User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
FDA Approves 10th Humira Biosimilar, With Interchangeability
The US Food and Drug Administration has approved the first interchangeable, high-concentration, citrate-free adalimumab biosimilar, adalimumab-ryvk (Simlandi).
This is the 10th adalimumab biosimilar approved by the regulatory agency and the first biosimilar approval in the US market for the Icelandic pharmaceutical company Alvotech in partnership with Teva Pharmaceuticals.
“An interchangeable citrate-free, high-concentration biosimilar adalimumab has the potential to change the market dynamics in a rapidly evolving environment for biosimilars in the U.S.,” said Robert Wessman, chairman and CEO of Alvotech, in a company press release on February 23.
Adalimumab-ryvk was approved in the European Union in 2021 and in Australia and Canada in 2022.
Adalimumab-ryvk is indicated for adults with rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, ulcerative colitis, Crohn’s disease, plaque psoriasis, hidradenitis suppurativa, and noninfectious intermediate and posterior uveitis and panuveitis. In pediatric patients, it is indicated for polyarticular juvenile idiopathic arthritis in children 2 years of age and older and Crohn’s disease in children 6 years of age and older.
Adalimumab-ryvk is the third Humira biosimilar overall granted interchangeability status, which allows pharmacists (depending on state law) to substitute the biosimilar for the reference product without involving the prescribing clinician. Adalimumab-adbm (Cyltezo), manufactured by Boehringer Ingelheim, and adalimumab-afzb (Abrilada), manufactured by Pfizer, were previously granted interchangeability status; however, they both are interchangeable with the low-concentration formulation of Humira, which make up only an estimated 15% of Humira prescriptions, according to a report by Goodroot.
Adalimumab-ryvk will be launched “imminently” in the United States, according to the press release, but no specific dates were provided. It is also not yet known how the biosimilar will be priced compared with Humira. Other adalimumab biosimilars have launched with discounts from 5% to 85% of Humira’s list price.
A version of this article appeared on Medscape.com.
The US Food and Drug Administration has approved the first interchangeable, high-concentration, citrate-free adalimumab biosimilar, adalimumab-ryvk (Simlandi).
This is the 10th adalimumab biosimilar approved by the regulatory agency and the first biosimilar approval in the US market for the Icelandic pharmaceutical company Alvotech in partnership with Teva Pharmaceuticals.
“An interchangeable citrate-free, high-concentration biosimilar adalimumab has the potential to change the market dynamics in a rapidly evolving environment for biosimilars in the U.S.,” said Robert Wessman, chairman and CEO of Alvotech, in a company press release on February 23.
Adalimumab-ryvk was approved in the European Union in 2021 and in Australia and Canada in 2022.
Adalimumab-ryvk is indicated for adults with rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, ulcerative colitis, Crohn’s disease, plaque psoriasis, hidradenitis suppurativa, and noninfectious intermediate and posterior uveitis and panuveitis. In pediatric patients, it is indicated for polyarticular juvenile idiopathic arthritis in children 2 years of age and older and Crohn’s disease in children 6 years of age and older.
Adalimumab-ryvk is the third Humira biosimilar overall granted interchangeability status, which allows pharmacists (depending on state law) to substitute the biosimilar for the reference product without involving the prescribing clinician. Adalimumab-adbm (Cyltezo), manufactured by Boehringer Ingelheim, and adalimumab-afzb (Abrilada), manufactured by Pfizer, were previously granted interchangeability status; however, they both are interchangeable with the low-concentration formulation of Humira, which make up only an estimated 15% of Humira prescriptions, according to a report by Goodroot.
Adalimumab-ryvk will be launched “imminently” in the United States, according to the press release, but no specific dates were provided. It is also not yet known how the biosimilar will be priced compared with Humira. Other adalimumab biosimilars have launched with discounts from 5% to 85% of Humira’s list price.
A version of this article appeared on Medscape.com.
The US Food and Drug Administration has approved the first interchangeable, high-concentration, citrate-free adalimumab biosimilar, adalimumab-ryvk (Simlandi).
This is the 10th adalimumab biosimilar approved by the regulatory agency and the first biosimilar approval in the US market for the Icelandic pharmaceutical company Alvotech in partnership with Teva Pharmaceuticals.
“An interchangeable citrate-free, high-concentration biosimilar adalimumab has the potential to change the market dynamics in a rapidly evolving environment for biosimilars in the U.S.,” said Robert Wessman, chairman and CEO of Alvotech, in a company press release on February 23.
Adalimumab-ryvk was approved in the European Union in 2021 and in Australia and Canada in 2022.
Adalimumab-ryvk is indicated for adults with rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, ulcerative colitis, Crohn’s disease, plaque psoriasis, hidradenitis suppurativa, and noninfectious intermediate and posterior uveitis and panuveitis. In pediatric patients, it is indicated for polyarticular juvenile idiopathic arthritis in children 2 years of age and older and Crohn’s disease in children 6 years of age and older.
Adalimumab-ryvk is the third Humira biosimilar overall granted interchangeability status, which allows pharmacists (depending on state law) to substitute the biosimilar for the reference product without involving the prescribing clinician. Adalimumab-adbm (Cyltezo), manufactured by Boehringer Ingelheim, and adalimumab-afzb (Abrilada), manufactured by Pfizer, were previously granted interchangeability status; however, they both are interchangeable with the low-concentration formulation of Humira, which make up only an estimated 15% of Humira prescriptions, according to a report by Goodroot.
Adalimumab-ryvk will be launched “imminently” in the United States, according to the press release, but no specific dates were provided. It is also not yet known how the biosimilar will be priced compared with Humira. Other adalimumab biosimilars have launched with discounts from 5% to 85% of Humira’s list price.
A version of this article appeared on Medscape.com.
Few Pediatricians Comfortable Treating Youth With OUD
An estimated 1 in 100 adolescents ages 12-17 years in the United States have an opioid use disorder (OUD). But fewer than 5% of adolescents with OUD get buprenorphine or naltrexone, though the treatments are recommended by the American Academy of Pediatrics (AAP), new data show.
Meanwhile, adolescent drug overdose deaths more than doubled between 2019 and 2021, with most involving opioids.
Scott E. Hadland, MD, MPH, with the Division of Adolescent and Young Adult Medicine at Mass General for Children in Boston, and colleagues detailed the extent of the treatment gap and barriers to prescribing and caring for youth with OUD in primary care in a research letter published February 26 in JAMA Pediatrics.
Dr. Hadland’s team mailed 1,681 US pediatricians a survey and the response rate was 43.0%. Researchers included in the sample 474 primary care pediatricians who care for adolescents.
Who Should Treat OUD?
Most respondents (average age, 49.5; 74.0% female) agreed or strongly agreed that it is their responsibility to identify substance use disorders (93.9%) and refer patients to treatment (97.4%).
Fewer agreed or strongly agreed that it is their responsibility to treat substance use disorders (20.3%) or prescribe medications (12.4%). Fewer than half of the respondents said they felt prepared or very prepared to counsel adolescents on opioid use (48.3%) compared with those comfortable counseling on alcohol (87.1%), cannabis (81.7%), and e-cigarette use (80.1%; P < .001).
Pediatricians were less likely to provide counseling (63.0%) and more likely to refer patients to care off-site (71.8%) for opioid use than for alcohol (87.7% and 51.7%); cannabis (88.9% and 45.4%); or e-cigarette use (91.6% and 26.5%) (P < .001 for all comparisons).
Training Lacking in Residency Programs
“These results reveal an opportunity for greater workforce training in line with a 2019 survey showing fewer than 1 in 3 US pediatric residency programs included education on prescribing OUD medications,” the authors wrote. Training focused on treating OUD in primary care, including prescribing medications and addressing possible misperceptions, may be needed,” they noted.
The survey predated the elimination in 2023 of the federal buprenorphine waiver requirement, which made prescribing buprenorphine easier, so these results do not reflect any changes from that elimination, they wrote.
Sharon Levy, MD, MPH, chief of the Division of Addiction Medicine at Boston Children’s Hospital and professor of pediatrics at Harvard Medical School in Boston, who was not part of this study, said more education on addiction medicine is needed for general pediatricians.
She said it’s time to push beyond the current framework of Screening, Brief Intervention, Referral to Treatment (SBIRT), because that doesn’t include “prescribing medications to manage withdrawal or suppress cravings or the use of lab testing, both of which could be accomplished in primary care.”
Dr. Levy said she considers substance use disorders the same way she considers other chronic conditions: Most patients can be treated in primary care. “Specialty care and higher levels of care need to be available for patients who are most complicated and/or having a flare of their condition.”
“In my opinion, most teens with opioid use disorder can and should be treated in primary care. I worry about referring teens with opioid use disorder to get medication somewhere else because there are few places that deliver this service to this age group. Additionally, teens and families are not always willing to pursue a referral, and many will get lost along the way.”
Promising Models
At Boston Children’s, she said, the Division of Addiction Medicine has created a consultation call line that primary care providers can call for help with any questions about teen substance use.
After running the consultation for about a year, she said, the program wanted to add ways to help patients directly and hired and trained social workers who can see pediatric patients with substance use problems for counseling via telemedicine. “The program also now supports group therapy for pediatric patients and parents, so that primary care providers can refer patients directly to group therapy,” Dr. Levy said.
The growth of telehealth since the pandemic may allow for new models of care.
“For example, now our Adolescent Substance Use and Addiction Program at Boston Children’s Hospital can provide services, including medication induction and follow-up, virtually,” Dr. Levy said. “This allows us to treat young people anywhere in the state. There have been instances in which a primary care provider referred us patients with OUD and then partnered with us, including performing physicals for teens who could not get to Boston to see us in person. At the end of the day, the more models we can come up with the better.”
Dr. Hadland reported honoraria from the AAP outside the submitted work. Two coauthors reported receiving salary support from the AAP during the conduct of the study. A coauthor reported serving as the chair of the AAP Committee on Substance Use and Prevention outside the submitted work. This work was supported by a grant from the Conrad N. Hilton Foundation via the AAP. Dr. Sharon Levy’s husband, Ofer Levy, MD, PhD, is director of the Precision Vaccines Program at Boston Children’s Hospital.
An estimated 1 in 100 adolescents ages 12-17 years in the United States have an opioid use disorder (OUD). But fewer than 5% of adolescents with OUD get buprenorphine or naltrexone, though the treatments are recommended by the American Academy of Pediatrics (AAP), new data show.
Meanwhile, adolescent drug overdose deaths more than doubled between 2019 and 2021, with most involving opioids.
Scott E. Hadland, MD, MPH, with the Division of Adolescent and Young Adult Medicine at Mass General for Children in Boston, and colleagues detailed the extent of the treatment gap and barriers to prescribing and caring for youth with OUD in primary care in a research letter published February 26 in JAMA Pediatrics.
Dr. Hadland’s team mailed 1,681 US pediatricians a survey and the response rate was 43.0%. Researchers included in the sample 474 primary care pediatricians who care for adolescents.
Who Should Treat OUD?
Most respondents (average age, 49.5; 74.0% female) agreed or strongly agreed that it is their responsibility to identify substance use disorders (93.9%) and refer patients to treatment (97.4%).
Fewer agreed or strongly agreed that it is their responsibility to treat substance use disorders (20.3%) or prescribe medications (12.4%). Fewer than half of the respondents said they felt prepared or very prepared to counsel adolescents on opioid use (48.3%) compared with those comfortable counseling on alcohol (87.1%), cannabis (81.7%), and e-cigarette use (80.1%; P < .001).
Pediatricians were less likely to provide counseling (63.0%) and more likely to refer patients to care off-site (71.8%) for opioid use than for alcohol (87.7% and 51.7%); cannabis (88.9% and 45.4%); or e-cigarette use (91.6% and 26.5%) (P < .001 for all comparisons).
Training Lacking in Residency Programs
“These results reveal an opportunity for greater workforce training in line with a 2019 survey showing fewer than 1 in 3 US pediatric residency programs included education on prescribing OUD medications,” the authors wrote. Training focused on treating OUD in primary care, including prescribing medications and addressing possible misperceptions, may be needed,” they noted.
The survey predated the elimination in 2023 of the federal buprenorphine waiver requirement, which made prescribing buprenorphine easier, so these results do not reflect any changes from that elimination, they wrote.
Sharon Levy, MD, MPH, chief of the Division of Addiction Medicine at Boston Children’s Hospital and professor of pediatrics at Harvard Medical School in Boston, who was not part of this study, said more education on addiction medicine is needed for general pediatricians.
She said it’s time to push beyond the current framework of Screening, Brief Intervention, Referral to Treatment (SBIRT), because that doesn’t include “prescribing medications to manage withdrawal or suppress cravings or the use of lab testing, both of which could be accomplished in primary care.”
Dr. Levy said she considers substance use disorders the same way she considers other chronic conditions: Most patients can be treated in primary care. “Specialty care and higher levels of care need to be available for patients who are most complicated and/or having a flare of their condition.”
“In my opinion, most teens with opioid use disorder can and should be treated in primary care. I worry about referring teens with opioid use disorder to get medication somewhere else because there are few places that deliver this service to this age group. Additionally, teens and families are not always willing to pursue a referral, and many will get lost along the way.”
Promising Models
At Boston Children’s, she said, the Division of Addiction Medicine has created a consultation call line that primary care providers can call for help with any questions about teen substance use.
After running the consultation for about a year, she said, the program wanted to add ways to help patients directly and hired and trained social workers who can see pediatric patients with substance use problems for counseling via telemedicine. “The program also now supports group therapy for pediatric patients and parents, so that primary care providers can refer patients directly to group therapy,” Dr. Levy said.
The growth of telehealth since the pandemic may allow for new models of care.
“For example, now our Adolescent Substance Use and Addiction Program at Boston Children’s Hospital can provide services, including medication induction and follow-up, virtually,” Dr. Levy said. “This allows us to treat young people anywhere in the state. There have been instances in which a primary care provider referred us patients with OUD and then partnered with us, including performing physicals for teens who could not get to Boston to see us in person. At the end of the day, the more models we can come up with the better.”
Dr. Hadland reported honoraria from the AAP outside the submitted work. Two coauthors reported receiving salary support from the AAP during the conduct of the study. A coauthor reported serving as the chair of the AAP Committee on Substance Use and Prevention outside the submitted work. This work was supported by a grant from the Conrad N. Hilton Foundation via the AAP. Dr. Sharon Levy’s husband, Ofer Levy, MD, PhD, is director of the Precision Vaccines Program at Boston Children’s Hospital.
An estimated 1 in 100 adolescents ages 12-17 years in the United States have an opioid use disorder (OUD). But fewer than 5% of adolescents with OUD get buprenorphine or naltrexone, though the treatments are recommended by the American Academy of Pediatrics (AAP), new data show.
Meanwhile, adolescent drug overdose deaths more than doubled between 2019 and 2021, with most involving opioids.
Scott E. Hadland, MD, MPH, with the Division of Adolescent and Young Adult Medicine at Mass General for Children in Boston, and colleagues detailed the extent of the treatment gap and barriers to prescribing and caring for youth with OUD in primary care in a research letter published February 26 in JAMA Pediatrics.
Dr. Hadland’s team mailed 1,681 US pediatricians a survey and the response rate was 43.0%. Researchers included in the sample 474 primary care pediatricians who care for adolescents.
Who Should Treat OUD?
Most respondents (average age, 49.5; 74.0% female) agreed or strongly agreed that it is their responsibility to identify substance use disorders (93.9%) and refer patients to treatment (97.4%).
Fewer agreed or strongly agreed that it is their responsibility to treat substance use disorders (20.3%) or prescribe medications (12.4%). Fewer than half of the respondents said they felt prepared or very prepared to counsel adolescents on opioid use (48.3%) compared with those comfortable counseling on alcohol (87.1%), cannabis (81.7%), and e-cigarette use (80.1%; P < .001).
Pediatricians were less likely to provide counseling (63.0%) and more likely to refer patients to care off-site (71.8%) for opioid use than for alcohol (87.7% and 51.7%); cannabis (88.9% and 45.4%); or e-cigarette use (91.6% and 26.5%) (P < .001 for all comparisons).
Training Lacking in Residency Programs
“These results reveal an opportunity for greater workforce training in line with a 2019 survey showing fewer than 1 in 3 US pediatric residency programs included education on prescribing OUD medications,” the authors wrote. Training focused on treating OUD in primary care, including prescribing medications and addressing possible misperceptions, may be needed,” they noted.
The survey predated the elimination in 2023 of the federal buprenorphine waiver requirement, which made prescribing buprenorphine easier, so these results do not reflect any changes from that elimination, they wrote.
Sharon Levy, MD, MPH, chief of the Division of Addiction Medicine at Boston Children’s Hospital and professor of pediatrics at Harvard Medical School in Boston, who was not part of this study, said more education on addiction medicine is needed for general pediatricians.
