User login
Migraine Linked with Increased Risk of Stroke, MI
Migraine headache was associated with an increased long-term risk of cardiovascular and cerebrovascular events, a recent study found. This effect was due to an increased risk of stroke (both ischemic and hemorrhagic) and myocardial infarction (MI). There was a moderate to severe degree of heterogeneity for the outcomes, which was partly explained by the presence of aura. A total of 16 cohort studies (18 study records) with 394,942 migraineurs and 757,465 non-migraineurs were analyzed. Researchers found:
- Migraine was associated with a higher risk of a major adverse cardiovascular and cerebrovascular event driven by a higher risk of stroke and MI.
- There was no difference in the risk of all-cause mortality, with a considerable degree of statistical heterogeneity between the studies.
- The presence of aura was an effect modifier for stroke and all-cause mortality.
Migraine and the risk of cardiovascular and cerebrovascular events: A meta-analysis of 16 cohort studies including 1,152,407 subjects. [Published online ahead of print March 27, 2018]. BMJ Open. doi:10.1136/bmjopen-2017-020498.
Migraine headache was associated with an increased long-term risk of cardiovascular and cerebrovascular events, a recent study found. This effect was due to an increased risk of stroke (both ischemic and hemorrhagic) and myocardial infarction (MI). There was a moderate to severe degree of heterogeneity for the outcomes, which was partly explained by the presence of aura. A total of 16 cohort studies (18 study records) with 394,942 migraineurs and 757,465 non-migraineurs were analyzed. Researchers found:
- Migraine was associated with a higher risk of a major adverse cardiovascular and cerebrovascular event driven by a higher risk of stroke and MI.
- There was no difference in the risk of all-cause mortality, with a considerable degree of statistical heterogeneity between the studies.
- The presence of aura was an effect modifier for stroke and all-cause mortality.
Migraine and the risk of cardiovascular and cerebrovascular events: A meta-analysis of 16 cohort studies including 1,152,407 subjects. [Published online ahead of print March 27, 2018]. BMJ Open. doi:10.1136/bmjopen-2017-020498.
Migraine headache was associated with an increased long-term risk of cardiovascular and cerebrovascular events, a recent study found. This effect was due to an increased risk of stroke (both ischemic and hemorrhagic) and myocardial infarction (MI). There was a moderate to severe degree of heterogeneity for the outcomes, which was partly explained by the presence of aura. A total of 16 cohort studies (18 study records) with 394,942 migraineurs and 757,465 non-migraineurs were analyzed. Researchers found:
- Migraine was associated with a higher risk of a major adverse cardiovascular and cerebrovascular event driven by a higher risk of stroke and MI.
- There was no difference in the risk of all-cause mortality, with a considerable degree of statistical heterogeneity between the studies.
- The presence of aura was an effect modifier for stroke and all-cause mortality.
Migraine and the risk of cardiovascular and cerebrovascular events: A meta-analysis of 16 cohort studies including 1,152,407 subjects. [Published online ahead of print March 27, 2018]. BMJ Open. doi:10.1136/bmjopen-2017-020498.
Migraine Common in Patients with Sarcoidosis
Migraine is a common comorbidity in patients with sarcoidosis, according to a recent study. As such, better recognition and targeted treatment of migraine has the potential to improve quality of life as part of a comprehensive care plan for those with sarcoidosis. The ID Migraine questionnaire was administered to a well-phenotyped observational cohort of patients with sarcoidosis (most of whom were seeking specialty care) and healthy controls. Predictors of migraine status were examined using univariate and multivariable logistic regression. Researchers found:
- Migraine was seen in 29% of 96 patients with sarcoidosis and 13% of 39 healthy controls.
- Among those with sarcoidosis, in univariate regression analysis only female sex was predictive of having migraine, and in a multivariable regression female sex remained significant.
There was no association between migraine and age, depression, dyspnea, immunosuppression use, or erythrocyte sedimentation rate.
Migraine is common in patients with sarcoidosis. [Published online ahead of print March 26, 2018]. Cephalalgia. doi:10.1177/0333102418768037.
Migraine is a common comorbidity in patients with sarcoidosis, according to a recent study. As such, better recognition and targeted treatment of migraine has the potential to improve quality of life as part of a comprehensive care plan for those with sarcoidosis. The ID Migraine questionnaire was administered to a well-phenotyped observational cohort of patients with sarcoidosis (most of whom were seeking specialty care) and healthy controls. Predictors of migraine status were examined using univariate and multivariable logistic regression. Researchers found:
- Migraine was seen in 29% of 96 patients with sarcoidosis and 13% of 39 healthy controls.
- Among those with sarcoidosis, in univariate regression analysis only female sex was predictive of having migraine, and in a multivariable regression female sex remained significant.
There was no association between migraine and age, depression, dyspnea, immunosuppression use, or erythrocyte sedimentation rate.
Migraine is common in patients with sarcoidosis. [Published online ahead of print March 26, 2018]. Cephalalgia. doi:10.1177/0333102418768037.
Migraine is a common comorbidity in patients with sarcoidosis, according to a recent study. As such, better recognition and targeted treatment of migraine has the potential to improve quality of life as part of a comprehensive care plan for those with sarcoidosis. The ID Migraine questionnaire was administered to a well-phenotyped observational cohort of patients with sarcoidosis (most of whom were seeking specialty care) and healthy controls. Predictors of migraine status were examined using univariate and multivariable logistic regression. Researchers found:
- Migraine was seen in 29% of 96 patients with sarcoidosis and 13% of 39 healthy controls.
- Among those with sarcoidosis, in univariate regression analysis only female sex was predictive of having migraine, and in a multivariable regression female sex remained significant.
There was no association between migraine and age, depression, dyspnea, immunosuppression use, or erythrocyte sedimentation rate.
Migraine is common in patients with sarcoidosis. [Published online ahead of print March 26, 2018]. Cephalalgia. doi:10.1177/0333102418768037.
Fibrosis-related genes are dysregulated in HCV-induced liver disease
Two major pathways exhibited high dysregulation in early liver fibrosis compared with controls or compared with late liver fibrosis – the transforming growth factor beta (TGF-beta)–related pathway genes and Matrix deposition–associated genes, according to an online report in the journal Gene.
The study examined 105 treatment naive HCV genotype 4–infected patients and 16 healthy subjects. The gene-regulation assays were done via PCR arrays on 84 fibrosis-related genes followed by customization of a smaller array consisting of 11 genes that were designed on the bases of results obtained from the larger array. Genes that displayed significant dysregulation at mRNA levels were validated at protein levels, according to the authors.
The researchers found that the expression at protein levels confirmed the RNA data, thereby excluding dysregulation at posttranscriptional levels.
“We assume that the overall expression pattern of ECM molecules described in the present study may be utilized for a prognostic transcriptomic or proteomic signatures for staging of liver fibrosis,” the authors concluded.
SOURCE: Dawood RM et al. Gene 2018 Apr 21. doi: 10.1016/j.gene.2018.04.032.
Two major pathways exhibited high dysregulation in early liver fibrosis compared with controls or compared with late liver fibrosis – the transforming growth factor beta (TGF-beta)–related pathway genes and Matrix deposition–associated genes, according to an online report in the journal Gene.
The study examined 105 treatment naive HCV genotype 4–infected patients and 16 healthy subjects. The gene-regulation assays were done via PCR arrays on 84 fibrosis-related genes followed by customization of a smaller array consisting of 11 genes that were designed on the bases of results obtained from the larger array. Genes that displayed significant dysregulation at mRNA levels were validated at protein levels, according to the authors.
The researchers found that the expression at protein levels confirmed the RNA data, thereby excluding dysregulation at posttranscriptional levels.
“We assume that the overall expression pattern of ECM molecules described in the present study may be utilized for a prognostic transcriptomic or proteomic signatures for staging of liver fibrosis,” the authors concluded.
SOURCE: Dawood RM et al. Gene 2018 Apr 21. doi: 10.1016/j.gene.2018.04.032.
Two major pathways exhibited high dysregulation in early liver fibrosis compared with controls or compared with late liver fibrosis – the transforming growth factor beta (TGF-beta)–related pathway genes and Matrix deposition–associated genes, according to an online report in the journal Gene.
The study examined 105 treatment naive HCV genotype 4–infected patients and 16 healthy subjects. The gene-regulation assays were done via PCR arrays on 84 fibrosis-related genes followed by customization of a smaller array consisting of 11 genes that were designed on the bases of results obtained from the larger array. Genes that displayed significant dysregulation at mRNA levels were validated at protein levels, according to the authors.
The researchers found that the expression at protein levels confirmed the RNA data, thereby excluding dysregulation at posttranscriptional levels.
“We assume that the overall expression pattern of ECM molecules described in the present study may be utilized for a prognostic transcriptomic or proteomic signatures for staging of liver fibrosis,” the authors concluded.
SOURCE: Dawood RM et al. Gene 2018 Apr 21. doi: 10.1016/j.gene.2018.04.032.
FROM GENE
New point-of-care HCV assay shows promise for developing world
Researchers have developed a new portable point-of-care (PoC) molecular test for hepatitis C virus (HCV), with sensitivity and specificity that fulfills the recent FIND/WHO Target Product Profile for HCV decentralized testing in low- and middle-income countries, according to an online report in the journal Gut.

The new assay identified all major HCV genotypes, with a limit of detection of 2,362 IU/mL. In the PoC-HCV Genedrive Viral Detection Assay Validation Study (NCT02992184), 422 patients chronically infected with HCV and 503 controls negative for anti-HCV and HCV RNA were assayed with the device. The Genedrive HCV assay showed 98.6% sensitivity and 100% specificity to detect HCV, the researchers reported. The test was further validated in a small clinical setting in a resource-limited country, they added.
“The next step with the Genedrive HCV assay requires prospective validation in real-life decentralized settings in low-income and middle-income countries,” the authors concluded.
SOURCE: Llibre A et al. Gut 2018 Apr 3. doi: 10.1136/gutjnl-2017-315783.
Researchers have developed a new portable point-of-care (PoC) molecular test for hepatitis C virus (HCV), with sensitivity and specificity that fulfills the recent FIND/WHO Target Product Profile for HCV decentralized testing in low- and middle-income countries, according to an online report in the journal Gut.
The new assay identified all major HCV genotypes, with a limit of detection of 2,362 IU/mL. In the PoC-HCV Genedrive Viral Detection Assay Validation Study (NCT02992184), 422 patients chronically infected with HCV and 503 controls negative for anti-HCV and HCV RNA were assayed with the device. The Genedrive HCV assay showed 98.6% sensitivity and 100% specificity to detect HCV, the researchers reported. The test was further validated in a small clinical setting in a resource-limited country, they added.
“The next step with the Genedrive HCV assay requires prospective validation in real-life decentralized settings in low-income and middle-income countries,” the authors concluded.
SOURCE: Llibre A et al. Gut 2018 Apr 3. doi: 10.1136/gutjnl-2017-315783.
Researchers have developed a new portable point-of-care (PoC) molecular test for hepatitis C virus (HCV), with sensitivity and specificity that fulfills the recent FIND/WHO Target Product Profile for HCV decentralized testing in low- and middle-income countries, according to an online report in the journal Gut.
The new assay identified all major HCV genotypes, with a limit of detection of 2,362 IU/mL. In the PoC-HCV Genedrive Viral Detection Assay Validation Study (NCT02992184), 422 patients chronically infected with HCV and 503 controls negative for anti-HCV and HCV RNA were assayed with the device. The Genedrive HCV assay showed 98.6% sensitivity and 100% specificity to detect HCV, the researchers reported. The test was further validated in a small clinical setting in a resource-limited country, they added.
“The next step with the Genedrive HCV assay requires prospective validation in real-life decentralized settings in low-income and middle-income countries,” the authors concluded.
SOURCE: Llibre A et al. Gut 2018 Apr 3. doi: 10.1136/gutjnl-2017-315783.
FROM GUT
MDedge Daily News: Which nonopioids are ripe for abuse?
There’s new clarity on multiple sclerosis therapy. How infections boost Sjogren’s syndrome risk. And bum kidneys shouldn’t stop dabigatran reversal.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
There’s new clarity on multiple sclerosis therapy. How infections boost Sjogren’s syndrome risk. And bum kidneys shouldn’t stop dabigatran reversal.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
There’s new clarity on multiple sclerosis therapy. How infections boost Sjogren’s syndrome risk. And bum kidneys shouldn’t stop dabigatran reversal.
Listen to the MDedge Daily News podcast for all the details on today’s top news.

Value-Based Purchasing for Hospital-Acquired Venous Thromboembolism: Too Much, Too Soon
As a hospital-acquired condition responsible for a significant share of preventable deaths in the United States,1 venous thromboembolism (VTE) prevention should remain a high priority for healthcare organizations. Pursuant to the goal of reducing the frequency of this and other hospital-acquired conditions, several performance measures have been developed by third-party payers in the United States to provide incentives for inpatients to receive prophylaxis measures appropriate to their specific level of risk. Perhaps the best known of these is the Hospital Value-Based Purchasing Program, initiated by the Center for Medicare and Medicaid Studies (CMS) in 2013 as a provision of the Affordable Care Act.2 The Joint Commission, as steward of the 6 VTE-related measures,3 dictates the criteria for assessing performance. However, recent adjustments to one of these measures have been performed in such a way that neglects real-world considerations faced by providers and threatens to delegitimize the important role that value-based purchasing should have in reimbursement.
Effective in 2017, the guidelines pertaining to abstraction-based reporting added a new component to the VTE-6 measure, which applies to those inpatients not ordered to receive mechanical or pharmacologic prophylaxis who go on to suffer VTE. Specifically, it is concerned with how accurately hospitals stratify such patients as low risk before the decision is made to not order either method of prophylaxis. With the update, to satisfy the measure, a formal assessment confirming a patient’s low-risk status must have been documented between arrival and the time the VTE diagnostic test was performed. The guidelines explicitly note that only 3 risk assessment models (RAMs) are accepted, including the Caprini DVT Prediction Score, Padua Prediction Score, and IMPROVE VTE Risk Score.4 The rationale for this addition to the measure clearly is to protect patients from being incorrectly designated as low risk and subsequently receiving inadequate prophylaxis that could increase their likelihood of developing preventable VTE. Unfortunately, in its current form, it imposes a substantial burden on providers and healthcare organizations, without much promise of significantly reducing rates of this pervasive threat to patient safety.
LIMITATIONS
Although the aim of reducing the incidence of VTE is laudable, this updated requirement for VTE-6 is problematic on several levels. First, there is considerable uncertainty regarding how to implement the RAMs clinically in a user-friendly way that is conducive to their intended use. Due to limitations in most computerized physician order entry systems, it is not feasible to mandate the RAMs for only those patients not ordered for VTE prophylaxis (nor would it be sensible to restrict performing the assessment to low-risk patients, as the point of RAMs is to help risk stratify and not simply validate whatever determinations were already made by other means). As virtually every class of inpatient has some risk of VTE development, these factors effectively require that a score be tabulated on all admitted patients, giving the measure an enormous footprint on clinical operations. This is important because the permissible RAMs can sometimes be quite burdensome to complete faithfully. For instance, the Caprini Score necessitates the fairly prodigious collection and input of up to 26 data points. Some of the questions require exceedingly granular data, such as whether there is any “history of unexplained stillborn infant, recurrent spontaneous abortion (more than 3), premature birth with toxemia or growth restricted infant.”5 This clearly is far outside the scope of most focused admission assessments. Already deluged with the number of clicks inherent to the workflow of most electronic health records,6 it seems likely that some providers default to selecting “no” for such prompts as a time-saving measure, potentially sabotaging the goal of linking patients with a risk-appropriate method of prophylaxis. Meanwhile, those who are diligent about completing the assessment honestly will find themselves rewarded with less time to dedicate to other critical aspects of patient care.7
The small number of RAMs accepted under the measure also fails to account for the breadth of clinical circumstances providers faced. Although the permitted models are validated in certain patient populations, they exclude some that might be better suited for many practice environments. The University of California San Diego “3 bucket” design, for instance, has been shown to result in high levels of risk-appropriate prophylaxis, has high inter-user agreement, and perhaps most importantly, is relatively quick and easy to use.8 Also critical, it is easier to integrate into the admission workflow for under-resourced hospitals that might not have the ability to incorporate a point-based risk score calculator into their electronic health records.
Finally, the relative abruptness with which the changes were made complicated the task for institutions to integrate the RAMs into their applicable order sets in a user-friendly fashion. The new guidelines were released only 6 months before taking effect,9 and the RAM requirement was not widely advertised. This left a fairly short window that does not seem to reflect an understanding by the Joint Commission of the process required by hospitals to make such a transition responsibly. This should involve obtaining inputs from multiple specialty stakeholders on which RAM to employ, working with information system specialists on how to restructure key order sets, and education of end-users on how to apply them correctly.10
RECOMMENDATIONS
For these reasons, the rollout of the VTE-6 update falls well short of its ambitions. Satisfying the measure necessitates a substantial investment of time and effort by providers and yet forcing the use of such decidedly imperfect RAMs could paradoxically worsen accurate risk stratification and appropriate use of prophylaxis. Also, while it represents only a small slice of pay-for-performance initiatives, its broader impact should not be underestimated. Unlike many of the more specific items, the VTE measures affect the workflow related to virtually all hospitalized patients. Therefore, it is imperative that regulators “get it right,” as it might only take one poorly conceived mandate of this type to risk permanently souring providers and hospitals on the idea of value-based purchasing. The Joint Commission and CMS ought to seriously consider retracting the new provisions until the role of RAMs for VTE prevention is better understood. This would buy time to reconfigure the measure in a way that is compatible with actual clinical care and for hospitals to thoughtfully design how new requirements can best be implemented.
Disclosurses
The author has nothing to disclose.
1. Clagett GP, Anderson FA Jr, Heit J, Levine MN, Wheeler HB. Prevention of venous thromboembolism. Chest. 1995;108(4 Suppl):312S-334S. PubMed
2. Center for Medicare and Medicaid Studies. Hospital value based purchasing. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/downloads/Hospital_VBPurchasing_Fact_Sheet_ICN907664.pdf. Accessed December 18, 2017.
