User login
Shortage reported of antibiotic commonly used for children
The liquid form of the antibiotic amoxicillin often used to treat ear infections and strep throat in children is in short supply, just as Americans head into the season when they use the bacteria-fighting drug the most.
The FDA officially listed the shortage Oct. 28, but pharmacists, hospitals, and a supply tracking database sounded alarms earlier this month.
“The scary part is, we’re coming into the time of the year where you have the greatest need,” independent pharmacy owner Hugh Chancy, PharmD, of Georgia, told NBC News.
Thus far, reports indicate the impact of the shortages is not widespread but does affect some pharmacies, and at least one hospital has published an algorithm for offering treatment alternatives.
CVS told Bloomberg News that some stores are experiencing shortages of certain doses of amoxicillin, but a Walmart spokesperson said its diverse supply chain meant none of its pharmacies were affected.
“Hypothetically, if amoxicillin doesn’t come into stock for some time, then we’re potentially having to use less effective antibiotics with more side effects,” said Ohio pediatrician Sean Gallagher, MD, according to Bloomberg.
The shortage impacts three of the four largest amoxicillin manufacturers worldwide, according to the Center for Infectious Disease Research and Policy (CIDRAP) at the University of Minnesota. The FDA listed the reason for the shortage as “demand increase for drug,” except in the case of manufacturer Sandoz, for which the reason listed read “information pending.”
A company spokesperson told Bloomberg the reasons were complex.
“The combination in rapid succession of the pandemic impact and consequent demand swings, manufacturing capacity constraints, scarcity of raw materials, and the current energy crisis means we face a uniquely difficult situation in the short term,” Sandoz spokesperson Leslie Pott told Bloomberg.
According to Bloomberg, other major manufacturers are still delivering the product, but limiting new orders.
The American Society of Health-System Pharmacists issued an alert for the shortage last week via its real time drug shortage database.
“Amoxicillin comes in many forms – including capsules, powders and chewable tablets – but the most common type children take is the liquid form, which makes up at least 19 products that are part of the” shortage, Becker’s Hospital Review summarized of the database reports.
The pediatric health system Children’s Minnesota told CIDRAP that supplies are low and that alternatives are being prescribed “when appropriate.”
“As a final step, we temporarily discontinued our standard procedure of dispensing the entire bottle of amoxicillin (which comes in multiple sizes),” a spokesperson told CIDRAP. “We are instead mixing and pouring the exact amount for each course of therapy, to eliminate waste.”
The Minnesota pediatric clinic and others are particularly on alert because of the surge nationwide of a respiratory virus that particularly impacts children known as RSV.
“We have certainly observed an increase in recent use most likely correlating with the surge in RSV and other respiratory viruses with concern for superimposed bacterial infection in our critically ill and hospitalized patient population,” Laura Bio, PharmD, a clinical pharmacy specialist at Stanford Medicine Children’s Health told CIDRAP.
A version of this article first appeared on WebMD.com.
The liquid form of the antibiotic amoxicillin often used to treat ear infections and strep throat in children is in short supply, just as Americans head into the season when they use the bacteria-fighting drug the most.
The FDA officially listed the shortage Oct. 28, but pharmacists, hospitals, and a supply tracking database sounded alarms earlier this month.
“The scary part is, we’re coming into the time of the year where you have the greatest need,” independent pharmacy owner Hugh Chancy, PharmD, of Georgia, told NBC News.
Thus far, reports indicate the impact of the shortages is not widespread but does affect some pharmacies, and at least one hospital has published an algorithm for offering treatment alternatives.
CVS told Bloomberg News that some stores are experiencing shortages of certain doses of amoxicillin, but a Walmart spokesperson said its diverse supply chain meant none of its pharmacies were affected.
“Hypothetically, if amoxicillin doesn’t come into stock for some time, then we’re potentially having to use less effective antibiotics with more side effects,” said Ohio pediatrician Sean Gallagher, MD, according to Bloomberg.
The shortage impacts three of the four largest amoxicillin manufacturers worldwide, according to the Center for Infectious Disease Research and Policy (CIDRAP) at the University of Minnesota. The FDA listed the reason for the shortage as “demand increase for drug,” except in the case of manufacturer Sandoz, for which the reason listed read “information pending.”
A company spokesperson told Bloomberg the reasons were complex.
“The combination in rapid succession of the pandemic impact and consequent demand swings, manufacturing capacity constraints, scarcity of raw materials, and the current energy crisis means we face a uniquely difficult situation in the short term,” Sandoz spokesperson Leslie Pott told Bloomberg.
According to Bloomberg, other major manufacturers are still delivering the product, but limiting new orders.
The American Society of Health-System Pharmacists issued an alert for the shortage last week via its real time drug shortage database.
“Amoxicillin comes in many forms – including capsules, powders and chewable tablets – but the most common type children take is the liquid form, which makes up at least 19 products that are part of the” shortage, Becker’s Hospital Review summarized of the database reports.
The pediatric health system Children’s Minnesota told CIDRAP that supplies are low and that alternatives are being prescribed “when appropriate.”
“As a final step, we temporarily discontinued our standard procedure of dispensing the entire bottle of amoxicillin (which comes in multiple sizes),” a spokesperson told CIDRAP. “We are instead mixing and pouring the exact amount for each course of therapy, to eliminate waste.”
The Minnesota pediatric clinic and others are particularly on alert because of the surge nationwide of a respiratory virus that particularly impacts children known as RSV.
“We have certainly observed an increase in recent use most likely correlating with the surge in RSV and other respiratory viruses with concern for superimposed bacterial infection in our critically ill and hospitalized patient population,” Laura Bio, PharmD, a clinical pharmacy specialist at Stanford Medicine Children’s Health told CIDRAP.
A version of this article first appeared on WebMD.com.
The liquid form of the antibiotic amoxicillin often used to treat ear infections and strep throat in children is in short supply, just as Americans head into the season when they use the bacteria-fighting drug the most.
The FDA officially listed the shortage Oct. 28, but pharmacists, hospitals, and a supply tracking database sounded alarms earlier this month.
“The scary part is, we’re coming into the time of the year where you have the greatest need,” independent pharmacy owner Hugh Chancy, PharmD, of Georgia, told NBC News.
Thus far, reports indicate the impact of the shortages is not widespread but does affect some pharmacies, and at least one hospital has published an algorithm for offering treatment alternatives.
CVS told Bloomberg News that some stores are experiencing shortages of certain doses of amoxicillin, but a Walmart spokesperson said its diverse supply chain meant none of its pharmacies were affected.
“Hypothetically, if amoxicillin doesn’t come into stock for some time, then we’re potentially having to use less effective antibiotics with more side effects,” said Ohio pediatrician Sean Gallagher, MD, according to Bloomberg.
The shortage impacts three of the four largest amoxicillin manufacturers worldwide, according to the Center for Infectious Disease Research and Policy (CIDRAP) at the University of Minnesota. The FDA listed the reason for the shortage as “demand increase for drug,” except in the case of manufacturer Sandoz, for which the reason listed read “information pending.”
A company spokesperson told Bloomberg the reasons were complex.
“The combination in rapid succession of the pandemic impact and consequent demand swings, manufacturing capacity constraints, scarcity of raw materials, and the current energy crisis means we face a uniquely difficult situation in the short term,” Sandoz spokesperson Leslie Pott told Bloomberg.
According to Bloomberg, other major manufacturers are still delivering the product, but limiting new orders.
The American Society of Health-System Pharmacists issued an alert for the shortage last week via its real time drug shortage database.
“Amoxicillin comes in many forms – including capsules, powders and chewable tablets – but the most common type children take is the liquid form, which makes up at least 19 products that are part of the” shortage, Becker’s Hospital Review summarized of the database reports.
The pediatric health system Children’s Minnesota told CIDRAP that supplies are low and that alternatives are being prescribed “when appropriate.”
“As a final step, we temporarily discontinued our standard procedure of dispensing the entire bottle of amoxicillin (which comes in multiple sizes),” a spokesperson told CIDRAP. “We are instead mixing and pouring the exact amount for each course of therapy, to eliminate waste.”
The Minnesota pediatric clinic and others are particularly on alert because of the surge nationwide of a respiratory virus that particularly impacts children known as RSV.
“We have certainly observed an increase in recent use most likely correlating with the surge in RSV and other respiratory viruses with concern for superimposed bacterial infection in our critically ill and hospitalized patient population,” Laura Bio, PharmD, a clinical pharmacy specialist at Stanford Medicine Children’s Health told CIDRAP.
A version of this article first appeared on WebMD.com.
Machine learning identifies childhood characteristics that predict bipolar disorder
This is the first quantitative approach to predict bipolar disorder, offering sensitivity and specificity of 75% and 76%, respectively, reported lead author Mai Uchida, MD, director of the pediatric depression program at Massachusetts General Hospital and assistant professor of psychiatry at Harvard Medical School, Boston, and colleagues. With further development, the model could be used to identify at-risk children via electronic medical records, enabling earlier monitoring and intervention.

“Although longitudinal studies have found the prognosis of early-onset mood disorders to be unfavorable, research has also shown there are effective treatments and therapies that could significantly alleviate the patients’ and their families’ struggles from the diagnoses,” the investigators wrote in the Journal of Psychiatric Research. “Thus, early identification of the risks and interventions for early symptoms of pediatric mood disorders is crucial.”
To this end, Dr. Uchida and colleagues teamed up with the Gabrieli Lab at MIT, who have published extensively in the realm of neurodevelopment. They sourced data from 492 children, 6-18 years at baseline, who were involved in two longitudinal case-control family studies focused on ADHD. Inputs included psychometric scales, structured diagnostic interviews, social and cognitive functioning assessments, and sociodemographic data.
At 10-year follow-up, 10% of these children had developed bipolar disorder, a notably higher rate than the 3%-4% prevalence in the general population.
“This is a population that’s overrepresented,” Dr. Uchida said in an interview.
She offered two primary reasons for this: First, the families involved in the study were probably willing to be followed for 10 years because they had ongoing concerns about their child’s mental health. Second, the studies enrolled children diagnosed with ADHD, a condition associated with increased risk of bipolar disorder.
Using machine learning algorithms that processed the baseline data while accounting for the skewed distribution, the investigators were able to predict which of the children in the population would go on to develop bipolar disorder. The final model offered a sensitivity of 75%, a specificity of 76%, and an area under the receiver operating characteristic curve of 75%.
“To the best of our knowledge, this represents the first study using machine-learning algorithms for this purpose in pediatric psychiatry,” the investigators wrote.
Integrating models into electronic medical records
In the future, this model, or one like it, could be incorporated into software that automatically analyzes electronic medical records and notifies physicians about high-risk patients, Dr. Uchida predicted.
“Not all patients would connect to intervention,” she said. “Maybe it just means that you invite them in for a visit, or you observe them a little bit more carefully. I think that’s where we are hoping that machine learning and medical practice will go.”
When asked about the potential bias posed by psychiatric evaluation, compared with something like blood work results, Dr. Uchida suggested that this subjectivity can be overcome.
“I’m not entirely bothered by that,” she said, offering a list of objective data points that could be harvested from records, such as number of referrals, medications, and hospitalizations. Narrative text in medical records could also be analyzed, she said, potentially detecting key words that are more often associated with high-risk patients.
“Risk prediction is never going to be 100% accurate,” Dr. Uchida said. “But I do think that there will be things [in electronic medical records] that could guide how worried we should be, or how quickly we should intervene.”
Opening doors to personalized care
Martin Gignac, MD, chief of psychiatry at Montreal Children’s Hospital and associate professor at McGill University, Montreal, said the present study offers further support for the existence of pediatric-onset bipolar disorder, which “remains controversial” despite “solid evidence.”

“I’m impressed that we have 10-year-long longitudinal follow-up studies that corroborate the importance of this disorder, and show strong predictors of who is at risk,” Dr. Gignac said in an interview. “Clinicians treating a pediatric population should be aware that some of those children with mental health problems might have severe mental health problems, and you have to have the appropriate tools to screen them.”
Advanced tools like the one developed by Dr. Uchida and colleagues should lead to more personalized care, he said.
“We’re going to be able to define what your individual risk is, and maybe most importantly, what you can do to prevent the development of certain disorders,” Dr. Gignac said. “Are there any risks that are dynamic in nature, and that we can act upon? Exposure to stress, for example.”
While more work is needed to bring machine learning into daily psychiatric practice, Dr. Gignac concluded on an optimistic note.
“These instruments should translate from research into clinical practice in order to make difference for the patients we care for,” he said. “This is the type of hope that I hold – that it’s going to be applicable in clinical practice, hopefully, in the near future.”
The investigators disclosed relationships with InCarda, Baylis Medical, Johnson & Johnson, and others. Dr. Gignac disclosed no relevant competing interests.
This is the first quantitative approach to predict bipolar disorder, offering sensitivity and specificity of 75% and 76%, respectively, reported lead author Mai Uchida, MD, director of the pediatric depression program at Massachusetts General Hospital and assistant professor of psychiatry at Harvard Medical School, Boston, and colleagues. With further development, the model could be used to identify at-risk children via electronic medical records, enabling earlier monitoring and intervention.
“Although longitudinal studies have found the prognosis of early-onset mood disorders to be unfavorable, research has also shown there are effective treatments and therapies that could significantly alleviate the patients’ and their families’ struggles from the diagnoses,” the investigators wrote in the Journal of Psychiatric Research. “Thus, early identification of the risks and interventions for early symptoms of pediatric mood disorders is crucial.”
To this end, Dr. Uchida and colleagues teamed up with the Gabrieli Lab at MIT, who have published extensively in the realm of neurodevelopment. They sourced data from 492 children, 6-18 years at baseline, who were involved in two longitudinal case-control family studies focused on ADHD. Inputs included psychometric scales, structured diagnostic interviews, social and cognitive functioning assessments, and sociodemographic data.
At 10-year follow-up, 10% of these children had developed bipolar disorder, a notably higher rate than the 3%-4% prevalence in the general population.
“This is a population that’s overrepresented,” Dr. Uchida said in an interview.
She offered two primary reasons for this: First, the families involved in the study were probably willing to be followed for 10 years because they had ongoing concerns about their child’s mental health. Second, the studies enrolled children diagnosed with ADHD, a condition associated with increased risk of bipolar disorder.
Using machine learning algorithms that processed the baseline data while accounting for the skewed distribution, the investigators were able to predict which of the children in the population would go on to develop bipolar disorder. The final model offered a sensitivity of 75%, a specificity of 76%, and an area under the receiver operating characteristic curve of 75%.
“To the best of our knowledge, this represents the first study using machine-learning algorithms for this purpose in pediatric psychiatry,” the investigators wrote.
Integrating models into electronic medical records
In the future, this model, or one like it, could be incorporated into software that automatically analyzes electronic medical records and notifies physicians about high-risk patients, Dr. Uchida predicted.
“Not all patients would connect to intervention,” she said. “Maybe it just means that you invite them in for a visit, or you observe them a little bit more carefully. I think that’s where we are hoping that machine learning and medical practice will go.”
When asked about the potential bias posed by psychiatric evaluation, compared with something like blood work results, Dr. Uchida suggested that this subjectivity can be overcome.
“I’m not entirely bothered by that,” she said, offering a list of objective data points that could be harvested from records, such as number of referrals, medications, and hospitalizations. Narrative text in medical records could also be analyzed, she said, potentially detecting key words that are more often associated with high-risk patients.
“Risk prediction is never going to be 100% accurate,” Dr. Uchida said. “But I do think that there will be things [in electronic medical records] that could guide how worried we should be, or how quickly we should intervene.”
Opening doors to personalized care
Martin Gignac, MD, chief of psychiatry at Montreal Children’s Hospital and associate professor at McGill University, Montreal, said the present study offers further support for the existence of pediatric-onset bipolar disorder, which “remains controversial” despite “solid evidence.”
“I’m impressed that we have 10-year-long longitudinal follow-up studies that corroborate the importance of this disorder, and show strong predictors of who is at risk,” Dr. Gignac said in an interview. “Clinicians treating a pediatric population should be aware that some of those children with mental health problems might have severe mental health problems, and you have to have the appropriate tools to screen them.”
Advanced tools like the one developed by Dr. Uchida and colleagues should lead to more personalized care, he said.
“We’re going to be able to define what your individual risk is, and maybe most importantly, what you can do to prevent the development of certain disorders,” Dr. Gignac said. “Are there any risks that are dynamic in nature, and that we can act upon? Exposure to stress, for example.”
While more work is needed to bring machine learning into daily psychiatric practice, Dr. Gignac concluded on an optimistic note.
“These instruments should translate from research into clinical practice in order to make difference for the patients we care for,” he said. “This is the type of hope that I hold – that it’s going to be applicable in clinical practice, hopefully, in the near future.”
The investigators disclosed relationships with InCarda, Baylis Medical, Johnson & Johnson, and others. Dr. Gignac disclosed no relevant competing interests.
This is the first quantitative approach to predict bipolar disorder, offering sensitivity and specificity of 75% and 76%, respectively, reported lead author Mai Uchida, MD, director of the pediatric depression program at Massachusetts General Hospital and assistant professor of psychiatry at Harvard Medical School, Boston, and colleagues. With further development, the model could be used to identify at-risk children via electronic medical records, enabling earlier monitoring and intervention.
“Although longitudinal studies have found the prognosis of early-onset mood disorders to be unfavorable, research has also shown there are effective treatments and therapies that could significantly alleviate the patients’ and their families’ struggles from the diagnoses,” the investigators wrote in the Journal of Psychiatric Research. “Thus, early identification of the risks and interventions for early symptoms of pediatric mood disorders is crucial.”
To this end, Dr. Uchida and colleagues teamed up with the Gabrieli Lab at MIT, who have published extensively in the realm of neurodevelopment. They sourced data from 492 children, 6-18 years at baseline, who were involved in two longitudinal case-control family studies focused on ADHD. Inputs included psychometric scales, structured diagnostic interviews, social and cognitive functioning assessments, and sociodemographic data.
