User login
Food for Thought
This special issue is dedicated to resident education on psoriasis. With that in mind, we hope to address many topics of interest to those in training. Over the years, diet has been a hot topic among psoriasis patients. They want to know how diet affects psoriasis and what changes can be made to their diet to improve their condition. Although they have expected specific answers, my response has usually been that they should, of course, eat an overall healthy and balanced diet, and lose weight if necessary. I have continued, however, that no specific diet has been recommended. However, now we have some information that may start to give us some answers.
The Mediterranean diet has been regarded as a healthy regimen.1 This diet emphasizes eating primarily plant-based foods, such as fruits and vegetables; whole grains; legumes; and nuts. Other recommendations include replacing butter with healthy fats such as olive oil and canola oil, using herbs and spices instead of salt to flavor foods, and limiting red meat to no more than a few times a month.1
As we know, psoriasis is a chronic inflammatory disease. The Mediterranean diet has been shown to reduce chronic inflammation and has a positive effect on the risk for metabolic syndrome and cardiovascular events.1 Phan et al1 hypothesized a positive effect of the Mediterranean diet on psoriasis. They performed a study to assess the association between a score that reflects the adhesion to a Mediterranean diet (MEDI-LITE) and the onset and/or severity of psoriasis.1
The NutriNet-Santé program is an ongoing, observational, web-based questionnaire cohort study launched in France in May 2009.1 Data were collected and analyzed between April 2017 and June 2017. Individuals with psoriasis were identified utilizing a validated online questionnaire and then categorized by disease severity into 1 of 3 groups: severe psoriasis, nonsevere psoriasis, and psoriasis free.1
During the initial 2 years of participation in the cohort, data on dietary intake (including alcohol) were gathered to calculate the MEDI-LITE score, ranging from 0 (no adherence) to 18 (maximum adherence).1 Of the 158,361 total web-based participants, 35,735 (23%) replied to the psoriasis questionnaire.1 Of the respondents, 3557 (10%) individuals reported having psoriasis. The condition was severe in 878 cases (24.7%), and 299 (8.4%) incident cases were recorded (cases occurring >2 years after participant inclusion in the cohort). After adjustment for confounding factors, the investigators found a significant inverse relationship between the MEDI-LITE score and having severe psoriasis (odds ratio [OR], 0.71; 95% CI, 0.55-0.92 for the MEDI-LITE score’s second tertile [score of 8 to 9]; and OR, 0.78; 95% CI, 0.59-1.01 for the third tertile [score of 10 to 18]).1
The authors noted that patients with severe psoriasis displayed low levels of adherence to the Mediterranean diet.1 They commented that this finding supports the hypothesis that the Mediterranean diet may slow the progression of psoriasis. If these findings are confirmed, adherence to a Mediterranean diet should be integrated into the routine management of moderate to severe psoriasis.1 These findings are by no means definitive, but it is a first step in helping us define more specific dietary recommendations for psoriasis.
This issue includes several articles looking at various facets of psoriasis important to residents, including the pathophysiology of psoriasis,2 treatment approach using biologic therapies,3 risk factors and triggers for psoriasis,4 and the psychosocial impact of psoriasis.5 We hope that you find this issue enjoyable and informative.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort [published online July 25, 2018]. JAMA Dermatol. doi:10.1001/jamadermatol.2018.2127.
- Hugh JM, Weinberg JM. Update on the pathophysiology of psoriasis. Cutis. 2018;102(suppl 5):6-12.
- McKay C, Kondratuk KE, Miller JP, et al. Biologic therapy in psoriasis: navigating the options. Cutis. 2018;102(suppl 5):13-17.
- Lee EB, Wu KK, Lee MP, et al. Psoriasis risk factors and triggers. Cutis. 2018;102(suppl 5):18-20.
- Kolli SS, Amin SD, Pona A, et al. Psychosocial impact of psoriasis: a review for dermatology residents. Cutis. 2018;102(suppl 5):21-25.
This special issue is dedicated to resident education on psoriasis. With that in mind, we hope to address many topics of interest to those in training. Over the years, diet has been a hot topic among psoriasis patients. They want to know how diet affects psoriasis and what changes can be made to their diet to improve their condition. Although they have expected specific answers, my response has usually been that they should, of course, eat an overall healthy and balanced diet, and lose weight if necessary. I have continued, however, that no specific diet has been recommended. However, now we have some information that may start to give us some answers.
The Mediterranean diet has been regarded as a healthy regimen.1 This diet emphasizes eating primarily plant-based foods, such as fruits and vegetables; whole grains; legumes; and nuts. Other recommendations include replacing butter with healthy fats such as olive oil and canola oil, using herbs and spices instead of salt to flavor foods, and limiting red meat to no more than a few times a month.1
As we know, psoriasis is a chronic inflammatory disease. The Mediterranean diet has been shown to reduce chronic inflammation and has a positive effect on the risk for metabolic syndrome and cardiovascular events.1 Phan et al1 hypothesized a positive effect of the Mediterranean diet on psoriasis. They performed a study to assess the association between a score that reflects the adhesion to a Mediterranean diet (MEDI-LITE) and the onset and/or severity of psoriasis.1
The NutriNet-Santé program is an ongoing, observational, web-based questionnaire cohort study launched in France in May 2009.1 Data were collected and analyzed between April 2017 and June 2017. Individuals with psoriasis were identified utilizing a validated online questionnaire and then categorized by disease severity into 1 of 3 groups: severe psoriasis, nonsevere psoriasis, and psoriasis free.1
During the initial 2 years of participation in the cohort, data on dietary intake (including alcohol) were gathered to calculate the MEDI-LITE score, ranging from 0 (no adherence) to 18 (maximum adherence).1 Of the 158,361 total web-based participants, 35,735 (23%) replied to the psoriasis questionnaire.1 Of the respondents, 3557 (10%) individuals reported having psoriasis. The condition was severe in 878 cases (24.7%), and 299 (8.4%) incident cases were recorded (cases occurring >2 years after participant inclusion in the cohort). After adjustment for confounding factors, the investigators found a significant inverse relationship between the MEDI-LITE score and having severe psoriasis (odds ratio [OR], 0.71; 95% CI, 0.55-0.92 for the MEDI-LITE score’s second tertile [score of 8 to 9]; and OR, 0.78; 95% CI, 0.59-1.01 for the third tertile [score of 10 to 18]).1
The authors noted that patients with severe psoriasis displayed low levels of adherence to the Mediterranean diet.1 They commented that this finding supports the hypothesis that the Mediterranean diet may slow the progression of psoriasis. If these findings are confirmed, adherence to a Mediterranean diet should be integrated into the routine management of moderate to severe psoriasis.1 These findings are by no means definitive, but it is a first step in helping us define more specific dietary recommendations for psoriasis.
This issue includes several articles looking at various facets of psoriasis important to residents, including the pathophysiology of psoriasis,2 treatment approach using biologic therapies,3 risk factors and triggers for psoriasis,4 and the psychosocial impact of psoriasis.5 We hope that you find this issue enjoyable and informative.
This special issue is dedicated to resident education on psoriasis. With that in mind, we hope to address many topics of interest to those in training. Over the years, diet has been a hot topic among psoriasis patients. They want to know how diet affects psoriasis and what changes can be made to their diet to improve their condition. Although they have expected specific answers, my response has usually been that they should, of course, eat an overall healthy and balanced diet, and lose weight if necessary. I have continued, however, that no specific diet has been recommended. However, now we have some information that may start to give us some answers.
The Mediterranean diet has been regarded as a healthy regimen.1 This diet emphasizes eating primarily plant-based foods, such as fruits and vegetables; whole grains; legumes; and nuts. Other recommendations include replacing butter with healthy fats such as olive oil and canola oil, using herbs and spices instead of salt to flavor foods, and limiting red meat to no more than a few times a month.1
As we know, psoriasis is a chronic inflammatory disease. The Mediterranean diet has been shown to reduce chronic inflammation and has a positive effect on the risk for metabolic syndrome and cardiovascular events.1 Phan et al1 hypothesized a positive effect of the Mediterranean diet on psoriasis. They performed a study to assess the association between a score that reflects the adhesion to a Mediterranean diet (MEDI-LITE) and the onset and/or severity of psoriasis.1
The NutriNet-Santé program is an ongoing, observational, web-based questionnaire cohort study launched in France in May 2009.1 Data were collected and analyzed between April 2017 and June 2017. Individuals with psoriasis were identified utilizing a validated online questionnaire and then categorized by disease severity into 1 of 3 groups: severe psoriasis, nonsevere psoriasis, and psoriasis free.1
During the initial 2 years of participation in the cohort, data on dietary intake (including alcohol) were gathered to calculate the MEDI-LITE score, ranging from 0 (no adherence) to 18 (maximum adherence).1 Of the 158,361 total web-based participants, 35,735 (23%) replied to the psoriasis questionnaire.1 Of the respondents, 3557 (10%) individuals reported having psoriasis. The condition was severe in 878 cases (24.7%), and 299 (8.4%) incident cases were recorded (cases occurring >2 years after participant inclusion in the cohort). After adjustment for confounding factors, the investigators found a significant inverse relationship between the MEDI-LITE score and having severe psoriasis (odds ratio [OR], 0.71; 95% CI, 0.55-0.92 for the MEDI-LITE score’s second tertile [score of 8 to 9]; and OR, 0.78; 95% CI, 0.59-1.01 for the third tertile [score of 10 to 18]).1
The authors noted that patients with severe psoriasis displayed low levels of adherence to the Mediterranean diet.1 They commented that this finding supports the hypothesis that the Mediterranean diet may slow the progression of psoriasis. If these findings are confirmed, adherence to a Mediterranean diet should be integrated into the routine management of moderate to severe psoriasis.1 These findings are by no means definitive, but it is a first step in helping us define more specific dietary recommendations for psoriasis.
This issue includes several articles looking at various facets of psoriasis important to residents, including the pathophysiology of psoriasis,2 treatment approach using biologic therapies,3 risk factors and triggers for psoriasis,4 and the psychosocial impact of psoriasis.5 We hope that you find this issue enjoyable and informative.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort [published online July 25, 2018]. JAMA Dermatol. doi:10.1001/jamadermatol.2018.2127.
- Hugh JM, Weinberg JM. Update on the pathophysiology of psoriasis. Cutis. 2018;102(suppl 5):6-12.
- McKay C, Kondratuk KE, Miller JP, et al. Biologic therapy in psoriasis: navigating the options. Cutis. 2018;102(suppl 5):13-17.
- Lee EB, Wu KK, Lee MP, et al. Psoriasis risk factors and triggers. Cutis. 2018;102(suppl 5):18-20.
- Kolli SS, Amin SD, Pona A, et al. Psychosocial impact of psoriasis: a review for dermatology residents. Cutis. 2018;102(suppl 5):21-25.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort [published online July 25, 2018]. JAMA Dermatol. doi:10.1001/jamadermatol.2018.2127.
- Hugh JM, Weinberg JM. Update on the pathophysiology of psoriasis. Cutis. 2018;102(suppl 5):6-12.
- McKay C, Kondratuk KE, Miller JP, et al. Biologic therapy in psoriasis: navigating the options. Cutis. 2018;102(suppl 5):13-17.
- Lee EB, Wu KK, Lee MP, et al. Psoriasis risk factors and triggers. Cutis. 2018;102(suppl 5):18-20.
- Kolli SS, Amin SD, Pona A, et al. Psychosocial impact of psoriasis: a review for dermatology residents. Cutis. 2018;102(suppl 5):21-25.
With one hand, administration boosts ACA marketplaces, weakens them with another
In the span of less than 12 hours, the Trump administration took two seemingly contradictory actions that could have profound effects on the insurance marketplaces set up by the Affordable Care Act.
First, officials issued guidance on the morning of Oct. 22 that could weaken the exchanges set up for people who buy their own insurance. The new approach makes it easier for states to get around some ACA requirements, including allowing the use of federal subsidies for skimpier plans that can reject people with preexisting conditions.
Yet, the other move – a proposed rule unveiled that evening – could bolster ACA marketplaces by sending millions of people with job-based coverage there, armed with tax-free money from their employers to buy individual plans.
Both efforts play into the parallel narratives dominating the bitter political debate over the ACA.
The administration, frustrated that Congress did not repeal the law, say some critics and policy experts, is working to undermine it by weakening the marketplaces and the law’s consumer protections. Those efforts make it easier for insurers to offer skimpier policies that bypass the law’s rules, such as its ban on annual or lifetime limits or its protections for people with preexisting conditions. Congress also zeroed out the tax penalty for not having coverage, effective next year. Combined, the moves could reduce enrollment in ACA plans, potentially driving up premiums for those who remain.
