrian University of Athens.
The primary endpoint, PFS in the intention-to-treat population, differed significantly between the study arms when the data were analyzed at a pre-planned interim analysis. For those receiving carfilzomib, PFS was 18.7 months, compared with 9.4 months in the bortezomib group, for a hazard ratio of 0.53 (P less than 0.0001). Data were drawn from a pre-specified interim analysis, and study participation is ongoing, with 28-day dosing cycles repeated until disease progression occurrs or the patients experience unacceptable toxicity or withdraw consent.
Investigators had pre-specified subgroup analyses to examine what populations fared better and worse in each arm. Prior bortezomib exposure did not significantly affect PFS for those receiving carfilzomib. Not enough patients had received carfilzomib prior to the study to permit analysis of this effect. Though carfilzomib arm patients with high-risk factors on cytogenetic analysis fared slightly better than those on bortezomib, “neither proteasome inhibitor appears to significantly overcome the adverse prognostic effect of high-risk cytogenetics,” wrote Dr. Dimopoulos and his co-authors.
Secondary outcome measures included the numbers of patients in each arm achieving complete response or better (with this group broken into “stringent complete response” as well as complete response). Significantly more carfilzomib than bortezomib patients reached this mark (58 patients [13%] vs 29 patients [6%], P equal to 0.001). A larger group of patients in each arm met the secondary outcome measure of experiencing very good partial response or better, with those on the carfelzomib arm faring significantly better than those receiving bortezomib (252 [54%] vs 133 [29%], P less than 0.0001).
Dr. Dimopoulos and his co-authors remarked on the importance of these findings, noting that “The finding that the proportion of patients with a complete response or better and very good partial response or better was higher in the carfilzomib group than in the bortezomib group is encouraging because studies have shown an association between depth of response and improved survival in patients with multiple myeloma.”
ENDEAVOR’s safety analyses included all patients who received at least one dose of study drug. Overall, the carfilzomib patients had a 48% rate of serious adverse events (224 of 463 patients for this analysis), compared to 36% of the bortezomib group (162 of 456 patients).
On-study death rates were similar between the two groups, with 18 of 464 patients (4%) in the carfilzomib group and 16 of 465 patients (3%) in the bortezomib group dying during the study period. Four deaths in each arm were judged related to disease progression.
A pre-planned substudy examined cardiac function for patients in both study arms, and did not find increased risk of left or right ventricular dysfunction or cumulative cardiac injury for carfilzomib when compared with bortezomib.
The open label nature of the study, an acknowledged limitation, was necessitated by the different dosing regimens of the two drugs, but the treatment allocation was hidden from the independent review committee that assessed participant disease status. The funder was also masked to per-group treatment results.
Carfilzomib’s irreversable proteosome inhibition may lead to more sustained proteasomal inhibition, Dr. Dimopoulous and his co-authors posited. Also, carfilzomib can overcome bortezomib resistance and is more potent against cell lines naive to this class of drugs in preclinical studies, they said.
“Taken together, the results from the ENDEAVOR study suggest an important role for carfilzomib-based regimens for patients with relapsed or refractory jultiple myeloma,” wrote Dr. Dimopoulos and his collaborators.
The study was supported by Onyx Pharmaceuticals, Inc, a subsidiary of Amgen. Dr. Dimopoulos reported non-financial support from Onyx Pharmaceuticals, Celgene Corporation, and Ortho-Biotech. Several co-authors reported multiple financial associations with pharmaceutical companies.
On Twitter @karioakes
In a head-to-head clinical trial, carfilzomib (Kyprolis) plus dexamethasone doubled the length of progression-free survival (PFS) when compared with bortezomib (Velcade) plus dexamethasone for individuals with relapsed or refractory multiple myeloma. Response rates were also significantly higher for those receiving carfilzomib, though neither medication could overcome high-risk cytogenetic factors.
The international multi-center phase 3 open-label ENDEAVOR study (clinicaltrials.gov ID: NCT01568866) compared outcomes for a total of 929 patients who were randomized 1:1 to receive one of the treatments. Randomization was stratified to account for prior treatments, including prior proteasome treatment, as well as disease severity by International Staging System stage.
