User login
Primary care involvement improves chance of meeting recommended surveillance intervals
CHICAGO – Only a small portion of patients with nondysplastic Barrett’s esophagus received appropriately timed endoscopic surveillance, a large database study showed.
But rather than being neglected, patients were more likely to be overassessed, with follow-up endoscopy performed more frequently than the recommended 3- to 5-year intervals, Anna Tavakkoli, MD, reported at the annual Digestive Disease Week.
“Very few patients entered our surveillance program with appropriate surveillance intervals,” said Dr. Tavakkoli, a gastroenterology fellow at the University of Michigan, Ann Arbor.

“We don’t have a formal program at the University of Michigan that drives coordination of care, but we do have great communication here between our primary care providers and our specialists. Our electronic medical records system makes quick messaging between providers easy, and primary care is very good about incorporating diagnoses into patients’ problem lists.”
Malignant transformation of nondysplastic Barrett’s is uncommon, with rates of no more than 4% per year. This understanding led three major societies – the American Gastroenterology Association, the American Society of Gastrointestinal Endoscopy, and the American College of Gastroenterology – to amend their surveillance recommendations in 2011 and 2012. All three societies now recommend a surveillance endoscopy every 3-5 years after the initial diagnosis of nondysplastic Barrett’s esophagus. In fact, the AGA has incorporated this suggestion into its five “Choosing Wisely” recommendations aimed at decreasing overutilization of testing and procedures.
Dr. Tavakkoli’s study examined surveillance timing in a cohort of 1,602 patients with nondysplastic Barrett’s who entered the University of Michigan Barrett’s Esophagus Registry from 1994 to 2016. All of these patents had at least three endoscopies or at least 5 years of follow-up data since their last endoscopy. The primary outcome was identification of trends in the appropriateness of surveillance of patients with nondysplastic Barrett’s esophagus at the University of Michigan. In her analysis, oversurveillance was defined as less than 3 years between the second and third endoscopy; undersurveillance was defined as more than 5 years between them. Dr. Tavakkoli and her colleagues also looked at patients who were lost to follow-up, defined as never receiving a second endoscopy after their initial diagnosis of nondysplastic Barrett’s esophagus and patients who were never surveilled, defined as never receiving their third endoscopy. All patients were compared with those who underwent appropriate surveillance, defined as 3-5 years between their second and third procedure.
The majority of patients were male, and the mean age was 59 years; 30% had long-segment Barrett’s, and 41% had a primary care provider in the university health care system. Most (90%) had their second endoscopy before 2012, when two of the three major societies issued their updated surveillance recommendations.
Of the entire cohort, 40% were lost to follow-up; 17% were never surveilled, and 3% were undersurveilled. Almost a third (31%) were oversurveilled, while just 8% had the appropriate surveillance, Dr. Tavakkoli said.
She then looked at several demographic and clinical factors associated with surveillance in each group, including sex, age, race, and income, comorbidities, length of Barrett’s, family history of esophageal cancer, and whether the patient had a University of Michigan primary care provider.
Having long-segment Barrett’s was associated with a 2.5-times increased risk of receiving a third endoscopy earlier than 3 years, which may be driven by studies that have shown that the risk of malignant transformation increases with Barrett’s length, she said.
The presence of a primary care physician significantly reduced the risk of inappropriate follow-up in every group, except patients who were undersurveilled, she said. The presence of a primary care physician at the University of Michigan decreased the risk of oversurveillance by 56%.
The positive influence of an in-system primary care physician was an important finding in this study, Dr Tavakkoli said. “The oncology data have shown us that poor coordination of care between oncologists and primary care providers contributes to avoidable patient morbidity and mortality, fragmented care, and increased costs. In 2005, the Institute of Medicine published a report emphasizing that coordination between specialists and primary care providers is one of the four key components to cancer survivorship care. There have been a number of GI studies looking at how primary care’s involvement in colorectal screening improves the rates of patients who undergo screening, but among Barrett’s patients, there have not been data showing that having a primary care physician at the center where endoscopic surveillance is done improves utilization patterns.”
Dr. Tavakkoli had no financial disclosures.
[email protected]
On Twitter @alz_gal
CHICAGO – Only a small portion of patients with nondysplastic Barrett’s esophagus received appropriately timed endoscopic surveillance, a large database study showed.
But rather than being neglected, patients were more likely to be overassessed, with follow-up endoscopy performed more frequently than the recommended 3- to 5-year intervals, Anna Tavakkoli, MD, reported at the annual Digestive Disease Week.
“Very few patients entered our surveillance program with appropriate surveillance intervals,” said Dr. Tavakkoli, a gastroenterology fellow at the University of Michigan, Ann Arbor.
“We don’t have a formal program at the University of Michigan that drives coordination of care, but we do have great communication here between our primary care providers and our specialists. Our electronic medical records system makes quick messaging between providers easy, and primary care is very good about incorporating diagnoses into patients’ problem lists.”
Malignant transformation of nondysplastic Barrett’s is uncommon, with rates of no more than 4% per year. This understanding led three major societies – the American Gastroenterology Association, the American Society of Gastrointestinal Endoscopy, and the American College of Gastroenterology – to amend their surveillance recommendations in 2011 and 2012. All three societies now recommend a surveillance endoscopy every 3-5 years after the initial diagnosis of nondysplastic Barrett’s esophagus. In fact, the AGA has incorporated this suggestion into its five “Choosing Wisely” recommendations aimed at decreasing overutilization of testing and procedures.
Dr. Tavakkoli’s study examined surveillance timing in a cohort of 1,602 patients with nondysplastic Barrett’s who entered the University of Michigan Barrett’s Esophagus Registry from 1994 to 2016. All of these patents had at least three endoscopies or at least 5 years of follow-up data since their last endoscopy. The primary outcome was identification of trends in the appropriateness of surveillance of patients with nondysplastic Barrett’s esophagus at the University of Michigan. In her analysis, oversurveillance was defined as less than 3 years between the second and third endoscopy; undersurveillance was defined as more than 5 years between them. Dr. Tavakkoli and her colleagues also looked at patients who were lost to follow-up, defined as never receiving a second endoscopy after their initial diagnosis of nondysplastic Barrett’s esophagus and patients who were never surveilled, defined as never receiving their third endoscopy. All patients were compared with those who underwent appropriate surveillance, defined as 3-5 years between their second and third procedure.
The majority of patients were male, and the mean age was 59 years; 30% had long-segment Barrett’s, and 41% had a primary care provider in the university health care system. Most (90%) had their second endoscopy before 2012, when two of the three major societies issued their updated surveillance recommendations.
Of the entire cohort, 40% were lost to follow-up; 17% were never surveilled, and 3% were undersurveilled. Almost a third (31%) were oversurveilled, while just 8% had the appropriate surveillance, Dr. Tavakkoli said.
She then looked at several demographic and clinical factors associated with surveillance in each group, including sex, age, race, and income, comorbidities, length of Barrett’s, family history of esophageal cancer, and whether the patient had a University of Michigan primary care provider.
Having long-segment Barrett’s was associated with a 2.5-times increased risk of receiving a third endoscopy earlier than 3 years, which may be driven by studies that have shown that the risk of malignant transformation increases with Barrett’s length, she said.
The presence of a primary care physician significantly reduced the risk of inappropriate follow-up in every group, except patients who were undersurveilled, she said. The presence of a primary care physician at the University of Michigan decreased the risk of oversurveillance by 56%.
The positive influence of an in-system primary care physician was an important finding in this study, Dr Tavakkoli said. “The oncology data have shown us that poor coordination of care between oncologists and primary care providers contributes to avoidable patient morbidity and mortality, fragmented care, and increased costs. In 2005, the Institute of Medicine published a report emphasizing that coordination between specialists and primary care providers is one of the four key components to cancer survivorship care. There have been a number of GI studies looking at how primary care’s involvement in colorectal screening improves the rates of patients who undergo screening, but among Barrett’s patients, there have not been data showing that having a primary care physician at the center where endoscopic surveillance is done improves utilization patterns.”
Dr. Tavakkoli had no financial disclosures.
[email protected]
On Twitter @alz_gal
CHICAGO – Only a small portion of patients with nondysplastic Barrett’s esophagus received appropriately timed endoscopic surveillance, a large database study showed.
But rather than being neglected, patients were more likely to be overassessed, with follow-up endoscopy performed more frequently than the recommended 3- to 5-year intervals, Anna Tavakkoli, MD, reported at the annual Digestive Disease Week.
“Very few patients entered our surveillance program with appropriate surveillance intervals,” said Dr. Tavakkoli, a gastroenterology fellow at the University of Michigan, Ann Arbor.
“We don’t have a formal program at the University of Michigan that drives coordination of care, but we do have great communication here between our primary care providers and our specialists. Our electronic medical records system makes quick messaging between providers easy, and primary care is very good about incorporating diagnoses into patients’ problem lists.”
Malignant transformation of nondysplastic Barrett’s is uncommon, with rates of no more than 4% per year. This understanding led three major societies – the American Gastroenterology Association, the American Society of Gastrointestinal Endoscopy, and the American College of Gastroenterology – to amend their surveillance recommendations in 2011 and 2012. All three societies now recommend a surveillance endoscopy every 3-5 years after the initial diagnosis of nondysplastic Barrett’s esophagus. In fact, the AGA has incorporated this suggestion into its five “Choosing Wisely” recommendations aimed at decreasing overutilization of testing and procedures.
Dr. Tavakkoli’s study examined surveillance timing in a cohort of 1,602 patients with nondysplastic Barrett’s who entered the University of Michigan Barrett’s Esophagus Registry from 1994 to 2016. All of these patents had at least three endoscopies or at least 5 years of follow-up data since their last endoscopy. The primary outcome was identification of trends in the appropriateness of surveillance of patients with nondysplastic Barrett’s esophagus at the University of Michigan. In her analysis, oversurveillance was defined as less than 3 years between the second and third endoscopy; undersurveillance was defined as more than 5 years between them. Dr. Tavakkoli and her colleagues also looked at patients who were lost to follow-up, defined as never receiving a second endoscopy after their initial diagnosis of nondysplastic Barrett’s esophagus and patients who were never surveilled, defined as never receiving their third endoscopy. All patients were compared with those who underwent appropriate surveillance, defined as 3-5 years between their second and third procedure.
The majority of patients were male, and the mean age was 59 years; 30% had long-segment Barrett’s, and 41% had a primary care provider in the university health care system. Most (90%) had their second endoscopy before 2012, when two of the three major societies issued their updated surveillance recommendations.
Of the entire cohort, 40% were lost to follow-up; 17% were never surveilled, and 3% were undersurveilled. Almost a third (31%) were oversurveilled, while just 8% had the appropriate surveillance, Dr. Tavakkoli said.
She then looked at several demographic and clinical factors associated with surveillance in each group, including sex, age, race, and income, comorbidities, length of Barrett’s, family history of esophageal cancer, and whether the patient had a University of Michigan primary care provider.
Having long-segment Barrett’s was associated with a 2.5-times increased risk of receiving a third endoscopy earlier than 3 years, which may be driven by studies that have shown that the risk of malignant transformation increases with Barrett’s length, she said.
The presence of a primary care physician significantly reduced the risk of inappropriate follow-up in every group, except patients who were undersurveilled, she said. The presence of a primary care physician at the University of Michigan decreased the risk of oversurveillance by 56%.
The positive influence of an in-system primary care physician was an important finding in this study, Dr Tavakkoli said. “The oncology data have shown us that poor coordination of care between oncologists and primary care providers contributes to avoidable patient morbidity and mortality, fragmented care, and increased costs. In 2005, the Institute of Medicine published a report emphasizing that coordination between specialists and primary care providers is one of the four key components to cancer survivorship care. There have been a number of GI studies looking at how primary care’s involvement in colorectal screening improves the rates of patients who undergo screening, but among Barrett’s patients, there have not been data showing that having a primary care physician at the center where endoscopic surveillance is done improves utilization patterns.”
Dr. Tavakkoli had no financial disclosures.
[email protected]
On Twitter @alz_gal
AT DDW
Key clinical point:
Major finding: Only 8% of patients had the recommended interval of 3-5 years between surveillance endoscopies.
Data source: A retrospective database cohort of 1,602 patients.
Disclosures: Dr. Tavakkoli had no relevant financial disclosures.
Later school start tied to more sleep
BOSTON – Teenagers slept more hours when they started school later, a new study found.
With 73% of high schoolers reporting receiving less than the recommended 8 hours of sleep per night, teens are among the most sleep-deprived members of society.
In this study, the later a teenager started school, the later he or she woke up, with average wake-up times having been just after 6:00 a.m. for those starting school between 7:00 and 7:30 a.m. and about 7:00 a.m. for those starting school after 8:30 a.m. But only those teens who started after 8:30 a.m. achieved the 8-hour recommended sleep duration, said Nicole Nahmod, during a presentation at the annual meeting of the Associated Professional Sleep Societies.
The students with the later school start times averaged 32 minutes of extra sleep, when compared with their early-rising colleagues, noted Ms. Nahmod, who is one of the study’s authors, a research technician, and study coordinator at Pennsylvania State University, Hershey. Specifically, adolescents who started school after 8:30 a.m. had a mean sleep duration of 8.1 hours, while those who started school earlier slept for only 7.5 hours a night.*

Acknowledging the importance of adequate sleep on teen health, mood, and school performance, the American Academy of Pediatrics recommends that middle schools and high schools begin at 8:30 a.m. or later. However, most school days start earlier than that, as evidenced by this dataset. While 72% of the study’s participants started school between 7:00 a.m. and 8:30 a.m., as many as 15% of the study participants began school during the narrower 7:00-7:30 a.m. window.
In this study, researchers from Penn State used data from 413 adolescents (mean age, 15.4 years; 46% male), who participated in a substudy of the Fragile Families & Child Wellbeing Study conducted across 20 large American cities. The study oversampled nonmarried pregnant mothers, resulting in a racially diverse sample and a high proportion of low-income families. The household income for 29% of the sample was below the poverty line.*
For this substudy, sleep duration was calculated from app-based daily diary reports of bed times, wake times, and school start times on days when a teen attended school.
The study was limited by its cross-sectional nature, but was enriched by a diverse range of school start times sampled from 20 U.S. cities, the high proportion of at-risk teens, and the use of a daily sleep diary.
“Current literature shows associations between later school start times and academic success, mood, and health. It also shows a decrease in motor vehicle accidents, tardiness, school dropout, and daytime sleepiness,” Ms. Nahmod noted.
Her continued research in this area involves analyzing the relationships between actigraphically assessed sleep measures in students and school start times.
Ms. Nahmod reported having no financial disclosures.
*This article was updated on June 6, 2017.
BOSTON – Teenagers slept more hours when they started school later, a new study found.
With 73% of high schoolers reporting receiving less than the recommended 8 hours of sleep per night, teens are among the most sleep-deprived members of society.
In this study, the later a teenager started school, the later he or she woke up, with average wake-up times having been just after 6:00 a.m. for those starting school between 7:00 and 7:30 a.m. and about 7:00 a.m. for those starting school after 8:30 a.m. But only those teens who started after 8:30 a.m. achieved the 8-hour recommended sleep duration, said Nicole Nahmod, during a presentation at the annual meeting of the Associated Professional Sleep Societies.
The students with the later school start times averaged 32 minutes of extra sleep, when compared with their early-rising colleagues, noted Ms. Nahmod, who is one of the study’s authors, a research technician, and study coordinator at Pennsylvania State University, Hershey. Specifically, adolescents who started school after 8:30 a.m. had a mean sleep duration of 8.1 hours, while those who started school earlier slept for only 7.5 hours a night.*
Acknowledging the importance of adequate sleep on teen health, mood, and school performance, the American Academy of Pediatrics recommends that middle schools and high schools begin at 8:30 a.m. or later. However, most school days start earlier than that, as evidenced by this dataset. While 72% of the study’s participants started school between 7:00 a.m. and 8:30 a.m., as many as 15% of the study participants began school during the narrower 7:00-7:30 a.m. window.
In this study, researchers from Penn State used data from 413 adolescents (mean age, 15.4 years; 46% male), who participated in a substudy of the Fragile Families & Child Wellbeing Study conducted across 20 large American cities. The study oversampled nonmarried pregnant mothers, resulting in a racially diverse sample and a high proportion of low-income families. The household income for 29% of the sample was below the poverty line.*
For this substudy, sleep duration was calculated from app-based daily diary reports of bed times, wake times, and school start times on days when a teen attended school.
The study was limited by its cross-sectional nature, but was enriched by a diverse range of school start times sampled from 20 U.S. cities, the high proportion of at-risk teens, and the use of a daily sleep diary.
“Current literature shows associations between later school start times and academic success, mood, and health. It also shows a decrease in motor vehicle accidents, tardiness, school dropout, and daytime sleepiness,” Ms. Nahmod noted.
Her continued research in this area involves analyzing the relationships between actigraphically assessed sleep measures in students and school start times.
Ms. Nahmod reported having no financial disclosures.
*This article was updated on June 6, 2017.
BOSTON – Teenagers slept more hours when they started school later, a new study found.
With 73% of high schoolers reporting receiving less than the recommended 8 hours of sleep per night, teens are among the most sleep-deprived members of society.
In this study, the later a teenager started school, the later he or she woke up, with average wake-up times having been just after 6:00 a.m. for those starting school between 7:00 and 7:30 a.m. and about 7:00 a.m. for those starting school after 8:30 a.m. But only those teens who started after 8:30 a.m. achieved the 8-hour recommended sleep duration, said Nicole Nahmod, during a presentation at the annual meeting of the Associated Professional Sleep Societies.
The students with the later school start times averaged 32 minutes of extra sleep, when compared with their early-rising colleagues, noted Ms. Nahmod, who is one of the study’s authors, a research technician, and study coordinator at Pennsylvania State University, Hershey. Specifically, adolescents who started school after 8:30 a.m. had a mean sleep duration of 8.1 hours, while those who started school earlier slept for only 7.5 hours a night.*
Acknowledging the importance of adequate sleep on teen health, mood, and school performance, the American Academy of Pediatrics recommends that middle schools and high schools begin at 8:30 a.m. or later. However, most school days start earlier than that, as evidenced by this dataset. While 72% of the study’s participants started school between 7:00 a.m. and 8:30 a.m., as many as 15% of the study participants began school during the narrower 7:00-7:30 a.m. window.
In this study, researchers from Penn State used data from 413 adolescents (mean age, 15.4 years; 46% male), who participated in a substudy of the Fragile Families & Child Wellbeing Study conducted across 20 large American cities. The study oversampled nonmarried pregnant mothers, resulting in a racially diverse sample and a high proportion of low-income families. The household income for 29% of the sample was below the poverty line.*
For this substudy, sleep duration was calculated from app-based daily diary reports of bed times, wake times, and school start times on days when a teen attended school.
The study was limited by its cross-sectional nature, but was enriched by a diverse range of school start times sampled from 20 U.S. cities, the high proportion of at-risk teens, and the use of a daily sleep diary.
“Current literature shows associations between later school start times and academic success, mood, and health. It also shows a decrease in motor vehicle accidents, tardiness, school dropout, and daytime sleepiness,” Ms. Nahmod noted.
Her continued research in this area involves analyzing the relationships between actigraphically assessed sleep measures in students and school start times.
Ms. Nahmod reported having no financial disclosures.
*This article was updated on June 6, 2017.
AT SLEEP 2017
Key clinical point: Teenage students who start school after 8:30 a.m. are more likely to get the recommended 8 hours of sleep than are their counterparts with earlier start times.
Major finding: Adolescents who started school after 8:30 a.m. had a mean sleep duration of 8.1 hours, compared with 7.5 hours for students who started school earlier.
Data source: A national longitudinal birth cohort study of 413 adolescents.
Disclosures: Ms. Nahmod reported having no financial disclosures.
Antiangiogenesis in Small-Cell Lung Cancer: Is There a Path Forward?
Study Overview
Objective. To evaluate efficacy of adding bevacizumab to first-line chemotherapy for treatment of extensive-disease small-cell lung cancer (ED-SCLC).
Design. Phase III prospective multicenter randomized clinical trial.
Setting and participants. The study was conducted at 29 Italian centers and was supported by the Agenzia Italiana del Farmaco. Study entry was limited to patients with histologically or cytologically documented ED-SCLC who were previously untreated with systemic therapy, were 18 years of age or older, and had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 to 2. Adequate bone marrow, renal, and liver functions were required. Patients with asymptomatic, treated brain metastases were eligible for trial participation. Exclusions included the following: mixed histologic diagnosis of SCLC and non–SCLC; history of grade 2 hemoptysis; evidence of lung tumor cavitation; significant traumatic injury within the 4 weeks before first dose of study treatment; other active malignancies (previous or current); and any underlying medical condition that might be aggravated by treatment.