The administration and Republicans in Congress say they are looking to assist those left behind by the ACA – people who don’t get subsidies to help them buy coverage and are desperate for less expensive options – even if that means purchasing less robust coverage.
“These are people who were buying insurance before [the law] and then the rules changed and they could not buy it because they could not afford it,” said Joe Antos, a resident scholar at the conservative American Enterprise Institute. “They have been slowly dropping out of insurance coverage altogether.”
The efforts are dramatically reshaping the ACA and the individual insurance market to one that looks more as it did before the 2010 law, when regulation, coverage, and consumer protections varied widely across the country.
“Some states will do everything they can to keep individual markets strong and stable. Others won’t,” said Sabrina Corlette, research professor at the Center on Health Insurance Reforms at Georgetown University.
So what expectations should consumers have? Here are three key takeaways:
Protections for preexisting health problems are uncertain
Polls show that keeping the ACA’s guarantees on coverage for people with medical problems is a top concern for Americans, and Democrats have made their defense of the health law a key part of their midterm election campaigns.
Republicans have gotten that message and even those who voted to repeal the ACA or joined a lawsuit by 20 red states to overturn it now say they want to protect people with preexisting conditions. Still, GOP lawmakers have not introduced any plan that would be as protective as the current law.
In August, the administration released a rule allowing expanded use of short-term plans, which are less expensive than ACA policies. To get those lower prices, most of these plans do not cover prescription drugs, maternity care, mental health, or substance abuse treatments.
The move is unlikely to benefit people with health problems, as short-term plans can reject people with preexisting conditions or decline to cover care for those medical problems.
Under the rule, insurers can sell them starting in 2019 for up to a year’s duration, with an option to renew for up to 3 years, reversing an Obama-era directive that limited them to 90 days.
Administration officials estimate such plans could draw 600,000 new enrollees next year, and others have estimated the numbers could be far higher. The concern is if many healthy people in 2019 switch out of the ACA market and choose short-term plans, premiums will rise for those who remain, including those with preexisting conditions, or make the ACA market less attractive for insurers.
Where you live matters more
One of the biggest changes ushered in with the ACA was a standard set of rules across all states.
Before the law took effect, consumers buying their own coverage saw tremendous variation in what was offered and what protections they had, depending on the state where they lived.
Most states, for example, allowed insurers to reject people with medical conditions. A few states required insurers to charge similar premiums across the board, but most allowed wide variations based on age, gender or health. Some skimpy plans didn’t cover prescription drugs, chemotherapy, or other medical services.
By standardizing the rules and benefits, the ACA barred insurers from rejecting applicants with medical conditions or charging them more. Women and men get the same premium rates and insurers could charge older people no more than three times what they charged younger ones.
Under the new guidance issued this week giving states more flexibility on what is offered, consumers could again see a wide variation on coverage, premium rules, and even subsidy eligibility.
“It shifts pressure to state politicians,” said Caroline Pearson, a senior fellow at NORC, a nonpartisan research institution at the University of Chicago. That could play into the calculus of whether a state will seek to make broad changes to help people who cannot afford ACA plans, even if the trade-off affects people with medical conditions.
“You risk making some worse off by threatening those markets,” said Pearson. “That is always going to be hard.”
Millions more will join the “buy-your-own” ranks
The proposed rule released Oct. 23 allows employers to fund tax-free accounts – called health reimbursement arrangements (HRAs) – that workers can use to buy their own coverage on the ACA marketplaces.
The administration estimates about 10 million people would do so by 2028 – a substantial boost for those exchanges, which policymakers say never hit the enrollment numbers needed to attract enough insurers and hold prices down.
John Barkett, senior director of policy affairs at Willis Towers Watson, a benefits consulting firm, said he expects employers to “seriously consider” the new market. The infusion of workers will improve options by attracting more insurers, he added.
“These people coming in will be employer-sponsored, they’ll have steady jobs,” Barkett noted, and will likely stick with coverage longer than those typically in the individual market.
Currently more than 14 million people buy their own insurance, with about 10 million of those using federal or state ACA marketplaces. The others buy private plans through brokers.
The proposed rule won’t be finalized for months, but it could result in new options by 2020.
If these workers seeking coverage are generally healthy, the infusion could slow premium increases in the overall ACA marketplace because it would improve the risk pool for insurers.
But, if employers with mainly higher-cost or older workers opt to move to the marketplaces, it could help drive up premiums.
In an odd twist, the administration notes in the proposed rule that the ACA has provisions that could protect the marketplace from that type of adverse selection, which can drive up prices. But most of the protective factors cited by the rule have been weakened, removed, or expired, such as the tax penalty for being uninsured and the federal subsidies for insurers to cover lower deductibles for certain low-income consumers.
Benefits consultants and policy experts are skeptical about how many companies will move to the HRA plan, given the tight labor market. Continued uncertainty about the fate of the ACA marketplace may keep them reluctant to send workers out on their own, they say.
Health benefits are a big factor in attracting and retaining workers, said Chris Condeluci, a Washington attorney who previously worked for Sen. Chuck Grassley (R-Iowa) and served as counsel to the Senate Finance Committee during the drafting of the ACA.
“Most employers believe their group health plan will provide better health coverage than an individual market plan,” he said.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
In the span of less than 12 hours, the Trump administration took two seemingly contradictory actions that could have profound effects on the insurance marketplaces set up by the Affordable Care Act.
First, officials issued guidance on the morning of Oct. 22 that could weaken the exchanges set up for people who buy their own insurance. The new approach makes it easier for states to get around some ACA requirements, including allowing the use of federal subsidies for skimpier plans that can reject people with preexisting conditions.
Yet, the other move – a proposed rule unveiled that evening – could bolster ACA marketplaces by sending millions of people with job-based coverage there, armed with tax-free money from their employers to buy individual plans.
Both efforts play into the parallel narratives dominating the bitter political debate over the ACA.
The administration, frustrated that Congress did not repeal the law, say some critics and policy experts, is working to undermine it by weakening the marketplaces and the law’s consumer protections. Those efforts make it easier for insurers to offer skimpier policies that bypass the law’s rules, such as its ban on annual or lifetime limits or its protections for people with preexisting conditions. Congress also zeroed out the tax penalty for not having coverage, effective next year. Combined, the moves could reduce enrollment in ACA plans, potentially driving up premiums for those who remain.
The administration and Republicans in Congress say they are looking to assist those left behind by the ACA – people who don’t get subsidies to help them buy coverage and are desperate for less expensive options – even if that means purchasing less robust coverage.
“These are people who were buying insurance before [the law] and then the rules changed and they could not buy it because they could not afford it,” said Joe Antos, a resident scholar at the conservative American Enterprise Institute. “They have been slowly dropping out of insurance coverage altogether.”
The efforts are dramatically reshaping the ACA and the individual insurance market to one that looks more as it did before the 2010 law, when regulation, coverage, and consumer protections varied widely across the country.
“Some states will do everything they can to keep individual markets strong and stable. Others won’t,” said Sabrina Corlette, research professor at the Center on Health Insurance Reforms at Georgetown University.
So what expectations should consumers have? Here are three key takeaways:
Protections for preexisting health problems are uncertain
Polls show that keeping the ACA’s guarantees on coverage for people with medical problems is a top concern for Americans, and Democrats have made their defense of the health law a key part of their midterm election campaigns.
Republicans have gotten that message and even those who voted to repeal the ACA or joined a lawsuit by 20 red states to overturn it now say they want to protect people with preexisting conditions. Still, GOP lawmakers have not introduced any plan that would be as protective as the current law.
In August, the administration released a rule allowing expanded use of short-term plans, which are less expensive than ACA policies. To get those lower prices, most of these plans do not cover prescription drugs, maternity care, mental health, or substance abuse treatments.
The move is unlikely to benefit people with health problems, as short-term plans can reject people with preexisting conditions or decline to cover care for those medical problems.
Under the rule, insurers can sell them starting in 2019 for up to a year’s duration, with an option to renew for up to 3 years, reversing an Obama-era directive that limited them to 90 days.
Administration officials estimate such plans could draw 600,000 new enrollees next year, and others have estimated the numbers could be far higher. The concern is if many healthy people in 2019 switch out of the ACA market and choose short-term plans, premiums will rise for those who remain, including those with preexisting conditions, or make the ACA market less attractive for insurers.
Where you live matters more
One of the biggest changes ushered in with the ACA was a standard set of rules across all states.
Before the law took effect, consumers buying their own coverage saw tremendous variation in what was offered and what protections they had, depending on the state where they lived.
Most states, for example, allowed insurers to reject people with medical conditions. A few states required insurers to charge similar premiums across the board, but most allowed wide variations based on age, gender or health. Some skimpy plans didn’t cover prescription drugs, chemotherapy, or other medical services.
By standardizing the rules and benefits, the ACA barred insurers from rejecting applicants with medical conditions or charging them more. Women and men get the same premium rates and insurers could charge older people no more than three times what they charged younger ones.
Under the new guidance issued this week giving states more flexibility on what is offered, consumers could again see a wide variation on coverage, premium rules, and even subsidy eligibility.
“It shifts pressure to state politicians,” said Caroline Pearson, a senior fellow at NORC, a nonpartisan research institution at the University of Chicago. That could play into the calculus of whether a state will seek to make broad changes to help people who cannot afford ACA plans, even if the trade-off affects people with medical conditions.
“You risk making some worse off by threatening those markets,” said Pearson. “That is always going to be hard.”
Millions more will join the “buy-your-own” ranks
The proposed rule released Oct. 23 allows employers to fund tax-free accounts – called health reimbursement arrangements (HRAs) – that workers can use to buy their own coverage on the ACA marketplaces.
The administration estimates about 10 million people would do so by 2028 – a substantial boost for those exchanges, which policymakers say never hit the enrollment numbers needed to attract enough insurers and hold prices down.
John Barkett, senior director of policy affairs at Willis Towers Watson, a benefits consulting firm, said he expects employers to “seriously consider” the new market. The infusion of workers will improve options by attracting more insurers, he added.
“These people coming in will be employer-sponsored, they’ll have steady jobs,” Barkett noted, and will likely stick with coverage longer than those typically in the individual market.
Currently more than 14 million people buy their own insurance, with about 10 million of those using federal or state ACA marketplaces. The others buy private plans through brokers.
The proposed rule won’t be finalized for months, but it could result in new options by 2020.
If these workers seeking coverage are generally healthy, the infusion could slow premium increases in the overall ACA marketplace because it would improve the risk pool for insurers.
But, if employers with mainly higher-cost or older workers opt to move to the marketplaces, it could help drive up premiums.
In an odd twist, the administration notes in the proposed rule that the ACA has provisions that could protect the marketplace from that type of adverse selection, which can drive up prices. But most of the protective factors cited by the rule have been weakened, removed, or expired, such as the tax penalty for being uninsured and the federal subsidies for insurers to cover lower deductibles for certain low-income consumers.
Benefits consultants and policy experts are skeptical about how many companies will move to the HRA plan, given the tight labor market. Continued uncertainty about the fate of the ACA marketplace may keep them reluctant to send workers out on their own, they say.
Health benefits are a big factor in attracting and retaining workers, said Chris Condeluci, a Washington attorney who previously worked for Sen. Chuck Grassley (R-Iowa) and served as counsel to the Senate Finance Committee during the drafting of the ACA.
“Most employers believe their group health plan will provide better health coverage than an individual market plan,” he said.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
In the span of less than 12 hours, the Trump administration took two seemingly contradictory actions that could have profound effects on the insurance marketplaces set up by the Affordable Care Act.
First, officials issued guidance on the morning of Oct. 22 that could weaken the exchanges set up for people who buy their own insurance. The new approach makes it easier for states to get around some ACA requirements, including allowing the use of federal subsidies for skimpier plans that can reject people with preexisting conditions.
Yet, the other move – a proposed rule unveiled that evening – could bolster ACA marketplaces by sending millions of people with job-based coverage there, armed with tax-free money from their employers to buy individual plans.
Both efforts play into the parallel narratives dominating the bitter political debate over the ACA.
The administration, frustrated that Congress did not repeal the law, say some critics and policy experts, is working to undermine it by weakening the marketplaces and the law’s consumer protections. Those efforts make it easier for insurers to offer skimpier policies that bypass the law’s rules, such as its ban on annual or lifetime limits or its protections for people with preexisting conditions. Congress also zeroed out the tax penalty for not having coverage, effective next year. Combined, the moves could reduce enrollment in ACA plans, potentially driving up premiums for those who remain.
