User login
Pediatric psoriasis linked to multiple psychiatric comorbidities
VIENNA – Psoriasis in children and adolescents is associated with significantly increased risk of a variety of psychiatric comorbidities, according to a large Danish national study.
This finding has important public health implications. Psoriasis is a common skin disease, and 30% of cases have their onset in childhood or adolescence, Tanja Todberg, MD, observed at the annual congress of the European Academy of Dermatology and Venereology.

Diagnosis of psoriasis was based upon medical records and documentation that at least a second prescription for a topical vitamin D derivative had been filled. Those agents are the overwhelming choice as first-line therapy in the pediatric population, explained Dr. Todberg of the University of Copenhagen.
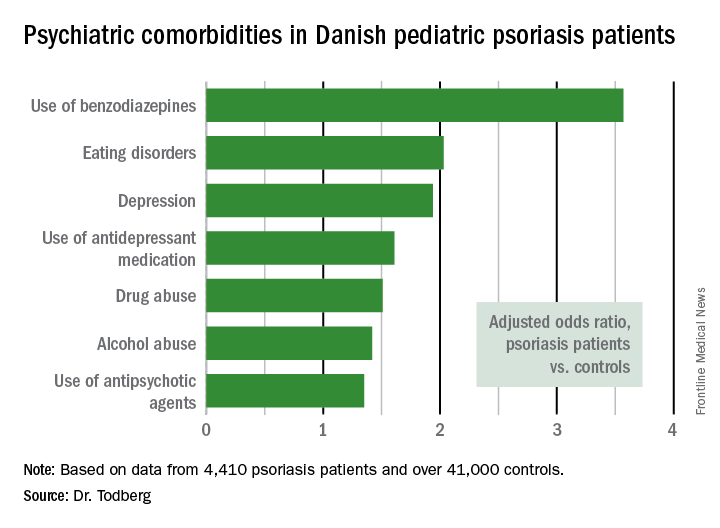
The pediatric psoriasis patients proved to be at significantly increased risk of being diagnosed with depression, eating disorders, drug abuse, and alcohol abuse. They were also more likely than controls to be prescribed antidepressants, antipsychotic agents, and benzodiazepines. That was every prespecified psychiatric outcome that Dr. Todberg and her coinvestigators included in the study except for one: anxiety disorders occurred at a similar rate in the pediatric psoriasis patients and controls.
Dr. Todberg reported having no financial conflicts of interest regarding this study, which was supported by Danish medical research funding.
[email protected]
VIENNA – Psoriasis in children and adolescents is associated with significantly increased risk of a variety of psychiatric comorbidities, according to a large Danish national study.
This finding has important public health implications. Psoriasis is a common skin disease, and 30% of cases have their onset in childhood or adolescence, Tanja Todberg, MD, observed at the annual congress of the European Academy of Dermatology and Venereology.
Diagnosis of psoriasis was based upon medical records and documentation that at least a second prescription for a topical vitamin D derivative had been filled. Those agents are the overwhelming choice as first-line therapy in the pediatric population, explained Dr. Todberg of the University of Copenhagen.
The pediatric psoriasis patients proved to be at significantly increased risk of being diagnosed with depression, eating disorders, drug abuse, and alcohol abuse. They were also more likely than controls to be prescribed antidepressants, antipsychotic agents, and benzodiazepines. That was every prespecified psychiatric outcome that Dr. Todberg and her coinvestigators included in the study except for one: anxiety disorders occurred at a similar rate in the pediatric psoriasis patients and controls.
Dr. Todberg reported having no financial conflicts of interest regarding this study, which was supported by Danish medical research funding.
[email protected]
VIENNA – Psoriasis in children and adolescents is associated with significantly increased risk of a variety of psychiatric comorbidities, according to a large Danish national study.
This finding has important public health implications. Psoriasis is a common skin disease, and 30% of cases have their onset in childhood or adolescence, Tanja Todberg, MD, observed at the annual congress of the European Academy of Dermatology and Venereology.
Diagnosis of psoriasis was based upon medical records and documentation that at least a second prescription for a topical vitamin D derivative had been filled. Those agents are the overwhelming choice as first-line therapy in the pediatric population, explained Dr. Todberg of the University of Copenhagen.
The pediatric psoriasis patients proved to be at significantly increased risk of being diagnosed with depression, eating disorders, drug abuse, and alcohol abuse. They were also more likely than controls to be prescribed antidepressants, antipsychotic agents, and benzodiazepines. That was every prespecified psychiatric outcome that Dr. Todberg and her coinvestigators included in the study except for one: anxiety disorders occurred at a similar rate in the pediatric psoriasis patients and controls.
Dr. Todberg reported having no financial conflicts of interest regarding this study, which was supported by Danish medical research funding.
[email protected]
AT THE EADV CONGRESS
Key clinical point:
Major finding: Danish pediatric patients with psoriasis were significantly more at risk of developing a range of psychiatric disorders than were control subjects.
Data source: This was a retrospective study of prospectively collected registry data on all 4,410 Danish children and adolescents diagnosed with psoriasis during 1997-2012 and more than 41,000 Danish controls matched for age, sex, and calendar year.
Disclosures: The presenter reported having no financial conflicts of interest regarding this study, which was supported by Danish medical research funds.
Study offers reassuring data on certolizumab use in pregnancy
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.

Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point:
Major finding: The rate of major congenital malformations in a large prospective series of pregnancies in women on certolizumab was reassuringly low at 4.2%, with no pattern of malformations being seen.
Data source: This was a report on maternal and fetal outcomes of 256 prospectively followed pregnancies in women on certolizumab.
Disclosures: The presenter reported receiving research funds from and serving as a consultant to UCB, which markets certolizumab and maintains the pregnancy registry.
Bioactive lipid shows promise in atopic dermatitis
VIENNA – A novel oral agent known as DS107 showed promise as a safe and effective treatment for moderate-to-severe atopic dermatitis in a phase IIa proof-of-concept study, Diamant Thaçi, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“Clear efficacy signals in the reduction of clinical symptoms of atopic dermatitis were detected within 2 weeks of treatment, with the maximum improvement in the endpoints observed between weeks 4 and 8,” said Dr. Thaçi, professor of dermatology and head of the Comprehensive Center for Inflammation Medicine at University Hospital Schleswig-Holstein, in Lübeck, Germany.

Dr. Thaçi presented the results of an 8-week, double-blind, randomized, placebo-controlled, multicenter phase IIa study that included 102 patients with moderate-to-severe AD. Participants averaged a baseline Investigator Global Assessment (IGA) score of 3.5 on the 0-5 scale. They were randomized to 2 g of oral DS107 once daily or an equal quantity of mineral oil as a placebo control.
Based upon the encouraging findings, a 300-patient phase IIb study will get underway soon. It will examine the effects of 1 g of DS107 per day as well as 2 g in the hope that the lower dose will cut down on the high rate of minor GI side effects seen at 2 g/day while preserving the efficacy of the higher dose.
The primary efficacy endpoint in the phase IIa study was at least a 2-point drop from baseline in IGA plus an end-of-treatment IGA of 0 or 1, meaning clear or almost clear. In the intent-to-treat analysis, this was achieved in 21.6% of the group assigned to DS107, compared with 11.8% of controls. In the 71 participants who actually completed 8 weeks of treatment – 35 in the DS107 arm, 36 controls – the composite efficacy endpoint was achieved in 31.4% of those on active treatment and 16.7% on mineral oil.
Significant separation between the active treatment and control arms in terms of itch visual analog scores was seen by week 4. This was a particularly encouraging finding, since patients report pruritis to be the most troublesome symptom of AD, Dr. Thaçi noted.
Significantly greater improvement in quality of life as measured by the Patient-Oriented Eczema Measure (POEM) was also seen by week 4 in the DS107 group as compared with controls.
No severe adverse events occurred in the study. However, more than one-quarter of subjects interrupted or discontinued participation because of mild nausea, loose stools, and/or abdominal pain. These issues were equally common in the DS107 and mineral oil groups, and Dr. Thaçi and his coinvestigators suspect that for many patients it was simply a matter of too much oil in the stomach. The GI symptoms resolved quickly without intervention after a brief halt of therapy, but some patients never returned to participation.
“This problem can be solved in the future with a different dosing design or even a different method of delivering the DGLA,” the dermatologist added.
Asked about the significant placebo response seen in the study, Dr. Thaçi shrugged it off as “quite understandable.”
“Placebo is not always a placebo. There is the feeling of fullness in the stomach, there is some emollient effect, the continuous contact with the physician. You see this in all the clinical trials in atopic dermatitis: in the beginning, the first 2-3 weeks, you have some influence of placebo,” he said.
The placebo effect was greatly diminished in patients with more severe AD. In the subset with a baseline IGA of 4 or 5, none of the control subjects achieved the primary efficacy endpoint, while more than 20% on DS107 did, Dr. Thaçi noted.
The mechanism of benefit of DGLA is not fully understood as yet. Animal studies point to an antibacterial effect, and DGLA also reduces levels of inflammatory cytokines. Eosinophilia was reduced to a much greater extent in the DS107 group than controls.
Dr. Thaçi reported serving as a consultant to DS Pharma, the privately held biopharmaceutical company that is developing oral DS107, as well as to numerous other pharmaceutical companies.
[email protected]
VIENNA – A novel oral agent known as DS107 showed promise as a safe and effective treatment for moderate-to-severe atopic dermatitis in a phase IIa proof-of-concept study, Diamant Thaçi, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“Clear efficacy signals in the reduction of clinical symptoms of atopic dermatitis were detected within 2 weeks of treatment, with the maximum improvement in the endpoints observed between weeks 4 and 8,” said Dr. Thaçi, professor of dermatology and head of the Comprehensive Center for Inflammation Medicine at University Hospital Schleswig-Holstein, in Lübeck, Germany.
Dr. Thaçi presented the results of an 8-week, double-blind, randomized, placebo-controlled, multicenter phase IIa study that included 102 patients with moderate-to-severe AD. Participants averaged a baseline Investigator Global Assessment (IGA) score of 3.5 on the 0-5 scale. They were randomized to 2 g of oral DS107 once daily or an equal quantity of mineral oil as a placebo control.
Based upon the encouraging findings, a 300-patient phase IIb study will get underway soon. It will examine the effects of 1 g of DS107 per day as well as 2 g in the hope that the lower dose will cut down on the high rate of minor GI side effects seen at 2 g/day while preserving the efficacy of the higher dose.
The primary efficacy endpoint in the phase IIa study was at least a 2-point drop from baseline in IGA plus an end-of-treatment IGA of 0 or 1, meaning clear or almost clear. In the intent-to-treat analysis, this was achieved in 21.6% of the group assigned to DS107, compared with 11.8% of controls. In the 71 participants who actually completed 8 weeks of treatment – 35 in the DS107 arm, 36 controls – the composite efficacy endpoint was achieved in 31.4% of those on active treatment and 16.7% on mineral oil.
Significant separation between the active treatment and control arms in terms of itch visual analog scores was seen by week 4. This was a particularly encouraging finding, since patients report pruritis to be the most troublesome symptom of AD, Dr. Thaçi noted.
Significantly greater improvement in quality of life as measured by the Patient-Oriented Eczema Measure (POEM) was also seen by week 4 in the DS107 group as compared with controls.
No severe adverse events occurred in the study. However, more than one-quarter of subjects interrupted or discontinued participation because of mild nausea, loose stools, and/or abdominal pain. These issues were equally common in the DS107 and mineral oil groups, and Dr. Thaçi and his coinvestigators suspect that for many patients it was simply a matter of too much oil in the stomach. The GI symptoms resolved quickly without intervention after a brief halt of therapy, but some patients never returned to participation.
“This problem can be solved in the future with a different dosing design or even a different method of delivering the DGLA,” the dermatologist added.
Asked about the significant placebo response seen in the study, Dr. Thaçi shrugged it off as “quite understandable.”
“Placebo is not always a placebo. There is the feeling of fullness in the stomach, there is some emollient effect, the continuous contact with the physician. You see this in all the clinical trials in atopic dermatitis: in the beginning, the first 2-3 weeks, you have some influence of placebo,” he said.
The placebo effect was greatly diminished in patients with more severe AD. In the subset with a baseline IGA of 4 or 5, none of the control subjects achieved the primary efficacy endpoint, while more than 20% on DS107 did, Dr. Thaçi noted.
The mechanism of benefit of DGLA is not fully understood as yet. Animal studies point to an antibacterial effect, and DGLA also reduces levels of inflammatory cytokines. Eosinophilia was reduced to a much greater extent in the DS107 group than controls.
