User login
New and Noteworthy Information—November
Hormone therapy may reduce the risk of Alzheimer’s disease for women who take the treatment at a time near menopause, but if hormone therapy is begun after menopause it may not reduce such risk, according to a study in the October 30 Neurology. Researchers followed 1,768 women who were part of a population-based study and found that 176 women developed Alzheimer’s disease between 1995 and 2006. Women who used any type of hormone therapy within five years of menopause had a 30% less risk of Alzheimer’s disease. However, those who began hormone therapy five or more years after menopause did not have a reduced disease risk. In addition, women who began opposed compounds in the three years prior to the baseline assessment had an increased risk of Alzheimer’s disease. The association of hormone therapy use and risk of Alzheimer’s disease may depend on the timing of use and deserves further study, the investigators concluded.
Engaging in physical activity may protect older adults from brain atrophy and white matter lesions, researchers reported in the October 23 Neurology. The study examined self-reported leisure and physical activity at age 70 among a sample of 691 adults. At age 73, participants were assessed for structural brain biomarkers, and the investigators found that a higher level of physical activity was significantly associated with higher fractional anisotropy, less atrophy, lower white matter load, and larger gray and normal-appearing white matter volumes. These associations remained significant after adjustments for age, social class, and health status. The researchers noted that although their results support the role of physical activity as a potential neuroprotective factor, “the direction of causation is unclear from this observational study.”
Poor physical performance is associated with greater odds of dementia in persons age 90 or older, according to a study published in the online October 22 Archives of Neurology. The 629 participants (72.5% women) were from The 90+ Study, a population-based, longitudinal, epidemiologic study of aging and dementia. Participants’ mean age was 94, and all-cause dementia was the main outcome measure. Measures of physical performance included a 4-m walk, five chair stands, standing balance, and grip strength. Researchers found that poor physical performance in all measures was significantly associated with an increased risk of dementia. “Our findings suggest that dementia is a complex neurodegenerative process that may affect physical performance and cognition,” the investigators concluded. “Additional research is necessary to determine the temporal relationship between poor physical activity and cognitive dysfunction.”
Exposure to selective serotonin reuptake inhibitors (SSRI) is associated with an increased risk of intracerebral and intracranial hemorrhage, though the absolute risk of those events is low, according to a study published in the October 30 Neurology. In this meta-analysis, investigators searched for controlled observational studies that compared SSRI users with a control group not receiving SSRIs. The researchers found that intracranial and intracerebral hemorrhage were related to SSRI exposure in unadjusted and adjusted analyses. A subset of five studies showed that SSRI exposure combined with oral anticoagulants was linked with an increased risk of bleeding, compared with use of oral anticoagulants alone. “When all studies were analyzed together, increased risk was seen across cohort studies, case-control studies, and case-crossover studies,” the study authors noted.
The herpes zoster vaccine is effective in preventing herpes zoster in older adults, according to research published in the online October 17 Cochrane Database of Systematic Reviews. The study authors conducted a meta-analysis of eight randomized controlled trials of adults who had a mean age older than 60. The trials had a total of 52,269 participants. Patients who received the vaccine had fewer confirmed cases of herpes zoster than those who received placebo. Analysis of age groups showed that vaccine benefits were greatest for patients ages 60 to 69, as well as for those 70 and older. However, persons ages 60 to 69 experienced more frequent side effects than did persons 70 and older. “In general, zoster vaccine is well tolerated; it produces few systemic adverse events and injection site adverse effects of mild to moderate intensity,” wrote the researchers.
Strokes are increasingly occurring in younger patients, researchers reported in the October 23 Neurology. Between 1993 and 1994 and between 1999 and 2005, strokes were recorded in an estimated population of 1.3 million. The investigators used a mixed-model approach to test for differences in age trends over time, and they found that the mean age at stroke decreased by a significant amount, from 71.2 years in 1993/1994 to 69.2 years in 2005. Furthermore, the proportion of all strokes in persons younger than 55 increased from 12.9% in 1993 to 18.6% in 2005. “This is of great public health significance because strokes in younger patients carry the potential for greater lifetime burden of disability and because some potential contributors identified for this trend are modifiable,” the researchers concluded.
The FDA has approved perampanel (Fycompa), an AMPA receptor agonist, as an adjunctive treatment for partial-onset seizures with or without secondarily generalized seizures in patients ages 12 and older with epilepsy. Perampanel is a novel agent that reduces neuronal hyperexcitation associated with seizures by inhibiting glutamate activity at postsynaptic AMPA receptors, and it is the first antiepileptic agent approved in the US to work in this manner. In three phase III, global, randomized, double-blind, placebo-controlled studies (1,480 patients), researchers concluded that perampanel significantly reduced seizure frequency in patients with partial-onset seizures with or without secondary generalized seizures. Patients experienced adverse events that included dizziness, somnolence, fatigue, irritability, falls, nausea, ataxia, balance disorder, gait disturbance, vertigo, and weight gain.
Persons who survive an ischemic stroke and continue smoking have a greater risk of heart attack, death, or another stroke, compared with those who have never smoked, researchers reported in the online October 25 Stroke. The study included 1,589 patients who experienced a first or recurrent ischemic stroke between 1996 and 1999. The investigators tracked the cohort for 10 years and found that patients who smoked when they had a stroke were 30% more likely to have a poor outcome and that current smokers who survived the first 28 days after a stroke had a 42% higher risk of poor outcome. In addition, former smokers had an 18% higher risk of poor outcomes. The authors also noted that smoking had the greatest effect on younger male patients, particularly those from a disadvantaged background.
For every 400 to 500 persons with an intermediate risk of cardiovascular disease who undergo screening for C-reactive protein or fibrinogen, one additional event in a period of 10 years may be prevented, researchers reported in the October 4 New England Journal of Medicine. In a meta-analysis of 52 prospective studies of persons without a history of cardiovascular disease, the investigators sought to determine whether assessing C-reactive protein or fibrinogen in addition to conventional cardiovascular risk factors leads to better prediction of cardiovascular risk.
Of 100,000 adults ages 40 and older, 15,025 would be classified as intermediate risk using conventional factors, and 13,199 would remain if statin therapy were initiated in accordance with guidelines. “Additional targeted assessment of C-reactive protein or fibrinogen levels in the 13,199 remaining participants at intermediate risk could help prevent approximately 30 additional cardiovascular events over the course of 10 years,” the researchers stated.
Extradural motor cortex stimulation for patients with Parkinson’s disease is a safe procedure that leads to moderate improvement of motor symptoms and in quality of life, according to a study published in the October Neurosurgery. Researchers assessed the safety and efficacy of one year of unilateral extradural motor cortex stimulation in nine patients with Parkinson’s disease. At baseline, participants were evaluated with the Unified Parkinson’s Disease Rating Scale and the Parkinson’s Disease Quality of Life Questionnaire. Quality of life scores increased at months three, six, and 12, and disease scores decreased from baseline during the year. Furthermore, bilateral motor effects were observed after three to four weeks. No surgical complications, adverse events, or cognitive and behavioral changes were observed, the study authors said.
The use of beta blockers is not associated with a lower risk of composite cardiovascular events in patients with either coronary artery disease (CAD) risk factors only, known prior myocardial infarction, or known CAD without myocardial infarction, according to an investigation published in the October 3 JAMA. In this longitudinal, observational study, 44,708 patients were categorized into three cohorts— 14,043 patients with known prior myocardial infarction, 12,012 patients with known CAD but without myocardial infarction, and 18,653 patients with CAD risk factors only. The primary outcome was a composite of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke. For all outcomes tested, investigators found that event rates were not significantly different in patients with beta-blocker use, compared with those without beta-blocker use, even among those in the prior myocardial infarction cohort.
—Lauren LeBano
Hormone therapy may reduce the risk of Alzheimer’s disease for women who take the treatment at a time near menopause, but if hormone therapy is begun after menopause it may not reduce such risk, according to a study in the October 30 Neurology. Researchers followed 1,768 women who were part of a population-based study and found that 176 women developed Alzheimer’s disease between 1995 and 2006. Women who used any type of hormone therapy within five years of menopause had a 30% less risk of Alzheimer’s disease. However, those who began hormone therapy five or more years after menopause did not have a reduced disease risk. In addition, women who began opposed compounds in the three years prior to the baseline assessment had an increased risk of Alzheimer’s disease. The association of hormone therapy use and risk of Alzheimer’s disease may depend on the timing of use and deserves further study, the investigators concluded.
Engaging in physical activity may protect older adults from brain atrophy and white matter lesions, researchers reported in the October 23 Neurology. The study examined self-reported leisure and physical activity at age 70 among a sample of 691 adults. At age 73, participants were assessed for structural brain biomarkers, and the investigators found that a higher level of physical activity was significantly associated with higher fractional anisotropy, less atrophy, lower white matter load, and larger gray and normal-appearing white matter volumes. These associations remained significant after adjustments for age, social class, and health status. The researchers noted that although their results support the role of physical activity as a potential neuroprotective factor, “the direction of causation is unclear from this observational study.”
Poor physical performance is associated with greater odds of dementia in persons age 90 or older, according to a study published in the online October 22 Archives of Neurology. The 629 participants (72.5% women) were from The 90+ Study, a population-based, longitudinal, epidemiologic study of aging and dementia. Participants’ mean age was 94, and all-cause dementia was the main outcome measure. Measures of physical performance included a 4-m walk, five chair stands, standing balance, and grip strength. Researchers found that poor physical performance in all measures was significantly associated with an increased risk of dementia. “Our findings suggest that dementia is a complex neurodegenerative process that may affect physical performance and cognition,” the investigators concluded. “Additional research is necessary to determine the temporal relationship between poor physical activity and cognitive dysfunction.”
Exposure to selective serotonin reuptake inhibitors (SSRI) is associated with an increased risk of intracerebral and intracranial hemorrhage, though the absolute risk of those events is low, according to a study published in the October 30 Neurology. In this meta-analysis, investigators searched for controlled observational studies that compared SSRI users with a control group not receiving SSRIs. The researchers found that intracranial and intracerebral hemorrhage were related to SSRI exposure in unadjusted and adjusted analyses. A subset of five studies showed that SSRI exposure combined with oral anticoagulants was linked with an increased risk of bleeding, compared with use of oral anticoagulants alone. “When all studies were analyzed together, increased risk was seen across cohort studies, case-control studies, and case-crossover studies,” the study authors noted.
The herpes zoster vaccine is effective in preventing herpes zoster in older adults, according to research published in the online October 17 Cochrane Database of Systematic Reviews. The study authors conducted a meta-analysis of eight randomized controlled trials of adults who had a mean age older than 60. The trials had a total of 52,269 participants. Patients who received the vaccine had fewer confirmed cases of herpes zoster than those who received placebo. Analysis of age groups showed that vaccine benefits were greatest for patients ages 60 to 69, as well as for those 70 and older. However, persons ages 60 to 69 experienced more frequent side effects than did persons 70 and older. “In general, zoster vaccine is well tolerated; it produces few systemic adverse events and injection site adverse effects of mild to moderate intensity,” wrote the researchers.
Strokes are increasingly occurring in younger patients, researchers reported in the October 23 Neurology. Between 1993 and 1994 and between 1999 and 2005, strokes were recorded in an estimated population of 1.3 million. The investigators used a mixed-model approach to test for differences in age trends over time, and they found that the mean age at stroke decreased by a significant amount, from 71.2 years in 1993/1994 to 69.2 years in 2005. Furthermore, the proportion of all strokes in persons younger than 55 increased from 12.9% in 1993 to 18.6% in 2005. “This is of great public health significance because strokes in younger patients carry the potential for greater lifetime burden of disability and because some potential contributors identified for this trend are modifiable,” the researchers concluded.
The FDA has approved perampanel (Fycompa), an AMPA receptor agonist, as an adjunctive treatment for partial-onset seizures with or without secondarily generalized seizures in patients ages 12 and older with epilepsy. Perampanel is a novel agent that reduces neuronal hyperexcitation associated with seizures by inhibiting glutamate activity at postsynaptic AMPA receptors, and it is the first antiepileptic agent approved in the US to work in this manner. In three phase III, global, randomized, double-blind, placebo-controlled studies (1,480 patients), researchers concluded that perampanel significantly reduced seizure frequency in patients with partial-onset seizures with or without secondary generalized seizures. Patients experienced adverse events that included dizziness, somnolence, fatigue, irritability, falls, nausea, ataxia, balance disorder, gait disturbance, vertigo, and weight gain.
Persons who survive an ischemic stroke and continue smoking have a greater risk of heart attack, death, or another stroke, compared with those who have never smoked, researchers reported in the online October 25 Stroke. The study included 1,589 patients who experienced a first or recurrent ischemic stroke between 1996 and 1999. The investigators tracked the cohort for 10 years and found that patients who smoked when they had a stroke were 30% more likely to have a poor outcome and that current smokers who survived the first 28 days after a stroke had a 42% higher risk of poor outcome. In addition, former smokers had an 18% higher risk of poor outcomes. The authors also noted that smoking had the greatest effect on younger male patients, particularly those from a disadvantaged background.
For every 400 to 500 persons with an intermediate risk of cardiovascular disease who undergo screening for C-reactive protein or fibrinogen, one additional event in a period of 10 years may be prevented, researchers reported in the October 4 New England Journal of Medicine. In a meta-analysis of 52 prospective studies of persons without a history of cardiovascular disease, the investigators sought to determine whether assessing C-reactive protein or fibrinogen in addition to conventional cardiovascular risk factors leads to better prediction of cardiovascular risk.
Of 100,000 adults ages 40 and older, 15,025 would be classified as intermediate risk using conventional factors, and 13,199 would remain if statin therapy were initiated in accordance with guidelines. “Additional targeted assessment of C-reactive protein or fibrinogen levels in the 13,199 remaining participants at intermediate risk could help prevent approximately 30 additional cardiovascular events over the course of 10 years,” the researchers stated.
Extradural motor cortex stimulation for patients with Parkinson’s disease is a safe procedure that leads to moderate improvement of motor symptoms and in quality of life, according to a study published in the October Neurosurgery. Researchers assessed the safety and efficacy of one year of unilateral extradural motor cortex stimulation in nine patients with Parkinson’s disease. At baseline, participants were evaluated with the Unified Parkinson’s Disease Rating Scale and the Parkinson’s Disease Quality of Life Questionnaire. Quality of life scores increased at months three, six, and 12, and disease scores decreased from baseline during the year. Furthermore, bilateral motor effects were observed after three to four weeks. No surgical complications, adverse events, or cognitive and behavioral changes were observed, the study authors said.
The use of beta blockers is not associated with a lower risk of composite cardiovascular events in patients with either coronary artery disease (CAD) risk factors only, known prior myocardial infarction, or known CAD without myocardial infarction, according to an investigation published in the October 3 JAMA. In this longitudinal, observational study, 44,708 patients were categorized into three cohorts— 14,043 patients with known prior myocardial infarction, 12,012 patients with known CAD but without myocardial infarction, and 18,653 patients with CAD risk factors only. The primary outcome was a composite of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke. For all outcomes tested, investigators found that event rates were not significantly different in patients with beta-blocker use, compared with those without beta-blocker use, even among those in the prior myocardial infarction cohort.
—Lauren LeBano
Hormone therapy may reduce the risk of Alzheimer’s disease for women who take the treatment at a time near menopause, but if hormone therapy is begun after menopause it may not reduce such risk, according to a study in the October 30 Neurology. Researchers followed 1,768 women who were part of a population-based study and found that 176 women developed Alzheimer’s disease between 1995 and 2006. Women who used any type of hormone therapy within five years of menopause had a 30% less risk of Alzheimer’s disease. However, those who began hormone therapy five or more years after menopause did not have a reduced disease risk. In addition, women who began opposed compounds in the three years prior to the baseline assessment had an increased risk of Alzheimer’s disease. The association of hormone therapy use and risk of Alzheimer’s disease may depend on the timing of use and deserves further study, the investigators concluded.
Engaging in physical activity may protect older adults from brain atrophy and white matter lesions, researchers reported in the October 23 Neurology. The study examined self-reported leisure and physical activity at age 70 among a sample of 691 adults. At age 73, participants were assessed for structural brain biomarkers, and the investigators found that a higher level of physical activity was significantly associated with higher fractional anisotropy, less atrophy, lower white matter load, and larger gray and normal-appearing white matter volumes. These associations remained significant after adjustments for age, social class, and health status. The researchers noted that although their results support the role of physical activity as a potential neuroprotective factor, “the direction of causation is unclear from this observational study.”
Poor physical performance is associated with greater odds of dementia in persons age 90 or older, according to a study published in the online October 22 Archives of Neurology. The 629 participants (72.5% women) were from The 90+ Study, a population-based, longitudinal, epidemiologic study of aging and dementia. Participants’ mean age was 94, and all-cause dementia was the main outcome measure. Measures of physical performance included a 4-m walk, five chair stands, standing balance, and grip strength. Researchers found that poor physical performance in all measures was significantly associated with an increased risk of dementia. “Our findings suggest that dementia is a complex neurodegenerative process that may affect physical performance and cognition,” the investigators concluded. “Additional research is necessary to determine the temporal relationship between poor physical activity and cognitive dysfunction.”
Exposure to selective serotonin reuptake inhibitors (SSRI) is associated with an increased risk of intracerebral and intracranial hemorrhage, though the absolute risk of those events is low, according to a study published in the October 30 Neurology. In this meta-analysis, investigators searched for controlled observational studies that compared SSRI users with a control group not receiving SSRIs. The researchers found that intracranial and intracerebral hemorrhage were related to SSRI exposure in unadjusted and adjusted analyses. A subset of five studies showed that SSRI exposure combined with oral anticoagulants was linked with an increased risk of bleeding, compared with use of oral anticoagulants alone. “When all studies were analyzed together, increased risk was seen across cohort studies, case-control studies, and case-crossover studies,” the study authors noted.
The herpes zoster vaccine is effective in preventing herpes zoster in older adults, according to research published in the online October 17 Cochrane Database of Systematic Reviews. The study authors conducted a meta-analysis of eight randomized controlled trials of adults who had a mean age older than 60. The trials had a total of 52,269 participants. Patients who received the vaccine had fewer confirmed cases of herpes zoster than those who received placebo. Analysis of age groups showed that vaccine benefits were greatest for patients ages 60 to 69, as well as for those 70 and older. However, persons ages 60 to 69 experienced more frequent side effects than did persons 70 and older. “In general, zoster vaccine is well tolerated; it produces few systemic adverse events and injection site adverse effects of mild to moderate intensity,” wrote the researchers.
Strokes are increasingly occurring in younger patients, researchers reported in the October 23 Neurology. Between 1993 and 1994 and between 1999 and 2005, strokes were recorded in an estimated population of 1.3 million. The investigators used a mixed-model approach to test for differences in age trends over time, and they found that the mean age at stroke decreased by a significant amount, from 71.2 years in 1993/1994 to 69.2 years in 2005. Furthermore, the proportion of all strokes in persons younger than 55 increased from 12.9% in 1993 to 18.6% in 2005. “This is of great public health significance because strokes in younger patients carry the potential for greater lifetime burden of disability and because some potential contributors identified for this trend are modifiable,” the researchers concluded.
The FDA has approved perampanel (Fycompa), an AMPA receptor agonist, as an adjunctive treatment for partial-onset seizures with or without secondarily generalized seizures in patients ages 12 and older with epilepsy. Perampanel is a novel agent that reduces neuronal hyperexcitation associated with seizures by inhibiting glutamate activity at postsynaptic AMPA receptors, and it is the first antiepileptic agent approved in the US to work in this manner. In three phase III, global, randomized, double-blind, placebo-controlled studies (1,480 patients), researchers concluded that perampanel significantly reduced seizure frequency in patients with partial-onset seizures with or without secondary generalized seizures. Patients experienced adverse events that included dizziness, somnolence, fatigue, irritability, falls, nausea, ataxia, balance disorder, gait disturbance, vertigo, and weight gain.
Persons who survive an ischemic stroke and continue smoking have a greater risk of heart attack, death, or another stroke, compared with those who have never smoked, researchers reported in the online October 25 Stroke. The study included 1,589 patients who experienced a first or recurrent ischemic stroke between 1996 and 1999. The investigators tracked the cohort for 10 years and found that patients who smoked when they had a stroke were 30% more likely to have a poor outcome and that current smokers who survived the first 28 days after a stroke had a 42% higher risk of poor outcome. In addition, former smokers had an 18% higher risk of poor outcomes. The authors also noted that smoking had the greatest effect on younger male patients, particularly those from a disadvantaged background.
For every 400 to 500 persons with an intermediate risk of cardiovascular disease who undergo screening for C-reactive protein or fibrinogen, one additional event in a period of 10 years may be prevented, researchers reported in the October 4 New England Journal of Medicine. In a meta-analysis of 52 prospective studies of persons without a history of cardiovascular disease, the investigators sought to determine whether assessing C-reactive protein or fibrinogen in addition to conventional cardiovascular risk factors leads to better prediction of cardiovascular risk.
Of 100,000 adults ages 40 and older, 15,025 would be classified as intermediate risk using conventional factors, and 13,199 would remain if statin therapy were initiated in accordance with guidelines. “Additional targeted assessment of C-reactive protein or fibrinogen levels in the 13,199 remaining participants at intermediate risk could help prevent approximately 30 additional cardiovascular events over the course of 10 years,” the researchers stated.
Extradural motor cortex stimulation for patients with Parkinson’s disease is a safe procedure that leads to moderate improvement of motor symptoms and in quality of life, according to a study published in the October Neurosurgery. Researchers assessed the safety and efficacy of one year of unilateral extradural motor cortex stimulation in nine patients with Parkinson’s disease. At baseline, participants were evaluated with the Unified Parkinson’s Disease Rating Scale and the Parkinson’s Disease Quality of Life Questionnaire. Quality of life scores increased at months three, six, and 12, and disease scores decreased from baseline during the year. Furthermore, bilateral motor effects were observed after three to four weeks. No surgical complications, adverse events, or cognitive and behavioral changes were observed, the study authors said.
The use of beta blockers is not associated with a lower risk of composite cardiovascular events in patients with either coronary artery disease (CAD) risk factors only, known prior myocardial infarction, or known CAD without myocardial infarction, according to an investigation published in the October 3 JAMA. In this longitudinal, observational study, 44,708 patients were categorized into three cohorts— 14,043 patients with known prior myocardial infarction, 12,012 patients with known CAD but without myocardial infarction, and 18,653 patients with CAD risk factors only. The primary outcome was a composite of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke. For all outcomes tested, investigators found that event rates were not significantly different in patients with beta-blocker use, compared with those without beta-blocker use, even among those in the prior myocardial infarction cohort.
—Lauren LeBano
How to Handle "Incidentalomas"
Maggie, 42, presents to the emergency department with chronic intermittent abdominal pain and bloating with constipation and occasional diarrhea. She denies fever, chills, nausea, vomiting, melana, bright red blood per rectum, or changes in stool caliper, and she says she otherwise feels well.
Relevant lab and study results include: a comprehensive metabolic panel, complete blood count with differential, beta hCG (human chorionic gonadotropin), urinalysis, and amylase and lipase, all within normal limits; pregnancy test, negative; abdominal x-ray, within normal limits except increased stool in distal colon; and abdominal CT, 2.3-cm right adrenal mass and a Hounsfield measurement of 4 units.
Maggie has a right adrenal incidentaloma (incidentally discovered adenoma that was not in the differential diagnosis). Such findings have become all too often the case, due to the immediate access to and overutilization of high-resolution CT, MRI, and ultrasound. We are now seeing a significantly increased number of incidental adrenal lesions/masses discovered on images not intended to look for adrenal-related diseases (eg, Cushing syndrome, pheochromocytomas, and aldosterone-producing adenomas).
Q: How common are adrenal adenomas, and what must I consider?
Incidental adrenal adenomas are found on 4.4% of abdominal CTs, and in one autopsy series were discovered in 8.7%. Prevalence increases with age, with occurrence of < 1% in patients younger than 30 and about 7% for patients 70 or older.
Evaluation is based on two concerns: First, is the adrenal mass benign or malignant? Second, is the mass secretory or nonsecretory (non-hormone secreting) in nature?
The fortunate news about adrenal incidentalomas is that 80% are benign and nonsecretory, which provides immediate reassuring news to the patient. Examples of benign adrenal masses are: adenoma, lipoma, cyst, ganglioneuroma, hematoma, and infection (eg, tuberculosis, fungal).
The other encouraging statistic is that only 1:4,000 adrenal incidentalomas are malignant. Examples of malignant adrenal masses are: adrenocortical carcinoma, metastatic neoplasm, lymphoma, and malignant pheochromocytoma.
Q: Does adrenal adenoma size matter?
Yes, the larger the size of the adenoma, the higher the association with malignancy. The guide below (based on CT findings) shows not only malignancy potential as it relates to size, but also the importance of Hounsfield units and when surgical intervention is recommended.
Imaging (CT scan)
< 4 cm: homogeneous mass with smooth borders and < 10 Hounsfield units; suggests benign mass (likelihood of malignancy, about 2%)
4 to 6 cm: follow closely, consider surgery (likelihood of malignancy, about 6%)
> 6 cm: surgery indicated (likelihood of malignancy, about 25%)
Some providers and patients inquire whether it is helpful or necessary to biopsy. It is generally not advisable to biopsy, especially if the findings are favorable for benign nonsecretory masses, since there is a high false-negative rate. An indication for biopsy is if the patient has a history of extra-adrenal malignancy; this will distinguish recurrence or metastatic disease from a benign mass. A final proviso: If biopsy is performed, make sure the adrenal mass is not a pheochromocytoma, as biopsy of a hormone-secreting neoplasm can lead to a hypertensive emergency.
