User login
QI enthusiast turns QI leader
Editor’s note: This new series highlights the professional pathways of quality improvement leaders. This month features the story of Kevin O’Leary, MD, MS, SFHM, chief of hospital medicine at Northwestern University Feinberg School of Medicine in Chicago.
Kevin O’Leary, MD, MS, SFHM, chose a career path in hospital medicine for the reasons that attract many to the specialty – a love of “a little bit of everything, clinically” and the opportunity to problem-solve a diverse range of professional challenges on a daily basis.

“I was frustrated with our internal inefficiencies, and motivated by wanting to provide optimal care to patients,” Dr. O’Leary said, recalling his entry into the world of quality improvement. “It was the first time as a physician that I felt like quality was a problem that I owned – and if anyone was going to address it, it would have to be a hospitalist.”
That epiphany 16 years ago led Dr. O’Leary, now chief of hospital medicine at the same institution, on a path of enacting change. He began volunteering on small improvement projects around the hospital, which led to an invitation to chair the Quality Management Committee in the hospital medicine department. He continued to build his skills by enrolling in Six Sigma training and in Northwestern University’s Master in Healthcare Quality and Patient Safety program.
“That was transformative,” Dr. O’Leary said. “The master’s program, coupled with performance training, changed the trajectory of my career in quality improvement.”
While he encourages anyone with an interest in QI to seek additional training opportunities, he says personal qualities – tenacity, curiosity, and a willingness to collaborate—are better predictors of success. For those wondering how to get started, “look for a niche, an unmet need that is valuable to your organization, and fill it,” he advised. “You don’t have to be an expert in that area, but you can become one.”
Making strong connections within the hospital system is essential. Reach out to the contacts you know, he said, and if they are not the ones to help you solve the problem, they often know who can.
“That’s key to quality improvement success, as well as career success,” he said. “Find a mentor. It might be someone who is more senior within the hospitalist group, in medicine, or even outside the hospital. Meet with them regularly and ask them for feedback on your ideas.”
Newcomers to QI should embrace opportunities to change care and not get discouraged when a project has unintended outcomes.
“Failure is when a team never gets to the point of implementing the intervention or when a team doesn’t know whether the intervention has actually changed results,” he said. “Learning why an intervention isn’t effective can be as valuable as implementing one that is. If every project is successful, it just means that you’re not taking enough risks.”
Dr. O’Leary spends about 25% of his professional time providing clinical care, and another 15% meeting his responsibilities as division chief. He uses the other protected time in his schedule to lead QI and teach QI skills in programs like Northwestern Medicine’s Academy for Quality and Safety Improvement (AQSI).
As a former faculty member in SHM’s Quality and Safety Educator’s Academy (QSEA), he has trained medical educators to develop curricula in quality improvement and patient safety. He says both AQSI and QSEA are especially effective because they encourage interaction, which is valuable to professionals at all levels looking to advance their skill in QI.
“Even in a teaching capacity,” he noted, “what I learned from other faculty and participants in QSEA was critical.”
Residents and junior hospitalists often have the impression that they lack the skills to lead quality initiatives, but Dr. O’Leary says medical school provides the nuts and bolts – analytical skills, statistical knowledge, critical thinking. He encouraged hospitalists to move ahead, even without formal QI training.
“If you have strong interpersonal skills – the willingness to make friends and build connections – you will be successful,” he said.
It’s also an excellent way to learn about the ins and outs of the hospital system and the work of other departments and specialties. Dr. O’Leary especially enjoys that aspect of his work, as well as the ability to address systemic issues that he values.
“I get the greatest fulfillment from the opportunity to be creative … and to implement projects that are important to me and help patients,” he said. “As long as the projects align with organizational goals, I can usually find the support we need to be successful.”
Claudia Stahl is a content manager for the Society of Hospital Medicine.
Editor’s note: This new series highlights the professional pathways of quality improvement leaders. This month features the story of Kevin O’Leary, MD, MS, SFHM, chief of hospital medicine at Northwestern University Feinberg School of Medicine in Chicago.
Kevin O’Leary, MD, MS, SFHM, chose a career path in hospital medicine for the reasons that attract many to the specialty – a love of “a little bit of everything, clinically” and the opportunity to problem-solve a diverse range of professional challenges on a daily basis.
“I was frustrated with our internal inefficiencies, and motivated by wanting to provide optimal care to patients,” Dr. O’Leary said, recalling his entry into the world of quality improvement. “It was the first time as a physician that I felt like quality was a problem that I owned – and if anyone was going to address it, it would have to be a hospitalist.”
That epiphany 16 years ago led Dr. O’Leary, now chief of hospital medicine at the same institution, on a path of enacting change. He began volunteering on small improvement projects around the hospital, which led to an invitation to chair the Quality Management Committee in the hospital medicine department. He continued to build his skills by enrolling in Six Sigma training and in Northwestern University’s Master in Healthcare Quality and Patient Safety program.
“That was transformative,” Dr. O’Leary said. “The master’s program, coupled with performance training, changed the trajectory of my career in quality improvement.”
While he encourages anyone with an interest in QI to seek additional training opportunities, he says personal qualities – tenacity, curiosity, and a willingness to collaborate—are better predictors of success. For those wondering how to get started, “look for a niche, an unmet need that is valuable to your organization, and fill it,” he advised. “You don’t have to be an expert in that area, but you can become one.”
Making strong connections within the hospital system is essential. Reach out to the contacts you know, he said, and if they are not the ones to help you solve the problem, they often know who can.
“That’s key to quality improvement success, as well as career success,” he said. “Find a mentor. It might be someone who is more senior within the hospitalist group, in medicine, or even outside the hospital. Meet with them regularly and ask them for feedback on your ideas.”
Newcomers to QI should embrace opportunities to change care and not get discouraged when a project has unintended outcomes.
“Failure is when a team never gets to the point of implementing the intervention or when a team doesn’t know whether the intervention has actually changed results,” he said. “Learning why an intervention isn’t effective can be as valuable as implementing one that is. If every project is successful, it just means that you’re not taking enough risks.”
Dr. O’Leary spends about 25% of his professional time providing clinical care, and another 15% meeting his responsibilities as division chief. He uses the other protected time in his schedule to lead QI and teach QI skills in programs like Northwestern Medicine’s Academy for Quality and Safety Improvement (AQSI).
As a former faculty member in SHM’s Quality and Safety Educator’s Academy (QSEA), he has trained medical educators to develop curricula in quality improvement and patient safety. He says both AQSI and QSEA are especially effective because they encourage interaction, which is valuable to professionals at all levels looking to advance their skill in QI.
“Even in a teaching capacity,” he noted, “what I learned from other faculty and participants in QSEA was critical.”
Residents and junior hospitalists often have the impression that they lack the skills to lead quality initiatives, but Dr. O’Leary says medical school provides the nuts and bolts – analytical skills, statistical knowledge, critical thinking. He encouraged hospitalists to move ahead, even without formal QI training.
“If you have strong interpersonal skills – the willingness to make friends and build connections – you will be successful,” he said.
It’s also an excellent way to learn about the ins and outs of the hospital system and the work of other departments and specialties. Dr. O’Leary especially enjoys that aspect of his work, as well as the ability to address systemic issues that he values.
“I get the greatest fulfillment from the opportunity to be creative … and to implement projects that are important to me and help patients,” he said. “As long as the projects align with organizational goals, I can usually find the support we need to be successful.”
Claudia Stahl is a content manager for the Society of Hospital Medicine.
Editor’s note: This new series highlights the professional pathways of quality improvement leaders. This month features the story of Kevin O’Leary, MD, MS, SFHM, chief of hospital medicine at Northwestern University Feinberg School of Medicine in Chicago.
Kevin O’Leary, MD, MS, SFHM, chose a career path in hospital medicine for the reasons that attract many to the specialty – a love of “a little bit of everything, clinically” and the opportunity to problem-solve a diverse range of professional challenges on a daily basis.
“I was frustrated with our internal inefficiencies, and motivated by wanting to provide optimal care to patients,” Dr. O’Leary said, recalling his entry into the world of quality improvement. “It was the first time as a physician that I felt like quality was a problem that I owned – and if anyone was going to address it, it would have to be a hospitalist.”
That epiphany 16 years ago led Dr. O’Leary, now chief of hospital medicine at the same institution, on a path of enacting change. He began volunteering on small improvement projects around the hospital, which led to an invitation to chair the Quality Management Committee in the hospital medicine department. He continued to build his skills by enrolling in Six Sigma training and in Northwestern University’s Master in Healthcare Quality and Patient Safety program.
“That was transformative,” Dr. O’Leary said. “The master’s program, coupled with performance training, changed the trajectory of my career in quality improvement.”
While he encourages anyone with an interest in QI to seek additional training opportunities, he says personal qualities – tenacity, curiosity, and a willingness to collaborate—are better predictors of success. For those wondering how to get started, “look for a niche, an unmet need that is valuable to your organization, and fill it,” he advised. “You don’t have to be an expert in that area, but you can become one.”
Making strong connections within the hospital system is essential. Reach out to the contacts you know, he said, and if they are not the ones to help you solve the problem, they often know who can.
“That’s key to quality improvement success, as well as career success,” he said. “Find a mentor. It might be someone who is more senior within the hospitalist group, in medicine, or even outside the hospital. Meet with them regularly and ask them for feedback on your ideas.”
Newcomers to QI should embrace opportunities to change care and not get discouraged when a project has unintended outcomes.
“Failure is when a team never gets to the point of implementing the intervention or when a team doesn’t know whether the intervention has actually changed results,” he said. “Learning why an intervention isn’t effective can be as valuable as implementing one that is. If every project is successful, it just means that you’re not taking enough risks.”
Dr. O’Leary spends about 25% of his professional time providing clinical care, and another 15% meeting his responsibilities as division chief. He uses the other protected time in his schedule to lead QI and teach QI skills in programs like Northwestern Medicine’s Academy for Quality and Safety Improvement (AQSI).
As a former faculty member in SHM’s Quality and Safety Educator’s Academy (QSEA), he has trained medical educators to develop curricula in quality improvement and patient safety. He says both AQSI and QSEA are especially effective because they encourage interaction, which is valuable to professionals at all levels looking to advance their skill in QI.
“Even in a teaching capacity,” he noted, “what I learned from other faculty and participants in QSEA was critical.”
Residents and junior hospitalists often have the impression that they lack the skills to lead quality initiatives, but Dr. O’Leary says medical school provides the nuts and bolts – analytical skills, statistical knowledge, critical thinking. He encouraged hospitalists to move ahead, even without formal QI training.
“If you have strong interpersonal skills – the willingness to make friends and build connections – you will be successful,” he said.
It’s also an excellent way to learn about the ins and outs of the hospital system and the work of other departments and specialties. Dr. O’Leary especially enjoys that aspect of his work, as well as the ability to address systemic issues that he values.
“I get the greatest fulfillment from the opportunity to be creative … and to implement projects that are important to me and help patients,” he said. “As long as the projects align with organizational goals, I can usually find the support we need to be successful.”
Claudia Stahl is a content manager for the Society of Hospital Medicine.
Marijuana abuse linked to increased MI risk
Washington – Marijuana abuse was independently associated with an eye-opening doubled risk of acute MI in a large, retrospective, age-matched cohort study, Ahmad Tarek Chami, MD, reported at the annual meeting of the American College of Cardiology.
The link was strongest by far in young adult marijuana abusers, with an adjusted 3.2-fold increased risk of MI in 25- to 29-year-olds with marijuana abuse noted in their medical records, compared with age-matched controls and a 4.56-fold greater risk among the 30- to 34-year-old cannabis abusers, according to Dr. Chami of Case Western Reserve University in Cleveland.

These data constitute a signal warranting further research. Public opinion regarding potheads has undergone a huge shift. Medical and/or recreational marijuana is now legal in 28 states and the District of Columbia. Surveys indicate that, in 2015, 8.3% of Americans aged 12 years and older had used marijuana during the previous month, and 13.5% had used it within the past year.
“Cardiologists and other physicians are more likely than ever before to encounter patients who use marijuana or even ask them to prescribe it,” Dr. Chami said.
The cannabis plant contains more than 60 cannabinoids. Although marijuana is widely prescribed for treatment of nausea, anorexia, neuropathic pain, glaucoma, seizure disorders, and other conditions, the long-term effects of marijuana on the cardiovascular system are largely unknown, he continued.
This ambiguity was the impetus for Dr. Chami’s study. In it, he utilized a database incorporating 26 health care systems across the United States with nearly 50 million patients, which is maintained by Explorys, an 8-year-old Cleveland-based company.
Dr. Chami identified 210,700 patients with cannabis abuse noted in their medical records, covering provider/patient encounters between October 2011 and September 2016. Their mean age was 36.8 years. The abusers were age-matched to 10,395,060 non–marijuana abuser controls.
The 5-year cumulative incidence of MI in this skewed–young patient population was significantly higher than in the marijuana abuser group: 1.28%, compared with 0.89%, for a 44% increase in relative risk.
However, the marijuana abusers also had a significantly higher burden of cardiovascular risk factors than did their non–cannabis abusing counterparts. They were 2.85 times more likely to have hypertension, 1.59 times more likely to be dyslipidemic, and 7.2 times more likely to be cigarette smokers, and they had a 2.8 times greater prevalence of diabetes. Of note, they were also 17.6 times more likely to have been diagnosed with alcohol abuse, and 61 times more likely to abuse cocaine.
In a multivariate analysis adjusted for these and other potential confounders, marijuana abuse remained independently associated with a 1.73-fold increased risk of acute MI. Moreover, after eliminating patients with known coronary artery disease, the strongest risk factor for MI, from the analysis, marijuana abuse was independently associated with a twofold increased risk of MI.
This was a retrospective study, one limitation of which was the standard caveat regarding the possibility of unrecognized confounders that couldn’t be taken into account.
Another study limitation is the uncertainty regarding the diagnosis of “cannabis abuser” in patients’ charts. The Explorys cloud-based database relies on ICD codes to capture data. It doesn’t include specific information on how much marijuana a patient who was labeled as an abuser was actually using. This limitation raises an unanswered question: Were young adults who abused marijuana at highest risk for MI because of heavier use, or are younger patients’ coronary arteries somehow more vulnerable to marijuana’s potential adverse cardiovascular effects?
Several audience members called Dr. Chami’s study “very provocative.” Aaron D. Kugelmass, MD, said that the fundamental question in his mind is whether the cardiovascular hazard of marijuana identified in this study is the result of the practice of smoking the raw product, usually associated with illicit marijuana abusers.
Today, legalized marijuana is often consumed in the form of edible products, tinctures, and other derivatives that don’t involve inhalation of smoke. Whether these alternative forms of consumption pose any cardiovascular risk is an important unresolved issue in this era of widespread decriminalization of cannabis, noted Dr. Kugelmass, chief of cardiology and medical director of the Heart and Vascular Center at Baystate Medical Center in Springfield, Mass.
Dr. Chami reported having no financial conflicts regarding his study.
Washington – Marijuana abuse was independently associated with an eye-opening doubled risk of acute MI in a large, retrospective, age-matched cohort study, Ahmad Tarek Chami, MD, reported at the annual meeting of the American College of Cardiology.
The link was strongest by far in young adult marijuana abusers, with an adjusted 3.2-fold increased risk of MI in 25- to 29-year-olds with marijuana abuse noted in their medical records, compared with age-matched controls and a 4.56-fold greater risk among the 30- to 34-year-old cannabis abusers, according to Dr. Chami of Case Western Reserve University in Cleveland.
These data constitute a signal warranting further research. Public opinion regarding potheads has undergone a huge shift. Medical and/or recreational marijuana is now legal in 28 states and the District of Columbia. Surveys indicate that, in 2015, 8.3% of Americans aged 12 years and older had used marijuana during the previous month, and 13.5% had used it within the past year.
“Cardiologists and other physicians are more likely than ever before to encounter patients who use marijuana or even ask them to prescribe it,” Dr. Chami said.
The cannabis plant contains more than 60 cannabinoids. Although marijuana is widely prescribed for treatment of nausea, anorexia, neuropathic pain, glaucoma, seizure disorders, and other conditions, the long-term effects of marijuana on the cardiovascular system are largely unknown, he continued.
This ambiguity was the impetus for Dr. Chami’s study. In it, he utilized a database incorporating 26 health care systems across the United States with nearly 50 million patients, which is maintained by Explorys, an 8-year-old Cleveland-based company.
Dr. Chami identified 210,700 patients with cannabis abuse noted in their medical records, covering provider/patient encounters between October 2011 and September 2016. Their mean age was 36.8 years. The abusers were age-matched to 10,395,060 non–marijuana abuser controls.
The 5-year cumulative incidence of MI in this skewed–young patient population was significantly higher than in the marijuana abuser group: 1.28%, compared with 0.89%, for a 44% increase in relative risk.
However, the marijuana abusers also had a significantly higher burden of cardiovascular risk factors than did their non–cannabis abusing counterparts. They were 2.85 times more likely to have hypertension, 1.59 times more likely to be dyslipidemic, and 7.2 times more likely to be cigarette smokers, and they had a 2.8 times greater prevalence of diabetes. Of note, they were also 17.6 times more likely to have been diagnosed with alcohol abuse, and 61 times more likely to abuse cocaine.
In a multivariate analysis adjusted for these and other potential confounders, marijuana abuse remained independently associated with a 1.73-fold increased risk of acute MI. Moreover, after eliminating patients with known coronary artery disease, the strongest risk factor for MI, from the analysis, marijuana abuse was independently associated with a twofold increased risk of MI.
This was a retrospective study, one limitation of which was the standard caveat regarding the possibility of unrecognized confounders that couldn’t be taken into account.
Another study limitation is the uncertainty regarding the diagnosis of “cannabis abuser” in patients’ charts. The Explorys cloud-based database relies on ICD codes to capture data. It doesn’t include specific information on how much marijuana a patient who was labeled as an abuser was actually using. This limitation raises an unanswered question: Were young adults who abused marijuana at highest risk for MI because of heavier use, or are younger patients’ coronary arteries somehow more vulnerable to marijuana’s potential adverse cardiovascular effects?
Several audience members called Dr. Chami’s study “very provocative.” Aaron D. Kugelmass, MD, said that the fundamental question in his mind is whether the cardiovascular hazard of marijuana identified in this study is the result of the practice of smoking the raw product, usually associated with illicit marijuana abusers.
Today, legalized marijuana is often consumed in the form of edible products, tinctures, and other derivatives that don’t involve inhalation of smoke. Whether these alternative forms of consumption pose any cardiovascular risk is an important unresolved issue in this era of widespread decriminalization of cannabis, noted Dr. Kugelmass, chief of cardiology and medical director of the Heart and Vascular Center at Baystate Medical Center in Springfield, Mass.
Dr. Chami reported having no financial conflicts regarding his study.
Washington – Marijuana abuse was independently associated with an eye-opening doubled risk of acute MI in a large, retrospective, age-matched cohort study, Ahmad Tarek Chami, MD, reported at the annual meeting of the American College of Cardiology.
The link was strongest by far in young adult marijuana abusers, with an adjusted 3.2-fold increased risk of MI in 25- to 29-year-olds with marijuana abuse noted in their medical records, compared with age-matched controls and a 4.56-fold greater risk among the 30- to 34-year-old cannabis abusers, according to Dr. Chami of Case Western Reserve University in Cleveland.
These data constitute a signal warranting further research. Public opinion regarding potheads has undergone a huge shift. Medical and/or recreational marijuana is now legal in 28 states and the District of Columbia. Surveys indicate that, in 2015, 8.3% of Americans aged 12 years and older had used marijuana during the previous month, and 13.5% had used it within the past year.
“Cardiologists and other physicians are more likely than ever before to encounter patients who use marijuana or even ask them to prescribe it,” Dr. Chami said.
The cannabis plant contains more than 60 cannabinoids. Although marijuana is widely prescribed for treatment of nausea, anorexia, neuropathic pain, glaucoma, seizure disorders, and other conditions, the long-term effects of marijuana on the cardiovascular system are largely unknown, he continued.
This ambiguity was the impetus for Dr. Chami’s study. In it, he utilized a database incorporating 26 health care systems across the United States with nearly 50 million patients, which is maintained by Explorys, an 8-year-old Cleveland-based company.
Dr. Chami identified 210,700 patients with cannabis abuse noted in their medical records, covering provider/patient encounters between October 2011 and September 2016. Their mean age was 36.8 years. The abusers were age-matched to 10,395,060 non–marijuana abuser controls.
