User login
-
EMA Greenlights Four Drugs for Bladder and Other Cancers
Balversa
The CHMP endorsed the approval of Balversa (erdafitinib, Janssen-Cilag International N.V.), intended for the treatment of urothelial carcinoma, a type of cancer affecting the bladder and urinary system.
As a monotherapy, Balversa is indicated for the treatment of adult patients with unresectable or metastatic urothelial carcinoma harboring susceptible FGFR3 genetic alterations. These patients must have previously received at least one line of therapy containing a programmed death receptor 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor in the unresectable or metastatic treatment setting.
Urothelial carcinoma is the most common form of bladder cancer, the ninth most frequently diagnosed cancer worldwide. In 2022, there were approximately 614,000 new cases of bladder cancer and 220,000 deaths globally.
The highest incidence rates in both men and women are found in Southern Europe. Greece had 5800 new cases and 1537 deaths in 2018. Spain has the highest incidence rate in men globally. Since the 1990s, bladder cancer incidence trends have diverged by sex, with rates decreasing or stabilizing in men but increasing among women in certain European countries.
The CHMP recommendation is based on data from cohort 1 of the phase 3 THOR trial, which compared erdafitinib with standard-of-care chemotherapy (investigator’s choice of docetaxel or vinflunine). Cohort 1 included 266 adults with advanced urothelial cancer harboring selected FGFR3 alterations.
All patients had disease progression after one or two prior treatments, at least one of which included a PD-1 or PD-L1 inhibitor. The major efficacy endpoints were overall survival, progression free survival, and objective response rate (ORR).
Treatment with erdafitinib reduced the risk for death by 36% compared with chemotherapy (hazard ratio [HR], 0.64; P = .005). Median overall survival was 12.1 months in the erdafitinib arm vs 7.8 months in the chemotherapy arm. Median progression-free survival was 5.6 months in the erdafitinib arm vs 2.7 months in the chemotherapy arm (HR, 0.58; P = .0002). ORR was 35.3% with erdafitinib compared with 8.5% with chemotherapy.
Balversa will be available as 3-mg, 4-mg, and 5-mg film-coated tablets. Erdafitinib, the active substance in Balversa, is an antineoplastic protein kinase inhibitor that suppresses fibroblast growth factor receptor (FGFR) tyrosine kinases. Deregulation of FGFR3 signaling is implicated in the pathogenesis of urothelial cancer, and FGFR inhibition has demonstrated antitumor activity in FGFR-expressing cells.
Ordspono
The committee adopted a positive opinion for Ordspono (odronextamab, Regeneron Ireland Designated Activity Company), indicated as a monotherapy for the treatment of adult patients with:
- Relapsed or refractory follicular lymphoma (rrFL), after two or more lines of systemic therapy.
- Relapsed or refractory diffuse large B-cell lymphoma (rrDLBCL), after two or more lines of systemic therapy.
The approval recommendation is based on phase 2 trials (NCT02290951, NCT03888105), which demonstrated high ORRs in patients with rrFL and rrDLBCL.
In the DLBCL cohort, a 49% ORR was achieved in heavily pretreated patients who had not received chimeric antigen receptor T-cell therapy. A total of 31% achieved a complete response.
The FL cohort showed an 82% response rate in patients with grades I-IIIA disease, with 75% of the overall population achieving a complete response.
Ordspono will be available as a 2-mg, 80-mg, and 320-mg concentrate for solution for infusion. The active substance of Ordspono is odronextamab, a bispecific antibody that targets CD20-expressing B cells and CD3-expressing T cells. By binding to both, it induces T-cell activation and generates a polyclonal cytotoxic T-cell response, leading to the lysis of malignant B cells.
Generics
The panel also adopted positive opinions for two generic cancer medicines.
Enzalutamide Viatris (enzalutamide) is indicated for the treatment of adult men with prostate cancer in several scenarios:
- As monotherapy or with androgen-deprivation therapy for high-risk biochemical recurrent nonmetastatic hormone-sensitive prostate cancer in men unsuitable for salvage-radiotherapy.
- In combination with androgen-deprivation therapy for metastatic hormone-sensitive prostate cancer.
- For high-risk nonmetastatic castration-resistant prostate cancer (CRPC).
- For metastatic CRPC in men who are asymptomatic or mildly symptomatic after failure of androgen-deprivation therapy, where chemotherapy is not yet indicated.
- For metastatic CRPC in men whose disease has progressed on or after docetaxel therapy.
Enzalutamide Viatris is a generic version of Xtandi, authorized in the European Union since June 2013. Studies have confirmed the satisfactory quality and bioequivalence of Enzalutamide Viatris to Xtandi.
Enzalutamide Viatris will be available as 40-mg and 80-mg film-coated tablets. The active substance of Enzalutamide Viatris is enzalutamide, a hormone antagonist that blocks multiple steps in the androgen receptor–signaling pathway.
Nilotinib Accord (nilotinib) is indicated for the treatment of Philadelphia chromosome–positive chronic myelogenous leukemia (CML).
It is used in adult and pediatric patients with newly diagnosed CML in the chronic phase, adult patients with chronic phase and accelerated phase CML with resistance or intolerance to prior therapy including imatinib, and pediatric patients with CML with resistance or intolerance to prior therapy including imatinib.
Nilotinib Accord is a generic of Tasigna, authorized in the European Union since November 2007. Studies have demonstrated the satisfactory quality and bioequivalence of Nilotinib Accord to Tasigna.
Nilotinib Accord will be available as 50-mg, 150-mg, and 200-mg hard capsules. The active substance of Nilotinib Accord is nilotinib, an antineoplastic protein kinase inhibitor that targets BCR-ABL kinase and other oncogenic kinases.
A version of this article appeared on Medscape.com.
Balversa
The CHMP endorsed the approval of Balversa (erdafitinib, Janssen-Cilag International N.V.), intended for the treatment of urothelial carcinoma, a type of cancer affecting the bladder and urinary system.
As a monotherapy, Balversa is indicated for the treatment of adult patients with unresectable or metastatic urothelial carcinoma harboring susceptible FGFR3 genetic alterations. These patients must have previously received at least one line of therapy containing a programmed death receptor 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor in the unresectable or metastatic treatment setting.
Urothelial carcinoma is the most common form of bladder cancer, the ninth most frequently diagnosed cancer worldwide. In 2022, there were approximately 614,000 new cases of bladder cancer and 220,000 deaths globally.
The highest incidence rates in both men and women are found in Southern Europe. Greece had 5800 new cases and 1537 deaths in 2018. Spain has the highest incidence rate in men globally. Since the 1990s, bladder cancer incidence trends have diverged by sex, with rates decreasing or stabilizing in men but increasing among women in certain European countries.
The CHMP recommendation is based on data from cohort 1 of the phase 3 THOR trial, which compared erdafitinib with standard-of-care chemotherapy (investigator’s choice of docetaxel or vinflunine). Cohort 1 included 266 adults with advanced urothelial cancer harboring selected FGFR3 alterations.
All patients had disease progression after one or two prior treatments, at least one of which included a PD-1 or PD-L1 inhibitor. The major efficacy endpoints were overall survival, progression free survival, and objective response rate (ORR).
Treatment with erdafitinib reduced the risk for death by 36% compared with chemotherapy (hazard ratio [HR], 0.64; P = .005). Median overall survival was 12.1 months in the erdafitinib arm vs 7.8 months in the chemotherapy arm. Median progression-free survival was 5.6 months in the erdafitinib arm vs 2.7 months in the chemotherapy arm (HR, 0.58; P = .0002). ORR was 35.3% with erdafitinib compared with 8.5% with chemotherapy.
Balversa will be available as 3-mg, 4-mg, and 5-mg film-coated tablets. Erdafitinib, the active substance in Balversa, is an antineoplastic protein kinase inhibitor that suppresses fibroblast growth factor receptor (FGFR) tyrosine kinases. Deregulation of FGFR3 signaling is implicated in the pathogenesis of urothelial cancer, and FGFR inhibition has demonstrated antitumor activity in FGFR-expressing cells.
Ordspono
The committee adopted a positive opinion for Ordspono (odronextamab, Regeneron Ireland Designated Activity Company), indicated as a monotherapy for the treatment of adult patients with:
- Relapsed or refractory follicular lymphoma (rrFL), after two or more lines of systemic therapy.
- Relapsed or refractory diffuse large B-cell lymphoma (rrDLBCL), after two or more lines of systemic therapy.
The approval recommendation is based on phase 2 trials (NCT02290951, NCT03888105), which demonstrated high ORRs in patients with rrFL and rrDLBCL.
In the DLBCL cohort, a 49% ORR was achieved in heavily pretreated patients who had not received chimeric antigen receptor T-cell therapy. A total of 31% achieved a complete response.
The FL cohort showed an 82% response rate in patients with grades I-IIIA disease, with 75% of the overall population achieving a complete response.
Ordspono will be available as a 2-mg, 80-mg, and 320-mg concentrate for solution for infusion. The active substance of Ordspono is odronextamab, a bispecific antibody that targets CD20-expressing B cells and CD3-expressing T cells. By binding to both, it induces T-cell activation and generates a polyclonal cytotoxic T-cell response, leading to the lysis of malignant B cells.
Generics
The panel also adopted positive opinions for two generic cancer medicines.
Enzalutamide Viatris (enzalutamide) is indicated for the treatment of adult men with prostate cancer in several scenarios:
- As monotherapy or with androgen-deprivation therapy for high-risk biochemical recurrent nonmetastatic hormone-sensitive prostate cancer in men unsuitable for salvage-radiotherapy.
- In combination with androgen-deprivation therapy for metastatic hormone-sensitive prostate cancer.
- For high-risk nonmetastatic castration-resistant prostate cancer (CRPC).
- For metastatic CRPC in men who are asymptomatic or mildly symptomatic after failure of androgen-deprivation therapy, where chemotherapy is not yet indicated.
- For metastatic CRPC in men whose disease has progressed on or after docetaxel therapy.
Enzalutamide Viatris is a generic version of Xtandi, authorized in the European Union since June 2013. Studies have confirmed the satisfactory quality and bioequivalence of Enzalutamide Viatris to Xtandi.
Enzalutamide Viatris will be available as 40-mg and 80-mg film-coated tablets. The active substance of Enzalutamide Viatris is enzalutamide, a hormone antagonist that blocks multiple steps in the androgen receptor–signaling pathway.
Nilotinib Accord (nilotinib) is indicated for the treatment of Philadelphia chromosome–positive chronic myelogenous leukemia (CML).
It is used in adult and pediatric patients with newly diagnosed CML in the chronic phase, adult patients with chronic phase and accelerated phase CML with resistance or intolerance to prior therapy including imatinib, and pediatric patients with CML with resistance or intolerance to prior therapy including imatinib.
Nilotinib Accord is a generic of Tasigna, authorized in the European Union since November 2007. Studies have demonstrated the satisfactory quality and bioequivalence of Nilotinib Accord to Tasigna.
Nilotinib Accord will be available as 50-mg, 150-mg, and 200-mg hard capsules. The active substance of Nilotinib Accord is nilotinib, an antineoplastic protein kinase inhibitor that targets BCR-ABL kinase and other oncogenic kinases.
A version of this article appeared on Medscape.com.
Balversa
The CHMP endorsed the approval of Balversa (erdafitinib, Janssen-Cilag International N.V.), intended for the treatment of urothelial carcinoma, a type of cancer affecting the bladder and urinary system.
As a monotherapy, Balversa is indicated for the treatment of adult patients with unresectable or metastatic urothelial carcinoma harboring susceptible FGFR3 genetic alterations. These patients must have previously received at least one line of therapy containing a programmed death receptor 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor in the unresectable or metastatic treatment setting.
Urothelial carcinoma is the most common form of bladder cancer, the ninth most frequently diagnosed cancer worldwide. In 2022, there were approximately 614,000 new cases of bladder cancer and 220,000 deaths globally.
The highest incidence rates in both men and women are found in Southern Europe. Greece had 5800 new cases and 1537 deaths in 2018. Spain has the highest incidence rate in men globally. Since the 1990s, bladder cancer incidence trends have diverged by sex, with rates decreasing or stabilizing in men but increasing among women in certain European countries.
The CHMP recommendation is based on data from cohort 1 of the phase 3 THOR trial, which compared erdafitinib with standard-of-care chemotherapy (investigator’s choice of docetaxel or vinflunine). Cohort 1 included 266 adults with advanced urothelial cancer harboring selected FGFR3 alterations.
All patients had disease progression after one or two prior treatments, at least one of which included a PD-1 or PD-L1 inhibitor. The major efficacy endpoints were overall survival, progression free survival, and objective response rate (ORR).
Treatment with erdafitinib reduced the risk for death by 36% compared with chemotherapy (hazard ratio [HR], 0.64; P = .005). Median overall survival was 12.1 months in the erdafitinib arm vs 7.8 months in the chemotherapy arm. Median progression-free survival was 5.6 months in the erdafitinib arm vs 2.7 months in the chemotherapy arm (HR, 0.58; P = .0002). ORR was 35.3% with erdafitinib compared with 8.5% with chemotherapy.
Balversa will be available as 3-mg, 4-mg, and 5-mg film-coated tablets. Erdafitinib, the active substance in Balversa, is an antineoplastic protein kinase inhibitor that suppresses fibroblast growth factor receptor (FGFR) tyrosine kinases. Deregulation of FGFR3 signaling is implicated in the pathogenesis of urothelial cancer, and FGFR inhibition has demonstrated antitumor activity in FGFR-expressing cells.
Ordspono
The committee adopted a positive opinion for Ordspono (odronextamab, Regeneron Ireland Designated Activity Company), indicated as a monotherapy for the treatment of adult patients with:
- Relapsed or refractory follicular lymphoma (rrFL), after two or more lines of systemic therapy.
- Relapsed or refractory diffuse large B-cell lymphoma (rrDLBCL), after two or more lines of systemic therapy.
The approval recommendation is based on phase 2 trials (NCT02290951, NCT03888105), which demonstrated high ORRs in patients with rrFL and rrDLBCL.
In the DLBCL cohort, a 49% ORR was achieved in heavily pretreated patients who had not received chimeric antigen receptor T-cell therapy. A total of 31% achieved a complete response.
The FL cohort showed an 82% response rate in patients with grades I-IIIA disease, with 75% of the overall population achieving a complete response.
Ordspono will be available as a 2-mg, 80-mg, and 320-mg concentrate for solution for infusion. The active substance of Ordspono is odronextamab, a bispecific antibody that targets CD20-expressing B cells and CD3-expressing T cells. By binding to both, it induces T-cell activation and generates a polyclonal cytotoxic T-cell response, leading to the lysis of malignant B cells.
Generics
The panel also adopted positive opinions for two generic cancer medicines.
Enzalutamide Viatris (enzalutamide) is indicated for the treatment of adult men with prostate cancer in several scenarios:
- As monotherapy or with androgen-deprivation therapy for high-risk biochemical recurrent nonmetastatic hormone-sensitive prostate cancer in men unsuitable for salvage-radiotherapy.
- In combination with androgen-deprivation therapy for metastatic hormone-sensitive prostate cancer.
- For high-risk nonmetastatic castration-resistant prostate cancer (CRPC).
- For metastatic CRPC in men who are asymptomatic or mildly symptomatic after failure of androgen-deprivation therapy, where chemotherapy is not yet indicated.
- For metastatic CRPC in men whose disease has progressed on or after docetaxel therapy.
Enzalutamide Viatris is a generic version of Xtandi, authorized in the European Union since June 2013. Studies have confirmed the satisfactory quality and bioequivalence of Enzalutamide Viatris to Xtandi.
Enzalutamide Viatris will be available as 40-mg and 80-mg film-coated tablets. The active substance of Enzalutamide Viatris is enzalutamide, a hormone antagonist that blocks multiple steps in the androgen receptor–signaling pathway.
Nilotinib Accord (nilotinib) is indicated for the treatment of Philadelphia chromosome–positive chronic myelogenous leukemia (CML).
It is used in adult and pediatric patients with newly diagnosed CML in the chronic phase, adult patients with chronic phase and accelerated phase CML with resistance or intolerance to prior therapy including imatinib, and pediatric patients with CML with resistance or intolerance to prior therapy including imatinib.
Nilotinib Accord is a generic of Tasigna, authorized in the European Union since November 2007. Studies have demonstrated the satisfactory quality and bioequivalence of Nilotinib Accord to Tasigna.
Nilotinib Accord will be available as 50-mg, 150-mg, and 200-mg hard capsules. The active substance of Nilotinib Accord is nilotinib, an antineoplastic protein kinase inhibitor that targets BCR-ABL kinase and other oncogenic kinases.
A version of this article appeared on Medscape.com.
ASCO 2024: Treating Myeloma Just Got More Complicated
For brevity’s sake, I’ll focus on trials about newly diagnosed MM and myeloma at first relapse. Here’s my take on how to interpret those studies in light of broader evidence, what I view as their key limitations, and how what came out of ASCO 2024 changes my approach.
