User login
Use of a Core Reamer for the Resection of a Central Distal Femoral Physeal Bone Bridge: A Novel Technique with 3-Year Follow-up
ABSTRACT
A central distal femoral physeal bone bridge in a boy aged 5 years and 7 months was resected with a fluoroscopically guided core reamer placed through a lateral parapatellar approach. At 3-year follow-up, the boy’s leg-length discrepancy was 3.0 cm (3.9 cm preoperatively), and the physeal bone bridge did not recur. The patient had full function and no pain or other patellofemoral complaints. This technique provided direct access to the physeal bone bridge, and complete resection was performed without injury to the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which is expected to grow normally in the absence of the bridge.
A physeal bone bridge is an osseous connection that forms across a physis. It may cause partial premature physeal arrest. Angular deformity and limb-length discrepancy are the main complications caused by physeal bone bridges.1-4 The indications for the treatment of physeal bridges are well documented.1-5 Trauma and infection are common causes of distal femoral physeal bone bridges. Arkader and colleagues6 showed that among different types of physeal bridges, the Salter-Harris type is significantly associated with complications, among which growth arrest is the most common and occurs in 27.4% of all patients.
The treatment of distal femoral physeal bone bridges is technically difficult and provides variable results. Poor results are reported in 13% to 40% of patients.7-10 Procedure failure has been attributed to incomplete resection with the persistent tethering and dislodgement of the graft.11 Methods with improved efficacy for the removal of central physeal bridges will help prevent reformation after treatment. We have used a novel technique that allows the direct resection of a central physeal bone bridge in the distal femur through the use of a fluoroscopically guided core reamer. This technique enables the complete removal of the bone bridge and the direct visual assessment of the remaining physis. The patient’s parents provided written informed consent for print and electronic publication of this case report.
CASE
A 3-year-old boy with a history of hemifacial microsomia presented for the evaluation of genu valgum and leg-length discrepancy. His intermalleolar distance at that time was 8 cm. A standing radiograph of his lower extremities demonstrated changes consistent with physiologic genu valgum. He had no history of knee trauma, infection, or pain.
At the age of 5 years and 7 months, the patient returned for a repeat evaluation and was noted to exhibit the progressive valgus deformity of the right leg and a leg-length discrepancy of 3.9 cm (Figure 1). 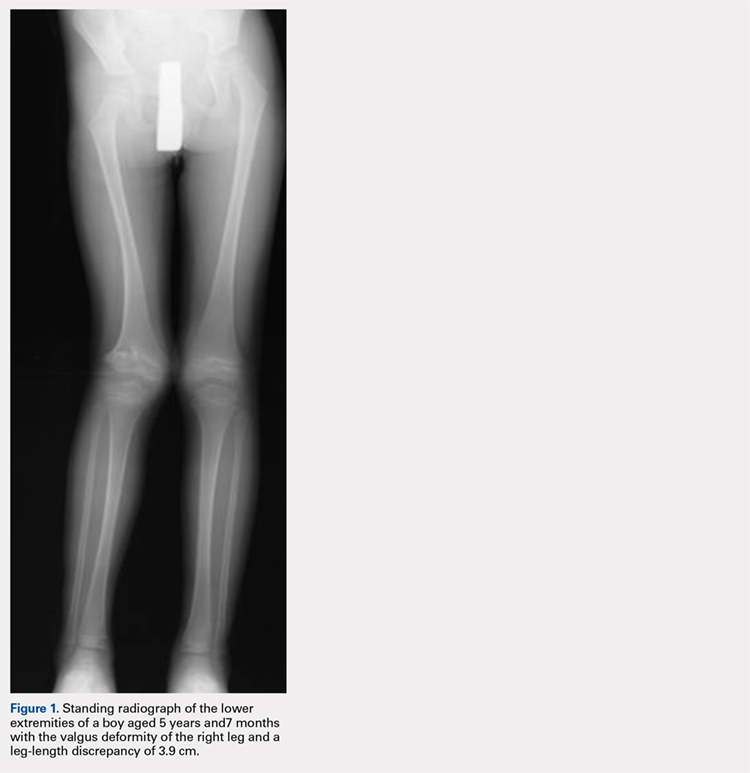
Continue to: With the patient supine on the operating...
OPERATIVE TECHNIQUE
With the patient supine on the operating table and after the administration of general anesthesia, 3-dimensional (3-D) fluoroscopy was used to localize the bone bridge, which confirmed the fluoroscopic location that was previously visualized through preoperative 3-D imaging. The leg was elevated, and a tourniquet was applied and inflated. A lateral parapatellar approach was used to isolate the distal femoral physis anteriorly because the bone bridge was centered just lateral to the central portion of the distal femoral physis. A Kirschner wire was placed in the center of the bridge under anteroposterior and lateral fluoroscopic imaging (Figures 3A-3E). 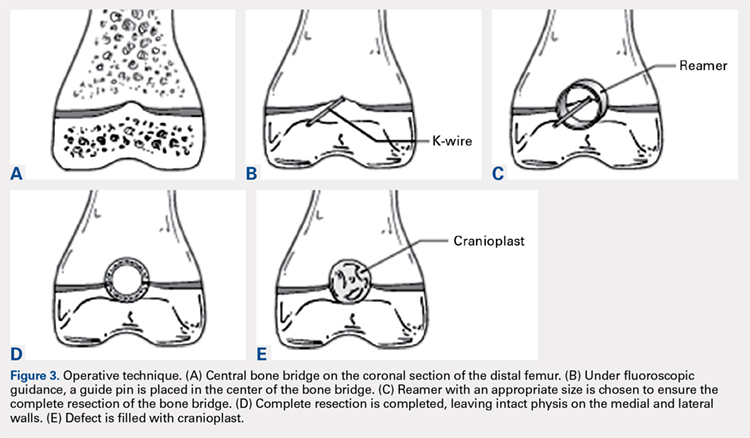
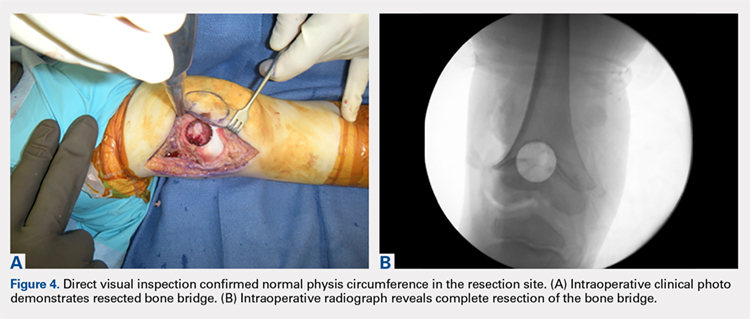
OUTCOME
The patient healed uneventfully, and early range-of-motion exercises were started 6 weeks postoperatively. At 6-month follow-up, his leg-length discrepancy was 2.7 cm, and the bone bridge did not recur. At 3-year follow-up, his leg-length discrepancy was 3.0 cm, and the bone bridge did not recur. Over the 3 years postoperatively, the patient exhibited 9.8 cm of growth on his operative side and 9.5 cm on his nonoperative side (Figure 5). 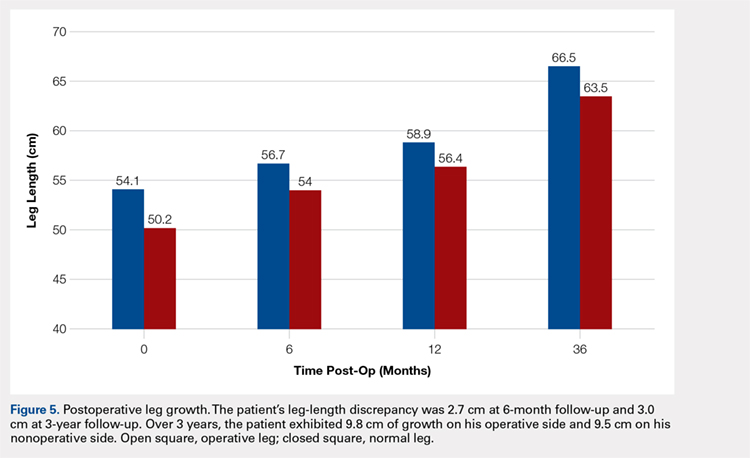
DISCUSSION
Given the considerable growth potential of the distal femoral physis,1,14-16 an injury to the distal femoral physis and the formation of a physeal bone bridge can have a profound effect on a young patient in terms of leg-length discrepancy and angular deformity. Fracture from trauma or infection is a common cause of physeal bone bridges.6,17-19 The etiology of our patient’s distal femoral physeal bone bridge is idiopathic, which is considerably less common than other etiologies, and the incidence of idiopathic physeal bone bridge formation is not well established in the literature. Hresko and Kasser21 identified atraumatic physeal bone bridge formations in 7 patients. Among the 13 patients with physeal bone bridges described by Broughton and colleagues,20 the cause of bridge formation is unknown in 1.
Physeal bone bridges that form centrally are particularly challenging because they are difficult to visualize through a peripheral approach. A number of methods for resecting central physeal bone bridges have been described. These methods have varying degrees of success. In 1981, Langenskiöld7 first described the creation of a metaphyseal mirror and the use of a dental mirror for visualization. This technique, however, yielded unfavorable results in 16% of patients. Williamson and Staheli9 reported poor results in 23% of patients. Loraas and Schmale4 described the use of an endoscope, termed an osteoscope, for visualization, citing advantages of superior illumination and potential for image magnification and capture. Marsh and Polzhofer8 also showed this technique to have low morbidity but poor results in 13% of patients, whereas Moreta and colleagues10 reported poor results in 2 out of 5 patients. The rate of poor results of these methods may be related to the technical difficulty of using dental mirrors and arthroscopes and can be improved by highly efficient direct methods with improved visualization, such as the method described in this article.
Continue to: Proper imaging is necessary for...
Proper imaging is necessary for the accurate quantification of bone bridges to determine resectability and to identify the best surgical approach to resection. MRI with software for the generation of 3-D physeal maps is a reproducible method with good interobserver reliability.22,23 Intraoperative computer-assisted imaging also is beneficial for determining the extent and location of the resection to ensure complete bone bridge removal.24
To our knowledge, a direct approach through parapatellar arthrotomy for the resection of a centrally located distal femoral physeal bone bridge has not been previously described. This novel technique provided direct access to the physeal bone bridge and was performed without injuring the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which may grow normally in the absence of the bridge. Instead of using a lateral or medial approach with a metaphyseal window,4 we directly approached this central bar through a parapatellar approach and were able to completely resect it under direct visualization. This obviated the need for an arthroscope or dental mirror. To remove the entire physeal bone bridge, we needed to resect completely from the anterior cortex to the posterior cortex. Although this technique potentially increased the risk of iatrogenic fracture, we believed that this risk would not differ greatly from that of disrupting the medial or lateral metaphysis and would be more stable with either axial and torsion load. At 3-year follow-up, the patient exhibited restored normal growth in his operative limb relative to that in his nonoperative limb, had not developed angular deformity, and had maintained his previously developed limb-length discrepancy that could be corrected with the epiphysiodesis of his opposite limb at a later date.
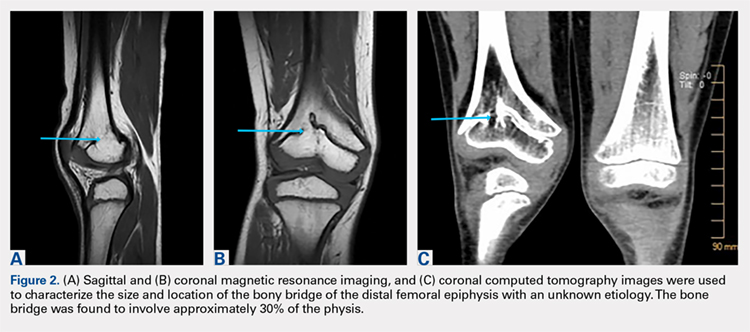
The limitations to this technique include the fact that it may be most effective with small-to moderate-sized central physeal bone bridges, although resection has shown good results with up to 70% physeal involvement.8 In this patient, the bone bridge was moderately sized (30% of the physis), centrally located, and clearly visible on fluoroscopy. These characteristics increased the technical safety and ease of the procedure. The resection of large, peripheral bridges may destabilize the distal femur. The destabilization of the distal femur, in turn, can lead to fracture. Patellofemoral mechanics may also be affected during the treatment of distal femoral physeal bone bridges. This patient has not experienced any patellofemoral dysfunction or symptoms. Given the patient’s age and significant amount of remaining growth, he will need close monitoring until he reaches skeletal maturity.
This paper will be judged for the Resident Writer’s Award.
1. Murphy GA. Disorders of tendons and fascia and adolescent and adult pes planus. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 12th edition. Philadelphia, PA: Mosby-Elsevier; 2013:3966-3972.
2. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
3. Stans AA. Excision of physeal bar. In: Wiesel SW, ed. Operative Techniques in Orthopaedic Surgery. Philadelphia, PA: Lippincott Williams & Wilkins; 2011:1244-1249.
4. Loraas EK, Schmale GA. Endoscopically aided physeal bar takedown and guided growth for the treatment of angular limb deformity. J Pediatr Orthop B. 2012;21(4):348-351. doi:10.1097/BPB.0b013e328346d308.
5. Inoue T, Naito M, Fuhii T, Akiyoshi Y, Yoshimura I, Takamura K. Partial physeal growth arrest treated by bridge resection and artificial dura substitute interposition. J Pediatr Orthop B. 2006;15(1):65-69. doi:10.1097/01202412-200601000-00014.
6. Arkader A, Warner WC Jr, Horn BD, Shaw RN, Wells L. Predicting the outcome of physeal fractures of the distal femur. J Pediatr Orthop. 2007;27(6):703-708. doi:10.1097/BPO.0b013e3180dca0e5.
7. Langenskiöld A. Surgical treatment of partial closure of the growth plate. J Pediatr Orthop. 1981;1(1):3-11. doi:10.1097/01241398-198101010-00002.
8. Marsh JS, Polzhofer GK. Arthroscopically assisted central physeal bar resection. J Pediatr Orthop. 2006;26(2):255-259. doi:10.1097/01.bpo.0000218533.43986.e1.
9. Williamson RV, Staheli LT. Partial physeal growth arrest: treatment by bridge resection and fat interposition. J Pediatr Orthop. 1990;10(6):769-776. doi:10.1097/01241398-199011000-00012.
10. Moreta J, Abril JC, Miranda C. Arthroscopy-assisted resection-interposition of post-traumatic central physeal bridges. Rev Esp Cir Orthop Traumatol. 2013;57(5):333-339. doi:10.1016/j.recot.2013.07.004.
11. Hasler CC, Foster BK. Secondary tethers after physeal bar resection: a common source of failure? Clin Orthop Relat Res. 2002;405:242-249.
12. Paley D, Bhave A, Herzenberg JE, Bowen JR. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82(10):1432-1446. doi:10.2106/00004623-200010000-00010.
13. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
14. Rathjen KE, Kim HKW. Physeal injuries and growth disturbances. In: Flynn JM, Skaggs DL, Waters PM, eds. Rockwood and Wilkins’ Fractures in Children. 8th edition. Philadelphia, PA: Wolters-Kluwer; 2015:135-137.
15. Peterson CA, Peterson HA. Analysis of the incidence of injuries to the epiphyseal growth plate. J Trauma. 1972;12(4):275-281. doi:10.1097/00005373-197204000-00002.
16. Pritchett JW. Longitudinal growth and growth-plate activity in the lower extremity. Clin Orthop Relat Res. 1992;275:274-279.
17. Cassebaum WH, Patterson AH. Fracture of the distal femoral epiphysis. Clin Orthop Relat Res. 1965;41:79-91. doi:10.1097/00003086-196500410-00009.
18. Dahl WJ, Silva S, Vanderhave KL. Distal femoral physeal fixation: are smooth pins really safe? J Pedatir Orthop. 2014;34(2):134-138. doi:10.1097/BPO.0000000000000083.
19. Roberts J. Fracture separation of the distal femoral epiphyseal growth line. J Bone Joint Surg Am. 1973;55:1324.
20. Broughton NS, Dickens DR, Cole WG, Menelaus MB. Epiphyseolysis for partial growth plate arrest. Results after four years or at maturity. J Bone Joint Surg Br. 1989;71(1):13-16. doi:10.1302/0301-620X.71B1.2914983.
21. Hresko MT, Kasser JR. Physeal arrest about the knee associated with non-physeal fractures in the lower extremity. J Bone Joint Surg Am. 1989;71(5):698-703. doi:10.2106/00004623-198971050-00009.
22. Lurie B, Koff MF, Shah P, et al. Three-dimensional magnetic resonance imaging of physeal injury: reliability and clinical utility. J Pediatr Orthop. 2014;34(3):239-245. doi:10.1097/BPO.0000000000000104.
23. Sailhan F, Chotel F, Guibal AL, et al. Three-dimensional MR imaging in the assessment of physeal growth arrest. Eur Radiol. 2004;14(9):1600-1608. doi:10.1007/s00330-004-2319-z.
24. Kang HG, Yoon SJ, Kim JR. Resection of a physeal bar under computer-assisted guidance. J Bone Joint Surg Br. 2010;92(10):1452-1455. doi:10.1302/0301-620X.92B10.24587.
ABSTRACT
A central distal femoral physeal bone bridge in a boy aged 5 years and 7 months was resected with a fluoroscopically guided core reamer placed through a lateral parapatellar approach. At 3-year follow-up, the boy’s leg-length discrepancy was 3.0 cm (3.9 cm preoperatively), and the physeal bone bridge did not recur. The patient had full function and no pain or other patellofemoral complaints. This technique provided direct access to the physeal bone bridge, and complete resection was performed without injury to the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which is expected to grow normally in the absence of the bridge.
A physeal bone bridge is an osseous connection that forms across a physis. It may cause partial premature physeal arrest. Angular deformity and limb-length discrepancy are the main complications caused by physeal bone bridges.1-4 The indications for the treatment of physeal bridges are well documented.1-5 Trauma and infection are common causes of distal femoral physeal bone bridges. Arkader and colleagues6 showed that among different types of physeal bridges, the Salter-Harris type is significantly associated with complications, among which growth arrest is the most common and occurs in 27.4% of all patients.
The treatment of distal femoral physeal bone bridges is technically difficult and provides variable results. Poor results are reported in 13% to 40% of patients.7-10 Procedure failure has been attributed to incomplete resection with the persistent tethering and dislodgement of the graft.11 Methods with improved efficacy for the removal of central physeal bridges will help prevent reformation after treatment. We have used a novel technique that allows the direct resection of a central physeal bone bridge in the distal femur through the use of a fluoroscopically guided core reamer. This technique enables the complete removal of the bone bridge and the direct visual assessment of the remaining physis. The patient’s parents provided written informed consent for print and electronic publication of this case report.
CASE
A 3-year-old boy with a history of hemifacial microsomia presented for the evaluation of genu valgum and leg-length discrepancy. His intermalleolar distance at that time was 8 cm. A standing radiograph of his lower extremities demonstrated changes consistent with physiologic genu valgum. He had no history of knee trauma, infection, or pain.
At the age of 5 years and 7 months, the patient returned for a repeat evaluation and was noted to exhibit the progressive valgus deformity of the right leg and a leg-length discrepancy of 3.9 cm (Figure 1).
Continue to: With the patient supine on the operating...
OPERATIVE TECHNIQUE
With the patient supine on the operating table and after the administration of general anesthesia, 3-dimensional (3-D) fluoroscopy was used to localize the bone bridge, which confirmed the fluoroscopic location that was previously visualized through preoperative 3-D imaging. The leg was elevated, and a tourniquet was applied and inflated. A lateral parapatellar approach was used to isolate the distal femoral physis anteriorly because the bone bridge was centered just lateral to the central portion of the distal femoral physis. A Kirschner wire was placed in the center of the bridge under anteroposterior and lateral fluoroscopic imaging (Figures 3A-3E).
OUTCOME
The patient healed uneventfully, and early range-of-motion exercises were started 6 weeks postoperatively. At 6-month follow-up, his leg-length discrepancy was 2.7 cm, and the bone bridge did not recur. At 3-year follow-up, his leg-length discrepancy was 3.0 cm, and the bone bridge did not recur. Over the 3 years postoperatively, the patient exhibited 9.8 cm of growth on his operative side and 9.5 cm on his nonoperative side (Figure 5).
DISCUSSION
Given the considerable growth potential of the distal femoral physis,1,14-16 an injury to the distal femoral physis and the formation of a physeal bone bridge can have a profound effect on a young patient in terms of leg-length discrepancy and angular deformity. Fracture from trauma or infection is a common cause of physeal bone bridges.6,17-19 The etiology of our patient’s distal femoral physeal bone bridge is idiopathic, which is considerably less common than other etiologies, and the incidence of idiopathic physeal bone bridge formation is not well established in the literature. Hresko and Kasser21 identified atraumatic physeal bone bridge formations in 7 patients. Among the 13 patients with physeal bone bridges described by Broughton and colleagues,20 the cause of bridge formation is unknown in 1.
Physeal bone bridges that form centrally are particularly challenging because they are difficult to visualize through a peripheral approach. A number of methods for resecting central physeal bone bridges have been described. These methods have varying degrees of success. In 1981, Langenskiöld7 first described the creation of a metaphyseal mirror and the use of a dental mirror for visualization. This technique, however, yielded unfavorable results in 16% of patients. Williamson and Staheli9 reported poor results in 23% of patients. Loraas and Schmale4 described the use of an endoscope, termed an osteoscope, for visualization, citing advantages of superior illumination and potential for image magnification and capture. Marsh and Polzhofer8 also showed this technique to have low morbidity but poor results in 13% of patients, whereas Moreta and colleagues10 reported poor results in 2 out of 5 patients. The rate of poor results of these methods may be related to the technical difficulty of using dental mirrors and arthroscopes and can be improved by highly efficient direct methods with improved visualization, such as the method described in this article.
Continue to: Proper imaging is necessary for...
Proper imaging is necessary for the accurate quantification of bone bridges to determine resectability and to identify the best surgical approach to resection. MRI with software for the generation of 3-D physeal maps is a reproducible method with good interobserver reliability.22,23 Intraoperative computer-assisted imaging also is beneficial for determining the extent and location of the resection to ensure complete bone bridge removal.24
To our knowledge, a direct approach through parapatellar arthrotomy for the resection of a centrally located distal femoral physeal bone bridge has not been previously described. This novel technique provided direct access to the physeal bone bridge and was performed without injuring the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which may grow normally in the absence of the bridge. Instead of using a lateral or medial approach with a metaphyseal window,4 we directly approached this central bar through a parapatellar approach and were able to completely resect it under direct visualization. This obviated the need for an arthroscope or dental mirror. To remove the entire physeal bone bridge, we needed to resect completely from the anterior cortex to the posterior cortex. Although this technique potentially increased the risk of iatrogenic fracture, we believed that this risk would not differ greatly from that of disrupting the medial or lateral metaphysis and would be more stable with either axial and torsion load. At 3-year follow-up, the patient exhibited restored normal growth in his operative limb relative to that in his nonoperative limb, had not developed angular deformity, and had maintained his previously developed limb-length discrepancy that could be corrected with the epiphysiodesis of his opposite limb at a later date.
The limitations to this technique include the fact that it may be most effective with small-to moderate-sized central physeal bone bridges, although resection has shown good results with up to 70% physeal involvement.8 In this patient, the bone bridge was moderately sized (30% of the physis), centrally located, and clearly visible on fluoroscopy. These characteristics increased the technical safety and ease of the procedure. The resection of large, peripheral bridges may destabilize the distal femur. The destabilization of the distal femur, in turn, can lead to fracture. Patellofemoral mechanics may also be affected during the treatment of distal femoral physeal bone bridges. This patient has not experienced any patellofemoral dysfunction or symptoms. Given the patient’s age and significant amount of remaining growth, he will need close monitoring until he reaches skeletal maturity.
This paper will be judged for the Resident Writer’s Award.
ABSTRACT
A central distal femoral physeal bone bridge in a boy aged 5 years and 7 months was resected with a fluoroscopically guided core reamer placed through a lateral parapatellar approach. At 3-year follow-up, the boy’s leg-length discrepancy was 3.0 cm (3.9 cm preoperatively), and the physeal bone bridge did not recur. The patient had full function and no pain or other patellofemoral complaints. This technique provided direct access to the physeal bone bridge, and complete resection was performed without injury to the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which is expected to grow normally in the absence of the bridge.
A physeal bone bridge is an osseous connection that forms across a physis. It may cause partial premature physeal arrest. Angular deformity and limb-length discrepancy are the main complications caused by physeal bone bridges.1-4 The indications for the treatment of physeal bridges are well documented.1-5 Trauma and infection are common causes of distal femoral physeal bone bridges. Arkader and colleagues6 showed that among different types of physeal bridges, the Salter-Harris type is significantly associated with complications, among which growth arrest is the most common and occurs in 27.4% of all patients.
The treatment of distal femoral physeal bone bridges is technically difficult and provides variable results. Poor results are reported in 13% to 40% of patients.7-10 Procedure failure has been attributed to incomplete resection with the persistent tethering and dislodgement of the graft.11 Methods with improved efficacy for the removal of central physeal bridges will help prevent reformation after treatment. We have used a novel technique that allows the direct resection of a central physeal bone bridge in the distal femur through the use of a fluoroscopically guided core reamer. This technique enables the complete removal of the bone bridge and the direct visual assessment of the remaining physis. The patient’s parents provided written informed consent for print and electronic publication of this case report.
CASE
A 3-year-old boy with a history of hemifacial microsomia presented for the evaluation of genu valgum and leg-length discrepancy. His intermalleolar distance at that time was 8 cm. A standing radiograph of his lower extremities demonstrated changes consistent with physiologic genu valgum. He had no history of knee trauma, infection, or pain.
At the age of 5 years and 7 months, the patient returned for a repeat evaluation and was noted to exhibit the progressive valgus deformity of the right leg and a leg-length discrepancy of 3.9 cm (Figure 1).
Continue to: With the patient supine on the operating...
OPERATIVE TECHNIQUE
With the patient supine on the operating table and after the administration of general anesthesia, 3-dimensional (3-D) fluoroscopy was used to localize the bone bridge, which confirmed the fluoroscopic location that was previously visualized through preoperative 3-D imaging. The leg was elevated, and a tourniquet was applied and inflated. A lateral parapatellar approach was used to isolate the distal femoral physis anteriorly because the bone bridge was centered just lateral to the central portion of the distal femoral physis. A Kirschner wire was placed in the center of the bridge under anteroposterior and lateral fluoroscopic imaging (Figures 3A-3E).
OUTCOME
The patient healed uneventfully, and early range-of-motion exercises were started 6 weeks postoperatively. At 6-month follow-up, his leg-length discrepancy was 2.7 cm, and the bone bridge did not recur. At 3-year follow-up, his leg-length discrepancy was 3.0 cm, and the bone bridge did not recur. Over the 3 years postoperatively, the patient exhibited 9.8 cm of growth on his operative side and 9.5 cm on his nonoperative side (Figure 5).
DISCUSSION
Given the considerable growth potential of the distal femoral physis,1,14-16 an injury to the distal femoral physis and the formation of a physeal bone bridge can have a profound effect on a young patient in terms of leg-length discrepancy and angular deformity. Fracture from trauma or infection is a common cause of physeal bone bridges.6,17-19 The etiology of our patient’s distal femoral physeal bone bridge is idiopathic, which is considerably less common than other etiologies, and the incidence of idiopathic physeal bone bridge formation is not well established in the literature. Hresko and Kasser21 identified atraumatic physeal bone bridge formations in 7 patients. Among the 13 patients with physeal bone bridges described by Broughton and colleagues,20 the cause of bridge formation is unknown in 1.
Physeal bone bridges that form centrally are particularly challenging because they are difficult to visualize through a peripheral approach. A number of methods for resecting central physeal bone bridges have been described. These methods have varying degrees of success. In 1981, Langenskiöld7 first described the creation of a metaphyseal mirror and the use of a dental mirror for visualization. This technique, however, yielded unfavorable results in 16% of patients. Williamson and Staheli9 reported poor results in 23% of patients. Loraas and Schmale4 described the use of an endoscope, termed an osteoscope, for visualization, citing advantages of superior illumination and potential for image magnification and capture. Marsh and Polzhofer8 also showed this technique to have low morbidity but poor results in 13% of patients, whereas Moreta and colleagues10 reported poor results in 2 out of 5 patients. The rate of poor results of these methods may be related to the technical difficulty of using dental mirrors and arthroscopes and can be improved by highly efficient direct methods with improved visualization, such as the method described in this article.
Continue to: Proper imaging is necessary for...
Proper imaging is necessary for the accurate quantification of bone bridges to determine resectability and to identify the best surgical approach to resection. MRI with software for the generation of 3-D physeal maps is a reproducible method with good interobserver reliability.22,23 Intraoperative computer-assisted imaging also is beneficial for determining the extent and location of the resection to ensure complete bone bridge removal.24
To our knowledge, a direct approach through parapatellar arthrotomy for the resection of a centrally located distal femoral physeal bone bridge has not been previously described. This novel technique provided direct access to the physeal bone bridge and was performed without injuring the adjacent physeal cartilage in the medial and lateral columns of the distal femur, which may grow normally in the absence of the bridge. Instead of using a lateral or medial approach with a metaphyseal window,4 we directly approached this central bar through a parapatellar approach and were able to completely resect it under direct visualization. This obviated the need for an arthroscope or dental mirror. To remove the entire physeal bone bridge, we needed to resect completely from the anterior cortex to the posterior cortex. Although this technique potentially increased the risk of iatrogenic fracture, we believed that this risk would not differ greatly from that of disrupting the medial or lateral metaphysis and would be more stable with either axial and torsion load. At 3-year follow-up, the patient exhibited restored normal growth in his operative limb relative to that in his nonoperative limb, had not developed angular deformity, and had maintained his previously developed limb-length discrepancy that could be corrected with the epiphysiodesis of his opposite limb at a later date.
The limitations to this technique include the fact that it may be most effective with small-to moderate-sized central physeal bone bridges, although resection has shown good results with up to 70% physeal involvement.8 In this patient, the bone bridge was moderately sized (30% of the physis), centrally located, and clearly visible on fluoroscopy. These characteristics increased the technical safety and ease of the procedure. The resection of large, peripheral bridges may destabilize the distal femur. The destabilization of the distal femur, in turn, can lead to fracture. Patellofemoral mechanics may also be affected during the treatment of distal femoral physeal bone bridges. This patient has not experienced any patellofemoral dysfunction or symptoms. Given the patient’s age and significant amount of remaining growth, he will need close monitoring until he reaches skeletal maturity.
This paper will be judged for the Resident Writer’s Award.
1. Murphy GA. Disorders of tendons and fascia and adolescent and adult pes planus. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 12th edition. Philadelphia, PA: Mosby-Elsevier; 2013:3966-3972.
2. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
3. Stans AA. Excision of physeal bar. In: Wiesel SW, ed. Operative Techniques in Orthopaedic Surgery. Philadelphia, PA: Lippincott Williams & Wilkins; 2011:1244-1249.
4. Loraas EK, Schmale GA. Endoscopically aided physeal bar takedown and guided growth for the treatment of angular limb deformity. J Pediatr Orthop B. 2012;21(4):348-351. doi:10.1097/BPB.0b013e328346d308.
5. Inoue T, Naito M, Fuhii T, Akiyoshi Y, Yoshimura I, Takamura K. Partial physeal growth arrest treated by bridge resection and artificial dura substitute interposition. J Pediatr Orthop B. 2006;15(1):65-69. doi:10.1097/01202412-200601000-00014.
6. Arkader A, Warner WC Jr, Horn BD, Shaw RN, Wells L. Predicting the outcome of physeal fractures of the distal femur. J Pediatr Orthop. 2007;27(6):703-708. doi:10.1097/BPO.0b013e3180dca0e5.
7. Langenskiöld A. Surgical treatment of partial closure of the growth plate. J Pediatr Orthop. 1981;1(1):3-11. doi:10.1097/01241398-198101010-00002.
8. Marsh JS, Polzhofer GK. Arthroscopically assisted central physeal bar resection. J Pediatr Orthop. 2006;26(2):255-259. doi:10.1097/01.bpo.0000218533.43986.e1.
9. Williamson RV, Staheli LT. Partial physeal growth arrest: treatment by bridge resection and fat interposition. J Pediatr Orthop. 1990;10(6):769-776. doi:10.1097/01241398-199011000-00012.
10. Moreta J, Abril JC, Miranda C. Arthroscopy-assisted resection-interposition of post-traumatic central physeal bridges. Rev Esp Cir Orthop Traumatol. 2013;57(5):333-339. doi:10.1016/j.recot.2013.07.004.
11. Hasler CC, Foster BK. Secondary tethers after physeal bar resection: a common source of failure? Clin Orthop Relat Res. 2002;405:242-249.
12. Paley D, Bhave A, Herzenberg JE, Bowen JR. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82(10):1432-1446. doi:10.2106/00004623-200010000-00010.
13. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
14. Rathjen KE, Kim HKW. Physeal injuries and growth disturbances. In: Flynn JM, Skaggs DL, Waters PM, eds. Rockwood and Wilkins’ Fractures in Children. 8th edition. Philadelphia, PA: Wolters-Kluwer; 2015:135-137.
15. Peterson CA, Peterson HA. Analysis of the incidence of injuries to the epiphyseal growth plate. J Trauma. 1972;12(4):275-281. doi:10.1097/00005373-197204000-00002.
16. Pritchett JW. Longitudinal growth and growth-plate activity in the lower extremity. Clin Orthop Relat Res. 1992;275:274-279.
17. Cassebaum WH, Patterson AH. Fracture of the distal femoral epiphysis. Clin Orthop Relat Res. 1965;41:79-91. doi:10.1097/00003086-196500410-00009.
18. Dahl WJ, Silva S, Vanderhave KL. Distal femoral physeal fixation: are smooth pins really safe? J Pedatir Orthop. 2014;34(2):134-138. doi:10.1097/BPO.0000000000000083.
19. Roberts J. Fracture separation of the distal femoral epiphyseal growth line. J Bone Joint Surg Am. 1973;55:1324.
20. Broughton NS, Dickens DR, Cole WG, Menelaus MB. Epiphyseolysis for partial growth plate arrest. Results after four years or at maturity. J Bone Joint Surg Br. 1989;71(1):13-16. doi:10.1302/0301-620X.71B1.2914983.
21. Hresko MT, Kasser JR. Physeal arrest about the knee associated with non-physeal fractures in the lower extremity. J Bone Joint Surg Am. 1989;71(5):698-703. doi:10.2106/00004623-198971050-00009.
22. Lurie B, Koff MF, Shah P, et al. Three-dimensional magnetic resonance imaging of physeal injury: reliability and clinical utility. J Pediatr Orthop. 2014;34(3):239-245. doi:10.1097/BPO.0000000000000104.
23. Sailhan F, Chotel F, Guibal AL, et al. Three-dimensional MR imaging in the assessment of physeal growth arrest. Eur Radiol. 2004;14(9):1600-1608. doi:10.1007/s00330-004-2319-z.
24. Kang HG, Yoon SJ, Kim JR. Resection of a physeal bar under computer-assisted guidance. J Bone Joint Surg Br. 2010;92(10):1452-1455. doi:10.1302/0301-620X.92B10.24587.
1. Murphy GA. Disorders of tendons and fascia and adolescent and adult pes planus. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 12th edition. Philadelphia, PA: Mosby-Elsevier; 2013:3966-3972.
2. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
3. Stans AA. Excision of physeal bar. In: Wiesel SW, ed. Operative Techniques in Orthopaedic Surgery. Philadelphia, PA: Lippincott Williams & Wilkins; 2011:1244-1249.
4. Loraas EK, Schmale GA. Endoscopically aided physeal bar takedown and guided growth for the treatment of angular limb deformity. J Pediatr Orthop B. 2012;21(4):348-351. doi:10.1097/BPB.0b013e328346d308.
5. Inoue T, Naito M, Fuhii T, Akiyoshi Y, Yoshimura I, Takamura K. Partial physeal growth arrest treated by bridge resection and artificial dura substitute interposition. J Pediatr Orthop B. 2006;15(1):65-69. doi:10.1097/01202412-200601000-00014.
6. Arkader A, Warner WC Jr, Horn BD, Shaw RN, Wells L. Predicting the outcome of physeal fractures of the distal femur. J Pediatr Orthop. 2007;27(6):703-708. doi:10.1097/BPO.0b013e3180dca0e5.
7. Langenskiöld A. Surgical treatment of partial closure of the growth plate. J Pediatr Orthop. 1981;1(1):3-11. doi:10.1097/01241398-198101010-00002.
8. Marsh JS, Polzhofer GK. Arthroscopically assisted central physeal bar resection. J Pediatr Orthop. 2006;26(2):255-259. doi:10.1097/01.bpo.0000218533.43986.e1.
9. Williamson RV, Staheli LT. Partial physeal growth arrest: treatment by bridge resection and fat interposition. J Pediatr Orthop. 1990;10(6):769-776. doi:10.1097/01241398-199011000-00012.
10. Moreta J, Abril JC, Miranda C. Arthroscopy-assisted resection-interposition of post-traumatic central physeal bridges. Rev Esp Cir Orthop Traumatol. 2013;57(5):333-339. doi:10.1016/j.recot.2013.07.004.
