User login
Preventing thrombosis without increasing bleeding risk

It may be possible to disrupt thrombosis without increasing the risk of bleeding, according to preclinical research published in Nature Communications.
“We have found a new thrombosis target that does not increase bleeding risk,” said study author Daniel I. Simon, MD, of University Hospitals Cleveland Medical Center in Cleveland, Ohio.
“Our discovery indicates that you can identify a new pathway and target that mediates blood clotting but does not affect our body’s natural processes to stop bleeding.”
The new pathway centers around a pair of protein receptors. One—Mac-1—is found on the surface of leukocytes recruited to sites of blood vessel injury, and the other—GPIbα—resides on the surface of platelets.
When the receptors interact, they trigger cascades of signals that amplify both inflammation and clotting.
The researchers found that genetically engineered mice, either without the Mac-1 receptor or with a mutant form of the receptor, could not bind GPIbα on platelets. As a result, the mice had delayed clot formation in response to artery injury.
However, these mice had similar platelet counts, platelet activation, plasma coagulation activity, and bleeding time as wild-type mice.
Additional experiments in mice showed that an antibody targeting Mac-1:GPIba inhibits thrombus formation.
And glucosamine, a small-molecule inhibitor of Mac-1:GPIba binding, inhibits thrombus formation without increasing bleeding risk.
Mice exposed to glucosamine were still able to successfully stop minor bleeding, like tail cuts, and maintain normal coagulation and platelet function.
The researchers believe these findings could lead to the development of better antithrombotic agents, as “the interaction between leukocyte Mac-1 and platelet GPIba is positioned as a novel and targetable mediator of thrombosis but not hemostasis.”
“Current anticlotting drugs and antiplatelet agents are effective in reducing heart attack and stroke but are associated with increased bleeding and transfusion,” Dr Simon said. “We have learned that bleeding and transfusion complications are equally as bad from a prognosis standpoint as heart attack or stroke.” ![]()
It may be possible to disrupt thrombosis without increasing the risk of bleeding, according to preclinical research published in Nature Communications.
“We have found a new thrombosis target that does not increase bleeding risk,” said study author Daniel I. Simon, MD, of University Hospitals Cleveland Medical Center in Cleveland, Ohio.
“Our discovery indicates that you can identify a new pathway and target that mediates blood clotting but does not affect our body’s natural processes to stop bleeding.”
The new pathway centers around a pair of protein receptors. One—Mac-1—is found on the surface of leukocytes recruited to sites of blood vessel injury, and the other—GPIbα—resides on the surface of platelets.
When the receptors interact, they trigger cascades of signals that amplify both inflammation and clotting.
The researchers found that genetically engineered mice, either without the Mac-1 receptor or with a mutant form of the receptor, could not bind GPIbα on platelets. As a result, the mice had delayed clot formation in response to artery injury.
However, these mice had similar platelet counts, platelet activation, plasma coagulation activity, and bleeding time as wild-type mice.
Additional experiments in mice showed that an antibody targeting Mac-1:GPIba inhibits thrombus formation.
And glucosamine, a small-molecule inhibitor of Mac-1:GPIba binding, inhibits thrombus formation without increasing bleeding risk.
Mice exposed to glucosamine were still able to successfully stop minor bleeding, like tail cuts, and maintain normal coagulation and platelet function.
The researchers believe these findings could lead to the development of better antithrombotic agents, as “the interaction between leukocyte Mac-1 and platelet GPIba is positioned as a novel and targetable mediator of thrombosis but not hemostasis.”
“Current anticlotting drugs and antiplatelet agents are effective in reducing heart attack and stroke but are associated with increased bleeding and transfusion,” Dr Simon said. “We have learned that bleeding and transfusion complications are equally as bad from a prognosis standpoint as heart attack or stroke.” ![]()
It may be possible to disrupt thrombosis without increasing the risk of bleeding, according to preclinical research published in Nature Communications.
“We have found a new thrombosis target that does not increase bleeding risk,” said study author Daniel I. Simon, MD, of University Hospitals Cleveland Medical Center in Cleveland, Ohio.
“Our discovery indicates that you can identify a new pathway and target that mediates blood clotting but does not affect our body’s natural processes to stop bleeding.”
The new pathway centers around a pair of protein receptors. One—Mac-1—is found on the surface of leukocytes recruited to sites of blood vessel injury, and the other—GPIbα—resides on the surface of platelets.
When the receptors interact, they trigger cascades of signals that amplify both inflammation and clotting.
The researchers found that genetically engineered mice, either without the Mac-1 receptor or with a mutant form of the receptor, could not bind GPIbα on platelets. As a result, the mice had delayed clot formation in response to artery injury.
However, these mice had similar platelet counts, platelet activation, plasma coagulation activity, and bleeding time as wild-type mice.
Additional experiments in mice showed that an antibody targeting Mac-1:GPIba inhibits thrombus formation.
And glucosamine, a small-molecule inhibitor of Mac-1:GPIba binding, inhibits thrombus formation without increasing bleeding risk.
Mice exposed to glucosamine were still able to successfully stop minor bleeding, like tail cuts, and maintain normal coagulation and platelet function.
The researchers believe these findings could lead to the development of better antithrombotic agents, as “the interaction between leukocyte Mac-1 and platelet GPIba is positioned as a novel and targetable mediator of thrombosis but not hemostasis.”
“Current anticlotting drugs and antiplatelet agents are effective in reducing heart attack and stroke but are associated with increased bleeding and transfusion,” Dr Simon said. “We have learned that bleeding and transfusion complications are equally as bad from a prognosis standpoint as heart attack or stroke.” ![]()
Gene plays key role in iron homeostasis

A gene known to prevent autoimmune diseases is a key regulator in iron uptake, according to research published in Cell Reports.
“We found previously that, when mice lack the gene Regnase-1, they suffer from severe autoimmune diseases and anemia,” said study author Masanori Yoshinaga, MD, of Kyoto University in Japan.
“At first, we assumed that anemia was a secondary effect, but, after detailed analysis, we found that the 2 symptoms develop independently.”
Continued study of mice with a Regnase-1 mutation revealed a functional defect in the principal site for iron absorption in the body, the duodenum.
“The next step was to find the role of Regnase-1 in iron-uptake maintenance,” Dr Yoshinaga said. “We started by looking at the most important iron-uptake gene, Transferrin Receptor 1, or TfR1.”
“Our results showed that Regnase-1 degrades the mRNA of TfR1, thereby inhibiting the synthesis of the TfR1 protein and, additionally, that it likely regulates other important iron-controlling genes.”
“Further analysis of Regnase-1 in iron-related homeostasis may provide insight into the mechanisms causing anemia and other iron-related disorders, perhaps eventually leading to new methods of treatment,” said study author Osamu Takeuchi, MD, PhD, of Kyoto University. ![]()
A gene known to prevent autoimmune diseases is a key regulator in iron uptake, according to research published in Cell Reports.
“We found previously that, when mice lack the gene Regnase-1, they suffer from severe autoimmune diseases and anemia,” said study author Masanori Yoshinaga, MD, of Kyoto University in Japan.
“At first, we assumed that anemia was a secondary effect, but, after detailed analysis, we found that the 2 symptoms develop independently.”
Continued study of mice with a Regnase-1 mutation revealed a functional defect in the principal site for iron absorption in the body, the duodenum.
“The next step was to find the role of Regnase-1 in iron-uptake maintenance,” Dr Yoshinaga said. “We started by looking at the most important iron-uptake gene, Transferrin Receptor 1, or TfR1.”
“Our results showed that Regnase-1 degrades the mRNA of TfR1, thereby inhibiting the synthesis of the TfR1 protein and, additionally, that it likely regulates other important iron-controlling genes.”
“Further analysis of Regnase-1 in iron-related homeostasis may provide insight into the mechanisms causing anemia and other iron-related disorders, perhaps eventually leading to new methods of treatment,” said study author Osamu Takeuchi, MD, PhD, of Kyoto University. ![]()
A gene known to prevent autoimmune diseases is a key regulator in iron uptake, according to research published in Cell Reports.
“We found previously that, when mice lack the gene Regnase-1, they suffer from severe autoimmune diseases and anemia,” said study author Masanori Yoshinaga, MD, of Kyoto University in Japan.
“At first, we assumed that anemia was a secondary effect, but, after detailed analysis, we found that the 2 symptoms develop independently.”
Continued study of mice with a Regnase-1 mutation revealed a functional defect in the principal site for iron absorption in the body, the duodenum.
“The next step was to find the role of Regnase-1 in iron-uptake maintenance,” Dr Yoshinaga said. “We started by looking at the most important iron-uptake gene, Transferrin Receptor 1, or TfR1.”
“Our results showed that Regnase-1 degrades the mRNA of TfR1, thereby inhibiting the synthesis of the TfR1 protein and, additionally, that it likely regulates other important iron-controlling genes.”
“Further analysis of Regnase-1 in iron-related homeostasis may provide insight into the mechanisms causing anemia and other iron-related disorders, perhaps eventually leading to new methods of treatment,” said study author Osamu Takeuchi, MD, PhD, of Kyoto University. ![]()
Stroke: A road map for subacute management
CASE › A 68-year-old woman with hypertension and hyperlipidemia comes into your office for evaluation of a 30-minute episode of sudden-onset right-hand weakness and difficulty speaking that occurred 4 days earlier. The patient, who is also a smoker, has come in at the insistence of her daughter. On examination, her blood pressure (BP) is 145/88 mm Hg and her heart rate is 76 beats/minute and regular. She appears well and her language function is normal. The rest of her examination is normal. How would you proceed?
Stroke—the death of nerve cells due to a lack of blood supply from either infarction or hemorrhage—strikes nearly 800,000 people in the United States every year.1,2 Of these events, 130,000 are fatal, making stroke the fifth leading cause of death.3 Effective, early evaluation and cause-specific treatment are crucial parts of stroke care.
Research has helped to clarify the critical role primary care physicians play in recognizing, triaging, and managing stroke and transient ischemic attacks (TIA). This article reviews what we know about the different ways that a stroke and a TIA can present, the appropriate diagnostic work-up for patients presenting with symptoms of either event, and management strategies for subacute care (24 hours to up to 14 days after a stroke has occurred).4,5 Unless otherwise specified, this review will focus on ischemic stroke because 87% of strokes are attributable to ischemia.1
A follow-up to this article on secondary stroke prevention will appear in the journal next month.
Look to onset more than type of symptoms for clues
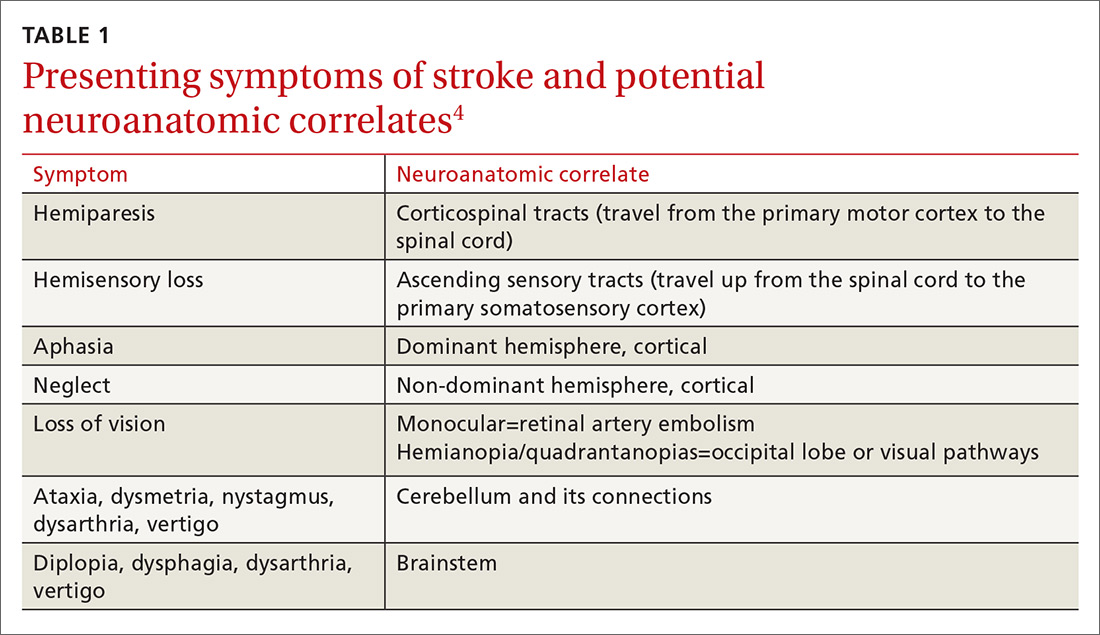
Stroke presents as a sudden onset of neurologic deficits (language, motor, sensory, cerebellar, or brainstem functions) (TABLE 14). Because presenting symptoms can vary widely, sudden onset, rather than particular symptoms, should raise a red flag for potential stroke.
The differential diagnosis includes: seizure, complex migraine, medication effect (eg, slurred speech or confusion after taking a central nervous system [CNS] depressant), toxin exposure, electrolyte abnormalities (especially hypoglycemia), concussion/trauma, infection of the CNS, peripheral vertigo, demyelination, intracranial mass, Bell’s palsy, and psychogenic disorders. The history and physical, along with laboratory findings and brain imaging (detailed later in this article), will guide the FP toward (or away from) these various etiologies.
Optimal triage is a subject of ongoing interest and research
If stroke or TIA remains a possibility after an initial assessment, it’s time to stratify patients by risk.
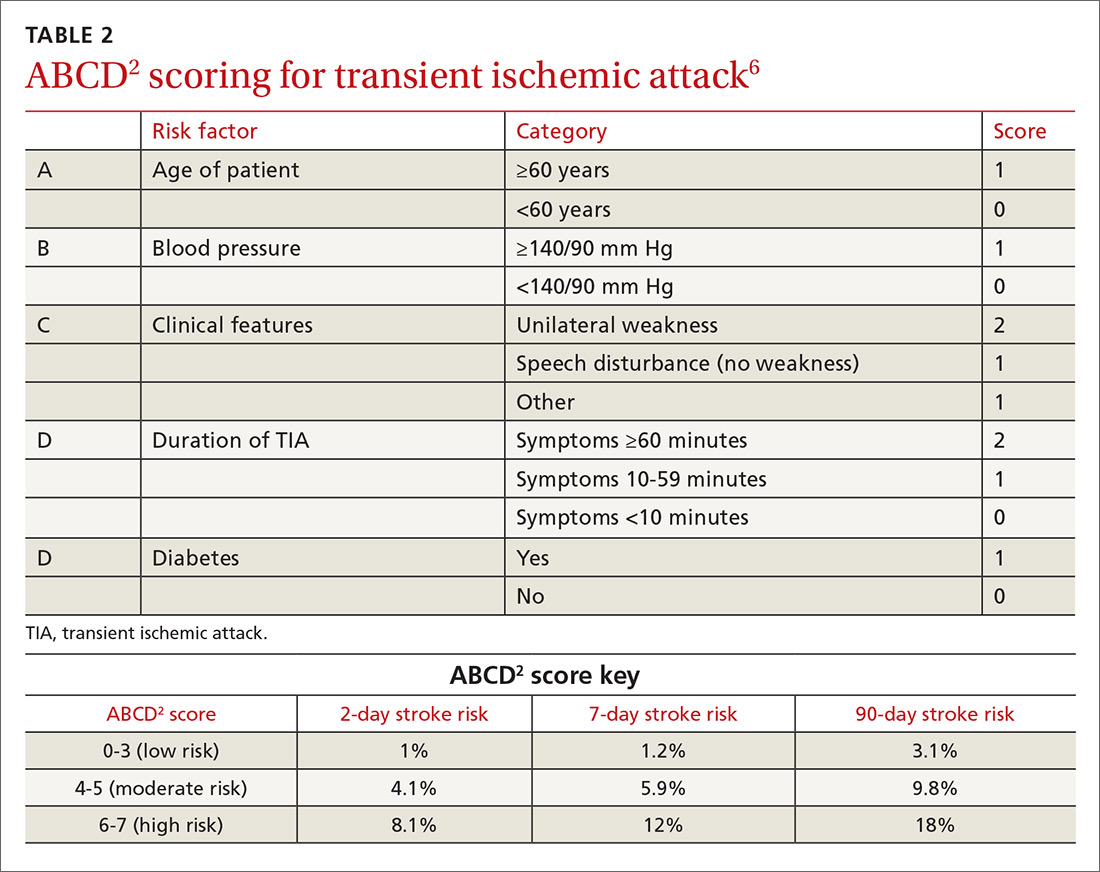
One of the most widely accepted tools is the ABCD2 score (see TABLE 26). Clinicians can employ the ABCD2 risk stratification tool when trying to determine whether it is reasonable to pursue an expedited work-up (ie, <1 day) in the outpatient setting or recommend that the patient be evaluated in an emergency department (ED). The 90-day stroke rate following a TIA ranges from 3% with an ABCD2 score of 0 to 3 to 18% with a score of 6 or 7. A score of 0 to 3 is considered relatively low risk; in the absence of other compelling factors, rapid outpatient evaluation is appropriate. For patients with an ABCD2 score ≥4, referral to the ED or direct admission to the hospital is advised.
The validity of the ABCD2 score for risk stratification has been studied extensively with conflicting results.7-10 As with any assessment tool, it should be used as a guide, and should not supplant a full assessment of the patient or the judgment of the examining physician. In making the decision regarding inpatient or outpatient evaluation, it’s also important to consider available resources, access to specialists, and patient preference.
In a 2016 population-based study, the 30-day recurrent stroke/TIA rate for patients hospitalized for TIA was 3% compared with 10.7% for those discharged from the ED with referral to a stroke clinic and 10.6% for those discharged from the ED without a referral to a stroke clinic.11 These data suggest that only patients for whom you have a low clinical suspicion of stroke/TIA should be worked up as outpatients, and that hospital admission is advised in moderate- and high-risk cases. The findings also highlight the critical role that primary care physicians can play in triaging and managing these patients for secondary prevention.
CASE › This patient’s recent history of sudden-onset right-sided weakness and expressive language dysfunction is suspicious for left hemispheric ischemia. She has several risk factors for stroke, and her ABCD2 score is 5 (hypertension, age ≥60 years, unilateral weakness, and duration 10-59 min), which places her at moderate risk. Thus, the recommendation would be to have her go directly to an ED for rapid evaluation.
The diagnostic work-up
Even when a patient is sent to the ED, the FP plays a critical role in his or her continuing care. FPs will often coordinate with inpatient care and manage transition of care to the outpatient setting. (And in many communities, the ED or hospital physicians may themselves be family practitioners.)
In terms of care, not even an aspirin should be administered in a case like this because the patient has not yet had any neuroimaging, and differentiation of ischemic from hemorrhagic stroke cannot be made on clinical grounds alone. Once an ischemic stroke is confirmed, determining the etiology is critical given the significant management differences between the different types of stroke (atherosclerotic, cardioembolic, lacunar, or other).
Which imaging method, and when?
While a computerized tomography (CT) scan is the preferred initial imaging strategy for acute stroke to discern the ischemic type from the hemorrhagic, MRI is preferred for the evaluation of acute ischemic stroke because the method has a higher sensitivity for infarction and a greater ability to identify findings (such as demyelination) that would suggest an alternative diagnosis.
In addition to evaluating the brain parenchyma, physicians must also assess the cerebral vasculature. CT angiography (CTA) or MR angiography (MRA) of the head and neck are preferred over carotid ultrasound because they are capable of evaluating the entire cerebrovascular system12,13 and can be instrumental in identifying potential causes of stroke, as well as guiding therapeutic decisions. Carotid ultrasound is a reasonable alternative for patients presenting with symptoms indicative of anterior circulation involvement when CTA and MRA are unavailable or contraindicated, but it will not identify intracranial vascular disease, proximal common carotid disease, or vertebrobasilar disease.
Getting to the cause of suspected stroke: Labs and other diagnostic tests
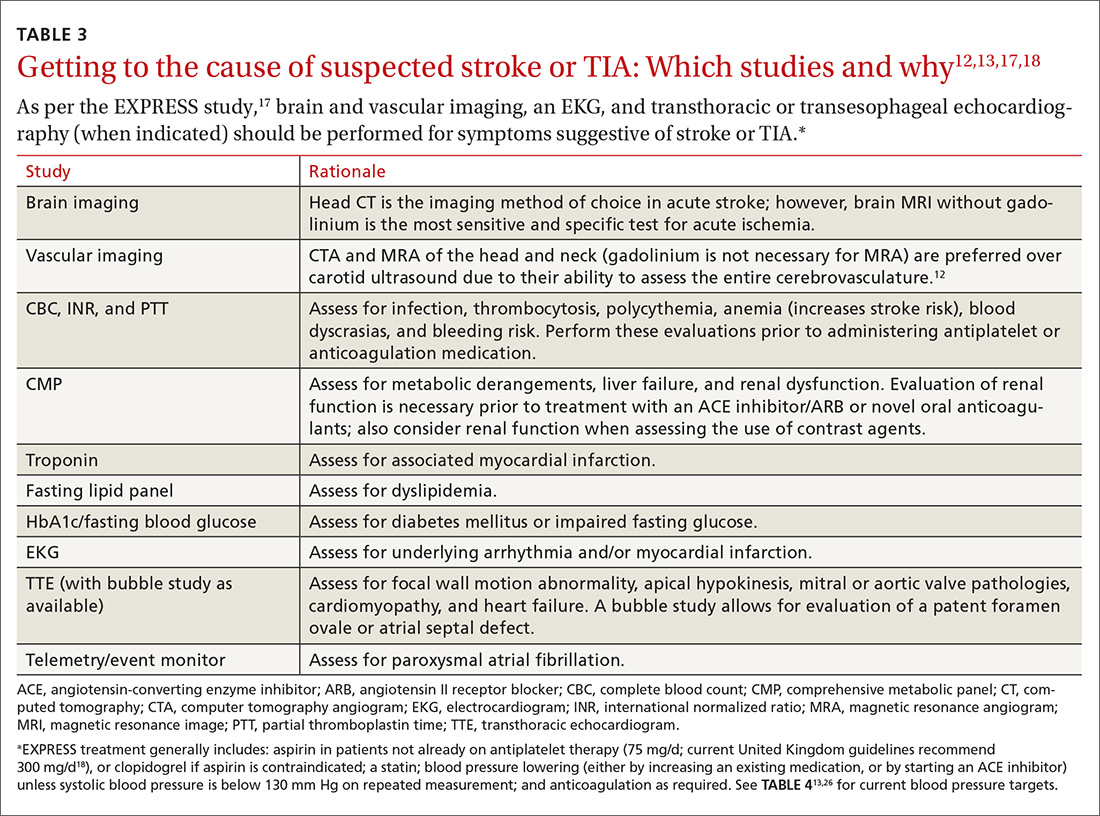
A routine work-up includes BP checks, routine labs (complete blood count, complete metabolic panel, coagulation profile, and troponin), an electrocardiogram (EKG), a transthoracic echocardiogram (TTE) with bubble study if possible, and a minimum of 24 to 48 hours of cardiac rhythm monitoring. Cardiac rhythm monitoring should be extended in the setting of clinical concern for unidentified paroxysmal atrial fibrillation, such as an embolism without a proximal vascular source, multiple embolic infarcts in different vascular territories, a dilated left atrium, or other risk factors for atrial fibrillation that include smoking, systolic hypertension, diabetes, and heart failure (see TABLE 312,13,17,18).14-16 This standard diagnostic work-up will identify the cause of stroke in 70% to 80% of patients.19
Additional investigations to consider if the etiology is not yet elucidated include a transesophageal echocardiogram (TEE), cerebral angiography, a coagulopathy evaluation, a lumbar puncture, and a vasculitis work-up. If available, consultation with a neurologist is appropriate for any patient who has had a stroke or TIA. Patients with unclear etiologies or for whom there are questions concerning strategies for preventing secondary stroke should be referred to Neurology and preferably a stroke specialist.
Timing matters, even when symptoms have resolved (ie, TIA).11,20 The EXPRESS trial17 (the Early use of eXisting PREventive Strategies for Stroke) looked at the effect of urgent assessment and treatment (≤1 day) of patients presenting with a TIA or minor stroke on the risk of recurrent stroke within 90 days. The diagnostic work-up included brain and vascular imaging together with an EKG. This intensive approach led to an absolute risk reduction of 8.2% (from 10.3% to 2.1%) in the risk of recurrent stroke at 90 days (number needed to treat [NNT]=12).17
Expedited work-up and treatment was also recently evaluated in a non-trial, real-world setting and was associated with reducing recurrent stroke by more than half the rate reported in older studies.20 Overall, the data suggest that evaluation within 24 hours confers substantial benefit, and that this evaluation can happen in an outpatient setting.21-23
Acute management: Use of tPA
Once imaging rules out intracranial hemorrhage, patients should be treated with tissue plasminogen activator (tPA) or an endovascular intervention as per guidelines.24 For patients with ischemic stroke ineligible for tPA or endovascular treatments, the initial focus is to determine the etiology of the symptoms so that the best strategies for prevention of secondary stroke may be employed.
Aspirin should be provided within 24 to 48 hours to all patients after intracranial hemorrhage is ruled out. Aspirin should be delayed for 24 hours in those given thrombolytics. The initial recommended dose of aspirin is 325 mg with continued low-dose (81 mg) aspirin daily.13 The addition of clopidogrel to aspirin within 24 hours of an event and continued for 21 days, followed by aspirin alone, was shown to be beneficial in a Chinese population with high-risk TIA (ABCD2 score ≥4) or minor stroke (National Institutes of Health Stroke Scale [NIHSS] ≤3).25 Anticoagulation with heparin, warfarin, or a novel oral anticoagulant is generally not indicated in the acute setting due to the risk of hemorrhagic transformation.
Acute BP management depends upon the type of stroke (ischemic or hemorrhagic), eligibility for thrombolytics, timing of presentation, and possible comorbidities such as myocardial infarction or aortic dissection (see TABLE 413,26). In the absence of contraindications, high-intensity statins should be initiated in all patients able to take oral medications.

CASE › You appropriately referred your patient to the local ED. A head CT with head and neck CTA was performed. While the head CT did not show any abnormalities, the CTA demonstrated high-grade left internal carotid artery stenosis. The patient was given an initial dose of aspirin 325 mg and a high-intensity statin and admitted for further management. An MRI revealed a small shower of emboli in the left hemisphere, confirming the diagnosis of stroke over TIA. Labs were marginally remarkable with a low-density lipoprotein level of 115 mg/dL and an HbA1c of 6.2. Telemetry monitoring did not reveal any arrhythmias, and TTE was normal. BP remained in the high-normal to low-hypertensive range.
A Vascular Surgery consultation was obtained and the patient underwent a left carotid endarterectomy the following day. She did well without surgical complications. Her BP medications were adjusted; a combination of an angiotensin-converting enzyme inhibitor and a thiazide diuretic achieved a goal BP <140/90 mm Hg.
Permissive hypertension was not indicated due to her presentation >48 hours beyond the acute event. Low-dose aspirin and a high-intensity statin were continued, for secondary stroke prevention in the setting of atherosclerotic disease. She received smoking cessation counseling, which will continue.