The Return of Belantamab
Belantamab, a BCMA targeting antibody-drug conjugate, previously had shown a response rate of 34% in a single-arm, heavily pretreated population, albeit with modest progression free survival (PFS), only to fail its confirmatory randomized study against pomalidomide/dexamethasone. Given the ocular toxicity associated with belantamab, many — including myself — had written off this drug (save in exceptional/unique circumstances), especially with the rise of novel immunotherapies targeting BCMA, such as chimeric antigen receptor (CAR T-cell) therapy and bispecific antibodies.

However, this year at ASCO, two key randomized trials were presented with concurrent publications, a trial of belantamab/bortezomib/dexamethasone versus daratumumab/bortezomib/dexamethasone (DVd) (DREAMM-7), and a trial of belantamab/pomalidomide/dexamethasone versus bortezomib/pomalidomide/dexamethasone (DREAMM-8). Both trials evaluated patients with myeloma who had relapsed disease and had received at least one prior line of therapy.
In both trials, the belantamab triplet beat the other triplets for the endpoint of PFS (median PFS 36.6 vs 13 months for DREAMM-7, and 12 months PFS 71% vs 51% for DREAMM-8). We must commend the bold three-versus-three design and a convincing result.
What are the caveats? Some censoring of information happened in DREAMM-7, which helped make the intervention arm look better than reality and the control arm look even worse than reality. To illustrate this point: the control arm of DVd (PFS 13 months) underperformed, compared to the CASTOR trial, where DVd led to a PFS of 16.7 months. The drug remains toxic, with high rates of keratopathy and vision problems in its current dosing schema. (Perhaps the future lies in less frequent dosing.) This toxicity is almost always reversible, but it is a huge problem to deal with, and our current quality-of-life instruments fail miserably at capturing this.
Furthermore, DVd is now emerging as perhaps the weakest daratumumab triplet that exists. Almost all patients in this trial had disease sensitivity to lenalidomide, and daratumumab/lenalidomide/dexamethasone (PFS of 45 months in the POLLUX trial) is unequivocally easier to use and handle (in my opinion) than this belantamab triplet--which is quite literally “an eyesore.” Would belantamab-based triplets beat dara/len/dex for patients with lenalidomide sensitive disease? Or, for that matter, would belantamab combos beat anti-CD38+carfilzomib+dex combinations, or cilta-cel (which is also now approved for first relapse)?
How do I foresee the future of belantamab? Despite these unequivocally positive results, I am not enthused about using it for most patients at first relapse. When trials for bispecifics at first relapse read out, my enthusiasm will likely wane even more. Still, it is useful to have belantamab in the armamentarium. For some patients perceived to be at very high risk of infection, belantamab-based triplets may indeed prove to be a better option than bispecifics. However, I suspect that with better dosing strategies for bispecifics, perhaps even that trend may be mitigated. Since we do not yet have bispecifics available in this line, my suggested algorithm for first relapse is as follows:
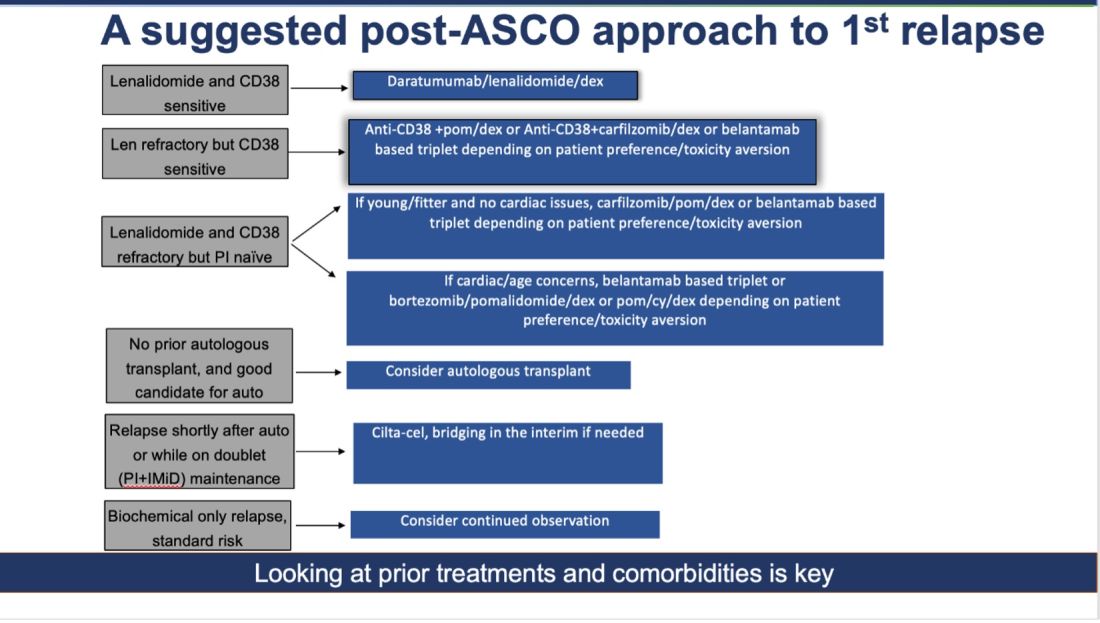
Newly Diagnosed MM: The Era of Quads Solidifies
At ASCO 2024, two key trials with concurrent publications assessed the role of quadruplets (without the use of transplant): the IMROZ trial of a quadruplet of isatuximab/bortezomib/lenalidomide/dexamethasone versus bortezomib/lenalidomide/dexamethasone (VRd), and the BENEFIT trial (isatuximab/lenalidomide/bortezomib/dexamethasone versus isatuximab/lenalidomide/dexamethasone).
The IMROZ trial tested the addition of an anti-CD38 antibody to a triplet backbone, and the results are compelling. The PFS was not reached for the quad vs 54 months for VRd. Unlike in the belantamab trial (where the control arm underperformed), here the control arm really overperformed. In this case, we have never seen such a compelling PFS of 54 months for VRd before. (Based on other trials, VRd PFS has been more in the ballpark of 35-43 months.) This speaks to the fitness and biology of the patients enrolled in this trial, and perhaps to how we will not see such stellar results with this quad recreated in real life.
The addition of isatuximab did not seem to impair quality of life, and although there were more treatment-related deaths with isatuximab, those higher numbers seem to have been driven by longer treatment durations. For this study, the upper age limit was 80 years, and most patients enrolled had an excellent functional status--making it clear that frail patients were greatly underrepresented.
What can we conclude from this study? For fit, older patients (who would have been transplant-eligible in the United States), this study provides excellent proof of concept that very good outcomes can be obtained without the use of transplantation. In treating frail patients, we do not know if quads are safe (or even necessary, compared to gentler sequencing), so these data are not applicable.
High-risk cytogenetics were underrepresented, and although the subgroup analysis for such patients did not show a benefit, it is hard to draw conclusions either way. For me, this trial is further evidence that for many older patients with MM, even if you “can” do a transplant, you probably “shouldn’t, they will experience increasingly better outcomes.
The standard for newly diagnosed MM in older patients for whom transplant is not intended is currently dara/len/dex. Is isa/bort/len/dex better? I do not know. It may give a better PFS, but the addition of bortezomib will lead to more neuropathy: 60% of patients developed neuropathy here, with 7% developing Grade III/IV peripheral neuropathy.
To resolve this issue, highly individualized discussions with patients will be needed. The BENEFIT trial evaluated this question more directly, with a randomized comparison of Isa-VRd versus Isa-Rd (the role of bortezomib being the main variable assessed here) with a primary endpoint of MRD negativity at 10-5 at 18 months. Although MRD negativity allows for a quick read-out, having MRD as an endpoint is a foregone conclusion. Adding another drug will almost certainly lead to deeper responses. But is it worth it?
In the BENEFIT trial, the MRD negativity at 10-5 was 26% versus 53% with the quad. However, peripheral neuropathy rates were much higher with the quad (28% vs 52%). Without longer-term data such as PFS and OS, I do not know whether it is worth the extra risks of neuropathy for older patients. Their priority may not be eradication of cancer cells at all costs. Instead, it may be better quality of life and functioning while preserving survival.
To sum up: Post-ASCO 2024, the approach to newly diagnosed MM just got a lot more complicated. For fit, older patients willing to endure extra toxicities of neuropathy (and acknowledging that we do not know whether survival will be any better with this approach), a quad is a very reasonable option to offer while forgoing transplant, in resource-rich areas of the world, such as the United States. Omitting a transplant now seems very reasonable for most older adults. However, a nuanced and individualized approach remains paramount. And given the speed of new developments, even this suggested approach will be outdated soon!
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
For brevity’s sake, I’ll focus on trials about newly diagnosed MM and myeloma at first relapse. Here’s my take on how to interpret those studies in light of broader evidence, what I view as their key limitations, and how what came out of ASCO 2024 changes my approach.
The Return of Belantamab
Belantamab, a BCMA targeting antibody-drug conjugate, previously had shown a response rate of 34% in a single-arm, heavily pretreated population, albeit with modest progression free survival (PFS), only to fail its confirmatory randomized study against pomalidomide/dexamethasone. Given the ocular toxicity associated with belantamab, many — including myself — had written off this drug (save in exceptional/unique circumstances), especially with the rise of novel immunotherapies targeting BCMA, such as chimeric antigen receptor (CAR T-cell) therapy and bispecific antibodies.
However, this year at ASCO, two key randomized trials were presented with concurrent publications, a trial of belantamab/bortezomib/dexamethasone versus daratumumab/bortezomib/dexamethasone (DVd) (DREAMM-7), and a trial of belantamab/pomalidomide/dexamethasone versus bortezomib/pomalidomide/dexamethasone (DREAMM-8). Both trials evaluated patients with myeloma who had relapsed disease and had received at least one prior line of therapy.
In both trials, the belantamab triplet beat the other triplets for the endpoint of PFS (median PFS 36.6 vs 13 months for DREAMM-7, and 12 months PFS 71% vs 51% for DREAMM-8). We must commend the bold three-versus-three design and a convincing result.
What are the caveats? Some censoring of information happened in DREAMM-7, which helped make the intervention arm look better than reality and the control arm look even worse than reality. To illustrate this point: the control arm of DVd (PFS 13 months) underperformed, compared to the CASTOR trial, where DVd led to a PFS of 16.7 months. The drug remains toxic, with high rates of keratopathy and vision problems in its current dosing schema. (Perhaps the future lies in less frequent dosing.) This toxicity is almost always reversible, but it is a huge problem to deal with, and our current quality-of-life instruments fail miserably at capturing this.
Furthermore, DVd is now emerging as perhaps the weakest daratumumab triplet that exists. Almost all patients in this trial had disease sensitivity to lenalidomide, and daratumumab/lenalidomide/dexamethasone (PFS of 45 months in the POLLUX trial) is unequivocally easier to use and handle (in my opinion) than this belantamab triplet--which is quite literally “an eyesore.” Would belantamab-based triplets beat dara/len/dex for patients with lenalidomide sensitive disease? Or, for that matter, would belantamab combos beat anti-CD38+carfilzomib+dex combinations, or cilta-cel (which is also now approved for first relapse)?
How do I foresee the future of belantamab? Despite these unequivocally positive results, I am not enthused about using it for most patients at first relapse. When trials for bispecifics at first relapse read out, my enthusiasm will likely wane even more. Still, it is useful to have belantamab in the armamentarium. For some patients perceived to be at very high risk of infection, belantamab-based triplets may indeed prove to be a better option than bispecifics. However, I suspect that with better dosing strategies for bispecifics, perhaps even that trend may be mitigated. Since we do not yet have bispecifics available in this line, my suggested algorithm for first relapse is as follows:
Newly Diagnosed MM: The Era of Quads Solidifies
At ASCO 2024, two key trials with concurrent publications assessed the role of quadruplets (without the use of transplant): the IMROZ trial of a quadruplet of isatuximab/bortezomib/lenalidomide/dexamethasone versus bortezomib/lenalidomide/dexamethasone (VRd), and the BENEFIT trial (isatuximab/lenalidomide/bortezomib/dexamethasone versus isatuximab/lenalidomide/dexamethasone).
The IMROZ trial tested the addition of an anti-CD38 antibody to a triplet backbone, and the results are compelling. The PFS was not reached for the quad vs 54 months for VRd. Unlike in the belantamab trial (where the control arm underperformed), here the control arm really overperformed. In this case, we have never seen such a compelling PFS of 54 months for VRd before. (Based on other trials, VRd PFS has been more in the ballpark of 35-43 months.) This speaks to the fitness and biology of the patients enrolled in this trial, and perhaps to how we will not see such stellar results with this quad recreated in real life.
The addition of isatuximab did not seem to impair quality of life, and although there were more treatment-related deaths with isatuximab, those higher numbers seem to have been driven by longer treatment durations. For this study, the upper age limit was 80 years, and most patients enrolled had an excellent functional status--making it clear that frail patients were greatly underrepresented.
What can we conclude from this study? For fit, older patients (who would have been transplant-eligible in the United States), this study provides excellent proof of concept that very good outcomes can be obtained without the use of transplantation. In treating frail patients, we do not know if quads are safe (or even necessary, compared to gentler sequencing), so these data are not applicable.
High-risk cytogenetics were underrepresented, and although the subgroup analysis for such patients did not show a benefit, it is hard to draw conclusions either way. For me, this trial is further evidence that for many older patients with MM, even if you “can” do a transplant, you probably “shouldn’t, they will experience increasingly better outcomes.
The standard for newly diagnosed MM in older patients for whom transplant is not intended is currently dara/len/dex. Is isa/bort/len/dex better? I do not know. It may give a better PFS, but the addition of bortezomib will lead to more neuropathy: 60% of patients developed neuropathy here, with 7% developing Grade III/IV peripheral neuropathy.
To resolve this issue, highly individualized discussions with patients will be needed. The BENEFIT trial evaluated this question more directly, with a randomized comparison of Isa-VRd versus Isa-Rd (the role of bortezomib being the main variable assessed here) with a primary endpoint of MRD negativity at 10-5 at 18 months. Although MRD negativity allows for a quick read-out, having MRD as an endpoint is a foregone conclusion. Adding another drug will almost certainly lead to deeper responses. But is it worth it?
In the BENEFIT trial, the MRD negativity at 10-5 was 26% versus 53% with the quad. However, peripheral neuropathy rates were much higher with the quad (28% vs 52%). Without longer-term data such as PFS and OS, I do not know whether it is worth the extra risks of neuropathy for older patients. Their priority may not be eradication of cancer cells at all costs. Instead, it may be better quality of life and functioning while preserving survival.
To sum up: Post-ASCO 2024, the approach to newly diagnosed MM just got a lot more complicated. For fit, older patients willing to endure extra toxicities of neuropathy (and acknowledging that we do not know whether survival will be any better with this approach), a quad is a very reasonable option to offer while forgoing transplant, in resource-rich areas of the world, such as the United States. Omitting a transplant now seems very reasonable for most older adults. However, a nuanced and individualized approach remains paramount. And given the speed of new developments, even this suggested approach will be outdated soon!
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
For brevity’s sake, I’ll focus on trials about newly diagnosed MM and myeloma at first relapse. Here’s my take on how to interpret those studies in light of broader evidence, what I view as their key limitations, and how what came out of ASCO 2024 changes my approach.
The Return of Belantamab
Belantamab, a BCMA targeting antibody-drug conjugate, previously had shown a response rate of 34% in a single-arm, heavily pretreated population, albeit with modest progression free survival (PFS), only to fail its confirmatory randomized study against pomalidomide/dexamethasone. Given the ocular toxicity associated with belantamab, many — including myself — had written off this drug (save in exceptional/unique circumstances), especially with the rise of novel immunotherapies targeting BCMA, such as chimeric antigen receptor (CAR T-cell) therapy and bispecific antibodies.
However, this year at ASCO, two key randomized trials were presented with concurrent publications, a trial of belantamab/bortezomib/dexamethasone versus daratumumab/bortezomib/dexamethasone (DVd) (DREAMM-7), and a trial of belantamab/pomalidomide/dexamethasone versus bortezomib/pomalidomide/dexamethasone (DREAMM-8). Both trials evaluated patients with myeloma who had relapsed disease and had received at least one prior line of therapy.
In both trials, the belantamab triplet beat the other triplets for the endpoint of PFS (median PFS 36.6 vs 13 months for DREAMM-7, and 12 months PFS 71% vs 51% for DREAMM-8). We must commend the bold three-versus-three design and a convincing result.
What are the caveats? Some censoring of information happened in DREAMM-7, which helped make the intervention arm look better than reality and the control arm look even worse than reality. To illustrate this point: the control arm of DVd (PFS 13 months) underperformed, compared to the CASTOR trial, where DVd led to a PFS of 16.7 months. The drug remains toxic, with high rates of keratopathy and vision problems in its current dosing schema. (Perhaps the future lies in less frequent dosing.) This toxicity is almost always reversible, but it is a huge problem to deal with, and our current quality-of-life instruments fail miserably at capturing this.
