User login
Locus Minoris Resistentiae: Mycobacterium chelonae in Striae Distensae
To the Editor:
Immunosuppressed patients are at particular risk for disseminated mycobacterial infections. A locus minoris resistentiae offers less resistance to the infectious spread of these microorganisms. We present a case of Mycobacterium chelonae infection preferentially involving striae distensae.
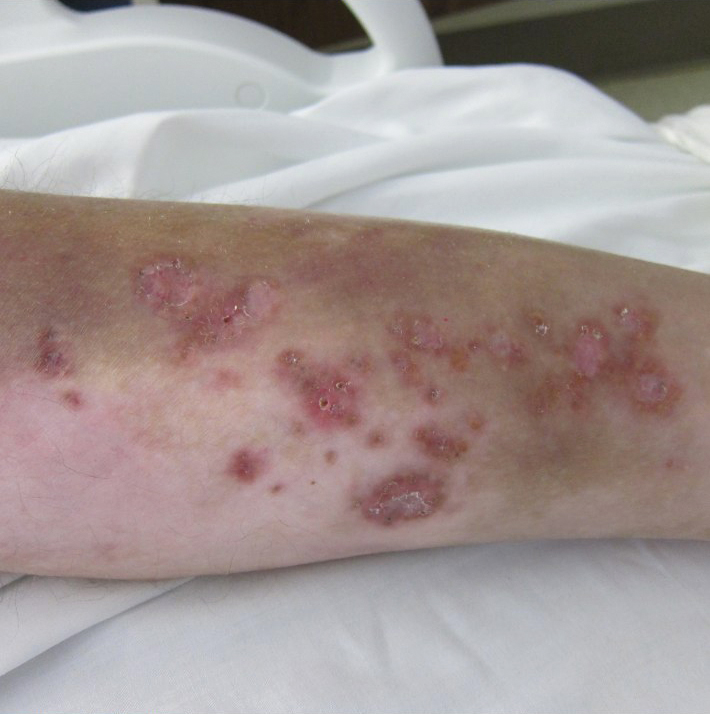
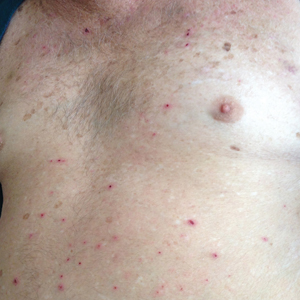
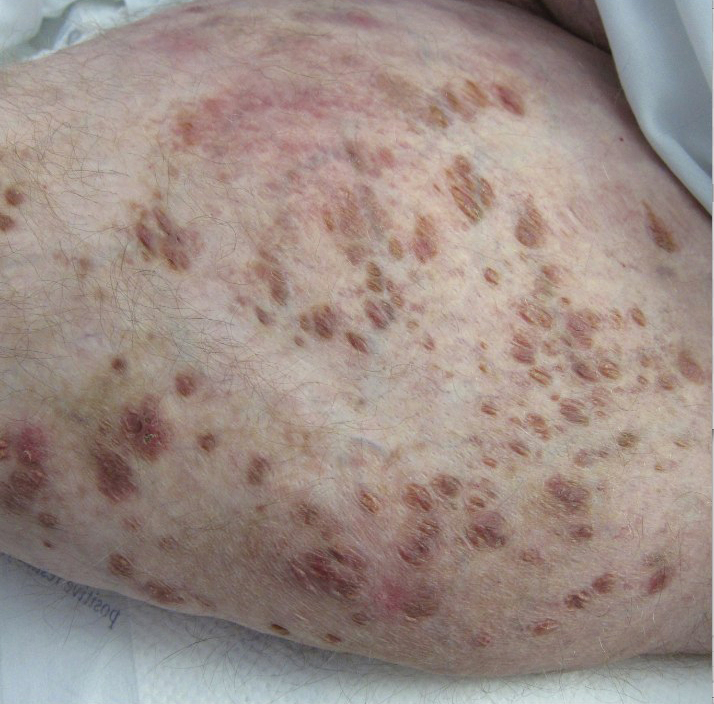
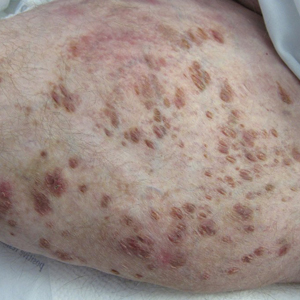
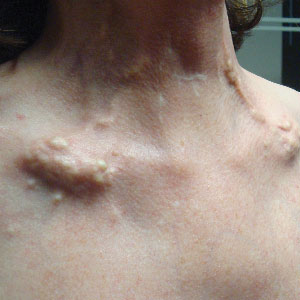
A 30-year-old man with chronic eosinophilic pneumonia requiring high-dose corticosteroid therapy presented with widespread skin lesions. He reported no history of cutaneous trauma or aquatic activities. Physical examination revealed the patient was markedly cushingoid with generalized cutaneous atrophy and widespread striae. Multiple erythematous papules surrounded a large ulceration on the dorsal aspect of the left hand. Depressed erythematous plaques and several small crusted erosions extended up the left lower leg (Figure 1) to the knee. Strikingly, numerous brown and pink papules and small plaques on the left thigh were primarily confined within striae (Figure 2).
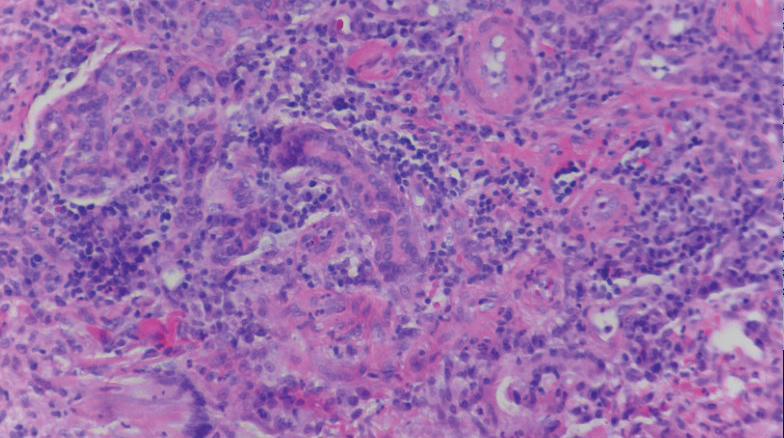
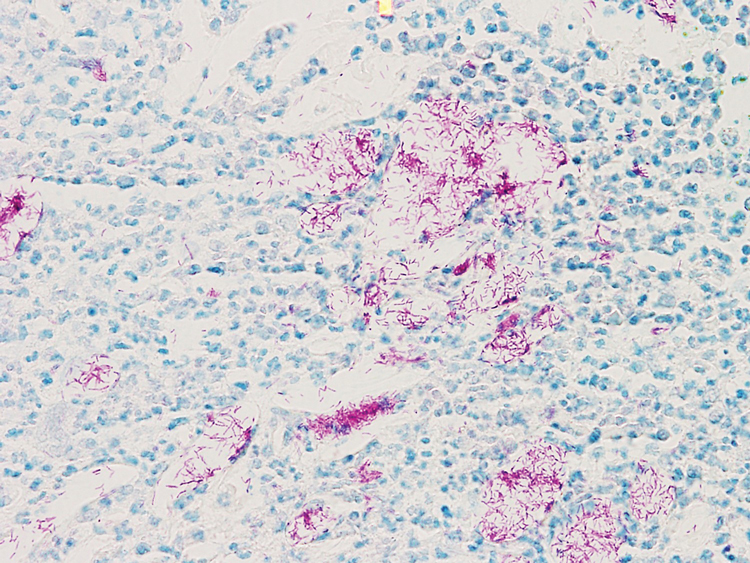
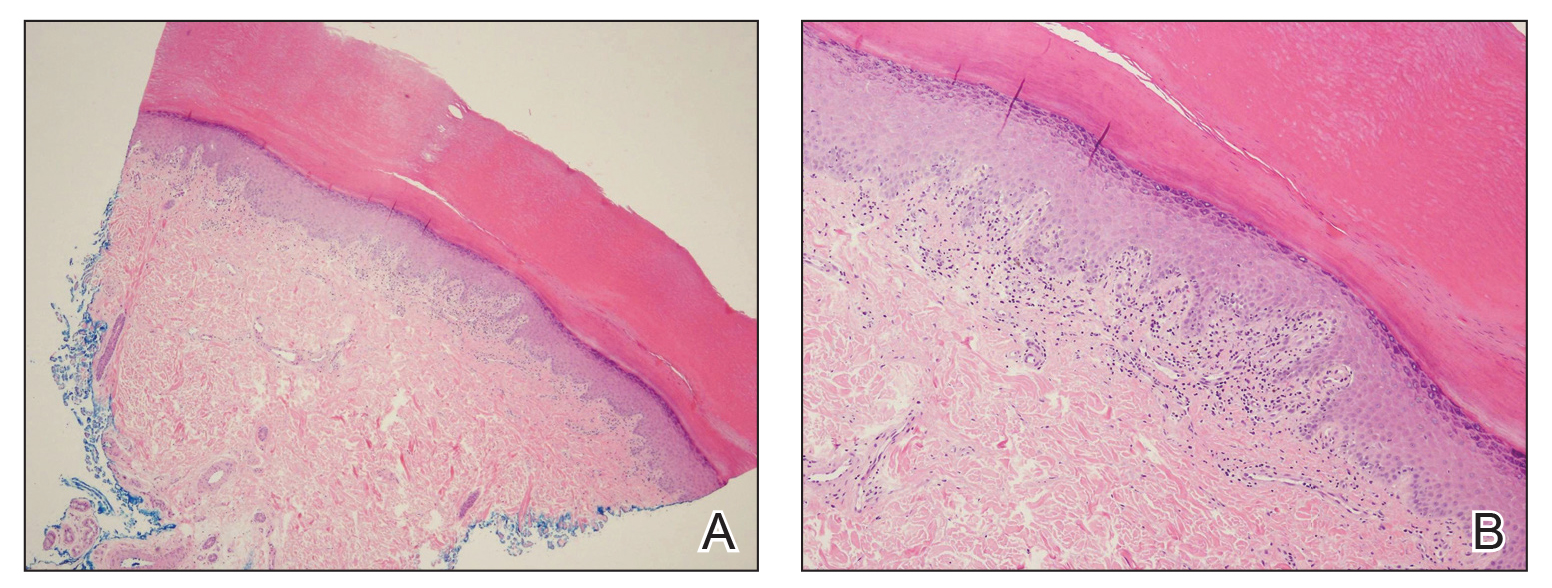
A biopsy of the left thigh revealed granulomatous inflammation (Figure 3) with numerous acid-fast bacilli (Figure 4). Broad-spectrum coverage for fast-growing acid-fast bacilli with amikacin, ceftriaxone, levofloxacin, and clarithromycin was initiated with steady improvement of the eruption. Tissue culture subsequently grew Mycobacterium abscessus-chelonae, and therapy was narrowed to clarithromycin and moxifloxacin.
Mycobacterium chelonae is a rapidly growing mycobacteria isolated from soil and water worldwide, and human skin is a commensal organism. Cutaneous infections have been associated with traumatic injury, tattooing, surgery, cosmetic procedures, vascular access sites, and acupuncture.1 Most cases of cutaneous M chelonae infection begin as a violaceous nodule. Over weeks to months, the localized infection progresses to multiple papules, nodules, draining abscesses, or ulcers. Infections tend to disseminate in immunosuppressed patients, and granulomatous inflammation may not be seen.1
Atypical mycobacterial infection occurring within striae distensae is an example of locus minoris resistentiae—a place of less resistance. Wolf et al2 hypothesized that locus minoris resistentiae could explain the occurrence of an isotopic response or the occurrence of a new skin disorder at the site of a previously healed skin condition. They suggested that the same site could be affected by 2 unrelated diseases at different times due to an inherited or acquired susceptibility in the area.2 Herpes zoster serves as a primary example of Wolf phenomenon, as numerous conditions including granuloma annulare, pseudolymphoma, Bowen disease, and acne have reportedly emerged in its wake.3
Although locus minoris resistentiae does not specifically involve traumatized skin, it must be distinguished from the Koebner phenomenon, characterized by the appearance of isomorphic lesions in areas of otherwise healthy skin subjected to cutaneous injury, as well as the pseudo-Koebner phenomenon, a similar process involving infectious agents.3
In our patient, striae distensae represented areas of increased predisposition to infection. The catabolic effect of high corticosteroid levels on fibroblast activity decreased collagen deposition in the dermal matrix, leading to the formation of linear bands of atrophic skin.4 The elastic fiber network in striae distensae is reduced and reorganized compared to normal skin, in which an intertwining elastic system forms a continuum from the dermoepidermal junction to the deep dermis.5 The number of vertically oriented fibrillin microfibrils subjacent to the dermoepidermal junction and elastin fibers in the papillary dermis is comparatively diminished such that the elastin and fibrillin fibers in the deep dermis run more horizontally compared to normal skin.4 Consequently, collagen alignment in striae distensae demonstrates more anisotropy, or directionally dependent variability, and the dermal matrix is looser and more floccular than the surrounding skin.6 These alterations of dermal architecture likely provide a mechanical advantage for intradermal spread of M chelonae within striae.
Other dermatoses have been observed to occur within striae distensae, specifically leukemia cutis, urticarial vasculitis, lupus erythematosus, keloids, linear focal elastosis, chronic graft-vs-host disease, psoriasis, gestational pemphigoid, and vitiligo.7 Given the dissimilarities of these conditions, the distinctive milieu of striae must provide an invitation—a locus minoris resistentiae—for secondary pathology.
Chronic corticosteroid use leads to both immunosuppression and striae distensae, effectively creating a perfect storm for an atypical mycobacterial skin infection demonstrating locus minoris resistentiae. The immunosuppressed state makes patients more susceptible to infection, and striae distensae may serve as a conduit for the offending organisms.
Acknowledgments—We are indebted to Letty Peterson, MD (Vidalia, Georgia), for her referral of this case, and to Stephen Mullins, MD (Augusta, Georgia), for his dermatopathology services.
- Hay RJ. Mycobacterium chelonae—a growing problem in soft tissue infection. Cur Opin Infect Dis. 2009;22:99-101.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Medeiros do Santos Camargo C, Brotas AM, Ramos-e-Silva M, et al. Isomorphic phenomenon of Koebner: facts and controversies. Clin Dermatol. 2013;31:741-749.
- Watson REB, Parry EJ, Humphries JD, et al. Fibrillin microfibrils are reduced in skin exhibiting striae distensae. Br J Dermatol. 1998;138:931-397.
- Bertin C, A Lopes-DaCunha, Nkengne A, et al. Striae distensae are characterized by distinct microstructural features as measured by non-invasive methods in vivo. Skin Res Technol. 2014;20:81-86.
- Elsaie ML, Baumann LS, Elsaaiee LT. Striae distensae (stretch marks) and different modalities of therapy: an update. Dermatol Surg. 2009;35:563-573.
- Liu CI, Hsu CH. Leukemia cutis at the site of striae distensae: an isotopic response? Int J Dermatol. 1995;34:341-348.
To the Editor:
Immunosuppressed patients are at particular risk for disseminated mycobacterial infections. A locus minoris resistentiae offers less resistance to the infectious spread of these microorganisms. We present a case of Mycobacterium chelonae infection preferentially involving striae distensae.
A 30-year-old man with chronic eosinophilic pneumonia requiring high-dose corticosteroid therapy presented with widespread skin lesions. He reported no history of cutaneous trauma or aquatic activities. Physical examination revealed the patient was markedly cushingoid with generalized cutaneous atrophy and widespread striae. Multiple erythematous papules surrounded a large ulceration on the dorsal aspect of the left hand. Depressed erythematous plaques and several small crusted erosions extended up the left lower leg (Figure 1) to the knee. Strikingly, numerous brown and pink papules and small plaques on the left thigh were primarily confined within striae (Figure 2).
A biopsy of the left thigh revealed granulomatous inflammation (Figure 3) with numerous acid-fast bacilli (Figure 4). Broad-spectrum coverage for fast-growing acid-fast bacilli with amikacin, ceftriaxone, levofloxacin, and clarithromycin was initiated with steady improvement of the eruption. Tissue culture subsequently grew Mycobacterium abscessus-chelonae, and therapy was narrowed to clarithromycin and moxifloxacin.
Mycobacterium chelonae is a rapidly growing mycobacteria isolated from soil and water worldwide, and human skin is a commensal organism. Cutaneous infections have been associated with traumatic injury, tattooing, surgery, cosmetic procedures, vascular access sites, and acupuncture.1 Most cases of cutaneous M chelonae infection begin as a violaceous nodule. Over weeks to months, the localized infection progresses to multiple papules, nodules, draining abscesses, or ulcers. Infections tend to disseminate in immunosuppressed patients, and granulomatous inflammation may not be seen.1
Atypical mycobacterial infection occurring within striae distensae is an example of locus minoris resistentiae—a place of less resistance. Wolf et al2 hypothesized that locus minoris resistentiae could explain the occurrence of an isotopic response or the occurrence of a new skin disorder at the site of a previously healed skin condition. They suggested that the same site could be affected by 2 unrelated diseases at different times due to an inherited or acquired susceptibility in the area.2 Herpes zoster serves as a primary example of Wolf phenomenon, as numerous conditions including granuloma annulare, pseudolymphoma, Bowen disease, and acne have reportedly emerged in its wake.3
Although locus minoris resistentiae does not specifically involve traumatized skin, it must be distinguished from the Koebner phenomenon, characterized by the appearance of isomorphic lesions in areas of otherwise healthy skin subjected to cutaneous injury, as well as the pseudo-Koebner phenomenon, a similar process involving infectious agents.3
In our patient, striae distensae represented areas of increased predisposition to infection. The catabolic effect of high corticosteroid levels on fibroblast activity decreased collagen deposition in the dermal matrix, leading to the formation of linear bands of atrophic skin.4 The elastic fiber network in striae distensae is reduced and reorganized compared to normal skin, in which an intertwining elastic system forms a continuum from the dermoepidermal junction to the deep dermis.5 The number of vertically oriented fibrillin microfibrils subjacent to the dermoepidermal junction and elastin fibers in the papillary dermis is comparatively diminished such that the elastin and fibrillin fibers in the deep dermis run more horizontally compared to normal skin.4 Consequently, collagen alignment in striae distensae demonstrates more anisotropy, or directionally dependent variability, and the dermal matrix is looser and more floccular than the surrounding skin.6 These alterations of dermal architecture likely provide a mechanical advantage for intradermal spread of M chelonae within striae.
Other dermatoses have been observed to occur within striae distensae, specifically leukemia cutis, urticarial vasculitis, lupus erythematosus, keloids, linear focal elastosis, chronic graft-vs-host disease, psoriasis, gestational pemphigoid, and vitiligo.7 Given the dissimilarities of these conditions, the distinctive milieu of striae must provide an invitation—a locus minoris resistentiae—for secondary pathology.
Chronic corticosteroid use leads to both immunosuppression and striae distensae, effectively creating a perfect storm for an atypical mycobacterial skin infection demonstrating locus minoris resistentiae. The immunosuppressed state makes patients more susceptible to infection, and striae distensae may serve as a conduit for the offending organisms.
Acknowledgments—We are indebted to Letty Peterson, MD (Vidalia, Georgia), for her referral of this case, and to Stephen Mullins, MD (Augusta, Georgia), for his dermatopathology services.
To the Editor:
Immunosuppressed patients are at particular risk for disseminated mycobacterial infections. A locus minoris resistentiae offers less resistance to the infectious spread of these microorganisms. We present a case of Mycobacterium chelonae infection preferentially involving striae distensae.
A 30-year-old man with chronic eosinophilic pneumonia requiring high-dose corticosteroid therapy presented with widespread skin lesions. He reported no history of cutaneous trauma or aquatic activities. Physical examination revealed the patient was markedly cushingoid with generalized cutaneous atrophy and widespread striae. Multiple erythematous papules surrounded a large ulceration on the dorsal aspect of the left hand. Depressed erythematous plaques and several small crusted erosions extended up the left lower leg (Figure 1) to the knee. Strikingly, numerous brown and pink papules and small plaques on the left thigh were primarily confined within striae (Figure 2).
A biopsy of the left thigh revealed granulomatous inflammation (Figure 3) with numerous acid-fast bacilli (Figure 4). Broad-spectrum coverage for fast-growing acid-fast bacilli with amikacin, ceftriaxone, levofloxacin, and clarithromycin was initiated with steady improvement of the eruption. Tissue culture subsequently grew Mycobacterium abscessus-chelonae, and therapy was narrowed to clarithromycin and moxifloxacin.
Mycobacterium chelonae is a rapidly growing mycobacteria isolated from soil and water worldwide, and human skin is a commensal organism. Cutaneous infections have been associated with traumatic injury, tattooing, surgery, cosmetic procedures, vascular access sites, and acupuncture.1 Most cases of cutaneous M chelonae infection begin as a violaceous nodule. Over weeks to months, the localized infection progresses to multiple papules, nodules, draining abscesses, or ulcers. Infections tend to disseminate in immunosuppressed patients, and granulomatous inflammation may not be seen.1
Atypical mycobacterial infection occurring within striae distensae is an example of locus minoris resistentiae—a place of less resistance. Wolf et al2 hypothesized that locus minoris resistentiae could explain the occurrence of an isotopic response or the occurrence of a new skin disorder at the site of a previously healed skin condition. They suggested that the same site could be affected by 2 unrelated diseases at different times due to an inherited or acquired susceptibility in the area.2 Herpes zoster serves as a primary example of Wolf phenomenon, as numerous conditions including granuloma annulare, pseudolymphoma, Bowen disease, and acne have reportedly emerged in its wake.3
Although locus minoris resistentiae does not specifically involve traumatized skin, it must be distinguished from the Koebner phenomenon, characterized by the appearance of isomorphic lesions in areas of otherwise healthy skin subjected to cutaneous injury, as well as the pseudo-Koebner phenomenon, a similar process involving infectious agents.3
In our patient, striae distensae represented areas of increased predisposition to infection. The catabolic effect of high corticosteroid levels on fibroblast activity decreased collagen deposition in the dermal matrix, leading to the formation of linear bands of atrophic skin.4 The elastic fiber network in striae distensae is reduced and reorganized compared to normal skin, in which an intertwining elastic system forms a continuum from the dermoepidermal junction to the deep dermis.5 The number of vertically oriented fibrillin microfibrils subjacent to the dermoepidermal junction and elastin fibers in the papillary dermis is comparatively diminished such that the elastin and fibrillin fibers in the deep dermis run more horizontally compared to normal skin.4 Consequently, collagen alignment in striae distensae demonstrates more anisotropy, or directionally dependent variability, and the dermal matrix is looser and more floccular than the surrounding skin.6 These alterations of dermal architecture likely provide a mechanical advantage for intradermal spread of M chelonae within striae.
Other dermatoses have been observed to occur within striae distensae, specifically leukemia cutis, urticarial vasculitis, lupus erythematosus, keloids, linear focal elastosis, chronic graft-vs-host disease, psoriasis, gestational pemphigoid, and vitiligo.7 Given the dissimilarities of these conditions, the distinctive milieu of striae must provide an invitation—a locus minoris resistentiae—for secondary pathology.
Chronic corticosteroid use leads to both immunosuppression and striae distensae, effectively creating a perfect storm for an atypical mycobacterial skin infection demonstrating locus minoris resistentiae. The immunosuppressed state makes patients more susceptible to infection, and striae distensae may serve as a conduit for the offending organisms.
Acknowledgments—We are indebted to Letty Peterson, MD (Vidalia, Georgia), for her referral of this case, and to Stephen Mullins, MD (Augusta, Georgia), for his dermatopathology services.
- Hay RJ. Mycobacterium chelonae—a growing problem in soft tissue infection. Cur Opin Infect Dis. 2009;22:99-101.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Medeiros do Santos Camargo C, Brotas AM, Ramos-e-Silva M, et al. Isomorphic phenomenon of Koebner: facts and controversies. Clin Dermatol. 2013;31:741-749.
- Watson REB, Parry EJ, Humphries JD, et al. Fibrillin microfibrils are reduced in skin exhibiting striae distensae. Br J Dermatol. 1998;138:931-397.
- Bertin C, A Lopes-DaCunha, Nkengne A, et al. Striae distensae are characterized by distinct microstructural features as measured by non-invasive methods in vivo. Skin Res Technol. 2014;20:81-86.
- Elsaie ML, Baumann LS, Elsaaiee LT. Striae distensae (stretch marks) and different modalities of therapy: an update. Dermatol Surg. 2009;35:563-573.
- Liu CI, Hsu CH. Leukemia cutis at the site of striae distensae: an isotopic response? Int J Dermatol. 1995;34:341-348.
- Hay RJ. Mycobacterium chelonae—a growing problem in soft tissue infection. Cur Opin Infect Dis. 2009;22:99-101.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Medeiros do Santos Camargo C, Brotas AM, Ramos-e-Silva M, et al. Isomorphic phenomenon of Koebner: facts and controversies. Clin Dermatol. 2013;31:741-749.