Dr. Thaçi reported serving as a consultant to DS Pharma, the privately held biopharmaceutical company that is developing oral DS107, as well as to numerous other pharmaceutical companies.
[email protected]
VIENNA – A novel oral agent known as DS107 showed promise as a safe and effective treatment for moderate-to-severe atopic dermatitis in a phase IIa proof-of-concept study, Diamant Thaçi, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“Clear efficacy signals in the reduction of clinical symptoms of atopic dermatitis were detected within 2 weeks of treatment, with the maximum improvement in the endpoints observed between weeks 4 and 8,” said Dr. Thaçi, professor of dermatology and head of the Comprehensive Center for Inflammation Medicine at University Hospital Schleswig-Holstein, in Lübeck, Germany.
Dr. Thaçi presented the results of an 8-week, double-blind, randomized, placebo-controlled, multicenter phase IIa study that included 102 patients with moderate-to-severe AD. Participants averaged a baseline Investigator Global Assessment (IGA) score of 3.5 on the 0-5 scale. They were randomized to 2 g of oral DS107 once daily or an equal quantity of mineral oil as a placebo control.
Based upon the encouraging findings, a 300-patient phase IIb study will get underway soon. It will examine the effects of 1 g of DS107 per day as well as 2 g in the hope that the lower dose will cut down on the high rate of minor GI side effects seen at 2 g/day while preserving the efficacy of the higher dose.
The primary efficacy endpoint in the phase IIa study was at least a 2-point drop from baseline in IGA plus an end-of-treatment IGA of 0 or 1, meaning clear or almost clear. In the intent-to-treat analysis, this was achieved in 21.6% of the group assigned to DS107, compared with 11.8% of controls. In the 71 participants who actually completed 8 weeks of treatment – 35 in the DS107 arm, 36 controls – the composite efficacy endpoint was achieved in 31.4% of those on active treatment and 16.7% on mineral oil.
Significant separation between the active treatment and control arms in terms of itch visual analog scores was seen by week 4. This was a particularly encouraging finding, since patients report pruritis to be the most troublesome symptom of AD, Dr. Thaçi noted.
Significantly greater improvement in quality of life as measured by the Patient-Oriented Eczema Measure (POEM) was also seen by week 4 in the DS107 group as compared with controls.
No severe adverse events occurred in the study. However, more than one-quarter of subjects interrupted or discontinued participation because of mild nausea, loose stools, and/or abdominal pain. These issues were equally common in the DS107 and mineral oil groups, and Dr. Thaçi and his coinvestigators suspect that for many patients it was simply a matter of too much oil in the stomach. The GI symptoms resolved quickly without intervention after a brief halt of therapy, but some patients never returned to participation.
“This problem can be solved in the future with a different dosing design or even a different method of delivering the DGLA,” the dermatologist added.
Asked about the significant placebo response seen in the study, Dr. Thaçi shrugged it off as “quite understandable.”
“Placebo is not always a placebo. There is the feeling of fullness in the stomach, there is some emollient effect, the continuous contact with the physician. You see this in all the clinical trials in atopic dermatitis: in the beginning, the first 2-3 weeks, you have some influence of placebo,” he said.
The placebo effect was greatly diminished in patients with more severe AD. In the subset with a baseline IGA of 4 or 5, none of the control subjects achieved the primary efficacy endpoint, while more than 20% on DS107 did, Dr. Thaçi noted.
The mechanism of benefit of DGLA is not fully understood as yet. Animal studies point to an antibacterial effect, and DGLA also reduces levels of inflammatory cytokines. Eosinophilia was reduced to a much greater extent in the DS107 group than controls.
Dr. Thaçi reported serving as a consultant to DS Pharma, the privately held biopharmaceutical company that is developing oral DS107, as well as to numerous other pharmaceutical companies.
[email protected]
AT THE EADV CONGRESS
Key clinical point:
Major finding: Once-daily oral DS107, which contains a bioactive lipid, achieved significant improvement in moderate-to-severe atopic dermatitis in 31.4% of patients, compared with 16.7% on placebo.
Data source: This phase IIa, double-blind, randomized, placebo-controlled, multicenter 8-week trial included 102 adults with moderate-to-severe atopic dermatitis.
Disclosures: The study presenter reported serving as a consultant to DS Pharma, the privately held biopharmaceutical company that is developing oral DS107, as well as to numerous other pharmaceutical companies.
Topical crisaborole boosts quality of life in atopic dermatitis
VIENNA – Topical crisaborole 2% ointment administered twice a day was consistently associated with clinically meaningful quality of life improvement scores on multiple measures in the two pivotal phase III, randomized, controlled trials of atopic dermatitis (AD) patients aged 2 years old through adulthood, Amy S. Paller, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
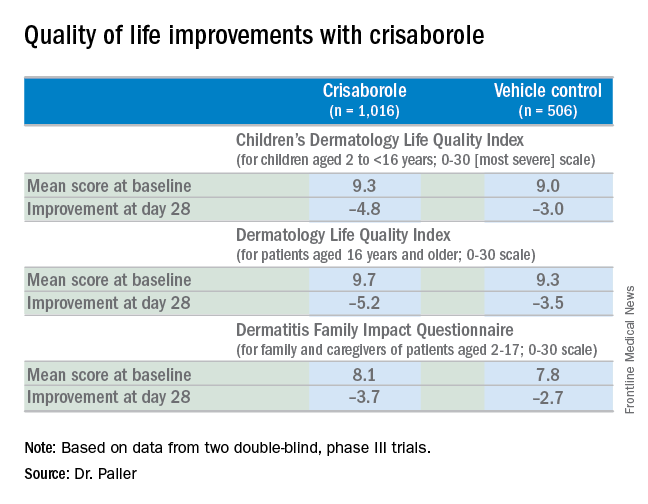
AD in children and adolescents is infamous for the adverse quality of life impact it imposes upon the patients’ parents, family, and caregivers. So the significant improvement seen with crisaborole, compared with its vehicle on the Dermatitis Family Impact (DFI) Questionnaire was particularly gratifying. The DFI questionnaire assesses quality of life in key domains, including family, parent, and caregiver sleep, emotional distress, relationships, family leisure, and ability to do housework or go shopping.

“If approved, crisaborole ... could improve the quality of life for patients with mild to moderate atopic dermatitis and, very importantly, for their families as well,” declared Dr. Paller, professor of dermatology and chair of the department of dermatology and professor of pediatrics at Northwestern University, Chicago.
Crisaborole’s developer, Anacor Pharmaceuticals, has filed an application for approval for treating mild to moderate AD in patients aged 2 years and older, now under review at the Food and Drug Administration.
In a separate presentation at the EADV congress, Lawrence F. Eichenfield, MD, presented the results of a long-term, open-label crisaborole safety study of 48-52 weeks duration. The long-term study involved 517 participants in the two pivotal phase III trials. There were no serious adverse events and no long-term cutaneous adverse events such as the skin atrophy or telangiectasias that can occur with topical steroids. The safety profile was favorable for long-term treatment of patients 2 years of age or older with mild to moderate AD.

Crisaborole 2% topical ointment is a novel, boron-based, nonsteroidal inhibitor of phosphodiesterase 4 (PDE-4). AD is marked by overactivity of PDE-4, which results in decreased levels of cyclic AMP and resultant increased release of inflammatory cytokines.
Dr. Paller noted that, in the previously reported efficacy results of the two pivotal, double-blind, 28-day, phase III trials, crisaborole treatment reduced global disease severity and provided early and sustained improvement in itch severity. She presented the prespecified quality of life results for the two identically designed, parallel pivotal trials, which totaled 1,016 patients on crisaborole and 506 on its vehicle. At baseline, 39% of subjects had mild AD, and 61% had moderate AD. The mean body surface area affected was 18%. Participants’ mean age was 12.3 years, and 14% were aged 8 years old or older.
The structure of the long-term safety study suggests how crisaborole might be used in clinical practice. During the year-long, open-label study, patients were evaluated every 28 days. If their skin was deemed clear or almost clear on the basis of an Investigator’s Static Global Assessment (ISGA) score of 0 or 1, they were taken off crisaborole and could use only emollients for the next 28 days, at which time they would be reevaluated. At that point, if they had an ISGA of 2 or more, they went back on crisaborole twice a day for 28 days until their next evaluation.
Dr. Eichenfield reported that 10.2% of participants in the long-term safety study reported treatment-related adverse events, which were mild to moderate. The most frequently reported of these were mild to moderate flares of AD during a 28-day off-treatment period in 1.3% of patients, application site pain in 2.3%, and application site infection in 1.2%.
Dr. Paller and Dr. Eichenfield reported serving as consultants to Anacor Pharmaceuticals.
The FDA review of crisaborole is expected to be completed by early January 2017, according to Anacor.
VIENNA – Topical crisaborole 2% ointment administered twice a day was consistently associated with clinically meaningful quality of life improvement scores on multiple measures in the two pivotal phase III, randomized, controlled trials of atopic dermatitis (AD) patients aged 2 years old through adulthood, Amy S. Paller, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
AD in children and adolescents is infamous for the adverse quality of life impact it imposes upon the patients’ parents, family, and caregivers. So the significant improvement seen with crisaborole, compared with its vehicle on the Dermatitis Family Impact (DFI) Questionnaire was particularly gratifying. The DFI questionnaire assesses quality of life in key domains, including family, parent, and caregiver sleep, emotional distress, relationships, family leisure, and ability to do housework or go shopping.
“If approved, crisaborole ... could improve the quality of life for patients with mild to moderate atopic dermatitis and, very importantly, for their families as well,” declared Dr. Paller, professor of dermatology and chair of the department of dermatology and professor of pediatrics at Northwestern University, Chicago.
Crisaborole’s developer, Anacor Pharmaceuticals, has filed an application for approval for treating mild to moderate AD in patients aged 2 years and older, now under review at the Food and Drug Administration.
In a separate presentation at the EADV congress, Lawrence F. Eichenfield, MD, presented the results of a long-term, open-label crisaborole safety study of 48-52 weeks duration. The long-term study involved 517 participants in the two pivotal phase III trials. There were no serious adverse events and no long-term cutaneous adverse events such as the skin atrophy or telangiectasias that can occur with topical steroids. The safety profile was favorable for long-term treatment of patients 2 years of age or older with mild to moderate AD.
Crisaborole 2% topical ointment is a novel, boron-based, nonsteroidal inhibitor of phosphodiesterase 4 (PDE-4). AD is marked by overactivity of PDE-4, which results in decreased levels of cyclic AMP and resultant increased release of inflammatory cytokines.
Dr. Paller noted that, in the previously reported efficacy results of the two pivotal, double-blind, 28-day, phase III trials, crisaborole treatment reduced global disease severity and provided early and sustained improvement in itch severity. She presented the prespecified quality of life results for the two identically designed, parallel pivotal trials, which totaled 1,016 patients on crisaborole and 506 on its vehicle. At baseline, 39% of subjects had mild AD, and 61% had moderate AD. The mean body surface area affected was 18%. Participants’ mean age was 12.3 years, and 14% were aged 8 years old or older.
The structure of the long-term safety study suggests how crisaborole might be used in clinical practice. During the year-long, open-label study, patients were evaluated every 28 days. If their skin was deemed clear or almost clear on the basis of an Investigator’s Static Global Assessment (ISGA) score of 0 or 1, they were taken off crisaborole and could use only emollients for the next 28 days, at which time they would be reevaluated. At that point, if they had an ISGA of 2 or more, they went back on crisaborole twice a day for 28 days until their next evaluation.
Dr. Eichenfield reported that 10.2% of participants in the long-term safety study reported treatment-related adverse events, which were mild to moderate. The most frequently reported of these were mild to moderate flares of AD during a 28-day off-treatment period in 1.3% of patients, application site pain in 2.3%, and application site infection in 1.2%.
Dr. Paller and Dr. Eichenfield reported serving as consultants to Anacor Pharmaceuticals.
The FDA review of crisaborole is expected to be completed by early January 2017, according to Anacor.
VIENNA – Topical crisaborole 2% ointment administered twice a day was consistently associated with clinically meaningful quality of life improvement scores on multiple measures in the two pivotal phase III, randomized, controlled trials of atopic dermatitis (AD) patients aged 2 years old through adulthood, Amy S. Paller, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
AD in children and adolescents is infamous for the adverse quality of life impact it imposes upon the patients’ parents, family, and caregivers. So the significant improvement seen with crisaborole, compared with its vehicle on the Dermatitis Family Impact (DFI) Questionnaire was particularly gratifying. The DFI questionnaire assesses quality of life in key domains, including family, parent, and caregiver sleep, emotional distress, relationships, family leisure, and ability to do housework or go shopping.
