User login
A Not-So-Old Football Injury
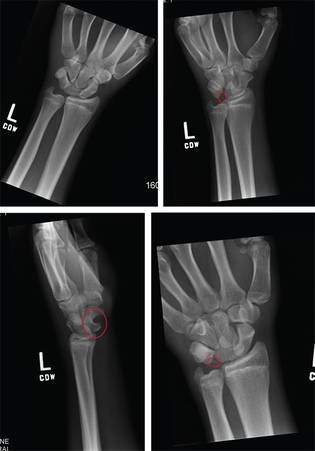
ANSWER
Imaging shows a ventral dislocation of the lunate. There is also a tiny avulsion fracture on the ulnar aspect of the adjacent triquetrum.
The patient was referred to orthopedics for a perilunate dislocation of the left wrist. He underwent successful closed reduction and was placed in a short arm cast for four weeks.
ANSWER
Imaging shows a ventral dislocation of the lunate. There is also a tiny avulsion fracture on the ulnar aspect of the adjacent triquetrum.
The patient was referred to orthopedics for a perilunate dislocation of the left wrist. He underwent successful closed reduction and was placed in a short arm cast for four weeks.
ANSWER
Imaging shows a ventral dislocation of the lunate. There is also a tiny avulsion fracture on the ulnar aspect of the adjacent triquetrum.
The patient was referred to orthopedics for a perilunate dislocation of the left wrist. He underwent successful closed reduction and was placed in a short arm cast for four weeks.
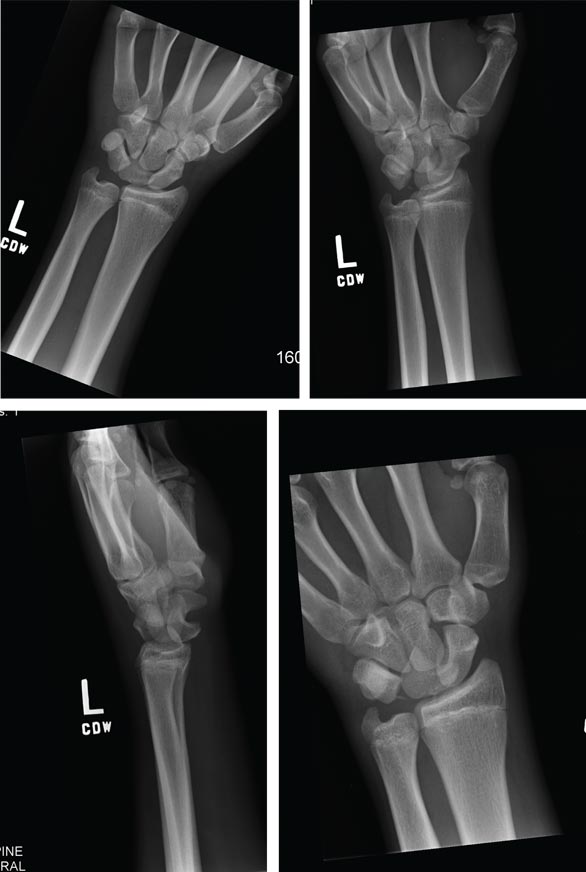
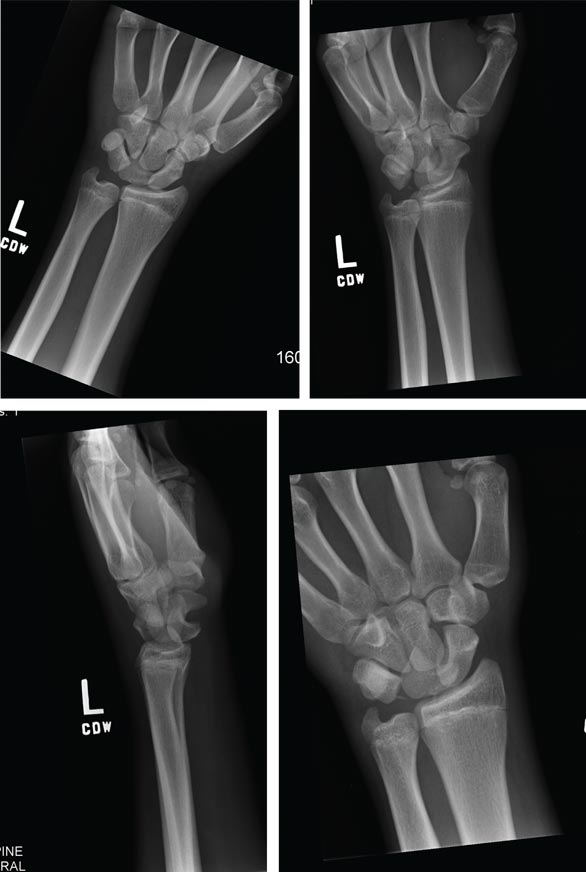
A 15-year-old boy presents for evaluation of left wrist pain. He says that two days ago, while playing football, he fell onto his outstretched left hand, which twisted upon impact with the ground. Immediately after the fall, he experienced severe pain. Since then, the pain has been constant, although it lessens to a moderate dull ache at rest and sharpens with activity. He denies any numbness or tingling in the affected hand and wrist. There are no other areas of injury from the fall, nor is there significant medical history. Physical exam identifies moderate left wrist swelling with focal tenderness over the volar aspect of the distal radius, extending to the wrist. The patient has limited active and passive flexion and extension of the left hand and wrist, along with reduced grip strength due to pain. There is mild navicular tenderness. Radial pulse is 2+, the hand is warm to the touch, and the skin is intact. Sensation is intact in all of the digits, which also demonstrate brisk capillary refill. Radiographs of the left wrist are obtained. What is your impression?
Postoperative Patient Suddenly Worsens
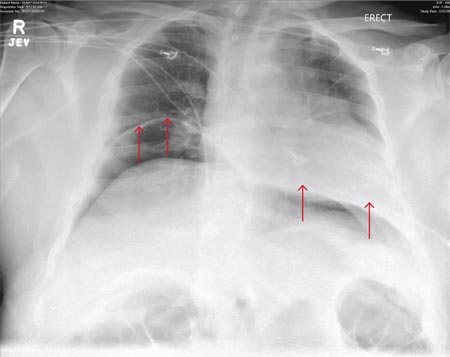
ANSWER
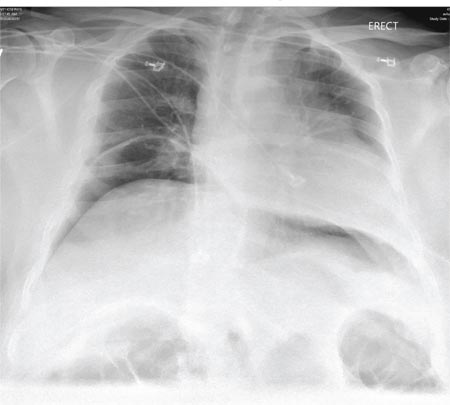
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
ANSWER
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
ANSWER
The radiograph demonstrates bilateral elevated diaphragm with a moderate amount of visible free air. With no history of recent abdominal procedures, the primary concern is a perforated viscus.
Urgent surgical consultation, as well as CT of the abdomen and pelvis, was obtained. The imaging confirmed the free air but provided no clear etiology. The patient underwent emergent laparotomy later that day and was found to have a perforated colon.
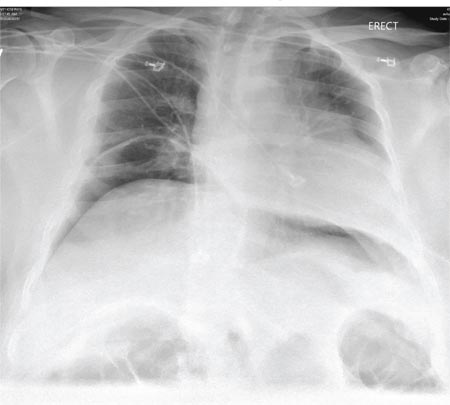
A 55-year-old man undergoes an elective craniotomy for tumor resection, with uneventful preoperative and intraoperative stages. Immediately postoperative, however, he experiences seizures. Noncontrast CT of the head is negative except for postoperative changes. The patient is placed in the ICU for close monitoring. He is slowly improving when, on the fifth postoperative day, tachypnea and dyspnea are observed. The patient is afebrile. His blood pressure is 116/70 mm Hg; pulse, 90 beats/min; respiratory rate, 30 breaths/min; and O2 saturation, 98%. A stat portable chest radiograph is obtained. What is your impression?
Leg Swelling Accompanied by Weight Gain
In the past two weeks, a 59-year-old postmenopausal woman has noticed swelling in her legs and a 10-lb weight gain. For the past three days, she has also had a vague, aching pain in the right upper abdominal quadrant, which surprises her, since her gall bladder was removed long ago. There is no prior history of chest pain, dyspnea, or systemic hypertension.