- Watson REB, Parry EJ, Humphries JD, et al. Fibrillin microfibrils are reduced in skin exhibiting striae distensae. Br J Dermatol. 1998;138:931-397.
- Bertin C, A Lopes-DaCunha, Nkengne A, et al. Striae distensae are characterized by distinct microstructural features as measured by non-invasive methods in vivo. Skin Res Technol. 2014;20:81-86.
- Elsaie ML, Baumann LS, Elsaaiee LT. Striae distensae (stretch marks) and different modalities of therapy: an update. Dermatol Surg. 2009;35:563-573.
- Liu CI, Hsu CH. Leukemia cutis at the site of striae distensae: an isotopic response? Int J Dermatol. 1995;34:341-348.
Practice Points
- Striae distensae, seen frequently in the setting of chronic corticosteroid use, are at an increased risk for localized infection, particularly in immunocompromised patients. There should be a low threshold to biopsy striae distensae that demonstrate morphologic evolution.
- The Koebner reaction, also known as an isomorphic response, refers to the appearance of certain dermatoses in previously healthy skin subjected to cutaneous injury.
- Locus minoris resistentiae is an isotropic response that characterizes the presentation of a new dermatosis within an area previously affected by an unrelated skin condition that has healed.
Dystrophic Calcinosis Cutis: Treatment With Intravenous Sodium Thiosulfate
To the Editor:
Severe dystrophic calcinosis cutis is a debilitating disease with no universally accepted therapeutic options. This case demonstrates the benefit of intravenous (IV) sodium thiosulfate in alleviating the calcified lesions as well as the associated pain and disability. This application of IV sodium thiosulfate with a favorable outcome is new and should be considered for the treatment of generalized dystrophic calcinosis cutis, especially when topical, procedural, or surgical options are not feasible.
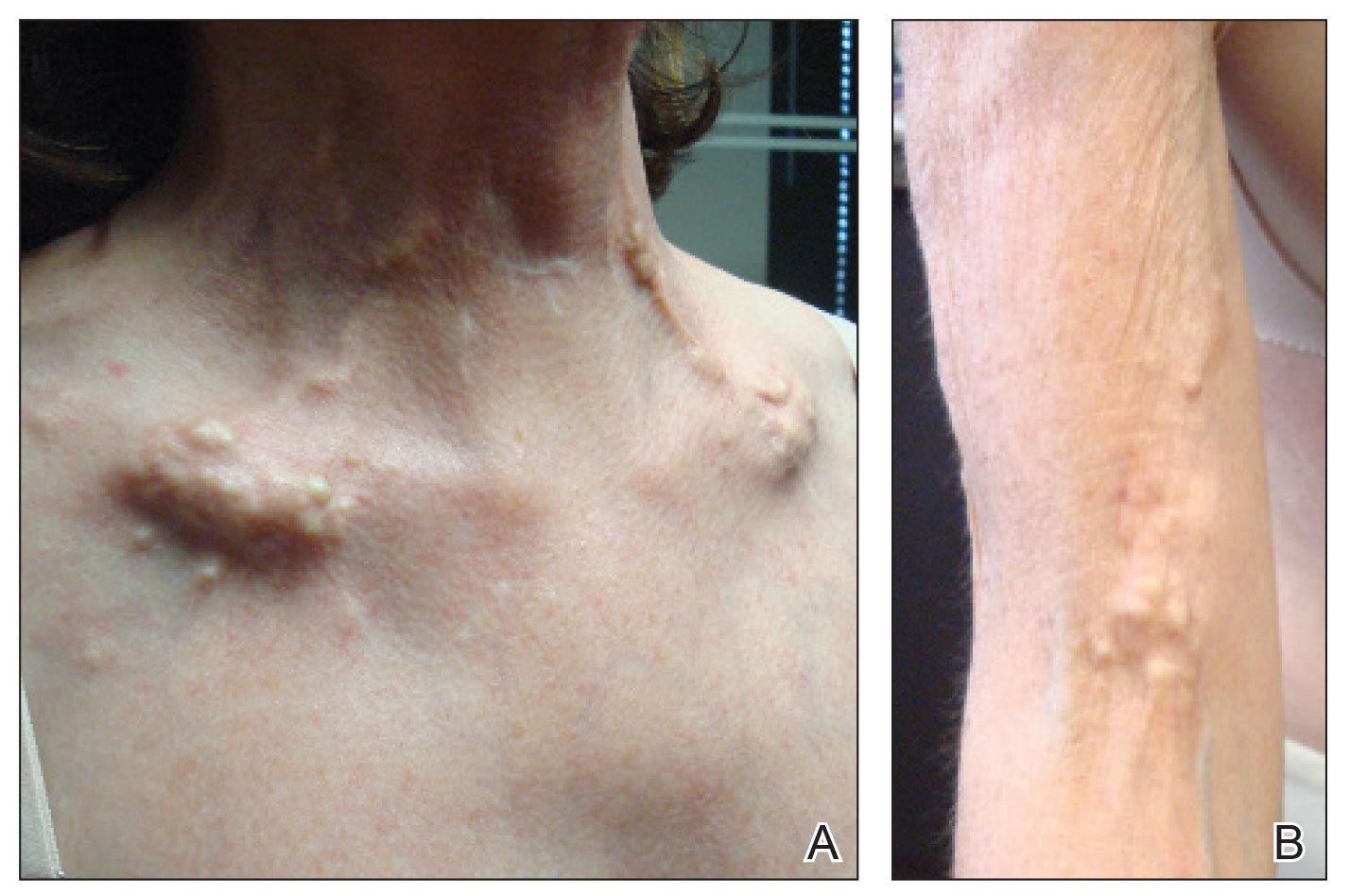
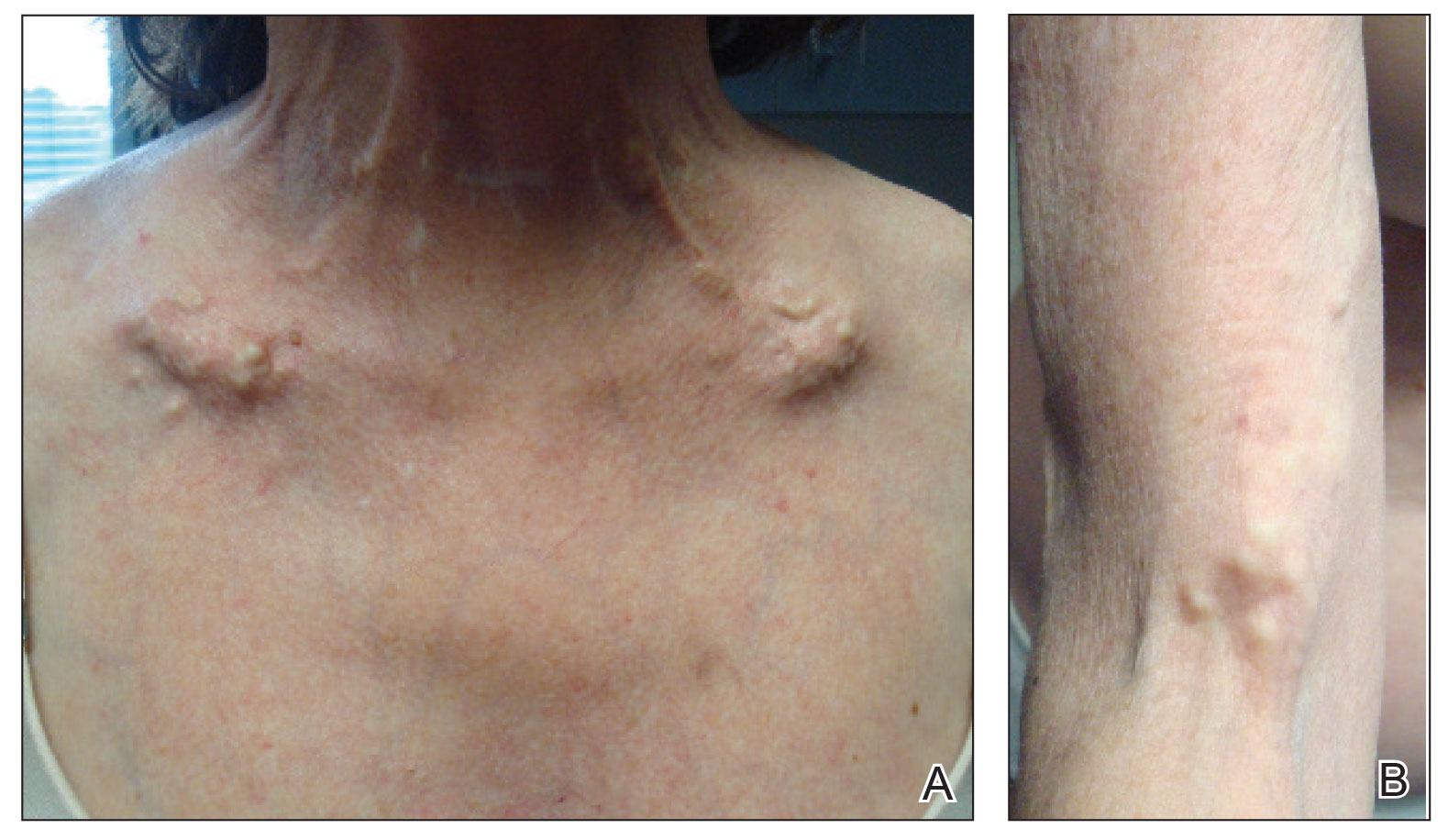
A 54-year-old woman with a history of well-controlled dermatomyositis and systemic lupus erythematosus presented with diffuse, hard, calcified lesions on the legs, arms, clavicular region, and neck that had slowly progressed over at least a 10-year period (Figure 1). The lesions were consistent with dystrophic calcinosis cutis. The patient was started on 12.5 g of IV sodium thiosulfate 3 times weekly infused over 30 minutes. Drastic diminution of the cutaneous calcification was observed at 3-month follow-up (Figure 2). She reported decreased pain and burning as well as increased overall functionality and improved sleep. The patient completed 8 months of therapy, but the treatment was stopped secondary to suspicion of a lupuslike flare, and the lesions recurred with more widespread involvement, including the trunk, tendons, bony prominences, and supraclavicular soft tissue. The patient reported burning pain and pruritus that resulted in impairment of daily activities such as getting dressed. Sodium thiosulfate was restarted once weekly, which again resulted in reduction of the dystrophic calcinosis cutis.
Dystrophic calcinosis cutis is a debilitating disease that results in considerable morbidity and pain with major implications on quality of life. The pathophysiology is unclear; calcium and phosphate serum levels generally are normal. A proposed mechanism is that chronic inflammation causes tissue damage and defective collagen synthesis, resulting in a distorted architecture that facilitates calcium deposition in the skin and subcutaneous tissues.1 Dystrophic calcinosis cutis most commonly is associated with systemic sclerosis and dermatomyositis but also can be seen in systemic lupus erythematosus, panniculitis, and other connective tissue diseases. It also can occur with skin neoplasms, collagen and elastin disorders, porphyria cutanea tarda, and pancreatic panniculitis.1 Progression of dystrophic calcinosis cutis usually is independent of the associated disease status.
Treatment is based on anecdotal evidence from case reports, as there is no universally accepted pharmacologic or procedural intervention available for dystrophic calcinosis cutis. Medications that have been reported to be helpful to varying degrees include diltiazem, colchicine, minocycline, IV immunoglobulin, ceftriaxone, aluminum hydroxide, probenecid, alendronic acid, etidronate disodium, warfarin, intralesional corticosteroids, and sodium thiosulfate. Procedural interventions also have been reported, such as surgical excision, extracorporeal shock wave lithotripsy, and CO2 and erbium: YAG lasers.1 Surgical excision of dystrophic calcinosis cutis is widely implemented but outcomes are poor. Moreover, in patients with widely diffuse calcinosis, targeted procedural therapy is impractical.
Intravenous sodium thiosulfate has been widely used for the treatment of calciphylaxis secondary to end-stage renal failure and tumoral calcinosis.2 It also has been reported to be effective in iatrogenic calcinosis cutis secondary to extravasation of calcium-containing solutions in a patient with T-cell acute lymphoblastic leukemia.3 However, reports of its use in treating dystrophic calcinosis cutis are limited. Intravenous sodium thiosulfate—10 g 3 times weekly for 2 weeks, followed by 15 g twice weekly for the next 3 months—was used with abatacept for treatment of dystrophic calcinosis cutis in a patient with juvenile dermatomyositis.4 Other formulations of sodium thiosulfate have been reported to result in clearance of calcified lesions, including a topical application compounded in zinc oxide5 and intradermal injection at the base of a nodule.6 We used 12.5 g over 30 minutes 3 times weekly; however, the dose can be increased to 25 g over 60 minutes if 3 to 4 treatments are tolerated, with nausea being the only notable side effect. Its mechanism of action in treating dystrophic calcinosis cutis is unclear, but it likely is due to its ability to chelate and dissolve calcium deposits. Topical and intradermal therapy is impractical for widespread, dystrophic calcinosis cutis as in our patient.
Our case highlights the successful use of IV sodium thiosulfate as a stand-alone treatment modality for generalized dystrophic calcinosis cutis in an adult patient. Both our patient and a child in a previously reported case who received the same treatment4 had dermatomyositis, but we suspect IV sodium thiosulfate also may be effective for dystrophic calcinosis cutis associated with other diseases. Sodium thiosulfate should be considered as a treatment for patients who experience tremendous pain and disability. It is safe, inexpensive, and easy to administer and is especially helpful in patients for whom topical, intradermal, or procedural therapy is not possible.
- Gutierrez A Jr, Wetter DA. Calcinosis cutis in autoimmune connective tissue diseases. Dermatol Ther. 2012;25:195-206.
- Mageau A, Guigonis V, Ratzimbasafy V, et al. Intravenous sodium thiosulfate for treating tumoral calcinosis associated with systemic disorders: report of four cases. Joint Bone Spine. 2017;84:341-344.
Raffaella C, Annapaola C, Tullio I, et al. Successful treatment of severe iatrogenic calcinosis cutis with intravenous sodium thiosulfate in a child affected by T-acute lymphoblastic leukemia. Pediatr Dermatol. 2009;26:311-315. - Arabshahi B, Silverman RA, Jones OY, et al. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatrics. 2012;160:520-522.
- Bair B, Fivenson D. A novel treatment for ulcerative calcinosis cutis. J Drugs Dermatol. 2011;10:1042-1044.
- Smith GP. Intradermal sodium thiosulfate for exophytic calcinosis cutis of connective tissue disease. J Am Acad Dermatol. 2013;69:E146-E147.
To the Editor:
Severe dystrophic calcinosis cutis is a debilitating disease with no universally accepted therapeutic options. This case demonstrates the benefit of intravenous (IV) sodium thiosulfate in alleviating the calcified lesions as well as the associated pain and disability. This application of IV sodium thiosulfate with a favorable outcome is new and should be considered for the treatment of generalized dystrophic calcinosis cutis, especially when topical, procedural, or surgical options are not feasible.
A 54-year-old woman with a history of well-controlled dermatomyositis and systemic lupus erythematosus presented with diffuse, hard, calcified lesions on the legs, arms, clavicular region, and neck that had slowly progressed over at least a 10-year period (Figure 1). The lesions were consistent with dystrophic calcinosis cutis. The patient was started on 12.5 g of IV sodium thiosulfate 3 times weekly infused over 30 minutes. Drastic diminution of the cutaneous calcification was observed at 3-month follow-up (Figure 2). She reported decreased pain and burning as well as increased overall functionality and improved sleep. The patient completed 8 months of therapy, but the treatment was stopped secondary to suspicion of a lupuslike flare, and the lesions recurred with more widespread involvement, including the trunk, tendons, bony prominences, and supraclavicular soft tissue. The patient reported burning pain and pruritus that resulted in impairment of daily activities such as getting dressed. Sodium thiosulfate was restarted once weekly, which again resulted in reduction of the dystrophic calcinosis cutis.
Dystrophic calcinosis cutis is a debilitating disease that results in considerable morbidity and pain with major implications on quality of life. The pathophysiology is unclear; calcium and phosphate serum levels generally are normal. A proposed mechanism is that chronic inflammation causes tissue damage and defective collagen synthesis, resulting in a distorted architecture that facilitates calcium deposition in the skin and subcutaneous tissues.1 Dystrophic calcinosis cutis most commonly is associated with systemic sclerosis and dermatomyositis but also can be seen in systemic lupus erythematosus, panniculitis, and other connective tissue diseases. It also can occur with skin neoplasms, collagen and elastin disorders, porphyria cutanea tarda, and pancreatic panniculitis.1 Progression of dystrophic calcinosis cutis usually is independent of the associated disease status.
Treatment is based on anecdotal evidence from case reports, as there is no universally accepted pharmacologic or procedural intervention available for dystrophic calcinosis cutis. Medications that have been reported to be helpful to varying degrees include diltiazem, colchicine, minocycline, IV immunoglobulin, ceftriaxone, aluminum hydroxide, probenecid, alendronic acid, etidronate disodium, warfarin, intralesional corticosteroids, and sodium thiosulfate. Procedural interventions also have been reported, such as surgical excision, extracorporeal shock wave lithotripsy, and CO2 and erbium: YAG lasers.1 Surgical excision of dystrophic calcinosis cutis is widely implemented but outcomes are poor. Moreover, in patients with widely diffuse calcinosis, targeted procedural therapy is impractical.
Intravenous sodium thiosulfate has been widely used for the treatment of calciphylaxis secondary to end-stage renal failure and tumoral calcinosis.2 It also has been reported to be effective in iatrogenic calcinosis cutis secondary to extravasation of calcium-containing solutions in a patient with T-cell acute lymphoblastic leukemia.3 However, reports of its use in treating dystrophic calcinosis cutis are limited. Intravenous sodium thiosulfate—10 g 3 times weekly for 2 weeks, followed by 15 g twice weekly for the next 3 months—was used with abatacept for treatment of dystrophic calcinosis cutis in a patient with juvenile dermatomyositis.4 Other formulations of sodium thiosulfate have been reported to result in clearance of calcified lesions, including a topical application compounded in zinc oxide5 and intradermal injection at the base of a nodule.6 We used 12.5 g over 30 minutes 3 times weekly; however, the dose can be increased to 25 g over 60 minutes if 3 to 4 treatments are tolerated, with nausea being the only notable side effect. Its mechanism of action in treating dystrophic calcinosis cutis is unclear, but it likely is due to its ability to chelate and dissolve calcium deposits. Topical and intradermal therapy is impractical for widespread, dystrophic calcinosis cutis as in our patient.
Our case highlights the successful use of IV sodium thiosulfate as a stand-alone treatment modality for generalized dystrophic calcinosis cutis in an adult patient. Both our patient and a child in a previously reported case who received the same treatment4 had dermatomyositis, but we suspect IV sodium thiosulfate also may be effective for dystrophic calcinosis cutis associated with other diseases. Sodium thiosulfate should be considered as a treatment for patients who experience tremendous pain and disability. It is safe, inexpensive, and easy to administer and is especially helpful in patients for whom topical, intradermal, or procedural therapy is not possible.
To the Editor:
Severe dystrophic calcinosis cutis is a debilitating disease with no universally accepted therapeutic options. This case demonstrates the benefit of intravenous (IV) sodium thiosulfate in alleviating the calcified lesions as well as the associated pain and disability. This application of IV sodium thiosulfate with a favorable outcome is new and should be considered for the treatment of generalized dystrophic calcinosis cutis, especially when topical, procedural, or surgical options are not feasible.
A 54-year-old woman with a history of well-controlled dermatomyositis and systemic lupus erythematosus presented with diffuse, hard, calcified lesions on the legs, arms, clavicular region, and neck that had slowly progressed over at least a 10-year period (Figure 1). The lesions were consistent with dystrophic calcinosis cutis. The patient was started on 12.5 g of IV sodium thiosulfate 3 times weekly infused over 30 minutes. Drastic diminution of the cutaneous calcification was observed at 3-month follow-up (Figure 2). She reported decreased pain and burning as well as increased overall functionality and improved sleep. The patient completed 8 months of therapy, but the treatment was stopped secondary to suspicion of a lupuslike flare, and the lesions recurred with more widespread involvement, including the trunk, tendons, bony prominences, and supraclavicular soft tissue. The patient reported burning pain and pruritus that resulted in impairment of daily activities such as getting dressed. Sodium thiosulfate was restarted once weekly, which again resulted in reduction of the dystrophic calcinosis cutis.
Dystrophic calcinosis cutis is a debilitating disease that results in considerable morbidity and pain with major implications on quality of life. The pathophysiology is unclear; calcium and phosphate serum levels generally are normal. A proposed mechanism is that chronic inflammation causes tissue damage and defective collagen synthesis, resulting in a distorted architecture that facilitates calcium deposition in the skin and subcutaneous tissues.1 Dystrophic calcinosis cutis most commonly is associated with systemic sclerosis and dermatomyositis but also can be seen in systemic lupus erythematosus, panniculitis, and other connective tissue diseases. It also can occur with skin neoplasms, collagen and elastin disorders, porphyria cutanea tarda, and pancreatic panniculitis.1 Progression of dystrophic calcinosis cutis usually is independent of the associated disease status.
Treatment is based on anecdotal evidence from case reports, as there is no universally accepted pharmacologic or procedural intervention available for dystrophic calcinosis cutis. Medications that have been reported to be helpful to varying degrees include diltiazem, colchicine, minocycline, IV immunoglobulin, ceftriaxone, aluminum hydroxide, probenecid, alendronic acid, etidronate disodium, warfarin, intralesional corticosteroids, and sodium thiosulfate. Procedural interventions also have been reported, such as surgical excision, extracorporeal shock wave lithotripsy, and CO2 and erbium: YAG lasers.1 Surgical excision of dystrophic calcinosis cutis is widely implemented but outcomes are poor. Moreover, in patients with widely diffuse calcinosis, targeted procedural therapy is impractical.
Intravenous sodium thiosulfate has been widely used for the treatment of calciphylaxis secondary to end-stage renal failure and tumoral calcinosis.2 It also has been reported to be effective in iatrogenic calcinosis cutis secondary to extravasation of calcium-containing solutions in a patient with T-cell acute lymphoblastic leukemia.3 However, reports of its use in treating dystrophic calcinosis cutis are limited. Intravenous sodium thiosulfate—10 g 3 times weekly for 2 weeks, followed by 15 g twice weekly for the next 3 months—was used with abatacept for treatment of dystrophic calcinosis cutis in a patient with juvenile dermatomyositis.4 Other formulations of sodium thiosulfate have been reported to result in clearance of calcified lesions, including a topical application compounded in zinc oxide5 and intradermal injection at the base of a nodule.6 We used 12.5 g over 30 minutes 3 times weekly; however, the dose can be increased to 25 g over 60 minutes if 3 to 4 treatments are tolerated, with nausea being the only notable side effect. Its mechanism of action in treating dystrophic calcinosis cutis is unclear, but it likely is due to its ability to chelate and dissolve calcium deposits. Topical and intradermal therapy is impractical for widespread, dystrophic calcinosis cutis as in our patient.
Our case highlights the successful use of IV sodium thiosulfate as a stand-alone treatment modality for generalized dystrophic calcinosis cutis in an adult patient. Both our patient and a child in a previously reported case who received the same treatment4 had dermatomyositis, but we suspect IV sodium thiosulfate also may be effective for dystrophic calcinosis cutis associated with other diseases. Sodium thiosulfate should be considered as a treatment for patients who experience tremendous pain and disability. It is safe, inexpensive, and easy to administer and is especially helpful in patients for whom topical, intradermal, or procedural therapy is not possible.
- Gutierrez A Jr, Wetter DA. Calcinosis cutis in autoimmune connective tissue diseases. Dermatol Ther. 2012;25:195-206.
- Mageau A, Guigonis V, Ratzimbasafy V, et al. Intravenous sodium thiosulfate for treating tumoral calcinosis associated with systemic disorders: report of four cases. Joint Bone Spine. 2017;84:341-344.
Raffaella C, Annapaola C, Tullio I, et al. Successful treatment of severe iatrogenic calcinosis cutis with intravenous sodium thiosulfate in a child affected by T-acute lymphoblastic leukemia. Pediatr Dermatol. 2009;26:311-315. - Arabshahi B, Silverman RA, Jones OY, et al. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatrics. 2012;160:520-522.
- Bair B, Fivenson D. A novel treatment for ulcerative calcinosis cutis. J Drugs Dermatol. 2011;10:1042-1044.
- Smith GP. Intradermal sodium thiosulfate for exophytic calcinosis cutis of connective tissue disease. J Am Acad Dermatol. 2013;69:E146-E147.
- Gutierrez A Jr, Wetter DA. Calcinosis cutis in autoimmune connective tissue diseases. Dermatol Ther. 2012;25:195-206.
- Mageau A, Guigonis V, Ratzimbasafy V, et al. Intravenous sodium thiosulfate for treating tumoral calcinosis associated with systemic disorders: report of four cases. Joint Bone Spine. 2017;84:341-344.