The 5-year cumulative incidence of MI in this skewed–young patient population was significantly higher than in the marijuana abuser group: 1.28%, compared with 0.89%, for a 44% increase in relative risk.
However, the marijuana abusers also had a significantly higher burden of cardiovascular risk factors than did their non–cannabis abusing counterparts. They were 2.85 times more likely to have hypertension, 1.59 times more likely to be dyslipidemic, and 7.2 times more likely to be cigarette smokers, and they had a 2.8 times greater prevalence of diabetes. Of note, they were also 17.6 times more likely to have been diagnosed with alcohol abuse, and 61 times more likely to abuse cocaine.
In a multivariate analysis adjusted for these and other potential confounders, marijuana abuse remained independently associated with a 1.73-fold increased risk of acute MI. Moreover, after eliminating patients with known coronary artery disease, the strongest risk factor for MI, from the analysis, marijuana abuse was independently associated with a twofold increased risk of MI.
This was a retrospective study, one limitation of which was the standard caveat regarding the possibility of unrecognized confounders that couldn’t be taken into account.
Another study limitation is the uncertainty regarding the diagnosis of “cannabis abuser” in patients’ charts. The Explorys cloud-based database relies on ICD codes to capture data. It doesn’t include specific information on how much marijuana a patient who was labeled as an abuser was actually using. This limitation raises an unanswered question: Were young adults who abused marijuana at highest risk for MI because of heavier use, or are younger patients’ coronary arteries somehow more vulnerable to marijuana’s potential adverse cardiovascular effects?
Several audience members called Dr. Chami’s study “very provocative.” Aaron D. Kugelmass, MD, said that the fundamental question in his mind is whether the cardiovascular hazard of marijuana identified in this study is the result of the practice of smoking the raw product, usually associated with illicit marijuana abusers.
Today, legalized marijuana is often consumed in the form of edible products, tinctures, and other derivatives that don’t involve inhalation of smoke. Whether these alternative forms of consumption pose any cardiovascular risk is an important unresolved issue in this era of widespread decriminalization of cannabis, noted Dr. Kugelmass, chief of cardiology and medical director of the Heart and Vascular Center at Baystate Medical Center in Springfield, Mass.
Dr. Chami reported having no financial conflicts regarding his study.
At ACC 17
Key clinical point:
Major finding: Marijuana abuse was associated with a twofold increased risk of acute MI independent of cardiovascular risk factor levels.
Data source: A retrospective cohort study including 210,700 patients with cannabis abuse noted in their medical record and 10,395,060 age-matched controls.
Disclosures: The study presenter reported having no financial conflicts.
A Game for All Seasons
Sports and sports figures provide both a welcome relief from the stress of dealing with life and death in the ED and memorable ways of characterizing serious health care issues. When the Institute of Medicine issued its 2006 report “Hospital-Based Emergency Care: At the Breaking Point,” we thought that a quote about a popular restaurant by the late, great New York Yankees catcher Yogi Berra better described the severely overcrowded EDs and ambulance diversion: “Nobody goes there anymore—it’s too crowded.”
In late winter and early spring of this year, after vigorous attempts to repeal/revise the 2010 Affordable Care Act (ACA), a replacement bill was withdrawn immediately prior to a Congressional vote on March 24 due to a lack of support. Seven years earlier, when President Obama also seemed to have little chance of getting the ACA through Congress, we thought that there would be “many more balks before the president and Congress finally pitched a viable health care package to the nation.” Only a month later, however, with the ACA now the law, we suggested that our erroneous prediction was similar to that of “a father who convinces his son to leave for the parking lot during the bottom of the ninth inning of a 3-0 game only to hear the roar of the crowd from the exit ramp as the rookie batter hits a grand-slam home run to win the game.”
In June 2012, when the Supreme Court ruled on the constitutionality of the ACA, many reporters quickly read the Court’s rejection of the first two arguments defending the ACA, and rushed to report that it was dead, without considering that the government had “one more out to go…[the one] casting ACA as a tax—considered to be the weakest player in the lineup—[which] managed to score the winning run to uphold ACA. Game over. Final score: ACA wins 5 to 4.”
But with the 2017 baseball season finally underway, it is a recent football game that provides the perfect paradigm for emergency medicine (EM) and emergency physicians (EPs). The New England Patriots were slight favorites to win Super Bowl 51 over the Atlanta Falcons on February 5,and the first quarter ended with no score. But by halftime, Atlanta was leading 21-3. In 50 years of Super Bowls, no team had ever overcome more than a 10-point deficit to win the game, and with a little over 8 minutes left in the third quarter, the deficit had widened even further to 28-3. Then the Patriots began to turn things around. Though the Patriots never led during regulation play, and no Super Bowl had ever gone into overtime, the fourth quarter ended in a 28-28 tie, and the Patriots went on to win 34-28 in overtime.
Coming out of the locker room to play the second half of that game in front of over 111 million viewers must have been a daunting experience for the Patriots, but no more so than the experience depicted in EP/cinematographer Ryan McGarry’s award-winning documentary “Code Black,” in which he shows young EM residents walking through a packed waiting room to begin their shift, realizing that in the next 12 hours, they could never treat all of the ill patients waiting to be seen. But the young residents proceeded to treat one patient after another without ever giving up or losing their idealism, until in the end, they, too, had won the game against all odds.
Many patients arrive in EDs so ill that there is no reasonable expectation any intervention can save them, but we nevertheless try and sometimes succeed in doing the seemingly impossible. It is the type of medicine we have chosen to devote our careers to, and we are no less heroes than were the Patriots on February 5, 2017. Each time we go out to “play ball” in our overcrowded EDs, it is worth remembering another famous Yogi Berra quote: “It ain’t over till it’s over.”
Sports and sports figures provide both a welcome relief from the stress of dealing with life and death in the ED and memorable ways of characterizing serious health care issues. When the Institute of Medicine issued its 2006 report “Hospital-Based Emergency Care: At the Breaking Point,” we thought that a quote about a popular restaurant by the late, great New York Yankees catcher Yogi Berra better described the severely overcrowded EDs and ambulance diversion: “Nobody goes there anymore—it’s too crowded.”
In late winter and early spring of this year, after vigorous attempts to repeal/revise the 2010 Affordable Care Act (ACA), a replacement bill was withdrawn immediately prior to a Congressional vote on March 24 due to a lack of support. Seven years earlier, when President Obama also seemed to have little chance of getting the ACA through Congress, we thought that there would be “many more balks before the president and Congress finally pitched a viable health care package to the nation.” Only a month later, however, with the ACA now the law, we suggested that our erroneous prediction was similar to that of “a father who convinces his son to leave for the parking lot during the bottom of the ninth inning of a 3-0 game only to hear the roar of the crowd from the exit ramp as the rookie batter hits a grand-slam home run to win the game.”
In June 2012, when the Supreme Court ruled on the constitutionality of the ACA, many reporters quickly read the Court’s rejection of the first two arguments defending the ACA, and rushed to report that it was dead, without considering that the government had “one more out to go…[the one] casting ACA as a tax—considered to be the weakest player in the lineup—[which] managed to score the winning run to uphold ACA. Game over. Final score: ACA wins 5 to 4.”
But with the 2017 baseball season finally underway, it is a recent football game that provides the perfect paradigm for emergency medicine (EM) and emergency physicians (EPs). The New England Patriots were slight favorites to win Super Bowl 51 over the Atlanta Falcons on February 5,and the first quarter ended with no score. But by halftime, Atlanta was leading 21-3. In 50 years of Super Bowls, no team had ever overcome more than a 10-point deficit to win the game, and with a little over 8 minutes left in the third quarter, the deficit had widened even further to 28-3. Then the Patriots began to turn things around. Though the Patriots never led during regulation play, and no Super Bowl had ever gone into overtime, the fourth quarter ended in a 28-28 tie, and the Patriots went on to win 34-28 in overtime.
Coming out of the locker room to play the second half of that game in front of over 111 million viewers must have been a daunting experience for the Patriots, but no more so than the experience depicted in EP/cinematographer Ryan McGarry’s award-winning documentary “Code Black,” in which he shows young EM residents walking through a packed waiting room to begin their shift, realizing that in the next 12 hours, they could never treat all of the ill patients waiting to be seen. But the young residents proceeded to treat one patient after another without ever giving up or losing their idealism, until in the end, they, too, had won the game against all odds.
Many patients arrive in EDs so ill that there is no reasonable expectation any intervention can save them, but we nevertheless try and sometimes succeed in doing the seemingly impossible. It is the type of medicine we have chosen to devote our careers to, and we are no less heroes than were the Patriots on February 5, 2017. Each time we go out to “play ball” in our overcrowded EDs, it is worth remembering another famous Yogi Berra quote: “It ain’t over till it’s over.”
Sports and sports figures provide both a welcome relief from the stress of dealing with life and death in the ED and memorable ways of characterizing serious health care issues. When the Institute of Medicine issued its 2006 report “Hospital-Based Emergency Care: At the Breaking Point,” we thought that a quote about a popular restaurant by the late, great New York Yankees catcher Yogi Berra better described the severely overcrowded EDs and ambulance diversion: “Nobody goes there anymore—it’s too crowded.”
In late winter and early spring of this year, after vigorous attempts to repeal/revise the 2010 Affordable Care Act (ACA), a replacement bill was withdrawn immediately prior to a Congressional vote on March 24 due to a lack of support. Seven years earlier, when President Obama also seemed to have little chance of getting the ACA through Congress, we thought that there would be “many more balks before the president and Congress finally pitched a viable health care package to the nation.” Only a month later, however, with the ACA now the law, we suggested that our erroneous prediction was similar to that of “a father who convinces his son to leave for the parking lot during the bottom of the ninth inning of a 3-0 game only to hear the roar of the crowd from the exit ramp as the rookie batter hits a grand-slam home run to win the game.”
In June 2012, when the Supreme Court ruled on the constitutionality of the ACA, many reporters quickly read the Court’s rejection of the first two arguments defending the ACA, and rushed to report that it was dead, without considering that the government had “one more out to go…[the one] casting ACA as a tax—considered to be the weakest player in the lineup—[which] managed to score the winning run to uphold ACA. Game over. Final score: ACA wins 5 to 4.”
But with the 2017 baseball season finally underway, it is a recent football game that provides the perfect paradigm for emergency medicine (EM) and emergency physicians (EPs). The New England Patriots were slight favorites to win Super Bowl 51 over the Atlanta Falcons on February 5,and the first quarter ended with no score. But by halftime, Atlanta was leading 21-3. In 50 years of Super Bowls, no team had ever overcome more than a 10-point deficit to win the game, and with a little over 8 minutes left in the third quarter, the deficit had widened even further to 28-3. Then the Patriots began to turn things around. Though the Patriots never led during regulation play, and no Super Bowl had ever gone into overtime, the fourth quarter ended in a 28-28 tie, and the Patriots went on to win 34-28 in overtime.
Coming out of the locker room to play the second half of that game in front of over 111 million viewers must have been a daunting experience for the Patriots, but no more so than the experience depicted in EP/cinematographer Ryan McGarry’s award-winning documentary “Code Black,” in which he shows young EM residents walking through a packed waiting room to begin their shift, realizing that in the next 12 hours, they could never treat all of the ill patients waiting to be seen. But the young residents proceeded to treat one patient after another without ever giving up or losing their idealism, until in the end, they, too, had won the game against all odds.
Many patients arrive in EDs so ill that there is no reasonable expectation any intervention can save them, but we nevertheless try and sometimes succeed in doing the seemingly impossible. It is the type of medicine we have chosen to devote our careers to, and we are no less heroes than were the Patriots on February 5, 2017. Each time we go out to “play ball” in our overcrowded EDs, it is worth remembering another famous Yogi Berra quote: “It ain’t over till it’s over.”

Paraspinous Cervical Nerve Block for Primary Headache
Headaches—pain or discomfort in the head, scalp, or neck—are a very common reason for ED visits.1 In 2011, the World Health Organization estimated that 46.5% of the population in North and South America aged 18 to 65 years old experienced at least one headache within the previous year.1
Migraine is a recurrent headache disorder that afflicts 18% of US women and 9% of US men,2 resulting in at least 1.2 million visits to US EDs annually.1 The economic cost resulting from migraine-related loss of productive time in the US workforce is more than $13 billion per year, most of which is in the form of reduced work productivity.3 Management and treatment for migraine headache in the ED commonly include intravenous (IV) or intramuscular (IM) medications, fluids, or oxygen. While ultimately effective, these methods require nursing care and additional time for posttreatment monitoring, both of which adversely affect patient flow.
In 2006, Mellick et al4 described the safety and effectiveness of paraspinous cervical nerve block (PCNB) to abort migraine headaches. Despite its demonstrated efficacy and safety, a decade later, PCNB is still rarely used. Friedman et al5 ranked peripheral nerve blocks as the fourth step in management suggestions for primary headache.
Case Reports of Headache Patients
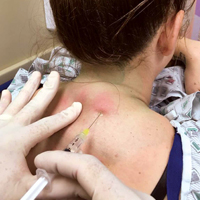
We report on seven headache patients we treated in our ED with PCNB who had good-to-complete resolution of pain, suggesting that PCNB is efficacious and can potentially shorten the ED length of stay. This series of seven patients (six female, one male) was a convenience sample of primary headache patients who presented over a 10-month period and were safely and rapidly treated with PCNB (Table).

In each case, the PCNB procedure was explained to the patient and consent was obtained. Each patient was treated with a total of 3 cc of 0.5% bupivacaine with epinephrine injected into the posterior neck according to the method described by Mellick et al.4 Our seven patients achieved an average 5-point reduction in pain on a 10-point pain scale, with 0 = no pain and 10 = worse possible pain.
Other than the provision of medications, no nursing assistance was required. Only one of the patients required further treatment after the PCNB, and none had an adverse reaction. All of the patients reported that their headaches were similar in nature to past headaches. Based on their history and physical examination, none were diagnosed to be experiencing a secondary, more serious cause of headache, and none subsequently returned to our institution with a secondary type of headache.
The Paraspinal Cervical Nerve Block
Paraspinous cervical nerve block requires less time to administer and recovery is shorter than that from IM or IV opioids, sedatives, or neuroleptics. It is an easy technique to teach since it requires bilateral injections.
Technique
Prior to the procedure, cleanse the bilateral paravertebral zones surrounding C6 and C7 with chlorhexidine. Next, fill a 3 cc syringe using 0.5% bupivacaine with epinephrine.

Once the injection is complete, withdraw the needle completely, and compress and massage the injection site to facilitate anesthetic diffusion to surrounding tissues.

Indications
Paraspinous cervical nerve block is an appropriate treatment only for patients who are having a typical episode of chronic, recurring headaches, whose history and physical examination do not suggest the need for any further diagnostic work-up, and who, in the judgment of the treating clinician, require only pain relief.
Contraindications
A patient should not be considered for PCNB if he or she has a new-onset headache, fever, altered mental status, focal neurological deficits, meningismus, findings suggestive of meningitis, papilledema, increased intracranial pressure from a space-occupying lesion, recent head trauma with concern for intracranial hemorrhage, or suspicion of an alternate diagnosis.
Efficacy and Patient Response
Paraspinous cervical nerve block has been shown to decrease pain in patients who had failed standard migraine therapy and patients reported no complications. Of the seven patients in this case report, only one patient received opioids in the ED and none received prescriptions for opioids upon discharge for outpatient use.
Mellick and Mellick6 have postulated that pain may be modified due to the PCNB effect on the convergence of the trigeminal nerve with sensory fibers from the upper cervical roots. Since cervical innervation provides feeling to the head and upper neck, blocking this input can ameliorate pain.6
Summary
This series of seven patients provides further evidence of the effectiveness of PCNB in relieving headache symptoms for patients with recurrent, primary headaches when a secondary, more serious cause has been clinically excluded. Each of the seven patients had marked improvement of their pain and required only minimal nursing attention; moreover, all stated they would willingly undergo the procedure for future painful episodes.
Although there were no reported complications, this series is too small to demonstrate complete safety of the procedure. While this report is limited by a small sample size, it demonstrates that this is a quick, effective, and easily learned method of addressing a common ED complaint that obviates the need for parenteral medications and offers a potentially decreased patient length of stay.
Paraspinous cervical nerve block is a promising modality of treatment of ED patients who present with headache and migraine symptoms who do not respond to their outpatient “rescue” therapy. This procedure should be considered as an early treatment for migraine and other primary headaches unless contraindicated.
1. World Health Organization. Atlas of headache disorders and resources in the world 2011. http://www.who.int/mental_health/management/who_atlas_headache_disorders_results.pdf. Accessed February 9, 2017.
2. Victor TW, Hu X, Campbell JC, Buse DC, Lipton RB. Migraine prevalence by age and sex in the United States: a life-span study. Cephalalgia. 2010;30(9):1065-1072. doi:10.1177/0333102409355601.
3. Chawla J. Migraine headache. http://emedicine.medscape.com/article/1142556-overview. Accessed February 9, 2017.
4. Mellick LB, McIlrath ST, Mellick GA. Treatment of headaches in the ED with lower cervical intramuscular bupivacaine injections: a 1-year retrospective review of 417 patients. Headache. 2006;46(9):1441-1449.
5. Friedman BW, West J, Vinson DR, et al. Current management of migraine in US emergency departments: an analysis of the National Hospital Ambulatory Medical Care Survey. Cephalalgia. 2015;35:301-309.
6. Mellick GA, Mellick LB. Lower cervical intramuscular bupivacaine injections—another treatment option for headaches. http://www.neurologist-doctor.com/images/Mellick_Headache_injections.pdf. Accessed February 9, 2017.
Headaches—pain or discomfort in the head, scalp, or neck—are a very common reason for ED visits.1 In 2011, the World Health Organization estimated that 46.5% of the population in North and South America aged 18 to 65 years old experienced at least one headache within the previous year.1
Migraine is a recurrent headache disorder that afflicts 18% of US women and 9% of US men,2 resulting in at least 1.2 million visits to US EDs annually.1 The economic cost resulting from migraine-related loss of productive time in the US workforce is more than $13 billion per year, most of which is in the form of reduced work productivity.3 Management and treatment for migraine headache in the ED commonly include intravenous (IV) or intramuscular (IM) medications, fluids, or oxygen. While ultimately effective, these methods require nursing care and additional time for posttreatment monitoring, both of which adversely affect patient flow.
In 2006, Mellick et al4 described the safety and effectiveness of paraspinous cervical nerve block (PCNB) to abort migraine headaches. Despite its demonstrated efficacy and safety, a decade later, PCNB is still rarely used. Friedman et al5 ranked peripheral nerve blocks as the fourth step in management suggestions for primary headache.
Case Reports of Headache Patients
We report on seven headache patients we treated in our ED with PCNB who had good-to-complete resolution of pain, suggesting that PCNB is efficacious and can potentially shorten the ED length of stay. This series of seven patients (six female, one male) was a convenience sample of primary headache patients who presented over a 10-month period and were safely and rapidly treated with PCNB (Table).
In each case, the PCNB procedure was explained to the patient and consent was obtained. Each patient was treated with a total of 3 cc of 0.5% bupivacaine with epinephrine injected into the posterior neck according to the method described by Mellick et al.4 Our seven patients achieved an average 5-point reduction in pain on a 10-point pain scale, with 0 = no pain and 10 = worse possible pain.
Other than the provision of medications, no nursing assistance was required. Only one of the patients required further treatment after the PCNB, and none had an adverse reaction. All of the patients reported that their headaches were similar in nature to past headaches. Based on their history and physical examination, none were diagnosed to be experiencing a secondary, more serious cause of headache, and none subsequently returned to our institution with a secondary type of headache.
The Paraspinal Cervical Nerve Block
Paraspinous cervical nerve block requires less time to administer and recovery is shorter than that from IM or IV opioids, sedatives, or neuroleptics. It is an easy technique to teach since it requires bilateral injections.
Technique
Prior to the procedure, cleanse the bilateral paravertebral zones surrounding C6 and C7 with chlorhexidine. Next, fill a 3 cc syringe using 0.5% bupivacaine with epinephrine.
Once the injection is complete, withdraw the needle completely, and compress and massage the injection site to facilitate anesthetic diffusion to surrounding tissues.
Indications
Paraspinous cervical nerve block is an appropriate treatment only for patients who are having a typical episode of chronic, recurring headaches, whose history and physical examination do not suggest the need for any further diagnostic work-up, and who, in the judgment of the treating clinician, require only pain relief.