Intervention. Patients received a combination of intravenous cisplatin (25 mg/m2 on days 1 to 3), etoposide (100 mg/m2 on days 1 to 3), and bevacizumab (7.5 mg/kg intravenously on day 1) administered every 3 weeks (experimental arm); or the same cisplatin and etoposide chemotherapy regimen alone given every 3 weeks (control arm). Carboplatin (area under the curve 5 on day 1) could be substituted for cisplatin in case of cisplatin contraindications or cisplatin-associated toxicity. Tumor response, on the basis of investigator-assessed Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1), was evaluated every 3 cycles during chemotherapy treatment. After 6 cycles of chemotherapy, tumor assessment was performed every 9 weeks in both arms. In the absence of progression, patients in the treatment arm continued bevacizumab alone until disease progression or for a maximum of 18 courses. Survival follow-up information was collected every 6 months after treatment termination or last dose of study drug, until death or loss to follow-up.
Main outcome measure. The primary end point was overall survival (OS). Response rate, toxicity, and progression-free survival (PFS) were secondary end points.
Main results. 205 patients were randomized between November 2009 and October 2015. 204 patients were considered in the intent-to-treat analysis (103 in the control arm and 101 in the treatment arm). Most patients were male with ECOG PS of 0 to 1. Median age was 64 years. The median number of chemotherapy courses administered was 6 in both arms. Cisplatin was used in majority of the patients. Average relative dose intensities for all drugs were well balanced between 2 groups. A lower percentage of patients in the treatment arm (14.7%) than in the control arm (22.3%) discontinued treatment because of radiologic disease progression, which was the main reason for treatment discontinuation.
At a median follow-up of 34.9 months, the median PFS was 5.7 in the control arm and 6.7 months in the treatment arm (hazard ratio [HR], 0.72; 95% CI, 0.54 to 0.97; P = 0.30). Median OS times were 8.9 months and 9.8 months, and 1-year survival rates were 25% and 37% (HR, 0.78; 95% CI, 0.58 to 1.06; P = 0.113) in the control arm and treatment arm, respectively. A significant effect of the maintenance treatment on OS (HR, 0.60; 95% CI, 0.40 to 0.91, P = 0.011) was observed. A subgroup analysis revealed a statistically significant interaction for OS between treatment and sex; the addition of bevacizumab led to a significant survival benefit in men (HR, 0.55) and to a possible detrimental effect in women (HR, 1.55; interaction test, P = 0.003).
Addition of bevacizumab did not result in increase in hematologic toxicity such as anemia, neutropenia, or thrombocytopenia. Concerning the nonhematologic toxicity, only hypertension was more frequent in the bevacizumab arm (6.3%) compared to chemotherapy alone arm (1%). The rates of proteinuria and thrombosis were similar in both arms.
Conclusion. The addition of bevacizumab to cisplatin and etoposide in the first-line treatment of ED-SCLC had an acceptable toxicity profile and led to a statistically significant improvement in PFS, which, however, did not translate into a statistically significant increase in OS.
Commentary
SCLC currently accounts for approximately 12% to 15% of all lung cancers [1]. It is characterized by a rapid growth rate, metastasis at the time of diagnosis, sensitivity to first-line platinum-based chemotherapy, and invariable recurrence and progressive resistance to subsequent lines of therapy. A number of clinical trials over the past 2 decades have failed to produce outcomes superior to platinum-based doublet chemotherapy, leaving a significant unmet need [2]. Vascular endothelial growth factor (VEGF) is the most important proangiogenic factor, and it is implicated in tumor growth [3]. Bevacizumab, a humanized monoclonal antibody directed against VEGF, is now indicated in the treatment of several tumor types including non–SCLC and breast, colorectal, kidney, and ovarian cancer. Positive signal with bevacizumab was seen in phase II studies, providing rationale for this phase III trial [4,5] .
The study by Tiseo and colleagues reported the outcomes of a randomized study that added bevacizumab to standard combination therapy with platinum and etoposide for the treatment of ED-SCLC. A small statistically significant improvement was seen in PFS (6.7 months vs. 5.7 months, favoring the bevacizumab group). However, the study failed to meet the primary end point of improved OS.
So where do antiangiogenesis agents go from here? Alternative angiogenesis inhibitors with broader mechanism of action are being explored in clinical trials. One such trial (ClinicalTrials.gov identifier: NCT02945852) is evaluating the role of the tyrosine kinase inhibitor apatinib in combination with chemotherapy in ED-SCLC. Apatinib selectively inhibits the vascular growth factor receptor-2 (VEGFR2). In addition, this agent also inhibits c-kit and c-SRC tyrosine kinase. It would be interesting to see if antiangiogenic agents with broader mechanisms would be more effective in SCLC. Immunotherapy with checkpoint inhibitors such as nivolumab and pembrolizumab have revolutionized the lung cancer treatment paradigm. It would be interesting to see if bevacizumab could be safely added to these immunotherapy agents. The ongoing CheckMate 370 (ClinicalTrials.gov identifier: NCT02574078) is addressing this question, evaluating the safety of combining nivolumab with bevacizumab in non-SCLC.
Applications for Clinical Practice
The current study does not support the addition of bevacizumab as a standard therapeutic option in the first-line treatment of ED-SCLC. However, given that there was a trend towards improved OS, alternative strategies of incorporating antiangiogenesis agents should be considered in future clinical trials.
—Deval Rajyaguru, MD
1. Neal JW, Gubens MA, Wakelee HA. Current management of small cell lung cancer. Clin Chest Med 2011;32:853–63.
2. Bunn PA Jr, Minna JD, Augustyn A, et al. Small cell lung cancer. Can recent advances in biology and molecular biology be translated into improved outcomes? J Thorac Oncol 2016;11:453–74.
3. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003:9:669–676.
4. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27:6006–11.
5. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: Results from the SALUTE trial. J Clin Oncol 2011;29:2215–22.
Study Overview
Objective. To evaluate efficacy of adding bevacizumab to first-line chemotherapy for treatment of extensive-disease small-cell lung cancer (ED-SCLC).
Design. Phase III prospective multicenter randomized clinical trial.
Setting and participants. The study was conducted at 29 Italian centers and was supported by the Agenzia Italiana del Farmaco. Study entry was limited to patients with histologically or cytologically documented ED-SCLC who were previously untreated with systemic therapy, were 18 years of age or older, and had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 to 2. Adequate bone marrow, renal, and liver functions were required. Patients with asymptomatic, treated brain metastases were eligible for trial participation. Exclusions included the following: mixed histologic diagnosis of SCLC and non–SCLC; history of grade 2 hemoptysis; evidence of lung tumor cavitation; significant traumatic injury within the 4 weeks before first dose of study treatment; other active malignancies (previous or current); and any underlying medical condition that might be aggravated by treatment.
Intervention. Patients received a combination of intravenous cisplatin (25 mg/m2 on days 1 to 3), etoposide (100 mg/m2 on days 1 to 3), and bevacizumab (7.5 mg/kg intravenously on day 1) administered every 3 weeks (experimental arm); or the same cisplatin and etoposide chemotherapy regimen alone given every 3 weeks (control arm). Carboplatin (area under the curve 5 on day 1) could be substituted for cisplatin in case of cisplatin contraindications or cisplatin-associated toxicity. Tumor response, on the basis of investigator-assessed Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1), was evaluated every 3 cycles during chemotherapy treatment. After 6 cycles of chemotherapy, tumor assessment was performed every 9 weeks in both arms. In the absence of progression, patients in the treatment arm continued bevacizumab alone until disease progression or for a maximum of 18 courses. Survival follow-up information was collected every 6 months after treatment termination or last dose of study drug, until death or loss to follow-up.
Main outcome measure. The primary end point was overall survival (OS). Response rate, toxicity, and progression-free survival (PFS) were secondary end points.
Main results. 205 patients were randomized between November 2009 and October 2015. 204 patients were considered in the intent-to-treat analysis (103 in the control arm and 101 in the treatment arm). Most patients were male with ECOG PS of 0 to 1. Median age was 64 years. The median number of chemotherapy courses administered was 6 in both arms. Cisplatin was used in majority of the patients. Average relative dose intensities for all drugs were well balanced between 2 groups. A lower percentage of patients in the treatment arm (14.7%) than in the control arm (22.3%) discontinued treatment because of radiologic disease progression, which was the main reason for treatment discontinuation.
At a median follow-up of 34.9 months, the median PFS was 5.7 in the control arm and 6.7 months in the treatment arm (hazard ratio [HR], 0.72; 95% CI, 0.54 to 0.97; P = 0.30). Median OS times were 8.9 months and 9.8 months, and 1-year survival rates were 25% and 37% (HR, 0.78; 95% CI, 0.58 to 1.06; P = 0.113) in the control arm and treatment arm, respectively. A significant effect of the maintenance treatment on OS (HR, 0.60; 95% CI, 0.40 to 0.91, P = 0.011) was observed. A subgroup analysis revealed a statistically significant interaction for OS between treatment and sex; the addition of bevacizumab led to a significant survival benefit in men (HR, 0.55) and to a possible detrimental effect in women (HR, 1.55; interaction test, P = 0.003).
Addition of bevacizumab did not result in increase in hematologic toxicity such as anemia, neutropenia, or thrombocytopenia. Concerning the nonhematologic toxicity, only hypertension was more frequent in the bevacizumab arm (6.3%) compared to chemotherapy alone arm (1%). The rates of proteinuria and thrombosis were similar in both arms.
Conclusion. The addition of bevacizumab to cisplatin and etoposide in the first-line treatment of ED-SCLC had an acceptable toxicity profile and led to a statistically significant improvement in PFS, which, however, did not translate into a statistically significant increase in OS.
Commentary
SCLC currently accounts for approximately 12% to 15% of all lung cancers [1]. It is characterized by a rapid growth rate, metastasis at the time of diagnosis, sensitivity to first-line platinum-based chemotherapy, and invariable recurrence and progressive resistance to subsequent lines of therapy. A number of clinical trials over the past 2 decades have failed to produce outcomes superior to platinum-based doublet chemotherapy, leaving a significant unmet need [2]. Vascular endothelial growth factor (VEGF) is the most important proangiogenic factor, and it is implicated in tumor growth [3]. Bevacizumab, a humanized monoclonal antibody directed against VEGF, is now indicated in the treatment of several tumor types including non–SCLC and breast, colorectal, kidney, and ovarian cancer. Positive signal with bevacizumab was seen in phase II studies, providing rationale for this phase III trial [4,5] .
The study by Tiseo and colleagues reported the outcomes of a randomized study that added bevacizumab to standard combination therapy with platinum and etoposide for the treatment of ED-SCLC. A small statistically significant improvement was seen in PFS (6.7 months vs. 5.7 months, favoring the bevacizumab group). However, the study failed to meet the primary end point of improved OS.
So where do antiangiogenesis agents go from here? Alternative angiogenesis inhibitors with broader mechanism of action are being explored in clinical trials. One such trial (ClinicalTrials.gov identifier: NCT02945852) is evaluating the role of the tyrosine kinase inhibitor apatinib in combination with chemotherapy in ED-SCLC. Apatinib selectively inhibits the vascular growth factor receptor-2 (VEGFR2). In addition, this agent also inhibits c-kit and c-SRC tyrosine kinase. It would be interesting to see if antiangiogenic agents with broader mechanisms would be more effective in SCLC. Immunotherapy with checkpoint inhibitors such as nivolumab and pembrolizumab have revolutionized the lung cancer treatment paradigm. It would be interesting to see if bevacizumab could be safely added to these immunotherapy agents. The ongoing CheckMate 370 (ClinicalTrials.gov identifier: NCT02574078) is addressing this question, evaluating the safety of combining nivolumab with bevacizumab in non-SCLC.
Applications for Clinical Practice
The current study does not support the addition of bevacizumab as a standard therapeutic option in the first-line treatment of ED-SCLC. However, given that there was a trend towards improved OS, alternative strategies of incorporating antiangiogenesis agents should be considered in future clinical trials.
—Deval Rajyaguru, MD
Study Overview
Objective. To evaluate efficacy of adding bevacizumab to first-line chemotherapy for treatment of extensive-disease small-cell lung cancer (ED-SCLC).
Design. Phase III prospective multicenter randomized clinical trial.
Setting and participants. The study was conducted at 29 Italian centers and was supported by the Agenzia Italiana del Farmaco. Study entry was limited to patients with histologically or cytologically documented ED-SCLC who were previously untreated with systemic therapy, were 18 years of age or older, and had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 to 2. Adequate bone marrow, renal, and liver functions were required. Patients with asymptomatic, treated brain metastases were eligible for trial participation. Exclusions included the following: mixed histologic diagnosis of SCLC and non–SCLC; history of grade 2 hemoptysis; evidence of lung tumor cavitation; significant traumatic injury within the 4 weeks before first dose of study treatment; other active malignancies (previous or current); and any underlying medical condition that might be aggravated by treatment.
Intervention. Patients received a combination of intravenous cisplatin (25 mg/m2 on days 1 to 3), etoposide (100 mg/m2 on days 1 to 3), and bevacizumab (7.5 mg/kg intravenously on day 1) administered every 3 weeks (experimental arm); or the same cisplatin and etoposide chemotherapy regimen alone given every 3 weeks (control arm). Carboplatin (area under the curve 5 on day 1) could be substituted for cisplatin in case of cisplatin contraindications or cisplatin-associated toxicity. Tumor response, on the basis of investigator-assessed Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1), was evaluated every 3 cycles during chemotherapy treatment. After 6 cycles of chemotherapy, tumor assessment was performed every 9 weeks in both arms. In the absence of progression, patients in the treatment arm continued bevacizumab alone until disease progression or for a maximum of 18 courses. Survival follow-up information was collected every 6 months after treatment termination or last dose of study drug, until death or loss to follow-up.
Main outcome measure. The primary end point was overall survival (OS). Response rate, toxicity, and progression-free survival (PFS) were secondary end points.
Main results. 205 patients were randomized between November 2009 and October 2015. 204 patients were considered in the intent-to-treat analysis (103 in the control arm and 101 in the treatment arm). Most patients were male with ECOG PS of 0 to 1. Median age was 64 years. The median number of chemotherapy courses administered was 6 in both arms. Cisplatin was used in majority of the patients. Average relative dose intensities for all drugs were well balanced between 2 groups. A lower percentage of patients in the treatment arm (14.7%) than in the control arm (22.3%) discontinued treatment because of radiologic disease progression, which was the main reason for treatment discontinuation.
At a median follow-up of 34.9 months, the median PFS was 5.7 in the control arm and 6.7 months in the treatment arm (hazard ratio [HR], 0.72; 95% CI, 0.54 to 0.97; P = 0.30). Median OS times were 8.9 months and 9.8 months, and 1-year survival rates were 25% and 37% (HR, 0.78; 95% CI, 0.58 to 1.06; P = 0.113) in the control arm and treatment arm, respectively. A significant effect of the maintenance treatment on OS (HR, 0.60; 95% CI, 0.40 to 0.91, P = 0.011) was observed. A subgroup analysis revealed a statistically significant interaction for OS between treatment and sex; the addition of bevacizumab led to a significant survival benefit in men (HR, 0.55) and to a possible detrimental effect in women (HR, 1.55; interaction test, P = 0.003).
Addition of bevacizumab did not result in increase in hematologic toxicity such as anemia, neutropenia, or thrombocytopenia. Concerning the nonhematologic toxicity, only hypertension was more frequent in the bevacizumab arm (6.3%) compared to chemotherapy alone arm (1%). The rates of proteinuria and thrombosis were similar in both arms.
Conclusion. The addition of bevacizumab to cisplatin and etoposide in the first-line treatment of ED-SCLC had an acceptable toxicity profile and led to a statistically significant improvement in PFS, which, however, did not translate into a statistically significant increase in OS.
Commentary
SCLC currently accounts for approximately 12% to 15% of all lung cancers [1]. It is characterized by a rapid growth rate, metastasis at the time of diagnosis, sensitivity to first-line platinum-based chemotherapy, and invariable recurrence and progressive resistance to subsequent lines of therapy. A number of clinical trials over the past 2 decades have failed to produce outcomes superior to platinum-based doublet chemotherapy, leaving a significant unmet need [2]. Vascular endothelial growth factor (VEGF) is the most important proangiogenic factor, and it is implicated in tumor growth [3]. Bevacizumab, a humanized monoclonal antibody directed against VEGF, is now indicated in the treatment of several tumor types including non–SCLC and breast, colorectal, kidney, and ovarian cancer. Positive signal with bevacizumab was seen in phase II studies, providing rationale for this phase III trial [4,5] .
The study by Tiseo and colleagues reported the outcomes of a randomized study that added bevacizumab to standard combination therapy with platinum and etoposide for the treatment of ED-SCLC. A small statistically significant improvement was seen in PFS (6.7 months vs. 5.7 months, favoring the bevacizumab group). However, the study failed to meet the primary end point of improved OS.
So where do antiangiogenesis agents go from here? Alternative angiogenesis inhibitors with broader mechanism of action are being explored in clinical trials. One such trial (ClinicalTrials.gov identifier: NCT02945852) is evaluating the role of the tyrosine kinase inhibitor apatinib in combination with chemotherapy in ED-SCLC. Apatinib selectively inhibits the vascular growth factor receptor-2 (VEGFR2). In addition, this agent also inhibits c-kit and c-SRC tyrosine kinase. It would be interesting to see if antiangiogenic agents with broader mechanisms would be more effective in SCLC. Immunotherapy with checkpoint inhibitors such as nivolumab and pembrolizumab have revolutionized the lung cancer treatment paradigm. It would be interesting to see if bevacizumab could be safely added to these immunotherapy agents. The ongoing CheckMate 370 (ClinicalTrials.gov identifier: NCT02574078) is addressing this question, evaluating the safety of combining nivolumab with bevacizumab in non-SCLC.
Applications for Clinical Practice
The current study does not support the addition of bevacizumab as a standard therapeutic option in the first-line treatment of ED-SCLC. However, given that there was a trend towards improved OS, alternative strategies of incorporating antiangiogenesis agents should be considered in future clinical trials.
—Deval Rajyaguru, MD
1. Neal JW, Gubens MA, Wakelee HA. Current management of small cell lung cancer. Clin Chest Med 2011;32:853–63.
2. Bunn PA Jr, Minna JD, Augustyn A, et al. Small cell lung cancer. Can recent advances in biology and molecular biology be translated into improved outcomes? J Thorac Oncol 2016;11:453–74.
3. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003:9:669–676.
4. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27:6006–11.
5. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: Results from the SALUTE trial. J Clin Oncol 2011;29:2215–22.
1. Neal JW, Gubens MA, Wakelee HA. Current management of small cell lung cancer. Clin Chest Med 2011;32:853–63.
2. Bunn PA Jr, Minna JD, Augustyn A, et al. Small cell lung cancer. Can recent advances in biology and molecular biology be translated into improved outcomes? J Thorac Oncol 2016;11:453–74.
3. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003:9:669–676.
4. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27:6006–11.
5. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: Results from the SALUTE trial. J Clin Oncol 2011;29:2215–22.
Vigorous Physical Activity Associated with Greater Arterial Compliance in Both Large and Small Arteries
Study Overview
Objective. To investigate the association between habitually high levels of physical activity and the compliance of the large and small arteries in men and women throughout the life span.
Design. Cross-sectional study.
Setting and participants. 83 healthy men (n = 44) and women (n = 39) aged between 18 and 78 years were recruited to participate in the study. Potential participants were recruited via flyers designed to elicit responses from either very highly active (participate in regular, vigorous exercise more than 5 times per week) or less active/sedentary individuals (participate in light to moderate exercise less than 3 times per week or none at all). Both groups subjectively reported maintaining the specified activity level for at least the past 5 years. The highly active subjects performed regular vigorous swimming as their primary mode of exercise training as most were members of a varsity or masters swim team. All subjects were free of overt chronic diseases, nonsmokers, and none were taking vasoactive medications as assessed by a medical history questionnaire. All subjects provided written informed consent to participate. The study was reviewed and approved by the institutional review board at Indiana University.
Physical activity was self-assessed in all subject groups with a log detailing their activity over the previous 7 days. To ensure the older highly active population performed vigorous physical activity ≥ 5 days per week, the subjective activity log was verified by a 7-day previously validated, commercially available heart rate monitor and accelerometer (Actiheart, CamNtech, Cambridge, UK).
Main outcome measure. Compliance of the small and large arteries (inverse of stiffness) measured using a commercial pulse wave analyzer (Model CR-2000, Hypertension Diagnositics, Eagen, MN), which according to the manufacturer measures proximal capacitive compliance (C1, or estimate of large artery compliance) and distal oscillatory compliance (C2, or small artery compliance) [1].
Results. The study found a positive association between routine vigorous physical activity and arterial compliance. Specifically, the results suggest that vigorous physical activity is associated with greater compliance of the small and large arteries in both younger and older adults (P < 0.05). In addition, both the highly active and less active younger groups as well as the highly active older group demonstrated greater large arterial compliance compared to the less active older group (P < 0.008). No significant differences were found between men and women.
Conclusion. Researchers concluded that participation in habitual vigorous physical activity is associated with benefits to the compliance of the small and large arteries. Habitual vigorous physical activity over time may attenuate age-associated cardiovascular impairments.
