User login
Does measuring natriuretic peptides have a role in patients with chronic kidney disease?
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.
What is the role of probiotics in the treatment of acute Clostridium difficile-associated diarrhea?
Overall, the evidence does not support using probiotics to treat Clostridium difficile-associated diarrhea (CDAD). More studies are needed to determine if they are helpful and, if so, which ones and at what dosages.
WHAT ARE PROBIOTICS?
Probiotics are live bacteria or fungi that carry health benefits when ingested. There is great interest in using these agents to treat and prevent gastrointestinal disorders, as they have been said to inhibit the growth or invasion of pathogenic bacteria, enhance the intestinal barrier, and augment the immune system by regulating cytokines. Their proposed use in treating and preventing CDAD is based on their presumed mechanisms of action and effectiveness in other disorders of the gastrointestinal tract. Given that these readily available bacteria and fungi appear to be safe and well tolerated, their potential use in CDAD is of substantial interest.
LIMITED STUDIES AVAILABLE
Few clinical trials have tested probiotics in CDAD. Two recent systematic reviews did not find a clear benefit to adding probiotics to antibiotics to treat CDAD.1,2 Six trials of various probiotics were included in a 2006 meta-analysis.3 Overall, the analysis did find a benefit, but this was mostly derived from two trials of Saccharomyces boulardii.4,5 This yeast has a mechanism other probiotics do not have: a protease that it produces can degrade the exotoxins produced by C difficile.6
McFarland et al4 gave either S boulardii or placebo to 124 patients who were having either a first episode or a recurrence of CDAD. All patients also received either vancomycin (Vancocin) or metronidazole (Flagyl) in doses chosen by their physician. Patients taking S boulardii were more likely to have their diarrhea resolve and not recur, though post hoc analysis found that this benefit was limited to those with recurrent CDAD.4
Surawicz et al5 gave either S boulardii or placebo to 168 patients with recurrent CDAD who were also participating in a trial comparing vancomycin in a high dose, vancomycin in a low dose, and metronidazole. The probiotic was beneficial, but only in patients on high-dose vancomycin (2 g/day). These patients tended to have a more severe form of CDAD with colitis.
YOGURT, OVER-THE-COUNTER PRODUCTS MAY NOT CONTAIN ACTIVE BACTERIA
The efficacy of over-the-counter probiotic preparations and probiotic-containing foods, such as yogurt, is difficult to determine. For example, in the case of yogurts with “live and active cultures,” the inocula must remain stable from the factory to the grocery store shelf to the table and then through the gastrointestinal tract to the colon. The number of bacteria that survive this long journey is variable.
Another issue is whether probiotic products contain the species and quantities of organisms listed on their labels. In studies that have attempted to examine this issue, many of the products contained species not listed on the label. Most products that did contain viable cells of the stated therapeutic agent did so at a lower number than listed.7,8 The contents and dosages of these over-the-counter products are not regulated and may vary even within the same brand.
The US Food and Drug Administration (FDA) classifies these products as dietary supplements and therefore does not test them for efficacy or safety, though it does have the ability to remove them from the market if they are proven harmful.
FEW ADVERSE EFFECTS
Adverse effects seem to be uncommon with probiotics. Untoward symptoms include flatulence, bloating, and thirst. There are reports of invasive disease, including Lactobacillus bacteremia and Saccharomyces fungemia, occurring after these probiotics were given to patients with severe comorbidities.9,10,11
BENIGN STRAINS OF C DIFFICILE MAY PROTECT AGAINST CDAD
Interestingly, C difficile itself may serve as a probiotic, preventing future episodes of CDAD. Several studies in hamsters showed that colonization with nontoxigenic strains of C difficile can prevent infection with toxigenic strains. In these studies, hamsters receiving clindamycin (Cleocin) or cefoxitin (Mefoxin) were given nontoxigenic strains of C difficile that were either susceptible or resistant to the antibiotic, followed by a toxigenic strain. Those given resistant nontoxigenic strains were significantly less likely to develop CDAD. One study, for example, found that 100% of hamsters given a clindamycin-resistant, nontoxigenic strain of C difficile were protected from CDAD.12
INFECTION CONTROL IS KEY
Novel treatments for CDAD and ways to prevent it are constantly being sought as C difficile has reemerged in hospitals across North America and Europe. However, CDAD is fundamentally a hospital-acquired infection transmitted from patient to patient via the hands of health care workers. The most common predisposing factor is antibiotic use. While new therapeutic advances would be welcome, hand hygiene, basic infection control practice, and judicious use of antimicrobials are essential to decreasing the incidence of this disease.
- Dendukuri N, Costa V, McGregor M, Brophy JM. Probiotic therapy for the prevention and treatment of Clostridium difficile-associated diarrhea: a systematic review. CMAJ 2005; 173:167–170.
- Pillai A, Nelson R. Probiotics for treatment of Clostridium difficile-associated colitis in adults. Cochrane Database Syst Rev 2008;CD004611.
- McFarland LV. Meta-analysis of probiotics for the prevention of antibiotic associated diarrhea and the treatment of Clostridium difficile disease. Am J Gastroenterol 2006; 101:812–822.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Surawicz CM, McFarland LV, Greenberg RN, et al. The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin Infect Dis 2000; 31:1012–1017.
- Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect Immun 1999; 67:302–307.
- Huff BA. Caveat emptor. “Probiotics” might not be what they seem. Can Fam Physician 2004; 50:583–587.
- Coeuret V, Gueguen M, Vernoux J. Numbers and strains of lactobacilli in some probiotic products. Int J Food Microbiol 2004; 97:147–156.
- Land MH, Rouster-Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK. Lactobacillus sepsis associated with probiotic therapy. Pediatrics 2005; 115;178–181.
- Lherm T, Monet C, Nougière B, et al. Seven cases of fungemia with Saccharomyces boulardii in critically ill patients. Intensive Care Med 2002; 28:797–801.
- Salminen MK, Rautelin H, Tynkkynen S, et al. Lactobacillus bacteremia, clinical significance, and patient outcome, with special focus on probiotic L. rhamnosus GG. Clin Infect Dis 2004; 38:62–69.
- Merrigan MM, Sambol SP, Johnson S, Gerding DN. Prevention of fatal Clostridium difficile-associated disease during continuous administration of clindamycin in hamsters. J Infect Dis 2003; 188:1922–1927.
Overall, the evidence does not support using probiotics to treat Clostridium difficile-associated diarrhea (CDAD). More studies are needed to determine if they are helpful and, if so, which ones and at what dosages.
WHAT ARE PROBIOTICS?
Probiotics are live bacteria or fungi that carry health benefits when ingested. There is great interest in using these agents to treat and prevent gastrointestinal disorders, as they have been said to inhibit the growth or invasion of pathogenic bacteria, enhance the intestinal barrier, and augment the immune system by regulating cytokines. Their proposed use in treating and preventing CDAD is based on their presumed mechanisms of action and effectiveness in other disorders of the gastrointestinal tract. Given that these readily available bacteria and fungi appear to be safe and well tolerated, their potential use in CDAD is of substantial interest.
LIMITED STUDIES AVAILABLE
Few clinical trials have tested probiotics in CDAD. Two recent systematic reviews did not find a clear benefit to adding probiotics to antibiotics to treat CDAD.1,2 Six trials of various probiotics were included in a 2006 meta-analysis.3 Overall, the analysis did find a benefit, but this was mostly derived from two trials of Saccharomyces boulardii.4,5 This yeast has a mechanism other probiotics do not have: a protease that it produces can degrade the exotoxins produced by C difficile.6
McFarland et al4 gave either S boulardii or placebo to 124 patients who were having either a first episode or a recurrence of CDAD. All patients also received either vancomycin (Vancocin) or metronidazole (Flagyl) in doses chosen by their physician. Patients taking S boulardii were more likely to have their diarrhea resolve and not recur, though post hoc analysis found that this benefit was limited to those with recurrent CDAD.4
Surawicz et al5 gave either S boulardii or placebo to 168 patients with recurrent CDAD who were also participating in a trial comparing vancomycin in a high dose, vancomycin in a low dose, and metronidazole. The probiotic was beneficial, but only in patients on high-dose vancomycin (2 g/day). These patients tended to have a more severe form of CDAD with colitis.
YOGURT, OVER-THE-COUNTER PRODUCTS MAY NOT CONTAIN ACTIVE BACTERIA
The efficacy of over-the-counter probiotic preparations and probiotic-containing foods, such as yogurt, is difficult to determine. For example, in the case of yogurts with “live and active cultures,” the inocula must remain stable from the factory to the grocery store shelf to the table and then through the gastrointestinal tract to the colon. The number of bacteria that survive this long journey is variable.
Another issue is whether probiotic products contain the species and quantities of organisms listed on their labels. In studies that have attempted to examine this issue, many of the products contained species not listed on the label. Most products that did contain viable cells of the stated therapeutic agent did so at a lower number than listed.7,8 The contents and dosages of these over-the-counter products are not regulated and may vary even within the same brand.
The US Food and Drug Administration (FDA) classifies these products as dietary supplements and therefore does not test them for efficacy or safety, though it does have the ability to remove them from the market if they are proven harmful.
FEW ADVERSE EFFECTS
Adverse effects seem to be uncommon with probiotics. Untoward symptoms include flatulence, bloating, and thirst. There are reports of invasive disease, including Lactobacillus bacteremia and Saccharomyces fungemia, occurring after these probiotics were given to patients with severe comorbidities.9,10,11
BENIGN STRAINS OF C DIFFICILE MAY PROTECT AGAINST CDAD
Interestingly, C difficile itself may serve as a probiotic, preventing future episodes of CDAD. Several studies in hamsters showed that colonization with nontoxigenic strains of C difficile can prevent infection with toxigenic strains. In these studies, hamsters receiving clindamycin (Cleocin) or cefoxitin (Mefoxin) were given nontoxigenic strains of C difficile that were either susceptible or resistant to the antibiotic, followed by a toxigenic strain. Those given resistant nontoxigenic strains were significantly less likely to develop CDAD. One study, for example, found that 100% of hamsters given a clindamycin-resistant, nontoxigenic strain of C difficile were protected from CDAD.12
INFECTION CONTROL IS KEY
Novel treatments for CDAD and ways to prevent it are constantly being sought as C difficile has reemerged in hospitals across North America and Europe. However, CDAD is fundamentally a hospital-acquired infection transmitted from patient to patient via the hands of health care workers. The most common predisposing factor is antibiotic use. While new therapeutic advances would be welcome, hand hygiene, basic infection control practice, and judicious use of antimicrobials are essential to decreasing the incidence of this disease.
Overall, the evidence does not support using probiotics to treat Clostridium difficile-associated diarrhea (CDAD). More studies are needed to determine if they are helpful and, if so, which ones and at what dosages.
WHAT ARE PROBIOTICS?
Probiotics are live bacteria or fungi that carry health benefits when ingested. There is great interest in using these agents to treat and prevent gastrointestinal disorders, as they have been said to inhibit the growth or invasion of pathogenic bacteria, enhance the intestinal barrier, and augment the immune system by regulating cytokines. Their proposed use in treating and preventing CDAD is based on their presumed mechanisms of action and effectiveness in other disorders of the gastrointestinal tract. Given that these readily available bacteria and fungi appear to be safe and well tolerated, their potential use in CDAD is of substantial interest.
LIMITED STUDIES AVAILABLE
Few clinical trials have tested probiotics in CDAD. Two recent systematic reviews did not find a clear benefit to adding probiotics to antibiotics to treat CDAD.1,2 Six trials of various probiotics were included in a 2006 meta-analysis.3 Overall, the analysis did find a benefit, but this was mostly derived from two trials of Saccharomyces boulardii.4,5 This yeast has a mechanism other probiotics do not have: a protease that it produces can degrade the exotoxins produced by C difficile.6
McFarland et al4 gave either S boulardii or placebo to 124 patients who were having either a first episode or a recurrence of CDAD. All patients also received either vancomycin (Vancocin) or metronidazole (Flagyl) in doses chosen by their physician. Patients taking S boulardii were more likely to have their diarrhea resolve and not recur, though post hoc analysis found that this benefit was limited to those with recurrent CDAD.4
Surawicz et al5 gave either S boulardii or placebo to 168 patients with recurrent CDAD who were also participating in a trial comparing vancomycin in a high dose, vancomycin in a low dose, and metronidazole. The probiotic was beneficial, but only in patients on high-dose vancomycin (2 g/day). These patients tended to have a more severe form of CDAD with colitis.
YOGURT, OVER-THE-COUNTER PRODUCTS MAY NOT CONTAIN ACTIVE BACTERIA
The efficacy of over-the-counter probiotic preparations and probiotic-containing foods, such as yogurt, is difficult to determine. For example, in the case of yogurts with “live and active cultures,” the inocula must remain stable from the factory to the grocery store shelf to the table and then through the gastrointestinal tract to the colon. The number of bacteria that survive this long journey is variable.
Another issue is whether probiotic products contain the species and quantities of organisms listed on their labels. In studies that have attempted to examine this issue, many of the products contained species not listed on the label. Most products that did contain viable cells of the stated therapeutic agent did so at a lower number than listed.7,8 The contents and dosages of these over-the-counter products are not regulated and may vary even within the same brand.
The US Food and Drug Administration (FDA) classifies these products as dietary supplements and therefore does not test them for efficacy or safety, though it does have the ability to remove them from the market if they are proven harmful.
FEW ADVERSE EFFECTS
Adverse effects seem to be uncommon with probiotics. Untoward symptoms include flatulence, bloating, and thirst. There are reports of invasive disease, including Lactobacillus bacteremia and Saccharomyces fungemia, occurring after these probiotics were given to patients with severe comorbidities.9,10,11
BENIGN STRAINS OF C DIFFICILE MAY PROTECT AGAINST CDAD
Interestingly, C difficile itself may serve as a probiotic, preventing future episodes of CDAD. Several studies in hamsters showed that colonization with nontoxigenic strains of C difficile can prevent infection with toxigenic strains. In these studies, hamsters receiving clindamycin (Cleocin) or cefoxitin (Mefoxin) were given nontoxigenic strains of C difficile that were either susceptible or resistant to the antibiotic, followed by a toxigenic strain. Those given resistant nontoxigenic strains were significantly less likely to develop CDAD. One study, for example, found that 100% of hamsters given a clindamycin-resistant, nontoxigenic strain of C difficile were protected from CDAD.12
INFECTION CONTROL IS KEY
Novel treatments for CDAD and ways to prevent it are constantly being sought as C difficile has reemerged in hospitals across North America and Europe. However, CDAD is fundamentally a hospital-acquired infection transmitted from patient to patient via the hands of health care workers. The most common predisposing factor is antibiotic use. While new therapeutic advances would be welcome, hand hygiene, basic infection control practice, and judicious use of antimicrobials are essential to decreasing the incidence of this disease.
- Dendukuri N, Costa V, McGregor M, Brophy JM. Probiotic therapy for the prevention and treatment of Clostridium difficile-associated diarrhea: a systematic review. CMAJ 2005; 173:167–170.
- Pillai A, Nelson R. Probiotics for treatment of Clostridium difficile-associated colitis in adults. Cochrane Database Syst Rev 2008;CD004611.
- McFarland LV. Meta-analysis of probiotics for the prevention of antibiotic associated diarrhea and the treatment of Clostridium difficile disease. Am J Gastroenterol 2006; 101:812–822.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Surawicz CM, McFarland LV, Greenberg RN, et al. The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin Infect Dis 2000; 31:1012–1017.
- Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect Immun 1999; 67:302–307.
- Huff BA. Caveat emptor. “Probiotics” might not be what they seem. Can Fam Physician 2004; 50:583–587.
- Coeuret V, Gueguen M, Vernoux J. Numbers and strains of lactobacilli in some probiotic products. Int J Food Microbiol 2004; 97:147–156.
- Land MH, Rouster-Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK. Lactobacillus sepsis associated with probiotic therapy. Pediatrics 2005; 115;178–181.
- Lherm T, Monet C, Nougière B, et al. Seven cases of fungemia with Saccharomyces boulardii in critically ill patients. Intensive Care Med 2002; 28:797–801.
- Salminen MK, Rautelin H, Tynkkynen S, et al. Lactobacillus bacteremia, clinical significance, and patient outcome, with special focus on probiotic L. rhamnosus GG. Clin Infect Dis 2004; 38:62–69.
- Merrigan MM, Sambol SP, Johnson S, Gerding DN. Prevention of fatal Clostridium difficile-associated disease during continuous administration of clindamycin in hamsters. J Infect Dis 2003; 188:1922–1927.
- Dendukuri N, Costa V, McGregor M, Brophy JM. Probiotic therapy for the prevention and treatment of Clostridium difficile-associated diarrhea: a systematic review. CMAJ 2005; 173:167–170.
- Pillai A, Nelson R. Probiotics for treatment of Clostridium difficile-associated colitis in adults. Cochrane Database Syst Rev 2008;CD004611.
- McFarland LV. Meta-analysis of probiotics for the prevention of antibiotic associated diarrhea and the treatment of Clostridium difficile disease. Am J Gastroenterol 2006; 101:812–822.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Surawicz CM, McFarland LV, Greenberg RN, et al. The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin Infect Dis 2000; 31:1012–1017.
- Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect Immun 1999; 67:302–307.
- Huff BA. Caveat emptor. “Probiotics” might not be what they seem. Can Fam Physician 2004; 50:583–587.
- Coeuret V, Gueguen M, Vernoux J. Numbers and strains of lactobacilli in some probiotic products. Int J Food Microbiol 2004; 97:147–156.
- Land MH, Rouster-Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK. Lactobacillus sepsis associated with probiotic therapy. Pediatrics 2005; 115;178–181.
- Lherm T, Monet C, Nougière B, et al. Seven cases of fungemia with Saccharomyces boulardii in critically ill patients. Intensive Care Med 2002; 28:797–801.
- Salminen MK, Rautelin H, Tynkkynen S, et al. Lactobacillus bacteremia, clinical significance, and patient outcome, with special focus on probiotic L. rhamnosus GG. Clin Infect Dis 2004; 38:62–69.
- Merrigan MM, Sambol SP, Johnson S, Gerding DN. Prevention of fatal Clostridium difficile-associated disease during continuous administration of clindamycin in hamsters. J Infect Dis 2003; 188:1922–1927.
What is cell phone elbow, and what should we tell our patients?
With prolonged cellular telephone use, people may note the onset of aching, burning, numbness, or tingling in the ulnar forearm and hand. This constellation of symptoms, termed “cell phone elbow” by the lay press, is known medically as cubital tunnel syndrome—the second most common nerve compression syndrome in the upper extremities after carpal tunnel syndrome.
In most cases, treatment consists simply of modifying the activity and avoiding activities that aggravate the symptoms. Switching hands frequently while talking on the phone or using a hands-free headset can help. Other daily activities that produce cubital tunnel syndrome include leaning on an elbow while driving or working, and sitting at a computer workstation that requires elbow flexion greater than 90 degrees. Making ergonomic adjustments to these activities is beneficial.
For patients who have nocturnal symptoms, a simple elbow pad worn anteriorly or a towel wrapped around the elbow to prevent flexion while sleeping can be very efficacious. Occasionally, anti-inflammatory injections can be given to quiet an inflamed ulnar nerve and reduce symptoms.1 Surgical interventions, discussed below, are available for patients with severe, persistent symptoms.
WHAT IS CUBITAL TUNNEL SYNDROME?
Cellular telephone use has increased exponentially, with 3.3 billion service contracts active worldwide—or about one for every two people on the planet. The exact incidence of cell phone elbow is not known, but anecdotal reports and our own clinical experience indicate that its incidence parallels the rise in the use of cell phones and computer workstations.
Cubital tunnel syndrome is caused by compression of the ulnar nerve as it traverses the posterior elbow, wrapping around the medial condyle of the humerus. When people hold their elbow flexed for a prolonged period, such as when speaking on the phone or sleeping at night, the ulnar nerve is placed in tension; the nerve itself can elongate 4.5 to 8 mm with elbow flexion.2 Additionally, flexion of the elbow narrows the space available for the nerve2 and can cause a sevenfold to 20-fold increase in the pressure within the cubital tunnel, depending on muscle contraction.3 This can be compounded by compression on the nerve, either from various fascial bands surrounding the nerve or from extrinsic sources of compression, such as leaning on one’s elbow while driving or talking. This increased pressure on the nerve leads to decreased blood flow and nerve ischemia; this in turn causes increased permeability of the epineurial vessels and nerve edema, enlarging the nerve and continuing the cycle. Less frequently, cubital tunnel symptoms can be caused by the ulnar nerve subluxing in and out of its groove in the posterior elbow, leading to nerve inflammation and swelling from the repetitive friction.
THE CLINICAL PRESENTATION
The clinical picture of cubital tunnel syndrome consists of numbness or paresthesias in the small and ring fingers. Dorsal ulnar hand numbness, which is not present if the ulnar nerve is compressed at Guyon’s canal, helps the clinician differentiate cubital tunnel nerve compression from distal ulnar nerve compression.
If ulnar nerve compression persists, symptoms may progress to hand fatigue and weakness, including difficulty opening bottles or jars. Chronic and severe compression may lead to permanent motor deficits, including an inability to adduct the small finger (Wartenberg sign) and severe clawing of the ring and small fingers (a hand posture of metacarpophalangeal extension and flexion of the proximal and distal interphalangeal joints due to dysfunction of the ulnar-innervated intrinsic hand musculature). Patients may be unable to grasp things in a key-pinch grip, using a fingertip grip instead (Froment sign).
THE DIAGNOSIS IS USUALLY CLINICAL
The diagnosis of cubital tunnel syndrome is first and foremost a clinical one based on a thorough history, including symptoms, duration, and aggravating activities and factors.
The physical examination should include evaluation of sensibility of the hand, including the Semmes-Weinstein monofilament test and vibratory perception test, which will be affected before the Weber two-point discrimination test. Sensibility of the entire hand should be assessed to differentiate focal ulnar deficits from more widespread peripheral neuropathies.
Motor function can be evaluated by asking the patient to hold the fingers abducted, testing key-pinch grip, or asking the patient to cross the middle finger over the index finger. This crossed-finger test is quite reliable, as it is difficult to “fake out” with other muscles.4
The examination should also evaluate the cervical spine and vascularity. Provocative maneuvers can be performed to elicit symptoms, including the Hoffman-Tinel test (tapping the ulnar nerve in its groove at the posterior medial elbow, eliciting electric shocks or tingling radiating into the small finger). The equivalent of the Phalen maneuver for carpal tunnel syndrome can be performed by having the patient sit with the elbow fully flexed for 30 seconds to see if symptoms are reproduced; this may be positive in 10% of normal individuals. 5 One can combine elbow flexion with compression over the proximal ulnar nerve; this maneuver has good sensitivity and specificity. 6 Early in the disease, these provocative maneuvers may be the only examination findings, since sensation and motor function are usually normal.
Ruling out other entities that can cause numbness in the distribution of the medial hand and forearm is also important. These entities include cervical spine conditions such as herniated disk impinging on the C8 nerve root, or a space-occupying lesion of the cervical spine such as a tumor or syrinx.
The neck should be examined for loss of motion. Also, a Spurling test of the cervical spine checks for foraminal nerve impingement: with the patient seated, the clinician extends the patient’s neck and rotates it toward the involved side, then presses down on the top of the patient’s head and asks if this reproduces or worsens the symptoms in the patient’s arm. Hyperreflexia of the upper extremities or the presence of a Hoffman sign should alert the clinician to a more central process. In unclear cases or in patients with known cervical disease, electromyography should be able to differentiate ulnar neuropathy from a C8 nerveroot impingement or confirm the presence of both conditions (a so-called “double crush” phenomenon).
Other less common entities that can present with hand tingling include an apical lung tumor compressing the lower brachial plexus, thoracic outlet syndrome, or peripheral neuropathy (diabetes, vitamin B12 deficiency, hypothyroidism, alcoholism). Other conditions that can cause medial-sided elbow pain include elbow instability or medial epicondylitis (golfer’s elbow); however, these are not associated with numbness or tingling by themselves.
DIAGNOSTIC TESTS
Advanced diagnostic studies may help in certain cases, although they are not essential if the diagnosis is obvious on clinical examination.
Imaging studies may include plain radiography to look for osteophytes or bone fragments, which may impinge on the ulnar nerve, particularly in an arthritic or previously traumatized elbow. Magnetic resonance imaging is only indicated if a space-occupying lesion is suspected. Electrodiagnostic studies may help when findings are equivocal, when the site of compression is unclear, or when coexisting conditions such as diabetes or cervical spine disease make the diagnosis unclear. Nerve conduction studies may be unreliable early in cubital tunnel syndrome, as nondiseased nerve fibers may be tested, creating a false-negative result. Performing the study with the patient’s elbow flexed may increase the sensitivity of the test. Electromyography generally does not become positive until later in the disease, when more profound changes have occurred.
TREATMENT OF CELL PHONE ELBOW
As mentioned, changing how one uses a cell phone often helps, as does avoiding activities that require the elbow to remain flexed more than 90 degrees for extended periods. But when nonoperative means fail to reduce symptoms, surgery may be warranted.
Operative interventions include simple decompression or transposing the nerve from its usual course around the posterior elbow to a path anterior to the elbow, thus decreasing the tension on the nerve. This can be done either subcutaneously or by embedding the nerve in or under the muscles of the forearm.
In patients with coexisting medial epicondylitis or a subluxing nerve, the medial epicondyle can be excised. Techniques for minimally invasive or endoscopic ulnar nerve decompression have been recently introduced, but the long-term results with these are not yet known.
Overall, treatment for persistent paresthesias is successful even when patients present late, but those who present early have a better chance of full sensory and motor recovery.
- Pechan J, Kredba J. Treatment of cubital tunnel syndrome by means of local administration of cortisonoids. Acta Univ Carol [Med] (Praha) 1980; 26:125–133.
- Apfelberg DB, Larson SJ. Dynamic anatomy of the ulnar nerve at the elbow. Plast Reconstr Surg 1973; 51:79–81.
- Werner CO, Ohlin P, Elmqvist D. Pressures recorded in ulnar neuropathy. Acta Orthop Scand 1985; 56:404–406.
- Earle AS, Vlastou C. Crossed fingers and other tests of ulnar nerve motor function. J Hand Surg [Am] 1980; 5:560–565.
- Rayann GM, Jensen C, Duke J. Elbow flexion test in the normal population. J Hand Surg [Am] 1992; 17:86–89.
- Novak CB, Lee GW, Mackinnon SE, Lay L. Provocative testing for cubital tunnel syndrome. J Hand Surg [Am] 1994; 19:817–820.
With prolonged cellular telephone use, people may note the onset of aching, burning, numbness, or tingling in the ulnar forearm and hand. This constellation of symptoms, termed “cell phone elbow” by the lay press, is known medically as cubital tunnel syndrome—the second most common nerve compression syndrome in the upper extremities after carpal tunnel syndrome.
In most cases, treatment consists simply of modifying the activity and avoiding activities that aggravate the symptoms. Switching hands frequently while talking on the phone or using a hands-free headset can help. Other daily activities that produce cubital tunnel syndrome include leaning on an elbow while driving or working, and sitting at a computer workstation that requires elbow flexion greater than 90 degrees. Making ergonomic adjustments to these activities is beneficial.
For patients who have nocturnal symptoms, a simple elbow pad worn anteriorly or a towel wrapped around the elbow to prevent flexion while sleeping can be very efficacious. Occasionally, anti-inflammatory injections can be given to quiet an inflamed ulnar nerve and reduce symptoms.1 Surgical interventions, discussed below, are available for patients with severe, persistent symptoms.