Q: How do I determine whether the mass is hormone-secreting?
Although 80% are nonsecretory, you must still maintain a high index of suspicion so as not to miss a potentially problematic and fully treatable adenoma. A thorough history is essential in screening for hormonal excess arising from adrenal adenomas, since the signs and symptoms can be insidious. The three hormones secreted by adrenal adenomas are cortisol, aldosterone, and catecholamines (seen in Cushing syndrome, aldosterone-producing adenoma [APA], and pheochromocytoma, respectively).
It is important to note that Cushing syndrome has an insidious onset and can be easily missed. Hyperaldosteronism presents with hypertension (requiring several medications) and commonly hypokalemia. And pheochromocytoma can be “written off as” anxiety disorder, panic attack, or even hypoglycemia symptoms (especially if patients are treated for diabetes with agents that cause hypoglycemia). To help in your differential diagnosis of secretory adenomas, know that APA accounts for only 1%, and therefore the majority will secrete cortisol and (far less likely) catecholamines.
Q: What is the appropriate laboratory work-up?
The best simple screening test for hypercortisolemia is a 1-mg overnight dexamethasone suppression test. If this value is increased to ≥ 3 µg/dL, it should be followed up with a more sensitive test (a 24-hour urine for creatinine and free cortisol) to further assess for hypercortisolemia.
Patients thought to have a potential pheochromocytoma should undergo measurement of plasma fractionated metanephrines and normetanephrines or 24-hour urine for total metanephrines and fractionated catecholamines.
Finally, for patients with hypokalemia and hypertension or refractory hypertension requiring multiple (> 3) antihypertensive medications, plasma renin activity (PRA) and plasma aldosterone concentration (PAC) should be obtained. A low PRA and a PAC > 15 ng/dL, along with a PAC/PRA ratio of > 20, is highly suggestive of an APA.
Q: What is the treatment and follow-up?
Here is a quick reference guide regarding surgical treatment and medical follow-up and surveillance:
• Adrenalectomy (pheochromocytoma, APA, Cushing syndrome): for masses 4 to 6 cm, consider surgery, especially if > 10 Hounsfield units; for masses > 6 cm, there is an increased risk for malignancy and surgery is required.
• Follow-up for low-suspicion, nonsecretory masses: abdominal CT 3 to 6 months after the initial scan, then annually for 1 to 2 years; hormonal evaluation and follow-up annually for 5 years, to evaluate for signs and symptoms of hormonal excess.
SUGGESTED READING
American Association of Clinical Endocrinologists/American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas. Endocr Pract. 2009;15(Suppl 1).
Management of the Clinically Inapparent Adrenal Mass (Incidentaloma). NIH State-of-the-Science Conference Statement; February 4-6, 2002.
Slawik M, Reincke M. Adrenal incidentalomas (Chapter 20). EndoText.com. www.endotext.org/adrenal/adrenal20/adrenal20.htm. Accessed October 12, 2012.
Fitzgerald PA, Goldfien A. Adrenal medulla. In: Greenspan F, Gardner D, eds. Basic and Clinical Endocrinology. 7th ed. McGraw-Hill: 2003;453-473.
The Washington Manual Endocrinology Specialty Consult. 2005;57-61, 71-84.
Endocrine Secrets. 4th ed. 2005;197-204, 241-252, 257-265.
Cleveland Clinic Endocrine & Metabolism Board Review. www.clevelandclinicmeded.com/live/courses/ann/endoreview/default.asp. Accessed October 12, 2012.
| Clinician Reviews in partnership with |
| Clinician Reviews in partnership with |
| Clinician Reviews in partnership with |
Maggie, 42, presents to the emergency department with chronic intermittent abdominal pain and bloating with constipation and occasional diarrhea. She denies fever, chills, nausea, vomiting, melana, bright red blood per rectum, or changes in stool caliper, and she says she otherwise feels well.
Relevant lab and study results include: a comprehensive metabolic panel, complete blood count with differential, beta hCG (human chorionic gonadotropin), urinalysis, and amylase and lipase, all within normal limits; pregnancy test, negative; abdominal x-ray, within normal limits except increased stool in distal colon; and abdominal CT, 2.3-cm right adrenal mass and a Hounsfield measurement of 4 units.
Maggie has a right adrenal incidentaloma (incidentally discovered adenoma that was not in the differential diagnosis). Such findings have become all too often the case, due to the immediate access to and overutilization of high-resolution CT, MRI, and ultrasound. We are now seeing a significantly increased number of incidental adrenal lesions/masses discovered on images not intended to look for adrenal-related diseases (eg, Cushing syndrome, pheochromocytomas, and aldosterone-producing adenomas).
Q: How common are adrenal adenomas, and what must I consider?
Incidental adrenal adenomas are found on 4.4% of abdominal CTs, and in one autopsy series were discovered in 8.7%. Prevalence increases with age, with occurrence of < 1% in patients younger than 30 and about 7% for patients 70 or older.
Evaluation is based on two concerns: First, is the adrenal mass benign or malignant? Second, is the mass secretory or nonsecretory (non-hormone secreting) in nature?
The fortunate news about adrenal incidentalomas is that 80% are benign and nonsecretory, which provides immediate reassuring news to the patient. Examples of benign adrenal masses are: adenoma, lipoma, cyst, ganglioneuroma, hematoma, and infection (eg, tuberculosis, fungal).
The other encouraging statistic is that only 1:4,000 adrenal incidentalomas are malignant. Examples of malignant adrenal masses are: adrenocortical carcinoma, metastatic neoplasm, lymphoma, and malignant pheochromocytoma.
Q: Does adrenal adenoma size matter?
Yes, the larger the size of the adenoma, the higher the association with malignancy. The guide below (based on CT findings) shows not only malignancy potential as it relates to size, but also the importance of Hounsfield units and when surgical intervention is recommended.
Imaging (CT scan)
< 4 cm: homogeneous mass with smooth borders and < 10 Hounsfield units; suggests benign mass (likelihood of malignancy, about 2%)
4 to 6 cm: follow closely, consider surgery (likelihood of malignancy, about 6%)
> 6 cm: surgery indicated (likelihood of malignancy, about 25%)
Some providers and patients inquire whether it is helpful or necessary to biopsy. It is generally not advisable to biopsy, especially if the findings are favorable for benign nonsecretory masses, since there is a high false-negative rate. An indication for biopsy is if the patient has a history of extra-adrenal malignancy; this will distinguish recurrence or metastatic disease from a benign mass. A final proviso: If biopsy is performed, make sure the adrenal mass is not a pheochromocytoma, as biopsy of a hormone-secreting neoplasm can lead to a hypertensive emergency.
Q: How do I determine whether the mass is hormone-secreting?
Although 80% are nonsecretory, you must still maintain a high index of suspicion so as not to miss a potentially problematic and fully treatable adenoma. A thorough history is essential in screening for hormonal excess arising from adrenal adenomas, since the signs and symptoms can be insidious. The three hormones secreted by adrenal adenomas are cortisol, aldosterone, and catecholamines (seen in Cushing syndrome, aldosterone-producing adenoma [APA], and pheochromocytoma, respectively).
It is important to note that Cushing syndrome has an insidious onset and can be easily missed. Hyperaldosteronism presents with hypertension (requiring several medications) and commonly hypokalemia. And pheochromocytoma can be “written off as” anxiety disorder, panic attack, or even hypoglycemia symptoms (especially if patients are treated for diabetes with agents that cause hypoglycemia). To help in your differential diagnosis of secretory adenomas, know that APA accounts for only 1%, and therefore the majority will secrete cortisol and (far less likely) catecholamines.
Q: What is the appropriate laboratory work-up?
The best simple screening test for hypercortisolemia is a 1-mg overnight dexamethasone suppression test. If this value is increased to ≥ 3 µg/dL, it should be followed up with a more sensitive test (a 24-hour urine for creatinine and free cortisol) to further assess for hypercortisolemia.
Patients thought to have a potential pheochromocytoma should undergo measurement of plasma fractionated metanephrines and normetanephrines or 24-hour urine for total metanephrines and fractionated catecholamines.
Finally, for patients with hypokalemia and hypertension or refractory hypertension requiring multiple (> 3) antihypertensive medications, plasma renin activity (PRA) and plasma aldosterone concentration (PAC) should be obtained. A low PRA and a PAC > 15 ng/dL, along with a PAC/PRA ratio of > 20, is highly suggestive of an APA.
Q: What is the treatment and follow-up?
Here is a quick reference guide regarding surgical treatment and medical follow-up and surveillance:
• Adrenalectomy (pheochromocytoma, APA, Cushing syndrome): for masses 4 to 6 cm, consider surgery, especially if > 10 Hounsfield units; for masses > 6 cm, there is an increased risk for malignancy and surgery is required.
• Follow-up for low-suspicion, nonsecretory masses: abdominal CT 3 to 6 months after the initial scan, then annually for 1 to 2 years; hormonal evaluation and follow-up annually for 5 years, to evaluate for signs and symptoms of hormonal excess.
SUGGESTED READING
American Association of Clinical Endocrinologists/American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas. Endocr Pract. 2009;15(Suppl 1).
Management of the Clinically Inapparent Adrenal Mass (Incidentaloma). NIH State-of-the-Science Conference Statement; February 4-6, 2002.
Slawik M, Reincke M. Adrenal incidentalomas (Chapter 20). EndoText.com. www.endotext.org/adrenal/adrenal20/adrenal20.htm. Accessed October 12, 2012.
Fitzgerald PA, Goldfien A. Adrenal medulla. In: Greenspan F, Gardner D, eds. Basic and Clinical Endocrinology. 7th ed. McGraw-Hill: 2003;453-473.
The Washington Manual Endocrinology Specialty Consult. 2005;57-61, 71-84.
Endocrine Secrets. 4th ed. 2005;197-204, 241-252, 257-265.
Cleveland Clinic Endocrine & Metabolism Board Review. www.clevelandclinicmeded.com/live/courses/ann/endoreview/default.asp. Accessed October 12, 2012.
Maggie, 42, presents to the emergency department with chronic intermittent abdominal pain and bloating with constipation and occasional diarrhea. She denies fever, chills, nausea, vomiting, melana, bright red blood per rectum, or changes in stool caliper, and she says she otherwise feels well.
Relevant lab and study results include: a comprehensive metabolic panel, complete blood count with differential, beta hCG (human chorionic gonadotropin), urinalysis, and amylase and lipase, all within normal limits; pregnancy test, negative; abdominal x-ray, within normal limits except increased stool in distal colon; and abdominal CT, 2.3-cm right adrenal mass and a Hounsfield measurement of 4 units.
Maggie has a right adrenal incidentaloma (incidentally discovered adenoma that was not in the differential diagnosis). Such findings have become all too often the case, due to the immediate access to and overutilization of high-resolution CT, MRI, and ultrasound. We are now seeing a significantly increased number of incidental adrenal lesions/masses discovered on images not intended to look for adrenal-related diseases (eg, Cushing syndrome, pheochromocytomas, and aldosterone-producing adenomas).
Q: How common are adrenal adenomas, and what must I consider?
Incidental adrenal adenomas are found on 4.4% of abdominal CTs, and in one autopsy series were discovered in 8.7%. Prevalence increases with age, with occurrence of < 1% in patients younger than 30 and about 7% for patients 70 or older.
Evaluation is based on two concerns: First, is the adrenal mass benign or malignant? Second, is the mass secretory or nonsecretory (non-hormone secreting) in nature?
The fortunate news about adrenal incidentalomas is that 80% are benign and nonsecretory, which provides immediate reassuring news to the patient. Examples of benign adrenal masses are: adenoma, lipoma, cyst, ganglioneuroma, hematoma, and infection (eg, tuberculosis, fungal).
The other encouraging statistic is that only 1:4,000 adrenal incidentalomas are malignant. Examples of malignant adrenal masses are: adrenocortical carcinoma, metastatic neoplasm, lymphoma, and malignant pheochromocytoma.
Q: Does adrenal adenoma size matter?
Yes, the larger the size of the adenoma, the higher the association with malignancy. The guide below (based on CT findings) shows not only malignancy potential as it relates to size, but also the importance of Hounsfield units and when surgical intervention is recommended.
Imaging (CT scan)
< 4 cm: homogeneous mass with smooth borders and < 10 Hounsfield units; suggests benign mass (likelihood of malignancy, about 2%)
4 to 6 cm: follow closely, consider surgery (likelihood of malignancy, about 6%)
> 6 cm: surgery indicated (likelihood of malignancy, about 25%)
Some providers and patients inquire whether it is helpful or necessary to biopsy. It is generally not advisable to biopsy, especially if the findings are favorable for benign nonsecretory masses, since there is a high false-negative rate. An indication for biopsy is if the patient has a history of extra-adrenal malignancy; this will distinguish recurrence or metastatic disease from a benign mass. A final proviso: If biopsy is performed, make sure the adrenal mass is not a pheochromocytoma, as biopsy of a hormone-secreting neoplasm can lead to a hypertensive emergency.
Q: How do I determine whether the mass is hormone-secreting?
Although 80% are nonsecretory, you must still maintain a high index of suspicion so as not to miss a potentially problematic and fully treatable adenoma. A thorough history is essential in screening for hormonal excess arising from adrenal adenomas, since the signs and symptoms can be insidious. The three hormones secreted by adrenal adenomas are cortisol, aldosterone, and catecholamines (seen in Cushing syndrome, aldosterone-producing adenoma [APA], and pheochromocytoma, respectively).
It is important to note that Cushing syndrome has an insidious onset and can be easily missed. Hyperaldosteronism presents with hypertension (requiring several medications) and commonly hypokalemia. And pheochromocytoma can be “written off as” anxiety disorder, panic attack, or even hypoglycemia symptoms (especially if patients are treated for diabetes with agents that cause hypoglycemia). To help in your differential diagnosis of secretory adenomas, know that APA accounts for only 1%, and therefore the majority will secrete cortisol and (far less likely) catecholamines.
Q: What is the appropriate laboratory work-up?
The best simple screening test for hypercortisolemia is a 1-mg overnight dexamethasone suppression test. If this value is increased to ≥ 3 µg/dL, it should be followed up with a more sensitive test (a 24-hour urine for creatinine and free cortisol) to further assess for hypercortisolemia.
Patients thought to have a potential pheochromocytoma should undergo measurement of plasma fractionated metanephrines and normetanephrines or 24-hour urine for total metanephrines and fractionated catecholamines.
Finally, for patients with hypokalemia and hypertension or refractory hypertension requiring multiple (> 3) antihypertensive medications, plasma renin activity (PRA) and plasma aldosterone concentration (PAC) should be obtained. A low PRA and a PAC > 15 ng/dL, along with a PAC/PRA ratio of > 20, is highly suggestive of an APA.
Q: What is the treatment and follow-up?
Here is a quick reference guide regarding surgical treatment and medical follow-up and surveillance:
• Adrenalectomy (pheochromocytoma, APA, Cushing syndrome): for masses 4 to 6 cm, consider surgery, especially if > 10 Hounsfield units; for masses > 6 cm, there is an increased risk for malignancy and surgery is required.
• Follow-up for low-suspicion, nonsecretory masses: abdominal CT 3 to 6 months after the initial scan, then annually for 1 to 2 years; hormonal evaluation and follow-up annually for 5 years, to evaluate for signs and symptoms of hormonal excess.
SUGGESTED READING
American Association of Clinical Endocrinologists/American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas. Endocr Pract. 2009;15(Suppl 1).
Management of the Clinically Inapparent Adrenal Mass (Incidentaloma). NIH State-of-the-Science Conference Statement; February 4-6, 2002.
Slawik M, Reincke M. Adrenal incidentalomas (Chapter 20). EndoText.com. www.endotext.org/adrenal/adrenal20/adrenal20.htm. Accessed October 12, 2012.
Fitzgerald PA, Goldfien A. Adrenal medulla. In: Greenspan F, Gardner D, eds. Basic and Clinical Endocrinology. 7th ed. McGraw-Hill: 2003;453-473.
The Washington Manual Endocrinology Specialty Consult. 2005;57-61, 71-84.
Endocrine Secrets. 4th ed. 2005;197-204, 241-252, 257-265.
Cleveland Clinic Endocrine & Metabolism Board Review. www.clevelandclinicmeded.com/live/courses/ann/endoreview/default.asp. Accessed October 12, 2012.
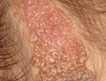
Prevalence of Scalp Disorders and Hair Loss in Children
Aquatic Antagonists: Bluegill (Lepomis macrochirus)
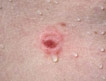
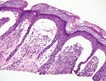
Dermatitis Herpetiformis
Inattention to history dooms patient to repeat it ... Persistent breast lumps but no biopsy ... more
When an atypical presentation is missed
A 50-YEAR-OLD MORBIDLY OBESE MAN went to his family physician with complaints of back pain radiating to the chest, episodic shortness of breath, and diaphoresis. He had a history of uncontrolled high cholesterol. An electrocardiogram showed a Q wave in an inferior lead, which the physician attributed to an old infarct. The doctor didn’t order cardiac enzymes because his office couldn’t do the test.
The physician discharged the patient with a diagnosis of chest pain and a prescription for acetaminophen and hydrocodone. He was scheduled to see a cardiologist in 10 days, but no further cardiology workup was done.
The man died an hour later.
PLAINTIFF’S CLAIM The doctor was negligent in failing to recognize acute coronary syndrome resulting from obstructive coronary artery disease.
THE DEFENSE The patient was discharged in stable condition; cardiac arrest so soon after discharge increased the likelihood that the patient would have suffered sudden cardiac death even if he’d received emergency treatment.
VERDICT $825,000 Virginia settlement.
COMMENT Common, serious problems can present in atypical ways. A high index of suspicion for coronary artery disease in high-risk patients with thoracic pain and shortness of breath—as well as a rapid, thorough evaluation—should keep you out of court (and your patients alive).
Treatment delayed while infection spins out of control
VOMITING, DIARRHEA, AND PAIN AND SWELLING IN THE RIGHT HAND led to an ambulance trip to the emergency department (ED) for a 31-year-old woman. The ED physician diagnosed cellulitis and sepsis. Later that day, the patient was admitted to the intensive care unit, where the admitting physician noted lethargy and confusion, tachycardia, and blueness of the middle and ring fingers on the woman’s right hand. Her medical record suggested that she might have been bitten by a spider.
The patient spent the next 3 days in the ICU in deteriorating condition. She was then transferred to another hospital for treatment of necrotizing fasciitis. She underwent a number of surgeries, including amputation of her right middle and ring fingers, which resulted in significant scarring and deformity of her right hand and forearm.
PLAINTIFF’S CLAIM The defendants were negligent in failing to diagnose necrotizing fasciitis promptly.
THE DEFENSE The defendants who didn’t settle denied any negligence.
VERDICT $80,000 Indiana settlement with the defendant hospital and 1 physician; Indiana defense verdict for the other defendants.
COMMENT When serious infections don’t resolve in a timely manner, expert consultation is imperative.
Inattention to history dooms patient to repeat it
HEADACHES, FEVER, CHILLS, AND JOINT AND MUSCLE PAIN prompted a 42-year-old man to visit his medical group. He told the nurse practitioner (NP) who examined him that his mother had died of a ruptured cerebral aneurysm. The NP diagnosed a viral syndrome, ordered blood tests, and sent the patient home with prescriptions for antibiotics and pain medication. The patient didn’t undergo a neurologic examination.
About 2 weeks later, while continuing to suffer from headaches, the man collapsed and was found unresponsive. A computed tomography scan of his brain showed a subarachnoid hemorrhage and intercerebral hematoma. Further tests revealed a ruptured complex aneurysm, the cause of the hemorrhage. Despite aggressive treatment, the patient fell into a coma and died 3 months later.
PLAINTIFF’S CLAIM The NP should have realized that the patient was at high risk of an aneurysm.
THE DEFENSE No information about the defense is available.
VERDICT $1.5 million New Jersey settlement.
COMMENT I provided expert opinion in a similar case a couple of years ago. The lesson: Pay attention to the family history!
Persistent breast lumps, but no biopsy
ABOUT 3 YEARS AFTER GIVING BIRTH, a 38-year-old woman, who was still breastfeeding, went to her primary care physician complaining of pain, a dime-sized lump in her breast, and discharge from the nipple. The patient’s 2-year-old breast implants limited examination by the nurse practitioner (NP) who saw her. Galactorrhea was diagnosed and the woman was told to stop breastfeeding, apply ice packs, and come back in 2 weeks.
When the patient returned, her only remaining complaint was the lump, which the primary care physician attributed to mastitis. At a routine check-up 5 months later, the patient continued to complain of breast lumps. No breast exam was done, but the woman was referred to a gynecologist. An appointment for a breast ultrasound was scheduled for later in the month, but the patient said she didn’t receive notification of the date.
Metastatic breast cancer was subsequently diagnosed, and the woman died about 3 years later.
PLAINTIFF’S CLAIM The NP and primary care physician should have recommended a biopsy sooner.
THE DEFENSE The care given was proper; an earlier diagnosis wouldn’t have changed the outcome.
VERDICT $750,000 Massachusetts settlement.
COMMENT Failure to recommend biopsy of breast lumps is a leading cause of malpractice cases against family physicians. All persistent breast lumps require referral for biopsy— regardless of the patient’s age.
A red flag that was ignored for too long
A MAN IN HIS EARLY 30S consulted an orthopedist for mid-back pain. The doctor took radiographs of the man’s lower back and reported that he saw nothing amiss. When the man returned 3 months later complaining of the same kind of pain, the orthopedist examined him, prescribed a muscle relaxant, and sent him for physical therapy. The physician did not take any radiographs.
Four months later, the patient came back with pain in his mid-back and ribs. The orthopedist ordered radiographs of the ribs, which were read as normal.
After 18 months, the patient consulted another orthopedist, who ordered a magnetic resonance imaging scan and diagnosed a spinal plasmacytoma at levels T9 to T11. The tumor had destroyed some vertebrae and was compressing the spinal cord.
The patient underwent surgery to remove the tumor and insert screws from T6 to L2 to stabilize the spine. He wore a brace around his torso for months and had a bone marrow transplant. The patient couldn’t return to work.
PLAINTIFF’S CLAIM The tumor was clearly visible on the radiographs taken at the patient’s third visit to the first orthopedist; thoracic spine radiographs should have been taken at the previous 2 visits.
THE DEFENSE No information about the defense is available.
VERDICT $875,000 New Jersey settlement.
COMMENT Current guidelines recommend a red flags approach to imaging. This patient had a red flag—unremitting pain. When back pain persists unabated, it’s time for a thorough evaluation.
When an atypical presentation is missed
A 50-YEAR-OLD MORBIDLY OBESE MAN went to his family physician with complaints of back pain radiating to the chest, episodic shortness of breath, and diaphoresis. He had a history of uncontrolled high cholesterol. An electrocardiogram showed a Q wave in an inferior lead, which the physician attributed to an old infarct. The doctor didn’t order cardiac enzymes because his office couldn’t do the test.
The physician discharged the patient with a diagnosis of chest pain and a prescription for acetaminophen and hydrocodone. He was scheduled to see a cardiologist in 10 days, but no further cardiology workup was done.
The man died an hour later.
PLAINTIFF’S CLAIM The doctor was negligent in failing to recognize acute coronary syndrome resulting from obstructive coronary artery disease.
THE DEFENSE The patient was discharged in stable condition; cardiac arrest so soon after discharge increased the likelihood that the patient would have suffered sudden cardiac death even if he’d received emergency treatment.
VERDICT $825,000 Virginia settlement.
COMMENT Common, serious problems can present in atypical ways. A high index of suspicion for coronary artery disease in high-risk patients with thoracic pain and shortness of breath—as well as a rapid, thorough evaluation—should keep you out of court (and your patients alive).
Treatment delayed while infection spins out of control
VOMITING, DIARRHEA, AND PAIN AND SWELLING IN THE RIGHT HAND led to an ambulance trip to the emergency department (ED) for a 31-year-old woman. The ED physician diagnosed cellulitis and sepsis. Later that day, the patient was admitted to the intensive care unit, where the admitting physician noted lethargy and confusion, tachycardia, and blueness of the middle and ring fingers on the woman’s right hand. Her medical record suggested that she might have been bitten by a spider.
The patient spent the next 3 days in the ICU in deteriorating condition. She was then transferred to another hospital for treatment of necrotizing fasciitis. She underwent a number of surgeries, including amputation of her right middle and ring fingers, which resulted in significant scarring and deformity of her right hand and forearm.
PLAINTIFF’S CLAIM The defendants were negligent in failing to diagnose necrotizing fasciitis promptly.
THE DEFENSE The defendants who didn’t settle denied any negligence.
VERDICT $80,000 Indiana settlement with the defendant hospital and 1 physician; Indiana defense verdict for the other defendants.
COMMENT When serious infections don’t resolve in a timely manner, expert consultation is imperative.
Inattention to history dooms patient to repeat it
HEADACHES, FEVER, CHILLS, AND JOINT AND MUSCLE PAIN prompted a 42-year-old man to visit his medical group. He told the nurse practitioner (NP) who examined him that his mother had died of a ruptured cerebral aneurysm. The NP diagnosed a viral syndrome, ordered blood tests, and sent the patient home with prescriptions for antibiotics and pain medication. The patient didn’t undergo a neurologic examination.
