User login
Emergency Imaging: Atraumatic Leg Pain
Case
A 96-year-old woman with a medical history of sciatica, vertigo, osteoporosis, and dementia presented with atraumatic right leg pain. She stated that the pain, which began 4 weeks prior to presentation, started in her right groin. The patient’s primary care physician diagnosed her with tendonitis, and prescribed acetaminophen/codeine and naproxen sodium for the pain. However, the patient’s pain progressively worsened to the point where she was no longer able to ambulate or bear weight on her right hip, prompting this visit to the ED.
On physical examination, the patient’s right hip was tender to palpation without any signs of physical deformity of the lower extremity. Upon hip flexion, she grimaced and communicated her pain.
Radiographs and computed tomography images taken of the right hip, femur, and pelvis demonstrated low-bone mineral density without fracture.
What is the diagnosis?
Answer
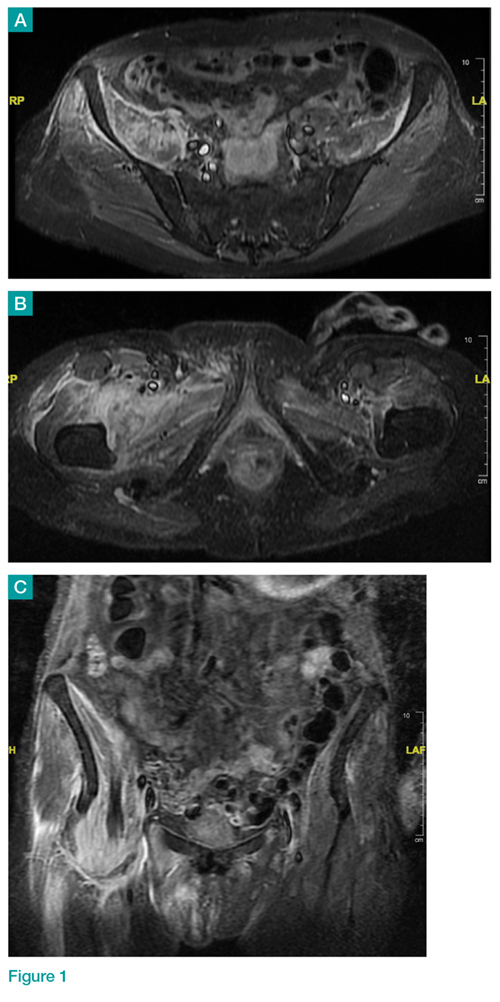
Axial and coronal edema-sensitive images of the pelvis demonstrated edema (increased signal) within the right psoas, iliacus, and iliopsoas muscles (red arrows, Figures 2a-2c), which were in contrast to the normal pelvic muscles on the left side (white arrows, Figures 2a-2c).
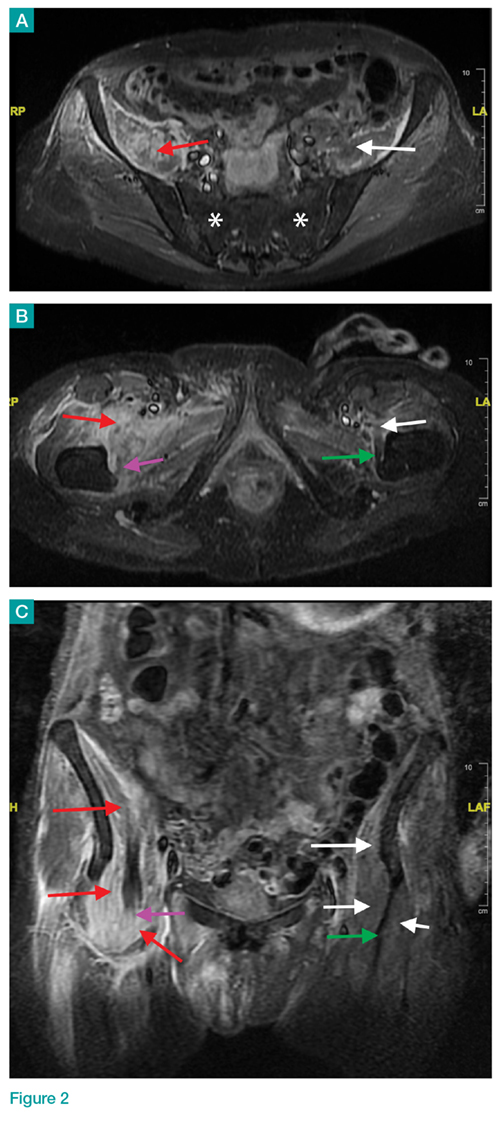
Iliopsoas Musculotendinous Unit
The iliopsoas musculotendinous unit consists of the psoas major, the psoas minor, and the iliacus, with the psoas minor absent in 40% to 50% of cases.1,2 The iliacus muscle arises from the iliac wing and inserts with the psoas tendon onto the lesser trochanter of the femur. These muscles function as primary flexors of the thigh and trunk, as well as lateral flexors of the lower vertebral column.2
Signs and Symptoms
In non-sports-related injuries, iliopsoas tendon tears typically occur in elderly female patients—even in the absence of any trauma or known predisposing factors. Patients with iliopsoas tears typically present with hip or groin pain, and weakness with hip flexion, which clinically may mimic hip or sacral fracture. An anterior thigh mass or ecchymosis may also be present. Complete tear of the iliopsoas tendon usually occurs at or near the distal insertion at the lesser trochanter, and is often associated with proximal retraction of the tendon to the level of the femoral head.1
Imaging Studies
Iliopsoas tendon injury is best evaluated with MRI, particularly with fluid-sensitive sequences. Patients with iliopsoas tendon tears have abnormal signal in the muscle belly, likely related to edema and hemorrhage, and hematoma or fluid around the torn tendon and at the site of retraction. In pediatric patients, iliopsoas injury is typically an avulsion of the lesser trochanter prior to fusion of the apophysis.3,4 In adult patients with avulsion of the lesser trochanter, this injury is regarded as a sign of metastatic disease until proven otherwise.5
Treatment
Patients with iliopsoas tendon rupture are treated conservatively with rest, ice, and physical therapy (PT). Preservation of the distal muscular insertion of the lateral portion of the iliacus muscle is thought to play a role in positive clinical outcomes.3
The patient in this case was admitted to the hospital and treated for pain with standing acetaminophen, tramadol as needed, and a lidocaine patch. After attending multiple inpatient PT sessions, she was discharged to a subacute rehabilitation facility.
1. Bergman G. MRI Web clinic – October 2015: Iliopsoas tendinopathy. Radsource. http://radsource.us/iliopsoas-tendinopathy/. Accessed November 22, 2017.
2. Van Dyke JA, Holley HC, Anderson SD. Review of iliopsoas anatomy and pathology. Radiographics. 1987;7(1):53-84. doi:10.1148/radiographics.7.1.3448631.
3. Lecouvet FE, Demondion X, Leemrijse T, Vande Berg BC, Devogelaer JP, Malghem J. Spontaneous rupture of the distal iliopsoas tendon: clinical and imaging findings, with anatomic correlations. Eur Radiol. 2005;15(11):2341-2346. doi:10.1007/s00330-005-2811-0.
4. Bui KL, Ilaslan H, Recht M, Sundaram M. Iliopsoas injury: an MRI study of patterns and prevalence correlated with clinical findings. Skeletal Radiol. 2008;37(3):245-249. doi:10.1007/s00256-007-0414-3.
5. James SL, Davies AM. Atraumatic avulsion of the lesser trochanter as an indicator of tumour infiltration. Eur Radiol. 2006;16(2):512-514.
Case
A 96-year-old woman with a medical history of sciatica, vertigo, osteoporosis, and dementia presented with atraumatic right leg pain. She stated that the pain, which began 4 weeks prior to presentation, started in her right groin. The patient’s primary care physician diagnosed her with tendonitis, and prescribed acetaminophen/codeine and naproxen sodium for the pain. However, the patient’s pain progressively worsened to the point where she was no longer able to ambulate or bear weight on her right hip, prompting this visit to the ED.
On physical examination, the patient’s right hip was tender to palpation without any signs of physical deformity of the lower extremity. Upon hip flexion, she grimaced and communicated her pain.
Radiographs and computed tomography images taken of the right hip, femur, and pelvis demonstrated low-bone mineral density without fracture.
What is the diagnosis?
Answer
Axial and coronal edema-sensitive images of the pelvis demonstrated edema (increased signal) within the right psoas, iliacus, and iliopsoas muscles (red arrows, Figures 2a-2c), which were in contrast to the normal pelvic muscles on the left side (white arrows, Figures 2a-2c).
Iliopsoas Musculotendinous Unit
The iliopsoas musculotendinous unit consists of the psoas major, the psoas minor, and the iliacus, with the psoas minor absent in 40% to 50% of cases.1,2 The iliacus muscle arises from the iliac wing and inserts with the psoas tendon onto the lesser trochanter of the femur. These muscles function as primary flexors of the thigh and trunk, as well as lateral flexors of the lower vertebral column.2
Signs and Symptoms
In non-sports-related injuries, iliopsoas tendon tears typically occur in elderly female patients—even in the absence of any trauma or known predisposing factors. Patients with iliopsoas tears typically present with hip or groin pain, and weakness with hip flexion, which clinically may mimic hip or sacral fracture. An anterior thigh mass or ecchymosis may also be present. Complete tear of the iliopsoas tendon usually occurs at or near the distal insertion at the lesser trochanter, and is often associated with proximal retraction of the tendon to the level of the femoral head.1
Imaging Studies
Iliopsoas tendon injury is best evaluated with MRI, particularly with fluid-sensitive sequences. Patients with iliopsoas tendon tears have abnormal signal in the muscle belly, likely related to edema and hemorrhage, and hematoma or fluid around the torn tendon and at the site of retraction. In pediatric patients, iliopsoas injury is typically an avulsion of the lesser trochanter prior to fusion of the apophysis.3,4 In adult patients with avulsion of the lesser trochanter, this injury is regarded as a sign of metastatic disease until proven otherwise.5
Treatment
Patients with iliopsoas tendon rupture are treated conservatively with rest, ice, and physical therapy (PT). Preservation of the distal muscular insertion of the lateral portion of the iliacus muscle is thought to play a role in positive clinical outcomes.3
The patient in this case was admitted to the hospital and treated for pain with standing acetaminophen, tramadol as needed, and a lidocaine patch. After attending multiple inpatient PT sessions, she was discharged to a subacute rehabilitation facility.
Case
A 96-year-old woman with a medical history of sciatica, vertigo, osteoporosis, and dementia presented with atraumatic right leg pain. She stated that the pain, which began 4 weeks prior to presentation, started in her right groin. The patient’s primary care physician diagnosed her with tendonitis, and prescribed acetaminophen/codeine and naproxen sodium for the pain. However, the patient’s pain progressively worsened to the point where she was no longer able to ambulate or bear weight on her right hip, prompting this visit to the ED.
On physical examination, the patient’s right hip was tender to palpation without any signs of physical deformity of the lower extremity. Upon hip flexion, she grimaced and communicated her pain.
Radiographs and computed tomography images taken of the right hip, femur, and pelvis demonstrated low-bone mineral density without fracture.
What is the diagnosis?
Answer
Axial and coronal edema-sensitive images of the pelvis demonstrated edema (increased signal) within the right psoas, iliacus, and iliopsoas muscles (red arrows, Figures 2a-2c), which were in contrast to the normal pelvic muscles on the left side (white arrows, Figures 2a-2c).
Iliopsoas Musculotendinous Unit
The iliopsoas musculotendinous unit consists of the psoas major, the psoas minor, and the iliacus, with the psoas minor absent in 40% to 50% of cases.1,2 The iliacus muscle arises from the iliac wing and inserts with the psoas tendon onto the lesser trochanter of the femur. These muscles function as primary flexors of the thigh and trunk, as well as lateral flexors of the lower vertebral column.2
Signs and Symptoms
In non-sports-related injuries, iliopsoas tendon tears typically occur in elderly female patients—even in the absence of any trauma or known predisposing factors. Patients with iliopsoas tears typically present with hip or groin pain, and weakness with hip flexion, which clinically may mimic hip or sacral fracture. An anterior thigh mass or ecchymosis may also be present. Complete tear of the iliopsoas tendon usually occurs at or near the distal insertion at the lesser trochanter, and is often associated with proximal retraction of the tendon to the level of the femoral head.1
Imaging Studies
Iliopsoas tendon injury is best evaluated with MRI, particularly with fluid-sensitive sequences. Patients with iliopsoas tendon tears have abnormal signal in the muscle belly, likely related to edema and hemorrhage, and hematoma or fluid around the torn tendon and at the site of retraction. In pediatric patients, iliopsoas injury is typically an avulsion of the lesser trochanter prior to fusion of the apophysis.3,4 In adult patients with avulsion of the lesser trochanter, this injury is regarded as a sign of metastatic disease until proven otherwise.5
Treatment
Patients with iliopsoas tendon rupture are treated conservatively with rest, ice, and physical therapy (PT). Preservation of the distal muscular insertion of the lateral portion of the iliacus muscle is thought to play a role in positive clinical outcomes.3
The patient in this case was admitted to the hospital and treated for pain with standing acetaminophen, tramadol as needed, and a lidocaine patch. After attending multiple inpatient PT sessions, she was discharged to a subacute rehabilitation facility.
1. Bergman G. MRI Web clinic – October 2015: Iliopsoas tendinopathy. Radsource. http://radsource.us/iliopsoas-tendinopathy/. Accessed November 22, 2017.
2. Van Dyke JA, Holley HC, Anderson SD. Review of iliopsoas anatomy and pathology. Radiographics. 1987;7(1):53-84. doi:10.1148/radiographics.7.1.3448631.
3. Lecouvet FE, Demondion X, Leemrijse T, Vande Berg BC, Devogelaer JP, Malghem J. Spontaneous rupture of the distal iliopsoas tendon: clinical and imaging findings, with anatomic correlations. Eur Radiol. 2005;15(11):2341-2346. doi:10.1007/s00330-005-2811-0.
4. Bui KL, Ilaslan H, Recht M, Sundaram M. Iliopsoas injury: an MRI study of patterns and prevalence correlated with clinical findings. Skeletal Radiol. 2008;37(3):245-249. doi:10.1007/s00256-007-0414-3.
5. James SL, Davies AM. Atraumatic avulsion of the lesser trochanter as an indicator of tumour infiltration. Eur Radiol. 2006;16(2):512-514.
1. Bergman G. MRI Web clinic – October 2015: Iliopsoas tendinopathy. Radsource. http://radsource.us/iliopsoas-tendinopathy/. Accessed November 22, 2017.
2. Van Dyke JA, Holley HC, Anderson SD. Review of iliopsoas anatomy and pathology. Radiographics. 1987;7(1):53-84. doi:10.1148/radiographics.7.1.3448631.
3. Lecouvet FE, Demondion X, Leemrijse T, Vande Berg BC, Devogelaer JP, Malghem J. Spontaneous rupture of the distal iliopsoas tendon: clinical and imaging findings, with anatomic correlations. Eur Radiol. 2005;15(11):2341-2346. doi:10.1007/s00330-005-2811-0.
4. Bui KL, Ilaslan H, Recht M, Sundaram M. Iliopsoas injury: an MRI study of patterns and prevalence correlated with clinical findings. Skeletal Radiol. 2008;37(3):245-249. doi:10.1007/s00256-007-0414-3.
5. James SL, Davies AM. Atraumatic avulsion of the lesser trochanter as an indicator of tumour infiltration. Eur Radiol. 2006;16(2):512-514.
Case Studies in Toxicology: Start Low and Go Slow
Case
A woman in her third decade with no known medical history was dropped off at the waiting area of the ED for evaluation of depressed mental status. Upon arrival, the patient was unresponsive and cyanotic, with a pulse oximetry of 65% on room air. Bag-valve mask (BVM) ventilation rapidly improved oxygen saturation to 90%. The patient’s other vital signs were: heart rate, 141 beats/min; blood pressure (BP), 117/65 mm Hg; and temperature, afebrile.
Upon examination, the patient’s pupils were pinpoint and her ventilatory effort was shallow, leading the emergency physician (EP) to suspect the patient’s depressed mental status was due to an opioid overdose.
The patient was given 2 mg of intravenous (IV) naloxone, after which she became more alert and responsive, with improved respiratory effort. After receiving naloxone, the patient vomited copiously. Pulmonary examination revealed diffuse rales, most prominently at the right lung base, and a cough productive of thick sputum.
During the patient’s course in the ED, she became increasingly hypotensive with systolic BP readings around 70 mm Hg; tachycardia, fluctuating at around 120 beats/min; and persistent hypoxia of 90% saturation on a nonrebreather mask. A chest X-ray demonstrated pulmonary edema with a continuous diaphragm sign suggesting pneumomediastinum. A computed tomography (CT) scan of the chest confirmed pulmonary edema with extensive pneumomediastinum, and the patient was admitted to the intensive care unit (ICU).
What is naloxone and why is it used?
Naloxone is a nonselective, short-acting, pure opioid antagonist that works at the mu, kappa, and sigma receptors, with the highest affinity for the mu receptor. It is a competitive opioid receptor antagonist that has an elimination half-life of approximately 30 minutes. Though naloxone was originally developed to reverse the effects of anesthesia postoperatively,1 today it is more commonly used to treat ventilatory depression in patients whose clinical findings are most likely due to an opioid overdose.
Opioid-dependent individuals who abstain from use for more than a few hours generally develop opioid withdrawal syndrome (OWS). The effects of OWS include mild-to-moderate tachycardia and hypertension, nausea, vomiting, piloerection, rhinorrhea, and agitated behavior. However, when opioid-dependent patients receive naloxone, OWS develops at a much faster rate (ie, seconds after naloxone administration) and is often more severe.
Findings of naloxone-precipitated OWS include pronounced vital sign abnormalities, seizures,pulmonary edema, and cardiac arrhythmias such as ventricular tachycardia.2 These latter findings are primarily due to the sudden release of catecholamines.3 In addition, patients suffer the psychological pangs of withdrawal, including dysphoria and drug craving, which often leads to poor decision-making as they search for additional opioids to alleviate these troubling effects.
What determines response to naloxone and development of OWS?
The severity of precipitated OWS following naloxone administration is determined by both the degree of the patient’s opioid dependency and the dosage and rate at which naloxone is given. The depth of opioid dependence is determined to a large extent by the quantity of opioid regularly used and the frequency of exposure. For example, a patient who takes 30 mg of oxycodone daily will likely demonstrate mild OWS, while one who uses 300 mg daily will demonstrate more severe OWS—whether due to abstinence or naloxone.
In addition, longer exposure time of the patient’s brain to opioids increases the dependency level. Continuous use of extended-release opioids or methadone, which are both of long duration, essentially “bathe” the brain receptors in opioid around the clock, whereas short-acting opioids, such as fentanyl or heroin, cause peaks and troughs in brain concentrations throughout the day. These trough periods reduce dependency, but increase the abuse liability of the opioid. Patients who only use opioids on the weekend, for example, will have minimal or no OWS following naloxone administration, nor will the toddler with an exploratory ingestion of an opioid medication found in the home. It is therefore important to gauge the extent of a patient’s opioid use to improve the safe use of naloxone in the ED.
What is the optimal dosing of naloxone and proper patient management?
It is essential for clinicians to remember that the ultimate goal of naloxone administration in the ED is to reverse ventilatory depression—not to restore a patient to a normal mental status.4 In fact, full awakening, in addition to precipitating OWS, may lead to difficult interpersonal situations in the ED, since such patients often insist on leaving the ED before the effects of naloxone wear off. This situation places the EP in the undesirable position of discharging a patient who may predictably relapse—though unlikely to die—after release.5
Management in the Hospital Setting. Given the advanced medical care environment in a hospital, the approach to opioid overdose patients can be metered. This means providing temporary noninvasive mechanical ventilatory support through BVM or laryngeal mask airways, which allow both oxygenation and ventilation (reducing the patient’s partial pressure of carbon dioxide), prior to giving naloxone.6 Studies on animal models have shown that lowering the partial pressure of carbon dioxide reduces the catecholamine response to naloxone.7
Although recent literature and textbook recommendations regarding naloxone dosages vary,1 the safest initial dose of naloxone in the hospital setting is 0.04 mg (40 mcg) IV, or 0.08 mg (80 mcg) intramuscularly (IM).8 Whether given by IV or IM route, frequent reassessment of the adequacy of spontaneous ventilatory effort and oxygenation are required.
While the rate of opioid reversal is slower when giving lower doses of naloxone, this approach reduces the severity of precipitated OWS. In fact, in most patients who receive low-dose naloxone administration will not awaken but will develop life-sustaining spontaneous ventilation.8
By monitoring of the patient’s ventilatory rate and depth, along with capnometry and pulse oximetry (without providing exogenous oxygen), the EP can identify the need for additional naloxone. Since the half-life of naloxone is shorter than that of many opioids, proper ventilatory monitoring is essential to assess for the waning of naloxone’s effects and return of respiratory depression.
Treatment in the Nonhospital Setting. Emergency medical service (EMS) workers typically, and often by situational necessity, approach opioid overdose patients more aggressively than do EPs in the ED. Although some EMS systems utilize the IV route, most EMS workers, like laypersons, administer an initial naloxone dose of 0.4 mg IM or 2 or 4 mg intranasally (IN). Due to the slower rate of absorption and lower bioavailability (with IN administration), both IM and IN naloxone equate to roughly 0.08 mg IV.
For patients in whom there is no risk for opioid dependence, the initial dose of naloxone is relatively inconsequential, and higher doses can be safely administered. However, for most patients, including those in the ED setting, in whom one cannot be certain of their depth of dependence, the safest approach is to “start low and go slow” with naloxone administration, while providing supportive care.
Case Conclusion
The patient was not opioid-naïve, explaining the catecholamine surge and related cardiovascular dysfunction and pulmonary edema. The pneumomediastinum and pulmonary aspiration were due to the violent retching and vomiting. After being admitted to the ICU, the patient was started on vancomycin and piperacillin/tazobactam for empiric coverage for mediastinal emphysema. She was kept NPO, assessed by cardiothoracic surgery, and treated with gentle fluid hydration.
A repeat CT showed a stable pneumomediastinum. Her hypoxia, tachycardia, and hypotension gradually improved over about 6 hours. The following day, the patient’s mental status normalized, and she discharged herself from the hospital against medical advice.
1. Connors NJ, Nelson LS. The evolution of recommended naloxone dosing for opioid overdose by medical specialty. J Med Toxicol. 2016;12(3):276-281. doi:10.1007/s13181-016-0559-3.
2. Lameijer, H, Azizi N, Ligtenberg JJ, Ter Maaten JC. Ventricular tachycardia after naloxone administration: a drug related complication? Case report and literature review. Drug Saf Case Rep. 2014;1(1):2. doi:10.1007/s40800-014-0002-0.
3. Kienbaum P, Thürauf N, Michel MC, Scherbaum N, Gastpar M, Peters J. Profound increase in epinephrine concentration in plasma and cardiovascular stimulation after mu-opioid receptor blockade in opioid-addicted patients during barbiturate-induced anesthesia for acute detoxification. Anesthesiology. 1998;88(5):1154-1161.
4. Kim HK, Nelson LS. Reducing the harm of opioid overdose with the safe use of naloxone: a pharmacologic review. Expert Opin Drug Saf. 2015;14 (7 ):1137-1146. doi:10.1517/14740338.2015.1037274.
5. Willman MW, Liss DB, Schwarz ES, Mullins ME. Do heroin overdose patients require observation after receiving naloxone? Clin Toxicol (Phila). 2017;55(2):81-87. doi:10.1080/15563650.2016.1253846.
6. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367(2):146-155. doi:10.1056/NEJMra1202561.
7. Mills CA, Flacke JW, Miller JD, Davis LJ, Bloor BC, Flacke WE. Cardiovascular effects of fentanyl reversal by naloxone at varying arterial carbon dioxide tensions in dogs. Anesth Analg. 1988;67(8):730-736.
8. Kim HK, Nelson LS. Reversal of opioid-induced ventilatory depression using low-dose naloxone (0.04 mg): a case series. J Med Toxicol. 2015;12(1):107-110. doi:10.1007/s13181-015-0499-3.
Case
A woman in her third decade with no known medical history was dropped off at the waiting area of the ED for evaluation of depressed mental status. Upon arrival, the patient was unresponsive and cyanotic, with a pulse oximetry of 65% on room air. Bag-valve mask (BVM) ventilation rapidly improved oxygen saturation to 90%. The patient’s other vital signs were: heart rate, 141 beats/min; blood pressure (BP), 117/65 mm Hg; and temperature, afebrile.
Upon examination, the patient’s pupils were pinpoint and her ventilatory effort was shallow, leading the emergency physician (EP) to suspect the patient’s depressed mental status was due to an opioid overdose.
The patient was given 2 mg of intravenous (IV) naloxone, after which she became more alert and responsive, with improved respiratory effort. After receiving naloxone, the patient vomited copiously. Pulmonary examination revealed diffuse rales, most prominently at the right lung base, and a cough productive of thick sputum.
During the patient’s course in the ED, she became increasingly hypotensive with systolic BP readings around 70 mm Hg; tachycardia, fluctuating at around 120 beats/min; and persistent hypoxia of 90% saturation on a nonrebreather mask. A chest X-ray demonstrated pulmonary edema with a continuous diaphragm sign suggesting pneumomediastinum. A computed tomography (CT) scan of the chest confirmed pulmonary edema with extensive pneumomediastinum, and the patient was admitted to the intensive care unit (ICU).
What is naloxone and why is it used?
Naloxone is a nonselective, short-acting, pure opioid antagonist that works at the mu, kappa, and sigma receptors, with the highest affinity for the mu receptor. It is a competitive opioid receptor antagonist that has an elimination half-life of approximately 30 minutes. Though naloxone was originally developed to reverse the effects of anesthesia postoperatively,1 today it is more commonly used to treat ventilatory depression in patients whose clinical findings are most likely due to an opioid overdose.
Opioid-dependent individuals who abstain from use for more than a few hours generally develop opioid withdrawal syndrome (OWS). The effects of OWS include mild-to-moderate tachycardia and hypertension, nausea, vomiting, piloerection, rhinorrhea, and agitated behavior. However, when opioid-dependent patients receive naloxone, OWS develops at a much faster rate (ie, seconds after naloxone administration) and is often more severe.
Findings of naloxone-precipitated OWS include pronounced vital sign abnormalities, seizures,pulmonary edema, and cardiac arrhythmias such as ventricular tachycardia.2 These latter findings are primarily due to the sudden release of catecholamines.3 In addition, patients suffer the psychological pangs of withdrawal, including dysphoria and drug craving, which often leads to poor decision-making as they search for additional opioids to alleviate these troubling effects.
What determines response to naloxone and development of OWS?
The severity of precipitated OWS following naloxone administration is determined by both the degree of the patient’s opioid dependency and the dosage and rate at which naloxone is given. The depth of opioid dependence is determined to a large extent by the quantity of opioid regularly used and the frequency of exposure. For example, a patient who takes 30 mg of oxycodone daily will likely demonstrate mild OWS, while one who uses 300 mg daily will demonstrate more severe OWS—whether due to abstinence or naloxone.
In addition, longer exposure time of the patient’s brain to opioids increases the dependency level. Continuous use of extended-release opioids or methadone, which are both of long duration, essentially “bathe” the brain receptors in opioid around the clock, whereas short-acting opioids, such as fentanyl or heroin, cause peaks and troughs in brain concentrations throughout the day. These trough periods reduce dependency, but increase the abuse liability of the opioid. Patients who only use opioids on the weekend, for example, will have minimal or no OWS following naloxone administration, nor will the toddler with an exploratory ingestion of an opioid medication found in the home. It is therefore important to gauge the extent of a patient’s opioid use to improve the safe use of naloxone in the ED.
What is the optimal dosing of naloxone and proper patient management?
It is essential for clinicians to remember that the ultimate goal of naloxone administration in the ED is to reverse ventilatory depression—not to restore a patient to a normal mental status.4 In fact, full awakening, in addition to precipitating OWS, may lead to difficult interpersonal situations in the ED, since such patients often insist on leaving the ED before the effects of naloxone wear off. This situation places the EP in the undesirable position of discharging a patient who may predictably relapse—though unlikely to die—after release.5
Management in the Hospital Setting. Given the advanced medical care environment in a hospital, the approach to opioid overdose patients can be metered. This means providing temporary noninvasive mechanical ventilatory support through BVM or laryngeal mask airways, which allow both oxygenation and ventilation (reducing the patient’s partial pressure of carbon dioxide), prior to giving naloxone.6 Studies on animal models have shown that lowering the partial pressure of carbon dioxide reduces the catecholamine response to naloxone.7
Although recent literature and textbook recommendations regarding naloxone dosages vary,1 the safest initial dose of naloxone in the hospital setting is 0.04 mg (40 mcg) IV, or 0.08 mg (80 mcg) intramuscularly (IM).8 Whether given by IV or IM route, frequent reassessment of the adequacy of spontaneous ventilatory effort and oxygenation are required.
While the rate of opioid reversal is slower when giving lower doses of naloxone, this approach reduces the severity of precipitated OWS. In fact, in most patients who receive low-dose naloxone administration will not awaken but will develop life-sustaining spontaneous ventilation.8
By monitoring of the patient’s ventilatory rate and depth, along with capnometry and pulse oximetry (without providing exogenous oxygen), the EP can identify the need for additional naloxone. Since the half-life of naloxone is shorter than that of many opioids, proper ventilatory monitoring is essential to assess for the waning of naloxone’s effects and return of respiratory depression.
Treatment in the Nonhospital Setting. Emergency medical service (EMS) workers typically, and often by situational necessity, approach opioid overdose patients more aggressively than do EPs in the ED. Although some EMS systems utilize the IV route, most EMS workers, like laypersons, administer an initial naloxone dose of 0.4 mg IM or 2 or 4 mg intranasally (IN). Due to the slower rate of absorption and lower bioavailability (with IN administration), both IM and IN naloxone equate to roughly 0.08 mg IV.
For patients in whom there is no risk for opioid dependence, the initial dose of naloxone is relatively inconsequential, and higher doses can be safely administered. However, for most patients, including those in the ED setting, in whom one cannot be certain of their depth of dependence, the safest approach is to “start low and go slow” with naloxone administration, while providing supportive care.
Case Conclusion
The patient was not opioid-naïve, explaining the catecholamine surge and related cardiovascular dysfunction and pulmonary edema. The pneumomediastinum and pulmonary aspiration were due to the violent retching and vomiting. After being admitted to the ICU, the patient was started on vancomycin and piperacillin/tazobactam for empiric coverage for mediastinal emphysema. She was kept NPO, assessed by cardiothoracic surgery, and treated with gentle fluid hydration.
A repeat CT showed a stable pneumomediastinum. Her hypoxia, tachycardia, and hypotension gradually improved over about 6 hours. The following day, the patient’s mental status normalized, and she discharged herself from the hospital against medical advice.
Case
A woman in her third decade with no known medical history was dropped off at the waiting area of the ED for evaluation of depressed mental status. Upon arrival, the patient was unresponsive and cyanotic, with a pulse oximetry of 65% on room air. Bag-valve mask (BVM) ventilation rapidly improved oxygen saturation to 90%. The patient’s other vital signs were: heart rate, 141 beats/min; blood pressure (BP), 117/65 mm Hg; and temperature, afebrile.
Upon examination, the patient’s pupils were pinpoint and her ventilatory effort was shallow, leading the emergency physician (EP) to suspect the patient’s depressed mental status was due to an opioid overdose.
The patient was given 2 mg of intravenous (IV) naloxone, after which she became more alert and responsive, with improved respiratory effort. After receiving naloxone, the patient vomited copiously. Pulmonary examination revealed diffuse rales, most prominently at the right lung base, and a cough productive of thick sputum.
During the patient’s course in the ED, she became increasingly hypotensive with systolic BP readings around 70 mm Hg; tachycardia, fluctuating at around 120 beats/min; and persistent hypoxia of 90% saturation on a nonrebreather mask. A chest X-ray demonstrated pulmonary edema with a continuous diaphragm sign suggesting pneumomediastinum. A computed tomography (CT) scan of the chest confirmed pulmonary edema with extensive pneumomediastinum, and the patient was admitted to the intensive care unit (ICU).
What is naloxone and why is it used?
Naloxone is a nonselective, short-acting, pure opioid antagonist that works at the mu, kappa, and sigma receptors, with the highest affinity for the mu receptor. It is a competitive opioid receptor antagonist that has an elimination half-life of approximately 30 minutes. Though naloxone was originally developed to reverse the effects of anesthesia postoperatively,1 today it is more commonly used to treat ventilatory depression in patients whose clinical findings are most likely due to an opioid overdose.
Opioid-dependent individuals who abstain from use for more than a few hours generally develop opioid withdrawal syndrome (OWS). The effects of OWS include mild-to-moderate tachycardia and hypertension, nausea, vomiting, piloerection, rhinorrhea, and agitated behavior. However, when opioid-dependent patients receive naloxone, OWS develops at a much faster rate (ie, seconds after naloxone administration) and is often more severe.
Findings of naloxone-precipitated OWS include pronounced vital sign abnormalities, seizures,pulmonary edema, and cardiac arrhythmias such as ventricular tachycardia.2 These latter findings are primarily due to the sudden release of catecholamines.3 In addition, patients suffer the psychological pangs of withdrawal, including dysphoria and drug craving, which often leads to poor decision-making as they search for additional opioids to alleviate these troubling effects.
What determines response to naloxone and development of OWS?
The severity of precipitated OWS following naloxone administration is determined by both the degree of the patient’s opioid dependency and the dosage and rate at which naloxone is given. The depth of opioid dependence is determined to a large extent by the quantity of opioid regularly used and the frequency of exposure. For example, a patient who takes 30 mg of oxycodone daily will likely demonstrate mild OWS, while one who uses 300 mg daily will demonstrate more severe OWS—whether due to abstinence or naloxone.
In addition, longer exposure time of the patient’s brain to opioids increases the dependency level. Continuous use of extended-release opioids or methadone, which are both of long duration, essentially “bathe” the brain receptors in opioid around the clock, whereas short-acting opioids, such as fentanyl or heroin, cause peaks and troughs in brain concentrations throughout the day. These trough periods reduce dependency, but increase the abuse liability of the opioid. Patients who only use opioids on the weekend, for example, will have minimal or no OWS following naloxone administration, nor will the toddler with an exploratory ingestion of an opioid medication found in the home. It is therefore important to gauge the extent of a patient’s opioid use to improve the safe use of naloxone in the ED.
What is the optimal dosing of naloxone and proper patient management?
It is essential for clinicians to remember that the ultimate goal of naloxone administration in the ED is to reverse ventilatory depression—not to restore a patient to a normal mental status.4 In fact, full awakening, in addition to precipitating OWS, may lead to difficult interpersonal situations in the ED, since such patients often insist on leaving the ED before the effects of naloxone wear off. This situation places the EP in the undesirable position of discharging a patient who may predictably relapse—though unlikely to die—after release.5
Management in the Hospital Setting. Given the advanced medical care environment in a hospital, the approach to opioid overdose patients can be metered. This means providing temporary noninvasive mechanical ventilatory support through BVM or laryngeal mask airways, which allow both oxygenation and ventilation (reducing the patient’s partial pressure of carbon dioxide), prior to giving naloxone.6 Studies on animal models have shown that lowering the partial pressure of carbon dioxide reduces the catecholamine response to naloxone.7
Although recent literature and textbook recommendations regarding naloxone dosages vary,1 the safest initial dose of naloxone in the hospital setting is 0.04 mg (40 mcg) IV, or 0.08 mg (80 mcg) intramuscularly (IM).8 Whether given by IV or IM route, frequent reassessment of the adequacy of spontaneous ventilatory effort and oxygenation are required.
While the rate of opioid reversal is slower when giving lower doses of naloxone, this approach reduces the severity of precipitated OWS. In fact, in most patients who receive low-dose naloxone administration will not awaken but will develop life-sustaining spontaneous ventilation.8
By monitoring of the patient’s ventilatory rate and depth, along with capnometry and pulse oximetry (without providing exogenous oxygen), the EP can identify the need for additional naloxone. Since the half-life of naloxone is shorter than that of many opioids, proper ventilatory monitoring is essential to assess for the waning of naloxone’s effects and return of respiratory depression.
Treatment in the Nonhospital Setting. Emergency medical service (EMS) workers typically, and often by situational necessity, approach opioid overdose patients more aggressively than do EPs in the ED. Although some EMS systems utilize the IV route, most EMS workers, like laypersons, administer an initial naloxone dose of 0.4 mg IM or 2 or 4 mg intranasally (IN). Due to the slower rate of absorption and lower bioavailability (with IN administration), both IM and IN naloxone equate to roughly 0.08 mg IV.
For patients in whom there is no risk for opioid dependence, the initial dose of naloxone is relatively inconsequential, and higher doses can be safely administered. However, for most patients, including those in the ED setting, in whom one cannot be certain of their depth of dependence, the safest approach is to “start low and go slow” with naloxone administration, while providing supportive care.
Case Conclusion
The patient was not opioid-naïve, explaining the catecholamine surge and related cardiovascular dysfunction and pulmonary edema. The pneumomediastinum and pulmonary aspiration were due to the violent retching and vomiting. After being admitted to the ICU, the patient was started on vancomycin and piperacillin/tazobactam for empiric coverage for mediastinal emphysema. She was kept NPO, assessed by cardiothoracic surgery, and treated with gentle fluid hydration.
A repeat CT showed a stable pneumomediastinum. Her hypoxia, tachycardia, and hypotension gradually improved over about 6 hours. The following day, the patient’s mental status normalized, and she discharged herself from the hospital against medical advice.
1. Connors NJ, Nelson LS. The evolution of recommended naloxone dosing for opioid overdose by medical specialty. J Med Toxicol. 2016;12(3):276-281. doi:10.1007/s13181-016-0559-3.
2. Lameijer, H, Azizi N, Ligtenberg JJ, Ter Maaten JC. Ventricular tachycardia after naloxone administration: a drug related complication? Case report and literature review. Drug Saf Case Rep. 2014;1(1):2. doi:10.1007/s40800-014-0002-0.
3. Kienbaum P, Thürauf N, Michel MC, Scherbaum N, Gastpar M, Peters J. Profound increase in epinephrine concentration in plasma and cardiovascular stimulation after mu-opioid receptor blockade in opioid-addicted patients during barbiturate-induced anesthesia for acute detoxification. Anesthesiology. 1998;88(5):1154-1161.
4. Kim HK, Nelson LS. Reducing the harm of opioid overdose with the safe use of naloxone: a pharmacologic review. Expert Opin Drug Saf. 2015;14 (7 ):1137-1146. doi:10.1517/14740338.2015.1037274.
5. Willman MW, Liss DB, Schwarz ES, Mullins ME. Do heroin overdose patients require observation after receiving naloxone? Clin Toxicol (Phila). 2017;55(2):81-87. doi:10.1080/15563650.2016.1253846.
6. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367(2):146-155. doi:10.1056/NEJMra1202561.
7. Mills CA, Flacke JW, Miller JD, Davis LJ, Bloor BC, Flacke WE. Cardiovascular effects of fentanyl reversal by naloxone at varying arterial carbon dioxide tensions in dogs. Anesth Analg. 1988;67(8):730-736.
8. Kim HK, Nelson LS. Reversal of opioid-induced ventilatory depression using low-dose naloxone (0.04 mg): a case series. J Med Toxicol. 2015;12(1):107-110. doi:10.1007/s13181-015-0499-3.
1. Connors NJ, Nelson LS. The evolution of recommended naloxone dosing for opioid overdose by medical specialty. J Med Toxicol. 2016;12(3):276-281. doi:10.1007/s13181-016-0559-3.