She said it’s time to push beyond the current framework of Screening, Brief Intervention, Referral to Treatment (SBIRT), because that doesn’t include “prescribing medications to manage withdrawal or suppress cravings or the use of lab testing, both of which could be accomplished in primary care.”
Dr. Levy said she considers substance use disorders the same way she considers other chronic conditions: Most patients can be treated in primary care. “Specialty care and higher levels of care need to be available for patients who are most complicated and/or having a flare of their condition.”
“In my opinion, most teens with opioid use disorder can and should be treated in primary care. I worry about referring teens with opioid use disorder to get medication somewhere else because there are few places that deliver this service to this age group. Additionally, teens and families are not always willing to pursue a referral, and many will get lost along the way.”
Promising Models
At Boston Children’s, she said, the Division of Addiction Medicine has created a consultation call line that primary care providers can call for help with any questions about teen substance use.
After running the consultation for about a year, she said, the program wanted to add ways to help patients directly and hired and trained social workers who can see pediatric patients with substance use problems for counseling via telemedicine. “The program also now supports group therapy for pediatric patients and parents, so that primary care providers can refer patients directly to group therapy,” Dr. Levy said.
The growth of telehealth since the pandemic may allow for new models of care.
“For example, now our Adolescent Substance Use and Addiction Program at Boston Children’s Hospital can provide services, including medication induction and follow-up, virtually,” Dr. Levy said. “This allows us to treat young people anywhere in the state. There have been instances in which a primary care provider referred us patients with OUD and then partnered with us, including performing physicals for teens who could not get to Boston to see us in person. At the end of the day, the more models we can come up with the better.”
Dr. Hadland reported honoraria from the AAP outside the submitted work. Two coauthors reported receiving salary support from the AAP during the conduct of the study. A coauthor reported serving as the chair of the AAP Committee on Substance Use and Prevention outside the submitted work. This work was supported by a grant from the Conrad N. Hilton Foundation via the AAP. Dr. Sharon Levy’s husband, Ofer Levy, MD, PhD, is director of the Precision Vaccines Program at Boston Children’s Hospital.
FROM JAMA PEDIATRICS
Communicating Bad News to Patients
Communicating bad news to patients is one of the most stressful and challenging clinical tasks for any physician, regardless of his or her specialty. This task is more frequent for physicians caring for oncology patients and can also affect the physician’s emotional state.
The manner in which bad news is communicated plays a significant role in the psychological burden on the patient, and various communication techniques and guidelines have been developed to enable physicians to perform this difficult task effectively.
Revealing bad news in person whenever possible, to address the emotional responses of patients or relatives, is part of the prevailing expert recommendations. However, it has been acknowledged that in certain situations, communicating bad news over the phone is more feasible.
Since the beginning of the COVID-19 pandemic, the disclosure of bad news over the phone has become a necessary substitute for in-person visits and an integral part of clinical practice worldwide. It remains to be clarified what the real psychological impact on patients and their closest relatives is when delivering bad news over the phone compared with delivering it in person.
Right and Wrong Ways
The most popular guideline for communicating bad news is SPIKES, a six-phase protocol with a special application for cancer patients. It is used in various countries (eg, the United States, France, and Germany) as a guide for this sensitive practice and for training in communication skills in this context. The SPIKES acronym refers to the following six recommended steps for delivering bad news:
- Setting: Set up the conversation.
- Perception: Assess the patient’s perception.
- Invitation: Ask the patient what he or she would like to know.
- Knowledge: Provide the patient with knowledge and information, breaking it down into small parts.
- Emotions: Acknowledge and empathetically address the patient’s emotions.
- Strategy and Summary: Summarize and define a medical action plan.
The lesson from SPIKES is that when a person experiences strong emotions, it is difficult to continue discussing anything, and they will struggle to hear anything. Allowing for silence is fundamental. In addition, empathy allows the patient to express his or her feelings and concerns, as well as provide support. The aim is not to argue but to allow the expression of emotions without criticism. However, these recommendations are primarily based on expert opinion and less on empirical evidence, due to the difficulty of studies in assessing patient outcomes in various phases of these protocols.
A recent study analyzed the differences in psychological distress between patients who received bad news over the phone vs those who received it in person. The study was a systematic review and meta-analysis.
The investigators examined 5944 studies, including 11 qualitative analysis studies, nine meta-analyses, and four randomized controlled trials.
In a set of studies ranging from moderate to good quality, no difference in psychological distress was found when bad news was disclosed over the phone compared with in person, regarding anxiety, depression, and posttraumatic stress disorder.
There was no average difference in patient satisfaction levels when bad news was delivered over the phone compared with in person. The risk for dissatisfaction was similar between groups.
Clinical Practice Guidelines
The demand for telemedicine, including the disclosure of bad news, is growing despite the limited knowledge of potential adverse effects. The results of existing studies suggest that the mode of disclosure may play a secondary role, and the manner in which bad news is communicated may be more important.
Therefore, it is paramount to prepare patients or their families for the possibility of receiving bad news well in advance and, during the conversation, to ensure first and foremost that they are in an appropriate environment. The structure and content of the conversation may be relevant, and adhering to dedicated communication strategies can be a wise choice for the physician and the interlocutor.
This story was translated from Univadis Italy, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Communicating bad news to patients is one of the most stressful and challenging clinical tasks for any physician, regardless of his or her specialty. This task is more frequent for physicians caring for oncology patients and can also affect the physician’s emotional state.
The manner in which bad news is communicated plays a significant role in the psychological burden on the patient, and various communication techniques and guidelines have been developed to enable physicians to perform this difficult task effectively.
Revealing bad news in person whenever possible, to address the emotional responses of patients or relatives, is part of the prevailing expert recommendations. However, it has been acknowledged that in certain situations, communicating bad news over the phone is more feasible.
Since the beginning of the COVID-19 pandemic, the disclosure of bad news over the phone has become a necessary substitute for in-person visits and an integral part of clinical practice worldwide. It remains to be clarified what the real psychological impact on patients and their closest relatives is when delivering bad news over the phone compared with delivering it in person.
Right and Wrong Ways
The most popular guideline for communicating bad news is SPIKES, a six-phase protocol with a special application for cancer patients. It is used in various countries (eg, the United States, France, and Germany) as a guide for this sensitive practice and for training in communication skills in this context. The SPIKES acronym refers to the following six recommended steps for delivering bad news:
- Setting: Set up the conversation.
- Perception: Assess the patient’s perception.
- Invitation: Ask the patient what he or she would like to know.
- Knowledge: Provide the patient with knowledge and information, breaking it down into small parts.
- Emotions: Acknowledge and empathetically address the patient’s emotions.
- Strategy and Summary: Summarize and define a medical action plan.
The lesson from SPIKES is that when a person experiences strong emotions, it is difficult to continue discussing anything, and they will struggle to hear anything. Allowing for silence is fundamental. In addition, empathy allows the patient to express his or her feelings and concerns, as well as provide support. The aim is not to argue but to allow the expression of emotions without criticism. However, these recommendations are primarily based on expert opinion and less on empirical evidence, due to the difficulty of studies in assessing patient outcomes in various phases of these protocols.
A recent study analyzed the differences in psychological distress between patients who received bad news over the phone vs those who received it in person. The study was a systematic review and meta-analysis.
The investigators examined 5944 studies, including 11 qualitative analysis studies, nine meta-analyses, and four randomized controlled trials.
In a set of studies ranging from moderate to good quality, no difference in psychological distress was found when bad news was disclosed over the phone compared with in person, regarding anxiety, depression, and posttraumatic stress disorder.
There was no average difference in patient satisfaction levels when bad news was delivered over the phone compared with in person. The risk for dissatisfaction was similar between groups.
Clinical Practice Guidelines
The demand for telemedicine, including the disclosure of bad news, is growing despite the limited knowledge of potential adverse effects. The results of existing studies suggest that the mode of disclosure may play a secondary role, and the manner in which bad news is communicated may be more important.
Therefore, it is paramount to prepare patients or their families for the possibility of receiving bad news well in advance and, during the conversation, to ensure first and foremost that they are in an appropriate environment. The structure and content of the conversation may be relevant, and adhering to dedicated communication strategies can be a wise choice for the physician and the interlocutor.
This story was translated from Univadis Italy, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Communicating bad news to patients is one of the most stressful and challenging clinical tasks for any physician, regardless of his or her specialty. This task is more frequent for physicians caring for oncology patients and can also affect the physician’s emotional state.
The manner in which bad news is communicated plays a significant role in the psychological burden on the patient, and various communication techniques and guidelines have been developed to enable physicians to perform this difficult task effectively.
Revealing bad news in person whenever possible, to address the emotional responses of patients or relatives, is part of the prevailing expert recommendations. However, it has been acknowledged that in certain situations, communicating bad news over the phone is more feasible.
Since the beginning of the COVID-19 pandemic, the disclosure of bad news over the phone has become a necessary substitute for in-person visits and an integral part of clinical practice worldwide. It remains to be clarified what the real psychological impact on patients and their closest relatives is when delivering bad news over the phone compared with delivering it in person.
Right and Wrong Ways
The most popular guideline for communicating bad news is SPIKES, a six-phase protocol with a special application for cancer patients. It is used in various countries (eg, the United States, France, and Germany) as a guide for this sensitive practice and for training in communication skills in this context. The SPIKES acronym refers to the following six recommended steps for delivering bad news:
- Setting: Set up the conversation.
- Perception: Assess the patient’s perception.
- Invitation: Ask the patient what he or she would like to know.
- Knowledge: Provide the patient with knowledge and information, breaking it down into small parts.
- Emotions: Acknowledge and empathetically address the patient’s emotions.
- Strategy and Summary: Summarize and define a medical action plan.
The lesson from SPIKES is that when a person experiences strong emotions, it is difficult to continue discussing anything, and they will struggle to hear anything. Allowing for silence is fundamental. In addition, empathy allows the patient to express his or her feelings and concerns, as well as provide support. The aim is not to argue but to allow the expression of emotions without criticism. However, these recommendations are primarily based on expert opinion and less on empirical evidence, due to the difficulty of studies in assessing patient outcomes in various phases of these protocols.
A recent study analyzed the differences in psychological distress between patients who received bad news over the phone vs those who received it in person. The study was a systematic review and meta-analysis.
The investigators examined 5944 studies, including 11 qualitative analysis studies, nine meta-analyses, and four randomized controlled trials.
In a set of studies ranging from moderate to good quality, no difference in psychological distress was found when bad news was disclosed over the phone compared with in person, regarding anxiety, depression, and posttraumatic stress disorder.
There was no average difference in patient satisfaction levels when bad news was delivered over the phone compared with in person. The risk for dissatisfaction was similar between groups.
Clinical Practice Guidelines
The demand for telemedicine, including the disclosure of bad news, is growing despite the limited knowledge of potential adverse effects. The results of existing studies suggest that the mode of disclosure may play a secondary role, and the manner in which bad news is communicated may be more important.
Therefore, it is paramount to prepare patients or their families for the possibility of receiving bad news well in advance and, during the conversation, to ensure first and foremost that they are in an appropriate environment. The structure and content of the conversation may be relevant, and adhering to dedicated communication strategies can be a wise choice for the physician and the interlocutor.
This story was translated from Univadis Italy, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
The Ghost Research Haunting Nordic Medical Trials
Campaigners for greater transparency in medical science have reiterated calls for more to be done to avoid “medical research waste” after an investigation found that results from more than a fifth of clinical trials across five Nordic countries have never been made public.
Nonpublication of clinical trial results wastes public money, harms patients, and undermines public health, the researchers said.
There is already a well-defined ethical responsibility to publish trial results. Article 36 of the Declaration of Helsinki on Ethical Principles for Medical Research Involving Human Subjects states that “researchers have a duty to make publicly available the results of their research on human subjects,” and World Health Organization best practice protocols call for results to be uploaded onto trial registries within 12 months of trial completion.
Research Waste Is a ‘Pervasive Problem’
So, how and why do so many trials end up gathering dust in a drawer? The latest study, published February 5 as a preprint, evaluated the reporting outcomes of 2113 clinical trials at medical universities and university hospitals in Nordic countries between 2016 and 2019. It found that across the five countries, 22% of all clinical trial results had not been shared. Furthermore, only 27% of all trial results were made public, either on registries or in journals, within 12 months. Even 2 years after trials ended, only around half of results (51.7%) had been put into the public domain.
The authors concluded that missing and delayed results from academically-led clinical trials was a “pervasive problem” in Nordic countries and that institutions, funding bodies, and policymakers needed to ensure that regulations around reporting results were adhered to so that important findings are not lost.
Study first author, Gustav Nilsonne, MD, PHD, from the Department of Clinical Neuroscience at the Karolinska Institutet, Sweden, told this news organization: “Most people I talk to — most colleagues who are clinical scientists — tend to think that the main reason is that negative results are not as interesting to publish and therefore they get lower priority, and they get published later and sometimes not at all.”
Experts stressed that the problem is not confined to Nordic countries and that wasted medical research persists elsewhere in Europe and remains a global problem. For instance, a report published in the Journal of Clinical Epidemiology found that 30% of German trials completed between 2014 and 2017 remained unpublished 5 years after completion.
The Case for Laws, Monitoring, and Fines
Till Bruckner, PHD, from TranspariMED, which campaigns to end evidence distortion in medicine, told this news organization: “What is needed to comprehensively fix the problem is a national legal requirement to make all trial results public, coupled with effective monitoring, and followed by sanctions in the rare cases where institutions refuse to comply.”
Dr. Nilsonne added: “We have argued that the sponsors need to take greater responsibility, but also that there needs to be somebody whose job it is to monitor clinical trials reporting. It shouldn’t have to be that we do this as researchers on a shoestring with no dedicated resources. It should be somebody’s job.”
Since January 31, 2023, all initial clinical trial applications in the European Union must be submitted through the EU Clinical Trials Information System. Dr. Bruckner said that “the picture is not yet clear” in Europe, as the first trial results under the system are not expected until later this year. Even then, enforcement lies with regulators in individual countries. And while Denmark has already indicated it will enforce the regulations, he warned that other countries “might turn a blind eye.”
He pointed out that existing laws don’t apply to all types of trials. “That means that for many trials, nobody is legally responsible for ensuring that results are made public, and no government agency has any oversight or mandate,” he said.
Outside the EU, the United Kingdom has helped lead the way through the NHS Health Research Authority (HRA), which registers trials run in the country. One year after a trial has been completed, the HRA checks to see if the results have been uploaded to the registry and issues reminders if they haven’t.
In an update of its work in January, the authority said that compliance had hovered at just below 90% between 2018 and 2021 but that it was working to increase this to 100% by working with stakeholders across the research sector.
Dr. Nilsonne considers the UK system of central registration and follow-up an attractive option. “I would love to see something along those lines in other countries too,” he said.
‘Rampant Noncompliance’ in the United States
In the United States, a requirement to make trial results public is backed by law. Despite this, there’s evidence of “rampant noncompliance” and minimal government action, according to Megan Curtin from Universities Allied for Essential Medicines (UAEM), which has been tracking the issue in the United States and working to push universities and others to make their findings available.
The US Food and Drug Administration (FDA) shares responsibility with the National Institutes of Health for enforcement of clinical trial results reporting, but the UAEM says nearly 4000 trials are currently out of compliance with reporting requirements. In January last year, the UAEM copublished a report with the National Center for Health Research and TranspariMED, which found that 3627 American children participated in clinical trials whose results remain unreported.
The FDA can levy a fine of up to $10,000 USD for a violation of the law, but UAEM said that, as of January 2023, the FDA had sent only 92 preliminary notices of noncompliance and four notices of noncompliance. “A clear difference between the EU field of clinical trial operation and US clinical trials is that there are clear laws for reporting within 12 months, which can be enforced, but they’re not being enforced by the FDA,” Ms. Curtin told this news organization.
The UAEM is pushing the FDA to issue a minimum of 250 preliminary notices of noncompliance each year to noncompliant trial sponsors.
Dr. Nilsonne said: “I do believe we have a great responsibility to the patients that do contribute. We need to make sure that the harms and risks that a clinical trial entails are really balanced by knowledge gain, and if the results are never reported, then we can’t have a knowledge gain.”
A version of this article appeared on Medscape.com.
Campaigners for greater transparency in medical science have reiterated calls for more to be done to avoid “medical research waste” after an investigation found that results from more than a fifth of clinical trials across five Nordic countries have never been made public.
Nonpublication of clinical trial results wastes public money, harms patients, and undermines public health, the researchers said.
There is already a well-defined ethical responsibility to publish trial results. Article 36 of the Declaration of Helsinki on Ethical Principles for Medical Research Involving Human Subjects states that “researchers have a duty to make publicly available the results of their research on human subjects,” and World Health Organization best practice protocols call for results to be uploaded onto trial registries within 12 months of trial completion.