About 2 weeks later, while continuing to suffer from headaches, the man collapsed and was found unresponsive. A computed tomography scan of his brain showed a subarachnoid hemorrhage and intercerebral hematoma. Further tests revealed a ruptured complex aneurysm, the cause of the hemorrhage. Despite aggressive treatment, the patient fell into a coma and died 3 months later.
PLAINTIFF’S CLAIM The NP should have realized that the patient was at high risk of an aneurysm.
THE DEFENSE No information about the defense is available.
VERDICT $1.5 million New Jersey settlement.
COMMENT I provided expert opinion in a similar case a couple of years ago. The lesson: Pay attention to the family history!
Persistent breast lumps, but no biopsy
ABOUT 3 YEARS AFTER GIVING BIRTH, a 38-year-old woman, who was still breastfeeding, went to her primary care physician complaining of pain, a dime-sized lump in her breast, and discharge from the nipple. The patient’s 2-year-old breast implants limited examination by the nurse practitioner (NP) who saw her. Galactorrhea was diagnosed and the woman was told to stop breastfeeding, apply ice packs, and come back in 2 weeks.
When the patient returned, her only remaining complaint was the lump, which the primary care physician attributed to mastitis. At a routine check-up 5 months later, the patient continued to complain of breast lumps. No breast exam was done, but the woman was referred to a gynecologist. An appointment for a breast ultrasound was scheduled for later in the month, but the patient said she didn’t receive notification of the date.
Metastatic breast cancer was subsequently diagnosed, and the woman died about 3 years later.
PLAINTIFF’S CLAIM The NP and primary care physician should have recommended a biopsy sooner.
THE DEFENSE The care given was proper; an earlier diagnosis wouldn’t have changed the outcome.
VERDICT $750,000 Massachusetts settlement.
COMMENT Failure to recommend biopsy of breast lumps is a leading cause of malpractice cases against family physicians. All persistent breast lumps require referral for biopsy— regardless of the patient’s age.
A red flag that was ignored for too long
A MAN IN HIS EARLY 30S consulted an orthopedist for mid-back pain. The doctor took radiographs of the man’s lower back and reported that he saw nothing amiss. When the man returned 3 months later complaining of the same kind of pain, the orthopedist examined him, prescribed a muscle relaxant, and sent him for physical therapy. The physician did not take any radiographs.
Four months later, the patient came back with pain in his mid-back and ribs. The orthopedist ordered radiographs of the ribs, which were read as normal.
After 18 months, the patient consulted another orthopedist, who ordered a magnetic resonance imaging scan and diagnosed a spinal plasmacytoma at levels T9 to T11. The tumor had destroyed some vertebrae and was compressing the spinal cord.
The patient underwent surgery to remove the tumor and insert screws from T6 to L2 to stabilize the spine. He wore a brace around his torso for months and had a bone marrow transplant. The patient couldn’t return to work.
PLAINTIFF’S CLAIM The tumor was clearly visible on the radiographs taken at the patient’s third visit to the first orthopedist; thoracic spine radiographs should have been taken at the previous 2 visits.
THE DEFENSE No information about the defense is available.
VERDICT $875,000 New Jersey settlement.
COMMENT Current guidelines recommend a red flags approach to imaging. This patient had a red flag—unremitting pain. When back pain persists unabated, it’s time for a thorough evaluation.
When an atypical presentation is missed
A 50-YEAR-OLD MORBIDLY OBESE MAN went to his family physician with complaints of back pain radiating to the chest, episodic shortness of breath, and diaphoresis. He had a history of uncontrolled high cholesterol. An electrocardiogram showed a Q wave in an inferior lead, which the physician attributed to an old infarct. The doctor didn’t order cardiac enzymes because his office couldn’t do the test.
The physician discharged the patient with a diagnosis of chest pain and a prescription for acetaminophen and hydrocodone. He was scheduled to see a cardiologist in 10 days, but no further cardiology workup was done.
The man died an hour later.
PLAINTIFF’S CLAIM The doctor was negligent in failing to recognize acute coronary syndrome resulting from obstructive coronary artery disease.
THE DEFENSE The patient was discharged in stable condition; cardiac arrest so soon after discharge increased the likelihood that the patient would have suffered sudden cardiac death even if he’d received emergency treatment.
VERDICT $825,000 Virginia settlement.
COMMENT Common, serious problems can present in atypical ways. A high index of suspicion for coronary artery disease in high-risk patients with thoracic pain and shortness of breath—as well as a rapid, thorough evaluation—should keep you out of court (and your patients alive).
Treatment delayed while infection spins out of control
VOMITING, DIARRHEA, AND PAIN AND SWELLING IN THE RIGHT HAND led to an ambulance trip to the emergency department (ED) for a 31-year-old woman. The ED physician diagnosed cellulitis and sepsis. Later that day, the patient was admitted to the intensive care unit, where the admitting physician noted lethargy and confusion, tachycardia, and blueness of the middle and ring fingers on the woman’s right hand. Her medical record suggested that she might have been bitten by a spider.
The patient spent the next 3 days in the ICU in deteriorating condition. She was then transferred to another hospital for treatment of necrotizing fasciitis. She underwent a number of surgeries, including amputation of her right middle and ring fingers, which resulted in significant scarring and deformity of her right hand and forearm.
PLAINTIFF’S CLAIM The defendants were negligent in failing to diagnose necrotizing fasciitis promptly.
THE DEFENSE The defendants who didn’t settle denied any negligence.
VERDICT $80,000 Indiana settlement with the defendant hospital and 1 physician; Indiana defense verdict for the other defendants.
COMMENT When serious infections don’t resolve in a timely manner, expert consultation is imperative.
Inattention to history dooms patient to repeat it
HEADACHES, FEVER, CHILLS, AND JOINT AND MUSCLE PAIN prompted a 42-year-old man to visit his medical group. He told the nurse practitioner (NP) who examined him that his mother had died of a ruptured cerebral aneurysm. The NP diagnosed a viral syndrome, ordered blood tests, and sent the patient home with prescriptions for antibiotics and pain medication. The patient didn’t undergo a neurologic examination.
About 2 weeks later, while continuing to suffer from headaches, the man collapsed and was found unresponsive. A computed tomography scan of his brain showed a subarachnoid hemorrhage and intercerebral hematoma. Further tests revealed a ruptured complex aneurysm, the cause of the hemorrhage. Despite aggressive treatment, the patient fell into a coma and died 3 months later.
PLAINTIFF’S CLAIM The NP should have realized that the patient was at high risk of an aneurysm.
THE DEFENSE No information about the defense is available.
VERDICT $1.5 million New Jersey settlement.
COMMENT I provided expert opinion in a similar case a couple of years ago. The lesson: Pay attention to the family history!
Persistent breast lumps, but no biopsy
ABOUT 3 YEARS AFTER GIVING BIRTH, a 38-year-old woman, who was still breastfeeding, went to her primary care physician complaining of pain, a dime-sized lump in her breast, and discharge from the nipple. The patient’s 2-year-old breast implants limited examination by the nurse practitioner (NP) who saw her. Galactorrhea was diagnosed and the woman was told to stop breastfeeding, apply ice packs, and come back in 2 weeks.
When the patient returned, her only remaining complaint was the lump, which the primary care physician attributed to mastitis. At a routine check-up 5 months later, the patient continued to complain of breast lumps. No breast exam was done, but the woman was referred to a gynecologist. An appointment for a breast ultrasound was scheduled for later in the month, but the patient said she didn’t receive notification of the date.
Metastatic breast cancer was subsequently diagnosed, and the woman died about 3 years later.
PLAINTIFF’S CLAIM The NP and primary care physician should have recommended a biopsy sooner.
THE DEFENSE The care given was proper; an earlier diagnosis wouldn’t have changed the outcome.
VERDICT $750,000 Massachusetts settlement.
COMMENT Failure to recommend biopsy of breast lumps is a leading cause of malpractice cases against family physicians. All persistent breast lumps require referral for biopsy— regardless of the patient’s age.
A red flag that was ignored for too long
A MAN IN HIS EARLY 30S consulted an orthopedist for mid-back pain. The doctor took radiographs of the man’s lower back and reported that he saw nothing amiss. When the man returned 3 months later complaining of the same kind of pain, the orthopedist examined him, prescribed a muscle relaxant, and sent him for physical therapy. The physician did not take any radiographs.
Four months later, the patient came back with pain in his mid-back and ribs. The orthopedist ordered radiographs of the ribs, which were read as normal.
After 18 months, the patient consulted another orthopedist, who ordered a magnetic resonance imaging scan and diagnosed a spinal plasmacytoma at levels T9 to T11. The tumor had destroyed some vertebrae and was compressing the spinal cord.
The patient underwent surgery to remove the tumor and insert screws from T6 to L2 to stabilize the spine. He wore a brace around his torso for months and had a bone marrow transplant. The patient couldn’t return to work.
PLAINTIFF’S CLAIM The tumor was clearly visible on the radiographs taken at the patient’s third visit to the first orthopedist; thoracic spine radiographs should have been taken at the previous 2 visits.
THE DEFENSE No information about the defense is available.
VERDICT $875,000 New Jersey settlement.
COMMENT Current guidelines recommend a red flags approach to imaging. This patient had a red flag—unremitting pain. When back pain persists unabated, it’s time for a thorough evaluation.
Patient overusing antianxiety meds? Say so (in a letter)
Express your concern about long-term use of benzodiazepines in a letter—a simple intervention that patients often respond to by reducing or eliminating their use of the drug.1
STRENGTH OF RECOMMENDATION
A: Based on a well-done meta-analysis with few clinical trials.
Mugunthan K, McGuire T, Glasziou P. Minimal interventions to decrease long-term use of benzodiazepines in primary care: a systematic review and meta-analysis. Br J Gen Pract. 2011;61:e573-e578.
ILLUSTRATIVE CASE
A 65-year-old patient has been taking lorazepam for insomnia for more than a year. You are concerned about her ongoing use of the benzodiazepine and want to wean her from the medication. What strategies can you use to decrease, or eliminate, her use of the drug?
Benzodiazepines are commonly used medications, with an estimated 12-month prevalence of use of 8.6% in the United States.2 While short-term use of these antianxiety medications can be effective, long-term use (defined as regular use for >3 months) is associated with significant risk.
Abuse linked to chronic use
Prescription drug abuse has recently become the nation’s leading cause of accidental death, overtaking motor vehicle accidents.3 And tranquilizers, including benzodiazepines, are the second most abused prescription medication, after pain relievers.4 In addition to dependence and withdrawal, chronic use of benzodiazepines is associated with daytime somnolence, blunted reflexes, memory loss, cognitive impairment, and an increased risk of falls and fractures—particularly in older patients.5
Reducing long-term use of benzodiazepines in a primary care setting is important but challenging. Until recently, most of the successful strategies reported were resource intensive and required multiple office visits.6
STUDY SUMMARY: Brief interventions are often effective
This study was a meta-analysis of randomized controlled trials in which “minimal interventions” were compared with usual care for their effectiveness in reducing or eliminating benzodiazepine use in primary care patients. A minimal intervention was defined as a letter, self-help information, or short consultation with a primary care provider. In each case, the message to the patient included (a) an expression of concern about the patient’s long-term use of the medication, (b) information about the potential adverse effects of the medication, and (c) advice on how to gradually reduce or stop using it.
Three studies met the inclusion criteria for randomization, blinding, and analysis by intention-to-treat.7-9 All 3 (n=615) had a 6-month follow-up period, a higher proportion of women (>60%), and participants with a mean age >60 years. Few patients were lost to follow-up; withdrawal rates were low and similar in all 3 studies. Each study compared a letter with usual care; 2 of the 3 had a third arm that included both a letter and a short consultation.
Pooled results from the studies showed twice the reduction in benzodiazepine use in the intervention groups compared with the control groups (risk ratio [RR]=2.04; 95% confidence interval [CI], 1.5-2.8; P< .001). The RR for cessation of benzodiazepine use was 2.3 (95% CI, 1.3-4.2; P= .003). The number needed to treat for a reduction or cessation of use was 12. The studies reported benzodiazepine reduction rates of 20% to 35% in the intervention groups vs 6% to 15% in the usual care groups.7-9 There appeared to be no additional benefit to adding the brief consultation compared with the letter alone.
WHAT’S NEW?: This strategy is easy to implement
While many methods to reduce benzodiazepine use have been studied, most involved levels of skill and resources that are not feasible for widespread use. This study found that a letter, stating the risks of continued use of the medication and providing a weaning schedule and tips for handling withdrawal, can be effective in reducing chronic use in a small but significant part of the population.
CAVEATS: Effects of withdrawal went unaddressed
The study did not adequately address the adverse effects of withdrawal from benzodiazepines, with one of the studies reporting significantly worse qualitative (but not quantitative) withdrawal symptoms at 6 months.7 This is of particular concern, as withdrawal symptoms are associated with the potential for relapse and concomitant abuse of other drugs and alcohol. We recommend that primary care physicians screen for substance abuse prior to the intervention and arrange for adequate follow-up.
All 3 studies in the meta-analysis lasted 6 months; no longer-term outcomes were reported. In addition, the study did not yield enough information to identify patients who would be most likely to respond to this brief intervention.
CHALLENGES TO IMPLEMENTATION: Determining which patients to target
Identifying patients who are appropriate candidates for this brief intervention and providing adequate monitoring for adverse effects of withdrawal are the main challenges of this practice changer. Nonetheless, chronic benzodiazepine use is of considerable concern, and we believe that this is a useful, and manageable intervention.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
1. Mugunthan K, McGuire T, Glasziou P. Minimal interventions to decrease long-term use of benzodiazepines in primary care: a systematic review and meta-analysis. Br J Gen Pract. 2011;61:e573-e578.
2. Tyrer PJ. Benzodiazepines on trial. Br Med J. 1984;288:1101-1102.
3. Centers for Disease Control and Prevention. Deaths: Leading causes for 2008. June 6, 2012. Available at: http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_06.pdf. Accessed October 10, 2012.
4. National Institute on Drug Abuse. Topics in brief: Prescription drug abuse. Available at: http://www.drugabuse.gov/publications/topics-in-brief/prescription-drug-abuse. Accessed October 11, 2012.
5. Morin CM, Bastien C, Guay B, et al. Randomized clinical trail of supervised tapering and cognitive behavior therapy to facilitate benzodiazepine discontinuation in older adults with chronic insomnia. Am J Psychiatry. 2004;161:332-342.
6. Oude Voshaar RC, Couvee JE, van Balkorn AJ, et al. Strategies for discontinuing long-term benzodiazepine use-meta-analysis. Br J Psychiatr. 2006;189:213-220.
7. Bashir K, King M, Ashworth M. Controlled evaluation of brief intervention by general practitioners to reduce chronic use of benzodiazepines. Br J Gen Pract. 1994;44:408-412.
8. Cormack MA, Sweeney KG, Hughes-Jones H, et al. Evaluation of an easy, cost-effective strategy to cut benzodiazepine use in general practice. Br J Gen Pract. 1994;44:5-8
9. Heather NA, Bowie A, Ashton H, et al. Randomized controlled trial of two brief interventions against long-term benzodiazepine use: outcome of intervention. Addict Res Theory. 2004;12:141-145.
Express your concern about long-term use of benzodiazepines in a letter—a simple intervention that patients often respond to by reducing or eliminating their use of the drug.1
STRENGTH OF RECOMMENDATION
A: Based on a well-done meta-analysis with few clinical trials.
Mugunthan K, McGuire T, Glasziou P. Minimal interventions to decrease long-term use of benzodiazepines in primary care: a systematic review and meta-analysis. Br J Gen Pract. 2011;61:e573-e578.
ILLUSTRATIVE CASE
A 65-year-old patient has been taking lorazepam for insomnia for more than a year. You are concerned about her ongoing use of the benzodiazepine and want to wean her from the medication. What strategies can you use to decrease, or eliminate, her use of the drug?
Benzodiazepines are commonly used medications, with an estimated 12-month prevalence of use of 8.6% in the United States.2 While short-term use of these antianxiety medications can be effective, long-term use (defined as regular use for >3 months) is associated with significant risk.
Abuse linked to chronic use
Prescription drug abuse has recently become the nation’s leading cause of accidental death, overtaking motor vehicle accidents.3 And tranquilizers, including benzodiazepines, are the second most abused prescription medication, after pain relievers.4 In addition to dependence and withdrawal, chronic use of benzodiazepines is associated with daytime somnolence, blunted reflexes, memory loss, cognitive impairment, and an increased risk of falls and fractures—particularly in older patients.5
Reducing long-term use of benzodiazepines in a primary care setting is important but challenging. Until recently, most of the successful strategies reported were resource intensive and required multiple office visits.6
STUDY SUMMARY: Brief interventions are often effective
This study was a meta-analysis of randomized controlled trials in which “minimal interventions” were compared with usual care for their effectiveness in reducing or eliminating benzodiazepine use in primary care patients. A minimal intervention was defined as a letter, self-help information, or short consultation with a primary care provider. In each case, the message to the patient included (a) an expression of concern about the patient’s long-term use of the medication, (b) information about the potential adverse effects of the medication, and (c) advice on how to gradually reduce or stop using it.
Three studies met the inclusion criteria for randomization, blinding, and analysis by intention-to-treat.7-9 All 3 (n=615) had a 6-month follow-up period, a higher proportion of women (>60%), and participants with a mean age >60 years. Few patients were lost to follow-up; withdrawal rates were low and similar in all 3 studies. Each study compared a letter with usual care; 2 of the 3 had a third arm that included both a letter and a short consultation.
Pooled results from the studies showed twice the reduction in benzodiazepine use in the intervention groups compared with the control groups (risk ratio [RR]=2.04; 95% confidence interval [CI], 1.5-2.8; P< .001). The RR for cessation of benzodiazepine use was 2.3 (95% CI, 1.3-4.2; P= .003). The number needed to treat for a reduction or cessation of use was 12. The studies reported benzodiazepine reduction rates of 20% to 35% in the intervention groups vs 6% to 15% in the usual care groups.7-9 There appeared to be no additional benefit to adding the brief consultation compared with the letter alone.
WHAT’S NEW?: This strategy is easy to implement
While many methods to reduce benzodiazepine use have been studied, most involved levels of skill and resources that are not feasible for widespread use. This study found that a letter, stating the risks of continued use of the medication and providing a weaning schedule and tips for handling withdrawal, can be effective in reducing chronic use in a small but significant part of the population.
CAVEATS: Effects of withdrawal went unaddressed
The study did not adequately address the adverse effects of withdrawal from benzodiazepines, with one of the studies reporting significantly worse qualitative (but not quantitative) withdrawal symptoms at 6 months.7 This is of particular concern, as withdrawal symptoms are associated with the potential for relapse and concomitant abuse of other drugs and alcohol. We recommend that primary care physicians screen for substance abuse prior to the intervention and arrange for adequate follow-up.
All 3 studies in the meta-analysis lasted 6 months; no longer-term outcomes were reported. In addition, the study did not yield enough information to identify patients who would be most likely to respond to this brief intervention.
CHALLENGES TO IMPLEMENTATION: Determining which patients to target
Identifying patients who are appropriate candidates for this brief intervention and providing adequate monitoring for adverse effects of withdrawal are the main challenges of this practice changer. Nonetheless, chronic benzodiazepine use is of considerable concern, and we believe that this is a useful, and manageable intervention.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Express your concern about long-term use of benzodiazepines in a letter—a simple intervention that patients often respond to by reducing or eliminating their use of the drug.1
STRENGTH OF RECOMMENDATION
A: Based on a well-done meta-analysis with few clinical trials.
Mugunthan K, McGuire T, Glasziou P. Minimal interventions to decrease long-term use of benzodiazepines in primary care: a systematic review and meta-analysis. Br J Gen Pract. 2011;61:e573-e578.
ILLUSTRATIVE CASE
A 65-year-old patient has been taking lorazepam for insomnia for more than a year. You are concerned about her ongoing use of the benzodiazepine and want to wean her from the medication. What strategies can you use to decrease, or eliminate, her use of the drug?
Benzodiazepines are commonly used medications, with an estimated 12-month prevalence of use of 8.6% in the United States.2 While short-term use of these antianxiety medications can be effective, long-term use (defined as regular use for >3 months) is associated with significant risk.
Abuse linked to chronic use
Prescription drug abuse has recently become the nation’s leading cause of accidental death, overtaking motor vehicle accidents.3 And tranquilizers, including benzodiazepines, are the second most abused prescription medication, after pain relievers.4 In addition to dependence and withdrawal, chronic use of benzodiazepines is associated with daytime somnolence, blunted reflexes, memory loss, cognitive impairment, and an increased risk of falls and fractures—particularly in older patients.5
Reducing long-term use of benzodiazepines in a primary care setting is important but challenging. Until recently, most of the successful strategies reported were resource intensive and required multiple office visits.6
STUDY SUMMARY: Brief interventions are often effective
This study was a meta-analysis of randomized controlled trials in which “minimal interventions” were compared with usual care for their effectiveness in reducing or eliminating benzodiazepine use in primary care patients. A minimal intervention was defined as a letter, self-help information, or short consultation with a primary care provider. In each case, the message to the patient included (a) an expression of concern about the patient’s long-term use of the medication, (b) information about the potential adverse effects of the medication, and (c) advice on how to gradually reduce or stop using it.
Three studies met the inclusion criteria for randomization, blinding, and analysis by intention-to-treat.7-9 All 3 (n=615) had a 6-month follow-up period, a higher proportion of women (>60%), and participants with a mean age >60 years. Few patients were lost to follow-up; withdrawal rates were low and similar in all 3 studies. Each study compared a letter with usual care; 2 of the 3 had a third arm that included both a letter and a short consultation.
Pooled results from the studies showed twice the reduction in benzodiazepine use in the intervention groups compared with the control groups (risk ratio [RR]=2.04; 95% confidence interval [CI], 1.5-2.8; P< .001). The RR for cessation of benzodiazepine use was 2.3 (95% CI, 1.3-4.2; P= .003). The number needed to treat for a reduction or cessation of use was 12. The studies reported benzodiazepine reduction rates of 20% to 35% in the intervention groups vs 6% to 15% in the usual care groups.7-9 There appeared to be no additional benefit to adding the brief consultation compared with the letter alone.
WHAT’S NEW?: This strategy is easy to implement
While many methods to reduce benzodiazepine use have been studied, most involved levels of skill and resources that are not feasible for widespread use. This study found that a letter, stating the risks of continued use of the medication and providing a weaning schedule and tips for handling withdrawal, can be effective in reducing chronic use in a small but significant part of the population.
CAVEATS: Effects of withdrawal went unaddressed
The study did not adequately address the adverse effects of withdrawal from benzodiazepines, with one of the studies reporting significantly worse qualitative (but not quantitative) withdrawal symptoms at 6 months.7 This is of particular concern, as withdrawal symptoms are associated with the potential for relapse and concomitant abuse of other drugs and alcohol. We recommend that primary care physicians screen for substance abuse prior to the intervention and arrange for adequate follow-up.
All 3 studies in the meta-analysis lasted 6 months; no longer-term outcomes were reported. In addition, the study did not yield enough information to identify patients who would be most likely to respond to this brief intervention.
CHALLENGES TO IMPLEMENTATION: Determining which patients to target
Identifying patients who are appropriate candidates for this brief intervention and providing adequate monitoring for adverse effects of withdrawal are the main challenges of this practice changer. Nonetheless, chronic benzodiazepine use is of considerable concern, and we believe that this is a useful, and manageable intervention.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
1. Mugunthan K, McGuire T, Glasziou P. Minimal interventions to decrease long-term use of benzodiazepines in primary care: a systematic review and meta-analysis. Br J Gen Pract. 2011;61:e573-e578.
2. Tyrer PJ. Benzodiazepines on trial. Br Med J. 1984;288:1101-1102.
3. Centers for Disease Control and Prevention. Deaths: Leading causes for 2008. June 6, 2012. Available at: http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_06.pdf. Accessed October 10, 2012.
4. National Institute on Drug Abuse. Topics in brief: Prescription drug abuse. Available at: http://www.drugabuse.gov/publications/topics-in-brief/prescription-drug-abuse. Accessed October 11, 2012.
5. Morin CM, Bastien C, Guay B, et al. Randomized clinical trail of supervised tapering and cognitive behavior therapy to facilitate benzodiazepine discontinuation in older adults with chronic insomnia. Am J Psychiatry. 2004;161:332-342.
6. Oude Voshaar RC, Couvee JE, van Balkorn AJ, et al. Strategies for discontinuing long-term benzodiazepine use-meta-analysis. Br J Psychiatr. 2006;189:213-220.
7. Bashir K, King M, Ashworth M. Controlled evaluation of brief intervention by general practitioners to reduce chronic use of benzodiazepines. Br J Gen Pract. 1994;44:408-412.
8. Cormack MA, Sweeney KG, Hughes-Jones H, et al. Evaluation of an easy, cost-effective strategy to cut benzodiazepine use in general practice. Br J Gen Pract. 1994;44:5-8
9. Heather NA, Bowie A, Ashton H, et al. Randomized controlled trial of two brief interventions against long-term benzodiazepine use: outcome of intervention. Addict Res Theory. 2004;12:141-145.
1. Mugunthan K, McGuire T, Glasziou P. Minimal interventions to decrease long-term use of benzodiazepines in primary care: a systematic review and meta-analysis. Br J Gen Pract. 2011;61:e573-e578.