2. Lameijer, H, Azizi N, Ligtenberg JJ, Ter Maaten JC. Ventricular tachycardia after naloxone administration: a drug related complication? Case report and literature review. Drug Saf Case Rep. 2014;1(1):2. doi:10.1007/s40800-014-0002-0.
3. Kienbaum P, Thürauf N, Michel MC, Scherbaum N, Gastpar M, Peters J. Profound increase in epinephrine concentration in plasma and cardiovascular stimulation after mu-opioid receptor blockade in opioid-addicted patients during barbiturate-induced anesthesia for acute detoxification. Anesthesiology. 1998;88(5):1154-1161.
4. Kim HK, Nelson LS. Reducing the harm of opioid overdose with the safe use of naloxone: a pharmacologic review. Expert Opin Drug Saf. 2015;14 (7 ):1137-1146. doi:10.1517/14740338.2015.1037274.
5. Willman MW, Liss DB, Schwarz ES, Mullins ME. Do heroin overdose patients require observation after receiving naloxone? Clin Toxicol (Phila). 2017;55(2):81-87. doi:10.1080/15563650.2016.1253846.
6. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367(2):146-155. doi:10.1056/NEJMra1202561.
7. Mills CA, Flacke JW, Miller JD, Davis LJ, Bloor BC, Flacke WE. Cardiovascular effects of fentanyl reversal by naloxone at varying arterial carbon dioxide tensions in dogs. Anesth Analg. 1988;67(8):730-736.
8. Kim HK, Nelson LS. Reversal of opioid-induced ventilatory depression using low-dose naloxone (0.04 mg): a case series. J Med Toxicol. 2015;12(1):107-110. doi:10.1007/s13181-015-0499-3.

Peroneus Quartus Muscle
Take-Home Points
- PQ is easily mistaken for a PB tear.
- PQ is best identified on MRI, but commonly missed.
- For symptomatic cases, excision is the best treatment.
- Consider PQ in patients with chronic ankle pain, swelling, and/or instability.
The peroneus quartus (PQ) is an accessory muscle arising from the leg’s lateral compartment, which typically contains the peroneus longus (PL) and the peroneus brevis (PB). The many cadaveric studies that have been conducted indicate a general population prevalence ranging from 6.6% to 23%.1 Radiographic studies, including magnetic resonance imaging (MRI) and ultrasonography, have shown a similar prevalence.2 Although the PQ is asymptomatic in most cases, it may compromise the space of the superior peroneal tunnel and cause problems, including ankle pain, PB tear, subluxation of peroneal tendons, tendinous calcification, painful hypertrophy of retrotrochlear eminence, and recurrent hematomas.1,3-5 Given its differing anatomy, the PQ variously has been referred to as peroneocalcaneus externum, peroneocuboideus, long peroneal tendon, and peroneoperoneolongus.1
Although the PQ’s origin and insertion differ between subjects, the most common origin is the muscle fibers of the PB, and the most common insertion is the retrotrochlear eminence of the calcaneum.3
We report a case of peroneal tendon pathology that was initially thought to be caused primarily by impingement of a large osteochondroma on the tendons, but was later thought to be caused in part by a PQ and a split PB tendon seen only at the time of the second operation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 16-year-old boy with an osteochondroma of the right distal fibula presented to clinic with the chief complaint of lateral right ankle pain. A “sharp” pain accompanied by audible “popping” occurred with ankle motion. Medical history was significant only for non-Hodgkin lymphoma treated with bone marrow transplantation and whole body radiation at a young age. Physical examination revealed a palpable exostosis of the distal right fibula and associated ankle swelling.
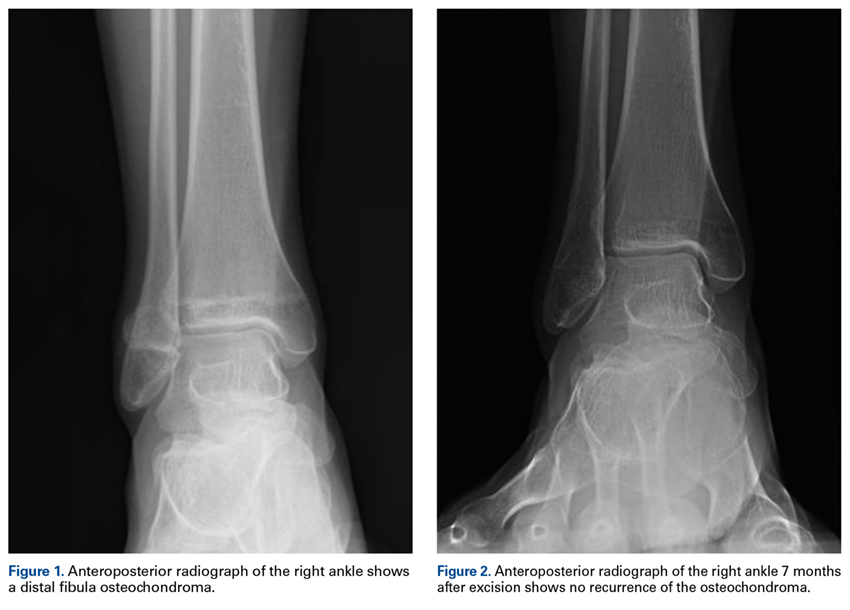
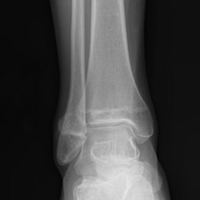
One year after surgery, the patient returned with recurring right ankle pain and audible popping during ankle movement. There was no appreciable peroneal tendon subluxation on physical examination. Repeat imaging of the ankle showed no recurrence of the osteochondroma (Figure 2).

Discussion
Absence of a PQ muscle in simian and prosimian species suggests that the PQ represents an evolutionary adaptation to evert the lateral foot and improve bipedal gait. Although the 3 peroneal (PL, PB, peroneus tertius [PT]) muscles evert the middle part of the lateral border of the foot, the PQ inserts on the retrotrochlear eminence, which everts the posterior part of the lateral border of the foot.1,6 The PQ has often been described as a variation of the PB. The PQ may also stabilize the ankle and reduce the energy required for walking. A similar functional adaptation has been proposed for the PT, which dorsiflexes at the ankle. Although presence of a PT also varies in the population, its occurrence does not correlate with presence of a PQ. In people with PQ muscles, there is an 83% to 95% incidence of also having PT muscles.7
PQ prevalence has ranged from 6.6% to 23% in cadaver studies2 and from 7% to 17% in radiologic studies.1 To better evaluate prevalence, Yammine2 performed a meta-analysis of data from 46 studies (cadaveric dissection, MRI, ultrasonography) and 3928 legs and found an overall incidence of 10.2% and a higher incidence in the Indian population than in other races. Another study found no correlation between PQ presence and sex.7
MRI is the best imaging modality for assessing for PQ but must be performed specifically for this anatomical variation. Axial images may show a fat pad separating the PQ muscle from the PB muscle.8 On imaging, a PQ muscle can be mistaken for a peroneal tendon tear. A feature that helps in distinguishing the 2 is location; the PQ typically is found posterior and medial to the PL and PB tendons, whereas PB tears are anterior to the retromalleolar groove.2 Presence of a PQ muscle may be missed on initial MRI, as occurred in our patient’s case. Zammit and Singh3 reviewed 80 leg MRIs and found 6 PQs. Only 1 of the 6 reports described the PQ as an “atypical appearance of peroneus brevis [that] appears to be made up of more than one tendon.”
Surgical excision is often adequate treatment for a symptomatic PQ. If the PQ muscle is small and symptomatic from pressure to the muscle mass, a short fasciotomy may be performed.9 More commonly, complete excision of the accessory muscle is required. Although the PQ muscle is usually asymptomatic, it should be considered in cases of chronic ankle pain, swelling, or instability; recurrent hematomas; and peroneal tendon subluxation or tears.5,7
Our patient’s diagnosis was initially overlooked because of an osteochondroma in the region of interest. It remains unclear whether his pain was caused by the PQ itself or, more likely, from the PB tear. It is thought that the accessory muscle adds bulk to the peroneal tunnel, predisposing to peroneal pathology, such as muscle tears and tendon subluxation. Regardless, advanced imaging performed before the index procedure, along with a general understanding of the PQ and its classic MRI findings, may have prevented the repeat operation in this case.
The PQ muscle is a rare but sometimes missed potential etiology of ankle pain and tendon subluxation. In our patient’s case, the most obvious abnormality, an osteochondroma, may have masked the true cause.
1. Athavale SA, Gupta V, Kotgirwar S, Singh V. The peroneus quartus muscle: clinical correlation with evolutionary importance. Anat Sci Int. 2012;87(2):106-110.
2. Yammine K. The accessory peroneal (fibular) muscles: peroneus quartus and peroneus digiti quinti. A systematic review and meta-analysis. Surg Radiol Anat. 2015;37(6):617-627.
3. Zammit J, Singh D. The peroneus quartus muscle. Anatomy and clinical relevance. J Bone Joint Surg Br. 2003;85(8):1134-1137.
4. Kulshreshtha R, Kadri S, Rajan DT. A case of unusual combination of injuries around the lateral malleolus. Foot. 2006;16(1):51-53.
5. Donley BG, Leyes M. Peroneus quartus muscle. A rare cause of chronic lateral ankle pain. Am J Sports Med. 2001;29(3):373-375.
6. Hecker P. Study on the peroneus of the tarsus. Anat Rec. 1923;26(1):79-82.
7. Rios Nascimento SR, Watanabe Costa R, Ruiz CR, Wafae N. Analysis on the incidence of the fibularis quartus muscle using magnetic resonance imaging. Anat Res Int. 2012;(2012):485149.
8. Wang XT, Rosenberg ZS, Mechlin MB, Schweitzer ME. Normal variants and diseases of the peroneal tendons and superior peroneal retinaculum: MR imaging features. Radiographics. 2005;25(3):587-602.
9. Martinelli B, Bernobi S. Peroneus quartus muscle and ankle pain. Foot Ankle Surg. 2002;8(3):223-225.
Take-Home Points
- PQ is easily mistaken for a PB tear.
- PQ is best identified on MRI, but commonly missed.
- For symptomatic cases, excision is the best treatment.
- Consider PQ in patients with chronic ankle pain, swelling, and/or instability.
The peroneus quartus (PQ) is an accessory muscle arising from the leg’s lateral compartment, which typically contains the peroneus longus (PL) and the peroneus brevis (PB). The many cadaveric studies that have been conducted indicate a general population prevalence ranging from 6.6% to 23%.1 Radiographic studies, including magnetic resonance imaging (MRI) and ultrasonography, have shown a similar prevalence.2 Although the PQ is asymptomatic in most cases, it may compromise the space of the superior peroneal tunnel and cause problems, including ankle pain, PB tear, subluxation of peroneal tendons, tendinous calcification, painful hypertrophy of retrotrochlear eminence, and recurrent hematomas.1,3-5 Given its differing anatomy, the PQ variously has been referred to as peroneocalcaneus externum, peroneocuboideus, long peroneal tendon, and peroneoperoneolongus.1
Although the PQ’s origin and insertion differ between subjects, the most common origin is the muscle fibers of the PB, and the most common insertion is the retrotrochlear eminence of the calcaneum.3
We report a case of peroneal tendon pathology that was initially thought to be caused primarily by impingement of a large osteochondroma on the tendons, but was later thought to be caused in part by a PQ and a split PB tendon seen only at the time of the second operation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 16-year-old boy with an osteochondroma of the right distal fibula presented to clinic with the chief complaint of lateral right ankle pain. A “sharp” pain accompanied by audible “popping” occurred with ankle motion. Medical history was significant only for non-Hodgkin lymphoma treated with bone marrow transplantation and whole body radiation at a young age. Physical examination revealed a palpable exostosis of the distal right fibula and associated ankle swelling.
One year after surgery, the patient returned with recurring right ankle pain and audible popping during ankle movement. There was no appreciable peroneal tendon subluxation on physical examination. Repeat imaging of the ankle showed no recurrence of the osteochondroma (Figure 2).
Discussion
Absence of a PQ muscle in simian and prosimian species suggests that the PQ represents an evolutionary adaptation to evert the lateral foot and improve bipedal gait. Although the 3 peroneal (PL, PB, peroneus tertius [PT]) muscles evert the middle part of the lateral border of the foot, the PQ inserts on the retrotrochlear eminence, which everts the posterior part of the lateral border of the foot.1,6 The PQ has often been described as a variation of the PB. The PQ may also stabilize the ankle and reduce the energy required for walking. A similar functional adaptation has been proposed for the PT, which dorsiflexes at the ankle. Although presence of a PT also varies in the population, its occurrence does not correlate with presence of a PQ. In people with PQ muscles, there is an 83% to 95% incidence of also having PT muscles.7
PQ prevalence has ranged from 6.6% to 23% in cadaver studies2 and from 7% to 17% in radiologic studies.1 To better evaluate prevalence, Yammine2 performed a meta-analysis of data from 46 studies (cadaveric dissection, MRI, ultrasonography) and 3928 legs and found an overall incidence of 10.2% and a higher incidence in the Indian population than in other races. Another study found no correlation between PQ presence and sex.7
MRI is the best imaging modality for assessing for PQ but must be performed specifically for this anatomical variation. Axial images may show a fat pad separating the PQ muscle from the PB muscle.8 On imaging, a PQ muscle can be mistaken for a peroneal tendon tear. A feature that helps in distinguishing the 2 is location; the PQ typically is found posterior and medial to the PL and PB tendons, whereas PB tears are anterior to the retromalleolar groove.2 Presence of a PQ muscle may be missed on initial MRI, as occurred in our patient’s case. Zammit and Singh3 reviewed 80 leg MRIs and found 6 PQs. Only 1 of the 6 reports described the PQ as an “atypical appearance of peroneus brevis [that] appears to be made up of more than one tendon.”
Surgical excision is often adequate treatment for a symptomatic PQ. If the PQ muscle is small and symptomatic from pressure to the muscle mass, a short fasciotomy may be performed.9 More commonly, complete excision of the accessory muscle is required. Although the PQ muscle is usually asymptomatic, it should be considered in cases of chronic ankle pain, swelling, or instability; recurrent hematomas; and peroneal tendon subluxation or tears.5,7
Our patient’s diagnosis was initially overlooked because of an osteochondroma in the region of interest. It remains unclear whether his pain was caused by the PQ itself or, more likely, from the PB tear. It is thought that the accessory muscle adds bulk to the peroneal tunnel, predisposing to peroneal pathology, such as muscle tears and tendon subluxation. Regardless, advanced imaging performed before the index procedure, along with a general understanding of the PQ and its classic MRI findings, may have prevented the repeat operation in this case.
The PQ muscle is a rare but sometimes missed potential etiology of ankle pain and tendon subluxation. In our patient’s case, the most obvious abnormality, an osteochondroma, may have masked the true cause.
Take-Home Points
- PQ is easily mistaken for a PB tear.
- PQ is best identified on MRI, but commonly missed.
- For symptomatic cases, excision is the best treatment.
- Consider PQ in patients with chronic ankle pain, swelling, and/or instability.
The peroneus quartus (PQ) is an accessory muscle arising from the leg’s lateral compartment, which typically contains the peroneus longus (PL) and the peroneus brevis (PB). The many cadaveric studies that have been conducted indicate a general population prevalence ranging from 6.6% to 23%.1 Radiographic studies, including magnetic resonance imaging (MRI) and ultrasonography, have shown a similar prevalence.2 Although the PQ is asymptomatic in most cases, it may compromise the space of the superior peroneal tunnel and cause problems, including ankle pain, PB tear, subluxation of peroneal tendons, tendinous calcification, painful hypertrophy of retrotrochlear eminence, and recurrent hematomas.1,3-5 Given its differing anatomy, the PQ variously has been referred to as peroneocalcaneus externum, peroneocuboideus, long peroneal tendon, and peroneoperoneolongus.1
Although the PQ’s origin and insertion differ between subjects, the most common origin is the muscle fibers of the PB, and the most common insertion is the retrotrochlear eminence of the calcaneum.3
We report a case of peroneal tendon pathology that was initially thought to be caused primarily by impingement of a large osteochondroma on the tendons, but was later thought to be caused in part by a PQ and a split PB tendon seen only at the time of the second operation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 16-year-old boy with an osteochondroma of the right distal fibula presented to clinic with the chief complaint of lateral right ankle pain. A “sharp” pain accompanied by audible “popping” occurred with ankle motion. Medical history was significant only for non-Hodgkin lymphoma treated with bone marrow transplantation and whole body radiation at a young age. Physical examination revealed a palpable exostosis of the distal right fibula and associated ankle swelling.
One year after surgery, the patient returned with recurring right ankle pain and audible popping during ankle movement. There was no appreciable peroneal tendon subluxation on physical examination. Repeat imaging of the ankle showed no recurrence of the osteochondroma (Figure 2).
Discussion
Absence of a PQ muscle in simian and prosimian species suggests that the PQ represents an evolutionary adaptation to evert the lateral foot and improve bipedal gait. Although the 3 peroneal (PL, PB, peroneus tertius [PT]) muscles evert the middle part of the lateral border of the foot, the PQ inserts on the retrotrochlear eminence, which everts the posterior part of the lateral border of the foot.1,6 The PQ has often been described as a variation of the PB. The PQ may also stabilize the ankle and reduce the energy required for walking. A similar functional adaptation has been proposed for the PT, which dorsiflexes at the ankle. Although presence of a PT also varies in the population, its occurrence does not correlate with presence of a PQ. In people with PQ muscles, there is an 83% to 95% incidence of also having PT muscles.7
PQ prevalence has ranged from 6.6% to 23% in cadaver studies2 and from 7% to 17% in radiologic studies.1 To better evaluate prevalence, Yammine2 performed a meta-analysis of data from 46 studies (cadaveric dissection, MRI, ultrasonography) and 3928 legs and found an overall incidence of 10.2% and a higher incidence in the Indian population than in other races. Another study found no correlation between PQ presence and sex.7
MRI is the best imaging modality for assessing for PQ but must be performed specifically for this anatomical variation. Axial images may show a fat pad separating the PQ muscle from the PB muscle.8 On imaging, a PQ muscle can be mistaken for a peroneal tendon tear. A feature that helps in distinguishing the 2 is location; the PQ typically is found posterior and medial to the PL and PB tendons, whereas PB tears are anterior to the retromalleolar groove.2 Presence of a PQ muscle may be missed on initial MRI, as occurred in our patient’s case. Zammit and Singh3 reviewed 80 leg MRIs and found 6 PQs. Only 1 of the 6 reports described the PQ as an “atypical appearance of peroneus brevis [that] appears to be made up of more than one tendon.”
Surgical excision is often adequate treatment for a symptomatic PQ. If the PQ muscle is small and symptomatic from pressure to the muscle mass, a short fasciotomy may be performed.9 More commonly, complete excision of the accessory muscle is required. Although the PQ muscle is usually asymptomatic, it should be considered in cases of chronic ankle pain, swelling, or instability; recurrent hematomas; and peroneal tendon subluxation or tears.5,7
Our patient’s diagnosis was initially overlooked because of an osteochondroma in the region of interest. It remains unclear whether his pain was caused by the PQ itself or, more likely, from the PB tear. It is thought that the accessory muscle adds bulk to the peroneal tunnel, predisposing to peroneal pathology, such as muscle tears and tendon subluxation. Regardless, advanced imaging performed before the index procedure, along with a general understanding of the PQ and its classic MRI findings, may have prevented the repeat operation in this case.
The PQ muscle is a rare but sometimes missed potential etiology of ankle pain and tendon subluxation. In our patient’s case, the most obvious abnormality, an osteochondroma, may have masked the true cause.
1. Athavale SA, Gupta V, Kotgirwar S, Singh V. The peroneus quartus muscle: clinical correlation with evolutionary importance. Anat Sci Int. 2012;87(2):106-110.
2. Yammine K. The accessory peroneal (fibular) muscles: peroneus quartus and peroneus digiti quinti. A systematic review and meta-analysis. Surg Radiol Anat. 2015;37(6):617-627.
3. Zammit J, Singh D. The peroneus quartus muscle. Anatomy and clinical relevance. J Bone Joint Surg Br. 2003;85(8):1134-1137.
4. Kulshreshtha R, Kadri S, Rajan DT. A case of unusual combination of injuries around the lateral malleolus. Foot. 2006;16(1):51-53.
5. Donley BG, Leyes M. Peroneus quartus muscle. A rare cause of chronic lateral ankle pain. Am J Sports Med. 2001;29(3):373-375.
6. Hecker P. Study on the peroneus of the tarsus. Anat Rec. 1923;26(1):79-82.
7. Rios Nascimento SR, Watanabe Costa R, Ruiz CR, Wafae N. Analysis on the incidence of the fibularis quartus muscle using magnetic resonance imaging. Anat Res Int. 2012;(2012):485149.
8. Wang XT, Rosenberg ZS, Mechlin MB, Schweitzer ME. Normal variants and diseases of the peroneal tendons and superior peroneal retinaculum: MR imaging features. Radiographics. 2005;25(3):587-602.
9. Martinelli B, Bernobi S. Peroneus quartus muscle and ankle pain. Foot Ankle Surg. 2002;8(3):223-225.
1. Athavale SA, Gupta V, Kotgirwar S, Singh V. The peroneus quartus muscle: clinical correlation with evolutionary importance. Anat Sci Int. 2012;87(2):106-110.
2. Yammine K. The accessory peroneal (fibular) muscles: peroneus quartus and peroneus digiti quinti. A systematic review and meta-analysis. Surg Radiol Anat. 2015;37(6):617-627.
3. Zammit J, Singh D. The peroneus quartus muscle. Anatomy and clinical relevance. J Bone Joint Surg Br. 2003;85(8):1134-1137.
4. Kulshreshtha R, Kadri S, Rajan DT. A case of unusual combination of injuries around the lateral malleolus. Foot. 2006;16(1):51-53.
5. Donley BG, Leyes M. Peroneus quartus muscle. A rare cause of chronic lateral ankle pain. Am J Sports Med. 2001;29(3):373-375.
6. Hecker P. Study on the peroneus of the tarsus. Anat Rec. 1923;26(1):79-82.
7. Rios Nascimento SR, Watanabe Costa R, Ruiz CR, Wafae N. Analysis on the incidence of the fibularis quartus muscle using magnetic resonance imaging. Anat Res Int. 2012;(2012):485149.
8. Wang XT, Rosenberg ZS, Mechlin MB, Schweitzer ME. Normal variants and diseases of the peroneal tendons and superior peroneal retinaculum: MR imaging features. Radiographics. 2005;25(3):587-602.
9. Martinelli B, Bernobi S. Peroneus quartus muscle and ankle pain. Foot Ankle Surg. 2002;8(3):223-225.
An Atypical Syphilis Presentation
Syphilis is a chronic systemic infection that has been allotted the epithet “the great imitator” for its gross and histologic similarity to numerous other skin pathologies. Well-characterized for centuries, syphilis features diverse clinical manifestations including a number of cutaneous symptoms.1
RELATED AUDIOCAST: The Syphilis Epidemic: Dermatologists on the Frontline of Treatment and Diagnosis
The primary stage of infection is classically defined by an asymptomatic chancre at the inoculation site. The secondary stage results from the systemic dissemination of the infection and typically is characterized by cutaneous eruptions, regional lymphadenopathy, and flulike symptoms. This stage gained its notoriety as the great imitator owing to its ability to present with a variety of papulosquamous eruptions. The secondary stage is followed by an asymptomatic latent period that may last months to years, followed by the tertiary stage, which is characterized by the neurologic, cardiovascular, and/or gummatous manifestations that represent the major sources of morbidity and mortality associated with syphilis. It is during the primary, secondary, and early latent stages that the infection is communicable.1
Case Report
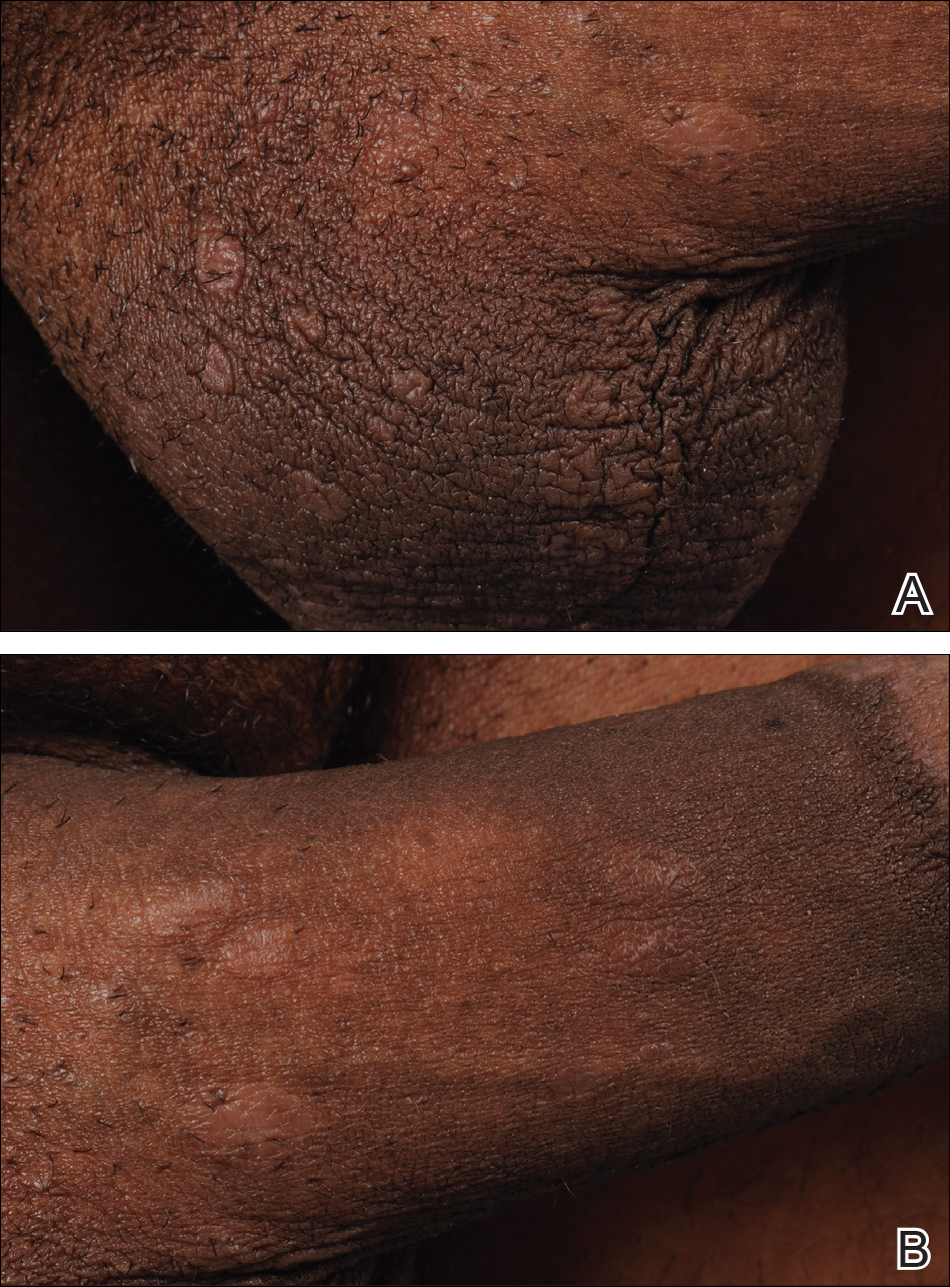
A 40-year-old man presented with multiple intensely pruritic, scattered, erythematous and slightly violaceous, flat-topped papules on the scrotum (Figure 1A) and penile shaft (Figure 1B) of 1 week’s duration. Some of these lesions were annular in appearance. The patient denied any other dermatologic concerns and showed no other skin lesions. A shave biopsy of the right side of the penile shaft was performed, revealing minimal papillary dermis and superficial perivascular dermatitis with substantial perivascular plasmalymphocytic infiltration. The epidermal layer was mildly acanthotic with parakeratosis. A tentative diagnosis of secondary syphilis of unknown latency was made and confirmatory laboratory studies were ordered.
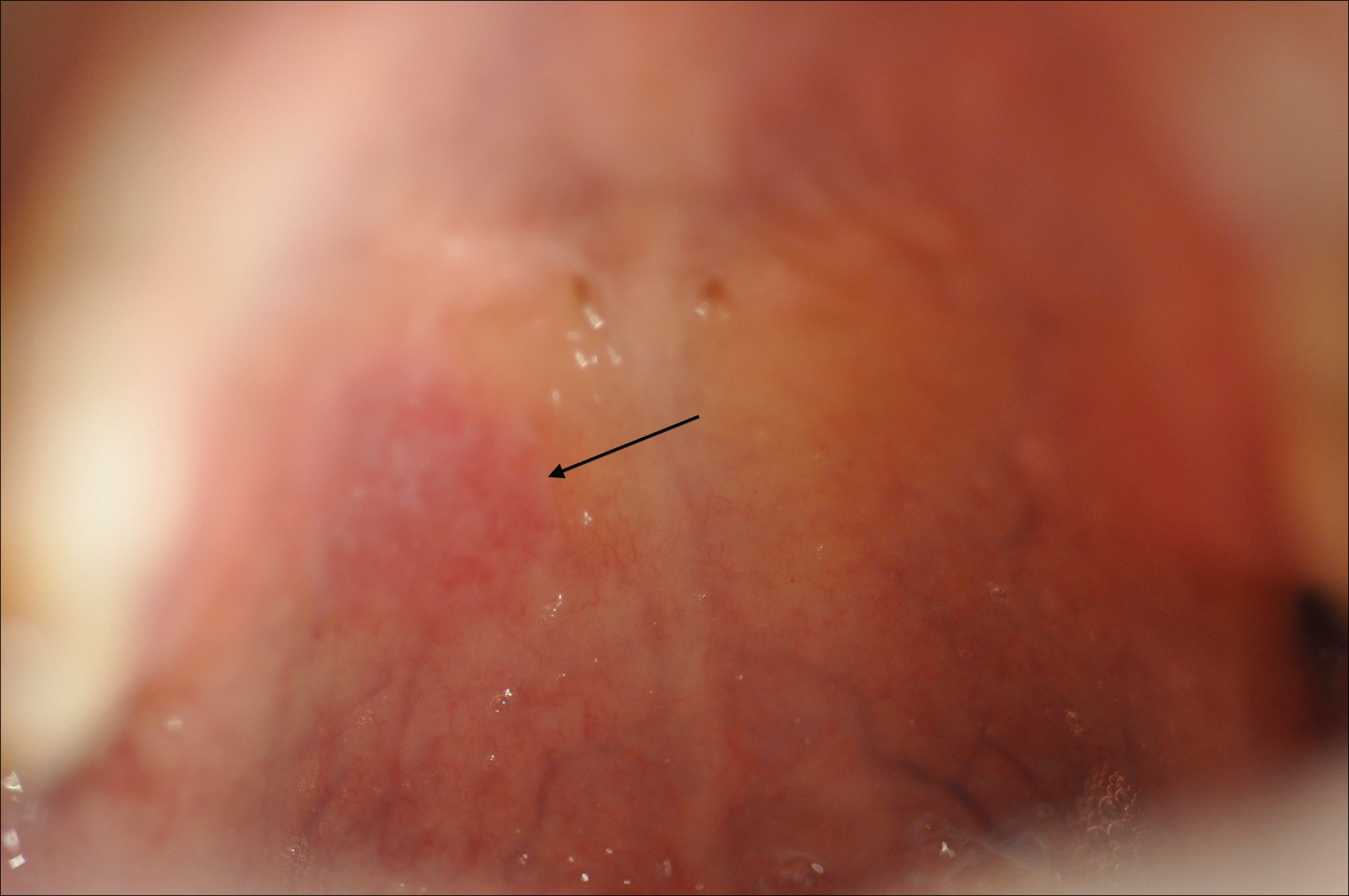
Within weeks, the patient developed a painful 7-mm white patch on the right lower mucosal lip followed several days later by the appearance of a painful lesion on the hard palate (Figure 2 [arrow indicates palatal lesion]) and odynophagia. He presented to the emergency department roughly 3 weeks from the time of index presentation and was started empirically on amoxicillin 500 mg 3 times daily for 10 days for suspicion of strep throat. At a scheduled follow-up with his dermatologist 1 week later, physical examination showed complete resolution of the mucosal lip patch and genital lesions. A round erythematous patch on the right hard palate consistent with a resolving mucosal patch also was noted. A diagnosis of secondary syphilitic infection was made with a rapid plasma reagin (RPR) titer of 1:32 (reference range, <1:1) and positive Treponema antibodies. The patient was treated with a single dose of intramuscular benzathine penicillin G 2.4 million U to prevent the development of tertiary syphilis.
Comment
Incidence
Syphilis has been well characterized since the early 15th century, though its geographic origin remains a topic of controversy.2 Although acquired syphilis infections represented a major source of morbidity and mortality in the early 20th century, the prevalence of syphilis in the United States declined substantially thereafter due to improved public health management.2 Syphilis was relatively rare in the United States by the year 1956, with fewer than 7000 cases of primary and secondary disease reported annually.3 The incidence of primary and secondary syphilis infections in the United States increased gradually until 1990 before declining precipitously and reaching an unprecedented low of 2.2 cases per 100,000 individuals in 2000.4 These shifts ultimately have resulted in decreased clinical familiarity with the disease presentation of syphilis among many health care providers. Since 2000, the incidence of syphilis infection has increased in the United States, with the greatest increases seen in men who have sex with men, intravenous drug users, and human immunodeficiency virus–infected individuals.5-7
RELATED ARTICLE: Syphilis and the Dermatologist
Pathogenesis and Transmission
The causative agent in syphilis infection is the bacterium Treponema pallidum, a member of the family Spirochaetaceae, which is distinguished by its thin, regularly coiled form and distinctive corkscrew motility.8 Syphilis is communicated primarily by sexual contact or in utero exposure during the primary and secondary stages of maternal infection.9 At the time of presentation, our patient denied having any new sexual partners or practices. He reported a monogamous heterosexual relationship within the months preceding presentation, suggesting historical inaccuracy on the part of the patient or probable infidelity in the reported relationship as an alternative means of infection transmission. Untreated individuals may be contagious for longer than 1 year,9 making transmission patterns difficult to track clinically.
Presentation
The clinical presentation of infection with T pallidum results from dual humoral and cell-mediated inflammatory responses in the host. The primary stage is classically defined by a single chancre, which develops at the inoculation site(s) 9 to 90 days following exposure. The chancre typically begins as a small papule that rapidly develops into a painless ulcer characterized by an indurated border, red base, bordering edema, and a diameter of 2 cm or less. Indolent regional lymphadenopathy often is observed in conjunction with the primary chancre.10 Our case is notable for the absence of a primary syphilitic lesion and lack of adenopathy. The primary chancre of syphilis typically resolves within 3 to 6 weeks of onset regardless of whether the patient is treated,4 thus suggesting the rare possibility that our patient developed a painless primary chancre without realizing it.
The secondary stage of syphilis infection arises weeks to months after resolution of the primary chancre and is triggered by hematogenous and lymphatic dissemination of the bacteria. The symptoms of secondary syphilis are primarily flulike and may include headache, malaise, fatigue, sore throat, arthralgia, and low-grade fever.9 Nontender regional lymphadenopathy and splenomegaly also have been reported.11 Our patient denied any systemic concerns throughout the duration of his illness, with the exception of odynophagia in association with ulceration of the oral mucosa. Abnormal laboratory findings in secondary syphilis are nonspecific and may include an elevated erythrocyte sedimentation rate and/or an increased white blood cell count with absolute lymphocytosis.12 Laboratory studies drawn at the time of presentation showed no such abnormalities in our patient.
The cutaneous signs of secondary syphilis arise concurrently with systemic manifestations and are a common finding, with lesions of the skin or oral mucosa present in up to 80% of patients,13 as in our case. Oral lesions classically involve ulcerations at the tip and sides of the tongue,12 which is distinct from our patient who developed oral lesions of the mucosal lip and hard palate.
Secondary syphilis classically features a copper-colored maculopapular rash with sharply delineated margins typically present on the palmar and plantar surfaces.14 Verrucous lesions appearing as moist exophytic plaques on the genitals, intertriginous areas, and/or perineum also have been described and are referred to as condyloma lata in the setting of secondary syphilis.15 In contrast to these classic findings, our patient demonstrated lichenoid lesions on the genitalia and white mucosal patches on the oral mucosa. Our case also was highly unusual because of the intense pruritus associated with the genital lesions, which starkly contrasts most secondary-stage cutaneous lesions that are classically asymptomatic.14 Additionally, our case was distinctive due to the lack of palmar or plantar involvement, which is considered a characteristic feature of secondary cutaneous syphilis.1 Finally, our case was notable for the presence of multiple annular cutaneous lesions, which indicated a late secondary-stage infection during which involution of the lesions produced endarteritis as deeper vessels became involved. A 20-year retrospective study by Abell et al11 demonstrated that 40% of syphilitic rashes are macular, 40% are maculopapular, 10% are papular (as in our case), and the remaining 10% are not easily grouped within these categories.
Differential Diagnosis
It has been estimated that approximately 8% of cutaneous syphilitic lesions demonstrate morphology and distributions suggestive of other dermatologic conditions, including atopic dermatitis, pityriasis rosea, psoriasis, drug-induced eruptions, erythema multiforme, mycosis fungoides, and far more uncommonly lichenoid lesions,16,17 as in our case.
Histopathology
It has been demonstrated that the gross appearance of the secondary syphilitic lesion depends both on the degree of inflammatory infiltrate and the extent of vascular involvement producing ischemia of the skin.1 Our case presented with small, flat-topped, papular lesions that grossly resembled lichen planus and were ultimately shown to be the product of dense lymphomononuclear infiltration extending perivascularly and throughout the superficial and deep dermis.
Biopsy of a lesion is one means of diagnosis, though the histologic appearance of secondary syphilis can mimic many other diseases. In primary and secondary syphilis, skin biopsy characteristically shows central thinning or ulceration of the epidermal layer with heavy dermal lymphocyte infiltration, lymphovascular proliferation with endarteritis, small-vessel thrombosis, and dermal necrosis. Lichen planus–type dermatitis is histologically characterized by hyperkeratosis, irregular epidermal hyperplasia, and a dermoepidermal junction that may be obscured by a dense lymphomononuclear infiltrate.9 The specimen taken from our patient showed minimal infiltrate in the papillary dermis, suggesting a diagnosis of secondary syphilis with lichenoid features. Despite a gross appearance consistent with lichen planus, the biopsy lacked the hydropic degeneration of the basal layer and keratinocyte necrosis that typically characterize this condition.
Diagnosis
Serologic testing for syphilis infection is comprised of nontreponemal and treponemal studies. Nontreponemal testing, which includes the RPR and VDRL test, detects antibodies to cardiolipin-lecithin antigen, a lipid component of the cell membranes of T pallidum. Because the specificity of these tests is fairly low, they typically are used only for screening and monitoring of disease progression and/or response to treatment. Approximately 25% of cases in the United States of primary syphilis are not detected by nontreponemal testing, whereas a nonreactive test nearly always excludes a diagnosis of secondary or latent-stage syphilitic infection.9 Indeed, nontreponemal studies show the highest antibody titers during the late secondary and early latent stages of infection with declining titers thereafter, even in the absence of antibiotic treatment. In our case, diagnosis was made by biopsy and RPR was used for staging; RPR was reactive at a dilution of 1:32, indicative of secondary or early latent infection.
Treponemal testing, which includes the fluorescent treponemal antibody absorption test, and multiplex flow immunoassay detects antibodies that are specific to syphilis infection. Treponemal antibodies are detectable earlier in the course of infection than nontreponemal antibodies and remain permanently detectable even following treatment. Because of its high specificity, treponemal testing often is used to confirm diagnosis after positive screening with nontreponemal tests.4 Positive fluorescent treponemal antibody absorption testing and positive multiplex flow immunoassay may be used to confirm the diagnosis of T pallidum infection.