At 10-year follow-up, 10% of these children had developed bipolar disorder, a notably higher rate than the 3%-4% prevalence in the general population.
“This is a population that’s overrepresented,” Dr. Uchida said in an interview.
She offered two primary reasons for this: First, the families involved in the study were probably willing to be followed for 10 years because they had ongoing concerns about their child’s mental health. Second, the studies enrolled children diagnosed with ADHD, a condition associated with increased risk of bipolar disorder.
Using machine learning algorithms that processed the baseline data while accounting for the skewed distribution, the investigators were able to predict which of the children in the population would go on to develop bipolar disorder. The final model offered a sensitivity of 75%, a specificity of 76%, and an area under the receiver operating characteristic curve of 75%.
“To the best of our knowledge, this represents the first study using machine-learning algorithms for this purpose in pediatric psychiatry,” the investigators wrote.
Integrating models into electronic medical records
In the future, this model, or one like it, could be incorporated into software that automatically analyzes electronic medical records and notifies physicians about high-risk patients, Dr. Uchida predicted.
“Not all patients would connect to intervention,” she said. “Maybe it just means that you invite them in for a visit, or you observe them a little bit more carefully. I think that’s where we are hoping that machine learning and medical practice will go.”
When asked about the potential bias posed by psychiatric evaluation, compared with something like blood work results, Dr. Uchida suggested that this subjectivity can be overcome.
“I’m not entirely bothered by that,” she said, offering a list of objective data points that could be harvested from records, such as number of referrals, medications, and hospitalizations. Narrative text in medical records could also be analyzed, she said, potentially detecting key words that are more often associated with high-risk patients.
“Risk prediction is never going to be 100% accurate,” Dr. Uchida said. “But I do think that there will be things [in electronic medical records] that could guide how worried we should be, or how quickly we should intervene.”
Opening doors to personalized care
Martin Gignac, MD, chief of psychiatry at Montreal Children’s Hospital and associate professor at McGill University, Montreal, said the present study offers further support for the existence of pediatric-onset bipolar disorder, which “remains controversial” despite “solid evidence.”
“I’m impressed that we have 10-year-long longitudinal follow-up studies that corroborate the importance of this disorder, and show strong predictors of who is at risk,” Dr. Gignac said in an interview. “Clinicians treating a pediatric population should be aware that some of those children with mental health problems might have severe mental health problems, and you have to have the appropriate tools to screen them.”
Advanced tools like the one developed by Dr. Uchida and colleagues should lead to more personalized care, he said.
“We’re going to be able to define what your individual risk is, and maybe most importantly, what you can do to prevent the development of certain disorders,” Dr. Gignac said. “Are there any risks that are dynamic in nature, and that we can act upon? Exposure to stress, for example.”
While more work is needed to bring machine learning into daily psychiatric practice, Dr. Gignac concluded on an optimistic note.
“These instruments should translate from research into clinical practice in order to make difference for the patients we care for,” he said. “This is the type of hope that I hold – that it’s going to be applicable in clinical practice, hopefully, in the near future.”
The investigators disclosed relationships with InCarda, Baylis Medical, Johnson & Johnson, and others. Dr. Gignac disclosed no relevant competing interests.
FROM THE JOURNAL OF PSYCHIATRIC RESEARCH
The truth of alcohol consequences
Bad drinking consequence No. 87: Joining the LOTME team
Alcohol and college students go together like peanut butter and jelly. Or peanut butter and chocolate. Or peanut butter and toothpaste. Peanut butter goes with a lot of things.
Naturally, when you combine alcohol and college students, bad decisions are sure to follow. But have you ever wondered just how many bad decisions alcohol causes? A team of researchers from Penn State University, the undisputed champion of poor drinking decisions (trust us, we know), sure has. They’ve even conducted a 4-year study of 1,700 students as they carved a drunken swath through the many fine local drinking establishments, such as East Halls or that one frat house that hosts medieval battle–style ping pong tournaments.
The students were surveyed twice a year throughout the study, and the researchers compiled a list of all the various consequences their subjects experienced. Ultimately, college students will experience an average of 102 consequences from drinking during their 4-year college careers, which is an impressive number. Try thinking up a hundred consequences for anything.
Some consequences are less common than others – we imagine “missing the Renaissance Faire because you felt drunker the morning after than while you were drinking” is pretty low on the list – but more than 96% of students reported that they’d experienced a hangover and that drinking had caused them to say or do embarrassing things. Also, more than 70% said they needed additional alcohol to feel any effect, a potential sign of alcohol use disorder.
Once they had their list, the researchers focused on 12 of the more common and severe consequences, such as blacking out, hangovers, and missing work/class, and asked the study participants how their parents would react to their drinking and those specific consequences. Students who believed their parents would disapprove of alcohol-related consequences actually experienced fewer consequences overall.
College students, it seems, really do care what their parents think, even if they don’t express it, the researchers said. That gives space for parents to offer advice about the consequences of hard drinking, making decisions while drunk, or bringing godawful Fireball whiskey to parties. Seriously, don’t do that. Stuff’s bad, and you should feel bad for bringing it. Your parents raised you better than that.
COVID ‘expert’ discusses data sharing
We interrupt our regularly scheduled programming to bring you this special news event. Elon Musk, the world’s second-most annoying human, is holding a press conference to discuss, of all things, COVID-19.
Reporter: Hey, Mr. Musketeer, what qualifies you to talk about a global pandemic?
EM: As the official king of the Twitterverse, I’m pretty much an expert on any topic.
Reporter: Okay then, Mr. Muskmelon, what can you tell us about the new study in Agricultural Economics, which looked at consumers’ knowledge of local COVID infection rates and their willingness to eat at restaurants?

EM: Well, I know that one of the investigators, Rigoberto Lopez, PhD, of the University of Connecticut, said “no news is bad news.” Restaurants located in cities where local regulations required COVID tracking recovered faster than those in areas that did not, according to data from 87 restaurants in 10 Chinese cities that were gathered between Dec. 1, 2019, and March 27, 2020. Having access to local infection rate data made customers more comfortable going out to eat, the investigators explained.
Second reporter: Interesting, Mr. Muskox, but how about this headline from CNN: “Workers flee China’s biggest iPhone factory over Covid outbreak”? Do you agree with analysts, who said that “the chaos at Zhengzhou could jeopardize Apple and Foxconn’s output in the coming weeks,” as CNN put it?
EM: I did see that a manager at Foxconn, which owns the factory and is known to its friends as Hon Hai Precision Industry, told a Chinese media outlet that “workers are panicking over the spread of the virus at the factory and lack of access to official information.” As we’ve already discussed, no news is bad news.
That’s all the time I have to chat with you today. I’m off to fire some more Twitter employees.
In case you hadn’t already guessed, Vlad Putin is officially more annoying than Elon Musk. We now return to this week’s typical LOTME shenanigans, already in progress.
The deadliest month
With climate change making the world hotter, leading to more heat stroke and organ failure, you would think the summer months would be the most deadly. In reality, though, it’s quite the opposite.

There are multiple factors that make January the most deadly month out of the year, as LiveScience discovered in a recent analysis.
Let’s go through them, shall we?
Respiratory viruses: Robert Glatter, MD, of Lenox Hill Hospital in New York, told LiveScence that winter is the time for illnesses like the flu, bacterial pneumonia, and RSV. Millions of people worldwide die from the flu, according to the CDC. And the World Health Organization reported lower respiratory infections as the fourth-leading cause of death worldwide before COVID came along.
Heart disease: Heart conditions are actually more fatal in the winter months, according to a study published in Circulation. The cold puts more stress on the heart to keep the body warm, which can be a challenge for people who already have preexisting heart conditions.
Space heaters: Dr. Glatter also told Live Science that the use of space heaters could be a factor in the cold winter months since they can lead to carbon monoxide poisoning and even fires. Silent killers.
Holiday season: A time for joy and merriment, certainly, but Christmas et al. have their downsides. By January we’re coming off a 3-month food and alcohol binge, which leads to cardiac stress. There’s also the psychological stress that comes with the season. Sometimes the most wonderful time of the year just isn’t.
So even though summer is hot, fall has hurricanes, and spring tends to have the highest suicide rate, winter still ends up being the deadliest season.
Bad drinking consequence No. 87: Joining the LOTME team
Alcohol and college students go together like peanut butter and jelly. Or peanut butter and chocolate. Or peanut butter and toothpaste. Peanut butter goes with a lot of things.
Naturally, when you combine alcohol and college students, bad decisions are sure to follow. But have you ever wondered just how many bad decisions alcohol causes? A team of researchers from Penn State University, the undisputed champion of poor drinking decisions (trust us, we know), sure has. They’ve even conducted a 4-year study of 1,700 students as they carved a drunken swath through the many fine local drinking establishments, such as East Halls or that one frat house that hosts medieval battle–style ping pong tournaments.
The students were surveyed twice a year throughout the study, and the researchers compiled a list of all the various consequences their subjects experienced. Ultimately, college students will experience an average of 102 consequences from drinking during their 4-year college careers, which is an impressive number. Try thinking up a hundred consequences for anything.
Some consequences are less common than others – we imagine “missing the Renaissance Faire because you felt drunker the morning after than while you were drinking” is pretty low on the list – but more than 96% of students reported that they’d experienced a hangover and that drinking had caused them to say or do embarrassing things. Also, more than 70% said they needed additional alcohol to feel any effect, a potential sign of alcohol use disorder.
Once they had their list, the researchers focused on 12 of the more common and severe consequences, such as blacking out, hangovers, and missing work/class, and asked the study participants how their parents would react to their drinking and those specific consequences. Students who believed their parents would disapprove of alcohol-related consequences actually experienced fewer consequences overall.
College students, it seems, really do care what their parents think, even if they don’t express it, the researchers said. That gives space for parents to offer advice about the consequences of hard drinking, making decisions while drunk, or bringing godawful Fireball whiskey to parties. Seriously, don’t do that. Stuff’s bad, and you should feel bad for bringing it. Your parents raised you better than that.
COVID ‘expert’ discusses data sharing
We interrupt our regularly scheduled programming to bring you this special news event. Elon Musk, the world’s second-most annoying human, is holding a press conference to discuss, of all things, COVID-19.
Reporter: Hey, Mr. Musketeer, what qualifies you to talk about a global pandemic?
EM: As the official king of the Twitterverse, I’m pretty much an expert on any topic.
Reporter: Okay then, Mr. Muskmelon, what can you tell us about the new study in Agricultural Economics, which looked at consumers’ knowledge of local COVID infection rates and their willingness to eat at restaurants?
EM: Well, I know that one of the investigators, Rigoberto Lopez, PhD, of the University of Connecticut, said “no news is bad news.” Restaurants located in cities where local regulations required COVID tracking recovered faster than those in areas that did not, according to data from 87 restaurants in 10 Chinese cities that were gathered between Dec. 1, 2019, and March 27, 2020. Having access to local infection rate data made customers more comfortable going out to eat, the investigators explained.
Second reporter: Interesting, Mr. Muskox, but how about this headline from CNN: “Workers flee China’s biggest iPhone factory over Covid outbreak”? Do you agree with analysts, who said that “the chaos at Zhengzhou could jeopardize Apple and Foxconn’s output in the coming weeks,” as CNN put it?
EM: I did see that a manager at Foxconn, which owns the factory and is known to its friends as Hon Hai Precision Industry, told a Chinese media outlet that “workers are panicking over the spread of the virus at the factory and lack of access to official information.” As we’ve already discussed, no news is bad news.
That’s all the time I have to chat with you today. I’m off to fire some more Twitter employees.
In case you hadn’t already guessed, Vlad Putin is officially more annoying than Elon Musk. We now return to this week’s typical LOTME shenanigans, already in progress.
The deadliest month
With climate change making the world hotter, leading to more heat stroke and organ failure, you would think the summer months would be the most deadly. In reality, though, it’s quite the opposite.
There are multiple factors that make January the most deadly month out of the year, as LiveScience discovered in a recent analysis.
Let’s go through them, shall we?
Respiratory viruses: Robert Glatter, MD, of Lenox Hill Hospital in New York, told LiveScence that winter is the time for illnesses like the flu, bacterial pneumonia, and RSV. Millions of people worldwide die from the flu, according to the CDC. And the World Health Organization reported lower respiratory infections as the fourth-leading cause of death worldwide before COVID came along.
Heart disease: Heart conditions are actually more fatal in the winter months, according to a study published in Circulation. The cold puts more stress on the heart to keep the body warm, which can be a challenge for people who already have preexisting heart conditions.
Space heaters: Dr. Glatter also told Live Science that the use of space heaters could be a factor in the cold winter months since they can lead to carbon monoxide poisoning and even fires. Silent killers.
Holiday season: A time for joy and merriment, certainly, but Christmas et al. have their downsides. By January we’re coming off a 3-month food and alcohol binge, which leads to cardiac stress. There’s also the psychological stress that comes with the season. Sometimes the most wonderful time of the year just isn’t.
So even though summer is hot, fall has hurricanes, and spring tends to have the highest suicide rate, winter still ends up being the deadliest season.
Bad drinking consequence No. 87: Joining the LOTME team
Alcohol and college students go together like peanut butter and jelly. Or peanut butter and chocolate. Or peanut butter and toothpaste. Peanut butter goes with a lot of things.
Naturally, when you combine alcohol and college students, bad decisions are sure to follow. But have you ever wondered just how many bad decisions alcohol causes? A team of researchers from Penn State University, the undisputed champion of poor drinking decisions (trust us, we know), sure has. They’ve even conducted a 4-year study of 1,700 students as they carved a drunken swath through the many fine local drinking establishments, such as East Halls or that one frat house that hosts medieval battle–style ping pong tournaments.
The students were surveyed twice a year throughout the study, and the researchers compiled a list of all the various consequences their subjects experienced. Ultimately, college students will experience an average of 102 consequences from drinking during their 4-year college careers, which is an impressive number. Try thinking up a hundred consequences for anything.
Some consequences are less common than others – we imagine “missing the Renaissance Faire because you felt drunker the morning after than while you were drinking” is pretty low on the list – but more than 96% of students reported that they’d experienced a hangover and that drinking had caused them to say or do embarrassing things. Also, more than 70% said they needed additional alcohol to feel any effect, a potential sign of alcohol use disorder.
Once they had their list, the researchers focused on 12 of the more common and severe consequences, such as blacking out, hangovers, and missing work/class, and asked the study participants how their parents would react to their drinking and those specific consequences. Students who believed their parents would disapprove of alcohol-related consequences actually experienced fewer consequences overall.
College students, it seems, really do care what their parents think, even if they don’t express it, the researchers said. That gives space for parents to offer advice about the consequences of hard drinking, making decisions while drunk, or bringing godawful Fireball whiskey to parties. Seriously, don’t do that. Stuff’s bad, and you should feel bad for bringing it. Your parents raised you better than that.
COVID ‘expert’ discusses data sharing
We interrupt our regularly scheduled programming to bring you this special news event. Elon Musk, the world’s second-most annoying human, is holding a press conference to discuss, of all things, COVID-19.
Reporter: Hey, Mr. Musketeer, what qualifies you to talk about a global pandemic?
EM: As the official king of the Twitterverse, I’m pretty much an expert on any topic.
Reporter: Okay then, Mr. Muskmelon, what can you tell us about the new study in Agricultural Economics, which looked at consumers’ knowledge of local COVID infection rates and their willingness to eat at restaurants?
EM: Well, I know that one of the investigators, Rigoberto Lopez, PhD, of the University of Connecticut, said “no news is bad news.” Restaurants located in cities where local regulations required COVID tracking recovered faster than those in areas that did not, according to data from 87 restaurants in 10 Chinese cities that were gathered between Dec. 1, 2019, and March 27, 2020. Having access to local infection rate data made customers more comfortable going out to eat, the investigators explained.
Second reporter: Interesting, Mr. Muskox, but how about this headline from CNN: “Workers flee China’s biggest iPhone factory over Covid outbreak”? Do you agree with analysts, who said that “the chaos at Zhengzhou could jeopardize Apple and Foxconn’s output in the coming weeks,” as CNN put it?
EM: I did see that a manager at Foxconn, which owns the factory and is known to its friends as Hon Hai Precision Industry, told a Chinese media outlet that “workers are panicking over the spread of the virus at the factory and lack of access to official information.” As we’ve already discussed, no news is bad news.
That’s all the time I have to chat with you today. I’m off to fire some more Twitter employees.
In case you hadn’t already guessed, Vlad Putin is officially more annoying than Elon Musk. We now return to this week’s typical LOTME shenanigans, already in progress.
The deadliest month
With climate change making the world hotter, leading to more heat stroke and organ failure, you would think the summer months would be the most deadly. In reality, though, it’s quite the opposite.
There are multiple factors that make January the most deadly month out of the year, as LiveScience discovered in a recent analysis.
Let’s go through them, shall we?
Respiratory viruses: Robert Glatter, MD, of Lenox Hill Hospital in New York, told LiveScence that winter is the time for illnesses like the flu, bacterial pneumonia, and RSV. Millions of people worldwide die from the flu, according to the CDC. And the World Health Organization reported lower respiratory infections as the fourth-leading cause of death worldwide before COVID came along.
Heart disease: Heart conditions are actually more fatal in the winter months, according to a study published in Circulation. The cold puts more stress on the heart to keep the body warm, which can be a challenge for people who already have preexisting heart conditions.
Space heaters: Dr. Glatter also told Live Science that the use of space heaters could be a factor in the cold winter months since they can lead to carbon monoxide poisoning and even fires. Silent killers.
Holiday season: A time for joy and merriment, certainly, but Christmas et al. have their downsides. By January we’re coming off a 3-month food and alcohol binge, which leads to cardiac stress. There’s also the psychological stress that comes with the season. Sometimes the most wonderful time of the year just isn’t.
So even though summer is hot, fall has hurricanes, and spring tends to have the highest suicide rate, winter still ends up being the deadliest season.