Raffaella C, Annapaola C, Tullio I, et al. Successful treatment of severe iatrogenic calcinosis cutis with intravenous sodium thiosulfate in a child affected by T-acute lymphoblastic leukemia. Pediatr Dermatol. 2009;26:311-315. - Arabshahi B, Silverman RA, Jones OY, et al. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatrics. 2012;160:520-522.
- Bair B, Fivenson D. A novel treatment for ulcerative calcinosis cutis. J Drugs Dermatol. 2011;10:1042-1044.
- Smith GP. Intradermal sodium thiosulfate for exophytic calcinosis cutis of connective tissue disease. J Am Acad Dermatol. 2013;69:E146-E147.
Practice Points
- Dystrophic calcinosis cutis is a potentially debilitating condition with limited effective therapies.
- Consider intravenous sodium thiosulfate in patients with diffuse and severe dystrophic calcinosis cutis.
Field Cancerization With Multiple Keratoacanthomas Successfully Treated With Topical and Intralesional 5-Fluorouracil
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
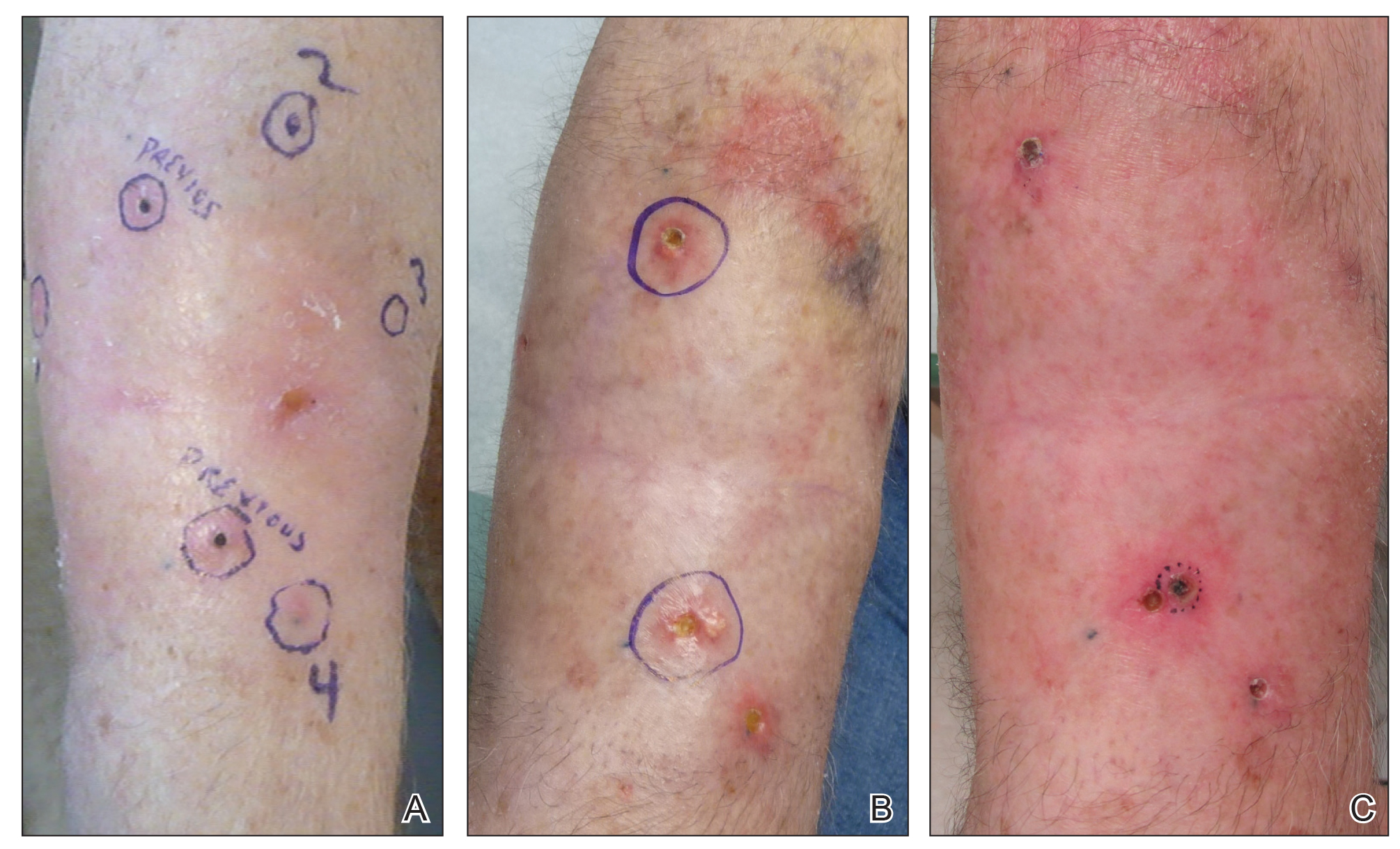
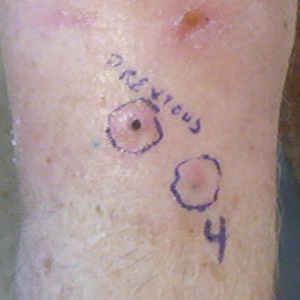
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
Methotrexate as a Treatment of Palmoplantar Lichen Planus
To the Editor:
Palmoplantar lichen planus (LP) is an uncommon variant of LP that involves the palms and soles. The prevalence of LP is approximately 0.1% to 2% in the general population. It can affect both mucosal and cutaneous surfaces.1 A study of 36 patients with LP showed that 25% (9/36) had palmar and/or plantar involvement.2 Palmoplantar LP is more commonly found in men than women, with an average age of onset of 38 to 65 years.3 It tends to affect the soles more often than the palms, with the most common site being the plantar arch. Itching generally is the most common symptom reported. Lesions often resolve over a few months, but relapses can occur in 10% to 29% of patients.2 The clinical morphology commonly is characterized as erythematous scaly plaques, hyperkeratotic plaques, or ulcerations.4 Due to its rare occurrence, palmoplantar LP often is misdiagnosed as psoriasis, eczematous dermatitis, tinea nigra, or secondary syphilis, making pathology extremely helpful in making the diagnosis.1 Darker skin types can obscure defining characteristics, further impeding a timely diagnosis. We describe a novel case of palmoplantar LP that was successfully treated with methotrexate.
A 38-year-old man with no notable medical history presented for dermatologic evaluation of a palmar and plantar rash of 4 months' duration. The rash was accompanied by intense burning pain and pruritus. Prior to presentation, he had been treated with multiple prednisone tapers starting at 40 mg daily as well as combination therapy of a 2-week course of minocycline 100 mg twice daily and clobetasol ointment twice daily for 4 months, with no notable improvement. Workup prior to presentation included a negative potassium hydroxide fungal preparation and a normal antinuclear antibody titer. A review of symptoms was negative for arthralgia, myalgia, photosensitivity, malar rash, Raynaud phenomenon, pleuritic pain, seizures, and psychosis.
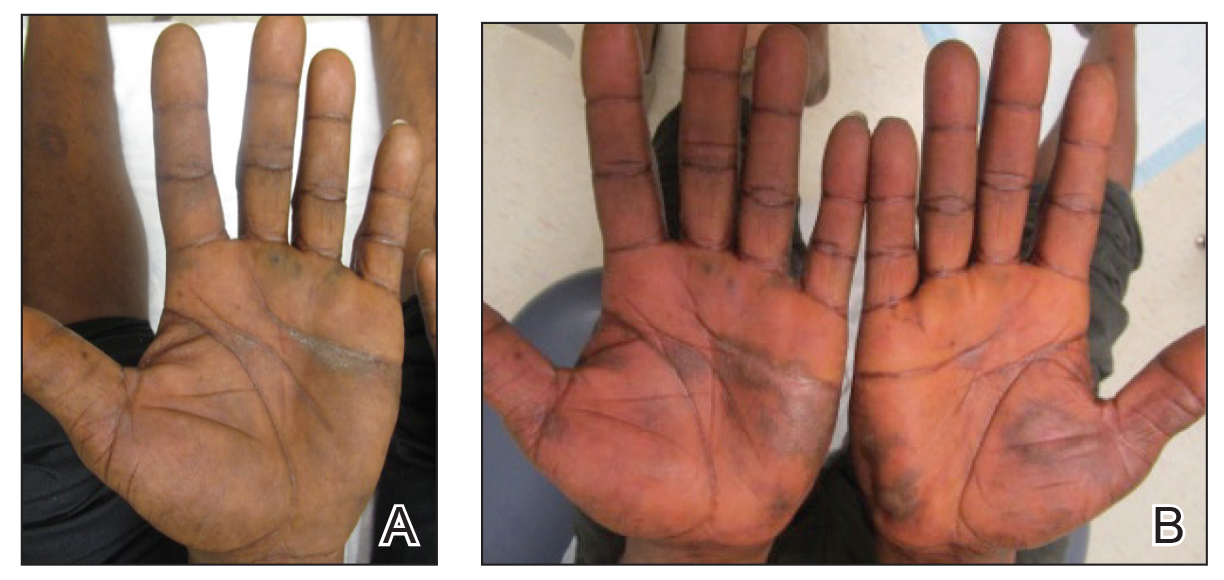
Physical examination revealed focal areas of mildly thick, hyperkeratotic scale with desquamation on the plantar and palmar surfaces of the feet and hands. The underlying skin of the feet consisted of dyspigmented patches of dark brown and hypopigmented skin with erythema, profound scaling, and sparing of the internal plantar arches (Figure 1A). On the palms, thin hyperkeratotic plaques with desquamation and erythematous maceration of the surrounding skin were observed (Figure 2A). Thin white plaques of the posterior bilateral buccal mucosa were appreciated as well as an erosion that extended to the lower lip.
The differential diagnosis included LP, psoriasis, acquired palmoplantar keratoderma, and discoid lupus erythematosus. Tinea pedis and tinea manuum were less likely in the setting of a negative potassium hydroxide fungal preparation.

A biopsy of the lateral aspect of the left foot showed a cell-poor interface dermatitis that could resemble partially treated LP or a lichenoid hypersensitivity reaction (Figure 3). Given the clinical and pathologic findings, a diagnosis of palmoplantar LP was favored. The patient was on no medications or over-the-counter supplements prior to the appearance of the rash, making a lichenoid hypersensitivity rash less likely. The histology findings likely were muted, as they were done at the end of the prednisone taper.
Minocycline and clobetasol ointment were discontinued, and the prednisone taper was completed as originally prescribed. The patient was started on 25 mg daily of acitretin for 4 weeks, then increased to 35 mg daily. Notable improvement in the palmar and plantar lesions was noted after the initial 4 weeks of therapy; however, acitretin treatment was discontinued due to lack of adequate insurance coverage for the medication. The patient became symptomatic several weeks following acitretin cessation and was started on methotrexate 15 mg weekly with triamcinolone acetonide paste 0.1% for the oral lesions. Once again, improvement was seen on both the palmar and plantar surfaces after 4 weeks of therapy (Figures 1B and 2B).
Evidence for treatment of palmoplantar LP is limited to a few case reports and case series. Documented treatments for palmoplantar LP include topical and systemic steroids, tazarotene, acitretin, and immunosuppressive medications.4 One case report described a patient who responded well to prednisone therapy (1 mg/kg daily for 3 weeks, then reduced to 5 mg daily).5 Another report described a patient who responded favorably to cyclosporine 3.5 mg/kg daily for 4 weeks, then tapered over another 4 weeks for a total of 8 weeks of treatment.4 Although the most common treatments described in the literature consist of acitretin as well as topical and systemic steroids, few have discussed the efficacy of methotrexate. In one study, acitretin did not result in clearance, but the patient saw profound improvement with methotrexate (titrated up to 25 mg weekly) over 2 months.1
In our case, treatment with methotrexate was proven successful in a patient who responded to acitretin but was unable to afford treatment. This case highlights a rare variant of a common disease and the possibility of methotrexate as a cost-effective and useful treatment option for LP.
- Rieder E, Hale CS, Meehan SA, et al. Palmoplantar lichen planus. Dermatol Online J. 2015;20:13030/qt1vn9s55z.
- Sánchez-Pérez J, Rios Buceta L, Fraga J, et al. Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients. Br J Dermatol. 2000;142:310-314.
- Gutte R, Khopkar U. Predominant palmoplantar lichen planus: a diagnostic challenge. Indian J Dermatol. 2014;59:343-347.
- Karakatsanis G, Patsatsi A, Kastoridou C, et al Palmoplantar lichen planus with umbilicated papules: an atypical case with rapid therapeutic response to cyclosporin. J Eur Acad Dermatol Venereol. 2007;21:1006-1007.
- Goucha S, Khaled A, Bennani Z, et al. Erosive lichen planus of the soles: Effective response to prednisone. Dermatol Ther. 2011;1:20-24.
To the Editor:
Palmoplantar lichen planus (LP) is an uncommon variant of LP that involves the palms and soles. The prevalence of LP is approximately 0.1% to 2% in the general population. It can affect both mucosal and cutaneous surfaces.1 A study of 36 patients with LP showed that 25% (9/36) had palmar and/or plantar involvement.2 Palmoplantar LP is more commonly found in men than women, with an average age of onset of 38 to 65 years.3 It tends to affect the soles more often than the palms, with the most common site being the plantar arch. Itching generally is the most common symptom reported. Lesions often resolve over a few months, but relapses can occur in 10% to 29% of patients.2 The clinical morphology commonly is characterized as erythematous scaly plaques, hyperkeratotic plaques, or ulcerations.4 Due to its rare occurrence, palmoplantar LP often is misdiagnosed as psoriasis, eczematous dermatitis, tinea nigra, or secondary syphilis, making pathology extremely helpful in making the diagnosis.1 Darker skin types can obscure defining characteristics, further impeding a timely diagnosis. We describe a novel case of palmoplantar LP that was successfully treated with methotrexate.
A 38-year-old man with no notable medical history presented for dermatologic evaluation of a palmar and plantar rash of 4 months' duration. The rash was accompanied by intense burning pain and pruritus. Prior to presentation, he had been treated with multiple prednisone tapers starting at 40 mg daily as well as combination therapy of a 2-week course of minocycline 100 mg twice daily and clobetasol ointment twice daily for 4 months, with no notable improvement. Workup prior to presentation included a negative potassium hydroxide fungal preparation and a normal antinuclear antibody titer. A review of symptoms was negative for arthralgia, myalgia, photosensitivity, malar rash, Raynaud phenomenon, pleuritic pain, seizures, and psychosis.
Physical examination revealed focal areas of mildly thick, hyperkeratotic scale with desquamation on the plantar and palmar surfaces of the feet and hands. The underlying skin of the feet consisted of dyspigmented patches of dark brown and hypopigmented skin with erythema, profound scaling, and sparing of the internal plantar arches (Figure 1A). On the palms, thin hyperkeratotic plaques with desquamation and erythematous maceration of the surrounding skin were observed (Figure 2A). Thin white plaques of the posterior bilateral buccal mucosa were appreciated as well as an erosion that extended to the lower lip.
The differential diagnosis included LP, psoriasis, acquired palmoplantar keratoderma, and discoid lupus erythematosus. Tinea pedis and tinea manuum were less likely in the setting of a negative potassium hydroxide fungal preparation.
A biopsy of the lateral aspect of the left foot showed a cell-poor interface dermatitis that could resemble partially treated LP or a lichenoid hypersensitivity reaction (Figure 3). Given the clinical and pathologic findings, a diagnosis of palmoplantar LP was favored. The patient was on no medications or over-the-counter supplements prior to the appearance of the rash, making a lichenoid hypersensitivity rash less likely. The histology findings likely were muted, as they were done at the end of the prednisone taper.
Minocycline and clobetasol ointment were discontinued, and the prednisone taper was completed as originally prescribed. The patient was started on 25 mg daily of acitretin for 4 weeks, then increased to 35 mg daily. Notable improvement in the palmar and plantar lesions was noted after the initial 4 weeks of therapy; however, acitretin treatment was discontinued due to lack of adequate insurance coverage for the medication. The patient became symptomatic several weeks following acitretin cessation and was started on methotrexate 15 mg weekly with triamcinolone acetonide paste 0.1% for the oral lesions. Once again, improvement was seen on both the palmar and plantar surfaces after 4 weeks of therapy (Figures 1B and 2B).
Evidence for treatment of palmoplantar LP is limited to a few case reports and case series. Documented treatments for palmoplantar LP include topical and systemic steroids, tazarotene, acitretin, and immunosuppressive medications.4 One case report described a patient who responded well to prednisone therapy (1 mg/kg daily for 3 weeks, then reduced to 5 mg daily).5 Another report described a patient who responded favorably to cyclosporine 3.5 mg/kg daily for 4 weeks, then tapered over another 4 weeks for a total of 8 weeks of treatment.4 Although the most common treatments described in the literature consist of acitretin as well as topical and systemic steroids, few have discussed the efficacy of methotrexate. In one study, acitretin did not result in clearance, but the patient saw profound improvement with methotrexate (titrated up to 25 mg weekly) over 2 months.1
In our case, treatment with methotrexate was proven successful in a patient who responded to acitretin but was unable to afford treatment. This case highlights a rare variant of a common disease and the possibility of methotrexate as a cost-effective and useful treatment option for LP.
To the Editor:
Palmoplantar lichen planus (LP) is an uncommon variant of LP that involves the palms and soles. The prevalence of LP is approximately 0.1% to 2% in the general population. It can affect both mucosal and cutaneous surfaces.1 A study of 36 patients with LP showed that 25% (9/36) had palmar and/or plantar involvement.2 Palmoplantar LP is more commonly found in men than women, with an average age of onset of 38 to 65 years.3 It tends to affect the soles more often than the palms, with the most common site being the plantar arch. Itching generally is the most common symptom reported. Lesions often resolve over a few months, but relapses can occur in 10% to 29% of patients.2 The clinical morphology commonly is characterized as erythematous scaly plaques, hyperkeratotic plaques, or ulcerations.4 Due to its rare occurrence, palmoplantar LP often is misdiagnosed as psoriasis, eczematous dermatitis, tinea nigra, or secondary syphilis, making pathology extremely helpful in making the diagnosis.1 Darker skin types can obscure defining characteristics, further impeding a timely diagnosis. We describe a novel case of palmoplantar LP that was successfully treated with methotrexate.
A 38-year-old man with no notable medical history presented for dermatologic evaluation of a palmar and plantar rash of 4 months' duration. The rash was accompanied by intense burning pain and pruritus. Prior to presentation, he had been treated with multiple prednisone tapers starting at 40 mg daily as well as combination therapy of a 2-week course of minocycline 100 mg twice daily and clobetasol ointment twice daily for 4 months, with no notable improvement. Workup prior to presentation included a negative potassium hydroxide fungal preparation and a normal antinuclear antibody titer. A review of symptoms was negative for arthralgia, myalgia, photosensitivity, malar rash, Raynaud phenomenon, pleuritic pain, seizures, and psychosis.
Physical examination revealed focal areas of mildly thick, hyperkeratotic scale with desquamation on the plantar and palmar surfaces of the feet and hands. The underlying skin of the feet consisted of dyspigmented patches of dark brown and hypopigmented skin with erythema, profound scaling, and sparing of the internal plantar arches (Figure 1A). On the palms, thin hyperkeratotic plaques with desquamation and erythematous maceration of the surrounding skin were observed (Figure 2A). Thin white plaques of the posterior bilateral buccal mucosa were appreciated as well as an erosion that extended to the lower lip.
The differential diagnosis included LP, psoriasis, acquired palmoplantar keratoderma, and discoid lupus erythematosus. Tinea pedis and tinea manuum were less likely in the setting of a negative potassium hydroxide fungal preparation.
A biopsy of the lateral aspect of the left foot showed a cell-poor interface dermatitis that could resemble partially treated LP or a lichenoid hypersensitivity reaction (Figure 3). Given the clinical and pathologic findings, a diagnosis of palmoplantar LP was favored. The patient was on no medications or over-the-counter supplements prior to the appearance of the rash, making a lichenoid hypersensitivity rash less likely. The histology findings likely were muted, as they were done at the end of the prednisone taper.
Minocycline and clobetasol ointment were discontinued, and the prednisone taper was completed as originally prescribed. The patient was started on 25 mg daily of acitretin for 4 weeks, then increased to 35 mg daily. Notable improvement in the palmar and plantar lesions was noted after the initial 4 weeks of therapy; however, acitretin treatment was discontinued due to lack of adequate insurance coverage for the medication. The patient became symptomatic several weeks following acitretin cessation and was started on methotrexate 15 mg weekly with triamcinolone acetonide paste 0.1% for the oral lesions. Once again, improvement was seen on both the palmar and plantar surfaces after 4 weeks of therapy (Figures 1B and 2B).
Evidence for treatment of palmoplantar LP is limited to a few case reports and case series. Documented treatments for palmoplantar LP include topical and systemic steroids, tazarotene, acitretin, and immunosuppressive medications.4 One case report described a patient who responded well to prednisone therapy (1 mg/kg daily for 3 weeks, then reduced to 5 mg daily).5 Another report described a patient who responded favorably to cyclosporine 3.5 mg/kg daily for 4 weeks, then tapered over another 4 weeks for a total of 8 weeks of treatment.4 Although the most common treatments described in the literature consist of acitretin as well as topical and systemic steroids, few have discussed the efficacy of methotrexate. In one study, acitretin did not result in clearance, but the patient saw profound improvement with methotrexate (titrated up to 25 mg weekly) over 2 months.1
In our case, treatment with methotrexate was proven successful in a patient who responded to acitretin but was unable to afford treatment. This case highlights a rare variant of a common disease and the possibility of methotrexate as a cost-effective and useful treatment option for LP.
- Rieder E, Hale CS, Meehan SA, et al. Palmoplantar lichen planus. Dermatol Online J. 2015;20:13030/qt1vn9s55z.
- Sánchez-Pérez J, Rios Buceta L, Fraga J, et al. Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients. Br J Dermatol. 2000;142:310-314.
- Gutte R, Khopkar U. Predominant palmoplantar lichen planus: a diagnostic challenge. Indian J Dermatol. 2014;59:343-347.
- Karakatsanis G, Patsatsi A, Kastoridou C, et al Palmoplantar lichen planus with umbilicated papules: an atypical case with rapid therapeutic response to cyclosporin. J Eur Acad Dermatol Venereol. 2007;21:1006-1007.
- Goucha S, Khaled A, Bennani Z, et al. Erosive lichen planus of the soles: Effective response to prednisone. Dermatol Ther. 2011;1:20-24.
- Rieder E, Hale CS, Meehan SA, et al. Palmoplantar lichen planus. Dermatol Online J. 2015;20:13030/qt1vn9s55z.
- Sánchez-Pérez J, Rios Buceta L, Fraga J, et al. Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients. Br J Dermatol. 2000;142:310-314.
- Gutte R, Khopkar U. Predominant palmoplantar lichen planus: a diagnostic challenge. Indian J Dermatol. 2014;59:343-347.
- Karakatsanis G, Patsatsi A, Kastoridou C, et al Palmoplantar lichen planus with umbilicated papules: an atypical case with rapid therapeutic response to cyclosporin. J Eur Acad Dermatol Venereol. 2007;21:1006-1007.
- Goucha S, Khaled A, Bennani Z, et al. Erosive lichen planus of the soles: Effective response to prednisone. Dermatol Ther. 2011;1:20-24.
Practice Points
- Palmoplantar lichen planus (LP) is a rare variant of LP that is resistant to most treatments.
- Methotrexate may be a cost-effective option in patients who cannot tolerate systemic retinoids.
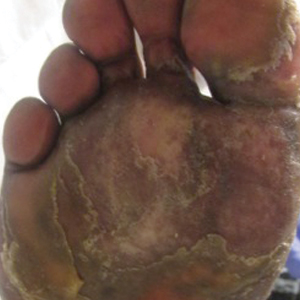
Apremilast and Systemic Retinoid Combination Treatment for Moderate to Severe Palmoplantar Psoriasis
To the Editor:
Psoriasis is a chronic inflammatory papulosquamous skin disease affecting 2% to 3% of the population.1 Its pathogenesis is multifactorial, consisting of a disrupted skin barrier and dysregulated immune activation.2
A wide armamentarium of topical and systemic treatments targeting different aspects of the disease pathogenesis have been developed over the years.3,4 Psoriasis was once considered a skin disease exclusively, but accumulating evidence suggests that it is accompanied by a multitude of systemic inflammatory comorbidities.5 This insight supports the concept of systemic treatment for patients with moderate to severe psoriasis. As a chronic disease, psoriasis requires continuous therapy. The treatment approach should focus on achieving efficacy and minimizing side effects. These goals can be achieved by combination, rotational, and sequential treatment approaches.6 Many therapeutic combinations have proven effective, using beneficially different mechanisms of action (MOAs) and toxicity profiles.7 We present a patient with moderate to severe recalcitrant palmoplantar psoriasis who demonstrated improvement with combination therapy.