Contraindications
A patient should not be considered for PCNB if he or she has a new-onset headache, fever, altered mental status, focal neurological deficits, meningismus, findings suggestive of meningitis, papilledema, increased intracranial pressure from a space-occupying lesion, recent head trauma with concern for intracranial hemorrhage, or suspicion of an alternate diagnosis.
Efficacy and Patient Response
Paraspinous cervical nerve block has been shown to decrease pain in patients who had failed standard migraine therapy and patients reported no complications. Of the seven patients in this case report, only one patient received opioids in the ED and none received prescriptions for opioids upon discharge for outpatient use.
Mellick and Mellick6 have postulated that pain may be modified due to the PCNB effect on the convergence of the trigeminal nerve with sensory fibers from the upper cervical roots. Since cervical innervation provides feeling to the head and upper neck, blocking this input can ameliorate pain.6
Summary
This series of seven patients provides further evidence of the effectiveness of PCNB in relieving headache symptoms for patients with recurrent, primary headaches when a secondary, more serious cause has been clinically excluded. Each of the seven patients had marked improvement of their pain and required only minimal nursing attention; moreover, all stated they would willingly undergo the procedure for future painful episodes.
Although there were no reported complications, this series is too small to demonstrate complete safety of the procedure. While this report is limited by a small sample size, it demonstrates that this is a quick, effective, and easily learned method of addressing a common ED complaint that obviates the need for parenteral medications and offers a potentially decreased patient length of stay.
Paraspinous cervical nerve block is a promising modality of treatment of ED patients who present with headache and migraine symptoms who do not respond to their outpatient “rescue” therapy. This procedure should be considered as an early treatment for migraine and other primary headaches unless contraindicated.
Headaches—pain or discomfort in the head, scalp, or neck—are a very common reason for ED visits.1 In 2011, the World Health Organization estimated that 46.5% of the population in North and South America aged 18 to 65 years old experienced at least one headache within the previous year.1
Migraine is a recurrent headache disorder that afflicts 18% of US women and 9% of US men,2 resulting in at least 1.2 million visits to US EDs annually.1 The economic cost resulting from migraine-related loss of productive time in the US workforce is more than $13 billion per year, most of which is in the form of reduced work productivity.3 Management and treatment for migraine headache in the ED commonly include intravenous (IV) or intramuscular (IM) medications, fluids, or oxygen. While ultimately effective, these methods require nursing care and additional time for posttreatment monitoring, both of which adversely affect patient flow.
In 2006, Mellick et al4 described the safety and effectiveness of paraspinous cervical nerve block (PCNB) to abort migraine headaches. Despite its demonstrated efficacy and safety, a decade later, PCNB is still rarely used. Friedman et al5 ranked peripheral nerve blocks as the fourth step in management suggestions for primary headache.
Case Reports of Headache Patients
We report on seven headache patients we treated in our ED with PCNB who had good-to-complete resolution of pain, suggesting that PCNB is efficacious and can potentially shorten the ED length of stay. This series of seven patients (six female, one male) was a convenience sample of primary headache patients who presented over a 10-month period and were safely and rapidly treated with PCNB (Table).
In each case, the PCNB procedure was explained to the patient and consent was obtained. Each patient was treated with a total of 3 cc of 0.5% bupivacaine with epinephrine injected into the posterior neck according to the method described by Mellick et al.4 Our seven patients achieved an average 5-point reduction in pain on a 10-point pain scale, with 0 = no pain and 10 = worse possible pain.
Other than the provision of medications, no nursing assistance was required. Only one of the patients required further treatment after the PCNB, and none had an adverse reaction. All of the patients reported that their headaches were similar in nature to past headaches. Based on their history and physical examination, none were diagnosed to be experiencing a secondary, more serious cause of headache, and none subsequently returned to our institution with a secondary type of headache.
The Paraspinal Cervical Nerve Block
Paraspinous cervical nerve block requires less time to administer and recovery is shorter than that from IM or IV opioids, sedatives, or neuroleptics. It is an easy technique to teach since it requires bilateral injections.
Technique
Prior to the procedure, cleanse the bilateral paravertebral zones surrounding C6 and C7 with chlorhexidine. Next, fill a 3 cc syringe using 0.5% bupivacaine with epinephrine.
Once the injection is complete, withdraw the needle completely, and compress and massage the injection site to facilitate anesthetic diffusion to surrounding tissues.
Indications
Paraspinous cervical nerve block is an appropriate treatment only for patients who are having a typical episode of chronic, recurring headaches, whose history and physical examination do not suggest the need for any further diagnostic work-up, and who, in the judgment of the treating clinician, require only pain relief.
Contraindications
A patient should not be considered for PCNB if he or she has a new-onset headache, fever, altered mental status, focal neurological deficits, meningismus, findings suggestive of meningitis, papilledema, increased intracranial pressure from a space-occupying lesion, recent head trauma with concern for intracranial hemorrhage, or suspicion of an alternate diagnosis.
Efficacy and Patient Response
Paraspinous cervical nerve block has been shown to decrease pain in patients who had failed standard migraine therapy and patients reported no complications. Of the seven patients in this case report, only one patient received opioids in the ED and none received prescriptions for opioids upon discharge for outpatient use.
Mellick and Mellick6 have postulated that pain may be modified due to the PCNB effect on the convergence of the trigeminal nerve with sensory fibers from the upper cervical roots. Since cervical innervation provides feeling to the head and upper neck, blocking this input can ameliorate pain.6
Summary
This series of seven patients provides further evidence of the effectiveness of PCNB in relieving headache symptoms for patients with recurrent, primary headaches when a secondary, more serious cause has been clinically excluded. Each of the seven patients had marked improvement of their pain and required only minimal nursing attention; moreover, all stated they would willingly undergo the procedure for future painful episodes.
Although there were no reported complications, this series is too small to demonstrate complete safety of the procedure. While this report is limited by a small sample size, it demonstrates that this is a quick, effective, and easily learned method of addressing a common ED complaint that obviates the need for parenteral medications and offers a potentially decreased patient length of stay.
Paraspinous cervical nerve block is a promising modality of treatment of ED patients who present with headache and migraine symptoms who do not respond to their outpatient “rescue” therapy. This procedure should be considered as an early treatment for migraine and other primary headaches unless contraindicated.
1. World Health Organization. Atlas of headache disorders and resources in the world 2011. http://www.who.int/mental_health/management/who_atlas_headache_disorders_results.pdf. Accessed February 9, 2017.
2. Victor TW, Hu X, Campbell JC, Buse DC, Lipton RB. Migraine prevalence by age and sex in the United States: a life-span study. Cephalalgia. 2010;30(9):1065-1072. doi:10.1177/0333102409355601.
3. Chawla J. Migraine headache. http://emedicine.medscape.com/article/1142556-overview. Accessed February 9, 2017.
4. Mellick LB, McIlrath ST, Mellick GA. Treatment of headaches in the ED with lower cervical intramuscular bupivacaine injections: a 1-year retrospective review of 417 patients. Headache. 2006;46(9):1441-1449.
5. Friedman BW, West J, Vinson DR, et al. Current management of migraine in US emergency departments: an analysis of the National Hospital Ambulatory Medical Care Survey. Cephalalgia. 2015;35:301-309.
6. Mellick GA, Mellick LB. Lower cervical intramuscular bupivacaine injections—another treatment option for headaches. http://www.neurologist-doctor.com/images/Mellick_Headache_injections.pdf. Accessed February 9, 2017.
1. World Health Organization. Atlas of headache disorders and resources in the world 2011. http://www.who.int/mental_health/management/who_atlas_headache_disorders_results.pdf. Accessed February 9, 2017.
2. Victor TW, Hu X, Campbell JC, Buse DC, Lipton RB. Migraine prevalence by age and sex in the United States: a life-span study. Cephalalgia. 2010;30(9):1065-1072. doi:10.1177/0333102409355601.
3. Chawla J. Migraine headache. http://emedicine.medscape.com/article/1142556-overview. Accessed February 9, 2017.
4. Mellick LB, McIlrath ST, Mellick GA. Treatment of headaches in the ED with lower cervical intramuscular bupivacaine injections: a 1-year retrospective review of 417 patients. Headache. 2006;46(9):1441-1449.
5. Friedman BW, West J, Vinson DR, et al. Current management of migraine in US emergency departments: an analysis of the National Hospital Ambulatory Medical Care Survey. Cephalalgia. 2015;35:301-309.
6. Mellick GA, Mellick LB. Lower cervical intramuscular bupivacaine injections—another treatment option for headaches. http://www.neurologist-doctor.com/images/Mellick_Headache_injections.pdf. Accessed February 9, 2017.
Hypothyroidism-Induced Stercoral Sigmoid Colonic Perforation
According to the Centers for Disease Control and Prevention, abdominal pain is the leading reason for ED visits in the United States, with approximately 10 million visits per year.1 Though a large number of presentations are due to nontraumatic causes of abdominal pain, one etiology is among the most time-sensitive and critical diagnoses: acute colonic perforation.
Colonic perforations can be caused by diverticulitis, trauma, malignancy, ulcerative colitis, and other etiologies.2 A rare, yet life-threatening cause of colonic perforation, of which only a few cases have been documented in the literature, is stercoral colonic perforation.2
Regardless of the etiology, the critical actions for any colonic perforation are quick recognition, medical stabilization, and surgical evaluation. This case report highlights the diagnosis and treatment of acute stercoral colonic perforation with peritonitis secondary to hypothyroidism.
Case
A 49-year-old woman with a medical history significant for hypothyroidism presented to the ED for evaluation of diffuse abdominal pain, nausea, and nonbilious, nonbloody vomiting that started in the early evening of presentation. The patient denied any previous pain or associated symptoms, and said she had a small, hard bowel movement 1 day prior to arrival. She began experiencing mild abdominal pain on the morning of presentation. Her symptoms acutely worsened at approximately 5:00
On physical examination, her vital signs were: heart rate, 156 beats/min; blood pressure, 134/84 mm Hg; respiratory rate, 20 breaths/min, and temperature, 97.4°F. The patient appeared ill and diaphoretic, writhing on the stretcher. Abdominal examination was significant for diminished bowel sounds, diffuse abdominal distension, rigidity, and tenderness with light palpation.
Laboratory evaluation showed an elevated lactic acid level of 7.7 mmol/L, a white blood cell count of 7,200 cells/mm3 (segment form, 69.5%), and the following abnormal blood chemistry results: creatinine, 2.08 mg/dL; aspartate aminotransferase, 176 U/L; alanine aminotransferase, 138 U/L; and thyroid-stimulating hormone (TSH), 225.3 mcIU/mL. Other laboratory results were within normal range. Her electrocardiogram showed sinus tachycardia with a rate of 154 beats/min, a QTc within normal limits, and no ST elevations or depressions.
An abdominopelvic computed tomography (CT) scan revealed free air, free fluid, and possibly stool within the abdomen and pelvis. The findings were consistent with a ruptured hollow viscus, possibly a sigmoid colonic perforation. The radiologist also noted hepatomegaly and significant hepatic steatosis. A surgeon was immediately notified and evaluated the patient in the ED. The working diagnosis was stercoral colonic perforation secondary to severe hypothyroidism, and the patient was taken emergently to the operating room for repair.
Intraoperatively, the patient underwent exploratory laparotomy, which revealed gross fecal contamination of the abdomen. The surgeon noted that there was fecal staining along the serosal surface of the small bowel and throughout the pelvis. There were also large, hard stool balls outside of the colon. The perforation was along the mesenteric surface of the sigmoid just above the rectosigmoid junction.
The abdomen was copiously irrigated, the perforated segment was resected, and a Hartmann colostomy was created. The diagnosis was stercoral sigmoid perforation with peritonitis, and the patient was transferred to the intensive care unit for antibiotic treatment and further medical care, including intravenous (IV) levothyroxine.
She was extubated uneventfully on postoperative day 2, and the acute renal failure improved with supportive care only. Her bowel function slowly returned without complication. She was switched to oral levothyroxine on postoperative day 3. On day 13, she was given strict instructions for continuation of her thyroid medication and close monitoring for postsurgical complications, and was discharged home with appropriate follow-up.
Discussion
Multiple contributing factors can lead to bowel perforation. In this case, severe hypothyroidism with constipation caused a colonic perforation. Our patient had severe constipation that increased intraluminal pressure, causing the bowel wall to become ischemic and subsequently perforate.3 Any disease that causes significant constipation or obstruction of transit could lead to the same catastrophic result.
According to Huang et al,4 as of 2002, fewer than 90 cases of general stercoral bowel perforation had been reported, with no clear age range. However, patients in their mid-50s to mid-60s appear to be the most commonly affected age group.4 Our patient was younger than this age group, making identification of the problem by age alone difficult.
Hypothyroidism
The incidence of hypothyroidism in iodine-replete communities varies between 1% to 2% of the general population.5 The condition is more common in older women, affecting approximately 10% of those over age 65 years. In the United States, the prevalence of biochemical hypothyroidism is 4.6%; however, clinically evident hypothyroidism is present in only 0.3%.6 Common causes for hypothyroidism are listed in the Table.7,8

Myxedema Coma
Untreated, hypothyroidism can lead to potentially fatal conditions, such as myxedema coma, which is characterized by hypothermia, hypotension, bradycardia, respiratory depression, and altered mental status.7 Severe myxedema coma can result in cardiovascular collapse, and eventual death. Electrocardiography findings of severe hypothyroidism include bradycardia, low-voltage QRS, and widespread T-wave inversions.7 Our patient was tachycardic and did not have any acute findings to suggest myxedema coma.
Treatment for myxedema coma includes supportive care with ventilatory support and pressor support if necessary. Patients should be given IV hydrocortisone, 100 mg, to treat possible adrenal insufficiency and T4, 4 mcg/kg by slow IV infusion.7 Caution should be taken if giving a patient T3 due to the risk of dysrhythmias and myocardial infarction (MI).7 As our patient was not displaying myxedema coma, the surgeon elected not to start IV thyroid replacement to avoid exacerbating the patient’s tachycardia and possibly precipitating an MI intraoperatively.
Conclusion
Our case underscores the importance of promptly recognizing the signs and symptoms of stercoral colonic perforation in patients who present with nontraumatic abdominal pain accompanied by nausea and nonbilious, nonbloody vomiting. Although stercoral colonic perforation is a rare cause of nontraumatic abdominal pain, as with any type of colonic perforation, it constitutes a life-threatening medical emergency. As our case illustrates, prompt diagnosis through a thorough history taking, physical examination, and laboratory and imaging studies is critical to ensure medical stabilization and surgical management to reduce morbidity and mortality.
1. Centers for Disease Control and Prevention. Table 10. Ten leading principal reasons for emergency department visits, by patient age and sex: United States, 2013. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed March 3, 2017.
2. Nam JK, Kim BS, Kim KS, Moon DJ. Clinical analysis of stercoral perforation of the colon. Korean J Gastroenterol. 2010;55:46-51.
3. Heffernan C, Pachter HL, Megibow AJ, Macari M. Stercoral colitis leading to fatal peritonitis: CT findings. AJR Am J Roentgenol. 2005;184(4):1189-1193. doi:10.2214/ajr.184.4.01841189.
4. Huang WS, Wang CS, Hsieh CC, Lin PY, Chin CC, Wang JY. Management of patients with stercoral perforation of the sigmoid colon: Report of five cases. World J Gastroenterol. 2006;12(3):500-503.
5. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160(4):526-534.
6. Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
7. Idrose AM. Hypothyroidism. In: Tintinalli JE, Stapczynski JS, Ma OJ, Yealy DM, Meckler GD, Cline DM. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 8th Edition. New York, NY: McGraw-Hill Education; 2016:1469-1472.
8. Skugor M. Hypothyroidism and Hyperthyroidism. Cleveland Clinic Center for Continuing Education. August 2014. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/endocrinology/hypothyroidism-and-hyperthyroidism/. Accessed March 3, 2017.
According to the Centers for Disease Control and Prevention, abdominal pain is the leading reason for ED visits in the United States, with approximately 10 million visits per year.1 Though a large number of presentations are due to nontraumatic causes of abdominal pain, one etiology is among the most time-sensitive and critical diagnoses: acute colonic perforation.
Colonic perforations can be caused by diverticulitis, trauma, malignancy, ulcerative colitis, and other etiologies.2 A rare, yet life-threatening cause of colonic perforation, of which only a few cases have been documented in the literature, is stercoral colonic perforation.2
Regardless of the etiology, the critical actions for any colonic perforation are quick recognition, medical stabilization, and surgical evaluation. This case report highlights the diagnosis and treatment of acute stercoral colonic perforation with peritonitis secondary to hypothyroidism.
Case
A 49-year-old woman with a medical history significant for hypothyroidism presented to the ED for evaluation of diffuse abdominal pain, nausea, and nonbilious, nonbloody vomiting that started in the early evening of presentation. The patient denied any previous pain or associated symptoms, and said she had a small, hard bowel movement 1 day prior to arrival. She began experiencing mild abdominal pain on the morning of presentation. Her symptoms acutely worsened at approximately 5:00
On physical examination, her vital signs were: heart rate, 156 beats/min; blood pressure, 134/84 mm Hg; respiratory rate, 20 breaths/min, and temperature, 97.4°F. The patient appeared ill and diaphoretic, writhing on the stretcher. Abdominal examination was significant for diminished bowel sounds, diffuse abdominal distension, rigidity, and tenderness with light palpation.
Laboratory evaluation showed an elevated lactic acid level of 7.7 mmol/L, a white blood cell count of 7,200 cells/mm3 (segment form, 69.5%), and the following abnormal blood chemistry results: creatinine, 2.08 mg/dL; aspartate aminotransferase, 176 U/L; alanine aminotransferase, 138 U/L; and thyroid-stimulating hormone (TSH), 225.3 mcIU/mL. Other laboratory results were within normal range. Her electrocardiogram showed sinus tachycardia with a rate of 154 beats/min, a QTc within normal limits, and no ST elevations or depressions.
An abdominopelvic computed tomography (CT) scan revealed free air, free fluid, and possibly stool within the abdomen and pelvis. The findings were consistent with a ruptured hollow viscus, possibly a sigmoid colonic perforation. The radiologist also noted hepatomegaly and significant hepatic steatosis. A surgeon was immediately notified and evaluated the patient in the ED. The working diagnosis was stercoral colonic perforation secondary to severe hypothyroidism, and the patient was taken emergently to the operating room for repair.
Intraoperatively, the patient underwent exploratory laparotomy, which revealed gross fecal contamination of the abdomen. The surgeon noted that there was fecal staining along the serosal surface of the small bowel and throughout the pelvis. There were also large, hard stool balls outside of the colon. The perforation was along the mesenteric surface of the sigmoid just above the rectosigmoid junction.
The abdomen was copiously irrigated, the perforated segment was resected, and a Hartmann colostomy was created. The diagnosis was stercoral sigmoid perforation with peritonitis, and the patient was transferred to the intensive care unit for antibiotic treatment and further medical care, including intravenous (IV) levothyroxine.
She was extubated uneventfully on postoperative day 2, and the acute renal failure improved with supportive care only. Her bowel function slowly returned without complication. She was switched to oral levothyroxine on postoperative day 3. On day 13, she was given strict instructions for continuation of her thyroid medication and close monitoring for postsurgical complications, and was discharged home with appropriate follow-up.
Discussion
Multiple contributing factors can lead to bowel perforation. In this case, severe hypothyroidism with constipation caused a colonic perforation. Our patient had severe constipation that increased intraluminal pressure, causing the bowel wall to become ischemic and subsequently perforate.3 Any disease that causes significant constipation or obstruction of transit could lead to the same catastrophic result.
According to Huang et al,4 as of 2002, fewer than 90 cases of general stercoral bowel perforation had been reported, with no clear age range. However, patients in their mid-50s to mid-60s appear to be the most commonly affected age group.4 Our patient was younger than this age group, making identification of the problem by age alone difficult.
Hypothyroidism
The incidence of hypothyroidism in iodine-replete communities varies between 1% to 2% of the general population.5 The condition is more common in older women, affecting approximately 10% of those over age 65 years. In the United States, the prevalence of biochemical hypothyroidism is 4.6%; however, clinically evident hypothyroidism is present in only 0.3%.6 Common causes for hypothyroidism are listed in the Table.7,8
Myxedema Coma
Untreated, hypothyroidism can lead to potentially fatal conditions, such as myxedema coma, which is characterized by hypothermia, hypotension, bradycardia, respiratory depression, and altered mental status.7 Severe myxedema coma can result in cardiovascular collapse, and eventual death. Electrocardiography findings of severe hypothyroidism include bradycardia, low-voltage QRS, and widespread T-wave inversions.7 Our patient was tachycardic and did not have any acute findings to suggest myxedema coma.