3. The Joint Commission. Specifications manual for national hospital inpatient quality measures. https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
4. The Joint Commission. Specifications manual for national hospital inpatient quality measures. https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
5. Venous Resource Center. Caprini score: DVT risk assessment. https://venousdisease.com/dvt-risk-assessment-online. Accessed December 19, 2017.
6. Hill RG, Sears LM, Melanson SW. 4000 Clicks: A productivity analysis of electronic medical records in a community hospital ED. Am J Emerg Med. 2013;31(11):1591-1594. PubMed
7. Clynch N, Kellett J. Medical documentation: Part of the solution, or part of the problem? A narrative review of the literature on the time spent on and value of medical documentation. Int J Med Inform. 2015;84(4):221-228. PubMed
8. Maynard GA, Morris TA, Jenkins IH, et al. Optimizing prevention of hospital-acquired venous thromboembolism (VTE): Prospective validation of a VTE risk assessment model. J Hosp Med. 2010;5(1):10-18. PubMed
9. The Joint Commission. Specifications manual for national hospital inpatient quality measures release notes v5.2. Available at: https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
10. Agency for Healthcare Quality and Research. Preventing hospital acquired venous thromboembolism: A guide for effective quality improvement. Available at: https://www.ahrq.gov/sites/default/files/publications/files/vteguide.pdf. Accessed December 18, 2017.
As a hospital-acquired condition responsible for a significant share of preventable deaths in the United States,1 venous thromboembolism (VTE) prevention should remain a high priority for healthcare organizations. Pursuant to the goal of reducing the frequency of this and other hospital-acquired conditions, several performance measures have been developed by third-party payers in the United States to provide incentives for inpatients to receive prophylaxis measures appropriate to their specific level of risk. Perhaps the best known of these is the Hospital Value-Based Purchasing Program, initiated by the Center for Medicare and Medicaid Studies (CMS) in 2013 as a provision of the Affordable Care Act.2 The Joint Commission, as steward of the 6 VTE-related measures,3 dictates the criteria for assessing performance. However, recent adjustments to one of these measures have been performed in such a way that neglects real-world considerations faced by providers and threatens to delegitimize the important role that value-based purchasing should have in reimbursement.
Effective in 2017, the guidelines pertaining to abstraction-based reporting added a new component to the VTE-6 measure, which applies to those inpatients not ordered to receive mechanical or pharmacologic prophylaxis who go on to suffer VTE. Specifically, it is concerned with how accurately hospitals stratify such patients as low risk before the decision is made to not order either method of prophylaxis. With the update, to satisfy the measure, a formal assessment confirming a patient’s low-risk status must have been documented between arrival and the time the VTE diagnostic test was performed. The guidelines explicitly note that only 3 risk assessment models (RAMs) are accepted, including the Caprini DVT Prediction Score, Padua Prediction Score, and IMPROVE VTE Risk Score.4 The rationale for this addition to the measure clearly is to protect patients from being incorrectly designated as low risk and subsequently receiving inadequate prophylaxis that could increase their likelihood of developing preventable VTE. Unfortunately, in its current form, it imposes a substantial burden on providers and healthcare organizations, without much promise of significantly reducing rates of this pervasive threat to patient safety.
LIMITATIONS
Although the aim of reducing the incidence of VTE is laudable, this updated requirement for VTE-6 is problematic on several levels. First, there is considerable uncertainty regarding how to implement the RAMs clinically in a user-friendly way that is conducive to their intended use. Due to limitations in most computerized physician order entry systems, it is not feasible to mandate the RAMs for only those patients not ordered for VTE prophylaxis (nor would it be sensible to restrict performing the assessment to low-risk patients, as the point of RAMs is to help risk stratify and not simply validate whatever determinations were already made by other means). As virtually every class of inpatient has some risk of VTE development, these factors effectively require that a score be tabulated on all admitted patients, giving the measure an enormous footprint on clinical operations. This is important because the permissible RAMs can sometimes be quite burdensome to complete faithfully. For instance, the Caprini Score necessitates the fairly prodigious collection and input of up to 26 data points. Some of the questions require exceedingly granular data, such as whether there is any “history of unexplained stillborn infant, recurrent spontaneous abortion (more than 3), premature birth with toxemia or growth restricted infant.”5 This clearly is far outside the scope of most focused admission assessments. Already deluged with the number of clicks inherent to the workflow of most electronic health records,6 it seems likely that some providers default to selecting “no” for such prompts as a time-saving measure, potentially sabotaging the goal of linking patients with a risk-appropriate method of prophylaxis. Meanwhile, those who are diligent about completing the assessment honestly will find themselves rewarded with less time to dedicate to other critical aspects of patient care.7
The small number of RAMs accepted under the measure also fails to account for the breadth of clinical circumstances providers faced. Although the permitted models are validated in certain patient populations, they exclude some that might be better suited for many practice environments. The University of California San Diego “3 bucket” design, for instance, has been shown to result in high levels of risk-appropriate prophylaxis, has high inter-user agreement, and perhaps most importantly, is relatively quick and easy to use.8 Also critical, it is easier to integrate into the admission workflow for under-resourced hospitals that might not have the ability to incorporate a point-based risk score calculator into their electronic health records.
Finally, the relative abruptness with which the changes were made complicated the task for institutions to integrate the RAMs into their applicable order sets in a user-friendly fashion. The new guidelines were released only 6 months before taking effect,9 and the RAM requirement was not widely advertised. This left a fairly short window that does not seem to reflect an understanding by the Joint Commission of the process required by hospitals to make such a transition responsibly. This should involve obtaining inputs from multiple specialty stakeholders on which RAM to employ, working with information system specialists on how to restructure key order sets, and education of end-users on how to apply them correctly.10
RECOMMENDATIONS
For these reasons, the rollout of the VTE-6 update falls well short of its ambitions. Satisfying the measure necessitates a substantial investment of time and effort by providers and yet forcing the use of such decidedly imperfect RAMs could paradoxically worsen accurate risk stratification and appropriate use of prophylaxis. Also, while it represents only a small slice of pay-for-performance initiatives, its broader impact should not be underestimated. Unlike many of the more specific items, the VTE measures affect the workflow related to virtually all hospitalized patients. Therefore, it is imperative that regulators “get it right,” as it might only take one poorly conceived mandate of this type to risk permanently souring providers and hospitals on the idea of value-based purchasing. The Joint Commission and CMS ought to seriously consider retracting the new provisions until the role of RAMs for VTE prevention is better understood. This would buy time to reconfigure the measure in a way that is compatible with actual clinical care and for hospitals to thoughtfully design how new requirements can best be implemented.
Disclosurses
The author has nothing to disclose.
As a hospital-acquired condition responsible for a significant share of preventable deaths in the United States,1 venous thromboembolism (VTE) prevention should remain a high priority for healthcare organizations. Pursuant to the goal of reducing the frequency of this and other hospital-acquired conditions, several performance measures have been developed by third-party payers in the United States to provide incentives for inpatients to receive prophylaxis measures appropriate to their specific level of risk. Perhaps the best known of these is the Hospital Value-Based Purchasing Program, initiated by the Center for Medicare and Medicaid Studies (CMS) in 2013 as a provision of the Affordable Care Act.2 The Joint Commission, as steward of the 6 VTE-related measures,3 dictates the criteria for assessing performance. However, recent adjustments to one of these measures have been performed in such a way that neglects real-world considerations faced by providers and threatens to delegitimize the important role that value-based purchasing should have in reimbursement.
Effective in 2017, the guidelines pertaining to abstraction-based reporting added a new component to the VTE-6 measure, which applies to those inpatients not ordered to receive mechanical or pharmacologic prophylaxis who go on to suffer VTE. Specifically, it is concerned with how accurately hospitals stratify such patients as low risk before the decision is made to not order either method of prophylaxis. With the update, to satisfy the measure, a formal assessment confirming a patient’s low-risk status must have been documented between arrival and the time the VTE diagnostic test was performed. The guidelines explicitly note that only 3 risk assessment models (RAMs) are accepted, including the Caprini DVT Prediction Score, Padua Prediction Score, and IMPROVE VTE Risk Score.4 The rationale for this addition to the measure clearly is to protect patients from being incorrectly designated as low risk and subsequently receiving inadequate prophylaxis that could increase their likelihood of developing preventable VTE. Unfortunately, in its current form, it imposes a substantial burden on providers and healthcare organizations, without much promise of significantly reducing rates of this pervasive threat to patient safety.
LIMITATIONS
Although the aim of reducing the incidence of VTE is laudable, this updated requirement for VTE-6 is problematic on several levels. First, there is considerable uncertainty regarding how to implement the RAMs clinically in a user-friendly way that is conducive to their intended use. Due to limitations in most computerized physician order entry systems, it is not feasible to mandate the RAMs for only those patients not ordered for VTE prophylaxis (nor would it be sensible to restrict performing the assessment to low-risk patients, as the point of RAMs is to help risk stratify and not simply validate whatever determinations were already made by other means). As virtually every class of inpatient has some risk of VTE development, these factors effectively require that a score be tabulated on all admitted patients, giving the measure an enormous footprint on clinical operations. This is important because the permissible RAMs can sometimes be quite burdensome to complete faithfully. For instance, the Caprini Score necessitates the fairly prodigious collection and input of up to 26 data points. Some of the questions require exceedingly granular data, such as whether there is any “history of unexplained stillborn infant, recurrent spontaneous abortion (more than 3), premature birth with toxemia or growth restricted infant.”5 This clearly is far outside the scope of most focused admission assessments. Already deluged with the number of clicks inherent to the workflow of most electronic health records,6 it seems likely that some providers default to selecting “no” for such prompts as a time-saving measure, potentially sabotaging the goal of linking patients with a risk-appropriate method of prophylaxis. Meanwhile, those who are diligent about completing the assessment honestly will find themselves rewarded with less time to dedicate to other critical aspects of patient care.7
The small number of RAMs accepted under the measure also fails to account for the breadth of clinical circumstances providers faced. Although the permitted models are validated in certain patient populations, they exclude some that might be better suited for many practice environments. The University of California San Diego “3 bucket” design, for instance, has been shown to result in high levels of risk-appropriate prophylaxis, has high inter-user agreement, and perhaps most importantly, is relatively quick and easy to use.8 Also critical, it is easier to integrate into the admission workflow for under-resourced hospitals that might not have the ability to incorporate a point-based risk score calculator into their electronic health records.
Finally, the relative abruptness with which the changes were made complicated the task for institutions to integrate the RAMs into their applicable order sets in a user-friendly fashion. The new guidelines were released only 6 months before taking effect,9 and the RAM requirement was not widely advertised. This left a fairly short window that does not seem to reflect an understanding by the Joint Commission of the process required by hospitals to make such a transition responsibly. This should involve obtaining inputs from multiple specialty stakeholders on which RAM to employ, working with information system specialists on how to restructure key order sets, and education of end-users on how to apply them correctly.10
RECOMMENDATIONS
For these reasons, the rollout of the VTE-6 update falls well short of its ambitions. Satisfying the measure necessitates a substantial investment of time and effort by providers and yet forcing the use of such decidedly imperfect RAMs could paradoxically worsen accurate risk stratification and appropriate use of prophylaxis. Also, while it represents only a small slice of pay-for-performance initiatives, its broader impact should not be underestimated. Unlike many of the more specific items, the VTE measures affect the workflow related to virtually all hospitalized patients. Therefore, it is imperative that regulators “get it right,” as it might only take one poorly conceived mandate of this type to risk permanently souring providers and hospitals on the idea of value-based purchasing. The Joint Commission and CMS ought to seriously consider retracting the new provisions until the role of RAMs for VTE prevention is better understood. This would buy time to reconfigure the measure in a way that is compatible with actual clinical care and for hospitals to thoughtfully design how new requirements can best be implemented.
Disclosurses
The author has nothing to disclose.
1. Clagett GP, Anderson FA Jr, Heit J, Levine MN, Wheeler HB. Prevention of venous thromboembolism. Chest. 1995;108(4 Suppl):312S-334S. PubMed
2. Center for Medicare and Medicaid Studies. Hospital value based purchasing. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/downloads/Hospital_VBPurchasing_Fact_Sheet_ICN907664.pdf. Accessed December 18, 2017.
3. The Joint Commission. Specifications manual for national hospital inpatient quality measures. https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
4. The Joint Commission. Specifications manual for national hospital inpatient quality measures. https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
5. Venous Resource Center. Caprini score: DVT risk assessment. https://venousdisease.com/dvt-risk-assessment-online. Accessed December 19, 2017.
6. Hill RG, Sears LM, Melanson SW. 4000 Clicks: A productivity analysis of electronic medical records in a community hospital ED. Am J Emerg Med. 2013;31(11):1591-1594. PubMed
7. Clynch N, Kellett J. Medical documentation: Part of the solution, or part of the problem? A narrative review of the literature on the time spent on and value of medical documentation. Int J Med Inform. 2015;84(4):221-228. PubMed
8. Maynard GA, Morris TA, Jenkins IH, et al. Optimizing prevention of hospital-acquired venous thromboembolism (VTE): Prospective validation of a VTE risk assessment model. J Hosp Med. 2010;5(1):10-18. PubMed
9. The Joint Commission. Specifications manual for national hospital inpatient quality measures release notes v5.2. Available at: https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
10. Agency for Healthcare Quality and Research. Preventing hospital acquired venous thromboembolism: A guide for effective quality improvement. Available at: https://www.ahrq.gov/sites/default/files/publications/files/vteguide.pdf. Accessed December 18, 2017.
1. Clagett GP, Anderson FA Jr, Heit J, Levine MN, Wheeler HB. Prevention of venous thromboembolism. Chest. 1995;108(4 Suppl):312S-334S. PubMed
2. Center for Medicare and Medicaid Studies. Hospital value based purchasing. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/downloads/Hospital_VBPurchasing_Fact_Sheet_ICN907664.pdf. Accessed December 18, 2017.
3. The Joint Commission. Specifications manual for national hospital inpatient quality measures. https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
4. The Joint Commission. Specifications manual for national hospital inpatient quality measures. https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
5. Venous Resource Center. Caprini score: DVT risk assessment. https://venousdisease.com/dvt-risk-assessment-online. Accessed December 19, 2017.
6. Hill RG, Sears LM, Melanson SW. 4000 Clicks: A productivity analysis of electronic medical records in a community hospital ED. Am J Emerg Med. 2013;31(11):1591-1594. PubMed
7. Clynch N, Kellett J. Medical documentation: Part of the solution, or part of the problem? A narrative review of the literature on the time spent on and value of medical documentation. Int J Med Inform. 2015;84(4):221-228. PubMed
8. Maynard GA, Morris TA, Jenkins IH, et al. Optimizing prevention of hospital-acquired venous thromboembolism (VTE): Prospective validation of a VTE risk assessment model. J Hosp Med. 2010;5(1):10-18. PubMed
9. The Joint Commission. Specifications manual for national hospital inpatient quality measures release notes v5.2. Available at: https://www.jointcommission.org/specifications_manual_for_national_hospital_inpatient_quality_measures.aspx. Accessed December 18, 2017.
10. Agency for Healthcare Quality and Research. Preventing hospital acquired venous thromboembolism: A guide for effective quality improvement. Available at: https://www.ahrq.gov/sites/default/files/publications/files/vteguide.pdf. Accessed December 18, 2017.
© 2018 Society of Hospital Medicine
Hospital Readmissions in Patients with Cirrhosis: A Systematic Review
Cirrhosis is a morbid condition characterized by complications such as ascites, gastrointestinal bleeding, and hepatic encephalopathy. These complications frequently require hospitalization, which is a substantial burden to the healthcare system. In 2012, liver disease was responsible for nearly 250,000 admissions across the United States, costing $3 billion.1 Despite this substantial resource utilization, outcomes remain poor, with an inpatient mortality of 6%. For those that survive, many experience hospital readmission.
More generally, early readmission reflects poor quality of care in the US. In 2004, 30-day readmissions occurred in nearly 20% of Medicare beneficiaries and costed over $17 billion.2 In response to this problem, the Affordable Care Act established the Hospital Readmissions Reduction Program (HRRP), which reduces Centers for Medicare & Medicaid Services (CMS) payments to hospitals with excess 30-day readmissions for high-risk conditions, including pneumonia and heart failure.3 Heart failure, in particular, has been the subject of numerous studies detailing risk factors and interventions to predict and prevent readmission.4-6 Based on this extensive evidence, guidelines recommend disease management programs to reduce readmissions in this population.7 In contrast, readmission in the cirrhosis population has received limited attention.
We therefore conducted a systematic review aiming to examine the range of readmission risk noted in the literature, with a focus on the model for end-stage liver disease (MELD) score as a risk factor for readmission.
METHODS
Search Strategy
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines for conducting and reporting systematic reviews.8 A literature search was performed by a medical librarian using the following databases: Ovid MEDLINE, PubMed, EMBASE, CINAHL, the full Cochrane Library, Scopus, Google Scholar, and ClinicalTrials.gov. All the databases were searched from 2000 to May 2017. We did not include older reports because the review focused on contemporary care; earlier studies may not reflect current cirrhosis management. To ensure literature saturation, included articles’ reference lists were reviewed.
Search strategies were developed by combining database-specific subject headings and keywords for readmissions with those for cirrhosis or its complications (Supplementary Material). Google Scholar and ClinicalTrials.gov were searched using keywords only. All results were limited to the English language and those published in 2000 or later, but no other limits were applied.
Identified records were reviewed based on strict criteria. We excluded case reports, case series, reviews, editorials, letters, and meeting abstracts without final peer-reviewed publication. We also excluded studies of pediatric populations (age < 18 years), patients without cirrhosis, and patients with liver transplants. We excluded studies in which patients were not hospitalized at study onset and those where the index admission was for an elective procedure. Because our interest was to identify factors associated with early readmission, we excluded studies that did not report readmissions within 90 days or those with a mean or median follow-up of less than 30 days. We also excluded studies that did not examine the association between readmission and at least 1 independent variable or intervention. Duplicate reports of a common sample were excluded unless the duplicate provided additional information, and such reports were examined together in our synthesis.