CORRESPONDENCE
Stephen A. Martin, MD, EdM, Barre Family Health Center, 151 Worcester Road, Barre, MA 01005; [email protected].
1. Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135:e146-e603.
2. Sacco RL, Kasner SE, Broderick JP, et al. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:2064-2089.
3. Kochanek KD, Murphy SL, Xu J, et al. Mortality in the United States, 2013. NCHS Data Brief. 2014:1-8. Available at: https://www.cdc.gov/nchs/data/databriefs/db178.pdf. Accessed June 5, 2016.
4. Flossmann E, Redgrave JN, Briley D, et al. Reliability of clinical diagnosis of the symptomatic vascular territory in patients with recent transient ischemic attack or minor stroke. Stroke. 2008;39:2457-2460.
5. Josephson SA, Sidney S, Pham TN, et al. Higher ABCD2 score predicts patients most likely to have true transient ischemic attack. Stroke. 2008;39:3096-3098. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18688003. Accessed June 5, 2016.
6. Hankey GJ. The ABCD, California, and unified ABCD2 risk scores predicted stroke within 2, 7, and 90 days after TIA. Evid Based Med. 2007;12:88.
7. Sheehan OC, Kyne L, Kelly LA, et al. Population-based study of ABCD2 score, carotid stenosis, and atrial fibrillation for early stroke prediction after transient ischemic attack: the North Dublin TIA study. Stroke. 2010;41:844-850.
8. Rothwell PM, Giles MF, Flossmann E, et al. A simple score (ABCD) to identify individuals at high early risk of stroke after transient ischaemic attack. Lancet. 2005; 366:29-36.
9. Tsivgoulis G, Spengos K, Manta P, et al. Validation of the ABCD score in identifying individuals at high early risk of stroke after a transient ischemic attack: a hospital-based case series study. Stroke. 2006;37:2892-2897.
10. Kiyohara T, Kamouchi M, Kumai Y, et al. ABCD3 and ABCD3-I scores are superior to ABCD2 score in the prediction of short- and long-term risks of stroke after transient ischemic attack. Stroke. 2014;45:418-425.
11. Sacco RL, Rundek T. The value of urgent specialized care for TIA and minor stroke. N Engl J Med. 2016;374:1577-1579.
12. Demchuk AM, Menon BK, Goyal M. Comparing vessel imaging: noncontrast computed tomography/computed tomographic angiography should be the new minimum standard in acute disabling stroke. Stroke. 2016;47:273-281.
13. Jauch EC, Saver JL, Adams HP, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:870-947.
14. Gladstone DJ, Spring M, Dorian P, et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370:2467-2477.
15. Sanna T, Diener HC, Passman RS, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. 2014;370:2478-2486.
16. Christophersen IE, Yin X, Larson MG, et al. A comparison of the CHARGE-AF and the CHA2DS2-VASc risk scores for prediction of atrial fibrillation ni the Framingham Heart Study. Am Heart J. 2016;178:45-54.
17. Rothwell PM, Giles MF, Chandratheva A, et al. Effect of urgent treatment of transient ischaemic attack and minor stroke on early recurrent stroke (EXPRESS study): a prospective population-based sequential comparison. Lancet. 2007;370:1432-1442.
18. National Institute for Health and Care Excellence. Stroke and transient ischaemic attack in over 16s: diagnosis and initial management. Available at: https://www.nice.org.uk/guidance/cg68. Published 2008. Accessed February 5, 2017.
19. Hart RG, Diener HC, Coutts SB, et al. Embolic strokes of undetermined source: the case for a new clinical construct. Lancet Neurol. 2014;13:429-438.
20. Amarenco P, Lavallée PC, Labreuche J, et al. One-year risk of stroke after transient ischemic attack or minor stroke. N Engl J Med. 2016;374:1533-1542.
21. Joshi JK, Ouyang B, Prabhakaran S. Should TIA patients be hospitalized or referred to a same-day clinic? A decision analysis. Neurology. 2011;77:2082-2088.
22. Mijalski C, Silver B. TIA management: should TIA patients be admitted? should TIA patients get combination antiplatelet therapy? The Neurohospitalist. 2015;5:151-160.
23. Silver B, Adeoye O. Management of patients with transient ischemic attack in the emergency department. Neurology. 2016;86:1568-1569.
24. Demaerschalk BM, Kleindorfer DO, Adeoye OM, et al. Scientific rationale for the inclusion and exclusion criteria for intravenous alteplase in acute ischemic stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016;47:581-641.
25. Wang Y, Wang Y, Zhao X, et al. Clopidogrel with aspirin in acute minor stroke or transient ischemic attack. N Engl J Med. 2013;369:11-19.
26. Hemphill JC, Greenberg SM, Anderson CS, et al. Guidelines for the management of spontaneous intracerebral hemorrhage. Stroke. 2015;46:2032-2060.
CASE › A 68-year-old woman with hypertension and hyperlipidemia comes into your office for evaluation of a 30-minute episode of sudden-onset right-hand weakness and difficulty speaking that occurred 4 days earlier. The patient, who is also a smoker, has come in at the insistence of her daughter. On examination, her blood pressure (BP) is 145/88 mm Hg and her heart rate is 76 beats/minute and regular. She appears well and her language function is normal. The rest of her examination is normal. How would you proceed?
Stroke—the death of nerve cells due to a lack of blood supply from either infarction or hemorrhage—strikes nearly 800,000 people in the United States every year.1,2 Of these events, 130,000 are fatal, making stroke the fifth leading cause of death.3 Effective, early evaluation and cause-specific treatment are crucial parts of stroke care.
Research has helped to clarify the critical role primary care physicians play in recognizing, triaging, and managing stroke and transient ischemic attacks (TIA). This article reviews what we know about the different ways that a stroke and a TIA can present, the appropriate diagnostic work-up for patients presenting with symptoms of either event, and management strategies for subacute care (24 hours to up to 14 days after a stroke has occurred).4,5 Unless otherwise specified, this review will focus on ischemic stroke because 87% of strokes are attributable to ischemia.1
A follow-up to this article on secondary stroke prevention will appear in the journal next month.
Look to onset more than type of symptoms for clues
Stroke presents as a sudden onset of neurologic deficits (language, motor, sensory, cerebellar, or brainstem functions) (TABLE 14). Because presenting symptoms can vary widely, sudden onset, rather than particular symptoms, should raise a red flag for potential stroke.
The differential diagnosis includes: seizure, complex migraine, medication effect (eg, slurred speech or confusion after taking a central nervous system [CNS] depressant), toxin exposure, electrolyte abnormalities (especially hypoglycemia), concussion/trauma, infection of the CNS, peripheral vertigo, demyelination, intracranial mass, Bell’s palsy, and psychogenic disorders. The history and physical, along with laboratory findings and brain imaging (detailed later in this article), will guide the FP toward (or away from) these various etiologies.
Optimal triage is a subject of ongoing interest and research
If stroke or TIA remains a possibility after an initial assessment, it’s time to stratify patients by risk.
One of the most widely accepted tools is the ABCD2 score (see TABLE 26). Clinicians can employ the ABCD2 risk stratification tool when trying to determine whether it is reasonable to pursue an expedited work-up (ie, <1 day) in the outpatient setting or recommend that the patient be evaluated in an emergency department (ED). The 90-day stroke rate following a TIA ranges from 3% with an ABCD2 score of 0 to 3 to 18% with a score of 6 or 7. A score of 0 to 3 is considered relatively low risk; in the absence of other compelling factors, rapid outpatient evaluation is appropriate. For patients with an ABCD2 score ≥4, referral to the ED or direct admission to the hospital is advised.
The validity of the ABCD2 score for risk stratification has been studied extensively with conflicting results.7-10 As with any assessment tool, it should be used as a guide, and should not supplant a full assessment of the patient or the judgment of the examining physician. In making the decision regarding inpatient or outpatient evaluation, it’s also important to consider available resources, access to specialists, and patient preference.
In a 2016 population-based study, the 30-day recurrent stroke/TIA rate for patients hospitalized for TIA was 3% compared with 10.7% for those discharged from the ED with referral to a stroke clinic and 10.6% for those discharged from the ED without a referral to a stroke clinic.11 These data suggest that only patients for whom you have a low clinical suspicion of stroke/TIA should be worked up as outpatients, and that hospital admission is advised in moderate- and high-risk cases. The findings also highlight the critical role that primary care physicians can play in triaging and managing these patients for secondary prevention.
CASE › This patient’s recent history of sudden-onset right-sided weakness and expressive language dysfunction is suspicious for left hemispheric ischemia. She has several risk factors for stroke, and her ABCD2 score is 5 (hypertension, age ≥60 years, unilateral weakness, and duration 10-59 min), which places her at moderate risk. Thus, the recommendation would be to have her go directly to an ED for rapid evaluation.
The diagnostic work-up
Even when a patient is sent to the ED, the FP plays a critical role in his or her continuing care. FPs will often coordinate with inpatient care and manage transition of care to the outpatient setting. (And in many communities, the ED or hospital physicians may themselves be family practitioners.)
In terms of care, not even an aspirin should be administered in a case like this because the patient has not yet had any neuroimaging, and differentiation of ischemic from hemorrhagic stroke cannot be made on clinical grounds alone. Once an ischemic stroke is confirmed, determining the etiology is critical given the significant management differences between the different types of stroke (atherosclerotic, cardioembolic, lacunar, or other).
Which imaging method, and when?
While a computerized tomography (CT) scan is the preferred initial imaging strategy for acute stroke to discern the ischemic type from the hemorrhagic, MRI is preferred for the evaluation of acute ischemic stroke because the method has a higher sensitivity for infarction and a greater ability to identify findings (such as demyelination) that would suggest an alternative diagnosis.
In addition to evaluating the brain parenchyma, physicians must also assess the cerebral vasculature. CT angiography (CTA) or MR angiography (MRA) of the head and neck are preferred over carotid ultrasound because they are capable of evaluating the entire cerebrovascular system12,13 and can be instrumental in identifying potential causes of stroke, as well as guiding therapeutic decisions. Carotid ultrasound is a reasonable alternative for patients presenting with symptoms indicative of anterior circulation involvement when CTA and MRA are unavailable or contraindicated, but it will not identify intracranial vascular disease, proximal common carotid disease, or vertebrobasilar disease.
Getting to the cause of suspected stroke: Labs and other diagnostic tests
A routine work-up includes BP checks, routine labs (complete blood count, complete metabolic panel, coagulation profile, and troponin), an electrocardiogram (EKG), a transthoracic echocardiogram (TTE) with bubble study if possible, and a minimum of 24 to 48 hours of cardiac rhythm monitoring. Cardiac rhythm monitoring should be extended in the setting of clinical concern for unidentified paroxysmal atrial fibrillation, such as an embolism without a proximal vascular source, multiple embolic infarcts in different vascular territories, a dilated left atrium, or other risk factors for atrial fibrillation that include smoking, systolic hypertension, diabetes, and heart failure (see TABLE 312,13,17,18).14-16 This standard diagnostic work-up will identify the cause of stroke in 70% to 80% of patients.19
Additional investigations to consider if the etiology is not yet elucidated include a transesophageal echocardiogram (TEE), cerebral angiography, a coagulopathy evaluation, a lumbar puncture, and a vasculitis work-up. If available, consultation with a neurologist is appropriate for any patient who has had a stroke or TIA. Patients with unclear etiologies or for whom there are questions concerning strategies for preventing secondary stroke should be referred to Neurology and preferably a stroke specialist.
Timing matters, even when symptoms have resolved (ie, TIA).11,20 The EXPRESS trial17 (the Early use of eXisting PREventive Strategies for Stroke) looked at the effect of urgent assessment and treatment (≤1 day) of patients presenting with a TIA or minor stroke on the risk of recurrent stroke within 90 days. The diagnostic work-up included brain and vascular imaging together with an EKG. This intensive approach led to an absolute risk reduction of 8.2% (from 10.3% to 2.1%) in the risk of recurrent stroke at 90 days (number needed to treat [NNT]=12).17
Expedited work-up and treatment was also recently evaluated in a non-trial, real-world setting and was associated with reducing recurrent stroke by more than half the rate reported in older studies.20 Overall, the data suggest that evaluation within 24 hours confers substantial benefit, and that this evaluation can happen in an outpatient setting.21-23
Acute management: Use of tPA
Once imaging rules out intracranial hemorrhage, patients should be treated with tissue plasminogen activator (tPA) or an endovascular intervention as per guidelines.24 For patients with ischemic stroke ineligible for tPA or endovascular treatments, the initial focus is to determine the etiology of the symptoms so that the best strategies for prevention of secondary stroke may be employed.
Aspirin should be provided within 24 to 48 hours to all patients after intracranial hemorrhage is ruled out. Aspirin should be delayed for 24 hours in those given thrombolytics. The initial recommended dose of aspirin is 325 mg with continued low-dose (81 mg) aspirin daily.13 The addition of clopidogrel to aspirin within 24 hours of an event and continued for 21 days, followed by aspirin alone, was shown to be beneficial in a Chinese population with high-risk TIA (ABCD2 score ≥4) or minor stroke (National Institutes of Health Stroke Scale [NIHSS] ≤3).25 Anticoagulation with heparin, warfarin, or a novel oral anticoagulant is generally not indicated in the acute setting due to the risk of hemorrhagic transformation.
Acute BP management depends upon the type of stroke (ischemic or hemorrhagic), eligibility for thrombolytics, timing of presentation, and possible comorbidities such as myocardial infarction or aortic dissection (see TABLE 413,26). In the absence of contraindications, high-intensity statins should be initiated in all patients able to take oral medications.
CASE › You appropriately referred your patient to the local ED. A head CT with head and neck CTA was performed. While the head CT did not show any abnormalities, the CTA demonstrated high-grade left internal carotid artery stenosis. The patient was given an initial dose of aspirin 325 mg and a high-intensity statin and admitted for further management. An MRI revealed a small shower of emboli in the left hemisphere, confirming the diagnosis of stroke over TIA. Labs were marginally remarkable with a low-density lipoprotein level of 115 mg/dL and an HbA1c of 6.2. Telemetry monitoring did not reveal any arrhythmias, and TTE was normal. BP remained in the high-normal to low-hypertensive range.
A Vascular Surgery consultation was obtained and the patient underwent a left carotid endarterectomy the following day. She did well without surgical complications. Her BP medications were adjusted; a combination of an angiotensin-converting enzyme inhibitor and a thiazide diuretic achieved a goal BP <140/90 mm Hg.
Permissive hypertension was not indicated due to her presentation >48 hours beyond the acute event. Low-dose aspirin and a high-intensity statin were continued, for secondary stroke prevention in the setting of atherosclerotic disease. She received smoking cessation counseling, which will continue.
CORRESPONDENCE
Stephen A. Martin, MD, EdM, Barre Family Health Center, 151 Worcester Road, Barre, MA 01005; [email protected].
CASE › A 68-year-old woman with hypertension and hyperlipidemia comes into your office for evaluation of a 30-minute episode of sudden-onset right-hand weakness and difficulty speaking that occurred 4 days earlier. The patient, who is also a smoker, has come in at the insistence of her daughter. On examination, her blood pressure (BP) is 145/88 mm Hg and her heart rate is 76 beats/minute and regular. She appears well and her language function is normal. The rest of her examination is normal. How would you proceed?
Stroke—the death of nerve cells due to a lack of blood supply from either infarction or hemorrhage—strikes nearly 800,000 people in the United States every year.1,2 Of these events, 130,000 are fatal, making stroke the fifth leading cause of death.3 Effective, early evaluation and cause-specific treatment are crucial parts of stroke care.
Research has helped to clarify the critical role primary care physicians play in recognizing, triaging, and managing stroke and transient ischemic attacks (TIA). This article reviews what we know about the different ways that a stroke and a TIA can present, the appropriate diagnostic work-up for patients presenting with symptoms of either event, and management strategies for subacute care (24 hours to up to 14 days after a stroke has occurred).4,5 Unless otherwise specified, this review will focus on ischemic stroke because 87% of strokes are attributable to ischemia.1
A follow-up to this article on secondary stroke prevention will appear in the journal next month.
Look to onset more than type of symptoms for clues
Stroke presents as a sudden onset of neurologic deficits (language, motor, sensory, cerebellar, or brainstem functions) (TABLE 14). Because presenting symptoms can vary widely, sudden onset, rather than particular symptoms, should raise a red flag for potential stroke.
The differential diagnosis includes: seizure, complex migraine, medication effect (eg, slurred speech or confusion after taking a central nervous system [CNS] depressant), toxin exposure, electrolyte abnormalities (especially hypoglycemia), concussion/trauma, infection of the CNS, peripheral vertigo, demyelination, intracranial mass, Bell’s palsy, and psychogenic disorders. The history and physical, along with laboratory findings and brain imaging (detailed later in this article), will guide the FP toward (or away from) these various etiologies.
Optimal triage is a subject of ongoing interest and research
If stroke or TIA remains a possibility after an initial assessment, it’s time to stratify patients by risk.
One of the most widely accepted tools is the ABCD2 score (see TABLE 26). Clinicians can employ the ABCD2 risk stratification tool when trying to determine whether it is reasonable to pursue an expedited work-up (ie, <1 day) in the outpatient setting or recommend that the patient be evaluated in an emergency department (ED). The 90-day stroke rate following a TIA ranges from 3% with an ABCD2 score of 0 to 3 to 18% with a score of 6 or 7. A score of 0 to 3 is considered relatively low risk; in the absence of other compelling factors, rapid outpatient evaluation is appropriate. For patients with an ABCD2 score ≥4, referral to the ED or direct admission to the hospital is advised.
The validity of the ABCD2 score for risk stratification has been studied extensively with conflicting results.7-10 As with any assessment tool, it should be used as a guide, and should not supplant a full assessment of the patient or the judgment of the examining physician. In making the decision regarding inpatient or outpatient evaluation, it’s also important to consider available resources, access to specialists, and patient preference.
In a 2016 population-based study, the 30-day recurrent stroke/TIA rate for patients hospitalized for TIA was 3% compared with 10.7% for those discharged from the ED with referral to a stroke clinic and 10.6% for those discharged from the ED without a referral to a stroke clinic.11 These data suggest that only patients for whom you have a low clinical suspicion of stroke/TIA should be worked up as outpatients, and that hospital admission is advised in moderate- and high-risk cases. The findings also highlight the critical role that primary care physicians can play in triaging and managing these patients for secondary prevention.
CASE › This patient’s recent history of sudden-onset right-sided weakness and expressive language dysfunction is suspicious for left hemispheric ischemia. She has several risk factors for stroke, and her ABCD2 score is 5 (hypertension, age ≥60 years, unilateral weakness, and duration 10-59 min), which places her at moderate risk. Thus, the recommendation would be to have her go directly to an ED for rapid evaluation.
The diagnostic work-up
Even when a patient is sent to the ED, the FP plays a critical role in his or her continuing care. FPs will often coordinate with inpatient care and manage transition of care to the outpatient setting. (And in many communities, the ED or hospital physicians may themselves be family practitioners.)
In terms of care, not even an aspirin should be administered in a case like this because the patient has not yet had any neuroimaging, and differentiation of ischemic from hemorrhagic stroke cannot be made on clinical grounds alone. Once an ischemic stroke is confirmed, determining the etiology is critical given the significant management differences between the different types of stroke (atherosclerotic, cardioembolic, lacunar, or other).
Which imaging method, and when?
While a computerized tomography (CT) scan is the preferred initial imaging strategy for acute stroke to discern the ischemic type from the hemorrhagic, MRI is preferred for the evaluation of acute ischemic stroke because the method has a higher sensitivity for infarction and a greater ability to identify findings (such as demyelination) that would suggest an alternative diagnosis.
In addition to evaluating the brain parenchyma, physicians must also assess the cerebral vasculature. CT angiography (CTA) or MR angiography (MRA) of the head and neck are preferred over carotid ultrasound because they are capable of evaluating the entire cerebrovascular system12,13 and can be instrumental in identifying potential causes of stroke, as well as guiding therapeutic decisions. Carotid ultrasound is a reasonable alternative for patients presenting with symptoms indicative of anterior circulation involvement when CTA and MRA are unavailable or contraindicated, but it will not identify intracranial vascular disease, proximal common carotid disease, or vertebrobasilar disease.
Getting to the cause of suspected stroke: Labs and other diagnostic tests
A routine work-up includes BP checks, routine labs (complete blood count, complete metabolic panel, coagulation profile, and troponin), an electrocardiogram (EKG), a transthoracic echocardiogram (TTE) with bubble study if possible, and a minimum of 24 to 48 hours of cardiac rhythm monitoring. Cardiac rhythm monitoring should be extended in the setting of clinical concern for unidentified paroxysmal atrial fibrillation, such as an embolism without a proximal vascular source, multiple embolic infarcts in different vascular territories, a dilated left atrium, or other risk factors for atrial fibrillation that include smoking, systolic hypertension, diabetes, and heart failure (see TABLE 312,13,17,18).14-16 This standard diagnostic work-up will identify the cause of stroke in 70% to 80% of patients.19
Additional investigations to consider if the etiology is not yet elucidated include a transesophageal echocardiogram (TEE), cerebral angiography, a coagulopathy evaluation, a lumbar puncture, and a vasculitis work-up. If available, consultation with a neurologist is appropriate for any patient who has had a stroke or TIA. Patients with unclear etiologies or for whom there are questions concerning strategies for preventing secondary stroke should be referred to Neurology and preferably a stroke specialist.
Timing matters, even when symptoms have resolved (ie, TIA).11,20 The EXPRESS trial17 (the Early use of eXisting PREventive Strategies for Stroke) looked at the effect of urgent assessment and treatment (≤1 day) of patients presenting with a TIA or minor stroke on the risk of recurrent stroke within 90 days. The diagnostic work-up included brain and vascular imaging together with an EKG. This intensive approach led to an absolute risk reduction of 8.2% (from 10.3% to 2.1%) in the risk of recurrent stroke at 90 days (number needed to treat [NNT]=12).17
Expedited work-up and treatment was also recently evaluated in a non-trial, real-world setting and was associated with reducing recurrent stroke by more than half the rate reported in older studies.20 Overall, the data suggest that evaluation within 24 hours confers substantial benefit, and that this evaluation can happen in an outpatient setting.21-23
Acute management: Use of tPA
Once imaging rules out intracranial hemorrhage, patients should be treated with tissue plasminogen activator (tPA) or an endovascular intervention as per guidelines.24 For patients with ischemic stroke ineligible for tPA or endovascular treatments, the initial focus is to determine the etiology of the symptoms so that the best strategies for prevention of secondary stroke may be employed.
Aspirin should be provided within 24 to 48 hours to all patients after intracranial hemorrhage is ruled out. Aspirin should be delayed for 24 hours in those given thrombolytics. The initial recommended dose of aspirin is 325 mg with continued low-dose (81 mg) aspirin daily.13 The addition of clopidogrel to aspirin within 24 hours of an event and continued for 21 days, followed by aspirin alone, was shown to be beneficial in a Chinese population with high-risk TIA (ABCD2 score ≥4) or minor stroke (National Institutes of Health Stroke Scale [NIHSS] ≤3).25 Anticoagulation with heparin, warfarin, or a novel oral anticoagulant is generally not indicated in the acute setting due to the risk of hemorrhagic transformation.
Acute BP management depends upon the type of stroke (ischemic or hemorrhagic), eligibility for thrombolytics, timing of presentation, and possible comorbidities such as myocardial infarction or aortic dissection (see TABLE 413,26). In the absence of contraindications, high-intensity statins should be initiated in all patients able to take oral medications.
CASE › You appropriately referred your patient to the local ED. A head CT with head and neck CTA was performed. While the head CT did not show any abnormalities, the CTA demonstrated high-grade left internal carotid artery stenosis. The patient was given an initial dose of aspirin 325 mg and a high-intensity statin and admitted for further management. An MRI revealed a small shower of emboli in the left hemisphere, confirming the diagnosis of stroke over TIA. Labs were marginally remarkable with a low-density lipoprotein level of 115 mg/dL and an HbA1c of 6.2. Telemetry monitoring did not reveal any arrhythmias, and TTE was normal. BP remained in the high-normal to low-hypertensive range.
A Vascular Surgery consultation was obtained and the patient underwent a left carotid endarterectomy the following day. She did well without surgical complications. Her BP medications were adjusted; a combination of an angiotensin-converting enzyme inhibitor and a thiazide diuretic achieved a goal BP <140/90 mm Hg.
Permissive hypertension was not indicated due to her presentation >48 hours beyond the acute event. Low-dose aspirin and a high-intensity statin were continued, for secondary stroke prevention in the setting of atherosclerotic disease. She received smoking cessation counseling, which will continue.
CORRESPONDENCE
Stephen A. Martin, MD, EdM, Barre Family Health Center, 151 Worcester Road, Barre, MA 01005; [email protected].
1. Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135:e146-e603.
2. Sacco RL, Kasner SE, Broderick JP, et al. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:2064-2089.
3. Kochanek KD, Murphy SL, Xu J, et al. Mortality in the United States, 2013. NCHS Data Brief. 2014:1-8. Available at: https://www.cdc.gov/nchs/data/databriefs/db178.pdf. Accessed June 5, 2016.
4. Flossmann E, Redgrave JN, Briley D, et al. Reliability of clinical diagnosis of the symptomatic vascular territory in patients with recent transient ischemic attack or minor stroke. Stroke. 2008;39:2457-2460.
5. Josephson SA, Sidney S, Pham TN, et al. Higher ABCD2 score predicts patients most likely to have true transient ischemic attack. Stroke. 2008;39:3096-3098. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18688003. Accessed June 5, 2016.
6. Hankey GJ. The ABCD, California, and unified ABCD2 risk scores predicted stroke within 2, 7, and 90 days after TIA. Evid Based Med. 2007;12:88.
7. Sheehan OC, Kyne L, Kelly LA, et al. Population-based study of ABCD2 score, carotid stenosis, and atrial fibrillation for early stroke prediction after transient ischemic attack: the North Dublin TIA study. Stroke. 2010;41:844-850.
8. Rothwell PM, Giles MF, Flossmann E, et al. A simple score (ABCD) to identify individuals at high early risk of stroke after transient ischaemic attack. Lancet. 2005; 366:29-36.
9. Tsivgoulis G, Spengos K, Manta P, et al. Validation of the ABCD score in identifying individuals at high early risk of stroke after a transient ischemic attack: a hospital-based case series study. Stroke. 2006;37:2892-2897.
10. Kiyohara T, Kamouchi M, Kumai Y, et al. ABCD3 and ABCD3-I scores are superior to ABCD2 score in the prediction of short- and long-term risks of stroke after transient ischemic attack. Stroke. 2014;45:418-425.
11. Sacco RL, Rundek T. The value of urgent specialized care for TIA and minor stroke. N Engl J Med. 2016;374:1577-1579.
12. Demchuk AM, Menon BK, Goyal M. Comparing vessel imaging: noncontrast computed tomography/computed tomographic angiography should be the new minimum standard in acute disabling stroke. Stroke. 2016;47:273-281.