Research Waste Is a ‘Pervasive Problem’
So, how and why do so many trials end up gathering dust in a drawer? The latest study, published February 5 as a preprint, evaluated the reporting outcomes of 2113 clinical trials at medical universities and university hospitals in Nordic countries between 2016 and 2019. It found that across the five countries, 22% of all clinical trial results had not been shared. Furthermore, only 27% of all trial results were made public, either on registries or in journals, within 12 months. Even 2 years after trials ended, only around half of results (51.7%) had been put into the public domain.
The authors concluded that missing and delayed results from academically-led clinical trials was a “pervasive problem” in Nordic countries and that institutions, funding bodies, and policymakers needed to ensure that regulations around reporting results were adhered to so that important findings are not lost.
Study first author, Gustav Nilsonne, MD, PHD, from the Department of Clinical Neuroscience at the Karolinska Institutet, Sweden, told this news organization: “Most people I talk to — most colleagues who are clinical scientists — tend to think that the main reason is that negative results are not as interesting to publish and therefore they get lower priority, and they get published later and sometimes not at all.”
Experts stressed that the problem is not confined to Nordic countries and that wasted medical research persists elsewhere in Europe and remains a global problem. For instance, a report published in the Journal of Clinical Epidemiology found that 30% of German trials completed between 2014 and 2017 remained unpublished 5 years after completion.
The Case for Laws, Monitoring, and Fines
Till Bruckner, PHD, from TranspariMED, which campaigns to end evidence distortion in medicine, told this news organization: “What is needed to comprehensively fix the problem is a national legal requirement to make all trial results public, coupled with effective monitoring, and followed by sanctions in the rare cases where institutions refuse to comply.”
Dr. Nilsonne added: “We have argued that the sponsors need to take greater responsibility, but also that there needs to be somebody whose job it is to monitor clinical trials reporting. It shouldn’t have to be that we do this as researchers on a shoestring with no dedicated resources. It should be somebody’s job.”
Since January 31, 2023, all initial clinical trial applications in the European Union must be submitted through the EU Clinical Trials Information System. Dr. Bruckner said that “the picture is not yet clear” in Europe, as the first trial results under the system are not expected until later this year. Even then, enforcement lies with regulators in individual countries. And while Denmark has already indicated it will enforce the regulations, he warned that other countries “might turn a blind eye.”
He pointed out that existing laws don’t apply to all types of trials. “That means that for many trials, nobody is legally responsible for ensuring that results are made public, and no government agency has any oversight or mandate,” he said.
Outside the EU, the United Kingdom has helped lead the way through the NHS Health Research Authority (HRA), which registers trials run in the country. One year after a trial has been completed, the HRA checks to see if the results have been uploaded to the registry and issues reminders if they haven’t.
In an update of its work in January, the authority said that compliance had hovered at just below 90% between 2018 and 2021 but that it was working to increase this to 100% by working with stakeholders across the research sector.
Dr. Nilsonne considers the UK system of central registration and follow-up an attractive option. “I would love to see something along those lines in other countries too,” he said.
‘Rampant Noncompliance’ in the United States
In the United States, a requirement to make trial results public is backed by law. Despite this, there’s evidence of “rampant noncompliance” and minimal government action, according to Megan Curtin from Universities Allied for Essential Medicines (UAEM), which has been tracking the issue in the United States and working to push universities and others to make their findings available.
The US Food and Drug Administration (FDA) shares responsibility with the National Institutes of Health for enforcement of clinical trial results reporting, but the UAEM says nearly 4000 trials are currently out of compliance with reporting requirements. In January last year, the UAEM copublished a report with the National Center for Health Research and TranspariMED, which found that 3627 American children participated in clinical trials whose results remain unreported.
The FDA can levy a fine of up to $10,000 USD for a violation of the law, but UAEM said that, as of January 2023, the FDA had sent only 92 preliminary notices of noncompliance and four notices of noncompliance. “A clear difference between the EU field of clinical trial operation and US clinical trials is that there are clear laws for reporting within 12 months, which can be enforced, but they’re not being enforced by the FDA,” Ms. Curtin told this news organization.
The UAEM is pushing the FDA to issue a minimum of 250 preliminary notices of noncompliance each year to noncompliant trial sponsors.
Dr. Nilsonne said: “I do believe we have a great responsibility to the patients that do contribute. We need to make sure that the harms and risks that a clinical trial entails are really balanced by knowledge gain, and if the results are never reported, then we can’t have a knowledge gain.”
A version of this article appeared on Medscape.com.
Campaigners for greater transparency in medical science have reiterated calls for more to be done to avoid “medical research waste” after an investigation found that results from more than a fifth of clinical trials across five Nordic countries have never been made public.
Nonpublication of clinical trial results wastes public money, harms patients, and undermines public health, the researchers said.
There is already a well-defined ethical responsibility to publish trial results. Article 36 of the Declaration of Helsinki on Ethical Principles for Medical Research Involving Human Subjects states that “researchers have a duty to make publicly available the results of their research on human subjects,” and World Health Organization best practice protocols call for results to be uploaded onto trial registries within 12 months of trial completion.
Research Waste Is a ‘Pervasive Problem’
So, how and why do so many trials end up gathering dust in a drawer? The latest study, published February 5 as a preprint, evaluated the reporting outcomes of 2113 clinical trials at medical universities and university hospitals in Nordic countries between 2016 and 2019. It found that across the five countries, 22% of all clinical trial results had not been shared. Furthermore, only 27% of all trial results were made public, either on registries or in journals, within 12 months. Even 2 years after trials ended, only around half of results (51.7%) had been put into the public domain.
The authors concluded that missing and delayed results from academically-led clinical trials was a “pervasive problem” in Nordic countries and that institutions, funding bodies, and policymakers needed to ensure that regulations around reporting results were adhered to so that important findings are not lost.
Study first author, Gustav Nilsonne, MD, PHD, from the Department of Clinical Neuroscience at the Karolinska Institutet, Sweden, told this news organization: “Most people I talk to — most colleagues who are clinical scientists — tend to think that the main reason is that negative results are not as interesting to publish and therefore they get lower priority, and they get published later and sometimes not at all.”
Experts stressed that the problem is not confined to Nordic countries and that wasted medical research persists elsewhere in Europe and remains a global problem. For instance, a report published in the Journal of Clinical Epidemiology found that 30% of German trials completed between 2014 and 2017 remained unpublished 5 years after completion.
The Case for Laws, Monitoring, and Fines
Till Bruckner, PHD, from TranspariMED, which campaigns to end evidence distortion in medicine, told this news organization: “What is needed to comprehensively fix the problem is a national legal requirement to make all trial results public, coupled with effective monitoring, and followed by sanctions in the rare cases where institutions refuse to comply.”
Dr. Nilsonne added: “We have argued that the sponsors need to take greater responsibility, but also that there needs to be somebody whose job it is to monitor clinical trials reporting. It shouldn’t have to be that we do this as researchers on a shoestring with no dedicated resources. It should be somebody’s job.”
Since January 31, 2023, all initial clinical trial applications in the European Union must be submitted through the EU Clinical Trials Information System. Dr. Bruckner said that “the picture is not yet clear” in Europe, as the first trial results under the system are not expected until later this year. Even then, enforcement lies with regulators in individual countries. And while Denmark has already indicated it will enforce the regulations, he warned that other countries “might turn a blind eye.”
He pointed out that existing laws don’t apply to all types of trials. “That means that for many trials, nobody is legally responsible for ensuring that results are made public, and no government agency has any oversight or mandate,” he said.
Outside the EU, the United Kingdom has helped lead the way through the NHS Health Research Authority (HRA), which registers trials run in the country. One year after a trial has been completed, the HRA checks to see if the results have been uploaded to the registry and issues reminders if they haven’t.
In an update of its work in January, the authority said that compliance had hovered at just below 90% between 2018 and 2021 but that it was working to increase this to 100% by working with stakeholders across the research sector.
Dr. Nilsonne considers the UK system of central registration and follow-up an attractive option. “I would love to see something along those lines in other countries too,” he said.
‘Rampant Noncompliance’ in the United States
In the United States, a requirement to make trial results public is backed by law. Despite this, there’s evidence of “rampant noncompliance” and minimal government action, according to Megan Curtin from Universities Allied for Essential Medicines (UAEM), which has been tracking the issue in the United States and working to push universities and others to make their findings available.
The US Food and Drug Administration (FDA) shares responsibility with the National Institutes of Health for enforcement of clinical trial results reporting, but the UAEM says nearly 4000 trials are currently out of compliance with reporting requirements. In January last year, the UAEM copublished a report with the National Center for Health Research and TranspariMED, which found that 3627 American children participated in clinical trials whose results remain unreported.
The FDA can levy a fine of up to $10,000 USD for a violation of the law, but UAEM said that, as of January 2023, the FDA had sent only 92 preliminary notices of noncompliance and four notices of noncompliance. “A clear difference between the EU field of clinical trial operation and US clinical trials is that there are clear laws for reporting within 12 months, which can be enforced, but they’re not being enforced by the FDA,” Ms. Curtin told this news organization.
The UAEM is pushing the FDA to issue a minimum of 250 preliminary notices of noncompliance each year to noncompliant trial sponsors.
Dr. Nilsonne said: “I do believe we have a great responsibility to the patients that do contribute. We need to make sure that the harms and risks that a clinical trial entails are really balanced by knowledge gain, and if the results are never reported, then we can’t have a knowledge gain.”
A version of this article appeared on Medscape.com.
Use of Biologics for Psoriasis Found to Confer a Survival Benefit
Among patients with psoriasis, the risk of mortality was strongly associated with hepatic injury, cardiovascular disease, and psychiatric affective disorders, but was reduced among those who received systemic therapy with biologics, researchers from Canada report.
Those are key findings from a large retrospective registry study of patients with psoriasis, published in The Journal of the American Academy of Dermatology.
“Psoriasis, a chronic inflammatory condition affecting approximately 3% of the western populations, bears a higher risk of mortality compared to healthy individuals, possibly by inducing systemic inflammation associated with numerous comorbidities, especially cardiovascular diseases, metabolic syndrome, and others,” wrote corresponding author Robert Gniadecki, MD, PhD, of the division of dermatology at the University of Alberta, Canada, and colleagues. “It has been argued that the use of systemic immunomodulatory agents quenches systemic inflammation and potentially improves patient survival. However, the evidence to support this hypothesis is limited.”
To investigate the impact of comorbidities and systemic therapies on all-cause mortality in psoriasis, the researchers used the Alberta Health Services Data Repository of Reporting database from January 1, 2012, to June 1, 2019, which represents a population base of 4.47 million individuals. They extracted data on 18,618 psoriasis cases and 55,854 controls, stratified cases according to the Charlson Comorbidity Index (CCI), a surrogate measure for comorbidity burden, and by the type of therapy received, and conducted statistical analyses including Cox proportional hazards regression to determine absolute hazard ratios representing relative effects of specific demographic and comorbidity factors on mortality within groups.
The median age in both cohorts was 48 years, and 51% were male. The researchers observed that mortality in the psoriasis cohort was significantly higher than in the controls (5.7% vs. 3.8%, respectively; P < .05), with a median age at the time of death of 72 vs. 74.4 years.
The CCI and comorbidities strongly predicted mortality, especially drug-induced liver injury (hazard ratio [HR], 1.78), bipolar disorder and suicidal ideation (HR, 1.24-1.58), and major cardiovascular diseases, which included myocardial infarction (MI), congestive heart failure (CHF), and cerebrovascular disease (CVA) (HR, 1.2-1.4).
Among patients in the psoriasis cohort, survival of those treated with biologic agents was higher than in controls, even after matching for CCI (3.2% vs. 4.4%, respectively, P < .05). “These patients also exhibit reduced overall mortality compared to those treated with methotrexate or topical agents,” Dr. Gniadecki and colleagues wrote. “There was no difference in mortality between methotrexate patients and the topical therapy patients, but any of those treatment groups had superior survival compared to the no-treatment cohort.”
They added that despite better survival among patients treated with biologic agents, no significant improvements were detected in their comorbidity profiles. “Notably, the frequency of major cardiovascular disease (MI, CHF, CVA) was the same as in the controls, and overall, the frequency of diseases coded as cardiovascular was slightly increased,” they wrote.
The fact that some factors could not be measured, including the type and severity of psoriasis, response to treatment, smoking history, and alcohol intake, was a study limitation, they noted.
Joel M. Gelfand, MD, director of the psoriasis and phototherapy treatment center at the University of Pennsylvania, Philadelphia, who was asked to comment on the analysis, said the study confirms prior work indicating that having psoriasis is a predictor of mortality. In addition, “there is a strong healthy user affect among patients who take and stay on biologics for psoriasis,” he told this news organization.

“The results are encouraging but are not able to establish a causal relationship between treating psoriasis with biologics and lowering mortality risk. Ultimately, randomized comparative trials will be needed to determine which approach or approaches to treating psoriasis, if any, lower the risk of psoriatic arthritis, cardiovascular disease, and mortality,” said Dr. Gelfand, who was not involved with the study.
Asked to comment on the results, Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was not involved with the study, said that “data such as these enable us to rationalize the cost of our fleet of biologics, as managing the outpatient/inpatient burden of many of these comorbidities will actually drain the healthcare system, more so than managing psoriasis in the first place. Certainly other interventions to address the well known comorbidities, such as cardiovascular and hepatic, are warranted, but what if you could prevent the problem in the first place? To be continued for that answer.”

The study was funded by Canadian Dermatology Foundation, Alberta Innovates, and by a Health Sciences TD Bank Studentship Award. Dr. Gniadecki reported conducting clinical trials for Bausch Health, AbbVie and Janssen, and he has received honoraria as consultant and/or speaker from AbbVie, Bausch Health, Eli Lilly, Janssen, Mallinckrodt, Novartis, Kyowa Kirin, Sun Pharma and Sanofi. The other authors had no disclosures. Dr. Gelfand reported serving as a consultant for AbbVie, Artax, Bristol-Myers Squibb, GlaxoSmithKline, and other companies. He is on the board of directors for the International Psoriasis Council and the Medical Dermatology Society. Dr. Friedman disclosed that he is a speaker for Janssen and Bristol Myers Squibb. He has received grants from Janssen, Pfizer, Bristol Myers Squibb, and Lilly, and has served as an advisor for Arcutis, Dermavant, and Janssen.
Among patients with psoriasis, the risk of mortality was strongly associated with hepatic injury, cardiovascular disease, and psychiatric affective disorders, but was reduced among those who received systemic therapy with biologics, researchers from Canada report.
Those are key findings from a large retrospective registry study of patients with psoriasis, published in The Journal of the American Academy of Dermatology.
“Psoriasis, a chronic inflammatory condition affecting approximately 3% of the western populations, bears a higher risk of mortality compared to healthy individuals, possibly by inducing systemic inflammation associated with numerous comorbidities, especially cardiovascular diseases, metabolic syndrome, and others,” wrote corresponding author Robert Gniadecki, MD, PhD, of the division of dermatology at the University of Alberta, Canada, and colleagues. “It has been argued that the use of systemic immunomodulatory agents quenches systemic inflammation and potentially improves patient survival. However, the evidence to support this hypothesis is limited.”
To investigate the impact of comorbidities and systemic therapies on all-cause mortality in psoriasis, the researchers used the Alberta Health Services Data Repository of Reporting database from January 1, 2012, to June 1, 2019, which represents a population base of 4.47 million individuals. They extracted data on 18,618 psoriasis cases and 55,854 controls, stratified cases according to the Charlson Comorbidity Index (CCI), a surrogate measure for comorbidity burden, and by the type of therapy received, and conducted statistical analyses including Cox proportional hazards regression to determine absolute hazard ratios representing relative effects of specific demographic and comorbidity factors on mortality within groups.
The median age in both cohorts was 48 years, and 51% were male. The researchers observed that mortality in the psoriasis cohort was significantly higher than in the controls (5.7% vs. 3.8%, respectively; P < .05), with a median age at the time of death of 72 vs. 74.4 years.
The CCI and comorbidities strongly predicted mortality, especially drug-induced liver injury (hazard ratio [HR], 1.78), bipolar disorder and suicidal ideation (HR, 1.24-1.58), and major cardiovascular diseases, which included myocardial infarction (MI), congestive heart failure (CHF), and cerebrovascular disease (CVA) (HR, 1.2-1.4).
Among patients in the psoriasis cohort, survival of those treated with biologic agents was higher than in controls, even after matching for CCI (3.2% vs. 4.4%, respectively, P < .05). “These patients also exhibit reduced overall mortality compared to those treated with methotrexate or topical agents,” Dr. Gniadecki and colleagues wrote. “There was no difference in mortality between methotrexate patients and the topical therapy patients, but any of those treatment groups had superior survival compared to the no-treatment cohort.”
They added that despite better survival among patients treated with biologic agents, no significant improvements were detected in their comorbidity profiles. “Notably, the frequency of major cardiovascular disease (MI, CHF, CVA) was the same as in the controls, and overall, the frequency of diseases coded as cardiovascular was slightly increased,” they wrote.
