User login
More on stigma
I just finished reading your editorial “A PSYCHIATRIC MANIFESTO: Stigma is hate speech and a hate crime” (
Our son went from an honor roll student before the pandemic to a child I barely recognized. Approximately 6 months into the pandemic, he was using drugs, vaping nicotine, destroying our property, and eloping at night. The journey of watching his decline and getting him help was agonizing. But the stigma around what was happening to him was an entirely separate animal.
Our society vilifies, ridicules, dismisses, and often makes fun of those with mental health issues. I experience it daily with my son and am on constant guard to shoot down any comments and to calmly teach those who say such cruel things. But the shame my son feels is the most devastating part. Although we keep reminding him that his condition is a medical condition like diabetes or heart disease, for a teenage boy, that makes no sense. He just wants to be “normal.” And living in a world that rarely represents mental illness this way, it’s almost a lost cause to get him to let go of this shame. All we can do is love him, be there for him, support him, and do what we can to educate those around us about the stigma of mental illness.
What a powerful and accurate article. Thank you for putting into words what I have been thinking and feeling, and for being as outraged as we are at how this vulnerable population is treated. My husband is a psychiatrist and we live in an affluent urban area, so we are not in the middle of nowhere with no knowledge of what is happening to our son. And despite that, we still suffer from the stigma.
Thank you, Dr. Nasrallah.
Name withheld
I need to take a moment to thank you for your editorial about stigma being hate speech and a hate crime. I really agree with you, and I think the way you formulated and articulated this message is very compelling.
I have focused on normalizing mental health differences among entrepreneurs as a destigmatization strategy (see https://www.sciencedirect.com/science/article/pii/S0883902622000027 and https://link.springer.com/article/10.1007/s11187-018-0059-8). Entrepreneurs clearly illustrate the fallacy of stigma. As a simple example, Elon Musk—the wealthiest person in the world—talks openly about being autistic, and possibly bipolar. These mental health differences help him create jobs and contribute to our shared prosperity. Nothing to be ashamed of there.
Thanks again for being such an effective advocate.
Michael A. Freeman, MD
Kentfield, California
Continue to: Thank you...
Thank you so much for your “Psychiatric Manifesto.” I will do my best to disseminate it amongst colleagues, patients, friends, family, and as many others as possible.
Daniel N. Pistone, MD
San Francisco, California
Once again, your words hit the pin on the head.
Robert W. Pollack, MD, ABPN, DLFAPA
Fort Myers, Florida
I just finished reading your editorial “A PSYCHIATRIC MANIFESTO: Stigma is hate speech and a hate crime” (
Our son went from an honor roll student before the pandemic to a child I barely recognized. Approximately 6 months into the pandemic, he was using drugs, vaping nicotine, destroying our property, and eloping at night. The journey of watching his decline and getting him help was agonizing. But the stigma around what was happening to him was an entirely separate animal.
Our society vilifies, ridicules, dismisses, and often makes fun of those with mental health issues. I experience it daily with my son and am on constant guard to shoot down any comments and to calmly teach those who say such cruel things. But the shame my son feels is the most devastating part. Although we keep reminding him that his condition is a medical condition like diabetes or heart disease, for a teenage boy, that makes no sense. He just wants to be “normal.” And living in a world that rarely represents mental illness this way, it’s almost a lost cause to get him to let go of this shame. All we can do is love him, be there for him, support him, and do what we can to educate those around us about the stigma of mental illness.
What a powerful and accurate article. Thank you for putting into words what I have been thinking and feeling, and for being as outraged as we are at how this vulnerable population is treated. My husband is a psychiatrist and we live in an affluent urban area, so we are not in the middle of nowhere with no knowledge of what is happening to our son. And despite that, we still suffer from the stigma.
Thank you, Dr. Nasrallah.
Name withheld
I need to take a moment to thank you for your editorial about stigma being hate speech and a hate crime. I really agree with you, and I think the way you formulated and articulated this message is very compelling.
I have focused on normalizing mental health differences among entrepreneurs as a destigmatization strategy (see https://www.sciencedirect.com/science/article/pii/S0883902622000027 and https://link.springer.com/article/10.1007/s11187-018-0059-8). Entrepreneurs clearly illustrate the fallacy of stigma. As a simple example, Elon Musk—the wealthiest person in the world—talks openly about being autistic, and possibly bipolar. These mental health differences help him create jobs and contribute to our shared prosperity. Nothing to be ashamed of there.
Thanks again for being such an effective advocate.
Michael A. Freeman, MD
Kentfield, California
Continue to: Thank you...
Thank you so much for your “Psychiatric Manifesto.” I will do my best to disseminate it amongst colleagues, patients, friends, family, and as many others as possible.
Daniel N. Pistone, MD
San Francisco, California
Once again, your words hit the pin on the head.
Robert W. Pollack, MD, ABPN, DLFAPA
Fort Myers, Florida
I just finished reading your editorial “A PSYCHIATRIC MANIFESTO: Stigma is hate speech and a hate crime” (
Our son went from an honor roll student before the pandemic to a child I barely recognized. Approximately 6 months into the pandemic, he was using drugs, vaping nicotine, destroying our property, and eloping at night. The journey of watching his decline and getting him help was agonizing. But the stigma around what was happening to him was an entirely separate animal.
Our society vilifies, ridicules, dismisses, and often makes fun of those with mental health issues. I experience it daily with my son and am on constant guard to shoot down any comments and to calmly teach those who say such cruel things. But the shame my son feels is the most devastating part. Although we keep reminding him that his condition is a medical condition like diabetes or heart disease, for a teenage boy, that makes no sense. He just wants to be “normal.” And living in a world that rarely represents mental illness this way, it’s almost a lost cause to get him to let go of this shame. All we can do is love him, be there for him, support him, and do what we can to educate those around us about the stigma of mental illness.
What a powerful and accurate article. Thank you for putting into words what I have been thinking and feeling, and for being as outraged as we are at how this vulnerable population is treated. My husband is a psychiatrist and we live in an affluent urban area, so we are not in the middle of nowhere with no knowledge of what is happening to our son. And despite that, we still suffer from the stigma.
Thank you, Dr. Nasrallah.
Name withheld
I need to take a moment to thank you for your editorial about stigma being hate speech and a hate crime. I really agree with you, and I think the way you formulated and articulated this message is very compelling.
I have focused on normalizing mental health differences among entrepreneurs as a destigmatization strategy (see https://www.sciencedirect.com/science/article/pii/S0883902622000027 and https://link.springer.com/article/10.1007/s11187-018-0059-8). Entrepreneurs clearly illustrate the fallacy of stigma. As a simple example, Elon Musk—the wealthiest person in the world—talks openly about being autistic, and possibly bipolar. These mental health differences help him create jobs and contribute to our shared prosperity. Nothing to be ashamed of there.
Thanks again for being such an effective advocate.
Michael A. Freeman, MD
Kentfield, California
Continue to: Thank you...
Thank you so much for your “Psychiatric Manifesto.” I will do my best to disseminate it amongst colleagues, patients, friends, family, and as many others as possible.
Daniel N. Pistone, MD
San Francisco, California
Once again, your words hit the pin on the head.
Robert W. Pollack, MD, ABPN, DLFAPA
Fort Myers, Florida
Malpractice lawyer gloats at win, then puts foot in mouth
During the closing arguments in a $10 million malpractice trial, attorney Robert McKenna III told jurors the claims against his client, a gastroenterologist, were baseless and equivalent to “extortion.” The patient’s family blamed the gastroenterologist for their father’s death, alleging the doctor perforated his colon during insertion of a feeding tube.
“I take pride in what I do, and I’ve got to tell you, in the 30 years I have been doing this, I have never seen a more insulting, factually devoid presentation in my entire career,” Mr. McKenna said, according to court transcripts. “On the strength of this evidence, they want you to award them $10 million. Welcome to America. Welcome to the personal injury machine, the personal injury industrial complex.”
After less than 30 minutes of deliberation, jurors returned a 12-0 verdict in favor of the physician.
However, Mr. McKenna, from Huntington Beach, Calif., described the case very differently to his staff in a celebration video, which he never expected to become public.
In the video, posted on Twitter and Instagram, Mr. McKenna bragged about how his legal team convinced jurors to doubt the patient’s official cause of death. He said the lawsuit involved a guy “that was probably negligently killed, but we kind of made it look like other people did it.”
“We actually had a death certificate that said he died the very way the plaintiff said he died, and we had to say, ‘No, you really shouldn’t believe what that death certificate says, or the coroner from the Orange County coroner’s office ... who says that it’s right,’” Mr. McKenna said in the video.
The 26-minute verdict was the fastest he’s ever received, Mr. McKenna says in the video, encouraging his partner to ring the firm’s victory bell.
“Overcoming all of those hurdles, we managed to sock three lawyers in the face,” Mr. McKenna said, referring to the plaintiffs’ lawyers.
The video of Mr. McKenna’s remarks is now in wide circulation after having been posted to online attorney forums, Instagram, where it’s been viewed more than 8,000 times, and Twitter, where views have reached over 3,000.
Jorge Ledezma, an Orange County, Calif., attorney who represented the patient’s family in the case, said the remarks make it appear as if Mr. McKenna tricked the jury.
“It was a drastic change from the comments he made to the jury during his closing arguments,” Mr. Ledezma said. “But the video is more important for what he doesn’t say. He doesn’t say his client did everything properly. He doesn’t say our case didn’t have any merit. He doesn’t say his client was a good doctor. Clearly, what he told the jury and what he believes are the exact opposite of each other.”
Mr. McKenna did not return multiple messages seeking comment for this story. In a statement to the LA Times, Mr. McKenna said his remarks were “intended purely as an internal briefing to our staff, using shorthand phrases which might understandably cause confusion for a lay audience unfamiliar with the case at hand, and the law in general.”
“I have expressed my apologies to my client, opposing counsel, and both the medical and legal communities,” Mr. McKenna said in the statement to the LA Times. “However, nothing about my remarks should call into question our very transparent trial strategy or the jury’s verdict in favor of my client.”
What happened to the patient?
Enrique Garcia Sanchez, 49, arrived at the critical care unit at South Coast Global Medical Center in Santa Ana, Calif., on Nov. 5, 2017, complaining of abdominal pain. He was diagnosed with acute pancreatitis, acute hypokalemia, and alcohol abuse, and transferred to the ICU, according to the family’s legal complaint.
Mr. Sanchez had a positive D-Dimer test, indicating a probable blood clot, and he appeared to be experiencing septic shock caused by pancreatitis, according to the complaint. By Nov. 17, Mr. Sanchez was suffering from respiratory failure and severe hypoxemia, and as a result, he was sedated. In addition, his abdomen was described as distended with decreased bowel sounds, according to court documents.
On. Nov. 18, a gastrointestinal specialist was consulted because of Mr. Sanchez’s prolonged intubation and oropharyngeal dysphagia, according to the lawsuit. On Nov. 21, air was leaking from Mr. Sanchez’s breathing tube with diffuse infiltration noted on the right side, and pneumonia.
Mr. Sanchez was eventually unable to swallow, and the gastroenterologist inserted a percutaneous endoscopic gastrostomy (PEG) tube, according to court records.
Mr. Sanchez’s condition worsened, and he developed respiratory distress, hypotension, and weakness during dialysis. On Dec. 9, 2017, physicians noted he had a bacterial infection, and he was later intubated on vent support because of progressive respiratory failure. Additionally, an internist reported that “fecal material” was observed per the PEG tube. Mr. Sanchez’s white blood cell count continued to rise, and his condition deteriorated. Mr. Sanchez died on Dec. 31, 2017.
A death certificate concluded that Mr. Sanchez died from complications of a PEG tube that perforated his colon, according to Mr. Ledezma. The plaintiffs’ legal team argued the gastroenterologist breached the standard of care by failing to ensure the tube was placed properly and failing to remedy the error after leakage was noted.
“Mr. Garcia died because of a misplaced PEG tube that perforated the colon, resulting in peritonitis and sepsis,” attorney Jose Robles said during his closing arguments. “Mr. Garcia had ascites, a contraindication for PEG tube placement. He had ileus, a contraindication for PEG tube placement. The standard of care requires that [the gastroenterologist] conduct a proper workup to confirm that a PEG tube placement can be done appropriately and safely.”
Mr. McKenna argued the gastroenterologist was not at fault for the patient’s death, and that complications from his pancreatitis ultimately killed him. During the trial, physicians who cared for Mr. Sanchez testified the patient had a less than 50% chance of survival.
“What he had was end-stage catastrophic [pancreatitis] that was affecting his organ system and aspiration pneumonia that made it impossible for him to try to breathe on his own,” Mr. McKenna said during closing arguments. “The man ... had a catastrophic injury that ate most of his pancreas. That is not a survivable event.”
Attorney faces backlash from legal community
Since his celebratory remarks were posted online, Mr. McKenna has faced much backlash, particularly from the legal community.
@mgvolada tweeted, “As an attorney I am revolted and I hope sanctions follow ... this is why people hate attorneys.”
@stevewieland, who identified himself as a trial lawyer, wrote he would not feel good about winning such a case.
“No wonder we get no love from the public,” he tweeted.
“Let’s see how the Court of Appeals thinks about your braggadocio and how this makes lawyers appear to the public,” tweeted @Stephen60134955, a self-identified attorney.
Mr. McKenna’s license remains active and in good standing with no disciplinary actions, according to the State Bar of California website.
Mr. Ledezma has filed a motion for a new trial, and a hearing on the motion is scheduled for Aug. 4, 2022. The motion was filed primarily because of issues during the trial, what Mr. Ledezma described as “inflammatory closing arguments,” and in small part, Mr. McKenna’s video remarks, he said.
If the motion is denied, the plaintiffs will move forward with an appeal, he said.
A version of this article first appeared on Medscape.com.
During the closing arguments in a $10 million malpractice trial, attorney Robert McKenna III told jurors the claims against his client, a gastroenterologist, were baseless and equivalent to “extortion.” The patient’s family blamed the gastroenterologist for their father’s death, alleging the doctor perforated his colon during insertion of a feeding tube.
“I take pride in what I do, and I’ve got to tell you, in the 30 years I have been doing this, I have never seen a more insulting, factually devoid presentation in my entire career,” Mr. McKenna said, according to court transcripts. “On the strength of this evidence, they want you to award them $10 million. Welcome to America. Welcome to the personal injury machine, the personal injury industrial complex.”
After less than 30 minutes of deliberation, jurors returned a 12-0 verdict in favor of the physician.
However, Mr. McKenna, from Huntington Beach, Calif., described the case very differently to his staff in a celebration video, which he never expected to become public.
In the video, posted on Twitter and Instagram, Mr. McKenna bragged about how his legal team convinced jurors to doubt the patient’s official cause of death. He said the lawsuit involved a guy “that was probably negligently killed, but we kind of made it look like other people did it.”
“We actually had a death certificate that said he died the very way the plaintiff said he died, and we had to say, ‘No, you really shouldn’t believe what that death certificate says, or the coroner from the Orange County coroner’s office ... who says that it’s right,’” Mr. McKenna said in the video.
The 26-minute verdict was the fastest he’s ever received, Mr. McKenna says in the video, encouraging his partner to ring the firm’s victory bell.
“Overcoming all of those hurdles, we managed to sock three lawyers in the face,” Mr. McKenna said, referring to the plaintiffs’ lawyers.
The video of Mr. McKenna’s remarks is now in wide circulation after having been posted to online attorney forums, Instagram, where it’s been viewed more than 8,000 times, and Twitter, where views have reached over 3,000.
Jorge Ledezma, an Orange County, Calif., attorney who represented the patient’s family in the case, said the remarks make it appear as if Mr. McKenna tricked the jury.
“It was a drastic change from the comments he made to the jury during his closing arguments,” Mr. Ledezma said. “But the video is more important for what he doesn’t say. He doesn’t say his client did everything properly. He doesn’t say our case didn’t have any merit. He doesn’t say his client was a good doctor. Clearly, what he told the jury and what he believes are the exact opposite of each other.”
Mr. McKenna did not return multiple messages seeking comment for this story. In a statement to the LA Times, Mr. McKenna said his remarks were “intended purely as an internal briefing to our staff, using shorthand phrases which might understandably cause confusion for a lay audience unfamiliar with the case at hand, and the law in general.”
“I have expressed my apologies to my client, opposing counsel, and both the medical and legal communities,” Mr. McKenna said in the statement to the LA Times. “However, nothing about my remarks should call into question our very transparent trial strategy or the jury’s verdict in favor of my client.”
What happened to the patient?
Enrique Garcia Sanchez, 49, arrived at the critical care unit at South Coast Global Medical Center in Santa Ana, Calif., on Nov. 5, 2017, complaining of abdominal pain. He was diagnosed with acute pancreatitis, acute hypokalemia, and alcohol abuse, and transferred to the ICU, according to the family’s legal complaint.
Mr. Sanchez had a positive D-Dimer test, indicating a probable blood clot, and he appeared to be experiencing septic shock caused by pancreatitis, according to the complaint. By Nov. 17, Mr. Sanchez was suffering from respiratory failure and severe hypoxemia, and as a result, he was sedated. In addition, his abdomen was described as distended with decreased bowel sounds, according to court documents.
On. Nov. 18, a gastrointestinal specialist was consulted because of Mr. Sanchez’s prolonged intubation and oropharyngeal dysphagia, according to the lawsuit. On Nov. 21, air was leaking from Mr. Sanchez’s breathing tube with diffuse infiltration noted on the right side, and pneumonia.
Mr. Sanchez was eventually unable to swallow, and the gastroenterologist inserted a percutaneous endoscopic gastrostomy (PEG) tube, according to court records.
Mr. Sanchez’s condition worsened, and he developed respiratory distress, hypotension, and weakness during dialysis. On Dec. 9, 2017, physicians noted he had a bacterial infection, and he was later intubated on vent support because of progressive respiratory failure. Additionally, an internist reported that “fecal material” was observed per the PEG tube. Mr. Sanchez’s white blood cell count continued to rise, and his condition deteriorated. Mr. Sanchez died on Dec. 31, 2017.
