User login
Perianal Basal Cell Carcinoma Treated With Mohs Micrographic Surgery
Basal cell carcinoma (BCC) is the most common skin cancer in the United States1 and most commonly occurs in sun-exposed areas. Although BCCs can and do develop on other non–sun-exposed areas of the body, BCCs of the perianal or genital regions are very rare (0.27% of cases). It is estimated that perianal BCCs account for less than 0.08% of all BCCs.2
We present a case of a superficial nodular perianal BCC that was discovered following an annual total-body skin examination and was treated with Mohs micrographic surgery (MMS).
Case Report
A 76-year-old man presented to the dermatology clinic for an annual total-body skin examination as well as evaluation of a new submental skin lesion. The patient’s medical history included successfully treated malignant melanoma in situ, multiple actinic keratoses, and an eccrine carcinoma. His family history was noncontributory. Inspection of the submental lesion revealed a pearly, 1.8-cm, telangiectatic, nodular plaque that was highly suspected to be a BCC. During the examination, a 1-cm pinkish-red plaque was found on the skin in the left perianal region (Figure 1). The patient was unaware of the lesion and did not report any symptoms upon questioning.
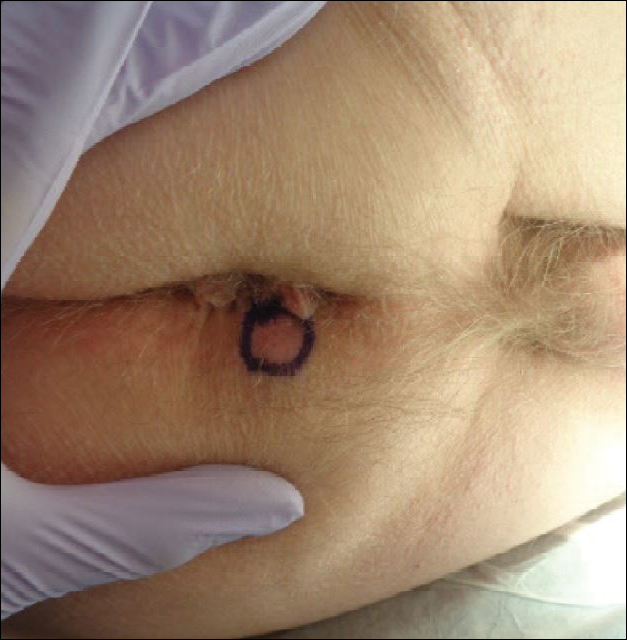
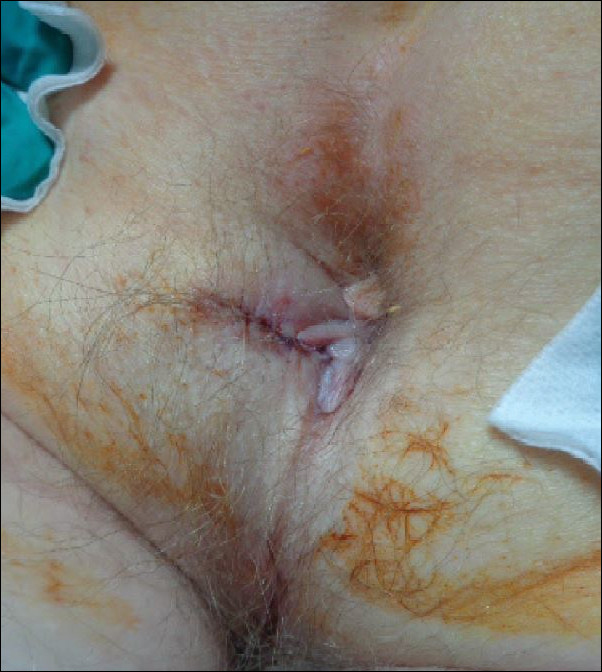
A shave biopsy of the submental lesion confirmed a diagnosis of micronodular BCC, and the patient was referred for MMS. It was decided to reevaluate the perianal lesion clinically at a follow-up appointment 2 months later and biopsy if it had not resolved. However, the patient did not attend the 2-month follow-up visit as scheduled, and it was not until the following year at his next annual total-body skin examination that the perianal lesion was rechecked. The lesion was unchanged at the time and was similar to the previous findings in both appearance and size. A punch biopsy was performed, and the pathology showed a superficial nodular perianal BCC (Figure 2). The perianal BCC was excised during a 2-stage MMS procedure with no recurrence at 6-month follow-up (Figure 3).

Comment
At the time of the patient’s initial visit, the differential diagnosis for this perianal lesion included an inflammatory or infectious dermatosis. Its asymptomatic nature made it difficult to determine how long it had been present. The lack of resolution on reevaluation of the lesion 1 year later raised the possibilities of amelanotic melanoma, squamous cell carcinoma, and lichen planus. Basal cell carcinoma was much lower in the differential diagnosis, as BCCs rarely are found in this area of the body; in fact, BCCs account for 0.2% of all anorectal neoplasms,3 and less than 0.08% of BCCs will occur in the perianal region.2
This challenging presentation is common for BCCs found in the perianal and perineal regions, as they are difficult to diagnose and often are overlooked as inflammatory dermatoses.4,5 The infrequency of perianal BCC reported in the literature as well as the predominance of BCC in sun-exposed areas makes it difficult for dermatologists to diagnose perianal BCC without biopsy. Another feature indicative of this diagnostic difficulty is that the average size of perianal and perineal BCCs has been found to be 1.95 cm.2 Without thorough and routine total-body skin examinations, there is no reliable way to catch asymptomatic BCCs in the perianal region until they have progressed far enough to become symptomatic. When possible, we recommend that dermatologists check the genital and anal regions during skin examinations and biopsy any suspicious lesions.
This case also highlights the challenge of missed appointments, which dermatologists also consistently face. Nonattendance rates in US dermatology clinics have been estimated at 17%,6 18.6%,7 19.4%,8 and 23.9%9 and present a challenge for even the best-run practices. Among patients with missed appointments, the most frequently stated reason in one survey was forgetting, and 24% of those contacted reported that they had not been reminded of their appointment.8 Many of the patients surveyed also expressed that they had preferred methods of receiving reminders such as e-mail or text message, which fell outside of traditional contact methods (eg, phone calls, voicemails). Confirming appointments ahead of time can reduce the number of missed appointments due to patient forgetfulness, and incorporating multiple communication modalities may lead to more effective appointment reminders.
Conclusion
Perianal BCC is challenging to diagnose and easy to overlook. Basal cell carcinoma is rarely found in the perianal regions and accounts for a fraction of all anorectal neoplasms. We recommend thorough total-body skin examinations that include the genital region and gluteal cleft when possible and encourage physicians to biopsy suspicious lesions in these regions. Routine, thorough total-body skin examinations can reveal neoplasms when they are smaller and asymptomatic. When surgical excision is indicated, MMS is an effective way to preserve as much tissue as possible and minimize recurrence.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatology. 2015;151:1081-1086.
- Gibson GE, Ahmed I. Perianal and genital basal cell carcinoma: a clinicopathologic review of 51 cases. J Am Acad Dermatol. 2001;45:68-71.
- Leonard D, Beddy D, Dozois EJ. Neoplasms of anal canal and perianal skin. Clin Colon Rectal Surg. 2011;24:54-63.
- Bulur I, Boyuk E, Saracoglu ZN, et al. Perianal basal cell carcinoma. Case Rep Dermatol. 2015;7:25-28.
- Collins PS, Farber GA, Hegre AM. Basal-cell carcinoma of the vulva. J Dermatol Surg Oncol. 1981;7:711-714.
- Penneys NS, Glaser DA. The incidence of cancellation and nonattendance at a dermatology clinic. J Am Acad Dermatol. 1990;40:714-718.
- Cronin P, DeCoste L, Kimball A. A multivariate analysis of dermatology missed appointment predictors. JAMA Dermatology. 2013;149:1435-1437.
- Moustafa FA, Ramsey L, Huang KE, et al. Factors associated with missed dermatology appointments. Cutis. 2015;96:E20-E23.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
Basal cell carcinoma (BCC) is the most common skin cancer in the United States1 and most commonly occurs in sun-exposed areas. Although BCCs can and do develop on other non–sun-exposed areas of the body, BCCs of the perianal or genital regions are very rare (0.27% of cases). It is estimated that perianal BCCs account for less than 0.08% of all BCCs.2
We present a case of a superficial nodular perianal BCC that was discovered following an annual total-body skin examination and was treated with Mohs micrographic surgery (MMS).
Case Report
A 76-year-old man presented to the dermatology clinic for an annual total-body skin examination as well as evaluation of a new submental skin lesion. The patient’s medical history included successfully treated malignant melanoma in situ, multiple actinic keratoses, and an eccrine carcinoma. His family history was noncontributory. Inspection of the submental lesion revealed a pearly, 1.8-cm, telangiectatic, nodular plaque that was highly suspected to be a BCC. During the examination, a 1-cm pinkish-red plaque was found on the skin in the left perianal region (Figure 1). The patient was unaware of the lesion and did not report any symptoms upon questioning.
A shave biopsy of the submental lesion confirmed a diagnosis of micronodular BCC, and the patient was referred for MMS. It was decided to reevaluate the perianal lesion clinically at a follow-up appointment 2 months later and biopsy if it had not resolved. However, the patient did not attend the 2-month follow-up visit as scheduled, and it was not until the following year at his next annual total-body skin examination that the perianal lesion was rechecked. The lesion was unchanged at the time and was similar to the previous findings in both appearance and size. A punch biopsy was performed, and the pathology showed a superficial nodular perianal BCC (Figure 2). The perianal BCC was excised during a 2-stage MMS procedure with no recurrence at 6-month follow-up (Figure 3).
Comment
At the time of the patient’s initial visit, the differential diagnosis for this perianal lesion included an inflammatory or infectious dermatosis. Its asymptomatic nature made it difficult to determine how long it had been present. The lack of resolution on reevaluation of the lesion 1 year later raised the possibilities of amelanotic melanoma, squamous cell carcinoma, and lichen planus. Basal cell carcinoma was much lower in the differential diagnosis, as BCCs rarely are found in this area of the body; in fact, BCCs account for 0.2% of all anorectal neoplasms,3 and less than 0.08% of BCCs will occur in the perianal region.2
This challenging presentation is common for BCCs found in the perianal and perineal regions, as they are difficult to diagnose and often are overlooked as inflammatory dermatoses.4,5 The infrequency of perianal BCC reported in the literature as well as the predominance of BCC in sun-exposed areas makes it difficult for dermatologists to diagnose perianal BCC without biopsy. Another feature indicative of this diagnostic difficulty is that the average size of perianal and perineal BCCs has been found to be 1.95 cm.2 Without thorough and routine total-body skin examinations, there is no reliable way to catch asymptomatic BCCs in the perianal region until they have progressed far enough to become symptomatic. When possible, we recommend that dermatologists check the genital and anal regions during skin examinations and biopsy any suspicious lesions.
This case also highlights the challenge of missed appointments, which dermatologists also consistently face. Nonattendance rates in US dermatology clinics have been estimated at 17%,6 18.6%,7 19.4%,8 and 23.9%9 and present a challenge for even the best-run practices. Among patients with missed appointments, the most frequently stated reason in one survey was forgetting, and 24% of those contacted reported that they had not been reminded of their appointment.8 Many of the patients surveyed also expressed that they had preferred methods of receiving reminders such as e-mail or text message, which fell outside of traditional contact methods (eg, phone calls, voicemails). Confirming appointments ahead of time can reduce the number of missed appointments due to patient forgetfulness, and incorporating multiple communication modalities may lead to more effective appointment reminders.
Conclusion
Perianal BCC is challenging to diagnose and easy to overlook. Basal cell carcinoma is rarely found in the perianal regions and accounts for a fraction of all anorectal neoplasms. We recommend thorough total-body skin examinations that include the genital region and gluteal cleft when possible and encourage physicians to biopsy suspicious lesions in these regions. Routine, thorough total-body skin examinations can reveal neoplasms when they are smaller and asymptomatic. When surgical excision is indicated, MMS is an effective way to preserve as much tissue as possible and minimize recurrence.
Basal cell carcinoma (BCC) is the most common skin cancer in the United States1 and most commonly occurs in sun-exposed areas. Although BCCs can and do develop on other non–sun-exposed areas of the body, BCCs of the perianal or genital regions are very rare (0.27% of cases). It is estimated that perianal BCCs account for less than 0.08% of all BCCs.2
We present a case of a superficial nodular perianal BCC that was discovered following an annual total-body skin examination and was treated with Mohs micrographic surgery (MMS).
Case Report
A 76-year-old man presented to the dermatology clinic for an annual total-body skin examination as well as evaluation of a new submental skin lesion. The patient’s medical history included successfully treated malignant melanoma in situ, multiple actinic keratoses, and an eccrine carcinoma. His family history was noncontributory. Inspection of the submental lesion revealed a pearly, 1.8-cm, telangiectatic, nodular plaque that was highly suspected to be a BCC. During the examination, a 1-cm pinkish-red plaque was found on the skin in the left perianal region (Figure 1). The patient was unaware of the lesion and did not report any symptoms upon questioning.
A shave biopsy of the submental lesion confirmed a diagnosis of micronodular BCC, and the patient was referred for MMS. It was decided to reevaluate the perianal lesion clinically at a follow-up appointment 2 months later and biopsy if it had not resolved. However, the patient did not attend the 2-month follow-up visit as scheduled, and it was not until the following year at his next annual total-body skin examination that the perianal lesion was rechecked. The lesion was unchanged at the time and was similar to the previous findings in both appearance and size. A punch biopsy was performed, and the pathology showed a superficial nodular perianal BCC (Figure 2). The perianal BCC was excised during a 2-stage MMS procedure with no recurrence at 6-month follow-up (Figure 3).
Comment
At the time of the patient’s initial visit, the differential diagnosis for this perianal lesion included an inflammatory or infectious dermatosis. Its asymptomatic nature made it difficult to determine how long it had been present. The lack of resolution on reevaluation of the lesion 1 year later raised the possibilities of amelanotic melanoma, squamous cell carcinoma, and lichen planus. Basal cell carcinoma was much lower in the differential diagnosis, as BCCs rarely are found in this area of the body; in fact, BCCs account for 0.2% of all anorectal neoplasms,3 and less than 0.08% of BCCs will occur in the perianal region.2
This challenging presentation is common for BCCs found in the perianal and perineal regions, as they are difficult to diagnose and often are overlooked as inflammatory dermatoses.4,5 The infrequency of perianal BCC reported in the literature as well as the predominance of BCC in sun-exposed areas makes it difficult for dermatologists to diagnose perianal BCC without biopsy. Another feature indicative of this diagnostic difficulty is that the average size of perianal and perineal BCCs has been found to be 1.95 cm.2 Without thorough and routine total-body skin examinations, there is no reliable way to catch asymptomatic BCCs in the perianal region until they have progressed far enough to become symptomatic. When possible, we recommend that dermatologists check the genital and anal regions during skin examinations and biopsy any suspicious lesions.
This case also highlights the challenge of missed appointments, which dermatologists also consistently face. Nonattendance rates in US dermatology clinics have been estimated at 17%,6 18.6%,7 19.4%,8 and 23.9%9 and present a challenge for even the best-run practices. Among patients with missed appointments, the most frequently stated reason in one survey was forgetting, and 24% of those contacted reported that they had not been reminded of their appointment.8 Many of the patients surveyed also expressed that they had preferred methods of receiving reminders such as e-mail or text message, which fell outside of traditional contact methods (eg, phone calls, voicemails). Confirming appointments ahead of time can reduce the number of missed appointments due to patient forgetfulness, and incorporating multiple communication modalities may lead to more effective appointment reminders.
Conclusion
Perianal BCC is challenging to diagnose and easy to overlook. Basal cell carcinoma is rarely found in the perianal regions and accounts for a fraction of all anorectal neoplasms. We recommend thorough total-body skin examinations that include the genital region and gluteal cleft when possible and encourage physicians to biopsy suspicious lesions in these regions. Routine, thorough total-body skin examinations can reveal neoplasms when they are smaller and asymptomatic. When surgical excision is indicated, MMS is an effective way to preserve as much tissue as possible and minimize recurrence.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatology. 2015;151:1081-1086.
- Gibson GE, Ahmed I. Perianal and genital basal cell carcinoma: a clinicopathologic review of 51 cases. J Am Acad Dermatol. 2001;45:68-71.
- Leonard D, Beddy D, Dozois EJ. Neoplasms of anal canal and perianal skin. Clin Colon Rectal Surg. 2011;24:54-63.
- Bulur I, Boyuk E, Saracoglu ZN, et al. Perianal basal cell carcinoma. Case Rep Dermatol. 2015;7:25-28.
- Collins PS, Farber GA, Hegre AM. Basal-cell carcinoma of the vulva. J Dermatol Surg Oncol. 1981;7:711-714.
