User login
Product increases FIX levels in hemophilia B
MADRID—The recombinant factor IX (FIX) product CB 2679d/ISU304 can increase FIX levels in patients with severe hemophilia B, according to a phase 1/2 trial.
Results showed a continuous linear increase in FIX activity levels following daily subcutaneous (SQ) dosing of CB 2679d for 6 days.
Adverse events were mild to moderate, and none of the patients have developed inhibitors to CB 2679d or FIX.
Howard Levy, MB ChB, PhD, chief medical officer of Catalyst Biosciences, Inc., presented these results at the 11th Annual Congress of The European Association for Haemophilia and Allied Disorders (EAHAD).
The research was sponsored by Catalyst Biosciences, Inc.
This phase 1/2 trial was divided into 5 cohorts.
In cohort 1, researchers compared single doses of intravenous (IV) CB 2679d and IV BeneFIX (recombinant FIX). CB 2679d proved 22 times more potent than BeneFIX. The products’ half-lives were 27.0 hours and 21.0 hours, respectively.
In cohorts 2 and 3, researchers compared single, ascending doses of IV CB 2679d to SQ CB 2679d. The bioavailability of SQ CB 2679d was 18.5%, and the half-life was 98.7 hours.
Cohort 4 was dropped, as it was another comparison of IV and SQ CB 2679d, which was considered unnecessary.
Cohort 5 included 5 patients who received daily doses of SQ CB 2679d. For 6 days, patients received CB 2679d at a dose of 140 IU/kg SQ.
The patients’ FIX activity levels increased from a median of <1% at baseline to a median of 15.7% (interquartile range, 14.9% to 16.6%) after all 6 doses.
The increase in FIX activity levels after the daily dosing was linear, indicating that continued SQ dosing may increase FIX activity further.
The median half-life was 63.2 hours (interquartile range, 60.2 to 64 hours), with the result that activity levels were still at 4% to 6.4% five days after the last dose.
No inhibitors to CB 2679d or FIX were observed in any of the cohorts in this trial.
In cohort 5, adverse events included pain, erythema, redness, and bruising after injections. Bruising occurred only with initial injections, and the severity of pain, erythema, and redness decreased over time (from moderate to mild).
“Existing IV therapies have FIX trough levels that can drop as low as 1% to 3% before repeat dosing,” Dr Levey said. “Daily subcutaneous dosing of CB 2679d has the potential to minimize the variability in FIX activity levels observed between IV doses and maintain individuals in the mild or even normal hemophilia range. This study demonstrates that, even after just 6 days of treatment, CB 2679d compares favorably to currently approved therapies for hemophilia B, all of which are infused IV.” ![]()
MADRID—The recombinant factor IX (FIX) product CB 2679d/ISU304 can increase FIX levels in patients with severe hemophilia B, according to a phase 1/2 trial.
Results showed a continuous linear increase in FIX activity levels following daily subcutaneous (SQ) dosing of CB 2679d for 6 days.
Adverse events were mild to moderate, and none of the patients have developed inhibitors to CB 2679d or FIX.
Howard Levy, MB ChB, PhD, chief medical officer of Catalyst Biosciences, Inc., presented these results at the 11th Annual Congress of The European Association for Haemophilia and Allied Disorders (EAHAD).
The research was sponsored by Catalyst Biosciences, Inc.
This phase 1/2 trial was divided into 5 cohorts.
In cohort 1, researchers compared single doses of intravenous (IV) CB 2679d and IV BeneFIX (recombinant FIX). CB 2679d proved 22 times more potent than BeneFIX. The products’ half-lives were 27.0 hours and 21.0 hours, respectively.
In cohorts 2 and 3, researchers compared single, ascending doses of IV CB 2679d to SQ CB 2679d. The bioavailability of SQ CB 2679d was 18.5%, and the half-life was 98.7 hours.
Cohort 4 was dropped, as it was another comparison of IV and SQ CB 2679d, which was considered unnecessary.
Cohort 5 included 5 patients who received daily doses of SQ CB 2679d. For 6 days, patients received CB 2679d at a dose of 140 IU/kg SQ.
The patients’ FIX activity levels increased from a median of <1% at baseline to a median of 15.7% (interquartile range, 14.9% to 16.6%) after all 6 doses.
The increase in FIX activity levels after the daily dosing was linear, indicating that continued SQ dosing may increase FIX activity further.
The median half-life was 63.2 hours (interquartile range, 60.2 to 64 hours), with the result that activity levels were still at 4% to 6.4% five days after the last dose.
No inhibitors to CB 2679d or FIX were observed in any of the cohorts in this trial.
In cohort 5, adverse events included pain, erythema, redness, and bruising after injections. Bruising occurred only with initial injections, and the severity of pain, erythema, and redness decreased over time (from moderate to mild).
“Existing IV therapies have FIX trough levels that can drop as low as 1% to 3% before repeat dosing,” Dr Levey said. “Daily subcutaneous dosing of CB 2679d has the potential to minimize the variability in FIX activity levels observed between IV doses and maintain individuals in the mild or even normal hemophilia range. This study demonstrates that, even after just 6 days of treatment, CB 2679d compares favorably to currently approved therapies for hemophilia B, all of which are infused IV.” ![]()
MADRID—The recombinant factor IX (FIX) product CB 2679d/ISU304 can increase FIX levels in patients with severe hemophilia B, according to a phase 1/2 trial.
Results showed a continuous linear increase in FIX activity levels following daily subcutaneous (SQ) dosing of CB 2679d for 6 days.
Adverse events were mild to moderate, and none of the patients have developed inhibitors to CB 2679d or FIX.
Howard Levy, MB ChB, PhD, chief medical officer of Catalyst Biosciences, Inc., presented these results at the 11th Annual Congress of The European Association for Haemophilia and Allied Disorders (EAHAD).
The research was sponsored by Catalyst Biosciences, Inc.
This phase 1/2 trial was divided into 5 cohorts.
In cohort 1, researchers compared single doses of intravenous (IV) CB 2679d and IV BeneFIX (recombinant FIX). CB 2679d proved 22 times more potent than BeneFIX. The products’ half-lives were 27.0 hours and 21.0 hours, respectively.
In cohorts 2 and 3, researchers compared single, ascending doses of IV CB 2679d to SQ CB 2679d. The bioavailability of SQ CB 2679d was 18.5%, and the half-life was 98.7 hours.
Cohort 4 was dropped, as it was another comparison of IV and SQ CB 2679d, which was considered unnecessary.
Cohort 5 included 5 patients who received daily doses of SQ CB 2679d. For 6 days, patients received CB 2679d at a dose of 140 IU/kg SQ.
The patients’ FIX activity levels increased from a median of <1% at baseline to a median of 15.7% (interquartile range, 14.9% to 16.6%) after all 6 doses.
The increase in FIX activity levels after the daily dosing was linear, indicating that continued SQ dosing may increase FIX activity further.
The median half-life was 63.2 hours (interquartile range, 60.2 to 64 hours), with the result that activity levels were still at 4% to 6.4% five days after the last dose.
No inhibitors to CB 2679d or FIX were observed in any of the cohorts in this trial.
In cohort 5, adverse events included pain, erythema, redness, and bruising after injections. Bruising occurred only with initial injections, and the severity of pain, erythema, and redness decreased over time (from moderate to mild).
“Existing IV therapies have FIX trough levels that can drop as low as 1% to 3% before repeat dosing,” Dr Levey said. “Daily subcutaneous dosing of CB 2679d has the potential to minimize the variability in FIX activity levels observed between IV doses and maintain individuals in the mild or even normal hemophilia range. This study demonstrates that, even after just 6 days of treatment, CB 2679d compares favorably to currently approved therapies for hemophilia B, all of which are infused IV.” ![]()
Agent can decrease GI toxicity in MM patients
Results of a case-control study suggest a cytoprotective agent can reduce treatment-related gastrointestinal (GI) toxicity in patients with multiple myeloma (MM).
Use of this agent, amifostine, was associated with significantly lower rates of grade 2 or higher oral mucositis, nausea, vomiting, and diarrhea.
Additionally, amifostine did not appear to compromise the anti-myeloma activity of treatment, which consisted of high-dose melphalan (HDM) and autologous hematopoietic stem cell transplant (auto-HSCT).
Ehsan Malek, MD, of Case Western Reserve University in Cleveland, Ohio, and his colleagues reported these findings in Leukemia & Lymphoma.
The researchers compared HDM plus auto-HSCT, with or without pre-treatment amifostine, in previously treated MM patients.
There were 107 patients who received amifostine and 114 who did not. The 107 patients received amifostine at 740 mg/m2, given as a bolus infusion at 24 hours and 15 minutes before HDM.
Baseline characteristics were largely similar in the amifostine and control groups. However, more patients in the amifostine group received a tandem HSCT (17 vs 0), and more patients in the control group had an ECOG performance status of 0 (64.3% vs 43%).
Patients in the amifostine group had a longer median time from diagnosis to first HSCT—10 months (range, 4-39) vs 7 months (range, 1-95).
A majority of patients in both groups were in partial response or better at baseline. However, more patients in the control group had stable disease (6.2% vs 1%) or progressive disease (8% vs 0%).
Results
For all-grade GI toxicities, there was largely no significant difference between the amifostine and control groups. However, patients in the amifostine group had significantly lower rates of grade 2 or higher GI toxicities.
Rates of all-grade GI toxicities in the amifostine and control groups, respectively, were:
- Oral mucositis—53.3% vs 64.0%, P=0.104
- Nausea—90.7% vs 95.6%, P=0.143
- Vomiting—65.4% vs 75.4%, P=0.102
- Diarrhea—93.5% vs 84.2%,P=0.030.
Rates of grade 2 or higher GI toxicities in the amifostine and control groups, respectively, were:
- Oral mucositis—27.1% vs 47.4%, P=0.002
- Nausea—31.8% vs 86.0%, P<0.0001
- Vomiting—18.7% vs 52.6%, P<0.0001
- Diarrhea—56.1% vs 73.7%, P=0.006.
The researchers said amifostine was well tolerated and produced no significant adverse effects.
They also said amifostine had “no discernable effect” on engraftment, progression-free survival, or overall survival.
The median time to neutrophil engraftment was 11 days (range, 9-16) in the control group and 10 days (range, 6-21) in the amifostine group (P=0.011). The median time to platelet engraftment was 18 days (range, 0-26) and 19 days (range, 8-71), respectively (P<0.21).
The median progression-free survival was 40 months in the amifostine group and 32 months in the control group (P=0.012). The median overall survival was 70 months and 67 months, respectively (P=0.84). ![]()
Results of a case-control study suggest a cytoprotective agent can reduce treatment-related gastrointestinal (GI) toxicity in patients with multiple myeloma (MM).
Use of this agent, amifostine, was associated with significantly lower rates of grade 2 or higher oral mucositis, nausea, vomiting, and diarrhea.
Additionally, amifostine did not appear to compromise the anti-myeloma activity of treatment, which consisted of high-dose melphalan (HDM) and autologous hematopoietic stem cell transplant (auto-HSCT).
Ehsan Malek, MD, of Case Western Reserve University in Cleveland, Ohio, and his colleagues reported these findings in Leukemia & Lymphoma.
The researchers compared HDM plus auto-HSCT, with or without pre-treatment amifostine, in previously treated MM patients.
There were 107 patients who received amifostine and 114 who did not. The 107 patients received amifostine at 740 mg/m2, given as a bolus infusion at 24 hours and 15 minutes before HDM.
Baseline characteristics were largely similar in the amifostine and control groups. However, more patients in the amifostine group received a tandem HSCT (17 vs 0), and more patients in the control group had an ECOG performance status of 0 (64.3% vs 43%).
Patients in the amifostine group had a longer median time from diagnosis to first HSCT—10 months (range, 4-39) vs 7 months (range, 1-95).
A majority of patients in both groups were in partial response or better at baseline. However, more patients in the control group had stable disease (6.2% vs 1%) or progressive disease (8% vs 0%).