Commentary
Arterial compliance declines with age, and increased arterial stiffness is associated with an increased risk of cardiovascular events [2]. Evidence suggests that physical activity may delay or prevent age-related increases in arterial stiffness [3]. Previous research regarding age-related arterial stiffness and exercise has focused primarily on the large arteries. For example, Tanaka found that regular aerobic-endurance exercise attenuates age-related reductions in central arterial compliance and restores levels in previously sedentary healthy middle-aged and older men [3]. More recently, a study by Duprez [4] found that small artery elasticity was superior to large artery elasticity with regard to predicting future CHD, stroke, and heart failure.
In this study, researchers cross-sectionally investigated the relationship of intense and continuous physical activity in young and older adults. The form of vigorous activity in this study was competitive swimming, as participants were recruited from a collegiate varsity and masters swim team. The study found a statistically strong association between routine vigorous physical activity and arterial compliance. These findings agree with several studies showing the benefits of vigorous exercise, but go beyond these by presenting findings on small artery compliance.
Methodologically, this study has some limitations. With the small sample, the study may not have been adequately powered. Further, physical activity assessment was by self-report in the main. Even though researchers had the participants keep a log, self-report measures may be inaccurate. Another limitation was the indirect method of measuring compliance, in which the radial waveform is calibrated to brachial blood pressure values. However, the researchers followed a valid model using the same BP level–based procedures reported in previous studies [1].
Applications for Clinical Practice
CVD is a major cause of disability and mortality in the United States. Health care professionals have a significant role to play in reducing cardiovascular risk factors in their patients, including encouraging aerobic exercise. The American Heart Association recommends at least 30 minutes of moderate-intensity aerobic activity at least 5 days per week or at least 25 minutes of vigorous aerobic activity at least 3 days per week, or a combination of moderate- and vigorous-intensity aerobic activity [4]. Patients can also be reminded that even modest levels of physical activity are associated with health benefits.
—Paloma Cesar de Sales, BS, RN, MS
1. Cohn JN, Finkelstein S, McVeigh G, et al. Noninvasive pulse wave analysis for the early detection of vascular disease. Hypertension 1995;26:503–8.
2. Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Failure Clin 2012;8:143–64.
3. Tanaka H, Dinenno FA, Monahan KD, et al. Aging, habitual exercise, and dynamic arterial compliance. Circulation 2000;102:1270–5.
4. Duprez DA, Jacobs DR Jr, Lutsey PL, et al. Association of small artery elasticity with incident cardiovascular disease in older adults: the multi-ethnic study of atherosclerosis. Am J Epidemiol 2011;174:528–36.
5. American Heart Association. Recommendations for physical activity in adults. Accessed at www.heart.org/HEARTORG/HealthyLiving/PhysicalActivity/FitnessBasics/American-Heart-Association-Recommendations-for-Physical-Activity-in-Adults_UCM_307976_Article.jsp#.WQx6ird77IU.
Study Overview
Objective. To investigate the association between habitually high levels of physical activity and the compliance of the large and small arteries in men and women throughout the life span.
Design. Cross-sectional study.
Setting and participants. 83 healthy men (n = 44) and women (n = 39) aged between 18 and 78 years were recruited to participate in the study. Potential participants were recruited via flyers designed to elicit responses from either very highly active (participate in regular, vigorous exercise more than 5 times per week) or less active/sedentary individuals (participate in light to moderate exercise less than 3 times per week or none at all). Both groups subjectively reported maintaining the specified activity level for at least the past 5 years. The highly active subjects performed regular vigorous swimming as their primary mode of exercise training as most were members of a varsity or masters swim team. All subjects were free of overt chronic diseases, nonsmokers, and none were taking vasoactive medications as assessed by a medical history questionnaire. All subjects provided written informed consent to participate. The study was reviewed and approved by the institutional review board at Indiana University.
Physical activity was self-assessed in all subject groups with a log detailing their activity over the previous 7 days. To ensure the older highly active population performed vigorous physical activity ≥ 5 days per week, the subjective activity log was verified by a 7-day previously validated, commercially available heart rate monitor and accelerometer (Actiheart, CamNtech, Cambridge, UK).
Main outcome measure. Compliance of the small and large arteries (inverse of stiffness) measured using a commercial pulse wave analyzer (Model CR-2000, Hypertension Diagnositics, Eagen, MN), which according to the manufacturer measures proximal capacitive compliance (C1, or estimate of large artery compliance) and distal oscillatory compliance (C2, or small artery compliance) [1].
Results. The study found a positive association between routine vigorous physical activity and arterial compliance. Specifically, the results suggest that vigorous physical activity is associated with greater compliance of the small and large arteries in both younger and older adults (P < 0.05). In addition, both the highly active and less active younger groups as well as the highly active older group demonstrated greater large arterial compliance compared to the less active older group (P < 0.008). No significant differences were found between men and women.
Conclusion. Researchers concluded that participation in habitual vigorous physical activity is associated with benefits to the compliance of the small and large arteries. Habitual vigorous physical activity over time may attenuate age-associated cardiovascular impairments.
Commentary
Arterial compliance declines with age, and increased arterial stiffness is associated with an increased risk of cardiovascular events [2]. Evidence suggests that physical activity may delay or prevent age-related increases in arterial stiffness [3]. Previous research regarding age-related arterial stiffness and exercise has focused primarily on the large arteries. For example, Tanaka found that regular aerobic-endurance exercise attenuates age-related reductions in central arterial compliance and restores levels in previously sedentary healthy middle-aged and older men [3]. More recently, a study by Duprez [4] found that small artery elasticity was superior to large artery elasticity with regard to predicting future CHD, stroke, and heart failure.
In this study, researchers cross-sectionally investigated the relationship of intense and continuous physical activity in young and older adults. The form of vigorous activity in this study was competitive swimming, as participants were recruited from a collegiate varsity and masters swim team. The study found a statistically strong association between routine vigorous physical activity and arterial compliance. These findings agree with several studies showing the benefits of vigorous exercise, but go beyond these by presenting findings on small artery compliance.
Methodologically, this study has some limitations. With the small sample, the study may not have been adequately powered. Further, physical activity assessment was by self-report in the main. Even though researchers had the participants keep a log, self-report measures may be inaccurate. Another limitation was the indirect method of measuring compliance, in which the radial waveform is calibrated to brachial blood pressure values. However, the researchers followed a valid model using the same BP level–based procedures reported in previous studies [1].
Applications for Clinical Practice
CVD is a major cause of disability and mortality in the United States. Health care professionals have a significant role to play in reducing cardiovascular risk factors in their patients, including encouraging aerobic exercise. The American Heart Association recommends at least 30 minutes of moderate-intensity aerobic activity at least 5 days per week or at least 25 minutes of vigorous aerobic activity at least 3 days per week, or a combination of moderate- and vigorous-intensity aerobic activity [4]. Patients can also be reminded that even modest levels of physical activity are associated with health benefits.
—Paloma Cesar de Sales, BS, RN, MS
Study Overview
Objective. To investigate the association between habitually high levels of physical activity and the compliance of the large and small arteries in men and women throughout the life span.
Design. Cross-sectional study.
Setting and participants. 83 healthy men (n = 44) and women (n = 39) aged between 18 and 78 years were recruited to participate in the study. Potential participants were recruited via flyers designed to elicit responses from either very highly active (participate in regular, vigorous exercise more than 5 times per week) or less active/sedentary individuals (participate in light to moderate exercise less than 3 times per week or none at all). Both groups subjectively reported maintaining the specified activity level for at least the past 5 years. The highly active subjects performed regular vigorous swimming as their primary mode of exercise training as most were members of a varsity or masters swim team. All subjects were free of overt chronic diseases, nonsmokers, and none were taking vasoactive medications as assessed by a medical history questionnaire. All subjects provided written informed consent to participate. The study was reviewed and approved by the institutional review board at Indiana University.
Physical activity was self-assessed in all subject groups with a log detailing their activity over the previous 7 days. To ensure the older highly active population performed vigorous physical activity ≥ 5 days per week, the subjective activity log was verified by a 7-day previously validated, commercially available heart rate monitor and accelerometer (Actiheart, CamNtech, Cambridge, UK).
Main outcome measure. Compliance of the small and large arteries (inverse of stiffness) measured using a commercial pulse wave analyzer (Model CR-2000, Hypertension Diagnositics, Eagen, MN), which according to the manufacturer measures proximal capacitive compliance (C1, or estimate of large artery compliance) and distal oscillatory compliance (C2, or small artery compliance) [1].
Results. The study found a positive association between routine vigorous physical activity and arterial compliance. Specifically, the results suggest that vigorous physical activity is associated with greater compliance of the small and large arteries in both younger and older adults (P < 0.05). In addition, both the highly active and less active younger groups as well as the highly active older group demonstrated greater large arterial compliance compared to the less active older group (P < 0.008). No significant differences were found between men and women.
Conclusion. Researchers concluded that participation in habitual vigorous physical activity is associated with benefits to the compliance of the small and large arteries. Habitual vigorous physical activity over time may attenuate age-associated cardiovascular impairments.
Commentary
Arterial compliance declines with age, and increased arterial stiffness is associated with an increased risk of cardiovascular events [2]. Evidence suggests that physical activity may delay or prevent age-related increases in arterial stiffness [3]. Previous research regarding age-related arterial stiffness and exercise has focused primarily on the large arteries. For example, Tanaka found that regular aerobic-endurance exercise attenuates age-related reductions in central arterial compliance and restores levels in previously sedentary healthy middle-aged and older men [3]. More recently, a study by Duprez [4] found that small artery elasticity was superior to large artery elasticity with regard to predicting future CHD, stroke, and heart failure.
In this study, researchers cross-sectionally investigated the relationship of intense and continuous physical activity in young and older adults. The form of vigorous activity in this study was competitive swimming, as participants were recruited from a collegiate varsity and masters swim team. The study found a statistically strong association between routine vigorous physical activity and arterial compliance. These findings agree with several studies showing the benefits of vigorous exercise, but go beyond these by presenting findings on small artery compliance.
Methodologically, this study has some limitations. With the small sample, the study may not have been adequately powered. Further, physical activity assessment was by self-report in the main. Even though researchers had the participants keep a log, self-report measures may be inaccurate. Another limitation was the indirect method of measuring compliance, in which the radial waveform is calibrated to brachial blood pressure values. However, the researchers followed a valid model using the same BP level–based procedures reported in previous studies [1].
Applications for Clinical Practice
CVD is a major cause of disability and mortality in the United States. Health care professionals have a significant role to play in reducing cardiovascular risk factors in their patients, including encouraging aerobic exercise. The American Heart Association recommends at least 30 minutes of moderate-intensity aerobic activity at least 5 days per week or at least 25 minutes of vigorous aerobic activity at least 3 days per week, or a combination of moderate- and vigorous-intensity aerobic activity [4]. Patients can also be reminded that even modest levels of physical activity are associated with health benefits.
—Paloma Cesar de Sales, BS, RN, MS
1. Cohn JN, Finkelstein S, McVeigh G, et al. Noninvasive pulse wave analysis for the early detection of vascular disease. Hypertension 1995;26:503–8.
2. Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Failure Clin 2012;8:143–64.
3. Tanaka H, Dinenno FA, Monahan KD, et al. Aging, habitual exercise, and dynamic arterial compliance. Circulation 2000;102:1270–5.
4. Duprez DA, Jacobs DR Jr, Lutsey PL, et al. Association of small artery elasticity with incident cardiovascular disease in older adults: the multi-ethnic study of atherosclerosis. Am J Epidemiol 2011;174:528–36.
5. American Heart Association. Recommendations for physical activity in adults. Accessed at www.heart.org/HEARTORG/HealthyLiving/PhysicalActivity/FitnessBasics/American-Heart-Association-Recommendations-for-Physical-Activity-in-Adults_UCM_307976_Article.jsp#.WQx6ird77IU.
1. Cohn JN, Finkelstein S, McVeigh G, et al. Noninvasive pulse wave analysis for the early detection of vascular disease. Hypertension 1995;26:503–8.
2. Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Failure Clin 2012;8:143–64.
3. Tanaka H, Dinenno FA, Monahan KD, et al. Aging, habitual exercise, and dynamic arterial compliance. Circulation 2000;102:1270–5.
4. Duprez DA, Jacobs DR Jr, Lutsey PL, et al. Association of small artery elasticity with incident cardiovascular disease in older adults: the multi-ethnic study of atherosclerosis. Am J Epidemiol 2011;174:528–36.
5. American Heart Association. Recommendations for physical activity in adults. Accessed at www.heart.org/HEARTORG/HealthyLiving/PhysicalActivity/FitnessBasics/American-Heart-Association-Recommendations-for-Physical-Activity-in-Adults_UCM_307976_Article.jsp#.WQx6ird77IU.
Views of Primary Care Physicians Regarding the Promotion of Healthy Lifestyles and Weight Management Among Their Patients
From the University of Florida (Dr. Tucker, Ms. Ukonu, Ms. Kang, Ms. Good), Gainesville, FL; the University of Florida–Jacksonville (Dr. Shah, Dr. Bilello), Jacksonville, FL; and Ball State University (Dr. Arthur), Muncie, IN.
Abstracts
- Objective: To assess primary care physicians’ practices, knowledge, and beliefs regarding their efforts to promote healthy lifestyles and weight management among their patients.
- Methods: Study participants consisted of 25 primary care physicians from a regional primary care practice-based research network that includes 37 university-affiliated patient-centered medical homes and 2 nearby unaffiliated primary care sites. Participating physicians completed an online modified version of the Physician Survey of Practices on Diet, Physical Activity, and Weight Control–Adult Questionnaire.
- Results: The majority (88%) of participating physicians strongly believed it was their responsibility to promote a healthy diet, physical activity, and healthy weight loss and weight maintenance among patients. The 3 most commonly endorsed barriers were (a) not enough time, (b) minimal patient interest in improving his/her weight, and (c) lack of adequate weight-loss referral resources. The top 3 physician-perceived practice improvements that would be helpful with these practices were (a) better tools to communicate diet, physical activity, or weight problems to patients or family; (b) better mechanisms to connect patients to weight-loss referral resources; and (c) better counseling tools to guide patients regarding lifestyle modifications. 76% of the participating physicians correctly identified the BMI cutoff ranges for adult obesity, but only 32% did so for childhood obesity.
- Conclusion: It is important to provide primary care physicians with knowledge, effective tools, and resources to promote healthy lifestyles and weight loss and weight management among their patients.
Key words: obesity; primary care physicians; weight loss; weight management.
More than two-thirds of adults in the United States are overweight, with approximately 35% considered obese (defined as a body mass index ≥ 30) [1]. Obesity is associated with many of the leading causes of death in the United States (ie, diabetes, heart disease, stroke, and some types of cancer) and with poor mental health outcomes and reduced quality of life [2]. Racial/ethnic minorities and individuals with low incomes are disproportionately impacted by obesity and obesity-related diseases and negative health outcomes [3–5].
The US Preventive Services Task Force (USPSTF) recommends screening for obesity and intensive behavioral counseling, which are often the responsibilities of primary care providers [6]. Despite these recommendations, research suggests that primary care providers rarely screen their patients for obesity or refer them for intensive behavioral counseling despite evidence that doing so would improve patient health outcomes [5–7]. Lack of time to address weight issues during clinical visits, lack of training in weight management counseling, and lack of availability of intensive weight loss programs to which they can refer their patients are some of the reasons cited for not counseling patients about weight management [8].
Primary care providers deliver more hours of patient care than other providers, yet these providers have been unable to deliver medical interventions capable of producing even modest weight loss [10]. Obesity treatment options delivered in primary care settings have limited success, likely due to the low intensity of these treatment options. Many studies have shown that most obesity treatments in health care settings typically consist of scheduled monthly or quarterly visits that are 10 to 15 minutes in duration [11], despite evidence that more intense treatments are needed. Specifically, a systematic review of the obesity treatment literature performed by the USPSTF revealed that high-intensity, multicomponent behavioral interventions that include face-to-face counseling on diet and physical activity and behavioral therapy more than once a month for 3 months are needed to produce significant weight loss (8–15 lb) among adult patients in primary care settings [12].
Since many of the characteristics of multicomponent behavioral interventions for treating obesity are both patient-centered and involve self-management, the patient-centered medical home (PCMH) seems to be the ideal setting to deliver these interventions [13]. Specifically, PCMHs provide patient-centered care that is wide-ranging, team-based, and coordinated across all elements of the health care system and the patient’s community [14]. These sites specifically provide primary care, which is the type of care that obesity disparity patient groups such as racial/ethnic minorities, sexual minorities, groups with low incomes, and the medically underserved are more likely to utilize [15].
Providing multicomponent behavioral interventions for obesity in PCMHs and other primary care sites will increase the likelihood of participation among the aforementioned obesity disparity groups. Despite the potential benefits of obesity treatment interventions offered in primary care settings, particularly for obesity disparity groups, the role of primary care providers in providing such treatment interventions is not clear [16]. We surveyed primary care physicians who primarily worked in PCMHs to assess their practices, knowledge, views/beliefs, perceived barriers, and perceived needed clinic practice improvements relative to promoting healthy lifestyles and weight management among their patients.
Methods
Participants
Primary care physicians were recruited from among a regional primary care practice-based research network that includes 37 PCMHs affiliated with an academic health center and 2 nearby primary care sites not affiliated with an academic health center. Fifty-two physicians at these centers received an invitation via email to participate in our online survey study. The invitation email included (a) a study endorsement note from the chair of the Community Health and Family Medicine Department affiliated with the PCMHs, (b) instructions about how to participate in the study, and (c) a link to the study. Participation inclusion criteria specified in the online informed consent form were: (a) working as a physician affiliated with the practice-based research network, (b) having access to a computer with internet connection, (c) being able to communicate in written English, and (d) providing written consent to participate in the study. Physicians were not provided compensation for participating in the study.
Survey Instrument
To assess physicians’ views and practices, we used a modified version of the Physician Survey of Practices on Diet, Physical Activity, and Weight Control–Adult Questionnaire [17]. The survey was sponsored by the National Cancer Institute in collaboration with several other NIH institutes and the CDC for evaluating current clinical practices among physicians, including the degree to which physicians evaluate their patients for obesity and offer them guidance designed to increase adherence to a health-promoting lifestyle (eg, recommendations on diet, weight, and physical activity). Additionally, the questionnaire assesses physicians’ perceived barriers to patient assessment, evaluation, and management. It also includes questions about physicians’ healthy lifestyle–related knowledge. In 2010, Smith and colleagues utilized the questionnaire with a nationally representative sample of primary care physicians (n = 1211) to investigate primary care physicians’ clinical practices in relation to overweight and obesity [18]. To our knowledge, no other physician survey has been developed to assess current engagement in recommended clinical practices, barriers to engaging in recommended practices, as well as beliefs and knowledge regarding helping patients follow a health-promoting lifestyle. The original survey also includes questions regarding the physicians’ personal health status and health behaviors.
For our study, we modified the survey by removing questions regarding the physicians’ (a) perceived general health and well-being, (b) current dietary practices, (c) current level of engagement in physical activity, and (d) current engagement in professional activities unrelated to patient care (eg, research, teaching). Our modified survey included 7 questions asking about current practices regarding screening for obesity and referral of patients to weight management interventions. Two questions asked about physicians’ perceived barriers to helping patients adhere to a health-promoting lifestyle and maintain a healthy weight. Physicians were asked to rate their top 3 barriers from among a list of 11 pre-identified barriers and to rate their top 3 desired practice-related improvements from among a list of 10 pre-identified improvements. Physicians were given the option to provide additional barriers or improvements that were not already pre-identified. Seven questions assessed physicians’ views/beliefs related to helping patients achieve and maintain a health-promoting lifestyle and a healthy weight. These questions utilize a rating scale where 1 = strongly agree, 2 = agree somewhat, 3 = neither agree nor disagree, 4 = disagree somewhat, and 5 = strongly disagree. Four questions assessed physicians’ healthy lifestyle–related knowledge (BMI ranges/percentiles for adults/children, diet and exercise guideline recommendations [recommended amounts of moderate physical activity and servings of fruits and vegetables for adults]) and 11 questions ask about the physician (height, weight, demographics, and practice population).
Survey Administration
The survey was administered anonymously through Qualtrics, a secure, online survey platform. The survey was administered online to increase anonymity, increase response rate, and diminish potential physician-perceived barriers to participating in the study. The participating physicians were provided with a link that enabled them to access the survey. The survey excluded questions that required disclosure of identifying information. Survey data from Qualtrics were exported to an SPSS file that was stored on a password protected, secured computer in the research lab of the principal investigator for this study.
Data Analysis
Frequency analyses were applied to survey responses to determine the participating physicians’ endorsed barriers to and views regarding evaluating and managing patients’ weight, healthy eating, and physical activity; physicians’ views related to helping patients achieve and maintain a health-promoting lifestyle and a healthy weight; and physicians’ healthy lifestyle–related knowledge. Nonparametric t tests were conducted to examine differences in survey responses of the participating physicians in association with their sex (male or female), race (Asian vs. white/Caucasian), and BMI (BMI < 25 and BMI ≥ 25).
Approval for the study was obtained through the institutional review board of the University of Florida Health Science Center.