Two authors identified potentially eligible records by independently screening titles and abstracts. At this stage, records that did not meet the eligibility criteria were excluded, and the reasons for exclusion were not recorded. Records with disagreement were retained for full-text review. After this initial exclusion of records, the remaining full-text records were reviewed independently. For this full-text review, we recorded exclusion reasons and disagreements were resolved through discussion.
Data Collection
Data were abstracted from each study by 2 authors independently and recorded in a REDCap database.9 Discrepancies were resolved through discussion. We recorded study characteristics, including study design, setting, population (including the inclusion/exclusion criteria, sample size, and patient and hospitalization characteristics), interventions, and comparisons. To facilitate comparisons across studies, we employed validated methods to approximate means and standard deviations (SD).10 We recorded detailed information on outcomes including readmissions, preventability, independent variables, and mortality. Studies that focused on a single independent factor or intervention were classified as “focused,” while those that examined multiple factors were classified as “broad.” We used the Newcastle–Ottawa Scale to assess the risk of bias in each study.11 This instrument uses a 9-point scale to gauge methodological quality based on selection, group comparability, and exposure/outcome assessment.
Statistical Analysis
Analyses were performed using Stata 13.1 (StataCorp LP, College Station, Texas). We determined the pooled proportion of patients with 30-day readmission using a random-effects model, with the Freeman–Tukey double-arcsine transformation for meta-analysis of proportions.12 We investigated the heterogeneity by stratifying analyses according to prespecified study characteristics, including “broad” versus “focused.” However, the readmission risk was not different in the stratified analysis; therefore, we chose to pool the findings. For point estimates, 95% confidence intervals (CIs) were calculated, and a P-value < .05 was considered statistically significant.
RESULTS
Search Results
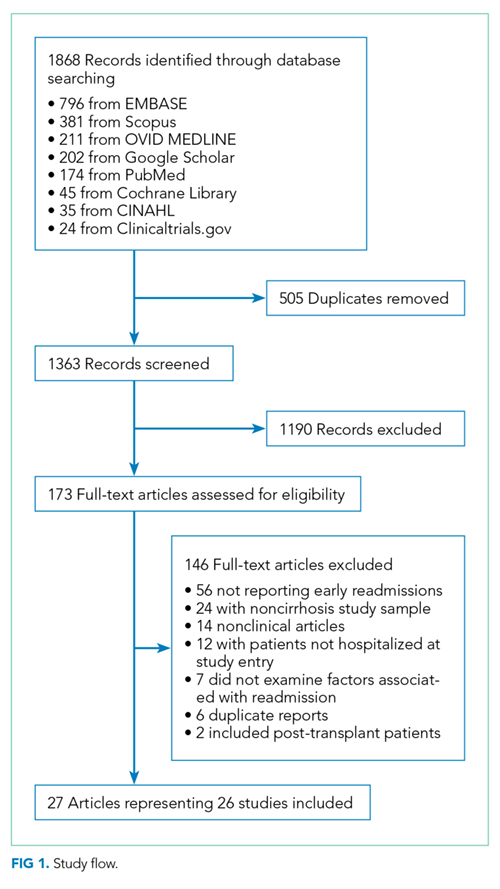
Study Characteristics
Two studies were performed in Australia, 4 in Europe, and the remainder in North America. Twenty one of the 26 studies were retrospective cohort studies (Table 1). Twenty studies were single-center studies (of which half were performed at transplant centers), and 4 of the 6 multicenter studies were based on administrative data with large samples (173,254 patients). The inclusion/exclusion criteria varied widely (Supplementary Material). Some studies only included patients admitted for specific cirrhosis complications, while others included those admitted for any reason. Two studies excluded patients admitted in the prior 30 days, and 6 excluded patients discharged to hospice. The mean risk of bias score was 7.5 (SD 1.3) out of a possible 9 points, with most lacking an adequate description of follow-up and several lacking adjustment for confounders.
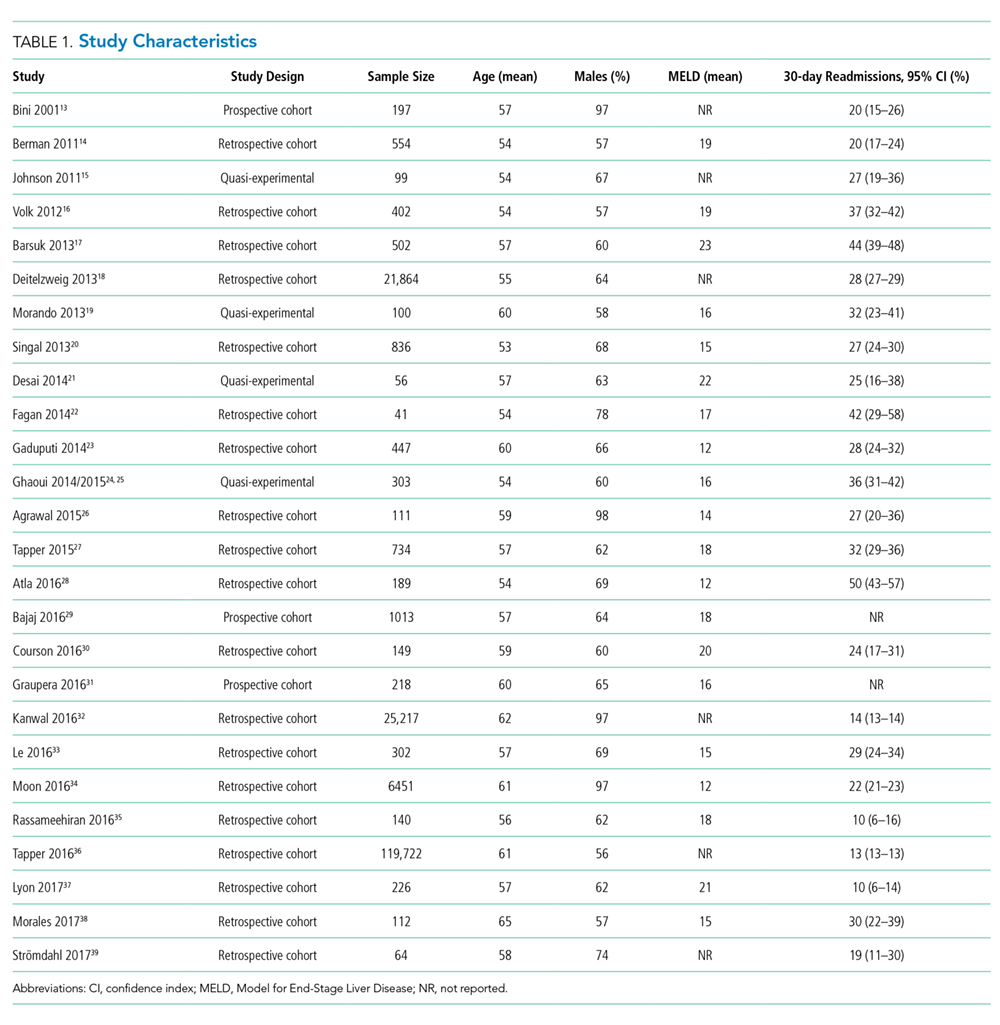
Outcomes
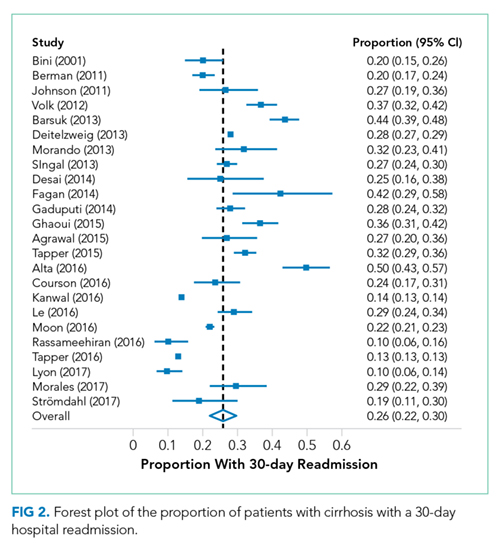
Readmission and MELD
DISCUSSION
Hospital readmission is a costly and common problem in the US.2 In addition to the negative impact that readmissions have on patients’ lives,40 readmissions are increasingly being used to measure quality. Unplanned 30-day readmissions are posted publicly, and excess readmissions for high-risk conditions are penalized through HRRP.3 Although HRRP does not currently include cirrhosis, the program has expanded to include several conditions that were not included in the initial iteration. Whether cirrhosis will be included in future iterations remains to be seen; however, increasing scrutiny is likely to continue. Of specific populations at risk, patients with cirrhosis are particularly vulnerable due to several features. Ascites management often requires hospitalization due to diuretic titration and poor access to paracentesis, and hepatic encephalopathy treatment requires complex lactulose titration.16 Other features of cirrhosis, such as gastrointestinal bleeding, infections, and renal failure, also place patients at risk of poor outcomes. The resulting readmission burden is high, with a pooled 30-day readmission rate of 26%. Other associated outcomes are also poor, with a consistent relationship between readmission and subsequent mortality.
We found striking heterogeneity in various aspects. First, the inclusion/exclusion criteria varied widely, both cirrhosis-specific (eg, spontaneous bacterial peritonitis) and more general (patients admitted within the prior 30 days). Some of these criteria may bias readmission estimates; the risk of readmission may be reduced in those on hospice, as patients forgo curative therapy. Additionally, an established risk factor for readmission is prior hospitalization41; excluding patients with prior admissions prohibits analysis of this variable. Another aspect is the capture of readmissions: readmissions outside of the index hospital were not included in most studies. In those that did include outside readmissions, the burden was sizeable: 17% in 1 single-center study and 23% in a multistate administrative database.16,36 These outside readmissions must be included in future studies; they are as important as same-center readmissions both to patients and CMS.3 Despite this heterogeneity, the studies scored relatively high on the Newcastle–Ottawa risk of bias scale, with the only common deficiency being an inadequate description of follow-up.
Building on the findings of this review, an important step will be the design of interventions to reduce readmissions. Such interventions require a full understanding of this population’s characteristics and needs. Critically, we found a lack of data on social determinants of health. Impairments in these factors are well-established contributors to readmission risk in other populations,4,40 and are highly prevalent in cirrhosis.42 Indeed, CMS has focused resources toward social determinants of health in the effort to reduce utilization and improve outcomes. This lack of data on social determinants of health, as well as other understudied factors, represents an important opportunity for future research efforts to better define the modifiable features that could be targeted in the future to prevent readmissions. Such research is urgently needed and will likely require prospective studies to gather these important factors. Notably, most studies in this systematic review were retrospective and therefore unable to examine many of these understudied factors. Another important aspect that has received little attention is readmission preventability: only 2 studies assessed preventability, both through unstructured chart review. Preventability assessments in noncirrhotic populations have used wide-ranging methodologies, yielding inconsistent results.43 This variability prompted recommendations that preventability should be assessed by multiple reviewers guided by explicit parameters.43 Such detailed attention to preventability is urgently needed to better inform interventions.
In contrast to the lack of data on social factors, we found that the MELD score was examined in most studies and was frequently associated with readmission. Despite this consistent association, differences in the MELD scores between studies limit inferences into specific cutoff values that could identify the highest risk patients. Because of its existing widespread clinical use, the MELD score may prove to be important in readmission risk stratification. Efforts to develop a useful model including the MELD score are needed to target interventions to the highest risk patients.
This review has several limitations. Although we used a broad search strategy to capture studies, some may not have been included due to our selection criteria. For instance, 1 retrospective paper described factors associated with high admission density during 1 year but did not specifically report the frequency of early readmissions.44 Similarly, a randomized trial of a disease management program did not specifically examine early readmissions.45 Another quasi-experimental study of a quality improvement initiative was not included because a large proportion of their subjects was post liver transplant.46 However, the inclusion of these papers is unlikely to change our conclusions; the retrospective study identified factors similar to those in the included studies, and the quasi-experimental study overlapped with the included study that assessed frailty.27 Another potential limitation is the exclusion of studies published in abstract form only. Such studies may be important, as the field of cirrhosis readmissions is relatively young. However, including only full-paper publications ensures the inclusion of only higher quality studies scrutinized during the peer-review process. Similarly, newer published studies may have been missed due to the abundant interest in this topic and ongoing research. Lastly, the significant heterogeneity of the studies limits conclusions that can be made regarding the pooled readmission rates.
In summary, we found that patients with cirrhosis experience a high incidence of hospital readmissions. Several processes of care may be associated with readmissions, suggesting room for improvement in caring for this population and reducing readmissions. However, we identified several gaps in the literature, which does not adequately describe social factors and is lacking details on readmission preventability assessment. Future studies should attempt to address these issues so that interventions can be targeted to the highest risk patients and designed to best meet the needs of patients with cirrhosis.
Disclosures
Dr. Orman, Dr. Ghabril, and Dr. Emmett report no potential conflicts of interest. Dr. Chalasani reports personal fees from Lilly, personal fees from Abbvie, personal fees from Tobira/Allergan, personal fees from Ardelyx, personal fees from Amarin, personal fees from Shire, personal fees from Madrigal, personal fees from DS Biopharma (Afimmune), personal fees from Cempra, personal fees from NuSirt, grants from Galectin, grants from Gilead, grants from Intercept, grants from Cumberland, grants from Conatus, personal fees from Immuron, and personal fees from Axovant, outside the submitted work.
Funding Information
This work was supported, in part, by the National Institutes of Health, KL2 TR001106 and K23 DK109202
1. Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149(7):1731-1741.e3. DOI: 10.1053/j.gastro.2015.08.045. PubMed
13. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34(6):1089-1095. DOI: 10.1053/jhep.2001.29204. PubMed
14. Berman K, Tandra S, Forssell K, et al. Incidence and predictors of 30-day readmission among patients hospitalized for advanced liver disease. Clin Gastroenterol Hepatol. 2011;9(3):254-259. DOI: 10.1016/j.cgh.2010.10.035. PubMed
15. Johnson EA, Spier BJ, Leff JA, Lucey MR, Said A. Optimising the care of patients with cirrhosis and gastrointestinal haemorrhage: a quality improvement study. Aliment Pharmacol Ther. 2011;34(1):76-82. DOI: 10.1111/j.1365-2036.2011.04692.x. PubMed
16. Volk ML, Tocco RS, Bazick J, Rakoski MO, Lok AS. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107(2):247-252. DOI: 10.1038/ajg.2011.314. PubMed
17. Barsuk JH, Cohen ER, Feinglass J, McGaghie WC, Wayne DB. Clinical outcomes after bedside and interventional radiology paracentesis procedures. Am J Med. 2013;126(4):349-356. DOI: 10.1016/j.amjmed.2012.09.016. PubMed
18. Deitelzweig S, Amin A, Christian R, Friend K, Lin J, Lowe TJ. Hyponatremia-associated healthcare burden among US patients hospitalized for cirrhosis. Adv Ther. 2013;30(1):71-80. DOI: 10.1007/s12325-012-0073-1. PubMed
19. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59(2):257-264. DOI: 10.1016/j.jhep.2013.03.010. PubMed
20. Singal AG, Rahimi RS, Clark C, et al. An automated model using electronic medical record data identifies patients with cirrhosis at high risk for readmission. Clin Gastroenterol Hepatol. 2013;11(10):1335-1341.e1. DOI: 10.1016/j.cgh.2013.03.022. PubMed
21. Desai AP, Satoskar R, Appannagari A, et al. Co-management between hospitalist and hepatologist improves the quality of care of inpatients with chronic liver disease. J Clin Gastroenterol. 2014;48(4):e30-e36. DOI: 10.1097/MCG.0b013e3182a87f70. PubMed
22. Fagan KJ, Zhao EY, Horsfall LU, et al. Burden of decompensated cirrhosis and ascites on hospital services in a tertiary care facility: time for change? Intern Med J. 2014;44(9):865-872. DOI: 10.1111/imj.12491. PubMed
23. Gaduputi V, Chandrala C, Abbas N, Tariq H, Chilimuri S, Balar B. Prognostic significance of hypokalemia in hepatic encephalopathy. Hepatogastroenterology. 2014;61(133):1170-1174. PubMed
26. Agrawal K, Kumar P, Markert R, Agrawal S. Risk factors for 30-day readmissions of individuals with decompensated cirrhosis. South Med J. 2015;108(11):682-687. DOI: 10.14423/SMJ.0000000000000371. PubMed
27. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Lai M. Standard assessments of frailty are validated predictors of mortality in hospitalized patients with cirrhosis. Hepatology. 2015;62(2):584-590. DOI: 10.1002/hep.27830. PubMed
28. Atla PR, Sheikh MY, Gill F, Kundu R, Choudhury J. Predictors of hospital re-admissions among Hispanics with hepatitis C-related cirrhosis. Ann Gastroenterol. 2016;29(4):515-520. DOI: 10.20524/aog.2016.0072. PubMed
29. Bajaj JS, Reddy KR, Tandon P, et al. The 3-month readmission rate remains unacceptably high in a large North American cohort of patients with cirrhosis. Hepatology. 2016;64(1):200-208. DOI: 10.1002/hep.28414. PubMed
30. Courson A, Jones GM, Twilla JD. Treatment of acute hepatic encephalopathy: comparing the effects of adding rifaximin to lactulose on patient outcomes. J Pharm Pract. 2016;29(3):212-217. DOI: 10.1177/0897190014566312. PubMed
31. Graupera I, Solà E, Fabrellas N, et al. Urine monocyte chemoattractant protein-1 is an independent predictive factor of hospital readmission and survival in cirrhosis. PLOS ONE. 2016;11(6):e0157371. DOI: 10.1371/journal.pone.0157371. PubMed
32. Kanwal F, Asch SM, Kramer JR, Cao Y, Asrani S, El-Serag HB. Early outpatient follow-up and 30-day outcomes in patients hospitalized with cirrhosis. Hepatology. 2016;64(2):569-581. DOI: 10.1002/hep.28558. PubMed
46. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Chang M, Lai M. A quality improvement initiative reduces 30-day rate of readmission for patients with cirrhosis. Clin Gastroenterol Hepatol. 2016;14(5):753-759. DOI: 10.1016/j.cgh.2015.08.041. PubMed
45. Wigg AJ, McCormick R, Wundke R, Woodman RJ. Efficacy of a chronic disease management model for patients with chronic liver failure. Clin Gastroenterol Hepatol. 2013;11(7):850-8.e1. DOI: 10.1016/j.cgh.2013.01.014. PubMed
44. Ganesh S, Rogal SS, Yadav D, Humar A, Behari J. Risk factors for frequent readmissions and barriers to transplantation in patients with cirrhosis. PLOS ONE. 2013;8(1):e55140. DOI: 10.1371/journal.pone.0055140. PubMed
43. van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391-E402. DOI: 10.1503/cmaj.101860. PubMed
42. Bajaj JS, Wade JB, Gibson DP, et al. The multi-dimensional burden of cirrhosis and hepatic encephalopathy on patients and caregivers. Am J Gastroenterol. 2011;106(9):1646-1653. DOI: 10.1038/ajg.2011.157. PubMed
41. van Walraven C, Dhalla IA, Bell C, et al. Derivation and validation of an index to predict early death or unplanned readmission after discharge from hospital to the community. CMAJ. 2010;182(6):551-557. DOI: 10.1503/cmaj.091117. PubMed
40. Rodríguez-Artalejo F, Guallar-Castillón P, Pascual CR, et al. Health-related quality of life as a predictor of hospital readmission and death among patients with heart failure. Arch Intern Med. 2005;165(11):1274-1279. DOI: 10.1001/archinte.165.11.1274. PubMed
39. Strömdahl M, Helgeson J, Kalaitzakis E. Emergency readmission following acute upper gastrointestinal bleeding. Eur J Gastroenterol Hepatol. 2017;29(1):73-77. DOI: 10.1097/MEG.0000000000000746. PubMed
38. Morales BP, Planas R, Bartoli R, et al. Early hospital readmission in decompensated cirrhosis: incidence, impact on mortality, and predictive factors. Dig Liver Dis. 2017;49(8):903-909. DOI: 10.1016/j.dld.2017.03.005. PubMed
37. Lyon KC, Likar E, Martello JL, Regier M. Retrospective cross-sectional pilot study of rifaximin dosing for the prevention of recurrent hepatic encephalopathy. J Gastroenterol Hepatol. 2017;32(9):1548-1552. DOI: 10.1111/jgh.13759. PubMed
36. Tapper EB, Halbert B, Mellinger J. Rates of and reasons for hospital readmissions in patients with cirrhosis: a multistate population-based cohort study. Clin Gastroenterol Hepatol. 2016;14(8):1181-1188.e2. DOI: 10.1016/j.cgh.2016.04.009. PubMed
35. Rassameehiran S, Mankongpaisarnrung C, Sutamtewagul G, Klomjit S, Rakvit A. Predictor of 90-day readmission rate for hepatic encephalopathy. South Med J. 2016;109(6):365-369. DOI: 10.14423/SMJ.0000000000000475. PubMed
34. Moon AM, Dominitz JA, Ioannou GN, Lowy E, Beste LA. Use of antibiotics among patients with cirrhosis and upper gastrointestinal bleeding is associated with reduced mortality. Clin Gastroenterol Hepatol. 2016;14(11):1629-1637.e1. DOI: 10.1016/j.cgh.2016.05.040. PubMed
33. Le S, Spelman T, Chong CP, et al. Could adherence to quality of care indicators for hospitalized patients with cirrhosis-related ascites improve clinical outcomes? Am J Gastroenterol. 2016;111(1):87-92. DOI: .10.1038/ajg.2015.402. PubMed
Cirrhosis is a morbid condition characterized by complications such as ascites, gastrointestinal bleeding, and hepatic encephalopathy. These complications frequently require hospitalization, which is a substantial burden to the healthcare system. In 2012, liver disease was responsible for nearly 250,000 admissions across the United States, costing $3 billion.1 Despite this substantial resource utilization, outcomes remain poor, with an inpatient mortality of 6%. For those that survive, many experience hospital readmission.
