User login
Teaching Value Project, Choosing Wisely Competition Accepting Applications for 2015
Costs of Care (www.costsofcare.org), a nonprofit dedicated to empowering patients and their caregivers to deflate medical bills, and the American Board of Internal Medicine (ABIM) Foundation plan to launch their second annual Teaching Value and Choosing Wisely Competition this fall to highlight healthcare innovations promoting value. The submission deadline is Feb. 1, 2015.
The Teaching Value Project, the main educational arm of Costs of Care, has developed web-based video training modules based on actual cases, along with other educational materials to help residents and medical students learn to make clinical decisions optimizing quality and cost, says Christopher Moriates, MD, hospitalist and co-chair of the University of California-San Francisco’s High-Value Care Committee.
Details of the contest will be posted at www.teachingvalue.org. For more information, send an e-mail to [email protected].
Larry Beresford is a freelance writer in Alameda, Calif.
Costs of Care (www.costsofcare.org), a nonprofit dedicated to empowering patients and their caregivers to deflate medical bills, and the American Board of Internal Medicine (ABIM) Foundation plan to launch their second annual Teaching Value and Choosing Wisely Competition this fall to highlight healthcare innovations promoting value. The submission deadline is Feb. 1, 2015.
The Teaching Value Project, the main educational arm of Costs of Care, has developed web-based video training modules based on actual cases, along with other educational materials to help residents and medical students learn to make clinical decisions optimizing quality and cost, says Christopher Moriates, MD, hospitalist and co-chair of the University of California-San Francisco’s High-Value Care Committee.
Details of the contest will be posted at www.teachingvalue.org. For more information, send an e-mail to [email protected].
Larry Beresford is a freelance writer in Alameda, Calif.
Costs of Care (www.costsofcare.org), a nonprofit dedicated to empowering patients and their caregivers to deflate medical bills, and the American Board of Internal Medicine (ABIM) Foundation plan to launch their second annual Teaching Value and Choosing Wisely Competition this fall to highlight healthcare innovations promoting value. The submission deadline is Feb. 1, 2015.
The Teaching Value Project, the main educational arm of Costs of Care, has developed web-based video training modules based on actual cases, along with other educational materials to help residents and medical students learn to make clinical decisions optimizing quality and cost, says Christopher Moriates, MD, hospitalist and co-chair of the University of California-San Francisco’s High-Value Care Committee.
Details of the contest will be posted at www.teachingvalue.org. For more information, send an e-mail to [email protected].
Larry Beresford is a freelance writer in Alameda, Calif.
Nonclinical Factors Influence Hospital Readmissions

The role of nonclinical factors in shaping rates of rehospitalization has been explored in several recent studies—and targeted through new legislation endorsed by the Society of Hospital Medicine. A study in Health Affairs compared hospital performance on 30-day readmissions for the first three diagnoses included in penalty calculations for CMS’ Hospital Readmissions Reduction Program (HRRP) and found that adjusting for patients’ socioeconomic status significantly reduced the rates of variation in readmissions between hospitals across the state of Missouri.1
For patients discharged between 2009 and 2012, analysis using a model enriched with census tract socioeconomic data found that the range of variation in readmissions between hospitals decreased to 1.8% from 6.5% for patients with acute myocardial infarction; to 7.4% from 14.0% for congestive heart failure; and to 3.7% from 7.4% for pneumonia, compared with rates unadjusted for these socioeconomic factors. Another study in the same journal by researchers at an urban teaching hospital found that patients living in high-poverty neighborhoods were 24% more likely to be readmitted to the hospital within 30 days, after adjusting for demographic and clinical characteristics.2
For a factor that may be more amenable to intervention by hospitalists, a standardized rehabilitation medicine test measuring patients’ ability to perform everyday tasks of living, such as the ability to move independently from bed to chair, wheelchair, or toilet was found to be a good predictor of readmissions.3 Few hospitals currently require assessment of their patients’ functional ability, notes the study’s lead author Erik Hoyer, MD, assistant professor in the department of physical medicine and rehabilitation at the Johns Hopkins University School of Medicine in Baltimore. But the score “is a direct reflection of the patient’s ability to heal [outside of the hospital].”
The Functional Independence Measure used in this study and in inpatient rehabilitation facilities nationwide is probably not the right tool for hospitalists because of its length and the training required to administer it, Dr. Hoyer says.
“There are other, easier tools that are available or in development that may also serve a similar purpose,” he says. “The main point is that routine functional assessment is important in the hospital setting, and developing strategies to improve patient function is likely an important way to improve outcomes such as hospital readmissions.”
The documented role of socioeconomic status in determining readmissions also is addressed by legislation introduced by Rep. Jim Renacci (R-Ohio) and supported by both the Society of Hospital Medicine and the American Hospital Association. The Establishing Beneficiary Equity in the Hospital Readmission Program Act (HR-4188) would adjust HRRP readmissions penalties to reflect “certain socioeconomic and health factors that increase the patient’s risk of readmissions.”
Larry Beresford is a freelance writer in Alameda, Calif.
The role of nonclinical factors in shaping rates of rehospitalization has been explored in several recent studies—and targeted through new legislation endorsed by the Society of Hospital Medicine. A study in Health Affairs compared hospital performance on 30-day readmissions for the first three diagnoses included in penalty calculations for CMS’ Hospital Readmissions Reduction Program (HRRP) and found that adjusting for patients’ socioeconomic status significantly reduced the rates of variation in readmissions between hospitals across the state of Missouri.1
For patients discharged between 2009 and 2012, analysis using a model enriched with census tract socioeconomic data found that the range of variation in readmissions between hospitals decreased to 1.8% from 6.5% for patients with acute myocardial infarction; to 7.4% from 14.0% for congestive heart failure; and to 3.7% from 7.4% for pneumonia, compared with rates unadjusted for these socioeconomic factors. Another study in the same journal by researchers at an urban teaching hospital found that patients living in high-poverty neighborhoods were 24% more likely to be readmitted to the hospital within 30 days, after adjusting for demographic and clinical characteristics.2
For a factor that may be more amenable to intervention by hospitalists, a standardized rehabilitation medicine test measuring patients’ ability to perform everyday tasks of living, such as the ability to move independently from bed to chair, wheelchair, or toilet was found to be a good predictor of readmissions.3 Few hospitals currently require assessment of their patients’ functional ability, notes the study’s lead author Erik Hoyer, MD, assistant professor in the department of physical medicine and rehabilitation at the Johns Hopkins University School of Medicine in Baltimore. But the score “is a direct reflection of the patient’s ability to heal [outside of the hospital].”
The Functional Independence Measure used in this study and in inpatient rehabilitation facilities nationwide is probably not the right tool for hospitalists because of its length and the training required to administer it, Dr. Hoyer says.
“There are other, easier tools that are available or in development that may also serve a similar purpose,” he says. “The main point is that routine functional assessment is important in the hospital setting, and developing strategies to improve patient function is likely an important way to improve outcomes such as hospital readmissions.”
The documented role of socioeconomic status in determining readmissions also is addressed by legislation introduced by Rep. Jim Renacci (R-Ohio) and supported by both the Society of Hospital Medicine and the American Hospital Association. The Establishing Beneficiary Equity in the Hospital Readmission Program Act (HR-4188) would adjust HRRP readmissions penalties to reflect “certain socioeconomic and health factors that increase the patient’s risk of readmissions.”
Larry Beresford is a freelance writer in Alameda, Calif.
The role of nonclinical factors in shaping rates of rehospitalization has been explored in several recent studies—and targeted through new legislation endorsed by the Society of Hospital Medicine. A study in Health Affairs compared hospital performance on 30-day readmissions for the first three diagnoses included in penalty calculations for CMS’ Hospital Readmissions Reduction Program (HRRP) and found that adjusting for patients’ socioeconomic status significantly reduced the rates of variation in readmissions between hospitals across the state of Missouri.1
For patients discharged between 2009 and 2012, analysis using a model enriched with census tract socioeconomic data found that the range of variation in readmissions between hospitals decreased to 1.8% from 6.5% for patients with acute myocardial infarction; to 7.4% from 14.0% for congestive heart failure; and to 3.7% from 7.4% for pneumonia, compared with rates unadjusted for these socioeconomic factors. Another study in the same journal by researchers at an urban teaching hospital found that patients living in high-poverty neighborhoods were 24% more likely to be readmitted to the hospital within 30 days, after adjusting for demographic and clinical characteristics.2
For a factor that may be more amenable to intervention by hospitalists, a standardized rehabilitation medicine test measuring patients’ ability to perform everyday tasks of living, such as the ability to move independently from bed to chair, wheelchair, or toilet was found to be a good predictor of readmissions.3 Few hospitals currently require assessment of their patients’ functional ability, notes the study’s lead author Erik Hoyer, MD, assistant professor in the department of physical medicine and rehabilitation at the Johns Hopkins University School of Medicine in Baltimore. But the score “is a direct reflection of the patient’s ability to heal [outside of the hospital].”
The Functional Independence Measure used in this study and in inpatient rehabilitation facilities nationwide is probably not the right tool for hospitalists because of its length and the training required to administer it, Dr. Hoyer says.
“There are other, easier tools that are available or in development that may also serve a similar purpose,” he says. “The main point is that routine functional assessment is important in the hospital setting, and developing strategies to improve patient function is likely an important way to improve outcomes such as hospital readmissions.”
The documented role of socioeconomic status in determining readmissions also is addressed by legislation introduced by Rep. Jim Renacci (R-Ohio) and supported by both the Society of Hospital Medicine and the American Hospital Association. The Establishing Beneficiary Equity in the Hospital Readmission Program Act (HR-4188) would adjust HRRP readmissions penalties to reflect “certain socioeconomic and health factors that increase the patient’s risk of readmissions.”
Larry Beresford is a freelance writer in Alameda, Calif.
Once-Weekly Antibiotic Might Be Effective for Treatment of Acute Bacterial Skin Infections
Clinical question: Is once-weekly intravenous dalbavancin as effective as conventional therapy for the treatment of acute bacterial skin infections?
Background: Acute bacterial skin infections are common and often require hospitalization for intravenous antibiotic administration. Treatment covering gram-positive bacteria usually is indicated. Dalbavancin is effective against gram-positives, including MRSA. Its long half-life makes it an attractive alternative to other commonly used antibiotics, which require more frequent dosing.
Study design: Phase 3, double-blinded RCT.
Setting: Multiple international centers.
Synopsis: Researchers randomized 1,312 patients with acute bacterial skin and skin-structure infections with signs of systemic infection requiring intravenous antibiotics to receive dalbavancin on days one and eight, with placebo on other days, or several doses of vancomycin with an option to switch to oral linezolid. The primary endpoint was cessation of spread of erythema and temperature of ≤37.6°C at 48-72 hours. Secondary endpoints included a decrease in lesion area of ≥20% at 48-72 hours and clinical success at end of therapy (determined by clinical and historical features).
Results of the primary endpoint were similar with dalbavancin and vancomycin-linezolid groups (79.7% and 79.8%, respectively) and were within 10 percentage points of noninferiority. The secondary endpoints were similar between both groups.
Limitations of the study were the early primary endpoint, lack of noninferiority analysis of the secondary endpoints, and cost-effective analysis.
Bottom line: Once-weekly dalbavancin appears to be similarly efficacious to intravenous vancomycin in the treatment of acute bacterial skin infections in terms of outcomes within 48-72 hours of therapy and might provide an alternative to continued inpatient hospitalization for intravenous antibiotics in stable patients.
Citation: Boucher HW, Wilcox M, Talbot GH, Puttagunta S, Das AF, Dunne MW. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370(23):2169-2179.
Clinical question: Is once-weekly intravenous dalbavancin as effective as conventional therapy for the treatment of acute bacterial skin infections?
Background: Acute bacterial skin infections are common and often require hospitalization for intravenous antibiotic administration. Treatment covering gram-positive bacteria usually is indicated. Dalbavancin is effective against gram-positives, including MRSA. Its long half-life makes it an attractive alternative to other commonly used antibiotics, which require more frequent dosing.
Study design: Phase 3, double-blinded RCT.
Setting: Multiple international centers.
Synopsis: Researchers randomized 1,312 patients with acute bacterial skin and skin-structure infections with signs of systemic infection requiring intravenous antibiotics to receive dalbavancin on days one and eight, with placebo on other days, or several doses of vancomycin with an option to switch to oral linezolid. The primary endpoint was cessation of spread of erythema and temperature of ≤37.6°C at 48-72 hours. Secondary endpoints included a decrease in lesion area of ≥20% at 48-72 hours and clinical success at end of therapy (determined by clinical and historical features).
Results of the primary endpoint were similar with dalbavancin and vancomycin-linezolid groups (79.7% and 79.8%, respectively) and were within 10 percentage points of noninferiority. The secondary endpoints were similar between both groups.
Limitations of the study were the early primary endpoint, lack of noninferiority analysis of the secondary endpoints, and cost-effective analysis.
Bottom line: Once-weekly dalbavancin appears to be similarly efficacious to intravenous vancomycin in the treatment of acute bacterial skin infections in terms of outcomes within 48-72 hours of therapy and might provide an alternative to continued inpatient hospitalization for intravenous antibiotics in stable patients.
Citation: Boucher HW, Wilcox M, Talbot GH, Puttagunta S, Das AF, Dunne MW. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370(23):2169-2179.
Clinical question: Is once-weekly intravenous dalbavancin as effective as conventional therapy for the treatment of acute bacterial skin infections?
Background: Acute bacterial skin infections are common and often require hospitalization for intravenous antibiotic administration. Treatment covering gram-positive bacteria usually is indicated. Dalbavancin is effective against gram-positives, including MRSA. Its long half-life makes it an attractive alternative to other commonly used antibiotics, which require more frequent dosing.
Study design: Phase 3, double-blinded RCT.
Setting: Multiple international centers.
Synopsis: Researchers randomized 1,312 patients with acute bacterial skin and skin-structure infections with signs of systemic infection requiring intravenous antibiotics to receive dalbavancin on days one and eight, with placebo on other days, or several doses of vancomycin with an option to switch to oral linezolid. The primary endpoint was cessation of spread of erythema and temperature of ≤37.6°C at 48-72 hours. Secondary endpoints included a decrease in lesion area of ≥20% at 48-72 hours and clinical success at end of therapy (determined by clinical and historical features).
Results of the primary endpoint were similar with dalbavancin and vancomycin-linezolid groups (79.7% and 79.8%, respectively) and were within 10 percentage points of noninferiority. The secondary endpoints were similar between both groups.
Limitations of the study were the early primary endpoint, lack of noninferiority analysis of the secondary endpoints, and cost-effective analysis.
Bottom line: Once-weekly dalbavancin appears to be similarly efficacious to intravenous vancomycin in the treatment of acute bacterial skin infections in terms of outcomes within 48-72 hours of therapy and might provide an alternative to continued inpatient hospitalization for intravenous antibiotics in stable patients.
Citation: Boucher HW, Wilcox M, Talbot GH, Puttagunta S, Das AF, Dunne MW. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370(23):2169-2179.
Continuous Positive Airway Pressure Outperforms Noctural Oxygen for Blood Pressure Reduction
Clinical question: What is the effect of continuous positive airway pressure (CPAP) or supplemental oxygen on ambulatory blood pressures and markers of cardiovascular risk when combined with sleep hygiene education in patients with obstructive sleep apnea (OSA) and coronary artery disease or cardiac risk factors?
Background: OSA is considered a risk factor for the development of hypertension. One meta-analysis showed reduction of mean arterial pressure (MAP) with CPAP therapy, but randomized controlled data on blood pressure reduction with treatment of OSA is lacking.
Study design: Randomized, parallel-group trial.
Setting: Four outpatient cardiology practices.
Synopsis: Patients ages 45-75 with OSA were randomized to receive nocturnal CPAP and healthy lifestyle and sleep education (HLSE), nocturnal oxygen therapy and HSLE, or HSLE alone. The primary outcome was 24-hour MAP. Secondary outcomes included fasting blood glucose, lipid panel, insulin level, erythrocyte sedimentation rate, C-reactive protein (CRP), and N-terminal pro-brain naturetic peptide.
Participants had high rates of diabetes, hypertension, and coronary artery disease. At 12 weeks, the CPAP arm experienced greater reductions in 24-hour MAP compared to both the nocturnal oxygen and HSLE arms (-2.8 mmHg [P=0.02] and -2.4 mmHg [P=0.04], respectively). No significant decrease in MAP was identified in the nocturnal oxygen arm when compared to the HSLE arm. The only significant difference in secondary outcomes was a decrease in CRP in the CPAP arm when compared to the HSLE arm, the clinical significance of which is unclear.
Bottom line: CPAP therapy with sleep hygiene education appears superior to nocturnal oxygen therapy with sleep hygiene education and sleep hygiene education alone in decreasing 24-hour MAP in patients with OSA and coronary artery disease or cardiac risk factors.
Citation: Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med. 2014;370(24):2276-2285.
Clinical question: What is the effect of continuous positive airway pressure (CPAP) or supplemental oxygen on ambulatory blood pressures and markers of cardiovascular risk when combined with sleep hygiene education in patients with obstructive sleep apnea (OSA) and coronary artery disease or cardiac risk factors?
Background: OSA is considered a risk factor for the development of hypertension. One meta-analysis showed reduction of mean arterial pressure (MAP) with CPAP therapy, but randomized controlled data on blood pressure reduction with treatment of OSA is lacking.
Study design: Randomized, parallel-group trial.
Setting: Four outpatient cardiology practices.
Synopsis: Patients ages 45-75 with OSA were randomized to receive nocturnal CPAP and healthy lifestyle and sleep education (HLSE), nocturnal oxygen therapy and HSLE, or HSLE alone. The primary outcome was 24-hour MAP. Secondary outcomes included fasting blood glucose, lipid panel, insulin level, erythrocyte sedimentation rate, C-reactive protein (CRP), and N-terminal pro-brain naturetic peptide.
Participants had high rates of diabetes, hypertension, and coronary artery disease. At 12 weeks, the CPAP arm experienced greater reductions in 24-hour MAP compared to both the nocturnal oxygen and HSLE arms (-2.8 mmHg [P=0.02] and -2.4 mmHg [P=0.04], respectively). No significant decrease in MAP was identified in the nocturnal oxygen arm when compared to the HSLE arm. The only significant difference in secondary outcomes was a decrease in CRP in the CPAP arm when compared to the HSLE arm, the clinical significance of which is unclear.
Bottom line: CPAP therapy with sleep hygiene education appears superior to nocturnal oxygen therapy with sleep hygiene education and sleep hygiene education alone in decreasing 24-hour MAP in patients with OSA and coronary artery disease or cardiac risk factors.
Citation: Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med. 2014;370(24):2276-2285.
Clinical question: What is the effect of continuous positive airway pressure (CPAP) or supplemental oxygen on ambulatory blood pressures and markers of cardiovascular risk when combined with sleep hygiene education in patients with obstructive sleep apnea (OSA) and coronary artery disease or cardiac risk factors?
Background: OSA is considered a risk factor for the development of hypertension. One meta-analysis showed reduction of mean arterial pressure (MAP) with CPAP therapy, but randomized controlled data on blood pressure reduction with treatment of OSA is lacking.
Study design: Randomized, parallel-group trial.
Setting: Four outpatient cardiology practices.
Synopsis: Patients ages 45-75 with OSA were randomized to receive nocturnal CPAP and healthy lifestyle and sleep education (HLSE), nocturnal oxygen therapy and HSLE, or HSLE alone. The primary outcome was 24-hour MAP. Secondary outcomes included fasting blood glucose, lipid panel, insulin level, erythrocyte sedimentation rate, C-reactive protein (CRP), and N-terminal pro-brain naturetic peptide.