“If approved, crisaborole ... could improve the quality of life for patients with mild to moderate atopic dermatitis and, very importantly, for their families as well,” declared Dr. Paller, professor of dermatology and chair of the department of dermatology and professor of pediatrics at Northwestern University, Chicago.
Crisaborole’s developer, Anacor Pharmaceuticals, has filed an application for approval for treating mild to moderate AD in patients aged 2 years and older, now under review at the Food and Drug Administration.
In a separate presentation at the EADV congress, Lawrence F. Eichenfield, MD, presented the results of a long-term, open-label crisaborole safety study of 48-52 weeks duration. The long-term study involved 517 participants in the two pivotal phase III trials. There were no serious adverse events and no long-term cutaneous adverse events such as the skin atrophy or telangiectasias that can occur with topical steroids. The safety profile was favorable for long-term treatment of patients 2 years of age or older with mild to moderate AD.
Crisaborole 2% topical ointment is a novel, boron-based, nonsteroidal inhibitor of phosphodiesterase 4 (PDE-4). AD is marked by overactivity of PDE-4, which results in decreased levels of cyclic AMP and resultant increased release of inflammatory cytokines.
Dr. Paller noted that, in the previously reported efficacy results of the two pivotal, double-blind, 28-day, phase III trials, crisaborole treatment reduced global disease severity and provided early and sustained improvement in itch severity. She presented the prespecified quality of life results for the two identically designed, parallel pivotal trials, which totaled 1,016 patients on crisaborole and 506 on its vehicle. At baseline, 39% of subjects had mild AD, and 61% had moderate AD. The mean body surface area affected was 18%. Participants’ mean age was 12.3 years, and 14% were aged 8 years old or older.
The structure of the long-term safety study suggests how crisaborole might be used in clinical practice. During the year-long, open-label study, patients were evaluated every 28 days. If their skin was deemed clear or almost clear on the basis of an Investigator’s Static Global Assessment (ISGA) score of 0 or 1, they were taken off crisaborole and could use only emollients for the next 28 days, at which time they would be reevaluated. At that point, if they had an ISGA of 2 or more, they went back on crisaborole twice a day for 28 days until their next evaluation.
Dr. Eichenfield reported that 10.2% of participants in the long-term safety study reported treatment-related adverse events, which were mild to moderate. The most frequently reported of these were mild to moderate flares of AD during a 28-day off-treatment period in 1.3% of patients, application site pain in 2.3%, and application site infection in 1.2%.
Dr. Paller and Dr. Eichenfield reported serving as consultants to Anacor Pharmaceuticals.
The FDA review of crisaborole is expected to be completed by early January 2017, according to Anacor.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Tildrakizumab for psoriasis scores high marks in phase III
VIENNA – The investigational interleukin-23 p-19 subunit inhibitor tildrakizumab achieved PASI 90 improvement rates approaching 60% in patients with moderate to severe plaque psoriasis at week 28 of the pivotal phase III reSURFACE 1 and reSURFACE 2 trials, Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Moreover, the PASI 100 rate at week 28 in the two clinical trials was 24% with tildrakizumab, a humanized monoclonal antibody, at the 100-mg dose and 30% at 200 mg.
It was at these highest efficacy thresholds that the p-19 inhibitor really separated itself from etanercept (Enbrel) in reSURFACE 2, where the two biologics went head-to-head in randomized fashion. Patients on etanercept had a PASI 90 rate at week 28 of 31%, roughly only half that of tildrakizumab at the higher dose.

Guselkumab was the other IL-23 p-19 inhibitor that was a featured attraction at the EADV congress, with 48-week outcomes presented from the 837-patient, pivotal phase III VOYAGE 1 trial. Although caution is always warranted in comparing results across clinical trials because of differences in study populations, guselkumab achieved better top-end efficacy numbers than did tildrakizumab: a PASI 90 of 73.3% at 16 weeks and 80.2% at 24 weeks, along with PASI 100 responses of 34.4% at 16 weeks and 44.4% at 24 weeks.
“I believe there will be characteristics of the new drugs beyond efficacy that will come into play when making treatment decisions: Is dosing every 8 weeks or every 12 weeks? What is the price? What is the outcome after 1 year? I think it’s too early to close the book in trying to understand what these different drugs do, but these phase III results do give us the insight that IL-23 p-19 is actually the sweet spot in psoriasis. By targeting it we are able to keep the disease under control with drugs that are very convenient to use,” Dr. Reich said.
He added that his psoriasis patients really appreciate the convenience of quarterly as opposed to more frequent dosing of biologics, and he does, too.
reSURFACE 1 is a 64-week, randomized, phase III trial conducted in the United States, Canada, and Europe in which 772 patients were randomized 2:2:1 to tildrakizumab at 100 mg or 200 mg or to placebo. reSURFACE 2 is a 64-week trial in which 1,090 patients were randomized 2:2:1:2 to tildrakizumab at 100 mg or 200 mg, placebo, or etanercept at 50 mg twice weekly for the first 12 weeks and once weekly thereafter. At week 12 in both trials, patients on placebo were rerandomized to tildrakizumab at 100 or 200 mg for the duration. Participants averaged a baseline Psoriasis Area and Severity index score of 20, a body weight of 88 kg, and disease involvement over 31% of their body surface area.
Tildrakizumab was dosed in a regimen that’s the same as for ustekinumab (Stelara), which inhibits IL-12 as well as IL-23: one subcutaneous injection at baseline, another 1 month later, and every 12 weeks thereafter.
Dr. Reich presented results of the two pivotal trials through week 28. The coprimary efficacy endpoints in both studies were the PASI 75 response and proportion of subjects with a Physician’s Global Assessment (PGA) score of 0 or 1, meaning clear or minimal disease, compared with placebo at week 12. In hindsight, he said, those were not the best endpoints to have employed.
“We have here a drug that takes a little while to get to full throttle. The primary endpoint selected here at week 12 does not show efficacy data that really separates tildrakizumab from a drug like Stelara. But at week 28 you move toward levels of PASI 90 and PASI 100 response that we really want to see,” the dermatologist said.
Combining the results of reSURFACE 1 and 2, the PASI 75 response rate at week 12 – after only two doses – was 63% in the 100-mg arm and 64% at 200 mg. But the rates kept climbing thereafter such that by week 28 the PASI 75 rates were 77% and 78%.
Fifty-seven percent of patients on tildrakizumab at 100 mg had a PGA score of 0 or 1 at week 12, as did 6% of placebo-treated controls. By week 28, 66% of patients on the lower dose of the p-19 inhibitor had a PGA of 0 or 1. Rates in patients on tildrakizumab at 200 mg were 57% and 66% at 12 and 28 weeks, respectively.
Rates of adverse events of special interest in new biologic agents, including severe infections, malignancies, and major cardiovascular events, were similarly low across all study arms.
“My feeling is that looking at week 12 and week 28 safety data is of limited value. All I can say here is that through week 28 in these two studies I don’t see a safety signal. But for me, the real insight will have to come from larger studies with longer follow-up,” Dr. Reich said.
Asked why he thinks tildrakizumab is a slow starter, with only middling efficacy at the 12-week mark before subsequently picking up steam, he said it’s probably not a matter of the wrong doses being selected for reSURFACE 1 and 2, since the outcomes with 100 and 200 mg are fairly similar. More likely, the monoclonal antibody takes a bit longer to bind to its target and neutralize it than do some of the other biologics.
“It could be that if you dosed tildrakizumab at weeks 0, 2, and 8 as induction therapy you’d hit the mark at 12 weeks,” he added.
The reSURFACE trials are funded by Sun Pharma and Merck. Dr. Reich reported having received research grants from and serving as a consultant to Merck and numerous other pharmaceutical companies interested in new treatments for inflammatory skin diseases.
VIENNA – The investigational interleukin-23 p-19 subunit inhibitor tildrakizumab achieved PASI 90 improvement rates approaching 60% in patients with moderate to severe plaque psoriasis at week 28 of the pivotal phase III reSURFACE 1 and reSURFACE 2 trials, Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Moreover, the PASI 100 rate at week 28 in the two clinical trials was 24% with tildrakizumab, a humanized monoclonal antibody, at the 100-mg dose and 30% at 200 mg.
It was at these highest efficacy thresholds that the p-19 inhibitor really separated itself from etanercept (Enbrel) in reSURFACE 2, where the two biologics went head-to-head in randomized fashion. Patients on etanercept had a PASI 90 rate at week 28 of 31%, roughly only half that of tildrakizumab at the higher dose.
Guselkumab was the other IL-23 p-19 inhibitor that was a featured attraction at the EADV congress, with 48-week outcomes presented from the 837-patient, pivotal phase III VOYAGE 1 trial. Although caution is always warranted in comparing results across clinical trials because of differences in study populations, guselkumab achieved better top-end efficacy numbers than did tildrakizumab: a PASI 90 of 73.3% at 16 weeks and 80.2% at 24 weeks, along with PASI 100 responses of 34.4% at 16 weeks and 44.4% at 24 weeks.
“I believe there will be characteristics of the new drugs beyond efficacy that will come into play when making treatment decisions: Is dosing every 8 weeks or every 12 weeks? What is the price? What is the outcome after 1 year? I think it’s too early to close the book in trying to understand what these different drugs do, but these phase III results do give us the insight that IL-23 p-19 is actually the sweet spot in psoriasis. By targeting it we are able to keep the disease under control with drugs that are very convenient to use,” Dr. Reich said.
He added that his psoriasis patients really appreciate the convenience of quarterly as opposed to more frequent dosing of biologics, and he does, too.
reSURFACE 1 is a 64-week, randomized, phase III trial conducted in the United States, Canada, and Europe in which 772 patients were randomized 2:2:1 to tildrakizumab at 100 mg or 200 mg or to placebo. reSURFACE 2 is a 64-week trial in which 1,090 patients were randomized 2:2:1:2 to tildrakizumab at 100 mg or 200 mg, placebo, or etanercept at 50 mg twice weekly for the first 12 weeks and once weekly thereafter. At week 12 in both trials, patients on placebo were rerandomized to tildrakizumab at 100 or 200 mg for the duration. Participants averaged a baseline Psoriasis Area and Severity index score of 20, a body weight of 88 kg, and disease involvement over 31% of their body surface area.
Tildrakizumab was dosed in a regimen that’s the same as for ustekinumab (Stelara), which inhibits IL-12 as well as IL-23: one subcutaneous injection at baseline, another 1 month later, and every 12 weeks thereafter.
Dr. Reich presented results of the two pivotal trials through week 28. The coprimary efficacy endpoints in both studies were the PASI 75 response and proportion of subjects with a Physician’s Global Assessment (PGA) score of 0 or 1, meaning clear or minimal disease, compared with placebo at week 12. In hindsight, he said, those were not the best endpoints to have employed.
“We have here a drug that takes a little while to get to full throttle. The primary endpoint selected here at week 12 does not show efficacy data that really separates tildrakizumab from a drug like Stelara. But at week 28 you move toward levels of PASI 90 and PASI 100 response that we really want to see,” the dermatologist said.
Combining the results of reSURFACE 1 and 2, the PASI 75 response rate at week 12 – after only two doses – was 63% in the 100-mg arm and 64% at 200 mg. But the rates kept climbing thereafter such that by week 28 the PASI 75 rates were 77% and 78%.
Fifty-seven percent of patients on tildrakizumab at 100 mg had a PGA score of 0 or 1 at week 12, as did 6% of placebo-treated controls. By week 28, 66% of patients on the lower dose of the p-19 inhibitor had a PGA of 0 or 1. Rates in patients on tildrakizumab at 200 mg were 57% and 66% at 12 and 28 weeks, respectively.
Rates of adverse events of special interest in new biologic agents, including severe infections, malignancies, and major cardiovascular events, were similarly low across all study arms.
“My feeling is that looking at week 12 and week 28 safety data is of limited value. All I can say here is that through week 28 in these two studies I don’t see a safety signal. But for me, the real insight will have to come from larger studies with longer follow-up,” Dr. Reich said.
Asked why he thinks tildrakizumab is a slow starter, with only middling efficacy at the 12-week mark before subsequently picking up steam, he said it’s probably not a matter of the wrong doses being selected for reSURFACE 1 and 2, since the outcomes with 100 and 200 mg are fairly similar. More likely, the monoclonal antibody takes a bit longer to bind to its target and neutralize it than do some of the other biologics.
“It could be that if you dosed tildrakizumab at weeks 0, 2, and 8 as induction therapy you’d hit the mark at 12 weeks,” he added.
