User login
-
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
Wait, a Health Worker Surplus? Workforce Report Projects Big Surprises
A surprising new report by the Mercer consulting firm projects that the American healthcare workforce will face a small shortfall in 2028 — a shortage of less than 1% of all employees.
Mercer’s projections are rosier than federal workforce projections, which paint a grimmer picture of impending shortages.
“The labor market is a little more stabilized right now, and most healthcare systems are seeing less turnover,” Dan Lezotte, PhD, a partner with Mercer, said in an interview. But he noted “critical shortages” are still expected in some areas.
Mercer last projected workforce numbers in a 2020-2021 report released during the height of the COVID-19 pandemic. Now, “the labor market is drastically different,” Dr. Lezotte said. Health workforce shortages and surpluses have long varied significantly by region across the country.
The report forecasts a small surplus of physicians in 2028 but not in states such as California, New York, and Texas. The upper Midwest states will largely see doctor surpluses while Southern states face shortages. Some states with general physician surpluses may still experience shortages of specialists.
A surplus of nearly 30,000 registered nurses is expected, but New York, New Jersey, and Connecticut are projected to have a combined shortage of 16,000 nurses.
Overall, the report projects a shortage of more than 100,000 healthcare workers nationally by 2028. That’s less than 1% of the entire healthcare workforce of 18.6 million expected by then.
The report also predicts a shortage of nurse practitioners, especially in California and New York, and a shortage of 73,000 nursing assistants, especially in California, New York, and Texas.
“Healthcare systems are having the most difficulty hiring and hanging on to those workers who are supposed to take up the load off physicians and nurses,” Dr. Lezotte said. “They’re competing not only with other healthcare systems but with other industries like Amazon warehouses or McDonald’s in California paying $20 an hour. Healthcare was a little slow to keep up with that. In a lot of healthcare systems, that’s their biggest headache right now.”
On the other hand, the report projects a national surplus of 48,000 home health/personal care aides.
That surprised Bianca K. Frogner, PhD, director of the Center for Health Workforce Studies at the University of Washington, Seattle.
“We are seeing increasing movement of investments toward moving patients out of skilled nursing facilities and keeping them in the home and community, which requires many more home health aides,” Dr. Frogner said. “Given such high turnover in this occupation, it’s hard to know if the surplus is really a surplus or if they will quickly be employed.”
Dr. Frogner receives grants and contracts from not-for-profit entities to investigate issues related to the health workforce.
Dr. Lezotte said the report’s findings are based on data from sources such as public and private databases and job postings. According to the report, “projections were made up to 2028 based on historical data up to 2023,” and “supply projections were derived using a linear autoregressive model based on historical supply within each occupation and geography.”
It’s not clear why some states like New York are expected to have huge shortages, but migration might be a factor, along with a lack of nearby nursing schools, Dr. Lezotte said.
As for shortages, Dr. Lezotte said healthcare systems will have to understand their local workforce situation and adapt. “They’ll need to be more proactive about their employee value proposition” via competitive pay and benefits Flexibility regarding scheduling is also important.
“They’re going to have to figure out how to up their game,” he said.
What about states with surpluses? They might be target-rich environments for states facing shortages, he said.
Positive Outlook Not Shared by Other Researchers
Other workforce projections conflict with Mercer’s, according to Jean Moore, DrPH, and Gaetano Forte, MS, director and assistant director of the Center for Health Workforce Studies, School of Public Health, University at Albany, New York.
The National Center for Health Workforce Analysis projects a 10% shortage of registered nurses and a 13% shortage of physicians in 2031. The agency didn’t make projections for home health aides because that workforce is in flux.
Why are Mercer’s projections so different? Dr. Lezotte said other projections assume that equity efforts will bring healthcare to everyone who needs it. The report assumes this won’t happen, he said. As a result, it expects there will be fewer patients who need to be served by workers.
Other projections expect a shortage of 300,000 registered nurses by 2035, Mr. Forte said. But the number of nurse practitioners in New York is growing quickly, Dr. Moore said.
Dr. Moore said it’s difficult to interpret Mercer’s findings because the company doesn’t provide enough information about its methodology.
“At some level, it’s not particularly useful regarding what the next steps are,” she said. “Projections should make you think about what you should do to change and improve, to create more of what you need.”
The Center for Health Workforce Studies at the University of Albany has provided consulting services to multiple companies that provide healthcare workforce projections. It has no relationship with Mercer.
A version of this article first appeared on Medscape.com.
A surprising new report by the Mercer consulting firm projects that the American healthcare workforce will face a small shortfall in 2028 — a shortage of less than 1% of all employees.
Mercer’s projections are rosier than federal workforce projections, which paint a grimmer picture of impending shortages.
“The labor market is a little more stabilized right now, and most healthcare systems are seeing less turnover,” Dan Lezotte, PhD, a partner with Mercer, said in an interview. But he noted “critical shortages” are still expected in some areas.
Mercer last projected workforce numbers in a 2020-2021 report released during the height of the COVID-19 pandemic. Now, “the labor market is drastically different,” Dr. Lezotte said. Health workforce shortages and surpluses have long varied significantly by region across the country.
The report forecasts a small surplus of physicians in 2028 but not in states such as California, New York, and Texas. The upper Midwest states will largely see doctor surpluses while Southern states face shortages. Some states with general physician surpluses may still experience shortages of specialists.
A surplus of nearly 30,000 registered nurses is expected, but New York, New Jersey, and Connecticut are projected to have a combined shortage of 16,000 nurses.
Overall, the report projects a shortage of more than 100,000 healthcare workers nationally by 2028. That’s less than 1% of the entire healthcare workforce of 18.6 million expected by then.
The report also predicts a shortage of nurse practitioners, especially in California and New York, and a shortage of 73,000 nursing assistants, especially in California, New York, and Texas.
“Healthcare systems are having the most difficulty hiring and hanging on to those workers who are supposed to take up the load off physicians and nurses,” Dr. Lezotte said. “They’re competing not only with other healthcare systems but with other industries like Amazon warehouses or McDonald’s in California paying $20 an hour. Healthcare was a little slow to keep up with that. In a lot of healthcare systems, that’s their biggest headache right now.”
On the other hand, the report projects a national surplus of 48,000 home health/personal care aides.
That surprised Bianca K. Frogner, PhD, director of the Center for Health Workforce Studies at the University of Washington, Seattle.
“We are seeing increasing movement of investments toward moving patients out of skilled nursing facilities and keeping them in the home and community, which requires many more home health aides,” Dr. Frogner said. “Given such high turnover in this occupation, it’s hard to know if the surplus is really a surplus or if they will quickly be employed.”
Dr. Frogner receives grants and contracts from not-for-profit entities to investigate issues related to the health workforce.
Dr. Lezotte said the report’s findings are based on data from sources such as public and private databases and job postings. According to the report, “projections were made up to 2028 based on historical data up to 2023,” and “supply projections were derived using a linear autoregressive model based on historical supply within each occupation and geography.”
It’s not clear why some states like New York are expected to have huge shortages, but migration might be a factor, along with a lack of nearby nursing schools, Dr. Lezotte said.
As for shortages, Dr. Lezotte said healthcare systems will have to understand their local workforce situation and adapt. “They’ll need to be more proactive about their employee value proposition” via competitive pay and benefits Flexibility regarding scheduling is also important.
“They’re going to have to figure out how to up their game,” he said.
What about states with surpluses? They might be target-rich environments for states facing shortages, he said.
Positive Outlook Not Shared by Other Researchers
Other workforce projections conflict with Mercer’s, according to Jean Moore, DrPH, and Gaetano Forte, MS, director and assistant director of the Center for Health Workforce Studies, School of Public Health, University at Albany, New York.
The National Center for Health Workforce Analysis projects a 10% shortage of registered nurses and a 13% shortage of physicians in 2031. The agency didn’t make projections for home health aides because that workforce is in flux.
Why are Mercer’s projections so different? Dr. Lezotte said other projections assume that equity efforts will bring healthcare to everyone who needs it. The report assumes this won’t happen, he said. As a result, it expects there will be fewer patients who need to be served by workers.
Other projections expect a shortage of 300,000 registered nurses by 2035, Mr. Forte said. But the number of nurse practitioners in New York is growing quickly, Dr. Moore said.
Dr. Moore said it’s difficult to interpret Mercer’s findings because the company doesn’t provide enough information about its methodology.
“At some level, it’s not particularly useful regarding what the next steps are,” she said. “Projections should make you think about what you should do to change and improve, to create more of what you need.”
The Center for Health Workforce Studies at the University of Albany has provided consulting services to multiple companies that provide healthcare workforce projections. It has no relationship with Mercer.
A version of this article first appeared on Medscape.com.
A surprising new report by the Mercer consulting firm projects that the American healthcare workforce will face a small shortfall in 2028 — a shortage of less than 1% of all employees.
Mercer’s projections are rosier than federal workforce projections, which paint a grimmer picture of impending shortages.
“The labor market is a little more stabilized right now, and most healthcare systems are seeing less turnover,” Dan Lezotte, PhD, a partner with Mercer, said in an interview. But he noted “critical shortages” are still expected in some areas.
Mercer last projected workforce numbers in a 2020-2021 report released during the height of the COVID-19 pandemic. Now, “the labor market is drastically different,” Dr. Lezotte said. Health workforce shortages and surpluses have long varied significantly by region across the country.
The report forecasts a small surplus of physicians in 2028 but not in states such as California, New York, and Texas. The upper Midwest states will largely see doctor surpluses while Southern states face shortages. Some states with general physician surpluses may still experience shortages of specialists.
A surplus of nearly 30,000 registered nurses is expected, but New York, New Jersey, and Connecticut are projected to have a combined shortage of 16,000 nurses.
Overall, the report projects a shortage of more than 100,000 healthcare workers nationally by 2028. That’s less than 1% of the entire healthcare workforce of 18.6 million expected by then.
The report also predicts a shortage of nurse practitioners, especially in California and New York, and a shortage of 73,000 nursing assistants, especially in California, New York, and Texas.
“Healthcare systems are having the most difficulty hiring and hanging on to those workers who are supposed to take up the load off physicians and nurses,” Dr. Lezotte said. “They’re competing not only with other healthcare systems but with other industries like Amazon warehouses or McDonald’s in California paying $20 an hour. Healthcare was a little slow to keep up with that. In a lot of healthcare systems, that’s their biggest headache right now.”
On the other hand, the report projects a national surplus of 48,000 home health/personal care aides.
That surprised Bianca K. Frogner, PhD, director of the Center for Health Workforce Studies at the University of Washington, Seattle.
“We are seeing increasing movement of investments toward moving patients out of skilled nursing facilities and keeping them in the home and community, which requires many more home health aides,” Dr. Frogner said. “Given such high turnover in this occupation, it’s hard to know if the surplus is really a surplus or if they will quickly be employed.”
Dr. Frogner receives grants and contracts from not-for-profit entities to investigate issues related to the health workforce.
Dr. Lezotte said the report’s findings are based on data from sources such as public and private databases and job postings. According to the report, “projections were made up to 2028 based on historical data up to 2023,” and “supply projections were derived using a linear autoregressive model based on historical supply within each occupation and geography.”
It’s not clear why some states like New York are expected to have huge shortages, but migration might be a factor, along with a lack of nearby nursing schools, Dr. Lezotte said.
As for shortages, Dr. Lezotte said healthcare systems will have to understand their local workforce situation and adapt. “They’ll need to be more proactive about their employee value proposition” via competitive pay and benefits Flexibility regarding scheduling is also important.
“They’re going to have to figure out how to up their game,” he said.
What about states with surpluses? They might be target-rich environments for states facing shortages, he said.
Positive Outlook Not Shared by Other Researchers
Other workforce projections conflict with Mercer’s, according to Jean Moore, DrPH, and Gaetano Forte, MS, director and assistant director of the Center for Health Workforce Studies, School of Public Health, University at Albany, New York.
The National Center for Health Workforce Analysis projects a 10% shortage of registered nurses and a 13% shortage of physicians in 2031. The agency didn’t make projections for home health aides because that workforce is in flux.
Why are Mercer’s projections so different? Dr. Lezotte said other projections assume that equity efforts will bring healthcare to everyone who needs it. The report assumes this won’t happen, he said. As a result, it expects there will be fewer patients who need to be served by workers.
Other projections expect a shortage of 300,000 registered nurses by 2035, Mr. Forte said. But the number of nurse practitioners in New York is growing quickly, Dr. Moore said.
Dr. Moore said it’s difficult to interpret Mercer’s findings because the company doesn’t provide enough information about its methodology.
“At some level, it’s not particularly useful regarding what the next steps are,” she said. “Projections should make you think about what you should do to change and improve, to create more of what you need.”
The Center for Health Workforce Studies at the University of Albany has provided consulting services to multiple companies that provide healthcare workforce projections. It has no relationship with Mercer.
A version of this article first appeared on Medscape.com.
Cell Phone Use Linked to Higher Heart Disease Risk
“We found that a poor sleep pattern, psychological distress, and neuroticism significantly mediated the positive association between weekly mobile phone usage time and the risk for incident CVD, with a mediating proportion of 5.11%, 11.50%, and 2.25%, respectively,” said principal investigator Xianhui Qin, MD, professor of nephrology at Southern Medical University, Guangzhou, China.
Poor sleep patterns and poor mental health could disrupt circadian rhythms and endocrine and metabolic functions, as well as increase inflammation, he explained.
In addition, chronic exposure to radiofrequency electromagnetic fields (RF-EMF) emitted from cell phones could lead to oxidative stress and an inflammatory response. Combined with smoking and diabetes, this exposure “may have a synergistic effect in increasing CVD risk,” Dr. Qin suggested.
The study was published online in the Canadian Journal of Cardiology.
Risk Underestimated?
The researchers aimed to examine the association of regular cell phone use with incident CVD and explore the mediating effects of sleep and mental health using linked hospital and mortality records.
Their analysis included 444,027 participants (mean age, 56 years; 44% men) without a history of CVD from the UK Biobank. A total of 378,161 participants were regular cell phone users.
Regular cell phone use was defined as at least one call per week. Weekly use was self-reported as the average time of calls per week during the previous 3 months.
The primary outcome was incident CVD. Secondary outcomes were each component of CVD (ie, coronary heart disease, stroke, atrial fibrillation, and heart failure) and increased carotid intima media thickness (CIMT).
Compared with nonregular cell phone users, regular users were younger, had higher proportions of current smokers and urban residents, and had lower proportions of history of hypertension and diabetes. They also had higher income, Townsend deprivation index, and body mass index, and lower education levels.
During a median follow-up of 12.3 years, 56,181 participants developed incident CVD. Compared with nonregular cell phone users, regular users had a significantly higher risk for incident CVD (hazard ratio, 1.04) and increased CIMT (odds ratio, 1.11).
Among regular cell phone users, the duration of cell phone use and hands-free device/speakerphone use during calls was not significantly associated with incident CVD. Yet a significant and positive dose-response relationship was seen between weekly cell phone usage time and the risk for CVD. The positive association was stronger in current vs noncurrent smokers and people with vs without diabetes.
To different extents, sleep patterns (5.11%), psychologic distress (11.5%), and neuroticism (2.25%) mediated the relationship between weekly cell phone usage time and the risk for incident CVD.
“Our study suggests that despite the advantages of mobile phone use, we should also pay attention to the potential harm of mobile phone use to cardiovascular health,” Dr. Qin said. “Future studies to assess the risk-benefit balance will help promote mobile phone use patterns that are conducive to cardiovascular health.”
Meanwhile, he added, “We encourage measures to reduce time spent on mobile phones to promote the primary prevention of CVD. On the other hand, improving sleep and mental health status may help reduce the higher risk of CVD associated with mobile phone use.”
There are several limitations to the study in addition to its observational nature, which cannot show cause and effect. The questionnaires on cell phone use were restricted to phone calls; other use patterns of cell phones (eg, messaging, watching videos, and browsing the web) were not considered. Although the researchers adjusted for many potential confounders, unmeasured confounding bias (eg, the type of cell phone used and other sources of RF-EMF) cannot be eliminated.
Weak Link?
In a comment, Nicholas Grubic, MSc, a PhD student in epidemiology at the University of Toronto, Ontario, Canada, and coauthor of a related editorial, said, “I found it interesting that there was a connection observed between mobile phone use and CVD. However, it is crucial to understand that this link appeared to be much weaker compared with other well-known cardiovascular risk factors, such as smoking, diabetes, and high blood pressure. For now, mobile phone use should not be a major concern for most people.”
Nevertheless, clinicians should encourage patients to practice healthy habits around their screen time, he advised. “This could include limiting mobile phone use before bedtime and taking regular breaks to engage in activities that promote heart health, such as exercising or spending time outdoors.
“For the time being, we probably won’t see mobile phone use included in standard assessments for cardiovascular risk or as a focal point of cardiovascular health promotion initiatives,” he added. Instead, clinicians should “focus on established risk factors that have a stronger impact on patients’ cardiovascular health.”
Nieca Goldberg, MD, a clinical associate professor of medicine at NYU Grossman School of Medicine in New York City and American Heart Association volunteer expert, had a similar message. “You don’t have to go back to using a landline,” she said. “Instead, patients should be more mindful of how much phone use is taking away from their physical activity, keeping them from sleeping, and causing them stress.” Clinicians should also remember to counsel smokers on smoking cessation.
“It would be important for future studies to look at time spent on the phone and the type of activities patients are doing on their phones, such as social media, calls, texts, movies, or streaming TV shows,” she said. “It would be important to see how phone use is leading to a sedentary lifestyle” and what that means for a larger, more diverse population.
The study was supported by the National Key R&D Program, the National Natural Science Foundation of China, and the Outstanding Youth Development Scheme of Nanfang Hospital, Southern Medical University. Dr. Qin, Dr. Grubic, and Dr. Goldberg reported having no relevant financial relationships.
A version of this article first appeared on Medscape.com.
“We found that a poor sleep pattern, psychological distress, and neuroticism significantly mediated the positive association between weekly mobile phone usage time and the risk for incident CVD, with a mediating proportion of 5.11%, 11.50%, and 2.25%, respectively,” said principal investigator Xianhui Qin, MD, professor of nephrology at Southern Medical University, Guangzhou, China.
Poor sleep patterns and poor mental health could disrupt circadian rhythms and endocrine and metabolic functions, as well as increase inflammation, he explained.