2. Tyrer PJ. Benzodiazepines on trial. Br Med J. 1984;288:1101-1102.
3. Centers for Disease Control and Prevention. Deaths: Leading causes for 2008. June 6, 2012. Available at: http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_06.pdf. Accessed October 10, 2012.
4. National Institute on Drug Abuse. Topics in brief: Prescription drug abuse. Available at: http://www.drugabuse.gov/publications/topics-in-brief/prescription-drug-abuse. Accessed October 11, 2012.
5. Morin CM, Bastien C, Guay B, et al. Randomized clinical trail of supervised tapering and cognitive behavior therapy to facilitate benzodiazepine discontinuation in older adults with chronic insomnia. Am J Psychiatry. 2004;161:332-342.
6. Oude Voshaar RC, Couvee JE, van Balkorn AJ, et al. Strategies for discontinuing long-term benzodiazepine use-meta-analysis. Br J Psychiatr. 2006;189:213-220.
7. Bashir K, King M, Ashworth M. Controlled evaluation of brief intervention by general practitioners to reduce chronic use of benzodiazepines. Br J Gen Pract. 1994;44:408-412.
8. Cormack MA, Sweeney KG, Hughes-Jones H, et al. Evaluation of an easy, cost-effective strategy to cut benzodiazepine use in general practice. Br J Gen Pract. 1994;44:5-8
9. Heather NA, Bowie A, Ashton H, et al. Randomized controlled trial of two brief interventions against long-term benzodiazepine use: outcome of intervention. Addict Res Theory. 2004;12:141-145.
Copyright © 2012 The Family Physicians Inquiries Network. All rights reserved.
Let’s put a stop to the prescribing cascade
I am delighted by the commonsense approach Drs. Weiss and Lee have taken in advising us to be wary of prescribing—or continuing—too many medications for our older patients (“Is your patient taking too many pills?”). Frankly, this advice applies to all patients, regardless of their age, and to virtually all family physicians. We all have stories about medication overuse. I’d like to tell you 2 of mine.
When Mrs. S, a 68-year-old patient, came to see me for the first time, I scanned her medication list. It included a nasal steroid for allergic rhinitis, a PPI for reflux, and 2 asthma inhalers—albuterol and an inhaled corticosteroid.
I asked her if she had hay fever. She didn’t think so. Heartburn? She said No. A history of asthma? No. So why was she taking these medications? To treat a chronic cough, the patient said. Was the cough better? No.
In the past 12 months, Mrs. S had seen an allergist, a gastroenterologist, and an otolaryngologist. The result? All 3 specialists added their favorite medication. I scanned the patient’s medication list again and noticed that she was taking amitriptyline 25 mg as a sleep aid. Because of the drug’s anticholinergic adverse effects, I had a hunch, and asked her to go one week without the amitriptyline. She agreed.
You can guess the happy ending. Mrs. S’s cough vanished, along with 4 medications she never needed in the first place. She was a victim of the prescribing cascade.
The other story is even more dramatic.
A friend who’s both an FP and a geriatrician became medical director of a local nursing home. To his chagrin, the average number of prescription drugs per resident when he took over was 9.6. Systematically, he went about reevaluating what residents really required. After a year and a half, the average had fallen to 5.4. The residents were no more depressed or agitated, and were generally more alert.
But here’s the catch: I checked back at the nursing home a couple of years after my friend left, and the average number of meds was back up to 10. It takes constant attention to not overprescribe. In fact, I now spend about as much time stopping meds as starting them.
Our health care system is the land of excess. It is up to family physicians—indeed, to all primary care clinicians—to ensure that we only prescribe or continue prescriptions when it’s the right patient, the right medication, at the right time.
Now it’s your turn. Send me your favorite, or most dramatic, medication overtreatment stories for our Letters column. We’ll continue the dialogue there.

John Hickner, MD, MSc
Editor-in-Chief
[email protected]
John Hickner, MD, MSc
Editor-in-Chief
[email protected]
John Hickner, MD, MSc
Editor-in-Chief
[email protected]
I am delighted by the commonsense approach Drs. Weiss and Lee have taken in advising us to be wary of prescribing—or continuing—too many medications for our older patients (“Is your patient taking too many pills?”). Frankly, this advice applies to all patients, regardless of their age, and to virtually all family physicians. We all have stories about medication overuse. I’d like to tell you 2 of mine.
When Mrs. S, a 68-year-old patient, came to see me for the first time, I scanned her medication list. It included a nasal steroid for allergic rhinitis, a PPI for reflux, and 2 asthma inhalers—albuterol and an inhaled corticosteroid.
I asked her if she had hay fever. She didn’t think so. Heartburn? She said No. A history of asthma? No. So why was she taking these medications? To treat a chronic cough, the patient said. Was the cough better? No.
In the past 12 months, Mrs. S had seen an allergist, a gastroenterologist, and an otolaryngologist. The result? All 3 specialists added their favorite medication. I scanned the patient’s medication list again and noticed that she was taking amitriptyline 25 mg as a sleep aid. Because of the drug’s anticholinergic adverse effects, I had a hunch, and asked her to go one week without the amitriptyline. She agreed.
You can guess the happy ending. Mrs. S’s cough vanished, along with 4 medications she never needed in the first place. She was a victim of the prescribing cascade.
The other story is even more dramatic.
A friend who’s both an FP and a geriatrician became medical director of a local nursing home. To his chagrin, the average number of prescription drugs per resident when he took over was 9.6. Systematically, he went about reevaluating what residents really required. After a year and a half, the average had fallen to 5.4. The residents were no more depressed or agitated, and were generally more alert.
But here’s the catch: I checked back at the nursing home a couple of years after my friend left, and the average number of meds was back up to 10. It takes constant attention to not overprescribe. In fact, I now spend about as much time stopping meds as starting them.
Our health care system is the land of excess. It is up to family physicians—indeed, to all primary care clinicians—to ensure that we only prescribe or continue prescriptions when it’s the right patient, the right medication, at the right time.
Now it’s your turn. Send me your favorite, or most dramatic, medication overtreatment stories for our Letters column. We’ll continue the dialogue there.
I am delighted by the commonsense approach Drs. Weiss and Lee have taken in advising us to be wary of prescribing—or continuing—too many medications for our older patients (“Is your patient taking too many pills?”). Frankly, this advice applies to all patients, regardless of their age, and to virtually all family physicians. We all have stories about medication overuse. I’d like to tell you 2 of mine.
When Mrs. S, a 68-year-old patient, came to see me for the first time, I scanned her medication list. It included a nasal steroid for allergic rhinitis, a PPI for reflux, and 2 asthma inhalers—albuterol and an inhaled corticosteroid.
I asked her if she had hay fever. She didn’t think so. Heartburn? She said No. A history of asthma? No. So why was she taking these medications? To treat a chronic cough, the patient said. Was the cough better? No.
In the past 12 months, Mrs. S had seen an allergist, a gastroenterologist, and an otolaryngologist. The result? All 3 specialists added their favorite medication. I scanned the patient’s medication list again and noticed that she was taking amitriptyline 25 mg as a sleep aid. Because of the drug’s anticholinergic adverse effects, I had a hunch, and asked her to go one week without the amitriptyline. She agreed.
You can guess the happy ending. Mrs. S’s cough vanished, along with 4 medications she never needed in the first place. She was a victim of the prescribing cascade.
The other story is even more dramatic.
A friend who’s both an FP and a geriatrician became medical director of a local nursing home. To his chagrin, the average number of prescription drugs per resident when he took over was 9.6. Systematically, he went about reevaluating what residents really required. After a year and a half, the average had fallen to 5.4. The residents were no more depressed or agitated, and were generally more alert.
But here’s the catch: I checked back at the nursing home a couple of years after my friend left, and the average number of meds was back up to 10. It takes constant attention to not overprescribe. In fact, I now spend about as much time stopping meds as starting them.
Our health care system is the land of excess. It is up to family physicians—indeed, to all primary care clinicians—to ensure that we only prescribe or continue prescriptions when it’s the right patient, the right medication, at the right time.
Now it’s your turn. Send me your favorite, or most dramatic, medication overtreatment stories for our Letters column. We’ll continue the dialogue there.
AGING: Are these 4 pain myths complicating care?
Beliefs about aging itself can also have dramatic consequences, both positive and negative. In one longitudinal study, those who had positive self-perceptions of aging when they were 50 had better health during 2 decades of follow-up and lived, on average, 7½ years longer than those who had negative self-perceptions at the age of 50.4
Although little research has focused specifically on pain-related stereotypes held by older adults, their importance has long been recognized.
Twenty years ago, a review found that the failure to incorporate older patients’ beliefs about pain could have a negative effect on pain management.5 And in 2011, an Institute of Medicine report found a critical need for public education to counter the myths, misunderstandings, stereotypes, and stigma that hinder pain management in patients across the lifespan.6
We set out to identify widely held stereotypes that older adults and physicians have about pain—and to report on primary studies that support or refute them. We focused on noncancer pain. In the pages that follow, we identify 4 key stereotypes that misrepresent the experience of older adults with regard to pain, and present evidence to debunk them.
Stereotype #1: Pain is a natural part of getting older
Chronic pain is often perceived as an age-related condition. In in-depth interviews, older adults with osteoarthritis reported pain as a normal, even essential, part of life. As one patient put it, “That’s how you know you’re alive … you ache.”7
Among primary care patients with osteoarthritis, those older than 70 years were more likely than younger patients to believe that people should expect to live with pain as they get older.8 And more than half of older adults who responded to a community-based survey considered arthritis to be a natural part of getting old.9
Physicians, too, often view pain as an inevitable part of the aging process, giving patients feedback such as “What do you expect? You’re just getting older.”10
Are they right?
Is pain inevitable? No
In fact, chronic pain is common in older adults, occurring in more than half of those assessed, according to some studies.11 In addition, some epidemiological studies have found an age-related increase in the prevalence of pain,12-14 with older age predicting a more likely onset of, and failure to recover from, persistent pain.15 But numerous studies have failed to find a direct relationship between pain and age.
A National Center for Health Statistics report found that 29% of adults between the ages of 45 and 64 years vs 21% of those 65 or older reported pain lasting >24 hours in the month before the survey.16 And a meta-analysis comparing age-related differences in pain perception found that the highest prevalence of chronic pain occurred at about age 65; a slight decline with advancing age followed, even beyond the age of 85.17
Chronic pain disorders are less frequent. In fact, many chronic pain disorders occur less frequently with advancing age. Population-based studies have found a lower prevalence of low back, neck, and face pain among older adults compared with their younger counterparts;16 evidence has also found lower rates of headache and abdominal pain.18 Other epidemiological studies suggest that the prevalence of musculoskeletal pain generally declines with advancing age,19 and a study of patients in their last 2 years of life found pain to be inversely correlated with age.20 These findings refute the stereotype that advancing age inexorably involves pain, and challenge the notion that pain in later life is normal and expected, and unworthy of treatment.
Stereotype #2: Pain worsens
over time
Some patients and physicians expect that as people age, their pain will increase in intensity. In one study of community-dwelling older adults, 87% of those surveyed rated the belief that more aches and pains are an accepted part of aging as definitely or somewhat true.21 Indeed, patients of all ages have expressed the belief that older age confers greater susceptibility to, and suffering from, painful conditions like arthritis.22 Many common causes of pain in older adults, especially osteoarthritis, are seen as resulting from degenerative changes, which worsen over time.23
Does pain intensify? Not necessarily
Some studies have linked older age to a worse prognosis for patients with musculoskeletal pain, but a greater number have found that aging has no effect on it.24
Pain does not always progress. In a large cohort of patients with peripheral joint osteoarthritis, radiographic joint space narrowing worsened over 3 years, but this did not correlate consistently with worsening pain.25 When the same cohort was assessed after 8 years, there was significant variability in pain, with no clear progression.26
In another study involving older patients with restrictive back pain, the pain was frequently short-lived and episodic and did not increase with age.27 And in a population sample in Norway, the mean number of pain sites decreased slightly over 14 years in those older than 60 years, while increasing in those aged 44 to 60.28 Another study of patients with knee osteoarthritis identified factors that were protective against a decline in pain-related function: These included good mental health, self-efficacy, social support, and greater activity—but not younger age.29 The enormous heterogeneity in both the experience and the course of pain suggests that age-related pain progression is neither universal nor expected—and contradicts a purely biological paradigm in which pain inevitably worsens over time.
Stereotype #3: Stoicism leads to pain tolerance
Some patients believe that the inability to deal with pain is a sign of being soft or weak, and that a “tough it out” approach makes pain easier to tolerate.7 In one survey, older adults were more likely than their younger counterparts to express such stoicism, frequently agreeing with statements like, “I maintain my pride and keep a stiff upper lip when in pain,” “I go on as if nothing had happened …,” and “Pain is something that should be ignored.” 30
Unfortunately, some physicians reinforce such attitudes, telling older patients, in effect, that they’d better “get used to it.”10 And family and friends may make it worse. Patients taking opioids reported that it wasn’t unusual for those close to them to view their use of these analgesics as a sign of weakness.31
Does stoicism help? Probably not
Older adults seem less likely than younger adults to label a sensation as painful, suggesting a more stoic approach in general.30 While some research has found that nociception—the perception of pain in response to painful stimuli—decreases with advancing age,32 other studies have found the opposite.33 And population-based studies focusing on the consequences of pain indicate that it continues to have powerful negative effects, especially depression and insomnia, in older patients.
The degree of pain experienced is more strongly associated with depression in older patients compared with younger adults,34 and greater pain reduces the likelihood that depression will improve with treatment.35 Pain also continues to interfere with sleep. In one national sample, 25% of those with arthritis said they suffered from insomnia, roughly twice the prevalence of insomnia found in those without arthritis.36 In another study, individuals with arthritis were 3 times more likely to have sleep problems compared with individuals without arthritis37—an association independent of age. Being stoic about pain, it appears, does not diminish its consequences over time or help patients better tolerate it.
Stereotype #4: Prescription analgesics are highly addictive
Patients often think that prescription analgesics, especially opioids, are highly addictive or harmful—and older adults may refuse to take them for fear of becoming addicted.7 The stereotype is often shared by family and friends, as well as clinicians.
In one study, one-third of physicians said they hesitated to prescribe opioid medications to older adults because of the risk of addiction (a concern that no clinician with training in geriatrics shared).38 What’s more, 16% of the physicians estimated that about one in 4 older patients receiving chronic opioid therapy becomes addicted. The actual risk is far lower. (More on that below.) News reports of an epidemic of prescription opioid addictions and fatalities,39 including the assertion that opioids are replacing heroin as the primary drug of choice on the street,40 may reinforce such stereotypes.
How great is the risk of addiction? For older adults, it’s very low
While rates of aberrant opioid use vary widely depending on the context, one consistent theme is that older age is associated with decreased risk.41 In one retrospective cohort study of older patients who had recently been started on an opioid medication for the treatment of chronic pain, only 3% showed evidence of behaviors associated with abuse or misuse.42
What’s more, long-term opioid use among older patients with painful conditions is relatively uncommon, and prescription patterns suggest that most older adults discontinue opioids after one or 2 prescriptions.42-44 Decades of research have found that, although opioid medications can cause physiological dependence, addiction is rare in patients treated with them.45,46 (To learn more, see “Diagnosing and treating opioid dependence,” J Fam Pract. 2012;61: 588-597.)
Debunking myths: Implications for practice
Our findings—that pain is not a natural part of aging and often improves or remains stable over time, stoicism does not lead to acclimation, and pain medications are not highly addictive in older adults—make it clear that the stereotypes we identified are misconceptions of pain in later life. Debunking these stereotypes has several implications for clinical practice. We recommend the following:
Identify and counter these stereotypes. Avoid reinforcing stereotypes; counter them by summarizing these evidence-based findings for older patients. We believe patients would be receptive.
In one study, more than 80% of patients with osteoarthritis said they wanted prognostic information about the course of the disease, but only about one-third had received it.47 Presenting the research findings would challenge patients’ stereotypes and help them reframe their expectations.
Elicit patients’ perspectives. Ask patients about age- and pain-related stereotypes and their expectations and perspectives of what constitutes successful treatment. Research shows that patients often wish to discuss lifestyle changes and nonmedical approaches to pain, for example, but that clinicians typically focus on medications instead.48
Emphasize the positive. Frame discussions of pain and aging in a positive light, offering encouragement rather than supporting stoicism or resignation. Attention to protective factors, including good mental health, self-efficacy, social support, and greater activity, may enable older patients to adapt better to any pain they experience.
CORRESPONDENCE
Stephen Thielke, MD, MSPH, MA, University of Washington, Psychiatry and Behavioral Sciences, Box 356560, Seattle, WA 98195; [email protected]
1. Herr K. Pain in the older adult: an imperative across all health care settings. Pain Manag Nurs. 2010;11(2 suppl):S1-S10.
2. Pitkala KH, Strandberg TE, Tilvis RS. Management of nonmalignant pain in home-dwelling older people: a population-based survey. J Am Geriatr Soc. 2002;50:1861-1865.
3. Levy B. Stereotype embodiment: a psychosocial approach to aging. Curr Dir Psychol Sci. 2009;18:332-336.
4. Levy BR, Slade MD, Kasl SV. Longitudinal benefit of positive self-perceptions of aging on functional health. J Gerontol B Psychol Sci Soc Sci. 2002;57:409-417.
5. Hofland SL. Elder beliefs: blocks to pain management. J Gerontol Nurs. 1992;18:19-23.
6. Institute of Medicine. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: National Academies Press; 2011.
7. Sale J, Gignac M, Hawker G. How “bad” does the pain have to be? A qualitative study examining adherence to pain medication in older adults with osteoarthritis. Arthritis Rheum. 2006;55:272-278.
8. Appelt CJ, Burant BC, Siminoff LA, et al. Health beliefs related to aging among older male patients with knee and/or hip osteoarthritis. J Gerontol A Biol Sci Med Sci. 2007;62:184-190.
9. Goodwin JS, Black SA, Satish S. Aging versus disease: the opinions of older black, Hispanic, and non-Hispanic white Americans about the causes and treatment of common medical conditions. J Am Geriatr Soc. 1999;47:973-979.
10. Gignac M, Davis A, Hawker G, et al. “What do you expect? You’re just getting older”: a comparison of perceived osteoarthritis-related and aging-related health experiences in middle- and older-age adults. Arthritis Rheum. 2006;55:905-912.
11. Helme RD, Gibson SJ. Pain in the elderly. In: Jensen TS, Turner JA, Weisenfeld-Hallin Z, eds. Progress in Pain Research and Management. Proceedings of the 8th World Congress on Pain. Vol 8. Seattle, Wash: IASP Press; 1997:919–944.
12. Badley EM, Tennant A. Changing profile of joint disorders with age: findings from a postal survey of the population of Calderdale, West Yorkshire, United Kingdom. Ann Rheumatic Dis. 1992;51:366-371.
13. Brattberg G, Parker MG, Thorslund M. A longitudinal study of pain: reported pain from middle age to old age. Clin J Pain. 1997;13:144-149.
14. Crook J, Rideout E, Browne G. The prevalence of pain complaints in a general population. Pain. 1984;18:299-314.
15. Gureje O, Simon GE, Von Korff M. A cross-national study of the course of persistent pain in primary care. Pain. 2001;92:195-200.
16. National Center for Health Statistics. Special feature: pain. In: Health, United States, 2006 with Chartbook on Trends in the Health of Americans. Hyattsville, Md: Centers for Disease Control and Prevention; 2006:68–87. Available at: http://www.cdc.gov/nchs/data/hus/hus06.pdf. Accessed October 16, 2012.
17. Gibson SJ, Helme RD. Age differences in pain perception and report: a review of physiological, psychological, laboratory and clinical studies. Pain Rev. 1995;2:111-137.
18. Gallagher RM, Verma S, Mossey J. Chronic pain. Sources of late-life pain and risk factors for disability. Geriatrics. 2000;55:40-44, 47.
19. Picavet HS, Schouten JS. Musculoskeletal pain in the Netherlands: prevalences, consequences and risk groups, the DMC(3)-study. Pain. 2003;102:167-178.
20. Smith AK, Cenzer IS, Knight SJ, et al. The epidemiology of pain during the last 2 years of life. Ann Intern Med. 2010;153:563-569.
21. Sarkisian CA, Hays RD, Mangione CM. Do older adults expect to age successfully? The association between expectations regarding aging and beliefs regarding healthcare seeking among older adults. J Am Geriatr Soc. 2002;50:1837-1843.
22. Keller ML, Leventhal H, Prohaska TR, et al. Beliefs about aging and illness in a community sample. Res Nurs Health. 1989;12:247-255.
23. Dougados M, Gueguen A, Nguyen M, et al. Longitudinal radiologic evaluation of osteoarthritis of the knee. J Rheumatology. 1992;19:378-384.
24. Mallen CD, Peat G, Thomas E, et al. Prognostic factors for musculoskeletal pain in primary care: a systematic review. Br J Gen Pract. 2007;57:655-661.
25. Dieppe PA, Cushnaghan J, Shepstone L. The Bristol ‘OA500’ study: progression of osteoarthritis (OA) over 3 years and the relationship between clinical and radiographic changes at the knee joint. Osteoarthritis Cartilage. 1997;5:87-97.
26. Dieppe P, Cushnaghan J, Tucker M, et al. The Bristol ‘OA500 study’: progression and impact of the disease after 8 years. Osteoarthritis Cartilage. 2000;8:63-68.
27. Makris UE, Fraenkel L, Han L, et al. Epidemiology of restricting back pain in community-living older persons. J Am Geriatr Soc. 2011;59:610-614.
28. Kamaleri Y, Natvig B, Ihlebaek CM, et al. Change in the number of musculoskeletal pain sites: a 14-year prospective study. Pain. 2009;141:25-30.
29. Sharma L, Cahue S, Song J, et al. Physical functioning over three years in knee osteoarthritis: role of psychosocial, local mechanical, and neuromuscular factors. Arthritis Rheum. 2003;48:3359-3370.
30. Yong HH, Gibson SJ, Horne DJ, et al. Development of a pin attitudes questionnaire to assess stoicism and cautiousness for possible age differences. J Gerontol B Psychol Sci Soc Sci. 2001;56:279-284.
31. Vallerand A, Nowak L. Chronic opioid therapy for nonmalignant pain: the patient’s perspective. Part II—barriers to chronic opioid therapy. Pain manag nurs. 2010;11:126-131.
32. Gibson SJ, Farrell M. A review of age differences in the neurophysiology of nociception and the perceptual experience of pain. Clin J Pain. 2004;20:227-239.
33. Woodrow KM, Friedman GD, Siegelaub AB, et al. Pain tolerance: differences according to age, sex and race. Psychosom Med. 1972;34:548-556.
34. Turk DC, Okifuji A, Scharff L. Chronic pain and depression: role of perceived impact and perceived control in different age cohorts. Pain. 1995;61:93-101.
35. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15:699-707.
36. Power JD, Perruccio AV, Badley EM. Pain as a mediator of sleep problems in arthritis and other chronic conditions. Arthritis Rheum. 2005;53:911-919.
37. Louie GH, Tektonidou MG, Caban-Martizen AJ, et al. Sleep disturbances in adults with arthritis: prevalence, mediators, and subgroups at greatest risk. Arthritis Care Res. 2011;63:247-260.
38. Lin JJ, Alfandre D, Moore C. Physician attitudes toward opioid prescribing for patients with persistent noncancer pain. Clin J Pain. 2007;23:799-803.
39. Hall AJ, Logan JE, Toblin RL, et al. Patterns of abuse among unintentional pharmaceutical overdose fatalities. JAMA. 2008;300:2613-2620.
40. Fischer B, Gittins J, Kendall P, et al. Thinking the unthinkable: could the increasing misuse of prescription opioids among street drug users offer benefits for public health? Public Health. 2009;123:145-146.
41. Fleming MF, Davis J, Passik SD. Reported lifetime aberrant drug-taking behaviors are predictive of current substance use and mental health problems in primary care patients. Pain Med. 2008;9:1098-1106.
42. Reid MC, Henderson CR, Jr, Papaleontiou M, et al. Characteristics of older adults receiving opioids in primary care: treatment duration and outcomes. Pain Med. 2010;11:1063-1071.
43. Solomon DH, Rassen JA, Glynn RJ, et al. The comparative safety of opioids for nonmalignant pain in older adults. Arch Intern Med. 2010;170:1979-1986.
44. Thielke SM, Simoni-Wastila L, Edlund MJ, et al. Age and sex trends in long-term opioid use in two large American health systems between 2000 and 2005. Pain Med. 2010;11:248-256.
45. Soden K, Ali S, Alloway L, et al. How do nurses assess and manage breakthrough pain in specialist palliative care inpatient units? A multicentre study. Palliat Med. 2010;24:294-298.
46. Papaleontiou M, Henderson CR, Jr, Turner BJ, et al. Outcomes associated with opioid use in the treatment of chronic noncancer pain in older adults: a systematic review and meta-analysis. J Am Geriatr Soc. 2010;58:1353-1369.