Furthermore, DVd is now emerging as perhaps the weakest daratumumab triplet that exists. Almost all patients in this trial had disease sensitivity to lenalidomide, and daratumumab/lenalidomide/dexamethasone (PFS of 45 months in the POLLUX trial) is unequivocally easier to use and handle (in my opinion) than this belantamab triplet--which is quite literally “an eyesore.” Would belantamab-based triplets beat dara/len/dex for patients with lenalidomide sensitive disease? Or, for that matter, would belantamab combos beat anti-CD38+carfilzomib+dex combinations, or cilta-cel (which is also now approved for first relapse)?
How do I foresee the future of belantamab? Despite these unequivocally positive results, I am not enthused about using it for most patients at first relapse. When trials for bispecifics at first relapse read out, my enthusiasm will likely wane even more. Still, it is useful to have belantamab in the armamentarium. For some patients perceived to be at very high risk of infection, belantamab-based triplets may indeed prove to be a better option than bispecifics. However, I suspect that with better dosing strategies for bispecifics, perhaps even that trend may be mitigated. Since we do not yet have bispecifics available in this line, my suggested algorithm for first relapse is as follows:
Newly Diagnosed MM: The Era of Quads Solidifies
At ASCO 2024, two key trials with concurrent publications assessed the role of quadruplets (without the use of transplant): the IMROZ trial of a quadruplet of isatuximab/bortezomib/lenalidomide/dexamethasone versus bortezomib/lenalidomide/dexamethasone (VRd), and the BENEFIT trial (isatuximab/lenalidomide/bortezomib/dexamethasone versus isatuximab/lenalidomide/dexamethasone).
The IMROZ trial tested the addition of an anti-CD38 antibody to a triplet backbone, and the results are compelling. The PFS was not reached for the quad vs 54 months for VRd. Unlike in the belantamab trial (where the control arm underperformed), here the control arm really overperformed. In this case, we have never seen such a compelling PFS of 54 months for VRd before. (Based on other trials, VRd PFS has been more in the ballpark of 35-43 months.) This speaks to the fitness and biology of the patients enrolled in this trial, and perhaps to how we will not see such stellar results with this quad recreated in real life.
The addition of isatuximab did not seem to impair quality of life, and although there were more treatment-related deaths with isatuximab, those higher numbers seem to have been driven by longer treatment durations. For this study, the upper age limit was 80 years, and most patients enrolled had an excellent functional status--making it clear that frail patients were greatly underrepresented.
What can we conclude from this study? For fit, older patients (who would have been transplant-eligible in the United States), this study provides excellent proof of concept that very good outcomes can be obtained without the use of transplantation. In treating frail patients, we do not know if quads are safe (or even necessary, compared to gentler sequencing), so these data are not applicable.
High-risk cytogenetics were underrepresented, and although the subgroup analysis for such patients did not show a benefit, it is hard to draw conclusions either way. For me, this trial is further evidence that for many older patients with MM, even if you “can” do a transplant, you probably “shouldn’t, they will experience increasingly better outcomes.
The standard for newly diagnosed MM in older patients for whom transplant is not intended is currently dara/len/dex. Is isa/bort/len/dex better? I do not know. It may give a better PFS, but the addition of bortezomib will lead to more neuropathy: 60% of patients developed neuropathy here, with 7% developing Grade III/IV peripheral neuropathy.
To resolve this issue, highly individualized discussions with patients will be needed. The BENEFIT trial evaluated this question more directly, with a randomized comparison of Isa-VRd versus Isa-Rd (the role of bortezomib being the main variable assessed here) with a primary endpoint of MRD negativity at 10-5 at 18 months. Although MRD negativity allows for a quick read-out, having MRD as an endpoint is a foregone conclusion. Adding another drug will almost certainly lead to deeper responses. But is it worth it?
In the BENEFIT trial, the MRD negativity at 10-5 was 26% versus 53% with the quad. However, peripheral neuropathy rates were much higher with the quad (28% vs 52%). Without longer-term data such as PFS and OS, I do not know whether it is worth the extra risks of neuropathy for older patients. Their priority may not be eradication of cancer cells at all costs. Instead, it may be better quality of life and functioning while preserving survival.
To sum up: Post-ASCO 2024, the approach to newly diagnosed MM just got a lot more complicated. For fit, older patients willing to endure extra toxicities of neuropathy (and acknowledging that we do not know whether survival will be any better with this approach), a quad is a very reasonable option to offer while forgoing transplant, in resource-rich areas of the world, such as the United States. Omitting a transplant now seems very reasonable for most older adults. However, a nuanced and individualized approach remains paramount. And given the speed of new developments, even this suggested approach will be outdated soon!
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
AML: Shorter Venetoclax Course Shows Promise for Some
However, the azacitidine plus venetoclax therapy — the “7+7” regimen — was associated with lower platelet transfusion requirements and lower 8-week mortality, suggesting the regimen might be preferable in certain patient populations, Alexandre Bazinet, MD, of the University of Texas MD Anderson Cancer Center, Houston, reported at the American Society of Clinical Oncology (ASCO) annual meeting.
The composite complete remission (CRc) rate, including complete remission with or without complete count recovery, was identical at 72% among 82 patients treated with the 7+7 regimen and 166 treated with standard therapy, and the complete remission (CR) rate was 57% and 55%, respectively, Dr. Bazinet said.
The median number of cycles to first response was one in both groups, but 42% of responders in the 7+7 group required more than one cycle to achieve their first response, compared with just 1% of those in the standard therapy group, he noted, adding that the median number of cycles to achieve best response was two in the 7+7 group and one in the standard therapy group.
The mortality rate at 4 weeks was similar in the groups (2% vs 5% for 7+7 vs standard therapy), but at 8 weeks, the mortality rate was significantly higher in the standard therapy group (6% vs 16%, respectively). Median overall survival (OS) was 11.2 months versus 10.3 months, and median 2-year survival was 27.7% versus 33.6% in the groups, respectively.
Event-free survival was 6.5 versus 7.4 months, and 2-year event-free survival was 24.5% versus 27.0%, respectively.
Of note, fewer patients in the 7+7 group required platelet transfusions during cycle 1 (62% vs 77%) and the cycle 1 rates of neutropenic fever and red cell transfusion requirements were similar in the two treatment groups, Dr. Bazinet said.
Study participants were 82 adults from seven centers in France who received the 7+7 regimen, and 166 adults from MD Anderson who received standard therapy with a hypomethylating agent plus venetoclax doublets given for 21-28 days during induction. Preliminary data on the 7+7 regimen in patients from the French centers were reported previously and “suggested preserved efficacy with potentially less toxicity,” he noted.
“A hypomethylating agent plus venetoclax doublets are standard-of-care in patients with AML who are older or ineligible for chemotherapy due to comorbidities,” Dr. Bazinet explained, adding that although the venetoclax label calls for 28 days of drug per cycle, shorter courses of 14 to 21 days are commonly used.
These findings are limited by the retrospective study design and by small patient numbers in many subgroups, he said.
“In addition, the cohorts were heterogeneous, consisting of patients treated with a variety of different regimens and across multiple centers and countries. The distribution of FLT3-ITD and NRAS/KRAS mutations differed significantly between cohorts,” he explained, also noting that prophylactic azole use differed across the cohort. “Furthermore, analysis of the toxicity results was also limited by likely differing transfusion polices in different centers.”
Overall, however, the findings suggest that reducing the duration of venetoclax is safe and results in similar CRc rates, although responses may be faster with standard dosing, he said, adding that “7+7 is potentially less toxic and is attractive in patients who are more frail or at risk for complications.”
“Our data support further study of shorter venetoclax duration, within emerging triplet regimens in patients with intermediate or low predictive benefit to mitigate toxicity,” he concluded.
Dr. Bazinet reported having no disclosures.
However, the azacitidine plus venetoclax therapy — the “7+7” regimen — was associated with lower platelet transfusion requirements and lower 8-week mortality, suggesting the regimen might be preferable in certain patient populations, Alexandre Bazinet, MD, of the University of Texas MD Anderson Cancer Center, Houston, reported at the American Society of Clinical Oncology (ASCO) annual meeting.
The composite complete remission (CRc) rate, including complete remission with or without complete count recovery, was identical at 72% among 82 patients treated with the 7+7 regimen and 166 treated with standard therapy, and the complete remission (CR) rate was 57% and 55%, respectively, Dr. Bazinet said.
The median number of cycles to first response was one in both groups, but 42% of responders in the 7+7 group required more than one cycle to achieve their first response, compared with just 1% of those in the standard therapy group, he noted, adding that the median number of cycles to achieve best response was two in the 7+7 group and one in the standard therapy group.
The mortality rate at 4 weeks was similar in the groups (2% vs 5% for 7+7 vs standard therapy), but at 8 weeks, the mortality rate was significantly higher in the standard therapy group (6% vs 16%, respectively). Median overall survival (OS) was 11.2 months versus 10.3 months, and median 2-year survival was 27.7% versus 33.6% in the groups, respectively.
Event-free survival was 6.5 versus 7.4 months, and 2-year event-free survival was 24.5% versus 27.0%, respectively.
Of note, fewer patients in the 7+7 group required platelet transfusions during cycle 1 (62% vs 77%) and the cycle 1 rates of neutropenic fever and red cell transfusion requirements were similar in the two treatment groups, Dr. Bazinet said.
Study participants were 82 adults from seven centers in France who received the 7+7 regimen, and 166 adults from MD Anderson who received standard therapy with a hypomethylating agent plus venetoclax doublets given for 21-28 days during induction. Preliminary data on the 7+7 regimen in patients from the French centers were reported previously and “suggested preserved efficacy with potentially less toxicity,” he noted.
“A hypomethylating agent plus venetoclax doublets are standard-of-care in patients with AML who are older or ineligible for chemotherapy due to comorbidities,” Dr. Bazinet explained, adding that although the venetoclax label calls for 28 days of drug per cycle, shorter courses of 14 to 21 days are commonly used.
These findings are limited by the retrospective study design and by small patient numbers in many subgroups, he said.
“In addition, the cohorts were heterogeneous, consisting of patients treated with a variety of different regimens and across multiple centers and countries. The distribution of FLT3-ITD and NRAS/KRAS mutations differed significantly between cohorts,” he explained, also noting that prophylactic azole use differed across the cohort. “Furthermore, analysis of the toxicity results was also limited by likely differing transfusion polices in different centers.”
Overall, however, the findings suggest that reducing the duration of venetoclax is safe and results in similar CRc rates, although responses may be faster with standard dosing, he said, adding that “7+7 is potentially less toxic and is attractive in patients who are more frail or at risk for complications.”
“Our data support further study of shorter venetoclax duration, within emerging triplet regimens in patients with intermediate or low predictive benefit to mitigate toxicity,” he concluded.
Dr. Bazinet reported having no disclosures.
However, the azacitidine plus venetoclax therapy — the “7+7” regimen — was associated with lower platelet transfusion requirements and lower 8-week mortality, suggesting the regimen might be preferable in certain patient populations, Alexandre Bazinet, MD, of the University of Texas MD Anderson Cancer Center, Houston, reported at the American Society of Clinical Oncology (ASCO) annual meeting.
The composite complete remission (CRc) rate, including complete remission with or without complete count recovery, was identical at 72% among 82 patients treated with the 7+7 regimen and 166 treated with standard therapy, and the complete remission (CR) rate was 57% and 55%, respectively, Dr. Bazinet said.
The median number of cycles to first response was one in both groups, but 42% of responders in the 7+7 group required more than one cycle to achieve their first response, compared with just 1% of those in the standard therapy group, he noted, adding that the median number of cycles to achieve best response was two in the 7+7 group and one in the standard therapy group.
The mortality rate at 4 weeks was similar in the groups (2% vs 5% for 7+7 vs standard therapy), but at 8 weeks, the mortality rate was significantly higher in the standard therapy group (6% vs 16%, respectively). Median overall survival (OS) was 11.2 months versus 10.3 months, and median 2-year survival was 27.7% versus 33.6% in the groups, respectively.
Event-free survival was 6.5 versus 7.4 months, and 2-year event-free survival was 24.5% versus 27.0%, respectively.
Of note, fewer patients in the 7+7 group required platelet transfusions during cycle 1 (62% vs 77%) and the cycle 1 rates of neutropenic fever and red cell transfusion requirements were similar in the two treatment groups, Dr. Bazinet said.
Study participants were 82 adults from seven centers in France who received the 7+7 regimen, and 166 adults from MD Anderson who received standard therapy with a hypomethylating agent plus venetoclax doublets given for 21-28 days during induction. Preliminary data on the 7+7 regimen in patients from the French centers were reported previously and “suggested preserved efficacy with potentially less toxicity,” he noted.
“A hypomethylating agent plus venetoclax doublets are standard-of-care in patients with AML who are older or ineligible for chemotherapy due to comorbidities,” Dr. Bazinet explained, adding that although the venetoclax label calls for 28 days of drug per cycle, shorter courses of 14 to 21 days are commonly used.
These findings are limited by the retrospective study design and by small patient numbers in many subgroups, he said.
“In addition, the cohorts were heterogeneous, consisting of patients treated with a variety of different regimens and across multiple centers and countries. The distribution of FLT3-ITD and NRAS/KRAS mutations differed significantly between cohorts,” he explained, also noting that prophylactic azole use differed across the cohort. “Furthermore, analysis of the toxicity results was also limited by likely differing transfusion polices in different centers.”
Overall, however, the findings suggest that reducing the duration of venetoclax is safe and results in similar CRc rates, although responses may be faster with standard dosing, he said, adding that “7+7 is potentially less toxic and is attractive in patients who are more frail or at risk for complications.”
“Our data support further study of shorter venetoclax duration, within emerging triplet regimens in patients with intermediate or low predictive benefit to mitigate toxicity,” he concluded.
Dr. Bazinet reported having no disclosures.
FROM ASCO 2024
FDA Proposes that Interchangeability Status for Biosimilars Doesn’t Need Switching Studies
The Food and Drug Administration (FDA) has issued new draft guidance that does not require additional switching studies for biosimilars seeking interchangeability. These studies were previously recommended to demonstrate that switching between the biosimilar and its reference product showed no greater risk than using the reference product alone.
“The recommendations in today’s draft guidance, when finalized, will provide clarity and transparency about the FDA’s thinking and align the review and approval process with existing and emerging science,” said Sarah Yim, MD, director of the FDA’s Office of Therapeutic Biologics and Biosimilars in a statement on June 20. “We have gained valuable experience reviewing both biosimilar and interchangeable biosimilar medications over the past 10 years. Both biosimilars and interchangeable biosimilars meet the same high standard of biosimilarity for FDA approval and both are as safe and effective as the reference product.”
An interchangeable status allows a biosimilar product to be swapped with the reference product without involvement from the prescribing provider, depending on state law.
While switching studies were not required under previous FDA guidance, the 2019 document did state that the agency “expects that applications generally will include data from a switching study or studies in one or more appropriate conditions of use.”
However, of the 13 biosimilars that received interchangeability status, 9 did not include switching study data.
“Experience has shown that, for the products approved as biosimilars to date, the risk in terms of safety or diminished efficacy is insignificant following single or multiple switches between a reference product and a biosimilar product,” the FDA stated. The agency’s investigators also conducted a systematic review of switching studies, which found no differences in risk for death, serious adverse events, and treatment discontinuations in participants switched between biosimilars and reference products and those that remained on reference products.
“Additionally, today’s analytical tools can accurately evaluate the structure and effects [of] biologic products, both in the lab (in vitro) and in living organisms (in vivo) with more precision and sensitivity than switching studies,” the agency noted.
The FDA is now calling for commentary on these draft recommendations to be submitted by Aug. 20, 2024.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) has issued new draft guidance that does not require additional switching studies for biosimilars seeking interchangeability. These studies were previously recommended to demonstrate that switching between the biosimilar and its reference product showed no greater risk than using the reference product alone.
“The recommendations in today’s draft guidance, when finalized, will provide clarity and transparency about the FDA’s thinking and align the review and approval process with existing and emerging science,” said Sarah Yim, MD, director of the FDA’s Office of Therapeutic Biologics and Biosimilars in a statement on June 20. “We have gained valuable experience reviewing both biosimilar and interchangeable biosimilar medications over the past 10 years. Both biosimilars and interchangeable biosimilars meet the same high standard of biosimilarity for FDA approval and both are as safe and effective as the reference product.”
An interchangeable status allows a biosimilar product to be swapped with the reference product without involvement from the prescribing provider, depending on state law.
While switching studies were not required under previous FDA guidance, the 2019 document did state that the agency “expects that applications generally will include data from a switching study or studies in one or more appropriate conditions of use.”
However, of the 13 biosimilars that received interchangeability status, 9 did not include switching study data.
“Experience has shown that, for the products approved as biosimilars to date, the risk in terms of safety or diminished efficacy is insignificant following single or multiple switches between a reference product and a biosimilar product,” the FDA stated. The agency’s investigators also conducted a systematic review of switching studies, which found no differences in risk for death, serious adverse events, and treatment discontinuations in participants switched between biosimilars and reference products and those that remained on reference products.
“Additionally, today’s analytical tools can accurately evaluate the structure and effects [of] biologic products, both in the lab (in vitro) and in living organisms (in vivo) with more precision and sensitivity than switching studies,” the agency noted.