11. Hasler CC, Foster BK. Secondary tethers after physeal bar resection: a common source of failure? Clin Orthop Relat Res. 2002;405:242-249.
12. Paley D, Bhave A, Herzenberg JE, Bowen JR. Multiplier method for predicting limb-length discrepancy. J Bone Joint Surg Am. 2000;82(10):1432-1446. doi:10.2106/00004623-200010000-00010.
13. Khoshhal KI, Kiefer GN. Physeal bridge resection. J Am Acad Orthop Surg. 2005;13(1):47-58. doi:10.5435/00124635-200501000-00007.
14. Rathjen KE, Kim HKW. Physeal injuries and growth disturbances. In: Flynn JM, Skaggs DL, Waters PM, eds. Rockwood and Wilkins’ Fractures in Children. 8th edition. Philadelphia, PA: Wolters-Kluwer; 2015:135-137.
15. Peterson CA, Peterson HA. Analysis of the incidence of injuries to the epiphyseal growth plate. J Trauma. 1972;12(4):275-281. doi:10.1097/00005373-197204000-00002.
16. Pritchett JW. Longitudinal growth and growth-plate activity in the lower extremity. Clin Orthop Relat Res. 1992;275:274-279.
17. Cassebaum WH, Patterson AH. Fracture of the distal femoral epiphysis. Clin Orthop Relat Res. 1965;41:79-91. doi:10.1097/00003086-196500410-00009.
18. Dahl WJ, Silva S, Vanderhave KL. Distal femoral physeal fixation: are smooth pins really safe? J Pedatir Orthop. 2014;34(2):134-138. doi:10.1097/BPO.0000000000000083.
19. Roberts J. Fracture separation of the distal femoral epiphyseal growth line. J Bone Joint Surg Am. 1973;55:1324.
20. Broughton NS, Dickens DR, Cole WG, Menelaus MB. Epiphyseolysis for partial growth plate arrest. Results after four years or at maturity. J Bone Joint Surg Br. 1989;71(1):13-16. doi:10.1302/0301-620X.71B1.2914983.
21. Hresko MT, Kasser JR. Physeal arrest about the knee associated with non-physeal fractures in the lower extremity. J Bone Joint Surg Am. 1989;71(5):698-703. doi:10.2106/00004623-198971050-00009.
22. Lurie B, Koff MF, Shah P, et al. Three-dimensional magnetic resonance imaging of physeal injury: reliability and clinical utility. J Pediatr Orthop. 2014;34(3):239-245. doi:10.1097/BPO.0000000000000104.
23. Sailhan F, Chotel F, Guibal AL, et al. Three-dimensional MR imaging in the assessment of physeal growth arrest. Eur Radiol. 2004;14(9):1600-1608. doi:10.1007/s00330-004-2319-z.
24. Kang HG, Yoon SJ, Kim JR. Resection of a physeal bar under computer-assisted guidance. J Bone Joint Surg Br. 2010;92(10):1452-1455. doi:10.1302/0301-620X.92B10.24587.
TAKE-HOME POINTS
- Central physeal arrest of the distal femur is challenging, but this surgical technique provides an option for treatment.
- Partial bone bridges can be resected, but advanced imaging with MRI or CT, or both, is helpful in preoperative planning.
- Regardless of the type of physeal bar resection that is chosen, it is unlikely that complete, normal bone growth will be restored and closed follow up will be needed.
Recurrence of Extranodal Natural Killer/T-cell Lymphoma Presenting as Tarsal Tunnel Syndrome
ABSTRACT
This case report is a rare form of lymphoma recurrence which presented as tarsal tunnel syndrome. The patient had been previously treated for the malignancy and was presumed to be in remission; however, standard radiology imaging protocols failed to include the distal extremities on these scans. The patient presented to the orthopedic clinic with tarsal tunnel symptoms and a mass in the tarsal tunnel. A complete evaluation resulted in a diagnosis of recurrence of the malignancy. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body fluorodeoxyglucose positron emission tomography computed tomography when evaluating for recurrence in patients.
Nasal-type, extranodal natural killer/T-cell lymphoma (ENKTL) is a rare form of non-Hodgkin lymphoma (NHL). Malignancies account for only 10% of NHL in Asian and South American populations. However, in Caucasians, it represents <1% of all cases. In addition, at 3:1 male to female ratio, the disease most commonly affects male patients who are 50 to 59 years old.1-3 The etiology of this malignancy is strongly related to prior infection with Epstein-Barr virus (EBV) as EBV-encoded early small ribonucleic acid on in situ hybridization of lymphoma cells is positive in 95% of cases.4-6
Typical sites of involvement include the nasal cavity, nasopharynx, and sinuses, causing patients to present with nasal obstruction, chronic sinusitis, or epistaxis. Additionally, ENKTL can occur primarily in the skin, gastrointestinal tract, spleen, and testis, whereas the bone marrow may be involved in 10% of cases. Although rare, unusual sites, including muscle, adrenals, and ovaries, have been published.7,8
Staging is best performed using the T-staging system, which accounts for the extent of local tumor involvement. Higher stages, such as T3 /T4, equate to locally advanced disease and imply a worse prognosis.9,10 Computed tomography (CT) and magnetic resonance imaging (MRI) help define local soft tissues and bony involvement. Furthermore, CT of the chest, abdomen, and pelvis as well as bone marrow biopsy are performed as part of the staging process. Lastly, fluorine-18 fluorodeoxyglucose positron emission tomography CT (18-FDG PET-CT) is often used to detect extranodal spread, define the extent of involvement, differentiate between lymphoma and inflammatory masses, and monitor for recurrence.11
Treatment for local ENKTL involves concurrent chemoradiotherapy followed by 3 cycles of etoposide, ifosfamide, cisplatin, and dexamethasone, which results in a complete response rate of 80%, and is the most favorable when comparing treatment modalities.12 Unfortunately, recurrence rates reach as high as 50%, whereas the 5-year survival rate is 59%.13,14 For recurrent or disseminated disease, high-dose chemotherapy and hematopoietic stem cell transplantation remain as alternative treatments for patients who have undergone 2 complete remissions and can be curative in some instances.13,15
Continue to: In summary, ENKTL is a rare form...
In summary, ENKTL is a rare form of NHL which classically presents in the nasal cavity; however, this type of lymphoma may present in a variety of extranodal sites.7,8 Despite the numerous published reports on ENKTL, no study has reported either primary or recurrent ENKTL in the feet or hands. To our knowledge, this is one of the first published cases of a patient who developed a rare and recurring ENKTL in the foot and ankle. The patient provided written informed consent for print and electronic publication of this case report.
CASE
A 59-year-old Caucasian woman was referred to the orthopedic foot and ankle clinic by her primary care physician for right medial ankle pain, skin ulceration, and numbness over the plantar aspect of her right foot. Upon questioning, the patient noted that the pain and numbness were present for almost 6 months. She denied trauma to the concerned area. Previously, the patient was observed and treated elsewhere for plantar fasciitis and was prescribed a brace before being immobilized in a controlled ankle motion (CAM) boot for 6 weeks. At follow-up with her outside provider, the patient had developed skin breakdown over the medial aspect of the right ankle, and this condition was presumed to be caused by the boot. After local wound care failed to improve her skin ulceration, she returned to her primary care physician, who ordered an MRI of the area and referred her to our specialty clinic.
Upon review, the patient’s past medical history included a diagnosis of nasal-type ENKTL. Her malignancy was treated with chemoradiotherapy 2 years prior to her consultation with the foot and ankle clinic.
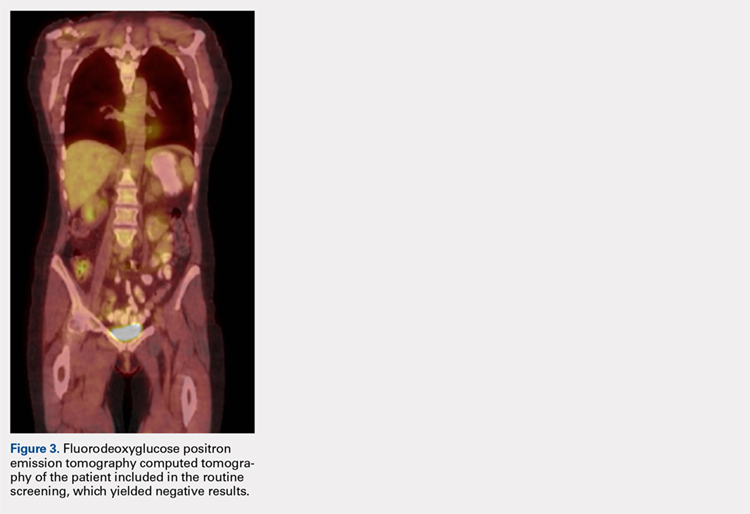
The patient was noted by her medical oncologist and interventional radiologist to be in complete stage 4 remission since being treated. She underwent routine MRI and CT scans of the head and neck at 6-month intervals and FDG PET-CT scans at 3-month intervals, as per institutional protocol. The examinations showed no evidence of malignancy or metabolically active disease. The last imaging study occurred 2 months prior to admission to the foot and ankle clinic.
The patient consulted her medical oncologist 1 month prior to presenting to our clinic and was noted to exhibit an “excellent response to chemoradiotherapy” and “continues to remain disease free at 2 years.” She was instructed to continue routine follow-up. However, the office notes mentioned no ankle pain and non-healing wounds.
During physical examination, the patient presented an antalgic gait on the right side. Inspection demonstrated an increased circumference of the right ankle compared with the left, with a soft, palpable mass over the medial aspect of her right ankle. A 3 cm × 2 cm, grade 2 abrasion of the skin was observed over the medial mass just posterior to her medial malleolus. Range of motion was within normal limits. The patient exhibited a palpable posterior tibial artery pulse and full strength upon muscle testing of the lower extremities. She featured a positive Tinel’s sign and discomfort over the mass itself, with the pain radiating down to the plantar aspect of her foot and diffuse numbness over the plantar aspect of the foot.
Continue to: Review of her plain radiographs...
Review of her plain radiographs demonstrated no bony abnormalities, fractures, nor visible deformity (Figures 1A, 1B).
At presentation, our differential diagnosis included recurrence of the malignancy, secondary malignancy, infection, and inflammatory disease. After a lengthy discussion with the patient and consultation with our institution’s musculoskeletal oncologist, the decision was made to perform a right-ankle mass biopsy and marginal excision with wound irrigation and débridement and tarsal tunnel release.
The patient was placed in the supine position with standard prepping and draping. The medial eschar was excised in an elliptical fashion, and a curvilinear, longitudinal approach was performed within the compartment to access the mass along the posteromedial aspect of the ankle. Although no evidence of infection was observed, the tissue was thickened with areas of necrosis down to the flexor retinaculum. Once the flexor retinaculum was opened, a fibrous, plaque-like mass was observed, and it was encased with flexor tendons and neurovascular structures of the tarsal tunnel. After mass excision, a complete tarsal tunnel release was performed until the neurovascular bundle was free. Irrigation and débridement of the ulcer were performed along with complicated wound closure, and the patient was placed in a well-padded postoperative splint.
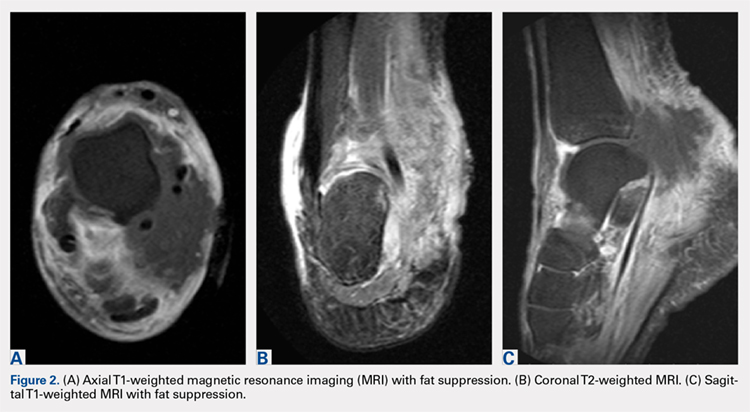
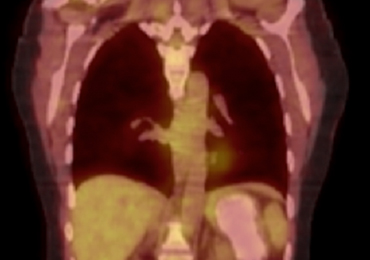
Pathology was finalized as a recurrent, EBV-positive, and nasal-type ENKTL. The patient underwent bone marrow biopsy, which yielded negative results. CT of the chest, abdomen, and pelvis were negative for the disease. FDG PET-CT, which included the extremities, was performed and demonstrated increased uptake in the right ankle, consistent with the malignancy (Figure 4). 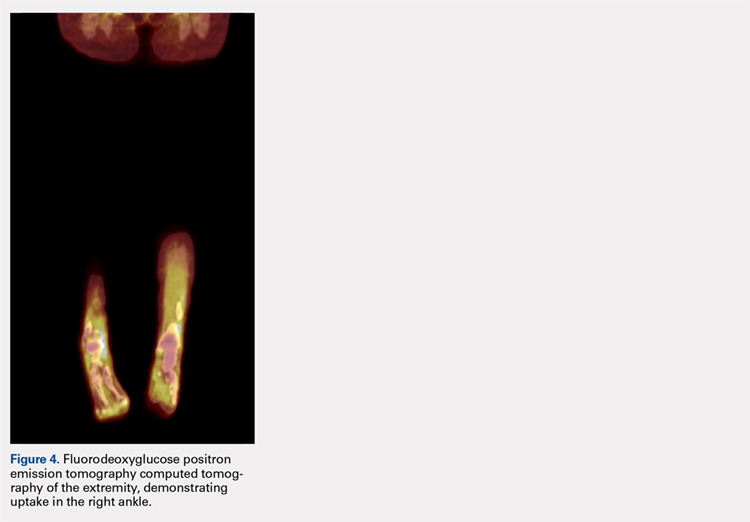
DISCUSSION
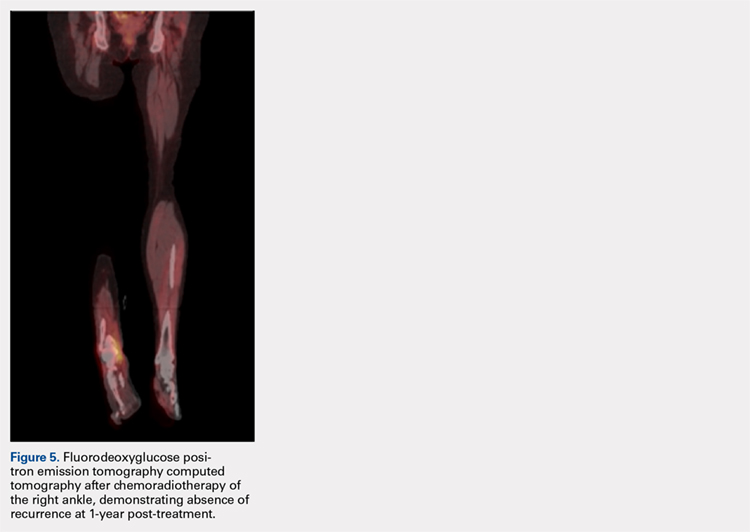
ENKTL is an uncommon form of lymphoma and is exceedingly rare in Caucasian females.1-3 Although the patient’s primary occurrence was in the nasal cavity, recurrence in the foot and ankle must still be described.7,8 To our knowledge, this article is one of the first published cases of a patient who developed a rare-recurrence ENKTL about the foot and ankle. Occurrence in extremities is extremely rare that the staging protocol does not include FDG PET-CT of these areas. The patient’s “negative” scans led many providers to neglect the symptoms in her right ankle until the lesion had ulcerated through the skin. If one would have relied on imaging reports and outside records alone, the diagnosis would have been delayed longer or missed all together. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body FDG PET-CT when evaluating for recurrence in patients.
1. Quintanilla-Martinez L, Kremer M, Keller G, et al. p53 mutations in nasal natural killer/T-cell lymphoma from Mexico: association with large cell morphology and advanced disease. Am J Pathol. 2001;159(6):2095-2105. doi:10.1016/S0002-9440(10)63061-1.
2. Au WY, Ma SY, Chim CS, et al. Clinicopathologic features and treatment outcome of mature T-cell and natural killer-cell lymphomas diagnosed according to the World Health Organization classification scheme: a single center experience of 10 years. Ann Oncol. 2005;16(2):206-214. doi:10.1093/annonc/mdi037.
3. Armitage JO. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood. 1997;89(11):3909-3918.
4. Medeiros LJ, Peiper SC, Elwood L, Yano T, Raffeld M, Jaffe ES. Angiocentric immunoproliferative lesions: a molecular analysis of eight cases. Hum Pathol. 1991;22(11):1150-1157. doi:10.1016/0046-8177(91)90269-U.
5. Ho FC, Srivastava G, Loke SL, et al. Presence of Epstein-Barr virus DNA in nasal lymphomas of B and ‘T’ cell type. Hematol Oncol. 1990;8(5):271-281. doi:10.1002/hon.2900080505.
6. Gelb AB, van de Rijn M, Regula DP Jr, et al. Epstein-Barr virus-associated natural killer-large granular lymphocyte leukemia. Hum Pathol. 1994;25(9):953-960. doi:10.1016/0046-8177(94)90018-3.
7. Petrella T, Delfau-Larue MH, Caillot D, et al. Nasopharyngeal lymphomas: further evidence for a natural killer cell origin. Hum Pathol. 1996;27(8):827-833. doi:10.1016/S0046-8177(96)90457-8.
8. Hasserjian RP, Harris NL. NK-cell lymphomas and leukemias: a spectrum of tumors with variable manifestations and immunophenotype. Am J Clin Pathol. 2007;127(6):860-868. doi:10.1309/2F39NX1AL3L54WU8.
9. Robbins KT, Fuller LM, Vlasak M. Primary lymphomas of the nasal cavity and paranasal sinuses. Cancer. 1985;56(4):814-819. doi:10.1002/1097-0142(19850815)56.
10. Ooi GC, Chim CS, Liang R, Tsang KW, Kwong YL. Nasal T-cell/natural killer cell lymphoma: CT and MR imaging features of a new clinicopathologic entity. Am J Roentgenol. 2000;174(4):1141-1145. doi:10.2214/ajr.174.4.1741141.
11. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621. doi:10.1007/s00277-008-0494-8.
12. Kim SJ, Kim K, Kim BS, et al. Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-cell lymphoma: consortium for improving survival of lymphoma study. J Clin Oncol. 2009;27(35):6027-6032. doi:10.1200/JCO.2009.23.8592.
13. Kwong YL. Natural killer-cell malignancies: diagnosis and treatment. Leukemia. 2005;19(12):2186-2194. doi:10.1038/sj.leu.2403955.
14. Liang R. Advances in the management and monitoring of extranodal NK/T-cell lymphoma, nasal type. Br J Haematol. 2009;147(1):13-21. doi:10.1111/j.1365-2141.2009.07802.x.
15. Yokoyama H, Yamamoto J, Tohmiya Y, et al. Allogeneic hematopoietic stem cell transplant following chemotherapy containing l-asparaginase as a promising treatment for patients with relapsed or refractory extranodal natural killer/T cell lymphoma, nasal type. Leuk Lymphoma. 2010;51(8):1509-1512. doi:10.3109/10428194.2010.487958.
ABSTRACT
This case report is a rare form of lymphoma recurrence which presented as tarsal tunnel syndrome. The patient had been previously treated for the malignancy and was presumed to be in remission; however, standard radiology imaging protocols failed to include the distal extremities on these scans. The patient presented to the orthopedic clinic with tarsal tunnel symptoms and a mass in the tarsal tunnel. A complete evaluation resulted in a diagnosis of recurrence of the malignancy. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body fluorodeoxyglucose positron emission tomography computed tomography when evaluating for recurrence in patients.
Nasal-type, extranodal natural killer/T-cell lymphoma (ENKTL) is a rare form of non-Hodgkin lymphoma (NHL). Malignancies account for only 10% of NHL in Asian and South American populations. However, in Caucasians, it represents <1% of all cases. In addition, at 3:1 male to female ratio, the disease most commonly affects male patients who are 50 to 59 years old.1-3 The etiology of this malignancy is strongly related to prior infection with Epstein-Barr virus (EBV) as EBV-encoded early small ribonucleic acid on in situ hybridization of lymphoma cells is positive in 95% of cases.4-6
Typical sites of involvement include the nasal cavity, nasopharynx, and sinuses, causing patients to present with nasal obstruction, chronic sinusitis, or epistaxis. Additionally, ENKTL can occur primarily in the skin, gastrointestinal tract, spleen, and testis, whereas the bone marrow may be involved in 10% of cases. Although rare, unusual sites, including muscle, adrenals, and ovaries, have been published.7,8
Staging is best performed using the T-staging system, which accounts for the extent of local tumor involvement. Higher stages, such as T3 /T4, equate to locally advanced disease and imply a worse prognosis.9,10 Computed tomography (CT) and magnetic resonance imaging (MRI) help define local soft tissues and bony involvement. Furthermore, CT of the chest, abdomen, and pelvis as well as bone marrow biopsy are performed as part of the staging process. Lastly, fluorine-18 fluorodeoxyglucose positron emission tomography CT (18-FDG PET-CT) is often used to detect extranodal spread, define the extent of involvement, differentiate between lymphoma and inflammatory masses, and monitor for recurrence.11
Treatment for local ENKTL involves concurrent chemoradiotherapy followed by 3 cycles of etoposide, ifosfamide, cisplatin, and dexamethasone, which results in a complete response rate of 80%, and is the most favorable when comparing treatment modalities.12 Unfortunately, recurrence rates reach as high as 50%, whereas the 5-year survival rate is 59%.13,14 For recurrent or disseminated disease, high-dose chemotherapy and hematopoietic stem cell transplantation remain as alternative treatments for patients who have undergone 2 complete remissions and can be curative in some instances.13,15
Continue to: In summary, ENKTL is a rare form...
In summary, ENKTL is a rare form of NHL which classically presents in the nasal cavity; however, this type of lymphoma may present in a variety of extranodal sites.7,8 Despite the numerous published reports on ENKTL, no study has reported either primary or recurrent ENKTL in the feet or hands. To our knowledge, this is one of the first published cases of a patient who developed a rare and recurring ENKTL in the foot and ankle. The patient provided written informed consent for print and electronic publication of this case report.
CASE
A 59-year-old Caucasian woman was referred to the orthopedic foot and ankle clinic by her primary care physician for right medial ankle pain, skin ulceration, and numbness over the plantar aspect of her right foot. Upon questioning, the patient noted that the pain and numbness were present for almost 6 months. She denied trauma to the concerned area. Previously, the patient was observed and treated elsewhere for plantar fasciitis and was prescribed a brace before being immobilized in a controlled ankle motion (CAM) boot for 6 weeks. At follow-up with her outside provider, the patient had developed skin breakdown over the medial aspect of the right ankle, and this condition was presumed to be caused by the boot. After local wound care failed to improve her skin ulceration, she returned to her primary care physician, who ordered an MRI of the area and referred her to our specialty clinic.
Upon review, the patient’s past medical history included a diagnosis of nasal-type ENKTL. Her malignancy was treated with chemoradiotherapy 2 years prior to her consultation with the foot and ankle clinic.
The patient was noted by her medical oncologist and interventional radiologist to be in complete stage 4 remission since being treated. She underwent routine MRI and CT scans of the head and neck at 6-month intervals and FDG PET-CT scans at 3-month intervals, as per institutional protocol. The examinations showed no evidence of malignancy or metabolically active disease. The last imaging study occurred 2 months prior to admission to the foot and ankle clinic.
The patient consulted her medical oncologist 1 month prior to presenting to our clinic and was noted to exhibit an “excellent response to chemoradiotherapy” and “continues to remain disease free at 2 years.” She was instructed to continue routine follow-up. However, the office notes mentioned no ankle pain and non-healing wounds.
During physical examination, the patient presented an antalgic gait on the right side. Inspection demonstrated an increased circumference of the right ankle compared with the left, with a soft, palpable mass over the medial aspect of her right ankle. A 3 cm × 2 cm, grade 2 abrasion of the skin was observed over the medial mass just posterior to her medial malleolus. Range of motion was within normal limits. The patient exhibited a palpable posterior tibial artery pulse and full strength upon muscle testing of the lower extremities. She featured a positive Tinel’s sign and discomfort over the mass itself, with the pain radiating down to the plantar aspect of her foot and diffuse numbness over the plantar aspect of the foot.
Continue to: Review of her plain radiographs...
Review of her plain radiographs demonstrated no bony abnormalities, fractures, nor visible deformity (Figures 1A, 1B).
At presentation, our differential diagnosis included recurrence of the malignancy, secondary malignancy, infection, and inflammatory disease. After a lengthy discussion with the patient and consultation with our institution’s musculoskeletal oncologist, the decision was made to perform a right-ankle mass biopsy and marginal excision with wound irrigation and débridement and tarsal tunnel release.
The patient was placed in the supine position with standard prepping and draping. The medial eschar was excised in an elliptical fashion, and a curvilinear, longitudinal approach was performed within the compartment to access the mass along the posteromedial aspect of the ankle. Although no evidence of infection was observed, the tissue was thickened with areas of necrosis down to the flexor retinaculum. Once the flexor retinaculum was opened, a fibrous, plaque-like mass was observed, and it was encased with flexor tendons and neurovascular structures of the tarsal tunnel. After mass excision, a complete tarsal tunnel release was performed until the neurovascular bundle was free. Irrigation and débridement of the ulcer were performed along with complicated wound closure, and the patient was placed in a well-padded postoperative splint.
Pathology was finalized as a recurrent, EBV-positive, and nasal-type ENKTL. The patient underwent bone marrow biopsy, which yielded negative results. CT of the chest, abdomen, and pelvis were negative for the disease. FDG PET-CT, which included the extremities, was performed and demonstrated increased uptake in the right ankle, consistent with the malignancy (Figure 4).
DISCUSSION
ENKTL is an uncommon form of lymphoma and is exceedingly rare in Caucasian females.1-3 Although the patient’s primary occurrence was in the nasal cavity, recurrence in the foot and ankle must still be described.7,8 To our knowledge, this article is one of the first published cases of a patient who developed a rare-recurrence ENKTL about the foot and ankle. Occurrence in extremities is extremely rare that the staging protocol does not include FDG PET-CT of these areas. The patient’s “negative” scans led many providers to neglect the symptoms in her right ankle until the lesion had ulcerated through the skin. If one would have relied on imaging reports and outside records alone, the diagnosis would have been delayed longer or missed all together. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body FDG PET-CT when evaluating for recurrence in patients.
ABSTRACT
This case report is a rare form of lymphoma recurrence which presented as tarsal tunnel syndrome. The patient had been previously treated for the malignancy and was presumed to be in remission; however, standard radiology imaging protocols failed to include the distal extremities on these scans. The patient presented to the orthopedic clinic with tarsal tunnel symptoms and a mass in the tarsal tunnel. A complete evaluation resulted in a diagnosis of recurrence of the malignancy. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body fluorodeoxyglucose positron emission tomography computed tomography when evaluating for recurrence in patients.
Nasal-type, extranodal natural killer/T-cell lymphoma (ENKTL) is a rare form of non-Hodgkin lymphoma (NHL). Malignancies account for only 10% of NHL in Asian and South American populations. However, in Caucasians, it represents <1% of all cases. In addition, at 3:1 male to female ratio, the disease most commonly affects male patients who are 50 to 59 years old.1-3 The etiology of this malignancy is strongly related to prior infection with Epstein-Barr virus (EBV) as EBV-encoded early small ribonucleic acid on in situ hybridization of lymphoma cells is positive in 95% of cases.4-6
Typical sites of involvement include the nasal cavity, nasopharynx, and sinuses, causing patients to present with nasal obstruction, chronic sinusitis, or epistaxis. Additionally, ENKTL can occur primarily in the skin, gastrointestinal tract, spleen, and testis, whereas the bone marrow may be involved in 10% of cases. Although rare, unusual sites, including muscle, adrenals, and ovaries, have been published.7,8
Staging is best performed using the T-staging system, which accounts for the extent of local tumor involvement. Higher stages, such as T3 /T4, equate to locally advanced disease and imply a worse prognosis.9,10 Computed tomography (CT) and magnetic resonance imaging (MRI) help define local soft tissues and bony involvement. Furthermore, CT of the chest, abdomen, and pelvis as well as bone marrow biopsy are performed as part of the staging process. Lastly, fluorine-18 fluorodeoxyglucose positron emission tomography CT (18-FDG PET-CT) is often used to detect extranodal spread, define the extent of involvement, differentiate between lymphoma and inflammatory masses, and monitor for recurrence.11
Treatment for local ENKTL involves concurrent chemoradiotherapy followed by 3 cycles of etoposide, ifosfamide, cisplatin, and dexamethasone, which results in a complete response rate of 80%, and is the most favorable when comparing treatment modalities.12 Unfortunately, recurrence rates reach as high as 50%, whereas the 5-year survival rate is 59%.13,14 For recurrent or disseminated disease, high-dose chemotherapy and hematopoietic stem cell transplantation remain as alternative treatments for patients who have undergone 2 complete remissions and can be curative in some instances.13,15
Continue to: In summary, ENKTL is a rare form...
In summary, ENKTL is a rare form of NHL which classically presents in the nasal cavity; however, this type of lymphoma may present in a variety of extranodal sites.7,8 Despite the numerous published reports on ENKTL, no study has reported either primary or recurrent ENKTL in the feet or hands. To our knowledge, this is one of the first published cases of a patient who developed a rare and recurring ENKTL in the foot and ankle. The patient provided written informed consent for print and electronic publication of this case report.
CASE
A 59-year-old Caucasian woman was referred to the orthopedic foot and ankle clinic by her primary care physician for right medial ankle pain, skin ulceration, and numbness over the plantar aspect of her right foot. Upon questioning, the patient noted that the pain and numbness were present for almost 6 months. She denied trauma to the concerned area. Previously, the patient was observed and treated elsewhere for plantar fasciitis and was prescribed a brace before being immobilized in a controlled ankle motion (CAM) boot for 6 weeks. At follow-up with her outside provider, the patient had developed skin breakdown over the medial aspect of the right ankle, and this condition was presumed to be caused by the boot. After local wound care failed to improve her skin ulceration, she returned to her primary care physician, who ordered an MRI of the area and referred her to our specialty clinic.
Upon review, the patient’s past medical history included a diagnosis of nasal-type ENKTL. Her malignancy was treated with chemoradiotherapy 2 years prior to her consultation with the foot and ankle clinic.
The patient was noted by her medical oncologist and interventional radiologist to be in complete stage 4 remission since being treated. She underwent routine MRI and CT scans of the head and neck at 6-month intervals and FDG PET-CT scans at 3-month intervals, as per institutional protocol. The examinations showed no evidence of malignancy or metabolically active disease. The last imaging study occurred 2 months prior to admission to the foot and ankle clinic.
The patient consulted her medical oncologist 1 month prior to presenting to our clinic and was noted to exhibit an “excellent response to chemoradiotherapy” and “continues to remain disease free at 2 years.” She was instructed to continue routine follow-up. However, the office notes mentioned no ankle pain and non-healing wounds.
During physical examination, the patient presented an antalgic gait on the right side. Inspection demonstrated an increased circumference of the right ankle compared with the left, with a soft, palpable mass over the medial aspect of her right ankle. A 3 cm × 2 cm, grade 2 abrasion of the skin was observed over the medial mass just posterior to her medial malleolus. Range of motion was within normal limits. The patient exhibited a palpable posterior tibial artery pulse and full strength upon muscle testing of the lower extremities. She featured a positive Tinel’s sign and discomfort over the mass itself, with the pain radiating down to the plantar aspect of her foot and diffuse numbness over the plantar aspect of the foot.
Continue to: Review of her plain radiographs...
Review of her plain radiographs demonstrated no bony abnormalities, fractures, nor visible deformity (Figures 1A, 1B).
At presentation, our differential diagnosis included recurrence of the malignancy, secondary malignancy, infection, and inflammatory disease. After a lengthy discussion with the patient and consultation with our institution’s musculoskeletal oncologist, the decision was made to perform a right-ankle mass biopsy and marginal excision with wound irrigation and débridement and tarsal tunnel release.
The patient was placed in the supine position with standard prepping and draping. The medial eschar was excised in an elliptical fashion, and a curvilinear, longitudinal approach was performed within the compartment to access the mass along the posteromedial aspect of the ankle. Although no evidence of infection was observed, the tissue was thickened with areas of necrosis down to the flexor retinaculum. Once the flexor retinaculum was opened, a fibrous, plaque-like mass was observed, and it was encased with flexor tendons and neurovascular structures of the tarsal tunnel. After mass excision, a complete tarsal tunnel release was performed until the neurovascular bundle was free. Irrigation and débridement of the ulcer were performed along with complicated wound closure, and the patient was placed in a well-padded postoperative splint.
Pathology was finalized as a recurrent, EBV-positive, and nasal-type ENKTL. The patient underwent bone marrow biopsy, which yielded negative results. CT of the chest, abdomen, and pelvis were negative for the disease. FDG PET-CT, which included the extremities, was performed and demonstrated increased uptake in the right ankle, consistent with the malignancy (Figure 4).
DISCUSSION
ENKTL is an uncommon form of lymphoma and is exceedingly rare in Caucasian females.1-3 Although the patient’s primary occurrence was in the nasal cavity, recurrence in the foot and ankle must still be described.7,8 To our knowledge, this article is one of the first published cases of a patient who developed a rare-recurrence ENKTL about the foot and ankle. Occurrence in extremities is extremely rare that the staging protocol does not include FDG PET-CT of these areas. The patient’s “negative” scans led many providers to neglect the symptoms in her right ankle until the lesion had ulcerated through the skin. If one would have relied on imaging reports and outside records alone, the diagnosis would have been delayed longer or missed all together. This case illustrates the importance of a thorough medical history and personal review of imaging studies, and how a systematic approach can produce the correct diagnosis for any unknown lesion. Furthermore, this case may prompt oncologists to consider obtaining whole-body FDG PET-CT when evaluating for recurrence in patients.
1. Quintanilla-Martinez L, Kremer M, Keller G, et al. p53 mutations in nasal natural killer/T-cell lymphoma from Mexico: association with large cell morphology and advanced disease. Am J Pathol. 2001;159(6):2095-2105. doi:10.1016/S0002-9440(10)63061-1.
2. Au WY, Ma SY, Chim CS, et al. Clinicopathologic features and treatment outcome of mature T-cell and natural killer-cell lymphomas diagnosed according to the World Health Organization classification scheme: a single center experience of 10 years. Ann Oncol. 2005;16(2):206-214. doi:10.1093/annonc/mdi037.
3. Armitage JO. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood. 1997;89(11):3909-3918.
4. Medeiros LJ, Peiper SC, Elwood L, Yano T, Raffeld M, Jaffe ES. Angiocentric immunoproliferative lesions: a molecular analysis of eight cases. Hum Pathol. 1991;22(11):1150-1157. doi:10.1016/0046-8177(91)90269-U.
5. Ho FC, Srivastava G, Loke SL, et al. Presence of Epstein-Barr virus DNA in nasal lymphomas of B and ‘T’ cell type. Hematol Oncol. 1990;8(5):271-281. doi:10.1002/hon.2900080505.
6. Gelb AB, van de Rijn M, Regula DP Jr, et al. Epstein-Barr virus-associated natural killer-large granular lymphocyte leukemia. Hum Pathol. 1994;25(9):953-960. doi:10.1016/0046-8177(94)90018-3.
7. Petrella T, Delfau-Larue MH, Caillot D, et al. Nasopharyngeal lymphomas: further evidence for a natural killer cell origin. Hum Pathol. 1996;27(8):827-833. doi:10.1016/S0046-8177(96)90457-8.
8. Hasserjian RP, Harris NL. NK-cell lymphomas and leukemias: a spectrum of tumors with variable manifestations and immunophenotype. Am J Clin Pathol. 2007;127(6):860-868. doi:10.1309/2F39NX1AL3L54WU8.
9. Robbins KT, Fuller LM, Vlasak M. Primary lymphomas of the nasal cavity and paranasal sinuses. Cancer. 1985;56(4):814-819. doi:10.1002/1097-0142(19850815)56.
10. Ooi GC, Chim CS, Liang R, Tsang KW, Kwong YL. Nasal T-cell/natural killer cell lymphoma: CT and MR imaging features of a new clinicopathologic entity. Am J Roentgenol. 2000;174(4):1141-1145. doi:10.2214/ajr.174.4.1741141.
11. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621. doi:10.1007/s00277-008-0494-8.
12. Kim SJ, Kim K, Kim BS, et al. Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-cell lymphoma: consortium for improving survival of lymphoma study. J Clin Oncol. 2009;27(35):6027-6032. doi:10.1200/JCO.2009.23.8592.
13. Kwong YL. Natural killer-cell malignancies: diagnosis and treatment. Leukemia. 2005;19(12):2186-2194. doi:10.1038/sj.leu.2403955.