The reSURFACE trials are funded by Sun Pharma and Merck. Dr. Reich reported having received research grants from and serving as a consultant to Merck and numerous other pharmaceutical companies interested in new treatments for inflammatory skin diseases.
VIENNA – The investigational interleukin-23 p-19 subunit inhibitor tildrakizumab achieved PASI 90 improvement rates approaching 60% in patients with moderate to severe plaque psoriasis at week 28 of the pivotal phase III reSURFACE 1 and reSURFACE 2 trials, Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Moreover, the PASI 100 rate at week 28 in the two clinical trials was 24% with tildrakizumab, a humanized monoclonal antibody, at the 100-mg dose and 30% at 200 mg.
It was at these highest efficacy thresholds that the p-19 inhibitor really separated itself from etanercept (Enbrel) in reSURFACE 2, where the two biologics went head-to-head in randomized fashion. Patients on etanercept had a PASI 90 rate at week 28 of 31%, roughly only half that of tildrakizumab at the higher dose.
Guselkumab was the other IL-23 p-19 inhibitor that was a featured attraction at the EADV congress, with 48-week outcomes presented from the 837-patient, pivotal phase III VOYAGE 1 trial. Although caution is always warranted in comparing results across clinical trials because of differences in study populations, guselkumab achieved better top-end efficacy numbers than did tildrakizumab: a PASI 90 of 73.3% at 16 weeks and 80.2% at 24 weeks, along with PASI 100 responses of 34.4% at 16 weeks and 44.4% at 24 weeks.
“I believe there will be characteristics of the new drugs beyond efficacy that will come into play when making treatment decisions: Is dosing every 8 weeks or every 12 weeks? What is the price? What is the outcome after 1 year? I think it’s too early to close the book in trying to understand what these different drugs do, but these phase III results do give us the insight that IL-23 p-19 is actually the sweet spot in psoriasis. By targeting it we are able to keep the disease under control with drugs that are very convenient to use,” Dr. Reich said.
He added that his psoriasis patients really appreciate the convenience of quarterly as opposed to more frequent dosing of biologics, and he does, too.
reSURFACE 1 is a 64-week, randomized, phase III trial conducted in the United States, Canada, and Europe in which 772 patients were randomized 2:2:1 to tildrakizumab at 100 mg or 200 mg or to placebo. reSURFACE 2 is a 64-week trial in which 1,090 patients were randomized 2:2:1:2 to tildrakizumab at 100 mg or 200 mg, placebo, or etanercept at 50 mg twice weekly for the first 12 weeks and once weekly thereafter. At week 12 in both trials, patients on placebo were rerandomized to tildrakizumab at 100 or 200 mg for the duration. Participants averaged a baseline Psoriasis Area and Severity index score of 20, a body weight of 88 kg, and disease involvement over 31% of their body surface area.
Tildrakizumab was dosed in a regimen that’s the same as for ustekinumab (Stelara), which inhibits IL-12 as well as IL-23: one subcutaneous injection at baseline, another 1 month later, and every 12 weeks thereafter.
Dr. Reich presented results of the two pivotal trials through week 28. The coprimary efficacy endpoints in both studies were the PASI 75 response and proportion of subjects with a Physician’s Global Assessment (PGA) score of 0 or 1, meaning clear or minimal disease, compared with placebo at week 12. In hindsight, he said, those were not the best endpoints to have employed.
“We have here a drug that takes a little while to get to full throttle. The primary endpoint selected here at week 12 does not show efficacy data that really separates tildrakizumab from a drug like Stelara. But at week 28 you move toward levels of PASI 90 and PASI 100 response that we really want to see,” the dermatologist said.
Combining the results of reSURFACE 1 and 2, the PASI 75 response rate at week 12 – after only two doses – was 63% in the 100-mg arm and 64% at 200 mg. But the rates kept climbing thereafter such that by week 28 the PASI 75 rates were 77% and 78%.
Fifty-seven percent of patients on tildrakizumab at 100 mg had a PGA score of 0 or 1 at week 12, as did 6% of placebo-treated controls. By week 28, 66% of patients on the lower dose of the p-19 inhibitor had a PGA of 0 or 1. Rates in patients on tildrakizumab at 200 mg were 57% and 66% at 12 and 28 weeks, respectively.
Rates of adverse events of special interest in new biologic agents, including severe infections, malignancies, and major cardiovascular events, were similarly low across all study arms.
“My feeling is that looking at week 12 and week 28 safety data is of limited value. All I can say here is that through week 28 in these two studies I don’t see a safety signal. But for me, the real insight will have to come from larger studies with longer follow-up,” Dr. Reich said.
Asked why he thinks tildrakizumab is a slow starter, with only middling efficacy at the 12-week mark before subsequently picking up steam, he said it’s probably not a matter of the wrong doses being selected for reSURFACE 1 and 2, since the outcomes with 100 and 200 mg are fairly similar. More likely, the monoclonal antibody takes a bit longer to bind to its target and neutralize it than do some of the other biologics.
“It could be that if you dosed tildrakizumab at weeks 0, 2, and 8 as induction therapy you’d hit the mark at 12 weeks,” he added.
The reSURFACE trials are funded by Sun Pharma and Merck. Dr. Reich reported having received research grants from and serving as a consultant to Merck and numerous other pharmaceutical companies interested in new treatments for inflammatory skin diseases.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Tildrakizumab dosed quarterly at 100 or 200 mg achieved PASI 75 response rates of 63%-64% at week 12 and 77%-78% at week 28.
Data source: reSURFACE 1 and 2: two pivotal, phase III, randomized clinical trials comprising 1,862 patients with moderate to severe plaque psoriasis.
Disclosures: The reSURFACE trials are funded by Sun Pharma and Merck. The presenter reported having received research grants from and serving as a consultant to Merck and numerous other pharmaceutical companies developing treatments for inflammatory skin diseases.
Secukinumab for psoriasis at 4 years: undiminished efficacy and safety
VIENNA – Four-year follow-up of patients on secukinumab for psoriasis shows sustained very high efficacy, with almost 100% of patients who had a Psoriasis Area and Severity Index (PASI) 90 or 100 response at 1 year maintaining it through 4 years, Robert Bissonnette, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“I must warn you that my presentation will be very boring as compared to what I’ve seen earlier at this meeting, the very cutting edge phase II and phase III data being presented. My presentation doesn’t contain any surprises. However, as a clinician who is using interleukin-17A inhibition in my practice to treat psoriasis patients, that’s probably what I want,” said Dr. Bissonnette, president of Innovaderm Research in Montreal.
“This is the longest-term safety and efficacy data available to date for patients treated with an IL-17 antagonist using an approved dose,” he noted.
Dr. Bissonnette presented 4-year results in the 165 participants who took the approved regimen from the start of the study. These were patients at the serious end of the disease severity spectrum. Their mean baseline PASI score was 23.5, with 33% of their body surface area being affected. Their mean Dermatology Life Quality Index (DLQI) score was 13.1. The mean body mass index was 28.7 kg/m2. A total of 71% of subjects had previously been on systemic therapy. One-third of participants had been on other biologics.
At 1 year, 88.9% of subjects had a PASI 75 response; at 4 years, the PASI 75 rate was 88.5%. Similarly, the PASI 90 rate was 68.5% at 1 year and 66.4% after 4 years. The PASI 100 rate was 43.8% at 1 year and 43.5% at year 4.
After 1 year on secukinumab, patients showed a mean 91.1% improvement, compared with their baseline PASI score. At 4 years, the figure was 90.8%.
Bearing in mind that the average baseline DLQI score at baseline was 13.1, it’s noteworthy that after 1 year on secukinumab, 72.7% of patients had a DLQI of 0 or 1, indicating psoriasis had no impact on their life. At year 4, the rate was 70.8%, Dr. Bissonnette continued.
As an audience member observed, however, the study population decreased from 165 patients to 131 over the course of 4 years. And since this was an “as observed” analysis, outcomes were counted only in those patients still in the study. It’s accepted as a legitimate statistical method, but it casts outcomes in a particularly favorable light.
“The main reason for dropouts was for personal reasons,” Dr. Bissonnette explained in response. “Number two was lack of or loss of efficacy. Loss of efficacy over time occurred at an absolute rate of 4%-8% per year.”
Overall, adverse event rates declined over the course of 4 years of follow-up.
“This is reassuring, but I don’t think it’s evidence that adverse events actually decrease over time because of longer use of secukinumab. I think it’s probably due to something we usually see in long-term clinical trials: a phenomenon of underreporting. When patients are treated with a new agent they tend to be very, very conscientious about what’s going on with their well-being. They will report a slight sore throat, a slight congestion. But once they’ve been on treatment for a longer time they’re less likely to report those very minor adverse events,” according to the dermatologist.
The Food and Drug Administration requires clinical trialists to keep careful track of selected adverse events in studies of biologic agents. In 4 years on secukinumab, there were no cases of tuberculosis, neutropenia, major adverse cardiovascular events, or Crohn’s disease. There were two cases of ulcerative colitis in year 2; however, one involved an exacerbation of preexisting disease. Also, two patients developed cancer other than nonmelanoma skin cancer in year 2. The incidence of vulvovaginal candidiasis was 1.8% during years 1 and 2, 0.6% in year 3, and zero in year 4.
Thus, the safety profile was favorable, with no pattern of increasing adverse events with longer medication use, Dr. Bissonnette concluded.
The study was sponsored by Novartis. Dr. Bissonnette reported serving as an investigator for and consultant to Novartis and 16 other pharmaceutical companies.
VIENNA – Four-year follow-up of patients on secukinumab for psoriasis shows sustained very high efficacy, with almost 100% of patients who had a Psoriasis Area and Severity Index (PASI) 90 or 100 response at 1 year maintaining it through 4 years, Robert Bissonnette, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“I must warn you that my presentation will be very boring as compared to what I’ve seen earlier at this meeting, the very cutting edge phase II and phase III data being presented. My presentation doesn’t contain any surprises. However, as a clinician who is using interleukin-17A inhibition in my practice to treat psoriasis patients, that’s probably what I want,” said Dr. Bissonnette, president of Innovaderm Research in Montreal.
“This is the longest-term safety and efficacy data available to date for patients treated with an IL-17 antagonist using an approved dose,” he noted.
Dr. Bissonnette presented 4-year results in the 165 participants who took the approved regimen from the start of the study. These were patients at the serious end of the disease severity spectrum. Their mean baseline PASI score was 23.5, with 33% of their body surface area being affected. Their mean Dermatology Life Quality Index (DLQI) score was 13.1. The mean body mass index was 28.7 kg/m2. A total of 71% of subjects had previously been on systemic therapy. One-third of participants had been on other biologics.
At 1 year, 88.9% of subjects had a PASI 75 response; at 4 years, the PASI 75 rate was 88.5%. Similarly, the PASI 90 rate was 68.5% at 1 year and 66.4% after 4 years. The PASI 100 rate was 43.8% at 1 year and 43.5% at year 4.
After 1 year on secukinumab, patients showed a mean 91.1% improvement, compared with their baseline PASI score. At 4 years, the figure was 90.8%.
Bearing in mind that the average baseline DLQI score at baseline was 13.1, it’s noteworthy that after 1 year on secukinumab, 72.7% of patients had a DLQI of 0 or 1, indicating psoriasis had no impact on their life. At year 4, the rate was 70.8%, Dr. Bissonnette continued.
As an audience member observed, however, the study population decreased from 165 patients to 131 over the course of 4 years. And since this was an “as observed” analysis, outcomes were counted only in those patients still in the study. It’s accepted as a legitimate statistical method, but it casts outcomes in a particularly favorable light.
“The main reason for dropouts was for personal reasons,” Dr. Bissonnette explained in response. “Number two was lack of or loss of efficacy. Loss of efficacy over time occurred at an absolute rate of 4%-8% per year.”
Overall, adverse event rates declined over the course of 4 years of follow-up.
“This is reassuring, but I don’t think it’s evidence that adverse events actually decrease over time because of longer use of secukinumab. I think it’s probably due to something we usually see in long-term clinical trials: a phenomenon of underreporting. When patients are treated with a new agent they tend to be very, very conscientious about what’s going on with their well-being. They will report a slight sore throat, a slight congestion. But once they’ve been on treatment for a longer time they’re less likely to report those very minor adverse events,” according to the dermatologist.
The Food and Drug Administration requires clinical trialists to keep careful track of selected adverse events in studies of biologic agents. In 4 years on secukinumab, there were no cases of tuberculosis, neutropenia, major adverse cardiovascular events, or Crohn’s disease. There were two cases of ulcerative colitis in year 2; however, one involved an exacerbation of preexisting disease. Also, two patients developed cancer other than nonmelanoma skin cancer in year 2. The incidence of vulvovaginal candidiasis was 1.8% during years 1 and 2, 0.6% in year 3, and zero in year 4.