A death certificate concluded that Mr. Sanchez died from complications of a PEG tube that perforated his colon, according to Mr. Ledezma. The plaintiffs’ legal team argued the gastroenterologist breached the standard of care by failing to ensure the tube was placed properly and failing to remedy the error after leakage was noted.
“Mr. Garcia died because of a misplaced PEG tube that perforated the colon, resulting in peritonitis and sepsis,” attorney Jose Robles said during his closing arguments. “Mr. Garcia had ascites, a contraindication for PEG tube placement. He had ileus, a contraindication for PEG tube placement. The standard of care requires that [the gastroenterologist] conduct a proper workup to confirm that a PEG tube placement can be done appropriately and safely.”
Mr. McKenna argued the gastroenterologist was not at fault for the patient’s death, and that complications from his pancreatitis ultimately killed him. During the trial, physicians who cared for Mr. Sanchez testified the patient had a less than 50% chance of survival.
“What he had was end-stage catastrophic [pancreatitis] that was affecting his organ system and aspiration pneumonia that made it impossible for him to try to breathe on his own,” Mr. McKenna said during closing arguments. “The man ... had a catastrophic injury that ate most of his pancreas. That is not a survivable event.”
Attorney faces backlash from legal community
Since his celebratory remarks were posted online, Mr. McKenna has faced much backlash, particularly from the legal community.
@mgvolada tweeted, “As an attorney I am revolted and I hope sanctions follow ... this is why people hate attorneys.”
@stevewieland, who identified himself as a trial lawyer, wrote he would not feel good about winning such a case.
“No wonder we get no love from the public,” he tweeted.
“Let’s see how the Court of Appeals thinks about your braggadocio and how this makes lawyers appear to the public,” tweeted @Stephen60134955, a self-identified attorney.
Mr. McKenna’s license remains active and in good standing with no disciplinary actions, according to the State Bar of California website.
Mr. Ledezma has filed a motion for a new trial, and a hearing on the motion is scheduled for Aug. 4, 2022. The motion was filed primarily because of issues during the trial, what Mr. Ledezma described as “inflammatory closing arguments,” and in small part, Mr. McKenna’s video remarks, he said.
If the motion is denied, the plaintiffs will move forward with an appeal, he said.
A version of this article first appeared on Medscape.com.
During the closing arguments in a $10 million malpractice trial, attorney Robert McKenna III told jurors the claims against his client, a gastroenterologist, were baseless and equivalent to “extortion.” The patient’s family blamed the gastroenterologist for their father’s death, alleging the doctor perforated his colon during insertion of a feeding tube.
“I take pride in what I do, and I’ve got to tell you, in the 30 years I have been doing this, I have never seen a more insulting, factually devoid presentation in my entire career,” Mr. McKenna said, according to court transcripts. “On the strength of this evidence, they want you to award them $10 million. Welcome to America. Welcome to the personal injury machine, the personal injury industrial complex.”
After less than 30 minutes of deliberation, jurors returned a 12-0 verdict in favor of the physician.
However, Mr. McKenna, from Huntington Beach, Calif., described the case very differently to his staff in a celebration video, which he never expected to become public.
In the video, posted on Twitter and Instagram, Mr. McKenna bragged about how his legal team convinced jurors to doubt the patient’s official cause of death. He said the lawsuit involved a guy “that was probably negligently killed, but we kind of made it look like other people did it.”
“We actually had a death certificate that said he died the very way the plaintiff said he died, and we had to say, ‘No, you really shouldn’t believe what that death certificate says, or the coroner from the Orange County coroner’s office ... who says that it’s right,’” Mr. McKenna said in the video.
The 26-minute verdict was the fastest he’s ever received, Mr. McKenna says in the video, encouraging his partner to ring the firm’s victory bell.
“Overcoming all of those hurdles, we managed to sock three lawyers in the face,” Mr. McKenna said, referring to the plaintiffs’ lawyers.
The video of Mr. McKenna’s remarks is now in wide circulation after having been posted to online attorney forums, Instagram, where it’s been viewed more than 8,000 times, and Twitter, where views have reached over 3,000.
Jorge Ledezma, an Orange County, Calif., attorney who represented the patient’s family in the case, said the remarks make it appear as if Mr. McKenna tricked the jury.
“It was a drastic change from the comments he made to the jury during his closing arguments,” Mr. Ledezma said. “But the video is more important for what he doesn’t say. He doesn’t say his client did everything properly. He doesn’t say our case didn’t have any merit. He doesn’t say his client was a good doctor. Clearly, what he told the jury and what he believes are the exact opposite of each other.”
Mr. McKenna did not return multiple messages seeking comment for this story. In a statement to the LA Times, Mr. McKenna said his remarks were “intended purely as an internal briefing to our staff, using shorthand phrases which might understandably cause confusion for a lay audience unfamiliar with the case at hand, and the law in general.”
“I have expressed my apologies to my client, opposing counsel, and both the medical and legal communities,” Mr. McKenna said in the statement to the LA Times. “However, nothing about my remarks should call into question our very transparent trial strategy or the jury’s verdict in favor of my client.”
What happened to the patient?
Enrique Garcia Sanchez, 49, arrived at the critical care unit at South Coast Global Medical Center in Santa Ana, Calif., on Nov. 5, 2017, complaining of abdominal pain. He was diagnosed with acute pancreatitis, acute hypokalemia, and alcohol abuse, and transferred to the ICU, according to the family’s legal complaint.
Mr. Sanchez had a positive D-Dimer test, indicating a probable blood clot, and he appeared to be experiencing septic shock caused by pancreatitis, according to the complaint. By Nov. 17, Mr. Sanchez was suffering from respiratory failure and severe hypoxemia, and as a result, he was sedated. In addition, his abdomen was described as distended with decreased bowel sounds, according to court documents.
On. Nov. 18, a gastrointestinal specialist was consulted because of Mr. Sanchez’s prolonged intubation and oropharyngeal dysphagia, according to the lawsuit. On Nov. 21, air was leaking from Mr. Sanchez’s breathing tube with diffuse infiltration noted on the right side, and pneumonia.
Mr. Sanchez was eventually unable to swallow, and the gastroenterologist inserted a percutaneous endoscopic gastrostomy (PEG) tube, according to court records.
Mr. Sanchez’s condition worsened, and he developed respiratory distress, hypotension, and weakness during dialysis. On Dec. 9, 2017, physicians noted he had a bacterial infection, and he was later intubated on vent support because of progressive respiratory failure. Additionally, an internist reported that “fecal material” was observed per the PEG tube. Mr. Sanchez’s white blood cell count continued to rise, and his condition deteriorated. Mr. Sanchez died on Dec. 31, 2017.
A death certificate concluded that Mr. Sanchez died from complications of a PEG tube that perforated his colon, according to Mr. Ledezma. The plaintiffs’ legal team argued the gastroenterologist breached the standard of care by failing to ensure the tube was placed properly and failing to remedy the error after leakage was noted.
“Mr. Garcia died because of a misplaced PEG tube that perforated the colon, resulting in peritonitis and sepsis,” attorney Jose Robles said during his closing arguments. “Mr. Garcia had ascites, a contraindication for PEG tube placement. He had ileus, a contraindication for PEG tube placement. The standard of care requires that [the gastroenterologist] conduct a proper workup to confirm that a PEG tube placement can be done appropriately and safely.”
Mr. McKenna argued the gastroenterologist was not at fault for the patient’s death, and that complications from his pancreatitis ultimately killed him. During the trial, physicians who cared for Mr. Sanchez testified the patient had a less than 50% chance of survival.
“What he had was end-stage catastrophic [pancreatitis] that was affecting his organ system and aspiration pneumonia that made it impossible for him to try to breathe on his own,” Mr. McKenna said during closing arguments. “The man ... had a catastrophic injury that ate most of his pancreas. That is not a survivable event.”
Attorney faces backlash from legal community
Since his celebratory remarks were posted online, Mr. McKenna has faced much backlash, particularly from the legal community.
@mgvolada tweeted, “As an attorney I am revolted and I hope sanctions follow ... this is why people hate attorneys.”
@stevewieland, who identified himself as a trial lawyer, wrote he would not feel good about winning such a case.
“No wonder we get no love from the public,” he tweeted.
“Let’s see how the Court of Appeals thinks about your braggadocio and how this makes lawyers appear to the public,” tweeted @Stephen60134955, a self-identified attorney.
Mr. McKenna’s license remains active and in good standing with no disciplinary actions, according to the State Bar of California website.
Mr. Ledezma has filed a motion for a new trial, and a hearing on the motion is scheduled for Aug. 4, 2022. The motion was filed primarily because of issues during the trial, what Mr. Ledezma described as “inflammatory closing arguments,” and in small part, Mr. McKenna’s video remarks, he said.
If the motion is denied, the plaintiffs will move forward with an appeal, he said.
A version of this article first appeared on Medscape.com.
Episodes of visual disturbance
On the basis of the history, examination, and investigations, retinal migraine was diagnosed according to the International Classification of Headache Disorders, third edition (1.2 migraine with aura; 1.2.4 retinal migraine). This classification system describes retinal migraine as a subtype of migraine with aura.
Retinal migraine (also called ophthalmic or ocular migraine) is relatively rare but is sometimes a cause of transient monocular blindness in young adults. It manifests as recurrent attacks of unilateral visual disturbance (positive symptoms) or blindness (negative symptoms) lasting from minutes to 1 hour, associated with minimal or no headache.
Some patients describe a positive visual symptom/disturbance in a mosaic pattern of scotomata that gradually enlarge, producing total or near-total unilateral visual loss. Precipitating factors may include emotional stress, hypertension, and hormonal contraceptive pills, as well as exercise, high altitude, dehydration, smoking, hypoglycemia, and hyperthermia.
Retinal migraine is believed to result from transient vasospasm of the choroidal or retinal arteries. A history of recurrent attacks of transient monocular visual disturbance or blindness, with or without a headache and without other neurologic symptoms, can suggest retinal migraine. A personal or family history of migraine can confirm the diagnosis.
Ruling out eye disease or vascular causes, especially when risk factors for arteriosclerosis exist, is important; that is, the condition must be differentiated from ocular or vascular causes of transient monocular blindness, mainly carotid artery disease.
Carotid duplex ultrasonography, transcranial Doppler ultrasonography, magnetic resonance angiography, or CT angiography of the brain may be helpful. Fluorescein or cerebral angiography is rarely necessary. A hypercoagulability workup and evaluation of the erythrocyte sedimentation rate may be useful in excluding other coagulation disorders associated with retinal vasculopathy.
Regarding management, calcium-channel blockers have shown some efficacy. Even in patients with low blood pressure, nifedipine 10-20 mg/d is generally tolerated. From the available literature on treatment of this condition, it is recommended that triptans, ergots, and beta-blockers be used with caution or avoided in patients with retinal migraine owing to the potential for exacerbating vasoconstriction of the retinal artery. Transient vision loss in retinal migraine has been associated with future onset of permanent vision loss from occlusive conditions such as central retinal artery occlusion and branch retinal artery occlusion.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the history, examination, and investigations, retinal migraine was diagnosed according to the International Classification of Headache Disorders, third edition (1.2 migraine with aura; 1.2.4 retinal migraine). This classification system describes retinal migraine as a subtype of migraine with aura.
Retinal migraine (also called ophthalmic or ocular migraine) is relatively rare but is sometimes a cause of transient monocular blindness in young adults. It manifests as recurrent attacks of unilateral visual disturbance (positive symptoms) or blindness (negative symptoms) lasting from minutes to 1 hour, associated with minimal or no headache.
Some patients describe a positive visual symptom/disturbance in a mosaic pattern of scotomata that gradually enlarge, producing total or near-total unilateral visual loss. Precipitating factors may include emotional stress, hypertension, and hormonal contraceptive pills, as well as exercise, high altitude, dehydration, smoking, hypoglycemia, and hyperthermia.
Retinal migraine is believed to result from transient vasospasm of the choroidal or retinal arteries. A history of recurrent attacks of transient monocular visual disturbance or blindness, with or without a headache and without other neurologic symptoms, can suggest retinal migraine. A personal or family history of migraine can confirm the diagnosis.
Ruling out eye disease or vascular causes, especially when risk factors for arteriosclerosis exist, is important; that is, the condition must be differentiated from ocular or vascular causes of transient monocular blindness, mainly carotid artery disease.
Carotid duplex ultrasonography, transcranial Doppler ultrasonography, magnetic resonance angiography, or CT angiography of the brain may be helpful. Fluorescein or cerebral angiography is rarely necessary. A hypercoagulability workup and evaluation of the erythrocyte sedimentation rate may be useful in excluding other coagulation disorders associated with retinal vasculopathy.
Regarding management, calcium-channel blockers have shown some efficacy. Even in patients with low blood pressure, nifedipine 10-20 mg/d is generally tolerated. From the available literature on treatment of this condition, it is recommended that triptans, ergots, and beta-blockers be used with caution or avoided in patients with retinal migraine owing to the potential for exacerbating vasoconstriction of the retinal artery. Transient vision loss in retinal migraine has been associated with future onset of permanent vision loss from occlusive conditions such as central retinal artery occlusion and branch retinal artery occlusion.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the history, examination, and investigations, retinal migraine was diagnosed according to the International Classification of Headache Disorders, third edition (1.2 migraine with aura; 1.2.4 retinal migraine). This classification system describes retinal migraine as a subtype of migraine with aura.
Retinal migraine (also called ophthalmic or ocular migraine) is relatively rare but is sometimes a cause of transient monocular blindness in young adults. It manifests as recurrent attacks of unilateral visual disturbance (positive symptoms) or blindness (negative symptoms) lasting from minutes to 1 hour, associated with minimal or no headache.
Some patients describe a positive visual symptom/disturbance in a mosaic pattern of scotomata that gradually enlarge, producing total or near-total unilateral visual loss. Precipitating factors may include emotional stress, hypertension, and hormonal contraceptive pills, as well as exercise, high altitude, dehydration, smoking, hypoglycemia, and hyperthermia.
Retinal migraine is believed to result from transient vasospasm of the choroidal or retinal arteries. A history of recurrent attacks of transient monocular visual disturbance or blindness, with or without a headache and without other neurologic symptoms, can suggest retinal migraine. A personal or family history of migraine can confirm the diagnosis.
Ruling out eye disease or vascular causes, especially when risk factors for arteriosclerosis exist, is important; that is, the condition must be differentiated from ocular or vascular causes of transient monocular blindness, mainly carotid artery disease.
Carotid duplex ultrasonography, transcranial Doppler ultrasonography, magnetic resonance angiography, or CT angiography of the brain may be helpful. Fluorescein or cerebral angiography is rarely necessary. A hypercoagulability workup and evaluation of the erythrocyte sedimentation rate may be useful in excluding other coagulation disorders associated with retinal vasculopathy.
Regarding management, calcium-channel blockers have shown some efficacy. Even in patients with low blood pressure, nifedipine 10-20 mg/d is generally tolerated. From the available literature on treatment of this condition, it is recommended that triptans, ergots, and beta-blockers be used with caution or avoided in patients with retinal migraine owing to the potential for exacerbating vasoconstriction of the retinal artery. Transient vision loss in retinal migraine has been associated with future onset of permanent vision loss from occlusive conditions such as central retinal artery occlusion and branch retinal artery occlusion.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.

A 23-year-old woman presents with sudden recurrent episodes of visual disturbance (extreme blurriness and partial blindness) in her right eye. She had seven or eight episodes over 30 hours; each episode lasted for 5-7 minutes, with spontaneous and full recovery. These were not associated with flashes of light, tingling, numbness, fever, or headache. She was asymptomatic between episodes.
She had normal vision in her left eye during these episodes, which she checked by covering both eyes alternately with her hands. The only significant history was four episodes of migraine with aura 3 years ago, which resolved spontaneously and did not recur. Family history was noncontributory. She had no history of illicit drug use or alcohol use.
On examination, her vital signs were normal. Blood pressure was 110/80 mm Hg, pulse 85 beats/min, and respiratory rate 16 breaths/min. There was no lymphadenopathy, and jugular venous pressure was not elevated. Visual acuity was 6/6, with normal visual fields and perimetry. Fundoscopy was normal. Complete blood count, liver function tests, renal function tests, erythrocyte sedimentation rate, antineutrophil antibodies, electrocardiography, transthoracic echocardiography, carotid Doppler, and MRI of the brain with contrast were all normal. She is taking no medications.

Ezetimibe plus statin: Attractive bypass to high-dose monotherapy
More patients with established atherosclerotic cardiovascular disease (ASCVD) achieved a low-density lipoprotein (LDL) cholesterol of less than 70 mg/dL, and fewer discontinued treatment with ezetimibe plus a moderate-dose statin, than did those on high-intensity statin monotherapy, a noninferiority trial shows.
While it’s now established that drug combinations can achieve better efficacy with lower risks than high-dose monotherapy, the study is the first to show the benefits of the strategy for ASCVD in a randomized trial with long-term follow-up.
The primary endpoint – 3-year composite of cardiovascular death, major cardiovascular events, or nonfatal stroke – occurred in about 9% of patients in each group, showing non-inferiority.
Furthermore, the authors suggest that ezetimibe combination therapy be considered earlier in the treatment of those at high risk of adverse events, rather than doubling the statin dose.
The study was published online in The Lancet.
Less intolerance, less discontinuations
The open-label study, dubbed RACING, randomized 3,780 patients with ASCVD to receive moderate-intensity rosuvastatin 10 mg plus ezetimibe 10 mg or high-intensity 20 mg rosuvastatin monotherapy. Participants’ average age was 64 and 75% were men.
