User login
Remission, low lupus activity predict lower risk of organ damage
Fulfilling definitions for prolonged lupus remission and reaching a low disease activity state are both associated with a lower risk of damage accrual in lupus patients, new research confirms.
“Identifying predictors of organ damage holds great importance for improving outcomes in SLE [systemic lupus erythematosus]” in light of the strength of organ damage in predicting mortality in people with SLE, wrote the investigators of the new study, led by Michael W. P. Tsang-A-Sjoe of the Amsterdam Rheumatology and Immunology Center, VU University Medical Center, the Netherlands.
The investigators prospectively assessed 183 patients from the Amsterdam SLE cohort for flares and once a year for retrospectively determined remission as defined in a 2015 study (Ann Rheum Dis. 2015;74:2117-22) and in Lupus Low Disease Activity State (LLDAS) criteria set by the Asia-Pacific Lupus Collaboration (Ann Rheum Dis. 2016;75:1615-21). The research team developed a prediction model for damage accrual during the 5-year follow-up through the use of backward logistic regression analyses that took these definitions into account (Rheumatology [Oxford]. 2016 Nov 1. doi: 10.1093/rheumatology/kew377).
The occurrence of at least one flare and the average daily prednisone dose during follow-up were significant predictors of damage accrual (Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index increase of 5 or more during follow-up) in the cohort of patients with or without prolonged remission and with or without LLDAS in half or more of the observations. For example, patients had a higher risk of damage accrual with both a mean daily oral prednisone dose greater than 7.5 mg (odds ratio, 3.6; 95% confidence interval, 1.8-7.2) and greater than 5 mg (OR, 2.8; 95% CI, 1.5-5.3), compared with those taking lower doses.
The researchers also identified a history of nephrological manifestations at baseline as a significant predictor of damage accrual, despite not being part of the criteria for remission or LLDAS.
Overall, 38 (32.5%) of 117 patients met the prolonged remission criteria, whereas LLDAS in half or more of the available observations occurred in 118 (64.5%) of 183 patients.
Prolonged remission during 5 years of follow-up and LLDAS in 50% or more of observations were associated with a reduced risk of damage accrual (OR, 0.20; 95% CI, 0.07-0.53; P = .001; and OR, 0.52; 95% CI, 0.28-0.99, P = .046, respectively).
The effects of both remission and LLDAS, according to the definitions used in the study, don’t settle the matter of which might be used in a treat-to-target strategy. Neither the remission criteria nor the LLDAS definition showed clear superiority over the other, although patients in prolonged remission seemed to accrue less damage, compared with patients classified as being in LLDAS. However, the study did not prove this point, and the investigators noted that “future studies with sufficient power are needed in order to directly compare both sets of criteria for superiority.”
The results might also point to the possibility of lengthening the interval between disease activity assessments. The current study assessed LLDAS and remission at 1 year, compared with other studies that have generally assessed patients at quarterly intervals.
“This finding is interesting because it suggests that in clinical practice, yearly – rather than quarterly – assessments of LLDAS and remission might be sufficient to identify patients at risk of damage accrual, although future studies are needed to confirm this finding,” they wrote.
The study had no specific funding source, and the authors declared having no conflicts of interest.
Fulfilling definitions for prolonged lupus remission and reaching a low disease activity state are both associated with a lower risk of damage accrual in lupus patients, new research confirms.
“Identifying predictors of organ damage holds great importance for improving outcomes in SLE [systemic lupus erythematosus]” in light of the strength of organ damage in predicting mortality in people with SLE, wrote the investigators of the new study, led by Michael W. P. Tsang-A-Sjoe of the Amsterdam Rheumatology and Immunology Center, VU University Medical Center, the Netherlands.
The investigators prospectively assessed 183 patients from the Amsterdam SLE cohort for flares and once a year for retrospectively determined remission as defined in a 2015 study (Ann Rheum Dis. 2015;74:2117-22) and in Lupus Low Disease Activity State (LLDAS) criteria set by the Asia-Pacific Lupus Collaboration (Ann Rheum Dis. 2016;75:1615-21). The research team developed a prediction model for damage accrual during the 5-year follow-up through the use of backward logistic regression analyses that took these definitions into account (Rheumatology [Oxford]. 2016 Nov 1. doi: 10.1093/rheumatology/kew377).
The occurrence of at least one flare and the average daily prednisone dose during follow-up were significant predictors of damage accrual (Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index increase of 5 or more during follow-up) in the cohort of patients with or without prolonged remission and with or without LLDAS in half or more of the observations. For example, patients had a higher risk of damage accrual with both a mean daily oral prednisone dose greater than 7.5 mg (odds ratio, 3.6; 95% confidence interval, 1.8-7.2) and greater than 5 mg (OR, 2.8; 95% CI, 1.5-5.3), compared with those taking lower doses.
The researchers also identified a history of nephrological manifestations at baseline as a significant predictor of damage accrual, despite not being part of the criteria for remission or LLDAS.
Overall, 38 (32.5%) of 117 patients met the prolonged remission criteria, whereas LLDAS in half or more of the available observations occurred in 118 (64.5%) of 183 patients.
Prolonged remission during 5 years of follow-up and LLDAS in 50% or more of observations were associated with a reduced risk of damage accrual (OR, 0.20; 95% CI, 0.07-0.53; P = .001; and OR, 0.52; 95% CI, 0.28-0.99, P = .046, respectively).
The effects of both remission and LLDAS, according to the definitions used in the study, don’t settle the matter of which might be used in a treat-to-target strategy. Neither the remission criteria nor the LLDAS definition showed clear superiority over the other, although patients in prolonged remission seemed to accrue less damage, compared with patients classified as being in LLDAS. However, the study did not prove this point, and the investigators noted that “future studies with sufficient power are needed in order to directly compare both sets of criteria for superiority.”
The results might also point to the possibility of lengthening the interval between disease activity assessments. The current study assessed LLDAS and remission at 1 year, compared with other studies that have generally assessed patients at quarterly intervals.
“This finding is interesting because it suggests that in clinical practice, yearly – rather than quarterly – assessments of LLDAS and remission might be sufficient to identify patients at risk of damage accrual, although future studies are needed to confirm this finding,” they wrote.
The study had no specific funding source, and the authors declared having no conflicts of interest.
Fulfilling definitions for prolonged lupus remission and reaching a low disease activity state are both associated with a lower risk of damage accrual in lupus patients, new research confirms.
“Identifying predictors of organ damage holds great importance for improving outcomes in SLE [systemic lupus erythematosus]” in light of the strength of organ damage in predicting mortality in people with SLE, wrote the investigators of the new study, led by Michael W. P. Tsang-A-Sjoe of the Amsterdam Rheumatology and Immunology Center, VU University Medical Center, the Netherlands.
The investigators prospectively assessed 183 patients from the Amsterdam SLE cohort for flares and once a year for retrospectively determined remission as defined in a 2015 study (Ann Rheum Dis. 2015;74:2117-22) and in Lupus Low Disease Activity State (LLDAS) criteria set by the Asia-Pacific Lupus Collaboration (Ann Rheum Dis. 2016;75:1615-21). The research team developed a prediction model for damage accrual during the 5-year follow-up through the use of backward logistic regression analyses that took these definitions into account (Rheumatology [Oxford]. 2016 Nov 1. doi: 10.1093/rheumatology/kew377).
The occurrence of at least one flare and the average daily prednisone dose during follow-up were significant predictors of damage accrual (Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index increase of 5 or more during follow-up) in the cohort of patients with or without prolonged remission and with or without LLDAS in half or more of the observations. For example, patients had a higher risk of damage accrual with both a mean daily oral prednisone dose greater than 7.5 mg (odds ratio, 3.6; 95% confidence interval, 1.8-7.2) and greater than 5 mg (OR, 2.8; 95% CI, 1.5-5.3), compared with those taking lower doses.
The researchers also identified a history of nephrological manifestations at baseline as a significant predictor of damage accrual, despite not being part of the criteria for remission or LLDAS.
Overall, 38 (32.5%) of 117 patients met the prolonged remission criteria, whereas LLDAS in half or more of the available observations occurred in 118 (64.5%) of 183 patients.
Prolonged remission during 5 years of follow-up and LLDAS in 50% or more of observations were associated with a reduced risk of damage accrual (OR, 0.20; 95% CI, 0.07-0.53; P = .001; and OR, 0.52; 95% CI, 0.28-0.99, P = .046, respectively).
The effects of both remission and LLDAS, according to the definitions used in the study, don’t settle the matter of which might be used in a treat-to-target strategy. Neither the remission criteria nor the LLDAS definition showed clear superiority over the other, although patients in prolonged remission seemed to accrue less damage, compared with patients classified as being in LLDAS. However, the study did not prove this point, and the investigators noted that “future studies with sufficient power are needed in order to directly compare both sets of criteria for superiority.”
The results might also point to the possibility of lengthening the interval between disease activity assessments. The current study assessed LLDAS and remission at 1 year, compared with other studies that have generally assessed patients at quarterly intervals.
“This finding is interesting because it suggests that in clinical practice, yearly – rather than quarterly – assessments of LLDAS and remission might be sufficient to identify patients at risk of damage accrual, although future studies are needed to confirm this finding,” they wrote.
The study had no specific funding source, and the authors declared having no conflicts of interest.
FROM RHEUMATOLOGY
Key clinical finding:
Major finding: At least one flare, average daily prednisone dose during follow-up, and nephrological manifestations at baseline were the most significant predictors of damage accrual in lupus patients.
Data source: Retrospective, longitudinal study of 183 patients from the Amsterdam SLE cohort with a median follow-up of 5 years.
Disclosures: The study had no specific funding source, and the authors declared having no conflicts of interest.
POEM procedure effective over the long term in achalasia
The therapeutic endoscopic procedure per-oral endoscopic myotomy (POEM) is safe and has a high clinical success rate at 2 years in patients with the rare esophageal motility disorder achalasia, according to results of the longest follow-up study to date.
The research team said that achalasia is characterized by the loss of enteric neurons, which results in impaired relaxation of the lower esophageal sphincter and absence of esophageal peristalsis.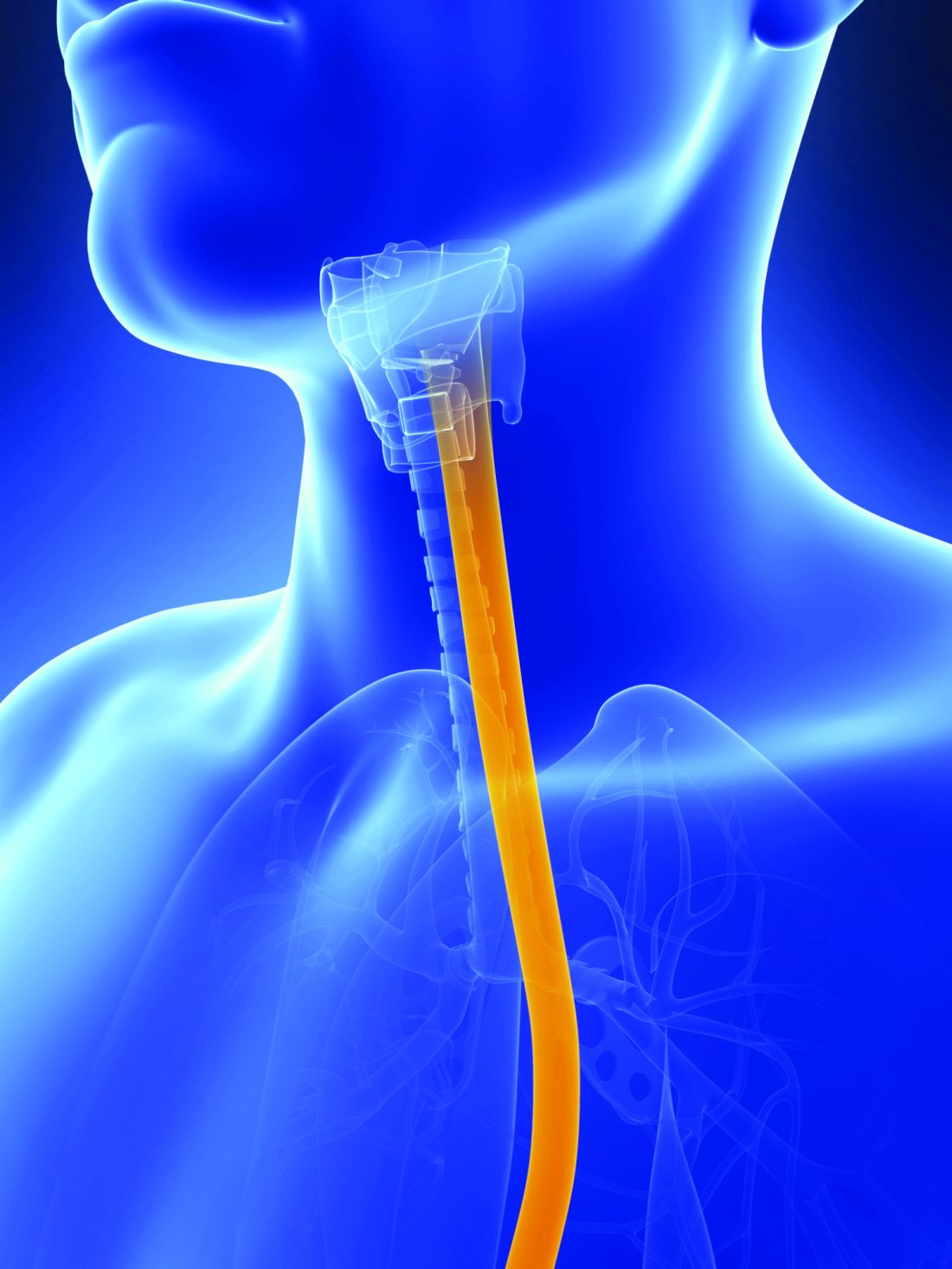
The newer procedure, called POEM, is based on a technique of submucosal endoscopy and endoscopic myotomy. However, while short-term studies have shown it to be safe and effective for achalasia, long-term data have been limited.