More generally, early readmission reflects poor quality of care in the US. In 2004, 30-day readmissions occurred in nearly 20% of Medicare beneficiaries and costed over $17 billion.2 In response to this problem, the Affordable Care Act established the Hospital Readmissions Reduction Program (HRRP), which reduces Centers for Medicare & Medicaid Services (CMS) payments to hospitals with excess 30-day readmissions for high-risk conditions, including pneumonia and heart failure.3 Heart failure, in particular, has been the subject of numerous studies detailing risk factors and interventions to predict and prevent readmission.4-6 Based on this extensive evidence, guidelines recommend disease management programs to reduce readmissions in this population.7 In contrast, readmission in the cirrhosis population has received limited attention.
We therefore conducted a systematic review aiming to examine the range of readmission risk noted in the literature, with a focus on the model for end-stage liver disease (MELD) score as a risk factor for readmission.
METHODS
Search Strategy
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines for conducting and reporting systematic reviews.8 A literature search was performed by a medical librarian using the following databases: Ovid MEDLINE, PubMed, EMBASE, CINAHL, the full Cochrane Library, Scopus, Google Scholar, and ClinicalTrials.gov. All the databases were searched from 2000 to May 2017. We did not include older reports because the review focused on contemporary care; earlier studies may not reflect current cirrhosis management. To ensure literature saturation, included articles’ reference lists were reviewed.
Search strategies were developed by combining database-specific subject headings and keywords for readmissions with those for cirrhosis or its complications (Supplementary Material). Google Scholar and ClinicalTrials.gov were searched using keywords only. All results were limited to the English language and those published in 2000 or later, but no other limits were applied.
Identified records were reviewed based on strict criteria. We excluded case reports, case series, reviews, editorials, letters, and meeting abstracts without final peer-reviewed publication. We also excluded studies of pediatric populations (age < 18 years), patients without cirrhosis, and patients with liver transplants. We excluded studies in which patients were not hospitalized at study onset and those where the index admission was for an elective procedure. Because our interest was to identify factors associated with early readmission, we excluded studies that did not report readmissions within 90 days or those with a mean or median follow-up of less than 30 days. We also excluded studies that did not examine the association between readmission and at least 1 independent variable or intervention. Duplicate reports of a common sample were excluded unless the duplicate provided additional information, and such reports were examined together in our synthesis.
Two authors identified potentially eligible records by independently screening titles and abstracts. At this stage, records that did not meet the eligibility criteria were excluded, and the reasons for exclusion were not recorded. Records with disagreement were retained for full-text review. After this initial exclusion of records, the remaining full-text records were reviewed independently. For this full-text review, we recorded exclusion reasons and disagreements were resolved through discussion.
Data Collection
Data were abstracted from each study by 2 authors independently and recorded in a REDCap database.9 Discrepancies were resolved through discussion. We recorded study characteristics, including study design, setting, population (including the inclusion/exclusion criteria, sample size, and patient and hospitalization characteristics), interventions, and comparisons. To facilitate comparisons across studies, we employed validated methods to approximate means and standard deviations (SD).10 We recorded detailed information on outcomes including readmissions, preventability, independent variables, and mortality. Studies that focused on a single independent factor or intervention were classified as “focused,” while those that examined multiple factors were classified as “broad.” We used the Newcastle–Ottawa Scale to assess the risk of bias in each study.11 This instrument uses a 9-point scale to gauge methodological quality based on selection, group comparability, and exposure/outcome assessment.
Statistical Analysis
Analyses were performed using Stata 13.1 (StataCorp LP, College Station, Texas). We determined the pooled proportion of patients with 30-day readmission using a random-effects model, with the Freeman–Tukey double-arcsine transformation for meta-analysis of proportions.12 We investigated the heterogeneity by stratifying analyses according to prespecified study characteristics, including “broad” versus “focused.” However, the readmission risk was not different in the stratified analysis; therefore, we chose to pool the findings. For point estimates, 95% confidence intervals (CIs) were calculated, and a P-value < .05 was considered statistically significant.
RESULTS
Search Results
Study Characteristics
Two studies were performed in Australia, 4 in Europe, and the remainder in North America. Twenty one of the 26 studies were retrospective cohort studies (Table 1). Twenty studies were single-center studies (of which half were performed at transplant centers), and 4 of the 6 multicenter studies were based on administrative data with large samples (173,254 patients). The inclusion/exclusion criteria varied widely (Supplementary Material). Some studies only included patients admitted for specific cirrhosis complications, while others included those admitted for any reason. Two studies excluded patients admitted in the prior 30 days, and 6 excluded patients discharged to hospice. The mean risk of bias score was 7.5 (SD 1.3) out of a possible 9 points, with most lacking an adequate description of follow-up and several lacking adjustment for confounders.
Outcomes
Readmission and MELD
DISCUSSION
Hospital readmission is a costly and common problem in the US.2 In addition to the negative impact that readmissions have on patients’ lives,40 readmissions are increasingly being used to measure quality. Unplanned 30-day readmissions are posted publicly, and excess readmissions for high-risk conditions are penalized through HRRP.3 Although HRRP does not currently include cirrhosis, the program has expanded to include several conditions that were not included in the initial iteration. Whether cirrhosis will be included in future iterations remains to be seen; however, increasing scrutiny is likely to continue. Of specific populations at risk, patients with cirrhosis are particularly vulnerable due to several features. Ascites management often requires hospitalization due to diuretic titration and poor access to paracentesis, and hepatic encephalopathy treatment requires complex lactulose titration.16 Other features of cirrhosis, such as gastrointestinal bleeding, infections, and renal failure, also place patients at risk of poor outcomes. The resulting readmission burden is high, with a pooled 30-day readmission rate of 26%. Other associated outcomes are also poor, with a consistent relationship between readmission and subsequent mortality.
We found striking heterogeneity in various aspects. First, the inclusion/exclusion criteria varied widely, both cirrhosis-specific (eg, spontaneous bacterial peritonitis) and more general (patients admitted within the prior 30 days). Some of these criteria may bias readmission estimates; the risk of readmission may be reduced in those on hospice, as patients forgo curative therapy. Additionally, an established risk factor for readmission is prior hospitalization41; excluding patients with prior admissions prohibits analysis of this variable. Another aspect is the capture of readmissions: readmissions outside of the index hospital were not included in most studies. In those that did include outside readmissions, the burden was sizeable: 17% in 1 single-center study and 23% in a multistate administrative database.16,36 These outside readmissions must be included in future studies; they are as important as same-center readmissions both to patients and CMS.3 Despite this heterogeneity, the studies scored relatively high on the Newcastle–Ottawa risk of bias scale, with the only common deficiency being an inadequate description of follow-up.
Building on the findings of this review, an important step will be the design of interventions to reduce readmissions. Such interventions require a full understanding of this population’s characteristics and needs. Critically, we found a lack of data on social determinants of health. Impairments in these factors are well-established contributors to readmission risk in other populations,4,40 and are highly prevalent in cirrhosis.42 Indeed, CMS has focused resources toward social determinants of health in the effort to reduce utilization and improve outcomes. This lack of data on social determinants of health, as well as other understudied factors, represents an important opportunity for future research efforts to better define the modifiable features that could be targeted in the future to prevent readmissions. Such research is urgently needed and will likely require prospective studies to gather these important factors. Notably, most studies in this systematic review were retrospective and therefore unable to examine many of these understudied factors. Another important aspect that has received little attention is readmission preventability: only 2 studies assessed preventability, both through unstructured chart review. Preventability assessments in noncirrhotic populations have used wide-ranging methodologies, yielding inconsistent results.43 This variability prompted recommendations that preventability should be assessed by multiple reviewers guided by explicit parameters.43 Such detailed attention to preventability is urgently needed to better inform interventions.
In contrast to the lack of data on social factors, we found that the MELD score was examined in most studies and was frequently associated with readmission. Despite this consistent association, differences in the MELD scores between studies limit inferences into specific cutoff values that could identify the highest risk patients. Because of its existing widespread clinical use, the MELD score may prove to be important in readmission risk stratification. Efforts to develop a useful model including the MELD score are needed to target interventions to the highest risk patients.
This review has several limitations. Although we used a broad search strategy to capture studies, some may not have been included due to our selection criteria. For instance, 1 retrospective paper described factors associated with high admission density during 1 year but did not specifically report the frequency of early readmissions.44 Similarly, a randomized trial of a disease management program did not specifically examine early readmissions.45 Another quasi-experimental study of a quality improvement initiative was not included because a large proportion of their subjects was post liver transplant.46 However, the inclusion of these papers is unlikely to change our conclusions; the retrospective study identified factors similar to those in the included studies, and the quasi-experimental study overlapped with the included study that assessed frailty.27 Another potential limitation is the exclusion of studies published in abstract form only. Such studies may be important, as the field of cirrhosis readmissions is relatively young. However, including only full-paper publications ensures the inclusion of only higher quality studies scrutinized during the peer-review process. Similarly, newer published studies may have been missed due to the abundant interest in this topic and ongoing research. Lastly, the significant heterogeneity of the studies limits conclusions that can be made regarding the pooled readmission rates.
In summary, we found that patients with cirrhosis experience a high incidence of hospital readmissions. Several processes of care may be associated with readmissions, suggesting room for improvement in caring for this population and reducing readmissions. However, we identified several gaps in the literature, which does not adequately describe social factors and is lacking details on readmission preventability assessment. Future studies should attempt to address these issues so that interventions can be targeted to the highest risk patients and designed to best meet the needs of patients with cirrhosis.
Disclosures
Dr. Orman, Dr. Ghabril, and Dr. Emmett report no potential conflicts of interest. Dr. Chalasani reports personal fees from Lilly, personal fees from Abbvie, personal fees from Tobira/Allergan, personal fees from Ardelyx, personal fees from Amarin, personal fees from Shire, personal fees from Madrigal, personal fees from DS Biopharma (Afimmune), personal fees from Cempra, personal fees from NuSirt, grants from Galectin, grants from Gilead, grants from Intercept, grants from Cumberland, grants from Conatus, personal fees from Immuron, and personal fees from Axovant, outside the submitted work.
Funding Information
This work was supported, in part, by the National Institutes of Health, KL2 TR001106 and K23 DK109202
Cirrhosis is a morbid condition characterized by complications such as ascites, gastrointestinal bleeding, and hepatic encephalopathy. These complications frequently require hospitalization, which is a substantial burden to the healthcare system. In 2012, liver disease was responsible for nearly 250,000 admissions across the United States, costing $3 billion.1 Despite this substantial resource utilization, outcomes remain poor, with an inpatient mortality of 6%. For those that survive, many experience hospital readmission.
More generally, early readmission reflects poor quality of care in the US. In 2004, 30-day readmissions occurred in nearly 20% of Medicare beneficiaries and costed over $17 billion.2 In response to this problem, the Affordable Care Act established the Hospital Readmissions Reduction Program (HRRP), which reduces Centers for Medicare & Medicaid Services (CMS) payments to hospitals with excess 30-day readmissions for high-risk conditions, including pneumonia and heart failure.3 Heart failure, in particular, has been the subject of numerous studies detailing risk factors and interventions to predict and prevent readmission.4-6 Based on this extensive evidence, guidelines recommend disease management programs to reduce readmissions in this population.7 In contrast, readmission in the cirrhosis population has received limited attention.
We therefore conducted a systematic review aiming to examine the range of readmission risk noted in the literature, with a focus on the model for end-stage liver disease (MELD) score as a risk factor for readmission.
METHODS
Search Strategy
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines for conducting and reporting systematic reviews.8 A literature search was performed by a medical librarian using the following databases: Ovid MEDLINE, PubMed, EMBASE, CINAHL, the full Cochrane Library, Scopus, Google Scholar, and ClinicalTrials.gov. All the databases were searched from 2000 to May 2017. We did not include older reports because the review focused on contemporary care; earlier studies may not reflect current cirrhosis management. To ensure literature saturation, included articles’ reference lists were reviewed.
Search strategies were developed by combining database-specific subject headings and keywords for readmissions with those for cirrhosis or its complications (Supplementary Material). Google Scholar and ClinicalTrials.gov were searched using keywords only. All results were limited to the English language and those published in 2000 or later, but no other limits were applied.
Identified records were reviewed based on strict criteria. We excluded case reports, case series, reviews, editorials, letters, and meeting abstracts without final peer-reviewed publication. We also excluded studies of pediatric populations (age < 18 years), patients without cirrhosis, and patients with liver transplants. We excluded studies in which patients were not hospitalized at study onset and those where the index admission was for an elective procedure. Because our interest was to identify factors associated with early readmission, we excluded studies that did not report readmissions within 90 days or those with a mean or median follow-up of less than 30 days. We also excluded studies that did not examine the association between readmission and at least 1 independent variable or intervention. Duplicate reports of a common sample were excluded unless the duplicate provided additional information, and such reports were examined together in our synthesis.
Two authors identified potentially eligible records by independently screening titles and abstracts. At this stage, records that did not meet the eligibility criteria were excluded, and the reasons for exclusion were not recorded. Records with disagreement were retained for full-text review. After this initial exclusion of records, the remaining full-text records were reviewed independently. For this full-text review, we recorded exclusion reasons and disagreements were resolved through discussion.
Data Collection
Data were abstracted from each study by 2 authors independently and recorded in a REDCap database.9 Discrepancies were resolved through discussion. We recorded study characteristics, including study design, setting, population (including the inclusion/exclusion criteria, sample size, and patient and hospitalization characteristics), interventions, and comparisons. To facilitate comparisons across studies, we employed validated methods to approximate means and standard deviations (SD).10 We recorded detailed information on outcomes including readmissions, preventability, independent variables, and mortality. Studies that focused on a single independent factor or intervention were classified as “focused,” while those that examined multiple factors were classified as “broad.” We used the Newcastle–Ottawa Scale to assess the risk of bias in each study.11 This instrument uses a 9-point scale to gauge methodological quality based on selection, group comparability, and exposure/outcome assessment.