The primary endpoint occurred in 9.1% of patients in the combination therapy group and 9.9% in the high-intensity monotherapy group. The absolute between-group difference was −0.78% (90% confidence interval [CI], −2.39 to 0.83), well below the 2% noninferiority margin.
In the combination therapy group, LDL cholesterol concentrations of less than 70 mg/dL were achieved in 73% of patients at 1 year, 75% at 2 years, and 72% at 3 years. By contrast, in the monotherapy group, the lower concentrations were seen in 55% at 1 year, 60% at 2 years, and 58% at 3 years.
Further, a post hoc analysis showed LDL concentrations of less than 55 mg/dL at 1, 2, and 3 years in 42%, 45%, and 42% of patients in the combination therapy group versus 25%, 29%, and 25% of those in the high-intensity statin monotherapy group.
Eighty-eight patients (4.8%) on combination therapy discontinued medication or received a dose reduction, versus 150 patients (8.2%) on monotherapy.
Rates of myonecrosis were similar in the combination therapy and high-intensity statin groups (11 vs. 13), whereas myalgia was more common with high-intensity statins (29 vs. 17). The open-label design could have led to bias in reporting of patient symptoms, the authors noted. All clinical events, however, were adjudicated by an independent committee masked to treatment assignment.
There might be “some level of difference” when extending the findings to other populations because the trial involved only Koreans, coauthor Myeong-Ki Hong, MD, Yonsei University, Seoul, South Korea, acknowledged in response to a query from this news organization. He thinks the findings can be applied broadly nonetheless, and his team is currently investigating whether certain patients might benefit more than others from the combination.
Various options for patients
“The field of hypertension changed [its] guidelines almost 20 years ago to consider the initial use of combination therapy in hard-to-treat patients,” Christie Mitchell Ballantyne, MD, Baylor College of Medicine, Houston, said in an interview. He coauthored an accompanying editorial with Baylor colleague Layla A. Abushamat, MD.
“We now have enough evidence of the efficacy and safety of combination therapy to consider early initiation of this approach in patients with challenging lipid disorders who are at increased risk of ASCVD events,” affirmed Dr. Ballantyne.
“This study reinforces important principles in the management and secondary prevention of cardiovascular disease, namely that LDL reduction and associated risk reduction can be achieved in various ways,” said Daniel Muñoz, MD, MPA, executive medical director of the Vanderbilt Heart & Vascular Institute, Vanderbilt University Medical Center, Nashville, Tenn.
However, he noted, “The high-intensity statin dose used as a comparator in this study was rosuvastatin 20 mg. In clinical practice, we often target maximally aggressive reduction of LDL via higher doses – that is, rosuvastatin 40 mg or atorvastatin 80 mg.”
The bottom line, said Dr. Muñoz, who was not involved in the study: “There are different ways to achieve LDL-lowering and associated risk reduction in patients with CVD. For patients who warrant but might not tolerate high-intensity statin therapy, this study supports the use of a moderate-intensity statin in combination with ezetimibe.”
The study was funded by Hanmi Pharmaceutical, Seoul, South Korea. One study coauthor received an institutional research grant from the company. No other authors reported relevant financial relationships, nor did Dr. Ballantyne, Dr. Abushamat, or Dr. Muñoz.
A version of this article first appeared on Medscape.com.
More patients with established atherosclerotic cardiovascular disease (ASCVD) achieved a low-density lipoprotein (LDL) cholesterol of less than 70 mg/dL, and fewer discontinued treatment with ezetimibe plus a moderate-dose statin, than did those on high-intensity statin monotherapy, a noninferiority trial shows.
While it’s now established that drug combinations can achieve better efficacy with lower risks than high-dose monotherapy, the study is the first to show the benefits of the strategy for ASCVD in a randomized trial with long-term follow-up.
The primary endpoint – 3-year composite of cardiovascular death, major cardiovascular events, or nonfatal stroke – occurred in about 9% of patients in each group, showing non-inferiority.
Furthermore, the authors suggest that ezetimibe combination therapy be considered earlier in the treatment of those at high risk of adverse events, rather than doubling the statin dose.
The study was published online in The Lancet.
Less intolerance, less discontinuations
The open-label study, dubbed RACING, randomized 3,780 patients with ASCVD to receive moderate-intensity rosuvastatin 10 mg plus ezetimibe 10 mg or high-intensity 20 mg rosuvastatin monotherapy. Participants’ average age was 64 and 75% were men.
The primary endpoint occurred in 9.1% of patients in the combination therapy group and 9.9% in the high-intensity monotherapy group. The absolute between-group difference was −0.78% (90% confidence interval [CI], −2.39 to 0.83), well below the 2% noninferiority margin.
In the combination therapy group, LDL cholesterol concentrations of less than 70 mg/dL were achieved in 73% of patients at 1 year, 75% at 2 years, and 72% at 3 years. By contrast, in the monotherapy group, the lower concentrations were seen in 55% at 1 year, 60% at 2 years, and 58% at 3 years.
Further, a post hoc analysis showed LDL concentrations of less than 55 mg/dL at 1, 2, and 3 years in 42%, 45%, and 42% of patients in the combination therapy group versus 25%, 29%, and 25% of those in the high-intensity statin monotherapy group.
Eighty-eight patients (4.8%) on combination therapy discontinued medication or received a dose reduction, versus 150 patients (8.2%) on monotherapy.
Rates of myonecrosis were similar in the combination therapy and high-intensity statin groups (11 vs. 13), whereas myalgia was more common with high-intensity statins (29 vs. 17). The open-label design could have led to bias in reporting of patient symptoms, the authors noted. All clinical events, however, were adjudicated by an independent committee masked to treatment assignment.
There might be “some level of difference” when extending the findings to other populations because the trial involved only Koreans, coauthor Myeong-Ki Hong, MD, Yonsei University, Seoul, South Korea, acknowledged in response to a query from this news organization. He thinks the findings can be applied broadly nonetheless, and his team is currently investigating whether certain patients might benefit more than others from the combination.
Various options for patients
“The field of hypertension changed [its] guidelines almost 20 years ago to consider the initial use of combination therapy in hard-to-treat patients,” Christie Mitchell Ballantyne, MD, Baylor College of Medicine, Houston, said in an interview. He coauthored an accompanying editorial with Baylor colleague Layla A. Abushamat, MD.
“We now have enough evidence of the efficacy and safety of combination therapy to consider early initiation of this approach in patients with challenging lipid disorders who are at increased risk of ASCVD events,” affirmed Dr. Ballantyne.
“This study reinforces important principles in the management and secondary prevention of cardiovascular disease, namely that LDL reduction and associated risk reduction can be achieved in various ways,” said Daniel Muñoz, MD, MPA, executive medical director of the Vanderbilt Heart & Vascular Institute, Vanderbilt University Medical Center, Nashville, Tenn.
However, he noted, “The high-intensity statin dose used as a comparator in this study was rosuvastatin 20 mg. In clinical practice, we often target maximally aggressive reduction of LDL via higher doses – that is, rosuvastatin 40 mg or atorvastatin 80 mg.”
The bottom line, said Dr. Muñoz, who was not involved in the study: “There are different ways to achieve LDL-lowering and associated risk reduction in patients with CVD. For patients who warrant but might not tolerate high-intensity statin therapy, this study supports the use of a moderate-intensity statin in combination with ezetimibe.”
The study was funded by Hanmi Pharmaceutical, Seoul, South Korea. One study coauthor received an institutional research grant from the company. No other authors reported relevant financial relationships, nor did Dr. Ballantyne, Dr. Abushamat, or Dr. Muñoz.
A version of this article first appeared on Medscape.com.
More patients with established atherosclerotic cardiovascular disease (ASCVD) achieved a low-density lipoprotein (LDL) cholesterol of less than 70 mg/dL, and fewer discontinued treatment with ezetimibe plus a moderate-dose statin, than did those on high-intensity statin monotherapy, a noninferiority trial shows.
While it’s now established that drug combinations can achieve better efficacy with lower risks than high-dose monotherapy, the study is the first to show the benefits of the strategy for ASCVD in a randomized trial with long-term follow-up.
The primary endpoint – 3-year composite of cardiovascular death, major cardiovascular events, or nonfatal stroke – occurred in about 9% of patients in each group, showing non-inferiority.
Furthermore, the authors suggest that ezetimibe combination therapy be considered earlier in the treatment of those at high risk of adverse events, rather than doubling the statin dose.
The study was published online in The Lancet.
Less intolerance, less discontinuations
The open-label study, dubbed RACING, randomized 3,780 patients with ASCVD to receive moderate-intensity rosuvastatin 10 mg plus ezetimibe 10 mg or high-intensity 20 mg rosuvastatin monotherapy. Participants’ average age was 64 and 75% were men.
The primary endpoint occurred in 9.1% of patients in the combination therapy group and 9.9% in the high-intensity monotherapy group. The absolute between-group difference was −0.78% (90% confidence interval [CI], −2.39 to 0.83), well below the 2% noninferiority margin.
In the combination therapy group, LDL cholesterol concentrations of less than 70 mg/dL were achieved in 73% of patients at 1 year, 75% at 2 years, and 72% at 3 years. By contrast, in the monotherapy group, the lower concentrations were seen in 55% at 1 year, 60% at 2 years, and 58% at 3 years.
Further, a post hoc analysis showed LDL concentrations of less than 55 mg/dL at 1, 2, and 3 years in 42%, 45%, and 42% of patients in the combination therapy group versus 25%, 29%, and 25% of those in the high-intensity statin monotherapy group.
Eighty-eight patients (4.8%) on combination therapy discontinued medication or received a dose reduction, versus 150 patients (8.2%) on monotherapy.
Rates of myonecrosis were similar in the combination therapy and high-intensity statin groups (11 vs. 13), whereas myalgia was more common with high-intensity statins (29 vs. 17). The open-label design could have led to bias in reporting of patient symptoms, the authors noted. All clinical events, however, were adjudicated by an independent committee masked to treatment assignment.
There might be “some level of difference” when extending the findings to other populations because the trial involved only Koreans, coauthor Myeong-Ki Hong, MD, Yonsei University, Seoul, South Korea, acknowledged in response to a query from this news organization. He thinks the findings can be applied broadly nonetheless, and his team is currently investigating whether certain patients might benefit more than others from the combination.
Various options for patients
“The field of hypertension changed [its] guidelines almost 20 years ago to consider the initial use of combination therapy in hard-to-treat patients,” Christie Mitchell Ballantyne, MD, Baylor College of Medicine, Houston, said in an interview. He coauthored an accompanying editorial with Baylor colleague Layla A. Abushamat, MD.
“We now have enough evidence of the efficacy and safety of combination therapy to consider early initiation of this approach in patients with challenging lipid disorders who are at increased risk of ASCVD events,” affirmed Dr. Ballantyne.
“This study reinforces important principles in the management and secondary prevention of cardiovascular disease, namely that LDL reduction and associated risk reduction can be achieved in various ways,” said Daniel Muñoz, MD, MPA, executive medical director of the Vanderbilt Heart & Vascular Institute, Vanderbilt University Medical Center, Nashville, Tenn.
However, he noted, “The high-intensity statin dose used as a comparator in this study was rosuvastatin 20 mg. In clinical practice, we often target maximally aggressive reduction of LDL via higher doses – that is, rosuvastatin 40 mg or atorvastatin 80 mg.”
The bottom line, said Dr. Muñoz, who was not involved in the study: “There are different ways to achieve LDL-lowering and associated risk reduction in patients with CVD. For patients who warrant but might not tolerate high-intensity statin therapy, this study supports the use of a moderate-intensity statin in combination with ezetimibe.”
The study was funded by Hanmi Pharmaceutical, Seoul, South Korea. One study coauthor received an institutional research grant from the company. No other authors reported relevant financial relationships, nor did Dr. Ballantyne, Dr. Abushamat, or Dr. Muñoz.
A version of this article first appeared on Medscape.com.
How doctors are weighing the legal risks of abortion care
The names of the doctors in this story have been changed at their request because of fear of legal repercussions and/or professional retaliation.
When an Ohio ob.gyn. had a patient in need of an abortion in July 2022, he knew he had to move quickly.
Daniel, who also sees patients at an abortion clinic, was treating a woman who came in for an abortion around 5 weeks into her pregnancy. And after going through the mandatory waiting periods, the required ultrasounds at each appointment, the consent process, and the options counseling, she was set for a surgical abortion the following Monday.
But on Monday, pre-op tests showed that her blood pressure was very high, posing a serious health risk if Daniel proceeded with the surgery.
Before the Supreme Court overturned Roe v. Wade in June, Daniel would have sent the patient home with instructions on how to lower her blood pressure over time. But the patient now had just four days to show the necessary improvement.
In this case, everything worked out. The patient returned Thursday and was able to have the procedure. But this is just one of the many day-to-day medical decisions abortion providers are now having to make with the changing legal risks being as top-of-mind to doctors as the safety of their patients.
Daniel said he doesn’t want the Ohio abortion law to change the way he communicates with his patients. As far as he knows, it’s still legal to talk to patients about self-managed abortions, as long as everything is unbiased and clearly stated, he says.
“But I don’t think I would get a lot of institutional support to have those conversations with patients because of the perceived legal liability,” says Daniel. “I will still have those conversations, but I’m not going to tell my employer that I’m having them and I’m not going to document them in the chart.”
Daniel is aware that having these kinds of discussions, or entertaining the possibility of omitting certain information from patient records, runs the risk of legal and professional consequences. Enforcement of these rules is foggy, too.
Under the Ohio law, if a fellow staff member suspects you of violating a law, you could be reported to a supervisor or licensing body. Abortion providers are aware they must be cautious about what they say because anti-abortion activitists, posing as patients, have secretly recorded conversations in the past, Daniel says.
Enforcement: The past, present, and future legal risks
Before Roe, enforcement of illegal abortion was spotty, says Mary Ziegler, JD, a professor at Florida State University College of Law, who specializes in the legal history of reproductive rights. At the start of the late 19th century, the doctors who provided illegal abortions would, in most cases, be prosecuted if a patient died as a result of the procedure.
A doctor in Ashland, Pa., named Robert Spencer was known for providing abortions in the small mining town where he practiced in the 1920s. He was reportedly arrested three times – once after a patient died as a result of abortion complications – but was ultimately acquitted.
For many doctors performing abortions at the time, “it was very much a kind of roll of the dice,” Ms. Ziegler says. “There was a sense that these laws were not enforced very much.”
Carole Joffe, PhD, a sociologist with expertise in reproductive health, recalls that there were very few doctors arrested, given the sheer number of abortions that were performed. The American College of Obstetricians and Gynecologists estimates that, in the years leading up to the original Roe decision, about 1.2 million women in the U.S. had illegal abortions – a number that exceeds today’s estimates.
Among the most notable cases of a doctor being detained was the arrest of gynecologist Jane Hodgson, MD, in 1970. Dr. Hodgson intentionally violated Minnesota law, which prohibited all abortions except in cases that were life-threatening to the patient.
After performing an abortion on a patient who had contracted rubella, also known as German measles, Dr. Hodgson was arrested, sentenced to 30 days in jail, and put on a year-long probation. She did not end up serving any time in jail, and her conviction was overturned after the Roe decision in 1973.
Now, the abortion restrictions being passed in many states have authorized much more sweeping penalties than those that existed in the pre-Roe era. According to Joffe, there is one key reason why we can anticipate more doctor arrests now.
“There simply was not the modern anti-abortion movement that we have come to know,” she says. “In the old days, there was not that much legal surveillance, and things were very unsafe. Fast forward to the present, we have much safer options now – like medication abortion pills – but we have a very different legal environment.”
Carmel Shachar, JD, MPH, a law and health policy expert at Harvard Law School, also expects that we will see more frequent prosecutions of doctors who provide abortion.
“There’s so much more data available through medical record-keeping and information generated by our phones and internet searches, that I think it would be much harder for a physician to fly under the radar,” Ms. Shachar says.
Also, Ms. Shachar emphasizes the power of prosecutorial discretion in abortion cases, where one prosecutor may choose to apply a law much more aggressively than another prosecutor in the next county over. Such has been seen in DeKalb County, Ga., which includes parts of Atlanta, where District Attorney Sherry Boston says she plans to use her prosecutorial discretion to address crimes like rape and murder, rather than “potentially investigat[ing] women and doctors for medical decisions,” Bloomberg Law reported. State Sen. Jen Jordan, the Democratic nominee for Georgia attorney general, has also said that, if elected, she would not enforce the state’s new 6-week abortion ban.
Is there a legal path forward for abortion care in states that forbid it?
Robin, an ob.gyn., became a complex family planning fellow in Utah to seek out further medical training and education in abortion care. Her plan was to solidify this as an area of expertise, so that, upon completing her fellowship, she could move back to her home state of Arizona to provide services there.
In Utah, where she currently practices, abortion is banned after 18 weeks. In Arizona, abortion is still allowed up to 24-26 weeks, until a pregnancy reaches “viability” (when a fetus is developed enough that it is able to survive outside the uterus with medical assistance). But new restrictions in Arizona may go into effect as early as September which would prohibit abortions after 15 weeks.
Despite the uncertain future of abortion access in Arizona, Robin still plans on moving there after her fellowship, but she hopes to travel to surrounding states to help provide abortion care where it’s less restricted. Even if she isn’t able to provide abortions at all, she says that there are still ways to help patients get safe, above-board abortions so as not to repeat the dangerous and often gruesome outcomes of self-induced abortions or those done by illegitimate practitioners before Roe.
“One of the roles that I think I can have as a physician is helping people with wraparound care for self-managed abortion,” says Robin. “If they can get the [abortion] pills online, then I can do the ultrasound beforehand, I can do the ultrasound after, I can talk them through it. I can help them with all the aspects of this care, I just can’t give them the pills myself.”
Whether a doctor can be penalized for “aiding and abetting” abortions that happen in different states remains an open question. In Texas, for example, Senate Bill 8 – which took effect Sept. 1, 2021 – not only established a fetal heartbeat law but added language that would allow private citizens to sue anyone who “knowingly engages in conduct that aids or abets the performance or inducement of an abortion” or anyone who even intends to do so.