Results
For all-grade GI toxicities, there was largely no significant difference between the amifostine and control groups. However, patients in the amifostine group had significantly lower rates of grade 2 or higher GI toxicities.
Rates of all-grade GI toxicities in the amifostine and control groups, respectively, were:
- Oral mucositis—53.3% vs 64.0%, P=0.104
- Nausea—90.7% vs 95.6%, P=0.143
- Vomiting—65.4% vs 75.4%, P=0.102
- Diarrhea—93.5% vs 84.2%,P=0.030.
Rates of grade 2 or higher GI toxicities in the amifostine and control groups, respectively, were:
- Oral mucositis—27.1% vs 47.4%, P=0.002
- Nausea—31.8% vs 86.0%, P<0.0001
- Vomiting—18.7% vs 52.6%, P<0.0001
- Diarrhea—56.1% vs 73.7%, P=0.006.
The researchers said amifostine was well tolerated and produced no significant adverse effects.
They also said amifostine had “no discernable effect” on engraftment, progression-free survival, or overall survival.
The median time to neutrophil engraftment was 11 days (range, 9-16) in the control group and 10 days (range, 6-21) in the amifostine group (P=0.011). The median time to platelet engraftment was 18 days (range, 0-26) and 19 days (range, 8-71), respectively (P<0.21).
The median progression-free survival was 40 months in the amifostine group and 32 months in the control group (P=0.012). The median overall survival was 70 months and 67 months, respectively (P=0.84). ![]()
Results of a case-control study suggest a cytoprotective agent can reduce treatment-related gastrointestinal (GI) toxicity in patients with multiple myeloma (MM).
Use of this agent, amifostine, was associated with significantly lower rates of grade 2 or higher oral mucositis, nausea, vomiting, and diarrhea.
Additionally, amifostine did not appear to compromise the anti-myeloma activity of treatment, which consisted of high-dose melphalan (HDM) and autologous hematopoietic stem cell transplant (auto-HSCT).
Ehsan Malek, MD, of Case Western Reserve University in Cleveland, Ohio, and his colleagues reported these findings in Leukemia & Lymphoma.
The researchers compared HDM plus auto-HSCT, with or without pre-treatment amifostine, in previously treated MM patients.
There were 107 patients who received amifostine and 114 who did not. The 107 patients received amifostine at 740 mg/m2, given as a bolus infusion at 24 hours and 15 minutes before HDM.
Baseline characteristics were largely similar in the amifostine and control groups. However, more patients in the amifostine group received a tandem HSCT (17 vs 0), and more patients in the control group had an ECOG performance status of 0 (64.3% vs 43%).
Patients in the amifostine group had a longer median time from diagnosis to first HSCT—10 months (range, 4-39) vs 7 months (range, 1-95).
A majority of patients in both groups were in partial response or better at baseline. However, more patients in the control group had stable disease (6.2% vs 1%) or progressive disease (8% vs 0%).
Results
For all-grade GI toxicities, there was largely no significant difference between the amifostine and control groups. However, patients in the amifostine group had significantly lower rates of grade 2 or higher GI toxicities.
Rates of all-grade GI toxicities in the amifostine and control groups, respectively, were:
- Oral mucositis—53.3% vs 64.0%, P=0.104
- Nausea—90.7% vs 95.6%, P=0.143
- Vomiting—65.4% vs 75.4%, P=0.102
- Diarrhea—93.5% vs 84.2%,P=0.030.
Rates of grade 2 or higher GI toxicities in the amifostine and control groups, respectively, were:
- Oral mucositis—27.1% vs 47.4%, P=0.002
- Nausea—31.8% vs 86.0%, P<0.0001
- Vomiting—18.7% vs 52.6%, P<0.0001
- Diarrhea—56.1% vs 73.7%, P=0.006.
The researchers said amifostine was well tolerated and produced no significant adverse effects.
They also said amifostine had “no discernable effect” on engraftment, progression-free survival, or overall survival.
The median time to neutrophil engraftment was 11 days (range, 9-16) in the control group and 10 days (range, 6-21) in the amifostine group (P=0.011). The median time to platelet engraftment was 18 days (range, 0-26) and 19 days (range, 8-71), respectively (P<0.21).
The median progression-free survival was 40 months in the amifostine group and 32 months in the control group (P=0.012). The median overall survival was 70 months and 67 months, respectively (P=0.84). ![]()
The Deer Stand Strikes Back
ANSWER
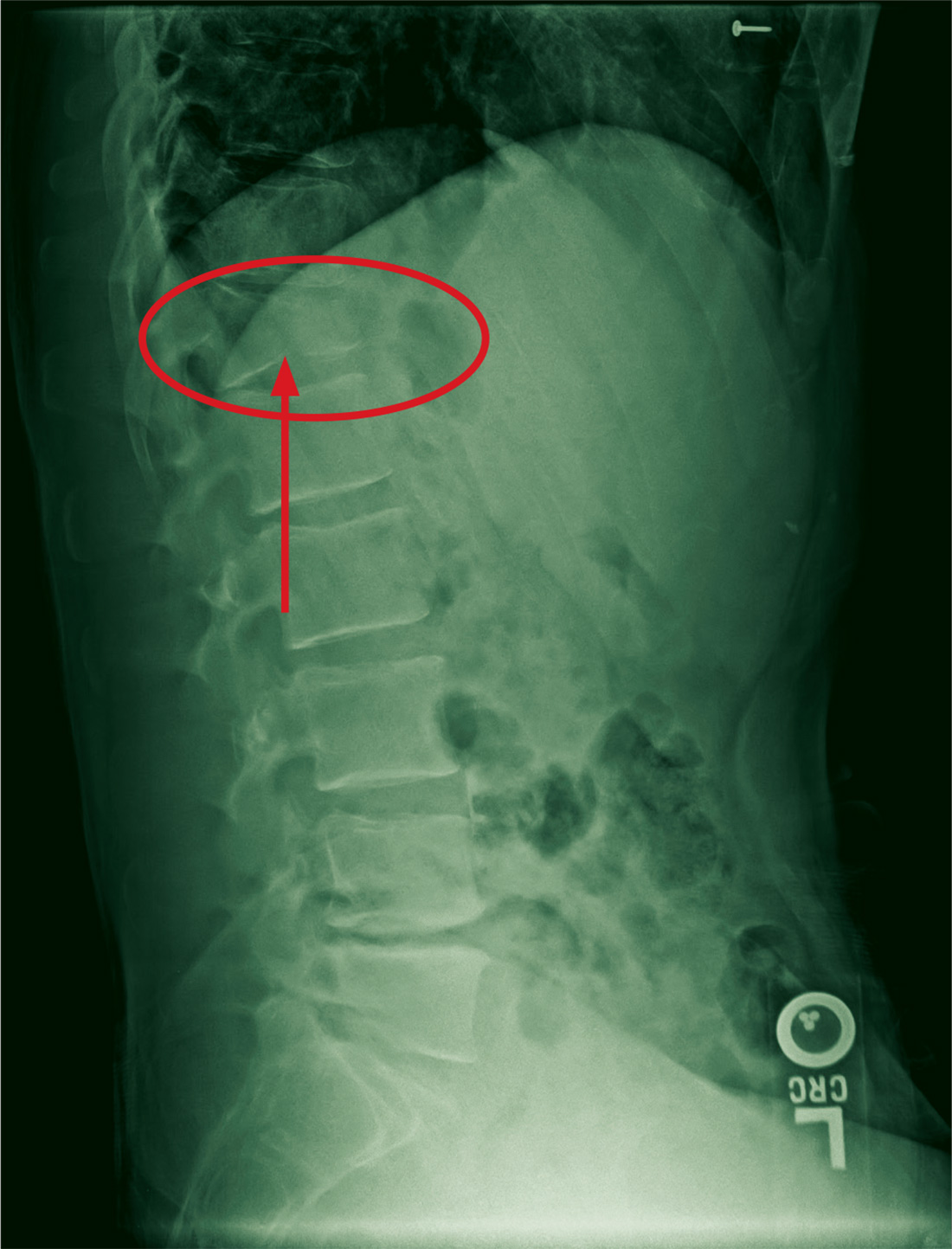
The radiograph demonstrates a compression fracture of T12 with moderate loss of height approaching 50% anteriorly. In addition, there is a vertically oriented fracture within the middle of the vertebral body, causing the back portion to be posteriorly displaced.
This type of burst fracture is potentially unstable and should be treated as such. The patient was placed on strict spine precautions and transferred to a facility where trauma and neurosurgical services were available.
ANSWER
The radiograph demonstrates a compression fracture of T12 with moderate loss of height approaching 50% anteriorly. In addition, there is a vertically oriented fracture within the middle of the vertebral body, causing the back portion to be posteriorly displaced.
This type of burst fracture is potentially unstable and should be treated as such. The patient was placed on strict spine precautions and transferred to a facility where trauma and neurosurgical services were available.
ANSWER
The radiograph demonstrates a compression fracture of T12 with moderate loss of height approaching 50% anteriorly. In addition, there is a vertically oriented fracture within the middle of the vertebral body, causing the back portion to be posteriorly displaced.
This type of burst fracture is potentially unstable and should be treated as such. The patient was placed on strict spine precautions and transferred to a facility where trauma and neurosurgical services were available.
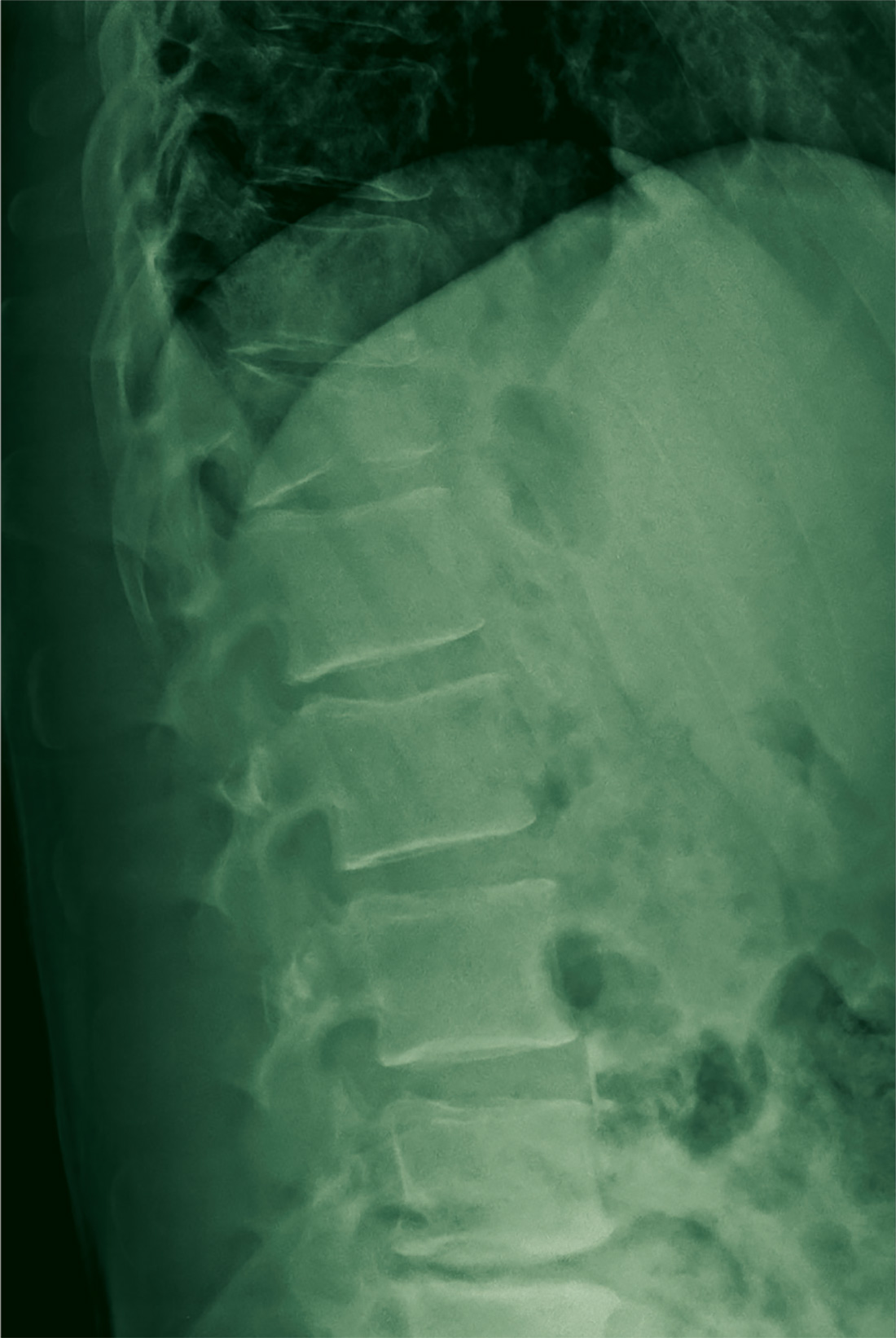
A 45-year-old man presents to your facility for evaluation of ongoing back pain. He reports that he fell out of a deer stand from an approximate height of 15 to 20 ft. He landed on his back but was able to get up, walk a short distance to his car, and drive home. Persistent pain is what brings him to the emergency department to seek treatment.