Reporting for the ENDEAVOR investigators, Dr. Meletios Dimopoulos and his co-authors published their results in The Lancet simultaneously with a presentation at the annual meeting of the American Society of Hematology [Lancet Oncol, http://dx.doi.org/10.1016/S1470-2045(15)00464-7] . Dr. Dimopoulos is a professor in the department of clinical therapeutics at the school of medicine of the National and Kapodistrian University of Athens.
The primary endpoint, PFS in the intention-to-treat population, differed significantly between the study arms when the data were analyzed at a pre-planned interim analysis. For those receiving carfilzomib, PFS was 18.7 months, compared with 9.4 months in the bortezomib group, for a hazard ratio of 0.53 (P less than 0.0001). Data were drawn from a pre-specified interim analysis, and study participation is ongoing, with 28-day dosing cycles repeated until disease progression occurrs or the patients experience unacceptable toxicity or withdraw consent.
Investigators had pre-specified subgroup analyses to examine what populations fared better and worse in each arm. Prior bortezomib exposure did not significantly affect PFS for those receiving carfilzomib. Not enough patients had received carfilzomib prior to the study to permit analysis of this effect. Though carfilzomib arm patients with high-risk factors on cytogenetic analysis fared slightly better than those on bortezomib, “neither proteasome inhibitor appears to significantly overcome the adverse prognostic effect of high-risk cytogenetics,” wrote Dr. Dimopoulos and his co-authors.
Secondary outcome measures included the numbers of patients in each arm achieving complete response or better (with this group broken into “stringent complete response” as well as complete response). Significantly more carfilzomib than bortezomib patients reached this mark (58 patients [13%] vs 29 patients [6%], P equal to 0.001). A larger group of patients in each arm met the secondary outcome measure of experiencing very good partial response or better, with those on the carfelzomib arm faring significantly better than those receiving bortezomib (252 [54%] vs 133 [29%], P less than 0.0001).
Dr. Dimopoulos and his co-authors remarked on the importance of these findings, noting that “The finding that the proportion of patients with a complete response or better and very good partial response or better was higher in the carfilzomib group than in the bortezomib group is encouraging because studies have shown an association between depth of response and improved survival in patients with multiple myeloma.”
ENDEAVOR’s safety analyses included all patients who received at least one dose of study drug. Overall, the carfilzomib patients had a 48% rate of serious adverse events (224 of 463 patients for this analysis), compared to 36% of the bortezomib group (162 of 456 patients).
On-study death rates were similar between the two groups, with 18 of 464 patients (4%) in the carfilzomib group and 16 of 465 patients (3%) in the bortezomib group dying during the study period. Four deaths in each arm were judged related to disease progression.
A pre-planned substudy examined cardiac function for patients in both study arms, and did not find increased risk of left or right ventricular dysfunction or cumulative cardiac injury for carfilzomib when compared with bortezomib.
The open label nature of the study, an acknowledged limitation, was necessitated by the different dosing regimens of the two drugs, but the treatment allocation was hidden from the independent review committee that assessed participant disease status. The funder was also masked to per-group treatment results.
Carfilzomib’s irreversable proteosome inhibition may lead to more sustained proteasomal inhibition, Dr. Dimopoulous and his co-authors posited. Also, carfilzomib can overcome bortezomib resistance and is more potent against cell lines naive to this class of drugs in preclinical studies, they said.
“Taken together, the results from the ENDEAVOR study suggest an important role for carfilzomib-based regimens for patients with relapsed or refractory jultiple myeloma,” wrote Dr. Dimopoulos and his collaborators.
The study was supported by Onyx Pharmaceuticals, Inc, a subsidiary of Amgen. Dr. Dimopoulos reported non-financial support from Onyx Pharmaceuticals, Celgene Corporation, and Ortho-Biotech. Several co-authors reported multiple financial associations with pharmaceutical companies.
On Twitter @karioakes
In a head-to-head clinical trial, carfilzomib (Kyprolis) plus dexamethasone doubled the length of progression-free survival (PFS) when compared with bortezomib (Velcade) plus dexamethasone for individuals with relapsed or refractory multiple myeloma. Response rates were also significantly higher for those receiving carfilzomib, though neither medication could overcome high-risk cytogenetic factors.
The international multi-center phase 3 open-label ENDEAVOR study (clinicaltrials.gov ID: NCT01568866) compared outcomes for a total of 929 patients who were randomized 1:1 to receive one of the treatments. Randomization was stratified to account for prior treatments, including prior proteasome treatment, as well as disease severity by International Staging System stage.