That’s what happened to Alan Braid, MD, an ob.gyn. based in San Antonio. He confessed in a Washington Post op-ed that he had performed an abortion after cardiac activity had been detected in the pregnancy. Aware of the legal risks, he has since been sued by three people, and those cases are still underway.
But Ms. Ziegler says the chances of a doctor from a progressive state actually getting extradited and prosecuted by a state with restrictive abortion laws is pretty low – not zero, but low.
Like Robin, Natalie – an ob.gyn. in her early 30s – is a complex family planning fellow in Massachusetts. After her fellowship, she wants to return to Texas, where she completed her residency training.
“I’m at the point in my training where everyone starts looking for jobs and figuring out their next steps,” says Natalie. “The Dobbs decision introduced a ton of chaos due to the vagueness in the laws and how they get enforced, and then there’s chaos within institutions themselves and what kind of risk tolerance they have.”
Looking towards her future career path, Natalie says that she would not consider a job at an institution that didn’t allow her to teach abortion care to students, speak publicly about abortion rights, or let her travel outside of Texas to continue providing abortion care. She’s also preemptively seeking legal counsel and general guidance – advice that Ms. Ziegler strongly urges doctors to heed, sooner rather than later.
In states that have strict abortion bans with exceptions for life-threatening cases, there is still a lack of clarity around what is actually considered life-threatening enough to pass as an exception.
“Is it life-threatening in the next 6 hours? 24 hours? Seven days? One month?” Robin asks. “In medicine, we don’t necessarily talk about if something is life-threatening or not, we just say that there’s a high risk of X thing happening in X period of time. What’s the threshold at which that meets legal criteria? Nobody has an answer for that.”
Robin explains that, in her patients who have cancer, a pregnancy wouldn’t “necessarily kill them within the span of the next 9 months, but it could certainly accelerate their disease that could kill them within the next year or two.”
Right now, she says she doesn’t know what she would do if and when she is put in that position as a doctor.
“I didn’t go to medical school and become a doctor to become a felon,” says Robin. “Our goal is to make as many legal changes as we can to protect our patients and then practice as much harm reduction and as much care as we can within the letter of the law.”
A version of this article first appeared on WebMD.com.
The names of the doctors in this story have been changed at their request because of fear of legal repercussions and/or professional retaliation.
When an Ohio ob.gyn. had a patient in need of an abortion in July 2022, he knew he had to move quickly.
Daniel, who also sees patients at an abortion clinic, was treating a woman who came in for an abortion around 5 weeks into her pregnancy. And after going through the mandatory waiting periods, the required ultrasounds at each appointment, the consent process, and the options counseling, she was set for a surgical abortion the following Monday.
But on Monday, pre-op tests showed that her blood pressure was very high, posing a serious health risk if Daniel proceeded with the surgery.
Before the Supreme Court overturned Roe v. Wade in June, Daniel would have sent the patient home with instructions on how to lower her blood pressure over time. But the patient now had just four days to show the necessary improvement.
In this case, everything worked out. The patient returned Thursday and was able to have the procedure. But this is just one of the many day-to-day medical decisions abortion providers are now having to make with the changing legal risks being as top-of-mind to doctors as the safety of their patients.
Daniel said he doesn’t want the Ohio abortion law to change the way he communicates with his patients. As far as he knows, it’s still legal to talk to patients about self-managed abortions, as long as everything is unbiased and clearly stated, he says.
“But I don’t think I would get a lot of institutional support to have those conversations with patients because of the perceived legal liability,” says Daniel. “I will still have those conversations, but I’m not going to tell my employer that I’m having them and I’m not going to document them in the chart.”
Daniel is aware that having these kinds of discussions, or entertaining the possibility of omitting certain information from patient records, runs the risk of legal and professional consequences. Enforcement of these rules is foggy, too.
Under the Ohio law, if a fellow staff member suspects you of violating a law, you could be reported to a supervisor or licensing body. Abortion providers are aware they must be cautious about what they say because anti-abortion activitists, posing as patients, have secretly recorded conversations in the past, Daniel says.
Enforcement: The past, present, and future legal risks
Before Roe, enforcement of illegal abortion was spotty, says Mary Ziegler, JD, a professor at Florida State University College of Law, who specializes in the legal history of reproductive rights. At the start of the late 19th century, the doctors who provided illegal abortions would, in most cases, be prosecuted if a patient died as a result of the procedure.
A doctor in Ashland, Pa., named Robert Spencer was known for providing abortions in the small mining town where he practiced in the 1920s. He was reportedly arrested three times – once after a patient died as a result of abortion complications – but was ultimately acquitted.
For many doctors performing abortions at the time, “it was very much a kind of roll of the dice,” Ms. Ziegler says. “There was a sense that these laws were not enforced very much.”
Carole Joffe, PhD, a sociologist with expertise in reproductive health, recalls that there were very few doctors arrested, given the sheer number of abortions that were performed. The American College of Obstetricians and Gynecologists estimates that, in the years leading up to the original Roe decision, about 1.2 million women in the U.S. had illegal abortions – a number that exceeds today’s estimates.
Among the most notable cases of a doctor being detained was the arrest of gynecologist Jane Hodgson, MD, in 1970. Dr. Hodgson intentionally violated Minnesota law, which prohibited all abortions except in cases that were life-threatening to the patient.
After performing an abortion on a patient who had contracted rubella, also known as German measles, Dr. Hodgson was arrested, sentenced to 30 days in jail, and put on a year-long probation. She did not end up serving any time in jail, and her conviction was overturned after the Roe decision in 1973.
Now, the abortion restrictions being passed in many states have authorized much more sweeping penalties than those that existed in the pre-Roe era. According to Joffe, there is one key reason why we can anticipate more doctor arrests now.
“There simply was not the modern anti-abortion movement that we have come to know,” she says. “In the old days, there was not that much legal surveillance, and things were very unsafe. Fast forward to the present, we have much safer options now – like medication abortion pills – but we have a very different legal environment.”
Carmel Shachar, JD, MPH, a law and health policy expert at Harvard Law School, also expects that we will see more frequent prosecutions of doctors who provide abortion.
“There’s so much more data available through medical record-keeping and information generated by our phones and internet searches, that I think it would be much harder for a physician to fly under the radar,” Ms. Shachar says.
Also, Ms. Shachar emphasizes the power of prosecutorial discretion in abortion cases, where one prosecutor may choose to apply a law much more aggressively than another prosecutor in the next county over. Such has been seen in DeKalb County, Ga., which includes parts of Atlanta, where District Attorney Sherry Boston says she plans to use her prosecutorial discretion to address crimes like rape and murder, rather than “potentially investigat[ing] women and doctors for medical decisions,” Bloomberg Law reported. State Sen. Jen Jordan, the Democratic nominee for Georgia attorney general, has also said that, if elected, she would not enforce the state’s new 6-week abortion ban.
Is there a legal path forward for abortion care in states that forbid it?
Robin, an ob.gyn., became a complex family planning fellow in Utah to seek out further medical training and education in abortion care. Her plan was to solidify this as an area of expertise, so that, upon completing her fellowship, she could move back to her home state of Arizona to provide services there.
In Utah, where she currently practices, abortion is banned after 18 weeks. In Arizona, abortion is still allowed up to 24-26 weeks, until a pregnancy reaches “viability” (when a fetus is developed enough that it is able to survive outside the uterus with medical assistance). But new restrictions in Arizona may go into effect as early as September which would prohibit abortions after 15 weeks.
Despite the uncertain future of abortion access in Arizona, Robin still plans on moving there after her fellowship, but she hopes to travel to surrounding states to help provide abortion care where it’s less restricted. Even if she isn’t able to provide abortions at all, she says that there are still ways to help patients get safe, above-board abortions so as not to repeat the dangerous and often gruesome outcomes of self-induced abortions or those done by illegitimate practitioners before Roe.
“One of the roles that I think I can have as a physician is helping people with wraparound care for self-managed abortion,” says Robin. “If they can get the [abortion] pills online, then I can do the ultrasound beforehand, I can do the ultrasound after, I can talk them through it. I can help them with all the aspects of this care, I just can’t give them the pills myself.”
Whether a doctor can be penalized for “aiding and abetting” abortions that happen in different states remains an open question. In Texas, for example, Senate Bill 8 – which took effect Sept. 1, 2021 – not only established a fetal heartbeat law but added language that would allow private citizens to sue anyone who “knowingly engages in conduct that aids or abets the performance or inducement of an abortion” or anyone who even intends to do so.
That’s what happened to Alan Braid, MD, an ob.gyn. based in San Antonio. He confessed in a Washington Post op-ed that he had performed an abortion after cardiac activity had been detected in the pregnancy. Aware of the legal risks, he has since been sued by three people, and those cases are still underway.
But Ms. Ziegler says the chances of a doctor from a progressive state actually getting extradited and prosecuted by a state with restrictive abortion laws is pretty low – not zero, but low.
Like Robin, Natalie – an ob.gyn. in her early 30s – is a complex family planning fellow in Massachusetts. After her fellowship, she wants to return to Texas, where she completed her residency training.
“I’m at the point in my training where everyone starts looking for jobs and figuring out their next steps,” says Natalie. “The Dobbs decision introduced a ton of chaos due to the vagueness in the laws and how they get enforced, and then there’s chaos within institutions themselves and what kind of risk tolerance they have.”
Looking towards her future career path, Natalie says that she would not consider a job at an institution that didn’t allow her to teach abortion care to students, speak publicly about abortion rights, or let her travel outside of Texas to continue providing abortion care. She’s also preemptively seeking legal counsel and general guidance – advice that Ms. Ziegler strongly urges doctors to heed, sooner rather than later.
In states that have strict abortion bans with exceptions for life-threatening cases, there is still a lack of clarity around what is actually considered life-threatening enough to pass as an exception.
“Is it life-threatening in the next 6 hours? 24 hours? Seven days? One month?” Robin asks. “In medicine, we don’t necessarily talk about if something is life-threatening or not, we just say that there’s a high risk of X thing happening in X period of time. What’s the threshold at which that meets legal criteria? Nobody has an answer for that.”
Robin explains that, in her patients who have cancer, a pregnancy wouldn’t “necessarily kill them within the span of the next 9 months, but it could certainly accelerate their disease that could kill them within the next year or two.”
Right now, she says she doesn’t know what she would do if and when she is put in that position as a doctor.
“I didn’t go to medical school and become a doctor to become a felon,” says Robin. “Our goal is to make as many legal changes as we can to protect our patients and then practice as much harm reduction and as much care as we can within the letter of the law.”
A version of this article first appeared on WebMD.com.
The names of the doctors in this story have been changed at their request because of fear of legal repercussions and/or professional retaliation.
When an Ohio ob.gyn. had a patient in need of an abortion in July 2022, he knew he had to move quickly.
Daniel, who also sees patients at an abortion clinic, was treating a woman who came in for an abortion around 5 weeks into her pregnancy. And after going through the mandatory waiting periods, the required ultrasounds at each appointment, the consent process, and the options counseling, she was set for a surgical abortion the following Monday.
But on Monday, pre-op tests showed that her blood pressure was very high, posing a serious health risk if Daniel proceeded with the surgery.
Before the Supreme Court overturned Roe v. Wade in June, Daniel would have sent the patient home with instructions on how to lower her blood pressure over time. But the patient now had just four days to show the necessary improvement.
In this case, everything worked out. The patient returned Thursday and was able to have the procedure. But this is just one of the many day-to-day medical decisions abortion providers are now having to make with the changing legal risks being as top-of-mind to doctors as the safety of their patients.
Daniel said he doesn’t want the Ohio abortion law to change the way he communicates with his patients. As far as he knows, it’s still legal to talk to patients about self-managed abortions, as long as everything is unbiased and clearly stated, he says.
“But I don’t think I would get a lot of institutional support to have those conversations with patients because of the perceived legal liability,” says Daniel. “I will still have those conversations, but I’m not going to tell my employer that I’m having them and I’m not going to document them in the chart.”
Daniel is aware that having these kinds of discussions, or entertaining the possibility of omitting certain information from patient records, runs the risk of legal and professional consequences. Enforcement of these rules is foggy, too.
Under the Ohio law, if a fellow staff member suspects you of violating a law, you could be reported to a supervisor or licensing body. Abortion providers are aware they must be cautious about what they say because anti-abortion activitists, posing as patients, have secretly recorded conversations in the past, Daniel says.
Enforcement: The past, present, and future legal risks
Before Roe, enforcement of illegal abortion was spotty, says Mary Ziegler, JD, a professor at Florida State University College of Law, who specializes in the legal history of reproductive rights. At the start of the late 19th century, the doctors who provided illegal abortions would, in most cases, be prosecuted if a patient died as a result of the procedure.
A doctor in Ashland, Pa., named Robert Spencer was known for providing abortions in the small mining town where he practiced in the 1920s. He was reportedly arrested three times – once after a patient died as a result of abortion complications – but was ultimately acquitted.
For many doctors performing abortions at the time, “it was very much a kind of roll of the dice,” Ms. Ziegler says. “There was a sense that these laws were not enforced very much.”
Carole Joffe, PhD, a sociologist with expertise in reproductive health, recalls that there were very few doctors arrested, given the sheer number of abortions that were performed. The American College of Obstetricians and Gynecologists estimates that, in the years leading up to the original Roe decision, about 1.2 million women in the U.S. had illegal abortions – a number that exceeds today’s estimates.
Among the most notable cases of a doctor being detained was the arrest of gynecologist Jane Hodgson, MD, in 1970. Dr. Hodgson intentionally violated Minnesota law, which prohibited all abortions except in cases that were life-threatening to the patient.
After performing an abortion on a patient who had contracted rubella, also known as German measles, Dr. Hodgson was arrested, sentenced to 30 days in jail, and put on a year-long probation. She did not end up serving any time in jail, and her conviction was overturned after the Roe decision in 1973.
Now, the abortion restrictions being passed in many states have authorized much more sweeping penalties than those that existed in the pre-Roe era. According to Joffe, there is one key reason why we can anticipate more doctor arrests now.
“There simply was not the modern anti-abortion movement that we have come to know,” she says. “In the old days, there was not that much legal surveillance, and things were very unsafe. Fast forward to the present, we have much safer options now – like medication abortion pills – but we have a very different legal environment.”
Carmel Shachar, JD, MPH, a law and health policy expert at Harvard Law School, also expects that we will see more frequent prosecutions of doctors who provide abortion.
“There’s so much more data available through medical record-keeping and information generated by our phones and internet searches, that I think it would be much harder for a physician to fly under the radar,” Ms. Shachar says.
Also, Ms. Shachar emphasizes the power of prosecutorial discretion in abortion cases, where one prosecutor may choose to apply a law much more aggressively than another prosecutor in the next county over. Such has been seen in DeKalb County, Ga., which includes parts of Atlanta, where District Attorney Sherry Boston says she plans to use her prosecutorial discretion to address crimes like rape and murder, rather than “potentially investigat[ing] women and doctors for medical decisions,” Bloomberg Law reported. State Sen. Jen Jordan, the Democratic nominee for Georgia attorney general, has also said that, if elected, she would not enforce the state’s new 6-week abortion ban.
Is there a legal path forward for abortion care in states that forbid it?
Robin, an ob.gyn., became a complex family planning fellow in Utah to seek out further medical training and education in abortion care. Her plan was to solidify this as an area of expertise, so that, upon completing her fellowship, she could move back to her home state of Arizona to provide services there.
In Utah, where she currently practices, abortion is banned after 18 weeks. In Arizona, abortion is still allowed up to 24-26 weeks, until a pregnancy reaches “viability” (when a fetus is developed enough that it is able to survive outside the uterus with medical assistance). But new restrictions in Arizona may go into effect as early as September which would prohibit abortions after 15 weeks.
Despite the uncertain future of abortion access in Arizona, Robin still plans on moving there after her fellowship, but she hopes to travel to surrounding states to help provide abortion care where it’s less restricted. Even if she isn’t able to provide abortions at all, she says that there are still ways to help patients get safe, above-board abortions so as not to repeat the dangerous and often gruesome outcomes of self-induced abortions or those done by illegitimate practitioners before Roe.
“One of the roles that I think I can have as a physician is helping people with wraparound care for self-managed abortion,” says Robin. “If they can get the [abortion] pills online, then I can do the ultrasound beforehand, I can do the ultrasound after, I can talk them through it. I can help them with all the aspects of this care, I just can’t give them the pills myself.”
Whether a doctor can be penalized for “aiding and abetting” abortions that happen in different states remains an open question. In Texas, for example, Senate Bill 8 – which took effect Sept. 1, 2021 – not only established a fetal heartbeat law but added language that would allow private citizens to sue anyone who “knowingly engages in conduct that aids or abets the performance or inducement of an abortion” or anyone who even intends to do so.
That’s what happened to Alan Braid, MD, an ob.gyn. based in San Antonio. He confessed in a Washington Post op-ed that he had performed an abortion after cardiac activity had been detected in the pregnancy. Aware of the legal risks, he has since been sued by three people, and those cases are still underway.
But Ms. Ziegler says the chances of a doctor from a progressive state actually getting extradited and prosecuted by a state with restrictive abortion laws is pretty low – not zero, but low.
Like Robin, Natalie – an ob.gyn. in her early 30s – is a complex family planning fellow in Massachusetts. After her fellowship, she wants to return to Texas, where she completed her residency training.
“I’m at the point in my training where everyone starts looking for jobs and figuring out their next steps,” says Natalie. “The Dobbs decision introduced a ton of chaos due to the vagueness in the laws and how they get enforced, and then there’s chaos within institutions themselves and what kind of risk tolerance they have.”