13. Jauch EC, Saver JL, Adams HP, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:870-947.
14. Gladstone DJ, Spring M, Dorian P, et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370:2467-2477.
15. Sanna T, Diener HC, Passman RS, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. 2014;370:2478-2486.
16. Christophersen IE, Yin X, Larson MG, et al. A comparison of the CHARGE-AF and the CHA2DS2-VASc risk scores for prediction of atrial fibrillation ni the Framingham Heart Study. Am Heart J. 2016;178:45-54.
17. Rothwell PM, Giles MF, Chandratheva A, et al. Effect of urgent treatment of transient ischaemic attack and minor stroke on early recurrent stroke (EXPRESS study): a prospective population-based sequential comparison. Lancet. 2007;370:1432-1442.
18. National Institute for Health and Care Excellence. Stroke and transient ischaemic attack in over 16s: diagnosis and initial management. Available at: https://www.nice.org.uk/guidance/cg68. Published 2008. Accessed February 5, 2017.
19. Hart RG, Diener HC, Coutts SB, et al. Embolic strokes of undetermined source: the case for a new clinical construct. Lancet Neurol. 2014;13:429-438.
20. Amarenco P, Lavallée PC, Labreuche J, et al. One-year risk of stroke after transient ischemic attack or minor stroke. N Engl J Med. 2016;374:1533-1542.
21. Joshi JK, Ouyang B, Prabhakaran S. Should TIA patients be hospitalized or referred to a same-day clinic? A decision analysis. Neurology. 2011;77:2082-2088.
22. Mijalski C, Silver B. TIA management: should TIA patients be admitted? should TIA patients get combination antiplatelet therapy? The Neurohospitalist. 2015;5:151-160.
23. Silver B, Adeoye O. Management of patients with transient ischemic attack in the emergency department. Neurology. 2016;86:1568-1569.
24. Demaerschalk BM, Kleindorfer DO, Adeoye OM, et al. Scientific rationale for the inclusion and exclusion criteria for intravenous alteplase in acute ischemic stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016;47:581-641.
25. Wang Y, Wang Y, Zhao X, et al. Clopidogrel with aspirin in acute minor stroke or transient ischemic attack. N Engl J Med. 2013;369:11-19.
26. Hemphill JC, Greenberg SM, Anderson CS, et al. Guidelines for the management of spontaneous intracerebral hemorrhage. Stroke. 2015;46:2032-2060.
1. Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135:e146-e603.
2. Sacco RL, Kasner SE, Broderick JP, et al. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:2064-2089.
3. Kochanek KD, Murphy SL, Xu J, et al. Mortality in the United States, 2013. NCHS Data Brief. 2014:1-8. Available at: https://www.cdc.gov/nchs/data/databriefs/db178.pdf. Accessed June 5, 2016.
4. Flossmann E, Redgrave JN, Briley D, et al. Reliability of clinical diagnosis of the symptomatic vascular territory in patients with recent transient ischemic attack or minor stroke. Stroke. 2008;39:2457-2460.
5. Josephson SA, Sidney S, Pham TN, et al. Higher ABCD2 score predicts patients most likely to have true transient ischemic attack. Stroke. 2008;39:3096-3098. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18688003. Accessed June 5, 2016.
6. Hankey GJ. The ABCD, California, and unified ABCD2 risk scores predicted stroke within 2, 7, and 90 days after TIA. Evid Based Med. 2007;12:88.
7. Sheehan OC, Kyne L, Kelly LA, et al. Population-based study of ABCD2 score, carotid stenosis, and atrial fibrillation for early stroke prediction after transient ischemic attack: the North Dublin TIA study. Stroke. 2010;41:844-850.
8. Rothwell PM, Giles MF, Flossmann E, et al. A simple score (ABCD) to identify individuals at high early risk of stroke after transient ischaemic attack. Lancet. 2005; 366:29-36.
9. Tsivgoulis G, Spengos K, Manta P, et al. Validation of the ABCD score in identifying individuals at high early risk of stroke after a transient ischemic attack: a hospital-based case series study. Stroke. 2006;37:2892-2897.
10. Kiyohara T, Kamouchi M, Kumai Y, et al. ABCD3 and ABCD3-I scores are superior to ABCD2 score in the prediction of short- and long-term risks of stroke after transient ischemic attack. Stroke. 2014;45:418-425.
11. Sacco RL, Rundek T. The value of urgent specialized care for TIA and minor stroke. N Engl J Med. 2016;374:1577-1579.
12. Demchuk AM, Menon BK, Goyal M. Comparing vessel imaging: noncontrast computed tomography/computed tomographic angiography should be the new minimum standard in acute disabling stroke. Stroke. 2016;47:273-281.
13. Jauch EC, Saver JL, Adams HP, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:870-947.
14. Gladstone DJ, Spring M, Dorian P, et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370:2467-2477.
15. Sanna T, Diener HC, Passman RS, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. 2014;370:2478-2486.
16. Christophersen IE, Yin X, Larson MG, et al. A comparison of the CHARGE-AF and the CHA2DS2-VASc risk scores for prediction of atrial fibrillation ni the Framingham Heart Study. Am Heart J. 2016;178:45-54.
17. Rothwell PM, Giles MF, Chandratheva A, et al. Effect of urgent treatment of transient ischaemic attack and minor stroke on early recurrent stroke (EXPRESS study): a prospective population-based sequential comparison. Lancet. 2007;370:1432-1442.
18. National Institute for Health and Care Excellence. Stroke and transient ischaemic attack in over 16s: diagnosis and initial management. Available at: https://www.nice.org.uk/guidance/cg68. Published 2008. Accessed February 5, 2017.
19. Hart RG, Diener HC, Coutts SB, et al. Embolic strokes of undetermined source: the case for a new clinical construct. Lancet Neurol. 2014;13:429-438.
20. Amarenco P, Lavallée PC, Labreuche J, et al. One-year risk of stroke after transient ischemic attack or minor stroke. N Engl J Med. 2016;374:1533-1542.
21. Joshi JK, Ouyang B, Prabhakaran S. Should TIA patients be hospitalized or referred to a same-day clinic? A decision analysis. Neurology. 2011;77:2082-2088.
22. Mijalski C, Silver B. TIA management: should TIA patients be admitted? should TIA patients get combination antiplatelet therapy? The Neurohospitalist. 2015;5:151-160.
23. Silver B, Adeoye O. Management of patients with transient ischemic attack in the emergency department. Neurology. 2016;86:1568-1569.
24. Demaerschalk BM, Kleindorfer DO, Adeoye OM, et al. Scientific rationale for the inclusion and exclusion criteria for intravenous alteplase in acute ischemic stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016;47:581-641.
25. Wang Y, Wang Y, Zhao X, et al. Clopidogrel with aspirin in acute minor stroke or transient ischemic attack. N Engl J Med. 2013;369:11-19.
26. Hemphill JC, Greenberg SM, Anderson CS, et al. Guidelines for the management of spontaneous intracerebral hemorrhage. Stroke. 2015;46:2032-2060.
PRACTICE RECOMMENDATIONS
› Perform an urgent work-up on patients presenting with symptoms of a transient ischemic attack or stroke. A
› Employ the ABCD2 risk stratification tool when determining whether it is reasonable to pursue an expedited work-up in the outpatient setting or recommend that a patient be evaluated in an emergency department. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
EMA recommends orphan designation for hemophilia B product

The European Medicines Agency’s (EMA’s) Committee for Orphan Medicinal Products has issued a positive opinion recommending orphan designation for CB 2679d/ISU304 for the treatment of hemophilia B.
CB 2679d is a coagulation factor IX variant that has demonstrated, in preclinical studies, the potential to normalize factor IX levels via a daily subcutaneous injection.
The product is being developed by Catalyst Biosciences and ISU Abxis. ISU Abxis plans to initiate a phase 1/2 study of CB 2679d in individuals with severe hemophilia B this month in South Korea.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
The European Medicines Agency’s (EMA’s) Committee for Orphan Medicinal Products has issued a positive opinion recommending orphan designation for CB 2679d/ISU304 for the treatment of hemophilia B.
CB 2679d is a coagulation factor IX variant that has demonstrated, in preclinical studies, the potential to normalize factor IX levels via a daily subcutaneous injection.
The product is being developed by Catalyst Biosciences and ISU Abxis. ISU Abxis plans to initiate a phase 1/2 study of CB 2679d in individuals with severe hemophilia B this month in South Korea.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
The European Medicines Agency’s (EMA’s) Committee for Orphan Medicinal Products has issued a positive opinion recommending orphan designation for CB 2679d/ISU304 for the treatment of hemophilia B.
CB 2679d is a coagulation factor IX variant that has demonstrated, in preclinical studies, the potential to normalize factor IX levels via a daily subcutaneous injection.
The product is being developed by Catalyst Biosciences and ISU Abxis. ISU Abxis plans to initiate a phase 1/2 study of CB 2679d in individuals with severe hemophilia B this month in South Korea.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
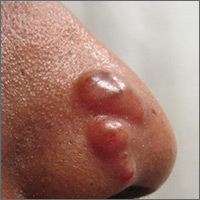
Nodules on nose and tattoos
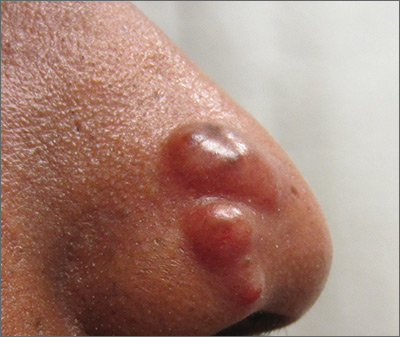
Based on the clinical presentation and skin biopsy results, the patient was given a diagnosis of cutaneous sarcoidosis. The biopsy from the right side of his nose demonstrated sarcoidal granulomas. A biopsy of one of the tattoo nodules showed sarcoidal granulomas, and close inspection revealed red tattoo pigment within the granulomatous inflammation. X-rays showed bilateral hilar lymphadenopathy, which was consistent with pulmonary sarcoidosis, and the lace-like appearance of the middle and distal phalanges was consistent with skeletal sarcoidosis.
Systemic sarcoidosis is an idiopathic, granulomatous disease that affects multiple organ systems, but primarily the lungs and lymphatic system. Cutaneous sarcoidosis can occur as a manifestation of systemic sarcoidosis. It may present as asymptomatic red or skin-colored papules and firm nodules within tattoos, old scars, or permanent makeup. Sarcoidosis usually occurs in red, black, or blue-black areas of tattoos, in which the pigment acts as a nidus for granuloma formation.
The first-line treatment for limited papules is a high-potency topical corticosteroid (eg, clobetasol 0.05% ointment applied twice weekly) and an intralesional corticosteroid (eg, triamcinolone, one 5-10 mg/mL injection every 4 weeks). Antimalarials such as hydroxychloroquine or methotrexate can also be helpful. Midpotency topical corticosteroids such as triamcinolone 0.1% cream and doxycycline hyclate have been reported to clear cutaneous lesions in tattoos. Oral corticosteroids are often effective for severe cutaneous sarcoidosis, but their multiple adverse effects (eg, diabetes and adrenal suppression) prevent prolonged use except in very low doses in conjunction with other therapies.
The nodules on this patient’s nose were successfully treated with intralesional triamcinolone 5 mg/mL. No treatment was initiated for the tattoo nodules because they were asymptomatic and the patient wasn’t bothered by their appearance. The patient’s hand swelling improved with a treatment of prednisone 10 mg/d. The rheumatologist considered a steroid-sparing immunosuppressive agent such as methotrexate; however, the patient was lost to follow-up.
Adapted from: Zhang J, Jansen R, Lim HW. Nodules on nose and tattoos. J Fam Pract. 2015;64:241-243.
Based on the clinical presentation and skin biopsy results, the patient was given a diagnosis of cutaneous sarcoidosis. The biopsy from the right side of his nose demonstrated sarcoidal granulomas. A biopsy of one of the tattoo nodules showed sarcoidal granulomas, and close inspection revealed red tattoo pigment within the granulomatous inflammation. X-rays showed bilateral hilar lymphadenopathy, which was consistent with pulmonary sarcoidosis, and the lace-like appearance of the middle and distal phalanges was consistent with skeletal sarcoidosis.
Systemic sarcoidosis is an idiopathic, granulomatous disease that affects multiple organ systems, but primarily the lungs and lymphatic system. Cutaneous sarcoidosis can occur as a manifestation of systemic sarcoidosis. It may present as asymptomatic red or skin-colored papules and firm nodules within tattoos, old scars, or permanent makeup. Sarcoidosis usually occurs in red, black, or blue-black areas of tattoos, in which the pigment acts as a nidus for granuloma formation.
The first-line treatment for limited papules is a high-potency topical corticosteroid (eg, clobetasol 0.05% ointment applied twice weekly) and an intralesional corticosteroid (eg, triamcinolone, one 5-10 mg/mL injection every 4 weeks). Antimalarials such as hydroxychloroquine or methotrexate can also be helpful. Midpotency topical corticosteroids such as triamcinolone 0.1% cream and doxycycline hyclate have been reported to clear cutaneous lesions in tattoos. Oral corticosteroids are often effective for severe cutaneous sarcoidosis, but their multiple adverse effects (eg, diabetes and adrenal suppression) prevent prolonged use except in very low doses in conjunction with other therapies.
The nodules on this patient’s nose were successfully treated with intralesional triamcinolone 5 mg/mL. No treatment was initiated for the tattoo nodules because they were asymptomatic and the patient wasn’t bothered by their appearance. The patient’s hand swelling improved with a treatment of prednisone 10 mg/d. The rheumatologist considered a steroid-sparing immunosuppressive agent such as methotrexate; however, the patient was lost to follow-up.
Adapted from: Zhang J, Jansen R, Lim HW. Nodules on nose and tattoos. J Fam Pract. 2015;64:241-243.
Based on the clinical presentation and skin biopsy results, the patient was given a diagnosis of cutaneous sarcoidosis. The biopsy from the right side of his nose demonstrated sarcoidal granulomas. A biopsy of one of the tattoo nodules showed sarcoidal granulomas, and close inspection revealed red tattoo pigment within the granulomatous inflammation. X-rays showed bilateral hilar lymphadenopathy, which was consistent with pulmonary sarcoidosis, and the lace-like appearance of the middle and distal phalanges was consistent with skeletal sarcoidosis.
Systemic sarcoidosis is an idiopathic, granulomatous disease that affects multiple organ systems, but primarily the lungs and lymphatic system. Cutaneous sarcoidosis can occur as a manifestation of systemic sarcoidosis. It may present as asymptomatic red or skin-colored papules and firm nodules within tattoos, old scars, or permanent makeup. Sarcoidosis usually occurs in red, black, or blue-black areas of tattoos, in which the pigment acts as a nidus for granuloma formation.
The first-line treatment for limited papules is a high-potency topical corticosteroid (eg, clobetasol 0.05% ointment applied twice weekly) and an intralesional corticosteroid (eg, triamcinolone, one 5-10 mg/mL injection every 4 weeks). Antimalarials such as hydroxychloroquine or methotrexate can also be helpful. Midpotency topical corticosteroids such as triamcinolone 0.1% cream and doxycycline hyclate have been reported to clear cutaneous lesions in tattoos. Oral corticosteroids are often effective for severe cutaneous sarcoidosis, but their multiple adverse effects (eg, diabetes and adrenal suppression) prevent prolonged use except in very low doses in conjunction with other therapies.
The nodules on this patient’s nose were successfully treated with intralesional triamcinolone 5 mg/mL. No treatment was initiated for the tattoo nodules because they were asymptomatic and the patient wasn’t bothered by their appearance. The patient’s hand swelling improved with a treatment of prednisone 10 mg/d. The rheumatologist considered a steroid-sparing immunosuppressive agent such as methotrexate; however, the patient was lost to follow-up.
Adapted from: Zhang J, Jansen R, Lim HW. Nodules on nose and tattoos. J Fam Pract. 2015;64:241-243.
Do oral decongestants have a clinically significant effect on BP in patients with hypertension?
EVIDENCE SUMMARY
A meta-analysis of 24 RCTs examined the effect of pseudoephedrine on BP and heart rate.1 Just 5 of the 24 studies specifically included hypertensive patients. In the population of patients with hypertension, the meta-analysis showed a small (1.2 mm Hg) rise in systolic BP with pseudoephedrine that was statistically significant (95% confidence interval [CI], 0.56-1.84 mm Hg), but the slight changes in diastolic BP and heart rate were not significant. No patient-oriented outcomes were measured.
The highest quality study within this group was a randomized, double-blind, placebo-controlled crossover study with 28 patients given sustained-release pseudoephedrine 120 mg twice daily for 72 hours, with BP measurements taken at 48 and 72 hours.2 The study was powered to identify an increase in systolic BP of 11 mm Hg, but the results showed just a 3.1 mm Hg rise in systolic BP at 48 hours (see TABLE1-7 for CI and other data).
In another double-blind, placebo-controlled RCT of 29 adults with hypertension (only 25 were included in the data analysis), there was no significant elevation in BP when oral pseudoephedrine was administered over the course of 3 days.3
Across the 5 studies in the meta-analysis, immediate-release and sustained-release forms of pseudoephedrine were included, hypertension was described as controlled but definitions of control were not always specified, and study length varied from 2 hours to 4 weeks.2-6 Patients on antihypertensive medications were included in some of the studies; patients who had active cardiovascular disease, peripheral vascular disease, and/or cerebrovascular disease were excluded.
One study specifically looked at the effects of a single dose of pseudoephedrine on BP in patients treated with 2 different beta-blockers and found no significant change from baseline, but this study was not powered to show differences less than 5 mm Hg.6 The study did show a change of 1 to 2 mm Hg in systolic BP, but this was not statistically significant.
An absence of information on older patients
There is a paucity of literature on treating older adults and medically complex patients (eg, those with uncontrolled or secondary causes of hypertension, cerebrovascular disease, coronary artery disease) with decongestants, as they were excluded in all studies. And the available evidence does not include reports of adverse events other than changes in BP.
1. Salerno SM, Jackson JL, Berbano EP. Effect of oral pseudoephedrine on blood pressure and heart rate. Arch Intern Med. 2005;165:1686-1694.
2. Beck RA, Mercado DL, Seguin SM, et al. Cardiovascular effects of pseudoephedrine in medically controlled hypertensive patients. Arch Int Med. 1992;152:1242-1245.
3. Bradley JG, Kallail KJ, Dorsch JN, et al. The effects of pseudoephedrine on blood pressure in patients with controlled, uncomplicated hypertension: a randomized, double-blind, placebo-controlled trial. J Am Board Fam Pract. 1991;4:201-206.
4. Chua SS, Benrimoj SI, Gordon RD, et al. A controlled clinical trial on the cardiovascular effects of single doses of pseudoephedrine in hypertensive patients. Br J Clin Pharmacol. 1989;28:369-372.
5. Coates ML, Rembold CM, Farr BM. Does pseudoephedrine increase blood pressure in patients with controlled hypertension? J Fam Pract. 1995;40:22-26.
6. Mores N, Campia U, Navarra P, et al. No cardiovascular effects of single-dose pseudoephedrine in patients with essential hypertension treated with beta-blockers. Eur J Clin Pharmacol. 1999;55:251-254.
7. Salerno SM, Jackson JL, Berbano EP. The impact of oral phenylpropanolamine on blood pressure: a meta-analysis and review of the literature. J Hum Hypertens. 2005;19:643-652.
EVIDENCE SUMMARY
A meta-analysis of 24 RCTs examined the effect of pseudoephedrine on BP and heart rate.1 Just 5 of the 24 studies specifically included hypertensive patients. In the population of patients with hypertension, the meta-analysis showed a small (1.2 mm Hg) rise in systolic BP with pseudoephedrine that was statistically significant (95% confidence interval [CI], 0.56-1.84 mm Hg), but the slight changes in diastolic BP and heart rate were not significant. No patient-oriented outcomes were measured.
The highest quality study within this group was a randomized, double-blind, placebo-controlled crossover study with 28 patients given sustained-release pseudoephedrine 120 mg twice daily for 72 hours, with BP measurements taken at 48 and 72 hours.2 The study was powered to identify an increase in systolic BP of 11 mm Hg, but the results showed just a 3.1 mm Hg rise in systolic BP at 48 hours (see TABLE1-7 for CI and other data).
In another double-blind, placebo-controlled RCT of 29 adults with hypertension (only 25 were included in the data analysis), there was no significant elevation in BP when oral pseudoephedrine was administered over the course of 3 days.3
Across the 5 studies in the meta-analysis, immediate-release and sustained-release forms of pseudoephedrine were included, hypertension was described as controlled but definitions of control were not always specified, and study length varied from 2 hours to 4 weeks.2-6 Patients on antihypertensive medications were included in some of the studies; patients who had active cardiovascular disease, peripheral vascular disease, and/or cerebrovascular disease were excluded.
One study specifically looked at the effects of a single dose of pseudoephedrine on BP in patients treated with 2 different beta-blockers and found no significant change from baseline, but this study was not powered to show differences less than 5 mm Hg.6 The study did show a change of 1 to 2 mm Hg in systolic BP, but this was not statistically significant.
An absence of information on older patients
There is a paucity of literature on treating older adults and medically complex patients (eg, those with uncontrolled or secondary causes of hypertension, cerebrovascular disease, coronary artery disease) with decongestants, as they were excluded in all studies. And the available evidence does not include reports of adverse events other than changes in BP.
EVIDENCE SUMMARY
A meta-analysis of 24 RCTs examined the effect of pseudoephedrine on BP and heart rate.1 Just 5 of the 24 studies specifically included hypertensive patients. In the population of patients with hypertension, the meta-analysis showed a small (1.2 mm Hg) rise in systolic BP with pseudoephedrine that was statistically significant (95% confidence interval [CI], 0.56-1.84 mm Hg), but the slight changes in diastolic BP and heart rate were not significant. No patient-oriented outcomes were measured.
The highest quality study within this group was a randomized, double-blind, placebo-controlled crossover study with 28 patients given sustained-release pseudoephedrine 120 mg twice daily for 72 hours, with BP measurements taken at 48 and 72 hours.2 The study was powered to identify an increase in systolic BP of 11 mm Hg, but the results showed just a 3.1 mm Hg rise in systolic BP at 48 hours (see TABLE1-7 for CI and other data).
In another double-blind, placebo-controlled RCT of 29 adults with hypertension (only 25 were included in the data analysis), there was no significant elevation in BP when oral pseudoephedrine was administered over the course of 3 days.3
Across the 5 studies in the meta-analysis, immediate-release and sustained-release forms of pseudoephedrine were included, hypertension was described as controlled but definitions of control were not always specified, and study length varied from 2 hours to 4 weeks.2-6 Patients on antihypertensive medications were included in some of the studies; patients who had active cardiovascular disease, peripheral vascular disease, and/or cerebrovascular disease were excluded.
One study specifically looked at the effects of a single dose of pseudoephedrine on BP in patients treated with 2 different beta-blockers and found no significant change from baseline, but this study was not powered to show differences less than 5 mm Hg.6 The study did show a change of 1 to 2 mm Hg in systolic BP, but this was not statistically significant.
An absence of information on older patients
There is a paucity of literature on treating older adults and medically complex patients (eg, those with uncontrolled or secondary causes of hypertension, cerebrovascular disease, coronary artery disease) with decongestants, as they were excluded in all studies. And the available evidence does not include reports of adverse events other than changes in BP.
1. Salerno SM, Jackson JL, Berbano EP. Effect of oral pseudoephedrine on blood pressure and heart rate. Arch Intern Med. 2005;165:1686-1694.
2. Beck RA, Mercado DL, Seguin SM, et al. Cardiovascular effects of pseudoephedrine in medically controlled hypertensive patients. Arch Int Med. 1992;152:1242-1245.
3. Bradley JG, Kallail KJ, Dorsch JN, et al. The effects of pseudoephedrine on blood pressure in patients with controlled, uncomplicated hypertension: a randomized, double-blind, placebo-controlled trial. J Am Board Fam Pract. 1991;4:201-206.
4. Chua SS, Benrimoj SI, Gordon RD, et al. A controlled clinical trial on the cardiovascular effects of single doses of pseudoephedrine in hypertensive patients. Br J Clin Pharmacol. 1989;28:369-372.
5. Coates ML, Rembold CM, Farr BM. Does pseudoephedrine increase blood pressure in patients with controlled hypertension? J Fam Pract. 1995;40:22-26.
6. Mores N, Campia U, Navarra P, et al. No cardiovascular effects of single-dose pseudoephedrine in patients with essential hypertension treated with beta-blockers. Eur J Clin Pharmacol. 1999;55:251-254.
7. Salerno SM, Jackson JL, Berbano EP. The impact of oral phenylpropanolamine on blood pressure: a meta-analysis and review of the literature. J Hum Hypertens. 2005;19:643-652.
1. Salerno SM, Jackson JL, Berbano EP. Effect of oral pseudoephedrine on blood pressure and heart rate. Arch Intern Med. 2005;165:1686-1694.
2. Beck RA, Mercado DL, Seguin SM, et al. Cardiovascular effects of pseudoephedrine in medically controlled hypertensive patients. Arch Int Med. 1992;152:1242-1245.
3. Bradley JG, Kallail KJ, Dorsch JN, et al. The effects of pseudoephedrine on blood pressure in patients with controlled, uncomplicated hypertension: a randomized, double-blind, placebo-controlled trial. J Am Board Fam Pract. 1991;4:201-206.
4. Chua SS, Benrimoj SI, Gordon RD, et al. A controlled clinical trial on the cardiovascular effects of single doses of pseudoephedrine in hypertensive patients. Br J Clin Pharmacol. 1989;28:369-372.
5. Coates ML, Rembold CM, Farr BM. Does pseudoephedrine increase blood pressure in patients with controlled hypertension? J Fam Pract. 1995;40:22-26.
6. Mores N, Campia U, Navarra P, et al. No cardiovascular effects of single-dose pseudoephedrine in patients with essential hypertension treated with beta-blockers. Eur J Clin Pharmacol. 1999;55:251-254.
7. Salerno SM, Jackson JL, Berbano EP. The impact of oral phenylpropanolamine on blood pressure: a meta-analysis and review of the literature. J Hum Hypertens. 2005;19:643-652.
Evidence-based answers from the Family Physicians Inquiries Network
EVIDENCE-BASED ANSWER:
It is unclear. Pseudoephedrine causes an average increase of 1.2 mm Hg in systolic blood pressure (BP) in patients with controlled hypertension. However, the studies are not adequately powered to provide evidence about whether this rise in systolic BP is linked to patient-oriented outcomes (strength of recommendation [SOR]: C, multiple randomized controlled trials [RCTs] supporting disease-oriented evidence). Significant variations in BP are defined differently among studies (TABLE1-7). In addition, we do not have data on chronic use of oral decongestants; the longest time on medication in these trials was 4 weeks.