47. Mallen CD, Peat G. Discussing prognosis with older people with musculoskeletal pain: a cross-sectional study in general practice. BMC Fam Pract. 2009;10:50.-
48. Rosemann T, Wensing M, Joest K, et al. Problems and needs for improving primary care of osteoarthritis patients: the views of patients, general practitioners and practice nurses. BMC Musculoskelet Disord. 2006;7:48.
| Surprise patients with the truth about pain & aging Stephen Thielke, MD, MSPH, MA |
Stephen Thielke, MD, MSPH, MA
University of Washington, Seattle
[email protected]
Joanna Sale, PhD
Li Ka Shing Knowledge Institute, St. Michael’s Hospital, Toronto, Canada
M. Carrington Reid, MD, PhD
Weill Cornell Medical College, New York
The authors reported no potential conflict of interest relevant to this article. This article was made possible by grant number 5P30 AG022845-10-10 from the National Institutes of Health/National Institute on Aging.
| Surprise patients with the truth about pain & aging Stephen Thielke, MD, MSPH, MA |
Stephen Thielke, MD, MSPH, MA
University of Washington, Seattle
[email protected]
Joanna Sale, PhD
Li Ka Shing Knowledge Institute, St. Michael’s Hospital, Toronto, Canada
M. Carrington Reid, MD, PhD
Weill Cornell Medical College, New York
The authors reported no potential conflict of interest relevant to this article. This article was made possible by grant number 5P30 AG022845-10-10 from the National Institutes of Health/National Institute on Aging.
| Surprise patients with the truth about pain & aging Stephen Thielke, MD, MSPH, MA |
Stephen Thielke, MD, MSPH, MA
University of Washington, Seattle
[email protected]
Joanna Sale, PhD
Li Ka Shing Knowledge Institute, St. Michael’s Hospital, Toronto, Canada
M. Carrington Reid, MD, PhD
Weill Cornell Medical College, New York
The authors reported no potential conflict of interest relevant to this article. This article was made possible by grant number 5P30 AG022845-10-10 from the National Institutes of Health/National Institute on Aging.
Beliefs about aging itself can also have dramatic consequences, both positive and negative. In one longitudinal study, those who had positive self-perceptions of aging when they were 50 had better health during 2 decades of follow-up and lived, on average, 7½ years longer than those who had negative self-perceptions at the age of 50.4
Although little research has focused specifically on pain-related stereotypes held by older adults, their importance has long been recognized.
Twenty years ago, a review found that the failure to incorporate older patients’ beliefs about pain could have a negative effect on pain management.5 And in 2011, an Institute of Medicine report found a critical need for public education to counter the myths, misunderstandings, stereotypes, and stigma that hinder pain management in patients across the lifespan.6
We set out to identify widely held stereotypes that older adults and physicians have about pain—and to report on primary studies that support or refute them. We focused on noncancer pain. In the pages that follow, we identify 4 key stereotypes that misrepresent the experience of older adults with regard to pain, and present evidence to debunk them.
Stereotype #1: Pain is a natural part of getting older
Chronic pain is often perceived as an age-related condition. In in-depth interviews, older adults with osteoarthritis reported pain as a normal, even essential, part of life. As one patient put it, “That’s how you know you’re alive … you ache.”7
Among primary care patients with osteoarthritis, those older than 70 years were more likely than younger patients to believe that people should expect to live with pain as they get older.8 And more than half of older adults who responded to a community-based survey considered arthritis to be a natural part of getting old.9
Physicians, too, often view pain as an inevitable part of the aging process, giving patients feedback such as “What do you expect? You’re just getting older.”10
Are they right?
Is pain inevitable? No
In fact, chronic pain is common in older adults, occurring in more than half of those assessed, according to some studies.11 In addition, some epidemiological studies have found an age-related increase in the prevalence of pain,12-14 with older age predicting a more likely onset of, and failure to recover from, persistent pain.15 But numerous studies have failed to find a direct relationship between pain and age.
A National Center for Health Statistics report found that 29% of adults between the ages of 45 and 64 years vs 21% of those 65 or older reported pain lasting >24 hours in the month before the survey.16 And a meta-analysis comparing age-related differences in pain perception found that the highest prevalence of chronic pain occurred at about age 65; a slight decline with advancing age followed, even beyond the age of 85.17
Chronic pain disorders are less frequent. In fact, many chronic pain disorders occur less frequently with advancing age. Population-based studies have found a lower prevalence of low back, neck, and face pain among older adults compared with their younger counterparts;16 evidence has also found lower rates of headache and abdominal pain.18 Other epidemiological studies suggest that the prevalence of musculoskeletal pain generally declines with advancing age,19 and a study of patients in their last 2 years of life found pain to be inversely correlated with age.20 These findings refute the stereotype that advancing age inexorably involves pain, and challenge the notion that pain in later life is normal and expected, and unworthy of treatment.
Stereotype #2: Pain worsens
over time
Some patients and physicians expect that as people age, their pain will increase in intensity. In one study of community-dwelling older adults, 87% of those surveyed rated the belief that more aches and pains are an accepted part of aging as definitely or somewhat true.21 Indeed, patients of all ages have expressed the belief that older age confers greater susceptibility to, and suffering from, painful conditions like arthritis.22 Many common causes of pain in older adults, especially osteoarthritis, are seen as resulting from degenerative changes, which worsen over time.23
Does pain intensify? Not necessarily
Some studies have linked older age to a worse prognosis for patients with musculoskeletal pain, but a greater number have found that aging has no effect on it.24
Pain does not always progress. In a large cohort of patients with peripheral joint osteoarthritis, radiographic joint space narrowing worsened over 3 years, but this did not correlate consistently with worsening pain.25 When the same cohort was assessed after 8 years, there was significant variability in pain, with no clear progression.26
In another study involving older patients with restrictive back pain, the pain was frequently short-lived and episodic and did not increase with age.27 And in a population sample in Norway, the mean number of pain sites decreased slightly over 14 years in those older than 60 years, while increasing in those aged 44 to 60.28 Another study of patients with knee osteoarthritis identified factors that were protective against a decline in pain-related function: These included good mental health, self-efficacy, social support, and greater activity—but not younger age.29 The enormous heterogeneity in both the experience and the course of pain suggests that age-related pain progression is neither universal nor expected—and contradicts a purely biological paradigm in which pain inevitably worsens over time.
Stereotype #3: Stoicism leads to pain tolerance
Some patients believe that the inability to deal with pain is a sign of being soft or weak, and that a “tough it out” approach makes pain easier to tolerate.7 In one survey, older adults were more likely than their younger counterparts to express such stoicism, frequently agreeing with statements like, “I maintain my pride and keep a stiff upper lip when in pain,” “I go on as if nothing had happened …,” and “Pain is something that should be ignored.” 30
Unfortunately, some physicians reinforce such attitudes, telling older patients, in effect, that they’d better “get used to it.”10 And family and friends may make it worse. Patients taking opioids reported that it wasn’t unusual for those close to them to view their use of these analgesics as a sign of weakness.31
Does stoicism help? Probably not
Older adults seem less likely than younger adults to label a sensation as painful, suggesting a more stoic approach in general.30 While some research has found that nociception—the perception of pain in response to painful stimuli—decreases with advancing age,32 other studies have found the opposite.33 And population-based studies focusing on the consequences of pain indicate that it continues to have powerful negative effects, especially depression and insomnia, in older patients.
The degree of pain experienced is more strongly associated with depression in older patients compared with younger adults,34 and greater pain reduces the likelihood that depression will improve with treatment.35 Pain also continues to interfere with sleep. In one national sample, 25% of those with arthritis said they suffered from insomnia, roughly twice the prevalence of insomnia found in those without arthritis.36 In another study, individuals with arthritis were 3 times more likely to have sleep problems compared with individuals without arthritis37—an association independent of age. Being stoic about pain, it appears, does not diminish its consequences over time or help patients better tolerate it.
Stereotype #4: Prescription analgesics are highly addictive
Patients often think that prescription analgesics, especially opioids, are highly addictive or harmful—and older adults may refuse to take them for fear of becoming addicted.7 The stereotype is often shared by family and friends, as well as clinicians.
In one study, one-third of physicians said they hesitated to prescribe opioid medications to older adults because of the risk of addiction (a concern that no clinician with training in geriatrics shared).38 What’s more, 16% of the physicians estimated that about one in 4 older patients receiving chronic opioid therapy becomes addicted. The actual risk is far lower. (More on that below.) News reports of an epidemic of prescription opioid addictions and fatalities,39 including the assertion that opioids are replacing heroin as the primary drug of choice on the street,40 may reinforce such stereotypes.
How great is the risk of addiction? For older adults, it’s very low
While rates of aberrant opioid use vary widely depending on the context, one consistent theme is that older age is associated with decreased risk.41 In one retrospective cohort study of older patients who had recently been started on an opioid medication for the treatment of chronic pain, only 3% showed evidence of behaviors associated with abuse or misuse.42
What’s more, long-term opioid use among older patients with painful conditions is relatively uncommon, and prescription patterns suggest that most older adults discontinue opioids after one or 2 prescriptions.42-44 Decades of research have found that, although opioid medications can cause physiological dependence, addiction is rare in patients treated with them.45,46 (To learn more, see “Diagnosing and treating opioid dependence,” J Fam Pract. 2012;61: 588-597.)
Debunking myths: Implications for practice
Our findings—that pain is not a natural part of aging and often improves or remains stable over time, stoicism does not lead to acclimation, and pain medications are not highly addictive in older adults—make it clear that the stereotypes we identified are misconceptions of pain in later life. Debunking these stereotypes has several implications for clinical practice. We recommend the following:
Identify and counter these stereotypes. Avoid reinforcing stereotypes; counter them by summarizing these evidence-based findings for older patients. We believe patients would be receptive.
In one study, more than 80% of patients with osteoarthritis said they wanted prognostic information about the course of the disease, but only about one-third had received it.47 Presenting the research findings would challenge patients’ stereotypes and help them reframe their expectations.
Elicit patients’ perspectives. Ask patients about age- and pain-related stereotypes and their expectations and perspectives of what constitutes successful treatment. Research shows that patients often wish to discuss lifestyle changes and nonmedical approaches to pain, for example, but that clinicians typically focus on medications instead.48
Emphasize the positive. Frame discussions of pain and aging in a positive light, offering encouragement rather than supporting stoicism or resignation. Attention to protective factors, including good mental health, self-efficacy, social support, and greater activity, may enable older patients to adapt better to any pain they experience.
CORRESPONDENCE
Stephen Thielke, MD, MSPH, MA, University of Washington, Psychiatry and Behavioral Sciences, Box 356560, Seattle, WA 98195; [email protected]
Beliefs about aging itself can also have dramatic consequences, both positive and negative. In one longitudinal study, those who had positive self-perceptions of aging when they were 50 had better health during 2 decades of follow-up and lived, on average, 7½ years longer than those who had negative self-perceptions at the age of 50.4
Although little research has focused specifically on pain-related stereotypes held by older adults, their importance has long been recognized.
Twenty years ago, a review found that the failure to incorporate older patients’ beliefs about pain could have a negative effect on pain management.5 And in 2011, an Institute of Medicine report found a critical need for public education to counter the myths, misunderstandings, stereotypes, and stigma that hinder pain management in patients across the lifespan.6
We set out to identify widely held stereotypes that older adults and physicians have about pain—and to report on primary studies that support or refute them. We focused on noncancer pain. In the pages that follow, we identify 4 key stereotypes that misrepresent the experience of older adults with regard to pain, and present evidence to debunk them.
Stereotype #1: Pain is a natural part of getting older
Chronic pain is often perceived as an age-related condition. In in-depth interviews, older adults with osteoarthritis reported pain as a normal, even essential, part of life. As one patient put it, “That’s how you know you’re alive … you ache.”7
Among primary care patients with osteoarthritis, those older than 70 years were more likely than younger patients to believe that people should expect to live with pain as they get older.8 And more than half of older adults who responded to a community-based survey considered arthritis to be a natural part of getting old.9
Physicians, too, often view pain as an inevitable part of the aging process, giving patients feedback such as “What do you expect? You’re just getting older.”10
Are they right?
Is pain inevitable? No
In fact, chronic pain is common in older adults, occurring in more than half of those assessed, according to some studies.11 In addition, some epidemiological studies have found an age-related increase in the prevalence of pain,12-14 with older age predicting a more likely onset of, and failure to recover from, persistent pain.15 But numerous studies have failed to find a direct relationship between pain and age.
A National Center for Health Statistics report found that 29% of adults between the ages of 45 and 64 years vs 21% of those 65 or older reported pain lasting >24 hours in the month before the survey.16 And a meta-analysis comparing age-related differences in pain perception found that the highest prevalence of chronic pain occurred at about age 65; a slight decline with advancing age followed, even beyond the age of 85.17
Chronic pain disorders are less frequent. In fact, many chronic pain disorders occur less frequently with advancing age. Population-based studies have found a lower prevalence of low back, neck, and face pain among older adults compared with their younger counterparts;16 evidence has also found lower rates of headache and abdominal pain.18 Other epidemiological studies suggest that the prevalence of musculoskeletal pain generally declines with advancing age,19 and a study of patients in their last 2 years of life found pain to be inversely correlated with age.20 These findings refute the stereotype that advancing age inexorably involves pain, and challenge the notion that pain in later life is normal and expected, and unworthy of treatment.
Stereotype #2: Pain worsens
over time
Some patients and physicians expect that as people age, their pain will increase in intensity. In one study of community-dwelling older adults, 87% of those surveyed rated the belief that more aches and pains are an accepted part of aging as definitely or somewhat true.21 Indeed, patients of all ages have expressed the belief that older age confers greater susceptibility to, and suffering from, painful conditions like arthritis.22 Many common causes of pain in older adults, especially osteoarthritis, are seen as resulting from degenerative changes, which worsen over time.23
Does pain intensify? Not necessarily
Some studies have linked older age to a worse prognosis for patients with musculoskeletal pain, but a greater number have found that aging has no effect on it.24
Pain does not always progress. In a large cohort of patients with peripheral joint osteoarthritis, radiographic joint space narrowing worsened over 3 years, but this did not correlate consistently with worsening pain.25 When the same cohort was assessed after 8 years, there was significant variability in pain, with no clear progression.26
In another study involving older patients with restrictive back pain, the pain was frequently short-lived and episodic and did not increase with age.27 And in a population sample in Norway, the mean number of pain sites decreased slightly over 14 years in those older than 60 years, while increasing in those aged 44 to 60.28 Another study of patients with knee osteoarthritis identified factors that were protective against a decline in pain-related function: These included good mental health, self-efficacy, social support, and greater activity—but not younger age.29 The enormous heterogeneity in both the experience and the course of pain suggests that age-related pain progression is neither universal nor expected—and contradicts a purely biological paradigm in which pain inevitably worsens over time.
Stereotype #3: Stoicism leads to pain tolerance
Some patients believe that the inability to deal with pain is a sign of being soft or weak, and that a “tough it out” approach makes pain easier to tolerate.7 In one survey, older adults were more likely than their younger counterparts to express such stoicism, frequently agreeing with statements like, “I maintain my pride and keep a stiff upper lip when in pain,” “I go on as if nothing had happened …,” and “Pain is something that should be ignored.” 30
Unfortunately, some physicians reinforce such attitudes, telling older patients, in effect, that they’d better “get used to it.”10 And family and friends may make it worse. Patients taking opioids reported that it wasn’t unusual for those close to them to view their use of these analgesics as a sign of weakness.31
Does stoicism help? Probably not
Older adults seem less likely than younger adults to label a sensation as painful, suggesting a more stoic approach in general.30 While some research has found that nociception—the perception of pain in response to painful stimuli—decreases with advancing age,32 other studies have found the opposite.33 And population-based studies focusing on the consequences of pain indicate that it continues to have powerful negative effects, especially depression and insomnia, in older patients.
The degree of pain experienced is more strongly associated with depression in older patients compared with younger adults,34 and greater pain reduces the likelihood that depression will improve with treatment.35 Pain also continues to interfere with sleep. In one national sample, 25% of those with arthritis said they suffered from insomnia, roughly twice the prevalence of insomnia found in those without arthritis.36 In another study, individuals with arthritis were 3 times more likely to have sleep problems compared with individuals without arthritis37—an association independent of age. Being stoic about pain, it appears, does not diminish its consequences over time or help patients better tolerate it.
Stereotype #4: Prescription analgesics are highly addictive
Patients often think that prescription analgesics, especially opioids, are highly addictive or harmful—and older adults may refuse to take them for fear of becoming addicted.7 The stereotype is often shared by family and friends, as well as clinicians.
In one study, one-third of physicians said they hesitated to prescribe opioid medications to older adults because of the risk of addiction (a concern that no clinician with training in geriatrics shared).38 What’s more, 16% of the physicians estimated that about one in 4 older patients receiving chronic opioid therapy becomes addicted. The actual risk is far lower. (More on that below.) News reports of an epidemic of prescription opioid addictions and fatalities,39 including the assertion that opioids are replacing heroin as the primary drug of choice on the street,40 may reinforce such stereotypes.
How great is the risk of addiction? For older adults, it’s very low
While rates of aberrant opioid use vary widely depending on the context, one consistent theme is that older age is associated with decreased risk.41 In one retrospective cohort study of older patients who had recently been started on an opioid medication for the treatment of chronic pain, only 3% showed evidence of behaviors associated with abuse or misuse.42
What’s more, long-term opioid use among older patients with painful conditions is relatively uncommon, and prescription patterns suggest that most older adults discontinue opioids after one or 2 prescriptions.42-44 Decades of research have found that, although opioid medications can cause physiological dependence, addiction is rare in patients treated with them.45,46 (To learn more, see “Diagnosing and treating opioid dependence,” J Fam Pract. 2012;61: 588-597.)
Debunking myths: Implications for practice
Our findings—that pain is not a natural part of aging and often improves or remains stable over time, stoicism does not lead to acclimation, and pain medications are not highly addictive in older adults—make it clear that the stereotypes we identified are misconceptions of pain in later life. Debunking these stereotypes has several implications for clinical practice. We recommend the following:
Identify and counter these stereotypes. Avoid reinforcing stereotypes; counter them by summarizing these evidence-based findings for older patients. We believe patients would be receptive.
In one study, more than 80% of patients with osteoarthritis said they wanted prognostic information about the course of the disease, but only about one-third had received it.47 Presenting the research findings would challenge patients’ stereotypes and help them reframe their expectations.
Elicit patients’ perspectives. Ask patients about age- and pain-related stereotypes and their expectations and perspectives of what constitutes successful treatment. Research shows that patients often wish to discuss lifestyle changes and nonmedical approaches to pain, for example, but that clinicians typically focus on medications instead.48
Emphasize the positive. Frame discussions of pain and aging in a positive light, offering encouragement rather than supporting stoicism or resignation. Attention to protective factors, including good mental health, self-efficacy, social support, and greater activity, may enable older patients to adapt better to any pain they experience.
CORRESPONDENCE
Stephen Thielke, MD, MSPH, MA, University of Washington, Psychiatry and Behavioral Sciences, Box 356560, Seattle, WA 98195; [email protected]
1. Herr K. Pain in the older adult: an imperative across all health care settings. Pain Manag Nurs. 2010;11(2 suppl):S1-S10.
2. Pitkala KH, Strandberg TE, Tilvis RS. Management of nonmalignant pain in home-dwelling older people: a population-based survey. J Am Geriatr Soc. 2002;50:1861-1865.
3. Levy B. Stereotype embodiment: a psychosocial approach to aging. Curr Dir Psychol Sci. 2009;18:332-336.
4. Levy BR, Slade MD, Kasl SV. Longitudinal benefit of positive self-perceptions of aging on functional health. J Gerontol B Psychol Sci Soc Sci. 2002;57:409-417.
5. Hofland SL. Elder beliefs: blocks to pain management. J Gerontol Nurs. 1992;18:19-23.
6. Institute of Medicine. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: National Academies Press; 2011.
7. Sale J, Gignac M, Hawker G. How “bad” does the pain have to be? A qualitative study examining adherence to pain medication in older adults with osteoarthritis. Arthritis Rheum. 2006;55:272-278.
8. Appelt CJ, Burant BC, Siminoff LA, et al. Health beliefs related to aging among older male patients with knee and/or hip osteoarthritis. J Gerontol A Biol Sci Med Sci. 2007;62:184-190.
9. Goodwin JS, Black SA, Satish S. Aging versus disease: the opinions of older black, Hispanic, and non-Hispanic white Americans about the causes and treatment of common medical conditions. J Am Geriatr Soc. 1999;47:973-979.
10. Gignac M, Davis A, Hawker G, et al. “What do you expect? You’re just getting older”: a comparison of perceived osteoarthritis-related and aging-related health experiences in middle- and older-age adults. Arthritis Rheum. 2006;55:905-912.
11. Helme RD, Gibson SJ. Pain in the elderly. In: Jensen TS, Turner JA, Weisenfeld-Hallin Z, eds. Progress in Pain Research and Management. Proceedings of the 8th World Congress on Pain. Vol 8. Seattle, Wash: IASP Press; 1997:919–944.
12. Badley EM, Tennant A. Changing profile of joint disorders with age: findings from a postal survey of the population of Calderdale, West Yorkshire, United Kingdom. Ann Rheumatic Dis. 1992;51:366-371.
13. Brattberg G, Parker MG, Thorslund M. A longitudinal study of pain: reported pain from middle age to old age. Clin J Pain. 1997;13:144-149.
14. Crook J, Rideout E, Browne G. The prevalence of pain complaints in a general population. Pain. 1984;18:299-314.
15. Gureje O, Simon GE, Von Korff M. A cross-national study of the course of persistent pain in primary care. Pain. 2001;92:195-200.
16. National Center for Health Statistics. Special feature: pain. In: Health, United States, 2006 with Chartbook on Trends in the Health of Americans. Hyattsville, Md: Centers for Disease Control and Prevention; 2006:68–87. Available at: http://www.cdc.gov/nchs/data/hus/hus06.pdf. Accessed October 16, 2012.
17. Gibson SJ, Helme RD. Age differences in pain perception and report: a review of physiological, psychological, laboratory and clinical studies. Pain Rev. 1995;2:111-137.
18. Gallagher RM, Verma S, Mossey J. Chronic pain. Sources of late-life pain and risk factors for disability. Geriatrics. 2000;55:40-44, 47.
19. Picavet HS, Schouten JS. Musculoskeletal pain in the Netherlands: prevalences, consequences and risk groups, the DMC(3)-study. Pain. 2003;102:167-178.
20. Smith AK, Cenzer IS, Knight SJ, et al. The epidemiology of pain during the last 2 years of life. Ann Intern Med. 2010;153:563-569.
21. Sarkisian CA, Hays RD, Mangione CM. Do older adults expect to age successfully? The association between expectations regarding aging and beliefs regarding healthcare seeking among older adults. J Am Geriatr Soc. 2002;50:1837-1843.
22. Keller ML, Leventhal H, Prohaska TR, et al. Beliefs about aging and illness in a community sample. Res Nurs Health. 1989;12:247-255.
23. Dougados M, Gueguen A, Nguyen M, et al. Longitudinal radiologic evaluation of osteoarthritis of the knee. J Rheumatology. 1992;19:378-384.
24. Mallen CD, Peat G, Thomas E, et al. Prognostic factors for musculoskeletal pain in primary care: a systematic review. Br J Gen Pract. 2007;57:655-661.
25. Dieppe PA, Cushnaghan J, Shepstone L. The Bristol ‘OA500’ study: progression of osteoarthritis (OA) over 3 years and the relationship between clinical and radiographic changes at the knee joint. Osteoarthritis Cartilage. 1997;5:87-97.
26. Dieppe P, Cushnaghan J, Tucker M, et al. The Bristol ‘OA500 study’: progression and impact of the disease after 8 years. Osteoarthritis Cartilage. 2000;8:63-68.
27. Makris UE, Fraenkel L, Han L, et al. Epidemiology of restricting back pain in community-living older persons. J Am Geriatr Soc. 2011;59:610-614.
28. Kamaleri Y, Natvig B, Ihlebaek CM, et al. Change in the number of musculoskeletal pain sites: a 14-year prospective study. Pain. 2009;141:25-30.
29. Sharma L, Cahue S, Song J, et al. Physical functioning over three years in knee osteoarthritis: role of psychosocial, local mechanical, and neuromuscular factors. Arthritis Rheum. 2003;48:3359-3370.
30. Yong HH, Gibson SJ, Horne DJ, et al. Development of a pin attitudes questionnaire to assess stoicism and cautiousness for possible age differences. J Gerontol B Psychol Sci Soc Sci. 2001;56:279-284.
31. Vallerand A, Nowak L. Chronic opioid therapy for nonmalignant pain: the patient’s perspective. Part II—barriers to chronic opioid therapy. Pain manag nurs. 2010;11:126-131.
32. Gibson SJ, Farrell M. A review of age differences in the neurophysiology of nociception and the perceptual experience of pain. Clin J Pain. 2004;20:227-239.
33. Woodrow KM, Friedman GD, Siegelaub AB, et al. Pain tolerance: differences according to age, sex and race. Psychosom Med. 1972;34:548-556.
34. Turk DC, Okifuji A, Scharff L. Chronic pain and depression: role of perceived impact and perceived control in different age cohorts. Pain. 1995;61:93-101.
35. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15:699-707.
36. Power JD, Perruccio AV, Badley EM. Pain as a mediator of sleep problems in arthritis and other chronic conditions. Arthritis Rheum. 2005;53:911-919.
37. Louie GH, Tektonidou MG, Caban-Martizen AJ, et al. Sleep disturbances in adults with arthritis: prevalence, mediators, and subgroups at greatest risk. Arthritis Care Res. 2011;63:247-260.
38. Lin JJ, Alfandre D, Moore C. Physician attitudes toward opioid prescribing for patients with persistent noncancer pain. Clin J Pain. 2007;23:799-803.
39. Hall AJ, Logan JE, Toblin RL, et al. Patterns of abuse among unintentional pharmaceutical overdose fatalities. JAMA. 2008;300:2613-2620.