The administration and Republicans in Congress say they are looking to assist those left behind by the ACA – people who don’t get subsidies to help them buy coverage and are desperate for less expensive options – even if that means purchasing less robust coverage.
“These are people who were buying insurance before [the law] and then the rules changed and they could not buy it because they could not afford it,” said Joe Antos, a resident scholar at the conservative American Enterprise Institute. “They have been slowly dropping out of insurance coverage altogether.”
The efforts are dramatically reshaping the ACA and the individual insurance market to one that looks more as it did before the 2010 law, when regulation, coverage, and consumer protections varied widely across the country.
“Some states will do everything they can to keep individual markets strong and stable. Others won’t,” said Sabrina Corlette, research professor at the Center on Health Insurance Reforms at Georgetown University.
So what expectations should consumers have? Here are three key takeaways:
Protections for preexisting health problems are uncertain
Polls show that keeping the ACA’s guarantees on coverage for people with medical problems is a top concern for Americans, and Democrats have made their defense of the health law a key part of their midterm election campaigns.
Republicans have gotten that message and even those who voted to repeal the ACA or joined a lawsuit by 20 red states to overturn it now say they want to protect people with preexisting conditions. Still, GOP lawmakers have not introduced any plan that would be as protective as the current law.
In August, the administration released a rule allowing expanded use of short-term plans, which are less expensive than ACA policies. To get those lower prices, most of these plans do not cover prescription drugs, maternity care, mental health, or substance abuse treatments.
The move is unlikely to benefit people with health problems, as short-term plans can reject people with preexisting conditions or decline to cover care for those medical problems.
Under the rule, insurers can sell them starting in 2019 for up to a year’s duration, with an option to renew for up to 3 years, reversing an Obama-era directive that limited them to 90 days.
Administration officials estimate such plans could draw 600,000 new enrollees next year, and others have estimated the numbers could be far higher. The concern is if many healthy people in 2019 switch out of the ACA market and choose short-term plans, premiums will rise for those who remain, including those with preexisting conditions, or make the ACA market less attractive for insurers.
Where you live matters more
One of the biggest changes ushered in with the ACA was a standard set of rules across all states.
Before the law took effect, consumers buying their own coverage saw tremendous variation in what was offered and what protections they had, depending on the state where they lived.
Most states, for example, allowed insurers to reject people with medical conditions. A few states required insurers to charge similar premiums across the board, but most allowed wide variations based on age, gender or health. Some skimpy plans didn’t cover prescription drugs, chemotherapy, or other medical services.
By standardizing the rules and benefits, the ACA barred insurers from rejecting applicants with medical conditions or charging them more. Women and men get the same premium rates and insurers could charge older people no more than three times what they charged younger ones.
Under the new guidance issued this week giving states more flexibility on what is offered, consumers could again see a wide variation on coverage, premium rules, and even subsidy eligibility.
“It shifts pressure to state politicians,” said Caroline Pearson, a senior fellow at NORC, a nonpartisan research institution at the University of Chicago. That could play into the calculus of whether a state will seek to make broad changes to help people who cannot afford ACA plans, even if the trade-off affects people with medical conditions.
“You risk making some worse off by threatening those markets,” said Pearson. “That is always going to be hard.”
Millions more will join the “buy-your-own” ranks
The proposed rule released Oct. 23 allows employers to fund tax-free accounts – called health reimbursement arrangements (HRAs) – that workers can use to buy their own coverage on the ACA marketplaces.
The administration estimates about 10 million people would do so by 2028 – a substantial boost for those exchanges, which policymakers say never hit the enrollment numbers needed to attract enough insurers and hold prices down.
John Barkett, senior director of policy affairs at Willis Towers Watson, a benefits consulting firm, said he expects employers to “seriously consider” the new market. The infusion of workers will improve options by attracting more insurers, he added.
“These people coming in will be employer-sponsored, they’ll have steady jobs,” Barkett noted, and will likely stick with coverage longer than those typically in the individual market.
Currently more than 14 million people buy their own insurance, with about 10 million of those using federal or state ACA marketplaces. The others buy private plans through brokers.
The proposed rule won’t be finalized for months, but it could result in new options by 2020.
If these workers seeking coverage are generally healthy, the infusion could slow premium increases in the overall ACA marketplace because it would improve the risk pool for insurers.
But, if employers with mainly higher-cost or older workers opt to move to the marketplaces, it could help drive up premiums.
In an odd twist, the administration notes in the proposed rule that the ACA has provisions that could protect the marketplace from that type of adverse selection, which can drive up prices. But most of the protective factors cited by the rule have been weakened, removed, or expired, such as the tax penalty for being uninsured and the federal subsidies for insurers to cover lower deductibles for certain low-income consumers.
Benefits consultants and policy experts are skeptical about how many companies will move to the HRA plan, given the tight labor market. Continued uncertainty about the fate of the ACA marketplace may keep them reluctant to send workers out on their own, they say.
Health benefits are a big factor in attracting and retaining workers, said Chris Condeluci, a Washington attorney who previously worked for Sen. Chuck Grassley (R-Iowa) and served as counsel to the Senate Finance Committee during the drafting of the ACA.
“Most employers believe their group health plan will provide better health coverage than an individual market plan,” he said.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Rapid bacterial testing of platelets saves money, reduces waste
BOSTON – Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, investigators said.

Rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 per year and cut the rate of platelet wastage from expiration by more than half, reported Adam L. Booth, MD, chief resident in the department of pathology at the University of Texas, Galveston, and his colleagues.
“When a person takes all this time to come in and donate, they do it under the impression that they’re going to help somebody, or several people, and you hate to see those platelets wasted. You want them to be used,” he said in an interview at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
Platelets typically have a shelf life of just 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted in a poster presentation.
A recently published Food and Drug Administration draft guidance for blood banks and transfusion services proposed changing regulations regarding bacterial control of blood products to allow for extended dating if the platelets are collected in an FDA-approved 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
To see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets, the investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
They looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, 332 units at a mean per-unit cost of $516.96 were wasted, for an annual cost of more than $171,000. After the start of testing, however, the annualized rate of waste dropped to 117 units, for an annualized cost of more than $60,000. The difference – minus the cost of rapid bacterial testing – resulted in an annual savings for the institution of nearly $89,000.
Prior to rapid testing, the annual wastage rate was 24%; after testing, it dropped to an annualized 10% rate, the investigators reported.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
“Our findings suggest that rapid bacterial testing can simultaneously enhance the safety of apheresis platelet transfusions and contribute to significant cost savings,” Dr. Booth and his colleagues wrote.
The study was internally funded. The authors reported having no conflicts of interest.
SOURCE: Booth AL et al. AABB18, Abstract INV4.
BOSTON – Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, investigators said.
Rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 per year and cut the rate of platelet wastage from expiration by more than half, reported Adam L. Booth, MD, chief resident in the department of pathology at the University of Texas, Galveston, and his colleagues.
“When a person takes all this time to come in and donate, they do it under the impression that they’re going to help somebody, or several people, and you hate to see those platelets wasted. You want them to be used,” he said in an interview at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
Platelets typically have a shelf life of just 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted in a poster presentation.
A recently published Food and Drug Administration draft guidance for blood banks and transfusion services proposed changing regulations regarding bacterial control of blood products to allow for extended dating if the platelets are collected in an FDA-approved 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
To see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets, the investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
They looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, 332 units at a mean per-unit cost of $516.96 were wasted, for an annual cost of more than $171,000. After the start of testing, however, the annualized rate of waste dropped to 117 units, for an annualized cost of more than $60,000. The difference – minus the cost of rapid bacterial testing – resulted in an annual savings for the institution of nearly $89,000.
Prior to rapid testing, the annual wastage rate was 24%; after testing, it dropped to an annualized 10% rate, the investigators reported.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
“Our findings suggest that rapid bacterial testing can simultaneously enhance the safety of apheresis platelet transfusions and contribute to significant cost savings,” Dr. Booth and his colleagues wrote.
The study was internally funded. The authors reported having no conflicts of interest.
SOURCE: Booth AL et al. AABB18, Abstract INV4.
BOSTON – Rapid bacterial testing of platelets in a hospital blood bank can result in both significant cost savings and reduced wastage of blood products, investigators said.
Rapid bacterial testing of 6- or 7-day-old apheresis platelets resulted in projected annual cost savings of nearly $89,000 per year and cut the rate of platelet wastage from expiration by more than half, reported Adam L. Booth, MD, chief resident in the department of pathology at the University of Texas, Galveston, and his colleagues.
“When a person takes all this time to come in and donate, they do it under the impression that they’re going to help somebody, or several people, and you hate to see those platelets wasted. You want them to be used,” he said in an interview at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
Platelets typically have a shelf life of just 5 days because longer storage increases the risk for bacterial growth and the potential for transfusion-transmitted infections, Dr. Booth and his colleagues noted in a poster presentation.
A recently published Food and Drug Administration draft guidance for blood banks and transfusion services proposed changing regulations regarding bacterial control of blood products to allow for extended dating if the platelets are collected in an FDA-approved 7-day storage container with labeling that requires testing every product with a bacterial detection device, or if the platelets are individually tested for bacterial detection using an approved device.
To see what effect the regulations, if implemented as expected, might have on acquisition costs and wastage of apheresis platelets, the investigators reviewed their center’s platelet acquisition costs and wastage from expiration 12 months before and 6 months after implementation of a rapid bacterial testing protocol, with 6-month results projected out to 1 year for comparison purposes.
They looked at data on bacterial testing of 6-day and 7-day-old apheresis platelets, and excluded data on platelet units that were due to expire on day 5 because they were not stored in FDA-approved containers.
Prior to testing, 332 units at a mean per-unit cost of $516.96 were wasted, for an annual cost of more than $171,000. After the start of testing, however, the annualized rate of waste dropped to 117 units, for an annualized cost of more than $60,000. The difference – minus the cost of rapid bacterial testing – resulted in an annual savings for the institution of nearly $89,000.
Prior to rapid testing, the annual wastage rate was 24%; after testing, it dropped to an annualized 10% rate, the investigators reported.
The number of units transfused and the associated costs of transfusions were similar between the time periods studied.
“Our findings suggest that rapid bacterial testing can simultaneously enhance the safety of apheresis platelet transfusions and contribute to significant cost savings,” Dr. Booth and his colleagues wrote.
The study was internally funded. The authors reported having no conflicts of interest.
SOURCE: Booth AL et al. AABB18, Abstract INV4.
REPORTING FROM AABB18
Key clinical point:
Major finding: Annualized cost savings with rapid bacterial testing were nearly $89,000; platelet wastage decreased from 24% to 10%.
Study details: A retrospective analysis of costs and product wastage before and after implementation of rapid bacterial testing.
Disclosures: The study was internally funded. The authors reported having no conflicts of interest.
Source: Booth AL et al. AABB18, Abstract INV4.
Antiphospholipid antibodies are surprisingly common in first-MI patients
CHICAGO – Patients with a first MI were nearly nine times more likely to have detectable IgG antiphospholipid antibodies than were matched controls in a cross-sectional cohort study, Elisabet Svenungsson, MD, PhD, reported at the annual meeting of the American College of Rheumatology.
Her case-control study included 805 Swedish patients tested for antiphospholipid antibodies 6-10 weeks after experiencing their first MI and an equal number of age-, sex-, and location-matched controls. Prior to their MIs, none of the patients had been diagnosed with antiphospholipid syndrome, which requires both positive antiphospholipid antibodies and a vascular thrombotic event or obstetric morbidity.
A positive test for IgG anti-cardiolipin antibody was present in 10.9% of the first-MI patients, compared with 0.9% of controls. Similarly, 10.4% of acute MI patients and 0.9% of controls were positive for anti-beta2-glycoprotein-1 antibodies. Most patients who tested positive for one were positive for both. Thus, it’s possible that IgG antiphospholipid antibody positivity is an important silent risk factor that’s present in 1 in 10 MI patients, according to Dr. Svenungsson, professor of rheumatology at the Karolinska Institute in Stockholm.
If these results are confirmed and expanded upon in additional studies, testing for antiphospholipid antibodies could become part of the routine care in patients with an acute MI. Those who test positive would meet the criteria for antiphospholipid syndrome and qualify for long-term oral anticoagulation to reduce their elevated risk of further vascular events, she explained in this video interview.
The study was published in Annals of Internal Medicine simultaneously with the presentation at the ACR annual meeting (Ann Int Med. 2018 Oct 23. doi: 10.7326/M18-2130).