A 50-year-old man presented with palmoplantar psoriasis of 7 years’ duration. His medical history included mild hyperlipidemia treated with atorvastatin. Prior topical treatments including calcipotriene, betamethasone dipropionate, and tacrolimus ointment did not result in improvement. Persistent acral involvement required further intervention, and the excimer laser was added to the therapeutic regimen with a minor additive therapeutic value. Acitretin (25 mg/d) was initiated; however, the disease flared up soon after. Acitretin was discontinued, and the patient was treated with apremilast (30 mg twice daily) for 9 months with a slight improvement. Physical examination revealed erythematous, fissured, scaly plaques involving both the palms and soles. Acitretin (25 mg/d) was reintroduced to the therapeutic regimen, and the acitretin-apremilast combination was used for 2 months. With this regimen, the patient experienced 90% improvement (Figures 1 and 2).
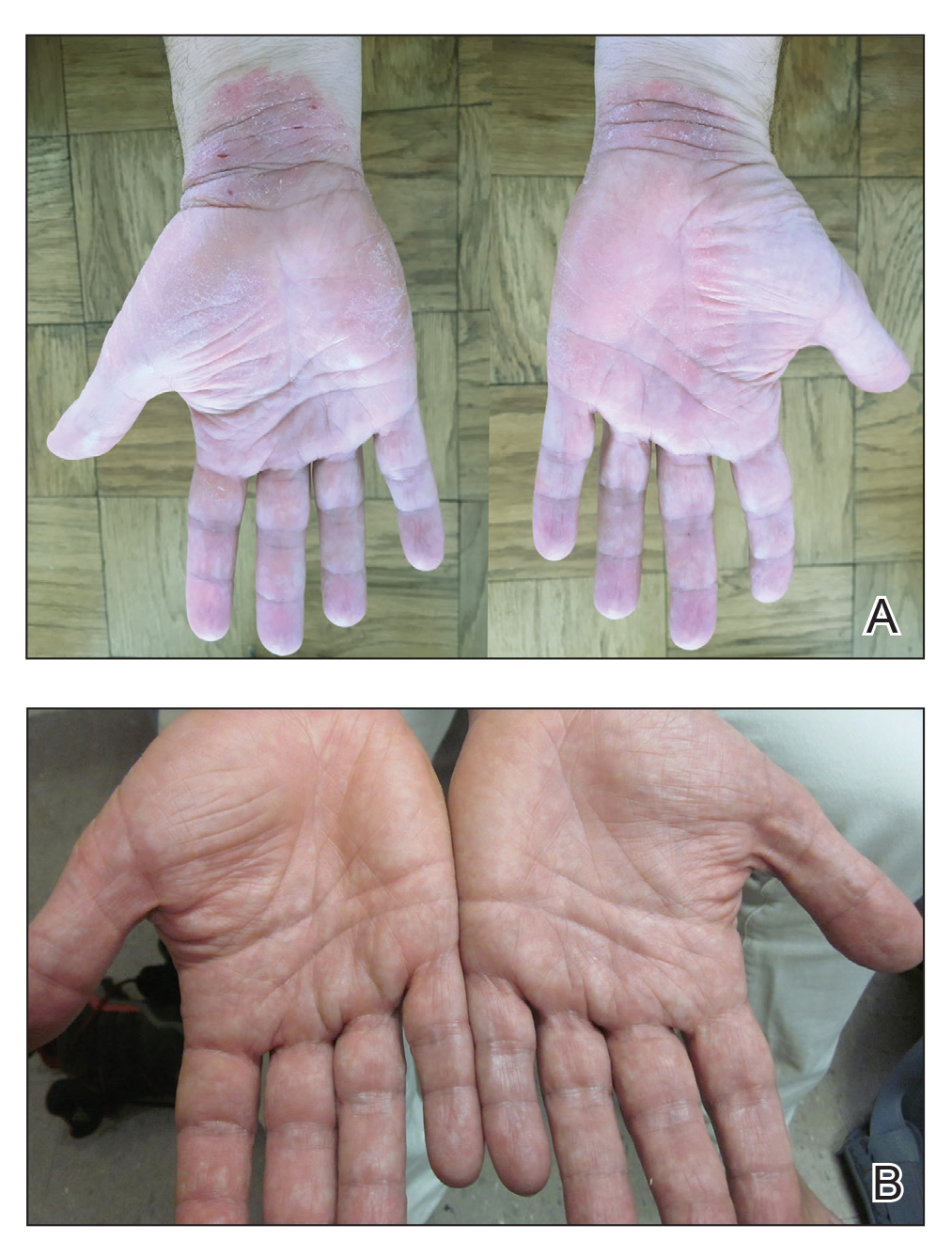

Palmoplantar psoriasis is a debilitating dermatosis that is extremely challenging to treat and is unresponsive to many modalities.8 Increased understanding of psoriasis mechanisms paved the path for the development of highly targeted biologic therapies9 with fewer side effects than drugs such as cyclosporine that indiscriminately neutralize multiple components of the immune system. Although highly specific, these targeted approaches are not without side effects10 and lead to diverse therapeutic outcomes, particularly when prescribed for palmoplantar psoriasis.11,12
The small-molecule inhibitor of phosphodiesterase 4—apremilast—was approved for plaque psoriasis treatment in late 2014. Although not fully elucidated, its MOA involves interfering with intracellular signaling, leading to increased intracellular cyclic adenosine monophosphate levels in inflammatory cells and keratinocytes.13 Proximal interruption of the pathologic cascade leads to the reduction of multiple proinflammatory cytokines with a simultaneous increase in anti-inflammatory mediators.13 Its efficacy and safety in the treatment of psoriasis have been shown in phase 2 and 3 clinical trials.14,15 In contrast to traditional oral therapies for psoriasis (ie, methotrexate, cyclosporine, acitretin), no laboratory test monitoring is needed and the safety profile is notably better.16
Acitretin, the active metabolite of etretinate, modulates epidermal differentiation and has immunomodulating activities.17 It commonly is used for treating palmoplantar psoriasis.8 Until recently, it was the only nonimmunosuppressive systemic treatment for psoriasis, and its combination with other systemic treatments, particularly biologics, has been advocated.18 Prior reports showed remarkable disease improvement when combining acitretin with alefacept, etanercept, infliximab, adalimumab, and ustekinumab.19 The optimal combination should include modalities with different MOAs without overlapping toxicities.19 Apremilast and acitretin have different MOAs and side-effect profiles, but another theoretical advantage is that they both interfere with intracellular signaling on the transcription level rather than affecting extracellular targets.13
Our patient with moderate to severe recalcitrant palmoplantar psoriasis demonstrated approximately 90% improvement following apremilast and acitretin combination therapy. This treatment regimen should be considered in cases of persistent acral disease resistant to other therapeutic efforts.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Nograles KE, Davidovici B, Krueger JG. New insights in the immunologic basis of psoriasis. Semin Cutan Med Surg. 2010;29:3-9.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 3. guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol. 2009;60:643-659.
- Ryan C, Kirby B. Psoriasis is a systemic disease with multiple cardiovascular and metabolic comorbidities. Dermatol Clin. 2015;33:41-44.
- Lebwohl M, Menter A, Koo J, et al. Combination therapy to treat moderate to severe psoriasis. J Am Acad Dermatol. 2004;50:416-430.
- Cather JC, Menter A. Combining traditional agents and biologics for the treatment of psoriasis. Semin Cutan Med Surg. 2005;24:37-45.
- Janagond AB, Kanwar AJ, Handa S. Efficacy and safety of systemic methotrexate vs. acitretin in psoriasis patients with significant palmoplantar involvement: a prospective, randomized study. J Eur Acad Dermatol Venereol. 2013;27:E384-E389.
- Campa M, Mansouri B, Warren R, et al. A review of biologic therapies targeting IL-23 and IL-17 for use in moderate-to-severe plaque psoriasis [published online December 29, 2015]. Dermatol Ther (Heidelb). 2015;6:1-12.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Jacobi A, Schuler G, Hertl M. Differential clinical response to alefacept in combination with methotrexate in two patients with refractory palmar psoriasis. Br J Dermatol. 2007;156:178-180.
- Meyer V, Goerge T, Luger TA, et al. Successful treatment of palmoplantar hyperkeratotic psoriasis with a combination of etanercept and alitretinoin. J Clin Aesthet Dermatol. 2011;4:45-46.
- Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83:1583-1590.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Zerilli T, Ocheretyaner E. Apremilast (Otezla): a new oral treatment for adults with psoriasis and psoriatic arthritis. P T. 2015;40:495-500.
- Pilkington T, Brogden RN. Acitretin—a review of its pharmacology and therapeutic use. Drugs. 1992;43:597-627.
- Lebwohl M. Combining the new biologic agents with our current psoriasis armamentarium. J Am Acad Dermatol. 2003;49:S118-S124.
- Heinecke GM, Luber AJ, Levitt JO, et al. Combination use of ustekinumab with other systemic therapies: a retrospective study in a tertiary referral center. J Drugs Dermatol. 2013;12:1098-1102.
To the Editor:
Psoriasis is a chronic inflammatory papulosquamous skin disease affecting 2% to 3% of the population.1 Its pathogenesis is multifactorial, consisting of a disrupted skin barrier and dysregulated immune activation.2
A wide armamentarium of topical and systemic treatments targeting different aspects of the disease pathogenesis have been developed over the years.3,4 Psoriasis was once considered a skin disease exclusively, but accumulating evidence suggests that it is accompanied by a multitude of systemic inflammatory comorbidities.5 This insight supports the concept of systemic treatment for patients with moderate to severe psoriasis. As a chronic disease, psoriasis requires continuous therapy. The treatment approach should focus on achieving efficacy and minimizing side effects. These goals can be achieved by combination, rotational, and sequential treatment approaches.6 Many therapeutic combinations have proven effective, using beneficially different mechanisms of action (MOAs) and toxicity profiles.7 We present a patient with moderate to severe recalcitrant palmoplantar psoriasis who demonstrated improvement with combination therapy.
A 50-year-old man presented with palmoplantar psoriasis of 7 years’ duration. His medical history included mild hyperlipidemia treated with atorvastatin. Prior topical treatments including calcipotriene, betamethasone dipropionate, and tacrolimus ointment did not result in improvement. Persistent acral involvement required further intervention, and the excimer laser was added to the therapeutic regimen with a minor additive therapeutic value. Acitretin (25 mg/d) was initiated; however, the disease flared up soon after. Acitretin was discontinued, and the patient was treated with apremilast (30 mg twice daily) for 9 months with a slight improvement. Physical examination revealed erythematous, fissured, scaly plaques involving both the palms and soles. Acitretin (25 mg/d) was reintroduced to the therapeutic regimen, and the acitretin-apremilast combination was used for 2 months. With this regimen, the patient experienced 90% improvement (Figures 1 and 2).
Palmoplantar psoriasis is a debilitating dermatosis that is extremely challenging to treat and is unresponsive to many modalities.8 Increased understanding of psoriasis mechanisms paved the path for the development of highly targeted biologic therapies9 with fewer side effects than drugs such as cyclosporine that indiscriminately neutralize multiple components of the immune system. Although highly specific, these targeted approaches are not without side effects10 and lead to diverse therapeutic outcomes, particularly when prescribed for palmoplantar psoriasis.11,12
The small-molecule inhibitor of phosphodiesterase 4—apremilast—was approved for plaque psoriasis treatment in late 2014. Although not fully elucidated, its MOA involves interfering with intracellular signaling, leading to increased intracellular cyclic adenosine monophosphate levels in inflammatory cells and keratinocytes.13 Proximal interruption of the pathologic cascade leads to the reduction of multiple proinflammatory cytokines with a simultaneous increase in anti-inflammatory mediators.13 Its efficacy and safety in the treatment of psoriasis have been shown in phase 2 and 3 clinical trials.14,15 In contrast to traditional oral therapies for psoriasis (ie, methotrexate, cyclosporine, acitretin), no laboratory test monitoring is needed and the safety profile is notably better.16
Acitretin, the active metabolite of etretinate, modulates epidermal differentiation and has immunomodulating activities.17 It commonly is used for treating palmoplantar psoriasis.8 Until recently, it was the only nonimmunosuppressive systemic treatment for psoriasis, and its combination with other systemic treatments, particularly biologics, has been advocated.18 Prior reports showed remarkable disease improvement when combining acitretin with alefacept, etanercept, infliximab, adalimumab, and ustekinumab.19 The optimal combination should include modalities with different MOAs without overlapping toxicities.19 Apremilast and acitretin have different MOAs and side-effect profiles, but another theoretical advantage is that they both interfere with intracellular signaling on the transcription level rather than affecting extracellular targets.13
Our patient with moderate to severe recalcitrant palmoplantar psoriasis demonstrated approximately 90% improvement following apremilast and acitretin combination therapy. This treatment regimen should be considered in cases of persistent acral disease resistant to other therapeutic efforts.
To the Editor:
Psoriasis is a chronic inflammatory papulosquamous skin disease affecting 2% to 3% of the population.1 Its pathogenesis is multifactorial, consisting of a disrupted skin barrier and dysregulated immune activation.2
A wide armamentarium of topical and systemic treatments targeting different aspects of the disease pathogenesis have been developed over the years.3,4 Psoriasis was once considered a skin disease exclusively, but accumulating evidence suggests that it is accompanied by a multitude of systemic inflammatory comorbidities.5 This insight supports the concept of systemic treatment for patients with moderate to severe psoriasis. As a chronic disease, psoriasis requires continuous therapy. The treatment approach should focus on achieving efficacy and minimizing side effects. These goals can be achieved by combination, rotational, and sequential treatment approaches.6 Many therapeutic combinations have proven effective, using beneficially different mechanisms of action (MOAs) and toxicity profiles.7 We present a patient with moderate to severe recalcitrant palmoplantar psoriasis who demonstrated improvement with combination therapy.
A 50-year-old man presented with palmoplantar psoriasis of 7 years’ duration. His medical history included mild hyperlipidemia treated with atorvastatin. Prior topical treatments including calcipotriene, betamethasone dipropionate, and tacrolimus ointment did not result in improvement. Persistent acral involvement required further intervention, and the excimer laser was added to the therapeutic regimen with a minor additive therapeutic value. Acitretin (25 mg/d) was initiated; however, the disease flared up soon after. Acitretin was discontinued, and the patient was treated with apremilast (30 mg twice daily) for 9 months with a slight improvement. Physical examination revealed erythematous, fissured, scaly plaques involving both the palms and soles. Acitretin (25 mg/d) was reintroduced to the therapeutic regimen, and the acitretin-apremilast combination was used for 2 months. With this regimen, the patient experienced 90% improvement (Figures 1 and 2).
Palmoplantar psoriasis is a debilitating dermatosis that is extremely challenging to treat and is unresponsive to many modalities.8 Increased understanding of psoriasis mechanisms paved the path for the development of highly targeted biologic therapies9 with fewer side effects than drugs such as cyclosporine that indiscriminately neutralize multiple components of the immune system. Although highly specific, these targeted approaches are not without side effects10 and lead to diverse therapeutic outcomes, particularly when prescribed for palmoplantar psoriasis.11,12
The small-molecule inhibitor of phosphodiesterase 4—apremilast—was approved for plaque psoriasis treatment in late 2014. Although not fully elucidated, its MOA involves interfering with intracellular signaling, leading to increased intracellular cyclic adenosine monophosphate levels in inflammatory cells and keratinocytes.13 Proximal interruption of the pathologic cascade leads to the reduction of multiple proinflammatory cytokines with a simultaneous increase in anti-inflammatory mediators.13 Its efficacy and safety in the treatment of psoriasis have been shown in phase 2 and 3 clinical trials.14,15 In contrast to traditional oral therapies for psoriasis (ie, methotrexate, cyclosporine, acitretin), no laboratory test monitoring is needed and the safety profile is notably better.16
Acitretin, the active metabolite of etretinate, modulates epidermal differentiation and has immunomodulating activities.17 It commonly is used for treating palmoplantar psoriasis.8 Until recently, it was the only nonimmunosuppressive systemic treatment for psoriasis, and its combination with other systemic treatments, particularly biologics, has been advocated.18 Prior reports showed remarkable disease improvement when combining acitretin with alefacept, etanercept, infliximab, adalimumab, and ustekinumab.19 The optimal combination should include modalities with different MOAs without overlapping toxicities.19 Apremilast and acitretin have different MOAs and side-effect profiles, but another theoretical advantage is that they both interfere with intracellular signaling on the transcription level rather than affecting extracellular targets.13
Our patient with moderate to severe recalcitrant palmoplantar psoriasis demonstrated approximately 90% improvement following apremilast and acitretin combination therapy. This treatment regimen should be considered in cases of persistent acral disease resistant to other therapeutic efforts.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Nograles KE, Davidovici B, Krueger JG. New insights in the immunologic basis of psoriasis. Semin Cutan Med Surg. 2010;29:3-9.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 3. guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol. 2009;60:643-659.
- Ryan C, Kirby B. Psoriasis is a systemic disease with multiple cardiovascular and metabolic comorbidities. Dermatol Clin. 2015;33:41-44.
- Lebwohl M, Menter A, Koo J, et al. Combination therapy to treat moderate to severe psoriasis. J Am Acad Dermatol. 2004;50:416-430.
- Cather JC, Menter A. Combining traditional agents and biologics for the treatment of psoriasis. Semin Cutan Med Surg. 2005;24:37-45.
- Janagond AB, Kanwar AJ, Handa S. Efficacy and safety of systemic methotrexate vs. acitretin in psoriasis patients with significant palmoplantar involvement: a prospective, randomized study. J Eur Acad Dermatol Venereol. 2013;27:E384-E389.
- Campa M, Mansouri B, Warren R, et al. A review of biologic therapies targeting IL-23 and IL-17 for use in moderate-to-severe plaque psoriasis [published online December 29, 2015]. Dermatol Ther (Heidelb). 2015;6:1-12.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Jacobi A, Schuler G, Hertl M. Differential clinical response to alefacept in combination with methotrexate in two patients with refractory palmar psoriasis. Br J Dermatol. 2007;156:178-180.
- Meyer V, Goerge T, Luger TA, et al. Successful treatment of palmoplantar hyperkeratotic psoriasis with a combination of etanercept and alitretinoin. J Clin Aesthet Dermatol. 2011;4:45-46.
- Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83:1583-1590.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Zerilli T, Ocheretyaner E. Apremilast (Otezla): a new oral treatment for adults with psoriasis and psoriatic arthritis. P T. 2015;40:495-500.
- Pilkington T, Brogden RN. Acitretin—a review of its pharmacology and therapeutic use. Drugs. 1992;43:597-627.
- Lebwohl M. Combining the new biologic agents with our current psoriasis armamentarium. J Am Acad Dermatol. 2003;49:S118-S124.
- Heinecke GM, Luber AJ, Levitt JO, et al. Combination use of ustekinumab with other systemic therapies: a retrospective study in a tertiary referral center. J Drugs Dermatol. 2013;12:1098-1102.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Nograles KE, Davidovici B, Krueger JG. New insights in the immunologic basis of psoriasis. Semin Cutan Med Surg. 2010;29:3-9.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 3. guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol. 2009;60:643-659.
- Ryan C, Kirby B. Psoriasis is a systemic disease with multiple cardiovascular and metabolic comorbidities. Dermatol Clin. 2015;33:41-44.
- Lebwohl M, Menter A, Koo J, et al. Combination therapy to treat moderate to severe psoriasis. J Am Acad Dermatol. 2004;50:416-430.
- Cather JC, Menter A. Combining traditional agents and biologics for the treatment of psoriasis. Semin Cutan Med Surg. 2005;24:37-45.
- Janagond AB, Kanwar AJ, Handa S. Efficacy and safety of systemic methotrexate vs. acitretin in psoriasis patients with significant palmoplantar involvement: a prospective, randomized study. J Eur Acad Dermatol Venereol. 2013;27:E384-E389.
- Campa M, Mansouri B, Warren R, et al. A review of biologic therapies targeting IL-23 and IL-17 for use in moderate-to-severe plaque psoriasis [published online December 29, 2015]. Dermatol Ther (Heidelb). 2015;6:1-12.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Jacobi A, Schuler G, Hertl M. Differential clinical response to alefacept in combination with methotrexate in two patients with refractory palmar psoriasis. Br J Dermatol. 2007;156:178-180.
- Meyer V, Goerge T, Luger TA, et al. Successful treatment of palmoplantar hyperkeratotic psoriasis with a combination of etanercept and alitretinoin. J Clin Aesthet Dermatol. 2011;4:45-46.
- Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83:1583-1590.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Zerilli T, Ocheretyaner E. Apremilast (Otezla): a new oral treatment for adults with psoriasis and psoriatic arthritis. P T. 2015;40:495-500.
- Pilkington T, Brogden RN. Acitretin—a review of its pharmacology and therapeutic use. Drugs. 1992;43:597-627.
- Lebwohl M. Combining the new biologic agents with our current psoriasis armamentarium. J Am Acad Dermatol. 2003;49:S118-S124.
- Heinecke GM, Luber AJ, Levitt JO, et al. Combination use of ustekinumab with other systemic therapies: a retrospective study in a tertiary referral center. J Drugs Dermatol. 2013;12:1098-1102.
Practice Points
- Palmoplantar psoriasis is challenging to treat and is unresponsive to many modalities.
- Combination, rotational, and sequential treatment approaches may minimize side effects and loss of efficacy as well as enhance treatment responses.
- Apremilast and acitretin combination therapy led to 90% skin improvement in a case of severe recalcitrant palmoplantar psoriasis.
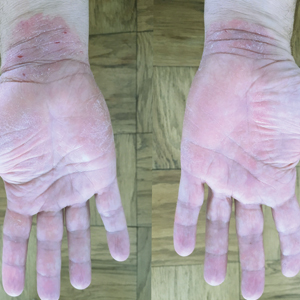
Atretic Cephalocele With Hypertrichosis
To the Editor:
A 2-week-old female infant presented to our dermatology clinic for evaluation of a 4.0×4.5-cm pink-red patch with a 1-cm central nodule and an overlying tuft of hair on the midline occipital region (Figure). The patient was born at 39 weeks’ gestation to nonconsanguineous parents via a normal spontaneous vaginal delivery and had an unremarkable prenatal course with no complications since birth. The red patch and tuft of hair were noted at birth, and the parents reported that the redness varied somewhat in size throughout the day and from day to day. An initial neurologic workup revealed no gross neurologic abnormalities. A head ultrasound revealed a soft-tissue hypervascular nodule that appeared separate from bony structures but showed evidence of a necklike extension from the nodule to the underlying soft tissues. The ultrasound could not definitively rule out intracranial extension; gross brain structures appeared normal. The initial differential diagnosis consisted of a congenital hemangioma (either a rapidly involuting or noninvoluting subtype), meningioma, or cephalocele.
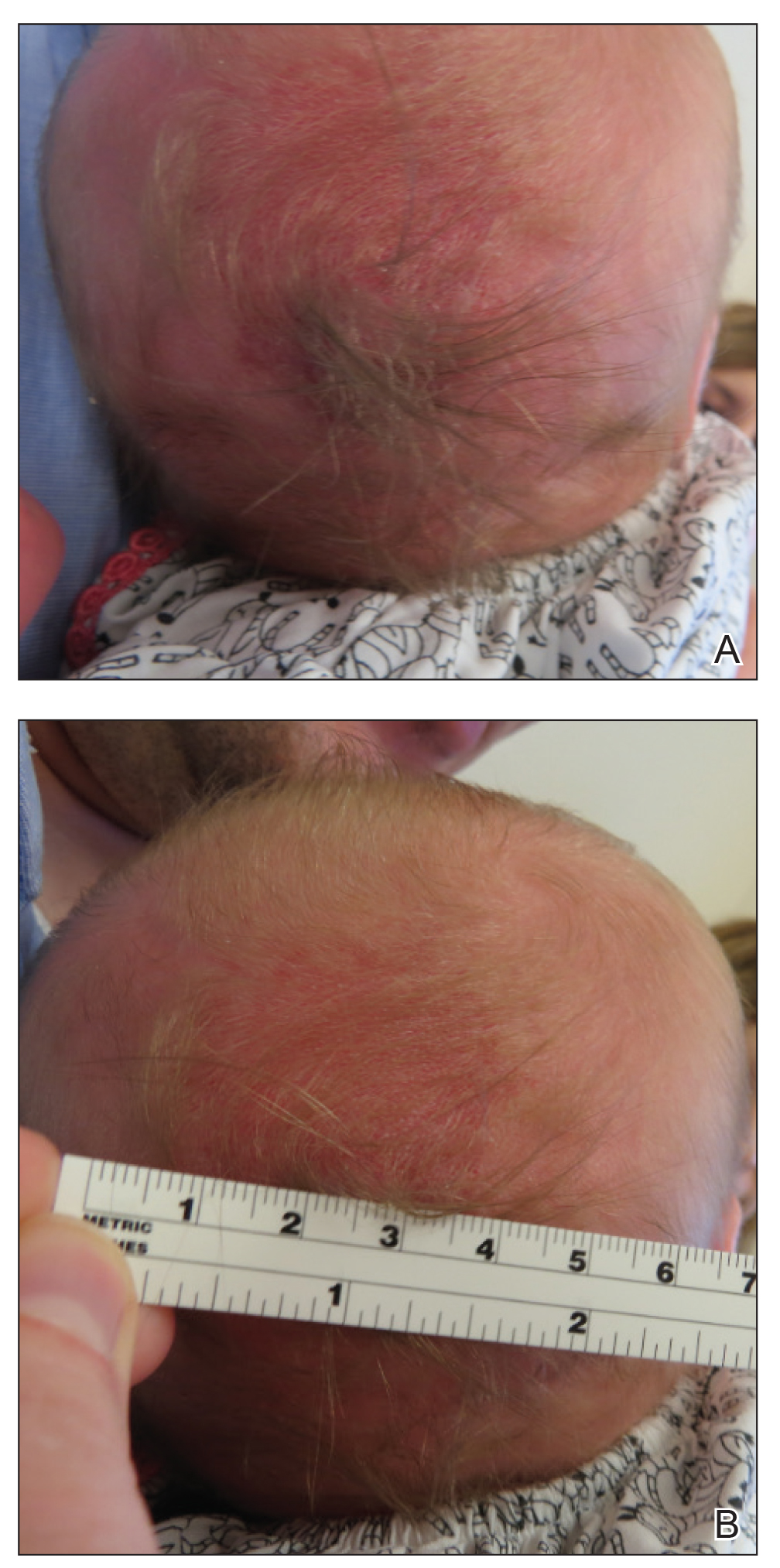
Consultation with the pediatric neurosurgery service was sought, and magnetic resonance imaging of the head was performed, which demonstrated a cystic lesion within the subcutaneous soft tissue in the midline posterior scalp approximately 2 cm above the torcula. There also was a thin stalk extending from the cyst and going through an osseous defect within the occipital bone and attaching to the falx cerebri. There was no evidence of any venous communication with the cerebral sinus tracts or intraparenchymal extension. No intracranial abnormalities were noted. Given the radiographic evidence, a presumptive diagnosis of an atretic cephalocele was made with the plan for surgical repair.
