User login
UV Light Beat Bleach for C. difficile Decontamination
SAN FRANCISCO – The M.D. Anderson Cancer Center is abandoning bleach for cleaning hospital rooms exposed to Clostridium difficile in favor of a new machine that kills the organism using ultraviolet light.
The machine reduced C. difficile counts as much as, or more than, bleach cleaning in a preliminary prospective trial in 30 hospital rooms previously occupied by patients infected with C. difficile. The machine is a bit more expensive than bleach at a cost of approximately $82,000 (or $3,000-$4,000 per month to lease), but it avoids damage to materials and the toxic environment for workers caused by the use of bleach or other corrosive chemicals, Dr. Shashank S. Ghantoji said in an interview at a poster presentation at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
Bleach treatment reduced the average number of colony-forming units of C. difficile from 2.39 before cleaning to 0.71, a 70% reduction in the contamination level. Treatment with the Pulsed Xenon UV machine (PX-UV) reduced the average number of colony-forming units from 22.97 to 1.10, a 95% reduction.
The postcleaning contamination levels were not statistically different between the bleach and PX-UV rooms, Dr. Ghantoji and his associates found. However, PX-UV decontamination is faster than using bleach, Dr. Ghantoji said. "It takes at least 45 minutes to clean a room with bleach, and it’s not good for the patients or the health care professionals," plus admissions staff usually are clamoring for the room to be ready as soon as possible, he said. Cleaning a room using the PX-UV method takes perhaps 15 minutes.
The PX-UV machine has been available for some time, but its adoption depends on how proactive hospital infection control teams are, he added. He said he is aware of at least two medical centers beyond M.D. Anderson that are also using the machine.
In the study, 298 samples were taken before and after cleaning from high-touch surfaces – the bathroom handrail, the bed control panel, the bed rail, the top of the bedside table, and the IV pole control panel or other equipment control panel – and analyzed for C. difficile endospores. Fifteen rooms were cleaned by the conventional method using a 1:10 solution of sodium hypochlorite (bleach), and 15 underwent a visual, nonbleach cleaning of surfaces followed by 15 minutes of treatment with the PX-UV.
With the PX-UV method, housekeeping workers clean the bathroom and place the remote-operated PX-UV in the bathroom with the door shut while they finish cleaning the rest of the room. Then the machine is placed on each side of the bed for 4 minutes of operation with workers gone. Sensors stop the machine if any movement is detected.
It works by emitting ultraviolet C light, which kills C. difficile. And here’s a bonus – it also kills vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus, Dr. Ghantoji of M.D. Anderson, Houston, said at the meeting, sponsored by the American Society for Microbiology.

"The PX-UV method may be a promising alternative to the current standard of decontamination, bleach," he said. Future studies should look at whether the PX-UV method decreases not just endospore counts but transmission of C. difficile, he added.
C. difficile causes more than 300,000 health care–associated infections each year in the United States, incurring $2,500-$3,500 in costs per infection aside from any surgical costs, he estimated. Current guidelines recommend that rooms previously occupied by patients infected with C. difficile be cleaned with a disinfectant registered with the Environmental Protection Agency as effective against the organism.
Xenex Healthcare Services, which markets the PX-UV machine, funded the study, and two of the investigators are employees of the company. Dr. Ghantoji reported having no other relevant financial disclosures.
SAN FRANCISCO – The M.D. Anderson Cancer Center is abandoning bleach for cleaning hospital rooms exposed to Clostridium difficile in favor of a new machine that kills the organism using ultraviolet light.
The machine reduced C. difficile counts as much as, or more than, bleach cleaning in a preliminary prospective trial in 30 hospital rooms previously occupied by patients infected with C. difficile. The machine is a bit more expensive than bleach at a cost of approximately $82,000 (or $3,000-$4,000 per month to lease), but it avoids damage to materials and the toxic environment for workers caused by the use of bleach or other corrosive chemicals, Dr. Shashank S. Ghantoji said in an interview at a poster presentation at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
Bleach treatment reduced the average number of colony-forming units of C. difficile from 2.39 before cleaning to 0.71, a 70% reduction in the contamination level. Treatment with the Pulsed Xenon UV machine (PX-UV) reduced the average number of colony-forming units from 22.97 to 1.10, a 95% reduction.
The postcleaning contamination levels were not statistically different between the bleach and PX-UV rooms, Dr. Ghantoji and his associates found. However, PX-UV decontamination is faster than using bleach, Dr. Ghantoji said. "It takes at least 45 minutes to clean a room with bleach, and it’s not good for the patients or the health care professionals," plus admissions staff usually are clamoring for the room to be ready as soon as possible, he said. Cleaning a room using the PX-UV method takes perhaps 15 minutes.
The PX-UV machine has been available for some time, but its adoption depends on how proactive hospital infection control teams are, he added. He said he is aware of at least two medical centers beyond M.D. Anderson that are also using the machine.
In the study, 298 samples were taken before and after cleaning from high-touch surfaces – the bathroom handrail, the bed control panel, the bed rail, the top of the bedside table, and the IV pole control panel or other equipment control panel – and analyzed for C. difficile endospores. Fifteen rooms were cleaned by the conventional method using a 1:10 solution of sodium hypochlorite (bleach), and 15 underwent a visual, nonbleach cleaning of surfaces followed by 15 minutes of treatment with the PX-UV.
With the PX-UV method, housekeeping workers clean the bathroom and place the remote-operated PX-UV in the bathroom with the door shut while they finish cleaning the rest of the room. Then the machine is placed on each side of the bed for 4 minutes of operation with workers gone. Sensors stop the machine if any movement is detected.
It works by emitting ultraviolet C light, which kills C. difficile. And here’s a bonus – it also kills vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus, Dr. Ghantoji of M.D. Anderson, Houston, said at the meeting, sponsored by the American Society for Microbiology.
"The PX-UV method may be a promising alternative to the current standard of decontamination, bleach," he said. Future studies should look at whether the PX-UV method decreases not just endospore counts but transmission of C. difficile, he added.
C. difficile causes more than 300,000 health care–associated infections each year in the United States, incurring $2,500-$3,500 in costs per infection aside from any surgical costs, he estimated. Current guidelines recommend that rooms previously occupied by patients infected with C. difficile be cleaned with a disinfectant registered with the Environmental Protection Agency as effective against the organism.
Xenex Healthcare Services, which markets the PX-UV machine, funded the study, and two of the investigators are employees of the company. Dr. Ghantoji reported having no other relevant financial disclosures.
SAN FRANCISCO – The M.D. Anderson Cancer Center is abandoning bleach for cleaning hospital rooms exposed to Clostridium difficile in favor of a new machine that kills the organism using ultraviolet light.
The machine reduced C. difficile counts as much as, or more than, bleach cleaning in a preliminary prospective trial in 30 hospital rooms previously occupied by patients infected with C. difficile. The machine is a bit more expensive than bleach at a cost of approximately $82,000 (or $3,000-$4,000 per month to lease), but it avoids damage to materials and the toxic environment for workers caused by the use of bleach or other corrosive chemicals, Dr. Shashank S. Ghantoji said in an interview at a poster presentation at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy.
Bleach treatment reduced the average number of colony-forming units of C. difficile from 2.39 before cleaning to 0.71, a 70% reduction in the contamination level. Treatment with the Pulsed Xenon UV machine (PX-UV) reduced the average number of colony-forming units from 22.97 to 1.10, a 95% reduction.
The postcleaning contamination levels were not statistically different between the bleach and PX-UV rooms, Dr. Ghantoji and his associates found. However, PX-UV decontamination is faster than using bleach, Dr. Ghantoji said. "It takes at least 45 minutes to clean a room with bleach, and it’s not good for the patients or the health care professionals," plus admissions staff usually are clamoring for the room to be ready as soon as possible, he said. Cleaning a room using the PX-UV method takes perhaps 15 minutes.
The PX-UV machine has been available for some time, but its adoption depends on how proactive hospital infection control teams are, he added. He said he is aware of at least two medical centers beyond M.D. Anderson that are also using the machine.
In the study, 298 samples were taken before and after cleaning from high-touch surfaces – the bathroom handrail, the bed control panel, the bed rail, the top of the bedside table, and the IV pole control panel or other equipment control panel – and analyzed for C. difficile endospores. Fifteen rooms were cleaned by the conventional method using a 1:10 solution of sodium hypochlorite (bleach), and 15 underwent a visual, nonbleach cleaning of surfaces followed by 15 minutes of treatment with the PX-UV.
With the PX-UV method, housekeeping workers clean the bathroom and place the remote-operated PX-UV in the bathroom with the door shut while they finish cleaning the rest of the room. Then the machine is placed on each side of the bed for 4 minutes of operation with workers gone. Sensors stop the machine if any movement is detected.
It works by emitting ultraviolet C light, which kills C. difficile. And here’s a bonus – it also kills vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus, Dr. Ghantoji of M.D. Anderson, Houston, said at the meeting, sponsored by the American Society for Microbiology.
"The PX-UV method may be a promising alternative to the current standard of decontamination, bleach," he said. Future studies should look at whether the PX-UV method decreases not just endospore counts but transmission of C. difficile, he added.
C. difficile causes more than 300,000 health care–associated infections each year in the United States, incurring $2,500-$3,500 in costs per infection aside from any surgical costs, he estimated. Current guidelines recommend that rooms previously occupied by patients infected with C. difficile be cleaned with a disinfectant registered with the Environmental Protection Agency as effective against the organism.
Xenex Healthcare Services, which markets the PX-UV machine, funded the study, and two of the investigators are employees of the company. Dr. Ghantoji reported having no other relevant financial disclosures.
Major Finding: Bleach killed 70% of C. difficile spores in hospital rooms compared with 95% decontamination using nonbleach cleaning plus UV light treatment. The difference between groups was not statistically significant.
Data Source: A prospective comparison was performed of the two cleaning methods in 30 rooms after discharge of patients infected with C. difficile.
Disclosures: Xenex Healthcare Services, which markets the PX-UV machine, funded the study, and two of the investigators are employees of the company. Dr. Ghantoji reported having no other relevant financial disclosures.
UV-C Light Blasts 'Bad Bugs' in Hospital Rooms
SAN DIEGO – A portable device that emits ultraviolet C light destroyed vancomycin-resistant enterococci, Acinetobacter, and Clostridium difficile from hospital rooms where patients infected with those bacteria had been housed, results from a small study demonstrated.
"There is growing evidence that the environment can be a source for acquisition of bad bugs," lead study investigator Dr. Deverick J. Anderson said in an interview prior to IDWeek 2012, where the research was presented during a poster session.
"Our study further strengthens the data that no-touch systems like UV-C light kill important bacteria and can potentially help with current cleaning strategies. While several groups have demonstrated that UV-C light work in experimental conditions we are demonstrating that it works in a real-world hospital environment."
Dr. Anderson of the department of medicine in the division of infectious diseases at Duke University, Durham, N.C., and his associates analyzed 39 rooms at two tertiary care hospitals that had just housed a patient with one of the different bad bugs: vancomycin-resistant enterococci (VRE), Acinetobacter, and C. difficile. After the patient was discharged but prior to the regular cleaning, the investigators obtained 15 or more cultures from several different locations in the hospital rooms, including bed rails, remote controls, and toilets. Then they wheeled in the TRU-D, an automated mobile disinfection system manufactured by Lumalier that is about 6 feet tall and is equipped with 8 sensors and 16 bulbs that emit UV-C light.
"Each room was irradiated between 25 and 45 minutes in order to eradicate both bacteria and bacterial spores," Dr. Anderson explained during a premeeting telephone press conference. "We then went back into the rooms and cultured the environment from the same locations."
After comparing the number of colony-forming units (CFUs) before and after irradiation "we were able to demonstrate that we could achieve well over 90% reduction in each of those three bad bugs after using the UV light," said Dr. Anderson, who also chairs the antimicrobial stewardship and evaluation team at Duke University Medical Center. "This occurred in all locations sampled, in both direct and indirect light."
Specifically, the UV-C irradiation reduced CFUs of VRE by 98%, C. difficile by 93%, and Acinetobacter by 98%.
"Based on these results we came to the conclusion that UV-C light is indeed effective in killing VRE, C. difficile, and Acinetobacter from the real-world hospital environment," Dr. Anderson said during the telephone press conference. "The idea behind achieving bacterial irradiation in shadow is actually taking advantage of the reflective properties of UV light. It literally bounces around the room and ends up hitting areas in shadow. That’s how bacterial reduction occurs."
He acknowledged certain limitations of the study, including the fact that the researchers were able to evaluate onlytwo hospital rooms with Acinetobacter "because of how infrequently this organism causes infections. Regardless, we reduced the amount of Acinetobacter in both of those rooms."
The study was sponsored by the Centers for Disease Control and Prevention. Lumalier donated the machines used in the study but had no role in the trial design or in review of the data. Dr. Anderson said that he had no relevant financial conflicts to disclose.
IDWeek 2012 is the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
SAN DIEGO – A portable device that emits ultraviolet C light destroyed vancomycin-resistant enterococci, Acinetobacter, and Clostridium difficile from hospital rooms where patients infected with those bacteria had been housed, results from a small study demonstrated.
"There is growing evidence that the environment can be a source for acquisition of bad bugs," lead study investigator Dr. Deverick J. Anderson said in an interview prior to IDWeek 2012, where the research was presented during a poster session.
"Our study further strengthens the data that no-touch systems like UV-C light kill important bacteria and can potentially help with current cleaning strategies. While several groups have demonstrated that UV-C light work in experimental conditions we are demonstrating that it works in a real-world hospital environment."
Dr. Anderson of the department of medicine in the division of infectious diseases at Duke University, Durham, N.C., and his associates analyzed 39 rooms at two tertiary care hospitals that had just housed a patient with one of the different bad bugs: vancomycin-resistant enterococci (VRE), Acinetobacter, and C. difficile. After the patient was discharged but prior to the regular cleaning, the investigators obtained 15 or more cultures from several different locations in the hospital rooms, including bed rails, remote controls, and toilets. Then they wheeled in the TRU-D, an automated mobile disinfection system manufactured by Lumalier that is about 6 feet tall and is equipped with 8 sensors and 16 bulbs that emit UV-C light.
"Each room was irradiated between 25 and 45 minutes in order to eradicate both bacteria and bacterial spores," Dr. Anderson explained during a premeeting telephone press conference. "We then went back into the rooms and cultured the environment from the same locations."
After comparing the number of colony-forming units (CFUs) before and after irradiation "we were able to demonstrate that we could achieve well over 90% reduction in each of those three bad bugs after using the UV light," said Dr. Anderson, who also chairs the antimicrobial stewardship and evaluation team at Duke University Medical Center. "This occurred in all locations sampled, in both direct and indirect light."
Specifically, the UV-C irradiation reduced CFUs of VRE by 98%, C. difficile by 93%, and Acinetobacter by 98%.
"Based on these results we came to the conclusion that UV-C light is indeed effective in killing VRE, C. difficile, and Acinetobacter from the real-world hospital environment," Dr. Anderson said during the telephone press conference. "The idea behind achieving bacterial irradiation in shadow is actually taking advantage of the reflective properties of UV light. It literally bounces around the room and ends up hitting areas in shadow. That’s how bacterial reduction occurs."
He acknowledged certain limitations of the study, including the fact that the researchers were able to evaluate onlytwo hospital rooms with Acinetobacter "because of how infrequently this organism causes infections. Regardless, we reduced the amount of Acinetobacter in both of those rooms."
The study was sponsored by the Centers for Disease Control and Prevention. Lumalier donated the machines used in the study but had no role in the trial design or in review of the data. Dr. Anderson said that he had no relevant financial conflicts to disclose.
IDWeek 2012 is the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
SAN DIEGO – A portable device that emits ultraviolet C light destroyed vancomycin-resistant enterococci, Acinetobacter, and Clostridium difficile from hospital rooms where patients infected with those bacteria had been housed, results from a small study demonstrated.
"There is growing evidence that the environment can be a source for acquisition of bad bugs," lead study investigator Dr. Deverick J. Anderson said in an interview prior to IDWeek 2012, where the research was presented during a poster session.
"Our study further strengthens the data that no-touch systems like UV-C light kill important bacteria and can potentially help with current cleaning strategies. While several groups have demonstrated that UV-C light work in experimental conditions we are demonstrating that it works in a real-world hospital environment."
Dr. Anderson of the department of medicine in the division of infectious diseases at Duke University, Durham, N.C., and his associates analyzed 39 rooms at two tertiary care hospitals that had just housed a patient with one of the different bad bugs: vancomycin-resistant enterococci (VRE), Acinetobacter, and C. difficile. After the patient was discharged but prior to the regular cleaning, the investigators obtained 15 or more cultures from several different locations in the hospital rooms, including bed rails, remote controls, and toilets. Then they wheeled in the TRU-D, an automated mobile disinfection system manufactured by Lumalier that is about 6 feet tall and is equipped with 8 sensors and 16 bulbs that emit UV-C light.
"Each room was irradiated between 25 and 45 minutes in order to eradicate both bacteria and bacterial spores," Dr. Anderson explained during a premeeting telephone press conference. "We then went back into the rooms and cultured the environment from the same locations."
After comparing the number of colony-forming units (CFUs) before and after irradiation "we were able to demonstrate that we could achieve well over 90% reduction in each of those three bad bugs after using the UV light," said Dr. Anderson, who also chairs the antimicrobial stewardship and evaluation team at Duke University Medical Center. "This occurred in all locations sampled, in both direct and indirect light."
Specifically, the UV-C irradiation reduced CFUs of VRE by 98%, C. difficile by 93%, and Acinetobacter by 98%.
"Based on these results we came to the conclusion that UV-C light is indeed effective in killing VRE, C. difficile, and Acinetobacter from the real-world hospital environment," Dr. Anderson said during the telephone press conference. "The idea behind achieving bacterial irradiation in shadow is actually taking advantage of the reflective properties of UV light. It literally bounces around the room and ends up hitting areas in shadow. That’s how bacterial reduction occurs."
He acknowledged certain limitations of the study, including the fact that the researchers were able to evaluate onlytwo hospital rooms with Acinetobacter "because of how infrequently this organism causes infections. Regardless, we reduced the amount of Acinetobacter in both of those rooms."
The study was sponsored by the Centers for Disease Control and Prevention. Lumalier donated the machines used in the study but had no role in the trial design or in review of the data. Dr. Anderson said that he had no relevant financial conflicts to disclose.
IDWeek 2012 is the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
Major Finding: UV-C irradiation of hospital rooms with a portable disinfection system reduced colony-forming units of vancomycin-resistant enterococci by 98%, C. difficile by 93%, and Acinetobacter by 98%.
Data Source: Results were taken from a study conducted in 39 hospital rooms at two tertiary medical centers.
Disclosures: The study was sponsored by the Centers for Disease Control and Prevention. Lumalier donated the machines used in the study but had no role in the trial design or in review of the data. Dr. Anderson said that he had no relevant financial conflicts to disclose.
Pharmacologic and nonpharmacologic treatment options for panic disorder
Promising C. difficile Antibiotic in Pipeline
SAN FRANCISCO – Those desperate for new treatments for Clostridium difficile infection may want to keep an eye on the experimental oral antibiotic cadazolid, which looked promising in an early-phase trial, according to Daniela Baldoni, Pharm.D.
Cadazolid is in the oxazolidinone class of antibiotics. Its mechanism of action consists mainly of bacterial protein-synthesis inhibition.
Cadazolid produced low systemic exposure with high concentrations at the desired site – the colon – and was well tolerated in 64 healthy men who received up to 3,000 mg b.i.d. for 10 days, she reported in a poster presentation at the conference. Dr. Baldoni is employed by Actelion Pharmaceuticals, the company that is developing cadazolid.
The study randomized nonsmoking men aged 45-60 years and a body mass index of 18-32 kg/m2 to single or multiple doses of cadazolid or placebo.
In the single-dose group, 30 fasting subjects received a single dose of 30, 100, 300, 1,000, or 3,000 mg cadazolid and 10 subjects received matching placebo. After a wash-out period of 8-15 days, the six subjects who had taken 300 mg received a second dose of 300 mg after eating instead of after fasting. In the multiple-dose group, 18 subjects took 300, 1,000, or 3,000 mg of cadazolid twice a day and 6 received matching placebo for 10 days.
Taking cadazolid with food appeared to increase the rate and extent of drug absorption by two- to fivefold. Blood samples showed low systemic exposure after single or multiple doses, with a minor, twofold increase in cadazolid in plasma after 10 days for all doses in the twice-a-day group, Dr. Baldoni reported at the meeting, sponsored by the American Society for Microbiology.
The dose or duration of treatment did not seem to affect the number of adverse events (none of which were serious). They occurred in 27%-39% of cadazolid-treated subjects and in 17%-40% taking placebo and were mostly headache or diarrhea.
All subjects completed the study except one man in the 100-mg single-dose subgroup who withdrew consent for reasons unrelated to adverse events.
SAN FRANCISCO – Those desperate for new treatments for Clostridium difficile infection may want to keep an eye on the experimental oral antibiotic cadazolid, which looked promising in an early-phase trial, according to Daniela Baldoni, Pharm.D.
Cadazolid is in the oxazolidinone class of antibiotics. Its mechanism of action consists mainly of bacterial protein-synthesis inhibition.
Cadazolid produced low systemic exposure with high concentrations at the desired site – the colon – and was well tolerated in 64 healthy men who received up to 3,000 mg b.i.d. for 10 days, she reported in a poster presentation at the conference. Dr. Baldoni is employed by Actelion Pharmaceuticals, the company that is developing cadazolid.
The study randomized nonsmoking men aged 45-60 years and a body mass index of 18-32 kg/m2 to single or multiple doses of cadazolid or placebo.
In the single-dose group, 30 fasting subjects received a single dose of 30, 100, 300, 1,000, or 3,000 mg cadazolid and 10 subjects received matching placebo. After a wash-out period of 8-15 days, the six subjects who had taken 300 mg received a second dose of 300 mg after eating instead of after fasting. In the multiple-dose group, 18 subjects took 300, 1,000, or 3,000 mg of cadazolid twice a day and 6 received matching placebo for 10 days.
Taking cadazolid with food appeared to increase the rate and extent of drug absorption by two- to fivefold. Blood samples showed low systemic exposure after single or multiple doses, with a minor, twofold increase in cadazolid in plasma after 10 days for all doses in the twice-a-day group, Dr. Baldoni reported at the meeting, sponsored by the American Society for Microbiology.
The dose or duration of treatment did not seem to affect the number of adverse events (none of which were serious). They occurred in 27%-39% of cadazolid-treated subjects and in 17%-40% taking placebo and were mostly headache or diarrhea.
All subjects completed the study except one man in the 100-mg single-dose subgroup who withdrew consent for reasons unrelated to adverse events.
SAN FRANCISCO – Those desperate for new treatments for Clostridium difficile infection may want to keep an eye on the experimental oral antibiotic cadazolid, which looked promising in an early-phase trial, according to Daniela Baldoni, Pharm.D.
Cadazolid is in the oxazolidinone class of antibiotics. Its mechanism of action consists mainly of bacterial protein-synthesis inhibition.
Cadazolid produced low systemic exposure with high concentrations at the desired site – the colon – and was well tolerated in 64 healthy men who received up to 3,000 mg b.i.d. for 10 days, she reported in a poster presentation at the conference. Dr. Baldoni is employed by Actelion Pharmaceuticals, the company that is developing cadazolid.
The study randomized nonsmoking men aged 45-60 years and a body mass index of 18-32 kg/m2 to single or multiple doses of cadazolid or placebo.
In the single-dose group, 30 fasting subjects received a single dose of 30, 100, 300, 1,000, or 3,000 mg cadazolid and 10 subjects received matching placebo. After a wash-out period of 8-15 days, the six subjects who had taken 300 mg received a second dose of 300 mg after eating instead of after fasting. In the multiple-dose group, 18 subjects took 300, 1,000, or 3,000 mg of cadazolid twice a day and 6 received matching placebo for 10 days.
Taking cadazolid with food appeared to increase the rate and extent of drug absorption by two- to fivefold. Blood samples showed low systemic exposure after single or multiple doses, with a minor, twofold increase in cadazolid in plasma after 10 days for all doses in the twice-a-day group, Dr. Baldoni reported at the meeting, sponsored by the American Society for Microbiology.
The dose or duration of treatment did not seem to affect the number of adverse events (none of which were serious). They occurred in 27%-39% of cadazolid-treated subjects and in 17%-40% taking placebo and were mostly headache or diarrhea.
All subjects completed the study except one man in the 100-mg single-dose subgroup who withdrew consent for reasons unrelated to adverse events.
Major Finding: The experimental antibiotic cadazolid concentrated in feces with low systemic exposure and few side effects after single doses or twice-a-day dosing for 10 days.
Data Source: Data are from a randomized, placebo-controlled study in 64 healthy, nonsmoking men.
Disclosures: Dr. Baldoni and most of her coinvestigators are employees of Actelion Pharmaceuticals, which funded the study.
Higher Dose for Severe C. difficile Speeds Response
SAN FRANCISCO – Two small studies suggest that treating severe Clostridium difficile infection with a higher initial dose of vancomycin may work better than the recommended dose of 125 mg every 6 hours.
The most recent study, presented in a poster at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy, surprised the investigators.
"Pharmacodynamically, the concentrations in stool of the standard dose of 125 mg are about 500-1,000 times greater than the MIC [minimum inhibitory concentration]," Yleana T. Garcia, Pharm.D., said in an interview. "So, we have enough concentration in the stool. We wanted to see that we have similar outcomes with patients who are treated with standard doses as with higher doses."
Instead, the retrospective review of 62 patients with severe diarrhea who received oral vancomycin for at least 3 days found that symptoms resolved significantly quicker in 19 patients who got 250 mg every 6 hours compared with 43 patients who got 125 mg every 6 hours, she and her associates reported.
Symptoms resolved by day 3 in nine patients (47%) on the high dose and six patients (15%) on the conventional dose, said Dr. Garcia, a palliative care fellow at the James J. Peters Veterans Affairs Medical Center, Bronx, N.Y.
There also were statistically nonsignificant trends toward a higher likelihood of clinical cure, shorter length of stay, and reduced risk of recurrence in patients with the higher dose. An increased death rate in the higher-dose group also was not statistically significant, and might be due to greater severity of illness at baseline in patients who got the higher dose of vancomycin, she said at the meeting, sponsored by the American Society for Microbiology.
The findings support those of a small prospective study that analyzed levels of vancomycin in feces collected from 15 patients with presumed or confirmed C. difficile infection. Drug concentrations were high in patients who got 250 or 500 mg q.i.d. but were inadequate in at least one patient on the first day of treatment with 125 mg q.i.d. (BMC Infect. Dis. 2010;10:363).<< http://www.biomedcentral.com/1471-2334/10/363 >>
"Higher doses like 250-500 mg may be warranted to reach adequate concentrations in the stool in the first 24-48 hours," Dr. Garcia said. "I’m not saying to use 250 for the whole treatment course, but there may be a role for a loading dose of 250 mg q6 for the first 24-48 hours, and then switching to 125 mg q6 for the remainder of the treatment course. We know that 125 does have adequate fecal concentration; it just may not be adequate on day 1."
The 2010 update to clinical practice guidelines for C. difficile infection in adults recommends treating severe C. difficile infection with oral vancomycin 125 mg every 6 hours or using 500 mg every 6 hours for patients with severe disease complicated by ileus, megacolon, or hypotension (Infect. Control Hosp. Epidemiol. 2010;31:431-55).
Severe C. difficile infection generally is defined as the presence of the organism plus leukocytosis with a white blood cell count of 15,000 cells/microL or greater, or a serum creatinine level at least 1.5 times baseline. The study reviewed records of patients who received vancomycin for these indications or hypotension, shock, ileus, megacolon, or evidence of colitis. The study excluded patients who were treated with any other medication besides metronidazole.
The study is continuing in order to increase the number of patients reviewed and the power of the findings.
Dr. Garcia reported having no financial disclosures.
SAN FRANCISCO – Two small studies suggest that treating severe Clostridium difficile infection with a higher initial dose of vancomycin may work better than the recommended dose of 125 mg every 6 hours.
The most recent study, presented in a poster at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy, surprised the investigators.
"Pharmacodynamically, the concentrations in stool of the standard dose of 125 mg are about 500-1,000 times greater than the MIC [minimum inhibitory concentration]," Yleana T. Garcia, Pharm.D., said in an interview. "So, we have enough concentration in the stool. We wanted to see that we have similar outcomes with patients who are treated with standard doses as with higher doses."
Instead, the retrospective review of 62 patients with severe diarrhea who received oral vancomycin for at least 3 days found that symptoms resolved significantly quicker in 19 patients who got 250 mg every 6 hours compared with 43 patients who got 125 mg every 6 hours, she and her associates reported.
Symptoms resolved by day 3 in nine patients (47%) on the high dose and six patients (15%) on the conventional dose, said Dr. Garcia, a palliative care fellow at the James J. Peters Veterans Affairs Medical Center, Bronx, N.Y.
There also were statistically nonsignificant trends toward a higher likelihood of clinical cure, shorter length of stay, and reduced risk of recurrence in patients with the higher dose. An increased death rate in the higher-dose group also was not statistically significant, and might be due to greater severity of illness at baseline in patients who got the higher dose of vancomycin, she said at the meeting, sponsored by the American Society for Microbiology.