Participants had high rates of diabetes, hypertension, and coronary artery disease. At 12 weeks, the CPAP arm experienced greater reductions in 24-hour MAP compared to both the nocturnal oxygen and HSLE arms (-2.8 mmHg [P=0.02] and -2.4 mmHg [P=0.04], respectively). No significant decrease in MAP was identified in the nocturnal oxygen arm when compared to the HSLE arm. The only significant difference in secondary outcomes was a decrease in CRP in the CPAP arm when compared to the HSLE arm, the clinical significance of which is unclear.
Bottom line: CPAP therapy with sleep hygiene education appears superior to nocturnal oxygen therapy with sleep hygiene education and sleep hygiene education alone in decreasing 24-hour MAP in patients with OSA and coronary artery disease or cardiac risk factors.
Citation: Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med. 2014;370(24):2276-2285.
Inflammatory rheumatic diseases raise venous thromboembolism risk
Individuals with inflammatory rheumatic diseases such as inflammatory arthritis, vasculitis, and connective tissue diseases, have a threefold increase in the risk of venous thromboembolism, compared with the general population, according to a meta-analysis.
The meta-analysis of 25 studies – 10 of which included patients with rheumatoid arthritis (RA) – found those with RA were more than twice as likely to develop deep vein thrombosis or a pulmonary embolism, compared with an age- and sex-matched individuals who had other comorbidities such as diabetes, peripheral vascular disease/coronary artery disease, and malignancy (OR, 2.23; 95% confidence interval, 2.02-2.47). The RA patients had a cumulative venous thromboembolism (VTE) incidence of 2.18% (Arthritis Res. Ther. 2014;16:435 [doi:10.1186/s13075-014-0435-y]).

Ten studies comprising 54,697 patients with systemic lupus erythematosus showed a cumulative thrombosis incidence of 7.29% (95% CI, 5.82%-8.75%). Other diseases for which the investigators calculated cumulative incidence rates of VTE, based on four studies apiece, were Sjögren’s syndrome (2.18%; 95% CI, 0.79%-3.57%), inflammatory myositis (4.03%; 95% CI, 2.38%-5.67%), Antineutrophil cytoplasmic antibody vasculitis (7.97%; 95% CI, 5.67%-10.28%), and systemic sclerosis (3.13%; 95% CI, 1.73%-4.52%).
“We believe that the increased VTE risk is associated with the activity of the inflammatory diseases, rather than with the treatments used for controlling the disease,” wrote Dr. Jason Lee of the University of Western Ontario, London, and Dr. Janet Pope, of the division of rheumatology at St. Joseph’s Health Care, London, Ont.
The authors said that they had no conflicts of interest.
Individuals with inflammatory rheumatic diseases such as inflammatory arthritis, vasculitis, and connective tissue diseases, have a threefold increase in the risk of venous thromboembolism, compared with the general population, according to a meta-analysis.
The meta-analysis of 25 studies – 10 of which included patients with rheumatoid arthritis (RA) – found those with RA were more than twice as likely to develop deep vein thrombosis or a pulmonary embolism, compared with an age- and sex-matched individuals who had other comorbidities such as diabetes, peripheral vascular disease/coronary artery disease, and malignancy (OR, 2.23; 95% confidence interval, 2.02-2.47). The RA patients had a cumulative venous thromboembolism (VTE) incidence of 2.18% (Arthritis Res. Ther. 2014;16:435 [doi:10.1186/s13075-014-0435-y]).
Ten studies comprising 54,697 patients with systemic lupus erythematosus showed a cumulative thrombosis incidence of 7.29% (95% CI, 5.82%-8.75%). Other diseases for which the investigators calculated cumulative incidence rates of VTE, based on four studies apiece, were Sjögren’s syndrome (2.18%; 95% CI, 0.79%-3.57%), inflammatory myositis (4.03%; 95% CI, 2.38%-5.67%), Antineutrophil cytoplasmic antibody vasculitis (7.97%; 95% CI, 5.67%-10.28%), and systemic sclerosis (3.13%; 95% CI, 1.73%-4.52%).
“We believe that the increased VTE risk is associated with the activity of the inflammatory diseases, rather than with the treatments used for controlling the disease,” wrote Dr. Jason Lee of the University of Western Ontario, London, and Dr. Janet Pope, of the division of rheumatology at St. Joseph’s Health Care, London, Ont.
The authors said that they had no conflicts of interest.
Individuals with inflammatory rheumatic diseases such as inflammatory arthritis, vasculitis, and connective tissue diseases, have a threefold increase in the risk of venous thromboembolism, compared with the general population, according to a meta-analysis.
The meta-analysis of 25 studies – 10 of which included patients with rheumatoid arthritis (RA) – found those with RA were more than twice as likely to develop deep vein thrombosis or a pulmonary embolism, compared with an age- and sex-matched individuals who had other comorbidities such as diabetes, peripheral vascular disease/coronary artery disease, and malignancy (OR, 2.23; 95% confidence interval, 2.02-2.47). The RA patients had a cumulative venous thromboembolism (VTE) incidence of 2.18% (Arthritis Res. Ther. 2014;16:435 [doi:10.1186/s13075-014-0435-y]).
Ten studies comprising 54,697 patients with systemic lupus erythematosus showed a cumulative thrombosis incidence of 7.29% (95% CI, 5.82%-8.75%). Other diseases for which the investigators calculated cumulative incidence rates of VTE, based on four studies apiece, were Sjögren’s syndrome (2.18%; 95% CI, 0.79%-3.57%), inflammatory myositis (4.03%; 95% CI, 2.38%-5.67%), Antineutrophil cytoplasmic antibody vasculitis (7.97%; 95% CI, 5.67%-10.28%), and systemic sclerosis (3.13%; 95% CI, 1.73%-4.52%).
“We believe that the increased VTE risk is associated with the activity of the inflammatory diseases, rather than with the treatments used for controlling the disease,” wrote Dr. Jason Lee of the University of Western Ontario, London, and Dr. Janet Pope, of the division of rheumatology at St. Joseph’s Health Care, London, Ont.
The authors said that they had no conflicts of interest.
FROM ARTHRITIS RESEARCH & THERAPY
Key clinical point: There is strong evidence for an elevated baseline risk of VTE in patients with inflammatory rheumatic diseases.
Major finding: Patients with rheumatoid arthritis are more than twice as likely to develop deep vein thrombosis or a pulmonary embolism, compared with an age- and sex-matched patients.
Data source: Meta-analysis of 25 studies.
Disclosures: No conflicts of interest were declared.

Primary Care Medical Services for Homeless Veterans
On a single night in 2012, 62,619 veterans experienced homelessness.1 With high rates of illness, as well as alcohol, tobacco, and other drug (ATOD) use among homeless adults, ending veteran homelessness is a signature initiative of the VA.2-4 However, despite increasing efforts to improve health care for homeless individuals, little is known about best practices in homeless-focused primary care.2,5
With an age-adjusted mortality that is 2 to 10 times higher than that of their housed counterparts, homeless veterans pose a formidable challenge for primary care providers (PCPs).6,7 The complexity of homeless persons’ primary care needs is compounded by poor social support and the need to navigate priorities (eg, shelter) that compete with medical care.6,8,9 Moreover, veterans may face unique vulnerabilities conferred by military-specific experiences.2,10
As of 2012, only 2 VA facilities had primary care clinics tailored to the needs of homeless veterans. McGuire and colleagues built a system of colocated primary care, mental health, and homeless services for mentally ill veterans in Los Angeles, California.11 Over 18 months, this clinic facilitated greater primary and preventive care delivery, though the population’s physical health status did not improve.11 More recently, O’Toole and colleagues implemented a homeless-focused primary care clinic in Providence, Rhode Island.6 Compared with a historical sample of homeless veterans in traditional VA primary care, veterans in this homeless-tailored clinic had greater improvements in some chronic disease outcomes.6 This clinic also decreased nonacute emergency department (ED) use and hospitalizations for general medical conditions.6
Despite these promising outcomes, the VA lacked a nationwide homeless-focused primary care initiative. In 2012 the VA Office of Homeless Programs and the Office of Primary Care Operations funded a national demonstration project to create Homeless Patient-Aligned Care Teams (HPACTs)—primary care medical clinics for homeless veterans—at 32 facilities. This demonstration project guided HPACTs to tailor clinical and social services to homeless veterans’ needs, establish processes to identify and refer appropriate veterans, and integrate distinct services.
There were no explicit instructions that detailed HPACT structure. Because new VA programs must fit local contextual factors, including infrastructure, space, personnel, and institutional/community resources, different models of homeless-focused primary care have evolved.
This article is a case study of HPACTs at 3 of the 32 participating VA facilities, each reflecting a distinct community and organizational context. In light of projected HPACT expansion and concerns that current services are better tailored to sheltered homeless veterans than to their unsheltered peers, there is particular importance to detailed clinic descriptions that vividly portray the intricate relationships between service design and populations served.1
METHODS
VA HPACTs established in May 2012 at 3 facilities were examined: Birmingham VAMC in Alabama (BIR), West Los Angeles VAMC in California (WLA), and VA Pittsburgh Healthcare System in Pennsylvania (PIT). Prior to this demonstration project, each facility offered a range of housing/social services and traditional primary care for veterans. These sites are a geographically diverse convenience sample that emerged from existing homeless-focused collaborations among the authors and represent geographically diverse HPACTs.
The national director of VA Homeless Programs formally determined that this comparison constitutes a VA operations activity that is not research.12 This activity was exempt from Institutional Review Board review.
Study Design
Timed at an early stage of HPACT implementation, this project had 3 aims: (1) To identify noteworthy similarities and/or differences among the initial HPACT clinic structures; (2) To compare and contrast the patient characteristics of veterans enrolled in each of these clinics; and (3) To use these data to inform ongoing HPACT service design.
HPACT program evaluation data are not presented. Rather, a nascent system of care is illustrated that contributes to the limited literature concerning the design and implementation of homeless-focused primary care. Such organizational profiles inform novel program delivery and hold particular utility for heterogeneous populations who are difficult to engage in care.13,14
Authors at each site independently developed lists of variables that fell within the 3 guiding principles of this demonstration project. These variables were compiled and iteratively reduced to a consolidated table that assessed each clinic, including location, operating hours, methods of patient identification and referral, and linkages to distinct services (eg, primary care, mental health, addiction, and social services).
This table also became a guide for HPACT directors to generate narrative clinic descriptions. Characteristics of VA medical homes that are embraced regardless of patients’ housing status (eg, patient-centered, team-based care) were also incorporated into these descriptions.15
Patient Characteristics
The VA electronic health record (EHR) was used to identify all patients enrolled in HPACTs at BIR, WLA, and PIT from May 1, 2012 (clinic inception), through September 30, 2012. Authors developed a standardized template for EHR review and coined the first HPACT record as the patient’s index visit. This record was used to code initial housing status, demographics, and acute medical conditions diagnosed/treated. If housing status was not recorded at the index visit, the first preceding informative record was used.
Records for 6 months preceding the index visit were used to identify the presence or absence of common medical conditions, psychiatric diagnoses, and ATOD abuse or dependence. Medical conditions were identified from a list of common outpatient diagnoses from the National Ambulatory Medical Care Survey, supplemented by common conditions among homeless men.16-18 The EHR problem list (a list of diagnoses, by patient) was used to obtain diagnoses, supplemented by notes from inpatient admissions/discharges, as well as ED, primary care, and subspecialty consultations within 6 months preceding the index visit. Prior VA health care use was ascertained from the EHR review of the same 6-month window, reflecting ED visits, inpatient admissions, primary care visits, subspecialty visits, and individual/group therapy for ATOD or other mental health problems. Data were used to generate site-specific descriptive statistics.
RESULTS
Like all VA hospital-based clinics, a universal EHR captured all medical and social service notes, orders, medications, and administrative records. All facilities had on-site EDs, medical/mental health specialty care, and pharmacies. Social services, including benefits counseling and housing services, were available on site. Table 1 summarizes the HPACT structures at BIR, WLA, and PIT.
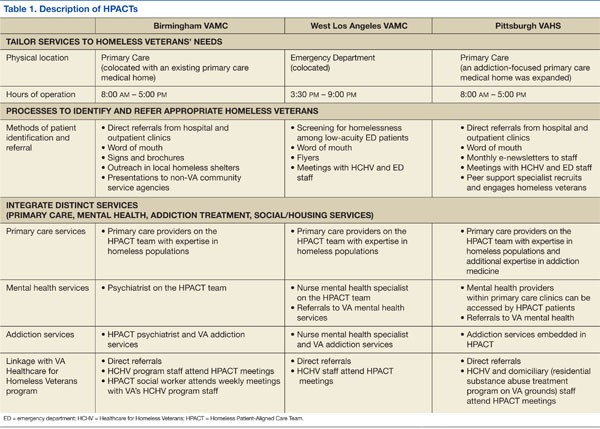
Birmingham
The BIR HPACT was devised as a new homeless-focused team located within a VA primary care clinic where other providers continued to see primary care patients. Staff and space were reallocated for this HPACT, which recruited patients in 4 ways: (1) the facility’s primary homeless program, Healthcare for Homeless Veterans, referred patients who previously sought VA housing but who required primary care; (2) an outreach specialist sought homeless veterans in shelters and on the streets; (3) HPACT staff marketed the clinic with presentations and flyers to other VA services and non-VA community agencies; and (4) a referral mechanism within the EHR.
A nurse practitioner was the PCP at this site, supervised by an academic internist experienced in the care of underserved populations, and the HPACT director, a physician certified in internal and addiction medicine. Patients at the BIR HPACT also received care from a social worker, registered nurse, licensed practical nurse, and psychiatrist who received all or a portion of their salaries from this demonstration project. This clinic accommodated walk-in appointments during business hours. Clinicians discussed clinical cases daily, and the full clinical and administrative team met weekly. To promote service integration, HPACT staff attended meetings of the BIR general primary care and Healthcare for Homeless Veterans programs, and vice versa.
West Los Angeles
At WLA, a previously established Homeless Screening Clinic that offered integrated social and medical services for homeless veterans with mental illness was already available during business hours and located within the site’s mental health program.11 However, because ED use for homeless veterans peaked after hours, the new WLA HPACT was established within the WLA ED.
During WLA HPACT hours (3 weekday evenings/week), routine nursing triage occurred for all patients who presented to the ED. However, distinct from other times of day, veterans who were triaged with low-acuity and who were appropriate for outpatient care were given a self-administered, 4-item questionnaire to identify patients who were homeless or at risk for becoming homeless. Veterans who were identified with this screening tool were offered the choice of an ED or HPACT visit. Veterans who chose the latter were assigned to the queue for an HPACT primary care visit instead of the ED.
A physician led the clinical team, with additional services from a mental health clinical nurse specialist and clerks who worked with homeless and/or mental health patients during business hours and provided part-time HPACT coverage. When needed, additional services were provided from colocated ED nurses and social workers. Providers were chosen for their aptitude in culturally responsive communication.
Patients who chose to be seen in HPACT received a primary care visit that also addressed the reason for ED presentation. HPACT staff worked collaboratively, with interdisciplinary team huddles that preceded each clinic session and ended each patient visit. Referrals and social service needs were tracked and monitored by the PCP. Specialty care referrals were also tracked and facilitated when possible with direct communication between HPACT providers and specialty services. Meetings with daytime Homeless Screening Clinic and ED staff facilitated cross-departmental collaborations that helped veterans prepare for and retain housing.
Pittsburgh
The HPACT at PIT evolved from an existing PACT that provided primary care-based addiction services.2 That team included an internist credentialed in addiction medicine and experienced in homeless health care, nurse practitioner, nurse care manager, nursing assistant, and clerks. At this site, HPACT providers had subspecialty expertise in the assessment and treatment of ATOD use, certification to prescribe buprenorphine for outpatient opioid detoxification and/or maintenance, and experience engaging homeless and other vulnerable veterans. The existing colocated clinic included 2 additional addiction medicine clinicians and a physician assistant experienced in ATOD use. The PIT HPACT was not restricted to patients with ATOD use.
At PIT, HPACT care was provided during business hours in a flexible model that allowed for walk-in visits. Providers from the existing addiction-focused PACT identified and referred homeless veterans, as well as patients deemed at-risk for becoming homeless. In addition, other homeless veterans who did not have a PCP could be referred through e-consults placed by any VA staff member. If a referred homeless veteran was already enrolled on a PCP’s panel, HPACT facilitated reengagement with the existing provider. Near the end of this data collection period, a peer support specialist also began community outreach to engage both enrolled and potential HPACT patients.
Patient Characteristics
Table 2 presents the demographic and housing characteristics of enrolled HPACT patients at BIR, WLA, and PIT from May 1, 2012, to September 30, 2012. Each site had a similar number of patients (n = 35/47/43 at BIR/WLA/PIT, respectively). Across sites, most patients were male and African American or white. The majority of patients at each site were housed in VA transitional housing/residential rehabilitation programs (33%/32%/40% at BIR/WLA/PIT, respectively). Fewer patients were unsheltered (on the streets or other places not meant for sleeping), though WLA had the most unsheltered patients (26%) compared with BIR (6%) and PIT (2%). BIR had more patients from emergency shelters (14%) than did the other 2 sites (4% at WLA and 0% at PIT). PIT had the most domiciled patients in houses/apartments (26%, compared with 6% each at BIR and WLA), but who were at risk for homelessness.
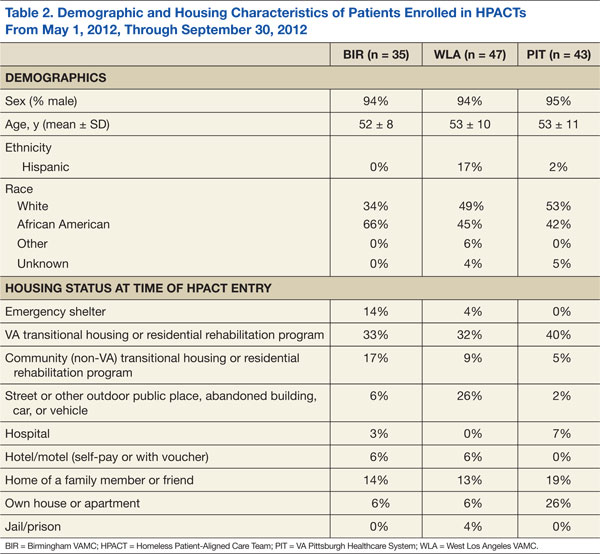
Table 3 summarizes the diagnoses and VA health care use patterns of these cohorts. In the first week of HPACT care, WLA addressed the most acute medical conditions (81% of patients had≥ 1 common condition), followed by BIR (54%) and PIT (23%). Among chronic conditions, chronic pain was diagnosed in 51% of patients at BIR, 30% of patients at WLA, and 12% of patients at PIT. Hepatitis C diagnoses were most common at WLA (36%), similar at PIT (33%), and lower at BIR (17%). Overall, chronic medical diagnoses were most common among patients at BIR (91%), followed by WLA (85%), and PIT (60%).