The patient does have a history of paroxysmal atrial fibrillation, palpitations, and pulmonary hypertension. She is chronically obese and has hypothyroidism. Surgical history is significant for cholecystectomy, hysterectomy, and left breast lumpectomy with axillary node dissection.
Her job at a local factory, assembling components for pressure washer pumps, requires her to sit for extended periods. She smokes 1.5 packs of cigarettes per day, a habit that began when she was 16. She drinks one or two beers daily and admits she has “many more” on the weekends. She has used marijuana in the recent past but not in the past month. She denies use of any other illicit drugs or homeopathic medications.
Her medication list includes levothyroxine sodium and ibuprofen. She says she’s “supposed to be taking some kind of heart medication” but hasn’t taken it for several months (and cannot remember the name). It was prescribed for her when she was on vacation in the Florida Keys and experienced similar symptoms. She sheepishly admits to trying her husband’s sildenafil, as she’s been told it works for pulmonary hypertension. She is allergic to sulfa and tetracycline.
Review of systems is remarkable for bilateral hip and ankle pain, which she attributes to her weight. She has had no change in bowel or bladder function. The remainder of the review is unremarkable.
Physical exam reveals a weight of 297 lb and height of 5’6”. Vital signs include a blood pressure of 128/88 mm Hg; pulse, 90 beats/min; respiratory rate, 14 breaths/min-1; temperature, 98.2°F; and O2 saturation, 98.2%.
She is morbidly obese and in no distress. Pertinent physical findings include elevated jugular venous return, bilateral rales in both lung bases, a soft, early diastolic murmur best heard at the left lower sternal border, and a regular rate and rhythm. She also has mild tenderness to deep palpation in the right upper abdominal quadrant. Her lower extremities demonstrate 3+ pitting edema to the level of the knees bilaterally. There are no skin lesions, and the neurologic exam is grossly intact.
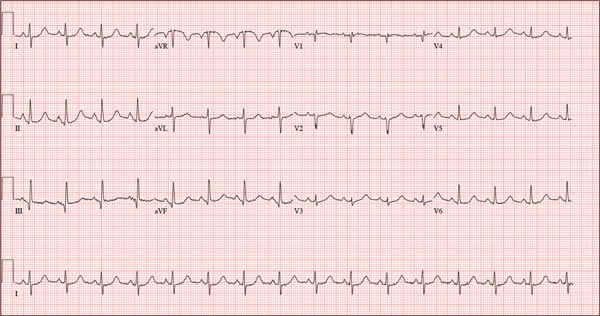
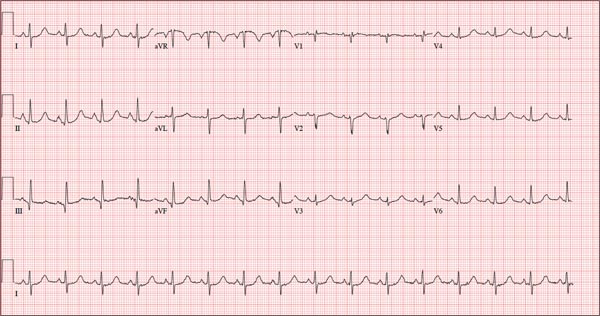
As part of her workup, you order an ECG, which reveals a ventricular rate of 94 beats/min; PR interval, 130 ms; QRS duration, 76 ms; QT/QTc interval, 394/492 ms; P axis, 50°; R axis, 80°; and T axis, 47°. What is your interpretation?
ANSWER
Pertinent findings on this ECG include normal sinus rhythm, right atrial enlargement, and a prolonged QT interval. Criteria for right atrial enlargement include P waves > 2.5 mm in leads II, III, and aVF and > 1.5 mm in leads V1 and V2. A prolonged QT interval is evidenced by a QTc > 470 ms using Bazett’s formula (QTc = QT divided by the square root of the RR interval).
The patient’s symptoms and ECG finding of right atrial enlargement coincide with pulmonary hypertension and right-sided heart failure. The prolonged QT interval may be due to her history of hypothyroidism; however, this has not been confirmed.
In the past two weeks, a 59-year-old postmenopausal woman has noticed swelling in her legs and a 10-lb weight gain. For the past three days, she has also had a vague, aching pain in the right upper abdominal quadrant, which surprises her, since her gall bladder was removed long ago. There is no prior history of chest pain, dyspnea, or systemic hypertension.
The patient does have a history of paroxysmal atrial fibrillation, palpitations, and pulmonary hypertension. She is chronically obese and has hypothyroidism. Surgical history is significant for cholecystectomy, hysterectomy, and left breast lumpectomy with axillary node dissection.
Her job at a local factory, assembling components for pressure washer pumps, requires her to sit for extended periods. She smokes 1.5 packs of cigarettes per day, a habit that began when she was 16. She drinks one or two beers daily and admits she has “many more” on the weekends. She has used marijuana in the recent past but not in the past month. She denies use of any other illicit drugs or homeopathic medications.
Her medication list includes levothyroxine sodium and ibuprofen. She says she’s “supposed to be taking some kind of heart medication” but hasn’t taken it for several months (and cannot remember the name). It was prescribed for her when she was on vacation in the Florida Keys and experienced similar symptoms. She sheepishly admits to trying her husband’s sildenafil, as she’s been told it works for pulmonary hypertension. She is allergic to sulfa and tetracycline.
Review of systems is remarkable for bilateral hip and ankle pain, which she attributes to her weight. She has had no change in bowel or bladder function. The remainder of the review is unremarkable.
Physical exam reveals a weight of 297 lb and height of 5’6”. Vital signs include a blood pressure of 128/88 mm Hg; pulse, 90 beats/min; respiratory rate, 14 breaths/min-1; temperature, 98.2°F; and O2 saturation, 98.2%.
She is morbidly obese and in no distress. Pertinent physical findings include elevated jugular venous return, bilateral rales in both lung bases, a soft, early diastolic murmur best heard at the left lower sternal border, and a regular rate and rhythm. She also has mild tenderness to deep palpation in the right upper abdominal quadrant. Her lower extremities demonstrate 3+ pitting edema to the level of the knees bilaterally. There are no skin lesions, and the neurologic exam is grossly intact.
As part of her workup, you order an ECG, which reveals a ventricular rate of 94 beats/min; PR interval, 130 ms; QRS duration, 76 ms; QT/QTc interval, 394/492 ms; P axis, 50°; R axis, 80°; and T axis, 47°. What is your interpretation?
ANSWER
Pertinent findings on this ECG include normal sinus rhythm, right atrial enlargement, and a prolonged QT interval. Criteria for right atrial enlargement include P waves > 2.5 mm in leads II, III, and aVF and > 1.5 mm in leads V1 and V2. A prolonged QT interval is evidenced by a QTc > 470 ms using Bazett’s formula (QTc = QT divided by the square root of the RR interval).
The patient’s symptoms and ECG finding of right atrial enlargement coincide with pulmonary hypertension and right-sided heart failure. The prolonged QT interval may be due to her history of hypothyroidism; however, this has not been confirmed.
In the past two weeks, a 59-year-old postmenopausal woman has noticed swelling in her legs and a 10-lb weight gain. For the past three days, she has also had a vague, aching pain in the right upper abdominal quadrant, which surprises her, since her gall bladder was removed long ago. There is no prior history of chest pain, dyspnea, or systemic hypertension.
The patient does have a history of paroxysmal atrial fibrillation, palpitations, and pulmonary hypertension. She is chronically obese and has hypothyroidism. Surgical history is significant for cholecystectomy, hysterectomy, and left breast lumpectomy with axillary node dissection.
Her job at a local factory, assembling components for pressure washer pumps, requires her to sit for extended periods. She smokes 1.5 packs of cigarettes per day, a habit that began when she was 16. She drinks one or two beers daily and admits she has “many more” on the weekends. She has used marijuana in the recent past but not in the past month. She denies use of any other illicit drugs or homeopathic medications.
Her medication list includes levothyroxine sodium and ibuprofen. She says she’s “supposed to be taking some kind of heart medication” but hasn’t taken it for several months (and cannot remember the name). It was prescribed for her when she was on vacation in the Florida Keys and experienced similar symptoms. She sheepishly admits to trying her husband’s sildenafil, as she’s been told it works for pulmonary hypertension. She is allergic to sulfa and tetracycline.
Review of systems is remarkable for bilateral hip and ankle pain, which she attributes to her weight. She has had no change in bowel or bladder function. The remainder of the review is unremarkable.
Physical exam reveals a weight of 297 lb and height of 5’6”. Vital signs include a blood pressure of 128/88 mm Hg; pulse, 90 beats/min; respiratory rate, 14 breaths/min-1; temperature, 98.2°F; and O2 saturation, 98.2%.