- Penneys NS, Glaser DA. The incidence of cancellation and nonattendance at a dermatology clinic. J Am Acad Dermatol. 1990;40:714-718.
- Cronin P, DeCoste L, Kimball A. A multivariate analysis of dermatology missed appointment predictors. JAMA Dermatology. 2013;149:1435-1437.
- Moustafa FA, Ramsey L, Huang KE, et al. Factors associated with missed dermatology appointments. Cutis. 2015;96:E20-E23.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatology. 2015;151:1081-1086.
- Gibson GE, Ahmed I. Perianal and genital basal cell carcinoma: a clinicopathologic review of 51 cases. J Am Acad Dermatol. 2001;45:68-71.
- Leonard D, Beddy D, Dozois EJ. Neoplasms of anal canal and perianal skin. Clin Colon Rectal Surg. 2011;24:54-63.
- Bulur I, Boyuk E, Saracoglu ZN, et al. Perianal basal cell carcinoma. Case Rep Dermatol. 2015;7:25-28.
- Collins PS, Farber GA, Hegre AM. Basal-cell carcinoma of the vulva. J Dermatol Surg Oncol. 1981;7:711-714.
- Penneys NS, Glaser DA. The incidence of cancellation and nonattendance at a dermatology clinic. J Am Acad Dermatol. 1990;40:714-718.
- Cronin P, DeCoste L, Kimball A. A multivariate analysis of dermatology missed appointment predictors. JAMA Dermatology. 2013;149:1435-1437.
- Moustafa FA, Ramsey L, Huang KE, et al. Factors associated with missed dermatology appointments. Cutis. 2015;96:E20-E23.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
Practice Points
- Basal cell carcinoma is less common in non–sun-exposed areas of the body and is exceptionally rare in the perineal and perianal regions.
- Thorough total-body skin examinations may lead to early detection of asymptomatic skin lesions, allowing for earlier and less invasive treatment.
- Appointment attendance and patient compliance are common challenges that dermatologists face. Patient reminders via their preferred method of communication may help reduce missed dermatology appointments.
Critical anemia • light-headedness • bilateral leg swelling • Dx?
THE CASE
A 40-year-old man was referred to the emergency department (ED) with critical anemia after routine blood work at an outside clinic showed a hemoglobin level of 3.5 g/dL. On presentation, he reported symptoms of fatigue, shortness of breath, bilateral leg swelling, dizziness (characterized as light-headedness), and frequent heartburn. He said that the symptoms began 5 weeks earlier, after he was exposed to a relative with hand, foot, and mouth disease.
Additionally, the patient reported an intentional 14-lb weight loss over the 6 months prior to presentation. He denied fever, rash, chest pain, loss of consciousness, headache, abdominal pain, hematemesis, melena, and hematochezia. His medical history was significant for peptic ulcer disease (diagnosed and treated at age 8). He did not recall the specifics, and he denied any related chronic symptoms or complications. His family history (paternal) was significant for colon cancer.
The physical exam revealed conjunctival pallor, skin pallor, jaundice, +1 bilateral lower extremity edema, tachycardia, and tachypnea. Stool Hemoccult was negative. On repeat complete blood count (performed in the ED), hemoglobin was found to be 3.1 g/dL with a mean corpuscular volume of 47 fL.
THE DIAGNOSIS
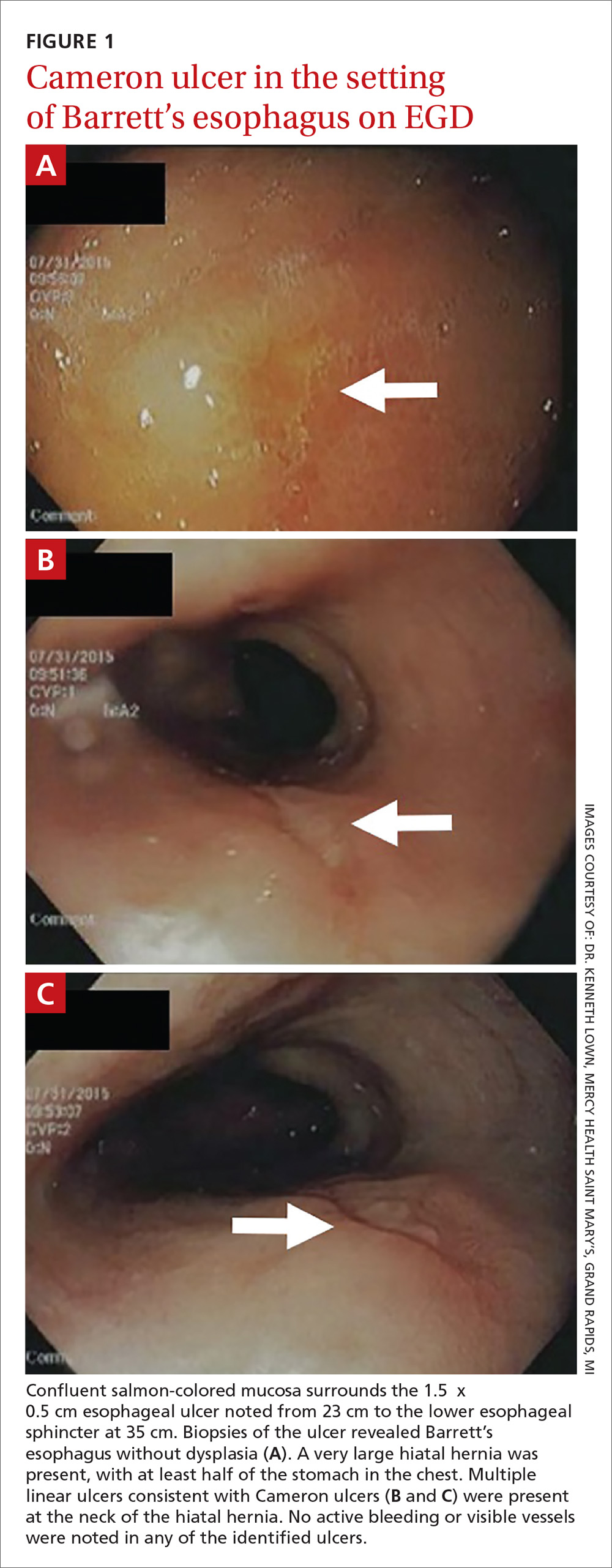
The patient was admitted to the family medicine service and received 4 units of packed red blood cells, which increased his hemoglobin to the target goal of >7 g/dL. A colonoscopy and an esophagogastroduodenoscopy (EGD) were performed (FIGURE 1A-1C), with results suggestive of diverticulosis, probable Barrett’s mucosa, esophageal ulcer, huge hiatal hernia (at least one-half of the stomach was in the chest), and Cameron ulcers. Esophageal biopsies showed cardiac mucosa with chronic inflammation. Esophageal ulcer biopsies revealed Barrett’s esophagus without dysplasia. Duodenal biopsies displayed normal mucosa.
DISCUSSION
Although rare, Cameron ulcers must be considered in the differential diagnosis of patients with chronic anemia of unknown origin. The potential for these lesions to result in chronic blood loss, which could over time manifest as severe anemia or hypovolemic shock, makes proper diagnosis and prompt treatment especially important.4,5
In our patient’s case, his severe anemia was likely the result of a combination of the esophageal and Cameron ulcers evidenced on EGD, rather than any single ulcer. In our review of the literature, we found no reports of any patients with anemia and Cameron ulcers who presented with hemoglobin levels as low as our patient had.
Treat with a PPI and iron supplementation
Multiple EGDs may be needed to properly diagnose Cameron ulcers, as they can be difficult to identify. Once a patient receives the diagnosis, he or she will typically be put on a daily proton pump inhibitor (PPI) regimen, such as omeprazole 20 mg bid. However, since many patients with Cameron ulcers also have acid-related problems (as was true in this case), a multifactorial acid suppression approach may be warranted.1 This may include recommending lifestyle modifications (eg, eating small meals, avoiding foods that provoke symptoms, or losing weight) and prescribing medications in addition to a PPI, such as an H2 blocker (eg, 300 mg qid, before meals and at bedtime).
In addition, iron sulfate (325 mg/d, in this case) and blood transfusions may be required to treat the anemia. In refractory cases, endoscopic or surgical interventions, such as hemoclipping, Nissen fundoplication, or laparoscopic gastropexy, may need to be performed.2
Our patient was given a prescription for ferrous sulfate 325 mg/d and omeprazole 20 mg bid. His symptoms improved with treatment, and he was discharged on Day 5; his hemoglobin remained >7 g/dL.
THE TAKEAWAY
The association between chronic iron deficiency anemia and Cameron ulcers has been established but is commonly overlooked in patients presenting with unexplained anemia or an undiagnosed hiatal hernia. This is likely due to their rarity as a cause of anemia, in general.
Furthermore, the lesions can be missed on EGD; multiple EGDs may be needed to make the diagnosis. Once diagnosed, Cameron ulcers typically respond well to twice daily PPI treatment. Patients with refractory, recurrent, or severe lesions, or large, symptomatic hiatal hernias should be referred for surgical assessment.
CORRESPONDENCE
Megan Yee, 801 Broadward Avenue NW, Grand Rapids, MI 49504; [email protected].
1. Maganty K, Smith RL. Cameron lesions: unusual cause of gastrointestinal bleeding and anemia. Digestion. 2008;77:214-217.
2. Camus M, Jensen DM, Ohning GV, et al. Severe upper gastrointestinal hemorrhage from linear gastric ulcers in large hiatal hernias: a large prospective case series of Cameron ulcers. Endoscopy. 2013;45:397-400.
3. Kimer N, Schmidt PN, Krag, A. Cameron lesions: an often overlooked cause of iron deficiency anaemia in patients with large hiatal hernias. BMJ Case Rep. 2010;2010.
4. Kapadia S, Jagroop S, Kumar, A. Cameron ulcers: an atypical source for a massive upper gastrointestinal bleed. World J Gastroenterol. 2012;18:4959-4961.
5. Gupta P, Suryadevara M, Das A, et al. Cameron ulcer causing severe anemia in a patient with diaphragmatic hernia. Am J Case Rep. 2015;16:733-736.
THE CASE
A 40-year-old man was referred to the emergency department (ED) with critical anemia after routine blood work at an outside clinic showed a hemoglobin level of 3.5 g/dL. On presentation, he reported symptoms of fatigue, shortness of breath, bilateral leg swelling, dizziness (characterized as light-headedness), and frequent heartburn. He said that the symptoms began 5 weeks earlier, after he was exposed to a relative with hand, foot, and mouth disease.
Additionally, the patient reported an intentional 14-lb weight loss over the 6 months prior to presentation. He denied fever, rash, chest pain, loss of consciousness, headache, abdominal pain, hematemesis, melena, and hematochezia. His medical history was significant for peptic ulcer disease (diagnosed and treated at age 8). He did not recall the specifics, and he denied any related chronic symptoms or complications. His family history (paternal) was significant for colon cancer.
The physical exam revealed conjunctival pallor, skin pallor, jaundice, +1 bilateral lower extremity edema, tachycardia, and tachypnea. Stool Hemoccult was negative. On repeat complete blood count (performed in the ED), hemoglobin was found to be 3.1 g/dL with a mean corpuscular volume of 47 fL.
THE DIAGNOSIS
The patient was admitted to the family medicine service and received 4 units of packed red blood cells, which increased his hemoglobin to the target goal of >7 g/dL. A colonoscopy and an esophagogastroduodenoscopy (EGD) were performed (FIGURE 1A-1C), with results suggestive of diverticulosis, probable Barrett’s mucosa, esophageal ulcer, huge hiatal hernia (at least one-half of the stomach was in the chest), and Cameron ulcers. Esophageal biopsies showed cardiac mucosa with chronic inflammation. Esophageal ulcer biopsies revealed Barrett’s esophagus without dysplasia. Duodenal biopsies displayed normal mucosa.
DISCUSSION
Although rare, Cameron ulcers must be considered in the differential diagnosis of patients with chronic anemia of unknown origin. The potential for these lesions to result in chronic blood loss, which could over time manifest as severe anemia or hypovolemic shock, makes proper diagnosis and prompt treatment especially important.4,5
In our patient’s case, his severe anemia was likely the result of a combination of the esophageal and Cameron ulcers evidenced on EGD, rather than any single ulcer. In our review of the literature, we found no reports of any patients with anemia and Cameron ulcers who presented with hemoglobin levels as low as our patient had.
Treat with a PPI and iron supplementation
Multiple EGDs may be needed to properly diagnose Cameron ulcers, as they can be difficult to identify. Once a patient receives the diagnosis, he or she will typically be put on a daily proton pump inhibitor (PPI) regimen, such as omeprazole 20 mg bid. However, since many patients with Cameron ulcers also have acid-related problems (as was true in this case), a multifactorial acid suppression approach may be warranted.1 This may include recommending lifestyle modifications (eg, eating small meals, avoiding foods that provoke symptoms, or losing weight) and prescribing medications in addition to a PPI, such as an H2 blocker (eg, 300 mg qid, before meals and at bedtime).
In addition, iron sulfate (325 mg/d, in this case) and blood transfusions may be required to treat the anemia. In refractory cases, endoscopic or surgical interventions, such as hemoclipping, Nissen fundoplication, or laparoscopic gastropexy, may need to be performed.2
Our patient was given a prescription for ferrous sulfate 325 mg/d and omeprazole 20 mg bid. His symptoms improved with treatment, and he was discharged on Day 5; his hemoglobin remained >7 g/dL.
THE TAKEAWAY
The association between chronic iron deficiency anemia and Cameron ulcers has been established but is commonly overlooked in patients presenting with unexplained anemia or an undiagnosed hiatal hernia. This is likely due to their rarity as a cause of anemia, in general.
Furthermore, the lesions can be missed on EGD; multiple EGDs may be needed to make the diagnosis. Once diagnosed, Cameron ulcers typically respond well to twice daily PPI treatment. Patients with refractory, recurrent, or severe lesions, or large, symptomatic hiatal hernias should be referred for surgical assessment.
CORRESPONDENCE
Megan Yee, 801 Broadward Avenue NW, Grand Rapids, MI 49504; [email protected].
THE CASE
A 40-year-old man was referred to the emergency department (ED) with critical anemia after routine blood work at an outside clinic showed a hemoglobin level of 3.5 g/dL. On presentation, he reported symptoms of fatigue, shortness of breath, bilateral leg swelling, dizziness (characterized as light-headedness), and frequent heartburn. He said that the symptoms began 5 weeks earlier, after he was exposed to a relative with hand, foot, and mouth disease.
Additionally, the patient reported an intentional 14-lb weight loss over the 6 months prior to presentation. He denied fever, rash, chest pain, loss of consciousness, headache, abdominal pain, hematemesis, melena, and hematochezia. His medical history was significant for peptic ulcer disease (diagnosed and treated at age 8). He did not recall the specifics, and he denied any related chronic symptoms or complications. His family history (paternal) was significant for colon cancer.
The physical exam revealed conjunctival pallor, skin pallor, jaundice, +1 bilateral lower extremity edema, tachycardia, and tachypnea. Stool Hemoccult was negative. On repeat complete blood count (performed in the ED), hemoglobin was found to be 3.1 g/dL with a mean corpuscular volume of 47 fL.
THE DIAGNOSIS
The patient was admitted to the family medicine service and received 4 units of packed red blood cells, which increased his hemoglobin to the target goal of >7 g/dL. A colonoscopy and an esophagogastroduodenoscopy (EGD) were performed (FIGURE 1A-1C), with results suggestive of diverticulosis, probable Barrett’s mucosa, esophageal ulcer, huge hiatal hernia (at least one-half of the stomach was in the chest), and Cameron ulcers. Esophageal biopsies showed cardiac mucosa with chronic inflammation. Esophageal ulcer biopsies revealed Barrett’s esophagus without dysplasia. Duodenal biopsies displayed normal mucosa.
DISCUSSION
Although rare, Cameron ulcers must be considered in the differential diagnosis of patients with chronic anemia of unknown origin. The potential for these lesions to result in chronic blood loss, which could over time manifest as severe anemia or hypovolemic shock, makes proper diagnosis and prompt treatment especially important.4,5
In our patient’s case, his severe anemia was likely the result of a combination of the esophageal and Cameron ulcers evidenced on EGD, rather than any single ulcer. In our review of the literature, we found no reports of any patients with anemia and Cameron ulcers who presented with hemoglobin levels as low as our patient had.
Treat with a PPI and iron supplementation
Multiple EGDs may be needed to properly diagnose Cameron ulcers, as they can be difficult to identify. Once a patient receives the diagnosis, he or she will typically be put on a daily proton pump inhibitor (PPI) regimen, such as omeprazole 20 mg bid. However, since many patients with Cameron ulcers also have acid-related problems (as was true in this case), a multifactorial acid suppression approach may be warranted.1 This may include recommending lifestyle modifications (eg, eating small meals, avoiding foods that provoke symptoms, or losing weight) and prescribing medications in addition to a PPI, such as an H2 blocker (eg, 300 mg qid, before meals and at bedtime).