Results
Participants
Twenty-five physicians out of 52 invited completed the survey (48% response rate). The vast majority of the study participants were PCMH-affiliated (92%–96%). Participating physicians ranged in age from 29 to 67 years old. Sixteen (64%) participating physicians identified as female, 7 (28%) participating physicians identified as male, and 2 (8%) participating physicians did not indicate a sex. Twenty (80%) participating physicians identified as being white, 3 (12%) participating physicians identified as being Asian/Asian American, and 2 (8%) did not indicate a race or ethnicity. Twenty-two (88%) participating physicians were employees of a large medical group affiliated with an academic medical center, 1 (4%) was employed in a physician-owned practice, and 2 (8%) did not indicate their main primary care practice location. Table 1 provides additional demographic data.
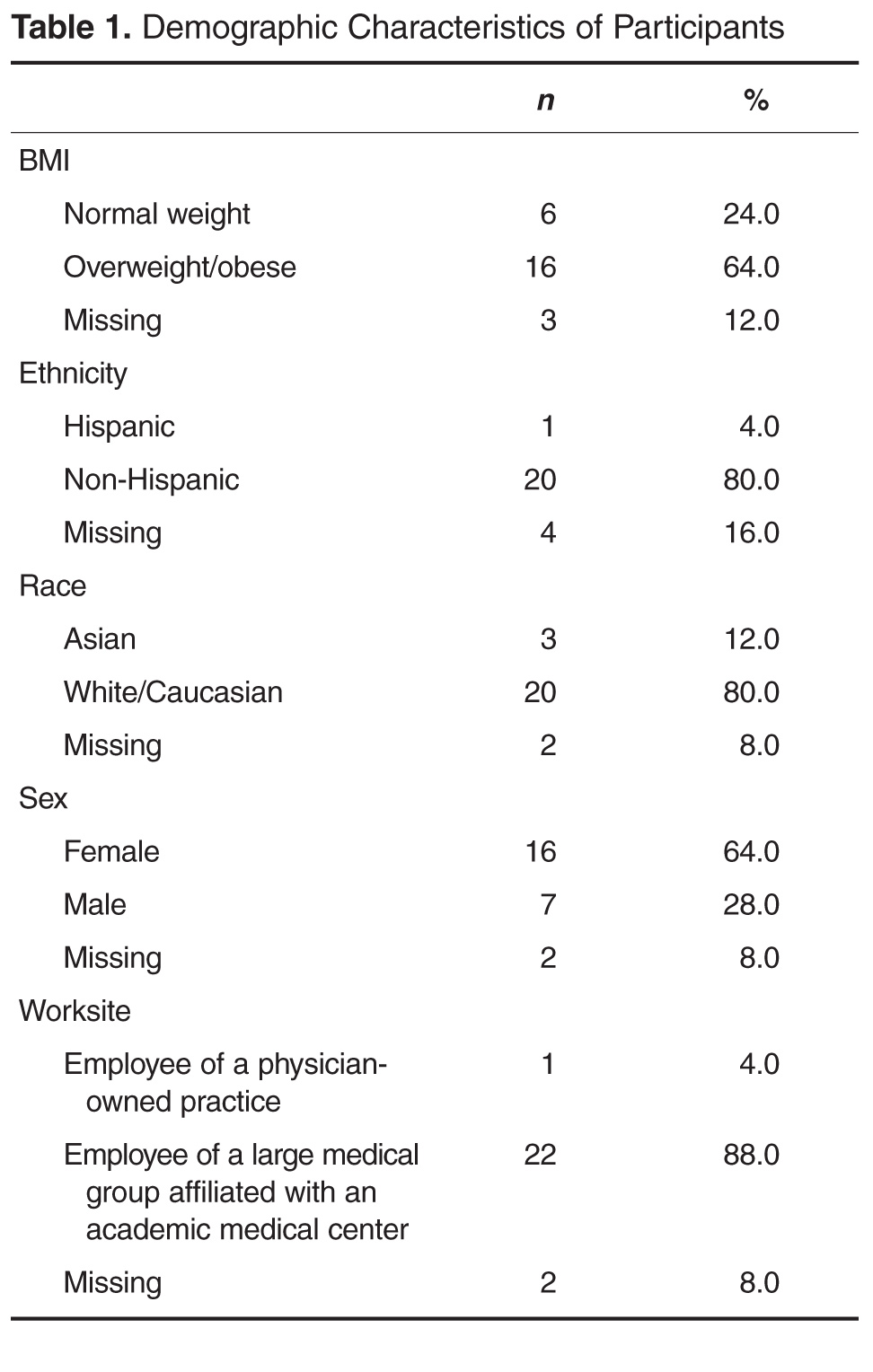
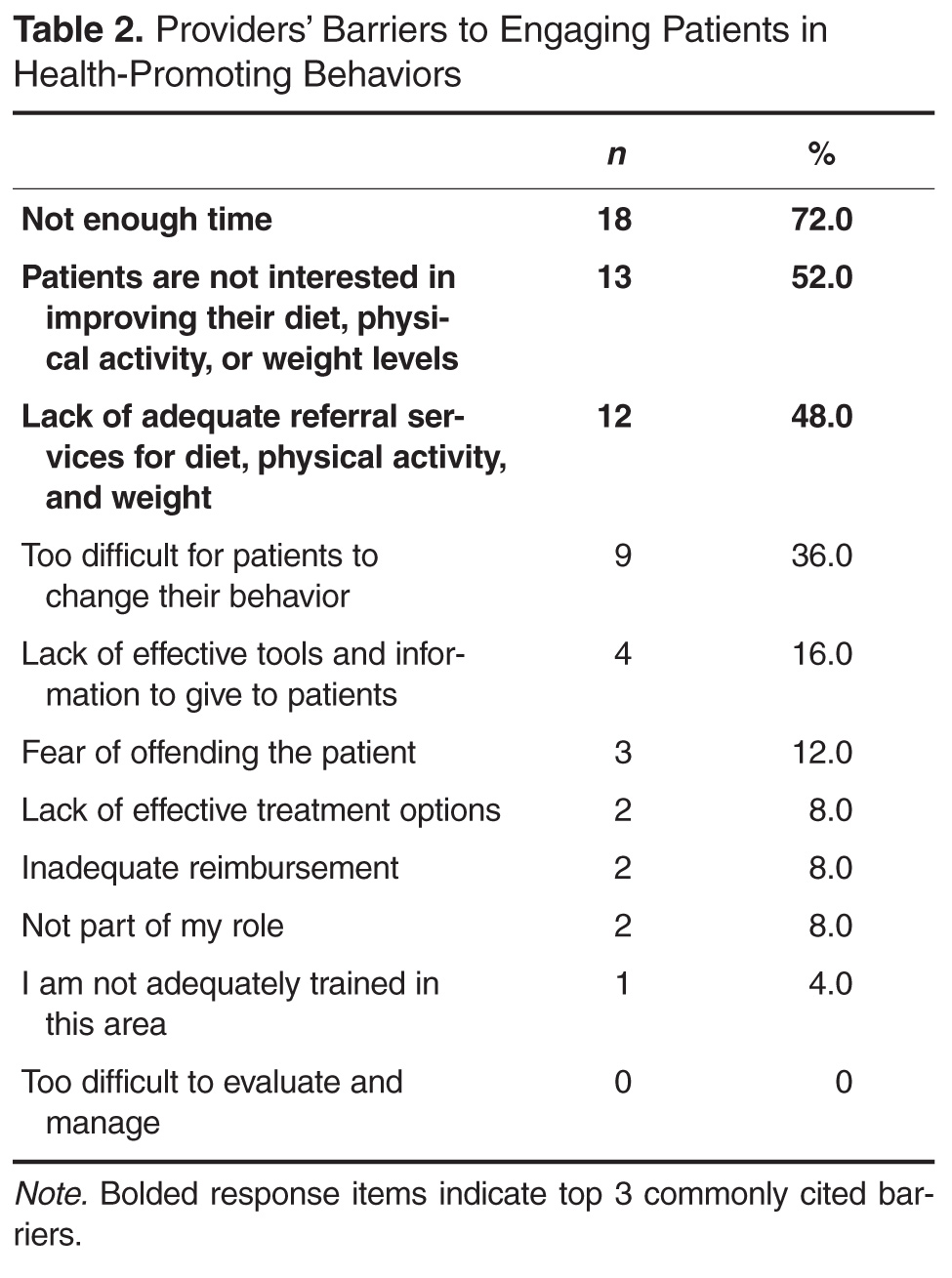
Approximately 88% of the participating physicians agreed that patients were more likely to adopt healthier lifestyles if their health care providers counseled them to do so (44% strongly agreed, 44% agreed somewhat). A majority of participating physicians endorsed the view that there are effective strategies and/or tools to (a) help patients eat a healthy diet (56% strongly agreed, 24% agreed somewhat), (b) engage in adequate amounts of physical activity (56% strongly agreed, 20% agreed somewhat), and (c) maintain a healthy weight or lose weight (48% strongly agreed, 
Many participating physicians expressed confidence in their ability to counsel their patients to (a) eat a healthy diet (64% strongly agreed, 28% agreed somewhat), (b) engage in adequate amounts of physical activity (68% strongly agreed, 24% agreed somewhat), and (c) maintain a healthy weight or lose weight (60% strongly agreed, 32% agreed somewhat). Most participating physicians at least somewhat agreed that they were effective at helping their patients (a) eat a healthy diet (24% strongly agreed, 52% agreed somewhat), (b) engage in adequate amounts of physical activity (20% strongly agreed, 56% agreed somewhat), and (c) maintain a healthy weight or lose weight (16% strongly agreed, 48% agreed somewhat). Some participating physicians expressed ambivalence about whether or not they were effective at helping their patients (a) eat a healthy diet (16% neither agreed nor disagreed), (b) engage in adequate amounts of physical activity (12% neither agreed nor disagreed), and (c) maintain a healthy weight or lose weight (20% neither agreed nor disagreed). A total of 8% of participating physicians did not endorse the belief that they were effective at helping their patients maintain a healthy weight or lose weight.
Most participating physicians at least somewhat agreed that they were effective in encouraging patients to engage in health-promoting activities (44% strongly agreed, and 44% agreed somewhat), whereas 4% neither agreed nor disagreed that they were effective in providing this encouragement. Interestingly, many participating physicians endorsed the view that they would be able to provide more credible and effective counseling to patients if they (the physicians themselves) ate a healthy diet (68% strongly agreed, 20% agreed somewhat) and engaged in adequate amounts of physical activity (68% strongly agreed, 20% agreed somewhat). A minority of participating physicians (4%) neither agreed or disagreed with this perspective.
In regards to participating physicians’ healthy lifestyle–related knowledge about current BMI ranges for adults or percentile ranges for children, most participating physicians were able to accurately identify the correct BMI cutoff ranges for overweight (80%) and obese (76%) adults. However, only 32% of participating physicians were able to correctly identify BMI percentile ranges for children; however, nearly all of the participating physicians saw mainly adult patients. Lastly, 76% of participating physicians were able to correctly identify the recommended amounts of moderate physical activity for adults 18 years of age and older, and only 56% were able to correctly identify the recommended amount of servings of fruits and vegetables.
There were no significant race-related differences in participating physicians views/beliefs, healthy lifestyle–related knowledge, and perceived barriers to helping patients engage in health promoting behaviors and weight management. There were no significant sex-related differences in these variables with the exception that women were more likely to respond that they did not know the BMI percentile range at which children or adolescents were considered to have a healthy weight (37.5% of women vs. 0% of men, P = 0.03). A similar percentage of men (66.7%) and women (64.7%) who chose among the 4 percentile range options (rather than endorsing “Don’t know”) chose an incorrect answer. Lastly, there were no significant self-reported BMI-related differences in participating physicians’ views/beliefs, healthy lifestyle–related knowledge, and perceived barriers to helping patients engage in health-promoting behaviors and weight management.
Discussion
Given the high percentage of adults in the United States who are overweight or obese and the associated health risks, it is paramount that primary care physicians advise their patients to manage their weight and adopt a health-promoting lifestyle. Research studies indicate that such advice is effective [18,19]. Furthermore, it has been found that most overweight and obese patients want more assistance with weight management than they are receiving from their primary care physicians [21]. This study thus explored primary care physicians’ knowledge, beliefs, and perceived barriers that may prevent them from providing such assistance. The primary care setting is the site where obesity disparity groups (eg, racial/ethnic minorities, groups with low household incomes) are most likely to receive care [22,23].
Most of the PCMH-affiliated physicians in this study agreed that they had the responsibility to promote weight-loss/management and healthy lifestyles among their patients. Consistent with prior research [9], the majority of the physicians in this study felt they were effective in their ability to counsel patients to eat a healthy diet and engage in physical activity. To illustrate, in a prior study [9], 77% of primary care providers thought that they could provide useful dieting tips to patients, and in this study, 80% believed they were effective in helping patients eat a healthy diet. However, despite this confidence in their ability to provide advice about healthy diets and physical activity, the providers in both this and in another prior study [25] were less confident in their ability to actually help patients lose weight. Only 64% of the providers in the present study felt they could be effective in assisting patients with losing weight or maintaining a healthy weight. Although this percentage is higher than the 44% of physicians found in a prior study [25] who felt confident in their ability to treat obesity, both studies clearly point to a need to decrease barriers that physicians face in helping clients lose weight.
A key finding of this study was the consensus among the participating physicians regarding what they perceived to be the common barriers to helping patients adhere to a health-promoting lifestyle. Consistent with past research [8,9], the 3 most common barriers cited by the participating physicians were that they did not have enough time, patients were not interested in improving their weight, and adequate referrals for diet, physical activity, and weight were lacking. Additional barriers endorsed included a lack of effective tools and information to give patients, and a fear of offending patients. Another barrier identified by the participating physicians is the perception that patients had difficulty in changing behaviors necessary for maintaining a healthier lifestyle.
When asked what would facilitate conversations with patients, the top 3 responses given were better tools to communicate diet, physical activity, or weight problems to patients or family members; better mechanisms to connect patients to specific referral sources; and better counseling tools to guide patients towards engagement in healthy lifestyles. Of note is the significant overlap between the perceived barriers and the needed facilitation tools. The clearest example of this overlap is that physicians noted a lack of adequate referral sources to be a barrier and that better mechanisms to connect patients to specific referral sources would facilitate their treatment of patients. Weight management referrals for patients in rural areas and for non-Hispanic black adults and Hispanic adults, among whom obesity is most prevalent in the United States [3,25], are particularly needed. Addressing this need is consistent with national calls to reduce/eliminate obesity and other disparities that plague the U.S. health care system. One promising avenue to facilitate weight management referrals is the development, evaluation, and wide dissemination of remote weight-loss support interventions, particularly in rural, racial/ethnic minority, and low-income communities. Indeed, several recent articles demonstrate the success of such weight management programs across diverse patient populations [27–29].
Many of the physicians who participated in the study (72%) endorsed lack of time as a significant barrier to discussing weight and weight-related behaviors with their patients. Therefore, finding time-efficient strategies to involve physicians in weight management interventions may prove particularly beneficial. One such evidence-based behavioral counseling framework—the 5As framework—has been endorsed by the Centers for Medicare and Medicaid Services and the USPSTF for use with obese patients during a typical 20-minute visit [28].
The second highest-rated barrier, perceived patient lack of motivation, warrants additional discussion. Despite over half of the physicians surveyed citing this as a barrier, previous studies have shown that the majority of overweight patients believe they should lose weight and are interested in losing weight [21]. This study highlights a potential discrepancy between physicians’ perceptions of patients’ interest in weight-loss and their patients’ actual interest. It is possible that this discrepancy can be avoided by training physicians on how to be culturally sensitive when addressing weight with their patients. Moreover, such cultural sensitivity training may be of great use, given that 12% of physicians in this study were apprehensive about discussing weight with their patients due to fear of offending them. Such training typically involves teaching physicians how to talk with patients in ways that enable patients to feel comfortable, trusting, and respectful in patient-physician/provider interactions [29].
Two other findings pertaining to providers deserve mention. Specifically, 88% of physicians believed that effectively encouraging patients to adhere to a healthy lifestyle included personally engaging in health-promoting activities. However, of the physicians surveyed, 64% were overweight/obese. Given the high percentage of physicians in this study that were overweight/obese and these physicians’ belief that their personal engagement in health-promoting activities is important to encourage patient engagement in a healthy lifestyle, it seems that future efforts are needed to facilitate health-promoting behaviors among physicians—efforts that may in turn aid them in encouraging their patients to adhere to a healthy lifestyle.
Finally, this study assessed physicians’ healthy lifestyle–related knowledge about current BMI ranges for adults and BMI percentile ranges for children, and recommended amounts of moderate physical activity and servings of fruits and vegetables for adults. Most physicians were able to correctly identify the adult BMI cutoff ranges for overweight and obesity and to identify the correct answers to questions about physical activity and fruits and vegetable consumption guidelines for adults. However, only 32% of physicians were able to correctly identify BMI percentile ranges for children and/or adolescents. This is understandable given that most of the physicians in this study provide care to adult patients. However, considering that in 2012 more than one-third of children and adolescents were overweight or obese [1], it is important that all physicians have knowledge of BMI percentile ranges for children and adolescents so that minimally they can convey this information to their adult patients who are parents. The USPSTF defines children and adolescent overweight as an age- and gender-specific BMI between the 85th and 94th percentiles, and children and adolescent obesity as an age- and gender-specific BMI ≥ 95th percentile [31]. Such knowledge of BMI cutoffs is needed in order for providers to comply with the USPSTF recommendation to screen all adults and children aged 6 years and older for obesity, and then offer or refer those with an obesity diagnosis to intensive multicomponent behavioral interventions [31–33].
While novel, the study also had several limitations. First, due to self-selection of participants, physicians who felt more confident in their abilities to address overweight or obesity with their patients might have been more likely to respond. Second, participating physicians may have given socially desirable responses to questions (ie, responses that present a favorable image of themselves) rather than true/accurate responses. Future studies could incorporate a social desirability scale in order to detect and control for any socially desirable responding [33]. Another limitation was the small sample size and the limited variability in geographic location of the participating physicians. Thus, the experiences of these physicians may not be generalizable to physicians in other geographic regions. Future similar studies to the present study are needed and such studies should use a larger and randomly selected sample of physicians that is racially/ethnically diverse. Finally, a limitation of this study is the 48% participation rate. Factors that may have contributed to this participation rate include lack of compensation for physicians and the likelihood that physicians may have extremely busy schedules that may discourage them from participating. However, it is important to note that the 48% participation rate of this study is better than the 25.6% participation rate in another similar study [25]. Future similar studies to the present study likely need to include strong incentives for physicians to be study participants.
Conclusion
Our study indicates that many primary care physicians may not talk with their patients about engaging in healthy eating, physical activity, and weight management because of perceived barriers that prevent them from doing so, rather than because of a lack of perceived responsibility to do so or a perception that counseling patients on these issues would be ineffective. This finding highlights the importance of providing physicians with the tools and resources needed to overcome the aforementioned barriers to fostering health-promoting lifestyles and a healthy weight among their patients and the importance of involving physicians in identifying these barriers and ways to overcome them.
Acknowledgement. We thank the patients and health care providers at the participating medical homes affiliated with University of Florida Health in Jacksonville, Florida, for making this research possible.
Corresponding author: Carolyn M. Tucker, PhD, University of Florida, [email protected].
Funding/support: Support for this research was provided by the Office of Research at UF–Gainesville, Florida, and by the National Institutes of Health and National Center for Research Resources CTSA grant UL1 TR000064.
Financial disclosures: None.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014;311:806–14.
2. Centers for Disease Control and Prevention. Adult obesity causes and consequences, 2016. Accessed 30 Apr 2017 at www.cdc.gov/obesity/adult/causes.html.
3. Centers for Disease Control and Prevention. CDC health disparities and inequalities report —United States, 2013. Accessed 30 Apr 2017 at www.cdc.gov/mmwr/pdf/other/su6203.pdf.
4. Wang Y, Beydoun MA. The obesity epidemic in the United States – gender, age, socioeconomic, racial/ethnic, and geographic characteristics: A systematic review and meta-regression analysis. Epidemiol Rev 2007;29:6–28.
5. Levine, JA. Poverty and obesity in the US. Diabetes 2011;60:2667–8.
6. Moyer VA. Screening for and management of obesity in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2012;157:373–8.
7. Aveyard P, Lewis A, Tearne S, et al. Screening and brief intervention for obesity in primary care: a parallel, two-arm, randomised trial. Lancet 2016;388:2492–500.
8. Jay M, Chintapalli S, Squires A, et al. Barriers and facilitators to providing primary care-based weight management services in a patient centered medical home for Veterans: a qualitative study. BMC Fam Pract 2015;16:167.
9. Ruelaz AR, Diefenbach, Simon B, et al. Perceived barriers to weight management. J Gen Intern Med 2007;22:518–22.
10. Carvajal R, Wadden TA, Tsai AG, et al. Managing obesity in primary care practice: A narrative review. Ann N Y Acad Sci 2013;1281:191–206.