Treatment for myxedema coma includes supportive care with ventilatory support and pressor support if necessary. Patients should be given IV hydrocortisone, 100 mg, to treat possible adrenal insufficiency and T4, 4 mcg/kg by slow IV infusion.7 Caution should be taken if giving a patient T3 due to the risk of dysrhythmias and myocardial infarction (MI).7 As our patient was not displaying myxedema coma, the surgeon elected not to start IV thyroid replacement to avoid exacerbating the patient’s tachycardia and possibly precipitating an MI intraoperatively.
Conclusion
Our case underscores the importance of promptly recognizing the signs and symptoms of stercoral colonic perforation in patients who present with nontraumatic abdominal pain accompanied by nausea and nonbilious, nonbloody vomiting. Although stercoral colonic perforation is a rare cause of nontraumatic abdominal pain, as with any type of colonic perforation, it constitutes a life-threatening medical emergency. As our case illustrates, prompt diagnosis through a thorough history taking, physical examination, and laboratory and imaging studies is critical to ensure medical stabilization and surgical management to reduce morbidity and mortality.
According to the Centers for Disease Control and Prevention, abdominal pain is the leading reason for ED visits in the United States, with approximately 10 million visits per year.1 Though a large number of presentations are due to nontraumatic causes of abdominal pain, one etiology is among the most time-sensitive and critical diagnoses: acute colonic perforation.
Colonic perforations can be caused by diverticulitis, trauma, malignancy, ulcerative colitis, and other etiologies.2 A rare, yet life-threatening cause of colonic perforation, of which only a few cases have been documented in the literature, is stercoral colonic perforation.2
Regardless of the etiology, the critical actions for any colonic perforation are quick recognition, medical stabilization, and surgical evaluation. This case report highlights the diagnosis and treatment of acute stercoral colonic perforation with peritonitis secondary to hypothyroidism.
Case
A 49-year-old woman with a medical history significant for hypothyroidism presented to the ED for evaluation of diffuse abdominal pain, nausea, and nonbilious, nonbloody vomiting that started in the early evening of presentation. The patient denied any previous pain or associated symptoms, and said she had a small, hard bowel movement 1 day prior to arrival. She began experiencing mild abdominal pain on the morning of presentation. Her symptoms acutely worsened at approximately 5:00
On physical examination, her vital signs were: heart rate, 156 beats/min; blood pressure, 134/84 mm Hg; respiratory rate, 20 breaths/min, and temperature, 97.4°F. The patient appeared ill and diaphoretic, writhing on the stretcher. Abdominal examination was significant for diminished bowel sounds, diffuse abdominal distension, rigidity, and tenderness with light palpation.
Laboratory evaluation showed an elevated lactic acid level of 7.7 mmol/L, a white blood cell count of 7,200 cells/mm3 (segment form, 69.5%), and the following abnormal blood chemistry results: creatinine, 2.08 mg/dL; aspartate aminotransferase, 176 U/L; alanine aminotransferase, 138 U/L; and thyroid-stimulating hormone (TSH), 225.3 mcIU/mL. Other laboratory results were within normal range. Her electrocardiogram showed sinus tachycardia with a rate of 154 beats/min, a QTc within normal limits, and no ST elevations or depressions.
An abdominopelvic computed tomography (CT) scan revealed free air, free fluid, and possibly stool within the abdomen and pelvis. The findings were consistent with a ruptured hollow viscus, possibly a sigmoid colonic perforation. The radiologist also noted hepatomegaly and significant hepatic steatosis. A surgeon was immediately notified and evaluated the patient in the ED. The working diagnosis was stercoral colonic perforation secondary to severe hypothyroidism, and the patient was taken emergently to the operating room for repair.
Intraoperatively, the patient underwent exploratory laparotomy, which revealed gross fecal contamination of the abdomen. The surgeon noted that there was fecal staining along the serosal surface of the small bowel and throughout the pelvis. There were also large, hard stool balls outside of the colon. The perforation was along the mesenteric surface of the sigmoid just above the rectosigmoid junction.
The abdomen was copiously irrigated, the perforated segment was resected, and a Hartmann colostomy was created. The diagnosis was stercoral sigmoid perforation with peritonitis, and the patient was transferred to the intensive care unit for antibiotic treatment and further medical care, including intravenous (IV) levothyroxine.
She was extubated uneventfully on postoperative day 2, and the acute renal failure improved with supportive care only. Her bowel function slowly returned without complication. She was switched to oral levothyroxine on postoperative day 3. On day 13, she was given strict instructions for continuation of her thyroid medication and close monitoring for postsurgical complications, and was discharged home with appropriate follow-up.
Discussion
Multiple contributing factors can lead to bowel perforation. In this case, severe hypothyroidism with constipation caused a colonic perforation. Our patient had severe constipation that increased intraluminal pressure, causing the bowel wall to become ischemic and subsequently perforate.3 Any disease that causes significant constipation or obstruction of transit could lead to the same catastrophic result.
According to Huang et al,4 as of 2002, fewer than 90 cases of general stercoral bowel perforation had been reported, with no clear age range. However, patients in their mid-50s to mid-60s appear to be the most commonly affected age group.4 Our patient was younger than this age group, making identification of the problem by age alone difficult.
Hypothyroidism
The incidence of hypothyroidism in iodine-replete communities varies between 1% to 2% of the general population.5 The condition is more common in older women, affecting approximately 10% of those over age 65 years. In the United States, the prevalence of biochemical hypothyroidism is 4.6%; however, clinically evident hypothyroidism is present in only 0.3%.6 Common causes for hypothyroidism are listed in the Table.7,8
Myxedema Coma
Untreated, hypothyroidism can lead to potentially fatal conditions, such as myxedema coma, which is characterized by hypothermia, hypotension, bradycardia, respiratory depression, and altered mental status.7 Severe myxedema coma can result in cardiovascular collapse, and eventual death. Electrocardiography findings of severe hypothyroidism include bradycardia, low-voltage QRS, and widespread T-wave inversions.7 Our patient was tachycardic and did not have any acute findings to suggest myxedema coma.
Treatment for myxedema coma includes supportive care with ventilatory support and pressor support if necessary. Patients should be given IV hydrocortisone, 100 mg, to treat possible adrenal insufficiency and T4, 4 mcg/kg by slow IV infusion.7 Caution should be taken if giving a patient T3 due to the risk of dysrhythmias and myocardial infarction (MI).7 As our patient was not displaying myxedema coma, the surgeon elected not to start IV thyroid replacement to avoid exacerbating the patient’s tachycardia and possibly precipitating an MI intraoperatively.
Conclusion
Our case underscores the importance of promptly recognizing the signs and symptoms of stercoral colonic perforation in patients who present with nontraumatic abdominal pain accompanied by nausea and nonbilious, nonbloody vomiting. Although stercoral colonic perforation is a rare cause of nontraumatic abdominal pain, as with any type of colonic perforation, it constitutes a life-threatening medical emergency. As our case illustrates, prompt diagnosis through a thorough history taking, physical examination, and laboratory and imaging studies is critical to ensure medical stabilization and surgical management to reduce morbidity and mortality.
1. Centers for Disease Control and Prevention. Table 10. Ten leading principal reasons for emergency department visits, by patient age and sex: United States, 2013. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed March 3, 2017.
2. Nam JK, Kim BS, Kim KS, Moon DJ. Clinical analysis of stercoral perforation of the colon. Korean J Gastroenterol. 2010;55:46-51.
3. Heffernan C, Pachter HL, Megibow AJ, Macari M. Stercoral colitis leading to fatal peritonitis: CT findings. AJR Am J Roentgenol. 2005;184(4):1189-1193. doi:10.2214/ajr.184.4.01841189.
4. Huang WS, Wang CS, Hsieh CC, Lin PY, Chin CC, Wang JY. Management of patients with stercoral perforation of the sigmoid colon: Report of five cases. World J Gastroenterol. 2006;12(3):500-503.
5. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160(4):526-534.
6. Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
7. Idrose AM. Hypothyroidism. In: Tintinalli JE, Stapczynski JS, Ma OJ, Yealy DM, Meckler GD, Cline DM. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 8th Edition. New York, NY: McGraw-Hill Education; 2016:1469-1472.
8. Skugor M. Hypothyroidism and Hyperthyroidism. Cleveland Clinic Center for Continuing Education. August 2014. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/endocrinology/hypothyroidism-and-hyperthyroidism/. Accessed March 3, 2017.
1. Centers for Disease Control and Prevention. Table 10. Ten leading principal reasons for emergency department visits, by patient age and sex: United States, 2013. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed March 3, 2017.
2. Nam JK, Kim BS, Kim KS, Moon DJ. Clinical analysis of stercoral perforation of the colon. Korean J Gastroenterol. 2010;55:46-51.
3. Heffernan C, Pachter HL, Megibow AJ, Macari M. Stercoral colitis leading to fatal peritonitis: CT findings. AJR Am J Roentgenol. 2005;184(4):1189-1193. doi:10.2214/ajr.184.4.01841189.
4. Huang WS, Wang CS, Hsieh CC, Lin PY, Chin CC, Wang JY. Management of patients with stercoral perforation of the sigmoid colon: Report of five cases. World J Gastroenterol. 2006;12(3):500-503.
5. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160(4):526-534.
6. Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
7. Idrose AM. Hypothyroidism. In: Tintinalli JE, Stapczynski JS, Ma OJ, Yealy DM, Meckler GD, Cline DM. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 8th Edition. New York, NY: McGraw-Hill Education; 2016:1469-1472.
8. Skugor M. Hypothyroidism and Hyperthyroidism. Cleveland Clinic Center for Continuing Education. August 2014. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/endocrinology/hypothyroidism-and-hyperthyroidism/. Accessed March 3, 2017.

Emergency Imaging: Multiple Comorbidities With Fever and Nonproductive Cough



Laboratory studies revealed leukocytosis with a left shift. Chest radiographs were negative for pneumonia. A magnetic resonance image (MRI) of the lumbar spine was obtained to evaluate for diskitis osteomyelitis. A radiograph of the pelvis was also obtained to evaluate the patient’s THAs, and a computed tomography scan (CT) of the abdomen and pelvis with contrast was obtained for further evaluation. Representative CT, radiographic, and MRI images are shown at left (Figures 1-3).
What is the suspected diagnosis?
Answer
The MRI of the lumbar spine demonstrated no evidence of diskitis osteomyelitis. However, T2-weighted axial images showed enlarged heterogeneous bilateral psoas muscles with bright signal, indicating the presence of fluid (white arrows, Figure 4).


On the pelvic radiographs, both femoral heads appeared off-center within the acetabular cups (red arrows, Figure 5). This eccentric positioning indicated wear of the polyethylene in the THAs that normally occupies the space between the acetabular cup and the femoral head. In addition, focal lucency in the right acetabulum indicated breakdown of the bone, a condition referred to as osteolysis (white asterisk, Figure 5).
An abdominopelvic CT scan with contrast was performed and confirmed the findings of polyethylene wear and osteolysis. The CT scan also demonstrated large bilateral hip joint effusions (white arrows, Figure 6), decompressed along distended bilateral iliopsoas bursae (red asterisks, Figure 6), and communicating with the bilateral psoas muscle collections (red arrows, Figure 6).
Osteolysis With Iliopsoas Bursitis
Bursae are fluid-filled sacs lined by synovial tissue located throughout the body to reduce friction at sites of movement between muscles, bones, and tendons. Bursitis develops when these sacs become inflamed and/or infected and fill with fluid. The iliopsoas bursa lies between the anterior capsule of the hip and the psoas tendon, iliacus tendon, and muscle fibers.1,2 This bursa frequently communicates with the hip joint.3,4 Iliopsoas bursal distension has been reported following THA in the setting of polyethylene wear,5 and aseptic bursitis is a commonly seen incidental finding at the time of revision surgery.6

In this patient, long-standing polyethylene-induced synovitis had markedly expanded the hip joints and iliopsoas bursae, eventually resulting in superinfection, which accounted for the patient’s symptoms.
Treatment
Based on the imaging findings, interventional radiology services were contacted. The interventional radiologist drained the bilateral psoas abscesses. Cultures of the fluid were positive for both methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible S aureus (MSSA). The patient was admitted to the hospital for treatment of MRSA and MSSA with intravenous antibiotic therapy. He recovered from the infection and was discharged on hospital day 2, with instructions to follow up with an orthopedic surgeon to discuss eventual revision of the bilateral THAs.
1. Chandler SB. The iliopsoas bursa in man. Anatom Record. 1934;58(3),235-240. doi:10.1002/ar.1090580304.
2. Tatu L, Parratte B, Vuillier F, Diop M, Monnier G. Descriptive anatomy of the femoral portion of the iliopsoas muscle. Anatomical basis of anterior snapping of the hip. Surg Radiol Anat. 2001;23(6):371-374.
3. Meaney JF, Cassar-Pullicino VN, Etherington R, Ritchie DA, McCall IW, Whitehouse GH. Ilio-psoas bursa enlargement. Clin Radiol. 1992;45(3):161-168.
4. Warren R, Kaye JJ, Salvati EA. Arthrographic demonstration of an enlarged iliopsoas bursa complicating osteoarthritis of the hip. A case report. J Bone Joint Surg Am. 1975;57(3):413-415.
5. Cheung YM, Gupte CM, Beverly MJ. Iliopsoas bursitis following total hip replacement. Arch Orthop Trauma Surg. 2004;124(10):720-723. Epub 2004 Oct 23. doi:10.1007/s00402-004-0751-9.
6. Howie DW, Cain CM, Cornish BL. Pseudo-abscess of the psoas bursa in failed double-cup arthroplasty of the hip. J Bone Joint Surg Br. 1991;73:29-32.
Laboratory studies revealed leukocytosis with a left shift. Chest radiographs were negative for pneumonia. A magnetic resonance image (MRI) of the lumbar spine was obtained to evaluate for diskitis osteomyelitis. A radiograph of the pelvis was also obtained to evaluate the patient’s THAs, and a computed tomography scan (CT) of the abdomen and pelvis with contrast was obtained for further evaluation. Representative CT, radiographic, and MRI images are shown at left (Figures 1-3).
What is the suspected diagnosis?
Answer
The MRI of the lumbar spine demonstrated no evidence of diskitis osteomyelitis. However, T2-weighted axial images showed enlarged heterogeneous bilateral psoas muscles with bright signal, indicating the presence of fluid (white arrows, Figure 4).
On the pelvic radiographs, both femoral heads appeared off-center within the acetabular cups (red arrows, Figure 5). This eccentric positioning indicated wear of the polyethylene in the THAs that normally occupies the space between the acetabular cup and the femoral head. In addition, focal lucency in the right acetabulum indicated breakdown of the bone, a condition referred to as osteolysis (white asterisk, Figure 5).
An abdominopelvic CT scan with contrast was performed and confirmed the findings of polyethylene wear and osteolysis. The CT scan also demonstrated large bilateral hip joint effusions (white arrows, Figure 6), decompressed along distended bilateral iliopsoas bursae (red asterisks, Figure 6), and communicating with the bilateral psoas muscle collections (red arrows, Figure 6).
Osteolysis With Iliopsoas Bursitis
Bursae are fluid-filled sacs lined by synovial tissue located throughout the body to reduce friction at sites of movement between muscles, bones, and tendons. Bursitis develops when these sacs become inflamed and/or infected and fill with fluid. The iliopsoas bursa lies between the anterior capsule of the hip and the psoas tendon, iliacus tendon, and muscle fibers.1,2 This bursa frequently communicates with the hip joint.3,4 Iliopsoas bursal distension has been reported following THA in the setting of polyethylene wear,5 and aseptic bursitis is a commonly seen incidental finding at the time of revision surgery.6
In this patient, long-standing polyethylene-induced synovitis had markedly expanded the hip joints and iliopsoas bursae, eventually resulting in superinfection, which accounted for the patient’s symptoms.
Treatment
Based on the imaging findings, interventional radiology services were contacted. The interventional radiologist drained the bilateral psoas abscesses. Cultures of the fluid were positive for both methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible S aureus (MSSA). The patient was admitted to the hospital for treatment of MRSA and MSSA with intravenous antibiotic therapy. He recovered from the infection and was discharged on hospital day 2, with instructions to follow up with an orthopedic surgeon to discuss eventual revision of the bilateral THAs.
Laboratory studies revealed leukocytosis with a left shift. Chest radiographs were negative for pneumonia. A magnetic resonance image (MRI) of the lumbar spine was obtained to evaluate for diskitis osteomyelitis. A radiograph of the pelvis was also obtained to evaluate the patient’s THAs, and a computed tomography scan (CT) of the abdomen and pelvis with contrast was obtained for further evaluation. Representative CT, radiographic, and MRI images are shown at left (Figures 1-3).
What is the suspected diagnosis?
Answer
The MRI of the lumbar spine demonstrated no evidence of diskitis osteomyelitis. However, T2-weighted axial images showed enlarged heterogeneous bilateral psoas muscles with bright signal, indicating the presence of fluid (white arrows, Figure 4).
On the pelvic radiographs, both femoral heads appeared off-center within the acetabular cups (red arrows, Figure 5). This eccentric positioning indicated wear of the polyethylene in the THAs that normally occupies the space between the acetabular cup and the femoral head. In addition, focal lucency in the right acetabulum indicated breakdown of the bone, a condition referred to as osteolysis (white asterisk, Figure 5).
An abdominopelvic CT scan with contrast was performed and confirmed the findings of polyethylene wear and osteolysis. The CT scan also demonstrated large bilateral hip joint effusions (white arrows, Figure 6), decompressed along distended bilateral iliopsoas bursae (red asterisks, Figure 6), and communicating with the bilateral psoas muscle collections (red arrows, Figure 6).
Osteolysis With Iliopsoas Bursitis
Bursae are fluid-filled sacs lined by synovial tissue located throughout the body to reduce friction at sites of movement between muscles, bones, and tendons. Bursitis develops when these sacs become inflamed and/or infected and fill with fluid. The iliopsoas bursa lies between the anterior capsule of the hip and the psoas tendon, iliacus tendon, and muscle fibers.1,2 This bursa frequently communicates with the hip joint.3,4 Iliopsoas bursal distension has been reported following THA in the setting of polyethylene wear,5 and aseptic bursitis is a commonly seen incidental finding at the time of revision surgery.6
In this patient, long-standing polyethylene-induced synovitis had markedly expanded the hip joints and iliopsoas bursae, eventually resulting in superinfection, which accounted for the patient’s symptoms.
Treatment
Based on the imaging findings, interventional radiology services were contacted. The interventional radiologist drained the bilateral psoas abscesses. Cultures of the fluid were positive for both methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible S aureus (MSSA). The patient was admitted to the hospital for treatment of MRSA and MSSA with intravenous antibiotic therapy. He recovered from the infection and was discharged on hospital day 2, with instructions to follow up with an orthopedic surgeon to discuss eventual revision of the bilateral THAs.
1. Chandler SB. The iliopsoas bursa in man. Anatom Record. 1934;58(3),235-240. doi:10.1002/ar.1090580304.
2. Tatu L, Parratte B, Vuillier F, Diop M, Monnier G. Descriptive anatomy of the femoral portion of the iliopsoas muscle. Anatomical basis of anterior snapping of the hip. Surg Radiol Anat. 2001;23(6):371-374.
3. Meaney JF, Cassar-Pullicino VN, Etherington R, Ritchie DA, McCall IW, Whitehouse GH. Ilio-psoas bursa enlargement. Clin Radiol. 1992;45(3):161-168.
4. Warren R, Kaye JJ, Salvati EA. Arthrographic demonstration of an enlarged iliopsoas bursa complicating osteoarthritis of the hip. A case report. J Bone Joint Surg Am. 1975;57(3):413-415.
5. Cheung YM, Gupte CM, Beverly MJ. Iliopsoas bursitis following total hip replacement. Arch Orthop Trauma Surg. 2004;124(10):720-723. Epub 2004 Oct 23. doi:10.1007/s00402-004-0751-9.
6. Howie DW, Cain CM, Cornish BL. Pseudo-abscess of the psoas bursa in failed double-cup arthroplasty of the hip. J Bone Joint Surg Br. 1991;73:29-32.
1. Chandler SB. The iliopsoas bursa in man. Anatom Record. 1934;58(3),235-240. doi:10.1002/ar.1090580304.
2. Tatu L, Parratte B, Vuillier F, Diop M, Monnier G. Descriptive anatomy of the femoral portion of the iliopsoas muscle. Anatomical basis of anterior snapping of the hip. Surg Radiol Anat. 2001;23(6):371-374.