The FDA is now calling for commentary on these draft recommendations to be submitted by Aug. 20, 2024.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) has issued new draft guidance that does not require additional switching studies for biosimilars seeking interchangeability. These studies were previously recommended to demonstrate that switching between the biosimilar and its reference product showed no greater risk than using the reference product alone.
“The recommendations in today’s draft guidance, when finalized, will provide clarity and transparency about the FDA’s thinking and align the review and approval process with existing and emerging science,” said Sarah Yim, MD, director of the FDA’s Office of Therapeutic Biologics and Biosimilars in a statement on June 20. “We have gained valuable experience reviewing both biosimilar and interchangeable biosimilar medications over the past 10 years. Both biosimilars and interchangeable biosimilars meet the same high standard of biosimilarity for FDA approval and both are as safe and effective as the reference product.”
An interchangeable status allows a biosimilar product to be swapped with the reference product without involvement from the prescribing provider, depending on state law.
While switching studies were not required under previous FDA guidance, the 2019 document did state that the agency “expects that applications generally will include data from a switching study or studies in one or more appropriate conditions of use.”
However, of the 13 biosimilars that received interchangeability status, 9 did not include switching study data.
“Experience has shown that, for the products approved as biosimilars to date, the risk in terms of safety or diminished efficacy is insignificant following single or multiple switches between a reference product and a biosimilar product,” the FDA stated. The agency’s investigators also conducted a systematic review of switching studies, which found no differences in risk for death, serious adverse events, and treatment discontinuations in participants switched between biosimilars and reference products and those that remained on reference products.
“Additionally, today’s analytical tools can accurately evaluate the structure and effects [of] biologic products, both in the lab (in vitro) and in living organisms (in vivo) with more precision and sensitivity than switching studies,” the agency noted.
The FDA is now calling for commentary on these draft recommendations to be submitted by Aug. 20, 2024.
A version of this article first appeared on Medscape.com.
B-ALL: New Findings Confirm Efficacy of CAR T Product
These findings also highlight the favorable impact of CAR T persistence on treatment outcomes, and suggest that consolidative stem cell transplant (SCT) in R/R B-ALL patients treated with obe-cel does not improve outcomes, Elias Jabbour, MD, of the University of Texas MD Anderson Cancer Center, Houston, reported at the American Society of Clinical Oncology (ASCO) annual meeting.
The overall complete remission or complete remission with incomplete count recovery rate was 78% among 127 patients enrolled in the open-label, single-arm study and infused with obe-cel. Among the 99 patients who responded, 18 proceeded to consolidative SCT while in remission, Dr. Jabbour said, noting that all 18 who received SCT were in minimal residual disease (MRD)–negative remission at the time of transplant.
Of those 18 patients, 10 had ongoing CAR T persistence prior to transplant, he said.
At median follow-up of 21.5 months, 40% of responders were in ongoing remission without the need for subsequent consolidation with SCT or other therapy, whereas SCT did not appear to improve outcomes.
The median event-free survival (EFS) after censoring for transplant was 11.9 months, and the 12-month EFS rate was 49.5%. Without censoring for transplant, the EFS and 12-month EFS rate were 9.0 months and 44%, respectively.
“I would like to highlight that the time to transplant was 100 days, and of those 18 patients, all in MRD-negative status ... 80% relapsed or died from transplant-related complications,” Dr. Jabbour said.
Median overall survival (OS) without censoring for transplant was 15.6 months, and the 12-month OS rate was 61.1%. After censoring for transplant, the median OS and 12-month OS rate 23.8 months 63.7%, respectively. The survival curves were fully overlapping, indicating that transplant did not improve OS outcomes.
“Furthermore, when you look at the EFS and [OS], both show a potential plateau for a long-term outcome, and this trend is similar to what was reported in a phase 1 trial with 2 years of follow up and more,” Dr. Jabbour said.
The investigators also assessed the impact of loss of CAR T-cell persistence and loss of B-cell aplasia and found that “both ongoing CAR T-cell persistence and ongoing B-cell aplasia, were correlated with better event-free survival,” he noted, explaining that the risk of relapse was 2.7 times greater in those who lost versus maintained CAR T-cell persistence, and 1.7 times greater in those who lost versus maintained B-cell aplasia.
Among those with ongoing remission at 6 months, median EFS was 15.1 months in those who lost CAR T-cell persistence, whereas the median EFS was not reached in those who maintained CAR T-cell persistence.
Obe-cel is an autologous CAR T-cell product with a fast off-rate CD19 binder designed to mitigate immunotoxicity and improve CAR T-cell expansion and persistence, Dr. Jabbour said, noting that pooled efficacy and safety results from the FELIX phase 1b and 2 trials of heavily pretreated patients have previously been reported.
The findings support the use of obe-cel as a standard treatment in this patient population, and demonstrate that ongoing CAR T-cell persistence and B-cell aplasia are associated with improved EFS — without further consolidation therapy after treatment, he concluded.
This study was funded by Autolus Therapeutics. Dr. Jabbour disclosed ties with Abbvie, Ascentage Pharma, Adaptive Biotechnologies, Amgen, Astellas Pharma, Bristol-Myers Squibb, Genentech, Incyte, Pfizer, and Takeda.
These findings also highlight the favorable impact of CAR T persistence on treatment outcomes, and suggest that consolidative stem cell transplant (SCT) in R/R B-ALL patients treated with obe-cel does not improve outcomes, Elias Jabbour, MD, of the University of Texas MD Anderson Cancer Center, Houston, reported at the American Society of Clinical Oncology (ASCO) annual meeting.
The overall complete remission or complete remission with incomplete count recovery rate was 78% among 127 patients enrolled in the open-label, single-arm study and infused with obe-cel. Among the 99 patients who responded, 18 proceeded to consolidative SCT while in remission, Dr. Jabbour said, noting that all 18 who received SCT were in minimal residual disease (MRD)–negative remission at the time of transplant.
Of those 18 patients, 10 had ongoing CAR T persistence prior to transplant, he said.
At median follow-up of 21.5 months, 40% of responders were in ongoing remission without the need for subsequent consolidation with SCT or other therapy, whereas SCT did not appear to improve outcomes.
The median event-free survival (EFS) after censoring for transplant was 11.9 months, and the 12-month EFS rate was 49.5%. Without censoring for transplant, the EFS and 12-month EFS rate were 9.0 months and 44%, respectively.
“I would like to highlight that the time to transplant was 100 days, and of those 18 patients, all in MRD-negative status ... 80% relapsed or died from transplant-related complications,” Dr. Jabbour said.
Median overall survival (OS) without censoring for transplant was 15.6 months, and the 12-month OS rate was 61.1%. After censoring for transplant, the median OS and 12-month OS rate 23.8 months 63.7%, respectively. The survival curves were fully overlapping, indicating that transplant did not improve OS outcomes.
“Furthermore, when you look at the EFS and [OS], both show a potential plateau for a long-term outcome, and this trend is similar to what was reported in a phase 1 trial with 2 years of follow up and more,” Dr. Jabbour said.
The investigators also assessed the impact of loss of CAR T-cell persistence and loss of B-cell aplasia and found that “both ongoing CAR T-cell persistence and ongoing B-cell aplasia, were correlated with better event-free survival,” he noted, explaining that the risk of relapse was 2.7 times greater in those who lost versus maintained CAR T-cell persistence, and 1.7 times greater in those who lost versus maintained B-cell aplasia.
Among those with ongoing remission at 6 months, median EFS was 15.1 months in those who lost CAR T-cell persistence, whereas the median EFS was not reached in those who maintained CAR T-cell persistence.
Obe-cel is an autologous CAR T-cell product with a fast off-rate CD19 binder designed to mitigate immunotoxicity and improve CAR T-cell expansion and persistence, Dr. Jabbour said, noting that pooled efficacy and safety results from the FELIX phase 1b and 2 trials of heavily pretreated patients have previously been reported.
The findings support the use of obe-cel as a standard treatment in this patient population, and demonstrate that ongoing CAR T-cell persistence and B-cell aplasia are associated with improved EFS — without further consolidation therapy after treatment, he concluded.
This study was funded by Autolus Therapeutics. Dr. Jabbour disclosed ties with Abbvie, Ascentage Pharma, Adaptive Biotechnologies, Amgen, Astellas Pharma, Bristol-Myers Squibb, Genentech, Incyte, Pfizer, and Takeda.
These findings also highlight the favorable impact of CAR T persistence on treatment outcomes, and suggest that consolidative stem cell transplant (SCT) in R/R B-ALL patients treated with obe-cel does not improve outcomes, Elias Jabbour, MD, of the University of Texas MD Anderson Cancer Center, Houston, reported at the American Society of Clinical Oncology (ASCO) annual meeting.
The overall complete remission or complete remission with incomplete count recovery rate was 78% among 127 patients enrolled in the open-label, single-arm study and infused with obe-cel. Among the 99 patients who responded, 18 proceeded to consolidative SCT while in remission, Dr. Jabbour said, noting that all 18 who received SCT were in minimal residual disease (MRD)–negative remission at the time of transplant.
Of those 18 patients, 10 had ongoing CAR T persistence prior to transplant, he said.
At median follow-up of 21.5 months, 40% of responders were in ongoing remission without the need for subsequent consolidation with SCT or other therapy, whereas SCT did not appear to improve outcomes.
The median event-free survival (EFS) after censoring for transplant was 11.9 months, and the 12-month EFS rate was 49.5%. Without censoring for transplant, the EFS and 12-month EFS rate were 9.0 months and 44%, respectively.
“I would like to highlight that the time to transplant was 100 days, and of those 18 patients, all in MRD-negative status ... 80% relapsed or died from transplant-related complications,” Dr. Jabbour said.
Median overall survival (OS) without censoring for transplant was 15.6 months, and the 12-month OS rate was 61.1%. After censoring for transplant, the median OS and 12-month OS rate 23.8 months 63.7%, respectively. The survival curves were fully overlapping, indicating that transplant did not improve OS outcomes.
“Furthermore, when you look at the EFS and [OS], both show a potential plateau for a long-term outcome, and this trend is similar to what was reported in a phase 1 trial with 2 years of follow up and more,” Dr. Jabbour said.
The investigators also assessed the impact of loss of CAR T-cell persistence and loss of B-cell aplasia and found that “both ongoing CAR T-cell persistence and ongoing B-cell aplasia, were correlated with better event-free survival,” he noted, explaining that the risk of relapse was 2.7 times greater in those who lost versus maintained CAR T-cell persistence, and 1.7 times greater in those who lost versus maintained B-cell aplasia.
Among those with ongoing remission at 6 months, median EFS was 15.1 months in those who lost CAR T-cell persistence, whereas the median EFS was not reached in those who maintained CAR T-cell persistence.
Obe-cel is an autologous CAR T-cell product with a fast off-rate CD19 binder designed to mitigate immunotoxicity and improve CAR T-cell expansion and persistence, Dr. Jabbour said, noting that pooled efficacy and safety results from the FELIX phase 1b and 2 trials of heavily pretreated patients have previously been reported.
The findings support the use of obe-cel as a standard treatment in this patient population, and demonstrate that ongoing CAR T-cell persistence and B-cell aplasia are associated with improved EFS — without further consolidation therapy after treatment, he concluded.
This study was funded by Autolus Therapeutics. Dr. Jabbour disclosed ties with Abbvie, Ascentage Pharma, Adaptive Biotechnologies, Amgen, Astellas Pharma, Bristol-Myers Squibb, Genentech, Incyte, Pfizer, and Takeda.
FROM ASCO 2024
FDA Approves Epcoritamab for R/R Follicular Lymphoma
This marks the second indication for the bispecific CD20-directed CD3 T-cell engager. The agent was first approved in 2023 for relapsed or refractory diffuse large B-cell lymphoma in adults.
The current approval was based on the single-arm EPCORE NHL-1 trial in 127 patients with follicular lymphoma who had received at least two lines of systemic therapy.
After a two step-up dosing regimen, the overall response rate was 82%, with 60% of patients achieving a complete response. At a median follow-up of 14.8 months, the median duration of response was not reached. The 12-month duration of response was 68.4%.
Efficacy was similar in the 86 patients who received a three step-up dosing schedule.
Labeling carries a black box warning of cytokine release syndrome and immune effector cell–associated neurotoxicity syndrome. Adverse events in 20% or more of patients included injection site reactions, cytokine release syndrome, COVID-19 infection, fatigue, upper respiratory tract infection, musculoskeletal pain, rash, diarrhea, pyrexia, cough, and headache.
Decreased lymphocyte count, neutrophil count, white blood cell count, and hemoglobin were the most common grade 3/4 laboratory abnormalities.
Three step-up dosing is the recommended regimen, with epcoritamab administered subcutaneously in 28-day cycles until disease progression or unacceptable toxicity. Dosing is increased by steps to the full 48 mg in cycle 1.
The price is $16,282.52 for 48 mg/0.8 mL, according to drugs.com.
A version of this article appeared on Medscape.com.
This marks the second indication for the bispecific CD20-directed CD3 T-cell engager. The agent was first approved in 2023 for relapsed or refractory diffuse large B-cell lymphoma in adults.
The current approval was based on the single-arm EPCORE NHL-1 trial in 127 patients with follicular lymphoma who had received at least two lines of systemic therapy.
After a two step-up dosing regimen, the overall response rate was 82%, with 60% of patients achieving a complete response. At a median follow-up of 14.8 months, the median duration of response was not reached. The 12-month duration of response was 68.4%.
Efficacy was similar in the 86 patients who received a three step-up dosing schedule.
Labeling carries a black box warning of cytokine release syndrome and immune effector cell–associated neurotoxicity syndrome. Adverse events in 20% or more of patients included injection site reactions, cytokine release syndrome, COVID-19 infection, fatigue, upper respiratory tract infection, musculoskeletal pain, rash, diarrhea, pyrexia, cough, and headache.
Decreased lymphocyte count, neutrophil count, white blood cell count, and hemoglobin were the most common grade 3/4 laboratory abnormalities.
Three step-up dosing is the recommended regimen, with epcoritamab administered subcutaneously in 28-day cycles until disease progression or unacceptable toxicity. Dosing is increased by steps to the full 48 mg in cycle 1.
The price is $16,282.52 for 48 mg/0.8 mL, according to drugs.com.
A version of this article appeared on Medscape.com.
This marks the second indication for the bispecific CD20-directed CD3 T-cell engager. The agent was first approved in 2023 for relapsed or refractory diffuse large B-cell lymphoma in adults.
The current approval was based on the single-arm EPCORE NHL-1 trial in 127 patients with follicular lymphoma who had received at least two lines of systemic therapy.
After a two step-up dosing regimen, the overall response rate was 82%, with 60% of patients achieving a complete response. At a median follow-up of 14.8 months, the median duration of response was not reached. The 12-month duration of response was 68.4%.
Efficacy was similar in the 86 patients who received a three step-up dosing schedule.
Labeling carries a black box warning of cytokine release syndrome and immune effector cell–associated neurotoxicity syndrome. Adverse events in 20% or more of patients included injection site reactions, cytokine release syndrome, COVID-19 infection, fatigue, upper respiratory tract infection, musculoskeletal pain, rash, diarrhea, pyrexia, cough, and headache.
Decreased lymphocyte count, neutrophil count, white blood cell count, and hemoglobin were the most common grade 3/4 laboratory abnormalities.
Three step-up dosing is the recommended regimen, with epcoritamab administered subcutaneously in 28-day cycles until disease progression or unacceptable toxicity. Dosing is increased by steps to the full 48 mg in cycle 1.
The price is $16,282.52 for 48 mg/0.8 mL, according to drugs.com.
A version of this article appeared on Medscape.com.
Hemophilia: Marstacimab Sustains Long-Term Bleeding Reduction
“In the long-term extension study treatment with marstacimab demonstrates sustained or improved efficacy for treated and total annualized bleeding rates (ABR) in adults and adolescents with hemophilia A or hemophilia B in this data set of patients without inhibitors,” first author Shamsah Kazani, MD, of Pfizer, Cambridge, Massachusetts, said in presenting the findings at the 2024 annual meeting of the European Hematology Association (EHA) in Madrid.
“The majority of the patients from the pivotal study chose to transition into the long-term extension, and we are finding that these patients are highly compliant with their weekly marstacimab dose, with more than 98% compliance,” Dr. Kazani said.
Marstacimab targets the tissue factor pathway inhibitor, a natural anticoagulation protein that prevents the formation of blood clots, and is administered as a once-weekly subcutaneous injection.
The therapy has been granted fast-track and orphan drug status in the United States, in addition to orphan drug status in the European Union for the prevention of hemophilia bleeding episodes.
If approved, the therapy would become the first once-weekly subcutaneous therapy for either hemophilia A or B. Emicizumab, which also is administered subcutaneously, is only approved to prevent or reduce bleeding in hemophilia A.
The latest findings are from an interim analysis of a long-term extension study involving 107 of 116 patients who were in the non-inhibitor cohort in the pivotal BASIS trial. Data from that trial, involving patients aged 12-75 previously showed favorable outcomes in the non-inhibitor cohort receiving marstacimab, and a cohort of patients with inhibitors is ongoing.