In addition, iron sulfate (325 mg/d, in this case) and blood transfusions may be required to treat the anemia. In refractory cases, endoscopic or surgical interventions, such as hemoclipping, Nissen fundoplication, or laparoscopic gastropexy, may need to be performed.2
Our patient was given a prescription for ferrous sulfate 325 mg/d and omeprazole 20 mg bid. His symptoms improved with treatment, and he was discharged on Day 5; his hemoglobin remained >7 g/dL.
THE TAKEAWAY
The association between chronic iron deficiency anemia and Cameron ulcers has been established but is commonly overlooked in patients presenting with unexplained anemia or an undiagnosed hiatal hernia. This is likely due to their rarity as a cause of anemia, in general.
Furthermore, the lesions can be missed on EGD; multiple EGDs may be needed to make the diagnosis. Once diagnosed, Cameron ulcers typically respond well to twice daily PPI treatment. Patients with refractory, recurrent, or severe lesions, or large, symptomatic hiatal hernias should be referred for surgical assessment.
CORRESPONDENCE
Megan Yee, 801 Broadward Avenue NW, Grand Rapids, MI 49504; [email protected].
1. Maganty K, Smith RL. Cameron lesions: unusual cause of gastrointestinal bleeding and anemia. Digestion. 2008;77:214-217.
2. Camus M, Jensen DM, Ohning GV, et al. Severe upper gastrointestinal hemorrhage from linear gastric ulcers in large hiatal hernias: a large prospective case series of Cameron ulcers. Endoscopy. 2013;45:397-400.
3. Kimer N, Schmidt PN, Krag, A. Cameron lesions: an often overlooked cause of iron deficiency anaemia in patients with large hiatal hernias. BMJ Case Rep. 2010;2010.
4. Kapadia S, Jagroop S, Kumar, A. Cameron ulcers: an atypical source for a massive upper gastrointestinal bleed. World J Gastroenterol. 2012;18:4959-4961.
5. Gupta P, Suryadevara M, Das A, et al. Cameron ulcer causing severe anemia in a patient with diaphragmatic hernia. Am J Case Rep. 2015;16:733-736.
1. Maganty K, Smith RL. Cameron lesions: unusual cause of gastrointestinal bleeding and anemia. Digestion. 2008;77:214-217.
2. Camus M, Jensen DM, Ohning GV, et al. Severe upper gastrointestinal hemorrhage from linear gastric ulcers in large hiatal hernias: a large prospective case series of Cameron ulcers. Endoscopy. 2013;45:397-400.
3. Kimer N, Schmidt PN, Krag, A. Cameron lesions: an often overlooked cause of iron deficiency anaemia in patients with large hiatal hernias. BMJ Case Rep. 2010;2010.
4. Kapadia S, Jagroop S, Kumar, A. Cameron ulcers: an atypical source for a massive upper gastrointestinal bleed. World J Gastroenterol. 2012;18:4959-4961.
5. Gupta P, Suryadevara M, Das A, et al. Cameron ulcer causing severe anemia in a patient with diaphragmatic hernia. Am J Case Rep. 2015;16:733-736.
Isolated ocular metastases from lung cancer
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
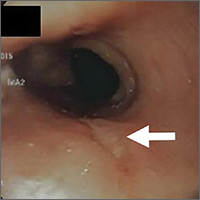
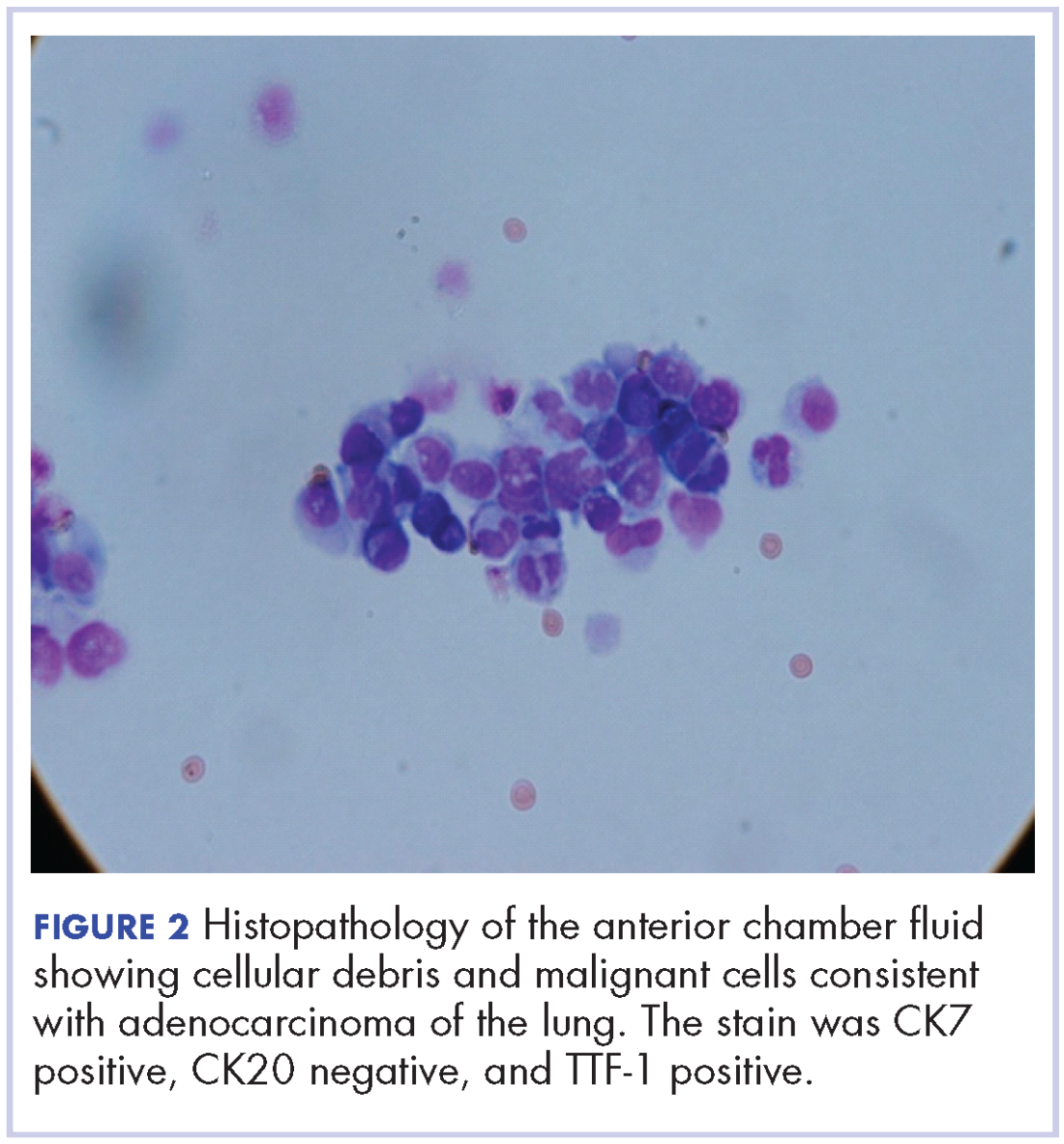
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.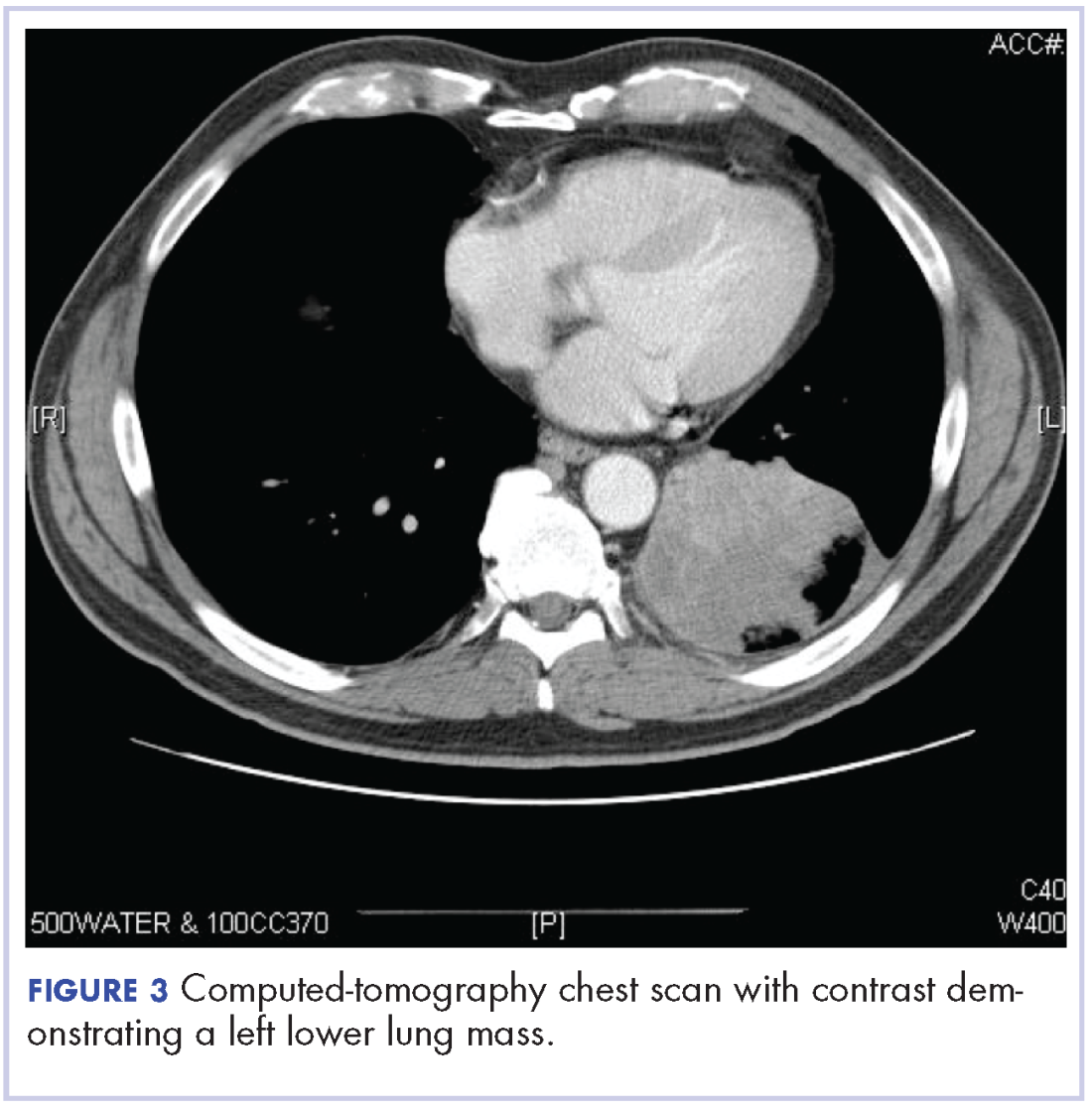

He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).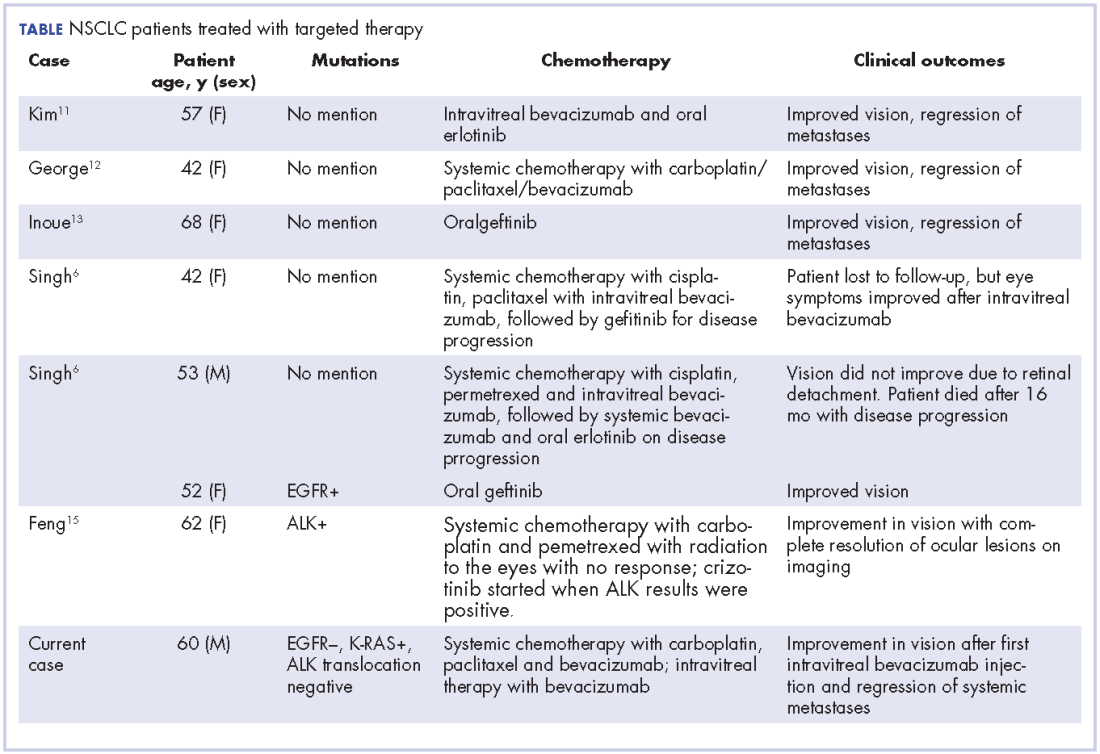
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.
He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.