Looking towards her future career path, Natalie says that she would not consider a job at an institution that didn’t allow her to teach abortion care to students, speak publicly about abortion rights, or let her travel outside of Texas to continue providing abortion care. She’s also preemptively seeking legal counsel and general guidance – advice that Ms. Ziegler strongly urges doctors to heed, sooner rather than later.
In states that have strict abortion bans with exceptions for life-threatening cases, there is still a lack of clarity around what is actually considered life-threatening enough to pass as an exception.
“Is it life-threatening in the next 6 hours? 24 hours? Seven days? One month?” Robin asks. “In medicine, we don’t necessarily talk about if something is life-threatening or not, we just say that there’s a high risk of X thing happening in X period of time. What’s the threshold at which that meets legal criteria? Nobody has an answer for that.”
Robin explains that, in her patients who have cancer, a pregnancy wouldn’t “necessarily kill them within the span of the next 9 months, but it could certainly accelerate their disease that could kill them within the next year or two.”
Right now, she says she doesn’t know what she would do if and when she is put in that position as a doctor.
“I didn’t go to medical school and become a doctor to become a felon,” says Robin. “Our goal is to make as many legal changes as we can to protect our patients and then practice as much harm reduction and as much care as we can within the letter of the law.”
A version of this article first appeared on WebMD.com.
Topical roflumilast approved for psoriasis in adults and adolescents
.
Roflumilast is a selective inhibitor of phosphodiesterase 4 (PDE4), the first approved for treating psoriasis, according to manufacturer Arcutis Biotherapeutics. The company announced the approval on July 29. Oral roflumilast (Daliresp ) was approved in 2011 for treating chronic obstructive pulmonary disease (COPD).“It’s a breakthrough topical therapy,” says Mark G. Lebwohl, MD, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York, and principal investigator in trials of topical roflumilast. In an interview, Dr. Lebwohl noted that the treatment significantly reduced psoriasis symptoms in both short- and long-term trials.

In addition, two features of this treatment set it apart from other topical psoriasis treatments, he said. Roflumilast is not a steroid, so does not have the risk for topical steroid-related side effects associated with chronic use, and, in clinical trials, topical roflumilast was effective in treating psoriasis in intertriginous areas, including the buttocks, underarms, and beneath the breasts, which are difficult to treat.
FDA approval is based on data from two phase 3 randomized, double-blind, vehicle-controlled trials, according to Arcutis. The primary endpoint was Investigator Global Assessment (IGA) success, defined as clear or almost clear with at least a two-grade improvement from baseline, and at least a two-grade IGA score improvement from baseline at 8 weeks.
At 8 weeks, 42.4% and 37.5% of the patients treated with topical roflumilast achieved an IGA success rate compared with 6.1% and 6.9% in the control groups, respectively (P < .0001 for both studies).
Treated patients also experienced significant improvements compared with those in the vehicle groups in secondary endpoints in the trials: Those included Intertriginous IGA (I-IGA) Success, Psoriasis Area Severity Index–75 (PASI-75), reductions in itch based on the Worst Itch–Numerical Rating Scale (WI-NRS), and self-reported psoriasis symptoms diary (PSD).
In the studies, 72% and 68% of patients treated with roflumilast met the I-IGA endpoint at 8 weeks versus 14% and 17%, respectively, of those on vehicle (P < .0001 for both studies).
In addition, by week 2, some participants treated with roflumilast had experienced reduced itchiness in both studies. At 8 weeks, among those with a WI-NRS score of 4 or more at baseline, 67% and 69% of the treated patients had at least a four-point reduction in the WI-NRS versus 26% and 33%, respectively, among those on vehicle (P < .0001 for both studies), according to the company.
In general, the cream was well tolerated. There were reports of diarrhea (3%), headache (2%), insomnia (1%), nausea (1%), application-site pain (1%), upper respiratory tract infections (1%), and urinary tract infections (1%). However, Dr. Lebwohl noted that these events were also observed in the control group.
“The study was unequivocal about the improvement in the intertriginous sites,” Dr. Lebwohl said. He contrasts that to the data from other nonsteroidal topicals, which he said can be associated with a rash or irritation in sensitive areas.
Dr. Lebwohl noted that PDE4 is an enzyme that increases inflammation and decreases anti-inflammatory mediators and that inhibiting PDE4 may interrupt some of the inflammation response responsible for psoriasis symptoms, as it has for other conditions such as atopic dermatitis. Data from the 8-week phase 3 trials and yearlong phase 2b, open-label studies support that hypothesis.
“I’m always excited for new psoriasis treatments to broaden our treatment armamentarium,” said Lauren E. Ploch, MD, a dermatologist who practices in Augusta, Ga., and Aiken, S.C., who was asked to comment on the approval.
Even a symptom that seems benign, like itching, Dr. Ploch added, can lead to reduced sleep and increased irritability. Referring to the data on the treatment in the sensitive, intertriginous areas, she noted that the skin in these areas is often thinner, so treatment with steroids can cause further thinning and damage to the skin. If roflumilast doesn’t cause burning, itching, or thinning, it will be a great option to treat these areas, she said in an interview. She was not involved in the trials.
Roflumilast cream will be marketed under the trade name Zoryve, and is expected to be available by mid-August, according to Arcutis.
Roflumilast cream is also under review in Canada for treatment of plaque psoriasis in adults and adolescents.
The studies were funded by Arcutis Biotherapeutics. Dr. Lebwohl reported receiving grant support and consulting fees from Arcutis. Dr. Ploch reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
.
Roflumilast is a selective inhibitor of phosphodiesterase 4 (PDE4), the first approved for treating psoriasis, according to manufacturer Arcutis Biotherapeutics. The company announced the approval on July 29. Oral roflumilast (Daliresp ) was approved in 2011 for treating chronic obstructive pulmonary disease (COPD).“It’s a breakthrough topical therapy,” says Mark G. Lebwohl, MD, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York, and principal investigator in trials of topical roflumilast. In an interview, Dr. Lebwohl noted that the treatment significantly reduced psoriasis symptoms in both short- and long-term trials.
In addition, two features of this treatment set it apart from other topical psoriasis treatments, he said. Roflumilast is not a steroid, so does not have the risk for topical steroid-related side effects associated with chronic use, and, in clinical trials, topical roflumilast was effective in treating psoriasis in intertriginous areas, including the buttocks, underarms, and beneath the breasts, which are difficult to treat.
FDA approval is based on data from two phase 3 randomized, double-blind, vehicle-controlled trials, according to Arcutis. The primary endpoint was Investigator Global Assessment (IGA) success, defined as clear or almost clear with at least a two-grade improvement from baseline, and at least a two-grade IGA score improvement from baseline at 8 weeks.
At 8 weeks, 42.4% and 37.5% of the patients treated with topical roflumilast achieved an IGA success rate compared with 6.1% and 6.9% in the control groups, respectively (P < .0001 for both studies).
Treated patients also experienced significant improvements compared with those in the vehicle groups in secondary endpoints in the trials: Those included Intertriginous IGA (I-IGA) Success, Psoriasis Area Severity Index–75 (PASI-75), reductions in itch based on the Worst Itch–Numerical Rating Scale (WI-NRS), and self-reported psoriasis symptoms diary (PSD).
In the studies, 72% and 68% of patients treated with roflumilast met the I-IGA endpoint at 8 weeks versus 14% and 17%, respectively, of those on vehicle (P < .0001 for both studies).
In addition, by week 2, some participants treated with roflumilast had experienced reduced itchiness in both studies. At 8 weeks, among those with a WI-NRS score of 4 or more at baseline, 67% and 69% of the treated patients had at least a four-point reduction in the WI-NRS versus 26% and 33%, respectively, among those on vehicle (P < .0001 for both studies), according to the company.
In general, the cream was well tolerated. There were reports of diarrhea (3%), headache (2%), insomnia (1%), nausea (1%), application-site pain (1%), upper respiratory tract infections (1%), and urinary tract infections (1%). However, Dr. Lebwohl noted that these events were also observed in the control group.
“The study was unequivocal about the improvement in the intertriginous sites,” Dr. Lebwohl said. He contrasts that to the data from other nonsteroidal topicals, which he said can be associated with a rash or irritation in sensitive areas.
Dr. Lebwohl noted that PDE4 is an enzyme that increases inflammation and decreases anti-inflammatory mediators and that inhibiting PDE4 may interrupt some of the inflammation response responsible for psoriasis symptoms, as it has for other conditions such as atopic dermatitis. Data from the 8-week phase 3 trials and yearlong phase 2b, open-label studies support that hypothesis.
“I’m always excited for new psoriasis treatments to broaden our treatment armamentarium,” said Lauren E. Ploch, MD, a dermatologist who practices in Augusta, Ga., and Aiken, S.C., who was asked to comment on the approval.
Even a symptom that seems benign, like itching, Dr. Ploch added, can lead to reduced sleep and increased irritability. Referring to the data on the treatment in the sensitive, intertriginous areas, she noted that the skin in these areas is often thinner, so treatment with steroids can cause further thinning and damage to the skin. If roflumilast doesn’t cause burning, itching, or thinning, it will be a great option to treat these areas, she said in an interview. She was not involved in the trials.
Roflumilast cream will be marketed under the trade name Zoryve, and is expected to be available by mid-August, according to Arcutis.
Roflumilast cream is also under review in Canada for treatment of plaque psoriasis in adults and adolescents.
The studies were funded by Arcutis Biotherapeutics. Dr. Lebwohl reported receiving grant support and consulting fees from Arcutis. Dr. Ploch reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
.
Roflumilast is a selective inhibitor of phosphodiesterase 4 (PDE4), the first approved for treating psoriasis, according to manufacturer Arcutis Biotherapeutics. The company announced the approval on July 29. Oral roflumilast (Daliresp ) was approved in 2011 for treating chronic obstructive pulmonary disease (COPD).“It’s a breakthrough topical therapy,” says Mark G. Lebwohl, MD, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York, and principal investigator in trials of topical roflumilast. In an interview, Dr. Lebwohl noted that the treatment significantly reduced psoriasis symptoms in both short- and long-term trials.
In addition, two features of this treatment set it apart from other topical psoriasis treatments, he said. Roflumilast is not a steroid, so does not have the risk for topical steroid-related side effects associated with chronic use, and, in clinical trials, topical roflumilast was effective in treating psoriasis in intertriginous areas, including the buttocks, underarms, and beneath the breasts, which are difficult to treat.
FDA approval is based on data from two phase 3 randomized, double-blind, vehicle-controlled trials, according to Arcutis. The primary endpoint was Investigator Global Assessment (IGA) success, defined as clear or almost clear with at least a two-grade improvement from baseline, and at least a two-grade IGA score improvement from baseline at 8 weeks.
At 8 weeks, 42.4% and 37.5% of the patients treated with topical roflumilast achieved an IGA success rate compared with 6.1% and 6.9% in the control groups, respectively (P < .0001 for both studies).
Treated patients also experienced significant improvements compared with those in the vehicle groups in secondary endpoints in the trials: Those included Intertriginous IGA (I-IGA) Success, Psoriasis Area Severity Index–75 (PASI-75), reductions in itch based on the Worst Itch–Numerical Rating Scale (WI-NRS), and self-reported psoriasis symptoms diary (PSD).
In the studies, 72% and 68% of patients treated with roflumilast met the I-IGA endpoint at 8 weeks versus 14% and 17%, respectively, of those on vehicle (P < .0001 for both studies).
In addition, by week 2, some participants treated with roflumilast had experienced reduced itchiness in both studies. At 8 weeks, among those with a WI-NRS score of 4 or more at baseline, 67% and 69% of the treated patients had at least a four-point reduction in the WI-NRS versus 26% and 33%, respectively, among those on vehicle (P < .0001 for both studies), according to the company.
In general, the cream was well tolerated. There were reports of diarrhea (3%), headache (2%), insomnia (1%), nausea (1%), application-site pain (1%), upper respiratory tract infections (1%), and urinary tract infections (1%). However, Dr. Lebwohl noted that these events were also observed in the control group.
“The study was unequivocal about the improvement in the intertriginous sites,” Dr. Lebwohl said. He contrasts that to the data from other nonsteroidal topicals, which he said can be associated with a rash or irritation in sensitive areas.
Dr. Lebwohl noted that PDE4 is an enzyme that increases inflammation and decreases anti-inflammatory mediators and that inhibiting PDE4 may interrupt some of the inflammation response responsible for psoriasis symptoms, as it has for other conditions such as atopic dermatitis. Data from the 8-week phase 3 trials and yearlong phase 2b, open-label studies support that hypothesis.
“I’m always excited for new psoriasis treatments to broaden our treatment armamentarium,” said Lauren E. Ploch, MD, a dermatologist who practices in Augusta, Ga., and Aiken, S.C., who was asked to comment on the approval.
Even a symptom that seems benign, like itching, Dr. Ploch added, can lead to reduced sleep and increased irritability. Referring to the data on the treatment in the sensitive, intertriginous areas, she noted that the skin in these areas is often thinner, so treatment with steroids can cause further thinning and damage to the skin. If roflumilast doesn’t cause burning, itching, or thinning, it will be a great option to treat these areas, she said in an interview. She was not involved in the trials.
Roflumilast cream will be marketed under the trade name Zoryve, and is expected to be available by mid-August, according to Arcutis.
Roflumilast cream is also under review in Canada for treatment of plaque psoriasis in adults and adolescents.
The studies were funded by Arcutis Biotherapeutics. Dr. Lebwohl reported receiving grant support and consulting fees from Arcutis. Dr. Ploch reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.

Flickering sensation in eyes
The American Diabetes Association (ADA) position statement on diabetic retinopathy states that hyperglycemia has been the most consistently associated risk factor for retinopathy. A large and consistent set of observational studies and clinical trials confirms the association of poor glucose control and retinopathy.
The Diabetes Control and Complications Trial (DCCT), a randomized controlled clinical trial of intensive glycemic control vs conventional glycemic control in people with type 1 diabetes (T1D), demonstrated that intensive therapy reduced the development or progression of diabetic retinopathy by 34%-76%. The DCCT also demonstrated a definitive relationship between hyperglycemia and diabetic microvascular complications, including retinopathy. Early treatment with intensive therapy was effective.
The UK Prospective Diabetes Study (UKPDS) of patients with newly diagnosed T2D conclusively demonstrated that improved blood glucose control reduced the risk for retinopathy and nephropathy and, possibly, neuropathy. The overall microvascular complication rate was decreased by 25% in patients receiving intensive therapy vs conventional therapy. Epidemiologic analysis of the UKPDS data showed a continuous relationship between the risk for microvascular complications and glycemia, such that every percentage-point decrease in A1c (eg, 9% to 8%) was associated with a 35% reduction in the risk for microvascular complications.
More recently, the ACCORD trial of medical therapies demonstrated that intensive glycemic control reduced the risk for progression of diabetic retinopathy in people with T2D of 10 years' duration. This study included 2856 ACCORD participants enrolled in the ACCORD Eye Study and followed for 4 years.
The ADA recommends screening by an ophthalmologist for diabetic retinopathy within 5 years of the diagnosis of T1D and at the time of diagnosis of T2D. Women with preexisting diabetes who are planning pregnancy or who have become pregnant should be screened before pregnancy or in the first trimester.
While optimization of blood glucose, blood pressure, and serum lipid levels in conjunction with appropriately scheduled dilated eye examinations can substantially decrease the risk for vision loss from diabetic retinopathy, a significant proportion of those affected with diabetes develop diabetic macular edema or proliferative changes that require intervention. ADA treatment recommendations are:
• Refer patients with any level of macular edema, severe nonproliferative diabetic retinopathy (a precursor of proliferative diabetic retinopathy), or proliferative diabetic retinopathy to an ophthalmologist knowledgeable and experienced in the management and treatment of diabetic retinopathy.
• Laser photocoagulation therapy reduces the risk for vision loss in patients with high-risk proliferative diabetic retinopathy and, in some cases, severe nonproliferative diabetic retinopathy.
• Intravitreous injections of anti–vascular endothelial growth factor are indicated for central-involved diabetic macular edema, which occurs beneath the foveal center and may threaten reading vision.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The American Diabetes Association (ADA) position statement on diabetic retinopathy states that hyperglycemia has been the most consistently associated risk factor for retinopathy. A large and consistent set of observational studies and clinical trials confirms the association of poor glucose control and retinopathy.
The Diabetes Control and Complications Trial (DCCT), a randomized controlled clinical trial of intensive glycemic control vs conventional glycemic control in people with type 1 diabetes (T1D), demonstrated that intensive therapy reduced the development or progression of diabetic retinopathy by 34%-76%. The DCCT also demonstrated a definitive relationship between hyperglycemia and diabetic microvascular complications, including retinopathy. Early treatment with intensive therapy was effective.
The UK Prospective Diabetes Study (UKPDS) of patients with newly diagnosed T2D conclusively demonstrated that improved blood glucose control reduced the risk for retinopathy and nephropathy and, possibly, neuropathy. The overall microvascular complication rate was decreased by 25% in patients receiving intensive therapy vs conventional therapy. Epidemiologic analysis of the UKPDS data showed a continuous relationship between the risk for microvascular complications and glycemia, such that every percentage-point decrease in A1c (eg, 9% to 8%) was associated with a 35% reduction in the risk for microvascular complications.
More recently, the ACCORD trial of medical therapies demonstrated that intensive glycemic control reduced the risk for progression of diabetic retinopathy in people with T2D of 10 years' duration. This study included 2856 ACCORD participants enrolled in the ACCORD Eye Study and followed for 4 years.
The ADA recommends screening by an ophthalmologist for diabetic retinopathy within 5 years of the diagnosis of T1D and at the time of diagnosis of T2D. Women with preexisting diabetes who are planning pregnancy or who have become pregnant should be screened before pregnancy or in the first trimester.