14. Liang R. Advances in the management and monitoring of extranodal NK/T-cell lymphoma, nasal type. Br J Haematol. 2009;147(1):13-21. doi:10.1111/j.1365-2141.2009.07802.x.
15. Yokoyama H, Yamamoto J, Tohmiya Y, et al. Allogeneic hematopoietic stem cell transplant following chemotherapy containing l-asparaginase as a promising treatment for patients with relapsed or refractory extranodal natural killer/T cell lymphoma, nasal type. Leuk Lymphoma. 2010;51(8):1509-1512. doi:10.3109/10428194.2010.487958.
1. Quintanilla-Martinez L, Kremer M, Keller G, et al. p53 mutations in nasal natural killer/T-cell lymphoma from Mexico: association with large cell morphology and advanced disease. Am J Pathol. 2001;159(6):2095-2105. doi:10.1016/S0002-9440(10)63061-1.
2. Au WY, Ma SY, Chim CS, et al. Clinicopathologic features and treatment outcome of mature T-cell and natural killer-cell lymphomas diagnosed according to the World Health Organization classification scheme: a single center experience of 10 years. Ann Oncol. 2005;16(2):206-214. doi:10.1093/annonc/mdi037.
3. Armitage JO. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood. 1997;89(11):3909-3918.
4. Medeiros LJ, Peiper SC, Elwood L, Yano T, Raffeld M, Jaffe ES. Angiocentric immunoproliferative lesions: a molecular analysis of eight cases. Hum Pathol. 1991;22(11):1150-1157. doi:10.1016/0046-8177(91)90269-U.
5. Ho FC, Srivastava G, Loke SL, et al. Presence of Epstein-Barr virus DNA in nasal lymphomas of B and ‘T’ cell type. Hematol Oncol. 1990;8(5):271-281. doi:10.1002/hon.2900080505.
6. Gelb AB, van de Rijn M, Regula DP Jr, et al. Epstein-Barr virus-associated natural killer-large granular lymphocyte leukemia. Hum Pathol. 1994;25(9):953-960. doi:10.1016/0046-8177(94)90018-3.
7. Petrella T, Delfau-Larue MH, Caillot D, et al. Nasopharyngeal lymphomas: further evidence for a natural killer cell origin. Hum Pathol. 1996;27(8):827-833. doi:10.1016/S0046-8177(96)90457-8.
8. Hasserjian RP, Harris NL. NK-cell lymphomas and leukemias: a spectrum of tumors with variable manifestations and immunophenotype. Am J Clin Pathol. 2007;127(6):860-868. doi:10.1309/2F39NX1AL3L54WU8.
9. Robbins KT, Fuller LM, Vlasak M. Primary lymphomas of the nasal cavity and paranasal sinuses. Cancer. 1985;56(4):814-819. doi:10.1002/1097-0142(19850815)56.
10. Ooi GC, Chim CS, Liang R, Tsang KW, Kwong YL. Nasal T-cell/natural killer cell lymphoma: CT and MR imaging features of a new clinicopathologic entity. Am J Roentgenol. 2000;174(4):1141-1145. doi:10.2214/ajr.174.4.1741141.
11. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621. doi:10.1007/s00277-008-0494-8.
12. Kim SJ, Kim K, Kim BS, et al. Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-cell lymphoma: consortium for improving survival of lymphoma study. J Clin Oncol. 2009;27(35):6027-6032. doi:10.1200/JCO.2009.23.8592.
13. Kwong YL. Natural killer-cell malignancies: diagnosis and treatment. Leukemia. 2005;19(12):2186-2194. doi:10.1038/sj.leu.2403955.
14. Liang R. Advances in the management and monitoring of extranodal NK/T-cell lymphoma, nasal type. Br J Haematol. 2009;147(1):13-21. doi:10.1111/j.1365-2141.2009.07802.x.
15. Yokoyama H, Yamamoto J, Tohmiya Y, et al. Allogeneic hematopoietic stem cell transplant following chemotherapy containing l-asparaginase as a promising treatment for patients with relapsed or refractory extranodal natural killer/T cell lymphoma, nasal type. Leuk Lymphoma. 2010;51(8):1509-1512. doi:10.3109/10428194.2010.487958.
TAKE-HOME POINTS
- A thorough review of systems, physical examination, and personal review of a patient’s advanced imaging is critical to avoid missed diagnosis or delays in diagnosis.
- Any mass lesion encountered in clinical practice, no matter how benign appearing, should be presumed malignant until proven otherwise.
- Fluorine-18 fluorodeoxyglucose positron emission tomography CT (18-FDG PET-CT) should include whole-body scans when evaluating patients for recurrence of malignancy.
Avulsion of the Anterior Lateral Meniscal Root Secondary to Tibial Eminence Fracture
ABSTRACT
The lateral tibial eminence shares a close relationship with the anterior root of the lateral meniscus. Limited studies have reported traumatic injury to the anterior meniscal roots in the setting of tibial eminence fractures, and reported rates of occurrence of concomitant meniscal and chondral injuries vary widely. The purpose of this article is to describe the case of a 28-year-old woman who had a complete avulsion of the anterolateral meniscal root caused by a tibial eminence fracture with resultant malunion and root displacement. The anterolateral meniscal root was anatomically repaired following arthroscopic resection of the malunited fragment.
The lateral tibial eminence is intimately associated with the root attachment of the anterior horn of the lateral meniscus.1-3 Previous studies have demonstrated both the close proximity of the anterior cruciate ligament (ACL) insertion to the meniscal roots and the potential for disruption in surgical interventions, such as tibial tunnel drilling in ACL reconstruction or placement of intramedullary tibial nails.4-6 The meniscal roots play a crucial role in force distribution, and disruption of these structures has been shown to significantly increase joint contact forces. Despite the deleterious effects of this injury, limited studies have reported on traumatic injury to the meniscal roots in the setting of tibial eminence fractures.
Reported rates of occurrence of concomitant meniscal and chondral injuries occurring with tibial eminence fractures vary widely, ranging from <5% to 40%.7,8 Although fractures to the tibial eminence are more common in children, an association between these injuries and concomitant soft tissue injuries, including meniscal, chondral, and collateral ligament injuries, in the adult population has been reported.7 Monto and Cameron-Donaldson8 used magnetic resonance imaging (MRI) to evaluate tibial eminence fractures in adults and found that 23% of study subjects had associated medial meniscus tears and 18% had lateral meniscus tears. In a similar study, Ishibashi and colleagues9 found that 25% of tibial eminence fractures were associated with lateral meniscus tears and 16% with medial meniscus tears.
These studies demonstrate the potential for meniscus injuries during tibial eminence fractures. However, the authors are unaware of any reports of complete tearing of the anterior horn of the lateral meniscus in association with this injury. This is an important injury to recognize and identify intraoperatively because an injury of this nature could potentially compromise the mechanical loading patterns and health of the articular cartilage of the lateral compartment of the knee. The purpose of this article is to describe a complete avulsion of the anterolateral meniscal root due to a tibial eminence fracture with resultant malunion and displacement of the root in a nonanatomical position. The patient provided written informed consent for print and electronic publication of this case report.
Continue to: A 28-year-old active woman...
CASE
A 28-year-old active woman presented to our clinic 22 months after sustaining a right knee tibial eminence fracture that was initially treated with extension immobilization, which resulted in a fibrous malunion. She subsequently sustained a second injury resulting in displacement of the malunion fracture fragment, and was treated at another institution 10 months prior to presentation at our clinic with arthroscopic reduction and internal fixation with a cannulated screw and washer of the tibial eminence fracture. This was followed by hardware removal 6 months prior to her office visit at our clinic. At presentation, she reported worsening right knee pain, mechanical symptoms, and loss of both flexion and extension compared with her uninjured knee. Conservative management, including activity modification, extensive physical therapy, and anti-inflammatory medication following her most recent procedure, had not resulted in improvement of her symptoms.
Physical examination revealed significantly reduced knee flexion and extension (+15°-120° on the affected side compared with 5° of hyperextension to 130° flexion of the contralateral knee). Ligamentous examination demonstrated no laxity with varus or valgus stress at 0° to 30° of flexion, negative posterior drawer, and a Grade 2 Lachman and positive pivot shift. She also exhibited pain with attempted right knee terminal extension. Radiographs and computed tomography scans were obtained and reviewed. They revealed a malunited tibial eminence fracture (Figures 1A-1D). 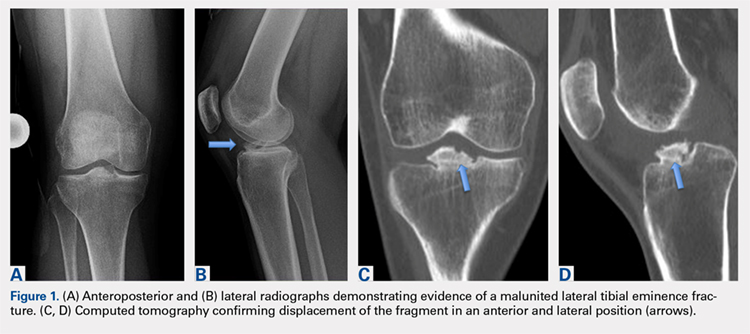
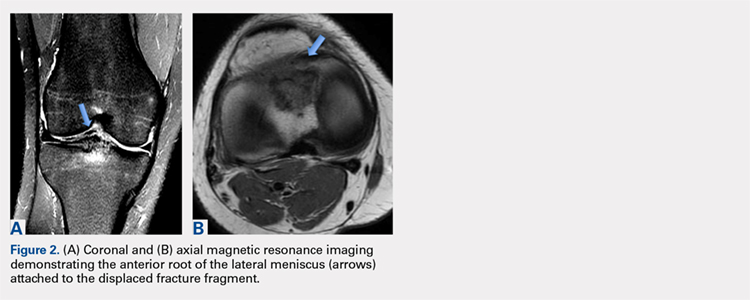
Arthroscopic assessment of the right knee demonstrated the large osseous fragment located in the anterolateral aspect of the joint with the displaced anterior horn of the lateral meniscus attached as well as significant anterior impingement limiting knee extension. Probing of the anterolateral meniscal root in the lateral compartment showed abundant surrounding scar tissue with an abnormal attachment, representing a chronic root avulsion. A mechanical shaver was used to débride the scar tissue and expose the malunited fragment, followed by complete osseous fragment excision with a high-speed burr (Figure 3). 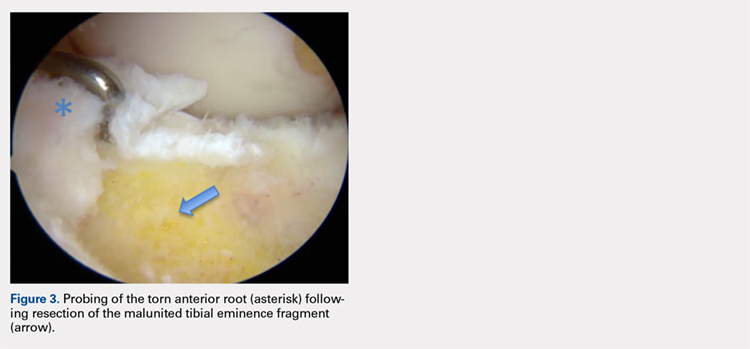
A soft tissue anterolateral meniscal root repair was performed by creating a 2-cm to 3-cm incision on the anterolateral tibia, just distal to the medial aspect of the Gerdy tubercle. To best restore the footprint of the repair and increase the potential for biologic healing, 2 transtibial tunnels were created at the location of the root attachment. An ACL aiming device with a cannulated sleeve was used to drill 2 bony tunnels approximately 5 mm apart, exiting at the anatomic root footprint. The drill pins were removed, leaving the 2 cannulas in place for later suture passage. A suture-passing device was used to pass 2 separate sutures through the detached meniscal root. 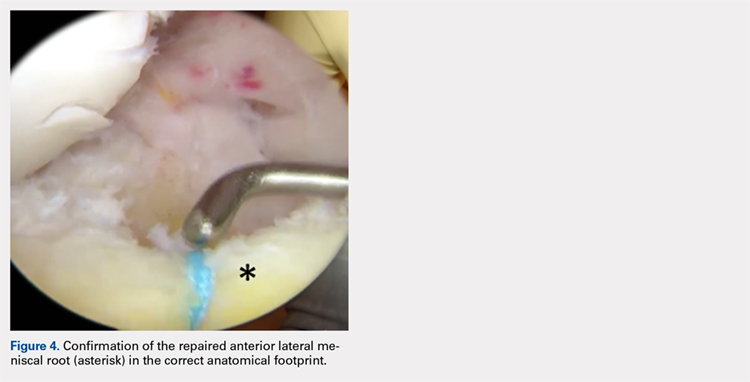
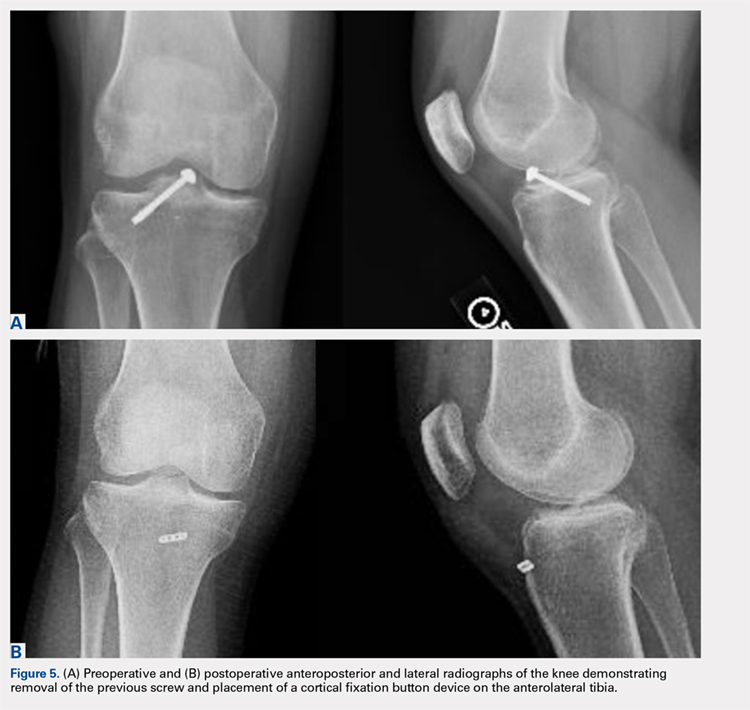
Continue to: Postoperatively, the patient was placed...
Postoperatively, the patient was placed on a non-weight-bearing protocol for her operative lower extremity for 6 weeks. A brace locked in extension was used for the same period of time (being removed only for physical therapy exercises). Enoxaparin was used for the first 2 weeks for deep vein thrombosis prophylaxis, followed by aspirin for an additional 4 weeks. Physical therapy was started on postoperative day 1 to begin working on early passive ROM exercises. Knee flexion was limited to 0° to 90° of flexion for the first 2 weeks and then progressed as tolerated.
DISCUSSION
This article describes a rare case of a patient with lateral meniscal anterior root avulsion in the setting of a tibial eminence fracture with subsequent malunion and root displacement. In a case such as this, delineation of the true extent of the injury is difficult because the anterior meniscal root can be torn, displaced, and nonanatomically scarred to surrounding soft tissues, making MRI interpretation challenging. Clinically, patients can present with a wide range of symptoms, including pain, mechanical symptoms, instability, and loss of knee motion.10
The anterior root of the lateral meniscus has been reported to be attached anterior to the lateral tibial eminence and adjacent to the insertion of the ACL. Fibrous connections extending from the anterior horn of the lateral meniscus attachment to the lateral tibial eminence are constant.11 Furumatsu and colleagues12 demonstrated the existence of dense fibers linking the anterior root of the lateral meniscus with the lateral aspect of the ACL tibial insertion. Acknowledging the close relationship of these structures is key to comprehending the importance of evaluating the anterior horn of the lateral meniscus in cases of tibial eminence fractures at the initial time of injury. Failure to diagnose this pathology can lead to poor clinical outcomes and early degenerative changes of the knee.
Tibial intercondylar eminence avulsion fractures are most likely to occur in children and adolescents, and are equivalent to an ACL tear in adults.13 When tibial eminence fractures occur in an older cohort, they are often combined with lesions of the menisci, capsule, or collateral ligaments.14 The initial injury in our patient demonstrated concomitant anterior root injury that progressed with time to nonanatomical healing of the root, leading to altered biomechanics. Surgical techniques available for meniscal root repair are broadly divided into transosseous suture repairs and suture anchor repairs.10 The transtibial pullout technique using 2 transtibial bone tunnels as described in this report is the senior author’s (RFL) preference because it provides a strong construct with minimal displacement of the repaired meniscus.15-17
This article describes a complete avulsion of the anterolateral meniscal root caused by a tibial eminence fracture with resultant malunion and displacement of the root in a nonanatomic position. Anterior meniscal root tears have been reported to result in altered biomechanics and force transmission across the knee, and therefore, anatomic repair of the anterior root is indicated.
1. James EW, LaPrade CM, Ellman MB, Wijdicks CA, Engebretsen L, LaPrade RF. Radiographic identification of the anterior and posterior root attachments of the medial and lateral menisci. Am J Sports Med. 2014;42(11):2707-2714. doi:10.1177/0363546514545863.
2. LaPrade CM, Foad A, Smith SD, et al. Biomechanical consequences of a nonanatomic posterior medial meniscal root repair. Am J Sports Med. 2015;43(4):912-920. doi:10.1177/0363546514566191.
3. LaPrade CM, James EW, Cram TR, Feagin JA, Engebretsen L, LaPrade RF. Meniscal root tears: a classification system based on tear morphology. Am J Sports Med. 2015;43(2):363-369. doi:10.1177/0363546514559684.
4. Ellman MB, James EW, LaPrade CM, LaPrade RF. Anterior meniscus root avulsion following intramedullary nailing for a tibial shaft fracture. Knee Surg Sports Traumatol Arthrosc. 2015;23(4):1188-1191. doi:10.1007/s00167-014-2941-5.
5. Padalecki JR, Jansson KS, Smith SD, et al. Biomechanical consequences of a complete radial tear adjacent to the medial meniscus posterior root attachment site: in situ pull-out repair restores derangement of joint mechanics. Am J Sports Med. 2014;42(3):699-707. doi:10.1177/0363546513499314.
6. LaPrade CM, Jisa KA, Cram TR, LaPrade RF. Posterior lateral meniscal root tear due to a malpositioned double-bundle anterior cruciate ligament reconstruction tibial tunnel. Knee Surg Sports Traumatol Arthrosc. 2015;23(12):3670-3673. doi:10.1007/s00167-014-3273-1.
7. Mitchell JJ, Sjostrom R, Mansour AA, et al. Incidence of meniscal injury and chondral pathology in anterior tibial spine fractures of children. J Pediatr Orthop. 2015;35(2):130-135. doi:10.1097/BPO.0000000000000249.
8. Monto RR, Cameron-Donaldson ML. Magnetic resonance imaging in the evaluation of tibial eminence fractures in adults. J Knee Surg. 2006;19(3):187-190.
9. Ishibashi Y, Tsuda E, Sasaki T, Toh S. Magnetic resonance imaging AIDS in detecting concomitant injuries in patients with tibial spine fractures. Clin Orthop Relat Res. 2005;(434):207-212.
10. Bhatia S, LaPrade CM, Ellman MB, LaPrade RF. Meniscal root tears significance, diagnosis, and treatment. Am J Sports Med. 2014;42(12):3016-3030. doi:10.1177/0363546514524162.
11. Ziegler CG, Pietrini SD, Westerhaus BD, et al. Arthroscopically pertinent landmarks for tunnel positioning in single-bundle and double-bundle anterior cruciate ligament reconstructions. Am J Sports Med. 2011;39(4):743-752. doi:10.1177/0363546510387511.
12. Furumatsu T, Kodama Y, Maehara A, et al. The anterior cruciate ligament-lateral meniscus complex: a histological study. Connect Tissue Res. 2016;57(2):91-98. doi:10.3109/03008207.2015.1081899.
13. Lubowitz JH, Grauer JD. Arthroscopic treatment of anterior cruciate ligament avulsion. Clin Orthop Rel Res. 1993;(294):242-246.
14. Falstie-Jensen S, Sondergard Petersen PE. Incarceration of the meniscus in fractures of the intercondylar eminence of the tibia in children. Injury. 1984;15(4):236-238.
15. LaPrade CM, LaPrade MD, Turnbull TL, Wijdicks CA, LaPrade RF. Biomechanical evaluation of the transtibial pull-out technique for posterior medial meniscal root repairs using 1 and 2 transtibial bone tunnels. Am J Sports Med. 2015;43(4):899-904. doi:10.1177/0363546514563278.
16. Menge TJ, Chahla J, Dean CS, Mitchell JJ, Moatshe G, LaPrade RF. Anterior meniscal root repair using a transtibial double-tunnel pullout technique. Arthrosc Tech. 2016;5(3):e679-e684. doi:10.1016/j.eats.2016.02.026.
17. Menge TJ, Dean CS, Chahla J, Mitchell JJ, LaPrade RF. Anterior horn meniscal repair using an outside-in suture technique. Arthrosc Tech. 2016;5(5):e1111-e1116. doi:10.1016/j.eats.2016.06.005.
ABSTRACT
The lateral tibial eminence shares a close relationship with the anterior root of the lateral meniscus. Limited studies have reported traumatic injury to the anterior meniscal roots in the setting of tibial eminence fractures, and reported rates of occurrence of concomitant meniscal and chondral injuries vary widely. The purpose of this article is to describe the case of a 28-year-old woman who had a complete avulsion of the anterolateral meniscal root caused by a tibial eminence fracture with resultant malunion and root displacement. The anterolateral meniscal root was anatomically repaired following arthroscopic resection of the malunited fragment.
The lateral tibial eminence is intimately associated with the root attachment of the anterior horn of the lateral meniscus.1-3 Previous studies have demonstrated both the close proximity of the anterior cruciate ligament (ACL) insertion to the meniscal roots and the potential for disruption in surgical interventions, such as tibial tunnel drilling in ACL reconstruction or placement of intramedullary tibial nails.4-6 The meniscal roots play a crucial role in force distribution, and disruption of these structures has been shown to significantly increase joint contact forces. Despite the deleterious effects of this injury, limited studies have reported on traumatic injury to the meniscal roots in the setting of tibial eminence fractures.
Reported rates of occurrence of concomitant meniscal and chondral injuries occurring with tibial eminence fractures vary widely, ranging from <5% to 40%.7,8 Although fractures to the tibial eminence are more common in children, an association between these injuries and concomitant soft tissue injuries, including meniscal, chondral, and collateral ligament injuries, in the adult population has been reported.7 Monto and Cameron-Donaldson8 used magnetic resonance imaging (MRI) to evaluate tibial eminence fractures in adults and found that 23% of study subjects had associated medial meniscus tears and 18% had lateral meniscus tears. In a similar study, Ishibashi and colleagues9 found that 25% of tibial eminence fractures were associated with lateral meniscus tears and 16% with medial meniscus tears.
These studies demonstrate the potential for meniscus injuries during tibial eminence fractures. However, the authors are unaware of any reports of complete tearing of the anterior horn of the lateral meniscus in association with this injury. This is an important injury to recognize and identify intraoperatively because an injury of this nature could potentially compromise the mechanical loading patterns and health of the articular cartilage of the lateral compartment of the knee. The purpose of this article is to describe a complete avulsion of the anterolateral meniscal root due to a tibial eminence fracture with resultant malunion and displacement of the root in a nonanatomical position. The patient provided written informed consent for print and electronic publication of this case report.
Continue to: A 28-year-old active woman...
CASE
A 28-year-old active woman presented to our clinic 22 months after sustaining a right knee tibial eminence fracture that was initially treated with extension immobilization, which resulted in a fibrous malunion. She subsequently sustained a second injury resulting in displacement of the malunion fracture fragment, and was treated at another institution 10 months prior to presentation at our clinic with arthroscopic reduction and internal fixation with a cannulated screw and washer of the tibial eminence fracture. This was followed by hardware removal 6 months prior to her office visit at our clinic. At presentation, she reported worsening right knee pain, mechanical symptoms, and loss of both flexion and extension compared with her uninjured knee. Conservative management, including activity modification, extensive physical therapy, and anti-inflammatory medication following her most recent procedure, had not resulted in improvement of her symptoms.
Physical examination revealed significantly reduced knee flexion and extension (+15°-120° on the affected side compared with 5° of hyperextension to 130° flexion of the contralateral knee). Ligamentous examination demonstrated no laxity with varus or valgus stress at 0° to 30° of flexion, negative posterior drawer, and a Grade 2 Lachman and positive pivot shift. She also exhibited pain with attempted right knee terminal extension. Radiographs and computed tomography scans were obtained and reviewed. They revealed a malunited tibial eminence fracture (Figures 1A-1D).
Arthroscopic assessment of the right knee demonstrated the large osseous fragment located in the anterolateral aspect of the joint with the displaced anterior horn of the lateral meniscus attached as well as significant anterior impingement limiting knee extension. Probing of the anterolateral meniscal root in the lateral compartment showed abundant surrounding scar tissue with an abnormal attachment, representing a chronic root avulsion. A mechanical shaver was used to débride the scar tissue and expose the malunited fragment, followed by complete osseous fragment excision with a high-speed burr (Figure 3).
A soft tissue anterolateral meniscal root repair was performed by creating a 2-cm to 3-cm incision on the anterolateral tibia, just distal to the medial aspect of the Gerdy tubercle. To best restore the footprint of the repair and increase the potential for biologic healing, 2 transtibial tunnels were created at the location of the root attachment. An ACL aiming device with a cannulated sleeve was used to drill 2 bony tunnels approximately 5 mm apart, exiting at the anatomic root footprint. The drill pins were removed, leaving the 2 cannulas in place for later suture passage. A suture-passing device was used to pass 2 separate sutures through the detached meniscal root.
Continue to: Postoperatively, the patient was placed...
Postoperatively, the patient was placed on a non-weight-bearing protocol for her operative lower extremity for 6 weeks. A brace locked in extension was used for the same period of time (being removed only for physical therapy exercises). Enoxaparin was used for the first 2 weeks for deep vein thrombosis prophylaxis, followed by aspirin for an additional 4 weeks. Physical therapy was started on postoperative day 1 to begin working on early passive ROM exercises. Knee flexion was limited to 0° to 90° of flexion for the first 2 weeks and then progressed as tolerated.
DISCUSSION
This article describes a rare case of a patient with lateral meniscal anterior root avulsion in the setting of a tibial eminence fracture with subsequent malunion and root displacement. In a case such as this, delineation of the true extent of the injury is difficult because the anterior meniscal root can be torn, displaced, and nonanatomically scarred to surrounding soft tissues, making MRI interpretation challenging. Clinically, patients can present with a wide range of symptoms, including pain, mechanical symptoms, instability, and loss of knee motion.10
The anterior root of the lateral meniscus has been reported to be attached anterior to the lateral tibial eminence and adjacent to the insertion of the ACL. Fibrous connections extending from the anterior horn of the lateral meniscus attachment to the lateral tibial eminence are constant.11 Furumatsu and colleagues12 demonstrated the existence of dense fibers linking the anterior root of the lateral meniscus with the lateral aspect of the ACL tibial insertion. Acknowledging the close relationship of these structures is key to comprehending the importance of evaluating the anterior horn of the lateral meniscus in cases of tibial eminence fractures at the initial time of injury. Failure to diagnose this pathology can lead to poor clinical outcomes and early degenerative changes of the knee.
Tibial intercondylar eminence avulsion fractures are most likely to occur in children and adolescents, and are equivalent to an ACL tear in adults.13 When tibial eminence fractures occur in an older cohort, they are often combined with lesions of the menisci, capsule, or collateral ligaments.14 The initial injury in our patient demonstrated concomitant anterior root injury that progressed with time to nonanatomical healing of the root, leading to altered biomechanics. Surgical techniques available for meniscal root repair are broadly divided into transosseous suture repairs and suture anchor repairs.10 The transtibial pullout technique using 2 transtibial bone tunnels as described in this report is the senior author’s (RFL) preference because it provides a strong construct with minimal displacement of the repaired meniscus.15-17
This article describes a complete avulsion of the anterolateral meniscal root caused by a tibial eminence fracture with resultant malunion and displacement of the root in a nonanatomic position. Anterior meniscal root tears have been reported to result in altered biomechanics and force transmission across the knee, and therefore, anatomic repair of the anterior root is indicated.
ABSTRACT
The lateral tibial eminence shares a close relationship with the anterior root of the lateral meniscus. Limited studies have reported traumatic injury to the anterior meniscal roots in the setting of tibial eminence fractures, and reported rates of occurrence of concomitant meniscal and chondral injuries vary widely. The purpose of this article is to describe the case of a 28-year-old woman who had a complete avulsion of the anterolateral meniscal root caused by a tibial eminence fracture with resultant malunion and root displacement. The anterolateral meniscal root was anatomically repaired following arthroscopic resection of the malunited fragment.
The lateral tibial eminence is intimately associated with the root attachment of the anterior horn of the lateral meniscus.1-3 Previous studies have demonstrated both the close proximity of the anterior cruciate ligament (ACL) insertion to the meniscal roots and the potential for disruption in surgical interventions, such as tibial tunnel drilling in ACL reconstruction or placement of intramedullary tibial nails.4-6 The meniscal roots play a crucial role in force distribution, and disruption of these structures has been shown to significantly increase joint contact forces. Despite the deleterious effects of this injury, limited studies have reported on traumatic injury to the meniscal roots in the setting of tibial eminence fractures.
Reported rates of occurrence of concomitant meniscal and chondral injuries occurring with tibial eminence fractures vary widely, ranging from <5% to 40%.7,8 Although fractures to the tibial eminence are more common in children, an association between these injuries and concomitant soft tissue injuries, including meniscal, chondral, and collateral ligament injuries, in the adult population has been reported.7 Monto and Cameron-Donaldson8 used magnetic resonance imaging (MRI) to evaluate tibial eminence fractures in adults and found that 23% of study subjects had associated medial meniscus tears and 18% had lateral meniscus tears. In a similar study, Ishibashi and colleagues9 found that 25% of tibial eminence fractures were associated with lateral meniscus tears and 16% with medial meniscus tears.
These studies demonstrate the potential for meniscus injuries during tibial eminence fractures. However, the authors are unaware of any reports of complete tearing of the anterior horn of the lateral meniscus in association with this injury. This is an important injury to recognize and identify intraoperatively because an injury of this nature could potentially compromise the mechanical loading patterns and health of the articular cartilage of the lateral compartment of the knee. The purpose of this article is to describe a complete avulsion of the anterolateral meniscal root due to a tibial eminence fracture with resultant malunion and displacement of the root in a nonanatomical position. The patient provided written informed consent for print and electronic publication of this case report.
Continue to: A 28-year-old active woman...
CASE
A 28-year-old active woman presented to our clinic 22 months after sustaining a right knee tibial eminence fracture that was initially treated with extension immobilization, which resulted in a fibrous malunion. She subsequently sustained a second injury resulting in displacement of the malunion fracture fragment, and was treated at another institution 10 months prior to presentation at our clinic with arthroscopic reduction and internal fixation with a cannulated screw and washer of the tibial eminence fracture. This was followed by hardware removal 6 months prior to her office visit at our clinic. At presentation, she reported worsening right knee pain, mechanical symptoms, and loss of both flexion and extension compared with her uninjured knee. Conservative management, including activity modification, extensive physical therapy, and anti-inflammatory medication following her most recent procedure, had not resulted in improvement of her symptoms.
Physical examination revealed significantly reduced knee flexion and extension (+15°-120° on the affected side compared with 5° of hyperextension to 130° flexion of the contralateral knee). Ligamentous examination demonstrated no laxity with varus or valgus stress at 0° to 30° of flexion, negative posterior drawer, and a Grade 2 Lachman and positive pivot shift. She also exhibited pain with attempted right knee terminal extension. Radiographs and computed tomography scans were obtained and reviewed. They revealed a malunited tibial eminence fracture (Figures 1A-1D).
Arthroscopic assessment of the right knee demonstrated the large osseous fragment located in the anterolateral aspect of the joint with the displaced anterior horn of the lateral meniscus attached as well as significant anterior impingement limiting knee extension. Probing of the anterolateral meniscal root in the lateral compartment showed abundant surrounding scar tissue with an abnormal attachment, representing a chronic root avulsion. A mechanical shaver was used to débride the scar tissue and expose the malunited fragment, followed by complete osseous fragment excision with a high-speed burr (Figure 3).
A soft tissue anterolateral meniscal root repair was performed by creating a 2-cm to 3-cm incision on the anterolateral tibia, just distal to the medial aspect of the Gerdy tubercle. To best restore the footprint of the repair and increase the potential for biologic healing, 2 transtibial tunnels were created at the location of the root attachment. An ACL aiming device with a cannulated sleeve was used to drill 2 bony tunnels approximately 5 mm apart, exiting at the anatomic root footprint. The drill pins were removed, leaving the 2 cannulas in place for later suture passage. A suture-passing device was used to pass 2 separate sutures through the detached meniscal root.
Continue to: Postoperatively, the patient was placed...
Postoperatively, the patient was placed on a non-weight-bearing protocol for her operative lower extremity for 6 weeks. A brace locked in extension was used for the same period of time (being removed only for physical therapy exercises). Enoxaparin was used for the first 2 weeks for deep vein thrombosis prophylaxis, followed by aspirin for an additional 4 weeks. Physical therapy was started on postoperative day 1 to begin working on early passive ROM exercises. Knee flexion was limited to 0° to 90° of flexion for the first 2 weeks and then progressed as tolerated.
DISCUSSION
This article describes a rare case of a patient with lateral meniscal anterior root avulsion in the setting of a tibial eminence fracture with subsequent malunion and root displacement. In a case such as this, delineation of the true extent of the injury is difficult because the anterior meniscal root can be torn, displaced, and nonanatomically scarred to surrounding soft tissues, making MRI interpretation challenging. Clinically, patients can present with a wide range of symptoms, including pain, mechanical symptoms, instability, and loss of knee motion.10
The anterior root of the lateral meniscus has been reported to be attached anterior to the lateral tibial eminence and adjacent to the insertion of the ACL. Fibrous connections extending from the anterior horn of the lateral meniscus attachment to the lateral tibial eminence are constant.11 Furumatsu and colleagues12 demonstrated the existence of dense fibers linking the anterior root of the lateral meniscus with the lateral aspect of the ACL tibial insertion. Acknowledging the close relationship of these structures is key to comprehending the importance of evaluating the anterior horn of the lateral meniscus in cases of tibial eminence fractures at the initial time of injury. Failure to diagnose this pathology can lead to poor clinical outcomes and early degenerative changes of the knee.
Tibial intercondylar eminence avulsion fractures are most likely to occur in children and adolescents, and are equivalent to an ACL tear in adults.13 When tibial eminence fractures occur in an older cohort, they are often combined with lesions of the menisci, capsule, or collateral ligaments.14 The initial injury in our patient demonstrated concomitant anterior root injury that progressed with time to nonanatomical healing of the root, leading to altered biomechanics. Surgical techniques available for meniscal root repair are broadly divided into transosseous suture repairs and suture anchor repairs.10 The transtibial pullout technique using 2 transtibial bone tunnels as described in this report is the senior author’s (RFL) preference because it provides a strong construct with minimal displacement of the repaired meniscus.15-17
This article describes a complete avulsion of the anterolateral meniscal root caused by a tibial eminence fracture with resultant malunion and displacement of the root in a nonanatomic position. Anterior meniscal root tears have been reported to result in altered biomechanics and force transmission across the knee, and therefore, anatomic repair of the anterior root is indicated.
1. James EW, LaPrade CM, Ellman MB, Wijdicks CA, Engebretsen L, LaPrade RF. Radiographic identification of the anterior and posterior root attachments of the medial and lateral menisci. Am J Sports Med. 2014;42(11):2707-2714. doi:10.1177/0363546514545863.
2. LaPrade CM, Foad A, Smith SD, et al. Biomechanical consequences of a nonanatomic posterior medial meniscal root repair. Am J Sports Med. 2015;43(4):912-920. doi:10.1177/0363546514566191.
3. LaPrade CM, James EW, Cram TR, Feagin JA, Engebretsen L, LaPrade RF. Meniscal root tears: a classification system based on tear morphology. Am J Sports Med. 2015;43(2):363-369. doi:10.1177/0363546514559684.
4. Ellman MB, James EW, LaPrade CM, LaPrade RF. Anterior meniscus root avulsion following intramedullary nailing for a tibial shaft fracture. Knee Surg Sports Traumatol Arthrosc. 2015;23(4):1188-1191. doi:10.1007/s00167-014-2941-5.
5. Padalecki JR, Jansson KS, Smith SD, et al. Biomechanical consequences of a complete radial tear adjacent to the medial meniscus posterior root attachment site: in situ pull-out repair restores derangement of joint mechanics. Am J Sports Med. 2014;42(3):699-707. doi:10.1177/0363546513499314.
6. LaPrade CM, Jisa KA, Cram TR, LaPrade RF. Posterior lateral meniscal root tear due to a malpositioned double-bundle anterior cruciate ligament reconstruction tibial tunnel. Knee Surg Sports Traumatol Arthrosc. 2015;23(12):3670-3673. doi:10.1007/s00167-014-3273-1.