He denies any weakness or numbness in his lower extremities. There are no bowel or bladder issues. His medical history is unremarkable.
On physical examination, you note a moderately uncomfortable male whose vital signs are normal. He is able to move all four extremities well, and his strength is intact throughout. He does have moderate tenderness within the thoracolumbar region, with no step-off appreciated. The paraspinous muscles are tender as well.
You order lumbosacral radiographs (lateral view shown). What is your impression?
Short Takes
Fluoroquinolone use tied to higher risk of aortic dissection and aneurysm
A meta-analysis of two observational studies found that current fluoroquinolone use was associated with modestly higher risk of aortic dissection (OR 2.79, 95% CI 2.31-3.37) and aortic aneurysm (OR 2.25, 95% CI 2.03-2.49).
Citation: Singh S, Nautiyal A. Aortic dissection and aortic aneurysms associated with fluoroquinolones: A systematic review and meta-analysis. Am J Med [online ahead of print, July 21, 2017].
Optimal lengths of postoperative opioid prescription range from 4 to 15 days

Citation: Scully RE, Schoenfeld AJ, Jiang W, et al. Defining optimal length of opioid pain medication prescription after common surgical procedures. JAMA Surg. Sept 2017.
Increased mortality in weekend and holiday hospital admissions
A meta-analysis of 97 studies showed a greater relative risk of mortality for patients admitted during the weekend or holidays compared to those admitted during the week. Subgroup analyses did not reveal significant effect modification by staffing level and other hospital factors. Further research to identify contributing factors and potential interventions is needed.
Citation: Pauls LA, Johnson-Paben R, McGready J, Murphy JD, Pronovost PJ, Wu CL. The weekend effect in hospitalized patients: a meta-analysis. J Hosp Med. 2017;12(9):760-6.
Kayexalate should not be taken at the same time as other medications
The Food and Drug Administration has released a drug safety communication on kayexalate (sodium polystyrene sulfonate). Given recent research showing that kayexalate may decrease the absorption and effectiveness of many oral medications, the FDA recommended that when prescribing kayexalate, it should be given at least 3 hours before or after administration of other oral medications. This time should be increased to 6 hours for those with conditions that result in delayed gastric emptying.
Citation: Food and Drug Administration. Kayexalate (sodium polystyrene sulfonate): Drug Safety Communication. 09/06/2017.
Most thrombophilia testing done in the hospital does not add value
A retrospective cohort study among emergency department and hospitalized patients at an academic medical center examined 163 patients and 1,451 thrombophilia tests; 63% of tests were of “minimal clinical utility.”
Citation: Cox N, Johnson SA, Vazquez S, et al. Patterns and appropriateness of thrombophilia testing in an academic medical center. J Hosp Med. 2017;12(9):705-709.
Fluoroquinolone use tied to higher risk of aortic dissection and aneurysm
A meta-analysis of two observational studies found that current fluoroquinolone use was associated with modestly higher risk of aortic dissection (OR 2.79, 95% CI 2.31-3.37) and aortic aneurysm (OR 2.25, 95% CI 2.03-2.49).
Citation: Singh S, Nautiyal A. Aortic dissection and aortic aneurysms associated with fluoroquinolones: A systematic review and meta-analysis. Am J Med [online ahead of print, July 21, 2017].
Optimal lengths of postoperative opioid prescription range from 4 to 15 days
Citation: Scully RE, Schoenfeld AJ, Jiang W, et al. Defining optimal length of opioid pain medication prescription after common surgical procedures. JAMA Surg. Sept 2017.
Increased mortality in weekend and holiday hospital admissions
A meta-analysis of 97 studies showed a greater relative risk of mortality for patients admitted during the weekend or holidays compared to those admitted during the week. Subgroup analyses did not reveal significant effect modification by staffing level and other hospital factors. Further research to identify contributing factors and potential interventions is needed.
Citation: Pauls LA, Johnson-Paben R, McGready J, Murphy JD, Pronovost PJ, Wu CL. The weekend effect in hospitalized patients: a meta-analysis. J Hosp Med. 2017;12(9):760-6.
Kayexalate should not be taken at the same time as other medications
The Food and Drug Administration has released a drug safety communication on kayexalate (sodium polystyrene sulfonate). Given recent research showing that kayexalate may decrease the absorption and effectiveness of many oral medications, the FDA recommended that when prescribing kayexalate, it should be given at least 3 hours before or after administration of other oral medications. This time should be increased to 6 hours for those with conditions that result in delayed gastric emptying.
Citation: Food and Drug Administration. Kayexalate (sodium polystyrene sulfonate): Drug Safety Communication. 09/06/2017.
Most thrombophilia testing done in the hospital does not add value
A retrospective cohort study among emergency department and hospitalized patients at an academic medical center examined 163 patients and 1,451 thrombophilia tests; 63% of tests were of “minimal clinical utility.”
Citation: Cox N, Johnson SA, Vazquez S, et al. Patterns and appropriateness of thrombophilia testing in an academic medical center. J Hosp Med. 2017;12(9):705-709.
Fluoroquinolone use tied to higher risk of aortic dissection and aneurysm
A meta-analysis of two observational studies found that current fluoroquinolone use was associated with modestly higher risk of aortic dissection (OR 2.79, 95% CI 2.31-3.37) and aortic aneurysm (OR 2.25, 95% CI 2.03-2.49).
Citation: Singh S, Nautiyal A. Aortic dissection and aortic aneurysms associated with fluoroquinolones: A systematic review and meta-analysis. Am J Med [online ahead of print, July 21, 2017].
Optimal lengths of postoperative opioid prescription range from 4 to 15 days
Citation: Scully RE, Schoenfeld AJ, Jiang W, et al. Defining optimal length of opioid pain medication prescription after common surgical procedures. JAMA Surg. Sept 2017.
Increased mortality in weekend and holiday hospital admissions
A meta-analysis of 97 studies showed a greater relative risk of mortality for patients admitted during the weekend or holidays compared to those admitted during the week. Subgroup analyses did not reveal significant effect modification by staffing level and other hospital factors. Further research to identify contributing factors and potential interventions is needed.
Citation: Pauls LA, Johnson-Paben R, McGready J, Murphy JD, Pronovost PJ, Wu CL. The weekend effect in hospitalized patients: a meta-analysis. J Hosp Med. 2017;12(9):760-6.
Kayexalate should not be taken at the same time as other medications
The Food and Drug Administration has released a drug safety communication on kayexalate (sodium polystyrene sulfonate). Given recent research showing that kayexalate may decrease the absorption and effectiveness of many oral medications, the FDA recommended that when prescribing kayexalate, it should be given at least 3 hours before or after administration of other oral medications. This time should be increased to 6 hours for those with conditions that result in delayed gastric emptying.
Citation: Food and Drug Administration. Kayexalate (sodium polystyrene sulfonate): Drug Safety Communication. 09/06/2017.
Most thrombophilia testing done in the hospital does not add value
A retrospective cohort study among emergency department and hospitalized patients at an academic medical center examined 163 patients and 1,451 thrombophilia tests; 63% of tests were of “minimal clinical utility.”
Citation: Cox N, Johnson SA, Vazquez S, et al. Patterns and appropriateness of thrombophilia testing in an academic medical center. J Hosp Med. 2017;12(9):705-709.
Large-vessel vasculitis: More severe in HIV-infected patients
Large-vessel vasculitis (LVV) associated with HIV-infected patients occurs more frequently in men and is significantly more severe, compared with the disease in their noninfected counterparts, according to a retrospective analysis of medical records during 2000-2015.
The study was conducted at Pitié-Salpêtrière Hospital in Paris to investigate the occurrence, characteristics, and treatment outcomes of LVV of patients infected with HIV, reported in the Journal of Vascular Surgery. Yasmina Ferfar, MD, and her colleagues diagnosed LVV using available imaging (computed tomography, positron emission tomography, and magnetic resonance angiography), histology, and Ishikawa or American College of Rheumatology (ACR) criteria of Takayasu arteritis (TA). All 93 patients selected for the study had LVV.

Vascular lesions were located in three areas; aorta (n = 7), supra-aortic trunks (7), and in digestive arteries (3). The Ishikawa or ACR criteria for TA diagnosis were met by 7 of the 11 (64%) patients.
Highly active antiretroviral therapy was prescribed for six patients at the time of vasculitis diagnosis and for four after diagnosis; HAART was delayed for the remaining patient whose HIV infection was under control.
Steroids, immunosuppressive, statins, and antiplatelet agents were provided in custom combinations to nine patients. Open surgical or endovascular repair was performed on 8 (73%) of the 11 patients with a mean follow-up at 95 months. As recorded at the last follow-up, 6 of the 11 patients had obtained stabilization of the vasculitis, improvement of vascular lesions had occurred in 4 of the 11 patients, and 1 patient had died from complications related to vascular surgery.
An age-matched control test comparison was performed between HIV-infected patients with large-vessel vasculitis and the HIV-negative patients with Takayasu arteritis that were identified in this study. The 82 patients in the LVV HIV-negative group had a median age of 42 years and 72 (88%) were women. Inflammatory syndrome was reported for 30 patients. Glucocorticosteroid treatments were given to 70 patients and immunosuppressive treatments were given to 57 patients.
Comparing LVV between the HIV-infected and their HIV-negative counterparts showed that vascular disease was significantly more severe for HIV-infected patients, according to the Ishikawa score (P = .017) and there was significantly greater use of vascular procedures (P = .047). LVV in association with HIV-infection also occurred more frequently with men (P = .014).
Dr. Ferfar and colleagues suggested that “HIV infection might be an accelerant for vasculitis,” based upon the increased severity of the disease they noted. They also indicated that glucocorticosteroids and immunosuppressive treatments were less often prescribed in HIV-positive patients (P = .001).
The effectiveness of steroids and immunosuppressant drugs seen with 7 of the 11 HIV-positive patients that met the Ishikawa or ACR criteria of TA suggests an overlap between HIV-associated LVV and TA, according to the researchers. Therefore, “HIV-infected patients with LVV should be treated with corticosteroids to avoid complications. Studies with a greater number of HIV-infected patients with LVV are required to confirm this hypothesis”.
The authors reported that they had no conflicts of interest.
SOURCE: Ferfar Y. et al. J Vasc Surg. 2018 Dec 11. doi. org/10.1016/j.jvs.2017.08.099.
Large-vessel vasculitis (LVV) associated with HIV-infected patients occurs more frequently in men and is significantly more severe, compared with the disease in their noninfected counterparts, according to a retrospective analysis of medical records during 2000-2015.
The study was conducted at Pitié-Salpêtrière Hospital in Paris to investigate the occurrence, characteristics, and treatment outcomes of LVV of patients infected with HIV, reported in the Journal of Vascular Surgery. Yasmina Ferfar, MD, and her colleagues diagnosed LVV using available imaging (computed tomography, positron emission tomography, and magnetic resonance angiography), histology, and Ishikawa or American College of Rheumatology (ACR) criteria of Takayasu arteritis (TA). All 93 patients selected for the study had LVV.