Participants entering the extension study were continuing on 150-mg subcutaneous doses of marstacimab, which had been administered in the BASIS study for 12 months after a loading dose of 300 mg.
Of the patients, 89 (83%) were adult and 18 (17%) were adolescents. Overall, they had a mean age of 29 years; 83 (76%) patients had hemophilia A, while 24 (22.4%) had hemophilia B.
Prior to switching to marstacimab treatment, 32 patients had been treated with factor replacement therapy on demand, while 75 received the therapy as routine prophylaxis.
With a mean additional duration of follow-up of 12.5 months in the extension study (range, 1-23.1 months), the overall rate of compliance was very high, at 98.9%.
In the pivotal and extension studies combined, 21% of patients had their marstacimab dose increased from 150 mg to 300 mg weekly, which was an option if patients had 2 or more spontaneous bleeds in a major joint while on the 150-mg dose.
In the hemophilia A and B groups combined, those previously treated with on-demand factor replacement therapy (n = 33) had substantial reductions in estimated ABR for treated bleeds from the baseline of 38.0 prior to initiating marstacimab, to 3.2 after 12 months of the treatment in the trial (P < .001).That reduction was sustained at an ABR of 3.7 after the mean additional 12.5 months in the extension study.
The corresponding estimated ABR rates in the routine prophylaxis group (n = 83) were 7.9 at baseline, 5.1 at the end of the trial, and 2.8 in the extension study analysis interim cutoff.
The authors then further stratified the results based on hemophilia A or B groups: Among patients with hemophilia A (n = 26), the on-demand subgroup had a baseline ABR of 40.6, which dropped substantially to just 3.6 after 12 months on marstacimab in the pivotal trial and was sustained at 5.3 in the extension study.
Similar trends were observed in the hemophilia A group who received routine prophylaxis (n = 65), with an ABR of 9.2 at baseline; 5.3 after the trial, and 3.1 at the extension study interim.
The trends were similar among those with hemophilia B, albeit with lower numbers of patients, consistent with hemophilia B being more rare.
The mean ABR at baseline in the on-demand group of those patients (n = 7) was 28.7, which was reduced to just 1.7 after the 12-months of active marstacimab treatment and sustained at 1.8 by the interim analysis of the extension study.
Of hemophilia B patients previously on routine prophylaxis (n = 18), the mean ABR at baseline was 3.3 and was at 4.7 at the end of the trial. The rate declined to 2.3 in the extension phase.
“We see that these trends of improvement with marstacimab are sustained into the long-term extension study, both in the on-demand group and in the routine prophylaxis groups,” Dr. Kazani said.
Importantly, she noted that marstacimab continued to be well tolerated and safe in the long-term extension study, with no reports of thromboembolic events, which had been a concern with the drug.
Commenting on the study, Margaret Ragni, MD, MPH, a professor of medicine and clinical and translational research in the Division of Hematology/Oncology at the University of Pittsburgh, Pittsburgh, Pennsylvania, noted that marstacimab could represent an important addition in the prevention of bleeds in hemophilia. “[If marstacimab is approved], hemophilia B patients [will] have a drug that can be given subcutaneously weekly to rebalance hemostasis, reducing bleeds, just as hemophilia A patients have with emicizumab.”
Dr. Ragni underscored, however, that caveats include the important point that “neither [marstacimab nor emicizumab] treats bleeds. For that, standard factor replacement therapy or bypass for inhibitors, would be required.”
Also, “a limitation with marstacimab is the lack of weight-dependent dosing. All use one dose [however, in the studies they did use 150 mg or 300 mg]. ... Furthermore, emicizumab can be given weekly, biweekly, or monthly, while that [variation in dosing] is not yet studied with marstacimab.”
And while no thromboembolic events occurred during the trial, Dr. Ragni underscored that “longer-term follow-up is needed.”
The marstacimab long-term extension study is designed to extend to 7 years of follow-up.
The study was sponsored by Pfizer, and Dr. Kazani is an employee of Pfizer. Dr. Ragni reported no disclosures.
“In the long-term extension study treatment with marstacimab demonstrates sustained or improved efficacy for treated and total annualized bleeding rates (ABR) in adults and adolescents with hemophilia A or hemophilia B in this data set of patients without inhibitors,” first author Shamsah Kazani, MD, of Pfizer, Cambridge, Massachusetts, said in presenting the findings at the 2024 annual meeting of the European Hematology Association (EHA) in Madrid.
“The majority of the patients from the pivotal study chose to transition into the long-term extension, and we are finding that these patients are highly compliant with their weekly marstacimab dose, with more than 98% compliance,” Dr. Kazani said.
Marstacimab targets the tissue factor pathway inhibitor, a natural anticoagulation protein that prevents the formation of blood clots, and is administered as a once-weekly subcutaneous injection.
The therapy has been granted fast-track and orphan drug status in the United States, in addition to orphan drug status in the European Union for the prevention of hemophilia bleeding episodes.
If approved, the therapy would become the first once-weekly subcutaneous therapy for either hemophilia A or B. Emicizumab, which also is administered subcutaneously, is only approved to prevent or reduce bleeding in hemophilia A.
The latest findings are from an interim analysis of a long-term extension study involving 107 of 116 patients who were in the non-inhibitor cohort in the pivotal BASIS trial. Data from that trial, involving patients aged 12-75 previously showed favorable outcomes in the non-inhibitor cohort receiving marstacimab, and a cohort of patients with inhibitors is ongoing.
Participants entering the extension study were continuing on 150-mg subcutaneous doses of marstacimab, which had been administered in the BASIS study for 12 months after a loading dose of 300 mg.
Of the patients, 89 (83%) were adult and 18 (17%) were adolescents. Overall, they had a mean age of 29 years; 83 (76%) patients had hemophilia A, while 24 (22.4%) had hemophilia B.
Prior to switching to marstacimab treatment, 32 patients had been treated with factor replacement therapy on demand, while 75 received the therapy as routine prophylaxis.
With a mean additional duration of follow-up of 12.5 months in the extension study (range, 1-23.1 months), the overall rate of compliance was very high, at 98.9%.
In the pivotal and extension studies combined, 21% of patients had their marstacimab dose increased from 150 mg to 300 mg weekly, which was an option if patients had 2 or more spontaneous bleeds in a major joint while on the 150-mg dose.
In the hemophilia A and B groups combined, those previously treated with on-demand factor replacement therapy (n = 33) had substantial reductions in estimated ABR for treated bleeds from the baseline of 38.0 prior to initiating marstacimab, to 3.2 after 12 months of the treatment in the trial (P < .001).That reduction was sustained at an ABR of 3.7 after the mean additional 12.5 months in the extension study.
The corresponding estimated ABR rates in the routine prophylaxis group (n = 83) were 7.9 at baseline, 5.1 at the end of the trial, and 2.8 in the extension study analysis interim cutoff.
The authors then further stratified the results based on hemophilia A or B groups: Among patients with hemophilia A (n = 26), the on-demand subgroup had a baseline ABR of 40.6, which dropped substantially to just 3.6 after 12 months on marstacimab in the pivotal trial and was sustained at 5.3 in the extension study.
Similar trends were observed in the hemophilia A group who received routine prophylaxis (n = 65), with an ABR of 9.2 at baseline; 5.3 after the trial, and 3.1 at the extension study interim.
The trends were similar among those with hemophilia B, albeit with lower numbers of patients, consistent with hemophilia B being more rare.
The mean ABR at baseline in the on-demand group of those patients (n = 7) was 28.7, which was reduced to just 1.7 after the 12-months of active marstacimab treatment and sustained at 1.8 by the interim analysis of the extension study.
Of hemophilia B patients previously on routine prophylaxis (n = 18), the mean ABR at baseline was 3.3 and was at 4.7 at the end of the trial. The rate declined to 2.3 in the extension phase.
“We see that these trends of improvement with marstacimab are sustained into the long-term extension study, both in the on-demand group and in the routine prophylaxis groups,” Dr. Kazani said.
Importantly, she noted that marstacimab continued to be well tolerated and safe in the long-term extension study, with no reports of thromboembolic events, which had been a concern with the drug.
Commenting on the study, Margaret Ragni, MD, MPH, a professor of medicine and clinical and translational research in the Division of Hematology/Oncology at the University of Pittsburgh, Pittsburgh, Pennsylvania, noted that marstacimab could represent an important addition in the prevention of bleeds in hemophilia. “[If marstacimab is approved], hemophilia B patients [will] have a drug that can be given subcutaneously weekly to rebalance hemostasis, reducing bleeds, just as hemophilia A patients have with emicizumab.”
Dr. Ragni underscored, however, that caveats include the important point that “neither [marstacimab nor emicizumab] treats bleeds. For that, standard factor replacement therapy or bypass for inhibitors, would be required.”
Also, “a limitation with marstacimab is the lack of weight-dependent dosing. All use one dose [however, in the studies they did use 150 mg or 300 mg]. ... Furthermore, emicizumab can be given weekly, biweekly, or monthly, while that [variation in dosing] is not yet studied with marstacimab.”
And while no thromboembolic events occurred during the trial, Dr. Ragni underscored that “longer-term follow-up is needed.”
The marstacimab long-term extension study is designed to extend to 7 years of follow-up.
The study was sponsored by Pfizer, and Dr. Kazani is an employee of Pfizer. Dr. Ragni reported no disclosures.
“In the long-term extension study treatment with marstacimab demonstrates sustained or improved efficacy for treated and total annualized bleeding rates (ABR) in adults and adolescents with hemophilia A or hemophilia B in this data set of patients without inhibitors,” first author Shamsah Kazani, MD, of Pfizer, Cambridge, Massachusetts, said in presenting the findings at the 2024 annual meeting of the European Hematology Association (EHA) in Madrid.
“The majority of the patients from the pivotal study chose to transition into the long-term extension, and we are finding that these patients are highly compliant with their weekly marstacimab dose, with more than 98% compliance,” Dr. Kazani said.
Marstacimab targets the tissue factor pathway inhibitor, a natural anticoagulation protein that prevents the formation of blood clots, and is administered as a once-weekly subcutaneous injection.
The therapy has been granted fast-track and orphan drug status in the United States, in addition to orphan drug status in the European Union for the prevention of hemophilia bleeding episodes.
If approved, the therapy would become the first once-weekly subcutaneous therapy for either hemophilia A or B. Emicizumab, which also is administered subcutaneously, is only approved to prevent or reduce bleeding in hemophilia A.
The latest findings are from an interim analysis of a long-term extension study involving 107 of 116 patients who were in the non-inhibitor cohort in the pivotal BASIS trial. Data from that trial, involving patients aged 12-75 previously showed favorable outcomes in the non-inhibitor cohort receiving marstacimab, and a cohort of patients with inhibitors is ongoing.
Participants entering the extension study were continuing on 150-mg subcutaneous doses of marstacimab, which had been administered in the BASIS study for 12 months after a loading dose of 300 mg.
Of the patients, 89 (83%) were adult and 18 (17%) were adolescents. Overall, they had a mean age of 29 years; 83 (76%) patients had hemophilia A, while 24 (22.4%) had hemophilia B.
Prior to switching to marstacimab treatment, 32 patients had been treated with factor replacement therapy on demand, while 75 received the therapy as routine prophylaxis.
With a mean additional duration of follow-up of 12.5 months in the extension study (range, 1-23.1 months), the overall rate of compliance was very high, at 98.9%.
In the pivotal and extension studies combined, 21% of patients had their marstacimab dose increased from 150 mg to 300 mg weekly, which was an option if patients had 2 or more spontaneous bleeds in a major joint while on the 150-mg dose.
In the hemophilia A and B groups combined, those previously treated with on-demand factor replacement therapy (n = 33) had substantial reductions in estimated ABR for treated bleeds from the baseline of 38.0 prior to initiating marstacimab, to 3.2 after 12 months of the treatment in the trial (P < .001).That reduction was sustained at an ABR of 3.7 after the mean additional 12.5 months in the extension study.
The corresponding estimated ABR rates in the routine prophylaxis group (n = 83) were 7.9 at baseline, 5.1 at the end of the trial, and 2.8 in the extension study analysis interim cutoff.
The authors then further stratified the results based on hemophilia A or B groups: Among patients with hemophilia A (n = 26), the on-demand subgroup had a baseline ABR of 40.6, which dropped substantially to just 3.6 after 12 months on marstacimab in the pivotal trial and was sustained at 5.3 in the extension study.
Similar trends were observed in the hemophilia A group who received routine prophylaxis (n = 65), with an ABR of 9.2 at baseline; 5.3 after the trial, and 3.1 at the extension study interim.
The trends were similar among those with hemophilia B, albeit with lower numbers of patients, consistent with hemophilia B being more rare.
The mean ABR at baseline in the on-demand group of those patients (n = 7) was 28.7, which was reduced to just 1.7 after the 12-months of active marstacimab treatment and sustained at 1.8 by the interim analysis of the extension study.
Of hemophilia B patients previously on routine prophylaxis (n = 18), the mean ABR at baseline was 3.3 and was at 4.7 at the end of the trial. The rate declined to 2.3 in the extension phase.
“We see that these trends of improvement with marstacimab are sustained into the long-term extension study, both in the on-demand group and in the routine prophylaxis groups,” Dr. Kazani said.
Importantly, she noted that marstacimab continued to be well tolerated and safe in the long-term extension study, with no reports of thromboembolic events, which had been a concern with the drug.
Commenting on the study, Margaret Ragni, MD, MPH, a professor of medicine and clinical and translational research in the Division of Hematology/Oncology at the University of Pittsburgh, Pittsburgh, Pennsylvania, noted that marstacimab could represent an important addition in the prevention of bleeds in hemophilia. “[If marstacimab is approved], hemophilia B patients [will] have a drug that can be given subcutaneously weekly to rebalance hemostasis, reducing bleeds, just as hemophilia A patients have with emicizumab.”
Dr. Ragni underscored, however, that caveats include the important point that “neither [marstacimab nor emicizumab] treats bleeds. For that, standard factor replacement therapy or bypass for inhibitors, would be required.”
Also, “a limitation with marstacimab is the lack of weight-dependent dosing. All use one dose [however, in the studies they did use 150 mg or 300 mg]. ... Furthermore, emicizumab can be given weekly, biweekly, or monthly, while that [variation in dosing] is not yet studied with marstacimab.”
And while no thromboembolic events occurred during the trial, Dr. Ragni underscored that “longer-term follow-up is needed.”
The marstacimab long-term extension study is designed to extend to 7 years of follow-up.
The study was sponsored by Pfizer, and Dr. Kazani is an employee of Pfizer. Dr. Ragni reported no disclosures.
FROM EHA 2024
Neurofilament Light Chain Detects Early Chemotherapy-Related Neurotoxicity
Investigators found Nfl levels increased in cancer patients following a first infusion of the medication paclitaxel and corresponded to neuropathy severity 6-12 months post-treatment, suggesting the blood protein may provide an early CIPN biomarker.
“Nfl after a single cycle could detect axonal degeneration,” said lead investigator Masarra Joda, a researcher and PhD candidate at the University of Sydney in Australia. She added that “quantification of Nfl may provide a clinically useful marker of emerging neurotoxicity in patients vulnerable to CIPN.”
The findings were presented at the Peripheral Nerve Society (PNS) 2024 annual meeting.
Common, Burdensome Side Effect
A common side effect of chemotherapy, CIPN manifests as sensory neuropathy and causes degeneration of the peripheral axons. A protein biomarker of axonal degeneration, Nfl has previously been investigated as a way of identifying patients at risk of CIPN.
The goal of the current study was to identify the potential link between Nfl with neurophysiological markers of axon degeneration in patients receiving the neurotoxin chemotherapy paclitaxel.
The study included 93 cancer patients. All were assessed at the beginning, middle, and end of treatment. CIPN was assessed using blood samples of Nfl and the Total Neuropathy Score (TNS), the Common Terminology Criteria for Adverse Events (CTCAE) neuropathy scale, and patient-reported measures using the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire–Chemotherapy-Induced Peripheral Neuropathy Module (EORTC-CIPN20).
Axonal degeneration was measured with neurophysiological tests including sural nerve compound sensory action potential (CSAP) for the lower limbs, and sensory median nerve CSAP, as well as stimulus threshold testing, for the upper limbs.
Almost all of study participants (97%) were female. The majority (66%) had breast cancer and 30% had gynecological cancer. Most (73%) were receiving a weekly regimen of paclitaxel, and the remainder were treated with taxanes plus platinum once every 3 weeks. By the end of treatment, 82% of the patients had developed CIPN, which was mild in 44% and moderate/severe in 38%.
Nfl levels increased significantly from baseline to after the first dose of chemotherapy (P < .001), “highlighting that nerve damage occurs from the very beginning of treatment,” senior investigator Susanna Park, PhD, told this news organization.
In addition, “patients with higher Nfl levels after a single paclitaxel treatment had greater neuropathy at the end of treatment (higher EORTC scores [P ≤ .026], and higher TNS scores [P ≤ .00]),” added Dr. Park, who is associate professor at the University of Sydney.