Race and gender: Tailoring treatment for sleep disorders is preferred and better
While trials of various interventions for obstructive sleep apnea and insomnia were effective, there was a strong suggestion that tailoring them according to the race/gender of the target populations strengthens engagement and improvements, according to a presentation by Dayna A. Johnson, PhD, MPH, at the annual meeting of the American College of Chest Physicians (CHEST).
Dr. Johnson, assistant professor at Emory University in Atlanta, stated that determinants of sleep disparities are multifactorial across the lifespan, from in utero to aging, but it was also important to focus on social determinants of poor sleep.
The complexity of factors, she said, calls for multilevel interventions beyond screening and treatment. In addition, neighborhood factors including safety, noise and light pollution, ventilation, and thermal comfort come into play.
Dr. Johnson cited the example of parents who work multiple jobs to provide for their families: “Minimum wage is not a livable wage, and parents may not be available to ensure that children have consistent bedtimes.” Interventions, she added, may have to be at the neighborhood level, including placing sleep specialists in the local neighborhood “where the need is.” Cleaning up a neighborhood reduces crime and overall health, while light shielding in public housing can lower light pollution.
Observing that African Americans have higher rates of obstructive sleep apnea, Dr. Johnson and colleagues designed a screening tool specifically for African Americans with five prediction models with increasing levels of factor measurements (from 4 to 10). The prediction accuracy across the models ascended in lockstep with the number of measures from 74.0% to 76.1%, with the simplest model including only age, body mass index, male sex, and snoring. The latter model added witnessed apneas, high depressive symptoms, two measures of waist and neck size, and sleepiness. Dr. Johnson pointed out that accuracy for well-established predictive models is notably lower: STOP-Bang score ranges from 56% to 66%; NoSAS ranges from 58% to 66% and the HCHS prediction model accuracy is 70%. Dr. Johnson said that a Latino model they developed was more accurate than the traditional models, but not as accurate as their model for African Americans.
Turning to specific interventions, and underscoring higher levels of stress and anxiety among African American and Hispanic populations, Dr. Johnson cited MINDS (Mindfulness Intervention to Improve Sleep and Reduce Diabetes Risk Among a Diverse Sample in Atlanta), her study at Emory University of mindfulness meditation. Although prior studies have confirmed sleep benefits of mindfulness meditation, studies tailored for African American or Hispanic populations have been lacking.
The MINDS pilot study investigators enrolled 17 individuals (mostly women, with a mixture of racial and ethnic groups comprising Black, White, Asian and Hispanic patients) with poor sleep quality as measured by the Pittsburgh Sleep Quality Index (PSQI). Most patients, Dr. Johnson said, were overweight. Because of COVID restrictions on clinic visits, the diabetes portion of the study was dropped. All participants received at least 3 days of instruction on mindfulness meditation, on dealing with stress and anxiety, and on optimum sleep health practices. While PSQI scores higher than 5 are considered to indicate poor sleep quality, the mean PSQI score at study outset in MINDS was 9.2, she stated.
After 30 days of the intervention, stress (on a perceived stress scale) was improved, as were PSQI scores and actigraphy measures of sleep duration, efficiency and wakefulness after sleep onset, Dr. Johnson reported. “Participants found the mindfulness app to be acceptable and appropriate, and to reduce time to falling asleep,” Dr. Johnson said.
Qualitative data gathered post intervention from four focus groups (two to six participants in each; 1-1.5 hours in length), revealed general acceptability of the MINDS app. It showed also that among those with 50% or more adherence to the intervention, time to falling asleep was reduced, as were sleep awakenings at night. The most striking finding, Dr. Johnson said, was that individuals from among racial/ethnic minorities expressed appreciation of the diversity of the meditation instructors, and said that they preferred instruction from a person of their own race and sex. Findings would be even more striking with a larger sample size, Dr. Johnson speculated.
Citing TASHE (Tailored Approach to Sleep Health Education), a further observational study on obstructive sleep apnea knowledge conducted at New York University, Dr. Johnson addressed the fact that current messages are not tailored to race/ethnic minorities with low-to-moderate symptom knowledge. Also, a 3-arm randomized clinical trial of Internet-delivered treatment (Sleep Healthy or SHUTI) with a version revised for Black women (SHUTI-BWHS) showed findings similar to those of other studies cited and suggested: “Tailoring may be necessary to increase uptake and sustainability and to improve sleep among racial/ethnic minorities.”
Dr. Johnson noted, in closing, that Black/African American individuals have higher risk for obstructive sleep apnea than that of their White counterparts and lower rates of screening for treatment.
Dr. Johnson’s research was funded by the National Institutes of Health; National Heart, Lung, and Blood Institute; Woodruff Health Sciences Center; Synergy Award; Rollins School of Public Health Dean’s Pilot and Innovation Award; and Georgia Center for Diabetes Translation Research Pilot and Feasibility award program. She reported no relevant conflicts.
While trials of various interventions for obstructive sleep apnea and insomnia were effective, there was a strong suggestion that tailoring them according to the race/gender of the target populations strengthens engagement and improvements, according to a presentation by Dayna A. Johnson, PhD, MPH, at the annual meeting of the American College of Chest Physicians (CHEST).
Dr. Johnson, assistant professor at Emory University in Atlanta, stated that determinants of sleep disparities are multifactorial across the lifespan, from in utero to aging, but it was also important to focus on social determinants of poor sleep.
The complexity of factors, she said, calls for multilevel interventions beyond screening and treatment. In addition, neighborhood factors including safety, noise and light pollution, ventilation, and thermal comfort come into play.
Dr. Johnson cited the example of parents who work multiple jobs to provide for their families: “Minimum wage is not a livable wage, and parents may not be available to ensure that children have consistent bedtimes.” Interventions, she added, may have to be at the neighborhood level, including placing sleep specialists in the local neighborhood “where the need is.” Cleaning up a neighborhood reduces crime and overall health, while light shielding in public housing can lower light pollution.
Observing that African Americans have higher rates of obstructive sleep apnea, Dr. Johnson and colleagues designed a screening tool specifically for African Americans with five prediction models with increasing levels of factor measurements (from 4 to 10). The prediction accuracy across the models ascended in lockstep with the number of measures from 74.0% to 76.1%, with the simplest model including only age, body mass index, male sex, and snoring. The latter model added witnessed apneas, high depressive symptoms, two measures of waist and neck size, and sleepiness. Dr. Johnson pointed out that accuracy for well-established predictive models is notably lower: STOP-Bang score ranges from 56% to 66%; NoSAS ranges from 58% to 66% and the HCHS prediction model accuracy is 70%. Dr. Johnson said that a Latino model they developed was more accurate than the traditional models, but not as accurate as their model for African Americans.
Turning to specific interventions, and underscoring higher levels of stress and anxiety among African American and Hispanic populations, Dr. Johnson cited MINDS (Mindfulness Intervention to Improve Sleep and Reduce Diabetes Risk Among a Diverse Sample in Atlanta), her study at Emory University of mindfulness meditation. Although prior studies have confirmed sleep benefits of mindfulness meditation, studies tailored for African American or Hispanic populations have been lacking.
The MINDS pilot study investigators enrolled 17 individuals (mostly women, with a mixture of racial and ethnic groups comprising Black, White, Asian and Hispanic patients) with poor sleep quality as measured by the Pittsburgh Sleep Quality Index (PSQI). Most patients, Dr. Johnson said, were overweight. Because of COVID restrictions on clinic visits, the diabetes portion of the study was dropped. All participants received at least 3 days of instruction on mindfulness meditation, on dealing with stress and anxiety, and on optimum sleep health practices. While PSQI scores higher than 5 are considered to indicate poor sleep quality, the mean PSQI score at study outset in MINDS was 9.2, she stated.
After 30 days of the intervention, stress (on a perceived stress scale) was improved, as were PSQI scores and actigraphy measures of sleep duration, efficiency and wakefulness after sleep onset, Dr. Johnson reported. “Participants found the mindfulness app to be acceptable and appropriate, and to reduce time to falling asleep,” Dr. Johnson said.
Qualitative data gathered post intervention from four focus groups (two to six participants in each; 1-1.5 hours in length), revealed general acceptability of the MINDS app. It showed also that among those with 50% or more adherence to the intervention, time to falling asleep was reduced, as were sleep awakenings at night. The most striking finding, Dr. Johnson said, was that individuals from among racial/ethnic minorities expressed appreciation of the diversity of the meditation instructors, and said that they preferred instruction from a person of their own race and sex. Findings would be even more striking with a larger sample size, Dr. Johnson speculated.
Citing TASHE (Tailored Approach to Sleep Health Education), a further observational study on obstructive sleep apnea knowledge conducted at New York University, Dr. Johnson addressed the fact that current messages are not tailored to race/ethnic minorities with low-to-moderate symptom knowledge. Also, a 3-arm randomized clinical trial of Internet-delivered treatment (Sleep Healthy or SHUTI) with a version revised for Black women (SHUTI-BWHS) showed findings similar to those of other studies cited and suggested: “Tailoring may be necessary to increase uptake and sustainability and to improve sleep among racial/ethnic minorities.”
Dr. Johnson noted, in closing, that Black/African American individuals have higher risk for obstructive sleep apnea than that of their White counterparts and lower rates of screening for treatment.
Dr. Johnson’s research was funded by the National Institutes of Health; National Heart, Lung, and Blood Institute; Woodruff Health Sciences Center; Synergy Award; Rollins School of Public Health Dean’s Pilot and Innovation Award; and Georgia Center for Diabetes Translation Research Pilot and Feasibility award program. She reported no relevant conflicts.
While trials of various interventions for obstructive sleep apnea and insomnia were effective, there was a strong suggestion that tailoring them according to the race/gender of the target populations strengthens engagement and improvements, according to a presentation by Dayna A. Johnson, PhD, MPH, at the annual meeting of the American College of Chest Physicians (CHEST).
Dr. Johnson, assistant professor at Emory University in Atlanta, stated that determinants of sleep disparities are multifactorial across the lifespan, from in utero to aging, but it was also important to focus on social determinants of poor sleep.
The complexity of factors, she said, calls for multilevel interventions beyond screening and treatment. In addition, neighborhood factors including safety, noise and light pollution, ventilation, and thermal comfort come into play.
Dr. Johnson cited the example of parents who work multiple jobs to provide for their families: “Minimum wage is not a livable wage, and parents may not be available to ensure that children have consistent bedtimes.” Interventions, she added, may have to be at the neighborhood level, including placing sleep specialists in the local neighborhood “where the need is.” Cleaning up a neighborhood reduces crime and overall health, while light shielding in public housing can lower light pollution.
Observing that African Americans have higher rates of obstructive sleep apnea, Dr. Johnson and colleagues designed a screening tool specifically for African Americans with five prediction models with increasing levels of factor measurements (from 4 to 10). The prediction accuracy across the models ascended in lockstep with the number of measures from 74.0% to 76.1%, with the simplest model including only age, body mass index, male sex, and snoring. The latter model added witnessed apneas, high depressive symptoms, two measures of waist and neck size, and sleepiness. Dr. Johnson pointed out that accuracy for well-established predictive models is notably lower: STOP-Bang score ranges from 56% to 66%; NoSAS ranges from 58% to 66% and the HCHS prediction model accuracy is 70%. Dr. Johnson said that a Latino model they developed was more accurate than the traditional models, but not as accurate as their model for African Americans.
Turning to specific interventions, and underscoring higher levels of stress and anxiety among African American and Hispanic populations, Dr. Johnson cited MINDS (Mindfulness Intervention to Improve Sleep and Reduce Diabetes Risk Among a Diverse Sample in Atlanta), her study at Emory University of mindfulness meditation. Although prior studies have confirmed sleep benefits of mindfulness meditation, studies tailored for African American or Hispanic populations have been lacking.
The MINDS pilot study investigators enrolled 17 individuals (mostly women, with a mixture of racial and ethnic groups comprising Black, White, Asian and Hispanic patients) with poor sleep quality as measured by the Pittsburgh Sleep Quality Index (PSQI). Most patients, Dr. Johnson said, were overweight. Because of COVID restrictions on clinic visits, the diabetes portion of the study was dropped. All participants received at least 3 days of instruction on mindfulness meditation, on dealing with stress and anxiety, and on optimum sleep health practices. While PSQI scores higher than 5 are considered to indicate poor sleep quality, the mean PSQI score at study outset in MINDS was 9.2, she stated.
After 30 days of the intervention, stress (on a perceived stress scale) was improved, as were PSQI scores and actigraphy measures of sleep duration, efficiency and wakefulness after sleep onset, Dr. Johnson reported. “Participants found the mindfulness app to be acceptable and appropriate, and to reduce time to falling asleep,” Dr. Johnson said.
Qualitative data gathered post intervention from four focus groups (two to six participants in each; 1-1.5 hours in length), revealed general acceptability of the MINDS app. It showed also that among those with 50% or more adherence to the intervention, time to falling asleep was reduced, as were sleep awakenings at night. The most striking finding, Dr. Johnson said, was that individuals from among racial/ethnic minorities expressed appreciation of the diversity of the meditation instructors, and said that they preferred instruction from a person of their own race and sex. Findings would be even more striking with a larger sample size, Dr. Johnson speculated.
Citing TASHE (Tailored Approach to Sleep Health Education), a further observational study on obstructive sleep apnea knowledge conducted at New York University, Dr. Johnson addressed the fact that current messages are not tailored to race/ethnic minorities with low-to-moderate symptom knowledge. Also, a 3-arm randomized clinical trial of Internet-delivered treatment (Sleep Healthy or SHUTI) with a version revised for Black women (SHUTI-BWHS) showed findings similar to those of other studies cited and suggested: “Tailoring may be necessary to increase uptake and sustainability and to improve sleep among racial/ethnic minorities.”
Dr. Johnson noted, in closing, that Black/African American individuals have higher risk for obstructive sleep apnea than that of their White counterparts and lower rates of screening for treatment.
Dr. Johnson’s research was funded by the National Institutes of Health; National Heart, Lung, and Blood Institute; Woodruff Health Sciences Center; Synergy Award; Rollins School of Public Health Dean’s Pilot and Innovation Award; and Georgia Center for Diabetes Translation Research Pilot and Feasibility award program. She reported no relevant conflicts.
FROM CHEST 2022
Gout too often treated only in emergency department
Only about one in three patients seen in the emergency department of an academic health system for acute gout had a follow-up visit that addressed this condition, Lesley Jackson, MD, of the University of Alabama at Birmingham, reported at the annual research symposium of the Gout, Hyperuricemia, and Crystal Associated Disease Network (G-CAN).
Dr. Jackson presented research done on patients seen within her university’s health system, looking at 72 patients seen in the ED between September 2021 and February 2022. Medications prescribed at discharge from the ED included corticosteroids (46 patients, or 64%), opioids (45 patients, 63%), NSAIDs (31 patients, 43%), and colchicine (23 patients, 32%).
Only 26 patients, or about 36%, had a subsequent outpatient visit in the UAB health system addressing gout, she said. Of 33 patients with any outpatient follow-up visit within the UAB system, 21 were within 1 month after the index ED visit, followed by 3 more prior to 3 months, and 9 more after 3 months.
The limitations of the study includes its collection of data from a single institution. But the results highlight the need for improved quality of care for gout, with too many people being treated for this condition primarily in the ED, she said.
In an email exchange arranged by the Arthritis Foundation, Herbert S. B. Baraf, MD, said he agreed that patients too often limit their treatment for gout to seeking care for acute attacks in the ED.
Because of competing demands, physicians working there are more to take a “Band-Aid” approach and not impress upon patients that gout is a lifelong condition that needs follow-up and monitoring, said Dr. Baraf, clinical professor of medicine at George Washington University, Washington, and an associate clinical professor at the University of Maryland, Baltimore. He retired from private practice in 2022.
“This problem is akin to the patient who has a hip fracture due to osteoporosis who gets a surgical repair but is never referred for osteoporotic management,” wrote Dr. Baraf, who is a former board member of the Arthritis Foundation.
He suggested viewing gout as a form of arthritis that has two components.
“The first, that which brings the patient to seek medical care, is the often exquisitely painful attack of pain and swelling in a joint or joints that comes on acutely,” he wrote. “Calming these attacks are the focus of the patient and the doctor, who does the evaluation as relief of pain and inflammation is the most pressing task at hand.”
But equally important is the second element, addressing the cause of these flare ups of arthritis, he wrote. Elevated uric acid leads to crystalline deposits of urate in the joints, particularly in the feet, ankles, knees, and hands. Over time, these deposits generate seemingly random flare ups of acute joint pain in one or more of these areas.
“Thus, when a patient presents to an emergency room with a first or second attack of gout, pain relief is the primary focus of the visit,” Dr. Baraf wrote. “But if over time that is the only focus, and the elevation of serum uric acid is not addressed, deposits will continue to mount and flare ups will occur with increasing frequency and severity.”
This study was supported by a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Jackson has no relevant financial disclosures.
Only about one in three patients seen in the emergency department of an academic health system for acute gout had a follow-up visit that addressed this condition, Lesley Jackson, MD, of the University of Alabama at Birmingham, reported at the annual research symposium of the Gout, Hyperuricemia, and Crystal Associated Disease Network (G-CAN).
Dr. Jackson presented research done on patients seen within her university’s health system, looking at 72 patients seen in the ED between September 2021 and February 2022. Medications prescribed at discharge from the ED included corticosteroids (46 patients, or 64%), opioids (45 patients, 63%), NSAIDs (31 patients, 43%), and colchicine (23 patients, 32%).
Only 26 patients, or about 36%, had a subsequent outpatient visit in the UAB health system addressing gout, she said. Of 33 patients with any outpatient follow-up visit within the UAB system, 21 were within 1 month after the index ED visit, followed by 3 more prior to 3 months, and 9 more after 3 months.
The limitations of the study includes its collection of data from a single institution. But the results highlight the need for improved quality of care for gout, with too many people being treated for this condition primarily in the ED, she said.
In an email exchange arranged by the Arthritis Foundation, Herbert S. B. Baraf, MD, said he agreed that patients too often limit their treatment for gout to seeking care for acute attacks in the ED.