The tertiary stage of syphilis infection can occur years after conclusion of the secondary stage and is comprised of one or more of the following: gummas, aortic dilatation or dissection, and neurosyphilitic manifestations such as tabes dorsalis or general paresis.1 It is of vital importance to identify syphilis infection prior to the onset of the tertiary stage to prevent substantial morbidity and mortality.
Treatment
Our patient’s symptoms abated after empiric treatment with amoxicillin for presumed streptococcal throat infection after he presented to the emergency department with odynophagia, which is not surprising given the moderate-spectrum coverage of this β-lactam antibiotic as well as the near-complete susceptibility of Treponema spirochetes to amoxicillin in primary and secondary syphilis with notably lower efficacy in latent or tertiary disease. It was essential to treat the patient with a single dose of intramuscular benzathine penicillin G 2.4 million U, which has been shown to reliably prevent recurrence of infection or progression to tertiary syphilis.18
Conclusion
We present a rare case of lichenoid secondary syphilis in the absence of lesions on the palmar and plantar surfaces. The patient lacked any other cutaneous or systemic manifestations, except for odynophagia in association with oral mucosal lesions. He denied any new sexual partners and did not recall having a primary chancre. Also strikingly unusual in this case was the intense pruritus associated with the genital eruption, which is unlike the classic lack of symptoms experienced in the great majority of eruptions due to secondary syphilis. A clinical appreciation of the many cutaneous manifestations of syphilis infection remains critical to early identification of the disease prior to progression to the tertiary stage and its devastating sequelae.
- Dourmishev LA, Assen L. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Kilmarx PH, St Louis ME. The evolving epidemiology of syphilis. Am J Public Health. 1995;85(8, pt 1):1053-1054.
- Patton ME, Su JR, Nelson R, et al. Primary and secondary syphilis—United States, 2005-2013. MMWR Morb Mortal Wkly Rep. 2014;63:402-406.
- Coffin LS, Newberry A, Hagan H, et al. Syphilis in drug users in low and middle income countries. Int J Drug Policy. 2010;21:20-27.
- Gao L, Zhang L, Jin Q. Meta-analysis: prevalence of HIV infection and syphilis among MSM in China. Sex Transm Infect. 2009;85:354-358.
- Karp G, Schlaeffer F, Jotkowitz A, et al. Syphilis and HIV co-infection. Eur J Int Med. 2009;20:9-13.
- Hol EL, Lukehart SA. Syphilis: using modern approaches to understand an old disease. J Clin Invest. 2011;121:4584-4592.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis. J Foot Ankle Surg. 2011;50:96-101.
- Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68:283-290.
- Abell E, Marks R, Jones W. Secondary syphilis: a clinicopathological review. Br J Dermatol. 1975;93:53-61.
- Fiumara N. The treponematoses. Int Dermatol. 1992;1:953-974.
- Martin DH, Mroczkowski TF. Dermatological manifestations of sexually transmitted diseases other than HIV. Infect Dis Clin North Am. 1994;8:533-583.
- Morton RS. The treponematoses. In: Champion RH, Bourton JL, Burns DA, et al. Rook’s Textbook of Dermatology. 6th ed. London, United Kingdom: Blackwell Science; 1998:1237-1275.
- Rosen T, Hwong H. Pedal interdigital condylomata lata: a rare sign of secondary syphilis. Sex Transm Dis. 2001;28:184-186.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Tang MBY, Yosipovitch G, Tan SH. Secondary syphilis presenting as a lichen planus-like rash. J Eur Acad Dermatol Venereol. 2004;18:185-187.
- Onoda Y. Clinical evaluation of amoxicillin in the treatment of syphilis. J Int Med. 1979;7:539-545.
Syphilis is a chronic systemic infection that has been allotted the epithet “the great imitator” for its gross and histologic similarity to numerous other skin pathologies. Well-characterized for centuries, syphilis features diverse clinical manifestations including a number of cutaneous symptoms.1
RELATED AUDIOCAST: The Syphilis Epidemic: Dermatologists on the Frontline of Treatment and Diagnosis
The primary stage of infection is classically defined by an asymptomatic chancre at the inoculation site. The secondary stage results from the systemic dissemination of the infection and typically is characterized by cutaneous eruptions, regional lymphadenopathy, and flulike symptoms. This stage gained its notoriety as the great imitator owing to its ability to present with a variety of papulosquamous eruptions. The secondary stage is followed by an asymptomatic latent period that may last months to years, followed by the tertiary stage, which is characterized by the neurologic, cardiovascular, and/or gummatous manifestations that represent the major sources of morbidity and mortality associated with syphilis. It is during the primary, secondary, and early latent stages that the infection is communicable.1
Case Report
A 40-year-old man presented with multiple intensely pruritic, scattered, erythematous and slightly violaceous, flat-topped papules on the scrotum (Figure 1A) and penile shaft (Figure 1B) of 1 week’s duration. Some of these lesions were annular in appearance. The patient denied any other dermatologic concerns and showed no other skin lesions. A shave biopsy of the right side of the penile shaft was performed, revealing minimal papillary dermis and superficial perivascular dermatitis with substantial perivascular plasmalymphocytic infiltration. The epidermal layer was mildly acanthotic with parakeratosis. A tentative diagnosis of secondary syphilis of unknown latency was made and confirmatory laboratory studies were ordered.
Within weeks, the patient developed a painful 7-mm white patch on the right lower mucosal lip followed several days later by the appearance of a painful lesion on the hard palate (Figure 2 [arrow indicates palatal lesion]) and odynophagia. He presented to the emergency department roughly 3 weeks from the time of index presentation and was started empirically on amoxicillin 500 mg 3 times daily for 10 days for suspicion of strep throat. At a scheduled follow-up with his dermatologist 1 week later, physical examination showed complete resolution of the mucosal lip patch and genital lesions. A round erythematous patch on the right hard palate consistent with a resolving mucosal patch also was noted. A diagnosis of secondary syphilitic infection was made with a rapid plasma reagin (RPR) titer of 1:32 (reference range, <1:1) and positive Treponema antibodies. The patient was treated with a single dose of intramuscular benzathine penicillin G 2.4 million U to prevent the development of tertiary syphilis.
Comment
Incidence
Syphilis has been well characterized since the early 15th century, though its geographic origin remains a topic of controversy.2 Although acquired syphilis infections represented a major source of morbidity and mortality in the early 20th century, the prevalence of syphilis in the United States declined substantially thereafter due to improved public health management.2 Syphilis was relatively rare in the United States by the year 1956, with fewer than 7000 cases of primary and secondary disease reported annually.3 The incidence of primary and secondary syphilis infections in the United States increased gradually until 1990 before declining precipitously and reaching an unprecedented low of 2.2 cases per 100,000 individuals in 2000.4 These shifts ultimately have resulted in decreased clinical familiarity with the disease presentation of syphilis among many health care providers. Since 2000, the incidence of syphilis infection has increased in the United States, with the greatest increases seen in men who have sex with men, intravenous drug users, and human immunodeficiency virus–infected individuals.5-7
RELATED ARTICLE: Syphilis and the Dermatologist
Pathogenesis and Transmission
The causative agent in syphilis infection is the bacterium Treponema pallidum, a member of the family Spirochaetaceae, which is distinguished by its thin, regularly coiled form and distinctive corkscrew motility.8 Syphilis is communicated primarily by sexual contact or in utero exposure during the primary and secondary stages of maternal infection.9 At the time of presentation, our patient denied having any new sexual partners or practices. He reported a monogamous heterosexual relationship within the months preceding presentation, suggesting historical inaccuracy on the part of the patient or probable infidelity in the reported relationship as an alternative means of infection transmission. Untreated individuals may be contagious for longer than 1 year,9 making transmission patterns difficult to track clinically.
Presentation
The clinical presentation of infection with T pallidum results from dual humoral and cell-mediated inflammatory responses in the host. The primary stage is classically defined by a single chancre, which develops at the inoculation site(s) 9 to 90 days following exposure. The chancre typically begins as a small papule that rapidly develops into a painless ulcer characterized by an indurated border, red base, bordering edema, and a diameter of 2 cm or less. Indolent regional lymphadenopathy often is observed in conjunction with the primary chancre.10 Our case is notable for the absence of a primary syphilitic lesion and lack of adenopathy. The primary chancre of syphilis typically resolves within 3 to 6 weeks of onset regardless of whether the patient is treated,4 thus suggesting the rare possibility that our patient developed a painless primary chancre without realizing it.
The secondary stage of syphilis infection arises weeks to months after resolution of the primary chancre and is triggered by hematogenous and lymphatic dissemination of the bacteria. The symptoms of secondary syphilis are primarily flulike and may include headache, malaise, fatigue, sore throat, arthralgia, and low-grade fever.9 Nontender regional lymphadenopathy and splenomegaly also have been reported.11 Our patient denied any systemic concerns throughout the duration of his illness, with the exception of odynophagia in association with ulceration of the oral mucosa. Abnormal laboratory findings in secondary syphilis are nonspecific and may include an elevated erythrocyte sedimentation rate and/or an increased white blood cell count with absolute lymphocytosis.12 Laboratory studies drawn at the time of presentation showed no such abnormalities in our patient.
The cutaneous signs of secondary syphilis arise concurrently with systemic manifestations and are a common finding, with lesions of the skin or oral mucosa present in up to 80% of patients,13 as in our case. Oral lesions classically involve ulcerations at the tip and sides of the tongue,12 which is distinct from our patient who developed oral lesions of the mucosal lip and hard palate.
Secondary syphilis classically features a copper-colored maculopapular rash with sharply delineated margins typically present on the palmar and plantar surfaces.14 Verrucous lesions appearing as moist exophytic plaques on the genitals, intertriginous areas, and/or perineum also have been described and are referred to as condyloma lata in the setting of secondary syphilis.15 In contrast to these classic findings, our patient demonstrated lichenoid lesions on the genitalia and white mucosal patches on the oral mucosa. Our case also was highly unusual because of the intense pruritus associated with the genital lesions, which starkly contrasts most secondary-stage cutaneous lesions that are classically asymptomatic.14 Additionally, our case was distinctive due to the lack of palmar or plantar involvement, which is considered a characteristic feature of secondary cutaneous syphilis.1 Finally, our case was notable for the presence of multiple annular cutaneous lesions, which indicated a late secondary-stage infection during which involution of the lesions produced endarteritis as deeper vessels became involved. A 20-year retrospective study by Abell et al11 demonstrated that 40% of syphilitic rashes are macular, 40% are maculopapular, 10% are papular (as in our case), and the remaining 10% are not easily grouped within these categories.
Differential Diagnosis
It has been estimated that approximately 8% of cutaneous syphilitic lesions demonstrate morphology and distributions suggestive of other dermatologic conditions, including atopic dermatitis, pityriasis rosea, psoriasis, drug-induced eruptions, erythema multiforme, mycosis fungoides, and far more uncommonly lichenoid lesions,16,17 as in our case.
Histopathology
It has been demonstrated that the gross appearance of the secondary syphilitic lesion depends both on the degree of inflammatory infiltrate and the extent of vascular involvement producing ischemia of the skin.1 Our case presented with small, flat-topped, papular lesions that grossly resembled lichen planus and were ultimately shown to be the product of dense lymphomononuclear infiltration extending perivascularly and throughout the superficial and deep dermis.
Biopsy of a lesion is one means of diagnosis, though the histologic appearance of secondary syphilis can mimic many other diseases. In primary and secondary syphilis, skin biopsy characteristically shows central thinning or ulceration of the epidermal layer with heavy dermal lymphocyte infiltration, lymphovascular proliferation with endarteritis, small-vessel thrombosis, and dermal necrosis. Lichen planus–type dermatitis is histologically characterized by hyperkeratosis, irregular epidermal hyperplasia, and a dermoepidermal junction that may be obscured by a dense lymphomononuclear infiltrate.9 The specimen taken from our patient showed minimal infiltrate in the papillary dermis, suggesting a diagnosis of secondary syphilis with lichenoid features. Despite a gross appearance consistent with lichen planus, the biopsy lacked the hydropic degeneration of the basal layer and keratinocyte necrosis that typically characterize this condition.
Diagnosis
Serologic testing for syphilis infection is comprised of nontreponemal and treponemal studies. Nontreponemal testing, which includes the RPR and VDRL test, detects antibodies to cardiolipin-lecithin antigen, a lipid component of the cell membranes of T pallidum. Because the specificity of these tests is fairly low, they typically are used only for screening and monitoring of disease progression and/or response to treatment. Approximately 25% of cases in the United States of primary syphilis are not detected by nontreponemal testing, whereas a nonreactive test nearly always excludes a diagnosis of secondary or latent-stage syphilitic infection.9 Indeed, nontreponemal studies show the highest antibody titers during the late secondary and early latent stages of infection with declining titers thereafter, even in the absence of antibiotic treatment. In our case, diagnosis was made by biopsy and RPR was used for staging; RPR was reactive at a dilution of 1:32, indicative of secondary or early latent infection.
Treponemal testing, which includes the fluorescent treponemal antibody absorption test, and multiplex flow immunoassay detects antibodies that are specific to syphilis infection. Treponemal antibodies are detectable earlier in the course of infection than nontreponemal antibodies and remain permanently detectable even following treatment. Because of its high specificity, treponemal testing often is used to confirm diagnosis after positive screening with nontreponemal tests.4 Positive fluorescent treponemal antibody absorption testing and positive multiplex flow immunoassay may be used to confirm the diagnosis of T pallidum infection.
The tertiary stage of syphilis infection can occur years after conclusion of the secondary stage and is comprised of one or more of the following: gummas, aortic dilatation or dissection, and neurosyphilitic manifestations such as tabes dorsalis or general paresis.1 It is of vital importance to identify syphilis infection prior to the onset of the tertiary stage to prevent substantial morbidity and mortality.
Treatment
Our patient’s symptoms abated after empiric treatment with amoxicillin for presumed streptococcal throat infection after he presented to the emergency department with odynophagia, which is not surprising given the moderate-spectrum coverage of this β-lactam antibiotic as well as the near-complete susceptibility of Treponema spirochetes to amoxicillin in primary and secondary syphilis with notably lower efficacy in latent or tertiary disease. It was essential to treat the patient with a single dose of intramuscular benzathine penicillin G 2.4 million U, which has been shown to reliably prevent recurrence of infection or progression to tertiary syphilis.18
Conclusion
We present a rare case of lichenoid secondary syphilis in the absence of lesions on the palmar and plantar surfaces. The patient lacked any other cutaneous or systemic manifestations, except for odynophagia in association with oral mucosal lesions. He denied any new sexual partners and did not recall having a primary chancre. Also strikingly unusual in this case was the intense pruritus associated with the genital eruption, which is unlike the classic lack of symptoms experienced in the great majority of eruptions due to secondary syphilis. A clinical appreciation of the many cutaneous manifestations of syphilis infection remains critical to early identification of the disease prior to progression to the tertiary stage and its devastating sequelae.
Syphilis is a chronic systemic infection that has been allotted the epithet “the great imitator” for its gross and histologic similarity to numerous other skin pathologies. Well-characterized for centuries, syphilis features diverse clinical manifestations including a number of cutaneous symptoms.1
RELATED AUDIOCAST: The Syphilis Epidemic: Dermatologists on the Frontline of Treatment and Diagnosis
The primary stage of infection is classically defined by an asymptomatic chancre at the inoculation site. The secondary stage results from the systemic dissemination of the infection and typically is characterized by cutaneous eruptions, regional lymphadenopathy, and flulike symptoms. This stage gained its notoriety as the great imitator owing to its ability to present with a variety of papulosquamous eruptions. The secondary stage is followed by an asymptomatic latent period that may last months to years, followed by the tertiary stage, which is characterized by the neurologic, cardiovascular, and/or gummatous manifestations that represent the major sources of morbidity and mortality associated with syphilis. It is during the primary, secondary, and early latent stages that the infection is communicable.1
Case Report
A 40-year-old man presented with multiple intensely pruritic, scattered, erythematous and slightly violaceous, flat-topped papules on the scrotum (Figure 1A) and penile shaft (Figure 1B) of 1 week’s duration. Some of these lesions were annular in appearance. The patient denied any other dermatologic concerns and showed no other skin lesions. A shave biopsy of the right side of the penile shaft was performed, revealing minimal papillary dermis and superficial perivascular dermatitis with substantial perivascular plasmalymphocytic infiltration. The epidermal layer was mildly acanthotic with parakeratosis. A tentative diagnosis of secondary syphilis of unknown latency was made and confirmatory laboratory studies were ordered.
Within weeks, the patient developed a painful 7-mm white patch on the right lower mucosal lip followed several days later by the appearance of a painful lesion on the hard palate (Figure 2 [arrow indicates palatal lesion]) and odynophagia. He presented to the emergency department roughly 3 weeks from the time of index presentation and was started empirically on amoxicillin 500 mg 3 times daily for 10 days for suspicion of strep throat. At a scheduled follow-up with his dermatologist 1 week later, physical examination showed complete resolution of the mucosal lip patch and genital lesions. A round erythematous patch on the right hard palate consistent with a resolving mucosal patch also was noted. A diagnosis of secondary syphilitic infection was made with a rapid plasma reagin (RPR) titer of 1:32 (reference range, <1:1) and positive Treponema antibodies. The patient was treated with a single dose of intramuscular benzathine penicillin G 2.4 million U to prevent the development of tertiary syphilis.
Comment
Incidence
Syphilis has been well characterized since the early 15th century, though its geographic origin remains a topic of controversy.2 Although acquired syphilis infections represented a major source of morbidity and mortality in the early 20th century, the prevalence of syphilis in the United States declined substantially thereafter due to improved public health management.2 Syphilis was relatively rare in the United States by the year 1956, with fewer than 7000 cases of primary and secondary disease reported annually.3 The incidence of primary and secondary syphilis infections in the United States increased gradually until 1990 before declining precipitously and reaching an unprecedented low of 2.2 cases per 100,000 individuals in 2000.4 These shifts ultimately have resulted in decreased clinical familiarity with the disease presentation of syphilis among many health care providers. Since 2000, the incidence of syphilis infection has increased in the United States, with the greatest increases seen in men who have sex with men, intravenous drug users, and human immunodeficiency virus–infected individuals.5-7
RELATED ARTICLE: Syphilis and the Dermatologist
Pathogenesis and Transmission
The causative agent in syphilis infection is the bacterium Treponema pallidum, a member of the family Spirochaetaceae, which is distinguished by its thin, regularly coiled form and distinctive corkscrew motility.8 Syphilis is communicated primarily by sexual contact or in utero exposure during the primary and secondary stages of maternal infection.9 At the time of presentation, our patient denied having any new sexual partners or practices. He reported a monogamous heterosexual relationship within the months preceding presentation, suggesting historical inaccuracy on the part of the patient or probable infidelity in the reported relationship as an alternative means of infection transmission. Untreated individuals may be contagious for longer than 1 year,9 making transmission patterns difficult to track clinically.
Presentation
The clinical presentation of infection with T pallidum results from dual humoral and cell-mediated inflammatory responses in the host. The primary stage is classically defined by a single chancre, which develops at the inoculation site(s) 9 to 90 days following exposure. The chancre typically begins as a small papule that rapidly develops into a painless ulcer characterized by an indurated border, red base, bordering edema, and a diameter of 2 cm or less. Indolent regional lymphadenopathy often is observed in conjunction with the primary chancre.10 Our case is notable for the absence of a primary syphilitic lesion and lack of adenopathy. The primary chancre of syphilis typically resolves within 3 to 6 weeks of onset regardless of whether the patient is treated,4 thus suggesting the rare possibility that our patient developed a painless primary chancre without realizing it.
The secondary stage of syphilis infection arises weeks to months after resolution of the primary chancre and is triggered by hematogenous and lymphatic dissemination of the bacteria. The symptoms of secondary syphilis are primarily flulike and may include headache, malaise, fatigue, sore throat, arthralgia, and low-grade fever.9 Nontender regional lymphadenopathy and splenomegaly also have been reported.11 Our patient denied any systemic concerns throughout the duration of his illness, with the exception of odynophagia in association with ulceration of the oral mucosa. Abnormal laboratory findings in secondary syphilis are nonspecific and may include an elevated erythrocyte sedimentation rate and/or an increased white blood cell count with absolute lymphocytosis.12 Laboratory studies drawn at the time of presentation showed no such abnormalities in our patient.
The cutaneous signs of secondary syphilis arise concurrently with systemic manifestations and are a common finding, with lesions of the skin or oral mucosa present in up to 80% of patients,13 as in our case. Oral lesions classically involve ulcerations at the tip and sides of the tongue,12 which is distinct from our patient who developed oral lesions of the mucosal lip and hard palate.
Secondary syphilis classically features a copper-colored maculopapular rash with sharply delineated margins typically present on the palmar and plantar surfaces.14 Verrucous lesions appearing as moist exophytic plaques on the genitals, intertriginous areas, and/or perineum also have been described and are referred to as condyloma lata in the setting of secondary syphilis.15 In contrast to these classic findings, our patient demonstrated lichenoid lesions on the genitalia and white mucosal patches on the oral mucosa. Our case also was highly unusual because of the intense pruritus associated with the genital lesions, which starkly contrasts most secondary-stage cutaneous lesions that are classically asymptomatic.14 Additionally, our case was distinctive due to the lack of palmar or plantar involvement, which is considered a characteristic feature of secondary cutaneous syphilis.1 Finally, our case was notable for the presence of multiple annular cutaneous lesions, which indicated a late secondary-stage infection during which involution of the lesions produced endarteritis as deeper vessels became involved. A 20-year retrospective study by Abell et al11 demonstrated that 40% of syphilitic rashes are macular, 40% are maculopapular, 10% are papular (as in our case), and the remaining 10% are not easily grouped within these categories.
Differential Diagnosis
It has been estimated that approximately 8% of cutaneous syphilitic lesions demonstrate morphology and distributions suggestive of other dermatologic conditions, including atopic dermatitis, pityriasis rosea, psoriasis, drug-induced eruptions, erythema multiforme, mycosis fungoides, and far more uncommonly lichenoid lesions,16,17 as in our case.
Histopathology
It has been demonstrated that the gross appearance of the secondary syphilitic lesion depends both on the degree of inflammatory infiltrate and the extent of vascular involvement producing ischemia of the skin.1 Our case presented with small, flat-topped, papular lesions that grossly resembled lichen planus and were ultimately shown to be the product of dense lymphomononuclear infiltration extending perivascularly and throughout the superficial and deep dermis.
Biopsy of a lesion is one means of diagnosis, though the histologic appearance of secondary syphilis can mimic many other diseases. In primary and secondary syphilis, skin biopsy characteristically shows central thinning or ulceration of the epidermal layer with heavy dermal lymphocyte infiltration, lymphovascular proliferation with endarteritis, small-vessel thrombosis, and dermal necrosis. Lichen planus–type dermatitis is histologically characterized by hyperkeratosis, irregular epidermal hyperplasia, and a dermoepidermal junction that may be obscured by a dense lymphomononuclear infiltrate.9 The specimen taken from our patient showed minimal infiltrate in the papillary dermis, suggesting a diagnosis of secondary syphilis with lichenoid features. Despite a gross appearance consistent with lichen planus, the biopsy lacked the hydropic degeneration of the basal layer and keratinocyte necrosis that typically characterize this condition.
Diagnosis
Serologic testing for syphilis infection is comprised of nontreponemal and treponemal studies. Nontreponemal testing, which includes the RPR and VDRL test, detects antibodies to cardiolipin-lecithin antigen, a lipid component of the cell membranes of T pallidum. Because the specificity of these tests is fairly low, they typically are used only for screening and monitoring of disease progression and/or response to treatment. Approximately 25% of cases in the United States of primary syphilis are not detected by nontreponemal testing, whereas a nonreactive test nearly always excludes a diagnosis of secondary or latent-stage syphilitic infection.9 Indeed, nontreponemal studies show the highest antibody titers during the late secondary and early latent stages of infection with declining titers thereafter, even in the absence of antibiotic treatment. In our case, diagnosis was made by biopsy and RPR was used for staging; RPR was reactive at a dilution of 1:32, indicative of secondary or early latent infection.
Treponemal testing, which includes the fluorescent treponemal antibody absorption test, and multiplex flow immunoassay detects antibodies that are specific to syphilis infection. Treponemal antibodies are detectable earlier in the course of infection than nontreponemal antibodies and remain permanently detectable even following treatment. Because of its high specificity, treponemal testing often is used to confirm diagnosis after positive screening with nontreponemal tests.4 Positive fluorescent treponemal antibody absorption testing and positive multiplex flow immunoassay may be used to confirm the diagnosis of T pallidum infection.
The tertiary stage of syphilis infection can occur years after conclusion of the secondary stage and is comprised of one or more of the following: gummas, aortic dilatation or dissection, and neurosyphilitic manifestations such as tabes dorsalis or general paresis.1 It is of vital importance to identify syphilis infection prior to the onset of the tertiary stage to prevent substantial morbidity and mortality.
Treatment
Our patient’s symptoms abated after empiric treatment with amoxicillin for presumed streptococcal throat infection after he presented to the emergency department with odynophagia, which is not surprising given the moderate-spectrum coverage of this β-lactam antibiotic as well as the near-complete susceptibility of Treponema spirochetes to amoxicillin in primary and secondary syphilis with notably lower efficacy in latent or tertiary disease. It was essential to treat the patient with a single dose of intramuscular benzathine penicillin G 2.4 million U, which has been shown to reliably prevent recurrence of infection or progression to tertiary syphilis.18
Conclusion
We present a rare case of lichenoid secondary syphilis in the absence of lesions on the palmar and plantar surfaces. The patient lacked any other cutaneous or systemic manifestations, except for odynophagia in association with oral mucosal lesions. He denied any new sexual partners and did not recall having a primary chancre. Also strikingly unusual in this case was the intense pruritus associated with the genital eruption, which is unlike the classic lack of symptoms experienced in the great majority of eruptions due to secondary syphilis. A clinical appreciation of the many cutaneous manifestations of syphilis infection remains critical to early identification of the disease prior to progression to the tertiary stage and its devastating sequelae.
- Dourmishev LA, Assen L. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Kilmarx PH, St Louis ME. The evolving epidemiology of syphilis. Am J Public Health. 1995;85(8, pt 1):1053-1054.
- Patton ME, Su JR, Nelson R, et al. Primary and secondary syphilis—United States, 2005-2013. MMWR Morb Mortal Wkly Rep. 2014;63:402-406.
- Coffin LS, Newberry A, Hagan H, et al. Syphilis in drug users in low and middle income countries. Int J Drug Policy. 2010;21:20-27.
- Gao L, Zhang L, Jin Q. Meta-analysis: prevalence of HIV infection and syphilis among MSM in China. Sex Transm Infect. 2009;85:354-358.
- Karp G, Schlaeffer F, Jotkowitz A, et al. Syphilis and HIV co-infection. Eur J Int Med. 2009;20:9-13.
- Hol EL, Lukehart SA. Syphilis: using modern approaches to understand an old disease. J Clin Invest. 2011;121:4584-4592.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis. J Foot Ankle Surg. 2011;50:96-101.
- Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68:283-290.
- Abell E, Marks R, Jones W. Secondary syphilis: a clinicopathological review. Br J Dermatol. 1975;93:53-61.
- Fiumara N. The treponematoses. Int Dermatol. 1992;1:953-974.
- Martin DH, Mroczkowski TF. Dermatological manifestations of sexually transmitted diseases other than HIV. Infect Dis Clin North Am. 1994;8:533-583.
- Morton RS. The treponematoses. In: Champion RH, Bourton JL, Burns DA, et al. Rook’s Textbook of Dermatology. 6th ed. London, United Kingdom: Blackwell Science; 1998:1237-1275.
- Rosen T, Hwong H. Pedal interdigital condylomata lata: a rare sign of secondary syphilis. Sex Transm Dis. 2001;28:184-186.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Tang MBY, Yosipovitch G, Tan SH. Secondary syphilis presenting as a lichen planus-like rash. J Eur Acad Dermatol Venereol. 2004;18:185-187.
- Onoda Y. Clinical evaluation of amoxicillin in the treatment of syphilis. J Int Med. 1979;7:539-545.
- Dourmishev LA, Assen L. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Kilmarx PH, St Louis ME. The evolving epidemiology of syphilis. Am J Public Health. 1995;85(8, pt 1):1053-1054.
- Patton ME, Su JR, Nelson R, et al. Primary and secondary syphilis—United States, 2005-2013. MMWR Morb Mortal Wkly Rep. 2014;63:402-406.
- Coffin LS, Newberry A, Hagan H, et al. Syphilis in drug users in low and middle income countries. Int J Drug Policy. 2010;21:20-27.
- Gao L, Zhang L, Jin Q. Meta-analysis: prevalence of HIV infection and syphilis among MSM in China. Sex Transm Infect. 2009;85:354-358.
- Karp G, Schlaeffer F, Jotkowitz A, et al. Syphilis and HIV co-infection. Eur J Int Med. 2009;20:9-13.
- Hol EL, Lukehart SA. Syphilis: using modern approaches to understand an old disease. J Clin Invest. 2011;121:4584-4592.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis. J Foot Ankle Surg. 2011;50:96-101.
- Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68:283-290.
- Abell E, Marks R, Jones W. Secondary syphilis: a clinicopathological review. Br J Dermatol. 1975;93:53-61.
- Fiumara N. The treponematoses. Int Dermatol. 1992;1:953-974.
- Martin DH, Mroczkowski TF. Dermatological manifestations of sexually transmitted diseases other than HIV. Infect Dis Clin North Am. 1994;8:533-583.
- Morton RS. The treponematoses. In: Champion RH, Bourton JL, Burns DA, et al. Rook’s Textbook of Dermatology. 6th ed. London, United Kingdom: Blackwell Science; 1998:1237-1275.
- Rosen T, Hwong H. Pedal interdigital condylomata lata: a rare sign of secondary syphilis. Sex Transm Dis. 2001;28:184-186.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Tang MBY, Yosipovitch G, Tan SH. Secondary syphilis presenting as a lichen planus-like rash. J Eur Acad Dermatol Venereol. 2004;18:185-187.
- Onoda Y. Clinical evaluation of amoxicillin in the treatment of syphilis. J Int Med. 1979;7:539-545.
Practice Points
- Syphilis retains its reputation as “the great imitator” due to its wide variability in clinical presentation and propensity for misdiagnosis.
- Lichenoid syphilis is a well-described cutaneous presentation of secondary syphilis, though the characteristics of these lesions remain highly variable and require a high degree of clinical suspicion.
- Treponema pallidum is partially susceptible to most β-lactam antibiotics in primary and early secondary stages of infection; thus, use of these medications can obscure symptoms without adequately treating the infection.
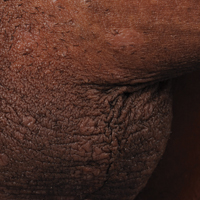
Sjögren-Larsson Syndrome: Definitive Diagnosis on Magnetic Resonance Spectroscopy
Sjögren-Larsson syndrome (SLS) is a rare autosomal-recessive neurocutaneous disorder comprising a triad of ichthyosis, mental retardation, and spastic diplegia or quadriplegia.1 The disorder was first described by Sjögren and Larsson2 in 1957. Early reports of SLS were mainly in white patients, with a particularly high prevalence of 8.3 cases per 100,000 individuals in the county of Västerbotten in Sweden.3 Reports of SLS in Asian and Indian populations are rare.4,5 We report a case of SLS in an Indian boy.
Case Report
A 12-year-old Indian boy born to nonconsanguineous parents after a full-term pregnancy with normal vaginal delivery presented with generalized dry scaly skin that had been present since 2 months of age. He had a history of delayed milestones (ie, facial recognition, sitting without support at 3 years of age), inability to walk, dysarthria, mental retardation). He had never attended school due to subnormal intellectual functioning. He had a single episode of a tonic-clonic seizure at 4 years of age but was not on any regular antiepileptic medication. There was a history of similar skin lesions in one male sibling of the patient and in 2 maternal uncles. None of them survived beyond early childhood, but detailed information regarding the cutaneous and neurologic manifestations in these family members was not available.
Cutaneous examination revealed lamellarlike ichthyosis on the dorsal aspects of the arms and legs (Figure 1A). Ichthyosis with lichenification was present on the neck, axillae, cubital and popliteal fossae, and abdomen (Figure 1B). The palms and soles showed keratoderma. Neurologic examination of the arms revealed mild rigidity and brisk reflexes. Examination of the legs showed marked rigidity, brisk knee jerks, ankle clonus, extensor plantar reflexes, flexion deformity with contractures, and scissor gait. A Goddard (Seguin) formboard test was performed and indicated a mental age of 4 years. The patient’s IQ was in the range of 25 to 30, indicating a severe degree of subnormality in intellectual functioning. The clinical presentation suggested a diagnosis of SLS.
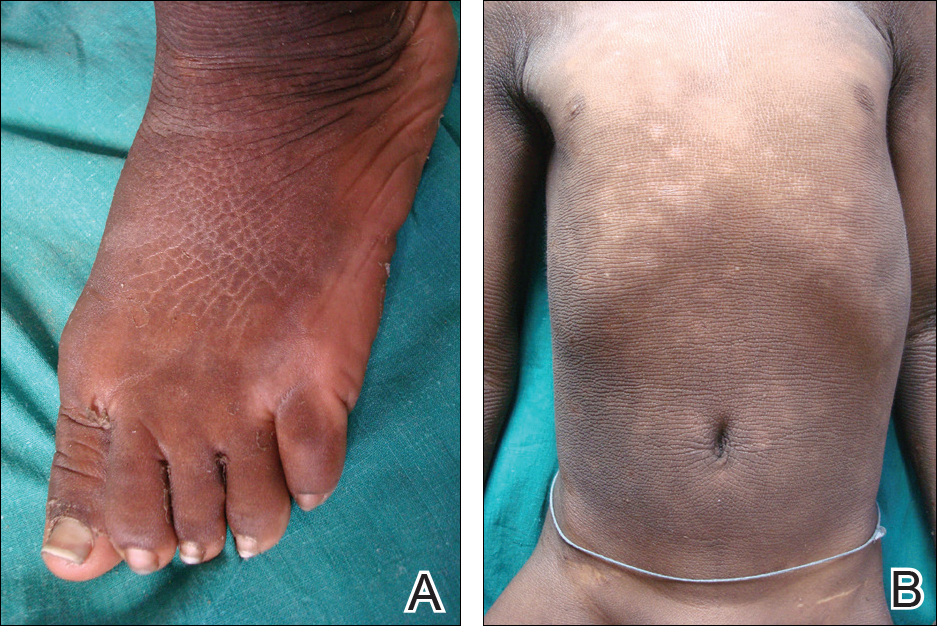
A skin biopsy from the ichthyotic lesion showed hyperkeratosis, acanthosis, and papillomatosis with sparse superficial perivascular lymphocytic infiltrate, thus confirming the diagnosis of lamellar ichthyosis. Fundus examination was normal. Magnetic resonance imaging (MRI) of the brain revealed confluent symmetrical signal abnormalities along the body of the lateral ventricles, white matter in the perioccipital horn, and in deep white matter of centrum semiovale (Figure 2). Magnetic resonance spectroscopy revealed a narrow lipid peak at approximately 1.3 ppm in the region of signal abnormality (Figure 3). Thus, the diagnosis of SLS was confirmed. Measurement of fatty aldehyde dehydrogenase (FALDH) activity and genetic analysis were not performed due to unavailability.
The patient was treated with topical emollients for the ichthyosis. To reduce his dietary intake of long-chain fatty acids and increase the intake of omega-3 and omega-6 fatty acids, the patient’s parents were advised to use canola, mustard, and/or coconut oil for cooking for the patient, and skim milk was recommended instead of whole milk. Neurodevelopmental techniques in the form of stretching exercises were given to maintain his range of movements. Gutter splints were given to maintain the knees in extension for physiological standing and to prevent osteoporosis. Subsequently, the patient also underwent a multilevel soft-tissue release (hip and knee joints) to relieve the contractures. These measures resulted in considerable improvement and the patient was able to walk with support.
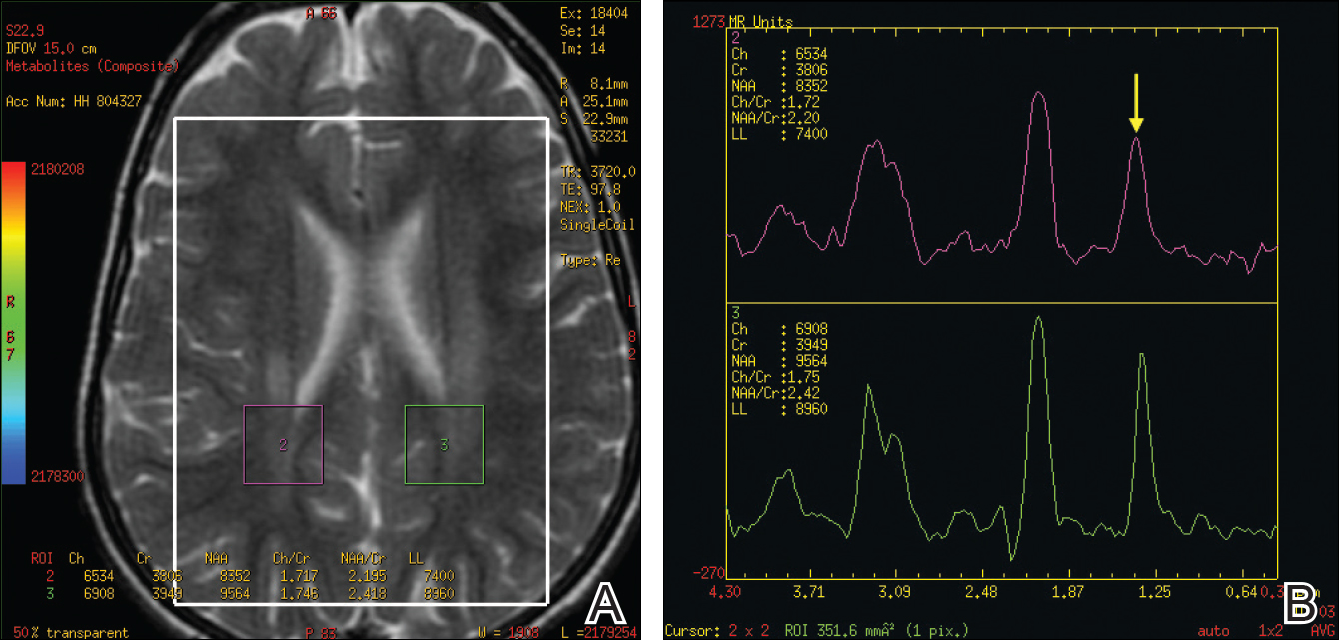
Comment
Presentation
The characteristic clinical features of SLS begin to develop during the intranatal period and infancy.1,6 Pathologic skin involvement can be detected as early as week 23 of gestation. Preterm births associated with SLS have commonly been described.3 Ichthyosis often is evident at birth, but collodion membrane is uncommon. Severe pruritus is a marked feature unlike most other types of ichthyosis. The ichthyosis often is generalized with prominent involvement of the flexural areas and nape of the neck, varying from fine furfuraceous to larger lamellarlike scales. Velvety orange or brown lichenification often is a predominant feature in the flexures of the arms, legs, neck, and mid abdomen. Mental retardation, developmental delay, and spasticity usually become apparent at 1 to 2 years of age and subsequently are nonprogressive.6,7 However, patients rarely have been described with normal intellectual functioning.7 Spasticity often is more severe in the lower limbs and may lead to contractures, kyphoscoliosis, hip dislocation, and short stature. Delayed speech and dysarthria are common. Parafoveal glistening white dots on the retina are a pathognomonic feature and typically appear in the first 2 years of life; however, they are seen in approximately 30% of patients and increase slightly in number with age.6,8 There may be associated decreased visual acuity, photophobia, myopia, and astigmatism. Other clinical features include enamel hypoplasia, metaphyseal dysplasia, and epilepsy.1,6
Gene Mutations
Sjögren-Larsson syndrome is caused by mutation in the aldehyde dehydrogenase 3 family member A2 gene, ALDH3A2 (17p11.2), which codes for FALDH.1,6,7 The ALDH3A2 gene is 11 exons long and gives rise to 2 protein isoforms that differ in their carboxy-terminal domains; the major isoform, composed of 485 amino acids, localizes to the endoplasmic reticulum. The minor protein isoform (FALDHv) is composed of 508 amino acids, possesses a longer carboxy-terminal, and appears to be targeted to the peroxisome. Several mutations have been reported throughout the ALDH3A2 gene, including missense mutations (most common [38% of cases of SLS6]), deletions, insertions, splicing errors, and complex rearrangements. Although several of these mutations are private, several common mutations may be indicative of founder effects (ie, shared ancestry), consanguinity, or recurrent mutational events (mutation hotspots).6,7 Despite the wide spectrum of mutations, there is very little phenotypic variation, with consistently severe cutaneous and neurological involvement occurring in a majority of patients.7 However, Lossos et al9 described remarkable phenotypic variation in 6 siblings of an Arab family and suggested that additional unknown genetic or environmental factors may compensate for the biochemical defect.