7. Mitchell JJ, Sjostrom R, Mansour AA, et al. Incidence of meniscal injury and chondral pathology in anterior tibial spine fractures of children. J Pediatr Orthop. 2015;35(2):130-135. doi:10.1097/BPO.0000000000000249.
8. Monto RR, Cameron-Donaldson ML. Magnetic resonance imaging in the evaluation of tibial eminence fractures in adults. J Knee Surg. 2006;19(3):187-190.
9. Ishibashi Y, Tsuda E, Sasaki T, Toh S. Magnetic resonance imaging AIDS in detecting concomitant injuries in patients with tibial spine fractures. Clin Orthop Relat Res. 2005;(434):207-212.
10. Bhatia S, LaPrade CM, Ellman MB, LaPrade RF. Meniscal root tears significance, diagnosis, and treatment. Am J Sports Med. 2014;42(12):3016-3030. doi:10.1177/0363546514524162.
11. Ziegler CG, Pietrini SD, Westerhaus BD, et al. Arthroscopically pertinent landmarks for tunnel positioning in single-bundle and double-bundle anterior cruciate ligament reconstructions. Am J Sports Med. 2011;39(4):743-752. doi:10.1177/0363546510387511.
12. Furumatsu T, Kodama Y, Maehara A, et al. The anterior cruciate ligament-lateral meniscus complex: a histological study. Connect Tissue Res. 2016;57(2):91-98. doi:10.3109/03008207.2015.1081899.
13. Lubowitz JH, Grauer JD. Arthroscopic treatment of anterior cruciate ligament avulsion. Clin Orthop Rel Res. 1993;(294):242-246.
14. Falstie-Jensen S, Sondergard Petersen PE. Incarceration of the meniscus in fractures of the intercondylar eminence of the tibia in children. Injury. 1984;15(4):236-238.
15. LaPrade CM, LaPrade MD, Turnbull TL, Wijdicks CA, LaPrade RF. Biomechanical evaluation of the transtibial pull-out technique for posterior medial meniscal root repairs using 1 and 2 transtibial bone tunnels. Am J Sports Med. 2015;43(4):899-904. doi:10.1177/0363546514563278.
16. Menge TJ, Chahla J, Dean CS, Mitchell JJ, Moatshe G, LaPrade RF. Anterior meniscal root repair using a transtibial double-tunnel pullout technique. Arthrosc Tech. 2016;5(3):e679-e684. doi:10.1016/j.eats.2016.02.026.
17. Menge TJ, Dean CS, Chahla J, Mitchell JJ, LaPrade RF. Anterior horn meniscal repair using an outside-in suture technique. Arthrosc Tech. 2016;5(5):e1111-e1116. doi:10.1016/j.eats.2016.06.005.
1. James EW, LaPrade CM, Ellman MB, Wijdicks CA, Engebretsen L, LaPrade RF. Radiographic identification of the anterior and posterior root attachments of the medial and lateral menisci. Am J Sports Med. 2014;42(11):2707-2714. doi:10.1177/0363546514545863.
2. LaPrade CM, Foad A, Smith SD, et al. Biomechanical consequences of a nonanatomic posterior medial meniscal root repair. Am J Sports Med. 2015;43(4):912-920. doi:10.1177/0363546514566191.
3. LaPrade CM, James EW, Cram TR, Feagin JA, Engebretsen L, LaPrade RF. Meniscal root tears: a classification system based on tear morphology. Am J Sports Med. 2015;43(2):363-369. doi:10.1177/0363546514559684.
4. Ellman MB, James EW, LaPrade CM, LaPrade RF. Anterior meniscus root avulsion following intramedullary nailing for a tibial shaft fracture. Knee Surg Sports Traumatol Arthrosc. 2015;23(4):1188-1191. doi:10.1007/s00167-014-2941-5.
5. Padalecki JR, Jansson KS, Smith SD, et al. Biomechanical consequences of a complete radial tear adjacent to the medial meniscus posterior root attachment site: in situ pull-out repair restores derangement of joint mechanics. Am J Sports Med. 2014;42(3):699-707. doi:10.1177/0363546513499314.
6. LaPrade CM, Jisa KA, Cram TR, LaPrade RF. Posterior lateral meniscal root tear due to a malpositioned double-bundle anterior cruciate ligament reconstruction tibial tunnel. Knee Surg Sports Traumatol Arthrosc. 2015;23(12):3670-3673. doi:10.1007/s00167-014-3273-1.
7. Mitchell JJ, Sjostrom R, Mansour AA, et al. Incidence of meniscal injury and chondral pathology in anterior tibial spine fractures of children. J Pediatr Orthop. 2015;35(2):130-135. doi:10.1097/BPO.0000000000000249.
8. Monto RR, Cameron-Donaldson ML. Magnetic resonance imaging in the evaluation of tibial eminence fractures in adults. J Knee Surg. 2006;19(3):187-190.
9. Ishibashi Y, Tsuda E, Sasaki T, Toh S. Magnetic resonance imaging AIDS in detecting concomitant injuries in patients with tibial spine fractures. Clin Orthop Relat Res. 2005;(434):207-212.
10. Bhatia S, LaPrade CM, Ellman MB, LaPrade RF. Meniscal root tears significance, diagnosis, and treatment. Am J Sports Med. 2014;42(12):3016-3030. doi:10.1177/0363546514524162.
11. Ziegler CG, Pietrini SD, Westerhaus BD, et al. Arthroscopically pertinent landmarks for tunnel positioning in single-bundle and double-bundle anterior cruciate ligament reconstructions. Am J Sports Med. 2011;39(4):743-752. doi:10.1177/0363546510387511.
12. Furumatsu T, Kodama Y, Maehara A, et al. The anterior cruciate ligament-lateral meniscus complex: a histological study. Connect Tissue Res. 2016;57(2):91-98. doi:10.3109/03008207.2015.1081899.
13. Lubowitz JH, Grauer JD. Arthroscopic treatment of anterior cruciate ligament avulsion. Clin Orthop Rel Res. 1993;(294):242-246.
14. Falstie-Jensen S, Sondergard Petersen PE. Incarceration of the meniscus in fractures of the intercondylar eminence of the tibia in children. Injury. 1984;15(4):236-238.
15. LaPrade CM, LaPrade MD, Turnbull TL, Wijdicks CA, LaPrade RF. Biomechanical evaluation of the transtibial pull-out technique for posterior medial meniscal root repairs using 1 and 2 transtibial bone tunnels. Am J Sports Med. 2015;43(4):899-904. doi:10.1177/0363546514563278.
16. Menge TJ, Chahla J, Dean CS, Mitchell JJ, Moatshe G, LaPrade RF. Anterior meniscal root repair using a transtibial double-tunnel pullout technique. Arthrosc Tech. 2016;5(3):e679-e684. doi:10.1016/j.eats.2016.02.026.
17. Menge TJ, Dean CS, Chahla J, Mitchell JJ, LaPrade RF. Anterior horn meniscal repair using an outside-in suture technique. Arthrosc Tech. 2016;5(5):e1111-e1116. doi:10.1016/j.eats.2016.06.005.
TAKE-HOME POINTS
- Root tears of all meniscal attachments have been described. A comprehensive anatomic understanding of the meniscal roots is of utmost importance to suspect root lesions.
- A detailed physical examination along with imaging methods should be performed to make the correct diagnosis. In cases of evident injuries, such as a tibial spine fracture, additional soft tissue pathology should also be assessed.
- It is important to restore all torn root attachments to restore joint loading and contact areas. An anatomical root repair is needed to yield optimal results.
- Progressive rehabilitation with early ROM starting on postoperative day 1 can help avoid loss of knee motion and arthrofibrosis.
Gone but Not Forgotten: Acute Appendicitis Postappendectomy
Acute appendicitis is a common condition emergency physicians (EPs) encounter in the ED, and it is also one of the most common general surgeries.1Although stump appendicitis is a rare, long-term complication of appendectomy, it should always be included in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy. Delays in diagnosing stump appendicitis can lead to perforation, gangrene, and sepsis.2
Case
A 33-year-old previously healthy man, whose medical history was significant for an appendectomy 6 months earlier, presented to the ED with progressive and worsening right lower quadrant abdominal pain that radiated to his right testicle. The patient stated that the pain started 3 days prior while he was lifting a bale of hay. He further noted having a fever of 102oF, nausea, and vomiting hours prior to his arrival at the ED.
Upon presentation, the patient’s vital signs were: heart rate, 89 beats/min; respiratory rate, 17 breaths/min; blood pressure, 132/84 mm Hg; and temperature, 98.9°F. Oxygen saturation was 98% on room air. Physical examination revealed exquisite tenderness in the right lower quadrant and suprapubic region. The testicular examination and the remainder of the physical examination were normal. Laboratory evaluation included a complete blood count and urinalysis, the results of which were significant for an elevated white blood cell count of 17 x 109/Lmicroscopic hematuria, trace leukocyte esterase, and ketones.

A computed tomography (CT) scan of the abdomen and pelvis with intravenous (IV) and oral contrast demonstrated a phlegmonous process surrounding the surgical site, which was concerning for stump appendicitis. The terminal ilium and colon were noted to be normal (Figures 1 and 2).

The patient was started on IV fluids and IV antibiotics, and received Zosyn in the ED. Surgical service was consulted, and the patient was admitted to the hospital where he continued nonoperative treatment with IV ciprofloxacin and metronidazole. The patient was discharged home on hospital day 3 without further complication. A repeat CT scan was taken of the abdomen and pelvis 3 weeks after discharge, and demonstrated complete resolution of the inflammatory process at the appendiceal stump with chronic scarring.
Discussion
Approximately 7% of patients who present to the ED with abdominal pain are diagnosed with appendicitis.3 Although appendectomy is one of the most common surgical procedures, stump appendicitis is a rare postsurgical complication, with a reported incidence of 1 in 50,000 cases.4,5
Stump appendicitis is an acute inflammation of the residual appendicular stump; the incidence of stump perforation is approximately 60% to 70%.4,6 Thus, stump appendicitis has a high morbidity and complication rate. Unfortunately, though stump appendicitis is a condition in which timely diagnosis and intervention are essential to prevent morbidity, due to its rarity and low occurrence, there is often a delay in diagnosis. It is therefore important that EPs include stump appendicitis in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy.
Stump appendicitis was initially described by Rose et al in 1945.2 This condition is underreported, and the exact causes are still unclear.Of the reported cases of stump appendicitis, approximately 66% developed following an open surgical appendectomy;5 therefore, complicated surgery or difficult dissection of the appendix is considered a risk factor for stump appendicitis. Conversely, adequate visualization of the appendiceal base during appendectomy and a stump measuring less than 3 to 5 mm1,4 are associated with a lower risk for stump appendicitis.
Stump appendicitis can develop as early as a few days postappendectomy or as late as 50 years postappendectomy. Patients with stump appendicitis present with signs and symptoms similar to that of acute appendicitis.2,4,7 Diagnosis can be made through ultrasound or CT studies, though CT is the preferred modality due to its higher specificity and ability to exclude other causes of right-sided abdominal pain.4
Management
Surgical intervention to remove the appendiceal stump is typically the preferred treatment. However, as with our patient, cases of successful and uncomplicated medical management have been reported.1,2,4
Conclusion
While stump appendicitis is rare, there has been a rise in the number of reported cases over the past few years due to the increasing use and availability of CT.4 The diagnosis of stump appendicitis is time-critical to prevent associated complications of stump perforation, gangrene, and sepsis. It is therefore imperative that EPs consider this
1. Shah T, Gupta RK, Karkee RJ, Agarwal CS. Recurrent pain abdomen following appendectomy: stump appendicitis, a surgeon’s dilemma. Clin Case Rep. 2017;5(3):215-217. doi:10.1002/ccr3.781.
2. Giwa A, Reyes M. Three times a charm…a case of repeat appendicitis status post two prior appendectomies. Am J Emerg Med. 2018;36(3):528.e1-528.e2. doi:10.1016/j.ajem.2017.12.024.
3. Addiss DG, Shaffer N, Fowler BS, Tauxe RV. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol. 1990;132(5):910-925.
4. Hendahewa R, Shekhar A, Ratnayake S. The dilemma of stump appendicitis—a case report and literature review. Int J Surg. Case Rep. 2015;14:101-103. doi:10.1016/j.ijscr.2015.07.017.
5. Liang MK, Lo HG, Marks JL. Stump appendicitis: a comprehensive review of literature. Am Surg. 2006;72(2):162-166.
6. Parthsarathi R, Jankar SV, Chittawadgi B, et al. Laraposcopic management of symptomatic residual appendicular tip: a rare case report. J Minim Access Surg. 2017;13(2):154-156. doi:10.4103/0972-9941.199610.
7. Kanona H, Al Samaraee A, Nice C, Bhattacharya V. Stump appendicitis: a review. Int J Surg. 2012;10(9):425-428. doi:10.1016/j.ijsu.2012.07.007.
Acute appendicitis is a common condition emergency physicians (EPs) encounter in the ED, and it is also one of the most common general surgeries.1Although stump appendicitis is a rare, long-term complication of appendectomy, it should always be included in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy. Delays in diagnosing stump appendicitis can lead to perforation, gangrene, and sepsis.2
Case
A 33-year-old previously healthy man, whose medical history was significant for an appendectomy 6 months earlier, presented to the ED with progressive and worsening right lower quadrant abdominal pain that radiated to his right testicle. The patient stated that the pain started 3 days prior while he was lifting a bale of hay. He further noted having a fever of 102oF, nausea, and vomiting hours prior to his arrival at the ED.
Upon presentation, the patient’s vital signs were: heart rate, 89 beats/min; respiratory rate, 17 breaths/min; blood pressure, 132/84 mm Hg; and temperature, 98.9°F. Oxygen saturation was 98% on room air. Physical examination revealed exquisite tenderness in the right lower quadrant and suprapubic region. The testicular examination and the remainder of the physical examination were normal. Laboratory evaluation included a complete blood count and urinalysis, the results of which were significant for an elevated white blood cell count of 17 x 109/Lmicroscopic hematuria, trace leukocyte esterase, and ketones.
A computed tomography (CT) scan of the abdomen and pelvis with intravenous (IV) and oral contrast demonstrated a phlegmonous process surrounding the surgical site, which was concerning for stump appendicitis. The terminal ilium and colon were noted to be normal (Figures 1 and 2).
The patient was started on IV fluids and IV antibiotics, and received Zosyn in the ED. Surgical service was consulted, and the patient was admitted to the hospital where he continued nonoperative treatment with IV ciprofloxacin and metronidazole. The patient was discharged home on hospital day 3 without further complication. A repeat CT scan was taken of the abdomen and pelvis 3 weeks after discharge, and demonstrated complete resolution of the inflammatory process at the appendiceal stump with chronic scarring.
Discussion
Approximately 7% of patients who present to the ED with abdominal pain are diagnosed with appendicitis.3 Although appendectomy is one of the most common surgical procedures, stump appendicitis is a rare postsurgical complication, with a reported incidence of 1 in 50,000 cases.4,5
Stump appendicitis is an acute inflammation of the residual appendicular stump; the incidence of stump perforation is approximately 60% to 70%.4,6 Thus, stump appendicitis has a high morbidity and complication rate. Unfortunately, though stump appendicitis is a condition in which timely diagnosis and intervention are essential to prevent morbidity, due to its rarity and low occurrence, there is often a delay in diagnosis. It is therefore important that EPs include stump appendicitis in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy.
Stump appendicitis was initially described by Rose et al in 1945.2 This condition is underreported, and the exact causes are still unclear.Of the reported cases of stump appendicitis, approximately 66% developed following an open surgical appendectomy;5 therefore, complicated surgery or difficult dissection of the appendix is considered a risk factor for stump appendicitis. Conversely, adequate visualization of the appendiceal base during appendectomy and a stump measuring less than 3 to 5 mm1,4 are associated with a lower risk for stump appendicitis.
Stump appendicitis can develop as early as a few days postappendectomy or as late as 50 years postappendectomy. Patients with stump appendicitis present with signs and symptoms similar to that of acute appendicitis.2,4,7 Diagnosis can be made through ultrasound or CT studies, though CT is the preferred modality due to its higher specificity and ability to exclude other causes of right-sided abdominal pain.4
Management
Surgical intervention to remove the appendiceal stump is typically the preferred treatment. However, as with our patient, cases of successful and uncomplicated medical management have been reported.1,2,4
Conclusion
While stump appendicitis is rare, there has been a rise in the number of reported cases over the past few years due to the increasing use and availability of CT.4 The diagnosis of stump appendicitis is time-critical to prevent associated complications of stump perforation, gangrene, and sepsis. It is therefore imperative that EPs consider this
Acute appendicitis is a common condition emergency physicians (EPs) encounter in the ED, and it is also one of the most common general surgeries.1Although stump appendicitis is a rare, long-term complication of appendectomy, it should always be included in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy. Delays in diagnosing stump appendicitis can lead to perforation, gangrene, and sepsis.2
Case
A 33-year-old previously healthy man, whose medical history was significant for an appendectomy 6 months earlier, presented to the ED with progressive and worsening right lower quadrant abdominal pain that radiated to his right testicle. The patient stated that the pain started 3 days prior while he was lifting a bale of hay. He further noted having a fever of 102oF, nausea, and vomiting hours prior to his arrival at the ED.
Upon presentation, the patient’s vital signs were: heart rate, 89 beats/min; respiratory rate, 17 breaths/min; blood pressure, 132/84 mm Hg; and temperature, 98.9°F. Oxygen saturation was 98% on room air. Physical examination revealed exquisite tenderness in the right lower quadrant and suprapubic region. The testicular examination and the remainder of the physical examination were normal. Laboratory evaluation included a complete blood count and urinalysis, the results of which were significant for an elevated white blood cell count of 17 x 109/Lmicroscopic hematuria, trace leukocyte esterase, and ketones.
A computed tomography (CT) scan of the abdomen and pelvis with intravenous (IV) and oral contrast demonstrated a phlegmonous process surrounding the surgical site, which was concerning for stump appendicitis. The terminal ilium and colon were noted to be normal (Figures 1 and 2).
The patient was started on IV fluids and IV antibiotics, and received Zosyn in the ED. Surgical service was consulted, and the patient was admitted to the hospital where he continued nonoperative treatment with IV ciprofloxacin and metronidazole. The patient was discharged home on hospital day 3 without further complication. A repeat CT scan was taken of the abdomen and pelvis 3 weeks after discharge, and demonstrated complete resolution of the inflammatory process at the appendiceal stump with chronic scarring.
Discussion
Approximately 7% of patients who present to the ED with abdominal pain are diagnosed with appendicitis.3 Although appendectomy is one of the most common surgical procedures, stump appendicitis is a rare postsurgical complication, with a reported incidence of 1 in 50,000 cases.4,5
Stump appendicitis is an acute inflammation of the residual appendicular stump; the incidence of stump perforation is approximately 60% to 70%.4,6 Thus, stump appendicitis has a high morbidity and complication rate. Unfortunately, though stump appendicitis is a condition in which timely diagnosis and intervention are essential to prevent morbidity, due to its rarity and low occurrence, there is often a delay in diagnosis. It is therefore important that EPs include stump appendicitis in the differential diagnosis of patients presenting with right-sided abdominal pain and a history of appendectomy.
Stump appendicitis was initially described by Rose et al in 1945.2 This condition is underreported, and the exact causes are still unclear.Of the reported cases of stump appendicitis, approximately 66% developed following an open surgical appendectomy;5 therefore, complicated surgery or difficult dissection of the appendix is considered a risk factor for stump appendicitis. Conversely, adequate visualization of the appendiceal base during appendectomy and a stump measuring less than 3 to 5 mm1,4 are associated with a lower risk for stump appendicitis.
Stump appendicitis can develop as early as a few days postappendectomy or as late as 50 years postappendectomy. Patients with stump appendicitis present with signs and symptoms similar to that of acute appendicitis.2,4,7 Diagnosis can be made through ultrasound or CT studies, though CT is the preferred modality due to its higher specificity and ability to exclude other causes of right-sided abdominal pain.4
Management
Surgical intervention to remove the appendiceal stump is typically the preferred treatment. However, as with our patient, cases of successful and uncomplicated medical management have been reported.1,2,4
Conclusion
While stump appendicitis is rare, there has been a rise in the number of reported cases over the past few years due to the increasing use and availability of CT.4 The diagnosis of stump appendicitis is time-critical to prevent associated complications of stump perforation, gangrene, and sepsis. It is therefore imperative that EPs consider this
1. Shah T, Gupta RK, Karkee RJ, Agarwal CS. Recurrent pain abdomen following appendectomy: stump appendicitis, a surgeon’s dilemma. Clin Case Rep. 2017;5(3):215-217. doi:10.1002/ccr3.781.
2. Giwa A, Reyes M. Three times a charm…a case of repeat appendicitis status post two prior appendectomies. Am J Emerg Med. 2018;36(3):528.e1-528.e2. doi:10.1016/j.ajem.2017.12.024.
3. Addiss DG, Shaffer N, Fowler BS, Tauxe RV. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol. 1990;132(5):910-925.
4. Hendahewa R, Shekhar A, Ratnayake S. The dilemma of stump appendicitis—a case report and literature review. Int J Surg. Case Rep. 2015;14:101-103. doi:10.1016/j.ijscr.2015.07.017.
5. Liang MK, Lo HG, Marks JL. Stump appendicitis: a comprehensive review of literature. Am Surg. 2006;72(2):162-166.
6. Parthsarathi R, Jankar SV, Chittawadgi B, et al. Laraposcopic management of symptomatic residual appendicular tip: a rare case report. J Minim Access Surg. 2017;13(2):154-156. doi:10.4103/0972-9941.199610.
7. Kanona H, Al Samaraee A, Nice C, Bhattacharya V. Stump appendicitis: a review. Int J Surg. 2012;10(9):425-428. doi:10.1016/j.ijsu.2012.07.007.
1. Shah T, Gupta RK, Karkee RJ, Agarwal CS. Recurrent pain abdomen following appendectomy: stump appendicitis, a surgeon’s dilemma. Clin Case Rep. 2017;5(3):215-217. doi:10.1002/ccr3.781.
2. Giwa A, Reyes M. Three times a charm…a case of repeat appendicitis status post two prior appendectomies. Am J Emerg Med. 2018;36(3):528.e1-528.e2. doi:10.1016/j.ajem.2017.12.024.
3. Addiss DG, Shaffer N, Fowler BS, Tauxe RV. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol. 1990;132(5):910-925.
4. Hendahewa R, Shekhar A, Ratnayake S. The dilemma of stump appendicitis—a case report and literature review. Int J Surg. Case Rep. 2015;14:101-103. doi:10.1016/j.ijscr.2015.07.017.
5. Liang MK, Lo HG, Marks JL. Stump appendicitis: a comprehensive review of literature. Am Surg. 2006;72(2):162-166.
6. Parthsarathi R, Jankar SV, Chittawadgi B, et al. Laraposcopic management of symptomatic residual appendicular tip: a rare case report. J Minim Access Surg. 2017;13(2):154-156. doi:10.4103/0972-9941.199610.
7. Kanona H, Al Samaraee A, Nice C, Bhattacharya V. Stump appendicitis: a review. Int J Surg. 2012;10(9):425-428. doi:10.1016/j.ijsu.2012.07.007.
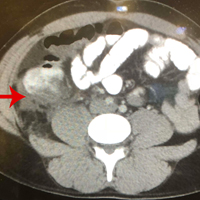
The Clinical Pathophysiology of Chronic Systemic Sclerosis
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF. 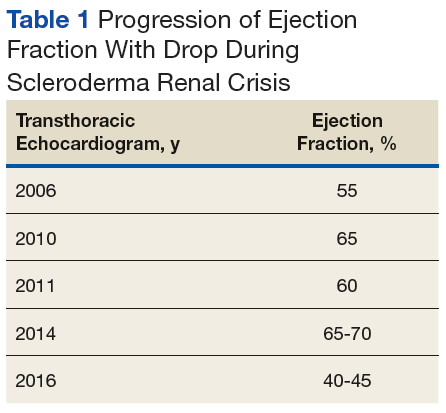
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1). 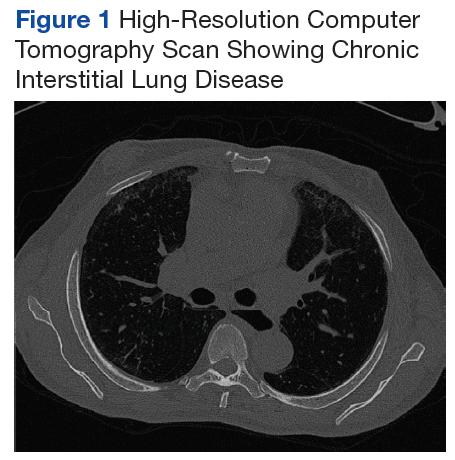
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc. 
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8 
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3). 
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4). 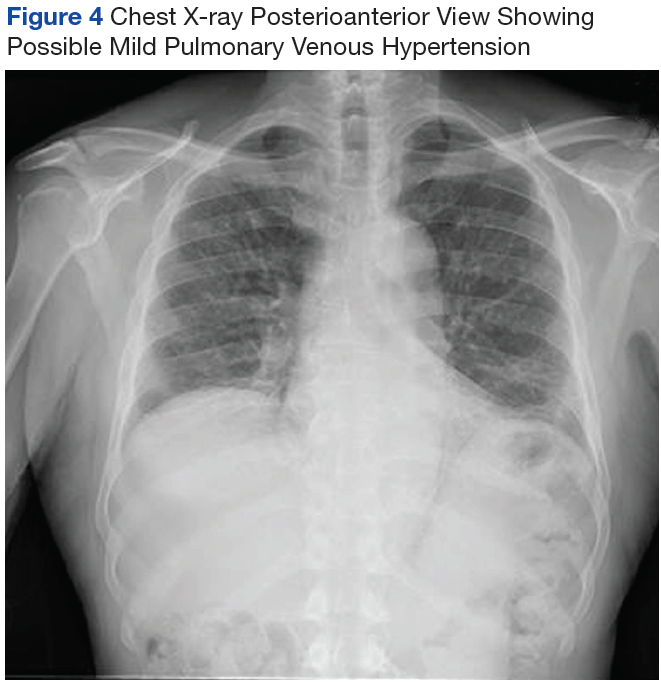
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10 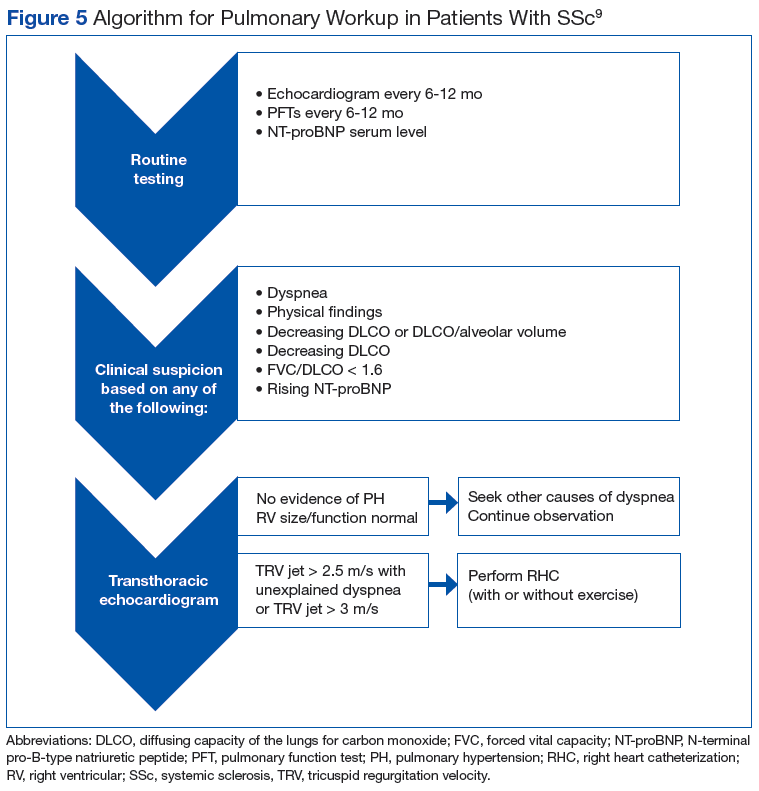
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF.
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1).
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF.
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1).
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
Metastatic Meningioma of the Scalp
Meningiomas generally present as slow-growing, expanding intracranial lesions and are the most common benign intracranial tumor in adults.1 Rarely, meningioma exhibits malignant potential and presents as an extracranial soft-tissue mass through extension or as a primary extracranial cutaneous neoplasm. The differential diagnosis of scalp neoplasms must be broadened to include uncommon tumors such as meningioma. We present a rare case of a 68-year-old woman with scalp metastasis of meningioma 11 years after initial resection of the primary tumor.
Case Report
A 68-year-old woman presented for evaluation of an asymptomatic nodule on the left parietal scalp of 2 years’ duration. She denied any headaches, difficulty with balance, vision changes, or changes in mentation. Her medical history was remarkable for a benign meningioma removed from the right parietal scalp 11 years prior without radiation therapy, as well as type 2 diabetes mellitus and arthritis. The patient’s son died from a brain tumor, but the exact tumor type and age at the time of death were unknown. Her current medications included metformin, insulin glargine, aspirin, and a daily multivitamin. She denied any allergies or history of smoking.
Physical examination of the scalp revealed 4 fixed, nontender, flesh-colored nodules: 2 on the left parietal scalp measuring 3.0 cm and 0.8 cm, respectively (Figure 1A); a 0.4-cm nodule on the right posterior occipital scalp; and a 1.6-cm sausage-shaped nodule on the right temple (Figure 1B). No positive lymph nodes were appreciated, and no additional lesions were noted. No additional atypical lesions were noted on full cutaneous examination.
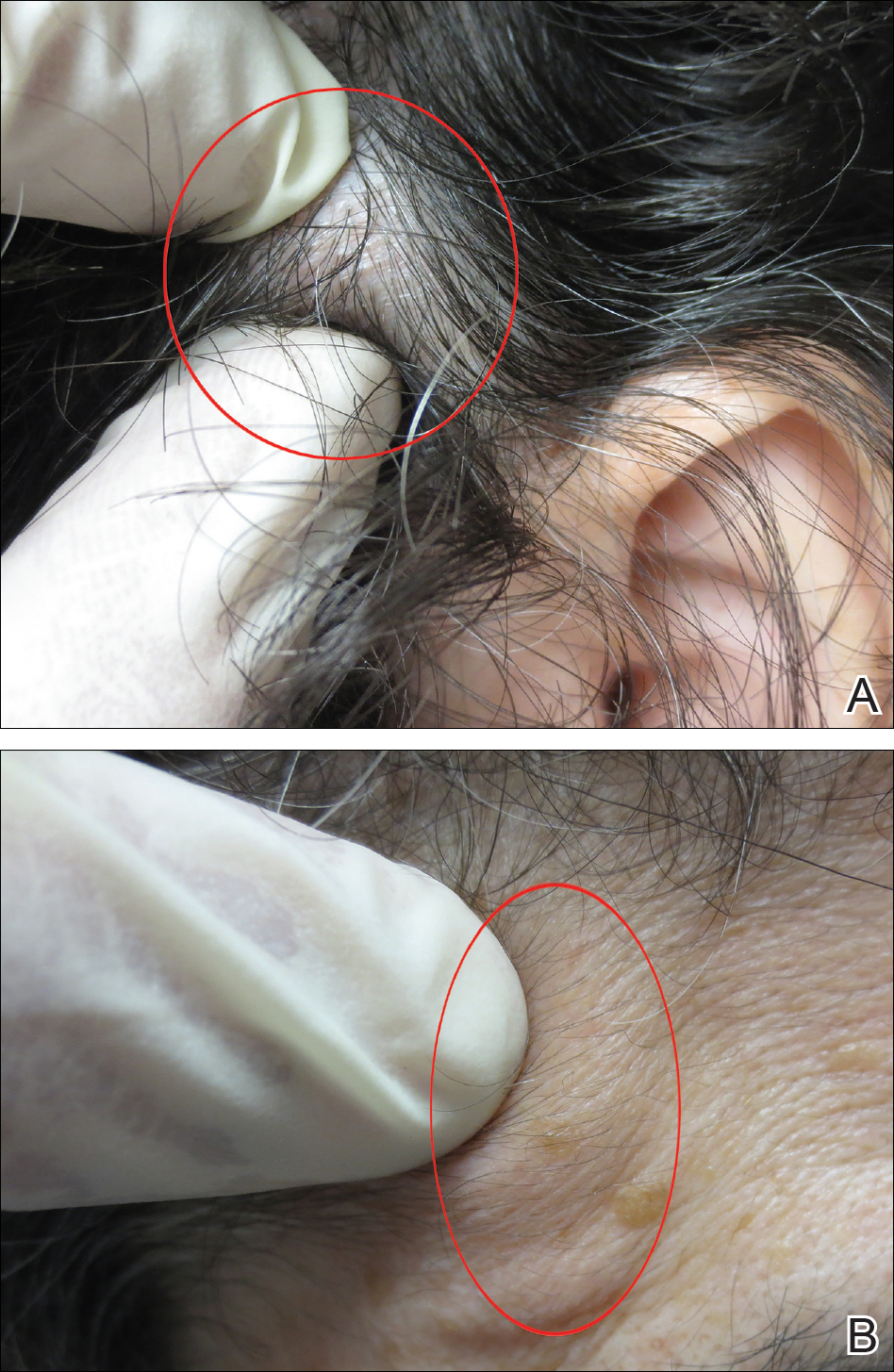
A diagnostic 6-mm punch biopsy of the largest nodule was performed. Intraoperatively, there was no apparent cyst wall, but coiled, loose, stringlike, pink-yellow tissue was removed from the base of the wound before closing with sutures.
The primary histologic finding was cells within fibrous tissue containing delicate round-oval nuclei, inconspicuous nucleoli, and lightly eosinophilic cytoplasm with an indistinct border (Figure 2). Immunohistochemical studies for S100 protein were focal and limited to the cytoplasm of a subset of neoplastic cells (Figure 3). Tumor cells stained positive for epithelial membrane antigen (EMA) and were focally positive for progesterone receptor (Figure 4). Tumor cells were negative for CD31 and CD34. Based on the clinical and histologic findings, a diagnosis of metastatic meningioma of the scalp was made.