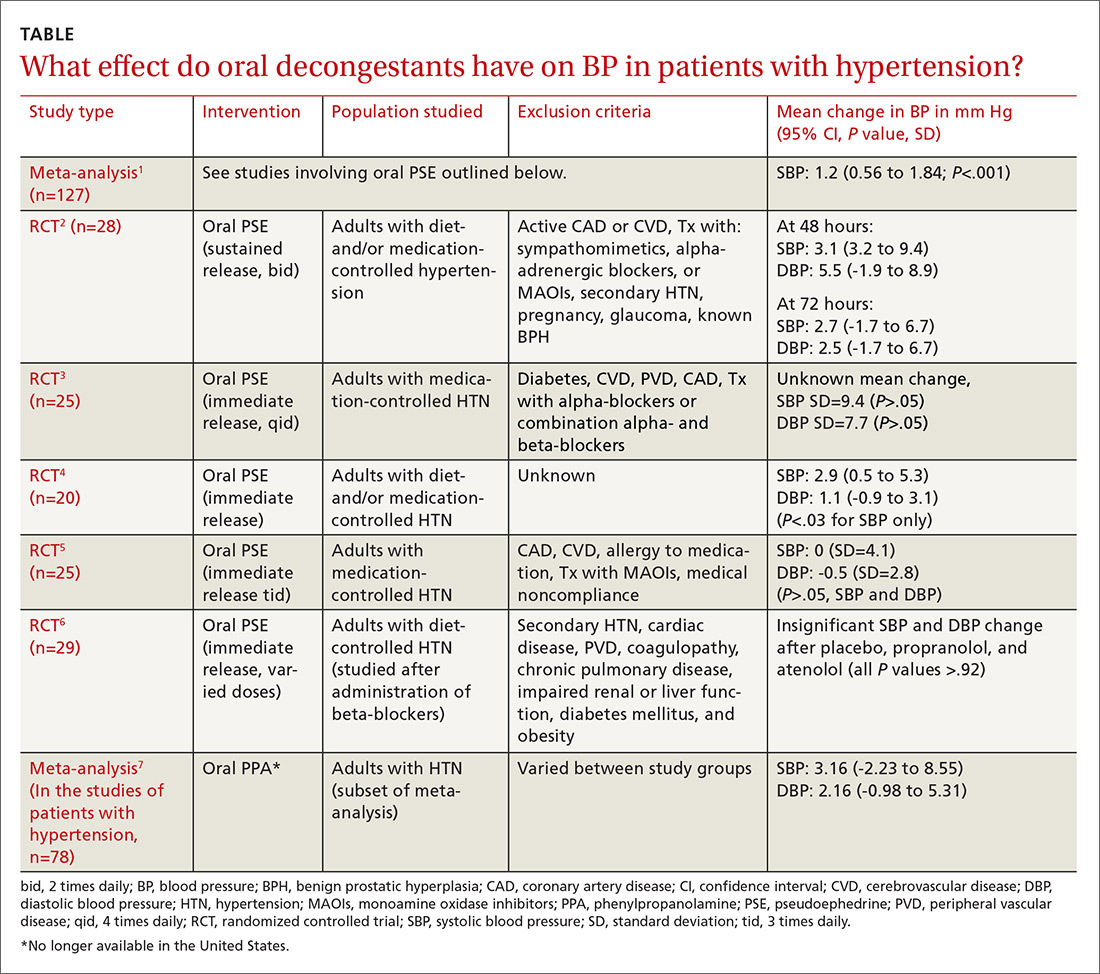
Neuropathic pain treatment provides unexpected benefit
THE CASE
A 57-year-old African American woman was being treated at our clinic for neurogenic urinary incontinence (UI). The UI, which occurred day and night, began 2 years earlier following a laminectomy of vertebrae C3 to C6 with spinal fusion of C3 to C7 for cervical spinal stenosis. The UI persisted despite physical therapy and trials of oxybutynin and imipramine. Since the surgery, the patient had also been experiencing chronic (debilitating) neuropathic pain in both legs, and the sensation of incomplete bladder emptying. She denied bowel incontinence or saddle anesthesia. Her prescription medications included hydrocodone-acetaminophen 7.5/325 mg every 6 hours as needed for pain and lisinopril 20 mg/d for essential hypertension. The patient’s body mass index (BMI) was 23.3.
A urine culture initially grew Klebsiella pneumoniae, which we successfully treated with ciprofloxacin. A urinalysis was unremarkable, and blood urea nitrogen and creatinine levels were within normal limits.
We started the patient on oral duloxetine 30 mg/d for her neuropathic pain. The patient hadn’t undergone a urologic evaluation before starting duloxetine, so no urodynamic studies or measurements had been conducted. At that point, we sent the patient to a urologist for an evaluation.
At a follow-up visit with one of our clinic providers <3 months later, the patient reported that the duloxetine was providing her with some pain relief and that she was “waking up dry” in the mornings and having fewer UI symptoms throughout the day, as well as at night. The patient denied any adverse effects such as nausea, gastrointestinal upset, weight changes, xerostomia, fatigue, insomnia, headaches, or dizziness. Duloxetine was titrated up to 60 mg/d for better control of her neuropathic pain. At the next follow-up visit at our clinic 3 months later, her UI was 80% to 90% improved and she was able to stop her opioid pain medications.
DISCUSSION
UI is a significant problem in the United States and around the world. For women, the prevalence of UI ranges from 15% to 69%; among men, the prevalence is 5% to 24%.1-3 The economic burden of UI includes both medical and nonmedical (eg, pads, diapers, laundry, and dry cleaning) care. The total national cost was estimated at $66 billion in 2007: $49 billion for direct medical costs, $2 billion for direct nonmedical costs, and $15 billion for indirect costs.4 And those costs are expected to increase 25% by 2020, mainly because of the aging population.
Risk factors for UI other than gender include advancing age, obesity, non-Hispanic white race, depression, hypertension, type 2 diabetes mellitus, neurologic disease, and functional limitations/general poor health.5-7 Comorbid depression and BMI >30, as well as the presence and duration of diabetes, increase the odds for developing UI.7,8
Risk factors for women include hysterectomy,7 increasing parity, and delivery of at least one infant >9.5 pounds; the risk is the same for both vaginal and cesarean-section delivery.6 Specific risk factors for men include prostate cancer, prostate surgery, and prostate radiation.5
Significant, chronic comorbidities of UI include depression and chronic pain. While quality of life is negatively affected by UI alone, the coexistence of depression and UI produces an additive negative effect on quality of life.9
Types and treatment of UI
There are 5 types of UI: urge, stress, overflow, functional, and mixed.10
- Urge incontinence is the leakage of urine following a sensation of sudden urgency to void.
- Stress incontinence is urine leakage associated with increased intra-abdominal pressure such as with coughing or sneezing and is typically associated with weakened pelvic floor musculature.
- Overflow incontinence is more common in men, and is typically caused by prostatic disease. The urethral outlet is obstructed leading to increased pressure within the bladder and subsequent leakage of urine.
- Functional incontinence is caused by physical or cognitive impairment leading to a decreased ability to get to a bathroom quickly enough to void.
- Mixed incontinence is when symptoms of stress and urgency incontinence are present.
There are 3 broad categories of treatment methods for urinary incontinence: behavioral, pharmacologic, and surgical. Behavioral interventions are subdivided into caregiver-dependent (prompted voiding, habit retraining, and timed voiding) and patient-directed (bladder training, pelvic floor muscle training, strategies for bladder control, education, and self-monitoring) techniques. Pharmacologic treatment typically consists of antimuscarinics (eg, oxybutynin, tolterodine, solifenacin) and tricyclic antidepressants (eg, imipramine).11 Injections of onabotulinumtoxinA into the detrusor muscle have also been shown to reduce the symptoms of urinary incontinence.12 Surgical options for treatment of UI include retro-pubic suspension, slings, and, in some instances, artificial urethral sphincters.13
A novel treatment for neurogenic UI?
Despite the many treatments available for UI, none comprehensively addresses UI and its common comorbidities.
The role of duloxetine. Normal micturition is regulated by the somatic nervous system and an autonomic reflex arc; the neurotransmitters serotonin and norepinephrine play an important role in the neural regulation of micturition and urinary continence. Duloxetine, alone or as an adjunctive treatment, is a potential novel therapy that treats 2 common comorbidities of UI—chronic pain and depression.
As a selective serotonin norepinephrine reuptake inhibitor (SNRI), duloxetine acts at the molecular level to block the reuptake of serotonin and norepinephrine from synaptic clefts. Specifically, the medication blocks the 5-hydroxytryptamine (5-HT) reuptake transporters, as well as the norepinephrine transporters, of pre-synaptic neurons.14 Thus, the concentrations of 5-HT and norepinephrine increase in the synaptic cleft.
Functionally, the accumulation of norepinephrine inhibits micturition by relaxing the detrusor muscle and constricting the urethral smooth muscle. In addition, a higher concentration of 5-HT at the neuromuscular junction leads to constriction of the external urethral sphincter.
Duloxetine has been shown to be effective in the treatment of other types of UI, such as stress UI15 and mixed UI.16 Additionally, it was found to be effective when compared with placebo in women with overactive bladder syndrome17 and in women with multiple sclerosis and depression.18 However, we are not aware of any cases using duloxetine for the treatment of neurogenic UI.
THE TAKEAWAY
Duloxetine is a potential novel drug choice for the treatment of neurogenic UI. Its effects on serotonin and norepinephrine at the synaptic cleft and neuromuscular junction could provide relief for those who have not found relief from other therapies. Further research—particularly a prospective, randomized controlled trial—is needed to determine if duloxetine is, in fact, more than just a theoretical candidate to treat UI and, if so, the most effective dosing.
Offering duloxetine for the treatment of neurogenic UI would potentially address coexisting conditions—such as pain or depression—thus improving patient compliance and reducing health care spending. Before beginning therapy, urodynamic studies to identify the type of UI should be completed, or, at a minimum, post-void residual volume should be measured.
ACKNOWLEDGEMENTS
The authors would like to thank Julie Hughbanks, MLS, Library Manager, Parkview Health Resource Library, for her assistance with the library searches used for this case report.
1. Markland AD, Richter HE, Fwu CW, et al. Prevalence and trends of urinary incontinence in adults in the United States, 2001 to 2008. J Urol. 2011;186:589-593.
2. Buckley BS, Lapitan MC; Epidemiology Committee of the Fourth International Consultation on Incontinence, Paris, 2008. Prevalence of urinary incontinence in men, women, and children—current evidence: findings of the Fourth International Consultation on Incontinence. Urology. 2010;76:265-270.
3. Gorina Y, Schappert S, Bercovitz A, et al. Prevalence of incontinence among older Americans. Vital Health Stat 3. 2014;1-33.
4. Coyne KS, Wein A, Nicholson S, et al. Economic burden of urgency urinary incontinence in the United States: a systematic review. J Manag Care Pharm. 2014;20:130-140.
5. Shamliyan TA, Wyman JF, Ping R, et al. Male urinary incontinence: prevalence, risk factors, and preventive interventions. Rev Urol. 2009;11:145-165.
6. Matthews CA, Whitehead WE, Townsend MK, et al. Risk factors for urinary, fecal, or dual incontinence in the Nurses’ Health Study. Obstet Gynecol. 2013;122:539-545.
7. Danforth KN, Townsend MK, Lifford K, et al. Risk factors for urinary incontinence among middle-aged women. Am J Obstet Gynecol. 2006;194:339-345.
8. Lifford KL, Curhan GC, Hu FB, et al. Type 2 diabetes mellitus and risk of developing urinary incontinence. J Am Geriatr Soc. 2005;53:1851-1857.
9. Avery JC, Stocks NP, Duggan P, et al. Identifying the quality of life effects of urinary incontinence with depression in an Australian population. BMC Urol. 2013;13:11.
10. National Kidney and Urologic Diseases Information Clearinghouse. Urinary incontinence in women. Available at: http://kidney.niddk.nih.gov/KUDISEASES/pubs/uiwomen/UI-Women_508.pdf. Accessed January 2, 2015.
11. Ontario Medical Advisory Secretariat. Behavioural interventions for urinary incontinence in community-dwelling seniors: an evidence-based analysis. Ontario Health Technology Assessment Series. 2008:8. Available at: http://www.hqontario.ca/Portals/0/Documents/evidence/reports/rev_aic_ui_20081002.pdf. Accessed November 30, 2015.
12. Cox L, Cameron A. OnabotulinumtoxinA for the treatment of overactive bladder. Res Rep Urol. 2014;6:79-89.
13. Dmochowski RR, Blaivas JM, Gormley EA, et al. Update of AUA guideline on the surgical management of female stress urinary incontinence. J Urol. 2010;183:1906-1914.
14. Duloxetine. US National Library of Medicine: National Center for Biotechnology Information. 2015. Available at: http://pubchem.ncbi.nlm.nih.gov/compound/duloxetine. Accessed October 20, 2015.
15. Li J, Yang L, Pu C, et al. The role of duloxetine in stress urinary incontinence: a systematic review and meta-analysis. Int Urol Nephrol. 2013;45:679-686.
16. Bent AE, Gousse AE, Hendrix SL, et al. Duloxetine compared with placebo for the treatment of women with mixed urinary incontinence. Neurourol Urodyn. 2008;27:212-221.
17. Steers WD, Herschorn S, Kreder KJ, et al; Duloxetine OAB Study Group. Duloxetine compared with placebo for treating women with symptoms of overactive bladder. BJU Int. 2007;100:337-345.
18. Di Rezze S, Frasca V, Inghilleri M, et al. Duloxetine for the treatment of overactive bladder syndrome in multiple sclerosis: a pilot study. Clin Neuropharmacol. 2012;35:231-234.
THE CASE
A 57-year-old African American woman was being treated at our clinic for neurogenic urinary incontinence (UI). The UI, which occurred day and night, began 2 years earlier following a laminectomy of vertebrae C3 to C6 with spinal fusion of C3 to C7 for cervical spinal stenosis. The UI persisted despite physical therapy and trials of oxybutynin and imipramine. Since the surgery, the patient had also been experiencing chronic (debilitating) neuropathic pain in both legs, and the sensation of incomplete bladder emptying. She denied bowel incontinence or saddle anesthesia. Her prescription medications included hydrocodone-acetaminophen 7.5/325 mg every 6 hours as needed for pain and lisinopril 20 mg/d for essential hypertension. The patient’s body mass index (BMI) was 23.3.
A urine culture initially grew Klebsiella pneumoniae, which we successfully treated with ciprofloxacin. A urinalysis was unremarkable, and blood urea nitrogen and creatinine levels were within normal limits.
We started the patient on oral duloxetine 30 mg/d for her neuropathic pain. The patient hadn’t undergone a urologic evaluation before starting duloxetine, so no urodynamic studies or measurements had been conducted. At that point, we sent the patient to a urologist for an evaluation.
At a follow-up visit with one of our clinic providers <3 months later, the patient reported that the duloxetine was providing her with some pain relief and that she was “waking up dry” in the mornings and having fewer UI symptoms throughout the day, as well as at night. The patient denied any adverse effects such as nausea, gastrointestinal upset, weight changes, xerostomia, fatigue, insomnia, headaches, or dizziness. Duloxetine was titrated up to 60 mg/d for better control of her neuropathic pain. At the next follow-up visit at our clinic 3 months later, her UI was 80% to 90% improved and she was able to stop her opioid pain medications.
DISCUSSION
UI is a significant problem in the United States and around the world. For women, the prevalence of UI ranges from 15% to 69%; among men, the prevalence is 5% to 24%.1-3 The economic burden of UI includes both medical and nonmedical (eg, pads, diapers, laundry, and dry cleaning) care. The total national cost was estimated at $66 billion in 2007: $49 billion for direct medical costs, $2 billion for direct nonmedical costs, and $15 billion for indirect costs.4 And those costs are expected to increase 25% by 2020, mainly because of the aging population.
Risk factors for UI other than gender include advancing age, obesity, non-Hispanic white race, depression, hypertension, type 2 diabetes mellitus, neurologic disease, and functional limitations/general poor health.5-7 Comorbid depression and BMI >30, as well as the presence and duration of diabetes, increase the odds for developing UI.7,8
Risk factors for women include hysterectomy,7 increasing parity, and delivery of at least one infant >9.5 pounds; the risk is the same for both vaginal and cesarean-section delivery.6 Specific risk factors for men include prostate cancer, prostate surgery, and prostate radiation.5
Significant, chronic comorbidities of UI include depression and chronic pain. While quality of life is negatively affected by UI alone, the coexistence of depression and UI produces an additive negative effect on quality of life.9
Types and treatment of UI
There are 5 types of UI: urge, stress, overflow, functional, and mixed.10
- Urge incontinence is the leakage of urine following a sensation of sudden urgency to void.
- Stress incontinence is urine leakage associated with increased intra-abdominal pressure such as with coughing or sneezing and is typically associated with weakened pelvic floor musculature.
- Overflow incontinence is more common in men, and is typically caused by prostatic disease. The urethral outlet is obstructed leading to increased pressure within the bladder and subsequent leakage of urine.
- Functional incontinence is caused by physical or cognitive impairment leading to a decreased ability to get to a bathroom quickly enough to void.
- Mixed incontinence is when symptoms of stress and urgency incontinence are present.
There are 3 broad categories of treatment methods for urinary incontinence: behavioral, pharmacologic, and surgical. Behavioral interventions are subdivided into caregiver-dependent (prompted voiding, habit retraining, and timed voiding) and patient-directed (bladder training, pelvic floor muscle training, strategies for bladder control, education, and self-monitoring) techniques. Pharmacologic treatment typically consists of antimuscarinics (eg, oxybutynin, tolterodine, solifenacin) and tricyclic antidepressants (eg, imipramine).11 Injections of onabotulinumtoxinA into the detrusor muscle have also been shown to reduce the symptoms of urinary incontinence.12 Surgical options for treatment of UI include retro-pubic suspension, slings, and, in some instances, artificial urethral sphincters.13
A novel treatment for neurogenic UI?
Despite the many treatments available for UI, none comprehensively addresses UI and its common comorbidities.
The role of duloxetine. Normal micturition is regulated by the somatic nervous system and an autonomic reflex arc; the neurotransmitters serotonin and norepinephrine play an important role in the neural regulation of micturition and urinary continence. Duloxetine, alone or as an adjunctive treatment, is a potential novel therapy that treats 2 common comorbidities of UI—chronic pain and depression.
As a selective serotonin norepinephrine reuptake inhibitor (SNRI), duloxetine acts at the molecular level to block the reuptake of serotonin and norepinephrine from synaptic clefts. Specifically, the medication blocks the 5-hydroxytryptamine (5-HT) reuptake transporters, as well as the norepinephrine transporters, of pre-synaptic neurons.14 Thus, the concentrations of 5-HT and norepinephrine increase in the synaptic cleft.
Functionally, the accumulation of norepinephrine inhibits micturition by relaxing the detrusor muscle and constricting the urethral smooth muscle. In addition, a higher concentration of 5-HT at the neuromuscular junction leads to constriction of the external urethral sphincter.
Duloxetine has been shown to be effective in the treatment of other types of UI, such as stress UI15 and mixed UI.16 Additionally, it was found to be effective when compared with placebo in women with overactive bladder syndrome17 and in women with multiple sclerosis and depression.18 However, we are not aware of any cases using duloxetine for the treatment of neurogenic UI.
THE TAKEAWAY
Duloxetine is a potential novel drug choice for the treatment of neurogenic UI. Its effects on serotonin and norepinephrine at the synaptic cleft and neuromuscular junction could provide relief for those who have not found relief from other therapies. Further research—particularly a prospective, randomized controlled trial—is needed to determine if duloxetine is, in fact, more than just a theoretical candidate to treat UI and, if so, the most effective dosing.
Offering duloxetine for the treatment of neurogenic UI would potentially address coexisting conditions—such as pain or depression—thus improving patient compliance and reducing health care spending. Before beginning therapy, urodynamic studies to identify the type of UI should be completed, or, at a minimum, post-void residual volume should be measured.
ACKNOWLEDGEMENTS
The authors would like to thank Julie Hughbanks, MLS, Library Manager, Parkview Health Resource Library, for her assistance with the library searches used for this case report.
THE CASE
A 57-year-old African American woman was being treated at our clinic for neurogenic urinary incontinence (UI). The UI, which occurred day and night, began 2 years earlier following a laminectomy of vertebrae C3 to C6 with spinal fusion of C3 to C7 for cervical spinal stenosis. The UI persisted despite physical therapy and trials of oxybutynin and imipramine. Since the surgery, the patient had also been experiencing chronic (debilitating) neuropathic pain in both legs, and the sensation of incomplete bladder emptying. She denied bowel incontinence or saddle anesthesia. Her prescription medications included hydrocodone-acetaminophen 7.5/325 mg every 6 hours as needed for pain and lisinopril 20 mg/d for essential hypertension. The patient’s body mass index (BMI) was 23.3.
A urine culture initially grew Klebsiella pneumoniae, which we successfully treated with ciprofloxacin. A urinalysis was unremarkable, and blood urea nitrogen and creatinine levels were within normal limits.
We started the patient on oral duloxetine 30 mg/d for her neuropathic pain. The patient hadn’t undergone a urologic evaluation before starting duloxetine, so no urodynamic studies or measurements had been conducted. At that point, we sent the patient to a urologist for an evaluation.
At a follow-up visit with one of our clinic providers <3 months later, the patient reported that the duloxetine was providing her with some pain relief and that she was “waking up dry” in the mornings and having fewer UI symptoms throughout the day, as well as at night. The patient denied any adverse effects such as nausea, gastrointestinal upset, weight changes, xerostomia, fatigue, insomnia, headaches, or dizziness. Duloxetine was titrated up to 60 mg/d for better control of her neuropathic pain. At the next follow-up visit at our clinic 3 months later, her UI was 80% to 90% improved and she was able to stop her opioid pain medications.
DISCUSSION
UI is a significant problem in the United States and around the world. For women, the prevalence of UI ranges from 15% to 69%; among men, the prevalence is 5% to 24%.1-3 The economic burden of UI includes both medical and nonmedical (eg, pads, diapers, laundry, and dry cleaning) care. The total national cost was estimated at $66 billion in 2007: $49 billion for direct medical costs, $2 billion for direct nonmedical costs, and $15 billion for indirect costs.4 And those costs are expected to increase 25% by 2020, mainly because of the aging population.
Risk factors for UI other than gender include advancing age, obesity, non-Hispanic white race, depression, hypertension, type 2 diabetes mellitus, neurologic disease, and functional limitations/general poor health.5-7 Comorbid depression and BMI >30, as well as the presence and duration of diabetes, increase the odds for developing UI.7,8
Risk factors for women include hysterectomy,7 increasing parity, and delivery of at least one infant >9.5 pounds; the risk is the same for both vaginal and cesarean-section delivery.6 Specific risk factors for men include prostate cancer, prostate surgery, and prostate radiation.5
Significant, chronic comorbidities of UI include depression and chronic pain. While quality of life is negatively affected by UI alone, the coexistence of depression and UI produces an additive negative effect on quality of life.9
Types and treatment of UI
There are 5 types of UI: urge, stress, overflow, functional, and mixed.10
- Urge incontinence is the leakage of urine following a sensation of sudden urgency to void.
- Stress incontinence is urine leakage associated with increased intra-abdominal pressure such as with coughing or sneezing and is typically associated with weakened pelvic floor musculature.
- Overflow incontinence is more common in men, and is typically caused by prostatic disease. The urethral outlet is obstructed leading to increased pressure within the bladder and subsequent leakage of urine.
- Functional incontinence is caused by physical or cognitive impairment leading to a decreased ability to get to a bathroom quickly enough to void.
- Mixed incontinence is when symptoms of stress and urgency incontinence are present.
There are 3 broad categories of treatment methods for urinary incontinence: behavioral, pharmacologic, and surgical. Behavioral interventions are subdivided into caregiver-dependent (prompted voiding, habit retraining, and timed voiding) and patient-directed (bladder training, pelvic floor muscle training, strategies for bladder control, education, and self-monitoring) techniques. Pharmacologic treatment typically consists of antimuscarinics (eg, oxybutynin, tolterodine, solifenacin) and tricyclic antidepressants (eg, imipramine).11 Injections of onabotulinumtoxinA into the detrusor muscle have also been shown to reduce the symptoms of urinary incontinence.12 Surgical options for treatment of UI include retro-pubic suspension, slings, and, in some instances, artificial urethral sphincters.13
A novel treatment for neurogenic UI?
Despite the many treatments available for UI, none comprehensively addresses UI and its common comorbidities.
The role of duloxetine. Normal micturition is regulated by the somatic nervous system and an autonomic reflex arc; the neurotransmitters serotonin and norepinephrine play an important role in the neural regulation of micturition and urinary continence. Duloxetine, alone or as an adjunctive treatment, is a potential novel therapy that treats 2 common comorbidities of UI—chronic pain and depression.
As a selective serotonin norepinephrine reuptake inhibitor (SNRI), duloxetine acts at the molecular level to block the reuptake of serotonin and norepinephrine from synaptic clefts. Specifically, the medication blocks the 5-hydroxytryptamine (5-HT) reuptake transporters, as well as the norepinephrine transporters, of pre-synaptic neurons.14 Thus, the concentrations of 5-HT and norepinephrine increase in the synaptic cleft.
Functionally, the accumulation of norepinephrine inhibits micturition by relaxing the detrusor muscle and constricting the urethral smooth muscle. In addition, a higher concentration of 5-HT at the neuromuscular junction leads to constriction of the external urethral sphincter.
Duloxetine has been shown to be effective in the treatment of other types of UI, such as stress UI15 and mixed UI.16 Additionally, it was found to be effective when compared with placebo in women with overactive bladder syndrome17 and in women with multiple sclerosis and depression.18 However, we are not aware of any cases using duloxetine for the treatment of neurogenic UI.
THE TAKEAWAY
Duloxetine is a potential novel drug choice for the treatment of neurogenic UI. Its effects on serotonin and norepinephrine at the synaptic cleft and neuromuscular junction could provide relief for those who have not found relief from other therapies. Further research—particularly a prospective, randomized controlled trial—is needed to determine if duloxetine is, in fact, more than just a theoretical candidate to treat UI and, if so, the most effective dosing.
Offering duloxetine for the treatment of neurogenic UI would potentially address coexisting conditions—such as pain or depression—thus improving patient compliance and reducing health care spending. Before beginning therapy, urodynamic studies to identify the type of UI should be completed, or, at a minimum, post-void residual volume should be measured.
ACKNOWLEDGEMENTS
The authors would like to thank Julie Hughbanks, MLS, Library Manager, Parkview Health Resource Library, for her assistance with the library searches used for this case report.
1. Markland AD, Richter HE, Fwu CW, et al. Prevalence and trends of urinary incontinence in adults in the United States, 2001 to 2008. J Urol. 2011;186:589-593.
2. Buckley BS, Lapitan MC; Epidemiology Committee of the Fourth International Consultation on Incontinence, Paris, 2008. Prevalence of urinary incontinence in men, women, and children—current evidence: findings of the Fourth International Consultation on Incontinence. Urology. 2010;76:265-270.
3. Gorina Y, Schappert S, Bercovitz A, et al. Prevalence of incontinence among older Americans. Vital Health Stat 3. 2014;1-33.
4. Coyne KS, Wein A, Nicholson S, et al. Economic burden of urgency urinary incontinence in the United States: a systematic review. J Manag Care Pharm. 2014;20:130-140.