Reporting for the ENDEAVOR investigators, Dr. Meletios Dimopoulos and his co-authors published their results in The Lancet simultaneously with a presentation at the annual meeting of the American Society of Hematology [Lancet Oncol, http://dx.doi.org/10.1016/S1470-2045(15)00464-7] . Dr. Dimopoulos is a professor in the department of clinical therapeutics at the school of medicine of the National and Kapodistrian University of Athens.
The primary endpoint, PFS in the intention-to-treat population, differed significantly between the study arms when the data were analyzed at a pre-planned interim analysis. For those receiving carfilzomib, PFS was 18.7 months, compared with 9.4 months in the bortezomib group, for a hazard ratio of 0.53 (P less than 0.0001). Data were drawn from a pre-specified interim analysis, and study participation is ongoing, with 28-day dosing cycles repeated until disease progression occurrs or the patients experience unacceptable toxicity or withdraw consent.
Investigators had pre-specified subgroup analyses to examine what populations fared better and worse in each arm. Prior bortezomib exposure did not significantly affect PFS for those receiving carfilzomib. Not enough patients had received carfilzomib prior to the study to permit analysis of this effect. Though carfilzomib arm patients with high-risk factors on cytogenetic analysis fared slightly better than those on bortezomib, “neither proteasome inhibitor appears to significantly overcome the adverse prognostic effect of high-risk cytogenetics,” wrote Dr. Dimopoulos and his co-authors.
Secondary outcome measures included the numbers of patients in each arm achieving complete response or better (with this group broken into “stringent complete response” as well as complete response). Significantly more carfilzomib than bortezomib patients reached this mark (58 patients [13%] vs 29 patients [6%], P equal to 0.001). A larger group of patients in each arm met the secondary outcome measure of experiencing very good partial response or better, with those on the carfelzomib arm faring significantly better than those receiving bortezomib (252 [54%] vs 133 [29%], P less than 0.0001).
Dr. Dimopoulos and his co-authors remarked on the importance of these findings, noting that “The finding that the proportion of patients with a complete response or better and very good partial response or better was higher in the carfilzomib group than in the bortezomib group is encouraging because studies have shown an association between depth of response and improved survival in patients with multiple myeloma.”
ENDEAVOR’s safety analyses included all patients who received at least one dose of study drug. Overall, the carfilzomib patients had a 48% rate of serious adverse events (224 of 463 patients for this analysis), compared to 36% of the bortezomib group (162 of 456 patients).
On-study death rates were similar between the two groups, with 18 of 464 patients (4%) in the carfilzomib group and 16 of 465 patients (3%) in the bortezomib group dying during the study period. Four deaths in each arm were judged related to disease progression.
A pre-planned substudy examined cardiac function for patients in both study arms, and did not find increased risk of left or right ventricular dysfunction or cumulative cardiac injury for carfilzomib when compared with bortezomib.
The open label nature of the study, an acknowledged limitation, was necessitated by the different dosing regimens of the two drugs, but the treatment allocation was hidden from the independent review committee that assessed participant disease status. The funder was also masked to per-group treatment results.
Carfilzomib’s irreversable proteosome inhibition may lead to more sustained proteasomal inhibition, Dr. Dimopoulous and his co-authors posited. Also, carfilzomib can overcome bortezomib resistance and is more potent against cell lines naive to this class of drugs in preclinical studies, they said.
“Taken together, the results from the ENDEAVOR study suggest an important role for carfilzomib-based regimens for patients with relapsed or refractory jultiple myeloma,” wrote Dr. Dimopoulos and his collaborators.
The study was supported by Onyx Pharmaceuticals, Inc, a subsidiary of Amgen. Dr. Dimopoulos reported non-financial support from Onyx Pharmaceuticals, Celgene Corporation, and Ortho-Biotech. Several co-authors reported multiple financial associations with pharmaceutical companies.
On Twitter @karioakes
FROM ASH 2015
Key clinical point: Carfilzomib nearly doubled progression-free survival (PFS) compared with bortezomib for relapsed and refractory multiple myeloma
Major finding: For those receiving carfilzomib, PFS was 18.7 months, compared with 9.4 months in the bortezomib group, for a hazard ratio of 0.53 (P less than .0001).
Data source: International randomized open-label clinical trial of 979 patients with refractory or relapsed multiple myeloma.