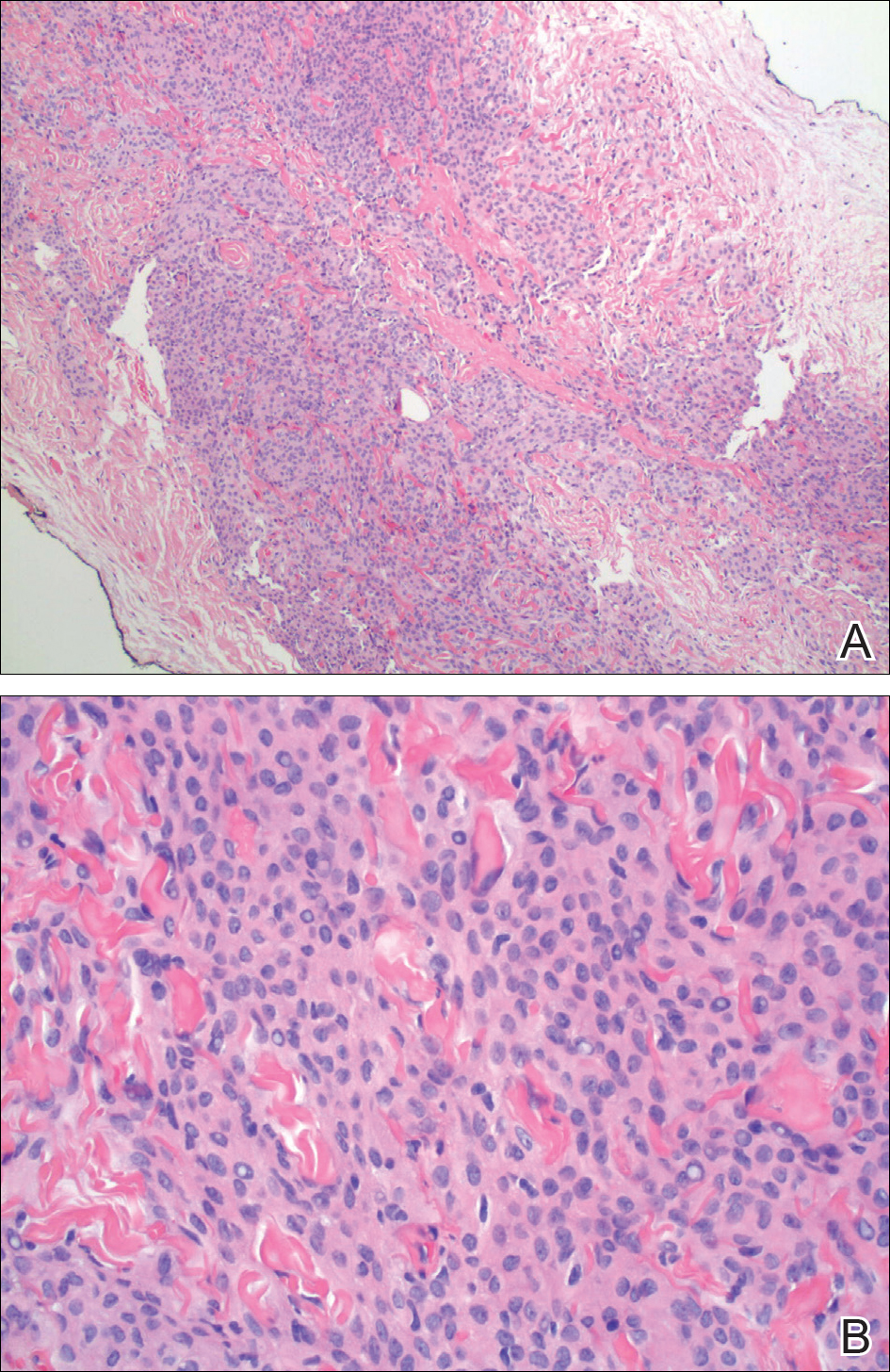
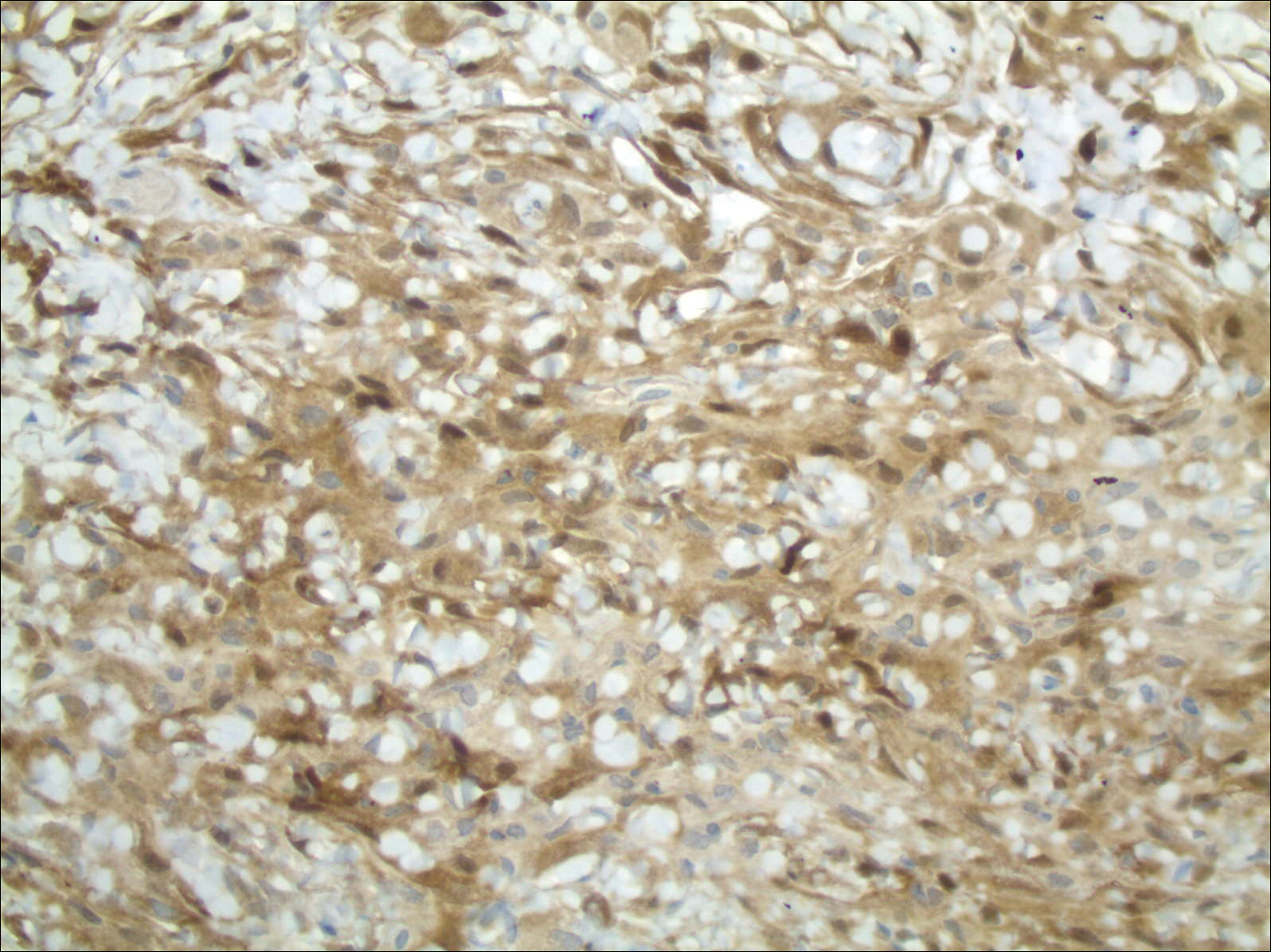
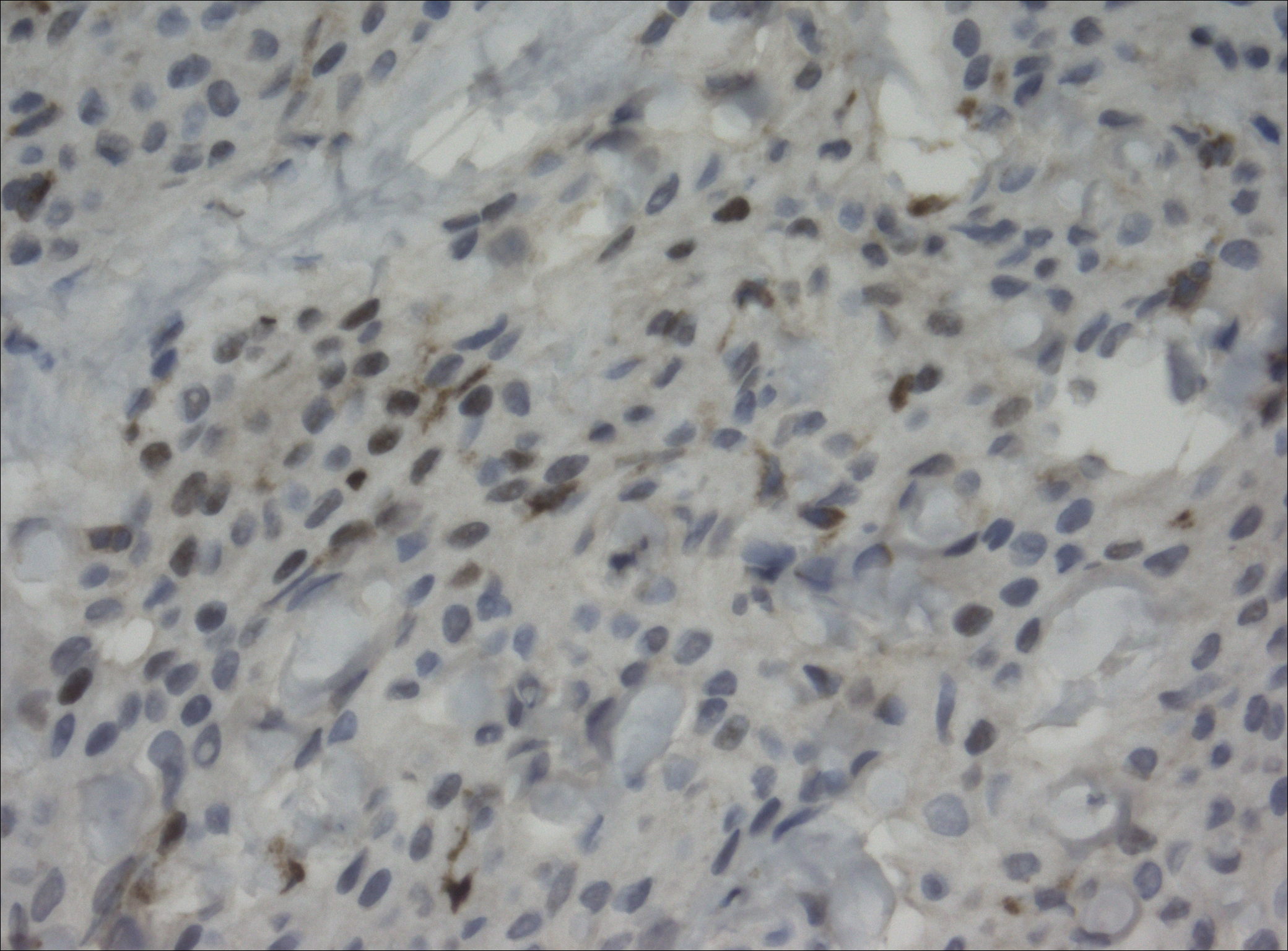
Magnetic resonance imaging and positron emission tomography of the head, neck, and chest demonstrated 3 residual subcutaneous nodules on the scalp and an indeterminate subcentimeter nodule in the right lung. The 0.4-cm nodule on the right posterior occipital scalp was removed without complication, and no radiation therapy was administered. The rest of the lesions were monitored. She remained under the close observation of a neurosurgeon and underwent repeat imaging of the scalp nodules and lungs, initially at 3 months and then routinely at the patient’s comfort. The patient currently denies any neurologic symptoms.
Comment
Meningiomas are derived from meningothelial cells found in the leptomeninges and in the choroid plexus of the ventricles of the brain.2 They are common intracranial neoplasms that generally are associated with a benign course and present during the fourth to sixth decades of life. Meningiomas constitute 13% to 30% of intracranial neoplasms and usually are female predominant (3:1).3,4 Rarely, malignant transformation can lead to local and distant metastasis to the lungs,5,6 liver,7 and skeletal system.8 In cases of metastatic spread, there is an increased incidence in males versus females.9-11
Risk Factors
Although many meningiomas are sporadic, numerous risk factors have been associated with the disease development. One study showed a link between exposure to ionizing radiation and subsequent development of meningioma.12 Another study found a population link between a higher incidence of meningioma and nuclear exposure in Hiroshima, Japan, after the atomic bomb blast in 1980.13 There is an increased incidence of meningioma in patients exposed to radiography from frequent dental imaging, particularly when older machines with higher levels of radiation exposure are used.14Another study demonstrated a correlation between meningioma and hormonal factors (eg, estrogen for hormone therapy) and exacerbation of symptoms during pregnancy.15 There also is an increased incidence of meningioma in breast cancer patients.4 Genetic alterations also have been implicated in the development of meningioma. It was found that 50% of patients with a mutation in the neurofibromatosis 2 gene (which codes for the merlin protein) had associated meningiomas.16,17 Scalp nodules in patients with neurofibromatosis type 2 increases suspicion of a scalp meningioma and necessitates biopsy.
Clinical Presentation
Cutaneous meningiomas typically present as firm, subcutaneous nodules. Scalp nodules ranging from alopecia18,19 to hypertrichosis20 have been reported. These neoplasms can be painless or painful, depending on mass effect and location.
Classification
The primary clinical classification system of metastatic meningioma was first described in 1974.21 Type 1 meningioma refers to congenital lesions that tend to cluster closer to the midline. Type 2 refers to ectopic soft-tissue lesions that extend to the skin from likely remnants of arachnoid cells. These lesions are more likely to be found around the eyes, ears, nose, and mouth. Type 3 meningiomas extend from intracranial tumors that secondarily involve the skin through proliferation through bone or anatomic defects. Type 3 is the result of direct extension and the location of the cutaneous presentation depends on the location of the intracranial lesion.4,22,23
Pathology
Meningiomas exhibit a range of morphologic appearances on histopathology. In almost all meningiomas, tumor cells are concentrically wrapped in tight whorls with round-oval nuclei and delicate chromatin, central clearing, and pale pseudonuclear inclusions. Lamellate calcifications known as psammoma bodies are a common finding. Immunohistochemical studies show that most meningiomas are positive for EMA, vimentin, and progesterone receptor. S100 protein expression, if present, usually is focal.
Differential Diagnosis
Asymptomatic nodules on the scalp may present a diagnostic challenge to physicians. Most common scalp lesions tend to be cystic or lipomatous. In children, a broad differential diagnosis should be considered, including dermoid and epidermoid tumors, dermal sinus tumors, hemangiomas, metastasis of another tumor, aplasia cutis congenita, pilomatricoma, and lipoma. In adults, the differential should focus on epidermoid cysts, lipomas, metastasis of other tumors, osteomas, arteriovenous fistulae, and heterotopic brain tissue. Often, microscopic examination is necessary, along with additional immunohistochemical staining (eg, EMA, vimentin).
Treatment
Treatment options for meningioma include observation, surgical resection, radiotherapy, and systemic therapy, as well as a combination of these modalities. The choice of therapy depends on such variables as patient age; performance status; comorbidities; presence or absence of symptoms (including focal neurologic deficits); and tumor location, size, and grade. It is important to note that there is limited knowledge looking at the results of various treatment modalities, and no consensus approach has been established.
Conclusion
Our patient’s medical history was remarkable for an intracranial meningioma 11 years prior to the current presentation, and she was found to have biopsy-proven metastatic meningioma without recurrence of the initial tumor. Patients presenting with a scalp nodule warrant a thorough medical history and consideration beyond common cysts and lipomas.
- Mackay B, Bruner JM, Luna MA. Malignant meningioma of the scalp. Ultrastruc Pathol. 1994;18:235-240.
- Whittle IR, Smith C, Navoo P, et al. Meningiomas. Lancet. 2004;363:1535-1543.
- Bauman G, Fisher B, Schild S, et al. Meningioma, ependymoma, and other adult brain tumors. In: Gunderson LL, Tepper JE, eds. Clinical Radiation Oncology. Philadelphia, PA: Elsevier Churchill Livingstone; 2007:539-566.
- Claus EB, Bondy ML, Schildkraut JM, et al. Epidemiology of intracranial meningioma. Neurosurgery. 2005;57:1088-1095.
- Tworek JA, Mikhail AA, Blaivas M. Meningioma: local recurrence and pulmonary metastasis diagnosed by fine needle aspiration. Acta Cytol. 1997;41:946-947.
- Shin MS, Holman WL, Herrera GA, et al. Extensive pulmonary metastasis of an intracranial meningioma with repeated recurrence: radiographic and pathologic features. South Med J. 1996;89:313-318.
- Ferguson JM, Flinn J. Intracranial meningioma with hepatic metastases and hypoglycaemia treated by selective hepatic arterial chemo-embolization. Australas Radiol.1995;39:97-99.
- Palmer JD, Cook PL, Ellison DW. Extracranial osseous metastases from intracranial meningioma. Br J Neurosurg. 1994;8:215-218.
- Glasauer FE, Yuan RH. Intracranial tumours with extracranial metastases. case report and review of the literature. J Neurosurg. 1963;20:474-493.
- Shuangshoti S, Hongsaprabhas C, Netsky MG. Metastasizing meningioma. Cancer. 1970;26:832-841.
- Ohta M, Iwaki T, Kitamoto T, et al. MIB-1 staining index and scoring of histological features in meningioma. Cancer. 1994;74:3176-3189.
- Wrensch M, Minn Y, Chew T, et al. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro Oncol. 2002;4:278-299.
- Shintani T, Hayakawa N, Hoshi M, et al. High incidence of meningioma among Hiroshima atomic bomb survivors. J Rad Res. 1999;40:49-57.
- Claus EB, Calvocoressi L, Bondy ML, et al. Dental x-rays and risk of meningioma. Cancer. 2012;118:4530-4537.
- Blitshteyn S, Crook JE, Jaeckle KA. Is there an association between meningioma and hormone replacement therapy? J Clin Oncol. 2008;26:279-282.
- Fontaine B, Rouleau GA, Seizinger BR, et al. Molecular genetics of neurofibromatosis 2 and related tumors (acoustic neuromas and meningioma). Ann N Y Acad Sci. 1991;615:338-343.
- Rabin BM, Meyer JR, Berlin JW, et al. Radiation-induced changes of the central nervous system and head and neck. Radiographics. 1996;16:1055-1072.
- Tanaka S, Okazaki M, Egusa G, et al. A case of pheochromocytoma associated with meningioma. J Intern Med. 1991;229:371-373.
- Zeikus P, Robinson-Bostom L, Stopa E. Primary cutaneous meningioma in association with a sinus pericranii. J Am Acad Dermatol. 2006;54(2 suppl):S49-S50.
- Junaid TA, Nkposong EO, Kolawole TM. Cutaneous meningiomas and an ovarian fibroma in a three-year-old girl. J Pathol. 1972;108:165-167.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningioma—a clinicopathologic study. Cancer. 1974;34:728-744.
- Shuangshoti S, Boonjunwetwat D, Kaoroptham S. Association of primary intraspinal meningiomas and subcutaneous meningioma of the cervical region: case report and review of literature. Surg Neurol. 1992;38:129-134.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol. 2012;136:208-211.
Meningiomas generally present as slow-growing, expanding intracranial lesions and are the most common benign intracranial tumor in adults.1 Rarely, meningioma exhibits malignant potential and presents as an extracranial soft-tissue mass through extension or as a primary extracranial cutaneous neoplasm. The differential diagnosis of scalp neoplasms must be broadened to include uncommon tumors such as meningioma. We present a rare case of a 68-year-old woman with scalp metastasis of meningioma 11 years after initial resection of the primary tumor.
Case Report
A 68-year-old woman presented for evaluation of an asymptomatic nodule on the left parietal scalp of 2 years’ duration. She denied any headaches, difficulty with balance, vision changes, or changes in mentation. Her medical history was remarkable for a benign meningioma removed from the right parietal scalp 11 years prior without radiation therapy, as well as type 2 diabetes mellitus and arthritis. The patient’s son died from a brain tumor, but the exact tumor type and age at the time of death were unknown. Her current medications included metformin, insulin glargine, aspirin, and a daily multivitamin. She denied any allergies or history of smoking.
Physical examination of the scalp revealed 4 fixed, nontender, flesh-colored nodules: 2 on the left parietal scalp measuring 3.0 cm and 0.8 cm, respectively (Figure 1A); a 0.4-cm nodule on the right posterior occipital scalp; and a 1.6-cm sausage-shaped nodule on the right temple (Figure 1B). No positive lymph nodes were appreciated, and no additional lesions were noted. No additional atypical lesions were noted on full cutaneous examination.
A diagnostic 6-mm punch biopsy of the largest nodule was performed. Intraoperatively, there was no apparent cyst wall, but coiled, loose, stringlike, pink-yellow tissue was removed from the base of the wound before closing with sutures.
The primary histologic finding was cells within fibrous tissue containing delicate round-oval nuclei, inconspicuous nucleoli, and lightly eosinophilic cytoplasm with an indistinct border (Figure 2). Immunohistochemical studies for S100 protein were focal and limited to the cytoplasm of a subset of neoplastic cells (Figure 3). Tumor cells stained positive for epithelial membrane antigen (EMA) and were focally positive for progesterone receptor (Figure 4). Tumor cells were negative for CD31 and CD34. Based on the clinical and histologic findings, a diagnosis of metastatic meningioma of the scalp was made.
Magnetic resonance imaging and positron emission tomography of the head, neck, and chest demonstrated 3 residual subcutaneous nodules on the scalp and an indeterminate subcentimeter nodule in the right lung. The 0.4-cm nodule on the right posterior occipital scalp was removed without complication, and no radiation therapy was administered. The rest of the lesions were monitored. She remained under the close observation of a neurosurgeon and underwent repeat imaging of the scalp nodules and lungs, initially at 3 months and then routinely at the patient’s comfort. The patient currently denies any neurologic symptoms.
Comment
Meningiomas are derived from meningothelial cells found in the leptomeninges and in the choroid plexus of the ventricles of the brain.2 They are common intracranial neoplasms that generally are associated with a benign course and present during the fourth to sixth decades of life. Meningiomas constitute 13% to 30% of intracranial neoplasms and usually are female predominant (3:1).3,4 Rarely, malignant transformation can lead to local and distant metastasis to the lungs,5,6 liver,7 and skeletal system.8 In cases of metastatic spread, there is an increased incidence in males versus females.9-11
Risk Factors
Although many meningiomas are sporadic, numerous risk factors have been associated with the disease development. One study showed a link between exposure to ionizing radiation and subsequent development of meningioma.12 Another study found a population link between a higher incidence of meningioma and nuclear exposure in Hiroshima, Japan, after the atomic bomb blast in 1980.13 There is an increased incidence of meningioma in patients exposed to radiography from frequent dental imaging, particularly when older machines with higher levels of radiation exposure are used.14Another study demonstrated a correlation between meningioma and hormonal factors (eg, estrogen for hormone therapy) and exacerbation of symptoms during pregnancy.15 There also is an increased incidence of meningioma in breast cancer patients.4 Genetic alterations also have been implicated in the development of meningioma. It was found that 50% of patients with a mutation in the neurofibromatosis 2 gene (which codes for the merlin protein) had associated meningiomas.16,17 Scalp nodules in patients with neurofibromatosis type 2 increases suspicion of a scalp meningioma and necessitates biopsy.
Clinical Presentation
Cutaneous meningiomas typically present as firm, subcutaneous nodules. Scalp nodules ranging from alopecia18,19 to hypertrichosis20 have been reported. These neoplasms can be painless or painful, depending on mass effect and location.
Classification
The primary clinical classification system of metastatic meningioma was first described in 1974.21 Type 1 meningioma refers to congenital lesions that tend to cluster closer to the midline. Type 2 refers to ectopic soft-tissue lesions that extend to the skin from likely remnants of arachnoid cells. These lesions are more likely to be found around the eyes, ears, nose, and mouth. Type 3 meningiomas extend from intracranial tumors that secondarily involve the skin through proliferation through bone or anatomic defects. Type 3 is the result of direct extension and the location of the cutaneous presentation depends on the location of the intracranial lesion.4,22,23
Pathology
Meningiomas exhibit a range of morphologic appearances on histopathology. In almost all meningiomas, tumor cells are concentrically wrapped in tight whorls with round-oval nuclei and delicate chromatin, central clearing, and pale pseudonuclear inclusions. Lamellate calcifications known as psammoma bodies are a common finding. Immunohistochemical studies show that most meningiomas are positive for EMA, vimentin, and progesterone receptor. S100 protein expression, if present, usually is focal.
Differential Diagnosis
Asymptomatic nodules on the scalp may present a diagnostic challenge to physicians. Most common scalp lesions tend to be cystic or lipomatous. In children, a broad differential diagnosis should be considered, including dermoid and epidermoid tumors, dermal sinus tumors, hemangiomas, metastasis of another tumor, aplasia cutis congenita, pilomatricoma, and lipoma. In adults, the differential should focus on epidermoid cysts, lipomas, metastasis of other tumors, osteomas, arteriovenous fistulae, and heterotopic brain tissue. Often, microscopic examination is necessary, along with additional immunohistochemical staining (eg, EMA, vimentin).
Treatment
Treatment options for meningioma include observation, surgical resection, radiotherapy, and systemic therapy, as well as a combination of these modalities. The choice of therapy depends on such variables as patient age; performance status; comorbidities; presence or absence of symptoms (including focal neurologic deficits); and tumor location, size, and grade. It is important to note that there is limited knowledge looking at the results of various treatment modalities, and no consensus approach has been established.
Conclusion
Our patient’s medical history was remarkable for an intracranial meningioma 11 years prior to the current presentation, and she was found to have biopsy-proven metastatic meningioma without recurrence of the initial tumor. Patients presenting with a scalp nodule warrant a thorough medical history and consideration beyond common cysts and lipomas.
Meningiomas generally present as slow-growing, expanding intracranial lesions and are the most common benign intracranial tumor in adults.1 Rarely, meningioma exhibits malignant potential and presents as an extracranial soft-tissue mass through extension or as a primary extracranial cutaneous neoplasm. The differential diagnosis of scalp neoplasms must be broadened to include uncommon tumors such as meningioma. We present a rare case of a 68-year-old woman with scalp metastasis of meningioma 11 years after initial resection of the primary tumor.
Case Report
A 68-year-old woman presented for evaluation of an asymptomatic nodule on the left parietal scalp of 2 years’ duration. She denied any headaches, difficulty with balance, vision changes, or changes in mentation. Her medical history was remarkable for a benign meningioma removed from the right parietal scalp 11 years prior without radiation therapy, as well as type 2 diabetes mellitus and arthritis. The patient’s son died from a brain tumor, but the exact tumor type and age at the time of death were unknown. Her current medications included metformin, insulin glargine, aspirin, and a daily multivitamin. She denied any allergies or history of smoking.
Physical examination of the scalp revealed 4 fixed, nontender, flesh-colored nodules: 2 on the left parietal scalp measuring 3.0 cm and 0.8 cm, respectively (Figure 1A); a 0.4-cm nodule on the right posterior occipital scalp; and a 1.6-cm sausage-shaped nodule on the right temple (Figure 1B). No positive lymph nodes were appreciated, and no additional lesions were noted. No additional atypical lesions were noted on full cutaneous examination.
A diagnostic 6-mm punch biopsy of the largest nodule was performed. Intraoperatively, there was no apparent cyst wall, but coiled, loose, stringlike, pink-yellow tissue was removed from the base of the wound before closing with sutures.
The primary histologic finding was cells within fibrous tissue containing delicate round-oval nuclei, inconspicuous nucleoli, and lightly eosinophilic cytoplasm with an indistinct border (Figure 2). Immunohistochemical studies for S100 protein were focal and limited to the cytoplasm of a subset of neoplastic cells (Figure 3). Tumor cells stained positive for epithelial membrane antigen (EMA) and were focally positive for progesterone receptor (Figure 4). Tumor cells were negative for CD31 and CD34. Based on the clinical and histologic findings, a diagnosis of metastatic meningioma of the scalp was made.
Magnetic resonance imaging and positron emission tomography of the head, neck, and chest demonstrated 3 residual subcutaneous nodules on the scalp and an indeterminate subcentimeter nodule in the right lung. The 0.4-cm nodule on the right posterior occipital scalp was removed without complication, and no radiation therapy was administered. The rest of the lesions were monitored. She remained under the close observation of a neurosurgeon and underwent repeat imaging of the scalp nodules and lungs, initially at 3 months and then routinely at the patient’s comfort. The patient currently denies any neurologic symptoms.
Comment
Meningiomas are derived from meningothelial cells found in the leptomeninges and in the choroid plexus of the ventricles of the brain.2 They are common intracranial neoplasms that generally are associated with a benign course and present during the fourth to sixth decades of life. Meningiomas constitute 13% to 30% of intracranial neoplasms and usually are female predominant (3:1).3,4 Rarely, malignant transformation can lead to local and distant metastasis to the lungs,5,6 liver,7 and skeletal system.8 In cases of metastatic spread, there is an increased incidence in males versus females.9-11
Risk Factors
Although many meningiomas are sporadic, numerous risk factors have been associated with the disease development. One study showed a link between exposure to ionizing radiation and subsequent development of meningioma.12 Another study found a population link between a higher incidence of meningioma and nuclear exposure in Hiroshima, Japan, after the atomic bomb blast in 1980.13 There is an increased incidence of meningioma in patients exposed to radiography from frequent dental imaging, particularly when older machines with higher levels of radiation exposure are used.14Another study demonstrated a correlation between meningioma and hormonal factors (eg, estrogen for hormone therapy) and exacerbation of symptoms during pregnancy.15 There also is an increased incidence of meningioma in breast cancer patients.4 Genetic alterations also have been implicated in the development of meningioma. It was found that 50% of patients with a mutation in the neurofibromatosis 2 gene (which codes for the merlin protein) had associated meningiomas.16,17 Scalp nodules in patients with neurofibromatosis type 2 increases suspicion of a scalp meningioma and necessitates biopsy.
Clinical Presentation
Cutaneous meningiomas typically present as firm, subcutaneous nodules. Scalp nodules ranging from alopecia18,19 to hypertrichosis20 have been reported. These neoplasms can be painless or painful, depending on mass effect and location.
Classification
The primary clinical classification system of metastatic meningioma was first described in 1974.21 Type 1 meningioma refers to congenital lesions that tend to cluster closer to the midline. Type 2 refers to ectopic soft-tissue lesions that extend to the skin from likely remnants of arachnoid cells. These lesions are more likely to be found around the eyes, ears, nose, and mouth. Type 3 meningiomas extend from intracranial tumors that secondarily involve the skin through proliferation through bone or anatomic defects. Type 3 is the result of direct extension and the location of the cutaneous presentation depends on the location of the intracranial lesion.4,22,23
Pathology
Meningiomas exhibit a range of morphologic appearances on histopathology. In almost all meningiomas, tumor cells are concentrically wrapped in tight whorls with round-oval nuclei and delicate chromatin, central clearing, and pale pseudonuclear inclusions. Lamellate calcifications known as psammoma bodies are a common finding. Immunohistochemical studies show that most meningiomas are positive for EMA, vimentin, and progesterone receptor. S100 protein expression, if present, usually is focal.
Differential Diagnosis
Asymptomatic nodules on the scalp may present a diagnostic challenge to physicians. Most common scalp lesions tend to be cystic or lipomatous. In children, a broad differential diagnosis should be considered, including dermoid and epidermoid tumors, dermal sinus tumors, hemangiomas, metastasis of another tumor, aplasia cutis congenita, pilomatricoma, and lipoma. In adults, the differential should focus on epidermoid cysts, lipomas, metastasis of other tumors, osteomas, arteriovenous fistulae, and heterotopic brain tissue. Often, microscopic examination is necessary, along with additional immunohistochemical staining (eg, EMA, vimentin).
Treatment
Treatment options for meningioma include observation, surgical resection, radiotherapy, and systemic therapy, as well as a combination of these modalities. The choice of therapy depends on such variables as patient age; performance status; comorbidities; presence or absence of symptoms (including focal neurologic deficits); and tumor location, size, and grade. It is important to note that there is limited knowledge looking at the results of various treatment modalities, and no consensus approach has been established.
Conclusion
Our patient’s medical history was remarkable for an intracranial meningioma 11 years prior to the current presentation, and she was found to have biopsy-proven metastatic meningioma without recurrence of the initial tumor. Patients presenting with a scalp nodule warrant a thorough medical history and consideration beyond common cysts and lipomas.
- Mackay B, Bruner JM, Luna MA. Malignant meningioma of the scalp. Ultrastruc Pathol. 1994;18:235-240.
- Whittle IR, Smith C, Navoo P, et al. Meningiomas. Lancet. 2004;363:1535-1543.
- Bauman G, Fisher B, Schild S, et al. Meningioma, ependymoma, and other adult brain tumors. In: Gunderson LL, Tepper JE, eds. Clinical Radiation Oncology. Philadelphia, PA: Elsevier Churchill Livingstone; 2007:539-566.
- Claus EB, Bondy ML, Schildkraut JM, et al. Epidemiology of intracranial meningioma. Neurosurgery. 2005;57:1088-1095.
- Tworek JA, Mikhail AA, Blaivas M. Meningioma: local recurrence and pulmonary metastasis diagnosed by fine needle aspiration. Acta Cytol. 1997;41:946-947.
- Shin MS, Holman WL, Herrera GA, et al. Extensive pulmonary metastasis of an intracranial meningioma with repeated recurrence: radiographic and pathologic features. South Med J. 1996;89:313-318.
- Ferguson JM, Flinn J. Intracranial meningioma with hepatic metastases and hypoglycaemia treated by selective hepatic arterial chemo-embolization. Australas Radiol.1995;39:97-99.
- Palmer JD, Cook PL, Ellison DW. Extracranial osseous metastases from intracranial meningioma. Br J Neurosurg. 1994;8:215-218.
- Glasauer FE, Yuan RH. Intracranial tumours with extracranial metastases. case report and review of the literature. J Neurosurg. 1963;20:474-493.
- Shuangshoti S, Hongsaprabhas C, Netsky MG. Metastasizing meningioma. Cancer. 1970;26:832-841.
- Ohta M, Iwaki T, Kitamoto T, et al. MIB-1 staining index and scoring of histological features in meningioma. Cancer. 1994;74:3176-3189.
- Wrensch M, Minn Y, Chew T, et al. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro Oncol. 2002;4:278-299.
- Shintani T, Hayakawa N, Hoshi M, et al. High incidence of meningioma among Hiroshima atomic bomb survivors. J Rad Res. 1999;40:49-57.
- Claus EB, Calvocoressi L, Bondy ML, et al. Dental x-rays and risk of meningioma. Cancer. 2012;118:4530-4537.
- Blitshteyn S, Crook JE, Jaeckle KA. Is there an association between meningioma and hormone replacement therapy? J Clin Oncol. 2008;26:279-282.
- Fontaine B, Rouleau GA, Seizinger BR, et al. Molecular genetics of neurofibromatosis 2 and related tumors (acoustic neuromas and meningioma). Ann N Y Acad Sci. 1991;615:338-343.
- Rabin BM, Meyer JR, Berlin JW, et al. Radiation-induced changes of the central nervous system and head and neck. Radiographics. 1996;16:1055-1072.
- Tanaka S, Okazaki M, Egusa G, et al. A case of pheochromocytoma associated with meningioma. J Intern Med. 1991;229:371-373.
- Zeikus P, Robinson-Bostom L, Stopa E. Primary cutaneous meningioma in association with a sinus pericranii. J Am Acad Dermatol. 2006;54(2 suppl):S49-S50.
- Junaid TA, Nkposong EO, Kolawole TM. Cutaneous meningiomas and an ovarian fibroma in a three-year-old girl. J Pathol. 1972;108:165-167.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningioma—a clinicopathologic study. Cancer. 1974;34:728-744.
- Shuangshoti S, Boonjunwetwat D, Kaoroptham S. Association of primary intraspinal meningiomas and subcutaneous meningioma of the cervical region: case report and review of literature. Surg Neurol. 1992;38:129-134.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol. 2012;136:208-211.
- Mackay B, Bruner JM, Luna MA. Malignant meningioma of the scalp. Ultrastruc Pathol. 1994;18:235-240.
- Whittle IR, Smith C, Navoo P, et al. Meningiomas. Lancet. 2004;363:1535-1543.
- Bauman G, Fisher B, Schild S, et al. Meningioma, ependymoma, and other adult brain tumors. In: Gunderson LL, Tepper JE, eds. Clinical Radiation Oncology. Philadelphia, PA: Elsevier Churchill Livingstone; 2007:539-566.
- Claus EB, Bondy ML, Schildkraut JM, et al. Epidemiology of intracranial meningioma. Neurosurgery. 2005;57:1088-1095.
- Tworek JA, Mikhail AA, Blaivas M. Meningioma: local recurrence and pulmonary metastasis diagnosed by fine needle aspiration. Acta Cytol. 1997;41:946-947.
- Shin MS, Holman WL, Herrera GA, et al. Extensive pulmonary metastasis of an intracranial meningioma with repeated recurrence: radiographic and pathologic features. South Med J. 1996;89:313-318.
- Ferguson JM, Flinn J. Intracranial meningioma with hepatic metastases and hypoglycaemia treated by selective hepatic arterial chemo-embolization. Australas Radiol.1995;39:97-99.
- Palmer JD, Cook PL, Ellison DW. Extracranial osseous metastases from intracranial meningioma. Br J Neurosurg. 1994;8:215-218.
- Glasauer FE, Yuan RH. Intracranial tumours with extracranial metastases. case report and review of the literature. J Neurosurg. 1963;20:474-493.
- Shuangshoti S, Hongsaprabhas C, Netsky MG. Metastasizing meningioma. Cancer. 1970;26:832-841.
- Ohta M, Iwaki T, Kitamoto T, et al. MIB-1 staining index and scoring of histological features in meningioma. Cancer. 1994;74:3176-3189.
- Wrensch M, Minn Y, Chew T, et al. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro Oncol. 2002;4:278-299.
- Shintani T, Hayakawa N, Hoshi M, et al. High incidence of meningioma among Hiroshima atomic bomb survivors. J Rad Res. 1999;40:49-57.
- Claus EB, Calvocoressi L, Bondy ML, et al. Dental x-rays and risk of meningioma. Cancer. 2012;118:4530-4537.
- Blitshteyn S, Crook JE, Jaeckle KA. Is there an association between meningioma and hormone replacement therapy? J Clin Oncol. 2008;26:279-282.
- Fontaine B, Rouleau GA, Seizinger BR, et al. Molecular genetics of neurofibromatosis 2 and related tumors (acoustic neuromas and meningioma). Ann N Y Acad Sci. 1991;615:338-343.
- Rabin BM, Meyer JR, Berlin JW, et al. Radiation-induced changes of the central nervous system and head and neck. Radiographics. 1996;16:1055-1072.
- Tanaka S, Okazaki M, Egusa G, et al. A case of pheochromocytoma associated with meningioma. J Intern Med. 1991;229:371-373.
- Zeikus P, Robinson-Bostom L, Stopa E. Primary cutaneous meningioma in association with a sinus pericranii. J Am Acad Dermatol. 2006;54(2 suppl):S49-S50.
- Junaid TA, Nkposong EO, Kolawole TM. Cutaneous meningiomas and an ovarian fibroma in a three-year-old girl. J Pathol. 1972;108:165-167.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningioma—a clinicopathologic study. Cancer. 1974;34:728-744.
- Shuangshoti S, Boonjunwetwat D, Kaoroptham S. Association of primary intraspinal meningiomas and subcutaneous meningioma of the cervical region: case report and review of literature. Surg Neurol. 1992;38:129-134.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol. 2012;136:208-211.