“As achalasia is a chronic disease with a risk of recurrence of symptoms after treatment, long-term efficacy, rather than short-term data, is important for decision making surrounding the optimal therapeutic modalities,” wrote the investigators, led by Saowanee Ngamruengphong, MD, of Johns Hopkins Hospital, Baltimore (Gastrointest Endosc. 2016. doi: 10.1016/j.gie.2016.09.017).
The study involved 205 patients with achalasia from 10 centers in the United States, Europe, and Asia. Patients were included in the study if they had at least 2 years of follow-up data available.
Results showed that the overall clinical success – defined by a decrease in Eckardt score to 3 or lower – for the procedure was 91% at 2 years. The Eckardt score (maximum 12) is a composite measure of frequency of symptoms and severity of weight loss.
Overall, 18 patients (8.8%) were considered to have clinical failure (Eckardt score greater than 3) after POEM.
A history of prior pneumatic dilation was associated with long-term treatment failure (odds ratio, 3.41; 95% confidence interval, 1.25-9.23).
The researchers noted that the therapeutic success of POEM decreased over time as the clinical success rate of the procedure at 6 months’ follow-up had been 98%.
This is consistent with findings from other studies and also with other procedures such as Heller myotomy and pneumatic dilation.
“After POEM or other standard therapies, patients with achalasia require ongoing follow-up to assess recurrent symptoms with additional evaluation and treatment when indicated,” they wrote.
In terms of safety, the researchers reported procedure-related adverse events in 8.2% of patients, with one patient requiring surgical intervention.
Abnormal esophageal acid exposure and reflux esophagitis were also documented in 37.5% and 18% of patients, respectively.
Multivariate analysis revealed that full-thickness myotomy was independently associated with postoperative reflux esophagitis (adjusted OR, 3.99; 95% CI, 1.16-13.68; P = .002).
The authors noted that while reflux was common after POEM, almost all patients could be successfully treated with proton pump inhibitors.
“Prospective randomized studies comparing POEM and current standard therapy are awaited, but in the meantime, it can be considered an effective alternative option for patients with achalasia where expertise is available,” they concluded.
The therapeutic endoscopic procedure per-oral endoscopic myotomy (POEM) is safe and has a high clinical success rate at 2 years in patients with the rare esophageal motility disorder achalasia, according to results of the longest follow-up study to date.
The research team said that achalasia is characterized by the loss of enteric neurons, which results in impaired relaxation of the lower esophageal sphincter and absence of esophageal peristalsis.
The newer procedure, called POEM, is based on a technique of submucosal endoscopy and endoscopic myotomy. However, while short-term studies have shown it to be safe and effective for achalasia, long-term data have been limited.
“As achalasia is a chronic disease with a risk of recurrence of symptoms after treatment, long-term efficacy, rather than short-term data, is important for decision making surrounding the optimal therapeutic modalities,” wrote the investigators, led by Saowanee Ngamruengphong, MD, of Johns Hopkins Hospital, Baltimore (Gastrointest Endosc. 2016. doi: 10.1016/j.gie.2016.09.017).
The study involved 205 patients with achalasia from 10 centers in the United States, Europe, and Asia. Patients were included in the study if they had at least 2 years of follow-up data available.
Results showed that the overall clinical success – defined by a decrease in Eckardt score to 3 or lower – for the procedure was 91% at 2 years. The Eckardt score (maximum 12) is a composite measure of frequency of symptoms and severity of weight loss.
Overall, 18 patients (8.8%) were considered to have clinical failure (Eckardt score greater than 3) after POEM.
A history of prior pneumatic dilation was associated with long-term treatment failure (odds ratio, 3.41; 95% confidence interval, 1.25-9.23).
The researchers noted that the therapeutic success of POEM decreased over time as the clinical success rate of the procedure at 6 months’ follow-up had been 98%.
This is consistent with findings from other studies and also with other procedures such as Heller myotomy and pneumatic dilation.
“After POEM or other standard therapies, patients with achalasia require ongoing follow-up to assess recurrent symptoms with additional evaluation and treatment when indicated,” they wrote.
In terms of safety, the researchers reported procedure-related adverse events in 8.2% of patients, with one patient requiring surgical intervention.
Abnormal esophageal acid exposure and reflux esophagitis were also documented in 37.5% and 18% of patients, respectively.
Multivariate analysis revealed that full-thickness myotomy was independently associated with postoperative reflux esophagitis (adjusted OR, 3.99; 95% CI, 1.16-13.68; P = .002).
The authors noted that while reflux was common after POEM, almost all patients could be successfully treated with proton pump inhibitors.
“Prospective randomized studies comparing POEM and current standard therapy are awaited, but in the meantime, it can be considered an effective alternative option for patients with achalasia where expertise is available,” they concluded.
The therapeutic endoscopic procedure per-oral endoscopic myotomy (POEM) is safe and has a high clinical success rate at 2 years in patients with the rare esophageal motility disorder achalasia, according to results of the longest follow-up study to date.
The research team said that achalasia is characterized by the loss of enteric neurons, which results in impaired relaxation of the lower esophageal sphincter and absence of esophageal peristalsis.
The newer procedure, called POEM, is based on a technique of submucosal endoscopy and endoscopic myotomy. However, while short-term studies have shown it to be safe and effective for achalasia, long-term data have been limited.
“As achalasia is a chronic disease with a risk of recurrence of symptoms after treatment, long-term efficacy, rather than short-term data, is important for decision making surrounding the optimal therapeutic modalities,” wrote the investigators, led by Saowanee Ngamruengphong, MD, of Johns Hopkins Hospital, Baltimore (Gastrointest Endosc. 2016. doi: 10.1016/j.gie.2016.09.017).
The study involved 205 patients with achalasia from 10 centers in the United States, Europe, and Asia. Patients were included in the study if they had at least 2 years of follow-up data available.
Results showed that the overall clinical success – defined by a decrease in Eckardt score to 3 or lower – for the procedure was 91% at 2 years. The Eckardt score (maximum 12) is a composite measure of frequency of symptoms and severity of weight loss.
Overall, 18 patients (8.8%) were considered to have clinical failure (Eckardt score greater than 3) after POEM.
A history of prior pneumatic dilation was associated with long-term treatment failure (odds ratio, 3.41; 95% confidence interval, 1.25-9.23).
The researchers noted that the therapeutic success of POEM decreased over time as the clinical success rate of the procedure at 6 months’ follow-up had been 98%.
This is consistent with findings from other studies and also with other procedures such as Heller myotomy and pneumatic dilation.
“After POEM or other standard therapies, patients with achalasia require ongoing follow-up to assess recurrent symptoms with additional evaluation and treatment when indicated,” they wrote.
In terms of safety, the researchers reported procedure-related adverse events in 8.2% of patients, with one patient requiring surgical intervention.
Abnormal esophageal acid exposure and reflux esophagitis were also documented in 37.5% and 18% of patients, respectively.
Multivariate analysis revealed that full-thickness myotomy was independently associated with postoperative reflux esophagitis (adjusted OR, 3.99; 95% CI, 1.16-13.68; P = .002).
The authors noted that while reflux was common after POEM, almost all patients could be successfully treated with proton pump inhibitors.
“Prospective randomized studies comparing POEM and current standard therapy are awaited, but in the meantime, it can be considered an effective alternative option for patients with achalasia where expertise is available,” they concluded.
FROM GASTROINTESTINAL ENDOSCOPY
Key clinical point: The therapeutic endoscopic procedure per-oral endoscopic myotomy (POEM) has a high clinical success rate at 2 years in patients with achalasia.
Main finding: Overall clinical success for the procedure – defined by a decrease in Eckardt score to 3 or lower – was 91% at 2 years’ follow-up.
Data source: Retrospective study of 205 patients with achalasia from 10 centers across the United States, Europe, and Asia.
Disclosures: Several of the authors are consultants for Medtronic, Boston Scientific, and Sandhill Scientific, but no conflicts of interest were declared in relation to the current paper.
PsA bone loss measurement: A surrogate for radiographic progression?
An advanced computer assisted digital x-ray radiogrammetry technique that measures bone thickness has the potential to be a surrogate marker of radiographic progression in psoriatic arthritis, according to a report in Arthritis Research & Therapy.
The method uses software called BoneXpert to sensitively differentiate between the different stages of disease manifestation affecting bone integrity. Digital x-ray radiogrammetry (DXR) with BoneXpert has a clinical advantage over standard techniques such as radiographs through its ability to be integrated into a picture archiving and communication system that allows direct image analysis and quantification of bone loss, according to the study authors, led by Alexander Pfeil, MD, of Jena (Germany) University Hospital – Friedrich Schiller University.
The researchers used the computer-assisted diagnosis software to measure the metacarpal index (MCI) and its cortical thickness score (MCI T-score) in the metacarpal bones of 104 psoriatic arthritis (PsA) patients who fulfilled the CASPAR criteria. All patients were treated either with nonsteroidal anti-inflammatory drugs or disease-modifying antirheumatic drugs (Arthritis Res Ther. 2016;18:248. doi: 10.1186/s13075-016-1145-4).
In the total PsA cohort, the MCI T-score showed a significantly reduced negative value of –1.289. “The reduced MCI T-score was clearly associated with a reduced bone mineral density of the metacarpal bones in PsA,” the investigators wrote.
For all scores, the researchers found a severity-dependent reduction for the BoneXpert parameters of MCI, MCI T-score, T, and Bone Health Index.
The strongest reductions were seen for MCI and T using the Proliferation Score (MCI: –28.3%; T: –31.9%) and the Destruction Score (MCI: –30.8%; T: –30.9%) of the Psoriatic Arthritis Ratingen Score.
A reduced MCI and T-score was directly associated with cortical thinning and the periarticular demineralization of the metacarpal bones, and highlighted a direct association with bone destruction and bone proliferation in PsA, the investigators said.
“The measurement of periarticular bone loss can be considered a complementary approach to verify PsA-related bony changes and a surrogate marker for PsA progression,” the researchers suggested.
The technique’s high reproducibility can also be used to optimize an appropriate individual therapeutic strategy, they added.
The study had no specific funding source, and the authors declared no conflicts of interest.
An advanced computer assisted digital x-ray radiogrammetry technique that measures bone thickness has the potential to be a surrogate marker of radiographic progression in psoriatic arthritis, according to a report in Arthritis Research & Therapy.
The method uses software called BoneXpert to sensitively differentiate between the different stages of disease manifestation affecting bone integrity. Digital x-ray radiogrammetry (DXR) with BoneXpert has a clinical advantage over standard techniques such as radiographs through its ability to be integrated into a picture archiving and communication system that allows direct image analysis and quantification of bone loss, according to the study authors, led by Alexander Pfeil, MD, of Jena (Germany) University Hospital – Friedrich Schiller University.
The researchers used the computer-assisted diagnosis software to measure the metacarpal index (MCI) and its cortical thickness score (MCI T-score) in the metacarpal bones of 104 psoriatic arthritis (PsA) patients who fulfilled the CASPAR criteria. All patients were treated either with nonsteroidal anti-inflammatory drugs or disease-modifying antirheumatic drugs (Arthritis Res Ther. 2016;18:248. doi: 10.1186/s13075-016-1145-4).
In the total PsA cohort, the MCI T-score showed a significantly reduced negative value of –1.289. “The reduced MCI T-score was clearly associated with a reduced bone mineral density of the metacarpal bones in PsA,” the investigators wrote.
For all scores, the researchers found a severity-dependent reduction for the BoneXpert parameters of MCI, MCI T-score, T, and Bone Health Index.