Thus, the safety profile was favorable, with no pattern of increasing adverse events with longer medication use, Dr. Bissonnette concluded.
The study was sponsored by Novartis. Dr. Bissonnette reported serving as an investigator for and consultant to Novartis and 16 other pharmaceutical companies.
VIENNA – Four-year follow-up of patients on secukinumab for psoriasis shows sustained very high efficacy, with almost 100% of patients who had a Psoriasis Area and Severity Index (PASI) 90 or 100 response at 1 year maintaining it through 4 years, Robert Bissonnette, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“I must warn you that my presentation will be very boring as compared to what I’ve seen earlier at this meeting, the very cutting edge phase II and phase III data being presented. My presentation doesn’t contain any surprises. However, as a clinician who is using interleukin-17A inhibition in my practice to treat psoriasis patients, that’s probably what I want,” said Dr. Bissonnette, president of Innovaderm Research in Montreal.
“This is the longest-term safety and efficacy data available to date for patients treated with an IL-17 antagonist using an approved dose,” he noted.
Dr. Bissonnette presented 4-year results in the 165 participants who took the approved regimen from the start of the study. These were patients at the serious end of the disease severity spectrum. Their mean baseline PASI score was 23.5, with 33% of their body surface area being affected. Their mean Dermatology Life Quality Index (DLQI) score was 13.1. The mean body mass index was 28.7 kg/m2. A total of 71% of subjects had previously been on systemic therapy. One-third of participants had been on other biologics.
At 1 year, 88.9% of subjects had a PASI 75 response; at 4 years, the PASI 75 rate was 88.5%. Similarly, the PASI 90 rate was 68.5% at 1 year and 66.4% after 4 years. The PASI 100 rate was 43.8% at 1 year and 43.5% at year 4.
After 1 year on secukinumab, patients showed a mean 91.1% improvement, compared with their baseline PASI score. At 4 years, the figure was 90.8%.
Bearing in mind that the average baseline DLQI score at baseline was 13.1, it’s noteworthy that after 1 year on secukinumab, 72.7% of patients had a DLQI of 0 or 1, indicating psoriasis had no impact on their life. At year 4, the rate was 70.8%, Dr. Bissonnette continued.
As an audience member observed, however, the study population decreased from 165 patients to 131 over the course of 4 years. And since this was an “as observed” analysis, outcomes were counted only in those patients still in the study. It’s accepted as a legitimate statistical method, but it casts outcomes in a particularly favorable light.
“The main reason for dropouts was for personal reasons,” Dr. Bissonnette explained in response. “Number two was lack of or loss of efficacy. Loss of efficacy over time occurred at an absolute rate of 4%-8% per year.”
Overall, adverse event rates declined over the course of 4 years of follow-up.
“This is reassuring, but I don’t think it’s evidence that adverse events actually decrease over time because of longer use of secukinumab. I think it’s probably due to something we usually see in long-term clinical trials: a phenomenon of underreporting. When patients are treated with a new agent they tend to be very, very conscientious about what’s going on with their well-being. They will report a slight sore throat, a slight congestion. But once they’ve been on treatment for a longer time they’re less likely to report those very minor adverse events,” according to the dermatologist.
The Food and Drug Administration requires clinical trialists to keep careful track of selected adverse events in studies of biologic agents. In 4 years on secukinumab, there were no cases of tuberculosis, neutropenia, major adverse cardiovascular events, or Crohn’s disease. There were two cases of ulcerative colitis in year 2; however, one involved an exacerbation of preexisting disease. Also, two patients developed cancer other than nonmelanoma skin cancer in year 2. The incidence of vulvovaginal candidiasis was 1.8% during years 1 and 2, 0.6% in year 3, and zero in year 4.
Thus, the safety profile was favorable, with no pattern of increasing adverse events with longer medication use, Dr. Bissonnette concluded.
The study was sponsored by Novartis. Dr. Bissonnette reported serving as an investigator for and consultant to Novartis and 16 other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point:
Major finding: After 1 year on secukinumab, 43.8% of psoriasis patients had a PASI 100 response. After 3 additional years on the interleukin-17A inhibitor, the rate was virtually unchanged at 43.5%.
Data source: This was analysis of 165 psoriasis patients on secukinumab at the approved dose prospectively followed for 4 years in an extension of a phase III clinical trial.
Disclosures: Novartis sponsored the study. The presenter reported serving as an investigator for and consultant to Novartis and 16 other pharmaceutical companies.
NOACs show benefit in calciphylaxis
VIENNA – The novel oral anticoagulants may provide effective adjunctive therapy in patients with calciphylaxis, Brian J. King, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
He presented a retrospective case series of 16 patients with a confirmed diagnosis of calciphylaxis who were treated with NOACs at the Mayo Clinic in Rochester, Minn., where he is a dermatology resident. The results were impressive, particularly given that the estimated 1-year survival following diagnosis of calciphylaxis is only 45%.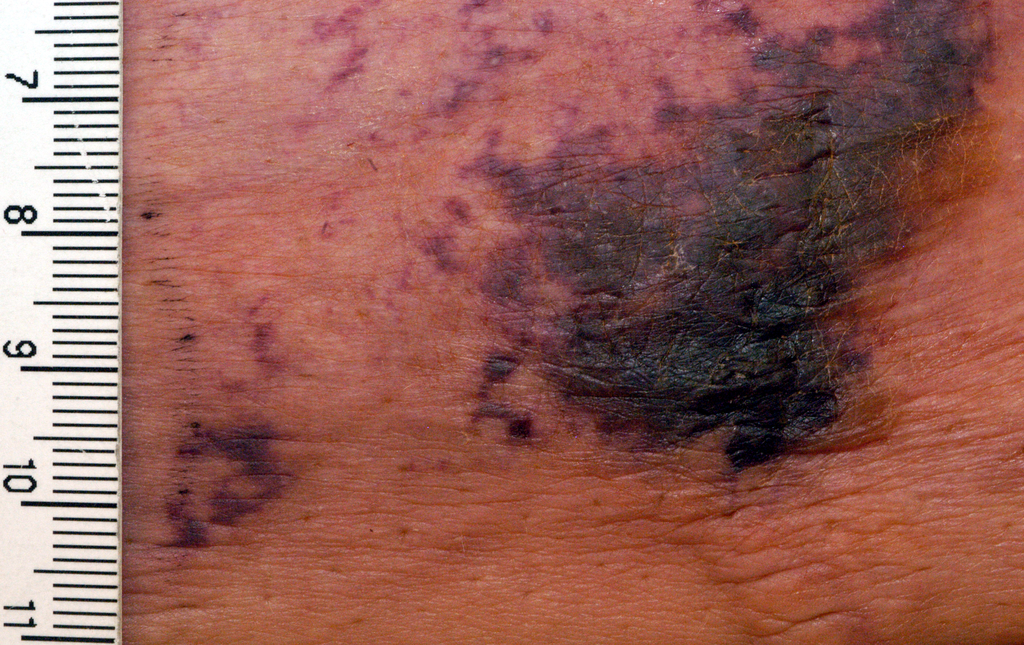
At a mean followup of 418 days, 9 of 16 patients were still alive. More remarkably, five of those nine experienced complete resolution of their clinical lesions and remained alive at a mean followup of 775 days.
Calciphylaxis is a cutaneous manifestation of arteriolar thrombosis. It is classically associated with end-stage renal disease, hyperparathyroidism, a variety of hypercoagulable states, diabetes, and/or obesity. Fifteen of the 16 patients in the Mayo series were women. Fourteen patients had proximal involvement. The lesions occurred most often in fatty tissue on the hips, abdomen, thighs, breasts, and buttocks.
“It’s important to know that this is a deep, incredibly painful process and should not be confused with superficial crusted ulcerations,” Dr. King said.
A variety of treatments have been utilized for calciphylaxis, including sodium thiosulfate, debridement, advanced wound care, hyperbaric oxygen, and parathyroidectomy. But they are often ineffective.
Why not simply use warfarin instead of a costlier NOAC in addressing the problem? Because warfarin has actually been implicated as a cause of the vascular calcification that leads to thrombosis of dermal and pannicular arterioles. Indeed, 12 of the 16 patients in this series were on warfarin at the time of diagnosis of calciphylaxis, either for deep venous thrombosis, pulmonary embolism, or stroke prevention in atrial fibrillation. All were transitioned to a NOAC.
One group of Belgian investigators has provided evidence that strongly suggests the mechanism by which warfarin causes vascular calcification is via inhibition of vitamin K-dependent activation of matrix GLA 1, an enzyme which prevents calcification of vascular endothelial cells (BMC Nephrol. 2014 Sep 4;15:145).
“It is possible and even likely that the vessel calcification we see in patients on warfarin predisposes to thrombosis,” according to Dr. King.
The pathologic diagnostic criteria for calciphylaxis utilized at the Mayo Clinic require skin biopsy evidence of medial calcification and intimal fibroplasia of pannicular arterioles with cutaneous necrosis. Extravascular calcium deposition or thrombosis of pannicular or dermal arterioles is also typically present.
The major clinical criteria are necrotic cutaneous ulcers over indurated plaques, or indurated plaques without ulceration in adipose-rich tissue. The minor criteria are livedo racemosa, hemorrhagic bullae, or hemorrhagic plaques.
Asked if the NOACs can be used interchangeably for treatment of calciphylaxis, Dr. King said the direct factor Xa inhibitor apixaban is the NOAC of choice for this condition at the Mayo Clinic because unlike rivaroxaban (Xarelto) it doesn’t require dosing adjustment in the setting of renal impairment, which is extremely common in patients with calciphylaxis. The direct thrombin inhibitor dabigatran (Pradaxa) is contraindicated in chronic renal failure. Edoxaban (Savaysa) is not on the Mayo Clinic’s formulary, but it is contraindicated in patients with a creatinine clearance of 95 mL/min or more.
Dr. King said he and his coinvestigators recognize that a retrospective case series such as this must be considered hypothesis-generating and nondefinitive. They have already begun a larger prospective comparative outcomes study.
Dr. King reported having no financial conflicts of interest.
VIENNA – The novel oral anticoagulants may provide effective adjunctive therapy in patients with calciphylaxis, Brian J. King, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
He presented a retrospective case series of 16 patients with a confirmed diagnosis of calciphylaxis who were treated with NOACs at the Mayo Clinic in Rochester, Minn., where he is a dermatology resident. The results were impressive, particularly given that the estimated 1-year survival following diagnosis of calciphylaxis is only 45%.
At a mean followup of 418 days, 9 of 16 patients were still alive. More remarkably, five of those nine experienced complete resolution of their clinical lesions and remained alive at a mean followup of 775 days.
Calciphylaxis is a cutaneous manifestation of arteriolar thrombosis. It is classically associated with end-stage renal disease, hyperparathyroidism, a variety of hypercoagulable states, diabetes, and/or obesity. Fifteen of the 16 patients in the Mayo series were women. Fourteen patients had proximal involvement. The lesions occurred most often in fatty tissue on the hips, abdomen, thighs, breasts, and buttocks.
“It’s important to know that this is a deep, incredibly painful process and should not be confused with superficial crusted ulcerations,” Dr. King said.
A variety of treatments have been utilized for calciphylaxis, including sodium thiosulfate, debridement, advanced wound care, hyperbaric oxygen, and parathyroidectomy. But they are often ineffective.
Why not simply use warfarin instead of a costlier NOAC in addressing the problem? Because warfarin has actually been implicated as a cause of the vascular calcification that leads to thrombosis of dermal and pannicular arterioles. Indeed, 12 of the 16 patients in this series were on warfarin at the time of diagnosis of calciphylaxis, either for deep venous thrombosis, pulmonary embolism, or stroke prevention in atrial fibrillation. All were transitioned to a NOAC.
One group of Belgian investigators has provided evidence that strongly suggests the mechanism by which warfarin causes vascular calcification is via inhibition of vitamin K-dependent activation of matrix GLA 1, an enzyme which prevents calcification of vascular endothelial cells (BMC Nephrol. 2014 Sep 4;15:145).
“It is possible and even likely that the vessel calcification we see in patients on warfarin predisposes to thrombosis,” according to Dr. King.
The pathologic diagnostic criteria for calciphylaxis utilized at the Mayo Clinic require skin biopsy evidence of medial calcification and intimal fibroplasia of pannicular arterioles with cutaneous necrosis. Extravascular calcium deposition or thrombosis of pannicular or dermal arterioles is also typically present.