The findings support those of a small prospective study that analyzed levels of vancomycin in feces collected from 15 patients with presumed or confirmed C. difficile infection. Drug concentrations were high in patients who got 250 or 500 mg q.i.d. but were inadequate in at least one patient on the first day of treatment with 125 mg q.i.d. (BMC Infect. Dis. 2010;10:363).<< http://www.biomedcentral.com/1471-2334/10/363 >>
"Higher doses like 250-500 mg may be warranted to reach adequate concentrations in the stool in the first 24-48 hours," Dr. Garcia said. "I’m not saying to use 250 for the whole treatment course, but there may be a role for a loading dose of 250 mg q6 for the first 24-48 hours, and then switching to 125 mg q6 for the remainder of the treatment course. We know that 125 does have adequate fecal concentration; it just may not be adequate on day 1."
The 2010 update to clinical practice guidelines for C. difficile infection in adults recommends treating severe C. difficile infection with oral vancomycin 125 mg every 6 hours or using 500 mg every 6 hours for patients with severe disease complicated by ileus, megacolon, or hypotension (Infect. Control Hosp. Epidemiol. 2010;31:431-55).
Severe C. difficile infection generally is defined as the presence of the organism plus leukocytosis with a white blood cell count of 15,000 cells/microL or greater, or a serum creatinine level at least 1.5 times baseline. The study reviewed records of patients who received vancomycin for these indications or hypotension, shock, ileus, megacolon, or evidence of colitis. The study excluded patients who were treated with any other medication besides metronidazole.
The study is continuing in order to increase the number of patients reviewed and the power of the findings.
Dr. Garcia reported having no financial disclosures.
SAN FRANCISCO – Two small studies suggest that treating severe Clostridium difficile infection with a higher initial dose of vancomycin may work better than the recommended dose of 125 mg every 6 hours.
The most recent study, presented in a poster at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy, surprised the investigators.
"Pharmacodynamically, the concentrations in stool of the standard dose of 125 mg are about 500-1,000 times greater than the MIC [minimum inhibitory concentration]," Yleana T. Garcia, Pharm.D., said in an interview. "So, we have enough concentration in the stool. We wanted to see that we have similar outcomes with patients who are treated with standard doses as with higher doses."
Instead, the retrospective review of 62 patients with severe diarrhea who received oral vancomycin for at least 3 days found that symptoms resolved significantly quicker in 19 patients who got 250 mg every 6 hours compared with 43 patients who got 125 mg every 6 hours, she and her associates reported.
Symptoms resolved by day 3 in nine patients (47%) on the high dose and six patients (15%) on the conventional dose, said Dr. Garcia, a palliative care fellow at the James J. Peters Veterans Affairs Medical Center, Bronx, N.Y.
There also were statistically nonsignificant trends toward a higher likelihood of clinical cure, shorter length of stay, and reduced risk of recurrence in patients with the higher dose. An increased death rate in the higher-dose group also was not statistically significant, and might be due to greater severity of illness at baseline in patients who got the higher dose of vancomycin, she said at the meeting, sponsored by the American Society for Microbiology.
The findings support those of a small prospective study that analyzed levels of vancomycin in feces collected from 15 patients with presumed or confirmed C. difficile infection. Drug concentrations were high in patients who got 250 or 500 mg q.i.d. but were inadequate in at least one patient on the first day of treatment with 125 mg q.i.d. (BMC Infect. Dis. 2010;10:363).<< http://www.biomedcentral.com/1471-2334/10/363 >>
"Higher doses like 250-500 mg may be warranted to reach adequate concentrations in the stool in the first 24-48 hours," Dr. Garcia said. "I’m not saying to use 250 for the whole treatment course, but there may be a role for a loading dose of 250 mg q6 for the first 24-48 hours, and then switching to 125 mg q6 for the remainder of the treatment course. We know that 125 does have adequate fecal concentration; it just may not be adequate on day 1."
The 2010 update to clinical practice guidelines for C. difficile infection in adults recommends treating severe C. difficile infection with oral vancomycin 125 mg every 6 hours or using 500 mg every 6 hours for patients with severe disease complicated by ileus, megacolon, or hypotension (Infect. Control Hosp. Epidemiol. 2010;31:431-55).
Severe C. difficile infection generally is defined as the presence of the organism plus leukocytosis with a white blood cell count of 15,000 cells/microL or greater, or a serum creatinine level at least 1.5 times baseline. The study reviewed records of patients who received vancomycin for these indications or hypotension, shock, ileus, megacolon, or evidence of colitis. The study excluded patients who were treated with any other medication besides metronidazole.
The study is continuing in order to increase the number of patients reviewed and the power of the findings.
Dr. Garcia reported having no financial disclosures.
Major Finding: Symptoms of severe C. difficile infection resolved by day 3 of oral vancomycin treatment in 9 of 19 patients treated with 250 mg every 6 hours (47%) compared with 6 of 43 patients on 125 mg every 6 hours (15%).
Data Source: Retrospective review of records on 62 adults at one institution treated for at least 3 days with oral vancomycin for severe C. difficile infection.
Disclosures: Dr. Garcia reported having no financial disclosures.
New Directions in Small-Vessel Vasculitis: ANCA, Target Organs, Treatment, and Beyond

Supplement Editor:
Carol Langford, MD, MHS
Associate Editors:
Leonard Calabrese, DO, and Gary Hoffman, MD
Contents
Diagnosis, ANCA testing, and disease activity
Clinical features and diagnosis of small-vessel vasculitis
Carol Langford, MD, MHS
Controversies in ANCA testing
Ulrich Specks, MD
Defining disease activity and damage in patients with small-vessel vasculitis
Peter A. Merkel, MD, MPH
Impact on individual organs
Upper airway manifestations of granulomatosis with polyangiitis
Daniel S. Alam, MD; Rahul Seth, MD; Raj Sindwani, MD; Erika A. Woodson, MD; and Karthik Rajasekaran, MD
Renal disease in small-vessel vasculitis
Kirsten de Groot, MD
Pulmonary disease in small-vessel vasculitis
Thomas R. Gildea, MD, MS
Ocular manifestations of small-vessel vasculitis
James A. Garrity, MD
Monitoring and safety
Monitoring patients with vasculitis
Alexandra Villa-Forte, MD, MPH
Safety issues in vasculitis: Infections and immunizations in the immunosuppressed host
Carlos M. Isada, MD, FCCP
Treatment considerations
Treating vasculitis with conventional immunosuppressive agents
David Jayne, MD, FRCP
Biologic agents in the treatment of granulomatosis with polyangiitis
Ulrich Specks, MD
Historical perspective
History of vasculitis: The life and work of Adolf Kussmaul
Eric L. Matteson, MD, MPH
Supplement Editor:
Carol Langford, MD, MHS
Associate Editors:
Leonard Calabrese, DO, and Gary Hoffman, MD
Contents
Diagnosis, ANCA testing, and disease activity
Clinical features and diagnosis of small-vessel vasculitis
Carol Langford, MD, MHS
Controversies in ANCA testing
Ulrich Specks, MD
Defining disease activity and damage in patients with small-vessel vasculitis
Peter A. Merkel, MD, MPH
Impact on individual organs
Upper airway manifestations of granulomatosis with polyangiitis
Daniel S. Alam, MD; Rahul Seth, MD; Raj Sindwani, MD; Erika A. Woodson, MD; and Karthik Rajasekaran, MD
Renal disease in small-vessel vasculitis
Kirsten de Groot, MD
Pulmonary disease in small-vessel vasculitis
Thomas R. Gildea, MD, MS
Ocular manifestations of small-vessel vasculitis
James A. Garrity, MD
Monitoring and safety
Monitoring patients with vasculitis
Alexandra Villa-Forte, MD, MPH
Safety issues in vasculitis: Infections and immunizations in the immunosuppressed host
Carlos M. Isada, MD, FCCP
Treatment considerations
Treating vasculitis with conventional immunosuppressive agents
David Jayne, MD, FRCP
Biologic agents in the treatment of granulomatosis with polyangiitis
Ulrich Specks, MD
Historical perspective
History of vasculitis: The life and work of Adolf Kussmaul
Eric L. Matteson, MD, MPH
Supplement Editor:
Carol Langford, MD, MHS
Associate Editors:
Leonard Calabrese, DO, and Gary Hoffman, MD
Contents
Diagnosis, ANCA testing, and disease activity
Clinical features and diagnosis of small-vessel vasculitis
Carol Langford, MD, MHS
Controversies in ANCA testing
Ulrich Specks, MD
Defining disease activity and damage in patients with small-vessel vasculitis
Peter A. Merkel, MD, MPH
Impact on individual organs
Upper airway manifestations of granulomatosis with polyangiitis
Daniel S. Alam, MD; Rahul Seth, MD; Raj Sindwani, MD; Erika A. Woodson, MD; and Karthik Rajasekaran, MD
Renal disease in small-vessel vasculitis
Kirsten de Groot, MD
Pulmonary disease in small-vessel vasculitis
Thomas R. Gildea, MD, MS
Ocular manifestations of small-vessel vasculitis
James A. Garrity, MD
Monitoring and safety
Monitoring patients with vasculitis
Alexandra Villa-Forte, MD, MPH
Safety issues in vasculitis: Infections and immunizations in the immunosuppressed host
Carlos M. Isada, MD, FCCP
Treatment considerations
Treating vasculitis with conventional immunosuppressive agents
David Jayne, MD, FRCP
Biologic agents in the treatment of granulomatosis with polyangiitis
Ulrich Specks, MD
Historical perspective
History of vasculitis: The life and work of Adolf Kussmaul
Eric L. Matteson, MD, MPH

Clinical features and diagnosis of small-vessel vasculitis
Vasculitis refers to inflammation of the blood vessel. This inflammation can cause vessel wall thickening that compromises or occludes the vessel lumen, ultimately resulting in organ ischemia. It also can cause vessel wall attenuation that predisposes to aneurysm formation or breach of the vessel integrity with resultant hemorrhage into the tissue.
Vasculitis can be thought of as a primary or secondary process. Primary vasculitides are unique disease entities without a currently identified underlying cause in which vasculitis forms the pathologic basis of tissue injury. Vasculitis can occur secondary to medication exposure or an underlying illness, including infections, malignancy, cryoglobulinemia, and rheumatic diseases (such as systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, or myositis).
Primary vasculitides may differ in epidemiology, such as the age at which they occur and the gender most likely to be affected, their clinical manifestations (including signs, symptoms, and patterns of organ involvement), the diagnostic approach (biopsy, arteriography, and laboratory investigation), treatment (supportive care, glucocorticoids alone, or in combination with other immunosuppressants), and the size of the vessels predominantly affected (large, medium, or small).
Small-vessel vasculitis affects the arteriole, capillary, and venule. An excellent example of small-vessel vasculitis and the one most commonly encountered in clinical practice is cutaneous vasculitis, in which extravasation of erythrocytes from disrupted small vessels is observed histologically, with the clinical sequelae of palpable purpura. Although categorization based on the predominant vessel size that is affected is a helpful way to view these diseases, this is not absolute and each disease has the potential to affect a diverse range of vessels.
This article explores the clinical features and diagnosis of three forms of vasculitis that predominantly affect the small vessels: granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]), microscopic polyangiitis (MPA), and eosinophilic GPA (Churg-Strauss syndrome, EGPA).
GRANULOMATOSIS WITH POLYANGIITIS
Granulomatosis with polyangiitis is characterized by granulomatous inflammation involving the respiratory tract and by vasculitis affecting small- to medium-sized vessels in which necrotizing glomerulonephritis is common.
Wide range of presentations, manifestations
Approximately 90% of patients with GPA have upper or lower airway involvement or both.1 Upper airway or ear symptoms affect 73% of patients initially and 92% overall.1 Direct inspection of the nasal membranes shows a cobblestoned or ulcerated appearance, and computed tomography reveals mucosal thickening of the sinuses. In some instances, sinus disease can compromise blood supply to the cartilaginous portion of the nasal septum, leading to nasal septum perforations or collapse of the nasal bridge. Another manifestation of upper airway disease and GPA is subglottic stenosis, a narrowing in the subglottic region located just below the vocal cords. The narrowing typically spans about 1 cm and rarely extends or involves the remainder of the trachea.
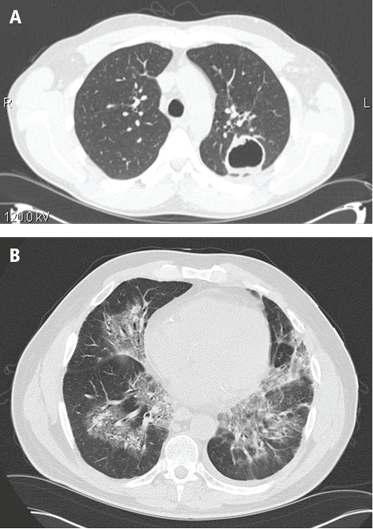
The lungs are involved in 85% of patients.1 Radiographic abnormalities can be diverse and include bilateral pulmonary nodular infiltrates, single or multiple cavities, and bilateral ground glass infiltrates as can be seen in pulmonary hemorrhage (Figure). Bronchoscopy may reveal endobronchial stenosis, and pleural disease can occur rarely.
Approximately 20% of patients with GPA may have glomerulonephritis when they first present for medical attention, but it eventually develops in nearly 80% of patients during the disease course.1 Despite its potential for rapid progression, glomerulonephritis presents a diagnostic challenge because it is asymptomatic. It is detected by evidence of proteinuria and an active urine sediment with dysmorphic red blood cells and red blood cell casts.
Ocular involvement occurs eventually in 52% of patients with GPA.1 Any ocular structure can be affected and ocular involvement can be visually threatening. The more prominent ocular manifestations include scleritis/episcleritis or orbital disease.
Cutaneous manifestations, observed in 46% of patients, include verrucous-appearing lesions on the elbow and infarctions in the skin and nail folds.1 Other rare manifestations can occur, such as pericarditis and cerebral vasculitis.
Although nearly all patients present with upper or lower airway symptoms, the multisystem nature of GPA explains the wide range of presentations and the varying degrees of disease severity.
Differential diagnosis
The differential diagnosis in GPA is varied. Particularly in the setting of isolated lung or sinus disease, infection is foremost in the differential diagnosis. Even in the nonimmunosuppressed host, unusual infections such as mycobacteria, histoplasmosis, and other fungal infections should be considered. Lymphadenopathy, rarely seen in GPA, should raise concern for other causes of disease. Lymphoproliferative processes and other neoplasms, other rheumatic diseases, granulomatous disease (ie, sarcoidosis), and other causes of glomerulonephritis (when present) also merit consideration. Differentiation of these entities from GPA is essential because the treatment differs in many instances.
The differential diagnosis for patients who present with midline destructive lesions must include other causes of collapse of the nasal bridge, nasal septum perforation, and possibly palate destruction. Erosions of the hard palate in particular should raise an immediate red flag for entities other than GPA, such as lymphoproliferative diseases; rare infections, particularly if the patient has studied or worked abroad; and cocaine exposure.
Diagnostic evaluation
A diagnosis of GPA is typically based on the presence of histologic features in a clinically compatible setting. Diagnostic features include necrosis, granulomatous inflammation, vasculitis, and special stains and cultures negative for microorganisms.
Biopsy sites are determined by evidence of clinical disease affecting a target organ and the likelihood of obtaining diagnostically meaningful findings from that site. One challenge is that biopsies are not always diagnostic. The changes tend to be patchy and the likelihood of a positive yield is associated with the amount of tissue that can be obtained. Tissues from the ear, nose, and throat have a yield of about 20%, depending upon the site and the biopsy size. The highest yield comes from radiographically abnormal pulmonary parenchyma. Although transbronchial biopsies are attractive because they are less invasive than open lung biopsy, they are also far less diagnostic, with fewer than 10% having a positive yield. Because cutaneous vasculitis is observed in many settings, its presence is usually insufficient evidence for diagnosis. The renal histologic appearance is a focal, segmental, crescentic, and necrotizing glomerulonephritis that has few to no immune complexes (pauciimmune glomerulonephritis).1–3
Chest imaging should be performed in any patient in whom GPA is part of the differential diagnosis, since up to one-third of patients may be asymptomatic yet have pulmonary radiographic findings.
Laboratory assessment should include serum chemistries to evaluate renal and hepatic function, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. If the urinalysis is positive for blood, microscopy should be performed on fresh urine to look for casts. In the setting of pulmonary-renal manifestations, testing for other causes, such as antiglomerular basement antibodies and antinuclear antibodies, should be considered.
Serologic testing for antineutrophil cytoplasmic antibodies (ANCA) has provided a useful tool in suggesting the diagnosis of GPA. Two forms of ANCA have been identified in patients with vasculitis: ANCA directed against the neutrophil serine protease proteinase-3 (PR3), which results in a cytoplasmic immunofluorescence (cANCA) pattern; and ANCA directed against the neutrophil enzyme myeloperoxidase (MPO), which causes a perinuclear immunofluorescence (pANCA) pattern.4 Approximately 80% to 95% of ANCA found in patients with active severe GPA are detectable PR3-cANCA, while 5% to 20% are MPO-pANCA.5 The predictive value of ANCA for the diagnosis depends on the spectrum of clinical features. As ANCA can be seen in other settings, ANCA as the basis for diagnosis in place of tissue biopsy should be used with caution and only in selected instances where their predictive value would equal that of biopsy. The presence of ANCA is not necessary to establish the diagnosis, as up to 20% of patients with GPA may be ANCA-negative.6
MICROSCOPIC POLYANGIITIS
The history of MPA dates to 1866, with the description of periarteritis nodosa.7 The term “microscopic polyarteritis” was introduced in 1948, when glomerular disease was recognized in some patients.8 In 1994, the Chapel Hill Consensus Conference defined MPA as a necrotizing vasculitis with few or no immune deposits that affects small vessels (ie, capillaries, venules, or arterioles). Necrotizing arteritis of small- and medium-sized arteries may be present. Necrotizing glomerulonephritis and pulmonary capillaritis commonly occur.9 MPA shares many clinical features with GPA and is currently said to be distinguished by the absence of granulomatous inflammation.9
Presentations and manifestations
In one assessment of organ system involvement in 85 patients with MPA, investigators observed glomerular syndrome in 82% of patients.10 They also found a high predilection for involvement of the skin, joints, and lungs. Pulmonary hemorrhage is a particularly important manifestation of MPA because it can be immediately life-threatening.
Differential diagnosis
The differential diagnosis for MPA is similar to GPA in the inclusion of other causes of classic pulmonary-renal syndromes, such as antiglomerular basement membrane disease and systemic lupus erythematosus. Poststreptococcal glomerulonephritis should be considered when the kidney is the predominant organ involved in the absence of lung disease. In the setting of pulmonary infiltrates, infections and neoplasms remain significant in the differential diagnosis.
Diagnostic evaluation
The diagnosis of MPA is based on consistent clinical features and compatible histologic findings. The histologic renal lesion is identical to that seen in GPA. Pulmonary disease typically includes capillaritis and is notable for the absence of evidence of immune deposition, in contrast to antiglomerular basement membrane disease.
Chest imaging is indicated when MPA is part of the differential diagnosis. Computed tomography is the preferred technique, as early alveolar hemorrhage that can occur in MPA may not be visualized on a chest radiograph.
Laboratory assessment should include serum chemistries, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. Additional testing should be pursued for other diseases as indicated by the clinical features.
Approximately 40% to 80% of patients with MPA have MPO-pANCA.5 Approximately 15% of patients are MPO-pANCA positive,6 and 0% to 20% are ANCA-negative. As with GPA, ANCA is useful to suggest—but not diagnose—disease in many instances. The absence of ANCA does not rule out MPA.
EOSINOPHILIC GPA
Eosinophilic GPA is a unique entity characterized by eosinophil-rich and granulomatous inflammation involving the respiratory tract and necrotizing vasculitis of small- to medium-size vessels. It is also associated with asthma and eosinophilia.
Different disease phases
Eosinophilic GPA is often thought of as having three phases: prodromal, eosinophilic, and vasculitic.11,12 Although helpful conceptually, these phases may not always be present and may not occur in sequence.
The prodromal phase is characterized by asthma associated with allergic rhinitis with or without polyposis. The eosinophilic phase is characterized by the presence of eosinophilia in the blood and tissue. Eosinophilia is a prominent feature, although accurate detection and assessment can be challenging in the setting of glucocorticoid use for asthma as this normalizes the eosinophil count.
The vasculitic phase distinguishes EGPA from other eosinophilic disorders. Features of vasculitis may occur in multiple organ sites, including the nerves, lungs, heart, gastrointestinal tract, and kidneys. In one series of 96 patients, nearly 100% had asthma, and peripheral nervous system involvement in the form of mononeuritis multiplex was present in 72%.12 Cardiac involvement is of particular importance as it is a prominent cause of disease-related mortality. Cardiac manifestations include myocarditis, pericarditis, endocarditis, valvulitis, and coronary vasculitis.
Differential diagnosis
The differential diagnosis of EGPA shares similarities with GPA and MPA, but also includes eosino philic disorders such as hypereosinophilic syndrome, eosinophilic leukemia, and parasitic diseases.
Diagnostic evaluation
Diagnosis is often based on the presence of asthma, a finding of peripheral eosinophilia (> 1,500 cells/mm3), and the presence of systemic vasculitis involving, ideally, two or more extrapulmonary organs. While histologic confirmation remains ideal, demonstration of characteristic findings on biopsy can be difficult. Glomerular involvement is far less common than in GPA and MPA, but, when present, the renal lesion is identical. Pulmonary histologic findings can be diverse and include the classic “allergic-granuloma” as originally described by Churg and Strauss, as well as isolated granulomatous inflammation, eosinophilic inflammation, or small-vessel vasculitis. Tissue eosinophilia is a prominent finding that typically is seen on biopsies of skin, nerve, and gastrointestinal tissues.
Chest imaging should be performed when EGPA is part of the differential diagnosis. Because of the potential for cardiac involvement, a baseline echocardiogram should be obtained. Pulmonary function tests may be useful, particularly in patients who have a strong asthmatic component.
Similar to GPA and MPA, laboratory assessment includes serum chemistries, complete blood count with differential to determine the eosinophil count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. With the allergic and asthmatic components, immunoglobulin E levels are frequently elevated. Additional testing for other eosinophilic diseases should be pursued as indicated by the clinical features.
Only about 40% of patients are ANCA-positive.13 Most of these are MPO-pANCA, with PR3-cANCA occurring less commonly. Although some reports have suggested differing clinical patterns of EGPA based on ANCA positivity, the presence or absence of ANCA is less helpful in the diagnosis.13
DIFFERENTIATION
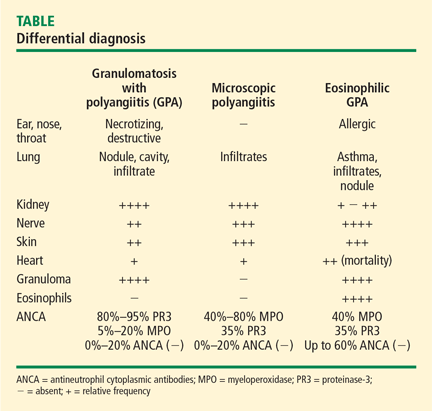
A key histologic difference between GPA and MPA is the presence of granulomatous inflammation in GPA and its absence in MPA under the current nomenclature system.9 Granulomatous inflammation can be seen in EGPA, but it is usually accompanied by eosinophils, which are less likely to be present in GPA and MPA.
The predominance of the ANCA immunofluorescence pattern and target antigen differs between GPA and MPA, with ANCA positivity occurring in 38% of patients with EGPA.13
SUMMARY
Conceptualizing vasculitic disease based on vessel size can be useful, but it is not an absolute definition. Although GPA, MPA, and EGPA predominantly affect small- to medium-sized vessels, these disease entities are phenotypically unique, with both shared features and differences. Common to all three entities is the potential for organ- and life-threatening manifestations, particularly involving the lungs, kidneys, nerves, gastrointestinal tract, and heart. All three entities need aggressive immuno suppression for severe disease. Recognition of these entities and the distinctions among them can guide the approach to diagnosis, treatment, and future outcomes.
- Hoffman GS, Kerr GS, Leavitt RY, et al Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis: review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Bosch X, Guilabert A, Font J. Antineutrophil cytoplasmic antibodies. Lancet 2006; 368:404–418.
- Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis Rheum 1998;1521–1537.
- Wiik A. What you should know about PR3-ANCA. An introduction. Arthritis Res 2000; 2:252–254.
- Kussmaul A, Maier R. Über eine bisher nicht beschriebene eigenthümliche Arterienerkrankung (Periarteritis nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskellähmung einhergeht. Dtsch Arch Klin Med 1866; 1:484–518.
- Davson J, Ball J, Platt R. The kidney in periarteritis nodosa. Q J Med 1948; 17:175–202.
- Jennette C, Falk RJ, Andrassy K, et al Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994; 37:187–192.
- Guillevin L, Durand-Gasselin B, Cevallos R, et al Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999; 42:421–430.
- Keogh KA, Specks U. Churg-Strauss syndrome. Semin Respir Crit Care Med 2006; 27:148–157.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome: clinical study and long-term follow-up of 96 patients. Medicine 1999; 78:26–37.
- Sablé-Fourtassou R, Cohen P, Mahr A, et al., for the French Vasculitis Study Group. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005; 143:632–638.
Vasculitis refers to inflammation of the blood vessel. This inflammation can cause vessel wall thickening that compromises or occludes the vessel lumen, ultimately resulting in organ ischemia. It also can cause vessel wall attenuation that predisposes to aneurysm formation or breach of the vessel integrity with resultant hemorrhage into the tissue.
Vasculitis can be thought of as a primary or secondary process. Primary vasculitides are unique disease entities without a currently identified underlying cause in which vasculitis forms the pathologic basis of tissue injury. Vasculitis can occur secondary to medication exposure or an underlying illness, including infections, malignancy, cryoglobulinemia, and rheumatic diseases (such as systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, or myositis).
Primary vasculitides may differ in epidemiology, such as the age at which they occur and the gender most likely to be affected, their clinical manifestations (including signs, symptoms, and patterns of organ involvement), the diagnostic approach (biopsy, arteriography, and laboratory investigation), treatment (supportive care, glucocorticoids alone, or in combination with other immunosuppressants), and the size of the vessels predominantly affected (large, medium, or small).
Small-vessel vasculitis affects the arteriole, capillary, and venule. An excellent example of small-vessel vasculitis and the one most commonly encountered in clinical practice is cutaneous vasculitis, in which extravasation of erythrocytes from disrupted small vessels is observed histologically, with the clinical sequelae of palpable purpura. Although categorization based on the predominant vessel size that is affected is a helpful way to view these diseases, this is not absolute and each disease has the potential to affect a diverse range of vessels.
This article explores the clinical features and diagnosis of three forms of vasculitis that predominantly affect the small vessels: granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]), microscopic polyangiitis (MPA), and eosinophilic GPA (Churg-Strauss syndrome, EGPA).
GRANULOMATOSIS WITH POLYANGIITIS
Granulomatosis with polyangiitis is characterized by granulomatous inflammation involving the respiratory tract and by vasculitis affecting small- to medium-sized vessels in which necrotizing glomerulonephritis is common.
Wide range of presentations, manifestations
Approximately 90% of patients with GPA have upper or lower airway involvement or both.1 Upper airway or ear symptoms affect 73% of patients initially and 92% overall.1 Direct inspection of the nasal membranes shows a cobblestoned or ulcerated appearance, and computed tomography reveals mucosal thickening of the sinuses. In some instances, sinus disease can compromise blood supply to the cartilaginous portion of the nasal septum, leading to nasal septum perforations or collapse of the nasal bridge. Another manifestation of upper airway disease and GPA is subglottic stenosis, a narrowing in the subglottic region located just below the vocal cords. The narrowing typically spans about 1 cm and rarely extends or involves the remainder of the trachea.
The lungs are involved in 85% of patients.1 Radiographic abnormalities can be diverse and include bilateral pulmonary nodular infiltrates, single or multiple cavities, and bilateral ground glass infiltrates as can be seen in pulmonary hemorrhage (Figure). Bronchoscopy may reveal endobronchial stenosis, and pleural disease can occur rarely.
Approximately 20% of patients with GPA may have glomerulonephritis when they first present for medical attention, but it eventually develops in nearly 80% of patients during the disease course.1 Despite its potential for rapid progression, glomerulonephritis presents a diagnostic challenge because it is asymptomatic. It is detected by evidence of proteinuria and an active urine sediment with dysmorphic red blood cells and red blood cell casts.
Ocular involvement occurs eventually in 52% of patients with GPA.1 Any ocular structure can be affected and ocular involvement can be visually threatening. The more prominent ocular manifestations include scleritis/episcleritis or orbital disease.
Cutaneous manifestations, observed in 46% of patients, include verrucous-appearing lesions on the elbow and infarctions in the skin and nail folds.1 Other rare manifestations can occur, such as pericarditis and cerebral vasculitis.
Although nearly all patients present with upper or lower airway symptoms, the multisystem nature of GPA explains the wide range of presentations and the varying degrees of disease severity.