Because of competing demands, physicians working there are more to take a “Band-Aid” approach and not impress upon patients that gout is a lifelong condition that needs follow-up and monitoring, said Dr. Baraf, clinical professor of medicine at George Washington University, Washington, and an associate clinical professor at the University of Maryland, Baltimore. He retired from private practice in 2022.
“This problem is akin to the patient who has a hip fracture due to osteoporosis who gets a surgical repair but is never referred for osteoporotic management,” wrote Dr. Baraf, who is a former board member of the Arthritis Foundation.
He suggested viewing gout as a form of arthritis that has two components.
“The first, that which brings the patient to seek medical care, is the often exquisitely painful attack of pain and swelling in a joint or joints that comes on acutely,” he wrote. “Calming these attacks are the focus of the patient and the doctor, who does the evaluation as relief of pain and inflammation is the most pressing task at hand.”
But equally important is the second element, addressing the cause of these flare ups of arthritis, he wrote. Elevated uric acid leads to crystalline deposits of urate in the joints, particularly in the feet, ankles, knees, and hands. Over time, these deposits generate seemingly random flare ups of acute joint pain in one or more of these areas.
“Thus, when a patient presents to an emergency room with a first or second attack of gout, pain relief is the primary focus of the visit,” Dr. Baraf wrote. “But if over time that is the only focus, and the elevation of serum uric acid is not addressed, deposits will continue to mount and flare ups will occur with increasing frequency and severity.”
This study was supported by a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Jackson has no relevant financial disclosures.
Only about one in three patients seen in the emergency department of an academic health system for acute gout had a follow-up visit that addressed this condition, Lesley Jackson, MD, of the University of Alabama at Birmingham, reported at the annual research symposium of the Gout, Hyperuricemia, and Crystal Associated Disease Network (G-CAN).
Dr. Jackson presented research done on patients seen within her university’s health system, looking at 72 patients seen in the ED between September 2021 and February 2022. Medications prescribed at discharge from the ED included corticosteroids (46 patients, or 64%), opioids (45 patients, 63%), NSAIDs (31 patients, 43%), and colchicine (23 patients, 32%).
Only 26 patients, or about 36%, had a subsequent outpatient visit in the UAB health system addressing gout, she said. Of 33 patients with any outpatient follow-up visit within the UAB system, 21 were within 1 month after the index ED visit, followed by 3 more prior to 3 months, and 9 more after 3 months.
The limitations of the study includes its collection of data from a single institution. But the results highlight the need for improved quality of care for gout, with too many people being treated for this condition primarily in the ED, she said.
In an email exchange arranged by the Arthritis Foundation, Herbert S. B. Baraf, MD, said he agreed that patients too often limit their treatment for gout to seeking care for acute attacks in the ED.
Because of competing demands, physicians working there are more to take a “Band-Aid” approach and not impress upon patients that gout is a lifelong condition that needs follow-up and monitoring, said Dr. Baraf, clinical professor of medicine at George Washington University, Washington, and an associate clinical professor at the University of Maryland, Baltimore. He retired from private practice in 2022.
“This problem is akin to the patient who has a hip fracture due to osteoporosis who gets a surgical repair but is never referred for osteoporotic management,” wrote Dr. Baraf, who is a former board member of the Arthritis Foundation.
He suggested viewing gout as a form of arthritis that has two components.
“The first, that which brings the patient to seek medical care, is the often exquisitely painful attack of pain and swelling in a joint or joints that comes on acutely,” he wrote. “Calming these attacks are the focus of the patient and the doctor, who does the evaluation as relief of pain and inflammation is the most pressing task at hand.”
But equally important is the second element, addressing the cause of these flare ups of arthritis, he wrote. Elevated uric acid leads to crystalline deposits of urate in the joints, particularly in the feet, ankles, knees, and hands. Over time, these deposits generate seemingly random flare ups of acute joint pain in one or more of these areas.
“Thus, when a patient presents to an emergency room with a first or second attack of gout, pain relief is the primary focus of the visit,” Dr. Baraf wrote. “But if over time that is the only focus, and the elevation of serum uric acid is not addressed, deposits will continue to mount and flare ups will occur with increasing frequency and severity.”
This study was supported by a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Jackson has no relevant financial disclosures.
FROM G-CAN 2022
Hyaluronidase for Skin Necrosis Induced by Amiodarone
To the Editor:
Amiodarone is an oral or intravenous (IV) drug commonly used to treat supraventricular and ventricular arrhythmia as well as atrial fibrillation.1 Adverse drug reactions associated with the use of amiodarone include pulmonary, gastrointestinal, thyroid, ocular, neurologic, and cutaneous reactions.1 Long-term use of amiodarone—typically more than 4 months—can lead to slate-gray skin discoloration and photosensitivity, both of which can be reversed with drug withdrawal.2,3 Phlebitis also has been described in less than 3% of patients who receive peripheral IV administration of amiodarone.4
Amiodarone-induced skin necrosis due to extravasation is a rare complication of this antiarrhythmic medication, with only 3 reported cases in the literature according to a PubMed search of articles indexed for MEDLINE using the search terms amiodarone and skin and (necrosis or ischemia or extravasation or reaction).5–7 Although hyaluronidase is a known therapy for extravasation of fluids, including parenteral nutrition and chemotherapy, its use for the treatment of extravasation from amiodarone is not well documented.6 We report a case of skin necrosis of the left dorsal forearm and the left dorsal and ventral hand following infusion of amiodarone through a peripheral IV line, which was treated with injections of hyaluronidase.
A 77-year-old man was admitted to the emergency department for sepsis secondary to cholangitis in the setting of an obstructive gallbladder stone. His medical history was notable for multivessel coronary artery disease and atrial flutter treated with ablation. One day after admission, endoscopic retrograde cholangiopancreatography was attempted and aborted due to atrial fibrillation with rapid ventricular response. A second endoscopic retrograde cholangiopancreatography attempt was made 4 days later, during which the patient underwent cardiac arrest. During this event, amiodarone was administered in a 200-mL solution (1.8 mg/mL) in 5% dextrose through a peripheral IV line in the left forearm. The patient was stabilized and transferred to the intensive care unit.
Twenty-four hours after amiodarone administration, erythema was noted on the left dorsal forearm. Within hours, the digits of the hand became a dark, dusky color, which spread to involve the forearm. Surgical debridement was not deemed necessary; the left arm was elevated, and warm compresses were applied regularly. Within the next week, the skin of the left hand and dorsal forearm had progressively worsened and took on a well-demarcated, dusky blue hue surrounded by an erythematous border involving the proximal forearm and upper arm (Figure 1A). The skin was fragile and had overlying bullae (Figure 1B).

Hyaluronidase (1000 U) was injected into the surrounding areas of erythema, which resolved from the left proximal forearm to the elbow within 2 days after injection (Figure 2). The dusky violaceous patches were persistent, and the necrotic bullae were unchanged. Hyaluronidase (1000 U) was injected into necrotic skin of the left dorsal forearm and dorsal and ventral hand. No improvement was noted on subsequent evaluations of this area. While still an inpatient, he received wound care and twice-daily Doppler ultrasounds in the areas of necrosis. The patient lost sensation in the left hand with increased soft tissue necrosis and developed an eschar on the left dorsal forearm. Due to the progressive loss of function and necrosis, a partial forearm amputation was performed that healed well, and the patient experienced improvement in range of motion of the left upper extremity.

Well-known adverse reactions of amiodarone treatment include pulmonary fibrosis, hepatic dysfunction, hypothyroidism and hyperthyroidism, peripheral neuropathy, and corneal deposits.1 Cutaneous adverse reactions include photosensitivity (phototoxic and photoallergic reactions), hyperpigmentation, pseudoporphyria, and linear IgA bullous dermatosis. Less commonly, it also can cause urticaria, pruritus, erythema nodosum, purpura, and toxic epidermal necrolysis.3 Amiodarone-induced skin necrosis is rare, first described by Russell and Saltissi5 in 2006 in a 60-year-old man who developed dark discoloration and edema of the forearm 24 hours after initiation of an amiodarone peripheral IV. The patient was treated with hot or cold packs and steroid cream per the pharmaceutical company’s recommendations; however, patient outcomes were not discussed.5 A 77-year-old man who received subcutaneous amiodarone due to misplaced vascular access developed edema and bullae of the forearm followed by tissue necrosis, resulting in notably reduced mobility.6 Fox et al7 described a 60-year-old man who developed atrial fibrillation after emergent spinal fusion and laminectomy. He received intradermal hyaluronidase administration within 24 hours of developing severe pain from extravasation induced by amiodarone with no adverse outcomes and full recovery.7
There are numerous properties of amiodarone that may have resulted in the skin necrosis seen in these cases. The acidic pH (3.5–4.5) of amiodarone can contribute to coagulative necrosis, cellular desiccation, eschar formation, and edema.8 It also can contain additives such as polysorbate and benzyl alcohol, which may contribute to the drug’s vesicant properties.9
Current recommendations for IV administration of amiodarone include delivery through a central vein with high concentrations (>2 mg/mL) because peripheral infusion is slower and may cause phlebitis.4 In-line filters also may be a potential method of preventing phlebitis with peripheral IV administration of amiodarone.10 Extravasation of amiodarone can be treated nonpharmacologically with limb elevation and warm compresses, as these methods may promote vasodilation and enhance drug removal.5-7 However, when extravasation leads to progressive erythema and skin necrosis or is refractory to these therapies, intradermal injection of hyaluronidase should be considered. Hyaluronidase mediates the degradation of hyaluronic acid in the extracellular matrix, allowing for increased permeability of injected fluids into tissues and diluting the concentration of toxins at the site of exposure.9,11 It has been used to treat extravasation of fluids such as parenteral nutrition, electrolyte infusion, antibiotics, aminophylline, mannitol, and chemotherapy.11 Although hyaluronidase has been recognized as therapeutic for extravasation, there is no established consistent dosing or proper technique. In the setting of infiltration of chemotherapy, doses of hyaluronidase ranging from 150 to 1500 U/mL can be subcutaneously or intradermally injected into the site within 1 hour of extravasation. Side effects of using hyaluronidase are rare, including local pruritus, allergic reactions, urticaria, and angioedema.12
The patient described by Fox et al7 who fully recovered from amiodarone extravasation after hyaluronidase injections likely benefited from quick intervention, as he received amiodarone within 24 hours of the care team identifying initial erythema. Although our patient did have improvement of the areas of erythema on the forearm, evidence of skin and subcutaneous tissue necrosis on the left hand and proximal forearm was already apparent and not reversible, most likely caused by late intervention of intradermal hyaluronidase almost a week after the extravasation event. It is important to identify amiodarone as the source of extravasation and administer intradermal hyaluronidase in a timely fashion for extravasation refractory to conventional measurements to prevent progression to severe tissue damage.
Our case draws attention to the risk for skin necrosis with peripheral IV administration of amiodarone. Interventions include limb elevation, warm compresses, and consideration of intradermal hyaluronidase within 24 hours of extravasation, as this may reduce the severity of subsequent tissue damage with minimal side effects.
- Epstein AE, Olshansky B, Naccarelli GV, et al. Practical management guide for clinicians who treat patients with amiodarone. Am J Med. 2016;129:468-475. doi:10.1016/j.amjmed.2015.08.039
- Harris L, McKenna WJ, Rowland E, et al. Side effects of long-term amiodarone therapy. Circulation. 1983;67:45-51. doi:10.1161/01.cir.67.1.45
- Jaworski K, Walecka I, Rudnicka L, et al. Cutaneous adverse reactions of amiodarone. Med Sci Monit. 2014;20:2369-2372. doi:10.12659/MSM.890881
- Kowey Peter R, Marinchak Roger A, Rials Seth J, et al. Intravenous amiodarone. J Am Coll Cardiol. 1997;29:1190-1198. doi:10.1016/S0735-1097(97)00069-7
- Russell SJ, Saltissi S. Amiodarone induced skin necrosis. Heart. 2006;92:1395. doi:10.1136/hrt.2005.086157
- Grove EL. Skin necrosis and consequences of accidental subcutaneous administration of amiodarone. Ugeskr Laeger. 2015;177:V66928.
- Fox AN, Villanueva R, Miller JL. Management of amiodarone extravasation with intradermal hyaluronidase. Am J Health Syst Pharm. 2017;74:1545-1548. doi:10.2146/ajhp160737
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632. doi:https://doi.org/10.1002/phar.1396
- Le A, Patel S. Extravasation of noncytotoxic drugs: a review of the literature. Ann Pharmacother. 2014;48:870-886. doi:10.1177/1060028014527820
- Slim AM, Roth JE, Duffy B, et al. The incidence of phlebitis with intravenous amiodarone at guideline dose recommendations. Mil Med. 2007;172:1279-1283.
- Girish KS, Kemparaju K. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 2007;80:1921-1943. doi:10.1016/j.lfs.2007.02.037
- Jung H. Hyaluronidase: an overview of its properties, applications, and side effects. Arch Plast Surg. 2020;47:297-300. doi:10.5999/aps.2020.00752
To the Editor:
Amiodarone is an oral or intravenous (IV) drug commonly used to treat supraventricular and ventricular arrhythmia as well as atrial fibrillation.1 Adverse drug reactions associated with the use of amiodarone include pulmonary, gastrointestinal, thyroid, ocular, neurologic, and cutaneous reactions.1 Long-term use of amiodarone—typically more than 4 months—can lead to slate-gray skin discoloration and photosensitivity, both of which can be reversed with drug withdrawal.2,3 Phlebitis also has been described in less than 3% of patients who receive peripheral IV administration of amiodarone.4
Amiodarone-induced skin necrosis due to extravasation is a rare complication of this antiarrhythmic medication, with only 3 reported cases in the literature according to a PubMed search of articles indexed for MEDLINE using the search terms amiodarone and skin and (necrosis or ischemia or extravasation or reaction).5–7 Although hyaluronidase is a known therapy for extravasation of fluids, including parenteral nutrition and chemotherapy, its use for the treatment of extravasation from amiodarone is not well documented.6 We report a case of skin necrosis of the left dorsal forearm and the left dorsal and ventral hand following infusion of amiodarone through a peripheral IV line, which was treated with injections of hyaluronidase.
A 77-year-old man was admitted to the emergency department for sepsis secondary to cholangitis in the setting of an obstructive gallbladder stone. His medical history was notable for multivessel coronary artery disease and atrial flutter treated with ablation. One day after admission, endoscopic retrograde cholangiopancreatography was attempted and aborted due to atrial fibrillation with rapid ventricular response. A second endoscopic retrograde cholangiopancreatography attempt was made 4 days later, during which the patient underwent cardiac arrest. During this event, amiodarone was administered in a 200-mL solution (1.8 mg/mL) in 5% dextrose through a peripheral IV line in the left forearm. The patient was stabilized and transferred to the intensive care unit.
Twenty-four hours after amiodarone administration, erythema was noted on the left dorsal forearm. Within hours, the digits of the hand became a dark, dusky color, which spread to involve the forearm. Surgical debridement was not deemed necessary; the left arm was elevated, and warm compresses were applied regularly. Within the next week, the skin of the left hand and dorsal forearm had progressively worsened and took on a well-demarcated, dusky blue hue surrounded by an erythematous border involving the proximal forearm and upper arm (Figure 1A). The skin was fragile and had overlying bullae (Figure 1B).
Hyaluronidase (1000 U) was injected into the surrounding areas of erythema, which resolved from the left proximal forearm to the elbow within 2 days after injection (Figure 2). The dusky violaceous patches were persistent, and the necrotic bullae were unchanged. Hyaluronidase (1000 U) was injected into necrotic skin of the left dorsal forearm and dorsal and ventral hand. No improvement was noted on subsequent evaluations of this area. While still an inpatient, he received wound care and twice-daily Doppler ultrasounds in the areas of necrosis. The patient lost sensation in the left hand with increased soft tissue necrosis and developed an eschar on the left dorsal forearm. Due to the progressive loss of function and necrosis, a partial forearm amputation was performed that healed well, and the patient experienced improvement in range of motion of the left upper extremity.
Well-known adverse reactions of amiodarone treatment include pulmonary fibrosis, hepatic dysfunction, hypothyroidism and hyperthyroidism, peripheral neuropathy, and corneal deposits.1 Cutaneous adverse reactions include photosensitivity (phototoxic and photoallergic reactions), hyperpigmentation, pseudoporphyria, and linear IgA bullous dermatosis. Less commonly, it also can cause urticaria, pruritus, erythema nodosum, purpura, and toxic epidermal necrolysis.3 Amiodarone-induced skin necrosis is rare, first described by Russell and Saltissi5 in 2006 in a 60-year-old man who developed dark discoloration and edema of the forearm 24 hours after initiation of an amiodarone peripheral IV. The patient was treated with hot or cold packs and steroid cream per the pharmaceutical company’s recommendations; however, patient outcomes were not discussed.5 A 77-year-old man who received subcutaneous amiodarone due to misplaced vascular access developed edema and bullae of the forearm followed by tissue necrosis, resulting in notably reduced mobility.6 Fox et al7 described a 60-year-old man who developed atrial fibrillation after emergent spinal fusion and laminectomy. He received intradermal hyaluronidase administration within 24 hours of developing severe pain from extravasation induced by amiodarone with no adverse outcomes and full recovery.7
There are numerous properties of amiodarone that may have resulted in the skin necrosis seen in these cases. The acidic pH (3.5–4.5) of amiodarone can contribute to coagulative necrosis, cellular desiccation, eschar formation, and edema.8 It also can contain additives such as polysorbate and benzyl alcohol, which may contribute to the drug’s vesicant properties.9
Current recommendations for IV administration of amiodarone include delivery through a central vein with high concentrations (>2 mg/mL) because peripheral infusion is slower and may cause phlebitis.4 In-line filters also may be a potential method of preventing phlebitis with peripheral IV administration of amiodarone.10 Extravasation of amiodarone can be treated nonpharmacologically with limb elevation and warm compresses, as these methods may promote vasodilation and enhance drug removal.5-7 However, when extravasation leads to progressive erythema and skin necrosis or is refractory to these therapies, intradermal injection of hyaluronidase should be considered. Hyaluronidase mediates the degradation of hyaluronic acid in the extracellular matrix, allowing for increased permeability of injected fluids into tissues and diluting the concentration of toxins at the site of exposure.9,11 It has been used to treat extravasation of fluids such as parenteral nutrition, electrolyte infusion, antibiotics, aminophylline, mannitol, and chemotherapy.11 Although hyaluronidase has been recognized as therapeutic for extravasation, there is no established consistent dosing or proper technique. In the setting of infiltration of chemotherapy, doses of hyaluronidase ranging from 150 to 1500 U/mL can be subcutaneously or intradermally injected into the site within 1 hour of extravasation. Side effects of using hyaluronidase are rare, including local pruritus, allergic reactions, urticaria, and angioedema.12
The patient described by Fox et al7 who fully recovered from amiodarone extravasation after hyaluronidase injections likely benefited from quick intervention, as he received amiodarone within 24 hours of the care team identifying initial erythema. Although our patient did have improvement of the areas of erythema on the forearm, evidence of skin and subcutaneous tissue necrosis on the left hand and proximal forearm was already apparent and not reversible, most likely caused by late intervention of intradermal hyaluronidase almost a week after the extravasation event. It is important to identify amiodarone as the source of extravasation and administer intradermal hyaluronidase in a timely fashion for extravasation refractory to conventional measurements to prevent progression to severe tissue damage.