In addition, chronic exposure to radiofrequency electromagnetic fields (RF-EMF) emitted from cell phones could lead to oxidative stress and an inflammatory response. Combined with smoking and diabetes, this exposure “may have a synergistic effect in increasing CVD risk,” Dr. Qin suggested.
The study was published online in the Canadian Journal of Cardiology.
Risk Underestimated?
The researchers aimed to examine the association of regular cell phone use with incident CVD and explore the mediating effects of sleep and mental health using linked hospital and mortality records.
Their analysis included 444,027 participants (mean age, 56 years; 44% men) without a history of CVD from the UK Biobank. A total of 378,161 participants were regular cell phone users.
Regular cell phone use was defined as at least one call per week. Weekly use was self-reported as the average time of calls per week during the previous 3 months.
The primary outcome was incident CVD. Secondary outcomes were each component of CVD (ie, coronary heart disease, stroke, atrial fibrillation, and heart failure) and increased carotid intima media thickness (CIMT).
Compared with nonregular cell phone users, regular users were younger, had higher proportions of current smokers and urban residents, and had lower proportions of history of hypertension and diabetes. They also had higher income, Townsend deprivation index, and body mass index, and lower education levels.
During a median follow-up of 12.3 years, 56,181 participants developed incident CVD. Compared with nonregular cell phone users, regular users had a significantly higher risk for incident CVD (hazard ratio, 1.04) and increased CIMT (odds ratio, 1.11).
Among regular cell phone users, the duration of cell phone use and hands-free device/speakerphone use during calls was not significantly associated with incident CVD. Yet a significant and positive dose-response relationship was seen between weekly cell phone usage time and the risk for CVD. The positive association was stronger in current vs noncurrent smokers and people with vs without diabetes.
To different extents, sleep patterns (5.11%), psychologic distress (11.5%), and neuroticism (2.25%) mediated the relationship between weekly cell phone usage time and the risk for incident CVD.
“Our study suggests that despite the advantages of mobile phone use, we should also pay attention to the potential harm of mobile phone use to cardiovascular health,” Dr. Qin said. “Future studies to assess the risk-benefit balance will help promote mobile phone use patterns that are conducive to cardiovascular health.”
Meanwhile, he added, “We encourage measures to reduce time spent on mobile phones to promote the primary prevention of CVD. On the other hand, improving sleep and mental health status may help reduce the higher risk of CVD associated with mobile phone use.”
There are several limitations to the study in addition to its observational nature, which cannot show cause and effect. The questionnaires on cell phone use were restricted to phone calls; other use patterns of cell phones (eg, messaging, watching videos, and browsing the web) were not considered. Although the researchers adjusted for many potential confounders, unmeasured confounding bias (eg, the type of cell phone used and other sources of RF-EMF) cannot be eliminated.
Weak Link?
In a comment, Nicholas Grubic, MSc, a PhD student in epidemiology at the University of Toronto, Ontario, Canada, and coauthor of a related editorial, said, “I found it interesting that there was a connection observed between mobile phone use and CVD. However, it is crucial to understand that this link appeared to be much weaker compared with other well-known cardiovascular risk factors, such as smoking, diabetes, and high blood pressure. For now, mobile phone use should not be a major concern for most people.”
Nevertheless, clinicians should encourage patients to practice healthy habits around their screen time, he advised. “This could include limiting mobile phone use before bedtime and taking regular breaks to engage in activities that promote heart health, such as exercising or spending time outdoors.
“For the time being, we probably won’t see mobile phone use included in standard assessments for cardiovascular risk or as a focal point of cardiovascular health promotion initiatives,” he added. Instead, clinicians should “focus on established risk factors that have a stronger impact on patients’ cardiovascular health.”
Nieca Goldberg, MD, a clinical associate professor of medicine at NYU Grossman School of Medicine in New York City and American Heart Association volunteer expert, had a similar message. “You don’t have to go back to using a landline,” she said. “Instead, patients should be more mindful of how much phone use is taking away from their physical activity, keeping them from sleeping, and causing them stress.” Clinicians should also remember to counsel smokers on smoking cessation.
“It would be important for future studies to look at time spent on the phone and the type of activities patients are doing on their phones, such as social media, calls, texts, movies, or streaming TV shows,” she said. “It would be important to see how phone use is leading to a sedentary lifestyle” and what that means for a larger, more diverse population.
The study was supported by the National Key R&D Program, the National Natural Science Foundation of China, and the Outstanding Youth Development Scheme of Nanfang Hospital, Southern Medical University. Dr. Qin, Dr. Grubic, and Dr. Goldberg reported having no relevant financial relationships.
A version of this article first appeared on Medscape.com.
“We found that a poor sleep pattern, psychological distress, and neuroticism significantly mediated the positive association between weekly mobile phone usage time and the risk for incident CVD, with a mediating proportion of 5.11%, 11.50%, and 2.25%, respectively,” said principal investigator Xianhui Qin, MD, professor of nephrology at Southern Medical University, Guangzhou, China.
Poor sleep patterns and poor mental health could disrupt circadian rhythms and endocrine and metabolic functions, as well as increase inflammation, he explained.
In addition, chronic exposure to radiofrequency electromagnetic fields (RF-EMF) emitted from cell phones could lead to oxidative stress and an inflammatory response. Combined with smoking and diabetes, this exposure “may have a synergistic effect in increasing CVD risk,” Dr. Qin suggested.
The study was published online in the Canadian Journal of Cardiology.
Risk Underestimated?
The researchers aimed to examine the association of regular cell phone use with incident CVD and explore the mediating effects of sleep and mental health using linked hospital and mortality records.
Their analysis included 444,027 participants (mean age, 56 years; 44% men) without a history of CVD from the UK Biobank. A total of 378,161 participants were regular cell phone users.
Regular cell phone use was defined as at least one call per week. Weekly use was self-reported as the average time of calls per week during the previous 3 months.
The primary outcome was incident CVD. Secondary outcomes were each component of CVD (ie, coronary heart disease, stroke, atrial fibrillation, and heart failure) and increased carotid intima media thickness (CIMT).
Compared with nonregular cell phone users, regular users were younger, had higher proportions of current smokers and urban residents, and had lower proportions of history of hypertension and diabetes. They also had higher income, Townsend deprivation index, and body mass index, and lower education levels.
During a median follow-up of 12.3 years, 56,181 participants developed incident CVD. Compared with nonregular cell phone users, regular users had a significantly higher risk for incident CVD (hazard ratio, 1.04) and increased CIMT (odds ratio, 1.11).
Among regular cell phone users, the duration of cell phone use and hands-free device/speakerphone use during calls was not significantly associated with incident CVD. Yet a significant and positive dose-response relationship was seen between weekly cell phone usage time and the risk for CVD. The positive association was stronger in current vs noncurrent smokers and people with vs without diabetes.
To different extents, sleep patterns (5.11%), psychologic distress (11.5%), and neuroticism (2.25%) mediated the relationship between weekly cell phone usage time and the risk for incident CVD.
“Our study suggests that despite the advantages of mobile phone use, we should also pay attention to the potential harm of mobile phone use to cardiovascular health,” Dr. Qin said. “Future studies to assess the risk-benefit balance will help promote mobile phone use patterns that are conducive to cardiovascular health.”
Meanwhile, he added, “We encourage measures to reduce time spent on mobile phones to promote the primary prevention of CVD. On the other hand, improving sleep and mental health status may help reduce the higher risk of CVD associated with mobile phone use.”
There are several limitations to the study in addition to its observational nature, which cannot show cause and effect. The questionnaires on cell phone use were restricted to phone calls; other use patterns of cell phones (eg, messaging, watching videos, and browsing the web) were not considered. Although the researchers adjusted for many potential confounders, unmeasured confounding bias (eg, the type of cell phone used and other sources of RF-EMF) cannot be eliminated.
Weak Link?
In a comment, Nicholas Grubic, MSc, a PhD student in epidemiology at the University of Toronto, Ontario, Canada, and coauthor of a related editorial, said, “I found it interesting that there was a connection observed between mobile phone use and CVD. However, it is crucial to understand that this link appeared to be much weaker compared with other well-known cardiovascular risk factors, such as smoking, diabetes, and high blood pressure. For now, mobile phone use should not be a major concern for most people.”
Nevertheless, clinicians should encourage patients to practice healthy habits around their screen time, he advised. “This could include limiting mobile phone use before bedtime and taking regular breaks to engage in activities that promote heart health, such as exercising or spending time outdoors.
“For the time being, we probably won’t see mobile phone use included in standard assessments for cardiovascular risk or as a focal point of cardiovascular health promotion initiatives,” he added. Instead, clinicians should “focus on established risk factors that have a stronger impact on patients’ cardiovascular health.”
Nieca Goldberg, MD, a clinical associate professor of medicine at NYU Grossman School of Medicine in New York City and American Heart Association volunteer expert, had a similar message. “You don’t have to go back to using a landline,” she said. “Instead, patients should be more mindful of how much phone use is taking away from their physical activity, keeping them from sleeping, and causing them stress.” Clinicians should also remember to counsel smokers on smoking cessation.
“It would be important for future studies to look at time spent on the phone and the type of activities patients are doing on their phones, such as social media, calls, texts, movies, or streaming TV shows,” she said. “It would be important to see how phone use is leading to a sedentary lifestyle” and what that means for a larger, more diverse population.
The study was supported by the National Key R&D Program, the National Natural Science Foundation of China, and the Outstanding Youth Development Scheme of Nanfang Hospital, Southern Medical University. Dr. Qin, Dr. Grubic, and Dr. Goldberg reported having no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE CANADIAN JOURNAL OF CARDIOLOGY
Nighttime Outdoor Light Pollution Linked to Alzheimer’s Risk
a new national study suggested.
Analyses of state and county light pollution data and Medicare claims showed that areas with higher average nighttime light intensity had a greater prevalence of Alzheimer’s disease.
Among people aged 65 years or older, Alzheimer’s disease prevalence was more strongly associated with nightly light pollution exposure than with alcohol misuse, chronic kidney disease, depression, or obesity.
In those younger than 65 years, greater nighttime light intensity had a stronger association with Alzheimer’s disease prevalence than any other risk factor included in the study.
“The results are pretty striking when you do these comparisons and it’s true for people of all ages,” said Robin Voigt-Zuwala, PhD, lead author and director, Circadian Rhythm Research Laboratory, Rush University, Chicago, Illinois.
The study was published online in Frontiers of Neuroscience.
Shining a Light
Exposure to artificial outdoor light at night has been associated with adverse health effects such as sleep disruption, obesity, atherosclerosis, and cancer, but this is the first study to look specifically at Alzheimer’s disease, investigators noted.
Two recent studies reported higher risks for mild cognitive impairment among Chinese veterans and late-onset dementia among Italian residents living in areas with brighter outdoor light at night.
For this study, Dr. Voigt-Zuwala and colleagues examined the relationship between Alzheimer’s disease prevalence and average nighttime light intensity in the lower 48 states using data from Medicare Part A and B, the Centers for Disease Control and Prevention, and NASA satellite–acquired radiance data.
The data were averaged for the years 2012-2018 and states divided into five groups based on average nighttime light intensity.
The darkest states were Montana, Wyoming, South Dakota, Idaho, Maine, New Mexico, Vermont, Oregon, Utah, and Nevada. The brightest states were Indiana, Illinois, Florida, Ohio, Massachusetts, Connecticut, Maryland, Delaware, Rhode Island, and New Jersey.
Analysis of variance revealed a significant difference in Alzheimer’s disease prevalence between state groups (P < .0001). Multiple comparisons testing also showed that states with the lowest average nighttime light had significantly different Alzheimer’s disease prevalence than those with higher intensity.
The same positive relationship was observed when each year was assessed individually and at the county level, using data from 45 counties and the District of Columbia.
Strong Association
The investigators also found that state average nighttime light intensity is significantly associated with Alzheimer’s disease prevalence (P = .006). This effect was seen across all ages, sexes, and races except Asian Pacific Island, the latter possibly related to statistical power, the authors said.
When known or proposed risk factors for Alzheimer’s disease were added to the model, atrial fibrillation, diabetes, hyperlipidemia, hypertension, and stroke had a stronger association with Alzheimer’s disease than average nighttime light intensity.
Nighttime light intensity, however, was more strongly associated with Alzheimer’s disease prevalence than alcohol abuse, chronic kidney disease, depression, heart failure, and obesity.
Moreover, in people younger than 65 years, nighttime light pollution had a stronger association with Alzheimer’s disease prevalence than all other risk factors (P = .007).
The mechanism behind this increased vulnerability is unclear, but there may be an interplay between genetic susceptibility of an individual and how they respond to light, Dr. Voigt-Zuwala suggested.
“APOE4 is the genotype most highly associated with Alzheimer’s disease risk, and maybe the people who have that genotype are just more sensitive to the effects of light exposure at night, more sensitive to circadian rhythm disruption,” she said.
The authors noted that additional research is needed but suggested light pollution may also influence Alzheimer’s disease through sleep disruption, which can promote inflammation, activate microglia and astrocytes, and negatively alter the clearance of amyloid beta, and by decreasing the levels of brain-derived neurotrophic factor.
Are We Measuring the Right Light?
“It’s a good article and it’s got a good message, but I have some caveats to that,” said George C. Brainard, PhD, director, Light Research Program, Thomas Jefferson University in Philadelphia, Pennsylvania, and a pioneer in the study of how light affects biology including breast cancer in night-shift workers.
The biggest caveat, and one acknowledged by the authors, is that the study didn’t measure indoor light exposure and relied instead on satellite imaging.
“They’re very striking images, but they may not be particularly relevant. And here’s why: People don’t live outdoors all night,” Dr. Brainard said.
Instead, people spend much of their time at night indoors where they’re exposed to lighting in the home and from smartphones, laptops, and television screens.
“It doesn’t invalidate their work. It’s an important advancement, an important observation,” Dr. Brainard said. “But the important thing really is to find out what is the population exposed to that triggers this response, and it’s probably indoor lighting related to the amount and physical characteristics of indoor lighting. It doesn’t mean outdoor lighting can’t play a role. It certainly can.”
Reached for comment, Erik Musiek, MD, PhD, a professor of neurology whose lab at Washington University School of Medicine in St. Louis, Missouri, has extensively studied circadian clock disruption and Alzheimer’s disease pathology in the brain, said the study provides a 10,000-foot view of the issue.
For example, the study was not designed to detect whether people living in high light pollution areas are actually experiencing more outdoor light at night and if risk factors such as air pollution and low socioeconomic status may correlate with these areas.
“Most of what we worry about is do people have lights on in the house, do they have their TV on, their screens up to their face late at night? This can’t tell us about that,” Dr. Musiek said. “But on the other hand, this kind of light exposure is something that public policy can affect.”
“It’s hard to control people’s personal habits nor should we probably, but we can control what types of bulbs you put into streetlights, how bright they are, and where you put lighting in a public place,” he added. “So I do think there’s value there.”
At least 19 states, the District of Columbia, and Puerto Rico have laws in place to reduce light pollution, with the majority doing so to promote energy conservation, public safety, aesthetic interests, or astronomical research, according to the National Conference of State Legislatures.
To respond to some of the limitations in this study, Dr. Voigt-Zuwala is writing a grant application for a new project to look at both indoor and outdoor light exposure on an individual level.
“This is what I’ve been wanting to study for a long time, and this study is just sort of the stepping stone, the proof of concept that this is something we need to be investigating,” she said.
Dr. Voigt-Zuwala reported RO1 and R24 grants from the National Institutes of Health (NIH), one coauthor reported an NIH R24 grant; another reported having no conflicts of interest. Dr. Brainard reported having no relevant conflicts of interest. Dr. Musiek reported research funding from Eisai Pharmaceuticals.
A version of this article first appeared on Medscape.com.
a new national study suggested.
Analyses of state and county light pollution data and Medicare claims showed that areas with higher average nighttime light intensity had a greater prevalence of Alzheimer’s disease.
Among people aged 65 years or older, Alzheimer’s disease prevalence was more strongly associated with nightly light pollution exposure than with alcohol misuse, chronic kidney disease, depression, or obesity.
In those younger than 65 years, greater nighttime light intensity had a stronger association with Alzheimer’s disease prevalence than any other risk factor included in the study.
“The results are pretty striking when you do these comparisons and it’s true for people of all ages,” said Robin Voigt-Zuwala, PhD, lead author and director, Circadian Rhythm Research Laboratory, Rush University, Chicago, Illinois.
The study was published online in Frontiers of Neuroscience.
Shining a Light
Exposure to artificial outdoor light at night has been associated with adverse health effects such as sleep disruption, obesity, atherosclerosis, and cancer, but this is the first study to look specifically at Alzheimer’s disease, investigators noted.
Two recent studies reported higher risks for mild cognitive impairment among Chinese veterans and late-onset dementia among Italian residents living in areas with brighter outdoor light at night.
For this study, Dr. Voigt-Zuwala and colleagues examined the relationship between Alzheimer’s disease prevalence and average nighttime light intensity in the lower 48 states using data from Medicare Part A and B, the Centers for Disease Control and Prevention, and NASA satellite–acquired radiance data.
The data were averaged for the years 2012-2018 and states divided into five groups based on average nighttime light intensity.
The darkest states were Montana, Wyoming, South Dakota, Idaho, Maine, New Mexico, Vermont, Oregon, Utah, and Nevada. The brightest states were Indiana, Illinois, Florida, Ohio, Massachusetts, Connecticut, Maryland, Delaware, Rhode Island, and New Jersey.
Analysis of variance revealed a significant difference in Alzheimer’s disease prevalence between state groups (P < .0001). Multiple comparisons testing also showed that states with the lowest average nighttime light had significantly different Alzheimer’s disease prevalence than those with higher intensity.
The same positive relationship was observed when each year was assessed individually and at the county level, using data from 45 counties and the District of Columbia.
Strong Association
The investigators also found that state average nighttime light intensity is significantly associated with Alzheimer’s disease prevalence (P = .006). This effect was seen across all ages, sexes, and races except Asian Pacific Island, the latter possibly related to statistical power, the authors said.
When known or proposed risk factors for Alzheimer’s disease were added to the model, atrial fibrillation, diabetes, hyperlipidemia, hypertension, and stroke had a stronger association with Alzheimer’s disease than average nighttime light intensity.
Nighttime light intensity, however, was more strongly associated with Alzheimer’s disease prevalence than alcohol abuse, chronic kidney disease, depression, heart failure, and obesity.