WHAT IS CUBITAL TUNNEL SYNDROME?
Cellular telephone use has increased exponentially, with 3.3 billion service contracts active worldwide—or about one for every two people on the planet. The exact incidence of cell phone elbow is not known, but anecdotal reports and our own clinical experience indicate that its incidence parallels the rise in the use of cell phones and computer workstations.
Cubital tunnel syndrome is caused by compression of the ulnar nerve as it traverses the posterior elbow, wrapping around the medial condyle of the humerus. When people hold their elbow flexed for a prolonged period, such as when speaking on the phone or sleeping at night, the ulnar nerve is placed in tension; the nerve itself can elongate 4.5 to 8 mm with elbow flexion.2 Additionally, flexion of the elbow narrows the space available for the nerve2 and can cause a sevenfold to 20-fold increase in the pressure within the cubital tunnel, depending on muscle contraction.3 This can be compounded by compression on the nerve, either from various fascial bands surrounding the nerve or from extrinsic sources of compression, such as leaning on one’s elbow while driving or talking. This increased pressure on the nerve leads to decreased blood flow and nerve ischemia; this in turn causes increased permeability of the epineurial vessels and nerve edema, enlarging the nerve and continuing the cycle. Less frequently, cubital tunnel symptoms can be caused by the ulnar nerve subluxing in and out of its groove in the posterior elbow, leading to nerve inflammation and swelling from the repetitive friction.
THE CLINICAL PRESENTATION
The clinical picture of cubital tunnel syndrome consists of numbness or paresthesias in the small and ring fingers. Dorsal ulnar hand numbness, which is not present if the ulnar nerve is compressed at Guyon’s canal, helps the clinician differentiate cubital tunnel nerve compression from distal ulnar nerve compression.
If ulnar nerve compression persists, symptoms may progress to hand fatigue and weakness, including difficulty opening bottles or jars. Chronic and severe compression may lead to permanent motor deficits, including an inability to adduct the small finger (Wartenberg sign) and severe clawing of the ring and small fingers (a hand posture of metacarpophalangeal extension and flexion of the proximal and distal interphalangeal joints due to dysfunction of the ulnar-innervated intrinsic hand musculature). Patients may be unable to grasp things in a key-pinch grip, using a fingertip grip instead (Froment sign).
THE DIAGNOSIS IS USUALLY CLINICAL
The diagnosis of cubital tunnel syndrome is first and foremost a clinical one based on a thorough history, including symptoms, duration, and aggravating activities and factors.
The physical examination should include evaluation of sensibility of the hand, including the Semmes-Weinstein monofilament test and vibratory perception test, which will be affected before the Weber two-point discrimination test. Sensibility of the entire hand should be assessed to differentiate focal ulnar deficits from more widespread peripheral neuropathies.
Motor function can be evaluated by asking the patient to hold the fingers abducted, testing key-pinch grip, or asking the patient to cross the middle finger over the index finger. This crossed-finger test is quite reliable, as it is difficult to “fake out” with other muscles.4
The examination should also evaluate the cervical spine and vascularity. Provocative maneuvers can be performed to elicit symptoms, including the Hoffman-Tinel test (tapping the ulnar nerve in its groove at the posterior medial elbow, eliciting electric shocks or tingling radiating into the small finger). The equivalent of the Phalen maneuver for carpal tunnel syndrome can be performed by having the patient sit with the elbow fully flexed for 30 seconds to see if symptoms are reproduced; this may be positive in 10% of normal individuals. 5 One can combine elbow flexion with compression over the proximal ulnar nerve; this maneuver has good sensitivity and specificity. 6 Early in the disease, these provocative maneuvers may be the only examination findings, since sensation and motor function are usually normal.
Ruling out other entities that can cause numbness in the distribution of the medial hand and forearm is also important. These entities include cervical spine conditions such as herniated disk impinging on the C8 nerve root, or a space-occupying lesion of the cervical spine such as a tumor or syrinx.
The neck should be examined for loss of motion. Also, a Spurling test of the cervical spine checks for foraminal nerve impingement: with the patient seated, the clinician extends the patient’s neck and rotates it toward the involved side, then presses down on the top of the patient’s head and asks if this reproduces or worsens the symptoms in the patient’s arm. Hyperreflexia of the upper extremities or the presence of a Hoffman sign should alert the clinician to a more central process. In unclear cases or in patients with known cervical disease, electromyography should be able to differentiate ulnar neuropathy from a C8 nerveroot impingement or confirm the presence of both conditions (a so-called “double crush” phenomenon).
Other less common entities that can present with hand tingling include an apical lung tumor compressing the lower brachial plexus, thoracic outlet syndrome, or peripheral neuropathy (diabetes, vitamin B12 deficiency, hypothyroidism, alcoholism). Other conditions that can cause medial-sided elbow pain include elbow instability or medial epicondylitis (golfer’s elbow); however, these are not associated with numbness or tingling by themselves.
DIAGNOSTIC TESTS
Advanced diagnostic studies may help in certain cases, although they are not essential if the diagnosis is obvious on clinical examination.
Imaging studies may include plain radiography to look for osteophytes or bone fragments, which may impinge on the ulnar nerve, particularly in an arthritic or previously traumatized elbow. Magnetic resonance imaging is only indicated if a space-occupying lesion is suspected. Electrodiagnostic studies may help when findings are equivocal, when the site of compression is unclear, or when coexisting conditions such as diabetes or cervical spine disease make the diagnosis unclear. Nerve conduction studies may be unreliable early in cubital tunnel syndrome, as nondiseased nerve fibers may be tested, creating a false-negative result. Performing the study with the patient’s elbow flexed may increase the sensitivity of the test. Electromyography generally does not become positive until later in the disease, when more profound changes have occurred.
TREATMENT OF CELL PHONE ELBOW
As mentioned, changing how one uses a cell phone often helps, as does avoiding activities that require the elbow to remain flexed more than 90 degrees for extended periods. But when nonoperative means fail to reduce symptoms, surgery may be warranted.
Operative interventions include simple decompression or transposing the nerve from its usual course around the posterior elbow to a path anterior to the elbow, thus decreasing the tension on the nerve. This can be done either subcutaneously or by embedding the nerve in or under the muscles of the forearm.
In patients with coexisting medial epicondylitis or a subluxing nerve, the medial epicondyle can be excised. Techniques for minimally invasive or endoscopic ulnar nerve decompression have been recently introduced, but the long-term results with these are not yet known.
Overall, treatment for persistent paresthesias is successful even when patients present late, but those who present early have a better chance of full sensory and motor recovery.
With prolonged cellular telephone use, people may note the onset of aching, burning, numbness, or tingling in the ulnar forearm and hand. This constellation of symptoms, termed “cell phone elbow” by the lay press, is known medically as cubital tunnel syndrome—the second most common nerve compression syndrome in the upper extremities after carpal tunnel syndrome.
In most cases, treatment consists simply of modifying the activity and avoiding activities that aggravate the symptoms. Switching hands frequently while talking on the phone or using a hands-free headset can help. Other daily activities that produce cubital tunnel syndrome include leaning on an elbow while driving or working, and sitting at a computer workstation that requires elbow flexion greater than 90 degrees. Making ergonomic adjustments to these activities is beneficial.
For patients who have nocturnal symptoms, a simple elbow pad worn anteriorly or a towel wrapped around the elbow to prevent flexion while sleeping can be very efficacious. Occasionally, anti-inflammatory injections can be given to quiet an inflamed ulnar nerve and reduce symptoms.1 Surgical interventions, discussed below, are available for patients with severe, persistent symptoms.
WHAT IS CUBITAL TUNNEL SYNDROME?
Cellular telephone use has increased exponentially, with 3.3 billion service contracts active worldwide—or about one for every two people on the planet. The exact incidence of cell phone elbow is not known, but anecdotal reports and our own clinical experience indicate that its incidence parallels the rise in the use of cell phones and computer workstations.
Cubital tunnel syndrome is caused by compression of the ulnar nerve as it traverses the posterior elbow, wrapping around the medial condyle of the humerus. When people hold their elbow flexed for a prolonged period, such as when speaking on the phone or sleeping at night, the ulnar nerve is placed in tension; the nerve itself can elongate 4.5 to 8 mm with elbow flexion.2 Additionally, flexion of the elbow narrows the space available for the nerve2 and can cause a sevenfold to 20-fold increase in the pressure within the cubital tunnel, depending on muscle contraction.3 This can be compounded by compression on the nerve, either from various fascial bands surrounding the nerve or from extrinsic sources of compression, such as leaning on one’s elbow while driving or talking. This increased pressure on the nerve leads to decreased blood flow and nerve ischemia; this in turn causes increased permeability of the epineurial vessels and nerve edema, enlarging the nerve and continuing the cycle. Less frequently, cubital tunnel symptoms can be caused by the ulnar nerve subluxing in and out of its groove in the posterior elbow, leading to nerve inflammation and swelling from the repetitive friction.
THE CLINICAL PRESENTATION
The clinical picture of cubital tunnel syndrome consists of numbness or paresthesias in the small and ring fingers. Dorsal ulnar hand numbness, which is not present if the ulnar nerve is compressed at Guyon’s canal, helps the clinician differentiate cubital tunnel nerve compression from distal ulnar nerve compression.
If ulnar nerve compression persists, symptoms may progress to hand fatigue and weakness, including difficulty opening bottles or jars. Chronic and severe compression may lead to permanent motor deficits, including an inability to adduct the small finger (Wartenberg sign) and severe clawing of the ring and small fingers (a hand posture of metacarpophalangeal extension and flexion of the proximal and distal interphalangeal joints due to dysfunction of the ulnar-innervated intrinsic hand musculature). Patients may be unable to grasp things in a key-pinch grip, using a fingertip grip instead (Froment sign).
THE DIAGNOSIS IS USUALLY CLINICAL
The diagnosis of cubital tunnel syndrome is first and foremost a clinical one based on a thorough history, including symptoms, duration, and aggravating activities and factors.
The physical examination should include evaluation of sensibility of the hand, including the Semmes-Weinstein monofilament test and vibratory perception test, which will be affected before the Weber two-point discrimination test. Sensibility of the entire hand should be assessed to differentiate focal ulnar deficits from more widespread peripheral neuropathies.
Motor function can be evaluated by asking the patient to hold the fingers abducted, testing key-pinch grip, or asking the patient to cross the middle finger over the index finger. This crossed-finger test is quite reliable, as it is difficult to “fake out” with other muscles.4
The examination should also evaluate the cervical spine and vascularity. Provocative maneuvers can be performed to elicit symptoms, including the Hoffman-Tinel test (tapping the ulnar nerve in its groove at the posterior medial elbow, eliciting electric shocks or tingling radiating into the small finger). The equivalent of the Phalen maneuver for carpal tunnel syndrome can be performed by having the patient sit with the elbow fully flexed for 30 seconds to see if symptoms are reproduced; this may be positive in 10% of normal individuals. 5 One can combine elbow flexion with compression over the proximal ulnar nerve; this maneuver has good sensitivity and specificity. 6 Early in the disease, these provocative maneuvers may be the only examination findings, since sensation and motor function are usually normal.
Ruling out other entities that can cause numbness in the distribution of the medial hand and forearm is also important. These entities include cervical spine conditions such as herniated disk impinging on the C8 nerve root, or a space-occupying lesion of the cervical spine such as a tumor or syrinx.
The neck should be examined for loss of motion. Also, a Spurling test of the cervical spine checks for foraminal nerve impingement: with the patient seated, the clinician extends the patient’s neck and rotates it toward the involved side, then presses down on the top of the patient’s head and asks if this reproduces or worsens the symptoms in the patient’s arm. Hyperreflexia of the upper extremities or the presence of a Hoffman sign should alert the clinician to a more central process. In unclear cases or in patients with known cervical disease, electromyography should be able to differentiate ulnar neuropathy from a C8 nerveroot impingement or confirm the presence of both conditions (a so-called “double crush” phenomenon).
Other less common entities that can present with hand tingling include an apical lung tumor compressing the lower brachial plexus, thoracic outlet syndrome, or peripheral neuropathy (diabetes, vitamin B12 deficiency, hypothyroidism, alcoholism). Other conditions that can cause medial-sided elbow pain include elbow instability or medial epicondylitis (golfer’s elbow); however, these are not associated with numbness or tingling by themselves.
DIAGNOSTIC TESTS
Advanced diagnostic studies may help in certain cases, although they are not essential if the diagnosis is obvious on clinical examination.
Imaging studies may include plain radiography to look for osteophytes or bone fragments, which may impinge on the ulnar nerve, particularly in an arthritic or previously traumatized elbow. Magnetic resonance imaging is only indicated if a space-occupying lesion is suspected. Electrodiagnostic studies may help when findings are equivocal, when the site of compression is unclear, or when coexisting conditions such as diabetes or cervical spine disease make the diagnosis unclear. Nerve conduction studies may be unreliable early in cubital tunnel syndrome, as nondiseased nerve fibers may be tested, creating a false-negative result. Performing the study with the patient’s elbow flexed may increase the sensitivity of the test. Electromyography generally does not become positive until later in the disease, when more profound changes have occurred.
TREATMENT OF CELL PHONE ELBOW
As mentioned, changing how one uses a cell phone often helps, as does avoiding activities that require the elbow to remain flexed more than 90 degrees for extended periods. But when nonoperative means fail to reduce symptoms, surgery may be warranted.
Operative interventions include simple decompression or transposing the nerve from its usual course around the posterior elbow to a path anterior to the elbow, thus decreasing the tension on the nerve. This can be done either subcutaneously or by embedding the nerve in or under the muscles of the forearm.
In patients with coexisting medial epicondylitis or a subluxing nerve, the medial epicondyle can be excised. Techniques for minimally invasive or endoscopic ulnar nerve decompression have been recently introduced, but the long-term results with these are not yet known.
Overall, treatment for persistent paresthesias is successful even when patients present late, but those who present early have a better chance of full sensory and motor recovery.
- Pechan J, Kredba J. Treatment of cubital tunnel syndrome by means of local administration of cortisonoids. Acta Univ Carol [Med] (Praha) 1980; 26:125–133.
- Apfelberg DB, Larson SJ. Dynamic anatomy of the ulnar nerve at the elbow. Plast Reconstr Surg 1973; 51:79–81.
- Werner CO, Ohlin P, Elmqvist D. Pressures recorded in ulnar neuropathy. Acta Orthop Scand 1985; 56:404–406.
- Earle AS, Vlastou C. Crossed fingers and other tests of ulnar nerve motor function. J Hand Surg [Am] 1980; 5:560–565.
- Rayann GM, Jensen C, Duke J. Elbow flexion test in the normal population. J Hand Surg [Am] 1992; 17:86–89.
- Novak CB, Lee GW, Mackinnon SE, Lay L. Provocative testing for cubital tunnel syndrome. J Hand Surg [Am] 1994; 19:817–820.
- Pechan J, Kredba J. Treatment of cubital tunnel syndrome by means of local administration of cortisonoids. Acta Univ Carol [Med] (Praha) 1980; 26:125–133.
- Apfelberg DB, Larson SJ. Dynamic anatomy of the ulnar nerve at the elbow. Plast Reconstr Surg 1973; 51:79–81.
- Werner CO, Ohlin P, Elmqvist D. Pressures recorded in ulnar neuropathy. Acta Orthop Scand 1985; 56:404–406.
- Earle AS, Vlastou C. Crossed fingers and other tests of ulnar nerve motor function. J Hand Surg [Am] 1980; 5:560–565.
- Rayann GM, Jensen C, Duke J. Elbow flexion test in the normal population. J Hand Surg [Am] 1992; 17:86–89.
- Novak CB, Lee GW, Mackinnon SE, Lay L. Provocative testing for cubital tunnel syndrome. J Hand Surg [Am] 1994; 19:817–820.
What is the utility of measuring the serum ammonia level in patients with altered mental status?
If you already know that the patient with altered mental status has decompensated liver disease, measuring the arterial or venous ammonia level has little utility. In these patients, one’s clinical suspicion is the main guide to diagnosing hepatic encephalopathy, and a normal or modestly elevated blood ammonia level does not rule out the diagnosis.
On the other hand, provided that it is appropriately performed, blood ammonia testing may be helpful if there is no clear evidence of underlying chronic liver disease. In these cases, an elevated blood ammonia level may have significant prognostic value (as in acute liver failure) or may prompt you to initiate further evaluation for uncommon but significant meta bolic disorders such as urea cycle disorders.
WHEN AMMONIA LEVELS RISE

Ammonia levels are elevated in several conditions in which its production is increased (eg, in convulsive seizures with increased muscle production) or its clearance is impaired (eg, in hepatocellular dysfunction, portosystemic shunting, or both, with subsequent impaired hepatic detoxification of ammonia).
Because the blood-brain barrier is highly permeable to ammonia, the brain is exposed to excessive concentrations of it in these circumstances. In the brain, ammonia is thought to cause both functional and structural abnormalities that could explain neuropsychiatric dysfunction, often manifested as an altered mental status of variable degree.1–3
DOES THE PATIENT HAVE DECOMPENSATED LIVER DISEASE?
Physicians often measure the venous (and less often, the arterial) ammonia level while evaluating patients presenting with altered mental status. However, in many cases, this test result may be of uncertain utility—it may not have a significant impact on a specific patient’s management and, worse, it can confuse the physician regarding diagnosis. Also, the test itself is a needless expense. Therefore, we need to carefully consider whether to obtain a blood ammonia test and how to interpret the results in patients with altered mental status.
The key initial question in such patients is whether the patient is known to have decompensated liver disease with a typical clinical picture of hepatic encephalopathy.
If the patient is known to have chronic liver disease
Hepatic encephalopathy is a common complication of end-stage liver disease and is also one of the diagnostic markers of acute liver failure. An accepted factor in its pathophysiology is that the liver fails to clear toxic products of bacterial metabolism brought via the portal venous system from the gut, owing to low detoxifying capacity, portosystemic shunts, or both.4 Although the exact neurotoxins involved remain poorly defined, ammonia is thought to play a central role.5–7
If the patient is known to have chronic liver disease, we usually do not need to measure the blood ammonia level because normal levels in these patients do not rule out hepatic encephalopathy. Multiple studies have shown that the ammonia level correlates to some extent with the severity of hepatic encephalopathy,8 but ammonia levels substantially overlap among patients with differing clinical grades of hepatic encephalopathy. Moreover, 69% of patients with no evidence of encephalopathy had ammonia levels higher than normal in a study by Ong et al.8
Therefore, hyperammonemia is neither sensitive nor specific for the presence or the degree of hepatic encephalopathy. In this respect, three related issues should be emphasized:
Altered mental status in cirrhotic patients does not always equal hepatic encephalopathy. Regardless of the degree of blood ammonia elevation, other relevant causes of altered mental status should be excluded on the basis of the clinical presentation.
Computed tomography of the head is usually obtained in cirrhotic patients:
- Who have changes in mental status but whose presentation is not typical of hepatic encephalopathy (such as those with focal neurologic signs);
- In cases of severe hepatic encephalopathy, suspected head trauma (especially given the commonly associated coagulopathy in cirrhotic patients), and hepatic encephalopathy resistant to standard therapy; and
- Without clear precipitating factors for hepatic encephalopathy, such as infection (eg, spontaneous bacterial peritonitis) and renal insufficiency.
Similarly, in alcoholic patients who present with altered mental status, we should always consider Wernicke encephalopathy.
In patients with established hepatic encephalopathy, monitoring the ammonia level during therapy is not as useful as ongoing clinical assessment.
In patients with acute liver failure, a blood ammonia level may have a special prognostic value. In hyperammonemic states that subsequently lead to elevated ammonia in the brain, astrocytes convert ammonia to glutamine. Glutamine is not toxic, but it is osmotically active, and as it accumulates, it leads to astrocyte swelling and brain edema. This pathologic process is very prominent in acute hyperammonemic states in which astrocytes do not have time to adapt osmotically by pumping in myoinositol.9 Clemmesen et al10 have shown that arterial ammonia levels higher than 200 μg/dL are strongly associated with cerebral herniation in patients with acute liver failure.
If the patient is not known to have chronic liver disease
Occasionally, the blood ammonia level is found to be high in a patient who presents with altered mental status but who does not have known liver disease. In these patients, undiagnosed or new-onset decompensated cirrhosis is still possible, and the possibility should be explored. Acute liver failure is another possibility, but it is usually obvious, with associated coagulopathy, hyperbilirubinemia, and other clinical and laboratory features.
The main diagnostic challenge is in patients who have altered mental status and hyperammonemia but no features to suggest the above possibilities. In this setting, three tasks should be approached simultaneously:
- Look for and aggressively manage cerebral edema and increased intracranial pressure with ammonia-lowering measures such as lactulose, renal replacement therapy, and other specific therapeutic agents if a urea cycle disorder is suspected.11
- Search for causes of elevated ammonia other than hepatic dysfunction. These causes can be classified into two major categories11: 1) causes of increased ammonia production such as total parenteral nutrition, gastrointestinal hemorrhage, and steroid use, and 2) causes of decreased ammonia excretion such as portosystemic shunts, medications that decrease ammonia metabolism, and inborn errors of metabolism such as urea cycle disorders. Portosystemic shunts have been well documented in patients with no underlying liver disease.12
Several drugs, such as glycine (used during transurethral prostate resection), salicylates, and valproate raise the ammonia level by altering the urea cycle.11 Although most severe inborn errors of metabolism become evident early in childhood, certain urea cycle disorders, especially ornithine transcarbamylase deficiency, may manifest later during adulthood when a precipitating event occurs, such as an increase in protein intake (eg, with total parenteral nutrition), use of certain medications, or infection.
- Explore concomitant or alternative causes of altered mental status based on the clinical setting, such as a cerebrovascular accident, infectious meningoencephalitis, drug intoxication, or other metabolic or systemic disorders.
IN AMMONIA TESTING, TECHNIQUE MATTERS
To obtain an accurate measurement, the blood sample for ammonia testing must be obtained and handled properly. Prolonged application of a tourniquet or fist-clenching while obtaining the blood sample or improper specimen handling can result in a falsely elevated blood ammonia level, which can lead you down the wrong diagnostic pathway.
Venous blood, if appropriately collected, transported in ice, and handled quickly for analysis, has been shown to be as useful as arterial blood in ammonia measurement.8
- Williams R. Bacterial flora and pathogenesis in hepatic encephalopathy. Aliment Pharmacol Ther 2007; 25(suppl 1):17–22.
- Lockwood AH. Positron emission tomography in the study of hepatic encephalopathy. Metab Brain Dis 2002; 17:431–435.
- Hazell AS, Butterworth RF. Hepatic encephalopathy: an update of pathophysiologic mechanisms. Proc Soc Exp Biol Med 1999; 222:99–112.
- Blei AT, Córdoba J; Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol 2001; 96:1968–1976.
- Abou-Assi S, Vlahcevic ZR. Hepatic encephalopathy. Metabolic consequence of cirrhosis often is reversible. Postgrad Med 2001; 109:52–54,57–60,63–65.
- Cordoba J, Blei AT. Treatment of hepatic encephalopathy. Am J Gastroenterol 1997; 92:1429–1439.
- Ong JP, Mullen KD. Hepatic encephalopathy. Eur J Gastroenterol Hepatol 2001; 13:325–334.
- Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med 2003; 114:188–193.
- Blei AT. The pathophysiology of brain edema in acute liver failure. Neurochem Int 2005; 47:71–77.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest 2007; 132:1368–1378.
- Watanabe A. Portal-systemic encephalopathy in non-cirrhotic patients: classification of clinical types, diagnosis and treatment. J Gastroenterol Hepatol 2000; 15:969–979.
If you already know that the patient with altered mental status has decompensated liver disease, measuring the arterial or venous ammonia level has little utility. In these patients, one’s clinical suspicion is the main guide to diagnosing hepatic encephalopathy, and a normal or modestly elevated blood ammonia level does not rule out the diagnosis.
On the other hand, provided that it is appropriately performed, blood ammonia testing may be helpful if there is no clear evidence of underlying chronic liver disease. In these cases, an elevated blood ammonia level may have significant prognostic value (as in acute liver failure) or may prompt you to initiate further evaluation for uncommon but significant meta bolic disorders such as urea cycle disorders.
WHEN AMMONIA LEVELS RISE
Ammonia levels are elevated in several conditions in which its production is increased (eg, in convulsive seizures with increased muscle production) or its clearance is impaired (eg, in hepatocellular dysfunction, portosystemic shunting, or both, with subsequent impaired hepatic detoxification of ammonia).
Because the blood-brain barrier is highly permeable to ammonia, the brain is exposed to excessive concentrations of it in these circumstances. In the brain, ammonia is thought to cause both functional and structural abnormalities that could explain neuropsychiatric dysfunction, often manifested as an altered mental status of variable degree.1–3
DOES THE PATIENT HAVE DECOMPENSATED LIVER DISEASE?
Physicians often measure the venous (and less often, the arterial) ammonia level while evaluating patients presenting with altered mental status. However, in many cases, this test result may be of uncertain utility—it may not have a significant impact on a specific patient’s management and, worse, it can confuse the physician regarding diagnosis. Also, the test itself is a needless expense. Therefore, we need to carefully consider whether to obtain a blood ammonia test and how to interpret the results in patients with altered mental status.
The key initial question in such patients is whether the patient is known to have decompensated liver disease with a typical clinical picture of hepatic encephalopathy.
If the patient is known to have chronic liver disease
Hepatic encephalopathy is a common complication of end-stage liver disease and is also one of the diagnostic markers of acute liver failure. An accepted factor in its pathophysiology is that the liver fails to clear toxic products of bacterial metabolism brought via the portal venous system from the gut, owing to low detoxifying capacity, portosystemic shunts, or both.4 Although the exact neurotoxins involved remain poorly defined, ammonia is thought to play a central role.5–7
If the patient is known to have chronic liver disease, we usually do not need to measure the blood ammonia level because normal levels in these patients do not rule out hepatic encephalopathy. Multiple studies have shown that the ammonia level correlates to some extent with the severity of hepatic encephalopathy,8 but ammonia levels substantially overlap among patients with differing clinical grades of hepatic encephalopathy. Moreover, 69% of patients with no evidence of encephalopathy had ammonia levels higher than normal in a study by Ong et al.8
Therefore, hyperammonemia is neither sensitive nor specific for the presence or the degree of hepatic encephalopathy. In this respect, three related issues should be emphasized:
Altered mental status in cirrhotic patients does not always equal hepatic encephalopathy. Regardless of the degree of blood ammonia elevation, other relevant causes of altered mental status should be excluded on the basis of the clinical presentation.
Computed tomography of the head is usually obtained in cirrhotic patients:
- Who have changes in mental status but whose presentation is not typical of hepatic encephalopathy (such as those with focal neurologic signs);
- In cases of severe hepatic encephalopathy, suspected head trauma (especially given the commonly associated coagulopathy in cirrhotic patients), and hepatic encephalopathy resistant to standard therapy; and
- Without clear precipitating factors for hepatic encephalopathy, such as infection (eg, spontaneous bacterial peritonitis) and renal insufficiency.
Similarly, in alcoholic patients who present with altered mental status, we should always consider Wernicke encephalopathy.
In patients with established hepatic encephalopathy, monitoring the ammonia level during therapy is not as useful as ongoing clinical assessment.