11. Wadden TA, Neiberg RH, Wing RR, et al; Look AHEAD Research Group. Four-year weight-losses in the Look AHEAD study: factors associated with long-term success. Obesity (Silver Spring) 2011;19:1987–98.
12. LeBlanc ES, O’Connor, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2011;155:434–47.
13. Rittenhouse DR, Shortell SM. The patient-centered medical home: will it stand the test of health reform? JAMA 2009;301:2038–40.
14. Scholle S, Torda P, Peikes D, et al. Engaging patients and families in the medical home (prepared by Mathematica Policy Research under contract no. HHSA290200900019ITO2.) AHRQ Pub No. 10-0083-EF. Rockville, MD: Agency for Healthcare Research and Quality; June 2010.
15. US Department of Health and Human Services. HHS action plan to reduce racial and ethnic health disparities: A nation free of disparities in health and health care. 2011.
16. Ard J. Obesity in the US: what is the best role for primary care? BMJ 2015;350:1–10.
17. National Cancer Institute. Physcian survey of practices on diet, physical activity, and weight control. 2010. Accessed 30 Apr 2017 at https://healthcaredelivery.cancer.gov/energy_balance/phys_pract_q_adult.pdf.
18. Smith AW, Borowski LA, Liu B, et al. US primary care physicians’ diet-, physical activity–, and weight-related care of adult patients. Am J Prev Med 2011;41:33–42.
19. Simons-Morton DG, Calfas KJ, et al. Effects of interventions in health care settings on physical activity or cardiorespiratory fitness. Am J Prev Med 1998;15:413–30.
20. Bowerman S, Bellman M, Saltsman P, et al. Implementation of a primary care physician network obesity management program. Obes Res 2001;9 Suppl 4:321S–5S.
21. Potter MB, Vu JD, Croughan-Minihane M. Weight management: what patients want from their primary care physician. J Fam Pract 2001;50:513–8.
22. National Association of Community Health Centers. Community health centers: The local prescription for better quality and lower costs. March 2011. Accessed at www.nachc.org.
23. Institute of Medicine. Unequal treatment: confronting racial and ethnic disparities in healthcare. Washington, DC: National Academy of Sciences Press; 2003.
24. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 2012;307:491–7.
25. Bleich SN, Bennett WL, Gudzune KA, Cooper LA. National survey of US primary care physicians’ perspectives about causes of obesity and solutions to improve care. BMJ Open 2012;2(6).
26. Patterson PD, Moore CG, Probst JC, Shinogle JA. Obesity and physical inactivity in rural America. J Rural Health 2004;20:151–9.
27. Shaikh U, Cole SL, Marcin JP, Nesbitt TS. Clinical management and patient outcomes among children and adolescents receiving telemedicine consultations for obesity. Telemed e-Health 2008;14:434–40.
28. Schlair S, Moore S, Mcmacken M, Jay M. How to deliver high-quality obesity counseling in primary care using the 5As framework. J Clin Outcomes Manag 2012;19:221–9.
29. Tucker CM, Arthur TM, Roncoroni J, et al. Patient-centered culturally sensitive health care. Am J Lifestyle Med 2013;9:63–77.
30. Ma J, Yank V, Xiao L, et al. Translating the diabetes prevention program lifestyle intervention for weight loss into primary care. JAMA Intern Med 2013;173:113–21.
31. U.S. Preventive Services Task Force. Screening for obesity in children and adolescents: U.S. Preventive Services Task Force recommendation statement. Pediatrics 2010;125:361–7.
32. Final recommendation statement: obesity in adults: screening and management. US Preventive Services Task Force. Oct 2014. Accessed at www.uspreventiveservicestaskforce.org.
33. van de Mortel, TF. Faking it: social desirability response bias in self-report research. Aust J Advanced Nurs 2008:25:40–8.
From the University of Florida (Dr. Tucker, Ms. Ukonu, Ms. Kang, Ms. Good), Gainesville, FL; the University of Florida–Jacksonville (Dr. Shah, Dr. Bilello), Jacksonville, FL; and Ball State University (Dr. Arthur), Muncie, IN.
Abstracts
- Objective: To assess primary care physicians’ practices, knowledge, and beliefs regarding their efforts to promote healthy lifestyles and weight management among their patients.
- Methods: Study participants consisted of 25 primary care physicians from a regional primary care practice-based research network that includes 37 university-affiliated patient-centered medical homes and 2 nearby unaffiliated primary care sites. Participating physicians completed an online modified version of the Physician Survey of Practices on Diet, Physical Activity, and Weight Control–Adult Questionnaire.
- Results: The majority (88%) of participating physicians strongly believed it was their responsibility to promote a healthy diet, physical activity, and healthy weight loss and weight maintenance among patients. The 3 most commonly endorsed barriers were (a) not enough time, (b) minimal patient interest in improving his/her weight, and (c) lack of adequate weight-loss referral resources. The top 3 physician-perceived practice improvements that would be helpful with these practices were (a) better tools to communicate diet, physical activity, or weight problems to patients or family; (b) better mechanisms to connect patients to weight-loss referral resources; and (c) better counseling tools to guide patients regarding lifestyle modifications. 76% of the participating physicians correctly identified the BMI cutoff ranges for adult obesity, but only 32% did so for childhood obesity.
- Conclusion: It is important to provide primary care physicians with knowledge, effective tools, and resources to promote healthy lifestyles and weight loss and weight management among their patients.
Key words: obesity; primary care physicians; weight loss; weight management.
More than two-thirds of adults in the United States are overweight, with approximately 35% considered obese (defined as a body mass index ≥ 30) [1]. Obesity is associated with many of the leading causes of death in the United States (ie, diabetes, heart disease, stroke, and some types of cancer) and with poor mental health outcomes and reduced quality of life [2]. Racial/ethnic minorities and individuals with low incomes are disproportionately impacted by obesity and obesity-related diseases and negative health outcomes [3–5].
The US Preventive Services Task Force (USPSTF) recommends screening for obesity and intensive behavioral counseling, which are often the responsibilities of primary care providers [6]. Despite these recommendations, research suggests that primary care providers rarely screen their patients for obesity or refer them for intensive behavioral counseling despite evidence that doing so would improve patient health outcomes [5–7]. Lack of time to address weight issues during clinical visits, lack of training in weight management counseling, and lack of availability of intensive weight loss programs to which they can refer their patients are some of the reasons cited for not counseling patients about weight management [8].
Primary care providers deliver more hours of patient care than other providers, yet these providers have been unable to deliver medical interventions capable of producing even modest weight loss [10]. Obesity treatment options delivered in primary care settings have limited success, likely due to the low intensity of these treatment options. Many studies have shown that most obesity treatments in health care settings typically consist of scheduled monthly or quarterly visits that are 10 to 15 minutes in duration [11], despite evidence that more intense treatments are needed. Specifically, a systematic review of the obesity treatment literature performed by the USPSTF revealed that high-intensity, multicomponent behavioral interventions that include face-to-face counseling on diet and physical activity and behavioral therapy more than once a month for 3 months are needed to produce significant weight loss (8–15 lb) among adult patients in primary care settings [12].
Since many of the characteristics of multicomponent behavioral interventions for treating obesity are both patient-centered and involve self-management, the patient-centered medical home (PCMH) seems to be the ideal setting to deliver these interventions [13]. Specifically, PCMHs provide patient-centered care that is wide-ranging, team-based, and coordinated across all elements of the health care system and the patient’s community [14]. These sites specifically provide primary care, which is the type of care that obesity disparity patient groups such as racial/ethnic minorities, sexual minorities, groups with low incomes, and the medically underserved are more likely to utilize [15].
Providing multicomponent behavioral interventions for obesity in PCMHs and other primary care sites will increase the likelihood of participation among the aforementioned obesity disparity groups. Despite the potential benefits of obesity treatment interventions offered in primary care settings, particularly for obesity disparity groups, the role of primary care providers in providing such treatment interventions is not clear [16]. We surveyed primary care physicians who primarily worked in PCMHs to assess their practices, knowledge, views/beliefs, perceived barriers, and perceived needed clinic practice improvements relative to promoting healthy lifestyles and weight management among their patients.
Methods
Participants
Primary care physicians were recruited from among a regional primary care practice-based research network that includes 37 PCMHs affiliated with an academic health center and 2 nearby primary care sites not affiliated with an academic health center. Fifty-two physicians at these centers received an invitation via email to participate in our online survey study. The invitation email included (a) a study endorsement note from the chair of the Community Health and Family Medicine Department affiliated with the PCMHs, (b) instructions about how to participate in the study, and (c) a link to the study. Participation inclusion criteria specified in the online informed consent form were: (a) working as a physician affiliated with the practice-based research network, (b) having access to a computer with internet connection, (c) being able to communicate in written English, and (d) providing written consent to participate in the study. Physicians were not provided compensation for participating in the study.
Survey Instrument
To assess physicians’ views and practices, we used a modified version of the Physician Survey of Practices on Diet, Physical Activity, and Weight Control–Adult Questionnaire [17]. The survey was sponsored by the National Cancer Institute in collaboration with several other NIH institutes and the CDC for evaluating current clinical practices among physicians, including the degree to which physicians evaluate their patients for obesity and offer them guidance designed to increase adherence to a health-promoting lifestyle (eg, recommendations on diet, weight, and physical activity). Additionally, the questionnaire assesses physicians’ perceived barriers to patient assessment, evaluation, and management. It also includes questions about physicians’ healthy lifestyle–related knowledge. In 2010, Smith and colleagues utilized the questionnaire with a nationally representative sample of primary care physicians (n = 1211) to investigate primary care physicians’ clinical practices in relation to overweight and obesity [18]. To our knowledge, no other physician survey has been developed to assess current engagement in recommended clinical practices, barriers to engaging in recommended practices, as well as beliefs and knowledge regarding helping patients follow a health-promoting lifestyle. The original survey also includes questions regarding the physicians’ personal health status and health behaviors.
For our study, we modified the survey by removing questions regarding the physicians’ (a) perceived general health and well-being, (b) current dietary practices, (c) current level of engagement in physical activity, and (d) current engagement in professional activities unrelated to patient care (eg, research, teaching). Our modified survey included 7 questions asking about current practices regarding screening for obesity and referral of patients to weight management interventions. Two questions asked about physicians’ perceived barriers to helping patients adhere to a health-promoting lifestyle and maintain a healthy weight. Physicians were asked to rate their top 3 barriers from among a list of 11 pre-identified barriers and to rate their top 3 desired practice-related improvements from among a list of 10 pre-identified improvements. Physicians were given the option to provide additional barriers or improvements that were not already pre-identified. Seven questions assessed physicians’ views/beliefs related to helping patients achieve and maintain a health-promoting lifestyle and a healthy weight. These questions utilize a rating scale where 1 = strongly agree, 2 = agree somewhat, 3 = neither agree nor disagree, 4 = disagree somewhat, and 5 = strongly disagree. Four questions assessed physicians’ healthy lifestyle–related knowledge (BMI ranges/percentiles for adults/children, diet and exercise guideline recommendations [recommended amounts of moderate physical activity and servings of fruits and vegetables for adults]) and 11 questions ask about the physician (height, weight, demographics, and practice population).
Survey Administration
The survey was administered anonymously through Qualtrics, a secure, online survey platform. The survey was administered online to increase anonymity, increase response rate, and diminish potential physician-perceived barriers to participating in the study. The participating physicians were provided with a link that enabled them to access the survey. The survey excluded questions that required disclosure of identifying information. Survey data from Qualtrics were exported to an SPSS file that was stored on a password protected, secured computer in the research lab of the principal investigator for this study.
Data Analysis
Frequency analyses were applied to survey responses to determine the participating physicians’ endorsed barriers to and views regarding evaluating and managing patients’ weight, healthy eating, and physical activity; physicians’ views related to helping patients achieve and maintain a health-promoting lifestyle and a healthy weight; and physicians’ healthy lifestyle–related knowledge. Nonparametric t tests were conducted to examine differences in survey responses of the participating physicians in association with their sex (male or female), race (Asian vs. white/Caucasian), and BMI (BMI < 25 and BMI ≥ 25).
Approval for the study was obtained through the institutional review board of the University of Florida Health Science Center.
Results
Participants
Twenty-five physicians out of 52 invited completed the survey (48% response rate). The vast majority of the study participants were PCMH-affiliated (92%–96%). Participating physicians ranged in age from 29 to 67 years old. Sixteen (64%) participating physicians identified as female, 7 (28%) participating physicians identified as male, and 2 (8%) participating physicians did not indicate a sex. Twenty (80%) participating physicians identified as being white, 3 (12%) participating physicians identified as being Asian/Asian American, and 2 (8%) did not indicate a race or ethnicity. Twenty-two (88%) participating physicians were employees of a large medical group affiliated with an academic medical center, 1 (4%) was employed in a physician-owned practice, and 2 (8%) did not indicate their main primary care practice location. Table 1 provides additional demographic data.
Approximately 88% of the participating physicians agreed that patients were more likely to adopt healthier lifestyles if their health care providers counseled them to do so (44% strongly agreed, 44% agreed somewhat). A majority of participating physicians endorsed the view that there are effective strategies and/or tools to (a) help patients eat a healthy diet (56% strongly agreed, 24% agreed somewhat), (b) engage in adequate amounts of physical activity (56% strongly agreed, 20% agreed somewhat), and (c) maintain a healthy weight or lose weight (48% strongly agreed,
Many participating physicians expressed confidence in their ability to counsel their patients to (a) eat a healthy diet (64% strongly agreed, 28% agreed somewhat), (b) engage in adequate amounts of physical activity (68% strongly agreed, 24% agreed somewhat), and (c) maintain a healthy weight or lose weight (60% strongly agreed, 32% agreed somewhat). Most participating physicians at least somewhat agreed that they were effective at helping their patients (a) eat a healthy diet (24% strongly agreed, 52% agreed somewhat), (b) engage in adequate amounts of physical activity (20% strongly agreed, 56% agreed somewhat), and (c) maintain a healthy weight or lose weight (16% strongly agreed, 48% agreed somewhat). Some participating physicians expressed ambivalence about whether or not they were effective at helping their patients (a) eat a healthy diet (16% neither agreed nor disagreed), (b) engage in adequate amounts of physical activity (12% neither agreed nor disagreed), and (c) maintain a healthy weight or lose weight (20% neither agreed nor disagreed). A total of 8% of participating physicians did not endorse the belief that they were effective at helping their patients maintain a healthy weight or lose weight.
Most participating physicians at least somewhat agreed that they were effective in encouraging patients to engage in health-promoting activities (44% strongly agreed, and 44% agreed somewhat), whereas 4% neither agreed nor disagreed that they were effective in providing this encouragement. Interestingly, many participating physicians endorsed the view that they would be able to provide more credible and effective counseling to patients if they (the physicians themselves) ate a healthy diet (68% strongly agreed, 20% agreed somewhat) and engaged in adequate amounts of physical activity (68% strongly agreed, 20% agreed somewhat). A minority of participating physicians (4%) neither agreed or disagreed with this perspective.
In regards to participating physicians’ healthy lifestyle–related knowledge about current BMI ranges for adults or percentile ranges for children, most participating physicians were able to accurately identify the correct BMI cutoff ranges for overweight (80%) and obese (76%) adults. However, only 32% of participating physicians were able to correctly identify BMI percentile ranges for children; however, nearly all of the participating physicians saw mainly adult patients. Lastly, 76% of participating physicians were able to correctly identify the recommended amounts of moderate physical activity for adults 18 years of age and older, and only 56% were able to correctly identify the recommended amount of servings of fruits and vegetables.
There were no significant race-related differences in participating physicians views/beliefs, healthy lifestyle–related knowledge, and perceived barriers to helping patients engage in health promoting behaviors and weight management. There were no significant sex-related differences in these variables with the exception that women were more likely to respond that they did not know the BMI percentile range at which children or adolescents were considered to have a healthy weight (37.5% of women vs. 0% of men, P = 0.03). A similar percentage of men (66.7%) and women (64.7%) who chose among the 4 percentile range options (rather than endorsing “Don’t know”) chose an incorrect answer. Lastly, there were no significant self-reported BMI-related differences in participating physicians’ views/beliefs, healthy lifestyle–related knowledge, and perceived barriers to helping patients engage in health-promoting behaviors and weight management.
Discussion
Given the high percentage of adults in the United States who are overweight or obese and the associated health risks, it is paramount that primary care physicians advise their patients to manage their weight and adopt a health-promoting lifestyle. Research studies indicate that such advice is effective [18,19]. Furthermore, it has been found that most overweight and obese patients want more assistance with weight management than they are receiving from their primary care physicians [21]. This study thus explored primary care physicians’ knowledge, beliefs, and perceived barriers that may prevent them from providing such assistance. The primary care setting is the site where obesity disparity groups (eg, racial/ethnic minorities, groups with low household incomes) are most likely to receive care [22,23].
Most of the PCMH-affiliated physicians in this study agreed that they had the responsibility to promote weight-loss/management and healthy lifestyles among their patients. Consistent with prior research [9], the majority of the physicians in this study felt they were effective in their ability to counsel patients to eat a healthy diet and engage in physical activity. To illustrate, in a prior study [9], 77% of primary care providers thought that they could provide useful dieting tips to patients, and in this study, 80% believed they were effective in helping patients eat a healthy diet. However, despite this confidence in their ability to provide advice about healthy diets and physical activity, the providers in both this and in another prior study [25] were less confident in their ability to actually help patients lose weight. Only 64% of the providers in the present study felt they could be effective in assisting patients with losing weight or maintaining a healthy weight. Although this percentage is higher than the 44% of physicians found in a prior study [25] who felt confident in their ability to treat obesity, both studies clearly point to a need to decrease barriers that physicians face in helping clients lose weight.
A key finding of this study was the consensus among the participating physicians regarding what they perceived to be the common barriers to helping patients adhere to a health-promoting lifestyle. Consistent with past research [8,9], the 3 most common barriers cited by the participating physicians were that they did not have enough time, patients were not interested in improving their weight, and adequate referrals for diet, physical activity, and weight were lacking. Additional barriers endorsed included a lack of effective tools and information to give patients, and a fear of offending patients. Another barrier identified by the participating physicians is the perception that patients had difficulty in changing behaviors necessary for maintaining a healthier lifestyle.
When asked what would facilitate conversations with patients, the top 3 responses given were better tools to communicate diet, physical activity, or weight problems to patients or family members; better mechanisms to connect patients to specific referral sources; and better counseling tools to guide patients towards engagement in healthy lifestyles. Of note is the significant overlap between the perceived barriers and the needed facilitation tools. The clearest example of this overlap is that physicians noted a lack of adequate referral sources to be a barrier and that better mechanisms to connect patients to specific referral sources would facilitate their treatment of patients. Weight management referrals for patients in rural areas and for non-Hispanic black adults and Hispanic adults, among whom obesity is most prevalent in the United States [3,25], are particularly needed. Addressing this need is consistent with national calls to reduce/eliminate obesity and other disparities that plague the U.S. health care system. One promising avenue to facilitate weight management referrals is the development, evaluation, and wide dissemination of remote weight-loss support interventions, particularly in rural, racial/ethnic minority, and low-income communities. Indeed, several recent articles demonstrate the success of such weight management programs across diverse patient populations [27–29].
Many of the physicians who participated in the study (72%) endorsed lack of time as a significant barrier to discussing weight and weight-related behaviors with their patients. Therefore, finding time-efficient strategies to involve physicians in weight management interventions may prove particularly beneficial. One such evidence-based behavioral counseling framework—the 5As framework—has been endorsed by the Centers for Medicare and Medicaid Services and the USPSTF for use with obese patients during a typical 20-minute visit [28].
The second highest-rated barrier, perceived patient lack of motivation, warrants additional discussion. Despite over half of the physicians surveyed citing this as a barrier, previous studies have shown that the majority of overweight patients believe they should lose weight and are interested in losing weight [21]. This study highlights a potential discrepancy between physicians’ perceptions of patients’ interest in weight-loss and their patients’ actual interest. It is possible that this discrepancy can be avoided by training physicians on how to be culturally sensitive when addressing weight with their patients. Moreover, such cultural sensitivity training may be of great use, given that 12% of physicians in this study were apprehensive about discussing weight with their patients due to fear of offending them. Such training typically involves teaching physicians how to talk with patients in ways that enable patients to feel comfortable, trusting, and respectful in patient-physician/provider interactions [29].
Two other findings pertaining to providers deserve mention. Specifically, 88% of physicians believed that effectively encouraging patients to adhere to a healthy lifestyle included personally engaging in health-promoting activities. However, of the physicians surveyed, 64% were overweight/obese. Given the high percentage of physicians in this study that were overweight/obese and these physicians’ belief that their personal engagement in health-promoting activities is important to encourage patient engagement in a healthy lifestyle, it seems that future efforts are needed to facilitate health-promoting behaviors among physicians—efforts that may in turn aid them in encouraging their patients to adhere to a healthy lifestyle.