Moreover, in people younger than 65 years, nighttime light pollution had a stronger association with Alzheimer’s disease prevalence than all other risk factors (P = .007).
The mechanism behind this increased vulnerability is unclear, but there may be an interplay between genetic susceptibility of an individual and how they respond to light, Dr. Voigt-Zuwala suggested.
“APOE4 is the genotype most highly associated with Alzheimer’s disease risk, and maybe the people who have that genotype are just more sensitive to the effects of light exposure at night, more sensitive to circadian rhythm disruption,” she said.
The authors noted that additional research is needed but suggested light pollution may also influence Alzheimer’s disease through sleep disruption, which can promote inflammation, activate microglia and astrocytes, and negatively alter the clearance of amyloid beta, and by decreasing the levels of brain-derived neurotrophic factor.
Are We Measuring the Right Light?
“It’s a good article and it’s got a good message, but I have some caveats to that,” said George C. Brainard, PhD, director, Light Research Program, Thomas Jefferson University in Philadelphia, Pennsylvania, and a pioneer in the study of how light affects biology including breast cancer in night-shift workers.
The biggest caveat, and one acknowledged by the authors, is that the study didn’t measure indoor light exposure and relied instead on satellite imaging.
“They’re very striking images, but they may not be particularly relevant. And here’s why: People don’t live outdoors all night,” Dr. Brainard said.
Instead, people spend much of their time at night indoors where they’re exposed to lighting in the home and from smartphones, laptops, and television screens.
“It doesn’t invalidate their work. It’s an important advancement, an important observation,” Dr. Brainard said. “But the important thing really is to find out what is the population exposed to that triggers this response, and it’s probably indoor lighting related to the amount and physical characteristics of indoor lighting. It doesn’t mean outdoor lighting can’t play a role. It certainly can.”
Reached for comment, Erik Musiek, MD, PhD, a professor of neurology whose lab at Washington University School of Medicine in St. Louis, Missouri, has extensively studied circadian clock disruption and Alzheimer’s disease pathology in the brain, said the study provides a 10,000-foot view of the issue.
For example, the study was not designed to detect whether people living in high light pollution areas are actually experiencing more outdoor light at night and if risk factors such as air pollution and low socioeconomic status may correlate with these areas.
“Most of what we worry about is do people have lights on in the house, do they have their TV on, their screens up to their face late at night? This can’t tell us about that,” Dr. Musiek said. “But on the other hand, this kind of light exposure is something that public policy can affect.”
“It’s hard to control people’s personal habits nor should we probably, but we can control what types of bulbs you put into streetlights, how bright they are, and where you put lighting in a public place,” he added. “So I do think there’s value there.”
At least 19 states, the District of Columbia, and Puerto Rico have laws in place to reduce light pollution, with the majority doing so to promote energy conservation, public safety, aesthetic interests, or astronomical research, according to the National Conference of State Legislatures.
To respond to some of the limitations in this study, Dr. Voigt-Zuwala is writing a grant application for a new project to look at both indoor and outdoor light exposure on an individual level.
“This is what I’ve been wanting to study for a long time, and this study is just sort of the stepping stone, the proof of concept that this is something we need to be investigating,” she said.
Dr. Voigt-Zuwala reported RO1 and R24 grants from the National Institutes of Health (NIH), one coauthor reported an NIH R24 grant; another reported having no conflicts of interest. Dr. Brainard reported having no relevant conflicts of interest. Dr. Musiek reported research funding from Eisai Pharmaceuticals.
A version of this article first appeared on Medscape.com.
a new national study suggested.
Analyses of state and county light pollution data and Medicare claims showed that areas with higher average nighttime light intensity had a greater prevalence of Alzheimer’s disease.
Among people aged 65 years or older, Alzheimer’s disease prevalence was more strongly associated with nightly light pollution exposure than with alcohol misuse, chronic kidney disease, depression, or obesity.
In those younger than 65 years, greater nighttime light intensity had a stronger association with Alzheimer’s disease prevalence than any other risk factor included in the study.
“The results are pretty striking when you do these comparisons and it’s true for people of all ages,” said Robin Voigt-Zuwala, PhD, lead author and director, Circadian Rhythm Research Laboratory, Rush University, Chicago, Illinois.
The study was published online in Frontiers of Neuroscience.
Shining a Light
Exposure to artificial outdoor light at night has been associated with adverse health effects such as sleep disruption, obesity, atherosclerosis, and cancer, but this is the first study to look specifically at Alzheimer’s disease, investigators noted.
Two recent studies reported higher risks for mild cognitive impairment among Chinese veterans and late-onset dementia among Italian residents living in areas with brighter outdoor light at night.
For this study, Dr. Voigt-Zuwala and colleagues examined the relationship between Alzheimer’s disease prevalence and average nighttime light intensity in the lower 48 states using data from Medicare Part A and B, the Centers for Disease Control and Prevention, and NASA satellite–acquired radiance data.
The data were averaged for the years 2012-2018 and states divided into five groups based on average nighttime light intensity.
The darkest states were Montana, Wyoming, South Dakota, Idaho, Maine, New Mexico, Vermont, Oregon, Utah, and Nevada. The brightest states were Indiana, Illinois, Florida, Ohio, Massachusetts, Connecticut, Maryland, Delaware, Rhode Island, and New Jersey.
Analysis of variance revealed a significant difference in Alzheimer’s disease prevalence between state groups (P < .0001). Multiple comparisons testing also showed that states with the lowest average nighttime light had significantly different Alzheimer’s disease prevalence than those with higher intensity.
The same positive relationship was observed when each year was assessed individually and at the county level, using data from 45 counties and the District of Columbia.
Strong Association
The investigators also found that state average nighttime light intensity is significantly associated with Alzheimer’s disease prevalence (P = .006). This effect was seen across all ages, sexes, and races except Asian Pacific Island, the latter possibly related to statistical power, the authors said.
When known or proposed risk factors for Alzheimer’s disease were added to the model, atrial fibrillation, diabetes, hyperlipidemia, hypertension, and stroke had a stronger association with Alzheimer’s disease than average nighttime light intensity.
Nighttime light intensity, however, was more strongly associated with Alzheimer’s disease prevalence than alcohol abuse, chronic kidney disease, depression, heart failure, and obesity.
Moreover, in people younger than 65 years, nighttime light pollution had a stronger association with Alzheimer’s disease prevalence than all other risk factors (P = .007).
The mechanism behind this increased vulnerability is unclear, but there may be an interplay between genetic susceptibility of an individual and how they respond to light, Dr. Voigt-Zuwala suggested.
“APOE4 is the genotype most highly associated with Alzheimer’s disease risk, and maybe the people who have that genotype are just more sensitive to the effects of light exposure at night, more sensitive to circadian rhythm disruption,” she said.
The authors noted that additional research is needed but suggested light pollution may also influence Alzheimer’s disease through sleep disruption, which can promote inflammation, activate microglia and astrocytes, and negatively alter the clearance of amyloid beta, and by decreasing the levels of brain-derived neurotrophic factor.
Are We Measuring the Right Light?
“It’s a good article and it’s got a good message, but I have some caveats to that,” said George C. Brainard, PhD, director, Light Research Program, Thomas Jefferson University in Philadelphia, Pennsylvania, and a pioneer in the study of how light affects biology including breast cancer in night-shift workers.
The biggest caveat, and one acknowledged by the authors, is that the study didn’t measure indoor light exposure and relied instead on satellite imaging.
“They’re very striking images, but they may not be particularly relevant. And here’s why: People don’t live outdoors all night,” Dr. Brainard said.
Instead, people spend much of their time at night indoors where they’re exposed to lighting in the home and from smartphones, laptops, and television screens.
“It doesn’t invalidate their work. It’s an important advancement, an important observation,” Dr. Brainard said. “But the important thing really is to find out what is the population exposed to that triggers this response, and it’s probably indoor lighting related to the amount and physical characteristics of indoor lighting. It doesn’t mean outdoor lighting can’t play a role. It certainly can.”
Reached for comment, Erik Musiek, MD, PhD, a professor of neurology whose lab at Washington University School of Medicine in St. Louis, Missouri, has extensively studied circadian clock disruption and Alzheimer’s disease pathology in the brain, said the study provides a 10,000-foot view of the issue.
For example, the study was not designed to detect whether people living in high light pollution areas are actually experiencing more outdoor light at night and if risk factors such as air pollution and low socioeconomic status may correlate with these areas.
“Most of what we worry about is do people have lights on in the house, do they have their TV on, their screens up to their face late at night? This can’t tell us about that,” Dr. Musiek said. “But on the other hand, this kind of light exposure is something that public policy can affect.”
“It’s hard to control people’s personal habits nor should we probably, but we can control what types of bulbs you put into streetlights, how bright they are, and where you put lighting in a public place,” he added. “So I do think there’s value there.”
At least 19 states, the District of Columbia, and Puerto Rico have laws in place to reduce light pollution, with the majority doing so to promote energy conservation, public safety, aesthetic interests, or astronomical research, according to the National Conference of State Legislatures.
To respond to some of the limitations in this study, Dr. Voigt-Zuwala is writing a grant application for a new project to look at both indoor and outdoor light exposure on an individual level.
“This is what I’ve been wanting to study for a long time, and this study is just sort of the stepping stone, the proof of concept that this is something we need to be investigating,” she said.
Dr. Voigt-Zuwala reported RO1 and R24 grants from the National Institutes of Health (NIH), one coauthor reported an NIH R24 grant; another reported having no conflicts of interest. Dr. Brainard reported having no relevant conflicts of interest. Dr. Musiek reported research funding from Eisai Pharmaceuticals.
A version of this article first appeared on Medscape.com.
FROM FRONTIERS OF NEUROSCIENCE
Do Clonal Hematopoiesis and Mosaic Chromosomal Alterations Increase Solid Tumor Risk?
Clonal hematopoiesis of indeterminate potential (CHIP) and mosaic chromosomal alterations (mCAs) are associated with an increased risk for breast cancer, and CHIP is associated with increased mortality in patients with colon cancer, according to the authors of new research.
These findings, drawn from almost 11,000 patients in the Women’s Health Initiative (WHI) study, add further evidence that CHIP and mCA drive solid tumor risk, alongside known associations with hematologic malignancies, reported lead author Pinkal Desai, MD, associate professor of medicine and clinical director of molecular aging at Englander Institute for Precision Medicine, Weill Cornell Medical College, New York City, and colleagues.
How This Study Differs From Others of Breast Cancer Risk Factors
“The independent effect of CHIP and mCA on risk and mortality from solid tumors has not been elucidated due to lack of detailed data on mortality outcomes and risk factors,” the investigators wrote in Cancer, although some previous studies have suggested a link.
In particular, the investigators highlighted a 2022 UK Biobank study, which reported an association between CHIP and lung cancer and a borderline association with breast cancer that did not quite reach statistical significance.
But the UK Biobank study was confined to a UK population, Dr. Desai noted in an interview, and the data were less detailed than those in the present investigation.
“In terms of risk, the part that was lacking in previous studies was a comprehensive assessment of risk factors that increase risk for all these cancers,” Dr. Desai said. “For example, for breast cancer, we had very detailed data on [participants’] Gail risk score, which is known to impact breast cancer risk. We also had mammogram data and colonoscopy data.”
In an accompanying editorial, Koichi Takahashi, MD, PhD , and Nehali Shah, BS, of The University of Texas MD Anderson Cancer Center, Houston, Texas, pointed out the same UK Biobank findings, then noted that CHIP has also been linked with worse overall survival in unselected cancer patients. Still, they wrote, “the impact of CH on cancer risk and mortality remains controversial due to conflicting data and context‐dependent effects,” necessitating studies like this one by Dr. Desai and colleagues.
How Was the Relationship Between CHIP, MCA, and Solid Tumor Risk Assessed?
To explore possible associations between CHIP, mCA, and solid tumors, the investigators analyzed whole genome sequencing data from 10,866 women in the WHI, a multi-study program that began in 1992 and involved 161,808 women in both observational and clinical trial cohorts.
In 2002, the first big data release from the WHI suggested that hormone replacement therapy (HRT) increased breast cancer risk, leading to widespread reduction in HRT use.
More recent reports continue to shape our understanding of these risks, suggesting differences across cancer types. For breast cancer, the WHI data suggested that HRT-associated risk was largely driven by formulations involving progesterone and estrogen, whereas estrogen-only formulations, now more common, are generally considered to present an acceptable risk profile for suitable patients.
The new study accounted for this potential HRT-associated risk, including by adjusting for patients who received HRT, type of HRT received, and duration of HRT received. According to Desai, this approach is commonly used when analyzing data from the WHI, nullifying concerns about the potentially deleterious effects of the hormones used in the study.
“Our question was not ‘does HRT cause cancer?’ ” Dr. Desai said in an interview. “But HRT can be linked to breast cancer risk and has a potential to be a confounder, and hence the above methodology.
“So I can say that the confounding/effect modification that HRT would have contributed to in the relationship between exposure (CH and mCA) and outcome (cancer) is well adjusted for as described above. This is standard in WHI analyses,” she continued.
“Every Women’s Health Initiative analysis that comes out — not just for our study — uses a standard method ... where you account for hormonal therapy,” Dr. Desai added, again noting that many other potential risk factors were considered, enabling a “detailed, robust” analysis.
Dr. Takahashi and Ms. Shah agreed. “A notable strength of this study is its adjustment for many confounding factors,” they wrote. “The cohort’s well‐annotated data on other known cancer risk factors allowed for a robust assessment of CH’s independent risk.”
How Do Findings Compare With Those of the UK Biobank Study?
CHIP was associated with a 30% increased risk for breast cancer (hazard ratio [HR], 1.30; 95% CI, 1.03-1.64; P = .02), strengthening the borderline association reported by the UK Biobank study.
In contrast with the UK Biobank study, CHIP was not associated with lung cancer risk, although this may have been caused by fewer cases of lung cancer and a lack of male patients, Dr. Desai suggested.
“The discrepancy between the studies lies in the risk of lung cancer, although the point estimate in the current study suggested a positive association,” wrote Dr. Takahashi and Ms. Shah.
As in the UK Biobank study, CHIP was not associated with increased risk of developing colorectal cancer.
Mortality analysis, however, which was not conducted in the UK Biobank study, offered a new insight: Patients with existing colorectal cancer and CHIP had a significantly higher mortality risk than those without CHIP. Before stage adjustment, risk for mortality among those with colorectal cancer and CHIP was fourfold higher than those without CHIP (HR, 3.99; 95% CI, 2.41-6.62; P < .001). After stage adjustment, CHIP was still associated with a twofold higher mortality risk (HR, 2.50; 95% CI, 1.32-4.72; P = .004).
The investigators’ first mCA analyses, which employed a cell fraction cutoff greater than 3%, were unfruitful. But raising the cell fraction threshold to 5% in an exploratory analysis showed that autosomal mCA was associated with a 39% increased risk for breast cancer (HR, 1.39; 95% CI, 1.06-1.83; P = .01). No such associations were found between mCA and colorectal or lung cancer, regardless of cell fraction threshold.
The original 3% cell fraction threshold was selected on the basis of previous studies reporting a link between mCA and hematologic malignancies at this cutoff, Dr. Desai said.
She and her colleagues said a higher 5% cutoff might be needed, as they suspected that the link between mCA and solid tumors may not be causal, requiring a higher mutation rate.
Why Do Results Differ Between These Types of Studies?
Dr. Takahashi and Ms. Shah suggested that one possible limitation of the new study, and an obstacle to comparing results with the UK Biobank study and others like it, goes beyond population heterogeneity; incongruent findings could also be explained by differences in whole genome sequencing (WGS) technique.
“Although WGS allows sensitive detection of mCA through broad genomic coverage, it is less effective at detecting CHIP with low variant allele frequency (VAF) due to its relatively shallow depth (30x),” they wrote. “Consequently, the prevalence of mCA (18.8%) was much higher than that of CHIP (8.3%) in this cohort, contrasting with other studies using deeper sequencing.” As a result, the present study may have underestimated CHIP prevalence because of shallow sequencing depth.
“This inconsistency is a common challenge in CH population studies due to the lack of standardized methodologies and the frequent reliance on preexisting data not originally intended for CH detection,” Dr. Takahashi and Ms. Shah said.
Even so, despite the “heavily context-dependent” nature of these reported risks, the body of evidence to date now offers a convincing biological rationale linking CH with cancer development and outcomes, they added.
How Do the CHIP- and mCA-associated Risks Differ Between Solid Tumors and Blood Cancers?
“[These solid tumor risks are] not causal in the way CHIP mutations are causal for blood cancers,” Dr. Desai said. “Here we are talking about solid tumor risk, and it’s kind of scattered. It’s not just breast cancer ... there’s also increased colon cancer mortality. So I feel these mutations are doing something different ... they are sort of an added factor.”
Specific mechanisms remain unclear, Dr. Desai said, although she speculated about possible impacts on the inflammatory state or alterations to the tumor microenvironment.
“These are blood cells, right?” Dr. Desai asked. “They’re everywhere, and they’re changing something inherently in these tumors.”
Future research and therapeutic development
Siddhartha Jaiswal, MD, PhD, assistant professor in the Department of Pathology at Stanford University in California, whose lab focuses on clonal hematopoiesis, said the causality question is central to future research.
“The key question is, are these mutations acting because they alter the function of blood cells in some way to promote cancer risk, or is it reflective of some sort of shared etiology that’s not causal?” Dr. Jaiswal said in an interview.
Available data support both possibilities.
On one side, “reasonable evidence” supports the noncausal view, Dr. Jaiswal noted, because telomere length is one of the most common genetic risk factors for clonal hematopoiesis and also for solid tumors, suggesting a shared genetic factor. On the other hand, CHIP and mCA could be directly protumorigenic via conferred disturbances of immune cell function.
When asked if both causal and noncausal factors could be at play, Dr. Jaiswal said, “yeah, absolutely.”
The presence of a causal association could be promising from a therapeutic standpoint.
“If it turns out that this association is driven by a direct causal effect of the mutations, perhaps related to immune cell function or dysfunction, then targeting that dysfunction could be a therapeutic path to improve outcomes in people, and there’s a lot of interest in this,” Dr. Jaiswal said. He went on to explain how a trial exploring this approach via interleukin-8 inhibition in lung cancer fell short.
Yet earlier intervention may still hold promise, according to experts.