In patients with acute liver failure, a blood ammonia level may have a special prognostic value. In hyperammonemic states that subsequently lead to elevated ammonia in the brain, astrocytes convert ammonia to glutamine. Glutamine is not toxic, but it is osmotically active, and as it accumulates, it leads to astrocyte swelling and brain edema. This pathologic process is very prominent in acute hyperammonemic states in which astrocytes do not have time to adapt osmotically by pumping in myoinositol.9 Clemmesen et al10 have shown that arterial ammonia levels higher than 200 μg/dL are strongly associated with cerebral herniation in patients with acute liver failure.
If the patient is not known to have chronic liver disease
Occasionally, the blood ammonia level is found to be high in a patient who presents with altered mental status but who does not have known liver disease. In these patients, undiagnosed or new-onset decompensated cirrhosis is still possible, and the possibility should be explored. Acute liver failure is another possibility, but it is usually obvious, with associated coagulopathy, hyperbilirubinemia, and other clinical and laboratory features.
The main diagnostic challenge is in patients who have altered mental status and hyperammonemia but no features to suggest the above possibilities. In this setting, three tasks should be approached simultaneously:
- Look for and aggressively manage cerebral edema and increased intracranial pressure with ammonia-lowering measures such as lactulose, renal replacement therapy, and other specific therapeutic agents if a urea cycle disorder is suspected.11
- Search for causes of elevated ammonia other than hepatic dysfunction. These causes can be classified into two major categories11: 1) causes of increased ammonia production such as total parenteral nutrition, gastrointestinal hemorrhage, and steroid use, and 2) causes of decreased ammonia excretion such as portosystemic shunts, medications that decrease ammonia metabolism, and inborn errors of metabolism such as urea cycle disorders. Portosystemic shunts have been well documented in patients with no underlying liver disease.12
Several drugs, such as glycine (used during transurethral prostate resection), salicylates, and valproate raise the ammonia level by altering the urea cycle.11 Although most severe inborn errors of metabolism become evident early in childhood, certain urea cycle disorders, especially ornithine transcarbamylase deficiency, may manifest later during adulthood when a precipitating event occurs, such as an increase in protein intake (eg, with total parenteral nutrition), use of certain medications, or infection.
- Explore concomitant or alternative causes of altered mental status based on the clinical setting, such as a cerebrovascular accident, infectious meningoencephalitis, drug intoxication, or other metabolic or systemic disorders.
IN AMMONIA TESTING, TECHNIQUE MATTERS
To obtain an accurate measurement, the blood sample for ammonia testing must be obtained and handled properly. Prolonged application of a tourniquet or fist-clenching while obtaining the blood sample or improper specimen handling can result in a falsely elevated blood ammonia level, which can lead you down the wrong diagnostic pathway.
Venous blood, if appropriately collected, transported in ice, and handled quickly for analysis, has been shown to be as useful as arterial blood in ammonia measurement.8
If you already know that the patient with altered mental status has decompensated liver disease, measuring the arterial or venous ammonia level has little utility. In these patients, one’s clinical suspicion is the main guide to diagnosing hepatic encephalopathy, and a normal or modestly elevated blood ammonia level does not rule out the diagnosis.
On the other hand, provided that it is appropriately performed, blood ammonia testing may be helpful if there is no clear evidence of underlying chronic liver disease. In these cases, an elevated blood ammonia level may have significant prognostic value (as in acute liver failure) or may prompt you to initiate further evaluation for uncommon but significant meta bolic disorders such as urea cycle disorders.
WHEN AMMONIA LEVELS RISE
Ammonia levels are elevated in several conditions in which its production is increased (eg, in convulsive seizures with increased muscle production) or its clearance is impaired (eg, in hepatocellular dysfunction, portosystemic shunting, or both, with subsequent impaired hepatic detoxification of ammonia).
Because the blood-brain barrier is highly permeable to ammonia, the brain is exposed to excessive concentrations of it in these circumstances. In the brain, ammonia is thought to cause both functional and structural abnormalities that could explain neuropsychiatric dysfunction, often manifested as an altered mental status of variable degree.1–3
DOES THE PATIENT HAVE DECOMPENSATED LIVER DISEASE?
Physicians often measure the venous (and less often, the arterial) ammonia level while evaluating patients presenting with altered mental status. However, in many cases, this test result may be of uncertain utility—it may not have a significant impact on a specific patient’s management and, worse, it can confuse the physician regarding diagnosis. Also, the test itself is a needless expense. Therefore, we need to carefully consider whether to obtain a blood ammonia test and how to interpret the results in patients with altered mental status.
The key initial question in such patients is whether the patient is known to have decompensated liver disease with a typical clinical picture of hepatic encephalopathy.
If the patient is known to have chronic liver disease
Hepatic encephalopathy is a common complication of end-stage liver disease and is also one of the diagnostic markers of acute liver failure. An accepted factor in its pathophysiology is that the liver fails to clear toxic products of bacterial metabolism brought via the portal venous system from the gut, owing to low detoxifying capacity, portosystemic shunts, or both.4 Although the exact neurotoxins involved remain poorly defined, ammonia is thought to play a central role.5–7
If the patient is known to have chronic liver disease, we usually do not need to measure the blood ammonia level because normal levels in these patients do not rule out hepatic encephalopathy. Multiple studies have shown that the ammonia level correlates to some extent with the severity of hepatic encephalopathy,8 but ammonia levels substantially overlap among patients with differing clinical grades of hepatic encephalopathy. Moreover, 69% of patients with no evidence of encephalopathy had ammonia levels higher than normal in a study by Ong et al.8
Therefore, hyperammonemia is neither sensitive nor specific for the presence or the degree of hepatic encephalopathy. In this respect, three related issues should be emphasized:
Altered mental status in cirrhotic patients does not always equal hepatic encephalopathy. Regardless of the degree of blood ammonia elevation, other relevant causes of altered mental status should be excluded on the basis of the clinical presentation.
Computed tomography of the head is usually obtained in cirrhotic patients:
- Who have changes in mental status but whose presentation is not typical of hepatic encephalopathy (such as those with focal neurologic signs);
- In cases of severe hepatic encephalopathy, suspected head trauma (especially given the commonly associated coagulopathy in cirrhotic patients), and hepatic encephalopathy resistant to standard therapy; and
- Without clear precipitating factors for hepatic encephalopathy, such as infection (eg, spontaneous bacterial peritonitis) and renal insufficiency.
Similarly, in alcoholic patients who present with altered mental status, we should always consider Wernicke encephalopathy.
In patients with established hepatic encephalopathy, monitoring the ammonia level during therapy is not as useful as ongoing clinical assessment.
In patients with acute liver failure, a blood ammonia level may have a special prognostic value. In hyperammonemic states that subsequently lead to elevated ammonia in the brain, astrocytes convert ammonia to glutamine. Glutamine is not toxic, but it is osmotically active, and as it accumulates, it leads to astrocyte swelling and brain edema. This pathologic process is very prominent in acute hyperammonemic states in which astrocytes do not have time to adapt osmotically by pumping in myoinositol.9 Clemmesen et al10 have shown that arterial ammonia levels higher than 200 μg/dL are strongly associated with cerebral herniation in patients with acute liver failure.
If the patient is not known to have chronic liver disease
Occasionally, the blood ammonia level is found to be high in a patient who presents with altered mental status but who does not have known liver disease. In these patients, undiagnosed or new-onset decompensated cirrhosis is still possible, and the possibility should be explored. Acute liver failure is another possibility, but it is usually obvious, with associated coagulopathy, hyperbilirubinemia, and other clinical and laboratory features.
The main diagnostic challenge is in patients who have altered mental status and hyperammonemia but no features to suggest the above possibilities. In this setting, three tasks should be approached simultaneously:
- Look for and aggressively manage cerebral edema and increased intracranial pressure with ammonia-lowering measures such as lactulose, renal replacement therapy, and other specific therapeutic agents if a urea cycle disorder is suspected.11
- Search for causes of elevated ammonia other than hepatic dysfunction. These causes can be classified into two major categories11: 1) causes of increased ammonia production such as total parenteral nutrition, gastrointestinal hemorrhage, and steroid use, and 2) causes of decreased ammonia excretion such as portosystemic shunts, medications that decrease ammonia metabolism, and inborn errors of metabolism such as urea cycle disorders. Portosystemic shunts have been well documented in patients with no underlying liver disease.12
Several drugs, such as glycine (used during transurethral prostate resection), salicylates, and valproate raise the ammonia level by altering the urea cycle.11 Although most severe inborn errors of metabolism become evident early in childhood, certain urea cycle disorders, especially ornithine transcarbamylase deficiency, may manifest later during adulthood when a precipitating event occurs, such as an increase in protein intake (eg, with total parenteral nutrition), use of certain medications, or infection.
- Explore concomitant or alternative causes of altered mental status based on the clinical setting, such as a cerebrovascular accident, infectious meningoencephalitis, drug intoxication, or other metabolic or systemic disorders.
IN AMMONIA TESTING, TECHNIQUE MATTERS
To obtain an accurate measurement, the blood sample for ammonia testing must be obtained and handled properly. Prolonged application of a tourniquet or fist-clenching while obtaining the blood sample or improper specimen handling can result in a falsely elevated blood ammonia level, which can lead you down the wrong diagnostic pathway.
Venous blood, if appropriately collected, transported in ice, and handled quickly for analysis, has been shown to be as useful as arterial blood in ammonia measurement.8
- Williams R. Bacterial flora and pathogenesis in hepatic encephalopathy. Aliment Pharmacol Ther 2007; 25(suppl 1):17–22.
- Lockwood AH. Positron emission tomography in the study of hepatic encephalopathy. Metab Brain Dis 2002; 17:431–435.
- Hazell AS, Butterworth RF. Hepatic encephalopathy: an update of pathophysiologic mechanisms. Proc Soc Exp Biol Med 1999; 222:99–112.
- Blei AT, Córdoba J; Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol 2001; 96:1968–1976.
- Abou-Assi S, Vlahcevic ZR. Hepatic encephalopathy. Metabolic consequence of cirrhosis often is reversible. Postgrad Med 2001; 109:52–54,57–60,63–65.
- Cordoba J, Blei AT. Treatment of hepatic encephalopathy. Am J Gastroenterol 1997; 92:1429–1439.
- Ong JP, Mullen KD. Hepatic encephalopathy. Eur J Gastroenterol Hepatol 2001; 13:325–334.
- Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med 2003; 114:188–193.
- Blei AT. The pathophysiology of brain edema in acute liver failure. Neurochem Int 2005; 47:71–77.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest 2007; 132:1368–1378.
- Watanabe A. Portal-systemic encephalopathy in non-cirrhotic patients: classification of clinical types, diagnosis and treatment. J Gastroenterol Hepatol 2000; 15:969–979.
- Williams R. Bacterial flora and pathogenesis in hepatic encephalopathy. Aliment Pharmacol Ther 2007; 25(suppl 1):17–22.
- Lockwood AH. Positron emission tomography in the study of hepatic encephalopathy. Metab Brain Dis 2002; 17:431–435.
- Hazell AS, Butterworth RF. Hepatic encephalopathy: an update of pathophysiologic mechanisms. Proc Soc Exp Biol Med 1999; 222:99–112.
- Blei AT, Córdoba J; Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol 2001; 96:1968–1976.
- Abou-Assi S, Vlahcevic ZR. Hepatic encephalopathy. Metabolic consequence of cirrhosis often is reversible. Postgrad Med 2001; 109:52–54,57–60,63–65.
- Cordoba J, Blei AT. Treatment of hepatic encephalopathy. Am J Gastroenterol 1997; 92:1429–1439.
- Ong JP, Mullen KD. Hepatic encephalopathy. Eur J Gastroenterol Hepatol 2001; 13:325–334.
- Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med 2003; 114:188–193.
- Blei AT. The pathophysiology of brain edema in acute liver failure. Neurochem Int 2005; 47:71–77.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest 2007; 132:1368–1378.
- Watanabe A. Portal-systemic encephalopathy in non-cirrhotic patients: classification of clinical types, diagnosis and treatment. J Gastroenterol Hepatol 2000; 15:969–979.
Who should receive the shingles vaccine?
The Advisory Committee on Immunization Practices (ACIP) recommends routinely giving a single dose of live zoster vaccine to immunocompetent patients age 60 and older at their first clinical encounter. The vaccine effectively prevents shingles and postherpetic neuralgia and their associated burden of illness. Although not all insurance companies pay for it yet, it should be offered to all patients for whom it is indicated to increase their health-related quality of life.
IMMUNITY WANES WITH AGE
Shingles, also known as zoster or herpes zoster, is caused by retrograde transport of the varicella zoster virus (VZV) from the ganglia to the skin in a host who had a primary varicella infection (chickenpox) in the past.1 Although shingles is not one of the diseases that must be reported to public health authorities, more than 1 million cases are estimated to occur each year in the United States. From 10% to 30% of people develop shingles during their lifetime.2,3
The elderly are at particular risk of shingles, because immunity to VZV wanes as a part of normal aging. As many as 50% of people who live to age 85 will have shingles at some point in their life.
Moreover, about 20% of patients with shingles develop postherpetic neuralgia,3,4 the pain and discomfort of which can be disabling and can diminish quality of life.5
Antiviral therapy reduces the severity and duration of an episode of shingles but does not prevent postherpetic neuralgia.2,6 Steroids provide additional relief of acute zoster pain, but they do not clearly prevent postherpetic neuralgia either and should be used only in combination with antiviral drugs. Preventing zoster and postherpetic neuralgia by routine vaccination should be a goal in our efforts to promote healthy aging, especially with the increasing number of elderly in our country.7
THE VACCINE IS EFFECTIVE: THE SHINGLES PREVENTION STUDY
The Shingles Prevention Study was a prospective, double-blind trial in more than 38,000 adults, median age 69 years (range 59–99), who were followed for a mean of 3.13 years (range 1 day to 4.9 years) after receiving the zoster vaccine or placebo.8–10
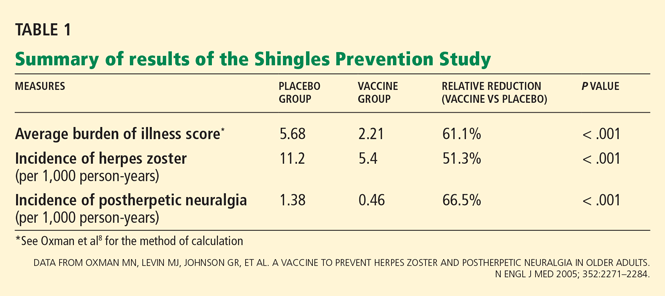
The virus in the vaccine did not elicit shingles in any patient. After vaccination, if lesions did occur, they were from the patient’s native strain, not the vaccine strain.8
ZOSTAVAX
The US Food and Drug Administration approved the zoster vaccine in May 2006 for prevention of herpes zoster in people age 60 and older. Zostavax, licensed by Merck, is the only vaccine available for this purpose.11,12 Zoster vaccine is not indicated for treating episodes of shingles or postherpetic neuralgia or for preventing primary varicella infection (chickenpox).
Zostavax does not contain thimerosal, a mercury-based preservative used in other vaccines. Therefore, it must be kept frozen at an average temperature of –15°C (5°F) and should not be used if its temperature rises above –5°C (23°F).13,14 Just before it is given, the vaccine is reconstituted with the supplied diluents and then injected subcutaneously in the deltoid region.12
No booster dose is recommended at present. Also, many cases of herpes zoster occur in people under age 60, for whom there is no recommendation.11,15 Although the vaccine would probably be safe and effective in this younger group, data are insufficient to recommend vaccinating them.16
ZOSTER VACCINE (ZOSTAVAX) IS NOT CHICKENPOX VACCINE (VARIVAX)
Both Zostavax and the chickenpox vaccine (Varivax) are live, attenuated vaccines from the same Oka/Merck strain of the virus, but Zostavax is about 14 times more potent than Varivax (Zostavax contains 8,700–60,000 plaque-forming units of virus, whereas Varivax contains 1,350), and they should not be used interchangeably.14
GIVING ZOSTER VACCINE WITH OTHER VACCINES IN THE ELDERLY
Zostavax can be given either simultaneously with or at any time before or after any inactivated vaccine (such as tetanus toxoid, influenza, pneumococcus). However, each vaccine must be given in a separate syringe at a different anatomic site.17
VACCINATE EVEN IF THE PATIENT DOESN’T RECALL HAVING CHICKENPOX
Even in people who do not recall ever having chickenpox, the rate of VZV seropositivity is very high (> 95% in those over age 60 in the United States).18 The ACIP recommends vaccination whether or not the patient reports having had chickenpox. Serologic testing to determine varicella immunity is not needed before vaccination, nor was it required for entry in the Shingles Prevention Study.
Furthermore, in VZV-seronegative adults, giving the zoster vaccine is thought to provide at least partial protection against varicella, and no data indicate any excessive adverse effects in this population.
VACCINATE EVEN IF THE PATIENT HAS HAD SHINGLES
The ACIP says that people with a history of zoster can be vaccinated. Recurrent zoster has been confirmed in immunocompetent patients soon after a previous episode. There is no test to confirm prior zoster episodes, and if the patient is immunocompetent, no different safety concerns are anticipated with vaccination in this group.16
ADVERSE EFFECTS ARE MILD
No significant safety concerns have been noted with zoster vaccine. Mild local reactions (erythema, swelling, pain, pruritus) and headache are the most common adverse events. There have been no differences in the numbers and types of serious adverse events during the 42 days after receipt of vaccine or placebo.
CONTRAINDICATIONS
Contraindications to zoster vaccine are:
- A history of anaphylactic or anaphylactoid reactions to gelatin, neomycin, or other components of the vaccine
- Acquired or primary immune deficiency states, including AIDS
- Cancer chemotherapy or radiotherapy
- Leukemia
- Lymphoma
- Organ transplantation
- Active untreated tuberculosis
- Pregnancy or breast-feeding.
However, patients with leukemia in remission who have not received chemotherapy (eg, alkylating drugs or antimetabolites) or radiation for at least 3 months can receive zoster vaccine.
Although zoster vaccine is contraindicated in conditions of cellular immune deficiency, patients with humoral immunodeficiency (eg, hypogammaglobulinemia or dysgammaglobulinemia) can receive it.
Diabetes, hypertension, chronic renal failure, coronary artery disease, chronic lung disease, rheumatoid arthritis, and other medical conditions are not considered contraindications to the vaccine.
The ACIP does not recommend any upper age limit for the vaccine, and preventing zoster is particularly important in the oldest elderly because they have the highest incidence of zoster and postherpetic neuralgia.16
Do not vaccinate during immunosuppressive treatment
If immunosuppressive treatment is planned (eg, with corticosteroids or anti-tumor necrosis factor agents), the vaccine should be given at least 14 days (preferably 1 month) before immunosuppression begins.
The safety and efficacy of zoster vaccine is unknown in patients receiving recombinant human immune mediators and immune modulators, especially anti-tumor necrosis factor agents such as adalimumab (Humira), infliximab (Remicade), or etanercept (Enbrel). These patients should be vaccinated 1 month before starting the treatment or 1 month after stopping it.16
Patients on corticosteroids in doses equivalent to prednisone 20 mg/day or more for 2 or more weeks should not be vaccinated against zoster unless the steroids have been stopped for at least 1 month.11
Low doses of methotrexate (< 0.4 mg/kg/week), azathioprine (Azasan) (< 3.0 mg/kg/day), or 6-mercaptopurine (Purinethol, 6-MP) (< 1.5 mg/kg/day) for the treatment of rheumatoid arthritis, psoriasis, polymyositis, sarcoidosis, inflammatory bowel disease, and other conditions are not contraindications to zoster vaccination.
Medications against herpes, such as acyclovir (Zovirax), famciclovir (Famvir), or valacyclovir (Valtrex) should be discontinued at least 24 hours before zoster vaccination and should not be started until 14 days afterward.
COSTLY AND EFFECTIVE? OR COST-EFFECTIVE?
The average cost associated with an acute episode of zoster ranges from $112 to $287 if treated on an outpatient basis and $3,221 to $7,206 if the patient is hospitalized (costs in 2006).19
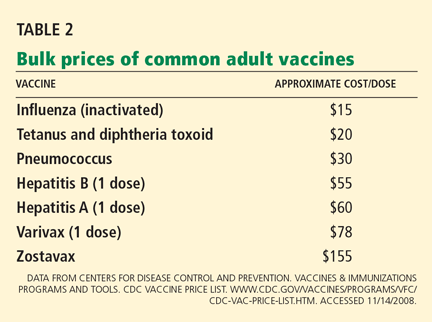
Although pharmacists are licensed to administer influenza and pneumococcal vaccines, several states do not specifically allow them to administer zoster vaccine. Moreover, one usually cannot provide the vaccine out of the office stock and get reimbursed for it (except by some private insurance companies). Instead, the patient needs to take a prescription to a local pharmacy, where the vaccine is placed on ice and then brought back to the physician’s office for administration.
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Gnann JW, Whitley RJ. Clinical practice. Herpes zoster. N Engl J Med 2002; 347:340–346.
- Katz J, Cooper EM, Walther RR, Sweeney EW, Dworkin RH. Acute pain in herpes zoster and its impact on health-related quality of life. Clin Infect Dis 2004; 39:342–348.
- Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med 1965; 58:9–20.
- Lydick E, Epstein RS, Himmelberger D, White CJ. Herpes zoster and quality of life: a self-limited disease with severe impact. Neurology 1995; 45:S52–S53.
- Kost RG, Straus SE. Postherpetic neuralgia—pathogenesis, treatment, and prevention. N Engl J Med 1996; 335:32–42.
- Johnson R, McElhaney J, Pedalino B, Levin M. Prevention of herpes zoster and its painful and debilitating complications. Int J Infect Dis 2007; 11( suppl 2):S43–S48.
- Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Burke MS. Herpes zoster vaccine: clinical trial evidence and implications for medical practice. J Am Osteopath Assoc 2007; 107( suppl 1):S14–S18.
- Betts RF. Vaccination strategies for the prevention of herpes zoster and postherpetic neuralgia. J Am Acad Dermatol 2007; 57( suppl):S143–S147.
- Kroger AT, Atkinson WL, Marcuse EK, Pickering LK. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–48.
- Merck. Zostavax zoster vaccine live suspension for subcutaneous injection. www.merck.com/product/usa/pi_circulars/z/zostavax_pi.pdf. Accessed 11/17/2008.
- Holodniy M. Prevention of shingles by varicella zoster virus vaccination. Expert Rev Vaccines 2006; 5:431–443.
- Herpes zoster vaccine (Zostavax). Med Lett Drugs Ther 2006; 48:73–74.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Bader MS. Immunization for the elderly. Am J Med Sci 2007; 334:481–486.
- Kilgore PE, Kruszon-Moran D, Seward JF, et al. Varicella in Americans from NHANES III: implications for control through routine immunization. J Med Virol 2003; 70( suppl 1):S111–S118.
- Dworkin RH, White R, O’Connor AB, Baser O, Hawkins K. Healthcare costs of acute and chronic pain associated with a diagnosis of herpes zoster. J Am Geriatr Soc 2007; 55:1168–1175.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
The Advisory Committee on Immunization Practices (ACIP) recommends routinely giving a single dose of live zoster vaccine to immunocompetent patients age 60 and older at their first clinical encounter. The vaccine effectively prevents shingles and postherpetic neuralgia and their associated burden of illness. Although not all insurance companies pay for it yet, it should be offered to all patients for whom it is indicated to increase their health-related quality of life.
IMMUNITY WANES WITH AGE
Shingles, also known as zoster or herpes zoster, is caused by retrograde transport of the varicella zoster virus (VZV) from the ganglia to the skin in a host who had a primary varicella infection (chickenpox) in the past.1 Although shingles is not one of the diseases that must be reported to public health authorities, more than 1 million cases are estimated to occur each year in the United States. From 10% to 30% of people develop shingles during their lifetime.2,3
The elderly are at particular risk of shingles, because immunity to VZV wanes as a part of normal aging. As many as 50% of people who live to age 85 will have shingles at some point in their life.
Moreover, about 20% of patients with shingles develop postherpetic neuralgia,3,4 the pain and discomfort of which can be disabling and can diminish quality of life.5
Antiviral therapy reduces the severity and duration of an episode of shingles but does not prevent postherpetic neuralgia.2,6 Steroids provide additional relief of acute zoster pain, but they do not clearly prevent postherpetic neuralgia either and should be used only in combination with antiviral drugs. Preventing zoster and postherpetic neuralgia by routine vaccination should be a goal in our efforts to promote healthy aging, especially with the increasing number of elderly in our country.7
THE VACCINE IS EFFECTIVE: THE SHINGLES PREVENTION STUDY
The Shingles Prevention Study was a prospective, double-blind trial in more than 38,000 adults, median age 69 years (range 59–99), who were followed for a mean of 3.13 years (range 1 day to 4.9 years) after receiving the zoster vaccine or placebo.8–10
The virus in the vaccine did not elicit shingles in any patient. After vaccination, if lesions did occur, they were from the patient’s native strain, not the vaccine strain.8
ZOSTAVAX
The US Food and Drug Administration approved the zoster vaccine in May 2006 for prevention of herpes zoster in people age 60 and older. Zostavax, licensed by Merck, is the only vaccine available for this purpose.11,12 Zoster vaccine is not indicated for treating episodes of shingles or postherpetic neuralgia or for preventing primary varicella infection (chickenpox).
Zostavax does not contain thimerosal, a mercury-based preservative used in other vaccines. Therefore, it must be kept frozen at an average temperature of –15°C (5°F) and should not be used if its temperature rises above –5°C (23°F).13,14 Just before it is given, the vaccine is reconstituted with the supplied diluents and then injected subcutaneously in the deltoid region.12
No booster dose is recommended at present. Also, many cases of herpes zoster occur in people under age 60, for whom there is no recommendation.11,15 Although the vaccine would probably be safe and effective in this younger group, data are insufficient to recommend vaccinating them.16
ZOSTER VACCINE (ZOSTAVAX) IS NOT CHICKENPOX VACCINE (VARIVAX)
Both Zostavax and the chickenpox vaccine (Varivax) are live, attenuated vaccines from the same Oka/Merck strain of the virus, but Zostavax is about 14 times more potent than Varivax (Zostavax contains 8,700–60,000 plaque-forming units of virus, whereas Varivax contains 1,350), and they should not be used interchangeably.14
GIVING ZOSTER VACCINE WITH OTHER VACCINES IN THE ELDERLY
Zostavax can be given either simultaneously with or at any time before or after any inactivated vaccine (such as tetanus toxoid, influenza, pneumococcus). However, each vaccine must be given in a separate syringe at a different anatomic site.17
VACCINATE EVEN IF THE PATIENT DOESN’T RECALL HAVING CHICKENPOX
Even in people who do not recall ever having chickenpox, the rate of VZV seropositivity is very high (> 95% in those over age 60 in the United States).18 The ACIP recommends vaccination whether or not the patient reports having had chickenpox. Serologic testing to determine varicella immunity is not needed before vaccination, nor was it required for entry in the Shingles Prevention Study.
Furthermore, in VZV-seronegative adults, giving the zoster vaccine is thought to provide at least partial protection against varicella, and no data indicate any excessive adverse effects in this population.
VACCINATE EVEN IF THE PATIENT HAS HAD SHINGLES
The ACIP says that people with a history of zoster can be vaccinated. Recurrent zoster has been confirmed in immunocompetent patients soon after a previous episode. There is no test to confirm prior zoster episodes, and if the patient is immunocompetent, no different safety concerns are anticipated with vaccination in this group.16
ADVERSE EFFECTS ARE MILD
No significant safety concerns have been noted with zoster vaccine. Mild local reactions (erythema, swelling, pain, pruritus) and headache are the most common adverse events. There have been no differences in the numbers and types of serious adverse events during the 42 days after receipt of vaccine or placebo.