The strongest reductions were seen for MCI and T using the Proliferation Score (MCI: –28.3%; T: –31.9%) and the Destruction Score (MCI: –30.8%; T: –30.9%) of the Psoriatic Arthritis Ratingen Score.
A reduced MCI and T-score was directly associated with cortical thinning and the periarticular demineralization of the metacarpal bones, and highlighted a direct association with bone destruction and bone proliferation in PsA, the investigators said.
“The measurement of periarticular bone loss can be considered a complementary approach to verify PsA-related bony changes and a surrogate marker for PsA progression,” the researchers suggested.
The technique’s high reproducibility can also be used to optimize an appropriate individual therapeutic strategy, they added.
The study had no specific funding source, and the authors declared no conflicts of interest.
An advanced computer assisted digital x-ray radiogrammetry technique that measures bone thickness has the potential to be a surrogate marker of radiographic progression in psoriatic arthritis, according to a report in Arthritis Research & Therapy.
The method uses software called BoneXpert to sensitively differentiate between the different stages of disease manifestation affecting bone integrity. Digital x-ray radiogrammetry (DXR) with BoneXpert has a clinical advantage over standard techniques such as radiographs through its ability to be integrated into a picture archiving and communication system that allows direct image analysis and quantification of bone loss, according to the study authors, led by Alexander Pfeil, MD, of Jena (Germany) University Hospital – Friedrich Schiller University.
The researchers used the computer-assisted diagnosis software to measure the metacarpal index (MCI) and its cortical thickness score (MCI T-score) in the metacarpal bones of 104 psoriatic arthritis (PsA) patients who fulfilled the CASPAR criteria. All patients were treated either with nonsteroidal anti-inflammatory drugs or disease-modifying antirheumatic drugs (Arthritis Res Ther. 2016;18:248. doi: 10.1186/s13075-016-1145-4).
In the total PsA cohort, the MCI T-score showed a significantly reduced negative value of –1.289. “The reduced MCI T-score was clearly associated with a reduced bone mineral density of the metacarpal bones in PsA,” the investigators wrote.
For all scores, the researchers found a severity-dependent reduction for the BoneXpert parameters of MCI, MCI T-score, T, and Bone Health Index.
The strongest reductions were seen for MCI and T using the Proliferation Score (MCI: –28.3%; T: –31.9%) and the Destruction Score (MCI: –30.8%; T: –30.9%) of the Psoriatic Arthritis Ratingen Score.
A reduced MCI and T-score was directly associated with cortical thinning and the periarticular demineralization of the metacarpal bones, and highlighted a direct association with bone destruction and bone proliferation in PsA, the investigators said.
“The measurement of periarticular bone loss can be considered a complementary approach to verify PsA-related bony changes and a surrogate marker for PsA progression,” the researchers suggested.
The technique’s high reproducibility can also be used to optimize an appropriate individual therapeutic strategy, they added.
The study had no specific funding source, and the authors declared no conflicts of interest.
FROM ARTHRITIS RESEARCH & THERAPY
Key clinical point:
Main finding: In the total PsA cohort, the MCI T-score showed a significantly reduced negative value of –1.289.
Data source: A cohort of 104 PsA patients fulfilling the CASPAR criteria who were taking nonsteroidal inflammatory drugs or disease-modifying antirheumatic drugs.
Disclosures: The study had no specific funding source, and the authors declared no conflicts of interest.
Disease severity, QOL outcome measures need standardizing in atopic dermatitis research
The number of outcome measures used to assess disease severity and quality of life (QOL) in randomized controlled trials of patients with atopic dermatitis (AD) has risen in recent years, according to a systematic review reports.
An overall lack of standardization of these outcome measures, however, is hindering the synthesis and translation of research into clinical practice, reported Mary K. Hill of the University of Colorado, Aurora, and her associates.
“Standardization of disease severity and QOL outcome instruments is essential for comparability among studies and improved quality of research evidence,” they wrote (J Am Acad Dermatol. 2016 Nov;75[5]:906-17. doi: 10.1016/j.jaad.2016.07.002).
Their systematic review of 135 randomized controlled trials (RCTs) identified 62 disease-severity and 28 quality-of-life instruments used in studies of patients with AD between July 2010 and July 2015.
This was a drastic increase from the 20 disease severity scales and 14 QOL indices identified in a previous systematic review of 382 RCTs of AD therapies conducted between 1985 and 2010, they noted.
In their review, the most frequently used disease severity scale was the Scoring Atopic Dermatitis (SCORAD) index, which was used in 79 studies. That was followed by the visual analogue scale (VAS) for pruritus, used in 30 studies; the Investigator’s Global Assessment (IGA) tool, used in 29 studies; and the Eczema Area and Severity Index (EASI), used in 28 studies.
But despite the well-documented burden of AD, the researchers noted that only 33% of the RCTs they reviewed assessed QOL. “This is up from the 18% of RCTs on AD that reported QOL outcomes between 1985 and July 2010, perhaps signifying gradually increased attention to patient emotional well-being,” Ms. Hill and her associates wrote.
A trend described by the authors, however, as “perhaps the most disconcerting” was that 75% of identified QOL instruments were used only once. “Continued increases in the reporting of QOL outcomes will be of limited benefit for interstudy comparisons if the diversity of measures used also continues to rise,” they said.
Adding to the confusion, the researchers found frequent overlap in the naming and content of instruments used in studies, making it even more challenging to identify meaningful comparisons.
There was no funding source, and the authors had no conflicts of interest to declare.
The number of outcome measures used to assess disease severity and quality of life (QOL) in randomized controlled trials of patients with atopic dermatitis (AD) has risen in recent years, according to a systematic review reports.
An overall lack of standardization of these outcome measures, however, is hindering the synthesis and translation of research into clinical practice, reported Mary K. Hill of the University of Colorado, Aurora, and her associates.
“Standardization of disease severity and QOL outcome instruments is essential for comparability among studies and improved quality of research evidence,” they wrote (J Am Acad Dermatol. 2016 Nov;75[5]:906-17. doi: 10.1016/j.jaad.2016.07.002).
Their systematic review of 135 randomized controlled trials (RCTs) identified 62 disease-severity and 28 quality-of-life instruments used in studies of patients with AD between July 2010 and July 2015.
This was a drastic increase from the 20 disease severity scales and 14 QOL indices identified in a previous systematic review of 382 RCTs of AD therapies conducted between 1985 and 2010, they noted.
In their review, the most frequently used disease severity scale was the Scoring Atopic Dermatitis (SCORAD) index, which was used in 79 studies. That was followed by the visual analogue scale (VAS) for pruritus, used in 30 studies; the Investigator’s Global Assessment (IGA) tool, used in 29 studies; and the Eczema Area and Severity Index (EASI), used in 28 studies.
But despite the well-documented burden of AD, the researchers noted that only 33% of the RCTs they reviewed assessed QOL. “This is up from the 18% of RCTs on AD that reported QOL outcomes between 1985 and July 2010, perhaps signifying gradually increased attention to patient emotional well-being,” Ms. Hill and her associates wrote.
A trend described by the authors, however, as “perhaps the most disconcerting” was that 75% of identified QOL instruments were used only once. “Continued increases in the reporting of QOL outcomes will be of limited benefit for interstudy comparisons if the diversity of measures used also continues to rise,” they said.
Adding to the confusion, the researchers found frequent overlap in the naming and content of instruments used in studies, making it even more challenging to identify meaningful comparisons.
There was no funding source, and the authors had no conflicts of interest to declare.
The number of outcome measures used to assess disease severity and quality of life (QOL) in randomized controlled trials of patients with atopic dermatitis (AD) has risen in recent years, according to a systematic review reports.
An overall lack of standardization of these outcome measures, however, is hindering the synthesis and translation of research into clinical practice, reported Mary K. Hill of the University of Colorado, Aurora, and her associates.
“Standardization of disease severity and QOL outcome instruments is essential for comparability among studies and improved quality of research evidence,” they wrote (J Am Acad Dermatol. 2016 Nov;75[5]:906-17. doi: 10.1016/j.jaad.2016.07.002).
Their systematic review of 135 randomized controlled trials (RCTs) identified 62 disease-severity and 28 quality-of-life instruments used in studies of patients with AD between July 2010 and July 2015.
This was a drastic increase from the 20 disease severity scales and 14 QOL indices identified in a previous systematic review of 382 RCTs of AD therapies conducted between 1985 and 2010, they noted.
In their review, the most frequently used disease severity scale was the Scoring Atopic Dermatitis (SCORAD) index, which was used in 79 studies. That was followed by the visual analogue scale (VAS) for pruritus, used in 30 studies; the Investigator’s Global Assessment (IGA) tool, used in 29 studies; and the Eczema Area and Severity Index (EASI), used in 28 studies.
But despite the well-documented burden of AD, the researchers noted that only 33% of the RCTs they reviewed assessed QOL. “This is up from the 18% of RCTs on AD that reported QOL outcomes between 1985 and July 2010, perhaps signifying gradually increased attention to patient emotional well-being,” Ms. Hill and her associates wrote.
A trend described by the authors, however, as “perhaps the most disconcerting” was that 75% of identified QOL instruments were used only once. “Continued increases in the reporting of QOL outcomes will be of limited benefit for interstudy comparisons if the diversity of measures used also continues to rise,” they said.
Adding to the confusion, the researchers found frequent overlap in the naming and content of instruments used in studies, making it even more challenging to identify meaningful comparisons.
There was no funding source, and the authors had no conflicts of interest to declare.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: Disease severity and quality-of-life measures used in randomized controlled trials (RCTs) of atopic dermatitis (AD) need to be standardized so that study outcomes can be meaningfully compared and translated into clinical practice.
Major finding: The use of outcome measures used in atopic dermatitis RCTs has increased recently but still fall short in measuring quality-of-life outcomes.
Data source: A systematic review of 135 RCTs published between 2010-2015 that involved patients with AD.
Disclosures: There was no funding source, and the authors had no conflicts of interest to declare.
MS relapse predictors identified after patients stop treatment
Patients with multiple sclerosis treated with interferon-beta or glatiramer acetate who are aged 45 and over, and have no evidence of clinical disease activity for more than 4 years, have a high likelihood of remaining relapse free after stopping treatment, research showed.
The study, conducted by Gabriel Bsteh, MD, of the department of neurology at the Medical University of Innsbruck (Austria) and his colleagues, provides some evidence in the absence of randomized trials that may help guide discussions with patients – particularly those who have not had a relapse in a while – when they ask if they could discontinue disease-modifying treatment (DMT).
After a median follow-up period of 3.8 years, 98 patients (44.3%) had a relapse. Confirmed disability progression occurred in 46 patients (20.8%), and 15 patients (6.8%) converted to secondary progressive multiple sclerosis.
The independent predictors of absence of relapse after discontinuing treatment included age 45 years or older at discontinuation (HR = 0.47; confidence interval, 0.23–0.95; P = .038), absence of relapses for 4 or more years on DMT before discontinuation (HR = 0.29; CI, 0.10–0.82; P = .020), and absence of contrast-enhancing lesions (HR = 0.46; CI, 0.28–0.78; P =.004). A combination of age 45 years or older and absence of relapses after 4 or more years on DMT was associated with a very low risk of having a relapse after discontinuation, regardless of MRI results (HR = 0.06; CI, 0.01-0.44; P less than .001).
Higher Expanded Disability Status Scale (EDSS) scores at discontinuation, age older than 45 years at discontinuation, and longer disease duration were the only significant independent predictors of disability progression after discontinuation, irrespective of the presence of relapses on DMT or gadolinium-enhancing lesions.
“This underlines the concept of a window of opportunity in the treatment of MS, in the sense that once a certain extent of disability is reached, the impact of relapses, and therefore the effect of anti-inflammatory treatment, are drastically reduced,” the investigators wrote.
The study is limited because of its observational retrospective nature, but the research team said their results emphasized the importance of regular, thorough clinical evaluation of patients with RRMS. “While MRI may have a role in aiding decision making regarding DMT discontinuation, our data clearly show that demographic factors and clinical monitoring go a long way in risk stratification,” they wrote.
The authors received no financial support for the study, but several of the authors reported participating in meetings or receiving honoraria from several pharmaceutical companies.
Patients with multiple sclerosis treated with interferon-beta or glatiramer acetate who are aged 45 and over, and have no evidence of clinical disease activity for more than 4 years, have a high likelihood of remaining relapse free after stopping treatment, research showed.
The study, conducted by Gabriel Bsteh, MD, of the department of neurology at the Medical University of Innsbruck (Austria) and his colleagues, provides some evidence in the absence of randomized trials that may help guide discussions with patients – particularly those who have not had a relapse in a while – when they ask if they could discontinue disease-modifying treatment (DMT).