Vascular lesions were located in three areas; aorta (n = 7), supra-aortic trunks (7), and in digestive arteries (3). The Ishikawa or ACR criteria for TA diagnosis were met by 7 of the 11 (64%) patients.
Highly active antiretroviral therapy was prescribed for six patients at the time of vasculitis diagnosis and for four after diagnosis; HAART was delayed for the remaining patient whose HIV infection was under control.
Steroids, immunosuppressive, statins, and antiplatelet agents were provided in custom combinations to nine patients. Open surgical or endovascular repair was performed on 8 (73%) of the 11 patients with a mean follow-up at 95 months. As recorded at the last follow-up, 6 of the 11 patients had obtained stabilization of the vasculitis, improvement of vascular lesions had occurred in 4 of the 11 patients, and 1 patient had died from complications related to vascular surgery.
An age-matched control test comparison was performed between HIV-infected patients with large-vessel vasculitis and the HIV-negative patients with Takayasu arteritis that were identified in this study. The 82 patients in the LVV HIV-negative group had a median age of 42 years and 72 (88%) were women. Inflammatory syndrome was reported for 30 patients. Glucocorticosteroid treatments were given to 70 patients and immunosuppressive treatments were given to 57 patients.
Comparing LVV between the HIV-infected and their HIV-negative counterparts showed that vascular disease was significantly more severe for HIV-infected patients, according to the Ishikawa score (P = .017) and there was significantly greater use of vascular procedures (P = .047). LVV in association with HIV-infection also occurred more frequently with men (P = .014).
Dr. Ferfar and colleagues suggested that “HIV infection might be an accelerant for vasculitis,” based upon the increased severity of the disease they noted. They also indicated that glucocorticosteroids and immunosuppressive treatments were less often prescribed in HIV-positive patients (P = .001).
The effectiveness of steroids and immunosuppressant drugs seen with 7 of the 11 HIV-positive patients that met the Ishikawa or ACR criteria of TA suggests an overlap between HIV-associated LVV and TA, according to the researchers. Therefore, “HIV-infected patients with LVV should be treated with corticosteroids to avoid complications. Studies with a greater number of HIV-infected patients with LVV are required to confirm this hypothesis”.
The authors reported that they had no conflicts of interest.
SOURCE: Ferfar Y. et al. J Vasc Surg. 2018 Dec 11. doi. org/10.1016/j.jvs.2017.08.099.
Large-vessel vasculitis (LVV) associated with HIV-infected patients occurs more frequently in men and is significantly more severe, compared with the disease in their noninfected counterparts, according to a retrospective analysis of medical records during 2000-2015.
The study was conducted at Pitié-Salpêtrière Hospital in Paris to investigate the occurrence, characteristics, and treatment outcomes of LVV of patients infected with HIV, reported in the Journal of Vascular Surgery. Yasmina Ferfar, MD, and her colleagues diagnosed LVV using available imaging (computed tomography, positron emission tomography, and magnetic resonance angiography), histology, and Ishikawa or American College of Rheumatology (ACR) criteria of Takayasu arteritis (TA). All 93 patients selected for the study had LVV.
Vascular lesions were located in three areas; aorta (n = 7), supra-aortic trunks (7), and in digestive arteries (3). The Ishikawa or ACR criteria for TA diagnosis were met by 7 of the 11 (64%) patients.
Highly active antiretroviral therapy was prescribed for six patients at the time of vasculitis diagnosis and for four after diagnosis; HAART was delayed for the remaining patient whose HIV infection was under control.
Steroids, immunosuppressive, statins, and antiplatelet agents were provided in custom combinations to nine patients. Open surgical or endovascular repair was performed on 8 (73%) of the 11 patients with a mean follow-up at 95 months. As recorded at the last follow-up, 6 of the 11 patients had obtained stabilization of the vasculitis, improvement of vascular lesions had occurred in 4 of the 11 patients, and 1 patient had died from complications related to vascular surgery.
An age-matched control test comparison was performed between HIV-infected patients with large-vessel vasculitis and the HIV-negative patients with Takayasu arteritis that were identified in this study. The 82 patients in the LVV HIV-negative group had a median age of 42 years and 72 (88%) were women. Inflammatory syndrome was reported for 30 patients. Glucocorticosteroid treatments were given to 70 patients and immunosuppressive treatments were given to 57 patients.
Comparing LVV between the HIV-infected and their HIV-negative counterparts showed that vascular disease was significantly more severe for HIV-infected patients, according to the Ishikawa score (P = .017) and there was significantly greater use of vascular procedures (P = .047). LVV in association with HIV-infection also occurred more frequently with men (P = .014).
Dr. Ferfar and colleagues suggested that “HIV infection might be an accelerant for vasculitis,” based upon the increased severity of the disease they noted. They also indicated that glucocorticosteroids and immunosuppressive treatments were less often prescribed in HIV-positive patients (P = .001).
The effectiveness of steroids and immunosuppressant drugs seen with 7 of the 11 HIV-positive patients that met the Ishikawa or ACR criteria of TA suggests an overlap between HIV-associated LVV and TA, according to the researchers. Therefore, “HIV-infected patients with LVV should be treated with corticosteroids to avoid complications. Studies with a greater number of HIV-infected patients with LVV are required to confirm this hypothesis”.
The authors reported that they had no conflicts of interest.
SOURCE: Ferfar Y. et al. J Vasc Surg. 2018 Dec 11. doi. org/10.1016/j.jvs.2017.08.099.
FROM THE JOURNAL OF VASCULAR SURGERY
Key clinical point: Corticosteroid treatments may reduce severity and avoid some complications of LVV in HIV-infected patients.
Major finding: LVV is significantly more severe in HIV-infected patients and is associated more frequently with men.
Study details: A Pitié-Salpêtrière Hospital retrospective study of 11 HIV-infected patients with LVV, compared with 82 uninfected age-matched patients with LVV.
Disclosures: The authors reported that they had no conflicts of interest.
Source: Ferfar Y et al. J Vasc Surg. 2018. doi. org/10.1016/j.jvs.2017.08.099.
Majority of lupus patients lack eye exam around the start of hydroxychloroquine
Only one-third of lupus patients on Medicaid had a baseline retinal exam before initiating treatment with hydroxychloroquine in an analysis of a claims database, despite the exam being recommended as the standard in multiple clinical guidelines.
The analysis of 12,755 patients with systemic lupus erythematosus (SLE) on Medicaid from 29 of the most populated U.S. states between 2001 and 2010 found that 32.5% received a baseline dilated eye exam 30 days before, and up to 1 year after, starting treatment with the anchor drug hydroxychloroquine (HCQ). This figure rose to 40% when Humphrey visual field tests and other “optional” eye exams were included, Tzu-Chieh Lin, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues reported in Arthritis Care & Research.
HCQ is known to cause retinal damage in some patients, binding to and accumulating in the retinal pigment epithelium melanin. If undetected, patients may gradually lose central vision. There is particular risk for HCQ-induced retinal damage in patients on higher doses (5 mg/kg or greater of actual weight), HCQ exposure over 5 years, or renal, hepatic, or retinal disease, the research team explained.
Baseline retinal examinations for first-time HCQ users were first recommended by American Academy of Ophthalmology guidelines in 2002, and in 2009 the American College of Rheumatology developed guidelines specifically for SLE that also recommended a funduscopic exam within 1 year of starting HCQ.
The proportion of patients beginning treatment with HCQ who received a retinal exam only slightly increased from 31% in 2001 to 34.4% in 2009 (P value for linear trend over time = .12).
Women were significantly more likely to have a baseline retinal exam (odds ratio, 1.30; 95% confidence interval, 1.08-1.55), and certain sociodemographic factors also predicted who received a baseline exam, likely reflecting problems with adherence and access to health care.
For example, the likelihood of having a baseline exam proved significantly lower for blacks and American Indian/Alaska natives (OR, 0.85; 95% CI, 0.77-0.93; and OR, 0.69; 95% CI, 0.47-1.02) than for whites, whereas Asian Americans had greater odds than whites (OR, 1.30; 95% CI, 1.06-1.60).
“We have previously observed that these sociodemographic factors have been associated with differences in both access to care and adherence in the SLE Medicaid population for indicated care, including infection prevention, adherence to indicated medications including HCQ, treatments for lupus nephritis, and choices in renal replacement therapies and use of erythropoietin-stimulating agents among SLE end-stage renal disease patients,” they wrote.
Patients who had received multiple SLE-related laboratory tests (OR, 1.33; 95% CI, 1.16-1.52) and liver function tests (OR, 1.16; 95% CI, 1.04-1.29) were also more likely to have an eye exam before commencing HCQ.
The researchers said both patient and physician factors likely played a role in adherence to preventive medical care.
“While adherence to daily dosing, by the latest recommendations less than 5 mg/kg actual weight, is a more important factor determining renal toxicity risk, baseline examinations are necessary to avoid treating patients with preexisting retinal disease and to allow early detection of changes from baseline,” the research team concluded.
The work was supported by grant awards from the Rheumatology Research Foundation and the National Institutes of Health. No disclosures were made.
SOURCE: Lin T et al. Arthritis Care Res. 2018 Feb 6. doi: 10.1002/acr.23530.
Only one-third of lupus patients on Medicaid had a baseline retinal exam before initiating treatment with hydroxychloroquine in an analysis of a claims database, despite the exam being recommended as the standard in multiple clinical guidelines.
The analysis of 12,755 patients with systemic lupus erythematosus (SLE) on Medicaid from 29 of the most populated U.S. states between 2001 and 2010 found that 32.5% received a baseline dilated eye exam 30 days before, and up to 1 year after, starting treatment with the anchor drug hydroxychloroquine (HCQ). This figure rose to 40% when Humphrey visual field tests and other “optional” eye exams were included, Tzu-Chieh Lin, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues reported in Arthritis Care & Research.
HCQ is known to cause retinal damage in some patients, binding to and accumulating in the retinal pigment epithelium melanin. If undetected, patients may gradually lose central vision. There is particular risk for HCQ-induced retinal damage in patients on higher doses (5 mg/kg or greater of actual weight), HCQ exposure over 5 years, or renal, hepatic, or retinal disease, the research team explained.
Baseline retinal examinations for first-time HCQ users were first recommended by American Academy of Ophthalmology guidelines in 2002, and in 2009 the American College of Rheumatology developed guidelines specifically for SLE that also recommended a funduscopic exam within 1 year of starting HCQ.
The proportion of patients beginning treatment with HCQ who received a retinal exam only slightly increased from 31% in 2001 to 34.4% in 2009 (P value for linear trend over time = .12).
Women were significantly more likely to have a baseline retinal exam (odds ratio, 1.30; 95% confidence interval, 1.08-1.55), and certain sociodemographic factors also predicted who received a baseline exam, likely reflecting problems with adherence and access to health care.
For example, the likelihood of having a baseline exam proved significantly lower for blacks and American Indian/Alaska natives (OR, 0.85; 95% CI, 0.77-0.93; and OR, 0.69; 95% CI, 0.47-1.02) than for whites, whereas Asian Americans had greater odds than whites (OR, 1.30; 95% CI, 1.06-1.60).
“We have previously observed that these sociodemographic factors have been associated with differences in both access to care and adherence in the SLE Medicaid population for indicated care, including infection prevention, adherence to indicated medications including HCQ, treatments for lupus nephritis, and choices in renal replacement therapies and use of erythropoietin-stimulating agents among SLE end-stage renal disease patients,” they wrote.
Patients who had received multiple SLE-related laboratory tests (OR, 1.33; 95% CI, 1.16-1.52) and liver function tests (OR, 1.16; 95% CI, 1.04-1.29) were also more likely to have an eye exam before commencing HCQ.
The researchers said both patient and physician factors likely played a role in adherence to preventive medical care.
“While adherence to daily dosing, by the latest recommendations less than 5 mg/kg actual weight, is a more important factor determining renal toxicity risk, baseline examinations are necessary to avoid treating patients with preexisting retinal disease and to allow early detection of changes from baseline,” the research team concluded.