Disclosures: The study was supported by Onyx Pharmaceuticals, Inc, a subsidiary of Amgen. Dr. Dimopoulos reported non-financial support from Onyx Pharmaceuticals, Celgene Corporation, and Ortho-Biotech. Several co-authors reported multiple financial associations with pharmaceutical companies.
Elotuzumab approved for multiple myeloma
Elotuzumab (Empliciti) has been approved as combination therapy for individuals with relapsed or refractory multiple myeloma, the Food and Drug Administration announced Nov. 30.
Elotuzumab’s approval was based on a randomized, open-label clinical trial of 646 individuals with relapsed or refractory multiple myeloma. The multicenter trial compared progression-free survival (PFS) and overall response rate (ORR) for 321 individuals receiving lenalidomide (Revlimid) and dexamethasone to the PFS and ORR for 325 individuals receiving elotuzumab, lenalidomide, and dexamethasone.
For individuals receiving elotuzumab together with lenalidomide and dexamethasone, the median PFS of 19.4 months was significantly longer than the median 14.9 months of PFS seen in those receiving just lenalidomide and dexamethasone. For the elotuzumab-containing arm, the ORR was also significantly greater at 78.5%, compared with an ORR of 65.5% for the arm not receiving elotuzumab.
Slightly more patients in the elotuzumab-containing arm experienced serious adverse events, at 65.4% vs. 56.5% for those not receiving elotuzumab. Of the adverse reactions seen in more than 20% of patients, those that occurred more frequently in patients receiving elotuzumab included fatigue, diarrhea, fever, constipation, cough, peripheral neuropathy, upper respiratory tract infection, decreased appetite, and pneumonia.
Elotuzumab is a monoclonal antibody given intravenously; it can interfere with determination of complete response, and infusion reactions, infections, second primary malignancies, and hepatotoxicity may also occur.
Elotuzumab’s target is the surface antigen signaling lymphocyte activation molecule family 7 (SLAMF7). This protein is present on malignant cells in multiple myeloma and is also present on human immune cells such as natural killer cells and monocytes. The recommended dosing is 10 mg/kg intravenously weekly for 2 weeks, then every 2 weeks until disease progression or unacceptable toxicity occurs. Acetaminophen and histamine1 (H1) and H2 blockade should be given preadministration.
“The approval of elotuzumab marks another important milestone in the treatment of multiple myeloma in a short period of time,” Walter M. Capone, president and CEO of the Multiple Myeloma Research Foundation, said in a statement.
Today’s approval is the third in November for multiple myeloma therapies, following the Nov. 16 approval of the monoclonal antibody daratumumab (Darzalex) and the Nov. 20 approval of the oral proteasome inhibitor ixazomib (Ninlaro).
Elotuzumab received priority review by the FDA as a breakthrough drug and also has orphan drug designation. Elotuzumab will be marketed as Empliciti by Bristol-Myers Squibb.
On Twitter @karioakes
Elotuzumab (Empliciti) has been approved as combination therapy for individuals with relapsed or refractory multiple myeloma, the Food and Drug Administration announced Nov. 30.
Elotuzumab’s approval was based on a randomized, open-label clinical trial of 646 individuals with relapsed or refractory multiple myeloma. The multicenter trial compared progression-free survival (PFS) and overall response rate (ORR) for 321 individuals receiving lenalidomide (Revlimid) and dexamethasone to the PFS and ORR for 325 individuals receiving elotuzumab, lenalidomide, and dexamethasone.
For individuals receiving elotuzumab together with lenalidomide and dexamethasone, the median PFS of 19.4 months was significantly longer than the median 14.9 months of PFS seen in those receiving just lenalidomide and dexamethasone. For the elotuzumab-containing arm, the ORR was also significantly greater at 78.5%, compared with an ORR of 65.5% for the arm not receiving elotuzumab.
Slightly more patients in the elotuzumab-containing arm experienced serious adverse events, at 65.4% vs. 56.5% for those not receiving elotuzumab. Of the adverse reactions seen in more than 20% of patients, those that occurred more frequently in patients receiving elotuzumab included fatigue, diarrhea, fever, constipation, cough, peripheral neuropathy, upper respiratory tract infection, decreased appetite, and pneumonia.
Elotuzumab is a monoclonal antibody given intravenously; it can interfere with determination of complete response, and infusion reactions, infections, second primary malignancies, and hepatotoxicity may also occur.