Our case draws attention to the risk for skin necrosis with peripheral IV administration of amiodarone. Interventions include limb elevation, warm compresses, and consideration of intradermal hyaluronidase within 24 hours of extravasation, as this may reduce the severity of subsequent tissue damage with minimal side effects.
To the Editor:
Amiodarone is an oral or intravenous (IV) drug commonly used to treat supraventricular and ventricular arrhythmia as well as atrial fibrillation.1 Adverse drug reactions associated with the use of amiodarone include pulmonary, gastrointestinal, thyroid, ocular, neurologic, and cutaneous reactions.1 Long-term use of amiodarone—typically more than 4 months—can lead to slate-gray skin discoloration and photosensitivity, both of which can be reversed with drug withdrawal.2,3 Phlebitis also has been described in less than 3% of patients who receive peripheral IV administration of amiodarone.4
Amiodarone-induced skin necrosis due to extravasation is a rare complication of this antiarrhythmic medication, with only 3 reported cases in the literature according to a PubMed search of articles indexed for MEDLINE using the search terms amiodarone and skin and (necrosis or ischemia or extravasation or reaction).5–7 Although hyaluronidase is a known therapy for extravasation of fluids, including parenteral nutrition and chemotherapy, its use for the treatment of extravasation from amiodarone is not well documented.6 We report a case of skin necrosis of the left dorsal forearm and the left dorsal and ventral hand following infusion of amiodarone through a peripheral IV line, which was treated with injections of hyaluronidase.
A 77-year-old man was admitted to the emergency department for sepsis secondary to cholangitis in the setting of an obstructive gallbladder stone. His medical history was notable for multivessel coronary artery disease and atrial flutter treated with ablation. One day after admission, endoscopic retrograde cholangiopancreatography was attempted and aborted due to atrial fibrillation with rapid ventricular response. A second endoscopic retrograde cholangiopancreatography attempt was made 4 days later, during which the patient underwent cardiac arrest. During this event, amiodarone was administered in a 200-mL solution (1.8 mg/mL) in 5% dextrose through a peripheral IV line in the left forearm. The patient was stabilized and transferred to the intensive care unit.
Twenty-four hours after amiodarone administration, erythema was noted on the left dorsal forearm. Within hours, the digits of the hand became a dark, dusky color, which spread to involve the forearm. Surgical debridement was not deemed necessary; the left arm was elevated, and warm compresses were applied regularly. Within the next week, the skin of the left hand and dorsal forearm had progressively worsened and took on a well-demarcated, dusky blue hue surrounded by an erythematous border involving the proximal forearm and upper arm (Figure 1A). The skin was fragile and had overlying bullae (Figure 1B).
Hyaluronidase (1000 U) was injected into the surrounding areas of erythema, which resolved from the left proximal forearm to the elbow within 2 days after injection (Figure 2). The dusky violaceous patches were persistent, and the necrotic bullae were unchanged. Hyaluronidase (1000 U) was injected into necrotic skin of the left dorsal forearm and dorsal and ventral hand. No improvement was noted on subsequent evaluations of this area. While still an inpatient, he received wound care and twice-daily Doppler ultrasounds in the areas of necrosis. The patient lost sensation in the left hand with increased soft tissue necrosis and developed an eschar on the left dorsal forearm. Due to the progressive loss of function and necrosis, a partial forearm amputation was performed that healed well, and the patient experienced improvement in range of motion of the left upper extremity.
Well-known adverse reactions of amiodarone treatment include pulmonary fibrosis, hepatic dysfunction, hypothyroidism and hyperthyroidism, peripheral neuropathy, and corneal deposits.1 Cutaneous adverse reactions include photosensitivity (phototoxic and photoallergic reactions), hyperpigmentation, pseudoporphyria, and linear IgA bullous dermatosis. Less commonly, it also can cause urticaria, pruritus, erythema nodosum, purpura, and toxic epidermal necrolysis.3 Amiodarone-induced skin necrosis is rare, first described by Russell and Saltissi5 in 2006 in a 60-year-old man who developed dark discoloration and edema of the forearm 24 hours after initiation of an amiodarone peripheral IV. The patient was treated with hot or cold packs and steroid cream per the pharmaceutical company’s recommendations; however, patient outcomes were not discussed.5 A 77-year-old man who received subcutaneous amiodarone due to misplaced vascular access developed edema and bullae of the forearm followed by tissue necrosis, resulting in notably reduced mobility.6 Fox et al7 described a 60-year-old man who developed atrial fibrillation after emergent spinal fusion and laminectomy. He received intradermal hyaluronidase administration within 24 hours of developing severe pain from extravasation induced by amiodarone with no adverse outcomes and full recovery.7
There are numerous properties of amiodarone that may have resulted in the skin necrosis seen in these cases. The acidic pH (3.5–4.5) of amiodarone can contribute to coagulative necrosis, cellular desiccation, eschar formation, and edema.8 It also can contain additives such as polysorbate and benzyl alcohol, which may contribute to the drug’s vesicant properties.9
Current recommendations for IV administration of amiodarone include delivery through a central vein with high concentrations (>2 mg/mL) because peripheral infusion is slower and may cause phlebitis.4 In-line filters also may be a potential method of preventing phlebitis with peripheral IV administration of amiodarone.10 Extravasation of amiodarone can be treated nonpharmacologically with limb elevation and warm compresses, as these methods may promote vasodilation and enhance drug removal.5-7 However, when extravasation leads to progressive erythema and skin necrosis or is refractory to these therapies, intradermal injection of hyaluronidase should be considered. Hyaluronidase mediates the degradation of hyaluronic acid in the extracellular matrix, allowing for increased permeability of injected fluids into tissues and diluting the concentration of toxins at the site of exposure.9,11 It has been used to treat extravasation of fluids such as parenteral nutrition, electrolyte infusion, antibiotics, aminophylline, mannitol, and chemotherapy.11 Although hyaluronidase has been recognized as therapeutic for extravasation, there is no established consistent dosing or proper technique. In the setting of infiltration of chemotherapy, doses of hyaluronidase ranging from 150 to 1500 U/mL can be subcutaneously or intradermally injected into the site within 1 hour of extravasation. Side effects of using hyaluronidase are rare, including local pruritus, allergic reactions, urticaria, and angioedema.12
The patient described by Fox et al7 who fully recovered from amiodarone extravasation after hyaluronidase injections likely benefited from quick intervention, as he received amiodarone within 24 hours of the care team identifying initial erythema. Although our patient did have improvement of the areas of erythema on the forearm, evidence of skin and subcutaneous tissue necrosis on the left hand and proximal forearm was already apparent and not reversible, most likely caused by late intervention of intradermal hyaluronidase almost a week after the extravasation event. It is important to identify amiodarone as the source of extravasation and administer intradermal hyaluronidase in a timely fashion for extravasation refractory to conventional measurements to prevent progression to severe tissue damage.
Our case draws attention to the risk for skin necrosis with peripheral IV administration of amiodarone. Interventions include limb elevation, warm compresses, and consideration of intradermal hyaluronidase within 24 hours of extravasation, as this may reduce the severity of subsequent tissue damage with minimal side effects.
- Epstein AE, Olshansky B, Naccarelli GV, et al. Practical management guide for clinicians who treat patients with amiodarone. Am J Med. 2016;129:468-475. doi:10.1016/j.amjmed.2015.08.039
- Harris L, McKenna WJ, Rowland E, et al. Side effects of long-term amiodarone therapy. Circulation. 1983;67:45-51. doi:10.1161/01.cir.67.1.45
- Jaworski K, Walecka I, Rudnicka L, et al. Cutaneous adverse reactions of amiodarone. Med Sci Monit. 2014;20:2369-2372. doi:10.12659/MSM.890881
- Kowey Peter R, Marinchak Roger A, Rials Seth J, et al. Intravenous amiodarone. J Am Coll Cardiol. 1997;29:1190-1198. doi:10.1016/S0735-1097(97)00069-7
- Russell SJ, Saltissi S. Amiodarone induced skin necrosis. Heart. 2006;92:1395. doi:10.1136/hrt.2005.086157
- Grove EL. Skin necrosis and consequences of accidental subcutaneous administration of amiodarone. Ugeskr Laeger. 2015;177:V66928.
- Fox AN, Villanueva R, Miller JL. Management of amiodarone extravasation with intradermal hyaluronidase. Am J Health Syst Pharm. 2017;74:1545-1548. doi:10.2146/ajhp160737
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632. doi:https://doi.org/10.1002/phar.1396
- Le A, Patel S. Extravasation of noncytotoxic drugs: a review of the literature. Ann Pharmacother. 2014;48:870-886. doi:10.1177/1060028014527820
- Slim AM, Roth JE, Duffy B, et al. The incidence of phlebitis with intravenous amiodarone at guideline dose recommendations. Mil Med. 2007;172:1279-1283.
- Girish KS, Kemparaju K. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 2007;80:1921-1943. doi:10.1016/j.lfs.2007.02.037
- Jung H. Hyaluronidase: an overview of its properties, applications, and side effects. Arch Plast Surg. 2020;47:297-300. doi:10.5999/aps.2020.00752
- Epstein AE, Olshansky B, Naccarelli GV, et al. Practical management guide for clinicians who treat patients with amiodarone. Am J Med. 2016;129:468-475. doi:10.1016/j.amjmed.2015.08.039
- Harris L, McKenna WJ, Rowland E, et al. Side effects of long-term amiodarone therapy. Circulation. 1983;67:45-51. doi:10.1161/01.cir.67.1.45
- Jaworski K, Walecka I, Rudnicka L, et al. Cutaneous adverse reactions of amiodarone. Med Sci Monit. 2014;20:2369-2372. doi:10.12659/MSM.890881
- Kowey Peter R, Marinchak Roger A, Rials Seth J, et al. Intravenous amiodarone. J Am Coll Cardiol. 1997;29:1190-1198. doi:10.1016/S0735-1097(97)00069-7
- Russell SJ, Saltissi S. Amiodarone induced skin necrosis. Heart. 2006;92:1395. doi:10.1136/hrt.2005.086157
- Grove EL. Skin necrosis and consequences of accidental subcutaneous administration of amiodarone. Ugeskr Laeger. 2015;177:V66928.
- Fox AN, Villanueva R, Miller JL. Management of amiodarone extravasation with intradermal hyaluronidase. Am J Health Syst Pharm. 2017;74:1545-1548. doi:10.2146/ajhp160737
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632. doi:https://doi.org/10.1002/phar.1396
- Le A, Patel S. Extravasation of noncytotoxic drugs: a review of the literature. Ann Pharmacother. 2014;48:870-886. doi:10.1177/1060028014527820
- Slim AM, Roth JE, Duffy B, et al. The incidence of phlebitis with intravenous amiodarone at guideline dose recommendations. Mil Med. 2007;172:1279-1283.
- Girish KS, Kemparaju K. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 2007;80:1921-1943. doi:10.1016/j.lfs.2007.02.037
- Jung H. Hyaluronidase: an overview of its properties, applications, and side effects. Arch Plast Surg. 2020;47:297-300. doi:10.5999/aps.2020.00752
Practice Points
- Intravenous amiodarone administered peripherally can induce skin extravasation, leading to necrosis.
- Dermatologists should be aware that early intervention with intradermal hyaluronidase may reduce the severity of tissue damage caused by amiodarone-induced skin necrosis.

Genital Lentiginosis: A Benign Pigmentary Abnormality Often Raising Concern for Melanoma
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.

Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
.")
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
.")
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.
Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
To the Editor:
Genital lentiginosis (also known as mucosal melanotic macules, vulvar melanosis, penile melanosis, and penile lentigines) occurs in men and women.1 Lesions present in adult life as multifocal, asymmetrical, pigmented patches that can have a mottled appearance or exhibit skip areas. The irregular appearance of the pigmented areas often raises concern for melanoma. Biopsy reveals increased pigmentation along the basal layer of the epidermis; the irregular distribution of single melanocytes and pagetoid spread typical of melanoma in situ is not identified.
Genital lentiginosis usually occurs as an isolated finding; however, the condition can be a manifestation of Laugier-Hunziker syndrome, Carney complex, and Bannayan-Riley-Ruvalcaba syndrome.1-3 When it occurs as an isolated finding, the patient can be reassured and treatment is unnecessary. Because genital lentiginosis may mimic the appearance of melanoma, it is important for physicians to differentiate the two and make a correct diagnosis. We present a case of genital lentiginosis that mimicked vulvar melanoma.
A 64-year-old woman was referred by her gynecologist to dermatology to rule out vulvar melanoma. The patient had a history of hypothyroidism and hypercholesterolemia but was otherwise in good health. Genital examination revealed asymptomatic pigmented macules and patches of unknown duration (Figure 1). Specimens were taken from 3 areas by punch biopsy to clarify the diagnosis. All 3 specimens showed identical features including basilar pigmentation, occasional melanophages in the papillary dermis, and no evidence of nests or pagetoid spread of atypical melanocytes (Figures 2 and 3). Histologic findings were diagnostic for genital lentiginosis. The patient was reassured, and no treatment was provided. At 6-month follow-up there was no change in clinical appearance.
Genital lentiginosis is characterized by brown lesions that can have a mottled appearance and often are associated with skip areas.1 Lesions can be strikingly irregular and darkly pigmented.
Although the lesions of genital lentiginosis most often are isolated findings, they can be a clue to several uncommon syndromes such as autosomal-dominant Bannayan-Riley-Ruvalcaba syndrome, which is associated with genital lentiginosis, intestinal polyposis, and macrocephaly.3 Vascular malformations, lipomatosis, verrucal keratoses, and acrochordons can occur. Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome may share genetic linkage; mutations in the tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) has been implicated in both syndromes.4 Underlying Carney complex should be excluded when genital lentiginosis is encountered.
Genital lentiginosis is idiopathic in most instances, but reports of lesions occurring after annular lichen planus suggest a possible mechanism.5 The disappearance of lentigines after imatinib therapy suggests a role for c-kit, a receptor tyrosine kinase that is involved in intracellular signaling, in some cases.6 At times, lesions can simulate trichrome vitiligo or have a reticulate pattern.7
Men and women present at different points in the course of disease. Men often present with penile lesions 14 years after onset, on average; they notice a gradual increase in the size of lesions. Because women can have greater difficulty self-examining the genital region, they tend to present much later in the course but often within a few months after initial inspection.1,8
Genital lentiginosis can mimic melanoma with nonhomogeneous pigmentation, asymmetry, and unilateral distribution, which makes dermoscopic assessment of colors helpful in narrowing the differential diagnosis. Melanoma is associated with combinations of gray, red, blue, and white, which are not found in genital lentiginosis.9
Biopsy of a genital lentigo is diagnostic, distinguishing the lesion from melanoma—failing to reveal the atypical melanocytes and pagetoid spread characteristic of melanoma in situ. Histologic findings can cause diagnostic difficulties when concurrent lichen sclerosus is associated with genital lentigines or nevi.10
Lentigines on sun-damaged skin or in the setting of xeroderma pigmentosum have been associated with melanoma,11-13 but genital lentigines are not considered a form of precancerous melanosis. In women, early diagnosis is important when there is concern for melanoma because the prognosis for vulvar melanoma is improved in thin lesions.14
Other entities in the differential include secondary syphilis, which commonly presents as macules and scaly papules and can be found on mucosal surfaces such as the oral cavity,15 as well as Kaposi sarcoma, which is characterized by purplish, brown, or black macules, plaques, and nodules, more commonly in immunosuppressed patients.16
To avoid unwarranted concern and unnecessary surgery, dermatologists should be aware of genital lentigines and their characteristic presentation in adults.