She is morbidly obese and in no distress. Pertinent physical findings include elevated jugular venous return, bilateral rales in both lung bases, a soft, early diastolic murmur best heard at the left lower sternal border, and a regular rate and rhythm. She also has mild tenderness to deep palpation in the right upper abdominal quadrant. Her lower extremities demonstrate 3+ pitting edema to the level of the knees bilaterally. There are no skin lesions, and the neurologic exam is grossly intact.
As part of her workup, you order an ECG, which reveals a ventricular rate of 94 beats/min; PR interval, 130 ms; QRS duration, 76 ms; QT/QTc interval, 394/492 ms; P axis, 50°; R axis, 80°; and T axis, 47°. What is your interpretation?
ANSWER
Pertinent findings on this ECG include normal sinus rhythm, right atrial enlargement, and a prolonged QT interval. Criteria for right atrial enlargement include P waves > 2.5 mm in leads II, III, and aVF and > 1.5 mm in leads V1 and V2. A prolonged QT interval is evidenced by a QTc > 470 ms using Bazett’s formula (QTc = QT divided by the square root of the RR interval).
The patient’s symptoms and ECG finding of right atrial enlargement coincide with pulmonary hypertension and right-sided heart failure. The prolonged QT interval may be due to her history of hypothyroidism; however, this has not been confirmed.
Those symptoms first appeared two weeks ago. Now, this woman also has abdominal pain. What does an ECG add to the clinical picture?
Universal bisphosphonates after wrist fracture prevent hip fractures but at a cost
Giving bone-strengthening medication routinely to all elderly women who sustain a wrist fracture would reduce subsequent hip fractures by about a quarter, but at a cost of over $200,000 per prevented fracture. Additionally, expanded bisphosphonate use could cause nearly 20,000 more atypical femur fractures in this population.
Dr. Suneel Bhat and his associates at Thomas Jefferson University, Philadelphia, used sophisticated modeling techniques to project costs and consequences of wider prescribing of bisphosphonates for bone fragility in women aged 65 years and older. Distal radius fracture is known to be associated with osteoporosis in women of this age, who are then at increased risk of subsequent fracture. Dr. Bhat presented his findings on March 24 at the annual meeting of the American Academy of Orthopaedic Surgeons in Las Vegas.
Study authors drew from the medical literature and publicly available Medicare data to obtain fracture incidence data and cost information for statistical modeling. Bhat and colleagues obtained age-specific incidence of distal radius fractures among women aged 65 and older, as well as rates of hip fracture following wrist fractures, both for those who did and did not receive the bisphosphonate risendronate. Atypical femur fracture is a known complication of bisphosphonate treatment for some patients; the risk of this complication and medication costs were drawn from the literature.
To assess the direct costs of hip fracture treatment, investigators used Medicare reimbursement data to price treatment components, including inpatient care as well as surgical and anesthesia services.
From these data, investigators used a modified Monte Carlo technique to obtain a cost and incidence model. This model predicted that 357,656 lifetime subsequent hip fractures would occur in elderly women with wrist fracture; this number would drop to 262,767 with universal bisphosphonate treatment. The cost for this savings, an aggregate $19.5 billion in drug costs, meant that each fracture prevented would cost $205,534. An additional 19,464 patients would sustain atypical femur fracture from risendronate treatment.
Average risendronate costs were estimated at $1,485/patient-year. This figure would have to fall to just $70/patient yearly to make risendronate treatment cost effective, Dr. Bhat calculated.
Giving bone-strengthening medication routinely to all elderly women who sustain a wrist fracture would reduce subsequent hip fractures by about a quarter, but at a cost of over $200,000 per prevented fracture. Additionally, expanded bisphosphonate use could cause nearly 20,000 more atypical femur fractures in this population.
Dr. Suneel Bhat and his associates at Thomas Jefferson University, Philadelphia, used sophisticated modeling techniques to project costs and consequences of wider prescribing of bisphosphonates for bone fragility in women aged 65 years and older. Distal radius fracture is known to be associated with osteoporosis in women of this age, who are then at increased risk of subsequent fracture. Dr. Bhat presented his findings on March 24 at the annual meeting of the American Academy of Orthopaedic Surgeons in Las Vegas.
Study authors drew from the medical literature and publicly available Medicare data to obtain fracture incidence data and cost information for statistical modeling. Bhat and colleagues obtained age-specific incidence of distal radius fractures among women aged 65 and older, as well as rates of hip fracture following wrist fractures, both for those who did and did not receive the bisphosphonate risendronate. Atypical femur fracture is a known complication of bisphosphonate treatment for some patients; the risk of this complication and medication costs were drawn from the literature.
To assess the direct costs of hip fracture treatment, investigators used Medicare reimbursement data to price treatment components, including inpatient care as well as surgical and anesthesia services.
From these data, investigators used a modified Monte Carlo technique to obtain a cost and incidence model. This model predicted that 357,656 lifetime subsequent hip fractures would occur in elderly women with wrist fracture; this number would drop to 262,767 with universal bisphosphonate treatment. The cost for this savings, an aggregate $19.5 billion in drug costs, meant that each fracture prevented would cost $205,534. An additional 19,464 patients would sustain atypical femur fracture from risendronate treatment.
Average risendronate costs were estimated at $1,485/patient-year. This figure would have to fall to just $70/patient yearly to make risendronate treatment cost effective, Dr. Bhat calculated.
Giving bone-strengthening medication routinely to all elderly women who sustain a wrist fracture would reduce subsequent hip fractures by about a quarter, but at a cost of over $200,000 per prevented fracture. Additionally, expanded bisphosphonate use could cause nearly 20,000 more atypical femur fractures in this population.
Dr. Suneel Bhat and his associates at Thomas Jefferson University, Philadelphia, used sophisticated modeling techniques to project costs and consequences of wider prescribing of bisphosphonates for bone fragility in women aged 65 years and older. Distal radius fracture is known to be associated with osteoporosis in women of this age, who are then at increased risk of subsequent fracture. Dr. Bhat presented his findings on March 24 at the annual meeting of the American Academy of Orthopaedic Surgeons in Las Vegas.
Study authors drew from the medical literature and publicly available Medicare data to obtain fracture incidence data and cost information for statistical modeling. Bhat and colleagues obtained age-specific incidence of distal radius fractures among women aged 65 and older, as well as rates of hip fracture following wrist fractures, both for those who did and did not receive the bisphosphonate risendronate. Atypical femur fracture is a known complication of bisphosphonate treatment for some patients; the risk of this complication and medication costs were drawn from the literature.
To assess the direct costs of hip fracture treatment, investigators used Medicare reimbursement data to price treatment components, including inpatient care as well as surgical and anesthesia services.
From these data, investigators used a modified Monte Carlo technique to obtain a cost and incidence model. This model predicted that 357,656 lifetime subsequent hip fractures would occur in elderly women with wrist fracture; this number would drop to 262,767 with universal bisphosphonate treatment. The cost for this savings, an aggregate $19.5 billion in drug costs, meant that each fracture prevented would cost $205,534. An additional 19,464 patients would sustain atypical femur fracture from risendronate treatment.
Average risendronate costs were estimated at $1,485/patient-year. This figure would have to fall to just $70/patient yearly to make risendronate treatment cost effective, Dr. Bhat calculated.
FROM AAOS 2015
Key clinical point: Savings from prevention of hip fractures with universal bisphosphonates after distal radius fractures in elderly women would be outweighed by drug costs.
Major findings: If all women 65 years of age and older who have had a distal radius fracture went on to receive bisphosphonate treatment, 94,888 lifetime hip fractures would be avoided, but at a cost of $205,534 per averted fracture, and with an additional 19,464 atypical femur fractures.
Data source: Modeling based on fracture incidence data and medication, surgical, and aftercare cost data drawn from literature review and publicly available Medicare databases.
Disclosures: Dr. Asif Ilyas reported publishing royalties and financial or material support from Jaypee Medical Publishers and is on the editorial or governing board of Orthopedic Clinics of North America. The other authors reported that they had no conflicts of interest.
As Problem Spreads, Man Seeks Help
ANSWER
The correct answer is Schamberg disease (choice “b”), a benign form of capillaritis; see Discussion for more information.
Scurvy patients can present with ecchymosis (among other findings that were missing in this case). But scurvy (choice “a”) is rare, and by the time the disease is evident, the patient is typically quite ill.
Cutaneous T-cell lymphoma (choice “c”) can manifest as purpuric annular lesions. However, these would be unlikely to take the distributive pattern seen in this case, and they usually have an atrophic surface.
Thrombocytopenia (choice “d”) and other coagulopathies, although rightly considered, would probably manifest in other ways as well (ie, not just cutaneously).