Elotuzumab’s target is the surface antigen signaling lymphocyte activation molecule family 7 (SLAMF7). This protein is present on malignant cells in multiple myeloma and is also present on human immune cells such as natural killer cells and monocytes. The recommended dosing is 10 mg/kg intravenously weekly for 2 weeks, then every 2 weeks until disease progression or unacceptable toxicity occurs. Acetaminophen and histamine1 (H1) and H2 blockade should be given preadministration.
“The approval of elotuzumab marks another important milestone in the treatment of multiple myeloma in a short period of time,” Walter M. Capone, president and CEO of the Multiple Myeloma Research Foundation, said in a statement.
Today’s approval is the third in November for multiple myeloma therapies, following the Nov. 16 approval of the monoclonal antibody daratumumab (Darzalex) and the Nov. 20 approval of the oral proteasome inhibitor ixazomib (Ninlaro).
Elotuzumab received priority review by the FDA as a breakthrough drug and also has orphan drug designation. Elotuzumab will be marketed as Empliciti by Bristol-Myers Squibb.
On Twitter @karioakes
Elotuzumab (Empliciti) has been approved as combination therapy for individuals with relapsed or refractory multiple myeloma, the Food and Drug Administration announced Nov. 30.
Elotuzumab’s approval was based on a randomized, open-label clinical trial of 646 individuals with relapsed or refractory multiple myeloma. The multicenter trial compared progression-free survival (PFS) and overall response rate (ORR) for 321 individuals receiving lenalidomide (Revlimid) and dexamethasone to the PFS and ORR for 325 individuals receiving elotuzumab, lenalidomide, and dexamethasone.
For individuals receiving elotuzumab together with lenalidomide and dexamethasone, the median PFS of 19.4 months was significantly longer than the median 14.9 months of PFS seen in those receiving just lenalidomide and dexamethasone. For the elotuzumab-containing arm, the ORR was also significantly greater at 78.5%, compared with an ORR of 65.5% for the arm not receiving elotuzumab.
Slightly more patients in the elotuzumab-containing arm experienced serious adverse events, at 65.4% vs. 56.5% for those not receiving elotuzumab. Of the adverse reactions seen in more than 20% of patients, those that occurred more frequently in patients receiving elotuzumab included fatigue, diarrhea, fever, constipation, cough, peripheral neuropathy, upper respiratory tract infection, decreased appetite, and pneumonia.
Elotuzumab is a monoclonal antibody given intravenously; it can interfere with determination of complete response, and infusion reactions, infections, second primary malignancies, and hepatotoxicity may also occur.
Elotuzumab’s target is the surface antigen signaling lymphocyte activation molecule family 7 (SLAMF7). This protein is present on malignant cells in multiple myeloma and is also present on human immune cells such as natural killer cells and monocytes. The recommended dosing is 10 mg/kg intravenously weekly for 2 weeks, then every 2 weeks until disease progression or unacceptable toxicity occurs. Acetaminophen and histamine1 (H1) and H2 blockade should be given preadministration.
“The approval of elotuzumab marks another important milestone in the treatment of multiple myeloma in a short period of time,” Walter M. Capone, president and CEO of the Multiple Myeloma Research Foundation, said in a statement.
Today’s approval is the third in November for multiple myeloma therapies, following the Nov. 16 approval of the monoclonal antibody daratumumab (Darzalex) and the Nov. 20 approval of the oral proteasome inhibitor ixazomib (Ninlaro).
Elotuzumab received priority review by the FDA as a breakthrough drug and also has orphan drug designation. Elotuzumab will be marketed as Empliciti by Bristol-Myers Squibb.
On Twitter @karioakes
Novel treatments for AKN include laser hair removal, UVB
LAS VEGAS – Bothersome and even disfiguring for some, acne keloidalis nuchae (AKN) is a mysterious but common skin disorder mostly affecting men of African descent.
But an accurate clinical diagnosis and a flexible treatment approach can boost treatment success, said Dr. Andrew F. Alexis, chair of the department of dermatology, and director of the Skin of Color Center, Mount Sinai St Luke’s and Mount Sinai Roosevelt, New York.