Lipid Metabolism
Fatty aldehyde dehydrogenase is expressed in almost all cells and tissues and catalyzes the oxidation of fatty aldehydes to fatty acids (eFigure 1). It also is a part of the fatty alcohol:NAD oxidoreductase (FAO) enzyme complex, which catalyzes fatty alcohol oxidation to fatty acid. Fatty aldehyde dehydrogenase deficiency leads to accumulation of long-chain alcohols (eg, hexadecanol, octadecanol, octadecenol) and diversion of fatty alcohol into alternate biosynthetic pathways such as wax esters and 1-O-alkyl-2,3-diacylglycerol.10 Other lipids that are increased are illustrated in eFigure 2. Accumulation of these lipids, toxic effects of abnormal lipids (especially fatty aldehydes and Schiff base protein-lipid adducts), and lack of essential lipids (eg, polyunsaturated fatty acids, ceramides 1 and 6, triglycerides) are responsible for the classical cutaneous, neurologic, and ophthalmologic features of SLS.

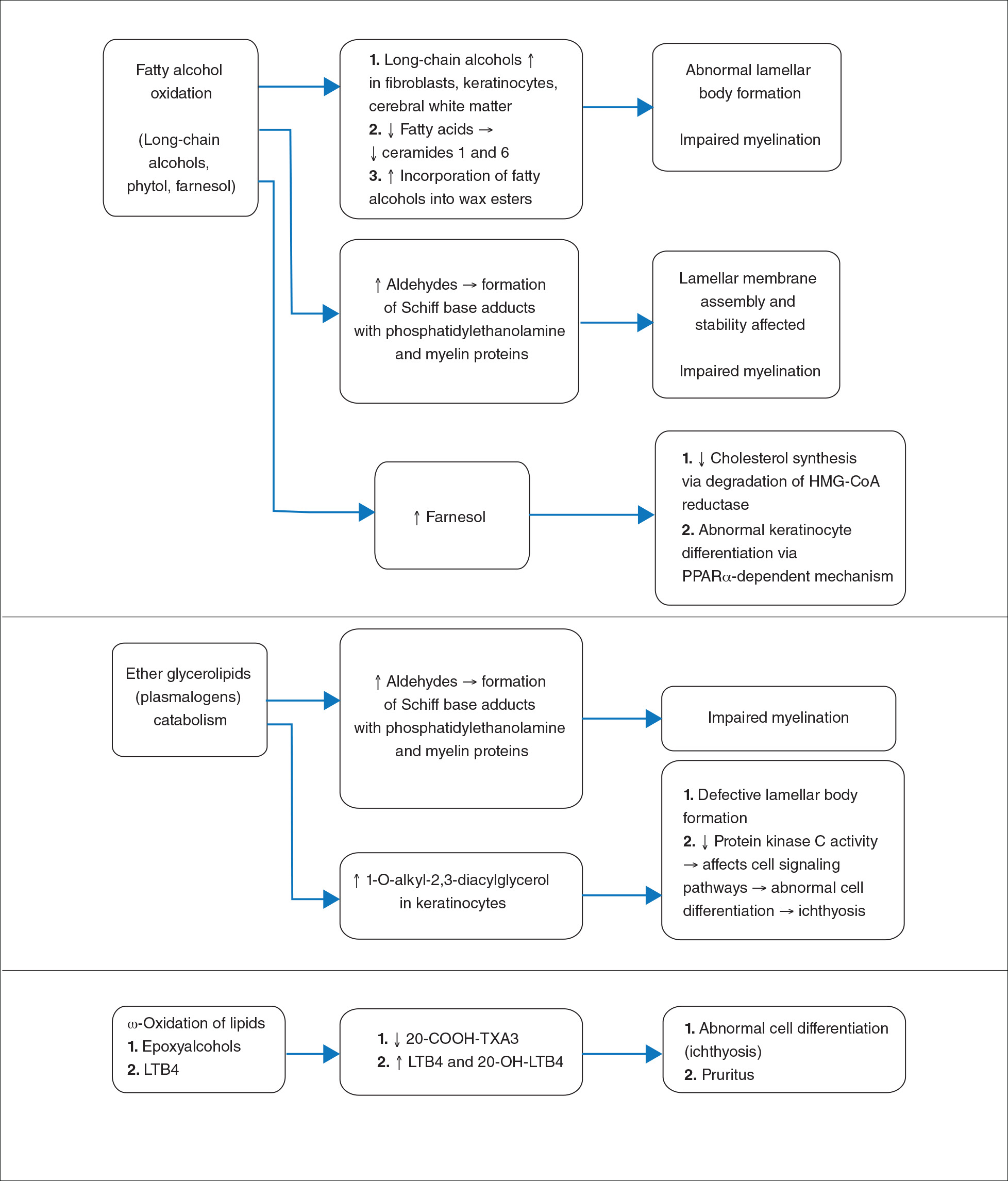
Histopathology
The epidermal permeability barrier is critically dependent on the appropriate lipid composition of the multilamellar stratum corneum intercellular membranes, an equimolar ratio of cholesterol, ceramides, and fatty acids. Histopathology of the skin in SLS generally shows hyperkeratosis, papillomatosis, acanthosis, and a mildly thickened granular layer. Ultrastructural studies of the skin reveal misshapen/empty lamellar bodies, abnormal cytoplasmic lamellar inclusions in the granular keratinocytes, lipid droplets in the stratum corneum with decreased lamellar bilayers, and lamellar/nonlamellar phase separation in the stratum corneum interstitium.11 These findings indicate that lipid metabolism dysfunction in SLS results in marked impairment in formation and secretion of lamellar bodies in the epidermis and consequent disorganization of the stratum corneum lamellar membranes. The resulting disruption of the skin barrier function leads to increased transepidermal water loss, resulting in ichthyosis.11,12 Another proposed mechanism for ichthyosis in SLS is disruption of the normal epidermal differentiation resulting from abnormal lipid metabolites (eFigure 2). Also, increased leukotriene B4 (LTB4) and 20-hydroxy-leukotriene B4 (20-OH-LTB4)(eFigure 1) may be responsible for the considerable pruritus seen in SLS.10
Neurologic Findings
Neurologic changes in SLS result from delayed and deficient myelination. Neuropathological studies have shown ballooning of myelin sheaths, extensive loss of myelin, axonal damage, and astrogliosis. The presence of lipoid material positive for periodic acid–Schiff that stains light rather than dark pink, dense distribution of round/ellipsoid bodies in the white matter of the cerebrum and brainstem positive for periodic acid–Schiff, and proliferation of perivascular macrophages containing lipofuscinlike pigments also have been described.13 Possibly, in the absence of FALDH, metabolism of plasmalogens (a major component of myelin) results in increased fatty aldehydes, which are either diverted to fatty alcohols or form adducts with phosphatidylethanolamine and myelin basic proteins (eFigure 1). Magnetic resonance imaging of the brain usually shows hypomyelination involving the periventricular white matter extending from the frontal to the occipital area.7,14 Mild ventricular enlargement may be an additional feature.14
A useful application of MRI is the proton magnetic resonance spectroscopy, which quantifies the brain metabolites noninvasively, displaying them as a spectrum on a graph. The spectrum comprises a set of resonances/peaks distributed along an x-axis. The resonances of these metabolites are obtained after suppressing the large signals from water protons. Proton magnetic resonance spectroscopy of the normal brain shows 3 prominent peaks: (1) N-acetylaspartate (NAA) at 2.02 ppm, (2) creatine at 3.02 ppm, and (3) choline at 3.22 ppm. In SLS, cerebral proton MRI spectroscopy reveals a characteristic abnormal, prominent, and narrow lipid peak at 1.3 ppm (corresponding to hexadecanol and octadecanol) and may offer a quantitative parameter for monitoring the effects of therapeutic interventions.7,14,15 The most intense lipid peaks are located in the periventricular regions in the anterior and posterior trigones. An abnormal but much smaller peak may be seen at 0.8 to 0.9 ppm, corresponding to phytol.14 Gradual emergence of these changes occurs in the first 2 years of life and then remains stable.15 Proton magnetic resonance spectroscopy also can be used for screening of SLS heterozygotes.16 Lipid peaks have been described in other disorders of lipid metabolism, but they are less intense, broader, and disappear on longer echo time sequences.14
Besides the characteristic parafoveal glistening white dots the retina, optical coherence tomography shows focal hyperreflectivitity in the perifoveal ganglion cell layer and inner plexiform layer of the retina as well as cystoid foveal degeneration.17 The intraretinal deposition of lipid metabolites probably leads to Müller cell degeneration with subsequent formation of cystoid spaces and atrophic changes in the fovea.
Measurement of FALDH or FAO activity in cultured skin fibroblasts and leukocytes using flurometric or gas chromatography mass spectrometry assays is a reliable biochemical test in cases of SLS as well as in heterozygotes.17 A decrease in FALDH/FAO activity also can be demonstrated by histochemical staining in skin biopsy.11 Pathologic urinary excretion of LTB4 and 20-OH-LTB4 also is a biochemical marker of SLS. Mutation analysis for a specific gene defect is diagnostic in cases of SLS as well as in heterozygotes. Prenatal diagnosis of SLS is possible by assessing FALDH activity or gene defects in cultured chorionic villus fibroblasts and amniocytes.18,19
Differential Diagnosis
The differential diagnosis of SLS includes congenital ichthyosiform erythroderma with neurological signs (Tay syndrome, Conradi-Hünermann-Happle syndrome) and neurocutaneous disorders such as neutral lipid storage disease and multiple sulfatase deficiency; however, the nature of the ichthyosis, presence of spastic diplegia/tetraplegia, characteristic parafoveal glistening white dots on the retina, and MRI and proton magnetic resonance spectroscopy findings help to easily differentiate SLS from these disorders.
Treatment
Treatment of SLS mainly is palliative. Ichthyosis can be treated with topical keratolytics, emollients, calcipotriol, and oral retinoids (acitretin).6 Zileuton, a 5-lipoxygenase inhibitor, inhibits synthesis of LTB4 and cysteinyl leukotrienes, thereby reducing the severity of pruritus and also has been shown to improve the speed of information processing.18 Similarly, montelukast, a leuko-triene antagonist, is helpful in relieving the agonizing pruritus.19 Experimental studies have shown that bezafibrate, a peroxisome proliferator-activated receptor α agonist, induces FALDH activity in fibroblasts of SLS patients that still have some residual FALDH activity, but further research is required to determine whether SLS patients could benefit from treatment.20 Physiotherapy helps in relieving the spasticity to some extent, such as in our case. Dietary intervention with reduced fat intake (up to 30% of total daily calorific requirement) and supplementation with omega-3 and omega-6 fatty acids has shown variable results in anecdotal reports.21-23 Gene therapy using recombinant adeno-associated virus 2 vectors to restore FALDH has been projected as a future treatment option.24 Despite lack of effective treatment options, most patients of SLS survive well into adulthood.
Conclusion
Because ichthyosis is one of the earliest and prominent symptoms of SLS, a dermatologist can play an important role in early diagnosis. Any child with the classical pattern of ichthyosis should be thoroughly examined for early neurologic signs and investigated to rule out SLS. Proton magnetic resonance spectroscopy serves as a useful adjunct in the diagnosis of SLS by confirming the accumulation of abnormal lipids in the periventricular white matter, especially when specific enzyme analysis and genetic analysis are not available in resource-restricted settings.
- Judge MR, McLean WHI, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, eds. Rook’s Textbook of Dermatology. 7th ed. West Sussex, United Kingdom: Wiley & Sons; 2004:34.37-34.39.
- Sjögren T, Larsson T. Oligophrenia in association with congenital ichthyosis and spastic disorders. Acta Psychiatr Neurol Scand. 1957;32:1-113.
- Jagell S, Gustavson KH, Holmgren G. Sjögren-Larsson syndrome in Sweden. a clinical, genetic and epidemiological study. Clin Genet. 1981;19:233-256.
- Sood M, Trehan A, Dinakaran J, et al. Sjögren-Larsson syndrome. Indian J Pediatr. 2002;69:193-194.
- Uppal M, Srinivas CR, Thowfeeq KT. Sjögren-Larsson syndrome: report of two cases. Indian J Dermatol Venereol Leprol. 2004;70:110-111.
- Rizzo WB. Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1-9.
- Willemsen MA, Ijlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain. 2001;124(pt 7):1426-1437.
- Willemsen MA, Cruysberg JR, Rotteveel JJ, et al. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjögren-Larsson syndrome. Am J Ophthalmol. 2000;130:782-789.
- Lossos A, Khoury M, Rizzo WB, et al. Phenotypic variability among adult siblings with Sjögren-Larsson syndrome. Arch Neurol. 2006;63:278-280.
- Rizzo WB, Craft DA, Somer T, et al. Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjögren-Larsson syndrome. J Lipid Res. 2008;49:410-419.
- Rizzo WB, S’Aulis D, Jennings MA, et al. Ichthyosis in Sjögren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443-451.
- Rizzo WB. The role of fatty aldehyde dehydrogenase in epidermal structure and function. Dermatoendocrinol. 2011;2:91-99.
- Yamaguchi K, Handa T. Sjögren-Larsson syndrome: postmortem brain abnormalities. Pediatr Neurol. 1998;18:338-341.
- Mano T, Ono J, Kaminaga T, et al. Proton MR spectroscopy of Sjögren-Larsson’s Syndrome. Am J Neuroradiol. 1999;20:1671-1673.
- Willemsen MA, van der Graf M, van der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjögren-Larsson syndrome: characterization of the leukoencephalopathy. Am J Neuroradiol. 2004;25:649-657.
- Kaminaga T, Mano T, Ono J, et al. Proton magnetic resonance spectroscopy of Sjögren-Larsson Syndrome. Magn Reson Med. 2001;45:1112-1115.
- Fuijkschot J, Cruysberg JR, Willemsen MA, et al. Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren-Larsson syndrome detected by optical coherence tomography. Ophthalmology. 2008;115:870-875.
- Willemsen MA, Lutt MA, Steijlen PM, et al. Clinical and biochemical effects of zileuton in patients with the Sjögren-Larsson syndrome. Eur J Pediatr. 2001;160:711-717.
- Pirgon O, Aydin K, Atabek ME. Proton magnetic resonance spectroscopy findings and clinical effects of montelukast sodium in a case with Sjögren-Larsson syndrome. J Child Neurol. 2006;21:1092-1095.
- Gloerich J, Ijlst L, Wanders RJ, et al. Bezafibrate induces FALDH in human fibroblasts; implications for Sjögren-Larsson syndrome Mol Genet Metab. 2006;89:111-115.
- Auada MP, Taube MB, Collares EF, et al. Sjögren-Larsson syndrome: biochemical defects and follow up in three cases. Eur J Dermatol. 2002;12:263-266.
- Taube B, Billeaud C, Labreze C, et al. Sjögren-Larsson syndrome: early diagnosis, dietary management and biochemical studies in two cases. Dermatology. 1999;198:340-345.
- Rizzo WB. Genetics and prospective therapeutic targets for Sjögren-Larsson Syndrome. Expert Opin Orphan Drugs. 2016;4:395-406.
- Haug S, Braun-Falco M. Restoration of fatty aldehyde dehydrogenase deficiency in Sjögren-Larsson syndrome. Gene Ther. 2006;13:1021-1026.
Sjögren-Larsson syndrome (SLS) is a rare autosomal-recessive neurocutaneous disorder comprising a triad of ichthyosis, mental retardation, and spastic diplegia or quadriplegia.1 The disorder was first described by Sjögren and Larsson2 in 1957. Early reports of SLS were mainly in white patients, with a particularly high prevalence of 8.3 cases per 100,000 individuals in the county of Västerbotten in Sweden.3 Reports of SLS in Asian and Indian populations are rare.4,5 We report a case of SLS in an Indian boy.
Case Report
A 12-year-old Indian boy born to nonconsanguineous parents after a full-term pregnancy with normal vaginal delivery presented with generalized dry scaly skin that had been present since 2 months of age. He had a history of delayed milestones (ie, facial recognition, sitting without support at 3 years of age), inability to walk, dysarthria, mental retardation). He had never attended school due to subnormal intellectual functioning. He had a single episode of a tonic-clonic seizure at 4 years of age but was not on any regular antiepileptic medication. There was a history of similar skin lesions in one male sibling of the patient and in 2 maternal uncles. None of them survived beyond early childhood, but detailed information regarding the cutaneous and neurologic manifestations in these family members was not available.
Cutaneous examination revealed lamellarlike ichthyosis on the dorsal aspects of the arms and legs (Figure 1A). Ichthyosis with lichenification was present on the neck, axillae, cubital and popliteal fossae, and abdomen (Figure 1B). The palms and soles showed keratoderma. Neurologic examination of the arms revealed mild rigidity and brisk reflexes. Examination of the legs showed marked rigidity, brisk knee jerks, ankle clonus, extensor plantar reflexes, flexion deformity with contractures, and scissor gait. A Goddard (Seguin) formboard test was performed and indicated a mental age of 4 years. The patient’s IQ was in the range of 25 to 30, indicating a severe degree of subnormality in intellectual functioning. The clinical presentation suggested a diagnosis of SLS.
A skin biopsy from the ichthyotic lesion showed hyperkeratosis, acanthosis, and papillomatosis with sparse superficial perivascular lymphocytic infiltrate, thus confirming the diagnosis of lamellar ichthyosis. Fundus examination was normal. Magnetic resonance imaging (MRI) of the brain revealed confluent symmetrical signal abnormalities along the body of the lateral ventricles, white matter in the perioccipital horn, and in deep white matter of centrum semiovale (Figure 2). Magnetic resonance spectroscopy revealed a narrow lipid peak at approximately 1.3 ppm in the region of signal abnormality (Figure 3). Thus, the diagnosis of SLS was confirmed. Measurement of fatty aldehyde dehydrogenase (FALDH) activity and genetic analysis were not performed due to unavailability.
The patient was treated with topical emollients for the ichthyosis. To reduce his dietary intake of long-chain fatty acids and increase the intake of omega-3 and omega-6 fatty acids, the patient’s parents were advised to use canola, mustard, and/or coconut oil for cooking for the patient, and skim milk was recommended instead of whole milk. Neurodevelopmental techniques in the form of stretching exercises were given to maintain his range of movements. Gutter splints were given to maintain the knees in extension for physiological standing and to prevent osteoporosis. Subsequently, the patient also underwent a multilevel soft-tissue release (hip and knee joints) to relieve the contractures. These measures resulted in considerable improvement and the patient was able to walk with support.
Comment
Presentation
The characteristic clinical features of SLS begin to develop during the intranatal period and infancy.1,6 Pathologic skin involvement can be detected as early as week 23 of gestation. Preterm births associated with SLS have commonly been described.3 Ichthyosis often is evident at birth, but collodion membrane is uncommon. Severe pruritus is a marked feature unlike most other types of ichthyosis. The ichthyosis often is generalized with prominent involvement of the flexural areas and nape of the neck, varying from fine furfuraceous to larger lamellarlike scales. Velvety orange or brown lichenification often is a predominant feature in the flexures of the arms, legs, neck, and mid abdomen. Mental retardation, developmental delay, and spasticity usually become apparent at 1 to 2 years of age and subsequently are nonprogressive.6,7 However, patients rarely have been described with normal intellectual functioning.7 Spasticity often is more severe in the lower limbs and may lead to contractures, kyphoscoliosis, hip dislocation, and short stature. Delayed speech and dysarthria are common. Parafoveal glistening white dots on the retina are a pathognomonic feature and typically appear in the first 2 years of life; however, they are seen in approximately 30% of patients and increase slightly in number with age.6,8 There may be associated decreased visual acuity, photophobia, myopia, and astigmatism. Other clinical features include enamel hypoplasia, metaphyseal dysplasia, and epilepsy.1,6
Gene Mutations
Sjögren-Larsson syndrome is caused by mutation in the aldehyde dehydrogenase 3 family member A2 gene, ALDH3A2 (17p11.2), which codes for FALDH.1,6,7 The ALDH3A2 gene is 11 exons long and gives rise to 2 protein isoforms that differ in their carboxy-terminal domains; the major isoform, composed of 485 amino acids, localizes to the endoplasmic reticulum. The minor protein isoform (FALDHv) is composed of 508 amino acids, possesses a longer carboxy-terminal, and appears to be targeted to the peroxisome. Several mutations have been reported throughout the ALDH3A2 gene, including missense mutations (most common [38% of cases of SLS6]), deletions, insertions, splicing errors, and complex rearrangements. Although several of these mutations are private, several common mutations may be indicative of founder effects (ie, shared ancestry), consanguinity, or recurrent mutational events (mutation hotspots).6,7 Despite the wide spectrum of mutations, there is very little phenotypic variation, with consistently severe cutaneous and neurological involvement occurring in a majority of patients.7 However, Lossos et al9 described remarkable phenotypic variation in 6 siblings of an Arab family and suggested that additional unknown genetic or environmental factors may compensate for the biochemical defect.
Lipid Metabolism
Fatty aldehyde dehydrogenase is expressed in almost all cells and tissues and catalyzes the oxidation of fatty aldehydes to fatty acids (eFigure 1). It also is a part of the fatty alcohol:NAD oxidoreductase (FAO) enzyme complex, which catalyzes fatty alcohol oxidation to fatty acid. Fatty aldehyde dehydrogenase deficiency leads to accumulation of long-chain alcohols (eg, hexadecanol, octadecanol, octadecenol) and diversion of fatty alcohol into alternate biosynthetic pathways such as wax esters and 1-O-alkyl-2,3-diacylglycerol.10 Other lipids that are increased are illustrated in eFigure 2. Accumulation of these lipids, toxic effects of abnormal lipids (especially fatty aldehydes and Schiff base protein-lipid adducts), and lack of essential lipids (eg, polyunsaturated fatty acids, ceramides 1 and 6, triglycerides) are responsible for the classical cutaneous, neurologic, and ophthalmologic features of SLS.
Histopathology
The epidermal permeability barrier is critically dependent on the appropriate lipid composition of the multilamellar stratum corneum intercellular membranes, an equimolar ratio of cholesterol, ceramides, and fatty acids. Histopathology of the skin in SLS generally shows hyperkeratosis, papillomatosis, acanthosis, and a mildly thickened granular layer. Ultrastructural studies of the skin reveal misshapen/empty lamellar bodies, abnormal cytoplasmic lamellar inclusions in the granular keratinocytes, lipid droplets in the stratum corneum with decreased lamellar bilayers, and lamellar/nonlamellar phase separation in the stratum corneum interstitium.11 These findings indicate that lipid metabolism dysfunction in SLS results in marked impairment in formation and secretion of lamellar bodies in the epidermis and consequent disorganization of the stratum corneum lamellar membranes. The resulting disruption of the skin barrier function leads to increased transepidermal water loss, resulting in ichthyosis.11,12 Another proposed mechanism for ichthyosis in SLS is disruption of the normal epidermal differentiation resulting from abnormal lipid metabolites (eFigure 2). Also, increased leukotriene B4 (LTB4) and 20-hydroxy-leukotriene B4 (20-OH-LTB4)(eFigure 1) may be responsible for the considerable pruritus seen in SLS.10
Neurologic Findings
Neurologic changes in SLS result from delayed and deficient myelination. Neuropathological studies have shown ballooning of myelin sheaths, extensive loss of myelin, axonal damage, and astrogliosis. The presence of lipoid material positive for periodic acid–Schiff that stains light rather than dark pink, dense distribution of round/ellipsoid bodies in the white matter of the cerebrum and brainstem positive for periodic acid–Schiff, and proliferation of perivascular macrophages containing lipofuscinlike pigments also have been described.13 Possibly, in the absence of FALDH, metabolism of plasmalogens (a major component of myelin) results in increased fatty aldehydes, which are either diverted to fatty alcohols or form adducts with phosphatidylethanolamine and myelin basic proteins (eFigure 1). Magnetic resonance imaging of the brain usually shows hypomyelination involving the periventricular white matter extending from the frontal to the occipital area.7,14 Mild ventricular enlargement may be an additional feature.14
A useful application of MRI is the proton magnetic resonance spectroscopy, which quantifies the brain metabolites noninvasively, displaying them as a spectrum on a graph. The spectrum comprises a set of resonances/peaks distributed along an x-axis. The resonances of these metabolites are obtained after suppressing the large signals from water protons. Proton magnetic resonance spectroscopy of the normal brain shows 3 prominent peaks: (1) N-acetylaspartate (NAA) at 2.02 ppm, (2) creatine at 3.02 ppm, and (3) choline at 3.22 ppm. In SLS, cerebral proton MRI spectroscopy reveals a characteristic abnormal, prominent, and narrow lipid peak at 1.3 ppm (corresponding to hexadecanol and octadecanol) and may offer a quantitative parameter for monitoring the effects of therapeutic interventions.7,14,15 The most intense lipid peaks are located in the periventricular regions in the anterior and posterior trigones. An abnormal but much smaller peak may be seen at 0.8 to 0.9 ppm, corresponding to phytol.14 Gradual emergence of these changes occurs in the first 2 years of life and then remains stable.15 Proton magnetic resonance spectroscopy also can be used for screening of SLS heterozygotes.16 Lipid peaks have been described in other disorders of lipid metabolism, but they are less intense, broader, and disappear on longer echo time sequences.14
Besides the characteristic parafoveal glistening white dots the retina, optical coherence tomography shows focal hyperreflectivitity in the perifoveal ganglion cell layer and inner plexiform layer of the retina as well as cystoid foveal degeneration.17 The intraretinal deposition of lipid metabolites probably leads to Müller cell degeneration with subsequent formation of cystoid spaces and atrophic changes in the fovea.
Measurement of FALDH or FAO activity in cultured skin fibroblasts and leukocytes using flurometric or gas chromatography mass spectrometry assays is a reliable biochemical test in cases of SLS as well as in heterozygotes.17 A decrease in FALDH/FAO activity also can be demonstrated by histochemical staining in skin biopsy.11 Pathologic urinary excretion of LTB4 and 20-OH-LTB4 also is a biochemical marker of SLS. Mutation analysis for a specific gene defect is diagnostic in cases of SLS as well as in heterozygotes. Prenatal diagnosis of SLS is possible by assessing FALDH activity or gene defects in cultured chorionic villus fibroblasts and amniocytes.18,19
Differential Diagnosis
The differential diagnosis of SLS includes congenital ichthyosiform erythroderma with neurological signs (Tay syndrome, Conradi-Hünermann-Happle syndrome) and neurocutaneous disorders such as neutral lipid storage disease and multiple sulfatase deficiency; however, the nature of the ichthyosis, presence of spastic diplegia/tetraplegia, characteristic parafoveal glistening white dots on the retina, and MRI and proton magnetic resonance spectroscopy findings help to easily differentiate SLS from these disorders.
Treatment
Treatment of SLS mainly is palliative. Ichthyosis can be treated with topical keratolytics, emollients, calcipotriol, and oral retinoids (acitretin).6 Zileuton, a 5-lipoxygenase inhibitor, inhibits synthesis of LTB4 and cysteinyl leukotrienes, thereby reducing the severity of pruritus and also has been shown to improve the speed of information processing.18 Similarly, montelukast, a leuko-triene antagonist, is helpful in relieving the agonizing pruritus.19 Experimental studies have shown that bezafibrate, a peroxisome proliferator-activated receptor α agonist, induces FALDH activity in fibroblasts of SLS patients that still have some residual FALDH activity, but further research is required to determine whether SLS patients could benefit from treatment.20 Physiotherapy helps in relieving the spasticity to some extent, such as in our case. Dietary intervention with reduced fat intake (up to 30% of total daily calorific requirement) and supplementation with omega-3 and omega-6 fatty acids has shown variable results in anecdotal reports.21-23 Gene therapy using recombinant adeno-associated virus 2 vectors to restore FALDH has been projected as a future treatment option.24 Despite lack of effective treatment options, most patients of SLS survive well into adulthood.
Conclusion
Because ichthyosis is one of the earliest and prominent symptoms of SLS, a dermatologist can play an important role in early diagnosis. Any child with the classical pattern of ichthyosis should be thoroughly examined for early neurologic signs and investigated to rule out SLS. Proton magnetic resonance spectroscopy serves as a useful adjunct in the diagnosis of SLS by confirming the accumulation of abnormal lipids in the periventricular white matter, especially when specific enzyme analysis and genetic analysis are not available in resource-restricted settings.
Sjögren-Larsson syndrome (SLS) is a rare autosomal-recessive neurocutaneous disorder comprising a triad of ichthyosis, mental retardation, and spastic diplegia or quadriplegia.1 The disorder was first described by Sjögren and Larsson2 in 1957. Early reports of SLS were mainly in white patients, with a particularly high prevalence of 8.3 cases per 100,000 individuals in the county of Västerbotten in Sweden.3 Reports of SLS in Asian and Indian populations are rare.4,5 We report a case of SLS in an Indian boy.
Case Report
A 12-year-old Indian boy born to nonconsanguineous parents after a full-term pregnancy with normal vaginal delivery presented with generalized dry scaly skin that had been present since 2 months of age. He had a history of delayed milestones (ie, facial recognition, sitting without support at 3 years of age), inability to walk, dysarthria, mental retardation). He had never attended school due to subnormal intellectual functioning. He had a single episode of a tonic-clonic seizure at 4 years of age but was not on any regular antiepileptic medication. There was a history of similar skin lesions in one male sibling of the patient and in 2 maternal uncles. None of them survived beyond early childhood, but detailed information regarding the cutaneous and neurologic manifestations in these family members was not available.
Cutaneous examination revealed lamellarlike ichthyosis on the dorsal aspects of the arms and legs (Figure 1A). Ichthyosis with lichenification was present on the neck, axillae, cubital and popliteal fossae, and abdomen (Figure 1B). The palms and soles showed keratoderma. Neurologic examination of the arms revealed mild rigidity and brisk reflexes. Examination of the legs showed marked rigidity, brisk knee jerks, ankle clonus, extensor plantar reflexes, flexion deformity with contractures, and scissor gait. A Goddard (Seguin) formboard test was performed and indicated a mental age of 4 years. The patient’s IQ was in the range of 25 to 30, indicating a severe degree of subnormality in intellectual functioning. The clinical presentation suggested a diagnosis of SLS.
A skin biopsy from the ichthyotic lesion showed hyperkeratosis, acanthosis, and papillomatosis with sparse superficial perivascular lymphocytic infiltrate, thus confirming the diagnosis of lamellar ichthyosis. Fundus examination was normal. Magnetic resonance imaging (MRI) of the brain revealed confluent symmetrical signal abnormalities along the body of the lateral ventricles, white matter in the perioccipital horn, and in deep white matter of centrum semiovale (Figure 2). Magnetic resonance spectroscopy revealed a narrow lipid peak at approximately 1.3 ppm in the region of signal abnormality (Figure 3). Thus, the diagnosis of SLS was confirmed. Measurement of fatty aldehyde dehydrogenase (FALDH) activity and genetic analysis were not performed due to unavailability.
The patient was treated with topical emollients for the ichthyosis. To reduce his dietary intake of long-chain fatty acids and increase the intake of omega-3 and omega-6 fatty acids, the patient’s parents were advised to use canola, mustard, and/or coconut oil for cooking for the patient, and skim milk was recommended instead of whole milk. Neurodevelopmental techniques in the form of stretching exercises were given to maintain his range of movements. Gutter splints were given to maintain the knees in extension for physiological standing and to prevent osteoporosis. Subsequently, the patient also underwent a multilevel soft-tissue release (hip and knee joints) to relieve the contractures. These measures resulted in considerable improvement and the patient was able to walk with support.
Comment
Presentation
The characteristic clinical features of SLS begin to develop during the intranatal period and infancy.1,6 Pathologic skin involvement can be detected as early as week 23 of gestation. Preterm births associated with SLS have commonly been described.3 Ichthyosis often is evident at birth, but collodion membrane is uncommon. Severe pruritus is a marked feature unlike most other types of ichthyosis. The ichthyosis often is generalized with prominent involvement of the flexural areas and nape of the neck, varying from fine furfuraceous to larger lamellarlike scales. Velvety orange or brown lichenification often is a predominant feature in the flexures of the arms, legs, neck, and mid abdomen. Mental retardation, developmental delay, and spasticity usually become apparent at 1 to 2 years of age and subsequently are nonprogressive.6,7 However, patients rarely have been described with normal intellectual functioning.7 Spasticity often is more severe in the lower limbs and may lead to contractures, kyphoscoliosis, hip dislocation, and short stature. Delayed speech and dysarthria are common. Parafoveal glistening white dots on the retina are a pathognomonic feature and typically appear in the first 2 years of life; however, they are seen in approximately 30% of patients and increase slightly in number with age.6,8 There may be associated decreased visual acuity, photophobia, myopia, and astigmatism. Other clinical features include enamel hypoplasia, metaphyseal dysplasia, and epilepsy.1,6
Gene Mutations
Sjögren-Larsson syndrome is caused by mutation in the aldehyde dehydrogenase 3 family member A2 gene, ALDH3A2 (17p11.2), which codes for FALDH.1,6,7 The ALDH3A2 gene is 11 exons long and gives rise to 2 protein isoforms that differ in their carboxy-terminal domains; the major isoform, composed of 485 amino acids, localizes to the endoplasmic reticulum. The minor protein isoform (FALDHv) is composed of 508 amino acids, possesses a longer carboxy-terminal, and appears to be targeted to the peroxisome. Several mutations have been reported throughout the ALDH3A2 gene, including missense mutations (most common [38% of cases of SLS6]), deletions, insertions, splicing errors, and complex rearrangements. Although several of these mutations are private, several common mutations may be indicative of founder effects (ie, shared ancestry), consanguinity, or recurrent mutational events (mutation hotspots).6,7 Despite the wide spectrum of mutations, there is very little phenotypic variation, with consistently severe cutaneous and neurological involvement occurring in a majority of patients.7 However, Lossos et al9 described remarkable phenotypic variation in 6 siblings of an Arab family and suggested that additional unknown genetic or environmental factors may compensate for the biochemical defect.
Lipid Metabolism
Fatty aldehyde dehydrogenase is expressed in almost all cells and tissues and catalyzes the oxidation of fatty aldehydes to fatty acids (eFigure 1). It also is a part of the fatty alcohol:NAD oxidoreductase (FAO) enzyme complex, which catalyzes fatty alcohol oxidation to fatty acid. Fatty aldehyde dehydrogenase deficiency leads to accumulation of long-chain alcohols (eg, hexadecanol, octadecanol, octadecenol) and diversion of fatty alcohol into alternate biosynthetic pathways such as wax esters and 1-O-alkyl-2,3-diacylglycerol.10 Other lipids that are increased are illustrated in eFigure 2. Accumulation of these lipids, toxic effects of abnormal lipids (especially fatty aldehydes and Schiff base protein-lipid adducts), and lack of essential lipids (eg, polyunsaturated fatty acids, ceramides 1 and 6, triglycerides) are responsible for the classical cutaneous, neurologic, and ophthalmologic features of SLS.
Histopathology
The epidermal permeability barrier is critically dependent on the appropriate lipid composition of the multilamellar stratum corneum intercellular membranes, an equimolar ratio of cholesterol, ceramides, and fatty acids. Histopathology of the skin in SLS generally shows hyperkeratosis, papillomatosis, acanthosis, and a mildly thickened granular layer. Ultrastructural studies of the skin reveal misshapen/empty lamellar bodies, abnormal cytoplasmic lamellar inclusions in the granular keratinocytes, lipid droplets in the stratum corneum with decreased lamellar bilayers, and lamellar/nonlamellar phase separation in the stratum corneum interstitium.11 These findings indicate that lipid metabolism dysfunction in SLS results in marked impairment in formation and secretion of lamellar bodies in the epidermis and consequent disorganization of the stratum corneum lamellar membranes. The resulting disruption of the skin barrier function leads to increased transepidermal water loss, resulting in ichthyosis.11,12 Another proposed mechanism for ichthyosis in SLS is disruption of the normal epidermal differentiation resulting from abnormal lipid metabolites (eFigure 2). Also, increased leukotriene B4 (LTB4) and 20-hydroxy-leukotriene B4 (20-OH-LTB4)(eFigure 1) may be responsible for the considerable pruritus seen in SLS.10
Neurologic Findings
Neurologic changes in SLS result from delayed and deficient myelination. Neuropathological studies have shown ballooning of myelin sheaths, extensive loss of myelin, axonal damage, and astrogliosis. The presence of lipoid material positive for periodic acid–Schiff that stains light rather than dark pink, dense distribution of round/ellipsoid bodies in the white matter of the cerebrum and brainstem positive for periodic acid–Schiff, and proliferation of perivascular macrophages containing lipofuscinlike pigments also have been described.13 Possibly, in the absence of FALDH, metabolism of plasmalogens (a major component of myelin) results in increased fatty aldehydes, which are either diverted to fatty alcohols or form adducts with phosphatidylethanolamine and myelin basic proteins (eFigure 1). Magnetic resonance imaging of the brain usually shows hypomyelination involving the periventricular white matter extending from the frontal to the occipital area.7,14 Mild ventricular enlargement may be an additional feature.14
A useful application of MRI is the proton magnetic resonance spectroscopy, which quantifies the brain metabolites noninvasively, displaying them as a spectrum on a graph. The spectrum comprises a set of resonances/peaks distributed along an x-axis. The resonances of these metabolites are obtained after suppressing the large signals from water protons. Proton magnetic resonance spectroscopy of the normal brain shows 3 prominent peaks: (1) N-acetylaspartate (NAA) at 2.02 ppm, (2) creatine at 3.02 ppm, and (3) choline at 3.22 ppm. In SLS, cerebral proton MRI spectroscopy reveals a characteristic abnormal, prominent, and narrow lipid peak at 1.3 ppm (corresponding to hexadecanol and octadecanol) and may offer a quantitative parameter for monitoring the effects of therapeutic interventions.7,14,15 The most intense lipid peaks are located in the periventricular regions in the anterior and posterior trigones. An abnormal but much smaller peak may be seen at 0.8 to 0.9 ppm, corresponding to phytol.14 Gradual emergence of these changes occurs in the first 2 years of life and then remains stable.15 Proton magnetic resonance spectroscopy also can be used for screening of SLS heterozygotes.16 Lipid peaks have been described in other disorders of lipid metabolism, but they are less intense, broader, and disappear on longer echo time sequences.14
Besides the characteristic parafoveal glistening white dots the retina, optical coherence tomography shows focal hyperreflectivitity in the perifoveal ganglion cell layer and inner plexiform layer of the retina as well as cystoid foveal degeneration.17 The intraretinal deposition of lipid metabolites probably leads to Müller cell degeneration with subsequent formation of cystoid spaces and atrophic changes in the fovea.