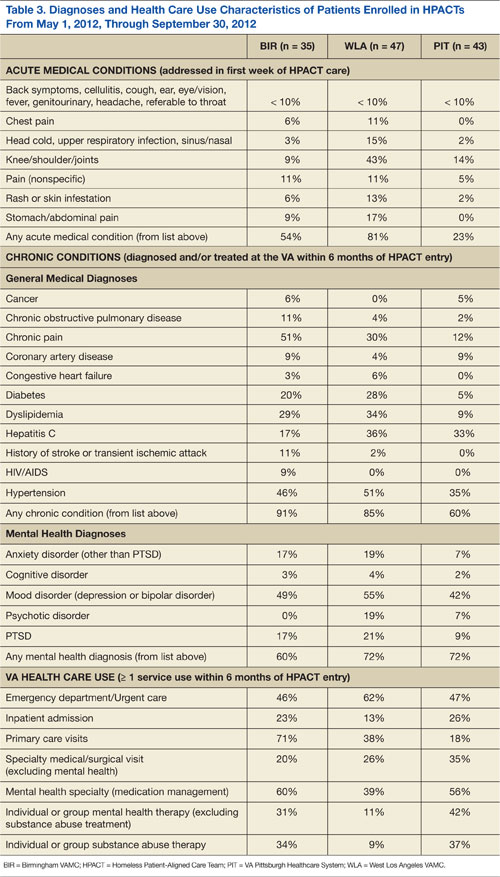
Among mental health diagnoses, mood disorders were the most prevalent across sites (49%/55%/42% at BIR/WLA/PIT, respectively), followed by posttraumatic stress disorder (17%/21%/9% at BIR/WLA/PIT, respectively). WLA had more patients with psychosis (19%) than did the other 2 sites (7% at PIT and 0% at BIR). Overall, mental illness was common among HPACT patients across sites (60%/72%/72% at BIR/WLA/PIT, respectively).
Health care use in the 6 months before HPACT enrollment differed between sites. BIR and PIT had similar rates of ED/urgent care use (46% and 47% sought care over the prior 6 months, respectively), but WLA had the highest rate (62%). However, inpatient admission rates were lowest at WLA (13%) and again similar at BIR (23%) and PIT (26%). BIR patients had the most VA primary care exposure before HPACT entry (71% obtained primary care in the past 6 months), followed by WLA (38%) and PIT (18%). Mental health specialty care was also highest at BIR (60%), followed by PIT (56%), then WLA (39%)
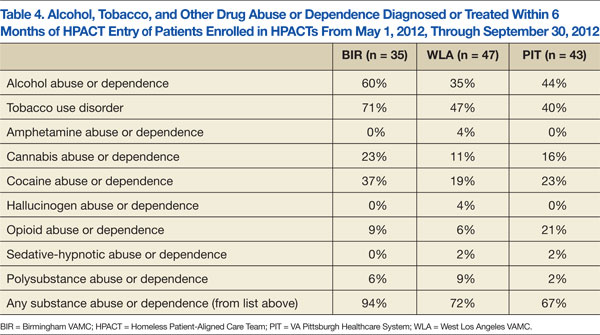
Table 4 presents the prevalence of ATOD abuse or dependence among HPACT patients in the6 months before clinic enrollment. The most commonly misused substances were alcohol (60%/35%/44% at BIR/WLA/PIT, respectively), tobacco (71%/47%/40% at BIR/WLA/PIT, respectively), and cocaine (37%/19%/23% at BIR/WLA/PIT, respectively). PIT had the highest percentages of patients with opioid misuse (21%), followed by BIR (9%) and WLA (6%). Overall, these disorders were prevalent across sites, highest at BIR (94%) and similar at WLA (72%) and PIT (67%).
DISCUSSION
In this case report of early HPACT implementation, strikingly different models of homeless-focused primary care at the geographically distinct facilities were found. The lack of a gold standard primary care medical home for homeless persons—compounded by contrasting local contextual features—led to distinct clinic designs. BIR capitalized on primary care needs among veterans who previously sought housing and relied on the available space within an operating primary care clinic. As WLA already offered primary care for homeless veterans that was colocated with mental health services, the HPACT at this site was devised as an after-hours clinic colocated with the ED.11 At PIT, an existing primary care team with addiction expertise expanded its role to include a focus on homelessness, without needing new space or staff.
The initial HPACT patient cohorts likely reflected these contrasting clinic structures. That is, at WLA, the higher rates of unsheltered patients, prevalence of acute medical conditions and psychotic disorders, and greater ED use was likely driven by ED colocation. The physical location of this site’s HPACT may also speak to greater future decreases in ED use. At BIR, the use of a dedicated HPACT community outreach worker likely led to greater recruitment from emergency shelters. However, the clinic mainly recruited veterans who had previously engaged in VA mainstream primary care services and individuals with ATOD use. At PIT, higher rates of psychotherapy were likely facilitated by the HPACT’s placement within an addiction treatment setting, which may favor psychosocial rehabilitation. The distinctly higher rate of patients with opioid misuse at PIT likely paralleled the ATOD expertise of its providers and/or buprenorphine availability.
The more challenging questions surround the implementation of the current clinic models to address the needs of these patient cohorts and possible avenues to improve each clinic. High rates of chronic medical illness, mental illness, and ATOD use are well known in the homeless veteran and general populations.2,9 Within these 3 HPACTs, the high rates of medical/mental illness and ATOD use speak favorably about the clinics’ respective recruitment strategies; ie, normative homeless populations with high rates of illness are enrolling in these clinics. However, current service integration practices may be enhanced with the specific knowledge gained from this examination. For example, the very high rates of mood and anxiety disorders at each site suggest a role for an embedded mental health provider with prescribing privileges (the model adopted by BIR) as opposed to mental health referrals used at WLA and PIT. There may also be a role for cognitive behavioral therapy services within these clinics. Similarly, the high rates of ATOD (especially alcohol, tobacco, and cocaine) misuse suggest a role for addiction medicine training among the PCPs (the PIT model) as well as psychosocial rehabilitation for ATOD use within the HPACTs. High rates of chronic medical conditions, such as diabetes, hepatitis C, and hypertension elucidate possible roles for specialty care integration and/or chronic disease management programs tailored to the homeless.
Comparing the housing status of these cohorts can help in the design of future homeless-tailored primary care operations and improve these HPACTs. Most patients across sites lived in VA transitional housing/residential rehabilitation programs. As such, current referral practices at these 3 HPACTs proved sufficient in recruiting this subpopulation of homeless veterans. However, in light of national data showing that the count of unsheltered homeless veterans has not declined as rapidly as the count of homeless veterans overall, the higher numbers of veterans recruited from interim sheltering arrangements suggest a need for enhanced outreach to unsheltered individuals.1 WLA data suggest that linkages to EDs can advance this objective. BIR data show that targeted outreach in shelters can engage this high-risk, transiently sheltered subpopulation in primary care. At the time of this project, PIT just began using a peer support specialist for outreach to unsheltered veterans. It will be important to evaluate the outcomes of this new referral strategy.
Limitations
These exploratory findings—though from a small convenience sample within a nascent, growing program—generated critical and detailed information to guide ongoing policies and service design. However, these findings have limitations. First, though this research contributes to limited existing literature about the operational design of homeless-focused primary care, no outcome data were included. Although a comprehensive evaluation of all HPACT sites is a distinct and useful endeavor, this project instead offers a rapid, detailed illustration of 3 early-stage clinics. Though smaller in scope, this effort informs other facilities developing homeless-focused primary care initiatives and the larger demonstration project.
Second, a convenience sample of 3 urban facilities with strong academic ties and community commitment to providing services for homeless persons was presented. It may be difficult to translate these findings to communities with fewer resources.
Third, EHR review was used to determine patient demographics, diagnoses, and patterns of health care use. Though EHR review offers detailed information that is unavailable from administrative data, EHR is subject to variations in documentation patterns.
Last, differing characteristics of the homeless veteran population in each city may interact with contrasting HPACT structures to influence the characteristics of patients served. For example, though the data suggest that linkages with the ED may facilitate greater recruitment of unsheltered veterans in WLA, Los Angeles is known to have particularly high rates of unsheltered individuals.1
CONCLUSIONS
Clinicians, administrators, and researchers in the safety net may benefit from the experience implementing new clinics to recruit and engage homeless veterans in primary care. In a relatively nascent field with few accepted models of care, this paper offers detailed descriptions of newly developed homeless-focused primary care clinics at 3 VA facilities, which can inform other sites undertaking similar initiatives. This study highlights the wide range of approaches to building such clinics, with important variations in structural characteristics such as clinic location and operating hours, as well as within the intricacies of service integration patterns.
Within primary care clinics for homeless adults, this study suggests a role for embedded mental health (medication management and psychotherapy) and substance abuse services, chronic disease management programs tailored to this vulnerable population, and the role of linkages to the ED or community-based outreach to recruit unsheltered homeless patients. To pave a path toward identifying an evidence-based model of homeless-focused primary care, future studies are needed to study nationwide HPACT outcomes, including health status, patient satisfaction, quality of life, housing, and cost-effectiveness.
Acknowledgements
This work was undertaken in part by the VA’s PACT Demonstration Laboratory initiative, supporting and evaluating VA’s transition to a patient-centered medical home. Funding for the PACT Demonstration Laboratory initiative was provided by the VA Office of Patient Care Services. This project received support from the VISN 22 VA Assessment and Improvement Lab for Patient Centered Care (VAIL-PCC) (XVA 65-018; PI: Rubenstein).
Dr. Gabrielian was supported in part by the VA Office of Academic Affiliations, Advanced Fellowship Program in Mental Illness Research and Treatment. Drs. Gelberg and Andersen were supported in part by NIDA DA 022445. Dr. Andersen received additional support from the UCLA/DREW Project EXPORT, National Center on Minority Health and Health Disparities, P20MD000148/P20MD000182. Dr. Broyles was supported by a Career Development Award (CDA 10–014) from the VA Health Services Research & Development service. Dr. Kertesz was supported through funding of VISN 7.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Cortes A, Henry M, de la Cruz RJ, Brown S. The 2012 Point-in-Time Estimates of Homelessness: Volume I of the 2012 Annual Homeless Assessment Report. Washington, DC: The U.S. Department of Housing and Urban Development, Office of Community Planning and Development; 2012.
2. Balshem H, Christensen V, Tuepker A, Kansagara D. A Critical Review of the Literature Regarding Homelessness Among Veterans. U.S. Department of Veterans Affairs, Veterans Health Administration, Health Services Research & Development Service; 2011. VA-ESP Project #05-225.
3. O’Toole TP, Pirraglia PA, Dosa D, et al; Primary Care-Special Populations Treatment Team. Building care systems to improve access for high-risk and vulnerable veteran populations. J Gen Intern Med. 2011;26(suppl 2):683-688.
4. Opening Doors: Federal Strategic Plan to Prevent and End Homelessness. Washington, DC: The United States Interagency Council on Homelessness; 2010.
5. Nakashima J, McGuire J, Berman S, Daniels W. Developing programs for homeless veterans: Understanding driving forces in implementation. Soc Work Health Care. 2004;40(2):1-12.
6. O’Toole TP, Buckel L, Bourgault C, et al. Applying the chronic care model to homeless veterans: Effect of a population approach to primary care on utilization and clinical outcomes. Am J Public Health. 2010;100(12):2493-2499.
7. Desai MM, Rosenheck RA, Kasprow WJ. Determinants of receipt of ambulatory medical care in a national sample of mentally ill homeless veterans. Med Care. 2003;41(2):275-287.
8. Irene Wong YL, Stanhope V. Conceptualizing community: A comparison of neighborhood characteristics of supportive housing for persons with psychiatric and developmental disabilities. Soc Sci Med. 2009;68(8):1376-1387.
9. Gelberg L, Gallagher TC, Andersen RM, Koegel P. Competing priorities as a barrier to medical care among homeless adults in Los Angeles. Am J Public Health. 1997;87(2):217-220.
10. Hamilton AB, Poza I, Washington DL. “Homelessness and trauma go hand-in-hand”: Pathways to homelessness among women veterans. Womens Health Issues. 2011;21(suppl 4):S203-S209.
11. McGuire J, Gelberg L, Blue-Howells J, Rosenheck RA. Access to primary care for homeless veterans with serious mental illness or substance abuse: A follow-up evaluation of co-located primary care and homeless social services. Adm Policy Ment Health. 2009;36(4):255-264.
12. VHA Operations Activities That May Constitute Research. Washington, DC: U.S. Department of Veterans Affairs, Veterans Health Administration; 2011. VHA Handbook 1058.05.
13. Resnick SG, Armstrong M, Sperrazza M, Harkness L, Rosenheck RA. A model of consumer-provider partnership: Vet-to-Vet. Psychiatr Rehabil J. 2004;28(2):185-187.
14. Hogan TP, Wakefield B, Nazi KM, Houston TK, Weaver FM. Promoting access through complementary eHealth technologies: Recommendations for VA’s Home Telehealth and personal health record programs. J Gen Intern Med. 2011;26(suppl 2):628-635.
15. Rosland AM, Nelson K, Sun H, et al. The patient-centered medical home in the Veterans Health Administration. Am J Manag Care. 2013;19(7):e263-e272.
16. Hsiao CJ, Cherry DK, Beatty PC, Rechtsteiner EA. National Ambulatory Medical Care Survey: 2007 summary. Natl Health Stat Report. 2010;3(27):1-32.
17. Hwang SW, Orav EJ, O’Connell JJ, Lebow JM, Brennan TA. Causes of death in homeless adults in Boston. Ann Intern Med. 1997;126(8):625-628.
18. Hwang SW, Tolomiczenko G, Kouyoumdjian FG, Garner RE. Interventions to improve the health of the homeless: A systematic review. Am J Prev Med. 2005;29(4):311-319.
On a single night in 2012, 62,619 veterans experienced homelessness.1 With high rates of illness, as well as alcohol, tobacco, and other drug (ATOD) use among homeless adults, ending veteran homelessness is a signature initiative of the VA.2-4 However, despite increasing efforts to improve health care for homeless individuals, little is known about best practices in homeless-focused primary care.2,5
With an age-adjusted mortality that is 2 to 10 times higher than that of their housed counterparts, homeless veterans pose a formidable challenge for primary care providers (PCPs).6,7 The complexity of homeless persons’ primary care needs is compounded by poor social support and the need to navigate priorities (eg, shelter) that compete with medical care.6,8,9 Moreover, veterans may face unique vulnerabilities conferred by military-specific experiences.2,10
As of 2012, only 2 VA facilities had primary care clinics tailored to the needs of homeless veterans. McGuire and colleagues built a system of colocated primary care, mental health, and homeless services for mentally ill veterans in Los Angeles, California.11 Over 18 months, this clinic facilitated greater primary and preventive care delivery, though the population’s physical health status did not improve.11 More recently, O’Toole and colleagues implemented a homeless-focused primary care clinic in Providence, Rhode Island.6 Compared with a historical sample of homeless veterans in traditional VA primary care, veterans in this homeless-tailored clinic had greater improvements in some chronic disease outcomes.6 This clinic also decreased nonacute emergency department (ED) use and hospitalizations for general medical conditions.6
Despite these promising outcomes, the VA lacked a nationwide homeless-focused primary care initiative. In 2012 the VA Office of Homeless Programs and the Office of Primary Care Operations funded a national demonstration project to create Homeless Patient-Aligned Care Teams (HPACTs)—primary care medical clinics for homeless veterans—at 32 facilities. This demonstration project guided HPACTs to tailor clinical and social services to homeless veterans’ needs, establish processes to identify and refer appropriate veterans, and integrate distinct services.
There were no explicit instructions that detailed HPACT structure. Because new VA programs must fit local contextual factors, including infrastructure, space, personnel, and institutional/community resources, different models of homeless-focused primary care have evolved.
This article is a case study of HPACTs at 3 of the 32 participating VA facilities, each reflecting a distinct community and organizational context. In light of projected HPACT expansion and concerns that current services are better tailored to sheltered homeless veterans than to their unsheltered peers, there is particular importance to detailed clinic descriptions that vividly portray the intricate relationships between service design and populations served.1
METHODS
VA HPACTs established in May 2012 at 3 facilities were examined: Birmingham VAMC in Alabama (BIR), West Los Angeles VAMC in California (WLA), and VA Pittsburgh Healthcare System in Pennsylvania (PIT). Prior to this demonstration project, each facility offered a range of housing/social services and traditional primary care for veterans. These sites are a geographically diverse convenience sample that emerged from existing homeless-focused collaborations among the authors and represent geographically diverse HPACTs.
The national director of VA Homeless Programs formally determined that this comparison constitutes a VA operations activity that is not research.12 This activity was exempt from Institutional Review Board review.
Study Design
Timed at an early stage of HPACT implementation, this project had 3 aims: (1) To identify noteworthy similarities and/or differences among the initial HPACT clinic structures; (2) To compare and contrast the patient characteristics of veterans enrolled in each of these clinics; and (3) To use these data to inform ongoing HPACT service design.
HPACT program evaluation data are not presented. Rather, a nascent system of care is illustrated that contributes to the limited literature concerning the design and implementation of homeless-focused primary care. Such organizational profiles inform novel program delivery and hold particular utility for heterogeneous populations who are difficult to engage in care.13,14
Authors at each site independently developed lists of variables that fell within the 3 guiding principles of this demonstration project. These variables were compiled and iteratively reduced to a consolidated table that assessed each clinic, including location, operating hours, methods of patient identification and referral, and linkages to distinct services (eg, primary care, mental health, addiction, and social services).
This table also became a guide for HPACT directors to generate narrative clinic descriptions. Characteristics of VA medical homes that are embraced regardless of patients’ housing status (eg, patient-centered, team-based care) were also incorporated into these descriptions.15
Patient Characteristics
The VA electronic health record (EHR) was used to identify all patients enrolled in HPACTs at BIR, WLA, and PIT from May 1, 2012 (clinic inception), through September 30, 2012. Authors developed a standardized template for EHR review and coined the first HPACT record as the patient’s index visit. This record was used to code initial housing status, demographics, and acute medical conditions diagnosed/treated. If housing status was not recorded at the index visit, the first preceding informative record was used.
Records for 6 months preceding the index visit were used to identify the presence or absence of common medical conditions, psychiatric diagnoses, and ATOD abuse or dependence. Medical conditions were identified from a list of common outpatient diagnoses from the National Ambulatory Medical Care Survey, supplemented by common conditions among homeless men.16-18 The EHR problem list (a list of diagnoses, by patient) was used to obtain diagnoses, supplemented by notes from inpatient admissions/discharges, as well as ED, primary care, and subspecialty consultations within 6 months preceding the index visit. Prior VA health care use was ascertained from the EHR review of the same 6-month window, reflecting ED visits, inpatient admissions, primary care visits, subspecialty visits, and individual/group therapy for ATOD or other mental health problems. Data were used to generate site-specific descriptive statistics.
RESULTS
Like all VA hospital-based clinics, a universal EHR captured all medical and social service notes, orders, medications, and administrative records. All facilities had on-site EDs, medical/mental health specialty care, and pharmacies. Social services, including benefits counseling and housing services, were available on site. Table 1 summarizes the HPACT structures at BIR, WLA, and PIT.
Birmingham
The BIR HPACT was devised as a new homeless-focused team located within a VA primary care clinic where other providers continued to see primary care patients. Staff and space were reallocated for this HPACT, which recruited patients in 4 ways: (1) the facility’s primary homeless program, Healthcare for Homeless Veterans, referred patients who previously sought VA housing but who required primary care; (2) an outreach specialist sought homeless veterans in shelters and on the streets; (3) HPACT staff marketed the clinic with presentations and flyers to other VA services and non-VA community agencies; and (4) a referral mechanism within the EHR.
A nurse practitioner was the PCP at this site, supervised by an academic internist experienced in the care of underserved populations, and the HPACT director, a physician certified in internal and addiction medicine. Patients at the BIR HPACT also received care from a social worker, registered nurse, licensed practical nurse, and psychiatrist who received all or a portion of their salaries from this demonstration project. This clinic accommodated walk-in appointments during business hours. Clinicians discussed clinical cases daily, and the full clinical and administrative team met weekly. To promote service integration, HPACT staff attended meetings of the BIR general primary care and Healthcare for Homeless Veterans programs, and vice versa.