He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
Resolution of refractory pruritus with aprepitant in a patient with microcystic adnexal carcinoma
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.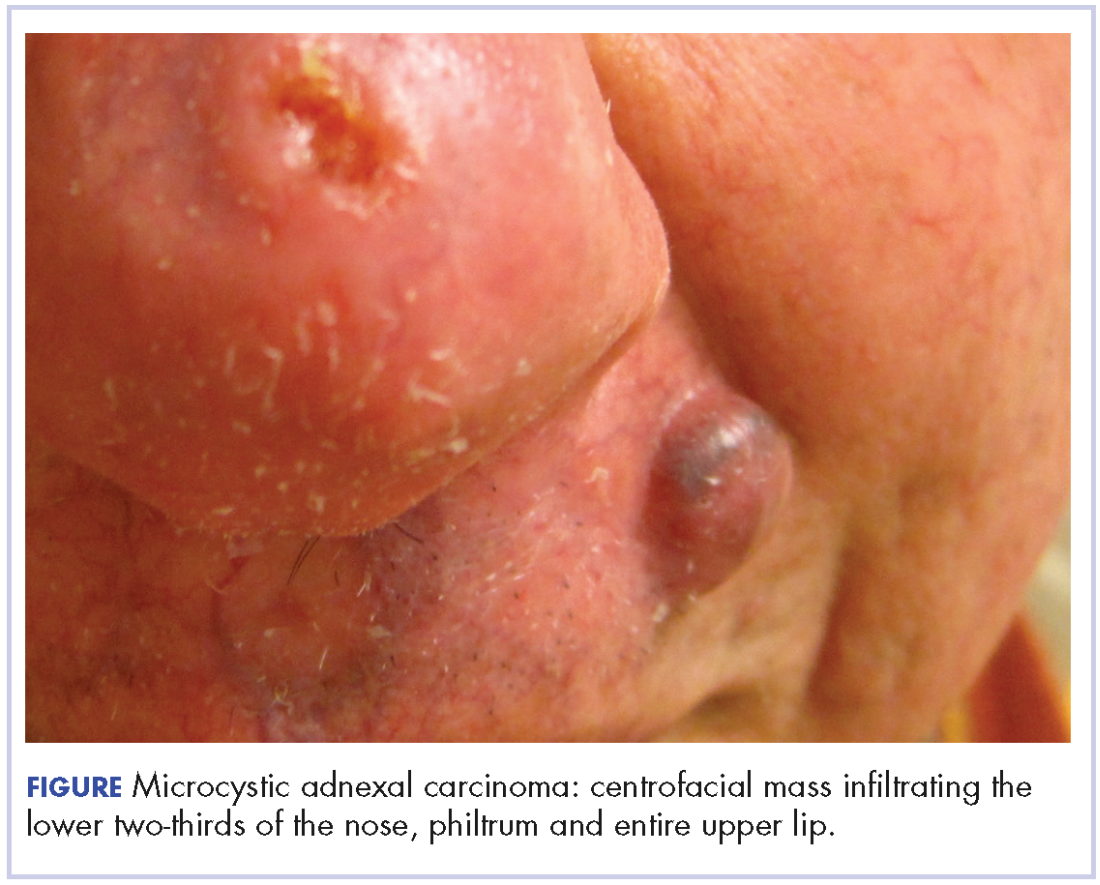
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
Rare paraneoplastic dermatomyositis secondary to high-grade bladder cancer
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).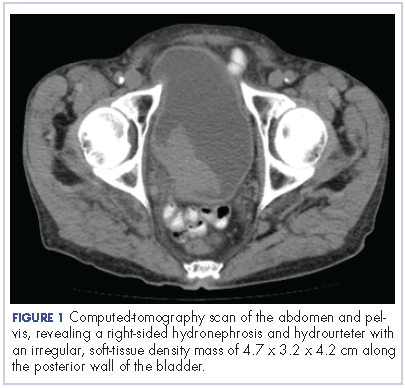
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
Rapid Development of Life-Threatening Emamectin Benzoate Poisoning
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
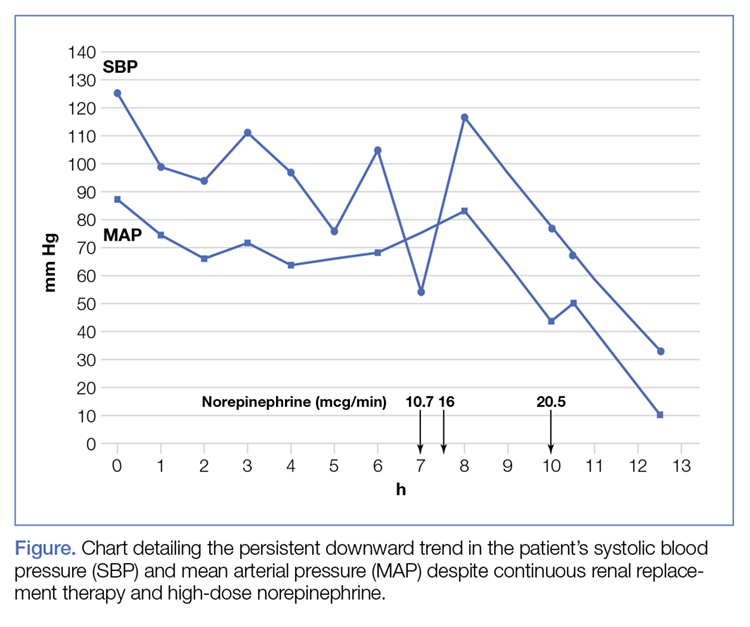
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
Intralymphatic Histiocytosis Treated With Intralesional Triamcinolone Acetonide and Pressure Bandage
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
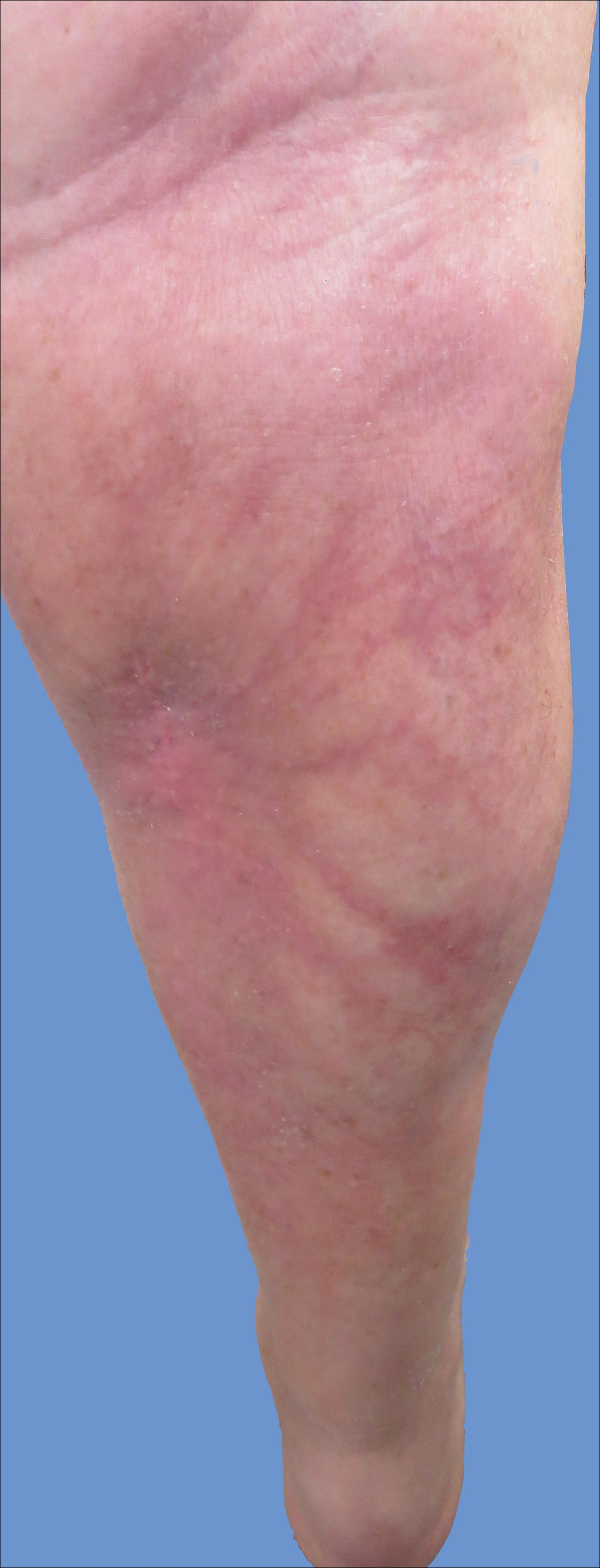
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
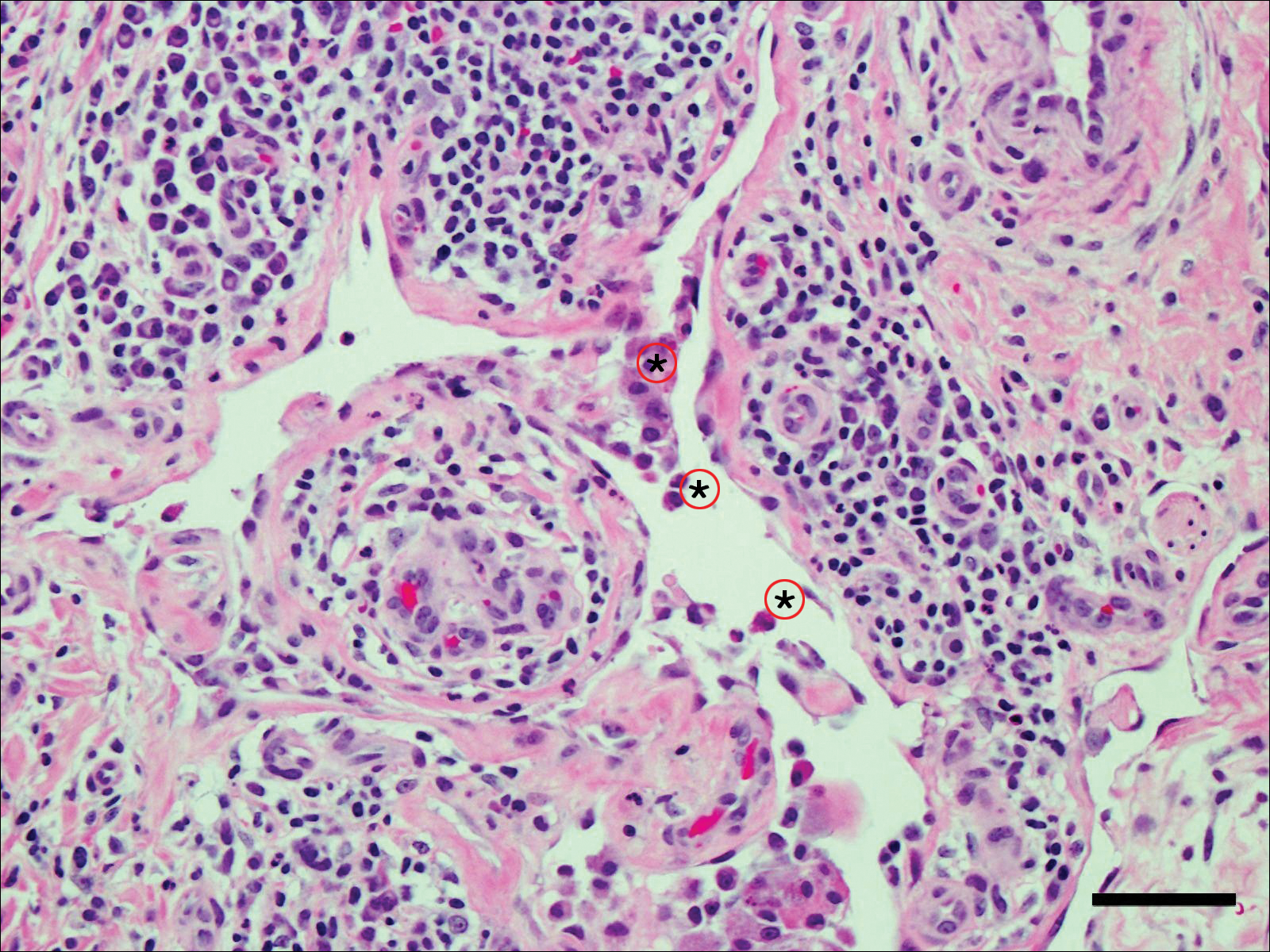
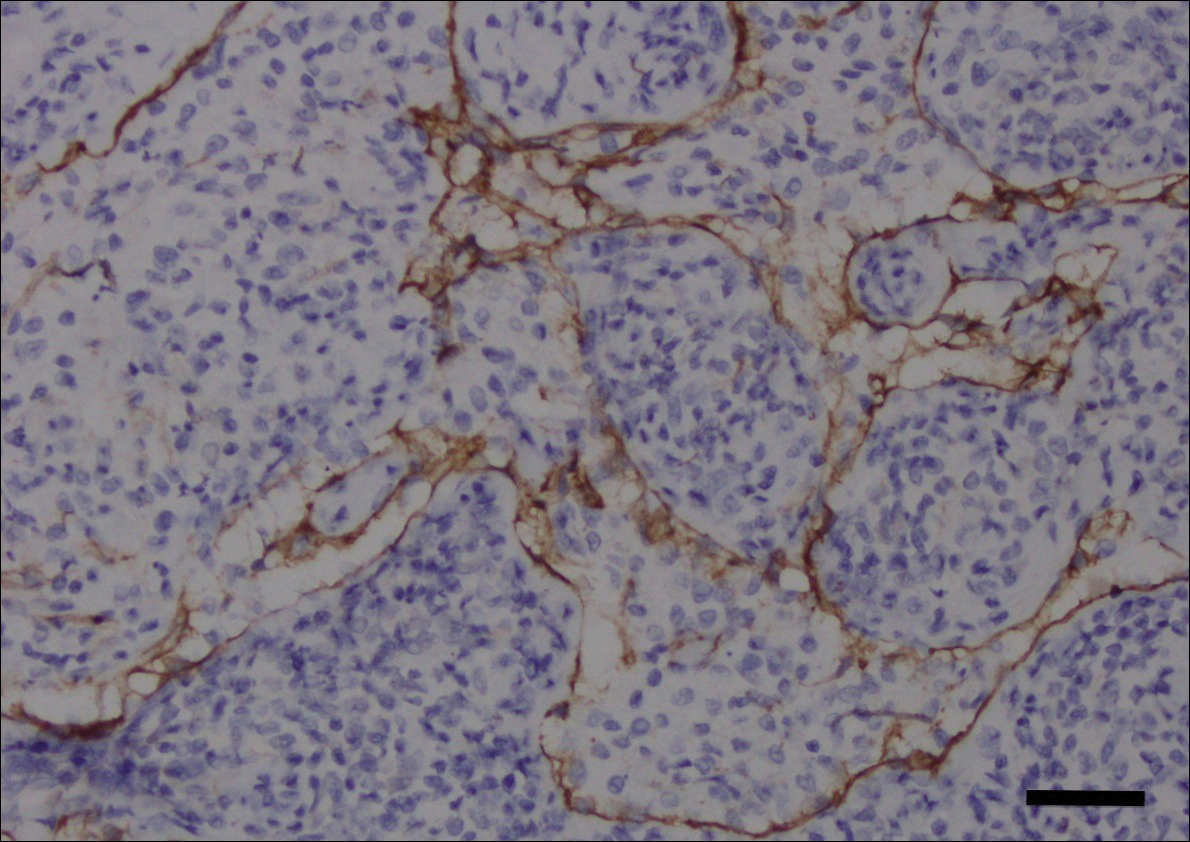
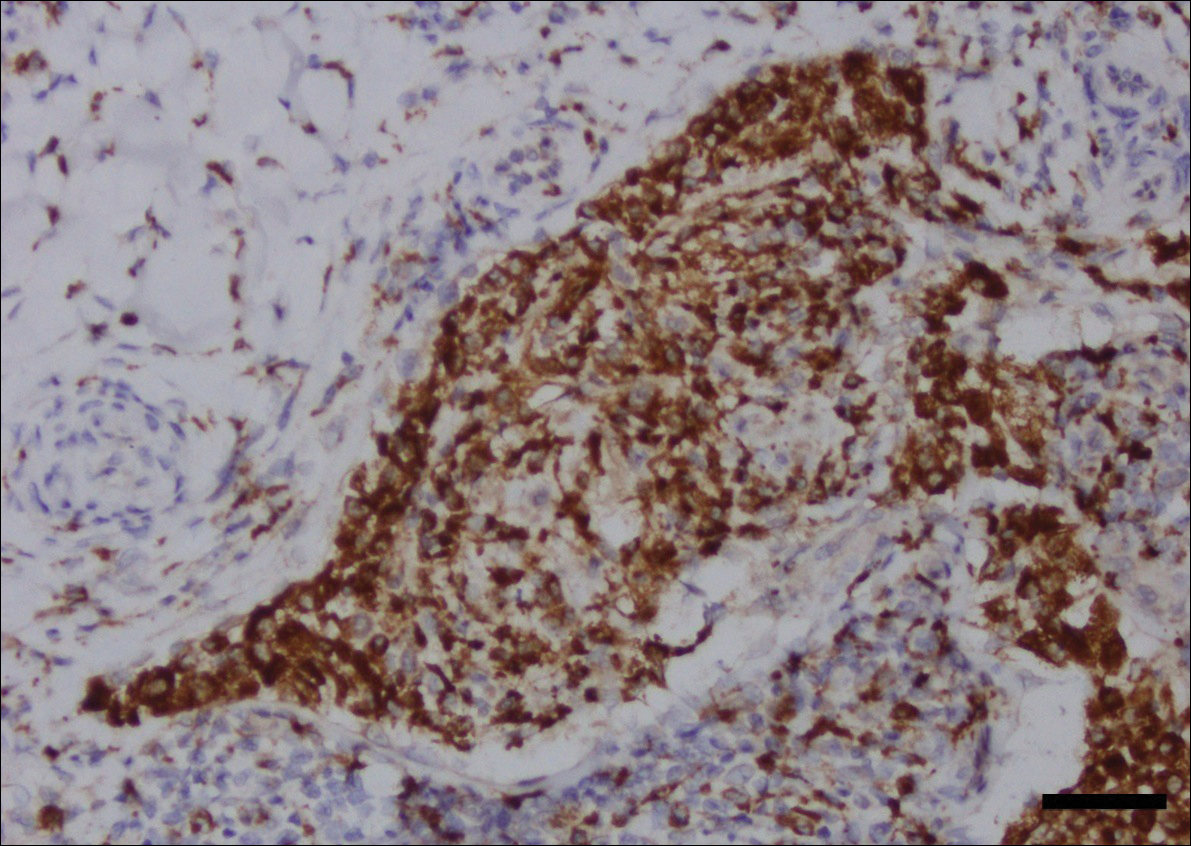
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
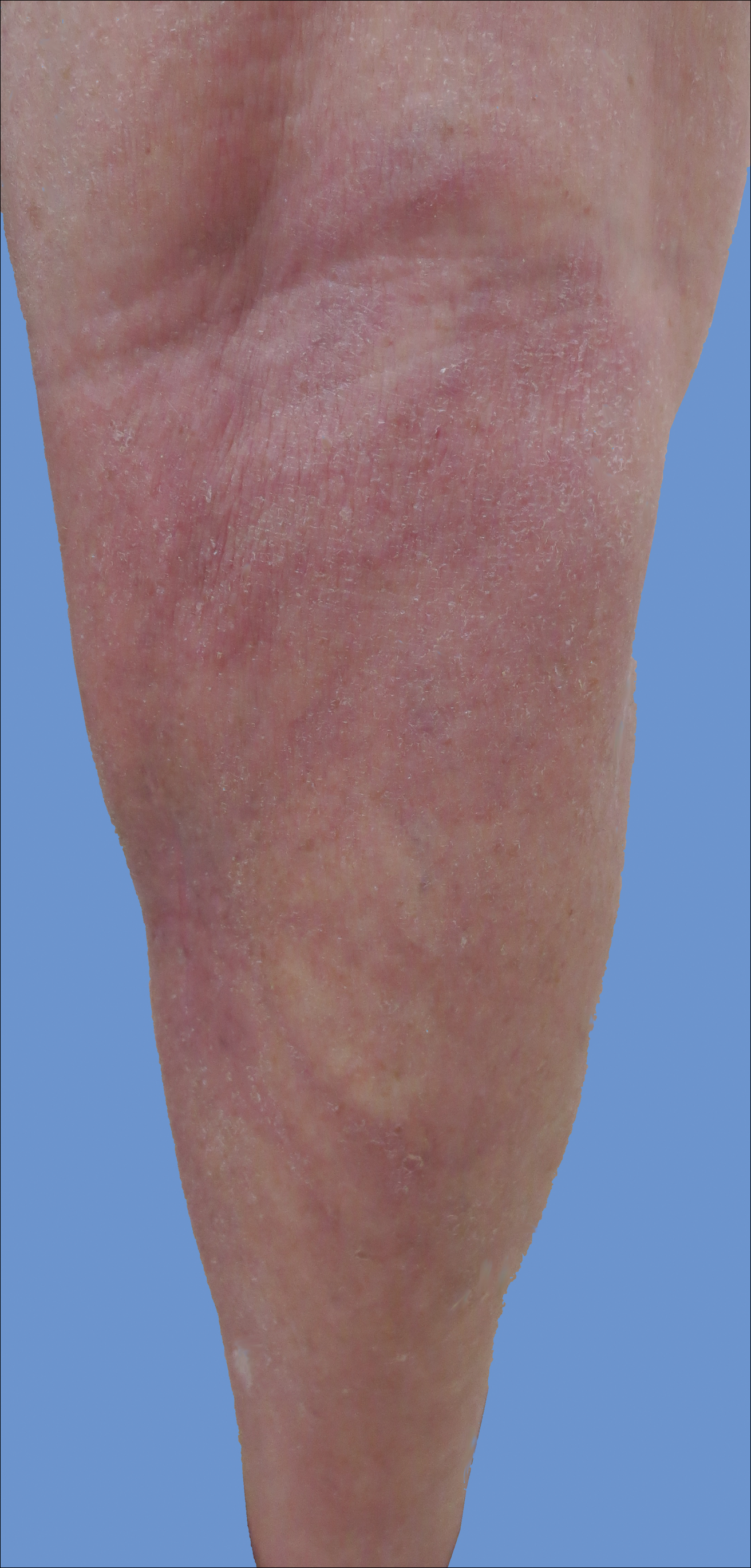
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
Practice Points
- Intralymphatic histiocytosis is a rare disorder often associated with rheumatic arthritis and joint prostheses.