SOURCE: Grosso G et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 855.
CHICAGO – Patients with a first MI were nearly nine times more likely to have detectable IgG antiphospholipid antibodies than were matched controls in a cross-sectional cohort study, Elisabet Svenungsson, MD, PhD, reported at the annual meeting of the American College of Rheumatology.
Her case-control study included 805 Swedish patients tested for antiphospholipid antibodies 6-10 weeks after experiencing their first MI and an equal number of age-, sex-, and location-matched controls. Prior to their MIs, none of the patients had been diagnosed with antiphospholipid syndrome, which requires both positive antiphospholipid antibodies and a vascular thrombotic event or obstetric morbidity.
A positive test for IgG anti-cardiolipin antibody was present in 10.9% of the first-MI patients, compared with 0.9% of controls. Similarly, 10.4% of acute MI patients and 0.9% of controls were positive for anti-beta2-glycoprotein-1 antibodies. Most patients who tested positive for one were positive for both. Thus, it’s possible that IgG antiphospholipid antibody positivity is an important silent risk factor that’s present in 1 in 10 MI patients, according to Dr. Svenungsson, professor of rheumatology at the Karolinska Institute in Stockholm.
If these results are confirmed and expanded upon in additional studies, testing for antiphospholipid antibodies could become part of the routine care in patients with an acute MI. Those who test positive would meet the criteria for antiphospholipid syndrome and qualify for long-term oral anticoagulation to reduce their elevated risk of further vascular events, she explained in this video interview.
The study was published in Annals of Internal Medicine simultaneously with the presentation at the ACR annual meeting (Ann Int Med. 2018 Oct 23. doi: 10.7326/M18-2130).
SOURCE: Grosso G et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 855.
CHICAGO – Patients with a first MI were nearly nine times more likely to have detectable IgG antiphospholipid antibodies than were matched controls in a cross-sectional cohort study, Elisabet Svenungsson, MD, PhD, reported at the annual meeting of the American College of Rheumatology.
Her case-control study included 805 Swedish patients tested for antiphospholipid antibodies 6-10 weeks after experiencing their first MI and an equal number of age-, sex-, and location-matched controls. Prior to their MIs, none of the patients had been diagnosed with antiphospholipid syndrome, which requires both positive antiphospholipid antibodies and a vascular thrombotic event or obstetric morbidity.
A positive test for IgG anti-cardiolipin antibody was present in 10.9% of the first-MI patients, compared with 0.9% of controls. Similarly, 10.4% of acute MI patients and 0.9% of controls were positive for anti-beta2-glycoprotein-1 antibodies. Most patients who tested positive for one were positive for both. Thus, it’s possible that IgG antiphospholipid antibody positivity is an important silent risk factor that’s present in 1 in 10 MI patients, according to Dr. Svenungsson, professor of rheumatology at the Karolinska Institute in Stockholm.
If these results are confirmed and expanded upon in additional studies, testing for antiphospholipid antibodies could become part of the routine care in patients with an acute MI. Those who test positive would meet the criteria for antiphospholipid syndrome and qualify for long-term oral anticoagulation to reduce their elevated risk of further vascular events, she explained in this video interview.
The study was published in Annals of Internal Medicine simultaneously with the presentation at the ACR annual meeting (Ann Int Med. 2018 Oct 23. doi: 10.7326/M18-2130).
SOURCE: Grosso G et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 855.
REPORTING FROM THE ACR ANNUAL MEETING

Is respiratory compromise the new “sepsis”?
Hospitalists can play a key role in prevention
Clinicians and even the general public are aware of the dangers of sepsis, the life-threatening illness caused by a body’s response to an infection. Irrespective of one’s perception of pharmaceutical marketing materials or the evidence-based medicine used, awareness about sepsis has led to earlier diagnosis and interventions that have likely saved countless patients’ lives.

Moreover, hospitalists have played a key role in sepsis prevention. In their research, “Improving Survival from Sepsis in Noncritical Units: Role of Hospitalists and Sepsis Team in Early Detection and Initial Treatment of Septic Patients,” Adriana Ducci, MD, and her colleagues showed that a hospitalist-managed sepsis protocol improved sepsis case notifications and patient outcomes.
Although sepsis and respiratory compromise are clearly very different conditions, I believe that greater awareness about respiratory compromise will lead to earlier diagnosis and interventions, which will theoretically improve patient outcomes. Moreover, as with the sepsis awareness campaign, hospitalists can play a key role in recognizing respiratory compromise and in the implementation of appropriate interventions.
As defined by the Respiratory Compromise Institute, “respiratory compromise” is defined as a state in which there is a high likelihood of decompensation into respiratory failure and/or death, but, in which specific interventions – be it therapeutic and/or monitoring – might prevent or mitigate this decompensation.
A significant segment of patients who may be at risk for respiratory compromise are those receiving opioids. The cost of opioid-related adverse events, in terms of both human life and hospital expenses, remains at the forefront of the public eye. It has been estimated that yearly costs in the United States associated with opioid-related postoperative respiratory failure were estimated at $2 billion.
Thomas W. Frederickson MD, FACP, SFHM, MBA, the lead author of the Society of Hospital Medicine guide for Reducing Adverse Drug Events Related to Opioids (RADEO), emphasized in a podcast with the Physician-Patient Alliance for Health & Safety the need to identify patient conditions that pose a greater risk of respiratory compromise.
In particular, Dr. Frederickson pointed out the need to screen for obstructive sleep apnea (OSA): “Patients with obstructive sleep apnea are dependent upon their arousal mechanism in order to avoid respiratory depression and eventual respiratory failure. When these patients receive opioid medication, it decreases this ability for arousal. That puts them at risk for a sudden spiral that includes respiratory insufficiency and respiratory arrest. This can happen very quickly and part of the risk is that the traditional monitoring for sedation that we use in the hospital – that is on a periodic basis and depends upon nursing interventions and questioning – really becomes much less effective in this patient population that can have a respiratory arrest, because of failure to arouse, very quickly. So, a monitoring regimen that takes place every 60 minutes is likely to be ineffective.”
Patient conditions such as OSA should be considered, along with other comorbidities. As the RADEO Guide states: “Before starting opioid therapy, either in surgical or non-surgical settings, it is important to identify any real or potential risks of respiratory depression or other opioid-related adverse effects. Patient comorbidities such as OSA, neurologic disorders, organ impairment, substance abuse history, and other medication use are important aspects to consider.”
Although we have clearly recognized a significant increase in respiratory complications associated with opioid administration, there are other areas, which are non–opioid related, that can create respiratory compromise. We view many patients with stable or underlying respiratory conditions, whether it be COPD, sleep apnea, or preexisting pathophysiology, where either due to sedative agents, or an acute illness – like pneumonia – they can go from a stable condition to respiratory compromise and become at risk for respiratory failure.
A classic example of that in my world of anesthesia has been the well-recognized area of non–operating room anesthesia – in particular, in endoscopy suites where numerous endoscopy procedures are performed under the administration of propofol or other anxiolytic-like drugs. There has been a well-recognized incidence of sentinel events related to oxygenation and ventilation, including death.
Many clinicians see sedation as a benign introduction of relatively limited-effect drugs, which isn’t always true. So, therefore, it is essential that clinicians understand three things:
1. The drugs we employ as sedative agents can have variable effects on individuals depending on their tolerance and their underlying medical condition.
2. The dosages and particular combination of drugs employed may cause an adverse event – for example, the combination of opioids and benzodiazepines.
3. There are factors that can distract from the clinical assessment of routine vital signs, such as respiratory rate, heart rate, and blood pressure. For example, when pulse oximetry is administered with oxygen therapy, there can often be a delay in the recognition of hypoventilation. Consequently, that’s why more and more clinicians are beginning to utilize capnography, or CO2 monitoring, in the expired gas to earlier detect depressed respiratory rate and/or apnea, as well as signs of hypoventilation or inadequate ventilation.
There clearly are obstacles to continuous patient monitoring, such as the associated cost, familiarity with the utilization, the benefit, as well as the limitations of specific monitors in different clinical situations, which mandates an educational process to employ these. However, currently, patient monitoring provides the best early indicator of a patient’s deterioration and the possibility of respiratory compromise.
In my field, we have become very comfortable with capnography and patient monitoring, because for decades it’s been a standard of care for monitoring in the operating room. The role for utilization of capnography for patients who are receiving an opioid or sedative agent outside of the operating room needs to be further assessed. However, technology is not a silver bullet and should be used as an adjunct to clinical judgment in at-risk populations.
Simple recognition and greater awareness of respiratory compromise, just as with sepsis awareness campaigns, will mean more patients are diagnosed earlier, more appropriate interventions are made, and hopefully more adverse events and patient deaths are averted.
Dr. Vender is the emeritus Harris Family Foundation chairman of the department of anesthesiology at NorthShore University Health System in Evanston, Ill. He is clinical professor at the University of Chicago Pritzker School of Medicine and chairman, Clinical Advisory Committee, Respiratory Compromise Institute. Dr. Vender has consulted with Medtronic.
Hospitalists can play a key role in prevention
Hospitalists can play a key role in prevention
Clinicians and even the general public are aware of the dangers of sepsis, the life-threatening illness caused by a body’s response to an infection. Irrespective of one’s perception of pharmaceutical marketing materials or the evidence-based medicine used, awareness about sepsis has led to earlier diagnosis and interventions that have likely saved countless patients’ lives.
Moreover, hospitalists have played a key role in sepsis prevention. In their research, “Improving Survival from Sepsis in Noncritical Units: Role of Hospitalists and Sepsis Team in Early Detection and Initial Treatment of Septic Patients,” Adriana Ducci, MD, and her colleagues showed that a hospitalist-managed sepsis protocol improved sepsis case notifications and patient outcomes.
Although sepsis and respiratory compromise are clearly very different conditions, I believe that greater awareness about respiratory compromise will lead to earlier diagnosis and interventions, which will theoretically improve patient outcomes. Moreover, as with the sepsis awareness campaign, hospitalists can play a key role in recognizing respiratory compromise and in the implementation of appropriate interventions.
As defined by the Respiratory Compromise Institute, “respiratory compromise” is defined as a state in which there is a high likelihood of decompensation into respiratory failure and/or death, but, in which specific interventions – be it therapeutic and/or monitoring – might prevent or mitigate this decompensation.
A significant segment of patients who may be at risk for respiratory compromise are those receiving opioids. The cost of opioid-related adverse events, in terms of both human life and hospital expenses, remains at the forefront of the public eye. It has been estimated that yearly costs in the United States associated with opioid-related postoperative respiratory failure were estimated at $2 billion.
Thomas W. Frederickson MD, FACP, SFHM, MBA, the lead author of the Society of Hospital Medicine guide for Reducing Adverse Drug Events Related to Opioids (RADEO), emphasized in a podcast with the Physician-Patient Alliance for Health & Safety the need to identify patient conditions that pose a greater risk of respiratory compromise.
In particular, Dr. Frederickson pointed out the need to screen for obstructive sleep apnea (OSA): “Patients with obstructive sleep apnea are dependent upon their arousal mechanism in order to avoid respiratory depression and eventual respiratory failure. When these patients receive opioid medication, it decreases this ability for arousal. That puts them at risk for a sudden spiral that includes respiratory insufficiency and respiratory arrest. This can happen very quickly and part of the risk is that the traditional monitoring for sedation that we use in the hospital – that is on a periodic basis and depends upon nursing interventions and questioning – really becomes much less effective in this patient population that can have a respiratory arrest, because of failure to arouse, very quickly. So, a monitoring regimen that takes place every 60 minutes is likely to be ineffective.”
Patient conditions such as OSA should be considered, along with other comorbidities. As the RADEO Guide states: “Before starting opioid therapy, either in surgical or non-surgical settings, it is important to identify any real or potential risks of respiratory depression or other opioid-related adverse effects. Patient comorbidities such as OSA, neurologic disorders, organ impairment, substance abuse history, and other medication use are important aspects to consider.”
Although we have clearly recognized a significant increase in respiratory complications associated with opioid administration, there are other areas, which are non–opioid related, that can create respiratory compromise. We view many patients with stable or underlying respiratory conditions, whether it be COPD, sleep apnea, or preexisting pathophysiology, where either due to sedative agents, or an acute illness – like pneumonia – they can go from a stable condition to respiratory compromise and become at risk for respiratory failure.