Differential diagnosis
The differential diagnosis in GPA is varied. Particularly in the setting of isolated lung or sinus disease, infection is foremost in the differential diagnosis. Even in the nonimmunosuppressed host, unusual infections such as mycobacteria, histoplasmosis, and other fungal infections should be considered. Lymphadenopathy, rarely seen in GPA, should raise concern for other causes of disease. Lymphoproliferative processes and other neoplasms, other rheumatic diseases, granulomatous disease (ie, sarcoidosis), and other causes of glomerulonephritis (when present) also merit consideration. Differentiation of these entities from GPA is essential because the treatment differs in many instances.
The differential diagnosis for patients who present with midline destructive lesions must include other causes of collapse of the nasal bridge, nasal septum perforation, and possibly palate destruction. Erosions of the hard palate in particular should raise an immediate red flag for entities other than GPA, such as lymphoproliferative diseases; rare infections, particularly if the patient has studied or worked abroad; and cocaine exposure.
Diagnostic evaluation
A diagnosis of GPA is typically based on the presence of histologic features in a clinically compatible setting. Diagnostic features include necrosis, granulomatous inflammation, vasculitis, and special stains and cultures negative for microorganisms.
Biopsy sites are determined by evidence of clinical disease affecting a target organ and the likelihood of obtaining diagnostically meaningful findings from that site. One challenge is that biopsies are not always diagnostic. The changes tend to be patchy and the likelihood of a positive yield is associated with the amount of tissue that can be obtained. Tissues from the ear, nose, and throat have a yield of about 20%, depending upon the site and the biopsy size. The highest yield comes from radiographically abnormal pulmonary parenchyma. Although transbronchial biopsies are attractive because they are less invasive than open lung biopsy, they are also far less diagnostic, with fewer than 10% having a positive yield. Because cutaneous vasculitis is observed in many settings, its presence is usually insufficient evidence for diagnosis. The renal histologic appearance is a focal, segmental, crescentic, and necrotizing glomerulonephritis that has few to no immune complexes (pauciimmune glomerulonephritis).1–3
Chest imaging should be performed in any patient in whom GPA is part of the differential diagnosis, since up to one-third of patients may be asymptomatic yet have pulmonary radiographic findings.
Laboratory assessment should include serum chemistries to evaluate renal and hepatic function, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. If the urinalysis is positive for blood, microscopy should be performed on fresh urine to look for casts. In the setting of pulmonary-renal manifestations, testing for other causes, such as antiglomerular basement antibodies and antinuclear antibodies, should be considered.
Serologic testing for antineutrophil cytoplasmic antibodies (ANCA) has provided a useful tool in suggesting the diagnosis of GPA. Two forms of ANCA have been identified in patients with vasculitis: ANCA directed against the neutrophil serine protease proteinase-3 (PR3), which results in a cytoplasmic immunofluorescence (cANCA) pattern; and ANCA directed against the neutrophil enzyme myeloperoxidase (MPO), which causes a perinuclear immunofluorescence (pANCA) pattern.4 Approximately 80% to 95% of ANCA found in patients with active severe GPA are detectable PR3-cANCA, while 5% to 20% are MPO-pANCA.5 The predictive value of ANCA for the diagnosis depends on the spectrum of clinical features. As ANCA can be seen in other settings, ANCA as the basis for diagnosis in place of tissue biopsy should be used with caution and only in selected instances where their predictive value would equal that of biopsy. The presence of ANCA is not necessary to establish the diagnosis, as up to 20% of patients with GPA may be ANCA-negative.6
MICROSCOPIC POLYANGIITIS
The history of MPA dates to 1866, with the description of periarteritis nodosa.7 The term “microscopic polyarteritis” was introduced in 1948, when glomerular disease was recognized in some patients.8 In 1994, the Chapel Hill Consensus Conference defined MPA as a necrotizing vasculitis with few or no immune deposits that affects small vessels (ie, capillaries, venules, or arterioles). Necrotizing arteritis of small- and medium-sized arteries may be present. Necrotizing glomerulonephritis and pulmonary capillaritis commonly occur.9 MPA shares many clinical features with GPA and is currently said to be distinguished by the absence of granulomatous inflammation.9
Presentations and manifestations
In one assessment of organ system involvement in 85 patients with MPA, investigators observed glomerular syndrome in 82% of patients.10 They also found a high predilection for involvement of the skin, joints, and lungs. Pulmonary hemorrhage is a particularly important manifestation of MPA because it can be immediately life-threatening.
Differential diagnosis
The differential diagnosis for MPA is similar to GPA in the inclusion of other causes of classic pulmonary-renal syndromes, such as antiglomerular basement membrane disease and systemic lupus erythematosus. Poststreptococcal glomerulonephritis should be considered when the kidney is the predominant organ involved in the absence of lung disease. In the setting of pulmonary infiltrates, infections and neoplasms remain significant in the differential diagnosis.
Diagnostic evaluation
The diagnosis of MPA is based on consistent clinical features and compatible histologic findings. The histologic renal lesion is identical to that seen in GPA. Pulmonary disease typically includes capillaritis and is notable for the absence of evidence of immune deposition, in contrast to antiglomerular basement membrane disease.
Chest imaging is indicated when MPA is part of the differential diagnosis. Computed tomography is the preferred technique, as early alveolar hemorrhage that can occur in MPA may not be visualized on a chest radiograph.
Laboratory assessment should include serum chemistries, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. Additional testing should be pursued for other diseases as indicated by the clinical features.
Approximately 40% to 80% of patients with MPA have MPO-pANCA.5 Approximately 15% of patients are MPO-pANCA positive,6 and 0% to 20% are ANCA-negative. As with GPA, ANCA is useful to suggest—but not diagnose—disease in many instances. The absence of ANCA does not rule out MPA.
EOSINOPHILIC GPA
Eosinophilic GPA is a unique entity characterized by eosinophil-rich and granulomatous inflammation involving the respiratory tract and necrotizing vasculitis of small- to medium-size vessels. It is also associated with asthma and eosinophilia.
Different disease phases
Eosinophilic GPA is often thought of as having three phases: prodromal, eosinophilic, and vasculitic.11,12 Although helpful conceptually, these phases may not always be present and may not occur in sequence.
The prodromal phase is characterized by asthma associated with allergic rhinitis with or without polyposis. The eosinophilic phase is characterized by the presence of eosinophilia in the blood and tissue. Eosinophilia is a prominent feature, although accurate detection and assessment can be challenging in the setting of glucocorticoid use for asthma as this normalizes the eosinophil count.
The vasculitic phase distinguishes EGPA from other eosinophilic disorders. Features of vasculitis may occur in multiple organ sites, including the nerves, lungs, heart, gastrointestinal tract, and kidneys. In one series of 96 patients, nearly 100% had asthma, and peripheral nervous system involvement in the form of mononeuritis multiplex was present in 72%.12 Cardiac involvement is of particular importance as it is a prominent cause of disease-related mortality. Cardiac manifestations include myocarditis, pericarditis, endocarditis, valvulitis, and coronary vasculitis.
Differential diagnosis
The differential diagnosis of EGPA shares similarities with GPA and MPA, but also includes eosino philic disorders such as hypereosinophilic syndrome, eosinophilic leukemia, and parasitic diseases.
Diagnostic evaluation
Diagnosis is often based on the presence of asthma, a finding of peripheral eosinophilia (> 1,500 cells/mm3), and the presence of systemic vasculitis involving, ideally, two or more extrapulmonary organs. While histologic confirmation remains ideal, demonstration of characteristic findings on biopsy can be difficult. Glomerular involvement is far less common than in GPA and MPA, but, when present, the renal lesion is identical. Pulmonary histologic findings can be diverse and include the classic “allergic-granuloma” as originally described by Churg and Strauss, as well as isolated granulomatous inflammation, eosinophilic inflammation, or small-vessel vasculitis. Tissue eosinophilia is a prominent finding that typically is seen on biopsies of skin, nerve, and gastrointestinal tissues.
Chest imaging should be performed when EGPA is part of the differential diagnosis. Because of the potential for cardiac involvement, a baseline echocardiogram should be obtained. Pulmonary function tests may be useful, particularly in patients who have a strong asthmatic component.
Similar to GPA and MPA, laboratory assessment includes serum chemistries, complete blood count with differential to determine the eosinophil count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. With the allergic and asthmatic components, immunoglobulin E levels are frequently elevated. Additional testing for other eosinophilic diseases should be pursued as indicated by the clinical features.
Only about 40% of patients are ANCA-positive.13 Most of these are MPO-pANCA, with PR3-cANCA occurring less commonly. Although some reports have suggested differing clinical patterns of EGPA based on ANCA positivity, the presence or absence of ANCA is less helpful in the diagnosis.13
DIFFERENTIATION
A key histologic difference between GPA and MPA is the presence of granulomatous inflammation in GPA and its absence in MPA under the current nomenclature system.9 Granulomatous inflammation can be seen in EGPA, but it is usually accompanied by eosinophils, which are less likely to be present in GPA and MPA.
The predominance of the ANCA immunofluorescence pattern and target antigen differs between GPA and MPA, with ANCA positivity occurring in 38% of patients with EGPA.13
SUMMARY
Conceptualizing vasculitic disease based on vessel size can be useful, but it is not an absolute definition. Although GPA, MPA, and EGPA predominantly affect small- to medium-sized vessels, these disease entities are phenotypically unique, with both shared features and differences. Common to all three entities is the potential for organ- and life-threatening manifestations, particularly involving the lungs, kidneys, nerves, gastrointestinal tract, and heart. All three entities need aggressive immuno suppression for severe disease. Recognition of these entities and the distinctions among them can guide the approach to diagnosis, treatment, and future outcomes.
Vasculitis refers to inflammation of the blood vessel. This inflammation can cause vessel wall thickening that compromises or occludes the vessel lumen, ultimately resulting in organ ischemia. It also can cause vessel wall attenuation that predisposes to aneurysm formation or breach of the vessel integrity with resultant hemorrhage into the tissue.
Vasculitis can be thought of as a primary or secondary process. Primary vasculitides are unique disease entities without a currently identified underlying cause in which vasculitis forms the pathologic basis of tissue injury. Vasculitis can occur secondary to medication exposure or an underlying illness, including infections, malignancy, cryoglobulinemia, and rheumatic diseases (such as systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, or myositis).
Primary vasculitides may differ in epidemiology, such as the age at which they occur and the gender most likely to be affected, their clinical manifestations (including signs, symptoms, and patterns of organ involvement), the diagnostic approach (biopsy, arteriography, and laboratory investigation), treatment (supportive care, glucocorticoids alone, or in combination with other immunosuppressants), and the size of the vessels predominantly affected (large, medium, or small).
Small-vessel vasculitis affects the arteriole, capillary, and venule. An excellent example of small-vessel vasculitis and the one most commonly encountered in clinical practice is cutaneous vasculitis, in which extravasation of erythrocytes from disrupted small vessels is observed histologically, with the clinical sequelae of palpable purpura. Although categorization based on the predominant vessel size that is affected is a helpful way to view these diseases, this is not absolute and each disease has the potential to affect a diverse range of vessels.
This article explores the clinical features and diagnosis of three forms of vasculitis that predominantly affect the small vessels: granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]), microscopic polyangiitis (MPA), and eosinophilic GPA (Churg-Strauss syndrome, EGPA).
GRANULOMATOSIS WITH POLYANGIITIS
Granulomatosis with polyangiitis is characterized by granulomatous inflammation involving the respiratory tract and by vasculitis affecting small- to medium-sized vessels in which necrotizing glomerulonephritis is common.
Wide range of presentations, manifestations
Approximately 90% of patients with GPA have upper or lower airway involvement or both.1 Upper airway or ear symptoms affect 73% of patients initially and 92% overall.1 Direct inspection of the nasal membranes shows a cobblestoned or ulcerated appearance, and computed tomography reveals mucosal thickening of the sinuses. In some instances, sinus disease can compromise blood supply to the cartilaginous portion of the nasal septum, leading to nasal septum perforations or collapse of the nasal bridge. Another manifestation of upper airway disease and GPA is subglottic stenosis, a narrowing in the subglottic region located just below the vocal cords. The narrowing typically spans about 1 cm and rarely extends or involves the remainder of the trachea.
The lungs are involved in 85% of patients.1 Radiographic abnormalities can be diverse and include bilateral pulmonary nodular infiltrates, single or multiple cavities, and bilateral ground glass infiltrates as can be seen in pulmonary hemorrhage (Figure). Bronchoscopy may reveal endobronchial stenosis, and pleural disease can occur rarely.
Approximately 20% of patients with GPA may have glomerulonephritis when they first present for medical attention, but it eventually develops in nearly 80% of patients during the disease course.1 Despite its potential for rapid progression, glomerulonephritis presents a diagnostic challenge because it is asymptomatic. It is detected by evidence of proteinuria and an active urine sediment with dysmorphic red blood cells and red blood cell casts.
Ocular involvement occurs eventually in 52% of patients with GPA.1 Any ocular structure can be affected and ocular involvement can be visually threatening. The more prominent ocular manifestations include scleritis/episcleritis or orbital disease.
Cutaneous manifestations, observed in 46% of patients, include verrucous-appearing lesions on the elbow and infarctions in the skin and nail folds.1 Other rare manifestations can occur, such as pericarditis and cerebral vasculitis.
Although nearly all patients present with upper or lower airway symptoms, the multisystem nature of GPA explains the wide range of presentations and the varying degrees of disease severity.
Differential diagnosis
The differential diagnosis in GPA is varied. Particularly in the setting of isolated lung or sinus disease, infection is foremost in the differential diagnosis. Even in the nonimmunosuppressed host, unusual infections such as mycobacteria, histoplasmosis, and other fungal infections should be considered. Lymphadenopathy, rarely seen in GPA, should raise concern for other causes of disease. Lymphoproliferative processes and other neoplasms, other rheumatic diseases, granulomatous disease (ie, sarcoidosis), and other causes of glomerulonephritis (when present) also merit consideration. Differentiation of these entities from GPA is essential because the treatment differs in many instances.
The differential diagnosis for patients who present with midline destructive lesions must include other causes of collapse of the nasal bridge, nasal septum perforation, and possibly palate destruction. Erosions of the hard palate in particular should raise an immediate red flag for entities other than GPA, such as lymphoproliferative diseases; rare infections, particularly if the patient has studied or worked abroad; and cocaine exposure.
Diagnostic evaluation
A diagnosis of GPA is typically based on the presence of histologic features in a clinically compatible setting. Diagnostic features include necrosis, granulomatous inflammation, vasculitis, and special stains and cultures negative for microorganisms.
Biopsy sites are determined by evidence of clinical disease affecting a target organ and the likelihood of obtaining diagnostically meaningful findings from that site. One challenge is that biopsies are not always diagnostic. The changes tend to be patchy and the likelihood of a positive yield is associated with the amount of tissue that can be obtained. Tissues from the ear, nose, and throat have a yield of about 20%, depending upon the site and the biopsy size. The highest yield comes from radiographically abnormal pulmonary parenchyma. Although transbronchial biopsies are attractive because they are less invasive than open lung biopsy, they are also far less diagnostic, with fewer than 10% having a positive yield. Because cutaneous vasculitis is observed in many settings, its presence is usually insufficient evidence for diagnosis. The renal histologic appearance is a focal, segmental, crescentic, and necrotizing glomerulonephritis that has few to no immune complexes (pauciimmune glomerulonephritis).1–3
Chest imaging should be performed in any patient in whom GPA is part of the differential diagnosis, since up to one-third of patients may be asymptomatic yet have pulmonary radiographic findings.
Laboratory assessment should include serum chemistries to evaluate renal and hepatic function, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. If the urinalysis is positive for blood, microscopy should be performed on fresh urine to look for casts. In the setting of pulmonary-renal manifestations, testing for other causes, such as antiglomerular basement antibodies and antinuclear antibodies, should be considered.
Serologic testing for antineutrophil cytoplasmic antibodies (ANCA) has provided a useful tool in suggesting the diagnosis of GPA. Two forms of ANCA have been identified in patients with vasculitis: ANCA directed against the neutrophil serine protease proteinase-3 (PR3), which results in a cytoplasmic immunofluorescence (cANCA) pattern; and ANCA directed against the neutrophil enzyme myeloperoxidase (MPO), which causes a perinuclear immunofluorescence (pANCA) pattern.4 Approximately 80% to 95% of ANCA found in patients with active severe GPA are detectable PR3-cANCA, while 5% to 20% are MPO-pANCA.5 The predictive value of ANCA for the diagnosis depends on the spectrum of clinical features. As ANCA can be seen in other settings, ANCA as the basis for diagnosis in place of tissue biopsy should be used with caution and only in selected instances where their predictive value would equal that of biopsy. The presence of ANCA is not necessary to establish the diagnosis, as up to 20% of patients with GPA may be ANCA-negative.6
MICROSCOPIC POLYANGIITIS
The history of MPA dates to 1866, with the description of periarteritis nodosa.7 The term “microscopic polyarteritis” was introduced in 1948, when glomerular disease was recognized in some patients.8 In 1994, the Chapel Hill Consensus Conference defined MPA as a necrotizing vasculitis with few or no immune deposits that affects small vessels (ie, capillaries, venules, or arterioles). Necrotizing arteritis of small- and medium-sized arteries may be present. Necrotizing glomerulonephritis and pulmonary capillaritis commonly occur.9 MPA shares many clinical features with GPA and is currently said to be distinguished by the absence of granulomatous inflammation.9
Presentations and manifestations
In one assessment of organ system involvement in 85 patients with MPA, investigators observed glomerular syndrome in 82% of patients.10 They also found a high predilection for involvement of the skin, joints, and lungs. Pulmonary hemorrhage is a particularly important manifestation of MPA because it can be immediately life-threatening.
Differential diagnosis
The differential diagnosis for MPA is similar to GPA in the inclusion of other causes of classic pulmonary-renal syndromes, such as antiglomerular basement membrane disease and systemic lupus erythematosus. Poststreptococcal glomerulonephritis should be considered when the kidney is the predominant organ involved in the absence of lung disease. In the setting of pulmonary infiltrates, infections and neoplasms remain significant in the differential diagnosis.
Diagnostic evaluation
The diagnosis of MPA is based on consistent clinical features and compatible histologic findings. The histologic renal lesion is identical to that seen in GPA. Pulmonary disease typically includes capillaritis and is notable for the absence of evidence of immune deposition, in contrast to antiglomerular basement membrane disease.
Chest imaging is indicated when MPA is part of the differential diagnosis. Computed tomography is the preferred technique, as early alveolar hemorrhage that can occur in MPA may not be visualized on a chest radiograph.
Laboratory assessment should include serum chemistries, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. Additional testing should be pursued for other diseases as indicated by the clinical features.
Approximately 40% to 80% of patients with MPA have MPO-pANCA.5 Approximately 15% of patients are MPO-pANCA positive,6 and 0% to 20% are ANCA-negative. As with GPA, ANCA is useful to suggest—but not diagnose—disease in many instances. The absence of ANCA does not rule out MPA.
EOSINOPHILIC GPA
Eosinophilic GPA is a unique entity characterized by eosinophil-rich and granulomatous inflammation involving the respiratory tract and necrotizing vasculitis of small- to medium-size vessels. It is also associated with asthma and eosinophilia.
Different disease phases
Eosinophilic GPA is often thought of as having three phases: prodromal, eosinophilic, and vasculitic.11,12 Although helpful conceptually, these phases may not always be present and may not occur in sequence.
The prodromal phase is characterized by asthma associated with allergic rhinitis with or without polyposis. The eosinophilic phase is characterized by the presence of eosinophilia in the blood and tissue. Eosinophilia is a prominent feature, although accurate detection and assessment can be challenging in the setting of glucocorticoid use for asthma as this normalizes the eosinophil count.
The vasculitic phase distinguishes EGPA from other eosinophilic disorders. Features of vasculitis may occur in multiple organ sites, including the nerves, lungs, heart, gastrointestinal tract, and kidneys. In one series of 96 patients, nearly 100% had asthma, and peripheral nervous system involvement in the form of mononeuritis multiplex was present in 72%.12 Cardiac involvement is of particular importance as it is a prominent cause of disease-related mortality. Cardiac manifestations include myocarditis, pericarditis, endocarditis, valvulitis, and coronary vasculitis.
Differential diagnosis
The differential diagnosis of EGPA shares similarities with GPA and MPA, but also includes eosino philic disorders such as hypereosinophilic syndrome, eosinophilic leukemia, and parasitic diseases.
Diagnostic evaluation
Diagnosis is often based on the presence of asthma, a finding of peripheral eosinophilia (> 1,500 cells/mm3), and the presence of systemic vasculitis involving, ideally, two or more extrapulmonary organs. While histologic confirmation remains ideal, demonstration of characteristic findings on biopsy can be difficult. Glomerular involvement is far less common than in GPA and MPA, but, when present, the renal lesion is identical. Pulmonary histologic findings can be diverse and include the classic “allergic-granuloma” as originally described by Churg and Strauss, as well as isolated granulomatous inflammation, eosinophilic inflammation, or small-vessel vasculitis. Tissue eosinophilia is a prominent finding that typically is seen on biopsies of skin, nerve, and gastrointestinal tissues.
Chest imaging should be performed when EGPA is part of the differential diagnosis. Because of the potential for cardiac involvement, a baseline echocardiogram should be obtained. Pulmonary function tests may be useful, particularly in patients who have a strong asthmatic component.
Similar to GPA and MPA, laboratory assessment includes serum chemistries, complete blood count with differential to determine the eosinophil count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. With the allergic and asthmatic components, immunoglobulin E levels are frequently elevated. Additional testing for other eosinophilic diseases should be pursued as indicated by the clinical features.
Only about 40% of patients are ANCA-positive.13 Most of these are MPO-pANCA, with PR3-cANCA occurring less commonly. Although some reports have suggested differing clinical patterns of EGPA based on ANCA positivity, the presence or absence of ANCA is less helpful in the diagnosis.13
DIFFERENTIATION
A key histologic difference between GPA and MPA is the presence of granulomatous inflammation in GPA and its absence in MPA under the current nomenclature system.9 Granulomatous inflammation can be seen in EGPA, but it is usually accompanied by eosinophils, which are less likely to be present in GPA and MPA.
The predominance of the ANCA immunofluorescence pattern and target antigen differs between GPA and MPA, with ANCA positivity occurring in 38% of patients with EGPA.13
SUMMARY
Conceptualizing vasculitic disease based on vessel size can be useful, but it is not an absolute definition. Although GPA, MPA, and EGPA predominantly affect small- to medium-sized vessels, these disease entities are phenotypically unique, with both shared features and differences. Common to all three entities is the potential for organ- and life-threatening manifestations, particularly involving the lungs, kidneys, nerves, gastrointestinal tract, and heart. All three entities need aggressive immuno suppression for severe disease. Recognition of these entities and the distinctions among them can guide the approach to diagnosis, treatment, and future outcomes.
- Hoffman GS, Kerr GS, Leavitt RY, et al Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis: review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Bosch X, Guilabert A, Font J. Antineutrophil cytoplasmic antibodies. Lancet 2006; 368:404–418.
- Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis Rheum 1998;1521–1537.
- Wiik A. What you should know about PR3-ANCA. An introduction. Arthritis Res 2000; 2:252–254.
- Kussmaul A, Maier R. Über eine bisher nicht beschriebene eigenthümliche Arterienerkrankung (Periarteritis nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskellähmung einhergeht. Dtsch Arch Klin Med 1866; 1:484–518.
- Davson J, Ball J, Platt R. The kidney in periarteritis nodosa. Q J Med 1948; 17:175–202.
- Jennette C, Falk RJ, Andrassy K, et al Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994; 37:187–192.
- Guillevin L, Durand-Gasselin B, Cevallos R, et al Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999; 42:421–430.
- Keogh KA, Specks U. Churg-Strauss syndrome. Semin Respir Crit Care Med 2006; 27:148–157.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome: clinical study and long-term follow-up of 96 patients. Medicine 1999; 78:26–37.
- Sablé-Fourtassou R, Cohen P, Mahr A, et al., for the French Vasculitis Study Group. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005; 143:632–638.
- Hoffman GS, Kerr GS, Leavitt RY, et al Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis: review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Bosch X, Guilabert A, Font J. Antineutrophil cytoplasmic antibodies. Lancet 2006; 368:404–418.
- Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis Rheum 1998;1521–1537.
- Wiik A. What you should know about PR3-ANCA. An introduction. Arthritis Res 2000; 2:252–254.
- Kussmaul A, Maier R. Über eine bisher nicht beschriebene eigenthümliche Arterienerkrankung (Periarteritis nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskellähmung einhergeht. Dtsch Arch Klin Med 1866; 1:484–518.
- Davson J, Ball J, Platt R. The kidney in periarteritis nodosa. Q J Med 1948; 17:175–202.
- Jennette C, Falk RJ, Andrassy K, et al Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994; 37:187–192.
- Guillevin L, Durand-Gasselin B, Cevallos R, et al Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999; 42:421–430.
- Keogh KA, Specks U. Churg-Strauss syndrome. Semin Respir Crit Care Med 2006; 27:148–157.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome: clinical study and long-term follow-up of 96 patients. Medicine 1999; 78:26–37.
- Sablé-Fourtassou R, Cohen P, Mahr A, et al., for the French Vasculitis Study Group. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005; 143:632–638.
Controversies in ANCA testing
Antineutrophil cytoplasmic antibody (ANCA) detection is a valuable tool for diagnosing small-vessel vasculitis,1 but measuring and interpreting ANCA levels is an inexact science. There is no single perfect ANCA test, and even a perfect test would not provide definitive clinical answers. The diagnostic utility of ANCA testing depends on the methodologic accuracy of the test and the appropriate ordering of testing in the right clinical setting. This article examines three important questions about this technology:
- What is the best ANCA test methodology?
- What is the prognostic value of serial ANCA testing?
- What is the clinical implication of ANCA type?
WHAT IS THE BEST ANCA TEST METHODOLOGY?
The diagnostic utility of ANCA testing depends on both the methodologic accuracy of the test and the appropriate ordering of tests. Methodologic accuracy comprises the analytic sensitivity and specificity of the test. Analytic sensitivity refers to the accurate identification of the presence of ANCA, and analytic specificity refers to measurement of only the entity in question (ANCA), not confounded by the presence of other entities (antibodies).
Equally as important as analytic accuracy is the appropriate ordering of the tests in the right clinical setting. Using a test that is sensitive to the presence of a specific ANCA type accurately identifies the presence of either proteinase-3 (PR3)- or myeloperoxidase (MPO)-ANCA. Once obtained, test results must be evaluated in terms of their relationship to the diagnosis being considered. If the tests are deemed diagnostically useful based on the results, the data can be used to assess the positive and negative predictive value of the tests.
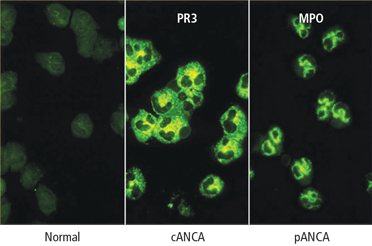
Immunofluorescence or antigen-specific testing—or both?
A definitive diagnosis is more likely if an immunofluorescence staining pattern of cANCA is paired with the antigen specificity of PR3-ANCA, for example, or a perinuclear immunofluorescence pattern (pANCA) is paired with a positive MPO-ANCA. When positive test pairings have been obtained and the patient’s antigen ANCA reactivity is known, subsequent serial ANCA testing with an antigen-specific assay alone may be indicated, because the ANCA types of patients with vasculitis are unlikely to switch between PR3 and MPO during the course of their disease. If matching pairings are not obtained, the diagnostic utility of the tests remains unconfirmed.
Antigen type (PR3 or MPO) is determined through antigen-specific methods that include solid-phase assays and other methods of bringing the specific antigen in contact with the specific antibody in question. There are two categories of solid-phase assays: the enzyme-linked immunoabsorbent assay (ELISA) and the capture ELISA. In the ELISA methodology, the antigen is directly coated to a plastic plate; in the capture ELISA, an anchor, usually a monoclonal antibody or combination of antibodies, captures the target antigen on the plate. In both ELISA and capture ELISA assays, ANCA contained in the serum sample subjected to testing bind to the immobilized antigen. The amount of ANCA bound to the antigen can then be detected by a secondary antibody that is conjugated with an enzyme that can elicit a color reaction. The intensity of the color reaction is proportional to the amount of ANCA bound to the immobilized antigen.
The ELISA methodology tends to trade off analytic sensitivity for specificity, since the antigen purification process (which allows the ELISA system to increase its specificity) can cause conformational changes to the antigen being bound to the plate. This, in turn, causes a loss of some recognition of the conformationally sensitive ANCA.
In capture ELISA, a specific antibody captures the antigen; this stabilizes the conformation, boosts the analytic sensitivity, and allows a gentler purification process because it only captures the antigen in question and then binds it to the plate. This process decreases false-positive test results caused by residual contaminants in the antigen preparation. Analytic sensitivity issues may come into play if the anchoring monoclonal antibody competes for the epitope on the antigen being recognized by the serum antibody in question (ANCA), causing occasional false-negative results.