The fact that some factors could not be measured, including the type and severity of psoriasis, response to treatment, smoking history, and alcohol intake, was a study limitation, they noted.
Joel M. Gelfand, MD, director of the psoriasis and phototherapy treatment center at the University of Pennsylvania, Philadelphia, who was asked to comment on the analysis, said the study confirms prior work indicating that having psoriasis is a predictor of mortality. In addition, “there is a strong healthy user affect among patients who take and stay on biologics for psoriasis,” he told this news organization.
“The results are encouraging but are not able to establish a causal relationship between treating psoriasis with biologics and lowering mortality risk. Ultimately, randomized comparative trials will be needed to determine which approach or approaches to treating psoriasis, if any, lower the risk of psoriatic arthritis, cardiovascular disease, and mortality,” said Dr. Gelfand, who was not involved with the study.
Asked to comment on the results, Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was not involved with the study, said that “data such as these enable us to rationalize the cost of our fleet of biologics, as managing the outpatient/inpatient burden of many of these comorbidities will actually drain the healthcare system, more so than managing psoriasis in the first place. Certainly other interventions to address the well known comorbidities, such as cardiovascular and hepatic, are warranted, but what if you could prevent the problem in the first place? To be continued for that answer.”
The study was funded by Canadian Dermatology Foundation, Alberta Innovates, and by a Health Sciences TD Bank Studentship Award. Dr. Gniadecki reported conducting clinical trials for Bausch Health, AbbVie and Janssen, and he has received honoraria as consultant and/or speaker from AbbVie, Bausch Health, Eli Lilly, Janssen, Mallinckrodt, Novartis, Kyowa Kirin, Sun Pharma and Sanofi. The other authors had no disclosures. Dr. Gelfand reported serving as a consultant for AbbVie, Artax, Bristol-Myers Squibb, GlaxoSmithKline, and other companies. He is on the board of directors for the International Psoriasis Council and the Medical Dermatology Society. Dr. Friedman disclosed that he is a speaker for Janssen and Bristol Myers Squibb. He has received grants from Janssen, Pfizer, Bristol Myers Squibb, and Lilly, and has served as an advisor for Arcutis, Dermavant, and Janssen.
Among patients with psoriasis, the risk of mortality was strongly associated with hepatic injury, cardiovascular disease, and psychiatric affective disorders, but was reduced among those who received systemic therapy with biologics, researchers from Canada report.
Those are key findings from a large retrospective registry study of patients with psoriasis, published in The Journal of the American Academy of Dermatology.
“Psoriasis, a chronic inflammatory condition affecting approximately 3% of the western populations, bears a higher risk of mortality compared to healthy individuals, possibly by inducing systemic inflammation associated with numerous comorbidities, especially cardiovascular diseases, metabolic syndrome, and others,” wrote corresponding author Robert Gniadecki, MD, PhD, of the division of dermatology at the University of Alberta, Canada, and colleagues. “It has been argued that the use of systemic immunomodulatory agents quenches systemic inflammation and potentially improves patient survival. However, the evidence to support this hypothesis is limited.”
To investigate the impact of comorbidities and systemic therapies on all-cause mortality in psoriasis, the researchers used the Alberta Health Services Data Repository of Reporting database from January 1, 2012, to June 1, 2019, which represents a population base of 4.47 million individuals. They extracted data on 18,618 psoriasis cases and 55,854 controls, stratified cases according to the Charlson Comorbidity Index (CCI), a surrogate measure for comorbidity burden, and by the type of therapy received, and conducted statistical analyses including Cox proportional hazards regression to determine absolute hazard ratios representing relative effects of specific demographic and comorbidity factors on mortality within groups.
The median age in both cohorts was 48 years, and 51% were male. The researchers observed that mortality in the psoriasis cohort was significantly higher than in the controls (5.7% vs. 3.8%, respectively; P < .05), with a median age at the time of death of 72 vs. 74.4 years.
The CCI and comorbidities strongly predicted mortality, especially drug-induced liver injury (hazard ratio [HR], 1.78), bipolar disorder and suicidal ideation (HR, 1.24-1.58), and major cardiovascular diseases, which included myocardial infarction (MI), congestive heart failure (CHF), and cerebrovascular disease (CVA) (HR, 1.2-1.4).
Among patients in the psoriasis cohort, survival of those treated with biologic agents was higher than in controls, even after matching for CCI (3.2% vs. 4.4%, respectively, P < .05). “These patients also exhibit reduced overall mortality compared to those treated with methotrexate or topical agents,” Dr. Gniadecki and colleagues wrote. “There was no difference in mortality between methotrexate patients and the topical therapy patients, but any of those treatment groups had superior survival compared to the no-treatment cohort.”
They added that despite better survival among patients treated with biologic agents, no significant improvements were detected in their comorbidity profiles. “Notably, the frequency of major cardiovascular disease (MI, CHF, CVA) was the same as in the controls, and overall, the frequency of diseases coded as cardiovascular was slightly increased,” they wrote.
The fact that some factors could not be measured, including the type and severity of psoriasis, response to treatment, smoking history, and alcohol intake, was a study limitation, they noted.
Joel M. Gelfand, MD, director of the psoriasis and phototherapy treatment center at the University of Pennsylvania, Philadelphia, who was asked to comment on the analysis, said the study confirms prior work indicating that having psoriasis is a predictor of mortality. In addition, “there is a strong healthy user affect among patients who take and stay on biologics for psoriasis,” he told this news organization.
“The results are encouraging but are not able to establish a causal relationship between treating psoriasis with biologics and lowering mortality risk. Ultimately, randomized comparative trials will be needed to determine which approach or approaches to treating psoriasis, if any, lower the risk of psoriatic arthritis, cardiovascular disease, and mortality,” said Dr. Gelfand, who was not involved with the study.
Asked to comment on the results, Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was not involved with the study, said that “data such as these enable us to rationalize the cost of our fleet of biologics, as managing the outpatient/inpatient burden of many of these comorbidities will actually drain the healthcare system, more so than managing psoriasis in the first place. Certainly other interventions to address the well known comorbidities, such as cardiovascular and hepatic, are warranted, but what if you could prevent the problem in the first place? To be continued for that answer.”
The study was funded by Canadian Dermatology Foundation, Alberta Innovates, and by a Health Sciences TD Bank Studentship Award. Dr. Gniadecki reported conducting clinical trials for Bausch Health, AbbVie and Janssen, and he has received honoraria as consultant and/or speaker from AbbVie, Bausch Health, Eli Lilly, Janssen, Mallinckrodt, Novartis, Kyowa Kirin, Sun Pharma and Sanofi. The other authors had no disclosures. Dr. Gelfand reported serving as a consultant for AbbVie, Artax, Bristol-Myers Squibb, GlaxoSmithKline, and other companies. He is on the board of directors for the International Psoriasis Council and the Medical Dermatology Society. Dr. Friedman disclosed that he is a speaker for Janssen and Bristol Myers Squibb. He has received grants from Janssen, Pfizer, Bristol Myers Squibb, and Lilly, and has served as an advisor for Arcutis, Dermavant, and Janssen.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Asparaginase in ALL: Innovative Ways to Manage Toxicity
The good news, hematologists note, is that new strategies have been developed to address side effects. “We’ve gotten better at managing them,” pediatric oncologist Birte Wistinghausen, MD, of Children’s National Hospital in Washington, DC, said in an interview.
According to her, key approaches include sensitivity testing and “pre-medication” to prevent adverse effects from appearing in the first place.
The American Cancer Society estimates that 6,550 new cases of ALL appear in the United States each year, and 1,330 people die from the disease.
“Most cases of ALL occur in children, but most deaths from ALL (about 4 out of 5) occur in adults,” the organization reports. Indeed, the 5-year survival rate in children is now at about 90%, a number that hematologists partially attribute to the power of asparaginase.
Researchers believe that asparaginase, an enzyme, works by breaking down a substance called asparagine, which ALL cells use to reproduce. The drug is “universally used throughout the treatment of ALL in children and adolescents,” Luke Maese, DO, associate professor of pediatrics at the University of Utah–Huntsman Cancer Institute, Salt Lake City, and director of Leukemia/Lymphoma at Primary Children’s Hospital, said in an interview. “It has become more and more adopted in the treatment of young adults as well.”
The formulations of available asparaginase have evolved over the years, Dr. Maese said. “Currently, the first-line asparaginase products delivered in the majority of patients throughout the world are pegylated, meaning they have an extended duration of action. There are non-pegylated asparaginase products that are used as well.”
The pegylated drugs are much easier on patients since they don’t require frequent injections, according to experts.
Treatment protocols vary, Dr. Maese said. “Some use the drug intermittently intermixed throughout therapy, and others have periods of continuous asparaginase use — i.e. 10-20 weeks of repeated doses of the drug.”
All patients are likely to experience side effects, he said, and about 5%-10% of standard-risk and 20%-25% of high-risk patients will experience clinically significant problems.
When asparaginase is given by IV, its rapid onset can lead to a condition called acute hyperammonemia, in which ammonia levels rise and patients develop flushing, anxiety, and low blood pressure, said Dr. Wistinghausen of Children’s National Hospital. “But that is not a reason to abandon asparaginase.”
It can be difficult to differentiate this effect from hypersensitivity — allergic reactions — which can range from hives to full anaphylactic shock that requires treatment with epinephrine, she said.
According to Dr. Maese, other major side effects other than hypersensitivity include pancreatitis, hepatotoxicity, and thrombosis. The most dangerous of these side effects are hypersensitivity and pancreatitis, which can lead to discontinuation of treatment, he said. Indeed, a 2017 study found that 2% of 465 patients with ALL who developed asparaginase-associated pancreatitis died, and 8% needed mechanical ventilation.
There’s no way to predict which patients may be susceptible to pancreatitis, Michael J. Burke, MD, professor of pediatrics and director of leukemia/lymphoma director at Children’s Wisconsin and Medical College of Wisconsin, said in an interview.
As for therapy options if pancreatitis develops, a 2022 review cowritten by Dr. Maese reported that clinicians have been leaning toward re-treating patients with asparaginase since it’s so crucial to treatment. This has worked about 50% of the time, the review reported, and “many groups consider it in the setting of all grade 2 pancreatitis and grade 3 pancreatitis without prolonged illness or severe complications.”
As for hypersensitivity, the most prevalent adverse effect, clinicians frequently administer anti-allergy medications prior to infusion. This approach, known as “pre-medication,” is controversial. Research has produced conflicting results, with a 2022 study in the journal Blood finding that pre-medication had no effect on hypersensitivity in children with ALL.
“Although there is mixed data, most institutions utilize this,” Dr. Maese said. “At our institution, we continue to use pre-medication prior to pegylated asparaginase but do not use it with non-pegylated asparaginase.”
Specifically, most institutions administer H2 and H1 blockers, Dr. Wistinghausen said. “Some institutions also use hydrocortisone” — a steroid — “but our institution only uses it if patients have a reaction.”
Other potential adverse effects to treatment include infusion reactions, which can mimic allergic reactions such as nausea, vomiting, abdominal pain, and flushing, Dr. Maese said.
Asked how to treat patients who cannot tolerate first-line treatment with asparaginase, Dr. Burke responded, “There are second-generation asparaginase formulations for once a patient develops an allergy.”
Dr. Maese said his institution switches patients when necessary to asparaginase Erwinia chrysanthemi (recombinant)-rywn, also known as Rylaze.
Another recent development in ALL treatment is the widespread use of drug monitoring to make sure asparaginase is reaching therapeutic levels. “Asparagine itself is difficult to measure so we use a surrogate of asparaginase levels to demonstrate efficacy of the drug,” he said. “There is conflicting literature as to what constitutes a therapeutic level, but the internationally accepted standard is a level of ≥ 0.1 IU/mL. We monitor asparaginase levels routinely with pegylated asparaginase but not with non-pegylated asparaginase.”
Tests can turn up “silent inactivation,” a term that refers to when the drug is “inactivated” and is not effective, Dr. Maese said. “There are several guidelines that have defined inactivation.” According to the 2022 report cowritten by Dr. Maese, Rylaze can be an alternative option if initial asparaginase treatment isn’t working.
With regard to cost, treatment with asparaginase can cost tens of thousands of dollars. However, insurers routinely pay for treatment plus pre-medication and testing, Dr. Burke said. “There’s no pushback. It seems to be accepted.”
What’s next on the horizon? “We need to understand better those patients who are at risk for toxicity,” Dr. Maese noted. “We understand obesity causes risk for certain toxicities, but have little else to go on. There has been some work with genomics and its relationship to risk of toxicity. However, it has been difficult to translate what has been found to patients.”
There’s work in progress that is exploring other preventive approaches to decrease toxicity, he said. Also, “optimizing the dosing of asparaginase has been explored more in Europe and within a smaller consortium in North America.”
In addition, he said, “as we begin to increase use of immunotherapy within our chemotherapy backbones, we need to understand the relationship these drugs have with asparaginase treatment.”
Dr. Burke and Dr. Wistinghausen have no disclosures. Dr. Maese discloses relationships with Jazz (advisory board, consultant, speakers bureau) and Servier (advisory board).
The good news, hematologists note, is that new strategies have been developed to address side effects. “We’ve gotten better at managing them,” pediatric oncologist Birte Wistinghausen, MD, of Children’s National Hospital in Washington, DC, said in an interview.
According to her, key approaches include sensitivity testing and “pre-medication” to prevent adverse effects from appearing in the first place.
The American Cancer Society estimates that 6,550 new cases of ALL appear in the United States each year, and 1,330 people die from the disease.
“Most cases of ALL occur in children, but most deaths from ALL (about 4 out of 5) occur in adults,” the organization reports. Indeed, the 5-year survival rate in children is now at about 90%, a number that hematologists partially attribute to the power of asparaginase.
Researchers believe that asparaginase, an enzyme, works by breaking down a substance called asparagine, which ALL cells use to reproduce. The drug is “universally used throughout the treatment of ALL in children and adolescents,” Luke Maese, DO, associate professor of pediatrics at the University of Utah–Huntsman Cancer Institute, Salt Lake City, and director of Leukemia/Lymphoma at Primary Children’s Hospital, said in an interview. “It has become more and more adopted in the treatment of young adults as well.”
The formulations of available asparaginase have evolved over the years, Dr. Maese said. “Currently, the first-line asparaginase products delivered in the majority of patients throughout the world are pegylated, meaning they have an extended duration of action. There are non-pegylated asparaginase products that are used as well.”
The pegylated drugs are much easier on patients since they don’t require frequent injections, according to experts.
Treatment protocols vary, Dr. Maese said. “Some use the drug intermittently intermixed throughout therapy, and others have periods of continuous asparaginase use — i.e. 10-20 weeks of repeated doses of the drug.”
All patients are likely to experience side effects, he said, and about 5%-10% of standard-risk and 20%-25% of high-risk patients will experience clinically significant problems.
When asparaginase is given by IV, its rapid onset can lead to a condition called acute hyperammonemia, in which ammonia levels rise and patients develop flushing, anxiety, and low blood pressure, said Dr. Wistinghausen of Children’s National Hospital. “But that is not a reason to abandon asparaginase.”
It can be difficult to differentiate this effect from hypersensitivity — allergic reactions — which can range from hives to full anaphylactic shock that requires treatment with epinephrine, she said.
According to Dr. Maese, other major side effects other than hypersensitivity include pancreatitis, hepatotoxicity, and thrombosis. The most dangerous of these side effects are hypersensitivity and pancreatitis, which can lead to discontinuation of treatment, he said. Indeed, a 2017 study found that 2% of 465 patients with ALL who developed asparaginase-associated pancreatitis died, and 8% needed mechanical ventilation.
There’s no way to predict which patients may be susceptible to pancreatitis, Michael J. Burke, MD, professor of pediatrics and director of leukemia/lymphoma director at Children’s Wisconsin and Medical College of Wisconsin, said in an interview.
As for therapy options if pancreatitis develops, a 2022 review cowritten by Dr. Maese reported that clinicians have been leaning toward re-treating patients with asparaginase since it’s so crucial to treatment. This has worked about 50% of the time, the review reported, and “many groups consider it in the setting of all grade 2 pancreatitis and grade 3 pancreatitis without prolonged illness or severe complications.”
As for hypersensitivity, the most prevalent adverse effect, clinicians frequently administer anti-allergy medications prior to infusion. This approach, known as “pre-medication,” is controversial. Research has produced conflicting results, with a 2022 study in the journal Blood finding that pre-medication had no effect on hypersensitivity in children with ALL.
“Although there is mixed data, most institutions utilize this,” Dr. Maese said. “At our institution, we continue to use pre-medication prior to pegylated asparaginase but do not use it with non-pegylated asparaginase.”
Specifically, most institutions administer H2 and H1 blockers, Dr. Wistinghausen said. “Some institutions also use hydrocortisone” — a steroid — “but our institution only uses it if patients have a reaction.”