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
- Hwang L, Wilson H, Orengo I. Off-center fold: irregular, pigmented genital macules. Arch Dermatol. 2000;136:1559-1564. doi:10.1001/archderm.136.12.1559-b
- Rhodes AR, Silverman RA, Harrist TJ, et al. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol. 1984;10:72-82. doi:10.1016/s0190-9622(84)80047-x
- Erkek E, Hizel S, Sanl C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53:639-643. doi:10.1016/j.jaad.2005.06.022
- Blum RR, Rahimizadeh A, Kardon N, et al. Genital lentigines in a 6-year-old boy with a family history of Cowden’s disease: clinical and genetic evidence of the linkage between Bannayan-Riley-Ruvalcaba syndrome and Cowden’s disease. J Cutan Med Surg. 2001;5:228-230. doi:10.1177/120347540100500307
- Isbary G, Dyall-Smith D, Coras-Stepanek B, et al. Penile lentigo (genital mucosal macule) following annular lichen planus: a possible association? Australas J Dermatol. 2014;55:159-161. doi:10.1111/ajd.12169
- Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313-1316. doi:10.1001/archdermatol.2009.263
- Romero- A, R, , et al. Reticulate genital pigmentation associated with localized vitiligo. Arch Dermatol. 2010; 146:574-575. doi:10.1001/archdermatol.2010.69
- Barnhill RL, Albert LS, Shama SK, et al. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. 1990;22:453-460. doi:10.1016/0190-9622(90)70064-o
- De Giorgi V, Gori A, Salvati L, et al. Clinical and dermoscopic features of vulvar melanosis over the last 20 years. JAMA Dermatol. 2020;156:1185–1191. doi:10.1001/jamadermatol.2020.2528
- El Shabrawi-Caelen L, Soyer HP, Schaeppi H, et al. Genital lentigines and melanocytic nevi with superimposed lichen sclerosus: a diagnostic challenge. J Am Acad Dermatol. 2004;50:690-694. doi:10.1016/j.jaad.2003.09.034
- Shatkin M, Helm MF, Muhlbauer A, et al. Solar lentigo evolving into fatal metastatic melanoma in a patient who initially refused surgery. N A J Med Sci. 2020;1:28-31. doi:10.7156/najms.2020.1301028
- Stern JB, Peck GL, Haupt HM, et al. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591-594. doi:10.1016/0190-9622(93)70079-9
- Byrom L, Barksdale S, Weedon D, et al. Unstable solar lentigo: a defined separate entity. Australas J Dermatol. 2016;57:229-234. doi:10.1111/ajd.12447
- Panizzon RG. Vulvar melanoma. Semin Dermatol. 1996;15:67-70. doi:10.1016/s1085-5629(96)80021-6
- Chapel TA. The signs and symptoms of secondary syphilis. Sex Transm Dis. 1980;7:161-164. doi:10.1097/00007435-198010000-00002
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151. doi:10.1002/jso.20090
Practice Points
- The irregular appearance of genital lentiginosis—multifocal, asymmetric, irregular, and darkly pigmented patches—often raises concern for melanoma but is benign.
- Certain genetic conditions can present with genital lentiginosis.
- Dermoscopic assessment of the lesion color is highly helpful in narrowing the differential diagnosis; seeing no gray, red, blue, or white makes melanoma less likely.
- Be aware of genital lentigines and their characteristic presentation in adulthood to avoid unwarranted concern and unneeded surgery.

Commentary: Novel Migraine Treatment Side Effects, November 2022
Calcitonin gene-related peptide (CGRP) antagonist medications are becoming more and more of a mainstay of both migraine prevention and acute treatment. Four CGRP monoclonal antibody medications and three oral CGRP antagonists are now available for prevention and acute treatment of migraine. When this class of medications was developed, many patients and providers were initially concerned regarding any potentially unanticipated long-term adverse events. The first CGRP antagonist medication, erenumab, was associated with constipation, and, although this has not been formally reported with other CGRP medications, there is some anecdotal evidence of gastrointestinal (GI) discomfort with both oral and other monoclonal antibody medications in this class. In 2021, erenumab was also reported to be associated with hypertension and a formal warning was issued for this.
Before CGRP was used for migraine prevention, it had been known to be a potent vasodilator. Although the randomized controlled trials (RCT) for all CGRP medications included a longitudinal review of blood pressure (BP) measures, none of those initial trials revealed an increased risk for hypertension or other cardiovascular events or concerns. Some of the initial trials even included patients at higher cardiovascular risk. Migraine itself is associated with an increased vascular risk; it is therefore extremely important to determine whether a potential treatment may be increasing this risk further.
In the study by de Vries and colleagues, all patients were started on either erenumab or fremanezumab and were included if they had a follow-up blood pressure measurement within 6 months. All patients had at least8 migraine days per month and had used four or more preventive medications. Patients taking erenumab were first started at 70 mg monthly and were optionally increased to 140 mg after 3 months. Fremanezumab was always given at a dose of 225 mg monthly (as opposed to 675 mg every 3 months). BP data were collected at baseline and followed up at every visit, with a maximum of 12 months of review. Patients were excluded if their baseline BP was measured while they were undergoing tapering an antihypertensive medication at the same time that they were starting the CGRP antagonist. Patients already treated for hypertension were included only if there were no changes to their hypertensive regimen.
Patients were also compared with a control group with a similar distribution in sex, age, and diagnosis, and who were not taking any migraine preventive medication that would affect their BP. Control group BP was also measured at least two different time points 1-3 months from baseline.
A total of 211 patients were enrolled: 109 in the erenumab group and 87 in the fremanezumab group. The erenumab group was also associated with an increase in both systolic BP (SBP) and diastolic BP (DBP) (SBP ≤ 9 mm Hg; DBP ≤ 6.3 mm Hg). The fremanezumab group had a lower increase in SBP (1 mm Hg) and no increase in DBP. Nine patients were started on antihypertensive medications, five of them after the baseline BP recording only. There was no change over time in the control group.
As noted, CGRP has a potent vasodilatory effect. Although not proven, a theory has been posited that CGRP receptor antagonist medications are somewhat more likely to lead to systemic adverse effects than are ligand-blocking medications. This may, at least in part, explain why erenumab appears to have more associated GI discomfort and constipation as well as a vascular effect with hypertension. In light of these findings, it is absolutely necessary for the additional CGRP medications to also be studied for these potential risk factors.
With the advent of novel acute treatments for migraine, many providers ask themselves what the potential risks and benefits are for each new class of drugs developed and approved over the past few years. Johnston and colleagues reviewed five recently published RCT to compare risk-benefit profiles for lasmiditan, rimegepant, and ubrogepant. Lasmiditan is the only medication in the ditan class, a serotonin (5HT-1F) receptor agonist medication approved for the acute treatment of migraine. Rimegepant and ubrogepant are oral CGRP antagonists.
Lasmiditan does have a potential risk for impairment while driving due to excessive sedation and dizziness; it is also more likely than the CGRP medications to lead to the development of medication overuse headache. The CGRP medications, however, are associated with a smaller responder rate for headache freedom at 2 hours. The investigators reviewed the data published in five RCT to develop a statistically based decision-making process that correlates with the number needed to treat vs the number needed to harm for all three of these medications. The number needed to treat is a statistically defined parameter that characterizes the number of patients that need to be treated with an intervention to achieve a positive event. The number needed to harm refers to an additional negative event relative to the reference treatment (placebo in the case of an RCT).
The reviewed studies compared multiple dosages of these medications. Efficacy outcomes were pain relief and pain freedom at 2 hours, sustained pain relief from 2 to 24 hours, and freedom from the most bothersome symptom at 2 hours. Safety outcomes were dizziness and nausea.
The number needed to treat was the lowest for 200 mg lasmiditan (twice the highest recommended acute dose), followed by 75 mg rimegepant. The number needed to harm was highest for 25 mg ubrogepant (half of the lowest recommended acute dose). Nausea was lowest for 75 mg rimegepant.
An individualized approach is always recommended when considering both preventive and acute treatments for migraine. It is definitely worth keeping these results in mind when discussing potential acute treatment options with patients. This is especially true when considering patients who may be more likely to experience either dizziness or nausea with other acute treatments. It is also worth individualizing a potential acute treatment when a patient experiences rapid-onset migraine symptoms. Further investigations into both acute and preventive treatments would enlighten and further individualize our clinical approaches.
Erenumab currently carries a prescriber warning for constipation. Although there has been some anecdotal evidence for constipation with other CGRP antagonists, this has never fully been investigated. Currently, the other CGRP medication options have fewer side effects and not all are associated with constipation. Kudrow and colleagues sought to review the incidence of constipation, and GI motility in general, with both erenumab and galcanezumab. Their hypothesis was that a single dose of erenumab would be associated with delayed GI motility and a single dose of galcanezumab would not be.
This study was conducted as a multicenter trial with single-blinding. A total of 65 patients were enrolled and given either 140 mg erenumab or 240 mg galcanezumab (the loading dose). GI motility was measured via a wireless motility capsule at baseline before treatment and repeated at 2 weeks. This test is approved by the US Food and Drug Administration for evaluation of gastric transit time in patients with suspected gastroparesis and for evaluation of colonic transit time in patients with chronic idiopathic constipation. Patients with prior GI symptoms were excluded, as were patients taking a tricyclic antidepressant or a calcium-channel blocker, owing to known constipation with these agents.
The primary endpoint in this study was change from a baseline colonic transit time 2 weeks after injection with the CGRP monoclonal antibody. Secondary endpoints included change from baseline in whole-gut transit time, gastric emptying time, small-bowel transfer time, and combined small- and large-bowel transfer time. The Gastrointestinal Symptom Rating Scale (GSRS) was also used, evaluating abdominal pain, reflux, indigestion, constipation, and diarrhea based on a 7-point response, ranging from no discomfort to very severe discomfort.
The primary endpoint of baseline change in colonic transit time was not statistically significant between the groups: A mean change of 5.8 hours was noted for erenumab and 5.4 hours for galcanezumab. Most secondary endpoints were also not statistically significantly different between the two groups. Small-bowel transit time was decreased in the galcanezumab group. When the patient-reported scales were reviewed, spontaneous bowel movements decreased significantly in the erenumab group, a finding that was not seen in the galcanezumab group and was statistically significant. The GSRS also showed a small but statistically significant change in the erenumab group.
This study does appear to show a significant difference in the two CGRP antagonist medications. The full side-effect profiles of the four CGRP monoclonal antibodies and three oral CGRP blocking medications available remain unknown. Further head-to-head comparisons will allow better differentiation of these options and better individualization of patient care.
Calcitonin gene-related peptide (CGRP) antagonist medications are becoming more and more of a mainstay of both migraine prevention and acute treatment. Four CGRP monoclonal antibody medications and three oral CGRP antagonists are now available for prevention and acute treatment of migraine. When this class of medications was developed, many patients and providers were initially concerned regarding any potentially unanticipated long-term adverse events. The first CGRP antagonist medication, erenumab, was associated with constipation, and, although this has not been formally reported with other CGRP medications, there is some anecdotal evidence of gastrointestinal (GI) discomfort with both oral and other monoclonal antibody medications in this class. In 2021, erenumab was also reported to be associated with hypertension and a formal warning was issued for this.
Before CGRP was used for migraine prevention, it had been known to be a potent vasodilator. Although the randomized controlled trials (RCT) for all CGRP medications included a longitudinal review of blood pressure (BP) measures, none of those initial trials revealed an increased risk for hypertension or other cardiovascular events or concerns. Some of the initial trials even included patients at higher cardiovascular risk. Migraine itself is associated with an increased vascular risk; it is therefore extremely important to determine whether a potential treatment may be increasing this risk further.
In the study by de Vries and colleagues, all patients were started on either erenumab or fremanezumab and were included if they had a follow-up blood pressure measurement within 6 months. All patients had at least8 migraine days per month and had used four or more preventive medications. Patients taking erenumab were first started at 70 mg monthly and were optionally increased to 140 mg after 3 months. Fremanezumab was always given at a dose of 225 mg monthly (as opposed to 675 mg every 3 months). BP data were collected at baseline and followed up at every visit, with a maximum of 12 months of review. Patients were excluded if their baseline BP was measured while they were undergoing tapering an antihypertensive medication at the same time that they were starting the CGRP antagonist. Patients already treated for hypertension were included only if there were no changes to their hypertensive regimen.
Patients were also compared with a control group with a similar distribution in sex, age, and diagnosis, and who were not taking any migraine preventive medication that would affect their BP. Control group BP was also measured at least two different time points 1-3 months from baseline.
A total of 211 patients were enrolled: 109 in the erenumab group and 87 in the fremanezumab group. The erenumab group was also associated with an increase in both systolic BP (SBP) and diastolic BP (DBP) (SBP ≤ 9 mm Hg; DBP ≤ 6.3 mm Hg). The fremanezumab group had a lower increase in SBP (1 mm Hg) and no increase in DBP. Nine patients were started on antihypertensive medications, five of them after the baseline BP recording only. There was no change over time in the control group.
As noted, CGRP has a potent vasodilatory effect. Although not proven, a theory has been posited that CGRP receptor antagonist medications are somewhat more likely to lead to systemic adverse effects than are ligand-blocking medications. This may, at least in part, explain why erenumab appears to have more associated GI discomfort and constipation as well as a vascular effect with hypertension. In light of these findings, it is absolutely necessary for the additional CGRP medications to also be studied for these potential risk factors.
With the advent of novel acute treatments for migraine, many providers ask themselves what the potential risks and benefits are for each new class of drugs developed and approved over the past few years. Johnston and colleagues reviewed five recently published RCT to compare risk-benefit profiles for lasmiditan, rimegepant, and ubrogepant. Lasmiditan is the only medication in the ditan class, a serotonin (5HT-1F) receptor agonist medication approved for the acute treatment of migraine. Rimegepant and ubrogepant are oral CGRP antagonists.
Lasmiditan does have a potential risk for impairment while driving due to excessive sedation and dizziness; it is also more likely than the CGRP medications to lead to the development of medication overuse headache. The CGRP medications, however, are associated with a smaller responder rate for headache freedom at 2 hours. The investigators reviewed the data published in five RCT to develop a statistically based decision-making process that correlates with the number needed to treat vs the number needed to harm for all three of these medications. The number needed to treat is a statistically defined parameter that characterizes the number of patients that need to be treated with an intervention to achieve a positive event. The number needed to harm refers to an additional negative event relative to the reference treatment (placebo in the case of an RCT).
The reviewed studies compared multiple dosages of these medications. Efficacy outcomes were pain relief and pain freedom at 2 hours, sustained pain relief from 2 to 24 hours, and freedom from the most bothersome symptom at 2 hours. Safety outcomes were dizziness and nausea.
The number needed to treat was the lowest for 200 mg lasmiditan (twice the highest recommended acute dose), followed by 75 mg rimegepant. The number needed to harm was highest for 25 mg ubrogepant (half of the lowest recommended acute dose). Nausea was lowest for 75 mg rimegepant.
An individualized approach is always recommended when considering both preventive and acute treatments for migraine. It is definitely worth keeping these results in mind when discussing potential acute treatment options with patients. This is especially true when considering patients who may be more likely to experience either dizziness or nausea with other acute treatments. It is also worth individualizing a potential acute treatment when a patient experiences rapid-onset migraine symptoms. Further investigations into both acute and preventive treatments would enlighten and further individualize our clinical approaches.
Erenumab currently carries a prescriber warning for constipation. Although there has been some anecdotal evidence for constipation with other CGRP antagonists, this has never fully been investigated. Currently, the other CGRP medication options have fewer side effects and not all are associated with constipation. Kudrow and colleagues sought to review the incidence of constipation, and GI motility in general, with both erenumab and galcanezumab. Their hypothesis was that a single dose of erenumab would be associated with delayed GI motility and a single dose of galcanezumab would not be.
This study was conducted as a multicenter trial with single-blinding. A total of 65 patients were enrolled and given either 140 mg erenumab or 240 mg galcanezumab (the loading dose). GI motility was measured via a wireless motility capsule at baseline before treatment and repeated at 2 weeks. This test is approved by the US Food and Drug Administration for evaluation of gastric transit time in patients with suspected gastroparesis and for evaluation of colonic transit time in patients with chronic idiopathic constipation. Patients with prior GI symptoms were excluded, as were patients taking a tricyclic antidepressant or a calcium-channel blocker, owing to known constipation with these agents.
The primary endpoint in this study was change from a baseline colonic transit time 2 weeks after injection with the CGRP monoclonal antibody. Secondary endpoints included change from baseline in whole-gut transit time, gastric emptying time, small-bowel transfer time, and combined small- and large-bowel transfer time. The Gastrointestinal Symptom Rating Scale (GSRS) was also used, evaluating abdominal pain, reflux, indigestion, constipation, and diarrhea based on a 7-point response, ranging from no discomfort to very severe discomfort.
The primary endpoint of baseline change in colonic transit time was not statistically significant between the groups: A mean change of 5.8 hours was noted for erenumab and 5.4 hours for galcanezumab. Most secondary endpoints were also not statistically significantly different between the two groups. Small-bowel transit time was decreased in the galcanezumab group. When the patient-reported scales were reviewed, spontaneous bowel movements decreased significantly in the erenumab group, a finding that was not seen in the galcanezumab group and was statistically significant. The GSRS also showed a small but statistically significant change in the erenumab group.
This study does appear to show a significant difference in the two CGRP antagonist medications. The full side-effect profiles of the four CGRP monoclonal antibodies and three oral CGRP blocking medications available remain unknown. Further head-to-head comparisons will allow better differentiation of these options and better individualization of patient care.
Calcitonin gene-related peptide (CGRP) antagonist medications are becoming more and more of a mainstay of both migraine prevention and acute treatment. Four CGRP monoclonal antibody medications and three oral CGRP antagonists are now available for prevention and acute treatment of migraine. When this class of medications was developed, many patients and providers were initially concerned regarding any potentially unanticipated long-term adverse events. The first CGRP antagonist medication, erenumab, was associated with constipation, and, although this has not been formally reported with other CGRP medications, there is some anecdotal evidence of gastrointestinal (GI) discomfort with both oral and other monoclonal antibody medications in this class. In 2021, erenumab was also reported to be associated with hypertension and a formal warning was issued for this.
Before CGRP was used for migraine prevention, it had been known to be a potent vasodilator. Although the randomized controlled trials (RCT) for all CGRP medications included a longitudinal review of blood pressure (BP) measures, none of those initial trials revealed an increased risk for hypertension or other cardiovascular events or concerns. Some of the initial trials even included patients at higher cardiovascular risk. Migraine itself is associated with an increased vascular risk; it is therefore extremely important to determine whether a potential treatment may be increasing this risk further.