After a median follow-up period of 3.8 years, 98 patients (44.3%) had a relapse. Confirmed disability progression occurred in 46 patients (20.8%), and 15 patients (6.8%) converted to secondary progressive multiple sclerosis.
The independent predictors of absence of relapse after discontinuing treatment included age 45 years or older at discontinuation (HR = 0.47; confidence interval, 0.23–0.95; P = .038), absence of relapses for 4 or more years on DMT before discontinuation (HR = 0.29; CI, 0.10–0.82; P = .020), and absence of contrast-enhancing lesions (HR = 0.46; CI, 0.28–0.78; P =.004). A combination of age 45 years or older and absence of relapses after 4 or more years on DMT was associated with a very low risk of having a relapse after discontinuation, regardless of MRI results (HR = 0.06; CI, 0.01-0.44; P less than .001).
Higher Expanded Disability Status Scale (EDSS) scores at discontinuation, age older than 45 years at discontinuation, and longer disease duration were the only significant independent predictors of disability progression after discontinuation, irrespective of the presence of relapses on DMT or gadolinium-enhancing lesions.
“This underlines the concept of a window of opportunity in the treatment of MS, in the sense that once a certain extent of disability is reached, the impact of relapses, and therefore the effect of anti-inflammatory treatment, are drastically reduced,” the investigators wrote.
The study is limited because of its observational retrospective nature, but the research team said their results emphasized the importance of regular, thorough clinical evaluation of patients with RRMS. “While MRI may have a role in aiding decision making regarding DMT discontinuation, our data clearly show that demographic factors and clinical monitoring go a long way in risk stratification,” they wrote.
The authors received no financial support for the study, but several of the authors reported participating in meetings or receiving honoraria from several pharmaceutical companies.
Patients with multiple sclerosis treated with interferon-beta or glatiramer acetate who are aged 45 and over, and have no evidence of clinical disease activity for more than 4 years, have a high likelihood of remaining relapse free after stopping treatment, research showed.
The study, conducted by Gabriel Bsteh, MD, of the department of neurology at the Medical University of Innsbruck (Austria) and his colleagues, provides some evidence in the absence of randomized trials that may help guide discussions with patients – particularly those who have not had a relapse in a while – when they ask if they could discontinue disease-modifying treatment (DMT).
After a median follow-up period of 3.8 years, 98 patients (44.3%) had a relapse. Confirmed disability progression occurred in 46 patients (20.8%), and 15 patients (6.8%) converted to secondary progressive multiple sclerosis.
The independent predictors of absence of relapse after discontinuing treatment included age 45 years or older at discontinuation (HR = 0.47; confidence interval, 0.23–0.95; P = .038), absence of relapses for 4 or more years on DMT before discontinuation (HR = 0.29; CI, 0.10–0.82; P = .020), and absence of contrast-enhancing lesions (HR = 0.46; CI, 0.28–0.78; P =.004). A combination of age 45 years or older and absence of relapses after 4 or more years on DMT was associated with a very low risk of having a relapse after discontinuation, regardless of MRI results (HR = 0.06; CI, 0.01-0.44; P less than .001).
Higher Expanded Disability Status Scale (EDSS) scores at discontinuation, age older than 45 years at discontinuation, and longer disease duration were the only significant independent predictors of disability progression after discontinuation, irrespective of the presence of relapses on DMT or gadolinium-enhancing lesions.
“This underlines the concept of a window of opportunity in the treatment of MS, in the sense that once a certain extent of disability is reached, the impact of relapses, and therefore the effect of anti-inflammatory treatment, are drastically reduced,” the investigators wrote.
The study is limited because of its observational retrospective nature, but the research team said their results emphasized the importance of regular, thorough clinical evaluation of patients with RRMS. “While MRI may have a role in aiding decision making regarding DMT discontinuation, our data clearly show that demographic factors and clinical monitoring go a long way in risk stratification,” they wrote.
The authors received no financial support for the study, but several of the authors reported participating in meetings or receiving honoraria from several pharmaceutical companies.
FROM MULTIPLE SCLEROSIS JOURNAL
Key clinical point: Age and time since last relapse can predict relapsing-remitting multiple sclerosis patients with a lower risk of relapse after they discontinue interferon-beta or glatiramer acetate.
Main finding: A combination of age 45 years or older and absence of relapses after 4 or more years on disease-modifying treatment was associated with a very low risk of having a relapse after discontinuation, regardless of MRI results (HR = 0.06; CI, 0.01–0.44; P less than .001).
Data source: An observational retrospective analysis of 221 patients from the Innsbruck MS database with relapsing-remitting multiple sclerosis who discontinued disease-modifying treatment after more than 12 months and had documented follow-up at 2 years.
Disclosures: The authors received no financial support for the study, but several of the authors reported participating in meetings or receiving honoraria from several pharmaceutical companies.
Amyloid PET scans may have clinical utility in patients with cognitive impairment
Amyloid PET scans can affect diagnostic thinking and patient management when used alongside routine diagnostic work up in adults suspected of having Alzheimer’s disease, results from the multicenter, open-label Assessment of the Incremental Diagnostic Value of Florbetapir 18F Imaging in Patients with Cognitive Impairment (INDIA-FBP) study reveal.
The investigators, led by Marina Boccardi, PhD, of the LANVIE-Laboratory of Neuroimaging of Aging at the University of Geneva, conducted the study to provide evidence of whether amyloid PET imaging has diagnostic utility in patients suspected of having Alzheimer’s disease because amyloid PET scanning in its current state has debatable clinical value because of a lack of disease-modifying drugs available to treat Alzheimer’s and the expense of the scans.
The trial was published in this week’s edition of JAMA Neurology and Dr. Boccardi and her colleagues found that amyloid PET scans in 228 cognitively impaired Italian adults improved diagnostic confidence and led to a change in diagnosis in 28% of patients whose prescan diagnosis was Alzheimer’s disease and in 25% (P = .02) of patients who had a non–Alzheimer’s disease–related dementia diagnosis. A positive scan resulted in a change in diagnosis to Alzheimer’s disease 72% of the time in patients with a prescan diagnosis of frontotemporal lobar degeneration (FTLD). Cognition-specific medications were initiated in 66% of previously untreated patients and were withdrawn in 33% of previously treated patients, results from the study showed (JAMA Neurol. 2016 Oct 31. doi: 10.1001/jamaneurol.2016.3751).
The scans’ ability to provide additional information to change a diagnosis justifies their use, especially when considering “reports of adverse events in patients with FTLD treated with cholinesterase inhibitors and of ineffectiveness for cognitive impairment of vascular etiology,” the investigators wrote.
The use of amyloid PET scans to change a diagnosis to Alzheimer’s disease or confirm its diagnosis also allowed for earlier detection and intervention, gave patients an opportunity to be included in clinical trials, and allowed patients to make residential and financial arrangements at a time when they were still able to express their preference.
Anticipated results from the Imaging Dementia—Evidence for Amyloid Scanning (IDEAS) and Amyloid Imaging to Prevent Alzheimer’s Disease (AMYPAD) studies will expand on the current observations on a larger scale as well as help to quantify the cost-effectiveness of amyloid PET in clinical routine practice, they said.
In the current study, a prescan diagnosis was made by clinicians who, at the time of the work-up, estimated diagnostic confidence and provided drug treatment. At the same time, an amyloid PET/CT scan was performed and the result was communicated to the clinicians upon work-up completion. Clinicians relied on the scan results for their final clinical diagnoses and identification of the most-appropriate treatment.
Amyloid PET scans are currently not reimbursed by the Centers for Medicare & Medicaid Services outside of their use in clinical trials.
This study was sponsored by Istituto di Ricovero e Cura a Carattere Scientifico Centro San Giovanni di Dio Fatebenefratelli in Brescia, Italy, and Avid Radiopharmaceuticals. Several of the authors reported serving on the scientific advisory boards of pharmaceutical companies.
While amyloid PET scans have a clear role in research and in the right clinical setting they can support a suspected diagnosis of Alzheimer’s disease or help a clinician to choose between Alzheimer’s and a non-Alzheimer’s condition, their use can have pitfalls even at a diagnostically simplified level.

There are at least four patient populations in whom amyloid PET scans could potentially have clinical utility, but these are not addressed in the current debate over their use or in this study: patients with early-onset dementia, patients with normal pressure hydrocephalus, patients with rapidly progressive dementia in which the differential diagnosis includes some reversible conditions that may require a diagnostic brain biopsy, and rare patients with cerebral amyloid angiopathy who develop tumorlike accumulations that necessitate a brain biopsy for diagnosis.

Richard J. Caselli, MD, and Bryan K. Woodruff, MD, are with the department of neurology at the Mayo Clinic Arizona, Scottsdale. Their comments are derived from an editorial accompanying Dr. Boccardi and colleagues’ report (JAMA Neurol. 2016 Oct 31. doi: 10.1001/jamaneurol.2016.3792). Dr. Caselli reported receiving research funding from Merck, and Dr. Woodruff reported receiving research funding from Genentech and Avid Pharmaceuticals.
While amyloid PET scans have a clear role in research and in the right clinical setting they can support a suspected diagnosis of Alzheimer’s disease or help a clinician to choose between Alzheimer’s and a non-Alzheimer’s condition, their use can have pitfalls even at a diagnostically simplified level.
There are at least four patient populations in whom amyloid PET scans could potentially have clinical utility, but these are not addressed in the current debate over their use or in this study: patients with early-onset dementia, patients with normal pressure hydrocephalus, patients with rapidly progressive dementia in which the differential diagnosis includes some reversible conditions that may require a diagnostic brain biopsy, and rare patients with cerebral amyloid angiopathy who develop tumorlike accumulations that necessitate a brain biopsy for diagnosis.
Richard J. Caselli, MD, and Bryan K. Woodruff, MD, are with the department of neurology at the Mayo Clinic Arizona, Scottsdale. Their comments are derived from an editorial accompanying Dr. Boccardi and colleagues’ report (JAMA Neurol. 2016 Oct 31. doi: 10.1001/jamaneurol.2016.3792). Dr. Caselli reported receiving research funding from Merck, and Dr. Woodruff reported receiving research funding from Genentech and Avid Pharmaceuticals.
While amyloid PET scans have a clear role in research and in the right clinical setting they can support a suspected diagnosis of Alzheimer’s disease or help a clinician to choose between Alzheimer’s and a non-Alzheimer’s condition, their use can have pitfalls even at a diagnostically simplified level.
There are at least four patient populations in whom amyloid PET scans could potentially have clinical utility, but these are not addressed in the current debate over their use or in this study: patients with early-onset dementia, patients with normal pressure hydrocephalus, patients with rapidly progressive dementia in which the differential diagnosis includes some reversible conditions that may require a diagnostic brain biopsy, and rare patients with cerebral amyloid angiopathy who develop tumorlike accumulations that necessitate a brain biopsy for diagnosis.
Richard J. Caselli, MD, and Bryan K. Woodruff, MD, are with the department of neurology at the Mayo Clinic Arizona, Scottsdale. Their comments are derived from an editorial accompanying Dr. Boccardi and colleagues’ report (JAMA Neurol. 2016 Oct 31. doi: 10.1001/jamaneurol.2016.3792). Dr. Caselli reported receiving research funding from Merck, and Dr. Woodruff reported receiving research funding from Genentech and Avid Pharmaceuticals.
Amyloid PET scans can affect diagnostic thinking and patient management when used alongside routine diagnostic work up in adults suspected of having Alzheimer’s disease, results from the multicenter, open-label Assessment of the Incremental Diagnostic Value of Florbetapir 18F Imaging in Patients with Cognitive Impairment (INDIA-FBP) study reveal.
The investigators, led by Marina Boccardi, PhD, of the LANVIE-Laboratory of Neuroimaging of Aging at the University of Geneva, conducted the study to provide evidence of whether amyloid PET imaging has diagnostic utility in patients suspected of having Alzheimer’s disease because amyloid PET scanning in its current state has debatable clinical value because of a lack of disease-modifying drugs available to treat Alzheimer’s and the expense of the scans.
The trial was published in this week’s edition of JAMA Neurology and Dr. Boccardi and her colleagues found that amyloid PET scans in 228 cognitively impaired Italian adults improved diagnostic confidence and led to a change in diagnosis in 28% of patients whose prescan diagnosis was Alzheimer’s disease and in 25% (P = .02) of patients who had a non–Alzheimer’s disease–related dementia diagnosis. A positive scan resulted in a change in diagnosis to Alzheimer’s disease 72% of the time in patients with a prescan diagnosis of frontotemporal lobar degeneration (FTLD). Cognition-specific medications were initiated in 66% of previously untreated patients and were withdrawn in 33% of previously treated patients, results from the study showed (JAMA Neurol. 2016 Oct 31. doi: 10.1001/jamaneurol.2016.3751).