Another method now applied to commercial ANCA testing involves bead-based multiplex assays. These assays are based on principles similar to the ELISA or capture ELISA methods. In multiplex microsphere technology, the purified antigen is bound to a polystyrene microsphere instead of a plate. The microsphere is then presented to the antibody in question. The bead is then introduced to a secondary antibody labeled with a fluorescent marker (instead of an enzyme) for detection of the antibody. One advantage of this system is that various beads containing different antigens can be introduced to the same serum sample, and then different color reactions can be measured for each bead. Because only one antigen is bound to each microsphere (eg, PR3-ANCA, MPOANCA or other specific autoantibodies), only specific antibodies will react to each bead of a specific color. If there is no MPO antibody in the sample, there will be no reaction against the MPO antigen bead; however, if PR3-ANCA is present in the sample, it would react with the PR3 antigen beads. Using this methodology, a single serum sample can be tested for a multitude of autoantibodies at the same time (see “Interpreting ANCA results: Accurate tests, appropriate orders,”2–10 above).

WHAT IS THE PROGNOSTIC VALUE OF SERIAL ANCA TESTING?
Persistent changes in ANCA levels in relapsing disease may have some value in predicting outcome. The issues to consider include the methodology used to determine serial ANCA levels, correlations between ANCA and disease activity, and the use of ANCA changes to guide treatment.
Does methodology matter when determining serial ANCA levels?
Methodology in serial ANCA testing is probably unimportant as long as the same method is used serially. Analysis of large groups of ANCA-positive patients show a statistically highly significant correlation among results obtained with different detection methods, including immunofluorescence, direct ELISA, or capture ELISA. However, at the individual patient level there is some variability.
Do ANCA levels correlate with disease activity?
In a prospective study, serial ANCA samples obtained during the Wegener’s Granulomatosis Etanercept Trial (WGET)11 were processed in the same manner (collected every 3 months, mean follow-up of 22 months, uniform handling of samples). All samples were analyzed by capture ELISA, and disease activity was measured by the Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis (BVAS/WG). The results indicated that an increase in PR3-ANCA levels was not a significant predictor of relapse. The frequency of a relapse within 1 year of an increase in PR3-ANCA levels was found to be approximately 50%,11 a result similar to that reported in several smaller studies of different design and methodology.
Should ANCA changes guide treatment?
The available data regarding serial ANCA testing are limited mostly to PR3-ANCA. Serial ANCA testing has limited value as a guide to treatment and, in general, changes in ANCA levels alone should not be used to guide treatment decisions. In new patients without documented serial ANCA level associations, an increase in PR3-ANCA levels has no reliable predictive value. The existing literature suggests that this lack of association is not dependent on the method used for ANCA detection. For individual patients in whom long-term serial ANCA testing has been performed and a relationship between PR3-ANCA levels and disease activity has been established, serial ANCA testing can have some predictive value and can be used to guide treatment. For example, when remission is achieved by depleting B cells in patients with chronically relapsing GPA, ANCA levels usually go down. After B-cell reconstitution, the ANCA level rises in most patients, and this rise is usually associated with a flare shortly thereafter. A flare can be preempted when this pattern is determined in a specific patient, and preemptive treatment is applied accordingly.12
WHAT IS THE IMPLICATION OF ANCA TYPE?
Available reports consistently suggest that PR3-ANCA is associated with a higher mortality than MPO-ANCA (relative risk [RR], 3.78),13 and a higher relapse rate.14,15 A more rapid loss of renal function among patients with glomerulonephritis and PR3-ANCA than those with MPO-ANCA has also been reported.16 Using remission as the starting point, the number of days from complete remission to first disease flare was plotted for patients with MPO- versus PR3-ANCA in an analysis of long-term data from the Rituximab in ANCA-Associated Vasculitis (RAVE) trial.17 The resulting curve demonstrated a divergence in the probability of remaining in remission, confirming that remission maintenance is clearly greater in patients with MPO-ANCA than in patients with PR3-ANCA.
The primary end point of the RAVE trial was remission of disease without the use of prednisone at 6 months. There was little difference in end point achieved based on comparison of diagnosis (microscopic polyangiitis or granulomatosis) or treatment arms (rituximab versus cyclophosphamide); however, an analysis of end point data separating the patients by ANCA type showed that the treatment response to rituximab was superior to that of cyclophosphamide among patients with PR3-ANCA, whereas in patients with MPO-ANCA, there was little difference in response associated with either treatment. Regarding the likelihood of attaining an ANCA-negative status after 6 months, again MPO-ANCA patients showed no difference in frequency on either treatment. Among PR3-ANCA–positive patients, 50% in the rituximab arm attained ANCA-negative status compared with only 17% in the cyclophosphamide arm.17
SUMMARY
Diagnostic utility of ANCA testing depends on the methodology and clinical setting. Only cANCA/PR3-ANCA and pANCA/MPO-ANCA pairings have positive predictive value for diagnosis of small-vessel vasculitis. Mismatches in results, findings of human neutrophil elastase–ANCA, or identification of multiple positive antigens should be considered in cases of cocaine or drug use.
The clinical utility of serial ANCA testing is unconfirmed. Good data currently exist only for PR3-ANCA, and different drugs may affect ANCA levels in different ways. ANCA type is significant in that PR3-ANCA portends a higher relapse rate and poorer patient outcomes compared with MPO-ANCA.
- Russell KA, Wiegert E, Schroeder DR, Homburger HA, Specks U. Detection of anti-neutrophil cytoplasmic antibodies under actual clinical testing conditions. Clin Immunol 2002; 103:196–203.
- Langford CA. The diagnostic utility of c-ANCA in Wegener’s granulomatosis. Cleve Clin J Med 1998; 65:135–140.
- Trimarchi M, Gregorini G, Facchetti F, et al. Cocaine-induced midline destructive lesions: clinical, radiographic, histopathologic, and serologic features and their differentiation from Wegener granulomatosis. Medicine 2001; 80:391–404.
- Wiesner O, Russell KA, Lee AS, et al. Antineutrophil cytoplasmic antibodies reacting with human neutrophil elastase as a diagnostic marker for cocaine-induced midline destructive lesions but not autoimmune vasculitis. Arthritis Rheum 2004; 50:2954–2965.
- Peikert T, Finkielman JD, Hummel AM, et al. Functional characterization of antineutrophil cytoplasmic antibodies in patients with cocaine-induced midline destructive lesions. Arthritis Rheum 2008; 58:1546–1551.
- Knowles L, Buxton JA, Skuridina N, et al. Levamisole tainted cocaine causing severe neutropenia in Alberta and British Columbia. Harm Reduct J 2009; 6 (Nov 17):30. doi: 10.1186/1477-7517-6-30.
- Zhu NY, LeGatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med 2009; 150:287–289.
- Bradford M, Rosenberg B, Moreno J, Dumyati G. Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole. Ann Intern Med 2010; 152:758–759.
- Waller JM, Feramisco JD, Alberta-Wszolek L, McCalmont TH, Fox LP. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit [published online ahead of print March 20, 2010]? J Am Acad Dermatol 2010; 63:530–535. doi: 10.1016/j.jaad.2010.01.055
- Chang A, Osterloh J, Thomas J. Levamisole: a dangerous new cocaine adulterant [published online ahead of print July 28, 2010]. Clin Pharmacol Ther 2010; 88:408–411. doi: 10.1038/clpt.2010.156
- Finkielman JD, Merkel PA, Schroeder D, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med 2007; 147:611–619.
- Cartin-Ceba R, Golbin J, Keogh KA, et al. Rituximab for remission induction and maintenance in granulomatosis with polyangiitis (Wegener’s): a single-center ten-year experience [published online ahead of print June 21, 2012]. Arthritis Rheum. doi: 10.1002/art.34584
- Hogan SL, Nachman PH, Wilkman AS, Jennette JC, Falk RJ; the Glomerular Disease Collaborative Network. Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 1996; 7:23–32.
- Booth AD, Almond MK, Burns A, et al. Outcome of ANCA-associated renal vasculitis: a 5-year retrospective study. Am J Kidney Dis 2003; 41:776–784.
- Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Franssen CFM, Gans ROB, Arends B, et al. Differences between anti-myeloperoxidase- and anti-proteinase 3-associated renal disease. Kidney Int 1995; 47:193–199.
- Stone JH, Merkel PA, Spiera R, et al; for the RAVE–ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010; 363:221–232.
Antineutrophil cytoplasmic antibody (ANCA) detection is a valuable tool for diagnosing small-vessel vasculitis,1 but measuring and interpreting ANCA levels is an inexact science. There is no single perfect ANCA test, and even a perfect test would not provide definitive clinical answers. The diagnostic utility of ANCA testing depends on the methodologic accuracy of the test and the appropriate ordering of testing in the right clinical setting. This article examines three important questions about this technology:
- What is the best ANCA test methodology?
- What is the prognostic value of serial ANCA testing?
- What is the clinical implication of ANCA type?
WHAT IS THE BEST ANCA TEST METHODOLOGY?
The diagnostic utility of ANCA testing depends on both the methodologic accuracy of the test and the appropriate ordering of tests. Methodologic accuracy comprises the analytic sensitivity and specificity of the test. Analytic sensitivity refers to the accurate identification of the presence of ANCA, and analytic specificity refers to measurement of only the entity in question (ANCA), not confounded by the presence of other entities (antibodies).
Equally as important as analytic accuracy is the appropriate ordering of the tests in the right clinical setting. Using a test that is sensitive to the presence of a specific ANCA type accurately identifies the presence of either proteinase-3 (PR3)- or myeloperoxidase (MPO)-ANCA. Once obtained, test results must be evaluated in terms of their relationship to the diagnosis being considered. If the tests are deemed diagnostically useful based on the results, the data can be used to assess the positive and negative predictive value of the tests.
Immunofluorescence or antigen-specific testing—or both?
A definitive diagnosis is more likely if an immunofluorescence staining pattern of cANCA is paired with the antigen specificity of PR3-ANCA, for example, or a perinuclear immunofluorescence pattern (pANCA) is paired with a positive MPO-ANCA. When positive test pairings have been obtained and the patient’s antigen ANCA reactivity is known, subsequent serial ANCA testing with an antigen-specific assay alone may be indicated, because the ANCA types of patients with vasculitis are unlikely to switch between PR3 and MPO during the course of their disease. If matching pairings are not obtained, the diagnostic utility of the tests remains unconfirmed.
Antigen type (PR3 or MPO) is determined through antigen-specific methods that include solid-phase assays and other methods of bringing the specific antigen in contact with the specific antibody in question. There are two categories of solid-phase assays: the enzyme-linked immunoabsorbent assay (ELISA) and the capture ELISA. In the ELISA methodology, the antigen is directly coated to a plastic plate; in the capture ELISA, an anchor, usually a monoclonal antibody or combination of antibodies, captures the target antigen on the plate. In both ELISA and capture ELISA assays, ANCA contained in the serum sample subjected to testing bind to the immobilized antigen. The amount of ANCA bound to the antigen can then be detected by a secondary antibody that is conjugated with an enzyme that can elicit a color reaction. The intensity of the color reaction is proportional to the amount of ANCA bound to the immobilized antigen.
The ELISA methodology tends to trade off analytic sensitivity for specificity, since the antigen purification process (which allows the ELISA system to increase its specificity) can cause conformational changes to the antigen being bound to the plate. This, in turn, causes a loss of some recognition of the conformationally sensitive ANCA.
In capture ELISA, a specific antibody captures the antigen; this stabilizes the conformation, boosts the analytic sensitivity, and allows a gentler purification process because it only captures the antigen in question and then binds it to the plate. This process decreases false-positive test results caused by residual contaminants in the antigen preparation. Analytic sensitivity issues may come into play if the anchoring monoclonal antibody competes for the epitope on the antigen being recognized by the serum antibody in question (ANCA), causing occasional false-negative results.
Another method now applied to commercial ANCA testing involves bead-based multiplex assays. These assays are based on principles similar to the ELISA or capture ELISA methods. In multiplex microsphere technology, the purified antigen is bound to a polystyrene microsphere instead of a plate. The microsphere is then presented to the antibody in question. The bead is then introduced to a secondary antibody labeled with a fluorescent marker (instead of an enzyme) for detection of the antibody. One advantage of this system is that various beads containing different antigens can be introduced to the same serum sample, and then different color reactions can be measured for each bead. Because only one antigen is bound to each microsphere (eg, PR3-ANCA, MPOANCA or other specific autoantibodies), only specific antibodies will react to each bead of a specific color. If there is no MPO antibody in the sample, there will be no reaction against the MPO antigen bead; however, if PR3-ANCA is present in the sample, it would react with the PR3 antigen beads. Using this methodology, a single serum sample can be tested for a multitude of autoantibodies at the same time (see “Interpreting ANCA results: Accurate tests, appropriate orders,”2–10 above).
WHAT IS THE PROGNOSTIC VALUE OF SERIAL ANCA TESTING?
Persistent changes in ANCA levels in relapsing disease may have some value in predicting outcome. The issues to consider include the methodology used to determine serial ANCA levels, correlations between ANCA and disease activity, and the use of ANCA changes to guide treatment.
Does methodology matter when determining serial ANCA levels?
Methodology in serial ANCA testing is probably unimportant as long as the same method is used serially. Analysis of large groups of ANCA-positive patients show a statistically highly significant correlation among results obtained with different detection methods, including immunofluorescence, direct ELISA, or capture ELISA. However, at the individual patient level there is some variability.
Do ANCA levels correlate with disease activity?
In a prospective study, serial ANCA samples obtained during the Wegener’s Granulomatosis Etanercept Trial (WGET)11 were processed in the same manner (collected every 3 months, mean follow-up of 22 months, uniform handling of samples). All samples were analyzed by capture ELISA, and disease activity was measured by the Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis (BVAS/WG). The results indicated that an increase in PR3-ANCA levels was not a significant predictor of relapse. The frequency of a relapse within 1 year of an increase in PR3-ANCA levels was found to be approximately 50%,11 a result similar to that reported in several smaller studies of different design and methodology.
Should ANCA changes guide treatment?
The available data regarding serial ANCA testing are limited mostly to PR3-ANCA. Serial ANCA testing has limited value as a guide to treatment and, in general, changes in ANCA levels alone should not be used to guide treatment decisions. In new patients without documented serial ANCA level associations, an increase in PR3-ANCA levels has no reliable predictive value. The existing literature suggests that this lack of association is not dependent on the method used for ANCA detection. For individual patients in whom long-term serial ANCA testing has been performed and a relationship between PR3-ANCA levels and disease activity has been established, serial ANCA testing can have some predictive value and can be used to guide treatment. For example, when remission is achieved by depleting B cells in patients with chronically relapsing GPA, ANCA levels usually go down. After B-cell reconstitution, the ANCA level rises in most patients, and this rise is usually associated with a flare shortly thereafter. A flare can be preempted when this pattern is determined in a specific patient, and preemptive treatment is applied accordingly.12
WHAT IS THE IMPLICATION OF ANCA TYPE?
Available reports consistently suggest that PR3-ANCA is associated with a higher mortality than MPO-ANCA (relative risk [RR], 3.78),13 and a higher relapse rate.14,15 A more rapid loss of renal function among patients with glomerulonephritis and PR3-ANCA than those with MPO-ANCA has also been reported.16 Using remission as the starting point, the number of days from complete remission to first disease flare was plotted for patients with MPO- versus PR3-ANCA in an analysis of long-term data from the Rituximab in ANCA-Associated Vasculitis (RAVE) trial.17 The resulting curve demonstrated a divergence in the probability of remaining in remission, confirming that remission maintenance is clearly greater in patients with MPO-ANCA than in patients with PR3-ANCA.
The primary end point of the RAVE trial was remission of disease without the use of prednisone at 6 months. There was little difference in end point achieved based on comparison of diagnosis (microscopic polyangiitis or granulomatosis) or treatment arms (rituximab versus cyclophosphamide); however, an analysis of end point data separating the patients by ANCA type showed that the treatment response to rituximab was superior to that of cyclophosphamide among patients with PR3-ANCA, whereas in patients with MPO-ANCA, there was little difference in response associated with either treatment. Regarding the likelihood of attaining an ANCA-negative status after 6 months, again MPO-ANCA patients showed no difference in frequency on either treatment. Among PR3-ANCA–positive patients, 50% in the rituximab arm attained ANCA-negative status compared with only 17% in the cyclophosphamide arm.17
SUMMARY
Diagnostic utility of ANCA testing depends on the methodology and clinical setting. Only cANCA/PR3-ANCA and pANCA/MPO-ANCA pairings have positive predictive value for diagnosis of small-vessel vasculitis. Mismatches in results, findings of human neutrophil elastase–ANCA, or identification of multiple positive antigens should be considered in cases of cocaine or drug use.
The clinical utility of serial ANCA testing is unconfirmed. Good data currently exist only for PR3-ANCA, and different drugs may affect ANCA levels in different ways. ANCA type is significant in that PR3-ANCA portends a higher relapse rate and poorer patient outcomes compared with MPO-ANCA.
Antineutrophil cytoplasmic antibody (ANCA) detection is a valuable tool for diagnosing small-vessel vasculitis,1 but measuring and interpreting ANCA levels is an inexact science. There is no single perfect ANCA test, and even a perfect test would not provide definitive clinical answers. The diagnostic utility of ANCA testing depends on the methodologic accuracy of the test and the appropriate ordering of testing in the right clinical setting. This article examines three important questions about this technology:
- What is the best ANCA test methodology?
- What is the prognostic value of serial ANCA testing?
- What is the clinical implication of ANCA type?
WHAT IS THE BEST ANCA TEST METHODOLOGY?
The diagnostic utility of ANCA testing depends on both the methodologic accuracy of the test and the appropriate ordering of tests. Methodologic accuracy comprises the analytic sensitivity and specificity of the test. Analytic sensitivity refers to the accurate identification of the presence of ANCA, and analytic specificity refers to measurement of only the entity in question (ANCA), not confounded by the presence of other entities (antibodies).
Equally as important as analytic accuracy is the appropriate ordering of the tests in the right clinical setting. Using a test that is sensitive to the presence of a specific ANCA type accurately identifies the presence of either proteinase-3 (PR3)- or myeloperoxidase (MPO)-ANCA. Once obtained, test results must be evaluated in terms of their relationship to the diagnosis being considered. If the tests are deemed diagnostically useful based on the results, the data can be used to assess the positive and negative predictive value of the tests.
Immunofluorescence or antigen-specific testing—or both?
A definitive diagnosis is more likely if an immunofluorescence staining pattern of cANCA is paired with the antigen specificity of PR3-ANCA, for example, or a perinuclear immunofluorescence pattern (pANCA) is paired with a positive MPO-ANCA. When positive test pairings have been obtained and the patient’s antigen ANCA reactivity is known, subsequent serial ANCA testing with an antigen-specific assay alone may be indicated, because the ANCA types of patients with vasculitis are unlikely to switch between PR3 and MPO during the course of their disease. If matching pairings are not obtained, the diagnostic utility of the tests remains unconfirmed.
Antigen type (PR3 or MPO) is determined through antigen-specific methods that include solid-phase assays and other methods of bringing the specific antigen in contact with the specific antibody in question. There are two categories of solid-phase assays: the enzyme-linked immunoabsorbent assay (ELISA) and the capture ELISA. In the ELISA methodology, the antigen is directly coated to a plastic plate; in the capture ELISA, an anchor, usually a monoclonal antibody or combination of antibodies, captures the target antigen on the plate. In both ELISA and capture ELISA assays, ANCA contained in the serum sample subjected to testing bind to the immobilized antigen. The amount of ANCA bound to the antigen can then be detected by a secondary antibody that is conjugated with an enzyme that can elicit a color reaction. The intensity of the color reaction is proportional to the amount of ANCA bound to the immobilized antigen.
The ELISA methodology tends to trade off analytic sensitivity for specificity, since the antigen purification process (which allows the ELISA system to increase its specificity) can cause conformational changes to the antigen being bound to the plate. This, in turn, causes a loss of some recognition of the conformationally sensitive ANCA.
In capture ELISA, a specific antibody captures the antigen; this stabilizes the conformation, boosts the analytic sensitivity, and allows a gentler purification process because it only captures the antigen in question and then binds it to the plate. This process decreases false-positive test results caused by residual contaminants in the antigen preparation. Analytic sensitivity issues may come into play if the anchoring monoclonal antibody competes for the epitope on the antigen being recognized by the serum antibody in question (ANCA), causing occasional false-negative results.
Another method now applied to commercial ANCA testing involves bead-based multiplex assays. These assays are based on principles similar to the ELISA or capture ELISA methods. In multiplex microsphere technology, the purified antigen is bound to a polystyrene microsphere instead of a plate. The microsphere is then presented to the antibody in question. The bead is then introduced to a secondary antibody labeled with a fluorescent marker (instead of an enzyme) for detection of the antibody. One advantage of this system is that various beads containing different antigens can be introduced to the same serum sample, and then different color reactions can be measured for each bead. Because only one antigen is bound to each microsphere (eg, PR3-ANCA, MPOANCA or other specific autoantibodies), only specific antibodies will react to each bead of a specific color. If there is no MPO antibody in the sample, there will be no reaction against the MPO antigen bead; however, if PR3-ANCA is present in the sample, it would react with the PR3 antigen beads. Using this methodology, a single serum sample can be tested for a multitude of autoantibodies at the same time (see “Interpreting ANCA results: Accurate tests, appropriate orders,”2–10 above).
WHAT IS THE PROGNOSTIC VALUE OF SERIAL ANCA TESTING?
Persistent changes in ANCA levels in relapsing disease may have some value in predicting outcome. The issues to consider include the methodology used to determine serial ANCA levels, correlations between ANCA and disease activity, and the use of ANCA changes to guide treatment.
Does methodology matter when determining serial ANCA levels?
Methodology in serial ANCA testing is probably unimportant as long as the same method is used serially. Analysis of large groups of ANCA-positive patients show a statistically highly significant correlation among results obtained with different detection methods, including immunofluorescence, direct ELISA, or capture ELISA. However, at the individual patient level there is some variability.
Do ANCA levels correlate with disease activity?
In a prospective study, serial ANCA samples obtained during the Wegener’s Granulomatosis Etanercept Trial (WGET)11 were processed in the same manner (collected every 3 months, mean follow-up of 22 months, uniform handling of samples). All samples were analyzed by capture ELISA, and disease activity was measured by the Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis (BVAS/WG). The results indicated that an increase in PR3-ANCA levels was not a significant predictor of relapse. The frequency of a relapse within 1 year of an increase in PR3-ANCA levels was found to be approximately 50%,11 a result similar to that reported in several smaller studies of different design and methodology.
Should ANCA changes guide treatment?
The available data regarding serial ANCA testing are limited mostly to PR3-ANCA. Serial ANCA testing has limited value as a guide to treatment and, in general, changes in ANCA levels alone should not be used to guide treatment decisions. In new patients without documented serial ANCA level associations, an increase in PR3-ANCA levels has no reliable predictive value. The existing literature suggests that this lack of association is not dependent on the method used for ANCA detection. For individual patients in whom long-term serial ANCA testing has been performed and a relationship between PR3-ANCA levels and disease activity has been established, serial ANCA testing can have some predictive value and can be used to guide treatment. For example, when remission is achieved by depleting B cells in patients with chronically relapsing GPA, ANCA levels usually go down. After B-cell reconstitution, the ANCA level rises in most patients, and this rise is usually associated with a flare shortly thereafter. A flare can be preempted when this pattern is determined in a specific patient, and preemptive treatment is applied accordingly.12
WHAT IS THE IMPLICATION OF ANCA TYPE?
Available reports consistently suggest that PR3-ANCA is associated with a higher mortality than MPO-ANCA (relative risk [RR], 3.78),13 and a higher relapse rate.14,15 A more rapid loss of renal function among patients with glomerulonephritis and PR3-ANCA than those with MPO-ANCA has also been reported.16 Using remission as the starting point, the number of days from complete remission to first disease flare was plotted for patients with MPO- versus PR3-ANCA in an analysis of long-term data from the Rituximab in ANCA-Associated Vasculitis (RAVE) trial.17 The resulting curve demonstrated a divergence in the probability of remaining in remission, confirming that remission maintenance is clearly greater in patients with MPO-ANCA than in patients with PR3-ANCA.
The primary end point of the RAVE trial was remission of disease without the use of prednisone at 6 months. There was little difference in end point achieved based on comparison of diagnosis (microscopic polyangiitis or granulomatosis) or treatment arms (rituximab versus cyclophosphamide); however, an analysis of end point data separating the patients by ANCA type showed that the treatment response to rituximab was superior to that of cyclophosphamide among patients with PR3-ANCA, whereas in patients with MPO-ANCA, there was little difference in response associated with either treatment. Regarding the likelihood of attaining an ANCA-negative status after 6 months, again MPO-ANCA patients showed no difference in frequency on either treatment. Among PR3-ANCA–positive patients, 50% in the rituximab arm attained ANCA-negative status compared with only 17% in the cyclophosphamide arm.17
SUMMARY
Diagnostic utility of ANCA testing depends on the methodology and clinical setting. Only cANCA/PR3-ANCA and pANCA/MPO-ANCA pairings have positive predictive value for diagnosis of small-vessel vasculitis. Mismatches in results, findings of human neutrophil elastase–ANCA, or identification of multiple positive antigens should be considered in cases of cocaine or drug use.
The clinical utility of serial ANCA testing is unconfirmed. Good data currently exist only for PR3-ANCA, and different drugs may affect ANCA levels in different ways. ANCA type is significant in that PR3-ANCA portends a higher relapse rate and poorer patient outcomes compared with MPO-ANCA.
- Russell KA, Wiegert E, Schroeder DR, Homburger HA, Specks U. Detection of anti-neutrophil cytoplasmic antibodies under actual clinical testing conditions. Clin Immunol 2002; 103:196–203.
- Langford CA. The diagnostic utility of c-ANCA in Wegener’s granulomatosis. Cleve Clin J Med 1998; 65:135–140.
- Trimarchi M, Gregorini G, Facchetti F, et al. Cocaine-induced midline destructive lesions: clinical, radiographic, histopathologic, and serologic features and their differentiation from Wegener granulomatosis. Medicine 2001; 80:391–404.
- Wiesner O, Russell KA, Lee AS, et al. Antineutrophil cytoplasmic antibodies reacting with human neutrophil elastase as a diagnostic marker for cocaine-induced midline destructive lesions but not autoimmune vasculitis. Arthritis Rheum 2004; 50:2954–2965.
- Peikert T, Finkielman JD, Hummel AM, et al. Functional characterization of antineutrophil cytoplasmic antibodies in patients with cocaine-induced midline destructive lesions. Arthritis Rheum 2008; 58:1546–1551.
- Knowles L, Buxton JA, Skuridina N, et al. Levamisole tainted cocaine causing severe neutropenia in Alberta and British Columbia. Harm Reduct J 2009; 6 (Nov 17):30. doi: 10.1186/1477-7517-6-30.
- Zhu NY, LeGatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med 2009; 150:287–289.
- Bradford M, Rosenberg B, Moreno J, Dumyati G. Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole. Ann Intern Med 2010; 152:758–759.
- Waller JM, Feramisco JD, Alberta-Wszolek L, McCalmont TH, Fox LP. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit [published online ahead of print March 20, 2010]? J Am Acad Dermatol 2010; 63:530–535. doi: 10.1016/j.jaad.2010.01.055
- Chang A, Osterloh J, Thomas J. Levamisole: a dangerous new cocaine adulterant [published online ahead of print July 28, 2010]. Clin Pharmacol Ther 2010; 88:408–411. doi: 10.1038/clpt.2010.156
- Finkielman JD, Merkel PA, Schroeder D, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med 2007; 147:611–619.
- Cartin-Ceba R, Golbin J, Keogh KA, et al. Rituximab for remission induction and maintenance in granulomatosis with polyangiitis (Wegener’s): a single-center ten-year experience [published online ahead of print June 21, 2012]. Arthritis Rheum. doi: 10.1002/art.34584
- Hogan SL, Nachman PH, Wilkman AS, Jennette JC, Falk RJ; the Glomerular Disease Collaborative Network. Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 1996; 7:23–32.
- Booth AD, Almond MK, Burns A, et al. Outcome of ANCA-associated renal vasculitis: a 5-year retrospective study. Am J Kidney Dis 2003; 41:776–784.
- Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Franssen CFM, Gans ROB, Arends B, et al. Differences between anti-myeloperoxidase- and anti-proteinase 3-associated renal disease. Kidney Int 1995; 47:193–199.
- Stone JH, Merkel PA, Spiera R, et al; for the RAVE–ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010; 363:221–232.
- Russell KA, Wiegert E, Schroeder DR, Homburger HA, Specks U. Detection of anti-neutrophil cytoplasmic antibodies under actual clinical testing conditions. Clin Immunol 2002; 103:196–203.
- Langford CA. The diagnostic utility of c-ANCA in Wegener’s granulomatosis. Cleve Clin J Med 1998; 65:135–140.
- Trimarchi M, Gregorini G, Facchetti F, et al. Cocaine-induced midline destructive lesions: clinical, radiographic, histopathologic, and serologic features and their differentiation from Wegener granulomatosis. Medicine 2001; 80:391–404.
- Wiesner O, Russell KA, Lee AS, et al. Antineutrophil cytoplasmic antibodies reacting with human neutrophil elastase as a diagnostic marker for cocaine-induced midline destructive lesions but not autoimmune vasculitis. Arthritis Rheum 2004; 50:2954–2965.
- Peikert T, Finkielman JD, Hummel AM, et al. Functional characterization of antineutrophil cytoplasmic antibodies in patients with cocaine-induced midline destructive lesions. Arthritis Rheum 2008; 58:1546–1551.