Squamoid Eccrine Ductal Carcinoma
Eccrine carcinomas are uncommon cutaneous neoplasms demonstrating nonuniform histologic features, behavior, and nomenclature. Given the rarity of these tumors, no known criteria by which to diagnose the tumor or guidelines for treatment have been proposed. We report a rare case of an immunocompromised patient with a primary squamoid eccrine ductal carcinoma (SEDC) who was subsequently treated with radical resection and axillary dissection. It was later determined that the patient had distant metastasis of SEDC. A review of the literature on the diagnosis, treatment, and surveillance of SEDC also is provided.
Case Report
A 77-year-old man whose medical history was remarkable for chronic lymphocytic leukemia (CLL) and numerous previous basal cell carcinomas and squamous cell carcinomas (SCCs) presented with a 5-cm, stellate, sclerotic plaque on the left chest of approximately 2 years’ duration (Figure 1) and a 3-mm pink papule on the right nasal sidewall of 2 months’ duration. Initial histology of both lesions revealed carcinoma with squamous and ductal differentiation extending from the undersurface of the epidermis, favoring a diagnosis of SEDC (Figure 2). At the time of initial presentation, the patient also had a 6-mm pink papule on the right chest of several months duration that was consistent with a well-differentiated sebaceous carcinoma on histology.
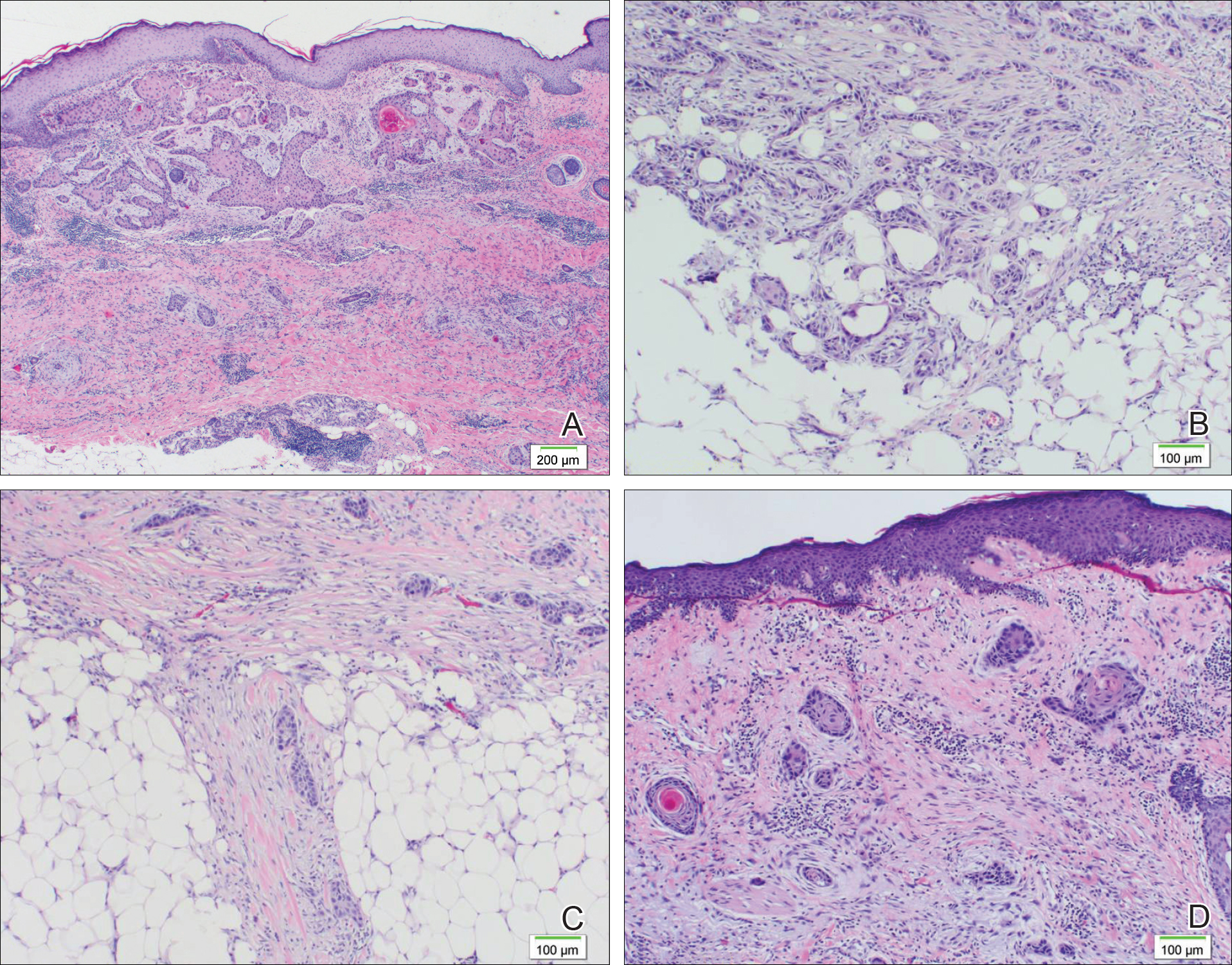
Further analysis of the lesion on the left chest revealed positive staining for cytokeratin (CK) 5/14 and p63, suggestive of a cutaneous malignancy. Staining for S100 protein highlighted rare cells in the basal layer of tumor aggregates. The immunohistochemical profile showed negative staining for CK7, CK5D3, epithelial membrane antigen (EMA), estrogen receptor, progesterone receptor, and human epidermal growth factor 2.
Diagnosis of SEDC of the chest and nasal lesions was based on the morphologic architecture, which included ductal formation noted within the tumor. The chest lesion also had prominent squamoid differentiation. Another histologic feature consistent with SEDC was poorly demarcated, infiltrative neoplastic cells extending into the dermis and subcutis. Although there was some positive focal staining for carcinoembryonic antigen (CEA), variegation within the tumor and the prominent squamoid component might have contributed to this unexpected staining pattern.
The patient was admitted to the hospital for excision of the lesion on the chest wall. Initial workup revealed macrocytic anemia, which required transfusion, and an incidental finding of non–small-cell lung cancer. The chest lesion was unrelated to the non–small-cell lung cancer based on the staining profile. Material from the lung stained positive for thyroid transcription factor 1 (TTF-1) and exhibited rare staining for p63; however, the chest lesion did not stain positive for TTF-1 and had strong staining affinity for p63, indicative of a cutaneous malignancy.
The lesion on the chest wall was definitively excised. Pathologic analysis revealed a dermal-based infiltrative tumor of irregular nests and cords of squamoid cells with focal ductal formation in a fibromyxoid background stroma, suggestive of an adnexal carcinoma with a considerable degree of squamous differentiation and favoring a diagnosis of SEDC. Focal perineural invasion was noted, but no lymphovascular spread was identified; however, metastasis was identified in 1 of 26 axillary lymph nodes. The patient underwent 9 sessions of radiation therapy for the lung cancer and also was given cetuximab.
Three months later, the nasal tumor was subsequently excised in an outpatient procedure, and the final biopsy report indicated a diagnosis of basal cell carcinoma. One-and-a-half years later, in follow-up with surgery after removal of the chest lesion, a 2×3-cm mass was excised from the left neck that demonstrated lymph nodes consistent with metastatic SEDC. Careful evaluation of this patient, including family history and genetic screening, was considered. Our patient continues to follow-up with the dermatology department every 3 months. He has been doing well and has had multiple additional primary SCCs in the subsequent 5 years of follow-up.
Comment
Eccrine carcinoma is the most common subtype of adnexal carcinoma, representing 0.01% of all cutaneous tumors.1 S
Eccrine carcinoma is observed clinically as a slow-growing, nodular plaque on the scalp, arms, legs, or trunk in middle-aged and elderly individuals.1 Squamoid eccrine ductal carcinoma also has been reported in a young woman.5 Another immunocompromised patient was identified in the literature with a great toe lesion that showed follicular differentiation along with the usual SEDC features of squamoid and ductal differentiation.6 The etiology of SEDC is controversial but is thought to be an SCC arising from eccrine glands, a subtype of eccrine carcinoma with extensive squamoid differentiation, or a biphenotypic carcinoma.1,7
Histologically, SEDC is poorly circumscribed with an infiltrative growth pattern and deep extension into the dermis and subcutaneous tissue. The lesion is characterized by prominent squamous epithelial proliferation superficially with cellular atypia, keratinous cyst formation, squamous eddies, and eccrine ductal differentiation.1
The differential diagnosis of SEDC includes SCC; metastatic carcinoma with squamoid features; and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma, and porocarcinoma with squamous differentiation.1
Immunohistochemistry has a role in the diagnosis of SEDC. Findings include positive staining for S100 protein, EMA, CKs, and CEA. Glandular tissue stains positive for EMA and CEA, supporting an adnexal origin.1 Positivity for p63 and CK5/6 supports the conclusion that this is a primary cutaneous malignancy, not a metastatic disease.1
Squamoid eccrine ductal carcinoma has an indeterminate malignant potential. There is a disparity of clinical behavior between SCC and eccrine cancers; however, because squamous differentiation sometimes dominates the histological picture, eccrine carcinomas can be misdiagnosed as SCC.1,8 Eccrine adnexal tumors are characterized by multiple local recurrences (70%–80% of cases); perineural invasion; and metastasis (50% of cases) to regional lymph nodes and viscera, including the lungs, liver, bones, and brain.1 Squamous cell carcinoma, however, has a markedly lower recurrence rate (3.1%–18.7% of cases) and rate of metastasis (5.2%–37.8%).1
Squamoid eccrine ductal carcinoma is classified as one of the less aggressive eccrine tumors, although the low number of cases makes it a controversial conclusion.1 To our knowledge, no cases of SEDC metastasis have been reported with SEDC. Recurrence of SEDC has been reported locally, and perineural or perivascular invasion (or both) has been demonstrated in 3 cases.1
Since SEDC has invasive and metastatic potential, as demonstrated in our case, along with elevated local recurrence rates, physicians must be able to properly diagnose this rare entity and recommend an appropriate surgical modality. Due to the low incidence of SEDC, there are no known randomized studies comparing treatment modalities.1 O
Surgical extirpation with complete margin examination is recommended, as SEDC tends to be underestimated in size, is aggressive in its infiltration, and is predisposed to perineural and perivascular invasion. T
Along with the rarity of SEDC in our patient, the simultaneous occurrence of 3 primary malignancies also is unusual. Patients with CLL have progressive defects of cell- and humoral-mediated immunity, causing immunosuppression. In a retrospective study, Tsimberidou et al9 reviewed the records of 2028 untreated CLL patients and determined that 27% had another primary malignancy, including skin (30%) and lung cancers (6%), which were two of the malignancies seen in our patient. The investigators concluded that patients with CLL have more than twice the risk of developing a second primary malignancy and an increased frequency of certain cancer types.9 Furthermore, treatment regimens for CLL have been considered to increase cell- and humoral-mediated immune defects at specific cancer sites,10 although the exact mechanism of this action is unknown. Development of a second primary malignancy (or even a third) in patients with SEDC is increasingly being reported in CLL patients.9,10
A high index of suspicion with SEDC in the differential diagnosis should be maintained in elderly men with slow-growing, solitary, nodular lesions of the scalp, nose, arms, legs, or trunk.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. Clin Aesthet Dermatol. 2013;6:33-36.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;916:799-802.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma)[published online April 15, 2015]. J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Kavand S, Cassarino DS. Squamoid eccrine ductal carcinoma: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Tsimberidou AM, Wen S, McLaughlin P, et al. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol. 2009;27:904-910.
- Dasanu CA, Alexandrescu DT. Risk for second nonlymphoid neoplasms in chronic lymphocytic leukemia. Med Gen Med. 2007;9:35.
Eccrine carcinomas are uncommon cutaneous neoplasms demonstrating nonuniform histologic features, behavior, and nomenclature. Given the rarity of these tumors, no known criteria by which to diagnose the tumor or guidelines for treatment have been proposed. We report a rare case of an immunocompromised patient with a primary squamoid eccrine ductal carcinoma (SEDC) who was subsequently treated with radical resection and axillary dissection. It was later determined that the patient had distant metastasis of SEDC. A review of the literature on the diagnosis, treatment, and surveillance of SEDC also is provided.
Case Report
A 77-year-old man whose medical history was remarkable for chronic lymphocytic leukemia (CLL) and numerous previous basal cell carcinomas and squamous cell carcinomas (SCCs) presented with a 5-cm, stellate, sclerotic plaque on the left chest of approximately 2 years’ duration (Figure 1) and a 3-mm pink papule on the right nasal sidewall of 2 months’ duration. Initial histology of both lesions revealed carcinoma with squamous and ductal differentiation extending from the undersurface of the epidermis, favoring a diagnosis of SEDC (Figure 2). At the time of initial presentation, the patient also had a 6-mm pink papule on the right chest of several months duration that was consistent with a well-differentiated sebaceous carcinoma on histology.
Further analysis of the lesion on the left chest revealed positive staining for cytokeratin (CK) 5/14 and p63, suggestive of a cutaneous malignancy. Staining for S100 protein highlighted rare cells in the basal layer of tumor aggregates. The immunohistochemical profile showed negative staining for CK7, CK5D3, epithelial membrane antigen (EMA), estrogen receptor, progesterone receptor, and human epidermal growth factor 2.
Diagnosis of SEDC of the chest and nasal lesions was based on the morphologic architecture, which included ductal formation noted within the tumor. The chest lesion also had prominent squamoid differentiation. Another histologic feature consistent with SEDC was poorly demarcated, infiltrative neoplastic cells extending into the dermis and subcutis. Although there was some positive focal staining for carcinoembryonic antigen (CEA), variegation within the tumor and the prominent squamoid component might have contributed to this unexpected staining pattern.
The patient was admitted to the hospital for excision of the lesion on the chest wall. Initial workup revealed macrocytic anemia, which required transfusion, and an incidental finding of non–small-cell lung cancer. The chest lesion was unrelated to the non–small-cell lung cancer based on the staining profile. Material from the lung stained positive for thyroid transcription factor 1 (TTF-1) and exhibited rare staining for p63; however, the chest lesion did not stain positive for TTF-1 and had strong staining affinity for p63, indicative of a cutaneous malignancy.
The lesion on the chest wall was definitively excised. Pathologic analysis revealed a dermal-based infiltrative tumor of irregular nests and cords of squamoid cells with focal ductal formation in a fibromyxoid background stroma, suggestive of an adnexal carcinoma with a considerable degree of squamous differentiation and favoring a diagnosis of SEDC. Focal perineural invasion was noted, but no lymphovascular spread was identified; however, metastasis was identified in 1 of 26 axillary lymph nodes. The patient underwent 9 sessions of radiation therapy for the lung cancer and also was given cetuximab.
Three months later, the nasal tumor was subsequently excised in an outpatient procedure, and the final biopsy report indicated a diagnosis of basal cell carcinoma. One-and-a-half years later, in follow-up with surgery after removal of the chest lesion, a 2×3-cm mass was excised from the left neck that demonstrated lymph nodes consistent with metastatic SEDC. Careful evaluation of this patient, including family history and genetic screening, was considered. Our patient continues to follow-up with the dermatology department every 3 months. He has been doing well and has had multiple additional primary SCCs in the subsequent 5 years of follow-up.
Comment
Eccrine carcinoma is the most common subtype of adnexal carcinoma, representing 0.01% of all cutaneous tumors.1 S
Eccrine carcinoma is observed clinically as a slow-growing, nodular plaque on the scalp, arms, legs, or trunk in middle-aged and elderly individuals.1 Squamoid eccrine ductal carcinoma also has been reported in a young woman.5 Another immunocompromised patient was identified in the literature with a great toe lesion that showed follicular differentiation along with the usual SEDC features of squamoid and ductal differentiation.6 The etiology of SEDC is controversial but is thought to be an SCC arising from eccrine glands, a subtype of eccrine carcinoma with extensive squamoid differentiation, or a biphenotypic carcinoma.1,7
Histologically, SEDC is poorly circumscribed with an infiltrative growth pattern and deep extension into the dermis and subcutaneous tissue. The lesion is characterized by prominent squamous epithelial proliferation superficially with cellular atypia, keratinous cyst formation, squamous eddies, and eccrine ductal differentiation.1
The differential diagnosis of SEDC includes SCC; metastatic carcinoma with squamoid features; and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma, and porocarcinoma with squamous differentiation.1
Immunohistochemistry has a role in the diagnosis of SEDC. Findings include positive staining for S100 protein, EMA, CKs, and CEA. Glandular tissue stains positive for EMA and CEA, supporting an adnexal origin.1 Positivity for p63 and CK5/6 supports the conclusion that this is a primary cutaneous malignancy, not a metastatic disease.1
Squamoid eccrine ductal carcinoma has an indeterminate malignant potential. There is a disparity of clinical behavior between SCC and eccrine cancers; however, because squamous differentiation sometimes dominates the histological picture, eccrine carcinomas can be misdiagnosed as SCC.1,8 Eccrine adnexal tumors are characterized by multiple local recurrences (70%–80% of cases); perineural invasion; and metastasis (50% of cases) to regional lymph nodes and viscera, including the lungs, liver, bones, and brain.1 Squamous cell carcinoma, however, has a markedly lower recurrence rate (3.1%–18.7% of cases) and rate of metastasis (5.2%–37.8%).1
Squamoid eccrine ductal carcinoma is classified as one of the less aggressive eccrine tumors, although the low number of cases makes it a controversial conclusion.1 To our knowledge, no cases of SEDC metastasis have been reported with SEDC. Recurrence of SEDC has been reported locally, and perineural or perivascular invasion (or both) has been demonstrated in 3 cases.1
Since SEDC has invasive and metastatic potential, as demonstrated in our case, along with elevated local recurrence rates, physicians must be able to properly diagnose this rare entity and recommend an appropriate surgical modality. Due to the low incidence of SEDC, there are no known randomized studies comparing treatment modalities.1 O
Surgical extirpation with complete margin examination is recommended, as SEDC tends to be underestimated in size, is aggressive in its infiltration, and is predisposed to perineural and perivascular invasion. T
Along with the rarity of SEDC in our patient, the simultaneous occurrence of 3 primary malignancies also is unusual. Patients with CLL have progressive defects of cell- and humoral-mediated immunity, causing immunosuppression. In a retrospective study, Tsimberidou et al9 reviewed the records of 2028 untreated CLL patients and determined that 27% had another primary malignancy, including skin (30%) and lung cancers (6%), which were two of the malignancies seen in our patient. The investigators concluded that patients with CLL have more than twice the risk of developing a second primary malignancy and an increased frequency of certain cancer types.9 Furthermore, treatment regimens for CLL have been considered to increase cell- and humoral-mediated immune defects at specific cancer sites,10 although the exact mechanism of this action is unknown. Development of a second primary malignancy (or even a third) in patients with SEDC is increasingly being reported in CLL patients.9,10
A high index of suspicion with SEDC in the differential diagnosis should be maintained in elderly men with slow-growing, solitary, nodular lesions of the scalp, nose, arms, legs, or trunk.
Eccrine carcinomas are uncommon cutaneous neoplasms demonstrating nonuniform histologic features, behavior, and nomenclature. Given the rarity of these tumors, no known criteria by which to diagnose the tumor or guidelines for treatment have been proposed. We report a rare case of an immunocompromised patient with a primary squamoid eccrine ductal carcinoma (SEDC) who was subsequently treated with radical resection and axillary dissection. It was later determined that the patient had distant metastasis of SEDC. A review of the literature on the diagnosis, treatment, and surveillance of SEDC also is provided.
Case Report
A 77-year-old man whose medical history was remarkable for chronic lymphocytic leukemia (CLL) and numerous previous basal cell carcinomas and squamous cell carcinomas (SCCs) presented with a 5-cm, stellate, sclerotic plaque on the left chest of approximately 2 years’ duration (Figure 1) and a 3-mm pink papule on the right nasal sidewall of 2 months’ duration. Initial histology of both lesions revealed carcinoma with squamous and ductal differentiation extending from the undersurface of the epidermis, favoring a diagnosis of SEDC (Figure 2). At the time of initial presentation, the patient also had a 6-mm pink papule on the right chest of several months duration that was consistent with a well-differentiated sebaceous carcinoma on histology.
Further analysis of the lesion on the left chest revealed positive staining for cytokeratin (CK) 5/14 and p63, suggestive of a cutaneous malignancy. Staining for S100 protein highlighted rare cells in the basal layer of tumor aggregates. The immunohistochemical profile showed negative staining for CK7, CK5D3, epithelial membrane antigen (EMA), estrogen receptor, progesterone receptor, and human epidermal growth factor 2.
Diagnosis of SEDC of the chest and nasal lesions was based on the morphologic architecture, which included ductal formation noted within the tumor. The chest lesion also had prominent squamoid differentiation. Another histologic feature consistent with SEDC was poorly demarcated, infiltrative neoplastic cells extending into the dermis and subcutis. Although there was some positive focal staining for carcinoembryonic antigen (CEA), variegation within the tumor and the prominent squamoid component might have contributed to this unexpected staining pattern.
The patient was admitted to the hospital for excision of the lesion on the chest wall. Initial workup revealed macrocytic anemia, which required transfusion, and an incidental finding of non–small-cell lung cancer. The chest lesion was unrelated to the non–small-cell lung cancer based on the staining profile. Material from the lung stained positive for thyroid transcription factor 1 (TTF-1) and exhibited rare staining for p63; however, the chest lesion did not stain positive for TTF-1 and had strong staining affinity for p63, indicative of a cutaneous malignancy.
The lesion on the chest wall was definitively excised. Pathologic analysis revealed a dermal-based infiltrative tumor of irregular nests and cords of squamoid cells with focal ductal formation in a fibromyxoid background stroma, suggestive of an adnexal carcinoma with a considerable degree of squamous differentiation and favoring a diagnosis of SEDC. Focal perineural invasion was noted, but no lymphovascular spread was identified; however, metastasis was identified in 1 of 26 axillary lymph nodes. The patient underwent 9 sessions of radiation therapy for the lung cancer and also was given cetuximab.
Three months later, the nasal tumor was subsequently excised in an outpatient procedure, and the final biopsy report indicated a diagnosis of basal cell carcinoma. One-and-a-half years later, in follow-up with surgery after removal of the chest lesion, a 2×3-cm mass was excised from the left neck that demonstrated lymph nodes consistent with metastatic SEDC. Careful evaluation of this patient, including family history and genetic screening, was considered. Our patient continues to follow-up with the dermatology department every 3 months. He has been doing well and has had multiple additional primary SCCs in the subsequent 5 years of follow-up.
Comment
Eccrine carcinoma is the most common subtype of adnexal carcinoma, representing 0.01% of all cutaneous tumors.1 S
Eccrine carcinoma is observed clinically as a slow-growing, nodular plaque on the scalp, arms, legs, or trunk in middle-aged and elderly individuals.1 Squamoid eccrine ductal carcinoma also has been reported in a young woman.5 Another immunocompromised patient was identified in the literature with a great toe lesion that showed follicular differentiation along with the usual SEDC features of squamoid and ductal differentiation.6 The etiology of SEDC is controversial but is thought to be an SCC arising from eccrine glands, a subtype of eccrine carcinoma with extensive squamoid differentiation, or a biphenotypic carcinoma.1,7
Histologically, SEDC is poorly circumscribed with an infiltrative growth pattern and deep extension into the dermis and subcutaneous tissue. The lesion is characterized by prominent squamous epithelial proliferation superficially with cellular atypia, keratinous cyst formation, squamous eddies, and eccrine ductal differentiation.1
The differential diagnosis of SEDC includes SCC; metastatic carcinoma with squamoid features; and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma, and porocarcinoma with squamous differentiation.1
Immunohistochemistry has a role in the diagnosis of SEDC. Findings include positive staining for S100 protein, EMA, CKs, and CEA. Glandular tissue stains positive for EMA and CEA, supporting an adnexal origin.1 Positivity for p63 and CK5/6 supports the conclusion that this is a primary cutaneous malignancy, not a metastatic disease.1
Squamoid eccrine ductal carcinoma has an indeterminate malignant potential. There is a disparity of clinical behavior between SCC and eccrine cancers; however, because squamous differentiation sometimes dominates the histological picture, eccrine carcinomas can be misdiagnosed as SCC.1,8 Eccrine adnexal tumors are characterized by multiple local recurrences (70%–80% of cases); perineural invasion; and metastasis (50% of cases) to regional lymph nodes and viscera, including the lungs, liver, bones, and brain.1 Squamous cell carcinoma, however, has a markedly lower recurrence rate (3.1%–18.7% of cases) and rate of metastasis (5.2%–37.8%).1
Squamoid eccrine ductal carcinoma is classified as one of the less aggressive eccrine tumors, although the low number of cases makes it a controversial conclusion.1 To our knowledge, no cases of SEDC metastasis have been reported with SEDC. Recurrence of SEDC has been reported locally, and perineural or perivascular invasion (or both) has been demonstrated in 3 cases.1
Since SEDC has invasive and metastatic potential, as demonstrated in our case, along with elevated local recurrence rates, physicians must be able to properly diagnose this rare entity and recommend an appropriate surgical modality. Due to the low incidence of SEDC, there are no known randomized studies comparing treatment modalities.1 O
Surgical extirpation with complete margin examination is recommended, as SEDC tends to be underestimated in size, is aggressive in its infiltration, and is predisposed to perineural and perivascular invasion. T
Along with the rarity of SEDC in our patient, the simultaneous occurrence of 3 primary malignancies also is unusual. Patients with CLL have progressive defects of cell- and humoral-mediated immunity, causing immunosuppression. In a retrospective study, Tsimberidou et al9 reviewed the records of 2028 untreated CLL patients and determined that 27% had another primary malignancy, including skin (30%) and lung cancers (6%), which were two of the malignancies seen in our patient. The investigators concluded that patients with CLL have more than twice the risk of developing a second primary malignancy and an increased frequency of certain cancer types.9 Furthermore, treatment regimens for CLL have been considered to increase cell- and humoral-mediated immune defects at specific cancer sites,10 although the exact mechanism of this action is unknown. Development of a second primary malignancy (or even a third) in patients with SEDC is increasingly being reported in CLL patients.9,10
A high index of suspicion with SEDC in the differential diagnosis should be maintained in elderly men with slow-growing, solitary, nodular lesions of the scalp, nose, arms, legs, or trunk.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. Clin Aesthet Dermatol. 2013;6:33-36.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;916:799-802.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma)[published online April 15, 2015]. J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Kavand S, Cassarino DS. Squamoid eccrine ductal carcinoma: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Tsimberidou AM, Wen S, McLaughlin P, et al. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol. 2009;27:904-910.
- Dasanu CA, Alexandrescu DT. Risk for second nonlymphoid neoplasms in chronic lymphocytic leukemia. Med Gen Med. 2007;9:35.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. Clin Aesthet Dermatol. 2013;6:33-36.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;916:799-802.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma)[published online April 15, 2015]. J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Kavand S, Cassarino DS. Squamoid eccrine ductal carcinoma: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Tsimberidou AM, Wen S, McLaughlin P, et al. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol. 2009;27:904-910.
- Dasanu CA, Alexandrescu DT. Risk for second nonlymphoid neoplasms in chronic lymphocytic leukemia. Med Gen Med. 2007;9:35.
Practice Points
- Squamoid eccrine ductal carcinoma (SEDC) is an extremely rare cutaneous tumor of unknown etiology.
- A high index of suspicion with SEDC in the differential diagnosis should be maintained in elderly men with slow-growing, solitary, nodular lesions of the scalp, nose, arms, legs, or trunk.
- Development of a second or even a third primary malignancy in patients with SEDC is increasingly being reported in CLL patients.
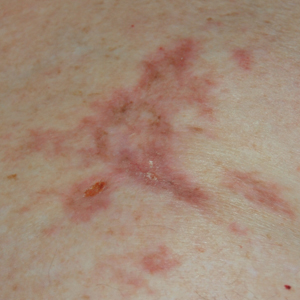
Pigmented Squamous Cell Carcinoma Presenting as Longitudinal Melanonychia in a Transplant Recipient
Case Report
A 62-year-old black man presented for examination of a dark longitudinal streak located adjacent to the lateral nail fold on the third finger of the left hand. The lesion had been present for several months, during which time it had slowly expanded in size. The fingertip had recently become tender, which interfered with the patient’s ability to work. His past medical history was remarkable for end-stage renal disease secondary to glomerulonephritis with nephrotic syndrome of unclear etiology. He initially was treated by an outside physician using peritoneal dialysis for 3 years until he underwent renal transplantation in 2004 with a cadaveric organ. Other remarkable medical conditions included posttransplantation diabetes, hyperlipidemia, and gout. His multidrug regimen included 2 immunosuppressive medications: oral cyclosporine 125 mg twice daily and oral mycophenolate mofetil 250 mg twice daily.
A broad, irregular, black, pigmented, subungual band was noted on the left third finger. The lesion appeared to emanate from below the nail cuticle and traveled along the nail longitudinally toward the distal tip. The band appeared darker at the edge adjacent to the lateral nail fold and grew lighter near the middle of the nail where its free edge was noted to be irregular. A slightly thickened lateral nail fold with an irregular, small, sawtoothlike hyperkeratosis and hyperpigmentation also was noted (Figure 1).
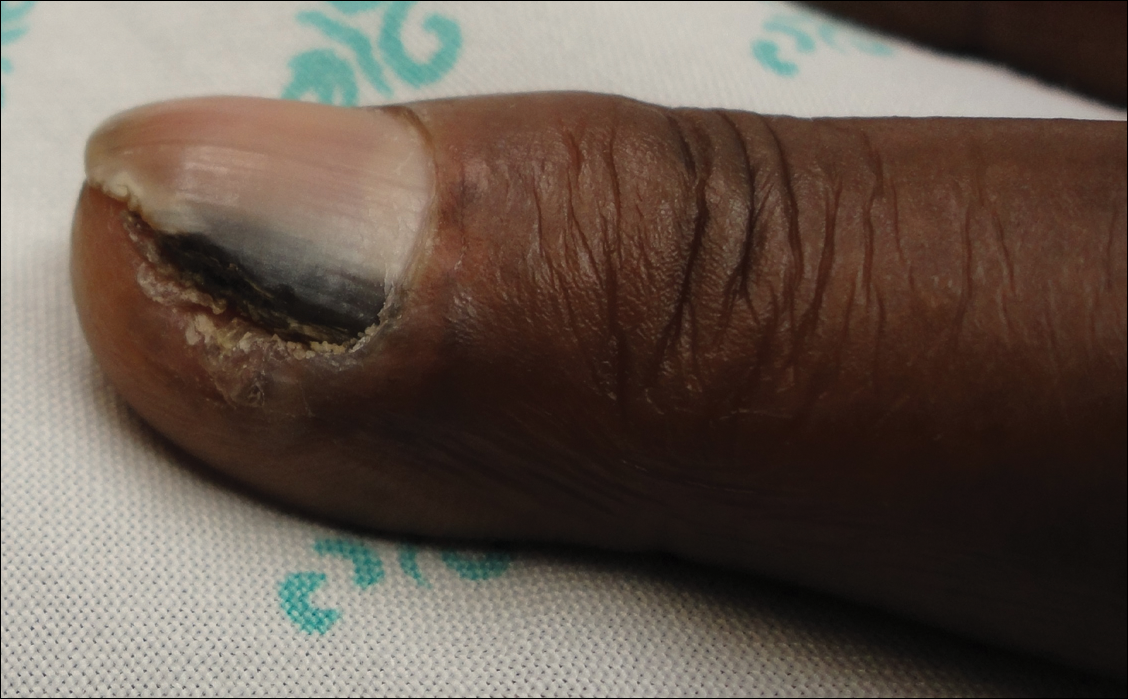
Subungual melanoma, onychomycosis, squamous cell carcinoma (SCC), and a verruca copresenting with onychomycosis were considered in the differential diagnosis. The patient underwent nail avulsion and biopsy of the nail bed as well as the nail matrix. Histopathology was notable for malignant dyskeratosis with a lack of nuclear maturation, occasional mitoses, multinucleation, and individual cell keratinization (Figure 2). Immunostaining for S100 was negative, while staining for cytokeratins AE1/AE3 was positive. Deposition of melanin pigment in the malignant dyskeratotic cells was noted. Periodic acid–Schiff staining identified pseudohyphae without invasion of the nail plate. A diagnosis of pigmented SCC (pSCC) was made. The patient’s nail also was sent for fungal cultures that later grew Candida glabrata and Candida parapsilosis.
The patient underwent Mohs micrographic surgery for removal of the pSCC, which was found to be more extensive than originally suspected and required en bloc excision of the nail repaired with a full-thickness skin graft from the left forearm. The area healed well with some hyperpigmentation (Figure 3).
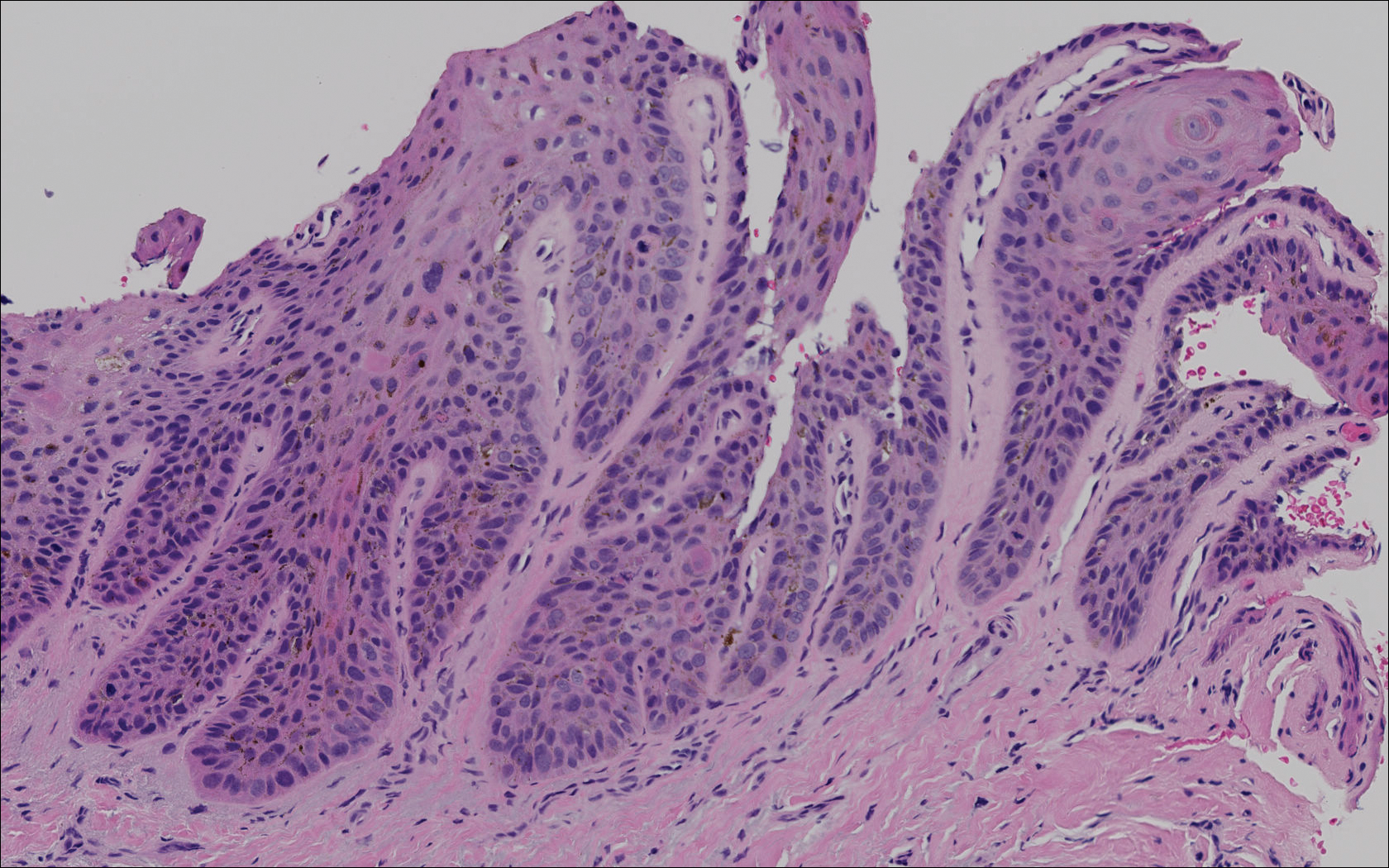
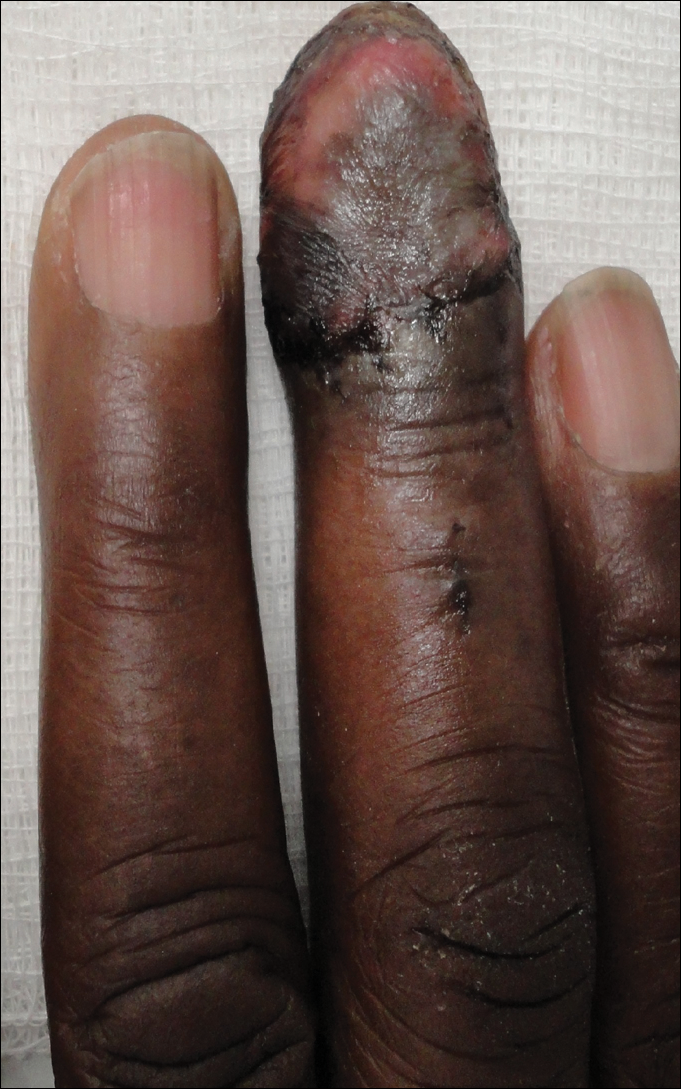
Comment
Among the various types of skin cancer, an estimated 700,000 patients are diagnosed with SCC annually, making it the second most common form of skin cancer in the United States.1 Basal cell carcinoma (BCC) is the most common skin cancer among whites in the United States, while in contrast SCC is the most common skin cancer in patients with skin of color.2 Only an estimated 2% to 5% of all SCCs are pigmented, and this variant is more commonly seen in patients with skin of color.3-5 One analysis of 52 cases of pSCC showed that common features included a flat or slightly raised appearance and hyperpigmentation with varying levels of scaling.6 Studies have shown an altered presentation of pSCC in black skin with increased melanin production and thickness of the stratum corneum in contrast with cases seen in white patients.7 Other potential features include scaling, erosive changes, and sharply demarcated borders. Squamous cell carcinoma typically occurs in sun-exposed areas, reflecting its association with UV light damage; however, SCC in skin of color patients has been noted to occur in sun-protected areas and in areas of chronic scarring.8 Pigmented SCC also appears to follow this distribution, as affected areas are not necessarily in direct exposure to the sun. Pigmented SCCs have been associated with pruritus and/or burning pain, which also was seen in our case when our patient complained of tenderness at the site.