Finally, this study assessed physicians’ healthy lifestyle–related knowledge about current BMI ranges for adults and BMI percentile ranges for children, and recommended amounts of moderate physical activity and servings of fruits and vegetables for adults. Most physicians were able to correctly identify the adult BMI cutoff ranges for overweight and obesity and to identify the correct answers to questions about physical activity and fruits and vegetable consumption guidelines for adults. However, only 32% of physicians were able to correctly identify BMI percentile ranges for children and/or adolescents. This is understandable given that most of the physicians in this study provide care to adult patients. However, considering that in 2012 more than one-third of children and adolescents were overweight or obese [1], it is important that all physicians have knowledge of BMI percentile ranges for children and adolescents so that minimally they can convey this information to their adult patients who are parents. The USPSTF defines children and adolescent overweight as an age- and gender-specific BMI between the 85th and 94th percentiles, and children and adolescent obesity as an age- and gender-specific BMI ≥ 95th percentile [31]. Such knowledge of BMI cutoffs is needed in order for providers to comply with the USPSTF recommendation to screen all adults and children aged 6 years and older for obesity, and then offer or refer those with an obesity diagnosis to intensive multicomponent behavioral interventions [31–33].
While novel, the study also had several limitations. First, due to self-selection of participants, physicians who felt more confident in their abilities to address overweight or obesity with their patients might have been more likely to respond. Second, participating physicians may have given socially desirable responses to questions (ie, responses that present a favorable image of themselves) rather than true/accurate responses. Future studies could incorporate a social desirability scale in order to detect and control for any socially desirable responding [33]. Another limitation was the small sample size and the limited variability in geographic location of the participating physicians. Thus, the experiences of these physicians may not be generalizable to physicians in other geographic regions. Future similar studies to the present study are needed and such studies should use a larger and randomly selected sample of physicians that is racially/ethnically diverse. Finally, a limitation of this study is the 48% participation rate. Factors that may have contributed to this participation rate include lack of compensation for physicians and the likelihood that physicians may have extremely busy schedules that may discourage them from participating. However, it is important to note that the 48% participation rate of this study is better than the 25.6% participation rate in another similar study [25]. Future similar studies to the present study likely need to include strong incentives for physicians to be study participants.
Conclusion
Our study indicates that many primary care physicians may not talk with their patients about engaging in healthy eating, physical activity, and weight management because of perceived barriers that prevent them from doing so, rather than because of a lack of perceived responsibility to do so or a perception that counseling patients on these issues would be ineffective. This finding highlights the importance of providing physicians with the tools and resources needed to overcome the aforementioned barriers to fostering health-promoting lifestyles and a healthy weight among their patients and the importance of involving physicians in identifying these barriers and ways to overcome them.
Acknowledgement. We thank the patients and health care providers at the participating medical homes affiliated with University of Florida Health in Jacksonville, Florida, for making this research possible.
Corresponding author: Carolyn M. Tucker, PhD, University of Florida, [email protected].
Funding/support: Support for this research was provided by the Office of Research at UF–Gainesville, Florida, and by the National Institutes of Health and National Center for Research Resources CTSA grant UL1 TR000064.
Financial disclosures: None.
From the University of Florida (Dr. Tucker, Ms. Ukonu, Ms. Kang, Ms. Good), Gainesville, FL; the University of Florida–Jacksonville (Dr. Shah, Dr. Bilello), Jacksonville, FL; and Ball State University (Dr. Arthur), Muncie, IN.
Abstracts
- Objective: To assess primary care physicians’ practices, knowledge, and beliefs regarding their efforts to promote healthy lifestyles and weight management among their patients.
- Methods: Study participants consisted of 25 primary care physicians from a regional primary care practice-based research network that includes 37 university-affiliated patient-centered medical homes and 2 nearby unaffiliated primary care sites. Participating physicians completed an online modified version of the Physician Survey of Practices on Diet, Physical Activity, and Weight Control–Adult Questionnaire.
- Results: The majority (88%) of participating physicians strongly believed it was their responsibility to promote a healthy diet, physical activity, and healthy weight loss and weight maintenance among patients. The 3 most commonly endorsed barriers were (a) not enough time, (b) minimal patient interest in improving his/her weight, and (c) lack of adequate weight-loss referral resources. The top 3 physician-perceived practice improvements that would be helpful with these practices were (a) better tools to communicate diet, physical activity, or weight problems to patients or family; (b) better mechanisms to connect patients to weight-loss referral resources; and (c) better counseling tools to guide patients regarding lifestyle modifications. 76% of the participating physicians correctly identified the BMI cutoff ranges for adult obesity, but only 32% did so for childhood obesity.
- Conclusion: It is important to provide primary care physicians with knowledge, effective tools, and resources to promote healthy lifestyles and weight loss and weight management among their patients.
Key words: obesity; primary care physicians; weight loss; weight management.
More than two-thirds of adults in the United States are overweight, with approximately 35% considered obese (defined as a body mass index ≥ 30) [1]. Obesity is associated with many of the leading causes of death in the United States (ie, diabetes, heart disease, stroke, and some types of cancer) and with poor mental health outcomes and reduced quality of life [2]. Racial/ethnic minorities and individuals with low incomes are disproportionately impacted by obesity and obesity-related diseases and negative health outcomes [3–5].
The US Preventive Services Task Force (USPSTF) recommends screening for obesity and intensive behavioral counseling, which are often the responsibilities of primary care providers [6]. Despite these recommendations, research suggests that primary care providers rarely screen their patients for obesity or refer them for intensive behavioral counseling despite evidence that doing so would improve patient health outcomes [5–7]. Lack of time to address weight issues during clinical visits, lack of training in weight management counseling, and lack of availability of intensive weight loss programs to which they can refer their patients are some of the reasons cited for not counseling patients about weight management [8].
Primary care providers deliver more hours of patient care than other providers, yet these providers have been unable to deliver medical interventions capable of producing even modest weight loss [10]. Obesity treatment options delivered in primary care settings have limited success, likely due to the low intensity of these treatment options. Many studies have shown that most obesity treatments in health care settings typically consist of scheduled monthly or quarterly visits that are 10 to 15 minutes in duration [11], despite evidence that more intense treatments are needed. Specifically, a systematic review of the obesity treatment literature performed by the USPSTF revealed that high-intensity, multicomponent behavioral interventions that include face-to-face counseling on diet and physical activity and behavioral therapy more than once a month for 3 months are needed to produce significant weight loss (8–15 lb) among adult patients in primary care settings [12].
Since many of the characteristics of multicomponent behavioral interventions for treating obesity are both patient-centered and involve self-management, the patient-centered medical home (PCMH) seems to be the ideal setting to deliver these interventions [13]. Specifically, PCMHs provide patient-centered care that is wide-ranging, team-based, and coordinated across all elements of the health care system and the patient’s community [14]. These sites specifically provide primary care, which is the type of care that obesity disparity patient groups such as racial/ethnic minorities, sexual minorities, groups with low incomes, and the medically underserved are more likely to utilize [15].
Providing multicomponent behavioral interventions for obesity in PCMHs and other primary care sites will increase the likelihood of participation among the aforementioned obesity disparity groups. Despite the potential benefits of obesity treatment interventions offered in primary care settings, particularly for obesity disparity groups, the role of primary care providers in providing such treatment interventions is not clear [16]. We surveyed primary care physicians who primarily worked in PCMHs to assess their practices, knowledge, views/beliefs, perceived barriers, and perceived needed clinic practice improvements relative to promoting healthy lifestyles and weight management among their patients.
Methods
Participants
Primary care physicians were recruited from among a regional primary care practice-based research network that includes 37 PCMHs affiliated with an academic health center and 2 nearby primary care sites not affiliated with an academic health center. Fifty-two physicians at these centers received an invitation via email to participate in our online survey study. The invitation email included (a) a study endorsement note from the chair of the Community Health and Family Medicine Department affiliated with the PCMHs, (b) instructions about how to participate in the study, and (c) a link to the study. Participation inclusion criteria specified in the online informed consent form were: (a) working as a physician affiliated with the practice-based research network, (b) having access to a computer with internet connection, (c) being able to communicate in written English, and (d) providing written consent to participate in the study. Physicians were not provided compensation for participating in the study.
Survey Instrument
To assess physicians’ views and practices, we used a modified version of the Physician Survey of Practices on Diet, Physical Activity, and Weight Control–Adult Questionnaire [17]. The survey was sponsored by the National Cancer Institute in collaboration with several other NIH institutes and the CDC for evaluating current clinical practices among physicians, including the degree to which physicians evaluate their patients for obesity and offer them guidance designed to increase adherence to a health-promoting lifestyle (eg, recommendations on diet, weight, and physical activity). Additionally, the questionnaire assesses physicians’ perceived barriers to patient assessment, evaluation, and management. It also includes questions about physicians’ healthy lifestyle–related knowledge. In 2010, Smith and colleagues utilized the questionnaire with a nationally representative sample of primary care physicians (n = 1211) to investigate primary care physicians’ clinical practices in relation to overweight and obesity [18]. To our knowledge, no other physician survey has been developed to assess current engagement in recommended clinical practices, barriers to engaging in recommended practices, as well as beliefs and knowledge regarding helping patients follow a health-promoting lifestyle. The original survey also includes questions regarding the physicians’ personal health status and health behaviors.
For our study, we modified the survey by removing questions regarding the physicians’ (a) perceived general health and well-being, (b) current dietary practices, (c) current level of engagement in physical activity, and (d) current engagement in professional activities unrelated to patient care (eg, research, teaching). Our modified survey included 7 questions asking about current practices regarding screening for obesity and referral of patients to weight management interventions. Two questions asked about physicians’ perceived barriers to helping patients adhere to a health-promoting lifestyle and maintain a healthy weight. Physicians were asked to rate their top 3 barriers from among a list of 11 pre-identified barriers and to rate their top 3 desired practice-related improvements from among a list of 10 pre-identified improvements. Physicians were given the option to provide additional barriers or improvements that were not already pre-identified. Seven questions assessed physicians’ views/beliefs related to helping patients achieve and maintain a health-promoting lifestyle and a healthy weight. These questions utilize a rating scale where 1 = strongly agree, 2 = agree somewhat, 3 = neither agree nor disagree, 4 = disagree somewhat, and 5 = strongly disagree. Four questions assessed physicians’ healthy lifestyle–related knowledge (BMI ranges/percentiles for adults/children, diet and exercise guideline recommendations [recommended amounts of moderate physical activity and servings of fruits and vegetables for adults]) and 11 questions ask about the physician (height, weight, demographics, and practice population).
Survey Administration
The survey was administered anonymously through Qualtrics, a secure, online survey platform. The survey was administered online to increase anonymity, increase response rate, and diminish potential physician-perceived barriers to participating in the study. The participating physicians were provided with a link that enabled them to access the survey. The survey excluded questions that required disclosure of identifying information. Survey data from Qualtrics were exported to an SPSS file that was stored on a password protected, secured computer in the research lab of the principal investigator for this study.
Data Analysis
Frequency analyses were applied to survey responses to determine the participating physicians’ endorsed barriers to and views regarding evaluating and managing patients’ weight, healthy eating, and physical activity; physicians’ views related to helping patients achieve and maintain a health-promoting lifestyle and a healthy weight; and physicians’ healthy lifestyle–related knowledge. Nonparametric t tests were conducted to examine differences in survey responses of the participating physicians in association with their sex (male or female), race (Asian vs. white/Caucasian), and BMI (BMI < 25 and BMI ≥ 25).
Approval for the study was obtained through the institutional review board of the University of Florida Health Science Center.
Results
Participants
Twenty-five physicians out of 52 invited completed the survey (48% response rate). The vast majority of the study participants were PCMH-affiliated (92%–96%). Participating physicians ranged in age from 29 to 67 years old. Sixteen (64%) participating physicians identified as female, 7 (28%) participating physicians identified as male, and 2 (8%) participating physicians did not indicate a sex. Twenty (80%) participating physicians identified as being white, 3 (12%) participating physicians identified as being Asian/Asian American, and 2 (8%) did not indicate a race or ethnicity. Twenty-two (88%) participating physicians were employees of a large medical group affiliated with an academic medical center, 1 (4%) was employed in a physician-owned practice, and 2 (8%) did not indicate their main primary care practice location. Table 1 provides additional demographic data.
Approximately 88% of the participating physicians agreed that patients were more likely to adopt healthier lifestyles if their health care providers counseled them to do so (44% strongly agreed, 44% agreed somewhat). A majority of participating physicians endorsed the view that there are effective strategies and/or tools to (a) help patients eat a healthy diet (56% strongly agreed, 24% agreed somewhat), (b) engage in adequate amounts of physical activity (56% strongly agreed, 20% agreed somewhat), and (c) maintain a healthy weight or lose weight (48% strongly agreed,
Many participating physicians expressed confidence in their ability to counsel their patients to (a) eat a healthy diet (64% strongly agreed, 28% agreed somewhat), (b) engage in adequate amounts of physical activity (68% strongly agreed, 24% agreed somewhat), and (c) maintain a healthy weight or lose weight (60% strongly agreed, 32% agreed somewhat). Most participating physicians at least somewhat agreed that they were effective at helping their patients (a) eat a healthy diet (24% strongly agreed, 52% agreed somewhat), (b) engage in adequate amounts of physical activity (20% strongly agreed, 56% agreed somewhat), and (c) maintain a healthy weight or lose weight (16% strongly agreed, 48% agreed somewhat). Some participating physicians expressed ambivalence about whether or not they were effective at helping their patients (a) eat a healthy diet (16% neither agreed nor disagreed), (b) engage in adequate amounts of physical activity (12% neither agreed nor disagreed), and (c) maintain a healthy weight or lose weight (20% neither agreed nor disagreed). A total of 8% of participating physicians did not endorse the belief that they were effective at helping their patients maintain a healthy weight or lose weight.
Most participating physicians at least somewhat agreed that they were effective in encouraging patients to engage in health-promoting activities (44% strongly agreed, and 44% agreed somewhat), whereas 4% neither agreed nor disagreed that they were effective in providing this encouragement. Interestingly, many participating physicians endorsed the view that they would be able to provide more credible and effective counseling to patients if they (the physicians themselves) ate a healthy diet (68% strongly agreed, 20% agreed somewhat) and engaged in adequate amounts of physical activity (68% strongly agreed, 20% agreed somewhat). A minority of participating physicians (4%) neither agreed or disagreed with this perspective.
In regards to participating physicians’ healthy lifestyle–related knowledge about current BMI ranges for adults or percentile ranges for children, most participating physicians were able to accurately identify the correct BMI cutoff ranges for overweight (80%) and obese (76%) adults. However, only 32% of participating physicians were able to correctly identify BMI percentile ranges for children; however, nearly all of the participating physicians saw mainly adult patients. Lastly, 76% of participating physicians were able to correctly identify the recommended amounts of moderate physical activity for adults 18 years of age and older, and only 56% were able to correctly identify the recommended amount of servings of fruits and vegetables.
There were no significant race-related differences in participating physicians views/beliefs, healthy lifestyle–related knowledge, and perceived barriers to helping patients engage in health promoting behaviors and weight management. There were no significant sex-related differences in these variables with the exception that women were more likely to respond that they did not know the BMI percentile range at which children or adolescents were considered to have a healthy weight (37.5% of women vs. 0% of men, P = 0.03). A similar percentage of men (66.7%) and women (64.7%) who chose among the 4 percentile range options (rather than endorsing “Don’t know”) chose an incorrect answer. Lastly, there were no significant self-reported BMI-related differences in participating physicians’ views/beliefs, healthy lifestyle–related knowledge, and perceived barriers to helping patients engage in health-promoting behaviors and weight management.
Discussion
Given the high percentage of adults in the United States who are overweight or obese and the associated health risks, it is paramount that primary care physicians advise their patients to manage their weight and adopt a health-promoting lifestyle. Research studies indicate that such advice is effective [18,19]. Furthermore, it has been found that most overweight and obese patients want more assistance with weight management than they are receiving from their primary care physicians [21]. This study thus explored primary care physicians’ knowledge, beliefs, and perceived barriers that may prevent them from providing such assistance. The primary care setting is the site where obesity disparity groups (eg, racial/ethnic minorities, groups with low household incomes) are most likely to receive care [22,23].
Most of the PCMH-affiliated physicians in this study agreed that they had the responsibility to promote weight-loss/management and healthy lifestyles among their patients. Consistent with prior research [9], the majority of the physicians in this study felt they were effective in their ability to counsel patients to eat a healthy diet and engage in physical activity. To illustrate, in a prior study [9], 77% of primary care providers thought that they could provide useful dieting tips to patients, and in this study, 80% believed they were effective in helping patients eat a healthy diet. However, despite this confidence in their ability to provide advice about healthy diets and physical activity, the providers in both this and in another prior study [25] were less confident in their ability to actually help patients lose weight. Only 64% of the providers in the present study felt they could be effective in assisting patients with losing weight or maintaining a healthy weight. Although this percentage is higher than the 44% of physicians found in a prior study [25] who felt confident in their ability to treat obesity, both studies clearly point to a need to decrease barriers that physicians face in helping clients lose weight.
A key finding of this study was the consensus among the participating physicians regarding what they perceived to be the common barriers to helping patients adhere to a health-promoting lifestyle. Consistent with past research [8,9], the 3 most common barriers cited by the participating physicians were that they did not have enough time, patients were not interested in improving their weight, and adequate referrals for diet, physical activity, and weight were lacking. Additional barriers endorsed included a lack of effective tools and information to give patients, and a fear of offending patients. Another barrier identified by the participating physicians is the perception that patients had difficulty in changing behaviors necessary for maintaining a healthier lifestyle.
When asked what would facilitate conversations with patients, the top 3 responses given were better tools to communicate diet, physical activity, or weight problems to patients or family members; better mechanisms to connect patients to specific referral sources; and better counseling tools to guide patients towards engagement in healthy lifestyles. Of note is the significant overlap between the perceived barriers and the needed facilitation tools. The clearest example of this overlap is that physicians noted a lack of adequate referral sources to be a barrier and that better mechanisms to connect patients to specific referral sources would facilitate their treatment of patients. Weight management referrals for patients in rural areas and for non-Hispanic black adults and Hispanic adults, among whom obesity is most prevalent in the United States [3,25], are particularly needed. Addressing this need is consistent with national calls to reduce/eliminate obesity and other disparities that plague the U.S. health care system. One promising avenue to facilitate weight management referrals is the development, evaluation, and wide dissemination of remote weight-loss support interventions, particularly in rural, racial/ethnic minority, and low-income communities. Indeed, several recent articles demonstrate the success of such weight management programs across diverse patient populations [27–29].
Many of the physicians who participated in the study (72%) endorsed lack of time as a significant barrier to discussing weight and weight-related behaviors with their patients. Therefore, finding time-efficient strategies to involve physicians in weight management interventions may prove particularly beneficial. One such evidence-based behavioral counseling framework—the 5As framework—has been endorsed by the Centers for Medicare and Medicaid Services and the USPSTF for use with obese patients during a typical 20-minute visit [28].
The second highest-rated barrier, perceived patient lack of motivation, warrants additional discussion. Despite over half of the physicians surveyed citing this as a barrier, previous studies have shown that the majority of overweight patients believe they should lose weight and are interested in losing weight [21]. This study highlights a potential discrepancy between physicians’ perceptions of patients’ interest in weight-loss and their patients’ actual interest. It is possible that this discrepancy can be avoided by training physicians on how to be culturally sensitive when addressing weight with their patients. Moreover, such cultural sensitivity training may be of great use, given that 12% of physicians in this study were apprehensive about discussing weight with their patients due to fear of offending them. Such training typically involves teaching physicians how to talk with patients in ways that enable patients to feel comfortable, trusting, and respectful in patient-physician/provider interactions [29].
Two other findings pertaining to providers deserve mention. Specifically, 88% of physicians believed that effectively encouraging patients to adhere to a healthy lifestyle included personally engaging in health-promoting activities. However, of the physicians surveyed, 64% were overweight/obese. Given the high percentage of physicians in this study that were overweight/obese and these physicians’ belief that their personal engagement in health-promoting activities is important to encourage patient engagement in a healthy lifestyle, it seems that future efforts are needed to facilitate health-promoting behaviors among physicians—efforts that may in turn aid them in encouraging their patients to adhere to a healthy lifestyle.
Finally, this study assessed physicians’ healthy lifestyle–related knowledge about current BMI ranges for adults and BMI percentile ranges for children, and recommended amounts of moderate physical activity and servings of fruits and vegetables for adults. Most physicians were able to correctly identify the adult BMI cutoff ranges for overweight and obesity and to identify the correct answers to questions about physical activity and fruits and vegetable consumption guidelines for adults. However, only 32% of physicians were able to correctly identify BMI percentile ranges for children and/or adolescents. This is understandable given that most of the physicians in this study provide care to adult patients. However, considering that in 2012 more than one-third of children and adolescents were overweight or obese [1], it is important that all physicians have knowledge of BMI percentile ranges for children and adolescents so that minimally they can convey this information to their adult patients who are parents. The USPSTF defines children and adolescent overweight as an age- and gender-specific BMI between the 85th and 94th percentiles, and children and adolescent obesity as an age- and gender-specific BMI ≥ 95th percentile [31]. Such knowledge of BMI cutoffs is needed in order for providers to comply with the USPSTF recommendation to screen all adults and children aged 6 years and older for obesity, and then offer or refer those with an obesity diagnosis to intensive multicomponent behavioral interventions [31–33].