Statistical Analysis
Analyses were performed using Stata 13.1 (StataCorp LP, College Station, Texas). We determined the pooled proportion of patients with 30-day readmission using a random-effects model, with the Freeman–Tukey double-arcsine transformation for meta-analysis of proportions.12 We investigated the heterogeneity by stratifying analyses according to prespecified study characteristics, including “broad” versus “focused.” However, the readmission risk was not different in the stratified analysis; therefore, we chose to pool the findings. For point estimates, 95% confidence intervals (CIs) were calculated, and a P-value < .05 was considered statistically significant.
RESULTS
Search Results
Study Characteristics
Two studies were performed in Australia, 4 in Europe, and the remainder in North America. Twenty one of the 26 studies were retrospective cohort studies (Table 1). Twenty studies were single-center studies (of which half were performed at transplant centers), and 4 of the 6 multicenter studies were based on administrative data with large samples (173,254 patients). The inclusion/exclusion criteria varied widely (Supplementary Material). Some studies only included patients admitted for specific cirrhosis complications, while others included those admitted for any reason. Two studies excluded patients admitted in the prior 30 days, and 6 excluded patients discharged to hospice. The mean risk of bias score was 7.5 (SD 1.3) out of a possible 9 points, with most lacking an adequate description of follow-up and several lacking adjustment for confounders.
Outcomes
Readmission and MELD
DISCUSSION
Hospital readmission is a costly and common problem in the US.2 In addition to the negative impact that readmissions have on patients’ lives,40 readmissions are increasingly being used to measure quality. Unplanned 30-day readmissions are posted publicly, and excess readmissions for high-risk conditions are penalized through HRRP.3 Although HRRP does not currently include cirrhosis, the program has expanded to include several conditions that were not included in the initial iteration. Whether cirrhosis will be included in future iterations remains to be seen; however, increasing scrutiny is likely to continue. Of specific populations at risk, patients with cirrhosis are particularly vulnerable due to several features. Ascites management often requires hospitalization due to diuretic titration and poor access to paracentesis, and hepatic encephalopathy treatment requires complex lactulose titration.16 Other features of cirrhosis, such as gastrointestinal bleeding, infections, and renal failure, also place patients at risk of poor outcomes. The resulting readmission burden is high, with a pooled 30-day readmission rate of 26%. Other associated outcomes are also poor, with a consistent relationship between readmission and subsequent mortality.
We found striking heterogeneity in various aspects. First, the inclusion/exclusion criteria varied widely, both cirrhosis-specific (eg, spontaneous bacterial peritonitis) and more general (patients admitted within the prior 30 days). Some of these criteria may bias readmission estimates; the risk of readmission may be reduced in those on hospice, as patients forgo curative therapy. Additionally, an established risk factor for readmission is prior hospitalization41; excluding patients with prior admissions prohibits analysis of this variable. Another aspect is the capture of readmissions: readmissions outside of the index hospital were not included in most studies. In those that did include outside readmissions, the burden was sizeable: 17% in 1 single-center study and 23% in a multistate administrative database.16,36 These outside readmissions must be included in future studies; they are as important as same-center readmissions both to patients and CMS.3 Despite this heterogeneity, the studies scored relatively high on the Newcastle–Ottawa risk of bias scale, with the only common deficiency being an inadequate description of follow-up.
Building on the findings of this review, an important step will be the design of interventions to reduce readmissions. Such interventions require a full understanding of this population’s characteristics and needs. Critically, we found a lack of data on social determinants of health. Impairments in these factors are well-established contributors to readmission risk in other populations,4,40 and are highly prevalent in cirrhosis.42 Indeed, CMS has focused resources toward social determinants of health in the effort to reduce utilization and improve outcomes. This lack of data on social determinants of health, as well as other understudied factors, represents an important opportunity for future research efforts to better define the modifiable features that could be targeted in the future to prevent readmissions. Such research is urgently needed and will likely require prospective studies to gather these important factors. Notably, most studies in this systematic review were retrospective and therefore unable to examine many of these understudied factors. Another important aspect that has received little attention is readmission preventability: only 2 studies assessed preventability, both through unstructured chart review. Preventability assessments in noncirrhotic populations have used wide-ranging methodologies, yielding inconsistent results.43 This variability prompted recommendations that preventability should be assessed by multiple reviewers guided by explicit parameters.43 Such detailed attention to preventability is urgently needed to better inform interventions.
In contrast to the lack of data on social factors, we found that the MELD score was examined in most studies and was frequently associated with readmission. Despite this consistent association, differences in the MELD scores between studies limit inferences into specific cutoff values that could identify the highest risk patients. Because of its existing widespread clinical use, the MELD score may prove to be important in readmission risk stratification. Efforts to develop a useful model including the MELD score are needed to target interventions to the highest risk patients.
This review has several limitations. Although we used a broad search strategy to capture studies, some may not have been included due to our selection criteria. For instance, 1 retrospective paper described factors associated with high admission density during 1 year but did not specifically report the frequency of early readmissions.44 Similarly, a randomized trial of a disease management program did not specifically examine early readmissions.45 Another quasi-experimental study of a quality improvement initiative was not included because a large proportion of their subjects was post liver transplant.46 However, the inclusion of these papers is unlikely to change our conclusions; the retrospective study identified factors similar to those in the included studies, and the quasi-experimental study overlapped with the included study that assessed frailty.27 Another potential limitation is the exclusion of studies published in abstract form only. Such studies may be important, as the field of cirrhosis readmissions is relatively young. However, including only full-paper publications ensures the inclusion of only higher quality studies scrutinized during the peer-review process. Similarly, newer published studies may have been missed due to the abundant interest in this topic and ongoing research. Lastly, the significant heterogeneity of the studies limits conclusions that can be made regarding the pooled readmission rates.
In summary, we found that patients with cirrhosis experience a high incidence of hospital readmissions. Several processes of care may be associated with readmissions, suggesting room for improvement in caring for this population and reducing readmissions. However, we identified several gaps in the literature, which does not adequately describe social factors and is lacking details on readmission preventability assessment. Future studies should attempt to address these issues so that interventions can be targeted to the highest risk patients and designed to best meet the needs of patients with cirrhosis.
Disclosures
Dr. Orman, Dr. Ghabril, and Dr. Emmett report no potential conflicts of interest. Dr. Chalasani reports personal fees from Lilly, personal fees from Abbvie, personal fees from Tobira/Allergan, personal fees from Ardelyx, personal fees from Amarin, personal fees from Shire, personal fees from Madrigal, personal fees from DS Biopharma (Afimmune), personal fees from Cempra, personal fees from NuSirt, grants from Galectin, grants from Gilead, grants from Intercept, grants from Cumberland, grants from Conatus, personal fees from Immuron, and personal fees from Axovant, outside the submitted work.
Funding Information
This work was supported, in part, by the National Institutes of Health, KL2 TR001106 and K23 DK109202
1. Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149(7):1731-1741.e3. DOI: 10.1053/j.gastro.2015.08.045. PubMed
13. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34(6):1089-1095. DOI: 10.1053/jhep.2001.29204. PubMed
14. Berman K, Tandra S, Forssell K, et al. Incidence and predictors of 30-day readmission among patients hospitalized for advanced liver disease. Clin Gastroenterol Hepatol. 2011;9(3):254-259. DOI: 10.1016/j.cgh.2010.10.035. PubMed
15. Johnson EA, Spier BJ, Leff JA, Lucey MR, Said A. Optimising the care of patients with cirrhosis and gastrointestinal haemorrhage: a quality improvement study. Aliment Pharmacol Ther. 2011;34(1):76-82. DOI: 10.1111/j.1365-2036.2011.04692.x. PubMed
16. Volk ML, Tocco RS, Bazick J, Rakoski MO, Lok AS. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107(2):247-252. DOI: 10.1038/ajg.2011.314. PubMed
17. Barsuk JH, Cohen ER, Feinglass J, McGaghie WC, Wayne DB. Clinical outcomes after bedside and interventional radiology paracentesis procedures. Am J Med. 2013;126(4):349-356. DOI: 10.1016/j.amjmed.2012.09.016. PubMed
18. Deitelzweig S, Amin A, Christian R, Friend K, Lin J, Lowe TJ. Hyponatremia-associated healthcare burden among US patients hospitalized for cirrhosis. Adv Ther. 2013;30(1):71-80. DOI: 10.1007/s12325-012-0073-1. PubMed
19. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59(2):257-264. DOI: 10.1016/j.jhep.2013.03.010. PubMed
20. Singal AG, Rahimi RS, Clark C, et al. An automated model using electronic medical record data identifies patients with cirrhosis at high risk for readmission. Clin Gastroenterol Hepatol. 2013;11(10):1335-1341.e1. DOI: 10.1016/j.cgh.2013.03.022. PubMed
21. Desai AP, Satoskar R, Appannagari A, et al. Co-management between hospitalist and hepatologist improves the quality of care of inpatients with chronic liver disease. J Clin Gastroenterol. 2014;48(4):e30-e36. DOI: 10.1097/MCG.0b013e3182a87f70. PubMed
22. Fagan KJ, Zhao EY, Horsfall LU, et al. Burden of decompensated cirrhosis and ascites on hospital services in a tertiary care facility: time for change? Intern Med J. 2014;44(9):865-872. DOI: 10.1111/imj.12491. PubMed
23. Gaduputi V, Chandrala C, Abbas N, Tariq H, Chilimuri S, Balar B. Prognostic significance of hypokalemia in hepatic encephalopathy. Hepatogastroenterology. 2014;61(133):1170-1174. PubMed
26. Agrawal K, Kumar P, Markert R, Agrawal S. Risk factors for 30-day readmissions of individuals with decompensated cirrhosis. South Med J. 2015;108(11):682-687. DOI: 10.14423/SMJ.0000000000000371. PubMed
27. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Lai M. Standard assessments of frailty are validated predictors of mortality in hospitalized patients with cirrhosis. Hepatology. 2015;62(2):584-590. DOI: 10.1002/hep.27830. PubMed
28. Atla PR, Sheikh MY, Gill F, Kundu R, Choudhury J. Predictors of hospital re-admissions among Hispanics with hepatitis C-related cirrhosis. Ann Gastroenterol. 2016;29(4):515-520. DOI: 10.20524/aog.2016.0072. PubMed
29. Bajaj JS, Reddy KR, Tandon P, et al. The 3-month readmission rate remains unacceptably high in a large North American cohort of patients with cirrhosis. Hepatology. 2016;64(1):200-208. DOI: 10.1002/hep.28414. PubMed
30. Courson A, Jones GM, Twilla JD. Treatment of acute hepatic encephalopathy: comparing the effects of adding rifaximin to lactulose on patient outcomes. J Pharm Pract. 2016;29(3):212-217. DOI: 10.1177/0897190014566312. PubMed
31. Graupera I, Solà E, Fabrellas N, et al. Urine monocyte chemoattractant protein-1 is an independent predictive factor of hospital readmission and survival in cirrhosis. PLOS ONE. 2016;11(6):e0157371. DOI: 10.1371/journal.pone.0157371. PubMed
32. Kanwal F, Asch SM, Kramer JR, Cao Y, Asrani S, El-Serag HB. Early outpatient follow-up and 30-day outcomes in patients hospitalized with cirrhosis. Hepatology. 2016;64(2):569-581. DOI: 10.1002/hep.28558. PubMed
46. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Chang M, Lai M. A quality improvement initiative reduces 30-day rate of readmission for patients with cirrhosis. Clin Gastroenterol Hepatol. 2016;14(5):753-759. DOI: 10.1016/j.cgh.2015.08.041. PubMed
45. Wigg AJ, McCormick R, Wundke R, Woodman RJ. Efficacy of a chronic disease management model for patients with chronic liver failure. Clin Gastroenterol Hepatol. 2013;11(7):850-8.e1. DOI: 10.1016/j.cgh.2013.01.014. PubMed
44. Ganesh S, Rogal SS, Yadav D, Humar A, Behari J. Risk factors for frequent readmissions and barriers to transplantation in patients with cirrhosis. PLOS ONE. 2013;8(1):e55140. DOI: 10.1371/journal.pone.0055140. PubMed
43. van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391-E402. DOI: 10.1503/cmaj.101860. PubMed
42. Bajaj JS, Wade JB, Gibson DP, et al. The multi-dimensional burden of cirrhosis and hepatic encephalopathy on patients and caregivers. Am J Gastroenterol. 2011;106(9):1646-1653. DOI: 10.1038/ajg.2011.157. PubMed
41. van Walraven C, Dhalla IA, Bell C, et al. Derivation and validation of an index to predict early death or unplanned readmission after discharge from hospital to the community. CMAJ. 2010;182(6):551-557. DOI: 10.1503/cmaj.091117. PubMed
40. Rodríguez-Artalejo F, Guallar-Castillón P, Pascual CR, et al. Health-related quality of life as a predictor of hospital readmission and death among patients with heart failure. Arch Intern Med. 2005;165(11):1274-1279. DOI: 10.1001/archinte.165.11.1274. PubMed
39. Strömdahl M, Helgeson J, Kalaitzakis E. Emergency readmission following acute upper gastrointestinal bleeding. Eur J Gastroenterol Hepatol. 2017;29(1):73-77. DOI: 10.1097/MEG.0000000000000746. PubMed
38. Morales BP, Planas R, Bartoli R, et al. Early hospital readmission in decompensated cirrhosis: incidence, impact on mortality, and predictive factors. Dig Liver Dis. 2017;49(8):903-909. DOI: 10.1016/j.dld.2017.03.005. PubMed
37. Lyon KC, Likar E, Martello JL, Regier M. Retrospective cross-sectional pilot study of rifaximin dosing for the prevention of recurrent hepatic encephalopathy. J Gastroenterol Hepatol. 2017;32(9):1548-1552. DOI: 10.1111/jgh.13759. PubMed
36. Tapper EB, Halbert B, Mellinger J. Rates of and reasons for hospital readmissions in patients with cirrhosis: a multistate population-based cohort study. Clin Gastroenterol Hepatol. 2016;14(8):1181-1188.e2. DOI: 10.1016/j.cgh.2016.04.009. PubMed
35. Rassameehiran S, Mankongpaisarnrung C, Sutamtewagul G, Klomjit S, Rakvit A. Predictor of 90-day readmission rate for hepatic encephalopathy. South Med J. 2016;109(6):365-369. DOI: 10.14423/SMJ.0000000000000475. PubMed
34. Moon AM, Dominitz JA, Ioannou GN, Lowy E, Beste LA. Use of antibiotics among patients with cirrhosis and upper gastrointestinal bleeding is associated with reduced mortality. Clin Gastroenterol Hepatol. 2016;14(11):1629-1637.e1. DOI: 10.1016/j.cgh.2016.05.040. PubMed
33. Le S, Spelman T, Chong CP, et al. Could adherence to quality of care indicators for hospitalized patients with cirrhosis-related ascites improve clinical outcomes? Am J Gastroenterol. 2016;111(1):87-92. DOI: .10.1038/ajg.2015.402. PubMed
1. Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149(7):1731-1741.e3. DOI: 10.1053/j.gastro.2015.08.045. PubMed
13. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34(6):1089-1095. DOI: 10.1053/jhep.2001.29204. PubMed
14. Berman K, Tandra S, Forssell K, et al. Incidence and predictors of 30-day readmission among patients hospitalized for advanced liver disease. Clin Gastroenterol Hepatol. 2011;9(3):254-259. DOI: 10.1016/j.cgh.2010.10.035. PubMed
15. Johnson EA, Spier BJ, Leff JA, Lucey MR, Said A. Optimising the care of patients with cirrhosis and gastrointestinal haemorrhage: a quality improvement study. Aliment Pharmacol Ther. 2011;34(1):76-82. DOI: 10.1111/j.1365-2036.2011.04692.x. PubMed
16. Volk ML, Tocco RS, Bazick J, Rakoski MO, Lok AS. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107(2):247-252. DOI: 10.1038/ajg.2011.314. PubMed
17. Barsuk JH, Cohen ER, Feinglass J, McGaghie WC, Wayne DB. Clinical outcomes after bedside and interventional radiology paracentesis procedures. Am J Med. 2013;126(4):349-356. DOI: 10.1016/j.amjmed.2012.09.016. PubMed
18. Deitelzweig S, Amin A, Christian R, Friend K, Lin J, Lowe TJ. Hyponatremia-associated healthcare burden among US patients hospitalized for cirrhosis. Adv Ther. 2013;30(1):71-80. DOI: 10.1007/s12325-012-0073-1. PubMed
19. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59(2):257-264. DOI: 10.1016/j.jhep.2013.03.010. PubMed
20. Singal AG, Rahimi RS, Clark C, et al. An automated model using electronic medical record data identifies patients with cirrhosis at high risk for readmission. Clin Gastroenterol Hepatol. 2013;11(10):1335-1341.e1. DOI: 10.1016/j.cgh.2013.03.022. PubMed
21. Desai AP, Satoskar R, Appannagari A, et al. Co-management between hospitalist and hepatologist improves the quality of care of inpatients with chronic liver disease. J Clin Gastroenterol. 2014;48(4):e30-e36. DOI: 10.1097/MCG.0b013e3182a87f70. PubMed
22. Fagan KJ, Zhao EY, Horsfall LU, et al. Burden of decompensated cirrhosis and ascites on hospital services in a tertiary care facility: time for change? Intern Med J. 2014;44(9):865-872. DOI: 10.1111/imj.12491. PubMed
23. Gaduputi V, Chandrala C, Abbas N, Tariq H, Chilimuri S, Balar B. Prognostic significance of hypokalemia in hepatic encephalopathy. Hepatogastroenterology. 2014;61(133):1170-1174. PubMed
26. Agrawal K, Kumar P, Markert R, Agrawal S. Risk factors for 30-day readmissions of individuals with decompensated cirrhosis. South Med J. 2015;108(11):682-687. DOI: 10.14423/SMJ.0000000000000371. PubMed
27. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Lai M. Standard assessments of frailty are validated predictors of mortality in hospitalized patients with cirrhosis. Hepatology. 2015;62(2):584-590. DOI: 10.1002/hep.27830. PubMed
28. Atla PR, Sheikh MY, Gill F, Kundu R, Choudhury J. Predictors of hospital re-admissions among Hispanics with hepatitis C-related cirrhosis. Ann Gastroenterol. 2016;29(4):515-520. DOI: 10.20524/aog.2016.0072. PubMed
29. Bajaj JS, Reddy KR, Tandon P, et al. The 3-month readmission rate remains unacceptably high in a large North American cohort of patients with cirrhosis. Hepatology. 2016;64(1):200-208. DOI: 10.1002/hep.28414. PubMed
30. Courson A, Jones GM, Twilla JD. Treatment of acute hepatic encephalopathy: comparing the effects of adding rifaximin to lactulose on patient outcomes. J Pharm Pract. 2016;29(3):212-217. DOI: 10.1177/0897190014566312. PubMed
31. Graupera I, Solà E, Fabrellas N, et al. Urine monocyte chemoattractant protein-1 is an independent predictive factor of hospital readmission and survival in cirrhosis. PLOS ONE. 2016;11(6):e0157371. DOI: 10.1371/journal.pone.0157371. PubMed
32. Kanwal F, Asch SM, Kramer JR, Cao Y, Asrani S, El-Serag HB. Early outpatient follow-up and 30-day outcomes in patients hospitalized with cirrhosis. Hepatology. 2016;64(2):569-581. DOI: 10.1002/hep.28558. PubMed
46. Tapper EB, Finkelstein D, Mittleman MA, Piatkowski G, Chang M, Lai M. A quality improvement initiative reduces 30-day rate of readmission for patients with cirrhosis. Clin Gastroenterol Hepatol. 2016;14(5):753-759. DOI: 10.1016/j.cgh.2015.08.041. PubMed
45. Wigg AJ, McCormick R, Wundke R, Woodman RJ. Efficacy of a chronic disease management model for patients with chronic liver failure. Clin Gastroenterol Hepatol. 2013;11(7):850-8.e1. DOI: 10.1016/j.cgh.2013.01.014. PubMed
44. Ganesh S, Rogal SS, Yadav D, Humar A, Behari J. Risk factors for frequent readmissions and barriers to transplantation in patients with cirrhosis. PLOS ONE. 2013;8(1):e55140. DOI: 10.1371/journal.pone.0055140. PubMed
43. van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391-E402. DOI: 10.1503/cmaj.101860. PubMed
42. Bajaj JS, Wade JB, Gibson DP, et al. The multi-dimensional burden of cirrhosis and hepatic encephalopathy on patients and caregivers. Am J Gastroenterol. 2011;106(9):1646-1653. DOI: 10.1038/ajg.2011.157. PubMed
41. van Walraven C, Dhalla IA, Bell C, et al. Derivation and validation of an index to predict early death or unplanned readmission after discharge from hospital to the community. CMAJ. 2010;182(6):551-557. DOI: 10.1503/cmaj.091117. PubMed
40. Rodríguez-Artalejo F, Guallar-Castillón P, Pascual CR, et al. Health-related quality of life as a predictor of hospital readmission and death among patients with heart failure. Arch Intern Med. 2005;165(11):1274-1279. DOI: 10.1001/archinte.165.11.1274. PubMed
39. Strömdahl M, Helgeson J, Kalaitzakis E. Emergency readmission following acute upper gastrointestinal bleeding. Eur J Gastroenterol Hepatol. 2017;29(1):73-77. DOI: 10.1097/MEG.0000000000000746. PubMed
38. Morales BP, Planas R, Bartoli R, et al. Early hospital readmission in decompensated cirrhosis: incidence, impact on mortality, and predictive factors. Dig Liver Dis. 2017;49(8):903-909. DOI: 10.1016/j.dld.2017.03.005. PubMed
37. Lyon KC, Likar E, Martello JL, Regier M. Retrospective cross-sectional pilot study of rifaximin dosing for the prevention of recurrent hepatic encephalopathy. J Gastroenterol Hepatol. 2017;32(9):1548-1552. DOI: 10.1111/jgh.13759. PubMed
36. Tapper EB, Halbert B, Mellinger J. Rates of and reasons for hospital readmissions in patients with cirrhosis: a multistate population-based cohort study. Clin Gastroenterol Hepatol. 2016;14(8):1181-1188.e2. DOI: 10.1016/j.cgh.2016.04.009. PubMed
35. Rassameehiran S, Mankongpaisarnrung C, Sutamtewagul G, Klomjit S, Rakvit A. Predictor of 90-day readmission rate for hepatic encephalopathy. South Med J. 2016;109(6):365-369. DOI: 10.14423/SMJ.0000000000000475. PubMed
34. Moon AM, Dominitz JA, Ioannou GN, Lowy E, Beste LA. Use of antibiotics among patients with cirrhosis and upper gastrointestinal bleeding is associated with reduced mortality. Clin Gastroenterol Hepatol. 2016;14(11):1629-1637.e1. DOI: 10.1016/j.cgh.2016.05.040. PubMed
33. Le S, Spelman T, Chong CP, et al. Could adherence to quality of care indicators for hospitalized patients with cirrhosis-related ascites improve clinical outcomes? Am J Gastroenterol. 2016;111(1):87-92. DOI: .10.1038/ajg.2015.402. PubMed
© 2018 Society of Hospital Medicine
Tissue Isn’t the Issue
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.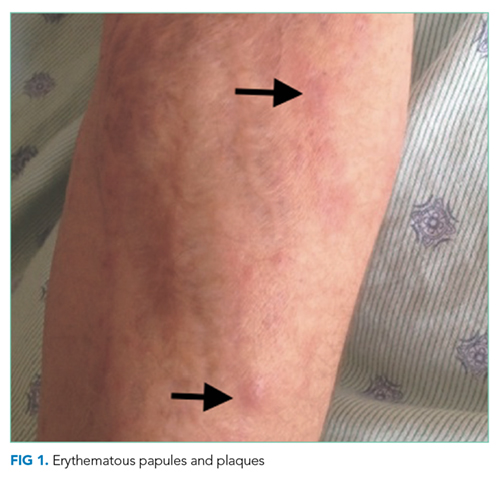
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower

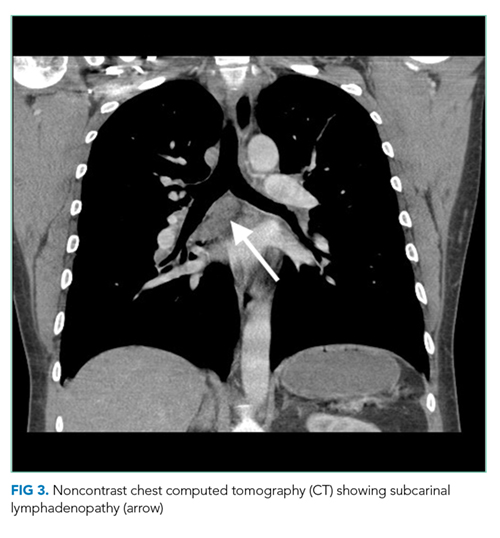
The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower
The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower
The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
© 2018 Society of Hospital Medicine
Continuous Physiologic Monitoring: False Alarms and Overdiagnosis
What is the most common intervention to which hospitalized children are exposed? Acetaminophen? IV access? Phlebotomy? Or is it being connected to a monitor?
In a study conducted in five children’s hospitals, Schondelmeyer et al found that exposure to continuous electronic physiologic monitoring was extremely common. During a selected 24-hour window of observation, nearly 100% of PICU and NICU patients and 26%-48% of medical–surgical patients were exposed to continuous monitoring.1 The latter is undoubtedly an underestimate given that monitoring periods less than 24 hours were not captured, patients may have been exposed before or after the 24-hour study window, and monitoring in the emergency department was not included.
The omnipresence of electronic physiologic monitoring in children’s hospitals is striking, particularly because we know very little about its benefits. Outside of the perioperative period, there is a dearth of evidence demonstrating improved outcomes for hospitalized children as a result of continuous physiologic monitoring. Guidelines for the most common inpatient pediatric conditions do not advocate for continuous physiologic monitoring. Presumably, this practice has become so pervasive in the absence of a strong evidence base and guideline recommendations because it is a passive, seemingly innocuous intervention that continuously collects important components of the physical examination (after all, they are known as “vital” signs). It is tempting to assume that providing clinicians with this information will make patients safer.
The danger of routinely exposing children to an intervention for which the benefits are unproven is that the net effect of the intervention may be harm. What could be harmful? The simple act of monitoring is distressing to children; sticky electrode pads stuck to their skin and a tangle of wires that restrict their movement–all impeding physical activity and contact with loved ones.
Then, there are the alarms. Schondelmeyer et al report a staggering number of them: between 42 and 152 alarms per monitored day on the floor; between 54 and 351 alarms in the intensive care units. The vast majority are false alarms, triggered by inappropriate preselected thresholds or displaced leads. This cacophony of noise only amplifies an already stressful environment for our patients–and their parents. Nurses and physicians are similarly stressed by alarms, not only by the noise but also by the frequent need to respond to them. The combination of frequent and largely unnecessary interruptions leads to alarm fatigue, whereby providers are desensitized to the alarms and may be slower to recognize a truly decompensating patient.
Continuous monitoring also risks overdiagnosis, the accurate detection of abnormalities that are not destined to cause problems, but nonetheless trigger interventions that can cause harm.2 Studies in adult populations have demonstrated that continuous monitoring can produce overdiagnosis. Repeated Cochrane reviews conclude that continuous electronic fetal monitoring during labor is associated with overdiagnosis of fetal distress—with attendant increase in cesarean sections without decreasing the risk for important neonatal outcomes such as cerebral palsy and mortality.3 A recent randomized trial of continuous pulmonary impedance monitoring intended to reduce readmission rates in patients with CHF instead found that continuous monitoring resulted in overdiagnosis of CHF exacerbations—paradoxically increasing hospital admission with no significant change in mortality (in fact, mortality was nominally higher in the monitoring group).4
Pediatric providers are probably no less susceptible to the impulse to act in the face of abnormalities detected by continuous monitoring. EKGs and electrolyte panels may be ordered in response to transient arrhythmias. Similarly, it is challenging for providers to watch a monitor flashing elevated respiratory rates in an otherwise healthy infant with bronchiolitis and not seek an escalation in care, including increased oxygen flow or transfer to a higher acuity unit. Although arrhythmia and respiratory rate alarms were common in Schondelmeyer et al’s study, low oxygen level was far and away the most common alarm. Indeed, the poster child of pediatric overdiagnosis in the setting of electronic physiologic monitoring is hypoxemia. The present body of literature suggests that overreliance on pulse oximetry among patients with bronchiolitis increases admission rates to the hospital and prolongs length of stay, without a measurable improvement in morbidity or mortality.5
Few patients cared for at American children’s hospitals will be discharged without exposure to prolonged periods of continuous physiologic monitoring. Undoubtedly, there are inpatients who benefit from this technology, such as children on mechanical ventilators. Unfortunately, there are also patients who are undoubtedly harmed by it. Greater understanding of which types of patients are more likely to benefit and which are more likely to be harmed is needed to determine whether continuous physiologic monitoring should remain our most common hospital intervention.
Disclosures
The authors have no financial relationships relevant to this article to disclose.
Funding
No external funding was secured for this study.
1. Schondelmeyer AC , Brady PW, Goel VV, et al. Physiologic monitor alarm rates at 5 children’s hospitals [published online ahead of print April 25, 2018}. J Hosp Med. 2018;13(6):396-398. PubMed
2. Welch HG, Schwartz L, Woloshin S. Overdiagnosed: Making people sick in the pursuit of health. Boston, Mass: Beacon Press; 2011.
3. Alfirevic Z, Devane D, Gyte GM, Cuthbert A. Continuous cardiotocography (CTG) as a form of electronic fetal monitoring (EFM) for fetal assessment during labour. Cochrane Database Syst Rev. 2017;2:Cd006066. PubMed
4. van Veldhuisen DJ, Braunschweig F, Conraads V, et al. Intrathoracic impedance monitoring, audible patient alerts, and outcome in patients with heart failure. Circulation. 2011;124:1719-1726. PubMed
5. Quinonez RA, Coon ER, Schroeder AR, Moyer VA. When technology creates uncertainty: pulse oximetry and overdiagnosis of hypoxaemia in bronchiolitis. BMJ. 2017;358:j3850. PubMed
What is the most common intervention to which hospitalized children are exposed? Acetaminophen? IV access? Phlebotomy? Or is it being connected to a monitor?
In a study conducted in five children’s hospitals, Schondelmeyer et al found that exposure to continuous electronic physiologic monitoring was extremely common. During a selected 24-hour window of observation, nearly 100% of PICU and NICU patients and 26%-48% of medical–surgical patients were exposed to continuous monitoring.1 The latter is undoubtedly an underestimate given that monitoring periods less than 24 hours were not captured, patients may have been exposed before or after the 24-hour study window, and monitoring in the emergency department was not included.
The omnipresence of electronic physiologic monitoring in children’s hospitals is striking, particularly because we know very little about its benefits. Outside of the perioperative period, there is a dearth of evidence demonstrating improved outcomes for hospitalized children as a result of continuous physiologic monitoring. Guidelines for the most common inpatient pediatric conditions do not advocate for continuous physiologic monitoring. Presumably, this practice has become so pervasive in the absence of a strong evidence base and guideline recommendations because it is a passive, seemingly innocuous intervention that continuously collects important components of the physical examination (after all, they are known as “vital” signs). It is tempting to assume that providing clinicians with this information will make patients safer.
The danger of routinely exposing children to an intervention for which the benefits are unproven is that the net effect of the intervention may be harm. What could be harmful? The simple act of monitoring is distressing to children; sticky electrode pads stuck to their skin and a tangle of wires that restrict their movement–all impeding physical activity and contact with loved ones.
Then, there are the alarms. Schondelmeyer et al report a staggering number of them: between 42 and 152 alarms per monitored day on the floor; between 54 and 351 alarms in the intensive care units. The vast majority are false alarms, triggered by inappropriate preselected thresholds or displaced leads. This cacophony of noise only amplifies an already stressful environment for our patients–and their parents. Nurses and physicians are similarly stressed by alarms, not only by the noise but also by the frequent need to respond to them. The combination of frequent and largely unnecessary interruptions leads to alarm fatigue, whereby providers are desensitized to the alarms and may be slower to recognize a truly decompensating patient.
Continuous monitoring also risks overdiagnosis, the accurate detection of abnormalities that are not destined to cause problems, but nonetheless trigger interventions that can cause harm.2 Studies in adult populations have demonstrated that continuous monitoring can produce overdiagnosis. Repeated Cochrane reviews conclude that continuous electronic fetal monitoring during labor is associated with overdiagnosis of fetal distress—with attendant increase in cesarean sections without decreasing the risk for important neonatal outcomes such as cerebral palsy and mortality.3 A recent randomized trial of continuous pulmonary impedance monitoring intended to reduce readmission rates in patients with CHF instead found that continuous monitoring resulted in overdiagnosis of CHF exacerbations—paradoxically increasing hospital admission with no significant change in mortality (in fact, mortality was nominally higher in the monitoring group).4
Pediatric providers are probably no less susceptible to the impulse to act in the face of abnormalities detected by continuous monitoring. EKGs and electrolyte panels may be ordered in response to transient arrhythmias. Similarly, it is challenging for providers to watch a monitor flashing elevated respiratory rates in an otherwise healthy infant with bronchiolitis and not seek an escalation in care, including increased oxygen flow or transfer to a higher acuity unit. Although arrhythmia and respiratory rate alarms were common in Schondelmeyer et al’s study, low oxygen level was far and away the most common alarm. Indeed, the poster child of pediatric overdiagnosis in the setting of electronic physiologic monitoring is hypoxemia. The present body of literature suggests that overreliance on pulse oximetry among patients with bronchiolitis increases admission rates to the hospital and prolongs length of stay, without a measurable improvement in morbidity or mortality.5
Few patients cared for at American children’s hospitals will be discharged without exposure to prolonged periods of continuous physiologic monitoring. Undoubtedly, there are inpatients who benefit from this technology, such as children on mechanical ventilators. Unfortunately, there are also patients who are undoubtedly harmed by it. Greater understanding of which types of patients are more likely to benefit and which are more likely to be harmed is needed to determine whether continuous physiologic monitoring should remain our most common hospital intervention.
Disclosures
The authors have no financial relationships relevant to this article to disclose.
Funding
No external funding was secured for this study.
What is the most common intervention to which hospitalized children are exposed? Acetaminophen? IV access? Phlebotomy? Or is it being connected to a monitor?
In a study conducted in five children’s hospitals, Schondelmeyer et al found that exposure to continuous electronic physiologic monitoring was extremely common. During a selected 24-hour window of observation, nearly 100% of PICU and NICU patients and 26%-48% of medical–surgical patients were exposed to continuous monitoring.1 The latter is undoubtedly an underestimate given that monitoring periods less than 24 hours were not captured, patients may have been exposed before or after the 24-hour study window, and monitoring in the emergency department was not included.
The omnipresence of electronic physiologic monitoring in children’s hospitals is striking, particularly because we know very little about its benefits. Outside of the perioperative period, there is a dearth of evidence demonstrating improved outcomes for hospitalized children as a result of continuous physiologic monitoring. Guidelines for the most common inpatient pediatric conditions do not advocate for continuous physiologic monitoring. Presumably, this practice has become so pervasive in the absence of a strong evidence base and guideline recommendations because it is a passive, seemingly innocuous intervention that continuously collects important components of the physical examination (after all, they are known as “vital” signs). It is tempting to assume that providing clinicians with this information will make patients safer.
The danger of routinely exposing children to an intervention for which the benefits are unproven is that the net effect of the intervention may be harm. What could be harmful? The simple act of monitoring is distressing to children; sticky electrode pads stuck to their skin and a tangle of wires that restrict their movement–all impeding physical activity and contact with loved ones.