3. Meaney JF, Cassar-Pullicino VN, Etherington R, Ritchie DA, McCall IW, Whitehouse GH. Ilio-psoas bursa enlargement. Clin Radiol. 1992;45(3):161-168.
4. Warren R, Kaye JJ, Salvati EA. Arthrographic demonstration of an enlarged iliopsoas bursa complicating osteoarthritis of the hip. A case report. J Bone Joint Surg Am. 1975;57(3):413-415.
5. Cheung YM, Gupte CM, Beverly MJ. Iliopsoas bursitis following total hip replacement. Arch Orthop Trauma Surg. 2004;124(10):720-723. Epub 2004 Oct 23. doi:10.1007/s00402-004-0751-9.
6. Howie DW, Cain CM, Cornish BL. Pseudo-abscess of the psoas bursa in failed double-cup arthroplasty of the hip. J Bone Joint Surg Br. 1991;73:29-32.

Malpractice Counsel: The Challenges of Cardioversion
Case
A 56-year-old woman presented to the ED with palpitations and lightheadedness, which began upon awakening that morning. The patient had a history of atrial fibrillation (AF), and believed this was the cause of her symptoms. Over the past 18 months, the patient had twice undergone successful cardioversion for AF with a rapid ventricular response (RVR); both cardioversions were performed by her cardiologist.
The patient denied experiencing any chest pain, shortness of breath, nausea, or vomiting. Her medical history was significant only for AF. Regarding her medication history, the patient had been prescribed metoprolol, but admitted that she frequently forgot to take it. She further stated that she was not taking aspirin or anticoagulation therapy for AF. She denied past or current alcohol consumption or tobacco use.
On physical examination, the patient’s vital signs were: heart rate (HR), 186 beats/min; blood pressure, 137/82 mm Hg; respiratory rate, 20 breaths/min, and temperature, afebrile. Oxygen saturation was 96% on room air. The head, eye, ears, nose, and throat examination was normal. Auscultation of the lungs revealed clear breath sounds bilaterally. On examination of the heart, the patient had an irregularly irregular rhythm that was tachycardic; no murmurs, rubs, or gallops were appreciated. The abdomen was soft and nontender. There was no edema or redness of the lower extremities.
The emergency physician (EP) placed the patient on a cardiac monitor and administered 2 L of oxygen via nasal cannula. An electrocardiogram (ECG), portable chest X-ray (CXR), and laboratory evaluation were ordered, and an intravenous (IV) line was established. The ECG revealed AF with RVR, without evidence of ischemia. The CXR was interpreted as normal. Laboratory studies, including complete blood count, basic metabolic profile, and serum troponin levels, were likewise within normal limits.
Based on the patient’s history and evaluation, the EP decided to cardiovert the patient rather than attempt rate control with IV medications. The patient consented to the cardioversion, based on the two previous successful cardioversions performed by her cardiologist. The EP gave the patient midazolam 2 mg IV and performed synchronized cardioversion at 200 joules. The patient converted to normal sinus rhythm with an HR of 86 beats/min. She was observed in the ED for 1 hour, given metoprolol 50 mg by mouth, and discharged home with instructions to follow up with her cardiologist the following week.
The next day, the patient suffered a large ischemic stroke in the distribution of the left middle cerebral artery, resulting in a dense right hemiparesis. The neurological deficit was significant, necessitating the patient’s placement in a nursing home.
The patient and her family sued the EP for malpractice for not anticoagulating the patient prior to and following cardioversion. A $3.3 million settlement was agreed upon prior to trial.
Discussion
Patients commonly present to the ED for complaints related to AF. In some cases, the EP is the first to diagnose the patient’s AF; in other cases, the patient has a history of AF and is presenting with a complication. The focus of this discussion is solely on the management of AF with RVR.
When managing a patient in AF with RVR, the EP must consider three issues: ventricular rate control (VRC), rhythm control, and anticoagulation. Selecting the best treatment strategy will depend on the patient’s hemodynamic stability, duration of her or his symptoms, local custom and preference, and the length of time the AF has been present.
Ventricular Rate Control and Cardioversion
For many stable patients, VRC is frequently the treatment of choice, with a goal HR of less than 100 beats/min. Intravenous diltiazem, esmolol, or metoprolol can be used to achieve VRC in patients in AF. Because these drugs only control ventricular rate and do not typically cardiovert, the risk of embolization is small.
Synchronized cardioversion has the benefit of providing both rate and rhythm control, but at the expense of the increased risk of arterial embolization. Some patients, including those with rheumatic heart disease, mitral stenosis, prosthetic heart valves, severe left ventricular dysfunction, or a history of thromboembolism, are at a constant high risk of developing a thromboembolism.1
Risk-Benefit Ratio and Anticoagulation Therapy
To help determine the risk-benefit ratio in patients without the risk factors mentioned above, the EP should calculate the CHADS2 (congestive heart failure [CHF], hypertension, age, diabetes mellitus [DM], prior stroke, transient ischemic attack [TIA], or thromboembolism [doubled]) score or CHA2DS2-VASC (CHF, hypertension, age 75 years or older [two scores], DM, previous stroke, TIA, or thromboembolism [doubled], vascular disease, age 65-74 years, sex [female]) score to help identify patients at risk for arterial embolic complications (Table).

For patients who have been in AF for less than 48 hours and who are at a very low-embolic risk (CHA2DS2-VASC score of 0), some experts suggest cardioversion without anticoagulation. However, other experts recommend anticoagulation prior to cardioversion—even in low-risk patients. Unfortunately, there is disagreement between professional organizations, with the American Heart Association/American College of Cardiology/Heart Rhythm Society stating that cardioversion may be performed with or without procedural anticoagulation,2 while the 2016 European Society of Cardiology guidelines recommend immediate initiation of anticoagulants in all such patients scheduled for cardioversion.3
The reasoning in favor of anticoagulation prior to cardioversion is supported by an observational study by Airaksinen et al4 of 2,481 patients undergoing cardioversion for AF of less than 48 hours duration. This study demonstrated a definite thromboembolic event in 38 (0.7%) of the patients within 30 days (median of 2 days). The thromboembolic event was stroke in 31 of the 38 patients.4 Airaksinen et al4 found that age older than 60 years, female sex, heart failure (HF), and DM were the strongest predictors of embolization. The risk of stroke in patients without HF and those younger than age 60 years was only 0.2%.4
In a similar observational study by Hansen et al5 of 16,274 patients in AF undergoing cardioversion with and without anticoagulation therapy, the absence of postcardioversion anticoagulation increased the risk of thromboembolism 2-fold—regardless of CHA2DS2-VASC scores.
Summary
While the management of AF with a duration of more than 48 hours should always include some type of anticoagulation therapy (pre- or postcardioversion, or both), the role of anticoagulation in low-risk patients with AF of less than 48 hours is not as clear. As this situation is not uncommon, the emergency medicine and cardiology physicians should consider developing a mutually agreed upon protocol on how best to manage these patients at their institution. When considering cardioversion without pre- or postanticoagulation in low-risk patients with AF, EPs should always involve the patient in the decision-making process.
1. Phang R, Manning WJ. Prevention of embolization prior to and after restoration of sinus rhythm in atrial fibrillation. UptoDate Web site. http://www.uptodate.com/contents/prevention-of-embolization-prior-to-and-after-restoration-of-sinus-rhythm-in-atrial-fibrillation. Updated October 10, 2016. Accessed March 6, 2017.
2. January CT, Wann LS, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. Erratum in Circulation. 2014;130(23):e272-e274. doi:10.1161/CIR.0000000000000041.
3. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016:37(38):2893-2962. doi:10.1093/eurheartj/ehw210.
4. Airaksinen KE, Grönberg T, Nuotio I, et al. Thromboembolic complications after cardioversion of acute atrial fibrillation: the FinCV (Finnish CardioVersion) study. J Am Coll Cardiol. 2013;62(13):1187-1192. doi:10.1016/j.jacc.2013.04.089.
5. Hansen ML, Jepsen RM, Olesen JB, et al. Thromboembolic risk in 16 274 atrial fibrillation patients undergoing direct current cardioversion with and without oral anticoagulant therapy. Europace. 2015;17(1):18-23. doi:10.1093/europace/euu189.
Case
A 56-year-old woman presented to the ED with palpitations and lightheadedness, which began upon awakening that morning. The patient had a history of atrial fibrillation (AF), and believed this was the cause of her symptoms. Over the past 18 months, the patient had twice undergone successful cardioversion for AF with a rapid ventricular response (RVR); both cardioversions were performed by her cardiologist.
The patient denied experiencing any chest pain, shortness of breath, nausea, or vomiting. Her medical history was significant only for AF. Regarding her medication history, the patient had been prescribed metoprolol, but admitted that she frequently forgot to take it. She further stated that she was not taking aspirin or anticoagulation therapy for AF. She denied past or current alcohol consumption or tobacco use.
On physical examination, the patient’s vital signs were: heart rate (HR), 186 beats/min; blood pressure, 137/82 mm Hg; respiratory rate, 20 breaths/min, and temperature, afebrile. Oxygen saturation was 96% on room air. The head, eye, ears, nose, and throat examination was normal. Auscultation of the lungs revealed clear breath sounds bilaterally. On examination of the heart, the patient had an irregularly irregular rhythm that was tachycardic; no murmurs, rubs, or gallops were appreciated. The abdomen was soft and nontender. There was no edema or redness of the lower extremities.
The emergency physician (EP) placed the patient on a cardiac monitor and administered 2 L of oxygen via nasal cannula. An electrocardiogram (ECG), portable chest X-ray (CXR), and laboratory evaluation were ordered, and an intravenous (IV) line was established. The ECG revealed AF with RVR, without evidence of ischemia. The CXR was interpreted as normal. Laboratory studies, including complete blood count, basic metabolic profile, and serum troponin levels, were likewise within normal limits.
Based on the patient’s history and evaluation, the EP decided to cardiovert the patient rather than attempt rate control with IV medications. The patient consented to the cardioversion, based on the two previous successful cardioversions performed by her cardiologist. The EP gave the patient midazolam 2 mg IV and performed synchronized cardioversion at 200 joules. The patient converted to normal sinus rhythm with an HR of 86 beats/min. She was observed in the ED for 1 hour, given metoprolol 50 mg by mouth, and discharged home with instructions to follow up with her cardiologist the following week.
The next day, the patient suffered a large ischemic stroke in the distribution of the left middle cerebral artery, resulting in a dense right hemiparesis. The neurological deficit was significant, necessitating the patient’s placement in a nursing home.
The patient and her family sued the EP for malpractice for not anticoagulating the patient prior to and following cardioversion. A $3.3 million settlement was agreed upon prior to trial.
Discussion
Patients commonly present to the ED for complaints related to AF. In some cases, the EP is the first to diagnose the patient’s AF; in other cases, the patient has a history of AF and is presenting with a complication. The focus of this discussion is solely on the management of AF with RVR.
When managing a patient in AF with RVR, the EP must consider three issues: ventricular rate control (VRC), rhythm control, and anticoagulation. Selecting the best treatment strategy will depend on the patient’s hemodynamic stability, duration of her or his symptoms, local custom and preference, and the length of time the AF has been present.
Ventricular Rate Control and Cardioversion
For many stable patients, VRC is frequently the treatment of choice, with a goal HR of less than 100 beats/min. Intravenous diltiazem, esmolol, or metoprolol can be used to achieve VRC in patients in AF. Because these drugs only control ventricular rate and do not typically cardiovert, the risk of embolization is small.
Synchronized cardioversion has the benefit of providing both rate and rhythm control, but at the expense of the increased risk of arterial embolization. Some patients, including those with rheumatic heart disease, mitral stenosis, prosthetic heart valves, severe left ventricular dysfunction, or a history of thromboembolism, are at a constant high risk of developing a thromboembolism.1
Risk-Benefit Ratio and Anticoagulation Therapy
To help determine the risk-benefit ratio in patients without the risk factors mentioned above, the EP should calculate the CHADS2 (congestive heart failure [CHF], hypertension, age, diabetes mellitus [DM], prior stroke, transient ischemic attack [TIA], or thromboembolism [doubled]) score or CHA2DS2-VASC (CHF, hypertension, age 75 years or older [two scores], DM, previous stroke, TIA, or thromboembolism [doubled], vascular disease, age 65-74 years, sex [female]) score to help identify patients at risk for arterial embolic complications (Table).
For patients who have been in AF for less than 48 hours and who are at a very low-embolic risk (CHA2DS2-VASC score of 0), some experts suggest cardioversion without anticoagulation. However, other experts recommend anticoagulation prior to cardioversion—even in low-risk patients. Unfortunately, there is disagreement between professional organizations, with the American Heart Association/American College of Cardiology/Heart Rhythm Society stating that cardioversion may be performed with or without procedural anticoagulation,2 while the 2016 European Society of Cardiology guidelines recommend immediate initiation of anticoagulants in all such patients scheduled for cardioversion.3
The reasoning in favor of anticoagulation prior to cardioversion is supported by an observational study by Airaksinen et al4 of 2,481 patients undergoing cardioversion for AF of less than 48 hours duration. This study demonstrated a definite thromboembolic event in 38 (0.7%) of the patients within 30 days (median of 2 days). The thromboembolic event was stroke in 31 of the 38 patients.4 Airaksinen et al4 found that age older than 60 years, female sex, heart failure (HF), and DM were the strongest predictors of embolization. The risk of stroke in patients without HF and those younger than age 60 years was only 0.2%.4
In a similar observational study by Hansen et al5 of 16,274 patients in AF undergoing cardioversion with and without anticoagulation therapy, the absence of postcardioversion anticoagulation increased the risk of thromboembolism 2-fold—regardless of CHA2DS2-VASC scores.
Summary
While the management of AF with a duration of more than 48 hours should always include some type of anticoagulation therapy (pre- or postcardioversion, or both), the role of anticoagulation in low-risk patients with AF of less than 48 hours is not as clear. As this situation is not uncommon, the emergency medicine and cardiology physicians should consider developing a mutually agreed upon protocol on how best to manage these patients at their institution. When considering cardioversion without pre- or postanticoagulation in low-risk patients with AF, EPs should always involve the patient in the decision-making process.
Case
A 56-year-old woman presented to the ED with palpitations and lightheadedness, which began upon awakening that morning. The patient had a history of atrial fibrillation (AF), and believed this was the cause of her symptoms. Over the past 18 months, the patient had twice undergone successful cardioversion for AF with a rapid ventricular response (RVR); both cardioversions were performed by her cardiologist.
The patient denied experiencing any chest pain, shortness of breath, nausea, or vomiting. Her medical history was significant only for AF. Regarding her medication history, the patient had been prescribed metoprolol, but admitted that she frequently forgot to take it. She further stated that she was not taking aspirin or anticoagulation therapy for AF. She denied past or current alcohol consumption or tobacco use.
On physical examination, the patient’s vital signs were: heart rate (HR), 186 beats/min; blood pressure, 137/82 mm Hg; respiratory rate, 20 breaths/min, and temperature, afebrile. Oxygen saturation was 96% on room air. The head, eye, ears, nose, and throat examination was normal. Auscultation of the lungs revealed clear breath sounds bilaterally. On examination of the heart, the patient had an irregularly irregular rhythm that was tachycardic; no murmurs, rubs, or gallops were appreciated. The abdomen was soft and nontender. There was no edema or redness of the lower extremities.
The emergency physician (EP) placed the patient on a cardiac monitor and administered 2 L of oxygen via nasal cannula. An electrocardiogram (ECG), portable chest X-ray (CXR), and laboratory evaluation were ordered, and an intravenous (IV) line was established. The ECG revealed AF with RVR, without evidence of ischemia. The CXR was interpreted as normal. Laboratory studies, including complete blood count, basic metabolic profile, and serum troponin levels, were likewise within normal limits.
Based on the patient’s history and evaluation, the EP decided to cardiovert the patient rather than attempt rate control with IV medications. The patient consented to the cardioversion, based on the two previous successful cardioversions performed by her cardiologist. The EP gave the patient midazolam 2 mg IV and performed synchronized cardioversion at 200 joules. The patient converted to normal sinus rhythm with an HR of 86 beats/min. She was observed in the ED for 1 hour, given metoprolol 50 mg by mouth, and discharged home with instructions to follow up with her cardiologist the following week.
The next day, the patient suffered a large ischemic stroke in the distribution of the left middle cerebral artery, resulting in a dense right hemiparesis. The neurological deficit was significant, necessitating the patient’s placement in a nursing home.
The patient and her family sued the EP for malpractice for not anticoagulating the patient prior to and following cardioversion. A $3.3 million settlement was agreed upon prior to trial.
Discussion
Patients commonly present to the ED for complaints related to AF. In some cases, the EP is the first to diagnose the patient’s AF; in other cases, the patient has a history of AF and is presenting with a complication. The focus of this discussion is solely on the management of AF with RVR.
When managing a patient in AF with RVR, the EP must consider three issues: ventricular rate control (VRC), rhythm control, and anticoagulation. Selecting the best treatment strategy will depend on the patient’s hemodynamic stability, duration of her or his symptoms, local custom and preference, and the length of time the AF has been present.
Ventricular Rate Control and Cardioversion
For many stable patients, VRC is frequently the treatment of choice, with a goal HR of less than 100 beats/min. Intravenous diltiazem, esmolol, or metoprolol can be used to achieve VRC in patients in AF. Because these drugs only control ventricular rate and do not typically cardiovert, the risk of embolization is small.
Synchronized cardioversion has the benefit of providing both rate and rhythm control, but at the expense of the increased risk of arterial embolization. Some patients, including those with rheumatic heart disease, mitral stenosis, prosthetic heart valves, severe left ventricular dysfunction, or a history of thromboembolism, are at a constant high risk of developing a thromboembolism.1
Risk-Benefit Ratio and Anticoagulation Therapy
To help determine the risk-benefit ratio in patients without the risk factors mentioned above, the EP should calculate the CHADS2 (congestive heart failure [CHF], hypertension, age, diabetes mellitus [DM], prior stroke, transient ischemic attack [TIA], or thromboembolism [doubled]) score or CHA2DS2-VASC (CHF, hypertension, age 75 years or older [two scores], DM, previous stroke, TIA, or thromboembolism [doubled], vascular disease, age 65-74 years, sex [female]) score to help identify patients at risk for arterial embolic complications (Table).
For patients who have been in AF for less than 48 hours and who are at a very low-embolic risk (CHA2DS2-VASC score of 0), some experts suggest cardioversion without anticoagulation. However, other experts recommend anticoagulation prior to cardioversion—even in low-risk patients. Unfortunately, there is disagreement between professional organizations, with the American Heart Association/American College of Cardiology/Heart Rhythm Society stating that cardioversion may be performed with or without procedural anticoagulation,2 while the 2016 European Society of Cardiology guidelines recommend immediate initiation of anticoagulants in all such patients scheduled for cardioversion.3
The reasoning in favor of anticoagulation prior to cardioversion is supported by an observational study by Airaksinen et al4 of 2,481 patients undergoing cardioversion for AF of less than 48 hours duration. This study demonstrated a definite thromboembolic event in 38 (0.7%) of the patients within 30 days (median of 2 days). The thromboembolic event was stroke in 31 of the 38 patients.4 Airaksinen et al4 found that age older than 60 years, female sex, heart failure (HF), and DM were the strongest predictors of embolization. The risk of stroke in patients without HF and those younger than age 60 years was only 0.2%.4
In a similar observational study by Hansen et al5 of 16,274 patients in AF undergoing cardioversion with and without anticoagulation therapy, the absence of postcardioversion anticoagulation increased the risk of thromboembolism 2-fold—regardless of CHA2DS2-VASC scores.
Summary
While the management of AF with a duration of more than 48 hours should always include some type of anticoagulation therapy (pre- or postcardioversion, or both), the role of anticoagulation in low-risk patients with AF of less than 48 hours is not as clear. As this situation is not uncommon, the emergency medicine and cardiology physicians should consider developing a mutually agreed upon protocol on how best to manage these patients at their institution. When considering cardioversion without pre- or postanticoagulation in low-risk patients with AF, EPs should always involve the patient in the decision-making process.
1. Phang R, Manning WJ. Prevention of embolization prior to and after restoration of sinus rhythm in atrial fibrillation. UptoDate Web site. http://www.uptodate.com/contents/prevention-of-embolization-prior-to-and-after-restoration-of-sinus-rhythm-in-atrial-fibrillation. Updated October 10, 2016. Accessed March 6, 2017.