CONTRAINDICATIONS
Contraindications to zoster vaccine are:
- A history of anaphylactic or anaphylactoid reactions to gelatin, neomycin, or other components of the vaccine
- Acquired or primary immune deficiency states, including AIDS
- Cancer chemotherapy or radiotherapy
- Leukemia
- Lymphoma
- Organ transplantation
- Active untreated tuberculosis
- Pregnancy or breast-feeding.
However, patients with leukemia in remission who have not received chemotherapy (eg, alkylating drugs or antimetabolites) or radiation for at least 3 months can receive zoster vaccine.
Although zoster vaccine is contraindicated in conditions of cellular immune deficiency, patients with humoral immunodeficiency (eg, hypogammaglobulinemia or dysgammaglobulinemia) can receive it.
Diabetes, hypertension, chronic renal failure, coronary artery disease, chronic lung disease, rheumatoid arthritis, and other medical conditions are not considered contraindications to the vaccine.
The ACIP does not recommend any upper age limit for the vaccine, and preventing zoster is particularly important in the oldest elderly because they have the highest incidence of zoster and postherpetic neuralgia.16
Do not vaccinate during immunosuppressive treatment
If immunosuppressive treatment is planned (eg, with corticosteroids or anti-tumor necrosis factor agents), the vaccine should be given at least 14 days (preferably 1 month) before immunosuppression begins.
The safety and efficacy of zoster vaccine is unknown in patients receiving recombinant human immune mediators and immune modulators, especially anti-tumor necrosis factor agents such as adalimumab (Humira), infliximab (Remicade), or etanercept (Enbrel). These patients should be vaccinated 1 month before starting the treatment or 1 month after stopping it.16
Patients on corticosteroids in doses equivalent to prednisone 20 mg/day or more for 2 or more weeks should not be vaccinated against zoster unless the steroids have been stopped for at least 1 month.11
Low doses of methotrexate (< 0.4 mg/kg/week), azathioprine (Azasan) (< 3.0 mg/kg/day), or 6-mercaptopurine (Purinethol, 6-MP) (< 1.5 mg/kg/day) for the treatment of rheumatoid arthritis, psoriasis, polymyositis, sarcoidosis, inflammatory bowel disease, and other conditions are not contraindications to zoster vaccination.
Medications against herpes, such as acyclovir (Zovirax), famciclovir (Famvir), or valacyclovir (Valtrex) should be discontinued at least 24 hours before zoster vaccination and should not be started until 14 days afterward.
COSTLY AND EFFECTIVE? OR COST-EFFECTIVE?
The average cost associated with an acute episode of zoster ranges from $112 to $287 if treated on an outpatient basis and $3,221 to $7,206 if the patient is hospitalized (costs in 2006).19
Although pharmacists are licensed to administer influenza and pneumococcal vaccines, several states do not specifically allow them to administer zoster vaccine. Moreover, one usually cannot provide the vaccine out of the office stock and get reimbursed for it (except by some private insurance companies). Instead, the patient needs to take a prescription to a local pharmacy, where the vaccine is placed on ice and then brought back to the physician’s office for administration.
The Advisory Committee on Immunization Practices (ACIP) recommends routinely giving a single dose of live zoster vaccine to immunocompetent patients age 60 and older at their first clinical encounter. The vaccine effectively prevents shingles and postherpetic neuralgia and their associated burden of illness. Although not all insurance companies pay for it yet, it should be offered to all patients for whom it is indicated to increase their health-related quality of life.
IMMUNITY WANES WITH AGE
Shingles, also known as zoster or herpes zoster, is caused by retrograde transport of the varicella zoster virus (VZV) from the ganglia to the skin in a host who had a primary varicella infection (chickenpox) in the past.1 Although shingles is not one of the diseases that must be reported to public health authorities, more than 1 million cases are estimated to occur each year in the United States. From 10% to 30% of people develop shingles during their lifetime.2,3
The elderly are at particular risk of shingles, because immunity to VZV wanes as a part of normal aging. As many as 50% of people who live to age 85 will have shingles at some point in their life.
Moreover, about 20% of patients with shingles develop postherpetic neuralgia,3,4 the pain and discomfort of which can be disabling and can diminish quality of life.5
Antiviral therapy reduces the severity and duration of an episode of shingles but does not prevent postherpetic neuralgia.2,6 Steroids provide additional relief of acute zoster pain, but they do not clearly prevent postherpetic neuralgia either and should be used only in combination with antiviral drugs. Preventing zoster and postherpetic neuralgia by routine vaccination should be a goal in our efforts to promote healthy aging, especially with the increasing number of elderly in our country.7
THE VACCINE IS EFFECTIVE: THE SHINGLES PREVENTION STUDY
The Shingles Prevention Study was a prospective, double-blind trial in more than 38,000 adults, median age 69 years (range 59–99), who were followed for a mean of 3.13 years (range 1 day to 4.9 years) after receiving the zoster vaccine or placebo.8–10
The virus in the vaccine did not elicit shingles in any patient. After vaccination, if lesions did occur, they were from the patient’s native strain, not the vaccine strain.8
ZOSTAVAX
The US Food and Drug Administration approved the zoster vaccine in May 2006 for prevention of herpes zoster in people age 60 and older. Zostavax, licensed by Merck, is the only vaccine available for this purpose.11,12 Zoster vaccine is not indicated for treating episodes of shingles or postherpetic neuralgia or for preventing primary varicella infection (chickenpox).
Zostavax does not contain thimerosal, a mercury-based preservative used in other vaccines. Therefore, it must be kept frozen at an average temperature of –15°C (5°F) and should not be used if its temperature rises above –5°C (23°F).13,14 Just before it is given, the vaccine is reconstituted with the supplied diluents and then injected subcutaneously in the deltoid region.12
No booster dose is recommended at present. Also, many cases of herpes zoster occur in people under age 60, for whom there is no recommendation.11,15 Although the vaccine would probably be safe and effective in this younger group, data are insufficient to recommend vaccinating them.16
ZOSTER VACCINE (ZOSTAVAX) IS NOT CHICKENPOX VACCINE (VARIVAX)
Both Zostavax and the chickenpox vaccine (Varivax) are live, attenuated vaccines from the same Oka/Merck strain of the virus, but Zostavax is about 14 times more potent than Varivax (Zostavax contains 8,700–60,000 plaque-forming units of virus, whereas Varivax contains 1,350), and they should not be used interchangeably.14
GIVING ZOSTER VACCINE WITH OTHER VACCINES IN THE ELDERLY
Zostavax can be given either simultaneously with or at any time before or after any inactivated vaccine (such as tetanus toxoid, influenza, pneumococcus). However, each vaccine must be given in a separate syringe at a different anatomic site.17
VACCINATE EVEN IF THE PATIENT DOESN’T RECALL HAVING CHICKENPOX
Even in people who do not recall ever having chickenpox, the rate of VZV seropositivity is very high (> 95% in those over age 60 in the United States).18 The ACIP recommends vaccination whether or not the patient reports having had chickenpox. Serologic testing to determine varicella immunity is not needed before vaccination, nor was it required for entry in the Shingles Prevention Study.
Furthermore, in VZV-seronegative adults, giving the zoster vaccine is thought to provide at least partial protection against varicella, and no data indicate any excessive adverse effects in this population.
VACCINATE EVEN IF THE PATIENT HAS HAD SHINGLES
The ACIP says that people with a history of zoster can be vaccinated. Recurrent zoster has been confirmed in immunocompetent patients soon after a previous episode. There is no test to confirm prior zoster episodes, and if the patient is immunocompetent, no different safety concerns are anticipated with vaccination in this group.16
ADVERSE EFFECTS ARE MILD
No significant safety concerns have been noted with zoster vaccine. Mild local reactions (erythema, swelling, pain, pruritus) and headache are the most common adverse events. There have been no differences in the numbers and types of serious adverse events during the 42 days after receipt of vaccine or placebo.
CONTRAINDICATIONS
Contraindications to zoster vaccine are:
- A history of anaphylactic or anaphylactoid reactions to gelatin, neomycin, or other components of the vaccine
- Acquired or primary immune deficiency states, including AIDS
- Cancer chemotherapy or radiotherapy
- Leukemia
- Lymphoma
- Organ transplantation
- Active untreated tuberculosis
- Pregnancy or breast-feeding.
However, patients with leukemia in remission who have not received chemotherapy (eg, alkylating drugs or antimetabolites) or radiation for at least 3 months can receive zoster vaccine.
Although zoster vaccine is contraindicated in conditions of cellular immune deficiency, patients with humoral immunodeficiency (eg, hypogammaglobulinemia or dysgammaglobulinemia) can receive it.
Diabetes, hypertension, chronic renal failure, coronary artery disease, chronic lung disease, rheumatoid arthritis, and other medical conditions are not considered contraindications to the vaccine.
The ACIP does not recommend any upper age limit for the vaccine, and preventing zoster is particularly important in the oldest elderly because they have the highest incidence of zoster and postherpetic neuralgia.16
Do not vaccinate during immunosuppressive treatment
If immunosuppressive treatment is planned (eg, with corticosteroids or anti-tumor necrosis factor agents), the vaccine should be given at least 14 days (preferably 1 month) before immunosuppression begins.
The safety and efficacy of zoster vaccine is unknown in patients receiving recombinant human immune mediators and immune modulators, especially anti-tumor necrosis factor agents such as adalimumab (Humira), infliximab (Remicade), or etanercept (Enbrel). These patients should be vaccinated 1 month before starting the treatment or 1 month after stopping it.16
Patients on corticosteroids in doses equivalent to prednisone 20 mg/day or more for 2 or more weeks should not be vaccinated against zoster unless the steroids have been stopped for at least 1 month.11
Low doses of methotrexate (< 0.4 mg/kg/week), azathioprine (Azasan) (< 3.0 mg/kg/day), or 6-mercaptopurine (Purinethol, 6-MP) (< 1.5 mg/kg/day) for the treatment of rheumatoid arthritis, psoriasis, polymyositis, sarcoidosis, inflammatory bowel disease, and other conditions are not contraindications to zoster vaccination.
Medications against herpes, such as acyclovir (Zovirax), famciclovir (Famvir), or valacyclovir (Valtrex) should be discontinued at least 24 hours before zoster vaccination and should not be started until 14 days afterward.
COSTLY AND EFFECTIVE? OR COST-EFFECTIVE?
The average cost associated with an acute episode of zoster ranges from $112 to $287 if treated on an outpatient basis and $3,221 to $7,206 if the patient is hospitalized (costs in 2006).19
Although pharmacists are licensed to administer influenza and pneumococcal vaccines, several states do not specifically allow them to administer zoster vaccine. Moreover, one usually cannot provide the vaccine out of the office stock and get reimbursed for it (except by some private insurance companies). Instead, the patient needs to take a prescription to a local pharmacy, where the vaccine is placed on ice and then brought back to the physician’s office for administration.
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Gnann JW, Whitley RJ. Clinical practice. Herpes zoster. N Engl J Med 2002; 347:340–346.
- Katz J, Cooper EM, Walther RR, Sweeney EW, Dworkin RH. Acute pain in herpes zoster and its impact on health-related quality of life. Clin Infect Dis 2004; 39:342–348.
- Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med 1965; 58:9–20.
- Lydick E, Epstein RS, Himmelberger D, White CJ. Herpes zoster and quality of life: a self-limited disease with severe impact. Neurology 1995; 45:S52–S53.
- Kost RG, Straus SE. Postherpetic neuralgia—pathogenesis, treatment, and prevention. N Engl J Med 1996; 335:32–42.
- Johnson R, McElhaney J, Pedalino B, Levin M. Prevention of herpes zoster and its painful and debilitating complications. Int J Infect Dis 2007; 11( suppl 2):S43–S48.
- Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Burke MS. Herpes zoster vaccine: clinical trial evidence and implications for medical practice. J Am Osteopath Assoc 2007; 107( suppl 1):S14–S18.
- Betts RF. Vaccination strategies for the prevention of herpes zoster and postherpetic neuralgia. J Am Acad Dermatol 2007; 57( suppl):S143–S147.
- Kroger AT, Atkinson WL, Marcuse EK, Pickering LK. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–48.
- Merck. Zostavax zoster vaccine live suspension for subcutaneous injection. www.merck.com/product/usa/pi_circulars/z/zostavax_pi.pdf. Accessed 11/17/2008.
- Holodniy M. Prevention of shingles by varicella zoster virus vaccination. Expert Rev Vaccines 2006; 5:431–443.
- Herpes zoster vaccine (Zostavax). Med Lett Drugs Ther 2006; 48:73–74.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Bader MS. Immunization for the elderly. Am J Med Sci 2007; 334:481–486.
- Kilgore PE, Kruszon-Moran D, Seward JF, et al. Varicella in Americans from NHANES III: implications for control through routine immunization. J Med Virol 2003; 70( suppl 1):S111–S118.
- Dworkin RH, White R, O’Connor AB, Baser O, Hawkins K. Healthcare costs of acute and chronic pain associated with a diagnosis of herpes zoster. J Am Geriatr Soc 2007; 55:1168–1175.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Gnann JW, Whitley RJ. Clinical practice. Herpes zoster. N Engl J Med 2002; 347:340–346.
- Katz J, Cooper EM, Walther RR, Sweeney EW, Dworkin RH. Acute pain in herpes zoster and its impact on health-related quality of life. Clin Infect Dis 2004; 39:342–348.
- Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med 1965; 58:9–20.
- Lydick E, Epstein RS, Himmelberger D, White CJ. Herpes zoster and quality of life: a self-limited disease with severe impact. Neurology 1995; 45:S52–S53.
- Kost RG, Straus SE. Postherpetic neuralgia—pathogenesis, treatment, and prevention. N Engl J Med 1996; 335:32–42.
- Johnson R, McElhaney J, Pedalino B, Levin M. Prevention of herpes zoster and its painful and debilitating complications. Int J Infect Dis 2007; 11( suppl 2):S43–S48.
- Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005; 352:2271–2284.
- Burke MS. Herpes zoster vaccine: clinical trial evidence and implications for medical practice. J Am Osteopath Assoc 2007; 107( suppl 1):S14–S18.
- Betts RF. Vaccination strategies for the prevention of herpes zoster and postherpetic neuralgia. J Am Acad Dermatol 2007; 57( suppl):S143–S147.
- Kroger AT, Atkinson WL, Marcuse EK, Pickering LK. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55:1–48.
- Merck. Zostavax zoster vaccine live suspension for subcutaneous injection. www.merck.com/product/usa/pi_circulars/z/zostavax_pi.pdf. Accessed 11/17/2008.
- Holodniy M. Prevention of shingles by varicella zoster virus vaccination. Expert Rev Vaccines 2006; 5:431–443.
- Herpes zoster vaccine (Zostavax). Med Lett Drugs Ther 2006; 48:73–74.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2008; 57:1–30.
- Bader MS. Immunization for the elderly. Am J Med Sci 2007; 334:481–486.
- Kilgore PE, Kruszon-Moran D, Seward JF, et al. Varicella in Americans from NHANES III: implications for control through routine immunization. J Med Virol 2003; 70( suppl 1):S111–S118.
- Dworkin RH, White R, O’Connor AB, Baser O, Hawkins K. Healthcare costs of acute and chronic pain associated with a diagnosis of herpes zoster. J Am Geriatr Soc 2007; 55:1168–1175.
- Dworkin RH, White R, O’Connor AB, Hawkins K. Health care expenditure burden of persisting herpes zoster pain. Pain Med 2008; 9:348–353.
Does noninvasive positive pressure ventilation have a role in managing hypercapnic respiratory failure due to an acute exacerbation of COPD?
Yes. In selected patients with hypercapnic respiratory failure due to an acute exacerbation of chronic obstructive pulmonary disease (COPD), noninvasive positive pressure ventilation (NIPPV) is an effective adjunct to usual medical therapy. In controlled trials, it reduced the need for endotracheal intubation, the length of hospital stay, and the risk of death.
Acute COPD exacerbations are responsible for more than 500,000 hospitalizations yearly in the United States, and 6% to 34% of patients die.1
Many patients need invasive ventilatory assistance via an endotracheal tube, but such therapy puts the patient at risk of ventilator-associated pneumonia, pneumothorax, and tracheal stenosis.
WHAT IS NONINVASIVE POSITIVE PRESSURE VENTILATION?
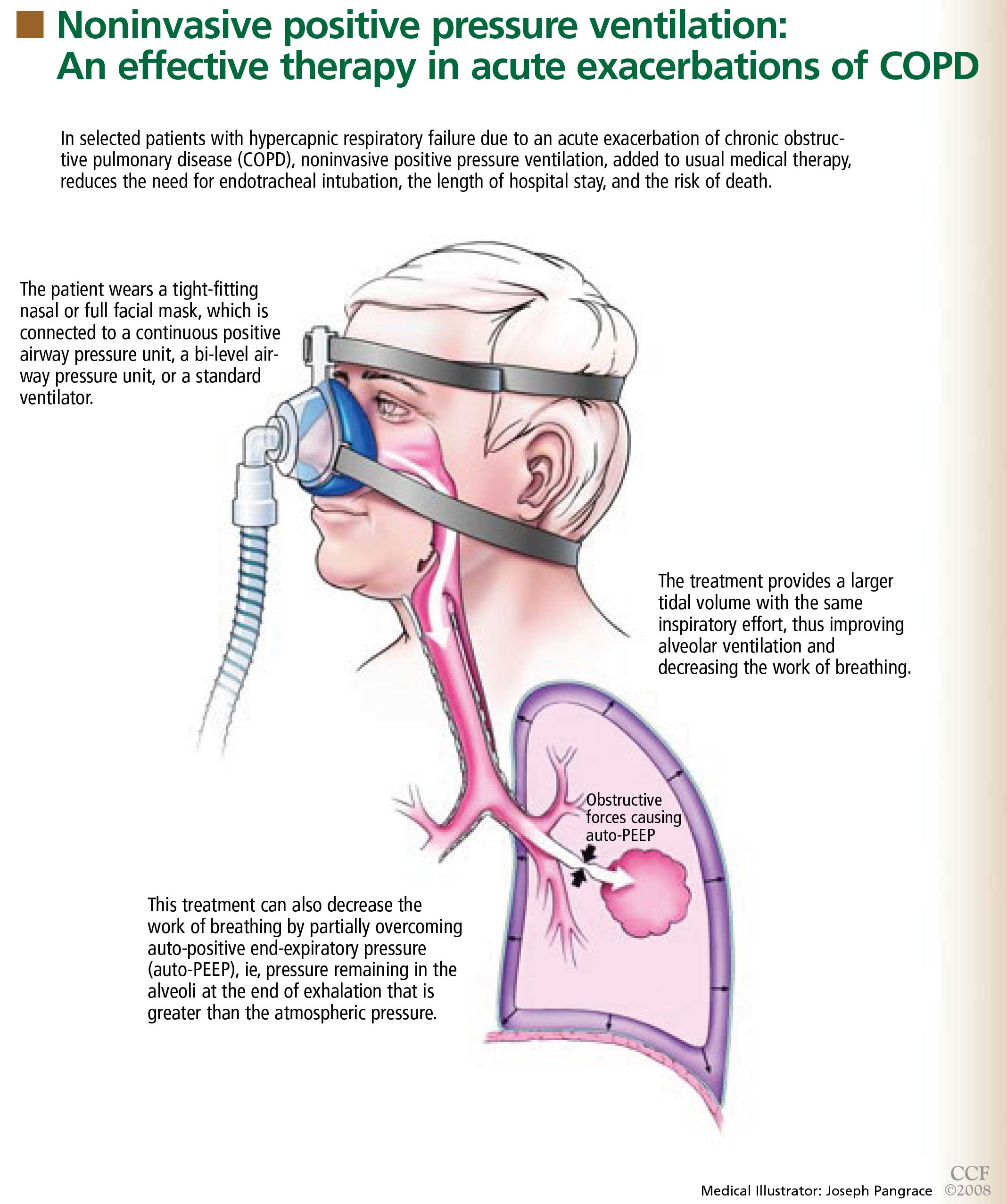
WHY IS IT BENEFICIAL?
Several mechanisms may explain why noninvasive positive pressure ventilation is beneficial in acute exacerbations of COPD.
Patients with decompensated respiratory failure lack sufficient alveolar ventilation, owing to abnormal respiratory mechanics and inspiratory muscle fatigue.10 For these patients, breathing faster does not fully compensate. Noninvasive positive pressure ventilation partially counteracts these factors by providing a larger tidal volume with the same inspiratory effort.10,11
Additionally, this treatment can decrease the work of breathing by partially overcoming auto-PEEP (positive end-expiratory pressure) in certain situations.2 Auto-PEEP is pressure greater than the atmospheric pressure remaining in the alveoli at the end of exhalation.12 This condition is related to limited expiratory flow and is common in those with severe COPD. Noninvasive positive pressure ventilation decreases the pressure difference between the atmosphere and the alveoli, thereby reducing the inspiratory force needed for initiation of inspiratory effort, which may reduce the work of breathing. However, caution should be used when using this therapy in tachypneic patients, in whom NIPPV may not fully overcome the auto-PEEP.
WHAT STUDIES SHOWED
Several randomized trials have shown NIPPV to be beneficial in acute hypercapnic COPD exacerbations. A recent meta-analysis of eight studies13 showed that, compared with usual care alone, this therapy was associated with:
- A lower mortality rate (relative risk 0.41; 95% confidence interval [CI] 0.26–0.64)
- Less need for endotracheal intubation (relative risk 0.42; 95% CI 0.31–0.59)
- A lower rate of treatment failure (relative risk 0.51; 95% CI 0.38–0.67)
- Greater improvements in the 1-hour post-treatment pH and PaCO2 levels
- A lower respiratory rate
- A shorter length of stay in the hospital.
WHICH PATIENTS SHOULD RECEIVE IT?

NIPPV is not suitable for all patients with hypercapnic respiratory failure. It should not be substituted for endotracheal intubation and mechanical ventilation if they are indicated, eg, in patients who are medically unstable because of hypotension, sepsis, hypoxia, or other life-threatening systemic illness. In addition, those who cannot protect the airway, who have had a worsening in mental status, or who have excessive secretions should not undergo NIPPV because they have a high risk of aspiration. Factors that predict that this therapy will fail include an Acute Physiology and Chronic Health Evaluation (APACHE) score of 29 or higher, a respiratory rate of 30 or higher, and a pH lower than 7.25 after 2 hours of this therapy.15
GENERAL WARD OR INTENSIVE CARE UNIT?
Mild to moderate COPD exacerbations (in which the pH is 7.30 or higher) can be effectively treated with NIPPV in a general ward if the staff has appropriate expertise.5,18 Keeping the patient in a general ward reduces cost and provides a favorable outcome in selected patients.5,19 However, if the patient’s hemodynamic or mental status deteriorates or if gas exchange, pH, respiratory rate, or dyspnea fail to improve, he or she should be transferred to an intensive care unit and endotracheal intubation should be considered.18 The use of NIPPV in general wards should always be approached with caution and should never be attempted without adequate patient supervision and an experienced respiratory therapy team.
TAKE-HOME MESSAGE
NIPPV has been shown to be an effective adjunct in the treatment of acute hypercapnic respiratory failure secondary to a COPD exacerbation, reducing the need for endotracheal intubation, the length of hospital stay, and the mortality rate. On the basis of controlled trials, NIPPV is now considered the ventilatory therapy of choice in selected patients with this condition. However, it should not be used as a substitute for intubation and mechanical ventilation if these are needed or if the patient is at risk of aspiration.
- Connors AF Jr, Dawson NV, Thomas C, et al. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and P for Outcomes and Risks of Treatments). Am J Respir Crit Care Med 1996; 154:959–967. (Erratum in: Am J Respir Crit Care Med 1997; 155:386).
- Mehta S, Hill NS. Noninvasive ventilation. Am J Respir Crit Care Med 2001; 163:540–577.
- Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med 1995; 333:817–822.
- Kramer N, Meyer TJ, Meharg J, Cece RD, Hill NS. Randomized, prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 1995; 151:1799–1806.
- Plant PK, Owen JL, Elliott MW. Early use of non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: a multicentre randomised controlled trial. Lancet 2000; 355:1931–1935.
- Wysocki M, Tric L, Wolff MA, Millet H, Herman B. Noninvasive pressure support ventilation in patients with acute respiratory failure. A randomized comparison with conventional therapy. Chest 1995; 107:761–768.
- Masip J, Betbesé AJ, Páez J, et al. Non-invasive pressure support ventilation versus conventional oxygen therapy in acute cardiogenic pulmonary oedema: a randomised trial. Lancet 2000; 356:2126–2132.
- Antonelli M, Conti G, Rocco M, et al. A comparison of noninvasive positive-pressure ventilation and conventional mechanical ventilation in patients with acute respiratory failure. N Engl J Med 1998; 339:429–435.
- Girault C, Daudenthun I, Chevron V, Tamion F, Leroy J, Bonmarchand G. Noninvasive ventilation as a systematic extubation and weaning technique in acute-on-chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med 1999; 160:86–92.
- Brochard L. Noninvasive ventilation for acute respiratory failure. JAMA 2002; 288:932–935.
- Brochard L, Isabey D, Piquet J, et al. Reversal of acute exacerbations of chronic obstructive lung disease by inspiratory assistance with a face mask. N Engl J Med 1990; 323:1523–1530.
- Mughal MM, Culver DA, Minai OA, Arroliga AC. Auto-positive end-expiratory pressure: mechanisms and treatment. Cleve Clin J Med 2005; 72:801–809.
- Lightowler JV, Wedzicha JA, Elliott MW, Ram FS. Non-invasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ 2003; 326:185.
- British Thoracic Society Standards of Care Committee. Non-invasive ventilation in acute respiratory failure. Thorax 2002; 57:192–211.
- Confalonieri M, Garuti G, Cattaruzza MS, et al. A chart of failure risk for noninvasive ventilation in patients with COPD exacerbation. Eur Respir J 2005; 25:348–355.
- American Respiratory Care Foundation Consensus Conference. Non-invasive positive pressure ventilation. Respir Care 1997; 42:364–369.
- Celikel T, Sungur M, Ceyhan B, Karakurt S. Comparison of noninvasive positive pressure ventilation with standard medical therapy in hypercapnic acute respiratory failure. Chest 1998; 114:1636–1642.
- Organized jointly by the American Thoracic Society, the European Respiratory Society, the European Society of Intensive Care Medicine, and the Société de Réanimation de Langue Francaise, and approved by ATS Board of Directors, December 2000. International Consensus Conferences in Intensive Care Medicine: noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 2001; 163:283–291.
- Plant PK, Owen JL, Parrott S, Elliott MW. Cost effectiveness of ward based non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease: economic analysis of randomised controlled trial. BMJ 2003; 326:956.
Yes. In selected patients with hypercapnic respiratory failure due to an acute exacerbation of chronic obstructive pulmonary disease (COPD), noninvasive positive pressure ventilation (NIPPV) is an effective adjunct to usual medical therapy. In controlled trials, it reduced the need for endotracheal intubation, the length of hospital stay, and the risk of death.
Acute COPD exacerbations are responsible for more than 500,000 hospitalizations yearly in the United States, and 6% to 34% of patients die.1
Many patients need invasive ventilatory assistance via an endotracheal tube, but such therapy puts the patient at risk of ventilator-associated pneumonia, pneumothorax, and tracheal stenosis.
WHAT IS NONINVASIVE POSITIVE PRESSURE VENTILATION?
WHY IS IT BENEFICIAL?