Then, there are the alarms. Schondelmeyer et al report a staggering number of them: between 42 and 152 alarms per monitored day on the floor; between 54 and 351 alarms in the intensive care units. The vast majority are false alarms, triggered by inappropriate preselected thresholds or displaced leads. This cacophony of noise only amplifies an already stressful environment for our patients–and their parents. Nurses and physicians are similarly stressed by alarms, not only by the noise but also by the frequent need to respond to them. The combination of frequent and largely unnecessary interruptions leads to alarm fatigue, whereby providers are desensitized to the alarms and may be slower to recognize a truly decompensating patient.
Continuous monitoring also risks overdiagnosis, the accurate detection of abnormalities that are not destined to cause problems, but nonetheless trigger interventions that can cause harm.2 Studies in adult populations have demonstrated that continuous monitoring can produce overdiagnosis. Repeated Cochrane reviews conclude that continuous electronic fetal monitoring during labor is associated with overdiagnosis of fetal distress—with attendant increase in cesarean sections without decreasing the risk for important neonatal outcomes such as cerebral palsy and mortality.3 A recent randomized trial of continuous pulmonary impedance monitoring intended to reduce readmission rates in patients with CHF instead found that continuous monitoring resulted in overdiagnosis of CHF exacerbations—paradoxically increasing hospital admission with no significant change in mortality (in fact, mortality was nominally higher in the monitoring group).4
Pediatric providers are probably no less susceptible to the impulse to act in the face of abnormalities detected by continuous monitoring. EKGs and electrolyte panels may be ordered in response to transient arrhythmias. Similarly, it is challenging for providers to watch a monitor flashing elevated respiratory rates in an otherwise healthy infant with bronchiolitis and not seek an escalation in care, including increased oxygen flow or transfer to a higher acuity unit. Although arrhythmia and respiratory rate alarms were common in Schondelmeyer et al’s study, low oxygen level was far and away the most common alarm. Indeed, the poster child of pediatric overdiagnosis in the setting of electronic physiologic monitoring is hypoxemia. The present body of literature suggests that overreliance on pulse oximetry among patients with bronchiolitis increases admission rates to the hospital and prolongs length of stay, without a measurable improvement in morbidity or mortality.5
Few patients cared for at American children’s hospitals will be discharged without exposure to prolonged periods of continuous physiologic monitoring. Undoubtedly, there are inpatients who benefit from this technology, such as children on mechanical ventilators. Unfortunately, there are also patients who are undoubtedly harmed by it. Greater understanding of which types of patients are more likely to benefit and which are more likely to be harmed is needed to determine whether continuous physiologic monitoring should remain our most common hospital intervention.
Disclosures
The authors have no financial relationships relevant to this article to disclose.
Funding
No external funding was secured for this study.
1. Schondelmeyer AC , Brady PW, Goel VV, et al. Physiologic monitor alarm rates at 5 children’s hospitals [published online ahead of print April 25, 2018}. J Hosp Med. 2018;13(6):396-398. PubMed
2. Welch HG, Schwartz L, Woloshin S. Overdiagnosed: Making people sick in the pursuit of health. Boston, Mass: Beacon Press; 2011.
3. Alfirevic Z, Devane D, Gyte GM, Cuthbert A. Continuous cardiotocography (CTG) as a form of electronic fetal monitoring (EFM) for fetal assessment during labour. Cochrane Database Syst Rev. 2017;2:Cd006066. PubMed
4. van Veldhuisen DJ, Braunschweig F, Conraads V, et al. Intrathoracic impedance monitoring, audible patient alerts, and outcome in patients with heart failure. Circulation. 2011;124:1719-1726. PubMed
5. Quinonez RA, Coon ER, Schroeder AR, Moyer VA. When technology creates uncertainty: pulse oximetry and overdiagnosis of hypoxaemia in bronchiolitis. BMJ. 2017;358:j3850. PubMed
1. Schondelmeyer AC , Brady PW, Goel VV, et al. Physiologic monitor alarm rates at 5 children’s hospitals [published online ahead of print April 25, 2018}. J Hosp Med. 2018;13(6):396-398. PubMed
2. Welch HG, Schwartz L, Woloshin S. Overdiagnosed: Making people sick in the pursuit of health. Boston, Mass: Beacon Press; 2011.
3. Alfirevic Z, Devane D, Gyte GM, Cuthbert A. Continuous cardiotocography (CTG) as a form of electronic fetal monitoring (EFM) for fetal assessment during labour. Cochrane Database Syst Rev. 2017;2:Cd006066. PubMed
4. van Veldhuisen DJ, Braunschweig F, Conraads V, et al. Intrathoracic impedance monitoring, audible patient alerts, and outcome in patients with heart failure. Circulation. 2011;124:1719-1726. PubMed
5. Quinonez RA, Coon ER, Schroeder AR, Moyer VA. When technology creates uncertainty: pulse oximetry and overdiagnosis of hypoxaemia in bronchiolitis. BMJ. 2017;358:j3850. PubMed
© 2018 Society of Hospital Medicine
Physiologic Monitor Alarm Rates at 5 Children’s Hospitals
Alarm fatigue is a patient safety hazard in hospitals1 that occurs when exposure to high rates of alarms leads clinicians to ignore or delay their responses to the alarms.2,3 To date, most studies of physiologic monitor alarms in hospitalized children have used data from single institutions and often only a few units within each institution.4 These limited studies have found that alarms in pediatric units are rarely actionable.2 They have also shown that physiologic monitor alarms occur frequently in children’s hospitals and that alarm rates can vary widely within a single institution,5 but the extent of variation between children’s hospitals is unknown. In this study, we aimed to describe and compare physiologic monitor alarm characteristics and the proportion of patients monitored in the inpatient units of 5 children’s hospitals.
METHODS
We performed a cross-sectional study using a point-prevalence design of physiologic monitor alarms and monitoring during a 24-hour period at 5 large, freestanding tertiary-care children’s hospitals. At the time of the study, each hospital had an alarm management committee in place and was working to address alarm fatigue. Each hospital’s institutional review board reviewed and approved the study.
We collected 24 consecutive hours of data from the inpatient units of each hospital between March 24, 2015, and May 1, 2015. Each hospital selected the data collection date within that window based on the availability of staff to perform data collection.6 We excluded emergency departments, procedural areas, and inpatient psychiatry and rehabilitation units. By using existing central alarm-collection software that interfaced with bedside physiologic monitors, we collected data on audible alarms generated for apnea, arrhythmia, low and high oxygen saturation, heart rate, respiratory rate, blood pressure, and exhaled carbon dioxide. Bedside alarm systems and alarm collection software differed between centers; therefore, alarm types that were not consistently collected at every institution (eg, alarms for electrode and device malfunction, ventilators, intracranial and central venous pressure monitors, and temperatures probes) were excluded. To estimate alarm rates and to account for fluctuations in hospital census throughout the day,7 we collected census (to calculate the number of alarms per patient day) and the number of monitored patients (to calculate the number of alarms per monitored-patient day, including only monitored patients in the denominator) on each unit at 3 time points, 8 hours apart. Patients were considered continuously monitored if they had presence of a waveform and data for pulse oximetry, respiratory rate, and/or heart rate at the time of data collection. We then determined the rate of alarms by unit type—medical-surgical unit (MSU), neonatal intensive care unit (NICU), or pediatric intensive care unit (PICU)—and the alarm types. Based on prior literature demonstrating up to 95% of alarms contributed by a minority of patients on a single unit,8 we also calculated the percentage of alarms contributed by beds in the highest quartile of alarms. We also assessed the percentage of patients monitored by unit type. The Supplementary Appendix shows the alarm parameter thresholds in use at the time of the study.
RESULTS
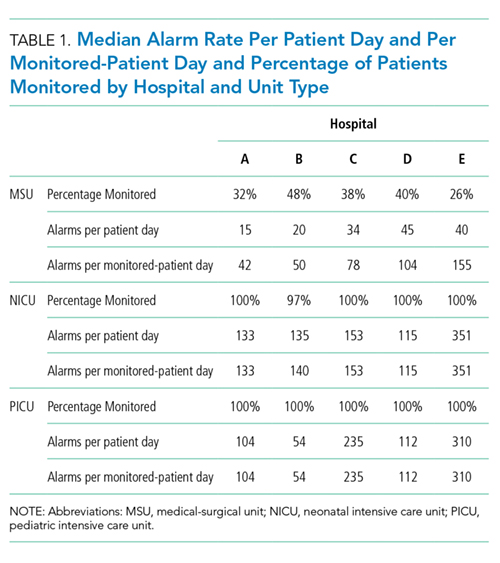
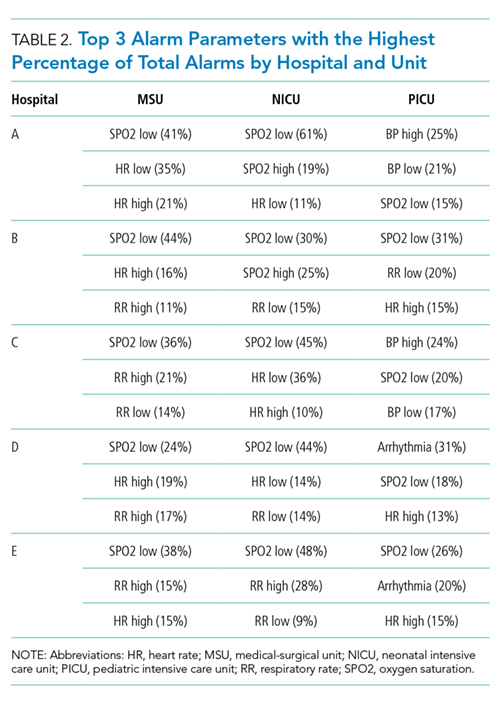
Averaged across study hospitals, one-quarter of the monitored beds were responsible for 71% of alarms in MSUs, 61% of alarms in NICUs, and 63% of alarms in PICUs.
DISCUSSION
Physiologic monitor alarm rates and the proportion of patients monitored varied widely between unit types and among the tertiary-care children’s hospitals in our study. We found that among MSUs, the hospital with the lowest proportion of beds monitored had the highest alarm rate, with over triple the rate seen at the hospital with the lowest alarm rate. Regardless of unit type, a small subgroup of patients at each hospital contributed a disproportionate share of alarms. These findings are concerning because of the patient morbidity and mortality associated with alarm fatigue1 and the studies suggesting that higher alarm rates may lead to delays in response to potentially critical alarms.2
We previously described alarm rates at a single children’s hospital and found that alarm rates were high both in and outside of the ICU areas.5 This study supports those findings and goes further to show that alarm rates on some MSUs approached rates seen in the ICU areas at other centers.4 However, our results should be considered in the context of several limitations. First, the 5 study hospitals utilized different bedside monitors, equipment, and software to collect alarm data. It is possible that this impacted how alarms were counted, though there were no technical specifications to suggest that results should have been biased in a specific way. Second, our data did not reflect alarm validity (ie, whether an alarm accurately reflected the physiologic state of the patient) or factors outside of the number of patients monitored—such as practices around ICU admission and transfer as well as monitor practices such as lead changes, the type of leads employed, and the degree to which alarm parameter thresholds could be customized, which may have also affected alarm rates. Finally, we excluded alarm types that were not consistently collected at all hospitals. We were also unable to capture alarms from other alarm-generating devices, including ventilators and infusion pumps, which have also been identified as sources of alarm-related safety issues in hospitals.9-11 This suggests that the alarm rates reported here underestimate the total number of audible alarms experienced by staff and by hospitalized patients and families.
While our data collection was limited in scope, the striking differences in alarm rates between hospitals and between similar units in the same hospitals suggest that unit- and hospital-level factors—including default alarm parameter threshold settings, types of monitors used, and monitoring practices such as the degree to which alarm parameters are customized to the patient’s physiologic state—likely contribute to the variability. It is also important to note that while there were clear outlier hospitals, no single hospital had the lowest alarm rate across all unit types. And while we found that a small number of patients contributed disproportionately to alarms, monitoring fewer patients overall was not consistently associated with lower alarm rates. While it is difficult to draw conclusions based on a limited study, these findings suggest that solutions to meaningfully lower alarm rates may be multifaceted. Standardization of care in multiple areas of medicine has shown the potential to decrease unnecessary utilization of testing and therapies while maintaining good patient outcomes.12-15 Our findings suggest that the concept of positive deviance,16 by which some organizations produce better outcomes than others despite similar limitations, may help identify successful alarm reduction strategies for further testing. Larger quantitative studies of alarm rates and ethnographic or qualitative studies of monitoring practices may reveal practices and policies that are associated with lower alarm rates with similar or improved monitoring outcomes.
CONCLUSION
We found wide variability in physiologic monitor alarm rates and the proportion of patients monitored across 5 children’s hospitals. Because alarm fatigue remains a pressing patient safety concern, further study of the features of high-performing (low-alarm) hospital systems may help identify barriers and facilitators of safe, effective monitoring and develop targeted interventions to reduce alarms.
ACKNOWLEDGEMENTS
The authors thank Melinda Egan, Matt MacMurchy, and Shannon Stemler for their assistance with data collection.
Disclosure
Dr. Bonafide is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number K23HL116427. Dr. Brady is supported by the Agency for Healthcare Research and Quality under Award Number K08HS23827. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Agency for Healthcare Research and Quality. There was no external funding obtained for this study. The authors have no conflicts of interest to disclose.
1. Sentinel Event Alert Issue 50: Medical device alarm safety in hospitals. The Joint Commission. April 8, 2013. www.jointcommission.org/sea_issue_50. Accessed December 16, 2017.
2. Bonafide CP, Lin R, Zander M, et al. Association between exposure to nonactionable physiologic monitor alarms and response time in a children’s hospital. J Hosp Med. 2015;10(6):345-351. PubMed
3. Voepel-Lewis T, Parker ML, Burke CN, et al. Pulse oximetry desaturation alarms on a general postoperative adult unit: A prospective observational study of nurse response time. Int J Nurs Stud. 2013;50(10):1351-1358. PubMed
4. Paine CW, Goel VV, Ely E, et al. Systematic review of physiologic monitor alarm characteristics and pragmatic interventions to reduce alarm frequency. J Hosp Med. 2016;11(2):136-144. PubMed
5. Schondelmeyer AC, Bonafide CP, Goel VV, et al. The frequency of physiologic monitor alarms in a children’s hospital. J Hosp Med. 2016;11(11):796-798. PubMed
6. Zingg W, Hopkins S, Gayet-Ageron A, et al. Health-care-associated infections in neonates, children, and adolescents: An analysis of paediatric data from the European Centre for Disease Prevention and Control point-prevalence survey. Lancet Infect Dis. 2017;17(4):381-389. PubMed
7. Fieldston E, Ragavan M, Jayaraman B, Metlay J, Pati S. Traditional measures of hospital utilization may not accurately reflect dynamic patient demand: Findings from a children’s hospital. Hosp Pediatr. 2012;2(1):10-18. PubMed
8. Cvach M, Kitchens M, Smith K, Harris P, Flack MN. Customizing alarm limits based on specific needs of patients. Biomed Instrum Technol. 2017;51(3):227-234. PubMed
9. Pham JC, Williams TL, Sparnon EM, Cillie TK, Scharen HF, Marella WM. Ventilator-related adverse events: A taxonomy and findings from 3 incident reporting systems. Respir Care. 2016;61(5):621-631. PubMed
10. Cho OM, Kim H, Lee YW, Cho I. Clinical alarms in intensive care units: Perceived obstacles of alarm management and alarm fatigue in nurses. Healthc Inform Res. 2016;22(1):46-53. PubMed
11. Edworthy J, Hellier E. Alarms and human behaviour: Implications for medical alarms. Br J Anaesth. 2006;97(1):12-17. PubMed
12. Fisher ES, Wennberg DE, Stukel TA, Gottlieb DJ, Lucas FL, Pinder EL. The implications of regional variations in medicare spending. Part 1: The content, quality, and accessibility of care. Ann Intern Med. 2003;138(4):273-287. PubMed
13. Fisher ES, Wennberg DE, Stukel TA, Gottlieb DJ, Lucas FL, Pinder EL. The implications of regional variations in medicare spending. Part 2: Health outcomes and satisfaction with care. Ann Intern Med. 2003;138(4):288-298. PubMed
14. Lion KC, Wright DR, Spencer S, Zhou C, Del Beccaro M, Mangione-Smith R. Standardized clinical pathways for hospitalized children and outcomes. Pediatrics. 2016;137(4) e20151202. PubMed
15. Goodman DC. Unwarranted variation in pediatric medical care. Pediatr Clin North Am. 2009;56(4):745-755. PubMed
16. Baxter R, Taylor N, Kellar I, Lawton R. What methods are used to apply positive deviance within healthcare organisations? A systematic review. BMJ Qual Saf. 2016;25(3):190-201. PubMed
Alarm fatigue is a patient safety hazard in hospitals1 that occurs when exposure to high rates of alarms leads clinicians to ignore or delay their responses to the alarms.2,3 To date, most studies of physiologic monitor alarms in hospitalized children have used data from single institutions and often only a few units within each institution.4 These limited studies have found that alarms in pediatric units are rarely actionable.2 They have also shown that physiologic monitor alarms occur frequently in children’s hospitals and that alarm rates can vary widely within a single institution,5 but the extent of variation between children’s hospitals is unknown. In this study, we aimed to describe and compare physiologic monitor alarm characteristics and the proportion of patients monitored in the inpatient units of 5 children’s hospitals.
METHODS
We performed a cross-sectional study using a point-prevalence design of physiologic monitor alarms and monitoring during a 24-hour period at 5 large, freestanding tertiary-care children’s hospitals. At the time of the study, each hospital had an alarm management committee in place and was working to address alarm fatigue. Each hospital’s institutional review board reviewed and approved the study.