The scans’ ability to provide additional information to change a diagnosis justifies their use, especially when considering “reports of adverse events in patients with FTLD treated with cholinesterase inhibitors and of ineffectiveness for cognitive impairment of vascular etiology,” the investigators wrote.
The use of amyloid PET scans to change a diagnosis to Alzheimer’s disease or confirm its diagnosis also allowed for earlier detection and intervention, gave patients an opportunity to be included in clinical trials, and allowed patients to make residential and financial arrangements at a time when they were still able to express their preference.
Anticipated results from the Imaging Dementia—Evidence for Amyloid Scanning (IDEAS) and Amyloid Imaging to Prevent Alzheimer’s Disease (AMYPAD) studies will expand on the current observations on a larger scale as well as help to quantify the cost-effectiveness of amyloid PET in clinical routine practice, they said.
In the current study, a prescan diagnosis was made by clinicians who, at the time of the work-up, estimated diagnostic confidence and provided drug treatment. At the same time, an amyloid PET/CT scan was performed and the result was communicated to the clinicians upon work-up completion. Clinicians relied on the scan results for their final clinical diagnoses and identification of the most-appropriate treatment.
Amyloid PET scans are currently not reimbursed by the Centers for Medicare & Medicaid Services outside of their use in clinical trials.
This study was sponsored by Istituto di Ricovero e Cura a Carattere Scientifico Centro San Giovanni di Dio Fatebenefratelli in Brescia, Italy, and Avid Radiopharmaceuticals. Several of the authors reported serving on the scientific advisory boards of pharmaceutical companies.
Amyloid PET scans can affect diagnostic thinking and patient management when used alongside routine diagnostic work up in adults suspected of having Alzheimer’s disease, results from the multicenter, open-label Assessment of the Incremental Diagnostic Value of Florbetapir 18F Imaging in Patients with Cognitive Impairment (INDIA-FBP) study reveal.
The investigators, led by Marina Boccardi, PhD, of the LANVIE-Laboratory of Neuroimaging of Aging at the University of Geneva, conducted the study to provide evidence of whether amyloid PET imaging has diagnostic utility in patients suspected of having Alzheimer’s disease because amyloid PET scanning in its current state has debatable clinical value because of a lack of disease-modifying drugs available to treat Alzheimer’s and the expense of the scans.
The trial was published in this week’s edition of JAMA Neurology and Dr. Boccardi and her colleagues found that amyloid PET scans in 228 cognitively impaired Italian adults improved diagnostic confidence and led to a change in diagnosis in 28% of patients whose prescan diagnosis was Alzheimer’s disease and in 25% (P = .02) of patients who had a non–Alzheimer’s disease–related dementia diagnosis. A positive scan resulted in a change in diagnosis to Alzheimer’s disease 72% of the time in patients with a prescan diagnosis of frontotemporal lobar degeneration (FTLD). Cognition-specific medications were initiated in 66% of previously untreated patients and were withdrawn in 33% of previously treated patients, results from the study showed (JAMA Neurol. 2016 Oct 31. doi: 10.1001/jamaneurol.2016.3751).
The scans’ ability to provide additional information to change a diagnosis justifies their use, especially when considering “reports of adverse events in patients with FTLD treated with cholinesterase inhibitors and of ineffectiveness for cognitive impairment of vascular etiology,” the investigators wrote.
The use of amyloid PET scans to change a diagnosis to Alzheimer’s disease or confirm its diagnosis also allowed for earlier detection and intervention, gave patients an opportunity to be included in clinical trials, and allowed patients to make residential and financial arrangements at a time when they were still able to express their preference.
Anticipated results from the Imaging Dementia—Evidence for Amyloid Scanning (IDEAS) and Amyloid Imaging to Prevent Alzheimer’s Disease (AMYPAD) studies will expand on the current observations on a larger scale as well as help to quantify the cost-effectiveness of amyloid PET in clinical routine practice, they said.
In the current study, a prescan diagnosis was made by clinicians who, at the time of the work-up, estimated diagnostic confidence and provided drug treatment. At the same time, an amyloid PET/CT scan was performed and the result was communicated to the clinicians upon work-up completion. Clinicians relied on the scan results for their final clinical diagnoses and identification of the most-appropriate treatment.
Amyloid PET scans are currently not reimbursed by the Centers for Medicare & Medicaid Services outside of their use in clinical trials.
This study was sponsored by Istituto di Ricovero e Cura a Carattere Scientifico Centro San Giovanni di Dio Fatebenefratelli in Brescia, Italy, and Avid Radiopharmaceuticals. Several of the authors reported serving on the scientific advisory boards of pharmaceutical companies.
Key clinical point:
Main finding: Amyloid PET scans led to a change in diagnosis in 28% of patients whose prescan diagnosis was Alzheimer’s disease (AD) and in 25% of patients who had a non–AD related dementia diagnosis.
Source: Multicenter, open-label study of the incremental diagnostic value of Florbetapir 18F imaging in 228 adult patients with cognitive impairment.
Disclosures: This study was sponsored by Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Centro San Giovanni di Dio Fatebenefratelli in Brescia, Italy, and Avid Radiopharmaceuticals. Several of the authors reported serving on the scientific advisory boards of pharmaceutical companies.
Two migraine prevention drugs prove no better than placebo in children
Two commonly used drugs to prevent migraine in adults, amitriptyline and topiramate, proved no better than placebo at preventing migraine in children and adolescents and also were associated with more adverse events in the double-blind, randomized Childhood and Adolescent Migraine Prevention (CHAMP) trial.
The findings of the trial, which was stopped early for futility following a recommendation from a data and safety monitoring board, suggest the adult model of migraine treatment in which both drugs have been effective may not apply to pediatric patients, according to the investigators (N Engl J Med 2016 Oct 27. doi: 10.1056/NEJMoa1610384). The study was published simultaneously with its presentation at the annual meeting of the Child Neurology Society in Vancouver.
The 24-week, multicenter trial involved 328 children and adolescents aged 8-17 years who were diagnosed with migraine with or without aura or chronic migraine without continuous headache. They were randomized on a 2:2:1 ratio to receive oral amitriptyline (n = 132), topiramate (n = 130), or placebo (n = 66) according to age (8-12 years vs. 13-17 years). The target dose in the study was 1 mg/kg of body weight per day for amitriptyline and 2 mg/kg for topiramate. Doses were escalated every 2 weeks over an 8-week period and dose modifications were made according to side effects.
Overall, 52% on amitriptyline, 55% on topiramate, and 61% on placebo reached the study’s primary endpoint of a relative reduction of 50% or more in the number of headache days from baseline to the last 28 days of the trial; the outcomes were not significantly different from one another. The participants recorded the number of headache days they experienced in a diary kept during and after the 28-day baseline period of the study.
No significant differences were seen between the groups in the secondary outcomes of headache-related disability, headache days, or the number of patients who completed the trial.
However, the authors observed higher rates of adverse events overall in the active treatment groups, compared with placebo. Amitriptyline users significantly more often reported fatigue (30% vs. 14%, P = .01) and dry mouth (25% vs. 12%, P = .03), whereas topiramate users significantly more often reported paresthesia (31% vs. 8%, P less than .001) and weight loss (8% vs. 0%, P = .02).
Other adverse events among topiramate users were fatigue (25%), dry mouth (18%), memory impairment (17%), aphasia (16%), cognitive disorder (16%), and upper respiratory tract infection (12%).
Serious adverse events included altered mood in three patients in the amitriptyline group and one suicide attempt in the topiramate group.
“Given the null outcome in this trial and the adverse events and serious adverse events reported in the amitriptyline and topiramate groups, the data do not show a favorable risk-benefit profile for the use of these therapies in pediatric migraine prevention, at least over the 24-week duration of the trial,” the study authors concluded.
They noted that during their trial, the Food and Drug Administration approved topiramate for the treatment of episodic migraine in adolescents aged 12-17 years.
“Although our trial included patients outside this age range and included those with either episodic or chronic migraine, the trial results suggest that prevention medication for pediatric migraine might be reexamined,” they wrote.
The high placebo response seen in the study suggested the placebo effect might be advantageous for children and adolescents with migraine, they added.
The trial was supported by grants from the National Institute of Neurological Disorders and Stroke and the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The authors had no relevant financial disclosures.
Two commonly used drugs to prevent migraine in adults, amitriptyline and topiramate, proved no better than placebo at preventing migraine in children and adolescents and also were associated with more adverse events in the double-blind, randomized Childhood and Adolescent Migraine Prevention (CHAMP) trial.
The findings of the trial, which was stopped early for futility following a recommendation from a data and safety monitoring board, suggest the adult model of migraine treatment in which both drugs have been effective may not apply to pediatric patients, according to the investigators (N Engl J Med 2016 Oct 27. doi: 10.1056/NEJMoa1610384). The study was published simultaneously with its presentation at the annual meeting of the Child Neurology Society in Vancouver.
The 24-week, multicenter trial involved 328 children and adolescents aged 8-17 years who were diagnosed with migraine with or without aura or chronic migraine without continuous headache. They were randomized on a 2:2:1 ratio to receive oral amitriptyline (n = 132), topiramate (n = 130), or placebo (n = 66) according to age (8-12 years vs. 13-17 years). The target dose in the study was 1 mg/kg of body weight per day for amitriptyline and 2 mg/kg for topiramate. Doses were escalated every 2 weeks over an 8-week period and dose modifications were made according to side effects.
Overall, 52% on amitriptyline, 55% on topiramate, and 61% on placebo reached the study’s primary endpoint of a relative reduction of 50% or more in the number of headache days from baseline to the last 28 days of the trial; the outcomes were not significantly different from one another. The participants recorded the number of headache days they experienced in a diary kept during and after the 28-day baseline period of the study.
No significant differences were seen between the groups in the secondary outcomes of headache-related disability, headache days, or the number of patients who completed the trial.
However, the authors observed higher rates of adverse events overall in the active treatment groups, compared with placebo. Amitriptyline users significantly more often reported fatigue (30% vs. 14%, P = .01) and dry mouth (25% vs. 12%, P = .03), whereas topiramate users significantly more often reported paresthesia (31% vs. 8%, P less than .001) and weight loss (8% vs. 0%, P = .02).
Other adverse events among topiramate users were fatigue (25%), dry mouth (18%), memory impairment (17%), aphasia (16%), cognitive disorder (16%), and upper respiratory tract infection (12%).
Serious adverse events included altered mood in three patients in the amitriptyline group and one suicide attempt in the topiramate group.
“Given the null outcome in this trial and the adverse events and serious adverse events reported in the amitriptyline and topiramate groups, the data do not show a favorable risk-benefit profile for the use of these therapies in pediatric migraine prevention, at least over the 24-week duration of the trial,” the study authors concluded.
They noted that during their trial, the Food and Drug Administration approved topiramate for the treatment of episodic migraine in adolescents aged 12-17 years.
“Although our trial included patients outside this age range and included those with either episodic or chronic migraine, the trial results suggest that prevention medication for pediatric migraine might be reexamined,” they wrote.
The high placebo response seen in the study suggested the placebo effect might be advantageous for children and adolescents with migraine, they added.
The trial was supported by grants from the National Institute of Neurological Disorders and Stroke and the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The authors had no relevant financial disclosures.
Two commonly used drugs to prevent migraine in adults, amitriptyline and topiramate, proved no better than placebo at preventing migraine in children and adolescents and also were associated with more adverse events in the double-blind, randomized Childhood and Adolescent Migraine Prevention (CHAMP) trial.
The findings of the trial, which was stopped early for futility following a recommendation from a data and safety monitoring board, suggest the adult model of migraine treatment in which both drugs have been effective may not apply to pediatric patients, according to the investigators (N Engl J Med 2016 Oct 27. doi: 10.1056/NEJMoa1610384). The study was published simultaneously with its presentation at the annual meeting of the Child Neurology Society in Vancouver.
The 24-week, multicenter trial involved 328 children and adolescents aged 8-17 years who were diagnosed with migraine with or without aura or chronic migraine without continuous headache. They were randomized on a 2:2:1 ratio to receive oral amitriptyline (n = 132), topiramate (n = 130), or placebo (n = 66) according to age (8-12 years vs. 13-17 years). The target dose in the study was 1 mg/kg of body weight per day for amitriptyline and 2 mg/kg for topiramate. Doses were escalated every 2 weeks over an 8-week period and dose modifications were made according to side effects.
Overall, 52% on amitriptyline, 55% on topiramate, and 61% on placebo reached the study’s primary endpoint of a relative reduction of 50% or more in the number of headache days from baseline to the last 28 days of the trial; the outcomes were not significantly different from one another. The participants recorded the number of headache days they experienced in a diary kept during and after the 28-day baseline period of the study.
No significant differences were seen between the groups in the secondary outcomes of headache-related disability, headache days, or the number of patients who completed the trial.