Several mechanisms may explain why noninvasive positive pressure ventilation is beneficial in acute exacerbations of COPD.
Patients with decompensated respiratory failure lack sufficient alveolar ventilation, owing to abnormal respiratory mechanics and inspiratory muscle fatigue.10 For these patients, breathing faster does not fully compensate. Noninvasive positive pressure ventilation partially counteracts these factors by providing a larger tidal volume with the same inspiratory effort.10,11
Additionally, this treatment can decrease the work of breathing by partially overcoming auto-PEEP (positive end-expiratory pressure) in certain situations.2 Auto-PEEP is pressure greater than the atmospheric pressure remaining in the alveoli at the end of exhalation.12 This condition is related to limited expiratory flow and is common in those with severe COPD. Noninvasive positive pressure ventilation decreases the pressure difference between the atmosphere and the alveoli, thereby reducing the inspiratory force needed for initiation of inspiratory effort, which may reduce the work of breathing. However, caution should be used when using this therapy in tachypneic patients, in whom NIPPV may not fully overcome the auto-PEEP.
WHAT STUDIES SHOWED
Several randomized trials have shown NIPPV to be beneficial in acute hypercapnic COPD exacerbations. A recent meta-analysis of eight studies13 showed that, compared with usual care alone, this therapy was associated with:
- A lower mortality rate (relative risk 0.41; 95% confidence interval [CI] 0.26–0.64)
- Less need for endotracheal intubation (relative risk 0.42; 95% CI 0.31–0.59)
- A lower rate of treatment failure (relative risk 0.51; 95% CI 0.38–0.67)
- Greater improvements in the 1-hour post-treatment pH and PaCO2 levels
- A lower respiratory rate
- A shorter length of stay in the hospital.
WHICH PATIENTS SHOULD RECEIVE IT?
NIPPV is not suitable for all patients with hypercapnic respiratory failure. It should not be substituted for endotracheal intubation and mechanical ventilation if they are indicated, eg, in patients who are medically unstable because of hypotension, sepsis, hypoxia, or other life-threatening systemic illness. In addition, those who cannot protect the airway, who have had a worsening in mental status, or who have excessive secretions should not undergo NIPPV because they have a high risk of aspiration. Factors that predict that this therapy will fail include an Acute Physiology and Chronic Health Evaluation (APACHE) score of 29 or higher, a respiratory rate of 30 or higher, and a pH lower than 7.25 after 2 hours of this therapy.15
GENERAL WARD OR INTENSIVE CARE UNIT?
Mild to moderate COPD exacerbations (in which the pH is 7.30 or higher) can be effectively treated with NIPPV in a general ward if the staff has appropriate expertise.5,18 Keeping the patient in a general ward reduces cost and provides a favorable outcome in selected patients.5,19 However, if the patient’s hemodynamic or mental status deteriorates or if gas exchange, pH, respiratory rate, or dyspnea fail to improve, he or she should be transferred to an intensive care unit and endotracheal intubation should be considered.18 The use of NIPPV in general wards should always be approached with caution and should never be attempted without adequate patient supervision and an experienced respiratory therapy team.
TAKE-HOME MESSAGE
NIPPV has been shown to be an effective adjunct in the treatment of acute hypercapnic respiratory failure secondary to a COPD exacerbation, reducing the need for endotracheal intubation, the length of hospital stay, and the mortality rate. On the basis of controlled trials, NIPPV is now considered the ventilatory therapy of choice in selected patients with this condition. However, it should not be used as a substitute for intubation and mechanical ventilation if these are needed or if the patient is at risk of aspiration.
Yes. In selected patients with hypercapnic respiratory failure due to an acute exacerbation of chronic obstructive pulmonary disease (COPD), noninvasive positive pressure ventilation (NIPPV) is an effective adjunct to usual medical therapy. In controlled trials, it reduced the need for endotracheal intubation, the length of hospital stay, and the risk of death.
Acute COPD exacerbations are responsible for more than 500,000 hospitalizations yearly in the United States, and 6% to 34% of patients die.1
Many patients need invasive ventilatory assistance via an endotracheal tube, but such therapy puts the patient at risk of ventilator-associated pneumonia, pneumothorax, and tracheal stenosis.
WHAT IS NONINVASIVE POSITIVE PRESSURE VENTILATION?
WHY IS IT BENEFICIAL?
Several mechanisms may explain why noninvasive positive pressure ventilation is beneficial in acute exacerbations of COPD.
Patients with decompensated respiratory failure lack sufficient alveolar ventilation, owing to abnormal respiratory mechanics and inspiratory muscle fatigue.10 For these patients, breathing faster does not fully compensate. Noninvasive positive pressure ventilation partially counteracts these factors by providing a larger tidal volume with the same inspiratory effort.10,11
Additionally, this treatment can decrease the work of breathing by partially overcoming auto-PEEP (positive end-expiratory pressure) in certain situations.2 Auto-PEEP is pressure greater than the atmospheric pressure remaining in the alveoli at the end of exhalation.12 This condition is related to limited expiratory flow and is common in those with severe COPD. Noninvasive positive pressure ventilation decreases the pressure difference between the atmosphere and the alveoli, thereby reducing the inspiratory force needed for initiation of inspiratory effort, which may reduce the work of breathing. However, caution should be used when using this therapy in tachypneic patients, in whom NIPPV may not fully overcome the auto-PEEP.
WHAT STUDIES SHOWED
Several randomized trials have shown NIPPV to be beneficial in acute hypercapnic COPD exacerbations. A recent meta-analysis of eight studies13 showed that, compared with usual care alone, this therapy was associated with:
- A lower mortality rate (relative risk 0.41; 95% confidence interval [CI] 0.26–0.64)
- Less need for endotracheal intubation (relative risk 0.42; 95% CI 0.31–0.59)
- A lower rate of treatment failure (relative risk 0.51; 95% CI 0.38–0.67)
- Greater improvements in the 1-hour post-treatment pH and PaCO2 levels
- A lower respiratory rate
- A shorter length of stay in the hospital.
WHICH PATIENTS SHOULD RECEIVE IT?
NIPPV is not suitable for all patients with hypercapnic respiratory failure. It should not be substituted for endotracheal intubation and mechanical ventilation if they are indicated, eg, in patients who are medically unstable because of hypotension, sepsis, hypoxia, or other life-threatening systemic illness. In addition, those who cannot protect the airway, who have had a worsening in mental status, or who have excessive secretions should not undergo NIPPV because they have a high risk of aspiration. Factors that predict that this therapy will fail include an Acute Physiology and Chronic Health Evaluation (APACHE) score of 29 or higher, a respiratory rate of 30 or higher, and a pH lower than 7.25 after 2 hours of this therapy.15
GENERAL WARD OR INTENSIVE CARE UNIT?
Mild to moderate COPD exacerbations (in which the pH is 7.30 or higher) can be effectively treated with NIPPV in a general ward if the staff has appropriate expertise.5,18 Keeping the patient in a general ward reduces cost and provides a favorable outcome in selected patients.5,19 However, if the patient’s hemodynamic or mental status deteriorates or if gas exchange, pH, respiratory rate, or dyspnea fail to improve, he or she should be transferred to an intensive care unit and endotracheal intubation should be considered.18 The use of NIPPV in general wards should always be approached with caution and should never be attempted without adequate patient supervision and an experienced respiratory therapy team.
TAKE-HOME MESSAGE
NIPPV has been shown to be an effective adjunct in the treatment of acute hypercapnic respiratory failure secondary to a COPD exacerbation, reducing the need for endotracheal intubation, the length of hospital stay, and the mortality rate. On the basis of controlled trials, NIPPV is now considered the ventilatory therapy of choice in selected patients with this condition. However, it should not be used as a substitute for intubation and mechanical ventilation if these are needed or if the patient is at risk of aspiration.
- Connors AF Jr, Dawson NV, Thomas C, et al. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and P for Outcomes and Risks of Treatments). Am J Respir Crit Care Med 1996; 154:959–967. (Erratum in: Am J Respir Crit Care Med 1997; 155:386).
- Mehta S, Hill NS. Noninvasive ventilation. Am J Respir Crit Care Med 2001; 163:540–577.
- Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med 1995; 333:817–822.
- Kramer N, Meyer TJ, Meharg J, Cece RD, Hill NS. Randomized, prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 1995; 151:1799–1806.
- Plant PK, Owen JL, Elliott MW. Early use of non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: a multicentre randomised controlled trial. Lancet 2000; 355:1931–1935.
- Wysocki M, Tric L, Wolff MA, Millet H, Herman B. Noninvasive pressure support ventilation in patients with acute respiratory failure. A randomized comparison with conventional therapy. Chest 1995; 107:761–768.
- Masip J, Betbesé AJ, Páez J, et al. Non-invasive pressure support ventilation versus conventional oxygen therapy in acute cardiogenic pulmonary oedema: a randomised trial. Lancet 2000; 356:2126–2132.
- Antonelli M, Conti G, Rocco M, et al. A comparison of noninvasive positive-pressure ventilation and conventional mechanical ventilation in patients with acute respiratory failure. N Engl J Med 1998; 339:429–435.
- Girault C, Daudenthun I, Chevron V, Tamion F, Leroy J, Bonmarchand G. Noninvasive ventilation as a systematic extubation and weaning technique in acute-on-chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med 1999; 160:86–92.
- Brochard L. Noninvasive ventilation for acute respiratory failure. JAMA 2002; 288:932–935.
- Brochard L, Isabey D, Piquet J, et al. Reversal of acute exacerbations of chronic obstructive lung disease by inspiratory assistance with a face mask. N Engl J Med 1990; 323:1523–1530.
- Mughal MM, Culver DA, Minai OA, Arroliga AC. Auto-positive end-expiratory pressure: mechanisms and treatment. Cleve Clin J Med 2005; 72:801–809.
- Lightowler JV, Wedzicha JA, Elliott MW, Ram FS. Non-invasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ 2003; 326:185.
- British Thoracic Society Standards of Care Committee. Non-invasive ventilation in acute respiratory failure. Thorax 2002; 57:192–211.
- Confalonieri M, Garuti G, Cattaruzza MS, et al. A chart of failure risk for noninvasive ventilation in patients with COPD exacerbation. Eur Respir J 2005; 25:348–355.
- American Respiratory Care Foundation Consensus Conference. Non-invasive positive pressure ventilation. Respir Care 1997; 42:364–369.
- Celikel T, Sungur M, Ceyhan B, Karakurt S. Comparison of noninvasive positive pressure ventilation with standard medical therapy in hypercapnic acute respiratory failure. Chest 1998; 114:1636–1642.
- Organized jointly by the American Thoracic Society, the European Respiratory Society, the European Society of Intensive Care Medicine, and the Société de Réanimation de Langue Francaise, and approved by ATS Board of Directors, December 2000. International Consensus Conferences in Intensive Care Medicine: noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 2001; 163:283–291.
- Plant PK, Owen JL, Parrott S, Elliott MW. Cost effectiveness of ward based non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease: economic analysis of randomised controlled trial. BMJ 2003; 326:956.
- Connors AF Jr, Dawson NV, Thomas C, et al. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and P for Outcomes and Risks of Treatments). Am J Respir Crit Care Med 1996; 154:959–967. (Erratum in: Am J Respir Crit Care Med 1997; 155:386).
- Mehta S, Hill NS. Noninvasive ventilation. Am J Respir Crit Care Med 2001; 163:540–577.
- Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med 1995; 333:817–822.
- Kramer N, Meyer TJ, Meharg J, Cece RD, Hill NS. Randomized, prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 1995; 151:1799–1806.
- Plant PK, Owen JL, Elliott MW. Early use of non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: a multicentre randomised controlled trial. Lancet 2000; 355:1931–1935.
- Wysocki M, Tric L, Wolff MA, Millet H, Herman B. Noninvasive pressure support ventilation in patients with acute respiratory failure. A randomized comparison with conventional therapy. Chest 1995; 107:761–768.
- Masip J, Betbesé AJ, Páez J, et al. Non-invasive pressure support ventilation versus conventional oxygen therapy in acute cardiogenic pulmonary oedema: a randomised trial. Lancet 2000; 356:2126–2132.
- Antonelli M, Conti G, Rocco M, et al. A comparison of noninvasive positive-pressure ventilation and conventional mechanical ventilation in patients with acute respiratory failure. N Engl J Med 1998; 339:429–435.
- Girault C, Daudenthun I, Chevron V, Tamion F, Leroy J, Bonmarchand G. Noninvasive ventilation as a systematic extubation and weaning technique in acute-on-chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med 1999; 160:86–92.
- Brochard L. Noninvasive ventilation for acute respiratory failure. JAMA 2002; 288:932–935.
- Brochard L, Isabey D, Piquet J, et al. Reversal of acute exacerbations of chronic obstructive lung disease by inspiratory assistance with a face mask. N Engl J Med 1990; 323:1523–1530.
- Mughal MM, Culver DA, Minai OA, Arroliga AC. Auto-positive end-expiratory pressure: mechanisms and treatment. Cleve Clin J Med 2005; 72:801–809.
- Lightowler JV, Wedzicha JA, Elliott MW, Ram FS. Non-invasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ 2003; 326:185.
- British Thoracic Society Standards of Care Committee. Non-invasive ventilation in acute respiratory failure. Thorax 2002; 57:192–211.
- Confalonieri M, Garuti G, Cattaruzza MS, et al. A chart of failure risk for noninvasive ventilation in patients with COPD exacerbation. Eur Respir J 2005; 25:348–355.
- American Respiratory Care Foundation Consensus Conference. Non-invasive positive pressure ventilation. Respir Care 1997; 42:364–369.
- Celikel T, Sungur M, Ceyhan B, Karakurt S. Comparison of noninvasive positive pressure ventilation with standard medical therapy in hypercapnic acute respiratory failure. Chest 1998; 114:1636–1642.
- Organized jointly by the American Thoracic Society, the European Respiratory Society, the European Society of Intensive Care Medicine, and the Société de Réanimation de Langue Francaise, and approved by ATS Board of Directors, December 2000. International Consensus Conferences in Intensive Care Medicine: noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 2001; 163:283–291.
- Plant PK, Owen JL, Parrott S, Elliott MW. Cost effectiveness of ward based non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease: economic analysis of randomised controlled trial. BMJ 2003; 326:956.
Should patients on long-term warfarin take aspirin for heart disease?
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
What are the caveats to using sodium phosphate agents for bowel preparation?
Sodium phosphate (NaP) agents were introduced to provide a gentler alternative to polyethylene glycol (PEG) bowel preparations, which require patients to drink up to 4 liters of fluid over a few hours.
However, in May 2006 the US Food and Drug Administration (FDA) issued an alert that NaP products for bowel cleansing may, in some patients, pose a risk of acute phosphate nephropathy, a rare form of acute renal failure.
Although NaP preparations are generally safe and well tolerated, they can cause significant fluid shifts and electrolyte abnormalities. As such, they should not be used in patients with baseline electrolyte imbalances, renal or hepatic dysfunction, or a number of other comorbidities.
CURRENT BOWEL-CLEANSING OPTIONS
For many years the standard preparation for bowel cleansing was a 4-liter or a 2-liter PEG electrolyte solution plus a laxative (eg, magnesium citrate, bisacodyl, or senna).1–3 The most frequent complaint heard from patients was that “the preparation is worse than the colonoscopy,” attributable to the taste and volume of the fluid they had to consume. Thus, compliance was often a significant issue with patients presenting for colonoscopy. In fact, inadequate bowel preparation is one of the most common reasons polyps are missed during colonoscopy.
Aqueous and tablet forms of NaP (sometimes with a laxative) have become a widely used alternative to PEG solutions because they require much less volume and as a result are more palatable, thereby improving compliance.4,5
NaP agents cleanse the colon by osmotically drawing plasma water into the bowel lumen. The patient must drink significant amounts of water or other oral solutions to prevent dehydration.
NaP-based bowel-cleansing agents are available in two forms: aqueous solution and tablet. Aqueous NaP (such as Fleet Phospho-soda) is a low-volume hyperosmotic solution containing 48 g of monobasic NaP and 18 g of dibasic NaP per 100 mL.6 An oral tablet form (such as Visicol and OsmoPrep) was developed to improve patient tolerance.7 Each 2-g tablet of Visicol contains 1,500 mg of active ingredients (monobasic and dibasic NaP) and 460 mg of microcrystalline cellulose, an inert polymer. Each OsmoPrep tablet contains 1,500 mg of the same active ingredients as Visicol, but the inert ingredients include PEG and magnesium stearate.
At first, the regimen was 40 tablets such as Visicol to be taken with water and bisacodyl. Subsequent regimens such as OsmoPrep with fewer tablets have been shown to be as effective and better tolerated.8 Microcrystalline cellulose in the tablet can produce a residue that may obscure the bowel mucosa. Newer preparations contain lower amounts of this inert ingredient, allowing for improved visualization of the colonic mucosa during colonoscopy.9
ADVANTAGES OF SODIUM PHOSPHATE BOWEL CLEANSERS
In a recent review article, Burke and Church10 noted that NaP cleansing regimens have been shown to be superior to PEG-electrolyte lavage solution with respect to tolerability and acceptance by patients, improved quality of bowel preparation, better mucosal visualization, and more efficient endoscopic examination. In addition, the volume of the preparation may also help decrease the risk of aspiration in some patients.2,3
DISADVANTAGES OF SODIUM PHOSPHATE AGENTS
Despite their comparable or better efficacy and their better tolerability, NaP agents have certain disadvantages.
Effects on the colonic mucosa
In rare cases NaP agents have been shown to alter the microscopic and macroscopic features of the colonic mucosa, and they can induce aphthoid erosions that may mimic those seen in inflammatory bowel disease and enteropathy or colopathy associated with nonsteroidal anti-inflammatory drugs (NSAIDs).11–13 Therefore, NaP agents should not be used prior to the initial endoscopic evaluation of patients with suspected inflammatory bowel disease, microscopic colitis, or NSAID-induced colonopathy.
Fluid and electrolyte shifts
Because NaP acts by drawing plasma water into the bowel lumen, significant volume and electrolyte shifts may occur.14,15 These can cause hypokalemia, hyperphosphatemia, hypocalcemia, hyponatremia or hypernatremia, hypomagnesemia, elevated blood urea nitrogen levels, decreased exercise capacity, increased plasma osmolarity,15–17 seizures,18 and acute renal failure with or without nephrocalcinosis.17,19–21
Thus, patients with significant comorbidities—such as a recent history of myocardial infarction, renal or hepatic insufficiency, or malnutrition—should not use NaP agents.22
Pivotal study of adverse events
In May 2006, the FDA issued an alert outlining the concerns of using oral NaP in specific patient populations. Of note were documented cases of acute phosphate nephropathy in 21 patients who used aqueous NaP (Fleet Phospho-Soda or Fleet Accu-Prep), and in 1 patient who used NaP tablets (Visicol).23 Acute renal injury was not limited to patients with preexisting renal insufficiency. It is uncertain whether this means that otherwise healthy people suffered renal injury or had risk factors besides renal insufficiency, since the data cited by the FDA report do not elucidate the possible risk factors for the development of nephropathy in patients with no preexisting renal insufficiency. So far, no cases of acute phosphate nephropathy or acute renal failure have been reported with OsmoPrep, a NaP tablet bowel preparation recently approved by the FDA.24 The long-term safety of OsmoPrep needs further evaluation.
PROCEED WITH CAUTION
Certain situations such as advanced age and cardiac, renal, and hepatic dysfunction call for extreme caution in the use of NaP bowel preparation agents. Therefore, it is recommended that patients with the following conditions should avoid using NaP agents for colon preparation:
- Hepatic or renal insufficiency (there are no data as to the degree of hepatic or renal insufficiency)
- Congestive heart failure
- Over age 65
- Dehydration or hypercalcemia
- Chronic use of drugs that affect renal perfusion, such as NSAIDs, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, or diuretics for hypertension.
Patients who take diuretics should not take them while they are using NaP for bowel preparation because of the risk of electrolyte abnormalities such as hypokalemia. In patients who have no alternative but to proceed with NaP preparation, our recommendation would be that the patient hold off taking diuretics, ACE inhibitors, and angiotensin receptor blockers while using the NaP prep. Given the importance of these medications in controlling diseases such as hypertension, the physician and the patient should jointly determine whether the benefits of using an NaP agent justify holding these drugs. We believe that patients taking these drugs should try using a PEG solution before considering NaP.
TASK FORCE GUIDELINES
Guidelines for using NaP bowel preparation agents, published by a task force of the American Society of Colon and Rectal Surgeons, the American Society for Gastrointestinal Endoscopy, and the Society of American Gastrointestinal and Endoscopic Surgeons,25 include the following caveats:
- Aqueous and tablet NaP colonic preparations are an alternative to PEG solutions, except in pediatric populations, patients over age 65, and those with bowel obstruction or other structural intestinal disorder, gut dysmotility, renal or hepatic insufficiency, congestive heart failure, or seizure disorder.
- Dosing should be 45 mL in divided doses, 10 to 12 hours apart, with at least one dose taken on the morning of the procedure.25
- The significant volume contraction and resulting dehydration seen in some patients using NaP preparations may be lessened by encouraging patients to drink fluids liberally during the days leading up to their procedure, and especially during NaP bowel preparation.26
- NaP tablets should be dosed as 32 to 40 tablets. On the evening before the procedure the patient should take 20 tablets and then 12 to 20 tablets approximately 3 to 5 hours before undergoing endoscopy. The tablets should be taken four at a time every 15 minutes with approximately 8 oz of clear liquid.25

- Sharma VK, Chockalingham SK, Ugheoke EA, et al. Prospective, randomized, controlled comparison of the use of polyethylene glycol electrolyte lavage solution in four-liter versus two-liter volumes and pretreatment with either magnesium citrate or bisacodyl for colonoscopy preparation. Gastrointest Endosc 1998; 47:167–171.
- Frommer D. Cleansing ability and tolerance of three bowel preparations for colonoscopy. Dis Colon Rectum 1997; 40:100–104.
- Hsu CW, Imperiale TF. Meta-analysis and cost comparison of polyethylene glycol lavage versus sodium phosphate for colonoscopy preparation. Gastrointest Endosc 1998; 48:276–282.
- Poon CM, Lee DWH, Mak SK, et al. Two liters of polyethylene glycol-electrolyte solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Afridi SA, Barthel JS, King PD, et al. Prospective, randomized trial comparing a new sodium phosphate-bisacodyl regimen with conventional PEG-ES lavage for outpatient colonoscopy preparation. Gastrointest Endosc 1995; 41:485–489.
- Schiller LR. Clinical pharmacology and use of laxatives and lavage solutions. J Clin Gastroenterol 1988; 28:11–18.
- Kastenberg D, Chasen R, Choudhary C, et al. Efficacy and safety of sodium phosphate tablets compared with PEG solution in colon cleansing. Two identically designed, randomized, controlled, parallel group multicenter phase III trials. Gastrointest Endosc 2001; 54:705–713.
- Rex DK, Chasen R, Pushpin MB. Safety and efficacy of two reduced dosing regimens of sodium phosphate tablets for preparation prior to colonoscopy. Aliment Pharmacol Ther 2002; 16:937–944.
- Rex DK, Khashab M. Efficacy and tolerability of a new formulation of sodium phosphate tablets and a reduced sodium phosphate dose, in colon cleansing: a single-center open-label pilot trial. Aliment Pharmacol Ther 2005; 21:465–468.
- Burke CA, Church JM. Enhancing the quality of colonoscopy: the importance of bowel purgatives. Gastrointest Endosc 2007; 66:565–573.
- Rejchrt S, Bures J, Siroky M, et al. A prospective, observational study of colonic mucosal abnormalities associated with orally administered sodium phosphate for colon cleansing before colonoscopy. Gastrointest Endosc 2004; 59:651–654.
- Hixson LJ. Colorectal ulcers associated with sodium phosphate catharsis. Gastrointest Endosc 1995; 42:101–102.
- Zwas FR, Cirillo NW, El-Serag HB, Eisen RN. Colonic mucosal abnormalities associated with oral sodium phosphate solution. Gastrointest Endosc 1996; 43:463–466.
- Clarkston WK, Tsen TN, Dies DF, Schratz CL, Vaswani SK, Bjerregaard P. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Kolts BE, Lyles WE, Achem SR, et al. A comparison of the effectiveness and patient tolerance of oral sodium phosphate, castor oil, and standard electrolyte lavage for colonoscopy or sigmoidoscopy preparations. Am J Gastroenterol 1993; 88:1218–1223.
- Holte K, Neilsen KG, Madsen JL, Kehlet H. Physiologic effects of bowel preparation. Dis Colon Rectum 2004; 47:1397–1402.
- Clarkston WK, Tsen TN, Dies DF, et al. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Frizelle FA, Colls BM. Hyponatremia and seizures after bowel preparation: report of three cases. Dis Colon Rectum 2005; 48:393–396.
- Markowitz GS, Nasr SH, Klein P, et al. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum Pathol 2004; 35:675–684.
- Lieberman DA, Ghormley J, Flora K. Effect of oral sodium phosphate colon preparation on serum electrolytes in patients with normal serum creatinine. Gastrointest Endosc 1996; 43:467–469.
- Gremse DA, Sacks AI, Raines S. Comparison of oral sodium phosphate to polyethylene-glycol-based solution for bowel preparation in children. J Pediatric Gastroenterol Nutr 1996; 23:586–590.
- Curran MP, Plosker GL. Oral sodium phosphate solution: a review of its use as a colonic cleanser. Drugs 2004; 64:1697–1714.
- Markowitz GS, Stokes MB, Radhakrishnan J, D’Agati VD. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol 2005; 16:3389–3396.
- FDA Alert. Patient information sheet. Oral sodium phosphate (OSP) products for bowel cleansing. 2006 May, Accessed January 8, 2008. www.fda.gov/CDER/drug/InfoSheets/patient/OSP_solutionPIS.htm.
- Wexner SD, Beck DE, Baron TH, et al. A consensus document on bowel preparation before colonoscopy prepared by a task force from the American Society of Colon and Rectal Surgeons (ASCRS), the American Society for Gastrointestinal Endoscopy (ASGE), and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES). Gastrointest Endosc 2006; 63:894–909.
- Huynh T, Vanner S, Paterson W. Safety profile of 5-h oral sodium phosphate regimen for colonoscopy cleansing: lack of clinically significant hypocalcemia or hypovolemia. Am J Gastroenterol 1995; 90:104–107.
Sodium phosphate (NaP) agents were introduced to provide a gentler alternative to polyethylene glycol (PEG) bowel preparations, which require patients to drink up to 4 liters of fluid over a few hours.
However, in May 2006 the US Food and Drug Administration (FDA) issued an alert that NaP products for bowel cleansing may, in some patients, pose a risk of acute phosphate nephropathy, a rare form of acute renal failure.
Although NaP preparations are generally safe and well tolerated, they can cause significant fluid shifts and electrolyte abnormalities. As such, they should not be used in patients with baseline electrolyte imbalances, renal or hepatic dysfunction, or a number of other comorbidities.
CURRENT BOWEL-CLEANSING OPTIONS
For many years the standard preparation for bowel cleansing was a 4-liter or a 2-liter PEG electrolyte solution plus a laxative (eg, magnesium citrate, bisacodyl, or senna).1–3 The most frequent complaint heard from patients was that “the preparation is worse than the colonoscopy,” attributable to the taste and volume of the fluid they had to consume. Thus, compliance was often a significant issue with patients presenting for colonoscopy. In fact, inadequate bowel preparation is one of the most common reasons polyps are missed during colonoscopy.