5. Shamliyan TA, Wyman JF, Ping R, et al. Male urinary incontinence: prevalence, risk factors, and preventive interventions. Rev Urol. 2009;11:145-165.
6. Matthews CA, Whitehead WE, Townsend MK, et al. Risk factors for urinary, fecal, or dual incontinence in the Nurses’ Health Study. Obstet Gynecol. 2013;122:539-545.
7. Danforth KN, Townsend MK, Lifford K, et al. Risk factors for urinary incontinence among middle-aged women. Am J Obstet Gynecol. 2006;194:339-345.
8. Lifford KL, Curhan GC, Hu FB, et al. Type 2 diabetes mellitus and risk of developing urinary incontinence. J Am Geriatr Soc. 2005;53:1851-1857.
9. Avery JC, Stocks NP, Duggan P, et al. Identifying the quality of life effects of urinary incontinence with depression in an Australian population. BMC Urol. 2013;13:11.
10. National Kidney and Urologic Diseases Information Clearinghouse. Urinary incontinence in women. Available at: http://kidney.niddk.nih.gov/KUDISEASES/pubs/uiwomen/UI-Women_508.pdf. Accessed January 2, 2015.
11. Ontario Medical Advisory Secretariat. Behavioural interventions for urinary incontinence in community-dwelling seniors: an evidence-based analysis. Ontario Health Technology Assessment Series. 2008:8. Available at: http://www.hqontario.ca/Portals/0/Documents/evidence/reports/rev_aic_ui_20081002.pdf. Accessed November 30, 2015.
12. Cox L, Cameron A. OnabotulinumtoxinA for the treatment of overactive bladder. Res Rep Urol. 2014;6:79-89.
13. Dmochowski RR, Blaivas JM, Gormley EA, et al. Update of AUA guideline on the surgical management of female stress urinary incontinence. J Urol. 2010;183:1906-1914.
14. Duloxetine. US National Library of Medicine: National Center for Biotechnology Information. 2015. Available at: http://pubchem.ncbi.nlm.nih.gov/compound/duloxetine. Accessed October 20, 2015.
15. Li J, Yang L, Pu C, et al. The role of duloxetine in stress urinary incontinence: a systematic review and meta-analysis. Int Urol Nephrol. 2013;45:679-686.
16. Bent AE, Gousse AE, Hendrix SL, et al. Duloxetine compared with placebo for the treatment of women with mixed urinary incontinence. Neurourol Urodyn. 2008;27:212-221.
17. Steers WD, Herschorn S, Kreder KJ, et al; Duloxetine OAB Study Group. Duloxetine compared with placebo for treating women with symptoms of overactive bladder. BJU Int. 2007;100:337-345.
18. Di Rezze S, Frasca V, Inghilleri M, et al. Duloxetine for the treatment of overactive bladder syndrome in multiple sclerosis: a pilot study. Clin Neuropharmacol. 2012;35:231-234.
1. Markland AD, Richter HE, Fwu CW, et al. Prevalence and trends of urinary incontinence in adults in the United States, 2001 to 2008. J Urol. 2011;186:589-593.
2. Buckley BS, Lapitan MC; Epidemiology Committee of the Fourth International Consultation on Incontinence, Paris, 2008. Prevalence of urinary incontinence in men, women, and children—current evidence: findings of the Fourth International Consultation on Incontinence. Urology. 2010;76:265-270.
3. Gorina Y, Schappert S, Bercovitz A, et al. Prevalence of incontinence among older Americans. Vital Health Stat 3. 2014;1-33.
4. Coyne KS, Wein A, Nicholson S, et al. Economic burden of urgency urinary incontinence in the United States: a systematic review. J Manag Care Pharm. 2014;20:130-140.
5. Shamliyan TA, Wyman JF, Ping R, et al. Male urinary incontinence: prevalence, risk factors, and preventive interventions. Rev Urol. 2009;11:145-165.
6. Matthews CA, Whitehead WE, Townsend MK, et al. Risk factors for urinary, fecal, or dual incontinence in the Nurses’ Health Study. Obstet Gynecol. 2013;122:539-545.
7. Danforth KN, Townsend MK, Lifford K, et al. Risk factors for urinary incontinence among middle-aged women. Am J Obstet Gynecol. 2006;194:339-345.
8. Lifford KL, Curhan GC, Hu FB, et al. Type 2 diabetes mellitus and risk of developing urinary incontinence. J Am Geriatr Soc. 2005;53:1851-1857.
9. Avery JC, Stocks NP, Duggan P, et al. Identifying the quality of life effects of urinary incontinence with depression in an Australian population. BMC Urol. 2013;13:11.
10. National Kidney and Urologic Diseases Information Clearinghouse. Urinary incontinence in women. Available at: http://kidney.niddk.nih.gov/KUDISEASES/pubs/uiwomen/UI-Women_508.pdf. Accessed January 2, 2015.
11. Ontario Medical Advisory Secretariat. Behavioural interventions for urinary incontinence in community-dwelling seniors: an evidence-based analysis. Ontario Health Technology Assessment Series. 2008:8. Available at: http://www.hqontario.ca/Portals/0/Documents/evidence/reports/rev_aic_ui_20081002.pdf. Accessed November 30, 2015.
12. Cox L, Cameron A. OnabotulinumtoxinA for the treatment of overactive bladder. Res Rep Urol. 2014;6:79-89.
13. Dmochowski RR, Blaivas JM, Gormley EA, et al. Update of AUA guideline on the surgical management of female stress urinary incontinence. J Urol. 2010;183:1906-1914.
14. Duloxetine. US National Library of Medicine: National Center for Biotechnology Information. 2015. Available at: http://pubchem.ncbi.nlm.nih.gov/compound/duloxetine. Accessed October 20, 2015.
15. Li J, Yang L, Pu C, et al. The role of duloxetine in stress urinary incontinence: a systematic review and meta-analysis. Int Urol Nephrol. 2013;45:679-686.
16. Bent AE, Gousse AE, Hendrix SL, et al. Duloxetine compared with placebo for the treatment of women with mixed urinary incontinence. Neurourol Urodyn. 2008;27:212-221.
17. Steers WD, Herschorn S, Kreder KJ, et al; Duloxetine OAB Study Group. Duloxetine compared with placebo for treating women with symptoms of overactive bladder. BJU Int. 2007;100:337-345.
18. Di Rezze S, Frasca V, Inghilleri M, et al. Duloxetine for the treatment of overactive bladder syndrome in multiple sclerosis: a pilot study. Clin Neuropharmacol. 2012;35:231-234.

The one thing that’s missing from the health care debate
The Affordable Care Act (aka Obamacare) may soon be out and the American Health Care Act (AHCA) may soon be in. Despite all of the rhetoric about making health care affordable by reducing insurance premiums, one thing has been conspicuously absent from the debate: how we are going to reduce the actual cost of health care. Yes, the AHCA may help reduce premiums, but what is most likely to result is not less expensive health care, but rather people paying less money on premiums and more out of their pockets for medicines and treatments. Especially troublesome is that older and sicker patients will be hit the hardest.
The American conundrum. Why do Americans pay twice what citizens of most other developed nations pay and get health care outcomes that are worse?1,2 Two reasons are that those who provide health care charge more in this country for services and medications, and physicians do a lot more testing and treatment here than their counterparts abroad.
One expert estimated that up to $700 billion could be saved by eliminating testing and treatments that provide marginal or no value to patients.3 For example, knee arthroscopy for moderate knee osteoarthritis produces no better outcomes than medical management.4 And many medications are much more expensive in the United States than in other countries. It seems that pharmaceutical companies are permitted greater profits here than elsewhere in the world, and these profits are at the expense of sick people and taxpayers.
How do we bend the cost curve downward? This is a tough question with no single correct answer, but we can all help. Some health care organizations have already reduced costs significantly without sacrificing quality by using team-based primary care as their foundation. Two examples are Nuka Health and Iora Health.5,6
As primary care physicians, we are in an ideal position to constrain unnecessary testing and treatments by establishing trusting relationships with patients, who will believe us when we tell them they don’t need an antibiotic for their chest cold or an MRI for their back pain.
If we control the cost of providing care, insurance premiums will follow suit.
1. The Commonwealth Fund. U.S. health care from a global perspective. Available at: http://www.commonwealthfund.org/publications/issue-briefs/2015/oct/us-health-care-from-a-global-perspective. Accessed May 14, 2017.
2. The Commonwealth Fund. US health system ranks last among eleven countries on measures of access, equity, quality, efficiency, and healthy lives. Available at: http://www.commonwealthfund.org/publications/press-releases/2014/jun/us-health-system-ranks-last. Accessed May 14, 2017.
3. Kelley R. Where can $700 billion in waste be cut annually from the U.S. healthcare system? Available at: http://www.hcca-info.org/Portals/0/PDFs/Resources/Conference_Handouts/Compliance_Institute/2010/P8handout6.pdf. Accessed May 14, 2017.
4. Kirkley A, Birmingham TB, Litchfield RB, et al. A randomized trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2008;359:1097-1107.
5. Gottlieb K. The Nuka System of Care: improving health through ownership and relationships. Int J Circumpolar Health. 2013;72. doi: 10.3402/ijch.v72i0.21118.
6. Iorahealth. Available at: www.iorahealth.com. Accessed May 14, 2017.
Editor-in-Chief

Editor-in-Chief
Editor-in-Chief
The Affordable Care Act (aka Obamacare) may soon be out and the American Health Care Act (AHCA) may soon be in. Despite all of the rhetoric about making health care affordable by reducing insurance premiums, one thing has been conspicuously absent from the debate: how we are going to reduce the actual cost of health care. Yes, the AHCA may help reduce premiums, but what is most likely to result is not less expensive health care, but rather people paying less money on premiums and more out of their pockets for medicines and treatments. Especially troublesome is that older and sicker patients will be hit the hardest.
The American conundrum. Why do Americans pay twice what citizens of most other developed nations pay and get health care outcomes that are worse?1,2 Two reasons are that those who provide health care charge more in this country for services and medications, and physicians do a lot more testing and treatment here than their counterparts abroad.
One expert estimated that up to $700 billion could be saved by eliminating testing and treatments that provide marginal or no value to patients.3 For example, knee arthroscopy for moderate knee osteoarthritis produces no better outcomes than medical management.4 And many medications are much more expensive in the United States than in other countries. It seems that pharmaceutical companies are permitted greater profits here than elsewhere in the world, and these profits are at the expense of sick people and taxpayers.
How do we bend the cost curve downward? This is a tough question with no single correct answer, but we can all help. Some health care organizations have already reduced costs significantly without sacrificing quality by using team-based primary care as their foundation. Two examples are Nuka Health and Iora Health.5,6
As primary care physicians, we are in an ideal position to constrain unnecessary testing and treatments by establishing trusting relationships with patients, who will believe us when we tell them they don’t need an antibiotic for their chest cold or an MRI for their back pain.
If we control the cost of providing care, insurance premiums will follow suit.
The Affordable Care Act (aka Obamacare) may soon be out and the American Health Care Act (AHCA) may soon be in. Despite all of the rhetoric about making health care affordable by reducing insurance premiums, one thing has been conspicuously absent from the debate: how we are going to reduce the actual cost of health care. Yes, the AHCA may help reduce premiums, but what is most likely to result is not less expensive health care, but rather people paying less money on premiums and more out of their pockets for medicines and treatments. Especially troublesome is that older and sicker patients will be hit the hardest.
The American conundrum. Why do Americans pay twice what citizens of most other developed nations pay and get health care outcomes that are worse?1,2 Two reasons are that those who provide health care charge more in this country for services and medications, and physicians do a lot more testing and treatment here than their counterparts abroad.
One expert estimated that up to $700 billion could be saved by eliminating testing and treatments that provide marginal or no value to patients.3 For example, knee arthroscopy for moderate knee osteoarthritis produces no better outcomes than medical management.4 And many medications are much more expensive in the United States than in other countries. It seems that pharmaceutical companies are permitted greater profits here than elsewhere in the world, and these profits are at the expense of sick people and taxpayers.
How do we bend the cost curve downward? This is a tough question with no single correct answer, but we can all help. Some health care organizations have already reduced costs significantly without sacrificing quality by using team-based primary care as their foundation. Two examples are Nuka Health and Iora Health.5,6
As primary care physicians, we are in an ideal position to constrain unnecessary testing and treatments by establishing trusting relationships with patients, who will believe us when we tell them they don’t need an antibiotic for their chest cold or an MRI for their back pain.
If we control the cost of providing care, insurance premiums will follow suit.
1. The Commonwealth Fund. U.S. health care from a global perspective. Available at: http://www.commonwealthfund.org/publications/issue-briefs/2015/oct/us-health-care-from-a-global-perspective. Accessed May 14, 2017.
2. The Commonwealth Fund. US health system ranks last among eleven countries on measures of access, equity, quality, efficiency, and healthy lives. Available at: http://www.commonwealthfund.org/publications/press-releases/2014/jun/us-health-system-ranks-last. Accessed May 14, 2017.
3. Kelley R. Where can $700 billion in waste be cut annually from the U.S. healthcare system? Available at: http://www.hcca-info.org/Portals/0/PDFs/Resources/Conference_Handouts/Compliance_Institute/2010/P8handout6.pdf. Accessed May 14, 2017.
4. Kirkley A, Birmingham TB, Litchfield RB, et al. A randomized trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2008;359:1097-1107.
5. Gottlieb K. The Nuka System of Care: improving health through ownership and relationships. Int J Circumpolar Health. 2013;72. doi: 10.3402/ijch.v72i0.21118.
6. Iorahealth. Available at: www.iorahealth.com. Accessed May 14, 2017.
1. The Commonwealth Fund. U.S. health care from a global perspective. Available at: http://www.commonwealthfund.org/publications/issue-briefs/2015/oct/us-health-care-from-a-global-perspective. Accessed May 14, 2017.
2. The Commonwealth Fund. US health system ranks last among eleven countries on measures of access, equity, quality, efficiency, and healthy lives. Available at: http://www.commonwealthfund.org/publications/press-releases/2014/jun/us-health-system-ranks-last. Accessed May 14, 2017.
3. Kelley R. Where can $700 billion in waste be cut annually from the U.S. healthcare system? Available at: http://www.hcca-info.org/Portals/0/PDFs/Resources/Conference_Handouts/Compliance_Institute/2010/P8handout6.pdf. Accessed May 14, 2017.
4. Kirkley A, Birmingham TB, Litchfield RB, et al. A randomized trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2008;359:1097-1107.
5. Gottlieb K. The Nuka System of Care: improving health through ownership and relationships. Int J Circumpolar Health. 2013;72. doi: 10.3402/ijch.v72i0.21118.
6. Iorahealth. Available at: www.iorahealth.com. Accessed May 14, 2017.
Post-bariatric surgery patients: Your role in their long-term care
More than one-third of American adults and approximately 17% of children and adolescents between the ages of 2 and 19 years are obese.1,2 Poor diet coupled with a sedentary lifestyle is the highest ranked cause of non-communicable disease and a leading cause of preventable death, according to the National Research Council.3
Bariatric surgery (BS) is a viable therapeutic option for obese patients who do not respond to conventional lifestyle interventions for losing weight. There are multiple gastrointestinal (GI) procedures available that are classified as either malabsorptive (Roux-en-Y gastric bypass [RYGB] and biliopancreatic diversion [BPD] with or without duodenal switch) or restrictive (laparoscopic adjustable gastric banding [LAGB] and vertical sleeve gastrectomy [VSG]).
Approximately half of the 196,000 bariatric procedures performed in the United States in 2015 were of the sleeve variety, another 23% were RYGB, and the remaining percentage was divided among the other types.4 Postoperative risks include nutritional deficiencies, decreased bone mineral density (BMD), dumping syndrome (when food rapidly dumps from the stomach to the intestine), and gastroesophageal reflux disease (GERD) with possible ulceration.
Despite these potential complications, a systematic review and meta-analysis found that obese people who underwent BS (gastric banding or gastric bypass) had significantly reduced risks of global, non-cardiovascular (CV), and CV mortality compared with obese controls.5 Helping patients to realize these benefits requires that the entire health care team—especially the family physician—is aware of the special considerations for this population.
To that end, this article reviews the details of diagnosing and managing post-surgical complications. It also addresses issues unique to managing certain subpopulations, such as post-BS patients who require revision surgery or who want to pursue body contouring surgery; adolescents who undergo BS surgery; and women who want to get pregnant postoperatively.
Monitor patients for these post-surgery complications
Postoperative BS follow-up varies depending on location, surgeon preference, and availability of multidisciplinary resources. At our institution, patients have a minimum of 3 follow-up visits with their surgeon (during hospitalization and 2 weeks and 2 months postoperatively). This is followed by visits with Endocrinology 6 months after surgery and annually thereafter. Given the variability of follow-up, family physicians should coordinate with specialists where appropriate and be aware of postoperative complications and monitoring since it is likely they will have the most frequent contact with these patients.
Nutritional deficiencies are common and require lifelong screening
Nutritional deficiencies are the most common complications of malabsorptive BS. Guidelines from the Endocrine Society, as well as guidelines from the American Association of Clinical Endocrinologists (AACE), The Obesity Society (TOS), and the American Society for Metabolic and Bariatric Surgery (ASMBS), recommend routine lifetime screening for deficiencies after surgery.6,7 Complete blood cell count, electrolytes, glucose, creatinine, and liver function tests should be obtained at one, 3, 6, 12, 18, and 24 months following surgery and annually thereafter.6
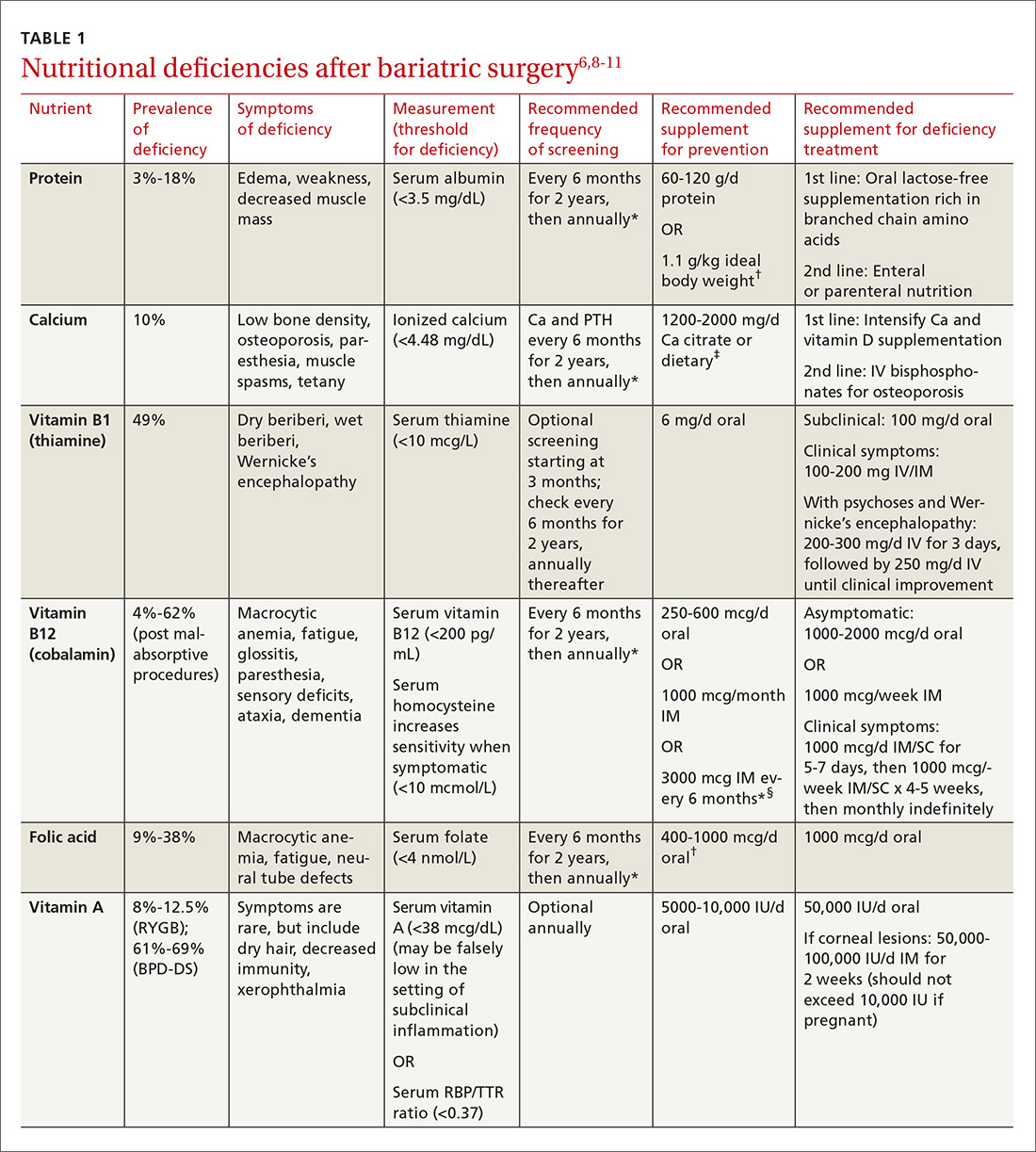
Multiple factors contribute to nutritional and micronutrient deficiencies, including reduced oral intake of food, decreased GI absorption, food intolerance, nausea/vomiting, and nonadherence with dietary supplements.8 Oral supplementation should be in chewable, powder, or liquid form because pill and capsule absorption may be altered.8,9 Over-the-counter multivitamins may not contain the requisite daily doses recommended after BS.9 Patients and physicians should evaluate supplements together to ensure appropriate nutritional and micronutrient supplementation (TABLE 16,8-11).
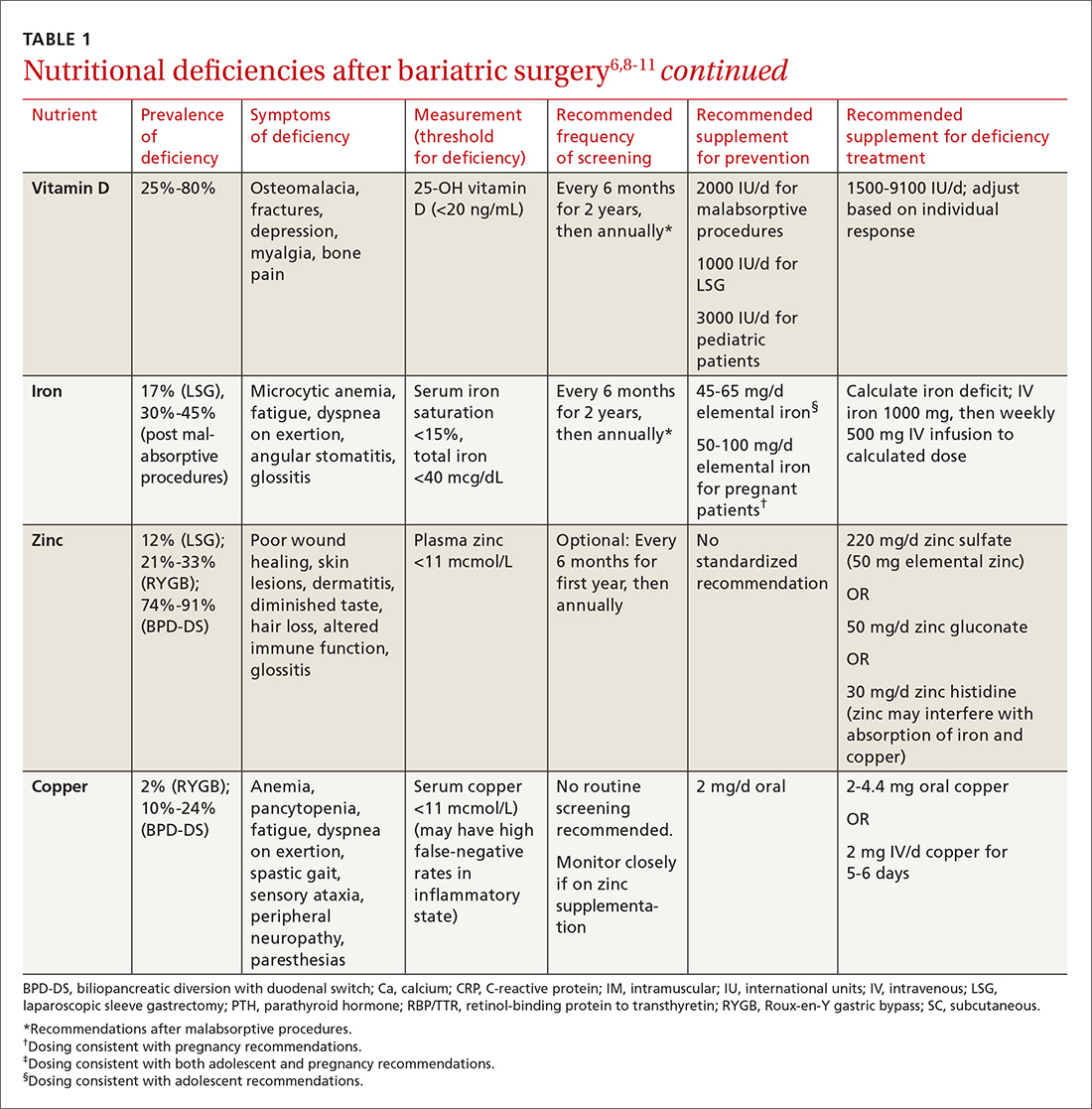
Bone mineral density can start to decrease soon after surgery
Studies evaluating BMD after BS have produced variable findings. In obese patients, dual-energy x-ray absorptiometry (DEXA) measurements may not be accurate due to adipose tissue artifact and table weight limits. In addition, limited data exist on the incidence of fractures after BS. Of 2 notable studies, only one, a population-based study involving 258 Minnesota residents who underwent a first bariatric surgery between 1985 and 2004, demonstrated a significantly increased incidence of fractures.12,13
In addition, studies show bone turnover markers, including C-terminal telopeptide, increase as early as 3 months after BS.14 Several guidelines recommend routine BMD screening after BS (TABLE 2).6,7 The mechanism of bone demineralization is likely multifactorial—a function of the magnitude of the weight loss and skeletal unloading, calcium and vitamin D deficiencies, and associated secondary hyperparathyroidism.15 Treatment for secondary hyperparathyroidism is adequate supplementation with vitamin D and calcium.

Optimal dosing for vitamin D has not been determined. One recent systematic review suggests routine prophylaxis with at least 2000 international units (IU)/d and found the greatest improvement for known deficiency with doses of 1500-9100 IU/d following malabsorptive surgeries.11 After laparoscopic sleeve gastrectomy, at least 1000 IU/d vitamin D is recommended.11
Overall, high variability exists among patients, and an individualized approach for dosing is recommended.11 Vitamin D levels should be monitored 2 and 4 weeks after initiation of treatment and every 3 months thereafter.11 Normal levels of serum calcium, 25-OH vitamin D, bone-specific alkaline phosphatase, and 24-hour urinary calcium excretion indicate adequate calcium and vitamin D supplementation.6
Dumping syndrome can lead to hypoglycemia
Dumping syndrome is a common complication following BS, with prevalence ranging from 25% to 75%, depending upon the type of procedure performed.16,17 There are 2 types: early and late. Early dumping syndrome occurs within 30 minutes of eating. Symptoms are related to the robust release of gastrointestinal hormones caused by rapid gastric emptying. Symptoms include nausea, abdominal pain, diarrhea, flushing, hypotension, and tachycardia.