Measurement of FALDH or FAO activity in cultured skin fibroblasts and leukocytes using flurometric or gas chromatography mass spectrometry assays is a reliable biochemical test in cases of SLS as well as in heterozygotes.17 A decrease in FALDH/FAO activity also can be demonstrated by histochemical staining in skin biopsy.11 Pathologic urinary excretion of LTB4 and 20-OH-LTB4 also is a biochemical marker of SLS. Mutation analysis for a specific gene defect is diagnostic in cases of SLS as well as in heterozygotes. Prenatal diagnosis of SLS is possible by assessing FALDH activity or gene defects in cultured chorionic villus fibroblasts and amniocytes.18,19
Differential Diagnosis
The differential diagnosis of SLS includes congenital ichthyosiform erythroderma with neurological signs (Tay syndrome, Conradi-Hünermann-Happle syndrome) and neurocutaneous disorders such as neutral lipid storage disease and multiple sulfatase deficiency; however, the nature of the ichthyosis, presence of spastic diplegia/tetraplegia, characteristic parafoveal glistening white dots on the retina, and MRI and proton magnetic resonance spectroscopy findings help to easily differentiate SLS from these disorders.
Treatment
Treatment of SLS mainly is palliative. Ichthyosis can be treated with topical keratolytics, emollients, calcipotriol, and oral retinoids (acitretin).6 Zileuton, a 5-lipoxygenase inhibitor, inhibits synthesis of LTB4 and cysteinyl leukotrienes, thereby reducing the severity of pruritus and also has been shown to improve the speed of information processing.18 Similarly, montelukast, a leuko-triene antagonist, is helpful in relieving the agonizing pruritus.19 Experimental studies have shown that bezafibrate, a peroxisome proliferator-activated receptor α agonist, induces FALDH activity in fibroblasts of SLS patients that still have some residual FALDH activity, but further research is required to determine whether SLS patients could benefit from treatment.20 Physiotherapy helps in relieving the spasticity to some extent, such as in our case. Dietary intervention with reduced fat intake (up to 30% of total daily calorific requirement) and supplementation with omega-3 and omega-6 fatty acids has shown variable results in anecdotal reports.21-23 Gene therapy using recombinant adeno-associated virus 2 vectors to restore FALDH has been projected as a future treatment option.24 Despite lack of effective treatment options, most patients of SLS survive well into adulthood.
Conclusion
Because ichthyosis is one of the earliest and prominent symptoms of SLS, a dermatologist can play an important role in early diagnosis. Any child with the classical pattern of ichthyosis should be thoroughly examined for early neurologic signs and investigated to rule out SLS. Proton magnetic resonance spectroscopy serves as a useful adjunct in the diagnosis of SLS by confirming the accumulation of abnormal lipids in the periventricular white matter, especially when specific enzyme analysis and genetic analysis are not available in resource-restricted settings.
- Judge MR, McLean WHI, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, eds. Rook’s Textbook of Dermatology. 7th ed. West Sussex, United Kingdom: Wiley & Sons; 2004:34.37-34.39.
- Sjögren T, Larsson T. Oligophrenia in association with congenital ichthyosis and spastic disorders. Acta Psychiatr Neurol Scand. 1957;32:1-113.
- Jagell S, Gustavson KH, Holmgren G. Sjögren-Larsson syndrome in Sweden. a clinical, genetic and epidemiological study. Clin Genet. 1981;19:233-256.
- Sood M, Trehan A, Dinakaran J, et al. Sjögren-Larsson syndrome. Indian J Pediatr. 2002;69:193-194.
- Uppal M, Srinivas CR, Thowfeeq KT. Sjögren-Larsson syndrome: report of two cases. Indian J Dermatol Venereol Leprol. 2004;70:110-111.
- Rizzo WB. Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1-9.
- Willemsen MA, Ijlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain. 2001;124(pt 7):1426-1437.
- Willemsen MA, Cruysberg JR, Rotteveel JJ, et al. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjögren-Larsson syndrome. Am J Ophthalmol. 2000;130:782-789.
- Lossos A, Khoury M, Rizzo WB, et al. Phenotypic variability among adult siblings with Sjögren-Larsson syndrome. Arch Neurol. 2006;63:278-280.
- Rizzo WB, Craft DA, Somer T, et al. Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjögren-Larsson syndrome. J Lipid Res. 2008;49:410-419.
- Rizzo WB, S’Aulis D, Jennings MA, et al. Ichthyosis in Sjögren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443-451.
- Rizzo WB. The role of fatty aldehyde dehydrogenase in epidermal structure and function. Dermatoendocrinol. 2011;2:91-99.
- Yamaguchi K, Handa T. Sjögren-Larsson syndrome: postmortem brain abnormalities. Pediatr Neurol. 1998;18:338-341.
- Mano T, Ono J, Kaminaga T, et al. Proton MR spectroscopy of Sjögren-Larsson’s Syndrome. Am J Neuroradiol. 1999;20:1671-1673.
- Willemsen MA, van der Graf M, van der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjögren-Larsson syndrome: characterization of the leukoencephalopathy. Am J Neuroradiol. 2004;25:649-657.
- Kaminaga T, Mano T, Ono J, et al. Proton magnetic resonance spectroscopy of Sjögren-Larsson Syndrome. Magn Reson Med. 2001;45:1112-1115.
- Fuijkschot J, Cruysberg JR, Willemsen MA, et al. Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren-Larsson syndrome detected by optical coherence tomography. Ophthalmology. 2008;115:870-875.
- Willemsen MA, Lutt MA, Steijlen PM, et al. Clinical and biochemical effects of zileuton in patients with the Sjögren-Larsson syndrome. Eur J Pediatr. 2001;160:711-717.
- Pirgon O, Aydin K, Atabek ME. Proton magnetic resonance spectroscopy findings and clinical effects of montelukast sodium in a case with Sjögren-Larsson syndrome. J Child Neurol. 2006;21:1092-1095.
- Gloerich J, Ijlst L, Wanders RJ, et al. Bezafibrate induces FALDH in human fibroblasts; implications for Sjögren-Larsson syndrome Mol Genet Metab. 2006;89:111-115.
- Auada MP, Taube MB, Collares EF, et al. Sjögren-Larsson syndrome: biochemical defects and follow up in three cases. Eur J Dermatol. 2002;12:263-266.
- Taube B, Billeaud C, Labreze C, et al. Sjögren-Larsson syndrome: early diagnosis, dietary management and biochemical studies in two cases. Dermatology. 1999;198:340-345.
- Rizzo WB. Genetics and prospective therapeutic targets for Sjögren-Larsson Syndrome. Expert Opin Orphan Drugs. 2016;4:395-406.
- Haug S, Braun-Falco M. Restoration of fatty aldehyde dehydrogenase deficiency in Sjögren-Larsson syndrome. Gene Ther. 2006;13:1021-1026.
- Judge MR, McLean WHI, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, eds. Rook’s Textbook of Dermatology. 7th ed. West Sussex, United Kingdom: Wiley & Sons; 2004:34.37-34.39.
- Sjögren T, Larsson T. Oligophrenia in association with congenital ichthyosis and spastic disorders. Acta Psychiatr Neurol Scand. 1957;32:1-113.
- Jagell S, Gustavson KH, Holmgren G. Sjögren-Larsson syndrome in Sweden. a clinical, genetic and epidemiological study. Clin Genet. 1981;19:233-256.
- Sood M, Trehan A, Dinakaran J, et al. Sjögren-Larsson syndrome. Indian J Pediatr. 2002;69:193-194.
- Uppal M, Srinivas CR, Thowfeeq KT. Sjögren-Larsson syndrome: report of two cases. Indian J Dermatol Venereol Leprol. 2004;70:110-111.
- Rizzo WB. Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1-9.
- Willemsen MA, Ijlst L, Steijlen PM, et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain. 2001;124(pt 7):1426-1437.
- Willemsen MA, Cruysberg JR, Rotteveel JJ, et al. Juvenile macular dystrophy associated with deficient activity of fatty aldehyde dehydrogenase in Sjögren-Larsson syndrome. Am J Ophthalmol. 2000;130:782-789.
- Lossos A, Khoury M, Rizzo WB, et al. Phenotypic variability among adult siblings with Sjögren-Larsson syndrome. Arch Neurol. 2006;63:278-280.
- Rizzo WB, Craft DA, Somer T, et al. Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjögren-Larsson syndrome. J Lipid Res. 2008;49:410-419.
- Rizzo WB, S’Aulis D, Jennings MA, et al. Ichthyosis in Sjögren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443-451.
- Rizzo WB. The role of fatty aldehyde dehydrogenase in epidermal structure and function. Dermatoendocrinol. 2011;2:91-99.
- Yamaguchi K, Handa T. Sjögren-Larsson syndrome: postmortem brain abnormalities. Pediatr Neurol. 1998;18:338-341.
- Mano T, Ono J, Kaminaga T, et al. Proton MR spectroscopy of Sjögren-Larsson’s Syndrome. Am J Neuroradiol. 1999;20:1671-1673.
- Willemsen MA, van der Graf M, van der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjögren-Larsson syndrome: characterization of the leukoencephalopathy. Am J Neuroradiol. 2004;25:649-657.
- Kaminaga T, Mano T, Ono J, et al. Proton magnetic resonance spectroscopy of Sjögren-Larsson Syndrome. Magn Reson Med. 2001;45:1112-1115.
- Fuijkschot J, Cruysberg JR, Willemsen MA, et al. Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren-Larsson syndrome detected by optical coherence tomography. Ophthalmology. 2008;115:870-875.
- Willemsen MA, Lutt MA, Steijlen PM, et al. Clinical and biochemical effects of zileuton in patients with the Sjögren-Larsson syndrome. Eur J Pediatr. 2001;160:711-717.
- Pirgon O, Aydin K, Atabek ME. Proton magnetic resonance spectroscopy findings and clinical effects of montelukast sodium in a case with Sjögren-Larsson syndrome. J Child Neurol. 2006;21:1092-1095.
- Gloerich J, Ijlst L, Wanders RJ, et al. Bezafibrate induces FALDH in human fibroblasts; implications for Sjögren-Larsson syndrome Mol Genet Metab. 2006;89:111-115.
- Auada MP, Taube MB, Collares EF, et al. Sjögren-Larsson syndrome: biochemical defects and follow up in three cases. Eur J Dermatol. 2002;12:263-266.
- Taube B, Billeaud C, Labreze C, et al. Sjögren-Larsson syndrome: early diagnosis, dietary management and biochemical studies in two cases. Dermatology. 1999;198:340-345.
- Rizzo WB. Genetics and prospective therapeutic targets for Sjögren-Larsson Syndrome. Expert Opin Orphan Drugs. 2016;4:395-406.
- Haug S, Braun-Falco M. Restoration of fatty aldehyde dehydrogenase deficiency in Sjögren-Larsson syndrome. Gene Ther. 2006;13:1021-1026.
Practice Points
- Sjögren-Larsson syndrome (SLS) is characterized by a clinical triad of ichthyosis, mental retardation, and spastic diplegia or quadriplegia.
- A characteristic lipid peak at 1.3 ppm on magnetic resonance spectroscopy is diagnostic of SLS.
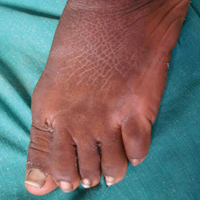
Long-term Pubic Dermatitis Diagnosed as White Piedra
Case Report
A 58-year-old man presented for evaluation of a pruritic rash involving the pubic area of 30 years’ duration. Multiple primary care physicians and dermatologists had evaluated the patient during this period, but he noted a specific diagnosis had not been rendered and multiple treatments had been unsuccessful. The patient described a rash, which was absent at the time of evaluation, as a self-remitting and exacerbating irritation typically induced by sweating and physical activity. The patient also stated that the irritation was associated with a strong, distinct, musty odor that severely interrupted his sex life and decreased his quality of life. Prior treatments included various topical corticosteroids, topical and oral antibiotics, and various homeopathic treatments that were minimally efficacious or nonefficacious. He was unsure if antifungals had previously been prescribed.
The patient’s medical history was notable for pulmonary interstitial fibrosis, anxiety, posttraumatic stress disorder, and mild glucose intolerance. The patient had no pertinent surgical history and no known drug allergies. Current medications included a bronchodilating inhaler, escitalopram, trazodone, buspirone, clonazepam, prazosin, gabapentin, and azithromycin for current upper respiratory tract infection. The patient was a former smoker and a social drinker.
On physical evaluation the pubic area displayed slight patchy erythema without a papular component and was otherwise unremarkable to the unaided eye. Upon palpation of the skin, there were no remarkable findings. Under dermoscopic evaluation, small white-yellow concretions along the hair shaft were noticed. Evaluation with a Wood lamp is shown in Figure 1.
The patient was treated empirically with ketoconazole cream 2% applied to the affected area once daily until follow-up 3 weeks later. The patient also was advised to shave the pubic area to remove potentially infected hairs, as white piedra (WP) was suspected. A diagnosis of WP was confirmed on histologic evaluation of pubic hair samples approximately 1 to 2 weeks later (Figure 2).
At 3-week follow-up, Wood lamp evaluation did not identify concretions along the pubic hair shafts. The patient was symptom free and extremely pleased. Of note, the patient did not shave the pubic area and was counseled on recurrence.
Comment
Piedra (meaning stone in Spanish) describes a group of fungal infections that present with gritty concretions on the hair shaft.1,2 In 1911, Horta3 classified piedra into 2 subtypes: black piedra, caused by Piedraia hortae, and WP, caused by Trichosporon species. Black piedra occurs more frequently in tropical countries and commonly affects hair shafts on the scalp.4 White piedra most commonly affects the pubic area, with rare cases in scalp and facial hair.1,5-7
Epidemiology
White piedra is seen worldwide, including Europe, South America, India, Southeast Asia, Africa, South America, and southern parts of the United States. The majority of cases occur in tropical and temperate regions.1 White piedra likely is underdiagnosed; for example, in a study of 166 young men with genital concerns in Houston, Texas, Trichosporon was isolated in 40% of cultured scrotal hairs.8
Species Identification
There are several species of WP; special techniques must be used to differentiate them, which is beyond the scope of this case. The known species include Trichosporon asahii, Trichosporon asteroides, Trichosporon cutaneum, Trichosporon inkin, Trichosporon mucoides, and Trichosporon ovoides.1Trichosporon asahii and T mucoides have been known to cause systemic infections in immunocompromised hosts known as trichosporonosis.1,9 As an example of a special technique used for species recognition, Sugita et al10 used sequence analysis of the ribosomal DNA intergenic spacer 1 regions to distinguish T asahii isolates. Identification of species may be warranted in the proper clinical scenario; however, histologic evaluation by an experienced dermatopathologist frequently is sufficient to identify the Trichosporon genus.
Transmission
The 2 most common causative organisms of WP are T inkin and T ovoides. Furthermore, T inkin causes the vast majority of WP in the pubic region.8
Diagnosis and Differential
White piedra is characterized by the presence of adherent tan to white nodules along the hair shafts. The concretions tend to be softer than black piedra and, unlike trichomycosis, normally do not fluoresce.1 They do not encircle the hair shaft as hair casts do and can be readily distinguished from Trichomycosis axillaris, black piedra, pediculosis, and trichorrhexis nodosa on microscopic examination.14 The hair shaft concretions of WP are difficult to visualize with the unaided eye. As a result, it is easily misdiagnosed.15 Upon palpation of the infected hair shafts, a grainy sensation is evident. Dermoscopy improves visualization, and fluorescence was useful in our case. Microscopic evaluation will identify the adherent organism and is readily cultured on Sabouraud agar.16 Although Trichosporon species typically do not fluoresce,1 Wood lamp examination occasionally may reveal the organism, such as in our case. A possible explanation for this finding is the synergistic relationship with Corynebacterium,17 some producing fluorescent chemicals. Growth of Trichosporon species may be enhanced by or even dependent on Corynebacterium; therefore, WP is likely a coinfection of fungus and bacteria.17,18 Studies also name a novel species of Brevibacterium in relationship with genital WP.19 This species was described as producing a foul odor, as in our patient.
Treatment
The American Academy of Dermatology’s Guidelines Committee recommends complete removal of the infected hairs.1 The recommendation traditionally is hair removal in conjunction with topical or oral medications,1 such as topical imidazoles, ciclopirox olamine, selenium sulfide 2%, chlorhexidine solution, zinc pyrithione, amphotericin B lotion, and oral itraconazole. Recurrence rates are high and spontaneous remission sometimes occurs.9,20 Triazole antifungals currently are preferred for treatment of Trichosporon infections.5 Patients should be counseled to dispose of undergarments, as the organism has been recovered from cotton fibers and are a source of reinfection.1,21
Conclusion
White piedra, though relatively uncommon, is likely underdiagnosed in the United States and should be suspected in any patient presenting with irritation and a foul odor in the genital area or multiple failed therapies for a nonspecific genital dermatitis. This clinical scenario warrants dermoscopic and Wood lamp examination of the affected skin and hair shafts in addition to microscopic examination of pubic hair shafts by a dermatopathologist. Fluorescence under Wood lamp may aid in diagnosis, and conflicting findings may be attributed to its synergistic relationship with Corynebacterium and Brevibacterium coinfection. Proper treatment includes shaving of the affected hair, oral or topical antifungal treatment, and disposal of affected clothing.
- Kiken DA, Sekaran A, Antaya RJ, et al. White piedra in children [published online September 18, 2006]. J Am Acad Dermatol. 2006;55:956-961.
- Walzam M, Leeming JG. White piedra and Trichosporon beigelii: the incidence in patients attending a clinic in genitourinary medicine. Genitourin Med. 1989;16:331-334.
- Horta P. Sobre una nova forma de piedra. Mem Inst Oswaldo Cruz. 1911;3:86-107.
- Fischman O. Black piedra in Brazil: a contribution to its study in Manaus (State of Amazonas). Mycopathol Mycol Appl. 1965;25:201-204.
- Benson PM, Lapins NA, Odom RB. White piedra. Arch Dermatol. 1983;119:602-604.
- Fischman O, Pires de Camargo Z, Meireles MCA. Genital white piedra: an emerging fungal disease? Fifth International Conference on Mycoses. PAHO Sci Publ. 1989;396:70-76.
- Tambe SA, Dhurat SR, Kumar CA, et al. Two cases of scalp piedra caused by Trichosporon ovoides. Indian J Dermatol Venereol Leprol. 2009;75:293-295.
- Kalter DC, Tschen JA, Cernoch PL, et al. Genital white piedra: epidemiology, microbiology, and therapy. J Am Acad Dermatol. 1986;14:982-993.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. United Kingdom: Saunders Elsevier; 2011.
- Sugita T, Nakajima M, Ikeda R, et al. Sequence analysis of the ribosomal DNA intergenic spacer 1 regions of Trichosporon species. J Clin Microbiol. 2002;40:1826-1830.
- Kaplan W. Piedra in lower animals: a case report of white piedra in a monkey and a review of the literature. J Am Vet Med Assoc. 1959;134:113-117.
- Carneiro JA, Assis FA, Filho JT. Piedra branca genital. An Bras Dermatol. 1971;46:265-269.
- Avram A, Buot G, Binet A, et al. Étude clinique et mycologique concernant 11 cas de trichosporie noueuse (piedra blanche) génito-pubienne. Ann Dermatol Venereol. 1987;114:819-827.
- So
bera JO, Elewski BE. Fungal diseases. In: Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. New York, NY: Mosby; 2003:1135-1138. - Gold I, Sommer B, Urson S, et al. White piedra: a frequently misdiagnosed infection of hair. Int J Dermatol. 1984;23:621-623.
- Smith JD, Murtishaw WA, McBride ME. White piedra (Trichosporosis). Arch Dermatol. 1973;107:439-442.
- Youker SR, Andreozzi RJ, Appelbaum PC, et al. White piedra: further evidence of a synergistic infection. J Am Acad Dermatol. 2003;49:746-749.
- Ellner KM, McBride ME, Kalter DC, et al. White piedra: evidence for a synergistic infection. Br J Dermatol. 1990;123:355-363.
- McBride ME, Ellner KM, Black HS, et al. A new Brevibacterium sp. isolated from infected genital hair of patients with white piedra. J Med Microbiol. 1993;39:255-261.
- Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for superficial mycotic infections of the skin: piedra. J Am Acad Dermatol. 1996;34:122-124.
- de Almeida HL Jr, Rivitti EA, Jaeger RG. White piedra: ultrastructure and a new microecological aspect. Mycoses. 1990;33:491-497.
Case Report
A 58-year-old man presented for evaluation of a pruritic rash involving the pubic area of 30 years’ duration. Multiple primary care physicians and dermatologists had evaluated the patient during this period, but he noted a specific diagnosis had not been rendered and multiple treatments had been unsuccessful. The patient described a rash, which was absent at the time of evaluation, as a self-remitting and exacerbating irritation typically induced by sweating and physical activity. The patient also stated that the irritation was associated with a strong, distinct, musty odor that severely interrupted his sex life and decreased his quality of life. Prior treatments included various topical corticosteroids, topical and oral antibiotics, and various homeopathic treatments that were minimally efficacious or nonefficacious. He was unsure if antifungals had previously been prescribed.
The patient’s medical history was notable for pulmonary interstitial fibrosis, anxiety, posttraumatic stress disorder, and mild glucose intolerance. The patient had no pertinent surgical history and no known drug allergies. Current medications included a bronchodilating inhaler, escitalopram, trazodone, buspirone, clonazepam, prazosin, gabapentin, and azithromycin for current upper respiratory tract infection. The patient was a former smoker and a social drinker.
On physical evaluation the pubic area displayed slight patchy erythema without a papular component and was otherwise unremarkable to the unaided eye. Upon palpation of the skin, there were no remarkable findings. Under dermoscopic evaluation, small white-yellow concretions along the hair shaft were noticed. Evaluation with a Wood lamp is shown in Figure 1.
The patient was treated empirically with ketoconazole cream 2% applied to the affected area once daily until follow-up 3 weeks later. The patient also was advised to shave the pubic area to remove potentially infected hairs, as white piedra (WP) was suspected. A diagnosis of WP was confirmed on histologic evaluation of pubic hair samples approximately 1 to 2 weeks later (Figure 2).
At 3-week follow-up, Wood lamp evaluation did not identify concretions along the pubic hair shafts. The patient was symptom free and extremely pleased. Of note, the patient did not shave the pubic area and was counseled on recurrence.
Comment
Piedra (meaning stone in Spanish) describes a group of fungal infections that present with gritty concretions on the hair shaft.1,2 In 1911, Horta3 classified piedra into 2 subtypes: black piedra, caused by Piedraia hortae, and WP, caused by Trichosporon species. Black piedra occurs more frequently in tropical countries and commonly affects hair shafts on the scalp.4 White piedra most commonly affects the pubic area, with rare cases in scalp and facial hair.1,5-7
Epidemiology
White piedra is seen worldwide, including Europe, South America, India, Southeast Asia, Africa, South America, and southern parts of the United States. The majority of cases occur in tropical and temperate regions.1 White piedra likely is underdiagnosed; for example, in a study of 166 young men with genital concerns in Houston, Texas, Trichosporon was isolated in 40% of cultured scrotal hairs.8
Species Identification
There are several species of WP; special techniques must be used to differentiate them, which is beyond the scope of this case. The known species include Trichosporon asahii, Trichosporon asteroides, Trichosporon cutaneum, Trichosporon inkin, Trichosporon mucoides, and Trichosporon ovoides.1Trichosporon asahii and T mucoides have been known to cause systemic infections in immunocompromised hosts known as trichosporonosis.1,9 As an example of a special technique used for species recognition, Sugita et al10 used sequence analysis of the ribosomal DNA intergenic spacer 1 regions to distinguish T asahii isolates. Identification of species may be warranted in the proper clinical scenario; however, histologic evaluation by an experienced dermatopathologist frequently is sufficient to identify the Trichosporon genus.
Transmission
The 2 most common causative organisms of WP are T inkin and T ovoides. Furthermore, T inkin causes the vast majority of WP in the pubic region.8
Diagnosis and Differential
White piedra is characterized by the presence of adherent tan to white nodules along the hair shafts. The concretions tend to be softer than black piedra and, unlike trichomycosis, normally do not fluoresce.1 They do not encircle the hair shaft as hair casts do and can be readily distinguished from Trichomycosis axillaris, black piedra, pediculosis, and trichorrhexis nodosa on microscopic examination.14 The hair shaft concretions of WP are difficult to visualize with the unaided eye. As a result, it is easily misdiagnosed.15 Upon palpation of the infected hair shafts, a grainy sensation is evident. Dermoscopy improves visualization, and fluorescence was useful in our case. Microscopic evaluation will identify the adherent organism and is readily cultured on Sabouraud agar.16 Although Trichosporon species typically do not fluoresce,1 Wood lamp examination occasionally may reveal the organism, such as in our case. A possible explanation for this finding is the synergistic relationship with Corynebacterium,17 some producing fluorescent chemicals. Growth of Trichosporon species may be enhanced by or even dependent on Corynebacterium; therefore, WP is likely a coinfection of fungus and bacteria.17,18 Studies also name a novel species of Brevibacterium in relationship with genital WP.19 This species was described as producing a foul odor, as in our patient.
Treatment
The American Academy of Dermatology’s Guidelines Committee recommends complete removal of the infected hairs.1 The recommendation traditionally is hair removal in conjunction with topical or oral medications,1 such as topical imidazoles, ciclopirox olamine, selenium sulfide 2%, chlorhexidine solution, zinc pyrithione, amphotericin B lotion, and oral itraconazole. Recurrence rates are high and spontaneous remission sometimes occurs.9,20 Triazole antifungals currently are preferred for treatment of Trichosporon infections.5 Patients should be counseled to dispose of undergarments, as the organism has been recovered from cotton fibers and are a source of reinfection.1,21
Conclusion
White piedra, though relatively uncommon, is likely underdiagnosed in the United States and should be suspected in any patient presenting with irritation and a foul odor in the genital area or multiple failed therapies for a nonspecific genital dermatitis. This clinical scenario warrants dermoscopic and Wood lamp examination of the affected skin and hair shafts in addition to microscopic examination of pubic hair shafts by a dermatopathologist. Fluorescence under Wood lamp may aid in diagnosis, and conflicting findings may be attributed to its synergistic relationship with Corynebacterium and Brevibacterium coinfection. Proper treatment includes shaving of the affected hair, oral or topical antifungal treatment, and disposal of affected clothing.
Case Report
A 58-year-old man presented for evaluation of a pruritic rash involving the pubic area of 30 years’ duration. Multiple primary care physicians and dermatologists had evaluated the patient during this period, but he noted a specific diagnosis had not been rendered and multiple treatments had been unsuccessful. The patient described a rash, which was absent at the time of evaluation, as a self-remitting and exacerbating irritation typically induced by sweating and physical activity. The patient also stated that the irritation was associated with a strong, distinct, musty odor that severely interrupted his sex life and decreased his quality of life. Prior treatments included various topical corticosteroids, topical and oral antibiotics, and various homeopathic treatments that were minimally efficacious or nonefficacious. He was unsure if antifungals had previously been prescribed.
The patient’s medical history was notable for pulmonary interstitial fibrosis, anxiety, posttraumatic stress disorder, and mild glucose intolerance. The patient had no pertinent surgical history and no known drug allergies. Current medications included a bronchodilating inhaler, escitalopram, trazodone, buspirone, clonazepam, prazosin, gabapentin, and azithromycin for current upper respiratory tract infection. The patient was a former smoker and a social drinker.
On physical evaluation the pubic area displayed slight patchy erythema without a papular component and was otherwise unremarkable to the unaided eye. Upon palpation of the skin, there were no remarkable findings. Under dermoscopic evaluation, small white-yellow concretions along the hair shaft were noticed. Evaluation with a Wood lamp is shown in Figure 1.
The patient was treated empirically with ketoconazole cream 2% applied to the affected area once daily until follow-up 3 weeks later. The patient also was advised to shave the pubic area to remove potentially infected hairs, as white piedra (WP) was suspected. A diagnosis of WP was confirmed on histologic evaluation of pubic hair samples approximately 1 to 2 weeks later (Figure 2).
At 3-week follow-up, Wood lamp evaluation did not identify concretions along the pubic hair shafts. The patient was symptom free and extremely pleased. Of note, the patient did not shave the pubic area and was counseled on recurrence.
Comment
Piedra (meaning stone in Spanish) describes a group of fungal infections that present with gritty concretions on the hair shaft.1,2 In 1911, Horta3 classified piedra into 2 subtypes: black piedra, caused by Piedraia hortae, and WP, caused by Trichosporon species. Black piedra occurs more frequently in tropical countries and commonly affects hair shafts on the scalp.4 White piedra most commonly affects the pubic area, with rare cases in scalp and facial hair.1,5-7
Epidemiology
White piedra is seen worldwide, including Europe, South America, India, Southeast Asia, Africa, South America, and southern parts of the United States. The majority of cases occur in tropical and temperate regions.1 White piedra likely is underdiagnosed; for example, in a study of 166 young men with genital concerns in Houston, Texas, Trichosporon was isolated in 40% of cultured scrotal hairs.8
Species Identification
There are several species of WP; special techniques must be used to differentiate them, which is beyond the scope of this case. The known species include Trichosporon asahii, Trichosporon asteroides, Trichosporon cutaneum, Trichosporon inkin, Trichosporon mucoides, and Trichosporon ovoides.1Trichosporon asahii and T mucoides have been known to cause systemic infections in immunocompromised hosts known as trichosporonosis.1,9 As an example of a special technique used for species recognition, Sugita et al10 used sequence analysis of the ribosomal DNA intergenic spacer 1 regions to distinguish T asahii isolates. Identification of species may be warranted in the proper clinical scenario; however, histologic evaluation by an experienced dermatopathologist frequently is sufficient to identify the Trichosporon genus.
Transmission
The 2 most common causative organisms of WP are T inkin and T ovoides. Furthermore, T inkin causes the vast majority of WP in the pubic region.8
Diagnosis and Differential
White piedra is characterized by the presence of adherent tan to white nodules along the hair shafts. The concretions tend to be softer than black piedra and, unlike trichomycosis, normally do not fluoresce.1 They do not encircle the hair shaft as hair casts do and can be readily distinguished from Trichomycosis axillaris, black piedra, pediculosis, and trichorrhexis nodosa on microscopic examination.14 The hair shaft concretions of WP are difficult to visualize with the unaided eye. As a result, it is easily misdiagnosed.15 Upon palpation of the infected hair shafts, a grainy sensation is evident. Dermoscopy improves visualization, and fluorescence was useful in our case. Microscopic evaluation will identify the adherent organism and is readily cultured on Sabouraud agar.16 Although Trichosporon species typically do not fluoresce,1 Wood lamp examination occasionally may reveal the organism, such as in our case. A possible explanation for this finding is the synergistic relationship with Corynebacterium,17 some producing fluorescent chemicals. Growth of Trichosporon species may be enhanced by or even dependent on Corynebacterium; therefore, WP is likely a coinfection of fungus and bacteria.17,18 Studies also name a novel species of Brevibacterium in relationship with genital WP.19 This species was described as producing a foul odor, as in our patient.
Treatment
The American Academy of Dermatology’s Guidelines Committee recommends complete removal of the infected hairs.1 The recommendation traditionally is hair removal in conjunction with topical or oral medications,1 such as topical imidazoles, ciclopirox olamine, selenium sulfide 2%, chlorhexidine solution, zinc pyrithione, amphotericin B lotion, and oral itraconazole. Recurrence rates are high and spontaneous remission sometimes occurs.9,20 Triazole antifungals currently are preferred for treatment of Trichosporon infections.5 Patients should be counseled to dispose of undergarments, as the organism has been recovered from cotton fibers and are a source of reinfection.1,21
Conclusion
White piedra, though relatively uncommon, is likely underdiagnosed in the United States and should be suspected in any patient presenting with irritation and a foul odor in the genital area or multiple failed therapies for a nonspecific genital dermatitis. This clinical scenario warrants dermoscopic and Wood lamp examination of the affected skin and hair shafts in addition to microscopic examination of pubic hair shafts by a dermatopathologist. Fluorescence under Wood lamp may aid in diagnosis, and conflicting findings may be attributed to its synergistic relationship with Corynebacterium and Brevibacterium coinfection. Proper treatment includes shaving of the affected hair, oral or topical antifungal treatment, and disposal of affected clothing.
- Kiken DA, Sekaran A, Antaya RJ, et al. White piedra in children [published online September 18, 2006]. J Am Acad Dermatol. 2006;55:956-961.
- Walzam M, Leeming JG. White piedra and Trichosporon beigelii: the incidence in patients attending a clinic in genitourinary medicine. Genitourin Med. 1989;16:331-334.
- Horta P. Sobre una nova forma de piedra. Mem Inst Oswaldo Cruz. 1911;3:86-107.
- Fischman O. Black piedra in Brazil: a contribution to its study in Manaus (State of Amazonas). Mycopathol Mycol Appl. 1965;25:201-204.
- Benson PM, Lapins NA, Odom RB. White piedra. Arch Dermatol. 1983;119:602-604.
- Fischman O, Pires de Camargo Z, Meireles MCA. Genital white piedra: an emerging fungal disease? Fifth International Conference on Mycoses. PAHO Sci Publ. 1989;396:70-76.
- Tambe SA, Dhurat SR, Kumar CA, et al. Two cases of scalp piedra caused by Trichosporon ovoides. Indian J Dermatol Venereol Leprol. 2009;75:293-295.
- Kalter DC, Tschen JA, Cernoch PL, et al. Genital white piedra: epidemiology, microbiology, and therapy. J Am Acad Dermatol. 1986;14:982-993.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. United Kingdom: Saunders Elsevier; 2011.
- Sugita T, Nakajima M, Ikeda R, et al. Sequence analysis of the ribosomal DNA intergenic spacer 1 regions of Trichosporon species. J Clin Microbiol. 2002;40:1826-1830.
- Kaplan W. Piedra in lower animals: a case report of white piedra in a monkey and a review of the literature. J Am Vet Med Assoc. 1959;134:113-117.
- Carneiro JA, Assis FA, Filho JT. Piedra branca genital. An Bras Dermatol. 1971;46:265-269.
- Avram A, Buot G, Binet A, et al. Étude clinique et mycologique concernant 11 cas de trichosporie noueuse (piedra blanche) génito-pubienne. Ann Dermatol Venereol. 1987;114:819-827.
- So
bera JO, Elewski BE. Fungal diseases. In: Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. New York, NY: Mosby; 2003:1135-1138. - Gold I, Sommer B, Urson S, et al. White piedra: a frequently misdiagnosed infection of hair. Int J Dermatol. 1984;23:621-623.
- Smith JD, Murtishaw WA, McBride ME. White piedra (Trichosporosis). Arch Dermatol. 1973;107:439-442.
- Youker SR, Andreozzi RJ, Appelbaum PC, et al. White piedra: further evidence of a synergistic infection. J Am Acad Dermatol. 2003;49:746-749.
- Ellner KM, McBride ME, Kalter DC, et al. White piedra: evidence for a synergistic infection. Br J Dermatol. 1990;123:355-363.
- McBride ME, Ellner KM, Black HS, et al. A new Brevibacterium sp. isolated from infected genital hair of patients with white piedra. J Med Microbiol. 1993;39:255-261.
- Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for superficial mycotic infections of the skin: piedra. J Am Acad Dermatol. 1996;34:122-124.
- de Almeida HL Jr, Rivitti EA, Jaeger RG. White piedra: ultrastructure and a new microecological aspect. Mycoses. 1990;33:491-497.
- Kiken DA, Sekaran A, Antaya RJ, et al. White piedra in children [published online September 18, 2006]. J Am Acad Dermatol. 2006;55:956-961.
- Walzam M, Leeming JG. White piedra and Trichosporon beigelii: the incidence in patients attending a clinic in genitourinary medicine. Genitourin Med. 1989;16:331-334.
- Horta P. Sobre una nova forma de piedra. Mem Inst Oswaldo Cruz. 1911;3:86-107.
- Fischman O. Black piedra in Brazil: a contribution to its study in Manaus (State of Amazonas). Mycopathol Mycol Appl. 1965;25:201-204.
- Benson PM, Lapins NA, Odom RB. White piedra. Arch Dermatol. 1983;119:602-604.
- Fischman O, Pires de Camargo Z, Meireles MCA. Genital white piedra: an emerging fungal disease? Fifth International Conference on Mycoses. PAHO Sci Publ. 1989;396:70-76.
- Tambe SA, Dhurat SR, Kumar CA, et al. Two cases of scalp piedra caused by Trichosporon ovoides. Indian J Dermatol Venereol Leprol. 2009;75:293-295.
- Kalter DC, Tschen JA, Cernoch PL, et al. Genital white piedra: epidemiology, microbiology, and therapy. J Am Acad Dermatol. 1986;14:982-993.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. United Kingdom: Saunders Elsevier; 2011.
- Sugita T, Nakajima M, Ikeda R, et al. Sequence analysis of the ribosomal DNA intergenic spacer 1 regions of Trichosporon species. J Clin Microbiol. 2002;40:1826-1830.
- Kaplan W. Piedra in lower animals: a case report of white piedra in a monkey and a review of the literature. J Am Vet Med Assoc. 1959;134:113-117.
- Carneiro JA, Assis FA, Filho JT. Piedra branca genital. An Bras Dermatol. 1971;46:265-269.
- Avram A, Buot G, Binet A, et al. Étude clinique et mycologique concernant 11 cas de trichosporie noueuse (piedra blanche) génito-pubienne. Ann Dermatol Venereol. 1987;114:819-827.
- So
bera JO, Elewski BE. Fungal diseases. In: Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. New York, NY: Mosby; 2003:1135-1138. - Gold I, Sommer B, Urson S, et al. White piedra: a frequently misdiagnosed infection of hair. Int J Dermatol. 1984;23:621-623.
- Smith JD, Murtishaw WA, McBride ME. White piedra (Trichosporosis). Arch Dermatol. 1973;107:439-442.
- Youker SR, Andreozzi RJ, Appelbaum PC, et al. White piedra: further evidence of a synergistic infection. J Am Acad Dermatol. 2003;49:746-749.
- Ellner KM, McBride ME, Kalter DC, et al. White piedra: evidence for a synergistic infection. Br J Dermatol. 1990;123:355-363.
- McBride ME, Ellner KM, Black HS, et al. A new Brevibacterium sp. isolated from infected genital hair of patients with white piedra. J Med Microbiol. 1993;39:255-261.
- Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for superficial mycotic infections of the skin: piedra. J Am Acad Dermatol. 1996;34:122-124.
- de Almeida HL Jr, Rivitti EA, Jaeger RG. White piedra: ultrastructure and a new microecological aspect. Mycoses. 1990;33:491-497.
- Although relatively uncommon, white piedra should be suspected in any patient presenting with irritation and foul odor in the genital area or multiple failed therapies for a nonspecific genital dermatitis.
- Wood lamp and dermoscopy should be used to evaluate for parasitic infections of the pubic hair shafts when nonspecific dermatitis presents in this area.