Aqueous and tablet forms of NaP (sometimes with a laxative) have become a widely used alternative to PEG solutions because they require much less volume and as a result are more palatable, thereby improving compliance.4,5
NaP agents cleanse the colon by osmotically drawing plasma water into the bowel lumen. The patient must drink significant amounts of water or other oral solutions to prevent dehydration.
NaP-based bowel-cleansing agents are available in two forms: aqueous solution and tablet. Aqueous NaP (such as Fleet Phospho-soda) is a low-volume hyperosmotic solution containing 48 g of monobasic NaP and 18 g of dibasic NaP per 100 mL.6 An oral tablet form (such as Visicol and OsmoPrep) was developed to improve patient tolerance.7 Each 2-g tablet of Visicol contains 1,500 mg of active ingredients (monobasic and dibasic NaP) and 460 mg of microcrystalline cellulose, an inert polymer. Each OsmoPrep tablet contains 1,500 mg of the same active ingredients as Visicol, but the inert ingredients include PEG and magnesium stearate.
At first, the regimen was 40 tablets such as Visicol to be taken with water and bisacodyl. Subsequent regimens such as OsmoPrep with fewer tablets have been shown to be as effective and better tolerated.8 Microcrystalline cellulose in the tablet can produce a residue that may obscure the bowel mucosa. Newer preparations contain lower amounts of this inert ingredient, allowing for improved visualization of the colonic mucosa during colonoscopy.9
ADVANTAGES OF SODIUM PHOSPHATE BOWEL CLEANSERS
In a recent review article, Burke and Church10 noted that NaP cleansing regimens have been shown to be superior to PEG-electrolyte lavage solution with respect to tolerability and acceptance by patients, improved quality of bowel preparation, better mucosal visualization, and more efficient endoscopic examination. In addition, the volume of the preparation may also help decrease the risk of aspiration in some patients.2,3
DISADVANTAGES OF SODIUM PHOSPHATE AGENTS
Despite their comparable or better efficacy and their better tolerability, NaP agents have certain disadvantages.
Effects on the colonic mucosa
In rare cases NaP agents have been shown to alter the microscopic and macroscopic features of the colonic mucosa, and they can induce aphthoid erosions that may mimic those seen in inflammatory bowel disease and enteropathy or colopathy associated with nonsteroidal anti-inflammatory drugs (NSAIDs).11–13 Therefore, NaP agents should not be used prior to the initial endoscopic evaluation of patients with suspected inflammatory bowel disease, microscopic colitis, or NSAID-induced colonopathy.
Fluid and electrolyte shifts
Because NaP acts by drawing plasma water into the bowel lumen, significant volume and electrolyte shifts may occur.14,15 These can cause hypokalemia, hyperphosphatemia, hypocalcemia, hyponatremia or hypernatremia, hypomagnesemia, elevated blood urea nitrogen levels, decreased exercise capacity, increased plasma osmolarity,15–17 seizures,18 and acute renal failure with or without nephrocalcinosis.17,19–21
Thus, patients with significant comorbidities—such as a recent history of myocardial infarction, renal or hepatic insufficiency, or malnutrition—should not use NaP agents.22
Pivotal study of adverse events
In May 2006, the FDA issued an alert outlining the concerns of using oral NaP in specific patient populations. Of note were documented cases of acute phosphate nephropathy in 21 patients who used aqueous NaP (Fleet Phospho-Soda or Fleet Accu-Prep), and in 1 patient who used NaP tablets (Visicol).23 Acute renal injury was not limited to patients with preexisting renal insufficiency. It is uncertain whether this means that otherwise healthy people suffered renal injury or had risk factors besides renal insufficiency, since the data cited by the FDA report do not elucidate the possible risk factors for the development of nephropathy in patients with no preexisting renal insufficiency. So far, no cases of acute phosphate nephropathy or acute renal failure have been reported with OsmoPrep, a NaP tablet bowel preparation recently approved by the FDA.24 The long-term safety of OsmoPrep needs further evaluation.
PROCEED WITH CAUTION
Certain situations such as advanced age and cardiac, renal, and hepatic dysfunction call for extreme caution in the use of NaP bowel preparation agents. Therefore, it is recommended that patients with the following conditions should avoid using NaP agents for colon preparation:
- Hepatic or renal insufficiency (there are no data as to the degree of hepatic or renal insufficiency)
- Congestive heart failure
- Over age 65
- Dehydration or hypercalcemia
- Chronic use of drugs that affect renal perfusion, such as NSAIDs, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, or diuretics for hypertension.
Patients who take diuretics should not take them while they are using NaP for bowel preparation because of the risk of electrolyte abnormalities such as hypokalemia. In patients who have no alternative but to proceed with NaP preparation, our recommendation would be that the patient hold off taking diuretics, ACE inhibitors, and angiotensin receptor blockers while using the NaP prep. Given the importance of these medications in controlling diseases such as hypertension, the physician and the patient should jointly determine whether the benefits of using an NaP agent justify holding these drugs. We believe that patients taking these drugs should try using a PEG solution before considering NaP.
TASK FORCE GUIDELINES
Guidelines for using NaP bowel preparation agents, published by a task force of the American Society of Colon and Rectal Surgeons, the American Society for Gastrointestinal Endoscopy, and the Society of American Gastrointestinal and Endoscopic Surgeons,25 include the following caveats:
- Aqueous and tablet NaP colonic preparations are an alternative to PEG solutions, except in pediatric populations, patients over age 65, and those with bowel obstruction or other structural intestinal disorder, gut dysmotility, renal or hepatic insufficiency, congestive heart failure, or seizure disorder.
- Dosing should be 45 mL in divided doses, 10 to 12 hours apart, with at least one dose taken on the morning of the procedure.25
- The significant volume contraction and resulting dehydration seen in some patients using NaP preparations may be lessened by encouraging patients to drink fluids liberally during the days leading up to their procedure, and especially during NaP bowel preparation.26
- NaP tablets should be dosed as 32 to 40 tablets. On the evening before the procedure the patient should take 20 tablets and then 12 to 20 tablets approximately 3 to 5 hours before undergoing endoscopy. The tablets should be taken four at a time every 15 minutes with approximately 8 oz of clear liquid.25
Sodium phosphate (NaP) agents were introduced to provide a gentler alternative to polyethylene glycol (PEG) bowel preparations, which require patients to drink up to 4 liters of fluid over a few hours.
However, in May 2006 the US Food and Drug Administration (FDA) issued an alert that NaP products for bowel cleansing may, in some patients, pose a risk of acute phosphate nephropathy, a rare form of acute renal failure.
Although NaP preparations are generally safe and well tolerated, they can cause significant fluid shifts and electrolyte abnormalities. As such, they should not be used in patients with baseline electrolyte imbalances, renal or hepatic dysfunction, or a number of other comorbidities.
CURRENT BOWEL-CLEANSING OPTIONS
For many years the standard preparation for bowel cleansing was a 4-liter or a 2-liter PEG electrolyte solution plus a laxative (eg, magnesium citrate, bisacodyl, or senna).1–3 The most frequent complaint heard from patients was that “the preparation is worse than the colonoscopy,” attributable to the taste and volume of the fluid they had to consume. Thus, compliance was often a significant issue with patients presenting for colonoscopy. In fact, inadequate bowel preparation is one of the most common reasons polyps are missed during colonoscopy.
Aqueous and tablet forms of NaP (sometimes with a laxative) have become a widely used alternative to PEG solutions because they require much less volume and as a result are more palatable, thereby improving compliance.4,5
NaP agents cleanse the colon by osmotically drawing plasma water into the bowel lumen. The patient must drink significant amounts of water or other oral solutions to prevent dehydration.
NaP-based bowel-cleansing agents are available in two forms: aqueous solution and tablet. Aqueous NaP (such as Fleet Phospho-soda) is a low-volume hyperosmotic solution containing 48 g of monobasic NaP and 18 g of dibasic NaP per 100 mL.6 An oral tablet form (such as Visicol and OsmoPrep) was developed to improve patient tolerance.7 Each 2-g tablet of Visicol contains 1,500 mg of active ingredients (monobasic and dibasic NaP) and 460 mg of microcrystalline cellulose, an inert polymer. Each OsmoPrep tablet contains 1,500 mg of the same active ingredients as Visicol, but the inert ingredients include PEG and magnesium stearate.
At first, the regimen was 40 tablets such as Visicol to be taken with water and bisacodyl. Subsequent regimens such as OsmoPrep with fewer tablets have been shown to be as effective and better tolerated.8 Microcrystalline cellulose in the tablet can produce a residue that may obscure the bowel mucosa. Newer preparations contain lower amounts of this inert ingredient, allowing for improved visualization of the colonic mucosa during colonoscopy.9
ADVANTAGES OF SODIUM PHOSPHATE BOWEL CLEANSERS
In a recent review article, Burke and Church10 noted that NaP cleansing regimens have been shown to be superior to PEG-electrolyte lavage solution with respect to tolerability and acceptance by patients, improved quality of bowel preparation, better mucosal visualization, and more efficient endoscopic examination. In addition, the volume of the preparation may also help decrease the risk of aspiration in some patients.2,3
DISADVANTAGES OF SODIUM PHOSPHATE AGENTS
Despite their comparable or better efficacy and their better tolerability, NaP agents have certain disadvantages.
Effects on the colonic mucosa
In rare cases NaP agents have been shown to alter the microscopic and macroscopic features of the colonic mucosa, and they can induce aphthoid erosions that may mimic those seen in inflammatory bowel disease and enteropathy or colopathy associated with nonsteroidal anti-inflammatory drugs (NSAIDs).11–13 Therefore, NaP agents should not be used prior to the initial endoscopic evaluation of patients with suspected inflammatory bowel disease, microscopic colitis, or NSAID-induced colonopathy.
Fluid and electrolyte shifts
Because NaP acts by drawing plasma water into the bowel lumen, significant volume and electrolyte shifts may occur.14,15 These can cause hypokalemia, hyperphosphatemia, hypocalcemia, hyponatremia or hypernatremia, hypomagnesemia, elevated blood urea nitrogen levels, decreased exercise capacity, increased plasma osmolarity,15–17 seizures,18 and acute renal failure with or without nephrocalcinosis.17,19–21
Thus, patients with significant comorbidities—such as a recent history of myocardial infarction, renal or hepatic insufficiency, or malnutrition—should not use NaP agents.22
Pivotal study of adverse events
In May 2006, the FDA issued an alert outlining the concerns of using oral NaP in specific patient populations. Of note were documented cases of acute phosphate nephropathy in 21 patients who used aqueous NaP (Fleet Phospho-Soda or Fleet Accu-Prep), and in 1 patient who used NaP tablets (Visicol).23 Acute renal injury was not limited to patients with preexisting renal insufficiency. It is uncertain whether this means that otherwise healthy people suffered renal injury or had risk factors besides renal insufficiency, since the data cited by the FDA report do not elucidate the possible risk factors for the development of nephropathy in patients with no preexisting renal insufficiency. So far, no cases of acute phosphate nephropathy or acute renal failure have been reported with OsmoPrep, a NaP tablet bowel preparation recently approved by the FDA.24 The long-term safety of OsmoPrep needs further evaluation.
PROCEED WITH CAUTION
Certain situations such as advanced age and cardiac, renal, and hepatic dysfunction call for extreme caution in the use of NaP bowel preparation agents. Therefore, it is recommended that patients with the following conditions should avoid using NaP agents for colon preparation:
- Hepatic or renal insufficiency (there are no data as to the degree of hepatic or renal insufficiency)
- Congestive heart failure
- Over age 65
- Dehydration or hypercalcemia
- Chronic use of drugs that affect renal perfusion, such as NSAIDs, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, or diuretics for hypertension.
Patients who take diuretics should not take them while they are using NaP for bowel preparation because of the risk of electrolyte abnormalities such as hypokalemia. In patients who have no alternative but to proceed with NaP preparation, our recommendation would be that the patient hold off taking diuretics, ACE inhibitors, and angiotensin receptor blockers while using the NaP prep. Given the importance of these medications in controlling diseases such as hypertension, the physician and the patient should jointly determine whether the benefits of using an NaP agent justify holding these drugs. We believe that patients taking these drugs should try using a PEG solution before considering NaP.
TASK FORCE GUIDELINES
Guidelines for using NaP bowel preparation agents, published by a task force of the American Society of Colon and Rectal Surgeons, the American Society for Gastrointestinal Endoscopy, and the Society of American Gastrointestinal and Endoscopic Surgeons,25 include the following caveats:
- Aqueous and tablet NaP colonic preparations are an alternative to PEG solutions, except in pediatric populations, patients over age 65, and those with bowel obstruction or other structural intestinal disorder, gut dysmotility, renal or hepatic insufficiency, congestive heart failure, or seizure disorder.
- Dosing should be 45 mL in divided doses, 10 to 12 hours apart, with at least one dose taken on the morning of the procedure.25
- The significant volume contraction and resulting dehydration seen in some patients using NaP preparations may be lessened by encouraging patients to drink fluids liberally during the days leading up to their procedure, and especially during NaP bowel preparation.26
- NaP tablets should be dosed as 32 to 40 tablets. On the evening before the procedure the patient should take 20 tablets and then 12 to 20 tablets approximately 3 to 5 hours before undergoing endoscopy. The tablets should be taken four at a time every 15 minutes with approximately 8 oz of clear liquid.25
- Sharma VK, Chockalingham SK, Ugheoke EA, et al. Prospective, randomized, controlled comparison of the use of polyethylene glycol electrolyte lavage solution in four-liter versus two-liter volumes and pretreatment with either magnesium citrate or bisacodyl for colonoscopy preparation. Gastrointest Endosc 1998; 47:167–171.
- Frommer D. Cleansing ability and tolerance of three bowel preparations for colonoscopy. Dis Colon Rectum 1997; 40:100–104.
- Hsu CW, Imperiale TF. Meta-analysis and cost comparison of polyethylene glycol lavage versus sodium phosphate for colonoscopy preparation. Gastrointest Endosc 1998; 48:276–282.
- Poon CM, Lee DWH, Mak SK, et al. Two liters of polyethylene glycol-electrolyte solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Afridi SA, Barthel JS, King PD, et al. Prospective, randomized trial comparing a new sodium phosphate-bisacodyl regimen with conventional PEG-ES lavage for outpatient colonoscopy preparation. Gastrointest Endosc 1995; 41:485–489.
- Schiller LR. Clinical pharmacology and use of laxatives and lavage solutions. J Clin Gastroenterol 1988; 28:11–18.
- Kastenberg D, Chasen R, Choudhary C, et al. Efficacy and safety of sodium phosphate tablets compared with PEG solution in colon cleansing. Two identically designed, randomized, controlled, parallel group multicenter phase III trials. Gastrointest Endosc 2001; 54:705–713.
- Rex DK, Chasen R, Pushpin MB. Safety and efficacy of two reduced dosing regimens of sodium phosphate tablets for preparation prior to colonoscopy. Aliment Pharmacol Ther 2002; 16:937–944.
- Rex DK, Khashab M. Efficacy and tolerability of a new formulation of sodium phosphate tablets and a reduced sodium phosphate dose, in colon cleansing: a single-center open-label pilot trial. Aliment Pharmacol Ther 2005; 21:465–468.
- Burke CA, Church JM. Enhancing the quality of colonoscopy: the importance of bowel purgatives. Gastrointest Endosc 2007; 66:565–573.
- Rejchrt S, Bures J, Siroky M, et al. A prospective, observational study of colonic mucosal abnormalities associated with orally administered sodium phosphate for colon cleansing before colonoscopy. Gastrointest Endosc 2004; 59:651–654.
- Hixson LJ. Colorectal ulcers associated with sodium phosphate catharsis. Gastrointest Endosc 1995; 42:101–102.
- Zwas FR, Cirillo NW, El-Serag HB, Eisen RN. Colonic mucosal abnormalities associated with oral sodium phosphate solution. Gastrointest Endosc 1996; 43:463–466.
- Clarkston WK, Tsen TN, Dies DF, Schratz CL, Vaswani SK, Bjerregaard P. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Kolts BE, Lyles WE, Achem SR, et al. A comparison of the effectiveness and patient tolerance of oral sodium phosphate, castor oil, and standard electrolyte lavage for colonoscopy or sigmoidoscopy preparations. Am J Gastroenterol 1993; 88:1218–1223.
- Holte K, Neilsen KG, Madsen JL, Kehlet H. Physiologic effects of bowel preparation. Dis Colon Rectum 2004; 47:1397–1402.
- Clarkston WK, Tsen TN, Dies DF, et al. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Frizelle FA, Colls BM. Hyponatremia and seizures after bowel preparation: report of three cases. Dis Colon Rectum 2005; 48:393–396.
- Markowitz GS, Nasr SH, Klein P, et al. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum Pathol 2004; 35:675–684.
- Lieberman DA, Ghormley J, Flora K. Effect of oral sodium phosphate colon preparation on serum electrolytes in patients with normal serum creatinine. Gastrointest Endosc 1996; 43:467–469.
- Gremse DA, Sacks AI, Raines S. Comparison of oral sodium phosphate to polyethylene-glycol-based solution for bowel preparation in children. J Pediatric Gastroenterol Nutr 1996; 23:586–590.
- Curran MP, Plosker GL. Oral sodium phosphate solution: a review of its use as a colonic cleanser. Drugs 2004; 64:1697–1714.
- Markowitz GS, Stokes MB, Radhakrishnan J, D’Agati VD. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol 2005; 16:3389–3396.
- FDA Alert. Patient information sheet. Oral sodium phosphate (OSP) products for bowel cleansing. 2006 May, Accessed January 8, 2008. www.fda.gov/CDER/drug/InfoSheets/patient/OSP_solutionPIS.htm.
- Wexner SD, Beck DE, Baron TH, et al. A consensus document on bowel preparation before colonoscopy prepared by a task force from the American Society of Colon and Rectal Surgeons (ASCRS), the American Society for Gastrointestinal Endoscopy (ASGE), and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES). Gastrointest Endosc 2006; 63:894–909.
- Huynh T, Vanner S, Paterson W. Safety profile of 5-h oral sodium phosphate regimen for colonoscopy cleansing: lack of clinically significant hypocalcemia or hypovolemia. Am J Gastroenterol 1995; 90:104–107.
- Sharma VK, Chockalingham SK, Ugheoke EA, et al. Prospective, randomized, controlled comparison of the use of polyethylene glycol electrolyte lavage solution in four-liter versus two-liter volumes and pretreatment with either magnesium citrate or bisacodyl for colonoscopy preparation. Gastrointest Endosc 1998; 47:167–171.
- Frommer D. Cleansing ability and tolerance of three bowel preparations for colonoscopy. Dis Colon Rectum 1997; 40:100–104.
- Hsu CW, Imperiale TF. Meta-analysis and cost comparison of polyethylene glycol lavage versus sodium phosphate for colonoscopy preparation. Gastrointest Endosc 1998; 48:276–282.
- Poon CM, Lee DWH, Mak SK, et al. Two liters of polyethylene glycol-electrolyte solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Afridi SA, Barthel JS, King PD, et al. Prospective, randomized trial comparing a new sodium phosphate-bisacodyl regimen with conventional PEG-ES lavage for outpatient colonoscopy preparation. Gastrointest Endosc 1995; 41:485–489.
- Schiller LR. Clinical pharmacology and use of laxatives and lavage solutions. J Clin Gastroenterol 1988; 28:11–18.
- Kastenberg D, Chasen R, Choudhary C, et al. Efficacy and safety of sodium phosphate tablets compared with PEG solution in colon cleansing. Two identically designed, randomized, controlled, parallel group multicenter phase III trials. Gastrointest Endosc 2001; 54:705–713.
- Rex DK, Chasen R, Pushpin MB. Safety and efficacy of two reduced dosing regimens of sodium phosphate tablets for preparation prior to colonoscopy. Aliment Pharmacol Ther 2002; 16:937–944.
- Rex DK, Khashab M. Efficacy and tolerability of a new formulation of sodium phosphate tablets and a reduced sodium phosphate dose, in colon cleansing: a single-center open-label pilot trial. Aliment Pharmacol Ther 2005; 21:465–468.
- Burke CA, Church JM. Enhancing the quality of colonoscopy: the importance of bowel purgatives. Gastrointest Endosc 2007; 66:565–573.
- Rejchrt S, Bures J, Siroky M, et al. A prospective, observational study of colonic mucosal abnormalities associated with orally administered sodium phosphate for colon cleansing before colonoscopy. Gastrointest Endosc 2004; 59:651–654.
- Hixson LJ. Colorectal ulcers associated with sodium phosphate catharsis. Gastrointest Endosc 1995; 42:101–102.
- Zwas FR, Cirillo NW, El-Serag HB, Eisen RN. Colonic mucosal abnormalities associated with oral sodium phosphate solution. Gastrointest Endosc 1996; 43:463–466.
- Clarkston WK, Tsen TN, Dies DF, Schratz CL, Vaswani SK, Bjerregaard P. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Kolts BE, Lyles WE, Achem SR, et al. A comparison of the effectiveness and patient tolerance of oral sodium phosphate, castor oil, and standard electrolyte lavage for colonoscopy or sigmoidoscopy preparations. Am J Gastroenterol 1993; 88:1218–1223.
- Holte K, Neilsen KG, Madsen JL, Kehlet H. Physiologic effects of bowel preparation. Dis Colon Rectum 2004; 47:1397–1402.
- Clarkston WK, Tsen TN, Dies DF, et al. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Frizelle FA, Colls BM. Hyponatremia and seizures after bowel preparation: report of three cases. Dis Colon Rectum 2005; 48:393–396.
- Markowitz GS, Nasr SH, Klein P, et al. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum Pathol 2004; 35:675–684.
- Lieberman DA, Ghormley J, Flora K. Effect of oral sodium phosphate colon preparation on serum electrolytes in patients with normal serum creatinine. Gastrointest Endosc 1996; 43:467–469.
- Gremse DA, Sacks AI, Raines S. Comparison of oral sodium phosphate to polyethylene-glycol-based solution for bowel preparation in children. J Pediatric Gastroenterol Nutr 1996; 23:586–590.
- Curran MP, Plosker GL. Oral sodium phosphate solution: a review of its use as a colonic cleanser. Drugs 2004; 64:1697–1714.
- Markowitz GS, Stokes MB, Radhakrishnan J, D’Agati VD. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol 2005; 16:3389–3396.
- FDA Alert. Patient information sheet. Oral sodium phosphate (OSP) products for bowel cleansing. 2006 May, Accessed January 8, 2008. www.fda.gov/CDER/drug/InfoSheets/patient/OSP_solutionPIS.htm.
- Wexner SD, Beck DE, Baron TH, et al. A consensus document on bowel preparation before colonoscopy prepared by a task force from the American Society of Colon and Rectal Surgeons (ASCRS), the American Society for Gastrointestinal Endoscopy (ASGE), and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES). Gastrointest Endosc 2006; 63:894–909.
- Huynh T, Vanner S, Paterson W. Safety profile of 5-h oral sodium phosphate regimen for colonoscopy cleansing: lack of clinically significant hypocalcemia or hypovolemia. Am J Gastroenterol 1995; 90:104–107.
Should all patients with chronic kidney disease take a statin?
We think some patients with chronic kidney disease should take a statin, particularly those in stages 1 through 4 (ie, not yet on dialysis1)* who have low-density lipoprotein cholesterol (LDL-C) levels higher than 100 mg/dL. However, few studies have addressed this question.
*Stages of chronic kidney disease1:
Stage 1—kidney damage with normal or high glomerular filtration rate (GFR ≥ 90 mL/min/1.73 m2)
Stage 2—kidney damage with mildly decreased GFR (60–89 mL/min/1.73 m2)
Stage 3—moderately decreased GFR (30–59 mL/min/1.73 m2)
Stage 4—severely decreased GFR (15–29 mL/min/1.73 m2)
Stage 5—kidney failure (GFR < 15 mL/min/1.73 m2 or dialysis)
The answer is murkier in patients on dialysis. Only one study has been done in this population, and it found no benefit from statin therapy. However, we would prescribe a statin for a dialysis patient who had known coronary artery disease and an LDL-C level higher than 100 mg/dL.
RATIONALE FOR STATIN USE: KIDNEY PATIENTS ARE AT RISK
Cardiovascular disease is common among patients with chronic kidney disease. While the risks of cardiovascular disease and death are highest among those requiring dialysis, earlier stages of chronic kidney disease also are associated with cardiovascular disease.2–4
The prevalence of traditional risk factors, particularly diabetes and hypertension, is high in all stages of kidney disease, and dyslipidemia is extremely common. Patients with chronic kidney disease who are not on dialysis tend to have lower levels of high-density lipoprotein cholesterol and higher levels of triglycerides, lipoprotein remnants, lipoprotein(a), and LDL-C. The lipid profile of dialysis patients is more complex, as malnutrition and inflammation in this population may lead to low cholesterol levels.
Since statins are effective for primary and secondary prevention of cardiovascular events in those in the general population with high LDL-C,5 we could expect that the same holds true for patients with chronic kidney disease. Furthermore, if kidney disease were considered a coronary heart disease equivalent, more than 85% of those with stage 3, 4, or 5 disease would qualify for lipid-lowering therapy by LDL-C criteria.6
However, compared with the large body of evidence in those without kidney disease, we have few data on the effect of statins on cardiovascular outcomes in those with kidney disease. Five of seven major trials of statins excluded patients with chronic kidney disease by using a creatinine cutoff or by excluding patients with known kidney disease.7
Renoprotective effects
Besides their cardiovascular effects, statins may slow the progression of kidney disease.
A subgroup analysis of the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) trial8 showed a 12% increase in creatinine clearance in the group receiving atorvastatin (Lipitor) (P = .0001). In comparison, creatinine clearance decreased by 4% in the placebo group.
A subgroup analysis of the Cholesterol and Recurrent Events (CARE) trial, a secondary prevention trial of pravastatin (Pravachol) vs placebo, showed a similar effect for patients with a glomerular filtration rate (GFR) less than 60 mL/min/1.73 m2 at baseline.9
A meta-analysis of 27 randomized trials (39,704 participants) concluded that, compared with no treatment, statins slowed the loss of GFR by a mean of 1.22 mL/min/year (95% confidence interval 0.44–2.0).10
Statins may confer this benefit independently of lipid-lowering. These drugs seem to decrease proteinuria, possibly by improving endothelial function or decreasing inflammation.11 A meta-analysis (1,384 patients) noted that 13 of 15 published studies found an antiproteinuric effect, with a greater effect in those with greater baseline proteinuria.12
The Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients With Progressive Renal Disease Trial (PLANET) will enroll 345 diabetic patients with protein-uria and hypercholesterolemia and examine the effects of rosuvastatin (Crestor) and atorvastatin on proteinuria and GFR.13
Cardioprotective effects in stages 1–4
Since patients with chronic kidney disease were excluded from most of the major statin trials, the best evidence in those with non-dialysis-dependent disease comes from post hoc analysis of data from the CARE study.14 While this trial excluded patients with more than 2+ proteinuria on dipstick analysis and those with creatinine values greater than 1.5 times the upper limit of normal, 1,711 of the initial 4,159 patients had a creatinine clearance of less than 75 mL/min; the mean creatinine clearance in this subgroup was 61. In this subgroup, pravastatin therapy was associated with a significantly lower risk of cardiovascular death or recurrent nonfatal myocardial infarction (MI) (hazard ratio 0.72, P < 0.05).