“[This study] provokes the hypothesis that CH‐targeted interventions could potentially reduce cancer risk in the future,” Dr. Takahashi and Ms. Shah said in their editorial.
The WHI program is funded by the National Heart, Lung, and Blood Institute; National Institutes of Health; and the Department of Health & Human Services. The investigators disclosed relationships with Eli Lilly, AbbVie, Celgene, and others. Dr. Jaiswal reported stock equity in a company that has an interest in clonal hematopoiesis.
A version of this article first appeared on Medscape.com.
Clonal hematopoiesis of indeterminate potential (CHIP) and mosaic chromosomal alterations (mCAs) are associated with an increased risk for breast cancer, and CHIP is associated with increased mortality in patients with colon cancer, according to the authors of new research.
These findings, drawn from almost 11,000 patients in the Women’s Health Initiative (WHI) study, add further evidence that CHIP and mCA drive solid tumor risk, alongside known associations with hematologic malignancies, reported lead author Pinkal Desai, MD, associate professor of medicine and clinical director of molecular aging at Englander Institute for Precision Medicine, Weill Cornell Medical College, New York City, and colleagues.
How This Study Differs From Others of Breast Cancer Risk Factors
“The independent effect of CHIP and mCA on risk and mortality from solid tumors has not been elucidated due to lack of detailed data on mortality outcomes and risk factors,” the investigators wrote in Cancer, although some previous studies have suggested a link.
In particular, the investigators highlighted a 2022 UK Biobank study, which reported an association between CHIP and lung cancer and a borderline association with breast cancer that did not quite reach statistical significance.
But the UK Biobank study was confined to a UK population, Dr. Desai noted in an interview, and the data were less detailed than those in the present investigation.
“In terms of risk, the part that was lacking in previous studies was a comprehensive assessment of risk factors that increase risk for all these cancers,” Dr. Desai said. “For example, for breast cancer, we had very detailed data on [participants’] Gail risk score, which is known to impact breast cancer risk. We also had mammogram data and colonoscopy data.”
In an accompanying editorial, Koichi Takahashi, MD, PhD , and Nehali Shah, BS, of The University of Texas MD Anderson Cancer Center, Houston, Texas, pointed out the same UK Biobank findings, then noted that CHIP has also been linked with worse overall survival in unselected cancer patients. Still, they wrote, “the impact of CH on cancer risk and mortality remains controversial due to conflicting data and context‐dependent effects,” necessitating studies like this one by Dr. Desai and colleagues.
How Was the Relationship Between CHIP, MCA, and Solid Tumor Risk Assessed?
To explore possible associations between CHIP, mCA, and solid tumors, the investigators analyzed whole genome sequencing data from 10,866 women in the WHI, a multi-study program that began in 1992 and involved 161,808 women in both observational and clinical trial cohorts.
In 2002, the first big data release from the WHI suggested that hormone replacement therapy (HRT) increased breast cancer risk, leading to widespread reduction in HRT use.
More recent reports continue to shape our understanding of these risks, suggesting differences across cancer types. For breast cancer, the WHI data suggested that HRT-associated risk was largely driven by formulations involving progesterone and estrogen, whereas estrogen-only formulations, now more common, are generally considered to present an acceptable risk profile for suitable patients.
The new study accounted for this potential HRT-associated risk, including by adjusting for patients who received HRT, type of HRT received, and duration of HRT received. According to Desai, this approach is commonly used when analyzing data from the WHI, nullifying concerns about the potentially deleterious effects of the hormones used in the study.
“Our question was not ‘does HRT cause cancer?’ ” Dr. Desai said in an interview. “But HRT can be linked to breast cancer risk and has a potential to be a confounder, and hence the above methodology.
“So I can say that the confounding/effect modification that HRT would have contributed to in the relationship between exposure (CH and mCA) and outcome (cancer) is well adjusted for as described above. This is standard in WHI analyses,” she continued.
“Every Women’s Health Initiative analysis that comes out — not just for our study — uses a standard method ... where you account for hormonal therapy,” Dr. Desai added, again noting that many other potential risk factors were considered, enabling a “detailed, robust” analysis.
Dr. Takahashi and Ms. Shah agreed. “A notable strength of this study is its adjustment for many confounding factors,” they wrote. “The cohort’s well‐annotated data on other known cancer risk factors allowed for a robust assessment of CH’s independent risk.”
How Do Findings Compare With Those of the UK Biobank Study?
CHIP was associated with a 30% increased risk for breast cancer (hazard ratio [HR], 1.30; 95% CI, 1.03-1.64; P = .02), strengthening the borderline association reported by the UK Biobank study.
In contrast with the UK Biobank study, CHIP was not associated with lung cancer risk, although this may have been caused by fewer cases of lung cancer and a lack of male patients, Dr. Desai suggested.
“The discrepancy between the studies lies in the risk of lung cancer, although the point estimate in the current study suggested a positive association,” wrote Dr. Takahashi and Ms. Shah.
As in the UK Biobank study, CHIP was not associated with increased risk of developing colorectal cancer.
Mortality analysis, however, which was not conducted in the UK Biobank study, offered a new insight: Patients with existing colorectal cancer and CHIP had a significantly higher mortality risk than those without CHIP. Before stage adjustment, risk for mortality among those with colorectal cancer and CHIP was fourfold higher than those without CHIP (HR, 3.99; 95% CI, 2.41-6.62; P < .001). After stage adjustment, CHIP was still associated with a twofold higher mortality risk (HR, 2.50; 95% CI, 1.32-4.72; P = .004).
The investigators’ first mCA analyses, which employed a cell fraction cutoff greater than 3%, were unfruitful. But raising the cell fraction threshold to 5% in an exploratory analysis showed that autosomal mCA was associated with a 39% increased risk for breast cancer (HR, 1.39; 95% CI, 1.06-1.83; P = .01). No such associations were found between mCA and colorectal or lung cancer, regardless of cell fraction threshold.
The original 3% cell fraction threshold was selected on the basis of previous studies reporting a link between mCA and hematologic malignancies at this cutoff, Dr. Desai said.
She and her colleagues said a higher 5% cutoff might be needed, as they suspected that the link between mCA and solid tumors may not be causal, requiring a higher mutation rate.
Why Do Results Differ Between These Types of Studies?
Dr. Takahashi and Ms. Shah suggested that one possible limitation of the new study, and an obstacle to comparing results with the UK Biobank study and others like it, goes beyond population heterogeneity; incongruent findings could also be explained by differences in whole genome sequencing (WGS) technique.
“Although WGS allows sensitive detection of mCA through broad genomic coverage, it is less effective at detecting CHIP with low variant allele frequency (VAF) due to its relatively shallow depth (30x),” they wrote. “Consequently, the prevalence of mCA (18.8%) was much higher than that of CHIP (8.3%) in this cohort, contrasting with other studies using deeper sequencing.” As a result, the present study may have underestimated CHIP prevalence because of shallow sequencing depth.
“This inconsistency is a common challenge in CH population studies due to the lack of standardized methodologies and the frequent reliance on preexisting data not originally intended for CH detection,” Dr. Takahashi and Ms. Shah said.
Even so, despite the “heavily context-dependent” nature of these reported risks, the body of evidence to date now offers a convincing biological rationale linking CH with cancer development and outcomes, they added.
How Do the CHIP- and mCA-associated Risks Differ Between Solid Tumors and Blood Cancers?
“[These solid tumor risks are] not causal in the way CHIP mutations are causal for blood cancers,” Dr. Desai said. “Here we are talking about solid tumor risk, and it’s kind of scattered. It’s not just breast cancer ... there’s also increased colon cancer mortality. So I feel these mutations are doing something different ... they are sort of an added factor.”
Specific mechanisms remain unclear, Dr. Desai said, although she speculated about possible impacts on the inflammatory state or alterations to the tumor microenvironment.
“These are blood cells, right?” Dr. Desai asked. “They’re everywhere, and they’re changing something inherently in these tumors.”
Future research and therapeutic development
Siddhartha Jaiswal, MD, PhD, assistant professor in the Department of Pathology at Stanford University in California, whose lab focuses on clonal hematopoiesis, said the causality question is central to future research.
“The key question is, are these mutations acting because they alter the function of blood cells in some way to promote cancer risk, or is it reflective of some sort of shared etiology that’s not causal?” Dr. Jaiswal said in an interview.
Available data support both possibilities.
On one side, “reasonable evidence” supports the noncausal view, Dr. Jaiswal noted, because telomere length is one of the most common genetic risk factors for clonal hematopoiesis and also for solid tumors, suggesting a shared genetic factor. On the other hand, CHIP and mCA could be directly protumorigenic via conferred disturbances of immune cell function.
When asked if both causal and noncausal factors could be at play, Dr. Jaiswal said, “yeah, absolutely.”
The presence of a causal association could be promising from a therapeutic standpoint.
“If it turns out that this association is driven by a direct causal effect of the mutations, perhaps related to immune cell function or dysfunction, then targeting that dysfunction could be a therapeutic path to improve outcomes in people, and there’s a lot of interest in this,” Dr. Jaiswal said. He went on to explain how a trial exploring this approach via interleukin-8 inhibition in lung cancer fell short.
Yet earlier intervention may still hold promise, according to experts.
“[This study] provokes the hypothesis that CH‐targeted interventions could potentially reduce cancer risk in the future,” Dr. Takahashi and Ms. Shah said in their editorial.
The WHI program is funded by the National Heart, Lung, and Blood Institute; National Institutes of Health; and the Department of Health & Human Services. The investigators disclosed relationships with Eli Lilly, AbbVie, Celgene, and others. Dr. Jaiswal reported stock equity in a company that has an interest in clonal hematopoiesis.
A version of this article first appeared on Medscape.com.
Clonal hematopoiesis of indeterminate potential (CHIP) and mosaic chromosomal alterations (mCAs) are associated with an increased risk for breast cancer, and CHIP is associated with increased mortality in patients with colon cancer, according to the authors of new research.
These findings, drawn from almost 11,000 patients in the Women’s Health Initiative (WHI) study, add further evidence that CHIP and mCA drive solid tumor risk, alongside known associations with hematologic malignancies, reported lead author Pinkal Desai, MD, associate professor of medicine and clinical director of molecular aging at Englander Institute for Precision Medicine, Weill Cornell Medical College, New York City, and colleagues.
How This Study Differs From Others of Breast Cancer Risk Factors
“The independent effect of CHIP and mCA on risk and mortality from solid tumors has not been elucidated due to lack of detailed data on mortality outcomes and risk factors,” the investigators wrote in Cancer, although some previous studies have suggested a link.
In particular, the investigators highlighted a 2022 UK Biobank study, which reported an association between CHIP and lung cancer and a borderline association with breast cancer that did not quite reach statistical significance.
But the UK Biobank study was confined to a UK population, Dr. Desai noted in an interview, and the data were less detailed than those in the present investigation.
“In terms of risk, the part that was lacking in previous studies was a comprehensive assessment of risk factors that increase risk for all these cancers,” Dr. Desai said. “For example, for breast cancer, we had very detailed data on [participants’] Gail risk score, which is known to impact breast cancer risk. We also had mammogram data and colonoscopy data.”
In an accompanying editorial, Koichi Takahashi, MD, PhD , and Nehali Shah, BS, of The University of Texas MD Anderson Cancer Center, Houston, Texas, pointed out the same UK Biobank findings, then noted that CHIP has also been linked with worse overall survival in unselected cancer patients. Still, they wrote, “the impact of CH on cancer risk and mortality remains controversial due to conflicting data and context‐dependent effects,” necessitating studies like this one by Dr. Desai and colleagues.
How Was the Relationship Between CHIP, MCA, and Solid Tumor Risk Assessed?
To explore possible associations between CHIP, mCA, and solid tumors, the investigators analyzed whole genome sequencing data from 10,866 women in the WHI, a multi-study program that began in 1992 and involved 161,808 women in both observational and clinical trial cohorts.
In 2002, the first big data release from the WHI suggested that hormone replacement therapy (HRT) increased breast cancer risk, leading to widespread reduction in HRT use.
More recent reports continue to shape our understanding of these risks, suggesting differences across cancer types. For breast cancer, the WHI data suggested that HRT-associated risk was largely driven by formulations involving progesterone and estrogen, whereas estrogen-only formulations, now more common, are generally considered to present an acceptable risk profile for suitable patients.
The new study accounted for this potential HRT-associated risk, including by adjusting for patients who received HRT, type of HRT received, and duration of HRT received. According to Desai, this approach is commonly used when analyzing data from the WHI, nullifying concerns about the potentially deleterious effects of the hormones used in the study.
“Our question was not ‘does HRT cause cancer?’ ” Dr. Desai said in an interview. “But HRT can be linked to breast cancer risk and has a potential to be a confounder, and hence the above methodology.
“So I can say that the confounding/effect modification that HRT would have contributed to in the relationship between exposure (CH and mCA) and outcome (cancer) is well adjusted for as described above. This is standard in WHI analyses,” she continued.
“Every Women’s Health Initiative analysis that comes out — not just for our study — uses a standard method ... where you account for hormonal therapy,” Dr. Desai added, again noting that many other potential risk factors were considered, enabling a “detailed, robust” analysis.
Dr. Takahashi and Ms. Shah agreed. “A notable strength of this study is its adjustment for many confounding factors,” they wrote. “The cohort’s well‐annotated data on other known cancer risk factors allowed for a robust assessment of CH’s independent risk.”
How Do Findings Compare With Those of the UK Biobank Study?
CHIP was associated with a 30% increased risk for breast cancer (hazard ratio [HR], 1.30; 95% CI, 1.03-1.64; P = .02), strengthening the borderline association reported by the UK Biobank study.
In contrast with the UK Biobank study, CHIP was not associated with lung cancer risk, although this may have been caused by fewer cases of lung cancer and a lack of male patients, Dr. Desai suggested.
“The discrepancy between the studies lies in the risk of lung cancer, although the point estimate in the current study suggested a positive association,” wrote Dr. Takahashi and Ms. Shah.
As in the UK Biobank study, CHIP was not associated with increased risk of developing colorectal cancer.
Mortality analysis, however, which was not conducted in the UK Biobank study, offered a new insight: Patients with existing colorectal cancer and CHIP had a significantly higher mortality risk than those without CHIP. Before stage adjustment, risk for mortality among those with colorectal cancer and CHIP was fourfold higher than those without CHIP (HR, 3.99; 95% CI, 2.41-6.62; P < .001). After stage adjustment, CHIP was still associated with a twofold higher mortality risk (HR, 2.50; 95% CI, 1.32-4.72; P = .004).
The investigators’ first mCA analyses, which employed a cell fraction cutoff greater than 3%, were unfruitful. But raising the cell fraction threshold to 5% in an exploratory analysis showed that autosomal mCA was associated with a 39% increased risk for breast cancer (HR, 1.39; 95% CI, 1.06-1.83; P = .01). No such associations were found between mCA and colorectal or lung cancer, regardless of cell fraction threshold.
The original 3% cell fraction threshold was selected on the basis of previous studies reporting a link between mCA and hematologic malignancies at this cutoff, Dr. Desai said.
She and her colleagues said a higher 5% cutoff might be needed, as they suspected that the link between mCA and solid tumors may not be causal, requiring a higher mutation rate.
Why Do Results Differ Between These Types of Studies?
Dr. Takahashi and Ms. Shah suggested that one possible limitation of the new study, and an obstacle to comparing results with the UK Biobank study and others like it, goes beyond population heterogeneity; incongruent findings could also be explained by differences in whole genome sequencing (WGS) technique.
“Although WGS allows sensitive detection of mCA through broad genomic coverage, it is less effective at detecting CHIP with low variant allele frequency (VAF) due to its relatively shallow depth (30x),” they wrote. “Consequently, the prevalence of mCA (18.8%) was much higher than that of CHIP (8.3%) in this cohort, contrasting with other studies using deeper sequencing.” As a result, the present study may have underestimated CHIP prevalence because of shallow sequencing depth.
“This inconsistency is a common challenge in CH population studies due to the lack of standardized methodologies and the frequent reliance on preexisting data not originally intended for CH detection,” Dr. Takahashi and Ms. Shah said.
Even so, despite the “heavily context-dependent” nature of these reported risks, the body of evidence to date now offers a convincing biological rationale linking CH with cancer development and outcomes, they added.
How Do the CHIP- and mCA-associated Risks Differ Between Solid Tumors and Blood Cancers?
“[These solid tumor risks are] not causal in the way CHIP mutations are causal for blood cancers,” Dr. Desai said. “Here we are talking about solid tumor risk, and it’s kind of scattered. It’s not just breast cancer ... there’s also increased colon cancer mortality. So I feel these mutations are doing something different ... they are sort of an added factor.”
Specific mechanisms remain unclear, Dr. Desai said, although she speculated about possible impacts on the inflammatory state or alterations to the tumor microenvironment.
“These are blood cells, right?” Dr. Desai asked. “They’re everywhere, and they’re changing something inherently in these tumors.”
Future research and therapeutic development
Siddhartha Jaiswal, MD, PhD, assistant professor in the Department of Pathology at Stanford University in California, whose lab focuses on clonal hematopoiesis, said the causality question is central to future research.
“The key question is, are these mutations acting because they alter the function of blood cells in some way to promote cancer risk, or is it reflective of some sort of shared etiology that’s not causal?” Dr. Jaiswal said in an interview.
Available data support both possibilities.
On one side, “reasonable evidence” supports the noncausal view, Dr. Jaiswal noted, because telomere length is one of the most common genetic risk factors for clonal hematopoiesis and also for solid tumors, suggesting a shared genetic factor. On the other hand, CHIP and mCA could be directly protumorigenic via conferred disturbances of immune cell function.
When asked if both causal and noncausal factors could be at play, Dr. Jaiswal said, “yeah, absolutely.”
The presence of a causal association could be promising from a therapeutic standpoint.
“If it turns out that this association is driven by a direct causal effect of the mutations, perhaps related to immune cell function or dysfunction, then targeting that dysfunction could be a therapeutic path to improve outcomes in people, and there’s a lot of interest in this,” Dr. Jaiswal said. He went on to explain how a trial exploring this approach via interleukin-8 inhibition in lung cancer fell short.
Yet earlier intervention may still hold promise, according to experts.
“[This study] provokes the hypothesis that CH‐targeted interventions could potentially reduce cancer risk in the future,” Dr. Takahashi and Ms. Shah said in their editorial.