However, the authors observed higher rates of adverse events overall in the active treatment groups, compared with placebo. Amitriptyline users significantly more often reported fatigue (30% vs. 14%, P = .01) and dry mouth (25% vs. 12%, P = .03), whereas topiramate users significantly more often reported paresthesia (31% vs. 8%, P less than .001) and weight loss (8% vs. 0%, P = .02).
Other adverse events among topiramate users were fatigue (25%), dry mouth (18%), memory impairment (17%), aphasia (16%), cognitive disorder (16%), and upper respiratory tract infection (12%).
Serious adverse events included altered mood in three patients in the amitriptyline group and one suicide attempt in the topiramate group.
“Given the null outcome in this trial and the adverse events and serious adverse events reported in the amitriptyline and topiramate groups, the data do not show a favorable risk-benefit profile for the use of these therapies in pediatric migraine prevention, at least over the 24-week duration of the trial,” the study authors concluded.
They noted that during their trial, the Food and Drug Administration approved topiramate for the treatment of episodic migraine in adolescents aged 12-17 years.
“Although our trial included patients outside this age range and included those with either episodic or chronic migraine, the trial results suggest that prevention medication for pediatric migraine might be reexamined,” they wrote.
The high placebo response seen in the study suggested the placebo effect might be advantageous for children and adolescents with migraine, they added.
The trial was supported by grants from the National Institute of Neurological Disorders and Stroke and the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The authors had no relevant financial disclosures.
FROM CNS 2016
Key clinical point:
Main finding: Fifty-two percent, 55%, and 61% of patients randomized to amitriptyline, topiramate, and placebo groups, respectively, reached the study primary endpoint of a relative reduction of 50% or more in the number of headache days from baseline to the last 28 days of the 24-week trial.
Data source: A phase III, randomized, placebo-controlled trial of 328 children and adolescents aged 8-17 years who were diagnosed with migraine with or without aura or chronic migraine without continuous headache.
Disclosures: The trial was supported by grants from the National Institute of Neurological Disorders and Stroke and the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The authors had no relevant financial disclosures.
Anti–nerve growth factor drug has long-term OA pain benefit, but unclear safety
Treatment for a year with the human anti–nerve growth factor monoclonal antibody fulranumab provided pain relief and functional benefits to patients with moderate to severe chronic osteoarthritis knee or hip pain but continued to show signs that the biologic may contribute to rapid progression of disease in a proportion of patients who came to need joint replacement, especially when used concurrently with nonsteroidal anti-inflammatory drugs.
The findings come from the double-blind extension phase of a 12-week, phase II, placebo-controlled, double-blind, randomized study that found significant reduction in the average pain intensity score (P less than or equal to .030) for patients who took fulranumab (Pain. 2013 Oct;154[10]:1910-9). The investigators wanted to determine the long-term safety of the biologic in light of the Food and Drug Administration’s 2010 “clinical hold” on studies of anti–nerve growth factor (anti-NGF) drugs such as fulranumab after concerns emerged that the class of anti-NGF antibodies may be associated with potential treatment-emergent adverse events (TEAEs) leading to joint destruction.
The long-term safety and efficacy results in the extension phase of the study when fulranumab was given as an adjunctive therapy to standard pain therapy revealed higher rates of serious TEAEs, including joint replacement, that were associated with fulranumab. While most of those TEAEs were independently adjudicated to stem from normal progression of OA, one-fifth of the patients on fulranumab who needed joint replacement had rapid progression of OA (RPOA).
Investigators led by Panna Sanga, MD, director of clinical research at Janssen, sought to determine the effects of the anti-NGF biologic as an adjunctive therapy to standard pain therapy over a 92-week period in 401 patients who had completed the 12-week efficacy study, as well as over a 26-week posttreatment follow-up. In the current extension phase of the trial, patients continued on their randomized dose in the original trial of placebo or subcutaneous fulranumab 1 mg or 3 mg every 4 weeks or fulranumab 3 mg, 6 mg, or 10 mg every 8 weeks, but they were permitted to change their concurrent pain medications as clinically needed.
Although the study intended for patients to receive 2 years of treatment (104 weeks), the FDA’s clinical hold meant that the median duration of exposure to fulranumab in the extension study was only 365-393 days across the dosing regimens, the authors explained (Arthritis Rheumatol. 2016 Oct 16 doi: 10.1002/art.39943).
Overall, 421 (90%) of 466 intent-to-treat patients experienced at least one TEAE during both study phases, with similar incidence between those randomized to placebo (n = 69, 88%) and the fulranumab groups (n = 352, 91%).
TEAEs that occurred with a frequency of 10% or more among all fulranumab-treated patients were arthralgia (21%), OA (18%), paresthesia, and upper respiratory tract infection (13% each), while these were arthralgia (15%), OA (14%), and sinusitis (12%) among patients randomized to placebo.
A total of 109 patients (23%) reported serious TEAEs, including 13 (17%) taking placebo and 96 (25%) taking fulranumab. Serious TEAEs that were reported in 5% or more of patients in the fulranumab groups were knee arthroplasty (n = 38, 10%) and hip arthroplasty (n = 26, 7%). Overall, 81 joint replacements occurred in 71 patients (placebo: n = , 11%; fulranumab: n = 63, 89%).
An independent adjudication committee that the study sponsor, Janssen, established after the study was put on hold ruled that the majority of these joint replacements resulted from the normal progression of OA (n = 56, 79%). However, the adjudication committee determined that 15 (21%) patients in the fulranumab treatment groups had RPOA.
The study authors pointed out that the patients with RPOA regularly used NSAIDs and had a prior history of OA in the affected joint. Nevertheless, because of the small number of RPOA cases per treatment group, a drug or dose effect for RPOA could not be evaluated, they said.
“Future studies are warranted to demonstrate whether limiting the use of concomitant chronic NSAIDs and using only lower doses of fulranumab may reduce the risk of RPOA,” they wrote.
The extension study also looked at the longer-term efficacy of fulranumab. Efficacy endpoints on the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain and physical function subscales and the Patient Global Assessment (PGA) scores that looked at changes from baseline showed that in comparison with placebo, dosing regimens of fulranumab 3 mg every 4 weeks and 10 mg every 8 weeks gave “continued effective relief of the pain associated with knee and hip OA as early as week 4 and was maintained up to week 53,” the researchers reported.
These results were further corroborated by improvements in physical function on the WOMAC physical function subscale and PGA scale scores, which suggested “sustained efficacy of long-term fulranumab treatment,” they said.
All authors except one were employees of Janssen.
Treatment for a year with the human anti–nerve growth factor monoclonal antibody fulranumab provided pain relief and functional benefits to patients with moderate to severe chronic osteoarthritis knee or hip pain but continued to show signs that the biologic may contribute to rapid progression of disease in a proportion of patients who came to need joint replacement, especially when used concurrently with nonsteroidal anti-inflammatory drugs.
The findings come from the double-blind extension phase of a 12-week, phase II, placebo-controlled, double-blind, randomized study that found significant reduction in the average pain intensity score (P less than or equal to .030) for patients who took fulranumab (Pain. 2013 Oct;154[10]:1910-9). The investigators wanted to determine the long-term safety of the biologic in light of the Food and Drug Administration’s 2010 “clinical hold” on studies of anti–nerve growth factor (anti-NGF) drugs such as fulranumab after concerns emerged that the class of anti-NGF antibodies may be associated with potential treatment-emergent adverse events (TEAEs) leading to joint destruction.
The long-term safety and efficacy results in the extension phase of the study when fulranumab was given as an adjunctive therapy to standard pain therapy revealed higher rates of serious TEAEs, including joint replacement, that were associated with fulranumab. While most of those TEAEs were independently adjudicated to stem from normal progression of OA, one-fifth of the patients on fulranumab who needed joint replacement had rapid progression of OA (RPOA).
Investigators led by Panna Sanga, MD, director of clinical research at Janssen, sought to determine the effects of the anti-NGF biologic as an adjunctive therapy to standard pain therapy over a 92-week period in 401 patients who had completed the 12-week efficacy study, as well as over a 26-week posttreatment follow-up. In the current extension phase of the trial, patients continued on their randomized dose in the original trial of placebo or subcutaneous fulranumab 1 mg or 3 mg every 4 weeks or fulranumab 3 mg, 6 mg, or 10 mg every 8 weeks, but they were permitted to change their concurrent pain medications as clinically needed.
Although the study intended for patients to receive 2 years of treatment (104 weeks), the FDA’s clinical hold meant that the median duration of exposure to fulranumab in the extension study was only 365-393 days across the dosing regimens, the authors explained (Arthritis Rheumatol. 2016 Oct 16 doi: 10.1002/art.39943).
Overall, 421 (90%) of 466 intent-to-treat patients experienced at least one TEAE during both study phases, with similar incidence between those randomized to placebo (n = 69, 88%) and the fulranumab groups (n = 352, 91%).
TEAEs that occurred with a frequency of 10% or more among all fulranumab-treated patients were arthralgia (21%), OA (18%), paresthesia, and upper respiratory tract infection (13% each), while these were arthralgia (15%), OA (14%), and sinusitis (12%) among patients randomized to placebo.
A total of 109 patients (23%) reported serious TEAEs, including 13 (17%) taking placebo and 96 (25%) taking fulranumab. Serious TEAEs that were reported in 5% or more of patients in the fulranumab groups were knee arthroplasty (n = 38, 10%) and hip arthroplasty (n = 26, 7%). Overall, 81 joint replacements occurred in 71 patients (placebo: n = , 11%; fulranumab: n = 63, 89%).
An independent adjudication committee that the study sponsor, Janssen, established after the study was put on hold ruled that the majority of these joint replacements resulted from the normal progression of OA (n = 56, 79%). However, the adjudication committee determined that 15 (21%) patients in the fulranumab treatment groups had RPOA.
The study authors pointed out that the patients with RPOA regularly used NSAIDs and had a prior history of OA in the affected joint. Nevertheless, because of the small number of RPOA cases per treatment group, a drug or dose effect for RPOA could not be evaluated, they said.
“Future studies are warranted to demonstrate whether limiting the use of concomitant chronic NSAIDs and using only lower doses of fulranumab may reduce the risk of RPOA,” they wrote.
The extension study also looked at the longer-term efficacy of fulranumab. Efficacy endpoints on the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain and physical function subscales and the Patient Global Assessment (PGA) scores that looked at changes from baseline showed that in comparison with placebo, dosing regimens of fulranumab 3 mg every 4 weeks and 10 mg every 8 weeks gave “continued effective relief of the pain associated with knee and hip OA as early as week 4 and was maintained up to week 53,” the researchers reported.
These results were further corroborated by improvements in physical function on the WOMAC physical function subscale and PGA scale scores, which suggested “sustained efficacy of long-term fulranumab treatment,” they said.
All authors except one were employees of Janssen.
Treatment for a year with the human anti–nerve growth factor monoclonal antibody fulranumab provided pain relief and functional benefits to patients with moderate to severe chronic osteoarthritis knee or hip pain but continued to show signs that the biologic may contribute to rapid progression of disease in a proportion of patients who came to need joint replacement, especially when used concurrently with nonsteroidal anti-inflammatory drugs.
The findings come from the double-blind extension phase of a 12-week, phase II, placebo-controlled, double-blind, randomized study that found significant reduction in the average pain intensity score (P less than or equal to .030) for patients who took fulranumab (Pain. 2013 Oct;154[10]:1910-9). The investigators wanted to determine the long-term safety of the biologic in light of the Food and Drug Administration’s 2010 “clinical hold” on studies of anti–nerve growth factor (anti-NGF) drugs such as fulranumab after concerns emerged that the class of anti-NGF antibodies may be associated with potential treatment-emergent adverse events (TEAEs) leading to joint destruction.
The long-term safety and efficacy results in the extension phase of the study when fulranumab was given as an adjunctive therapy to standard pain therapy revealed higher rates of serious TEAEs, including joint replacement, that were associated with fulranumab. While most of those TEAEs were independently adjudicated to stem from normal progression of OA, one-fifth of the patients on fulranumab who needed joint replacement had rapid progression of OA (RPOA).
Investigators led by Panna Sanga, MD, director of clinical research at Janssen, sought to determine the effects of the anti-NGF biologic as an adjunctive therapy to standard pain therapy over a 92-week period in 401 patients who had completed the 12-week efficacy study, as well as over a 26-week posttreatment follow-up. In the current extension phase of the trial, patients continued on their randomized dose in the original trial of placebo or subcutaneous fulranumab 1 mg or 3 mg every 4 weeks or fulranumab 3 mg, 6 mg, or 10 mg every 8 weeks, but they were permitted to change their concurrent pain medications as clinically needed.
Although the study intended for patients to receive 2 years of treatment (104 weeks), the FDA’s clinical hold meant that the median duration of exposure to fulranumab in the extension study was only 365-393 days across the dosing regimens, the authors explained (Arthritis Rheumatol. 2016 Oct 16 doi: 10.1002/art.39943).