Other potential adverse effects to treatment include infusion reactions, which can mimic allergic reactions such as nausea, vomiting, abdominal pain, and flushing, Dr. Maese said.
Asked how to treat patients who cannot tolerate first-line treatment with asparaginase, Dr. Burke responded, “There are second-generation asparaginase formulations for once a patient develops an allergy.”
Dr. Maese said his institution switches patients when necessary to asparaginase Erwinia chrysanthemi (recombinant)-rywn, also known as Rylaze.
Another recent development in ALL treatment is the widespread use of drug monitoring to make sure asparaginase is reaching therapeutic levels. “Asparagine itself is difficult to measure so we use a surrogate of asparaginase levels to demonstrate efficacy of the drug,” he said. “There is conflicting literature as to what constitutes a therapeutic level, but the internationally accepted standard is a level of ≥ 0.1 IU/mL. We monitor asparaginase levels routinely with pegylated asparaginase but not with non-pegylated asparaginase.”
Tests can turn up “silent inactivation,” a term that refers to when the drug is “inactivated” and is not effective, Dr. Maese said. “There are several guidelines that have defined inactivation.” According to the 2022 report cowritten by Dr. Maese, Rylaze can be an alternative option if initial asparaginase treatment isn’t working.
With regard to cost, treatment with asparaginase can cost tens of thousands of dollars. However, insurers routinely pay for treatment plus pre-medication and testing, Dr. Burke said. “There’s no pushback. It seems to be accepted.”
What’s next on the horizon? “We need to understand better those patients who are at risk for toxicity,” Dr. Maese noted. “We understand obesity causes risk for certain toxicities, but have little else to go on. There has been some work with genomics and its relationship to risk of toxicity. However, it has been difficult to translate what has been found to patients.”
There’s work in progress that is exploring other preventive approaches to decrease toxicity, he said. Also, “optimizing the dosing of asparaginase has been explored more in Europe and within a smaller consortium in North America.”
In addition, he said, “as we begin to increase use of immunotherapy within our chemotherapy backbones, we need to understand the relationship these drugs have with asparaginase treatment.”
Dr. Burke and Dr. Wistinghausen have no disclosures. Dr. Maese discloses relationships with Jazz (advisory board, consultant, speakers bureau) and Servier (advisory board).
The good news, hematologists note, is that new strategies have been developed to address side effects. “We’ve gotten better at managing them,” pediatric oncologist Birte Wistinghausen, MD, of Children’s National Hospital in Washington, DC, said in an interview.
According to her, key approaches include sensitivity testing and “pre-medication” to prevent adverse effects from appearing in the first place.
The American Cancer Society estimates that 6,550 new cases of ALL appear in the United States each year, and 1,330 people die from the disease.
“Most cases of ALL occur in children, but most deaths from ALL (about 4 out of 5) occur in adults,” the organization reports. Indeed, the 5-year survival rate in children is now at about 90%, a number that hematologists partially attribute to the power of asparaginase.
Researchers believe that asparaginase, an enzyme, works by breaking down a substance called asparagine, which ALL cells use to reproduce. The drug is “universally used throughout the treatment of ALL in children and adolescents,” Luke Maese, DO, associate professor of pediatrics at the University of Utah–Huntsman Cancer Institute, Salt Lake City, and director of Leukemia/Lymphoma at Primary Children’s Hospital, said in an interview. “It has become more and more adopted in the treatment of young adults as well.”
The formulations of available asparaginase have evolved over the years, Dr. Maese said. “Currently, the first-line asparaginase products delivered in the majority of patients throughout the world are pegylated, meaning they have an extended duration of action. There are non-pegylated asparaginase products that are used as well.”
The pegylated drugs are much easier on patients since they don’t require frequent injections, according to experts.
Treatment protocols vary, Dr. Maese said. “Some use the drug intermittently intermixed throughout therapy, and others have periods of continuous asparaginase use — i.e. 10-20 weeks of repeated doses of the drug.”
All patients are likely to experience side effects, he said, and about 5%-10% of standard-risk and 20%-25% of high-risk patients will experience clinically significant problems.
When asparaginase is given by IV, its rapid onset can lead to a condition called acute hyperammonemia, in which ammonia levels rise and patients develop flushing, anxiety, and low blood pressure, said Dr. Wistinghausen of Children’s National Hospital. “But that is not a reason to abandon asparaginase.”
It can be difficult to differentiate this effect from hypersensitivity — allergic reactions — which can range from hives to full anaphylactic shock that requires treatment with epinephrine, she said.
According to Dr. Maese, other major side effects other than hypersensitivity include pancreatitis, hepatotoxicity, and thrombosis. The most dangerous of these side effects are hypersensitivity and pancreatitis, which can lead to discontinuation of treatment, he said. Indeed, a 2017 study found that 2% of 465 patients with ALL who developed asparaginase-associated pancreatitis died, and 8% needed mechanical ventilation.
There’s no way to predict which patients may be susceptible to pancreatitis, Michael J. Burke, MD, professor of pediatrics and director of leukemia/lymphoma director at Children’s Wisconsin and Medical College of Wisconsin, said in an interview.
As for therapy options if pancreatitis develops, a 2022 review cowritten by Dr. Maese reported that clinicians have been leaning toward re-treating patients with asparaginase since it’s so crucial to treatment. This has worked about 50% of the time, the review reported, and “many groups consider it in the setting of all grade 2 pancreatitis and grade 3 pancreatitis without prolonged illness or severe complications.”
As for hypersensitivity, the most prevalent adverse effect, clinicians frequently administer anti-allergy medications prior to infusion. This approach, known as “pre-medication,” is controversial. Research has produced conflicting results, with a 2022 study in the journal Blood finding that pre-medication had no effect on hypersensitivity in children with ALL.
“Although there is mixed data, most institutions utilize this,” Dr. Maese said. “At our institution, we continue to use pre-medication prior to pegylated asparaginase but do not use it with non-pegylated asparaginase.”
Specifically, most institutions administer H2 and H1 blockers, Dr. Wistinghausen said. “Some institutions also use hydrocortisone” — a steroid — “but our institution only uses it if patients have a reaction.”
Other potential adverse effects to treatment include infusion reactions, which can mimic allergic reactions such as nausea, vomiting, abdominal pain, and flushing, Dr. Maese said.
Asked how to treat patients who cannot tolerate first-line treatment with asparaginase, Dr. Burke responded, “There are second-generation asparaginase formulations for once a patient develops an allergy.”
Dr. Maese said his institution switches patients when necessary to asparaginase Erwinia chrysanthemi (recombinant)-rywn, also known as Rylaze.
Another recent development in ALL treatment is the widespread use of drug monitoring to make sure asparaginase is reaching therapeutic levels. “Asparagine itself is difficult to measure so we use a surrogate of asparaginase levels to demonstrate efficacy of the drug,” he said. “There is conflicting literature as to what constitutes a therapeutic level, but the internationally accepted standard is a level of ≥ 0.1 IU/mL. We monitor asparaginase levels routinely with pegylated asparaginase but not with non-pegylated asparaginase.”
Tests can turn up “silent inactivation,” a term that refers to when the drug is “inactivated” and is not effective, Dr. Maese said. “There are several guidelines that have defined inactivation.” According to the 2022 report cowritten by Dr. Maese, Rylaze can be an alternative option if initial asparaginase treatment isn’t working.
With regard to cost, treatment with asparaginase can cost tens of thousands of dollars. However, insurers routinely pay for treatment plus pre-medication and testing, Dr. Burke said. “There’s no pushback. It seems to be accepted.”
What’s next on the horizon? “We need to understand better those patients who are at risk for toxicity,” Dr. Maese noted. “We understand obesity causes risk for certain toxicities, but have little else to go on. There has been some work with genomics and its relationship to risk of toxicity. However, it has been difficult to translate what has been found to patients.”
There’s work in progress that is exploring other preventive approaches to decrease toxicity, he said. Also, “optimizing the dosing of asparaginase has been explored more in Europe and within a smaller consortium in North America.”
In addition, he said, “as we begin to increase use of immunotherapy within our chemotherapy backbones, we need to understand the relationship these drugs have with asparaginase treatment.”
Dr. Burke and Dr. Wistinghausen have no disclosures. Dr. Maese discloses relationships with Jazz (advisory board, consultant, speakers bureau) and Servier (advisory board).
Stimulants for ADHD Not Linked to Prescription Drug Misuse
TOPLINE:
The use of stimulant therapy by adolescents with attention-deficit/hyperactivity disorder (ADHD) was not associated with later prescription drug misuse (PDM), a new study showed. However, misuse of prescription stimulants during adolescence was associated with significantly higher odds of later PDM.
METHODOLOGY:
- Data came from 11,066 participants in the ongoing Monitoring the Future panel study (baseline cohort years 2005-2017), a multicohort US national longitudinal study of adolescents followed into adulthood, in which procedures and measures are kept consistent across time.
- Participants (ages 17 and 18 years, 51.7% female, 11.2% Black, 15.7% Hispanic, and 59.6% White) completed self-administered questionnaires, with biennial follow-up during young adulthood (ages 19-24 years).
- The questionnaires asked about the number of occasions (if any) in which respondents used a prescription drug (benzodiazepine, opioid, or stimulant) on their own, without a physician’s order.
- Baseline covariates included sex, race, ethnicity, grade point average during high school, parental education, past 2-week binge drinking, past-month cigarette use, and past-year marijuana use, as well as demographic factors.
TAKEAWAY:
- Overall, 9.9% of participants reported lifetime stimulant therapy for ADHD, and 18.6% reported lifetime prescription stimulant misuse at baseline.
- Adolescents who received stimulant therapy for ADHD were less likely to report past-year prescription stimulant misuse as young adults compared with their same-age peers who did not receive stimulant therapy (adjusted odds ratio, 0.71; 95% CI, 0.52-0.99).
- The researchers found no significant differences between adolescents with or without lifetime stimulants in later incidence or prevalence of past-year PDM during young adulthood.
- The most robust predictor of prescription stimulant misuse during young adulthood was prescription stimulant misuse during adolescence; similarly, the most robust predictors of prescription opioid and prescription benzodiazepine misuse during young adulthood were prescription opioid and prescription benzodiazepine misuse (respectively) during adolescence.
IN PRACTICE:
“These findings amplify accumulating evidence suggesting that careful monitoring and screening during adolescence could identify individuals who are at relatively greater risk for PDM and need more comprehensive substance use assessment,” the authors wrote.
SOURCE:
Sean Esteban McCabe, PhD, professor and director, Center for the Study of Drugs, Alcohol, Smoking and Health, University of Michigan School of Nursing, Ann Arbor, was the lead and corresponding author of the study. It was published online on February 7 in Psychiatric Sciences.
LIMITATIONS:
Some subpopulations with higher rates of substance use, including youths who left school before completion and institutionalized populations, were excluded from the study, which may have led to an underestimation of PDM. Moreover, some potential confounders (eg, comorbid psychiatric conditions) were not assessed.
DISCLOSURES:
This study was supported by a research award from the US Food and Drug Administration and research awards from the National Institute on Drug Abuse of the NIH. Dr. McCabe reported no relevant financial relationships. The other authors’ disclosures are listed in the original paper.
A version of this article appeared on Medscape.com.
TOPLINE:
The use of stimulant therapy by adolescents with attention-deficit/hyperactivity disorder (ADHD) was not associated with later prescription drug misuse (PDM), a new study showed. However, misuse of prescription stimulants during adolescence was associated with significantly higher odds of later PDM.
METHODOLOGY:
- Data came from 11,066 participants in the ongoing Monitoring the Future panel study (baseline cohort years 2005-2017), a multicohort US national longitudinal study of adolescents followed into adulthood, in which procedures and measures are kept consistent across time.
- Participants (ages 17 and 18 years, 51.7% female, 11.2% Black, 15.7% Hispanic, and 59.6% White) completed self-administered questionnaires, with biennial follow-up during young adulthood (ages 19-24 years).
- The questionnaires asked about the number of occasions (if any) in which respondents used a prescription drug (benzodiazepine, opioid, or stimulant) on their own, without a physician’s order.
- Baseline covariates included sex, race, ethnicity, grade point average during high school, parental education, past 2-week binge drinking, past-month cigarette use, and past-year marijuana use, as well as demographic factors.
TAKEAWAY:
- Overall, 9.9% of participants reported lifetime stimulant therapy for ADHD, and 18.6% reported lifetime prescription stimulant misuse at baseline.
- Adolescents who received stimulant therapy for ADHD were less likely to report past-year prescription stimulant misuse as young adults compared with their same-age peers who did not receive stimulant therapy (adjusted odds ratio, 0.71; 95% CI, 0.52-0.99).
- The researchers found no significant differences between adolescents with or without lifetime stimulants in later incidence or prevalence of past-year PDM during young adulthood.
- The most robust predictor of prescription stimulant misuse during young adulthood was prescription stimulant misuse during adolescence; similarly, the most robust predictors of prescription opioid and prescription benzodiazepine misuse during young adulthood were prescription opioid and prescription benzodiazepine misuse (respectively) during adolescence.
IN PRACTICE:
“These findings amplify accumulating evidence suggesting that careful monitoring and screening during adolescence could identify individuals who are at relatively greater risk for PDM and need more comprehensive substance use assessment,” the authors wrote.
SOURCE:
Sean Esteban McCabe, PhD, professor and director, Center for the Study of Drugs, Alcohol, Smoking and Health, University of Michigan School of Nursing, Ann Arbor, was the lead and corresponding author of the study. It was published online on February 7 in Psychiatric Sciences.
LIMITATIONS:
Some subpopulations with higher rates of substance use, including youths who left school before completion and institutionalized populations, were excluded from the study, which may have led to an underestimation of PDM. Moreover, some potential confounders (eg, comorbid psychiatric conditions) were not assessed.
DISCLOSURES:
This study was supported by a research award from the US Food and Drug Administration and research awards from the National Institute on Drug Abuse of the NIH. Dr. McCabe reported no relevant financial relationships. The other authors’ disclosures are listed in the original paper.
A version of this article appeared on Medscape.com.
TOPLINE:
The use of stimulant therapy by adolescents with attention-deficit/hyperactivity disorder (ADHD) was not associated with later prescription drug misuse (PDM), a new study showed. However, misuse of prescription stimulants during adolescence was associated with significantly higher odds of later PDM.
METHODOLOGY:
- Data came from 11,066 participants in the ongoing Monitoring the Future panel study (baseline cohort years 2005-2017), a multicohort US national longitudinal study of adolescents followed into adulthood, in which procedures and measures are kept consistent across time.
- Participants (ages 17 and 18 years, 51.7% female, 11.2% Black, 15.7% Hispanic, and 59.6% White) completed self-administered questionnaires, with biennial follow-up during young adulthood (ages 19-24 years).
- The questionnaires asked about the number of occasions (if any) in which respondents used a prescription drug (benzodiazepine, opioid, or stimulant) on their own, without a physician’s order.
- Baseline covariates included sex, race, ethnicity, grade point average during high school, parental education, past 2-week binge drinking, past-month cigarette use, and past-year marijuana use, as well as demographic factors.
TAKEAWAY:
- Overall, 9.9% of participants reported lifetime stimulant therapy for ADHD, and 18.6% reported lifetime prescription stimulant misuse at baseline.
- Adolescents who received stimulant therapy for ADHD were less likely to report past-year prescription stimulant misuse as young adults compared with their same-age peers who did not receive stimulant therapy (adjusted odds ratio, 0.71; 95% CI, 0.52-0.99).
- The researchers found no significant differences between adolescents with or without lifetime stimulants in later incidence or prevalence of past-year PDM during young adulthood.
- The most robust predictor of prescription stimulant misuse during young adulthood was prescription stimulant misuse during adolescence; similarly, the most robust predictors of prescription opioid and prescription benzodiazepine misuse during young adulthood were prescription opioid and prescription benzodiazepine misuse (respectively) during adolescence.
IN PRACTICE:
“These findings amplify accumulating evidence suggesting that careful monitoring and screening during adolescence could identify individuals who are at relatively greater risk for PDM and need more comprehensive substance use assessment,” the authors wrote.
SOURCE:
Sean Esteban McCabe, PhD, professor and director, Center for the Study of Drugs, Alcohol, Smoking and Health, University of Michigan School of Nursing, Ann Arbor, was the lead and corresponding author of the study. It was published online on February 7 in Psychiatric Sciences.
LIMITATIONS:
Some subpopulations with higher rates of substance use, including youths who left school before completion and institutionalized populations, were excluded from the study, which may have led to an underestimation of PDM. Moreover, some potential confounders (eg, comorbid psychiatric conditions) were not assessed.
DISCLOSURES:
This study was supported by a research award from the US Food and Drug Administration and research awards from the National Institute on Drug Abuse of the NIH. Dr. McCabe reported no relevant financial relationships. The other authors’ disclosures are listed in the original paper.
A version of this article appeared on Medscape.com.