- The diagnosis is made by histopathology as well as D2-40 and CD68 immunostaining.
- While there is no gold standard of treatment for intralymphatic histiocytosis, intralesional triamcinolone proved efficacious in this case with prolonged results.
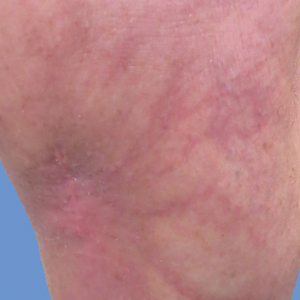
Synovial Chondromatosis: An Unusual Case of Knee Pain and Swelling
Joint mice or loose/rice bodies are infrequently encountered within joints. Usually, they are either fibrin or cartilaginous. The fibrin type, typically results from bleeding within a joint from synovitis, rheumatoid arthritis (RA), or tuberculosis, and the cartilaginous/osteocartilaginous type develop from trauma or osteoarthritis.1 A rare cause of osteocartilaginous joint mice is synovial chondromatosis(SC), which can produce multiple loose bodies that originate from the synovial membranes of joints, bursae, and tendon sheaths of large joints; the knee being the most common (50%-65 % of cases).1,2
A case of a male who had multiple years of left knee pain and swelling without a documented traumatic cause is presented
Case Presentation
A 34-year-old male veteran was evaluated and treated in a VA orthopedic outpatient clinic by a physician assistant for anterior left knee pain and swelling of insidious onset that had persisted for 1.5 years. The patient reported experiencing no trauma. His primary care provider already was treating the patient with nonsteroidal anti-inflammatory medications (NSAIDs), icing, and bracing. He had full motion in his knee with extension/flexion 0° to 130°. Collateral and cruciate ligaments were stable. He had a positive McMurray test. The X-rays showed no pathology. Due to the positive meniscal tear signs, a magnetic resonance image (MRI) was ordered.
The patient was intermittently nonadherent with follow-up care. The MRI results were available at a subsequent appointment 3 months after the index evaluation, which revealed a large joint effusion with rice bodies, small erosion of the posterior tibialplateau, and synovial proliferation of the anterior knee joint. A steroid injection to his affected knee was given. Concerns for possible RA led to a workup. The laboratory results included rheumatoid factor (weakly positive), antinuclear-antibodies (negative), human immunodeficiency virus (positivewith western blot negative), C-reactive protein (< 0.02 mg/L), erythrocyte sedimentation rate (5 mm/h), white blood cell count (4.6 µL), hepatitis B surface antigen (reactive), hepatitis A antibody (IgG reactive), synovial fluid cultures, and Gram stain (negative).
The patient saw a rheumatologist 7 months after a RA referral was processed. The consulting rheumatologist was unconcerned by a weakly positive rheumatoid factor, which was later repeated and was negative. The rheumatologist excluded the possibility of RA, and the patient was diagnosed with oligoarthritis. The treatment rendered was to continue NSAIDs and to return to the orthopedic clinic for continued care.
The patient had irregular follow-up visits where he received multiple methylprednisolone acetate intra-articular injections. His motion regressed until extension/flexion had decreased to 5°/85°. At this point the patient was forwarded to an operative orthopedic surgeon for evaluation for surgical intervention. Recent anteroposterior and lateral left knee X-rays showed faint intra-articular calcification, joint effusion, with mild arthritic changes of the patellofemoral joint (Figures 1A and 1B). 
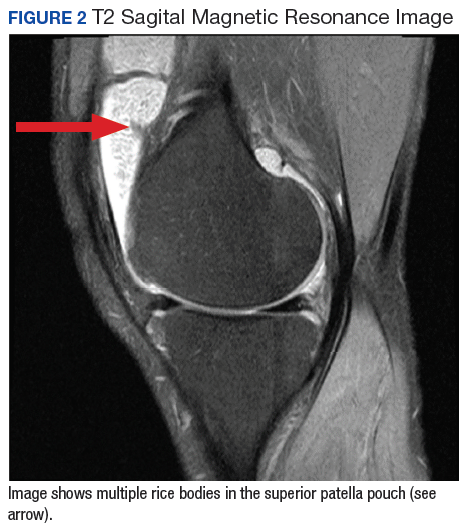
At surgery, on placing the infralateral portal, clear straw-colored fluid exited the cannula followed by copious small white rice bodies, which were sent to pathology for evaluation. The knee was surgically evaluated, and extensive rice bodies were encountered (Figure 3). These were extracted with a full radius shaver. The chondral surfaces were inspected. There were no arthritic changes, but the synovial lining of the joint was hypertrophied and reactive (Milgram phase 2). After all loose bodies were extracted, the patient’s incisions were closed with nylon suture, and he was placed in sterile dressings with a postoperative range of motion brace.
The patient presented for his routine postoperative visit 14 days after surgery. Pathology results showed synociocytes, and inflammatory cells were negative for malignancy. The patient was forwarded to a local hospital for further evaluation and treatment by an orthopedic oncologist due to a reported 5% chance of malignant transformation.1-3 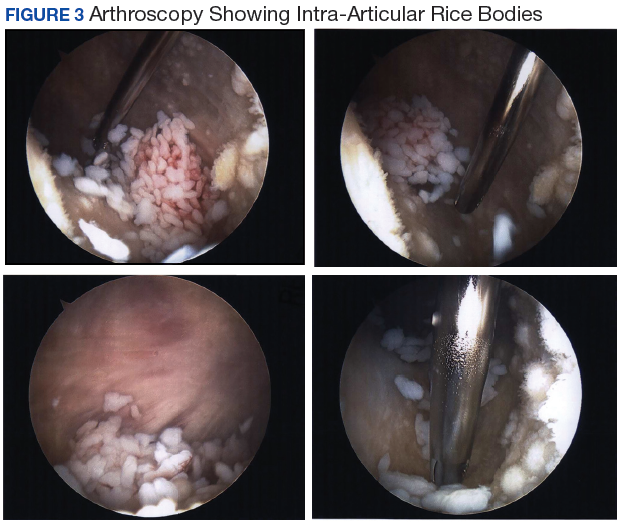
Discussion
Synovial chondromatosis or osteochondromatosis is a rare, benign, metaplastic, typically monoarticular disorder of the synovial lining of joints, bursae, and synovial sheaths, usually affecting large joints.1-5 Although any joint can be involved, such as metacarpalphalangeal joints, temporomandibular joints, distal radio-ulnar joints, and the hips, the knee is the most common with an occurrence rate 50% to 65%.3-5 Extra-articular proliferation can be seen in cases of osteochondromatosis.2 It is characterized by the formation of intra-articular nodules of the synovium that can detach and become loose bodies, which can secondarily become calcified/ossified.4,6 The differential diagnosis associated with SC should include synovial hemangioma, pigmented villonodular synovitis, synovial cyst, lipoma arborescence, and malignancies, such as synovial chondrosarcoma or synovial sarcoma.3
Men are affected twice as much as are women, usually in the fourth through sixth decades of life, and a mean age of 47.7 years.1,3-5,7,8 The SC occurrence rate in adults is 1:100,000.2 Patients typically present with insidious gradual mechanical symptoms, such as pain (> 85% of cases), swelling (42%-58%), and decreased motion (38%-55%) in the affected joint.2,3,6 Often there is crepitus with motion, diffuse tenderness, effusion, and occasionally nodules can be palpated.2,3 Histologically, the synovium exhibits condrocytic metaplasia of fibroblasts with influence from transforming growth factor-β and bone morphogenic proteins.1,4
Synovial chondromatosis can mimic osteoarthritis or meniscal pathology.3 Because of a chance of malignant transformation, any patient with rapid late deterioration of clinical features should be evaluated for chondrosarcoma or synovial sarcoma.1-4 Plain radiographs may help differentiate the cause showing calcific joint mice and peri-articular erosions. However, the intra-articular loose bodies are frequently radiolucent, and a MRI may be warranted to definitively differentiate the diagnosis.2,7,8
Loose bodies tend to exhibit a low signal on T1-weighted images and a high signal on T2-weighted images, although there may a be low signal on all images where there is extensive calcification of the loose bodies.2 Ultrasound also is a useful diagnostic tool that can show numerous echogenic bodies, effusion, and synovial hypertrophy.2
A classic article by Milgram discussed the phases of the proliferative changes associated with SC, where phase 1 shows active intrasynovial disease with no loose bodies.9 Phase 2 has transitional lesions with osteochondral nodules within the synovial membrane and free bodies within the joint cavity. Last, in phase 3, there are multiple osteochondral free bodies but quiescent intrasynovial disease. The patient in this case study exhibited intra-articular activity mimicking phase 2 with extensive intra-articular loose bodies and reactive synovial lining.3,9
In the early phase of the disease, conservative management may be trialed with NSAIDs, bracing, and injections, but typically surgical intervention is warranted after free bodies are found present, because they limit motion and cause recalcitrant swelling.2,8 There is a controversy whether arthroscopic removal of loose bodies or excision with synovectomy is the treatment of choice.6 Ogilvie-Harris and colleagues reviewed the results of both procedures and found that although removal of loose bodies alone may be sufficient, there is the potential for recurrence.9,10 In order to reduce potential recurrence, removal of loose bodies with anterior and posterior synovectomy is the treatment of choice.9
If arthroscopic removal of loose bodies without synovectomy is performed, then the patient should be followed closely for recurrence, which Jesalpura and colleagues reported to occur for 11.5% of patients.9,11 If there is a reappearance, then a synovectomy should be performed.10 A recommended treatment option for recalcitrant SC is radiation, but this carries the added risk of perpetuating malignant transformation.1,7
Unfortunately, osteoarthritis can be a significant long-term postoperative adverse effect.3,6-8 This typically is related to the amount of articular damage that is present at surgery. Many times, the arthritis becomes significant enough to require total joint arthroplasty.4 Close long-term follow-up is recommended, because although rare, there is a chance of malignant change.1-4
Conclusion
Synovial chondromatosis is an uncommon cause of knee pain and swelling and should be included in the differential diagnosis when evaluating any adult aged 30 years to 50 years with knee pain of insidious onset. Appropriate workup, intervention, and treatment will allow final diagnosis and correlating care to be administered to the patient.
1. Libbey NP, Mirrer F. Synovial chondromatosis. Med Health R I. 2011;94(9):274-275.
2. Giancane G, Tanturri de Horatio L, Buonuomo PS, Barbuti D, Lais G, Cortis E. Swollen knee due to primary synovial chondromatosis in pediatrics: a rare and possibly misdiagnosed condition. Rheumatol Int. 2013;33(8):2183-2185.
3. Serbest S, Tiftikçi U, Karaaslan F, Tosun HB, Sevinç HF, Balci M. A neglected case of giant synovial chondromatosis in knee joint. Pan Afr Med J. 2015;22:5.
4. Hallam P, Ashwood N, Cobb J, Fazal A, Heatley W. Malignant transformation in synovial chondromatosis of the knee? Knee. 2001;8(3):239-242.
5. Pimentel Cde Q, Hoff LS, de Sousa LF, Cordeiro RA, Pereira RM. Primary synovial osteochondromatosis of the knee. Rheumatol (Oxford). 2015;54(10):1815.
6. Damron TA, Sim FH. Soft-tissue tumors about the knee. J Am Acad Orthop Surg. 1997;5(3):141-152.
7. Krych A, Odland A, Rose P, et al. Onconlogic conditions that simulate common sports injuries. J Am Acad Orthop Surg. 2014;22(4):223-234.
8. Adelani MA, Wupperman RM, Holt GE. Benign synovial disorders. J Am Acad Orthop Surg. 2008;16(5):268-275.
9. Migram JW. Synovial osteochondromatosis: a histopathological study of thirty cases. J Bone Joint Surg Am. 1977;59(6):792-801.
10. Ogilvie-Harris DJ, Saleh K. Generalized synovial chondromatosis of the knee: a comparison of removal of the loose bodies alone with arthroscopic synovectomy. Arthroscopy.1994;10(2):166-170.
11. Jesalpura JP, Chung HW, Patnaik S, Choi HW, Kim JI, Nha KW. Athroscopic treatment of localized synovial chondromatosis of the posterior knee joint. Orthopedics. 2010;33(1):49
Joint mice or loose/rice bodies are infrequently encountered within joints. Usually, they are either fibrin or cartilaginous. The fibrin type, typically results from bleeding within a joint from synovitis, rheumatoid arthritis (RA), or tuberculosis, and the cartilaginous/osteocartilaginous type develop from trauma or osteoarthritis.1 A rare cause of osteocartilaginous joint mice is synovial chondromatosis(SC), which can produce multiple loose bodies that originate from the synovial membranes of joints, bursae, and tendon sheaths of large joints; the knee being the most common (50%-65 % of cases).1,2
A case of a male who had multiple years of left knee pain and swelling without a documented traumatic cause is presented
Case Presentation
A 34-year-old male veteran was evaluated and treated in a VA orthopedic outpatient clinic by a physician assistant for anterior left knee pain and swelling of insidious onset that had persisted for 1.5 years. The patient reported experiencing no trauma. His primary care provider already was treating the patient with nonsteroidal anti-inflammatory medications (NSAIDs), icing, and bracing. He had full motion in his knee with extension/flexion 0° to 130°. Collateral and cruciate ligaments were stable. He had a positive McMurray test. The X-rays showed no pathology. Due to the positive meniscal tear signs, a magnetic resonance image (MRI) was ordered.
The patient was intermittently nonadherent with follow-up care. The MRI results were available at a subsequent appointment 3 months after the index evaluation, which revealed a large joint effusion with rice bodies, small erosion of the posterior tibialplateau, and synovial proliferation of the anterior knee joint. A steroid injection to his affected knee was given. Concerns for possible RA led to a workup. The laboratory results included rheumatoid factor (weakly positive), antinuclear-antibodies (negative), human immunodeficiency virus (positivewith western blot negative), C-reactive protein (< 0.02 mg/L), erythrocyte sedimentation rate (5 mm/h), white blood cell count (4.6 µL), hepatitis B surface antigen (reactive), hepatitis A antibody (IgG reactive), synovial fluid cultures, and Gram stain (negative).