We describe the case of a subungual pSCC clinically presenting as longitudinal melanonychia. Pigmented SCC presenting as longitudinal melanonychia was first described by Baran and Simon in 1988.9 Since that time, it has been reported that approximately 10% of subungual pSCCs clinically present as longitudinal melanonychia.10,11 A retrospective study reviewing 35 cases of SCC of the nail apparatus found that 5 (14.3%) cases presented as longitudinal melanonychia.10 Another retrospective study found that 6 of 51 (11.8%) cases of SCCs affecting the nail unit presented as the warty type of SCC in association with longitudinal melanonychia.12 Cases of pSCC in situ appearing as longitudinal melanonychia also have been reported.13,14
Risk factors for the development of pSCC include advanced age, male sex, presence of human papilloma virus, and use of immunosuppressants.15 Male predominance and advanced age at the time of diagnosis (mean age, 67 years) have been observed in pSCC cases.16 It is now well established that renal transplant recipients have an increased risk of SCC, with a reported incidence rate of 5% to 6%.16 When these patients develop an SCC, they typically follow a more aggressive course. Renal transplantation has a higher ratio than cardiac transplantation for SCC development (2.37:1), whereas cardiac transplantation is associated with a higher risk of BCC development.17 A study of 384 transplant recipients found that 96 (25.0%) had a postsurgical nonmelanoma skin cancer (NMSC), with a ratio of SCC to BCC of 1.2:1.16 The calculated incidence of NMSC at 10 and 20 years posttransplantation was 24.2% and 54.4%, respectively. Another study also determined that SCC rates (50.0%) in postrenal transplant recipients were approximately twice that of BCC (27.0%).18
A daily regimen of immunosuppressive medications such as cyclosporine and mycophenolate mofetil showed an increased risk for development of NMSC.15 Immunosuppressive medications play an important role in the pathogenesis of SCC due to a direct oncogenic effect as well as impairment of the immune system’s ability to fight precancerous developments.15 A 4-year study of 100 renal transplant recipients using mycophenolate mofetil as part of an immunosuppressive regimen reported 22% NMSC findings among 9 patients.19 On average, patients developed an NMSC approximately 61 months posttransplantation, with a wide range from 2 to 120 months.
Advanced age was another important risk factor, with each decade of life producing a 60% increase in instantaneous risk of SCC development for transplant recipients.15 A steady increase in risk was related to the length of time adhering to an immunosuppressive regimen, especially from 2 to 6 years, and then remaining constant in subsequent years. For older patients on immunosuppressant regimens for more than 8 years, the calculated relative risk was noted to be over 200 times greater than the normal population’s development of skin cancers.18
Conclusion
Although cases of pSCC presenting as longitudinal melanonychia have previously been reported,9-14,20 our case is unique in that it describes pSCC in a renal transplant recipient. Our patient had many of the known risk factors for the development of pSCC including male sex, advanced age, skin of color, history of renal transplantation, and immunosuppressive therapy. Although regular full-body skin examinations are an accepted part of renal transplantation follow-up due to SCC risk, our case emphasizes the need to remain vigilant due to possible atypical presentations among the immunosuppressed. The nail unit should not be overlooked during the clinical examination of renal transplant recipients as demonstrated by our patient’s rare presentation of pSCC in the nail.
- Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012 [published online February 1, 2013]. J Am Acad Dermatol. 2013;68:957-966.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Krishna R, Lewis A, Orengo IF, et al. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
- Brinca A, Teixeira V, Goncalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-818.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease. J Am Acad Dermatol. 2010;62:597-604.
- Singh B, Bhaya M, Shaha A, et al. Presentation, course, and outcome of head and neck cancers in African Americans: a case-control study. Laryngoscope. 1998;108(8 pt 1):1159-1163.
- Cancer Facts and Figures 2006. Atlanta, GA: American Cancer Society; 2006.
- Baran R, Simon C. Longitudinal melanonychia: a symptom of Bowen’s disease. J Am Acad Dermatol. 1988;18:1359-1360.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Ishida M, Iwai M, Yoshida K, et al. Subungual pigmented squamous cell carcinoma presenting as longitudinal melanonychia: a case report with review of the literature. Int J Clin Exp Pathol. 2014;7:844-847.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Saito T, Uchi H, Moroi Y, et al. Subungual Bowen disease revealed by longitudinal melanonychia. J Am Acad Dermatol. 2012;67:E240-E241.
- Saxena A, Kasper DA, Campanelli CD, et al. Pigmented Bowen’s disease clinically mimicking melanoma on the nail. Dermatol Surg. 2006;32:1522-1525.
- Mackenzie KA, Wells JE, Lynn KL, et al. First and subsequent nonmelanoma skin cancers: incidence and predictors in a population of New Zealand renal transplant recipients. Nephrol Dial Transplant. 2010;25:300-306.
- Gutiérrez-Mendoza D, Narro-Llorente R, Karam-Orantes M, et al. Dermoscopy clues in pigmented Bowen’s disease [published online ahead of print September 16, 2010]. Dermatol Res Pract. 2010;2010.
- Euvards S, Kanitakis J, Pouteil-Noble C, et al. Comparative epidemiologic study of premalignant and malignant epithelial cutaneous lesions developing after kidney and heart transplantation. J Am Acad Dermatol. 1995;33(2 pt 1):222-229.
- Moloney FJ, Comber H, O’Lorcain P, et al. A population-based study of skin cancer incidence and prevalence in renal transplant patients. Br J Dermatol. 2006;154:498-504.
- Formicone F, Fargnoli MC, Pisani F, et al. Cutaneous manifestations in Italian kidney transplant recipients. Transplant Proc. 2005;37:2527-2528.
- Fernandes Massa A, Debarbieux S, Depaepe L, et al. Pigmented squamous cell carcinoma of the nail bed presenting as a melanonychia striata: diagnosis by perioperative reflectance confocal microscopy. Br J Dermatol. 2013;169:198-199.
Case Report
A 62-year-old black man presented for examination of a dark longitudinal streak located adjacent to the lateral nail fold on the third finger of the left hand. The lesion had been present for several months, during which time it had slowly expanded in size. The fingertip had recently become tender, which interfered with the patient’s ability to work. His past medical history was remarkable for end-stage renal disease secondary to glomerulonephritis with nephrotic syndrome of unclear etiology. He initially was treated by an outside physician using peritoneal dialysis for 3 years until he underwent renal transplantation in 2004 with a cadaveric organ. Other remarkable medical conditions included posttransplantation diabetes, hyperlipidemia, and gout. His multidrug regimen included 2 immunosuppressive medications: oral cyclosporine 125 mg twice daily and oral mycophenolate mofetil 250 mg twice daily.
A broad, irregular, black, pigmented, subungual band was noted on the left third finger. The lesion appeared to emanate from below the nail cuticle and traveled along the nail longitudinally toward the distal tip. The band appeared darker at the edge adjacent to the lateral nail fold and grew lighter near the middle of the nail where its free edge was noted to be irregular. A slightly thickened lateral nail fold with an irregular, small, sawtoothlike hyperkeratosis and hyperpigmentation also was noted (Figure 1).
Subungual melanoma, onychomycosis, squamous cell carcinoma (SCC), and a verruca copresenting with onychomycosis were considered in the differential diagnosis. The patient underwent nail avulsion and biopsy of the nail bed as well as the nail matrix. Histopathology was notable for malignant dyskeratosis with a lack of nuclear maturation, occasional mitoses, multinucleation, and individual cell keratinization (Figure 2). Immunostaining for S100 was negative, while staining for cytokeratins AE1/AE3 was positive. Deposition of melanin pigment in the malignant dyskeratotic cells was noted. Periodic acid–Schiff staining identified pseudohyphae without invasion of the nail plate. A diagnosis of pigmented SCC (pSCC) was made. The patient’s nail also was sent for fungal cultures that later grew Candida glabrata and Candida parapsilosis.
The patient underwent Mohs micrographic surgery for removal of the pSCC, which was found to be more extensive than originally suspected and required en bloc excision of the nail repaired with a full-thickness skin graft from the left forearm. The area healed well with some hyperpigmentation (Figure 3).
Comment
Among the various types of skin cancer, an estimated 700,000 patients are diagnosed with SCC annually, making it the second most common form of skin cancer in the United States.1 Basal cell carcinoma (BCC) is the most common skin cancer among whites in the United States, while in contrast SCC is the most common skin cancer in patients with skin of color.2 Only an estimated 2% to 5% of all SCCs are pigmented, and this variant is more commonly seen in patients with skin of color.3-5 One analysis of 52 cases of pSCC showed that common features included a flat or slightly raised appearance and hyperpigmentation with varying levels of scaling.6 Studies have shown an altered presentation of pSCC in black skin with increased melanin production and thickness of the stratum corneum in contrast with cases seen in white patients.7 Other potential features include scaling, erosive changes, and sharply demarcated borders. Squamous cell carcinoma typically occurs in sun-exposed areas, reflecting its association with UV light damage; however, SCC in skin of color patients has been noted to occur in sun-protected areas and in areas of chronic scarring.8 Pigmented SCC also appears to follow this distribution, as affected areas are not necessarily in direct exposure to the sun. Pigmented SCCs have been associated with pruritus and/or burning pain, which also was seen in our case when our patient complained of tenderness at the site.
We describe the case of a subungual pSCC clinically presenting as longitudinal melanonychia. Pigmented SCC presenting as longitudinal melanonychia was first described by Baran and Simon in 1988.9 Since that time, it has been reported that approximately 10% of subungual pSCCs clinically present as longitudinal melanonychia.10,11 A retrospective study reviewing 35 cases of SCC of the nail apparatus found that 5 (14.3%) cases presented as longitudinal melanonychia.10 Another retrospective study found that 6 of 51 (11.8%) cases of SCCs affecting the nail unit presented as the warty type of SCC in association with longitudinal melanonychia.12 Cases of pSCC in situ appearing as longitudinal melanonychia also have been reported.13,14
Risk factors for the development of pSCC include advanced age, male sex, presence of human papilloma virus, and use of immunosuppressants.15 Male predominance and advanced age at the time of diagnosis (mean age, 67 years) have been observed in pSCC cases.16 It is now well established that renal transplant recipients have an increased risk of SCC, with a reported incidence rate of 5% to 6%.16 When these patients develop an SCC, they typically follow a more aggressive course. Renal transplantation has a higher ratio than cardiac transplantation for SCC development (2.37:1), whereas cardiac transplantation is associated with a higher risk of BCC development.17 A study of 384 transplant recipients found that 96 (25.0%) had a postsurgical nonmelanoma skin cancer (NMSC), with a ratio of SCC to BCC of 1.2:1.16 The calculated incidence of NMSC at 10 and 20 years posttransplantation was 24.2% and 54.4%, respectively. Another study also determined that SCC rates (50.0%) in postrenal transplant recipients were approximately twice that of BCC (27.0%).18
A daily regimen of immunosuppressive medications such as cyclosporine and mycophenolate mofetil showed an increased risk for development of NMSC.15 Immunosuppressive medications play an important role in the pathogenesis of SCC due to a direct oncogenic effect as well as impairment of the immune system’s ability to fight precancerous developments.15 A 4-year study of 100 renal transplant recipients using mycophenolate mofetil as part of an immunosuppressive regimen reported 22% NMSC findings among 9 patients.19 On average, patients developed an NMSC approximately 61 months posttransplantation, with a wide range from 2 to 120 months.
Advanced age was another important risk factor, with each decade of life producing a 60% increase in instantaneous risk of SCC development for transplant recipients.15 A steady increase in risk was related to the length of time adhering to an immunosuppressive regimen, especially from 2 to 6 years, and then remaining constant in subsequent years. For older patients on immunosuppressant regimens for more than 8 years, the calculated relative risk was noted to be over 200 times greater than the normal population’s development of skin cancers.18
Conclusion
Although cases of pSCC presenting as longitudinal melanonychia have previously been reported,9-14,20 our case is unique in that it describes pSCC in a renal transplant recipient. Our patient had many of the known risk factors for the development of pSCC including male sex, advanced age, skin of color, history of renal transplantation, and immunosuppressive therapy. Although regular full-body skin examinations are an accepted part of renal transplantation follow-up due to SCC risk, our case emphasizes the need to remain vigilant due to possible atypical presentations among the immunosuppressed. The nail unit should not be overlooked during the clinical examination of renal transplant recipients as demonstrated by our patient’s rare presentation of pSCC in the nail.
Case Report
A 62-year-old black man presented for examination of a dark longitudinal streak located adjacent to the lateral nail fold on the third finger of the left hand. The lesion had been present for several months, during which time it had slowly expanded in size. The fingertip had recently become tender, which interfered with the patient’s ability to work. His past medical history was remarkable for end-stage renal disease secondary to glomerulonephritis with nephrotic syndrome of unclear etiology. He initially was treated by an outside physician using peritoneal dialysis for 3 years until he underwent renal transplantation in 2004 with a cadaveric organ. Other remarkable medical conditions included posttransplantation diabetes, hyperlipidemia, and gout. His multidrug regimen included 2 immunosuppressive medications: oral cyclosporine 125 mg twice daily and oral mycophenolate mofetil 250 mg twice daily.
A broad, irregular, black, pigmented, subungual band was noted on the left third finger. The lesion appeared to emanate from below the nail cuticle and traveled along the nail longitudinally toward the distal tip. The band appeared darker at the edge adjacent to the lateral nail fold and grew lighter near the middle of the nail where its free edge was noted to be irregular. A slightly thickened lateral nail fold with an irregular, small, sawtoothlike hyperkeratosis and hyperpigmentation also was noted (Figure 1).
Subungual melanoma, onychomycosis, squamous cell carcinoma (SCC), and a verruca copresenting with onychomycosis were considered in the differential diagnosis. The patient underwent nail avulsion and biopsy of the nail bed as well as the nail matrix. Histopathology was notable for malignant dyskeratosis with a lack of nuclear maturation, occasional mitoses, multinucleation, and individual cell keratinization (Figure 2). Immunostaining for S100 was negative, while staining for cytokeratins AE1/AE3 was positive. Deposition of melanin pigment in the malignant dyskeratotic cells was noted. Periodic acid–Schiff staining identified pseudohyphae without invasion of the nail plate. A diagnosis of pigmented SCC (pSCC) was made. The patient’s nail also was sent for fungal cultures that later grew Candida glabrata and Candida parapsilosis.
The patient underwent Mohs micrographic surgery for removal of the pSCC, which was found to be more extensive than originally suspected and required en bloc excision of the nail repaired with a full-thickness skin graft from the left forearm. The area healed well with some hyperpigmentation (Figure 3).
Comment
Among the various types of skin cancer, an estimated 700,000 patients are diagnosed with SCC annually, making it the second most common form of skin cancer in the United States.1 Basal cell carcinoma (BCC) is the most common skin cancer among whites in the United States, while in contrast SCC is the most common skin cancer in patients with skin of color.2 Only an estimated 2% to 5% of all SCCs are pigmented, and this variant is more commonly seen in patients with skin of color.3-5 One analysis of 52 cases of pSCC showed that common features included a flat or slightly raised appearance and hyperpigmentation with varying levels of scaling.6 Studies have shown an altered presentation of pSCC in black skin with increased melanin production and thickness of the stratum corneum in contrast with cases seen in white patients.7 Other potential features include scaling, erosive changes, and sharply demarcated borders. Squamous cell carcinoma typically occurs in sun-exposed areas, reflecting its association with UV light damage; however, SCC in skin of color patients has been noted to occur in sun-protected areas and in areas of chronic scarring.8 Pigmented SCC also appears to follow this distribution, as affected areas are not necessarily in direct exposure to the sun. Pigmented SCCs have been associated with pruritus and/or burning pain, which also was seen in our case when our patient complained of tenderness at the site.
We describe the case of a subungual pSCC clinically presenting as longitudinal melanonychia. Pigmented SCC presenting as longitudinal melanonychia was first described by Baran and Simon in 1988.9 Since that time, it has been reported that approximately 10% of subungual pSCCs clinically present as longitudinal melanonychia.10,11 A retrospective study reviewing 35 cases of SCC of the nail apparatus found that 5 (14.3%) cases presented as longitudinal melanonychia.10 Another retrospective study found that 6 of 51 (11.8%) cases of SCCs affecting the nail unit presented as the warty type of SCC in association with longitudinal melanonychia.12 Cases of pSCC in situ appearing as longitudinal melanonychia also have been reported.13,14
Risk factors for the development of pSCC include advanced age, male sex, presence of human papilloma virus, and use of immunosuppressants.15 Male predominance and advanced age at the time of diagnosis (mean age, 67 years) have been observed in pSCC cases.16 It is now well established that renal transplant recipients have an increased risk of SCC, with a reported incidence rate of 5% to 6%.16 When these patients develop an SCC, they typically follow a more aggressive course. Renal transplantation has a higher ratio than cardiac transplantation for SCC development (2.37:1), whereas cardiac transplantation is associated with a higher risk of BCC development.17 A study of 384 transplant recipients found that 96 (25.0%) had a postsurgical nonmelanoma skin cancer (NMSC), with a ratio of SCC to BCC of 1.2:1.16 The calculated incidence of NMSC at 10 and 20 years posttransplantation was 24.2% and 54.4%, respectively. Another study also determined that SCC rates (50.0%) in postrenal transplant recipients were approximately twice that of BCC (27.0%).18
A daily regimen of immunosuppressive medications such as cyclosporine and mycophenolate mofetil showed an increased risk for development of NMSC.15 Immunosuppressive medications play an important role in the pathogenesis of SCC due to a direct oncogenic effect as well as impairment of the immune system’s ability to fight precancerous developments.15 A 4-year study of 100 renal transplant recipients using mycophenolate mofetil as part of an immunosuppressive regimen reported 22% NMSC findings among 9 patients.19 On average, patients developed an NMSC approximately 61 months posttransplantation, with a wide range from 2 to 120 months.
Advanced age was another important risk factor, with each decade of life producing a 60% increase in instantaneous risk of SCC development for transplant recipients.15 A steady increase in risk was related to the length of time adhering to an immunosuppressive regimen, especially from 2 to 6 years, and then remaining constant in subsequent years. For older patients on immunosuppressant regimens for more than 8 years, the calculated relative risk was noted to be over 200 times greater than the normal population’s development of skin cancers.18
Conclusion
Although cases of pSCC presenting as longitudinal melanonychia have previously been reported,9-14,20 our case is unique in that it describes pSCC in a renal transplant recipient. Our patient had many of the known risk factors for the development of pSCC including male sex, advanced age, skin of color, history of renal transplantation, and immunosuppressive therapy. Although regular full-body skin examinations are an accepted part of renal transplantation follow-up due to SCC risk, our case emphasizes the need to remain vigilant due to possible atypical presentations among the immunosuppressed. The nail unit should not be overlooked during the clinical examination of renal transplant recipients as demonstrated by our patient’s rare presentation of pSCC in the nail.
- Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012 [published online February 1, 2013]. J Am Acad Dermatol. 2013;68:957-966.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Krishna R, Lewis A, Orengo IF, et al. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
- Brinca A, Teixeira V, Goncalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-818.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease. J Am Acad Dermatol. 2010;62:597-604.
- Singh B, Bhaya M, Shaha A, et al. Presentation, course, and outcome of head and neck cancers in African Americans: a case-control study. Laryngoscope. 1998;108(8 pt 1):1159-1163.
- Cancer Facts and Figures 2006. Atlanta, GA: American Cancer Society; 2006.
- Baran R, Simon C. Longitudinal melanonychia: a symptom of Bowen’s disease. J Am Acad Dermatol. 1988;18:1359-1360.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Ishida M, Iwai M, Yoshida K, et al. Subungual pigmented squamous cell carcinoma presenting as longitudinal melanonychia: a case report with review of the literature. Int J Clin Exp Pathol. 2014;7:844-847.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Saito T, Uchi H, Moroi Y, et al. Subungual Bowen disease revealed by longitudinal melanonychia. J Am Acad Dermatol. 2012;67:E240-E241.
- Saxena A, Kasper DA, Campanelli CD, et al. Pigmented Bowen’s disease clinically mimicking melanoma on the nail. Dermatol Surg. 2006;32:1522-1525.
- Mackenzie KA, Wells JE, Lynn KL, et al. First and subsequent nonmelanoma skin cancers: incidence and predictors in a population of New Zealand renal transplant recipients. Nephrol Dial Transplant. 2010;25:300-306.
- Gutiérrez-Mendoza D, Narro-Llorente R, Karam-Orantes M, et al. Dermoscopy clues in pigmented Bowen’s disease [published online ahead of print September 16, 2010]. Dermatol Res Pract. 2010;2010.
- Euvards S, Kanitakis J, Pouteil-Noble C, et al. Comparative epidemiologic study of premalignant and malignant epithelial cutaneous lesions developing after kidney and heart transplantation. J Am Acad Dermatol. 1995;33(2 pt 1):222-229.
- Moloney FJ, Comber H, O’Lorcain P, et al. A population-based study of skin cancer incidence and prevalence in renal transplant patients. Br J Dermatol. 2006;154:498-504.
- Formicone F, Fargnoli MC, Pisani F, et al. Cutaneous manifestations in Italian kidney transplant recipients. Transplant Proc. 2005;37:2527-2528.
- Fernandes Massa A, Debarbieux S, Depaepe L, et al. Pigmented squamous cell carcinoma of the nail bed presenting as a melanonychia striata: diagnosis by perioperative reflectance confocal microscopy. Br J Dermatol. 2013;169:198-199.
- Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012 [published online February 1, 2013]. J Am Acad Dermatol. 2013;68:957-966.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Krishna R, Lewis A, Orengo IF, et al. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
- Brinca A, Teixeira V, Goncalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-818.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease. J Am Acad Dermatol. 2010;62:597-604.
- Singh B, Bhaya M, Shaha A, et al. Presentation, course, and outcome of head and neck cancers in African Americans: a case-control study. Laryngoscope. 1998;108(8 pt 1):1159-1163.
- Cancer Facts and Figures 2006. Atlanta, GA: American Cancer Society; 2006.
- Baran R, Simon C. Longitudinal melanonychia: a symptom of Bowen’s disease. J Am Acad Dermatol. 1988;18:1359-1360.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Ishida M, Iwai M, Yoshida K, et al. Subungual pigmented squamous cell carcinoma presenting as longitudinal melanonychia: a case report with review of the literature. Int J Clin Exp Pathol. 2014;7:844-847.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Saito T, Uchi H, Moroi Y, et al. Subungual Bowen disease revealed by longitudinal melanonychia. J Am Acad Dermatol. 2012;67:E240-E241.
- Saxena A, Kasper DA, Campanelli CD, et al. Pigmented Bowen’s disease clinically mimicking melanoma on the nail. Dermatol Surg. 2006;32:1522-1525.
- Mackenzie KA, Wells JE, Lynn KL, et al. First and subsequent nonmelanoma skin cancers: incidence and predictors in a population of New Zealand renal transplant recipients. Nephrol Dial Transplant. 2010;25:300-306.
- Gutiérrez-Mendoza D, Narro-Llorente R, Karam-Orantes M, et al. Dermoscopy clues in pigmented Bowen’s disease [published online ahead of print September 16, 2010]. Dermatol Res Pract. 2010;2010.
- Euvards S, Kanitakis J, Pouteil-Noble C, et al. Comparative epidemiologic study of premalignant and malignant epithelial cutaneous lesions developing after kidney and heart transplantation. J Am Acad Dermatol. 1995;33(2 pt 1):222-229.
- Moloney FJ, Comber H, O’Lorcain P, et al. A population-based study of skin cancer incidence and prevalence in renal transplant patients. Br J Dermatol. 2006;154:498-504.
- Formicone F, Fargnoli MC, Pisani F, et al. Cutaneous manifestations in Italian kidney transplant recipients. Transplant Proc. 2005;37:2527-2528.
- Fernandes Massa A, Debarbieux S, Depaepe L, et al. Pigmented squamous cell carcinoma of the nail bed presenting as a melanonychia striata: diagnosis by perioperative reflectance confocal microscopy. Br J Dermatol. 2013;169:198-199.
Practice Points
- Risk factors for the development of pigmented squamous cell carcinoma (pSCC) include older age, male sex, and use of immunosuppressant medications.
- Subungual pSCC can present as longitudinal melanonychia and should be considered in the differential diagnosis for melanonychia in patients with skin of color or those who are immunosuppressed.
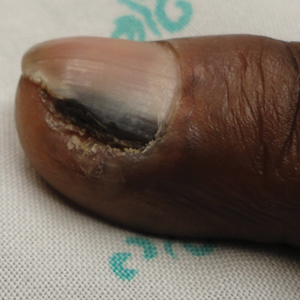
Discoid Lupus Erythematosus Following Herpes Zoster
Cutaneous manifestations of systemic lupus erythematosus (SLE) can be classified as lupus-specific or lupus-nonspecific skin lesions. Lupus-specific lesions commonly are photodistributed, with involvement of the malar region, arms, and trunk. The development of discoid lupus erythematosus (DLE) in areas of trauma, including sun-exposed skin, is not uncommon and may be associated with an isomorphic response. We present a rare case of an isomorphic response following herpes zoster (HZ) in a young woman undergoing treatment with immunosuppressive agents for SLE and DLE. Potential prophylactic therapy also is discussed.
Case Report
A 19-year-old woman initially presented to an outside dermatologist for evaluation of new-onset scarring alopecia, crusted erythematous plaques on the face and arms, and arthralgia. A punch biopsy of a lesion on the left arm demonstrated a lichenoid and perivascular lymphocytic infiltrate with scattered necrotic keratinocytes, perifollicular inflammation, and focally thickened basement membrane at the dermoepidermal junction consistent with discoid lupus erythematosus (DLE). A laboratory workup for SLE revealed 1:1280 antinuclear antibodies (reference range, negative <1:80) with elevated titers of double-stranded DNA, Smith, ribonucleoprotein, Sjögren syndrome A, and Sjögren syndrome B autoantibodies with low complement levels. Based on these findings, a diagnosis of SLE and DLE was made.
At that time, the patient was started on hydroxychloroquine 200 mg twice daily for SLE. Four days later she developed swelling in both hands and feet, and hydroxychloroquine was stopped due to a presumed adverse reaction; however, her symptoms subsequently were determined to be polyarthritis secondary to a lupus flare. Prednisone 10 mg once daily was then initiated. The patient was encouraged to restart hydroxychloroquine, but she declined.
Over the next 13 months, the patient developed severe photosensitivity, oral ulcers, Raynaud phenomenon, anemia, and nephrotic-range proteinuria. She ultimately was diagnosed by the nephrology department at our institution with mixed diffuse proliferative and membranous glomerulonephritis. Induction therapy with oral mycophenolate mofetil 1000 mg twice daily and prednisone 60 mg once daily was started, followed by the addition of tacrolimus 1 mg twice daily. Despite immunosuppressive therapy, she continued to develop new discoid lesions on the face, chest, and arms. Th
After 4 weeks of treatment with mycophenolate mofetil, prednisone, and tacrolimus, the patient developed a painful vesicular rash on the left breast with extension over the left axilla and scapula in a T3 to T4 dermatomal distribution. A clinical diagnosis of HZ was made, and she was started on intravenous acyclovir 10 mg/kg in dextrose 5% every 8 hours for 4 days followed by oral valacyclovir 1000 mg every 8 hours for 14 days, which led to resolution of the eruption.
Over the next 4 months, the patient continued to experience pain confined to the same dermatomal area as the HZ, which was consistent with postherpetic neuralgia. Mycophenolate mofetil was discontinued after she developed acute liver toxicity attributed to the drug. Upon discontinuation, the patient developed a new pruritic rash on both arms and the back. Physical examination by the dermatology department at our institution revealed diffuse, scaly, hyperpigmented papules and annular plaques with central pink hypopigmentation on the face, ears, anterior chest, arms, hands, and back. On the left anterior chest and back, the distribution was strikingly unilateral and multidermatomal (Figure 1). Upon further questioning, the patient confirmed that the areas of the new rash coincided with areas previously affected by HZ. Histologic examination of a representative lesion from the left lateral breast revealed hyperkeratosis, follicular plugging, a patchy lichenoid and perivascular mononuclear cell infiltrate, and pigment incontinence (Figure 2A). These histologic features were subtle and were not diagnostic for lupus; however, direct immunofluorescence demonstrated a continuous granular band of IgG and C3 along the dermoepidermal junction, confirming the diagnosis of DLE (Figure 2B). The histologic findings and clinical presentation were consistent with the development of DLE in areas of previous trauma from HZ. The patient continues to follow-up with the rheumatology and nephrology departments but was lost to dermatology follow-up.
Comment
The pathogenesis of DLE is poorly understood but is thought to be multifactorial, involving genetics, sun exposure, and immune dysregulation.1 Development of DLE lesions in skin traumatized by tattoos, scratches, scars, and prolonged heat exposure has been reported.2 Clarification of the mechanism(s) underlying these traumatized areas may provide insight into the pathophysiology of DLE.
The isomorphic response, also known as the Köbner phenomenon, is the development of a preexisting skin condition at a site of trauma. This phenomenon has been observed in several dermatologic conditions including psoriasis, lichen planus, systemic sclerosis, dermatomyositis, sarcoidosis, vitiligo, and DLE.3 Koebnerization may result from trauma to the skin caused by scratches, sun exposure, radiography, prolonged heat and cold exposure, pressure, tattoos, scars, and inflammatory dermatoses.2,4 Ueki4 suggested that localized trauma to the skin stimulates an immune response that makes the traumatized site a target for a preexisting skin condition. Inflammatory mediators such as IL-1, tumor necrosis factor α, IL-6, and interferon γ have been implicated in the pathophysiology of the isomorphic response.4
Wolf isotopic response is a similar entity that refers to the development of a novel skin condition at the site of a distinct, previously resolved skin disorder. This phenomenon was described by Wolf et al5 in 1995, and since then over 170 cases have been reported.5-7 In most cases the initial skin condition is HZ, although herpes simplex virus has also been implicated. The common resulting skin conditions include granulomatous reactions, malignant tumors, lichen planus, morphea, and infections. The notion that the antecedent skin disease alters the affected site and causes it to be more susceptible to autoimmunity has been proposed as a mechanism for the isotopic response.7,8 While one might consider our presentation of DLE following HZ to be an isotopic response, we believe this case is best classified as an isomorphic response, as the patient already had an established diagnosis of DLE.
The development of DLE at the site of a previous HZ eruption has been described in 2 other cases of young women with SLE.9,10 Unique to our case is the development of a multidermatomal eruption, which may be an indication of her degree of immunosuppression, as immunosuppressed patients are more likely to present with multidermatomal reactivation of varicella zoster virus and postherpetic neuralgia.11 The similarities between our case and the 2 prior reports—including the patients’ age, sex, history of SLE, and degree of immunosuppression—are noteworthy in that they may represent a subset of SLE patients who are predisposed to developing koebnerization following HZ. Physicians should be aware of this phenomenon and consider being proactive in preventing long-term damage.
When feasible, physicians should consider administering the HZ vaccine to reduce the course and severity of HZ before prescribing immunosuppressive agents. When HZ presents in young, immunosuppressed women with a history of SLE, we suggest monitoring the affected sites closely for any evidence of DLE. Topical corticosteroids should be applied to involved areas of the face or body at the earliest appearance of such lesions, which may prevent the isomorphic response and its potentially scarring DLE lesions. This will be our therapeutic approach if we encounter a similar clinical situation in the future. Fur
Acknowledgment
We thank Carolyn E. Grotkowski, MD, from the Department of Pathology, Cooper Medical School of Rowan University, Camden, New Jersey, for her assistance in photographing the pathology slides.
- Lin JH, Dutz JP, Sontheimer RD, et al. Pathophysiology of cutaneous lupus erythematosus. Clinic Rev Allerg Immunol. 2007;33:85-106.
- Ueki H. Köbner phenomenon in lupus erythematosus [in German]. Hautarzt. 1994;45:154-160.
- Boyd AS, Neldner KH. The isomorphic response of Koebner. Int J Dermatol. 1990;29:401-410.
- Ueki H. Koebner phenomenon in lupus erythematosus with special consideration of clinical findings. Autoimmun Rev. 2005;4:219-223.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-vs-host disease to sites of skin injury. Arch Dermatol. 2011;147:1081-1086.
- Lee NY, Daniel AS, Dasher DA, et al. Cutaneous lupus after herpes zoster: isomorphic, isotopic, or both [published online May 29, 2012]? Pediatr Dermatol. 2013;30:e110-e113.
- Longhi BS, Centeville M, Marini R, et al. Koebner’s phenomenon in systemic lupus erythematosus. Rheumatol Int. 2012;32:1403-1405.
- Failla V, Jacques J, Castronovo C, et al. Herpes zoster in patients treated with biologicals. Dermatology. 2012;224:251-256.
Cutaneous manifestations of systemic lupus erythematosus (SLE) can be classified as lupus-specific or lupus-nonspecific skin lesions. Lupus-specific lesions commonly are photodistributed, with involvement of the malar region, arms, and trunk. The development of discoid lupus erythematosus (DLE) in areas of trauma, including sun-exposed skin, is not uncommon and may be associated with an isomorphic response. We present a rare case of an isomorphic response following herpes zoster (HZ) in a young woman undergoing treatment with immunosuppressive agents for SLE and DLE. Potential prophylactic therapy also is discussed.
Case Report
A 19-year-old woman initially presented to an outside dermatologist for evaluation of new-onset scarring alopecia, crusted erythematous plaques on the face and arms, and arthralgia. A punch biopsy of a lesion on the left arm demonstrated a lichenoid and perivascular lymphocytic infiltrate with scattered necrotic keratinocytes, perifollicular inflammation, and focally thickened basement membrane at the dermoepidermal junction consistent with discoid lupus erythematosus (DLE). A laboratory workup for SLE revealed 1:1280 antinuclear antibodies (reference range, negative <1:80) with elevated titers of double-stranded DNA, Smith, ribonucleoprotein, Sjögren syndrome A, and Sjögren syndrome B autoantibodies with low complement levels. Based on these findings, a diagnosis of SLE and DLE was made.
At that time, the patient was started on hydroxychloroquine 200 mg twice daily for SLE. Four days later she developed swelling in both hands and feet, and hydroxychloroquine was stopped due to a presumed adverse reaction; however, her symptoms subsequently were determined to be polyarthritis secondary to a lupus flare. Prednisone 10 mg once daily was then initiated. The patient was encouraged to restart hydroxychloroquine, but she declined.