- Knowles L, Buxton JA, Skuridina N, et al. Levamisole tainted cocaine causing severe neutropenia in Alberta and British Columbia. Harm Reduct J 2009; 6 (Nov 17):30. doi: 10.1186/1477-7517-6-30.
- Zhu NY, LeGatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med 2009; 150:287–289.
- Bradford M, Rosenberg B, Moreno J, Dumyati G. Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole. Ann Intern Med 2010; 152:758–759.
- Waller JM, Feramisco JD, Alberta-Wszolek L, McCalmont TH, Fox LP. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit [published online ahead of print March 20, 2010]? J Am Acad Dermatol 2010; 63:530–535. doi: 10.1016/j.jaad.2010.01.055
- Chang A, Osterloh J, Thomas J. Levamisole: a dangerous new cocaine adulterant [published online ahead of print July 28, 2010]. Clin Pharmacol Ther 2010; 88:408–411. doi: 10.1038/clpt.2010.156
- Finkielman JD, Merkel PA, Schroeder D, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med 2007; 147:611–619.
- Cartin-Ceba R, Golbin J, Keogh KA, et al. Rituximab for remission induction and maintenance in granulomatosis with polyangiitis (Wegener’s): a single-center ten-year experience [published online ahead of print June 21, 2012]. Arthritis Rheum. doi: 10.1002/art.34584
- Hogan SL, Nachman PH, Wilkman AS, Jennette JC, Falk RJ; the Glomerular Disease Collaborative Network. Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 1996; 7:23–32.
- Booth AD, Almond MK, Burns A, et al. Outcome of ANCA-associated renal vasculitis: a 5-year retrospective study. Am J Kidney Dis 2003; 41:776–784.
- Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Franssen CFM, Gans ROB, Arends B, et al. Differences between anti-myeloperoxidase- and anti-proteinase 3-associated renal disease. Kidney Int 1995; 47:193–199.
- Stone JH, Merkel PA, Spiera R, et al; for the RAVE–ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010; 363:221–232.
Defining disease activity and damage in patients with small-vessel vasculitis
Small-vessel vasculitides are complex diseases with highly variable clinical features and are associated with considerable morbidity and mortality. These systemic, multisystem, multiorgan diseases often threaten vital organs with manifestations that include upper airway disease, pulmonary disease, glomerulonephritis, neuropathy, arthritis/arthralgias, malaise/fatigue, eye disease, skin/mucosa irregularities, vascular disease, cardiac disease, and gastrointestinal disease.
Accurate assessment of the patient with vasculitis is a challenge for the clinician and is critical for managing therapeutic interventions throughout the course of the disease. Effective management includes repeated evaluations of the activity and severity of the disease as well as the damage it has caused. These distinct yet overlapping concepts must be measured separately but evaluated as a whole. Additional categorizations of disease course, such as whether it is active (new-onset, persistent, or flare) or in remission, further define the disease and are routinely employed in guiding treatment choices.
The importance of accurately assessing a patient’s clinical status is clear, but it is also important to standardize and quantify vasculitis assessment tools for use in clinical trials. Standardized assessments are needed to:
- Guide clinical trial enrollment criteria
- Describe and compare study populations
- Quantify and measure treatment effectiveness
- Describe long-term outcomes
- Translate standardized assessment tools into clinical practice.
Over the past few decades, improvements in clinical research have resulted in increasingly accurate data obtained from well-designed randomized controlled trials, all of which are based on better clinical assessments. Improving the quality of the assessment tools has improved both the interpretation of trial results and translation of findings into clinical practice.
DISEASE ASSESSMENT
When assessing patients with vasculitis, whether clinically or in the context of a clinical trial, it is essential to differentiate among disease activity, damage, and severity:
Disease activity, such as active bleeding or mucosal inflammation, is treatable and potentially responsive to therapy.
Disease damage is generally irreversible and not improved by treating vasculitis. Damage may be caused by the disease itself, its treatment, or a comorbid condition. In general, once damage is identified, it is considered permanent if it remains unchanged for more than 6 months. In the Wegener’s Granulomatosis Etanercept Trial,1 damage that occurred in more than 10% of the cohort included hearing loss; proteinuria (≥ 0.5 g/24 hours); nasal blockage, chronic discharge, or crusting; nasal bridge collapse or septal perforation; glomerular filtration rate at least 50% lower than premorbid baseline; subglottic stenosis; and chronic sinusitis or radiologic damage. Disease-related damage can be addressed; a saddle-nose deformity can respond to plastic surgery, for example, but treating vasculitis will have no effect on the underlying established anatomic defect.
Disease severity assesses the intensity of the disease and guides the clinician in gauging how aggressive the therapy should be.
Vasculitis has two primary disease states: remission and active disease. In remission, there is no evidence of active disease. This is often qualified by describing the remission as either complete or partial; it is further defined by introducing an element of time, such as a “sustained” remission of more than 6 months. Active disease is the presence of any ongoing expression of vasculitis that is not caused by disease damage, comorbidity, or treatment. Active disease can be graded as low, medium, or high; if active disease lasts longer than 6 months, it is described as persistent or sustained. Flare, a manifestation of active disease, describes the transition from remission to active disease and is characterized by worsening of disease activity. Flares are graded as nonsevere or severe.
These descriptions of disease status can be further broken down into whether they are occurring “on” or “off” treatment. All of these elements are important and the subtleties and differences are critical in interpreting data for use in the clinical setting or in clinical trials.
CLINICAL ASSESSMENT
Assessing the status of disease for a patient with granulomatosis with polyangiitis (GPA, Wegener’s granulomatosis [WG]) or microscopic polyangiitis (MPA) begins with a detailed medical history and physical examination every time the patient is seen. Appropriate laboratory assessments include a complete blood count, tests of renal function, acute phase reactants (possibly as disease markers, but not necessarily to guide therapy), and other laboratory tests as needed. Controversy exists regarding the role of antineutrophil cytoplasmic antibody (ANCA) testing in assessment of disease activity.
Urinalyses are key for assessing activity; if a urine dipstick result is positive, a subsequent microscopic examination should be conducted. Microscopic review may demonstrate red cell casts that a routine laboratory check may not reveal. In addition to spotting de novo hematuria, looking for a change in dipstick results may prove valuable, since hematuria may increase in patients in whom persistent hematuria has already been noted. The change may be due to renal disease from the vasculitis, cyclophosphamide-induced bladder toxicity, a kidney stone, menses, or a variety of other causes, but if the hematuria is not monitored, a key assessment will be missed.
Disease staging through diagnostic imaging of the sinuses, neck, and chest should be performed on a regular basis as appropriate, beginning at the patient’s initial visit. Restaging, in much the same way as an oncologist restages cancer, should take place regularly, because this informs whether to make a major change in therapy (eg, from cyclophosphamide, azathioprine, or rituximab). Restaging will also allow benchmarking of old, new, and changed damage so that when the disease recurs, the existing damage can be differentiated from new lesions. Once the disease has stabilized, imaging can be discontinued.
Consultations with otolaryngologists, ophthalmologists, cardiologists, and other specialists should be sought as needed. Serial audiograms, laryngoscopy, echocardiograms, and other appropriate tests should be performed as required. Biopsies are useful for assessment of patients, particularly at diagnosis, but also when it becomes necessary to reassess the progress of a patient’s disease or to identify a potential infection versus a possible malignancy. Biopsy is particularly helpful for kidney disease. If of active disease, then repeat biopsy is appropriate to determine whether the deterioration is associated with persistent active disease, the natural history of declining kidney function, or another cause. Patients with vasculitis may develop new comorbidities, particularly infections, so vigilance is always required. Importantly, documentation and awareness of disease-related damage are crucial in order to avoid overtreatment; damage should not be treated if therapy will not improve it.
ASSESSING DISEASE ACTIVITY AND DAMAGE

Birmingham Vasculitis Activity Score
Introduced in 1994, the Birmingham Vasculitis Activity Score (BVAS) is a single-page checklist that records weighted data on more than 50 items and nine organ systems; the sum of the individual items provides the final score.2 There have been two revisions of the BVAS; one focuses on GPA (BVAS/WG)3 and the other, BVAS version 3 (v.3) is more simplified.4 For all of the BVAS tools, remission is defined as a score of 0. Any score greater than 0 defines active disease. Each system is evaluated as being active or not, with items characterized as more severe being weighted more heavily. The use of the BVAS/WG is illustrated in two patients (see “Assessment with the BVAS/WG,” below).

Every major randomized controlled trial in the past 15 years has used the BVAS or one of its derivatives to define outcomes, but primary outcomes were not defined strictly from the BVAS itself. There were important differences in the trials’ definitions of remission, which is the outcome of interest. For example, some trials allow for minor disease activity concurrent with partial remission, while others require full absence of disease activity to achieve “remission.”
Vasculitis Damage Index
The Vasculitis Damage Index (VDI) is a single-page catalog of damage items separated into 11 groupings. Limitations of the index include lack of attribution (to vasculitis, treatment, or comorbidities), gradation, weighting, and patient input (patient-reported outcomes).5 Revisions to the VDI have been made in the ANCA Vasculitis Index of Damage (AVID),6 which incorporates an expanded list of damage items, as well as an even more expanded version called the Combined Damage Assessment Index that combines the items from the VDI and AVID.7 While these tools provide a means to catalog damage by choosing whether an item is present or not, a more data-driven approach to damage assessment is needed that incorporates weighting into the tool.
Damage assessment may be the most important measure in evaluating the patient with vasculitis. In addition to keeping patients alive, one of the main purposes in treating active disease is to prevent damage, maintaining quality of life for the patient for the long term and improving outcomes.
PATIENT-REPORTED OUTCOMES
Patients have a different perspective on their disease than that provided by assessment tools or physicians. Because physician and patient ratings are often disparate, health-related quality of life (HRQOL) is an increasingly important outcome measure for patients as well as regulatory agencies. In a 2010 study, structured patient-reported assessments of burden of disease were obtained from 264 patients with vasculitis in the United States, Germany, and the United Kingdom. Patients ranked items in terms of most frequent burdens of disease. Across ages and countries, patients most commonly rated fatigue/energy loss, pain, musculoskeletal symptoms, and social manifestations as the most severe ramifications of their disease.11 None of the burdens of disease identified in this study are universally measured in the current assessment tools. Patients with active disease had more of the most commonly listed burden-of-disease items; however, patients still suffered from these burdens when the disease was inactive. These disease burden items are therefore mostly dynamic problems and not simply chronic issues.
Patient ratings differ considerably from physician ratings in terms of importance. For example, patients rate nasal manifestations, weight gain, and some chronic pain and fatigue items higher than renal insufficiency and stroke. There is a clear need to address not only physician-ranked issues, but also patient-ranked issues in assessing and treating vasculitis.11
When measuring HRQOL via the Medical Outcomes Study 36-item short-form health survey (SF-36) in patients with vasculitis, a correlation is noted between QOL and sustained remission. In a study by Tomasson et al, QOL was measured using the SF-36 upon treatment following a flare.12 In all patients, SF-36 increased dramatically immediately following treatment but then leveled off over time. In patients who achieved sustained remission, SF-36 scores continued to rise from baseline. In patients who did not achieve a sustained remission, the SF-36 scores did not improve. This QOL measure, therefore, captures a value that other assessments do not, further demonstrating its utility as part of the assessment process.
VALIDATED OUTCOME MEASURES
Outcome Measures in Rheumatology (OMERACT) is an international group of clinicians, trialists, epidemiologists, biostatisticians, health economists, industry executives, and FDA and European Medicines Agency officials who meet every 2 years to promote data-based validation of outcome measures for a variety of diseases. OMERACT endorses core sets of validated outcomes when data demonstrate veracity, discrimination, and feasibility.13 For each domain in the vasculitis arena, there is an associated validated instrument: for disease activity, the validated instruments are the BVAS, BVAS/WG, and BVAS v.3; for damage assessment, the instrument is the VDI; for patient-reported outcomes, the instrument is the SF-36; and finally, for mortality, the instrument is death.13 This core set of measures helps frame how future trials in vasculitis will be standardized and assists in comparing trials, which is particularly important to regulatory agencies.
The tools for disease assessment in vasculitis still need to be refined for activity and damage assessment in order to be more scalable and precise, thereby measuring smaller effects. Patient-reported outcomes and patient perspectives on disease need to be better captured, and reliable biomarkers need to be discovered or further developed. Improved outcome measures must be developed for other types of vasculitis, such as eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome), giant cell (temporal) arteritis, and Takayasu arteritis,14 in order to conduct and report trial results. These outcome measures could also translate into tools that can be used to assess patients and make treatment decisions, thereby helping the clinician at the bedside.
- Seo P, Min YI, Holbrook JT, et al. Damage caused by Wegener’s granulomatosis and its treatment: prospective data from the Wegener’s Granulomatosis Etanercept Trial (WGET). Arthritis Rheum 2005; 52:2168–2178.
- Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM 1994; 87:671–678.
- Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum 2001; 44:912–920.
- Mukhtyar C, Lee R, Brown D, et al. Modification and valid ation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis 2009; 68:1827–1832.
- Exley AR, Bacon PA, Luqmani RA, et al. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum 1997; 40:371–380.
- Seo P, Luqmani RA, Flossmann O, et al. The future of damage assessment in vasculitis. J Rheumatol 2007; 34:1357–1371.
- Seo P, Jayne D, Luqmani R, Merkel PA. Assessment of damage in vasculitis: expert ratings of damage. Rheumatology (Oxford) 2009; 48:823–827.
- de Groot K, Gross WL, Herlyn K, Reinhold-Keller E. Development and validation of a disease extent index for Wegener’s granulomatosis. Clin Nephrol 2001; 55:31–38.
- Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P; French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011; 90:19–27.
- Merkel PA, Cuthbertson DD, Hellmich B, et al. Comparison of disease activity measures for anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis. Ann Rheum Dis 2009; 68:103–106.
- Herlyn K, Hellmich B, Seo P, Merkel PA. Patient-reported outcome assessment in vasculitis may provide important data and a unique perspective. Arthritis Care Res (Hoboken) 2010; 62:1639–1645.
- Tomasson G, Boers M, Walsh M, et al. Assessment of health related quality of life as an outcome measure in granulomatosis with polyangiitis (Wegener’s). Arthritis Care Res (Hoboken) 2012; 64:273–279.
- Merkel PA, Aydin SZ, Boers M, et al .The OMERACT core set of outcome measures for use in clinical trials of ANCA-associated vasculitis. J Rheumatol 2011; 38:1480–1486.
- Direskeneli H, Aydin SZ, Kermani TA, et al. Development of outcome measures for large-vessel vasculitis for use in clinical trials: opportunities, challenges, and research agenda. J Rheumatol 2011; 38:1471–1479.
Small-vessel vasculitides are complex diseases with highly variable clinical features and are associated with considerable morbidity and mortality. These systemic, multisystem, multiorgan diseases often threaten vital organs with manifestations that include upper airway disease, pulmonary disease, glomerulonephritis, neuropathy, arthritis/arthralgias, malaise/fatigue, eye disease, skin/mucosa irregularities, vascular disease, cardiac disease, and gastrointestinal disease.
Accurate assessment of the patient with vasculitis is a challenge for the clinician and is critical for managing therapeutic interventions throughout the course of the disease. Effective management includes repeated evaluations of the activity and severity of the disease as well as the damage it has caused. These distinct yet overlapping concepts must be measured separately but evaluated as a whole. Additional categorizations of disease course, such as whether it is active (new-onset, persistent, or flare) or in remission, further define the disease and are routinely employed in guiding treatment choices.
The importance of accurately assessing a patient’s clinical status is clear, but it is also important to standardize and quantify vasculitis assessment tools for use in clinical trials. Standardized assessments are needed to:
- Guide clinical trial enrollment criteria
- Describe and compare study populations
- Quantify and measure treatment effectiveness
- Describe long-term outcomes
- Translate standardized assessment tools into clinical practice.
Over the past few decades, improvements in clinical research have resulted in increasingly accurate data obtained from well-designed randomized controlled trials, all of which are based on better clinical assessments. Improving the quality of the assessment tools has improved both the interpretation of trial results and translation of findings into clinical practice.
DISEASE ASSESSMENT
When assessing patients with vasculitis, whether clinically or in the context of a clinical trial, it is essential to differentiate among disease activity, damage, and severity:
Disease activity, such as active bleeding or mucosal inflammation, is treatable and potentially responsive to therapy.
Disease damage is generally irreversible and not improved by treating vasculitis. Damage may be caused by the disease itself, its treatment, or a comorbid condition. In general, once damage is identified, it is considered permanent if it remains unchanged for more than 6 months. In the Wegener’s Granulomatosis Etanercept Trial,1 damage that occurred in more than 10% of the cohort included hearing loss; proteinuria (≥ 0.5 g/24 hours); nasal blockage, chronic discharge, or crusting; nasal bridge collapse or septal perforation; glomerular filtration rate at least 50% lower than premorbid baseline; subglottic stenosis; and chronic sinusitis or radiologic damage. Disease-related damage can be addressed; a saddle-nose deformity can respond to plastic surgery, for example, but treating vasculitis will have no effect on the underlying established anatomic defect.
Disease severity assesses the intensity of the disease and guides the clinician in gauging how aggressive the therapy should be.
Vasculitis has two primary disease states: remission and active disease. In remission, there is no evidence of active disease. This is often qualified by describing the remission as either complete or partial; it is further defined by introducing an element of time, such as a “sustained” remission of more than 6 months. Active disease is the presence of any ongoing expression of vasculitis that is not caused by disease damage, comorbidity, or treatment. Active disease can be graded as low, medium, or high; if active disease lasts longer than 6 months, it is described as persistent or sustained. Flare, a manifestation of active disease, describes the transition from remission to active disease and is characterized by worsening of disease activity. Flares are graded as nonsevere or severe.
These descriptions of disease status can be further broken down into whether they are occurring “on” or “off” treatment. All of these elements are important and the subtleties and differences are critical in interpreting data for use in the clinical setting or in clinical trials.
CLINICAL ASSESSMENT
Assessing the status of disease for a patient with granulomatosis with polyangiitis (GPA, Wegener’s granulomatosis [WG]) or microscopic polyangiitis (MPA) begins with a detailed medical history and physical examination every time the patient is seen. Appropriate laboratory assessments include a complete blood count, tests of renal function, acute phase reactants (possibly as disease markers, but not necessarily to guide therapy), and other laboratory tests as needed. Controversy exists regarding the role of antineutrophil cytoplasmic antibody (ANCA) testing in assessment of disease activity.
Urinalyses are key for assessing activity; if a urine dipstick result is positive, a subsequent microscopic examination should be conducted. Microscopic review may demonstrate red cell casts that a routine laboratory check may not reveal. In addition to spotting de novo hematuria, looking for a change in dipstick results may prove valuable, since hematuria may increase in patients in whom persistent hematuria has already been noted. The change may be due to renal disease from the vasculitis, cyclophosphamide-induced bladder toxicity, a kidney stone, menses, or a variety of other causes, but if the hematuria is not monitored, a key assessment will be missed.
Disease staging through diagnostic imaging of the sinuses, neck, and chest should be performed on a regular basis as appropriate, beginning at the patient’s initial visit. Restaging, in much the same way as an oncologist restages cancer, should take place regularly, because this informs whether to make a major change in therapy (eg, from cyclophosphamide, azathioprine, or rituximab). Restaging will also allow benchmarking of old, new, and changed damage so that when the disease recurs, the existing damage can be differentiated from new lesions. Once the disease has stabilized, imaging can be discontinued.
Consultations with otolaryngologists, ophthalmologists, cardiologists, and other specialists should be sought as needed. Serial audiograms, laryngoscopy, echocardiograms, and other appropriate tests should be performed as required. Biopsies are useful for assessment of patients, particularly at diagnosis, but also when it becomes necessary to reassess the progress of a patient’s disease or to identify a potential infection versus a possible malignancy. Biopsy is particularly helpful for kidney disease. If of active disease, then repeat biopsy is appropriate to determine whether the deterioration is associated with persistent active disease, the natural history of declining kidney function, or another cause. Patients with vasculitis may develop new comorbidities, particularly infections, so vigilance is always required. Importantly, documentation and awareness of disease-related damage are crucial in order to avoid overtreatment; damage should not be treated if therapy will not improve it.
ASSESSING DISEASE ACTIVITY AND DAMAGE
Birmingham Vasculitis Activity Score
Introduced in 1994, the Birmingham Vasculitis Activity Score (BVAS) is a single-page checklist that records weighted data on more than 50 items and nine organ systems; the sum of the individual items provides the final score.2 There have been two revisions of the BVAS; one focuses on GPA (BVAS/WG)3 and the other, BVAS version 3 (v.3) is more simplified.4 For all of the BVAS tools, remission is defined as a score of 0. Any score greater than 0 defines active disease. Each system is evaluated as being active or not, with items characterized as more severe being weighted more heavily. The use of the BVAS/WG is illustrated in two patients (see “Assessment with the BVAS/WG,” below).
Every major randomized controlled trial in the past 15 years has used the BVAS or one of its derivatives to define outcomes, but primary outcomes were not defined strictly from the BVAS itself. There were important differences in the trials’ definitions of remission, which is the outcome of interest. For example, some trials allow for minor disease activity concurrent with partial remission, while others require full absence of disease activity to achieve “remission.”
Vasculitis Damage Index
The Vasculitis Damage Index (VDI) is a single-page catalog of damage items separated into 11 groupings. Limitations of the index include lack of attribution (to vasculitis, treatment, or comorbidities), gradation, weighting, and patient input (patient-reported outcomes).5 Revisions to the VDI have been made in the ANCA Vasculitis Index of Damage (AVID),6 which incorporates an expanded list of damage items, as well as an even more expanded version called the Combined Damage Assessment Index that combines the items from the VDI and AVID.7 While these tools provide a means to catalog damage by choosing whether an item is present or not, a more data-driven approach to damage assessment is needed that incorporates weighting into the tool.
Damage assessment may be the most important measure in evaluating the patient with vasculitis. In addition to keeping patients alive, one of the main purposes in treating active disease is to prevent damage, maintaining quality of life for the patient for the long term and improving outcomes.
PATIENT-REPORTED OUTCOMES
Patients have a different perspective on their disease than that provided by assessment tools or physicians. Because physician and patient ratings are often disparate, health-related quality of life (HRQOL) is an increasingly important outcome measure for patients as well as regulatory agencies. In a 2010 study, structured patient-reported assessments of burden of disease were obtained from 264 patients with vasculitis in the United States, Germany, and the United Kingdom. Patients ranked items in terms of most frequent burdens of disease. Across ages and countries, patients most commonly rated fatigue/energy loss, pain, musculoskeletal symptoms, and social manifestations as the most severe ramifications of their disease.11 None of the burdens of disease identified in this study are universally measured in the current assessment tools. Patients with active disease had more of the most commonly listed burden-of-disease items; however, patients still suffered from these burdens when the disease was inactive. These disease burden items are therefore mostly dynamic problems and not simply chronic issues.
Patient ratings differ considerably from physician ratings in terms of importance. For example, patients rate nasal manifestations, weight gain, and some chronic pain and fatigue items higher than renal insufficiency and stroke. There is a clear need to address not only physician-ranked issues, but also patient-ranked issues in assessing and treating vasculitis.11
When measuring HRQOL via the Medical Outcomes Study 36-item short-form health survey (SF-36) in patients with vasculitis, a correlation is noted between QOL and sustained remission. In a study by Tomasson et al, QOL was measured using the SF-36 upon treatment following a flare.12 In all patients, SF-36 increased dramatically immediately following treatment but then leveled off over time. In patients who achieved sustained remission, SF-36 scores continued to rise from baseline. In patients who did not achieve a sustained remission, the SF-36 scores did not improve. This QOL measure, therefore, captures a value that other assessments do not, further demonstrating its utility as part of the assessment process.
VALIDATED OUTCOME MEASURES
Outcome Measures in Rheumatology (OMERACT) is an international group of clinicians, trialists, epidemiologists, biostatisticians, health economists, industry executives, and FDA and European Medicines Agency officials who meet every 2 years to promote data-based validation of outcome measures for a variety of diseases. OMERACT endorses core sets of validated outcomes when data demonstrate veracity, discrimination, and feasibility.13 For each domain in the vasculitis arena, there is an associated validated instrument: for disease activity, the validated instruments are the BVAS, BVAS/WG, and BVAS v.3; for damage assessment, the instrument is the VDI; for patient-reported outcomes, the instrument is the SF-36; and finally, for mortality, the instrument is death.13 This core set of measures helps frame how future trials in vasculitis will be standardized and assists in comparing trials, which is particularly important to regulatory agencies.
The tools for disease assessment in vasculitis still need to be refined for activity and damage assessment in order to be more scalable and precise, thereby measuring smaller effects. Patient-reported outcomes and patient perspectives on disease need to be better captured, and reliable biomarkers need to be discovered or further developed. Improved outcome measures must be developed for other types of vasculitis, such as eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome), giant cell (temporal) arteritis, and Takayasu arteritis,14 in order to conduct and report trial results. These outcome measures could also translate into tools that can be used to assess patients and make treatment decisions, thereby helping the clinician at the bedside.
Small-vessel vasculitides are complex diseases with highly variable clinical features and are associated with considerable morbidity and mortality. These systemic, multisystem, multiorgan diseases often threaten vital organs with manifestations that include upper airway disease, pulmonary disease, glomerulonephritis, neuropathy, arthritis/arthralgias, malaise/fatigue, eye disease, skin/mucosa irregularities, vascular disease, cardiac disease, and gastrointestinal disease.
Accurate assessment of the patient with vasculitis is a challenge for the clinician and is critical for managing therapeutic interventions throughout the course of the disease. Effective management includes repeated evaluations of the activity and severity of the disease as well as the damage it has caused. These distinct yet overlapping concepts must be measured separately but evaluated as a whole. Additional categorizations of disease course, such as whether it is active (new-onset, persistent, or flare) or in remission, further define the disease and are routinely employed in guiding treatment choices.
The importance of accurately assessing a patient’s clinical status is clear, but it is also important to standardize and quantify vasculitis assessment tools for use in clinical trials. Standardized assessments are needed to:
- Guide clinical trial enrollment criteria
- Describe and compare study populations
- Quantify and measure treatment effectiveness
- Describe long-term outcomes
- Translate standardized assessment tools into clinical practice.
Over the past few decades, improvements in clinical research have resulted in increasingly accurate data obtained from well-designed randomized controlled trials, all of which are based on better clinical assessments. Improving the quality of the assessment tools has improved both the interpretation of trial results and translation of findings into clinical practice.
DISEASE ASSESSMENT
When assessing patients with vasculitis, whether clinically or in the context of a clinical trial, it is essential to differentiate among disease activity, damage, and severity:
Disease activity, such as active bleeding or mucosal inflammation, is treatable and potentially responsive to therapy.
Disease damage is generally irreversible and not improved by treating vasculitis. Damage may be caused by the disease itself, its treatment, or a comorbid condition. In general, once damage is identified, it is considered permanent if it remains unchanged for more than 6 months. In the Wegener’s Granulomatosis Etanercept Trial,1 damage that occurred in more than 10% of the cohort included hearing loss; proteinuria (≥ 0.5 g/24 hours); nasal blockage, chronic discharge, or crusting; nasal bridge collapse or septal perforation; glomerular filtration rate at least 50% lower than premorbid baseline; subglottic stenosis; and chronic sinusitis or radiologic damage. Disease-related damage can be addressed; a saddle-nose deformity can respond to plastic surgery, for example, but treating vasculitis will have no effect on the underlying established anatomic defect.
Disease severity assesses the intensity of the disease and guides the clinician in gauging how aggressive the therapy should be.
Vasculitis has two primary disease states: remission and active disease. In remission, there is no evidence of active disease. This is often qualified by describing the remission as either complete or partial; it is further defined by introducing an element of time, such as a “sustained” remission of more than 6 months. Active disease is the presence of any ongoing expression of vasculitis that is not caused by disease damage, comorbidity, or treatment. Active disease can be graded as low, medium, or high; if active disease lasts longer than 6 months, it is described as persistent or sustained. Flare, a manifestation of active disease, describes the transition from remission to active disease and is characterized by worsening of disease activity. Flares are graded as nonsevere or severe.
These descriptions of disease status can be further broken down into whether they are occurring “on” or “off” treatment. All of these elements are important and the subtleties and differences are critical in interpreting data for use in the clinical setting or in clinical trials.
CLINICAL ASSESSMENT
Assessing the status of disease for a patient with granulomatosis with polyangiitis (GPA, Wegener’s granulomatosis [WG]) or microscopic polyangiitis (MPA) begins with a detailed medical history and physical examination every time the patient is seen. Appropriate laboratory assessments include a complete blood count, tests of renal function, acute phase reactants (possibly as disease markers, but not necessarily to guide therapy), and other laboratory tests as needed. Controversy exists regarding the role of antineutrophil cytoplasmic antibody (ANCA) testing in assessment of disease activity.
Urinalyses are key for assessing activity; if a urine dipstick result is positive, a subsequent microscopic examination should be conducted. Microscopic review may demonstrate red cell casts that a routine laboratory check may not reveal. In addition to spotting de novo hematuria, looking for a change in dipstick results may prove valuable, since hematuria may increase in patients in whom persistent hematuria has already been noted. The change may be due to renal disease from the vasculitis, cyclophosphamide-induced bladder toxicity, a kidney stone, menses, or a variety of other causes, but if the hematuria is not monitored, a key assessment will be missed.