Overall, 421 (90%) of 466 intent-to-treat patients experienced at least one TEAE during both study phases, with similar incidence between those randomized to placebo (n = 69, 88%) and the fulranumab groups (n = 352, 91%).
TEAEs that occurred with a frequency of 10% or more among all fulranumab-treated patients were arthralgia (21%), OA (18%), paresthesia, and upper respiratory tract infection (13% each), while these were arthralgia (15%), OA (14%), and sinusitis (12%) among patients randomized to placebo.
A total of 109 patients (23%) reported serious TEAEs, including 13 (17%) taking placebo and 96 (25%) taking fulranumab. Serious TEAEs that were reported in 5% or more of patients in the fulranumab groups were knee arthroplasty (n = 38, 10%) and hip arthroplasty (n = 26, 7%). Overall, 81 joint replacements occurred in 71 patients (placebo: n = , 11%; fulranumab: n = 63, 89%).
An independent adjudication committee that the study sponsor, Janssen, established after the study was put on hold ruled that the majority of these joint replacements resulted from the normal progression of OA (n = 56, 79%). However, the adjudication committee determined that 15 (21%) patients in the fulranumab treatment groups had RPOA.
The study authors pointed out that the patients with RPOA regularly used NSAIDs and had a prior history of OA in the affected joint. Nevertheless, because of the small number of RPOA cases per treatment group, a drug or dose effect for RPOA could not be evaluated, they said.
“Future studies are warranted to demonstrate whether limiting the use of concomitant chronic NSAIDs and using only lower doses of fulranumab may reduce the risk of RPOA,” they wrote.
The extension study also looked at the longer-term efficacy of fulranumab. Efficacy endpoints on the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain and physical function subscales and the Patient Global Assessment (PGA) scores that looked at changes from baseline showed that in comparison with placebo, dosing regimens of fulranumab 3 mg every 4 weeks and 10 mg every 8 weeks gave “continued effective relief of the pain associated with knee and hip OA as early as week 4 and was maintained up to week 53,” the researchers reported.
These results were further corroborated by improvements in physical function on the WOMAC physical function subscale and PGA scale scores, which suggested “sustained efficacy of long-term fulranumab treatment,” they said.
All authors except one were employees of Janssen.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point:
Major finding: A total of 81 joint replacements occurred in 71 patients (placebo: n = 8, 11%; fulranumab: n = 63, 89%).
Data source: Phase II randomized double-blind extension study of 401 patients with moderate to severe chronic OA knee or hip pain.
Disclosures: All authors except one were employees of Janssen, which funded the study.
Younger age, inconsistency of care affect antiviral adherence in hepatitis B
Patients with chronic hepatitis B who are aged under 35 years or do not see the same clinician consistently are more likely to not take their antiviral medication, finds the largest study of its kind to date.
According to the authors of the research led by Nicole Allard, PhD, from the WHO collaborative Centre for Viral Hepatitis and the Peter Doherty Institute for Infection and Immunity in Melbourne, their findings illustrate the need for clinicians to reinforce the importance of medication adherence at each patient consultation.
Furthermore, “providing more flexible and convenient care arrangements, reminder systems, and working with the individual to maximize medication adherence should be a routine part of clinical practice,” they wrote in their paper published in the Journal of Viral Hepatitis. Previous research had shown that adherence to antiviral therapy in the treatment of hepatitis B was associated with improved patient outcomes. Suboptimal adherence could lead to antiviral resistance or antiviral breakthrough, potentially leading to liver cirrhosis and liver cancer.
Understanding poor adherence in clinical settings, together with the factors associated with lower adherence, was important in informing efforts for promoting adherence, the research team said (J Viral Hepat. 2016. doi: 10.1111/jvh.12582).
In their retrospective study they assessed the pharmacy records of 1,026 patients who were dispensed antiviral therapy for hepatitis B across four Australian public hospital outpatient clinics between 2010 and 2013.
They discovered that one in five patients were “nonadherent” to therapy based on a medication possession ratio of less than 0.90 – a cutoff shown in previous studies to be correlated with virologic breakthrough.
One significant factor that affected adherence to medication was being aged under 35 years (P = .002), the research team reported.
This finding was similar to other studies of chronic diseases that had shown younger patients were at a higher risk of nonadherence, the authors noted.
“Recognition of the challenges faced by a younger person regularly taking medication at a time in their lives when they feel well is important,” they wrote.
Poor adherence was also associated with seeing multiple clinicians over shorter periods of time.
“While hospitals accommodate trainees and staffing changes occur regularly, seeking to maximize the consistency of clinical care delivery may be an important strategy to support better adherence and optimizing care for individuals,” they said.
Factors such as variations in the amount of medication, duration of treatment, sex, and cultural background did not affect adherence to medication.
“Adherence is a dynamic process and needs to be addressed regularly in a nonjudgmental way working with individuals towards improved health,” they concluded.
Patients with chronic hepatitis B who are aged under 35 years or do not see the same clinician consistently are more likely to not take their antiviral medication, finds the largest study of its kind to date.
According to the authors of the research led by Nicole Allard, PhD, from the WHO collaborative Centre for Viral Hepatitis and the Peter Doherty Institute for Infection and Immunity in Melbourne, their findings illustrate the need for clinicians to reinforce the importance of medication adherence at each patient consultation.
Furthermore, “providing more flexible and convenient care arrangements, reminder systems, and working with the individual to maximize medication adherence should be a routine part of clinical practice,” they wrote in their paper published in the Journal of Viral Hepatitis. Previous research had shown that adherence to antiviral therapy in the treatment of hepatitis B was associated with improved patient outcomes. Suboptimal adherence could lead to antiviral resistance or antiviral breakthrough, potentially leading to liver cirrhosis and liver cancer.
Understanding poor adherence in clinical settings, together with the factors associated with lower adherence, was important in informing efforts for promoting adherence, the research team said (J Viral Hepat. 2016. doi: 10.1111/jvh.12582).
In their retrospective study they assessed the pharmacy records of 1,026 patients who were dispensed antiviral therapy for hepatitis B across four Australian public hospital outpatient clinics between 2010 and 2013.
They discovered that one in five patients were “nonadherent” to therapy based on a medication possession ratio of less than 0.90 – a cutoff shown in previous studies to be correlated with virologic breakthrough.
One significant factor that affected adherence to medication was being aged under 35 years (P = .002), the research team reported.
This finding was similar to other studies of chronic diseases that had shown younger patients were at a higher risk of nonadherence, the authors noted.
“Recognition of the challenges faced by a younger person regularly taking medication at a time in their lives when they feel well is important,” they wrote.
Poor adherence was also associated with seeing multiple clinicians over shorter periods of time.
“While hospitals accommodate trainees and staffing changes occur regularly, seeking to maximize the consistency of clinical care delivery may be an important strategy to support better adherence and optimizing care for individuals,” they said.
Factors such as variations in the amount of medication, duration of treatment, sex, and cultural background did not affect adherence to medication.
“Adherence is a dynamic process and needs to be addressed regularly in a nonjudgmental way working with individuals towards improved health,” they concluded.
Patients with chronic hepatitis B who are aged under 35 years or do not see the same clinician consistently are more likely to not take their antiviral medication, finds the largest study of its kind to date.
According to the authors of the research led by Nicole Allard, PhD, from the WHO collaborative Centre for Viral Hepatitis and the Peter Doherty Institute for Infection and Immunity in Melbourne, their findings illustrate the need for clinicians to reinforce the importance of medication adherence at each patient consultation.
Furthermore, “providing more flexible and convenient care arrangements, reminder systems, and working with the individual to maximize medication adherence should be a routine part of clinical practice,” they wrote in their paper published in the Journal of Viral Hepatitis. Previous research had shown that adherence to antiviral therapy in the treatment of hepatitis B was associated with improved patient outcomes. Suboptimal adherence could lead to antiviral resistance or antiviral breakthrough, potentially leading to liver cirrhosis and liver cancer.
Understanding poor adherence in clinical settings, together with the factors associated with lower adherence, was important in informing efforts for promoting adherence, the research team said (J Viral Hepat. 2016. doi: 10.1111/jvh.12582).
In their retrospective study they assessed the pharmacy records of 1,026 patients who were dispensed antiviral therapy for hepatitis B across four Australian public hospital outpatient clinics between 2010 and 2013.
They discovered that one in five patients were “nonadherent” to therapy based on a medication possession ratio of less than 0.90 – a cutoff shown in previous studies to be correlated with virologic breakthrough.
One significant factor that affected adherence to medication was being aged under 35 years (P = .002), the research team reported.
This finding was similar to other studies of chronic diseases that had shown younger patients were at a higher risk of nonadherence, the authors noted.
“Recognition of the challenges faced by a younger person regularly taking medication at a time in their lives when they feel well is important,” they wrote.
Poor adherence was also associated with seeing multiple clinicians over shorter periods of time.
“While hospitals accommodate trainees and staffing changes occur regularly, seeking to maximize the consistency of clinical care delivery may be an important strategy to support better adherence and optimizing care for individuals,” they said.
Factors such as variations in the amount of medication, duration of treatment, sex, and cultural background did not affect adherence to medication.
“Adherence is a dynamic process and needs to be addressed regularly in a nonjudgmental way working with individuals towards improved health,” they concluded.
FROM THE JOURNAL OF VIRAL HEPATITIS
Key clinical point: Adherence to antiviral medication is a dynamic process that clinicians need to address regularly in a nonjudgmental way at each consultation, particularly in younger patients. Providing flexible and convenient care arrangements that foster continuity of care will also improve adherence to medication.
Major finding: One in five patients with hepatitis B were nonadherent to antiviral medications. Younger age (under 35) (P = .002), and inconsistency of care predicted nonadherence.
Data Source: A retrospective study of the pharmacy records of 1,026 patients who were attending a public hospital clinic and were prescribed antiviral therapy for hepatitis B.
Disclosures: Dr. Nicole Allard is supported by an APA scholarship from the University of Melbourne. Funding for the project was provided by the Royal Melbourne Hospital lottery grant. Dr. Allard declared no conflicts of interest in the last 3 years. Several of the authors in the group reported financial ties to pharmaceutical companies.
Deutetrabenazine modestly reduces chorea of Huntington disease
A deuterated form of the vesicular monoamine transporter type 2 inhibitor tetrabenazine, called deutetrabenazine, modestly reduced chorea of Huntington disease over 12 weeks in a phase III, double-blind, randomized, placebo-controlled trial.
First author Samuel Frank, MD, of Beth Israel Deaconess Medical Center in Boston and his colleagues in the Huntington Study Group wrote that the observed treatment effect of 2.5 points on the primary end point of total maximal chorea score, “taken together with improvements in patient-centered end points, such as PGIC [Patient Global Impression of Change] and SF-36 [36-Item Short Form] physical functioning component scales, may be of clinical relevance, although this remains to be determined. The difference in total maximal chorea score associated with deutetrabenazine treatment that was observed in this study is notable given the progressive decline in total maximal chorea score and total motor score that has been previously described as part of the natural history of Huntington disease.”
The phase III trial is the first to use an active ingredient with deuterium substituted for hydrogen at key positions. The use of deuterium in the molecule, according to the investigators, prolongs “plasma half-life and reduce[s] metabolic variability, without changing target pharmacology,” and its “longer half-life and unique pharmacokinetic profile ... may enable less frequent and lower daily doses, thus achieving similar systemic exposure with lower peak concentrations and simplified dosing, compared with tetrabenazine.”
Tetrabenazine (Xenazine) is used worldwide and is the only U.S. Food and Drug Administration–approved therapy for treating chorea associated with Huntington disease.
The trial enrolled 90 patients with genetically diagnosed Huntington disease across 34 centers in the United States and Canada. The patients had a baseline total maximal chorea score of 8 or higher (range 0-28) and were randomized on a 1:1 ratio to receive deuterated tetrabenazine (deutetrabenazine) or placebo. The deutetrabenazine group had a mean age of 55 years and 49% were male. The placebo group had a mean age of 52 years and 62% were men. Patients on the active drug were commenced at a total daily dose of 6 mg (divided into two doses) for the first week. Doses were then increased weekly by 6 mg daily until either chorea was adequately controlled or the maximum dose of 48 mg a day was reached. The final dose was maintained for an additional 4 weeks for a total of 12 weeks, followed by a 1-week washout (JAMA 2016;316[1]:40-50. doi: 10.1001/jama.2016.8655).
The total maximal chorea score, in which higher scores represent worse chorea, that was measured from baseline to an average across the maintenance period during weeks 9 through 12 of therapy declined by a statistically significant 4.4 points – from a mean of 12.1 to 7.7 – in the deutetrabenazine group, compared with a decline of 1.9 points – from a mean of 13.2 to 11.3 – in the placebo group.