“Importantly, we also looked at long-term outcomes beyond the end of chemotherapy, because chronic neuropathy produces a significant burden in cancer survivors,” said Dr. Park.
“Among a total of 44 patients who completed the 6- to 12-month post-treatment follow-up, NfL levels after a single treatment were linked to severity of nerve damage quantified with neurophysiological tests, and greater Nfl levels at mid-treatment were correlated with worse patient and neurologically graded neuropathy at 6-12 months.”
Dr. Park said the results suggest that NfL may provide a biomarker of long-term axon damage and that Nfl assays “may enable clinicians to evaluate the risk of long-term toxicity early during paclitaxel treatment to hopefully provide clinically significant information to guide better treatment titration.”
Currently, she said, CIPN is a prominent cause of dose reduction and early chemotherapy cessation.
“For example, in early breast cancer around 25% of patients experience a dose reduction due to the severity of neuropathy symptoms.” But, she said, “there is no standardized way of identifying which patients are at risk of long-term neuropathy and therefore, may benefit more from dose reduction. In this setting, a biomarker such as Nfl could provide oncologists with more information about the risk of long-term toxicity and take that into account in dose decision-making.”
For some cancers, she added, there are multiple potential therapy options.
“A biomarker such as NfL could assist in determining risk-benefit profile in terms of switching to alternate therapies. However, further studies will be needed to fully define the utility of NfL as a biomarker of paclitaxel neuropathy.”
Promising Research
Commenting on the research for this news organization, Maryam Lustberg, MD, associate professor, director of the Center for Breast Cancer at Smilow Cancer Hospital and Yale Cancer Center, and chief of Breast Medical Oncology at Yale Cancer Center, in New Haven, Connecticut, said the study “builds on a body of work previously reported by others showing that neurofilament light chains as detected in the blood can be associated with early signs of neurotoxic injury.”
She added that the research “is promising, since existing clinical and patient-reported measures tend to under-detect chemotherapy-induced neuropathy until more permanent injury might have occurred.”
Dr. Lustberg, who is immediate past president of the Multinational Association of Supportive Care in Cancer, said future studies are needed before Nfl testing can be implemented in routine practice, but that “early detection will allow earlier initiation of supportive care strategies such as physical therapy and exercise, as well as dose modifications, which may be helpful for preventing permanent damage and improving quality of life.”
The investigators and Dr. Lustberg report no relevant financial relationships.
A version of this article appeared on Medscape.com.
Investigators found Nfl levels increased in cancer patients following a first infusion of the medication paclitaxel and corresponded to neuropathy severity 6-12 months post-treatment, suggesting the blood protein may provide an early CIPN biomarker.
“Nfl after a single cycle could detect axonal degeneration,” said lead investigator Masarra Joda, a researcher and PhD candidate at the University of Sydney in Australia. She added that “quantification of Nfl may provide a clinically useful marker of emerging neurotoxicity in patients vulnerable to CIPN.”
The findings were presented at the Peripheral Nerve Society (PNS) 2024 annual meeting.
Common, Burdensome Side Effect
A common side effect of chemotherapy, CIPN manifests as sensory neuropathy and causes degeneration of the peripheral axons. A protein biomarker of axonal degeneration, Nfl has previously been investigated as a way of identifying patients at risk of CIPN.
The goal of the current study was to identify the potential link between Nfl with neurophysiological markers of axon degeneration in patients receiving the neurotoxin chemotherapy paclitaxel.
The study included 93 cancer patients. All were assessed at the beginning, middle, and end of treatment. CIPN was assessed using blood samples of Nfl and the Total Neuropathy Score (TNS), the Common Terminology Criteria for Adverse Events (CTCAE) neuropathy scale, and patient-reported measures using the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire–Chemotherapy-Induced Peripheral Neuropathy Module (EORTC-CIPN20).
Axonal degeneration was measured with neurophysiological tests including sural nerve compound sensory action potential (CSAP) for the lower limbs, and sensory median nerve CSAP, as well as stimulus threshold testing, for the upper limbs.
Almost all of study participants (97%) were female. The majority (66%) had breast cancer and 30% had gynecological cancer. Most (73%) were receiving a weekly regimen of paclitaxel, and the remainder were treated with taxanes plus platinum once every 3 weeks. By the end of treatment, 82% of the patients had developed CIPN, which was mild in 44% and moderate/severe in 38%.
Nfl levels increased significantly from baseline to after the first dose of chemotherapy (P < .001), “highlighting that nerve damage occurs from the very beginning of treatment,” senior investigator Susanna Park, PhD, told this news organization.
In addition, “patients with higher Nfl levels after a single paclitaxel treatment had greater neuropathy at the end of treatment (higher EORTC scores [P ≤ .026], and higher TNS scores [P ≤ .00]),” added Dr. Park, who is associate professor at the University of Sydney.
“Importantly, we also looked at long-term outcomes beyond the end of chemotherapy, because chronic neuropathy produces a significant burden in cancer survivors,” said Dr. Park.
“Among a total of 44 patients who completed the 6- to 12-month post-treatment follow-up, NfL levels after a single treatment were linked to severity of nerve damage quantified with neurophysiological tests, and greater Nfl levels at mid-treatment were correlated with worse patient and neurologically graded neuropathy at 6-12 months.”
Dr. Park said the results suggest that NfL may provide a biomarker of long-term axon damage and that Nfl assays “may enable clinicians to evaluate the risk of long-term toxicity early during paclitaxel treatment to hopefully provide clinically significant information to guide better treatment titration.”
Currently, she said, CIPN is a prominent cause of dose reduction and early chemotherapy cessation.
“For example, in early breast cancer around 25% of patients experience a dose reduction due to the severity of neuropathy symptoms.” But, she said, “there is no standardized way of identifying which patients are at risk of long-term neuropathy and therefore, may benefit more from dose reduction. In this setting, a biomarker such as Nfl could provide oncologists with more information about the risk of long-term toxicity and take that into account in dose decision-making.”
For some cancers, she added, there are multiple potential therapy options.
“A biomarker such as NfL could assist in determining risk-benefit profile in terms of switching to alternate therapies. However, further studies will be needed to fully define the utility of NfL as a biomarker of paclitaxel neuropathy.”
Promising Research
Commenting on the research for this news organization, Maryam Lustberg, MD, associate professor, director of the Center for Breast Cancer at Smilow Cancer Hospital and Yale Cancer Center, and chief of Breast Medical Oncology at Yale Cancer Center, in New Haven, Connecticut, said the study “builds on a body of work previously reported by others showing that neurofilament light chains as detected in the blood can be associated with early signs of neurotoxic injury.”
She added that the research “is promising, since existing clinical and patient-reported measures tend to under-detect chemotherapy-induced neuropathy until more permanent injury might have occurred.”
Dr. Lustberg, who is immediate past president of the Multinational Association of Supportive Care in Cancer, said future studies are needed before Nfl testing can be implemented in routine practice, but that “early detection will allow earlier initiation of supportive care strategies such as physical therapy and exercise, as well as dose modifications, which may be helpful for preventing permanent damage and improving quality of life.”
The investigators and Dr. Lustberg report no relevant financial relationships.
A version of this article appeared on Medscape.com.
Investigators found Nfl levels increased in cancer patients following a first infusion of the medication paclitaxel and corresponded to neuropathy severity 6-12 months post-treatment, suggesting the blood protein may provide an early CIPN biomarker.
“Nfl after a single cycle could detect axonal degeneration,” said lead investigator Masarra Joda, a researcher and PhD candidate at the University of Sydney in Australia. She added that “quantification of Nfl may provide a clinically useful marker of emerging neurotoxicity in patients vulnerable to CIPN.”
The findings were presented at the Peripheral Nerve Society (PNS) 2024 annual meeting.
Common, Burdensome Side Effect
A common side effect of chemotherapy, CIPN manifests as sensory neuropathy and causes degeneration of the peripheral axons. A protein biomarker of axonal degeneration, Nfl has previously been investigated as a way of identifying patients at risk of CIPN.
The goal of the current study was to identify the potential link between Nfl with neurophysiological markers of axon degeneration in patients receiving the neurotoxin chemotherapy paclitaxel.
The study included 93 cancer patients. All were assessed at the beginning, middle, and end of treatment. CIPN was assessed using blood samples of Nfl and the Total Neuropathy Score (TNS), the Common Terminology Criteria for Adverse Events (CTCAE) neuropathy scale, and patient-reported measures using the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire–Chemotherapy-Induced Peripheral Neuropathy Module (EORTC-CIPN20).
Axonal degeneration was measured with neurophysiological tests including sural nerve compound sensory action potential (CSAP) for the lower limbs, and sensory median nerve CSAP, as well as stimulus threshold testing, for the upper limbs.
Almost all of study participants (97%) were female. The majority (66%) had breast cancer and 30% had gynecological cancer. Most (73%) were receiving a weekly regimen of paclitaxel, and the remainder were treated with taxanes plus platinum once every 3 weeks. By the end of treatment, 82% of the patients had developed CIPN, which was mild in 44% and moderate/severe in 38%.
Nfl levels increased significantly from baseline to after the first dose of chemotherapy (P < .001), “highlighting that nerve damage occurs from the very beginning of treatment,” senior investigator Susanna Park, PhD, told this news organization.
In addition, “patients with higher Nfl levels after a single paclitaxel treatment had greater neuropathy at the end of treatment (higher EORTC scores [P ≤ .026], and higher TNS scores [P ≤ .00]),” added Dr. Park, who is associate professor at the University of Sydney.
“Importantly, we also looked at long-term outcomes beyond the end of chemotherapy, because chronic neuropathy produces a significant burden in cancer survivors,” said Dr. Park.
“Among a total of 44 patients who completed the 6- to 12-month post-treatment follow-up, NfL levels after a single treatment were linked to severity of nerve damage quantified with neurophysiological tests, and greater Nfl levels at mid-treatment were correlated with worse patient and neurologically graded neuropathy at 6-12 months.”
Dr. Park said the results suggest that NfL may provide a biomarker of long-term axon damage and that Nfl assays “may enable clinicians to evaluate the risk of long-term toxicity early during paclitaxel treatment to hopefully provide clinically significant information to guide better treatment titration.”
Currently, she said, CIPN is a prominent cause of dose reduction and early chemotherapy cessation.
“For example, in early breast cancer around 25% of patients experience a dose reduction due to the severity of neuropathy symptoms.” But, she said, “there is no standardized way of identifying which patients are at risk of long-term neuropathy and therefore, may benefit more from dose reduction. In this setting, a biomarker such as Nfl could provide oncologists with more information about the risk of long-term toxicity and take that into account in dose decision-making.”
For some cancers, she added, there are multiple potential therapy options.
“A biomarker such as NfL could assist in determining risk-benefit profile in terms of switching to alternate therapies. However, further studies will be needed to fully define the utility of NfL as a biomarker of paclitaxel neuropathy.”
Promising Research
Commenting on the research for this news organization, Maryam Lustberg, MD, associate professor, director of the Center for Breast Cancer at Smilow Cancer Hospital and Yale Cancer Center, and chief of Breast Medical Oncology at Yale Cancer Center, in New Haven, Connecticut, said the study “builds on a body of work previously reported by others showing that neurofilament light chains as detected in the blood can be associated with early signs of neurotoxic injury.”
She added that the research “is promising, since existing clinical and patient-reported measures tend to under-detect chemotherapy-induced neuropathy until more permanent injury might have occurred.”
Dr. Lustberg, who is immediate past president of the Multinational Association of Supportive Care in Cancer, said future studies are needed before Nfl testing can be implemented in routine practice, but that “early detection will allow earlier initiation of supportive care strategies such as physical therapy and exercise, as well as dose modifications, which may be helpful for preventing permanent damage and improving quality of life.”
The investigators and Dr. Lustberg report no relevant financial relationships.
A version of this article appeared on Medscape.com.
AT PNS 2024
CMS Announces End to Cyberattack Relief Program
The Centers for Medicare & Medicaid Services (CMS) has announced the conclusion of a program that provided billions in early Medicare payments to those affected by the Change Healthcare/UnitedHealth Group cyberattack last winter.
CMS reported that the program advanced more than $2.55 billion in Medicare payments to > 4200 Part A providers, including hospitals, and more than $717.18 million in payments to Part B suppliers such as physicians, nonphysician practitioners, and durable medical equipment suppliers.
According to CMS, the Medicare billing system is now functioning properly, and 96% of the early payments have been recovered. The advances were to represent ≤ 30 days of typical claims payments in a 3-month period of 2023, with full repayment expected within 90 days through “automatic recoupment from Medicare claims” — no extensions allowed.
The agency took a victory lap regarding its response. “In the face of one of the most widespread cyberattacks on the US health care industry, CMS promptly took action to get providers and suppliers access to the funds they needed to continue providing patients with vital care,” CMS Administrator Chiquita Brooks-LaSure said in a statement. “Our efforts helped minimize the disruptive fallout from this incident, and we will remain vigilant to be ready to address future events.”
Ongoing Concerns from Health Care Organizations
Ben Teicher, an American Hospital Association spokesman, said that the organization hopes that CMS will be responsive if there’s more need for action after the advance payment program expires. The organization represents about 5000 hospitals, health care systems, and other providers.
“Our members report that the aftereffects of this event will likely be felt throughout the remainder of the year,” he said. According to Teicher, hospitals remain concerned about their ability to process claims and appeal denials, the safety of reconnecting to cyber services, and access to information needed to bill patients and reconcile payments.
In addition, hospitals are concerned about “financial support to mitigate the considerable costs incurred as a result of the cyberattack,” he said.
Charlene MacDonald, executive vice-president of public affairs at the Federation of American Hospitals, which represents more than 1000 for-profit hospitals, sent a statement to this news organization that said some providers “are still feeling the effects of care denials and delays caused by insurer inaction.
“We appreciate that the Administration acted within its authority to support providers during this unprecedented crisis and blunt these devastating impacts, especially because a vast majority of managed care companies failed to step up to the plate,” she said. “It is now time to shift our focus to holding plans accountable for using tactics to delay and deny needed patient care.”
Cyberattack Impact and Response
The ransom-based cyberattack against Change Healthcare/UnitedHealth Group targeted an electronic data interchange clearing house processing payer reimbursement systems, disrupting cash flows at hospitals and medical practices, and affecting patient access to prescriptions and life-saving therapy.
Change Healthcare — part of the UnitedHealth Group subsidiary Optum — processes half of all medical claims, according to a Department of Justice lawsuit. The American Hospital Association described the cyberattack as “the most significant and consequential incident of its kind” in US history.
By late March, UnitedHealth Group said nearly all medical and pharmacy claims were processing properly, while a deputy secretary of the US Department of Health & Human Services told clinicians that officials were focusing on the last group of clinicians who were facing cash-flow problems.
Still, a senior advisor with CMS told providers at that time that “we have heard from so many providers over the last several weeks who are really struggling to make ends meet right now or who are worried that they will not be able to make payroll in the weeks to come.”
Randy Dotinga is a freelance health/medical reporter and board member of the Association of Health Care Journalists.
A version of this article appeared on Medscape.com.
The Centers for Medicare & Medicaid Services (CMS) has announced the conclusion of a program that provided billions in early Medicare payments to those affected by the Change Healthcare/UnitedHealth Group cyberattack last winter.
CMS reported that the program advanced more than $2.55 billion in Medicare payments to > 4200 Part A providers, including hospitals, and more than $717.18 million in payments to Part B suppliers such as physicians, nonphysician practitioners, and durable medical equipment suppliers.
According to CMS, the Medicare billing system is now functioning properly, and 96% of the early payments have been recovered. The advances were to represent ≤ 30 days of typical claims payments in a 3-month period of 2023, with full repayment expected within 90 days through “automatic recoupment from Medicare claims” — no extensions allowed.
The agency took a victory lap regarding its response. “In the face of one of the most widespread cyberattacks on the US health care industry, CMS promptly took action to get providers and suppliers access to the funds they needed to continue providing patients with vital care,” CMS Administrator Chiquita Brooks-LaSure said in a statement. “Our efforts helped minimize the disruptive fallout from this incident, and we will remain vigilant to be ready to address future events.”
Ongoing Concerns from Health Care Organizations
Ben Teicher, an American Hospital Association spokesman, said that the organization hopes that CMS will be responsive if there’s more need for action after the advance payment program expires. The organization represents about 5000 hospitals, health care systems, and other providers.
“Our members report that the aftereffects of this event will likely be felt throughout the remainder of the year,” he said. According to Teicher, hospitals remain concerned about their ability to process claims and appeal denials, the safety of reconnecting to cyber services, and access to information needed to bill patients and reconcile payments.
In addition, hospitals are concerned about “financial support to mitigate the considerable costs incurred as a result of the cyberattack,” he said.
Charlene MacDonald, executive vice-president of public affairs at the Federation of American Hospitals, which represents more than 1000 for-profit hospitals, sent a statement to this news organization that said some providers “are still feeling the effects of care denials and delays caused by insurer inaction.
“We appreciate that the Administration acted within its authority to support providers during this unprecedented crisis and blunt these devastating impacts, especially because a vast majority of managed care companies failed to step up to the plate,” she said. “It is now time to shift our focus to holding plans accountable for using tactics to delay and deny needed patient care.”