Study Eyes Longer IV Ertapenem for Recalcitrant Hidradenitis Suppurativa
at the completion of treatment, a retrospective study showed.
“These findings suggest a course of 12 to 16 weeks of ertapenem may be appropriate as a new standard length of therapy in HS patients, which is at least twice the current recommendation of the North American treatment guidelines,” wrote corresponding author Steven R. Cohen, MD, MPH, of the departments of dermatology at Weill Cornell Medicine and Albert Einstein College of Medicine, New York, and his coauthors. The results were published online February 14, 2024, in JAMA Dermatology.
In an earlier study , some of the same researchers evaluated the efficacy of daily IV ertapenem for 6 weeks in seven patients with HS. The patients experienced “notable remediation of disease that was rapidly lost within 1 month of withdrawal.”
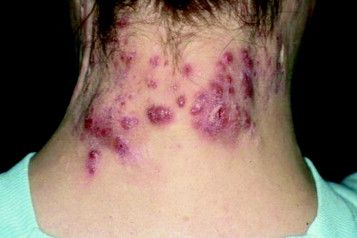
Treatment guidelines published in 2019 recommend ertapenem as a highly effective third-line therapy limited to one 6-week course “as rescue therapy or during surgical planning, given the practical barriers to home infusions and concerns about antibiotic resistance” .
For the current analysis, Dr. Cohen and colleagues explored the effects of a longer duration of treatment with ertapenem in this patient population. They retrospectively reviewed the medical records of 98 patients with HS who received care at Albert Einstein College of Medicine’s Montefiore HS Center between 2018 and 2022. Each patient used an elastomeric pump to self-administer 1 g IV ertapenem daily for 12-16 weeks.
Key outcome measures of interest were the HS Physician Global Assessment (PGA) score (a 6-point scale ranging from clear to very severe) and a numerical rating scale (NRS) for pain (an 11-point scale in which a score of 0 indicates no pain and a score of 10 indicates the worst possible pain) and markers of inflammation such as leukocytes, erythrocyte sedimentation rate, C-reactive protein (CRP), and interleukin (IL)-6. The researchers measured these outcomes at baseline, the midcourse of IV ertapenem treatment, at the end of the course, and post therapy.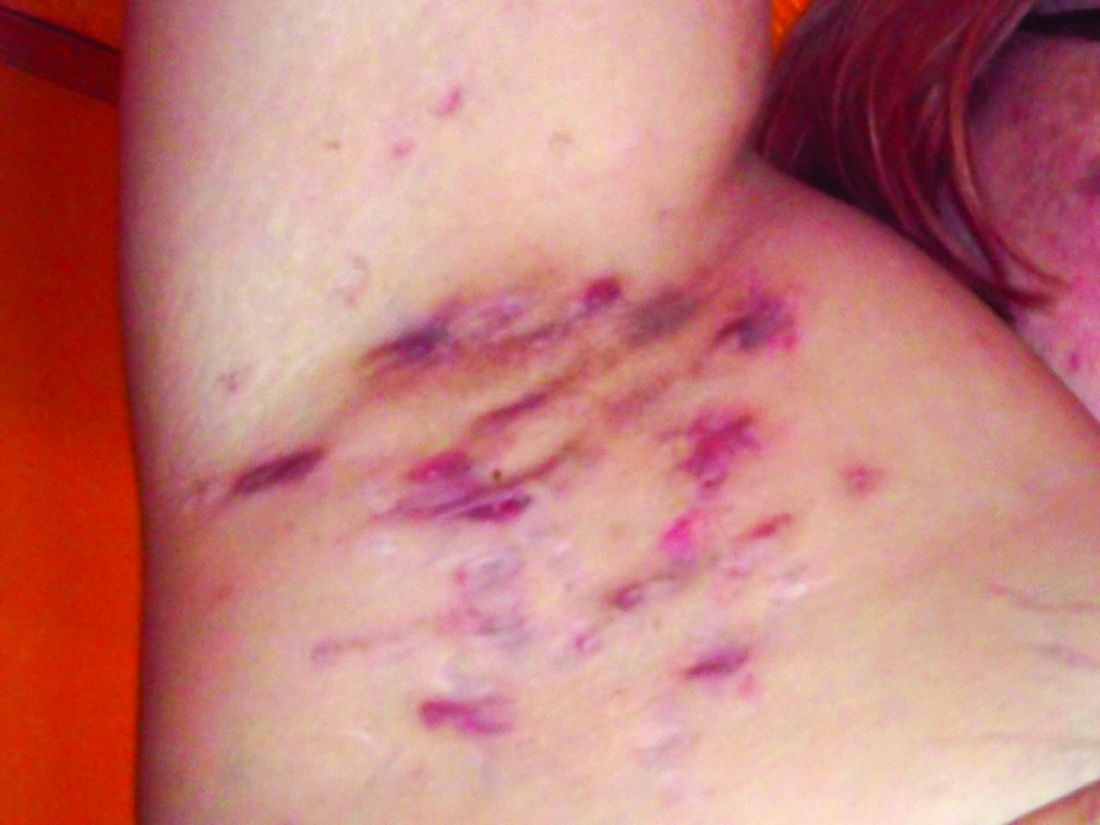
The mean age of the patients was 35.8 years, 62.2% were female, and 60.2% were Black. The mean treatment duration was 13.1 weeks and the mean posttherapy follow-up occurred after a mean of 7.8 weeks.
Between baseline and posttherapy follow-up, the HS PGA scores dropped from a mean of 3.9 to 2.7 and the NRS for pain dropped from 4.2 to 1.8 (P < .001 for both associations). Markers of inflammation also dropped between baseline and post therapy.
Specifically, values for CRP dropped from 5.4 to 2.4 mg/dL; IL-6 dropped from 25.2 to 13.7, and leukocytes dropped from 11.3 to 10.0 (P < .001 for all associations). Among the 76 patients who participated in a follow-up telephone survey, 63 (80.3%) reported medium to high satisfaction with their course of ertapenem, and 69 (90.8%) said they would recommend the treatment to other patients with HS.
The authors noted certain limitations of their study, including its retrospective, single-center design, the lack of a control group, and the fact that the HS-PGA scores at each visit did not meet the threshold of a 2-point decrease that is considered a clinically meaningful in the medical literature.
The definitive mechanism of ertapenem efficacy remains elusive, the authors pointed out. “Although oral antibiotics are generally accepted as a core therapeutic approach to HS, much less is known about the efficacy of IV antibiotics, especially ertapenem, a parenteral carbapenem possessing activity against many gram-positive bacteria, gram-negative bacteria, and anaerobic organisms,” they wrote.
In an accompanying editorial, Haley B. Naik, MD, MHSc, a dermatologist at the University of California, San Francisco, said that adopting prolonged courses of ertapenem treatment “comes with substantial individual and public health considerations”.

“Even though HS is a noninfectious disease, microbes might play a role in inciting HS immune dysregulation, prompting the inclusion of antimicrobial therapy in treatment regimens. However, broad-spectrum antibiotics for HS are associated with high levels of antibiotic resistance,” she wrote. Prolonged use of ertapenem and other carbapenems in HS treatment “will likely increase antimicrobial resistance, thereby limiting management of both HS and comorbid infections.”
Jennifer L. Hsiao, MD, a dermatologist who directs the HS clinic at the University of Southern California, Los Angeles, who was asked to comment on the study, said that, despite significant advances in the management of HS over the past decade, there are still patients who do not respond adequately to standard treatments.

For these patients, IV ertapenem can serve as a valuable bridge to a longer-term therapeutic option, “be it surgery or escalated immunomodulation,” such as dual biologic therapy, she said. “In my personal experience, IV ertapenem, which like the authors I also typically use for a 12-week course, delivers impressive and fast results even in the worst disease cases.
“It can be difficult to maintain the therapeutic benefit of ertapenem after it is discontinued, which is why patients should be on concomitant medications as they were in this study and have a post-ertapenem treatment plan in place,” said Dr. Hsiao, who was not involved with the study. “Hopefully, we will be able to one day understand why ertapenem is so effective for HS and be able to harness that benefit for patients without concern for antimicrobial resistance.”
Dr. Cohen reported receiving personal fees from Verrica Pharmaceuticals and belonging to the Board of Trustees of the American Skin Association outside the submitted work. No other disclosures were reported. Dr. Naik reported having received grants from AbbVie and the National Institutes of Health; personal fees from Novartis, UCB, Boehringer Ingelheim, 23andMe, Aristea Therapeutics, Medscape, Sonoma Biotherapeutics, DAVA Oncology, and Pfizer; and shares from Radera during the conduct of the study. She is a board member of the Hidradenitis Suppurativa Foundation. Dr. Hsiao disclosed that she is a member of the board of directors for the Hidradenitis Suppurativa Foundation. She has served as a consultant for AbbVie, Aclaris, Boehringer Ingelheim, Incyte, Novartis, UCB, as a speaker for AbbVie, Novartis, and UCB, and as an investigator for Amgen, Boehringer Ingelheim, and Incyte.
at the completion of treatment, a retrospective study showed.
“These findings suggest a course of 12 to 16 weeks of ertapenem may be appropriate as a new standard length of therapy in HS patients, which is at least twice the current recommendation of the North American treatment guidelines,” wrote corresponding author Steven R. Cohen, MD, MPH, of the departments of dermatology at Weill Cornell Medicine and Albert Einstein College of Medicine, New York, and his coauthors. The results were published online February 14, 2024, in JAMA Dermatology.
In an earlier study , some of the same researchers evaluated the efficacy of daily IV ertapenem for 6 weeks in seven patients with HS. The patients experienced “notable remediation of disease that was rapidly lost within 1 month of withdrawal.”
Treatment guidelines published in 2019 recommend ertapenem as a highly effective third-line therapy limited to one 6-week course “as rescue therapy or during surgical planning, given the practical barriers to home infusions and concerns about antibiotic resistance” .
For the current analysis, Dr. Cohen and colleagues explored the effects of a longer duration of treatment with ertapenem in this patient population. They retrospectively reviewed the medical records of 98 patients with HS who received care at Albert Einstein College of Medicine’s Montefiore HS Center between 2018 and 2022. Each patient used an elastomeric pump to self-administer 1 g IV ertapenem daily for 12-16 weeks.
Key outcome measures of interest were the HS Physician Global Assessment (PGA) score (a 6-point scale ranging from clear to very severe) and a numerical rating scale (NRS) for pain (an 11-point scale in which a score of 0 indicates no pain and a score of 10 indicates the worst possible pain) and markers of inflammation such as leukocytes, erythrocyte sedimentation rate, C-reactive protein (CRP), and interleukin (IL)-6. The researchers measured these outcomes at baseline, the midcourse of IV ertapenem treatment, at the end of the course, and post therapy.
The mean age of the patients was 35.8 years, 62.2% were female, and 60.2% were Black. The mean treatment duration was 13.1 weeks and the mean posttherapy follow-up occurred after a mean of 7.8 weeks.
Between baseline and posttherapy follow-up, the HS PGA scores dropped from a mean of 3.9 to 2.7 and the NRS for pain dropped from 4.2 to 1.8 (P < .001 for both associations). Markers of inflammation also dropped between baseline and post therapy.
Specifically, values for CRP dropped from 5.4 to 2.4 mg/dL; IL-6 dropped from 25.2 to 13.7, and leukocytes dropped from 11.3 to 10.0 (P < .001 for all associations). Among the 76 patients who participated in a follow-up telephone survey, 63 (80.3%) reported medium to high satisfaction with their course of ertapenem, and 69 (90.8%) said they would recommend the treatment to other patients with HS.
The authors noted certain limitations of their study, including its retrospective, single-center design, the lack of a control group, and the fact that the HS-PGA scores at each visit did not meet the threshold of a 2-point decrease that is considered a clinically meaningful in the medical literature.
The definitive mechanism of ertapenem efficacy remains elusive, the authors pointed out. “Although oral antibiotics are generally accepted as a core therapeutic approach to HS, much less is known about the efficacy of IV antibiotics, especially ertapenem, a parenteral carbapenem possessing activity against many gram-positive bacteria, gram-negative bacteria, and anaerobic organisms,” they wrote.
In an accompanying editorial, Haley B. Naik, MD, MHSc, a dermatologist at the University of California, San Francisco, said that adopting prolonged courses of ertapenem treatment “comes with substantial individual and public health considerations”.
“Even though HS is a noninfectious disease, microbes might play a role in inciting HS immune dysregulation, prompting the inclusion of antimicrobial therapy in treatment regimens. However, broad-spectrum antibiotics for HS are associated with high levels of antibiotic resistance,” she wrote. Prolonged use of ertapenem and other carbapenems in HS treatment “will likely increase antimicrobial resistance, thereby limiting management of both HS and comorbid infections.”
Jennifer L. Hsiao, MD, a dermatologist who directs the HS clinic at the University of Southern California, Los Angeles, who was asked to comment on the study, said that, despite significant advances in the management of HS over the past decade, there are still patients who do not respond adequately to standard treatments.
For these patients, IV ertapenem can serve as a valuable bridge to a longer-term therapeutic option, “be it surgery or escalated immunomodulation,” such as dual biologic therapy, she said. “In my personal experience, IV ertapenem, which like the authors I also typically use for a 12-week course, delivers impressive and fast results even in the worst disease cases.
“It can be difficult to maintain the therapeutic benefit of ertapenem after it is discontinued, which is why patients should be on concomitant medications as they were in this study and have a post-ertapenem treatment plan in place,” said Dr. Hsiao, who was not involved with the study. “Hopefully, we will be able to one day understand why ertapenem is so effective for HS and be able to harness that benefit for patients without concern for antimicrobial resistance.”
Dr. Cohen reported receiving personal fees from Verrica Pharmaceuticals and belonging to the Board of Trustees of the American Skin Association outside the submitted work. No other disclosures were reported. Dr. Naik reported having received grants from AbbVie and the National Institutes of Health; personal fees from Novartis, UCB, Boehringer Ingelheim, 23andMe, Aristea Therapeutics, Medscape, Sonoma Biotherapeutics, DAVA Oncology, and Pfizer; and shares from Radera during the conduct of the study. She is a board member of the Hidradenitis Suppurativa Foundation. Dr. Hsiao disclosed that she is a member of the board of directors for the Hidradenitis Suppurativa Foundation. She has served as a consultant for AbbVie, Aclaris, Boehringer Ingelheim, Incyte, Novartis, UCB, as a speaker for AbbVie, Novartis, and UCB, and as an investigator for Amgen, Boehringer Ingelheim, and Incyte.
at the completion of treatment, a retrospective study showed.
“These findings suggest a course of 12 to 16 weeks of ertapenem may be appropriate as a new standard length of therapy in HS patients, which is at least twice the current recommendation of the North American treatment guidelines,” wrote corresponding author Steven R. Cohen, MD, MPH, of the departments of dermatology at Weill Cornell Medicine and Albert Einstein College of Medicine, New York, and his coauthors. The results were published online February 14, 2024, in JAMA Dermatology.
In an earlier study , some of the same researchers evaluated the efficacy of daily IV ertapenem for 6 weeks in seven patients with HS. The patients experienced “notable remediation of disease that was rapidly lost within 1 month of withdrawal.”
Treatment guidelines published in 2019 recommend ertapenem as a highly effective third-line therapy limited to one 6-week course “as rescue therapy or during surgical planning, given the practical barriers to home infusions and concerns about antibiotic resistance” .
For the current analysis, Dr. Cohen and colleagues explored the effects of a longer duration of treatment with ertapenem in this patient population. They retrospectively reviewed the medical records of 98 patients with HS who received care at Albert Einstein College of Medicine’s Montefiore HS Center between 2018 and 2022. Each patient used an elastomeric pump to self-administer 1 g IV ertapenem daily for 12-16 weeks.
Key outcome measures of interest were the HS Physician Global Assessment (PGA) score (a 6-point scale ranging from clear to very severe) and a numerical rating scale (NRS) for pain (an 11-point scale in which a score of 0 indicates no pain and a score of 10 indicates the worst possible pain) and markers of inflammation such as leukocytes, erythrocyte sedimentation rate, C-reactive protein (CRP), and interleukin (IL)-6. The researchers measured these outcomes at baseline, the midcourse of IV ertapenem treatment, at the end of the course, and post therapy.
The mean age of the patients was 35.8 years, 62.2% were female, and 60.2% were Black. The mean treatment duration was 13.1 weeks and the mean posttherapy follow-up occurred after a mean of 7.8 weeks.
Between baseline and posttherapy follow-up, the HS PGA scores dropped from a mean of 3.9 to 2.7 and the NRS for pain dropped from 4.2 to 1.8 (P < .001 for both associations). Markers of inflammation also dropped between baseline and post therapy.
Specifically, values for CRP dropped from 5.4 to 2.4 mg/dL; IL-6 dropped from 25.2 to 13.7, and leukocytes dropped from 11.3 to 10.0 (P < .001 for all associations). Among the 76 patients who participated in a follow-up telephone survey, 63 (80.3%) reported medium to high satisfaction with their course of ertapenem, and 69 (90.8%) said they would recommend the treatment to other patients with HS.