In the study by de Vries and colleagues, all patients were started on either erenumab or fremanezumab and were included if they had a follow-up blood pressure measurement within 6 months. All patients had at least8 migraine days per month and had used four or more preventive medications. Patients taking erenumab were first started at 70 mg monthly and were optionally increased to 140 mg after 3 months. Fremanezumab was always given at a dose of 225 mg monthly (as opposed to 675 mg every 3 months). BP data were collected at baseline and followed up at every visit, with a maximum of 12 months of review. Patients were excluded if their baseline BP was measured while they were undergoing tapering an antihypertensive medication at the same time that they were starting the CGRP antagonist. Patients already treated for hypertension were included only if there were no changes to their hypertensive regimen.
Patients were also compared with a control group with a similar distribution in sex, age, and diagnosis, and who were not taking any migraine preventive medication that would affect their BP. Control group BP was also measured at least two different time points 1-3 months from baseline.
A total of 211 patients were enrolled: 109 in the erenumab group and 87 in the fremanezumab group. The erenumab group was also associated with an increase in both systolic BP (SBP) and diastolic BP (DBP) (SBP ≤ 9 mm Hg; DBP ≤ 6.3 mm Hg). The fremanezumab group had a lower increase in SBP (1 mm Hg) and no increase in DBP. Nine patients were started on antihypertensive medications, five of them after the baseline BP recording only. There was no change over time in the control group.
As noted, CGRP has a potent vasodilatory effect. Although not proven, a theory has been posited that CGRP receptor antagonist medications are somewhat more likely to lead to systemic adverse effects than are ligand-blocking medications. This may, at least in part, explain why erenumab appears to have more associated GI discomfort and constipation as well as a vascular effect with hypertension. In light of these findings, it is absolutely necessary for the additional CGRP medications to also be studied for these potential risk factors.
With the advent of novel acute treatments for migraine, many providers ask themselves what the potential risks and benefits are for each new class of drugs developed and approved over the past few years. Johnston and colleagues reviewed five recently published RCT to compare risk-benefit profiles for lasmiditan, rimegepant, and ubrogepant. Lasmiditan is the only medication in the ditan class, a serotonin (5HT-1F) receptor agonist medication approved for the acute treatment of migraine. Rimegepant and ubrogepant are oral CGRP antagonists.
Lasmiditan does have a potential risk for impairment while driving due to excessive sedation and dizziness; it is also more likely than the CGRP medications to lead to the development of medication overuse headache. The CGRP medications, however, are associated with a smaller responder rate for headache freedom at 2 hours. The investigators reviewed the data published in five RCT to develop a statistically based decision-making process that correlates with the number needed to treat vs the number needed to harm for all three of these medications. The number needed to treat is a statistically defined parameter that characterizes the number of patients that need to be treated with an intervention to achieve a positive event. The number needed to harm refers to an additional negative event relative to the reference treatment (placebo in the case of an RCT).
The reviewed studies compared multiple dosages of these medications. Efficacy outcomes were pain relief and pain freedom at 2 hours, sustained pain relief from 2 to 24 hours, and freedom from the most bothersome symptom at 2 hours. Safety outcomes were dizziness and nausea.
The number needed to treat was the lowest for 200 mg lasmiditan (twice the highest recommended acute dose), followed by 75 mg rimegepant. The number needed to harm was highest for 25 mg ubrogepant (half of the lowest recommended acute dose). Nausea was lowest for 75 mg rimegepant.
An individualized approach is always recommended when considering both preventive and acute treatments for migraine. It is definitely worth keeping these results in mind when discussing potential acute treatment options with patients. This is especially true when considering patients who may be more likely to experience either dizziness or nausea with other acute treatments. It is also worth individualizing a potential acute treatment when a patient experiences rapid-onset migraine symptoms. Further investigations into both acute and preventive treatments would enlighten and further individualize our clinical approaches.
Erenumab currently carries a prescriber warning for constipation. Although there has been some anecdotal evidence for constipation with other CGRP antagonists, this has never fully been investigated. Currently, the other CGRP medication options have fewer side effects and not all are associated with constipation. Kudrow and colleagues sought to review the incidence of constipation, and GI motility in general, with both erenumab and galcanezumab. Their hypothesis was that a single dose of erenumab would be associated with delayed GI motility and a single dose of galcanezumab would not be.
This study was conducted as a multicenter trial with single-blinding. A total of 65 patients were enrolled and given either 140 mg erenumab or 240 mg galcanezumab (the loading dose). GI motility was measured via a wireless motility capsule at baseline before treatment and repeated at 2 weeks. This test is approved by the US Food and Drug Administration for evaluation of gastric transit time in patients with suspected gastroparesis and for evaluation of colonic transit time in patients with chronic idiopathic constipation. Patients with prior GI symptoms were excluded, as were patients taking a tricyclic antidepressant or a calcium-channel blocker, owing to known constipation with these agents.
The primary endpoint in this study was change from a baseline colonic transit time 2 weeks after injection with the CGRP monoclonal antibody. Secondary endpoints included change from baseline in whole-gut transit time, gastric emptying time, small-bowel transfer time, and combined small- and large-bowel transfer time. The Gastrointestinal Symptom Rating Scale (GSRS) was also used, evaluating abdominal pain, reflux, indigestion, constipation, and diarrhea based on a 7-point response, ranging from no discomfort to very severe discomfort.
The primary endpoint of baseline change in colonic transit time was not statistically significant between the groups: A mean change of 5.8 hours was noted for erenumab and 5.4 hours for galcanezumab. Most secondary endpoints were also not statistically significantly different between the two groups. Small-bowel transit time was decreased in the galcanezumab group. When the patient-reported scales were reviewed, spontaneous bowel movements decreased significantly in the erenumab group, a finding that was not seen in the galcanezumab group and was statistically significant. The GSRS also showed a small but statistically significant change in the erenumab group.
This study does appear to show a significant difference in the two CGRP antagonist medications. The full side-effect profiles of the four CGRP monoclonal antibodies and three oral CGRP blocking medications available remain unknown. Further head-to-head comparisons will allow better differentiation of these options and better individualization of patient care.
How to prevent a feared complication after joint replacement
Knee and hip replacements can improve how well patients get around and can significantly increase their quality of life. But if a bone near the new joint breaks, the injury can be a major setback for the patient’s mobility, and the consequences can be life-threatening.
The proportion of patients who experience a periprosthetic fracture within 5 years of total hip arthroplasty is 0.9%. After total knee arthroplasty (TKA), the proportion is 0.6%, research shows.
Those rates might seem low. But given that more than a million of these joint replacement surgeries are performed each year in the United States – they are the most common inpatient surgical procedures among people aged 65 and older – thousands of revision surgeries due to periprosthetic fractures occur each year.
Primary care clinicians who make their patients’ bone health a priority early on – years before surgery, ideally – may help patients enjoy the benefits of new joints long term.

At the 2022 annual Santa Fe Bone Symposium this summer, Susan V. Bukata, MD, professor and chair of orthopedics at the University of California, San Diego, showed an image of “what we’re trying to avoid” – a patient with a broken bone and infection. Unfortunately, Dr. Bukata said, the patient’s clinicians had not adequately addressed her skeletal health before the injury.
“This is a complete disaster for this person who went in having a total hip to improve their function and now will probably never walk normally on that leg,” Dr. Bukata said at the meeting.
The patient eventually underwent total femur replacement. Five surgeries were required to clear the infection.
Medical and surgical advances have allowed more people – including older patients and those with other medical conditions – to undergo joint replacement surgery, including replacement of knees, hips, and shoulders.
The surgeries often are performed for adults whose bones are thinning. Sometimes surgeons don’t realize just how thin a patient’s bone is until they are operating.
Prioritizing bone health
In patients with osteoporosis, the bone surrounding the new joint is weaker than the metal of the prosthesis, and the metal can rip out of the bone, Dr. Bukata told this news organization. A periprosthetic fracture should be recognized as an osteoporotic fracture, too, although these fractures have not typically been categorized that way, she said.
People live with total joints in place for as long as 40 years, and fractures around the implants are “one of the fastest growing injuries that we are seeing in older patients,” Dr. Bukata said. “People don’t think of those as osteoporotic fractures. But a 90-year-old who falls and breaks next to their total knee, if they didn’t have that total knee in place, everybody would be, like, ‘Oh, that’s an osteoporotic fracture.’ ”
Periprosthetic fractures tend not to occur right after surgery but rather after the bone continues to lose density as the patient ages, Dr. Bukata said.
Missed chances
One approach to preventing periprosthetic fractures could involve prioritizing bone health earlier in life and diagnosing and treating osteoporosis well before a patient is scheduled for surgery.
A patient’s initial visit to their primary care doctor because of joint pain is an opportunity to check on and promote their bone health, given that they might be a candidate for surgery in the future, Dr. Bukata said.
Ahead of a scheduled surgery, patients can see endocrinologists or rheumatologists to receive medication to try to strengthen bones. Doctors may be limited in how much of a difference they can make in a matter of several weeks or months with these drugs, however. These patients still likely will need to be treated as if they have osteoporosis, Dr. Bukata said.
When surgeons realize that a patient has weaker bones while they are in the middle of an operation, they should emphasize the importance of bone health after the procedure, Dr. Bukata said.
Strengthening, maintaining, and protecting bone should be seen as a long-term investment in the patient’s success after a joint replacement. That said, “There is no clear evidence or protocol for us to follow,” she said. “The mantra at UCSD now is, let’s keep it simple. Get the patient on track. And then we can always refine things as we continue to treat the patient.”
Health systems should establish routines in which bone health is discussed before surgery in the way patient education programs address smoking cessation, nutrition, and weight management, Dr. Bukata said. Another step in the right direction could involve setting electronic medical records to automatically order assessments of bone health when a surgeon books a case.

Linda A. Russell, MD, rheumatologist and director of perioperative medicine at the Hospital for Special Surgery in New York, said periprosthetic fractures are a “complication we fear.”
“It’s a big deal to try to repair it,” Dr. Russell said. “Sometimes you need to revise the joint, or sometimes you need to put lots more hardware in.” Surgeons increasingly appreciate the need to pay attention to the quality of the bone before they operate, she said.
Nevertheless, Dr. Russell does not necessarily say that such cases call for alarm or particularly aggressive treatment regimens – just regular bone health evaluations before and after surgery to see whether patients have osteoporosis and are candidates for treatment.
Lifelong effort
In some ways, to address bone health at the time of surgery may be too late.
Bone health “is not something that you can have as an afterthought when you’re 75 years old,” said Elizabeth Matzkin, MD, chief of women’s sports medicine at Brigham and Women’s Hospital, in Boston.
The chance of being able to rebuild bone mass at that age is slim. If patients maximize bone density when they are young, they can afford to lose some bone mass each year as they age.
To that end, a healthy diet, exercise, not smoking, and cutting back on alcohol can help, she said.
For Dr. Matzkin, a fragility fracture is a red flag that the patient’s bone density is probably not optimal. In such cases, she prepares for various scenarios during surgery, such as a screw not holding in a low-density bone.
Recently published research reflects that prior fragility fractures are a significant risk factor for complications after surgery, including periprosthetic fractures.
Edward J. Testa, MD, of Brown University, Providence, R.I., and colleagues analyzed insurance claims to compare outcomes for 24,398 patients who had experienced a fragility fracture – that is, a break caused by low-velocity trauma such as a fall – during the 3 years before their TKA procedure and a matched group of patients who were similar in many respects but who had not had a fragility fracture in the 3 years before surgery.
Dr. Testa’s group found that a history of fragility fracture was associated with higher rates of complications in the year after surgery, including hospital readmissions (hazard ratio = 1.30; 95% CI, 1.22-1.38), periprosthetic fractures (odds ratio = 2.72; 95% CI, 1.89-3.99), and secondary fragility fractures (OR = 4.62; 95% CI, 4.19-5.12). Patients who had previously experienced fragility fractures also experienced dislocated prostheses (OR = 1.76; 95% CI, 1.22-2.56) and periprosthetic infections (OR = 1.49; 95% CI, 1.29-1.71) at higher rates.
The rates of complications were similar regardless of whether patients had filled a prescription for medications used to treat osteoporosis, including bisphosphonates, vitamin D replacement, raloxifene, and denosumab, the researchers reported.
The lack of a clear association between these treatments and patient outcomes could be related to an insufficient duration of pharmacotherapy before or after TKA, poor medication adherence, or small sample sizes, Dr. Testa said.
Given the findings, which were published online in the Journal of Arthroplasty, “patients with a history of fragility fracture should be identified and counseled appropriately for a possible increased risk of the aforementioned complications, and optimized when possible, prior to undergoing TKA,” Dr. Testa told this news organization. “Ultimately, the decision to move forward with surgery is far more complex than the identification of this sole, yet important, risk factor for certain postoperative, implant-related complications.”
Treatment gaps
Prior research has shown that women aged 70 years and older are at higher risk for periprosthetic fractures. Many women in this age group who could receive treatment for osteoporosis do not, and major treatment gaps exist worldwide, noted Neil Binkley, MD, with the University of Wisconsin–Madison, in a separate talk at the Santa Fe Bone Symposium.
Ensuring adequate protein intake and addressing the risk of falling are other measures that clinicians can take to promote healthy bones, apart from prescribing drugs, he said.
Unpublished data from one group show that nearly 90% of periprosthetic fractures may result from falls, while about 8% may be spontaneous. “We need to be thinking about falls,” Dr. Binkley said.
Dr. Bukata has consulted for Amgen, Radius, and Solarea Bio and has served on a speakers bureau for Radius. She also is a board member for the Orthopaedic Research Society and the American Academy of Orthopaedic Surgeons Board of Specialty Societies. Dr. Binkley has received research support from Radius and has consulted for Amgen.
A version of this article first appeared on Medscape.com.
Knee and hip replacements can improve how well patients get around and can significantly increase their quality of life. But if a bone near the new joint breaks, the injury can be a major setback for the patient’s mobility, and the consequences can be life-threatening.
The proportion of patients who experience a periprosthetic fracture within 5 years of total hip arthroplasty is 0.9%. After total knee arthroplasty (TKA), the proportion is 0.6%, research shows.
Those rates might seem low. But given that more than a million of these joint replacement surgeries are performed each year in the United States – they are the most common inpatient surgical procedures among people aged 65 and older – thousands of revision surgeries due to periprosthetic fractures occur each year.
Primary care clinicians who make their patients’ bone health a priority early on – years before surgery, ideally – may help patients enjoy the benefits of new joints long term.
At the 2022 annual Santa Fe Bone Symposium this summer, Susan V. Bukata, MD, professor and chair of orthopedics at the University of California, San Diego, showed an image of “what we’re trying to avoid” – a patient with a broken bone and infection. Unfortunately, Dr. Bukata said, the patient’s clinicians had not adequately addressed her skeletal health before the injury.
“This is a complete disaster for this person who went in having a total hip to improve their function and now will probably never walk normally on that leg,” Dr. Bukata said at the meeting.
The patient eventually underwent total femur replacement. Five surgeries were required to clear the infection.
Medical and surgical advances have allowed more people – including older patients and those with other medical conditions – to undergo joint replacement surgery, including replacement of knees, hips, and shoulders.
The surgeries often are performed for adults whose bones are thinning. Sometimes surgeons don’t realize just how thin a patient’s bone is until they are operating.
Prioritizing bone health
In patients with osteoporosis, the bone surrounding the new joint is weaker than the metal of the prosthesis, and the metal can rip out of the bone, Dr. Bukata told this news organization. A periprosthetic fracture should be recognized as an osteoporotic fracture, too, although these fractures have not typically been categorized that way, she said.
People live with total joints in place for as long as 40 years, and fractures around the implants are “one of the fastest growing injuries that we are seeing in older patients,” Dr. Bukata said. “People don’t think of those as osteoporotic fractures. But a 90-year-old who falls and breaks next to their total knee, if they didn’t have that total knee in place, everybody would be, like, ‘Oh, that’s an osteoporotic fracture.’ ”
Periprosthetic fractures tend not to occur right after surgery but rather after the bone continues to lose density as the patient ages, Dr. Bukata said.
Missed chances
One approach to preventing periprosthetic fractures could involve prioritizing bone health earlier in life and diagnosing and treating osteoporosis well before a patient is scheduled for surgery.
A patient’s initial visit to their primary care doctor because of joint pain is an opportunity to check on and promote their bone health, given that they might be a candidate for surgery in the future, Dr. Bukata said.
Ahead of a scheduled surgery, patients can see endocrinologists or rheumatologists to receive medication to try to strengthen bones. Doctors may be limited in how much of a difference they can make in a matter of several weeks or months with these drugs, however. These patients still likely will need to be treated as if they have osteoporosis, Dr. Bukata said.
When surgeons realize that a patient has weaker bones while they are in the middle of an operation, they should emphasize the importance of bone health after the procedure, Dr. Bukata said.
Strengthening, maintaining, and protecting bone should be seen as a long-term investment in the patient’s success after a joint replacement. That said, “There is no clear evidence or protocol for us to follow,” she said. “The mantra at UCSD now is, let’s keep it simple. Get the patient on track. And then we can always refine things as we continue to treat the patient.”
Health systems should establish routines in which bone health is discussed before surgery in the way patient education programs address smoking cessation, nutrition, and weight management, Dr. Bukata said. Another step in the right direction could involve setting electronic medical records to automatically order assessments of bone health when a surgeon books a case.
Linda A. Russell, MD, rheumatologist and director of perioperative medicine at the Hospital for Special Surgery in New York, said periprosthetic fractures are a “complication we fear.”
“It’s a big deal to try to repair it,” Dr. Russell said. “Sometimes you need to revise the joint, or sometimes you need to put lots more hardware in.” Surgeons increasingly appreciate the need to pay attention to the quality of the bone before they operate, she said.
Nevertheless, Dr. Russell does not necessarily say that such cases call for alarm or particularly aggressive treatment regimens – just regular bone health evaluations before and after surgery to see whether patients have osteoporosis and are candidates for treatment.
Lifelong effort
In some ways, to address bone health at the time of surgery may be too late.