The work was supported by grant awards from the Rheumatology Research Foundation and the National Institutes of Health. No disclosures were made.
SOURCE: Lin T et al. Arthritis Care Res. 2018 Feb 6. doi: 10.1002/acr.23530.
Only one-third of lupus patients on Medicaid had a baseline retinal exam before initiating treatment with hydroxychloroquine in an analysis of a claims database, despite the exam being recommended as the standard in multiple clinical guidelines.
The analysis of 12,755 patients with systemic lupus erythematosus (SLE) on Medicaid from 29 of the most populated U.S. states between 2001 and 2010 found that 32.5% received a baseline dilated eye exam 30 days before, and up to 1 year after, starting treatment with the anchor drug hydroxychloroquine (HCQ). This figure rose to 40% when Humphrey visual field tests and other “optional” eye exams were included, Tzu-Chieh Lin, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues reported in Arthritis Care & Research.
HCQ is known to cause retinal damage in some patients, binding to and accumulating in the retinal pigment epithelium melanin. If undetected, patients may gradually lose central vision. There is particular risk for HCQ-induced retinal damage in patients on higher doses (5 mg/kg or greater of actual weight), HCQ exposure over 5 years, or renal, hepatic, or retinal disease, the research team explained.
Baseline retinal examinations for first-time HCQ users were first recommended by American Academy of Ophthalmology guidelines in 2002, and in 2009 the American College of Rheumatology developed guidelines specifically for SLE that also recommended a funduscopic exam within 1 year of starting HCQ.
The proportion of patients beginning treatment with HCQ who received a retinal exam only slightly increased from 31% in 2001 to 34.4% in 2009 (P value for linear trend over time = .12).
Women were significantly more likely to have a baseline retinal exam (odds ratio, 1.30; 95% confidence interval, 1.08-1.55), and certain sociodemographic factors also predicted who received a baseline exam, likely reflecting problems with adherence and access to health care.
For example, the likelihood of having a baseline exam proved significantly lower for blacks and American Indian/Alaska natives (OR, 0.85; 95% CI, 0.77-0.93; and OR, 0.69; 95% CI, 0.47-1.02) than for whites, whereas Asian Americans had greater odds than whites (OR, 1.30; 95% CI, 1.06-1.60).
“We have previously observed that these sociodemographic factors have been associated with differences in both access to care and adherence in the SLE Medicaid population for indicated care, including infection prevention, adherence to indicated medications including HCQ, treatments for lupus nephritis, and choices in renal replacement therapies and use of erythropoietin-stimulating agents among SLE end-stage renal disease patients,” they wrote.
Patients who had received multiple SLE-related laboratory tests (OR, 1.33; 95% CI, 1.16-1.52) and liver function tests (OR, 1.16; 95% CI, 1.04-1.29) were also more likely to have an eye exam before commencing HCQ.
The researchers said both patient and physician factors likely played a role in adherence to preventive medical care.
“While adherence to daily dosing, by the latest recommendations less than 5 mg/kg actual weight, is a more important factor determining renal toxicity risk, baseline examinations are necessary to avoid treating patients with preexisting retinal disease and to allow early detection of changes from baseline,” the research team concluded.
The work was supported by grant awards from the Rheumatology Research Foundation and the National Institutes of Health. No disclosures were made.
SOURCE: Lin T et al. Arthritis Care Res. 2018 Feb 6. doi: 10.1002/acr.23530.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point:
Major finding: Of 12,755 Medicaid SLE patients, 32.5% received a baseline dilated-eye exam 30 days before, and up to 1 year after, starting treatment with HCQ.
Study details: An analysis of 12,755 Medicaid SLE patients from 29 of the most populated U.S. states between 2001 and 2010 who had initiated HCQ.
Disclosures: The work was supported by grant awards from the Rheumatology Research Foundation and the National Institutes of Health. No disclosures were made.
Source: Lin T et al. Arthritis Care Res. 2018 Feb 6. doi: 10.1002/acr.23530.
Monthly vs. biweekly ultrasounds to ID fetal growth and amniotic fluid abnormalities
DALLAS – Monthly ultrasound scans in the last trimester pick up just as many fetal growth and amniotic fluid problems as do scans done every 2 weeks, Suneet P. Chauhan, MD, reported at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“We found that about one in three of these high-risk pregnancies was complicated by an abnormality of fetal growth or amniotic fluid,” said Dr. Chauhan of the department of obstetrics, gynecology, and reproductive sciences at the University of Texas, Houston. “But more frequent scans did not increase the frequency of identifying these abnormalities. And since there are approximately 1.7 million such high-risk pregnancies each year, these findings have significant implications on personal, departmental, and societal levels.”

However, he added, the SUN trial, which randomized women to monthly or biweekly scans, wasn’t large enough to definitively determine the optimal timing of ultrasounds in this population. “We need larger trials to determine this.”
The trial results, published in the American Journal of Obstetrics & Gynecology, comprised 228 women with singleton pregnancies complicated by medical comorbidities, putting the fetus at risk of problems with growth or amniotic fluid volume. They were randomized to ultrasound scans every 2 weeks or every 4 weeks during the last trimester. The primary outcome was identification of four pregnancy complications: fetal growth restriction, large for gestational age, oligohydramnios, or polyhydramnios.
There were also secondary outcomes of composite maternal morbidity (chorioamnionitis, wound infection, transfusion, diabetic ketoacidosis, venous thromboembolism, ICU admission, and death) and neonatal morbidity (Apgar score of less than 5 at 5 minutes, umbilical artery pH less than 7, hyperbilirubinemia, intubation, high-grade intraventricular hemorrhage, necrotizing enterocolitis, and death).
Women were eligible for the study if they had autoimmune disease, a body mass index of 40 kg/m2 or higher, a history of delivering a small for gestational age or macrosomic infant, hypertensive disease, substance abuse, or sickle cell disease.
About half had multiple risk factors, the three most common being high body mass index, diabetes (either gestational or insulin-dependent), and hypertensive disease. Their gestational age at randomization was a mean of 23 weeks. The first scan was conducted at a mean of 29 weeks.
There were 28% more scans in the 2-week group than in the 4-week group (492 vs. 382). There were also more exams for potentially concerning findings, including more scans looking at growth (359 vs. 278), biophysical profiles (99 vs. 79), and umbilical artery Dopplers (34 vs. 27). Despite the increased scans and investigations, the primary outcome was detected at similar points (31.7 vs. 32.2 weeks).
A primary outcome finding was detected in 38% of the 2-week group and 32% of the 4-week group – not significantly different. There were no significant differences in the proportion of outcomes detected in each group, including fetal growth restriction (19% vs. 16%), large for gestational age (14% each group), oligohydramnios (4% each group) and polyhydramnios (9% each group).
Infants in each group were born at about the same time – 13.5 weeks after randomization. The mean gestational age in both groups was 37 weeks, with about 28.5% less than 37 weeks at birth. There were no significant differences in the rate of cesarean deliveries, or small or large for gestational age births.
The composite maternal morbidity outcome was similar in the 2-week and 4-week groups (26% vs. 22%). There were no significant differences in any of the individual components of the outcome. No mother died.
The composite neonatal outcome was also similar between the groups (14% vs. 12%). There were no significant differences in any of the individual components. No infant died.
The study was sponsored by the University of Texas Health Science Center, Houston. Dr. Chauhan had no financial disclosures.
SOURCE: Roberts RP et al. Am J Obstet Gynecol. 2018;218:S3.
DALLAS – Monthly ultrasound scans in the last trimester pick up just as many fetal growth and amniotic fluid problems as do scans done every 2 weeks, Suneet P. Chauhan, MD, reported at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“We found that about one in three of these high-risk pregnancies was complicated by an abnormality of fetal growth or amniotic fluid,” said Dr. Chauhan of the department of obstetrics, gynecology, and reproductive sciences at the University of Texas, Houston. “But more frequent scans did not increase the frequency of identifying these abnormalities. And since there are approximately 1.7 million such high-risk pregnancies each year, these findings have significant implications on personal, departmental, and societal levels.”
However, he added, the SUN trial, which randomized women to monthly or biweekly scans, wasn’t large enough to definitively determine the optimal timing of ultrasounds in this population. “We need larger trials to determine this.”
The trial results, published in the American Journal of Obstetrics & Gynecology, comprised 228 women with singleton pregnancies complicated by medical comorbidities, putting the fetus at risk of problems with growth or amniotic fluid volume. They were randomized to ultrasound scans every 2 weeks or every 4 weeks during the last trimester. The primary outcome was identification of four pregnancy complications: fetal growth restriction, large for gestational age, oligohydramnios, or polyhydramnios.
There were also secondary outcomes of composite maternal morbidity (chorioamnionitis, wound infection, transfusion, diabetic ketoacidosis, venous thromboembolism, ICU admission, and death) and neonatal morbidity (Apgar score of less than 5 at 5 minutes, umbilical artery pH less than 7, hyperbilirubinemia, intubation, high-grade intraventricular hemorrhage, necrotizing enterocolitis, and death).
Women were eligible for the study if they had autoimmune disease, a body mass index of 40 kg/m2 or higher, a history of delivering a small for gestational age or macrosomic infant, hypertensive disease, substance abuse, or sickle cell disease.
About half had multiple risk factors, the three most common being high body mass index, diabetes (either gestational or insulin-dependent), and hypertensive disease. Their gestational age at randomization was a mean of 23 weeks. The first scan was conducted at a mean of 29 weeks.
There were 28% more scans in the 2-week group than in the 4-week group (492 vs. 382). There were also more exams for potentially concerning findings, including more scans looking at growth (359 vs. 278), biophysical profiles (99 vs. 79), and umbilical artery Dopplers (34 vs. 27). Despite the increased scans and investigations, the primary outcome was detected at similar points (31.7 vs. 32.2 weeks).
A primary outcome finding was detected in 38% of the 2-week group and 32% of the 4-week group – not significantly different. There were no significant differences in the proportion of outcomes detected in each group, including fetal growth restriction (19% vs. 16%), large for gestational age (14% each group), oligohydramnios (4% each group) and polyhydramnios (9% each group).
Infants in each group were born at about the same time – 13.5 weeks after randomization. The mean gestational age in both groups was 37 weeks, with about 28.5% less than 37 weeks at birth. There were no significant differences in the rate of cesarean deliveries, or small or large for gestational age births.
The composite maternal morbidity outcome was similar in the 2-week and 4-week groups (26% vs. 22%). There were no significant differences in any of the individual components of the outcome. No mother died.
The composite neonatal outcome was also similar between the groups (14% vs. 12%). There were no significant differences in any of the individual components. No infant died.
The study was sponsored by the University of Texas Health Science Center, Houston. Dr. Chauhan had no financial disclosures.
SOURCE: Roberts RP et al. Am J Obstet Gynecol. 2018;218:S3.
DALLAS – Monthly ultrasound scans in the last trimester pick up just as many fetal growth and amniotic fluid problems as do scans done every 2 weeks, Suneet P. Chauhan, MD, reported at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“We found that about one in three of these high-risk pregnancies was complicated by an abnormality of fetal growth or amniotic fluid,” said Dr. Chauhan of the department of obstetrics, gynecology, and reproductive sciences at the University of Texas, Houston. “But more frequent scans did not increase the frequency of identifying these abnormalities. And since there are approximately 1.7 million such high-risk pregnancies each year, these findings have significant implications on personal, departmental, and societal levels.”
However, he added, the SUN trial, which randomized women to monthly or biweekly scans, wasn’t large enough to definitively determine the optimal timing of ultrasounds in this population. “We need larger trials to determine this.”
The trial results, published in the American Journal of Obstetrics & Gynecology, comprised 228 women with singleton pregnancies complicated by medical comorbidities, putting the fetus at risk of problems with growth or amniotic fluid volume. They were randomized to ultrasound scans every 2 weeks or every 4 weeks during the last trimester. The primary outcome was identification of four pregnancy complications: fetal growth restriction, large for gestational age, oligohydramnios, or polyhydramnios.