The patient was re-evaluated at 3 and 4 months of age; there were no changes in the size or appearance of the lesion, and she continued to meet all developmental milestones. At 9 months of age the patient underwent uncomplicated neurosurgery to repair the cephalocele. Histopathologic examination of the resected lesion was consistent with an atretic cephalocele and showed positive staining for epithelial membrane antigen, which further confirmed a meningothelial origin; no glial elements were identified. The postoperative course was uncomplicated, and the patient was healing well at a follow-up examination 2 weeks after the procedure.
This case highlights the importance of an extensive workup when a patient presents with a midline lesion and hypertrichosis. The patient’s red patch, excluding the hair tuft, was reminiscent of a vascular malformation or hemangioma precursor lesion given the hypervascularity, the history of the lesion being present since birth, the lack of neurologic symptomatology, and the history of meeting all developmental milestones. The differential diagnosis for this patient was extensive, as many neurologic conditions present with cutaneous findings. Having central nervous system (CNS) and cutaneous comorbidities coincide underscores their common neuroectodermal origin during embryogenesis.1,2
Atretic cephalocele is a rare diagnosis, with the prevalence of cephaloceles estimated to be 0.8 to 3.0 per 10,000 births.3 It typically occurs in either the parietal or occipital scalp as a skin nodule with a hair tuft or alopecic lesion with or without a hair collar. A cephalocele is defined as a skin-covered protrusion of intracranial contents through a bony defect. Central nervous system tissue, meninges, or cerebrospinal fluid can protrude outside the skull with this condition. An atretic cephalocele refers to a cephalocele that arrested in development and represents approximately 40% to 50% of all cephaloceles.4 Various hypotheses have explained the development of atretic cephaloceles: it represents a neural crest remnant, regression of a meningocele in utero, injury of multipotential mesenchymal cells, and either failure of the neural tube to close or reopening of the neural tube after closure.4-6 There is evidence of developmental defects in skin appendages including sweat and sebaceous glands, arrector pili muscles, and hair follicles in and around the skin overlying the cephalocele, suggesting that there is a developmental abnormality of not only the CNS but also the cutaneous tissue.5 Typical radiographic findings include a cystic lesion with underlying defect in the skull. A vertical positioning of the straight sinus also has been demonstrated to be a consistent finding that can aid in diagnosis.4
Imaging is of utmost importance when a patient presents with a tuft of hair on the scalp to rule out intracranial extension and associated abnormalities such as gray matter heterotopia, hypogenesis of the corpus callosum, hydrocephalus, and Dandy-Walker and Walker-Warburg syndromes, which have all been associated with atretic cephaloceles.4,7 The impact of location of the intracranial abnormality on prognosis has been contested, with some reporting a better prognosis with occipital cephalocele vs parietal cephalocele while others have found the opposite to be true.6,7
Cutaneous abnormalities presenting with hypertrichosis (ie, hair tuft, hair collar) and/or capillary malformations increase the likelihood of a cranial dysraphism, especially when these findings present together and occur in and around the midline. Clinical examination cannot rule out an underlying connection to the CNS; these findings require appropriate radiographic imaging assessment prior to any procedural intervention.
- Drolet BA, Clowry L, McTigue K, et al. The hair collar sign: marker for cranial dysraphism. Pediatrics. 1995;96(2, pt 1):309-313.
- Sewell MJ, Chiu YE, Drolet BA. Neural tube dysraphism: review of cutaneous markers and imaging. Pediatr Dermatol. 2015;32:161-170.
- Carvalho DR, Giuliani LR, Simão GN, et al. Autosomal dominant atretic cephalocele with phenotype variability: report of a Brazilian family with six affected in four generation. Am J Med Genet A. 2006;140:1458-1462.
- Bick DS, Brockland JJ, Scott AR. A scalp lesion with intracranial extension. atretic cephalocele. JAMA Otolaryngol Head Neck Surg. 2015;141:289-290.
- Fukuyama M, Tanese K, Yasuda F, et al. Two cases of atretic cephalocele, and histological evaluation of skin appendages in the surrounding skin. Clin Exp Dermatol. 2016;41:48-52.
- Martinez-Lage JF, Sola J, Casas C, et al. Atretic cephalocele: the tip of the iceberg. J Neurosurg. 1992;77:230-235.
- Yakota A, Kajiwara H, Kohchi M, et al. Parietal cephalocele: clinical importance of its atretic form and associated malformation. J Neurosurg. 1988;69:545-551.
To the Editor:
A 2-week-old female infant presented to our dermatology clinic for evaluation of a 4.0×4.5-cm pink-red patch with a 1-cm central nodule and an overlying tuft of hair on the midline occipital region (Figure). The patient was born at 39 weeks’ gestation to nonconsanguineous parents via a normal spontaneous vaginal delivery and had an unremarkable prenatal course with no complications since birth. The red patch and tuft of hair were noted at birth, and the parents reported that the redness varied somewhat in size throughout the day and from day to day. An initial neurologic workup revealed no gross neurologic abnormalities. A head ultrasound revealed a soft-tissue hypervascular nodule that appeared separate from bony structures but showed evidence of a necklike extension from the nodule to the underlying soft tissues. The ultrasound could not definitively rule out intracranial extension; gross brain structures appeared normal. The initial differential diagnosis consisted of a congenital hemangioma (either a rapidly involuting or noninvoluting subtype), meningioma, or cephalocele.
Consultation with the pediatric neurosurgery service was sought, and magnetic resonance imaging of the head was performed, which demonstrated a cystic lesion within the subcutaneous soft tissue in the midline posterior scalp approximately 2 cm above the torcula. There also was a thin stalk extending from the cyst and going through an osseous defect within the occipital bone and attaching to the falx cerebri. There was no evidence of any venous communication with the cerebral sinus tracts or intraparenchymal extension. No intracranial abnormalities were noted. Given the radiographic evidence, a presumptive diagnosis of an atretic cephalocele was made with the plan for surgical repair.
The patient was re-evaluated at 3 and 4 months of age; there were no changes in the size or appearance of the lesion, and she continued to meet all developmental milestones. At 9 months of age the patient underwent uncomplicated neurosurgery to repair the cephalocele. Histopathologic examination of the resected lesion was consistent with an atretic cephalocele and showed positive staining for epithelial membrane antigen, which further confirmed a meningothelial origin; no glial elements were identified. The postoperative course was uncomplicated, and the patient was healing well at a follow-up examination 2 weeks after the procedure.
This case highlights the importance of an extensive workup when a patient presents with a midline lesion and hypertrichosis. The patient’s red patch, excluding the hair tuft, was reminiscent of a vascular malformation or hemangioma precursor lesion given the hypervascularity, the history of the lesion being present since birth, the lack of neurologic symptomatology, and the history of meeting all developmental milestones. The differential diagnosis for this patient was extensive, as many neurologic conditions present with cutaneous findings. Having central nervous system (CNS) and cutaneous comorbidities coincide underscores their common neuroectodermal origin during embryogenesis.1,2
Atretic cephalocele is a rare diagnosis, with the prevalence of cephaloceles estimated to be 0.8 to 3.0 per 10,000 births.3 It typically occurs in either the parietal or occipital scalp as a skin nodule with a hair tuft or alopecic lesion with or without a hair collar. A cephalocele is defined as a skin-covered protrusion of intracranial contents through a bony defect. Central nervous system tissue, meninges, or cerebrospinal fluid can protrude outside the skull with this condition. An atretic cephalocele refers to a cephalocele that arrested in development and represents approximately 40% to 50% of all cephaloceles.4 Various hypotheses have explained the development of atretic cephaloceles: it represents a neural crest remnant, regression of a meningocele in utero, injury of multipotential mesenchymal cells, and either failure of the neural tube to close or reopening of the neural tube after closure.4-6 There is evidence of developmental defects in skin appendages including sweat and sebaceous glands, arrector pili muscles, and hair follicles in and around the skin overlying the cephalocele, suggesting that there is a developmental abnormality of not only the CNS but also the cutaneous tissue.5 Typical radiographic findings include a cystic lesion with underlying defect in the skull. A vertical positioning of the straight sinus also has been demonstrated to be a consistent finding that can aid in diagnosis.4
Imaging is of utmost importance when a patient presents with a tuft of hair on the scalp to rule out intracranial extension and associated abnormalities such as gray matter heterotopia, hypogenesis of the corpus callosum, hydrocephalus, and Dandy-Walker and Walker-Warburg syndromes, which have all been associated with atretic cephaloceles.4,7 The impact of location of the intracranial abnormality on prognosis has been contested, with some reporting a better prognosis with occipital cephalocele vs parietal cephalocele while others have found the opposite to be true.6,7
Cutaneous abnormalities presenting with hypertrichosis (ie, hair tuft, hair collar) and/or capillary malformations increase the likelihood of a cranial dysraphism, especially when these findings present together and occur in and around the midline. Clinical examination cannot rule out an underlying connection to the CNS; these findings require appropriate radiographic imaging assessment prior to any procedural intervention.
To the Editor:
A 2-week-old female infant presented to our dermatology clinic for evaluation of a 4.0×4.5-cm pink-red patch with a 1-cm central nodule and an overlying tuft of hair on the midline occipital region (Figure). The patient was born at 39 weeks’ gestation to nonconsanguineous parents via a normal spontaneous vaginal delivery and had an unremarkable prenatal course with no complications since birth. The red patch and tuft of hair were noted at birth, and the parents reported that the redness varied somewhat in size throughout the day and from day to day. An initial neurologic workup revealed no gross neurologic abnormalities. A head ultrasound revealed a soft-tissue hypervascular nodule that appeared separate from bony structures but showed evidence of a necklike extension from the nodule to the underlying soft tissues. The ultrasound could not definitively rule out intracranial extension; gross brain structures appeared normal. The initial differential diagnosis consisted of a congenital hemangioma (either a rapidly involuting or noninvoluting subtype), meningioma, or cephalocele.
Consultation with the pediatric neurosurgery service was sought, and magnetic resonance imaging of the head was performed, which demonstrated a cystic lesion within the subcutaneous soft tissue in the midline posterior scalp approximately 2 cm above the torcula. There also was a thin stalk extending from the cyst and going through an osseous defect within the occipital bone and attaching to the falx cerebri. There was no evidence of any venous communication with the cerebral sinus tracts or intraparenchymal extension. No intracranial abnormalities were noted. Given the radiographic evidence, a presumptive diagnosis of an atretic cephalocele was made with the plan for surgical repair.
The patient was re-evaluated at 3 and 4 months of age; there were no changes in the size or appearance of the lesion, and she continued to meet all developmental milestones. At 9 months of age the patient underwent uncomplicated neurosurgery to repair the cephalocele. Histopathologic examination of the resected lesion was consistent with an atretic cephalocele and showed positive staining for epithelial membrane antigen, which further confirmed a meningothelial origin; no glial elements were identified. The postoperative course was uncomplicated, and the patient was healing well at a follow-up examination 2 weeks after the procedure.
This case highlights the importance of an extensive workup when a patient presents with a midline lesion and hypertrichosis. The patient’s red patch, excluding the hair tuft, was reminiscent of a vascular malformation or hemangioma precursor lesion given the hypervascularity, the history of the lesion being present since birth, the lack of neurologic symptomatology, and the history of meeting all developmental milestones. The differential diagnosis for this patient was extensive, as many neurologic conditions present with cutaneous findings. Having central nervous system (CNS) and cutaneous comorbidities coincide underscores their common neuroectodermal origin during embryogenesis.1,2
Atretic cephalocele is a rare diagnosis, with the prevalence of cephaloceles estimated to be 0.8 to 3.0 per 10,000 births.3 It typically occurs in either the parietal or occipital scalp as a skin nodule with a hair tuft or alopecic lesion with or without a hair collar. A cephalocele is defined as a skin-covered protrusion of intracranial contents through a bony defect. Central nervous system tissue, meninges, or cerebrospinal fluid can protrude outside the skull with this condition. An atretic cephalocele refers to a cephalocele that arrested in development and represents approximately 40% to 50% of all cephaloceles.4 Various hypotheses have explained the development of atretic cephaloceles: it represents a neural crest remnant, regression of a meningocele in utero, injury of multipotential mesenchymal cells, and either failure of the neural tube to close or reopening of the neural tube after closure.4-6 There is evidence of developmental defects in skin appendages including sweat and sebaceous glands, arrector pili muscles, and hair follicles in and around the skin overlying the cephalocele, suggesting that there is a developmental abnormality of not only the CNS but also the cutaneous tissue.5 Typical radiographic findings include a cystic lesion with underlying defect in the skull. A vertical positioning of the straight sinus also has been demonstrated to be a consistent finding that can aid in diagnosis.4
Imaging is of utmost importance when a patient presents with a tuft of hair on the scalp to rule out intracranial extension and associated abnormalities such as gray matter heterotopia, hypogenesis of the corpus callosum, hydrocephalus, and Dandy-Walker and Walker-Warburg syndromes, which have all been associated with atretic cephaloceles.4,7 The impact of location of the intracranial abnormality on prognosis has been contested, with some reporting a better prognosis with occipital cephalocele vs parietal cephalocele while others have found the opposite to be true.6,7
Cutaneous abnormalities presenting with hypertrichosis (ie, hair tuft, hair collar) and/or capillary malformations increase the likelihood of a cranial dysraphism, especially when these findings present together and occur in and around the midline. Clinical examination cannot rule out an underlying connection to the CNS; these findings require appropriate radiographic imaging assessment prior to any procedural intervention.
- Drolet BA, Clowry L, McTigue K, et al. The hair collar sign: marker for cranial dysraphism. Pediatrics. 1995;96(2, pt 1):309-313.
- Sewell MJ, Chiu YE, Drolet BA. Neural tube dysraphism: review of cutaneous markers and imaging. Pediatr Dermatol. 2015;32:161-170.
- Carvalho DR, Giuliani LR, Simão GN, et al. Autosomal dominant atretic cephalocele with phenotype variability: report of a Brazilian family with six affected in four generation. Am J Med Genet A. 2006;140:1458-1462.
- Bick DS, Brockland JJ, Scott AR. A scalp lesion with intracranial extension. atretic cephalocele. JAMA Otolaryngol Head Neck Surg. 2015;141:289-290.
- Fukuyama M, Tanese K, Yasuda F, et al. Two cases of atretic cephalocele, and histological evaluation of skin appendages in the surrounding skin. Clin Exp Dermatol. 2016;41:48-52.
- Martinez-Lage JF, Sola J, Casas C, et al. Atretic cephalocele: the tip of the iceberg. J Neurosurg. 1992;77:230-235.
- Yakota A, Kajiwara H, Kohchi M, et al. Parietal cephalocele: clinical importance of its atretic form and associated malformation. J Neurosurg. 1988;69:545-551.
- Drolet BA, Clowry L, McTigue K, et al. The hair collar sign: marker for cranial dysraphism. Pediatrics. 1995;96(2, pt 1):309-313.
- Sewell MJ, Chiu YE, Drolet BA. Neural tube dysraphism: review of cutaneous markers and imaging. Pediatr Dermatol. 2015;32:161-170.
- Carvalho DR, Giuliani LR, Simão GN, et al. Autosomal dominant atretic cephalocele with phenotype variability: report of a Brazilian family with six affected in four generation. Am J Med Genet A. 2006;140:1458-1462.
- Bick DS, Brockland JJ, Scott AR. A scalp lesion with intracranial extension. atretic cephalocele. JAMA Otolaryngol Head Neck Surg. 2015;141:289-290.
- Fukuyama M, Tanese K, Yasuda F, et al. Two cases of atretic cephalocele, and histological evaluation of skin appendages in the surrounding skin. Clin Exp Dermatol. 2016;41:48-52.
- Martinez-Lage JF, Sola J, Casas C, et al. Atretic cephalocele: the tip of the iceberg. J Neurosurg. 1992;77:230-235.
- Yakota A, Kajiwara H, Kohchi M, et al. Parietal cephalocele: clinical importance of its atretic form and associated malformation. J Neurosurg. 1988;69:545-551.
Practice Points
- Atretic cephalocele is a rare diagnosis occurring on the scalp as a nodule with an overlying hair tuft or alopecia with or without a hair collar.
- Imaging is of utmost importance when presented with a tuft of hair on the midline to rule out intracranial extension and associated abnormalities.
Rupioid Psoriasis and Psoriatic Arthritis in a Patient With Skin of Color
To the Editor:
A 49-year-old black woman presented with multiple hyperkeratotic papules that progressed over the last 2 months to circular plaques with central thick black crust resembling eschar. She first noticed these lesions as firm, small, black papules on the legs and continued to develop new lesions that eventually evolved into large, coin-shaped, hyperkeratotic plaques. Her medical history was notable for stage III non-Hodgkin follicular lymphoma in remission after treatment with rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone 7 months earlier, and chronic hepatitis B infection being treated with entecavir. Her family history was not remarkable for psoriasis or inflammatory arthritis.
She initially was seen by internal medicine and was started on topical triamcinolone with no improvement of the lesions. At presentation to dermatology, physical examination revealed firm, small, black, hyperkeratotic papules (Figure 1A) and circular plaques with a rim of erythema and central thick, smooth, black crust resembling eschar (Figure 1B). No other skin changes were noted at the time. The bilateral metacarpophalangeal, bilateral proximal interphalangeal, left wrist, and bilateral ankle joints were remarkable for tenderness, swelling, and reduced range of motion. She noted concomitant arthralgia and stiffness but denied fever. She had no other systemic symptoms including night sweats, weight loss, fatigue, malaise, sun sensitivity, oral ulcers, or hair loss. A radiograph of the hand was negative for erosive changes but showed mild periarticular osteopenia and fusiform soft tissue swelling of the third digit. Given the central appearance of eschar in the larger lesions, the initial differential diagnosis included Sweet syndrome, invasive fungal infection, vasculitis, and recurrent lymphoma.
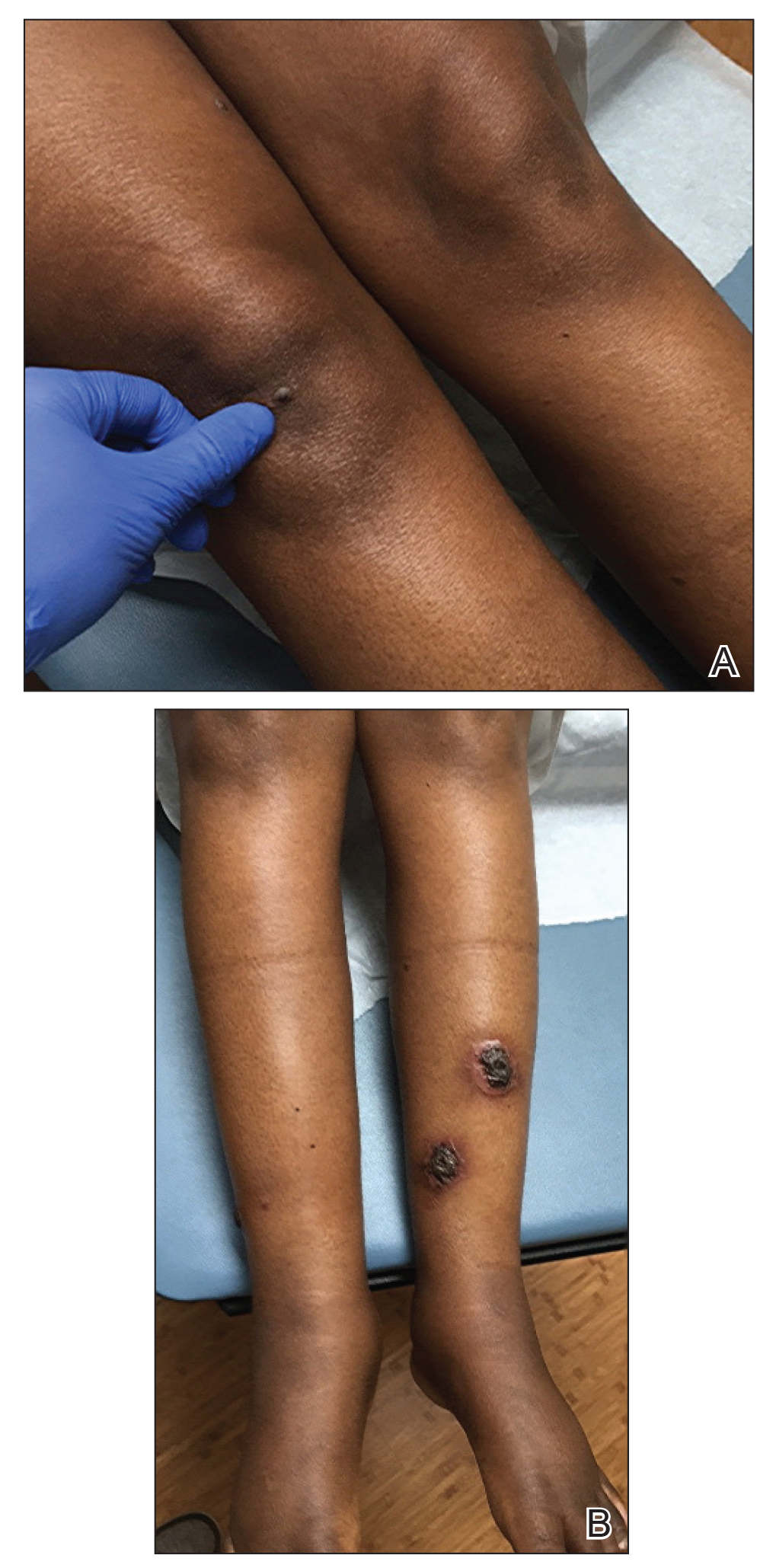
A 4-mm punch biopsy specimen of a representative lesion on the right leg revealed psoriasiform epidermal hyperplasia, parakeratosis, neutrophils in the stratum corneum and spinosum, elongation of the rete ridges, and superficial vascular ectasia, which favored a diagnosis of psoriasis (Figure 2). A periodic acid-Schiff stain was negative for fungal hyphae. Fungal culture, bacterial tissue culture, and acid-fast bacilli smear were negative. Absence of deep dermal inflammation precluded a diagnosis of Sweet syndrome. Further notable laboratory studies included negative human immunodeficiency virus (HIV) antibody, rapid plasma reagin, hepatitis C antibody, and rheumatoid factor.