While novel, the study also had several limitations. First, due to self-selection of participants, physicians who felt more confident in their abilities to address overweight or obesity with their patients might have been more likely to respond. Second, participating physicians may have given socially desirable responses to questions (ie, responses that present a favorable image of themselves) rather than true/accurate responses. Future studies could incorporate a social desirability scale in order to detect and control for any socially desirable responding [33]. Another limitation was the small sample size and the limited variability in geographic location of the participating physicians. Thus, the experiences of these physicians may not be generalizable to physicians in other geographic regions. Future similar studies to the present study are needed and such studies should use a larger and randomly selected sample of physicians that is racially/ethnically diverse. Finally, a limitation of this study is the 48% participation rate. Factors that may have contributed to this participation rate include lack of compensation for physicians and the likelihood that physicians may have extremely busy schedules that may discourage them from participating. However, it is important to note that the 48% participation rate of this study is better than the 25.6% participation rate in another similar study [25]. Future similar studies to the present study likely need to include strong incentives for physicians to be study participants.
Conclusion
Our study indicates that many primary care physicians may not talk with their patients about engaging in healthy eating, physical activity, and weight management because of perceived barriers that prevent them from doing so, rather than because of a lack of perceived responsibility to do so or a perception that counseling patients on these issues would be ineffective. This finding highlights the importance of providing physicians with the tools and resources needed to overcome the aforementioned barriers to fostering health-promoting lifestyles and a healthy weight among their patients and the importance of involving physicians in identifying these barriers and ways to overcome them.
Acknowledgement. We thank the patients and health care providers at the participating medical homes affiliated with University of Florida Health in Jacksonville, Florida, for making this research possible.
Corresponding author: Carolyn M. Tucker, PhD, University of Florida, [email protected].
Funding/support: Support for this research was provided by the Office of Research at UF–Gainesville, Florida, and by the National Institutes of Health and National Center for Research Resources CTSA grant UL1 TR000064.
Financial disclosures: None.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014;311:806–14.
2. Centers for Disease Control and Prevention. Adult obesity causes and consequences, 2016. Accessed 30 Apr 2017 at www.cdc.gov/obesity/adult/causes.html.
3. Centers for Disease Control and Prevention. CDC health disparities and inequalities report —United States, 2013. Accessed 30 Apr 2017 at www.cdc.gov/mmwr/pdf/other/su6203.pdf.
4. Wang Y, Beydoun MA. The obesity epidemic in the United States – gender, age, socioeconomic, racial/ethnic, and geographic characteristics: A systematic review and meta-regression analysis. Epidemiol Rev 2007;29:6–28.
5. Levine, JA. Poverty and obesity in the US. Diabetes 2011;60:2667–8.
6. Moyer VA. Screening for and management of obesity in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2012;157:373–8.
7. Aveyard P, Lewis A, Tearne S, et al. Screening and brief intervention for obesity in primary care: a parallel, two-arm, randomised trial. Lancet 2016;388:2492–500.
8. Jay M, Chintapalli S, Squires A, et al. Barriers and facilitators to providing primary care-based weight management services in a patient centered medical home for Veterans: a qualitative study. BMC Fam Pract 2015;16:167.
9. Ruelaz AR, Diefenbach, Simon B, et al. Perceived barriers to weight management. J Gen Intern Med 2007;22:518–22.
10. Carvajal R, Wadden TA, Tsai AG, et al. Managing obesity in primary care practice: A narrative review. Ann N Y Acad Sci 2013;1281:191–206.
11. Wadden TA, Neiberg RH, Wing RR, et al; Look AHEAD Research Group. Four-year weight-losses in the Look AHEAD study: factors associated with long-term success. Obesity (Silver Spring) 2011;19:1987–98.
12. LeBlanc ES, O’Connor, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2011;155:434–47.
13. Rittenhouse DR, Shortell SM. The patient-centered medical home: will it stand the test of health reform? JAMA 2009;301:2038–40.
14. Scholle S, Torda P, Peikes D, et al. Engaging patients and families in the medical home (prepared by Mathematica Policy Research under contract no. HHSA290200900019ITO2.) AHRQ Pub No. 10-0083-EF. Rockville, MD: Agency for Healthcare Research and Quality; June 2010.
15. US Department of Health and Human Services. HHS action plan to reduce racial and ethnic health disparities: A nation free of disparities in health and health care. 2011.
16. Ard J. Obesity in the US: what is the best role for primary care? BMJ 2015;350:1–10.
17. National Cancer Institute. Physcian survey of practices on diet, physical activity, and weight control. 2010. Accessed 30 Apr 2017 at https://healthcaredelivery.cancer.gov/energy_balance/phys_pract_q_adult.pdf.
18. Smith AW, Borowski LA, Liu B, et al. US primary care physicians’ diet-, physical activity–, and weight-related care of adult patients. Am J Prev Med 2011;41:33–42.
19. Simons-Morton DG, Calfas KJ, et al. Effects of interventions in health care settings on physical activity or cardiorespiratory fitness. Am J Prev Med 1998;15:413–30.
20. Bowerman S, Bellman M, Saltsman P, et al. Implementation of a primary care physician network obesity management program. Obes Res 2001;9 Suppl 4:321S–5S.
21. Potter MB, Vu JD, Croughan-Minihane M. Weight management: what patients want from their primary care physician. J Fam Pract 2001;50:513–8.
22. National Association of Community Health Centers. Community health centers: The local prescription for better quality and lower costs. March 2011. Accessed at www.nachc.org.
23. Institute of Medicine. Unequal treatment: confronting racial and ethnic disparities in healthcare. Washington, DC: National Academy of Sciences Press; 2003.
24. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 2012;307:491–7.
25. Bleich SN, Bennett WL, Gudzune KA, Cooper LA. National survey of US primary care physicians’ perspectives about causes of obesity and solutions to improve care. BMJ Open 2012;2(6).
26. Patterson PD, Moore CG, Probst JC, Shinogle JA. Obesity and physical inactivity in rural America. J Rural Health 2004;20:151–9.
27. Shaikh U, Cole SL, Marcin JP, Nesbitt TS. Clinical management and patient outcomes among children and adolescents receiving telemedicine consultations for obesity. Telemed e-Health 2008;14:434–40.
28. Schlair S, Moore S, Mcmacken M, Jay M. How to deliver high-quality obesity counseling in primary care using the 5As framework. J Clin Outcomes Manag 2012;19:221–9.
29. Tucker CM, Arthur TM, Roncoroni J, et al. Patient-centered culturally sensitive health care. Am J Lifestyle Med 2013;9:63–77.
30. Ma J, Yank V, Xiao L, et al. Translating the diabetes prevention program lifestyle intervention for weight loss into primary care. JAMA Intern Med 2013;173:113–21.
31. U.S. Preventive Services Task Force. Screening for obesity in children and adolescents: U.S. Preventive Services Task Force recommendation statement. Pediatrics 2010;125:361–7.
32. Final recommendation statement: obesity in adults: screening and management. US Preventive Services Task Force. Oct 2014. Accessed at www.uspreventiveservicestaskforce.org.
33. van de Mortel, TF. Faking it: social desirability response bias in self-report research. Aust J Advanced Nurs 2008:25:40–8.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014;311:806–14.
2. Centers for Disease Control and Prevention. Adult obesity causes and consequences, 2016. Accessed 30 Apr 2017 at www.cdc.gov/obesity/adult/causes.html.
3. Centers for Disease Control and Prevention. CDC health disparities and inequalities report —United States, 2013. Accessed 30 Apr 2017 at www.cdc.gov/mmwr/pdf/other/su6203.pdf.
4. Wang Y, Beydoun MA. The obesity epidemic in the United States – gender, age, socioeconomic, racial/ethnic, and geographic characteristics: A systematic review and meta-regression analysis. Epidemiol Rev 2007;29:6–28.
5. Levine, JA. Poverty and obesity in the US. Diabetes 2011;60:2667–8.
6. Moyer VA. Screening for and management of obesity in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2012;157:373–8.
7. Aveyard P, Lewis A, Tearne S, et al. Screening and brief intervention for obesity in primary care: a parallel, two-arm, randomised trial. Lancet 2016;388:2492–500.
8. Jay M, Chintapalli S, Squires A, et al. Barriers and facilitators to providing primary care-based weight management services in a patient centered medical home for Veterans: a qualitative study. BMC Fam Pract 2015;16:167.
9. Ruelaz AR, Diefenbach, Simon B, et al. Perceived barriers to weight management. J Gen Intern Med 2007;22:518–22.
10. Carvajal R, Wadden TA, Tsai AG, et al. Managing obesity in primary care practice: A narrative review. Ann N Y Acad Sci 2013;1281:191–206.
11. Wadden TA, Neiberg RH, Wing RR, et al; Look AHEAD Research Group. Four-year weight-losses in the Look AHEAD study: factors associated with long-term success. Obesity (Silver Spring) 2011;19:1987–98.
12. LeBlanc ES, O’Connor, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2011;155:434–47.
13. Rittenhouse DR, Shortell SM. The patient-centered medical home: will it stand the test of health reform? JAMA 2009;301:2038–40.
14. Scholle S, Torda P, Peikes D, et al. Engaging patients and families in the medical home (prepared by Mathematica Policy Research under contract no. HHSA290200900019ITO2.) AHRQ Pub No. 10-0083-EF. Rockville, MD: Agency for Healthcare Research and Quality; June 2010.
15. US Department of Health and Human Services. HHS action plan to reduce racial and ethnic health disparities: A nation free of disparities in health and health care. 2011.
16. Ard J. Obesity in the US: what is the best role for primary care? BMJ 2015;350:1–10.
17. National Cancer Institute. Physcian survey of practices on diet, physical activity, and weight control. 2010. Accessed 30 Apr 2017 at https://healthcaredelivery.cancer.gov/energy_balance/phys_pract_q_adult.pdf.
18. Smith AW, Borowski LA, Liu B, et al. US primary care physicians’ diet-, physical activity–, and weight-related care of adult patients. Am J Prev Med 2011;41:33–42.
19. Simons-Morton DG, Calfas KJ, et al. Effects of interventions in health care settings on physical activity or cardiorespiratory fitness. Am J Prev Med 1998;15:413–30.
20. Bowerman S, Bellman M, Saltsman P, et al. Implementation of a primary care physician network obesity management program. Obes Res 2001;9 Suppl 4:321S–5S.
21. Potter MB, Vu JD, Croughan-Minihane M. Weight management: what patients want from their primary care physician. J Fam Pract 2001;50:513–8.
22. National Association of Community Health Centers. Community health centers: The local prescription for better quality and lower costs. March 2011. Accessed at www.nachc.org.
23. Institute of Medicine. Unequal treatment: confronting racial and ethnic disparities in healthcare. Washington, DC: National Academy of Sciences Press; 2003.
24. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 2012;307:491–7.
25. Bleich SN, Bennett WL, Gudzune KA, Cooper LA. National survey of US primary care physicians’ perspectives about causes of obesity and solutions to improve care. BMJ Open 2012;2(6).
26. Patterson PD, Moore CG, Probst JC, Shinogle JA. Obesity and physical inactivity in rural America. J Rural Health 2004;20:151–9.
27. Shaikh U, Cole SL, Marcin JP, Nesbitt TS. Clinical management and patient outcomes among children and adolescents receiving telemedicine consultations for obesity. Telemed e-Health 2008;14:434–40.
28. Schlair S, Moore S, Mcmacken M, Jay M. How to deliver high-quality obesity counseling in primary care using the 5As framework. J Clin Outcomes Manag 2012;19:221–9.
29. Tucker CM, Arthur TM, Roncoroni J, et al. Patient-centered culturally sensitive health care. Am J Lifestyle Med 2013;9:63–77.
30. Ma J, Yank V, Xiao L, et al. Translating the diabetes prevention program lifestyle intervention for weight loss into primary care. JAMA Intern Med 2013;173:113–21.
31. U.S. Preventive Services Task Force. Screening for obesity in children and adolescents: U.S. Preventive Services Task Force recommendation statement. Pediatrics 2010;125:361–7.
32. Final recommendation statement: obesity in adults: screening and management. US Preventive Services Task Force. Oct 2014. Accessed at www.uspreventiveservicestaskforce.org.
33. van de Mortel, TF. Faking it: social desirability response bias in self-report research. Aust J Advanced Nurs 2008:25:40–8.
Erratum
The article "Handheld Reflectance Confocal Microscopy to Aid in the Management of Complex Facial Lentigo Maligna" (Cutis. 2017;99:346-352) contained an error in the author affiliations. The affiliations should have read:
All from the Dermatology Service, Memorial Sloan Kettering Cancer Center, New York, New York. Dr. Yélamos also is from the Dermatology Department, Hospital Clínic, Universitat de Barcelona, Spain. Dr. Rossi also is from the Department of Dermatology, Weill Cornell Medical College, New York.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
The article "Handheld Reflectance Confocal Microscopy to Aid in the Management of Complex Facial Lentigo Maligna" (Cutis. 2017;99:346-352) contained an error in the author affiliations. The affiliations should have read:
All from the Dermatology Service, Memorial Sloan Kettering Cancer Center, New York, New York. Dr. Yélamos also is from the Dermatology Department, Hospital Clínic, Universitat de Barcelona, Spain. Dr. Rossi also is from the Department of Dermatology, Weill Cornell Medical College, New York.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
The article "Handheld Reflectance Confocal Microscopy to Aid in the Management of Complex Facial Lentigo Maligna" (Cutis. 2017;99:346-352) contained an error in the author affiliations. The affiliations should have read:
All from the Dermatology Service, Memorial Sloan Kettering Cancer Center, New York, New York. Dr. Yélamos also is from the Dermatology Department, Hospital Clínic, Universitat de Barcelona, Spain. Dr. Rossi also is from the Department of Dermatology, Weill Cornell Medical College, New York.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
ABP 501 equivalent in efficacy to adalimumab for moderate to severe RA
The biosimilar ABP 501 is equally as effective and safe as adalimumab for the treatment of moderate to severe rheumatoid arthritis, according to results of a phase III clinical trial.
In a randomized, double-blind equivalence study across 100 medical centers in 12 countries, 526 patients with moderate to severe RA and inadequate response to methotrexate received either ABP 501 or adalimumab. Of the 526, 494 completed the study. Just over 80% of patients were women, and 95.1% were white, with a mean age of 55.9 years. Patients received either 40-mg ABP 501 or adalimumab subcutaneously on day 1 and every 2 weeks until week 22, with primary endpoint assessments conducted after 24 weeks, according to Dr. Stanley Cohen and his associates (Ann Rheum Dis. 2017 Jun 5. doi: 10.1136/annrheumdis-2016-210459).
Treatment-emergent adverse events occurred in 132 of the 264 patients in the ABP 501 group and in 143 of the 262 patients in the adalimumab group. Common treatment-emergent adverse events in both groups included nasopharyngitis, headache, arthralgia, cough, and upper respiratory infection. Serious adverse events occurred in 10 patients who received ABP 501 and in 13 patients who received adalimumab. Sepsis was the only serious adverse event that occurred in more than one patient.
Over the course of the study, 38.3% of patients who received ABP 501 and 38.2% of patients who received adalimumab tested positive for binding antidrug antibodies.
The study data “contribute to the totality-of-evidence–based requirements to demonstrate that ABP 501 is similar to adalimumab. The FDA has, thus, approved ABP 501 for use as a biosimilar to adalimumab, making it a valuable new therapeutic option for the treatment of moderate to severe RA,” Dr. Cohen and his associates concluded.
The study was funded by Amgen. Dr. Cohen and five of his associates reported conflicts of interest. Two investigators are employees of Amgen.
The biosimilar ABP 501 is equally as effective and safe as adalimumab for the treatment of moderate to severe rheumatoid arthritis, according to results of a phase III clinical trial.
In a randomized, double-blind equivalence study across 100 medical centers in 12 countries, 526 patients with moderate to severe RA and inadequate response to methotrexate received either ABP 501 or adalimumab. Of the 526, 494 completed the study. Just over 80% of patients were women, and 95.1% were white, with a mean age of 55.9 years. Patients received either 40-mg ABP 501 or adalimumab subcutaneously on day 1 and every 2 weeks until week 22, with primary endpoint assessments conducted after 24 weeks, according to Dr. Stanley Cohen and his associates (Ann Rheum Dis. 2017 Jun 5. doi: 10.1136/annrheumdis-2016-210459).
Treatment-emergent adverse events occurred in 132 of the 264 patients in the ABP 501 group and in 143 of the 262 patients in the adalimumab group. Common treatment-emergent adverse events in both groups included nasopharyngitis, headache, arthralgia, cough, and upper respiratory infection. Serious adverse events occurred in 10 patients who received ABP 501 and in 13 patients who received adalimumab. Sepsis was the only serious adverse event that occurred in more than one patient.
Over the course of the study, 38.3% of patients who received ABP 501 and 38.2% of patients who received adalimumab tested positive for binding antidrug antibodies.
The study data “contribute to the totality-of-evidence–based requirements to demonstrate that ABP 501 is similar to adalimumab. The FDA has, thus, approved ABP 501 for use as a biosimilar to adalimumab, making it a valuable new therapeutic option for the treatment of moderate to severe RA,” Dr. Cohen and his associates concluded.
The study was funded by Amgen. Dr. Cohen and five of his associates reported conflicts of interest. Two investigators are employees of Amgen.
The biosimilar ABP 501 is equally as effective and safe as adalimumab for the treatment of moderate to severe rheumatoid arthritis, according to results of a phase III clinical trial.
In a randomized, double-blind equivalence study across 100 medical centers in 12 countries, 526 patients with moderate to severe RA and inadequate response to methotrexate received either ABP 501 or adalimumab. Of the 526, 494 completed the study. Just over 80% of patients were women, and 95.1% were white, with a mean age of 55.9 years. Patients received either 40-mg ABP 501 or adalimumab subcutaneously on day 1 and every 2 weeks until week 22, with primary endpoint assessments conducted after 24 weeks, according to Dr. Stanley Cohen and his associates (Ann Rheum Dis. 2017 Jun 5. doi: 10.1136/annrheumdis-2016-210459).
Treatment-emergent adverse events occurred in 132 of the 264 patients in the ABP 501 group and in 143 of the 262 patients in the adalimumab group. Common treatment-emergent adverse events in both groups included nasopharyngitis, headache, arthralgia, cough, and upper respiratory infection. Serious adverse events occurred in 10 patients who received ABP 501 and in 13 patients who received adalimumab. Sepsis was the only serious adverse event that occurred in more than one patient.
Over the course of the study, 38.3% of patients who received ABP 501 and 38.2% of patients who received adalimumab tested positive for binding antidrug antibodies.
The study data “contribute to the totality-of-evidence–based requirements to demonstrate that ABP 501 is similar to adalimumab. The FDA has, thus, approved ABP 501 for use as a biosimilar to adalimumab, making it a valuable new therapeutic option for the treatment of moderate to severe RA,” Dr. Cohen and his associates concluded.
The study was funded by Amgen. Dr. Cohen and five of his associates reported conflicts of interest. Two investigators are employees of Amgen.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point:
Major finding: Adequate ACR 20 response occurred in 74.6% of patients receiving ABP 501 and in 72.4% of patients receiving adalimumab.
Data source: A randomized, double-blind equivalence study across 100 medical centers in 12 countries of 526 patients with moderate to severe RA.
Disclosures: The study was funded by Amgen. Dr. Cohen and five of his associated reported conflicts of interest. Two investigators are employees of Amgen.
Endometriosis detection by microRNA possible in early stages
VANCOUVER – A panel of 11 microRNA biomarkers was effective in predicting endometriosis in a small validation study conducted at a Belgium hospital, showing the progress researchers are making toward a noninvasive test.
The research team at Leuven University Hospital developed the model using a population of 120 women, 82 of whom had endometriosis. After extensive analysis, they identified 11 microRNAs, which varied significantly between endometriosis patients and healthy controls and could be used as part of a panel to detect endometriosis. The panel performed well in their validation sample of 60 women with endometriosis and 30 controls, with a sensitivity of 92% and a specificity of 96%.
That validation is good news, but it is still far from being clinically useful. First, the model needs to be tested further in their own predominantly Caucasian population. Then, it needs to be validated by outside researchers, and the model will almost certainly have to be altered to account for different genetic backgrounds, lifestyle habits, and other factors, Arne Vanhie, MD, of Leuven University Hospital said at the World Congress on Endometriosis.
“We may have to tweak it a bit,” he said.
Unfortunately, that tweaking is likely to be time consuming. That’s because RNA extraction and sequencing is labor intensive and expensive, though it could be readily automated if it becomes commercialized.
It’s also technically challenging because microRNA is present in very low concentrations in plasma so researchers must spot a rare signal surrounded by a sea of noise, and that can mean a lot of technical refinement along the way. “We did a lot of testing and trial and error to see what was the best protocol,” Dr. Vanhie said.