There were also secondary outcomes of composite maternal morbidity (chorioamnionitis, wound infection, transfusion, diabetic ketoacidosis, venous thromboembolism, ICU admission, and death) and neonatal morbidity (Apgar score of less than 5 at 5 minutes, umbilical artery pH less than 7, hyperbilirubinemia, intubation, high-grade intraventricular hemorrhage, necrotizing enterocolitis, and death).
Women were eligible for the study if they had autoimmune disease, a body mass index of 40 kg/m2 or higher, a history of delivering a small for gestational age or macrosomic infant, hypertensive disease, substance abuse, or sickle cell disease.
About half had multiple risk factors, the three most common being high body mass index, diabetes (either gestational or insulin-dependent), and hypertensive disease. Their gestational age at randomization was a mean of 23 weeks. The first scan was conducted at a mean of 29 weeks.
There were 28% more scans in the 2-week group than in the 4-week group (492 vs. 382). There were also more exams for potentially concerning findings, including more scans looking at growth (359 vs. 278), biophysical profiles (99 vs. 79), and umbilical artery Dopplers (34 vs. 27). Despite the increased scans and investigations, the primary outcome was detected at similar points (31.7 vs. 32.2 weeks).
A primary outcome finding was detected in 38% of the 2-week group and 32% of the 4-week group – not significantly different. There were no significant differences in the proportion of outcomes detected in each group, including fetal growth restriction (19% vs. 16%), large for gestational age (14% each group), oligohydramnios (4% each group) and polyhydramnios (9% each group).
Infants in each group were born at about the same time – 13.5 weeks after randomization. The mean gestational age in both groups was 37 weeks, with about 28.5% less than 37 weeks at birth. There were no significant differences in the rate of cesarean deliveries, or small or large for gestational age births.
The composite maternal morbidity outcome was similar in the 2-week and 4-week groups (26% vs. 22%). There were no significant differences in any of the individual components of the outcome. No mother died.
The composite neonatal outcome was also similar between the groups (14% vs. 12%). There were no significant differences in any of the individual components. No infant died.
The study was sponsored by the University of Texas Health Science Center, Houston. Dr. Chauhan had no financial disclosures.
SOURCE: Roberts RP et al. Am J Obstet Gynecol. 2018;218:S3.
REPORTING FROM THE PREGNANCY MEETING
Key clinical point: Ultrasound scans every 4 weeks identify just as many fetal growth of amniotic fluid problems as more frequent scans.
Major finding: A fetal or fluid abnormality was detected in 38% of the 2-week group and 32% of the 4-week group – not significantly different.
Study details: The randomized prospective trial comprised 228 women.
Disclosures: The University of Texas Medical Center, Houston, sponsored the study. Dr. Chauhan had no financial disclosures.
Source: Roberts RP et al. Am J Obstet Gynecol. 2018;218:S3.
TRAAP trial looks at tranexamic acid to prevent postpartum hemorrhage after vaginal delivery
DALLAS – An intravenous drug administered to women along with oxytocin after vaginal delivery did not reduce the risk of a large-volume hemorrhage, but it did significantly affect some other markers of postpartum blood loss.
The TRAAP trial failed to achieve its primary endpoint – reduction of postpartum hemorrhage of at least 500 ml, Loïc Sentilhes, MD, reported at the meeting sponsored by the Society for Maternal-Fetal Medicine. But tranexamic acid’s success in the prespecified secondary endpoints suggests there still may be a place for the drug in obstetrics, said Dr. Sentilhes of the University Hospital of Bordeaux, France.

TRAAP randomized 4,079 women in labor for a planned vaginal birth to either 1 g of tranexamic acid or placebo. The interventions were added to existing IV fluids within 2 minutes after vaginal birth, after the women had also received routine oxytocin.
The study’s primary outcome was the incidence of postpartum hemorrhage, defined as a blood loss of 500 ml or more. There were a number of prespecified secondary outcomes, including clinically significant blood loss, need for additional uterotonics, severe hemorrhage (more than 1,000 mL), estimated total blood loss, transfusion, and changes in hemoglobin and hematocrit.
The study also looked at these outcomes in prespecified subgroups: women who had an episiotomy, who had an operative delivery, and who had a history of prior postpartum hemorrhage.
Women in the study were a mean age of 30 years; about half were primiparous, and 5% were attempting a vaginal birth after cesarean section. Of the women, 4% had experienced a prior postpartum hemorrhage. Labor was induced in 20%, and about 60% needed labor augmentation.
An operative vaginal delivery was necessary for 17%. Forms of assistance included forceps (55%) and vacuum extraction (45%). About a quarter had an episiotomy, and 8% had a macrosomic neonate of more than 4,000 g.
The primary endpoint of at least 500 ml blood loss occurred in 8.1% of the active group and 9.8% of the placebo group – a nonsignificant difference (relative risk, 0.83; P = .07).
Of the secondary endpoints, only two – clinically significant bleeding and need for additional uterotonics – were significantly better in the tranexamic acid group. The drug reduced the risk of clinically significant bleeding by 26% (RR, 0.74; P = .004) and the need for additional uterotonics by 25% (RR, 0.75; P = .006).
While the primary endpoint was not realized in the overall patient group, tranexamic acid achieved barely significant risk reductions of 34% in the risk of hemorrhage among women who had an operative delivery (RR, 0.66; P = .0498) and 27% among those who had an episiotomy (RR, 0.73; P = .049).
It was no better than placebo in women with a history of postpartum hemorrhage.
Those who received the drug were twice as likely to experience nausea and vomiting after delivery as did those who received placebo. But tranexamic acid exerted no lingering prothrombotic effects at 3 months after delivery, with no signals in deep vein thrombosis, no pulmonary embolism, and no clots in the ovarian or superficial veins.
The TRAAP trial was funded by the French Ministry of Health under its clinical research hospital program. Dr. Sentilhes had no financial disclosures.
SOURCE: Sentilhes L et al. Am J Obstet Gynecol. 2018;218:S1.
DALLAS – An intravenous drug administered to women along with oxytocin after vaginal delivery did not reduce the risk of a large-volume hemorrhage, but it did significantly affect some other markers of postpartum blood loss.
The TRAAP trial failed to achieve its primary endpoint – reduction of postpartum hemorrhage of at least 500 ml, Loïc Sentilhes, MD, reported at the meeting sponsored by the Society for Maternal-Fetal Medicine. But tranexamic acid’s success in the prespecified secondary endpoints suggests there still may be a place for the drug in obstetrics, said Dr. Sentilhes of the University Hospital of Bordeaux, France.
TRAAP randomized 4,079 women in labor for a planned vaginal birth to either 1 g of tranexamic acid or placebo. The interventions were added to existing IV fluids within 2 minutes after vaginal birth, after the women had also received routine oxytocin.
The study’s primary outcome was the incidence of postpartum hemorrhage, defined as a blood loss of 500 ml or more. There were a number of prespecified secondary outcomes, including clinically significant blood loss, need for additional uterotonics, severe hemorrhage (more than 1,000 mL), estimated total blood loss, transfusion, and changes in hemoglobin and hematocrit.
The study also looked at these outcomes in prespecified subgroups: women who had an episiotomy, who had an operative delivery, and who had a history of prior postpartum hemorrhage.
Women in the study were a mean age of 30 years; about half were primiparous, and 5% were attempting a vaginal birth after cesarean section. Of the women, 4% had experienced a prior postpartum hemorrhage. Labor was induced in 20%, and about 60% needed labor augmentation.
An operative vaginal delivery was necessary for 17%. Forms of assistance included forceps (55%) and vacuum extraction (45%). About a quarter had an episiotomy, and 8% had a macrosomic neonate of more than 4,000 g.
The primary endpoint of at least 500 ml blood loss occurred in 8.1% of the active group and 9.8% of the placebo group – a nonsignificant difference (relative risk, 0.83; P = .07).
Of the secondary endpoints, only two – clinically significant bleeding and need for additional uterotonics – were significantly better in the tranexamic acid group. The drug reduced the risk of clinically significant bleeding by 26% (RR, 0.74; P = .004) and the need for additional uterotonics by 25% (RR, 0.75; P = .006).
While the primary endpoint was not realized in the overall patient group, tranexamic acid achieved barely significant risk reductions of 34% in the risk of hemorrhage among women who had an operative delivery (RR, 0.66; P = .0498) and 27% among those who had an episiotomy (RR, 0.73; P = .049).
It was no better than placebo in women with a history of postpartum hemorrhage.
Those who received the drug were twice as likely to experience nausea and vomiting after delivery as did those who received placebo. But tranexamic acid exerted no lingering prothrombotic effects at 3 months after delivery, with no signals in deep vein thrombosis, no pulmonary embolism, and no clots in the ovarian or superficial veins.
The TRAAP trial was funded by the French Ministry of Health under its clinical research hospital program. Dr. Sentilhes had no financial disclosures.
SOURCE: Sentilhes L et al. Am J Obstet Gynecol. 2018;218:S1.
DALLAS – An intravenous drug administered to women along with oxytocin after vaginal delivery did not reduce the risk of a large-volume hemorrhage, but it did significantly affect some other markers of postpartum blood loss.
The TRAAP trial failed to achieve its primary endpoint – reduction of postpartum hemorrhage of at least 500 ml, Loïc Sentilhes, MD, reported at the meeting sponsored by the Society for Maternal-Fetal Medicine. But tranexamic acid’s success in the prespecified secondary endpoints suggests there still may be a place for the drug in obstetrics, said Dr. Sentilhes of the University Hospital of Bordeaux, France.
TRAAP randomized 4,079 women in labor for a planned vaginal birth to either 1 g of tranexamic acid or placebo. The interventions were added to existing IV fluids within 2 minutes after vaginal birth, after the women had also received routine oxytocin.
The study’s primary outcome was the incidence of postpartum hemorrhage, defined as a blood loss of 500 ml or more. There were a number of prespecified secondary outcomes, including clinically significant blood loss, need for additional uterotonics, severe hemorrhage (more than 1,000 mL), estimated total blood loss, transfusion, and changes in hemoglobin and hematocrit.
The study also looked at these outcomes in prespecified subgroups: women who had an episiotomy, who had an operative delivery, and who had a history of prior postpartum hemorrhage.
Women in the study were a mean age of 30 years; about half were primiparous, and 5% were attempting a vaginal birth after cesarean section. Of the women, 4% had experienced a prior postpartum hemorrhage. Labor was induced in 20%, and about 60% needed labor augmentation.
An operative vaginal delivery was necessary for 17%. Forms of assistance included forceps (55%) and vacuum extraction (45%). About a quarter had an episiotomy, and 8% had a macrosomic neonate of more than 4,000 g.
The primary endpoint of at least 500 ml blood loss occurred in 8.1% of the active group and 9.8% of the placebo group – a nonsignificant difference (relative risk, 0.83; P = .07).
Of the secondary endpoints, only two – clinically significant bleeding and need for additional uterotonics – were significantly better in the tranexamic acid group. The drug reduced the risk of clinically significant bleeding by 26% (RR, 0.74; P = .004) and the need for additional uterotonics by 25% (RR, 0.75; P = .006).
While the primary endpoint was not realized in the overall patient group, tranexamic acid achieved barely significant risk reductions of 34% in the risk of hemorrhage among women who had an operative delivery (RR, 0.66; P = .0498) and 27% among those who had an episiotomy (RR, 0.73; P = .049).
It was no better than placebo in women with a history of postpartum hemorrhage.
Those who received the drug were twice as likely to experience nausea and vomiting after delivery as did those who received placebo. But tranexamic acid exerted no lingering prothrombotic effects at 3 months after delivery, with no signals in deep vein thrombosis, no pulmonary embolism, and no clots in the ovarian or superficial veins.
The TRAAP trial was funded by the French Ministry of Health under its clinical research hospital program. Dr. Sentilhes had no financial disclosures.
SOURCE: Sentilhes L et al. Am J Obstet Gynecol. 2018;218:S1.