Continue for the discussion >>
DISCUSSION
Schamberg disease is typical of a whole class of conditions in which red blood cells (RBCs) are extravasated from slightly damaged capillaries. This results in nonblanchable purpura and subsequent hemosiderin staining caused by phagocytosis of the RBCs by macrophages. Clinically, this family of diseases present as cayenne pepper–colored macules, most of which are annular in configuration.
Schamberg is, by far, the most common of these conditions. This presentation was typical: manifestation on the knees and ankles followed by upward spread (hence the condition’s other name, progressive pigmentary purpura). Usually resolving on their own within months, these lesions are almost always asymptomatic—but nonetheless alarming to the patient.
Other equally benign, self-limited forms of capillaritis include those in which lesions are pruritic (eg, purpura of Doucas and Kapetanakis) or lichenoid (purpura of Gougerot-Blum). Another example is lichen aureus, in which only one or two lesions, more gold than brown, appear on the legs of younger patients.
There are many theories as to these conditions’ cause, the most common of which is increased intravascular pressure secondary to dependence. However, if this were so, we’d likely see a great deal more cases, since many patients have problems related to venous insufficiency.
In some cases, skin biopsy (usually 4-mm punch) is necessary to rule out more serious diseases, such as an early form of cutaneous T-cell lymphoma. When a coagulopathy is suspected, blood work is necessary to confirm or rule out the diagnosis. In this case, there was no reason to suspect coagulopathy (or scurvy), since no other signs were seen.
This patient was educated about his diagnosis and provided Web-based resources he could consult. Various treatments—topical steroids, increased vitamin C intake, and increased exposure to UV light—have been tried but with disappointing results.
ANSWER
The correct answer is Schamberg disease (choice “b”), a benign form of capillaritis; see Discussion for more information.
Scurvy patients can present with ecchymosis (among other findings that were missing in this case). But scurvy (choice “a”) is rare, and by the time the disease is evident, the patient is typically quite ill.
Cutaneous T-cell lymphoma (choice “c”) can manifest as purpuric annular lesions. However, these would be unlikely to take the distributive pattern seen in this case, and they usually have an atrophic surface.
Thrombocytopenia (choice “d”) and other coagulopathies, although rightly considered, would probably manifest in other ways as well (ie, not just cutaneously).
Continue for the discussion >>
DISCUSSION
Schamberg disease is typical of a whole class of conditions in which red blood cells (RBCs) are extravasated from slightly damaged capillaries. This results in nonblanchable purpura and subsequent hemosiderin staining caused by phagocytosis of the RBCs by macrophages. Clinically, this family of diseases present as cayenne pepper–colored macules, most of which are annular in configuration.
Schamberg is, by far, the most common of these conditions. This presentation was typical: manifestation on the knees and ankles followed by upward spread (hence the condition’s other name, progressive pigmentary purpura). Usually resolving on their own within months, these lesions are almost always asymptomatic—but nonetheless alarming to the patient.
Other equally benign, self-limited forms of capillaritis include those in which lesions are pruritic (eg, purpura of Doucas and Kapetanakis) or lichenoid (purpura of Gougerot-Blum). Another example is lichen aureus, in which only one or two lesions, more gold than brown, appear on the legs of younger patients.
There are many theories as to these conditions’ cause, the most common of which is increased intravascular pressure secondary to dependence. However, if this were so, we’d likely see a great deal more cases, since many patients have problems related to venous insufficiency.
In some cases, skin biopsy (usually 4-mm punch) is necessary to rule out more serious diseases, such as an early form of cutaneous T-cell lymphoma. When a coagulopathy is suspected, blood work is necessary to confirm or rule out the diagnosis. In this case, there was no reason to suspect coagulopathy (or scurvy), since no other signs were seen.
This patient was educated about his diagnosis and provided Web-based resources he could consult. Various treatments—topical steroids, increased vitamin C intake, and increased exposure to UV light—have been tried but with disappointing results.
ANSWER
The correct answer is Schamberg disease (choice “b”), a benign form of capillaritis; see Discussion for more information.
Scurvy patients can present with ecchymosis (among other findings that were missing in this case). But scurvy (choice “a”) is rare, and by the time the disease is evident, the patient is typically quite ill.
Cutaneous T-cell lymphoma (choice “c”) can manifest as purpuric annular lesions. However, these would be unlikely to take the distributive pattern seen in this case, and they usually have an atrophic surface.
Thrombocytopenia (choice “d”) and other coagulopathies, although rightly considered, would probably manifest in other ways as well (ie, not just cutaneously).
Continue for the discussion >>
DISCUSSION
Schamberg disease is typical of a whole class of conditions in which red blood cells (RBCs) are extravasated from slightly damaged capillaries. This results in nonblanchable purpura and subsequent hemosiderin staining caused by phagocytosis of the RBCs by macrophages. Clinically, this family of diseases present as cayenne pepper–colored macules, most of which are annular in configuration.
Schamberg is, by far, the most common of these conditions. This presentation was typical: manifestation on the knees and ankles followed by upward spread (hence the condition’s other name, progressive pigmentary purpura). Usually resolving on their own within months, these lesions are almost always asymptomatic—but nonetheless alarming to the patient.
Other equally benign, self-limited forms of capillaritis include those in which lesions are pruritic (eg, purpura of Doucas and Kapetanakis) or lichenoid (purpura of Gougerot-Blum). Another example is lichen aureus, in which only one or two lesions, more gold than brown, appear on the legs of younger patients.
There are many theories as to these conditions’ cause, the most common of which is increased intravascular pressure secondary to dependence. However, if this were so, we’d likely see a great deal more cases, since many patients have problems related to venous insufficiency.
In some cases, skin biopsy (usually 4-mm punch) is necessary to rule out more serious diseases, such as an early form of cutaneous T-cell lymphoma. When a coagulopathy is suspected, blood work is necessary to confirm or rule out the diagnosis. In this case, there was no reason to suspect coagulopathy (or scurvy), since no other signs were seen.
This patient was educated about his diagnosis and provided Web-based resources he could consult. Various treatments—topical steroids, increased vitamin C intake, and increased exposure to UV light—have been tried but with disappointing results.
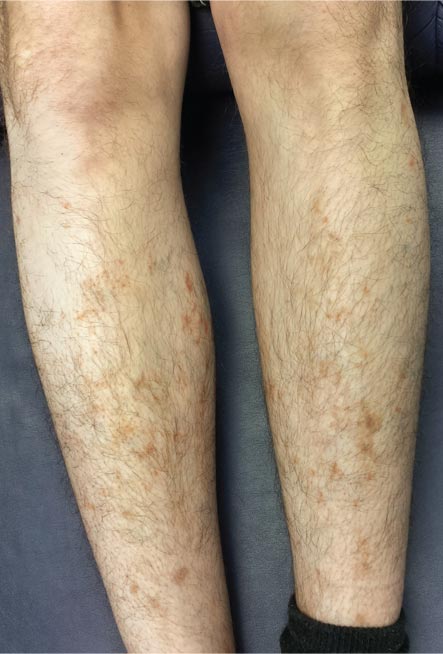
For several months, a 30-year-old man has had an asymptomatic rash on his legs. The lesions first appeared on his lower legs and ankles; over the subsequent months, they have spread upward. Now, the rash reaches to just below his knees. During this time, he has had two bouts of strep throat, both adequately treated. He denies any other skin problems and has no relevant family history. The patient denies alcohol or drug abuse and is not taking any prescription medications. Prior to referral to dermatology, he was seen in two urgent care clinics; at one, he received a diagnosis of fungal infection and at the other, of “vitamin deficiency.” He was given a month-long course of terbinafine (250 mg/d) that produced no change in his rash. He achieved the same (non)result from an increased intake of vitamins. Examination reveals annular reddish brown macules, measuring 1 to 3 cm, sparsely distributed from the knees to just above the ankles on both legs. The lesions are a bit more densely arrayed on the anterior legs. There is no palpable component to any of them and no discernable surface scale. Digital pressure fails to blanch the lesions. The hairs and follicles on the patient’s legs appear normal. There are no notable skin changes elsewhere, and the patient is alert, oriented, and in no distress.

Men Have a Higher Level of Function Before and After Total Knee Replacement Surgery
LAS VEGAS—While men and women have similar levels of improvement following total knee replacement (TKR) surgery, men have higher levels of function before and after TKR, according to new research presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
According to the Agency for Healthcare Research and Quality, more than 600,000 knee replacements are performed in the United States each year. In 2012, 393,345 women and 237,896 men underwent TKR, most often to alleviate the pain and immobility associated with late-stage arthritis. While research has looked at the anatomic differences between the knees of men and women, the higher levels of arthritis in women versus men, and the utilization of TKR among men and women, there has been little study on how gender influences the level of function before and after surgery.