The news isn’t all bad. With a well-funded, intensive effort, development could be swift. “There’s no intervention. You just need a tube of blood, so you could go forward with this to make it clinically relevant very easily,” Dr. Vanhie said.
The study was sponsored by the Belgian government. Dr. Vanhie reported having no financial disclosures.
VANCOUVER – A panel of 11 microRNA biomarkers was effective in predicting endometriosis in a small validation study conducted at a Belgium hospital, showing the progress researchers are making toward a noninvasive test.
The research team at Leuven University Hospital developed the model using a population of 120 women, 82 of whom had endometriosis. After extensive analysis, they identified 11 microRNAs, which varied significantly between endometriosis patients and healthy controls and could be used as part of a panel to detect endometriosis. The panel performed well in their validation sample of 60 women with endometriosis and 30 controls, with a sensitivity of 92% and a specificity of 96%.
That validation is good news, but it is still far from being clinically useful. First, the model needs to be tested further in their own predominantly Caucasian population. Then, it needs to be validated by outside researchers, and the model will almost certainly have to be altered to account for different genetic backgrounds, lifestyle habits, and other factors, Arne Vanhie, MD, of Leuven University Hospital said at the World Congress on Endometriosis.
“We may have to tweak it a bit,” he said.
Unfortunately, that tweaking is likely to be time consuming. That’s because RNA extraction and sequencing is labor intensive and expensive, though it could be readily automated if it becomes commercialized.
It’s also technically challenging because microRNA is present in very low concentrations in plasma so researchers must spot a rare signal surrounded by a sea of noise, and that can mean a lot of technical refinement along the way. “We did a lot of testing and trial and error to see what was the best protocol,” Dr. Vanhie said.
The news isn’t all bad. With a well-funded, intensive effort, development could be swift. “There’s no intervention. You just need a tube of blood, so you could go forward with this to make it clinically relevant very easily,” Dr. Vanhie said.
The study was sponsored by the Belgian government. Dr. Vanhie reported having no financial disclosures.
VANCOUVER – A panel of 11 microRNA biomarkers was effective in predicting endometriosis in a small validation study conducted at a Belgium hospital, showing the progress researchers are making toward a noninvasive test.
The research team at Leuven University Hospital developed the model using a population of 120 women, 82 of whom had endometriosis. After extensive analysis, they identified 11 microRNAs, which varied significantly between endometriosis patients and healthy controls and could be used as part of a panel to detect endometriosis. The panel performed well in their validation sample of 60 women with endometriosis and 30 controls, with a sensitivity of 92% and a specificity of 96%.
That validation is good news, but it is still far from being clinically useful. First, the model needs to be tested further in their own predominantly Caucasian population. Then, it needs to be validated by outside researchers, and the model will almost certainly have to be altered to account for different genetic backgrounds, lifestyle habits, and other factors, Arne Vanhie, MD, of Leuven University Hospital said at the World Congress on Endometriosis.
“We may have to tweak it a bit,” he said.
Unfortunately, that tweaking is likely to be time consuming. That’s because RNA extraction and sequencing is labor intensive and expensive, though it could be readily automated if it becomes commercialized.
It’s also technically challenging because microRNA is present in very low concentrations in plasma so researchers must spot a rare signal surrounded by a sea of noise, and that can mean a lot of technical refinement along the way. “We did a lot of testing and trial and error to see what was the best protocol,” Dr. Vanhie said.
The news isn’t all bad. With a well-funded, intensive effort, development could be swift. “There’s no intervention. You just need a tube of blood, so you could go forward with this to make it clinically relevant very easily,” Dr. Vanhie said.
The study was sponsored by the Belgian government. Dr. Vanhie reported having no financial disclosures.
AT WCE 2017
Key clinical point:
Major finding: The panel detected endometriosis with 92% sensitivity and 96% specificity.
Data source: The development sample included 120 women and a validation group of 90 women.
Disclosures: The study was sponsored by the Belgian government. Dr. Vanhie reported having no financial disclosures.
CBO: Demand for primary care services continues to grow
Demand for primary care services will rise by 18% between 2013 and 2023 under current law conditions, according to a report from Congressional Budget Office.
“That increase is caused primarily by growth in the size of the population,” CBO analysts said in the report. “Larger gains in health insurance coverage and more rapid aging of the population explain why the increase is expected to be larger than the 15.5% rise of the previous decade.”
While the report assumes continuation of Affordable Care Act programs, insurance coverage is the smallest component of the 18% rise, according to the CBO.
Drivers of the increase include:
- Population growth (8.7%).
- Growth in volume of care and intensity of care (4%).
- Population aging (3.2%).
- Gains in health insurance coverage (2.1%).
The analysis echoes modeling of primary care demand done by the American Academy of Family Physicians.
The CBO report does not offer a specific forecast on whether the supply of primary care physicians will meet the increased demand, but it does offer a number of policy options to increase the supply of primary care doctors.
“Such options include paying more for primary care through Medicare or Medicaid, subsidizing more residencies in primary care, helping repay loans held by medical students who agree to pursue primary care, and making it easier for foreign doctors to practice in the United States,” the report states.
Dr. Bazemore concurred with the options. “I think that is a very reasonable summary,” he said.
In particular, he noted that the payment discrepancy between primary and specialty care is a key concern when it comes to primary care shortage.
“If you have big gaps in income between subspecialists and primary care, you have more students making the former choice rather than the latter,” he said. The gap could be closed many ways, such as providing primary care bonus payments, revaluing the evaluation and management codes, blended payment models, or alternate payment models that are coming out that allow primary care providers to share in savings for improving population health.
“If you can’t narrow that gap, you won’t get more primary care physicians,” he said.
The CBO analysis is based on insurance coverage projections as of March 2016 and assumed continuation of Affordable Care Act programs. The authors noted that “changes to the ACA, including repeal or replacement, would yield different levels of insurance coverage and thus different effects on demand for primary care.”
Demand for primary care services will rise by 18% between 2013 and 2023 under current law conditions, according to a report from Congressional Budget Office.
“That increase is caused primarily by growth in the size of the population,” CBO analysts said in the report. “Larger gains in health insurance coverage and more rapid aging of the population explain why the increase is expected to be larger than the 15.5% rise of the previous decade.”
While the report assumes continuation of Affordable Care Act programs, insurance coverage is the smallest component of the 18% rise, according to the CBO.
Drivers of the increase include:
- Population growth (8.7%).
- Growth in volume of care and intensity of care (4%).
- Population aging (3.2%).
- Gains in health insurance coverage (2.1%).
The analysis echoes modeling of primary care demand done by the American Academy of Family Physicians.
The CBO report does not offer a specific forecast on whether the supply of primary care physicians will meet the increased demand, but it does offer a number of policy options to increase the supply of primary care doctors.
“Such options include paying more for primary care through Medicare or Medicaid, subsidizing more residencies in primary care, helping repay loans held by medical students who agree to pursue primary care, and making it easier for foreign doctors to practice in the United States,” the report states.
Dr. Bazemore concurred with the options. “I think that is a very reasonable summary,” he said.
In particular, he noted that the payment discrepancy between primary and specialty care is a key concern when it comes to primary care shortage.
“If you have big gaps in income between subspecialists and primary care, you have more students making the former choice rather than the latter,” he said. The gap could be closed many ways, such as providing primary care bonus payments, revaluing the evaluation and management codes, blended payment models, or alternate payment models that are coming out that allow primary care providers to share in savings for improving population health.
“If you can’t narrow that gap, you won’t get more primary care physicians,” he said.
The CBO analysis is based on insurance coverage projections as of March 2016 and assumed continuation of Affordable Care Act programs. The authors noted that “changes to the ACA, including repeal or replacement, would yield different levels of insurance coverage and thus different effects on demand for primary care.”
Demand for primary care services will rise by 18% between 2013 and 2023 under current law conditions, according to a report from Congressional Budget Office.
“That increase is caused primarily by growth in the size of the population,” CBO analysts said in the report. “Larger gains in health insurance coverage and more rapid aging of the population explain why the increase is expected to be larger than the 15.5% rise of the previous decade.”
While the report assumes continuation of Affordable Care Act programs, insurance coverage is the smallest component of the 18% rise, according to the CBO.
Drivers of the increase include:
- Population growth (8.7%).
- Growth in volume of care and intensity of care (4%).
- Population aging (3.2%).
- Gains in health insurance coverage (2.1%).
The analysis echoes modeling of primary care demand done by the American Academy of Family Physicians.
The CBO report does not offer a specific forecast on whether the supply of primary care physicians will meet the increased demand, but it does offer a number of policy options to increase the supply of primary care doctors.
“Such options include paying more for primary care through Medicare or Medicaid, subsidizing more residencies in primary care, helping repay loans held by medical students who agree to pursue primary care, and making it easier for foreign doctors to practice in the United States,” the report states.
Dr. Bazemore concurred with the options. “I think that is a very reasonable summary,” he said.
In particular, he noted that the payment discrepancy between primary and specialty care is a key concern when it comes to primary care shortage.
“If you have big gaps in income between subspecialists and primary care, you have more students making the former choice rather than the latter,” he said. The gap could be closed many ways, such as providing primary care bonus payments, revaluing the evaluation and management codes, blended payment models, or alternate payment models that are coming out that allow primary care providers to share in savings for improving population health.
“If you can’t narrow that gap, you won’t get more primary care physicians,” he said.
The CBO analysis is based on insurance coverage projections as of March 2016 and assumed continuation of Affordable Care Act programs. The authors noted that “changes to the ACA, including repeal or replacement, would yield different levels of insurance coverage and thus different effects on demand for primary care.”

Purpuric Lesions of the Scalp, Axillae, and Groin of an Infant
The Diagnosis: Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is a clonal proliferative disorder of Langerhans cells that can affect any organ, most commonly the skin and bones. It typically develops in children aged 1 to 3 years, with a male to female ratio of 2 to 1.1 Skin manifestations include purpuric papules, pustules, vesicles, erosions, and fissuring distributed predominantly on the scalp and flexural sites. Mucosal sites, particularly the oral mucosa, may be involved and usually present as erosions associated with underlying bone lesions.1 Langerhans cell histiocytosis should be considered in the differential diagnosis of recalcitrant diaper dermatitis in an infant, especially when there is purpura and erosions, as seen in our patient. Common conditions in infants such as cutaneous candidiasis (intense erythema with superficial erosions, peripheral scale and satellite pustules on flexural areas, potassium hydroxide microscopy revealing yeast forms and pseudohyphae) and seborrheic dermatitis (well-defined pink to red, moist, and often scaly patches favoring the folds) may be distinguished clinically from Hailey-Hailey disease (malodorous plaques with fissures and erosions favoring the folds), which is rare in infancy, and acrodermatitis enteropathica (erythema and erosions with scale-crust and desquamation on periorificial, acral, and intertriginous skin).
Histopathologic evaluation is instrumental in diagnosing the skin lesions of LCH. Further evaluation for systemic involvement is necessary once the diagnosis is made. Skin biopsy of the scalp and right inguinal fold revealed a wedge-shaped infiltrate of histiocytes with slightly folded nuclear contours in our patient (Figure 1). CD1a (Figure 2) and S-100 stains were markedly positive, which is characteristic of LCH. Complete blood cell count, renal function, liver function, urinalysis, and flow cytometry results were within reference range. A skeletal survey and echocardiogram were unremarkable; however, mild hepatosplenomegaly was noted on abdominal ultrasonography.
Treatment of LCH varies based on the extent of organ involvement. For isolated cutaneous disease, topical steroids, topical nitrogen mustard, phototherapy, and thalidomide may be employed.2 Multisystem disease requires chemotherapeutic agents including vinblastine and prednisone.2,3 Because more than half of patients with LCH have oncogenic BRAF V600E mutations,4 vemurafenib may have a therapeutic role in treatment. Rare case reports have documented disease response in patients with LCH and Erdheim-Chester disease.5,6
Prognosis varies based on age and extent of systemic involvement. Children younger than 2 years with multiorgan involvement have a poor prognosis (35%-55% mortality rate) compared to older children without hematopoietic, hepatosplenic, or lung involvement (100% survival rate). Additionally, response to treatment affects prognosis, as there is a 66% mortality rate in those who do not respond to treatment after 6 weeks.3 Long-term sequelae of LCH include endocrine dysfunction (ie, diabetes insipidus, growth hormone deficiencies), hearing impairment, orthopedic impairment, and neuropsychological disease; thus, multidisciplinary care often is neccessary.7
Given the multisystem involvement in our patient, he was treated with vinblastine, 6-mercaptopurine, and prednisolone with only partial and transient disease response. He was then treated with clofarabine with dramatic resolution of the mediastinal mass on follow-up positron emission tomography. The cutaneous lesions persisted and were managed with topical corticosteroids.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years [published online October 25, 2012]. Pediatr Blood Cancer. 2013;60:175-184.
- Gadner H, Grois N, Arico M, et al; Histiocyte Society. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138:728-734.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495-1500.
- Charles J, Beani JC, Fiandrino G, et al. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71:E97-E99.
- Martin A, Macmillan S, Murphy D, et al. Langerhans cell histiocytosis: 23 years' paediatric experience highlights severe long-term sequelae. Scott Med J. 2014;59:149-157.
The Diagnosis: Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is a clonal proliferative disorder of Langerhans cells that can affect any organ, most commonly the skin and bones. It typically develops in children aged 1 to 3 years, with a male to female ratio of 2 to 1.1 Skin manifestations include purpuric papules, pustules, vesicles, erosions, and fissuring distributed predominantly on the scalp and flexural sites. Mucosal sites, particularly the oral mucosa, may be involved and usually present as erosions associated with underlying bone lesions.1 Langerhans cell histiocytosis should be considered in the differential diagnosis of recalcitrant diaper dermatitis in an infant, especially when there is purpura and erosions, as seen in our patient. Common conditions in infants such as cutaneous candidiasis (intense erythema with superficial erosions, peripheral scale and satellite pustules on flexural areas, potassium hydroxide microscopy revealing yeast forms and pseudohyphae) and seborrheic dermatitis (well-defined pink to red, moist, and often scaly patches favoring the folds) may be distinguished clinically from Hailey-Hailey disease (malodorous plaques with fissures and erosions favoring the folds), which is rare in infancy, and acrodermatitis enteropathica (erythema and erosions with scale-crust and desquamation on periorificial, acral, and intertriginous skin).
Histopathologic evaluation is instrumental in diagnosing the skin lesions of LCH. Further evaluation for systemic involvement is necessary once the diagnosis is made. Skin biopsy of the scalp and right inguinal fold revealed a wedge-shaped infiltrate of histiocytes with slightly folded nuclear contours in our patient (Figure 1). CD1a (Figure 2) and S-100 stains were markedly positive, which is characteristic of LCH. Complete blood cell count, renal function, liver function, urinalysis, and flow cytometry results were within reference range. A skeletal survey and echocardiogram were unremarkable; however, mild hepatosplenomegaly was noted on abdominal ultrasonography.
Treatment of LCH varies based on the extent of organ involvement. For isolated cutaneous disease, topical steroids, topical nitrogen mustard, phototherapy, and thalidomide may be employed.2 Multisystem disease requires chemotherapeutic agents including vinblastine and prednisone.2,3 Because more than half of patients with LCH have oncogenic BRAF V600E mutations,4 vemurafenib may have a therapeutic role in treatment. Rare case reports have documented disease response in patients with LCH and Erdheim-Chester disease.5,6
Prognosis varies based on age and extent of systemic involvement. Children younger than 2 years with multiorgan involvement have a poor prognosis (35%-55% mortality rate) compared to older children without hematopoietic, hepatosplenic, or lung involvement (100% survival rate). Additionally, response to treatment affects prognosis, as there is a 66% mortality rate in those who do not respond to treatment after 6 weeks.3 Long-term sequelae of LCH include endocrine dysfunction (ie, diabetes insipidus, growth hormone deficiencies), hearing impairment, orthopedic impairment, and neuropsychological disease; thus, multidisciplinary care often is neccessary.7
Given the multisystem involvement in our patient, he was treated with vinblastine, 6-mercaptopurine, and prednisolone with only partial and transient disease response. He was then treated with clofarabine with dramatic resolution of the mediastinal mass on follow-up positron emission tomography. The cutaneous lesions persisted and were managed with topical corticosteroids.
The Diagnosis: Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is a clonal proliferative disorder of Langerhans cells that can affect any organ, most commonly the skin and bones. It typically develops in children aged 1 to 3 years, with a male to female ratio of 2 to 1.1 Skin manifestations include purpuric papules, pustules, vesicles, erosions, and fissuring distributed predominantly on the scalp and flexural sites. Mucosal sites, particularly the oral mucosa, may be involved and usually present as erosions associated with underlying bone lesions.1 Langerhans cell histiocytosis should be considered in the differential diagnosis of recalcitrant diaper dermatitis in an infant, especially when there is purpura and erosions, as seen in our patient. Common conditions in infants such as cutaneous candidiasis (intense erythema with superficial erosions, peripheral scale and satellite pustules on flexural areas, potassium hydroxide microscopy revealing yeast forms and pseudohyphae) and seborrheic dermatitis (well-defined pink to red, moist, and often scaly patches favoring the folds) may be distinguished clinically from Hailey-Hailey disease (malodorous plaques with fissures and erosions favoring the folds), which is rare in infancy, and acrodermatitis enteropathica (erythema and erosions with scale-crust and desquamation on periorificial, acral, and intertriginous skin).
Histopathologic evaluation is instrumental in diagnosing the skin lesions of LCH. Further evaluation for systemic involvement is necessary once the diagnosis is made. Skin biopsy of the scalp and right inguinal fold revealed a wedge-shaped infiltrate of histiocytes with slightly folded nuclear contours in our patient (Figure 1). CD1a (Figure 2) and S-100 stains were markedly positive, which is characteristic of LCH. Complete blood cell count, renal function, liver function, urinalysis, and flow cytometry results were within reference range. A skeletal survey and echocardiogram were unremarkable; however, mild hepatosplenomegaly was noted on abdominal ultrasonography.
Treatment of LCH varies based on the extent of organ involvement. For isolated cutaneous disease, topical steroids, topical nitrogen mustard, phototherapy, and thalidomide may be employed.2 Multisystem disease requires chemotherapeutic agents including vinblastine and prednisone.2,3 Because more than half of patients with LCH have oncogenic BRAF V600E mutations,4 vemurafenib may have a therapeutic role in treatment. Rare case reports have documented disease response in patients with LCH and Erdheim-Chester disease.5,6
Prognosis varies based on age and extent of systemic involvement. Children younger than 2 years with multiorgan involvement have a poor prognosis (35%-55% mortality rate) compared to older children without hematopoietic, hepatosplenic, or lung involvement (100% survival rate). Additionally, response to treatment affects prognosis, as there is a 66% mortality rate in those who do not respond to treatment after 6 weeks.3 Long-term sequelae of LCH include endocrine dysfunction (ie, diabetes insipidus, growth hormone deficiencies), hearing impairment, orthopedic impairment, and neuropsychological disease; thus, multidisciplinary care often is neccessary.7
Given the multisystem involvement in our patient, he was treated with vinblastine, 6-mercaptopurine, and prednisolone with only partial and transient disease response. He was then treated with clofarabine with dramatic resolution of the mediastinal mass on follow-up positron emission tomography. The cutaneous lesions persisted and were managed with topical corticosteroids.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years [published online October 25, 2012]. Pediatr Blood Cancer. 2013;60:175-184.
- Gadner H, Grois N, Arico M, et al; Histiocyte Society. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138:728-734.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495-1500.
- Charles J, Beani JC, Fiandrino G, et al. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71:E97-E99.
- Martin A, Macmillan S, Murphy D, et al. Langerhans cell histiocytosis: 23 years' paediatric experience highlights severe long-term sequelae. Scott Med J. 2014;59:149-157.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years [published online October 25, 2012]. Pediatr Blood Cancer. 2013;60:175-184.
- Gadner H, Grois N, Arico M, et al; Histiocyte Society. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001;138:728-734.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495-1500.
- Charles J, Beani JC, Fiandrino G, et al. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71:E97-E99.
- Martin A, Macmillan S, Murphy D, et al. Langerhans cell histiocytosis: 23 years' paediatric experience highlights severe long-term sequelae. Scott Med J. 2014;59:149-157.
A 7-month-old boy admitted to the hospital with new-onset respiratory stridor was found to have a rash of the scalp, axillae, and groin of 1 month's duration that was unresponsive to treatment with mineral oil. Bronchoscopy revealed tracheal compression, and urgent magnetic resonance imaging of the chest demonstrated an anterior mediastinal mass. Prior to presentation, the patient was otherwise healthy with normal growth and development. On physical examination, scattered red-brown and purpuric papules with hemorrhagic crust were noted on the scalp. There were well-defined pink erosive patches and purpuric papules in the inguinal folds bilaterally and similar erosive patches in the axillae. Numerous punched out ulcerations were noted on the lower gingiva. There was no palpable lymphadenopathy. The hands, feet, penis, scrotum, and perianal area were spared. Biopsies of the skin and mediastinal mass were performed.