West Los Angeles
At WLA, a previously established Homeless Screening Clinic that offered integrated social and medical services for homeless veterans with mental illness was already available during business hours and located within the site’s mental health program.11 However, because ED use for homeless veterans peaked after hours, the new WLA HPACT was established within the WLA ED.
During WLA HPACT hours (3 weekday evenings/week), routine nursing triage occurred for all patients who presented to the ED. However, distinct from other times of day, veterans who were triaged with low-acuity and who were appropriate for outpatient care were given a self-administered, 4-item questionnaire to identify patients who were homeless or at risk for becoming homeless. Veterans who were identified with this screening tool were offered the choice of an ED or HPACT visit. Veterans who chose the latter were assigned to the queue for an HPACT primary care visit instead of the ED.
A physician led the clinical team, with additional services from a mental health clinical nurse specialist and clerks who worked with homeless and/or mental health patients during business hours and provided part-time HPACT coverage. When needed, additional services were provided from colocated ED nurses and social workers. Providers were chosen for their aptitude in culturally responsive communication.
Patients who chose to be seen in HPACT received a primary care visit that also addressed the reason for ED presentation. HPACT staff worked collaboratively, with interdisciplinary team huddles that preceded each clinic session and ended each patient visit. Referrals and social service needs were tracked and monitored by the PCP. Specialty care referrals were also tracked and facilitated when possible with direct communication between HPACT providers and specialty services. Meetings with daytime Homeless Screening Clinic and ED staff facilitated cross-departmental collaborations that helped veterans prepare for and retain housing.
Pittsburgh
The HPACT at PIT evolved from an existing PACT that provided primary care-based addiction services.2 That team included an internist credentialed in addiction medicine and experienced in homeless health care, nurse practitioner, nurse care manager, nursing assistant, and clerks. At this site, HPACT providers had subspecialty expertise in the assessment and treatment of ATOD use, certification to prescribe buprenorphine for outpatient opioid detoxification and/or maintenance, and experience engaging homeless and other vulnerable veterans. The existing colocated clinic included 2 additional addiction medicine clinicians and a physician assistant experienced in ATOD use. The PIT HPACT was not restricted to patients with ATOD use.
At PIT, HPACT care was provided during business hours in a flexible model that allowed for walk-in visits. Providers from the existing addiction-focused PACT identified and referred homeless veterans, as well as patients deemed at-risk for becoming homeless. In addition, other homeless veterans who did not have a PCP could be referred through e-consults placed by any VA staff member. If a referred homeless veteran was already enrolled on a PCP’s panel, HPACT facilitated reengagement with the existing provider. Near the end of this data collection period, a peer support specialist also began community outreach to engage both enrolled and potential HPACT patients.
Patient Characteristics
Table 2 presents the demographic and housing characteristics of enrolled HPACT patients at BIR, WLA, and PIT from May 1, 2012, to September 30, 2012. Each site had a similar number of patients (n = 35/47/43 at BIR/WLA/PIT, respectively). Across sites, most patients were male and African American or white. The majority of patients at each site were housed in VA transitional housing/residential rehabilitation programs (33%/32%/40% at BIR/WLA/PIT, respectively). Fewer patients were unsheltered (on the streets or other places not meant for sleeping), though WLA had the most unsheltered patients (26%) compared with BIR (6%) and PIT (2%). BIR had more patients from emergency shelters (14%) than did the other 2 sites (4% at WLA and 0% at PIT). PIT had the most domiciled patients in houses/apartments (26%, compared with 6% each at BIR and WLA), but who were at risk for homelessness.
Table 3 summarizes the diagnoses and VA health care use patterns of these cohorts. In the first week of HPACT care, WLA addressed the most acute medical conditions (81% of patients had≥ 1 common condition), followed by BIR (54%) and PIT (23%). Among chronic conditions, chronic pain was diagnosed in 51% of patients at BIR, 30% of patients at WLA, and 12% of patients at PIT. Hepatitis C diagnoses were most common at WLA (36%), similar at PIT (33%), and lower at BIR (17%). Overall, chronic medical diagnoses were most common among patients at BIR (91%), followed by WLA (85%), and PIT (60%).
Among mental health diagnoses, mood disorders were the most prevalent across sites (49%/55%/42% at BIR/WLA/PIT, respectively), followed by posttraumatic stress disorder (17%/21%/9% at BIR/WLA/PIT, respectively). WLA had more patients with psychosis (19%) than did the other 2 sites (7% at PIT and 0% at BIR). Overall, mental illness was common among HPACT patients across sites (60%/72%/72% at BIR/WLA/PIT, respectively).
Health care use in the 6 months before HPACT enrollment differed between sites. BIR and PIT had similar rates of ED/urgent care use (46% and 47% sought care over the prior 6 months, respectively), but WLA had the highest rate (62%). However, inpatient admission rates were lowest at WLA (13%) and again similar at BIR (23%) and PIT (26%). BIR patients had the most VA primary care exposure before HPACT entry (71% obtained primary care in the past 6 months), followed by WLA (38%) and PIT (18%). Mental health specialty care was also highest at BIR (60%), followed by PIT (56%), then WLA (39%)
Table 4 presents the prevalence of ATOD abuse or dependence among HPACT patients in the6 months before clinic enrollment. The most commonly misused substances were alcohol (60%/35%/44% at BIR/WLA/PIT, respectively), tobacco (71%/47%/40% at BIR/WLA/PIT, respectively), and cocaine (37%/19%/23% at BIR/WLA/PIT, respectively). PIT had the highest percentages of patients with opioid misuse (21%), followed by BIR (9%) and WLA (6%). Overall, these disorders were prevalent across sites, highest at BIR (94%) and similar at WLA (72%) and PIT (67%).
DISCUSSION
In this case report of early HPACT implementation, strikingly different models of homeless-focused primary care at the geographically distinct facilities were found. The lack of a gold standard primary care medical home for homeless persons—compounded by contrasting local contextual features—led to distinct clinic designs. BIR capitalized on primary care needs among veterans who previously sought housing and relied on the available space within an operating primary care clinic. As WLA already offered primary care for homeless veterans that was colocated with mental health services, the HPACT at this site was devised as an after-hours clinic colocated with the ED.11 At PIT, an existing primary care team with addiction expertise expanded its role to include a focus on homelessness, without needing new space or staff.
The initial HPACT patient cohorts likely reflected these contrasting clinic structures. That is, at WLA, the higher rates of unsheltered patients, prevalence of acute medical conditions and psychotic disorders, and greater ED use was likely driven by ED colocation. The physical location of this site’s HPACT may also speak to greater future decreases in ED use. At BIR, the use of a dedicated HPACT community outreach worker likely led to greater recruitment from emergency shelters. However, the clinic mainly recruited veterans who had previously engaged in VA mainstream primary care services and individuals with ATOD use. At PIT, higher rates of psychotherapy were likely facilitated by the HPACT’s placement within an addiction treatment setting, which may favor psychosocial rehabilitation. The distinctly higher rate of patients with opioid misuse at PIT likely paralleled the ATOD expertise of its providers and/or buprenorphine availability.
The more challenging questions surround the implementation of the current clinic models to address the needs of these patient cohorts and possible avenues to improve each clinic. High rates of chronic medical illness, mental illness, and ATOD use are well known in the homeless veteran and general populations.2,9 Within these 3 HPACTs, the high rates of medical/mental illness and ATOD use speak favorably about the clinics’ respective recruitment strategies; ie, normative homeless populations with high rates of illness are enrolling in these clinics. However, current service integration practices may be enhanced with the specific knowledge gained from this examination. For example, the very high rates of mood and anxiety disorders at each site suggest a role for an embedded mental health provider with prescribing privileges (the model adopted by BIR) as opposed to mental health referrals used at WLA and PIT. There may also be a role for cognitive behavioral therapy services within these clinics. Similarly, the high rates of ATOD (especially alcohol, tobacco, and cocaine) misuse suggest a role for addiction medicine training among the PCPs (the PIT model) as well as psychosocial rehabilitation for ATOD use within the HPACTs. High rates of chronic medical conditions, such as diabetes, hepatitis C, and hypertension elucidate possible roles for specialty care integration and/or chronic disease management programs tailored to the homeless.
Comparing the housing status of these cohorts can help in the design of future homeless-tailored primary care operations and improve these HPACTs. Most patients across sites lived in VA transitional housing/residential rehabilitation programs. As such, current referral practices at these 3 HPACTs proved sufficient in recruiting this subpopulation of homeless veterans. However, in light of national data showing that the count of unsheltered homeless veterans has not declined as rapidly as the count of homeless veterans overall, the higher numbers of veterans recruited from interim sheltering arrangements suggest a need for enhanced outreach to unsheltered individuals.1 WLA data suggest that linkages to EDs can advance this objective. BIR data show that targeted outreach in shelters can engage this high-risk, transiently sheltered subpopulation in primary care. At the time of this project, PIT just began using a peer support specialist for outreach to unsheltered veterans. It will be important to evaluate the outcomes of this new referral strategy.
Limitations
These exploratory findings—though from a small convenience sample within a nascent, growing program—generated critical and detailed information to guide ongoing policies and service design. However, these findings have limitations. First, though this research contributes to limited existing literature about the operational design of homeless-focused primary care, no outcome data were included. Although a comprehensive evaluation of all HPACT sites is a distinct and useful endeavor, this project instead offers a rapid, detailed illustration of 3 early-stage clinics. Though smaller in scope, this effort informs other facilities developing homeless-focused primary care initiatives and the larger demonstration project.
Second, a convenience sample of 3 urban facilities with strong academic ties and community commitment to providing services for homeless persons was presented. It may be difficult to translate these findings to communities with fewer resources.
Third, EHR review was used to determine patient demographics, diagnoses, and patterns of health care use. Though EHR review offers detailed information that is unavailable from administrative data, EHR is subject to variations in documentation patterns.
Last, differing characteristics of the homeless veteran population in each city may interact with contrasting HPACT structures to influence the characteristics of patients served. For example, though the data suggest that linkages with the ED may facilitate greater recruitment of unsheltered veterans in WLA, Los Angeles is known to have particularly high rates of unsheltered individuals.1
CONCLUSIONS
Clinicians, administrators, and researchers in the safety net may benefit from the experience implementing new clinics to recruit and engage homeless veterans in primary care. In a relatively nascent field with few accepted models of care, this paper offers detailed descriptions of newly developed homeless-focused primary care clinics at 3 VA facilities, which can inform other sites undertaking similar initiatives. This study highlights the wide range of approaches to building such clinics, with important variations in structural characteristics such as clinic location and operating hours, as well as within the intricacies of service integration patterns.
Within primary care clinics for homeless adults, this study suggests a role for embedded mental health (medication management and psychotherapy) and substance abuse services, chronic disease management programs tailored to this vulnerable population, and the role of linkages to the ED or community-based outreach to recruit unsheltered homeless patients. To pave a path toward identifying an evidence-based model of homeless-focused primary care, future studies are needed to study nationwide HPACT outcomes, including health status, patient satisfaction, quality of life, housing, and cost-effectiveness.
Acknowledgements
This work was undertaken in part by the VA’s PACT Demonstration Laboratory initiative, supporting and evaluating VA’s transition to a patient-centered medical home. Funding for the PACT Demonstration Laboratory initiative was provided by the VA Office of Patient Care Services. This project received support from the VISN 22 VA Assessment and Improvement Lab for Patient Centered Care (VAIL-PCC) (XVA 65-018; PI: Rubenstein).
Dr. Gabrielian was supported in part by the VA Office of Academic Affiliations, Advanced Fellowship Program in Mental Illness Research and Treatment. Drs. Gelberg and Andersen were supported in part by NIDA DA 022445. Dr. Andersen received additional support from the UCLA/DREW Project EXPORT, National Center on Minority Health and Health Disparities, P20MD000148/P20MD000182. Dr. Broyles was supported by a Career Development Award (CDA 10–014) from the VA Health Services Research & Development service. Dr. Kertesz was supported through funding of VISN 7.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
On a single night in 2012, 62,619 veterans experienced homelessness.1 With high rates of illness, as well as alcohol, tobacco, and other drug (ATOD) use among homeless adults, ending veteran homelessness is a signature initiative of the VA.2-4 However, despite increasing efforts to improve health care for homeless individuals, little is known about best practices in homeless-focused primary care.2,5
With an age-adjusted mortality that is 2 to 10 times higher than that of their housed counterparts, homeless veterans pose a formidable challenge for primary care providers (PCPs).6,7 The complexity of homeless persons’ primary care needs is compounded by poor social support and the need to navigate priorities (eg, shelter) that compete with medical care.6,8,9 Moreover, veterans may face unique vulnerabilities conferred by military-specific experiences.2,10
As of 2012, only 2 VA facilities had primary care clinics tailored to the needs of homeless veterans. McGuire and colleagues built a system of colocated primary care, mental health, and homeless services for mentally ill veterans in Los Angeles, California.11 Over 18 months, this clinic facilitated greater primary and preventive care delivery, though the population’s physical health status did not improve.11 More recently, O’Toole and colleagues implemented a homeless-focused primary care clinic in Providence, Rhode Island.6 Compared with a historical sample of homeless veterans in traditional VA primary care, veterans in this homeless-tailored clinic had greater improvements in some chronic disease outcomes.6 This clinic also decreased nonacute emergency department (ED) use and hospitalizations for general medical conditions.6
Despite these promising outcomes, the VA lacked a nationwide homeless-focused primary care initiative. In 2012 the VA Office of Homeless Programs and the Office of Primary Care Operations funded a national demonstration project to create Homeless Patient-Aligned Care Teams (HPACTs)—primary care medical clinics for homeless veterans—at 32 facilities. This demonstration project guided HPACTs to tailor clinical and social services to homeless veterans’ needs, establish processes to identify and refer appropriate veterans, and integrate distinct services.
There were no explicit instructions that detailed HPACT structure. Because new VA programs must fit local contextual factors, including infrastructure, space, personnel, and institutional/community resources, different models of homeless-focused primary care have evolved.
This article is a case study of HPACTs at 3 of the 32 participating VA facilities, each reflecting a distinct community and organizational context. In light of projected HPACT expansion and concerns that current services are better tailored to sheltered homeless veterans than to their unsheltered peers, there is particular importance to detailed clinic descriptions that vividly portray the intricate relationships between service design and populations served.1
METHODS
VA HPACTs established in May 2012 at 3 facilities were examined: Birmingham VAMC in Alabama (BIR), West Los Angeles VAMC in California (WLA), and VA Pittsburgh Healthcare System in Pennsylvania (PIT). Prior to this demonstration project, each facility offered a range of housing/social services and traditional primary care for veterans. These sites are a geographically diverse convenience sample that emerged from existing homeless-focused collaborations among the authors and represent geographically diverse HPACTs.
The national director of VA Homeless Programs formally determined that this comparison constitutes a VA operations activity that is not research.12 This activity was exempt from Institutional Review Board review.
Study Design
Timed at an early stage of HPACT implementation, this project had 3 aims: (1) To identify noteworthy similarities and/or differences among the initial HPACT clinic structures; (2) To compare and contrast the patient characteristics of veterans enrolled in each of these clinics; and (3) To use these data to inform ongoing HPACT service design.
HPACT program evaluation data are not presented. Rather, a nascent system of care is illustrated that contributes to the limited literature concerning the design and implementation of homeless-focused primary care. Such organizational profiles inform novel program delivery and hold particular utility for heterogeneous populations who are difficult to engage in care.13,14
Authors at each site independently developed lists of variables that fell within the 3 guiding principles of this demonstration project. These variables were compiled and iteratively reduced to a consolidated table that assessed each clinic, including location, operating hours, methods of patient identification and referral, and linkages to distinct services (eg, primary care, mental health, addiction, and social services).
This table also became a guide for HPACT directors to generate narrative clinic descriptions. Characteristics of VA medical homes that are embraced regardless of patients’ housing status (eg, patient-centered, team-based care) were also incorporated into these descriptions.15
Patient Characteristics
The VA electronic health record (EHR) was used to identify all patients enrolled in HPACTs at BIR, WLA, and PIT from May 1, 2012 (clinic inception), through September 30, 2012. Authors developed a standardized template for EHR review and coined the first HPACT record as the patient’s index visit. This record was used to code initial housing status, demographics, and acute medical conditions diagnosed/treated. If housing status was not recorded at the index visit, the first preceding informative record was used.
Records for 6 months preceding the index visit were used to identify the presence or absence of common medical conditions, psychiatric diagnoses, and ATOD abuse or dependence. Medical conditions were identified from a list of common outpatient diagnoses from the National Ambulatory Medical Care Survey, supplemented by common conditions among homeless men.16-18 The EHR problem list (a list of diagnoses, by patient) was used to obtain diagnoses, supplemented by notes from inpatient admissions/discharges, as well as ED, primary care, and subspecialty consultations within 6 months preceding the index visit. Prior VA health care use was ascertained from the EHR review of the same 6-month window, reflecting ED visits, inpatient admissions, primary care visits, subspecialty visits, and individual/group therapy for ATOD or other mental health problems. Data were used to generate site-specific descriptive statistics.
RESULTS
Like all VA hospital-based clinics, a universal EHR captured all medical and social service notes, orders, medications, and administrative records. All facilities had on-site EDs, medical/mental health specialty care, and pharmacies. Social services, including benefits counseling and housing services, were available on site. Table 1 summarizes the HPACT structures at BIR, WLA, and PIT.
Birmingham
The BIR HPACT was devised as a new homeless-focused team located within a VA primary care clinic where other providers continued to see primary care patients. Staff and space were reallocated for this HPACT, which recruited patients in 4 ways: (1) the facility’s primary homeless program, Healthcare for Homeless Veterans, referred patients who previously sought VA housing but who required primary care; (2) an outreach specialist sought homeless veterans in shelters and on the streets; (3) HPACT staff marketed the clinic with presentations and flyers to other VA services and non-VA community agencies; and (4) a referral mechanism within the EHR.
A nurse practitioner was the PCP at this site, supervised by an academic internist experienced in the care of underserved populations, and the HPACT director, a physician certified in internal and addiction medicine. Patients at the BIR HPACT also received care from a social worker, registered nurse, licensed practical nurse, and psychiatrist who received all or a portion of their salaries from this demonstration project. This clinic accommodated walk-in appointments during business hours. Clinicians discussed clinical cases daily, and the full clinical and administrative team met weekly. To promote service integration, HPACT staff attended meetings of the BIR general primary care and Healthcare for Homeless Veterans programs, and vice versa.
West Los Angeles
At WLA, a previously established Homeless Screening Clinic that offered integrated social and medical services for homeless veterans with mental illness was already available during business hours and located within the site’s mental health program.11 However, because ED use for homeless veterans peaked after hours, the new WLA HPACT was established within the WLA ED.
During WLA HPACT hours (3 weekday evenings/week), routine nursing triage occurred for all patients who presented to the ED. However, distinct from other times of day, veterans who were triaged with low-acuity and who were appropriate for outpatient care were given a self-administered, 4-item questionnaire to identify patients who were homeless or at risk for becoming homeless. Veterans who were identified with this screening tool were offered the choice of an ED or HPACT visit. Veterans who chose the latter were assigned to the queue for an HPACT primary care visit instead of the ED.
A physician led the clinical team, with additional services from a mental health clinical nurse specialist and clerks who worked with homeless and/or mental health patients during business hours and provided part-time HPACT coverage. When needed, additional services were provided from colocated ED nurses and social workers. Providers were chosen for their aptitude in culturally responsive communication.