Improvements in secondary endpoints such as PGIC and SF-36 physical functioning component scales were also seen in the treatment group, although no improvement was seen in the Berg Balance Test.
Adverse events were similar between the deutetrabenazine and placebo groups, including depression, anxiety, and akathisia.
“Further research is needed to assess the clinical importance of the effect size, and to determine longer-term efficacy and safety,” the study authors concluded.
The study was supported by Auspex Pharmaceuticals, a wholly owned subsidiary of Teva Pharmaceutical Industries. Dr. Frank reported receiving grants from the Huntington Study Group, and many other authors in the group reported financial ties to pharmaceutical companies, including Auspex and Teva. Several authors are employees of Auspex.
The study demonstrated a proof of principle that deutetrabenazine, compared with placebo, provided improvement on chorea over 12 weeks and allowed less frequent drug dosing with fewer adverse effects, but it’s not possible to determine how the drug compares with tetrabenazine.
An ideal trial would involve three groups comparing deutetrabenazine, tetrabenazine, and placebo. A head-to-head, noninferiority comparison of deutetrabenazine against tetrabenazine, as was recommended by one of the developers of the compound, would also have been useful.
Nevertheless, tetrabenazine was approved by the FDA for the treatment of chorea in Huntington disease essentially based on efficacy data similar to the current study. Assuming deutetrabenazine was not priced significantly higher than tetrabenazine, its “favorable profile” would offer an additional option for patients and clinicians if and when the drug is approved.
The ongoing ARC-HD (Alternatives for Reducing Chorea in Huntington Disease) study is examining the safety and tolerability of patients with Huntington disease who switch from tetrabenazine to deutetrabenazine (and also includes patients from the current deutetrabenazine study who chose to switch to open-label deutetrabenazine). It should help to resolve whether the effect sizes are clinically meaningful and stable over a period longer than 12 weeks.
Michael D. Geschwind, MD, PhD, and Nick Paras, PhD, are in the department of neurology at the University of California, San Francisco. Dr. Geschwind reported receiving grants from the National Institute on Aging and from many sources in industry, including travel support from Teva. Dr. Paras reported receiving research support from Daiichi-Sankyo, the U.S. Department of Defense, and the Tau Consortium. Their comments are derived from an editorial accompanying the phase III trial’s paper (JAMA. 2016;316[1]:33-5).
The study demonstrated a proof of principle that deutetrabenazine, compared with placebo, provided improvement on chorea over 12 weeks and allowed less frequent drug dosing with fewer adverse effects, but it’s not possible to determine how the drug compares with tetrabenazine.
An ideal trial would involve three groups comparing deutetrabenazine, tetrabenazine, and placebo. A head-to-head, noninferiority comparison of deutetrabenazine against tetrabenazine, as was recommended by one of the developers of the compound, would also have been useful.
Nevertheless, tetrabenazine was approved by the FDA for the treatment of chorea in Huntington disease essentially based on efficacy data similar to the current study. Assuming deutetrabenazine was not priced significantly higher than tetrabenazine, its “favorable profile” would offer an additional option for patients and clinicians if and when the drug is approved.
The ongoing ARC-HD (Alternatives for Reducing Chorea in Huntington Disease) study is examining the safety and tolerability of patients with Huntington disease who switch from tetrabenazine to deutetrabenazine (and also includes patients from the current deutetrabenazine study who chose to switch to open-label deutetrabenazine). It should help to resolve whether the effect sizes are clinically meaningful and stable over a period longer than 12 weeks.
Michael D. Geschwind, MD, PhD, and Nick Paras, PhD, are in the department of neurology at the University of California, San Francisco. Dr. Geschwind reported receiving grants from the National Institute on Aging and from many sources in industry, including travel support from Teva. Dr. Paras reported receiving research support from Daiichi-Sankyo, the U.S. Department of Defense, and the Tau Consortium. Their comments are derived from an editorial accompanying the phase III trial’s paper (JAMA. 2016;316[1]:33-5).
The study demonstrated a proof of principle that deutetrabenazine, compared with placebo, provided improvement on chorea over 12 weeks and allowed less frequent drug dosing with fewer adverse effects, but it’s not possible to determine how the drug compares with tetrabenazine.
An ideal trial would involve three groups comparing deutetrabenazine, tetrabenazine, and placebo. A head-to-head, noninferiority comparison of deutetrabenazine against tetrabenazine, as was recommended by one of the developers of the compound, would also have been useful.
Nevertheless, tetrabenazine was approved by the FDA for the treatment of chorea in Huntington disease essentially based on efficacy data similar to the current study. Assuming deutetrabenazine was not priced significantly higher than tetrabenazine, its “favorable profile” would offer an additional option for patients and clinicians if and when the drug is approved.
The ongoing ARC-HD (Alternatives for Reducing Chorea in Huntington Disease) study is examining the safety and tolerability of patients with Huntington disease who switch from tetrabenazine to deutetrabenazine (and also includes patients from the current deutetrabenazine study who chose to switch to open-label deutetrabenazine). It should help to resolve whether the effect sizes are clinically meaningful and stable over a period longer than 12 weeks.
Michael D. Geschwind, MD, PhD, and Nick Paras, PhD, are in the department of neurology at the University of California, San Francisco. Dr. Geschwind reported receiving grants from the National Institute on Aging and from many sources in industry, including travel support from Teva. Dr. Paras reported receiving research support from Daiichi-Sankyo, the U.S. Department of Defense, and the Tau Consortium. Their comments are derived from an editorial accompanying the phase III trial’s paper (JAMA. 2016;316[1]:33-5).
A deuterated form of the vesicular monoamine transporter type 2 inhibitor tetrabenazine, called deutetrabenazine, modestly reduced chorea of Huntington disease over 12 weeks in a phase III, double-blind, randomized, placebo-controlled trial.
First author Samuel Frank, MD, of Beth Israel Deaconess Medical Center in Boston and his colleagues in the Huntington Study Group wrote that the observed treatment effect of 2.5 points on the primary end point of total maximal chorea score, “taken together with improvements in patient-centered end points, such as PGIC [Patient Global Impression of Change] and SF-36 [36-Item Short Form] physical functioning component scales, may be of clinical relevance, although this remains to be determined. The difference in total maximal chorea score associated with deutetrabenazine treatment that was observed in this study is notable given the progressive decline in total maximal chorea score and total motor score that has been previously described as part of the natural history of Huntington disease.”
The phase III trial is the first to use an active ingredient with deuterium substituted for hydrogen at key positions. The use of deuterium in the molecule, according to the investigators, prolongs “plasma half-life and reduce[s] metabolic variability, without changing target pharmacology,” and its “longer half-life and unique pharmacokinetic profile ... may enable less frequent and lower daily doses, thus achieving similar systemic exposure with lower peak concentrations and simplified dosing, compared with tetrabenazine.”
Tetrabenazine (Xenazine) is used worldwide and is the only U.S. Food and Drug Administration–approved therapy for treating chorea associated with Huntington disease.
The trial enrolled 90 patients with genetically diagnosed Huntington disease across 34 centers in the United States and Canada. The patients had a baseline total maximal chorea score of 8 or higher (range 0-28) and were randomized on a 1:1 ratio to receive deuterated tetrabenazine (deutetrabenazine) or placebo. The deutetrabenazine group had a mean age of 55 years and 49% were male. The placebo group had a mean age of 52 years and 62% were men. Patients on the active drug were commenced at a total daily dose of 6 mg (divided into two doses) for the first week. Doses were then increased weekly by 6 mg daily until either chorea was adequately controlled or the maximum dose of 48 mg a day was reached. The final dose was maintained for an additional 4 weeks for a total of 12 weeks, followed by a 1-week washout (JAMA 2016;316[1]:40-50. doi: 10.1001/jama.2016.8655).
The total maximal chorea score, in which higher scores represent worse chorea, that was measured from baseline to an average across the maintenance period during weeks 9 through 12 of therapy declined by a statistically significant 4.4 points – from a mean of 12.1 to 7.7 – in the deutetrabenazine group, compared with a decline of 1.9 points – from a mean of 13.2 to 11.3 – in the placebo group.
Improvements in secondary endpoints such as PGIC and SF-36 physical functioning component scales were also seen in the treatment group, although no improvement was seen in the Berg Balance Test.
Adverse events were similar between the deutetrabenazine and placebo groups, including depression, anxiety, and akathisia.
“Further research is needed to assess the clinical importance of the effect size, and to determine longer-term efficacy and safety,” the study authors concluded.
The study was supported by Auspex Pharmaceuticals, a wholly owned subsidiary of Teva Pharmaceutical Industries. Dr. Frank reported receiving grants from the Huntington Study Group, and many other authors in the group reported financial ties to pharmaceutical companies, including Auspex and Teva. Several authors are employees of Auspex.
A deuterated form of the vesicular monoamine transporter type 2 inhibitor tetrabenazine, called deutetrabenazine, modestly reduced chorea of Huntington disease over 12 weeks in a phase III, double-blind, randomized, placebo-controlled trial.
First author Samuel Frank, MD, of Beth Israel Deaconess Medical Center in Boston and his colleagues in the Huntington Study Group wrote that the observed treatment effect of 2.5 points on the primary end point of total maximal chorea score, “taken together with improvements in patient-centered end points, such as PGIC [Patient Global Impression of Change] and SF-36 [36-Item Short Form] physical functioning component scales, may be of clinical relevance, although this remains to be determined. The difference in total maximal chorea score associated with deutetrabenazine treatment that was observed in this study is notable given the progressive decline in total maximal chorea score and total motor score that has been previously described as part of the natural history of Huntington disease.”
The phase III trial is the first to use an active ingredient with deuterium substituted for hydrogen at key positions. The use of deuterium in the molecule, according to the investigators, prolongs “plasma half-life and reduce[s] metabolic variability, without changing target pharmacology,” and its “longer half-life and unique pharmacokinetic profile ... may enable less frequent and lower daily doses, thus achieving similar systemic exposure with lower peak concentrations and simplified dosing, compared with tetrabenazine.”
Tetrabenazine (Xenazine) is used worldwide and is the only U.S. Food and Drug Administration–approved therapy for treating chorea associated with Huntington disease.
The trial enrolled 90 patients with genetically diagnosed Huntington disease across 34 centers in the United States and Canada. The patients had a baseline total maximal chorea score of 8 or higher (range 0-28) and were randomized on a 1:1 ratio to receive deuterated tetrabenazine (deutetrabenazine) or placebo. The deutetrabenazine group had a mean age of 55 years and 49% were male. The placebo group had a mean age of 52 years and 62% were men. Patients on the active drug were commenced at a total daily dose of 6 mg (divided into two doses) for the first week. Doses were then increased weekly by 6 mg daily until either chorea was adequately controlled or the maximum dose of 48 mg a day was reached. The final dose was maintained for an additional 4 weeks for a total of 12 weeks, followed by a 1-week washout (JAMA 2016;316[1]:40-50. doi: 10.1001/jama.2016.8655).
The total maximal chorea score, in which higher scores represent worse chorea, that was measured from baseline to an average across the maintenance period during weeks 9 through 12 of therapy declined by a statistically significant 4.4 points – from a mean of 12.1 to 7.7 – in the deutetrabenazine group, compared with a decline of 1.9 points – from a mean of 13.2 to 11.3 – in the placebo group.
Improvements in secondary endpoints such as PGIC and SF-36 physical functioning component scales were also seen in the treatment group, although no improvement was seen in the Berg Balance Test.
Adverse events were similar between the deutetrabenazine and placebo groups, including depression, anxiety, and akathisia.
“Further research is needed to assess the clinical importance of the effect size, and to determine longer-term efficacy and safety,” the study authors concluded.
The study was supported by Auspex Pharmaceuticals, a wholly owned subsidiary of Teva Pharmaceutical Industries. Dr. Frank reported receiving grants from the Huntington Study Group, and many other authors in the group reported financial ties to pharmaceutical companies, including Auspex and Teva. Several authors are employees of Auspex.
FROM JAMA
Key clinical point: Use of a deuterated form of tetrabenazine resulted in modest reductions in chorea of Huntington disease over 12 weeks, compared with placebo.
Major finding: The total maximal chorea score measured from baseline to maintenance therapy was reduced by a statistically significant 4.4 points in patients taking deutetrabenazine, compared with 1.9 point in those taking placebo.
Data source: A double-blind, randomized, placebo-controlled trial involving 90 patients genetically diagnosed with Huntington disease.
Disclosures: The study was supported by Auspex Pharmaceuticals, a wholly owned subsidiary of Teva Pharmaceutical Industries. Dr. Frank reported receiving grants from the Huntington Study Group, and many other authors in the group reported financial ties to pharmaceutical companies, including Auspex and Teva. Several authors are employees of Auspex.