A classic example of that in my world of anesthesia has been the well-recognized area of non–operating room anesthesia – in particular, in endoscopy suites where numerous endoscopy procedures are performed under the administration of propofol or other anxiolytic-like drugs. There has been a well-recognized incidence of sentinel events related to oxygenation and ventilation, including death.
Many clinicians see sedation as a benign introduction of relatively limited-effect drugs, which isn’t always true. So, therefore, it is essential that clinicians understand three things:
1. The drugs we employ as sedative agents can have variable effects on individuals depending on their tolerance and their underlying medical condition.
2. The dosages and particular combination of drugs employed may cause an adverse event – for example, the combination of opioids and benzodiazepines.
3. There are factors that can distract from the clinical assessment of routine vital signs, such as respiratory rate, heart rate, and blood pressure. For example, when pulse oximetry is administered with oxygen therapy, there can often be a delay in the recognition of hypoventilation. Consequently, that’s why more and more clinicians are beginning to utilize capnography, or CO2 monitoring, in the expired gas to earlier detect depressed respiratory rate and/or apnea, as well as signs of hypoventilation or inadequate ventilation.
There clearly are obstacles to continuous patient monitoring, such as the associated cost, familiarity with the utilization, the benefit, as well as the limitations of specific monitors in different clinical situations, which mandates an educational process to employ these. However, currently, patient monitoring provides the best early indicator of a patient’s deterioration and the possibility of respiratory compromise.
In my field, we have become very comfortable with capnography and patient monitoring, because for decades it’s been a standard of care for monitoring in the operating room. The role for utilization of capnography for patients who are receiving an opioid or sedative agent outside of the operating room needs to be further assessed. However, technology is not a silver bullet and should be used as an adjunct to clinical judgment in at-risk populations.
Simple recognition and greater awareness of respiratory compromise, just as with sepsis awareness campaigns, will mean more patients are diagnosed earlier, more appropriate interventions are made, and hopefully more adverse events and patient deaths are averted.
Dr. Vender is the emeritus Harris Family Foundation chairman of the department of anesthesiology at NorthShore University Health System in Evanston, Ill. He is clinical professor at the University of Chicago Pritzker School of Medicine and chairman, Clinical Advisory Committee, Respiratory Compromise Institute. Dr. Vender has consulted with Medtronic.
Clinicians and even the general public are aware of the dangers of sepsis, the life-threatening illness caused by a body’s response to an infection. Irrespective of one’s perception of pharmaceutical marketing materials or the evidence-based medicine used, awareness about sepsis has led to earlier diagnosis and interventions that have likely saved countless patients’ lives.
Moreover, hospitalists have played a key role in sepsis prevention. In their research, “Improving Survival from Sepsis in Noncritical Units: Role of Hospitalists and Sepsis Team in Early Detection and Initial Treatment of Septic Patients,” Adriana Ducci, MD, and her colleagues showed that a hospitalist-managed sepsis protocol improved sepsis case notifications and patient outcomes.
Although sepsis and respiratory compromise are clearly very different conditions, I believe that greater awareness about respiratory compromise will lead to earlier diagnosis and interventions, which will theoretically improve patient outcomes. Moreover, as with the sepsis awareness campaign, hospitalists can play a key role in recognizing respiratory compromise and in the implementation of appropriate interventions.
As defined by the Respiratory Compromise Institute, “respiratory compromise” is defined as a state in which there is a high likelihood of decompensation into respiratory failure and/or death, but, in which specific interventions – be it therapeutic and/or monitoring – might prevent or mitigate this decompensation.
A significant segment of patients who may be at risk for respiratory compromise are those receiving opioids. The cost of opioid-related adverse events, in terms of both human life and hospital expenses, remains at the forefront of the public eye. It has been estimated that yearly costs in the United States associated with opioid-related postoperative respiratory failure were estimated at $2 billion.
Thomas W. Frederickson MD, FACP, SFHM, MBA, the lead author of the Society of Hospital Medicine guide for Reducing Adverse Drug Events Related to Opioids (RADEO), emphasized in a podcast with the Physician-Patient Alliance for Health & Safety the need to identify patient conditions that pose a greater risk of respiratory compromise.
In particular, Dr. Frederickson pointed out the need to screen for obstructive sleep apnea (OSA): “Patients with obstructive sleep apnea are dependent upon their arousal mechanism in order to avoid respiratory depression and eventual respiratory failure. When these patients receive opioid medication, it decreases this ability for arousal. That puts them at risk for a sudden spiral that includes respiratory insufficiency and respiratory arrest. This can happen very quickly and part of the risk is that the traditional monitoring for sedation that we use in the hospital – that is on a periodic basis and depends upon nursing interventions and questioning – really becomes much less effective in this patient population that can have a respiratory arrest, because of failure to arouse, very quickly. So, a monitoring regimen that takes place every 60 minutes is likely to be ineffective.”
Patient conditions such as OSA should be considered, along with other comorbidities. As the RADEO Guide states: “Before starting opioid therapy, either in surgical or non-surgical settings, it is important to identify any real or potential risks of respiratory depression or other opioid-related adverse effects. Patient comorbidities such as OSA, neurologic disorders, organ impairment, substance abuse history, and other medication use are important aspects to consider.”
Although we have clearly recognized a significant increase in respiratory complications associated with opioid administration, there are other areas, which are non–opioid related, that can create respiratory compromise. We view many patients with stable or underlying respiratory conditions, whether it be COPD, sleep apnea, or preexisting pathophysiology, where either due to sedative agents, or an acute illness – like pneumonia – they can go from a stable condition to respiratory compromise and become at risk for respiratory failure.
A classic example of that in my world of anesthesia has been the well-recognized area of non–operating room anesthesia – in particular, in endoscopy suites where numerous endoscopy procedures are performed under the administration of propofol or other anxiolytic-like drugs. There has been a well-recognized incidence of sentinel events related to oxygenation and ventilation, including death.
Many clinicians see sedation as a benign introduction of relatively limited-effect drugs, which isn’t always true. So, therefore, it is essential that clinicians understand three things:
1. The drugs we employ as sedative agents can have variable effects on individuals depending on their tolerance and their underlying medical condition.
2. The dosages and particular combination of drugs employed may cause an adverse event – for example, the combination of opioids and benzodiazepines.
3. There are factors that can distract from the clinical assessment of routine vital signs, such as respiratory rate, heart rate, and blood pressure. For example, when pulse oximetry is administered with oxygen therapy, there can often be a delay in the recognition of hypoventilation. Consequently, that’s why more and more clinicians are beginning to utilize capnography, or CO2 monitoring, in the expired gas to earlier detect depressed respiratory rate and/or apnea, as well as signs of hypoventilation or inadequate ventilation.
There clearly are obstacles to continuous patient monitoring, such as the associated cost, familiarity with the utilization, the benefit, as well as the limitations of specific monitors in different clinical situations, which mandates an educational process to employ these. However, currently, patient monitoring provides the best early indicator of a patient’s deterioration and the possibility of respiratory compromise.
In my field, we have become very comfortable with capnography and patient monitoring, because for decades it’s been a standard of care for monitoring in the operating room. The role for utilization of capnography for patients who are receiving an opioid or sedative agent outside of the operating room needs to be further assessed. However, technology is not a silver bullet and should be used as an adjunct to clinical judgment in at-risk populations.
Simple recognition and greater awareness of respiratory compromise, just as with sepsis awareness campaigns, will mean more patients are diagnosed earlier, more appropriate interventions are made, and hopefully more adverse events and patient deaths are averted.
Dr. Vender is the emeritus Harris Family Foundation chairman of the department of anesthesiology at NorthShore University Health System in Evanston, Ill. He is clinical professor at the University of Chicago Pritzker School of Medicine and chairman, Clinical Advisory Committee, Respiratory Compromise Institute. Dr. Vender has consulted with Medtronic.
Cancer and conference calls

You notice I said, “Ask the question.” I can always ask. I just can’t always answer.
Harriet listed her Chief Complaint as “psoriasis on the scalp.”
“My hairdresser says I have psoriasis,” she said.
I took a look. “You do,” I said. “Just one spot, though. Should be easy to control.”
I then ran through the list of what generally bothers people about scalp psoriasis. “It may come back now and then,” I said, “but you don’t have much of it and you haven’t had it long, so it shouldn’t take much effort to keep it under control. Psoriasis doesn’t cause permanent hair loss,” I added. “And you can color and condition your hair any way you want.”
Harriet smiled. That was what she wanted to hear. But it wasn’t all she wanted to hear.
“Why don’t I look you over completely?” I suggested. Harriet agreed. I found only lentigines and seborrheic keratoses all over, and I told her so.
“That’s wonderful,” said Harriet. “Just one more thing.”
“Sure.”
“That psoriasis on my head. It wouldn’t be cancer, would it?”
I opened my mouth to respond, but nothing came out. Sure, patients worry that anything they don’t understand might be cancer. But that’s to start with, not after a whole conversation about psoriasis. Right?
Maybe not.
“Not cancer,” I said. “Just some local inflammation.” Harriet was happy. I was perplexed. There’s always something new about patients to puzzle over.
Which I did for about 2 hours, until that puzzle was muscled out by another. I walked in to meet a very cheery Rory, who was punching his smartphone screen. “Wouldn’t you know it?” he said with a smile. “The same thing happened last time I came here. You walked in just as I was about to start a conference call.”
I thought of several responses, none of them appropriate.
“Last time you cauterized some of these milia thingies on my face,” said Rory. “I was hoping you could do that again.”
I peered at his face. “Sure,” I said, “if you want me to.”
“Just a sec,” said Rory, peering down at his phone. I assumed he was logging off the conference call.
“OK,” he said. “Go ahead.”
I revved up my Hyfrecator, which started to buzz.
“Wait, can they hear that?” Rory asked.
“Can who hear ... ?”
“This is Rory Stiefel,” he spoke into his phone. “Glad we could meet today. I wanted to talk to all of you about our plans to expand our network services into your Upper Midwestern territory.”
“Hold on,” I said (to myself), “You want me to desiccate your face while you’re expanding your network into the Upper Midwest??!!”
Rory motioned for me to continue. “Sure,” he said to his phone, “We can be up and running by the first of next month, no problem.” Apparently, the hum of the Hyfrecator wasn’t interrupting negotiations.
So I buzzed away, while Rory’s interlocutors responded with apparent enthusiasm. By the time he turned his other cheek, I figured he had occupied Minnesota.
“Did you get all the thingies?” Rory stage-whispered.
I nodded.
“Great!” he said, then turned back to his phone. “Well, this was a great meeting,” he said. “I’m glad we’re ready to go live. Talk to you guys next week to firm up logistics.” He punched the screen to sever the connection.
“Thanks for being so efficient,” he said, this time to me.
“No problem,” I said, now-silent Hyfrecator in hand.
“You’re sure you got them all?”
“I handed him a mirror. “Yes,” I said. “I got them all.”
“Well that’s terrific,” he said, jumping off the exam table and heading for the door. “Always a pleasure. See you next time!”
I don’t know exactly what he does, but Rory is one awesome multitasker.

As for me, I just have to consult the CPT code book to find the right designation for “Cautery of benign lesions during a corporate conference call, second episode.”
Any help, dear colleagues, with people or coding, will be appreciated.
I can always ask ...
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
You notice I said, “Ask the question.” I can always ask. I just can’t always answer.
Harriet listed her Chief Complaint as “psoriasis on the scalp.”
“My hairdresser says I have psoriasis,” she said.
I took a look. “You do,” I said. “Just one spot, though. Should be easy to control.”
I then ran through the list of what generally bothers people about scalp psoriasis. “It may come back now and then,” I said, “but you don’t have much of it and you haven’t had it long, so it shouldn’t take much effort to keep it under control. Psoriasis doesn’t cause permanent hair loss,” I added. “And you can color and condition your hair any way you want.”
Harriet smiled. That was what she wanted to hear. But it wasn’t all she wanted to hear.
“Why don’t I look you over completely?” I suggested. Harriet agreed. I found only lentigines and seborrheic keratoses all over, and I told her so.
“That’s wonderful,” said Harriet. “Just one more thing.”
“Sure.”
“That psoriasis on my head. It wouldn’t be cancer, would it?”
I opened my mouth to respond, but nothing came out. Sure, patients worry that anything they don’t understand might be cancer. But that’s to start with, not after a whole conversation about psoriasis. Right?
Maybe not.
“Not cancer,” I said. “Just some local inflammation.” Harriet was happy. I was perplexed. There’s always something new about patients to puzzle over.