Patients who chose to be seen in HPACT received a primary care visit that also addressed the reason for ED presentation. HPACT staff worked collaboratively, with interdisciplinary team huddles that preceded each clinic session and ended each patient visit. Referrals and social service needs were tracked and monitored by the PCP. Specialty care referrals were also tracked and facilitated when possible with direct communication between HPACT providers and specialty services. Meetings with daytime Homeless Screening Clinic and ED staff facilitated cross-departmental collaborations that helped veterans prepare for and retain housing.
Pittsburgh
The HPACT at PIT evolved from an existing PACT that provided primary care-based addiction services.2 That team included an internist credentialed in addiction medicine and experienced in homeless health care, nurse practitioner, nurse care manager, nursing assistant, and clerks. At this site, HPACT providers had subspecialty expertise in the assessment and treatment of ATOD use, certification to prescribe buprenorphine for outpatient opioid detoxification and/or maintenance, and experience engaging homeless and other vulnerable veterans. The existing colocated clinic included 2 additional addiction medicine clinicians and a physician assistant experienced in ATOD use. The PIT HPACT was not restricted to patients with ATOD use.
At PIT, HPACT care was provided during business hours in a flexible model that allowed for walk-in visits. Providers from the existing addiction-focused PACT identified and referred homeless veterans, as well as patients deemed at-risk for becoming homeless. In addition, other homeless veterans who did not have a PCP could be referred through e-consults placed by any VA staff member. If a referred homeless veteran was already enrolled on a PCP’s panel, HPACT facilitated reengagement with the existing provider. Near the end of this data collection period, a peer support specialist also began community outreach to engage both enrolled and potential HPACT patients.
Patient Characteristics
Table 2 presents the demographic and housing characteristics of enrolled HPACT patients at BIR, WLA, and PIT from May 1, 2012, to September 30, 2012. Each site had a similar number of patients (n = 35/47/43 at BIR/WLA/PIT, respectively). Across sites, most patients were male and African American or white. The majority of patients at each site were housed in VA transitional housing/residential rehabilitation programs (33%/32%/40% at BIR/WLA/PIT, respectively). Fewer patients were unsheltered (on the streets or other places not meant for sleeping), though WLA had the most unsheltered patients (26%) compared with BIR (6%) and PIT (2%). BIR had more patients from emergency shelters (14%) than did the other 2 sites (4% at WLA and 0% at PIT). PIT had the most domiciled patients in houses/apartments (26%, compared with 6% each at BIR and WLA), but who were at risk for homelessness.
Table 3 summarizes the diagnoses and VA health care use patterns of these cohorts. In the first week of HPACT care, WLA addressed the most acute medical conditions (81% of patients had≥ 1 common condition), followed by BIR (54%) and PIT (23%). Among chronic conditions, chronic pain was diagnosed in 51% of patients at BIR, 30% of patients at WLA, and 12% of patients at PIT. Hepatitis C diagnoses were most common at WLA (36%), similar at PIT (33%), and lower at BIR (17%). Overall, chronic medical diagnoses were most common among patients at BIR (91%), followed by WLA (85%), and PIT (60%).
Among mental health diagnoses, mood disorders were the most prevalent across sites (49%/55%/42% at BIR/WLA/PIT, respectively), followed by posttraumatic stress disorder (17%/21%/9% at BIR/WLA/PIT, respectively). WLA had more patients with psychosis (19%) than did the other 2 sites (7% at PIT and 0% at BIR). Overall, mental illness was common among HPACT patients across sites (60%/72%/72% at BIR/WLA/PIT, respectively).
Health care use in the 6 months before HPACT enrollment differed between sites. BIR and PIT had similar rates of ED/urgent care use (46% and 47% sought care over the prior 6 months, respectively), but WLA had the highest rate (62%). However, inpatient admission rates were lowest at WLA (13%) and again similar at BIR (23%) and PIT (26%). BIR patients had the most VA primary care exposure before HPACT entry (71% obtained primary care in the past 6 months), followed by WLA (38%) and PIT (18%). Mental health specialty care was also highest at BIR (60%), followed by PIT (56%), then WLA (39%)
Table 4 presents the prevalence of ATOD abuse or dependence among HPACT patients in the6 months before clinic enrollment. The most commonly misused substances were alcohol (60%/35%/44% at BIR/WLA/PIT, respectively), tobacco (71%/47%/40% at BIR/WLA/PIT, respectively), and cocaine (37%/19%/23% at BIR/WLA/PIT, respectively). PIT had the highest percentages of patients with opioid misuse (21%), followed by BIR (9%) and WLA (6%). Overall, these disorders were prevalent across sites, highest at BIR (94%) and similar at WLA (72%) and PIT (67%).
DISCUSSION
In this case report of early HPACT implementation, strikingly different models of homeless-focused primary care at the geographically distinct facilities were found. The lack of a gold standard primary care medical home for homeless persons—compounded by contrasting local contextual features—led to distinct clinic designs. BIR capitalized on primary care needs among veterans who previously sought housing and relied on the available space within an operating primary care clinic. As WLA already offered primary care for homeless veterans that was colocated with mental health services, the HPACT at this site was devised as an after-hours clinic colocated with the ED.11 At PIT, an existing primary care team with addiction expertise expanded its role to include a focus on homelessness, without needing new space or staff.
The initial HPACT patient cohorts likely reflected these contrasting clinic structures. That is, at WLA, the higher rates of unsheltered patients, prevalence of acute medical conditions and psychotic disorders, and greater ED use was likely driven by ED colocation. The physical location of this site’s HPACT may also speak to greater future decreases in ED use. At BIR, the use of a dedicated HPACT community outreach worker likely led to greater recruitment from emergency shelters. However, the clinic mainly recruited veterans who had previously engaged in VA mainstream primary care services and individuals with ATOD use. At PIT, higher rates of psychotherapy were likely facilitated by the HPACT’s placement within an addiction treatment setting, which may favor psychosocial rehabilitation. The distinctly higher rate of patients with opioid misuse at PIT likely paralleled the ATOD expertise of its providers and/or buprenorphine availability.
The more challenging questions surround the implementation of the current clinic models to address the needs of these patient cohorts and possible avenues to improve each clinic. High rates of chronic medical illness, mental illness, and ATOD use are well known in the homeless veteran and general populations.2,9 Within these 3 HPACTs, the high rates of medical/mental illness and ATOD use speak favorably about the clinics’ respective recruitment strategies; ie, normative homeless populations with high rates of illness are enrolling in these clinics. However, current service integration practices may be enhanced with the specific knowledge gained from this examination. For example, the very high rates of mood and anxiety disorders at each site suggest a role for an embedded mental health provider with prescribing privileges (the model adopted by BIR) as opposed to mental health referrals used at WLA and PIT. There may also be a role for cognitive behavioral therapy services within these clinics. Similarly, the high rates of ATOD (especially alcohol, tobacco, and cocaine) misuse suggest a role for addiction medicine training among the PCPs (the PIT model) as well as psychosocial rehabilitation for ATOD use within the HPACTs. High rates of chronic medical conditions, such as diabetes, hepatitis C, and hypertension elucidate possible roles for specialty care integration and/or chronic disease management programs tailored to the homeless.
Comparing the housing status of these cohorts can help in the design of future homeless-tailored primary care operations and improve these HPACTs. Most patients across sites lived in VA transitional housing/residential rehabilitation programs. As such, current referral practices at these 3 HPACTs proved sufficient in recruiting this subpopulation of homeless veterans. However, in light of national data showing that the count of unsheltered homeless veterans has not declined as rapidly as the count of homeless veterans overall, the higher numbers of veterans recruited from interim sheltering arrangements suggest a need for enhanced outreach to unsheltered individuals.1 WLA data suggest that linkages to EDs can advance this objective. BIR data show that targeted outreach in shelters can engage this high-risk, transiently sheltered subpopulation in primary care. At the time of this project, PIT just began using a peer support specialist for outreach to unsheltered veterans. It will be important to evaluate the outcomes of this new referral strategy.
Limitations
These exploratory findings—though from a small convenience sample within a nascent, growing program—generated critical and detailed information to guide ongoing policies and service design. However, these findings have limitations. First, though this research contributes to limited existing literature about the operational design of homeless-focused primary care, no outcome data were included. Although a comprehensive evaluation of all HPACT sites is a distinct and useful endeavor, this project instead offers a rapid, detailed illustration of 3 early-stage clinics. Though smaller in scope, this effort informs other facilities developing homeless-focused primary care initiatives and the larger demonstration project.
Second, a convenience sample of 3 urban facilities with strong academic ties and community commitment to providing services for homeless persons was presented. It may be difficult to translate these findings to communities with fewer resources.
Third, EHR review was used to determine patient demographics, diagnoses, and patterns of health care use. Though EHR review offers detailed information that is unavailable from administrative data, EHR is subject to variations in documentation patterns.
Last, differing characteristics of the homeless veteran population in each city may interact with contrasting HPACT structures to influence the characteristics of patients served. For example, though the data suggest that linkages with the ED may facilitate greater recruitment of unsheltered veterans in WLA, Los Angeles is known to have particularly high rates of unsheltered individuals.1
CONCLUSIONS
Clinicians, administrators, and researchers in the safety net may benefit from the experience implementing new clinics to recruit and engage homeless veterans in primary care. In a relatively nascent field with few accepted models of care, this paper offers detailed descriptions of newly developed homeless-focused primary care clinics at 3 VA facilities, which can inform other sites undertaking similar initiatives. This study highlights the wide range of approaches to building such clinics, with important variations in structural characteristics such as clinic location and operating hours, as well as within the intricacies of service integration patterns.
Within primary care clinics for homeless adults, this study suggests a role for embedded mental health (medication management and psychotherapy) and substance abuse services, chronic disease management programs tailored to this vulnerable population, and the role of linkages to the ED or community-based outreach to recruit unsheltered homeless patients. To pave a path toward identifying an evidence-based model of homeless-focused primary care, future studies are needed to study nationwide HPACT outcomes, including health status, patient satisfaction, quality of life, housing, and cost-effectiveness.
Acknowledgements
This work was undertaken in part by the VA’s PACT Demonstration Laboratory initiative, supporting and evaluating VA’s transition to a patient-centered medical home. Funding for the PACT Demonstration Laboratory initiative was provided by the VA Office of Patient Care Services. This project received support from the VISN 22 VA Assessment and Improvement Lab for Patient Centered Care (VAIL-PCC) (XVA 65-018; PI: Rubenstein).
Dr. Gabrielian was supported in part by the VA Office of Academic Affiliations, Advanced Fellowship Program in Mental Illness Research and Treatment. Drs. Gelberg and Andersen were supported in part by NIDA DA 022445. Dr. Andersen received additional support from the UCLA/DREW Project EXPORT, National Center on Minority Health and Health Disparities, P20MD000148/P20MD000182. Dr. Broyles was supported by a Career Development Award (CDA 10–014) from the VA Health Services Research & Development service. Dr. Kertesz was supported through funding of VISN 7.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Cortes A, Henry M, de la Cruz RJ, Brown S. The 2012 Point-in-Time Estimates of Homelessness: Volume I of the 2012 Annual Homeless Assessment Report. Washington, DC: The U.S. Department of Housing and Urban Development, Office of Community Planning and Development; 2012.
2. Balshem H, Christensen V, Tuepker A, Kansagara D. A Critical Review of the Literature Regarding Homelessness Among Veterans. U.S. Department of Veterans Affairs, Veterans Health Administration, Health Services Research & Development Service; 2011. VA-ESP Project #05-225.
3. O’Toole TP, Pirraglia PA, Dosa D, et al; Primary Care-Special Populations Treatment Team. Building care systems to improve access for high-risk and vulnerable veteran populations. J Gen Intern Med. 2011;26(suppl 2):683-688.
4. Opening Doors: Federal Strategic Plan to Prevent and End Homelessness. Washington, DC: The United States Interagency Council on Homelessness; 2010.
5. Nakashima J, McGuire J, Berman S, Daniels W. Developing programs for homeless veterans: Understanding driving forces in implementation. Soc Work Health Care. 2004;40(2):1-12.
6. O’Toole TP, Buckel L, Bourgault C, et al. Applying the chronic care model to homeless veterans: Effect of a population approach to primary care on utilization and clinical outcomes. Am J Public Health. 2010;100(12):2493-2499.
7. Desai MM, Rosenheck RA, Kasprow WJ. Determinants of receipt of ambulatory medical care in a national sample of mentally ill homeless veterans. Med Care. 2003;41(2):275-287.
8. Irene Wong YL, Stanhope V. Conceptualizing community: A comparison of neighborhood characteristics of supportive housing for persons with psychiatric and developmental disabilities. Soc Sci Med. 2009;68(8):1376-1387.
9. Gelberg L, Gallagher TC, Andersen RM, Koegel P. Competing priorities as a barrier to medical care among homeless adults in Los Angeles. Am J Public Health. 1997;87(2):217-220.
10. Hamilton AB, Poza I, Washington DL. “Homelessness and trauma go hand-in-hand”: Pathways to homelessness among women veterans. Womens Health Issues. 2011;21(suppl 4):S203-S209.
11. McGuire J, Gelberg L, Blue-Howells J, Rosenheck RA. Access to primary care for homeless veterans with serious mental illness or substance abuse: A follow-up evaluation of co-located primary care and homeless social services. Adm Policy Ment Health. 2009;36(4):255-264.
12. VHA Operations Activities That May Constitute Research. Washington, DC: U.S. Department of Veterans Affairs, Veterans Health Administration; 2011. VHA Handbook 1058.05.
13. Resnick SG, Armstrong M, Sperrazza M, Harkness L, Rosenheck RA. A model of consumer-provider partnership: Vet-to-Vet. Psychiatr Rehabil J. 2004;28(2):185-187.
14. Hogan TP, Wakefield B, Nazi KM, Houston TK, Weaver FM. Promoting access through complementary eHealth technologies: Recommendations for VA’s Home Telehealth and personal health record programs. J Gen Intern Med. 2011;26(suppl 2):628-635.
15. Rosland AM, Nelson K, Sun H, et al. The patient-centered medical home in the Veterans Health Administration. Am J Manag Care. 2013;19(7):e263-e272.
16. Hsiao CJ, Cherry DK, Beatty PC, Rechtsteiner EA. National Ambulatory Medical Care Survey: 2007 summary. Natl Health Stat Report. 2010;3(27):1-32.
17. Hwang SW, Orav EJ, O’Connell JJ, Lebow JM, Brennan TA. Causes of death in homeless adults in Boston. Ann Intern Med. 1997;126(8):625-628.
18. Hwang SW, Tolomiczenko G, Kouyoumdjian FG, Garner RE. Interventions to improve the health of the homeless: A systematic review. Am J Prev Med. 2005;29(4):311-319.
1. Cortes A, Henry M, de la Cruz RJ, Brown S. The 2012 Point-in-Time Estimates of Homelessness: Volume I of the 2012 Annual Homeless Assessment Report. Washington, DC: The U.S. Department of Housing and Urban Development, Office of Community Planning and Development; 2012.
2. Balshem H, Christensen V, Tuepker A, Kansagara D. A Critical Review of the Literature Regarding Homelessness Among Veterans. U.S. Department of Veterans Affairs, Veterans Health Administration, Health Services Research & Development Service; 2011. VA-ESP Project #05-225.
3. O’Toole TP, Pirraglia PA, Dosa D, et al; Primary Care-Special Populations Treatment Team. Building care systems to improve access for high-risk and vulnerable veteran populations. J Gen Intern Med. 2011;26(suppl 2):683-688.
4. Opening Doors: Federal Strategic Plan to Prevent and End Homelessness. Washington, DC: The United States Interagency Council on Homelessness; 2010.
5. Nakashima J, McGuire J, Berman S, Daniels W. Developing programs for homeless veterans: Understanding driving forces in implementation. Soc Work Health Care. 2004;40(2):1-12.
6. O’Toole TP, Buckel L, Bourgault C, et al. Applying the chronic care model to homeless veterans: Effect of a population approach to primary care on utilization and clinical outcomes. Am J Public Health. 2010;100(12):2493-2499.
7. Desai MM, Rosenheck RA, Kasprow WJ. Determinants of receipt of ambulatory medical care in a national sample of mentally ill homeless veterans. Med Care. 2003;41(2):275-287.
8. Irene Wong YL, Stanhope V. Conceptualizing community: A comparison of neighborhood characteristics of supportive housing for persons with psychiatric and developmental disabilities. Soc Sci Med. 2009;68(8):1376-1387.
9. Gelberg L, Gallagher TC, Andersen RM, Koegel P. Competing priorities as a barrier to medical care among homeless adults in Los Angeles. Am J Public Health. 1997;87(2):217-220.
10. Hamilton AB, Poza I, Washington DL. “Homelessness and trauma go hand-in-hand”: Pathways to homelessness among women veterans. Womens Health Issues. 2011;21(suppl 4):S203-S209.
11. McGuire J, Gelberg L, Blue-Howells J, Rosenheck RA. Access to primary care for homeless veterans with serious mental illness or substance abuse: A follow-up evaluation of co-located primary care and homeless social services. Adm Policy Ment Health. 2009;36(4):255-264.
12. VHA Operations Activities That May Constitute Research. Washington, DC: U.S. Department of Veterans Affairs, Veterans Health Administration; 2011. VHA Handbook 1058.05.
13. Resnick SG, Armstrong M, Sperrazza M, Harkness L, Rosenheck RA. A model of consumer-provider partnership: Vet-to-Vet. Psychiatr Rehabil J. 2004;28(2):185-187.
14. Hogan TP, Wakefield B, Nazi KM, Houston TK, Weaver FM. Promoting access through complementary eHealth technologies: Recommendations for VA’s Home Telehealth and personal health record programs. J Gen Intern Med. 2011;26(suppl 2):628-635.
15. Rosland AM, Nelson K, Sun H, et al. The patient-centered medical home in the Veterans Health Administration. Am J Manag Care. 2013;19(7):e263-e272.
16. Hsiao CJ, Cherry DK, Beatty PC, Rechtsteiner EA. National Ambulatory Medical Care Survey: 2007 summary. Natl Health Stat Report. 2010;3(27):1-32.
17. Hwang SW, Orav EJ, O’Connell JJ, Lebow JM, Brennan TA. Causes of death in homeless adults in Boston. Ann Intern Med. 1997;126(8):625-628.
18. Hwang SW, Tolomiczenko G, Kouyoumdjian FG, Garner RE. Interventions to improve the health of the homeless: A systematic review. Am J Prev Med. 2005;29(4):311-319.
How Effective Is Group Cognitive Behavioral Therapy to Treat PTSD?
Anxiety is a necessary and natural reaction to trauma, but, sometimes, anxiety symptoms become excessive and problematic, as experienced with posttraumatic stress disorder (PTSD). Some patients who struggle with PTSD endure a relentless apprehension so intense that it keeps them from participating in everyday activities, such as attending work and partaking in social activities. Associated anxiety symptoms severely impair everyday function and include increased heart rate, sweating, intrusive images, poor attention, fear, or insomnia. Posttraumatic stress disorder symptoms often lead to occupational dysfunction, relationship difficulty, and numerous other functional impairments.