We collected 24 consecutive hours of data from the inpatient units of each hospital between March 24, 2015, and May 1, 2015. Each hospital selected the data collection date within that window based on the availability of staff to perform data collection.6 We excluded emergency departments, procedural areas, and inpatient psychiatry and rehabilitation units. By using existing central alarm-collection software that interfaced with bedside physiologic monitors, we collected data on audible alarms generated for apnea, arrhythmia, low and high oxygen saturation, heart rate, respiratory rate, blood pressure, and exhaled carbon dioxide. Bedside alarm systems and alarm collection software differed between centers; therefore, alarm types that were not consistently collected at every institution (eg, alarms for electrode and device malfunction, ventilators, intracranial and central venous pressure monitors, and temperatures probes) were excluded. To estimate alarm rates and to account for fluctuations in hospital census throughout the day,7 we collected census (to calculate the number of alarms per patient day) and the number of monitored patients (to calculate the number of alarms per monitored-patient day, including only monitored patients in the denominator) on each unit at 3 time points, 8 hours apart. Patients were considered continuously monitored if they had presence of a waveform and data for pulse oximetry, respiratory rate, and/or heart rate at the time of data collection. We then determined the rate of alarms by unit type—medical-surgical unit (MSU), neonatal intensive care unit (NICU), or pediatric intensive care unit (PICU)—and the alarm types. Based on prior literature demonstrating up to 95% of alarms contributed by a minority of patients on a single unit,8 we also calculated the percentage of alarms contributed by beds in the highest quartile of alarms. We also assessed the percentage of patients monitored by unit type. The Supplementary Appendix shows the alarm parameter thresholds in use at the time of the study.
RESULTS
Averaged across study hospitals, one-quarter of the monitored beds were responsible for 71% of alarms in MSUs, 61% of alarms in NICUs, and 63% of alarms in PICUs.
DISCUSSION
Physiologic monitor alarm rates and the proportion of patients monitored varied widely between unit types and among the tertiary-care children’s hospitals in our study. We found that among MSUs, the hospital with the lowest proportion of beds monitored had the highest alarm rate, with over triple the rate seen at the hospital with the lowest alarm rate. Regardless of unit type, a small subgroup of patients at each hospital contributed a disproportionate share of alarms. These findings are concerning because of the patient morbidity and mortality associated with alarm fatigue1 and the studies suggesting that higher alarm rates may lead to delays in response to potentially critical alarms.2
We previously described alarm rates at a single children’s hospital and found that alarm rates were high both in and outside of the ICU areas.5 This study supports those findings and goes further to show that alarm rates on some MSUs approached rates seen in the ICU areas at other centers.4 However, our results should be considered in the context of several limitations. First, the 5 study hospitals utilized different bedside monitors, equipment, and software to collect alarm data. It is possible that this impacted how alarms were counted, though there were no technical specifications to suggest that results should have been biased in a specific way. Second, our data did not reflect alarm validity (ie, whether an alarm accurately reflected the physiologic state of the patient) or factors outside of the number of patients monitored—such as practices around ICU admission and transfer as well as monitor practices such as lead changes, the type of leads employed, and the degree to which alarm parameter thresholds could be customized, which may have also affected alarm rates. Finally, we excluded alarm types that were not consistently collected at all hospitals. We were also unable to capture alarms from other alarm-generating devices, including ventilators and infusion pumps, which have also been identified as sources of alarm-related safety issues in hospitals.9-11 This suggests that the alarm rates reported here underestimate the total number of audible alarms experienced by staff and by hospitalized patients and families.
While our data collection was limited in scope, the striking differences in alarm rates between hospitals and between similar units in the same hospitals suggest that unit- and hospital-level factors—including default alarm parameter threshold settings, types of monitors used, and monitoring practices such as the degree to which alarm parameters are customized to the patient’s physiologic state—likely contribute to the variability. It is also important to note that while there were clear outlier hospitals, no single hospital had the lowest alarm rate across all unit types. And while we found that a small number of patients contributed disproportionately to alarms, monitoring fewer patients overall was not consistently associated with lower alarm rates. While it is difficult to draw conclusions based on a limited study, these findings suggest that solutions to meaningfully lower alarm rates may be multifaceted. Standardization of care in multiple areas of medicine has shown the potential to decrease unnecessary utilization of testing and therapies while maintaining good patient outcomes.12-15 Our findings suggest that the concept of positive deviance,16 by which some organizations produce better outcomes than others despite similar limitations, may help identify successful alarm reduction strategies for further testing. Larger quantitative studies of alarm rates and ethnographic or qualitative studies of monitoring practices may reveal practices and policies that are associated with lower alarm rates with similar or improved monitoring outcomes.
CONCLUSION
We found wide variability in physiologic monitor alarm rates and the proportion of patients monitored across 5 children’s hospitals. Because alarm fatigue remains a pressing patient safety concern, further study of the features of high-performing (low-alarm) hospital systems may help identify barriers and facilitators of safe, effective monitoring and develop targeted interventions to reduce alarms.
ACKNOWLEDGEMENTS
The authors thank Melinda Egan, Matt MacMurchy, and Shannon Stemler for their assistance with data collection.
Disclosure
Dr. Bonafide is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number K23HL116427. Dr. Brady is supported by the Agency for Healthcare Research and Quality under Award Number K08HS23827. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Agency for Healthcare Research and Quality. There was no external funding obtained for this study. The authors have no conflicts of interest to disclose.
Alarm fatigue is a patient safety hazard in hospitals1 that occurs when exposure to high rates of alarms leads clinicians to ignore or delay their responses to the alarms.2,3 To date, most studies of physiologic monitor alarms in hospitalized children have used data from single institutions and often only a few units within each institution.4 These limited studies have found that alarms in pediatric units are rarely actionable.2 They have also shown that physiologic monitor alarms occur frequently in children’s hospitals and that alarm rates can vary widely within a single institution,5 but the extent of variation between children’s hospitals is unknown. In this study, we aimed to describe and compare physiologic monitor alarm characteristics and the proportion of patients monitored in the inpatient units of 5 children’s hospitals.
METHODS
We performed a cross-sectional study using a point-prevalence design of physiologic monitor alarms and monitoring during a 24-hour period at 5 large, freestanding tertiary-care children’s hospitals. At the time of the study, each hospital had an alarm management committee in place and was working to address alarm fatigue. Each hospital’s institutional review board reviewed and approved the study.
We collected 24 consecutive hours of data from the inpatient units of each hospital between March 24, 2015, and May 1, 2015. Each hospital selected the data collection date within that window based on the availability of staff to perform data collection.6 We excluded emergency departments, procedural areas, and inpatient psychiatry and rehabilitation units. By using existing central alarm-collection software that interfaced with bedside physiologic monitors, we collected data on audible alarms generated for apnea, arrhythmia, low and high oxygen saturation, heart rate, respiratory rate, blood pressure, and exhaled carbon dioxide. Bedside alarm systems and alarm collection software differed between centers; therefore, alarm types that were not consistently collected at every institution (eg, alarms for electrode and device malfunction, ventilators, intracranial and central venous pressure monitors, and temperatures probes) were excluded. To estimate alarm rates and to account for fluctuations in hospital census throughout the day,7 we collected census (to calculate the number of alarms per patient day) and the number of monitored patients (to calculate the number of alarms per monitored-patient day, including only monitored patients in the denominator) on each unit at 3 time points, 8 hours apart. Patients were considered continuously monitored if they had presence of a waveform and data for pulse oximetry, respiratory rate, and/or heart rate at the time of data collection. We then determined the rate of alarms by unit type—medical-surgical unit (MSU), neonatal intensive care unit (NICU), or pediatric intensive care unit (PICU)—and the alarm types. Based on prior literature demonstrating up to 95% of alarms contributed by a minority of patients on a single unit,8 we also calculated the percentage of alarms contributed by beds in the highest quartile of alarms. We also assessed the percentage of patients monitored by unit type. The Supplementary Appendix shows the alarm parameter thresholds in use at the time of the study.
RESULTS
Averaged across study hospitals, one-quarter of the monitored beds were responsible for 71% of alarms in MSUs, 61% of alarms in NICUs, and 63% of alarms in PICUs.
DISCUSSION
Physiologic monitor alarm rates and the proportion of patients monitored varied widely between unit types and among the tertiary-care children’s hospitals in our study. We found that among MSUs, the hospital with the lowest proportion of beds monitored had the highest alarm rate, with over triple the rate seen at the hospital with the lowest alarm rate. Regardless of unit type, a small subgroup of patients at each hospital contributed a disproportionate share of alarms. These findings are concerning because of the patient morbidity and mortality associated with alarm fatigue1 and the studies suggesting that higher alarm rates may lead to delays in response to potentially critical alarms.2
We previously described alarm rates at a single children’s hospital and found that alarm rates were high both in and outside of the ICU areas.5 This study supports those findings and goes further to show that alarm rates on some MSUs approached rates seen in the ICU areas at other centers.4 However, our results should be considered in the context of several limitations. First, the 5 study hospitals utilized different bedside monitors, equipment, and software to collect alarm data. It is possible that this impacted how alarms were counted, though there were no technical specifications to suggest that results should have been biased in a specific way. Second, our data did not reflect alarm validity (ie, whether an alarm accurately reflected the physiologic state of the patient) or factors outside of the number of patients monitored—such as practices around ICU admission and transfer as well as monitor practices such as lead changes, the type of leads employed, and the degree to which alarm parameter thresholds could be customized, which may have also affected alarm rates. Finally, we excluded alarm types that were not consistently collected at all hospitals. We were also unable to capture alarms from other alarm-generating devices, including ventilators and infusion pumps, which have also been identified as sources of alarm-related safety issues in hospitals.9-11 This suggests that the alarm rates reported here underestimate the total number of audible alarms experienced by staff and by hospitalized patients and families.
While our data collection was limited in scope, the striking differences in alarm rates between hospitals and between similar units in the same hospitals suggest that unit- and hospital-level factors—including default alarm parameter threshold settings, types of monitors used, and monitoring practices such as the degree to which alarm parameters are customized to the patient’s physiologic state—likely contribute to the variability. It is also important to note that while there were clear outlier hospitals, no single hospital had the lowest alarm rate across all unit types. And while we found that a small number of patients contributed disproportionately to alarms, monitoring fewer patients overall was not consistently associated with lower alarm rates. While it is difficult to draw conclusions based on a limited study, these findings suggest that solutions to meaningfully lower alarm rates may be multifaceted. Standardization of care in multiple areas of medicine has shown the potential to decrease unnecessary utilization of testing and therapies while maintaining good patient outcomes.12-15 Our findings suggest that the concept of positive deviance,16 by which some organizations produce better outcomes than others despite similar limitations, may help identify successful alarm reduction strategies for further testing. Larger quantitative studies of alarm rates and ethnographic or qualitative studies of monitoring practices may reveal practices and policies that are associated with lower alarm rates with similar or improved monitoring outcomes.
CONCLUSION
We found wide variability in physiologic monitor alarm rates and the proportion of patients monitored across 5 children’s hospitals. Because alarm fatigue remains a pressing patient safety concern, further study of the features of high-performing (low-alarm) hospital systems may help identify barriers and facilitators of safe, effective monitoring and develop targeted interventions to reduce alarms.
ACKNOWLEDGEMENTS
The authors thank Melinda Egan, Matt MacMurchy, and Shannon Stemler for their assistance with data collection.
Disclosure
Dr. Bonafide is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number K23HL116427. Dr. Brady is supported by the Agency for Healthcare Research and Quality under Award Number K08HS23827. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Agency for Healthcare Research and Quality. There was no external funding obtained for this study. The authors have no conflicts of interest to disclose.
1. Sentinel Event Alert Issue 50: Medical device alarm safety in hospitals. The Joint Commission. April 8, 2013. www.jointcommission.org/sea_issue_50. Accessed December 16, 2017.
2. Bonafide CP, Lin R, Zander M, et al. Association between exposure to nonactionable physiologic monitor alarms and response time in a children’s hospital. J Hosp Med. 2015;10(6):345-351. PubMed
3. Voepel-Lewis T, Parker ML, Burke CN, et al. Pulse oximetry desaturation alarms on a general postoperative adult unit: A prospective observational study of nurse response time. Int J Nurs Stud. 2013;50(10):1351-1358. PubMed
4. Paine CW, Goel VV, Ely E, et al. Systematic review of physiologic monitor alarm characteristics and pragmatic interventions to reduce alarm frequency. J Hosp Med. 2016;11(2):136-144. PubMed
5. Schondelmeyer AC, Bonafide CP, Goel VV, et al. The frequency of physiologic monitor alarms in a children’s hospital. J Hosp Med. 2016;11(11):796-798. PubMed
6. Zingg W, Hopkins S, Gayet-Ageron A, et al. Health-care-associated infections in neonates, children, and adolescents: An analysis of paediatric data from the European Centre for Disease Prevention and Control point-prevalence survey. Lancet Infect Dis. 2017;17(4):381-389. PubMed
7. Fieldston E, Ragavan M, Jayaraman B, Metlay J, Pati S. Traditional measures of hospital utilization may not accurately reflect dynamic patient demand: Findings from a children’s hospital. Hosp Pediatr. 2012;2(1):10-18. PubMed
8. Cvach M, Kitchens M, Smith K, Harris P, Flack MN. Customizing alarm limits based on specific needs of patients. Biomed Instrum Technol. 2017;51(3):227-234. PubMed
9. Pham JC, Williams TL, Sparnon EM, Cillie TK, Scharen HF, Marella WM. Ventilator-related adverse events: A taxonomy and findings from 3 incident reporting systems. Respir Care. 2016;61(5):621-631. PubMed
10. Cho OM, Kim H, Lee YW, Cho I. Clinical alarms in intensive care units: Perceived obstacles of alarm management and alarm fatigue in nurses. Healthc Inform Res. 2016;22(1):46-53. PubMed
11. Edworthy J, Hellier E. Alarms and human behaviour: Implications for medical alarms. Br J Anaesth. 2006;97(1):12-17. PubMed
12. Fisher ES, Wennberg DE, Stukel TA, Gottlieb DJ, Lucas FL, Pinder EL. The implications of regional variations in medicare spending. Part 1: The content, quality, and accessibility of care. Ann Intern Med. 2003;138(4):273-287. PubMed
13. Fisher ES, Wennberg DE, Stukel TA, Gottlieb DJ, Lucas FL, Pinder EL. The implications of regional variations in medicare spending. Part 2: Health outcomes and satisfaction with care. Ann Intern Med. 2003;138(4):288-298. PubMed
14. Lion KC, Wright DR, Spencer S, Zhou C, Del Beccaro M, Mangione-Smith R. Standardized clinical pathways for hospitalized children and outcomes. Pediatrics. 2016;137(4) e20151202. PubMed
15. Goodman DC. Unwarranted variation in pediatric medical care. Pediatr Clin North Am. 2009;56(4):745-755. PubMed
16. Baxter R, Taylor N, Kellar I, Lawton R. What methods are used to apply positive deviance within healthcare organisations? A systematic review. BMJ Qual Saf. 2016;25(3):190-201. PubMed
1. Sentinel Event Alert Issue 50: Medical device alarm safety in hospitals. The Joint Commission. April 8, 2013. www.jointcommission.org/sea_issue_50. Accessed December 16, 2017.
2. Bonafide CP, Lin R, Zander M, et al. Association between exposure to nonactionable physiologic monitor alarms and response time in a children’s hospital. J Hosp Med. 2015;10(6):345-351. PubMed
3. Voepel-Lewis T, Parker ML, Burke CN, et al. Pulse oximetry desaturation alarms on a general postoperative adult unit: A prospective observational study of nurse response time. Int J Nurs Stud. 2013;50(10):1351-1358. PubMed
4. Paine CW, Goel VV, Ely E, et al. Systematic review of physiologic monitor alarm characteristics and pragmatic interventions to reduce alarm frequency. J Hosp Med. 2016;11(2):136-144. PubMed
5. Schondelmeyer AC, Bonafide CP, Goel VV, et al. The frequency of physiologic monitor alarms in a children’s hospital. J Hosp Med. 2016;11(11):796-798. PubMed
6. Zingg W, Hopkins S, Gayet-Ageron A, et al. Health-care-associated infections in neonates, children, and adolescents: An analysis of paediatric data from the European Centre for Disease Prevention and Control point-prevalence survey. Lancet Infect Dis. 2017;17(4):381-389. PubMed
7. Fieldston E, Ragavan M, Jayaraman B, Metlay J, Pati S. Traditional measures of hospital utilization may not accurately reflect dynamic patient demand: Findings from a children’s hospital. Hosp Pediatr. 2012;2(1):10-18. PubMed
8. Cvach M, Kitchens M, Smith K, Harris P, Flack MN. Customizing alarm limits based on specific needs of patients. Biomed Instrum Technol. 2017;51(3):227-234. PubMed
9. Pham JC, Williams TL, Sparnon EM, Cillie TK, Scharen HF, Marella WM. Ventilator-related adverse events: A taxonomy and findings from 3 incident reporting systems. Respir Care. 2016;61(5):621-631. PubMed
10. Cho OM, Kim H, Lee YW, Cho I. Clinical alarms in intensive care units: Perceived obstacles of alarm management and alarm fatigue in nurses. Healthc Inform Res. 2016;22(1):46-53. PubMed
11. Edworthy J, Hellier E. Alarms and human behaviour: Implications for medical alarms. Br J Anaesth. 2006;97(1):12-17. PubMed
12. Fisher ES, Wennberg DE, Stukel TA, Gottlieb DJ, Lucas FL, Pinder EL. The implications of regional variations in medicare spending. Part 1: The content, quality, and accessibility of care. Ann Intern Med. 2003;138(4):273-287. PubMed
13. Fisher ES, Wennberg DE, Stukel TA, Gottlieb DJ, Lucas FL, Pinder EL. The implications of regional variations in medicare spending. Part 2: Health outcomes and satisfaction with care. Ann Intern Med. 2003;138(4):288-298. PubMed
14. Lion KC, Wright DR, Spencer S, Zhou C, Del Beccaro M, Mangione-Smith R. Standardized clinical pathways for hospitalized children and outcomes. Pediatrics. 2016;137(4) e20151202. PubMed
15. Goodman DC. Unwarranted variation in pediatric medical care. Pediatr Clin North Am. 2009;56(4):745-755. PubMed
16. Baxter R, Taylor N, Kellar I, Lawton R. What methods are used to apply positive deviance within healthcare organisations? A systematic review. BMJ Qual Saf. 2016;25(3):190-201. PubMed
© 2018 Society of Hospital Medicine