Which I did for about 2 hours, until that puzzle was muscled out by another. I walked in to meet a very cheery Rory, who was punching his smartphone screen. “Wouldn’t you know it?” he said with a smile. “The same thing happened last time I came here. You walked in just as I was about to start a conference call.”
I thought of several responses, none of them appropriate.
“Last time you cauterized some of these milia thingies on my face,” said Rory. “I was hoping you could do that again.”
I peered at his face. “Sure,” I said, “if you want me to.”
“Just a sec,” said Rory, peering down at his phone. I assumed he was logging off the conference call.
“OK,” he said. “Go ahead.”
I revved up my Hyfrecator, which started to buzz.
“Wait, can they hear that?” Rory asked.
“Can who hear ... ?”
“This is Rory Stiefel,” he spoke into his phone. “Glad we could meet today. I wanted to talk to all of you about our plans to expand our network services into your Upper Midwestern territory.”
“Hold on,” I said (to myself), “You want me to desiccate your face while you’re expanding your network into the Upper Midwest??!!”
Rory motioned for me to continue. “Sure,” he said to his phone, “We can be up and running by the first of next month, no problem.” Apparently, the hum of the Hyfrecator wasn’t interrupting negotiations.
So I buzzed away, while Rory’s interlocutors responded with apparent enthusiasm. By the time he turned his other cheek, I figured he had occupied Minnesota.
“Did you get all the thingies?” Rory stage-whispered.
I nodded.
“Great!” he said, then turned back to his phone. “Well, this was a great meeting,” he said. “I’m glad we’re ready to go live. Talk to you guys next week to firm up logistics.” He punched the screen to sever the connection.
“Thanks for being so efficient,” he said, this time to me.
“No problem,” I said, now-silent Hyfrecator in hand.
“You’re sure you got them all?”
“I handed him a mirror. “Yes,” I said. “I got them all.”
“Well that’s terrific,” he said, jumping off the exam table and heading for the door. “Always a pleasure. See you next time!”
I don’t know exactly what he does, but Rory is one awesome multitasker.
As for me, I just have to consult the CPT code book to find the right designation for “Cautery of benign lesions during a corporate conference call, second episode.”
Any help, dear colleagues, with people or coding, will be appreciated.
I can always ask ...
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
You notice I said, “Ask the question.” I can always ask. I just can’t always answer.
Harriet listed her Chief Complaint as “psoriasis on the scalp.”
“My hairdresser says I have psoriasis,” she said.
I took a look. “You do,” I said. “Just one spot, though. Should be easy to control.”
I then ran through the list of what generally bothers people about scalp psoriasis. “It may come back now and then,” I said, “but you don’t have much of it and you haven’t had it long, so it shouldn’t take much effort to keep it under control. Psoriasis doesn’t cause permanent hair loss,” I added. “And you can color and condition your hair any way you want.”
Harriet smiled. That was what she wanted to hear. But it wasn’t all she wanted to hear.
“Why don’t I look you over completely?” I suggested. Harriet agreed. I found only lentigines and seborrheic keratoses all over, and I told her so.
“That’s wonderful,” said Harriet. “Just one more thing.”
“Sure.”
“That psoriasis on my head. It wouldn’t be cancer, would it?”
I opened my mouth to respond, but nothing came out. Sure, patients worry that anything they don’t understand might be cancer. But that’s to start with, not after a whole conversation about psoriasis. Right?
Maybe not.
“Not cancer,” I said. “Just some local inflammation.” Harriet was happy. I was perplexed. There’s always something new about patients to puzzle over.
Which I did for about 2 hours, until that puzzle was muscled out by another. I walked in to meet a very cheery Rory, who was punching his smartphone screen. “Wouldn’t you know it?” he said with a smile. “The same thing happened last time I came here. You walked in just as I was about to start a conference call.”
I thought of several responses, none of them appropriate.
“Last time you cauterized some of these milia thingies on my face,” said Rory. “I was hoping you could do that again.”
I peered at his face. “Sure,” I said, “if you want me to.”
“Just a sec,” said Rory, peering down at his phone. I assumed he was logging off the conference call.
“OK,” he said. “Go ahead.”
I revved up my Hyfrecator, which started to buzz.
“Wait, can they hear that?” Rory asked.
“Can who hear ... ?”
“This is Rory Stiefel,” he spoke into his phone. “Glad we could meet today. I wanted to talk to all of you about our plans to expand our network services into your Upper Midwestern territory.”
“Hold on,” I said (to myself), “You want me to desiccate your face while you’re expanding your network into the Upper Midwest??!!”
Rory motioned for me to continue. “Sure,” he said to his phone, “We can be up and running by the first of next month, no problem.” Apparently, the hum of the Hyfrecator wasn’t interrupting negotiations.
So I buzzed away, while Rory’s interlocutors responded with apparent enthusiasm. By the time he turned his other cheek, I figured he had occupied Minnesota.
“Did you get all the thingies?” Rory stage-whispered.
I nodded.
“Great!” he said, then turned back to his phone. “Well, this was a great meeting,” he said. “I’m glad we’re ready to go live. Talk to you guys next week to firm up logistics.” He punched the screen to sever the connection.
“Thanks for being so efficient,” he said, this time to me.
“No problem,” I said, now-silent Hyfrecator in hand.
“You’re sure you got them all?”
“I handed him a mirror. “Yes,” I said. “I got them all.”
“Well that’s terrific,” he said, jumping off the exam table and heading for the door. “Always a pleasure. See you next time!”
I don’t know exactly what he does, but Rory is one awesome multitasker.
As for me, I just have to consult the CPT code book to find the right designation for “Cautery of benign lesions during a corporate conference call, second episode.”
Any help, dear colleagues, with people or coding, will be appreciated.
I can always ask ...
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
Initial screening not enough to catch all cases of preterm congenital hypothyroidism
reported Niamh McGrath, MD, of the department of pediatric endocrinology at Children’s University Hospital, Dublin, and her associates in the Journal of Pediatrics.
In a population-based prospective review of 898,424 records between 2004 and 2016, Dr. McGrath and her associates identified all preterm infants less than 33 weeks diagnosed with congenital hypothyroidism and receiving treatment with levothyroxine. Of the infants screened, just 53 were selected to participate in the study, including 26 who were diagnosed at the first thyroid-stimulating hormone (TSH) screening and 27 who had delayed TSH elevation.
Gestational age ranged from 23 to 33 weeks, median birth weight measured 1.2 kg, median serum TSH concentration at the time of diagnosis was 78.3 mU/L, and median free thyroxine concentration was 8.9 pmol/L.
For half of the infants ultimately diagnosed, congenital hypothyroidism was not detected during the initial newborn screening. The authors also noted that 25% of patients with delayed TSH elevation had been exposed to iodine while undergoing surgery for necrotizing enterocolitis; after age 28 days, four of these infants were found to have elevated TSH. They cautioned that while this finding emphasizes the need for close monitoring and repeat screening of infants who have been exposed to iodine for up to 1 month following exposure, they could not be certain that the iodine exposure was responsible for the transient hypothyroidism in these patients.
Dr. McGrath and her associates emphasized the importance of repeat TSH screening for all preterm infants, noting that standard protocol screenings conducted just once at age 2 weeks or 4 weeks are not sufficient to effectively identify all cases of congenital hypothyroidism in which TSH elevation is delayed. Instead, they recommended measuring TSH on days 3-5 and at 1 week, 2 weeks, 4 weeks, and term-corrected gestational age.
“Our data are consistent with studies showing a high incidence of delayed TSH rise, particularly in very-low-birth-weight infants,” the authors wrote. They speculated that delays in detecting primary congenital hypothyroidism could be caused by the suppression of TSH secretion as a result of “hypothalamic-pituitary immaturity, medication administration, and effects of serious neonatal illness.” In fact, had standard, recommended 2-week-only screening protocols been followed with their patient population, fully 48% of infants with delayed TSH elevation would have been overlooked; half of these patients were later found to have decompensated hypothyroidism.
Neurodevelopmental disability caused by congenital hypothyroidism, which affects roughly 1 in every 2,000-4,000 births, is increasingly being prevented with newborn screenings that identify the condition early, but the incidence of congenital hypothyroidism has increased considerably in the past 20 years. The authors attribute this increase to the gradual change in screening cutoff levels and the rise in number of preterm infants who are surviving.
The need for a second screening, as well as appropriate timing and optimal TSH cutoff, are “subjects of active debate,” the authors wrote. The latest European screening guidelines recommend second screenings for preterm and low-birth-weight infants at either 2 weeks of age or 2 weeks following preliminary screening. Although they make no recommendations regarding additional screenings, American Academy of Pediatrics guidelines, published in 2006, cite a “disproportionate incidence” of delayed increase in TSH and congenital hypothyroidism in infants with very low birth weight. Dr. McGrath and her associates speculated that not all screening programs have adopted repeat screening of preterm infants, perhaps because of the low yield results, “the transient nature of most cases” detected, as well as conflicting long-term data on neurodevelopmental outcomes.
Dr. McGrath receives funding from the Children’s Fund for Health. The authors declared no conflicts of interest.
SOURCE: McGrath N et al. J Pediatr. 2018 Oct 24. doi: 10.1016/j.jpeds.2018.09.044.
reported Niamh McGrath, MD, of the department of pediatric endocrinology at Children’s University Hospital, Dublin, and her associates in the Journal of Pediatrics.
In a population-based prospective review of 898,424 records between 2004 and 2016, Dr. McGrath and her associates identified all preterm infants less than 33 weeks diagnosed with congenital hypothyroidism and receiving treatment with levothyroxine. Of the infants screened, just 53 were selected to participate in the study, including 26 who were diagnosed at the first thyroid-stimulating hormone (TSH) screening and 27 who had delayed TSH elevation.
Gestational age ranged from 23 to 33 weeks, median birth weight measured 1.2 kg, median serum TSH concentration at the time of diagnosis was 78.3 mU/L, and median free thyroxine concentration was 8.9 pmol/L.
For half of the infants ultimately diagnosed, congenital hypothyroidism was not detected during the initial newborn screening. The authors also noted that 25% of patients with delayed TSH elevation had been exposed to iodine while undergoing surgery for necrotizing enterocolitis; after age 28 days, four of these infants were found to have elevated TSH. They cautioned that while this finding emphasizes the need for close monitoring and repeat screening of infants who have been exposed to iodine for up to 1 month following exposure, they could not be certain that the iodine exposure was responsible for the transient hypothyroidism in these patients.
Dr. McGrath and her associates emphasized the importance of repeat TSH screening for all preterm infants, noting that standard protocol screenings conducted just once at age 2 weeks or 4 weeks are not sufficient to effectively identify all cases of congenital hypothyroidism in which TSH elevation is delayed. Instead, they recommended measuring TSH on days 3-5 and at 1 week, 2 weeks, 4 weeks, and term-corrected gestational age.
“Our data are consistent with studies showing a high incidence of delayed TSH rise, particularly in very-low-birth-weight infants,” the authors wrote. They speculated that delays in detecting primary congenital hypothyroidism could be caused by the suppression of TSH secretion as a result of “hypothalamic-pituitary immaturity, medication administration, and effects of serious neonatal illness.” In fact, had standard, recommended 2-week-only screening protocols been followed with their patient population, fully 48% of infants with delayed TSH elevation would have been overlooked; half of these patients were later found to have decompensated hypothyroidism.
Neurodevelopmental disability caused by congenital hypothyroidism, which affects roughly 1 in every 2,000-4,000 births, is increasingly being prevented with newborn screenings that identify the condition early, but the incidence of congenital hypothyroidism has increased considerably in the past 20 years. The authors attribute this increase to the gradual change in screening cutoff levels and the rise in number of preterm infants who are surviving.
The need for a second screening, as well as appropriate timing and optimal TSH cutoff, are “subjects of active debate,” the authors wrote. The latest European screening guidelines recommend second screenings for preterm and low-birth-weight infants at either 2 weeks of age or 2 weeks following preliminary screening. Although they make no recommendations regarding additional screenings, American Academy of Pediatrics guidelines, published in 2006, cite a “disproportionate incidence” of delayed increase in TSH and congenital hypothyroidism in infants with very low birth weight. Dr. McGrath and her associates speculated that not all screening programs have adopted repeat screening of preterm infants, perhaps because of the low yield results, “the transient nature of most cases” detected, as well as conflicting long-term data on neurodevelopmental outcomes.
Dr. McGrath receives funding from the Children’s Fund for Health. The authors declared no conflicts of interest.
SOURCE: McGrath N et al. J Pediatr. 2018 Oct 24. doi: 10.1016/j.jpeds.2018.09.044.
reported Niamh McGrath, MD, of the department of pediatric endocrinology at Children’s University Hospital, Dublin, and her associates in the Journal of Pediatrics.