Speaking at the Skin Disease Education Foundation’s annual Las Vegas dermatology seminar, Dr. Alexis that the diagnosis can be made clinically, without the need for biopsies or skin scrapings. The condition is characterized by fibrotic, follicular papules, usually at the nape of the neck and on the occipital scalp. However, in more severe cases, the papules coalesce into a larger plaque. Occasionally, the characteristic scarring alopecia and keloidlike papules can be seen more toward the vertex of the scalp.
“We don’t really, truly know what the etiology of acne keloidalis nuchae is,” said Dr. Alexis, noting that many etiologies have been proposed. Some say it’s an epithelial elimination disorder, or a variant of lichen simplex chronicus. Many lay people may feel this is an ingrown hair disorder, but histopathologic studies have refuted this theory. These studies have also found no evidence of bacterial overgrowth in affected areas.
A newer and leading hypothesis is that AKN’s primary mechanism is fibrosis secondary to a mechanically induced folliculitis. At the very least, mechanical forces, even if they don’t cause the disorder, “certainly can exacerbate acne keloidalis nuchae, and short haircuts using electrical clippers may be a factor,” Dr. Alexis said. Support for this hypothesis can be found in two South African cross-sectional studies of nearly 2,000 men and boys: 86% of those with AKN reported receiving short haircuts with clippers. Among the adults reporting at least one episode of bleeding during a haircut, the odds ratio for having AKN was 3.45 (Int J Dermatol. 2011 Oct;50[10]:1212-6).
Therapy is multimodal, with some behavioral components, including avoidance of mechanical factors that can exacerbate AKN. In addition to low haircuts, tight-fitting caps and picking or scratching at the papules can worsen the condition.
Few studies exist to support one treatment regime over another for AKN, Dr. Alexis said. One small open label study of 20 individuals evaluated clobetasol 0.05% foam twice daily for two cycles of 2 weeks on and 2 weeks off treatment. Patients with persistent disease were then stepped down to 2 weeks of betamethasone valerate 0.12% foam twice daily. Overall, the mean papule/pustule count dropped by more than half, from 25 at baseline to 10 at 12 weeks, Dr. Alexis said (Cutis. 2005;75:317-21).
For larger papules and coalescent plaques, Dr. Alexis recommends intralesional triamcinolone acetonide at a concentration of 20-40mg/mL. “Don’t undertreat” and be sure to inject into, rather than under the lesion, he said. Concomitant topical class 1 or class 2 corticosteroids, such as clobetasol or fluocinonide, can be used along with injections. Addition of a topical antibiotic such as clindamycin can also accelerate resolution in his anecdotal experience.
Further recommendations based on practice experience include once-daily chlorhexidine washes of the affected area, as well as the addition of an oral tetracycline, such as doxycycline, when pustules are present or with more severe cases, Dr. Alexis said.
For some severely affected patients, surgical therapy may become the best treatment option. “You can inject all day long and still have disfiguring keloidlike plaque” in some patients with severe disease, he noted. Case series of both primary closure and healing by secondary intention have both shown good results. “Whichever camp you belong to, the key is to use a horizontal ellipse that includes the posterior hairline, and the depth of the excision must be down to the dermal-subcutaneous junction,” he said.
More recently, laser hair removal has been studied as a treatment alternative for AKN, Dr. Alexis pointed out. In a pilot study of 16 patients with AKN who received five sessions of laser hair removal, all had a marked reduction in papules and pustules at one year of follow-up (Eur J Dermatol. 2012 Sep-Oct;22[5]:645-50).
Another novel treatment approach used targeted UVB light with good results in a small split-scalp study. Pathology suggested the targeted skin was undergoing active matrix remodeling as a result of the UVB therapy (Br J Dermatol. 2014 Nov;171[5]:1156-63).
Dr. Alexis disclosed that he has received grants and research support from Allergan and Novartis, and speaker honoraria from Cipla. He has received consulting fees from Acclaris Therapeutics, Allergan, Amgen, Anacor, Bayer, Galderma, Johnson & Johnson, Leo, L’Oreal, Roche, Schick, Suneva, and Valeant.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
LAS VEGAS – Bothersome and even disfiguring for some, acne keloidalis nuchae (AKN) is a mysterious but common skin disorder mostly affecting men of African descent.
But an accurate clinical diagnosis and a flexible treatment approach can boost treatment success, said Dr. Andrew F. Alexis, chair of the department of dermatology, and director of the Skin of Color Center, Mount Sinai St Luke’s and Mount Sinai Roosevelt, New York.