Similarly, in the 4,491 patients with chronic kidney disease (mean GFR 55 mL/min/1.73 m2) in the Pravastatin Pooling Project, the hazard of new MI, cardiovascular death, or cardiac intervention was nearly 25% lower in the pravastatin group.15
The ongoing Study of Heart and Renal Protection (SHARP),16 a randomized trial of ezetimibe/simvastatin (Vytorin) that enrolled 6,000 people with stages 3 to 4 kidney disease and 3,000 dialysis patients, will help in determining whether statin therapy prevents new vascular events. The study was launched in 2003 and has now completed enrollment. The primary outcome measure will be the time to first vascular event; secondary analyses will address whether statins decrease proteinuria or slow the progression of kidney disease.
Cardioprotective effects in dialysis patients
The only major randomized trial of statins ever conducted in dialysis patients with diabetes, the German Diabetes and Dialysis Study (4D), did not find atorvastatin 20 mg to have any benefit compared with placebo in reducing a composite end point of death from cardiac causes, stroke, and nonfatal MI over a median of 4 years of follow-up, despite a decrease in LDL-C of over 40% in the treatment group.17 Adverse events were similar in the two groups. The lack of a detectable benefit may be due to differences in the cardiovascular milieu in dialysis patients, who may have more advanced disease, with preexisting cardiac remodeling and congestive heart failure, which may not be modified to the same extent by statin therapy. Alternatively, the dose of atorvastatin may have been too low, or 4 years of treatment may not be sufficient to detect a benefit in these patients.
An ongoing prospective, randomized, placebo-controlled trial in 3,000 hemodialysis patients, called A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Haemodialysis: an Assessment of Survival and Cardiovascular Events (AURORA),18 will help to clarify the role of statins in this population.
CONCLUSION
The National Kidney Foundation guidelines1,19 note that people with chronic kidney disease are at high risk of cardiovascular disease and therefore should be treated according to guidelines for treating traditional risk factors in high-risk groups. We believe that those with dyslipidemia who are in stages 1 through 4, particularly those with other risk factors for coronary heart disease, should receive a statin, with an LDL-C target of less than 100 mg/dL, even though we have few data from large trials focused on this population and even though LDL-C may not be the only reason to consider statin use. The pleiotropic effects of statins on proteinuria and progression of kidney function loss may be of benefit in this population as well, although we would not recommend starting a statin solely for these effects until more data are available.
Despite the negative results of the 4D trial, given the relative safety of statins and the lack of any trial data suggesting harm in patients with chronic kidney disease, in our practice we treat dialysis patients with known cardiovascular disease with a statin, with a target LDL-C level less than 100 mg/dL. In dialysis patients without known cardiovascular disease, the use of a statin is even more controversial, and decisions should be made on an individual basis.
Results from the SHARP, AURORA, and PLANET trials, each of which is focused on patients with chronic kidney disease, will help determine whether statins benefit patients at this stage of disease.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39 suppl 1:S1–S266.
- Shlipak MG, Sarnak MJ, Katz R, et al. Cystatin C and the risk of death and cardiovascular events among elderly persons. N Engl J Med 2005; 352:2049–2060.
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis 1998; 32 suppl 3:S112–S119.
- Manjunath G, Tighiouart H, Coresh J, et al. Level of kidney function as a risk factor for cardiovascular outcomes in the elderly. Kidney Int 2003; 63:1121–1129.
- Baigent C, Keech A, Kearney PM, et al Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Hyre AD, Fox CS, Astor BC, Cohen AJ, Muntner P. The impact of reclassifying moderate CKD as a coronary heart disease risk equivalent on the number of US adults recommended lipid-lowering treatment. Am J Kidney Dis 2007; 49:37–45.
- Coca SG, Krumholz HM, Garg AX, Parikh CR. Underrepresentation of renal disease in randomized controlled trials of cardiovascular disease. JAMA 2006; 296:1377–1384.
- Athyros VG, Mikhailidis DP, Papageorgiou AA, et al. The effect of statins versus untreated dyslipidaemia on renal function in patients with coronary heart disease. A subgroup analysis of the Greek atorvastatin and coronary heart disease evaluation (GREACE) study. J Clin Pathol 2004; 57:728–734.
- Tonelli M, Moye L, Sacks FM, Cole T, Curhan GC Cholesterol and Recurrent Events Trial Investigators. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol 2003; 14:1605–1613.
- Sandhu S, Wiebe N, Fried LF, Tonelli M. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol 2006; 17:2006–2016.
- Balk EM, Lau J, Goudas LC, et al. Effects of statins on nonlipid serum markers associated with cardiovascular disease: a systematic review. Ann Intern Med 2003; 139:670–682.
- Douglas K, O’Malley PG, Jackson JL. Meta-analysis: the effect of statins on albuminuria. Ann Intern Med 2006; 145:117–124.
- US National Institutes of Health. Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients with Progressive Renal Disease (PLANET 1). Accessed December 6, 2007. www.clinicaltrials.gov/ct/show/NCT00296374?order=1.
- Tonelli M, Moye L, Sacks FM, Kiberd B, Curhan G Cholesterol and Recurrent Events (CARE) Trial Investigators. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann Intern Med 2003; 138:98–104.
- Tonelli M, Isles C, Curhan GC, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation 2004; 110:1557–1563.
- Baigent C, Landry M. Study of Heart and Renal Protection (SHARP). Kidney Int Suppl 2003; 84:S207–S210.
- Wanner C, Krane V, Marz W, et al German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238–248.
- Fellstrom BC, Holdaas H, Jardine AG. Why do we need a statin trial in hemodialysis patients? Kidney Int 2003; 84 suppl:S204–S206.
- KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis 2007; 49 suppl 2:S12–154.
We think some patients with chronic kidney disease should take a statin, particularly those in stages 1 through 4 (ie, not yet on dialysis1)* who have low-density lipoprotein cholesterol (LDL-C) levels higher than 100 mg/dL. However, few studies have addressed this question.
*Stages of chronic kidney disease1:
Stage 1—kidney damage with normal or high glomerular filtration rate (GFR ≥ 90 mL/min/1.73 m2)
Stage 2—kidney damage with mildly decreased GFR (60–89 mL/min/1.73 m2)
Stage 3—moderately decreased GFR (30–59 mL/min/1.73 m2)
Stage 4—severely decreased GFR (15–29 mL/min/1.73 m2)
Stage 5—kidney failure (GFR < 15 mL/min/1.73 m2 or dialysis)
The answer is murkier in patients on dialysis. Only one study has been done in this population, and it found no benefit from statin therapy. However, we would prescribe a statin for a dialysis patient who had known coronary artery disease and an LDL-C level higher than 100 mg/dL.
RATIONALE FOR STATIN USE: KIDNEY PATIENTS ARE AT RISK
Cardiovascular disease is common among patients with chronic kidney disease. While the risks of cardiovascular disease and death are highest among those requiring dialysis, earlier stages of chronic kidney disease also are associated with cardiovascular disease.2–4
The prevalence of traditional risk factors, particularly diabetes and hypertension, is high in all stages of kidney disease, and dyslipidemia is extremely common. Patients with chronic kidney disease who are not on dialysis tend to have lower levels of high-density lipoprotein cholesterol and higher levels of triglycerides, lipoprotein remnants, lipoprotein(a), and LDL-C. The lipid profile of dialysis patients is more complex, as malnutrition and inflammation in this population may lead to low cholesterol levels.
Since statins are effective for primary and secondary prevention of cardiovascular events in those in the general population with high LDL-C,5 we could expect that the same holds true for patients with chronic kidney disease. Furthermore, if kidney disease were considered a coronary heart disease equivalent, more than 85% of those with stage 3, 4, or 5 disease would qualify for lipid-lowering therapy by LDL-C criteria.6
However, compared with the large body of evidence in those without kidney disease, we have few data on the effect of statins on cardiovascular outcomes in those with kidney disease. Five of seven major trials of statins excluded patients with chronic kidney disease by using a creatinine cutoff or by excluding patients with known kidney disease.7
Renoprotective effects
Besides their cardiovascular effects, statins may slow the progression of kidney disease.
A subgroup analysis of the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) trial8 showed a 12% increase in creatinine clearance in the group receiving atorvastatin (Lipitor) (P = .0001). In comparison, creatinine clearance decreased by 4% in the placebo group.
A subgroup analysis of the Cholesterol and Recurrent Events (CARE) trial, a secondary prevention trial of pravastatin (Pravachol) vs placebo, showed a similar effect for patients with a glomerular filtration rate (GFR) less than 60 mL/min/1.73 m2 at baseline.9
A meta-analysis of 27 randomized trials (39,704 participants) concluded that, compared with no treatment, statins slowed the loss of GFR by a mean of 1.22 mL/min/year (95% confidence interval 0.44–2.0).10
Statins may confer this benefit independently of lipid-lowering. These drugs seem to decrease proteinuria, possibly by improving endothelial function or decreasing inflammation.11 A meta-analysis (1,384 patients) noted that 13 of 15 published studies found an antiproteinuric effect, with a greater effect in those with greater baseline proteinuria.12
The Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients With Progressive Renal Disease Trial (PLANET) will enroll 345 diabetic patients with protein-uria and hypercholesterolemia and examine the effects of rosuvastatin (Crestor) and atorvastatin on proteinuria and GFR.13
Cardioprotective effects in stages 1–4
Since patients with chronic kidney disease were excluded from most of the major statin trials, the best evidence in those with non-dialysis-dependent disease comes from post hoc analysis of data from the CARE study.14 While this trial excluded patients with more than 2+ proteinuria on dipstick analysis and those with creatinine values greater than 1.5 times the upper limit of normal, 1,711 of the initial 4,159 patients had a creatinine clearance of less than 75 mL/min; the mean creatinine clearance in this subgroup was 61. In this subgroup, pravastatin therapy was associated with a significantly lower risk of cardiovascular death or recurrent nonfatal myocardial infarction (MI) (hazard ratio 0.72, P < 0.05).
Similarly, in the 4,491 patients with chronic kidney disease (mean GFR 55 mL/min/1.73 m2) in the Pravastatin Pooling Project, the hazard of new MI, cardiovascular death, or cardiac intervention was nearly 25% lower in the pravastatin group.15
The ongoing Study of Heart and Renal Protection (SHARP),16 a randomized trial of ezetimibe/simvastatin (Vytorin) that enrolled 6,000 people with stages 3 to 4 kidney disease and 3,000 dialysis patients, will help in determining whether statin therapy prevents new vascular events. The study was launched in 2003 and has now completed enrollment. The primary outcome measure will be the time to first vascular event; secondary analyses will address whether statins decrease proteinuria or slow the progression of kidney disease.
Cardioprotective effects in dialysis patients
The only major randomized trial of statins ever conducted in dialysis patients with diabetes, the German Diabetes and Dialysis Study (4D), did not find atorvastatin 20 mg to have any benefit compared with placebo in reducing a composite end point of death from cardiac causes, stroke, and nonfatal MI over a median of 4 years of follow-up, despite a decrease in LDL-C of over 40% in the treatment group.17 Adverse events were similar in the two groups. The lack of a detectable benefit may be due to differences in the cardiovascular milieu in dialysis patients, who may have more advanced disease, with preexisting cardiac remodeling and congestive heart failure, which may not be modified to the same extent by statin therapy. Alternatively, the dose of atorvastatin may have been too low, or 4 years of treatment may not be sufficient to detect a benefit in these patients.
An ongoing prospective, randomized, placebo-controlled trial in 3,000 hemodialysis patients, called A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Haemodialysis: an Assessment of Survival and Cardiovascular Events (AURORA),18 will help to clarify the role of statins in this population.
CONCLUSION
The National Kidney Foundation guidelines1,19 note that people with chronic kidney disease are at high risk of cardiovascular disease and therefore should be treated according to guidelines for treating traditional risk factors in high-risk groups. We believe that those with dyslipidemia who are in stages 1 through 4, particularly those with other risk factors for coronary heart disease, should receive a statin, with an LDL-C target of less than 100 mg/dL, even though we have few data from large trials focused on this population and even though LDL-C may not be the only reason to consider statin use. The pleiotropic effects of statins on proteinuria and progression of kidney function loss may be of benefit in this population as well, although we would not recommend starting a statin solely for these effects until more data are available.
Despite the negative results of the 4D trial, given the relative safety of statins and the lack of any trial data suggesting harm in patients with chronic kidney disease, in our practice we treat dialysis patients with known cardiovascular disease with a statin, with a target LDL-C level less than 100 mg/dL. In dialysis patients without known cardiovascular disease, the use of a statin is even more controversial, and decisions should be made on an individual basis.
Results from the SHARP, AURORA, and PLANET trials, each of which is focused on patients with chronic kidney disease, will help determine whether statins benefit patients at this stage of disease.
We think some patients with chronic kidney disease should take a statin, particularly those in stages 1 through 4 (ie, not yet on dialysis1)* who have low-density lipoprotein cholesterol (LDL-C) levels higher than 100 mg/dL. However, few studies have addressed this question.
*Stages of chronic kidney disease1:
Stage 1—kidney damage with normal or high glomerular filtration rate (GFR ≥ 90 mL/min/1.73 m2)
Stage 2—kidney damage with mildly decreased GFR (60–89 mL/min/1.73 m2)
Stage 3—moderately decreased GFR (30–59 mL/min/1.73 m2)
Stage 4—severely decreased GFR (15–29 mL/min/1.73 m2)
Stage 5—kidney failure (GFR < 15 mL/min/1.73 m2 or dialysis)
The answer is murkier in patients on dialysis. Only one study has been done in this population, and it found no benefit from statin therapy. However, we would prescribe a statin for a dialysis patient who had known coronary artery disease and an LDL-C level higher than 100 mg/dL.
RATIONALE FOR STATIN USE: KIDNEY PATIENTS ARE AT RISK
Cardiovascular disease is common among patients with chronic kidney disease. While the risks of cardiovascular disease and death are highest among those requiring dialysis, earlier stages of chronic kidney disease also are associated with cardiovascular disease.2–4
The prevalence of traditional risk factors, particularly diabetes and hypertension, is high in all stages of kidney disease, and dyslipidemia is extremely common. Patients with chronic kidney disease who are not on dialysis tend to have lower levels of high-density lipoprotein cholesterol and higher levels of triglycerides, lipoprotein remnants, lipoprotein(a), and LDL-C. The lipid profile of dialysis patients is more complex, as malnutrition and inflammation in this population may lead to low cholesterol levels.
Since statins are effective for primary and secondary prevention of cardiovascular events in those in the general population with high LDL-C,5 we could expect that the same holds true for patients with chronic kidney disease. Furthermore, if kidney disease were considered a coronary heart disease equivalent, more than 85% of those with stage 3, 4, or 5 disease would qualify for lipid-lowering therapy by LDL-C criteria.6
However, compared with the large body of evidence in those without kidney disease, we have few data on the effect of statins on cardiovascular outcomes in those with kidney disease. Five of seven major trials of statins excluded patients with chronic kidney disease by using a creatinine cutoff or by excluding patients with known kidney disease.7
Renoprotective effects
Besides their cardiovascular effects, statins may slow the progression of kidney disease.
A subgroup analysis of the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) trial8 showed a 12% increase in creatinine clearance in the group receiving atorvastatin (Lipitor) (P = .0001). In comparison, creatinine clearance decreased by 4% in the placebo group.
A subgroup analysis of the Cholesterol and Recurrent Events (CARE) trial, a secondary prevention trial of pravastatin (Pravachol) vs placebo, showed a similar effect for patients with a glomerular filtration rate (GFR) less than 60 mL/min/1.73 m2 at baseline.9
A meta-analysis of 27 randomized trials (39,704 participants) concluded that, compared with no treatment, statins slowed the loss of GFR by a mean of 1.22 mL/min/year (95% confidence interval 0.44–2.0).10
Statins may confer this benefit independently of lipid-lowering. These drugs seem to decrease proteinuria, possibly by improving endothelial function or decreasing inflammation.11 A meta-analysis (1,384 patients) noted that 13 of 15 published studies found an antiproteinuric effect, with a greater effect in those with greater baseline proteinuria.12
The Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients With Progressive Renal Disease Trial (PLANET) will enroll 345 diabetic patients with protein-uria and hypercholesterolemia and examine the effects of rosuvastatin (Crestor) and atorvastatin on proteinuria and GFR.13
Cardioprotective effects in stages 1–4
Since patients with chronic kidney disease were excluded from most of the major statin trials, the best evidence in those with non-dialysis-dependent disease comes from post hoc analysis of data from the CARE study.14 While this trial excluded patients with more than 2+ proteinuria on dipstick analysis and those with creatinine values greater than 1.5 times the upper limit of normal, 1,711 of the initial 4,159 patients had a creatinine clearance of less than 75 mL/min; the mean creatinine clearance in this subgroup was 61. In this subgroup, pravastatin therapy was associated with a significantly lower risk of cardiovascular death or recurrent nonfatal myocardial infarction (MI) (hazard ratio 0.72, P < 0.05).
Similarly, in the 4,491 patients with chronic kidney disease (mean GFR 55 mL/min/1.73 m2) in the Pravastatin Pooling Project, the hazard of new MI, cardiovascular death, or cardiac intervention was nearly 25% lower in the pravastatin group.15
The ongoing Study of Heart and Renal Protection (SHARP),16 a randomized trial of ezetimibe/simvastatin (Vytorin) that enrolled 6,000 people with stages 3 to 4 kidney disease and 3,000 dialysis patients, will help in determining whether statin therapy prevents new vascular events. The study was launched in 2003 and has now completed enrollment. The primary outcome measure will be the time to first vascular event; secondary analyses will address whether statins decrease proteinuria or slow the progression of kidney disease.
Cardioprotective effects in dialysis patients
The only major randomized trial of statins ever conducted in dialysis patients with diabetes, the German Diabetes and Dialysis Study (4D), did not find atorvastatin 20 mg to have any benefit compared with placebo in reducing a composite end point of death from cardiac causes, stroke, and nonfatal MI over a median of 4 years of follow-up, despite a decrease in LDL-C of over 40% in the treatment group.17 Adverse events were similar in the two groups. The lack of a detectable benefit may be due to differences in the cardiovascular milieu in dialysis patients, who may have more advanced disease, with preexisting cardiac remodeling and congestive heart failure, which may not be modified to the same extent by statin therapy. Alternatively, the dose of atorvastatin may have been too low, or 4 years of treatment may not be sufficient to detect a benefit in these patients.
An ongoing prospective, randomized, placebo-controlled trial in 3,000 hemodialysis patients, called A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Haemodialysis: an Assessment of Survival and Cardiovascular Events (AURORA),18 will help to clarify the role of statins in this population.
CONCLUSION
The National Kidney Foundation guidelines1,19 note that people with chronic kidney disease are at high risk of cardiovascular disease and therefore should be treated according to guidelines for treating traditional risk factors in high-risk groups. We believe that those with dyslipidemia who are in stages 1 through 4, particularly those with other risk factors for coronary heart disease, should receive a statin, with an LDL-C target of less than 100 mg/dL, even though we have few data from large trials focused on this population and even though LDL-C may not be the only reason to consider statin use. The pleiotropic effects of statins on proteinuria and progression of kidney function loss may be of benefit in this population as well, although we would not recommend starting a statin solely for these effects until more data are available.
Despite the negative results of the 4D trial, given the relative safety of statins and the lack of any trial data suggesting harm in patients with chronic kidney disease, in our practice we treat dialysis patients with known cardiovascular disease with a statin, with a target LDL-C level less than 100 mg/dL. In dialysis patients without known cardiovascular disease, the use of a statin is even more controversial, and decisions should be made on an individual basis.
Results from the SHARP, AURORA, and PLANET trials, each of which is focused on patients with chronic kidney disease, will help determine whether statins benefit patients at this stage of disease.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39 suppl 1:S1–S266.
- Shlipak MG, Sarnak MJ, Katz R, et al. Cystatin C and the risk of death and cardiovascular events among elderly persons. N Engl J Med 2005; 352:2049–2060.
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis 1998; 32 suppl 3:S112–S119.
- Manjunath G, Tighiouart H, Coresh J, et al. Level of kidney function as a risk factor for cardiovascular outcomes in the elderly. Kidney Int 2003; 63:1121–1129.
- Baigent C, Keech A, Kearney PM, et al Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Hyre AD, Fox CS, Astor BC, Cohen AJ, Muntner P. The impact of reclassifying moderate CKD as a coronary heart disease risk equivalent on the number of US adults recommended lipid-lowering treatment. Am J Kidney Dis 2007; 49:37–45.
- Coca SG, Krumholz HM, Garg AX, Parikh CR. Underrepresentation of renal disease in randomized controlled trials of cardiovascular disease. JAMA 2006; 296:1377–1384.
- Athyros VG, Mikhailidis DP, Papageorgiou AA, et al. The effect of statins versus untreated dyslipidaemia on renal function in patients with coronary heart disease. A subgroup analysis of the Greek atorvastatin and coronary heart disease evaluation (GREACE) study. J Clin Pathol 2004; 57:728–734.
- Tonelli M, Moye L, Sacks FM, Cole T, Curhan GC Cholesterol and Recurrent Events Trial Investigators. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol 2003; 14:1605–1613.
- Sandhu S, Wiebe N, Fried LF, Tonelli M. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol 2006; 17:2006–2016.
- Balk EM, Lau J, Goudas LC, et al. Effects of statins on nonlipid serum markers associated with cardiovascular disease: a systematic review. Ann Intern Med 2003; 139:670–682.
- Douglas K, O’Malley PG, Jackson JL. Meta-analysis: the effect of statins on albuminuria. Ann Intern Med 2006; 145:117–124.
- US National Institutes of Health. Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients with Progressive Renal Disease (PLANET 1). Accessed December 6, 2007. www.clinicaltrials.gov/ct/show/NCT00296374?order=1.
- Tonelli M, Moye L, Sacks FM, Kiberd B, Curhan G Cholesterol and Recurrent Events (CARE) Trial Investigators. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann Intern Med 2003; 138:98–104.
- Tonelli M, Isles C, Curhan GC, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation 2004; 110:1557–1563.
- Baigent C, Landry M. Study of Heart and Renal Protection (SHARP). Kidney Int Suppl 2003; 84:S207–S210.
- Wanner C, Krane V, Marz W, et al German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238–248.
- Fellstrom BC, Holdaas H, Jardine AG. Why do we need a statin trial in hemodialysis patients? Kidney Int 2003; 84 suppl:S204–S206.
- KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis 2007; 49 suppl 2:S12–154.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39 suppl 1:S1–S266.
- Shlipak MG, Sarnak MJ, Katz R, et al. Cystatin C and the risk of death and cardiovascular events among elderly persons. N Engl J Med 2005; 352:2049–2060.
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis 1998; 32 suppl 3:S112–S119.
- Manjunath G, Tighiouart H, Coresh J, et al. Level of kidney function as a risk factor for cardiovascular outcomes in the elderly. Kidney Int 2003; 63:1121–1129.
- Baigent C, Keech A, Kearney PM, et al Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Hyre AD, Fox CS, Astor BC, Cohen AJ, Muntner P. The impact of reclassifying moderate CKD as a coronary heart disease risk equivalent on the number of US adults recommended lipid-lowering treatment. Am J Kidney Dis 2007; 49:37–45.
- Coca SG, Krumholz HM, Garg AX, Parikh CR. Underrepresentation of renal disease in randomized controlled trials of cardiovascular disease. JAMA 2006; 296:1377–1384.
- Athyros VG, Mikhailidis DP, Papageorgiou AA, et al. The effect of statins versus untreated dyslipidaemia on renal function in patients with coronary heart disease. A subgroup analysis of the Greek atorvastatin and coronary heart disease evaluation (GREACE) study. J Clin Pathol 2004; 57:728–734.
- Tonelli M, Moye L, Sacks FM, Cole T, Curhan GC Cholesterol and Recurrent Events Trial Investigators. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol 2003; 14:1605–1613.
- Sandhu S, Wiebe N, Fried LF, Tonelli M. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol 2006; 17:2006–2016.
- Balk EM, Lau J, Goudas LC, et al. Effects of statins on nonlipid serum markers associated with cardiovascular disease: a systematic review. Ann Intern Med 2003; 139:670–682.
- Douglas K, O’Malley PG, Jackson JL. Meta-analysis: the effect of statins on albuminuria. Ann Intern Med 2006; 145:117–124.
- US National Institutes of Health. Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients with Progressive Renal Disease (PLANET 1). Accessed December 6, 2007. www.clinicaltrials.gov/ct/show/NCT00296374?order=1.
- Tonelli M, Moye L, Sacks FM, Kiberd B, Curhan G Cholesterol and Recurrent Events (CARE) Trial Investigators. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann Intern Med 2003; 138:98–104.
- Tonelli M, Isles C, Curhan GC, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation 2004; 110:1557–1563.
- Baigent C, Landry M. Study of Heart and Renal Protection (SHARP). Kidney Int Suppl 2003; 84:S207–S210.
- Wanner C, Krane V, Marz W, et al German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238–248.
- Fellstrom BC, Holdaas H, Jardine AG. Why do we need a statin trial in hemodialysis patients? Kidney Int 2003; 84 suppl:S204–S206.
- KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis 2007; 49 suppl 2:S12–154.
When should a methacholine challenge be ordered for a patient with suspected asthma?
The methacholine challenge test isused in several situations:
If the diagnosis of asthma is in question, eg, if the patient has symptoms that suggest asthma (either typical symptoms such as coughing, wheezing, and dyspnea or atypical symptoms) but normal results on regular spirometric testing and no response to a bronchodilator. Because the test has a high negative predictive value, it is more useful in ruling out asthma (if the result is negative) than in ruling it in (if the result is positive).1,2 A negative methacholine challenge test nearly rules out asthma; however, a positive test result needs to be interpreted cautiously if the patient is not experiencing symptoms.
In establishing a diagnosis of occupational asthma. For patients with remitting and relapsing symptoms suggestive of asthma associated with a particular work environment, a detailed history, physical examination, and methacholine challenge test can establish the diagnosis. Specific bronchial challenge testing with the suspected offending agent is possible, although this is more frequently used in research and in situations with significant legal or financial implications for the patient, such as workers’ compensation cases.3
Possibly, in managing asthma. In several clinical trials,4,5 outcomes were better when asthma management decisions were based on airway hyper responsiveness combined with conventional factors (symptoms and lung function) than with management based on conventional factors alone. These findings suggest that asthma management based on serial measurement of airway hyperresponsiveness may be useful in optimizing outcomes of care; however, adjustment in treatment according to response to serial methacholine challenge tests is currently not recommended for routine management of asthma.
In clinical research.
OBSTRUCTION CAN BE IMPROVED OR PROVOKED
Asthma is a chronic inflammatory disorder of the airways associated with characteristic clinical symptoms of wheezing, chest tightness, breathlessness, and cough. These symptoms may be associated with airflow limitation that is at least partially reversible, either spontaneously or with treatment.
Spirometry can confirm the diagnosis of asthma if lung function improves after a bronchodilator is given, as reflected by an increase in forced expiratory volume in 1 second (FEV1) of more than 12% and more than 0.2 L.6,7
Conversely, during bronchoprovocation testing, airflow obstruction is provoked by a stimulus known to elicit airway narrowing, such as inhaled methacholine. Bronchial hyperresponsiveness can reliably distinguish patients with asthma from those without asthma.
HOW THE TEST IS DONE
During the test, the patient inhales methacholine aerosols in increasing concentrations; various protocols can be used. Spirometry is performed before and after each dose, and the results are reported as a percent decrease in FEV1 from baseline for each step of the protocol.