While optimization of blood glucose, blood pressure, and serum lipid levels in conjunction with appropriately scheduled dilated eye examinations can substantially decrease the risk for vision loss from diabetic retinopathy, a significant proportion of those affected with diabetes develop diabetic macular edema or proliferative changes that require intervention. ADA treatment recommendations are:
• Refer patients with any level of macular edema, severe nonproliferative diabetic retinopathy (a precursor of proliferative diabetic retinopathy), or proliferative diabetic retinopathy to an ophthalmologist knowledgeable and experienced in the management and treatment of diabetic retinopathy.
• Laser photocoagulation therapy reduces the risk for vision loss in patients with high-risk proliferative diabetic retinopathy and, in some cases, severe nonproliferative diabetic retinopathy.
• Intravitreous injections of anti–vascular endothelial growth factor are indicated for central-involved diabetic macular edema, which occurs beneath the foveal center and may threaten reading vision.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The American Diabetes Association (ADA) position statement on diabetic retinopathy states that hyperglycemia has been the most consistently associated risk factor for retinopathy. A large and consistent set of observational studies and clinical trials confirms the association of poor glucose control and retinopathy.
The Diabetes Control and Complications Trial (DCCT), a randomized controlled clinical trial of intensive glycemic control vs conventional glycemic control in people with type 1 diabetes (T1D), demonstrated that intensive therapy reduced the development or progression of diabetic retinopathy by 34%-76%. The DCCT also demonstrated a definitive relationship between hyperglycemia and diabetic microvascular complications, including retinopathy. Early treatment with intensive therapy was effective.
The UK Prospective Diabetes Study (UKPDS) of patients with newly diagnosed T2D conclusively demonstrated that improved blood glucose control reduced the risk for retinopathy and nephropathy and, possibly, neuropathy. The overall microvascular complication rate was decreased by 25% in patients receiving intensive therapy vs conventional therapy. Epidemiologic analysis of the UKPDS data showed a continuous relationship between the risk for microvascular complications and glycemia, such that every percentage-point decrease in A1c (eg, 9% to 8%) was associated with a 35% reduction in the risk for microvascular complications.
More recently, the ACCORD trial of medical therapies demonstrated that intensive glycemic control reduced the risk for progression of diabetic retinopathy in people with T2D of 10 years' duration. This study included 2856 ACCORD participants enrolled in the ACCORD Eye Study and followed for 4 years.
The ADA recommends screening by an ophthalmologist for diabetic retinopathy within 5 years of the diagnosis of T1D and at the time of diagnosis of T2D. Women with preexisting diabetes who are planning pregnancy or who have become pregnant should be screened before pregnancy or in the first trimester.
While optimization of blood glucose, blood pressure, and serum lipid levels in conjunction with appropriately scheduled dilated eye examinations can substantially decrease the risk for vision loss from diabetic retinopathy, a significant proportion of those affected with diabetes develop diabetic macular edema or proliferative changes that require intervention. ADA treatment recommendations are:
• Refer patients with any level of macular edema, severe nonproliferative diabetic retinopathy (a precursor of proliferative diabetic retinopathy), or proliferative diabetic retinopathy to an ophthalmologist knowledgeable and experienced in the management and treatment of diabetic retinopathy.
• Laser photocoagulation therapy reduces the risk for vision loss in patients with high-risk proliferative diabetic retinopathy and, in some cases, severe nonproliferative diabetic retinopathy.
• Intravitreous injections of anti–vascular endothelial growth factor are indicated for central-involved diabetic macular edema, which occurs beneath the foveal center and may threaten reading vision.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
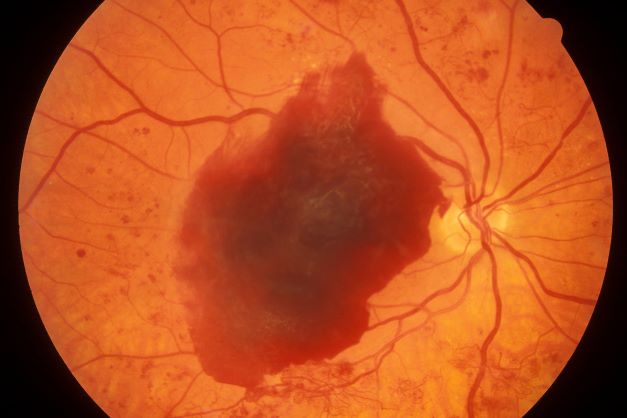
A 48-year-old Black man with type 2 diabetes (T2D) presented with complaints of a "flickering" sensation and a decrease in brightness of colors in both eyes as well as floaters in his left eye for several weeks. He reported that his symptoms fluctuate with changes in his blood glucose levels. His last eye examination was 2 years ago and his ocular history was unremarkable. His medical history was significant with a history of hypertension and T2D requiring insulin. His most recent glycated hemoglobin (A1c), 2 months ago, was 8.4%. His BMI was 31.2. The patient's medications were dulaglutide 0.75 mg injection pen, glargine insulin 42 units, losartan 100 mg, and amlodipine 10 mg.
On examination, his best-corrected visual acuity was 20/20 in the right eye and 20/30 in the left eye. Confrontation fields were intact, extraocular movements were full and extensive, and both pupils were equal, round, and reactive to light without afferent pupillary defects. Anterior segment examination was unremarkable in both eyes, without iris neovascularization. Intraocular pressures were 17 mm Hg in the right eye and 16 mm Hg in the left eye. On dilated fundus examination, the cup-to-disc ratio was 0.45 horizontally and vertically, with the presence of 1/4 disc diameters of neovascularization of the disc in the right eye and 2/3 disc diameters of neovascularization of the disc in the left eye.
Posterior segment findings were significant for scattered microaneurysms and dot/blot hemorrhages in the maculae. In the periphery of both eyes, there were tortuous vessels, scatter microaneurysms with dot/blot hemorrhages, and multiple areas of neovascularization elsewhere, with several foci of vitreous traction. There was no vitreous hemorrhage or tractional retinal detachment of either eye.
Spectral domain optical coherence tomography revealed an epiretinal membrane in the right eye and a blunted foveal contour with parafoveal cystic spaces, probably secondary to vitreomacular contraction. The left eye also had an epiretinal membrane and blunted foveal contour secondary to vitreomacular adhesion. The patient was diagnosed with bilateral high-risk proliferative diabetic retinopathy.
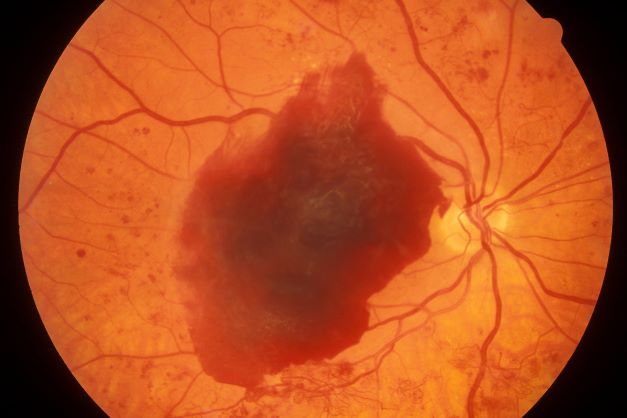
FDA panel urges caution with skin cancer–detecting tools
A and how to address longstanding issues of racial equity in this field of medicine.
The Food and Drug Administration has scheduled two meetings to gather expert feedback about managing an expected expansion in the use of skin lesion apps and devices. Outside of the United States, there are apps promoted as being able to help spot skin lesions that should trigger a medical visit.
The general and plastic surgery devices panel of the FDA’s Medical Devices Advisory Committee began work on this topic on July 28, with a wide-ranging discussion about potential expanded use of computer-aided, skin lesion analyzer (SLA) devices. On Friday, the panel is considering an FDA proposal to shift the designation for an approved device for aiding dermatologists in skin cancer diagnoses from the most stringent regulatory category, class III, to the less restrictive class II.
The FDA called the meeting amid growing interest in using technology to aid in finding cancers, with some of these products already marketed to consumers outside of the United States. There are presently no legally marketed, FDA-cleared or FDA-approved SLA devices indicated for use by clinicians other than dermatologists or the lay public, the agency said in a briefing memo for the meeting. There are two devices with FDA approval, though, for aiding dermatologists. The FDA approved SciBase’s Nevisense in 2017 and Mela Sciences’ MelaFind, which has fallen out of use, in 2012. Both are class III devices.
But some companies intend to offer products for consumers in the United States. The company SkinVision, for example, has developed an app of the same name, which is intended to detect suspicious-looking skin spots via smartphone photos. SkinVision’s website says the product has been offered to consumers in Australia for remote skin checks since 2015. People in the Netherlands and United Kingdom also can use SkinVision, according to the company’s website. SkinVision says the company is working on providing the app for U.S. customers, “but we are not quite there yet.”
During the meeting, FDA panelists repeatedly emphasized the potential risks of these devices in terms of sensitivity (how often a test correctly generates a positive result) and of specificity (how often a test correctly generates a negative result).
New tools intended to aid in detection of skin cancer might produce too many false positives and thus trigger floods of worried patients seeking care and often facing unnecessary biopsies, the FDA panelists said. But more worrisome would be FDA clearance of tools that delivered too many false negative results, leaving people unaware of their cancers.

The standards would have to be set very high for new products, especially those intended for consumers, said FDA panelist Murad Alam, MD, a dermatologist and vice chair of the department of dermatology at Northwestern University, Chicago. Current technologies for analyzing skin lesions are not yet up to that task. Dr. Alam likened the situation to the hopes for self-driving cars.
“It sounds great in principle. If you read the predictions from 20 years ago, it should already have happened,” Dr. Alam said. “But we’re still struggling with that because there are serious points of failure.”
FDA panelist Veronica Rotemberg, MD, PhD, a dermatologist at Memorial Sloan Kettering Cancer Center, New York, also argued for well-designed studies to understand how consumers and clinicians would react to new tools.
“We have to define what prospective information, in the intended use setting, we need to feel comfortable saying that these tools could be in a layperson’s hand or a primary care person’s hand,” Dr. Rotemberg said.
The studies would not need to be large, especially in the case of nonmelanoma skin cancer, which is common, she added.
“There’s too much nuance here for us to be able to say: ‘This is what would happen,’ without testing it,” Dr. Rotemberg said. “I do not think these prospective studies would be very burdensome, but they would help us understand what the burden would be and what the costs would be and what the potential harms would be.”
Because of rules against disclosing corporate information, the FDA cannot tell the public about the kinds of inquiries it already may have fielded from companies interested in selling skin cancer detection tools.
But in response to a question during the FDA meeting, Binita Ashar, MD, a top official in the FDA’s Center for Devices and Radiological Health, said there is interest in having these kinds of products sold in the United States as well.
“I can tell you that this a very timely discussion and questions that we’re posing to you are the questions that we’re encountering or that we have been grappling with,” Dr. Ashar said.
FDA panelists noted that many patients cannot get access easily to dermatology visits.
Companies seeking to develop SLA devices likely will market their tools as attempts to fill a gap that now exists in medical care.
But there will be challenges ahead in explaining to patients how to interpret readings from these tools, the FDA panelists said. Consumers should know these tools are meant to assist in diagnosis, and not to make it.
“I’m not sure the layperson will hear that,” said FDA panelist Paula E. Bourelly, MD, a dermatologist from Olney, Md.
As a result, use of SLA tools could create tension between physicians and patients, with consumers demanding biopsies after seeing readings they don’t understand.
“I do have great concerns about the layperson feeling overly confident and reducing the provider to a technician,” she said.
The FDA panelists were not asked to cast formal votes on any issues discussed during the meeting They instead engaged in broad discussions around questions posed by the FDA in three key areas:
- What standards should be used to confirm lesion diagnosis in clinical testing of the accuracy of SLA devices?
- What would be acceptable true false-positive and false-negative results (sensitivity and specificity) for different diagnoses and users?
- How can the FDA address health equity considerations based on variable incidence of skin lesions?
Developing standards
The FDA asked the panel to consider several scenarios for SLA devices and to discuss how standards might vary depending on the user of the device, whether it would be dermatologists, other clinicians, or consumers.
The agency sought comments in particular about using histological diagnosis (core specimen processing with a consensus diagnosis from an expert dermatopathologist panel). In the briefing document for the meeting, the FDA argued that this approach provides the greatest certainty in the diagnosis.
“Device developers, however, cite concerns, both practical and ethical, in requiring biopsy of all lesions, particularly those that appear benign,” the FDA said. “They have proposed alternate means of defining ground truth, including consensus opinion of experts (of visual or dermoscopic examination of the lesion[s]), opinion of one expert (visual or dermoscopic examination), or other methods.”
In summarizing the discussion on this question, the FDA panel chairman, Hobart W. Harris, MD, MPH, a surgeon from the University of California, San Francisco, noted that there was broad support for histological data in clinical trials of SLA devices, with some allowance for cases where more hybrid approaches would be used.
There were also suggestions offered about designing trials and the need for biopsies of lesions that are clearly benign, as this would help gather data to help in developing algorithms.
Dr. Alam said care should be taken in explaining to study participants that they might have to undergo biopsies that they didn’t need, as part of the larger effort to gather data. This should be detailed in the consent form, he said.
“But I also think this is a relatively minor risk,” Dr. Alam said, comparing these biopsies to the blood samples that patients in many clinical studies routinely give.
“Are all of those blood draws necessary to track the change in whatever parameters that are being tracked? Probably not,” Dr. Alam said. “I think it would be possible to explain to a reasonable patient what this entails.”
Dr. Alam noted that companies might face extra hurdles in enrolling study participants and keeping them in the trials if the FDA seeks this kind of biopsy data. “But I don’t think inconvenience to the study sponsor is a good argument” for not seeking this kind of data, he added.
Leaving a loophole where certain kinds of clearly benign lesions don’t require a biopsy would eventually erode the quality of the research done on these devices. “That bar will be moved to accommodate the convenience of the sponsor, to make the study feasible,” Dr. Alam said. “And pretty soon, you’ll be missing a lot of patients that really should have biopsies.”
Acceptable rates of false positives, false negatives
The FDA panel chair noted that his colleagues had strongly urged review standards that would require that the devices improve on the rates of successful catches of suspicious lesions and lower false positives. But they did not endorse specific targets regarding the sensitivity and specificity rates.
“No one seems to be comfortable with providing or preordaining” these targets, Dr. Harris said.
Panelist Deneen Hesser, MSHSA, RN, urged a deep recognition of the power of a FDA clearance in the view of consumers.
“We need to be cognizant of what the term ‘FDA approved’ means to the lay individual,” said Ms. Hesser, who served as the patient representative on the panel. “A patient who sees that those tools are FDA approved will assume that each of those is the gold standard” in terms of expectations for delivering accurate results.
Like many of the panelists, Dr. Rotemberg urged the FDA to gather data about how patients would react to different messages encoded in consumer-oriented products.

“If the device says: ‘You should see a dermatologist for this’ and no other information, that’s very different from [saying]: ‘That lesion is suspicious for melanoma,’ ” Dr. Rotemberg said.
Despite the likely difficulties in conducting trials, the FDA needs to have the data to answer key questions about patient and physician reactions to readings from new tools, Dr. Rotemberg said.
“We don’t know how many additional biopsies we would cause with a specificity of 80%” for a new SLA tool, Dr. Rotemberg said, giving an example. “We don’t know how confident a dermatologist might be to say: ‘Actually, I’m not suspicious about that lesion and we can just fudge it or not biopsy it.’ We don’t know any of that until we study it in real life.”
The panelists also urged the FDA to seek to ensure that new tools used in analyzing skin lesions improve the quality of diagnosis.
Addressing equity
The FDA also asked the panel to weigh in on whether the agency should clear SLA tools in cases where the existing study data is drawn heavily from people considered to be at higher risk for skin cancer.

“To ensure generalizability across the entire U.S. population, should FDA require SLAs indicated for use beyond cancerous lesions be tested in a representative U.S. population?” the FDA asked.
The three most common skin cancers – melanoma, basal cell carcinoma, and squamous cell carcinoma – are more prevalent in people with Fitzpatrick I and II skin types, who tend to get sunburns, not tans. But people of color are more likely to develop melanoma in areas that are not sun exposed, such as the sole of the foot or under fingernails or toenails.
“Due in part to lower expected risk and screening, these melanomas are often detected late,” the FDA said in the briefing document.
There was broad consensus among panelists that the FDA should encourage companies to enroll people with all skin types and tones.
But they also looked for ways that the FDA could clear devices based on initial studies conducted largely with people considered to be at higher risk, with the agency then requiring follow-up trials to see how these products would work for the general U.S. population.
A version of this article first appeared on Medscape.com.
A and how to address longstanding issues of racial equity in this field of medicine.
The Food and Drug Administration has scheduled two meetings to gather expert feedback about managing an expected expansion in the use of skin lesion apps and devices. Outside of the United States, there are apps promoted as being able to help spot skin lesions that should trigger a medical visit.
The general and plastic surgery devices panel of the FDA’s Medical Devices Advisory Committee began work on this topic on July 28, with a wide-ranging discussion about potential expanded use of computer-aided, skin lesion analyzer (SLA) devices. On Friday, the panel is considering an FDA proposal to shift the designation for an approved device for aiding dermatologists in skin cancer diagnoses from the most stringent regulatory category, class III, to the less restrictive class II.
The FDA called the meeting amid growing interest in using technology to aid in finding cancers, with some of these products already marketed to consumers outside of the United States. There are presently no legally marketed, FDA-cleared or FDA-approved SLA devices indicated for use by clinicians other than dermatologists or the lay public, the agency said in a briefing memo for the meeting. There are two devices with FDA approval, though, for aiding dermatologists. The FDA approved SciBase’s Nevisense in 2017 and Mela Sciences’ MelaFind, which has fallen out of use, in 2012. Both are class III devices.