Over the next 13 months, the patient developed severe photosensitivity, oral ulcers, Raynaud phenomenon, anemia, and nephrotic-range proteinuria. She ultimately was diagnosed by the nephrology department at our institution with mixed diffuse proliferative and membranous glomerulonephritis. Induction therapy with oral mycophenolate mofetil 1000 mg twice daily and prednisone 60 mg once daily was started, followed by the addition of tacrolimus 1 mg twice daily. Despite immunosuppressive therapy, she continued to develop new discoid lesions on the face, chest, and arms. Th
After 4 weeks of treatment with mycophenolate mofetil, prednisone, and tacrolimus, the patient developed a painful vesicular rash on the left breast with extension over the left axilla and scapula in a T3 to T4 dermatomal distribution. A clinical diagnosis of HZ was made, and she was started on intravenous acyclovir 10 mg/kg in dextrose 5% every 8 hours for 4 days followed by oral valacyclovir 1000 mg every 8 hours for 14 days, which led to resolution of the eruption.
Over the next 4 months, the patient continued to experience pain confined to the same dermatomal area as the HZ, which was consistent with postherpetic neuralgia. Mycophenolate mofetil was discontinued after she developed acute liver toxicity attributed to the drug. Upon discontinuation, the patient developed a new pruritic rash on both arms and the back. Physical examination by the dermatology department at our institution revealed diffuse, scaly, hyperpigmented papules and annular plaques with central pink hypopigmentation on the face, ears, anterior chest, arms, hands, and back. On the left anterior chest and back, the distribution was strikingly unilateral and multidermatomal (Figure 1). Upon further questioning, the patient confirmed that the areas of the new rash coincided with areas previously affected by HZ. Histologic examination of a representative lesion from the left lateral breast revealed hyperkeratosis, follicular plugging, a patchy lichenoid and perivascular mononuclear cell infiltrate, and pigment incontinence (Figure 2A). These histologic features were subtle and were not diagnostic for lupus; however, direct immunofluorescence demonstrated a continuous granular band of IgG and C3 along the dermoepidermal junction, confirming the diagnosis of DLE (Figure 2B). The histologic findings and clinical presentation were consistent with the development of DLE in areas of previous trauma from HZ. The patient continues to follow-up with the rheumatology and nephrology departments but was lost to dermatology follow-up.
Comment
The pathogenesis of DLE is poorly understood but is thought to be multifactorial, involving genetics, sun exposure, and immune dysregulation.1 Development of DLE lesions in skin traumatized by tattoos, scratches, scars, and prolonged heat exposure has been reported.2 Clarification of the mechanism(s) underlying these traumatized areas may provide insight into the pathophysiology of DLE.
The isomorphic response, also known as the Köbner phenomenon, is the development of a preexisting skin condition at a site of trauma. This phenomenon has been observed in several dermatologic conditions including psoriasis, lichen planus, systemic sclerosis, dermatomyositis, sarcoidosis, vitiligo, and DLE.3 Koebnerization may result from trauma to the skin caused by scratches, sun exposure, radiography, prolonged heat and cold exposure, pressure, tattoos, scars, and inflammatory dermatoses.2,4 Ueki4 suggested that localized trauma to the skin stimulates an immune response that makes the traumatized site a target for a preexisting skin condition. Inflammatory mediators such as IL-1, tumor necrosis factor α, IL-6, and interferon γ have been implicated in the pathophysiology of the isomorphic response.4
Wolf isotopic response is a similar entity that refers to the development of a novel skin condition at the site of a distinct, previously resolved skin disorder. This phenomenon was described by Wolf et al5 in 1995, and since then over 170 cases have been reported.5-7 In most cases the initial skin condition is HZ, although herpes simplex virus has also been implicated. The common resulting skin conditions include granulomatous reactions, malignant tumors, lichen planus, morphea, and infections. The notion that the antecedent skin disease alters the affected site and causes it to be more susceptible to autoimmunity has been proposed as a mechanism for the isotopic response.7,8 While one might consider our presentation of DLE following HZ to be an isotopic response, we believe this case is best classified as an isomorphic response, as the patient already had an established diagnosis of DLE.
The development of DLE at the site of a previous HZ eruption has been described in 2 other cases of young women with SLE.9,10 Unique to our case is the development of a multidermatomal eruption, which may be an indication of her degree of immunosuppression, as immunosuppressed patients are more likely to present with multidermatomal reactivation of varicella zoster virus and postherpetic neuralgia.11 The similarities between our case and the 2 prior reports—including the patients’ age, sex, history of SLE, and degree of immunosuppression—are noteworthy in that they may represent a subset of SLE patients who are predisposed to developing koebnerization following HZ. Physicians should be aware of this phenomenon and consider being proactive in preventing long-term damage.
When feasible, physicians should consider administering the HZ vaccine to reduce the course and severity of HZ before prescribing immunosuppressive agents. When HZ presents in young, immunosuppressed women with a history of SLE, we suggest monitoring the affected sites closely for any evidence of DLE. Topical corticosteroids should be applied to involved areas of the face or body at the earliest appearance of such lesions, which may prevent the isomorphic response and its potentially scarring DLE lesions. This will be our therapeutic approach if we encounter a similar clinical situation in the future. Fur
Acknowledgment
We thank Carolyn E. Grotkowski, MD, from the Department of Pathology, Cooper Medical School of Rowan University, Camden, New Jersey, for her assistance in photographing the pathology slides.
Cutaneous manifestations of systemic lupus erythematosus (SLE) can be classified as lupus-specific or lupus-nonspecific skin lesions. Lupus-specific lesions commonly are photodistributed, with involvement of the malar region, arms, and trunk. The development of discoid lupus erythematosus (DLE) in areas of trauma, including sun-exposed skin, is not uncommon and may be associated with an isomorphic response. We present a rare case of an isomorphic response following herpes zoster (HZ) in a young woman undergoing treatment with immunosuppressive agents for SLE and DLE. Potential prophylactic therapy also is discussed.
Case Report
A 19-year-old woman initially presented to an outside dermatologist for evaluation of new-onset scarring alopecia, crusted erythematous plaques on the face and arms, and arthralgia. A punch biopsy of a lesion on the left arm demonstrated a lichenoid and perivascular lymphocytic infiltrate with scattered necrotic keratinocytes, perifollicular inflammation, and focally thickened basement membrane at the dermoepidermal junction consistent with discoid lupus erythematosus (DLE). A laboratory workup for SLE revealed 1:1280 antinuclear antibodies (reference range, negative <1:80) with elevated titers of double-stranded DNA, Smith, ribonucleoprotein, Sjögren syndrome A, and Sjögren syndrome B autoantibodies with low complement levels. Based on these findings, a diagnosis of SLE and DLE was made.
At that time, the patient was started on hydroxychloroquine 200 mg twice daily for SLE. Four days later she developed swelling in both hands and feet, and hydroxychloroquine was stopped due to a presumed adverse reaction; however, her symptoms subsequently were determined to be polyarthritis secondary to a lupus flare. Prednisone 10 mg once daily was then initiated. The patient was encouraged to restart hydroxychloroquine, but she declined.
Over the next 13 months, the patient developed severe photosensitivity, oral ulcers, Raynaud phenomenon, anemia, and nephrotic-range proteinuria. She ultimately was diagnosed by the nephrology department at our institution with mixed diffuse proliferative and membranous glomerulonephritis. Induction therapy with oral mycophenolate mofetil 1000 mg twice daily and prednisone 60 mg once daily was started, followed by the addition of tacrolimus 1 mg twice daily. Despite immunosuppressive therapy, she continued to develop new discoid lesions on the face, chest, and arms. Th
After 4 weeks of treatment with mycophenolate mofetil, prednisone, and tacrolimus, the patient developed a painful vesicular rash on the left breast with extension over the left axilla and scapula in a T3 to T4 dermatomal distribution. A clinical diagnosis of HZ was made, and she was started on intravenous acyclovir 10 mg/kg in dextrose 5% every 8 hours for 4 days followed by oral valacyclovir 1000 mg every 8 hours for 14 days, which led to resolution of the eruption.
Over the next 4 months, the patient continued to experience pain confined to the same dermatomal area as the HZ, which was consistent with postherpetic neuralgia. Mycophenolate mofetil was discontinued after she developed acute liver toxicity attributed to the drug. Upon discontinuation, the patient developed a new pruritic rash on both arms and the back. Physical examination by the dermatology department at our institution revealed diffuse, scaly, hyperpigmented papules and annular plaques with central pink hypopigmentation on the face, ears, anterior chest, arms, hands, and back. On the left anterior chest and back, the distribution was strikingly unilateral and multidermatomal (Figure 1). Upon further questioning, the patient confirmed that the areas of the new rash coincided with areas previously affected by HZ. Histologic examination of a representative lesion from the left lateral breast revealed hyperkeratosis, follicular plugging, a patchy lichenoid and perivascular mononuclear cell infiltrate, and pigment incontinence (Figure 2A). These histologic features were subtle and were not diagnostic for lupus; however, direct immunofluorescence demonstrated a continuous granular band of IgG and C3 along the dermoepidermal junction, confirming the diagnosis of DLE (Figure 2B). The histologic findings and clinical presentation were consistent with the development of DLE in areas of previous trauma from HZ. The patient continues to follow-up with the rheumatology and nephrology departments but was lost to dermatology follow-up.
Comment
The pathogenesis of DLE is poorly understood but is thought to be multifactorial, involving genetics, sun exposure, and immune dysregulation.1 Development of DLE lesions in skin traumatized by tattoos, scratches, scars, and prolonged heat exposure has been reported.2 Clarification of the mechanism(s) underlying these traumatized areas may provide insight into the pathophysiology of DLE.
The isomorphic response, also known as the Köbner phenomenon, is the development of a preexisting skin condition at a site of trauma. This phenomenon has been observed in several dermatologic conditions including psoriasis, lichen planus, systemic sclerosis, dermatomyositis, sarcoidosis, vitiligo, and DLE.3 Koebnerization may result from trauma to the skin caused by scratches, sun exposure, radiography, prolonged heat and cold exposure, pressure, tattoos, scars, and inflammatory dermatoses.2,4 Ueki4 suggested that localized trauma to the skin stimulates an immune response that makes the traumatized site a target for a preexisting skin condition. Inflammatory mediators such as IL-1, tumor necrosis factor α, IL-6, and interferon γ have been implicated in the pathophysiology of the isomorphic response.4
Wolf isotopic response is a similar entity that refers to the development of a novel skin condition at the site of a distinct, previously resolved skin disorder. This phenomenon was described by Wolf et al5 in 1995, and since then over 170 cases have been reported.5-7 In most cases the initial skin condition is HZ, although herpes simplex virus has also been implicated. The common resulting skin conditions include granulomatous reactions, malignant tumors, lichen planus, morphea, and infections. The notion that the antecedent skin disease alters the affected site and causes it to be more susceptible to autoimmunity has been proposed as a mechanism for the isotopic response.7,8 While one might consider our presentation of DLE following HZ to be an isotopic response, we believe this case is best classified as an isomorphic response, as the patient already had an established diagnosis of DLE.
The development of DLE at the site of a previous HZ eruption has been described in 2 other cases of young women with SLE.9,10 Unique to our case is the development of a multidermatomal eruption, which may be an indication of her degree of immunosuppression, as immunosuppressed patients are more likely to present with multidermatomal reactivation of varicella zoster virus and postherpetic neuralgia.11 The similarities between our case and the 2 prior reports—including the patients’ age, sex, history of SLE, and degree of immunosuppression—are noteworthy in that they may represent a subset of SLE patients who are predisposed to developing koebnerization following HZ. Physicians should be aware of this phenomenon and consider being proactive in preventing long-term damage.
When feasible, physicians should consider administering the HZ vaccine to reduce the course and severity of HZ before prescribing immunosuppressive agents. When HZ presents in young, immunosuppressed women with a history of SLE, we suggest monitoring the affected sites closely for any evidence of DLE. Topical corticosteroids should be applied to involved areas of the face or body at the earliest appearance of such lesions, which may prevent the isomorphic response and its potentially scarring DLE lesions. This will be our therapeutic approach if we encounter a similar clinical situation in the future. Fur
Acknowledgment
We thank Carolyn E. Grotkowski, MD, from the Department of Pathology, Cooper Medical School of Rowan University, Camden, New Jersey, for her assistance in photographing the pathology slides.
- Lin JH, Dutz JP, Sontheimer RD, et al. Pathophysiology of cutaneous lupus erythematosus. Clinic Rev Allerg Immunol. 2007;33:85-106.
- Ueki H. Köbner phenomenon in lupus erythematosus [in German]. Hautarzt. 1994;45:154-160.
- Boyd AS, Neldner KH. The isomorphic response of Koebner. Int J Dermatol. 1990;29:401-410.
- Ueki H. Koebner phenomenon in lupus erythematosus with special consideration of clinical findings. Autoimmun Rev. 2005;4:219-223.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-vs-host disease to sites of skin injury. Arch Dermatol. 2011;147:1081-1086.
- Lee NY, Daniel AS, Dasher DA, et al. Cutaneous lupus after herpes zoster: isomorphic, isotopic, or both [published online May 29, 2012]? Pediatr Dermatol. 2013;30:e110-e113.
- Longhi BS, Centeville M, Marini R, et al. Koebner’s phenomenon in systemic lupus erythematosus. Rheumatol Int. 2012;32:1403-1405.
- Failla V, Jacques J, Castronovo C, et al. Herpes zoster in patients treated with biologicals. Dermatology. 2012;224:251-256.
- Lin JH, Dutz JP, Sontheimer RD, et al. Pathophysiology of cutaneous lupus erythematosus. Clinic Rev Allerg Immunol. 2007;33:85-106.
- Ueki H. Köbner phenomenon in lupus erythematosus [in German]. Hautarzt. 1994;45:154-160.
- Boyd AS, Neldner KH. The isomorphic response of Koebner. Int J Dermatol. 1990;29:401-410.
- Ueki H. Koebner phenomenon in lupus erythematosus with special consideration of clinical findings. Autoimmun Rev. 2005;4:219-223.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-vs-host disease to sites of skin injury. Arch Dermatol. 2011;147:1081-1086.
- Lee NY, Daniel AS, Dasher DA, et al. Cutaneous lupus after herpes zoster: isomorphic, isotopic, or both [published online May 29, 2012]? Pediatr Dermatol. 2013;30:e110-e113.
- Longhi BS, Centeville M, Marini R, et al. Koebner’s phenomenon in systemic lupus erythematosus. Rheumatol Int. 2012;32:1403-1405.
- Failla V, Jacques J, Castronovo C, et al. Herpes zoster in patients treated with biologicals. Dermatology. 2012;224:251-256.
Practice Points
- Discoid lupus erythematosus (DLE) most commonly presents as scaling and crusted plaques in sun-exposed areas of the face and arms. It also may present in skin traumatized by tattoos, scratches, scars, prolonged heat exposure, andherpes zoster (HZ).
- Patients with a history of DLE who subsequently develop HZ should be followed closely for the development of DLE in HZ-affected dermatomes.
- Following resolution of HZ, topical corticosteroids may have a role in prevention of DLE in HZ-affected dermatomes.
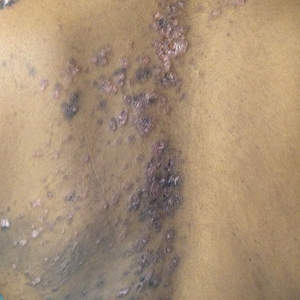
Secukinumab Emerges as a Rapidly Effective Therapy for Pityriasis Rubra Pilaris
Although there currently are no formal guidelines for the treatment of refractory pityriasis rubra pilaris (PRP), successful off-label treatment of the condition with multiple biologics approved for psoriasis has been reported.1,2 Secukinumab, an IL-17A antagonist, has shown particularly striking results in the treatment of PRP in 2 recent case reports.3,4 We report 2 additional cases of severe refractory PRP that responded rapidly to treatment with secukinumab. In both cases, the patients’ erythematous plaques resolved or had nearly resolved by week 4 of treatment. Our findings suggest that IL-17 plays an important role in PRP pathogenesis and support future clinical trials of anti–IL-17 agents for treatment of this entity.
Case Reports
Patient 1
A 60-year-old man with a history of biopsy-proven PRP presented with persistent generalized erythema, scattered patches of normal skin, and hyperkeratotic plaques on the bilateral palms of 1 year’s duration. Previous therapies included topical steroids, topical calcipotriene, adalimumab 40 mg once every other week, infliximab 5 mg/kg once every 8 weeks, ustekinumab 90 mg once every 12 weeks, acitretin 25 mg once daily, and most recently cyclosporine 200 mg twice daily. Of these treatments, infliximab was the
At 4 weeks’ follow-up, there was a marked decrease in erythema and scaling. The body surface area affected had decreased to 5%, and improvement of palmar keratoderma was noted. The patient continued with maintenance dosing of secukinumab 300 mg once every 4 weeks. By week 8, the erythema had fully resolved (Figure 1B), and he remained clear at week 24. No adverse events were noted since initiation of therapy.
Patient 2
A 74-year-old woman with a history of PRP that had previously been misdiagnosed as psoriasis by an outside physician presented for evaluation of palmoplantar keratoderma (Figure 2A), follicular hyperkeratosis, and erythematous plaques on the trunk and arms of 5 years’ duration. Previous therapies included topical steroids, topical urea, methotrexate 20 mg once weekly, adalimumab 40 mg once every other week, infliximab 10 mg/kg once every 4 weeks, ustekinumab 90 mg once every 12 weeks, and most recently acitretin 50 mg once daily.
The patient had been maintained on ustekinumab and acitretin for 2 years with only mild improvement. Ustekinumab was then discontinued, and after 3 months treatment with secukinumab was added to the once-daily acitretin. Similar to Patient 1, loading doses of secukinumab 300 mg were administered once weekly for 5 weeks. The plaques on the trunk and arms had resolved by week 4, but the palmoplantar keratoderma persisted. The patient continued with the maintenance dose of secukinumab 300 mg once every 4 weeks and reported an increase in peeling of the palms and soles at week 8.
By week 12 of treatment, the palmar keratoderma had resolved, and debridement of the soles revealed patches of normal skin (Figure 2B). By week 52, no adverse events had been noted. The patient continued to experience mild keratoderma of the soles, making us reluctant to discontinue acitretin; however, she has maintained her maximal response, and her quality of life has significantly improved. The patient was continued on acitretin and secukinumab, and her condition remained stable.
Comment
Because there are no formal treatment guidelines for refractory PRP, case reports play an important role in clinical decision-making. When a patient is unresponsive to topical medications and first-line traditional systemic therapies (eg, methotrexate, cyclosporine, acitretin), biologic drugs effective in the treatment of psoriasis are widely accepted as the next therapeutic step.1 The biologic medications that are most often reported in the treatment of PRP are the TNF-α antagonists, as they have been available the longest.1-2 In a systematic review of 15 patients with PRP who were treated with TNF-α antagonists,2 80% of patients achieved complete response (mean time to maximal response, 5 months). There also are a number of reports of successful treatment of PRP with the IL-12/23 antagonist ustekinumab, which has been commercially available since 2009.5-9 Although improvement was noted in most of these patients at the time of the second injection (week 4 of therapy), maximal response with ustekinumab typically occurs between weeks 12 and 28.10
In our cases of PRP treated with secukinumab as well as 2 others that were recently reported in the literature, resolution of erythema and plaques was rapid. This superiority of the response rate parallels the performance of secukinumab relative to ustekinumab in patients with psoriasis11 In one case of a 67-year-old man with PRP treated with secukinumab, scaling and pruritus were reduced by week 3 of treatment and erythema had cleared by week 8.3 In another case of a 33-year-old woman with PRP, pruritus resolved after 1 week of treatment and erythematous plaques and palmoplantar keratoderma improved by week 2.4 In both of our cases, plaques had resolved or nearly resolved by week 4 of follow-up. Patient 1 achieved complete response at week 8 of therapy. Patient 2 never attained complete response, but by week 12 she achieved maximal response, which still resulted in markedly increased quality of life. We do not intend to make additions to her treatment plan because she is currently the clearest she has been since onset of symptoms and is happy with her present condition.
Although it is difficult to predict the long-term prognosis in our 2 patients, we will continue their current regimens indefinitely—as long as the response persists and no adverse events are experienced. This approach is consistent with guidelines for management of plaque psoriasis with secukinumab.12
This accumulation of evidence suggests the importance of the role of IL-17 in the pathogenesis of PRP. The serum level of IL-17 was not evaluated in our patients, but elevation of IL-17 has been reported in a case of PRP.13 Further studies are needed to clarify the role of IL-17 in this disease entity.
Conclusion
Given the refractory nature of PRP and the relative safety of targeted immunotherapy, trials of new biologics and potent small molecules approved for psoriasis treatment are worth exploring for PRP. In light of our reports and those in the literature and given the relative safety of anti–IL-17 agents, it may be reasonable to consider such agents as a first-line therapy for this predictably refractory disease.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris. Am J Clin Dermatol. 2010;11:157-170.
- Petrof G, Almaani N, Archer CB, et al. A systematic review of the literature on the treatment of pityriasis rubra pilaris type 1 with TNF-antagonists. J Eur Acad Dermatol Venereol. 2013;27:E131-E135.
- Schuster D, Pfister-Wartha A, Bruckner-Tuderman L, et al. Successful treatment of refractory pityriasis rubra pilaris with secukinumab. JAMA Dermatol. 2016;152:1278-1280.
- Gauci ML, Jachiet M, Gottlieb J, et al. Successful treatment of type II pityriasis rubra pilaris with secukinumab. JAAD Case Rep. 2016;2:462-264.
- Chowdhary M, Davila U, Cohen DJ. Ustekinumab as an alternative treatment option for chronic pityriasis rubra pilaris. Case Rep Dermatol. 2015;7:46-50.
- Wohlrab J, Kreft B. Treatment of pityriasis rubra pilaris with ustekinumab. Br J Dermatol. 2010;163:655-656.
- Villaverde RR, Cano DS. Successful treatment of type 1 pityriasis rubra pilaris with ustekinumab therapy. Eur J Dermatol. 2010;20:630-631.
- Di Stefani A, Galluzzo M, Talamonti M, et al. Long-term ustekinumab treatment for refractory type I pityriasis rubra pilaris. J Dermatol Case Rep. 2013;7:5-9.
- Eytan O, Sarig O, Sprecher E, et al. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. 2014;171:420-422.
- Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371:1675-1684.
- Thaçi D, Blauvelt A, Reich K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73:400-409.
- van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Adnot-Desanlis L, Antonicelli F, Tabary T, et al. Effectiveness of infliximab in pityriasis rubra pilaris is associated with pro-inflammatory cytokine inhibition. Dermatology. 2013;226:41-46.
Although there currently are no formal guidelines for the treatment of refractory pityriasis rubra pilaris (PRP), successful off-label treatment of the condition with multiple biologics approved for psoriasis has been reported.1,2 Secukinumab, an IL-17A antagonist, has shown particularly striking results in the treatment of PRP in 2 recent case reports.3,4 We report 2 additional cases of severe refractory PRP that responded rapidly to treatment with secukinumab. In both cases, the patients’ erythematous plaques resolved or had nearly resolved by week 4 of treatment. Our findings suggest that IL-17 plays an important role in PRP pathogenesis and support future clinical trials of anti–IL-17 agents for treatment of this entity.
Case Reports
Patient 1
A 60-year-old man with a history of biopsy-proven PRP presented with persistent generalized erythema, scattered patches of normal skin, and hyperkeratotic plaques on the bilateral palms of 1 year’s duration. Previous therapies included topical steroids, topical calcipotriene, adalimumab 40 mg once every other week, infliximab 5 mg/kg once every 8 weeks, ustekinumab 90 mg once every 12 weeks, acitretin 25 mg once daily, and most recently cyclosporine 200 mg twice daily. Of these treatments, infliximab was the
At 4 weeks’ follow-up, there was a marked decrease in erythema and scaling. The body surface area affected had decreased to 5%, and improvement of palmar keratoderma was noted. The patient continued with maintenance dosing of secukinumab 300 mg once every 4 weeks. By week 8, the erythema had fully resolved (Figure 1B), and he remained clear at week 24. No adverse events were noted since initiation of therapy.
Patient 2
A 74-year-old woman with a history of PRP that had previously been misdiagnosed as psoriasis by an outside physician presented for evaluation of palmoplantar keratoderma (Figure 2A), follicular hyperkeratosis, and erythematous plaques on the trunk and arms of 5 years’ duration. Previous therapies included topical steroids, topical urea, methotrexate 20 mg once weekly, adalimumab 40 mg once every other week, infliximab 10 mg/kg once every 4 weeks, ustekinumab 90 mg once every 12 weeks, and most recently acitretin 50 mg once daily.
The patient had been maintained on ustekinumab and acitretin for 2 years with only mild improvement. Ustekinumab was then discontinued, and after 3 months treatment with secukinumab was added to the once-daily acitretin. Similar to Patient 1, loading doses of secukinumab 300 mg were administered once weekly for 5 weeks. The plaques on the trunk and arms had resolved by week 4, but the palmoplantar keratoderma persisted. The patient continued with the maintenance dose of secukinumab 300 mg once every 4 weeks and reported an increase in peeling of the palms and soles at week 8.
By week 12 of treatment, the palmar keratoderma had resolved, and debridement of the soles revealed patches of normal skin (Figure 2B). By week 52, no adverse events had been noted. The patient continued to experience mild keratoderma of the soles, making us reluctant to discontinue acitretin; however, she has maintained her maximal response, and her quality of life has significantly improved. The patient was continued on acitretin and secukinumab, and her condition remained stable.
Comment
Because there are no formal treatment guidelines for refractory PRP, case reports play an important role in clinical decision-making. When a patient is unresponsive to topical medications and first-line traditional systemic therapies (eg, methotrexate, cyclosporine, acitretin), biologic drugs effective in the treatment of psoriasis are widely accepted as the next therapeutic step.1 The biologic medications that are most often reported in the treatment of PRP are the TNF-α antagonists, as they have been available the longest.1-2 In a systematic review of 15 patients with PRP who were treated with TNF-α antagonists,2 80% of patients achieved complete response (mean time to maximal response, 5 months). There also are a number of reports of successful treatment of PRP with the IL-12/23 antagonist ustekinumab, which has been commercially available since 2009.5-9 Although improvement was noted in most of these patients at the time of the second injection (week 4 of therapy), maximal response with ustekinumab typically occurs between weeks 12 and 28.10
In our cases of PRP treated with secukinumab as well as 2 others that were recently reported in the literature, resolution of erythema and plaques was rapid. This superiority of the response rate parallels the performance of secukinumab relative to ustekinumab in patients with psoriasis11 In one case of a 67-year-old man with PRP treated with secukinumab, scaling and pruritus were reduced by week 3 of treatment and erythema had cleared by week 8.3 In another case of a 33-year-old woman with PRP, pruritus resolved after 1 week of treatment and erythematous plaques and palmoplantar keratoderma improved by week 2.4 In both of our cases, plaques had resolved or nearly resolved by week 4 of follow-up. Patient 1 achieved complete response at week 8 of therapy. Patient 2 never attained complete response, but by week 12 she achieved maximal response, which still resulted in markedly increased quality of life. We do not intend to make additions to her treatment plan because she is currently the clearest she has been since onset of symptoms and is happy with her present condition.
Although it is difficult to predict the long-term prognosis in our 2 patients, we will continue their current regimens indefinitely—as long as the response persists and no adverse events are experienced. This approach is consistent with guidelines for management of plaque psoriasis with secukinumab.12
This accumulation of evidence suggests the importance of the role of IL-17 in the pathogenesis of PRP. The serum level of IL-17 was not evaluated in our patients, but elevation of IL-17 has been reported in a case of PRP.13 Further studies are needed to clarify the role of IL-17 in this disease entity.
Conclusion
Given the refractory nature of PRP and the relative safety of targeted immunotherapy, trials of new biologics and potent small molecules approved for psoriasis treatment are worth exploring for PRP. In light of our reports and those in the literature and given the relative safety of anti–IL-17 agents, it may be reasonable to consider such agents as a first-line therapy for this predictably refractory disease.
Although there currently are no formal guidelines for the treatment of refractory pityriasis rubra pilaris (PRP), successful off-label treatment of the condition with multiple biologics approved for psoriasis has been reported.1,2 Secukinumab, an IL-17A antagonist, has shown particularly striking results in the treatment of PRP in 2 recent case reports.3,4 We report 2 additional cases of severe refractory PRP that responded rapidly to treatment with secukinumab. In both cases, the patients’ erythematous plaques resolved or had nearly resolved by week 4 of treatment. Our findings suggest that IL-17 plays an important role in PRP pathogenesis and support future clinical trials of anti–IL-17 agents for treatment of this entity.
Case Reports
Patient 1
A 60-year-old man with a history of biopsy-proven PRP presented with persistent generalized erythema, scattered patches of normal skin, and hyperkeratotic plaques on the bilateral palms of 1 year’s duration. Previous therapies included topical steroids, topical calcipotriene, adalimumab 40 mg once every other week, infliximab 5 mg/kg once every 8 weeks, ustekinumab 90 mg once every 12 weeks, acitretin 25 mg once daily, and most recently cyclosporine 200 mg twice daily. Of these treatments, infliximab was the
At 4 weeks’ follow-up, there was a marked decrease in erythema and scaling. The body surface area affected had decreased to 5%, and improvement of palmar keratoderma was noted. The patient continued with maintenance dosing of secukinumab 300 mg once every 4 weeks. By week 8, the erythema had fully resolved (Figure 1B), and he remained clear at week 24. No adverse events were noted since initiation of therapy.
Patient 2
A 74-year-old woman with a history of PRP that had previously been misdiagnosed as psoriasis by an outside physician presented for evaluation of palmoplantar keratoderma (Figure 2A), follicular hyperkeratosis, and erythematous plaques on the trunk and arms of 5 years’ duration. Previous therapies included topical steroids, topical urea, methotrexate 20 mg once weekly, adalimumab 40 mg once every other week, infliximab 10 mg/kg once every 4 weeks, ustekinumab 90 mg once every 12 weeks, and most recently acitretin 50 mg once daily.
The patient had been maintained on ustekinumab and acitretin for 2 years with only mild improvement. Ustekinumab was then discontinued, and after 3 months treatment with secukinumab was added to the once-daily acitretin. Similar to Patient 1, loading doses of secukinumab 300 mg were administered once weekly for 5 weeks. The plaques on the trunk and arms had resolved by week 4, but the palmoplantar keratoderma persisted. The patient continued with the maintenance dose of secukinumab 300 mg once every 4 weeks and reported an increase in peeling of the palms and soles at week 8.
By week 12 of treatment, the palmar keratoderma had resolved, and debridement of the soles revealed patches of normal skin (Figure 2B). By week 52, no adverse events had been noted. The patient continued to experience mild keratoderma of the soles, making us reluctant to discontinue acitretin; however, she has maintained her maximal response, and her quality of life has significantly improved. The patient was continued on acitretin and secukinumab, and her condition remained stable.
Comment
Because there are no formal treatment guidelines for refractory PRP, case reports play an important role in clinical decision-making. When a patient is unresponsive to topical medications and first-line traditional systemic therapies (eg, methotrexate, cyclosporine, acitretin), biologic drugs effective in the treatment of psoriasis are widely accepted as the next therapeutic step.1 The biologic medications that are most often reported in the treatment of PRP are the TNF-α antagonists, as they have been available the longest.1-2 In a systematic review of 15 patients with PRP who were treated with TNF-α antagonists,2 80% of patients achieved complete response (mean time to maximal response, 5 months). There also are a number of reports of successful treatment of PRP with the IL-12/23 antagonist ustekinumab, which has been commercially available since 2009.5-9 Although improvement was noted in most of these patients at the time of the second injection (week 4 of therapy), maximal response with ustekinumab typically occurs between weeks 12 and 28.10
In our cases of PRP treated with secukinumab as well as 2 others that were recently reported in the literature, resolution of erythema and plaques was rapid. This superiority of the response rate parallels the performance of secukinumab relative to ustekinumab in patients with psoriasis11 In one case of a 67-year-old man with PRP treated with secukinumab, scaling and pruritus were reduced by week 3 of treatment and erythema had cleared by week 8.3 In another case of a 33-year-old woman with PRP, pruritus resolved after 1 week of treatment and erythematous plaques and palmoplantar keratoderma improved by week 2.4 In both of our cases, plaques had resolved or nearly resolved by week 4 of follow-up. Patient 1 achieved complete response at week 8 of therapy. Patient 2 never attained complete response, but by week 12 she achieved maximal response, which still resulted in markedly increased quality of life. We do not intend to make additions to her treatment plan because she is currently the clearest she has been since onset of symptoms and is happy with her present condition.
Although it is difficult to predict the long-term prognosis in our 2 patients, we will continue their current regimens indefinitely—as long as the response persists and no adverse events are experienced. This approach is consistent with guidelines for management of plaque psoriasis with secukinumab.12
This accumulation of evidence suggests the importance of the role of IL-17 in the pathogenesis of PRP. The serum level of IL-17 was not evaluated in our patients, but elevation of IL-17 has been reported in a case of PRP.13 Further studies are needed to clarify the role of IL-17 in this disease entity.
Conclusion
Given the refractory nature of PRP and the relative safety of targeted immunotherapy, trials of new biologics and potent small molecules approved for psoriasis treatment are worth exploring for PRP. In light of our reports and those in the literature and given the relative safety of anti–IL-17 agents, it may be reasonable to consider such agents as a first-line therapy for this predictably refractory disease.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris. Am J Clin Dermatol. 2010;11:157-170.
- Petrof G, Almaani N, Archer CB, et al. A systematic review of the literature on the treatment of pityriasis rubra pilaris type 1 with TNF-antagonists. J Eur Acad Dermatol Venereol. 2013;27:E131-E135.
- Schuster D, Pfister-Wartha A, Bruckner-Tuderman L, et al. Successful treatment of refractory pityriasis rubra pilaris with secukinumab. JAMA Dermatol. 2016;152:1278-1280.
- Gauci ML, Jachiet M, Gottlieb J, et al. Successful treatment of type II pityriasis rubra pilaris with secukinumab. JAAD Case Rep. 2016;2:462-264.
- Chowdhary M, Davila U, Cohen DJ. Ustekinumab as an alternative treatment option for chronic pityriasis rubra pilaris. Case Rep Dermatol. 2015;7:46-50.
- Wohlrab J, Kreft B. Treatment of pityriasis rubra pilaris with ustekinumab. Br J Dermatol. 2010;163:655-656.
- Villaverde RR, Cano DS. Successful treatment of type 1 pityriasis rubra pilaris with ustekinumab therapy. Eur J Dermatol. 2010;20:630-631.
- Di Stefani A, Galluzzo M, Talamonti M, et al. Long-term ustekinumab treatment for refractory type I pityriasis rubra pilaris. J Dermatol Case Rep. 2013;7:5-9.
- Eytan O, Sarig O, Sprecher E, et al. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. 2014;171:420-422.
- Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371:1675-1684.
- Thaçi D, Blauvelt A, Reich K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73:400-409.
- van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Adnot-Desanlis L, Antonicelli F, Tabary T, et al. Effectiveness of infliximab in pityriasis rubra pilaris is associated with pro-inflammatory cytokine inhibition. Dermatology. 2013;226:41-46.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris. Am J Clin Dermatol. 2010;11:157-170.
- Petrof G, Almaani N, Archer CB, et al. A systematic review of the literature on the treatment of pityriasis rubra pilaris type 1 with TNF-antagonists. J Eur Acad Dermatol Venereol. 2013;27:E131-E135.
- Schuster D, Pfister-Wartha A, Bruckner-Tuderman L, et al. Successful treatment of refractory pityriasis rubra pilaris with secukinumab. JAMA Dermatol. 2016;152:1278-1280.
- Gauci ML, Jachiet M, Gottlieb J, et al. Successful treatment of type II pityriasis rubra pilaris with secukinumab. JAAD Case Rep. 2016;2:462-264.
- Chowdhary M, Davila U, Cohen DJ. Ustekinumab as an alternative treatment option for chronic pityriasis rubra pilaris. Case Rep Dermatol. 2015;7:46-50.
- Wohlrab J, Kreft B. Treatment of pityriasis rubra pilaris with ustekinumab. Br J Dermatol. 2010;163:655-656.
- Villaverde RR, Cano DS. Successful treatment of type 1 pityriasis rubra pilaris with ustekinumab therapy. Eur J Dermatol. 2010;20:630-631.
- Di Stefani A, Galluzzo M, Talamonti M, et al. Long-term ustekinumab treatment for refractory type I pityriasis rubra pilaris. J Dermatol Case Rep. 2013;7:5-9.
- Eytan O, Sarig O, Sprecher E, et al. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. 2014;171:420-422.
- Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371:1675-1684.
- Thaçi D, Blauvelt A, Reich K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73:400-409.
- van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Adnot-Desanlis L, Antonicelli F, Tabary T, et al. Effectiveness of infliximab in pityriasis rubra pilaris is associated with pro-inflammatory cytokine inhibition. Dermatology. 2013;226:41-46.
Practice Points
- In patients with pityriasis rubra pilaris (PRP) who have not responded to topical treatments, off-label treatment with systemic therapies approved for plaque psoriasis can be considered.
- Secukinumab, an IL-17A antagonist, has shown particularly striking results in the treatment of PRP.