40. Fischer B, Gittins J, Kendall P, et al. Thinking the unthinkable: could the increasing misuse of prescription opioids among street drug users offer benefits for public health? Public Health. 2009;123:145-146.
41. Fleming MF, Davis J, Passik SD. Reported lifetime aberrant drug-taking behaviors are predictive of current substance use and mental health problems in primary care patients. Pain Med. 2008;9:1098-1106.
42. Reid MC, Henderson CR, Jr, Papaleontiou M, et al. Characteristics of older adults receiving opioids in primary care: treatment duration and outcomes. Pain Med. 2010;11:1063-1071.
43. Solomon DH, Rassen JA, Glynn RJ, et al. The comparative safety of opioids for nonmalignant pain in older adults. Arch Intern Med. 2010;170:1979-1986.
44. Thielke SM, Simoni-Wastila L, Edlund MJ, et al. Age and sex trends in long-term opioid use in two large American health systems between 2000 and 2005. Pain Med. 2010;11:248-256.
45. Soden K, Ali S, Alloway L, et al. How do nurses assess and manage breakthrough pain in specialist palliative care inpatient units? A multicentre study. Palliat Med. 2010;24:294-298.
46. Papaleontiou M, Henderson CR, Jr, Turner BJ, et al. Outcomes associated with opioid use in the treatment of chronic noncancer pain in older adults: a systematic review and meta-analysis. J Am Geriatr Soc. 2010;58:1353-1369.
47. Mallen CD, Peat G. Discussing prognosis with older people with musculoskeletal pain: a cross-sectional study in general practice. BMC Fam Pract. 2009;10:50.-
48. Rosemann T, Wensing M, Joest K, et al. Problems and needs for improving primary care of osteoarthritis patients: the views of patients, general practitioners and practice nurses. BMC Musculoskelet Disord. 2006;7:48.
1. Herr K. Pain in the older adult: an imperative across all health care settings. Pain Manag Nurs. 2010;11(2 suppl):S1-S10.
2. Pitkala KH, Strandberg TE, Tilvis RS. Management of nonmalignant pain in home-dwelling older people: a population-based survey. J Am Geriatr Soc. 2002;50:1861-1865.
3. Levy B. Stereotype embodiment: a psychosocial approach to aging. Curr Dir Psychol Sci. 2009;18:332-336.
4. Levy BR, Slade MD, Kasl SV. Longitudinal benefit of positive self-perceptions of aging on functional health. J Gerontol B Psychol Sci Soc Sci. 2002;57:409-417.
5. Hofland SL. Elder beliefs: blocks to pain management. J Gerontol Nurs. 1992;18:19-23.
6. Institute of Medicine. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: National Academies Press; 2011.
7. Sale J, Gignac M, Hawker G. How “bad” does the pain have to be? A qualitative study examining adherence to pain medication in older adults with osteoarthritis. Arthritis Rheum. 2006;55:272-278.
8. Appelt CJ, Burant BC, Siminoff LA, et al. Health beliefs related to aging among older male patients with knee and/or hip osteoarthritis. J Gerontol A Biol Sci Med Sci. 2007;62:184-190.
9. Goodwin JS, Black SA, Satish S. Aging versus disease: the opinions of older black, Hispanic, and non-Hispanic white Americans about the causes and treatment of common medical conditions. J Am Geriatr Soc. 1999;47:973-979.
10. Gignac M, Davis A, Hawker G, et al. “What do you expect? You’re just getting older”: a comparison of perceived osteoarthritis-related and aging-related health experiences in middle- and older-age adults. Arthritis Rheum. 2006;55:905-912.
11. Helme RD, Gibson SJ. Pain in the elderly. In: Jensen TS, Turner JA, Weisenfeld-Hallin Z, eds. Progress in Pain Research and Management. Proceedings of the 8th World Congress on Pain. Vol 8. Seattle, Wash: IASP Press; 1997:919–944.
12. Badley EM, Tennant A. Changing profile of joint disorders with age: findings from a postal survey of the population of Calderdale, West Yorkshire, United Kingdom. Ann Rheumatic Dis. 1992;51:366-371.
13. Brattberg G, Parker MG, Thorslund M. A longitudinal study of pain: reported pain from middle age to old age. Clin J Pain. 1997;13:144-149.
14. Crook J, Rideout E, Browne G. The prevalence of pain complaints in a general population. Pain. 1984;18:299-314.
15. Gureje O, Simon GE, Von Korff M. A cross-national study of the course of persistent pain in primary care. Pain. 2001;92:195-200.
16. National Center for Health Statistics. Special feature: pain. In: Health, United States, 2006 with Chartbook on Trends in the Health of Americans. Hyattsville, Md: Centers for Disease Control and Prevention; 2006:68–87. Available at: http://www.cdc.gov/nchs/data/hus/hus06.pdf. Accessed October 16, 2012.
17. Gibson SJ, Helme RD. Age differences in pain perception and report: a review of physiological, psychological, laboratory and clinical studies. Pain Rev. 1995;2:111-137.
18. Gallagher RM, Verma S, Mossey J. Chronic pain. Sources of late-life pain and risk factors for disability. Geriatrics. 2000;55:40-44, 47.
19. Picavet HS, Schouten JS. Musculoskeletal pain in the Netherlands: prevalences, consequences and risk groups, the DMC(3)-study. Pain. 2003;102:167-178.
20. Smith AK, Cenzer IS, Knight SJ, et al. The epidemiology of pain during the last 2 years of life. Ann Intern Med. 2010;153:563-569.
21. Sarkisian CA, Hays RD, Mangione CM. Do older adults expect to age successfully? The association between expectations regarding aging and beliefs regarding healthcare seeking among older adults. J Am Geriatr Soc. 2002;50:1837-1843.
22. Keller ML, Leventhal H, Prohaska TR, et al. Beliefs about aging and illness in a community sample. Res Nurs Health. 1989;12:247-255.
23. Dougados M, Gueguen A, Nguyen M, et al. Longitudinal radiologic evaluation of osteoarthritis of the knee. J Rheumatology. 1992;19:378-384.
24. Mallen CD, Peat G, Thomas E, et al. Prognostic factors for musculoskeletal pain in primary care: a systematic review. Br J Gen Pract. 2007;57:655-661.
25. Dieppe PA, Cushnaghan J, Shepstone L. The Bristol ‘OA500’ study: progression of osteoarthritis (OA) over 3 years and the relationship between clinical and radiographic changes at the knee joint. Osteoarthritis Cartilage. 1997;5:87-97.
26. Dieppe P, Cushnaghan J, Tucker M, et al. The Bristol ‘OA500 study’: progression and impact of the disease after 8 years. Osteoarthritis Cartilage. 2000;8:63-68.
27. Makris UE, Fraenkel L, Han L, et al. Epidemiology of restricting back pain in community-living older persons. J Am Geriatr Soc. 2011;59:610-614.
28. Kamaleri Y, Natvig B, Ihlebaek CM, et al. Change in the number of musculoskeletal pain sites: a 14-year prospective study. Pain. 2009;141:25-30.
29. Sharma L, Cahue S, Song J, et al. Physical functioning over three years in knee osteoarthritis: role of psychosocial, local mechanical, and neuromuscular factors. Arthritis Rheum. 2003;48:3359-3370.
30. Yong HH, Gibson SJ, Horne DJ, et al. Development of a pin attitudes questionnaire to assess stoicism and cautiousness for possible age differences. J Gerontol B Psychol Sci Soc Sci. 2001;56:279-284.
31. Vallerand A, Nowak L. Chronic opioid therapy for nonmalignant pain: the patient’s perspective. Part II—barriers to chronic opioid therapy. Pain manag nurs. 2010;11:126-131.
32. Gibson SJ, Farrell M. A review of age differences in the neurophysiology of nociception and the perceptual experience of pain. Clin J Pain. 2004;20:227-239.
33. Woodrow KM, Friedman GD, Siegelaub AB, et al. Pain tolerance: differences according to age, sex and race. Psychosom Med. 1972;34:548-556.
34. Turk DC, Okifuji A, Scharff L. Chronic pain and depression: role of perceived impact and perceived control in different age cohorts. Pain. 1995;61:93-101.
35. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15:699-707.
36. Power JD, Perruccio AV, Badley EM. Pain as a mediator of sleep problems in arthritis and other chronic conditions. Arthritis Rheum. 2005;53:911-919.
37. Louie GH, Tektonidou MG, Caban-Martizen AJ, et al. Sleep disturbances in adults with arthritis: prevalence, mediators, and subgroups at greatest risk. Arthritis Care Res. 2011;63:247-260.
38. Lin JJ, Alfandre D, Moore C. Physician attitudes toward opioid prescribing for patients with persistent noncancer pain. Clin J Pain. 2007;23:799-803.
39. Hall AJ, Logan JE, Toblin RL, et al. Patterns of abuse among unintentional pharmaceutical overdose fatalities. JAMA. 2008;300:2613-2620.
40. Fischer B, Gittins J, Kendall P, et al. Thinking the unthinkable: could the increasing misuse of prescription opioids among street drug users offer benefits for public health? Public Health. 2009;123:145-146.
41. Fleming MF, Davis J, Passik SD. Reported lifetime aberrant drug-taking behaviors are predictive of current substance use and mental health problems in primary care patients. Pain Med. 2008;9:1098-1106.
42. Reid MC, Henderson CR, Jr, Papaleontiou M, et al. Characteristics of older adults receiving opioids in primary care: treatment duration and outcomes. Pain Med. 2010;11:1063-1071.
43. Solomon DH, Rassen JA, Glynn RJ, et al. The comparative safety of opioids for nonmalignant pain in older adults. Arch Intern Med. 2010;170:1979-1986.
44. Thielke SM, Simoni-Wastila L, Edlund MJ, et al. Age and sex trends in long-term opioid use in two large American health systems between 2000 and 2005. Pain Med. 2010;11:248-256.
45. Soden K, Ali S, Alloway L, et al. How do nurses assess and manage breakthrough pain in specialist palliative care inpatient units? A multicentre study. Palliat Med. 2010;24:294-298.
46. Papaleontiou M, Henderson CR, Jr, Turner BJ, et al. Outcomes associated with opioid use in the treatment of chronic noncancer pain in older adults: a systematic review and meta-analysis. J Am Geriatr Soc. 2010;58:1353-1369.
47. Mallen CD, Peat G. Discussing prognosis with older people with musculoskeletal pain: a cross-sectional study in general practice. BMC Fam Pract. 2009;10:50.-
48. Rosemann T, Wensing M, Joest K, et al. Problems and needs for improving primary care of osteoarthritis patients: the views of patients, general practitioners and practice nurses. BMC Musculoskelet Disord. 2006;7:48.
AGING: Is your patient taking too many pills?
• Consider the possibility that an adverse drug effect—rather than a new condition—is at play when a patient taking multiple medications develops a new symptom. C
• Use an online interaction checker, which can be accessed via a smart phone or tablet, to check for potential drug-drug interactions in patients on multiple medications. C
• Cross-check patients’ medications with a list of their medical problems, with the goal of discontinuing any drug that duplicates the action of another or is age-inappropriate, ineffective, or not indicated for the condition for which it was prescribed. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Older adults are taking more medications than ever before. Nearly 9 out of 10 US residents who are 60 years of age or older take at least one prescription drug, more than a third take 5 to 9 medications, and 12% take 10 or more.1
The increase is largely driven by newer medications to effectively treat a variety of medical conditions, and by practice guidelines that often recommend multidrug regimens.2
As a result, the term “polypharmacy,” which once referred to a specific number of medications, is now used more broadly to mean “a large number” of drugs.
From a safety standpoint, the number of medications a patient takes matters. The risk of adverse drug effects and dangerous drug-drug interactions increases significantly when an individual takes ≥5 medications.3
More than 4.5 million adverse drug effects occur each year in the United States, and nearly three quarters of them are initially evaluated in outpatient settings.4 Research suggests that about 80% of the time, these adverse effects are not recognized as such by the patient’s physician. So instead of discontinuing the offending medication, physicians treat the drug-related symptoms by adding yet another medication—a phenomenon known as “the prescribing cascade.”5
This review can help you safeguard older patients taking multiple medications by recognizing and responding to drug-related problems, identifying drugs that can be safely eliminated (or, in some cases, drugs that should be added), and checking regularly to ensure that the medication regimen is appropriate and up to date.
CASE Mrs. R, a 79-year-old woman who recently moved to town, is brought to your office by her daughter and son-in-law. The patient has a hard time reporting her medical history, but her daughter tells you her mother has chronic obstructive pulmonary disease (COPD), heart failure, type 2 diabetes, and mild urinary incontinence, and was recently diagnosed with early dementia.
Mrs. R’s daughter has brought in a bagful of medications, but she’s not sure which ones her mother takes regularly. The medications are an albuterol inhaler, alprazolam, digoxin, diphenhydramine, donepezil, furosemide, glargine insulin, guaifenesin, levothyroxine, metformin, extended-release metoprolol, naproxen, omeprazole, simvastatin, tolterodine, and zolpidem—a total of 16 different drugs.
If Mrs. R were your patient, how would you manage her multidrug regimen?
Start with a medication review
The first step in evaluating a patient’s medication regimen is to find out whether the drugs in the patient’s possession and/or in the medical record are the ones he or she is actually taking. Ask older patients who haven’t brought in their medications, or the caregiver of a confused patient, to bring them to the next visit.
The next step: Determine whether the medication regimen is right for the patient.
Polypharmacy may be indicated
Despite the risks associated with polypharmacy, do not assume that it is inappropriate. For some conditions, multiple medications are routinely recommended. Patients with heart failure, for example, have been shown to have better outcomes when they take 3 to 5 medications, including beta-blockers, angiotensin-converting enzyme (ACE) inhibitors, and diuretics.2
Some treatment guidelines also call for multiple medications. Achieving the more stringent blood pressure goals recommended in the Seventh Report of the Joint National Committee on Prevention, for instance, often requires 2 or more antihypertensive agents.6 In many cases, however, patients end up taking more drugs than necessary.
Is the patient taking the right drugs?
Medication reconciliation (determining whether the treatment regimen is appropriate for the patient’s diagnoses) is the way to find out.
The most widely recommended approach to medication reconciliation is to create a table and do a systematic review.7 List all the patient’s medical conditions in the first column and all current medications in the second column. Use the third column to note whether each medication is one the patient should be on, based not only on his or her medical conditions and other drugs being taken but also on current renal and hepatic function and body size, and contraindications.
A medication may be inappropriate if it duplicates, cancels out the action of, or otherwise interacts with another drug the patient is taking; is contraindicated in older patients; or is ineffective for the condition for which it was prescribed. In one key study of nearly 200 patients 65 years and older who took 5 or more medications, more than half had been prescribed at least one drug that was ineffective for the patient’s condition or that duplicated the action of another medication.8
In addition to finding drugs that the patient should not be taking, medication reconciliation may also reveal that the patient is not receiving optimal therapy and that one or more drugs should be added to his or her treatment regimen.
Check meds after transitions. A move from home to hospital, from emergency department to home, or any other transition relating to patient care should prompt a medication reconciliation. Medications are often added or inadvertently discontinued at such times,9,10 and instructions relating to medication are often misunderstood.11 In one study of 384 frail elderly patients being discharged from a hospital, for example, 44% were found to have been given at least one unnecessary prescription—most commonly for a medication that was neither indicated nor effective for any of the patient’s medical problems.12 It was also common for patients to be given drugs that duplicated the action of others they were already taking.
Even in the absence of such transitions, medication reconciliation should occur at regular intervals. Many physicians do a medication reconciliation at every visit to ensure that the medical record is accurate and the patient’s medication regimen is optimal.
Managing polypharmacy: These resources can help
Numerous tools are available to help you evaluate and monitor patients’ medication regimens, including some that were developed specifically for older patients.
START (Screening Tool to Alert doctors to Right Treatment) identifies drugs and drug classes that are underused with older patients.13 START criteria (TABLE 1)13-17 focus on medications that should be used yet are often omitted in older patients who have the appropriate indications.
TABLE 1
START criteria: Drug therapy that should be given to older patients13-17
Cardiovascular
|
Endocrine
|
Gastrointestinal
|
Musculoskeletal
|
Nervous system
|
Respiratory
|
| ACE, angiotensin-converting enzyme; BP, blood pressure; COPD, chronic obstructive pulmonary disease; CVD, cardiovascular disease; FEV1, forced expiratory volume in 1 second; GI, gastrointestinal; MI, myocardial infarction; PPI, proton-pump inhibitor; START, Screening Tool to alert doctors to right Treatment. |
In using START or any other drug-related tool, it is important to keep in mind that therapy should be individualized. Not all the medications in the START criteria are appropriate for every patient, and a medication that is indicated for a given medical condition may or may not provide real benefit for a particular patient. That would depend on the individual’s overall health and life expectancy, the goals of treatment, and how long it would take for the patient to realize any benefit from the drug in question.18 A vigorous 79-year-old might benefit from statin therapy for prevention of cardiovascular events, for instance, while a patient like Mrs. R, who is also 79 but has dementia and multiple other medical problems, would be unlikely to live long enough to realize such a benefit.
”Age” assessment tool. One criterion in deciding whether medication(s) are appropriate for an older patient is his or her “physiologic age”—calculated on the basis of the individual’s chronological age and self-reported health status (TABLE 2).19
TABLE 2
Calculating your patient’s “real” age19
| Actual age (y) | Physiologic age (y) | |||||||
|---|---|---|---|---|---|---|---|---|
| Self-reported health | ||||||||
| Excellent | Good | Fair | Poor | |||||
| Male | Female | Male | Female | Male | Female | Male | Female | |
| 65 | 58 | 60 | 64 | 64 | 68 | 66 | 73 | 72 |
| 70 | 62 | 65 | 69 | 69 | 73 | 71 | 78 | 77 |
| 75 | 67 | 70 | 74 | 74 | 78 | 76 | 83 | 82 |
| 80 | 72 | 75 | 79 | 79 | 83 | 81 | 85+ | 85+ |
Flagging drugs that may be inappropriate
Several tools have been developed to aid clinicians in identifying medications that are potentially inappropriate for older adults, although here, too, decisions about their use must be individualized. Two of the most widely used tools are the Beers criteria and STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions).
Beers criteria were developed by Mark Beers et al in 199120 and have been updated at regular intervals, most recently by the American Geriatrics Society in 2012.21 The drugs and drug classes included in the Beers criteria should not be prescribed for older patients in most cases, either because the risk of using them outweighs the benefit or because safer alternatives are available. Key components are listed in TABLE 3.21
TABLE 3
Beers criteria:* Drug classes that may be inappropriate for older adults21
| Drug class | Concern |
|---|---|
| Alpha-blockers with peripheral activity | Orthostatic hypotension |
| Anticholinergics | Cognitive impairment, urinary retention |
| Antipsychotics | Increased death rate when used for behavior control in patients with dementia |
| NSAIDs | Renal dysfunction, GI bleeding, fluid retention, exacerbation of heart failure |
| Sedative hypnotics | Cognitive impairment, delirium |
| Tricyclic antidepressants | Cognitive impairment, delirium, urinary retention |
| GI, gastrointestinal; NSAIDs, nonsteroidal anti-inflammatory drugs. *The full Beers criteria contains 53 drugs and drug classes that are generally inappropriate for older adults. The full list is available from the American Geriatrics Society at: www.americangeriatrics.org/files/documents/beers/2012BeersCriteria_JAGS.pdf. | |
One limitation of the Beers criteria has been its all-or-nothing approach, with many of the medications on the list deemed inappropriate for all older adults regardless of their circumstances. The 2012 update does a better job of individualizing recommendations: Medications are now categorized as those that should be avoided in older patients regardless of their diseases or conditions, those that should be avoided only in patients with certain diseases or conditions, and those that may be used for this patient population but require caution.21
STOPP is similar to the Beers criteria, but uses a different approach: Most medications on this list are considered in the context of specific medical problems.22 While the Beers criteria classify digoxin >0.125 mg/d as generally inappropriate for older adults, for example, STOPP criteria state that long-term dosing at that level is inappropriate only for those with impaired renal function.22 A list of medications identified by STOPP as contributing to hospitalization due to adverse drug effects is available at http://ageing.oxfordjournals.org/content/37/6/673.
Both tools address this drug category. Cumulative anticholinergic burden is a concept applied to the use of anticholinergic medications, which are included in both the Beers and STOPP criteria. Although isolated short-term exposure to a drug with anticholinergic properties may be tolerated by a healthy and cognitively intact older patient, repetitive exposure to such drugs, even if separated in time, has negative effects. One study evaluated more than 500 community-dwelling older adults and found that the more exposure an individual had to anticholinergic medications over the course of a year, the greater the impairment in short-term memory and activities of daily living.23 Another study, this one involving more than 13,000 community-dwelling and institutionalized patients, showed that the longer an older patient takes an anticholinergic medication, the more likely there is to be a measurable decline in performance on the Mini-Mental State Examination.24
Programs that flag potential interactions
Drug-drug interactions are a key concern of polypharmacy, and electronic medical records and prescribing systems that flag potential drug-drug interactions when a new medication is ordered are designed to help physicians avoid them. Unfortunately, clinicians only react to 3% to 9% of such notifications, overriding them because computerized systems often fail to distinguish between important and unimportant interactions.25-27 Thus, clinicians often must decide whether to react to or override warnings, an often difficult decision with patient safety and medicolegal implications. The best advice we can offer is to carefully evaluate drug interaction warnings using common sense, and seek consultation with a clinical pharmacist when uncertainty exists. This approach should prevent prescribing medications that have potentially harmful interactions with drugs the patient is already taking.
For physicians who do not have access to an electronic prescribing system that provides such notification, several online resources are available, some by subscription (eg, Lexicomp, www.lexi.com; Micromedex, www.micromedex.com/index.html; and Pepid, www.pepid.com) and others with free access (eg, AARP, healthtools.aarp.org/drug-interactions; Drugs.com (www.drugs.com/drug_interactions.php; and HealthLine, www.healthline.com/druginteractions).
CASE After doing a medication reconciliation for Mrs. R, you find that she is taking tolterodine, an anticholinergic medication for urge urinary incontinence, and donepezil, a procholinergic medication for dementia. This type of drug-drug interaction, in which the action of one drug effectively cancels out the effect of another, should not be ignored.
Overall, you identify 8 of her medications that could be discontinued: The list includes guaifenesin (a nonessential medication of questionable efficacy); naproxen (inappropriate per Beers criteria; inappropriate in patients with heart failure, according to STOPP); alprazolam, zolpidem, and diphenhydramine (duplicate medications that are all on the Beers criteria as inappropriate for chronic use and ill-advised in patients with cognitive impairment); and omeprazole and levothyroxine (for which nothing in the patient’s history suggests a need), as well as tolterodine. Depending on dose, digoxin is yet another candidate for discontinuation.
Discontinuing medications: Proceed carefully
Physicians are often reluctant to discontinue chronic medications in older patients—even in those with advanced disease who are not likely to benefit from treatment. Focus groups have identified a number of reasons for their hesitation, including:
- the assumption that patients have no problem taking large numbers of drugs
- the fear that patients may misinterpret a plan to discontinue medications as evidence that the physician is giving up on them
- the belief that physicians must comply with practice guidelines that recommend multiple drug treatments
- concern that proposing discontinuation of medications often leads to a discussion of life expectancy and end-of-life care.28
Physicians may also fear that discontinuation of certain drugs will increase the risk of adverse outcomes. More than 30 studies have evaluated discontinuation of chronic medications in older adults, however, and found that drugs as diverse as antihypertensives, antipsychotics, benzodiazepines, and selective serotonin reuptake inhibitors (SSRIs) can often be discontinued without adverse outcomes. In many cases, improvement in patient function results.29 Medications that present the most difficulty are those that patients often become physically or psychologically dependent on, such as benzodiazepines, guaifenesin, proton-pump inhibitors, nonsteroidal anti-inflammatory drugs, and SSRIs. Some (eg, benzodiazepines, SSRIs) require a gradual reduction; for others, no taper is required
(TABLE 4).30-37
TABLE 4
Recommendations for discontinuing hard-to-stop drugs
| Medication or drug class | Discontinuation regimen | Comments |
|---|---|---|
| Benzodiazepines30 | Taper dose by 25% q 2 wk | No withdrawal symptoms reported with this taper regimen. Subtle cognitive improvement noted over a period of months |
| Guaifenesin31 | Can be discontinued without tapering if not combined with opioids or other medications. Elimination half-life is approximately 1 hour | Guaifenesin is often marketed as a combination product with opioids; such combination products require tapering |
| PPIs32-34 | Decrease dose by 50% q 2 wk; supplement with H2 blocker if needed, but tapering of H2 blocker may be required | Abrupt discontinuation after long-term use causes rebound gastric acid hypersecretion and lowers rate of success. Higher success rates with taper regimen and in patients who do not have documented GERD |
| NSAIDs35 | No taper required | Short-term use (<3 mo) acceptable for patients with no contraindications |
| SSRIs36,37 | Gradual reduction in dose over 6-8 wk | Highest rate of success in patients without a clear diagnosis of depression |
| GERD, gastroesophageal reflux disease; NSAIDs, nonsteroidal anti-inflammatory drugs; PPIs, proton-pump inhibitors; SSRIs, selective serotonin reuptake inhibitors. | ||
CASE You trim down Mrs. R’s regimen by discontinuing each of the 8 drugs, one at a time, and carefully monitor the patient during the withdrawal period. Because she had been taking alprazolam daily, the dose is tapered slowly to avoid withdrawal. Omeprazole also requires a gradual taper to avoid rebound hyperacidity.3
After confirming that Mrs. R has heart failure and COPD, you identify 2 medications that should be added to her drug regimen—an ACE inhibitor for heart failure and an inhaled anticholinergic for COPD.