But some companies intend to offer products for consumers in the United States. The company SkinVision, for example, has developed an app of the same name, which is intended to detect suspicious-looking skin spots via smartphone photos. SkinVision’s website says the product has been offered to consumers in Australia for remote skin checks since 2015. People in the Netherlands and United Kingdom also can use SkinVision, according to the company’s website. SkinVision says the company is working on providing the app for U.S. customers, “but we are not quite there yet.”
During the meeting, FDA panelists repeatedly emphasized the potential risks of these devices in terms of sensitivity (how often a test correctly generates a positive result) and of specificity (how often a test correctly generates a negative result).
New tools intended to aid in detection of skin cancer might produce too many false positives and thus trigger floods of worried patients seeking care and often facing unnecessary biopsies, the FDA panelists said. But more worrisome would be FDA clearance of tools that delivered too many false negative results, leaving people unaware of their cancers.
The standards would have to be set very high for new products, especially those intended for consumers, said FDA panelist Murad Alam, MD, a dermatologist and vice chair of the department of dermatology at Northwestern University, Chicago. Current technologies for analyzing skin lesions are not yet up to that task. Dr. Alam likened the situation to the hopes for self-driving cars.
“It sounds great in principle. If you read the predictions from 20 years ago, it should already have happened,” Dr. Alam said. “But we’re still struggling with that because there are serious points of failure.”
FDA panelist Veronica Rotemberg, MD, PhD, a dermatologist at Memorial Sloan Kettering Cancer Center, New York, also argued for well-designed studies to understand how consumers and clinicians would react to new tools.
“We have to define what prospective information, in the intended use setting, we need to feel comfortable saying that these tools could be in a layperson’s hand or a primary care person’s hand,” Dr. Rotemberg said.
The studies would not need to be large, especially in the case of nonmelanoma skin cancer, which is common, she added.
“There’s too much nuance here for us to be able to say: ‘This is what would happen,’ without testing it,” Dr. Rotemberg said. “I do not think these prospective studies would be very burdensome, but they would help us understand what the burden would be and what the costs would be and what the potential harms would be.”
Because of rules against disclosing corporate information, the FDA cannot tell the public about the kinds of inquiries it already may have fielded from companies interested in selling skin cancer detection tools.
But in response to a question during the FDA meeting, Binita Ashar, MD, a top official in the FDA’s Center for Devices and Radiological Health, said there is interest in having these kinds of products sold in the United States as well.
“I can tell you that this a very timely discussion and questions that we’re posing to you are the questions that we’re encountering or that we have been grappling with,” Dr. Ashar said.
FDA panelists noted that many patients cannot get access easily to dermatology visits.
Companies seeking to develop SLA devices likely will market their tools as attempts to fill a gap that now exists in medical care.
But there will be challenges ahead in explaining to patients how to interpret readings from these tools, the FDA panelists said. Consumers should know these tools are meant to assist in diagnosis, and not to make it.
“I’m not sure the layperson will hear that,” said FDA panelist Paula E. Bourelly, MD, a dermatologist from Olney, Md.
As a result, use of SLA tools could create tension between physicians and patients, with consumers demanding biopsies after seeing readings they don’t understand.
“I do have great concerns about the layperson feeling overly confident and reducing the provider to a technician,” she said.
The FDA panelists were not asked to cast formal votes on any issues discussed during the meeting They instead engaged in broad discussions around questions posed by the FDA in three key areas:
- What standards should be used to confirm lesion diagnosis in clinical testing of the accuracy of SLA devices?
- What would be acceptable true false-positive and false-negative results (sensitivity and specificity) for different diagnoses and users?
- How can the FDA address health equity considerations based on variable incidence of skin lesions?
Developing standards
The FDA asked the panel to consider several scenarios for SLA devices and to discuss how standards might vary depending on the user of the device, whether it would be dermatologists, other clinicians, or consumers.
The agency sought comments in particular about using histological diagnosis (core specimen processing with a consensus diagnosis from an expert dermatopathologist panel). In the briefing document for the meeting, the FDA argued that this approach provides the greatest certainty in the diagnosis.
“Device developers, however, cite concerns, both practical and ethical, in requiring biopsy of all lesions, particularly those that appear benign,” the FDA said. “They have proposed alternate means of defining ground truth, including consensus opinion of experts (of visual or dermoscopic examination of the lesion[s]), opinion of one expert (visual or dermoscopic examination), or other methods.”
In summarizing the discussion on this question, the FDA panel chairman, Hobart W. Harris, MD, MPH, a surgeon from the University of California, San Francisco, noted that there was broad support for histological data in clinical trials of SLA devices, with some allowance for cases where more hybrid approaches would be used.
There were also suggestions offered about designing trials and the need for biopsies of lesions that are clearly benign, as this would help gather data to help in developing algorithms.
Dr. Alam said care should be taken in explaining to study participants that they might have to undergo biopsies that they didn’t need, as part of the larger effort to gather data. This should be detailed in the consent form, he said.
“But I also think this is a relatively minor risk,” Dr. Alam said, comparing these biopsies to the blood samples that patients in many clinical studies routinely give.
“Are all of those blood draws necessary to track the change in whatever parameters that are being tracked? Probably not,” Dr. Alam said. “I think it would be possible to explain to a reasonable patient what this entails.”
Dr. Alam noted that companies might face extra hurdles in enrolling study participants and keeping them in the trials if the FDA seeks this kind of biopsy data. “But I don’t think inconvenience to the study sponsor is a good argument” for not seeking this kind of data, he added.
Leaving a loophole where certain kinds of clearly benign lesions don’t require a biopsy would eventually erode the quality of the research done on these devices. “That bar will be moved to accommodate the convenience of the sponsor, to make the study feasible,” Dr. Alam said. “And pretty soon, you’ll be missing a lot of patients that really should have biopsies.”
Acceptable rates of false positives, false negatives
The FDA panel chair noted that his colleagues had strongly urged review standards that would require that the devices improve on the rates of successful catches of suspicious lesions and lower false positives. But they did not endorse specific targets regarding the sensitivity and specificity rates.
“No one seems to be comfortable with providing or preordaining” these targets, Dr. Harris said.
Panelist Deneen Hesser, MSHSA, RN, urged a deep recognition of the power of a FDA clearance in the view of consumers.
“We need to be cognizant of what the term ‘FDA approved’ means to the lay individual,” said Ms. Hesser, who served as the patient representative on the panel. “A patient who sees that those tools are FDA approved will assume that each of those is the gold standard” in terms of expectations for delivering accurate results.
Like many of the panelists, Dr. Rotemberg urged the FDA to gather data about how patients would react to different messages encoded in consumer-oriented products.
“If the device says: ‘You should see a dermatologist for this’ and no other information, that’s very different from [saying]: ‘That lesion is suspicious for melanoma,’ ” Dr. Rotemberg said.
Despite the likely difficulties in conducting trials, the FDA needs to have the data to answer key questions about patient and physician reactions to readings from new tools, Dr. Rotemberg said.
“We don’t know how many additional biopsies we would cause with a specificity of 80%” for a new SLA tool, Dr. Rotemberg said, giving an example. “We don’t know how confident a dermatologist might be to say: ‘Actually, I’m not suspicious about that lesion and we can just fudge it or not biopsy it.’ We don’t know any of that until we study it in real life.”
The panelists also urged the FDA to seek to ensure that new tools used in analyzing skin lesions improve the quality of diagnosis.
Addressing equity
The FDA also asked the panel to weigh in on whether the agency should clear SLA tools in cases where the existing study data is drawn heavily from people considered to be at higher risk for skin cancer.
“To ensure generalizability across the entire U.S. population, should FDA require SLAs indicated for use beyond cancerous lesions be tested in a representative U.S. population?” the FDA asked.
The three most common skin cancers – melanoma, basal cell carcinoma, and squamous cell carcinoma – are more prevalent in people with Fitzpatrick I and II skin types, who tend to get sunburns, not tans. But people of color are more likely to develop melanoma in areas that are not sun exposed, such as the sole of the foot or under fingernails or toenails.
“Due in part to lower expected risk and screening, these melanomas are often detected late,” the FDA said in the briefing document.
There was broad consensus among panelists that the FDA should encourage companies to enroll people with all skin types and tones.
But they also looked for ways that the FDA could clear devices based on initial studies conducted largely with people considered to be at higher risk, with the agency then requiring follow-up trials to see how these products would work for the general U.S. population.
A version of this article first appeared on Medscape.com.
A and how to address longstanding issues of racial equity in this field of medicine.
The Food and Drug Administration has scheduled two meetings to gather expert feedback about managing an expected expansion in the use of skin lesion apps and devices. Outside of the United States, there are apps promoted as being able to help spot skin lesions that should trigger a medical visit.
The general and plastic surgery devices panel of the FDA’s Medical Devices Advisory Committee began work on this topic on July 28, with a wide-ranging discussion about potential expanded use of computer-aided, skin lesion analyzer (SLA) devices. On Friday, the panel is considering an FDA proposal to shift the designation for an approved device for aiding dermatologists in skin cancer diagnoses from the most stringent regulatory category, class III, to the less restrictive class II.
The FDA called the meeting amid growing interest in using technology to aid in finding cancers, with some of these products already marketed to consumers outside of the United States. There are presently no legally marketed, FDA-cleared or FDA-approved SLA devices indicated for use by clinicians other than dermatologists or the lay public, the agency said in a briefing memo for the meeting. There are two devices with FDA approval, though, for aiding dermatologists. The FDA approved SciBase’s Nevisense in 2017 and Mela Sciences’ MelaFind, which has fallen out of use, in 2012. Both are class III devices.
But some companies intend to offer products for consumers in the United States. The company SkinVision, for example, has developed an app of the same name, which is intended to detect suspicious-looking skin spots via smartphone photos. SkinVision’s website says the product has been offered to consumers in Australia for remote skin checks since 2015. People in the Netherlands and United Kingdom also can use SkinVision, according to the company’s website. SkinVision says the company is working on providing the app for U.S. customers, “but we are not quite there yet.”
During the meeting, FDA panelists repeatedly emphasized the potential risks of these devices in terms of sensitivity (how often a test correctly generates a positive result) and of specificity (how often a test correctly generates a negative result).
New tools intended to aid in detection of skin cancer might produce too many false positives and thus trigger floods of worried patients seeking care and often facing unnecessary biopsies, the FDA panelists said. But more worrisome would be FDA clearance of tools that delivered too many false negative results, leaving people unaware of their cancers.
The standards would have to be set very high for new products, especially those intended for consumers, said FDA panelist Murad Alam, MD, a dermatologist and vice chair of the department of dermatology at Northwestern University, Chicago. Current technologies for analyzing skin lesions are not yet up to that task. Dr. Alam likened the situation to the hopes for self-driving cars.
“It sounds great in principle. If you read the predictions from 20 years ago, it should already have happened,” Dr. Alam said. “But we’re still struggling with that because there are serious points of failure.”
FDA panelist Veronica Rotemberg, MD, PhD, a dermatologist at Memorial Sloan Kettering Cancer Center, New York, also argued for well-designed studies to understand how consumers and clinicians would react to new tools.
“We have to define what prospective information, in the intended use setting, we need to feel comfortable saying that these tools could be in a layperson’s hand or a primary care person’s hand,” Dr. Rotemberg said.
The studies would not need to be large, especially in the case of nonmelanoma skin cancer, which is common, she added.
“There’s too much nuance here for us to be able to say: ‘This is what would happen,’ without testing it,” Dr. Rotemberg said. “I do not think these prospective studies would be very burdensome, but they would help us understand what the burden would be and what the costs would be and what the potential harms would be.”
Because of rules against disclosing corporate information, the FDA cannot tell the public about the kinds of inquiries it already may have fielded from companies interested in selling skin cancer detection tools.
But in response to a question during the FDA meeting, Binita Ashar, MD, a top official in the FDA’s Center for Devices and Radiological Health, said there is interest in having these kinds of products sold in the United States as well.
“I can tell you that this a very timely discussion and questions that we’re posing to you are the questions that we’re encountering or that we have been grappling with,” Dr. Ashar said.
FDA panelists noted that many patients cannot get access easily to dermatology visits.
Companies seeking to develop SLA devices likely will market their tools as attempts to fill a gap that now exists in medical care.
But there will be challenges ahead in explaining to patients how to interpret readings from these tools, the FDA panelists said. Consumers should know these tools are meant to assist in diagnosis, and not to make it.
“I’m not sure the layperson will hear that,” said FDA panelist Paula E. Bourelly, MD, a dermatologist from Olney, Md.
As a result, use of SLA tools could create tension between physicians and patients, with consumers demanding biopsies after seeing readings they don’t understand.
“I do have great concerns about the layperson feeling overly confident and reducing the provider to a technician,” she said.
The FDA panelists were not asked to cast formal votes on any issues discussed during the meeting They instead engaged in broad discussions around questions posed by the FDA in three key areas:
- What standards should be used to confirm lesion diagnosis in clinical testing of the accuracy of SLA devices?
- What would be acceptable true false-positive and false-negative results (sensitivity and specificity) for different diagnoses and users?
- How can the FDA address health equity considerations based on variable incidence of skin lesions?
Developing standards
The FDA asked the panel to consider several scenarios for SLA devices and to discuss how standards might vary depending on the user of the device, whether it would be dermatologists, other clinicians, or consumers.
The agency sought comments in particular about using histological diagnosis (core specimen processing with a consensus diagnosis from an expert dermatopathologist panel). In the briefing document for the meeting, the FDA argued that this approach provides the greatest certainty in the diagnosis.
“Device developers, however, cite concerns, both practical and ethical, in requiring biopsy of all lesions, particularly those that appear benign,” the FDA said. “They have proposed alternate means of defining ground truth, including consensus opinion of experts (of visual or dermoscopic examination of the lesion[s]), opinion of one expert (visual or dermoscopic examination), or other methods.”
In summarizing the discussion on this question, the FDA panel chairman, Hobart W. Harris, MD, MPH, a surgeon from the University of California, San Francisco, noted that there was broad support for histological data in clinical trials of SLA devices, with some allowance for cases where more hybrid approaches would be used.
There were also suggestions offered about designing trials and the need for biopsies of lesions that are clearly benign, as this would help gather data to help in developing algorithms.
Dr. Alam said care should be taken in explaining to study participants that they might have to undergo biopsies that they didn’t need, as part of the larger effort to gather data. This should be detailed in the consent form, he said.
“But I also think this is a relatively minor risk,” Dr. Alam said, comparing these biopsies to the blood samples that patients in many clinical studies routinely give.
“Are all of those blood draws necessary to track the change in whatever parameters that are being tracked? Probably not,” Dr. Alam said. “I think it would be possible to explain to a reasonable patient what this entails.”
Dr. Alam noted that companies might face extra hurdles in enrolling study participants and keeping them in the trials if the FDA seeks this kind of biopsy data. “But I don’t think inconvenience to the study sponsor is a good argument” for not seeking this kind of data, he added.
Leaving a loophole where certain kinds of clearly benign lesions don’t require a biopsy would eventually erode the quality of the research done on these devices. “That bar will be moved to accommodate the convenience of the sponsor, to make the study feasible,” Dr. Alam said. “And pretty soon, you’ll be missing a lot of patients that really should have biopsies.”
Acceptable rates of false positives, false negatives
The FDA panel chair noted that his colleagues had strongly urged review standards that would require that the devices improve on the rates of successful catches of suspicious lesions and lower false positives. But they did not endorse specific targets regarding the sensitivity and specificity rates.
“No one seems to be comfortable with providing or preordaining” these targets, Dr. Harris said.
Panelist Deneen Hesser, MSHSA, RN, urged a deep recognition of the power of a FDA clearance in the view of consumers.
“We need to be cognizant of what the term ‘FDA approved’ means to the lay individual,” said Ms. Hesser, who served as the patient representative on the panel. “A patient who sees that those tools are FDA approved will assume that each of those is the gold standard” in terms of expectations for delivering accurate results.
Like many of the panelists, Dr. Rotemberg urged the FDA to gather data about how patients would react to different messages encoded in consumer-oriented products.
“If the device says: ‘You should see a dermatologist for this’ and no other information, that’s very different from [saying]: ‘That lesion is suspicious for melanoma,’ ” Dr. Rotemberg said.
Despite the likely difficulties in conducting trials, the FDA needs to have the data to answer key questions about patient and physician reactions to readings from new tools, Dr. Rotemberg said.
“We don’t know how many additional biopsies we would cause with a specificity of 80%” for a new SLA tool, Dr. Rotemberg said, giving an example. “We don’t know how confident a dermatologist might be to say: ‘Actually, I’m not suspicious about that lesion and we can just fudge it or not biopsy it.’ We don’t know any of that until we study it in real life.”
The panelists also urged the FDA to seek to ensure that new tools used in analyzing skin lesions improve the quality of diagnosis.
Addressing equity
The FDA also asked the panel to weigh in on whether the agency should clear SLA tools in cases where the existing study data is drawn heavily from people considered to be at higher risk for skin cancer.
“To ensure generalizability across the entire U.S. population, should FDA require SLAs indicated for use beyond cancerous lesions be tested in a representative U.S. population?” the FDA asked.
The three most common skin cancers – melanoma, basal cell carcinoma, and squamous cell carcinoma – are more prevalent in people with Fitzpatrick I and II skin types, who tend to get sunburns, not tans. But people of color are more likely to develop melanoma in areas that are not sun exposed, such as the sole of the foot or under fingernails or toenails.
“Due in part to lower expected risk and screening, these melanomas are often detected late,” the FDA said in the briefing document.
There was broad consensus among panelists that the FDA should encourage companies to enroll people with all skin types and tones.
But they also looked for ways that the FDA could clear devices based on initial studies conducted largely with people considered to be at higher risk, with the agency then requiring follow-up trials to see how these products would work for the general U.S. population.
A version of this article first appeared on Medscape.com.