Disease staging through diagnostic imaging of the sinuses, neck, and chest should be performed on a regular basis as appropriate, beginning at the patient’s initial visit. Restaging, in much the same way as an oncologist restages cancer, should take place regularly, because this informs whether to make a major change in therapy (eg, from cyclophosphamide, azathioprine, or rituximab). Restaging will also allow benchmarking of old, new, and changed damage so that when the disease recurs, the existing damage can be differentiated from new lesions. Once the disease has stabilized, imaging can be discontinued.
Consultations with otolaryngologists, ophthalmologists, cardiologists, and other specialists should be sought as needed. Serial audiograms, laryngoscopy, echocardiograms, and other appropriate tests should be performed as required. Biopsies are useful for assessment of patients, particularly at diagnosis, but also when it becomes necessary to reassess the progress of a patient’s disease or to identify a potential infection versus a possible malignancy. Biopsy is particularly helpful for kidney disease. If of active disease, then repeat biopsy is appropriate to determine whether the deterioration is associated with persistent active disease, the natural history of declining kidney function, or another cause. Patients with vasculitis may develop new comorbidities, particularly infections, so vigilance is always required. Importantly, documentation and awareness of disease-related damage are crucial in order to avoid overtreatment; damage should not be treated if therapy will not improve it.
ASSESSING DISEASE ACTIVITY AND DAMAGE
Birmingham Vasculitis Activity Score
Introduced in 1994, the Birmingham Vasculitis Activity Score (BVAS) is a single-page checklist that records weighted data on more than 50 items and nine organ systems; the sum of the individual items provides the final score.2 There have been two revisions of the BVAS; one focuses on GPA (BVAS/WG)3 and the other, BVAS version 3 (v.3) is more simplified.4 For all of the BVAS tools, remission is defined as a score of 0. Any score greater than 0 defines active disease. Each system is evaluated as being active or not, with items characterized as more severe being weighted more heavily. The use of the BVAS/WG is illustrated in two patients (see “Assessment with the BVAS/WG,” below).
Every major randomized controlled trial in the past 15 years has used the BVAS or one of its derivatives to define outcomes, but primary outcomes were not defined strictly from the BVAS itself. There were important differences in the trials’ definitions of remission, which is the outcome of interest. For example, some trials allow for minor disease activity concurrent with partial remission, while others require full absence of disease activity to achieve “remission.”
Vasculitis Damage Index
The Vasculitis Damage Index (VDI) is a single-page catalog of damage items separated into 11 groupings. Limitations of the index include lack of attribution (to vasculitis, treatment, or comorbidities), gradation, weighting, and patient input (patient-reported outcomes).5 Revisions to the VDI have been made in the ANCA Vasculitis Index of Damage (AVID),6 which incorporates an expanded list of damage items, as well as an even more expanded version called the Combined Damage Assessment Index that combines the items from the VDI and AVID.7 While these tools provide a means to catalog damage by choosing whether an item is present or not, a more data-driven approach to damage assessment is needed that incorporates weighting into the tool.
Damage assessment may be the most important measure in evaluating the patient with vasculitis. In addition to keeping patients alive, one of the main purposes in treating active disease is to prevent damage, maintaining quality of life for the patient for the long term and improving outcomes.
PATIENT-REPORTED OUTCOMES
Patients have a different perspective on their disease than that provided by assessment tools or physicians. Because physician and patient ratings are often disparate, health-related quality of life (HRQOL) is an increasingly important outcome measure for patients as well as regulatory agencies. In a 2010 study, structured patient-reported assessments of burden of disease were obtained from 264 patients with vasculitis in the United States, Germany, and the United Kingdom. Patients ranked items in terms of most frequent burdens of disease. Across ages and countries, patients most commonly rated fatigue/energy loss, pain, musculoskeletal symptoms, and social manifestations as the most severe ramifications of their disease.11 None of the burdens of disease identified in this study are universally measured in the current assessment tools. Patients with active disease had more of the most commonly listed burden-of-disease items; however, patients still suffered from these burdens when the disease was inactive. These disease burden items are therefore mostly dynamic problems and not simply chronic issues.
Patient ratings differ considerably from physician ratings in terms of importance. For example, patients rate nasal manifestations, weight gain, and some chronic pain and fatigue items higher than renal insufficiency and stroke. There is a clear need to address not only physician-ranked issues, but also patient-ranked issues in assessing and treating vasculitis.11
When measuring HRQOL via the Medical Outcomes Study 36-item short-form health survey (SF-36) in patients with vasculitis, a correlation is noted between QOL and sustained remission. In a study by Tomasson et al, QOL was measured using the SF-36 upon treatment following a flare.12 In all patients, SF-36 increased dramatically immediately following treatment but then leveled off over time. In patients who achieved sustained remission, SF-36 scores continued to rise from baseline. In patients who did not achieve a sustained remission, the SF-36 scores did not improve. This QOL measure, therefore, captures a value that other assessments do not, further demonstrating its utility as part of the assessment process.
VALIDATED OUTCOME MEASURES
Outcome Measures in Rheumatology (OMERACT) is an international group of clinicians, trialists, epidemiologists, biostatisticians, health economists, industry executives, and FDA and European Medicines Agency officials who meet every 2 years to promote data-based validation of outcome measures for a variety of diseases. OMERACT endorses core sets of validated outcomes when data demonstrate veracity, discrimination, and feasibility.13 For each domain in the vasculitis arena, there is an associated validated instrument: for disease activity, the validated instruments are the BVAS, BVAS/WG, and BVAS v.3; for damage assessment, the instrument is the VDI; for patient-reported outcomes, the instrument is the SF-36; and finally, for mortality, the instrument is death.13 This core set of measures helps frame how future trials in vasculitis will be standardized and assists in comparing trials, which is particularly important to regulatory agencies.
The tools for disease assessment in vasculitis still need to be refined for activity and damage assessment in order to be more scalable and precise, thereby measuring smaller effects. Patient-reported outcomes and patient perspectives on disease need to be better captured, and reliable biomarkers need to be discovered or further developed. Improved outcome measures must be developed for other types of vasculitis, such as eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome), giant cell (temporal) arteritis, and Takayasu arteritis,14 in order to conduct and report trial results. These outcome measures could also translate into tools that can be used to assess patients and make treatment decisions, thereby helping the clinician at the bedside.
- Seo P, Min YI, Holbrook JT, et al. Damage caused by Wegener’s granulomatosis and its treatment: prospective data from the Wegener’s Granulomatosis Etanercept Trial (WGET). Arthritis Rheum 2005; 52:2168–2178.
- Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM 1994; 87:671–678.
- Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum 2001; 44:912–920.
- Mukhtyar C, Lee R, Brown D, et al. Modification and valid ation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis 2009; 68:1827–1832.
- Exley AR, Bacon PA, Luqmani RA, et al. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum 1997; 40:371–380.
- Seo P, Luqmani RA, Flossmann O, et al. The future of damage assessment in vasculitis. J Rheumatol 2007; 34:1357–1371.
- Seo P, Jayne D, Luqmani R, Merkel PA. Assessment of damage in vasculitis: expert ratings of damage. Rheumatology (Oxford) 2009; 48:823–827.
- de Groot K, Gross WL, Herlyn K, Reinhold-Keller E. Development and validation of a disease extent index for Wegener’s granulomatosis. Clin Nephrol 2001; 55:31–38.
- Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P; French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011; 90:19–27.
- Merkel PA, Cuthbertson DD, Hellmich B, et al. Comparison of disease activity measures for anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis. Ann Rheum Dis 2009; 68:103–106.
- Herlyn K, Hellmich B, Seo P, Merkel PA. Patient-reported outcome assessment in vasculitis may provide important data and a unique perspective. Arthritis Care Res (Hoboken) 2010; 62:1639–1645.
- Tomasson G, Boers M, Walsh M, et al. Assessment of health related quality of life as an outcome measure in granulomatosis with polyangiitis (Wegener’s). Arthritis Care Res (Hoboken) 2012; 64:273–279.
- Merkel PA, Aydin SZ, Boers M, et al .The OMERACT core set of outcome measures for use in clinical trials of ANCA-associated vasculitis. J Rheumatol 2011; 38:1480–1486.
- Direskeneli H, Aydin SZ, Kermani TA, et al. Development of outcome measures for large-vessel vasculitis for use in clinical trials: opportunities, challenges, and research agenda. J Rheumatol 2011; 38:1471–1479.
- Seo P, Min YI, Holbrook JT, et al. Damage caused by Wegener’s granulomatosis and its treatment: prospective data from the Wegener’s Granulomatosis Etanercept Trial (WGET). Arthritis Rheum 2005; 52:2168–2178.
- Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM 1994; 87:671–678.
- Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum 2001; 44:912–920.
- Mukhtyar C, Lee R, Brown D, et al. Modification and valid ation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis 2009; 68:1827–1832.
- Exley AR, Bacon PA, Luqmani RA, et al. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum 1997; 40:371–380.
- Seo P, Luqmani RA, Flossmann O, et al. The future of damage assessment in vasculitis. J Rheumatol 2007; 34:1357–1371.
- Seo P, Jayne D, Luqmani R, Merkel PA. Assessment of damage in vasculitis: expert ratings of damage. Rheumatology (Oxford) 2009; 48:823–827.
- de Groot K, Gross WL, Herlyn K, Reinhold-Keller E. Development and validation of a disease extent index for Wegener’s granulomatosis. Clin Nephrol 2001; 55:31–38.
- Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P; French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011; 90:19–27.
- Merkel PA, Cuthbertson DD, Hellmich B, et al. Comparison of disease activity measures for anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis. Ann Rheum Dis 2009; 68:103–106.
- Herlyn K, Hellmich B, Seo P, Merkel PA. Patient-reported outcome assessment in vasculitis may provide important data and a unique perspective. Arthritis Care Res (Hoboken) 2010; 62:1639–1645.
- Tomasson G, Boers M, Walsh M, et al. Assessment of health related quality of life as an outcome measure in granulomatosis with polyangiitis (Wegener’s). Arthritis Care Res (Hoboken) 2012; 64:273–279.
- Merkel PA, Aydin SZ, Boers M, et al .The OMERACT core set of outcome measures for use in clinical trials of ANCA-associated vasculitis. J Rheumatol 2011; 38:1480–1486.
- Direskeneli H, Aydin SZ, Kermani TA, et al. Development of outcome measures for large-vessel vasculitis for use in clinical trials: opportunities, challenges, and research agenda. J Rheumatol 2011; 38:1471–1479.
Upper airway manifestations of granulomatosis with polyangiitis
The head and neck are the most common sites of involvement at initial presentation of granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]). Head and neck manifestations occur initially in 73% of patients, and eventually, up to 92% of patients with GPA are affected.1 Many of these compromise the upper airway. Although treatment is multidisciplinary, the effects on the airway make it important to understand upper airway presentations and treatments. This article examines upper airway disease presentations, their assessment, and their advocated interventions.
DISEASE COURSE
Because head and neck involvement may be associated with a less aggressive form of GPA, outcomes for patients with predominantly head and neck involvement may be better compared with those who have involvement of other systems.2
The natural course of GPA may be indolent or rapidly progressive. Regardless, left untreated, it progresses to a generalized systemic disease that often leads to significant morbidity and likely mortality. Most patients (96%) achieve remission with immunosuppressive therapy, but nearly half (49%) have at least one relapse.1 For this reason, systemic immunosuppressive medications play a dominant role in systemic and localized head and neck disease control. Patients often require maintenance medications along with additional therapies during disease exacerbation.3 Therefore, key partnerships between internists, rheumatologists, and otolaryngologists are paramount in the treatment and follow-up of these patients.
DIAGNOSIS: MAINSTAY IS SEROLOGIC EVALUATION
The differential diagnosis of GPA includes infection, lymphoproliferative disease (T-cell lymphoma), systemic lupus erythematosus, rheumatoid arthritis, sarcoidosis, and other granulomatous diseases such as eosinophilic GPA (Churg-Strauss syndrome), polyarteritis nodosa, and microscopic polyangiitis. Appropriate diagnosis is critical because treatment of these entities varies drastically.
The mainstay of GPA diagnosis is serologic evaluation for a cytoplasmic pattern of antineutrophil cytoplasmic antibodies (cANCA), which are reactive toward proteinase-3 (PR3) or myeloperoxidase (MPO). Testing for cANCA yields a pooled sensitivity of 91% and specificity of 99%. Sensitivity falls significantly (63%) when the disease is in nonacute stages, while the specificity remains high.4 These cANCA test characteristics allow a high positive predictive value for this rare disease.
Biopsy is typically reserved for cases in which serologic ANCA testing is nondiagnostic. Biopsy tissue may be readily accessible from the head and neck, but these biopsies may bear significant false-negative rates.4–6 Diagnosis requires demonstration of palisading granulomas as vascular or extravascular lesions within the upper respiratory tract tissues. The specific site biopsied from within the head and neck has been shown to influence diagnostic yield, with sinonasal biopsies producing the highest yield.
SINONASAL MANIFESTATIONS
The nose and paranasal sinuses are the most frequently affected sites in the head and neck, noted in 64% to 80% of patients. Additionally, the nose is the only site of involvement in 30% of patients.7 Given the high frequency of sinonasal manifestations, GPA should be considered as a potential diagnosis among patients with persistent sinonasal disease.
Pathophysiology and disease course
The pathophysiologic mechanisms leading to the changes in the sinonasal tract in GPA have not been established. GPA is believed to be an immunologic disease that manifests as a vasculitis of small- and medium-sized vessels. Multiple potential causative factors have been identified, including fibrinoid necrosis of small blood vessels, epithelial granulomas, chronic inflammation, and prior surgical intervention.8,9 The acute and chronic inflammation, coupled with the epithelioid granuloma formation, damages adjacent small- to medium-sized vessels. The vasculitis leads to diminished blood flow and subsequent avascular necrosis, which may promote tissue necrosis and bone destruction. This destructive process typically starts in the midseptum supplied by Kiesselbach plexus and in the turbinates. The process then eventually spreads to the paranasal sinuses.8
Patient evaluation
Examination of the nasal cavities is typically performed by rigid or flexible nasal endoscopy and often reveals nasal crusting, friable erythematous mucosa, granulation, and even signs of sinusitis. All or part of the cartilaginous septum may be involved, leading to significant septal defects. As the degree of cartilage destruction increases, nasal dorsal support decreases, leading to a visible depression of the external nose known as a “saddle-nose” deformity, which is present in 23% of patients with GPA.7,10
Imaging assessment by computed tomography (CT) is needed to establish disease extent and involvement. Atypical findings may include bony erosion and destruction of the septum and turbinates; erosion of bony partitions within the ethmoid sinuses; neo-osteogenesis of the maxillary, frontal, and sphenoid sinuses; and complete bony obliteration of the maxillary, frontal, and sphenoid sinuses.9,11
Clinical presentation
Sinonasal disease indicates the degree of disease activity.12 Clinical findings may vary, but they have a significant impact on quality of life in these patients.13 Most patients with active disease present with nasal crusting (69%), chronic rhinosinusitis (CRS) symptoms (61%), nasal obstruction (58%), and serosanguinous nasal discharge (52%).10 Patients may also complain of foul-smelling rhinorrhea, recurrent epistaxis, hyposmia, anosmia, and epiphora (from granulomatous compression or obstruction of the lacrimal system). In a series of 120 patients with GPA, Cannady et al found that four (3.3%) patients had mucoceles and three (2.5%) had orbital pseudotumor.10
Any structure in the sinonasal cavity, including mucosa, septum, turbinates, and sinuses proper, may be affected because of the vasculitic involvement of mucosal blood vessels that causes diminished blood flow and subsequent necrosis. The area of the anterior septum supplied by Kiesselbach plexus is the most common site of active nasal disease, which can eventually lead to the common presentation of an anterior nasal septal perforation.
Otologic disease secondary to sinonasal GPA
Otologic involvement is observed in 19% to 38% of patients with GPA.14,15 Most patients with GPA who exhibit otologic symptoms have middle ear or mastoid disease. It typically appears as chronic otitis media (COM) with conductive hearing loss.16 In most cases, the otologic involvement is secondary to Eustachian tube dysfunction caused by the presence of extensive disease in the nasopharynx.
Additionally, chronic mastoiditis can result from direct mastoid involvement with GPA. Facial nerve palsy secondary to infective bony destruction is a rare but repeatedly reported complication of GPA.14,15
Inner ear involvement is a relatively common otologic presentation of GPA. Patients may experience sensorineural hearing loss (SNHL) as well as vertigo, which may mimic Cogan syndrome. Importantly, patients may exhibit inner ear involvement with or without middle ear and mastoid disease. The SNHL observed in patients with GPA may be responsive to steroid or immunosuppressive therapy.
Treatment
Refractory CRS in GPA is a complex problem for which aggressive surgical intervention is often counterproductive. Unfortunately, traditional medical therapies are also often inadequate to treat progressive sinonasal symptomatology. As the nasal tissue becomes devascularized, loss of normal mucociliary function aggravates the sinus pathology, and clinical symptoms may worsen. Simple antibiotic regimens used to manage uncomplicated sinusitis are often inadequate in these patients. The subsequent progression to frank necrosis in localized regions creates an intranasal foreign body, allowing bacterial colonization, which is often refractory to antibiotics because of the inability of drug tissue penetration into these devascularized nasal structures.12,17
Medical management must be tailored to be effective in this complex intranasal milieu. Successful treatment requires a multifaceted and often prolonged treatment course. A high index of suspicion should be maintained for Staphylococcus aureus. As a rule, endoscopically obtained cultures should be used to guide antibiotic selection. Several weeks of culture-directed antibiotics followed by topical antibiotic irrigations (eg, mupirocin irrigations) can be useful to reduce the frequency of sinonasal exacerbations.
Frequent saline irrigations using high-volume, high-flow irrigation devices (as opposed to low-volume, low-flow applicators such as nasal spray bottles) can be an excellent adjunct to maintenance therapy and are effective in clearing debris and augmenting mucociliary clearance in affected nasal cavities and those with septal perforations. Occasional in-office endoscopic debridement of large crusts adherent to intranasal structures or the edges of a septal perforation can also help to improve obstructive symptoms.
Surgery for refractory cases. Surgery should be reserved for refractory cases unresponsive to maximal medical efforts or those cases with impending complications (ie, mucoceles). Overall, only 16% of patients with sinonasal GPA required surgical intervention in a large series of 120 patients at our institution. In this series, one-third of all patients had undergone previous functional nasal surgery at an outside institution without resolution of symptoms. Anecdotal evidence suggests that surgery for GPA can contribute to additional scarring and lead to protracted sinonasal symptoms.10,18
The decision to perform surgery is individualized and based on severity of the disease process, patient expectations, and surgeon expertise. In our experience at Cleveland Clinic, functional endoscopic sinus surgery in the setting of GPA is a surgical challenge, given extensive alteration of the sinonasal anatomy from previous surgery, prior and ongoing inflammation, chronic crusting, and scarring. Consequently, it has been our practice to employ conservative efforts prior to consideration of surgery. A complete surgical cure is exceedingly rare, and the patient should be counseled about the possible need for revision surgery and ongoing nonsurgical therapies. Meticulous postoperative care with weekly postoperative debridement, saline or antibiotic irrigations, and culture-directed antibiotics, is essential during the early postoperative recovery phase.
Management of epiphora. The most common ophthalmologic findings in patients with GPA include chronic epiphora and orbital pseudotumor. With the advent of advanced endoscopic techniques, the otolaryngologist plays a greater role in the surgical management of these ophthalmologic disease entities. In a series reported by Cannady et al,10 endoscopic dacrocystorhinostomy was performed successfully in seven patients, including one revision.
Nasal reconstruction for saddle-nose deformity: effective in selected patients. The progressive loss of septal support that occurs with the enlarging anterior septal perforation often results in significant collapse of the cartilaginous midvault of the nose. The tip cartilages in turn also begin to lose projection, resulting in a shortened nose with the characteristic saddle-nose deformity. The psychologic impact of this disfiguring facial abnormality is significant. The loss of midvault support also results in worsening nasal obstruction and increases the incidence of anosmia as the superior nasal vault becomes obstructed. For these reasons, patients often seek referral for potential reconstruction.
Despite the potential benefits, the general consensus in the medical community has been that surgical procedures on the nose should be avoided in GPA patients.17 Most nasal destruction in these patients is the consequence of poor tissue perfusion from the active vasculitis. Poor wound healing, reconstructive graft resorption, and worsening necrosis have been observed in patients who have undergone ill-advised surgical procedures.
These poor outcomes do not, however, preclude the potential for safe and effective surgical intervention. In three small published series, good surgical outcomes were achieved but the procedures were done in very highly selected patients and were modified to address the specific clinical issues seen in GPA patients.19–21 The critical step in achieving a good outcome is working closely with the patient’s rheumatologist to identify an appropriate clinical window during which the patient’s disease process is in a period of relative remission. The second major factor is to modify the surgical techniques to take into account the very poor vascular framework of the recipient nasal bed.
Management of COM. Because the COM in patients with GPA is frequently secondary to nasopharyngeal disease, systemic control of GPA is the first priority. Systemic control is also the first-line treatment for patients with mixed or sensorineural hearing loss, or with vertigo. For continued or symptomatic middle ear effusions that do not resolve with systemic therapy, placement of a ventilation tube may be considered. In patients with significant hearing loss, hearing amplification devices may be warranted.15,22 Cochlear implant devices in GPA patients are experimental and may pose undue risks of meningitis.
SUBGLOTTIC STENOSIS AND TRACHEAL MANIFESTATIONS
Subglottic stenosis affects 10% to 20% of patients with GPA.1,23,24 Because of its potential life-threatening airway complications, patients should be carefully assessed for this disease manifestation. It may be the only manifestation of GPA or may be part of a spectrum of other disease manifestations. Therefore, the work-up for subglottic stenosis of unknown etiology should always include an evaluation for GPA.
Pathophysiology and disease course
The etiology of subglottic stenosis in GPA is not well understood. Theories primarily involve the vulnerability of the subglottic tissues to damage, chronic inflammation, and scarring.25 The combination of vasculitis in the setting of active inflammation may synergistically produce a hyperactive reparative mechanism in GPA patients that leads to cartilaginous fibrotic scarring and stenosis. Wound healing can be divided into the phases of inflammation, proliferation, and remodeling. An imbalance or exaggerated response of any of these levels (and likely all) produces an abnormal healing response.26 Similarly, each of these phases may be targeted to improve the healing process.
Patient evaluation
The presence of subglottic stenosis must be considered in a GPA patient with respiratory symptoms. As part of the routine initial evaluation, an office-based nasopharyngeal/laryngeal endoscopy using a flexible laryngoscope should be performed to assess the presence and severity of luminal airway narrowing. Flexible laryngoscopy reveals a circumferential narrowing of the subglottis. The stenotic tissue may vary from friable with erythematous and inflamed mucosa to a rigid mature fibrotic band, depending on the inflammatory state of the stenosis.18,27
Subclinical stenosis may be identified with routine endoscopy. An appropriate baseline is needed to follow the progression of disease and to adjust the timing of any potential intervention. The ability to digitally record a patient’s examination allows further tracking of disease and is commonly used in our practices.
Although flexible fiberoptic examination is critical in diagnosis and follow-up, intraoperative direct laryngoscopy using rigid laryngoscopes and telescopes provides the optimum view of the subglottis. In particular, this view provides greater information on the length and degree of the stenosis and allows evaluation of potential stenotic segments in the inferior trachea.
Spiral CT with 3-dimensional reconstruction of the laryngotracheal lumen and virtual bronchoscopy may provide information that complements laryngoscopy. CT may permit assessment of the entire tracheobronchial pathway. Because 15% to 55% of GPA patients have additional bronchial stenotic segments, assessment of the entire airway is important.28,29
Clinical presentation
Diagnosis of GPA in patients younger than 20 years is associated with the development of subglottic stenosis.23,30 The GPA patient with subglottic stenosis may or may not have other active systemic symptoms. The efficacy of systemic therapy often does not correlate with the degree of subglottic stenosis. Importantly, when systemic disease enters remission, the subglottic stenosis may remain due to residual scarring of the subglottis.31
Patients with subglottic stenosis may present with hoarseness, cough, wheeze, stridor, or dyspnea on exertion.27,32 The stridor and wheeze may be confused with the wheeze of asthma, often leading to misdiagnosis.17
Subglottic stenosis likely begins at a small degree and increases gradually, allowing the patient to adjust his or her breathing pattern until a critical stenotic airway area is reached. Typically, and dependent on their pulmonary health, patients are asymptomatic until about 75% airway stenosis (60% in children).33,34 At this point, symptoms may become evident and correlate with the degree of stenosis, ranging from cough and mild shortness of breath to life-threatening stridor and obstruction. Importantly, as the airway caliber narrows, mucous plugging becomes a greater concern, as it can cause acute stridulous exacerbations and airway obstruction.
A significant proportion of patients with GPA who have subclinical asymptomatic stenosis may not receive laryngeal examination. Patients who have suspicious clinical histories should be referred for evaluation of subglottic stenosis prior to symptom worsening.
Patients with significant (approximately 80%) stenosis can present with respiratory symptoms that may be life-threatening. Because airway management in this setting is substantially more difficult, the goal should be to obtain a diagnosis and perform intervention before this advanced presentation develops.
Pauzner et al described a possible association between GPA tracheal stenosis and pregnancy.35 Women of childbearing age who have GPA should be counseled about this possible association and the need for close follow-up during the partum and postpartum periods.
Treatment is controversial
The treatment of subglottic stenosis of GPA requires multidisciplinary management by the rheumatologist, otolaryngologist, and pulmonologist. Systemic manifestations of disease are managed by immunosuppressive therapy, but up to 80% of patients may require surgical management of subglottic stenosis, and the remaining 20% will respond to systemic medical therapy.22,23,36,37 Overall, the treatment of this disease is controversial and varies by center. The therapeutic arsenal consists of conventional immunosuppressive therapy, endoscopic dilation, endoscopic or laser excision, and surgical resection of the stenotic segment followed by reconstruction.
Tracheotomy. Historically, tracheotomies were performed in approximately one-half of patients with airway manifestations of GPA when the patient had active disease or when airway patency could not be adequately maintained. Most of these patients were eventually decannulated.23,25 At present, tracheotomy is performed infrequently and is reserved for patients who have either a severely tenuous airway (with tracheotomy the only safe option available to obtain a secure airway) or who express a preference for tracheotomy. In a recent study by Hoffman et al,38 tracheotomy was avoided in 21 patients through the use of stenosis dilation procedures.
Dilation. Endoscopic subglottic dilation is the currently advocated method of treatment, and has shown promising results. In two studies with a total of 41 GPA patients who were able to avoid tracheotomy and open surgical procedures, 24% underwent decannulation of previously placed tracheotomies and 24% required only one procedure at an average follow-up of 3.4 and 5 years per study. In these studies, the technique of intralesional corticosteroid with mechanical dilation (ILCD) was performed.31,36,38
Preferred: Dilation plus medical therapy
Because of the inflammatory etiology of this condition, surgical intervention has the risk of potentially worsening the stenosis. However, combining dilation of the stenosis with aggressive local medical treatment to prevent scar formation and cellular proliferation has been shown to be effective and safe. This treatment modality was recently recommended as the preferred therapy based on a number of relatively small clinical trials for subglottic stenosis, without the benefit of large controlled trials.
Our patient population consists of two subsets: (1) those who respond well to ILCD and systemic medical therapy, requiring a minimal number of dilations before no longer needing procedures because of a possible “burn out” of the subglottic disease, and (2) those who continue to have recurrence of stenosis, requiring repeat ILCD. The latter group requires close long-term observation.
To counter the effects of the exaggerated healing reaction of inflammation (early) and proliferation (late) following injury, two medications are applied to the area of repaired stenosis. The stenotic lesion is first injected submucosally with a long-acting corticosteroid suspension such as methylprednisolone. The solution is injected along the submucosal-perichondrial plane. Incisions are made in a star-like fashion, employing sharp metal microlaryngeal blades or, less commonly, the carbon dioxide laser. These incisions release the constricting stenotic ring and break it up, widening the diameter of the airway and simultaneously preserving islands of intact mucous membrane between the incisions. This epithelium is intended to regenerate and resurface the expanded lumen. Progressive serial dilations are performed using semirigid, flexible, smooth dilators or high-pressure balloon dilation. The next stage involves repeated topical applications of mitomycin-C to further inhibit fibrosis and restenosis by inhibiting cellular proliferation of the vigorous injury cycles of these lesions. Application of mitomycin-C to the dilated area of a laryngotracheal stenosis has been associated with a decreased rate of stenosis relapse.39
Our group at Cleveland Clinic has never used laser surgery alone without dilation on the subglottic stenosis caused by GPA. Incidentally, patients treated with laser surgery in other institutions prior to their referral to the Cleveland Clinic have developed complicating secondary stenoses that required more extensive surgical intervention to overcome the severe secondary superimposed damage. In theory, use of the laser may create unnecessary thermal injury that likely worsens local damage. These patients required laryngotracheal reconstructive procedures or had to undergo establishment of permanent tracheotomies.