The major clinical criteria are necrotic cutaneous ulcers over indurated plaques, or indurated plaques without ulceration in adipose-rich tissue. The minor criteria are livedo racemosa, hemorrhagic bullae, or hemorrhagic plaques.
Asked if the NOACs can be used interchangeably for treatment of calciphylaxis, Dr. King said the direct factor Xa inhibitor apixaban is the NOAC of choice for this condition at the Mayo Clinic because unlike rivaroxaban (Xarelto) it doesn’t require dosing adjustment in the setting of renal impairment, which is extremely common in patients with calciphylaxis. The direct thrombin inhibitor dabigatran (Pradaxa) is contraindicated in chronic renal failure. Edoxaban (Savaysa) is not on the Mayo Clinic’s formulary, but it is contraindicated in patients with a creatinine clearance of 95 mL/min or more.
Dr. King said he and his coinvestigators recognize that a retrospective case series such as this must be considered hypothesis-generating and nondefinitive. They have already begun a larger prospective comparative outcomes study.
Dr. King reported having no financial conflicts of interest.
VIENNA – The novel oral anticoagulants may provide effective adjunctive therapy in patients with calciphylaxis, Brian J. King, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
He presented a retrospective case series of 16 patients with a confirmed diagnosis of calciphylaxis who were treated with NOACs at the Mayo Clinic in Rochester, Minn., where he is a dermatology resident. The results were impressive, particularly given that the estimated 1-year survival following diagnosis of calciphylaxis is only 45%.
At a mean followup of 418 days, 9 of 16 patients were still alive. More remarkably, five of those nine experienced complete resolution of their clinical lesions and remained alive at a mean followup of 775 days.
Calciphylaxis is a cutaneous manifestation of arteriolar thrombosis. It is classically associated with end-stage renal disease, hyperparathyroidism, a variety of hypercoagulable states, diabetes, and/or obesity. Fifteen of the 16 patients in the Mayo series were women. Fourteen patients had proximal involvement. The lesions occurred most often in fatty tissue on the hips, abdomen, thighs, breasts, and buttocks.
“It’s important to know that this is a deep, incredibly painful process and should not be confused with superficial crusted ulcerations,” Dr. King said.
A variety of treatments have been utilized for calciphylaxis, including sodium thiosulfate, debridement, advanced wound care, hyperbaric oxygen, and parathyroidectomy. But they are often ineffective.
Why not simply use warfarin instead of a costlier NOAC in addressing the problem? Because warfarin has actually been implicated as a cause of the vascular calcification that leads to thrombosis of dermal and pannicular arterioles. Indeed, 12 of the 16 patients in this series were on warfarin at the time of diagnosis of calciphylaxis, either for deep venous thrombosis, pulmonary embolism, or stroke prevention in atrial fibrillation. All were transitioned to a NOAC.
One group of Belgian investigators has provided evidence that strongly suggests the mechanism by which warfarin causes vascular calcification is via inhibition of vitamin K-dependent activation of matrix GLA 1, an enzyme which prevents calcification of vascular endothelial cells (BMC Nephrol. 2014 Sep 4;15:145).
“It is possible and even likely that the vessel calcification we see in patients on warfarin predisposes to thrombosis,” according to Dr. King.
The pathologic diagnostic criteria for calciphylaxis utilized at the Mayo Clinic require skin biopsy evidence of medial calcification and intimal fibroplasia of pannicular arterioles with cutaneous necrosis. Extravascular calcium deposition or thrombosis of pannicular or dermal arterioles is also typically present.
The major clinical criteria are necrotic cutaneous ulcers over indurated plaques, or indurated plaques without ulceration in adipose-rich tissue. The minor criteria are livedo racemosa, hemorrhagic bullae, or hemorrhagic plaques.
Asked if the NOACs can be used interchangeably for treatment of calciphylaxis, Dr. King said the direct factor Xa inhibitor apixaban is the NOAC of choice for this condition at the Mayo Clinic because unlike rivaroxaban (Xarelto) it doesn’t require dosing adjustment in the setting of renal impairment, which is extremely common in patients with calciphylaxis. The direct thrombin inhibitor dabigatran (Pradaxa) is contraindicated in chronic renal failure. Edoxaban (Savaysa) is not on the Mayo Clinic’s formulary, but it is contraindicated in patients with a creatinine clearance of 95 mL/min or more.
Dr. King said he and his coinvestigators recognize that a retrospective case series such as this must be considered hypothesis-generating and nondefinitive. They have already begun a larger prospective comparative outcomes study.
Dr. King reported having no financial conflicts of interest.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Nine of 16 patients with calciphylaxis who were placed on adjunctive therapy with a novel oral anticoagulant responded with significant improvement in their cutaneous disease, four experienced disease stabilization, and three had progressive disease.
Data source: This was a retrospective case study of 16 patients with biopsy-confirmed calciphylaxis.
Disclosures: The study presenter reported having no financial conflicts of interest.
Atopic dermatitis prevention strategies under study
VIENNA – Diverse strategies aimed at preventing childhood atopic dermatitis (AD) now under study include installation of home water softeners, daily use of emollients starting at birth, and maternal consumption of probiotics beginning late in pregnancy, Carsten Flohr, PhD, said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
To date there is no effective method for preventing AD. Preventive strategies are needed sorely because the prevalence of pediatric AD worldwide is expected to increase substantially. It appears to have stabilized at roughly 20% in many affluent countries, but the global burden of the disease will climb as low-income countries – where AD is historically uncommon – become more developed and urbanized. This trend has been well documented via the International Study of Asthma and Allergies in Childhood (ISAAC), which in several phases has studied nearly 2 million children in more than 100 countries, noted Dr. Flohr of St. John’s Institute of Dermatology at King’s College London.

Dr. Flohr and coinvestigators in the Enquiring About Tolerance (EAT) study recently documented a significant association between water hardness and the risk of infant-onset AD. The investigators took advantage of the considerable variation in the amount of bedrock limestone across England, which enabled them to study the relationship between domestic water calcium carbonate concentrations and the presence of AD in 1,303 babies at 3 monthd of age drawn from the general population across the country. Filaggrin skin barrier gene mutation status was determined in all infants.
Infants whose water supply contained a calcium carbonate level above the median value were at an adjusted 46% greater risk of having visible AD at age 3 months than those whose household water calcium carbonate level was below the median. The AD risk rose by 1% for each 1 mg/L increase in calcium carbonate concentration above the median. This increased risk was confined to infants with a filaggrin skin barrier gene mutation; hard water didn’t increase early AD risk in children with the normal, wild-type version of the filaggrin gene (J Allergy Clin Immunol. 2016 Aug;138[2]:509-16).
As a result of these findings, a UK prevention trial is underway in which home water softeners are provided to families at high risk of having a baby with AD in water districts with high calcium carbonate concentrations. An earlier UK study found that installation of home water softeners didn’t reduce AD severity in children with established disease (PLoS Med. 2011 Feb 15;8[2]:e1000395), but disease prevention may be another story.
The role of the gut microbiota in development of childhood AD is an active area of investigation. Dr. Flohr said “there is a signal” that maternal intake of probiotics including lactobacilli and bifidobacteria in the third trimester and postnatally may reduce a child’s risk of developing AD by encouraging establishment of a more diverse gut microflora. He cited a meta-analysis of 14 published studies of probiotics which provided evidence of a 21% reduction in the incidence of AD in young children (Epidemiology 2012 May;23[3]:402-14). The studies have methodologic shortcomings, so multiple research groups are continuing to pursue the signal of an AD preventive effect.
Dr. Flor reported having no financial conflicts of interest regarding his presentation.
VIENNA – Diverse strategies aimed at preventing childhood atopic dermatitis (AD) now under study include installation of home water softeners, daily use of emollients starting at birth, and maternal consumption of probiotics beginning late in pregnancy, Carsten Flohr, PhD, said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
To date there is no effective method for preventing AD. Preventive strategies are needed sorely because the prevalence of pediatric AD worldwide is expected to increase substantially. It appears to have stabilized at roughly 20% in many affluent countries, but the global burden of the disease will climb as low-income countries – where AD is historically uncommon – become more developed and urbanized. This trend has been well documented via the International Study of Asthma and Allergies in Childhood (ISAAC), which in several phases has studied nearly 2 million children in more than 100 countries, noted Dr. Flohr of St. John’s Institute of Dermatology at King’s College London.
Dr. Flohr and coinvestigators in the Enquiring About Tolerance (EAT) study recently documented a significant association between water hardness and the risk of infant-onset AD. The investigators took advantage of the considerable variation in the amount of bedrock limestone across England, which enabled them to study the relationship between domestic water calcium carbonate concentrations and the presence of AD in 1,303 babies at 3 monthd of age drawn from the general population across the country. Filaggrin skin barrier gene mutation status was determined in all infants.
Infants whose water supply contained a calcium carbonate level above the median value were at an adjusted 46% greater risk of having visible AD at age 3 months than those whose household water calcium carbonate level was below the median. The AD risk rose by 1% for each 1 mg/L increase in calcium carbonate concentration above the median. This increased risk was confined to infants with a filaggrin skin barrier gene mutation; hard water didn’t increase early AD risk in children with the normal, wild-type version of the filaggrin gene (J Allergy Clin Immunol. 2016 Aug;138[2]:509-16).
As a result of these findings, a UK prevention trial is underway in which home water softeners are provided to families at high risk of having a baby with AD in water districts with high calcium carbonate concentrations. An earlier UK study found that installation of home water softeners didn’t reduce AD severity in children with established disease (PLoS Med. 2011 Feb 15;8[2]:e1000395), but disease prevention may be another story.
The role of the gut microbiota in development of childhood AD is an active area of investigation. Dr. Flohr said “there is a signal” that maternal intake of probiotics including lactobacilli and bifidobacteria in the third trimester and postnatally may reduce a child’s risk of developing AD by encouraging establishment of a more diverse gut microflora. He cited a meta-analysis of 14 published studies of probiotics which provided evidence of a 21% reduction in the incidence of AD in young children (Epidemiology 2012 May;23[3]:402-14). The studies have methodologic shortcomings, so multiple research groups are continuing to pursue the signal of an AD preventive effect.
Dr. Flor reported having no financial conflicts of interest regarding his presentation.
VIENNA – Diverse strategies aimed at preventing childhood atopic dermatitis (AD) now under study include installation of home water softeners, daily use of emollients starting at birth, and maternal consumption of probiotics beginning late in pregnancy, Carsten Flohr, PhD, said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
To date there is no effective method for preventing AD. Preventive strategies are needed sorely because the prevalence of pediatric AD worldwide is expected to increase substantially. It appears to have stabilized at roughly 20% in many affluent countries, but the global burden of the disease will climb as low-income countries – where AD is historically uncommon – become more developed and urbanized. This trend has been well documented via the International Study of Asthma and Allergies in Childhood (ISAAC), which in several phases has studied nearly 2 million children in more than 100 countries, noted Dr. Flohr of St. John’s Institute of Dermatology at King’s College London.
Dr. Flohr and coinvestigators in the Enquiring About Tolerance (EAT) study recently documented a significant association between water hardness and the risk of infant-onset AD. The investigators took advantage of the considerable variation in the amount of bedrock limestone across England, which enabled them to study the relationship between domestic water calcium carbonate concentrations and the presence of AD in 1,303 babies at 3 monthd of age drawn from the general population across the country. Filaggrin skin barrier gene mutation status was determined in all infants.
Infants whose water supply contained a calcium carbonate level above the median value were at an adjusted 46% greater risk of having visible AD at age 3 months than those whose household water calcium carbonate level was below the median. The AD risk rose by 1% for each 1 mg/L increase in calcium carbonate concentration above the median. This increased risk was confined to infants with a filaggrin skin barrier gene mutation; hard water didn’t increase early AD risk in children with the normal, wild-type version of the filaggrin gene (J Allergy Clin Immunol. 2016 Aug;138[2]:509-16).
As a result of these findings, a UK prevention trial is underway in which home water softeners are provided to families at high risk of having a baby with AD in water districts with high calcium carbonate concentrations. An earlier UK study found that installation of home water softeners didn’t reduce AD severity in children with established disease (PLoS Med. 2011 Feb 15;8[2]:e1000395), but disease prevention may be another story.
The role of the gut microbiota in development of childhood AD is an active area of investigation. Dr. Flohr said “there is a signal” that maternal intake of probiotics including lactobacilli and bifidobacteria in the third trimester and postnatally may reduce a child’s risk of developing AD by encouraging establishment of a more diverse gut microflora. He cited a meta-analysis of 14 published studies of probiotics which provided evidence of a 21% reduction in the incidence of AD in young children (Epidemiology 2012 May;23[3]:402-14). The studies have methodologic shortcomings, so multiple research groups are continuing to pursue the signal of an AD preventive effect.