Approximately 300,000 veterans meet the criteria for PTSD related to ongoing or recent wars.1 The veteran does not bear the personal and functional burden alone; however, the financial load is felt throughout society. One recent study suggests that for veterans diagnosed with PTSD, the first 2 years after deployment cost society an estimated $7,000 per individial.2 Current research suggests that this potentially debilitating disorder occurs in about 14% of Operation Iraqi Freedom/Operation Enduring Freedom combat troops, whereas the similar U.S. demographic population experiences PTSD at a rate of about 7%.1,3 The ongoing military trauma exposures are compelling the mental health community to establish efficient and effective treatment options.4,5
Several treatment strategies exist to reduce PTSD symptoms, but health care professionals must seek a balance between therapeutic benefit and cost. The treatment of PTSD is diverse and variable; however, in the most recent Clinical Practice Guideline (CPG) for PTSD, the VA and DoD specifically endorse some psychotherapeutic interventions while dissuading the use of others.6 Of note, the VA and DoD CPG strongly encourages Stress Inoculation Training (SIT) and similar cognitive therapies aimed at guiding patients through the process of consciously understanding the relationship between thoughts and feelings and then modifying thoughts to appropriately manage stressors.6 Meanwhile, group psychotherapy has been determined to be “somewhat helpful.”6 Even though cognitive- and group-based therapies have long been established as efficacious for numerous psychological disorders (depression, obsessive compulsive disorder, eating disorders, etc), neither the American Group Psychotherapy Association nor the VA and DoD CPG directly endorse the use of group cognitive behavioral therapy (GCBT) for the treatment of PTSD.6,7 However, both VA and DoD mental health providers commonly practice CBT and various group psychotherapies for the treatment of PTSD.
Despite the widespread use of CBT, there is a gap in the clinical understanding of the evidence supporting GCBT for PTSD. The goal of this synthesis was to understand the efficacy of treating PTSD symptoms with group psychotherapy. To begin this investigation, the following PICO (population, intervention, comparison, outcome) question was asked: In adults diagnosed with PTSD, how effective is group cognitive behavioral therapy in reducing PTSD-related symptoms?
Methods
Research articles addressing the use of GCBT in PTSD were obtained via database searches that took place during October 2012 (Table). Searched databases included the Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews Randomized Controlled Trials, Psychological Information (PsycINFO), and Public Medicine (PubMed).
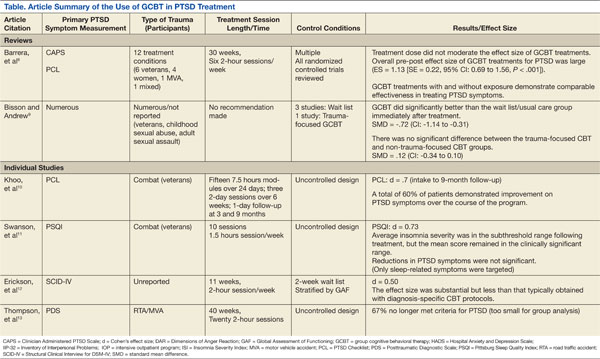
The PubMed database was searched using the following MeSH (medical subject heading) terms: “psychotherapy, group” and “stress disorders, post-traumatic” and “cognitive therapy.” Limitations were set to include only patients aged ≥ 18 years, results in English, those involving human subjects, and articles published within the past 5 years. A manual search of references was also conducted, and relevant articles were retained.
Articles that addressed primary substance abuse, other DSM Axis I disorders, intimate partner violence, or family issues were excluded from the evidence sample due to concerns of an alternate treatment focus. Articles with a focus on telehealth or alternative medicine were considered confounding to the scope of this review were also excluded. It was also noted that the term CBT is used collectively for an umbrella of treatments; however, treatments that focused on elements other than the components of CBT being delivered in a group were not included. To prevent duplication of the results, research from an inclusive review was not considered individually.
SUMMARY OF EVIDENCE
Six works fulfilled the PICO criteria and were of sufficient quality to be synthesized. Of the 6 articles retained for synthesis, 2 were high-level reviews. Both reviews supported the use of GCBT for PTSD treatment. Barrera and colleagues reported an overall large effect size regardless of the presence of exposure in-group among the 12 treatment conditions and 651 study participants.8 These researchers also reported that in-group exposure did not further traumatize other group members.
Similarly, although a notably older and smaller review, Bisson and Andrew reported a significant standard mean deviation between 4 GCBT treatment and wait list controls. These reviewers did not find a significant difference between trauma- and nontrauma-focused treatment groups. The reviews also noted that individual psychotherapy and/or pharmacotherapy was most often continued throughout the reviewed studies.8,9
The 4 other studies contribute substantively to this synthesis but arguably represent lower evidence quality. A large longitudinal study of 496 Australian veterans reported a large effect size that was sustained 9 months after treatment began.10 These researchers used an intensive outpatient program that included medication and other treatment modalities as the basis for GCBT delivery. They reported that the majority of the patients revealed improvement in PTSD symptoms.
Another study sampled a similar group of 10 combat veterans but focused particular attention on sleep-related PTSD symptoms of insomnia, nightmares, and sleep quality.11 Although these researchers were unable to report a significant difference in overall PTSD symptoms for the 8 subjects who completed the protocol, they did find a large effect size on insomnia severity and a medium effect size on sleep quality. Regular treatment, including medication, continued throughout this study.
Other researchers reported a medium effect size on PTSD symptoms while using GCBT in a heterogeneous group with various anxiety disorders, including obsessive compulsive disorder, generalized anxiety disorder, social phobia, panic disorders, and PTSD.12 Although reporting similar results as all other included studies, this study has some significant limitations, including a 26% dropout rate among the 152 participants. The final study included for synthesis reported a remarkable 67% elimination of the PTSD diagnosis among 6 motor vehicle accident survivors in the small, uncontrolled study.13 Concomitant treatments, including medications, were not reported in detail for these 4 studies except as mentioned.
As a whole, the 6 studies revealed some appreciable commonalities. Time since diagnosis did not seem to influence the results. Attrition was consistently found to be similar to other PTSD treatments. The reported session topics were loosely based on common CBT tenets (ie, education, challenging cognitions, and relaxation techniques) and were typically similar among treatment groups, including the use of homework.
DISCUSSION
As the diagnosis of PTSD increases to unfamiliar levels, GCBT has the potential to be helpful to clinicians and patients seeking alternatives to their current treatments.1,4,14 The reported results imply that GCBT can be useful in PTSD symptom reduction. This could be particularly useful to VA and military providers or rural providers operating with limited resources.
Treatment protocols are not well established and should be approached with care prior to the establishment of CBT treatment groups for those diagnosed with PTSD. Session overviews and descriptions, such as those mentioned in Thompson and colleagues, could provide a reference point for future use.13
Also worth considering, CBT can be an ambiguous term requiring deliberate definition within treatment protocols. As noted in the VA and DoD CPG, exposure- and trauma-focused treatment designs can be efficacious, but these elements do not seem to be required within the GCBT treatment setting.
The current research also suggests GCBT efficacy regardless of the index trauma. This does not suggest that heterogeneous groups were frequently studied nor can conclusions be drawn regarding heterogeneous treatment groups. Elements such as group size and session length are inconsistently reported and require specific consideration as well. There is a distinct lack of research directly comparing individual CBT with GCBT directly, which prohibits meaningful conclusions regarding PTSD symptom reduction. This research gap may well have influenced the recommendations within the VA and DoD CPG. Although some higher quality studies exist, many of the published reports on GCBT have noteworthy design flaw, such as inadequate controls and statistical analysis.
LIMITATIONS
There are some limitations to this literature synthesis. Although the search was limited to the past 5 years, the inclusion of reviews accounts for older evidence. As alluded to earlier, the lack of a standardized GCBT treatment protocol challenges results comparisons as well. The consequent treatment variations make direct interstudy comparison and synthesis difficult. Similarly, outcome measures varied between studies. Also, group psychotherapy is well established and accepted. Therefore, much of the supporting research was accomplished outside the parameters of this literature search. This empirical view of group psychotherapy among mental health providers may also contribute to the lack of available research.
It is also worth noting that studies finding neutral or negative results are often unpublished. This publication bias could account for the lack of available evidence. The research reports do not consistently report therapist qualifications; however, board certificates in group psychotherapy and CBT are undeniably variables available for debate. The inclusion of uncontrolled trials limits these findings as well. Although the above limitations are not exhaustive, they do provide necessary caveats to future generalizations.
FUTURE IMPLICATIONS
Perhaps the most important information to gain from future research is that of treatment outcomes. Studies that include a detailed outcome evaluation could reveal patient satisfaction, efficacy, and financial considerations. In the presence of adequate supportive data, GCBT could contribute outcome data regarding trauma survivor symptom normalization, peer support formation, access to care, treatment efficiency, and health care resources utilization. As noted in Barrera and colleagues, future analysis will require a greater volume of trials with an overall increase in methodological rigor.8
Current research has demonstrated a solid base from which to spawn specific treatment protocols. The available research is investigational in terms of treatment procedures. Replication of these studies could dictate treatment protocol and contribute substantively to future VA and DoD CPG updates. Future researchers should consider the use of a standard PTSD symptom assessment tool to make interstudy comparisons more meaningful. The length of treatment and exposure elements should be targeted specifically in future research as these components currently vary the most.
The military represents an obvious avenue for future research due to increased PTSD diagnosis in recent years. Although the etiology of the increase in PTSD is unclear and most likely multifactorial (decreased resilience, increased awareness, increased pursuit of secondary gains, etc), the need for treatment options is apparent.1 Group cohesion has been shown to be a core component of successful group psychotherapy, so military members who are accustomed to unit cohesion might represent a uniquely suitable population for this modality.15 Interestingly and for reasons not currently understood, veterans do not see effects of therapy as large as their civilian counterparts.8 This underscores the need for further evaluation of military-specific outcomes.
CONCLUSIONS
Although the available evidence is not robust, results do support the careful use of GCBT as an effective treatment for PTSD symptom reduction.8 Group psychotherapy has been generally regarded as an efficacious and cost-effective method to achieve similar outcomes to individual therapy. Increasing PTSD prevalence compels mental health care providers to explore all available treatment options. The potential for GCBT as an option is exciting, especially for mental health providers and those with limited resources. Rising health care standards and the current national fiscal situation is dictating a reevaluation of treatment options; so perhaps all health care providers will soon consider the use of GCBT.
As with any group assignment, the clinician should carefully consider the individual’s suitability and desire for group participation.16 With GCBT, providers could facilitate the relief of relentless apprehension and functional impairment for several patients simultaneously. Although there are many details left to explore regarding the use of GCBT for PTSD, the therapy’s foundation for use as a PTSD treatment is apparent.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Litz B, Schlenger W. Posttraumatic stress disorder in service members and new veterans of the Iraq and Afghanistan wars: A bibliography and critique. PTSD Res Q. 2009;20(1):1-3.
2. Tanielian T. Assessing combat exposure and post-traumatic stress disorder in troops and estimating the costs to society: Implications from the RAND Invisible Wounds of War Study. http://www.rand.org/pubs/testimonies/CT321.html. Published 2009. Accessed September 29, 2014.
3. Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
4. Cukor J, Spitalnick J, Difede J, Rizzo A, Rothbaum BO. Emerging treatments for PTSD. Clinical Psychol Rev. 2009;29(8):715-726.
5. Hoge CW. Interventions for war-related posttraumatic stress disorder: Meeting veterans where they are. JAMA. 2011;306(5):549-551.
6. Veterans Health Administration, Department of Defense. VA/DoD Clinical Practice Guideline: Management of Post-Traumatic Stress, Version 2.0. Washington, DC: Veterans Health Administration and Department of Defense; 2010.
7. Burlingame GM, Fuhriman A, Mosier J. The differential effectiveness of group psychotherapy: A meta-analytic perspective. Group Dyn. 2003;7(1):3-12.
8. Barrera TL, Mott JM, Hofstein RF, Teng EJ. A meta-analytic review of exposure in group cognitive behavioral therapy for posttraumatic stress disorder. Clin Psychol Rev. 2013;33(1):24-32.
9. Bisson J, Andrew M. Psychological treatment of post-traumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2007;(3):CD003388.
10. Khoo A, Dent MT, Oei TS. (2011). Group cognitive behaviour therapy for military service-related post-traumatic stress disorder: Effectiveness, sustainability and repeatability. Aust N Z J Psychiatry. 2011;45(8):663-672.
11. Swanson LM, Favorite TK, Horin E, Arnedt JT. A combined group treatment for nightmares and insomnia in combat veterans: A pilot study. J Trauma Stress. 2009;22(6):639-642.
12. Erickson DH, Janeck A, Tallman K. A cognitive-behavioral group for patients with various anxiety disorders. Psychiatr Serv. 2007;58(9):1205-1211.
13. Thompson AR, Wilde E, Boon K. The development of group CBT for the treatment of road-traffic-accident-related post-traumatic stress disorder: A pilot study. Cognitive Behav Therapist. 2010;2(1):32-42.
14. Slade T, Johnston A, Oakley-Browne MA, Andrews G, Whiteford H. 2007 National Survey of Mental Health and Wellbeing: Methods and key findings. Aust N Z J Psychiatry. 2009;43(7):594-605.
15. Crowe TP, Grenyer BF. Is therapist alliance or whole group cohesion more influential in group psychotherapy outcomes? Clin Psychol Psychother. 2008;15(4):239-246.
16. Leszcz M, Kobos JC. Evidence-based group psychotherapy: Using AGPA’s practice guidelines to enhance clinical effectiveness. J Clin Psychol. 2008;64(11):1238-1260.
Anxiety is a necessary and natural reaction to trauma, but, sometimes, anxiety symptoms become excessive and problematic, as experienced with posttraumatic stress disorder (PTSD). Some patients who struggle with PTSD endure a relentless apprehension so intense that it keeps them from participating in everyday activities, such as attending work and partaking in social activities. Associated anxiety symptoms severely impair everyday function and include increased heart rate, sweating, intrusive images, poor attention, fear, or insomnia. Posttraumatic stress disorder symptoms often lead to occupational dysfunction, relationship difficulty, and numerous other functional impairments.
Approximately 300,000 veterans meet the criteria for PTSD related to ongoing or recent wars.1 The veteran does not bear the personal and functional burden alone; however, the financial load is felt throughout society. One recent study suggests that for veterans diagnosed with PTSD, the first 2 years after deployment cost society an estimated $7,000 per individial.2 Current research suggests that this potentially debilitating disorder occurs in about 14% of Operation Iraqi Freedom/Operation Enduring Freedom combat troops, whereas the similar U.S. demographic population experiences PTSD at a rate of about 7%.1,3 The ongoing military trauma exposures are compelling the mental health community to establish efficient and effective treatment options.4,5
Several treatment strategies exist to reduce PTSD symptoms, but health care professionals must seek a balance between therapeutic benefit and cost. The treatment of PTSD is diverse and variable; however, in the most recent Clinical Practice Guideline (CPG) for PTSD, the VA and DoD specifically endorse some psychotherapeutic interventions while dissuading the use of others.6 Of note, the VA and DoD CPG strongly encourages Stress Inoculation Training (SIT) and similar cognitive therapies aimed at guiding patients through the process of consciously understanding the relationship between thoughts and feelings and then modifying thoughts to appropriately manage stressors.6 Meanwhile, group psychotherapy has been determined to be “somewhat helpful.”6 Even though cognitive- and group-based therapies have long been established as efficacious for numerous psychological disorders (depression, obsessive compulsive disorder, eating disorders, etc), neither the American Group Psychotherapy Association nor the VA and DoD CPG directly endorse the use of group cognitive behavioral therapy (GCBT) for the treatment of PTSD.6,7 However, both VA and DoD mental health providers commonly practice CBT and various group psychotherapies for the treatment of PTSD.
Despite the widespread use of CBT, there is a gap in the clinical understanding of the evidence supporting GCBT for PTSD. The goal of this synthesis was to understand the efficacy of treating PTSD symptoms with group psychotherapy. To begin this investigation, the following PICO (population, intervention, comparison, outcome) question was asked: In adults diagnosed with PTSD, how effective is group cognitive behavioral therapy in reducing PTSD-related symptoms?
Methods
Research articles addressing the use of GCBT in PTSD were obtained via database searches that took place during October 2012 (Table). Searched databases included the Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews Randomized Controlled Trials, Psychological Information (PsycINFO), and Public Medicine (PubMed).
The PubMed database was searched using the following MeSH (medical subject heading) terms: “psychotherapy, group” and “stress disorders, post-traumatic” and “cognitive therapy.” Limitations were set to include only patients aged ≥ 18 years, results in English, those involving human subjects, and articles published within the past 5 years. A manual search of references was also conducted, and relevant articles were retained.
Articles that addressed primary substance abuse, other DSM Axis I disorders, intimate partner violence, or family issues were excluded from the evidence sample due to concerns of an alternate treatment focus. Articles with a focus on telehealth or alternative medicine were considered confounding to the scope of this review were also excluded. It was also noted that the term CBT is used collectively for an umbrella of treatments; however, treatments that focused on elements other than the components of CBT being delivered in a group were not included. To prevent duplication of the results, research from an inclusive review was not considered individually.
SUMMARY OF EVIDENCE
Six works fulfilled the PICO criteria and were of sufficient quality to be synthesized. Of the 6 articles retained for synthesis, 2 were high-level reviews. Both reviews supported the use of GCBT for PTSD treatment. Barrera and colleagues reported an overall large effect size regardless of the presence of exposure in-group among the 12 treatment conditions and 651 study participants.8 These researchers also reported that in-group exposure did not further traumatize other group members.
Similarly, although a notably older and smaller review, Bisson and Andrew reported a significant standard mean deviation between 4 GCBT treatment and wait list controls. These reviewers did not find a significant difference between trauma- and nontrauma-focused treatment groups. The reviews also noted that individual psychotherapy and/or pharmacotherapy was most often continued throughout the reviewed studies.8,9
The 4 other studies contribute substantively to this synthesis but arguably represent lower evidence quality. A large longitudinal study of 496 Australian veterans reported a large effect size that was sustained 9 months after treatment began.10 These researchers used an intensive outpatient program that included medication and other treatment modalities as the basis for GCBT delivery. They reported that the majority of the patients revealed improvement in PTSD symptoms.
Another study sampled a similar group of 10 combat veterans but focused particular attention on sleep-related PTSD symptoms of insomnia, nightmares, and sleep quality.11 Although these researchers were unable to report a significant difference in overall PTSD symptoms for the 8 subjects who completed the protocol, they did find a large effect size on insomnia severity and a medium effect size on sleep quality. Regular treatment, including medication, continued throughout this study.
Other researchers reported a medium effect size on PTSD symptoms while using GCBT in a heterogeneous group with various anxiety disorders, including obsessive compulsive disorder, generalized anxiety disorder, social phobia, panic disorders, and PTSD.12 Although reporting similar results as all other included studies, this study has some significant limitations, including a 26% dropout rate among the 152 participants. The final study included for synthesis reported a remarkable 67% elimination of the PTSD diagnosis among 6 motor vehicle accident survivors in the small, uncontrolled study.13 Concomitant treatments, including medications, were not reported in detail for these 4 studies except as mentioned.
As a whole, the 6 studies revealed some appreciable commonalities. Time since diagnosis did not seem to influence the results. Attrition was consistently found to be similar to other PTSD treatments. The reported session topics were loosely based on common CBT tenets (ie, education, challenging cognitions, and relaxation techniques) and were typically similar among treatment groups, including the use of homework.
DISCUSSION
As the diagnosis of PTSD increases to unfamiliar levels, GCBT has the potential to be helpful to clinicians and patients seeking alternatives to their current treatments.1,4,14 The reported results imply that GCBT can be useful in PTSD symptom reduction. This could be particularly useful to VA and military providers or rural providers operating with limited resources.
Treatment protocols are not well established and should be approached with care prior to the establishment of CBT treatment groups for those diagnosed with PTSD. Session overviews and descriptions, such as those mentioned in Thompson and colleagues, could provide a reference point for future use.13
Also worth considering, CBT can be an ambiguous term requiring deliberate definition within treatment protocols. As noted in the VA and DoD CPG, exposure- and trauma-focused treatment designs can be efficacious, but these elements do not seem to be required within the GCBT treatment setting.