CONCLUSION
Granulomatosis with polyangiitis is a rare disease that may manifest in multiple areas of the head and neck. Careful attention to diagnosis and management is critical, as these patients tend to have progressive disease with debilitating sequelae. The rheumatologist, otolaryngologist, and internist should identify patients with any constellation of symptoms that may be typical of GPA. A collaborative effort to diagnose, treat, and follow these patients is paramount to successful disease management.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Mahr A, Girard T, Agher R, Guillevin L. Analysis of factors predictive of survival based on 49 patients with systemic Wegener’s granulomatosis and prospective follow-up. Rheumatology (Oxford) 2001; 40:492–498.
- Wung PK, Stone JH. Therapeutics of Wegener’s granulomatosis. Nat Clin Pract Rheumatol 2006; 2:192–200.
- Rao JK, Weinberger M, Oddone EZ, Allen NB, Landsman P, Feussner JR. The role of antineutrophil cytoplasmic antibody (c-ANCA) testing in the diagnosis of Wegener granulomatosis: a literature review and meta-analysis. Ann Intern Med 1995; 123:925–932.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Jennings CR, Jones NS, Dugar J, Powell RJ, Lowe J. Wegener’s granulomatosis—a review of diagnosis and treatment in 53 subjects. Rhinology 1998; 36:188–191.
- McDonald TJ, DeRemee RA. Wegener’s granulomatosis. Laryngoscope 1983; 93:220–231.
- Lloyd G, Lund VJ, Beale T, Howard D. Rhinologic changes in Wegener’s granulomatosis. J Laryngol Otol 2002; 116:565–569.
- Yang C, Talbot JM, Hwang PH. Bony abnormalities of the paranasal sinuses in patients with Wegener’s granulomatosis. Am J Rhinol 2001; 15:121–125.
- Cannady SB, Batra PS, Koening C, et al. Sinonasal Wegener granulomatosis: a single-institution experience with 120 cases. Laryngoscope 2009; 119:757–761.
- Grindler D, Cannady S, Batra PS. Computed tomography findings in sinonasal Wegener’s granulomatosis. Am J Rhinol Allergy 2009; 23:497–501.
- Hughes RG, Drake-Lee A. Nasal manifestations of granulomatous disease. Hosp Med 2001; 62:417–421.
- Srouji IA, Andrews P, Edwards C, Lund VJ. General and rhinosinusitis-related quality of life in patients with Wegener’s granulomatosis. Laryngoscope 2006; 116:1621–1625.
- Kornblut AD, Wolff SM, deFries HO, Fauci AS. Wegener’s granulomatosis. Laryngoscope 1980; 90:1453–1465.
- McCaffrey TV, McDonald TJ, Facer GW, DeRemee RA. Otologic manifestations of Wegener’s granulomatosis. Otolaryngol Head Neck Surg 1980; 88:586–593.
- Bradley PJ. Wegener’s granulomatosis of the ear. J Laryngol Otol 1983; 97:623–626.
- Rasmussen N. Management of the ear, nose, and throat manifestations of Wegener granulomatosis: an otorhinolaryngologist’s perspective. Curr Opin Rheumatol 2001; 13:3–11.
- Erickson VR, Hwang PH. Wegener’s granulomatosis: current trends in diagnosis and management. Curr Opin Otolaryngol Head Neck Surg 2007; 15:170–176.
- Congdon D, Sherris DA, Specks U, McDonald T. Long-term follow-up of repair of external nasal deformities in patients with Wegener’s granulomatosis. Laryngoscope 2002; 112:731–737.
- Duffy FJ, Rossi RM, Pribaz JJ. Reconstruction of Wegener’s nasal deformity using bilateral facial artery musculomucosal flaps. Plast Reconstr Surg 1998; 101:1330–1333.
- Shipchandler TZ, Chung BJ, Alam DS. Saddle nose deformity reconstruction with a split calvarial bone L-shaped strut. Arch Facial Plast Surg 2008; 10:305–311.
- Hernández-Rodríguez J, Hoffman GS, Koening CL. Surgical interventions and local therapy for Wegener’s granulomatosis. Curr Opin Rheumatol 2010; 22:29–36.
- Lebovics RS, Hoffman GS, Leavitt RY, et al. The management of subglottic stenosis in patients with Wegener’s granulomatosis. Laryngoscope 1992; 102:1341–1345.
- Waxman J, Bose WJ. Laryngeal manifestations of Wegener’s granulomatosis: case reports and review of the literature. J Rheumatol 1986; 13:408–411.
- Maronian NC, Azadeh H, Waugh P, Hillel A. Association of laryngopharyngeal reflux disease and subglottic stenosis. Ann Otol Rhinol Laryngol 2001; 110:606–612.
- Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci 2004; 9:283–289.
- Gubbels SP, Barkhuizen A, Hwang PH. Head and neck manifestations of Wegener’s granulomatosis. Otolaryngol Clin North Am 2003; 36:685–705.
- Polychronopoulos VS, Prakash UB, Golbin JM, Edell ES, Specks U. Airway involvement in Wegener’s granulomatosis. Rheum Dis Clin North Am 2007; 33:755–775.
- Daum TE, Specks U, Colby TV, et al. Tracheobronchial involvement in Wegener’s granulomatosis. Am J Respir Crit Care Med 1995; 151:522–526.
- Rottem M, Fauci AS, Hallahan CW, et al Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr 1993; 122:26–31.
- Eliachar I, Chan J, Akst L. New approaches to the management of subglottic stenosis in Wegener’s granulomatosis. Cleve Clin J Med 2002; 69( suppl 2):SII149–SII151.
- Solans-Laqué R, Bosch-Gil J, Canela M, Lorente J, Pallisa E, Vilardell-Tarrés M. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Lupus 2008; 17:832–836.
- Sandu K, Monnier P. Cricotracheal resection. Otolaryngol Clin North Am 2008; 41:981–998.
- Brouns M, Jayaraju ST, Lacor C, De Mey J, et al. Tracheal stenosis: a flow dynamics study [published online ahead of print November 30, 2006]. J Appl Physiol 2007; 102:1178–1184. doi: 10.1152/japplphysiol.01063.2006
- Pauzner R, Mayan H, Hershko E, Alcalay M, Farfel Z. Exacerbation of Wegener’s granulomatosis during pregnancy: report of a case with tracheal stenosis and literature review. J Rheumatol 1994; 21:1153–1156.
- Langford CA, Sneller MC, Hallahan CW, et al. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Arthritis Rheum 1996; 39:1754–1760.
- Schokkenbroek AA, Franssen CF, Dikkers FG. Dilatation tracheoscopy for laryngeal and tracheal stenosis in patients with Wegener’s granulomatosis [published online ahead of print November 14, 2007]. Eur Arch Otorhinolaryngol 2008; 265:549–555. doi: 10.1007/s00405-007-0518-3
- Hoffman GS, Thomas-Golbanov CK, Chan J, Akst LM, Eliachar I. Treatment of subglottic stenosis, due to Wegener’s granulomatosis, with intralesional corticosteroids and dilation. J Rheumatol 2003; 30:1017–1021.
- Smith ME, Elstad M. Mitomycin C and the endoscopic treatment of laryngotracheal stenosis: are two applications better than one? Laryngoscope 2009; 119:272–283.
The head and neck are the most common sites of involvement at initial presentation of granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]). Head and neck manifestations occur initially in 73% of patients, and eventually, up to 92% of patients with GPA are affected.1 Many of these compromise the upper airway. Although treatment is multidisciplinary, the effects on the airway make it important to understand upper airway presentations and treatments. This article examines upper airway disease presentations, their assessment, and their advocated interventions.
DISEASE COURSE
Because head and neck involvement may be associated with a less aggressive form of GPA, outcomes for patients with predominantly head and neck involvement may be better compared with those who have involvement of other systems.2
The natural course of GPA may be indolent or rapidly progressive. Regardless, left untreated, it progresses to a generalized systemic disease that often leads to significant morbidity and likely mortality. Most patients (96%) achieve remission with immunosuppressive therapy, but nearly half (49%) have at least one relapse.1 For this reason, systemic immunosuppressive medications play a dominant role in systemic and localized head and neck disease control. Patients often require maintenance medications along with additional therapies during disease exacerbation.3 Therefore, key partnerships between internists, rheumatologists, and otolaryngologists are paramount in the treatment and follow-up of these patients.
DIAGNOSIS: MAINSTAY IS SEROLOGIC EVALUATION
The differential diagnosis of GPA includes infection, lymphoproliferative disease (T-cell lymphoma), systemic lupus erythematosus, rheumatoid arthritis, sarcoidosis, and other granulomatous diseases such as eosinophilic GPA (Churg-Strauss syndrome), polyarteritis nodosa, and microscopic polyangiitis. Appropriate diagnosis is critical because treatment of these entities varies drastically.
The mainstay of GPA diagnosis is serologic evaluation for a cytoplasmic pattern of antineutrophil cytoplasmic antibodies (cANCA), which are reactive toward proteinase-3 (PR3) or myeloperoxidase (MPO). Testing for cANCA yields a pooled sensitivity of 91% and specificity of 99%. Sensitivity falls significantly (63%) when the disease is in nonacute stages, while the specificity remains high.4 These cANCA test characteristics allow a high positive predictive value for this rare disease.
Biopsy is typically reserved for cases in which serologic ANCA testing is nondiagnostic. Biopsy tissue may be readily accessible from the head and neck, but these biopsies may bear significant false-negative rates.4–6 Diagnosis requires demonstration of palisading granulomas as vascular or extravascular lesions within the upper respiratory tract tissues. The specific site biopsied from within the head and neck has been shown to influence diagnostic yield, with sinonasal biopsies producing the highest yield.
SINONASAL MANIFESTATIONS
The nose and paranasal sinuses are the most frequently affected sites in the head and neck, noted in 64% to 80% of patients. Additionally, the nose is the only site of involvement in 30% of patients.7 Given the high frequency of sinonasal manifestations, GPA should be considered as a potential diagnosis among patients with persistent sinonasal disease.
Pathophysiology and disease course
The pathophysiologic mechanisms leading to the changes in the sinonasal tract in GPA have not been established. GPA is believed to be an immunologic disease that manifests as a vasculitis of small- and medium-sized vessels. Multiple potential causative factors have been identified, including fibrinoid necrosis of small blood vessels, epithelial granulomas, chronic inflammation, and prior surgical intervention.8,9 The acute and chronic inflammation, coupled with the epithelioid granuloma formation, damages adjacent small- to medium-sized vessels. The vasculitis leads to diminished blood flow and subsequent avascular necrosis, which may promote tissue necrosis and bone destruction. This destructive process typically starts in the midseptum supplied by Kiesselbach plexus and in the turbinates. The process then eventually spreads to the paranasal sinuses.8
Patient evaluation
Examination of the nasal cavities is typically performed by rigid or flexible nasal endoscopy and often reveals nasal crusting, friable erythematous mucosa, granulation, and even signs of sinusitis. All or part of the cartilaginous septum may be involved, leading to significant septal defects. As the degree of cartilage destruction increases, nasal dorsal support decreases, leading to a visible depression of the external nose known as a “saddle-nose” deformity, which is present in 23% of patients with GPA.7,10
Imaging assessment by computed tomography (CT) is needed to establish disease extent and involvement. Atypical findings may include bony erosion and destruction of the septum and turbinates; erosion of bony partitions within the ethmoid sinuses; neo-osteogenesis of the maxillary, frontal, and sphenoid sinuses; and complete bony obliteration of the maxillary, frontal, and sphenoid sinuses.9,11
Clinical presentation
Sinonasal disease indicates the degree of disease activity.12 Clinical findings may vary, but they have a significant impact on quality of life in these patients.13 Most patients with active disease present with nasal crusting (69%), chronic rhinosinusitis (CRS) symptoms (61%), nasal obstruction (58%), and serosanguinous nasal discharge (52%).10 Patients may also complain of foul-smelling rhinorrhea, recurrent epistaxis, hyposmia, anosmia, and epiphora (from granulomatous compression or obstruction of the lacrimal system). In a series of 120 patients with GPA, Cannady et al found that four (3.3%) patients had mucoceles and three (2.5%) had orbital pseudotumor.10
Any structure in the sinonasal cavity, including mucosa, septum, turbinates, and sinuses proper, may be affected because of the vasculitic involvement of mucosal blood vessels that causes diminished blood flow and subsequent necrosis. The area of the anterior septum supplied by Kiesselbach plexus is the most common site of active nasal disease, which can eventually lead to the common presentation of an anterior nasal septal perforation.
Otologic disease secondary to sinonasal GPA
Otologic involvement is observed in 19% to 38% of patients with GPA.14,15 Most patients with GPA who exhibit otologic symptoms have middle ear or mastoid disease. It typically appears as chronic otitis media (COM) with conductive hearing loss.16 In most cases, the otologic involvement is secondary to Eustachian tube dysfunction caused by the presence of extensive disease in the nasopharynx.
Additionally, chronic mastoiditis can result from direct mastoid involvement with GPA. Facial nerve palsy secondary to infective bony destruction is a rare but repeatedly reported complication of GPA.14,15
Inner ear involvement is a relatively common otologic presentation of GPA. Patients may experience sensorineural hearing loss (SNHL) as well as vertigo, which may mimic Cogan syndrome. Importantly, patients may exhibit inner ear involvement with or without middle ear and mastoid disease. The SNHL observed in patients with GPA may be responsive to steroid or immunosuppressive therapy.
Treatment
Refractory CRS in GPA is a complex problem for which aggressive surgical intervention is often counterproductive. Unfortunately, traditional medical therapies are also often inadequate to treat progressive sinonasal symptomatology. As the nasal tissue becomes devascularized, loss of normal mucociliary function aggravates the sinus pathology, and clinical symptoms may worsen. Simple antibiotic regimens used to manage uncomplicated sinusitis are often inadequate in these patients. The subsequent progression to frank necrosis in localized regions creates an intranasal foreign body, allowing bacterial colonization, which is often refractory to antibiotics because of the inability of drug tissue penetration into these devascularized nasal structures.12,17
Medical management must be tailored to be effective in this complex intranasal milieu. Successful treatment requires a multifaceted and often prolonged treatment course. A high index of suspicion should be maintained for Staphylococcus aureus. As a rule, endoscopically obtained cultures should be used to guide antibiotic selection. Several weeks of culture-directed antibiotics followed by topical antibiotic irrigations (eg, mupirocin irrigations) can be useful to reduce the frequency of sinonasal exacerbations.
Frequent saline irrigations using high-volume, high-flow irrigation devices (as opposed to low-volume, low-flow applicators such as nasal spray bottles) can be an excellent adjunct to maintenance therapy and are effective in clearing debris and augmenting mucociliary clearance in affected nasal cavities and those with septal perforations. Occasional in-office endoscopic debridement of large crusts adherent to intranasal structures or the edges of a septal perforation can also help to improve obstructive symptoms.
Surgery for refractory cases. Surgery should be reserved for refractory cases unresponsive to maximal medical efforts or those cases with impending complications (ie, mucoceles). Overall, only 16% of patients with sinonasal GPA required surgical intervention in a large series of 120 patients at our institution. In this series, one-third of all patients had undergone previous functional nasal surgery at an outside institution without resolution of symptoms. Anecdotal evidence suggests that surgery for GPA can contribute to additional scarring and lead to protracted sinonasal symptoms.10,18
The decision to perform surgery is individualized and based on severity of the disease process, patient expectations, and surgeon expertise. In our experience at Cleveland Clinic, functional endoscopic sinus surgery in the setting of GPA is a surgical challenge, given extensive alteration of the sinonasal anatomy from previous surgery, prior and ongoing inflammation, chronic crusting, and scarring. Consequently, it has been our practice to employ conservative efforts prior to consideration of surgery. A complete surgical cure is exceedingly rare, and the patient should be counseled about the possible need for revision surgery and ongoing nonsurgical therapies. Meticulous postoperative care with weekly postoperative debridement, saline or antibiotic irrigations, and culture-directed antibiotics, is essential during the early postoperative recovery phase.
Management of epiphora. The most common ophthalmologic findings in patients with GPA include chronic epiphora and orbital pseudotumor. With the advent of advanced endoscopic techniques, the otolaryngologist plays a greater role in the surgical management of these ophthalmologic disease entities. In a series reported by Cannady et al,10 endoscopic dacrocystorhinostomy was performed successfully in seven patients, including one revision.
Nasal reconstruction for saddle-nose deformity: effective in selected patients. The progressive loss of septal support that occurs with the enlarging anterior septal perforation often results in significant collapse of the cartilaginous midvault of the nose. The tip cartilages in turn also begin to lose projection, resulting in a shortened nose with the characteristic saddle-nose deformity. The psychologic impact of this disfiguring facial abnormality is significant. The loss of midvault support also results in worsening nasal obstruction and increases the incidence of anosmia as the superior nasal vault becomes obstructed. For these reasons, patients often seek referral for potential reconstruction.
Despite the potential benefits, the general consensus in the medical community has been that surgical procedures on the nose should be avoided in GPA patients.17 Most nasal destruction in these patients is the consequence of poor tissue perfusion from the active vasculitis. Poor wound healing, reconstructive graft resorption, and worsening necrosis have been observed in patients who have undergone ill-advised surgical procedures.
These poor outcomes do not, however, preclude the potential for safe and effective surgical intervention. In three small published series, good surgical outcomes were achieved but the procedures were done in very highly selected patients and were modified to address the specific clinical issues seen in GPA patients.19–21 The critical step in achieving a good outcome is working closely with the patient’s rheumatologist to identify an appropriate clinical window during which the patient’s disease process is in a period of relative remission. The second major factor is to modify the surgical techniques to take into account the very poor vascular framework of the recipient nasal bed.
Management of COM. Because the COM in patients with GPA is frequently secondary to nasopharyngeal disease, systemic control of GPA is the first priority. Systemic control is also the first-line treatment for patients with mixed or sensorineural hearing loss, or with vertigo. For continued or symptomatic middle ear effusions that do not resolve with systemic therapy, placement of a ventilation tube may be considered. In patients with significant hearing loss, hearing amplification devices may be warranted.15,22 Cochlear implant devices in GPA patients are experimental and may pose undue risks of meningitis.
SUBGLOTTIC STENOSIS AND TRACHEAL MANIFESTATIONS
Subglottic stenosis affects 10% to 20% of patients with GPA.1,23,24 Because of its potential life-threatening airway complications, patients should be carefully assessed for this disease manifestation. It may be the only manifestation of GPA or may be part of a spectrum of other disease manifestations. Therefore, the work-up for subglottic stenosis of unknown etiology should always include an evaluation for GPA.
Pathophysiology and disease course
The etiology of subglottic stenosis in GPA is not well understood. Theories primarily involve the vulnerability of the subglottic tissues to damage, chronic inflammation, and scarring.25 The combination of vasculitis in the setting of active inflammation may synergistically produce a hyperactive reparative mechanism in GPA patients that leads to cartilaginous fibrotic scarring and stenosis. Wound healing can be divided into the phases of inflammation, proliferation, and remodeling. An imbalance or exaggerated response of any of these levels (and likely all) produces an abnormal healing response.26 Similarly, each of these phases may be targeted to improve the healing process.
Patient evaluation
The presence of subglottic stenosis must be considered in a GPA patient with respiratory symptoms. As part of the routine initial evaluation, an office-based nasopharyngeal/laryngeal endoscopy using a flexible laryngoscope should be performed to assess the presence and severity of luminal airway narrowing. Flexible laryngoscopy reveals a circumferential narrowing of the subglottis. The stenotic tissue may vary from friable with erythematous and inflamed mucosa to a rigid mature fibrotic band, depending on the inflammatory state of the stenosis.18,27
Subclinical stenosis may be identified with routine endoscopy. An appropriate baseline is needed to follow the progression of disease and to adjust the timing of any potential intervention. The ability to digitally record a patient’s examination allows further tracking of disease and is commonly used in our practices.
Although flexible fiberoptic examination is critical in diagnosis and follow-up, intraoperative direct laryngoscopy using rigid laryngoscopes and telescopes provides the optimum view of the subglottis. In particular, this view provides greater information on the length and degree of the stenosis and allows evaluation of potential stenotic segments in the inferior trachea.
Spiral CT with 3-dimensional reconstruction of the laryngotracheal lumen and virtual bronchoscopy may provide information that complements laryngoscopy. CT may permit assessment of the entire tracheobronchial pathway. Because 15% to 55% of GPA patients have additional bronchial stenotic segments, assessment of the entire airway is important.28,29
Clinical presentation
Diagnosis of GPA in patients younger than 20 years is associated with the development of subglottic stenosis.23,30 The GPA patient with subglottic stenosis may or may not have other active systemic symptoms. The efficacy of systemic therapy often does not correlate with the degree of subglottic stenosis. Importantly, when systemic disease enters remission, the subglottic stenosis may remain due to residual scarring of the subglottis.31
Patients with subglottic stenosis may present with hoarseness, cough, wheeze, stridor, or dyspnea on exertion.27,32 The stridor and wheeze may be confused with the wheeze of asthma, often leading to misdiagnosis.17
Subglottic stenosis likely begins at a small degree and increases gradually, allowing the patient to adjust his or her breathing pattern until a critical stenotic airway area is reached. Typically, and dependent on their pulmonary health, patients are asymptomatic until about 75% airway stenosis (60% in children).33,34 At this point, symptoms may become evident and correlate with the degree of stenosis, ranging from cough and mild shortness of breath to life-threatening stridor and obstruction. Importantly, as the airway caliber narrows, mucous plugging becomes a greater concern, as it can cause acute stridulous exacerbations and airway obstruction.
A significant proportion of patients with GPA who have subclinical asymptomatic stenosis may not receive laryngeal examination. Patients who have suspicious clinical histories should be referred for evaluation of subglottic stenosis prior to symptom worsening.
Patients with significant (approximately 80%) stenosis can present with respiratory symptoms that may be life-threatening. Because airway management in this setting is substantially more difficult, the goal should be to obtain a diagnosis and perform intervention before this advanced presentation develops.
Pauzner et al described a possible association between GPA tracheal stenosis and pregnancy.35 Women of childbearing age who have GPA should be counseled about this possible association and the need for close follow-up during the partum and postpartum periods.
Treatment is controversial
The treatment of subglottic stenosis of GPA requires multidisciplinary management by the rheumatologist, otolaryngologist, and pulmonologist. Systemic manifestations of disease are managed by immunosuppressive therapy, but up to 80% of patients may require surgical management of subglottic stenosis, and the remaining 20% will respond to systemic medical therapy.22,23,36,37 Overall, the treatment of this disease is controversial and varies by center. The therapeutic arsenal consists of conventional immunosuppressive therapy, endoscopic dilation, endoscopic or laser excision, and surgical resection of the stenotic segment followed by reconstruction.
Tracheotomy. Historically, tracheotomies were performed in approximately one-half of patients with airway manifestations of GPA when the patient had active disease or when airway patency could not be adequately maintained. Most of these patients were eventually decannulated.23,25 At present, tracheotomy is performed infrequently and is reserved for patients who have either a severely tenuous airway (with tracheotomy the only safe option available to obtain a secure airway) or who express a preference for tracheotomy. In a recent study by Hoffman et al,38 tracheotomy was avoided in 21 patients through the use of stenosis dilation procedures.
Dilation. Endoscopic subglottic dilation is the currently advocated method of treatment, and has shown promising results. In two studies with a total of 41 GPA patients who were able to avoid tracheotomy and open surgical procedures, 24% underwent decannulation of previously placed tracheotomies and 24% required only one procedure at an average follow-up of 3.4 and 5 years per study. In these studies, the technique of intralesional corticosteroid with mechanical dilation (ILCD) was performed.31,36,38
Preferred: Dilation plus medical therapy
Because of the inflammatory etiology of this condition, surgical intervention has the risk of potentially worsening the stenosis. However, combining dilation of the stenosis with aggressive local medical treatment to prevent scar formation and cellular proliferation has been shown to be effective and safe. This treatment modality was recently recommended as the preferred therapy based on a number of relatively small clinical trials for subglottic stenosis, without the benefit of large controlled trials.
Our patient population consists of two subsets: (1) those who respond well to ILCD and systemic medical therapy, requiring a minimal number of dilations before no longer needing procedures because of a possible “burn out” of the subglottic disease, and (2) those who continue to have recurrence of stenosis, requiring repeat ILCD. The latter group requires close long-term observation.
To counter the effects of the exaggerated healing reaction of inflammation (early) and proliferation (late) following injury, two medications are applied to the area of repaired stenosis. The stenotic lesion is first injected submucosally with a long-acting corticosteroid suspension such as methylprednisolone. The solution is injected along the submucosal-perichondrial plane. Incisions are made in a star-like fashion, employing sharp metal microlaryngeal blades or, less commonly, the carbon dioxide laser. These incisions release the constricting stenotic ring and break it up, widening the diameter of the airway and simultaneously preserving islands of intact mucous membrane between the incisions. This epithelium is intended to regenerate and resurface the expanded lumen. Progressive serial dilations are performed using semirigid, flexible, smooth dilators or high-pressure balloon dilation. The next stage involves repeated topical applications of mitomycin-C to further inhibit fibrosis and restenosis by inhibiting cellular proliferation of the vigorous injury cycles of these lesions. Application of mitomycin-C to the dilated area of a laryngotracheal stenosis has been associated with a decreased rate of stenosis relapse.39
Our group at Cleveland Clinic has never used laser surgery alone without dilation on the subglottic stenosis caused by GPA. Incidentally, patients treated with laser surgery in other institutions prior to their referral to the Cleveland Clinic have developed complicating secondary stenoses that required more extensive surgical intervention to overcome the severe secondary superimposed damage. In theory, use of the laser may create unnecessary thermal injury that likely worsens local damage. These patients required laryngotracheal reconstructive procedures or had to undergo establishment of permanent tracheotomies.
CONCLUSION
Granulomatosis with polyangiitis is a rare disease that may manifest in multiple areas of the head and neck. Careful attention to diagnosis and management is critical, as these patients tend to have progressive disease with debilitating sequelae. The rheumatologist, otolaryngologist, and internist should identify patients with any constellation of symptoms that may be typical of GPA. A collaborative effort to diagnose, treat, and follow these patients is paramount to successful disease management.
The head and neck are the most common sites of involvement at initial presentation of granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]). Head and neck manifestations occur initially in 73% of patients, and eventually, up to 92% of patients with GPA are affected.1 Many of these compromise the upper airway. Although treatment is multidisciplinary, the effects on the airway make it important to understand upper airway presentations and treatments. This article examines upper airway disease presentations, their assessment, and their advocated interventions.
DISEASE COURSE
Because head and neck involvement may be associated with a less aggressive form of GPA, outcomes for patients with predominantly head and neck involvement may be better compared with those who have involvement of other systems.2
The natural course of GPA may be indolent or rapidly progressive. Regardless, left untreated, it progresses to a generalized systemic disease that often leads to significant morbidity and likely mortality. Most patients (96%) achieve remission with immunosuppressive therapy, but nearly half (49%) have at least one relapse.1 For this reason, systemic immunosuppressive medications play a dominant role in systemic and localized head and neck disease control. Patients often require maintenance medications along with additional therapies during disease exacerbation.3 Therefore, key partnerships between internists, rheumatologists, and otolaryngologists are paramount in the treatment and follow-up of these patients.
DIAGNOSIS: MAINSTAY IS SEROLOGIC EVALUATION
The differential diagnosis of GPA includes infection, lymphoproliferative disease (T-cell lymphoma), systemic lupus erythematosus, rheumatoid arthritis, sarcoidosis, and other granulomatous diseases such as eosinophilic GPA (Churg-Strauss syndrome), polyarteritis nodosa, and microscopic polyangiitis. Appropriate diagnosis is critical because treatment of these entities varies drastically.
The mainstay of GPA diagnosis is serologic evaluation for a cytoplasmic pattern of antineutrophil cytoplasmic antibodies (cANCA), which are reactive toward proteinase-3 (PR3) or myeloperoxidase (MPO). Testing for cANCA yields a pooled sensitivity of 91% and specificity of 99%. Sensitivity falls significantly (63%) when the disease is in nonacute stages, while the specificity remains high.4 These cANCA test characteristics allow a high positive predictive value for this rare disease.
Biopsy is typically reserved for cases in which serologic ANCA testing is nondiagnostic. Biopsy tissue may be readily accessible from the head and neck, but these biopsies may bear significant false-negative rates.4–6 Diagnosis requires demonstration of palisading granulomas as vascular or extravascular lesions within the upper respiratory tract tissues. The specific site biopsied from within the head and neck has been shown to influence diagnostic yield, with sinonasal biopsies producing the highest yield.
SINONASAL MANIFESTATIONS
The nose and paranasal sinuses are the most frequently affected sites in the head and neck, noted in 64% to 80% of patients. Additionally, the nose is the only site of involvement in 30% of patients.7 Given the high frequency of sinonasal manifestations, GPA should be considered as a potential diagnosis among patients with persistent sinonasal disease.