In a population-based prospective review of 898,424 records between 2004 and 2016, Dr. McGrath and her associates identified all preterm infants less than 33 weeks diagnosed with congenital hypothyroidism and receiving treatment with levothyroxine. Of the infants screened, just 53 were selected to participate in the study, including 26 who were diagnosed at the first thyroid-stimulating hormone (TSH) screening and 27 who had delayed TSH elevation.
Gestational age ranged from 23 to 33 weeks, median birth weight measured 1.2 kg, median serum TSH concentration at the time of diagnosis was 78.3 mU/L, and median free thyroxine concentration was 8.9 pmol/L.
For half of the infants ultimately diagnosed, congenital hypothyroidism was not detected during the initial newborn screening. The authors also noted that 25% of patients with delayed TSH elevation had been exposed to iodine while undergoing surgery for necrotizing enterocolitis; after age 28 days, four of these infants were found to have elevated TSH. They cautioned that while this finding emphasizes the need for close monitoring and repeat screening of infants who have been exposed to iodine for up to 1 month following exposure, they could not be certain that the iodine exposure was responsible for the transient hypothyroidism in these patients.
Dr. McGrath and her associates emphasized the importance of repeat TSH screening for all preterm infants, noting that standard protocol screenings conducted just once at age 2 weeks or 4 weeks are not sufficient to effectively identify all cases of congenital hypothyroidism in which TSH elevation is delayed. Instead, they recommended measuring TSH on days 3-5 and at 1 week, 2 weeks, 4 weeks, and term-corrected gestational age.
“Our data are consistent with studies showing a high incidence of delayed TSH rise, particularly in very-low-birth-weight infants,” the authors wrote. They speculated that delays in detecting primary congenital hypothyroidism could be caused by the suppression of TSH secretion as a result of “hypothalamic-pituitary immaturity, medication administration, and effects of serious neonatal illness.” In fact, had standard, recommended 2-week-only screening protocols been followed with their patient population, fully 48% of infants with delayed TSH elevation would have been overlooked; half of these patients were later found to have decompensated hypothyroidism.
Neurodevelopmental disability caused by congenital hypothyroidism, which affects roughly 1 in every 2,000-4,000 births, is increasingly being prevented with newborn screenings that identify the condition early, but the incidence of congenital hypothyroidism has increased considerably in the past 20 years. The authors attribute this increase to the gradual change in screening cutoff levels and the rise in number of preterm infants who are surviving.
The need for a second screening, as well as appropriate timing and optimal TSH cutoff, are “subjects of active debate,” the authors wrote. The latest European screening guidelines recommend second screenings for preterm and low-birth-weight infants at either 2 weeks of age or 2 weeks following preliminary screening. Although they make no recommendations regarding additional screenings, American Academy of Pediatrics guidelines, published in 2006, cite a “disproportionate incidence” of delayed increase in TSH and congenital hypothyroidism in infants with very low birth weight. Dr. McGrath and her associates speculated that not all screening programs have adopted repeat screening of preterm infants, perhaps because of the low yield results, “the transient nature of most cases” detected, as well as conflicting long-term data on neurodevelopmental outcomes.
Dr. McGrath receives funding from the Children’s Fund for Health. The authors declared no conflicts of interest.
SOURCE: McGrath N et al. J Pediatr. 2018 Oct 24. doi: 10.1016/j.jpeds.2018.09.044.
FROM THE JOURNAL OF PEDIATRICS
Key clinical point: Periodic screenings are key to preventing permanent, decompensated hypothyroidism.
Major finding: High incidence of delayed TSH rise is common, especially in very-low-birth-weight infants.
Study details: A population-based prospective review of 898,424 records.
Disclosures: Dr. McGrath receives funding from the Children’s Fund for Health. The authors declared no conflicts of interest.
Source: McGrath N et al. J Pediatr. 2018 Oct 24. doi: 10.1016/j.jpeds.2018.09.044.
DoD and VA Sign Commitment to “Seamlessly” Sharing EHRs
Compatible electronic health record (EHR) systems at the US Department of Veterans Affairs (VA) and US Department of Defense (DoD) will ensure quality health care as service members transition to veterans, according to Defense Secretary James Mattis. The VA signed a contract with Cerner Corp last May to replace the 40-year-old Veterans Integrated System Technology Architecture (VistA) records system over the next 10 years with the new Cerner systems, which is in the pilot phase at DoD.
Mattis and VA Secretary Robert Wilkie signed a joint statement reinforcing the departments’ commitment to ensuring a successful transition from a legacy patient-data system to a modernized one. The statement represents “tangible evidence of our commitment to change how we deliver veteran-focused, provider-friendly care,” Wilkie said.
Both departments say the new EHR will be fully interoperable. Among the benefits: The collaboration will ensure that the VA understands the challenges encountered as DoD deploys Military Health System Genesis, its EHR system, the DoD says. It also will allow the VA to apply lessons learned to anticipate and mitigate known issues and assess prospective efficiencies to help deploy faster.
“The EHR will give health care providers a full picture of patient medical history, driving better clinical outcomes,” Wilkie said. “It will also help us identify veterans proactively who are at higher risk for issues, such as opioid addiction and suicide, so health care providers can intervene earlier and save lives.”
Compatible electronic health record (EHR) systems at the US Department of Veterans Affairs (VA) and US Department of Defense (DoD) will ensure quality health care as service members transition to veterans, according to Defense Secretary James Mattis. The VA signed a contract with Cerner Corp last May to replace the 40-year-old Veterans Integrated System Technology Architecture (VistA) records system over the next 10 years with the new Cerner systems, which is in the pilot phase at DoD.
Mattis and VA Secretary Robert Wilkie signed a joint statement reinforcing the departments’ commitment to ensuring a successful transition from a legacy patient-data system to a modernized one. The statement represents “tangible evidence of our commitment to change how we deliver veteran-focused, provider-friendly care,” Wilkie said.
Both departments say the new EHR will be fully interoperable. Among the benefits: The collaboration will ensure that the VA understands the challenges encountered as DoD deploys Military Health System Genesis, its EHR system, the DoD says. It also will allow the VA to apply lessons learned to anticipate and mitigate known issues and assess prospective efficiencies to help deploy faster.
“The EHR will give health care providers a full picture of patient medical history, driving better clinical outcomes,” Wilkie said. “It will also help us identify veterans proactively who are at higher risk for issues, such as opioid addiction and suicide, so health care providers can intervene earlier and save lives.”
Compatible electronic health record (EHR) systems at the US Department of Veterans Affairs (VA) and US Department of Defense (DoD) will ensure quality health care as service members transition to veterans, according to Defense Secretary James Mattis. The VA signed a contract with Cerner Corp last May to replace the 40-year-old Veterans Integrated System Technology Architecture (VistA) records system over the next 10 years with the new Cerner systems, which is in the pilot phase at DoD.
Mattis and VA Secretary Robert Wilkie signed a joint statement reinforcing the departments’ commitment to ensuring a successful transition from a legacy patient-data system to a modernized one. The statement represents “tangible evidence of our commitment to change how we deliver veteran-focused, provider-friendly care,” Wilkie said.
Both departments say the new EHR will be fully interoperable. Among the benefits: The collaboration will ensure that the VA understands the challenges encountered as DoD deploys Military Health System Genesis, its EHR system, the DoD says. It also will allow the VA to apply lessons learned to anticipate and mitigate known issues and assess prospective efficiencies to help deploy faster.
“The EHR will give health care providers a full picture of patient medical history, driving better clinical outcomes,” Wilkie said. “It will also help us identify veterans proactively who are at higher risk for issues, such as opioid addiction and suicide, so health care providers can intervene earlier and save lives.”
ACOG: First gynecologist visit between ages 13 and 15
The American College of Obstetricians and Gynecologists (ACOG) recommends that girls receive their first reproductive health visit between the ages of 13 and 15 years to discuss healthy relationships in addition to general reproductive health, according to a new committee opinion.
The recommendation, published online Oct. 24, emphasizes that such early visits provide opportunities for ob.gyns. to educate teenage girls and their guardians about age-appropriate health issues, such as sexual relationships, dating violence, and sexual coercion. Between the ages of 13 years and 15 years is an ideal window because middle school is a time that some adolescents develop their first romantic and sexual relationships (Obstet Gynecol. 2018; 132[5]:1317-18 doi: 10.1097/AOG.0000000000002946).
“Creating a nonjudgmental environment and educating staff on the unique concerns of adolescents are helpful ways to provide effective and appropriate care to this group of patients,” the authors wrote.
Ob.gyns. can use the early meeting to discuss key aspects of a healthy relationship with patients, including communication, self-respect, and mutual respect, while helping adolescents identify the characteristics of an unhealthy relationships such as dishonesty, intimidation, disrespect, and abuse, according to the opinion. As part of the discussion, ob.gyns. also may counsel patients to define their current relationship and their expectations for future relationships. Both relationships with and without sexual intimacy should be discussed, the opinion advises.
The recommendation reminds health care professionals to be mindful of federal and state confidentiality laws and that they be aware of mandatory reporting laws when intimate partner violence, teen dating violence, or statutory rape is suspected. In addition, the opinion notes that pregnant and parenting adolescents; lesbian, gay, bisexual, transgender, queer, or questioning (LGBTQ) individuals; and adolescents with physical and mental disabilities are at particular risk of disparities in the health care system.
“The promotion of healthy relationships in these groups requires the obstetrician-gynecologist to be aware of the unique barriers and hurdles to sexual and nonsexual expression, as well as to health care,” the opinion states. “Interventions to promote healthy relationships and a strong sexual health framework are more effective when started early and can affect indicators of long-term individual health and public health.”
The American College of Obstetricians and Gynecologists recommends that the first reproductive health visit occur between the ages of 13 and 15, and I agree with them. Often patients attending this appointment don’t have physical complaints, and we can focus on prevention and education. The visit can be about building the provider-patient relationship and may serve to ease fears and develop trust before visits for problem management.
There are a number of important health education topics to cover from puberty and menses to confidentiality and minor-access laws. Because many young people will begin to initiate romantic relationships during middle school, the topic of healthy relationships is critical. Unhealthy relationships, in their many forms, can have far reaching impacts on a young person’s health and wellness. For years, we’ve been talking with young people about preventing STIs or preventing unwanted pregnancy, but we’ve spent less energy working towards something.
I’m excited to see these recommendations and look forward to helping my younger patients think through relationships as important aspects of life and health, what they want from them, and how they can work towards them.
Melissa Kottke , MD is an obstetrician-gynecologist specializing in family planning and adolescent reproductive health at Emory University in Atlanta.
The American College of Obstetricians and Gynecologists recommends that the first reproductive health visit occur between the ages of 13 and 15, and I agree with them. Often patients attending this appointment don’t have physical complaints, and we can focus on prevention and education. The visit can be about building the provider-patient relationship and may serve to ease fears and develop trust before visits for problem management.
There are a number of important health education topics to cover from puberty and menses to confidentiality and minor-access laws. Because many young people will begin to initiate romantic relationships during middle school, the topic of healthy relationships is critical. Unhealthy relationships, in their many forms, can have far reaching impacts on a young person’s health and wellness. For years, we’ve been talking with young people about preventing STIs or preventing unwanted pregnancy, but we’ve spent less energy working towards something.
I’m excited to see these recommendations and look forward to helping my younger patients think through relationships as important aspects of life and health, what they want from them, and how they can work towards them.
Melissa Kottke , MD is an obstetrician-gynecologist specializing in family planning and adolescent reproductive health at Emory University in Atlanta.
The American College of Obstetricians and Gynecologists recommends that the first reproductive health visit occur between the ages of 13 and 15, and I agree with them. Often patients attending this appointment don’t have physical complaints, and we can focus on prevention and education. The visit can be about building the provider-patient relationship and may serve to ease fears and develop trust before visits for problem management.
There are a number of important health education topics to cover from puberty and menses to confidentiality and minor-access laws. Because many young people will begin to initiate romantic relationships during middle school, the topic of healthy relationships is critical. Unhealthy relationships, in their many forms, can have far reaching impacts on a young person’s health and wellness. For years, we’ve been talking with young people about preventing STIs or preventing unwanted pregnancy, but we’ve spent less energy working towards something.
I’m excited to see these recommendations and look forward to helping my younger patients think through relationships as important aspects of life and health, what they want from them, and how they can work towards them.
Melissa Kottke , MD is an obstetrician-gynecologist specializing in family planning and adolescent reproductive health at Emory University in Atlanta.