2. January CT, Wann LS, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. Erratum in Circulation. 2014;130(23):e272-e274. doi:10.1161/CIR.0000000000000041.
3. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016:37(38):2893-2962. doi:10.1093/eurheartj/ehw210.
4. Airaksinen KE, Grönberg T, Nuotio I, et al. Thromboembolic complications after cardioversion of acute atrial fibrillation: the FinCV (Finnish CardioVersion) study. J Am Coll Cardiol. 2013;62(13):1187-1192. doi:10.1016/j.jacc.2013.04.089.
5. Hansen ML, Jepsen RM, Olesen JB, et al. Thromboembolic risk in 16 274 atrial fibrillation patients undergoing direct current cardioversion with and without oral anticoagulant therapy. Europace. 2015;17(1):18-23. doi:10.1093/europace/euu189.
1. Phang R, Manning WJ. Prevention of embolization prior to and after restoration of sinus rhythm in atrial fibrillation. UptoDate Web site. http://www.uptodate.com/contents/prevention-of-embolization-prior-to-and-after-restoration-of-sinus-rhythm-in-atrial-fibrillation. Updated October 10, 2016. Accessed March 6, 2017.
2. January CT, Wann LS, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. Erratum in Circulation. 2014;130(23):e272-e274. doi:10.1161/CIR.0000000000000041.
3. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016:37(38):2893-2962. doi:10.1093/eurheartj/ehw210.
4. Airaksinen KE, Grönberg T, Nuotio I, et al. Thromboembolic complications after cardioversion of acute atrial fibrillation: the FinCV (Finnish CardioVersion) study. J Am Coll Cardiol. 2013;62(13):1187-1192. doi:10.1016/j.jacc.2013.04.089.
5. Hansen ML, Jepsen RM, Olesen JB, et al. Thromboembolic risk in 16 274 atrial fibrillation patients undergoing direct current cardioversion with and without oral anticoagulant therapy. Europace. 2015;17(1):18-23. doi:10.1093/europace/euu189.
First EDition: Mobile Stroke Units Becoming More Common, more
MITCHEL L. ZOLER
FRONTLINE MEDICAL NEWS
Mobile stroke units—specially equipped ambulances that bring a diagnostic computed tomography (CT) scanner and therapeutic thrombolysis directly to patients in the field—have begun to proliferate across the United States, although they remain investigational, with no clear proof of their incremental clinical value or cost-effectiveness.
The first US mobile stroke unit (MSU) launched in Houston, Texas in early 2014 (following the world’s first in Berlin, Germany, which began running in early 2011), and by early 2017, at least eight other US MSUs were in operation, most of them put into service during the prior 15 months. United States MSU locations now include Cleveland, Ohio; Denver, Colorado; Memphis, Tennessee; New York, New York; Toledo, Ohio; Trenton, New Jersey; and Northwestern Medicine and Rush University Medical Center in the western Chicago, Illinois region. A tenth MSU is slated to start operation at the University of California, Los Angeles later this year.
Early data collected at some of these sites show that initiating care of an acute ischemic stroke patient in an MSU shaves precious minutes off the time it takes to initiate thrombolytic therapy with tissue plasminogen activator (tPA), and findings from preliminary analyses suggest better functional outcomes for patients treated this way. However, leaders in the nascent field readily admit that the data needed to clearly prove the benefit patients receive from operating MSUs are still a few years off. This uncertainty about the added benefit to patients from MSUs couples with one clear fact: MSUs are expensive to start up, with a price tag of roughly $1 million to get an MSU on the road for the first time; they are also expensive to operate, with one estimate for the annual cost of keeping an MSU on the street at about $500,000 per year for staffing, supplies, and other expenses.
“Every US MSU I know of started with philanthropic gifts, but you need a business model” to keep the program running long-term, James C. Grotta, MD, said during a session focused on MSUs at the International Stroke Conference sponsored by the American Heart Association. “You can’t sustain an MSU with philanthropy,” said Dr Grotta, professor of neurology at the University of Texas Health Science Center in Houston, director and founder of the Houston MSU, and acknowledged “godfather” of all US MSUs.
“We believe that MSUs are very worthwhile and that the clinical and economic benefits of earlier stroke treatment [made possible with MSUs] could offset the costs, but we need to show this,” admitted May Nour, MD, a vascular and interventional neurologist at the University of California, Los Angeles (UCLA), and director of the soon-to-launch Los Angeles MSU.
The concept behind MSUs is simple: Each one carries a CT scanner on board so that once the vehicle’s staff identifies a patient with clinical signs of a significant-acute ischemic stroke in the field and confirms that the timing of the stroke onset suggests eligibility for tPA treatment, a CT scan can immediately be run on-site to finalize tPA eligibility. The MSU staff can then begin infusing the drug in the ambulance as it speeds the patient to an appropriate hospital.
In addition, many MSUs now carry a scanner that can perform a CT angiogram (CTA) to locate the occluding clot. If a large vessel occlusion is found, the crew can bring the patient directly to a comprehensive stroke center for a thrombectomy. If thrombectomy is not appropriate, the MSU crew may take the patient to a primary stroke center where thrombectomy is not available.
Another advantage to MSUs, in addition to quicker initiation of thrombolysis, is “getting patients to where they need to go faster and more directly,” said Dr Nour.
“Instead of bringing patients first to a hospital that’s unable to do thrombectomy and where treatment gets slowed down, with an MSU you can give tPA on the street and go straight to a thrombectomy center,” agreed Jeffrey L. Saver, MD, professor of neurology and director of the stroke unit at UCLA. “The MSU offers the tantalizing possibility that you can give tPA with no time hit because you can give it on the way directly to a comprehensive stroke center,” Dr Saver said during a session at the meeting.
Early Data on Effectiveness
Dr Nour reported some of the best evidence for the incremental clinical benefit of MSUs based on the reduced time for starting a tPA infusion. She used data the Berlin group published in September 2016 that compared the treatment courses and outcomes of patients managed with an MSU to similar patients managed by conventional ambulance transport for whom CT scan assessment and the start of tPA treatment did not begin until the patient reached a hospital. The German analysis showed that, in the observational Pre-hospital Acute Neurological Therapy and Optimization of Medical Care in Stroke Patients–Study (PHANTOM-S), among 353 patients treated by conventional transport, the median time from stroke onset to thrombolysis was 112 minutes, compared with a median of 73 minutes among 305 patients managed with an MSU, a statistically significant difference.1 However, the study found no significant difference for its primary endpoint: the percentage of patients with a modified Rankin Scale score of 1 or lower when measured 90 days after their respective strokes. This outcome occurred in 47% of the control patients managed conventionally and in 53% of those managed by an MSU, a difference that fell short of statistical significance
Dr Nour attributed the lack of statistical significance for this primary endpoint to the relatively small number of patients enrolled in PHANTOM-S. “The study was underpowered,” she said.
Dr Nour presented an analysis at the meeting that extrapolated the results out to 1,000 hypothetical patients and tallied the benefits that a larger number of patients could expect to receive if their outcomes paralleled those seen in the published results. It showed that among 1,000 stroke patients treated with an MSU, 58 were expected to be free from disability 90 days later, and an additional 124 patients would have some improvement in their 90-day clinical outcome based on their modified Rankin Scale scores when compared with patients undergoing conventional hospitalization.
“If this finding was confirmed in a larger, controlled study, it would suggest that MSU-based thrombolysis has substantial clinical benefit,” she concluded.
Another recent report looked at the first 100 stroke patients treated by the Cleveland MSU during 2014. Researchers at the Cleveland Clinic and Case Western Reserve University said that 16 of those 100 patients received tPA, and the median time from their emergency call to thrombolytic treatment was 38.5 minutes faster than for 53 stroke patients treated during the same period at EDs operated by the Cleveland Clinic, a statistically significant difference.2 However, this report included no data on clinical outcomes.
Running the Financial Numbers
Nailing down the incremental clinical benefit from MSUs is clearly a very important part of determining the value of this strategy, but another very practical concern is how much the service costs and whether it is financially sustainable.
“We did a cost-effectiveness analysis based on the PHANTOM-S data, and we were conservative by only looking at the benefit from early tPA treatment,” Heinrich J. Audebert, MD, professor of neurology at Charité Hospital in Berlin and head of the team running Berlin’s MSU, said during the MSU session at the meeting. “We did not take into account saving money by avoiding long-term stroke disability and just considered the cost of [immediate] care and the quality-adjusted life years. We calculated a cost of $35,000 per quality-adjusted life year, which is absolutely acceptable.”
He cautioned that this analysis was not based on actual outcomes but on the numbers needed to treat calculated from the PHANTOM-S results. “We need to now show this in controlled trials,” he admitted.
During his talk at the same session, Dr Grotta ran through the numbers for the Houston program. They spent $1.1 million to put their MSU into service in early 2014, and, based on the expenses accrued since then, he estimated an annual staffing cost of about $400,000 and an annual operating cost of about $100,000, for a total estimated 5-year cost of about $3.6 million. Staffing of the Houston MSU started with a registered nurse, CT technician, paramedic, and vascular neurologist, although, like most other US MSUs, the onboard neurologist has since been replaced by a second paramedic, and the neurological diagnostic consult is done via a telemedicine link.
Income from transport reimbursement, currently $500 per trip, and reimbursements of $17,000 above costs for administering tPA and of roughly $40,000 above costs for performing thrombectomy, are balancing these costs. Based on an estimated additional one thrombolysis case per month and one additional thrombectomy case per month, the MSU yields a potential incremental income to the hospital running the MSU of about $3.8 million over 5 years—enough to balance the operating cost, Dr Grotta said.
A key part of controlling costs is having the neurological consult done via a telemedicine link rather than by neurologist at the MSU. “Telemedicine reduces operational costs and improves efficiency,” noted M. Shazam Hussain, MD, interim director of the Cerebrovascular Center at the Cleveland Clinic. “Cost-effectiveness is a very important part of the concept” of MSUs, he said at the session.
The Houston group reported results from a study that directly compared the diagnostic performance of an onboard neurologist with that of a telemedicine neurologist linked-in remotely during MSU deployments for 174 patients. For these cases, the two neurologists each made an independent diagnosis that the researchers then compared. The two diagnoses concurred for 88% of the cases, Tzu-Ching Wu, MD, reported at the meeting. This rate of agreement matched the incidence of concordance between two neurologists who independently assessed the same patients at the hospital,3 said Dr Wu, a vascular neurologist and director of the telemedicine program at the University of Texas Health Science Center in Houston.
“The results support using telemedicine as the primary means of assessment on the MSU,” said Dr Wu. “This may enhance MSU efficiency and reduce costs.” His group’s next study of MSU telemedicine will compare the time needed to make a diagnostic decision using the two approaches, which Dr Wu reported was something not formally examined in the study.
However, telemedicine assessment of CT results gathered in an MSU has one major limitation: the time needed to transmit the huge amount of information from a CTA.
The MSU used by clinicians at the University of Tennessee, Memphis, incorporates an extremely powerful battery that enables “full CT scanner capability with a moving gantry,” said Andrei V. Alexandrov, MD, professor and chairman of neurology at the university. With this set up “we can do in-the-field multiphasic CT angiography from the aortic arch up within 4 minutes. The challenge of doing this is simple. It’s 1.7 gigabytes of data,” which would take a prohibitively long time to transmit from a remote site, he explained. As a result, the complete set of images from the field CTA is delivered on a memory stick to the attending hospital neurologist once the MSU returns.
Waiting for More Data
Despite these advances and the steady recent growth of MSUs, significant skepticism remains. “While mobile stroke units seem like a good idea and there is genuine hope that they will improve outcomes for selected stroke patients, there is not yet any evidence that this is the case,” wrote Bryan Bledsoe, DO, in a January 2017 editorial in the Journal of Emergency Medical Services. “They are expensive and financially nonsustainable. Without widespread deployment, they stand to benefit few, if any, patients. The money spent on these devices would be better spent on improving the current EMS system, including paramedic education, the availability of stroke centers, and on the early recognition of ELVO [emergent large vessel occlusion] strokes,” wrote Dr Bledsoe, professor of emergency medicine at the University of Nevada in Las Vegas.
Two other experts voiced concerns about MSUs in an editorial that accompanied a Cleveland Clinic report in March.4 “Even if MSUs meet an acceptable societal threshold for cost-effectiveness, cost-efficiency may prove a taller order to achieve return on investment for individual health systems and communities,” wrote Andrew M. Southerland, MD, and Ethan S. Brandler, MD. They cited the Cleveland report, which noted that the group’s first 100 MSU-treated patients came from a total of 317 MSU deployments and included 217 trips that were canceled prior to the MSU’s arrival at the patient’s location. In Berlin’s initial experience, more than 2,000 MSU deployments led to 200 tPA treatments and 349 cancellations before arrival, noted Dr Southerland, a neurologist at the University of Virginia in Charlottesville, and Dr Brandler, an emergency medicine physician at Stony Brook (NY) University.
“Hope remains that future trials may demonstrate the ultimate potential of mobile stroke units to improve long-term outcomes for more patients by treating them more quickly and effectively. In the meantime, ongoing efforts are needed to streamline MSU cost and efficiency,” they wrote.
Proponents of MSUs agree that what’s needed now are more data to prove efficacy and cost-effectiveness, as well as better integration into EMS programs. The first opportunity for documenting the clinical impact of MSUs on larger numbers of US patients may be from the BEnefits of Stroke Treatment Delivered using a Mobile Stroke Unit Compared to Standard Management by Emergency Medical Services (BEST-MSU) Study, funded by the Patient-Centered Outcomes Research Institute. This study is collecting data from the MSU programs in Denver, Houston, and Memphis. Although currently designed to enroll 697 patients, Dr Grotta said he hopes to bring the number up to 1,000 patients.
“We are following the health care use and its cost for every enrolled MSU and conventional patient for 1 year,” Dr Grotta explained in an interview. He hopes these results will provide the data needed to move MSUs from investigational status to routine and reimbursable care.
References
1. Kunz A, Ebinger M, Geisler F, et al. Functional outcomes of pre-hospital thrombolysis in a mobile stroke treatment unit compared with conventional care: an observational registry study. Lancet Neurol. 2016;15(10):1035-1043. doi:10.1016/S1474-4422(16)30129-6.
2. Taqui A, Cerejo R, Itrat A, et al; Cleveland Pre-Hospital Acute Stroke Treatment (PHAST) Group. Reduction in time to treatment in prehospital telemedicine evaluation and thrombolysis. Neurology. 2017 March 8. [Epub ahead of print]. doi:10.1212/WNL.0000000000003786.
3. Ramadan AR, Denny MC, Vahidy F, et al. Agreement among stroke faculty and fellows in treating ischemic stroke patients with tissue-type plasminogen activator and thrombectomy. Stroke. 2017;48(1):222-224. doi:10.1161/STROKEAHA.116.015214.
4. Southerland AM, Brandler ES. The cost-efficiency of mobile stroke units: Where the rubber meets the road. Neurology. 2017 Mar 8. [Epub ahead of print]. doi:10.1212/WNL.0000000000003833.
Pulmonary Embolism Common in Patients With Acute Exacerbations of COPD
JIM KLING
FRONTLINE MEDICAL NEWS
About 16% of patients with unexplained acute exacerbations of chronic obstructive pulmonary disease (AECOPD) had an accompanying pulmonary embolism (PE), usually in regions that could be targeted with anticoagulants, according to a new systematic review and meta-analysis.
Approximately 70% of AECOPD cases develop in response to an infection, but about 30% of the time, an AE has no clear cause, the authors said in a report on their research. There is a known biological link between inflammation and coagulation, which suggests that patients experiencing AECOPD may be at increased risk of PE.
The researchers reviewed and analyzed seven studies, comprising 880 patients. Among the authors’ reasons for conducting this research was to update the pooled prevalence of PE in AECOPD from a previous systematic review published in Chest in 2009.
The meta-analysis revealed that 16.1% of patients with AECOPD were also diagnosed with PE (95% confidence interval [CI], 8.3%-25.8%). There was a wide range of variation between individual studies (prevalence 3.3%-29.1%). In six studies that reported on deep vein thrombosis (DVT), the pooled prevalence of DVT was 10.5% (95% CI, 4.3%-19.0%).
Five of the studies identified the PE location. An analysis of those studies showed that 35% were in the main pulmonary artery, and 31.7% were in the lobar and interlobar arteries. Such findings “[suggest] that the majority of these embolisms have important clinical consequences,” the authors wrote.
The researchers also looked at clinical markers that accompanied AECOPD and found a potential signal with respect to pleuritic chest pain. One study found a strong association between pleuritic chest pain and AECOPD patients with PE (81% vs 40% in those without PE). A second study showed a similar association (24% in PE vs 11.5% in non-PE patients), and a third study found no significant difference.
The presence of PE was also linked to hypotension, syncope, and acute right failure on ultrasonography, suggesting that PE may be associated with heart failure.
Patients with PE were less likely to have symptoms consistent with a respiratory tract infection. They also tended to have higher mortality rates and longer hospitalization rates compared with those without PE.
The meta-analysis had some limitations, including the heterogeneity of findings in the included studies, as well as the potential for publication bias, since reports showing unusually low or high rates may be more likely to be published, the researchers noted. There was also a high proportion of male subjects in the included studies.
Overall, the researchers concluded that PE is more likely in patients with pleuritic chest pain and signs of heart failure, and less likely in patients with signs of a respiratory infection. That information “might add to the clinical decision-making in patients with an AECOPD, because it would be undesirable to perform [CT pulmonary angiography] in every patient with an AECOPD,” the researchers wrote.
Aleva FE, Voets LW, Simons SO, de Mast Q, van der Ven AJ, Heijdra YF. Prevalence and localization of pulmonary embolism in unexplained acute exacerbations of COPD: A systematic review and meta-analysis. Chest. 2017;151(3):544-554. doi:10.1016/j.chest.2016.07.034.
Norepinephrine Shortage Linked to Mortality in Patients With Septic Shock
AMY KARON
FRONTLINE MEDICAL NEWS
A national shortage of norepinephrine in the United States was associated with higher rates of mortality among patients hospitalized with septic shock, investigators reported.
Rates of in-hospital mortality in 2011 were 40% during quarters when hospitals were facing shortages and 36% when they were not, Emily Vail, MD, and her associates said at the International Symposium on Intensive Care and Emergency Medicine. The report was published simultaneously in JAMA.
The link between norepinephrine shortage and death from septic shock persisted even after the researchers accounted for numerous clinical and demographic factors (adjusted odds ratio, 1.2; 95% CI, 1.01 to 1.30; P = .03), wrote Dr Vail of Columbia University, New York.
Drug shortages are common in the United States, but few studies have explored their effects on patient outcomes. Investigators compared mortality rates among affected patients during 3-month intervals when hospitals were and were not using at least 20% less norepinephrine than baseline. The researchers used Premier Healthcare Database, which includes both standard claims and detailed, dated logs of all services billed to patients or insurance, with minimal missing data.
A total of 77% patients admitted with septic shock received norepinephrine before the shortage. During the lowest point of the shortage, 56% of patients received it, the researchers reported. Clinicians most often used phenylephrine instead, prescribing it to up to 54% of patients during the worst time of the shortage. The absolute increase in mortality during the quarters of shortage was 3.7% (95% CI, 1.5%-6.0%).
Several factors might explain the link between norepinephrine shortage and mortality, the investigators said. The vasopressors chosen to replace norepinephrine might result directly in worse outcomes, but a decrease in norepinephrine use also might be a proxy for relevant variables such as delayed use of vasopressors, lack of knowledge of how to optimally dose vasopressors besides norepinephrine, or the absence of a pharmacist dedicated to helping optimize the use of limited supplies.
The study did not uncover a dose-response association between greater decreases in norepinephrine use and increased mortality, the researchers noted. “This may be due to a threshold effect of vasopressor shortage on mortality, or lack of power due to relatively few hospital quarters at the extreme levels of vasopressor shortage,” they wrote.
Because the deaths captured included only those that occurred in-hospital, “the results may have underestimated mortality, particularly for hospitals that tend to transfer patients early to other skilled care facilities,” the researchers noted.
The cohort of patients was limited to those who received vasopressors for 2 or more days and excluded patients who died on the first day of vasopressor treatment, the researchers said.
Vail E, Gershengorn HB, Hua M, Walkey AJ, Rubenfeld G, Wunsch H. Association between US norepinephrine shortage and mortality among patients with septic shock. JAMA. 21 March 2017. [Epub ahead of print]. doi:10.1001/jama.2017.2841.