REPORTING FROM THE PREGNANCY MEETING
Key clinical point: Tranexamic acid didn’t significantly reduce the risk of a postpartum hemorrhage of 500 ml or more.
Major finding: Hemorrhage occurred in 8.1% of the active group and 9.8% of the placebo group – a nonsignificant difference (relative risk, 0.83; P = .07).
Study details: The randomized, placebo-controlled study comprised 4,079 women.
Disclosures: The TRAAP trial was funded by the French Ministry of Health under its clinical research hospital program. Dr. Sentilhes had no financial disclosures.
Source: Sentilhes L et al. Am J Obstet Gynecol. 2018;218:S1.
FIRST trial analysis shows more benefit for lenalidomide
Lenalidomide with low-dose dexamethasone showed a significant overall survival benefit, compared with melphalan with prednisone and thalidomide, for patients with transplant-ineligible newly diagnosed multiple myeloma, according to the final analysis of the phase 3 FIRST trial.
The prespecified final analysis evaluated overall survival (OS) at a follow-up of 60 months or longer, according to the study, which was published in Blood. The FIRST study included 1,623 patients randomized to receive lenalidomide with low-dose dexamethasone continuously until disease progression (Rd continuous), lenalidomide plus low-dose dexamethasone for 18 cycles instead of continuously (Rd18), or melphalan plus prednisone and thalidomide (MPT). Patients with high-risk cytogenetics were distributed evenly across the three treatment arms.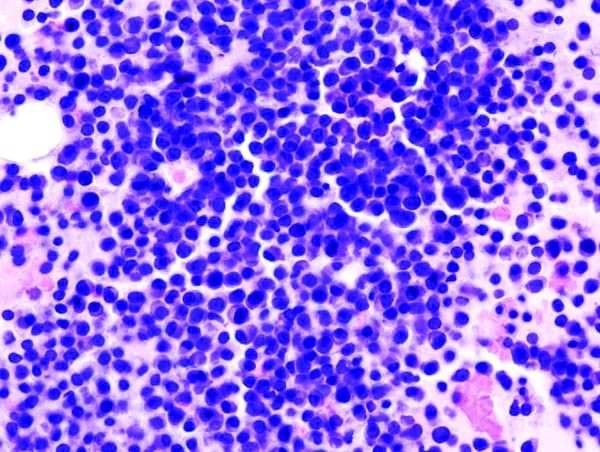
The Rd continuous cohort also experienced significantly longer PFS, compared with the MPT arm (HR, 0.69, 95% CI, 0.59-0.79, P less than .00001).
“Taken together, these findings suggest that Rd affords a clinical advantage in subsequent lines of therapy and highlight the importance of using Rd continuous and not MPT as first-line treatment of transplant-ineligible patients with [newly-diagnosed multiple myeloma],” the researchers wrote.
The OS benefit was similar for patients who received Rd continuous (59.1 months) and Rd18 (62.3 months). The researchers suggested that the similar survival could be due to a combination of factors, including the impact of subsequent lines of treatment and the older age of the patient population.
However, the benefit for Rd18 was not seen in the PFS analysis. Patients in the Rd18 group had a median PFS similar to that of patients in the MPT group (21 months vs. 21.9 months), both of which were shorter than the Rd continuous group (26 months).
The 4-year PFS more than doubled among patients in the Rd continuous arm (32.6%), compared with patients in both the Rd18 group (14.3%) and the MPT group (13.6%).
Patients in the Rd continuous group also experienced a decreased risk for progression or death, compared with patients in the Rd18 group (HR, 0.70, 95% CI, 0.60-0.81).
The researchers noted that no new safety concerns were observed in the final analysis. Second primary malignancies, including hematologic and solid tumors, were found in 36% of patients in the Rd continuous group, 38% of patients in the Rd18 group, and 46% of patients in the MPT group.
Celgene Corporation sponsored the study. Dr. Facon and his coauthors reported financial ties to various pharmaceutical companies, including Celgene.
SOURCE: Facon T et al. Blood. 2018 Jan 18;131(3):301-10.
Lenalidomide with low-dose dexamethasone showed a significant overall survival benefit, compared with melphalan with prednisone and thalidomide, for patients with transplant-ineligible newly diagnosed multiple myeloma, according to the final analysis of the phase 3 FIRST trial.
The prespecified final analysis evaluated overall survival (OS) at a follow-up of 60 months or longer, according to the study, which was published in Blood. The FIRST study included 1,623 patients randomized to receive lenalidomide with low-dose dexamethasone continuously until disease progression (Rd continuous), lenalidomide plus low-dose dexamethasone for 18 cycles instead of continuously (Rd18), or melphalan plus prednisone and thalidomide (MPT). Patients with high-risk cytogenetics were distributed evenly across the three treatment arms.
The Rd continuous cohort also experienced significantly longer PFS, compared with the MPT arm (HR, 0.69, 95% CI, 0.59-0.79, P less than .00001).
“Taken together, these findings suggest that Rd affords a clinical advantage in subsequent lines of therapy and highlight the importance of using Rd continuous and not MPT as first-line treatment of transplant-ineligible patients with [newly-diagnosed multiple myeloma],” the researchers wrote.
The OS benefit was similar for patients who received Rd continuous (59.1 months) and Rd18 (62.3 months). The researchers suggested that the similar survival could be due to a combination of factors, including the impact of subsequent lines of treatment and the older age of the patient population.
However, the benefit for Rd18 was not seen in the PFS analysis. Patients in the Rd18 group had a median PFS similar to that of patients in the MPT group (21 months vs. 21.9 months), both of which were shorter than the Rd continuous group (26 months).
The 4-year PFS more than doubled among patients in the Rd continuous arm (32.6%), compared with patients in both the Rd18 group (14.3%) and the MPT group (13.6%).
Patients in the Rd continuous group also experienced a decreased risk for progression or death, compared with patients in the Rd18 group (HR, 0.70, 95% CI, 0.60-0.81).
The researchers noted that no new safety concerns were observed in the final analysis. Second primary malignancies, including hematologic and solid tumors, were found in 36% of patients in the Rd continuous group, 38% of patients in the Rd18 group, and 46% of patients in the MPT group.
Celgene Corporation sponsored the study. Dr. Facon and his coauthors reported financial ties to various pharmaceutical companies, including Celgene.
SOURCE: Facon T et al. Blood. 2018 Jan 18;131(3):301-10.
Lenalidomide with low-dose dexamethasone showed a significant overall survival benefit, compared with melphalan with prednisone and thalidomide, for patients with transplant-ineligible newly diagnosed multiple myeloma, according to the final analysis of the phase 3 FIRST trial.
The prespecified final analysis evaluated overall survival (OS) at a follow-up of 60 months or longer, according to the study, which was published in Blood. The FIRST study included 1,623 patients randomized to receive lenalidomide with low-dose dexamethasone continuously until disease progression (Rd continuous), lenalidomide plus low-dose dexamethasone for 18 cycles instead of continuously (Rd18), or melphalan plus prednisone and thalidomide (MPT). Patients with high-risk cytogenetics were distributed evenly across the three treatment arms.
The Rd continuous cohort also experienced significantly longer PFS, compared with the MPT arm (HR, 0.69, 95% CI, 0.59-0.79, P less than .00001).
“Taken together, these findings suggest that Rd affords a clinical advantage in subsequent lines of therapy and highlight the importance of using Rd continuous and not MPT as first-line treatment of transplant-ineligible patients with [newly-diagnosed multiple myeloma],” the researchers wrote.
The OS benefit was similar for patients who received Rd continuous (59.1 months) and Rd18 (62.3 months). The researchers suggested that the similar survival could be due to a combination of factors, including the impact of subsequent lines of treatment and the older age of the patient population.
However, the benefit for Rd18 was not seen in the PFS analysis. Patients in the Rd18 group had a median PFS similar to that of patients in the MPT group (21 months vs. 21.9 months), both of which were shorter than the Rd continuous group (26 months).
The 4-year PFS more than doubled among patients in the Rd continuous arm (32.6%), compared with patients in both the Rd18 group (14.3%) and the MPT group (13.6%).
Patients in the Rd continuous group also experienced a decreased risk for progression or death, compared with patients in the Rd18 group (HR, 0.70, 95% CI, 0.60-0.81).
The researchers noted that no new safety concerns were observed in the final analysis. Second primary malignancies, including hematologic and solid tumors, were found in 36% of patients in the Rd continuous group, 38% of patients in the Rd18 group, and 46% of patients in the MPT group.
Celgene Corporation sponsored the study. Dr. Facon and his coauthors reported financial ties to various pharmaceutical companies, including Celgene.
SOURCE: Facon T et al. Blood. 2018 Jan 18;131(3):301-10.
FROM BLOOD
Key clinical point:
Major finding: Continuously administered lenalidomide with dexamethasone regimen improved PFS (HR, 0.69) and OS (HR, 0.78), compared with melphalan, prednisone, and thalidomide.
Study details: Final analysis of the phase 3 FIRST trial.
Disclosures: Celgene Corporation sponsored the study. Dr. Facon and his coauthors reported financial ties to various pharmaceutical companies, including Celgene.
Source: Facon T et al. Blood. 2018 Jan 18;131(3):301-10.
OSA patients report sleeping better with dronabinol
in a new study.
A paper published in the January edition of Sleep presents data from a phase 2, blinded, randomized controlled trial of the nonselective cannabinoid 1 and cannabinoid 2 receptor agonist, dronabinol, in 73 adults with moderate or severe obstructive sleep apnea (OSA). No approved drug treatments for OSA exist, and this study provides results “from the largest and longest randomized controlled trial to date of any putative drug treatment for OSA,” the researchers wrote.
Patients were randomized to 2.5 mg dronabinol or 10 mg dronabinol daily for up to 6 weeks, or placebo. At the end of treatment, researchers saw significant increases in the apnea-hypopnea index among the patients on placebo, while those who received dronabinol showed decreases in the number of apnea and hypopnea events per hour. Patients given the 2.5-mg dose of dronabinol had a mean decrease of 10.7 events per hour, and those on the 10-mg dose had a mean decrease of 12.9 events per hour compared with placebo.
The difference between the placebo and treatment arms was significant for both dosages, and the apnea-hypopnea index decreases were similar between the two dosages of dronabinol.
These effects were largely due to reductions in apnea events; the largest reduction was seen in the REM apnea index in patients treated with the 10-mg dose of dronabinol. However, there were few effects on the expression of hypopneas, except in the higher-dose group.
After adjustment for age, race, ethnicity, and baseline apnea-hypopnea index, the increases seen in the placebo group were no longer significant, but the decreases from baseline seen in the treatment arms were greater.
Dronabinol treatment was also associated with significant decreases, compared with placebo, in non-REM apnea-hypopnea index and REM apnea-hypopnea index.
Patients’ self-reported daytime sleepiness, measured by the Epworth Sleepiness Scale, remained similar compared with baseline in those who received placebo and the 2.5-mg/day dose of dronabinol, but decreased significantly by a mean of −2.3 points compared with placebo in those on the higher dose of dronabinol.
There were no significant changes from baseline in objective sleepiness, as measured by the maintenance of wakefulness test, in any of the study groups. Researchers also saw no significant changes in sleep architecture, oxygenation, or the duration of supine sleep in any of the study groups, although the patients on the higher dose of dronabinol showed a slight increase in REM sleep and those on placebo showed a slight decrease.
Younger patients and those with a greater preponderance of REM-related apnea/hypopnea, and shorter average event duration were both more likely to respond to treatment, but apart from these factors there were no other influences on likelihood of patients responding to dronabinol.
David W. Carley, PhD, of the University of Illinois at Chicago, and his coauthors noted that there was a great need for pharmacological treatments for obstructive sleep apnea because positive airway pressure – while effective – has poor long-term adherence rates.
“Based on a series of animal investigations, we proposed that drugs which dampen afferent vagal feedback to the medulla may be effective in stabilizing respiratory pattern generation and increasing activation of upper airway dilating muscles during sleep,” they wrote.