Going from 16 medications to 10 saves money, decreases the likelihood of adverse events and drug-drug interactions, and helps with adherence. Mrs. R’s new drug regimen is expected to lead to improvements in memory and overall quality of life, as well.
CORRESPONDENCE
Barry D. Weiss, MD, Department of Family and Community Medicine, University of Arizona College of Medicine, Tucson, AZ 85724; [email protected]
1. Gu Q, Dillon CF, Burt V. Prescription drug use continued to increase: US prescription drug data for 2007-2008. CDC/NCHS Data Brief. 2010;42:1-2.
2. Jessup K, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA guidelines for the diagnosis and management of heart failure in adults. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2009;119:1977-2016.
3. Johnell K, Klarin I. The relationship between number of drugs and potential drug-drug interactions in the elderly: a study of over 600,000 elderly patients from the Swedish Prescribed Drug Register. Drug Saf. 2007;30:911-918.
4. Sarkar U, Lopez A, Maselli JH, et al. Adverse drug events in US adult ambulatory medical care. Health Services Res. 2011;46:1517-1533.
5. Rollason V, Vogt N. Reduction of polypharmacy in the elderly. A systematic review of the role of the pharmacist. Drugs Aging. 2003;20:817-832.
6. National Heart, Lung, and Blood Institute. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Available at: www.nhlbi.nih.gov/guidelines/hypertension/jnc7full.pdf. Accessed October 11, 2012.
7. Steinman MA, Hanlon JT. Managing medications in clinically complex elders. JAMA. 2010;304:1592-1601.
8. Steinman MA, Landefeld CS, Rosenthal GE, et al. Polypharmacy and prescribing quality in older people. J Am Geriatr Soc. 2006;54:1516-23.
9. Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuations of medications for chronic disease. JAMA. 2011;306:840-847.
10. Moore C, Wisnivesky J, Williams S, et al. Medical errors related to discontinuity of care from an inpatient to an outpatient setting. J Gen Intern Med. 2003;18:646-651.
11. Ziaeian B, Arauho KL, Van Ness PH, et al. Medication reconciliation accuracy and patient understanding of intended medication changes on hospital discharge. J Gen Intern Med. 2012 July 12. ePub ahead of print.
12. Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc. 2005;53:1518-1523.
13. O’Mahony D, Gallagher P, Ryan C, et al. STOPP & START criteria: a new approach to detecting potentially inappropriate prescribing in old age. Eur Geriatr Med. 2010;1:45-51.
14. Denneboom W, Dautzenberg KGH, Grol R, et al. Analysis of polypharmacy in older patients in primary acre using a multidisciplinary expert panel. Br J Gen Pract. 2006;56:504-510.
15. Ko DT, Mamdani M, Alter DA. Lipid-lowering therapy with statins in high-risk elderly patients. JAMA. 2004;291:1864-70.
16. Wright RM, Sloane R, Pieper CF, et al. Underuse of indicated medications among physically frail older US veterans at the time of hospital discharge: results of a cross-sectional analysis of data from the Geriatric Evaluation and Management Drug Study. Am J Geriatr Pharmacother. 2009;7:271-280.
17. Garwood CL. Use of anticoagulation in elderly patients with atrial fibrillation who are risk for falls. Ann Pharmacother. 2008;42:523-532.
18. Holmes HM, Hayley DC, Alexander GC, et al. Reconsidering medication appropriateness for patients late in life. Arch Intern Med. 2006;166:605-609.
19. Simplified Methods for Estimating Life Expectancy. Available at: http://painconsortium.nih.gov/symptomresearch/chapter_14/Part_3/sec4/chspt3s4pg1.htm. Accessed October 9, 2012.
20. Beers MH, Ouslander JG, Rollingher I, et al. Explicit criteria for determining inappropriate medication use in nursing home residents. Arch Intern Med. 1991;151:1825-1832.
21. The American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society Update Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60:616-631.
22. Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Aging. 2008;37:673-379.
23. Han L, Agostini JV, Allore HG. Cumulative anticholinergic exposure is associated with poor memory and executive function in older men. J Am Geriatr Soc. 2008;56:2203-2210.
24. Fox C, Richardson K, Maidment ID, et al. Anticholinergic medication use and cognitive impairment in the older population: the medical research council cognitive function and ageing study. J Am Geriatr Soc. 2011;59:1477-1483.
25. Knight A, Falade O, Maygers J, et al. Factors associated with medication warning acceptance [abstract]. J Hosp Med. 2012;7(suppl 2):515.-
26. Isaac T, Weissman JS, Davis RB, et al. Overrides of medication alerts in ambulatory care. Arch Intern Med. 2009;169:305-311.
27. Van Der Sijs H, Aarts J, Vulto A, et al. Overriding of drug safety alerts in computerized physician order entry. J Am Med Inform Assoc. 2006;12:138-147.
28. Schuling J, Gebben H, Veehof LJG, et al. Deprescribing medication in very elderly patients with multimorbidity: the view of Dutch GPs. A qualitative study. BMC Family Practice. 2012;13:56. http://www.biomedcentral.com/1471-2296/13/56.
29. Iyer S, Naganathan V, McLachlan AJ, et al. Medication withdrawal trials in people aged 65 years and older. A systematic review. Drugs Aging. 2008;25:1021-1031.
30. Curran HV, Collins R, Fletcher S, et al. Older adults and withdrawal from benzodiazepine hypnotics in general practice: effects on cognitive function, sleep, mood and quality of life. Psychol Med. 2003;33:1223-1237.
31. Krinsky DL, Berardi RR, Ferreris SP, et al. Handbook of Nonprescription Drugs: An Interactive Approach to Self-Care. Washington, DC: American Pharmacists Association; 2012:209.
32. Bjornsson E, Abrahamsson H, Simren M, et al. Discontinuation of proton pump inhibitors in patients on long-term therapy: a double-blind, placebo-controlled trial. Aliment Pharmacol Ther. 2006;24:945-954.
33. Inadomi JM, Jamai R, Murata GH, et al. Step-down management of gastroesophageal reflux disease. Gastroenterology. 2001;131:1095-1100.
34. Hester SA. Proton pump inhibitors and rebound acid hypersecretion. Pharm Lett. 2009;25:250920.-
35. Taylor R, Jr, Lemtouni S, Weiss K, et al. Pain management in the elderly: an FDA safe use initiative expert panel’s view on preventable harm associated with NSAID therapy. Curr Gerontol Geriatr Res. 2012;196159.-
36. Ulfvarson J, Adami J, Wredling R, et al. Controlled withdrawal of selective serotonin reuptake inhibitor drugs in elderly patients in nursing homes with no indication of depression. Eur J Clin Pharmacol. 2003;59:735-740.
37. Lindstrom K, Ekedahl A, Carlsten A, et al. Can selective serotonin inhibitor drugs in elderly patients in nursing homes be reduced? Scand J Prim Health Care. 2007;25:3-8.
• Consider the possibility that an adverse drug effect—rather than a new condition—is at play when a patient taking multiple medications develops a new symptom. C
• Use an online interaction checker, which can be accessed via a smart phone or tablet, to check for potential drug-drug interactions in patients on multiple medications. C
• Cross-check patients’ medications with a list of their medical problems, with the goal of discontinuing any drug that duplicates the action of another or is age-inappropriate, ineffective, or not indicated for the condition for which it was prescribed. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Older adults are taking more medications than ever before. Nearly 9 out of 10 US residents who are 60 years of age or older take at least one prescription drug, more than a third take 5 to 9 medications, and 12% take 10 or more.1
The increase is largely driven by newer medications to effectively treat a variety of medical conditions, and by practice guidelines that often recommend multidrug regimens.2
As a result, the term “polypharmacy,” which once referred to a specific number of medications, is now used more broadly to mean “a large number” of drugs.
From a safety standpoint, the number of medications a patient takes matters. The risk of adverse drug effects and dangerous drug-drug interactions increases significantly when an individual takes ≥5 medications.3
More than 4.5 million adverse drug effects occur each year in the United States, and nearly three quarters of them are initially evaluated in outpatient settings.4 Research suggests that about 80% of the time, these adverse effects are not recognized as such by the patient’s physician. So instead of discontinuing the offending medication, physicians treat the drug-related symptoms by adding yet another medication—a phenomenon known as “the prescribing cascade.”5
This review can help you safeguard older patients taking multiple medications by recognizing and responding to drug-related problems, identifying drugs that can be safely eliminated (or, in some cases, drugs that should be added), and checking regularly to ensure that the medication regimen is appropriate and up to date.
CASE Mrs. R, a 79-year-old woman who recently moved to town, is brought to your office by her daughter and son-in-law. The patient has a hard time reporting her medical history, but her daughter tells you her mother has chronic obstructive pulmonary disease (COPD), heart failure, type 2 diabetes, and mild urinary incontinence, and was recently diagnosed with early dementia.
Mrs. R’s daughter has brought in a bagful of medications, but she’s not sure which ones her mother takes regularly. The medications are an albuterol inhaler, alprazolam, digoxin, diphenhydramine, donepezil, furosemide, glargine insulin, guaifenesin, levothyroxine, metformin, extended-release metoprolol, naproxen, omeprazole, simvastatin, tolterodine, and zolpidem—a total of 16 different drugs.
If Mrs. R were your patient, how would you manage her multidrug regimen?
Start with a medication review
The first step in evaluating a patient’s medication regimen is to find out whether the drugs in the patient’s possession and/or in the medical record are the ones he or she is actually taking. Ask older patients who haven’t brought in their medications, or the caregiver of a confused patient, to bring them to the next visit.
The next step: Determine whether the medication regimen is right for the patient.
Polypharmacy may be indicated
Despite the risks associated with polypharmacy, do not assume that it is inappropriate. For some conditions, multiple medications are routinely recommended. Patients with heart failure, for example, have been shown to have better outcomes when they take 3 to 5 medications, including beta-blockers, angiotensin-converting enzyme (ACE) inhibitors, and diuretics.2
Some treatment guidelines also call for multiple medications. Achieving the more stringent blood pressure goals recommended in the Seventh Report of the Joint National Committee on Prevention, for instance, often requires 2 or more antihypertensive agents.6 In many cases, however, patients end up taking more drugs than necessary.
Is the patient taking the right drugs?
Medication reconciliation (determining whether the treatment regimen is appropriate for the patient’s diagnoses) is the way to find out.
The most widely recommended approach to medication reconciliation is to create a table and do a systematic review.7 List all the patient’s medical conditions in the first column and all current medications in the second column. Use the third column to note whether each medication is one the patient should be on, based not only on his or her medical conditions and other drugs being taken but also on current renal and hepatic function and body size, and contraindications.
A medication may be inappropriate if it duplicates, cancels out the action of, or otherwise interacts with another drug the patient is taking; is contraindicated in older patients; or is ineffective for the condition for which it was prescribed. In one key study of nearly 200 patients 65 years and older who took 5 or more medications, more than half had been prescribed at least one drug that was ineffective for the patient’s condition or that duplicated the action of another medication.8
In addition to finding drugs that the patient should not be taking, medication reconciliation may also reveal that the patient is not receiving optimal therapy and that one or more drugs should be added to his or her treatment regimen.
Check meds after transitions. A move from home to hospital, from emergency department to home, or any other transition relating to patient care should prompt a medication reconciliation. Medications are often added or inadvertently discontinued at such times,9,10 and instructions relating to medication are often misunderstood.11 In one study of 384 frail elderly patients being discharged from a hospital, for example, 44% were found to have been given at least one unnecessary prescription—most commonly for a medication that was neither indicated nor effective for any of the patient’s medical problems.12 It was also common for patients to be given drugs that duplicated the action of others they were already taking.
Even in the absence of such transitions, medication reconciliation should occur at regular intervals. Many physicians do a medication reconciliation at every visit to ensure that the medical record is accurate and the patient’s medication regimen is optimal.
Managing polypharmacy: These resources can help
Numerous tools are available to help you evaluate and monitor patients’ medication regimens, including some that were developed specifically for older patients.
START (Screening Tool to Alert doctors to Right Treatment) identifies drugs and drug classes that are underused with older patients.13 START criteria (TABLE 1)13-17 focus on medications that should be used yet are often omitted in older patients who have the appropriate indications.
TABLE 1
START criteria: Drug therapy that should be given to older patients13-17
Cardiovascular
|
Endocrine
|
Gastrointestinal
|
Musculoskeletal
|
Nervous system
|
Respiratory
|
| ACE, angiotensin-converting enzyme; BP, blood pressure; COPD, chronic obstructive pulmonary disease; CVD, cardiovascular disease; FEV1, forced expiratory volume in 1 second; GI, gastrointestinal; MI, myocardial infarction; PPI, proton-pump inhibitor; START, Screening Tool to alert doctors to right Treatment. |
In using START or any other drug-related tool, it is important to keep in mind that therapy should be individualized. Not all the medications in the START criteria are appropriate for every patient, and a medication that is indicated for a given medical condition may or may not provide real benefit for a particular patient. That would depend on the individual’s overall health and life expectancy, the goals of treatment, and how long it would take for the patient to realize any benefit from the drug in question.18 A vigorous 79-year-old might benefit from statin therapy for prevention of cardiovascular events, for instance, while a patient like Mrs. R, who is also 79 but has dementia and multiple other medical problems, would be unlikely to live long enough to realize such a benefit.
”Age” assessment tool. One criterion in deciding whether medication(s) are appropriate for an older patient is his or her “physiologic age”—calculated on the basis of the individual’s chronological age and self-reported health status (TABLE 2).19
TABLE 2
Calculating your patient’s “real” age19
| Actual age (y) | Physiologic age (y) | |||||||
|---|---|---|---|---|---|---|---|---|
| Self-reported health | ||||||||
| Excellent | Good | Fair | Poor | |||||
| Male | Female | Male | Female | Male | Female | Male | Female | |
| 65 | 58 | 60 | 64 | 64 | 68 | 66 | 73 | 72 |
| 70 | 62 | 65 | 69 | 69 | 73 | 71 | 78 | 77 |
| 75 | 67 | 70 | 74 | 74 | 78 | 76 | 83 | 82 |
| 80 | 72 | 75 | 79 | 79 | 83 | 81 | 85+ | 85+ |
Flagging drugs that may be inappropriate
Several tools have been developed to aid clinicians in identifying medications that are potentially inappropriate for older adults, although here, too, decisions about their use must be individualized. Two of the most widely used tools are the Beers criteria and STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions).
Beers criteria were developed by Mark Beers et al in 199120 and have been updated at regular intervals, most recently by the American Geriatrics Society in 2012.21 The drugs and drug classes included in the Beers criteria should not be prescribed for older patients in most cases, either because the risk of using them outweighs the benefit or because safer alternatives are available. Key components are listed in TABLE 3.21
TABLE 3
Beers criteria:* Drug classes that may be inappropriate for older adults21
| Drug class | Concern |
|---|---|
| Alpha-blockers with peripheral activity | Orthostatic hypotension |
| Anticholinergics | Cognitive impairment, urinary retention |
| Antipsychotics | Increased death rate when used for behavior control in patients with dementia |
| NSAIDs | Renal dysfunction, GI bleeding, fluid retention, exacerbation of heart failure |
| Sedative hypnotics | Cognitive impairment, delirium |
| Tricyclic antidepressants | Cognitive impairment, delirium, urinary retention |
| GI, gastrointestinal; NSAIDs, nonsteroidal anti-inflammatory drugs. *The full Beers criteria contains 53 drugs and drug classes that are generally inappropriate for older adults. The full list is available from the American Geriatrics Society at: www.americangeriatrics.org/files/documents/beers/2012BeersCriteria_JAGS.pdf. | |
One limitation of the Beers criteria has been its all-or-nothing approach, with many of the medications on the list deemed inappropriate for all older adults regardless of their circumstances. The 2012 update does a better job of individualizing recommendations: Medications are now categorized as those that should be avoided in older patients regardless of their diseases or conditions, those that should be avoided only in patients with certain diseases or conditions, and those that may be used for this patient population but require caution.21
STOPP is similar to the Beers criteria, but uses a different approach: Most medications on this list are considered in the context of specific medical problems.22 While the Beers criteria classify digoxin >0.125 mg/d as generally inappropriate for older adults, for example, STOPP criteria state that long-term dosing at that level is inappropriate only for those with impaired renal function.22 A list of medications identified by STOPP as contributing to hospitalization due to adverse drug effects is available at http://ageing.oxfordjournals.org/content/37/6/673.
Both tools address this drug category. Cumulative anticholinergic burden is a concept applied to the use of anticholinergic medications, which are included in both the Beers and STOPP criteria. Although isolated short-term exposure to a drug with anticholinergic properties may be tolerated by a healthy and cognitively intact older patient, repetitive exposure to such drugs, even if separated in time, has negative effects. One study evaluated more than 500 community-dwelling older adults and found that the more exposure an individual had to anticholinergic medications over the course of a year, the greater the impairment in short-term memory and activities of daily living.23 Another study, this one involving more than 13,000 community-dwelling and institutionalized patients, showed that the longer an older patient takes an anticholinergic medication, the more likely there is to be a measurable decline in performance on the Mini-Mental State Examination.24
Programs that flag potential interactions
Drug-drug interactions are a key concern of polypharmacy, and electronic medical records and prescribing systems that flag potential drug-drug interactions when a new medication is ordered are designed to help physicians avoid them. Unfortunately, clinicians only react to 3% to 9% of such notifications, overriding them because computerized systems often fail to distinguish between important and unimportant interactions.25-27 Thus, clinicians often must decide whether to react to or override warnings, an often difficult decision with patient safety and medicolegal implications. The best advice we can offer is to carefully evaluate drug interaction warnings using common sense, and seek consultation with a clinical pharmacist when uncertainty exists. This approach should prevent prescribing medications that have potentially harmful interactions with drugs the patient is already taking.
For physicians who do not have access to an electronic prescribing system that provides such notification, several online resources are available, some by subscription (eg, Lexicomp, www.lexi.com; Micromedex, www.micromedex.com/index.html; and Pepid, www.pepid.com) and others with free access (eg, AARP, healthtools.aarp.org/drug-interactions; Drugs.com (www.drugs.com/drug_interactions.php; and HealthLine, www.healthline.com/druginteractions).
CASE After doing a medication reconciliation for Mrs. R, you find that she is taking tolterodine, an anticholinergic medication for urge urinary incontinence, and donepezil, a procholinergic medication for dementia. This type of drug-drug interaction, in which the action of one drug effectively cancels out the effect of another, should not be ignored.
Overall, you identify 8 of her medications that could be discontinued: The list includes guaifenesin (a nonessential medication of questionable efficacy); naproxen (inappropriate per Beers criteria; inappropriate in patients with heart failure, according to STOPP); alprazolam, zolpidem, and diphenhydramine (duplicate medications that are all on the Beers criteria as inappropriate for chronic use and ill-advised in patients with cognitive impairment); and omeprazole and levothyroxine (for which nothing in the patient’s history suggests a need), as well as tolterodine. Depending on dose, digoxin is yet another candidate for discontinuation.
Discontinuing medications: Proceed carefully
Physicians are often reluctant to discontinue chronic medications in older patients—even in those with advanced disease who are not likely to benefit from treatment. Focus groups have identified a number of reasons for their hesitation, including:
- the assumption that patients have no problem taking large numbers of drugs
- the fear that patients may misinterpret a plan to discontinue medications as evidence that the physician is giving up on them
- the belief that physicians must comply with practice guidelines that recommend multiple drug treatments
- concern that proposing discontinuation of medications often leads to a discussion of life expectancy and end-of-life care.28
Physicians may also fear that discontinuation of certain drugs will increase the risk of adverse outcomes. More than 30 studies have evaluated discontinuation of chronic medications in older adults, however, and found that drugs as diverse as antihypertensives, antipsychotics, benzodiazepines, and selective serotonin reuptake inhibitors (SSRIs) can often be discontinued without adverse outcomes. In many cases, improvement in patient function results.29 Medications that present the most difficulty are those that patients often become physically or psychologically dependent on, such as benzodiazepines, guaifenesin, proton-pump inhibitors, nonsteroidal anti-inflammatory drugs, and SSRIs. Some (eg, benzodiazepines, SSRIs) require a gradual reduction; for others, no taper is required
(TABLE 4).30-37
TABLE 4
Recommendations for discontinuing hard-to-stop drugs
| Medication or drug class | Discontinuation regimen | Comments |
|---|---|---|
| Benzodiazepines30 | Taper dose by 25% q 2 wk | No withdrawal symptoms reported with this taper regimen. Subtle cognitive improvement noted over a period of months |
| Guaifenesin31 | Can be discontinued without tapering if not combined with opioids or other medications. Elimination half-life is approximately 1 hour | Guaifenesin is often marketed as a combination product with opioids; such combination products require tapering |
| PPIs32-34 | Decrease dose by 50% q 2 wk; supplement with H2 blocker if needed, but tapering of H2 blocker may be required | Abrupt discontinuation after long-term use causes rebound gastric acid hypersecretion and lowers rate of success. Higher success rates with taper regimen and in patients who do not have documented GERD |
| NSAIDs35 | No taper required | Short-term use (<3 mo) acceptable for patients with no contraindications |
| SSRIs36,37 | Gradual reduction in dose over 6-8 wk | Highest rate of success in patients without a clear diagnosis of depression |
| GERD, gastroesophageal reflux disease; NSAIDs, nonsteroidal anti-inflammatory drugs; PPIs, proton-pump inhibitors; SSRIs, selective serotonin reuptake inhibitors. | ||
CASE You trim down Mrs. R’s regimen by discontinuing each of the 8 drugs, one at a time, and carefully monitor the patient during the withdrawal period. Because she had been taking alprazolam daily, the dose is tapered slowly to avoid withdrawal. Omeprazole also requires a gradual taper to avoid rebound hyperacidity.3
After confirming that Mrs. R has heart failure and COPD, you identify 2 medications that should be added to her drug regimen—an ACE inhibitor for heart failure and an inhaled anticholinergic for COPD.
Going from 16 medications to 10 saves money, decreases the likelihood of adverse events and drug-drug interactions, and helps with adherence. Mrs. R’s new drug regimen is expected to lead to improvements in memory and overall quality of life, as well.
CORRESPONDENCE
Barry D. Weiss, MD, Department of Family and Community Medicine, University of Arizona College of Medicine, Tucson, AZ 85724; [email protected]
• Consider the possibility that an adverse drug effect—rather than a new condition—is at play when a patient taking multiple medications develops a new symptom. C
• Use an online interaction checker, which can be accessed via a smart phone or tablet, to check for potential drug-drug interactions in patients on multiple medications. C
• Cross-check patients’ medications with a list of their medical problems, with the goal of discontinuing any drug that duplicates the action of another or is age-inappropriate, ineffective, or not indicated for the condition for which it was prescribed. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Older adults are taking more medications than ever before. Nearly 9 out of 10 US residents who are 60 years of age or older take at least one prescription drug, more than a third take 5 to 9 medications, and 12% take 10 or more.1
The increase is largely driven by newer medications to effectively treat a variety of medical conditions, and by practice guidelines that often recommend multidrug regimens.2
As a result, the term “polypharmacy,” which once referred to a specific number of medications, is now used more broadly to mean “a large number” of drugs.
From a safety standpoint, the number of medications a patient takes matters. The risk of adverse drug effects and dangerous drug-drug interactions increases significantly when an individual takes ≥5 medications.3
More than 4.5 million adverse drug effects occur each year in the United States, and nearly three quarters of them are initially evaluated in outpatient settings.4 Research suggests that about 80% of the time, these adverse effects are not recognized as such by the patient’s physician. So instead of discontinuing the offending medication, physicians treat the drug-related symptoms by adding yet another medication—a phenomenon known as “the prescribing cascade.”5
This review can help you safeguard older patients taking multiple medications by recognizing and responding to drug-related problems, identifying drugs that can be safely eliminated (or, in some cases, drugs that should be added), and checking regularly to ensure that the medication regimen is appropriate and up to date.
CASE Mrs. R, a 79-year-old woman who recently moved to town, is brought to your office by her daughter and son-in-law. The patient has a hard time reporting her medical history, but her daughter tells you her mother has chronic obstructive pulmonary disease (COPD), heart failure, type 2 diabetes, and mild urinary incontinence, and was recently diagnosed with early dementia.
Mrs. R’s daughter has brought in a bagful of medications, but she’s not sure which ones her mother takes regularly. The medications are an albuterol inhaler, alprazolam, digoxin, diphenhydramine, donepezil, furosemide, glargine insulin, guaifenesin, levothyroxine, metformin, extended-release metoprolol, naproxen, omeprazole, simvastatin, tolterodine, and zolpidem—a total of 16 different drugs.
If Mrs. R were your patient, how would you manage her multidrug regimen?
Start with a medication review
The first step in evaluating a patient’s medication regimen is to find out whether the drugs in the patient’s possession and/or in the medical record are the ones he or she is actually taking. Ask older patients who haven’t brought in their medications, or the caregiver of a confused patient, to bring them to the next visit.
The next step: Determine whether the medication regimen is right for the patient.