The authors noted certain limitations of their study, including its retrospective, single-center design, the lack of a control group, and the fact that the HS-PGA scores at each visit did not meet the threshold of a 2-point decrease that is considered a clinically meaningful in the medical literature.
The definitive mechanism of ertapenem efficacy remains elusive, the authors pointed out. “Although oral antibiotics are generally accepted as a core therapeutic approach to HS, much less is known about the efficacy of IV antibiotics, especially ertapenem, a parenteral carbapenem possessing activity against many gram-positive bacteria, gram-negative bacteria, and anaerobic organisms,” they wrote.
In an accompanying editorial, Haley B. Naik, MD, MHSc, a dermatologist at the University of California, San Francisco, said that adopting prolonged courses of ertapenem treatment “comes with substantial individual and public health considerations”.
“Even though HS is a noninfectious disease, microbes might play a role in inciting HS immune dysregulation, prompting the inclusion of antimicrobial therapy in treatment regimens. However, broad-spectrum antibiotics for HS are associated with high levels of antibiotic resistance,” she wrote. Prolonged use of ertapenem and other carbapenems in HS treatment “will likely increase antimicrobial resistance, thereby limiting management of both HS and comorbid infections.”
Jennifer L. Hsiao, MD, a dermatologist who directs the HS clinic at the University of Southern California, Los Angeles, who was asked to comment on the study, said that, despite significant advances in the management of HS over the past decade, there are still patients who do not respond adequately to standard treatments.
For these patients, IV ertapenem can serve as a valuable bridge to a longer-term therapeutic option, “be it surgery or escalated immunomodulation,” such as dual biologic therapy, she said. “In my personal experience, IV ertapenem, which like the authors I also typically use for a 12-week course, delivers impressive and fast results even in the worst disease cases.
“It can be difficult to maintain the therapeutic benefit of ertapenem after it is discontinued, which is why patients should be on concomitant medications as they were in this study and have a post-ertapenem treatment plan in place,” said Dr. Hsiao, who was not involved with the study. “Hopefully, we will be able to one day understand why ertapenem is so effective for HS and be able to harness that benefit for patients without concern for antimicrobial resistance.”
Dr. Cohen reported receiving personal fees from Verrica Pharmaceuticals and belonging to the Board of Trustees of the American Skin Association outside the submitted work. No other disclosures were reported. Dr. Naik reported having received grants from AbbVie and the National Institutes of Health; personal fees from Novartis, UCB, Boehringer Ingelheim, 23andMe, Aristea Therapeutics, Medscape, Sonoma Biotherapeutics, DAVA Oncology, and Pfizer; and shares from Radera during the conduct of the study. She is a board member of the Hidradenitis Suppurativa Foundation. Dr. Hsiao disclosed that she is a member of the board of directors for the Hidradenitis Suppurativa Foundation. She has served as a consultant for AbbVie, Aclaris, Boehringer Ingelheim, Incyte, Novartis, UCB, as a speaker for AbbVie, Novartis, and UCB, and as an investigator for Amgen, Boehringer Ingelheim, and Incyte.
FROM JAMA DERMATOLOGY
The Daycare Petri Dish
I can’t remember where I heard it. Maybe I made it up myself. But, one definition of a family is a group of folks with whom you share your genes and germs. In that same vein, one could define daycare as a group of germ-sharing children. Of course that’s news to almost no one. Parents who decide, or are forced, to send their children to daycare expect that those children will get more colds, “stomach flu,” and ear infections than the children who spend their days in isolation at home. Everyone from the pediatrician to the little old lady next door has warned parents of the inevitable reality of daycare. Of course there are upsides that parents can cling to, including increased socialization and the hope that getting sick young will build a more robust immunity in the long run.
However, there has been little research exploring the nuances of the germ sharing that we all know is happening in these and other social settings.
A team of evolutionary biologists at Harvard is working to better define the “social microbiome” and its “role in individuals’ susceptibility to, and resilience against, both communicable and noncommunicable diseases.” These researchers point out that while we harbor our own unique collection of microbes, we share those with the microbiomes of the people with whom we interact socially. They report that studies by other investigators have shown that residents of a household share a significant proportion of their gastrointestinal flora. There are other studies that have shown that villages can be identified by their own social biome. There are few social settings more intimately involved in microbe sharing than daycares. I have a friends who calls them “petri dishes.”

The biologists point out that antibiotic-resistant microbes can become part of an individual’s microbiome and can be shared with other individuals in their social group, who can then go on and share them in a different social environment. Imagine there is one popular physician in a community whose sense of antibiotic stewardship is, shall we say, somewhat lacking. By inappropriately prescribing antibiotics to a child or two in a daycare, he may be altering the social biome in that daycare, which could then jeopardize the health of all the children and eventually their own home-based social biomes, that may include an immune deficient individual.
The researchers also remind us that different cultures and countries may have different antibiotic usage patterns. Does this mean I am taking a risk by traveling in these “culture-dependent transmission landscapes”? Am I more likely to encounter an antibiotic-resistant microbe when I am visiting a country whose healthcare providers are less prudent prescribers?
However, as these evolutionary biologists point out, not all shared microbes are bad. There is some evidence in animals that individuals can share microbes that have been found to “increase resilience against colitis or improve their responsiveness to cancer therapy.” If a microbe can contribute to a disease that was once considered to be “noncommunicable,” we may need to redefine “communicable” in the light this more nuanced view of the social biome.
It became standard practice during the COVID pandemic to test dormitory and community sewage water to determine the level of infection. Can sewage water be used as a proxy for a social biome? If a parent is lucky enough to have a choice of daycares, would knowing each facility’s biome, as reflected in an analysis of its sewage effluent, help him or her decide? Should a daycare ask for a stool sample from each child before accepting him or her? Seems like this would raise some privacy issues, not to mention the logistical messiness of the process. As we learn more about social biomes, can we imagine a time when a daycare or country or region might proudly advertise itself as having the healthiest spectrum of microbes in its sewage system?
Communal living certainly has its benefits, not just for children but also adults as we realize how loneliness is eating its way into our society. However,
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
I can’t remember where I heard it. Maybe I made it up myself. But, one definition of a family is a group of folks with whom you share your genes and germs. In that same vein, one could define daycare as a group of germ-sharing children. Of course that’s news to almost no one. Parents who decide, or are forced, to send their children to daycare expect that those children will get more colds, “stomach flu,” and ear infections than the children who spend their days in isolation at home. Everyone from the pediatrician to the little old lady next door has warned parents of the inevitable reality of daycare. Of course there are upsides that parents can cling to, including increased socialization and the hope that getting sick young will build a more robust immunity in the long run.
However, there has been little research exploring the nuances of the germ sharing that we all know is happening in these and other social settings.
A team of evolutionary biologists at Harvard is working to better define the “social microbiome” and its “role in individuals’ susceptibility to, and resilience against, both communicable and noncommunicable diseases.” These researchers point out that while we harbor our own unique collection of microbes, we share those with the microbiomes of the people with whom we interact socially. They report that studies by other investigators have shown that residents of a household share a significant proportion of their gastrointestinal flora. There are other studies that have shown that villages can be identified by their own social biome. There are few social settings more intimately involved in microbe sharing than daycares. I have a friends who calls them “petri dishes.”
The biologists point out that antibiotic-resistant microbes can become part of an individual’s microbiome and can be shared with other individuals in their social group, who can then go on and share them in a different social environment. Imagine there is one popular physician in a community whose sense of antibiotic stewardship is, shall we say, somewhat lacking. By inappropriately prescribing antibiotics to a child or two in a daycare, he may be altering the social biome in that daycare, which could then jeopardize the health of all the children and eventually their own home-based social biomes, that may include an immune deficient individual.
The researchers also remind us that different cultures and countries may have different antibiotic usage patterns. Does this mean I am taking a risk by traveling in these “culture-dependent transmission landscapes”? Am I more likely to encounter an antibiotic-resistant microbe when I am visiting a country whose healthcare providers are less prudent prescribers?
However, as these evolutionary biologists point out, not all shared microbes are bad. There is some evidence in animals that individuals can share microbes that have been found to “increase resilience against colitis or improve their responsiveness to cancer therapy.” If a microbe can contribute to a disease that was once considered to be “noncommunicable,” we may need to redefine “communicable” in the light this more nuanced view of the social biome.
It became standard practice during the COVID pandemic to test dormitory and community sewage water to determine the level of infection. Can sewage water be used as a proxy for a social biome? If a parent is lucky enough to have a choice of daycares, would knowing each facility’s biome, as reflected in an analysis of its sewage effluent, help him or her decide? Should a daycare ask for a stool sample from each child before accepting him or her? Seems like this would raise some privacy issues, not to mention the logistical messiness of the process. As we learn more about social biomes, can we imagine a time when a daycare or country or region might proudly advertise itself as having the healthiest spectrum of microbes in its sewage system?
Communal living certainly has its benefits, not just for children but also adults as we realize how loneliness is eating its way into our society. However,
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
I can’t remember where I heard it. Maybe I made it up myself. But, one definition of a family is a group of folks with whom you share your genes and germs. In that same vein, one could define daycare as a group of germ-sharing children. Of course that’s news to almost no one. Parents who decide, or are forced, to send their children to daycare expect that those children will get more colds, “stomach flu,” and ear infections than the children who spend their days in isolation at home. Everyone from the pediatrician to the little old lady next door has warned parents of the inevitable reality of daycare. Of course there are upsides that parents can cling to, including increased socialization and the hope that getting sick young will build a more robust immunity in the long run.
However, there has been little research exploring the nuances of the germ sharing that we all know is happening in these and other social settings.
A team of evolutionary biologists at Harvard is working to better define the “social microbiome” and its “role in individuals’ susceptibility to, and resilience against, both communicable and noncommunicable diseases.” These researchers point out that while we harbor our own unique collection of microbes, we share those with the microbiomes of the people with whom we interact socially. They report that studies by other investigators have shown that residents of a household share a significant proportion of their gastrointestinal flora. There are other studies that have shown that villages can be identified by their own social biome. There are few social settings more intimately involved in microbe sharing than daycares. I have a friends who calls them “petri dishes.”
The biologists point out that antibiotic-resistant microbes can become part of an individual’s microbiome and can be shared with other individuals in their social group, who can then go on and share them in a different social environment. Imagine there is one popular physician in a community whose sense of antibiotic stewardship is, shall we say, somewhat lacking. By inappropriately prescribing antibiotics to a child or two in a daycare, he may be altering the social biome in that daycare, which could then jeopardize the health of all the children and eventually their own home-based social biomes, that may include an immune deficient individual.
The researchers also remind us that different cultures and countries may have different antibiotic usage patterns. Does this mean I am taking a risk by traveling in these “culture-dependent transmission landscapes”? Am I more likely to encounter an antibiotic-resistant microbe when I am visiting a country whose healthcare providers are less prudent prescribers?
However, as these evolutionary biologists point out, not all shared microbes are bad. There is some evidence in animals that individuals can share microbes that have been found to “increase resilience against colitis or improve their responsiveness to cancer therapy.” If a microbe can contribute to a disease that was once considered to be “noncommunicable,” we may need to redefine “communicable” in the light this more nuanced view of the social biome.
It became standard practice during the COVID pandemic to test dormitory and community sewage water to determine the level of infection. Can sewage water be used as a proxy for a social biome? If a parent is lucky enough to have a choice of daycares, would knowing each facility’s biome, as reflected in an analysis of its sewage effluent, help him or her decide? Should a daycare ask for a stool sample from each child before accepting him or her? Seems like this would raise some privacy issues, not to mention the logistical messiness of the process. As we learn more about social biomes, can we imagine a time when a daycare or country or region might proudly advertise itself as having the healthiest spectrum of microbes in its sewage system?
Communal living certainly has its benefits, not just for children but also adults as we realize how loneliness is eating its way into our society. However,
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
Mental Health Interventions for Refugee Children
In my previous article, “Mental Health Characteristics of Refugee Children,” we learned that in recent decades, refugeeism has become a growing problem that disproportionately affects children. Refugee children and their families experience a variety of traumas, often sustained across years and even decades, because of armed conflict, persecution, social upheavals, or environmental disasters. Refugees are at greater risks for PTSD and affective and psychotic disorders presumably due to increased traumatic life events before, during, and after migration. I used my own experience as a child refugee from Vietnam to elucidate the stressors evident in various phases of forced displacement.1

Risk Factors and Protective Factors
To a certain extent, the experiences of refugees are universal. All refugees experience some sort of humanitarian crisis that forces an emergent escape from their home across international borders to a new resettlement area. It is important to note that internally displaced people do not meet the United Nations’ (UN) official designation of refugee status; however, some agencies use a broader definition where they are designated as such.2,3 We will refer to those not meeting the UN criteria as displaced people, while refugees are those that do meet the UN criteria. Dr. Mina Fazel’s 2012 systematic review in The Lancet of mental health risk factors and protective factors for displaced and refugee children is the most comprehensive of its kind.4 It will be summarized in this section with some relevant personal reflection.
In terms of risk factors, external displacement likely results in additional stress and trauma, presumably from the lack of assess to one’s culture and the host country’s language. Understandably, this makes rebuilding of one’s life more difficult. Several studies show that displaced/refugee children experience more difficulty with psychosocial adaptation than non-displaced children. Violence, directly experienced or indirectly feared, both to the child and their parents, was the strongest predictor of mental health problems and withdrawn behavior. Children who were separated from their parents clearly fared worst in their mental health than those who did not, which is not surprising given the nature of their dependence on caregivers for protection and guidance. During resettlement, experienced or perceived discrimination from the host country was also a risk factor, as well as instability in housing and a drawn-out resettlement process. Female sex was a risk factor mainly for emotional problems. Poor financial support post-migration is associated with depression, but it is unclear whether pre-migration financial status was protective. From my own experience, it is likely not, given that once one becomes a refugee one does not have access to one’s wealth, except that which could be hidden on one’s body. Another risk factor was also if one’s parent had psychiatric problems or was single. Due to the migration, my mother was separated permanently from her husband, which caused her extraordinary isolation and loneliness, something that was palpably felt by myself as I grew up.
In terms of protective factors, family cohesion and cultural continuity appear critical. For myself, not only would I not have survived without my mother and aunt, but they constantly protected me from the harsh realities. My mother would distract me with seemingly trivial goals once we got to America, like finally tasting a hamburger, or talking about school and being reunited with my uncle. This is in line with another finding — that children have better mental health outcomes when their parents do not talk about their hardships. Once my family was resettled with my uncle and his family, they played a critical role in smoothing our transition, not only by providing us with housing, but also cultural knowledge. Cultural havens can restore some of the social position and way of life that refugees lose when they are able to reconnect with a society that recognizes their previous achievements and status. Finally, religion also seemed to be a protective factor.
Mental Health Interventions
In 2018, Dr. Fazel identified mental health interventions for refugee children in a narrative review.5 She acknowledged that these conclusions are limited by the paucity of preventive mental health research in children in general, as well as the mobile nature and complex cultural differences of refugee children. This is exacerbated by the small evidence base. Given that, she makes these recommendations for varying levels of interventions: individual, group, family, living circumstances, social interactions, and school.
On an individual level, effective interventions developed to address PTSD include narrative exposure therapy, trauma-focused cognitive behavior therapy, and eye-movement and desensitization therapy. Group-based interventions for trauma, for example school-based PTSD intervention programs in conflicted areas, have either been shown to not be effective, or only effective for reducing depression. The mental health of unaccompanied children separated from family fare better when placed in foster care, rather than other types of social support. This is further enhanced if the foster family is the same ethnicity.
On a family level, improvements in parenting style and parental mental health, family engagement with local culture and structures, and family-based mental health interventions all positively impact refugee children. Not surprisingly, refugee parents have a greater prevalence of mental health conditions. Several studies on refugeeism point out a greater occurrence of intimate partner violence (that negatively affects children) as well has harsher discipline and maltreatment of refugee children. Thus, mental health treatment for parents also directly improves the well-being of their children. Teaching parenting skills to mitigate the violent effect of their PTSD symptoms, as well as parenting classes that teach gentler styles, have been shown to reduce harsh parenting and mitigate aggressive behaviors in these children. These improvements are enhanced when these classes are taught by other refugees themselves.
School is key for helping refugee children since it is a site where they can access language proficiency, successful acculturation, and medical and mental health services. Several studies have identified the positive effects of better parental engagement with school, resulting in improved academic performance and reduced levels of depressive and PTSD symptoms. A review of learning problems in refugee children identified several factors for success. These include high academic and life ambition, parental involvement in education, accurate educational assessment and grade placement, teacher understanding of linguistic and cultural heritage, culturally appropriate school transition, supportive peer relationships, and successful acculturation. School certainly was key for my acculturation and language proficiency. When I arrived at 6 years old I was selectively mute for my year in first grade, namely because I did not know how to speak English and because I did not share the culture. However, my teacher correctly identified my deficiency and chose to place me in kindergarten, which allowed me the time to gain English proficiency. Though I was always the oldest one in class, that remediation was key in allowing eventual success in school leading up to my admission to UC Berkeley.