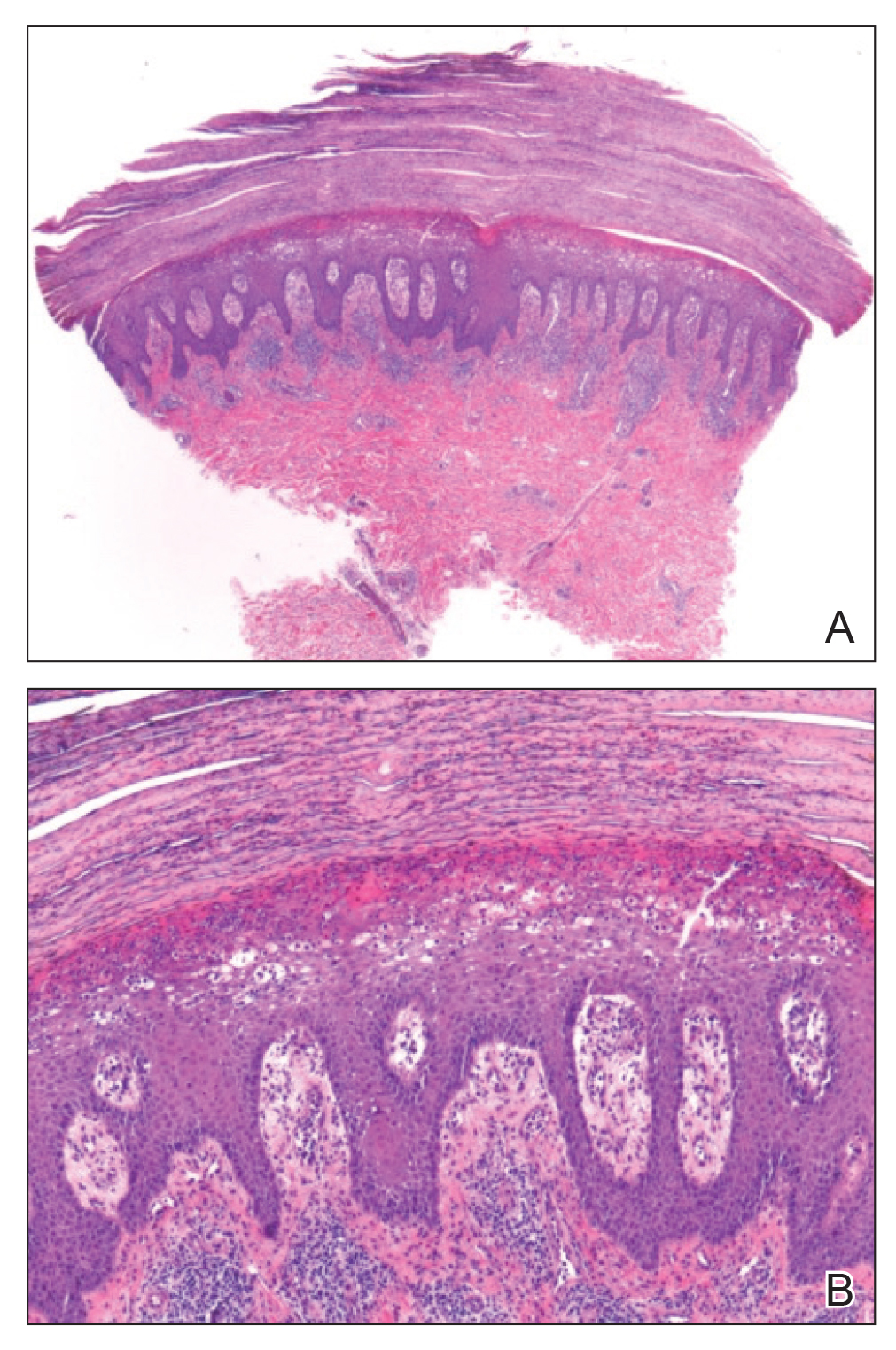
At follow-up 2 weeks later, the initial lesions were still present, and she had developed new widespread, well-demarcated, erythematous plaques with silver scale along the scalp, back, chest, and abdomen that were more typical of psoriasis. Oil spots were noted on several fingernails and toenails. Based on the clinicopathologic findings, nail changes, and asymmetric inflammatory arthritis, a diagnosis of rupioid psoriasis with psoriatic arthritis (PsA) was established. Treatment with clobetasol ointment 0.05% twice daily to active lesions was started. Initiation of systemic therapy with a steroid-sparing agent was deferred in anticipation of care coordination with rheumatology, hepatology, and hematology/oncology due to the patient's history of follicular lymphoma and chronic hepatitis B. Although attempts were made to avoid systemic corticosteroids due to the risk for a psoriasis flare upon discontinuation, because of the severity of arthralgia she was started on oral prednisone 20 mg daily by rheumatology with plans for a slow taper once an alternative systemic agent was started.1
At 10-week follow-up, the patient had marked improvement of psoriatic plaques with no active lesions while only on prednisone 20 mg daily. In consultation with her care team, she subsequently was started on methotrexate 10 mg weekly for 2 weeks followed by titration to 15 mg weekly. Plans were to start a prednisone taper after a month of methotrexate to allow her new treatment time for therapeutic effect. Notably, the patient chose to discontinue prednisone 2 weeks into methotrexate therapy after only two 10-mg doses of methotrexate weekly and well before therapeutic levels were achieved. Despite stopping prednisone early and without a taper, she did not experience a relapse in psoriatic skin lesions. Three months following initiation of methotrexate, she sustained resolution of the cutaneous lesions with only residual postinflammatory hyperpigmentation.
Psoriasis is a common chronic inflammatory skin disorder with multiple clinical presentations. There are several variants of psoriasis that are classified by their morphologic appearance including chronic plaque, guttate, erythrodermic, and pustular, with more than 90% of cases representing the plaque variant. Less common clinical presentations of psoriasis include rupioid, ostraceous, inverse, elephantine, and HIV associated.2 Rupioid psoriasis is a rare variant that presents with cone-shaped, limpetlike lesions.3,4 Similar to the limited epidemiological and clinical data pertaining to psoriasis in nonwhite racial groups, there also is a paucity of documented reports of rupioid psoriasis in skin of color.
Rupioid comes from the Greek word rhupos, meaning dirt or filth, and is used to describe well-demarcated lesions with thick, yellow, dirty-appearing, adherent crusts resembling oyster shells with a surrounding rim of erythema.5 Rupioid psoriasis initially was reported in 1948 and remains an uncommon and infrequently reported variant.6 The majority of reported cases have been associated with arthropathy, similar to our patient.3,4 Rupioid lesions also have been observed in an array of other diseases, such as secondary syphilis, crusted scabies, disseminated histoplasmosis, HIV, reactive arthritis, and aminoaciduria.7-11
Diagnosis of rupioid psoriasis can be confirmed with a skin biopsy, which demonstrates characteristic histopathologic findings of psoriasis.3 Laboratory analysis should be performed to rule out other causes of rupioid lesions, and PsA should be differentiated from rheumatoid arthritis if arthropathy is present. In our case, serum rapid plasma reagin, anti-HIV antibody, rheumatoid factor, and fungal cultures were negative. Usin0)g clinical findings, histopathology, laboratory analyses, and radiograph findings, the diagnosis of rupioid psoriasis with PsA was confirmed in our patient.
Psoriasis was not originally suspected in our patient due to the noncharacteristic lesions with smooth black crust--similar appearing to eschar--and the patient's complicated medical history. Variations in the presentation of psoriasis among white individuals and those with skin of color have been reported in the literature.12,13 Psoriatic lesions in darker skin tones may appear more violaceous or hyperpigmented with more conspicuous erythema and thicker plaques. Our patient lacked the classic rupioid appearance of concentric circular layers of dirty, yellow, oysterlike scale, and instead had thick, lamellate, black crust. A PubMed search of articles indexed for MEDLINE using the terms rupioid, coral reef psoriasis, rupioides, and rhupus revealed no other cases of rupioid psoriasis reported in black patients and no cases detailing the variations of rupioid lesions in skin of color. A case of rupioid psoriasis has been reported in a Hispanic patient, but the described psoriatic lesions were more characteristic of the dirty-appearing, conic plaques previously reported.14 Our case highlights a unique example of the variable presentations of cutaneous disorders in skin of color and black patients.
Our patient's case of rupioid psoriasis with PsA presented unique challenges for systemic treatment due to her multiple comorbidities. Rupioid psoriasis most often is treated with combination topical and systemic therapy, with agents such as methotrexate and cyclosporine having prior success.3,4 This variant of psoriasis is highly responsive to treatment, and marked improvement of lesions has been achieved with topical steroids alone with proper adherence.15 Our patient was started on clobetasol ointment 0.05% while a systemic agent was debated for her PsA. Although she did not have improvement with topical therapy alone, she experienced rapid resolution of the skin lesions after initiation of low-dose prednisone 20 mg daily. Interestingly, our patient did not experience a flare of the skin lesions upon discontinuation of systemic steroids despite the lack of an appropriate taper and methotrexate not having reached therapeutic levels.
The clinical nuances of rupioid psoriasis in skin of color have not yet been described and remain an important diagnostic consideration. Our patient achieved remission of skin lesions with sequential treatment of topical clobetasol, a low-dose systemic steroid, and methotrexate. Based on available reports, rupioid psoriasis may represent a variant of psoriasis that is highly responsive to treatment.
- Mrowietz U, Domm S. Systemic steroids in the treatment of psoriasis: what is fact, what is fiction? J Eur Acad Dermatol Venereol. 2013;27:1022-1025.
- Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: The McGraw-Hill Companies; 2012.
- Wang JL, Yang JH. Rupioid psoriasis associated with arthropathy. J Dermatol. 1997;24:46-49.
- Murakami T, Ohtsuki M, Nakagawa H. Rupioid psoriasis with arthropathy. Clin Exp Dermatol. 2000;25:409-412.
- Chung HJ, Marley-Kemp D, Keller M. Rupioid psoriasis and other skin diseases with rupioid manifestations. Cutis. 2014;94:119-121.
- Salamon M, Omulecki A, Sysa-Jedrzejowska A, et al. Psoriasisrupioides: a rare variant of a common disease. Cutis. 2011;88:135-137.
- Krase IZ, Cavanaugh K, Curiel-Lewandrowski C. A case of rupioid syphilis. JAAD Case Rep. 2016;2:141-143.
- Garofalo V, Saraceno R, Milana M, et al. Crusted scabies in a liver transplant patient mimicking rupioid psoriasis. Eur J Dermatol. 2016;26:495-496.
- Corti M, Villafane MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Sehgal VN, Koranne RV, Shyam Prasad AL. Unusual manifestations of Reiter's disease in a child. Dermatologica. 1985;170:77-79.
- Haim S, Gilhar A, Cohen A. Cutaneous manifestations associated with aminoaciduria. report of two cases. Dermatologica. 1978;156:244-250.
- McMichael AJ, Vachiramon V, Guzman-Sanchez DA, et al. Psoriasis in African-Americans: a caregivers' survey. J Drugs Dermatol. 2012;11:478-482.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Posligua A, Maldonado C, Gonzalez MG. Rupioid psoriasis preceded by varicella presenting as Koebner phenomenon. J Am Acad Dermatol. 2016;74(5 suppl 1):AB268.
- Feldman SR, Feldman S, Brown K, et al. "Coral reef" psoriasis: a marker of resistance to topical treatment. J Dermatolog Treat. 2008;19:257-258.
To the Editor:
A 49-year-old black woman presented with multiple hyperkeratotic papules that progressed over the last 2 months to circular plaques with central thick black crust resembling eschar. She first noticed these lesions as firm, small, black papules on the legs and continued to develop new lesions that eventually evolved into large, coin-shaped, hyperkeratotic plaques. Her medical history was notable for stage III non-Hodgkin follicular lymphoma in remission after treatment with rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone 7 months earlier, and chronic hepatitis B infection being treated with entecavir. Her family history was not remarkable for psoriasis or inflammatory arthritis.
She initially was seen by internal medicine and was started on topical triamcinolone with no improvement of the lesions. At presentation to dermatology, physical examination revealed firm, small, black, hyperkeratotic papules (Figure 1A) and circular plaques with a rim of erythema and central thick, smooth, black crust resembling eschar (Figure 1B). No other skin changes were noted at the time. The bilateral metacarpophalangeal, bilateral proximal interphalangeal, left wrist, and bilateral ankle joints were remarkable for tenderness, swelling, and reduced range of motion. She noted concomitant arthralgia and stiffness but denied fever. She had no other systemic symptoms including night sweats, weight loss, fatigue, malaise, sun sensitivity, oral ulcers, or hair loss. A radiograph of the hand was negative for erosive changes but showed mild periarticular osteopenia and fusiform soft tissue swelling of the third digit. Given the central appearance of eschar in the larger lesions, the initial differential diagnosis included Sweet syndrome, invasive fungal infection, vasculitis, and recurrent lymphoma.
A 4-mm punch biopsy specimen of a representative lesion on the right leg revealed psoriasiform epidermal hyperplasia, parakeratosis, neutrophils in the stratum corneum and spinosum, elongation of the rete ridges, and superficial vascular ectasia, which favored a diagnosis of psoriasis (Figure 2). A periodic acid-Schiff stain was negative for fungal hyphae. Fungal culture, bacterial tissue culture, and acid-fast bacilli smear were negative. Absence of deep dermal inflammation precluded a diagnosis of Sweet syndrome. Further notable laboratory studies included negative human immunodeficiency virus (HIV) antibody, rapid plasma reagin, hepatitis C antibody, and rheumatoid factor.
At follow-up 2 weeks later, the initial lesions were still present, and she had developed new widespread, well-demarcated, erythematous plaques with silver scale along the scalp, back, chest, and abdomen that were more typical of psoriasis. Oil spots were noted on several fingernails and toenails. Based on the clinicopathologic findings, nail changes, and asymmetric inflammatory arthritis, a diagnosis of rupioid psoriasis with psoriatic arthritis (PsA) was established. Treatment with clobetasol ointment 0.05% twice daily to active lesions was started. Initiation of systemic therapy with a steroid-sparing agent was deferred in anticipation of care coordination with rheumatology, hepatology, and hematology/oncology due to the patient's history of follicular lymphoma and chronic hepatitis B. Although attempts were made to avoid systemic corticosteroids due to the risk for a psoriasis flare upon discontinuation, because of the severity of arthralgia she was started on oral prednisone 20 mg daily by rheumatology with plans for a slow taper once an alternative systemic agent was started.1
At 10-week follow-up, the patient had marked improvement of psoriatic plaques with no active lesions while only on prednisone 20 mg daily. In consultation with her care team, she subsequently was started on methotrexate 10 mg weekly for 2 weeks followed by titration to 15 mg weekly. Plans were to start a prednisone taper after a month of methotrexate to allow her new treatment time for therapeutic effect. Notably, the patient chose to discontinue prednisone 2 weeks into methotrexate therapy after only two 10-mg doses of methotrexate weekly and well before therapeutic levels were achieved. Despite stopping prednisone early and without a taper, she did not experience a relapse in psoriatic skin lesions. Three months following initiation of methotrexate, she sustained resolution of the cutaneous lesions with only residual postinflammatory hyperpigmentation.
Psoriasis is a common chronic inflammatory skin disorder with multiple clinical presentations. There are several variants of psoriasis that are classified by their morphologic appearance including chronic plaque, guttate, erythrodermic, and pustular, with more than 90% of cases representing the plaque variant. Less common clinical presentations of psoriasis include rupioid, ostraceous, inverse, elephantine, and HIV associated.2 Rupioid psoriasis is a rare variant that presents with cone-shaped, limpetlike lesions.3,4 Similar to the limited epidemiological and clinical data pertaining to psoriasis in nonwhite racial groups, there also is a paucity of documented reports of rupioid psoriasis in skin of color.
Rupioid comes from the Greek word rhupos, meaning dirt or filth, and is used to describe well-demarcated lesions with thick, yellow, dirty-appearing, adherent crusts resembling oyster shells with a surrounding rim of erythema.5 Rupioid psoriasis initially was reported in 1948 and remains an uncommon and infrequently reported variant.6 The majority of reported cases have been associated with arthropathy, similar to our patient.3,4 Rupioid lesions also have been observed in an array of other diseases, such as secondary syphilis, crusted scabies, disseminated histoplasmosis, HIV, reactive arthritis, and aminoaciduria.7-11
Diagnosis of rupioid psoriasis can be confirmed with a skin biopsy, which demonstrates characteristic histopathologic findings of psoriasis.3 Laboratory analysis should be performed to rule out other causes of rupioid lesions, and PsA should be differentiated from rheumatoid arthritis if arthropathy is present. In our case, serum rapid plasma reagin, anti-HIV antibody, rheumatoid factor, and fungal cultures were negative. Usin0)g clinical findings, histopathology, laboratory analyses, and radiograph findings, the diagnosis of rupioid psoriasis with PsA was confirmed in our patient.
Psoriasis was not originally suspected in our patient due to the noncharacteristic lesions with smooth black crust--similar appearing to eschar--and the patient's complicated medical history. Variations in the presentation of psoriasis among white individuals and those with skin of color have been reported in the literature.12,13 Psoriatic lesions in darker skin tones may appear more violaceous or hyperpigmented with more conspicuous erythema and thicker plaques. Our patient lacked the classic rupioid appearance of concentric circular layers of dirty, yellow, oysterlike scale, and instead had thick, lamellate, black crust. A PubMed search of articles indexed for MEDLINE using the terms rupioid, coral reef psoriasis, rupioides, and rhupus revealed no other cases of rupioid psoriasis reported in black patients and no cases detailing the variations of rupioid lesions in skin of color. A case of rupioid psoriasis has been reported in a Hispanic patient, but the described psoriatic lesions were more characteristic of the dirty-appearing, conic plaques previously reported.14 Our case highlights a unique example of the variable presentations of cutaneous disorders in skin of color and black patients.
Our patient's case of rupioid psoriasis with PsA presented unique challenges for systemic treatment due to her multiple comorbidities. Rupioid psoriasis most often is treated with combination topical and systemic therapy, with agents such as methotrexate and cyclosporine having prior success.3,4 This variant of psoriasis is highly responsive to treatment, and marked improvement of lesions has been achieved with topical steroids alone with proper adherence.15 Our patient was started on clobetasol ointment 0.05% while a systemic agent was debated for her PsA. Although she did not have improvement with topical therapy alone, she experienced rapid resolution of the skin lesions after initiation of low-dose prednisone 20 mg daily. Interestingly, our patient did not experience a flare of the skin lesions upon discontinuation of systemic steroids despite the lack of an appropriate taper and methotrexate not having reached therapeutic levels.
The clinical nuances of rupioid psoriasis in skin of color have not yet been described and remain an important diagnostic consideration. Our patient achieved remission of skin lesions with sequential treatment of topical clobetasol, a low-dose systemic steroid, and methotrexate. Based on available reports, rupioid psoriasis may represent a variant of psoriasis that is highly responsive to treatment.
To the Editor:
A 49-year-old black woman presented with multiple hyperkeratotic papules that progressed over the last 2 months to circular plaques with central thick black crust resembling eschar. She first noticed these lesions as firm, small, black papules on the legs and continued to develop new lesions that eventually evolved into large, coin-shaped, hyperkeratotic plaques. Her medical history was notable for stage III non-Hodgkin follicular lymphoma in remission after treatment with rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone 7 months earlier, and chronic hepatitis B infection being treated with entecavir. Her family history was not remarkable for psoriasis or inflammatory arthritis.
She initially was seen by internal medicine and was started on topical triamcinolone with no improvement of the lesions. At presentation to dermatology, physical examination revealed firm, small, black, hyperkeratotic papules (Figure 1A) and circular plaques with a rim of erythema and central thick, smooth, black crust resembling eschar (Figure 1B). No other skin changes were noted at the time. The bilateral metacarpophalangeal, bilateral proximal interphalangeal, left wrist, and bilateral ankle joints were remarkable for tenderness, swelling, and reduced range of motion. She noted concomitant arthralgia and stiffness but denied fever. She had no other systemic symptoms including night sweats, weight loss, fatigue, malaise, sun sensitivity, oral ulcers, or hair loss. A radiograph of the hand was negative for erosive changes but showed mild periarticular osteopenia and fusiform soft tissue swelling of the third digit. Given the central appearance of eschar in the larger lesions, the initial differential diagnosis included Sweet syndrome, invasive fungal infection, vasculitis, and recurrent lymphoma.
A 4-mm punch biopsy specimen of a representative lesion on the right leg revealed psoriasiform epidermal hyperplasia, parakeratosis, neutrophils in the stratum corneum and spinosum, elongation of the rete ridges, and superficial vascular ectasia, which favored a diagnosis of psoriasis (Figure 2). A periodic acid-Schiff stain was negative for fungal hyphae. Fungal culture, bacterial tissue culture, and acid-fast bacilli smear were negative. Absence of deep dermal inflammation precluded a diagnosis of Sweet syndrome. Further notable laboratory studies included negative human immunodeficiency virus (HIV) antibody, rapid plasma reagin, hepatitis C antibody, and rheumatoid factor.
At follow-up 2 weeks later, the initial lesions were still present, and she had developed new widespread, well-demarcated, erythematous plaques with silver scale along the scalp, back, chest, and abdomen that were more typical of psoriasis. Oil spots were noted on several fingernails and toenails. Based on the clinicopathologic findings, nail changes, and asymmetric inflammatory arthritis, a diagnosis of rupioid psoriasis with psoriatic arthritis (PsA) was established. Treatment with clobetasol ointment 0.05% twice daily to active lesions was started. Initiation of systemic therapy with a steroid-sparing agent was deferred in anticipation of care coordination with rheumatology, hepatology, and hematology/oncology due to the patient's history of follicular lymphoma and chronic hepatitis B. Although attempts were made to avoid systemic corticosteroids due to the risk for a psoriasis flare upon discontinuation, because of the severity of arthralgia she was started on oral prednisone 20 mg daily by rheumatology with plans for a slow taper once an alternative systemic agent was started.1
At 10-week follow-up, the patient had marked improvement of psoriatic plaques with no active lesions while only on prednisone 20 mg daily. In consultation with her care team, she subsequently was started on methotrexate 10 mg weekly for 2 weeks followed by titration to 15 mg weekly. Plans were to start a prednisone taper after a month of methotrexate to allow her new treatment time for therapeutic effect. Notably, the patient chose to discontinue prednisone 2 weeks into methotrexate therapy after only two 10-mg doses of methotrexate weekly and well before therapeutic levels were achieved. Despite stopping prednisone early and without a taper, she did not experience a relapse in psoriatic skin lesions. Three months following initiation of methotrexate, she sustained resolution of the cutaneous lesions with only residual postinflammatory hyperpigmentation.
Psoriasis is a common chronic inflammatory skin disorder with multiple clinical presentations. There are several variants of psoriasis that are classified by their morphologic appearance including chronic plaque, guttate, erythrodermic, and pustular, with more than 90% of cases representing the plaque variant. Less common clinical presentations of psoriasis include rupioid, ostraceous, inverse, elephantine, and HIV associated.2 Rupioid psoriasis is a rare variant that presents with cone-shaped, limpetlike lesions.3,4 Similar to the limited epidemiological and clinical data pertaining to psoriasis in nonwhite racial groups, there also is a paucity of documented reports of rupioid psoriasis in skin of color.
Rupioid comes from the Greek word rhupos, meaning dirt or filth, and is used to describe well-demarcated lesions with thick, yellow, dirty-appearing, adherent crusts resembling oyster shells with a surrounding rim of erythema.5 Rupioid psoriasis initially was reported in 1948 and remains an uncommon and infrequently reported variant.6 The majority of reported cases have been associated with arthropathy, similar to our patient.3,4 Rupioid lesions also have been observed in an array of other diseases, such as secondary syphilis, crusted scabies, disseminated histoplasmosis, HIV, reactive arthritis, and aminoaciduria.7-11
Diagnosis of rupioid psoriasis can be confirmed with a skin biopsy, which demonstrates characteristic histopathologic findings of psoriasis.3 Laboratory analysis should be performed to rule out other causes of rupioid lesions, and PsA should be differentiated from rheumatoid arthritis if arthropathy is present. In our case, serum rapid plasma reagin, anti-HIV antibody, rheumatoid factor, and fungal cultures were negative. Usin0)g clinical findings, histopathology, laboratory analyses, and radiograph findings, the diagnosis of rupioid psoriasis with PsA was confirmed in our patient.
Psoriasis was not originally suspected in our patient due to the noncharacteristic lesions with smooth black crust--similar appearing to eschar--and the patient's complicated medical history. Variations in the presentation of psoriasis among white individuals and those with skin of color have been reported in the literature.12,13 Psoriatic lesions in darker skin tones may appear more violaceous or hyperpigmented with more conspicuous erythema and thicker plaques. Our patient lacked the classic rupioid appearance of concentric circular layers of dirty, yellow, oysterlike scale, and instead had thick, lamellate, black crust. A PubMed search of articles indexed for MEDLINE using the terms rupioid, coral reef psoriasis, rupioides, and rhupus revealed no other cases of rupioid psoriasis reported in black patients and no cases detailing the variations of rupioid lesions in skin of color. A case of rupioid psoriasis has been reported in a Hispanic patient, but the described psoriatic lesions were more characteristic of the dirty-appearing, conic plaques previously reported.14 Our case highlights a unique example of the variable presentations of cutaneous disorders in skin of color and black patients.