Pediatric Nevoid Basal Cell Carcinoma Syndrome
In 1960, Gorlin and Goltz1 first described nevoid basal cell carcinoma syndrome (NBCCS) as a distinct clinical entity with multiple basal cell carcinomas (BCCs), jaw cysts, and bifid ribs. This rare autosomal-dominant genodermatosis has a minimal prevalence of 1 case per 57,000 individuals2 and no sexual predilection.3 Nevoid basal cell carcinoma syndrome is caused by a mutation in the human homolog of a Drosophila gene, patched 1 (PTCH1), which is located on chromosome 9q22.3.4,5 The major clinical diagnostic criteria includes multiple BCCs, odontogenic keratocysts, palmar or plantar pits, ectopic calcification of the falx cerebri, and a family history of NBCCS.6 Basal cell carcinoma formation is affected by both skin pigmentation and sun exposure; 80% of white patients with NBCCS will develop at least 1 BCC compared to only 40% of black patients with NBCCS.7 Goldstein et al8 postulated that this disparity is associated with increased skin pigmentation providing UV radiation protection, thus decreasing the tumor burden. We report a case of an 11-year-old black boy with NBCCS to highlight the treatment considerations in pediatric cases of NBCCS.
Case Report
An 11-year-old boy with Fitzpatrick skin type V presented with a history of multiple facial lesions after undergoing excision of large keratocysts from the right maxilla, left maxilla, and right mandible. Physical examination revealed multiple light to dark brown facial papules (Figure 1), palmar and plantar pitting (Figure 2), and frontal bossing.
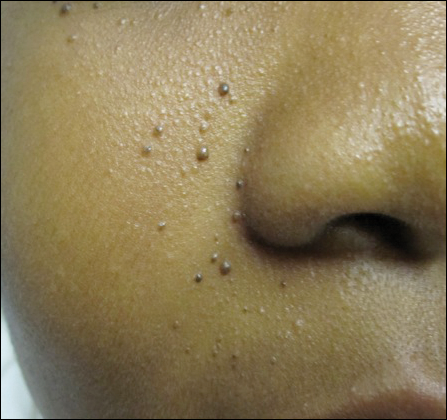
He was previously diagnosed with autism and his surgical history was notable only for excision of the keratocysts. The patient was not taking any medications and did not have any drug allergies. There was no maternal family history of skin cancer or related syndromes; his paternal family history was unknown. A shave biopsy was performed on a facial papule from the right nasolabial fold. Histopathologic evaluation revealed findings consistent with a pigmented nodular BCC (Figure 3). The patient was subsequently sent for magnetic resonance imaging of the brain, which demonstrated calcifications along the tentorium. Genetic consultation confirmed a heterozygous mutation of the PTCH1 gene.
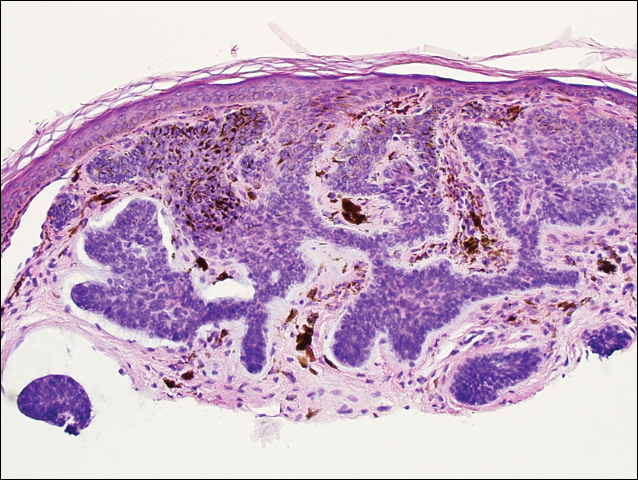
Over the next 12 months, the patient had multiple biopsy-proven pigmented BCCs. Initial management of these carcinomas located on cosmetically sensitive areas, including the upper eyelid and penis, were excised by a pediatric plastic surgeon. A truncal carcinoma was treated with electrodesiccation and curettage, which resulted in keloid formation. Early suspicious lesions were treated with imiquimod cream 5% 5 times weekly in combination with the prophylactic use of tretinoin cream 0.1%. Despite this treatment regimen, the patient continued to demonstrate multiple small clinical pigmented BCCs along the malar surfaces of the cheeks and dorsum of the nose. The patient’s mother deferred chemoprevention with an oral retinoid due to the extensive side-effect profile and long-term necessity of administration.
Management also encompassed BCC surveillance every 4 months; annual digital panorex of the jaw; routine dental screening; routine developmental screening; annual follow-up with a geneticist to ensure multidisciplinary care; and annual vision, hearing, and speech-screening examinations. Strict sun-protective measures were encouraged, including wearing a hat during physical education class.
Comment
Classification and Clinical Presentation
Nevoid basal cell carcinoma syndrome is a multisystem disorder that requires close monitoring under multidisciplinary care. Evans et al6 defined the diagnostic criteria of NBCCS to require the presence of 2 major criteria or 1 major and 2 minor criteria. The major criteria include multiple BCCs, an odontogenic keratocyst or polyostotic bone cyst, palmar or plantar pits, ectopic calcification of the falx cerebri, and family history of NBCCS. The minor criteria are defined as congenital skeletal anomalies; macrocephaly with frontal bossing; cardiac or ovarian fibromas; medulloblastoma; lymphomesenteric cysts; and congenital malformations such as cleft lip or palate, polydactyly, or eye anomalies.6 The mean age of initial BCC diagnosis is 21 years, with proliferation of cancers between puberty and 35 years of age.7,9 Our case is unique due to the patient’s young age at the time of diagnosis as well as his presentation with multiple BCCs with a darker skin type. Kimonis et al7 reported that approximately 20% of black patients develop their first BCC by the age of 21 years and 40% by 35 years. The presence of multiple BCCs is complicated by the limited treatment options in a pediatric patient. The patient’s inability to withstand multiple procedures contributed to our clinical decision to have multiple lesions removed under general anesthesia by a pediatric plastic surgeon.
Due to the patient’s young age of onset, we placed a great emphasis on close surveillance and management. A management protocol for pediatric patients with NBCCS was described by Bree and Shah; BCNS Colloquium Group10 (eTable). We closely followed this protocol for surveillance; however, we scheduled dermatologic examinations every 4 months due to his extensive history of BCCs.

Management
Our case presents a challenging therapeutic and management dilemma. The management of NBCCS utilizes a multitude of treatment modalities, but many of them posed cosmetic challenges in our patient such as postinflammatory hypopigmentation and the propensity for keloid formation. Although surgical excision or Mohs micrographic surgery is the standard of treatment of nodular BCCs, we were limited due to the patient’s inability to tolerate multiple surgical procedures without the use of general anesthesia.
Case reports have discussed the use of CO2 laser resurfacing for management of multiple facial BCCs in patients with NBCCS. Doctoroff et al11 treated a patient with 45 facial BCCs with full-face CO2 laser resurfacing, and in a 10-month follow-up period the patient developed 6 new BCCs on the face. Nouri et al12 described 3 cases of multiple BCCs on the face, trunk, and extremities treated with ultrapulse CO2 laser with postoperative Mohs sections verifying complete histologic clearance of tumors. All 3 patients had Fitzpatrick skin type IV; their ages were 2, 16, and 35 years. Local anesthesia was used in the 2-year-old patient and intravenous sedation in the 16-year-old patient.12 Although CO2 laser therapy may be a practical treatment option, it posed too many cosmetic concerns in our patient.
Photodynamic therapy (PDT) is an emerging treatment option for NBCCS patients. Itkin and Gilchrest13 treated 2 NBCCS patients with δ-aminolevulinic acid for 1 to 5 hours prior to treatment with blue light therapy. Complete clearance was documented in 89% (8/9) of superficial BCCs and 31% (5/16) of nodular BCCs on the face, indicating that blue light treatment may reduce the cutaneous tumor burden.13 Oseroff et al14 reported similar success in treating 3 children with NBCCS with 20% δ-aminolevulinic acid for 24 hours under occlusion followed by red light treatment. After 1 to 3 treatments, the children had 85% to 98% total clearance, demonstrating it as a viable treatment option in young patients that yields excellent cosmetic results and is well tolerated.14 Photodynamic therapy is reported to have a low risk of carcinogenicity15; however, there has been 1 reported case of melanoma developing at the site of multiple PDT treatments.16 Thus, the risk of carcinogenicity is increasingly bothersome in NBCCS patients due to their sensitivity to exposure. The limited number of studies using topical PDT on pediatric patients, the lack of treatment protocols for pediatric patients, and the need to use general anesthesia for pediatric patients all posed limitations to the use of PDT in our case.
Imiquimod cream 5% was shown in randomized, vehicle-controlled studies to be a safe and effective treatment of superficial BCCs when used 5 days weekly for 6 weeks.17 These studies excluded patients with NBCCS; however, other studies have been completed in patients with NBCCS. Kagy and Amonette18 successfully treated 3 nonfacial BCCs in a patient with NBCCS with imiquimod cream 5% daily for 18 weeks, with complete histologic resolution of the tumors. Micali et al19 also treated 4 patients with NBCCS using imiquimod cream 5% 3 to 5 times weekly for 8 to 14 weeks. Thirteen of 17 BCCs resolved, as confirmed with histologic evaluation.19 One case report revealed a child with NBCCS who was successfully managed with topical fluorouracil and topical tretinoin for more than 10 years.20 Our patient used imiquimod cream 5% 5 times weekly, which inhibited the growth of existing lesions but did not clear them entirely, as they were nodular in nature.
Chemoprevention with oral retinoids breaches a controversial treatment topic. In 1989, a case study of an NBCCS patient treated with surgical excision and oral etretinate for 12 months documented reduction of large tumors.21 A multicenter clinical trial reported that low-dose isotretinoin (10 mg daily) is ineffective in preventing the occurrence of new BCC formation in patients with a history of 2 or more sporadic BCCs.22 Chemoprevention with oral retinoids is well known for being effective for squamous cell carcinomas and actinic keratosis; however, the treatment is less effective for BCCs.22 Most importantly, the extensive side-effect profile and toxicity associated with long-term administration of oral retinoids prohibits many practitioners from routinely using them in pediatric NBCCS patients.
Nevoid basal cell carcinoma syndrome patients are exquisitely sensitive to ionizing radiation and the effects of UV exposure. Therefore, it is essential to emphasize the importance of sun-protective measures such as sun avoidance, broad-spectrum sunscreen use, and sun-protective clothing.
Conclusion
Nevoid basal cell carcinoma syndrome is a multisystem disorder with a notable predisposition for skin cancer. Our case demonstrates the treatment considerations in a pediatric patient with Fitzpatrick skin type V. Pediatric NBCCS patients develop BCCs at a young age and will continue to develop additional lesions throughout life; therefore, skin preservation is an important consideration when choosing the appropriate treatment regimen. Particularly in our patient, utilizing multiple strategic treatment modalities in combination with chemoprevention moving forward will be a continued management challenge. Strict adherence to a surveillance protocol is encouraged to closely monitor the systemic manifestations of the disorder.
- Gorlin RJ, Goltz R. Multiple nevoid basal cell epitheliomata, jaw cysts, bifid rib-a syndrome. N Engl J Med. 1960;262:908-911.
- Evans DGR, Farndon PA, Burnell LD, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959-961.
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530-539.
- Farndon PA, Del Mastro RG, Evans DG, et al. Location of gene for Gorlin Syndrome. Lancet. 1992;339:581-582.
- Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001;10:757-761.
- Evans DGR, Ladusans EJ, Rimmer S, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460-464.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299-308.
- Goldstein AM, Pastakia B, DiGiovanna JJ, et al. Clinical findings in two African-American families with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1994;50:272-281.
- Shanley S, Ratcliffe J, Hockey A, et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet. 1994;50:282-290.
- Bree AF, Shah MR; BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A. 2011;155:2091-2097.
- Doctoroff A, Oberlender SA, Purcell SM. Full-face carbon dioxide laser resurfacing in the management of a patient with the nevoid basal cell carcinoma syndrome. Dermatol Surg. 2003;29:1236-1240.
- Nouri K, Chang A, Trent JT, et al. Ultrapulse CO2 used for the successful treatment of basal cell carcinomas found in patients with basal cell nevus syndrome. Dermatol Surg. 2002;28:287-290.
- Itkin A, Gilchrest BA. δ-Aminolevulinic acid and blue light photodynamic therapy for treatment of multiple basal cell carcinomas in two patients with nevoid basal cell carcinoma syndrome. Dermatol Surg. 2004;30:1054-1061.
- Oseroff AR, Shieh S, Frawley NP, et al. Treatment of diffuse basal cell carcinomas and basaloid follicular hamartomas in nevoid basal cell carcinoma syndrome by wide-area 5-aminolevulinic acid photodynamic therapy. Arch Dermatol. 2005;141:60-67.
- Morton CA, Brown SB, Collins S, et al. Guidelines for topical photodynamic therapy: report of a workshop of the British Photodermatology Group. Br J Dermatol. 2002;146:552-567.
- Wolf P, Fink-Puches R, Reimann-Weber A, et al. Development of malignant melanoma after repeated topical photodynamic therapy with 5-aminolevulinic acid at the exposed site. Dermatology. 1997;194:53-54.
- Geisse J, Caro I, Lindholm J, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50:722-733.
- Kagy MK, Amonette R. The use of imiquimod 5% cream for the treatment of superficial basal cell carcinomas in a basal cell nevus syndrome patient. Dermatol Surg. 2000;26:577-579.
- Micali G, Lacarrubba F, Nasca MR, et al. The use of imiquimod 5% cream for the treatment of basal cell carcinoma as observed in Gorlin’s syndrome. Clin Exp Dermatol. 2003;28:19-23.
- Strange PR, Lang PG. Long-term management of basal cell nevus syndrome with topical tretinoin and 5-fluorouracil. J Am Acad Dermatol. 1992;27:842-845.
- Sanchez-Conejo-Mir J, Camacho F. Nevoid basal cell carcinoma syndrome: combined etretinate and surgical treatment. J Dermatol Surg Oncol. 1989;15:868-871.
- Tangrea JA, Edwards BK, Taylor PR, et al. Long-term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: a multicenter clinical trial. Isotretinoin-Basal Cell Carcinoma Study Group. J Natl Cancer Inst. 1992;84:328-332.
In 1960, Gorlin and Goltz1 first described nevoid basal cell carcinoma syndrome (NBCCS) as a distinct clinical entity with multiple basal cell carcinomas (BCCs), jaw cysts, and bifid ribs. This rare autosomal-dominant genodermatosis has a minimal prevalence of 1 case per 57,000 individuals2 and no sexual predilection.3 Nevoid basal cell carcinoma syndrome is caused by a mutation in the human homolog of a Drosophila gene, patched 1 (PTCH1), which is located on chromosome 9q22.3.4,5 The major clinical diagnostic criteria includes multiple BCCs, odontogenic keratocysts, palmar or plantar pits, ectopic calcification of the falx cerebri, and a family history of NBCCS.6 Basal cell carcinoma formation is affected by both skin pigmentation and sun exposure; 80% of white patients with NBCCS will develop at least 1 BCC compared to only 40% of black patients with NBCCS.7 Goldstein et al8 postulated that this disparity is associated with increased skin pigmentation providing UV radiation protection, thus decreasing the tumor burden. We report a case of an 11-year-old black boy with NBCCS to highlight the treatment considerations in pediatric cases of NBCCS.
Case Report
An 11-year-old boy with Fitzpatrick skin type V presented with a history of multiple facial lesions after undergoing excision of large keratocysts from the right maxilla, left maxilla, and right mandible. Physical examination revealed multiple light to dark brown facial papules (Figure 1), palmar and plantar pitting (Figure 2), and frontal bossing.
He was previously diagnosed with autism and his surgical history was notable only for excision of the keratocysts. The patient was not taking any medications and did not have any drug allergies. There was no maternal family history of skin cancer or related syndromes; his paternal family history was unknown. A shave biopsy was performed on a facial papule from the right nasolabial fold. Histopathologic evaluation revealed findings consistent with a pigmented nodular BCC (Figure 3). The patient was subsequently sent for magnetic resonance imaging of the brain, which demonstrated calcifications along the tentorium. Genetic consultation confirmed a heterozygous mutation of the PTCH1 gene.
Over the next 12 months, the patient had multiple biopsy-proven pigmented BCCs. Initial management of these carcinomas located on cosmetically sensitive areas, including the upper eyelid and penis, were excised by a pediatric plastic surgeon. A truncal carcinoma was treated with electrodesiccation and curettage, which resulted in keloid formation. Early suspicious lesions were treated with imiquimod cream 5% 5 times weekly in combination with the prophylactic use of tretinoin cream 0.1%. Despite this treatment regimen, the patient continued to demonstrate multiple small clinical pigmented BCCs along the malar surfaces of the cheeks and dorsum of the nose. The patient’s mother deferred chemoprevention with an oral retinoid due to the extensive side-effect profile and long-term necessity of administration.
Management also encompassed BCC surveillance every 4 months; annual digital panorex of the jaw; routine dental screening; routine developmental screening; annual follow-up with a geneticist to ensure multidisciplinary care; and annual vision, hearing, and speech-screening examinations. Strict sun-protective measures were encouraged, including wearing a hat during physical education class.
Comment
Classification and Clinical Presentation
Nevoid basal cell carcinoma syndrome is a multisystem disorder that requires close monitoring under multidisciplinary care. Evans et al6 defined the diagnostic criteria of NBCCS to require the presence of 2 major criteria or 1 major and 2 minor criteria. The major criteria include multiple BCCs, an odontogenic keratocyst or polyostotic bone cyst, palmar or plantar pits, ectopic calcification of the falx cerebri, and family history of NBCCS. The minor criteria are defined as congenital skeletal anomalies; macrocephaly with frontal bossing; cardiac or ovarian fibromas; medulloblastoma; lymphomesenteric cysts; and congenital malformations such as cleft lip or palate, polydactyly, or eye anomalies.6 The mean age of initial BCC diagnosis is 21 years, with proliferation of cancers between puberty and 35 years of age.7,9 Our case is unique due to the patient’s young age at the time of diagnosis as well as his presentation with multiple BCCs with a darker skin type. Kimonis et al7 reported that approximately 20% of black patients develop their first BCC by the age of 21 years and 40% by 35 years. The presence of multiple BCCs is complicated by the limited treatment options in a pediatric patient. The patient’s inability to withstand multiple procedures contributed to our clinical decision to have multiple lesions removed under general anesthesia by a pediatric plastic surgeon.
Due to the patient’s young age of onset, we placed a great emphasis on close surveillance and management. A management protocol for pediatric patients with NBCCS was described by Bree and Shah; BCNS Colloquium Group10 (eTable). We closely followed this protocol for surveillance; however, we scheduled dermatologic examinations every 4 months due to his extensive history of BCCs.
Management
Our case presents a challenging therapeutic and management dilemma. The management of NBCCS utilizes a multitude of treatment modalities, but many of them posed cosmetic challenges in our patient such as postinflammatory hypopigmentation and the propensity for keloid formation. Although surgical excision or Mohs micrographic surgery is the standard of treatment of nodular BCCs, we were limited due to the patient’s inability to tolerate multiple surgical procedures without the use of general anesthesia.
Case reports have discussed the use of CO2 laser resurfacing for management of multiple facial BCCs in patients with NBCCS. Doctoroff et al11 treated a patient with 45 facial BCCs with full-face CO2 laser resurfacing, and in a 10-month follow-up period the patient developed 6 new BCCs on the face. Nouri et al12 described 3 cases of multiple BCCs on the face, trunk, and extremities treated with ultrapulse CO2 laser with postoperative Mohs sections verifying complete histologic clearance of tumors. All 3 patients had Fitzpatrick skin type IV; their ages were 2, 16, and 35 years. Local anesthesia was used in the 2-year-old patient and intravenous sedation in the 16-year-old patient.12 Although CO2 laser therapy may be a practical treatment option, it posed too many cosmetic concerns in our patient.
Photodynamic therapy (PDT) is an emerging treatment option for NBCCS patients. Itkin and Gilchrest13 treated 2 NBCCS patients with δ-aminolevulinic acid for 1 to 5 hours prior to treatment with blue light therapy. Complete clearance was documented in 89% (8/9) of superficial BCCs and 31% (5/16) of nodular BCCs on the face, indicating that blue light treatment may reduce the cutaneous tumor burden.13 Oseroff et al14 reported similar success in treating 3 children with NBCCS with 20% δ-aminolevulinic acid for 24 hours under occlusion followed by red light treatment. After 1 to 3 treatments, the children had 85% to 98% total clearance, demonstrating it as a viable treatment option in young patients that yields excellent cosmetic results and is well tolerated.14 Photodynamic therapy is reported to have a low risk of carcinogenicity15; however, there has been 1 reported case of melanoma developing at the site of multiple PDT treatments.16 Thus, the risk of carcinogenicity is increasingly bothersome in NBCCS patients due to their sensitivity to exposure. The limited number of studies using topical PDT on pediatric patients, the lack of treatment protocols for pediatric patients, and the need to use general anesthesia for pediatric patients all posed limitations to the use of PDT in our case.
Imiquimod cream 5% was shown in randomized, vehicle-controlled studies to be a safe and effective treatment of superficial BCCs when used 5 days weekly for 6 weeks.17 These studies excluded patients with NBCCS; however, other studies have been completed in patients with NBCCS. Kagy and Amonette18 successfully treated 3 nonfacial BCCs in a patient with NBCCS with imiquimod cream 5% daily for 18 weeks, with complete histologic resolution of the tumors. Micali et al19 also treated 4 patients with NBCCS using imiquimod cream 5% 3 to 5 times weekly for 8 to 14 weeks. Thirteen of 17 BCCs resolved, as confirmed with histologic evaluation.19 One case report revealed a child with NBCCS who was successfully managed with topical fluorouracil and topical tretinoin for more than 10 years.20 Our patient used imiquimod cream 5% 5 times weekly, which inhibited the growth of existing lesions but did not clear them entirely, as they were nodular in nature.
Chemoprevention with oral retinoids breaches a controversial treatment topic. In 1989, a case study of an NBCCS patient treated with surgical excision and oral etretinate for 12 months documented reduction of large tumors.21 A multicenter clinical trial reported that low-dose isotretinoin (10 mg daily) is ineffective in preventing the occurrence of new BCC formation in patients with a history of 2 or more sporadic BCCs.22 Chemoprevention with oral retinoids is well known for being effective for squamous cell carcinomas and actinic keratosis; however, the treatment is less effective for BCCs.22 Most importantly, the extensive side-effect profile and toxicity associated with long-term administration of oral retinoids prohibits many practitioners from routinely using them in pediatric NBCCS patients.
Nevoid basal cell carcinoma syndrome patients are exquisitely sensitive to ionizing radiation and the effects of UV exposure. Therefore, it is essential to emphasize the importance of sun-protective measures such as sun avoidance, broad-spectrum sunscreen use, and sun-protective clothing.
Conclusion
Nevoid basal cell carcinoma syndrome is a multisystem disorder with a notable predisposition for skin cancer. Our case demonstrates the treatment considerations in a pediatric patient with Fitzpatrick skin type V. Pediatric NBCCS patients develop BCCs at a young age and will continue to develop additional lesions throughout life; therefore, skin preservation is an important consideration when choosing the appropriate treatment regimen. Particularly in our patient, utilizing multiple strategic treatment modalities in combination with chemoprevention moving forward will be a continued management challenge. Strict adherence to a surveillance protocol is encouraged to closely monitor the systemic manifestations of the disorder.
In 1960, Gorlin and Goltz1 first described nevoid basal cell carcinoma syndrome (NBCCS) as a distinct clinical entity with multiple basal cell carcinomas (BCCs), jaw cysts, and bifid ribs. This rare autosomal-dominant genodermatosis has a minimal prevalence of 1 case per 57,000 individuals2 and no sexual predilection.3 Nevoid basal cell carcinoma syndrome is caused by a mutation in the human homolog of a Drosophila gene, patched 1 (PTCH1), which is located on chromosome 9q22.3.4,5 The major clinical diagnostic criteria includes multiple BCCs, odontogenic keratocysts, palmar or plantar pits, ectopic calcification of the falx cerebri, and a family history of NBCCS.6 Basal cell carcinoma formation is affected by both skin pigmentation and sun exposure; 80% of white patients with NBCCS will develop at least 1 BCC compared to only 40% of black patients with NBCCS.7 Goldstein et al8 postulated that this disparity is associated with increased skin pigmentation providing UV radiation protection, thus decreasing the tumor burden. We report a case of an 11-year-old black boy with NBCCS to highlight the treatment considerations in pediatric cases of NBCCS.
Case Report
An 11-year-old boy with Fitzpatrick skin type V presented with a history of multiple facial lesions after undergoing excision of large keratocysts from the right maxilla, left maxilla, and right mandible. Physical examination revealed multiple light to dark brown facial papules (Figure 1), palmar and plantar pitting (Figure 2), and frontal bossing.
He was previously diagnosed with autism and his surgical history was notable only for excision of the keratocysts. The patient was not taking any medications and did not have any drug allergies. There was no maternal family history of skin cancer or related syndromes; his paternal family history was unknown. A shave biopsy was performed on a facial papule from the right nasolabial fold. Histopathologic evaluation revealed findings consistent with a pigmented nodular BCC (Figure 3). The patient was subsequently sent for magnetic resonance imaging of the brain, which demonstrated calcifications along the tentorium. Genetic consultation confirmed a heterozygous mutation of the PTCH1 gene.
Over the next 12 months, the patient had multiple biopsy-proven pigmented BCCs. Initial management of these carcinomas located on cosmetically sensitive areas, including the upper eyelid and penis, were excised by a pediatric plastic surgeon. A truncal carcinoma was treated with electrodesiccation and curettage, which resulted in keloid formation. Early suspicious lesions were treated with imiquimod cream 5% 5 times weekly in combination with the prophylactic use of tretinoin cream 0.1%. Despite this treatment regimen, the patient continued to demonstrate multiple small clinical pigmented BCCs along the malar surfaces of the cheeks and dorsum of the nose. The patient’s mother deferred chemoprevention with an oral retinoid due to the extensive side-effect profile and long-term necessity of administration.
Management also encompassed BCC surveillance every 4 months; annual digital panorex of the jaw; routine dental screening; routine developmental screening; annual follow-up with a geneticist to ensure multidisciplinary care; and annual vision, hearing, and speech-screening examinations. Strict sun-protective measures were encouraged, including wearing a hat during physical education class.
Comment
Classification and Clinical Presentation
Nevoid basal cell carcinoma syndrome is a multisystem disorder that requires close monitoring under multidisciplinary care. Evans et al6 defined the diagnostic criteria of NBCCS to require the presence of 2 major criteria or 1 major and 2 minor criteria. The major criteria include multiple BCCs, an odontogenic keratocyst or polyostotic bone cyst, palmar or plantar pits, ectopic calcification of the falx cerebri, and family history of NBCCS. The minor criteria are defined as congenital skeletal anomalies; macrocephaly with frontal bossing; cardiac or ovarian fibromas; medulloblastoma; lymphomesenteric cysts; and congenital malformations such as cleft lip or palate, polydactyly, or eye anomalies.6 The mean age of initial BCC diagnosis is 21 years, with proliferation of cancers between puberty and 35 years of age.7,9 Our case is unique due to the patient’s young age at the time of diagnosis as well as his presentation with multiple BCCs with a darker skin type. Kimonis et al7 reported that approximately 20% of black patients develop their first BCC by the age of 21 years and 40% by 35 years. The presence of multiple BCCs is complicated by the limited treatment options in a pediatric patient. The patient’s inability to withstand multiple procedures contributed to our clinical decision to have multiple lesions removed under general anesthesia by a pediatric plastic surgeon.
Due to the patient’s young age of onset, we placed a great emphasis on close surveillance and management. A management protocol for pediatric patients with NBCCS was described by Bree and Shah; BCNS Colloquium Group10 (eTable). We closely followed this protocol for surveillance; however, we scheduled dermatologic examinations every 4 months due to his extensive history of BCCs.
Management
Our case presents a challenging therapeutic and management dilemma. The management of NBCCS utilizes a multitude of treatment modalities, but many of them posed cosmetic challenges in our patient such as postinflammatory hypopigmentation and the propensity for keloid formation. Although surgical excision or Mohs micrographic surgery is the standard of treatment of nodular BCCs, we were limited due to the patient’s inability to tolerate multiple surgical procedures without the use of general anesthesia.
Case reports have discussed the use of CO2 laser resurfacing for management of multiple facial BCCs in patients with NBCCS. Doctoroff et al11 treated a patient with 45 facial BCCs with full-face CO2 laser resurfacing, and in a 10-month follow-up period the patient developed 6 new BCCs on the face. Nouri et al12 described 3 cases of multiple BCCs on the face, trunk, and extremities treated with ultrapulse CO2 laser with postoperative Mohs sections verifying complete histologic clearance of tumors. All 3 patients had Fitzpatrick skin type IV; their ages were 2, 16, and 35 years. Local anesthesia was used in the 2-year-old patient and intravenous sedation in the 16-year-old patient.12 Although CO2 laser therapy may be a practical treatment option, it posed too many cosmetic concerns in our patient.
Photodynamic therapy (PDT) is an emerging treatment option for NBCCS patients. Itkin and Gilchrest13 treated 2 NBCCS patients with δ-aminolevulinic acid for 1 to 5 hours prior to treatment with blue light therapy. Complete clearance was documented in 89% (8/9) of superficial BCCs and 31% (5/16) of nodular BCCs on the face, indicating that blue light treatment may reduce the cutaneous tumor burden.13 Oseroff et al14 reported similar success in treating 3 children with NBCCS with 20% δ-aminolevulinic acid for 24 hours under occlusion followed by red light treatment. After 1 to 3 treatments, the children had 85% to 98% total clearance, demonstrating it as a viable treatment option in young patients that yields excellent cosmetic results and is well tolerated.14 Photodynamic therapy is reported to have a low risk of carcinogenicity15; however, there has been 1 reported case of melanoma developing at the site of multiple PDT treatments.16 Thus, the risk of carcinogenicity is increasingly bothersome in NBCCS patients due to their sensitivity to exposure. The limited number of studies using topical PDT on pediatric patients, the lack of treatment protocols for pediatric patients, and the need to use general anesthesia for pediatric patients all posed limitations to the use of PDT in our case.
Imiquimod cream 5% was shown in randomized, vehicle-controlled studies to be a safe and effective treatment of superficial BCCs when used 5 days weekly for 6 weeks.17 These studies excluded patients with NBCCS; however, other studies have been completed in patients with NBCCS. Kagy and Amonette18 successfully treated 3 nonfacial BCCs in a patient with NBCCS with imiquimod cream 5% daily for 18 weeks, with complete histologic resolution of the tumors. Micali et al19 also treated 4 patients with NBCCS using imiquimod cream 5% 3 to 5 times weekly for 8 to 14 weeks. Thirteen of 17 BCCs resolved, as confirmed with histologic evaluation.19 One case report revealed a child with NBCCS who was successfully managed with topical fluorouracil and topical tretinoin for more than 10 years.20 Our patient used imiquimod cream 5% 5 times weekly, which inhibited the growth of existing lesions but did not clear them entirely, as they were nodular in nature.
Chemoprevention with oral retinoids breaches a controversial treatment topic. In 1989, a case study of an NBCCS patient treated with surgical excision and oral etretinate for 12 months documented reduction of large tumors.21 A multicenter clinical trial reported that low-dose isotretinoin (10 mg daily) is ineffective in preventing the occurrence of new BCC formation in patients with a history of 2 or more sporadic BCCs.22 Chemoprevention with oral retinoids is well known for being effective for squamous cell carcinomas and actinic keratosis; however, the treatment is less effective for BCCs.22 Most importantly, the extensive side-effect profile and toxicity associated with long-term administration of oral retinoids prohibits many practitioners from routinely using them in pediatric NBCCS patients.
Nevoid basal cell carcinoma syndrome patients are exquisitely sensitive to ionizing radiation and the effects of UV exposure. Therefore, it is essential to emphasize the importance of sun-protective measures such as sun avoidance, broad-spectrum sunscreen use, and sun-protective clothing.
Conclusion
Nevoid basal cell carcinoma syndrome is a multisystem disorder with a notable predisposition for skin cancer. Our case demonstrates the treatment considerations in a pediatric patient with Fitzpatrick skin type V. Pediatric NBCCS patients develop BCCs at a young age and will continue to develop additional lesions throughout life; therefore, skin preservation is an important consideration when choosing the appropriate treatment regimen. Particularly in our patient, utilizing multiple strategic treatment modalities in combination with chemoprevention moving forward will be a continued management challenge. Strict adherence to a surveillance protocol is encouraged to closely monitor the systemic manifestations of the disorder.
- Gorlin RJ, Goltz R. Multiple nevoid basal cell epitheliomata, jaw cysts, bifid rib-a syndrome. N Engl J Med. 1960;262:908-911.
- Evans DGR, Farndon PA, Burnell LD, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959-961.
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530-539.
- Farndon PA, Del Mastro RG, Evans DG, et al. Location of gene for Gorlin Syndrome. Lancet. 1992;339:581-582.
- Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001;10:757-761.
- Evans DGR, Ladusans EJ, Rimmer S, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460-464.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299-308.
- Goldstein AM, Pastakia B, DiGiovanna JJ, et al. Clinical findings in two African-American families with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1994;50:272-281.
- Shanley S, Ratcliffe J, Hockey A, et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet. 1994;50:282-290.
- Bree AF, Shah MR; BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A. 2011;155:2091-2097.
- Doctoroff A, Oberlender SA, Purcell SM. Full-face carbon dioxide laser resurfacing in the management of a patient with the nevoid basal cell carcinoma syndrome. Dermatol Surg. 2003;29:1236-1240.
- Nouri K, Chang A, Trent JT, et al. Ultrapulse CO2 used for the successful treatment of basal cell carcinomas found in patients with basal cell nevus syndrome. Dermatol Surg. 2002;28:287-290.
- Itkin A, Gilchrest BA. δ-Aminolevulinic acid and blue light photodynamic therapy for treatment of multiple basal cell carcinomas in two patients with nevoid basal cell carcinoma syndrome. Dermatol Surg. 2004;30:1054-1061.
- Oseroff AR, Shieh S, Frawley NP, et al. Treatment of diffuse basal cell carcinomas and basaloid follicular hamartomas in nevoid basal cell carcinoma syndrome by wide-area 5-aminolevulinic acid photodynamic therapy. Arch Dermatol. 2005;141:60-67.
- Morton CA, Brown SB, Collins S, et al. Guidelines for topical photodynamic therapy: report of a workshop of the British Photodermatology Group. Br J Dermatol. 2002;146:552-567.
- Wolf P, Fink-Puches R, Reimann-Weber A, et al. Development of malignant melanoma after repeated topical photodynamic therapy with 5-aminolevulinic acid at the exposed site. Dermatology. 1997;194:53-54.
- Geisse J, Caro I, Lindholm J, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50:722-733.
- Kagy MK, Amonette R. The use of imiquimod 5% cream for the treatment of superficial basal cell carcinomas in a basal cell nevus syndrome patient. Dermatol Surg. 2000;26:577-579.
- Micali G, Lacarrubba F, Nasca MR, et al. The use of imiquimod 5% cream for the treatment of basal cell carcinoma as observed in Gorlin’s syndrome. Clin Exp Dermatol. 2003;28:19-23.
- Strange PR, Lang PG. Long-term management of basal cell nevus syndrome with topical tretinoin and 5-fluorouracil. J Am Acad Dermatol. 1992;27:842-845.
- Sanchez-Conejo-Mir J, Camacho F. Nevoid basal cell carcinoma syndrome: combined etretinate and surgical treatment. J Dermatol Surg Oncol. 1989;15:868-871.
- Tangrea JA, Edwards BK, Taylor PR, et al. Long-term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: a multicenter clinical trial. Isotretinoin-Basal Cell Carcinoma Study Group. J Natl Cancer Inst. 1992;84:328-332.
- Gorlin RJ, Goltz R. Multiple nevoid basal cell epitheliomata, jaw cysts, bifid rib-a syndrome. N Engl J Med. 1960;262:908-911.
- Evans DGR, Farndon PA, Burnell LD, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959-961.
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530-539.
- Farndon PA, Del Mastro RG, Evans DG, et al. Location of gene for Gorlin Syndrome. Lancet. 1992;339:581-582.
- Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001;10:757-761.
- Evans DGR, Ladusans EJ, Rimmer S, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460-464.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299-308.
- Goldstein AM, Pastakia B, DiGiovanna JJ, et al. Clinical findings in two African-American families with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1994;50:272-281.
- Shanley S, Ratcliffe J, Hockey A, et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet. 1994;50:282-290.
- Bree AF, Shah MR; BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A. 2011;155:2091-2097.
- Doctoroff A, Oberlender SA, Purcell SM. Full-face carbon dioxide laser resurfacing in the management of a patient with the nevoid basal cell carcinoma syndrome. Dermatol Surg. 2003;29:1236-1240.
- Nouri K, Chang A, Trent JT, et al. Ultrapulse CO2 used for the successful treatment of basal cell carcinomas found in patients with basal cell nevus syndrome. Dermatol Surg. 2002;28:287-290.
- Itkin A, Gilchrest BA. δ-Aminolevulinic acid and blue light photodynamic therapy for treatment of multiple basal cell carcinomas in two patients with nevoid basal cell carcinoma syndrome. Dermatol Surg. 2004;30:1054-1061.
- Oseroff AR, Shieh S, Frawley NP, et al. Treatment of diffuse basal cell carcinomas and basaloid follicular hamartomas in nevoid basal cell carcinoma syndrome by wide-area 5-aminolevulinic acid photodynamic therapy. Arch Dermatol. 2005;141:60-67.
- Morton CA, Brown SB, Collins S, et al. Guidelines for topical photodynamic therapy: report of a workshop of the British Photodermatology Group. Br J Dermatol. 2002;146:552-567.
- Wolf P, Fink-Puches R, Reimann-Weber A, et al. Development of malignant melanoma after repeated topical photodynamic therapy with 5-aminolevulinic acid at the exposed site. Dermatology. 1997;194:53-54.
- Geisse J, Caro I, Lindholm J, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50:722-733.
- Kagy MK, Amonette R. The use of imiquimod 5% cream for the treatment of superficial basal cell carcinomas in a basal cell nevus syndrome patient. Dermatol Surg. 2000;26:577-579.
- Micali G, Lacarrubba F, Nasca MR, et al. The use of imiquimod 5% cream for the treatment of basal cell carcinoma as observed in Gorlin’s syndrome. Clin Exp Dermatol. 2003;28:19-23.
- Strange PR, Lang PG. Long-term management of basal cell nevus syndrome with topical tretinoin and 5-fluorouracil. J Am Acad Dermatol. 1992;27:842-845.
- Sanchez-Conejo-Mir J, Camacho F. Nevoid basal cell carcinoma syndrome: combined etretinate and surgical treatment. J Dermatol Surg Oncol. 1989;15:868-871.
- Tangrea JA, Edwards BK, Taylor PR, et al. Long-term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: a multicenter clinical trial. Isotretinoin-Basal Cell Carcinoma Study Group. J Natl Cancer Inst. 1992;84:328-332.
Practice Points
- Nevoid basal cell carcinoma syndrome (NBCCS) is a multisystem disorder that requires close monitoring under multidisciplinary care.
- The clinical manifestations of NBCCS include multiple basal cell carcinomas, odontogenic keratocysts, palmar or plantar pits, and calcification of the falx cerebri.
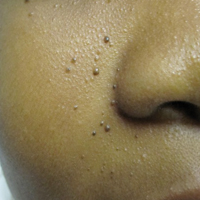
Lichen Planus Pemphigoides Treated With Ustekinumab
Case Report
A 71-year-old woman presented with pink to violaceous, flat-topped, polygonal papules consistent with lichen planus (LP) on the volar wrists, extensor elbows, and bilateral lower legs of 3 years’ duration. She also had erythematous, violaceous, infiltrated plaques with microvesiculation on the bilateral thighs of several months’ duration (Figure 1). She reported pruritus, burning, and discomfort. Her medical history included type 2 diabetes mellitus, hypertension, and asthma with no history of skin rashes. A complete physical examination was performed. Age-appropriate screening for malignancy was negative. Hepatitis B and C antibody serologies were negative. Her medications at the time included risedronate and atenolol, which she had been taking for several years.