The patient saw a rheumatologist 7 months after a RA referral was processed. The consulting rheumatologist was unconcerned by a weakly positive rheumatoid factor, which was later repeated and was negative. The rheumatologist excluded the possibility of RA, and the patient was diagnosed with oligoarthritis. The treatment rendered was to continue NSAIDs and to return to the orthopedic clinic for continued care.
The patient had irregular follow-up visits where he received multiple methylprednisolone acetate intra-articular injections. His motion regressed until extension/flexion had decreased to 5°/85°. At this point the patient was forwarded to an operative orthopedic surgeon for evaluation for surgical intervention. Recent anteroposterior and lateral left knee X-rays showed faint intra-articular calcification, joint effusion, with mild arthritic changes of the patellofemoral joint (Figures 1A and 1B).
At surgery, on placing the infralateral portal, clear straw-colored fluid exited the cannula followed by copious small white rice bodies, which were sent to pathology for evaluation. The knee was surgically evaluated, and extensive rice bodies were encountered (Figure 3). These were extracted with a full radius shaver. The chondral surfaces were inspected. There were no arthritic changes, but the synovial lining of the joint was hypertrophied and reactive (Milgram phase 2). After all loose bodies were extracted, the patient’s incisions were closed with nylon suture, and he was placed in sterile dressings with a postoperative range of motion brace.
The patient presented for his routine postoperative visit 14 days after surgery. Pathology results showed synociocytes, and inflammatory cells were negative for malignancy. The patient was forwarded to a local hospital for further evaluation and treatment by an orthopedic oncologist due to a reported 5% chance of malignant transformation.1-3
Discussion
Synovial chondromatosis or osteochondromatosis is a rare, benign, metaplastic, typically monoarticular disorder of the synovial lining of joints, bursae, and synovial sheaths, usually affecting large joints.1-5 Although any joint can be involved, such as metacarpalphalangeal joints, temporomandibular joints, distal radio-ulnar joints, and the hips, the knee is the most common with an occurrence rate 50% to 65%.3-5 Extra-articular proliferation can be seen in cases of osteochondromatosis.2 It is characterized by the formation of intra-articular nodules of the synovium that can detach and become loose bodies, which can secondarily become calcified/ossified.4,6 The differential diagnosis associated with SC should include synovial hemangioma, pigmented villonodular synovitis, synovial cyst, lipoma arborescence, and malignancies, such as synovial chondrosarcoma or synovial sarcoma.3
Men are affected twice as much as are women, usually in the fourth through sixth decades of life, and a mean age of 47.7 years.1,3-5,7,8 The SC occurrence rate in adults is 1:100,000.2 Patients typically present with insidious gradual mechanical symptoms, such as pain (> 85% of cases), swelling (42%-58%), and decreased motion (38%-55%) in the affected joint.2,3,6 Often there is crepitus with motion, diffuse tenderness, effusion, and occasionally nodules can be palpated.2,3 Histologically, the synovium exhibits condrocytic metaplasia of fibroblasts with influence from transforming growth factor-β and bone morphogenic proteins.1,4
Synovial chondromatosis can mimic osteoarthritis or meniscal pathology.3 Because of a chance of malignant transformation, any patient with rapid late deterioration of clinical features should be evaluated for chondrosarcoma or synovial sarcoma.1-4 Plain radiographs may help differentiate the cause showing calcific joint mice and peri-articular erosions. However, the intra-articular loose bodies are frequently radiolucent, and a MRI may be warranted to definitively differentiate the diagnosis.2,7,8
Loose bodies tend to exhibit a low signal on T1-weighted images and a high signal on T2-weighted images, although there may a be low signal on all images where there is extensive calcification of the loose bodies.2 Ultrasound also is a useful diagnostic tool that can show numerous echogenic bodies, effusion, and synovial hypertrophy.2
A classic article by Milgram discussed the phases of the proliferative changes associated with SC, where phase 1 shows active intrasynovial disease with no loose bodies.9 Phase 2 has transitional lesions with osteochondral nodules within the synovial membrane and free bodies within the joint cavity. Last, in phase 3, there are multiple osteochondral free bodies but quiescent intrasynovial disease. The patient in this case study exhibited intra-articular activity mimicking phase 2 with extensive intra-articular loose bodies and reactive synovial lining.3,9
In the early phase of the disease, conservative management may be trialed with NSAIDs, bracing, and injections, but typically surgical intervention is warranted after free bodies are found present, because they limit motion and cause recalcitrant swelling.2,8 There is a controversy whether arthroscopic removal of loose bodies or excision with synovectomy is the treatment of choice.6 Ogilvie-Harris and colleagues reviewed the results of both procedures and found that although removal of loose bodies alone may be sufficient, there is the potential for recurrence.9,10 In order to reduce potential recurrence, removal of loose bodies with anterior and posterior synovectomy is the treatment of choice.9
If arthroscopic removal of loose bodies without synovectomy is performed, then the patient should be followed closely for recurrence, which Jesalpura and colleagues reported to occur for 11.5% of patients.9,11 If there is a reappearance, then a synovectomy should be performed.10 A recommended treatment option for recalcitrant SC is radiation, but this carries the added risk of perpetuating malignant transformation.1,7
Unfortunately, osteoarthritis can be a significant long-term postoperative adverse effect.3,6-8 This typically is related to the amount of articular damage that is present at surgery. Many times, the arthritis becomes significant enough to require total joint arthroplasty.4 Close long-term follow-up is recommended, because although rare, there is a chance of malignant change.1-4
Conclusion
Synovial chondromatosis is an uncommon cause of knee pain and swelling and should be included in the differential diagnosis when evaluating any adult aged 30 years to 50 years with knee pain of insidious onset. Appropriate workup, intervention, and treatment will allow final diagnosis and correlating care to be administered to the patient.
Joint mice or loose/rice bodies are infrequently encountered within joints. Usually, they are either fibrin or cartilaginous. The fibrin type, typically results from bleeding within a joint from synovitis, rheumatoid arthritis (RA), or tuberculosis, and the cartilaginous/osteocartilaginous type develop from trauma or osteoarthritis.1 A rare cause of osteocartilaginous joint mice is synovial chondromatosis(SC), which can produce multiple loose bodies that originate from the synovial membranes of joints, bursae, and tendon sheaths of large joints; the knee being the most common (50%-65 % of cases).1,2
A case of a male who had multiple years of left knee pain and swelling without a documented traumatic cause is presented
Case Presentation
A 34-year-old male veteran was evaluated and treated in a VA orthopedic outpatient clinic by a physician assistant for anterior left knee pain and swelling of insidious onset that had persisted for 1.5 years. The patient reported experiencing no trauma. His primary care provider already was treating the patient with nonsteroidal anti-inflammatory medications (NSAIDs), icing, and bracing. He had full motion in his knee with extension/flexion 0° to 130°. Collateral and cruciate ligaments were stable. He had a positive McMurray test. The X-rays showed no pathology. Due to the positive meniscal tear signs, a magnetic resonance image (MRI) was ordered.
The patient was intermittently nonadherent with follow-up care. The MRI results were available at a subsequent appointment 3 months after the index evaluation, which revealed a large joint effusion with rice bodies, small erosion of the posterior tibialplateau, and synovial proliferation of the anterior knee joint. A steroid injection to his affected knee was given. Concerns for possible RA led to a workup. The laboratory results included rheumatoid factor (weakly positive), antinuclear-antibodies (negative), human immunodeficiency virus (positivewith western blot negative), C-reactive protein (< 0.02 mg/L), erythrocyte sedimentation rate (5 mm/h), white blood cell count (4.6 µL), hepatitis B surface antigen (reactive), hepatitis A antibody (IgG reactive), synovial fluid cultures, and Gram stain (negative).
The patient saw a rheumatologist 7 months after a RA referral was processed. The consulting rheumatologist was unconcerned by a weakly positive rheumatoid factor, which was later repeated and was negative. The rheumatologist excluded the possibility of RA, and the patient was diagnosed with oligoarthritis. The treatment rendered was to continue NSAIDs and to return to the orthopedic clinic for continued care.
The patient had irregular follow-up visits where he received multiple methylprednisolone acetate intra-articular injections. His motion regressed until extension/flexion had decreased to 5°/85°. At this point the patient was forwarded to an operative orthopedic surgeon for evaluation for surgical intervention. Recent anteroposterior and lateral left knee X-rays showed faint intra-articular calcification, joint effusion, with mild arthritic changes of the patellofemoral joint (Figures 1A and 1B).
At surgery, on placing the infralateral portal, clear straw-colored fluid exited the cannula followed by copious small white rice bodies, which were sent to pathology for evaluation. The knee was surgically evaluated, and extensive rice bodies were encountered (Figure 3). These were extracted with a full radius shaver. The chondral surfaces were inspected. There were no arthritic changes, but the synovial lining of the joint was hypertrophied and reactive (Milgram phase 2). After all loose bodies were extracted, the patient’s incisions were closed with nylon suture, and he was placed in sterile dressings with a postoperative range of motion brace.
The patient presented for his routine postoperative visit 14 days after surgery. Pathology results showed synociocytes, and inflammatory cells were negative for malignancy. The patient was forwarded to a local hospital for further evaluation and treatment by an orthopedic oncologist due to a reported 5% chance of malignant transformation.1-3
Discussion
Synovial chondromatosis or osteochondromatosis is a rare, benign, metaplastic, typically monoarticular disorder of the synovial lining of joints, bursae, and synovial sheaths, usually affecting large joints.1-5 Although any joint can be involved, such as metacarpalphalangeal joints, temporomandibular joints, distal radio-ulnar joints, and the hips, the knee is the most common with an occurrence rate 50% to 65%.3-5 Extra-articular proliferation can be seen in cases of osteochondromatosis.2 It is characterized by the formation of intra-articular nodules of the synovium that can detach and become loose bodies, which can secondarily become calcified/ossified.4,6 The differential diagnosis associated with SC should include synovial hemangioma, pigmented villonodular synovitis, synovial cyst, lipoma arborescence, and malignancies, such as synovial chondrosarcoma or synovial sarcoma.3
Men are affected twice as much as are women, usually in the fourth through sixth decades of life, and a mean age of 47.7 years.1,3-5,7,8 The SC occurrence rate in adults is 1:100,000.2 Patients typically present with insidious gradual mechanical symptoms, such as pain (> 85% of cases), swelling (42%-58%), and decreased motion (38%-55%) in the affected joint.2,3,6 Often there is crepitus with motion, diffuse tenderness, effusion, and occasionally nodules can be palpated.2,3 Histologically, the synovium exhibits condrocytic metaplasia of fibroblasts with influence from transforming growth factor-β and bone morphogenic proteins.1,4
Synovial chondromatosis can mimic osteoarthritis or meniscal pathology.3 Because of a chance of malignant transformation, any patient with rapid late deterioration of clinical features should be evaluated for chondrosarcoma or synovial sarcoma.1-4 Plain radiographs may help differentiate the cause showing calcific joint mice and peri-articular erosions. However, the intra-articular loose bodies are frequently radiolucent, and a MRI may be warranted to definitively differentiate the diagnosis.2,7,8
Loose bodies tend to exhibit a low signal on T1-weighted images and a high signal on T2-weighted images, although there may a be low signal on all images where there is extensive calcification of the loose bodies.2 Ultrasound also is a useful diagnostic tool that can show numerous echogenic bodies, effusion, and synovial hypertrophy.2
A classic article by Milgram discussed the phases of the proliferative changes associated with SC, where phase 1 shows active intrasynovial disease with no loose bodies.9 Phase 2 has transitional lesions with osteochondral nodules within the synovial membrane and free bodies within the joint cavity. Last, in phase 3, there are multiple osteochondral free bodies but quiescent intrasynovial disease. The patient in this case study exhibited intra-articular activity mimicking phase 2 with extensive intra-articular loose bodies and reactive synovial lining.3,9
In the early phase of the disease, conservative management may be trialed with NSAIDs, bracing, and injections, but typically surgical intervention is warranted after free bodies are found present, because they limit motion and cause recalcitrant swelling.2,8 There is a controversy whether arthroscopic removal of loose bodies or excision with synovectomy is the treatment of choice.6 Ogilvie-Harris and colleagues reviewed the results of both procedures and found that although removal of loose bodies alone may be sufficient, there is the potential for recurrence.9,10 In order to reduce potential recurrence, removal of loose bodies with anterior and posterior synovectomy is the treatment of choice.9
If arthroscopic removal of loose bodies without synovectomy is performed, then the patient should be followed closely for recurrence, which Jesalpura and colleagues reported to occur for 11.5% of patients.9,11 If there is a reappearance, then a synovectomy should be performed.10 A recommended treatment option for recalcitrant SC is radiation, but this carries the added risk of perpetuating malignant transformation.1,7
Unfortunately, osteoarthritis can be a significant long-term postoperative adverse effect.3,6-8 This typically is related to the amount of articular damage that is present at surgery. Many times, the arthritis becomes significant enough to require total joint arthroplasty.4 Close long-term follow-up is recommended, because although rare, there is a chance of malignant change.1-4
Conclusion
Synovial chondromatosis is an uncommon cause of knee pain and swelling and should be included in the differential diagnosis when evaluating any adult aged 30 years to 50 years with knee pain of insidious onset. Appropriate workup, intervention, and treatment will allow final diagnosis and correlating care to be administered to the patient.
1. Libbey NP, Mirrer F. Synovial chondromatosis. Med Health R I. 2011;94(9):274-275.
2. Giancane G, Tanturri de Horatio L, Buonuomo PS, Barbuti D, Lais G, Cortis E. Swollen knee due to primary synovial chondromatosis in pediatrics: a rare and possibly misdiagnosed condition. Rheumatol Int. 2013;33(8):2183-2185.
3. Serbest S, Tiftikçi U, Karaaslan F, Tosun HB, Sevinç HF, Balci M. A neglected case of giant synovial chondromatosis in knee joint. Pan Afr Med J. 2015;22:5.
4. Hallam P, Ashwood N, Cobb J, Fazal A, Heatley W. Malignant transformation in synovial chondromatosis of the knee? Knee. 2001;8(3):239-242.
5. Pimentel Cde Q, Hoff LS, de Sousa LF, Cordeiro RA, Pereira RM. Primary synovial osteochondromatosis of the knee. Rheumatol (Oxford). 2015;54(10):1815.
6. Damron TA, Sim FH. Soft-tissue tumors about the knee. J Am Acad Orthop Surg. 1997;5(3):141-152.
7. Krych A, Odland A, Rose P, et al. Onconlogic conditions that simulate common sports injuries. J Am Acad Orthop Surg. 2014;22(4):223-234.
8. Adelani MA, Wupperman RM, Holt GE. Benign synovial disorders. J Am Acad Orthop Surg. 2008;16(5):268-275.
9. Migram JW. Synovial osteochondromatosis: a histopathological study of thirty cases. J Bone Joint Surg Am. 1977;59(6):792-801.
10. Ogilvie-Harris DJ, Saleh K. Generalized synovial chondromatosis of the knee: a comparison of removal of the loose bodies alone with arthroscopic synovectomy. Arthroscopy.1994;10(2):166-170.
11. Jesalpura JP, Chung HW, Patnaik S, Choi HW, Kim JI, Nha KW. Athroscopic treatment of localized synovial chondromatosis of the posterior knee joint. Orthopedics. 2010;33(1):49
1. Libbey NP, Mirrer F. Synovial chondromatosis. Med Health R I. 2011;94(9):274-275.
2. Giancane G, Tanturri de Horatio L, Buonuomo PS, Barbuti D, Lais G, Cortis E. Swollen knee due to primary synovial chondromatosis in pediatrics: a rare and possibly misdiagnosed condition. Rheumatol Int. 2013;33(8):2183-2185.
3. Serbest S, Tiftikçi U, Karaaslan F, Tosun HB, Sevinç HF, Balci M. A neglected case of giant synovial chondromatosis in knee joint. Pan Afr Med J. 2015;22:5.
4. Hallam P, Ashwood N, Cobb J, Fazal A, Heatley W. Malignant transformation in synovial chondromatosis of the knee? Knee. 2001;8(3):239-242.
5. Pimentel Cde Q, Hoff LS, de Sousa LF, Cordeiro RA, Pereira RM. Primary synovial osteochondromatosis of the knee. Rheumatol (Oxford). 2015;54(10):1815.
6. Damron TA, Sim FH. Soft-tissue tumors about the knee. J Am Acad Orthop Surg. 1997;5(3):141-152.
7. Krych A, Odland A, Rose P, et al. Onconlogic conditions that simulate common sports injuries. J Am Acad Orthop Surg. 2014;22(4):223-234.