The WHI program is funded by the National Heart, Lung, and Blood Institute; National Institutes of Health; and the Department of Health & Human Services. The investigators disclosed relationships with Eli Lilly, AbbVie, Celgene, and others. Dr. Jaiswal reported stock equity in a company that has an interest in clonal hematopoiesis.
A version of this article first appeared on Medscape.com.
FROM CANCER
Stroke Risk from Atrial Fibrillation Rises in Presence of Rheumatoid Arthritis
TOPLINE:
Patients with both rheumatoid arthritis (RA) and atrial fibrillation (AF) have a higher risk for ischemic stroke than those with only AF. They are also less likely to receive oral anticoagulant treatment, which may contribute to this increased stroke risk.
METHODOLOGY:
- Researchers conducted a registry-based retrospective cohort study using the Norwegian Cardio-Rheuma Register to evaluate the risk for ischemic stroke following the diagnosis of AF in patients with or without RA.
- They included 163,595 patients with newly diagnosed AF between 2010 and 2017, of whom 2750 had RA. Patients had to be diagnosed with RA before the diagnosis of AF.
- They also assessed whether patients with RA were less likely to receive oral anticoagulants for stroke prevention within 3 months of AF diagnosis than those without RA.
- The median follow-up time was 2.5 years for patients with RA and 3.0 years for those without RA.
- The primary endpoint was ischemic stroke, which was identified through hospital admissions and visits.
TAKEAWAY:
- At 5 years, patients with both RA and AF showed a higher cumulative incidence of ischemic stroke than those with only AF (7.3% vs 5.0%).
- Among patients with AF, the risk of having a stroke was 25% higher in those with RA than in those without RA (adjusted hazard ratio, 1.25; 95% CI, 1.05-1.50).
- Patients with RA were also less likely to receive treatment with oral anticoagulants than those without RA, driven by concerns over potential interactions with RA medications, bleeding risk, or other factors (adjusted odds ratio, 0.88; 95% CI, 0.80-0.97).
IN PRACTICE:
“Our study prompts preventive measures such as meticulous cardiovascular risk factor control among patients with RA and AF and raises the question whether the presence of RA should be taken into account when considering OAC [oral anticoagulant] treatment for AF patients,” the authors wrote.
SOURCE:
This study was led by Anne M. Kerola, MD, PhD, Helsinki University Hospital and University of Helsinki in Finland. It was published online in Rheumatology.
LIMITATIONS:
This study lacked data on smoking, blood pressure measurements, alcohol use, and obesity, which may have affected the comprehensiveness of the findings. The study population was limited to Norway and may not be generalizable to other populations.
DISCLOSURES:
This study was supported by the Olav Thon Foundation, the Research Council of Norway, and the Foundation for Research in Rheumatology. Some authors received speaker fees, participated in advisory boards, served as consultants, or had other ties with some pharmaceutical companies and institutions.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Patients with both rheumatoid arthritis (RA) and atrial fibrillation (AF) have a higher risk for ischemic stroke than those with only AF. They are also less likely to receive oral anticoagulant treatment, which may contribute to this increased stroke risk.
METHODOLOGY:
- Researchers conducted a registry-based retrospective cohort study using the Norwegian Cardio-Rheuma Register to evaluate the risk for ischemic stroke following the diagnosis of AF in patients with or without RA.
- They included 163,595 patients with newly diagnosed AF between 2010 and 2017, of whom 2750 had RA. Patients had to be diagnosed with RA before the diagnosis of AF.
- They also assessed whether patients with RA were less likely to receive oral anticoagulants for stroke prevention within 3 months of AF diagnosis than those without RA.
- The median follow-up time was 2.5 years for patients with RA and 3.0 years for those without RA.
- The primary endpoint was ischemic stroke, which was identified through hospital admissions and visits.
TAKEAWAY:
- At 5 years, patients with both RA and AF showed a higher cumulative incidence of ischemic stroke than those with only AF (7.3% vs 5.0%).
- Among patients with AF, the risk of having a stroke was 25% higher in those with RA than in those without RA (adjusted hazard ratio, 1.25; 95% CI, 1.05-1.50).
- Patients with RA were also less likely to receive treatment with oral anticoagulants than those without RA, driven by concerns over potential interactions with RA medications, bleeding risk, or other factors (adjusted odds ratio, 0.88; 95% CI, 0.80-0.97).
IN PRACTICE:
“Our study prompts preventive measures such as meticulous cardiovascular risk factor control among patients with RA and AF and raises the question whether the presence of RA should be taken into account when considering OAC [oral anticoagulant] treatment for AF patients,” the authors wrote.
SOURCE:
This study was led by Anne M. Kerola, MD, PhD, Helsinki University Hospital and University of Helsinki in Finland. It was published online in Rheumatology.
LIMITATIONS:
This study lacked data on smoking, blood pressure measurements, alcohol use, and obesity, which may have affected the comprehensiveness of the findings. The study population was limited to Norway and may not be generalizable to other populations.
DISCLOSURES:
This study was supported by the Olav Thon Foundation, the Research Council of Norway, and the Foundation for Research in Rheumatology. Some authors received speaker fees, participated in advisory boards, served as consultants, or had other ties with some pharmaceutical companies and institutions.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Patients with both rheumatoid arthritis (RA) and atrial fibrillation (AF) have a higher risk for ischemic stroke than those with only AF. They are also less likely to receive oral anticoagulant treatment, which may contribute to this increased stroke risk.
METHODOLOGY:
- Researchers conducted a registry-based retrospective cohort study using the Norwegian Cardio-Rheuma Register to evaluate the risk for ischemic stroke following the diagnosis of AF in patients with or without RA.
- They included 163,595 patients with newly diagnosed AF between 2010 and 2017, of whom 2750 had RA. Patients had to be diagnosed with RA before the diagnosis of AF.
- They also assessed whether patients with RA were less likely to receive oral anticoagulants for stroke prevention within 3 months of AF diagnosis than those without RA.
- The median follow-up time was 2.5 years for patients with RA and 3.0 years for those without RA.
- The primary endpoint was ischemic stroke, which was identified through hospital admissions and visits.
TAKEAWAY:
- At 5 years, patients with both RA and AF showed a higher cumulative incidence of ischemic stroke than those with only AF (7.3% vs 5.0%).
- Among patients with AF, the risk of having a stroke was 25% higher in those with RA than in those without RA (adjusted hazard ratio, 1.25; 95% CI, 1.05-1.50).
- Patients with RA were also less likely to receive treatment with oral anticoagulants than those without RA, driven by concerns over potential interactions with RA medications, bleeding risk, or other factors (adjusted odds ratio, 0.88; 95% CI, 0.80-0.97).
IN PRACTICE:
“Our study prompts preventive measures such as meticulous cardiovascular risk factor control among patients with RA and AF and raises the question whether the presence of RA should be taken into account when considering OAC [oral anticoagulant] treatment for AF patients,” the authors wrote.
SOURCE:
This study was led by Anne M. Kerola, MD, PhD, Helsinki University Hospital and University of Helsinki in Finland. It was published online in Rheumatology.
LIMITATIONS:
This study lacked data on smoking, blood pressure measurements, alcohol use, and obesity, which may have affected the comprehensiveness of the findings. The study population was limited to Norway and may not be generalizable to other populations.
DISCLOSURES:
This study was supported by the Olav Thon Foundation, the Research Council of Norway, and the Foundation for Research in Rheumatology. Some authors received speaker fees, participated in advisory boards, served as consultants, or had other ties with some pharmaceutical companies and institutions.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
As Interest From Families Wanes, Pediatricians Scale Back on COVID Shots
When pediatrician Eric Ball, MD, opened a refrigerator full of childhood vaccines, all the expected shots were there — DTaP, polio, pneumococcal vaccine — except one.
“This is where we usually store our COVID vaccines, but we don’t have any right now because they all expired at the end of last year and we had to dispose of them,” said Dr. Ball, who is part of a pediatric practice in Orange County, California.
“We thought demand would be way higher than it was.”
Providers like Dr. Ball don’t want to waste money ordering doses that won’t be used, but they need enough on hand to vaccinate vulnerable children.
The Centers for Disease Control and Prevention recommends that anyone 6 months or older get the updated COVID vaccination, but in the 2023-24 vaccination season only about 15% of eligible children in the United States got a shot.
Dr. Ball said it was difficult to let vaccines go to waste in 2023. It was the first time the federal government was no longer picking up the tab for the shots, and providers had to pay upfront for the vaccines. Parents would often skip the COVID shot, which can have a very short shelf life, compared with other vaccines.
“Watching it sitting on our shelves expiring every 30 days, that’s like throwing away $150 repeatedly every day, multiple times a month,” Dr. Ball said.
in 2024, Dr. Ball slashed his fall vaccine order to the bare minimum to avoid another costly mistake.
“We took the number of flu vaccines that we order, and then we ordered 5% of that in COVID vaccines,” Dr. Ball said. “It’s a guess.”
That small vaccine order cost more than $63,000, he said.
Pharmacists, pharmacy interns, and techs are allowed to give COVID vaccines only to children age 3 and up, meaning babies and toddlers would need to visit a doctor’s office for inoculation.
It’s difficult to predict how parents will feel about the shots this fall, said Chicago pediatrician Scott Goldstein, MD. Unlike other vaccinations, COVID shots aren’t required for kids to attend school, and parental interest seems to wane with each new formulation. For a physician-owned practice such as Dr. Goldstein’s, the upfront cost of the vaccine can be a gamble.
“The cost of vaccines, that’s far and away our biggest expense. But it’s also the most important thing we do, you could argue, is vaccinating kids,” Dr. Goldstein said.
Insurance doesn’t necessarily cover vaccine storage accidents, which can put the practice at risk of financial ruin.
“We’ve had things happen like a refrigerator gets unplugged. And then we’re all of a sudden out $80,000 overnight,” Dr. Goldstein said.
South Carolina pediatrician Deborah Greenhouse, MD, said she would order more COVID vaccines for older children if the pharmaceutical companies that she buys from had a more forgiving return policy.
“Pfizer is creating that situation. If you’re only going to let us return 30%, we’re not going to buy much,” she said. “We can’t.”
Greenhouse owns her practice, so the remaining 70% of leftover shots would come out of her pocket.
Vaccine maker Pfizer will take back all unused COVID shots for young children, but only 30% of doses for people 12 and older.
Pfizer said in an Aug. 20 emailed statement, “The return policy was instituted as we recognize both the importance and the complexity of pediatric vaccination and wanted to ensure that pediatric offices did not have hurdles to providing vaccine to their young patients.”
Pfizer’s return policy is similar to policies from other drugmakers for pediatric flu vaccines, also recommended during the fall season. Physicians who are worried about unwanted COVID vaccines expiring on the shelves said flu shots cost them about $20 per dose, while COVID shots cost around $150 per dose.
“We run on a very thin margin. If we get stuck holding a ton of vaccine that we cannot return, we can’t absorb that kind of cost,” Dr. Greenhouse said.
Vaccine maker Moderna will accept COVID vaccine returns, but the amount depends on the individual contract with a provider. Novavax will accept the return of only unopened vaccines and doesn’t specify the amount they’ll accept.
Dr. Greenhouse wants to vaccinate as many children as possible but said she can’t afford to stock shots with a short shelf life. Once she runs out of the doses she’s ordered, Dr. Greenhouse plans to tell families to go to a pharmacy to get older children vaccinated. If pediatricians around the country are making the same calculations, doses for very small children could be harder to find at doctors’ offices.
“Frankly, it’s not an ideal situation, but it’s what we have to do to stay in business,” she said.
Dr. Ball worries that parents’ limited interest has caused pediatricians to minimize their vaccine orders, in turn making the newest COVID shots difficult to find once they become available.
“I think there’s just a misperception that it’s less of a big deal to get COVID, but I’m still sending babies to the hospital with COVID,” Dr. Ball said. “We’re still seeing kids with long COVID. This is with us forever.”
KFF Health News is a national newsroom that produces in-depth journalism about health issues and is one of the core operating programs at KFF — the independent source for health policy research, polling, and journalism.
When pediatrician Eric Ball, MD, opened a refrigerator full of childhood vaccines, all the expected shots were there — DTaP, polio, pneumococcal vaccine — except one.
“This is where we usually store our COVID vaccines, but we don’t have any right now because they all expired at the end of last year and we had to dispose of them,” said Dr. Ball, who is part of a pediatric practice in Orange County, California.
“We thought demand would be way higher than it was.”
Providers like Dr. Ball don’t want to waste money ordering doses that won’t be used, but they need enough on hand to vaccinate vulnerable children.
The Centers for Disease Control and Prevention recommends that anyone 6 months or older get the updated COVID vaccination, but in the 2023-24 vaccination season only about 15% of eligible children in the United States got a shot.
Dr. Ball said it was difficult to let vaccines go to waste in 2023. It was the first time the federal government was no longer picking up the tab for the shots, and providers had to pay upfront for the vaccines. Parents would often skip the COVID shot, which can have a very short shelf life, compared with other vaccines.
“Watching it sitting on our shelves expiring every 30 days, that’s like throwing away $150 repeatedly every day, multiple times a month,” Dr. Ball said.
in 2024, Dr. Ball slashed his fall vaccine order to the bare minimum to avoid another costly mistake.
“We took the number of flu vaccines that we order, and then we ordered 5% of that in COVID vaccines,” Dr. Ball said. “It’s a guess.”
That small vaccine order cost more than $63,000, he said.
Pharmacists, pharmacy interns, and techs are allowed to give COVID vaccines only to children age 3 and up, meaning babies and toddlers would need to visit a doctor’s office for inoculation.
It’s difficult to predict how parents will feel about the shots this fall, said Chicago pediatrician Scott Goldstein, MD. Unlike other vaccinations, COVID shots aren’t required for kids to attend school, and parental interest seems to wane with each new formulation. For a physician-owned practice such as Dr. Goldstein’s, the upfront cost of the vaccine can be a gamble.
“The cost of vaccines, that’s far and away our biggest expense. But it’s also the most important thing we do, you could argue, is vaccinating kids,” Dr. Goldstein said.
Insurance doesn’t necessarily cover vaccine storage accidents, which can put the practice at risk of financial ruin.
“We’ve had things happen like a refrigerator gets unplugged. And then we’re all of a sudden out $80,000 overnight,” Dr. Goldstein said.
South Carolina pediatrician Deborah Greenhouse, MD, said she would order more COVID vaccines for older children if the pharmaceutical companies that she buys from had a more forgiving return policy.
“Pfizer is creating that situation. If you’re only going to let us return 30%, we’re not going to buy much,” she said. “We can’t.”
Greenhouse owns her practice, so the remaining 70% of leftover shots would come out of her pocket.
Vaccine maker Pfizer will take back all unused COVID shots for young children, but only 30% of doses for people 12 and older.
Pfizer said in an Aug. 20 emailed statement, “The return policy was instituted as we recognize both the importance and the complexity of pediatric vaccination and wanted to ensure that pediatric offices did not have hurdles to providing vaccine to their young patients.”
Pfizer’s return policy is similar to policies from other drugmakers for pediatric flu vaccines, also recommended during the fall season. Physicians who are worried about unwanted COVID vaccines expiring on the shelves said flu shots cost them about $20 per dose, while COVID shots cost around $150 per dose.
“We run on a very thin margin. If we get stuck holding a ton of vaccine that we cannot return, we can’t absorb that kind of cost,” Dr. Greenhouse said.
Vaccine maker Moderna will accept COVID vaccine returns, but the amount depends on the individual contract with a provider. Novavax will accept the return of only unopened vaccines and doesn’t specify the amount they’ll accept.
Dr. Greenhouse wants to vaccinate as many children as possible but said she can’t afford to stock shots with a short shelf life. Once she runs out of the doses she’s ordered, Dr. Greenhouse plans to tell families to go to a pharmacy to get older children vaccinated. If pediatricians around the country are making the same calculations, doses for very small children could be harder to find at doctors’ offices.
“Frankly, it’s not an ideal situation, but it’s what we have to do to stay in business,” she said.
Dr. Ball worries that parents’ limited interest has caused pediatricians to minimize their vaccine orders, in turn making the newest COVID shots difficult to find once they become available.
“I think there’s just a misperception that it’s less of a big deal to get COVID, but I’m still sending babies to the hospital with COVID,” Dr. Ball said. “We’re still seeing kids with long COVID. This is with us forever.”
KFF Health News is a national newsroom that produces in-depth journalism about health issues and is one of the core operating programs at KFF — the independent source for health policy research, polling, and journalism.
When pediatrician Eric Ball, MD, opened a refrigerator full of childhood vaccines, all the expected shots were there — DTaP, polio, pneumococcal vaccine — except one.
“This is where we usually store our COVID vaccines, but we don’t have any right now because they all expired at the end of last year and we had to dispose of them,” said Dr. Ball, who is part of a pediatric practice in Orange County, California.
“We thought demand would be way higher than it was.”
Providers like Dr. Ball don’t want to waste money ordering doses that won’t be used, but they need enough on hand to vaccinate vulnerable children.
The Centers for Disease Control and Prevention recommends that anyone 6 months or older get the updated COVID vaccination, but in the 2023-24 vaccination season only about 15% of eligible children in the United States got a shot.
Dr. Ball said it was difficult to let vaccines go to waste in 2023. It was the first time the federal government was no longer picking up the tab for the shots, and providers had to pay upfront for the vaccines. Parents would often skip the COVID shot, which can have a very short shelf life, compared with other vaccines.
“Watching it sitting on our shelves expiring every 30 days, that’s like throwing away $150 repeatedly every day, multiple times a month,” Dr. Ball said.
in 2024, Dr. Ball slashed his fall vaccine order to the bare minimum to avoid another costly mistake.
“We took the number of flu vaccines that we order, and then we ordered 5% of that in COVID vaccines,” Dr. Ball said. “It’s a guess.”
That small vaccine order cost more than $63,000, he said.
Pharmacists, pharmacy interns, and techs are allowed to give COVID vaccines only to children age 3 and up, meaning babies and toddlers would need to visit a doctor’s office for inoculation.
It’s difficult to predict how parents will feel about the shots this fall, said Chicago pediatrician Scott Goldstein, MD. Unlike other vaccinations, COVID shots aren’t required for kids to attend school, and parental interest seems to wane with each new formulation. For a physician-owned practice such as Dr. Goldstein’s, the upfront cost of the vaccine can be a gamble.