Pathophysiology and disease course
The pathophysiologic mechanisms leading to the changes in the sinonasal tract in GPA have not been established. GPA is believed to be an immunologic disease that manifests as a vasculitis of small- and medium-sized vessels. Multiple potential causative factors have been identified, including fibrinoid necrosis of small blood vessels, epithelial granulomas, chronic inflammation, and prior surgical intervention.8,9 The acute and chronic inflammation, coupled with the epithelioid granuloma formation, damages adjacent small- to medium-sized vessels. The vasculitis leads to diminished blood flow and subsequent avascular necrosis, which may promote tissue necrosis and bone destruction. This destructive process typically starts in the midseptum supplied by Kiesselbach plexus and in the turbinates. The process then eventually spreads to the paranasal sinuses.8
Patient evaluation
Examination of the nasal cavities is typically performed by rigid or flexible nasal endoscopy and often reveals nasal crusting, friable erythematous mucosa, granulation, and even signs of sinusitis. All or part of the cartilaginous septum may be involved, leading to significant septal defects. As the degree of cartilage destruction increases, nasal dorsal support decreases, leading to a visible depression of the external nose known as a “saddle-nose” deformity, which is present in 23% of patients with GPA.7,10
Imaging assessment by computed tomography (CT) is needed to establish disease extent and involvement. Atypical findings may include bony erosion and destruction of the septum and turbinates; erosion of bony partitions within the ethmoid sinuses; neo-osteogenesis of the maxillary, frontal, and sphenoid sinuses; and complete bony obliteration of the maxillary, frontal, and sphenoid sinuses.9,11
Clinical presentation
Sinonasal disease indicates the degree of disease activity.12 Clinical findings may vary, but they have a significant impact on quality of life in these patients.13 Most patients with active disease present with nasal crusting (69%), chronic rhinosinusitis (CRS) symptoms (61%), nasal obstruction (58%), and serosanguinous nasal discharge (52%).10 Patients may also complain of foul-smelling rhinorrhea, recurrent epistaxis, hyposmia, anosmia, and epiphora (from granulomatous compression or obstruction of the lacrimal system). In a series of 120 patients with GPA, Cannady et al found that four (3.3%) patients had mucoceles and three (2.5%) had orbital pseudotumor.10
Any structure in the sinonasal cavity, including mucosa, septum, turbinates, and sinuses proper, may be affected because of the vasculitic involvement of mucosal blood vessels that causes diminished blood flow and subsequent necrosis. The area of the anterior septum supplied by Kiesselbach plexus is the most common site of active nasal disease, which can eventually lead to the common presentation of an anterior nasal septal perforation.
Otologic disease secondary to sinonasal GPA
Otologic involvement is observed in 19% to 38% of patients with GPA.14,15 Most patients with GPA who exhibit otologic symptoms have middle ear or mastoid disease. It typically appears as chronic otitis media (COM) with conductive hearing loss.16 In most cases, the otologic involvement is secondary to Eustachian tube dysfunction caused by the presence of extensive disease in the nasopharynx.
Additionally, chronic mastoiditis can result from direct mastoid involvement with GPA. Facial nerve palsy secondary to infective bony destruction is a rare but repeatedly reported complication of GPA.14,15
Inner ear involvement is a relatively common otologic presentation of GPA. Patients may experience sensorineural hearing loss (SNHL) as well as vertigo, which may mimic Cogan syndrome. Importantly, patients may exhibit inner ear involvement with or without middle ear and mastoid disease. The SNHL observed in patients with GPA may be responsive to steroid or immunosuppressive therapy.
Treatment
Refractory CRS in GPA is a complex problem for which aggressive surgical intervention is often counterproductive. Unfortunately, traditional medical therapies are also often inadequate to treat progressive sinonasal symptomatology. As the nasal tissue becomes devascularized, loss of normal mucociliary function aggravates the sinus pathology, and clinical symptoms may worsen. Simple antibiotic regimens used to manage uncomplicated sinusitis are often inadequate in these patients. The subsequent progression to frank necrosis in localized regions creates an intranasal foreign body, allowing bacterial colonization, which is often refractory to antibiotics because of the inability of drug tissue penetration into these devascularized nasal structures.12,17
Medical management must be tailored to be effective in this complex intranasal milieu. Successful treatment requires a multifaceted and often prolonged treatment course. A high index of suspicion should be maintained for Staphylococcus aureus. As a rule, endoscopically obtained cultures should be used to guide antibiotic selection. Several weeks of culture-directed antibiotics followed by topical antibiotic irrigations (eg, mupirocin irrigations) can be useful to reduce the frequency of sinonasal exacerbations.
Frequent saline irrigations using high-volume, high-flow irrigation devices (as opposed to low-volume, low-flow applicators such as nasal spray bottles) can be an excellent adjunct to maintenance therapy and are effective in clearing debris and augmenting mucociliary clearance in affected nasal cavities and those with septal perforations. Occasional in-office endoscopic debridement of large crusts adherent to intranasal structures or the edges of a septal perforation can also help to improve obstructive symptoms.
Surgery for refractory cases. Surgery should be reserved for refractory cases unresponsive to maximal medical efforts or those cases with impending complications (ie, mucoceles). Overall, only 16% of patients with sinonasal GPA required surgical intervention in a large series of 120 patients at our institution. In this series, one-third of all patients had undergone previous functional nasal surgery at an outside institution without resolution of symptoms. Anecdotal evidence suggests that surgery for GPA can contribute to additional scarring and lead to protracted sinonasal symptoms.10,18
The decision to perform surgery is individualized and based on severity of the disease process, patient expectations, and surgeon expertise. In our experience at Cleveland Clinic, functional endoscopic sinus surgery in the setting of GPA is a surgical challenge, given extensive alteration of the sinonasal anatomy from previous surgery, prior and ongoing inflammation, chronic crusting, and scarring. Consequently, it has been our practice to employ conservative efforts prior to consideration of surgery. A complete surgical cure is exceedingly rare, and the patient should be counseled about the possible need for revision surgery and ongoing nonsurgical therapies. Meticulous postoperative care with weekly postoperative debridement, saline or antibiotic irrigations, and culture-directed antibiotics, is essential during the early postoperative recovery phase.
Management of epiphora. The most common ophthalmologic findings in patients with GPA include chronic epiphora and orbital pseudotumor. With the advent of advanced endoscopic techniques, the otolaryngologist plays a greater role in the surgical management of these ophthalmologic disease entities. In a series reported by Cannady et al,10 endoscopic dacrocystorhinostomy was performed successfully in seven patients, including one revision.
Nasal reconstruction for saddle-nose deformity: effective in selected patients. The progressive loss of septal support that occurs with the enlarging anterior septal perforation often results in significant collapse of the cartilaginous midvault of the nose. The tip cartilages in turn also begin to lose projection, resulting in a shortened nose with the characteristic saddle-nose deformity. The psychologic impact of this disfiguring facial abnormality is significant. The loss of midvault support also results in worsening nasal obstruction and increases the incidence of anosmia as the superior nasal vault becomes obstructed. For these reasons, patients often seek referral for potential reconstruction.
Despite the potential benefits, the general consensus in the medical community has been that surgical procedures on the nose should be avoided in GPA patients.17 Most nasal destruction in these patients is the consequence of poor tissue perfusion from the active vasculitis. Poor wound healing, reconstructive graft resorption, and worsening necrosis have been observed in patients who have undergone ill-advised surgical procedures.
These poor outcomes do not, however, preclude the potential for safe and effective surgical intervention. In three small published series, good surgical outcomes were achieved but the procedures were done in very highly selected patients and were modified to address the specific clinical issues seen in GPA patients.19–21 The critical step in achieving a good outcome is working closely with the patient’s rheumatologist to identify an appropriate clinical window during which the patient’s disease process is in a period of relative remission. The second major factor is to modify the surgical techniques to take into account the very poor vascular framework of the recipient nasal bed.
Management of COM. Because the COM in patients with GPA is frequently secondary to nasopharyngeal disease, systemic control of GPA is the first priority. Systemic control is also the first-line treatment for patients with mixed or sensorineural hearing loss, or with vertigo. For continued or symptomatic middle ear effusions that do not resolve with systemic therapy, placement of a ventilation tube may be considered. In patients with significant hearing loss, hearing amplification devices may be warranted.15,22 Cochlear implant devices in GPA patients are experimental and may pose undue risks of meningitis.
SUBGLOTTIC STENOSIS AND TRACHEAL MANIFESTATIONS
Subglottic stenosis affects 10% to 20% of patients with GPA.1,23,24 Because of its potential life-threatening airway complications, patients should be carefully assessed for this disease manifestation. It may be the only manifestation of GPA or may be part of a spectrum of other disease manifestations. Therefore, the work-up for subglottic stenosis of unknown etiology should always include an evaluation for GPA.
Pathophysiology and disease course
The etiology of subglottic stenosis in GPA is not well understood. Theories primarily involve the vulnerability of the subglottic tissues to damage, chronic inflammation, and scarring.25 The combination of vasculitis in the setting of active inflammation may synergistically produce a hyperactive reparative mechanism in GPA patients that leads to cartilaginous fibrotic scarring and stenosis. Wound healing can be divided into the phases of inflammation, proliferation, and remodeling. An imbalance or exaggerated response of any of these levels (and likely all) produces an abnormal healing response.26 Similarly, each of these phases may be targeted to improve the healing process.
Patient evaluation
The presence of subglottic stenosis must be considered in a GPA patient with respiratory symptoms. As part of the routine initial evaluation, an office-based nasopharyngeal/laryngeal endoscopy using a flexible laryngoscope should be performed to assess the presence and severity of luminal airway narrowing. Flexible laryngoscopy reveals a circumferential narrowing of the subglottis. The stenotic tissue may vary from friable with erythematous and inflamed mucosa to a rigid mature fibrotic band, depending on the inflammatory state of the stenosis.18,27
Subclinical stenosis may be identified with routine endoscopy. An appropriate baseline is needed to follow the progression of disease and to adjust the timing of any potential intervention. The ability to digitally record a patient’s examination allows further tracking of disease and is commonly used in our practices.
Although flexible fiberoptic examination is critical in diagnosis and follow-up, intraoperative direct laryngoscopy using rigid laryngoscopes and telescopes provides the optimum view of the subglottis. In particular, this view provides greater information on the length and degree of the stenosis and allows evaluation of potential stenotic segments in the inferior trachea.
Spiral CT with 3-dimensional reconstruction of the laryngotracheal lumen and virtual bronchoscopy may provide information that complements laryngoscopy. CT may permit assessment of the entire tracheobronchial pathway. Because 15% to 55% of GPA patients have additional bronchial stenotic segments, assessment of the entire airway is important.28,29
Clinical presentation
Diagnosis of GPA in patients younger than 20 years is associated with the development of subglottic stenosis.23,30 The GPA patient with subglottic stenosis may or may not have other active systemic symptoms. The efficacy of systemic therapy often does not correlate with the degree of subglottic stenosis. Importantly, when systemic disease enters remission, the subglottic stenosis may remain due to residual scarring of the subglottis.31
Patients with subglottic stenosis may present with hoarseness, cough, wheeze, stridor, or dyspnea on exertion.27,32 The stridor and wheeze may be confused with the wheeze of asthma, often leading to misdiagnosis.17
Subglottic stenosis likely begins at a small degree and increases gradually, allowing the patient to adjust his or her breathing pattern until a critical stenotic airway area is reached. Typically, and dependent on their pulmonary health, patients are asymptomatic until about 75% airway stenosis (60% in children).33,34 At this point, symptoms may become evident and correlate with the degree of stenosis, ranging from cough and mild shortness of breath to life-threatening stridor and obstruction. Importantly, as the airway caliber narrows, mucous plugging becomes a greater concern, as it can cause acute stridulous exacerbations and airway obstruction.
A significant proportion of patients with GPA who have subclinical asymptomatic stenosis may not receive laryngeal examination. Patients who have suspicious clinical histories should be referred for evaluation of subglottic stenosis prior to symptom worsening.
Patients with significant (approximately 80%) stenosis can present with respiratory symptoms that may be life-threatening. Because airway management in this setting is substantially more difficult, the goal should be to obtain a diagnosis and perform intervention before this advanced presentation develops.
Pauzner et al described a possible association between GPA tracheal stenosis and pregnancy.35 Women of childbearing age who have GPA should be counseled about this possible association and the need for close follow-up during the partum and postpartum periods.
Treatment is controversial
The treatment of subglottic stenosis of GPA requires multidisciplinary management by the rheumatologist, otolaryngologist, and pulmonologist. Systemic manifestations of disease are managed by immunosuppressive therapy, but up to 80% of patients may require surgical management of subglottic stenosis, and the remaining 20% will respond to systemic medical therapy.22,23,36,37 Overall, the treatment of this disease is controversial and varies by center. The therapeutic arsenal consists of conventional immunosuppressive therapy, endoscopic dilation, endoscopic or laser excision, and surgical resection of the stenotic segment followed by reconstruction.
Tracheotomy. Historically, tracheotomies were performed in approximately one-half of patients with airway manifestations of GPA when the patient had active disease or when airway patency could not be adequately maintained. Most of these patients were eventually decannulated.23,25 At present, tracheotomy is performed infrequently and is reserved for patients who have either a severely tenuous airway (with tracheotomy the only safe option available to obtain a secure airway) or who express a preference for tracheotomy. In a recent study by Hoffman et al,38 tracheotomy was avoided in 21 patients through the use of stenosis dilation procedures.
Dilation. Endoscopic subglottic dilation is the currently advocated method of treatment, and has shown promising results. In two studies with a total of 41 GPA patients who were able to avoid tracheotomy and open surgical procedures, 24% underwent decannulation of previously placed tracheotomies and 24% required only one procedure at an average follow-up of 3.4 and 5 years per study. In these studies, the technique of intralesional corticosteroid with mechanical dilation (ILCD) was performed.31,36,38
Preferred: Dilation plus medical therapy
Because of the inflammatory etiology of this condition, surgical intervention has the risk of potentially worsening the stenosis. However, combining dilation of the stenosis with aggressive local medical treatment to prevent scar formation and cellular proliferation has been shown to be effective and safe. This treatment modality was recently recommended as the preferred therapy based on a number of relatively small clinical trials for subglottic stenosis, without the benefit of large controlled trials.
Our patient population consists of two subsets: (1) those who respond well to ILCD and systemic medical therapy, requiring a minimal number of dilations before no longer needing procedures because of a possible “burn out” of the subglottic disease, and (2) those who continue to have recurrence of stenosis, requiring repeat ILCD. The latter group requires close long-term observation.
To counter the effects of the exaggerated healing reaction of inflammation (early) and proliferation (late) following injury, two medications are applied to the area of repaired stenosis. The stenotic lesion is first injected submucosally with a long-acting corticosteroid suspension such as methylprednisolone. The solution is injected along the submucosal-perichondrial plane. Incisions are made in a star-like fashion, employing sharp metal microlaryngeal blades or, less commonly, the carbon dioxide laser. These incisions release the constricting stenotic ring and break it up, widening the diameter of the airway and simultaneously preserving islands of intact mucous membrane between the incisions. This epithelium is intended to regenerate and resurface the expanded lumen. Progressive serial dilations are performed using semirigid, flexible, smooth dilators or high-pressure balloon dilation. The next stage involves repeated topical applications of mitomycin-C to further inhibit fibrosis and restenosis by inhibiting cellular proliferation of the vigorous injury cycles of these lesions. Application of mitomycin-C to the dilated area of a laryngotracheal stenosis has been associated with a decreased rate of stenosis relapse.39
Our group at Cleveland Clinic has never used laser surgery alone without dilation on the subglottic stenosis caused by GPA. Incidentally, patients treated with laser surgery in other institutions prior to their referral to the Cleveland Clinic have developed complicating secondary stenoses that required more extensive surgical intervention to overcome the severe secondary superimposed damage. In theory, use of the laser may create unnecessary thermal injury that likely worsens local damage. These patients required laryngotracheal reconstructive procedures or had to undergo establishment of permanent tracheotomies.
CONCLUSION
Granulomatosis with polyangiitis is a rare disease that may manifest in multiple areas of the head and neck. Careful attention to diagnosis and management is critical, as these patients tend to have progressive disease with debilitating sequelae. The rheumatologist, otolaryngologist, and internist should identify patients with any constellation of symptoms that may be typical of GPA. A collaborative effort to diagnose, treat, and follow these patients is paramount to successful disease management.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Mahr A, Girard T, Agher R, Guillevin L. Analysis of factors predictive of survival based on 49 patients with systemic Wegener’s granulomatosis and prospective follow-up. Rheumatology (Oxford) 2001; 40:492–498.
- Wung PK, Stone JH. Therapeutics of Wegener’s granulomatosis. Nat Clin Pract Rheumatol 2006; 2:192–200.
- Rao JK, Weinberger M, Oddone EZ, Allen NB, Landsman P, Feussner JR. The role of antineutrophil cytoplasmic antibody (c-ANCA) testing in the diagnosis of Wegener granulomatosis: a literature review and meta-analysis. Ann Intern Med 1995; 123:925–932.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Jennings CR, Jones NS, Dugar J, Powell RJ, Lowe J. Wegener’s granulomatosis—a review of diagnosis and treatment in 53 subjects. Rhinology 1998; 36:188–191.
- McDonald TJ, DeRemee RA. Wegener’s granulomatosis. Laryngoscope 1983; 93:220–231.
- Lloyd G, Lund VJ, Beale T, Howard D. Rhinologic changes in Wegener’s granulomatosis. J Laryngol Otol 2002; 116:565–569.
- Yang C, Talbot JM, Hwang PH. Bony abnormalities of the paranasal sinuses in patients with Wegener’s granulomatosis. Am J Rhinol 2001; 15:121–125.
- Cannady SB, Batra PS, Koening C, et al. Sinonasal Wegener granulomatosis: a single-institution experience with 120 cases. Laryngoscope 2009; 119:757–761.
- Grindler D, Cannady S, Batra PS. Computed tomography findings in sinonasal Wegener’s granulomatosis. Am J Rhinol Allergy 2009; 23:497–501.
- Hughes RG, Drake-Lee A. Nasal manifestations of granulomatous disease. Hosp Med 2001; 62:417–421.
- Srouji IA, Andrews P, Edwards C, Lund VJ. General and rhinosinusitis-related quality of life in patients with Wegener’s granulomatosis. Laryngoscope 2006; 116:1621–1625.
- Kornblut AD, Wolff SM, deFries HO, Fauci AS. Wegener’s granulomatosis. Laryngoscope 1980; 90:1453–1465.
- McCaffrey TV, McDonald TJ, Facer GW, DeRemee RA. Otologic manifestations of Wegener’s granulomatosis. Otolaryngol Head Neck Surg 1980; 88:586–593.
- Bradley PJ. Wegener’s granulomatosis of the ear. J Laryngol Otol 1983; 97:623–626.
- Rasmussen N. Management of the ear, nose, and throat manifestations of Wegener granulomatosis: an otorhinolaryngologist’s perspective. Curr Opin Rheumatol 2001; 13:3–11.
- Erickson VR, Hwang PH. Wegener’s granulomatosis: current trends in diagnosis and management. Curr Opin Otolaryngol Head Neck Surg 2007; 15:170–176.
- Congdon D, Sherris DA, Specks U, McDonald T. Long-term follow-up of repair of external nasal deformities in patients with Wegener’s granulomatosis. Laryngoscope 2002; 112:731–737.
- Duffy FJ, Rossi RM, Pribaz JJ. Reconstruction of Wegener’s nasal deformity using bilateral facial artery musculomucosal flaps. Plast Reconstr Surg 1998; 101:1330–1333.
- Shipchandler TZ, Chung BJ, Alam DS. Saddle nose deformity reconstruction with a split calvarial bone L-shaped strut. Arch Facial Plast Surg 2008; 10:305–311.
- Hernández-Rodríguez J, Hoffman GS, Koening CL. Surgical interventions and local therapy for Wegener’s granulomatosis. Curr Opin Rheumatol 2010; 22:29–36.
- Lebovics RS, Hoffman GS, Leavitt RY, et al. The management of subglottic stenosis in patients with Wegener’s granulomatosis. Laryngoscope 1992; 102:1341–1345.
- Waxman J, Bose WJ. Laryngeal manifestations of Wegener’s granulomatosis: case reports and review of the literature. J Rheumatol 1986; 13:408–411.
- Maronian NC, Azadeh H, Waugh P, Hillel A. Association of laryngopharyngeal reflux disease and subglottic stenosis. Ann Otol Rhinol Laryngol 2001; 110:606–612.
- Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci 2004; 9:283–289.
- Gubbels SP, Barkhuizen A, Hwang PH. Head and neck manifestations of Wegener’s granulomatosis. Otolaryngol Clin North Am 2003; 36:685–705.
- Polychronopoulos VS, Prakash UB, Golbin JM, Edell ES, Specks U. Airway involvement in Wegener’s granulomatosis. Rheum Dis Clin North Am 2007; 33:755–775.
- Daum TE, Specks U, Colby TV, et al. Tracheobronchial involvement in Wegener’s granulomatosis. Am J Respir Crit Care Med 1995; 151:522–526.
- Rottem M, Fauci AS, Hallahan CW, et al Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr 1993; 122:26–31.
- Eliachar I, Chan J, Akst L. New approaches to the management of subglottic stenosis in Wegener’s granulomatosis. Cleve Clin J Med 2002; 69( suppl 2):SII149–SII151.
- Solans-Laqué R, Bosch-Gil J, Canela M, Lorente J, Pallisa E, Vilardell-Tarrés M. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Lupus 2008; 17:832–836.
- Sandu K, Monnier P. Cricotracheal resection. Otolaryngol Clin North Am 2008; 41:981–998.
- Brouns M, Jayaraju ST, Lacor C, De Mey J, et al. Tracheal stenosis: a flow dynamics study [published online ahead of print November 30, 2006]. J Appl Physiol 2007; 102:1178–1184. doi: 10.1152/japplphysiol.01063.2006
- Pauzner R, Mayan H, Hershko E, Alcalay M, Farfel Z. Exacerbation of Wegener’s granulomatosis during pregnancy: report of a case with tracheal stenosis and literature review. J Rheumatol 1994; 21:1153–1156.
- Langford CA, Sneller MC, Hallahan CW, et al. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Arthritis Rheum 1996; 39:1754–1760.
- Schokkenbroek AA, Franssen CF, Dikkers FG. Dilatation tracheoscopy for laryngeal and tracheal stenosis in patients with Wegener’s granulomatosis [published online ahead of print November 14, 2007]. Eur Arch Otorhinolaryngol 2008; 265:549–555. doi: 10.1007/s00405-007-0518-3
- Hoffman GS, Thomas-Golbanov CK, Chan J, Akst LM, Eliachar I. Treatment of subglottic stenosis, due to Wegener’s granulomatosis, with intralesional corticosteroids and dilation. J Rheumatol 2003; 30:1017–1021.
- Smith ME, Elstad M. Mitomycin C and the endoscopic treatment of laryngotracheal stenosis: are two applications better than one? Laryngoscope 2009; 119:272–283.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Mahr A, Girard T, Agher R, Guillevin L. Analysis of factors predictive of survival based on 49 patients with systemic Wegener’s granulomatosis and prospective follow-up. Rheumatology (Oxford) 2001; 40:492–498.
- Wung PK, Stone JH. Therapeutics of Wegener’s granulomatosis. Nat Clin Pract Rheumatol 2006; 2:192–200.
- Rao JK, Weinberger M, Oddone EZ, Allen NB, Landsman P, Feussner JR. The role of antineutrophil cytoplasmic antibody (c-ANCA) testing in the diagnosis of Wegener granulomatosis: a literature review and meta-analysis. Ann Intern Med 1995; 123:925–932.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Jennings CR, Jones NS, Dugar J, Powell RJ, Lowe J. Wegener’s granulomatosis—a review of diagnosis and treatment in 53 subjects. Rhinology 1998; 36:188–191.
- McDonald TJ, DeRemee RA. Wegener’s granulomatosis. Laryngoscope 1983; 93:220–231.
- Lloyd G, Lund VJ, Beale T, Howard D. Rhinologic changes in Wegener’s granulomatosis. J Laryngol Otol 2002; 116:565–569.
- Yang C, Talbot JM, Hwang PH. Bony abnormalities of the paranasal sinuses in patients with Wegener’s granulomatosis. Am J Rhinol 2001; 15:121–125.
- Cannady SB, Batra PS, Koening C, et al. Sinonasal Wegener granulomatosis: a single-institution experience with 120 cases. Laryngoscope 2009; 119:757–761.
- Grindler D, Cannady S, Batra PS. Computed tomography findings in sinonasal Wegener’s granulomatosis. Am J Rhinol Allergy 2009; 23:497–501.
- Hughes RG, Drake-Lee A. Nasal manifestations of granulomatous disease. Hosp Med 2001; 62:417–421.
- Srouji IA, Andrews P, Edwards C, Lund VJ. General and rhinosinusitis-related quality of life in patients with Wegener’s granulomatosis. Laryngoscope 2006; 116:1621–1625.
- Kornblut AD, Wolff SM, deFries HO, Fauci AS. Wegener’s granulomatosis. Laryngoscope 1980; 90:1453–1465.
- McCaffrey TV, McDonald TJ, Facer GW, DeRemee RA. Otologic manifestations of Wegener’s granulomatosis. Otolaryngol Head Neck Surg 1980; 88:586–593.
- Bradley PJ. Wegener’s granulomatosis of the ear. J Laryngol Otol 1983; 97:623–626.
- Rasmussen N. Management of the ear, nose, and throat manifestations of Wegener granulomatosis: an otorhinolaryngologist’s perspective. Curr Opin Rheumatol 2001; 13:3–11.
- Erickson VR, Hwang PH. Wegener’s granulomatosis: current trends in diagnosis and management. Curr Opin Otolaryngol Head Neck Surg 2007; 15:170–176.
- Congdon D, Sherris DA, Specks U, McDonald T. Long-term follow-up of repair of external nasal deformities in patients with Wegener’s granulomatosis. Laryngoscope 2002; 112:731–737.
- Duffy FJ, Rossi RM, Pribaz JJ. Reconstruction of Wegener’s nasal deformity using bilateral facial artery musculomucosal flaps. Plast Reconstr Surg 1998; 101:1330–1333.
- Shipchandler TZ, Chung BJ, Alam DS. Saddle nose deformity reconstruction with a split calvarial bone L-shaped strut. Arch Facial Plast Surg 2008; 10:305–311.
- Hernández-Rodríguez J, Hoffman GS, Koening CL. Surgical interventions and local therapy for Wegener’s granulomatosis. Curr Opin Rheumatol 2010; 22:29–36.
- Lebovics RS, Hoffman GS, Leavitt RY, et al. The management of subglottic stenosis in patients with Wegener’s granulomatosis. Laryngoscope 1992; 102:1341–1345.
- Waxman J, Bose WJ. Laryngeal manifestations of Wegener’s granulomatosis: case reports and review of the literature. J Rheumatol 1986; 13:408–411.
- Maronian NC, Azadeh H, Waugh P, Hillel A. Association of laryngopharyngeal reflux disease and subglottic stenosis. Ann Otol Rhinol Laryngol 2001; 110:606–612.
- Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci 2004; 9:283–289.
- Gubbels SP, Barkhuizen A, Hwang PH. Head and neck manifestations of Wegener’s granulomatosis. Otolaryngol Clin North Am 2003; 36:685–705.
- Polychronopoulos VS, Prakash UB, Golbin JM, Edell ES, Specks U. Airway involvement in Wegener’s granulomatosis. Rheum Dis Clin North Am 2007; 33:755–775.
- Daum TE, Specks U, Colby TV, et al. Tracheobronchial involvement in Wegener’s granulomatosis. Am J Respir Crit Care Med 1995; 151:522–526.
- Rottem M, Fauci AS, Hallahan CW, et al Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr 1993; 122:26–31.
- Eliachar I, Chan J, Akst L. New approaches to the management of subglottic stenosis in Wegener’s granulomatosis. Cleve Clin J Med 2002; 69( suppl 2):SII149–SII151.
- Solans-Laqué R, Bosch-Gil J, Canela M, Lorente J, Pallisa E, Vilardell-Tarrés M. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Lupus 2008; 17:832–836.
- Sandu K, Monnier P. Cricotracheal resection. Otolaryngol Clin North Am 2008; 41:981–998.
- Brouns M, Jayaraju ST, Lacor C, De Mey J, et al. Tracheal stenosis: a flow dynamics study [published online ahead of print November 30, 2006]. J Appl Physiol 2007; 102:1178–1184. doi: 10.1152/japplphysiol.01063.2006
- Pauzner R, Mayan H, Hershko E, Alcalay M, Farfel Z. Exacerbation of Wegener’s granulomatosis during pregnancy: report of a case with tracheal stenosis and literature review. J Rheumatol 1994; 21:1153–1156.
- Langford CA, Sneller MC, Hallahan CW, et al. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Arthritis Rheum 1996; 39:1754–1760.
- Schokkenbroek AA, Franssen CF, Dikkers FG. Dilatation tracheoscopy for laryngeal and tracheal stenosis in patients with Wegener’s granulomatosis [published online ahead of print November 14, 2007]. Eur Arch Otorhinolaryngol 2008; 265:549–555. doi: 10.1007/s00405-007-0518-3
- Hoffman GS, Thomas-Golbanov CK, Chan J, Akst LM, Eliachar I. Treatment of subglottic stenosis, due to Wegener’s granulomatosis, with intralesional corticosteroids and dilation. J Rheumatol 2003; 30:1017–1021.
- Smith ME, Elstad M. Mitomycin C and the endoscopic treatment of laryngotracheal stenosis: are two applications better than one? Laryngoscope 2009; 119:272–283.