MITCHEL L. ZOLER
FRONTLINE MEDICAL NEWS
Mobile stroke units—specially equipped ambulances that bring a diagnostic computed tomography (CT) scanner and therapeutic thrombolysis directly to patients in the field—have begun to proliferate across the United States, although they remain investigational, with no clear proof of their incremental clinical value or cost-effectiveness.
The first US mobile stroke unit (MSU) launched in Houston, Texas in early 2014 (following the world’s first in Berlin, Germany, which began running in early 2011), and by early 2017, at least eight other US MSUs were in operation, most of them put into service during the prior 15 months. United States MSU locations now include Cleveland, Ohio; Denver, Colorado; Memphis, Tennessee; New York, New York; Toledo, Ohio; Trenton, New Jersey; and Northwestern Medicine and Rush University Medical Center in the western Chicago, Illinois region. A tenth MSU is slated to start operation at the University of California, Los Angeles later this year.
Early data collected at some of these sites show that initiating care of an acute ischemic stroke patient in an MSU shaves precious minutes off the time it takes to initiate thrombolytic therapy with tissue plasminogen activator (tPA), and findings from preliminary analyses suggest better functional outcomes for patients treated this way. However, leaders in the nascent field readily admit that the data needed to clearly prove the benefit patients receive from operating MSUs are still a few years off. This uncertainty about the added benefit to patients from MSUs couples with one clear fact: MSUs are expensive to start up, with a price tag of roughly $1 million to get an MSU on the road for the first time; they are also expensive to operate, with one estimate for the annual cost of keeping an MSU on the street at about $500,000 per year for staffing, supplies, and other expenses.
“Every US MSU I know of started with philanthropic gifts, but you need a business model” to keep the program running long-term, James C. Grotta, MD, said during a session focused on MSUs at the International Stroke Conference sponsored by the American Heart Association. “You can’t sustain an MSU with philanthropy,” said Dr Grotta, professor of neurology at the University of Texas Health Science Center in Houston, director and founder of the Houston MSU, and acknowledged “godfather” of all US MSUs.
“We believe that MSUs are very worthwhile and that the clinical and economic benefits of earlier stroke treatment [made possible with MSUs] could offset the costs, but we need to show this,” admitted May Nour, MD, a vascular and interventional neurologist at the University of California, Los Angeles (UCLA), and director of the soon-to-launch Los Angeles MSU.
The concept behind MSUs is simple: Each one carries a CT scanner on board so that once the vehicle’s staff identifies a patient with clinical signs of a significant-acute ischemic stroke in the field and confirms that the timing of the stroke onset suggests eligibility for tPA treatment, a CT scan can immediately be run on-site to finalize tPA eligibility. The MSU staff can then begin infusing the drug in the ambulance as it speeds the patient to an appropriate hospital.
In addition, many MSUs now carry a scanner that can perform a CT angiogram (CTA) to locate the occluding clot. If a large vessel occlusion is found, the crew can bring the patient directly to a comprehensive stroke center for a thrombectomy. If thrombectomy is not appropriate, the MSU crew may take the patient to a primary stroke center where thrombectomy is not available.
Another advantage to MSUs, in addition to quicker initiation of thrombolysis, is “getting patients to where they need to go faster and more directly,” said Dr Nour.
“Instead of bringing patients first to a hospital that’s unable to do thrombectomy and where treatment gets slowed down, with an MSU you can give tPA on the street and go straight to a thrombectomy center,” agreed Jeffrey L. Saver, MD, professor of neurology and director of the stroke unit at UCLA. “The MSU offers the tantalizing possibility that you can give tPA with no time hit because you can give it on the way directly to a comprehensive stroke center,” Dr Saver said during a session at the meeting.
Early Data on Effectiveness
Dr Nour reported some of the best evidence for the incremental clinical benefit of MSUs based on the reduced time for starting a tPA infusion. She used data the Berlin group published in September 2016 that compared the treatment courses and outcomes of patients managed with an MSU to similar patients managed by conventional ambulance transport for whom CT scan assessment and the start of tPA treatment did not begin until the patient reached a hospital. The German analysis showed that, in the observational Pre-hospital Acute Neurological Therapy and Optimization of Medical Care in Stroke Patients–Study (PHANTOM-S), among 353 patients treated by conventional transport, the median time from stroke onset to thrombolysis was 112 minutes, compared with a median of 73 minutes among 305 patients managed with an MSU, a statistically significant difference.1 However, the study found no significant difference for its primary endpoint: the percentage of patients with a modified Rankin Scale score of 1 or lower when measured 90 days after their respective strokes. This outcome occurred in 47% of the control patients managed conventionally and in 53% of those managed by an MSU, a difference that fell short of statistical significance
Dr Nour attributed the lack of statistical significance for this primary endpoint to the relatively small number of patients enrolled in PHANTOM-S. “The study was underpowered,” she said.
Dr Nour presented an analysis at the meeting that extrapolated the results out to 1,000 hypothetical patients and tallied the benefits that a larger number of patients could expect to receive if their outcomes paralleled those seen in the published results. It showed that among 1,000 stroke patients treated with an MSU, 58 were expected to be free from disability 90 days later, and an additional 124 patients would have some improvement in their 90-day clinical outcome based on their modified Rankin Scale scores when compared with patients undergoing conventional hospitalization.
“If this finding was confirmed in a larger, controlled study, it would suggest that MSU-based thrombolysis has substantial clinical benefit,” she concluded.
Another recent report looked at the first 100 stroke patients treated by the Cleveland MSU during 2014. Researchers at the Cleveland Clinic and Case Western Reserve University said that 16 of those 100 patients received tPA, and the median time from their emergency call to thrombolytic treatment was 38.5 minutes faster than for 53 stroke patients treated during the same period at EDs operated by the Cleveland Clinic, a statistically significant difference.2 However, this report included no data on clinical outcomes.
Running the Financial Numbers
Nailing down the incremental clinical benefit from MSUs is clearly a very important part of determining the value of this strategy, but another very practical concern is how much the service costs and whether it is financially sustainable.
“We did a cost-effectiveness analysis based on the PHANTOM-S data, and we were conservative by only looking at the benefit from early tPA treatment,” Heinrich J. Audebert, MD, professor of neurology at Charité Hospital in Berlin and head of the team running Berlin’s MSU, said during the MSU session at the meeting. “We did not take into account saving money by avoiding long-term stroke disability and just considered the cost of [immediate] care and the quality-adjusted life years. We calculated a cost of $35,000 per quality-adjusted life year, which is absolutely acceptable.”
He cautioned that this analysis was not based on actual outcomes but on the numbers needed to treat calculated from the PHANTOM-S results. “We need to now show this in controlled trials,” he admitted.
During his talk at the same session, Dr Grotta ran through the numbers for the Houston program. They spent $1.1 million to put their MSU into service in early 2014, and, based on the expenses accrued since then, he estimated an annual staffing cost of about $400,000 and an annual operating cost of about $100,000, for a total estimated 5-year cost of about $3.6 million. Staffing of the Houston MSU started with a registered nurse, CT technician, paramedic, and vascular neurologist, although, like most other US MSUs, the onboard neurologist has since been replaced by a second paramedic, and the neurological diagnostic consult is done via a telemedicine link.
Income from transport reimbursement, currently $500 per trip, and reimbursements of $17,000 above costs for administering tPA and of roughly $40,000 above costs for performing thrombectomy, are balancing these costs. Based on an estimated additional one thrombolysis case per month and one additional thrombectomy case per month, the MSU yields a potential incremental income to the hospital running the MSU of about $3.8 million over 5 years—enough to balance the operating cost, Dr Grotta said.
A key part of controlling costs is having the neurological consult done via a telemedicine link rather than by neurologist at the MSU. “Telemedicine reduces operational costs and improves efficiency,” noted M. Shazam Hussain, MD, interim director of the Cerebrovascular Center at the Cleveland Clinic. “Cost-effectiveness is a very important part of the concept” of MSUs, he said at the session.
The Houston group reported results from a study that directly compared the diagnostic performance of an onboard neurologist with that of a telemedicine neurologist linked-in remotely during MSU deployments for 174 patients. For these cases, the two neurologists each made an independent diagnosis that the researchers then compared. The two diagnoses concurred for 88% of the cases, Tzu-Ching Wu, MD, reported at the meeting. This rate of agreement matched the incidence of concordance between two neurologists who independently assessed the same patients at the hospital,3 said Dr Wu, a vascular neurologist and director of the telemedicine program at the University of Texas Health Science Center in Houston.
“The results support using telemedicine as the primary means of assessment on the MSU,” said Dr Wu. “This may enhance MSU efficiency and reduce costs.” His group’s next study of MSU telemedicine will compare the time needed to make a diagnostic decision using the two approaches, which Dr Wu reported was something not formally examined in the study.
However, telemedicine assessment of CT results gathered in an MSU has one major limitation: the time needed to transmit the huge amount of information from a CTA.
The MSU used by clinicians at the University of Tennessee, Memphis, incorporates an extremely powerful battery that enables “full CT scanner capability with a moving gantry,” said Andrei V. Alexandrov, MD, professor and chairman of neurology at the university. With this set up “we can do in-the-field multiphasic CT angiography from the aortic arch up within 4 minutes. The challenge of doing this is simple. It’s 1.7 gigabytes of data,” which would take a prohibitively long time to transmit from a remote site, he explained. As a result, the complete set of images from the field CTA is delivered on a memory stick to the attending hospital neurologist once the MSU returns.
Waiting for More Data
Despite these advances and the steady recent growth of MSUs, significant skepticism remains. “While mobile stroke units seem like a good idea and there is genuine hope that they will improve outcomes for selected stroke patients, there is not yet any evidence that this is the case,” wrote Bryan Bledsoe, DO, in a January 2017 editorial in the Journal of Emergency Medical Services. “They are expensive and financially nonsustainable. Without widespread deployment, they stand to benefit few, if any, patients. The money spent on these devices would be better spent on improving the current EMS system, including paramedic education, the availability of stroke centers, and on the early recognition of ELVO [emergent large vessel occlusion] strokes,” wrote Dr Bledsoe, professor of emergency medicine at the University of Nevada in Las Vegas.
Two other experts voiced concerns about MSUs in an editorial that accompanied a Cleveland Clinic report in March.4 “Even if MSUs meet an acceptable societal threshold for cost-effectiveness, cost-efficiency may prove a taller order to achieve return on investment for individual health systems and communities,” wrote Andrew M. Southerland, MD, and Ethan S. Brandler, MD. They cited the Cleveland report, which noted that the group’s first 100 MSU-treated patients came from a total of 317 MSU deployments and included 217 trips that were canceled prior to the MSU’s arrival at the patient’s location. In Berlin’s initial experience, more than 2,000 MSU deployments led to 200 tPA treatments and 349 cancellations before arrival, noted Dr Southerland, a neurologist at the University of Virginia in Charlottesville, and Dr Brandler, an emergency medicine physician at Stony Brook (NY) University.
“Hope remains that future trials may demonstrate the ultimate potential of mobile stroke units to improve long-term outcomes for more patients by treating them more quickly and effectively. In the meantime, ongoing efforts are needed to streamline MSU cost and efficiency,” they wrote.
Proponents of MSUs agree that what’s needed now are more data to prove efficacy and cost-effectiveness, as well as better integration into EMS programs. The first opportunity for documenting the clinical impact of MSUs on larger numbers of US patients may be from the BEnefits of Stroke Treatment Delivered using a Mobile Stroke Unit Compared to Standard Management by Emergency Medical Services (BEST-MSU) Study, funded by the Patient-Centered Outcomes Research Institute. This study is collecting data from the MSU programs in Denver, Houston, and Memphis. Although currently designed to enroll 697 patients, Dr Grotta said he hopes to bring the number up to 1,000 patients.
“We are following the health care use and its cost for every enrolled MSU and conventional patient for 1 year,” Dr Grotta explained in an interview. He hopes these results will provide the data needed to move MSUs from investigational status to routine and reimbursable care.
References
1. Kunz A, Ebinger M, Geisler F, et al. Functional outcomes of pre-hospital thrombolysis in a mobile stroke treatment unit compared with conventional care: an observational registry study. Lancet Neurol. 2016;15(10):1035-1043. doi:10.1016/S1474-4422(16)30129-6.
2. Taqui A, Cerejo R, Itrat A, et al; Cleveland Pre-Hospital Acute Stroke Treatment (PHAST) Group. Reduction in time to treatment in prehospital telemedicine evaluation and thrombolysis. Neurology. 2017 March 8. [Epub ahead of print]. doi:10.1212/WNL.0000000000003786.
3. Ramadan AR, Denny MC, Vahidy F, et al. Agreement among stroke faculty and fellows in treating ischemic stroke patients with tissue-type plasminogen activator and thrombectomy. Stroke. 2017;48(1):222-224. doi:10.1161/STROKEAHA.116.015214.
4. Southerland AM, Brandler ES. The cost-efficiency of mobile stroke units: Where the rubber meets the road. Neurology. 2017 Mar 8. [Epub ahead of print]. doi:10.1212/WNL.0000000000003833.
Pulmonary Embolism Common in Patients With Acute Exacerbations of COPD
JIM KLING
FRONTLINE MEDICAL NEWS
About 16% of patients with unexplained acute exacerbations of chronic obstructive pulmonary disease (AECOPD) had an accompanying pulmonary embolism (PE), usually in regions that could be targeted with anticoagulants, according to a new systematic review and meta-analysis.
Approximately 70% of AECOPD cases develop in response to an infection, but about 30% of the time, an AE has no clear cause, the authors said in a report on their research. There is a known biological link between inflammation and coagulation, which suggests that patients experiencing AECOPD may be at increased risk of PE.
The researchers reviewed and analyzed seven studies, comprising 880 patients. Among the authors’ reasons for conducting this research was to update the pooled prevalence of PE in AECOPD from a previous systematic review published in Chest in 2009.
The meta-analysis revealed that 16.1% of patients with AECOPD were also diagnosed with PE (95% confidence interval [CI], 8.3%-25.8%). There was a wide range of variation between individual studies (prevalence 3.3%-29.1%). In six studies that reported on deep vein thrombosis (DVT), the pooled prevalence of DVT was 10.5% (95% CI, 4.3%-19.0%).
Five of the studies identified the PE location. An analysis of those studies showed that 35% were in the main pulmonary artery, and 31.7% were in the lobar and interlobar arteries. Such findings “[suggest] that the majority of these embolisms have important clinical consequences,” the authors wrote.
The researchers also looked at clinical markers that accompanied AECOPD and found a potential signal with respect to pleuritic chest pain. One study found a strong association between pleuritic chest pain and AECOPD patients with PE (81% vs 40% in those without PE). A second study showed a similar association (24% in PE vs 11.5% in non-PE patients), and a third study found no significant difference.
The presence of PE was also linked to hypotension, syncope, and acute right failure on ultrasonography, suggesting that PE may be associated with heart failure.
Patients with PE were less likely to have symptoms consistent with a respiratory tract infection. They also tended to have higher mortality rates and longer hospitalization rates compared with those without PE.
The meta-analysis had some limitations, including the heterogeneity of findings in the included studies, as well as the potential for publication bias, since reports showing unusually low or high rates may be more likely to be published, the researchers noted. There was also a high proportion of male subjects in the included studies.
Overall, the researchers concluded that PE is more likely in patients with pleuritic chest pain and signs of heart failure, and less likely in patients with signs of a respiratory infection. That information “might add to the clinical decision-making in patients with an AECOPD, because it would be undesirable to perform [CT pulmonary angiography] in every patient with an AECOPD,” the researchers wrote.
Aleva FE, Voets LW, Simons SO, de Mast Q, van der Ven AJ, Heijdra YF. Prevalence and localization of pulmonary embolism in unexplained acute exacerbations of COPD: A systematic review and meta-analysis. Chest. 2017;151(3):544-554. doi:10.1016/j.chest.2016.07.034.
Norepinephrine Shortage Linked to Mortality in Patients With Septic Shock
AMY KARON
FRONTLINE MEDICAL NEWS
A national shortage of norepinephrine in the United States was associated with higher rates of mortality among patients hospitalized with septic shock, investigators reported.
Rates of in-hospital mortality in 2011 were 40% during quarters when hospitals were facing shortages and 36% when they were not, Emily Vail, MD, and her associates said at the International Symposium on Intensive Care and Emergency Medicine. The report was published simultaneously in JAMA.
The link between norepinephrine shortage and death from septic shock persisted even after the researchers accounted for numerous clinical and demographic factors (adjusted odds ratio, 1.2; 95% CI, 1.01 to 1.30; P = .03), wrote Dr Vail of Columbia University, New York.
Drug shortages are common in the United States, but few studies have explored their effects on patient outcomes. Investigators compared mortality rates among affected patients during 3-month intervals when hospitals were and were not using at least 20% less norepinephrine than baseline. The researchers used Premier Healthcare Database, which includes both standard claims and detailed, dated logs of all services billed to patients or insurance, with minimal missing data.
A total of 77% patients admitted with septic shock received norepinephrine before the shortage. During the lowest point of the shortage, 56% of patients received it, the researchers reported. Clinicians most often used phenylephrine instead, prescribing it to up to 54% of patients during the worst time of the shortage. The absolute increase in mortality during the quarters of shortage was 3.7% (95% CI, 1.5%-6.0%).
Several factors might explain the link between norepinephrine shortage and mortality, the investigators said. The vasopressors chosen to replace norepinephrine might result directly in worse outcomes, but a decrease in norepinephrine use also might be a proxy for relevant variables such as delayed use of vasopressors, lack of knowledge of how to optimally dose vasopressors besides norepinephrine, or the absence of a pharmacist dedicated to helping optimize the use of limited supplies.
The study did not uncover a dose-response association between greater decreases in norepinephrine use and increased mortality, the researchers noted. “This may be due to a threshold effect of vasopressor shortage on mortality, or lack of power due to relatively few hospital quarters at the extreme levels of vasopressor shortage,” they wrote.
Because the deaths captured included only those that occurred in-hospital, “the results may have underestimated mortality, particularly for hospitals that tend to transfer patients early to other skilled care facilities,” the researchers noted.
The cohort of patients was limited to those who received vasopressors for 2 or more days and excluded patients who died on the first day of vasopressor treatment, the researchers said.
Vail E, Gershengorn HB, Hua M, Walkey AJ, Rubenfeld G, Wunsch H. Association between US norepinephrine shortage and mortality among patients with septic shock. JAMA. 21 March 2017. [Epub ahead of print]. doi:10.1001/jama.2017.2841.
MITCHEL L. ZOLER
FRONTLINE MEDICAL NEWS
Mobile stroke units—specially equipped ambulances that bring a diagnostic computed tomography (CT) scanner and therapeutic thrombolysis directly to patients in the field—have begun to proliferate across the United States, although they remain investigational, with no clear proof of their incremental clinical value or cost-effectiveness.
The first US mobile stroke unit (MSU) launched in Houston, Texas in early 2014 (following the world’s first in Berlin, Germany, which began running in early 2011), and by early 2017, at least eight other US MSUs were in operation, most of them put into service during the prior 15 months. United States MSU locations now include Cleveland, Ohio; Denver, Colorado; Memphis, Tennessee; New York, New York; Toledo, Ohio; Trenton, New Jersey; and Northwestern Medicine and Rush University Medical Center in the western Chicago, Illinois region. A tenth MSU is slated to start operation at the University of California, Los Angeles later this year.
Early data collected at some of these sites show that initiating care of an acute ischemic stroke patient in an MSU shaves precious minutes off the time it takes to initiate thrombolytic therapy with tissue plasminogen activator (tPA), and findings from preliminary analyses suggest better functional outcomes for patients treated this way. However, leaders in the nascent field readily admit that the data needed to clearly prove the benefit patients receive from operating MSUs are still a few years off. This uncertainty about the added benefit to patients from MSUs couples with one clear fact: MSUs are expensive to start up, with a price tag of roughly $1 million to get an MSU on the road for the first time; they are also expensive to operate, with one estimate for the annual cost of keeping an MSU on the street at about $500,000 per year for staffing, supplies, and other expenses.
“Every US MSU I know of started with philanthropic gifts, but you need a business model” to keep the program running long-term, James C. Grotta, MD, said during a session focused on MSUs at the International Stroke Conference sponsored by the American Heart Association. “You can’t sustain an MSU with philanthropy,” said Dr Grotta, professor of neurology at the University of Texas Health Science Center in Houston, director and founder of the Houston MSU, and acknowledged “godfather” of all US MSUs.