“The cost of vaccines, that’s far and away our biggest expense. But it’s also the most important thing we do, you could argue, is vaccinating kids,” Dr. Goldstein said.
Insurance doesn’t necessarily cover vaccine storage accidents, which can put the practice at risk of financial ruin.
“We’ve had things happen like a refrigerator gets unplugged. And then we’re all of a sudden out $80,000 overnight,” Dr. Goldstein said.
South Carolina pediatrician Deborah Greenhouse, MD, said she would order more COVID vaccines for older children if the pharmaceutical companies that she buys from had a more forgiving return policy.
“Pfizer is creating that situation. If you’re only going to let us return 30%, we’re not going to buy much,” she said. “We can’t.”
Greenhouse owns her practice, so the remaining 70% of leftover shots would come out of her pocket.
Vaccine maker Pfizer will take back all unused COVID shots for young children, but only 30% of doses for people 12 and older.
Pfizer said in an Aug. 20 emailed statement, “The return policy was instituted as we recognize both the importance and the complexity of pediatric vaccination and wanted to ensure that pediatric offices did not have hurdles to providing vaccine to their young patients.”
Pfizer’s return policy is similar to policies from other drugmakers for pediatric flu vaccines, also recommended during the fall season. Physicians who are worried about unwanted COVID vaccines expiring on the shelves said flu shots cost them about $20 per dose, while COVID shots cost around $150 per dose.
“We run on a very thin margin. If we get stuck holding a ton of vaccine that we cannot return, we can’t absorb that kind of cost,” Dr. Greenhouse said.
Vaccine maker Moderna will accept COVID vaccine returns, but the amount depends on the individual contract with a provider. Novavax will accept the return of only unopened vaccines and doesn’t specify the amount they’ll accept.
Dr. Greenhouse wants to vaccinate as many children as possible but said she can’t afford to stock shots with a short shelf life. Once she runs out of the doses she’s ordered, Dr. Greenhouse plans to tell families to go to a pharmacy to get older children vaccinated. If pediatricians around the country are making the same calculations, doses for very small children could be harder to find at doctors’ offices.
“Frankly, it’s not an ideal situation, but it’s what we have to do to stay in business,” she said.
Dr. Ball worries that parents’ limited interest has caused pediatricians to minimize their vaccine orders, in turn making the newest COVID shots difficult to find once they become available.
“I think there’s just a misperception that it’s less of a big deal to get COVID, but I’m still sending babies to the hospital with COVID,” Dr. Ball said. “We’re still seeing kids with long COVID. This is with us forever.”
KFF Health News is a national newsroom that produces in-depth journalism about health issues and is one of the core operating programs at KFF — the independent source for health policy research, polling, and journalism.
RSV Updates: Prophylaxis Approval and Hospitalization for Severe RSV
1. Pfizer announces positive top-line results from phase 3 study of ABRYSVO® in adults aged 18 to 59 at increased risk for RSV disease. Press release. Pfizer; April 9, 2024. Accessed May 22, 2024. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-positive-top-line-results-phase-3-study-1
2. Pfizer announces positive top-line data for full season two efficacy of ABRYSVO® for RSV in older adults. Press release. Pfizer; February 29, 2024. Accessed May 22, 2024. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-positive-top-line-data-full-season-two
3. CDC study shows effectiveness of RSV immunization for infants. Press release. US Centers for Disease Control and Prevention; March 7, 2024. Accessed May 22, 2024. https://www.cdc.gov/media/releases/2024/s0307-rsv-immunization.html
4. Moline HL, Tannis A, Toepfer AP, et al. Early estimate of nirsevimab effectiveness for prevention of respiratory syncytial virus–associated hospitalization among infants entering their first respiratory syncytial virus season — new vaccine surveillance network, October 2023–February 2024. MMWR Morb Mortal Wkly Rep. 2024;73(9):209-214. doi:10.15585/mmwr.mm7309a4
5. Havers FP, Whitaker M, Melgar M, et al; for the RSV-NET Surveillance Team. Characteristics and outcomes among adults aged ≥60 years hospitalized with laboratory-confirmed respiratory syncytial virus ─ RSV-NET, 12 states, July 2022–June 2023. MMWR Morb Mortal Wkly Rep. 2023;72(40):1075-1082. doi:10.15585/mmwr.mm7240a1
6. Walsh EE, Pérez Marc G, Zareba AM, et al; for the RENOIR Clinical Trial Group. Efficacy and safety of a bivalent RSV prefusion F vaccine in older adults. N Engl J Med. 2023;388(16):1465-1477. doi:10.1056/NEJMoa2213836
7. Fleming-Dutra KE, Jones JM, Roper LE, et al. Use of the Pfizer respiratory syncytial virus vaccine during pregnancy for the prevention of respiratory syncytial virus–associated lower respiratory tract disease in infants: recommendations of the Advisory Committee on Immunization Practices — United States, 2023. MMWR Morb Mortal Wkly Rep. 2023;72(41):1115-1122. doi:10.15585/mmwr.mm7241e1
8. Baker J, Aliabadi N, Munjal I, et al. Equivalent immunogenicity across three RSVpreF vaccine lots in healthy adults 18-49 years of age: results of a randomized phase 3 study. Vaccine. 2024;42(13):3172-3179. doi:10.1016/j.vaccine.2024.03.070
9. New data for AREXVY, GSK’s RSV vaccine, show potential to help protect adults aged 50 to 59 at increased risk for RSV disease. Press release. GSK; October 25, 2023. Accessed May 22, 2024. https://us.gsk.com/en-us/media/press-releases/new-data-for-arexvy/
1. Pfizer announces positive top-line results from phase 3 study of ABRYSVO® in adults aged 18 to 59 at increased risk for RSV disease. Press release. Pfizer; April 9, 2024. Accessed May 22, 2024. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-positive-top-line-results-phase-3-study-1
2. Pfizer announces positive top-line data for full season two efficacy of ABRYSVO® for RSV in older adults. Press release. Pfizer; February 29, 2024. Accessed May 22, 2024. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-positive-top-line-data-full-season-two
3. CDC study shows effectiveness of RSV immunization for infants. Press release. US Centers for Disease Control and Prevention; March 7, 2024. Accessed May 22, 2024. https://www.cdc.gov/media/releases/2024/s0307-rsv-immunization.html
4. Moline HL, Tannis A, Toepfer AP, et al. Early estimate of nirsevimab effectiveness for prevention of respiratory syncytial virus–associated hospitalization among infants entering their first respiratory syncytial virus season — new vaccine surveillance network, October 2023–February 2024. MMWR Morb Mortal Wkly Rep. 2024;73(9):209-214. doi:10.15585/mmwr.mm7309a4
5. Havers FP, Whitaker M, Melgar M, et al; for the RSV-NET Surveillance Team. Characteristics and outcomes among adults aged ≥60 years hospitalized with laboratory-confirmed respiratory syncytial virus ─ RSV-NET, 12 states, July 2022–June 2023. MMWR Morb Mortal Wkly Rep. 2023;72(40):1075-1082. doi:10.15585/mmwr.mm7240a1
6. Walsh EE, Pérez Marc G, Zareba AM, et al; for the RENOIR Clinical Trial Group. Efficacy and safety of a bivalent RSV prefusion F vaccine in older adults. N Engl J Med. 2023;388(16):1465-1477. doi:10.1056/NEJMoa2213836
7. Fleming-Dutra KE, Jones JM, Roper LE, et al. Use of the Pfizer respiratory syncytial virus vaccine during pregnancy for the prevention of respiratory syncytial virus–associated lower respiratory tract disease in infants: recommendations of the Advisory Committee on Immunization Practices — United States, 2023. MMWR Morb Mortal Wkly Rep. 2023;72(41):1115-1122. doi:10.15585/mmwr.mm7241e1
8. Baker J, Aliabadi N, Munjal I, et al. Equivalent immunogenicity across three RSVpreF vaccine lots in healthy adults 18-49 years of age: results of a randomized phase 3 study. Vaccine. 2024;42(13):3172-3179. doi:10.1016/j.vaccine.2024.03.070
9. New data for AREXVY, GSK’s RSV vaccine, show potential to help protect adults aged 50 to 59 at increased risk for RSV disease. Press release. GSK; October 25, 2023. Accessed May 22, 2024. https://us.gsk.com/en-us/media/press-releases/new-data-for-arexvy/
1. Pfizer announces positive top-line results from phase 3 study of ABRYSVO® in adults aged 18 to 59 at increased risk for RSV disease. Press release. Pfizer; April 9, 2024. Accessed May 22, 2024. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-positive-top-line-results-phase-3-study-1
2. Pfizer announces positive top-line data for full season two efficacy of ABRYSVO® for RSV in older adults. Press release. Pfizer; February 29, 2024. Accessed May 22, 2024. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-positive-top-line-data-full-season-two
3. CDC study shows effectiveness of RSV immunization for infants. Press release. US Centers for Disease Control and Prevention; March 7, 2024. Accessed May 22, 2024. https://www.cdc.gov/media/releases/2024/s0307-rsv-immunization.html
4. Moline HL, Tannis A, Toepfer AP, et al. Early estimate of nirsevimab effectiveness for prevention of respiratory syncytial virus–associated hospitalization among infants entering their first respiratory syncytial virus season — new vaccine surveillance network, October 2023–February 2024. MMWR Morb Mortal Wkly Rep. 2024;73(9):209-214. doi:10.15585/mmwr.mm7309a4
5. Havers FP, Whitaker M, Melgar M, et al; for the RSV-NET Surveillance Team. Characteristics and outcomes among adults aged ≥60 years hospitalized with laboratory-confirmed respiratory syncytial virus ─ RSV-NET, 12 states, July 2022–June 2023. MMWR Morb Mortal Wkly Rep. 2023;72(40):1075-1082. doi:10.15585/mmwr.mm7240a1
6. Walsh EE, Pérez Marc G, Zareba AM, et al; for the RENOIR Clinical Trial Group. Efficacy and safety of a bivalent RSV prefusion F vaccine in older adults. N Engl J Med. 2023;388(16):1465-1477. doi:10.1056/NEJMoa2213836
7. Fleming-Dutra KE, Jones JM, Roper LE, et al. Use of the Pfizer respiratory syncytial virus vaccine during pregnancy for the prevention of respiratory syncytial virus–associated lower respiratory tract disease in infants: recommendations of the Advisory Committee on Immunization Practices — United States, 2023. MMWR Morb Mortal Wkly Rep. 2023;72(41):1115-1122. doi:10.15585/mmwr.mm7241e1
8. Baker J, Aliabadi N, Munjal I, et al. Equivalent immunogenicity across three RSVpreF vaccine lots in healthy adults 18-49 years of age: results of a randomized phase 3 study. Vaccine. 2024;42(13):3172-3179. doi:10.1016/j.vaccine.2024.03.070