The current research also suggests GCBT efficacy regardless of the index trauma. This does not suggest that heterogeneous groups were frequently studied nor can conclusions be drawn regarding heterogeneous treatment groups. Elements such as group size and session length are inconsistently reported and require specific consideration as well. There is a distinct lack of research directly comparing individual CBT with GCBT directly, which prohibits meaningful conclusions regarding PTSD symptom reduction. This research gap may well have influenced the recommendations within the VA and DoD CPG. Although some higher quality studies exist, many of the published reports on GCBT have noteworthy design flaw, such as inadequate controls and statistical analysis.
LIMITATIONS
There are some limitations to this literature synthesis. Although the search was limited to the past 5 years, the inclusion of reviews accounts for older evidence. As alluded to earlier, the lack of a standardized GCBT treatment protocol challenges results comparisons as well. The consequent treatment variations make direct interstudy comparison and synthesis difficult. Similarly, outcome measures varied between studies. Also, group psychotherapy is well established and accepted. Therefore, much of the supporting research was accomplished outside the parameters of this literature search. This empirical view of group psychotherapy among mental health providers may also contribute to the lack of available research.
It is also worth noting that studies finding neutral or negative results are often unpublished. This publication bias could account for the lack of available evidence. The research reports do not consistently report therapist qualifications; however, board certificates in group psychotherapy and CBT are undeniably variables available for debate. The inclusion of uncontrolled trials limits these findings as well. Although the above limitations are not exhaustive, they do provide necessary caveats to future generalizations.
FUTURE IMPLICATIONS
Perhaps the most important information to gain from future research is that of treatment outcomes. Studies that include a detailed outcome evaluation could reveal patient satisfaction, efficacy, and financial considerations. In the presence of adequate supportive data, GCBT could contribute outcome data regarding trauma survivor symptom normalization, peer support formation, access to care, treatment efficiency, and health care resources utilization. As noted in Barrera and colleagues, future analysis will require a greater volume of trials with an overall increase in methodological rigor.8
Current research has demonstrated a solid base from which to spawn specific treatment protocols. The available research is investigational in terms of treatment procedures. Replication of these studies could dictate treatment protocol and contribute substantively to future VA and DoD CPG updates. Future researchers should consider the use of a standard PTSD symptom assessment tool to make interstudy comparisons more meaningful. The length of treatment and exposure elements should be targeted specifically in future research as these components currently vary the most.
The military represents an obvious avenue for future research due to increased PTSD diagnosis in recent years. Although the etiology of the increase in PTSD is unclear and most likely multifactorial (decreased resilience, increased awareness, increased pursuit of secondary gains, etc), the need for treatment options is apparent.1 Group cohesion has been shown to be a core component of successful group psychotherapy, so military members who are accustomed to unit cohesion might represent a uniquely suitable population for this modality.15 Interestingly and for reasons not currently understood, veterans do not see effects of therapy as large as their civilian counterparts.8 This underscores the need for further evaluation of military-specific outcomes.
CONCLUSIONS
Although the available evidence is not robust, results do support the careful use of GCBT as an effective treatment for PTSD symptom reduction.8 Group psychotherapy has been generally regarded as an efficacious and cost-effective method to achieve similar outcomes to individual therapy. Increasing PTSD prevalence compels mental health care providers to explore all available treatment options. The potential for GCBT as an option is exciting, especially for mental health providers and those with limited resources. Rising health care standards and the current national fiscal situation is dictating a reevaluation of treatment options; so perhaps all health care providers will soon consider the use of GCBT.
As with any group assignment, the clinician should carefully consider the individual’s suitability and desire for group participation.16 With GCBT, providers could facilitate the relief of relentless apprehension and functional impairment for several patients simultaneously. Although there are many details left to explore regarding the use of GCBT for PTSD, the therapy’s foundation for use as a PTSD treatment is apparent.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Anxiety is a necessary and natural reaction to trauma, but, sometimes, anxiety symptoms become excessive and problematic, as experienced with posttraumatic stress disorder (PTSD). Some patients who struggle with PTSD endure a relentless apprehension so intense that it keeps them from participating in everyday activities, such as attending work and partaking in social activities. Associated anxiety symptoms severely impair everyday function and include increased heart rate, sweating, intrusive images, poor attention, fear, or insomnia. Posttraumatic stress disorder symptoms often lead to occupational dysfunction, relationship difficulty, and numerous other functional impairments.
Approximately 300,000 veterans meet the criteria for PTSD related to ongoing or recent wars.1 The veteran does not bear the personal and functional burden alone; however, the financial load is felt throughout society. One recent study suggests that for veterans diagnosed with PTSD, the first 2 years after deployment cost society an estimated $7,000 per individial.2 Current research suggests that this potentially debilitating disorder occurs in about 14% of Operation Iraqi Freedom/Operation Enduring Freedom combat troops, whereas the similar U.S. demographic population experiences PTSD at a rate of about 7%.1,3 The ongoing military trauma exposures are compelling the mental health community to establish efficient and effective treatment options.4,5
Several treatment strategies exist to reduce PTSD symptoms, but health care professionals must seek a balance between therapeutic benefit and cost. The treatment of PTSD is diverse and variable; however, in the most recent Clinical Practice Guideline (CPG) for PTSD, the VA and DoD specifically endorse some psychotherapeutic interventions while dissuading the use of others.6 Of note, the VA and DoD CPG strongly encourages Stress Inoculation Training (SIT) and similar cognitive therapies aimed at guiding patients through the process of consciously understanding the relationship between thoughts and feelings and then modifying thoughts to appropriately manage stressors.6 Meanwhile, group psychotherapy has been determined to be “somewhat helpful.”6 Even though cognitive- and group-based therapies have long been established as efficacious for numerous psychological disorders (depression, obsessive compulsive disorder, eating disorders, etc), neither the American Group Psychotherapy Association nor the VA and DoD CPG directly endorse the use of group cognitive behavioral therapy (GCBT) for the treatment of PTSD.6,7 However, both VA and DoD mental health providers commonly practice CBT and various group psychotherapies for the treatment of PTSD.
Despite the widespread use of CBT, there is a gap in the clinical understanding of the evidence supporting GCBT for PTSD. The goal of this synthesis was to understand the efficacy of treating PTSD symptoms with group psychotherapy. To begin this investigation, the following PICO (population, intervention, comparison, outcome) question was asked: In adults diagnosed with PTSD, how effective is group cognitive behavioral therapy in reducing PTSD-related symptoms?
Methods
Research articles addressing the use of GCBT in PTSD were obtained via database searches that took place during October 2012 (Table). Searched databases included the Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews Randomized Controlled Trials, Psychological Information (PsycINFO), and Public Medicine (PubMed).
The PubMed database was searched using the following MeSH (medical subject heading) terms: “psychotherapy, group” and “stress disorders, post-traumatic” and “cognitive therapy.” Limitations were set to include only patients aged ≥ 18 years, results in English, those involving human subjects, and articles published within the past 5 years. A manual search of references was also conducted, and relevant articles were retained.
Articles that addressed primary substance abuse, other DSM Axis I disorders, intimate partner violence, or family issues were excluded from the evidence sample due to concerns of an alternate treatment focus. Articles with a focus on telehealth or alternative medicine were considered confounding to the scope of this review were also excluded. It was also noted that the term CBT is used collectively for an umbrella of treatments; however, treatments that focused on elements other than the components of CBT being delivered in a group were not included. To prevent duplication of the results, research from an inclusive review was not considered individually.
SUMMARY OF EVIDENCE
Six works fulfilled the PICO criteria and were of sufficient quality to be synthesized. Of the 6 articles retained for synthesis, 2 were high-level reviews. Both reviews supported the use of GCBT for PTSD treatment. Barrera and colleagues reported an overall large effect size regardless of the presence of exposure in-group among the 12 treatment conditions and 651 study participants.8 These researchers also reported that in-group exposure did not further traumatize other group members.
Similarly, although a notably older and smaller review, Bisson and Andrew reported a significant standard mean deviation between 4 GCBT treatment and wait list controls. These reviewers did not find a significant difference between trauma- and nontrauma-focused treatment groups. The reviews also noted that individual psychotherapy and/or pharmacotherapy was most often continued throughout the reviewed studies.8,9
The 4 other studies contribute substantively to this synthesis but arguably represent lower evidence quality. A large longitudinal study of 496 Australian veterans reported a large effect size that was sustained 9 months after treatment began.10 These researchers used an intensive outpatient program that included medication and other treatment modalities as the basis for GCBT delivery. They reported that the majority of the patients revealed improvement in PTSD symptoms.
Another study sampled a similar group of 10 combat veterans but focused particular attention on sleep-related PTSD symptoms of insomnia, nightmares, and sleep quality.11 Although these researchers were unable to report a significant difference in overall PTSD symptoms for the 8 subjects who completed the protocol, they did find a large effect size on insomnia severity and a medium effect size on sleep quality. Regular treatment, including medication, continued throughout this study.
Other researchers reported a medium effect size on PTSD symptoms while using GCBT in a heterogeneous group with various anxiety disorders, including obsessive compulsive disorder, generalized anxiety disorder, social phobia, panic disorders, and PTSD.12 Although reporting similar results as all other included studies, this study has some significant limitations, including a 26% dropout rate among the 152 participants. The final study included for synthesis reported a remarkable 67% elimination of the PTSD diagnosis among 6 motor vehicle accident survivors in the small, uncontrolled study.13 Concomitant treatments, including medications, were not reported in detail for these 4 studies except as mentioned.
As a whole, the 6 studies revealed some appreciable commonalities. Time since diagnosis did not seem to influence the results. Attrition was consistently found to be similar to other PTSD treatments. The reported session topics were loosely based on common CBT tenets (ie, education, challenging cognitions, and relaxation techniques) and were typically similar among treatment groups, including the use of homework.
DISCUSSION
As the diagnosis of PTSD increases to unfamiliar levels, GCBT has the potential to be helpful to clinicians and patients seeking alternatives to their current treatments.1,4,14 The reported results imply that GCBT can be useful in PTSD symptom reduction. This could be particularly useful to VA and military providers or rural providers operating with limited resources.
Treatment protocols are not well established and should be approached with care prior to the establishment of CBT treatment groups for those diagnosed with PTSD. Session overviews and descriptions, such as those mentioned in Thompson and colleagues, could provide a reference point for future use.13
Also worth considering, CBT can be an ambiguous term requiring deliberate definition within treatment protocols. As noted in the VA and DoD CPG, exposure- and trauma-focused treatment designs can be efficacious, but these elements do not seem to be required within the GCBT treatment setting.
The current research also suggests GCBT efficacy regardless of the index trauma. This does not suggest that heterogeneous groups were frequently studied nor can conclusions be drawn regarding heterogeneous treatment groups. Elements such as group size and session length are inconsistently reported and require specific consideration as well. There is a distinct lack of research directly comparing individual CBT with GCBT directly, which prohibits meaningful conclusions regarding PTSD symptom reduction. This research gap may well have influenced the recommendations within the VA and DoD CPG. Although some higher quality studies exist, many of the published reports on GCBT have noteworthy design flaw, such as inadequate controls and statistical analysis.
LIMITATIONS
There are some limitations to this literature synthesis. Although the search was limited to the past 5 years, the inclusion of reviews accounts for older evidence. As alluded to earlier, the lack of a standardized GCBT treatment protocol challenges results comparisons as well. The consequent treatment variations make direct interstudy comparison and synthesis difficult. Similarly, outcome measures varied between studies. Also, group psychotherapy is well established and accepted. Therefore, much of the supporting research was accomplished outside the parameters of this literature search. This empirical view of group psychotherapy among mental health providers may also contribute to the lack of available research.
It is also worth noting that studies finding neutral or negative results are often unpublished. This publication bias could account for the lack of available evidence. The research reports do not consistently report therapist qualifications; however, board certificates in group psychotherapy and CBT are undeniably variables available for debate. The inclusion of uncontrolled trials limits these findings as well. Although the above limitations are not exhaustive, they do provide necessary caveats to future generalizations.
FUTURE IMPLICATIONS
Perhaps the most important information to gain from future research is that of treatment outcomes. Studies that include a detailed outcome evaluation could reveal patient satisfaction, efficacy, and financial considerations. In the presence of adequate supportive data, GCBT could contribute outcome data regarding trauma survivor symptom normalization, peer support formation, access to care, treatment efficiency, and health care resources utilization. As noted in Barrera and colleagues, future analysis will require a greater volume of trials with an overall increase in methodological rigor.8
Current research has demonstrated a solid base from which to spawn specific treatment protocols. The available research is investigational in terms of treatment procedures. Replication of these studies could dictate treatment protocol and contribute substantively to future VA and DoD CPG updates. Future researchers should consider the use of a standard PTSD symptom assessment tool to make interstudy comparisons more meaningful. The length of treatment and exposure elements should be targeted specifically in future research as these components currently vary the most.
The military represents an obvious avenue for future research due to increased PTSD diagnosis in recent years. Although the etiology of the increase in PTSD is unclear and most likely multifactorial (decreased resilience, increased awareness, increased pursuit of secondary gains, etc), the need for treatment options is apparent.1 Group cohesion has been shown to be a core component of successful group psychotherapy, so military members who are accustomed to unit cohesion might represent a uniquely suitable population for this modality.15 Interestingly and for reasons not currently understood, veterans do not see effects of therapy as large as their civilian counterparts.8 This underscores the need for further evaluation of military-specific outcomes.
CONCLUSIONS
Although the available evidence is not robust, results do support the careful use of GCBT as an effective treatment for PTSD symptom reduction.8 Group psychotherapy has been generally regarded as an efficacious and cost-effective method to achieve similar outcomes to individual therapy. Increasing PTSD prevalence compels mental health care providers to explore all available treatment options. The potential for GCBT as an option is exciting, especially for mental health providers and those with limited resources. Rising health care standards and the current national fiscal situation is dictating a reevaluation of treatment options; so perhaps all health care providers will soon consider the use of GCBT.
As with any group assignment, the clinician should carefully consider the individual’s suitability and desire for group participation.16 With GCBT, providers could facilitate the relief of relentless apprehension and functional impairment for several patients simultaneously. Although there are many details left to explore regarding the use of GCBT for PTSD, the therapy’s foundation for use as a PTSD treatment is apparent.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Litz B, Schlenger W. Posttraumatic stress disorder in service members and new veterans of the Iraq and Afghanistan wars: A bibliography and critique. PTSD Res Q. 2009;20(1):1-3.
2. Tanielian T. Assessing combat exposure and post-traumatic stress disorder in troops and estimating the costs to society: Implications from the RAND Invisible Wounds of War Study. http://www.rand.org/pubs/testimonies/CT321.html. Published 2009. Accessed September 29, 2014.
3. Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
4. Cukor J, Spitalnick J, Difede J, Rizzo A, Rothbaum BO. Emerging treatments for PTSD. Clinical Psychol Rev. 2009;29(8):715-726.
5. Hoge CW. Interventions for war-related posttraumatic stress disorder: Meeting veterans where they are. JAMA. 2011;306(5):549-551.
6. Veterans Health Administration, Department of Defense. VA/DoD Clinical Practice Guideline: Management of Post-Traumatic Stress, Version 2.0. Washington, DC: Veterans Health Administration and Department of Defense; 2010.
7. Burlingame GM, Fuhriman A, Mosier J. The differential effectiveness of group psychotherapy: A meta-analytic perspective. Group Dyn. 2003;7(1):3-12.
8. Barrera TL, Mott JM, Hofstein RF, Teng EJ. A meta-analytic review of exposure in group cognitive behavioral therapy for posttraumatic stress disorder. Clin Psychol Rev. 2013;33(1):24-32.
9. Bisson J, Andrew M. Psychological treatment of post-traumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2007;(3):CD003388.
10. Khoo A, Dent MT, Oei TS. (2011). Group cognitive behaviour therapy for military service-related post-traumatic stress disorder: Effectiveness, sustainability and repeatability. Aust N Z J Psychiatry. 2011;45(8):663-672.
11. Swanson LM, Favorite TK, Horin E, Arnedt JT. A combined group treatment for nightmares and insomnia in combat veterans: A pilot study. J Trauma Stress. 2009;22(6):639-642.
12. Erickson DH, Janeck A, Tallman K. A cognitive-behavioral group for patients with various anxiety disorders. Psychiatr Serv. 2007;58(9):1205-1211.
13. Thompson AR, Wilde E, Boon K. The development of group CBT for the treatment of road-traffic-accident-related post-traumatic stress disorder: A pilot study. Cognitive Behav Therapist. 2010;2(1):32-42.
14. Slade T, Johnston A, Oakley-Browne MA, Andrews G, Whiteford H. 2007 National Survey of Mental Health and Wellbeing: Methods and key findings. Aust N Z J Psychiatry. 2009;43(7):594-605.
15. Crowe TP, Grenyer BF. Is therapist alliance or whole group cohesion more influential in group psychotherapy outcomes? Clin Psychol Psychother. 2008;15(4):239-246.
16. Leszcz M, Kobos JC. Evidence-based group psychotherapy: Using AGPA’s practice guidelines to enhance clinical effectiveness. J Clin Psychol. 2008;64(11):1238-1260.
1. Litz B, Schlenger W. Posttraumatic stress disorder in service members and new veterans of the Iraq and Afghanistan wars: A bibliography and critique. PTSD Res Q. 2009;20(1):1-3.
2. Tanielian T. Assessing combat exposure and post-traumatic stress disorder in troops and estimating the costs to society: Implications from the RAND Invisible Wounds of War Study. http://www.rand.org/pubs/testimonies/CT321.html. Published 2009. Accessed September 29, 2014.
3. Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-627.
4. Cukor J, Spitalnick J, Difede J, Rizzo A, Rothbaum BO. Emerging treatments for PTSD. Clinical Psychol Rev. 2009;29(8):715-726.
5. Hoge CW. Interventions for war-related posttraumatic stress disorder: Meeting veterans where they are. JAMA. 2011;306(5):549-551.
6. Veterans Health Administration, Department of Defense. VA/DoD Clinical Practice Guideline: Management of Post-Traumatic Stress, Version 2.0. Washington, DC: Veterans Health Administration and Department of Defense; 2010.
7. Burlingame GM, Fuhriman A, Mosier J. The differential effectiveness of group psychotherapy: A meta-analytic perspective. Group Dyn. 2003;7(1):3-12.
8. Barrera TL, Mott JM, Hofstein RF, Teng EJ. A meta-analytic review of exposure in group cognitive behavioral therapy for posttraumatic stress disorder. Clin Psychol Rev. 2013;33(1):24-32.
9. Bisson J, Andrew M. Psychological treatment of post-traumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2007;(3):CD003388.
10. Khoo A, Dent MT, Oei TS. (2011). Group cognitive behaviour therapy for military service-related post-traumatic stress disorder: Effectiveness, sustainability and repeatability. Aust N Z J Psychiatry. 2011;45(8):663-672.
11. Swanson LM, Favorite TK, Horin E, Arnedt JT. A combined group treatment for nightmares and insomnia in combat veterans: A pilot study. J Trauma Stress. 2009;22(6):639-642.
12. Erickson DH, Janeck A, Tallman K. A cognitive-behavioral group for patients with various anxiety disorders. Psychiatr Serv. 2007;58(9):1205-1211.
13. Thompson AR, Wilde E, Boon K. The development of group CBT for the treatment of road-traffic-accident-related post-traumatic stress disorder: A pilot study. Cognitive Behav Therapist. 2010;2(1):32-42.
14. Slade T, Johnston A, Oakley-Browne MA, Andrews G, Whiteford H. 2007 National Survey of Mental Health and Wellbeing: Methods and key findings. Aust N Z J Psychiatry. 2009;43(7):594-605.