Cyberattack Impact and Response
The ransom-based cyberattack against Change Healthcare/UnitedHealth Group targeted an electronic data interchange clearing house processing payer reimbursement systems, disrupting cash flows at hospitals and medical practices, and affecting patient access to prescriptions and life-saving therapy.
Change Healthcare — part of the UnitedHealth Group subsidiary Optum — processes half of all medical claims, according to a Department of Justice lawsuit. The American Hospital Association described the cyberattack as “the most significant and consequential incident of its kind” in US history.
By late March, UnitedHealth Group said nearly all medical and pharmacy claims were processing properly, while a deputy secretary of the US Department of Health & Human Services told clinicians that officials were focusing on the last group of clinicians who were facing cash-flow problems.
Still, a senior advisor with CMS told providers at that time that “we have heard from so many providers over the last several weeks who are really struggling to make ends meet right now or who are worried that they will not be able to make payroll in the weeks to come.”
Randy Dotinga is a freelance health/medical reporter and board member of the Association of Health Care Journalists.
A version of this article appeared on Medscape.com.
The Centers for Medicare & Medicaid Services (CMS) has announced the conclusion of a program that provided billions in early Medicare payments to those affected by the Change Healthcare/UnitedHealth Group cyberattack last winter.
CMS reported that the program advanced more than $2.55 billion in Medicare payments to > 4200 Part A providers, including hospitals, and more than $717.18 million in payments to Part B suppliers such as physicians, nonphysician practitioners, and durable medical equipment suppliers.
According to CMS, the Medicare billing system is now functioning properly, and 96% of the early payments have been recovered. The advances were to represent ≤ 30 days of typical claims payments in a 3-month period of 2023, with full repayment expected within 90 days through “automatic recoupment from Medicare claims” — no extensions allowed.
The agency took a victory lap regarding its response. “In the face of one of the most widespread cyberattacks on the US health care industry, CMS promptly took action to get providers and suppliers access to the funds they needed to continue providing patients with vital care,” CMS Administrator Chiquita Brooks-LaSure said in a statement. “Our efforts helped minimize the disruptive fallout from this incident, and we will remain vigilant to be ready to address future events.”
Ongoing Concerns from Health Care Organizations
Ben Teicher, an American Hospital Association spokesman, said that the organization hopes that CMS will be responsive if there’s more need for action after the advance payment program expires. The organization represents about 5000 hospitals, health care systems, and other providers.
“Our members report that the aftereffects of this event will likely be felt throughout the remainder of the year,” he said. According to Teicher, hospitals remain concerned about their ability to process claims and appeal denials, the safety of reconnecting to cyber services, and access to information needed to bill patients and reconcile payments.
In addition, hospitals are concerned about “financial support to mitigate the considerable costs incurred as a result of the cyberattack,” he said.
Charlene MacDonald, executive vice-president of public affairs at the Federation of American Hospitals, which represents more than 1000 for-profit hospitals, sent a statement to this news organization that said some providers “are still feeling the effects of care denials and delays caused by insurer inaction.
“We appreciate that the Administration acted within its authority to support providers during this unprecedented crisis and blunt these devastating impacts, especially because a vast majority of managed care companies failed to step up to the plate,” she said. “It is now time to shift our focus to holding plans accountable for using tactics to delay and deny needed patient care.”
Cyberattack Impact and Response
The ransom-based cyberattack against Change Healthcare/UnitedHealth Group targeted an electronic data interchange clearing house processing payer reimbursement systems, disrupting cash flows at hospitals and medical practices, and affecting patient access to prescriptions and life-saving therapy.
Change Healthcare — part of the UnitedHealth Group subsidiary Optum — processes half of all medical claims, according to a Department of Justice lawsuit. The American Hospital Association described the cyberattack as “the most significant and consequential incident of its kind” in US history.
By late March, UnitedHealth Group said nearly all medical and pharmacy claims were processing properly, while a deputy secretary of the US Department of Health & Human Services told clinicians that officials were focusing on the last group of clinicians who were facing cash-flow problems.
Still, a senior advisor with CMS told providers at that time that “we have heard from so many providers over the last several weeks who are really struggling to make ends meet right now or who are worried that they will not be able to make payroll in the weeks to come.”
Randy Dotinga is a freelance health/medical reporter and board member of the Association of Health Care Journalists.
A version of this article appeared on Medscape.com.
Is This Journal Legit? Predatory Publishers
This transcript has been edited for clarity.
Andrew N. Wilner, MD: My guest today is Dr. Jose Merino, editor in chief of the Neurology family of journals and professor of neurology and co-vice chair of education at Georgetown University in Washington, DC.
Our program today is a follow-up of Dr. Merino’s presentation at the recent American Academy of Neurology meeting in Denver, Colorado. Along with two other panelists, Dr. Merino discussed the role of open-access publication and the dangers of predatory journals.
Jose G. Merino, MD, MPhil: Thank you for having me here. It’s a pleasure.
Open Access Defined
Dr. Wilner: I remember when publication in neurology was pretty straightforward. It was either the green journal or the blue journal, but things have certainly changed. I think one topic that is not clear to everyone is this concept of open access. Could you define that for us?
Dr. Merino: Sure. Open access is a mode of publication that fosters more open or accessible science. The idea of open access is that it combines two main elements. One is that the papers that are published become immediately available to anybody with an internet connection anywhere in the world without any restrictions.
The second important element from open access, which makes it different from other models we can talk about, is the fact that the authors retain the copyright of their work, but they give the journal and readers a license to use, reproduce, and modify the content.
This is different, for example, from instances where we have funder mandates. For example, NIH papers have to become available 6 months after publication, so they’re available to everybody but not immediately.
Dr. Wilner: I remember that when a journal article was published, say, in Neurology, if you didn’t have a subscription to Neurology, you went to the library that hopefully had a subscription.
If they didn’t have it, you would write to the author and say, “Hey, I heard you have this great paper because the abstract was out there. Could you send me a reprint?” Has that whole universe evaporated?
Dr. Merino: It depends on how the paper is published. For example, in Neurology, some of the research we publish is open access. Basically, if you have an internet connection, you can access the paper.
That’s the case for papers published in our wholly open-access journals in the Neurology family like Neurology Neuroimmunology & Neuroinflammation, Neurology Genetics, or Neurology Education.
For other papers that are published in Neurology, not under open access, there is a paywall. For some of them, the paywall comes down after a few months based on funder mandates and so on. As I was mentioning, the NIH-funded papers are available 6 months later.
In the first 6 months, you may have to go to your library, and if your library has a subscription, you can download it directly. [This is also true for] those that always stay behind the paywall, where you have to have a subscription or your library has to have a subscription.
Is Pay to Publish a Red Flag?
Dr. Wilner: I’m a professional writer. With any luck, when I write something, I get paid to write it. There’s been a long tradition in academic medicine that when you submit an article to, say, Neurology, you don’t get paid as an author for the publication. Your reward is the honor of it being published.
Neurology supports itself in various ways, including advertising and so on. That’s been the contract: free publication for work that merits it, and the journal survives on its own.
With open access, one of the things that’s happened is that — and I’ve published open access myself — is that I get a notification that I need to pay to have my article that I’ve slaved over published. Explain that, please.
Dr. Merino: This is the issue with open access. As I mentioned, the paper gets published. You’re giving the journal a license to publish it. You’re retaining the copyright of your work. That means that the journal cannot make money or support itself by just publishing open access because they belong to you.
Typically, open-access journals are not in print and don’t have much in terms of advertising. The contract is you’re giving me a license to publish it, but it’s your journal, so you’re paying a fee for the journal expenses to basically produce your paper. That’s what’s happening with open access.
That’s been recognized with many funders, for example, with NIH funding or many of the European funders, they’re including open-access fees as part of their funding for research. Now, of course, this doesn’t help if you’re not a funded researcher or if you’re a fellow who’s doing work and so on.
Typically, most journals will have waived fees or lower fees for these situations. The reason for the open-access fee is the fact that you’re retaining the copyright. You’re not giving it to the journal who can then use it to generate its revenue for supporting itself, the editorial staff, and so on.
Dr. Wilner: This idea of charging for publication has created a satellite business of what are called predatory journals. How does one know if the open-access journal that I’m submitting to is really just in the business of wanting my $300 or my $900 to get published? How do I know if that’s a reasonable place to publish?
Predatory Journals
Dr. Merino: That’s a big challenge that has come with this whole idea of open access and the fact that now, many journals are online only, so you’re no longer seeing a physical copy. That has given rise to the predatory journals.
The predatory journal, by definition, is a journal that claims to be open access. They’ll take your paper and publish it, but they don’t provide all the other services that you would typically expect from the fact that you’re paying an open-access fee. This includes getting appropriate peer review, production of the manuscript, and long-term curation and storage of the manuscript.
Many predatory journals will take your open-access fee, accept any paper that you submit, regardless of the quality, because they’re charging the fees for that. They don’t send it to real peer review, and then in a few months, the journal disappears so there’s no way for anybody to actually find your paper anymore.
There are certain checklists. Dr. David Moher at the University of Toronto has produced some work trying to help us identify predatory journals.
One thing I typically suggest to people who ask me this question is: Have you ever heard of this journal before? Does the journal have a track record? How far back does the story of the journal go? Is it supported by a publisher that you know? Do you know anybody who has published there? Is it something you can easily access?
If in doubt, always ask your friendly medical librarian. There used to be lists that were kept in terms of predatory journals that were being constantly updated, but those had to be shut down. As far as I understand, there were legal issues in terms of how things got on that list.
I think that overall, if you’ve heard of it, if it’s relevant, if it’s known in your field, and if your librarian knows it, it’s probably a good legitimate open-access journal. There are many very good legitimate open-access journals.
I mentioned the two that we have in our family, but all the other major journals have their own open-access journal within their family. There are some, like BMC or PLOS, that are completely open-access and legitimate journals.
Impact Factor
Dr. Wilner: What about impact factor? Many journals boast about their impact factor. I’m not sure how to interpret that number.
Dr. Merino: Impact factor is very interesting. The impact factor was developed by medical librarians to try to identify the journals they should be subscribing to. It’s a measure of the average citations to an average paper in the journal.
It doesn’t tell you about specific papers. It tells you, on average, how many of the papers in this journal get cited so many times. It’s calculated by the number of articles that were cited divided by the number of articles that were published. Journals that publish many papers, like Neurology, have a hard time bringing up their impact factor beyond a certain level.
Similarly, very small journals with one or two very highly cited papers have a very high impact factor. It’s being used as a measure, perhaps inappropriately, of how good or how reputable a journal is. We all say we don’t care about journal impact factors, but we all know our journal impact factor and we used to know it to three decimals. Now, they changed the system, and there’s only one decimal point, which makes more sense.
This is more important, for example, for authors when deciding where to submit papers. I know that in some countries, particularly in Europe, the impact factor of the journal where you publish has an impact on your promotion decisions.
I would say what’s even more important than the impact factor, is to say, “Well, is this the journal that fits the scope of my paper? Is this the journal that reaches the audience that I want to reach when I write my paper?”
There are some papers, for example, that are very influential. The impact factor just captures citations. There are some papers that are very influential that may not get cited very often. There may be papers that change clinical practice.
If you read a paper that tells you that you should be changing how you treat your patients with myasthenia based on this paper, that may not get cited. It’s a very clinically focused paper, but it’s probably more impactful than one that gets cited very much in some respect, or they make it to public policy decisions, and so on.
I think it’s important to look more at the audience and the journal scope when you submit your papers.
Dr. Wilner: One other technical question. The journals also say they’re indexed in PubMed or Google Scholar. If I want to publish my paper and I want it indexed where the right people are going to find it, where does it need to be indexed?
Dr. Merino: I grew up using Index Medicus, MedlinePlus, and the Library of Science. I still do. If I need to find something, I go to PubMed. Ideally, papers are listed in MedlinePlus or can be found in PubMed. They’re not the same thing, but you can find them through them.
That would be an important thing. Nowadays, a lot more people are using Google Scholar or Google just to identify papers. It may be a little bit less relevant, but it’s still a measure of the quality of the journal before they get indexed in some of these. For example, if you get listed in MedlinePlus, it has gone through certain quality checks by the index itself to see whether they would accept the journal or not. That’s something you want to check.
Typically, most of the large journals or the journals you and I know about are listed in more than one place, right? They’re listed in Scopus and Web of Science. They’re listed in MedlinePlus and so on. Again, if you’re submitting your paper, go somewhere where you know the journal and you’ve heard about it.
Dr. Wilner: I’m not going to ask you about artificial intelligence. We can do that another time. I want to ask something closer to me, which is this question of publish or perish.
There seems to be, in academics, more emphasis on the number of papers that one has published rather than their quality. How does a younger academician or one who really needs to publish cope with that?
Dr. Merino: Many people are writing up research that may not be relevant or that may not be high quality just because you need to have a long list of papers to get promoted, for example, if you’re an academician.
Doug Altman, who was a very influential person in the field quality of not only medical statistics but also medical publishing, had the idea that we need less research, but we need better research.
We often receive papers where you say, well, what’s the rationale behind the question in this paper? It’s like they had a large amount of data and were trying to squeeze as much as they could out of that. I think, as a young academician, the important thing to think about is whether it is an important question that matters to you and to the field, from whatever perspective, whether it’s going to advance research, advance clinical care, or have public policy implications.
Is this one where the answer will be important no matter what the answer is? If you’re thinking of that, your work will be well recognized, people will know you, and you’ll get invited to collaborate. I think that’s the most important thing rather than just churning out a large number of papers.
The productivity will come from the fact that you start by saying, let me ask something that’s really meaningful to me and to the field, with a good question and using strong research methodology.
Dr. Wilner: Thanks for that, Dr. Merino. I think that’s very valuable for all of us. This has been a great discussion. Do you have any final comments before we wrap up?
Dr. Merino: I want to encourage people to continue reading medical journals all the time and submitting to us, again, good research and important questions with robust methodology. That’s what we’re looking for in Neurology and most serious medical journals.
Dr. Wilner is an associate professor of neurology at the University of Tennessee Health Science Center, Memphis. Dr. Merino is a professor in the department of neurology at Georgetown University Medical Center, Washington, DC. Dr. Wilner reported conflicts of interest with Accordant Health Services and Lulu Publishing. Dr. Merino reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Andrew N. Wilner, MD: My guest today is Dr. Jose Merino, editor in chief of the Neurology family of journals and professor of neurology and co-vice chair of education at Georgetown University in Washington, DC.
Our program today is a follow-up of Dr. Merino’s presentation at the recent American Academy of Neurology meeting in Denver, Colorado. Along with two other panelists, Dr. Merino discussed the role of open-access publication and the dangers of predatory journals.
Jose G. Merino, MD, MPhil: Thank you for having me here. It’s a pleasure.
Open Access Defined
Dr. Wilner: I remember when publication in neurology was pretty straightforward. It was either the green journal or the blue journal, but things have certainly changed. I think one topic that is not clear to everyone is this concept of open access. Could you define that for us?
Dr. Merino: Sure. Open access is a mode of publication that fosters more open or accessible science. The idea of open access is that it combines two main elements. One is that the papers that are published become immediately available to anybody with an internet connection anywhere in the world without any restrictions.
The second important element from open access, which makes it different from other models we can talk about, is the fact that the authors retain the copyright of their work, but they give the journal and readers a license to use, reproduce, and modify the content.
This is different, for example, from instances where we have funder mandates. For example, NIH papers have to become available 6 months after publication, so they’re available to everybody but not immediately.
Dr. Wilner: I remember that when a journal article was published, say, in Neurology, if you didn’t have a subscription to Neurology, you went to the library that hopefully had a subscription.
If they didn’t have it, you would write to the author and say, “Hey, I heard you have this great paper because the abstract was out there. Could you send me a reprint?” Has that whole universe evaporated?
Dr. Merino: It depends on how the paper is published. For example, in Neurology, some of the research we publish is open access. Basically, if you have an internet connection, you can access the paper.
That’s the case for papers published in our wholly open-access journals in the Neurology family like Neurology Neuroimmunology & Neuroinflammation, Neurology Genetics, or Neurology Education.
For other papers that are published in Neurology, not under open access, there is a paywall. For some of them, the paywall comes down after a few months based on funder mandates and so on. As I was mentioning, the NIH-funded papers are available 6 months later.
In the first 6 months, you may have to go to your library, and if your library has a subscription, you can download it directly. [This is also true for] those that always stay behind the paywall, where you have to have a subscription or your library has to have a subscription.
Is Pay to Publish a Red Flag?
Dr. Wilner: I’m a professional writer. With any luck, when I write something, I get paid to write it. There’s been a long tradition in academic medicine that when you submit an article to, say, Neurology, you don’t get paid as an author for the publication. Your reward is the honor of it being published.
Neurology supports itself in various ways, including advertising and so on. That’s been the contract: free publication for work that merits it, and the journal survives on its own.
With open access, one of the things that’s happened is that — and I’ve published open access myself — is that I get a notification that I need to pay to have my article that I’ve slaved over published. Explain that, please.