Late dumping is characterized as postprandial hypoglycemia occurring one to 3 hours after eating. Late dumping is likely caused by a combination of changes within the pancreatic beta cells and abnormal insulin response to glucose.16-18 Rapid gastric emptying leads to rapid release of glucose in the gut, which, in turn, leads to brisk insulin secretion. Since glucose is absorbed faster than insulin’s half-life, the resulting (relatively) high levels of insulin may cause hypoglycemia.16-18
Sigstad’s scoring system can be used to confirm suspected cases of dumping syndrome (TABLE 316,17,19). A diagnosis can also be made with an oral glucose challenge in which pulse, blood pressure, glucose, and hematocrit are measured after ingestion of 50 g glucose. The test is positive if heart rate increases by 10 beats per minute, hematocrit increases by 3% 30 minutes after ingestion, or glucose falls below 60 mg/dL 2 to 3 hours after ingestion.17
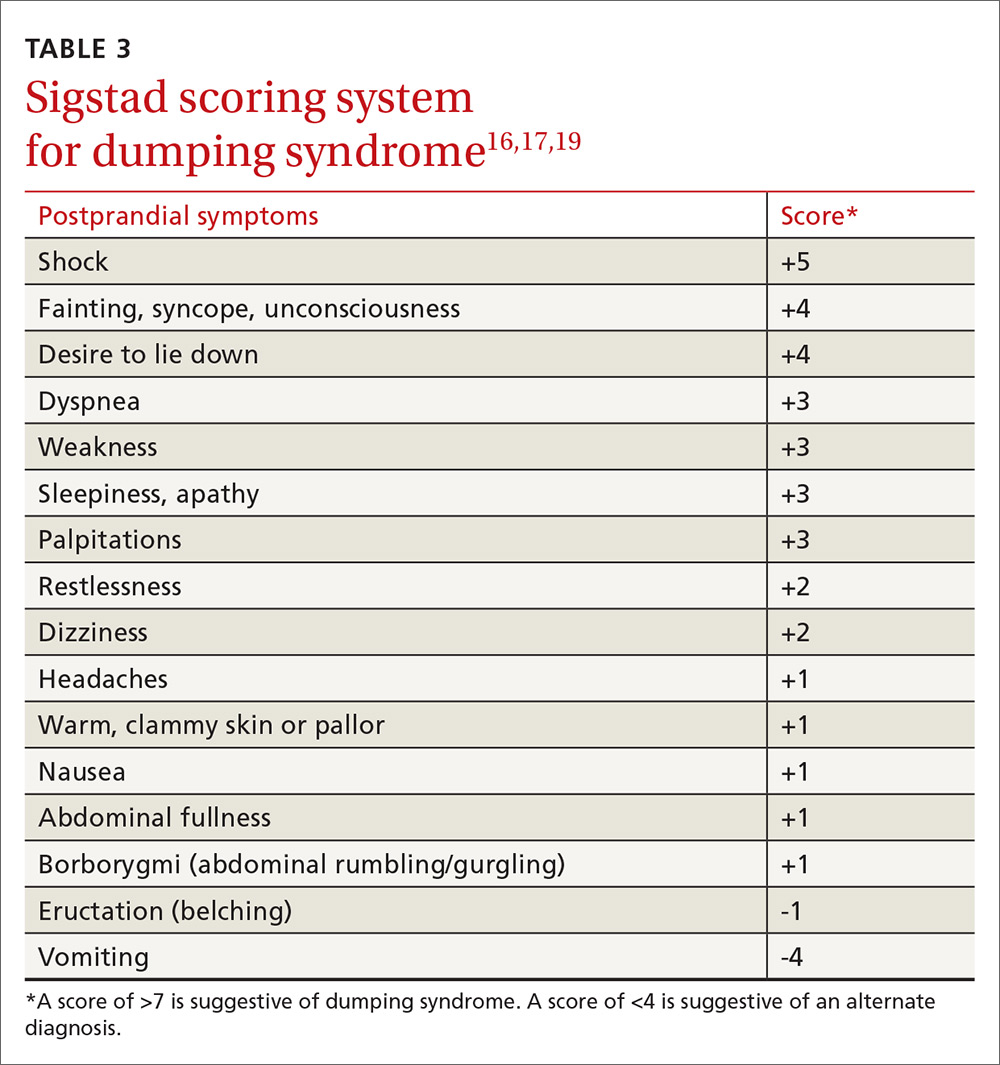
First-line treatment of dumping syndrome consists of dietary modifications. The goal is to slow the rate of gastric emptying by eating smaller, more frequent meals; separating beverages from food; decreasing carbohydrates; and increasing fiber and protein content.
If results are suboptimal after dietary changes, medications can be prescribed including acarbose to prevent postprandial hypoglycemia; anticholinergics such as dicyclomine to slow gastric emptying; and somatostatin to decrease gastric emptying and inhibit GI hormone release.17 Lastly, for resistant and severe postprandial hypoglycemia, a few patients have undergone pancreatectomy, but only about 65% experienced improvement in symptoms and 12% developed diabetes post-surgically.20
Gout attacks may initially increase, but then decrease
BS affects the incidence of gout attacks in patients with a history of gout. One comparative study of approximately 150 patients demonstrated that those with a history of gout had significantly more gout attacks in the first month after BS compared with obese patients with a history of gout undergoing other upper GI surgeries.21 There was no difference between malabsorptive and restrictive procedures. But after the first month, BS patients had significantly fewer gout attacks and lower uric acid levels than their obese counterparts.21
Protein rich diets, catabolism potentiated by aggressive caloric restriction following BS, and dehydration contribute to the initial increase. Therefore, patients who have had at least one gout attack in the year prior to surgery or who are on hypouricemic medication may benefit from at least one month of prophylactic therapy (eg, allopurinol and colchicine) after surgery.
GERD and ulceration: How to respond
Obesity is a known risk factor for GERD, but the effect of BS on GERD is uncertain and seems to vary with the procedure performed. RYGB decreases GERD and is, therefore, used as both a secondary treatment in those not responding to medications and a revision treatment for fundoplication and other types of BSs. Sleeve gastrectomy and adjustable gastric banding have mixed effects on GERD. A systematic review by de Jong et al revealed a decreased prevalence of reflux symptoms and GERD medication use after LAGB; however, during longer follow-up, 15% of previously unaffected patients reported experiencing GERD.22 The 2011 International Sleeve Gastrectomy Expert Panel Consensus Statement retrospectively noted a postoperative incidence of GERD as high as 31%.23
BS patients with GERD should be treated with a proton pump inhibitor. If this fails, refer patients to a gastroenterologist for further evaluation.24
Ulcers after BS may be an indication for revision surgery. Data are mixed regarding increased risk of marginal ulceration from nonsteroidal anti-inflammatory drug (NSAID) use, but NSAIDs have been linked to an increased risk of anastomotic leakage.25-28 Thus, it seems prudent to avoid NSAIDs in people who have undergone BS.
Keeping watch over psychiatric comorbidities
A recent meta-analysis by Dawes et al29 showed that about 23% of patients pursuing BS have a comorbid mood disorder. Specifically, the preoperative prevalence of depression (19%) and binge-eating disorder (17%) were found to be higher than rates in the general population.29 The meta-analysis found improvement in the prevalence of depression with fewer symptoms and less antidepressant medication use in the first 3 years after surgery and a decrease in the rate of binge-eating disorder, although there were fewer supporting data for the latter. These findings were observed with both restrictive and malabsorptive procedures.
The data are mixed regarding rates of alcohol abuse and suicide. Further research is necessary in this field. Patients who have had BS should receive ongoing psychiatric and psychological care from a multidisciplinary team as a matter of course.
Will a second surgery be needed?
Revision surgery. In 2015, about 14% of the almost 200,000 BSs performed were revisions.4 Revision surgery is indicated in BS patients with weight regain, recurrent comorbid diseases (eg, diabetes, hypertension), or complications of primary BS. Restrictive procedures have a higher revision rate than malabsorptive procedures, primarily due to a higher rate of weight regain.6,30
Because revision surgery is associated with more complications and possibly longer hospital stays than primary BS, it should be performed by a bariatric surgeon with extensive experience.30,31 Restrictive revisions are typically converted to malabsorptive procedures. Cost is a limiting factor as many patients’ insurance coverage is limited to one BS per lifetime.
Body contouring. Body contouring surgery (BCS) can improve physical and mental well-being and may be a protective factor for weight regain after bariatric surgery.32 Despite its desirability—particularly to women, adolescents, and those with large decreases in body mass index (BMI)—few patients can afford BCS since it is rarely covered by insurance.
Complications of BCS vary, but are most commonly infection and wound dehiscence. This is, in part, due to poorer wound healing in BS patients compared to those with nonsurgical massive weight loss. The cause of poor wound healing is thought to be secondary to nutritional deficiencies and the catabolic state induced by post-surgical weight loss. Recommendations for BCS include weight stability for more than one year after BS, age >16 years, excess skin causing significant functional impairment, non-smoking status, and presence of good social support.33
Bariatric surgery in adolescents is on the rise
Children in the highest body mass index quartile have more than twice the death rate of those in the lowest BMI quartile.34 Thus, it is not surprising that the rate of BS in adolescents is increasing.7 BS in this age group is successful for weight loss and improvement of comorbid conditions, with relatively low complication rates.35 Options include malabsorptive and restrictive procedures, although gastric banding has not been approved by the US Food and Drug Administration for patients under the age of 18 years.
After BS, adolescent girls should be counseled regarding the possibility of pregnancy (restoration of fertility) and appropriate contraception. Adolescent patients require nutritional supplementation after BS as indicated in TABLE 1.6,8-11
When determining which adolescents to refer for BS, we recommend the following criteria: 35-38
- failure of a minimum 6-month trial of a staged treatment approach, as recommended by Barlow et al,36 including diet, exercise, and pharmacologic treatment
- BMI ≥35 with type 2 diabetes or severe sleep apnea (apnea hypopnea index [AHI] >15)37
- BMI ≥40 with mild sleep apnea (AHI >5), hypertension, or pre-diabetes37
- Tanner stage IV or V
- at least 95% skeletal growth (for malabsorptive surgery).37 This can be determined using an estimated adult height from mid-parental height formula and assessing growth plate closure with hand radiographs for bone age
- appropriate maturity level permitting adherence
- good psychological support
- a multidisciplinary team for postoperative and long-term follow-up care.
Planning for the future: Exploring the possibility of pregnancy
Obesity is the primary cause of maternal and fetal morbidity during pregnancy. It is associated with increased rates of early miscarriage, congenital defects, macrosomia, and fetal death. Maternal risks of obesity include: gestational hypertension, gestational diabetes mellitus (GDM), and pre-eclampsia. Obese mothers also have a higher incidence of failed induction, caesarean section, and breastfeeding failure.10,39 Given that half of all BSs are performed in women of reproductive age, this population deserves special consideration.10
A recent meta-analysis by Galazis et al40 concluded that BS performed prior to pregnancy led to decreased rates of preeclampsia, GDM, large neonates, preterm birth, and neonatal intensive care unit admission. Perinatal mortality did not increase after BS. However, BS led to higher rates of maternal anemia. There was no significant difference between groups in incidence of cesarean section.
The post BS female patient should be advised to use a reliable form of contraception for a minimum of 12 to 18 months after surgery.6,10,39 Involve high-risk obstetric specialists during pregnancies. Diet should be supplemented as indicated in TABLE 1.6,8-11
CORRESPONDENCE
Amy Rothberg, MD, PhD, Domino’s Farms, Lobby G, Suite 1500, 24 Frank Lloyd Wright Drive, Ann Arbor, MI 48106; [email protected].
1. Centers for Disease Control and Prevention. Overweight and obesity. Adult obesity facts. Available at: https://www.cdc.gov/obesity/data/adult.html. Accessed April 5, 2017.
2. Centers for Disease Control and Prevention. Overweight and obesity. Childhood obesity facts. Available at: https://www.cdc.gov/obesity/data/childhood.html. Accessed April 5, 2017.
3. McGinnis JM. Actual causes of death, 1990-2010. Workshop on Determinants of Premature Mortality, September 18, 2013, National Research Council, Washington, DC.
4. American Society of Metabolic and Bariatric Surgery. Estimate of bariatric surgery numbers, 2011-2015. Available at: https://asmbs.org/resources/estimate-of-bariatric-surgery-numbers. Accessed April 5, 2017.
5. Pontiroli AE, Morabito A. Long-term prevention of mortality in morbid obesity through bariatric surgery. a systematic review and meta-analysis of trials performed with gastric banding and gastric bypass. Ann Surg. 2011;253:484-487.
6. Heber D, Greenway FL, Kaplan LM, et al. Endocrine and nutritional management of the post-bariatric surgery patient: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4823-4843.
7. Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient—2013 update: cosponsored by American Association of Clinical Endocrinologists, The Obesity Society, and American Society for Metabolic & Bariatric Surgery. Obesity. 2013;21:S1-S27.
8. Stein J, Stier C, Raab H, et al. Review article: the nutritional and pharmacological consequences of obesity surgery. Aliment Pharmacol Ther. 2014;40:582-609.
9. Boyce SG, Goriparthi R, Clark J, et al. Can composite nutritional supplement based on the current guidelines prevent vitamin and mineral deficiency after weight loss surgery? Obes Surg. 2016;26:966-971.
10. Beard JH, Bell RL, Duffy AJ. Reproductive considerations and pregnancy after bariatric surgery: current evidence and recommendations. Obes Surg. 2008;18:1023-1027.
11. Chakhtoura MT, Nakhoul NN, Shawwa K, et al. Hypovitaminosis D in bariatric surgery: a systematic review of observational studies. Metabolism. 2016;65:574-585.
12. Nakamura KM, Haglind EG, Clowes JA, et al. Fracture risk following bariatric surgery: a population-based study. Osteoporosis Int. 2014;25:151-158.
13. Lalmohamed A, de Vries F, Bazelier MT, et al. Risk of fracture after bariatric surgery in the United Kingdom: population-based, retrospective cohort study. BMJ. 2012;345:e5085.
14. Coates PS, Fernstrom JD, Fernstrom MH, et al. Gastric bypass surgery for morbid obesity leads to an increase in bone turnover and a decrease in bone mass. J Clin Endocrinol Metab. 2004;89:1061-1065.
15. Stein EM, Silverberg SJ. Bone loss after bariatric surgery: causes, consequences and management. Lancet Diabetes Endocrinol. 2014;2:165-174.
16. Tack J, Deloose E. Complications of bariatric surgery: dumping syndrome, reflux and vitamin deficiencies. Best Pract Res Clin Gastroenterol. 2014;28:741-749.
17. Berg P, McCallum R. Dumping syndrome: a review of the current concepts of pathophysiology, diagnosis, and treatment. Dig Dis Sci. 2016;61:11-18.
18. Ritz P, Vaurs C, Barigou M, et al. Hypoglycaemia after gastric bypass: mechanisms and treatment. Diabetes Obes Metab. 2016;18:217-223.
19. Sigstad H. A clinical diagnostic index in the diagnosis of the dumping syndrome. Changes in plasma volume and blood sugar after a test meal. Acta Med Scand. 1970;188:479-486.
20. Mala T. Postprandial hyperinsulinemic hypoglycaemia after gastric bypass surgical treatment. Surg Obes Relat Dis. 2014;10:1220-1225.
21. Romero-Talamás H, Daigle CR, Aminian A. The effect of bariatric surgery on gout: a comparative study. Surg Obes Relat Dis. 2014;10:1161-1165.
22. de Jong JR, Besselink MG, van Ramshorst B, et al. Effects of adjustable gastric banding on gastroesophageal reflux and esophageal motility: a systematic review. Obes Rev. 2010;11:297-305.
23. Rosenthal RJ; International Sleeve Gastrectomy Expert Panel. International Sleeve Gastrectomy Expert Panel Consensus Statement: best practice guidelines based on experience of >12,000 cases. Surg Obes Relat Dis. 2012;8:8-19.
24. Altieri MS, Pryor AD. Gastroesophageal reflux disease after bariatric procedures. Surg Clin North Am. 2015;95:579-591.
25. Hakkarainen TW, Steele SR, Bastaworous A, et al. Nonsteroidal anti-inflammatory drugs and the risk for anastomotic failure: a report from Washington State’s Surgical Care and Outcomes Assessment Program (SCOAP). JAMA Surg. 2015;150:223-228.
26. El-Hayek K, Timratana P, Shimizu H, et al. Marginal ulcer after Roux-en-Y gastric bypass: what have we really learned? Surg Endosc. 2012;26:2789-2796.
27. Sverdén E, Mattsson F, Sondén AM, et al. Risk factors for marginal ulcer after gastric bypass surgery for obesity: a population-based cohort study. Ann Surg. 2016;263:733-737.
28. Azagury DE, Abu Dayyeh BK, Greenwalt IT, et al. Marginal ulceration after Roux-en-Y gastric bypass surgery: characteristics, risk factors, treatment, and outcomes. Endoscopy. 2011;43:950-954.
29. Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: A meta-analysis. JAMA. 2016;315:150-163.
30. Ferrer-Márquez MP, Belda-Lozano R, Solvas-Salmerón MJ, et al. Revisional surgery after laparoscopic sleeve gastrectomy. Surg Laparosc Endosc Percutan Tech. 2015;25:6-9.
31. Sanchez H, Cabrera A, Cabrera K, et al. Laparoscopic Roux-en-Y gastric bypass as a revision procedure after restrictive bariatric surgery. Obes Surg. 2008;18:1539-1543.
32. van der Beek ES, Te Riele W, Specken TF, et al. The impact of reconstructive procedures following bariatric surgery on patient well-being and quality of life. Obes Surg. 2010;20:36-41.
33. Ellison JM, Steffen KJ, Sarwer DB. Body contouring after bariatric surgery. Eur Eat Disord Rev. 2015;23:479-487.
34. Franks PW, Hanson RL, Knowler WC, et al. Childhood obesity, other cardiovascular risk factors, and premature death. NEJM. 2010;362:485-489.
35. Gravelle BL, Broyles M. Interventions of weight reduction and prevention in children and adolescents: update. Am J Ther. 2015;22:159-166.
36. Barlow SE. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics. 2007;120:S164-S192.
37. Pratt JS, Lenders CM, Dionne EA, et al. Best practice updates for pediatric/adolescent weight loss surgery. Obesity. 2009;17:901-910.
38. Nogueira I, Hrovat K. Adolescent bariatric surgery: review on nutrition considerations. Nutr Clin Pract. 2014;29:740-746.
39. Nicklas JM, Barbour LA. Optimizing weight for maternal and infant health: tenable, or too late? Expert Rev Endocrinol Metab. 2015;10:227-242.
40. Galazis N, Docheva N, Simillis C, et al. Maternal and neonatal outcomes in women undergoing bariatric surgery: a systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2014;181:45-53.
More than one-third of American adults and approximately 17% of children and adolescents between the ages of 2 and 19 years are obese.1,2 Poor diet coupled with a sedentary lifestyle is the highest ranked cause of non-communicable disease and a leading cause of preventable death, according to the National Research Council.3
Bariatric surgery (BS) is a viable therapeutic option for obese patients who do not respond to conventional lifestyle interventions for losing weight. There are multiple gastrointestinal (GI) procedures available that are classified as either malabsorptive (Roux-en-Y gastric bypass [RYGB] and biliopancreatic diversion [BPD] with or without duodenal switch) or restrictive (laparoscopic adjustable gastric banding [LAGB] and vertical sleeve gastrectomy [VSG]).
Approximately half of the 196,000 bariatric procedures performed in the United States in 2015 were of the sleeve variety, another 23% were RYGB, and the remaining percentage was divided among the other types.4 Postoperative risks include nutritional deficiencies, decreased bone mineral density (BMD), dumping syndrome (when food rapidly dumps from the stomach to the intestine), and gastroesophageal reflux disease (GERD) with possible ulceration.
Despite these potential complications, a systematic review and meta-analysis found that obese people who underwent BS (gastric banding or gastric bypass) had significantly reduced risks of global, non-cardiovascular (CV), and CV mortality compared with obese controls.5 Helping patients to realize these benefits requires that the entire health care team—especially the family physician—is aware of the special considerations for this population.
To that end, this article reviews the details of diagnosing and managing post-surgical complications. It also addresses issues unique to managing certain subpopulations, such as post-BS patients who require revision surgery or who want to pursue body contouring surgery; adolescents who undergo BS surgery; and women who want to get pregnant postoperatively.
Monitor patients for these post-surgery complications
Postoperative BS follow-up varies depending on location, surgeon preference, and availability of multidisciplinary resources. At our institution, patients have a minimum of 3 follow-up visits with their surgeon (during hospitalization and 2 weeks and 2 months postoperatively). This is followed by visits with Endocrinology 6 months after surgery and annually thereafter. Given the variability of follow-up, family physicians should coordinate with specialists where appropriate and be aware of postoperative complications and monitoring since it is likely they will have the most frequent contact with these patients.
Nutritional deficiencies are common and require lifelong screening
Nutritional deficiencies are the most common complications of malabsorptive BS. Guidelines from the Endocrine Society, as well as guidelines from the American Association of Clinical Endocrinologists (AACE), The Obesity Society (TOS), and the American Society for Metabolic and Bariatric Surgery (ASMBS), recommend routine lifetime screening for deficiencies after surgery.6,7 Complete blood cell count, electrolytes, glucose, creatinine, and liver function tests should be obtained at one, 3, 6, 12, 18, and 24 months following surgery and annually thereafter.6
Multiple factors contribute to nutritional and micronutrient deficiencies, including reduced oral intake of food, decreased GI absorption, food intolerance, nausea/vomiting, and nonadherence with dietary supplements.8 Oral supplementation should be in chewable, powder, or liquid form because pill and capsule absorption may be altered.8,9 Over-the-counter multivitamins may not contain the requisite daily doses recommended after BS.9 Patients and physicians should evaluate supplements together to ensure appropriate nutritional and micronutrient supplementation (TABLE 16,8-11).
Bone mineral density can start to decrease soon after surgery
Studies evaluating BMD after BS have produced variable findings. In obese patients, dual-energy x-ray absorptiometry (DEXA) measurements may not be accurate due to adipose tissue artifact and table weight limits. In addition, limited data exist on the incidence of fractures after BS. Of 2 notable studies, only one, a population-based study involving 258 Minnesota residents who underwent a first bariatric surgery between 1985 and 2004, demonstrated a significantly increased incidence of fractures.12,13
In addition, studies show bone turnover markers, including C-terminal telopeptide, increase as early as 3 months after BS.14 Several guidelines recommend routine BMD screening after BS (TABLE 2).6,7 The mechanism of bone demineralization is likely multifactorial—a function of the magnitude of the weight loss and skeletal unloading, calcium and vitamin D deficiencies, and associated secondary hyperparathyroidism.15 Treatment for secondary hyperparathyroidism is adequate supplementation with vitamin D and calcium.
Optimal dosing for vitamin D has not been determined. One recent systematic review suggests routine prophylaxis with at least 2000 international units (IU)/d and found the greatest improvement for known deficiency with doses of 1500-9100 IU/d following malabsorptive surgeries.11 After laparoscopic sleeve gastrectomy, at least 1000 IU/d vitamin D is recommended.11
Overall, high variability exists among patients, and an individualized approach for dosing is recommended.11 Vitamin D levels should be monitored 2 and 4 weeks after initiation of treatment and every 3 months thereafter.11 Normal levels of serum calcium, 25-OH vitamin D, bone-specific alkaline phosphatase, and 24-hour urinary calcium excretion indicate adequate calcium and vitamin D supplementation.6
Dumping syndrome can lead to hypoglycemia
Dumping syndrome is a common complication following BS, with prevalence ranging from 25% to 75%, depending upon the type of procedure performed.16,17 There are 2 types: early and late. Early dumping syndrome occurs within 30 minutes of eating. Symptoms are related to the robust release of gastrointestinal hormones caused by rapid gastric emptying. Symptoms include nausea, abdominal pain, diarrhea, flushing, hypotension, and tachycardia.
Late dumping is characterized as postprandial hypoglycemia occurring one to 3 hours after eating. Late dumping is likely caused by a combination of changes within the pancreatic beta cells and abnormal insulin response to glucose.16-18 Rapid gastric emptying leads to rapid release of glucose in the gut, which, in turn, leads to brisk insulin secretion. Since glucose is absorbed faster than insulin’s half-life, the resulting (relatively) high levels of insulin may cause hypoglycemia.16-18
Sigstad’s scoring system can be used to confirm suspected cases of dumping syndrome (TABLE 316,17,19). A diagnosis can also be made with an oral glucose challenge in which pulse, blood pressure, glucose, and hematocrit are measured after ingestion of 50 g glucose. The test is positive if heart rate increases by 10 beats per minute, hematocrit increases by 3% 30 minutes after ingestion, or glucose falls below 60 mg/dL 2 to 3 hours after ingestion.17
First-line treatment of dumping syndrome consists of dietary modifications. The goal is to slow the rate of gastric emptying by eating smaller, more frequent meals; separating beverages from food; decreasing carbohydrates; and increasing fiber and protein content.
If results are suboptimal after dietary changes, medications can be prescribed including acarbose to prevent postprandial hypoglycemia; anticholinergics such as dicyclomine to slow gastric emptying; and somatostatin to decrease gastric emptying and inhibit GI hormone release.17 Lastly, for resistant and severe postprandial hypoglycemia, a few patients have undergone pancreatectomy, but only about 65% experienced improvement in symptoms and 12% developed diabetes post-surgically.20
Gout attacks may initially increase, but then decrease
BS affects the incidence of gout attacks in patients with a history of gout. One comparative study of approximately 150 patients demonstrated that those with a history of gout had significantly more gout attacks in the first month after BS compared with obese patients with a history of gout undergoing other upper GI surgeries.21 There was no difference between malabsorptive and restrictive procedures. But after the first month, BS patients had significantly fewer gout attacks and lower uric acid levels than their obese counterparts.21
Protein rich diets, catabolism potentiated by aggressive caloric restriction following BS, and dehydration contribute to the initial increase. Therefore, patients who have had at least one gout attack in the year prior to surgery or who are on hypouricemic medication may benefit from at least one month of prophylactic therapy (eg, allopurinol and colchicine) after surgery.