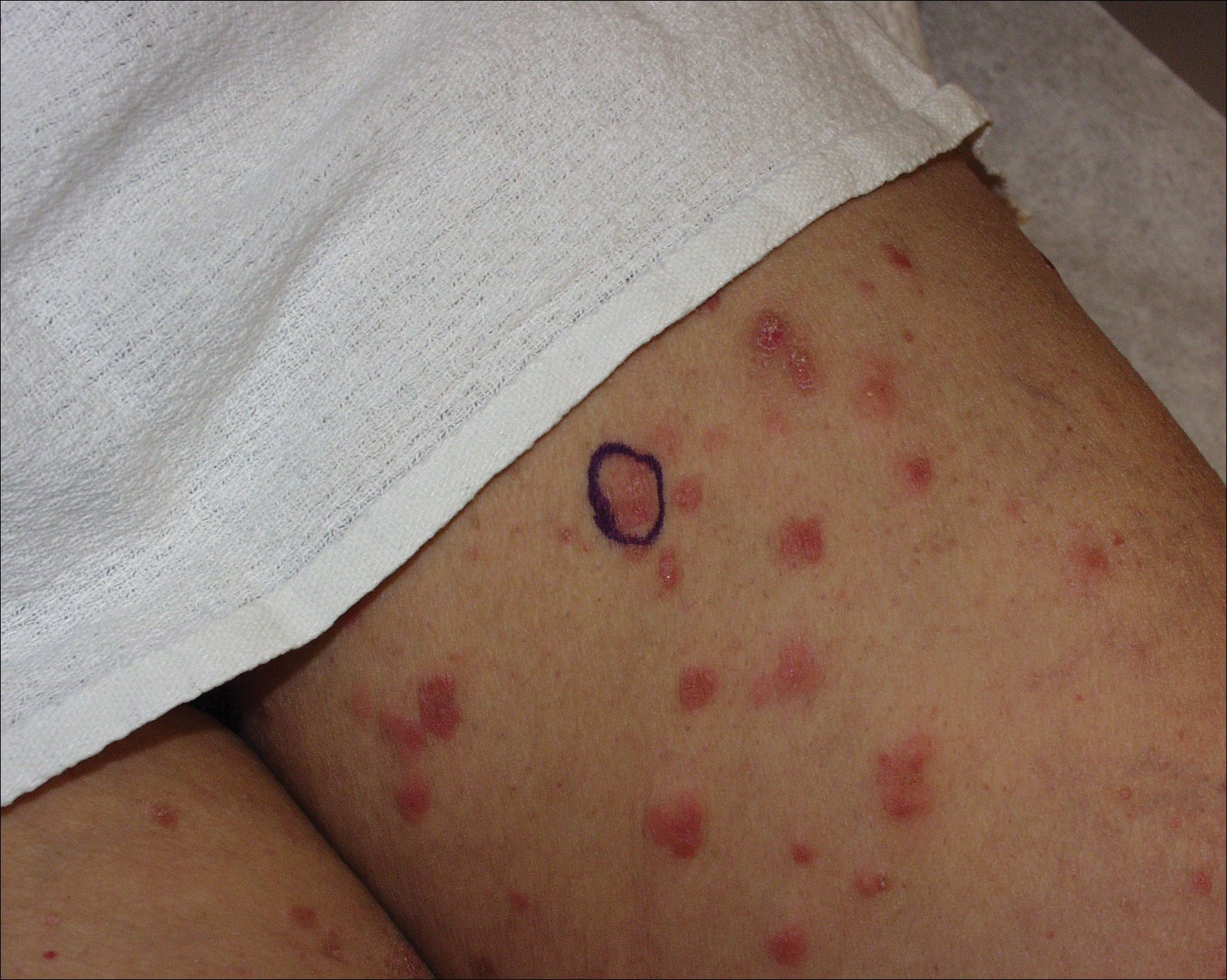
Punch biopsies from perilesional skin were submitted for hematoxylin and eosin staining and direct immunofluorescence (DIF). Histopathology showed a subepidermal blistering disease with tissue eosinophilia consistent with lichen planus pemphigoides (LPP)(Figure 2); direct immunofluorescence was positive for IgG, C3, and type IV collagen at the dermoepidermal junction. Serum BP180 was positive at 51 U/mL (reference range, <14 U/mL) and BP230 was negative. She was then started on tetracycline (500 mg twice daily), nicotinamide (500 mg twice daily), prednisone (5 mg daily), and dapsone (100 mg daily).
After 3 months without improvement, tetracycline and nicotinamide were discontinued, prednisone was increased to 10 mg daily, and dapsone was continued. A repeat biopsy was taken from a new area of involvement on the left lower leg, which revealed a psoriasiform dermatitis with interface changes. The DIF was positive for IgG and C3 along the basement membrane. A serum indirect immunofluorescence for BP180 also was positive.
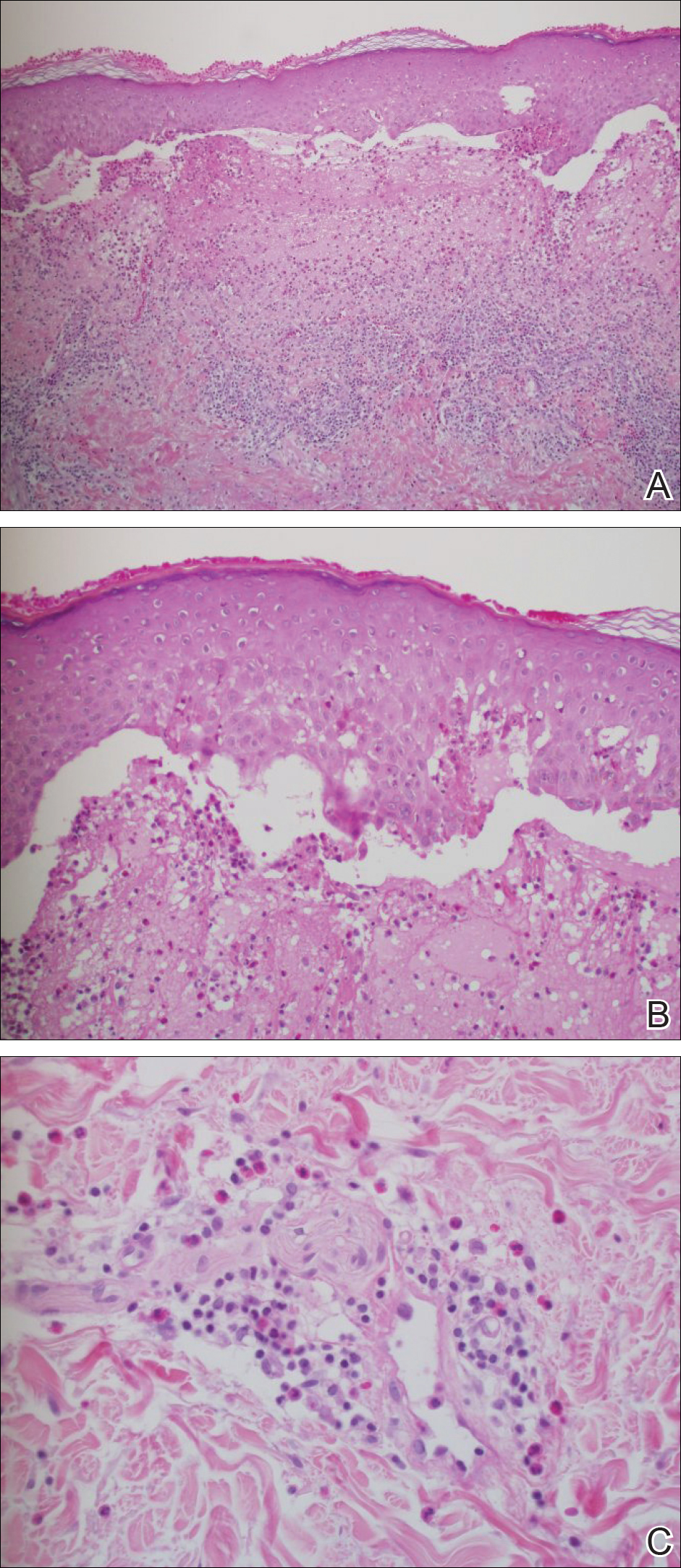
The patient developed mild hemolytic anemia on dapsone; the medication was eventually discontinued. Subsequent treatments included adequate trials of azathioprine, mycophenolate mofetil, and hydroxychloroquine. Azathioprine (150 mg daily) and hydroxychloroquine (400 mg daily) treatment failed. She initially improved on mycophenolate mofetil (500 mg in the morning and 1000 mg in the evening) with flattening of the papules on the arms and legs and decreased erythema. However, mycophenolate mofetil eventually lost its efficacy and was discontinued.
Because several medications failed (ie, tetracycline, nicotinamide, prednisone, dapsone, azathioprine, mycophenolate mofetil, hydroxychloroquine), she was started on ustekinumab (45 mg) initial loading dose by subcutaneous injection (patient’s weight, 63 kg). At 4 weeks, the patient was given the second subcutaneous injection of ustekinumab (45 mg). She experienced marked improvement with no new lesions. The prior lesions also had decreased in size and were only slightly pink. The prednisone dose was tapered to 5 mg daily.
She had near-complete resolution of the skin lesions 12 weeks after the second dose of ustekinumab. Since then, she has had some recrudescence of the papulosquamous lesions but no vesicles or bullae. With the exception of occasional scattered pink papules on the forearms, her condition greatly improved on ustekinumab. She is no longer taking any of the other medications with the exception of prednisone (down to 1 mg daily) with a plan to gradually taper completely off of it.
Comment
Clinical Presentation
Lichen planus pemphigoides is a rare autoimmune subepidermal blistering disease with few cases reported in the literature. It is considered a clinical variation of bullous pemphigoid (BP) or a coexistence of LP and BP.1,2 It is characterized by bullous lesions developing on LP papules as well as on clinically uninvolved areas of the skin. It has been reported that LPP is provoked by several medications including cinnarizine, captopril, ramipril, simvastatin, psoralen plus UVA, and antituberculous medications (eg, isoniazid, rifampin, ethambutol, pyrazinamide).1 Risedronate or atenolol have not been reported to cause LPP, LP, or BP; however, according to Litt,3 a lichenoid drug eruption has been associated with atenolol. Furthermore, some cases of LPP demonstrate overlapping characteristics with paraneoplastic pemphigus and have been associated with internal malignancy. Hamada et al4 described a case of LPP coupled with colon adenocarcinoma and numerous keratoacanthomas. The earliest depiction of the coexistence of a case of mainstream LP complicated by an extensive bullous eruption was by Kaposi5 in 1892. He coined the term lichen ruber pemphigoides.5
Compared to BP, LPP is believed to affect a younger age group and have a less serious clinical course. The mean age of onset of LPP is in the third to fourth decades of life, while BP typically presents in the sixth decade. When comparing the location of bullae in LPP versus BP, the lesions of LPP tend to occur on the limbs, while BP tends to occur on the trunk.6
Clinically, LPP is distinguished by the existence of bullous lesions developing atop of the lesions of LP as well as on normal skin, with the latter being more commonplace. A classic example of LPP is characterized by an initial episode of traditional LP lesions often having severe pruritus, with or without patches of erythema, with the sudden eruption of tense bullae. These bullae commonly appear on the extremities and can appear over the normal skin, erythematous patches, or preexisting papules.7 In the atypical clinical presentations of this dubious skin condition, the bullae may only be seen on the lesions of LP.8 There also could be a lichenoid erythrodermic manifestation of a bullous eruption.9
Oral lesions of LPP have been described but had not been studied immunopathologically until Allen et al10 portrayed a 59-year-old man with cutaneous and oral lesions of LPP. They performed biopsies on the oral lesions and examined them by routine light microscopy and immunofluorescent techniques. The fine keratotic striae on the anterior buccal mucosal lesions were clinically consistent with oral LP. Perilesional tissue in conjunction with ulceration of the posterior buccal mucosa demonstrated histologic and immunopathologic alterations consistent with BP.10
Histopathology
Histopathologically, the lesions of LP show a bandlike lymphohistiocytic infiltrate, colloid bodies in the dermis, irregular acanthosis with saw-toothed rete ridges, orthokeratosis, wedge-shaped hypergranulosis, and liquefaction degeneration of the basal layer. Direct immunofluorescence shows mainly IgM and C3 deposited on colloid bodies, fibrin, and fibrinogen.11 The histopathology of the bullous lesion of LPP depicts a subepidermal bulla with variable diffuse or sparse lymphohistiocytic infiltrate and frequent eosinophils with or without neutrophils in the upper dermis. The existence of C3 alone or with IgG along the dermoepidermal junction gives confirmation on DIF.7
Autoantibodies
The expression of IgG autoantibodies directed against the basement membrane zone distinguishes LPP from bullous LP.2 IgG autoantibodies to either one or both the 230-kDa and 180-kDa BP (type XVII collagen) antigens has been demonstrated with LPP.4,12-14 Hamada et al4 described a histologic pattern more consistent with paraneoplastic pemphigus. It has been suggested that injury to the basal cells in LP or damage due to other courses of therapy such as psoralen plus UVA unveil suppressed antigenic determinants or produce new antigens, leading to antibody development and production of BP.12,15
Zillikens et al2 performed a study to identify the target antigen of LPP autoantibodies. They used sera from patients with LPP (n=4) and stained the epidermal side of salt-split human skin in a configuration identical to BP sera. In BP, the autoimmune response is directed against BP180, a hemidesmosomal transmembrane collagenous glycoprotein. They demonstrated that sera from BP patients largely reacted with a set of 4 epitopes (MCW-0 through MCW-3) grouped within a 45 amino acid stretch of the major noncollagenous extracellular domain (NC16A) of BP180. By immunoblotting and enzyme-linked immunosorbent assay, LPP sera also were compellingly reactive with recombinant BP180 NC16A. Lichen planus pemphigoides epitopes were additionally mapped using a series of overlapping recombinant segments of the NC16A domain. The authors demonstrated that all LPP sera reacted with amino acids 46 through 59 of domain NC16A, a protein portion that was previously shown to be unreactive with BP sera. In addition, they showed that 2 LPP sera reacted with the immunodominant antigenic region related to BP. Furthermore, they identified a unique epitope within the BP180 NC16A domain—MCW-4—which was distinctively recognized by sera from patients with LPP.2
Pathogenesis
The pathogenesis of both LP and BP has been linked to multiple cytokines that induce apoptosis in basal keratinocytes. Implicated cytokines include IFN-γ, tumor necrosis factor α (TNF-α), IL-1, IL-6, and IL-8, as well as other apoptosis-related molecules, such as Fas/Apo-1 and Bcl-2 in LP.16-18 Soluble E-selectin, vascular endothelial growth factor, IL-1β, IL-8, IL-5, transforming growth factor β1, and TNF-α were found to be elevated in either blister fluid or sera of BP patients.15-17
Management
Lichen planus pemphigoides usually responds well to traditional therapies, with systemic steroids being the most efficacious treatment of extensive disease.12,13 Other options include tetracycline and nicotinamide, isotretinoin, dapsone, and immunosuppressive drugs such as systemic cortico-steroids.12 Demirçay et al12 described a patient with skin lesions that rapidly cleared after the administration of oral methylprednisolone (48 mg/d) and oral dapsone (100 mg/d). The methylprednisolone and dapsone were withdrawn after 12 and 16 weeks, respectively. There was no recurrence during the 1-year follow-up period.12 et al19 described a patient who was treated with pulsed intravenous corticosteroids and continued to develop new papular and vesicular skin lesions. However, when oral acitretin was added to the patient’s regimen, the skin lesions cleared.19 There are several case reports of the successful use of hydroxychloroquine in LP.20,21
Cutaneous, nail, and oral LP also can be treated with TNF-α inhibitors (eg, adalimumab, etanercept) with resolution of lesions.22-25 However, we have not been able to find any reports of treating LPP with biologic medications in a search of PubMed articles indexed for MEDLINE using the terms lichen planus pemphigoides and biologic treatments/therapies. Given the fact that TNF-α and other inflammatory cytokines are involved in the pathogenesis of BP and LP, it is feasible that they also may be involved in the pathogenesis of LPP.
In our patient with cutaneous LPP, we chose to use ustekinumab instead of a primary TNF-α inhibitor because ustekinumab indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22, which also could have played a role in the patient’s disease. Our goal was to use ustekinumab as a potential corticosteroid-sparing agent. Ustekinumab greatly improved her skin condition and allowed us to discontinue other medications.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Litt J. Litt’s Drug Eruptions and Reactions Manual. 18th Ed. London, England: Informa Healthcare; 2011.
- Hamada T, Fujimoto W, Okazaki F, et al. Lichen planus pemphigoides and multiple keratoacanthomas associated with colon adenocarcinoma. Br J Dermatol. 2004;151:252-254.
- Kaposi M. Lichen ruber pemphigoides. Arch Derm Syph. 1892;343-346.
- Swale VJ, Black MM, Bhogal BS. Lichen planus pemphigoides: two case reports. Clin Exp Dermatol. 1998;23:132-135.
- Okochi H, Nashiro K, Tsuchida T, et al. Lichen planus pemphigoides: case reports and results of immunofluorescence and immunoelectron microscopic study. J Am Acad Dermatol. 1990;22:626-631.
- Mendiratta V, Asati DP, Koranne RV. Lichen planus pemphigoides in an Indian female. Indian J Dermatol. 2005;50:224-226.
- Joly P, Tanasescu S, Wolkenstein P, et al. Lichenoid erythrodermic bullous pemphigoid of the African patient. J Am Acad Dermatol. 1998;39:691-697.
- Allen , , R. Lichen planus pemphigoides: report of a case with oral lesions. Oral Surg Oral Med Oral Pathol. 1987;63:184-188.
- Rapini RP. Practical Dermatopathology. Philadelphia, PA: Mosby Elsevier; 2005.
- Demirçay Z, Baykal C, Demirkesen C. Lichen planus pemphigoides: report of two cases. Int J Dermatol. 2001;40:757-759.
- Sakuma-Oyama Y, Powell AM, Albert S, et al. Lichen planus pemphigoides evolving into pemphigoid nodularis. Clin Exp Dermatol. 2004;28:613-616.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Ameglio F, D’Auria L, Cordiali-Fei P, et al. Bullous pemphigoid and pemphigus vulgaris: correlated behaviour of serum VEGF, sE-selectin and TNF-alpha levels. J Biol Regul Homeost Agents. 1997;11:148-153.
- Ameglio F, D’auria L, Bonifati C, et al. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. 1998;138:611-614.
- D’Auria L, Mussi A, Bonifati C, et al. Increased serum IL-6, TNF-alpha and IL-10 levels in patients with bullous pemphigoid: relationships with disease activity. J Eur Acad Dermatol Venereol. 1999;12:11-15.
- , ,, . Treatment of lichen planus pemphigoides with acitretin and pulsed corticosteroids. Hautarzt. 2003;54:268-273.
- Eisen D. Hydroxychloroquine sulfate (Plaquenil) improves oral lichen planus: an open trial. J Am Acad Dermatol. 1993;28:609-612.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 11th ed. Philadelphia, PA: Mosby Elsevier; 2011.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Yarom N. Etanercept for the management of oral lichen planus. Am J Clin Dermatol. 2007;8:121.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Irla N, Schneiter T, Haneke E, et al. Nail lichen planus: successful treatment with etanercept. Case Rep Dermatol. 2010;2:173-176.
Case Report
A 71-year-old woman presented with pink to violaceous, flat-topped, polygonal papules consistent with lichen planus (LP) on the volar wrists, extensor elbows, and bilateral lower legs of 3 years’ duration. She also had erythematous, violaceous, infiltrated plaques with microvesiculation on the bilateral thighs of several months’ duration (Figure 1). She reported pruritus, burning, and discomfort. Her medical history included type 2 diabetes mellitus, hypertension, and asthma with no history of skin rashes. A complete physical examination was performed. Age-appropriate screening for malignancy was negative. Hepatitis B and C antibody serologies were negative. Her medications at the time included risedronate and atenolol, which she had been taking for several years.
Punch biopsies from perilesional skin were submitted for hematoxylin and eosin staining and direct immunofluorescence (DIF). Histopathology showed a subepidermal blistering disease with tissue eosinophilia consistent with lichen planus pemphigoides (LPP)(Figure 2); direct immunofluorescence was positive for IgG, C3, and type IV collagen at the dermoepidermal junction. Serum BP180 was positive at 51 U/mL (reference range, <14 U/mL) and BP230 was negative. She was then started on tetracycline (500 mg twice daily), nicotinamide (500 mg twice daily), prednisone (5 mg daily), and dapsone (100 mg daily).
After 3 months without improvement, tetracycline and nicotinamide were discontinued, prednisone was increased to 10 mg daily, and dapsone was continued. A repeat biopsy was taken from a new area of involvement on the left lower leg, which revealed a psoriasiform dermatitis with interface changes. The DIF was positive for IgG and C3 along the basement membrane. A serum indirect immunofluorescence for BP180 also was positive.
The patient developed mild hemolytic anemia on dapsone; the medication was eventually discontinued. Subsequent treatments included adequate trials of azathioprine, mycophenolate mofetil, and hydroxychloroquine. Azathioprine (150 mg daily) and hydroxychloroquine (400 mg daily) treatment failed. She initially improved on mycophenolate mofetil (500 mg in the morning and 1000 mg in the evening) with flattening of the papules on the arms and legs and decreased erythema. However, mycophenolate mofetil eventually lost its efficacy and was discontinued.
Because several medications failed (ie, tetracycline, nicotinamide, prednisone, dapsone, azathioprine, mycophenolate mofetil, hydroxychloroquine), she was started on ustekinumab (45 mg) initial loading dose by subcutaneous injection (patient’s weight, 63 kg). At 4 weeks, the patient was given the second subcutaneous injection of ustekinumab (45 mg). She experienced marked improvement with no new lesions. The prior lesions also had decreased in size and were only slightly pink. The prednisone dose was tapered to 5 mg daily.
She had near-complete resolution of the skin lesions 12 weeks after the second dose of ustekinumab. Since then, she has had some recrudescence of the papulosquamous lesions but no vesicles or bullae. With the exception of occasional scattered pink papules on the forearms, her condition greatly improved on ustekinumab. She is no longer taking any of the other medications with the exception of prednisone (down to 1 mg daily) with a plan to gradually taper completely off of it.
Comment
Clinical Presentation
Lichen planus pemphigoides is a rare autoimmune subepidermal blistering disease with few cases reported in the literature. It is considered a clinical variation of bullous pemphigoid (BP) or a coexistence of LP and BP.1,2 It is characterized by bullous lesions developing on LP papules as well as on clinically uninvolved areas of the skin. It has been reported that LPP is provoked by several medications including cinnarizine, captopril, ramipril, simvastatin, psoralen plus UVA, and antituberculous medications (eg, isoniazid, rifampin, ethambutol, pyrazinamide).1 Risedronate or atenolol have not been reported to cause LPP, LP, or BP; however, according to Litt,3 a lichenoid drug eruption has been associated with atenolol. Furthermore, some cases of LPP demonstrate overlapping characteristics with paraneoplastic pemphigus and have been associated with internal malignancy. Hamada et al4 described a case of LPP coupled with colon adenocarcinoma and numerous keratoacanthomas. The earliest depiction of the coexistence of a case of mainstream LP complicated by an extensive bullous eruption was by Kaposi5 in 1892. He coined the term lichen ruber pemphigoides.5
Compared to BP, LPP is believed to affect a younger age group and have a less serious clinical course. The mean age of onset of LPP is in the third to fourth decades of life, while BP typically presents in the sixth decade. When comparing the location of bullae in LPP versus BP, the lesions of LPP tend to occur on the limbs, while BP tends to occur on the trunk.6
Clinically, LPP is distinguished by the existence of bullous lesions developing atop of the lesions of LP as well as on normal skin, with the latter being more commonplace. A classic example of LPP is characterized by an initial episode of traditional LP lesions often having severe pruritus, with or without patches of erythema, with the sudden eruption of tense bullae. These bullae commonly appear on the extremities and can appear over the normal skin, erythematous patches, or preexisting papules.7 In the atypical clinical presentations of this dubious skin condition, the bullae may only be seen on the lesions of LP.8 There also could be a lichenoid erythrodermic manifestation of a bullous eruption.9
Oral lesions of LPP have been described but had not been studied immunopathologically until Allen et al10 portrayed a 59-year-old man with cutaneous and oral lesions of LPP. They performed biopsies on the oral lesions and examined them by routine light microscopy and immunofluorescent techniques. The fine keratotic striae on the anterior buccal mucosal lesions were clinically consistent with oral LP. Perilesional tissue in conjunction with ulceration of the posterior buccal mucosa demonstrated histologic and immunopathologic alterations consistent with BP.10
Histopathology
Histopathologically, the lesions of LP show a bandlike lymphohistiocytic infiltrate, colloid bodies in the dermis, irregular acanthosis with saw-toothed rete ridges, orthokeratosis, wedge-shaped hypergranulosis, and liquefaction degeneration of the basal layer. Direct immunofluorescence shows mainly IgM and C3 deposited on colloid bodies, fibrin, and fibrinogen.11 The histopathology of the bullous lesion of LPP depicts a subepidermal bulla with variable diffuse or sparse lymphohistiocytic infiltrate and frequent eosinophils with or without neutrophils in the upper dermis. The existence of C3 alone or with IgG along the dermoepidermal junction gives confirmation on DIF.7
Autoantibodies
The expression of IgG autoantibodies directed against the basement membrane zone distinguishes LPP from bullous LP.2 IgG autoantibodies to either one or both the 230-kDa and 180-kDa BP (type XVII collagen) antigens has been demonstrated with LPP.4,12-14 Hamada et al4 described a histologic pattern more consistent with paraneoplastic pemphigus. It has been suggested that injury to the basal cells in LP or damage due to other courses of therapy such as psoralen plus UVA unveil suppressed antigenic determinants or produce new antigens, leading to antibody development and production of BP.12,15
Zillikens et al2 performed a study to identify the target antigen of LPP autoantibodies. They used sera from patients with LPP (n=4) and stained the epidermal side of salt-split human skin in a configuration identical to BP sera. In BP, the autoimmune response is directed against BP180, a hemidesmosomal transmembrane collagenous glycoprotein. They demonstrated that sera from BP patients largely reacted with a set of 4 epitopes (MCW-0 through MCW-3) grouped within a 45 amino acid stretch of the major noncollagenous extracellular domain (NC16A) of BP180. By immunoblotting and enzyme-linked immunosorbent assay, LPP sera also were compellingly reactive with recombinant BP180 NC16A. Lichen planus pemphigoides epitopes were additionally mapped using a series of overlapping recombinant segments of the NC16A domain. The authors demonstrated that all LPP sera reacted with amino acids 46 through 59 of domain NC16A, a protein portion that was previously shown to be unreactive with BP sera. In addition, they showed that 2 LPP sera reacted with the immunodominant antigenic region related to BP. Furthermore, they identified a unique epitope within the BP180 NC16A domain—MCW-4—which was distinctively recognized by sera from patients with LPP.2
Pathogenesis
The pathogenesis of both LP and BP has been linked to multiple cytokines that induce apoptosis in basal keratinocytes. Implicated cytokines include IFN-γ, tumor necrosis factor α (TNF-α), IL-1, IL-6, and IL-8, as well as other apoptosis-related molecules, such as Fas/Apo-1 and Bcl-2 in LP.16-18 Soluble E-selectin, vascular endothelial growth factor, IL-1β, IL-8, IL-5, transforming growth factor β1, and TNF-α were found to be elevated in either blister fluid or sera of BP patients.15-17
Management
Lichen planus pemphigoides usually responds well to traditional therapies, with systemic steroids being the most efficacious treatment of extensive disease.12,13 Other options include tetracycline and nicotinamide, isotretinoin, dapsone, and immunosuppressive drugs such as systemic cortico-steroids.12 Demirçay et al12 described a patient with skin lesions that rapidly cleared after the administration of oral methylprednisolone (48 mg/d) and oral dapsone (100 mg/d). The methylprednisolone and dapsone were withdrawn after 12 and 16 weeks, respectively. There was no recurrence during the 1-year follow-up period.12 et al19 described a patient who was treated with pulsed intravenous corticosteroids and continued to develop new papular and vesicular skin lesions. However, when oral acitretin was added to the patient’s regimen, the skin lesions cleared.19 There are several case reports of the successful use of hydroxychloroquine in LP.20,21
Cutaneous, nail, and oral LP also can be treated with TNF-α inhibitors (eg, adalimumab, etanercept) with resolution of lesions.22-25 However, we have not been able to find any reports of treating LPP with biologic medications in a search of PubMed articles indexed for MEDLINE using the terms lichen planus pemphigoides and biologic treatments/therapies. Given the fact that TNF-α and other inflammatory cytokines are involved in the pathogenesis of BP and LP, it is feasible that they also may be involved in the pathogenesis of LPP.
In our patient with cutaneous LPP, we chose to use ustekinumab instead of a primary TNF-α inhibitor because ustekinumab indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22, which also could have played a role in the patient’s disease. Our goal was to use ustekinumab as a potential corticosteroid-sparing agent. Ustekinumab greatly improved her skin condition and allowed us to discontinue other medications.
Case Report
A 71-year-old woman presented with pink to violaceous, flat-topped, polygonal papules consistent with lichen planus (LP) on the volar wrists, extensor elbows, and bilateral lower legs of 3 years’ duration. She also had erythematous, violaceous, infiltrated plaques with microvesiculation on the bilateral thighs of several months’ duration (Figure 1). She reported pruritus, burning, and discomfort. Her medical history included type 2 diabetes mellitus, hypertension, and asthma with no history of skin rashes. A complete physical examination was performed. Age-appropriate screening for malignancy was negative. Hepatitis B and C antibody serologies were negative. Her medications at the time included risedronate and atenolol, which she had been taking for several years.
Punch biopsies from perilesional skin were submitted for hematoxylin and eosin staining and direct immunofluorescence (DIF). Histopathology showed a subepidermal blistering disease with tissue eosinophilia consistent with lichen planus pemphigoides (LPP)(Figure 2); direct immunofluorescence was positive for IgG, C3, and type IV collagen at the dermoepidermal junction. Serum BP180 was positive at 51 U/mL (reference range, <14 U/mL) and BP230 was negative. She was then started on tetracycline (500 mg twice daily), nicotinamide (500 mg twice daily), prednisone (5 mg daily), and dapsone (100 mg daily).
After 3 months without improvement, tetracycline and nicotinamide were discontinued, prednisone was increased to 10 mg daily, and dapsone was continued. A repeat biopsy was taken from a new area of involvement on the left lower leg, which revealed a psoriasiform dermatitis with interface changes. The DIF was positive for IgG and C3 along the basement membrane. A serum indirect immunofluorescence for BP180 also was positive.
The patient developed mild hemolytic anemia on dapsone; the medication was eventually discontinued. Subsequent treatments included adequate trials of azathioprine, mycophenolate mofetil, and hydroxychloroquine. Azathioprine (150 mg daily) and hydroxychloroquine (400 mg daily) treatment failed. She initially improved on mycophenolate mofetil (500 mg in the morning and 1000 mg in the evening) with flattening of the papules on the arms and legs and decreased erythema. However, mycophenolate mofetil eventually lost its efficacy and was discontinued.
Because several medications failed (ie, tetracycline, nicotinamide, prednisone, dapsone, azathioprine, mycophenolate mofetil, hydroxychloroquine), she was started on ustekinumab (45 mg) initial loading dose by subcutaneous injection (patient’s weight, 63 kg). At 4 weeks, the patient was given the second subcutaneous injection of ustekinumab (45 mg). She experienced marked improvement with no new lesions. The prior lesions also had decreased in size and were only slightly pink. The prednisone dose was tapered to 5 mg daily.
She had near-complete resolution of the skin lesions 12 weeks after the second dose of ustekinumab. Since then, she has had some recrudescence of the papulosquamous lesions but no vesicles or bullae. With the exception of occasional scattered pink papules on the forearms, her condition greatly improved on ustekinumab. She is no longer taking any of the other medications with the exception of prednisone (down to 1 mg daily) with a plan to gradually taper completely off of it.
Comment
Clinical Presentation
Lichen planus pemphigoides is a rare autoimmune subepidermal blistering disease with few cases reported in the literature. It is considered a clinical variation of bullous pemphigoid (BP) or a coexistence of LP and BP.1,2 It is characterized by bullous lesions developing on LP papules as well as on clinically uninvolved areas of the skin. It has been reported that LPP is provoked by several medications including cinnarizine, captopril, ramipril, simvastatin, psoralen plus UVA, and antituberculous medications (eg, isoniazid, rifampin, ethambutol, pyrazinamide).1 Risedronate or atenolol have not been reported to cause LPP, LP, or BP; however, according to Litt,3 a lichenoid drug eruption has been associated with atenolol. Furthermore, some cases of LPP demonstrate overlapping characteristics with paraneoplastic pemphigus and have been associated with internal malignancy. Hamada et al4 described a case of LPP coupled with colon adenocarcinoma and numerous keratoacanthomas. The earliest depiction of the coexistence of a case of mainstream LP complicated by an extensive bullous eruption was by Kaposi5 in 1892. He coined the term lichen ruber pemphigoides.5
Compared to BP, LPP is believed to affect a younger age group and have a less serious clinical course. The mean age of onset of LPP is in the third to fourth decades of life, while BP typically presents in the sixth decade. When comparing the location of bullae in LPP versus BP, the lesions of LPP tend to occur on the limbs, while BP tends to occur on the trunk.6
Clinically, LPP is distinguished by the existence of bullous lesions developing atop of the lesions of LP as well as on normal skin, with the latter being more commonplace. A classic example of LPP is characterized by an initial episode of traditional LP lesions often having severe pruritus, with or without patches of erythema, with the sudden eruption of tense bullae. These bullae commonly appear on the extremities and can appear over the normal skin, erythematous patches, or preexisting papules.7 In the atypical clinical presentations of this dubious skin condition, the bullae may only be seen on the lesions of LP.8 There also could be a lichenoid erythrodermic manifestation of a bullous eruption.9
Oral lesions of LPP have been described but had not been studied immunopathologically until Allen et al10 portrayed a 59-year-old man with cutaneous and oral lesions of LPP. They performed biopsies on the oral lesions and examined them by routine light microscopy and immunofluorescent techniques. The fine keratotic striae on the anterior buccal mucosal lesions were clinically consistent with oral LP. Perilesional tissue in conjunction with ulceration of the posterior buccal mucosa demonstrated histologic and immunopathologic alterations consistent with BP.10
Histopathology
Histopathologically, the lesions of LP show a bandlike lymphohistiocytic infiltrate, colloid bodies in the dermis, irregular acanthosis with saw-toothed rete ridges, orthokeratosis, wedge-shaped hypergranulosis, and liquefaction degeneration of the basal layer. Direct immunofluorescence shows mainly IgM and C3 deposited on colloid bodies, fibrin, and fibrinogen.11 The histopathology of the bullous lesion of LPP depicts a subepidermal bulla with variable diffuse or sparse lymphohistiocytic infiltrate and frequent eosinophils with or without neutrophils in the upper dermis. The existence of C3 alone or with IgG along the dermoepidermal junction gives confirmation on DIF.7
Autoantibodies
The expression of IgG autoantibodies directed against the basement membrane zone distinguishes LPP from bullous LP.2 IgG autoantibodies to either one or both the 230-kDa and 180-kDa BP (type XVII collagen) antigens has been demonstrated with LPP.4,12-14 Hamada et al4 described a histologic pattern more consistent with paraneoplastic pemphigus. It has been suggested that injury to the basal cells in LP or damage due to other courses of therapy such as psoralen plus UVA unveil suppressed antigenic determinants or produce new antigens, leading to antibody development and production of BP.12,15
Zillikens et al2 performed a study to identify the target antigen of LPP autoantibodies. They used sera from patients with LPP (n=4) and stained the epidermal side of salt-split human skin in a configuration identical to BP sera. In BP, the autoimmune response is directed against BP180, a hemidesmosomal transmembrane collagenous glycoprotein. They demonstrated that sera from BP patients largely reacted with a set of 4 epitopes (MCW-0 through MCW-3) grouped within a 45 amino acid stretch of the major noncollagenous extracellular domain (NC16A) of BP180. By immunoblotting and enzyme-linked immunosorbent assay, LPP sera also were compellingly reactive with recombinant BP180 NC16A. Lichen planus pemphigoides epitopes were additionally mapped using a series of overlapping recombinant segments of the NC16A domain. The authors demonstrated that all LPP sera reacted with amino acids 46 through 59 of domain NC16A, a protein portion that was previously shown to be unreactive with BP sera. In addition, they showed that 2 LPP sera reacted with the immunodominant antigenic region related to BP. Furthermore, they identified a unique epitope within the BP180 NC16A domain—MCW-4—which was distinctively recognized by sera from patients with LPP.2
Pathogenesis
The pathogenesis of both LP and BP has been linked to multiple cytokines that induce apoptosis in basal keratinocytes. Implicated cytokines include IFN-γ, tumor necrosis factor α (TNF-α), IL-1, IL-6, and IL-8, as well as other apoptosis-related molecules, such as Fas/Apo-1 and Bcl-2 in LP.16-18 Soluble E-selectin, vascular endothelial growth factor, IL-1β, IL-8, IL-5, transforming growth factor β1, and TNF-α were found to be elevated in either blister fluid or sera of BP patients.15-17
Management
Lichen planus pemphigoides usually responds well to traditional therapies, with systemic steroids being the most efficacious treatment of extensive disease.12,13 Other options include tetracycline and nicotinamide, isotretinoin, dapsone, and immunosuppressive drugs such as systemic cortico-steroids.12 Demirçay et al12 described a patient with skin lesions that rapidly cleared after the administration of oral methylprednisolone (48 mg/d) and oral dapsone (100 mg/d). The methylprednisolone and dapsone were withdrawn after 12 and 16 weeks, respectively. There was no recurrence during the 1-year follow-up period.12 et al19 described a patient who was treated with pulsed intravenous corticosteroids and continued to develop new papular and vesicular skin lesions. However, when oral acitretin was added to the patient’s regimen, the skin lesions cleared.19 There are several case reports of the successful use of hydroxychloroquine in LP.20,21
Cutaneous, nail, and oral LP also can be treated with TNF-α inhibitors (eg, adalimumab, etanercept) with resolution of lesions.22-25 However, we have not been able to find any reports of treating LPP with biologic medications in a search of PubMed articles indexed for MEDLINE using the terms lichen planus pemphigoides and biologic treatments/therapies. Given the fact that TNF-α and other inflammatory cytokines are involved in the pathogenesis of BP and LP, it is feasible that they also may be involved in the pathogenesis of LPP.
In our patient with cutaneous LPP, we chose to use ustekinumab instead of a primary TNF-α inhibitor because ustekinumab indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22, which also could have played a role in the patient’s disease. Our goal was to use ustekinumab as a potential corticosteroid-sparing agent. Ustekinumab greatly improved her skin condition and allowed us to discontinue other medications.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Litt J. Litt’s Drug Eruptions and Reactions Manual. 18th Ed. London, England: Informa Healthcare; 2011.
- Hamada T, Fujimoto W, Okazaki F, et al. Lichen planus pemphigoides and multiple keratoacanthomas associated with colon adenocarcinoma. Br J Dermatol. 2004;151:252-254.
- Kaposi M. Lichen ruber pemphigoides. Arch Derm Syph. 1892;343-346.
- Swale VJ, Black MM, Bhogal BS. Lichen planus pemphigoides: two case reports. Clin Exp Dermatol. 1998;23:132-135.