8. Adelani MA, Wupperman RM, Holt GE. Benign synovial disorders. J Am Acad Orthop Surg. 2008;16(5):268-275.
9. Migram JW. Synovial osteochondromatosis: a histopathological study of thirty cases. J Bone Joint Surg Am. 1977;59(6):792-801.
10. Ogilvie-Harris DJ, Saleh K. Generalized synovial chondromatosis of the knee: a comparison of removal of the loose bodies alone with arthroscopic synovectomy. Arthroscopy.1994;10(2):166-170.
11. Jesalpura JP, Chung HW, Patnaik S, Choi HW, Kim JI, Nha KW. Athroscopic treatment of localized synovial chondromatosis of the posterior knee joint. Orthopedics. 2010;33(1):49
Acute Exertional Upper-Extremity Rhabdomyolysis in 3 Female Trainees
Acute exertional rhabdomyolysis (AER) is the breakdown/destruction of muscle tissue from extreme physical exertion. Risks that lead to AER include exercise in hot and humid conditions, improper hydration, inadequate recovery between bouts of exercise, intense physical training, and inadequate fitness levels. Other risk factors include sickle cell trait, ingestion of performance enhancing agents, anabolic steroids, and previous history of AER. This article, describes 3 cases of AER after a vigorous, upper body, organized, and supervised training session.
Rhabdomyolysis is not uncommon in competitive athletics,1-3 military training,4-8 and individual training.9-12 It is more common in the lower extremities after intense training or marathons. Creatine kinase (CK) levels rise within 12 hours of muscle injury, peak in 24 to 36 hours, and decrease at a rate of 30% to 40% per day.13 The serum half-life of CK is about 36 hours. The CK levels decline 3 to 5 days after resolution of muscle injury.14 Failure of CK levels to decrease suggests ongoing muscle injury or development of a compartment syndrome. The peak CK level, especially when it is > 15,000 U/L, may be predictive of renal failure.15
Total CK elevation is a sensitive but nonspecific marker for rhabdomyolysis. A CK level that is 1.5 × above the reference range suggests rhabdomyolysis, although CK levels in rhabdomyolysis often are as high as 100 times the reference range or more.12 Health care providers (HCPs) should suspect early rhabdomyolysis and initiate a full laboratory workup for patients with serum CK levels > 2 × the reference range and risk factors for rhabdomyolysis. Because the total CK may increase from the initial values, draw it is important to repeat total CK levels every 6 to 12 hours until a peak level is established.
Case Presentations
An upper body physical training session was conducted with a group of trainees (men and women, aged 24-35 years) 12 weeks into a 21-week program. The training session consisted of 125 push-ups and 85 pull-ups (assisted) performed within a 12-minute period in an indoor, climate-controlled facility. Liberal hydration with water or sports drinks was not allowed. On day 3 following the training session, 1 woman, and on day 4, 2 additional women presented to the clinic with extreme bilateral upper extremity muscle weakness, pain, and marked swelling of their upper-extremities from the shoulder to the forearm. None had firm compartments of the arm or forearm. Further, none had signs of compartment syndrome. There were trace blood and protein in their urine, and their serum CK ranged from 10,000 to 78,000 U/L.
Case 1
The first case to present to the clinic was a 26-year-old white female without any underlying disease. She was taking oral contraceptive medication. She usually exercised by running 10 to 12 miles/week with upper body workouts as required by the training program and was in excellent physical health at the halfway point in the training program.
Following the workout of 125 push-ups and 85 pull-ups, the patient experienced muscle soreness and fatigue and took ibuprofen 800 mg that evening. Over the next 2 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 3 after the workout, she presented to the clinic in moderate distress, unable to raise her arms above chest level. Her urine showed trace blood and protein, and her serum CK level was 73,044 U/L (Table 1). 
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 8 days, and had no sequelae.
Case 2
The second case involved a 27-year-old white female without any underlying disease who was not taking any medications and was in excellent physical health. Following the workout of 125 push-ups and 85 pull-ups, she experienced muscle soreness and fatigue. Over the next 3 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 4 postworkout, she presented to the clinic in moderate distress. Her urine showed trace blood and protein, and her serum CK level was > 8,000 U/L (the hospital stopped dilutions when the CK level exceeded 8,000 U/L) (Table 2). 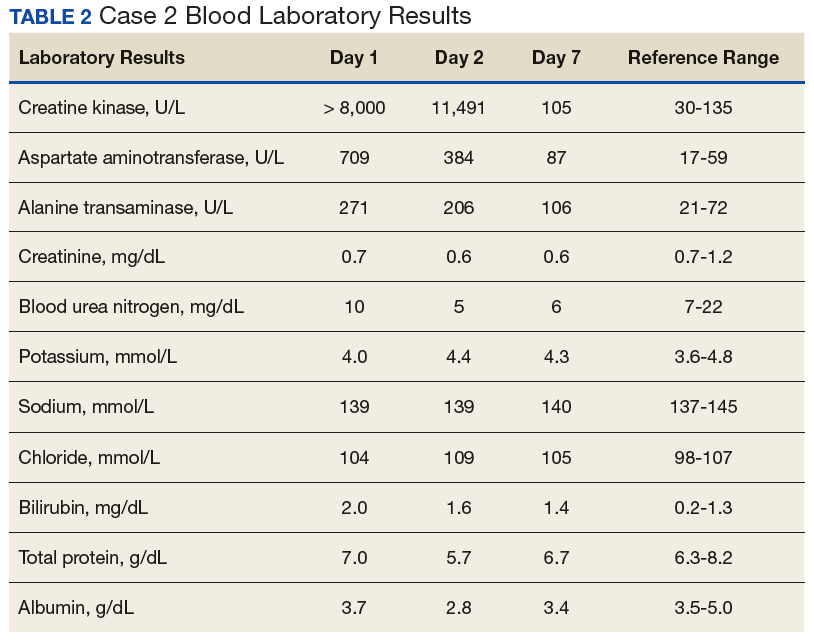
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 7 days, and had no sequelae.
Case 3
The third case involved a 33-year-old white female without any underlying disease who was taking oral contraceptive medication and was in excellent physical health. Following the 125 push-ups and 85 pull-ups workout, she experienced muscle soreness and fatigue. Over the next 3 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 4 postworkout, she presented to the clinic in moderate distress. Her urine showed trace blood and protein, and her serum CK level was 10,971 U/L (Table 3). 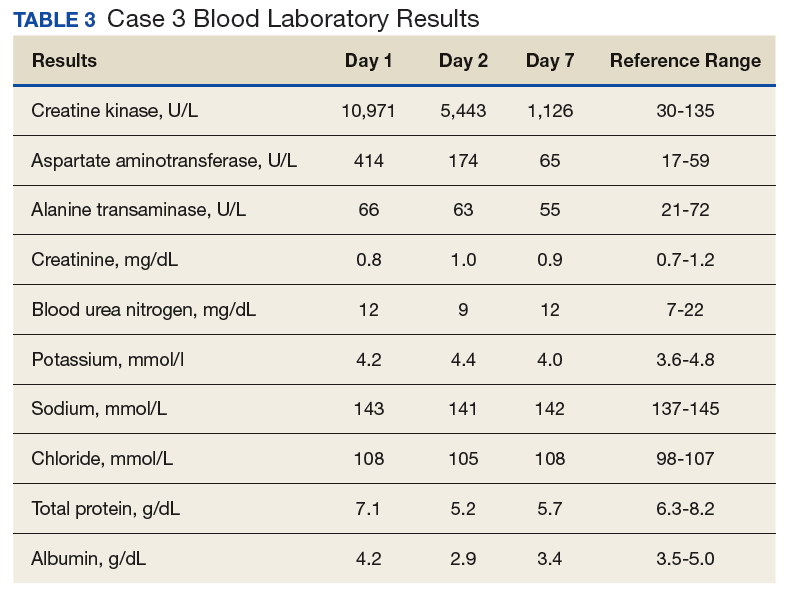
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 7 days, and had no sequelae.
Clinical Observation
The importance of brown urine is stressed as a key factor in the diagnosis of AER in much of the literature on the condition.1-10 It is suggested that brown urine is pathognomonic for this condition. However, the urine for the individuals presented here with significant AER was not brown or even tinged. In a field evaluation, an inexperienced HCP might miss an AER diagnosis in the absence of urine findings. In addition, given that AER can occur in stages, it is essential that the HCP have a situational awareness of this condition in today’s culture of fitness and exercise.
Other Causes
Cocaine is a common cause of rhabdomyolysis, namely, in urban patient populations. Prolonged vasoconstriction of intramuscular arteries may result in muscle ischemia and acute rhabdomyolysis, but there also is a direct toxic effect that can produce acute skeletal myofibrillar degeneration.
A number of prescription drugs have been implicated in cases of rhabdomyolysis, including colchicine, zidovudine, isoniazid, benzodiazepines, opiates, corticosteroids, statins, and fibric acid derivatives. One particular interaction that is clinically significant is the interaction between statins and fibrates.16
The pathogenesis of rhabdomyolysis precipitated by infections (whether bacterial, viral, or fungal) is thought to be the result of direct cell invasion of striated muscle and cellular degeneration by the pathogen. Substantial morbidity (57% of cases with acute renal failure) and mortality (death in almost 40% of cases) are linked to bacterial causes of rhabdomyolysis.
In adult patients, Legionella species most often are associated with rhabdomyolysis. Other bacteria linked to rhabdomyolysis include group A β-hemolytic streptococci, Salmonella species, Francisella tularensis and Escherichia coli. Viruses, such as influenza, parainfluenza, coxsackievirus, Epstein-Barr virus, adenovirus, HIV, and cytomegalovirus also have been associated with this condition.
Rhabdomyolysis also can be observed in septic patients without direct muscle infection when the damage is caused by a toxin, associated fever, dehydration, and rigors. Electrolyte disorders, such as hyponatremia or hypernatremia, hypokalemia, and hypophosphatemia can result in rhabdomyolysis, distorting the permeability and the functions of sarcolemma in the muscles. Some endocrine disorders (ie, pheochromocytoma and thyrotoxicosis) also are able to potentiate rhabdomyolysis due to hypermetabolism.
Prevention
A workout program should progress gradually according to the individual’s current level of fitness, whether it’s cardiovascular, circuit training, or weight training. Fluid intake should be monitored, particularly when the workout is long, intense, or hot and especially when the workout meets all 3 conditions. Fluid replacement is important, and for strenuous and longer training evolutions, electrolyte replacement should be considered. Hard exercise while maintaining a low-calorie diet or after long fasting periods should be avoided. Sufficient caloric and fluid intake to allow muscles to work efficiently during strenuous workout period is needed. Recreational drugs, including alcohol should be limited before exercise, and illicit recreational or performance-enhancing drugs should be avoided.
Conclusion
Acute exertional rhabdomyolysis is more common in the lower extremities and in males. Extremely rigorous upper-extremity training can result in AER. The presentation usually is clear with an inciting event and muscle pain in the extremity. Besides identifying associated risk factors and performing a thorough examination, the patient should be examined for compartment syndrome. Early diagnosis and comprehensive management are crucial to ensure full recovery and avoidance of complications, such as acute tubular necrosis, renal failure, cardiac arrhythmias from hyperkalemia, and death. As in most cases with early diagnosis and aggressive management, these patients fully recovered and experienced no sequelae at 8 weeks postevent.
1. Galvez R, Stacy J, Howley A. Exertional rhabdomyolysis in seven division-1 swimming athletes. Clin J Sport Med. 2008;18(4):366-368.
2. Smoot MK, Amendola A, Cramer E, et al. A cluster of exertional rhabdomyolysis affecting a division 1 football team. Clin J Sport Med. 2013;23(5):365-372.
3. Kahanov L, Eberman LE, Wasik M, Alvey T. Exertional rhabdomyolysis in a collegiate American football player after preventive cold-water immersion: a case report. J Athl Train. 2012;47(2):228-232.
4. Aizawa H, Morita K, Minami H, Sasaki N, Tobise K. Exertional rhabdomyolysis as a result of strenuous military training. J Neurol Sci. 1995;132(2):239-240.
5. Tietjen DP, Guzzi LM. Exertional rhabdomyolysis and acute renal failure following the Army Physical Fitness Test. Mil Med. 154(1):23-25.
6. Gardner JW, Kark JA. Fatal rhabdomyolysis presenting as mild heat illness in military training. Mil Med. 159(2):160-163.
7. Armed Forces Health Surveillance Center (AFHSC). Exertional rhabdomyolysis among U.S. military members, 2004-2007. MSMR. 2008;15(2):8-11.
8. Gitin EL, Demos MA. Acute exertional rhabdomyolysis: a syndrome of increasing importance to the military physician. Mil Med.1974;139(1):33-36.
9. Springer BL, Clarkson PM. Two cases of exertional rhabdomyolysis precipitated by personal trainers. Med Sci Sports Exerc. 2003;35(9):1499-1502.
10. Lin A, Lin C, Wang T, Leu J. Rhabdomyolysis in 119 students after repetitive exercise. Br J Sports Med. 2005;39(1):e3.
11. Hamer R. When exercise goes awry: exertional rhabdomyolysis. South Med J. 1997;90(5):548-551.
12. Soni SN, McDonald E, Marino C. Rhabdomyolysis after exercise. Postgrad Med. 1993;94(6):128-132.
13. Lappalainen H, Tiula E, Uotila L, Mänttäri M. Elimination kinetics of myoglobin and creatine kinase in rhabdomyolysis: implications for follow-up. Crit Care Med. 2002;30(10):2212-2215.
14. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
15. Minnema BJ, Neligan PC, Quraishi NA, Fehlings MG, Prakash S. A case of occult compartment syndrome and nonresolving rhabdomyolysis. J Gen Intern Med. 2008;23(6):871-874.
16. Efstratiadis G, Voulgaridou A, Nikiforou D, Kyventidis A, Kourkouni E, Vergoulas G. Rhabdomyolysis updated. Hippokratia. 2007;11(3):129-137.
Acute exertional rhabdomyolysis (AER) is the breakdown/destruction of muscle tissue from extreme physical exertion. Risks that lead to AER include exercise in hot and humid conditions, improper hydration, inadequate recovery between bouts of exercise, intense physical training, and inadequate fitness levels. Other risk factors include sickle cell trait, ingestion of performance enhancing agents, anabolic steroids, and previous history of AER. This article, describes 3 cases of AER after a vigorous, upper body, organized, and supervised training session.
Rhabdomyolysis is not uncommon in competitive athletics,1-3 military training,4-8 and individual training.9-12 It is more common in the lower extremities after intense training or marathons. Creatine kinase (CK) levels rise within 12 hours of muscle injury, peak in 24 to 36 hours, and decrease at a rate of 30% to 40% per day.13 The serum half-life of CK is about 36 hours. The CK levels decline 3 to 5 days after resolution of muscle injury.14 Failure of CK levels to decrease suggests ongoing muscle injury or development of a compartment syndrome. The peak CK level, especially when it is > 15,000 U/L, may be predictive of renal failure.15
Total CK elevation is a sensitive but nonspecific marker for rhabdomyolysis. A CK level that is 1.5 × above the reference range suggests rhabdomyolysis, although CK levels in rhabdomyolysis often are as high as 100 times the reference range or more.12 Health care providers (HCPs) should suspect early rhabdomyolysis and initiate a full laboratory workup for patients with serum CK levels > 2 × the reference range and risk factors for rhabdomyolysis. Because the total CK may increase from the initial values, draw it is important to repeat total CK levels every 6 to 12 hours until a peak level is established.
Case Presentations
An upper body physical training session was conducted with a group of trainees (men and women, aged 24-35 years) 12 weeks into a 21-week program. The training session consisted of 125 push-ups and 85 pull-ups (assisted) performed within a 12-minute period in an indoor, climate-controlled facility. Liberal hydration with water or sports drinks was not allowed. On day 3 following the training session, 1 woman, and on day 4, 2 additional women presented to the clinic with extreme bilateral upper extremity muscle weakness, pain, and marked swelling of their upper-extremities from the shoulder to the forearm. None had firm compartments of the arm or forearm. Further, none had signs of compartment syndrome. There were trace blood and protein in their urine, and their serum CK ranged from 10,000 to 78,000 U/L.
Case 1
The first case to present to the clinic was a 26-year-old white female without any underlying disease. She was taking oral contraceptive medication. She usually exercised by running 10 to 12 miles/week with upper body workouts as required by the training program and was in excellent physical health at the halfway point in the training program.
Following the workout of 125 push-ups and 85 pull-ups, the patient experienced muscle soreness and fatigue and took ibuprofen 800 mg that evening. Over the next 2 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 3 after the workout, she presented to the clinic in moderate distress, unable to raise her arms above chest level. Her urine showed trace blood and protein, and her serum CK level was 73,044 U/L (Table 1).
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 8 days, and had no sequelae.