15. Crowe TP, Grenyer BF. Is therapist alliance or whole group cohesion more influential in group psychotherapy outcomes? Clin Psychol Psychother. 2008;15(4):239-246.
16. Leszcz M, Kobos JC. Evidence-based group psychotherapy: Using AGPA’s practice guidelines to enhance clinical effectiveness. J Clin Psychol. 2008;64(11):1238-1260.
CAR T cells serve as bridge to HSCT in ALL

Credit: Charles Haymond
An anti-CD19 chimeric antigen receptor (CAR) T-cell therapy can elicit complete responses (CRs) in heavily pretreated patients with acute lymphoblastic leukemia (ALL), results of a phase 1 trial suggest.
More than half of the 21 patients enrolled achieved a CR, and most of those patients went on to hematopoietic stem cell transplant (HSCT).
All 10 patients who underwent HSCT remain leukemia-free at a median follow-up of 10 months.
The CAR T cells did prompt some serious adverse effects, but all effects were fully reversible.
Crystal L. Mackall, MD, of the National Cancer Institute in Bethesda, Maryland, and her colleagues reported these results in The Lancet.
The phase 1 study enrolled patients ages 1 to 30 years who had relapsed or refractory ALL or non-Hodgkin lymphoma. Twenty patients had B-cell ALL, and 1 had diffuse large B-cell lymphoma (DLBCL). All of the patients had been heavily pretreated, and 8 had received a prior HSCT.
Patients received a conditioning regimen of cyclophosphamide and fludarabine, followed by a single infusion of CAR T cells: either 1×10⁶ CAR-transduced T cells per kg or 3×10⁶ CAR-transduced T cells per kg.
The CAR T cells were produced from each patient’s own peripheral blood mononuclear cells, modified using a gammaretroviral vector encoding the CAR, as well as a CD28 costimulatory moiety. After the dose-escalation phase, an expansion cohort was treated at the maximum-tolerated dose.
Twenty-one patients were enrolled and infused, but 2 of them did not receive the prescribed dose of CAR T cells, as the assigned dose could not be generated. The maximum-tolerated dose was 1×10⁶ CAR T cells per kg.
All toxicities were fully reversible. The most common non-hematologic grade 3 adverse events were fever (n=9), hypokalemia (n=9), fever and neutropenia (n=8), and cytokine release syndrome (n=3). Grade 4 cytokine release syndrome occurred in 3 patients.
At day 28, 12 patients had achieved a minimal residual disease (MRD)-negative CR. One patient had an MRD-positive CR, 1 had an MRD-positive CR with incomplete count recovery, and 3 patients had stable disease. Four patients, including the one with DLBCL, progressed.
Ten of the patients with an MRD-negative CR subsequently underwent HSCT, and all 10 remained disease-free with a median follow-up of 10 months.
“The results show that this treatment is feasible in many patients with ALL and can eradicate chemoresistant disease with an acceptable toxicity profile,” said Gary Schiller, MD, of the University of California, Los Angeles, who was not involved in this study.
“Further, the findings demonstrate substantially higher response rates than seen in the literature for the most recently approved agent for refractory ALL. CD19-CAR therapy represents a potentially important new tool to address the urgent need for new treatment modalities in these patients.”
Researchers previously reported positive results with this therapy in patients with chemotherapy-refractory DLBCL. ![]()
Credit: Charles Haymond
An anti-CD19 chimeric antigen receptor (CAR) T-cell therapy can elicit complete responses (CRs) in heavily pretreated patients with acute lymphoblastic leukemia (ALL), results of a phase 1 trial suggest.
More than half of the 21 patients enrolled achieved a CR, and most of those patients went on to hematopoietic stem cell transplant (HSCT).
All 10 patients who underwent HSCT remain leukemia-free at a median follow-up of 10 months.
The CAR T cells did prompt some serious adverse effects, but all effects were fully reversible.
Crystal L. Mackall, MD, of the National Cancer Institute in Bethesda, Maryland, and her colleagues reported these results in The Lancet.
The phase 1 study enrolled patients ages 1 to 30 years who had relapsed or refractory ALL or non-Hodgkin lymphoma. Twenty patients had B-cell ALL, and 1 had diffuse large B-cell lymphoma (DLBCL). All of the patients had been heavily pretreated, and 8 had received a prior HSCT.
Patients received a conditioning regimen of cyclophosphamide and fludarabine, followed by a single infusion of CAR T cells: either 1×10⁶ CAR-transduced T cells per kg or 3×10⁶ CAR-transduced T cells per kg.
The CAR T cells were produced from each patient’s own peripheral blood mononuclear cells, modified using a gammaretroviral vector encoding the CAR, as well as a CD28 costimulatory moiety. After the dose-escalation phase, an expansion cohort was treated at the maximum-tolerated dose.
Twenty-one patients were enrolled and infused, but 2 of them did not receive the prescribed dose of CAR T cells, as the assigned dose could not be generated. The maximum-tolerated dose was 1×10⁶ CAR T cells per kg.
All toxicities were fully reversible. The most common non-hematologic grade 3 adverse events were fever (n=9), hypokalemia (n=9), fever and neutropenia (n=8), and cytokine release syndrome (n=3). Grade 4 cytokine release syndrome occurred in 3 patients.
At day 28, 12 patients had achieved a minimal residual disease (MRD)-negative CR. One patient had an MRD-positive CR, 1 had an MRD-positive CR with incomplete count recovery, and 3 patients had stable disease. Four patients, including the one with DLBCL, progressed.
Ten of the patients with an MRD-negative CR subsequently underwent HSCT, and all 10 remained disease-free with a median follow-up of 10 months.
“The results show that this treatment is feasible in many patients with ALL and can eradicate chemoresistant disease with an acceptable toxicity profile,” said Gary Schiller, MD, of the University of California, Los Angeles, who was not involved in this study.
“Further, the findings demonstrate substantially higher response rates than seen in the literature for the most recently approved agent for refractory ALL. CD19-CAR therapy represents a potentially important new tool to address the urgent need for new treatment modalities in these patients.”
Researchers previously reported positive results with this therapy in patients with chemotherapy-refractory DLBCL. ![]()
Credit: Charles Haymond
An anti-CD19 chimeric antigen receptor (CAR) T-cell therapy can elicit complete responses (CRs) in heavily pretreated patients with acute lymphoblastic leukemia (ALL), results of a phase 1 trial suggest.
More than half of the 21 patients enrolled achieved a CR, and most of those patients went on to hematopoietic stem cell transplant (HSCT).
All 10 patients who underwent HSCT remain leukemia-free at a median follow-up of 10 months.
The CAR T cells did prompt some serious adverse effects, but all effects were fully reversible.
Crystal L. Mackall, MD, of the National Cancer Institute in Bethesda, Maryland, and her colleagues reported these results in The Lancet.
The phase 1 study enrolled patients ages 1 to 30 years who had relapsed or refractory ALL or non-Hodgkin lymphoma. Twenty patients had B-cell ALL, and 1 had diffuse large B-cell lymphoma (DLBCL). All of the patients had been heavily pretreated, and 8 had received a prior HSCT.
Patients received a conditioning regimen of cyclophosphamide and fludarabine, followed by a single infusion of CAR T cells: either 1×10⁶ CAR-transduced T cells per kg or 3×10⁶ CAR-transduced T cells per kg.
The CAR T cells were produced from each patient’s own peripheral blood mononuclear cells, modified using a gammaretroviral vector encoding the CAR, as well as a CD28 costimulatory moiety. After the dose-escalation phase, an expansion cohort was treated at the maximum-tolerated dose.
Twenty-one patients were enrolled and infused, but 2 of them did not receive the prescribed dose of CAR T cells, as the assigned dose could not be generated. The maximum-tolerated dose was 1×10⁶ CAR T cells per kg.
All toxicities were fully reversible. The most common non-hematologic grade 3 adverse events were fever (n=9), hypokalemia (n=9), fever and neutropenia (n=8), and cytokine release syndrome (n=3). Grade 4 cytokine release syndrome occurred in 3 patients.
At day 28, 12 patients had achieved a minimal residual disease (MRD)-negative CR. One patient had an MRD-positive CR, 1 had an MRD-positive CR with incomplete count recovery, and 3 patients had stable disease. Four patients, including the one with DLBCL, progressed.
Ten of the patients with an MRD-negative CR subsequently underwent HSCT, and all 10 remained disease-free with a median follow-up of 10 months.
“The results show that this treatment is feasible in many patients with ALL and can eradicate chemoresistant disease with an acceptable toxicity profile,” said Gary Schiller, MD, of the University of California, Los Angeles, who was not involved in this study.
“Further, the findings demonstrate substantially higher response rates than seen in the literature for the most recently approved agent for refractory ALL. CD19-CAR therapy represents a potentially important new tool to address the urgent need for new treatment modalities in these patients.”
Researchers previously reported positive results with this therapy in patients with chemotherapy-refractory DLBCL. ![]()
Lactate Clearance Portends Better Outcomes after Cardiac Arrest
Clinical question: Is greater lactate clearance following resuscitation from cardiac arrest associated with lower mortality and better neurologic outcomes?
Background: Recommendations from the International Liaison Committee on Resuscitation for monitoring serial lactate levels in post-resuscitation patients are based primarily on extrapolation from other conditions such as sepsis. Two single-retrospective analyses found effective lactate clearance was associated with decreased mortality. This association had not previously been validated in a multicenter, prospective study.
Study design: Multicenter, prospective, observational study.
Setting: Four urban, tertiary-care teaching hospitals.
Synopsis: Absolute lactate levels and the differences in the percent lactate change over 24 hours were compared in 100 patients who suffered out-of-hospital cardiac arrest. Ninety-seven percent received therapeutic hypothermia, and overall survival was 46%. Survivors and patients with a good neurologic outcome had lower lactate levels at zero hours (4.1 vs. 7.3), 12 hours (2.2 vs. 6.0), and 24 hours (1.6 vs. 4.4) compared with nonsurvivors and patients with bad neurologic outcomes.
The percent lactate decreased was greater in survivors and in those with good neurologic outcomes (odds ratio, 2.2; 95% confidence interval, 1.1–4.4).
Nonsurvivors or those with poor neurologic outcomes were less likely to have received bystander CPR, to have suffered a witnessed arrest, or to have had a shockable rhythm at presentation. Superior lactate clearance in survivors and those with good neurologic outcomes suggests a potential role in developing markers of effective resuscitation.
Bottom line: Lower lactate levels and more effective clearance of lactate in patients following cardiac arrest are associated with improved survival and good neurologic outcome.
Citation: Donnino MW, Andersen LW, Giberson T, et al. Initial lactate and lactate change in post-cardiac arrest: a multicenter validation study. Crit Care Med. 2014;42(8):1804-1811.
Clinical question: Is greater lactate clearance following resuscitation from cardiac arrest associated with lower mortality and better neurologic outcomes?
Background: Recommendations from the International Liaison Committee on Resuscitation for monitoring serial lactate levels in post-resuscitation patients are based primarily on extrapolation from other conditions such as sepsis. Two single-retrospective analyses found effective lactate clearance was associated with decreased mortality. This association had not previously been validated in a multicenter, prospective study.
Study design: Multicenter, prospective, observational study.
Setting: Four urban, tertiary-care teaching hospitals.
Synopsis: Absolute lactate levels and the differences in the percent lactate change over 24 hours were compared in 100 patients who suffered out-of-hospital cardiac arrest. Ninety-seven percent received therapeutic hypothermia, and overall survival was 46%. Survivors and patients with a good neurologic outcome had lower lactate levels at zero hours (4.1 vs. 7.3), 12 hours (2.2 vs. 6.0), and 24 hours (1.6 vs. 4.4) compared with nonsurvivors and patients with bad neurologic outcomes.
The percent lactate decreased was greater in survivors and in those with good neurologic outcomes (odds ratio, 2.2; 95% confidence interval, 1.1–4.4).
Nonsurvivors or those with poor neurologic outcomes were less likely to have received bystander CPR, to have suffered a witnessed arrest, or to have had a shockable rhythm at presentation. Superior lactate clearance in survivors and those with good neurologic outcomes suggests a potential role in developing markers of effective resuscitation.
Bottom line: Lower lactate levels and more effective clearance of lactate in patients following cardiac arrest are associated with improved survival and good neurologic outcome.
Citation: Donnino MW, Andersen LW, Giberson T, et al. Initial lactate and lactate change in post-cardiac arrest: a multicenter validation study. Crit Care Med. 2014;42(8):1804-1811.
Clinical question: Is greater lactate clearance following resuscitation from cardiac arrest associated with lower mortality and better neurologic outcomes?
Background: Recommendations from the International Liaison Committee on Resuscitation for monitoring serial lactate levels in post-resuscitation patients are based primarily on extrapolation from other conditions such as sepsis. Two single-retrospective analyses found effective lactate clearance was associated with decreased mortality. This association had not previously been validated in a multicenter, prospective study.
Study design: Multicenter, prospective, observational study.
Setting: Four urban, tertiary-care teaching hospitals.
Synopsis: Absolute lactate levels and the differences in the percent lactate change over 24 hours were compared in 100 patients who suffered out-of-hospital cardiac arrest. Ninety-seven percent received therapeutic hypothermia, and overall survival was 46%. Survivors and patients with a good neurologic outcome had lower lactate levels at zero hours (4.1 vs. 7.3), 12 hours (2.2 vs. 6.0), and 24 hours (1.6 vs. 4.4) compared with nonsurvivors and patients with bad neurologic outcomes.
The percent lactate decreased was greater in survivors and in those with good neurologic outcomes (odds ratio, 2.2; 95% confidence interval, 1.1–4.4).
Nonsurvivors or those with poor neurologic outcomes were less likely to have received bystander CPR, to have suffered a witnessed arrest, or to have had a shockable rhythm at presentation. Superior lactate clearance in survivors and those with good neurologic outcomes suggests a potential role in developing markers of effective resuscitation.
Bottom line: Lower lactate levels and more effective clearance of lactate in patients following cardiac arrest are associated with improved survival and good neurologic outcome.
Citation: Donnino MW, Andersen LW, Giberson T, et al. Initial lactate and lactate change in post-cardiac arrest: a multicenter validation study. Crit Care Med. 2014;42(8):1804-1811.
Time to Meds Matters for Patients with Cardiac Arrest Due to Nonshockable Rhythms
Clinical question: Is earlier administration of epinephrine in patients with cardiac arrest due to nonshockable rhythms associated with increased return of spontaneous circulation, survival, and neurologically intact survival?
Background: About 200,000 hospitalized patients in the U.S. have a cardiac arrest, commonly due to nonshockable rhythms. Cardiopulmonary resuscitation has been the only efficacious intervention. There are no well-controlled trials of the use of epinephrine on survival and neurological outcomes.
Study design: Prospective cohort from a large multicenter registry of in-hospital cardiac arrests.
Setting: Data from 570 hospitals from 2000 to 2009.
Synopsis: Authors included 25,095 adults from 570 hospitals who had cardiac arrests in hospital with asystole or pulseless electrical activity as the initial rhythm. Time to first administration of epinephrine was recorded and then separated into quartiles, and odds ratios were evaluated using one to three minutes as the reference group. Outcomes of survival to hospital discharge (10%), return of spontaneous circulation (47%), and survival to hospital discharge with favorable neurologic status (7%) were assessed.
Survival to discharge decreased as the time to administration of the first dose of epinephrine increased. Of those patients receiving epinephrine in one minute, 12% survived. This dropped to 7% for those first receiving epinephrine after seven minutes. Return of spontaneous circulation and survival to discharge with favorable neurologic status showed a similar stepwise decrease with longer times to first administration of epinephrine.
Bottom line: Earlier administration of epinephrine to patients with cardiac arrest due to nonshockable rhythms is associated with improved survival to discharge, return of spontaneous circulation, and neurologically intact survival.
Citation: Donnino MW, Salciccioli JD, Howell MD, et al. Time to administration of epinephrine and outcome after in-hospital cardiac arrest with non-shockable rhythms: restrospective analysis of large in-hospital data registry. BMJ. 2014;348:g3028.
Clinical question: Is earlier administration of epinephrine in patients with cardiac arrest due to nonshockable rhythms associated with increased return of spontaneous circulation, survival, and neurologically intact survival?
Background: About 200,000 hospitalized patients in the U.S. have a cardiac arrest, commonly due to nonshockable rhythms. Cardiopulmonary resuscitation has been the only efficacious intervention. There are no well-controlled trials of the use of epinephrine on survival and neurological outcomes.
Study design: Prospective cohort from a large multicenter registry of in-hospital cardiac arrests.
Setting: Data from 570 hospitals from 2000 to 2009.
Synopsis: Authors included 25,095 adults from 570 hospitals who had cardiac arrests in hospital with asystole or pulseless electrical activity as the initial rhythm. Time to first administration of epinephrine was recorded and then separated into quartiles, and odds ratios were evaluated using one to three minutes as the reference group. Outcomes of survival to hospital discharge (10%), return of spontaneous circulation (47%), and survival to hospital discharge with favorable neurologic status (7%) were assessed.
Survival to discharge decreased as the time to administration of the first dose of epinephrine increased. Of those patients receiving epinephrine in one minute, 12% survived. This dropped to 7% for those first receiving epinephrine after seven minutes. Return of spontaneous circulation and survival to discharge with favorable neurologic status showed a similar stepwise decrease with longer times to first administration of epinephrine.
Bottom line: Earlier administration of epinephrine to patients with cardiac arrest due to nonshockable rhythms is associated with improved survival to discharge, return of spontaneous circulation, and neurologically intact survival.
Citation: Donnino MW, Salciccioli JD, Howell MD, et al. Time to administration of epinephrine and outcome after in-hospital cardiac arrest with non-shockable rhythms: restrospective analysis of large in-hospital data registry. BMJ. 2014;348:g3028.
Clinical question: Is earlier administration of epinephrine in patients with cardiac arrest due to nonshockable rhythms associated with increased return of spontaneous circulation, survival, and neurologically intact survival?
Background: About 200,000 hospitalized patients in the U.S. have a cardiac arrest, commonly due to nonshockable rhythms. Cardiopulmonary resuscitation has been the only efficacious intervention. There are no well-controlled trials of the use of epinephrine on survival and neurological outcomes.
Study design: Prospective cohort from a large multicenter registry of in-hospital cardiac arrests.
Setting: Data from 570 hospitals from 2000 to 2009.
Synopsis: Authors included 25,095 adults from 570 hospitals who had cardiac arrests in hospital with asystole or pulseless electrical activity as the initial rhythm. Time to first administration of epinephrine was recorded and then separated into quartiles, and odds ratios were evaluated using one to three minutes as the reference group. Outcomes of survival to hospital discharge (10%), return of spontaneous circulation (47%), and survival to hospital discharge with favorable neurologic status (7%) were assessed.
Survival to discharge decreased as the time to administration of the first dose of epinephrine increased. Of those patients receiving epinephrine in one minute, 12% survived. This dropped to 7% for those first receiving epinephrine after seven minutes. Return of spontaneous circulation and survival to discharge with favorable neurologic status showed a similar stepwise decrease with longer times to first administration of epinephrine.
Bottom line: Earlier administration of epinephrine to patients with cardiac arrest due to nonshockable rhythms is associated with improved survival to discharge, return of spontaneous circulation, and neurologically intact survival.
Citation: Donnino MW, Salciccioli JD, Howell MD, et al. Time to administration of epinephrine and outcome after in-hospital cardiac arrest with non-shockable rhythms: restrospective analysis of large in-hospital data registry. BMJ. 2014;348:g3028.