In this study, researchers identified and studied 287 TKR patients at 7 different institutions between 2005 and 2007. All of the patients were between the ages of 21 and 80 years at the time of surgery and had a body mass index less than 40 g/m². All of the patients except 2 had a diagnosis of end-stage arthritis. The patient group included 108 men (112 knees) with a mean age of 67 years, and 170 women with a mean age of 66 years. All of the patients were evaluated preoperatively and at the following 6 points following surgery: 6 weeks, 3 months, 1 year, 2 years, 5 years, and 7 years. A Kaplan-Meier assessment gauged implant survival, and quality-of-life measurements were taken at 3 and 4 years post-surgery. During each evaluation, researchers measured knee function, range of motion, extremity activity, and overall health.
At 5 years post-surgery, implant survival was 100% for men and 99.1% for women. Range of motion also was nearly identical between genders. Functional scores were consistently higher for the men versus women: preoperatively, 57.1 versus 51; postoperatively at 6 weeks, 63.7 versus 51.5; at 3 months, 83.1 versus 74.3; at 2 years, 90 versus 81.6; at 5 years, 90.1 versus 82.9; and at 7 years, 96 versus 79.5. Men also recovered faster within the 6-week recovery time after surgery; however, both genders had almost identical improvement in mean knee score function (improvement from presurgical levels) at 5 years.
“Our data supports that while both genders benefit from TKR, men have higher levels of function and activity both prior to and after TKR compared to women,” said investigators Jeffrey J. Cherian, DO, and Michael A. Mont, MD. “These functional outcome differences are most likely due to many factors, including biologic/genetic, and highlight the need for further research related to the role of gender of both the patient and the surgeon in the decision making process of TKR, sex-based biological differences in functional recovery capacity, and whether sex/gender based pre- and postoperative rehabilitation protocols are warranted.”
LAS VEGAS—While men and women have similar levels of improvement following total knee replacement (TKR) surgery, men have higher levels of function before and after TKR, according to new research presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
According to the Agency for Healthcare Research and Quality, more than 600,000 knee replacements are performed in the United States each year. In 2012, 393,345 women and 237,896 men underwent TKR, most often to alleviate the pain and immobility associated with late-stage arthritis. While research has looked at the anatomic differences between the knees of men and women, the higher levels of arthritis in women versus men, and the utilization of TKR among men and women, there has been little study on how gender influences the level of function before and after surgery.
In this study, researchers identified and studied 287 TKR patients at 7 different institutions between 2005 and 2007. All of the patients were between the ages of 21 and 80 years at the time of surgery and had a body mass index less than 40 g/m². All of the patients except 2 had a diagnosis of end-stage arthritis. The patient group included 108 men (112 knees) with a mean age of 67 years, and 170 women with a mean age of 66 years. All of the patients were evaluated preoperatively and at the following 6 points following surgery: 6 weeks, 3 months, 1 year, 2 years, 5 years, and 7 years. A Kaplan-Meier assessment gauged implant survival, and quality-of-life measurements were taken at 3 and 4 years post-surgery. During each evaluation, researchers measured knee function, range of motion, extremity activity, and overall health.
At 5 years post-surgery, implant survival was 100% for men and 99.1% for women. Range of motion also was nearly identical between genders. Functional scores were consistently higher for the men versus women: preoperatively, 57.1 versus 51; postoperatively at 6 weeks, 63.7 versus 51.5; at 3 months, 83.1 versus 74.3; at 2 years, 90 versus 81.6; at 5 years, 90.1 versus 82.9; and at 7 years, 96 versus 79.5. Men also recovered faster within the 6-week recovery time after surgery; however, both genders had almost identical improvement in mean knee score function (improvement from presurgical levels) at 5 years.
“Our data supports that while both genders benefit from TKR, men have higher levels of function and activity both prior to and after TKR compared to women,” said investigators Jeffrey J. Cherian, DO, and Michael A. Mont, MD. “These functional outcome differences are most likely due to many factors, including biologic/genetic, and highlight the need for further research related to the role of gender of both the patient and the surgeon in the decision making process of TKR, sex-based biological differences in functional recovery capacity, and whether sex/gender based pre- and postoperative rehabilitation protocols are warranted.”
LAS VEGAS—While men and women have similar levels of improvement following total knee replacement (TKR) surgery, men have higher levels of function before and after TKR, according to new research presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
According to the Agency for Healthcare Research and Quality, more than 600,000 knee replacements are performed in the United States each year. In 2012, 393,345 women and 237,896 men underwent TKR, most often to alleviate the pain and immobility associated with late-stage arthritis. While research has looked at the anatomic differences between the knees of men and women, the higher levels of arthritis in women versus men, and the utilization of TKR among men and women, there has been little study on how gender influences the level of function before and after surgery.
In this study, researchers identified and studied 287 TKR patients at 7 different institutions between 2005 and 2007. All of the patients were between the ages of 21 and 80 years at the time of surgery and had a body mass index less than 40 g/m². All of the patients except 2 had a diagnosis of end-stage arthritis. The patient group included 108 men (112 knees) with a mean age of 67 years, and 170 women with a mean age of 66 years. All of the patients were evaluated preoperatively and at the following 6 points following surgery: 6 weeks, 3 months, 1 year, 2 years, 5 years, and 7 years. A Kaplan-Meier assessment gauged implant survival, and quality-of-life measurements were taken at 3 and 4 years post-surgery. During each evaluation, researchers measured knee function, range of motion, extremity activity, and overall health.
At 5 years post-surgery, implant survival was 100% for men and 99.1% for women. Range of motion also was nearly identical between genders. Functional scores were consistently higher for the men versus women: preoperatively, 57.1 versus 51; postoperatively at 6 weeks, 63.7 versus 51.5; at 3 months, 83.1 versus 74.3; at 2 years, 90 versus 81.6; at 5 years, 90.1 versus 82.9; and at 7 years, 96 versus 79.5. Men also recovered faster within the 6-week recovery time after surgery; however, both genders had almost identical improvement in mean knee score function (improvement from presurgical levels) at 5 years.
“Our data supports that while both genders benefit from TKR, men have higher levels of function and activity both prior to and after TKR compared to women,” said investigators Jeffrey J. Cherian, DO, and Michael A. Mont, MD. “These functional outcome differences are most likely due to many factors, including biologic/genetic, and highlight the need for further research related to the role of gender of both the patient and the surgeon in the decision making process of TKR, sex-based biological differences in functional recovery capacity, and whether sex/gender based pre- and postoperative rehabilitation protocols are warranted.”
Routine Bisphosphonate Treatment for Women Older Than 65 Years Who Sustain a Wrist Fracture Could Prevent Nearly 95,000 Hip Fractures, But at a Significant Cost
LAS VEGAS—Routine bisphosphonate treatment of women older than 65 years who sustain a distal radius fracture could significantly reduce the risk for additional fractures, primarily hip fractures, but at an estimated cost of more than $2 billion annually, according to a study presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
More than 50% of men and women older than 80 years meet diagnostic criteria for osteoporosis, placing them at increased risk for bone fractures, including hip fractures, which cause an estimated 300,000 unplanned hospital admissions in the United States each year. The lifetime cost of a hip fracture is estimated at $81,300, of which approximately 44% of the costs are associated with nursing facility expenses. Bisphosphonates, a drug known to increase bone mass and prevent fractures, have been associated with atypical femur fractures in a small, but significant number of patients.
Researchers reviewed existing literature and Medicare data to determine distal radius fracture incidence and age-specific hip fracture rates after distal radius fracture with and without bisphosphonate treatment. A model was then created to determine future fracture rates with and without treatment and related costs.
The model predicted 357,656 lifetime hip fractures following distal radius fracture in all females age 65 years and older in the US. If these patients received regular bisphosphonate treatment following a distal radius fracture, the number of hip fractures would drop to 262,767 over the lifetime of these patients; however, an estimated 19,464 patients would suffer an atypical femur fracture as a result of the treatment.
The cost of routine bisphosphonate treatment, including the cost for treating associated atypical femur fractures, comes to a lifetime total of $19.5 billion, or approximately $205,534 per avoided hip fracture.
“Our study suggests that routine universal utilization of bisphosphonates in elderly women after distal radius fracture would not be economically advantageous despite the cost savings associated with reduction of the hip fracture burden in that population,” said lead study author, Suneel B. Bhat, MD, an orthopedic surgery resident at the Rothman Institute in Philadelphia.