Case 2
The second case involved a 27-year-old white female without any underlying disease who was not taking any medications and was in excellent physical health. Following the workout of 125 push-ups and 85 pull-ups, she experienced muscle soreness and fatigue. Over the next 3 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 4 postworkout, she presented to the clinic in moderate distress. Her urine showed trace blood and protein, and her serum CK level was > 8,000 U/L (the hospital stopped dilutions when the CK level exceeded 8,000 U/L) (Table 2).
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 7 days, and had no sequelae.
Case 3
The third case involved a 33-year-old white female without any underlying disease who was taking oral contraceptive medication and was in excellent physical health. Following the 125 push-ups and 85 pull-ups workout, she experienced muscle soreness and fatigue. Over the next 3 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 4 postworkout, she presented to the clinic in moderate distress. Her urine showed trace blood and protein, and her serum CK level was 10,971 U/L (Table 3).
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 7 days, and had no sequelae.
Clinical Observation
The importance of brown urine is stressed as a key factor in the diagnosis of AER in much of the literature on the condition.1-10 It is suggested that brown urine is pathognomonic for this condition. However, the urine for the individuals presented here with significant AER was not brown or even tinged. In a field evaluation, an inexperienced HCP might miss an AER diagnosis in the absence of urine findings. In addition, given that AER can occur in stages, it is essential that the HCP have a situational awareness of this condition in today’s culture of fitness and exercise.
Other Causes
Cocaine is a common cause of rhabdomyolysis, namely, in urban patient populations. Prolonged vasoconstriction of intramuscular arteries may result in muscle ischemia and acute rhabdomyolysis, but there also is a direct toxic effect that can produce acute skeletal myofibrillar degeneration.
A number of prescription drugs have been implicated in cases of rhabdomyolysis, including colchicine, zidovudine, isoniazid, benzodiazepines, opiates, corticosteroids, statins, and fibric acid derivatives. One particular interaction that is clinically significant is the interaction between statins and fibrates.16
The pathogenesis of rhabdomyolysis precipitated by infections (whether bacterial, viral, or fungal) is thought to be the result of direct cell invasion of striated muscle and cellular degeneration by the pathogen. Substantial morbidity (57% of cases with acute renal failure) and mortality (death in almost 40% of cases) are linked to bacterial causes of rhabdomyolysis.
In adult patients, Legionella species most often are associated with rhabdomyolysis. Other bacteria linked to rhabdomyolysis include group A β-hemolytic streptococci, Salmonella species, Francisella tularensis and Escherichia coli. Viruses, such as influenza, parainfluenza, coxsackievirus, Epstein-Barr virus, adenovirus, HIV, and cytomegalovirus also have been associated with this condition.
Rhabdomyolysis also can be observed in septic patients without direct muscle infection when the damage is caused by a toxin, associated fever, dehydration, and rigors. Electrolyte disorders, such as hyponatremia or hypernatremia, hypokalemia, and hypophosphatemia can result in rhabdomyolysis, distorting the permeability and the functions of sarcolemma in the muscles. Some endocrine disorders (ie, pheochromocytoma and thyrotoxicosis) also are able to potentiate rhabdomyolysis due to hypermetabolism.
Prevention
A workout program should progress gradually according to the individual’s current level of fitness, whether it’s cardiovascular, circuit training, or weight training. Fluid intake should be monitored, particularly when the workout is long, intense, or hot and especially when the workout meets all 3 conditions. Fluid replacement is important, and for strenuous and longer training evolutions, electrolyte replacement should be considered. Hard exercise while maintaining a low-calorie diet or after long fasting periods should be avoided. Sufficient caloric and fluid intake to allow muscles to work efficiently during strenuous workout period is needed. Recreational drugs, including alcohol should be limited before exercise, and illicit recreational or performance-enhancing drugs should be avoided.
Conclusion
Acute exertional rhabdomyolysis is more common in the lower extremities and in males. Extremely rigorous upper-extremity training can result in AER. The presentation usually is clear with an inciting event and muscle pain in the extremity. Besides identifying associated risk factors and performing a thorough examination, the patient should be examined for compartment syndrome. Early diagnosis and comprehensive management are crucial to ensure full recovery and avoidance of complications, such as acute tubular necrosis, renal failure, cardiac arrhythmias from hyperkalemia, and death. As in most cases with early diagnosis and aggressive management, these patients fully recovered and experienced no sequelae at 8 weeks postevent.
Acute exertional rhabdomyolysis (AER) is the breakdown/destruction of muscle tissue from extreme physical exertion. Risks that lead to AER include exercise in hot and humid conditions, improper hydration, inadequate recovery between bouts of exercise, intense physical training, and inadequate fitness levels. Other risk factors include sickle cell trait, ingestion of performance enhancing agents, anabolic steroids, and previous history of AER. This article, describes 3 cases of AER after a vigorous, upper body, organized, and supervised training session.
Rhabdomyolysis is not uncommon in competitive athletics,1-3 military training,4-8 and individual training.9-12 It is more common in the lower extremities after intense training or marathons. Creatine kinase (CK) levels rise within 12 hours of muscle injury, peak in 24 to 36 hours, and decrease at a rate of 30% to 40% per day.13 The serum half-life of CK is about 36 hours. The CK levels decline 3 to 5 days after resolution of muscle injury.14 Failure of CK levels to decrease suggests ongoing muscle injury or development of a compartment syndrome. The peak CK level, especially when it is > 15,000 U/L, may be predictive of renal failure.15
Total CK elevation is a sensitive but nonspecific marker for rhabdomyolysis. A CK level that is 1.5 × above the reference range suggests rhabdomyolysis, although CK levels in rhabdomyolysis often are as high as 100 times the reference range or more.12 Health care providers (HCPs) should suspect early rhabdomyolysis and initiate a full laboratory workup for patients with serum CK levels > 2 × the reference range and risk factors for rhabdomyolysis. Because the total CK may increase from the initial values, draw it is important to repeat total CK levels every 6 to 12 hours until a peak level is established.
Case Presentations
An upper body physical training session was conducted with a group of trainees (men and women, aged 24-35 years) 12 weeks into a 21-week program. The training session consisted of 125 push-ups and 85 pull-ups (assisted) performed within a 12-minute period in an indoor, climate-controlled facility. Liberal hydration with water or sports drinks was not allowed. On day 3 following the training session, 1 woman, and on day 4, 2 additional women presented to the clinic with extreme bilateral upper extremity muscle weakness, pain, and marked swelling of their upper-extremities from the shoulder to the forearm. None had firm compartments of the arm or forearm. Further, none had signs of compartment syndrome. There were trace blood and protein in their urine, and their serum CK ranged from 10,000 to 78,000 U/L.
Case 1
The first case to present to the clinic was a 26-year-old white female without any underlying disease. She was taking oral contraceptive medication. She usually exercised by running 10 to 12 miles/week with upper body workouts as required by the training program and was in excellent physical health at the halfway point in the training program.
Following the workout of 125 push-ups and 85 pull-ups, the patient experienced muscle soreness and fatigue and took ibuprofen 800 mg that evening. Over the next 2 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 3 after the workout, she presented to the clinic in moderate distress, unable to raise her arms above chest level. Her urine showed trace blood and protein, and her serum CK level was 73,044 U/L (Table 1).
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 8 days, and had no sequelae.
Case 2
The second case involved a 27-year-old white female without any underlying disease who was not taking any medications and was in excellent physical health. Following the workout of 125 push-ups and 85 pull-ups, she experienced muscle soreness and fatigue. Over the next 3 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 4 postworkout, she presented to the clinic in moderate distress. Her urine showed trace blood and protein, and her serum CK level was > 8,000 U/L (the hospital stopped dilutions when the CK level exceeded 8,000 U/L) (Table 2).
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 7 days, and had no sequelae.
Case 3
The third case involved a 33-year-old white female without any underlying disease who was taking oral contraceptive medication and was in excellent physical health. Following the 125 push-ups and 85 pull-ups workout, she experienced muscle soreness and fatigue. Over the next 3 days, she experienced continued muscle soreness/pain, increasing weakness, and marked swelling of her arms. On day 4 postworkout, she presented to the clinic in moderate distress. Her urine showed trace blood and protein, and her serum CK level was 10,971 U/L (Table 3).
The patient was diagnosed with rhabdomyolysis and admitted to the hospital. She received IV therapy, improved, and was discharged after 7 days, and had no sequelae.
Clinical Observation
The importance of brown urine is stressed as a key factor in the diagnosis of AER in much of the literature on the condition.1-10 It is suggested that brown urine is pathognomonic for this condition. However, the urine for the individuals presented here with significant AER was not brown or even tinged. In a field evaluation, an inexperienced HCP might miss an AER diagnosis in the absence of urine findings. In addition, given that AER can occur in stages, it is essential that the HCP have a situational awareness of this condition in today’s culture of fitness and exercise.
Other Causes
Cocaine is a common cause of rhabdomyolysis, namely, in urban patient populations. Prolonged vasoconstriction of intramuscular arteries may result in muscle ischemia and acute rhabdomyolysis, but there also is a direct toxic effect that can produce acute skeletal myofibrillar degeneration.
A number of prescription drugs have been implicated in cases of rhabdomyolysis, including colchicine, zidovudine, isoniazid, benzodiazepines, opiates, corticosteroids, statins, and fibric acid derivatives. One particular interaction that is clinically significant is the interaction between statins and fibrates.16
The pathogenesis of rhabdomyolysis precipitated by infections (whether bacterial, viral, or fungal) is thought to be the result of direct cell invasion of striated muscle and cellular degeneration by the pathogen. Substantial morbidity (57% of cases with acute renal failure) and mortality (death in almost 40% of cases) are linked to bacterial causes of rhabdomyolysis.
In adult patients, Legionella species most often are associated with rhabdomyolysis. Other bacteria linked to rhabdomyolysis include group A β-hemolytic streptococci, Salmonella species, Francisella tularensis and Escherichia coli. Viruses, such as influenza, parainfluenza, coxsackievirus, Epstein-Barr virus, adenovirus, HIV, and cytomegalovirus also have been associated with this condition.
Rhabdomyolysis also can be observed in septic patients without direct muscle infection when the damage is caused by a toxin, associated fever, dehydration, and rigors. Electrolyte disorders, such as hyponatremia or hypernatremia, hypokalemia, and hypophosphatemia can result in rhabdomyolysis, distorting the permeability and the functions of sarcolemma in the muscles. Some endocrine disorders (ie, pheochromocytoma and thyrotoxicosis) also are able to potentiate rhabdomyolysis due to hypermetabolism.
Prevention
A workout program should progress gradually according to the individual’s current level of fitness, whether it’s cardiovascular, circuit training, or weight training. Fluid intake should be monitored, particularly when the workout is long, intense, or hot and especially when the workout meets all 3 conditions. Fluid replacement is important, and for strenuous and longer training evolutions, electrolyte replacement should be considered. Hard exercise while maintaining a low-calorie diet or after long fasting periods should be avoided. Sufficient caloric and fluid intake to allow muscles to work efficiently during strenuous workout period is needed. Recreational drugs, including alcohol should be limited before exercise, and illicit recreational or performance-enhancing drugs should be avoided.
Conclusion
Acute exertional rhabdomyolysis is more common in the lower extremities and in males. Extremely rigorous upper-extremity training can result in AER. The presentation usually is clear with an inciting event and muscle pain in the extremity. Besides identifying associated risk factors and performing a thorough examination, the patient should be examined for compartment syndrome. Early diagnosis and comprehensive management are crucial to ensure full recovery and avoidance of complications, such as acute tubular necrosis, renal failure, cardiac arrhythmias from hyperkalemia, and death. As in most cases with early diagnosis and aggressive management, these patients fully recovered and experienced no sequelae at 8 weeks postevent.
1. Galvez R, Stacy J, Howley A. Exertional rhabdomyolysis in seven division-1 swimming athletes. Clin J Sport Med. 2008;18(4):366-368.
2. Smoot MK, Amendola A, Cramer E, et al. A cluster of exertional rhabdomyolysis affecting a division 1 football team. Clin J Sport Med. 2013;23(5):365-372.
3. Kahanov L, Eberman LE, Wasik M, Alvey T. Exertional rhabdomyolysis in a collegiate American football player after preventive cold-water immersion: a case report. J Athl Train. 2012;47(2):228-232.
4. Aizawa H, Morita K, Minami H, Sasaki N, Tobise K. Exertional rhabdomyolysis as a result of strenuous military training. J Neurol Sci. 1995;132(2):239-240.
5. Tietjen DP, Guzzi LM. Exertional rhabdomyolysis and acute renal failure following the Army Physical Fitness Test. Mil Med. 154(1):23-25.
6. Gardner JW, Kark JA. Fatal rhabdomyolysis presenting as mild heat illness in military training. Mil Med. 159(2):160-163.
7. Armed Forces Health Surveillance Center (AFHSC). Exertional rhabdomyolysis among U.S. military members, 2004-2007. MSMR. 2008;15(2):8-11.
8. Gitin EL, Demos MA. Acute exertional rhabdomyolysis: a syndrome of increasing importance to the military physician. Mil Med.1974;139(1):33-36.
9. Springer BL, Clarkson PM. Two cases of exertional rhabdomyolysis precipitated by personal trainers. Med Sci Sports Exerc. 2003;35(9):1499-1502.
10. Lin A, Lin C, Wang T, Leu J. Rhabdomyolysis in 119 students after repetitive exercise. Br J Sports Med. 2005;39(1):e3.
11. Hamer R. When exercise goes awry: exertional rhabdomyolysis. South Med J. 1997;90(5):548-551.
12. Soni SN, McDonald E, Marino C. Rhabdomyolysis after exercise. Postgrad Med. 1993;94(6):128-132.
13. Lappalainen H, Tiula E, Uotila L, Mänttäri M. Elimination kinetics of myoglobin and creatine kinase in rhabdomyolysis: implications for follow-up. Crit Care Med. 2002;30(10):2212-2215.
14. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
15. Minnema BJ, Neligan PC, Quraishi NA, Fehlings MG, Prakash S. A case of occult compartment syndrome and nonresolving rhabdomyolysis. J Gen Intern Med. 2008;23(6):871-874.
16. Efstratiadis G, Voulgaridou A, Nikiforou D, Kyventidis A, Kourkouni E, Vergoulas G. Rhabdomyolysis updated. Hippokratia. 2007;11(3):129-137.
1. Galvez R, Stacy J, Howley A. Exertional rhabdomyolysis in seven division-1 swimming athletes. Clin J Sport Med. 2008;18(4):366-368.
2. Smoot MK, Amendola A, Cramer E, et al. A cluster of exertional rhabdomyolysis affecting a division 1 football team. Clin J Sport Med. 2013;23(5):365-372.
3. Kahanov L, Eberman LE, Wasik M, Alvey T. Exertional rhabdomyolysis in a collegiate American football player after preventive cold-water immersion: a case report. J Athl Train. 2012;47(2):228-232.
4. Aizawa H, Morita K, Minami H, Sasaki N, Tobise K. Exertional rhabdomyolysis as a result of strenuous military training. J Neurol Sci. 1995;132(2):239-240.
5. Tietjen DP, Guzzi LM. Exertional rhabdomyolysis and acute renal failure following the Army Physical Fitness Test. Mil Med. 154(1):23-25.
6. Gardner JW, Kark JA. Fatal rhabdomyolysis presenting as mild heat illness in military training. Mil Med. 159(2):160-163.
7. Armed Forces Health Surveillance Center (AFHSC). Exertional rhabdomyolysis among U.S. military members, 2004-2007. MSMR. 2008;15(2):8-11.
8. Gitin EL, Demos MA. Acute exertional rhabdomyolysis: a syndrome of increasing importance to the military physician. Mil Med.1974;139(1):33-36.
9. Springer BL, Clarkson PM. Two cases of exertional rhabdomyolysis precipitated by personal trainers. Med Sci Sports Exerc. 2003;35(9):1499-1502.
10. Lin A, Lin C, Wang T, Leu J. Rhabdomyolysis in 119 students after repetitive exercise. Br J Sports Med. 2005;39(1):e3.
11. Hamer R. When exercise goes awry: exertional rhabdomyolysis. South Med J. 1997;90(5):548-551.
12. Soni SN, McDonald E, Marino C. Rhabdomyolysis after exercise. Postgrad Med. 1993;94(6):128-132.
13. Lappalainen H, Tiula E, Uotila L, Mänttäri M. Elimination kinetics of myoglobin and creatine kinase in rhabdomyolysis: implications for follow-up. Crit Care Med. 2002;30(10):2212-2215.
14. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
15. Minnema BJ, Neligan PC, Quraishi NA, Fehlings MG, Prakash S. A case of occult compartment syndrome and nonresolving rhabdomyolysis. J Gen Intern Med. 2008;23(6):871-874.
16. Efstratiadis G, Voulgaridou A, Nikiforou D, Kyventidis A, Kourkouni E, Vergoulas G. Rhabdomyolysis updated. Hippokratia. 2007;11(3):129-137.