Polypharmacy may be indicated
Despite the risks associated with polypharmacy, do not assume that it is inappropriate. For some conditions, multiple medications are routinely recommended. Patients with heart failure, for example, have been shown to have better outcomes when they take 3 to 5 medications, including beta-blockers, angiotensin-converting enzyme (ACE) inhibitors, and diuretics.2
Some treatment guidelines also call for multiple medications. Achieving the more stringent blood pressure goals recommended in the Seventh Report of the Joint National Committee on Prevention, for instance, often requires 2 or more antihypertensive agents.6 In many cases, however, patients end up taking more drugs than necessary.
Is the patient taking the right drugs?
Medication reconciliation (determining whether the treatment regimen is appropriate for the patient’s diagnoses) is the way to find out.
The most widely recommended approach to medication reconciliation is to create a table and do a systematic review.7 List all the patient’s medical conditions in the first column and all current medications in the second column. Use the third column to note whether each medication is one the patient should be on, based not only on his or her medical conditions and other drugs being taken but also on current renal and hepatic function and body size, and contraindications.
A medication may be inappropriate if it duplicates, cancels out the action of, or otherwise interacts with another drug the patient is taking; is contraindicated in older patients; or is ineffective for the condition for which it was prescribed. In one key study of nearly 200 patients 65 years and older who took 5 or more medications, more than half had been prescribed at least one drug that was ineffective for the patient’s condition or that duplicated the action of another medication.8
In addition to finding drugs that the patient should not be taking, medication reconciliation may also reveal that the patient is not receiving optimal therapy and that one or more drugs should be added to his or her treatment regimen.
Check meds after transitions. A move from home to hospital, from emergency department to home, or any other transition relating to patient care should prompt a medication reconciliation. Medications are often added or inadvertently discontinued at such times,9,10 and instructions relating to medication are often misunderstood.11 In one study of 384 frail elderly patients being discharged from a hospital, for example, 44% were found to have been given at least one unnecessary prescription—most commonly for a medication that was neither indicated nor effective for any of the patient’s medical problems.12 It was also common for patients to be given drugs that duplicated the action of others they were already taking.
Even in the absence of such transitions, medication reconciliation should occur at regular intervals. Many physicians do a medication reconciliation at every visit to ensure that the medical record is accurate and the patient’s medication regimen is optimal.
Managing polypharmacy: These resources can help
Numerous tools are available to help you evaluate and monitor patients’ medication regimens, including some that were developed specifically for older patients.
START (Screening Tool to Alert doctors to Right Treatment) identifies drugs and drug classes that are underused with older patients.13 START criteria (TABLE 1)13-17 focus on medications that should be used yet are often omitted in older patients who have the appropriate indications.
TABLE 1
START criteria: Drug therapy that should be given to older patients13-17
Cardiovascular
|
Endocrine
|
Gastrointestinal
|
Musculoskeletal
|
Nervous system
|
Respiratory
|
| ACE, angiotensin-converting enzyme; BP, blood pressure; COPD, chronic obstructive pulmonary disease; CVD, cardiovascular disease; FEV1, forced expiratory volume in 1 second; GI, gastrointestinal; MI, myocardial infarction; PPI, proton-pump inhibitor; START, Screening Tool to alert doctors to right Treatment. |
In using START or any other drug-related tool, it is important to keep in mind that therapy should be individualized. Not all the medications in the START criteria are appropriate for every patient, and a medication that is indicated for a given medical condition may or may not provide real benefit for a particular patient. That would depend on the individual’s overall health and life expectancy, the goals of treatment, and how long it would take for the patient to realize any benefit from the drug in question.18 A vigorous 79-year-old might benefit from statin therapy for prevention of cardiovascular events, for instance, while a patient like Mrs. R, who is also 79 but has dementia and multiple other medical problems, would be unlikely to live long enough to realize such a benefit.
”Age” assessment tool. One criterion in deciding whether medication(s) are appropriate for an older patient is his or her “physiologic age”—calculated on the basis of the individual’s chronological age and self-reported health status (TABLE 2).19
TABLE 2
Calculating your patient’s “real” age19
| Actual age (y) | Physiologic age (y) | |||||||
|---|---|---|---|---|---|---|---|---|
| Self-reported health | ||||||||
| Excellent | Good | Fair | Poor | |||||
| Male | Female | Male | Female | Male | Female | Male | Female | |
| 65 | 58 | 60 | 64 | 64 | 68 | 66 | 73 | 72 |
| 70 | 62 | 65 | 69 | 69 | 73 | 71 | 78 | 77 |
| 75 | 67 | 70 | 74 | 74 | 78 | 76 | 83 | 82 |
| 80 | 72 | 75 | 79 | 79 | 83 | 81 | 85+ | 85+ |
Flagging drugs that may be inappropriate
Several tools have been developed to aid clinicians in identifying medications that are potentially inappropriate for older adults, although here, too, decisions about their use must be individualized. Two of the most widely used tools are the Beers criteria and STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions).
Beers criteria were developed by Mark Beers et al in 199120 and have been updated at regular intervals, most recently by the American Geriatrics Society in 2012.21 The drugs and drug classes included in the Beers criteria should not be prescribed for older patients in most cases, either because the risk of using them outweighs the benefit or because safer alternatives are available. Key components are listed in TABLE 3.21
TABLE 3
Beers criteria:* Drug classes that may be inappropriate for older adults21
| Drug class | Concern |
|---|---|
| Alpha-blockers with peripheral activity | Orthostatic hypotension |
| Anticholinergics | Cognitive impairment, urinary retention |
| Antipsychotics | Increased death rate when used for behavior control in patients with dementia |
| NSAIDs | Renal dysfunction, GI bleeding, fluid retention, exacerbation of heart failure |
| Sedative hypnotics | Cognitive impairment, delirium |
| Tricyclic antidepressants | Cognitive impairment, delirium, urinary retention |
| GI, gastrointestinal; NSAIDs, nonsteroidal anti-inflammatory drugs. *The full Beers criteria contains 53 drugs and drug classes that are generally inappropriate for older adults. The full list is available from the American Geriatrics Society at: www.americangeriatrics.org/files/documents/beers/2012BeersCriteria_JAGS.pdf. | |
One limitation of the Beers criteria has been its all-or-nothing approach, with many of the medications on the list deemed inappropriate for all older adults regardless of their circumstances. The 2012 update does a better job of individualizing recommendations: Medications are now categorized as those that should be avoided in older patients regardless of their diseases or conditions, those that should be avoided only in patients with certain diseases or conditions, and those that may be used for this patient population but require caution.21
STOPP is similar to the Beers criteria, but uses a different approach: Most medications on this list are considered in the context of specific medical problems.22 While the Beers criteria classify digoxin >0.125 mg/d as generally inappropriate for older adults, for example, STOPP criteria state that long-term dosing at that level is inappropriate only for those with impaired renal function.22 A list of medications identified by STOPP as contributing to hospitalization due to adverse drug effects is available at http://ageing.oxfordjournals.org/content/37/6/673.
Both tools address this drug category. Cumulative anticholinergic burden is a concept applied to the use of anticholinergic medications, which are included in both the Beers and STOPP criteria. Although isolated short-term exposure to a drug with anticholinergic properties may be tolerated by a healthy and cognitively intact older patient, repetitive exposure to such drugs, even if separated in time, has negative effects. One study evaluated more than 500 community-dwelling older adults and found that the more exposure an individual had to anticholinergic medications over the course of a year, the greater the impairment in short-term memory and activities of daily living.23 Another study, this one involving more than 13,000 community-dwelling and institutionalized patients, showed that the longer an older patient takes an anticholinergic medication, the more likely there is to be a measurable decline in performance on the Mini-Mental State Examination.24
Programs that flag potential interactions
Drug-drug interactions are a key concern of polypharmacy, and electronic medical records and prescribing systems that flag potential drug-drug interactions when a new medication is ordered are designed to help physicians avoid them. Unfortunately, clinicians only react to 3% to 9% of such notifications, overriding them because computerized systems often fail to distinguish between important and unimportant interactions.25-27 Thus, clinicians often must decide whether to react to or override warnings, an often difficult decision with patient safety and medicolegal implications. The best advice we can offer is to carefully evaluate drug interaction warnings using common sense, and seek consultation with a clinical pharmacist when uncertainty exists. This approach should prevent prescribing medications that have potentially harmful interactions with drugs the patient is already taking.
For physicians who do not have access to an electronic prescribing system that provides such notification, several online resources are available, some by subscription (eg, Lexicomp, www.lexi.com; Micromedex, www.micromedex.com/index.html; and Pepid, www.pepid.com) and others with free access (eg, AARP, healthtools.aarp.org/drug-interactions; Drugs.com (www.drugs.com/drug_interactions.php; and HealthLine, www.healthline.com/druginteractions).
CASE After doing a medication reconciliation for Mrs. R, you find that she is taking tolterodine, an anticholinergic medication for urge urinary incontinence, and donepezil, a procholinergic medication for dementia. This type of drug-drug interaction, in which the action of one drug effectively cancels out the effect of another, should not be ignored.
Overall, you identify 8 of her medications that could be discontinued: The list includes guaifenesin (a nonessential medication of questionable efficacy); naproxen (inappropriate per Beers criteria; inappropriate in patients with heart failure, according to STOPP); alprazolam, zolpidem, and diphenhydramine (duplicate medications that are all on the Beers criteria as inappropriate for chronic use and ill-advised in patients with cognitive impairment); and omeprazole and levothyroxine (for which nothing in the patient’s history suggests a need), as well as tolterodine. Depending on dose, digoxin is yet another candidate for discontinuation.
Discontinuing medications: Proceed carefully
Physicians are often reluctant to discontinue chronic medications in older patients—even in those with advanced disease who are not likely to benefit from treatment. Focus groups have identified a number of reasons for their hesitation, including:
- the assumption that patients have no problem taking large numbers of drugs
- the fear that patients may misinterpret a plan to discontinue medications as evidence that the physician is giving up on them
- the belief that physicians must comply with practice guidelines that recommend multiple drug treatments
- concern that proposing discontinuation of medications often leads to a discussion of life expectancy and end-of-life care.28
Physicians may also fear that discontinuation of certain drugs will increase the risk of adverse outcomes. More than 30 studies have evaluated discontinuation of chronic medications in older adults, however, and found that drugs as diverse as antihypertensives, antipsychotics, benzodiazepines, and selective serotonin reuptake inhibitors (SSRIs) can often be discontinued without adverse outcomes. In many cases, improvement in patient function results.29 Medications that present the most difficulty are those that patients often become physically or psychologically dependent on, such as benzodiazepines, guaifenesin, proton-pump inhibitors, nonsteroidal anti-inflammatory drugs, and SSRIs. Some (eg, benzodiazepines, SSRIs) require a gradual reduction; for others, no taper is required
(TABLE 4).30-37
TABLE 4
Recommendations for discontinuing hard-to-stop drugs
| Medication or drug class | Discontinuation regimen | Comments |
|---|---|---|
| Benzodiazepines30 | Taper dose by 25% q 2 wk | No withdrawal symptoms reported with this taper regimen. Subtle cognitive improvement noted over a period of months |
| Guaifenesin31 | Can be discontinued without tapering if not combined with opioids or other medications. Elimination half-life is approximately 1 hour | Guaifenesin is often marketed as a combination product with opioids; such combination products require tapering |
| PPIs32-34 | Decrease dose by 50% q 2 wk; supplement with H2 blocker if needed, but tapering of H2 blocker may be required | Abrupt discontinuation after long-term use causes rebound gastric acid hypersecretion and lowers rate of success. Higher success rates with taper regimen and in patients who do not have documented GERD |
| NSAIDs35 | No taper required | Short-term use (<3 mo) acceptable for patients with no contraindications |
| SSRIs36,37 | Gradual reduction in dose over 6-8 wk | Highest rate of success in patients without a clear diagnosis of depression |
| GERD, gastroesophageal reflux disease; NSAIDs, nonsteroidal anti-inflammatory drugs; PPIs, proton-pump inhibitors; SSRIs, selective serotonin reuptake inhibitors. | ||
CASE You trim down Mrs. R’s regimen by discontinuing each of the 8 drugs, one at a time, and carefully monitor the patient during the withdrawal period. Because she had been taking alprazolam daily, the dose is tapered slowly to avoid withdrawal. Omeprazole also requires a gradual taper to avoid rebound hyperacidity.3
After confirming that Mrs. R has heart failure and COPD, you identify 2 medications that should be added to her drug regimen—an ACE inhibitor for heart failure and an inhaled anticholinergic for COPD.
Going from 16 medications to 10 saves money, decreases the likelihood of adverse events and drug-drug interactions, and helps with adherence. Mrs. R’s new drug regimen is expected to lead to improvements in memory and overall quality of life, as well.
CORRESPONDENCE
Barry D. Weiss, MD, Department of Family and Community Medicine, University of Arizona College of Medicine, Tucson, AZ 85724; [email protected]
1. Gu Q, Dillon CF, Burt V. Prescription drug use continued to increase: US prescription drug data for 2007-2008. CDC/NCHS Data Brief. 2010;42:1-2.
2. Jessup K, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA guidelines for the diagnosis and management of heart failure in adults. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2009;119:1977-2016.
3. Johnell K, Klarin I. The relationship between number of drugs and potential drug-drug interactions in the elderly: a study of over 600,000 elderly patients from the Swedish Prescribed Drug Register. Drug Saf. 2007;30:911-918.
4. Sarkar U, Lopez A, Maselli JH, et al. Adverse drug events in US adult ambulatory medical care. Health Services Res. 2011;46:1517-1533.
5. Rollason V, Vogt N. Reduction of polypharmacy in the elderly. A systematic review of the role of the pharmacist. Drugs Aging. 2003;20:817-832.
6. National Heart, Lung, and Blood Institute. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Available at: www.nhlbi.nih.gov/guidelines/hypertension/jnc7full.pdf. Accessed October 11, 2012.
7. Steinman MA, Hanlon JT. Managing medications in clinically complex elders. JAMA. 2010;304:1592-1601.
8. Steinman MA, Landefeld CS, Rosenthal GE, et al. Polypharmacy and prescribing quality in older people. J Am Geriatr Soc. 2006;54:1516-23.
9. Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuations of medications for chronic disease. JAMA. 2011;306:840-847.
10. Moore C, Wisnivesky J, Williams S, et al. Medical errors related to discontinuity of care from an inpatient to an outpatient setting. J Gen Intern Med. 2003;18:646-651.
11. Ziaeian B, Arauho KL, Van Ness PH, et al. Medication reconciliation accuracy and patient understanding of intended medication changes on hospital discharge. J Gen Intern Med. 2012 July 12. ePub ahead of print.
12. Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc. 2005;53:1518-1523.
13. O’Mahony D, Gallagher P, Ryan C, et al. STOPP & START criteria: a new approach to detecting potentially inappropriate prescribing in old age. Eur Geriatr Med. 2010;1:45-51.
14. Denneboom W, Dautzenberg KGH, Grol R, et al. Analysis of polypharmacy in older patients in primary acre using a multidisciplinary expert panel. Br J Gen Pract. 2006;56:504-510.
15. Ko DT, Mamdani M, Alter DA. Lipid-lowering therapy with statins in high-risk elderly patients. JAMA. 2004;291:1864-70.
16. Wright RM, Sloane R, Pieper CF, et al. Underuse of indicated medications among physically frail older US veterans at the time of hospital discharge: results of a cross-sectional analysis of data from the Geriatric Evaluation and Management Drug Study. Am J Geriatr Pharmacother. 2009;7:271-280.
17. Garwood CL. Use of anticoagulation in elderly patients with atrial fibrillation who are risk for falls. Ann Pharmacother. 2008;42:523-532.
18. Holmes HM, Hayley DC, Alexander GC, et al. Reconsidering medication appropriateness for patients late in life. Arch Intern Med. 2006;166:605-609.
19. Simplified Methods for Estimating Life Expectancy. Available at: http://painconsortium.nih.gov/symptomresearch/chapter_14/Part_3/sec4/chspt3s4pg1.htm. Accessed October 9, 2012.
20. Beers MH, Ouslander JG, Rollingher I, et al. Explicit criteria for determining inappropriate medication use in nursing home residents. Arch Intern Med. 1991;151:1825-1832.
21. The American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society Update Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60:616-631.
22. Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Aging. 2008;37:673-379.
23. Han L, Agostini JV, Allore HG. Cumulative anticholinergic exposure is associated with poor memory and executive function in older men. J Am Geriatr Soc. 2008;56:2203-2210.
24. Fox C, Richardson K, Maidment ID, et al. Anticholinergic medication use and cognitive impairment in the older population: the medical research council cognitive function and ageing study. J Am Geriatr Soc. 2011;59:1477-1483.
25. Knight A, Falade O, Maygers J, et al. Factors associated with medication warning acceptance [abstract]. J Hosp Med. 2012;7(suppl 2):515.-
26. Isaac T, Weissman JS, Davis RB, et al. Overrides of medication alerts in ambulatory care. Arch Intern Med. 2009;169:305-311.
27. Van Der Sijs H, Aarts J, Vulto A, et al. Overriding of drug safety alerts in computerized physician order entry. J Am Med Inform Assoc. 2006;12:138-147.
28. Schuling J, Gebben H, Veehof LJG, et al. Deprescribing medication in very elderly patients with multimorbidity: the view of Dutch GPs. A qualitative study. BMC Family Practice. 2012;13:56. http://www.biomedcentral.com/1471-2296/13/56.
29. Iyer S, Naganathan V, McLachlan AJ, et al. Medication withdrawal trials in people aged 65 years and older. A systematic review. Drugs Aging. 2008;25:1021-1031.
30. Curran HV, Collins R, Fletcher S, et al. Older adults and withdrawal from benzodiazepine hypnotics in general practice: effects on cognitive function, sleep, mood and quality of life. Psychol Med. 2003;33:1223-1237.
31. Krinsky DL, Berardi RR, Ferreris SP, et al. Handbook of Nonprescription Drugs: An Interactive Approach to Self-Care. Washington, DC: American Pharmacists Association; 2012:209.
32. Bjornsson E, Abrahamsson H, Simren M, et al. Discontinuation of proton pump inhibitors in patients on long-term therapy: a double-blind, placebo-controlled trial. Aliment Pharmacol Ther. 2006;24:945-954.
33. Inadomi JM, Jamai R, Murata GH, et al. Step-down management of gastroesophageal reflux disease. Gastroenterology. 2001;131:1095-1100.
34. Hester SA. Proton pump inhibitors and rebound acid hypersecretion. Pharm Lett. 2009;25:250920.-
35. Taylor R, Jr, Lemtouni S, Weiss K, et al. Pain management in the elderly: an FDA safe use initiative expert panel’s view on preventable harm associated with NSAID therapy. Curr Gerontol Geriatr Res. 2012;196159.-
36. Ulfvarson J, Adami J, Wredling R, et al. Controlled withdrawal of selective serotonin reuptake inhibitor drugs in elderly patients in nursing homes with no indication of depression. Eur J Clin Pharmacol. 2003;59:735-740.
37. Lindstrom K, Ekedahl A, Carlsten A, et al. Can selective serotonin inhibitor drugs in elderly patients in nursing homes be reduced? Scand J Prim Health Care. 2007;25:3-8.
1. Gu Q, Dillon CF, Burt V. Prescription drug use continued to increase: US prescription drug data for 2007-2008. CDC/NCHS Data Brief. 2010;42:1-2.
2. Jessup K, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA guidelines for the diagnosis and management of heart failure in adults. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2009;119:1977-2016.
3. Johnell K, Klarin I. The relationship between number of drugs and potential drug-drug interactions in the elderly: a study of over 600,000 elderly patients from the Swedish Prescribed Drug Register. Drug Saf. 2007;30:911-918.
4. Sarkar U, Lopez A, Maselli JH, et al. Adverse drug events in US adult ambulatory medical care. Health Services Res. 2011;46:1517-1533.
5. Rollason V, Vogt N. Reduction of polypharmacy in the elderly. A systematic review of the role of the pharmacist. Drugs Aging. 2003;20:817-832.
6. National Heart, Lung, and Blood Institute. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Available at: www.nhlbi.nih.gov/guidelines/hypertension/jnc7full.pdf. Accessed October 11, 2012.
7. Steinman MA, Hanlon JT. Managing medications in clinically complex elders. JAMA. 2010;304:1592-1601.
8. Steinman MA, Landefeld CS, Rosenthal GE, et al. Polypharmacy and prescribing quality in older people. J Am Geriatr Soc. 2006;54:1516-23.
9. Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuations of medications for chronic disease. JAMA. 2011;306:840-847.
10. Moore C, Wisnivesky J, Williams S, et al. Medical errors related to discontinuity of care from an inpatient to an outpatient setting. J Gen Intern Med. 2003;18:646-651.
11. Ziaeian B, Arauho KL, Van Ness PH, et al. Medication reconciliation accuracy and patient understanding of intended medication changes on hospital discharge. J Gen Intern Med. 2012 July 12. ePub ahead of print.
12. Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc. 2005;53:1518-1523.
13. O’Mahony D, Gallagher P, Ryan C, et al. STOPP & START criteria: a new approach to detecting potentially inappropriate prescribing in old age. Eur Geriatr Med. 2010;1:45-51.
14. Denneboom W, Dautzenberg KGH, Grol R, et al. Analysis of polypharmacy in older patients in primary acre using a multidisciplinary expert panel. Br J Gen Pract. 2006;56:504-510.
15. Ko DT, Mamdani M, Alter DA. Lipid-lowering therapy with statins in high-risk elderly patients. JAMA. 2004;291:1864-70.
16. Wright RM, Sloane R, Pieper CF, et al. Underuse of indicated medications among physically frail older US veterans at the time of hospital discharge: results of a cross-sectional analysis of data from the Geriatric Evaluation and Management Drug Study. Am J Geriatr Pharmacother. 2009;7:271-280.
17. Garwood CL. Use of anticoagulation in elderly patients with atrial fibrillation who are risk for falls. Ann Pharmacother. 2008;42:523-532.
18. Holmes HM, Hayley DC, Alexander GC, et al. Reconsidering medication appropriateness for patients late in life. Arch Intern Med. 2006;166:605-609.
19. Simplified Methods for Estimating Life Expectancy. Available at: http://painconsortium.nih.gov/symptomresearch/chapter_14/Part_3/sec4/chspt3s4pg1.htm. Accessed October 9, 2012.
20. Beers MH, Ouslander JG, Rollingher I, et al. Explicit criteria for determining inappropriate medication use in nursing home residents. Arch Intern Med. 1991;151:1825-1832.
21. The American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society Update Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60:616-631.
22. Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Aging. 2008;37:673-379.
23. Han L, Agostini JV, Allore HG. Cumulative anticholinergic exposure is associated with poor memory and executive function in older men. J Am Geriatr Soc. 2008;56:2203-2210.
24. Fox C, Richardson K, Maidment ID, et al. Anticholinergic medication use and cognitive impairment in the older population: the medical research council cognitive function and ageing study. J Am Geriatr Soc. 2011;59:1477-1483.
25. Knight A, Falade O, Maygers J, et al. Factors associated with medication warning acceptance [abstract]. J Hosp Med. 2012;7(suppl 2):515.-
26. Isaac T, Weissman JS, Davis RB, et al. Overrides of medication alerts in ambulatory care. Arch Intern Med. 2009;169:305-311.
27. Van Der Sijs H, Aarts J, Vulto A, et al. Overriding of drug safety alerts in computerized physician order entry. J Am Med Inform Assoc. 2006;12:138-147.
28. Schuling J, Gebben H, Veehof LJG, et al. Deprescribing medication in very elderly patients with multimorbidity: the view of Dutch GPs. A qualitative study. BMC Family Practice. 2012;13:56. http://www.biomedcentral.com/1471-2296/13/56.
29. Iyer S, Naganathan V, McLachlan AJ, et al. Medication withdrawal trials in people aged 65 years and older. A systematic review. Drugs Aging. 2008;25:1021-1031.
30. Curran HV, Collins R, Fletcher S, et al. Older adults and withdrawal from benzodiazepine hypnotics in general practice: effects on cognitive function, sleep, mood and quality of life. Psychol Med. 2003;33:1223-1237.
31. Krinsky DL, Berardi RR, Ferreris SP, et al. Handbook of Nonprescription Drugs: An Interactive Approach to Self-Care. Washington, DC: American Pharmacists Association; 2012:209.
32. Bjornsson E, Abrahamsson H, Simren M, et al. Discontinuation of proton pump inhibitors in patients on long-term therapy: a double-blind, placebo-controlled trial. Aliment Pharmacol Ther. 2006;24:945-954.
33. Inadomi JM, Jamai R, Murata GH, et al. Step-down management of gastroesophageal reflux disease. Gastroenterology. 2001;131:1095-1100.
34. Hester SA. Proton pump inhibitors and rebound acid hypersecretion. Pharm Lett. 2009;25:250920.-
35. Taylor R, Jr, Lemtouni S, Weiss K, et al. Pain management in the elderly: an FDA safe use initiative expert panel’s view on preventable harm associated with NSAID therapy. Curr Gerontol Geriatr Res. 2012;196159.-
36. Ulfvarson J, Adami J, Wredling R, et al. Controlled withdrawal of selective serotonin reuptake inhibitor drugs in elderly patients in nursing homes with no indication of depression. Eur J Clin Pharmacol. 2003;59:735-740.
37. Lindstrom K, Ekedahl A, Carlsten A, et al. Can selective serotonin inhibitor drugs in elderly patients in nursing homes be reduced? Scand J Prim Health Care. 2007;25:3-8.