9. New data for AREXVY, GSK’s RSV vaccine, show potential to help protect adults aged 50 to 59 at increased risk for RSV disease. Press release. GSK; October 25, 2023. Accessed May 22, 2024. https://us.gsk.com/en-us/media/press-releases/new-data-for-arexvy/
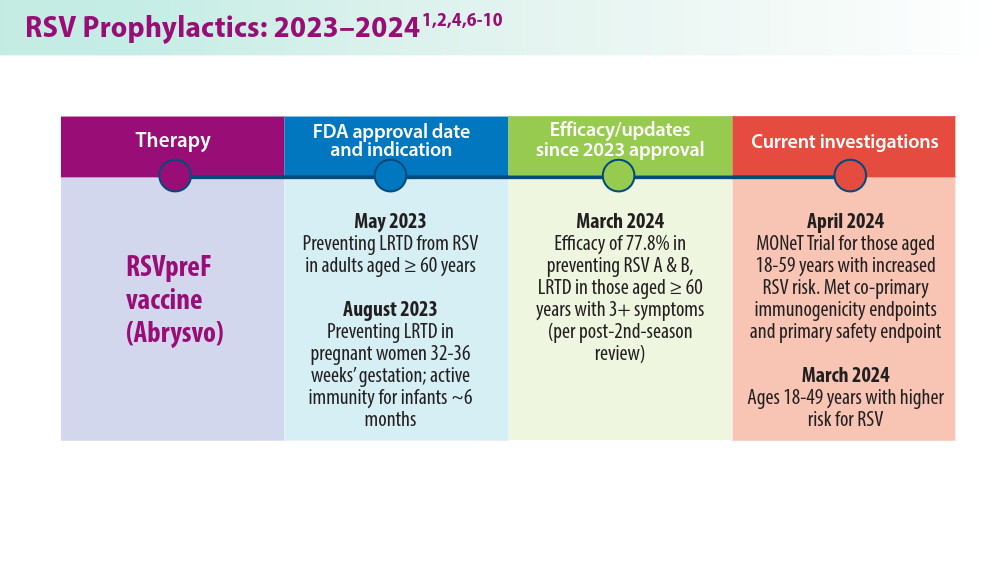
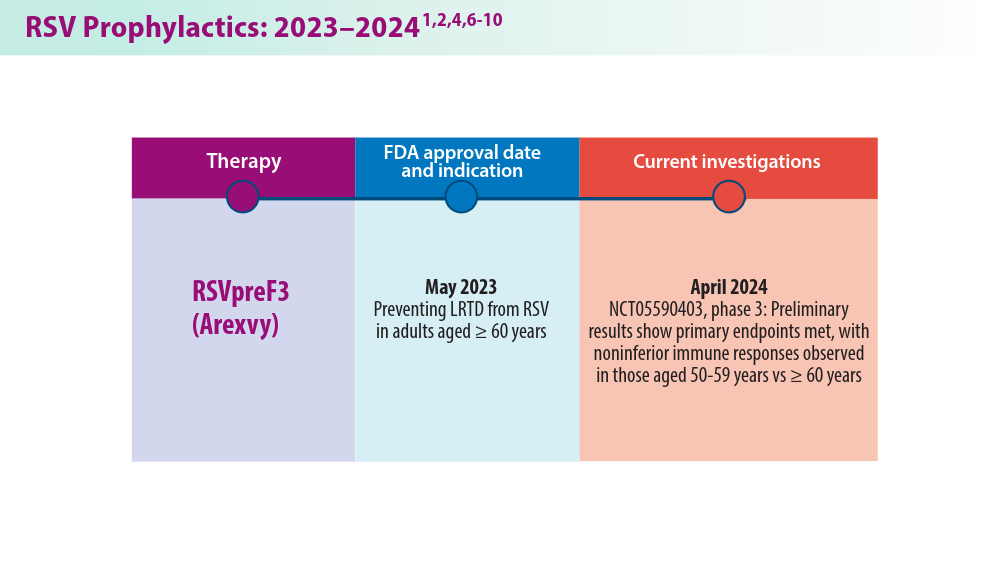
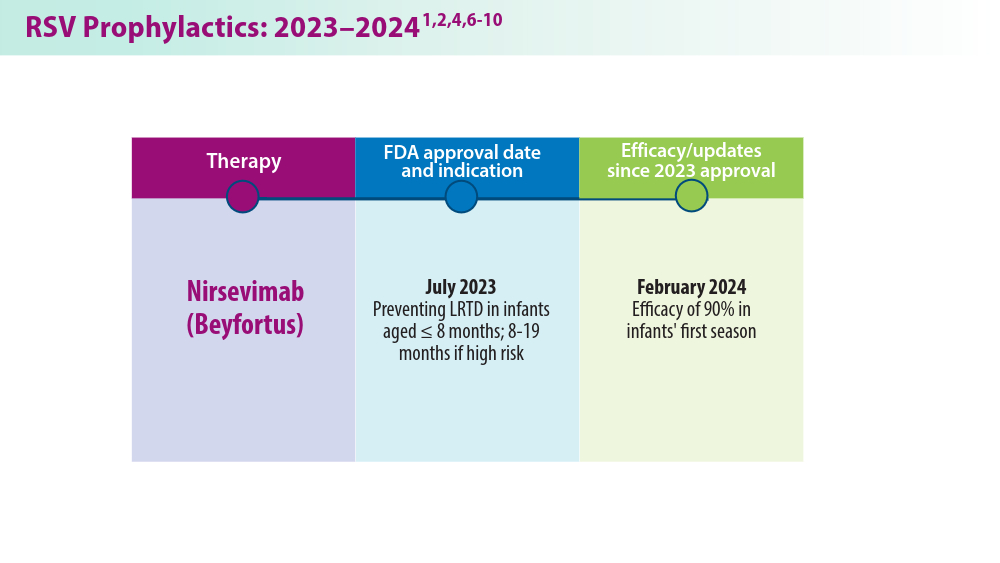
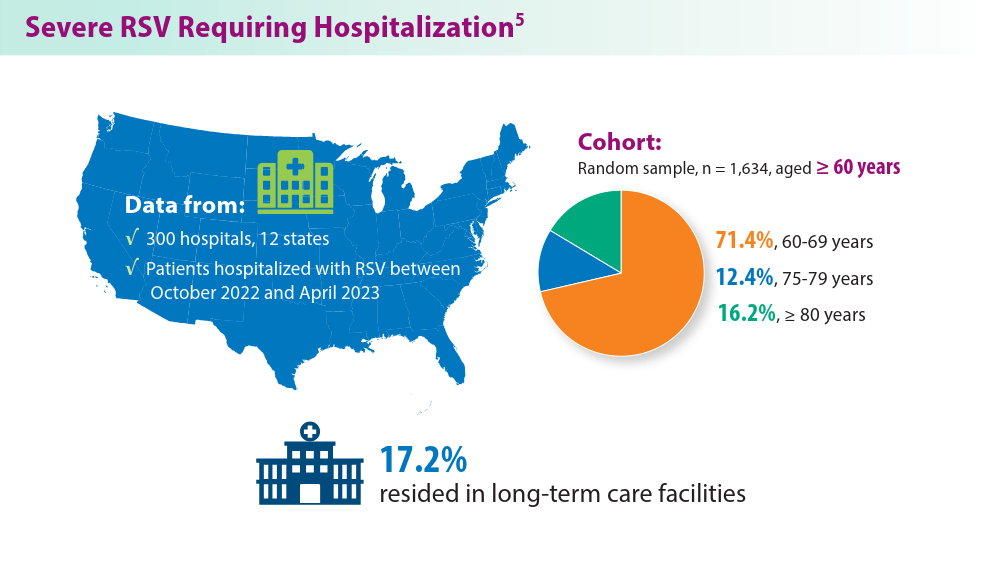
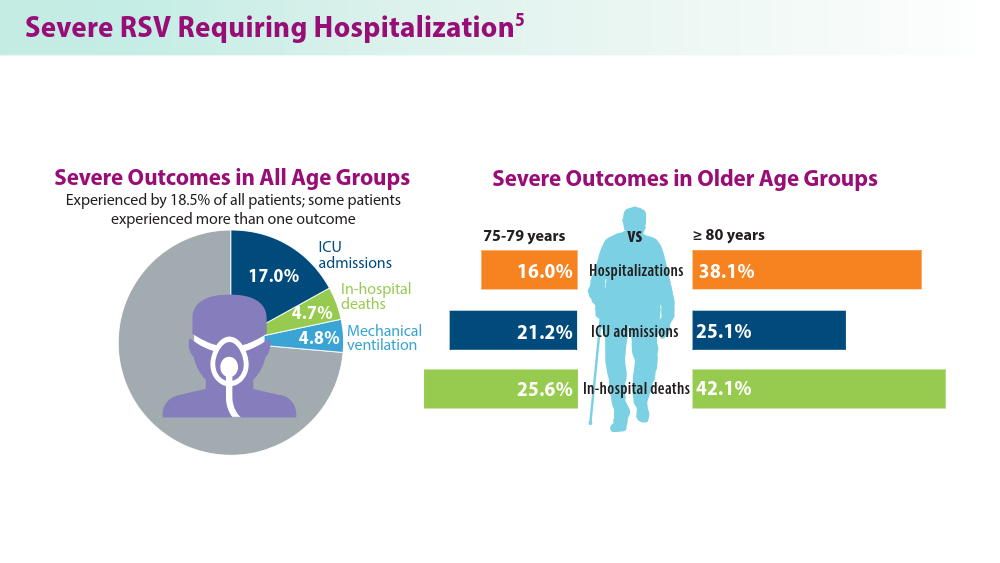
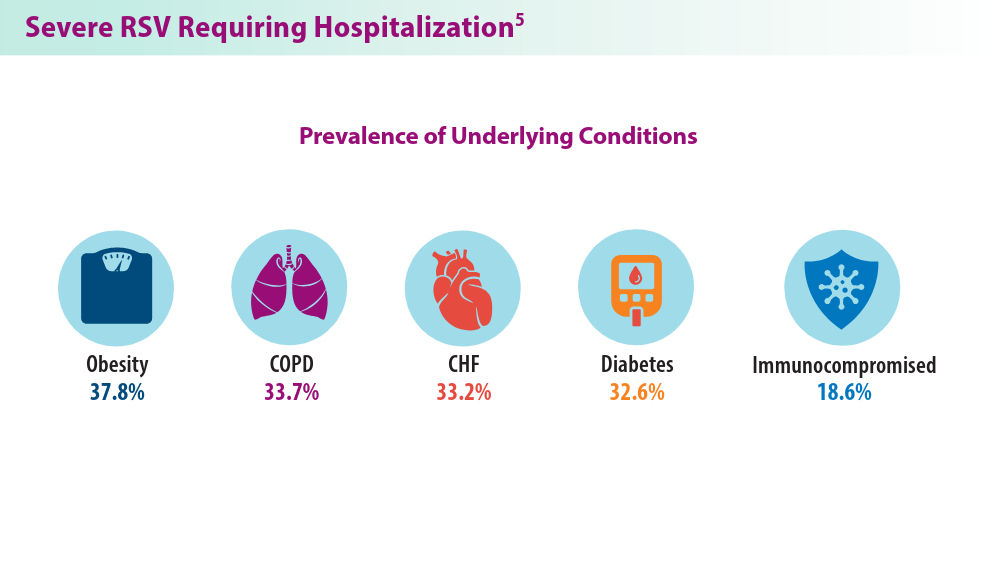
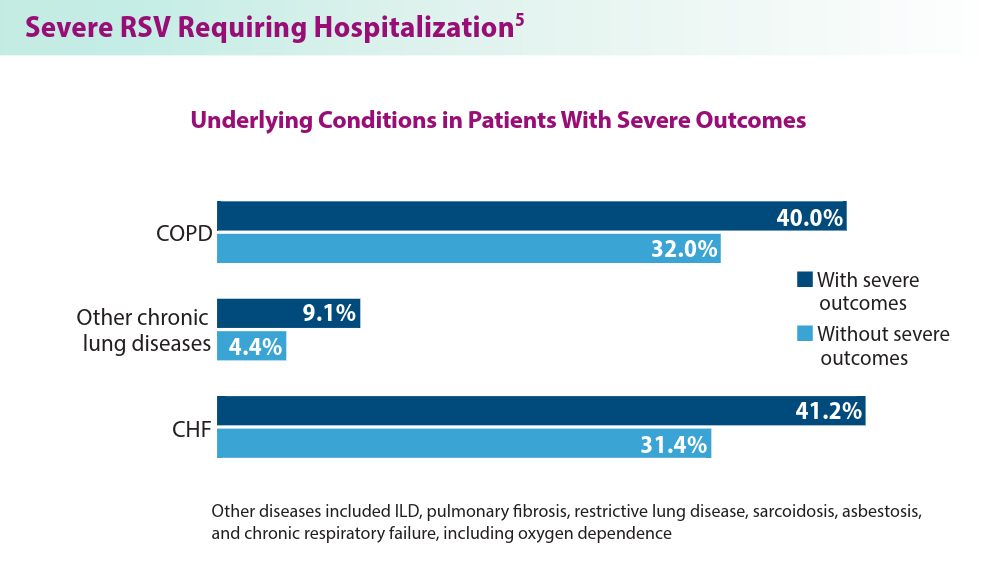
Targeted Therapies and Surgical Resection for Lung Cancer: Evolving Treatment Options
- American Cancer Society. Key statistics for lung cancer. Revised January 29, 2024. Accessed June 10, 2024. https://www.cancer.org/cancer/types/lung-cancer/about/key-statistics.html
- Drilon A, Camidge DR, Lin JJ, et al; for the TRIDENT-1 Investigators. Repotrectinib in ROS1 fusion-positive non-small-cell lung cancer. N Engl J Med. 2024;390(2):118-131. doi:10.1056/NEJMoa2302299
- Wu YL, Dziadziuszko R, Ahn JS, et al; for the ALINA Investigators. Alectinib in resected ALK-positive non-small-cell lung cancer. N Engl J Med. 2024;390(14):1265-1276.
- Mulligan L. Selective RET kinase inhibitors and lung cancer. N Engl J Med. 2023;389(20):1913-1916. doi:10.1056/NEJMe2311295
- Zhou C, Soloman B, Loong HH, et al; for the LIBRETTO-432 Trial Investigators. First-line selpercatinib or chemotherapy and pembrolizumab in RET fusion-positive NSCLC. N Engl J Med. 2023:389(20):1839-1850. doi:10.1056/NEJMoa239457
- Vaccaro K, Allen J, Whitfield TW, et al. Targeted therapies prime oncogene-driven lung cancers for macrophage-mediated destruction. bioRxiv. Preprint posted online March 6, 2023. doi:10.1101/2023.03.03.531059
- Liu M, Hu S, Yan N, Popowski KD, Cheng K. Inhalable extracellular vesicle delivery of IL-12 mRNA to treat lung cancer and promote systemic immunity. Nat Nanotechnol. 2024;19(4):565-575. doi:10.1038/s41565-023-01580-3
- Altorki N, Wang X, Kozono D, et al. Lobar or sublobar resection for peripheral stage IA non-small-cell lung cancer. N Engl J Med. 2023;388(6):489-498. doi:10.1056/NEJMoa2212083
- Koike T, Hasebe T, Nakamura M, Shimizu Y, Goto T, Tsuchida M. Towards better outcomes: segmentectomy for ground-glass opacity-dominant non-small cell lung cancer 3 cm or less─insights form JCOG1211 [editorial commentary]. AME Clin Trials Rev. 2023;1:5. doi:10.21037/actr-23-10
- Aokage K, Suzuki K, Saji H, et al; for the Japan Clinical Oncology Group. Segmentectomy for ground-glass-dominant lung cancer with a tumour diameter of 3 cm or less including groundglass opacity (JCOG1211): a multicentre, single-arm, confirmatory phase 3 trial. Lancet Respir Med. 2023;11(6):540-549. doi:10.1016/S2213-2600(23)00041-3
- Mandula JK, Sierra-Mondragon RA, Jimenez RV, et al. Jagged2 targeting in lung cancer activates anti-tumor immunity via Notch-induced functional reprogramming of tumor-associated macrophages. Immunity. 2024;57(5):1124-1140.e9. doi:10.1016/j.immuni.2024.03.020
- American Cancer Society. Key statistics for lung cancer. Revised January 29, 2024. Accessed June 10, 2024. https://www.cancer.org/cancer/types/lung-cancer/about/key-statistics.html
- Drilon A, Camidge DR, Lin JJ, et al; for the TRIDENT-1 Investigators. Repotrectinib in ROS1 fusion-positive non-small-cell lung cancer. N Engl J Med. 2024;390(2):118-131. doi:10.1056/NEJMoa2302299
- Wu YL, Dziadziuszko R, Ahn JS, et al; for the ALINA Investigators. Alectinib in resected ALK-positive non-small-cell lung cancer. N Engl J Med. 2024;390(14):1265-1276.
- Mulligan L. Selective RET kinase inhibitors and lung cancer. N Engl J Med. 2023;389(20):1913-1916. doi:10.1056/NEJMe2311295
- Zhou C, Soloman B, Loong HH, et al; for the LIBRETTO-432 Trial Investigators. First-line selpercatinib or chemotherapy and pembrolizumab in RET fusion-positive NSCLC. N Engl J Med. 2023:389(20):1839-1850. doi:10.1056/NEJMoa239457
- Vaccaro K, Allen J, Whitfield TW, et al. Targeted therapies prime oncogene-driven lung cancers for macrophage-mediated destruction. bioRxiv. Preprint posted online March 6, 2023. doi:10.1101/2023.03.03.531059
- Liu M, Hu S, Yan N, Popowski KD, Cheng K. Inhalable extracellular vesicle delivery of IL-12 mRNA to treat lung cancer and promote systemic immunity. Nat Nanotechnol. 2024;19(4):565-575. doi:10.1038/s41565-023-01580-3
- Altorki N, Wang X, Kozono D, et al. Lobar or sublobar resection for peripheral stage IA non-small-cell lung cancer. N Engl J Med. 2023;388(6):489-498. doi:10.1056/NEJMoa2212083
- Koike T, Hasebe T, Nakamura M, Shimizu Y, Goto T, Tsuchida M. Towards better outcomes: segmentectomy for ground-glass opacity-dominant non-small cell lung cancer 3 cm or less─insights form JCOG1211 [editorial commentary]. AME Clin Trials Rev. 2023;1:5. doi:10.21037/actr-23-10
- Aokage K, Suzuki K, Saji H, et al; for the Japan Clinical Oncology Group. Segmentectomy for ground-glass-dominant lung cancer with a tumour diameter of 3 cm or less including groundglass opacity (JCOG1211): a multicentre, single-arm, confirmatory phase 3 trial. Lancet Respir Med. 2023;11(6):540-549. doi:10.1016/S2213-2600(23)00041-3
- Mandula JK, Sierra-Mondragon RA, Jimenez RV, et al. Jagged2 targeting in lung cancer activates anti-tumor immunity via Notch-induced functional reprogramming of tumor-associated macrophages. Immunity. 2024;57(5):1124-1140.e9. doi:10.1016/j.immuni.2024.03.020
- American Cancer Society. Key statistics for lung cancer. Revised January 29, 2024. Accessed June 10, 2024. https://www.cancer.org/cancer/types/lung-cancer/about/key-statistics.html
- Drilon A, Camidge DR, Lin JJ, et al; for the TRIDENT-1 Investigators. Repotrectinib in ROS1 fusion-positive non-small-cell lung cancer. N Engl J Med. 2024;390(2):118-131. doi:10.1056/NEJMoa2302299
- Wu YL, Dziadziuszko R, Ahn JS, et al; for the ALINA Investigators. Alectinib in resected ALK-positive non-small-cell lung cancer. N Engl J Med. 2024;390(14):1265-1276.
- Mulligan L. Selective RET kinase inhibitors and lung cancer. N Engl J Med. 2023;389(20):1913-1916. doi:10.1056/NEJMe2311295
- Zhou C, Soloman B, Loong HH, et al; for the LIBRETTO-432 Trial Investigators. First-line selpercatinib or chemotherapy and pembrolizumab in RET fusion-positive NSCLC. N Engl J Med. 2023:389(20):1839-1850. doi:10.1056/NEJMoa239457
- Vaccaro K, Allen J, Whitfield TW, et al. Targeted therapies prime oncogene-driven lung cancers for macrophage-mediated destruction. bioRxiv. Preprint posted online March 6, 2023. doi:10.1101/2023.03.03.531059
- Liu M, Hu S, Yan N, Popowski KD, Cheng K. Inhalable extracellular vesicle delivery of IL-12 mRNA to treat lung cancer and promote systemic immunity. Nat Nanotechnol. 2024;19(4):565-575. doi:10.1038/s41565-023-01580-3
- Altorki N, Wang X, Kozono D, et al. Lobar or sublobar resection for peripheral stage IA non-small-cell lung cancer. N Engl J Med. 2023;388(6):489-498. doi:10.1056/NEJMoa2212083
- Koike T, Hasebe T, Nakamura M, Shimizu Y, Goto T, Tsuchida M. Towards better outcomes: segmentectomy for ground-glass opacity-dominant non-small cell lung cancer 3 cm or less─insights form JCOG1211 [editorial commentary]. AME Clin Trials Rev. 2023;1:5. doi:10.21037/actr-23-10
- Aokage K, Suzuki K, Saji H, et al; for the Japan Clinical Oncology Group. Segmentectomy for ground-glass-dominant lung cancer with a tumour diameter of 3 cm or less including groundglass opacity (JCOG1211): a multicentre, single-arm, confirmatory phase 3 trial. Lancet Respir Med. 2023;11(6):540-549. doi:10.1016/S2213-2600(23)00041-3
- Mandula JK, Sierra-Mondragon RA, Jimenez RV, et al. Jagged2 targeting in lung cancer activates anti-tumor immunity via Notch-induced functional reprogramming of tumor-associated macrophages. Immunity. 2024;57(5):1124-1140.e9. doi:10.1016/j.immuni.2024.03.020

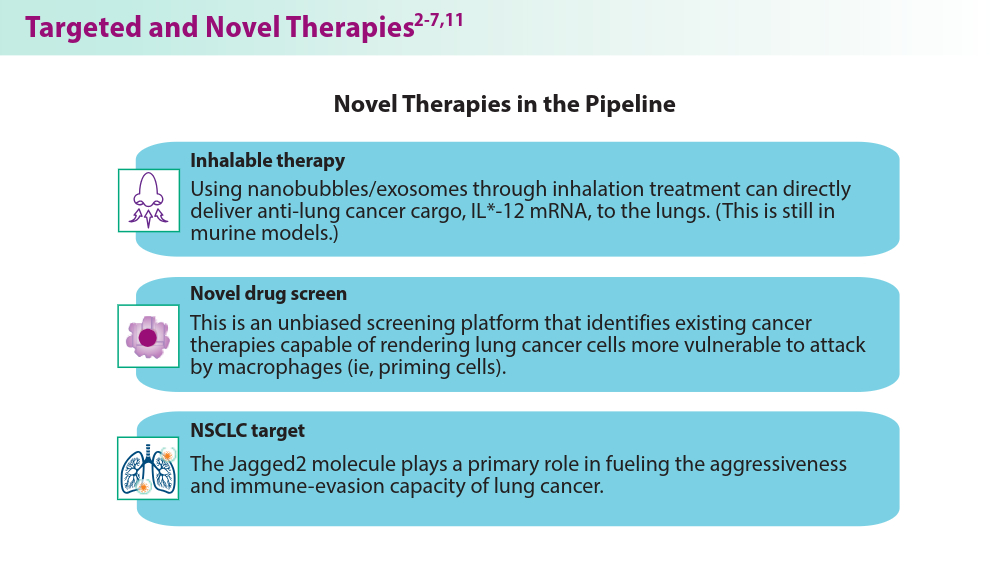
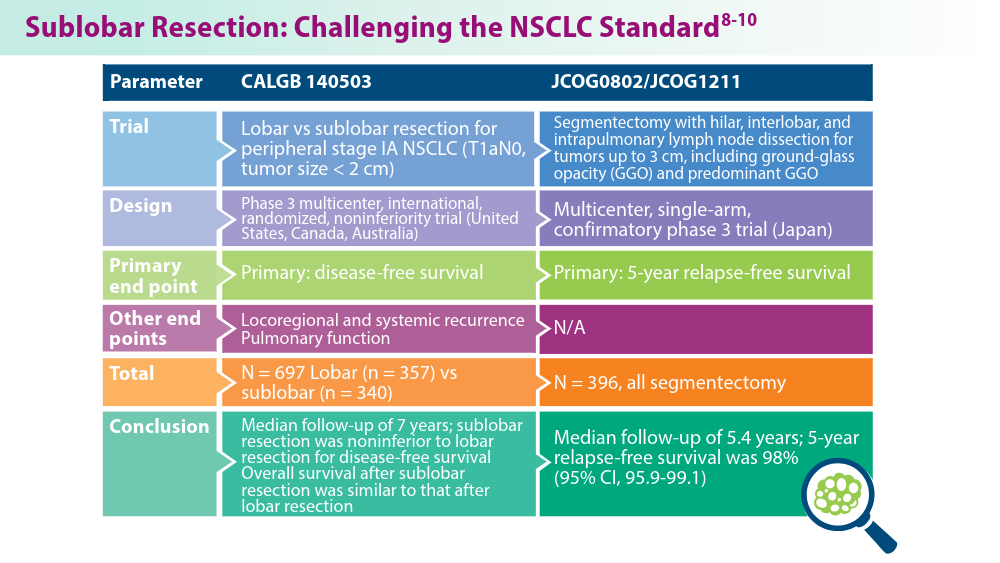

Closing the GAP in Idiopathic Pulmonary Fibrosis
5 things you should know about IPF. American Lung Association. April 12, 2023. Accessed June 21, 2024. https://www.lung.org/blog/idiopathic-pulmonary-fibrosis-things-to-know
Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566-572. doi:10.1016/S2213-2600(14)70101-8
Morrow T. Improving outcomes and managing costs in idiopathic pulmonary fibrosis. Am J Manag Care. 2019;25(11 suppl):S204-S209. PMID: 31419090
Man RK, Gogikar A, Nanda A, et al. A comparison of the effectiveness of nintedanib and pirfenidone in treating idiopathic pulmonary fibrosis: a systematic review. Cureus. 2024;16(2):e54268. doi:10.7759/cureus.54268
Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684-691. doi:10.7326/0003-4819-156-10-201205150-00004
Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST
Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group report. Am J Respir Crit Care Med. 2016;194(3):265-275. doi:10.1164/rccm.201604-0801CI
Abuserewa ST, Duff R, Becker G. Treatment of idiopathic pulmonary fibrosis. Cureus. 2021;13(5):e15360. doi:10.7759/cureus.15360
Lee JH, Jang JH, Jang HJ, et al. New prognostic scoring system for mortality in idiopathic pulmonary fibrosis by modifying the gender, age, and physiology model with desaturation during the six-minute walk test. Front Med (Lausanne). 2023;10:1052129. doi:10.3389/fmed.2023.1052129
Chandel A, Pastre J, Valery S, King CS, Nathan SD. Derivation and validation of a simple multidimensional index incorporating exercise capacity parameters for survival prediction in idiopathic pulmonary fibrosis. Thorax. 2023;78(4):368-375. doi:10.1136/thoraxjnl-2021-218440
Chandel A, King CS, Ignacio RV, et al. External validation and longitudinal application of the DO-GAP index to individualise survival prediction in idiopathic pulmonary fibrosis. ERJ Open Res. 2023;9(3):00124-2023. doi:10.1183/23120541.00124-2023
Suzuki Y, Mori K, Aono Y, et al. Combined assessment of the GAP index and body mass index at antifibrotic therapy initiation for prognosis of idiopathic pulmonary fibrosis. Sci Rep. 2021;11(1):18579. doi:10.1038/s41598-021-98161-y
Lacedonia D, De Pace CC, Rea G, et al. Machine learning and BMI improve the prognostic value of GAP index in treated IPF patients. Bioengineering (Basel). 2023;10(2):251. doi:10.3390/bioengineering10020251
Fujii H, Hara Y, Saigusa Y, et al. ILD-GAP combined with the Charlson Comorbidity Index score (ILD-GAPC) as a prognostic prediction model in patients with interstitial lung disease. Can Respir J. 2023;2023:5088207. doi:10.1155/2023/5088207
Ley B, Bradford WZ, Weycker D, Vittinghoff E, du Bois RM, Collard HR. Unified baseline and longitudinal mortality prediction in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1374-1381. doi:10.1183/09031936.00146314
Kreuter M, Lee JS, Tzouvelekis A, et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2021;204(1):74-81. doi:10.1164/rccm.202003-0669OC