Dr. Flor reported having no financial conflicts of interest regarding his presentation.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Lebrikizumab opens new door in atopic dermatitis therapy
VIENNA – The success of the interleukin-13 blocker lebrikizumab in the TREBLE trial provides a new avenue in the treatment of moderate-to-severe atopic dermatitis.
“This is another promising molecule in atopic dermatitis,” Eric L. Simpson, MD, declared in presenting the phase II TREBLE data at the annual congress of the European Academy of Dermatology and Venereology.

Interleukin-13 is known to be an especially potent promoter of type 1, IgE-mediated inflammation. For this reason, lebrikizumab is also under investigation in the treatment of severe asthma, where large phase III trials have been completed, as well as in idiopathic pulmonary fibrosis.
In mouse models of AD, topical anti-IL-13 therapy markedly reduces skin inflammation. This observation helped provide the rationale for investigating lebrikizumab as a novel therapy for AD.
TREBLE was a double-blind, dose-ranging study involving 209 adults with moderate-to-severe AD despite intensive topical corticosteroid therapy. Indeed, enrollment was restricted to patients who still had moderate or severe disease after 2 weeks of triamcinolone 0.1% cream BID. Continuation of that twice-daily topical steroid regimen was mandatory for the full 12-week study period that followed. Patients were randomized 1:1:1:1 to triamcinolone 0.1% BID plus either a single 125-mg subcutaneous dose of lebrikizumab at week 0, a single 250-mg dose of the biologic, 125 mg every 4 weeks, or placebo injections.
At baseline, after their 2 weeks of topical steroid monotherapy, patients had a median Eczema Area and Severity Index (EASI) score of 22 and a median SCORing Atopic Dermatitis (SCORAD) of about 56. They averaged 36 years of age. Slightly over 40% of their body surface area was affected.
The primary endpoint in TREBLE was the percentage of patients who achieved at least a 50% reduction from baseline on the EASI, or EASI 50. A dose-response effect was apparent: the EASI 50 rate was 62.3% in patients on placebo plus daily topical steroids, 69.2% with a single 125-mg dose of lebrikizumab, 69.8% with a single 250-mg dose, and 82.4% with 125 mg of lebrikizumab at weeks 0, 4, 8, and 12. Only the group with monthly dosing of the biologic plus daily triamcinolone 0.1% BID had an EASI 50 response rate significantly better than the controls on placebo plus topical steroid therapy.
A key secondary endpoint was the SCORAD 50 response. The rate was 26.4% in the control group, 34.6% in patients who received one 125-mg dose of lebrikizumab, 47.2% in those who got a single 250-mg dose, and 51% with monthly dosing. The SCORAD 50 rate was significantly greater than placebo in the latter two lebrikizumab arms.
The EASI 75 rate was significantly greater than placebo plus triamcinolone only in the group on monthly lebrikizumab plus topical steroids.
The EASI 50 and 75 response rates in the group on 125 mg of lebrikizumab every 4 weeks was still climbing with no plateau evident when the trial ended at 12 weeks. This suggests greater benefit might be achieved with longer treatment duration and/or a higher dosage, Dr. Simpson observed.
Turning to safety issues, he pointed out that the total number of adverse events and serious adverse events were similar across all four treatment arms. Herpes infections occurred in 2%-6% of patients who received lebrikizumab but in no controls. There was also a nonsignificant trend for more cases of conjunctivitis in the groups that received the biologic.
Audience member Andrew Blauvelt, MD, rose to take issue with the study design.
“This study is almost an advertisement for not using topical steroids in a biologic trial for atopic dermatitis. I think it just confuses everything. It especially doesn’t make sense when one of the inclusion criteria is inadequate control with topical steroids. Shouldn’t we be doing monotherapy trials in the new era of biologics for atopic dermatitis?” commented Dr. Blauvelt, president of the Oregon Medical Research Center in Portland.
Dr. Simpson replied that when TREBLE was designed, the thinking was that in real-world clinical practice, physicians often prescribe a biologic for AD on top of topical therapy. The goal was to conduct a trial that mimics that situation. That being said, he agreed with Dr. Blauvelt’s critique.
“I think we’re realizing that topical steroids have a very strong effect, especially if you use them all the way through a trial or ad lib,” he said. “Topical steroids bring too many confounders. If you can remove potent topical steroid therapy I think you’re going to have a clearer result.”
“We’re not where psoriasis is,” Dr. Simpson added. “We don’t have any standardized methodologies in atopic dermatitis clinical trials yet, but I think we’re getting there.”
The TREBLE trial was sponsored by Genentech/Roche. Dr. Simpson reported serving as an investigator for and consultant to that organization and numerous other pharmaceutical companies.
VIENNA – The success of the interleukin-13 blocker lebrikizumab in the TREBLE trial provides a new avenue in the treatment of moderate-to-severe atopic dermatitis.
“This is another promising molecule in atopic dermatitis,” Eric L. Simpson, MD, declared in presenting the phase II TREBLE data at the annual congress of the European Academy of Dermatology and Venereology.
Interleukin-13 is known to be an especially potent promoter of type 1, IgE-mediated inflammation. For this reason, lebrikizumab is also under investigation in the treatment of severe asthma, where large phase III trials have been completed, as well as in idiopathic pulmonary fibrosis.
In mouse models of AD, topical anti-IL-13 therapy markedly reduces skin inflammation. This observation helped provide the rationale for investigating lebrikizumab as a novel therapy for AD.
TREBLE was a double-blind, dose-ranging study involving 209 adults with moderate-to-severe AD despite intensive topical corticosteroid therapy. Indeed, enrollment was restricted to patients who still had moderate or severe disease after 2 weeks of triamcinolone 0.1% cream BID. Continuation of that twice-daily topical steroid regimen was mandatory for the full 12-week study period that followed. Patients were randomized 1:1:1:1 to triamcinolone 0.1% BID plus either a single 125-mg subcutaneous dose of lebrikizumab at week 0, a single 250-mg dose of the biologic, 125 mg every 4 weeks, or placebo injections.
At baseline, after their 2 weeks of topical steroid monotherapy, patients had a median Eczema Area and Severity Index (EASI) score of 22 and a median SCORing Atopic Dermatitis (SCORAD) of about 56. They averaged 36 years of age. Slightly over 40% of their body surface area was affected.
The primary endpoint in TREBLE was the percentage of patients who achieved at least a 50% reduction from baseline on the EASI, or EASI 50. A dose-response effect was apparent: the EASI 50 rate was 62.3% in patients on placebo plus daily topical steroids, 69.2% with a single 125-mg dose of lebrikizumab, 69.8% with a single 250-mg dose, and 82.4% with 125 mg of lebrikizumab at weeks 0, 4, 8, and 12. Only the group with monthly dosing of the biologic plus daily triamcinolone 0.1% BID had an EASI 50 response rate significantly better than the controls on placebo plus topical steroid therapy.
A key secondary endpoint was the SCORAD 50 response. The rate was 26.4% in the control group, 34.6% in patients who received one 125-mg dose of lebrikizumab, 47.2% in those who got a single 250-mg dose, and 51% with monthly dosing. The SCORAD 50 rate was significantly greater than placebo in the latter two lebrikizumab arms.
The EASI 75 rate was significantly greater than placebo plus triamcinolone only in the group on monthly lebrikizumab plus topical steroids.
The EASI 50 and 75 response rates in the group on 125 mg of lebrikizumab every 4 weeks was still climbing with no plateau evident when the trial ended at 12 weeks. This suggests greater benefit might be achieved with longer treatment duration and/or a higher dosage, Dr. Simpson observed.
Turning to safety issues, he pointed out that the total number of adverse events and serious adverse events were similar across all four treatment arms. Herpes infections occurred in 2%-6% of patients who received lebrikizumab but in no controls. There was also a nonsignificant trend for more cases of conjunctivitis in the groups that received the biologic.
Audience member Andrew Blauvelt, MD, rose to take issue with the study design.
“This study is almost an advertisement for not using topical steroids in a biologic trial for atopic dermatitis. I think it just confuses everything. It especially doesn’t make sense when one of the inclusion criteria is inadequate control with topical steroids. Shouldn’t we be doing monotherapy trials in the new era of biologics for atopic dermatitis?” commented Dr. Blauvelt, president of the Oregon Medical Research Center in Portland.
Dr. Simpson replied that when TREBLE was designed, the thinking was that in real-world clinical practice, physicians often prescribe a biologic for AD on top of topical therapy. The goal was to conduct a trial that mimics that situation. That being said, he agreed with Dr. Blauvelt’s critique.
“I think we’re realizing that topical steroids have a very strong effect, especially if you use them all the way through a trial or ad lib,” he said. “Topical steroids bring too many confounders. If you can remove potent topical steroid therapy I think you’re going to have a clearer result.”
“We’re not where psoriasis is,” Dr. Simpson added. “We don’t have any standardized methodologies in atopic dermatitis clinical trials yet, but I think we’re getting there.”
The TREBLE trial was sponsored by Genentech/Roche. Dr. Simpson reported serving as an investigator for and consultant to that organization and numerous other pharmaceutical companies.
VIENNA – The success of the interleukin-13 blocker lebrikizumab in the TREBLE trial provides a new avenue in the treatment of moderate-to-severe atopic dermatitis.
“This is another promising molecule in atopic dermatitis,” Eric L. Simpson, MD, declared in presenting the phase II TREBLE data at the annual congress of the European Academy of Dermatology and Venereology.
Interleukin-13 is known to be an especially potent promoter of type 1, IgE-mediated inflammation. For this reason, lebrikizumab is also under investigation in the treatment of severe asthma, where large phase III trials have been completed, as well as in idiopathic pulmonary fibrosis.
In mouse models of AD, topical anti-IL-13 therapy markedly reduces skin inflammation. This observation helped provide the rationale for investigating lebrikizumab as a novel therapy for AD.
TREBLE was a double-blind, dose-ranging study involving 209 adults with moderate-to-severe AD despite intensive topical corticosteroid therapy. Indeed, enrollment was restricted to patients who still had moderate or severe disease after 2 weeks of triamcinolone 0.1% cream BID. Continuation of that twice-daily topical steroid regimen was mandatory for the full 12-week study period that followed. Patients were randomized 1:1:1:1 to triamcinolone 0.1% BID plus either a single 125-mg subcutaneous dose of lebrikizumab at week 0, a single 250-mg dose of the biologic, 125 mg every 4 weeks, or placebo injections.
At baseline, after their 2 weeks of topical steroid monotherapy, patients had a median Eczema Area and Severity Index (EASI) score of 22 and a median SCORing Atopic Dermatitis (SCORAD) of about 56. They averaged 36 years of age. Slightly over 40% of their body surface area was affected.
The primary endpoint in TREBLE was the percentage of patients who achieved at least a 50% reduction from baseline on the EASI, or EASI 50. A dose-response effect was apparent: the EASI 50 rate was 62.3% in patients on placebo plus daily topical steroids, 69.2% with a single 125-mg dose of lebrikizumab, 69.8% with a single 250-mg dose, and 82.4% with 125 mg of lebrikizumab at weeks 0, 4, 8, and 12. Only the group with monthly dosing of the biologic plus daily triamcinolone 0.1% BID had an EASI 50 response rate significantly better than the controls on placebo plus topical steroid therapy.
A key secondary endpoint was the SCORAD 50 response. The rate was 26.4% in the control group, 34.6% in patients who received one 125-mg dose of lebrikizumab, 47.2% in those who got a single 250-mg dose, and 51% with monthly dosing. The SCORAD 50 rate was significantly greater than placebo in the latter two lebrikizumab arms.
The EASI 75 rate was significantly greater than placebo plus triamcinolone only in the group on monthly lebrikizumab plus topical steroids.
The EASI 50 and 75 response rates in the group on 125 mg of lebrikizumab every 4 weeks was still climbing with no plateau evident when the trial ended at 12 weeks. This suggests greater benefit might be achieved with longer treatment duration and/or a higher dosage, Dr. Simpson observed.
Turning to safety issues, he pointed out that the total number of adverse events and serious adverse events were similar across all four treatment arms. Herpes infections occurred in 2%-6% of patients who received lebrikizumab but in no controls. There was also a nonsignificant trend for more cases of conjunctivitis in the groups that received the biologic.
Audience member Andrew Blauvelt, MD, rose to take issue with the study design.