Vitamins and CVD, cancer prevention: USPSTF says evidence isn’t there
The leading causes of death in the United States are cardiovascular disease (CVD) and cancer. CVD causes about 800,00 deaths per year (30% of all deaths), and cancer is responsible for about 600,000 deaths annually (21%).1 Many adults—more than 50%—report regular use of dietary supplements, including multivitamins and minerals, to improve their overall health, believing that these supplements’ anti-inflammatory and antioxidative properties help prevent CVD and cancer.2 However, this belief is not supported by evidence, according to the US Preventive Services Task Force.
What did the Task Force find? After a recent reassessment of the topic of vitamin and mineral supplementation, the Task Force reaffirmed its position from 2014: There is insufficient evidence to recommend the use of vitamins and minerals to prevent CVD and cancer.3
For most of the vitamins and minerals included in the systematic review—vitamins A, C, D, E, and K; B vitamins; calcium; iron; zinc; and selenium—the Task Force could not find sufficient evidence to make a recommendation. It is important to note that if any of these are taken at the recommended levels, there is no evidence of serious harm from using them.
However, there is good evidence to recommend against the use of beta carotene and vitamin E.3 For beta carotene, the harms outweigh the benefits: Its use is associated with an increase in the risk of CVD death, as well as an increased incidence of lung cancer in smokers and those with occupational exposure to asbestos. The evidence on vitamin E indicates that it simply does not provide any cancer or CVD mortality benefit.
How to advise patients. Both the US Department of Health and Human Services (DHHS) and the American Heart Association state that most nutritional needs can be met through the consumption of nutritional foods and beverages; supplements do not add any benefit for those who consume a healthy diet.4 The updated USPSTF recommendations support this approach for community-dwelling, nonpregnant adults.
For those adults who want to supplement their diets—or who need to do so because of an inability to achieve an adequate diet—urge them to follow the recommendations found in DHHS’s Dietary Guidelines for Americans, 2020-2025.4
1. Murphy SL, Xu J, Kochanek KD, et al. Deaths: final data for 2018. Natl Vital Stat Rep. 2021;69:1-83.
2. Cowan AE, Jun S, Gahche JJ, et al. Dietary supplement use differs by socioeconomic and health-related characteristics among US adults, NHANES 2011-2014. Nutrients. 2018;10:1114. doi: 10.3390/nu10081114
3. USPSTF. Vitamin, mineral, and multivitamin supplementation to prevent cardiovascular disease and cancer: US Preventive Services Task Force recommendation statement. JAMA. 2022;327:2326-2333. doi: 10.1001/jama.2022.8970
4. US Department of Agriculture and US Department of Health and Human Services. Dietary Guidelines for Americans, 2020-2025. Published December 2020. Accessed July 13, 2022. www.dietaryguidelines.gov/current-dietary-guidelines
The leading causes of death in the United States are cardiovascular disease (CVD) and cancer. CVD causes about 800,00 deaths per year (30% of all deaths), and cancer is responsible for about 600,000 deaths annually (21%).1 Many adults—more than 50%—report regular use of dietary supplements, including multivitamins and minerals, to improve their overall health, believing that these supplements’ anti-inflammatory and antioxidative properties help prevent CVD and cancer.2 However, this belief is not supported by evidence, according to the US Preventive Services Task Force.
What did the Task Force find? After a recent reassessment of the topic of vitamin and mineral supplementation, the Task Force reaffirmed its position from 2014: There is insufficient evidence to recommend the use of vitamins and minerals to prevent CVD and cancer.3
For most of the vitamins and minerals included in the systematic review—vitamins A, C, D, E, and K; B vitamins; calcium; iron; zinc; and selenium—the Task Force could not find sufficient evidence to make a recommendation. It is important to note that if any of these are taken at the recommended levels, there is no evidence of serious harm from using them.
However, there is good evidence to recommend against the use of beta carotene and vitamin E.3 For beta carotene, the harms outweigh the benefits: Its use is associated with an increase in the risk of CVD death, as well as an increased incidence of lung cancer in smokers and those with occupational exposure to asbestos. The evidence on vitamin E indicates that it simply does not provide any cancer or CVD mortality benefit.
How to advise patients. Both the US Department of Health and Human Services (DHHS) and the American Heart Association state that most nutritional needs can be met through the consumption of nutritional foods and beverages; supplements do not add any benefit for those who consume a healthy diet.4 The updated USPSTF recommendations support this approach for community-dwelling, nonpregnant adults.
For those adults who want to supplement their diets—or who need to do so because of an inability to achieve an adequate diet—urge them to follow the recommendations found in DHHS’s Dietary Guidelines for Americans, 2020-2025.4
The leading causes of death in the United States are cardiovascular disease (CVD) and cancer. CVD causes about 800,00 deaths per year (30% of all deaths), and cancer is responsible for about 600,000 deaths annually (21%).1 Many adults—more than 50%—report regular use of dietary supplements, including multivitamins and minerals, to improve their overall health, believing that these supplements’ anti-inflammatory and antioxidative properties help prevent CVD and cancer.2 However, this belief is not supported by evidence, according to the US Preventive Services Task Force.
What did the Task Force find? After a recent reassessment of the topic of vitamin and mineral supplementation, the Task Force reaffirmed its position from 2014: There is insufficient evidence to recommend the use of vitamins and minerals to prevent CVD and cancer.3
For most of the vitamins and minerals included in the systematic review—vitamins A, C, D, E, and K; B vitamins; calcium; iron; zinc; and selenium—the Task Force could not find sufficient evidence to make a recommendation. It is important to note that if any of these are taken at the recommended levels, there is no evidence of serious harm from using them.
However, there is good evidence to recommend against the use of beta carotene and vitamin E.3 For beta carotene, the harms outweigh the benefits: Its use is associated with an increase in the risk of CVD death, as well as an increased incidence of lung cancer in smokers and those with occupational exposure to asbestos. The evidence on vitamin E indicates that it simply does not provide any cancer or CVD mortality benefit.
How to advise patients. Both the US Department of Health and Human Services (DHHS) and the American Heart Association state that most nutritional needs can be met through the consumption of nutritional foods and beverages; supplements do not add any benefit for those who consume a healthy diet.4 The updated USPSTF recommendations support this approach for community-dwelling, nonpregnant adults.
For those adults who want to supplement their diets—or who need to do so because of an inability to achieve an adequate diet—urge them to follow the recommendations found in DHHS’s Dietary Guidelines for Americans, 2020-2025.4
1. Murphy SL, Xu J, Kochanek KD, et al. Deaths: final data for 2018. Natl Vital Stat Rep. 2021;69:1-83.
2. Cowan AE, Jun S, Gahche JJ, et al. Dietary supplement use differs by socioeconomic and health-related characteristics among US adults, NHANES 2011-2014. Nutrients. 2018;10:1114. doi: 10.3390/nu10081114
3. USPSTF. Vitamin, mineral, and multivitamin supplementation to prevent cardiovascular disease and cancer: US Preventive Services Task Force recommendation statement. JAMA. 2022;327:2326-2333. doi: 10.1001/jama.2022.8970
4. US Department of Agriculture and US Department of Health and Human Services. Dietary Guidelines for Americans, 2020-2025. Published December 2020. Accessed July 13, 2022. www.dietaryguidelines.gov/current-dietary-guidelines
1. Murphy SL, Xu J, Kochanek KD, et al. Deaths: final data for 2018. Natl Vital Stat Rep. 2021;69:1-83.
2. Cowan AE, Jun S, Gahche JJ, et al. Dietary supplement use differs by socioeconomic and health-related characteristics among US adults, NHANES 2011-2014. Nutrients. 2018;10:1114. doi: 10.3390/nu10081114
3. USPSTF. Vitamin, mineral, and multivitamin supplementation to prevent cardiovascular disease and cancer: US Preventive Services Task Force recommendation statement. JAMA. 2022;327:2326-2333. doi: 10.1001/jama.2022.8970
4. US Department of Agriculture and US Department of Health and Human Services. Dietary Guidelines for Americans, 2020-2025. Published December 2020. Accessed July 13, 2022. www.dietaryguidelines.gov/current-dietary-guidelines

Then and now: Inflammatory bowel diseases
(IBD) creating a whole new landscape for the disease.

In 2007, IBD seemed to be primarily a disease of Caucasian and Jewish ancestry. While prevalence of IBD is still highest in the Western world, there is now increasing incidence, even accounting for detection bias, in people of all other ancestries globally. Incidence of IBD in children under the age of 18 years is also rising. Patients with IBD are living longer and, despite the notion that IBD is a disease primarily of younger adults, nearly one-third of Americans with IBD are 60 years and older.
“Adalimumab aids in Crohn’s disease” read the front page of the inaugural issue of GI & Hepatology News in January 2007. The article highlighted the GAIN study, which demonstrated that patients who lost response to infliximab responded to adalimumab, the second anti-tumor necrosis factor (TNF) agent approved for the treatment of Crohn’s disease and subsequently ulcerative colitis. Over the subsequent 15 years, the armamentarium of treatment options for Crohn’s disease and ulcerative colitis have rapidly proliferated: there are now four anti–tumor necrosis factor (TNF) agents, two anti-integrin agents, two anti-interleukin agents, two Janus kinase inhibitors and a sphingosine-1 receptor modulator approved for the treatment of IBD. Many more promising treatment options are in trials. Other mechanisms are under investigation as well, including antimicrobial therapies for ulcerative colitis and stem cell therapeutics for the treatment of refractory perianal fistulizing Crohn’s disease. Perhaps even more novel – dietary therapies are more rigorously under investigation.
“Ulcerative colitis guidelines endorse combined therapy” reads another headline from the inaugural GI & Hepatology News issue. The article discusses the European Crohn’s and Colitis Organisation’s consensus guideline that topical and systemic agents used together are superior to either used alone, referring specifically to mesalamine, both systemic and topical as well as the additional of topical corticosteroid to systemic mesalamine. Combination therapy has a completely new meaning in modern times. With the publication of the SONIC trial in 2010, combination therapy referred to an anti-TNF in combination with an immunomodulator for the ensuing decade. However, in this new era of IBD treatment, combination therapy could also mean a biologic with a small molecule or even combination biologics, for which there is an ongoing randomized controlled trial. On the topic of treatment strategies, one of the biggest shifts in the IBD treatment paradigm is the bottom-up versus top-down approach of treatment, with increasing evidence to suggest that early biologic initiation is more effective, especially in patients with Crohn’s disease. Therapeutic drug monitoring is mainstream. Treat-to-target strategies to achieve more stringent outcomes, such as biomarker, endoscopic and histologic normalization, especially in ulcerative colitis, have evolved to become the norm in 2022.
The combination of increased treatment options, decreased reliance on corticosteroids and stringent treatment strategies have resulted in improved outcomes: IBD-related hospitalizations, surgeries, and even mortality have declined since 2007. The growing recognition and focus on extra-intestinal manifestations, including fatigue, and the gut-brain axis are important steps to improving the overall quality of life of patients with IBD. Beyond treating the disease, we are now learning how to treat the patient. We will be developing personalized strategies to identify the right patient for the right treatment, including patient-level clinical and biologic markers. We need to identify those who are at risk for IBD to prevent the disease at a preclinical phase. Concomitantly, we must continue the quest to cure the disease!
Dr. Kochar is a gastroenterologist and inflammatory bowel disease specialist at Massachusetts General Hospital and a physician investigator in the clinical and translational epidemiology unit at The Mongan Institute, both in Boston. She has no relevant disclosures.
(IBD) creating a whole new landscape for the disease.
In 2007, IBD seemed to be primarily a disease of Caucasian and Jewish ancestry. While prevalence of IBD is still highest in the Western world, there is now increasing incidence, even accounting for detection bias, in people of all other ancestries globally. Incidence of IBD in children under the age of 18 years is also rising. Patients with IBD are living longer and, despite the notion that IBD is a disease primarily of younger adults, nearly one-third of Americans with IBD are 60 years and older.
“Adalimumab aids in Crohn’s disease” read the front page of the inaugural issue of GI & Hepatology News in January 2007. The article highlighted the GAIN study, which demonstrated that patients who lost response to infliximab responded to adalimumab, the second anti-tumor necrosis factor (TNF) agent approved for the treatment of Crohn’s disease and subsequently ulcerative colitis. Over the subsequent 15 years, the armamentarium of treatment options for Crohn’s disease and ulcerative colitis have rapidly proliferated: there are now four anti–tumor necrosis factor (TNF) agents, two anti-integrin agents, two anti-interleukin agents, two Janus kinase inhibitors and a sphingosine-1 receptor modulator approved for the treatment of IBD. Many more promising treatment options are in trials. Other mechanisms are under investigation as well, including antimicrobial therapies for ulcerative colitis and stem cell therapeutics for the treatment of refractory perianal fistulizing Crohn’s disease. Perhaps even more novel – dietary therapies are more rigorously under investigation.
“Ulcerative colitis guidelines endorse combined therapy” reads another headline from the inaugural GI & Hepatology News issue. The article discusses the European Crohn’s and Colitis Organisation’s consensus guideline that topical and systemic agents used together are superior to either used alone, referring specifically to mesalamine, both systemic and topical as well as the additional of topical corticosteroid to systemic mesalamine. Combination therapy has a completely new meaning in modern times. With the publication of the SONIC trial in 2010, combination therapy referred to an anti-TNF in combination with an immunomodulator for the ensuing decade. However, in this new era of IBD treatment, combination therapy could also mean a biologic with a small molecule or even combination biologics, for which there is an ongoing randomized controlled trial. On the topic of treatment strategies, one of the biggest shifts in the IBD treatment paradigm is the bottom-up versus top-down approach of treatment, with increasing evidence to suggest that early biologic initiation is more effective, especially in patients with Crohn’s disease. Therapeutic drug monitoring is mainstream. Treat-to-target strategies to achieve more stringent outcomes, such as biomarker, endoscopic and histologic normalization, especially in ulcerative colitis, have evolved to become the norm in 2022.
The combination of increased treatment options, decreased reliance on corticosteroids and stringent treatment strategies have resulted in improved outcomes: IBD-related hospitalizations, surgeries, and even mortality have declined since 2007. The growing recognition and focus on extra-intestinal manifestations, including fatigue, and the gut-brain axis are important steps to improving the overall quality of life of patients with IBD. Beyond treating the disease, we are now learning how to treat the patient. We will be developing personalized strategies to identify the right patient for the right treatment, including patient-level clinical and biologic markers. We need to identify those who are at risk for IBD to prevent the disease at a preclinical phase. Concomitantly, we must continue the quest to cure the disease!
Dr. Kochar is a gastroenterologist and inflammatory bowel disease specialist at Massachusetts General Hospital and a physician investigator in the clinical and translational epidemiology unit at The Mongan Institute, both in Boston. She has no relevant disclosures.
(IBD) creating a whole new landscape for the disease.
In 2007, IBD seemed to be primarily a disease of Caucasian and Jewish ancestry. While prevalence of IBD is still highest in the Western world, there is now increasing incidence, even accounting for detection bias, in people of all other ancestries globally. Incidence of IBD in children under the age of 18 years is also rising. Patients with IBD are living longer and, despite the notion that IBD is a disease primarily of younger adults, nearly one-third of Americans with IBD are 60 years and older.
“Adalimumab aids in Crohn’s disease” read the front page of the inaugural issue of GI & Hepatology News in January 2007. The article highlighted the GAIN study, which demonstrated that patients who lost response to infliximab responded to adalimumab, the second anti-tumor necrosis factor (TNF) agent approved for the treatment of Crohn’s disease and subsequently ulcerative colitis. Over the subsequent 15 years, the armamentarium of treatment options for Crohn’s disease and ulcerative colitis have rapidly proliferated: there are now four anti–tumor necrosis factor (TNF) agents, two anti-integrin agents, two anti-interleukin agents, two Janus kinase inhibitors and a sphingosine-1 receptor modulator approved for the treatment of IBD. Many more promising treatment options are in trials. Other mechanisms are under investigation as well, including antimicrobial therapies for ulcerative colitis and stem cell therapeutics for the treatment of refractory perianal fistulizing Crohn’s disease. Perhaps even more novel – dietary therapies are more rigorously under investigation.
“Ulcerative colitis guidelines endorse combined therapy” reads another headline from the inaugural GI & Hepatology News issue. The article discusses the European Crohn’s and Colitis Organisation’s consensus guideline that topical and systemic agents used together are superior to either used alone, referring specifically to mesalamine, both systemic and topical as well as the additional of topical corticosteroid to systemic mesalamine. Combination therapy has a completely new meaning in modern times. With the publication of the SONIC trial in 2010, combination therapy referred to an anti-TNF in combination with an immunomodulator for the ensuing decade. However, in this new era of IBD treatment, combination therapy could also mean a biologic with a small molecule or even combination biologics, for which there is an ongoing randomized controlled trial. On the topic of treatment strategies, one of the biggest shifts in the IBD treatment paradigm is the bottom-up versus top-down approach of treatment, with increasing evidence to suggest that early biologic initiation is more effective, especially in patients with Crohn’s disease. Therapeutic drug monitoring is mainstream. Treat-to-target strategies to achieve more stringent outcomes, such as biomarker, endoscopic and histologic normalization, especially in ulcerative colitis, have evolved to become the norm in 2022.
The combination of increased treatment options, decreased reliance on corticosteroids and stringent treatment strategies have resulted in improved outcomes: IBD-related hospitalizations, surgeries, and even mortality have declined since 2007. The growing recognition and focus on extra-intestinal manifestations, including fatigue, and the gut-brain axis are important steps to improving the overall quality of life of patients with IBD. Beyond treating the disease, we are now learning how to treat the patient. We will be developing personalized strategies to identify the right patient for the right treatment, including patient-level clinical and biologic markers. We need to identify those who are at risk for IBD to prevent the disease at a preclinical phase. Concomitantly, we must continue the quest to cure the disease!
Dr. Kochar is a gastroenterologist and inflammatory bowel disease specialist at Massachusetts General Hospital and a physician investigator in the clinical and translational epidemiology unit at The Mongan Institute, both in Boston. She has no relevant disclosures.