The American College of Obstetricians and Gynecologists (ACOG) recommends that girls receive their first reproductive health visit between the ages of 13 and 15 years to discuss healthy relationships in addition to general reproductive health, according to a new committee opinion.
The recommendation, published online Oct. 24, emphasizes that such early visits provide opportunities for ob.gyns. to educate teenage girls and their guardians about age-appropriate health issues, such as sexual relationships, dating violence, and sexual coercion. Between the ages of 13 years and 15 years is an ideal window because middle school is a time that some adolescents develop their first romantic and sexual relationships (Obstet Gynecol. 2018; 132[5]:1317-18 doi: 10.1097/AOG.0000000000002946).
“Creating a nonjudgmental environment and educating staff on the unique concerns of adolescents are helpful ways to provide effective and appropriate care to this group of patients,” the authors wrote.
Ob.gyns. can use the early meeting to discuss key aspects of a healthy relationship with patients, including communication, self-respect, and mutual respect, while helping adolescents identify the characteristics of an unhealthy relationships such as dishonesty, intimidation, disrespect, and abuse, according to the opinion. As part of the discussion, ob.gyns. also may counsel patients to define their current relationship and their expectations for future relationships. Both relationships with and without sexual intimacy should be discussed, the opinion advises.
The recommendation reminds health care professionals to be mindful of federal and state confidentiality laws and that they be aware of mandatory reporting laws when intimate partner violence, teen dating violence, or statutory rape is suspected. In addition, the opinion notes that pregnant and parenting adolescents; lesbian, gay, bisexual, transgender, queer, or questioning (LGBTQ) individuals; and adolescents with physical and mental disabilities are at particular risk of disparities in the health care system.
“The promotion of healthy relationships in these groups requires the obstetrician-gynecologist to be aware of the unique barriers and hurdles to sexual and nonsexual expression, as well as to health care,” the opinion states. “Interventions to promote healthy relationships and a strong sexual health framework are more effective when started early and can affect indicators of long-term individual health and public health.”
The American College of Obstetricians and Gynecologists (ACOG) recommends that girls receive their first reproductive health visit between the ages of 13 and 15 years to discuss healthy relationships in addition to general reproductive health, according to a new committee opinion.
The recommendation, published online Oct. 24, emphasizes that such early visits provide opportunities for ob.gyns. to educate teenage girls and their guardians about age-appropriate health issues, such as sexual relationships, dating violence, and sexual coercion. Between the ages of 13 years and 15 years is an ideal window because middle school is a time that some adolescents develop their first romantic and sexual relationships (Obstet Gynecol. 2018; 132[5]:1317-18 doi: 10.1097/AOG.0000000000002946).
“Creating a nonjudgmental environment and educating staff on the unique concerns of adolescents are helpful ways to provide effective and appropriate care to this group of patients,” the authors wrote.
Ob.gyns. can use the early meeting to discuss key aspects of a healthy relationship with patients, including communication, self-respect, and mutual respect, while helping adolescents identify the characteristics of an unhealthy relationships such as dishonesty, intimidation, disrespect, and abuse, according to the opinion. As part of the discussion, ob.gyns. also may counsel patients to define their current relationship and their expectations for future relationships. Both relationships with and without sexual intimacy should be discussed, the opinion advises.
The recommendation reminds health care professionals to be mindful of federal and state confidentiality laws and that they be aware of mandatory reporting laws when intimate partner violence, teen dating violence, or statutory rape is suspected. In addition, the opinion notes that pregnant and parenting adolescents; lesbian, gay, bisexual, transgender, queer, or questioning (LGBTQ) individuals; and adolescents with physical and mental disabilities are at particular risk of disparities in the health care system.
“The promotion of healthy relationships in these groups requires the obstetrician-gynecologist to be aware of the unique barriers and hurdles to sexual and nonsexual expression, as well as to health care,” the opinion states. “Interventions to promote healthy relationships and a strong sexual health framework are more effective when started early and can affect indicators of long-term individual health and public health.”
FROM OBSTETRICS & GYNECOLOGY
Full-dose quadrivalent flu vaccine shows increased efficacy in children
according to data from a randomized trial of nearly 2,000 children aged 6-35 months.
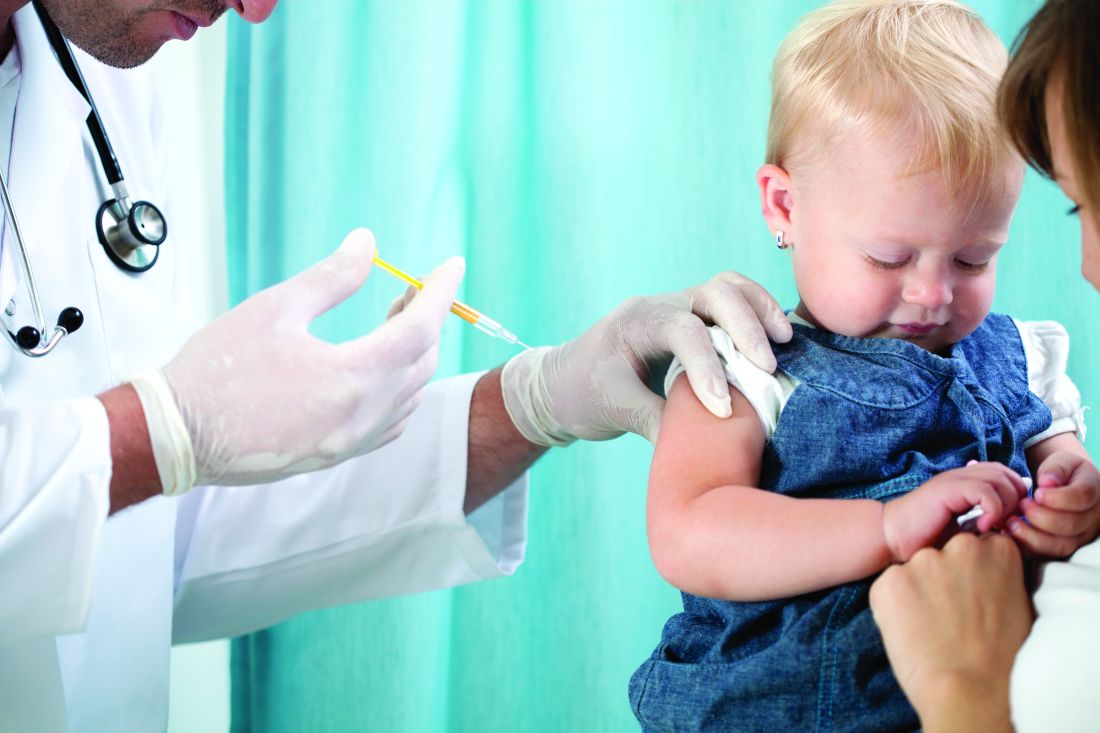
Data from previous studies have suggested that a full dose of vaccine may be more immunogenic in young children compared with a half dose, and Sanofi Pasteur has submitted a supplemental Biologics License Application to the Food and Drug Administration to allow use of the full 0.5-mL dose in children as young as 6 months, Monica Mercer, MD, of Sanofi Pasteur, said at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices in Atlanta.
Dr. Mercer presented findings from a phase IV randomized, observer-blinded study, in which the researchers assigned healthy children aged 6-35 months to receive Fluzone quadrivalent vaccine at a dose of 0.25 mL or 0.5 mL.
A total of 1,941 children (949 for the 0.25-mL dose and 992 for the 0.5-mL dose) were included in the safety analysis.
The most important safety outcome was to compare the rate of any fever, Dr. Mercer said at the meeting.
Overall, at 7 days after vaccination, the rate of fever was 11% for the half dose and 12% for the full dose, she said. The resulting difference of 0.84% met the criteria for noninferiority (less than 5%), she added.
In terms of safety, tenderness was the most frequently reported injection site reaction, noted in 47% of the half-dose group and 50% of the full-dose group. The rates of unsolicited adverse events were similar in both groups, the most common included diarrhea and cough, Dr. Mercer said.
No subjects in the full-dose group and three in the half-dose group discontinued the study because of adverse events. The only reported serious adverse event was one case of chronic urticaria in the half-dose group; no deaths were reported in either group.
As for efficacy, the full dose demonstrated noninferiority, compared with the half dose, against each of four strains: influenza A H1N1, influenza A H3N2, influenza B Victoria, and influenza B Yamagata. The geometric mean titers of the full and half doses for each of the four strains were, respectively, 310 and 214, 332 and 221, 348 and 261, and 349 and 243.
The potential action date for the supplemental Biologics License Application is January 2019, noted Dr. Mercer, who is employed by Sanofi Pasteur.
according to data from a randomized trial of nearly 2,000 children aged 6-35 months.
Data from previous studies have suggested that a full dose of vaccine may be more immunogenic in young children compared with a half dose, and Sanofi Pasteur has submitted a supplemental Biologics License Application to the Food and Drug Administration to allow use of the full 0.5-mL dose in children as young as 6 months, Monica Mercer, MD, of Sanofi Pasteur, said at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices in Atlanta.
Dr. Mercer presented findings from a phase IV randomized, observer-blinded study, in which the researchers assigned healthy children aged 6-35 months to receive Fluzone quadrivalent vaccine at a dose of 0.25 mL or 0.5 mL.
A total of 1,941 children (949 for the 0.25-mL dose and 992 for the 0.5-mL dose) were included in the safety analysis.
The most important safety outcome was to compare the rate of any fever, Dr. Mercer said at the meeting.
Overall, at 7 days after vaccination, the rate of fever was 11% for the half dose and 12% for the full dose, she said. The resulting difference of 0.84% met the criteria for noninferiority (less than 5%), she added.
In terms of safety, tenderness was the most frequently reported injection site reaction, noted in 47% of the half-dose group and 50% of the full-dose group. The rates of unsolicited adverse events were similar in both groups, the most common included diarrhea and cough, Dr. Mercer said.
No subjects in the full-dose group and three in the half-dose group discontinued the study because of adverse events. The only reported serious adverse event was one case of chronic urticaria in the half-dose group; no deaths were reported in either group.
As for efficacy, the full dose demonstrated noninferiority, compared with the half dose, against each of four strains: influenza A H1N1, influenza A H3N2, influenza B Victoria, and influenza B Yamagata. The geometric mean titers of the full and half doses for each of the four strains were, respectively, 310 and 214, 332 and 221, 348 and 261, and 349 and 243.
The potential action date for the supplemental Biologics License Application is January 2019, noted Dr. Mercer, who is employed by Sanofi Pasteur.
according to data from a randomized trial of nearly 2,000 children aged 6-35 months.
Data from previous studies have suggested that a full dose of vaccine may be more immunogenic in young children compared with a half dose, and Sanofi Pasteur has submitted a supplemental Biologics License Application to the Food and Drug Administration to allow use of the full 0.5-mL dose in children as young as 6 months, Monica Mercer, MD, of Sanofi Pasteur, said at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices in Atlanta.
Dr. Mercer presented findings from a phase IV randomized, observer-blinded study, in which the researchers assigned healthy children aged 6-35 months to receive Fluzone quadrivalent vaccine at a dose of 0.25 mL or 0.5 mL.
A total of 1,941 children (949 for the 0.25-mL dose and 992 for the 0.5-mL dose) were included in the safety analysis.
The most important safety outcome was to compare the rate of any fever, Dr. Mercer said at the meeting.
Overall, at 7 days after vaccination, the rate of fever was 11% for the half dose and 12% for the full dose, she said. The resulting difference of 0.84% met the criteria for noninferiority (less than 5%), she added.
In terms of safety, tenderness was the most frequently reported injection site reaction, noted in 47% of the half-dose group and 50% of the full-dose group. The rates of unsolicited adverse events were similar in both groups, the most common included diarrhea and cough, Dr. Mercer said.
No subjects in the full-dose group and three in the half-dose group discontinued the study because of adverse events. The only reported serious adverse event was one case of chronic urticaria in the half-dose group; no deaths were reported in either group.
As for efficacy, the full dose demonstrated noninferiority, compared with the half dose, against each of four strains: influenza A H1N1, influenza A H3N2, influenza B Victoria, and influenza B Yamagata. The geometric mean titers of the full and half doses for each of the four strains were, respectively, 310 and 214, 332 and 221, 348 and 261, and 349 and 243.
The potential action date for the supplemental Biologics License Application is January 2019, noted Dr. Mercer, who is employed by Sanofi Pasteur.
REPORTING FROM AN ACIP MEETING