Speaking at the Skin Disease Education Foundation’s annual Las Vegas dermatology seminar, Dr. Alexis that the diagnosis can be made clinically, without the need for biopsies or skin scrapings. The condition is characterized by fibrotic, follicular papules, usually at the nape of the neck and on the occipital scalp. However, in more severe cases, the papules coalesce into a larger plaque. Occasionally, the characteristic scarring alopecia and keloidlike papules can be seen more toward the vertex of the scalp.
“We don’t really, truly know what the etiology of acne keloidalis nuchae is,” said Dr. Alexis, noting that many etiologies have been proposed. Some say it’s an epithelial elimination disorder, or a variant of lichen simplex chronicus. Many lay people may feel this is an ingrown hair disorder, but histopathologic studies have refuted this theory. These studies have also found no evidence of bacterial overgrowth in affected areas.
A newer and leading hypothesis is that AKN’s primary mechanism is fibrosis secondary to a mechanically induced folliculitis. At the very least, mechanical forces, even if they don’t cause the disorder, “certainly can exacerbate acne keloidalis nuchae, and short haircuts using electrical clippers may be a factor,” Dr. Alexis said. Support for this hypothesis can be found in two South African cross-sectional studies of nearly 2,000 men and boys: 86% of those with AKN reported receiving short haircuts with clippers. Among the adults reporting at least one episode of bleeding during a haircut, the odds ratio for having AKN was 3.45 (Int J Dermatol. 2011 Oct;50[10]:1212-6).
Therapy is multimodal, with some behavioral components, including avoidance of mechanical factors that can exacerbate AKN. In addition to low haircuts, tight-fitting caps and picking or scratching at the papules can worsen the condition.
Few studies exist to support one treatment regime over another for AKN, Dr. Alexis said. One small open label study of 20 individuals evaluated clobetasol 0.05% foam twice daily for two cycles of 2 weeks on and 2 weeks off treatment. Patients with persistent disease were then stepped down to 2 weeks of betamethasone valerate 0.12% foam twice daily. Overall, the mean papule/pustule count dropped by more than half, from 25 at baseline to 10 at 12 weeks, Dr. Alexis said (Cutis. 2005;75:317-21).
For larger papules and coalescent plaques, Dr. Alexis recommends intralesional triamcinolone acetonide at a concentration of 20-40mg/mL. “Don’t undertreat” and be sure to inject into, rather than under the lesion, he said. Concomitant topical class 1 or class 2 corticosteroids, such as clobetasol or fluocinonide, can be used along with injections. Addition of a topical antibiotic such as clindamycin can also accelerate resolution in his anecdotal experience.
Further recommendations based on practice experience include once-daily chlorhexidine washes of the affected area, as well as the addition of an oral tetracycline, such as doxycycline, when pustules are present or with more severe cases, Dr. Alexis said.
For some severely affected patients, surgical therapy may become the best treatment option. “You can inject all day long and still have disfiguring keloidlike plaque” in some patients with severe disease, he noted. Case series of both primary closure and healing by secondary intention have both shown good results. “Whichever camp you belong to, the key is to use a horizontal ellipse that includes the posterior hairline, and the depth of the excision must be down to the dermal-subcutaneous junction,” he said.
More recently, laser hair removal has been studied as a treatment alternative for AKN, Dr. Alexis pointed out. In a pilot study of 16 patients with AKN who received five sessions of laser hair removal, all had a marked reduction in papules and pustules at one year of follow-up (Eur J Dermatol. 2012 Sep-Oct;22[5]:645-50).
Another novel treatment approach used targeted UVB light with good results in a small split-scalp study. Pathology suggested the targeted skin was undergoing active matrix remodeling as a result of the UVB therapy (Br J Dermatol. 2014 Nov;171[5]:1156-63).
Dr. Alexis disclosed that he has received grants and research support from Allergan and Novartis, and speaker honoraria from Cipla. He has received consulting fees from Acclaris Therapeutics, Allergan, Amgen, Anacor, Bayer, Galderma, Johnson & Johnson, Leo, L’Oreal, Roche, Schick, Suneva, and Valeant.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
LAS VEGAS – Bothersome and even disfiguring for some, acne keloidalis nuchae (AKN) is a mysterious but common skin disorder mostly affecting men of African descent.