Kreuter M, Lee JS, Tzouvelekis A, et al. A modified blood cell GAP (cGAP) to prognosticate outcomes in IPF. Poster presented at: European Respiratory Society International Congress; September 4-6, 2022. https://medically.gene.com/global/en/unrestricted/respiratory/ERS-2022/ers-2022-poster-kreuter-a-modified-blood-cell-gap.html
Nishikiori H, Chiba H, Lee SH, et al. A modified GAP model for East-Asian populations with idiopathic pulmonary fibrosis. Respir Investig. 2020;58(5):395-402. doi:10.1016/j.resinv.2020.04.001
5 things you should know about IPF. American Lung Association. April 12, 2023. Accessed June 21, 2024. https://www.lung.org/blog/idiopathic-pulmonary-fibrosis-things-to-know
Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566-572. doi:10.1016/S2213-2600(14)70101-8
Morrow T. Improving outcomes and managing costs in idiopathic pulmonary fibrosis. Am J Manag Care. 2019;25(11 suppl):S204-S209. PMID: 31419090
Man RK, Gogikar A, Nanda A, et al. A comparison of the effectiveness of nintedanib and pirfenidone in treating idiopathic pulmonary fibrosis: a systematic review. Cureus. 2024;16(2):e54268. doi:10.7759/cureus.54268
Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684-691. doi:10.7326/0003-4819-156-10-201205150-00004
Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST
Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group report. Am J Respir Crit Care Med. 2016;194(3):265-275. doi:10.1164/rccm.201604-0801CI
Abuserewa ST, Duff R, Becker G. Treatment of idiopathic pulmonary fibrosis. Cureus. 2021;13(5):e15360. doi:10.7759/cureus.15360
Lee JH, Jang JH, Jang HJ, et al. New prognostic scoring system for mortality in idiopathic pulmonary fibrosis by modifying the gender, age, and physiology model with desaturation during the six-minute walk test. Front Med (Lausanne). 2023;10:1052129. doi:10.3389/fmed.2023.1052129
Chandel A, Pastre J, Valery S, King CS, Nathan SD. Derivation and validation of a simple multidimensional index incorporating exercise capacity parameters for survival prediction in idiopathic pulmonary fibrosis. Thorax. 2023;78(4):368-375. doi:10.1136/thoraxjnl-2021-218440
Chandel A, King CS, Ignacio RV, et al. External validation and longitudinal application of the DO-GAP index to individualise survival prediction in idiopathic pulmonary fibrosis. ERJ Open Res. 2023;9(3):00124-2023. doi:10.1183/23120541.00124-2023
Suzuki Y, Mori K, Aono Y, et al. Combined assessment of the GAP index and body mass index at antifibrotic therapy initiation for prognosis of idiopathic pulmonary fibrosis. Sci Rep. 2021;11(1):18579. doi:10.1038/s41598-021-98161-y
Lacedonia D, De Pace CC, Rea G, et al. Machine learning and BMI improve the prognostic value of GAP index in treated IPF patients. Bioengineering (Basel). 2023;10(2):251. doi:10.3390/bioengineering10020251
Fujii H, Hara Y, Saigusa Y, et al. ILD-GAP combined with the Charlson Comorbidity Index score (ILD-GAPC) as a prognostic prediction model in patients with interstitial lung disease. Can Respir J. 2023;2023:5088207. doi:10.1155/2023/5088207
Ley B, Bradford WZ, Weycker D, Vittinghoff E, du Bois RM, Collard HR. Unified baseline and longitudinal mortality prediction in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1374-1381. doi:10.1183/09031936.00146314
Kreuter M, Lee JS, Tzouvelekis A, et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2021;204(1):74-81. doi:10.1164/rccm.202003-0669OC
Kreuter M, Lee JS, Tzouvelekis A, et al. A modified blood cell GAP (cGAP) to prognosticate outcomes in IPF. Poster presented at: European Respiratory Society International Congress; September 4-6, 2022. https://medically.gene.com/global/en/unrestricted/respiratory/ERS-2022/ers-2022-poster-kreuter-a-modified-blood-cell-gap.html
Nishikiori H, Chiba H, Lee SH, et al. A modified GAP model for East-Asian populations with idiopathic pulmonary fibrosis. Respir Investig. 2020;58(5):395-402. doi:10.1016/j.resinv.2020.04.001
5 things you should know about IPF. American Lung Association. April 12, 2023. Accessed June 21, 2024. https://www.lung.org/blog/idiopathic-pulmonary-fibrosis-things-to-know
Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566-572. doi:10.1016/S2213-2600(14)70101-8
Morrow T. Improving outcomes and managing costs in idiopathic pulmonary fibrosis. Am J Manag Care. 2019;25(11 suppl):S204-S209. PMID: 31419090
Man RK, Gogikar A, Nanda A, et al. A comparison of the effectiveness of nintedanib and pirfenidone in treating idiopathic pulmonary fibrosis: a systematic review. Cureus. 2024;16(2):e54268. doi:10.7759/cureus.54268
Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684-691. doi:10.7326/0003-4819-156-10-201205150-00004
Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST
Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group report. Am J Respir Crit Care Med. 2016;194(3):265-275. doi:10.1164/rccm.201604-0801CI
Abuserewa ST, Duff R, Becker G. Treatment of idiopathic pulmonary fibrosis. Cureus. 2021;13(5):e15360. doi:10.7759/cureus.15360
Lee JH, Jang JH, Jang HJ, et al. New prognostic scoring system for mortality in idiopathic pulmonary fibrosis by modifying the gender, age, and physiology model with desaturation during the six-minute walk test. Front Med (Lausanne). 2023;10:1052129. doi:10.3389/fmed.2023.1052129
Chandel A, Pastre J, Valery S, King CS, Nathan SD. Derivation and validation of a simple multidimensional index incorporating exercise capacity parameters for survival prediction in idiopathic pulmonary fibrosis. Thorax. 2023;78(4):368-375. doi:10.1136/thoraxjnl-2021-218440
Chandel A, King CS, Ignacio RV, et al. External validation and longitudinal application of the DO-GAP index to individualise survival prediction in idiopathic pulmonary fibrosis. ERJ Open Res. 2023;9(3):00124-2023. doi:10.1183/23120541.00124-2023
Suzuki Y, Mori K, Aono Y, et al. Combined assessment of the GAP index and body mass index at antifibrotic therapy initiation for prognosis of idiopathic pulmonary fibrosis. Sci Rep. 2021;11(1):18579. doi:10.1038/s41598-021-98161-y
Lacedonia D, De Pace CC, Rea G, et al. Machine learning and BMI improve the prognostic value of GAP index in treated IPF patients. Bioengineering (Basel). 2023;10(2):251. doi:10.3390/bioengineering10020251
Fujii H, Hara Y, Saigusa Y, et al. ILD-GAP combined with the Charlson Comorbidity Index score (ILD-GAPC) as a prognostic prediction model in patients with interstitial lung disease. Can Respir J. 2023;2023:5088207. doi:10.1155/2023/5088207
Ley B, Bradford WZ, Weycker D, Vittinghoff E, du Bois RM, Collard HR. Unified baseline and longitudinal mortality prediction in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1374-1381. doi:10.1183/09031936.00146314
Kreuter M, Lee JS, Tzouvelekis A, et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2021;204(1):74-81. doi:10.1164/rccm.202003-0669OC
Kreuter M, Lee JS, Tzouvelekis A, et al. A modified blood cell GAP (cGAP) to prognosticate outcomes in IPF. Poster presented at: European Respiratory Society International Congress; September 4-6, 2022. https://medically.gene.com/global/en/unrestricted/respiratory/ERS-2022/ers-2022-poster-kreuter-a-modified-blood-cell-gap.html
Nishikiori H, Chiba H, Lee SH, et al. A modified GAP model for East-Asian populations with idiopathic pulmonary fibrosis. Respir Investig. 2020;58(5):395-402. doi:10.1016/j.resinv.2020.04.001
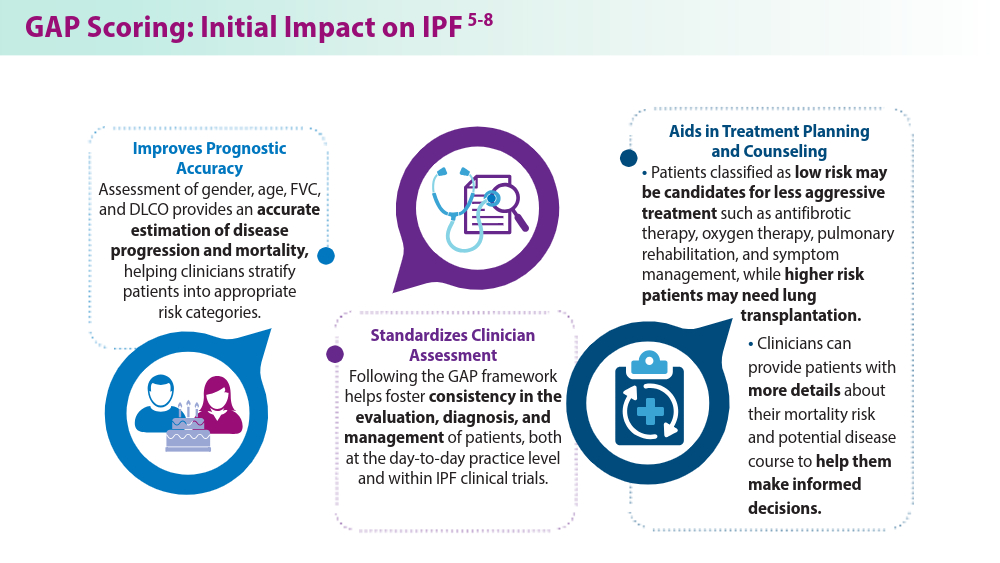
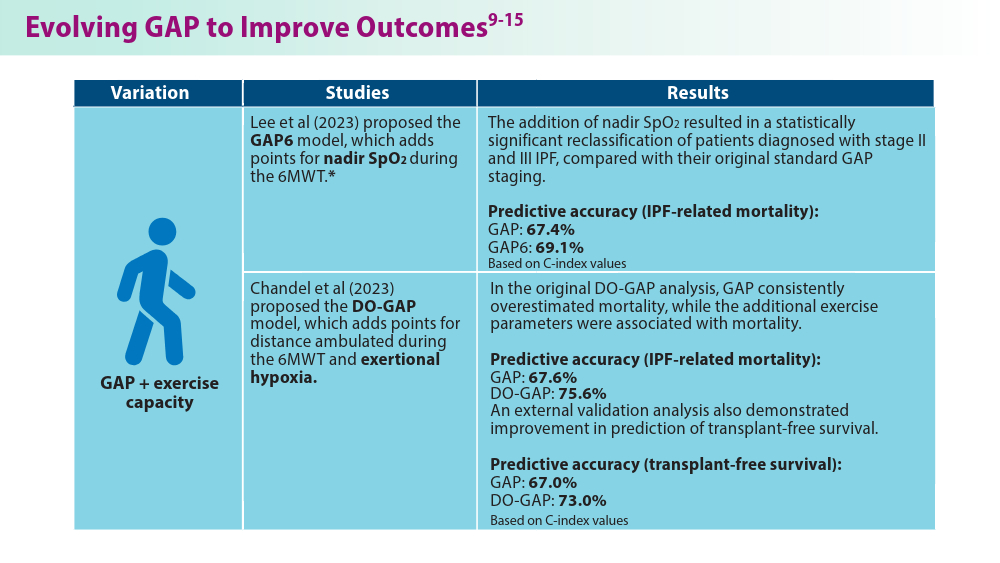


Pulmonology Data Trends 2024
Pulmonology Data Trends 2024 is a supplement to CHEST Physician highlighting the latest breakthroughs in pulmonology research and treatments through a series of infographics.
Read more:

Artificial Intelligence in Sleep Apnea
Ritwick Agrawal, MD, MS, FCCP
RSV Updates: Prophylaxis Approval and Hospitalization for Severe RSV
Riddhi Upadhyay, MD
Biologics in Asthma: Changing the Severe Asthma Paradigm
Shyam Subramanian, MD, FCCP
Updates in COPD Guidelines and Treatment
Dharani K. Narendra, MD, FCCP
Targeted Therapies and Surgical Resection for Lung Cancer: Evolving Treatment Options
Saadia A. Faiz, MD, FCCP
Closing the GAP in Idiopathic Pulmonary Fibrosis
Humayun Anjum, MD, FCCP
Severe Community-Acquired Pneumonia: Diagnostic Criteria, Treatment, and COVID-19
Sujith V. Cherian, MD, FCCP
Pulmonary Hypertension: Comorbidities and Novel Therapies
Mary Jo S. Farmer, MD, PhD, FCCP
The Genetic Side of Interstitial Lung Disease
Priya Balakrishnan, MD, MS, FCCP
Noninvasive Ventilation in Neuromuscular Disease
Sreelatha Naik, MD, FCCP, and Kelly Lobrutto, CRNP
Pulmonology Data Trends 2024 is a supplement to CHEST Physician highlighting the latest breakthroughs in pulmonology research and treatments through a series of infographics.
Read more:
Artificial Intelligence in Sleep Apnea
Ritwick Agrawal, MD, MS, FCCP
RSV Updates: Prophylaxis Approval and Hospitalization for Severe RSV
Riddhi Upadhyay, MD
Biologics in Asthma: Changing the Severe Asthma Paradigm
Shyam Subramanian, MD, FCCP
Updates in COPD Guidelines and Treatment
Dharani K. Narendra, MD, FCCP
Targeted Therapies and Surgical Resection for Lung Cancer: Evolving Treatment Options
Saadia A. Faiz, MD, FCCP
Closing the GAP in Idiopathic Pulmonary Fibrosis
Humayun Anjum, MD, FCCP
Severe Community-Acquired Pneumonia: Diagnostic Criteria, Treatment, and COVID-19
Sujith V. Cherian, MD, FCCP
Pulmonary Hypertension: Comorbidities and Novel Therapies
Mary Jo S. Farmer, MD, PhD, FCCP
The Genetic Side of Interstitial Lung Disease
Priya Balakrishnan, MD, MS, FCCP
Noninvasive Ventilation in Neuromuscular Disease
Sreelatha Naik, MD, FCCP, and Kelly Lobrutto, CRNP
Pulmonology Data Trends 2024 is a supplement to CHEST Physician highlighting the latest breakthroughs in pulmonology research and treatments through a series of infographics.
Read more:
Artificial Intelligence in Sleep Apnea
Ritwick Agrawal, MD, MS, FCCP
RSV Updates: Prophylaxis Approval and Hospitalization for Severe RSV
Riddhi Upadhyay, MD
Biologics in Asthma: Changing the Severe Asthma Paradigm
Shyam Subramanian, MD, FCCP
Updates in COPD Guidelines and Treatment
Dharani K. Narendra, MD, FCCP
Targeted Therapies and Surgical Resection for Lung Cancer: Evolving Treatment Options
Saadia A. Faiz, MD, FCCP
Closing the GAP in Idiopathic Pulmonary Fibrosis
Humayun Anjum, MD, FCCP
Severe Community-Acquired Pneumonia: Diagnostic Criteria, Treatment, and COVID-19
Sujith V. Cherian, MD, FCCP
Pulmonary Hypertension: Comorbidities and Novel Therapies
Mary Jo S. Farmer, MD, PhD, FCCP
The Genetic Side of Interstitial Lung Disease
Priya Balakrishnan, MD, MS, FCCP
Noninvasive Ventilation in Neuromuscular Disease
Sreelatha Naik, MD, FCCP, and Kelly Lobrutto, CRNP