The study authors also hypothesize that the cost of bisphosphonates would need to drop to $70 per patient each year, from the current average annual wholesale cost of $1,485 per patient, to make the treatment affordable to every patient age 65 years and older following a wrist fracture. In addition, selecting patients at lower risk for atypical femur fractures for treatment may reduce the number of bisphosphonate-related fractures. Confirming patient osteoporosis and fracture risk through a DEXA Scan (dual x-ray absorptiometry) before prescribing bisphosphonates remains the most cost-effective method for treating osteoporosis and avoiding subsequent fractures.
LAS VEGAS—Routine bisphosphonate treatment of women older than 65 years who sustain a distal radius fracture could significantly reduce the risk for additional fractures, primarily hip fractures, but at an estimated cost of more than $2 billion annually, according to a study presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
More than 50% of men and women older than 80 years meet diagnostic criteria for osteoporosis, placing them at increased risk for bone fractures, including hip fractures, which cause an estimated 300,000 unplanned hospital admissions in the United States each year. The lifetime cost of a hip fracture is estimated at $81,300, of which approximately 44% of the costs are associated with nursing facility expenses. Bisphosphonates, a drug known to increase bone mass and prevent fractures, have been associated with atypical femur fractures in a small, but significant number of patients.
Researchers reviewed existing literature and Medicare data to determine distal radius fracture incidence and age-specific hip fracture rates after distal radius fracture with and without bisphosphonate treatment. A model was then created to determine future fracture rates with and without treatment and related costs.
The model predicted 357,656 lifetime hip fractures following distal radius fracture in all females age 65 years and older in the US. If these patients received regular bisphosphonate treatment following a distal radius fracture, the number of hip fractures would drop to 262,767 over the lifetime of these patients; however, an estimated 19,464 patients would suffer an atypical femur fracture as a result of the treatment.
The cost of routine bisphosphonate treatment, including the cost for treating associated atypical femur fractures, comes to a lifetime total of $19.5 billion, or approximately $205,534 per avoided hip fracture.
“Our study suggests that routine universal utilization of bisphosphonates in elderly women after distal radius fracture would not be economically advantageous despite the cost savings associated with reduction of the hip fracture burden in that population,” said lead study author, Suneel B. Bhat, MD, an orthopedic surgery resident at the Rothman Institute in Philadelphia.
The study authors also hypothesize that the cost of bisphosphonates would need to drop to $70 per patient each year, from the current average annual wholesale cost of $1,485 per patient, to make the treatment affordable to every patient age 65 years and older following a wrist fracture. In addition, selecting patients at lower risk for atypical femur fractures for treatment may reduce the number of bisphosphonate-related fractures. Confirming patient osteoporosis and fracture risk through a DEXA Scan (dual x-ray absorptiometry) before prescribing bisphosphonates remains the most cost-effective method for treating osteoporosis and avoiding subsequent fractures.
LAS VEGAS—Routine bisphosphonate treatment of women older than 65 years who sustain a distal radius fracture could significantly reduce the risk for additional fractures, primarily hip fractures, but at an estimated cost of more than $2 billion annually, according to a study presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
More than 50% of men and women older than 80 years meet diagnostic criteria for osteoporosis, placing them at increased risk for bone fractures, including hip fractures, which cause an estimated 300,000 unplanned hospital admissions in the United States each year. The lifetime cost of a hip fracture is estimated at $81,300, of which approximately 44% of the costs are associated with nursing facility expenses. Bisphosphonates, a drug known to increase bone mass and prevent fractures, have been associated with atypical femur fractures in a small, but significant number of patients.
Researchers reviewed existing literature and Medicare data to determine distal radius fracture incidence and age-specific hip fracture rates after distal radius fracture with and without bisphosphonate treatment. A model was then created to determine future fracture rates with and without treatment and related costs.
The model predicted 357,656 lifetime hip fractures following distal radius fracture in all females age 65 years and older in the US. If these patients received regular bisphosphonate treatment following a distal radius fracture, the number of hip fractures would drop to 262,767 over the lifetime of these patients; however, an estimated 19,464 patients would suffer an atypical femur fracture as a result of the treatment.
The cost of routine bisphosphonate treatment, including the cost for treating associated atypical femur fractures, comes to a lifetime total of $19.5 billion, or approximately $205,534 per avoided hip fracture.
“Our study suggests that routine universal utilization of bisphosphonates in elderly women after distal radius fracture would not be economically advantageous despite the cost savings associated with reduction of the hip fracture burden in that population,” said lead study author, Suneel B. Bhat, MD, an orthopedic surgery resident at the Rothman Institute in Philadelphia.
The study authors also hypothesize that the cost of bisphosphonates would need to drop to $70 per patient each year, from the current average annual wholesale cost of $1,485 per patient, to make the treatment affordable to every patient age 65 years and older following a wrist fracture. In addition, selecting patients at lower risk for atypical femur fractures for treatment may reduce the number of bisphosphonate-related fractures. Confirming patient osteoporosis and fracture risk through a DEXA Scan (dual x-ray absorptiometry) before prescribing bisphosphonates remains the most cost-effective method for treating osteoporosis and avoiding subsequent fractures.
Nearly Half of Patients Have Delirium Before and After Hip Fracture Surgery, Diminishing Outcomes and Increasing Health Care Costs
LAS VEGAS─Nearly 50% of hip fracture patients, age 65 years and older, had delirium before, during, and after surgery, resulting in significantly longer hospital stays and higher costs for care, according to data presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS). Delirium was associated with 7.4 additional hospital days and approximately $8,000 more in hospital costs.
Approximately 300,000 Americans are hospitalized with hip fractures each year. The risk is particularly high in post-menopausal women who face an increased risk for osteoporosis. Delirium is common among older hip fracture patients, and multiple studies have found that patients with postoperative delirium are more likely to have complications, including infections, and less likely to return to their pre-injury level of function. Delirium patients also are more frequently placed in nursing homes following surgery and have an increased rate of mortality.
In this study, researchers at the University of Toronto sought to determine the economic implications of perioperative delirium in older orthopedic patients by reviewing hip fracture records between January 2011 and December 2012. A total of 242 hip fracture patients with a mean age of 82 years (ages 65 to 103 years) were studied. Demographic, clinical, surgical, and adverse events data were analyzed. Perioperative delirium was assessed using the Confusion Assessment Method (CAM).
The study found that 116 patients (48%) experienced delirium during hospital admission. The patients with delirium were significantly older (mean age 85 years), and were more likely to have a higher American Society of Anesthesiologists (ASA) score (1 represents a completely healthy fit patient, and 5 represents a patient not expected to live beyond 24 hours without surgery). After controlling for these differences, perioperative delirium was associated with 7.4 additional hospital days and $8,282 ($8,649 in US dollars) in additional hospital costs (1.5 times the cost of patients who did not experience delirium).
There were no differences in mean time between triage or admission and surgery, length of surgery, or anesthesia type between groups. A significantly greater proportion of patients who experienced perioperative delirium required long-term and/or skilled care facility admission following their hospital stay than did those who did not experience delirium (8% versus 0%).
“Older patients are at high risk of developing delirium during hospitalization for a hip fracture, which is associated with worse outcomes,” said orthopedic surgeon and lead study author Michael G. Zywiel, MD. “Our work demonstrates that delirium also markedly increases the cost of elderly patient care while in the hospital. Given the high number of patients hospitalized every year with a hip fracture, there is a real need to develop and fund improved interventions to prevent in-hospital delirium in these patients.
“Our research suggests that reducing the rate of delirium would simultaneously increase the quality of care while decreasing costs, presenting hospitals, surgeons, and other stakeholders with promising opportunities to improve the value of hip fracture care,” said Dr. Zywiel.
The AAOS’s new clinical practice guideline, “Management of Hip Fractures in the Elderly,” makes a series of recommendations to reduce delirium in older hip fracture patients. They include:
• Preoperative regional analgesia to reduce pain.
• Hip fracture surgery within 48 hours of hospital admission.
• Intensive physical therapy following hospital discharge to improve functional outcomes.
• An osteoporosis evaluation, as well as vitamin D and calcium supplements, for patients following a hip fracture.
LAS VEGAS─Nearly 50% of hip fracture patients, age 65 years and older, had delirium before, during, and after surgery, resulting in significantly longer hospital stays and higher costs for care, according to data presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS). Delirium was associated with 7.4 additional hospital days and approximately $8,000 more in hospital costs.
Approximately 300,000 Americans are hospitalized with hip fractures each year. The risk is particularly high in post-menopausal women who face an increased risk for osteoporosis. Delirium is common among older hip fracture patients, and multiple studies have found that patients with postoperative delirium are more likely to have complications, including infections, and less likely to return to their pre-injury level of function. Delirium patients also are more frequently placed in nursing homes following surgery and have an increased rate of mortality.