“This study is almost an advertisement for not using topical steroids in a biologic trial for atopic dermatitis. I think it just confuses everything. It especially doesn’t make sense when one of the inclusion criteria is inadequate control with topical steroids. Shouldn’t we be doing monotherapy trials in the new era of biologics for atopic dermatitis?” commented Dr. Blauvelt, president of the Oregon Medical Research Center in Portland.
Dr. Simpson replied that when TREBLE was designed, the thinking was that in real-world clinical practice, physicians often prescribe a biologic for AD on top of topical therapy. The goal was to conduct a trial that mimics that situation. That being said, he agreed with Dr. Blauvelt’s critique.
“I think we’re realizing that topical steroids have a very strong effect, especially if you use them all the way through a trial or ad lib,” he said. “Topical steroids bring too many confounders. If you can remove potent topical steroid therapy I think you’re going to have a clearer result.”
“We’re not where psoriasis is,” Dr. Simpson added. “We don’t have any standardized methodologies in atopic dermatitis clinical trials yet, but I think we’re getting there.”
The TREBLE trial was sponsored by Genentech/Roche. Dr. Simpson reported serving as an investigator for and consultant to that organization and numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Of atopic dermatitis patients randomized to 125 mg of lebrikizumab every 4 weeks on top of twice-daily potent topical steroids, 82% experienced at least a 50% reduction in Eczema Area and Severity Index scores at 12 weeks, compared with 62% on the same topical steroid regimen plus placebo injections.
Data source: The TREBLE trial was a phase II, double-blind, placebo-controlled, 12-week study including 209 adults with moderate-to-severe atopic dermatitis inadequately controlled by intensive topical steroid therapy.
Disclosures: The study was sponsored by Genentech/Roche. The presenter reported serving as an investigator for and consultant to that organization and numerous other pharmaceutical companies.
Topical anticholinergic for axillary hyperhidrosis hits marks in phase III
VIENNA – An investigational topical cholinergic receptor antagonist known as DRM04 achieved its efficacy and safety endpoints for the treatment of primary axillary hyperhidrosis in the pivotal phase III ATMOS-1 and ATMOS-2 trials.
“We haven’t had any good new treatment options for patients with hyperhidrosis for a long time,” Dr. David M. Pariser said, in presenting the pivotal trial outcomes data at the annual congress of the European Academy of Dermatology and Venereology.

ATMOS-1 and ATMOS-2 were identically designed 4-week, double-blind studies involving 697 patients with excessive underarm sweating who were randomized 2:1 to once daily use of DRM04 3.75% wipes or a vehicle control. The patients averaged 33 years of age, although as Dr. Pariser noted, primary axillary hyperhidrosis often begins in adolescence and patients as young as age 9 were enrolled. The majority of participants were female. Roughly two-thirds of subjects were grade 3 on the 4-point Hyperhidrosis Disease Severity Scale. The rest were grade 4.
The coprimary endpoints in ATMOS-1 and ATMOS-2 were a 4-point or greater improvement on the Axillary Sweating Daily Diary (ASDD) between baseline and week 4, and the absolute change from baseline in axillary sweat production measured gravimetrically.
The ASDD is a new patient-reported outcome measure developed specifically for the ATMOS trials. At baseline, participants scored a mean of 7.2 points on the 0-10 scale. A 4-point or greater improvement was seen at week 4 in 52.8% of ATMOS-1 participants on DRM04, compared with 28.3% of controls. In ATMOS-2, the spread was 66.1% vs. 26.9%.
Mean baseline sweat production was roughly 175 mg/5 min per armpit, a prodigious rate given that 50 mg/5 min is considered excessive. In ATMOS-1, the rate dropped by an adjusted average of 96.2 mg/5 min per armpit with active therapy, compared with 90.6 mg/5 min in the control group. In ATMOS-2, the DRM04 users had a mean drop in sweat production of 110.3 mg/5 min, compared with a reduction of 92.2 mg/5 min in the control group. Both of these differences were statistically significant and clinically meaningful, Dr. Pariser said.
The secondary endpoint in the studies was change in the Dermatology Life Quality Index from baseline to week 4. The improvement in DRM04 users averaged 8.1 points in ATMOS-1 and 8.6 points in ATMOS-2, both significantly greater than the 4.3- and 5.0-point improvements in the control arms.
In ATMOS-1, 9.2% of patients in the DRM04 arm dropped out of the study, in many cases because of anticholinergic side effects, the most common of which included dry mouth, dry eyes, urinary hesitation or less frequently retention, and constipation. These were mostly mild to moderate in nature and were generally responsive to temporary treatment discontinuation, which the study protocol allowed. The dropout rate in the DRM04 arm of ATMOS-2 was 6.8%.
Seven percent of subjects in the DRM04 study arms experienced mydriasis, most often unilaterally. Dr. Pariser said this might be due to patients touching an eye while they still had DRM04 on their hands, a problem that can be readily addressed in the medication use instructions.
The dropout rates in the control groups were 2.6% and 5%.
More than 80% of ATMOS-1 and ATMOS-2 participants enrolled in ARIDO, the 48-week, open label, phase III extension study of DRM04. Dermira, which is developing DRM04, has announced it plans to file for marketing approval by the Food and Drug Administration in the second half of 2017.
Dr. Pariser reported serving as an investigator for and consultant to Dermira, Brickell Biotech, and TheraVida.
VIENNA – An investigational topical cholinergic receptor antagonist known as DRM04 achieved its efficacy and safety endpoints for the treatment of primary axillary hyperhidrosis in the pivotal phase III ATMOS-1 and ATMOS-2 trials.
“We haven’t had any good new treatment options for patients with hyperhidrosis for a long time,” Dr. David M. Pariser said, in presenting the pivotal trial outcomes data at the annual congress of the European Academy of Dermatology and Venereology.
ATMOS-1 and ATMOS-2 were identically designed 4-week, double-blind studies involving 697 patients with excessive underarm sweating who were randomized 2:1 to once daily use of DRM04 3.75% wipes or a vehicle control. The patients averaged 33 years of age, although as Dr. Pariser noted, primary axillary hyperhidrosis often begins in adolescence and patients as young as age 9 were enrolled. The majority of participants were female. Roughly two-thirds of subjects were grade 3 on the 4-point Hyperhidrosis Disease Severity Scale. The rest were grade 4.
The coprimary endpoints in ATMOS-1 and ATMOS-2 were a 4-point or greater improvement on the Axillary Sweating Daily Diary (ASDD) between baseline and week 4, and the absolute change from baseline in axillary sweat production measured gravimetrically.
The ASDD is a new patient-reported outcome measure developed specifically for the ATMOS trials. At baseline, participants scored a mean of 7.2 points on the 0-10 scale. A 4-point or greater improvement was seen at week 4 in 52.8% of ATMOS-1 participants on DRM04, compared with 28.3% of controls. In ATMOS-2, the spread was 66.1% vs. 26.9%.
Mean baseline sweat production was roughly 175 mg/5 min per armpit, a prodigious rate given that 50 mg/5 min is considered excessive. In ATMOS-1, the rate dropped by an adjusted average of 96.2 mg/5 min per armpit with active therapy, compared with 90.6 mg/5 min in the control group. In ATMOS-2, the DRM04 users had a mean drop in sweat production of 110.3 mg/5 min, compared with a reduction of 92.2 mg/5 min in the control group. Both of these differences were statistically significant and clinically meaningful, Dr. Pariser said.
The secondary endpoint in the studies was change in the Dermatology Life Quality Index from baseline to week 4. The improvement in DRM04 users averaged 8.1 points in ATMOS-1 and 8.6 points in ATMOS-2, both significantly greater than the 4.3- and 5.0-point improvements in the control arms.
In ATMOS-1, 9.2% of patients in the DRM04 arm dropped out of the study, in many cases because of anticholinergic side effects, the most common of which included dry mouth, dry eyes, urinary hesitation or less frequently retention, and constipation. These were mostly mild to moderate in nature and were generally responsive to temporary treatment discontinuation, which the study protocol allowed. The dropout rate in the DRM04 arm of ATMOS-2 was 6.8%.
Seven percent of subjects in the DRM04 study arms experienced mydriasis, most often unilaterally. Dr. Pariser said this might be due to patients touching an eye while they still had DRM04 on their hands, a problem that can be readily addressed in the medication use instructions.
The dropout rates in the control groups were 2.6% and 5%.
More than 80% of ATMOS-1 and ATMOS-2 participants enrolled in ARIDO, the 48-week, open label, phase III extension study of DRM04. Dermira, which is developing DRM04, has announced it plans to file for marketing approval by the Food and Drug Administration in the second half of 2017.
Dr. Pariser reported serving as an investigator for and consultant to Dermira, Brickell Biotech, and TheraVida.
VIENNA – An investigational topical cholinergic receptor antagonist known as DRM04 achieved its efficacy and safety endpoints for the treatment of primary axillary hyperhidrosis in the pivotal phase III ATMOS-1 and ATMOS-2 trials.
“We haven’t had any good new treatment options for patients with hyperhidrosis for a long time,” Dr. David M. Pariser said, in presenting the pivotal trial outcomes data at the annual congress of the European Academy of Dermatology and Venereology.
ATMOS-1 and ATMOS-2 were identically designed 4-week, double-blind studies involving 697 patients with excessive underarm sweating who were randomized 2:1 to once daily use of DRM04 3.75% wipes or a vehicle control. The patients averaged 33 years of age, although as Dr. Pariser noted, primary axillary hyperhidrosis often begins in adolescence and patients as young as age 9 were enrolled. The majority of participants were female. Roughly two-thirds of subjects were grade 3 on the 4-point Hyperhidrosis Disease Severity Scale. The rest were grade 4.
The coprimary endpoints in ATMOS-1 and ATMOS-2 were a 4-point or greater improvement on the Axillary Sweating Daily Diary (ASDD) between baseline and week 4, and the absolute change from baseline in axillary sweat production measured gravimetrically.
The ASDD is a new patient-reported outcome measure developed specifically for the ATMOS trials. At baseline, participants scored a mean of 7.2 points on the 0-10 scale. A 4-point or greater improvement was seen at week 4 in 52.8% of ATMOS-1 participants on DRM04, compared with 28.3% of controls. In ATMOS-2, the spread was 66.1% vs. 26.9%.
Mean baseline sweat production was roughly 175 mg/5 min per armpit, a prodigious rate given that 50 mg/5 min is considered excessive. In ATMOS-1, the rate dropped by an adjusted average of 96.2 mg/5 min per armpit with active therapy, compared with 90.6 mg/5 min in the control group. In ATMOS-2, the DRM04 users had a mean drop in sweat production of 110.3 mg/5 min, compared with a reduction of 92.2 mg/5 min in the control group. Both of these differences were statistically significant and clinically meaningful, Dr. Pariser said.
The secondary endpoint in the studies was change in the Dermatology Life Quality Index from baseline to week 4. The improvement in DRM04 users averaged 8.1 points in ATMOS-1 and 8.6 points in ATMOS-2, both significantly greater than the 4.3- and 5.0-point improvements in the control arms.
In ATMOS-1, 9.2% of patients in the DRM04 arm dropped out of the study, in many cases because of anticholinergic side effects, the most common of which included dry mouth, dry eyes, urinary hesitation or less frequently retention, and constipation. These were mostly mild to moderate in nature and were generally responsive to temporary treatment discontinuation, which the study protocol allowed. The dropout rate in the DRM04 arm of ATMOS-2 was 6.8%.
Seven percent of subjects in the DRM04 study arms experienced mydriasis, most often unilaterally. Dr. Pariser said this might be due to patients touching an eye while they still had DRM04 on their hands, a problem that can be readily addressed in the medication use instructions.
The dropout rates in the control groups were 2.6% and 5%.
More than 80% of ATMOS-1 and ATMOS-2 participants enrolled in ARIDO, the 48-week, open label, phase III extension study of DRM04. Dermira, which is developing DRM04, has announced it plans to file for marketing approval by the Food and Drug Administration in the second half of 2017.
Dr. Pariser reported serving as an investigator for and consultant to Dermira, Brickell Biotech, and TheraVida.
AT THE EADV CONGRESS
Key clinical point: A once-daily topical anticholinergic agent called DRM04 achieved positive outcomes for the treatment of primary axillary hyperhidrosis in two pivotal phase III trials.
Major finding: 53% and 66% of subjects on DRM04 in two large studies achieved clinically meaningful improvement in axillary sweating, compared with 28% and 27%, respectively, of controls.
Data source: Based on findings from ATMOS-1 and ATMOS-2, identically designed, 4-week, double-blind, vehicle-controlled clinical trials including a total of 687 patients with primary axillary hyperhidrosis.
Disclosures: The studies were funded by Dermira. The presenter reported serving as an investigator for and consultant to the company.