“We believe that MSUs are very worthwhile and that the clinical and economic benefits of earlier stroke treatment [made possible with MSUs] could offset the costs, but we need to show this,” admitted May Nour, MD, a vascular and interventional neurologist at the University of California, Los Angeles (UCLA), and director of the soon-to-launch Los Angeles MSU.
The concept behind MSUs is simple: Each one carries a CT scanner on board so that once the vehicle’s staff identifies a patient with clinical signs of a significant-acute ischemic stroke in the field and confirms that the timing of the stroke onset suggests eligibility for tPA treatment, a CT scan can immediately be run on-site to finalize tPA eligibility. The MSU staff can then begin infusing the drug in the ambulance as it speeds the patient to an appropriate hospital.
In addition, many MSUs now carry a scanner that can perform a CT angiogram (CTA) to locate the occluding clot. If a large vessel occlusion is found, the crew can bring the patient directly to a comprehensive stroke center for a thrombectomy. If thrombectomy is not appropriate, the MSU crew may take the patient to a primary stroke center where thrombectomy is not available.
Another advantage to MSUs, in addition to quicker initiation of thrombolysis, is “getting patients to where they need to go faster and more directly,” said Dr Nour.
“Instead of bringing patients first to a hospital that’s unable to do thrombectomy and where treatment gets slowed down, with an MSU you can give tPA on the street and go straight to a thrombectomy center,” agreed Jeffrey L. Saver, MD, professor of neurology and director of the stroke unit at UCLA. “The MSU offers the tantalizing possibility that you can give tPA with no time hit because you can give it on the way directly to a comprehensive stroke center,” Dr Saver said during a session at the meeting.
Early Data on Effectiveness
Dr Nour reported some of the best evidence for the incremental clinical benefit of MSUs based on the reduced time for starting a tPA infusion. She used data the Berlin group published in September 2016 that compared the treatment courses and outcomes of patients managed with an MSU to similar patients managed by conventional ambulance transport for whom CT scan assessment and the start of tPA treatment did not begin until the patient reached a hospital. The German analysis showed that, in the observational Pre-hospital Acute Neurological Therapy and Optimization of Medical Care in Stroke Patients–Study (PHANTOM-S), among 353 patients treated by conventional transport, the median time from stroke onset to thrombolysis was 112 minutes, compared with a median of 73 minutes among 305 patients managed with an MSU, a statistically significant difference.1 However, the study found no significant difference for its primary endpoint: the percentage of patients with a modified Rankin Scale score of 1 or lower when measured 90 days after their respective strokes. This outcome occurred in 47% of the control patients managed conventionally and in 53% of those managed by an MSU, a difference that fell short of statistical significance
Dr Nour attributed the lack of statistical significance for this primary endpoint to the relatively small number of patients enrolled in PHANTOM-S. “The study was underpowered,” she said.
Dr Nour presented an analysis at the meeting that extrapolated the results out to 1,000 hypothetical patients and tallied the benefits that a larger number of patients could expect to receive if their outcomes paralleled those seen in the published results. It showed that among 1,000 stroke patients treated with an MSU, 58 were expected to be free from disability 90 days later, and an additional 124 patients would have some improvement in their 90-day clinical outcome based on their modified Rankin Scale scores when compared with patients undergoing conventional hospitalization.
“If this finding was confirmed in a larger, controlled study, it would suggest that MSU-based thrombolysis has substantial clinical benefit,” she concluded.
Another recent report looked at the first 100 stroke patients treated by the Cleveland MSU during 2014. Researchers at the Cleveland Clinic and Case Western Reserve University said that 16 of those 100 patients received tPA, and the median time from their emergency call to thrombolytic treatment was 38.5 minutes faster than for 53 stroke patients treated during the same period at EDs operated by the Cleveland Clinic, a statistically significant difference.2 However, this report included no data on clinical outcomes.
Running the Financial Numbers
Nailing down the incremental clinical benefit from MSUs is clearly a very important part of determining the value of this strategy, but another very practical concern is how much the service costs and whether it is financially sustainable.
“We did a cost-effectiveness analysis based on the PHANTOM-S data, and we were conservative by only looking at the benefit from early tPA treatment,” Heinrich J. Audebert, MD, professor of neurology at Charité Hospital in Berlin and head of the team running Berlin’s MSU, said during the MSU session at the meeting. “We did not take into account saving money by avoiding long-term stroke disability and just considered the cost of [immediate] care and the quality-adjusted life years. We calculated a cost of $35,000 per quality-adjusted life year, which is absolutely acceptable.”
He cautioned that this analysis was not based on actual outcomes but on the numbers needed to treat calculated from the PHANTOM-S results. “We need to now show this in controlled trials,” he admitted.
During his talk at the same session, Dr Grotta ran through the numbers for the Houston program. They spent $1.1 million to put their MSU into service in early 2014, and, based on the expenses accrued since then, he estimated an annual staffing cost of about $400,000 and an annual operating cost of about $100,000, for a total estimated 5-year cost of about $3.6 million. Staffing of the Houston MSU started with a registered nurse, CT technician, paramedic, and vascular neurologist, although, like most other US MSUs, the onboard neurologist has since been replaced by a second paramedic, and the neurological diagnostic consult is done via a telemedicine link.
Income from transport reimbursement, currently $500 per trip, and reimbursements of $17,000 above costs for administering tPA and of roughly $40,000 above costs for performing thrombectomy, are balancing these costs. Based on an estimated additional one thrombolysis case per month and one additional thrombectomy case per month, the MSU yields a potential incremental income to the hospital running the MSU of about $3.8 million over 5 years—enough to balance the operating cost, Dr Grotta said.
A key part of controlling costs is having the neurological consult done via a telemedicine link rather than by neurologist at the MSU. “Telemedicine reduces operational costs and improves efficiency,” noted M. Shazam Hussain, MD, interim director of the Cerebrovascular Center at the Cleveland Clinic. “Cost-effectiveness is a very important part of the concept” of MSUs, he said at the session.
The Houston group reported results from a study that directly compared the diagnostic performance of an onboard neurologist with that of a telemedicine neurologist linked-in remotely during MSU deployments for 174 patients. For these cases, the two neurologists each made an independent diagnosis that the researchers then compared. The two diagnoses concurred for 88% of the cases, Tzu-Ching Wu, MD, reported at the meeting. This rate of agreement matched the incidence of concordance between two neurologists who independently assessed the same patients at the hospital,3 said Dr Wu, a vascular neurologist and director of the telemedicine program at the University of Texas Health Science Center in Houston.
“The results support using telemedicine as the primary means of assessment on the MSU,” said Dr Wu. “This may enhance MSU efficiency and reduce costs.” His group’s next study of MSU telemedicine will compare the time needed to make a diagnostic decision using the two approaches, which Dr Wu reported was something not formally examined in the study.
However, telemedicine assessment of CT results gathered in an MSU has one major limitation: the time needed to transmit the huge amount of information from a CTA.
The MSU used by clinicians at the University of Tennessee, Memphis, incorporates an extremely powerful battery that enables “full CT scanner capability with a moving gantry,” said Andrei V. Alexandrov, MD, professor and chairman of neurology at the university. With this set up “we can do in-the-field multiphasic CT angiography from the aortic arch up within 4 minutes. The challenge of doing this is simple. It’s 1.7 gigabytes of data,” which would take a prohibitively long time to transmit from a remote site, he explained. As a result, the complete set of images from the field CTA is delivered on a memory stick to the attending hospital neurologist once the MSU returns.
Waiting for More Data
Despite these advances and the steady recent growth of MSUs, significant skepticism remains. “While mobile stroke units seem like a good idea and there is genuine hope that they will improve outcomes for selected stroke patients, there is not yet any evidence that this is the case,” wrote Bryan Bledsoe, DO, in a January 2017 editorial in the Journal of Emergency Medical Services. “They are expensive and financially nonsustainable. Without widespread deployment, they stand to benefit few, if any, patients. The money spent on these devices would be better spent on improving the current EMS system, including paramedic education, the availability of stroke centers, and on the early recognition of ELVO [emergent large vessel occlusion] strokes,” wrote Dr Bledsoe, professor of emergency medicine at the University of Nevada in Las Vegas.
Two other experts voiced concerns about MSUs in an editorial that accompanied a Cleveland Clinic report in March.4 “Even if MSUs meet an acceptable societal threshold for cost-effectiveness, cost-efficiency may prove a taller order to achieve return on investment for individual health systems and communities,” wrote Andrew M. Southerland, MD, and Ethan S. Brandler, MD. They cited the Cleveland report, which noted that the group’s first 100 MSU-treated patients came from a total of 317 MSU deployments and included 217 trips that were canceled prior to the MSU’s arrival at the patient’s location. In Berlin’s initial experience, more than 2,000 MSU deployments led to 200 tPA treatments and 349 cancellations before arrival, noted Dr Southerland, a neurologist at the University of Virginia in Charlottesville, and Dr Brandler, an emergency medicine physician at Stony Brook (NY) University.
“Hope remains that future trials may demonstrate the ultimate potential of mobile stroke units to improve long-term outcomes for more patients by treating them more quickly and effectively. In the meantime, ongoing efforts are needed to streamline MSU cost and efficiency,” they wrote.
Proponents of MSUs agree that what’s needed now are more data to prove efficacy and cost-effectiveness, as well as better integration into EMS programs. The first opportunity for documenting the clinical impact of MSUs on larger numbers of US patients may be from the BEnefits of Stroke Treatment Delivered using a Mobile Stroke Unit Compared to Standard Management by Emergency Medical Services (BEST-MSU) Study, funded by the Patient-Centered Outcomes Research Institute. This study is collecting data from the MSU programs in Denver, Houston, and Memphis. Although currently designed to enroll 697 patients, Dr Grotta said he hopes to bring the number up to 1,000 patients.
“We are following the health care use and its cost for every enrolled MSU and conventional patient for 1 year,” Dr Grotta explained in an interview. He hopes these results will provide the data needed to move MSUs from investigational status to routine and reimbursable care.
References
1. Kunz A, Ebinger M, Geisler F, et al. Functional outcomes of pre-hospital thrombolysis in a mobile stroke treatment unit compared with conventional care: an observational registry study. Lancet Neurol. 2016;15(10):1035-1043. doi:10.1016/S1474-4422(16)30129-6.
2. Taqui A, Cerejo R, Itrat A, et al; Cleveland Pre-Hospital Acute Stroke Treatment (PHAST) Group. Reduction in time to treatment in prehospital telemedicine evaluation and thrombolysis. Neurology. 2017 March 8. [Epub ahead of print]. doi:10.1212/WNL.0000000000003786.
3. Ramadan AR, Denny MC, Vahidy F, et al. Agreement among stroke faculty and fellows in treating ischemic stroke patients with tissue-type plasminogen activator and thrombectomy. Stroke. 2017;48(1):222-224. doi:10.1161/STROKEAHA.116.015214.
4. Southerland AM, Brandler ES. The cost-efficiency of mobile stroke units: Where the rubber meets the road. Neurology. 2017 Mar 8. [Epub ahead of print]. doi:10.1212/WNL.0000000000003833.
Pulmonary Embolism Common in Patients With Acute Exacerbations of COPD
JIM KLING
FRONTLINE MEDICAL NEWS
About 16% of patients with unexplained acute exacerbations of chronic obstructive pulmonary disease (AECOPD) had an accompanying pulmonary embolism (PE), usually in regions that could be targeted with anticoagulants, according to a new systematic review and meta-analysis.
Approximately 70% of AECOPD cases develop in response to an infection, but about 30% of the time, an AE has no clear cause, the authors said in a report on their research. There is a known biological link between inflammation and coagulation, which suggests that patients experiencing AECOPD may be at increased risk of PE.
The researchers reviewed and analyzed seven studies, comprising 880 patients. Among the authors’ reasons for conducting this research was to update the pooled prevalence of PE in AECOPD from a previous systematic review published in Chest in 2009.
The meta-analysis revealed that 16.1% of patients with AECOPD were also diagnosed with PE (95% confidence interval [CI], 8.3%-25.8%). There was a wide range of variation between individual studies (prevalence 3.3%-29.1%). In six studies that reported on deep vein thrombosis (DVT), the pooled prevalence of DVT was 10.5% (95% CI, 4.3%-19.0%).
Five of the studies identified the PE location. An analysis of those studies showed that 35% were in the main pulmonary artery, and 31.7% were in the lobar and interlobar arteries. Such findings “[suggest] that the majority of these embolisms have important clinical consequences,” the authors wrote.
The researchers also looked at clinical markers that accompanied AECOPD and found a potential signal with respect to pleuritic chest pain. One study found a strong association between pleuritic chest pain and AECOPD patients with PE (81% vs 40% in those without PE). A second study showed a similar association (24% in PE vs 11.5% in non-PE patients), and a third study found no significant difference.
The presence of PE was also linked to hypotension, syncope, and acute right failure on ultrasonography, suggesting that PE may be associated with heart failure.
Patients with PE were less likely to have symptoms consistent with a respiratory tract infection. They also tended to have higher mortality rates and longer hospitalization rates compared with those without PE.
The meta-analysis had some limitations, including the heterogeneity of findings in the included studies, as well as the potential for publication bias, since reports showing unusually low or high rates may be more likely to be published, the researchers noted. There was also a high proportion of male subjects in the included studies.
Overall, the researchers concluded that PE is more likely in patients with pleuritic chest pain and signs of heart failure, and less likely in patients with signs of a respiratory infection. That information “might add to the clinical decision-making in patients with an AECOPD, because it would be undesirable to perform [CT pulmonary angiography] in every patient with an AECOPD,” the researchers wrote.
Aleva FE, Voets LW, Simons SO, de Mast Q, van der Ven AJ, Heijdra YF. Prevalence and localization of pulmonary embolism in unexplained acute exacerbations of COPD: A systematic review and meta-analysis. Chest. 2017;151(3):544-554. doi:10.1016/j.chest.2016.07.034.
Norepinephrine Shortage Linked to Mortality in Patients With Septic Shock
AMY KARON
FRONTLINE MEDICAL NEWS
A national shortage of norepinephrine in the United States was associated with higher rates of mortality among patients hospitalized with septic shock, investigators reported.
Rates of in-hospital mortality in 2011 were 40% during quarters when hospitals were facing shortages and 36% when they were not, Emily Vail, MD, and her associates said at the International Symposium on Intensive Care and Emergency Medicine. The report was published simultaneously in JAMA.
The link between norepinephrine shortage and death from septic shock persisted even after the researchers accounted for numerous clinical and demographic factors (adjusted odds ratio, 1.2; 95% CI, 1.01 to 1.30; P = .03), wrote Dr Vail of Columbia University, New York.
Drug shortages are common in the United States, but few studies have explored their effects on patient outcomes. Investigators compared mortality rates among affected patients during 3-month intervals when hospitals were and were not using at least 20% less norepinephrine than baseline. The researchers used Premier Healthcare Database, which includes both standard claims and detailed, dated logs of all services billed to patients or insurance, with minimal missing data.
A total of 77% patients admitted with septic shock received norepinephrine before the shortage. During the lowest point of the shortage, 56% of patients received it, the researchers reported. Clinicians most often used phenylephrine instead, prescribing it to up to 54% of patients during the worst time of the shortage. The absolute increase in mortality during the quarters of shortage was 3.7% (95% CI, 1.5%-6.0%).
Several factors might explain the link between norepinephrine shortage and mortality, the investigators said. The vasopressors chosen to replace norepinephrine might result directly in worse outcomes, but a decrease in norepinephrine use also might be a proxy for relevant variables such as delayed use of vasopressors, lack of knowledge of how to optimally dose vasopressors besides norepinephrine, or the absence of a pharmacist dedicated to helping optimize the use of limited supplies.
The study did not uncover a dose-response association between greater decreases in norepinephrine use and increased mortality, the researchers noted. “This may be due to a threshold effect of vasopressor shortage on mortality, or lack of power due to relatively few hospital quarters at the extreme levels of vasopressor shortage,” they wrote.
Because the deaths captured included only those that occurred in-hospital, “the results may have underestimated mortality, particularly for hospitals that tend to transfer patients early to other skilled care facilities,” the researchers noted.
The cohort of patients was limited to those who received vasopressors for 2 or more days and excluded patients who died on the first day of vasopressor treatment, the researchers said.
Vail E, Gershengorn HB, Hua M, Walkey AJ, Rubenfeld G, Wunsch H. Association between US norepinephrine shortage and mortality among patients with septic shock. JAMA. 21 March 2017. [Epub ahead of print]. doi:10.1001/jama.2017.2841.

Osimertinib receives full approval for advanced EGFR-mutated NSCLC
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
VIDEO: Compassionate care, decriminalization crucial to mitigating addiction epidemic
SAN DIEGO – Health care providers need to practice compassionate care to achieve the best results when managing patients who are dealing with opioid addiction, according to a panel of experts who spoke at the annual meeting of the American College of Physicians.
Caring “compassionately is not enabling, [it’s] doing the right thing by the patient,” explained Chwen-Yuen Angie Chen, MD, of Stanford (Calif.) University. “You can practice compassionate care if you have a knowledge base. Knowledge is extremely powerful and enables you to follow evidence-based medicine, which is truly compassionate care.”
Dr. Chen spoke at length about addiction medicine during a press conference outlining the ACP’s new position paper on preventing and treating substance abuse, where she was joined by ACP President Nitin S. Damle, MD, and ACP Board of Regents Chair Thomas G. Tape, MD. All three emphasized the need for decriminalization and destigmatization of opioid abuse, and they called on physicians to guide patients through resources and compassionate care to help them overcome the affliction.
“We know that we need to either taper, detoxify, or reduce opioid dosing. We know that we ought not to coprescribe with sedatives. We know that, if you need addiction treatment, you get referred, and you don’t just get cut off,” explained Dr. Chen.
In a video interview, Dr. Chen talked about the key take-home messages of the position paper, and she explained other aspects of substance abuse that requires provider’s awareness.
Dr. Chen did not report any relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – Health care providers need to practice compassionate care to achieve the best results when managing patients who are dealing with opioid addiction, according to a panel of experts who spoke at the annual meeting of the American College of Physicians.
Caring “compassionately is not enabling, [it’s] doing the right thing by the patient,” explained Chwen-Yuen Angie Chen, MD, of Stanford (Calif.) University. “You can practice compassionate care if you have a knowledge base. Knowledge is extremely powerful and enables you to follow evidence-based medicine, which is truly compassionate care.”
Dr. Chen spoke at length about addiction medicine during a press conference outlining the ACP’s new position paper on preventing and treating substance abuse, where she was joined by ACP President Nitin S. Damle, MD, and ACP Board of Regents Chair Thomas G. Tape, MD. All three emphasized the need for decriminalization and destigmatization of opioid abuse, and they called on physicians to guide patients through resources and compassionate care to help them overcome the affliction.
“We know that we need to either taper, detoxify, or reduce opioid dosing. We know that we ought not to coprescribe with sedatives. We know that, if you need addiction treatment, you get referred, and you don’t just get cut off,” explained Dr. Chen.
In a video interview, Dr. Chen talked about the key take-home messages of the position paper, and she explained other aspects of substance abuse that requires provider’s awareness.
Dr. Chen did not report any relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – Health care providers need to practice compassionate care to achieve the best results when managing patients who are dealing with opioid addiction, according to a panel of experts who spoke at the annual meeting of the American College of Physicians.
Caring “compassionately is not enabling, [it’s] doing the right thing by the patient,” explained Chwen-Yuen Angie Chen, MD, of Stanford (Calif.) University. “You can practice compassionate care if you have a knowledge base. Knowledge is extremely powerful and enables you to follow evidence-based medicine, which is truly compassionate care.”
Dr. Chen spoke at length about addiction medicine during a press conference outlining the ACP’s new position paper on preventing and treating substance abuse, where she was joined by ACP President Nitin S. Damle, MD, and ACP Board of Regents Chair Thomas G. Tape, MD. All three emphasized the need for decriminalization and destigmatization of opioid abuse, and they called on physicians to guide patients through resources and compassionate care to help them overcome the affliction.
“We know that we need to either taper, detoxify, or reduce opioid dosing. We know that we ought not to coprescribe with sedatives. We know that, if you need addiction treatment, you get referred, and you don’t just get cut off,” explained Dr. Chen.
In a video interview, Dr. Chen talked about the key take-home messages of the position paper, and she explained other aspects of substance abuse that requires provider’s awareness.
Dr. Chen did not report any relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ACP INTERNAL MEDICINE