Our patient's case of rupioid psoriasis with PsA presented unique challenges for systemic treatment due to her multiple comorbidities. Rupioid psoriasis most often is treated with combination topical and systemic therapy, with agents such as methotrexate and cyclosporine having prior success.3,4 This variant of psoriasis is highly responsive to treatment, and marked improvement of lesions has been achieved with topical steroids alone with proper adherence.15 Our patient was started on clobetasol ointment 0.05% while a systemic agent was debated for her PsA. Although she did not have improvement with topical therapy alone, she experienced rapid resolution of the skin lesions after initiation of low-dose prednisone 20 mg daily. Interestingly, our patient did not experience a flare of the skin lesions upon discontinuation of systemic steroids despite the lack of an appropriate taper and methotrexate not having reached therapeutic levels.
The clinical nuances of rupioid psoriasis in skin of color have not yet been described and remain an important diagnostic consideration. Our patient achieved remission of skin lesions with sequential treatment of topical clobetasol, a low-dose systemic steroid, and methotrexate. Based on available reports, rupioid psoriasis may represent a variant of psoriasis that is highly responsive to treatment.
- Mrowietz U, Domm S. Systemic steroids in the treatment of psoriasis: what is fact, what is fiction? J Eur Acad Dermatol Venereol. 2013;27:1022-1025.
- Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: The McGraw-Hill Companies; 2012.
- Wang JL, Yang JH. Rupioid psoriasis associated with arthropathy. J Dermatol. 1997;24:46-49.
- Murakami T, Ohtsuki M, Nakagawa H. Rupioid psoriasis with arthropathy. Clin Exp Dermatol. 2000;25:409-412.
- Chung HJ, Marley-Kemp D, Keller M. Rupioid psoriasis and other skin diseases with rupioid manifestations. Cutis. 2014;94:119-121.
- Salamon M, Omulecki A, Sysa-Jedrzejowska A, et al. Psoriasisrupioides: a rare variant of a common disease. Cutis. 2011;88:135-137.
- Krase IZ, Cavanaugh K, Curiel-Lewandrowski C. A case of rupioid syphilis. JAAD Case Rep. 2016;2:141-143.
- Garofalo V, Saraceno R, Milana M, et al. Crusted scabies in a liver transplant patient mimicking rupioid psoriasis. Eur J Dermatol. 2016;26:495-496.
- Corti M, Villafane MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Sehgal VN, Koranne RV, Shyam Prasad AL. Unusual manifestations of Reiter's disease in a child. Dermatologica. 1985;170:77-79.
- Haim S, Gilhar A, Cohen A. Cutaneous manifestations associated with aminoaciduria. report of two cases. Dermatologica. 1978;156:244-250.
- McMichael AJ, Vachiramon V, Guzman-Sanchez DA, et al. Psoriasis in African-Americans: a caregivers' survey. J Drugs Dermatol. 2012;11:478-482.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Posligua A, Maldonado C, Gonzalez MG. Rupioid psoriasis preceded by varicella presenting as Koebner phenomenon. J Am Acad Dermatol. 2016;74(5 suppl 1):AB268.
- Feldman SR, Feldman S, Brown K, et al. "Coral reef" psoriasis: a marker of resistance to topical treatment. J Dermatolog Treat. 2008;19:257-258.
- Mrowietz U, Domm S. Systemic steroids in the treatment of psoriasis: what is fact, what is fiction? J Eur Acad Dermatol Venereol. 2013;27:1022-1025.
- Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: The McGraw-Hill Companies; 2012.
- Wang JL, Yang JH. Rupioid psoriasis associated with arthropathy. J Dermatol. 1997;24:46-49.
- Murakami T, Ohtsuki M, Nakagawa H. Rupioid psoriasis with arthropathy. Clin Exp Dermatol. 2000;25:409-412.
- Chung HJ, Marley-Kemp D, Keller M. Rupioid psoriasis and other skin diseases with rupioid manifestations. Cutis. 2014;94:119-121.
- Salamon M, Omulecki A, Sysa-Jedrzejowska A, et al. Psoriasisrupioides: a rare variant of a common disease. Cutis. 2011;88:135-137.
- Krase IZ, Cavanaugh K, Curiel-Lewandrowski C. A case of rupioid syphilis. JAAD Case Rep. 2016;2:141-143.
- Garofalo V, Saraceno R, Milana M, et al. Crusted scabies in a liver transplant patient mimicking rupioid psoriasis. Eur J Dermatol. 2016;26:495-496.
- Corti M, Villafane MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Sehgal VN, Koranne RV, Shyam Prasad AL. Unusual manifestations of Reiter's disease in a child. Dermatologica. 1985;170:77-79.
- Haim S, Gilhar A, Cohen A. Cutaneous manifestations associated with aminoaciduria. report of two cases. Dermatologica. 1978;156:244-250.
- McMichael AJ, Vachiramon V, Guzman-Sanchez DA, et al. Psoriasis in African-Americans: a caregivers' survey. J Drugs Dermatol. 2012;11:478-482.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Posligua A, Maldonado C, Gonzalez MG. Rupioid psoriasis preceded by varicella presenting as Koebner phenomenon. J Am Acad Dermatol. 2016;74(5 suppl 1):AB268.
- Feldman SR, Feldman S, Brown K, et al. "Coral reef" psoriasis: a marker of resistance to topical treatment. J Dermatolog Treat. 2008;19:257-258.
Practice Points
- Rupioid psoriasis in skin of color may present a diagnostic challenge for health care providers.
- Rupioid psoriasis may represent a psoriasis variant that is highly responsive to treatment.

Granular Parakeratosis
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
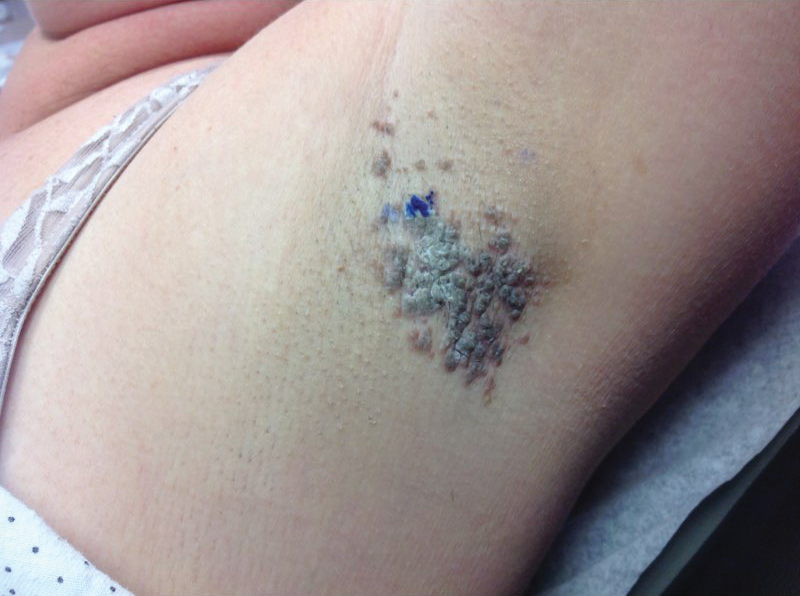
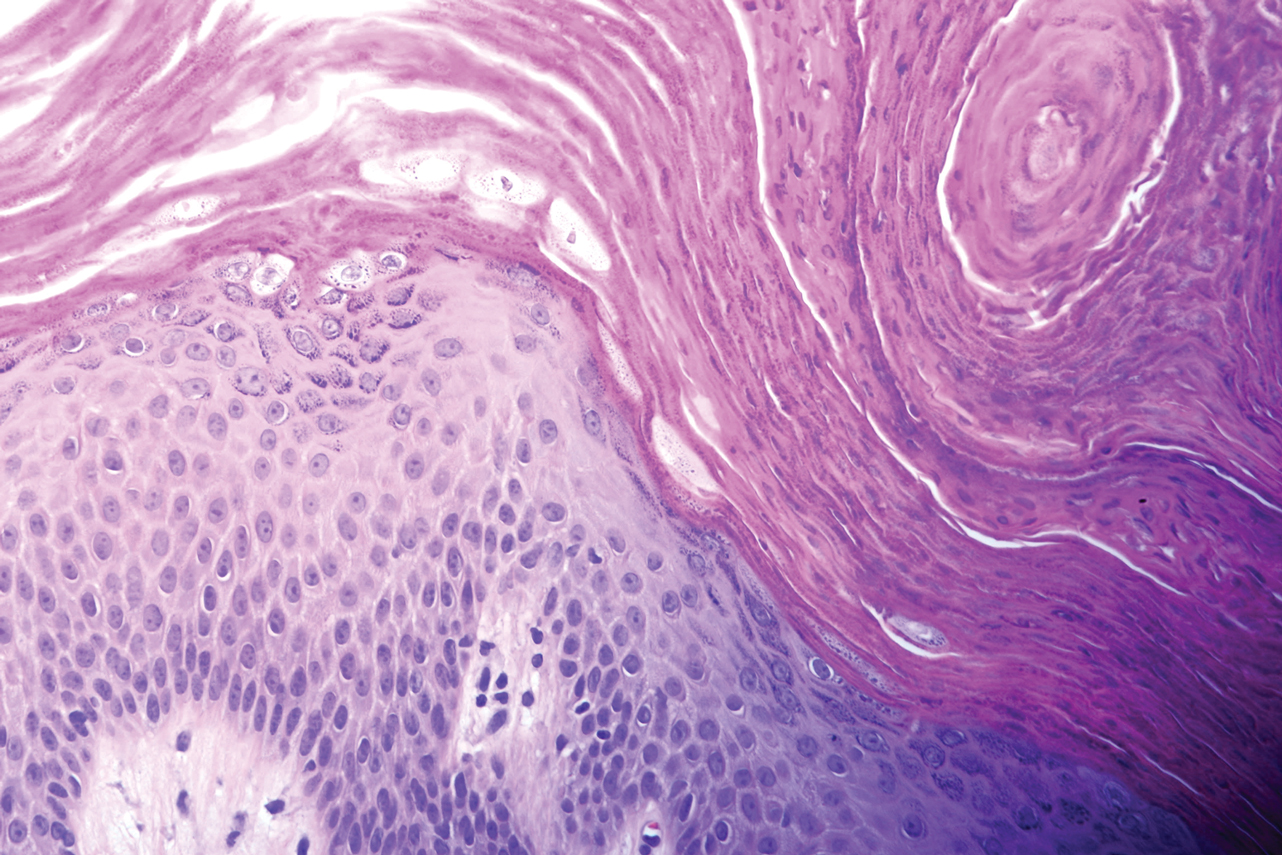
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
Practice Points
- Granular parakeratosis most commonly presents in middle-aged women in the axillae.
- The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.
- Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products.
Eczema Herpeticum in a Patient With Hailey-Hailey Disease Confounded by Coexistent Psoriasis
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
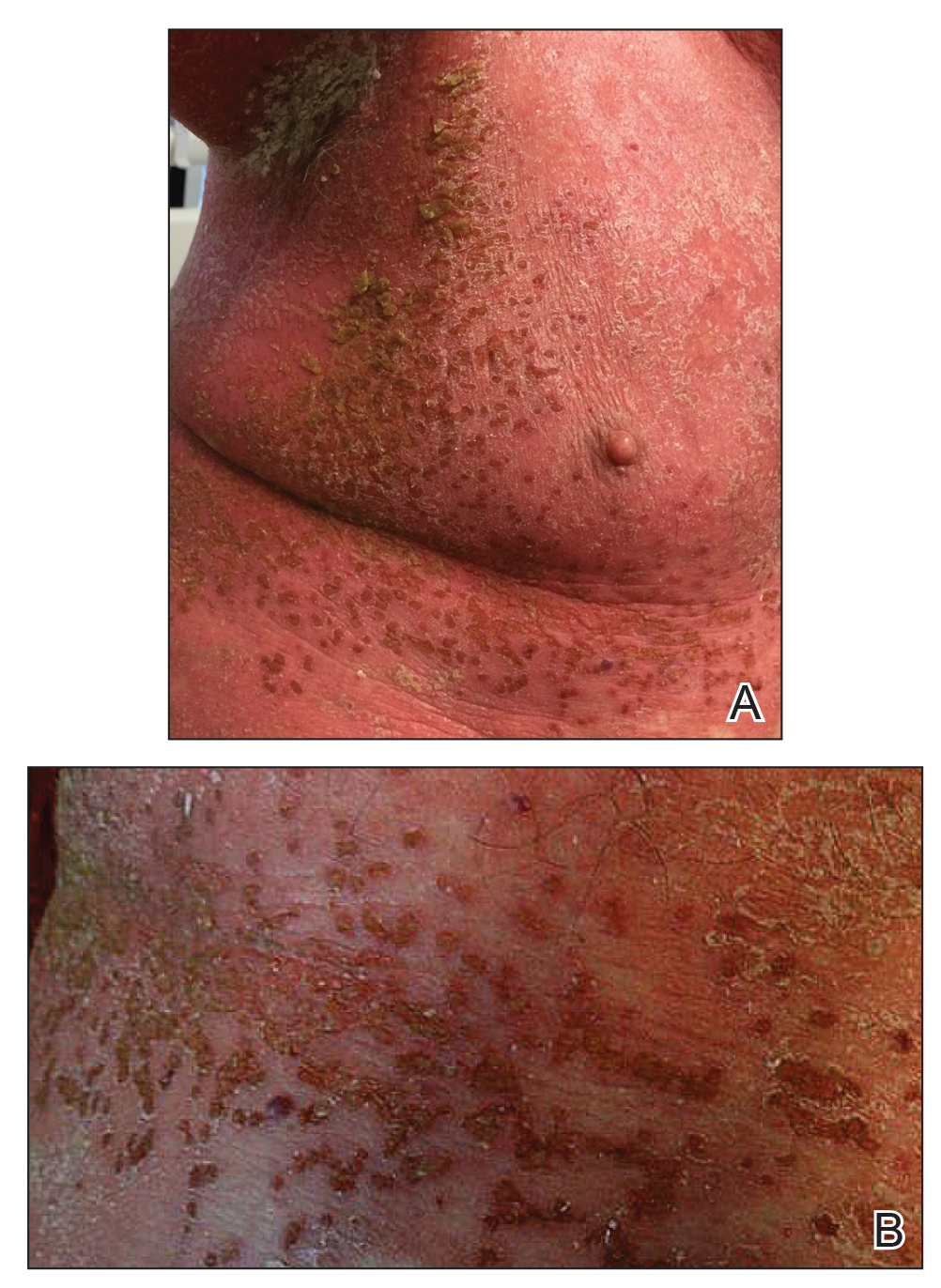
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
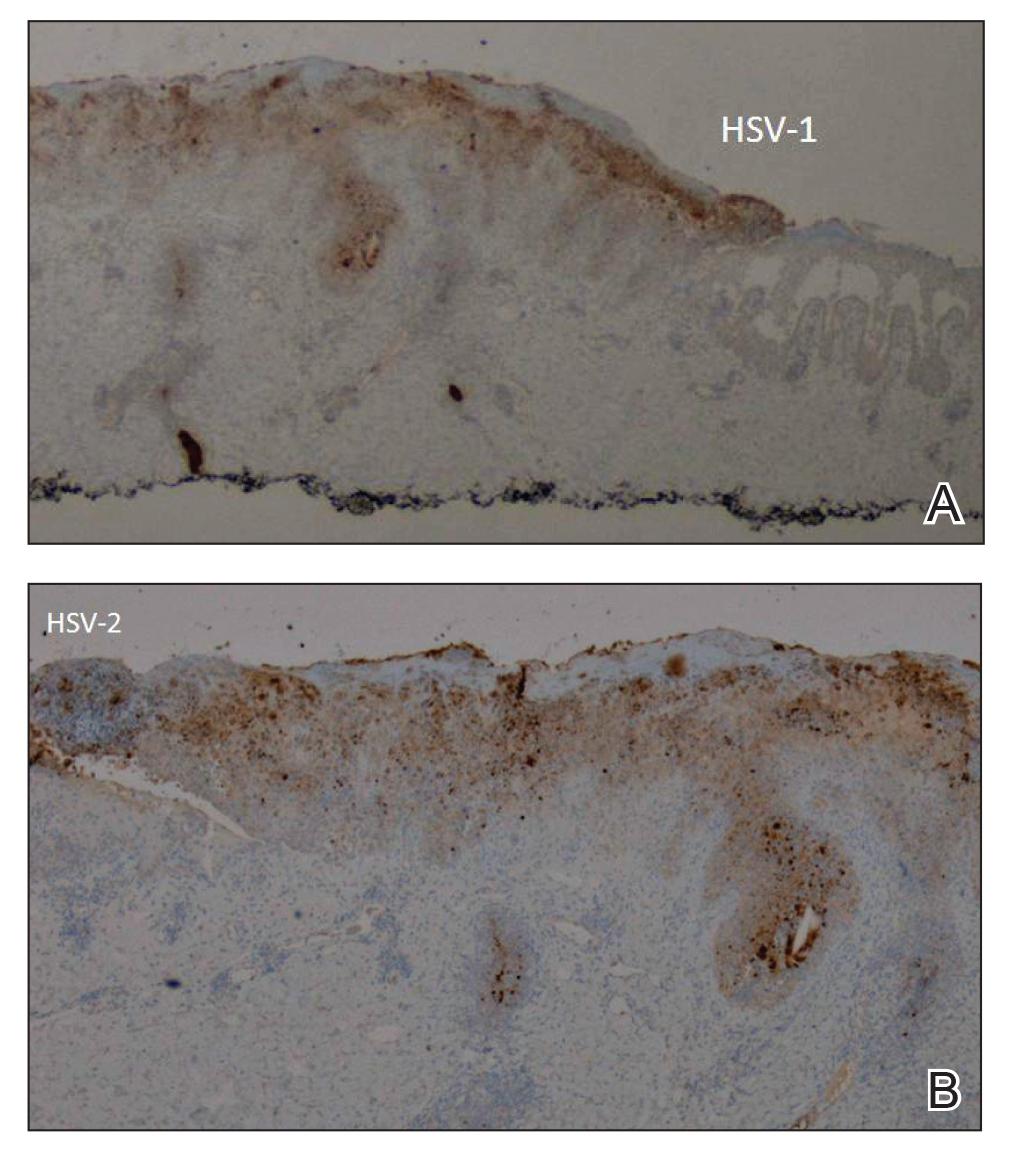
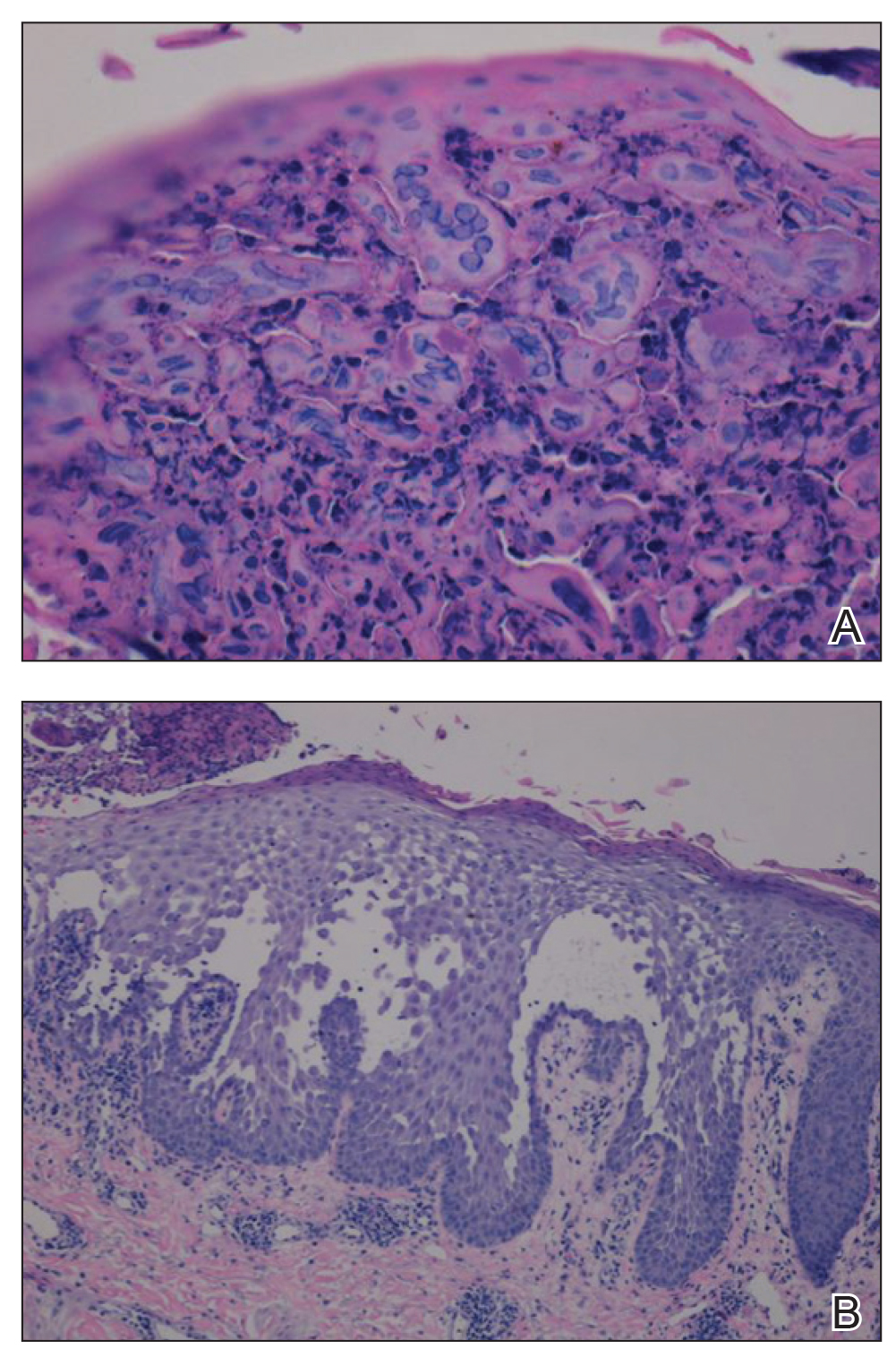
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
Practice Points
- Misdiagnosis of Hailey-Hailey disease (HHD) occurs because of a wide spectrum of presentations.
- Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.
- Clinicians should remain suspicious and evaluate for herpes simplex virus infection in refractory or sudden exacerbation of HHD.
Don't Let the Bedbugs Bite: An Unusual Presentation of Bedbug Infestation Resulting in Life-Threatening Anemia
To the Editor:
A 61-year-old man presented to the emergency department with a rash on the right leg, generalized pruritus, and chest pain. The patient described intermittent exertional pressure-like chest pain over the last few days but had no known prior cardiac history. He also noted worsening edema of the right leg with erythema. Three months prior he had been hospitalized for a similar presentation and was diagnosed with cellulitis of the right leg. The patient was treated with a course of trimethoprim-sulfamethoxazole and permethrin cream for presumed scabies and followed up with dermatology for the persistent generalized pruritic rash and cellulitis. At that time, he was diagnosed with stasis dermatitis with dermatitis neglecta and excoriations. He was educated on general hygiene and treated with triamcinolone, hydrophilic ointment, and pramoxine lotion for pruritus. He also was empirically treated again for scabies.
At the current presentation, preliminary investigation showed profound anemia with a hemoglobin level of 6.2 g/dL (baseline hemoglobin level 3 months prior, 13.1 g/dL). He was subsequently admitted to the general medicine ward for further investigation of severe symptomatic anemia. A medical history revealed moderate chronic obstructive pulmonary disease, hypertension, gastroesophageal reflux disease, xerosis, and fracture of the right ankle following open reduction internal fixation 6 years prior to admission. There was no history of blood loss, antiplatelet agents, or anticoagulants. He was on disability and lived in a single-room occupancy hotel. He did not report any high-risk sexual behaviors or abuse of alcohol or drugs. He actively smoked 1.5 packs of cigarettes per day for the last 30 years. He denied any allergies.
Physical examination revealed the patient was afebrile, nontoxic, disheveled, and in no acute distress. He had anicteric sclera and pale conjunctiva. The right leg appeared more erythematous and edematous compared to the left leg but without warmth or tenderness to palpation. He had innumerable 4- to 5-mm, erythematous, excoriated papules on the skin (Figure). His bed sheets were noted to have multiple rusty-black specks thought to be related to the crusted lesions. Physical examination was otherwise unremarkable.