Summary
In recent decades, refugeeism has become a growing problem that disproportionately affects children leading to traumas sustained across years and even decades, and greater risks for PTSD, as well as affective and psychotic disorders. Risk factors include the experience of violence, the separation from family, female gender, discrimination in the host country, unstable housing, and a drawn-out resettlement process. Protective factors consist of family cohesion, cultural continuity, support at schools, being protected from the truth of their harsh reality, stable housing, language acquisition, and quick resettlement. From these factors, effective mental interventions have been found to be the promotion of these protective factors as well as support for parental mental health and parenting skills, better parental engagement at school, and schools that correctly identify and address these children’s educational needs.
Dr. Nguyen is a second-year resident at UCSF Fresno Psychiatry Residency. He was a public high school English teacher for 15 years previously.*
References
1. Nguyen D. Mental Health Characteristics of Refugee Children. Pediatric News. 2023 Nov. 14. https://www.mdedge.com/pediatrics/article/266518/mental-health/mental-health-characteristics-refugee-children.
2. Office of the United Nations High Commissioner for Refugees. The Refugee Concept Under International Law. Global Compact for Safe, Orderly and Regular Migration. 2018 March 8. https://www.unhcr.org/sites/default/files/legacy-pdf/5aa290937.pdf.
3. Winer JP. Mental Health Practice with Immigrant and Refugee Youth [Power Point Slides]. Michigan Medicine. 2021 June 24. https://www.youtube.com/watch?v=ICkg4132SQY
4. Fazel M et al. Mental Health of Displaced and Refugee Children Resettled in High-Income Countries: Risk and Protective Factors. Lancet. 2012 Jan 21;379(9812):266-282. doi: 10.1016/S0140-6736(11)60051-2.
5. Fazel M, Betancourt TS. Preventive Mental Health Interventions for Refugee Children and Adolescents in High-Income Settings. Lancet Child Adolesc Health. 2018 Feb;2(2):121-132. doi: 10.1016/S2352-4642(17)30147-5.
*Correction, 2/27: An earlier version of this article misstated Dr. Nguyen's affiliation.
In my previous article, “Mental Health Characteristics of Refugee Children,” we learned that in recent decades, refugeeism has become a growing problem that disproportionately affects children. Refugee children and their families experience a variety of traumas, often sustained across years and even decades, because of armed conflict, persecution, social upheavals, or environmental disasters. Refugees are at greater risks for PTSD and affective and psychotic disorders presumably due to increased traumatic life events before, during, and after migration. I used my own experience as a child refugee from Vietnam to elucidate the stressors evident in various phases of forced displacement.1
Risk Factors and Protective Factors
To a certain extent, the experiences of refugees are universal. All refugees experience some sort of humanitarian crisis that forces an emergent escape from their home across international borders to a new resettlement area. It is important to note that internally displaced people do not meet the United Nations’ (UN) official designation of refugee status; however, some agencies use a broader definition where they are designated as such.2,3 We will refer to those not meeting the UN criteria as displaced people, while refugees are those that do meet the UN criteria. Dr. Mina Fazel’s 2012 systematic review in The Lancet of mental health risk factors and protective factors for displaced and refugee children is the most comprehensive of its kind.4 It will be summarized in this section with some relevant personal reflection.
In terms of risk factors, external displacement likely results in additional stress and trauma, presumably from the lack of assess to one’s culture and the host country’s language. Understandably, this makes rebuilding of one’s life more difficult. Several studies show that displaced/refugee children experience more difficulty with psychosocial adaptation than non-displaced children. Violence, directly experienced or indirectly feared, both to the child and their parents, was the strongest predictor of mental health problems and withdrawn behavior. Children who were separated from their parents clearly fared worst in their mental health than those who did not, which is not surprising given the nature of their dependence on caregivers for protection and guidance. During resettlement, experienced or perceived discrimination from the host country was also a risk factor, as well as instability in housing and a drawn-out resettlement process. Female sex was a risk factor mainly for emotional problems. Poor financial support post-migration is associated with depression, but it is unclear whether pre-migration financial status was protective. From my own experience, it is likely not, given that once one becomes a refugee one does not have access to one’s wealth, except that which could be hidden on one’s body. Another risk factor was also if one’s parent had psychiatric problems or was single. Due to the migration, my mother was separated permanently from her husband, which caused her extraordinary isolation and loneliness, something that was palpably felt by myself as I grew up.
In terms of protective factors, family cohesion and cultural continuity appear critical. For myself, not only would I not have survived without my mother and aunt, but they constantly protected me from the harsh realities. My mother would distract me with seemingly trivial goals once we got to America, like finally tasting a hamburger, or talking about school and being reunited with my uncle. This is in line with another finding — that children have better mental health outcomes when their parents do not talk about their hardships. Once my family was resettled with my uncle and his family, they played a critical role in smoothing our transition, not only by providing us with housing, but also cultural knowledge. Cultural havens can restore some of the social position and way of life that refugees lose when they are able to reconnect with a society that recognizes their previous achievements and status. Finally, religion also seemed to be a protective factor.
Mental Health Interventions
In 2018, Dr. Fazel identified mental health interventions for refugee children in a narrative review.5 She acknowledged that these conclusions are limited by the paucity of preventive mental health research in children in general, as well as the mobile nature and complex cultural differences of refugee children. This is exacerbated by the small evidence base. Given that, she makes these recommendations for varying levels of interventions: individual, group, family, living circumstances, social interactions, and school.
On an individual level, effective interventions developed to address PTSD include narrative exposure therapy, trauma-focused cognitive behavior therapy, and eye-movement and desensitization therapy. Group-based interventions for trauma, for example school-based PTSD intervention programs in conflicted areas, have either been shown to not be effective, or only effective for reducing depression. The mental health of unaccompanied children separated from family fare better when placed in foster care, rather than other types of social support. This is further enhanced if the foster family is the same ethnicity.
On a family level, improvements in parenting style and parental mental health, family engagement with local culture and structures, and family-based mental health interventions all positively impact refugee children. Not surprisingly, refugee parents have a greater prevalence of mental health conditions. Several studies on refugeeism point out a greater occurrence of intimate partner violence (that negatively affects children) as well has harsher discipline and maltreatment of refugee children. Thus, mental health treatment for parents also directly improves the well-being of their children. Teaching parenting skills to mitigate the violent effect of their PTSD symptoms, as well as parenting classes that teach gentler styles, have been shown to reduce harsh parenting and mitigate aggressive behaviors in these children. These improvements are enhanced when these classes are taught by other refugees themselves.
School is key for helping refugee children since it is a site where they can access language proficiency, successful acculturation, and medical and mental health services. Several studies have identified the positive effects of better parental engagement with school, resulting in improved academic performance and reduced levels of depressive and PTSD symptoms. A review of learning problems in refugee children identified several factors for success. These include high academic and life ambition, parental involvement in education, accurate educational assessment and grade placement, teacher understanding of linguistic and cultural heritage, culturally appropriate school transition, supportive peer relationships, and successful acculturation. School certainly was key for my acculturation and language proficiency. When I arrived at 6 years old I was selectively mute for my year in first grade, namely because I did not know how to speak English and because I did not share the culture. However, my teacher correctly identified my deficiency and chose to place me in kindergarten, which allowed me the time to gain English proficiency. Though I was always the oldest one in class, that remediation was key in allowing eventual success in school leading up to my admission to UC Berkeley.
Summary
In recent decades, refugeeism has become a growing problem that disproportionately affects children leading to traumas sustained across years and even decades, and greater risks for PTSD, as well as affective and psychotic disorders. Risk factors include the experience of violence, the separation from family, female gender, discrimination in the host country, unstable housing, and a drawn-out resettlement process. Protective factors consist of family cohesion, cultural continuity, support at schools, being protected from the truth of their harsh reality, stable housing, language acquisition, and quick resettlement. From these factors, effective mental interventions have been found to be the promotion of these protective factors as well as support for parental mental health and parenting skills, better parental engagement at school, and schools that correctly identify and address these children’s educational needs.
Dr. Nguyen is a second-year resident at UCSF Fresno Psychiatry Residency. He was a public high school English teacher for 15 years previously.*
References
1. Nguyen D. Mental Health Characteristics of Refugee Children. Pediatric News. 2023 Nov. 14. https://www.mdedge.com/pediatrics/article/266518/mental-health/mental-health-characteristics-refugee-children.
2. Office of the United Nations High Commissioner for Refugees. The Refugee Concept Under International Law. Global Compact for Safe, Orderly and Regular Migration. 2018 March 8. https://www.unhcr.org/sites/default/files/legacy-pdf/5aa290937.pdf.
3. Winer JP. Mental Health Practice with Immigrant and Refugee Youth [Power Point Slides]. Michigan Medicine. 2021 June 24. https://www.youtube.com/watch?v=ICkg4132SQY
4. Fazel M et al. Mental Health of Displaced and Refugee Children Resettled in High-Income Countries: Risk and Protective Factors. Lancet. 2012 Jan 21;379(9812):266-282. doi: 10.1016/S0140-6736(11)60051-2.
5. Fazel M, Betancourt TS. Preventive Mental Health Interventions for Refugee Children and Adolescents in High-Income Settings. Lancet Child Adolesc Health. 2018 Feb;2(2):121-132. doi: 10.1016/S2352-4642(17)30147-5.
*Correction, 2/27: An earlier version of this article misstated Dr. Nguyen's affiliation.
In my previous article, “Mental Health Characteristics of Refugee Children,” we learned that in recent decades, refugeeism has become a growing problem that disproportionately affects children. Refugee children and their families experience a variety of traumas, often sustained across years and even decades, because of armed conflict, persecution, social upheavals, or environmental disasters. Refugees are at greater risks for PTSD and affective and psychotic disorders presumably due to increased traumatic life events before, during, and after migration. I used my own experience as a child refugee from Vietnam to elucidate the stressors evident in various phases of forced displacement.1
Risk Factors and Protective Factors
To a certain extent, the experiences of refugees are universal. All refugees experience some sort of humanitarian crisis that forces an emergent escape from their home across international borders to a new resettlement area. It is important to note that internally displaced people do not meet the United Nations’ (UN) official designation of refugee status; however, some agencies use a broader definition where they are designated as such.2,3 We will refer to those not meeting the UN criteria as displaced people, while refugees are those that do meet the UN criteria. Dr. Mina Fazel’s 2012 systematic review in The Lancet of mental health risk factors and protective factors for displaced and refugee children is the most comprehensive of its kind.4 It will be summarized in this section with some relevant personal reflection.
In terms of risk factors, external displacement likely results in additional stress and trauma, presumably from the lack of assess to one’s culture and the host country’s language. Understandably, this makes rebuilding of one’s life more difficult. Several studies show that displaced/refugee children experience more difficulty with psychosocial adaptation than non-displaced children. Violence, directly experienced or indirectly feared, both to the child and their parents, was the strongest predictor of mental health problems and withdrawn behavior. Children who were separated from their parents clearly fared worst in their mental health than those who did not, which is not surprising given the nature of their dependence on caregivers for protection and guidance. During resettlement, experienced or perceived discrimination from the host country was also a risk factor, as well as instability in housing and a drawn-out resettlement process. Female sex was a risk factor mainly for emotional problems. Poor financial support post-migration is associated with depression, but it is unclear whether pre-migration financial status was protective. From my own experience, it is likely not, given that once one becomes a refugee one does not have access to one’s wealth, except that which could be hidden on one’s body. Another risk factor was also if one’s parent had psychiatric problems or was single. Due to the migration, my mother was separated permanently from her husband, which caused her extraordinary isolation and loneliness, something that was palpably felt by myself as I grew up.
In terms of protective factors, family cohesion and cultural continuity appear critical. For myself, not only would I not have survived without my mother and aunt, but they constantly protected me from the harsh realities. My mother would distract me with seemingly trivial goals once we got to America, like finally tasting a hamburger, or talking about school and being reunited with my uncle. This is in line with another finding — that children have better mental health outcomes when their parents do not talk about their hardships. Once my family was resettled with my uncle and his family, they played a critical role in smoothing our transition, not only by providing us with housing, but also cultural knowledge. Cultural havens can restore some of the social position and way of life that refugees lose when they are able to reconnect with a society that recognizes their previous achievements and status. Finally, religion also seemed to be a protective factor.
Mental Health Interventions
In 2018, Dr. Fazel identified mental health interventions for refugee children in a narrative review.5 She acknowledged that these conclusions are limited by the paucity of preventive mental health research in children in general, as well as the mobile nature and complex cultural differences of refugee children. This is exacerbated by the small evidence base. Given that, she makes these recommendations for varying levels of interventions: individual, group, family, living circumstances, social interactions, and school.
On an individual level, effective interventions developed to address PTSD include narrative exposure therapy, trauma-focused cognitive behavior therapy, and eye-movement and desensitization therapy. Group-based interventions for trauma, for example school-based PTSD intervention programs in conflicted areas, have either been shown to not be effective, or only effective for reducing depression. The mental health of unaccompanied children separated from family fare better when placed in foster care, rather than other types of social support. This is further enhanced if the foster family is the same ethnicity.
On a family level, improvements in parenting style and parental mental health, family engagement with local culture and structures, and family-based mental health interventions all positively impact refugee children. Not surprisingly, refugee parents have a greater prevalence of mental health conditions. Several studies on refugeeism point out a greater occurrence of intimate partner violence (that negatively affects children) as well has harsher discipline and maltreatment of refugee children. Thus, mental health treatment for parents also directly improves the well-being of their children. Teaching parenting skills to mitigate the violent effect of their PTSD symptoms, as well as parenting classes that teach gentler styles, have been shown to reduce harsh parenting and mitigate aggressive behaviors in these children. These improvements are enhanced when these classes are taught by other refugees themselves.
School is key for helping refugee children since it is a site where they can access language proficiency, successful acculturation, and medical and mental health services. Several studies have identified the positive effects of better parental engagement with school, resulting in improved academic performance and reduced levels of depressive and PTSD symptoms. A review of learning problems in refugee children identified several factors for success. These include high academic and life ambition, parental involvement in education, accurate educational assessment and grade placement, teacher understanding of linguistic and cultural heritage, culturally appropriate school transition, supportive peer relationships, and successful acculturation. School certainly was key for my acculturation and language proficiency. When I arrived at 6 years old I was selectively mute for my year in first grade, namely because I did not know how to speak English and because I did not share the culture. However, my teacher correctly identified my deficiency and chose to place me in kindergarten, which allowed me the time to gain English proficiency. Though I was always the oldest one in class, that remediation was key in allowing eventual success in school leading up to my admission to UC Berkeley.
Summary
In recent decades, refugeeism has become a growing problem that disproportionately affects children leading to traumas sustained across years and even decades, and greater risks for PTSD, as well as affective and psychotic disorders. Risk factors include the experience of violence, the separation from family, female gender, discrimination in the host country, unstable housing, and a drawn-out resettlement process. Protective factors consist of family cohesion, cultural continuity, support at schools, being protected from the truth of their harsh reality, stable housing, language acquisition, and quick resettlement. From these factors, effective mental interventions have been found to be the promotion of these protective factors as well as support for parental mental health and parenting skills, better parental engagement at school, and schools that correctly identify and address these children’s educational needs.
Dr. Nguyen is a second-year resident at UCSF Fresno Psychiatry Residency. He was a public high school English teacher for 15 years previously.*
References
1. Nguyen D. Mental Health Characteristics of Refugee Children. Pediatric News. 2023 Nov. 14. https://www.mdedge.com/pediatrics/article/266518/mental-health/mental-health-characteristics-refugee-children.
2. Office of the United Nations High Commissioner for Refugees. The Refugee Concept Under International Law. Global Compact for Safe, Orderly and Regular Migration. 2018 March 8. https://www.unhcr.org/sites/default/files/legacy-pdf/5aa290937.pdf.
3. Winer JP. Mental Health Practice with Immigrant and Refugee Youth [Power Point Slides]. Michigan Medicine. 2021 June 24. https://www.youtube.com/watch?v=ICkg4132SQY
4. Fazel M et al. Mental Health of Displaced and Refugee Children Resettled in High-Income Countries: Risk and Protective Factors. Lancet. 2012 Jan 21;379(9812):266-282. doi: 10.1016/S0140-6736(11)60051-2.
5. Fazel M, Betancourt TS. Preventive Mental Health Interventions for Refugee Children and Adolescents in High-Income Settings. Lancet Child Adolesc Health. 2018 Feb;2(2):121-132. doi: 10.1016/S2352-4642(17)30147-5.
*Correction, 2/27: An earlier version of this article misstated Dr. Nguyen's affiliation.