GERD and ulceration: How to respond
Obesity is a known risk factor for GERD, but the effect of BS on GERD is uncertain and seems to vary with the procedure performed. RYGB decreases GERD and is, therefore, used as both a secondary treatment in those not responding to medications and a revision treatment for fundoplication and other types of BSs. Sleeve gastrectomy and adjustable gastric banding have mixed effects on GERD. A systematic review by de Jong et al revealed a decreased prevalence of reflux symptoms and GERD medication use after LAGB; however, during longer follow-up, 15% of previously unaffected patients reported experiencing GERD.22 The 2011 International Sleeve Gastrectomy Expert Panel Consensus Statement retrospectively noted a postoperative incidence of GERD as high as 31%.23
BS patients with GERD should be treated with a proton pump inhibitor. If this fails, refer patients to a gastroenterologist for further evaluation.24
Ulcers after BS may be an indication for revision surgery. Data are mixed regarding increased risk of marginal ulceration from nonsteroidal anti-inflammatory drug (NSAID) use, but NSAIDs have been linked to an increased risk of anastomotic leakage.25-28 Thus, it seems prudent to avoid NSAIDs in people who have undergone BS.
Keeping watch over psychiatric comorbidities
A recent meta-analysis by Dawes et al29 showed that about 23% of patients pursuing BS have a comorbid mood disorder. Specifically, the preoperative prevalence of depression (19%) and binge-eating disorder (17%) were found to be higher than rates in the general population.29 The meta-analysis found improvement in the prevalence of depression with fewer symptoms and less antidepressant medication use in the first 3 years after surgery and a decrease in the rate of binge-eating disorder, although there were fewer supporting data for the latter. These findings were observed with both restrictive and malabsorptive procedures.
The data are mixed regarding rates of alcohol abuse and suicide. Further research is necessary in this field. Patients who have had BS should receive ongoing psychiatric and psychological care from a multidisciplinary team as a matter of course.
Will a second surgery be needed?
Revision surgery. In 2015, about 14% of the almost 200,000 BSs performed were revisions.4 Revision surgery is indicated in BS patients with weight regain, recurrent comorbid diseases (eg, diabetes, hypertension), or complications of primary BS. Restrictive procedures have a higher revision rate than malabsorptive procedures, primarily due to a higher rate of weight regain.6,30
Because revision surgery is associated with more complications and possibly longer hospital stays than primary BS, it should be performed by a bariatric surgeon with extensive experience.30,31 Restrictive revisions are typically converted to malabsorptive procedures. Cost is a limiting factor as many patients’ insurance coverage is limited to one BS per lifetime.
Body contouring. Body contouring surgery (BCS) can improve physical and mental well-being and may be a protective factor for weight regain after bariatric surgery.32 Despite its desirability—particularly to women, adolescents, and those with large decreases in body mass index (BMI)—few patients can afford BCS since it is rarely covered by insurance.
Complications of BCS vary, but are most commonly infection and wound dehiscence. This is, in part, due to poorer wound healing in BS patients compared to those with nonsurgical massive weight loss. The cause of poor wound healing is thought to be secondary to nutritional deficiencies and the catabolic state induced by post-surgical weight loss. Recommendations for BCS include weight stability for more than one year after BS, age >16 years, excess skin causing significant functional impairment, non-smoking status, and presence of good social support.33
Bariatric surgery in adolescents is on the rise
Children in the highest body mass index quartile have more than twice the death rate of those in the lowest BMI quartile.34 Thus, it is not surprising that the rate of BS in adolescents is increasing.7 BS in this age group is successful for weight loss and improvement of comorbid conditions, with relatively low complication rates.35 Options include malabsorptive and restrictive procedures, although gastric banding has not been approved by the US Food and Drug Administration for patients under the age of 18 years.
After BS, adolescent girls should be counseled regarding the possibility of pregnancy (restoration of fertility) and appropriate contraception. Adolescent patients require nutritional supplementation after BS as indicated in TABLE 1.6,8-11
When determining which adolescents to refer for BS, we recommend the following criteria: 35-38
- failure of a minimum 6-month trial of a staged treatment approach, as recommended by Barlow et al,36 including diet, exercise, and pharmacologic treatment
- BMI ≥35 with type 2 diabetes or severe sleep apnea (apnea hypopnea index [AHI] >15)37
- BMI ≥40 with mild sleep apnea (AHI >5), hypertension, or pre-diabetes37
- Tanner stage IV or V
- at least 95% skeletal growth (for malabsorptive surgery).37 This can be determined using an estimated adult height from mid-parental height formula and assessing growth plate closure with hand radiographs for bone age
- appropriate maturity level permitting adherence
- good psychological support
- a multidisciplinary team for postoperative and long-term follow-up care.
Planning for the future: Exploring the possibility of pregnancy
Obesity is the primary cause of maternal and fetal morbidity during pregnancy. It is associated with increased rates of early miscarriage, congenital defects, macrosomia, and fetal death. Maternal risks of obesity include: gestational hypertension, gestational diabetes mellitus (GDM), and pre-eclampsia. Obese mothers also have a higher incidence of failed induction, caesarean section, and breastfeeding failure.10,39 Given that half of all BSs are performed in women of reproductive age, this population deserves special consideration.10
A recent meta-analysis by Galazis et al40 concluded that BS performed prior to pregnancy led to decreased rates of preeclampsia, GDM, large neonates, preterm birth, and neonatal intensive care unit admission. Perinatal mortality did not increase after BS. However, BS led to higher rates of maternal anemia. There was no significant difference between groups in incidence of cesarean section.
The post BS female patient should be advised to use a reliable form of contraception for a minimum of 12 to 18 months after surgery.6,10,39 Involve high-risk obstetric specialists during pregnancies. Diet should be supplemented as indicated in TABLE 1.6,8-11
CORRESPONDENCE
Amy Rothberg, MD, PhD, Domino’s Farms, Lobby G, Suite 1500, 24 Frank Lloyd Wright Drive, Ann Arbor, MI 48106; [email protected].
More than one-third of American adults and approximately 17% of children and adolescents between the ages of 2 and 19 years are obese.1,2 Poor diet coupled with a sedentary lifestyle is the highest ranked cause of non-communicable disease and a leading cause of preventable death, according to the National Research Council.3
Bariatric surgery (BS) is a viable therapeutic option for obese patients who do not respond to conventional lifestyle interventions for losing weight. There are multiple gastrointestinal (GI) procedures available that are classified as either malabsorptive (Roux-en-Y gastric bypass [RYGB] and biliopancreatic diversion [BPD] with or without duodenal switch) or restrictive (laparoscopic adjustable gastric banding [LAGB] and vertical sleeve gastrectomy [VSG]).
Approximately half of the 196,000 bariatric procedures performed in the United States in 2015 were of the sleeve variety, another 23% were RYGB, and the remaining percentage was divided among the other types.4 Postoperative risks include nutritional deficiencies, decreased bone mineral density (BMD), dumping syndrome (when food rapidly dumps from the stomach to the intestine), and gastroesophageal reflux disease (GERD) with possible ulceration.
Despite these potential complications, a systematic review and meta-analysis found that obese people who underwent BS (gastric banding or gastric bypass) had significantly reduced risks of global, non-cardiovascular (CV), and CV mortality compared with obese controls.5 Helping patients to realize these benefits requires that the entire health care team—especially the family physician—is aware of the special considerations for this population.
To that end, this article reviews the details of diagnosing and managing post-surgical complications. It also addresses issues unique to managing certain subpopulations, such as post-BS patients who require revision surgery or who want to pursue body contouring surgery; adolescents who undergo BS surgery; and women who want to get pregnant postoperatively.
Monitor patients for these post-surgery complications
Postoperative BS follow-up varies depending on location, surgeon preference, and availability of multidisciplinary resources. At our institution, patients have a minimum of 3 follow-up visits with their surgeon (during hospitalization and 2 weeks and 2 months postoperatively). This is followed by visits with Endocrinology 6 months after surgery and annually thereafter. Given the variability of follow-up, family physicians should coordinate with specialists where appropriate and be aware of postoperative complications and monitoring since it is likely they will have the most frequent contact with these patients.
Nutritional deficiencies are common and require lifelong screening
Nutritional deficiencies are the most common complications of malabsorptive BS. Guidelines from the Endocrine Society, as well as guidelines from the American Association of Clinical Endocrinologists (AACE), The Obesity Society (TOS), and the American Society for Metabolic and Bariatric Surgery (ASMBS), recommend routine lifetime screening for deficiencies after surgery.6,7 Complete blood cell count, electrolytes, glucose, creatinine, and liver function tests should be obtained at one, 3, 6, 12, 18, and 24 months following surgery and annually thereafter.6
Multiple factors contribute to nutritional and micronutrient deficiencies, including reduced oral intake of food, decreased GI absorption, food intolerance, nausea/vomiting, and nonadherence with dietary supplements.8 Oral supplementation should be in chewable, powder, or liquid form because pill and capsule absorption may be altered.8,9 Over-the-counter multivitamins may not contain the requisite daily doses recommended after BS.9 Patients and physicians should evaluate supplements together to ensure appropriate nutritional and micronutrient supplementation (TABLE 16,8-11).
Bone mineral density can start to decrease soon after surgery
Studies evaluating BMD after BS have produced variable findings. In obese patients, dual-energy x-ray absorptiometry (DEXA) measurements may not be accurate due to adipose tissue artifact and table weight limits. In addition, limited data exist on the incidence of fractures after BS. Of 2 notable studies, only one, a population-based study involving 258 Minnesota residents who underwent a first bariatric surgery between 1985 and 2004, demonstrated a significantly increased incidence of fractures.12,13
In addition, studies show bone turnover markers, including C-terminal telopeptide, increase as early as 3 months after BS.14 Several guidelines recommend routine BMD screening after BS (TABLE 2).6,7 The mechanism of bone demineralization is likely multifactorial—a function of the magnitude of the weight loss and skeletal unloading, calcium and vitamin D deficiencies, and associated secondary hyperparathyroidism.15 Treatment for secondary hyperparathyroidism is adequate supplementation with vitamin D and calcium.
Optimal dosing for vitamin D has not been determined. One recent systematic review suggests routine prophylaxis with at least 2000 international units (IU)/d and found the greatest improvement for known deficiency with doses of 1500-9100 IU/d following malabsorptive surgeries.11 After laparoscopic sleeve gastrectomy, at least 1000 IU/d vitamin D is recommended.11
Overall, high variability exists among patients, and an individualized approach for dosing is recommended.11 Vitamin D levels should be monitored 2 and 4 weeks after initiation of treatment and every 3 months thereafter.11 Normal levels of serum calcium, 25-OH vitamin D, bone-specific alkaline phosphatase, and 24-hour urinary calcium excretion indicate adequate calcium and vitamin D supplementation.6
Dumping syndrome can lead to hypoglycemia
Dumping syndrome is a common complication following BS, with prevalence ranging from 25% to 75%, depending upon the type of procedure performed.16,17 There are 2 types: early and late. Early dumping syndrome occurs within 30 minutes of eating. Symptoms are related to the robust release of gastrointestinal hormones caused by rapid gastric emptying. Symptoms include nausea, abdominal pain, diarrhea, flushing, hypotension, and tachycardia.
Late dumping is characterized as postprandial hypoglycemia occurring one to 3 hours after eating. Late dumping is likely caused by a combination of changes within the pancreatic beta cells and abnormal insulin response to glucose.16-18 Rapid gastric emptying leads to rapid release of glucose in the gut, which, in turn, leads to brisk insulin secretion. Since glucose is absorbed faster than insulin’s half-life, the resulting (relatively) high levels of insulin may cause hypoglycemia.16-18
Sigstad’s scoring system can be used to confirm suspected cases of dumping syndrome (TABLE 316,17,19). A diagnosis can also be made with an oral glucose challenge in which pulse, blood pressure, glucose, and hematocrit are measured after ingestion of 50 g glucose. The test is positive if heart rate increases by 10 beats per minute, hematocrit increases by 3% 30 minutes after ingestion, or glucose falls below 60 mg/dL 2 to 3 hours after ingestion.17
First-line treatment of dumping syndrome consists of dietary modifications. The goal is to slow the rate of gastric emptying by eating smaller, more frequent meals; separating beverages from food; decreasing carbohydrates; and increasing fiber and protein content.
If results are suboptimal after dietary changes, medications can be prescribed including acarbose to prevent postprandial hypoglycemia; anticholinergics such as dicyclomine to slow gastric emptying; and somatostatin to decrease gastric emptying and inhibit GI hormone release.17 Lastly, for resistant and severe postprandial hypoglycemia, a few patients have undergone pancreatectomy, but only about 65% experienced improvement in symptoms and 12% developed diabetes post-surgically.20
Gout attacks may initially increase, but then decrease
BS affects the incidence of gout attacks in patients with a history of gout. One comparative study of approximately 150 patients demonstrated that those with a history of gout had significantly more gout attacks in the first month after BS compared with obese patients with a history of gout undergoing other upper GI surgeries.21 There was no difference between malabsorptive and restrictive procedures. But after the first month, BS patients had significantly fewer gout attacks and lower uric acid levels than their obese counterparts.21
Protein rich diets, catabolism potentiated by aggressive caloric restriction following BS, and dehydration contribute to the initial increase. Therefore, patients who have had at least one gout attack in the year prior to surgery or who are on hypouricemic medication may benefit from at least one month of prophylactic therapy (eg, allopurinol and colchicine) after surgery.
GERD and ulceration: How to respond
Obesity is a known risk factor for GERD, but the effect of BS on GERD is uncertain and seems to vary with the procedure performed. RYGB decreases GERD and is, therefore, used as both a secondary treatment in those not responding to medications and a revision treatment for fundoplication and other types of BSs. Sleeve gastrectomy and adjustable gastric banding have mixed effects on GERD. A systematic review by de Jong et al revealed a decreased prevalence of reflux symptoms and GERD medication use after LAGB; however, during longer follow-up, 15% of previously unaffected patients reported experiencing GERD.22 The 2011 International Sleeve Gastrectomy Expert Panel Consensus Statement retrospectively noted a postoperative incidence of GERD as high as 31%.23
BS patients with GERD should be treated with a proton pump inhibitor. If this fails, refer patients to a gastroenterologist for further evaluation.24
Ulcers after BS may be an indication for revision surgery. Data are mixed regarding increased risk of marginal ulceration from nonsteroidal anti-inflammatory drug (NSAID) use, but NSAIDs have been linked to an increased risk of anastomotic leakage.25-28 Thus, it seems prudent to avoid NSAIDs in people who have undergone BS.
Keeping watch over psychiatric comorbidities
A recent meta-analysis by Dawes et al29 showed that about 23% of patients pursuing BS have a comorbid mood disorder. Specifically, the preoperative prevalence of depression (19%) and binge-eating disorder (17%) were found to be higher than rates in the general population.29 The meta-analysis found improvement in the prevalence of depression with fewer symptoms and less antidepressant medication use in the first 3 years after surgery and a decrease in the rate of binge-eating disorder, although there were fewer supporting data for the latter. These findings were observed with both restrictive and malabsorptive procedures.
The data are mixed regarding rates of alcohol abuse and suicide. Further research is necessary in this field. Patients who have had BS should receive ongoing psychiatric and psychological care from a multidisciplinary team as a matter of course.
Will a second surgery be needed?
Revision surgery. In 2015, about 14% of the almost 200,000 BSs performed were revisions.4 Revision surgery is indicated in BS patients with weight regain, recurrent comorbid diseases (eg, diabetes, hypertension), or complications of primary BS. Restrictive procedures have a higher revision rate than malabsorptive procedures, primarily due to a higher rate of weight regain.6,30
Because revision surgery is associated with more complications and possibly longer hospital stays than primary BS, it should be performed by a bariatric surgeon with extensive experience.30,31 Restrictive revisions are typically converted to malabsorptive procedures. Cost is a limiting factor as many patients’ insurance coverage is limited to one BS per lifetime.
Body contouring. Body contouring surgery (BCS) can improve physical and mental well-being and may be a protective factor for weight regain after bariatric surgery.32 Despite its desirability—particularly to women, adolescents, and those with large decreases in body mass index (BMI)—few patients can afford BCS since it is rarely covered by insurance.
Complications of BCS vary, but are most commonly infection and wound dehiscence. This is, in part, due to poorer wound healing in BS patients compared to those with nonsurgical massive weight loss. The cause of poor wound healing is thought to be secondary to nutritional deficiencies and the catabolic state induced by post-surgical weight loss. Recommendations for BCS include weight stability for more than one year after BS, age >16 years, excess skin causing significant functional impairment, non-smoking status, and presence of good social support.33
Bariatric surgery in adolescents is on the rise
Children in the highest body mass index quartile have more than twice the death rate of those in the lowest BMI quartile.34 Thus, it is not surprising that the rate of BS in adolescents is increasing.7 BS in this age group is successful for weight loss and improvement of comorbid conditions, with relatively low complication rates.35 Options include malabsorptive and restrictive procedures, although gastric banding has not been approved by the US Food and Drug Administration for patients under the age of 18 years.
After BS, adolescent girls should be counseled regarding the possibility of pregnancy (restoration of fertility) and appropriate contraception. Adolescent patients require nutritional supplementation after BS as indicated in TABLE 1.6,8-11
When determining which adolescents to refer for BS, we recommend the following criteria: 35-38
- failure of a minimum 6-month trial of a staged treatment approach, as recommended by Barlow et al,36 including diet, exercise, and pharmacologic treatment
- BMI ≥35 with type 2 diabetes or severe sleep apnea (apnea hypopnea index [AHI] >15)37
- BMI ≥40 with mild sleep apnea (AHI >5), hypertension, or pre-diabetes37
- Tanner stage IV or V
- at least 95% skeletal growth (for malabsorptive surgery).37 This can be determined using an estimated adult height from mid-parental height formula and assessing growth plate closure with hand radiographs for bone age
- appropriate maturity level permitting adherence
- good psychological support
- a multidisciplinary team for postoperative and long-term follow-up care.
Planning for the future: Exploring the possibility of pregnancy
Obesity is the primary cause of maternal and fetal morbidity during pregnancy. It is associated with increased rates of early miscarriage, congenital defects, macrosomia, and fetal death. Maternal risks of obesity include: gestational hypertension, gestational diabetes mellitus (GDM), and pre-eclampsia. Obese mothers also have a higher incidence of failed induction, caesarean section, and breastfeeding failure.10,39 Given that half of all BSs are performed in women of reproductive age, this population deserves special consideration.10
A recent meta-analysis by Galazis et al40 concluded that BS performed prior to pregnancy led to decreased rates of preeclampsia, GDM, large neonates, preterm birth, and neonatal intensive care unit admission. Perinatal mortality did not increase after BS. However, BS led to higher rates of maternal anemia. There was no significant difference between groups in incidence of cesarean section.
The post BS female patient should be advised to use a reliable form of contraception for a minimum of 12 to 18 months after surgery.6,10,39 Involve high-risk obstetric specialists during pregnancies. Diet should be supplemented as indicated in TABLE 1.6,8-11
CORRESPONDENCE
Amy Rothberg, MD, PhD, Domino’s Farms, Lobby G, Suite 1500, 24 Frank Lloyd Wright Drive, Ann Arbor, MI 48106; [email protected].
1. Centers for Disease Control and Prevention. Overweight and obesity. Adult obesity facts. Available at: https://www.cdc.gov/obesity/data/adult.html. Accessed April 5, 2017.
2. Centers for Disease Control and Prevention. Overweight and obesity. Childhood obesity facts. Available at: https://www.cdc.gov/obesity/data/childhood.html. Accessed April 5, 2017.
3. McGinnis JM. Actual causes of death, 1990-2010. Workshop on Determinants of Premature Mortality, September 18, 2013, National Research Council, Washington, DC.
4. American Society of Metabolic and Bariatric Surgery. Estimate of bariatric surgery numbers, 2011-2015. Available at: https://asmbs.org/resources/estimate-of-bariatric-surgery-numbers. Accessed April 5, 2017.
5. Pontiroli AE, Morabito A. Long-term prevention of mortality in morbid obesity through bariatric surgery. a systematic review and meta-analysis of trials performed with gastric banding and gastric bypass. Ann Surg. 2011;253:484-487.
6. Heber D, Greenway FL, Kaplan LM, et al. Endocrine and nutritional management of the post-bariatric surgery patient: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4823-4843.
7. Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient—2013 update: cosponsored by American Association of Clinical Endocrinologists, The Obesity Society, and American Society for Metabolic & Bariatric Surgery. Obesity. 2013;21:S1-S27.
8. Stein J, Stier C, Raab H, et al. Review article: the nutritional and pharmacological consequences of obesity surgery. Aliment Pharmacol Ther. 2014;40:582-609.
9. Boyce SG, Goriparthi R, Clark J, et al. Can composite nutritional supplement based on the current guidelines prevent vitamin and mineral deficiency after weight loss surgery? Obes Surg. 2016;26:966-971.
10. Beard JH, Bell RL, Duffy AJ. Reproductive considerations and pregnancy after bariatric surgery: current evidence and recommendations. Obes Surg. 2008;18:1023-1027.
11. Chakhtoura MT, Nakhoul NN, Shawwa K, et al. Hypovitaminosis D in bariatric surgery: a systematic review of observational studies. Metabolism. 2016;65:574-585.
12. Nakamura KM, Haglind EG, Clowes JA, et al. Fracture risk following bariatric surgery: a population-based study. Osteoporosis Int. 2014;25:151-158.
13. Lalmohamed A, de Vries F, Bazelier MT, et al. Risk of fracture after bariatric surgery in the United Kingdom: population-based, retrospective cohort study. BMJ. 2012;345:e5085.
14. Coates PS, Fernstrom JD, Fernstrom MH, et al. Gastric bypass surgery for morbid obesity leads to an increase in bone turnover and a decrease in bone mass. J Clin Endocrinol Metab. 2004;89:1061-1065.
15. Stein EM, Silverberg SJ. Bone loss after bariatric surgery: causes, consequences and management. Lancet Diabetes Endocrinol. 2014;2:165-174.
16. Tack J, Deloose E. Complications of bariatric surgery: dumping syndrome, reflux and vitamin deficiencies. Best Pract Res Clin Gastroenterol. 2014;28:741-749.
17. Berg P, McCallum R. Dumping syndrome: a review of the current concepts of pathophysiology, diagnosis, and treatment. Dig Dis Sci. 2016;61:11-18.
18. Ritz P, Vaurs C, Barigou M, et al. Hypoglycaemia after gastric bypass: mechanisms and treatment. Diabetes Obes Metab. 2016;18:217-223.
19. Sigstad H. A clinical diagnostic index in the diagnosis of the dumping syndrome. Changes in plasma volume and blood sugar after a test meal. Acta Med Scand. 1970;188:479-486.
20. Mala T. Postprandial hyperinsulinemic hypoglycaemia after gastric bypass surgical treatment. Surg Obes Relat Dis. 2014;10:1220-1225.
21. Romero-Talamás H, Daigle CR, Aminian A. The effect of bariatric surgery on gout: a comparative study. Surg Obes Relat Dis. 2014;10:1161-1165.
22. de Jong JR, Besselink MG, van Ramshorst B, et al. Effects of adjustable gastric banding on gastroesophageal reflux and esophageal motility: a systematic review. Obes Rev. 2010;11:297-305.
23. Rosenthal RJ; International Sleeve Gastrectomy Expert Panel. International Sleeve Gastrectomy Expert Panel Consensus Statement: best practice guidelines based on experience of >12,000 cases. Surg Obes Relat Dis. 2012;8:8-19.
24. Altieri MS, Pryor AD. Gastroesophageal reflux disease after bariatric procedures. Surg Clin North Am. 2015;95:579-591.
25. Hakkarainen TW, Steele SR, Bastaworous A, et al. Nonsteroidal anti-inflammatory drugs and the risk for anastomotic failure: a report from Washington State’s Surgical Care and Outcomes Assessment Program (SCOAP). JAMA Surg. 2015;150:223-228.
26. El-Hayek K, Timratana P, Shimizu H, et al. Marginal ulcer after Roux-en-Y gastric bypass: what have we really learned? Surg Endosc. 2012;26:2789-2796.
27. Sverdén E, Mattsson F, Sondén AM, et al. Risk factors for marginal ulcer after gastric bypass surgery for obesity: a population-based cohort study. Ann Surg. 2016;263:733-737.
28. Azagury DE, Abu Dayyeh BK, Greenwalt IT, et al. Marginal ulceration after Roux-en-Y gastric bypass surgery: characteristics, risk factors, treatment, and outcomes. Endoscopy. 2011;43:950-954.
29. Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: A meta-analysis. JAMA. 2016;315:150-163.
30. Ferrer-Márquez MP, Belda-Lozano R, Solvas-Salmerón MJ, et al. Revisional surgery after laparoscopic sleeve gastrectomy. Surg Laparosc Endosc Percutan Tech. 2015;25:6-9.
31. Sanchez H, Cabrera A, Cabrera K, et al. Laparoscopic Roux-en-Y gastric bypass as a revision procedure after restrictive bariatric surgery. Obes Surg. 2008;18:1539-1543.
32. van der Beek ES, Te Riele W, Specken TF, et al. The impact of reconstructive procedures following bariatric surgery on patient well-being and quality of life. Obes Surg. 2010;20:36-41.
33. Ellison JM, Steffen KJ, Sarwer DB. Body contouring after bariatric surgery. Eur Eat Disord Rev. 2015;23:479-487.
34. Franks PW, Hanson RL, Knowler WC, et al. Childhood obesity, other cardiovascular risk factors, and premature death. NEJM. 2010;362:485-489.
35. Gravelle BL, Broyles M. Interventions of weight reduction and prevention in children and adolescents: update. Am J Ther. 2015;22:159-166.
36. Barlow SE. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics. 2007;120:S164-S192.
37. Pratt JS, Lenders CM, Dionne EA, et al. Best practice updates for pediatric/adolescent weight loss surgery. Obesity. 2009;17:901-910.
38. Nogueira I, Hrovat K. Adolescent bariatric surgery: review on nutrition considerations. Nutr Clin Pract. 2014;29:740-746.
39. Nicklas JM, Barbour LA. Optimizing weight for maternal and infant health: tenable, or too late? Expert Rev Endocrinol Metab. 2015;10:227-242.
40. Galazis N, Docheva N, Simillis C, et al. Maternal and neonatal outcomes in women undergoing bariatric surgery: a systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2014;181:45-53.
1. Centers for Disease Control and Prevention. Overweight and obesity. Adult obesity facts. Available at: https://www.cdc.gov/obesity/data/adult.html. Accessed April 5, 2017.
2. Centers for Disease Control and Prevention. Overweight and obesity. Childhood obesity facts. Available at: https://www.cdc.gov/obesity/data/childhood.html. Accessed April 5, 2017.
3. McGinnis JM. Actual causes of death, 1990-2010. Workshop on Determinants of Premature Mortality, September 18, 2013, National Research Council, Washington, DC.
4. American Society of Metabolic and Bariatric Surgery. Estimate of bariatric surgery numbers, 2011-2015. Available at: https://asmbs.org/resources/estimate-of-bariatric-surgery-numbers. Accessed April 5, 2017.
5. Pontiroli AE, Morabito A. Long-term prevention of mortality in morbid obesity through bariatric surgery. a systematic review and meta-analysis of trials performed with gastric banding and gastric bypass. Ann Surg. 2011;253:484-487.
6. Heber D, Greenway FL, Kaplan LM, et al. Endocrine and nutritional management of the post-bariatric surgery patient: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4823-4843.
7. Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient—2013 update: cosponsored by American Association of Clinical Endocrinologists, The Obesity Society, and American Society for Metabolic & Bariatric Surgery. Obesity. 2013;21:S1-S27.