A positive reaction is a 20% fall in FEV1, and the provocative concentration that causes a positive reaction (the PC20) is used to indicate the level of airway hyperresponsiveness. If the FEV1 does not fall by at least 20% with the highest concentration of methacholine, the testis interpreted as negative and the PC20 is reported as “more than 16 mg/mL” or “more than 25 mg/mL,” depending on the highest dose given.
The maximum dose of methacholine varies among pulmonary function testing laboratories and asthma specialists; final doses of 16, 25, and 32 mg/mL are commonly used. Studies have defined a range of 8 to 16 mg/mL as an optimal cutoff point to separate patients with asthma from those without asthma.2,6,7
The response to methacholine can also be expressed in terms of specific airway conductance;however, this is more complicated and requires body plethysmography.
Other stimuli that can be used as bronchoprovocation challenges to diagnose asthma include inhaled histamine, exposure to cold air, or eucapneic hyperventilation.Compared with these alternative stimuli, methacholine is the most feasible as it does not require extensive equipment and is better tolerated than histamine.8
POTENTIAL COMPLICATIONS
Methacholine elicits airway narrowing in susceptible people and can cause severe bronchoconstriction, hyperinflation, or severe coughing. However, this procedure is generally well tolerated, and respiratory symptoms inpatients who react to methacholine typically reverse promptly in response to bronchodilators.
Nevertheless, the test should be performed in a pulmonary function laboratory or doctor’s office with available personnel trained to treat acute bronchospasm and to use resuscitation equipment if needed. Informed consent should be obtained and recorded in the medical record after a detailed explanation of the risks and benefits of this procedure and alternatives to it.
CONTRAINDICATIONS

Baseline obstruction. A ratio of FEV1 to forced vital capacity less than 70% on baseline spirometry defines airway obstruction, and methacholine challenge for diagnostic purposes would not be indicated.
Furthermore, patients with low baseline lung function, who may not be able to compensate for a further decline in lung function due to methacholine-induced bronchospasm, are at increased risk of a serious respiratory reaction. For this reason, an FEV1 less than 50% of predicted or less than 1.0 L is an absolute contraindication to methacholine challenge testing, and an FEV1 less than 60% of predicted or less than 1.5 L must be evaluated on an individual basis.9
Myocardial infarction or stroke within the previous 3 months, uncontrolled hypertension, and aortic or cerebral aneurysm are absolute contraindications to this procedure, since induced bronchospasm may cause ventilation-perfusion mismatching resulting in arterial hypoxemia and compensatory changes in blood pressure, cardiac output, and heart rate. There is no increased risk of cardiac arrhythmia during methacholine challenge.10
Pregnancy is a relative contraindication to methacholine challenge testing; metha- choline is classified in pregnancy category C.
Inability to perform spirometry correctly is also a relative contraindication, and therefore this test is not recommended for preschool-age children.
SOME DRUGS SHOULD BE HELD
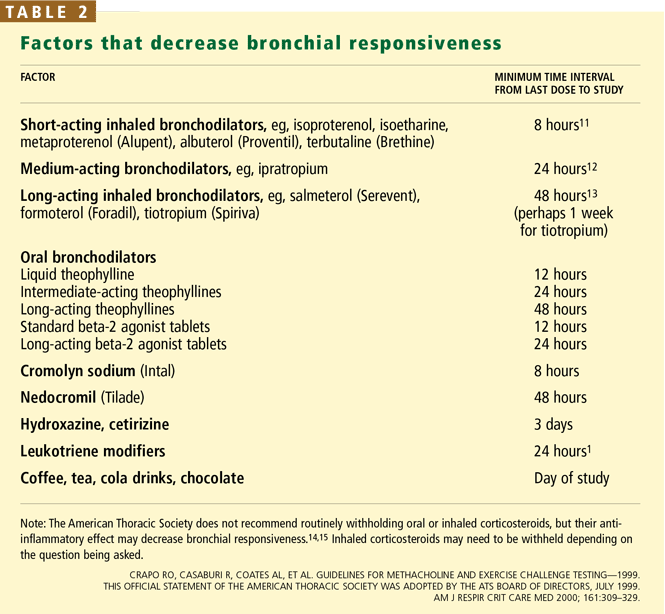
Other factors that can confound the results include smoking,16 respiratory infection, exercise, and consumption of caffeine (coffee, tea, chocolate, or cola drinks) on the day of the test. Airway responsiveness may worsen due to exposure to allergen or upper airway viral infections. Vigorous exercise could induce bronchoconstriction; therefore, performing other bronchial challenge procedures or exercise testing immediately before methacholine challenge may affect the results.17,18
Bronchial hyperresponsiveness is seen in a variety of disorders other than asthma, such as smoking-induced chronic airflow limitation, congestive heart failure, sarcoidosis, cysticfibrosis, and bronchiectasis, as well as in siblings of asthmatics and in people with allergic rhinitis.19 In these situations, the methacholine test can be falsely positive, and one should interpret the results in the context of the clinical history.
- Gilbert R, Auchincloss JH. Post-test probability of asthma following methacholine challenge. Chest 1990; 97:562–565.
- Perpina M, Pellicer C, de Diego A, Compte L, Macian V. Diagnostic value of the bronchial provocation test with methacholine in asthma: a Bayesian analysis approach. Chest 1993; 104:149–154.
- Tan RA, Spector SL. Provocation studies in the diagnosis of occupational asthma. Immunol Allergy Clin North Am 2003; 23:251–267.
- Sont JK, Willems LN, Bel EH, van Krieken JH, Vandenbroucke JP, Sterk PJ. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med 1999; 159:1043–1051.
- Green RH, Brightling CE, McKenna S, et al. Asthma exacerbations and sputum eosinophil counts: a randomized controlled trial. Lancet 2002; 360:1715–1721.
- Crapo RO, Casaburi R, Coates AL, et al. Guidelines for methacholine and exercise challenge testing—1999. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med 2000; 161:309–329.
- Miller MR, Hankinson J, Brusasco V, et al; ATS/ERS Task Force. Standardisation of spirometry. Eur Respir J 2005; 26:319–338.
- Fish JE, Kelly JF. Measurements of responsiveness in bronchoprovocation testing. J Allergy Clin Immunol 1979; 64:592–596.
- Martin RJ, Wanger JS, Irwin CG, Bucher Bartelson B, Cherniac RM. Methacholine challenge testing: safety of low starting FEV1. Asthma Clinical Research Network (ACRN). Chest 1997; 112:53–56.
- Malerba M, Radaeli A, Politi A, Ceriani L, Zulli R, Grassi V. Cardiac arrhythmia monitoring during bronchial provocation test with methacholine. Chest 2003; 124:813–818.
- Cockcroft DW, Swystun VA, Bhagat R. Interaction of inhaled beta 2 agonist and inhaled corticosteroid on airway responsiveness to allergen and methacholine. Am J Respir Crit Care Med 1995; 152:1485–1489.
- Reid JK, Davis BE, Cockcroft DW. The effect of ipratropium nasal spray on bronchial methacholine challenge. Chest 2005; 128:1245–1247.
- O’Connor BJ, Towse LJ, Barnes PJ. Prolonged effect of tiotropium bromide on methacholine-induced bronchoconstriction in asthma. Am J Respir Crit Care Med 1996; 154:876–880.
- Juniper EF, Kline PA, Vanzieleghem MA, Ramsdale EH, O’Byrne PM, Hargreave FE. Effect of long-term treatment with an inhaled corticosteroid (budesonide) on airway hyperresponsiveness and clinical asthma in nonsteroid-dependent asthmatics. Am Rev Respir Dis 1990; 142:832–836.
- Freezer NJ, Croasdell H, Doull IJ, Holgate ST. Effect of regular inhaled beclomethasone on exercise and methacholine airway responses in school children with recurrent wheeze. Eur Respir J 1995; 8:1488–1493.
- Jensen EJ, Dahl R, Steffensen F. Bronchial reactivity to cigarette smoke in smokers: repeatability, relationship to methacholine reactivity, smoking and atopy. Eu rRespir J 1998; 11:670–676.
- Cheung D, Dick EC, Timmers MC, de Klerk EP, Spaan WJ, Sterk PJ. Rhinovirus inhalation causes longlasting excessive airway narrowing in response to methacholine in asthmatic subjects in vivo. Am J Respir Crit Care Med 1995; 152:1490–1496.
- Dinh Xuan AT, Lockart A. Use of non-specific bronchial challenges in the assessment of anti-asthmatic drugs. Eur Respir Rev 1991; 1:19–24.
- Ramsdell JW, Nachtwey FJ, Moser KM. Bronchia lhyperreactivity in chronic obstructive bronchitis. Am Rev Respir Dis 1982; 126:829–832.
The methacholine challenge test isused in several situations:
If the diagnosis of asthma is in question, eg, if the patient has symptoms that suggest asthma (either typical symptoms such as coughing, wheezing, and dyspnea or atypical symptoms) but normal results on regular spirometric testing and no response to a bronchodilator. Because the test has a high negative predictive value, it is more useful in ruling out asthma (if the result is negative) than in ruling it in (if the result is positive).1,2 A negative methacholine challenge test nearly rules out asthma; however, a positive test result needs to be interpreted cautiously if the patient is not experiencing symptoms.
In establishing a diagnosis of occupational asthma. For patients with remitting and relapsing symptoms suggestive of asthma associated with a particular work environment, a detailed history, physical examination, and methacholine challenge test can establish the diagnosis. Specific bronchial challenge testing with the suspected offending agent is possible, although this is more frequently used in research and in situations with significant legal or financial implications for the patient, such as workers’ compensation cases.3
Possibly, in managing asthma. In several clinical trials,4,5 outcomes were better when asthma management decisions were based on airway hyper responsiveness combined with conventional factors (symptoms and lung function) than with management based on conventional factors alone. These findings suggest that asthma management based on serial measurement of airway hyperresponsiveness may be useful in optimizing outcomes of care; however, adjustment in treatment according to response to serial methacholine challenge tests is currently not recommended for routine management of asthma.
In clinical research.
OBSTRUCTION CAN BE IMPROVED OR PROVOKED
Asthma is a chronic inflammatory disorder of the airways associated with characteristic clinical symptoms of wheezing, chest tightness, breathlessness, and cough. These symptoms may be associated with airflow limitation that is at least partially reversible, either spontaneously or with treatment.
Spirometry can confirm the diagnosis of asthma if lung function improves after a bronchodilator is given, as reflected by an increase in forced expiratory volume in 1 second (FEV1) of more than 12% and more than 0.2 L.6,7
Conversely, during bronchoprovocation testing, airflow obstruction is provoked by a stimulus known to elicit airway narrowing, such as inhaled methacholine. Bronchial hyperresponsiveness can reliably distinguish patients with asthma from those without asthma.
HOW THE TEST IS DONE
During the test, the patient inhales methacholine aerosols in increasing concentrations; various protocols can be used. Spirometry is performed before and after each dose, and the results are reported as a percent decrease in FEV1 from baseline for each step of the protocol.
A positive reaction is a 20% fall in FEV1, and the provocative concentration that causes a positive reaction (the PC20) is used to indicate the level of airway hyperresponsiveness. If the FEV1 does not fall by at least 20% with the highest concentration of methacholine, the testis interpreted as negative and the PC20 is reported as “more than 16 mg/mL” or “more than 25 mg/mL,” depending on the highest dose given.
The maximum dose of methacholine varies among pulmonary function testing laboratories and asthma specialists; final doses of 16, 25, and 32 mg/mL are commonly used. Studies have defined a range of 8 to 16 mg/mL as an optimal cutoff point to separate patients with asthma from those without asthma.2,6,7
The response to methacholine can also be expressed in terms of specific airway conductance;however, this is more complicated and requires body plethysmography.
Other stimuli that can be used as bronchoprovocation challenges to diagnose asthma include inhaled histamine, exposure to cold air, or eucapneic hyperventilation.Compared with these alternative stimuli, methacholine is the most feasible as it does not require extensive equipment and is better tolerated than histamine.8
POTENTIAL COMPLICATIONS
Methacholine elicits airway narrowing in susceptible people and can cause severe bronchoconstriction, hyperinflation, or severe coughing. However, this procedure is generally well tolerated, and respiratory symptoms inpatients who react to methacholine typically reverse promptly in response to bronchodilators.
Nevertheless, the test should be performed in a pulmonary function laboratory or doctor’s office with available personnel trained to treat acute bronchospasm and to use resuscitation equipment if needed. Informed consent should be obtained and recorded in the medical record after a detailed explanation of the risks and benefits of this procedure and alternatives to it.
CONTRAINDICATIONS
Baseline obstruction. A ratio of FEV1 to forced vital capacity less than 70% on baseline spirometry defines airway obstruction, and methacholine challenge for diagnostic purposes would not be indicated.
Furthermore, patients with low baseline lung function, who may not be able to compensate for a further decline in lung function due to methacholine-induced bronchospasm, are at increased risk of a serious respiratory reaction. For this reason, an FEV1 less than 50% of predicted or less than 1.0 L is an absolute contraindication to methacholine challenge testing, and an FEV1 less than 60% of predicted or less than 1.5 L must be evaluated on an individual basis.9
Myocardial infarction or stroke within the previous 3 months, uncontrolled hypertension, and aortic or cerebral aneurysm are absolute contraindications to this procedure, since induced bronchospasm may cause ventilation-perfusion mismatching resulting in arterial hypoxemia and compensatory changes in blood pressure, cardiac output, and heart rate. There is no increased risk of cardiac arrhythmia during methacholine challenge.10
Pregnancy is a relative contraindication to methacholine challenge testing; metha- choline is classified in pregnancy category C.
Inability to perform spirometry correctly is also a relative contraindication, and therefore this test is not recommended for preschool-age children.
SOME DRUGS SHOULD BE HELD
Other factors that can confound the results include smoking,16 respiratory infection, exercise, and consumption of caffeine (coffee, tea, chocolate, or cola drinks) on the day of the test. Airway responsiveness may worsen due to exposure to allergen or upper airway viral infections. Vigorous exercise could induce bronchoconstriction; therefore, performing other bronchial challenge procedures or exercise testing immediately before methacholine challenge may affect the results.17,18
Bronchial hyperresponsiveness is seen in a variety of disorders other than asthma, such as smoking-induced chronic airflow limitation, congestive heart failure, sarcoidosis, cysticfibrosis, and bronchiectasis, as well as in siblings of asthmatics and in people with allergic rhinitis.19 In these situations, the methacholine test can be falsely positive, and one should interpret the results in the context of the clinical history.
The methacholine challenge test isused in several situations:
If the diagnosis of asthma is in question, eg, if the patient has symptoms that suggest asthma (either typical symptoms such as coughing, wheezing, and dyspnea or atypical symptoms) but normal results on regular spirometric testing and no response to a bronchodilator. Because the test has a high negative predictive value, it is more useful in ruling out asthma (if the result is negative) than in ruling it in (if the result is positive).1,2 A negative methacholine challenge test nearly rules out asthma; however, a positive test result needs to be interpreted cautiously if the patient is not experiencing symptoms.
In establishing a diagnosis of occupational asthma. For patients with remitting and relapsing symptoms suggestive of asthma associated with a particular work environment, a detailed history, physical examination, and methacholine challenge test can establish the diagnosis. Specific bronchial challenge testing with the suspected offending agent is possible, although this is more frequently used in research and in situations with significant legal or financial implications for the patient, such as workers’ compensation cases.3
Possibly, in managing asthma. In several clinical trials,4,5 outcomes were better when asthma management decisions were based on airway hyper responsiveness combined with conventional factors (symptoms and lung function) than with management based on conventional factors alone. These findings suggest that asthma management based on serial measurement of airway hyperresponsiveness may be useful in optimizing outcomes of care; however, adjustment in treatment according to response to serial methacholine challenge tests is currently not recommended for routine management of asthma.
In clinical research.
OBSTRUCTION CAN BE IMPROVED OR PROVOKED
Asthma is a chronic inflammatory disorder of the airways associated with characteristic clinical symptoms of wheezing, chest tightness, breathlessness, and cough. These symptoms may be associated with airflow limitation that is at least partially reversible, either spontaneously or with treatment.
Spirometry can confirm the diagnosis of asthma if lung function improves after a bronchodilator is given, as reflected by an increase in forced expiratory volume in 1 second (FEV1) of more than 12% and more than 0.2 L.6,7
Conversely, during bronchoprovocation testing, airflow obstruction is provoked by a stimulus known to elicit airway narrowing, such as inhaled methacholine. Bronchial hyperresponsiveness can reliably distinguish patients with asthma from those without asthma.
HOW THE TEST IS DONE
During the test, the patient inhales methacholine aerosols in increasing concentrations; various protocols can be used. Spirometry is performed before and after each dose, and the results are reported as a percent decrease in FEV1 from baseline for each step of the protocol.
A positive reaction is a 20% fall in FEV1, and the provocative concentration that causes a positive reaction (the PC20) is used to indicate the level of airway hyperresponsiveness. If the FEV1 does not fall by at least 20% with the highest concentration of methacholine, the testis interpreted as negative and the PC20 is reported as “more than 16 mg/mL” or “more than 25 mg/mL,” depending on the highest dose given.
The maximum dose of methacholine varies among pulmonary function testing laboratories and asthma specialists; final doses of 16, 25, and 32 mg/mL are commonly used. Studies have defined a range of 8 to 16 mg/mL as an optimal cutoff point to separate patients with asthma from those without asthma.2,6,7
The response to methacholine can also be expressed in terms of specific airway conductance;however, this is more complicated and requires body plethysmography.
Other stimuli that can be used as bronchoprovocation challenges to diagnose asthma include inhaled histamine, exposure to cold air, or eucapneic hyperventilation.Compared with these alternative stimuli, methacholine is the most feasible as it does not require extensive equipment and is better tolerated than histamine.8
POTENTIAL COMPLICATIONS
Methacholine elicits airway narrowing in susceptible people and can cause severe bronchoconstriction, hyperinflation, or severe coughing. However, this procedure is generally well tolerated, and respiratory symptoms inpatients who react to methacholine typically reverse promptly in response to bronchodilators.
Nevertheless, the test should be performed in a pulmonary function laboratory or doctor’s office with available personnel trained to treat acute bronchospasm and to use resuscitation equipment if needed. Informed consent should be obtained and recorded in the medical record after a detailed explanation of the risks and benefits of this procedure and alternatives to it.
CONTRAINDICATIONS
Baseline obstruction. A ratio of FEV1 to forced vital capacity less than 70% on baseline spirometry defines airway obstruction, and methacholine challenge for diagnostic purposes would not be indicated.
Furthermore, patients with low baseline lung function, who may not be able to compensate for a further decline in lung function due to methacholine-induced bronchospasm, are at increased risk of a serious respiratory reaction. For this reason, an FEV1 less than 50% of predicted or less than 1.0 L is an absolute contraindication to methacholine challenge testing, and an FEV1 less than 60% of predicted or less than 1.5 L must be evaluated on an individual basis.9
Myocardial infarction or stroke within the previous 3 months, uncontrolled hypertension, and aortic or cerebral aneurysm are absolute contraindications to this procedure, since induced bronchospasm may cause ventilation-perfusion mismatching resulting in arterial hypoxemia and compensatory changes in blood pressure, cardiac output, and heart rate. There is no increased risk of cardiac arrhythmia during methacholine challenge.10
Pregnancy is a relative contraindication to methacholine challenge testing; metha- choline is classified in pregnancy category C.
Inability to perform spirometry correctly is also a relative contraindication, and therefore this test is not recommended for preschool-age children.
SOME DRUGS SHOULD BE HELD
Other factors that can confound the results include smoking,16 respiratory infection, exercise, and consumption of caffeine (coffee, tea, chocolate, or cola drinks) on the day of the test. Airway responsiveness may worsen due to exposure to allergen or upper airway viral infections. Vigorous exercise could induce bronchoconstriction; therefore, performing other bronchial challenge procedures or exercise testing immediately before methacholine challenge may affect the results.17,18
Bronchial hyperresponsiveness is seen in a variety of disorders other than asthma, such as smoking-induced chronic airflow limitation, congestive heart failure, sarcoidosis, cysticfibrosis, and bronchiectasis, as well as in siblings of asthmatics and in people with allergic rhinitis.19 In these situations, the methacholine test can be falsely positive, and one should interpret the results in the context of the clinical history.
- Gilbert R, Auchincloss JH. Post-test probability of asthma following methacholine challenge. Chest 1990; 97:562–565.
- Perpina M, Pellicer C, de Diego A, Compte L, Macian V. Diagnostic value of the bronchial provocation test with methacholine in asthma: a Bayesian analysis approach. Chest 1993; 104:149–154.
- Tan RA, Spector SL. Provocation studies in the diagnosis of occupational asthma. Immunol Allergy Clin North Am 2003; 23:251–267.
- Sont JK, Willems LN, Bel EH, van Krieken JH, Vandenbroucke JP, Sterk PJ. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med 1999; 159:1043–1051.
- Green RH, Brightling CE, McKenna S, et al. Asthma exacerbations and sputum eosinophil counts: a randomized controlled trial. Lancet 2002; 360:1715–1721.
- Crapo RO, Casaburi R, Coates AL, et al. Guidelines for methacholine and exercise challenge testing—1999. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med 2000; 161:309–329.
- Miller MR, Hankinson J, Brusasco V, et al; ATS/ERS Task Force. Standardisation of spirometry. Eur Respir J 2005; 26:319–338.
- Fish JE, Kelly JF. Measurements of responsiveness in bronchoprovocation testing. J Allergy Clin Immunol 1979; 64:592–596.
- Martin RJ, Wanger JS, Irwin CG, Bucher Bartelson B, Cherniac RM. Methacholine challenge testing: safety of low starting FEV1. Asthma Clinical Research Network (ACRN). Chest 1997; 112:53–56.
- Malerba M, Radaeli A, Politi A, Ceriani L, Zulli R, Grassi V. Cardiac arrhythmia monitoring during bronchial provocation test with methacholine. Chest 2003; 124:813–818.
- Cockcroft DW, Swystun VA, Bhagat R. Interaction of inhaled beta 2 agonist and inhaled corticosteroid on airway responsiveness to allergen and methacholine. Am J Respir Crit Care Med 1995; 152:1485–1489.
- Reid JK, Davis BE, Cockcroft DW. The effect of ipratropium nasal spray on bronchial methacholine challenge. Chest 2005; 128:1245–1247.
- O’Connor BJ, Towse LJ, Barnes PJ. Prolonged effect of tiotropium bromide on methacholine-induced bronchoconstriction in asthma. Am J Respir Crit Care Med 1996; 154:876–880.
- Juniper EF, Kline PA, Vanzieleghem MA, Ramsdale EH, O’Byrne PM, Hargreave FE. Effect of long-term treatment with an inhaled corticosteroid (budesonide) on airway hyperresponsiveness and clinical asthma in nonsteroid-dependent asthmatics. Am Rev Respir Dis 1990; 142:832–836.
- Freezer NJ, Croasdell H, Doull IJ, Holgate ST. Effect of regular inhaled beclomethasone on exercise and methacholine airway responses in school children with recurrent wheeze. Eur Respir J 1995; 8:1488–1493.
- Jensen EJ, Dahl R, Steffensen F. Bronchial reactivity to cigarette smoke in smokers: repeatability, relationship to methacholine reactivity, smoking and atopy. Eu rRespir J 1998; 11:670–676.
- Cheung D, Dick EC, Timmers MC, de Klerk EP, Spaan WJ, Sterk PJ. Rhinovirus inhalation causes longlasting excessive airway narrowing in response to methacholine in asthmatic subjects in vivo. Am J Respir Crit Care Med 1995; 152:1490–1496.
- Dinh Xuan AT, Lockart A. Use of non-specific bronchial challenges in the assessment of anti-asthmatic drugs. Eur Respir Rev 1991; 1:19–24.
- Ramsdell JW, Nachtwey FJ, Moser KM. Bronchia lhyperreactivity in chronic obstructive bronchitis. Am Rev Respir Dis 1982; 126:829–832.
- Gilbert R, Auchincloss JH. Post-test probability of asthma following methacholine challenge. Chest 1990; 97:562–565.
- Perpina M, Pellicer C, de Diego A, Compte L, Macian V. Diagnostic value of the bronchial provocation test with methacholine in asthma: a Bayesian analysis approach. Chest 1993; 104:149–154.
- Tan RA, Spector SL. Provocation studies in the diagnosis of occupational asthma. Immunol Allergy Clin North Am 2003; 23:251–267.
- Sont JK, Willems LN, Bel EH, van Krieken JH, Vandenbroucke JP, Sterk PJ. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med 1999; 159:1043–1051.
- Green RH, Brightling CE, McKenna S, et al. Asthma exacerbations and sputum eosinophil counts: a randomized controlled trial. Lancet 2002; 360:1715–1721.
- Crapo RO, Casaburi R, Coates AL, et al. Guidelines for methacholine and exercise challenge testing—1999. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med 2000; 161:309–329.
- Miller MR, Hankinson J, Brusasco V, et al; ATS/ERS Task Force. Standardisation of spirometry. Eur Respir J 2005; 26:319–338.
- Fish JE, Kelly JF. Measurements of responsiveness in bronchoprovocation testing. J Allergy Clin Immunol 1979; 64:592–596.
- Martin RJ, Wanger JS, Irwin CG, Bucher Bartelson B, Cherniac RM. Methacholine challenge testing: safety of low starting FEV1. Asthma Clinical Research Network (ACRN). Chest 1997; 112:53–56.
- Malerba M, Radaeli A, Politi A, Ceriani L, Zulli R, Grassi V. Cardiac arrhythmia monitoring during bronchial provocation test with methacholine. Chest 2003; 124:813–818.
- Cockcroft DW, Swystun VA, Bhagat R. Interaction of inhaled beta 2 agonist and inhaled corticosteroid on airway responsiveness to allergen and methacholine. Am J Respir Crit Care Med 1995; 152:1485–1489.
- Reid JK, Davis BE, Cockcroft DW. The effect of ipratropium nasal spray on bronchial methacholine challenge. Chest 2005; 128:1245–1247.
- O’Connor BJ, Towse LJ, Barnes PJ. Prolonged effect of tiotropium bromide on methacholine-induced bronchoconstriction in asthma. Am J Respir Crit Care Med 1996; 154:876–880.
- Juniper EF, Kline PA, Vanzieleghem MA, Ramsdale EH, O’Byrne PM, Hargreave FE. Effect of long-term treatment with an inhaled corticosteroid (budesonide) on airway hyperresponsiveness and clinical asthma in nonsteroid-dependent asthmatics. Am Rev Respir Dis 1990; 142:832–836.
- Freezer NJ, Croasdell H, Doull IJ, Holgate ST. Effect of regular inhaled beclomethasone on exercise and methacholine airway responses in school children with recurrent wheeze. Eur Respir J 1995; 8:1488–1493.
- Jensen EJ, Dahl R, Steffensen F. Bronchial reactivity to cigarette smoke in smokers: repeatability, relationship to methacholine reactivity, smoking and atopy. Eu rRespir J 1998; 11:670–676.
- Cheung D, Dick EC, Timmers MC, de Klerk EP, Spaan WJ, Sterk PJ. Rhinovirus inhalation causes longlasting excessive airway narrowing in response to methacholine in asthmatic subjects in vivo. Am J Respir Crit Care Med 1995; 152:1490–1496.
- Dinh Xuan AT, Lockart A. Use of non-specific bronchial challenges in the assessment of anti-asthmatic drugs. Eur Respir Rev 1991; 1:19–24.
- Ramsdell JW, Nachtwey FJ, Moser KM. Bronchia lhyperreactivity in chronic obstructive bronchitis. Am Rev Respir Dis 1982; 126:829–832.