In this study, researchers at the University of Toronto sought to determine the economic implications of perioperative delirium in older orthopedic patients by reviewing hip fracture records between January 2011 and December 2012. A total of 242 hip fracture patients with a mean age of 82 years (ages 65 to 103 years) were studied. Demographic, clinical, surgical, and adverse events data were analyzed. Perioperative delirium was assessed using the Confusion Assessment Method (CAM).
The study found that 116 patients (48%) experienced delirium during hospital admission. The patients with delirium were significantly older (mean age 85 years), and were more likely to have a higher American Society of Anesthesiologists (ASA) score (1 represents a completely healthy fit patient, and 5 represents a patient not expected to live beyond 24 hours without surgery). After controlling for these differences, perioperative delirium was associated with 7.4 additional hospital days and $8,282 ($8,649 in US dollars) in additional hospital costs (1.5 times the cost of patients who did not experience delirium).
There were no differences in mean time between triage or admission and surgery, length of surgery, or anesthesia type between groups. A significantly greater proportion of patients who experienced perioperative delirium required long-term and/or skilled care facility admission following their hospital stay than did those who did not experience delirium (8% versus 0%).
“Older patients are at high risk of developing delirium during hospitalization for a hip fracture, which is associated with worse outcomes,” said orthopedic surgeon and lead study author Michael G. Zywiel, MD. “Our work demonstrates that delirium also markedly increases the cost of elderly patient care while in the hospital. Given the high number of patients hospitalized every year with a hip fracture, there is a real need to develop and fund improved interventions to prevent in-hospital delirium in these patients.
“Our research suggests that reducing the rate of delirium would simultaneously increase the quality of care while decreasing costs, presenting hospitals, surgeons, and other stakeholders with promising opportunities to improve the value of hip fracture care,” said Dr. Zywiel.
The AAOS’s new clinical practice guideline, “Management of Hip Fractures in the Elderly,” makes a series of recommendations to reduce delirium in older hip fracture patients. They include:
• Preoperative regional analgesia to reduce pain.
• Hip fracture surgery within 48 hours of hospital admission.
• Intensive physical therapy following hospital discharge to improve functional outcomes.
• An osteoporosis evaluation, as well as vitamin D and calcium supplements, for patients following a hip fracture.
LAS VEGAS─Nearly 50% of hip fracture patients, age 65 years and older, had delirium before, during, and after surgery, resulting in significantly longer hospital stays and higher costs for care, according to data presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS). Delirium was associated with 7.4 additional hospital days and approximately $8,000 more in hospital costs.
Approximately 300,000 Americans are hospitalized with hip fractures each year. The risk is particularly high in post-menopausal women who face an increased risk for osteoporosis. Delirium is common among older hip fracture patients, and multiple studies have found that patients with postoperative delirium are more likely to have complications, including infections, and less likely to return to their pre-injury level of function. Delirium patients also are more frequently placed in nursing homes following surgery and have an increased rate of mortality.
In this study, researchers at the University of Toronto sought to determine the economic implications of perioperative delirium in older orthopedic patients by reviewing hip fracture records between January 2011 and December 2012. A total of 242 hip fracture patients with a mean age of 82 years (ages 65 to 103 years) were studied. Demographic, clinical, surgical, and adverse events data were analyzed. Perioperative delirium was assessed using the Confusion Assessment Method (CAM).
The study found that 116 patients (48%) experienced delirium during hospital admission. The patients with delirium were significantly older (mean age 85 years), and were more likely to have a higher American Society of Anesthesiologists (ASA) score (1 represents a completely healthy fit patient, and 5 represents a patient not expected to live beyond 24 hours without surgery). After controlling for these differences, perioperative delirium was associated with 7.4 additional hospital days and $8,282 ($8,649 in US dollars) in additional hospital costs (1.5 times the cost of patients who did not experience delirium).
There were no differences in mean time between triage or admission and surgery, length of surgery, or anesthesia type between groups. A significantly greater proportion of patients who experienced perioperative delirium required long-term and/or skilled care facility admission following their hospital stay than did those who did not experience delirium (8% versus 0%).
“Older patients are at high risk of developing delirium during hospitalization for a hip fracture, which is associated with worse outcomes,” said orthopedic surgeon and lead study author Michael G. Zywiel, MD. “Our work demonstrates that delirium also markedly increases the cost of elderly patient care while in the hospital. Given the high number of patients hospitalized every year with a hip fracture, there is a real need to develop and fund improved interventions to prevent in-hospital delirium in these patients.
“Our research suggests that reducing the rate of delirium would simultaneously increase the quality of care while decreasing costs, presenting hospitals, surgeons, and other stakeholders with promising opportunities to improve the value of hip fracture care,” said Dr. Zywiel.
The AAOS’s new clinical practice guideline, “Management of Hip Fractures in the Elderly,” makes a series of recommendations to reduce delirium in older hip fracture patients. They include:
• Preoperative regional analgesia to reduce pain.
• Hip fracture surgery within 48 hours of hospital admission.
• Intensive physical therapy following hospital discharge to improve functional outcomes.
• An osteoporosis evaluation, as well as vitamin D and calcium supplements, for patients following a hip fracture.
Running Barefoot May Increase Injury Risk in Older, More Experienced Athletes
LAS VEGAS─In recent years there has been an explosion in barefoot running, as well as the purchase and use of “minimalist” running shoes that more closely resemble barefoot running by encouraging the balls of the feet, between the arch and toes, to hit the pavement first. A new study presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS) found that a significant number of experienced runners, age 30 years and older (40% of men and 20% of women), maintained a heel-first running pattern—which naturally occurs when wearing a shoe with an elevated heel—when running without shoes. Maintaining a heel-toe pattern while running barefoot or in a minimalist shoe may lead to more frequent injuries.
“Previous studies have demonstrated that an adolescent runner’s foot strike is heavily influenced by their running shoe,” said orthopedic surgeon Scott Mullen, MD, the lead author of the study. “Young runners quickly adapt to a forefoot strike pattern when running barefoot, whereas a heel strike is normally associated with wearing large-heeled training shoes.”
In this study, a team of researchers from the University of Kansas Department of Orthopedics and Sports Medicine measured the heel-to-toe drop of 26 runners, all age 30 years or older with at least 10 years of running experience, when each ran in a traditional running shoe, and again when barefoot. The heel and forefoot thickness was measured at running speeds of 6, 7, and 8 miles per hour (mph) for women, and 7, 8, and 9 mph for men. A motion capture system was utilized to analyze foot strikes by a single blinded examiner skilled in the use of the camera system and running mechanics.
Heel-to-toe thickness of the running shoe did not significantly correlate with a change in heel strike, nor did alterations in speed. Running barefoot resulted in a significant drop in percent heel strike at all speeds; however, 40% of the men and 20% of the women persisted with consistent strike patterns across all speeds with and without shoes.
“Our study indicates that older runners (age 30 years and older) are not able to adapt as quickly to running barefoot,” said Dr. Mullen. “The inability to adapt the foot strike to the change in shoe type may put these runners at increased risk of injury. Older runners should be cautious when transitioning to a more minimalist type of shoe.”
LAS VEGAS─In recent years there has been an explosion in barefoot running, as well as the purchase and use of “minimalist” running shoes that more closely resemble barefoot running by encouraging the balls of the feet, between the arch and toes, to hit the pavement first. A new study presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS) found that a significant number of experienced runners, age 30 years and older (40% of men and 20% of women), maintained a heel-first running pattern—which naturally occurs when wearing a shoe with an elevated heel—when running without shoes. Maintaining a heel-toe pattern while running barefoot or in a minimalist shoe may lead to more frequent injuries.
“Previous studies have demonstrated that an adolescent runner’s foot strike is heavily influenced by their running shoe,” said orthopedic surgeon Scott Mullen, MD, the lead author of the study. “Young runners quickly adapt to a forefoot strike pattern when running barefoot, whereas a heel strike is normally associated with wearing large-heeled training shoes.”
In this study, a team of researchers from the University of Kansas Department of Orthopedics and Sports Medicine measured the heel-to-toe drop of 26 runners, all age 30 years or older with at least 10 years of running experience, when each ran in a traditional running shoe, and again when barefoot. The heel and forefoot thickness was measured at running speeds of 6, 7, and 8 miles per hour (mph) for women, and 7, 8, and 9 mph for men. A motion capture system was utilized to analyze foot strikes by a single blinded examiner skilled in the use of the camera system and running mechanics.
Heel-to-toe thickness of the running shoe did not significantly correlate with a change in heel strike, nor did alterations in speed. Running barefoot resulted in a significant drop in percent heel strike at all speeds; however, 40% of the men and 20% of the women persisted with consistent strike patterns across all speeds with and without shoes.
“Our study indicates that older runners (age 30 years and older) are not able to adapt as quickly to running barefoot,” said Dr. Mullen. “The inability to adapt the foot strike to the change in shoe type may put these runners at increased risk of injury. Older runners should be cautious when transitioning to a more minimalist type of shoe.”