8. Stein J, Stier C, Raab H, et al. Review article: the nutritional and pharmacological consequences of obesity surgery. Aliment Pharmacol Ther. 2014;40:582-609.
9. Boyce SG, Goriparthi R, Clark J, et al. Can composite nutritional supplement based on the current guidelines prevent vitamin and mineral deficiency after weight loss surgery? Obes Surg. 2016;26:966-971.
10. Beard JH, Bell RL, Duffy AJ. Reproductive considerations and pregnancy after bariatric surgery: current evidence and recommendations. Obes Surg. 2008;18:1023-1027.
11. Chakhtoura MT, Nakhoul NN, Shawwa K, et al. Hypovitaminosis D in bariatric surgery: a systematic review of observational studies. Metabolism. 2016;65:574-585.
12. Nakamura KM, Haglind EG, Clowes JA, et al. Fracture risk following bariatric surgery: a population-based study. Osteoporosis Int. 2014;25:151-158.
13. Lalmohamed A, de Vries F, Bazelier MT, et al. Risk of fracture after bariatric surgery in the United Kingdom: population-based, retrospective cohort study. BMJ. 2012;345:e5085.
14. Coates PS, Fernstrom JD, Fernstrom MH, et al. Gastric bypass surgery for morbid obesity leads to an increase in bone turnover and a decrease in bone mass. J Clin Endocrinol Metab. 2004;89:1061-1065.
15. Stein EM, Silverberg SJ. Bone loss after bariatric surgery: causes, consequences and management. Lancet Diabetes Endocrinol. 2014;2:165-174.
16. Tack J, Deloose E. Complications of bariatric surgery: dumping syndrome, reflux and vitamin deficiencies. Best Pract Res Clin Gastroenterol. 2014;28:741-749.
17. Berg P, McCallum R. Dumping syndrome: a review of the current concepts of pathophysiology, diagnosis, and treatment. Dig Dis Sci. 2016;61:11-18.
18. Ritz P, Vaurs C, Barigou M, et al. Hypoglycaemia after gastric bypass: mechanisms and treatment. Diabetes Obes Metab. 2016;18:217-223.
19. Sigstad H. A clinical diagnostic index in the diagnosis of the dumping syndrome. Changes in plasma volume and blood sugar after a test meal. Acta Med Scand. 1970;188:479-486.
20. Mala T. Postprandial hyperinsulinemic hypoglycaemia after gastric bypass surgical treatment. Surg Obes Relat Dis. 2014;10:1220-1225.
21. Romero-Talamás H, Daigle CR, Aminian A. The effect of bariatric surgery on gout: a comparative study. Surg Obes Relat Dis. 2014;10:1161-1165.
22. de Jong JR, Besselink MG, van Ramshorst B, et al. Effects of adjustable gastric banding on gastroesophageal reflux and esophageal motility: a systematic review. Obes Rev. 2010;11:297-305.
23. Rosenthal RJ; International Sleeve Gastrectomy Expert Panel. International Sleeve Gastrectomy Expert Panel Consensus Statement: best practice guidelines based on experience of >12,000 cases. Surg Obes Relat Dis. 2012;8:8-19.
24. Altieri MS, Pryor AD. Gastroesophageal reflux disease after bariatric procedures. Surg Clin North Am. 2015;95:579-591.
25. Hakkarainen TW, Steele SR, Bastaworous A, et al. Nonsteroidal anti-inflammatory drugs and the risk for anastomotic failure: a report from Washington State’s Surgical Care and Outcomes Assessment Program (SCOAP). JAMA Surg. 2015;150:223-228.
26. El-Hayek K, Timratana P, Shimizu H, et al. Marginal ulcer after Roux-en-Y gastric bypass: what have we really learned? Surg Endosc. 2012;26:2789-2796.
27. Sverdén E, Mattsson F, Sondén AM, et al. Risk factors for marginal ulcer after gastric bypass surgery for obesity: a population-based cohort study. Ann Surg. 2016;263:733-737.
28. Azagury DE, Abu Dayyeh BK, Greenwalt IT, et al. Marginal ulceration after Roux-en-Y gastric bypass surgery: characteristics, risk factors, treatment, and outcomes. Endoscopy. 2011;43:950-954.
29. Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: A meta-analysis. JAMA. 2016;315:150-163.
30. Ferrer-Márquez MP, Belda-Lozano R, Solvas-Salmerón MJ, et al. Revisional surgery after laparoscopic sleeve gastrectomy. Surg Laparosc Endosc Percutan Tech. 2015;25:6-9.
31. Sanchez H, Cabrera A, Cabrera K, et al. Laparoscopic Roux-en-Y gastric bypass as a revision procedure after restrictive bariatric surgery. Obes Surg. 2008;18:1539-1543.
32. van der Beek ES, Te Riele W, Specken TF, et al. The impact of reconstructive procedures following bariatric surgery on patient well-being and quality of life. Obes Surg. 2010;20:36-41.
33. Ellison JM, Steffen KJ, Sarwer DB. Body contouring after bariatric surgery. Eur Eat Disord Rev. 2015;23:479-487.
34. Franks PW, Hanson RL, Knowler WC, et al. Childhood obesity, other cardiovascular risk factors, and premature death. NEJM. 2010;362:485-489.
35. Gravelle BL, Broyles M. Interventions of weight reduction and prevention in children and adolescents: update. Am J Ther. 2015;22:159-166.
36. Barlow SE. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics. 2007;120:S164-S192.
37. Pratt JS, Lenders CM, Dionne EA, et al. Best practice updates for pediatric/adolescent weight loss surgery. Obesity. 2009;17:901-910.
38. Nogueira I, Hrovat K. Adolescent bariatric surgery: review on nutrition considerations. Nutr Clin Pract. 2014;29:740-746.
39. Nicklas JM, Barbour LA. Optimizing weight for maternal and infant health: tenable, or too late? Expert Rev Endocrinol Metab. 2015;10:227-242.
40. Galazis N, Docheva N, Simillis C, et al. Maternal and neonatal outcomes in women undergoing bariatric surgery: a systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2014;181:45-53.
From The Journal of Family Practice | 2017;66(6):356-363.
PRACTICE RECOMMENDATIONS
› Routinely screen bariatric surgery patients for nutritional deficiencies throughout their life. A
› Avoid the use of nonsteroidal anti-inflammatory medications in patients who have had bariatric surgery becauseof the risk of anastomotic ulceration and leakage. A
› Consider revision surgery for bariatric surgery patients with weight regain, recurrent comorbid diseases, or surgical complications. B
› Counsel obese women who want to become pregnant that bariatric surgery decreases rates of future pregnancy complications. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Oral agent offers relief from generalized hyperhidrosis
ILLUSTRATIVE CASE
A 34-year-old woman presents to your office for unbearable sweating. She notes that the sweating occurs nearly daily on her hands, face, and in her axillary regions, causing social embarrassment. She has tried multiple antiperspirants to no avail. Is there anything she can take to reduce the sweating?
Hyperhidrosis is a common, self-limiting problem affecting 2% to 3% of the population in the United States.2 Patients may complain of localized sweating of the hands, feet, face, or underarms or more systemic, generalized sweating in multiple locations. Either way, patients always note a significant impact on their quality of life.
Treatment of hyperhidrosis has traditionally focused on topical therapies to the affected areas. Research has shown that localized treatment with antiperspirants containing aluminum salt is effective by both subjective report and objective measurements at reducing sweating—particularly in the axilla, hands, and feet.3,4 Additionally, a systematic review of observational and experimental studies found topical glycopyrrolate to be efficacious for craniofacial hyperhidrosis with minimal adverse effects.5 The availability of low-cost prescription and over-the-counter aluminum-based antiperspirant agents makes topicals the first-line choice.
More invasive treatments are available for hyperhidrosis that is refractory to topicals. In a double-blind, randomized controlled trial, researchers injected either botulinum toxin type A (BTX-A) 50 U or placebo in patients with bilateral primary axillary hyperhidrosis.6 Of the 207 patients who received treatment injections, 96.1% had at least a 50% reduction of axillary sweating at 4 weeks after one injection, as measured by gravimetric assessment. The BTX-A injections also produced a prolonged effect; mean duration between injections was 30.6 weeks.
Other invasive treatments include iontophoresis, surgery, and laser therapy; however, these methods are not suitable for body-wide application and are, thus, not appropriate for patients with generalized hyperhidrosis.
Oxybutynin is the first oral agent to emerge as a treatment option for hyperhidrosis. This cholinergic antagonist had historically been used to treat overactive bladder. As a cholinergic antagonist, oxybutynin not only reduces urinary frequency, but also decreases secretions in various locations and, thus, can cause dry mouth and reduce perspiration.
In one prospective placebo-controlled trial, 50 patients with generalized hyperhidrosis were randomized to receive either oxybutynin titrated from 2.5 mg orally once daily to 5 mg orally twice daily or placebo for 6 weeks.7 Seventeen (73.9%) patients receiving oxybutynin for palmar or axillary hyperhidrosis reported moderate to “great” resolution of their symptoms compared with 6 (27.3%) patients in the placebo group. Dry mouth was reported in 34.8% of patients receiving oxybutynin vs 9.1% of those who received placebo (P=.038); however, no patients dropped out of the study due to this adverse effect.7
STUDY SUMMARY
This multicenter, randomized controlled trial compared oxybutynin to placebo in 62 adults with localized or generalized hyperhidrosis from 12 outpatient dermatology practices in France. It is the first study to include patients with a localized, as well as a generalized form of the condition.
Patients were included if they were >18 years of age, enrolled in the National Health Insurance system in France, and reported a Hyperhidrosis Disease Severity Scale (HDSS) score ≥2. The HDSS is a validated, one-question tool (“How would you rate the severity of your sweating?”). Patients provide a score of 1 (no perceptible sweating and no interference with everyday life) to 4 (intolerable sweating with constant interference with everyday life).8 Patients were excluded if they had any contraindications to the use of an anticholinergic medication.
Patients randomized to oxybutynin took 2.5 mg/d orally initially and increased gradually over 8 days until reaching an effective dose that was not more than 7.5 mg/d. They then continued at that dose for 6 weeks. The primary outcome was improvement on the HDSS by one or more points measured at the beginning of the trial and at 6 weeks. Secondary outcomes included change in quality of life, as measured by the Dermatology Life Quality Index (DLQI) and reported adverse effects. The DLQI is a dermatology-specific quality-of-life measure consisting of 10 questions. Scores range from 0 (where their disease has no impact on their quality of life) to 30 (maximum impact of their disease on their quality of life).9
Improved HDSS and DLQI scores. Most patients (83%) in the study had generalized hyperhidrosis. Patients were in their mid-thirties. Sixty percent of the patients in the oxybutynin group had an improvement of one point or more on the 4-point HDSS compared to 27% in the placebo group (P<.01). DLQI scores improved by 6.9 points in the oxybutynin group and 2.3 points in the placebo group (P<.01).
The most common adverse effect was dry mouth, which occurred in 13 patients (43%) in the oxybutynin group and in 3 patients (11%) in the placebo group (P<.01); it did not cause any patients to drop out of the study. The second most common adverse effect was blurred vision, which only occurred in the oxybutynin group (4 patients; 13%).
WHAT'S NEW
This is the first randomized controlled trial to demonstrate the efficacy of an oral agent for generalized primary hyperhidrosis. This trial used a relatively low dose of oxybutynin, which produced significant benefit while minimizing anticholinergic adverse effects.
CAVEATS
There are many situations for which anticholinergic medications are inappropriate, including use by geriatric patients and those with gastrointestinal disorders, urinary retention, or glaucoma.
CHALLENGES TO IMPLEMENTATION
Few if any challenges exist to the utilization of oxybutynin; inexpensive generic versions are widely available.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Schollhammer M, Brenaut E, Menard-Andivot N, et al. Oxybutynin as a treatment for generalized hyperhidrosis: a randomized, placebo-controlled trial. Br J Dermatol. 2015;173:1163-1168.
2. Grabell DA, Hebert AA. Current and emerging medical therapies for primary hyperhidrosis. Dermatol Ther (Heidelb). 2017;7:25-36.
3. Innocenzi D, Lupi F, Bruni F, et al. Efficacy of a new aluminium salt thermophobic foam in the treatment of axillary and palmar primary hyperhidrosis: a pilot exploratory trial. Curr Med Res Opin. 2005;21:1949-1953.
4. Goh CL. Aluminum chloride hexahydrate versus palmar hyperhidrosis. Evaporimeter assessment. Int J Dermatol. 1990;29:368-370.
5. Nicholas R, Quddus A, Baker DM. Treatment of primary craniofacial hyperhidrosis: a systematic review. Am J Clin Dermatol. 2015;16:361-370.
6. Naumann M, Lowe NJ, Kumar CR, et al. Botulinum toxin type a is a safe and effective treatment for axillary hyperhidrosis over 16 months: a prospective study. Arch Dermatol. 2003;139:731-736.
7. Wolosker N, de Campos JR, Kauffman P, et al. A randomized placebo-controlled trial of oxybutynin for the initial treatment of palmar and axillary hyperhidrosis. J Vasc Surg. 2012;55:1696-1700.
8. Varella AY, Fukuda JM, Teivelis MP, et al. Translation and validation of Hyperhidrosis Disease Severity Scale. Rev Assoc Med Bras. 2016;62:843-847.
9. Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)—a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19:210-216.
ILLUSTRATIVE CASE
A 34-year-old woman presents to your office for unbearable sweating. She notes that the sweating occurs nearly daily on her hands, face, and in her axillary regions, causing social embarrassment. She has tried multiple antiperspirants to no avail. Is there anything she can take to reduce the sweating?
Hyperhidrosis is a common, self-limiting problem affecting 2% to 3% of the population in the United States.2 Patients may complain of localized sweating of the hands, feet, face, or underarms or more systemic, generalized sweating in multiple locations. Either way, patients always note a significant impact on their quality of life.
Treatment of hyperhidrosis has traditionally focused on topical therapies to the affected areas. Research has shown that localized treatment with antiperspirants containing aluminum salt is effective by both subjective report and objective measurements at reducing sweating—particularly in the axilla, hands, and feet.3,4 Additionally, a systematic review of observational and experimental studies found topical glycopyrrolate to be efficacious for craniofacial hyperhidrosis with minimal adverse effects.5 The availability of low-cost prescription and over-the-counter aluminum-based antiperspirant agents makes topicals the first-line choice.
More invasive treatments are available for hyperhidrosis that is refractory to topicals. In a double-blind, randomized controlled trial, researchers injected either botulinum toxin type A (BTX-A) 50 U or placebo in patients with bilateral primary axillary hyperhidrosis.6 Of the 207 patients who received treatment injections, 96.1% had at least a 50% reduction of axillary sweating at 4 weeks after one injection, as measured by gravimetric assessment. The BTX-A injections also produced a prolonged effect; mean duration between injections was 30.6 weeks.
Other invasive treatments include iontophoresis, surgery, and laser therapy; however, these methods are not suitable for body-wide application and are, thus, not appropriate for patients with generalized hyperhidrosis.
Oxybutynin is the first oral agent to emerge as a treatment option for hyperhidrosis. This cholinergic antagonist had historically been used to treat overactive bladder. As a cholinergic antagonist, oxybutynin not only reduces urinary frequency, but also decreases secretions in various locations and, thus, can cause dry mouth and reduce perspiration.
In one prospective placebo-controlled trial, 50 patients with generalized hyperhidrosis were randomized to receive either oxybutynin titrated from 2.5 mg orally once daily to 5 mg orally twice daily or placebo for 6 weeks.7 Seventeen (73.9%) patients receiving oxybutynin for palmar or axillary hyperhidrosis reported moderate to “great” resolution of their symptoms compared with 6 (27.3%) patients in the placebo group. Dry mouth was reported in 34.8% of patients receiving oxybutynin vs 9.1% of those who received placebo (P=.038); however, no patients dropped out of the study due to this adverse effect.7
STUDY SUMMARY
This multicenter, randomized controlled trial compared oxybutynin to placebo in 62 adults with localized or generalized hyperhidrosis from 12 outpatient dermatology practices in France. It is the first study to include patients with a localized, as well as a generalized form of the condition.
Patients were included if they were >18 years of age, enrolled in the National Health Insurance system in France, and reported a Hyperhidrosis Disease Severity Scale (HDSS) score ≥2. The HDSS is a validated, one-question tool (“How would you rate the severity of your sweating?”). Patients provide a score of 1 (no perceptible sweating and no interference with everyday life) to 4 (intolerable sweating with constant interference with everyday life).8 Patients were excluded if they had any contraindications to the use of an anticholinergic medication.
Patients randomized to oxybutynin took 2.5 mg/d orally initially and increased gradually over 8 days until reaching an effective dose that was not more than 7.5 mg/d. They then continued at that dose for 6 weeks. The primary outcome was improvement on the HDSS by one or more points measured at the beginning of the trial and at 6 weeks. Secondary outcomes included change in quality of life, as measured by the Dermatology Life Quality Index (DLQI) and reported adverse effects. The DLQI is a dermatology-specific quality-of-life measure consisting of 10 questions. Scores range from 0 (where their disease has no impact on their quality of life) to 30 (maximum impact of their disease on their quality of life).9
Improved HDSS and DLQI scores. Most patients (83%) in the study had generalized hyperhidrosis. Patients were in their mid-thirties. Sixty percent of the patients in the oxybutynin group had an improvement of one point or more on the 4-point HDSS compared to 27% in the placebo group (P<.01). DLQI scores improved by 6.9 points in the oxybutynin group and 2.3 points in the placebo group (P<.01).
The most common adverse effect was dry mouth, which occurred in 13 patients (43%) in the oxybutynin group and in 3 patients (11%) in the placebo group (P<.01); it did not cause any patients to drop out of the study. The second most common adverse effect was blurred vision, which only occurred in the oxybutynin group (4 patients; 13%).
WHAT'S NEW
This is the first randomized controlled trial to demonstrate the efficacy of an oral agent for generalized primary hyperhidrosis. This trial used a relatively low dose of oxybutynin, which produced significant benefit while minimizing anticholinergic adverse effects.
CAVEATS
There are many situations for which anticholinergic medications are inappropriate, including use by geriatric patients and those with gastrointestinal disorders, urinary retention, or glaucoma.
CHALLENGES TO IMPLEMENTATION
Few if any challenges exist to the utilization of oxybutynin; inexpensive generic versions are widely available.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 34-year-old woman presents to your office for unbearable sweating. She notes that the sweating occurs nearly daily on her hands, face, and in her axillary regions, causing social embarrassment. She has tried multiple antiperspirants to no avail. Is there anything she can take to reduce the sweating?
Hyperhidrosis is a common, self-limiting problem affecting 2% to 3% of the population in the United States.2 Patients may complain of localized sweating of the hands, feet, face, or underarms or more systemic, generalized sweating in multiple locations. Either way, patients always note a significant impact on their quality of life.
Treatment of hyperhidrosis has traditionally focused on topical therapies to the affected areas. Research has shown that localized treatment with antiperspirants containing aluminum salt is effective by both subjective report and objective measurements at reducing sweating—particularly in the axilla, hands, and feet.3,4 Additionally, a systematic review of observational and experimental studies found topical glycopyrrolate to be efficacious for craniofacial hyperhidrosis with minimal adverse effects.5 The availability of low-cost prescription and over-the-counter aluminum-based antiperspirant agents makes topicals the first-line choice.
More invasive treatments are available for hyperhidrosis that is refractory to topicals. In a double-blind, randomized controlled trial, researchers injected either botulinum toxin type A (BTX-A) 50 U or placebo in patients with bilateral primary axillary hyperhidrosis.6 Of the 207 patients who received treatment injections, 96.1% had at least a 50% reduction of axillary sweating at 4 weeks after one injection, as measured by gravimetric assessment. The BTX-A injections also produced a prolonged effect; mean duration between injections was 30.6 weeks.
Other invasive treatments include iontophoresis, surgery, and laser therapy; however, these methods are not suitable for body-wide application and are, thus, not appropriate for patients with generalized hyperhidrosis.
Oxybutynin is the first oral agent to emerge as a treatment option for hyperhidrosis. This cholinergic antagonist had historically been used to treat overactive bladder. As a cholinergic antagonist, oxybutynin not only reduces urinary frequency, but also decreases secretions in various locations and, thus, can cause dry mouth and reduce perspiration.
In one prospective placebo-controlled trial, 50 patients with generalized hyperhidrosis were randomized to receive either oxybutynin titrated from 2.5 mg orally once daily to 5 mg orally twice daily or placebo for 6 weeks.7 Seventeen (73.9%) patients receiving oxybutynin for palmar or axillary hyperhidrosis reported moderate to “great” resolution of their symptoms compared with 6 (27.3%) patients in the placebo group. Dry mouth was reported in 34.8% of patients receiving oxybutynin vs 9.1% of those who received placebo (P=.038); however, no patients dropped out of the study due to this adverse effect.7
STUDY SUMMARY
This multicenter, randomized controlled trial compared oxybutynin to placebo in 62 adults with localized or generalized hyperhidrosis from 12 outpatient dermatology practices in France. It is the first study to include patients with a localized, as well as a generalized form of the condition.
Patients were included if they were >18 years of age, enrolled in the National Health Insurance system in France, and reported a Hyperhidrosis Disease Severity Scale (HDSS) score ≥2. The HDSS is a validated, one-question tool (“How would you rate the severity of your sweating?”). Patients provide a score of 1 (no perceptible sweating and no interference with everyday life) to 4 (intolerable sweating with constant interference with everyday life).8 Patients were excluded if they had any contraindications to the use of an anticholinergic medication.
Patients randomized to oxybutynin took 2.5 mg/d orally initially and increased gradually over 8 days until reaching an effective dose that was not more than 7.5 mg/d. They then continued at that dose for 6 weeks. The primary outcome was improvement on the HDSS by one or more points measured at the beginning of the trial and at 6 weeks. Secondary outcomes included change in quality of life, as measured by the Dermatology Life Quality Index (DLQI) and reported adverse effects. The DLQI is a dermatology-specific quality-of-life measure consisting of 10 questions. Scores range from 0 (where their disease has no impact on their quality of life) to 30 (maximum impact of their disease on their quality of life).9
Improved HDSS and DLQI scores. Most patients (83%) in the study had generalized hyperhidrosis. Patients were in their mid-thirties. Sixty percent of the patients in the oxybutynin group had an improvement of one point or more on the 4-point HDSS compared to 27% in the placebo group (P<.01). DLQI scores improved by 6.9 points in the oxybutynin group and 2.3 points in the placebo group (P<.01).
The most common adverse effect was dry mouth, which occurred in 13 patients (43%) in the oxybutynin group and in 3 patients (11%) in the placebo group (P<.01); it did not cause any patients to drop out of the study. The second most common adverse effect was blurred vision, which only occurred in the oxybutynin group (4 patients; 13%).
WHAT'S NEW
This is the first randomized controlled trial to demonstrate the efficacy of an oral agent for generalized primary hyperhidrosis. This trial used a relatively low dose of oxybutynin, which produced significant benefit while minimizing anticholinergic adverse effects.
CAVEATS
There are many situations for which anticholinergic medications are inappropriate, including use by geriatric patients and those with gastrointestinal disorders, urinary retention, or glaucoma.
CHALLENGES TO IMPLEMENTATION
Few if any challenges exist to the utilization of oxybutynin; inexpensive generic versions are widely available.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Schollhammer M, Brenaut E, Menard-Andivot N, et al. Oxybutynin as a treatment for generalized hyperhidrosis: a randomized, placebo-controlled trial. Br J Dermatol. 2015;173:1163-1168.
2. Grabell DA, Hebert AA. Current and emerging medical therapies for primary hyperhidrosis. Dermatol Ther (Heidelb). 2017;7:25-36.
3. Innocenzi D, Lupi F, Bruni F, et al. Efficacy of a new aluminium salt thermophobic foam in the treatment of axillary and palmar primary hyperhidrosis: a pilot exploratory trial. Curr Med Res Opin. 2005;21:1949-1953.
4. Goh CL. Aluminum chloride hexahydrate versus palmar hyperhidrosis. Evaporimeter assessment. Int J Dermatol. 1990;29:368-370.
5. Nicholas R, Quddus A, Baker DM. Treatment of primary craniofacial hyperhidrosis: a systematic review. Am J Clin Dermatol. 2015;16:361-370.
6. Naumann M, Lowe NJ, Kumar CR, et al. Botulinum toxin type a is a safe and effective treatment for axillary hyperhidrosis over 16 months: a prospective study. Arch Dermatol. 2003;139:731-736.
7. Wolosker N, de Campos JR, Kauffman P, et al. A randomized placebo-controlled trial of oxybutynin for the initial treatment of palmar and axillary hyperhidrosis. J Vasc Surg. 2012;55:1696-1700.
8. Varella AY, Fukuda JM, Teivelis MP, et al. Translation and validation of Hyperhidrosis Disease Severity Scale. Rev Assoc Med Bras. 2016;62:843-847.
9. Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)—a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19:210-216.
1. Schollhammer M, Brenaut E, Menard-Andivot N, et al. Oxybutynin as a treatment for generalized hyperhidrosis: a randomized, placebo-controlled trial. Br J Dermatol. 2015;173:1163-1168.
2. Grabell DA, Hebert AA. Current and emerging medical therapies for primary hyperhidrosis. Dermatol Ther (Heidelb). 2017;7:25-36.
3. Innocenzi D, Lupi F, Bruni F, et al. Efficacy of a new aluminium salt thermophobic foam in the treatment of axillary and palmar primary hyperhidrosis: a pilot exploratory trial. Curr Med Res Opin. 2005;21:1949-1953.
4. Goh CL. Aluminum chloride hexahydrate versus palmar hyperhidrosis. Evaporimeter assessment. Int J Dermatol. 1990;29:368-370.
5. Nicholas R, Quddus A, Baker DM. Treatment of primary craniofacial hyperhidrosis: a systematic review. Am J Clin Dermatol. 2015;16:361-370.
6. Naumann M, Lowe NJ, Kumar CR, et al. Botulinum toxin type a is a safe and effective treatment for axillary hyperhidrosis over 16 months: a prospective study. Arch Dermatol. 2003;139:731-736.
7. Wolosker N, de Campos JR, Kauffman P, et al. A randomized placebo-controlled trial of oxybutynin for the initial treatment of palmar and axillary hyperhidrosis. J Vasc Surg. 2012;55:1696-1700.
8. Varella AY, Fukuda JM, Teivelis MP, et al. Translation and validation of Hyperhidrosis Disease Severity Scale. Rev Assoc Med Bras. 2016;62:843-847.
9. Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)—a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19:210-216.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
PRACTICE CHANGER
Use low-dose oxybutynin as a first-line treatment option for patients with primary hyperhidrosis to improve symptoms and quality of life.1
STRENGTH OF RECOMMENDATION
B: Based on a single, good quality, randomized controlled trial.
Schollhammer M, Brenaut E, Menard-Andivot N, et al. Oxybutynin as a treatment for generalized hyperhidrosis: a randomized, placebo-controlled trial. Br J Dermatol. 2015;173:1163-1168.