Bone health “is not something that you can have as an afterthought when you’re 75 years old,” said Elizabeth Matzkin, MD, chief of women’s sports medicine at Brigham and Women’s Hospital, in Boston.
The chance of being able to rebuild bone mass at that age is slim. If patients maximize bone density when they are young, they can afford to lose some bone mass each year as they age.
To that end, a healthy diet, exercise, not smoking, and cutting back on alcohol can help, she said.
For Dr. Matzkin, a fragility fracture is a red flag that the patient’s bone density is probably not optimal. In such cases, she prepares for various scenarios during surgery, such as a screw not holding in a low-density bone.
Recently published research reflects that prior fragility fractures are a significant risk factor for complications after surgery, including periprosthetic fractures.
Edward J. Testa, MD, of Brown University, Providence, R.I., and colleagues analyzed insurance claims to compare outcomes for 24,398 patients who had experienced a fragility fracture – that is, a break caused by low-velocity trauma such as a fall – during the 3 years before their TKA procedure and a matched group of patients who were similar in many respects but who had not had a fragility fracture in the 3 years before surgery.
Dr. Testa’s group found that a history of fragility fracture was associated with higher rates of complications in the year after surgery, including hospital readmissions (hazard ratio = 1.30; 95% CI, 1.22-1.38), periprosthetic fractures (odds ratio = 2.72; 95% CI, 1.89-3.99), and secondary fragility fractures (OR = 4.62; 95% CI, 4.19-5.12). Patients who had previously experienced fragility fractures also experienced dislocated prostheses (OR = 1.76; 95% CI, 1.22-2.56) and periprosthetic infections (OR = 1.49; 95% CI, 1.29-1.71) at higher rates.
The rates of complications were similar regardless of whether patients had filled a prescription for medications used to treat osteoporosis, including bisphosphonates, vitamin D replacement, raloxifene, and denosumab, the researchers reported.
The lack of a clear association between these treatments and patient outcomes could be related to an insufficient duration of pharmacotherapy before or after TKA, poor medication adherence, or small sample sizes, Dr. Testa said.
Given the findings, which were published online in the Journal of Arthroplasty, “patients with a history of fragility fracture should be identified and counseled appropriately for a possible increased risk of the aforementioned complications, and optimized when possible, prior to undergoing TKA,” Dr. Testa told this news organization. “Ultimately, the decision to move forward with surgery is far more complex than the identification of this sole, yet important, risk factor for certain postoperative, implant-related complications.”
Treatment gaps
Prior research has shown that women aged 70 years and older are at higher risk for periprosthetic fractures. Many women in this age group who could receive treatment for osteoporosis do not, and major treatment gaps exist worldwide, noted Neil Binkley, MD, with the University of Wisconsin–Madison, in a separate talk at the Santa Fe Bone Symposium.
Ensuring adequate protein intake and addressing the risk of falling are other measures that clinicians can take to promote healthy bones, apart from prescribing drugs, he said.
Unpublished data from one group show that nearly 90% of periprosthetic fractures may result from falls, while about 8% may be spontaneous. “We need to be thinking about falls,” Dr. Binkley said.
Dr. Bukata has consulted for Amgen, Radius, and Solarea Bio and has served on a speakers bureau for Radius. She also is a board member for the Orthopaedic Research Society and the American Academy of Orthopaedic Surgeons Board of Specialty Societies. Dr. Binkley has received research support from Radius and has consulted for Amgen.
A version of this article first appeared on Medscape.com.
Knee and hip replacements can improve how well patients get around and can significantly increase their quality of life. But if a bone near the new joint breaks, the injury can be a major setback for the patient’s mobility, and the consequences can be life-threatening.
The proportion of patients who experience a periprosthetic fracture within 5 years of total hip arthroplasty is 0.9%. After total knee arthroplasty (TKA), the proportion is 0.6%, research shows.
Those rates might seem low. But given that more than a million of these joint replacement surgeries are performed each year in the United States – they are the most common inpatient surgical procedures among people aged 65 and older – thousands of revision surgeries due to periprosthetic fractures occur each year.
Primary care clinicians who make their patients’ bone health a priority early on – years before surgery, ideally – may help patients enjoy the benefits of new joints long term.
At the 2022 annual Santa Fe Bone Symposium this summer, Susan V. Bukata, MD, professor and chair of orthopedics at the University of California, San Diego, showed an image of “what we’re trying to avoid” – a patient with a broken bone and infection. Unfortunately, Dr. Bukata said, the patient’s clinicians had not adequately addressed her skeletal health before the injury.
“This is a complete disaster for this person who went in having a total hip to improve their function and now will probably never walk normally on that leg,” Dr. Bukata said at the meeting.
The patient eventually underwent total femur replacement. Five surgeries were required to clear the infection.
Medical and surgical advances have allowed more people – including older patients and those with other medical conditions – to undergo joint replacement surgery, including replacement of knees, hips, and shoulders.
The surgeries often are performed for adults whose bones are thinning. Sometimes surgeons don’t realize just how thin a patient’s bone is until they are operating.
Prioritizing bone health
In patients with osteoporosis, the bone surrounding the new joint is weaker than the metal of the prosthesis, and the metal can rip out of the bone, Dr. Bukata told this news organization. A periprosthetic fracture should be recognized as an osteoporotic fracture, too, although these fractures have not typically been categorized that way, she said.
People live with total joints in place for as long as 40 years, and fractures around the implants are “one of the fastest growing injuries that we are seeing in older patients,” Dr. Bukata said. “People don’t think of those as osteoporotic fractures. But a 90-year-old who falls and breaks next to their total knee, if they didn’t have that total knee in place, everybody would be, like, ‘Oh, that’s an osteoporotic fracture.’ ”
Periprosthetic fractures tend not to occur right after surgery but rather after the bone continues to lose density as the patient ages, Dr. Bukata said.
Missed chances
One approach to preventing periprosthetic fractures could involve prioritizing bone health earlier in life and diagnosing and treating osteoporosis well before a patient is scheduled for surgery.
A patient’s initial visit to their primary care doctor because of joint pain is an opportunity to check on and promote their bone health, given that they might be a candidate for surgery in the future, Dr. Bukata said.
Ahead of a scheduled surgery, patients can see endocrinologists or rheumatologists to receive medication to try to strengthen bones. Doctors may be limited in how much of a difference they can make in a matter of several weeks or months with these drugs, however. These patients still likely will need to be treated as if they have osteoporosis, Dr. Bukata said.
When surgeons realize that a patient has weaker bones while they are in the middle of an operation, they should emphasize the importance of bone health after the procedure, Dr. Bukata said.
Strengthening, maintaining, and protecting bone should be seen as a long-term investment in the patient’s success after a joint replacement. That said, “There is no clear evidence or protocol for us to follow,” she said. “The mantra at UCSD now is, let’s keep it simple. Get the patient on track. And then we can always refine things as we continue to treat the patient.”
Health systems should establish routines in which bone health is discussed before surgery in the way patient education programs address smoking cessation, nutrition, and weight management, Dr. Bukata said. Another step in the right direction could involve setting electronic medical records to automatically order assessments of bone health when a surgeon books a case.
Linda A. Russell, MD, rheumatologist and director of perioperative medicine at the Hospital for Special Surgery in New York, said periprosthetic fractures are a “complication we fear.”
“It’s a big deal to try to repair it,” Dr. Russell said. “Sometimes you need to revise the joint, or sometimes you need to put lots more hardware in.” Surgeons increasingly appreciate the need to pay attention to the quality of the bone before they operate, she said.
Nevertheless, Dr. Russell does not necessarily say that such cases call for alarm or particularly aggressive treatment regimens – just regular bone health evaluations before and after surgery to see whether patients have osteoporosis and are candidates for treatment.
Lifelong effort
In some ways, to address bone health at the time of surgery may be too late.
Bone health “is not something that you can have as an afterthought when you’re 75 years old,” said Elizabeth Matzkin, MD, chief of women’s sports medicine at Brigham and Women’s Hospital, in Boston.
The chance of being able to rebuild bone mass at that age is slim. If patients maximize bone density when they are young, they can afford to lose some bone mass each year as they age.
To that end, a healthy diet, exercise, not smoking, and cutting back on alcohol can help, she said.
For Dr. Matzkin, a fragility fracture is a red flag that the patient’s bone density is probably not optimal. In such cases, she prepares for various scenarios during surgery, such as a screw not holding in a low-density bone.
Recently published research reflects that prior fragility fractures are a significant risk factor for complications after surgery, including periprosthetic fractures.
Edward J. Testa, MD, of Brown University, Providence, R.I., and colleagues analyzed insurance claims to compare outcomes for 24,398 patients who had experienced a fragility fracture – that is, a break caused by low-velocity trauma such as a fall – during the 3 years before their TKA procedure and a matched group of patients who were similar in many respects but who had not had a fragility fracture in the 3 years before surgery.
Dr. Testa’s group found that a history of fragility fracture was associated with higher rates of complications in the year after surgery, including hospital readmissions (hazard ratio = 1.30; 95% CI, 1.22-1.38), periprosthetic fractures (odds ratio = 2.72; 95% CI, 1.89-3.99), and secondary fragility fractures (OR = 4.62; 95% CI, 4.19-5.12). Patients who had previously experienced fragility fractures also experienced dislocated prostheses (OR = 1.76; 95% CI, 1.22-2.56) and periprosthetic infections (OR = 1.49; 95% CI, 1.29-1.71) at higher rates.
The rates of complications were similar regardless of whether patients had filled a prescription for medications used to treat osteoporosis, including bisphosphonates, vitamin D replacement, raloxifene, and denosumab, the researchers reported.
The lack of a clear association between these treatments and patient outcomes could be related to an insufficient duration of pharmacotherapy before or after TKA, poor medication adherence, or small sample sizes, Dr. Testa said.
Given the findings, which were published online in the Journal of Arthroplasty, “patients with a history of fragility fracture should be identified and counseled appropriately for a possible increased risk of the aforementioned complications, and optimized when possible, prior to undergoing TKA,” Dr. Testa told this news organization. “Ultimately, the decision to move forward with surgery is far more complex than the identification of this sole, yet important, risk factor for certain postoperative, implant-related complications.”
Treatment gaps
Prior research has shown that women aged 70 years and older are at higher risk for periprosthetic fractures. Many women in this age group who could receive treatment for osteoporosis do not, and major treatment gaps exist worldwide, noted Neil Binkley, MD, with the University of Wisconsin–Madison, in a separate talk at the Santa Fe Bone Symposium.
Ensuring adequate protein intake and addressing the risk of falling are other measures that clinicians can take to promote healthy bones, apart from prescribing drugs, he said.
Unpublished data from one group show that nearly 90% of periprosthetic fractures may result from falls, while about 8% may be spontaneous. “We need to be thinking about falls,” Dr. Binkley said.
Dr. Bukata has consulted for Amgen, Radius, and Solarea Bio and has served on a speakers bureau for Radius. She also is a board member for the Orthopaedic Research Society and the American Academy of Orthopaedic Surgeons Board of Specialty Societies. Dr. Binkley has received research support from Radius and has consulted for Amgen.
A version of this article first appeared on Medscape.com.
Mid-October flulike illness cases higher than past 5 years
Outpatient visits for influenzalike illness (ILI), which includes influenza, SARS-CoV-2, and RSV, were higher after 3 weeks than for any of the previous five flu seasons: 3.3% of visits reported through the CDC’s Outpatient Influenza-like Illness Surveillance Network involved ILI as of Oct. 22. The highest comparable rate in the previous 5 years was the 1.9% recorded in late October of 2021, shortly after the definition of ILI was changed to also include illnesses other than influenza.
This season’s higher flu activity is in contrast to the previous two, which were unusually mild. The change, however, is not unexpected, as William Schaffner, MD, an infectious disease expert and professor of preventive medicine at Vanderbilt University, recently told CNN.
“Here we are in the middle of October – not the middle of November – we’re already seeing scattered influenza cases, even hospitalized influenza cases, around the country,” he said. “So we know that this virus is now spreading out in the community already. It’s gathering speed already. It looks to me to be about a month early.”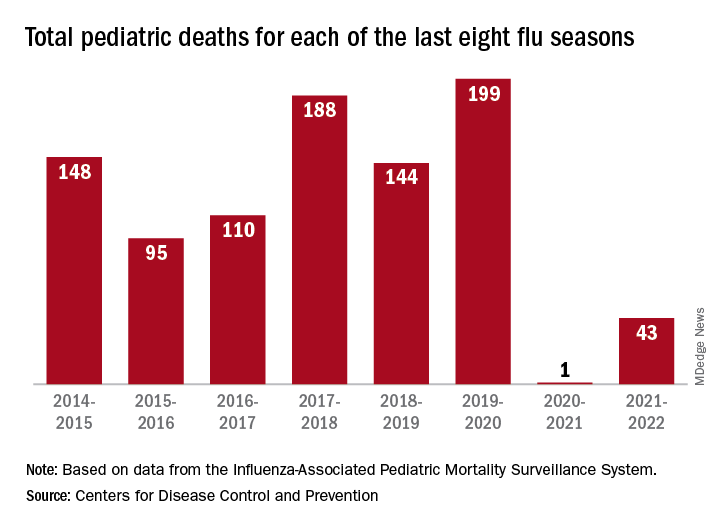
One indication of the mildness of the previous two flu seasons was the number of deaths, both pediatric and overall. Influenza-associated pediatric deaths had averaged about 110 per season over the previous eight seasons, compared with just 1 for 2020-2021 and 43 in 2021-2022. Overall flu deaths never reached 1% of all weekly deaths for either season, well below baseline levels for the flu, which range from 5.5% to 6.8%, CDC data show.
Other indicators of early severity
This season’s early rise in viral activity also can be seen in hospitalizations. The cumulative rate of flu-related admissions was 1.5 per 100,000 population as of Oct. 22, higher than the rate observed in the comparable week of previous seasons going back to 2010-2011, according to the CDC’s Influenza Hospitalization Surveillance Network.
A look at state reports of ILI outpatient visit rates shows that the District of Columbia and South Carolina are already in the very high range of the CDC’s severity scale, while 11 states are in the high range. Again going back to 2010-2011, no jurisdiction has ever been in the very high range this early in the season, based on data from the Outpatient Influenza-like Illness Surveillance Network.
Outpatient visits for influenzalike illness (ILI), which includes influenza, SARS-CoV-2, and RSV, were higher after 3 weeks than for any of the previous five flu seasons: 3.3% of visits reported through the CDC’s Outpatient Influenza-like Illness Surveillance Network involved ILI as of Oct. 22. The highest comparable rate in the previous 5 years was the 1.9% recorded in late October of 2021, shortly after the definition of ILI was changed to also include illnesses other than influenza.
This season’s higher flu activity is in contrast to the previous two, which were unusually mild. The change, however, is not unexpected, as William Schaffner, MD, an infectious disease expert and professor of preventive medicine at Vanderbilt University, recently told CNN.
“Here we are in the middle of October – not the middle of November – we’re already seeing scattered influenza cases, even hospitalized influenza cases, around the country,” he said. “So we know that this virus is now spreading out in the community already. It’s gathering speed already. It looks to me to be about a month early.”
One indication of the mildness of the previous two flu seasons was the number of deaths, both pediatric and overall. Influenza-associated pediatric deaths had averaged about 110 per season over the previous eight seasons, compared with just 1 for 2020-2021 and 43 in 2021-2022. Overall flu deaths never reached 1% of all weekly deaths for either season, well below baseline levels for the flu, which range from 5.5% to 6.8%, CDC data show.
Other indicators of early severity
This season’s early rise in viral activity also can be seen in hospitalizations. The cumulative rate of flu-related admissions was 1.5 per 100,000 population as of Oct. 22, higher than the rate observed in the comparable week of previous seasons going back to 2010-2011, according to the CDC’s Influenza Hospitalization Surveillance Network.
A look at state reports of ILI outpatient visit rates shows that the District of Columbia and South Carolina are already in the very high range of the CDC’s severity scale, while 11 states are in the high range. Again going back to 2010-2011, no jurisdiction has ever been in the very high range this early in the season, based on data from the Outpatient Influenza-like Illness Surveillance Network.
Outpatient visits for influenzalike illness (ILI), which includes influenza, SARS-CoV-2, and RSV, were higher after 3 weeks than for any of the previous five flu seasons: 3.3% of visits reported through the CDC’s Outpatient Influenza-like Illness Surveillance Network involved ILI as of Oct. 22. The highest comparable rate in the previous 5 years was the 1.9% recorded in late October of 2021, shortly after the definition of ILI was changed to also include illnesses other than influenza.
This season’s higher flu activity is in contrast to the previous two, which were unusually mild. The change, however, is not unexpected, as William Schaffner, MD, an infectious disease expert and professor of preventive medicine at Vanderbilt University, recently told CNN.
“Here we are in the middle of October – not the middle of November – we’re already seeing scattered influenza cases, even hospitalized influenza cases, around the country,” he said. “So we know that this virus is now spreading out in the community already. It’s gathering speed already. It looks to me to be about a month early.”
One indication of the mildness of the previous two flu seasons was the number of deaths, both pediatric and overall. Influenza-associated pediatric deaths had averaged about 110 per season over the previous eight seasons, compared with just 1 for 2020-2021 and 43 in 2021-2022. Overall flu deaths never reached 1% of all weekly deaths for either season, well below baseline levels for the flu, which range from 5.5% to 6.8%, CDC data show.
Other indicators of early severity
This season’s early rise in viral activity also can be seen in hospitalizations. The cumulative rate of flu-related admissions was 1.5 per 100,000 population as of Oct. 22, higher than the rate observed in the comparable week of previous seasons going back to 2010-2011, according to the CDC’s Influenza Hospitalization Surveillance Network.
A look at state reports of ILI outpatient visit rates shows that the District of Columbia and South Carolina are already in the very high range of the CDC’s severity scale, while 11 states are in the high range. Again going back to 2010-2011, no jurisdiction has ever been in the very high range this early in the season, based on data from the Outpatient Influenza-like Illness Surveillance Network.