LAS VEGAS─In recent years there has been an explosion in barefoot running, as well as the purchase and use of “minimalist” running shoes that more closely resemble barefoot running by encouraging the balls of the feet, between the arch and toes, to hit the pavement first. A new study presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS) found that a significant number of experienced runners, age 30 years and older (40% of men and 20% of women), maintained a heel-first running pattern—which naturally occurs when wearing a shoe with an elevated heel—when running without shoes. Maintaining a heel-toe pattern while running barefoot or in a minimalist shoe may lead to more frequent injuries.
“Previous studies have demonstrated that an adolescent runner’s foot strike is heavily influenced by their running shoe,” said orthopedic surgeon Scott Mullen, MD, the lead author of the study. “Young runners quickly adapt to a forefoot strike pattern when running barefoot, whereas a heel strike is normally associated with wearing large-heeled training shoes.”
In this study, a team of researchers from the University of Kansas Department of Orthopedics and Sports Medicine measured the heel-to-toe drop of 26 runners, all age 30 years or older with at least 10 years of running experience, when each ran in a traditional running shoe, and again when barefoot. The heel and forefoot thickness was measured at running speeds of 6, 7, and 8 miles per hour (mph) for women, and 7, 8, and 9 mph for men. A motion capture system was utilized to analyze foot strikes by a single blinded examiner skilled in the use of the camera system and running mechanics.
Heel-to-toe thickness of the running shoe did not significantly correlate with a change in heel strike, nor did alterations in speed. Running barefoot resulted in a significant drop in percent heel strike at all speeds; however, 40% of the men and 20% of the women persisted with consistent strike patterns across all speeds with and without shoes.
“Our study indicates that older runners (age 30 years and older) are not able to adapt as quickly to running barefoot,” said Dr. Mullen. “The inability to adapt the foot strike to the change in shoe type may put these runners at increased risk of injury. Older runners should be cautious when transitioning to a more minimalist type of shoe.”
Hip Replacement Patients May Safely Drive as Early As Two Weeks Following Surgery
LAS VEGAS—Improved surgical, pain management, and rehabilitation procedures can allow patients who undergo a total hip replacement (THR) to safely drive as early as 2 weeks following surgery, according to new research presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
Each year, more than 322,000 patients undergo hip replacement surgery in the United States. Previous studies, conducted more than a decade ago, recommended between 6 and 8 weeks of recovery before driving; however, recent advances in surgical treatment and care may have shortened this time frame. A shorter driving ban would allow patients to more quickly resume daily activities and return to work.
In this study, which appeared online November 2014 in the Journal of Arthroplasty, researchers evaluated 38 patients who underwent right THR between 2013 and 2014. Driving performance was evaluated using the Brake Reaction Test (BRT), which measures brake time reaction after a stimulus. All patients underwent preoperative assessment to establish a baseline reaction time, and then agreed to be retested at 2, 4, and 6 weeks after surgery. Patients were allowed to drive when their postoperative reaction time was equal to or less than their preoperative baseline reaction time. At each testing session patients were asked if they felt ready to drive again.
Of the 38 patients, 33 (87%) reached their baseline time within 2 weeks. The remaining patients (13%) reached their baseline at 4 weeks. Among the other findings of the study:
• There were no differences with respect to age, gender, or the use of assistance devices in terms of driving readiness.
• Of the 33 patients who tested ready to drive at 2 weeks, 24 (73%) stated that they felt ready to drive while 5 (15%) were not sure. Four patients (12%) reported that they did not feel ready to drive.
• Of the 5 patients who returned to driving at 4 weeks, 3 agreed that they were not able to drive at the 2-week mark, and the other 2 thought they were able to drive by 2 weeks.
“We found that brake reaction time returned to baseline or better in the vast majority of patients undergoing contemporary THR by 2 weeks following surgery, and all patients achieved a safe brake reaction time according to nationally recognized guidelines,” said lead study author and orthopedic surgeon Victor Hugo Hernandez, MD.
Dr. Hernandez said the “findings have allowed us to encourage patients to re-evaluate their driving ability as soon as 2 weeks after THR,” but warned that the study results “are based on our particular population, and caution should be taken in translating these results to the regular population.” In addition, patients should never drive if they are still taking narcotic pain medication.
LAS VEGAS—Improved surgical, pain management, and rehabilitation procedures can allow patients who undergo a total hip replacement (THR) to safely drive as early as 2 weeks following surgery, according to new research presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
Each year, more than 322,000 patients undergo hip replacement surgery in the United States. Previous studies, conducted more than a decade ago, recommended between 6 and 8 weeks of recovery before driving; however, recent advances in surgical treatment and care may have shortened this time frame. A shorter driving ban would allow patients to more quickly resume daily activities and return to work.
In this study, which appeared online November 2014 in the Journal of Arthroplasty, researchers evaluated 38 patients who underwent right THR between 2013 and 2014. Driving performance was evaluated using the Brake Reaction Test (BRT), which measures brake time reaction after a stimulus. All patients underwent preoperative assessment to establish a baseline reaction time, and then agreed to be retested at 2, 4, and 6 weeks after surgery. Patients were allowed to drive when their postoperative reaction time was equal to or less than their preoperative baseline reaction time. At each testing session patients were asked if they felt ready to drive again.
Of the 38 patients, 33 (87%) reached their baseline time within 2 weeks. The remaining patients (13%) reached their baseline at 4 weeks. Among the other findings of the study:
• There were no differences with respect to age, gender, or the use of assistance devices in terms of driving readiness.
• Of the 33 patients who tested ready to drive at 2 weeks, 24 (73%) stated that they felt ready to drive while 5 (15%) were not sure. Four patients (12%) reported that they did not feel ready to drive.
• Of the 5 patients who returned to driving at 4 weeks, 3 agreed that they were not able to drive at the 2-week mark, and the other 2 thought they were able to drive by 2 weeks.
“We found that brake reaction time returned to baseline or better in the vast majority of patients undergoing contemporary THR by 2 weeks following surgery, and all patients achieved a safe brake reaction time according to nationally recognized guidelines,” said lead study author and orthopedic surgeon Victor Hugo Hernandez, MD.
Dr. Hernandez said the “findings have allowed us to encourage patients to re-evaluate their driving ability as soon as 2 weeks after THR,” but warned that the study results “are based on our particular population, and caution should be taken in translating these results to the regular population.” In addition, patients should never drive if they are still taking narcotic pain medication.
LAS VEGAS—Improved surgical, pain management, and rehabilitation procedures can allow patients who undergo a total hip replacement (THR) to safely drive as early as 2 weeks following surgery, according to new research presented at the 2015 Annual Meeting of the American Academy of Orthopaedic Surgeons (AAOS).
Each year, more than 322,000 patients undergo hip replacement surgery in the United States. Previous studies, conducted more than a decade ago, recommended between 6 and 8 weeks of recovery before driving; however, recent advances in surgical treatment and care may have shortened this time frame. A shorter driving ban would allow patients to more quickly resume daily activities and return to work.
In this study, which appeared online November 2014 in the Journal of Arthroplasty, researchers evaluated 38 patients who underwent right THR between 2013 and 2014. Driving performance was evaluated using the Brake Reaction Test (BRT), which measures brake time reaction after a stimulus. All patients underwent preoperative assessment to establish a baseline reaction time, and then agreed to be retested at 2, 4, and 6 weeks after surgery. Patients were allowed to drive when their postoperative reaction time was equal to or less than their preoperative baseline reaction time. At each testing session patients were asked if they felt ready to drive again.
Of the 38 patients, 33 (87%) reached their baseline time within 2 weeks. The remaining patients (13%) reached their baseline at 4 weeks. Among the other findings of the study:
• There were no differences with respect to age, gender, or the use of assistance devices in terms of driving readiness.
• Of the 33 patients who tested ready to drive at 2 weeks, 24 (73%) stated that they felt ready to drive while 5 (15%) were not sure. Four patients (12%) reported that they did not feel ready to drive.
• Of the 5 patients who returned to driving at 4 weeks, 3 agreed that they were not able to drive at the 2-week mark, and the other 2 thought they were able to drive by 2 weeks.
“We found that brake reaction time returned to baseline or better in the vast majority of patients undergoing contemporary THR by 2 weeks following surgery, and all patients achieved a safe brake reaction time according to nationally recognized guidelines,” said lead study author and orthopedic surgeon Victor Hugo Hernandez, MD.
Dr. Hernandez said the “findings have allowed us to encourage patients to re-evaluate their driving ability as soon as 2 weeks after THR,” but warned that the study results “are based on our particular population, and caution should be taken in translating these results to the regular population.” In addition, patients should never drive if they are still taking narcotic pain medication.