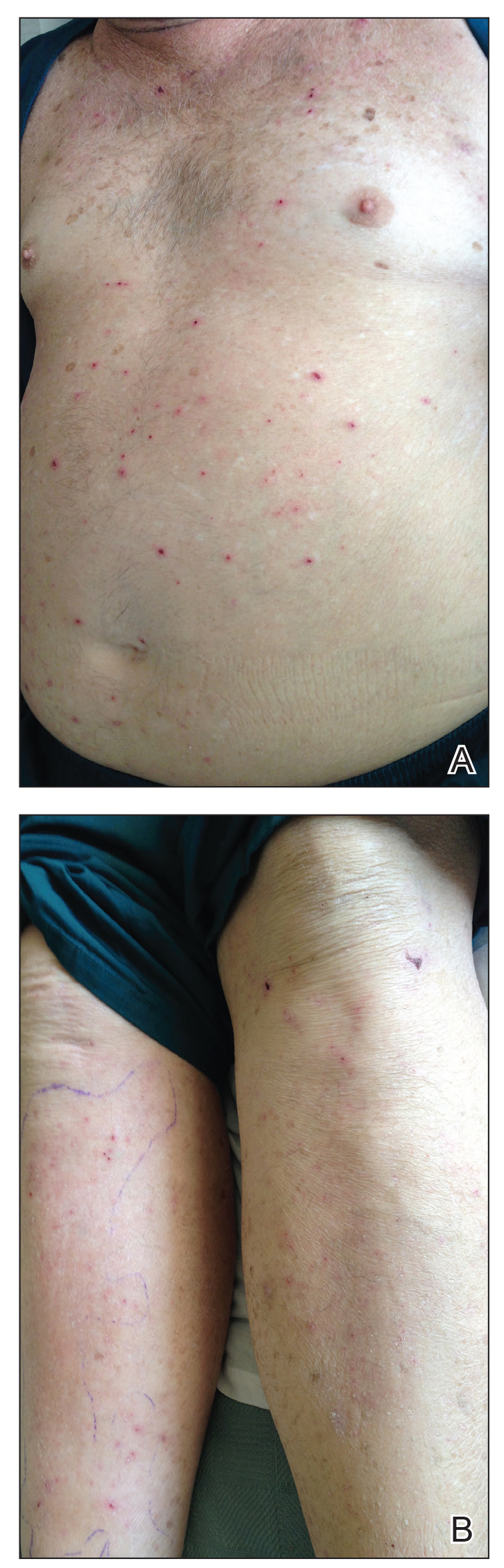
Laboratory workup revealed severe iron-deficiency anemia without any evidence of hemolysis, marrow suppression, infection, or immune compromise (Table). He had a vitamin B12 deficiency (197 pg/mL [reference range, 239-931 pg/mL]), but we felt it was very unlikely to be responsible for his profound, sudden-onset microcytic anemia. Further evaluation for occult bleeding revealed an unremarkable upper endoscopy with push enteroscopy and colonoscopy. An alternate etiology of the anemia could not be identified.
Subsequently, he reported multiple pruritic bug bites sustained at the hotel room where he resided and continued to note pruritus while hospitalized. Pest control inspected the hospital room and identified bedbugs, Cimex lectularius, among his belongings. Upon further review, his clothes and walker were found to be completely infested with these organisms in different stages of development. Treatment included blood transfusions, iron supplementation, and environmental control of the infested living space both in the hospital and at his residence, with subsequent resolution of symptoms and anemia. Two weeks following discharge, the patient no longer reported pruritus, and his hemoglobin level had returned to baseline.
Over the last decade there has been an exponential resurgence in C lectularius infestations in developed countries attributed to increasing global travel, growing pesticide resistance, lack of public awareness, and inadequate pest control programs. This re-emergence has resulted in a public health problem. Although bedbugs are not known to transmit infectious diseases, severe infestation can result in notable dermatitis, iron-deficiency anemia from chronic blood loss, superinfection, allergic reactions including anaphylaxis in rare cases, and psychologic distress.
Iron-deficiency anemia caused by excessive bedbug biting in infants and children has been documented as early as the 1960s.1 Our knowledge of severe anemia due to bedbug infestation is limited to only 4 cases in the literature, according to a PubMed search of articles indexed for MEDLINE using the terms bedbugs anemia and cimex anemia.1-4 All cases reported bedbug infestations involving personal clothing, belongings, and/or living spaces. Patient concerns at presentation ranged from lethargy and fatigue with pruritic rash to chest pain and syncope with findings of severe microcytic or normocytic anemia (hemoglobin level, 5-8 g/dL). All cases were treated supportively with blood transfusion and iron supplementation, with hemoglobin recovery after several weeks. Environmental extermination also was required to prevent recurrence.1-4 Given that each bedbug blood meal is on average 7 mm3, one would have to incur a minimum of 143,000 bites to experience a blood loss of 1 L.3
The differential diagnosis for a patient with generalized pruritus should be broad and includes dermatologic conditions (eg, xerosis, atopic dermatitis, contact dermatitis, urticaria, dermatophytosis, lichen simplex chronicus, psoriasis, scabies, pediculosis corporis and pubis, other arthropod bites, bullous pemphigoid), systemic disorders (eg, renal disease, diabetes mellitus, thyroid disease, cholestasis, human immunodeficiency virus), malignancy, connective tissue disease, medication side effects, and psychogenic and neuropathic itch.
The diagnosis of C lectularius infestation is confirmed by finding the wingless, reddish brown, flat and ovular arthropod, with adult lengths of 4 to 7 mm, approximately the size of an apple seed.5-11 Bedbugs typically are active at night and feed for 3 to 10 minutes. After their feed or during the day, bedbugs will return to their nest in furniture, mattresses, beds, walls, and floors. Bedbug bites appear as small clusters or lines of pruritic erythematous papules with a central hemorrhagic puncta. Other cutaneous symptoms include isolated pruritus, papules, nodules, and bullous eruptions.7 Additional signs of bedbug infestation include black fecal stains in areas of inhabitation as well as actual bedbugs feeding during the day due to overcrowding.
Treatment of pruritic localized cutaneous reactions is supportive and includes antipruritic agents, topical steroids, topical anesthetics, antihistamines, or topical or systemic antibiotics for secondary infections.5-11 Systemic reactions, including anaphylaxis, are treated with epinephrine, antihistamines, and/or corticosteroids, while severe anemia is treated supportively with blood transfusions and iron supplementation.5-11 To prevent reoccurrence, environmental control in the form of nonchemical and chemical treatments is crucial in controlling bedbug infestations.5-11
This case highlights the relevance of a rare but notable morbidity associated with bedbug infestation and the adverse effects of bedbugs on public health. This patient's living situation in a single-room occupancy hotel, poor hygiene, and possible cognitive impairment from his multiple medical conditions may have increased his risk for extreme bedbug infestation. With a good history, physical examination, proper inspection of the patient's belongings, and provider awareness of this epidemic, the severity of this patient's anemia may have been circumvented on the prior hospital admission and follow-up office visit. Once such an infestation is confirmed, a multidisciplinary approach including social work assistance, health services, and pest control is needed to appropriately treat the patient and the environment. Methods in preventing and managing this growing public health problem include improving hygiene, avoiding secondhand goods, and increasing awareness in the identification and proper elimination of bedbugs.5-7
- Venkatachalam PS, Belavady B. Loss of haemoglobin iron due to excessive biting by bed bugs. a possible aetiological factor in the iron deficiency anaemia of infants and children. Trans R Soc Trop Med Hyg. 1962;56:218-221.
- Pritchard MJ, Hwang SW. Severe anemia from bedbugs. CMAJ. 2009;181:287-288.
- Paulke-Korinek M, Széll M, Laferl H, et al. Bed bugs can cause severe anaemia in adults. Parasitol Res. 2012;110:2577-2579.
- Sabou M, Imperiale DG, Andrés E, et al. Bed bugs reproductive life cycle in the clothes of a patient suffering from Alzheimer's disease results in iron deficiency anemia. Parasite. 2013;20:16.
- Studdiford JS, Conniff KM, Trayes KP, et al. Bedbug infestation. Am Fam Physician. 2012;86:653-658.
- Goddard J, deShazo R. Bed bugs (Cimex lectularis) and clinical consequences of their bites. JAMA. 2009;301:1358-1366.
- Bernardeschi C, Le Cleach L, Delaunay P, et al. Bed bug infestation. BMJ. 2013;346:f138.
- Silvia Munoz-Price L, Safdar N, Beier JC, et al. Bed bugs inhealthcare settings. Infect Control Hosp Epidemiol. 2012;33:1137-1142.
- Huntington MK. When bed bugs bite. J Fam Pract. 2012;61:384-388.
- Delaunay P, Blanc V, Del Giudice P, et al. Bedbugs and infectious diseases. Clin Infect Dis. 2011;52:200-212.
- Doggett SL, Dwyer DE, Penas PF, et al. Bed bugs: clinical relevance and control options. Clin Microbiol Rev. 2012;25:164-192.
To the Editor:
A 61-year-old man presented to the emergency department with a rash on the right leg, generalized pruritus, and chest pain. The patient described intermittent exertional pressure-like chest pain over the last few days but had no known prior cardiac history. He also noted worsening edema of the right leg with erythema. Three months prior he had been hospitalized for a similar presentation and was diagnosed with cellulitis of the right leg. The patient was treated with a course of trimethoprim-sulfamethoxazole and permethrin cream for presumed scabies and followed up with dermatology for the persistent generalized pruritic rash and cellulitis. At that time, he was diagnosed with stasis dermatitis with dermatitis neglecta and excoriations. He was educated on general hygiene and treated with triamcinolone, hydrophilic ointment, and pramoxine lotion for pruritus. He also was empirically treated again for scabies.
At the current presentation, preliminary investigation showed profound anemia with a hemoglobin level of 6.2 g/dL (baseline hemoglobin level 3 months prior, 13.1 g/dL). He was subsequently admitted to the general medicine ward for further investigation of severe symptomatic anemia. A medical history revealed moderate chronic obstructive pulmonary disease, hypertension, gastroesophageal reflux disease, xerosis, and fracture of the right ankle following open reduction internal fixation 6 years prior to admission. There was no history of blood loss, antiplatelet agents, or anticoagulants. He was on disability and lived in a single-room occupancy hotel. He did not report any high-risk sexual behaviors or abuse of alcohol or drugs. He actively smoked 1.5 packs of cigarettes per day for the last 30 years. He denied any allergies.
Physical examination revealed the patient was afebrile, nontoxic, disheveled, and in no acute distress. He had anicteric sclera and pale conjunctiva. The right leg appeared more erythematous and edematous compared to the left leg but without warmth or tenderness to palpation. He had innumerable 4- to 5-mm, erythematous, excoriated papules on the skin (Figure). His bed sheets were noted to have multiple rusty-black specks thought to be related to the crusted lesions. Physical examination was otherwise unremarkable.
Laboratory workup revealed severe iron-deficiency anemia without any evidence of hemolysis, marrow suppression, infection, or immune compromise (Table). He had a vitamin B12 deficiency (197 pg/mL [reference range, 239-931 pg/mL]), but we felt it was very unlikely to be responsible for his profound, sudden-onset microcytic anemia. Further evaluation for occult bleeding revealed an unremarkable upper endoscopy with push enteroscopy and colonoscopy. An alternate etiology of the anemia could not be identified.
Subsequently, he reported multiple pruritic bug bites sustained at the hotel room where he resided and continued to note pruritus while hospitalized. Pest control inspected the hospital room and identified bedbugs, Cimex lectularius, among his belongings. Upon further review, his clothes and walker were found to be completely infested with these organisms in different stages of development. Treatment included blood transfusions, iron supplementation, and environmental control of the infested living space both in the hospital and at his residence, with subsequent resolution of symptoms and anemia. Two weeks following discharge, the patient no longer reported pruritus, and his hemoglobin level had returned to baseline.
Over the last decade there has been an exponential resurgence in C lectularius infestations in developed countries attributed to increasing global travel, growing pesticide resistance, lack of public awareness, and inadequate pest control programs. This re-emergence has resulted in a public health problem. Although bedbugs are not known to transmit infectious diseases, severe infestation can result in notable dermatitis, iron-deficiency anemia from chronic blood loss, superinfection, allergic reactions including anaphylaxis in rare cases, and psychologic distress.
Iron-deficiency anemia caused by excessive bedbug biting in infants and children has been documented as early as the 1960s.1 Our knowledge of severe anemia due to bedbug infestation is limited to only 4 cases in the literature, according to a PubMed search of articles indexed for MEDLINE using the terms bedbugs anemia and cimex anemia.1-4 All cases reported bedbug infestations involving personal clothing, belongings, and/or living spaces. Patient concerns at presentation ranged from lethargy and fatigue with pruritic rash to chest pain and syncope with findings of severe microcytic or normocytic anemia (hemoglobin level, 5-8 g/dL). All cases were treated supportively with blood transfusion and iron supplementation, with hemoglobin recovery after several weeks. Environmental extermination also was required to prevent recurrence.1-4 Given that each bedbug blood meal is on average 7 mm3, one would have to incur a minimum of 143,000 bites to experience a blood loss of 1 L.3
The differential diagnosis for a patient with generalized pruritus should be broad and includes dermatologic conditions (eg, xerosis, atopic dermatitis, contact dermatitis, urticaria, dermatophytosis, lichen simplex chronicus, psoriasis, scabies, pediculosis corporis and pubis, other arthropod bites, bullous pemphigoid), systemic disorders (eg, renal disease, diabetes mellitus, thyroid disease, cholestasis, human immunodeficiency virus), malignancy, connective tissue disease, medication side effects, and psychogenic and neuropathic itch.
The diagnosis of C lectularius infestation is confirmed by finding the wingless, reddish brown, flat and ovular arthropod, with adult lengths of 4 to 7 mm, approximately the size of an apple seed.5-11 Bedbugs typically are active at night and feed for 3 to 10 minutes. After their feed or during the day, bedbugs will return to their nest in furniture, mattresses, beds, walls, and floors. Bedbug bites appear as small clusters or lines of pruritic erythematous papules with a central hemorrhagic puncta. Other cutaneous symptoms include isolated pruritus, papules, nodules, and bullous eruptions.7 Additional signs of bedbug infestation include black fecal stains in areas of inhabitation as well as actual bedbugs feeding during the day due to overcrowding.
Treatment of pruritic localized cutaneous reactions is supportive and includes antipruritic agents, topical steroids, topical anesthetics, antihistamines, or topical or systemic antibiotics for secondary infections.5-11 Systemic reactions, including anaphylaxis, are treated with epinephrine, antihistamines, and/or corticosteroids, while severe anemia is treated supportively with blood transfusions and iron supplementation.5-11 To prevent reoccurrence, environmental control in the form of nonchemical and chemical treatments is crucial in controlling bedbug infestations.5-11
This case highlights the relevance of a rare but notable morbidity associated with bedbug infestation and the adverse effects of bedbugs on public health. This patient's living situation in a single-room occupancy hotel, poor hygiene, and possible cognitive impairment from his multiple medical conditions may have increased his risk for extreme bedbug infestation. With a good history, physical examination, proper inspection of the patient's belongings, and provider awareness of this epidemic, the severity of this patient's anemia may have been circumvented on the prior hospital admission and follow-up office visit. Once such an infestation is confirmed, a multidisciplinary approach including social work assistance, health services, and pest control is needed to appropriately treat the patient and the environment. Methods in preventing and managing this growing public health problem include improving hygiene, avoiding secondhand goods, and increasing awareness in the identification and proper elimination of bedbugs.5-7
To the Editor:
A 61-year-old man presented to the emergency department with a rash on the right leg, generalized pruritus, and chest pain. The patient described intermittent exertional pressure-like chest pain over the last few days but had no known prior cardiac history. He also noted worsening edema of the right leg with erythema. Three months prior he had been hospitalized for a similar presentation and was diagnosed with cellulitis of the right leg. The patient was treated with a course of trimethoprim-sulfamethoxazole and permethrin cream for presumed scabies and followed up with dermatology for the persistent generalized pruritic rash and cellulitis. At that time, he was diagnosed with stasis dermatitis with dermatitis neglecta and excoriations. He was educated on general hygiene and treated with triamcinolone, hydrophilic ointment, and pramoxine lotion for pruritus. He also was empirically treated again for scabies.
At the current presentation, preliminary investigation showed profound anemia with a hemoglobin level of 6.2 g/dL (baseline hemoglobin level 3 months prior, 13.1 g/dL). He was subsequently admitted to the general medicine ward for further investigation of severe symptomatic anemia. A medical history revealed moderate chronic obstructive pulmonary disease, hypertension, gastroesophageal reflux disease, xerosis, and fracture of the right ankle following open reduction internal fixation 6 years prior to admission. There was no history of blood loss, antiplatelet agents, or anticoagulants. He was on disability and lived in a single-room occupancy hotel. He did not report any high-risk sexual behaviors or abuse of alcohol or drugs. He actively smoked 1.5 packs of cigarettes per day for the last 30 years. He denied any allergies.
Physical examination revealed the patient was afebrile, nontoxic, disheveled, and in no acute distress. He had anicteric sclera and pale conjunctiva. The right leg appeared more erythematous and edematous compared to the left leg but without warmth or tenderness to palpation. He had innumerable 4- to 5-mm, erythematous, excoriated papules on the skin (Figure). His bed sheets were noted to have multiple rusty-black specks thought to be related to the crusted lesions. Physical examination was otherwise unremarkable.
Laboratory workup revealed severe iron-deficiency anemia without any evidence of hemolysis, marrow suppression, infection, or immune compromise (Table). He had a vitamin B12 deficiency (197 pg/mL [reference range, 239-931 pg/mL]), but we felt it was very unlikely to be responsible for his profound, sudden-onset microcytic anemia. Further evaluation for occult bleeding revealed an unremarkable upper endoscopy with push enteroscopy and colonoscopy. An alternate etiology of the anemia could not be identified.
Subsequently, he reported multiple pruritic bug bites sustained at the hotel room where he resided and continued to note pruritus while hospitalized. Pest control inspected the hospital room and identified bedbugs, Cimex lectularius, among his belongings. Upon further review, his clothes and walker were found to be completely infested with these organisms in different stages of development. Treatment included blood transfusions, iron supplementation, and environmental control of the infested living space both in the hospital and at his residence, with subsequent resolution of symptoms and anemia. Two weeks following discharge, the patient no longer reported pruritus, and his hemoglobin level had returned to baseline.
Over the last decade there has been an exponential resurgence in C lectularius infestations in developed countries attributed to increasing global travel, growing pesticide resistance, lack of public awareness, and inadequate pest control programs. This re-emergence has resulted in a public health problem. Although bedbugs are not known to transmit infectious diseases, severe infestation can result in notable dermatitis, iron-deficiency anemia from chronic blood loss, superinfection, allergic reactions including anaphylaxis in rare cases, and psychologic distress.
Iron-deficiency anemia caused by excessive bedbug biting in infants and children has been documented as early as the 1960s.1 Our knowledge of severe anemia due to bedbug infestation is limited to only 4 cases in the literature, according to a PubMed search of articles indexed for MEDLINE using the terms bedbugs anemia and cimex anemia.1-4 All cases reported bedbug infestations involving personal clothing, belongings, and/or living spaces. Patient concerns at presentation ranged from lethargy and fatigue with pruritic rash to chest pain and syncope with findings of severe microcytic or normocytic anemia (hemoglobin level, 5-8 g/dL). All cases were treated supportively with blood transfusion and iron supplementation, with hemoglobin recovery after several weeks. Environmental extermination also was required to prevent recurrence.1-4 Given that each bedbug blood meal is on average 7 mm3, one would have to incur a minimum of 143,000 bites to experience a blood loss of 1 L.3
The differential diagnosis for a patient with generalized pruritus should be broad and includes dermatologic conditions (eg, xerosis, atopic dermatitis, contact dermatitis, urticaria, dermatophytosis, lichen simplex chronicus, psoriasis, scabies, pediculosis corporis and pubis, other arthropod bites, bullous pemphigoid), systemic disorders (eg, renal disease, diabetes mellitus, thyroid disease, cholestasis, human immunodeficiency virus), malignancy, connective tissue disease, medication side effects, and psychogenic and neuropathic itch.
The diagnosis of C lectularius infestation is confirmed by finding the wingless, reddish brown, flat and ovular arthropod, with adult lengths of 4 to 7 mm, approximately the size of an apple seed.5-11 Bedbugs typically are active at night and feed for 3 to 10 minutes. After their feed or during the day, bedbugs will return to their nest in furniture, mattresses, beds, walls, and floors. Bedbug bites appear as small clusters or lines of pruritic erythematous papules with a central hemorrhagic puncta. Other cutaneous symptoms include isolated pruritus, papules, nodules, and bullous eruptions.7 Additional signs of bedbug infestation include black fecal stains in areas of inhabitation as well as actual bedbugs feeding during the day due to overcrowding.
Treatment of pruritic localized cutaneous reactions is supportive and includes antipruritic agents, topical steroids, topical anesthetics, antihistamines, or topical or systemic antibiotics for secondary infections.5-11 Systemic reactions, including anaphylaxis, are treated with epinephrine, antihistamines, and/or corticosteroids, while severe anemia is treated supportively with blood transfusions and iron supplementation.5-11 To prevent reoccurrence, environmental control in the form of nonchemical and chemical treatments is crucial in controlling bedbug infestations.5-11
This case highlights the relevance of a rare but notable morbidity associated with bedbug infestation and the adverse effects of bedbugs on public health. This patient's living situation in a single-room occupancy hotel, poor hygiene, and possible cognitive impairment from his multiple medical conditions may have increased his risk for extreme bedbug infestation. With a good history, physical examination, proper inspection of the patient's belongings, and provider awareness of this epidemic, the severity of this patient's anemia may have been circumvented on the prior hospital admission and follow-up office visit. Once such an infestation is confirmed, a multidisciplinary approach including social work assistance, health services, and pest control is needed to appropriately treat the patient and the environment. Methods in preventing and managing this growing public health problem include improving hygiene, avoiding secondhand goods, and increasing awareness in the identification and proper elimination of bedbugs.5-7
- Venkatachalam PS, Belavady B. Loss of haemoglobin iron due to excessive biting by bed bugs. a possible aetiological factor in the iron deficiency anaemia of infants and children. Trans R Soc Trop Med Hyg. 1962;56:218-221.
- Pritchard MJ, Hwang SW. Severe anemia from bedbugs. CMAJ. 2009;181:287-288.
- Paulke-Korinek M, Széll M, Laferl H, et al. Bed bugs can cause severe anaemia in adults. Parasitol Res. 2012;110:2577-2579.
- Sabou M, Imperiale DG, Andrés E, et al. Bed bugs reproductive life cycle in the clothes of a patient suffering from Alzheimer's disease results in iron deficiency anemia. Parasite. 2013;20:16.
- Studdiford JS, Conniff KM, Trayes KP, et al. Bedbug infestation. Am Fam Physician. 2012;86:653-658.
- Goddard J, deShazo R. Bed bugs (Cimex lectularis) and clinical consequences of their bites. JAMA. 2009;301:1358-1366.
- Bernardeschi C, Le Cleach L, Delaunay P, et al. Bed bug infestation. BMJ. 2013;346:f138.
- Silvia Munoz-Price L, Safdar N, Beier JC, et al. Bed bugs inhealthcare settings. Infect Control Hosp Epidemiol. 2012;33:1137-1142.
- Huntington MK. When bed bugs bite. J Fam Pract. 2012;61:384-388.
- Delaunay P, Blanc V, Del Giudice P, et al. Bedbugs and infectious diseases. Clin Infect Dis. 2011;52:200-212.
- Doggett SL, Dwyer DE, Penas PF, et al. Bed bugs: clinical relevance and control options. Clin Microbiol Rev. 2012;25:164-192.
- Venkatachalam PS, Belavady B. Loss of haemoglobin iron due to excessive biting by bed bugs. a possible aetiological factor in the iron deficiency anaemia of infants and children. Trans R Soc Trop Med Hyg. 1962;56:218-221.
- Pritchard MJ, Hwang SW. Severe anemia from bedbugs. CMAJ. 2009;181:287-288.
- Paulke-Korinek M, Széll M, Laferl H, et al. Bed bugs can cause severe anaemia in adults. Parasitol Res. 2012;110:2577-2579.
- Sabou M, Imperiale DG, Andrés E, et al. Bed bugs reproductive life cycle in the clothes of a patient suffering from Alzheimer's disease results in iron deficiency anemia. Parasite. 2013;20:16.
- Studdiford JS, Conniff KM, Trayes KP, et al. Bedbug infestation. Am Fam Physician. 2012;86:653-658.
- Goddard J, deShazo R. Bed bugs (Cimex lectularis) and clinical consequences of their bites. JAMA. 2009;301:1358-1366.
- Bernardeschi C, Le Cleach L, Delaunay P, et al. Bed bug infestation. BMJ. 2013;346:f138.
- Silvia Munoz-Price L, Safdar N, Beier JC, et al. Bed bugs inhealthcare settings. Infect Control Hosp Epidemiol. 2012;33:1137-1142.
- Huntington MK. When bed bugs bite. J Fam Pract. 2012;61:384-388.
- Delaunay P, Blanc V, Del Giudice P, et al. Bedbugs and infectious diseases. Clin Infect Dis. 2011;52:200-212.
- Doggett SL, Dwyer DE, Penas PF, et al. Bed bugs: clinical relevance and control options. Clin Microbiol Rev. 2012;25:164-192.
Practice Points
- There has been a resurgence in bedbug (Cimex lectularius) infestations in developed countries.
- Although rare, anemia due to bedbug infestation should be considered in patients presenting with anemia and a widespread pruritic papular eruption.
- A thorough history and physical examination are essential to prevent a delay in diagnosis and avoid a costly and unnecessary workup.
- Successful treatment requires a multidisciplinary approach, which includes medical management, social services, and pest control.