Dr. Merino: This is the issue with open access. As I mentioned, the paper gets published. You’re giving the journal a license to publish it. You’re retaining the copyright of your work. That means that the journal cannot make money or support itself by just publishing open access because they belong to you.
Typically, open-access journals are not in print and don’t have much in terms of advertising. The contract is you’re giving me a license to publish it, but it’s your journal, so you’re paying a fee for the journal expenses to basically produce your paper. That’s what’s happening with open access.
That’s been recognized with many funders, for example, with NIH funding or many of the European funders, they’re including open-access fees as part of their funding for research. Now, of course, this doesn’t help if you’re not a funded researcher or if you’re a fellow who’s doing work and so on.
Typically, most journals will have waived fees or lower fees for these situations. The reason for the open-access fee is the fact that you’re retaining the copyright. You’re not giving it to the journal who can then use it to generate its revenue for supporting itself, the editorial staff, and so on.
Dr. Wilner: This idea of charging for publication has created a satellite business of what are called predatory journals. How does one know if the open-access journal that I’m submitting to is really just in the business of wanting my $300 or my $900 to get published? How do I know if that’s a reasonable place to publish?
Predatory Journals
Dr. Merino: That’s a big challenge that has come with this whole idea of open access and the fact that now, many journals are online only, so you’re no longer seeing a physical copy. That has given rise to the predatory journals.
The predatory journal, by definition, is a journal that claims to be open access. They’ll take your paper and publish it, but they don’t provide all the other services that you would typically expect from the fact that you’re paying an open-access fee. This includes getting appropriate peer review, production of the manuscript, and long-term curation and storage of the manuscript.
Many predatory journals will take your open-access fee, accept any paper that you submit, regardless of the quality, because they’re charging the fees for that. They don’t send it to real peer review, and then in a few months, the journal disappears so there’s no way for anybody to actually find your paper anymore.
There are certain checklists. Dr. David Moher at the University of Toronto has produced some work trying to help us identify predatory journals.
One thing I typically suggest to people who ask me this question is: Have you ever heard of this journal before? Does the journal have a track record? How far back does the story of the journal go? Is it supported by a publisher that you know? Do you know anybody who has published there? Is it something you can easily access?
If in doubt, always ask your friendly medical librarian. There used to be lists that were kept in terms of predatory journals that were being constantly updated, but those had to be shut down. As far as I understand, there were legal issues in terms of how things got on that list.
I think that overall, if you’ve heard of it, if it’s relevant, if it’s known in your field, and if your librarian knows it, it’s probably a good legitimate open-access journal. There are many very good legitimate open-access journals.
I mentioned the two that we have in our family, but all the other major journals have their own open-access journal within their family. There are some, like BMC or PLOS, that are completely open-access and legitimate journals.
Impact Factor
Dr. Wilner: What about impact factor? Many journals boast about their impact factor. I’m not sure how to interpret that number.
Dr. Merino: Impact factor is very interesting. The impact factor was developed by medical librarians to try to identify the journals they should be subscribing to. It’s a measure of the average citations to an average paper in the journal.
It doesn’t tell you about specific papers. It tells you, on average, how many of the papers in this journal get cited so many times. It’s calculated by the number of articles that were cited divided by the number of articles that were published. Journals that publish many papers, like Neurology, have a hard time bringing up their impact factor beyond a certain level.
Similarly, very small journals with one or two very highly cited papers have a very high impact factor. It’s being used as a measure, perhaps inappropriately, of how good or how reputable a journal is. We all say we don’t care about journal impact factors, but we all know our journal impact factor and we used to know it to three decimals. Now, they changed the system, and there’s only one decimal point, which makes more sense.
This is more important, for example, for authors when deciding where to submit papers. I know that in some countries, particularly in Europe, the impact factor of the journal where you publish has an impact on your promotion decisions.
I would say what’s even more important than the impact factor, is to say, “Well, is this the journal that fits the scope of my paper? Is this the journal that reaches the audience that I want to reach when I write my paper?”
There are some papers, for example, that are very influential. The impact factor just captures citations. There are some papers that are very influential that may not get cited very often. There may be papers that change clinical practice.
If you read a paper that tells you that you should be changing how you treat your patients with myasthenia based on this paper, that may not get cited. It’s a very clinically focused paper, but it’s probably more impactful than one that gets cited very much in some respect, or they make it to public policy decisions, and so on.
I think it’s important to look more at the audience and the journal scope when you submit your papers.
Dr. Wilner: One other technical question. The journals also say they’re indexed in PubMed or Google Scholar. If I want to publish my paper and I want it indexed where the right people are going to find it, where does it need to be indexed?
Dr. Merino: I grew up using Index Medicus, MedlinePlus, and the Library of Science. I still do. If I need to find something, I go to PubMed. Ideally, papers are listed in MedlinePlus or can be found in PubMed. They’re not the same thing, but you can find them through them.
That would be an important thing. Nowadays, a lot more people are using Google Scholar or Google just to identify papers. It may be a little bit less relevant, but it’s still a measure of the quality of the journal before they get indexed in some of these. For example, if you get listed in MedlinePlus, it has gone through certain quality checks by the index itself to see whether they would accept the journal or not. That’s something you want to check.
Typically, most of the large journals or the journals you and I know about are listed in more than one place, right? They’re listed in Scopus and Web of Science. They’re listed in MedlinePlus and so on. Again, if you’re submitting your paper, go somewhere where you know the journal and you’ve heard about it.
Dr. Wilner: I’m not going to ask you about artificial intelligence. We can do that another time. I want to ask something closer to me, which is this question of publish or perish.
There seems to be, in academics, more emphasis on the number of papers that one has published rather than their quality. How does a younger academician or one who really needs to publish cope with that?
Dr. Merino: Many people are writing up research that may not be relevant or that may not be high quality just because you need to have a long list of papers to get promoted, for example, if you’re an academician.
Doug Altman, who was a very influential person in the field quality of not only medical statistics but also medical publishing, had the idea that we need less research, but we need better research.
We often receive papers where you say, well, what’s the rationale behind the question in this paper? It’s like they had a large amount of data and were trying to squeeze as much as they could out of that. I think, as a young academician, the important thing to think about is whether it is an important question that matters to you and to the field, from whatever perspective, whether it’s going to advance research, advance clinical care, or have public policy implications.
Is this one where the answer will be important no matter what the answer is? If you’re thinking of that, your work will be well recognized, people will know you, and you’ll get invited to collaborate. I think that’s the most important thing rather than just churning out a large number of papers.
The productivity will come from the fact that you start by saying, let me ask something that’s really meaningful to me and to the field, with a good question and using strong research methodology.
Dr. Wilner: Thanks for that, Dr. Merino. I think that’s very valuable for all of us. This has been a great discussion. Do you have any final comments before we wrap up?
Dr. Merino: I want to encourage people to continue reading medical journals all the time and submitting to us, again, good research and important questions with robust methodology. That’s what we’re looking for in Neurology and most serious medical journals.
Dr. Wilner is an associate professor of neurology at the University of Tennessee Health Science Center, Memphis. Dr. Merino is a professor in the department of neurology at Georgetown University Medical Center, Washington, DC. Dr. Wilner reported conflicts of interest with Accordant Health Services and Lulu Publishing. Dr. Merino reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Andrew N. Wilner, MD: My guest today is Dr. Jose Merino, editor in chief of the Neurology family of journals and professor of neurology and co-vice chair of education at Georgetown University in Washington, DC.
Our program today is a follow-up of Dr. Merino’s presentation at the recent American Academy of Neurology meeting in Denver, Colorado. Along with two other panelists, Dr. Merino discussed the role of open-access publication and the dangers of predatory journals.
Jose G. Merino, MD, MPhil: Thank you for having me here. It’s a pleasure.
Open Access Defined
Dr. Wilner: I remember when publication in neurology was pretty straightforward. It was either the green journal or the blue journal, but things have certainly changed. I think one topic that is not clear to everyone is this concept of open access. Could you define that for us?
Dr. Merino: Sure. Open access is a mode of publication that fosters more open or accessible science. The idea of open access is that it combines two main elements. One is that the papers that are published become immediately available to anybody with an internet connection anywhere in the world without any restrictions.
The second important element from open access, which makes it different from other models we can talk about, is the fact that the authors retain the copyright of their work, but they give the journal and readers a license to use, reproduce, and modify the content.
This is different, for example, from instances where we have funder mandates. For example, NIH papers have to become available 6 months after publication, so they’re available to everybody but not immediately.
Dr. Wilner: I remember that when a journal article was published, say, in Neurology, if you didn’t have a subscription to Neurology, you went to the library that hopefully had a subscription.
If they didn’t have it, you would write to the author and say, “Hey, I heard you have this great paper because the abstract was out there. Could you send me a reprint?” Has that whole universe evaporated?
Dr. Merino: It depends on how the paper is published. For example, in Neurology, some of the research we publish is open access. Basically, if you have an internet connection, you can access the paper.
That’s the case for papers published in our wholly open-access journals in the Neurology family like Neurology Neuroimmunology & Neuroinflammation, Neurology Genetics, or Neurology Education.
For other papers that are published in Neurology, not under open access, there is a paywall. For some of them, the paywall comes down after a few months based on funder mandates and so on. As I was mentioning, the NIH-funded papers are available 6 months later.
In the first 6 months, you may have to go to your library, and if your library has a subscription, you can download it directly. [This is also true for] those that always stay behind the paywall, where you have to have a subscription or your library has to have a subscription.
Is Pay to Publish a Red Flag?
Dr. Wilner: I’m a professional writer. With any luck, when I write something, I get paid to write it. There’s been a long tradition in academic medicine that when you submit an article to, say, Neurology, you don’t get paid as an author for the publication. Your reward is the honor of it being published.
Neurology supports itself in various ways, including advertising and so on. That’s been the contract: free publication for work that merits it, and the journal survives on its own.
With open access, one of the things that’s happened is that — and I’ve published open access myself — is that I get a notification that I need to pay to have my article that I’ve slaved over published. Explain that, please.
Dr. Merino: This is the issue with open access. As I mentioned, the paper gets published. You’re giving the journal a license to publish it. You’re retaining the copyright of your work. That means that the journal cannot make money or support itself by just publishing open access because they belong to you.
Typically, open-access journals are not in print and don’t have much in terms of advertising. The contract is you’re giving me a license to publish it, but it’s your journal, so you’re paying a fee for the journal expenses to basically produce your paper. That’s what’s happening with open access.
That’s been recognized with many funders, for example, with NIH funding or many of the European funders, they’re including open-access fees as part of their funding for research. Now, of course, this doesn’t help if you’re not a funded researcher or if you’re a fellow who’s doing work and so on.
Typically, most journals will have waived fees or lower fees for these situations. The reason for the open-access fee is the fact that you’re retaining the copyright. You’re not giving it to the journal who can then use it to generate its revenue for supporting itself, the editorial staff, and so on.
Dr. Wilner: This idea of charging for publication has created a satellite business of what are called predatory journals. How does one know if the open-access journal that I’m submitting to is really just in the business of wanting my $300 or my $900 to get published? How do I know if that’s a reasonable place to publish?
Predatory Journals
Dr. Merino: That’s a big challenge that has come with this whole idea of open access and the fact that now, many journals are online only, so you’re no longer seeing a physical copy. That has given rise to the predatory journals.
The predatory journal, by definition, is a journal that claims to be open access. They’ll take your paper and publish it, but they don’t provide all the other services that you would typically expect from the fact that you’re paying an open-access fee. This includes getting appropriate peer review, production of the manuscript, and long-term curation and storage of the manuscript.
Many predatory journals will take your open-access fee, accept any paper that you submit, regardless of the quality, because they’re charging the fees for that. They don’t send it to real peer review, and then in a few months, the journal disappears so there’s no way for anybody to actually find your paper anymore.
There are certain checklists. Dr. David Moher at the University of Toronto has produced some work trying to help us identify predatory journals.
One thing I typically suggest to people who ask me this question is: Have you ever heard of this journal before? Does the journal have a track record? How far back does the story of the journal go? Is it supported by a publisher that you know? Do you know anybody who has published there? Is it something you can easily access?
If in doubt, always ask your friendly medical librarian. There used to be lists that were kept in terms of predatory journals that were being constantly updated, but those had to be shut down. As far as I understand, there were legal issues in terms of how things got on that list.
I think that overall, if you’ve heard of it, if it’s relevant, if it’s known in your field, and if your librarian knows it, it’s probably a good legitimate open-access journal. There are many very good legitimate open-access journals.
I mentioned the two that we have in our family, but all the other major journals have their own open-access journal within their family. There are some, like BMC or PLOS, that are completely open-access and legitimate journals.
Impact Factor
Dr. Wilner: What about impact factor? Many journals boast about their impact factor. I’m not sure how to interpret that number.
Dr. Merino: Impact factor is very interesting. The impact factor was developed by medical librarians to try to identify the journals they should be subscribing to. It’s a measure of the average citations to an average paper in the journal.
It doesn’t tell you about specific papers. It tells you, on average, how many of the papers in this journal get cited so many times. It’s calculated by the number of articles that were cited divided by the number of articles that were published. Journals that publish many papers, like Neurology, have a hard time bringing up their impact factor beyond a certain level.
Similarly, very small journals with one or two very highly cited papers have a very high impact factor. It’s being used as a measure, perhaps inappropriately, of how good or how reputable a journal is. We all say we don’t care about journal impact factors, but we all know our journal impact factor and we used to know it to three decimals. Now, they changed the system, and there’s only one decimal point, which makes more sense.
This is more important, for example, for authors when deciding where to submit papers. I know that in some countries, particularly in Europe, the impact factor of the journal where you publish has an impact on your promotion decisions.
I would say what’s even more important than the impact factor, is to say, “Well, is this the journal that fits the scope of my paper? Is this the journal that reaches the audience that I want to reach when I write my paper?”
There are some papers, for example, that are very influential. The impact factor just captures citations. There are some papers that are very influential that may not get cited very often. There may be papers that change clinical practice.
If you read a paper that tells you that you should be changing how you treat your patients with myasthenia based on this paper, that may not get cited. It’s a very clinically focused paper, but it’s probably more impactful than one that gets cited very much in some respect, or they make it to public policy decisions, and so on.
I think it’s important to look more at the audience and the journal scope when you submit your papers.
Dr. Wilner: One other technical question. The journals also say they’re indexed in PubMed or Google Scholar. If I want to publish my paper and I want it indexed where the right people are going to find it, where does it need to be indexed?
Dr. Merino: I grew up using Index Medicus, MedlinePlus, and the Library of Science. I still do. If I need to find something, I go to PubMed. Ideally, papers are listed in MedlinePlus or can be found in PubMed. They’re not the same thing, but you can find them through them.
That would be an important thing. Nowadays, a lot more people are using Google Scholar or Google just to identify papers. It may be a little bit less relevant, but it’s still a measure of the quality of the journal before they get indexed in some of these. For example, if you get listed in MedlinePlus, it has gone through certain quality checks by the index itself to see whether they would accept the journal or not. That’s something you want to check.
Typically, most of the large journals or the journals you and I know about are listed in more than one place, right? They’re listed in Scopus and Web of Science. They’re listed in MedlinePlus and so on. Again, if you’re submitting your paper, go somewhere where you know the journal and you’ve heard about it.
Dr. Wilner: I’m not going to ask you about artificial intelligence. We can do that another time. I want to ask something closer to me, which is this question of publish or perish.
There seems to be, in academics, more emphasis on the number of papers that one has published rather than their quality. How does a younger academician or one who really needs to publish cope with that?
Dr. Merino: Many people are writing up research that may not be relevant or that may not be high quality just because you need to have a long list of papers to get promoted, for example, if you’re an academician.
Doug Altman, who was a very influential person in the field quality of not only medical statistics but also medical publishing, had the idea that we need less research, but we need better research.
We often receive papers where you say, well, what’s the rationale behind the question in this paper? It’s like they had a large amount of data and were trying to squeeze as much as they could out of that. I think, as a young academician, the important thing to think about is whether it is an important question that matters to you and to the field, from whatever perspective, whether it’s going to advance research, advance clinical care, or have public policy implications.
Is this one where the answer will be important no matter what the answer is? If you’re thinking of that, your work will be well recognized, people will know you, and you’ll get invited to collaborate. I think that’s the most important thing rather than just churning out a large number of papers.
The productivity will come from the fact that you start by saying, let me ask something that’s really meaningful to me and to the field, with a good question and using strong research methodology.
Dr. Wilner: Thanks for that, Dr. Merino. I think that’s very valuable for all of us. This has been a great discussion. Do you have any final comments before we wrap up?
Dr. Merino: I want to encourage people to continue reading medical journals all the time and submitting to us, again, good research and important questions with robust methodology. That’s what we’re looking for in Neurology and most serious medical journals.
Dr. Wilner is an associate professor of neurology at the University of Tennessee Health Science Center, Memphis. Dr. Merino is a professor in the department of neurology at Georgetown University Medical Center, Washington, DC. Dr. Wilner reported conflicts of interest with Accordant Health Services and Lulu Publishing. Dr. Merino reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.