One patient experienced diarrhea and vomiting that required admission to hospital, and which was judged as possibly related to the study medication. There were six other withdrawals due to adverse events including dizziness and vision changes, vertigo, ECG arrhythmias, and headache with dizziness and vomiting. Overall, nearly 90% of patients reported at least one adverse event, but the rates did not differ significantly between the treatment and placebo arms.
The researchers noted that significantly higher satisfaction scores were seen among patients receiving the higher dose of dronabinol.
“All of these observations argue that dronabinol, at doses from 2.5 to 10 mg/day, is safe for use by medically stable patients with moderate or severe OSA,” the authors wrote. “Participants also tolerated and adhered well to daily self-administration of dronabinol.”
The National Institutes of Health, National Heart, Lung, and Blood Institute, and National Center for Advancing Translational Sciences funded the study. One author declared grants from the National Institutes of Health for the study, and patents related to treatment of sleep-related breathing disorders by cannabinoid drugs. He also holds stock in RespireRx Pharmaceuticals, which holds an exclusive license to these and other related patents.
SOURCE: Carley D, et al. Sleep. 2018 Jan 1. doi: 10.1093/sleep/zsx184
This study has found a small overall effect on the apnea-hypopnea index with treatment, but a strong beneficial effect on subjective sleepiness. In addition, participants who received the higher dose of the drug showed significant satisfaction with their therapy. It is therefore intriguing that there was no impact on objective wakefulness or sleep architecture with this treatment.
This suggests that perhaps sleepiness and subjective wellbeing may be improved without necessarily seeing major improvements in the apnea-hypopnea index, which calls into question our use of this index as a primary end-point.
Sigrid C. Veasey, MD, is with the Center for Sleep and Circadian Neurobiology at the Perelman School of Medicine, University of Pennsylvania, Philadelphia. These comments are taken from an accompanying (Sleep 2018 Jan 1. doi: 10.1093/sleep/zsy014). No conflicts of interest were declared.
This study has found a small overall effect on the apnea-hypopnea index with treatment, but a strong beneficial effect on subjective sleepiness. In addition, participants who received the higher dose of the drug showed significant satisfaction with their therapy. It is therefore intriguing that there was no impact on objective wakefulness or sleep architecture with this treatment.
This suggests that perhaps sleepiness and subjective wellbeing may be improved without necessarily seeing major improvements in the apnea-hypopnea index, which calls into question our use of this index as a primary end-point.
Sigrid C. Veasey, MD, is with the Center for Sleep and Circadian Neurobiology at the Perelman School of Medicine, University of Pennsylvania, Philadelphia. These comments are taken from an accompanying (Sleep 2018 Jan 1. doi: 10.1093/sleep/zsy014). No conflicts of interest were declared.
This study has found a small overall effect on the apnea-hypopnea index with treatment, but a strong beneficial effect on subjective sleepiness. In addition, participants who received the higher dose of the drug showed significant satisfaction with their therapy. It is therefore intriguing that there was no impact on objective wakefulness or sleep architecture with this treatment.
This suggests that perhaps sleepiness and subjective wellbeing may be improved without necessarily seeing major improvements in the apnea-hypopnea index, which calls into question our use of this index as a primary end-point.
Sigrid C. Veasey, MD, is with the Center for Sleep and Circadian Neurobiology at the Perelman School of Medicine, University of Pennsylvania, Philadelphia. These comments are taken from an accompanying (Sleep 2018 Jan 1. doi: 10.1093/sleep/zsy014). No conflicts of interest were declared.
in a new study.
A paper published in the January edition of Sleep presents data from a phase 2, blinded, randomized controlled trial of the nonselective cannabinoid 1 and cannabinoid 2 receptor agonist, dronabinol, in 73 adults with moderate or severe obstructive sleep apnea (OSA). No approved drug treatments for OSA exist, and this study provides results “from the largest and longest randomized controlled trial to date of any putative drug treatment for OSA,” the researchers wrote.
Patients were randomized to 2.5 mg dronabinol or 10 mg dronabinol daily for up to 6 weeks, or placebo. At the end of treatment, researchers saw significant increases in the apnea-hypopnea index among the patients on placebo, while those who received dronabinol showed decreases in the number of apnea and hypopnea events per hour. Patients given the 2.5-mg dose of dronabinol had a mean decrease of 10.7 events per hour, and those on the 10-mg dose had a mean decrease of 12.9 events per hour compared with placebo.
The difference between the placebo and treatment arms was significant for both dosages, and the apnea-hypopnea index decreases were similar between the two dosages of dronabinol.
These effects were largely due to reductions in apnea events; the largest reduction was seen in the REM apnea index in patients treated with the 10-mg dose of dronabinol. However, there were few effects on the expression of hypopneas, except in the higher-dose group.
After adjustment for age, race, ethnicity, and baseline apnea-hypopnea index, the increases seen in the placebo group were no longer significant, but the decreases from baseline seen in the treatment arms were greater.
Dronabinol treatment was also associated with significant decreases, compared with placebo, in non-REM apnea-hypopnea index and REM apnea-hypopnea index.
Patients’ self-reported daytime sleepiness, measured by the Epworth Sleepiness Scale, remained similar compared with baseline in those who received placebo and the 2.5-mg/day dose of dronabinol, but decreased significantly by a mean of −2.3 points compared with placebo in those on the higher dose of dronabinol.
There were no significant changes from baseline in objective sleepiness, as measured by the maintenance of wakefulness test, in any of the study groups. Researchers also saw no significant changes in sleep architecture, oxygenation, or the duration of supine sleep in any of the study groups, although the patients on the higher dose of dronabinol showed a slight increase in REM sleep and those on placebo showed a slight decrease.
Younger patients and those with a greater preponderance of REM-related apnea/hypopnea, and shorter average event duration were both more likely to respond to treatment, but apart from these factors there were no other influences on likelihood of patients responding to dronabinol.
David W. Carley, PhD, of the University of Illinois at Chicago, and his coauthors noted that there was a great need for pharmacological treatments for obstructive sleep apnea because positive airway pressure – while effective – has poor long-term adherence rates.
“Based on a series of animal investigations, we proposed that drugs which dampen afferent vagal feedback to the medulla may be effective in stabilizing respiratory pattern generation and increasing activation of upper airway dilating muscles during sleep,” they wrote.
One patient experienced diarrhea and vomiting that required admission to hospital, and which was judged as possibly related to the study medication. There were six other withdrawals due to adverse events including dizziness and vision changes, vertigo, ECG arrhythmias, and headache with dizziness and vomiting. Overall, nearly 90% of patients reported at least one adverse event, but the rates did not differ significantly between the treatment and placebo arms.
The researchers noted that significantly higher satisfaction scores were seen among patients receiving the higher dose of dronabinol.
“All of these observations argue that dronabinol, at doses from 2.5 to 10 mg/day, is safe for use by medically stable patients with moderate or severe OSA,” the authors wrote. “Participants also tolerated and adhered well to daily self-administration of dronabinol.”
The National Institutes of Health, National Heart, Lung, and Blood Institute, and National Center for Advancing Translational Sciences funded the study. One author declared grants from the National Institutes of Health for the study, and patents related to treatment of sleep-related breathing disorders by cannabinoid drugs. He also holds stock in RespireRx Pharmaceuticals, which holds an exclusive license to these and other related patents.
SOURCE: Carley D, et al. Sleep. 2018 Jan 1. doi: 10.1093/sleep/zsx184
in a new study.
A paper published in the January edition of Sleep presents data from a phase 2, blinded, randomized controlled trial of the nonselective cannabinoid 1 and cannabinoid 2 receptor agonist, dronabinol, in 73 adults with moderate or severe obstructive sleep apnea (OSA). No approved drug treatments for OSA exist, and this study provides results “from the largest and longest randomized controlled trial to date of any putative drug treatment for OSA,” the researchers wrote.
Patients were randomized to 2.5 mg dronabinol or 10 mg dronabinol daily for up to 6 weeks, or placebo. At the end of treatment, researchers saw significant increases in the apnea-hypopnea index among the patients on placebo, while those who received dronabinol showed decreases in the number of apnea and hypopnea events per hour. Patients given the 2.5-mg dose of dronabinol had a mean decrease of 10.7 events per hour, and those on the 10-mg dose had a mean decrease of 12.9 events per hour compared with placebo.
The difference between the placebo and treatment arms was significant for both dosages, and the apnea-hypopnea index decreases were similar between the two dosages of dronabinol.
These effects were largely due to reductions in apnea events; the largest reduction was seen in the REM apnea index in patients treated with the 10-mg dose of dronabinol. However, there were few effects on the expression of hypopneas, except in the higher-dose group.
After adjustment for age, race, ethnicity, and baseline apnea-hypopnea index, the increases seen in the placebo group were no longer significant, but the decreases from baseline seen in the treatment arms were greater.
Dronabinol treatment was also associated with significant decreases, compared with placebo, in non-REM apnea-hypopnea index and REM apnea-hypopnea index.
Patients’ self-reported daytime sleepiness, measured by the Epworth Sleepiness Scale, remained similar compared with baseline in those who received placebo and the 2.5-mg/day dose of dronabinol, but decreased significantly by a mean of −2.3 points compared with placebo in those on the higher dose of dronabinol.
There were no significant changes from baseline in objective sleepiness, as measured by the maintenance of wakefulness test, in any of the study groups. Researchers also saw no significant changes in sleep architecture, oxygenation, or the duration of supine sleep in any of the study groups, although the patients on the higher dose of dronabinol showed a slight increase in REM sleep and those on placebo showed a slight decrease.
Younger patients and those with a greater preponderance of REM-related apnea/hypopnea, and shorter average event duration were both more likely to respond to treatment, but apart from these factors there were no other influences on likelihood of patients responding to dronabinol.
David W. Carley, PhD, of the University of Illinois at Chicago, and his coauthors noted that there was a great need for pharmacological treatments for obstructive sleep apnea because positive airway pressure – while effective – has poor long-term adherence rates.
“Based on a series of animal investigations, we proposed that drugs which dampen afferent vagal feedback to the medulla may be effective in stabilizing respiratory pattern generation and increasing activation of upper airway dilating muscles during sleep,” they wrote.
One patient experienced diarrhea and vomiting that required admission to hospital, and which was judged as possibly related to the study medication. There were six other withdrawals due to adverse events including dizziness and vision changes, vertigo, ECG arrhythmias, and headache with dizziness and vomiting. Overall, nearly 90% of patients reported at least one adverse event, but the rates did not differ significantly between the treatment and placebo arms.
The researchers noted that significantly higher satisfaction scores were seen among patients receiving the higher dose of dronabinol.
“All of these observations argue that dronabinol, at doses from 2.5 to 10 mg/day, is safe for use by medically stable patients with moderate or severe OSA,” the authors wrote. “Participants also tolerated and adhered well to daily self-administration of dronabinol.”
The National Institutes of Health, National Heart, Lung, and Blood Institute, and National Center for Advancing Translational Sciences funded the study. One author declared grants from the National Institutes of Health for the study, and patents related to treatment of sleep-related breathing disorders by cannabinoid drugs. He also holds stock in RespireRx Pharmaceuticals, which holds an exclusive license to these and other related patents.
SOURCE: Carley D, et al. Sleep. 2018 Jan 1. doi: 10.1093/sleep/zsx184
FROM SLEEP
Key clinical point: A cannabinoid receptor agonist may significantly reduce apnea and hypopnea events in patients with obstructive sleep apnea.
Major finding: Patients who received either low dose or high-dose dronabinol showed significant decreases in apnea and hypopnea events compared to those on placebo.
Data source: Randomized controlled, blinded phase II trial in 73 patients with obstructive sleep apnea.
Disclosures: The National Institutes of Health, National Heart Lung and Blood Institute, and National Center for Advancing Translational Sciences funded the study. One author declared grants from the National Institutes of Health for the study, and patents related to treatment of sleep-related breathing disorders by cannabinoid drugs. He also holds stock in RespireRx Pharmaceuticals, which holds an exclusive license to these and other related patents.
Source: Carley D, et al. Sleep. 2018 Jan 1. doi: 10.1093/sleep/zsx184.