But an accurate clinical diagnosis and a flexible treatment approach can boost treatment success, said Dr. Andrew F. Alexis, chair of the department of dermatology, and director of the Skin of Color Center, Mount Sinai St Luke’s and Mount Sinai Roosevelt, New York.
Speaking at the Skin Disease Education Foundation’s annual Las Vegas dermatology seminar, Dr. Alexis that the diagnosis can be made clinically, without the need for biopsies or skin scrapings. The condition is characterized by fibrotic, follicular papules, usually at the nape of the neck and on the occipital scalp. However, in more severe cases, the papules coalesce into a larger plaque. Occasionally, the characteristic scarring alopecia and keloidlike papules can be seen more toward the vertex of the scalp.
“We don’t really, truly know what the etiology of acne keloidalis nuchae is,” said Dr. Alexis, noting that many etiologies have been proposed. Some say it’s an epithelial elimination disorder, or a variant of lichen simplex chronicus. Many lay people may feel this is an ingrown hair disorder, but histopathologic studies have refuted this theory. These studies have also found no evidence of bacterial overgrowth in affected areas.
A newer and leading hypothesis is that AKN’s primary mechanism is fibrosis secondary to a mechanically induced folliculitis. At the very least, mechanical forces, even if they don’t cause the disorder, “certainly can exacerbate acne keloidalis nuchae, and short haircuts using electrical clippers may be a factor,” Dr. Alexis said. Support for this hypothesis can be found in two South African cross-sectional studies of nearly 2,000 men and boys: 86% of those with AKN reported receiving short haircuts with clippers. Among the adults reporting at least one episode of bleeding during a haircut, the odds ratio for having AKN was 3.45 (Int J Dermatol. 2011 Oct;50[10]:1212-6).
Therapy is multimodal, with some behavioral components, including avoidance of mechanical factors that can exacerbate AKN. In addition to low haircuts, tight-fitting caps and picking or scratching at the papules can worsen the condition.
Few studies exist to support one treatment regime over another for AKN, Dr. Alexis said. One small open label study of 20 individuals evaluated clobetasol 0.05% foam twice daily for two cycles of 2 weeks on and 2 weeks off treatment. Patients with persistent disease were then stepped down to 2 weeks of betamethasone valerate 0.12% foam twice daily. Overall, the mean papule/pustule count dropped by more than half, from 25 at baseline to 10 at 12 weeks, Dr. Alexis said (Cutis. 2005;75:317-21).
For larger papules and coalescent plaques, Dr. Alexis recommends intralesional triamcinolone acetonide at a concentration of 20-40mg/mL. “Don’t undertreat” and be sure to inject into, rather than under the lesion, he said. Concomitant topical class 1 or class 2 corticosteroids, such as clobetasol or fluocinonide, can be used along with injections. Addition of a topical antibiotic such as clindamycin can also accelerate resolution in his anecdotal experience.
Further recommendations based on practice experience include once-daily chlorhexidine washes of the affected area, as well as the addition of an oral tetracycline, such as doxycycline, when pustules are present or with more severe cases, Dr. Alexis said.
For some severely affected patients, surgical therapy may become the best treatment option. “You can inject all day long and still have disfiguring keloidlike plaque” in some patients with severe disease, he noted. Case series of both primary closure and healing by secondary intention have both shown good results. “Whichever camp you belong to, the key is to use a horizontal ellipse that includes the posterior hairline, and the depth of the excision must be down to the dermal-subcutaneous junction,” he said.
More recently, laser hair removal has been studied as a treatment alternative for AKN, Dr. Alexis pointed out. In a pilot study of 16 patients with AKN who received five sessions of laser hair removal, all had a marked reduction in papules and pustules at one year of follow-up (Eur J Dermatol. 2012 Sep-Oct;22[5]:645-50).
Another novel treatment approach used targeted UVB light with good results in a small split-scalp study. Pathology suggested the targeted skin was undergoing active matrix remodeling as a result of the UVB therapy (Br J Dermatol. 2014 Nov;171[5]:1156-63).
Dr. Alexis disclosed that he has received grants and research support from Allergan and Novartis, and speaker honoraria from Cipla. He has received consulting fees from Acclaris Therapeutics, Allergan, Amgen, Anacor, Bayer, Galderma, Johnson & Johnson, Leo, L’Oreal, Roche, Schick, Suneva, and Valeant.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
EXPERT ANALYSIS FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