- Okochi H, Nashiro K, Tsuchida T, et al. Lichen planus pemphigoides: case reports and results of immunofluorescence and immunoelectron microscopic study. J Am Acad Dermatol. 1990;22:626-631.
- Mendiratta V, Asati DP, Koranne RV. Lichen planus pemphigoides in an Indian female. Indian J Dermatol. 2005;50:224-226.
- Joly P, Tanasescu S, Wolkenstein P, et al. Lichenoid erythrodermic bullous pemphigoid of the African patient. J Am Acad Dermatol. 1998;39:691-697.
- Allen , , R. Lichen planus pemphigoides: report of a case with oral lesions. Oral Surg Oral Med Oral Pathol. 1987;63:184-188.
- Rapini RP. Practical Dermatopathology. Philadelphia, PA: Mosby Elsevier; 2005.
- Demirçay Z, Baykal C, Demirkesen C. Lichen planus pemphigoides: report of two cases. Int J Dermatol. 2001;40:757-759.
- Sakuma-Oyama Y, Powell AM, Albert S, et al. Lichen planus pemphigoides evolving into pemphigoid nodularis. Clin Exp Dermatol. 2004;28:613-616.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Ameglio F, D’Auria L, Cordiali-Fei P, et al. Bullous pemphigoid and pemphigus vulgaris: correlated behaviour of serum VEGF, sE-selectin and TNF-alpha levels. J Biol Regul Homeost Agents. 1997;11:148-153.
- Ameglio F, D’auria L, Bonifati C, et al. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. 1998;138:611-614.
- D’Auria L, Mussi A, Bonifati C, et al. Increased serum IL-6, TNF-alpha and IL-10 levels in patients with bullous pemphigoid: relationships with disease activity. J Eur Acad Dermatol Venereol. 1999;12:11-15.
- , ,, . Treatment of lichen planus pemphigoides with acitretin and pulsed corticosteroids. Hautarzt. 2003;54:268-273.
- Eisen D. Hydroxychloroquine sulfate (Plaquenil) improves oral lichen planus: an open trial. J Am Acad Dermatol. 1993;28:609-612.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 11th ed. Philadelphia, PA: Mosby Elsevier; 2011.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Yarom N. Etanercept for the management of oral lichen planus. Am J Clin Dermatol. 2007;8:121.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Irla N, Schneiter T, Haneke E, et al. Nail lichen planus: successful treatment with etanercept. Case Rep Dermatol. 2010;2:173-176.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Litt J. Litt’s Drug Eruptions and Reactions Manual. 18th Ed. London, England: Informa Healthcare; 2011.
- Hamada T, Fujimoto W, Okazaki F, et al. Lichen planus pemphigoides and multiple keratoacanthomas associated with colon adenocarcinoma. Br J Dermatol. 2004;151:252-254.
- Kaposi M. Lichen ruber pemphigoides. Arch Derm Syph. 1892;343-346.
- Swale VJ, Black MM, Bhogal BS. Lichen planus pemphigoides: two case reports. Clin Exp Dermatol. 1998;23:132-135.
- Okochi H, Nashiro K, Tsuchida T, et al. Lichen planus pemphigoides: case reports and results of immunofluorescence and immunoelectron microscopic study. J Am Acad Dermatol. 1990;22:626-631.
- Mendiratta V, Asati DP, Koranne RV. Lichen planus pemphigoides in an Indian female. Indian J Dermatol. 2005;50:224-226.
- Joly P, Tanasescu S, Wolkenstein P, et al. Lichenoid erythrodermic bullous pemphigoid of the African patient. J Am Acad Dermatol. 1998;39:691-697.
- Allen , , R. Lichen planus pemphigoides: report of a case with oral lesions. Oral Surg Oral Med Oral Pathol. 1987;63:184-188.
- Rapini RP. Practical Dermatopathology. Philadelphia, PA: Mosby Elsevier; 2005.
- Demirçay Z, Baykal C, Demirkesen C. Lichen planus pemphigoides: report of two cases. Int J Dermatol. 2001;40:757-759.
- Sakuma-Oyama Y, Powell AM, Albert S, et al. Lichen planus pemphigoides evolving into pemphigoid nodularis. Clin Exp Dermatol. 2004;28:613-616.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Ameglio F, D’Auria L, Cordiali-Fei P, et al. Bullous pemphigoid and pemphigus vulgaris: correlated behaviour of serum VEGF, sE-selectin and TNF-alpha levels. J Biol Regul Homeost Agents. 1997;11:148-153.
- Ameglio F, D’auria L, Bonifati C, et al. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. 1998;138:611-614.
- D’Auria L, Mussi A, Bonifati C, et al. Increased serum IL-6, TNF-alpha and IL-10 levels in patients with bullous pemphigoid: relationships with disease activity. J Eur Acad Dermatol Venereol. 1999;12:11-15.
- , ,, . Treatment of lichen planus pemphigoides with acitretin and pulsed corticosteroids. Hautarzt. 2003;54:268-273.
- Eisen D. Hydroxychloroquine sulfate (Plaquenil) improves oral lichen planus: an open trial. J Am Acad Dermatol. 1993;28:609-612.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 11th ed. Philadelphia, PA: Mosby Elsevier; 2011.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Yarom N. Etanercept for the management of oral lichen planus. Am J Clin Dermatol. 2007;8:121.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Irla N, Schneiter T, Haneke E, et al. Nail lichen planus: successful treatment with etanercept. Case Rep Dermatol. 2010;2:173-176.
- Lichen planus pemphigoides (LPP) is a rare autoimmune subepidermal blistering disease with few cases reported in the literature.
- Because tumor necrosis factor 11α (TNF-11α) and other inflammatory cytokines are involved in the pathogenesis of bullous pemphigoid and lichen planus, it is feasible that they also may be involved in the pathogenesis of LPP.
- Ustekinumab may be used to treat LPP as a potential corticosteroid-sparing agent because it indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22.

Multinucleate Cell Angiohistiocytoma
Multinucleate cell angiohistiocytoma (MCAH) is a rare benign, soft-tissue tumor first described in 1985 by Smith and Jones1 that presents clinically as erythematous to violaceous papules most commonly affecting females on the dorsal aspect of the hands and face.2 Multinucleate cell angiohistiocytoma is histologically characterized by vascular and histiocytic proliferations with dermal fibrosis. Few cases have been reported of lesions affecting the lower extremities. We report a case of MCAH affecting the legs.
Case Report
An 83-year-old white man with a history of basal cell carcinoma presented for evaluation of grouped, well-circumscribed, soft, red-violet, painless papules on the right anterior thigh that had been present for 8 months (Figure 1A). A review of symptoms was negative for immunologic, respiratory, and hematologic changes. The patient’s medical history also was remarkable for prostate cancer treated with radiation 18 years prior as well as right hip and left knee implants. The initial clinical impression was Kaposi sarcoma or a granulomatous disorder.
Histopathologic evaluation of a deep shave biopsy initially determined the lesion to be scar tissue without other pathologic findings. The patient returned to the clinic 12 months later for a complete skin examination given his history of skin cancer. Compared to clinical photographs taken a year prior, new violaceous papules were noted on the right thigh (Figure 1B) and left calf. Furthermore, there was no recurrence of the lesion at the prior biopsy site. Shave biopsies of the papules on the right thigh and left calf demonstrated similar histologic findings to each other. There was a mild increase in the number of small blood vessels in the superficial dermis (Figure 2A). A mild perivascular lymphocytic infiltrate surrounded some of these blood vessels. The endothelial cells had small nuclei with no evidence of nuclear pleomorphism. Careful examination of the interstitial dermis revealed scattered multinucleate cells with angulated cytoplasm (Figure 2B). Immunostaining for CD31 and human herpesvirus 8 were negative, excluding an infiltrative vascular tumor and Kaposi sarcoma, respectively. The diagnosis of MCAH was made based on the histopathologic findings.
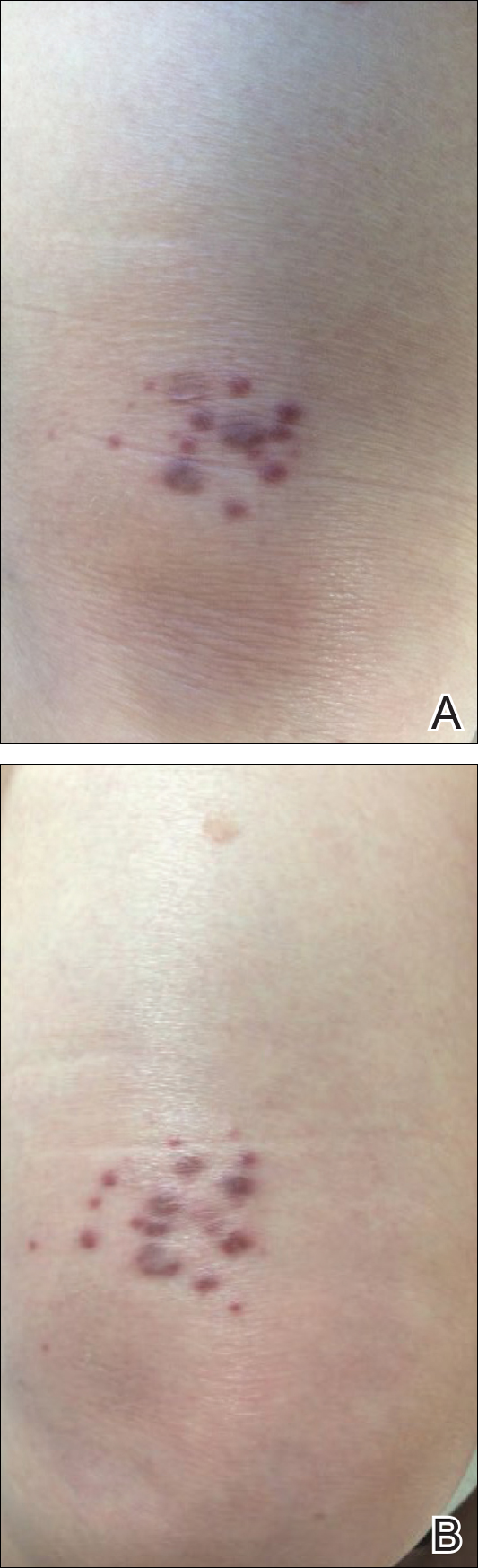
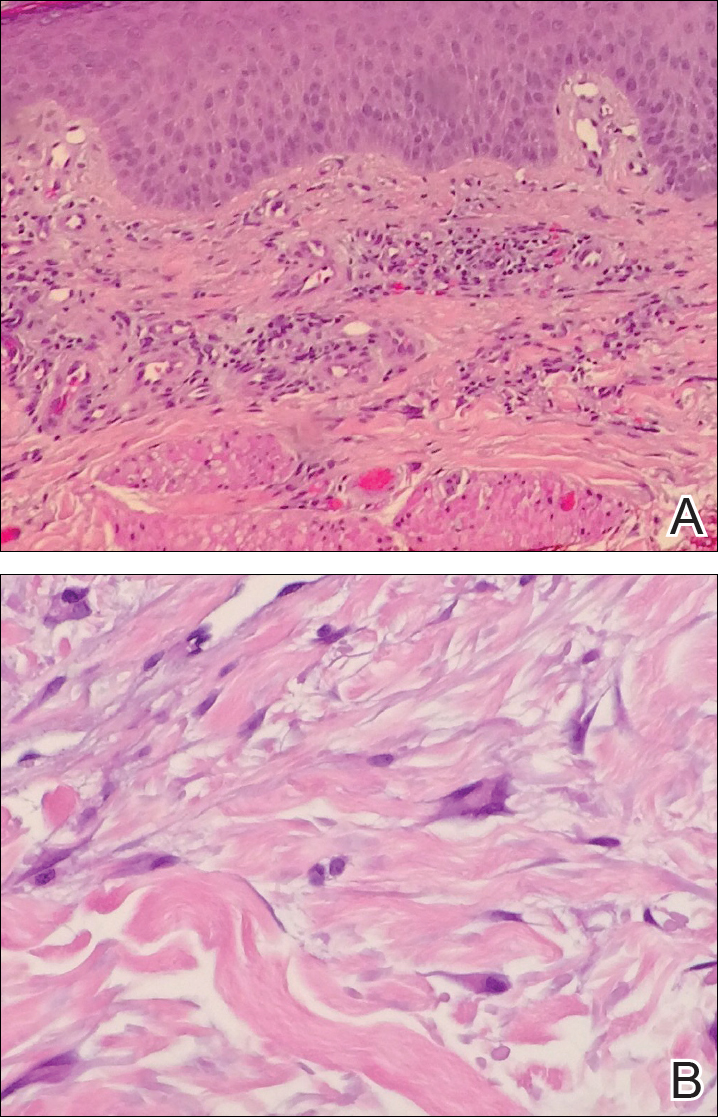
At 1-year follow-up, the condition was stable with no gross changes in the lesions based on prior photographs. Once again, there was no recurrence of the excised lesions at both biopsy sites.
Comment
Presentation
A systematic review of published reports determined that 79% of MCAH cases occur in females, with an average age of onset of 50.1 years.2 However, MCAH likely is underreported due to the overall lack of knowledge regarding this condition by physicians and pathologists. The hands and face are the most commonly affected areas, though other sites of involvement have been reported, including the lower extremities,3,4 oral mucosa and upper lip,5,6 and trunk,7,8 as well as generalized distribution.9-12 Additionally, 1 case presented as a single plaque on the trunk rather than having papular or nodular morphology.8 Multinucleate cell angiohistiocytoma lesions generally are asymptomatic, though pruritus may be present.13 The condition is regarded as benign, though a minority of cases have exhibited spontaneous resolution.14-16
Histopathology
Multinucleate cell angiohistiocytoma histology demonstrates full-thickness dermal microvessel proliferation and fibrosis with characteristic multinucleate giant cells.2,3 Vascular endothelial cells stained positive for CD68 in 60% of cases2 as well as the normal endothelial markers (ie, factor VIII, CD31, CD34). The multinucleate giant cells exhibit immunoreactivity for macrophage/histiocytic markers factor XIIIa and CD68.
Etiology
The pathogenesis of MCAH is not yet fully understood, but it is considered to be a benign vascular or fibrohistiocytic neoplasm.17 Calderaro et al18 described a series of 8 patients who developed MCAH either within a cutaneous neoplastic process or in conjunction with various cutaneous reactive conditions, including hidradenitis suppurativa and chronic radiation dermatitis, as well as overlying a bone prosthesis placed due to degenerative arthritis. These cases suggest that MCAH, or possibly a subset of the disease, is a reactive process.
Differential Diagnosis
The differential diagnosis for MCAH includes Kaposi sarcoma clinically and dermatofibroma and fibrous papules histologically. Sass et al21 determined the in vitro behavior of cultured MCAH cells to contrast markedly with Kaposi sarcoma–derived cells. Although Kaposi sarcoma–derived cells exhibited invasive behavior, cells isolated from MCAH lesions were less elongated and were unable to traverse basement membranes.
Treatment
Surgical excision or cryotherapy appear to be definitive treatments of MCAH; however, a number of cases have reported light and laser modalities as successful alternatives to excision. One case of MCAH affecting the face was treated with pulsed dye laser monotherapy.22 This modality allowed selective coagulation of the vascular structures in MCAH. At 8-month follow-up, the initial lesion was noted to be completely cleared, though similar lesions had recently appeared elsewhere on the face.22 Another case of MCAH affecting the leg was treated with pulsed dye laser and both topical and intralesional corticosteroid combination therapy. In this case, the lesion failed to respond to treatment, which may suggest that facial localization could influence response in pulsed dye laser treatment.3
Intense pulsed light also has been reported as a definitive treatment in 2 cases.2,13 Slight erythema and transient pruritus have been reported immediately following treatment. In this case, complete resolution with only residual hyperpigmentation was reported at 2-month follow-up, with no recurrence during 12 months of follow-up.13
Argon laser therapy has been used in 2 cases. After a single session, lesions were no longer palpable, with no scarring noted at 8 weeks follow-up.23 Lastly, 2 cases of MCAH have been successfully treated with the CO2 laser, with no relapse noted at 2.5- or 5-month follow-up, respectively.24
Conclusion
Multinucleate cell angiohistiocytoma is a rare and likely underdiagnosed dermatologic condition that is believed to be a reactive process. Characteristic histology of MCAH demonstrates microvascular proliferations of the dermis with multinucleate giant cells amidst a fibrous background. Although surgical excision is curative, there are reports in which laser and light therapies were used to effectively treat MCAH.
- Smith NP, Jones EW. Multinucleate cell angiohistiocytoma—a new entity. Br J Dermatol. 1985;113:15.
- Frew JW. Multinucleate cell angiohistiocytoma: clinicopathological correlation of 142 cases with insights into etiology and pathogenesis. Am J Dermatopathol. 2015;37:222-228.
- Applebaum DS, Shuja F, Hicks L, et al. Multinucleate cell angiohistiocytoma: a case report and review of the literature. Dermatol Online J. 2014;20:22610.
- Sagdeo A, Chu EY, Elenitsas R, et al. Multiple asymptomatic violaceous macules on the thigh. Multinucleate cell angiohistiocytoma (MCAH). JAMA Dermatol. 2013;149:357-363.
- Rawal YB, Anderson KM, Rawal SY. Multinucleate cell angiohistiocytoma: an uncommon mucosal tumour. Clin Exp Dermatol. 2009;34:333-336.
- Jones AC, Mullins D, Jimenez F. Multinucleate cell angiohistiocytoma of the upper lip. Oral Surg Oral Med Oral Pathol. 1994;78:743-747.
- Doshi-Chougule BN, Gust A, Mentzel T, et al. Multinucleate cell angiohistiocytoma with hypertrophic nerves. J Cutan Pathol. 2013;40:1048-1053.
- Issa AA, Lui H, Shapiro J, et al. Plaque-type multinucleate cell angiohistiocytoma. J Cutan Med Surg. 1998;3:112-114.
- Doane JA, Purdy K, Pasternak S. Generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2015;19:323-325.
- Marti N, Monteagudo C, Revert A, et al. Multiple papules on the trunk and extremities. generalized multinucleate cell angiohistiocytoma. Int J Dermatol. 2013;52:544-546.
- O’Blenes CA, Walsh NM, Green PJ, et al. Novel case of generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2010;14:178-180.
- Chang SN, Kim HS, Kim SC, et al. Generalized multinucleate cell angiohistiocytoma. J Am Acad Dermatol. 1996;35:320-322.
- Fernández-Jorge B, Del Pozo J, García-Silva J, et al. Multinucleate cell angiohistiocytoma: treatment using intense pulsed light. Dermatol Surg. 2009;35:1141-1143.
- Perez LP, Zulaica A, Rodriguez L, et al. Multinucleate cell angiohistiocytoma. report of five cases. J Cutan Pathol. 2006;33:349-352.
- Shapiro PE, Nova MP, Rosmarin LA, et al. Multinucleate cell angiohistiocytoma: a distinct entity diagnosable by clinical and histologic features. J Am Acad Dermatol. 1994;30:417-422.
- Jaconelli L, Kanitakis J, Ktiouet S, et al. Multinucleate cell angiohistiocytoma: report of three new cases and literature review. Dermatol Online J. 2009;15:4.
- Jones WE, Cerio R, Smith NP. Multinucleate cell angiohistiocytoma: an acquired vascular anomaly to be distinguished from Kaposi’s sarcoma. Br J Dermatol. 1990;122:651-663.
- Calderaro J, Rethers L, Ortonne N. Multinucleated cells angiohistiocytoma: a reactive lesion? Am J Dermatopathol. 2010;32:415-417.
- Cesinaro AM, Roncati L, Maiorana A. Estrogen receptor alpha overexpression in multinucleate cell angiohistiocytoma: new insights into the pathogenesis of a reactive process. Am J Dermatopathol. 2010;32:655-659.
- Losordo DW, Isner JM. Estrogen and angiogenesis: a review. Arterioscler Thromb Vasc Biol. 2001;21:6-12.
- Sass U, Noel JC, Andre J, et al. Multinucleate cell angiohistiocytoma: report of two cases with no evidence of human herpesvirus-8 infection. J Cutan Pathol. 2000;27:258-261.
- Richer V, Lui H. Facial multinucleate cell angiohistiocytoma: long-term remission with 585 nm pulsed dye laser. Clin Exp Dermatol. 2016;41:312-313.
- Kopera D, Smolle J, Kerl H. Multinucleate cell angiohistiocytoma: treatment with argon laser. Br J Dermatol. 1995;133:308-310.
- Väkevä L, Saksela O, Kariniemi AL. Multinucleate cell angiohistiocytoma: a report of four cases in Finland. Acta Derm Venereol. 2003;83:222-223.
Multinucleate cell angiohistiocytoma (MCAH) is a rare benign, soft-tissue tumor first described in 1985 by Smith and Jones1 that presents clinically as erythematous to violaceous papules most commonly affecting females on the dorsal aspect of the hands and face.2 Multinucleate cell angiohistiocytoma is histologically characterized by vascular and histiocytic proliferations with dermal fibrosis. Few cases have been reported of lesions affecting the lower extremities. We report a case of MCAH affecting the legs.
Case Report
An 83-year-old white man with a history of basal cell carcinoma presented for evaluation of grouped, well-circumscribed, soft, red-violet, painless papules on the right anterior thigh that had been present for 8 months (Figure 1A). A review of symptoms was negative for immunologic, respiratory, and hematologic changes. The patient’s medical history also was remarkable for prostate cancer treated with radiation 18 years prior as well as right hip and left knee implants. The initial clinical impression was Kaposi sarcoma or a granulomatous disorder.
Histopathologic evaluation of a deep shave biopsy initially determined the lesion to be scar tissue without other pathologic findings. The patient returned to the clinic 12 months later for a complete skin examination given his history of skin cancer. Compared to clinical photographs taken a year prior, new violaceous papules were noted on the right thigh (Figure 1B) and left calf. Furthermore, there was no recurrence of the lesion at the prior biopsy site. Shave biopsies of the papules on the right thigh and left calf demonstrated similar histologic findings to each other. There was a mild increase in the number of small blood vessels in the superficial dermis (Figure 2A). A mild perivascular lymphocytic infiltrate surrounded some of these blood vessels. The endothelial cells had small nuclei with no evidence of nuclear pleomorphism. Careful examination of the interstitial dermis revealed scattered multinucleate cells with angulated cytoplasm (Figure 2B). Immunostaining for CD31 and human herpesvirus 8 were negative, excluding an infiltrative vascular tumor and Kaposi sarcoma, respectively. The diagnosis of MCAH was made based on the histopathologic findings.
At 1-year follow-up, the condition was stable with no gross changes in the lesions based on prior photographs. Once again, there was no recurrence of the excised lesions at both biopsy sites.
Comment
Presentation
A systematic review of published reports determined that 79% of MCAH cases occur in females, with an average age of onset of 50.1 years.2 However, MCAH likely is underreported due to the overall lack of knowledge regarding this condition by physicians and pathologists. The hands and face are the most commonly affected areas, though other sites of involvement have been reported, including the lower extremities,3,4 oral mucosa and upper lip,5,6 and trunk,7,8 as well as generalized distribution.9-12 Additionally, 1 case presented as a single plaque on the trunk rather than having papular or nodular morphology.8 Multinucleate cell angiohistiocytoma lesions generally are asymptomatic, though pruritus may be present.13 The condition is regarded as benign, though a minority of cases have exhibited spontaneous resolution.14-16
Histopathology
Multinucleate cell angiohistiocytoma histology demonstrates full-thickness dermal microvessel proliferation and fibrosis with characteristic multinucleate giant cells.2,3 Vascular endothelial cells stained positive for CD68 in 60% of cases2 as well as the normal endothelial markers (ie, factor VIII, CD31, CD34). The multinucleate giant cells exhibit immunoreactivity for macrophage/histiocytic markers factor XIIIa and CD68.
Etiology
The pathogenesis of MCAH is not yet fully understood, but it is considered to be a benign vascular or fibrohistiocytic neoplasm.17 Calderaro et al18 described a series of 8 patients who developed MCAH either within a cutaneous neoplastic process or in conjunction with various cutaneous reactive conditions, including hidradenitis suppurativa and chronic radiation dermatitis, as well as overlying a bone prosthesis placed due to degenerative arthritis. These cases suggest that MCAH, or possibly a subset of the disease, is a reactive process.
Differential Diagnosis
The differential diagnosis for MCAH includes Kaposi sarcoma clinically and dermatofibroma and fibrous papules histologically. Sass et al21 determined the in vitro behavior of cultured MCAH cells to contrast markedly with Kaposi sarcoma–derived cells. Although Kaposi sarcoma–derived cells exhibited invasive behavior, cells isolated from MCAH lesions were less elongated and were unable to traverse basement membranes.
Treatment
Surgical excision or cryotherapy appear to be definitive treatments of MCAH; however, a number of cases have reported light and laser modalities as successful alternatives to excision. One case of MCAH affecting the face was treated with pulsed dye laser monotherapy.22 This modality allowed selective coagulation of the vascular structures in MCAH. At 8-month follow-up, the initial lesion was noted to be completely cleared, though similar lesions had recently appeared elsewhere on the face.22 Another case of MCAH affecting the leg was treated with pulsed dye laser and both topical and intralesional corticosteroid combination therapy. In this case, the lesion failed to respond to treatment, which may suggest that facial localization could influence response in pulsed dye laser treatment.3
Intense pulsed light also has been reported as a definitive treatment in 2 cases.2,13 Slight erythema and transient pruritus have been reported immediately following treatment. In this case, complete resolution with only residual hyperpigmentation was reported at 2-month follow-up, with no recurrence during 12 months of follow-up.13
Argon laser therapy has been used in 2 cases. After a single session, lesions were no longer palpable, with no scarring noted at 8 weeks follow-up.23 Lastly, 2 cases of MCAH have been successfully treated with the CO2 laser, with no relapse noted at 2.5- or 5-month follow-up, respectively.24
Conclusion
Multinucleate cell angiohistiocytoma is a rare and likely underdiagnosed dermatologic condition that is believed to be a reactive process. Characteristic histology of MCAH demonstrates microvascular proliferations of the dermis with multinucleate giant cells amidst a fibrous background. Although surgical excision is curative, there are reports in which laser and light therapies were used to effectively treat MCAH.
Multinucleate cell angiohistiocytoma (MCAH) is a rare benign, soft-tissue tumor first described in 1985 by Smith and Jones1 that presents clinically as erythematous to violaceous papules most commonly affecting females on the dorsal aspect of the hands and face.2 Multinucleate cell angiohistiocytoma is histologically characterized by vascular and histiocytic proliferations with dermal fibrosis. Few cases have been reported of lesions affecting the lower extremities. We report a case of MCAH affecting the legs.
Case Report
An 83-year-old white man with a history of basal cell carcinoma presented for evaluation of grouped, well-circumscribed, soft, red-violet, painless papules on the right anterior thigh that had been present for 8 months (Figure 1A). A review of symptoms was negative for immunologic, respiratory, and hematologic changes. The patient’s medical history also was remarkable for prostate cancer treated with radiation 18 years prior as well as right hip and left knee implants. The initial clinical impression was Kaposi sarcoma or a granulomatous disorder.
Histopathologic evaluation of a deep shave biopsy initially determined the lesion to be scar tissue without other pathologic findings. The patient returned to the clinic 12 months later for a complete skin examination given his history of skin cancer. Compared to clinical photographs taken a year prior, new violaceous papules were noted on the right thigh (Figure 1B) and left calf. Furthermore, there was no recurrence of the lesion at the prior biopsy site. Shave biopsies of the papules on the right thigh and left calf demonstrated similar histologic findings to each other. There was a mild increase in the number of small blood vessels in the superficial dermis (Figure 2A). A mild perivascular lymphocytic infiltrate surrounded some of these blood vessels. The endothelial cells had small nuclei with no evidence of nuclear pleomorphism. Careful examination of the interstitial dermis revealed scattered multinucleate cells with angulated cytoplasm (Figure 2B). Immunostaining for CD31 and human herpesvirus 8 were negative, excluding an infiltrative vascular tumor and Kaposi sarcoma, respectively. The diagnosis of MCAH was made based on the histopathologic findings.
At 1-year follow-up, the condition was stable with no gross changes in the lesions based on prior photographs. Once again, there was no recurrence of the excised lesions at both biopsy sites.
Comment
Presentation
A systematic review of published reports determined that 79% of MCAH cases occur in females, with an average age of onset of 50.1 years.2 However, MCAH likely is underreported due to the overall lack of knowledge regarding this condition by physicians and pathologists. The hands and face are the most commonly affected areas, though other sites of involvement have been reported, including the lower extremities,3,4 oral mucosa and upper lip,5,6 and trunk,7,8 as well as generalized distribution.9-12 Additionally, 1 case presented as a single plaque on the trunk rather than having papular or nodular morphology.8 Multinucleate cell angiohistiocytoma lesions generally are asymptomatic, though pruritus may be present.13 The condition is regarded as benign, though a minority of cases have exhibited spontaneous resolution.14-16
Histopathology
Multinucleate cell angiohistiocytoma histology demonstrates full-thickness dermal microvessel proliferation and fibrosis with characteristic multinucleate giant cells.2,3 Vascular endothelial cells stained positive for CD68 in 60% of cases2 as well as the normal endothelial markers (ie, factor VIII, CD31, CD34). The multinucleate giant cells exhibit immunoreactivity for macrophage/histiocytic markers factor XIIIa and CD68.
Etiology
The pathogenesis of MCAH is not yet fully understood, but it is considered to be a benign vascular or fibrohistiocytic neoplasm.17 Calderaro et al18 described a series of 8 patients who developed MCAH either within a cutaneous neoplastic process or in conjunction with various cutaneous reactive conditions, including hidradenitis suppurativa and chronic radiation dermatitis, as well as overlying a bone prosthesis placed due to degenerative arthritis. These cases suggest that MCAH, or possibly a subset of the disease, is a reactive process.
Differential Diagnosis
The differential diagnosis for MCAH includes Kaposi sarcoma clinically and dermatofibroma and fibrous papules histologically. Sass et al21 determined the in vitro behavior of cultured MCAH cells to contrast markedly with Kaposi sarcoma–derived cells. Although Kaposi sarcoma–derived cells exhibited invasive behavior, cells isolated from MCAH lesions were less elongated and were unable to traverse basement membranes.
Treatment
Surgical excision or cryotherapy appear to be definitive treatments of MCAH; however, a number of cases have reported light and laser modalities as successful alternatives to excision. One case of MCAH affecting the face was treated with pulsed dye laser monotherapy.22 This modality allowed selective coagulation of the vascular structures in MCAH. At 8-month follow-up, the initial lesion was noted to be completely cleared, though similar lesions had recently appeared elsewhere on the face.22 Another case of MCAH affecting the leg was treated with pulsed dye laser and both topical and intralesional corticosteroid combination therapy. In this case, the lesion failed to respond to treatment, which may suggest that facial localization could influence response in pulsed dye laser treatment.3
Intense pulsed light also has been reported as a definitive treatment in 2 cases.2,13 Slight erythema and transient pruritus have been reported immediately following treatment. In this case, complete resolution with only residual hyperpigmentation was reported at 2-month follow-up, with no recurrence during 12 months of follow-up.13
Argon laser therapy has been used in 2 cases. After a single session, lesions were no longer palpable, with no scarring noted at 8 weeks follow-up.23 Lastly, 2 cases of MCAH have been successfully treated with the CO2 laser, with no relapse noted at 2.5- or 5-month follow-up, respectively.24
Conclusion
Multinucleate cell angiohistiocytoma is a rare and likely underdiagnosed dermatologic condition that is believed to be a reactive process. Characteristic histology of MCAH demonstrates microvascular proliferations of the dermis with multinucleate giant cells amidst a fibrous background. Although surgical excision is curative, there are reports in which laser and light therapies were used to effectively treat MCAH.
- Smith NP, Jones EW. Multinucleate cell angiohistiocytoma—a new entity. Br J Dermatol. 1985;113:15.
- Frew JW. Multinucleate cell angiohistiocytoma: clinicopathological correlation of 142 cases with insights into etiology and pathogenesis. Am J Dermatopathol. 2015;37:222-228.
- Applebaum DS, Shuja F, Hicks L, et al. Multinucleate cell angiohistiocytoma: a case report and review of the literature. Dermatol Online J. 2014;20:22610.
- Sagdeo A, Chu EY, Elenitsas R, et al. Multiple asymptomatic violaceous macules on the thigh. Multinucleate cell angiohistiocytoma (MCAH). JAMA Dermatol. 2013;149:357-363.
- Rawal YB, Anderson KM, Rawal SY. Multinucleate cell angiohistiocytoma: an uncommon mucosal tumour. Clin Exp Dermatol. 2009;34:333-336.
- Jones AC, Mullins D, Jimenez F. Multinucleate cell angiohistiocytoma of the upper lip. Oral Surg Oral Med Oral Pathol. 1994;78:743-747.
- Doshi-Chougule BN, Gust A, Mentzel T, et al. Multinucleate cell angiohistiocytoma with hypertrophic nerves. J Cutan Pathol. 2013;40:1048-1053.
- Issa AA, Lui H, Shapiro J, et al. Plaque-type multinucleate cell angiohistiocytoma. J Cutan Med Surg. 1998;3:112-114.
- Doane JA, Purdy K, Pasternak S. Generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2015;19:323-325.
- Marti N, Monteagudo C, Revert A, et al. Multiple papules on the trunk and extremities. generalized multinucleate cell angiohistiocytoma. Int J Dermatol. 2013;52:544-546.
- O’Blenes CA, Walsh NM, Green PJ, et al. Novel case of generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2010;14:178-180.
- Chang SN, Kim HS, Kim SC, et al. Generalized multinucleate cell angiohistiocytoma. J Am Acad Dermatol. 1996;35:320-322.
- Fernández-Jorge B, Del Pozo J, García-Silva J, et al. Multinucleate cell angiohistiocytoma: treatment using intense pulsed light. Dermatol Surg. 2009;35:1141-1143.
- Perez LP, Zulaica A, Rodriguez L, et al. Multinucleate cell angiohistiocytoma. report of five cases. J Cutan Pathol. 2006;33:349-352.
- Shapiro PE, Nova MP, Rosmarin LA, et al. Multinucleate cell angiohistiocytoma: a distinct entity diagnosable by clinical and histologic features. J Am Acad Dermatol. 1994;30:417-422.
- Jaconelli L, Kanitakis J, Ktiouet S, et al. Multinucleate cell angiohistiocytoma: report of three new cases and literature review. Dermatol Online J. 2009;15:4.
- Jones WE, Cerio R, Smith NP. Multinucleate cell angiohistiocytoma: an acquired vascular anomaly to be distinguished from Kaposi’s sarcoma. Br J Dermatol. 1990;122:651-663.
- Calderaro J, Rethers L, Ortonne N. Multinucleated cells angiohistiocytoma: a reactive lesion? Am J Dermatopathol. 2010;32:415-417.
- Cesinaro AM, Roncati L, Maiorana A. Estrogen receptor alpha overexpression in multinucleate cell angiohistiocytoma: new insights into the pathogenesis of a reactive process. Am J Dermatopathol. 2010;32:655-659.
- Losordo DW, Isner JM. Estrogen and angiogenesis: a review. Arterioscler Thromb Vasc Biol. 2001;21:6-12.
- Sass U, Noel JC, Andre J, et al. Multinucleate cell angiohistiocytoma: report of two cases with no evidence of human herpesvirus-8 infection. J Cutan Pathol. 2000;27:258-261.
- Richer V, Lui H. Facial multinucleate cell angiohistiocytoma: long-term remission with 585 nm pulsed dye laser. Clin Exp Dermatol. 2016;41:312-313.
- Kopera D, Smolle J, Kerl H. Multinucleate cell angiohistiocytoma: treatment with argon laser. Br J Dermatol. 1995;133:308-310.
- Väkevä L, Saksela O, Kariniemi AL. Multinucleate cell angiohistiocytoma: a report of four cases in Finland. Acta Derm Venereol. 2003;83:222-223.
- Smith NP, Jones EW. Multinucleate cell angiohistiocytoma—a new entity. Br J Dermatol. 1985;113:15.
- Frew JW. Multinucleate cell angiohistiocytoma: clinicopathological correlation of 142 cases with insights into etiology and pathogenesis. Am J Dermatopathol. 2015;37:222-228.
- Applebaum DS, Shuja F, Hicks L, et al. Multinucleate cell angiohistiocytoma: a case report and review of the literature. Dermatol Online J. 2014;20:22610.
- Sagdeo A, Chu EY, Elenitsas R, et al. Multiple asymptomatic violaceous macules on the thigh. Multinucleate cell angiohistiocytoma (MCAH). JAMA Dermatol. 2013;149:357-363.
- Rawal YB, Anderson KM, Rawal SY. Multinucleate cell angiohistiocytoma: an uncommon mucosal tumour. Clin Exp Dermatol. 2009;34:333-336.
- Jones AC, Mullins D, Jimenez F. Multinucleate cell angiohistiocytoma of the upper lip. Oral Surg Oral Med Oral Pathol. 1994;78:743-747.
- Doshi-Chougule BN, Gust A, Mentzel T, et al. Multinucleate cell angiohistiocytoma with hypertrophic nerves. J Cutan Pathol. 2013;40:1048-1053.
- Issa AA, Lui H, Shapiro J, et al. Plaque-type multinucleate cell angiohistiocytoma. J Cutan Med Surg. 1998;3:112-114.
- Doane JA, Purdy K, Pasternak S. Generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2015;19:323-325.
- Marti N, Monteagudo C, Revert A, et al. Multiple papules on the trunk and extremities. generalized multinucleate cell angiohistiocytoma. Int J Dermatol. 2013;52:544-546.
- O’Blenes CA, Walsh NM, Green PJ, et al. Novel case of generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2010;14:178-180.
- Chang SN, Kim HS, Kim SC, et al. Generalized multinucleate cell angiohistiocytoma. J Am Acad Dermatol. 1996;35:320-322.
- Fernández-Jorge B, Del Pozo J, García-Silva J, et al. Multinucleate cell angiohistiocytoma: treatment using intense pulsed light. Dermatol Surg. 2009;35:1141-1143.
- Perez LP, Zulaica A, Rodriguez L, et al. Multinucleate cell angiohistiocytoma. report of five cases. J Cutan Pathol. 2006;33:349-352.
- Shapiro PE, Nova MP, Rosmarin LA, et al. Multinucleate cell angiohistiocytoma: a distinct entity diagnosable by clinical and histologic features. J Am Acad Dermatol. 1994;30:417-422.
- Jaconelli L, Kanitakis J, Ktiouet S, et al. Multinucleate cell angiohistiocytoma: report of three new cases and literature review. Dermatol Online J. 2009;15:4.
- Jones WE, Cerio R, Smith NP. Multinucleate cell angiohistiocytoma: an acquired vascular anomaly to be distinguished from Kaposi’s sarcoma. Br J Dermatol. 1990;122:651-663.
- Calderaro J, Rethers L, Ortonne N. Multinucleated cells angiohistiocytoma: a reactive lesion? Am J Dermatopathol. 2010;32:415-417.
- Cesinaro AM, Roncati L, Maiorana A. Estrogen receptor alpha overexpression in multinucleate cell angiohistiocytoma: new insights into the pathogenesis of a reactive process. Am J Dermatopathol. 2010;32:655-659.
- Losordo DW, Isner JM. Estrogen and angiogenesis: a review. Arterioscler Thromb Vasc Biol. 2001;21:6-12.
- Sass U, Noel JC, Andre J, et al. Multinucleate cell angiohistiocytoma: report of two cases with no evidence of human herpesvirus-8 infection. J Cutan Pathol. 2000;27:258-261.
- Richer V, Lui H. Facial multinucleate cell angiohistiocytoma: long-term remission with 585 nm pulsed dye laser. Clin Exp Dermatol. 2016;41:312-313.
- Kopera D, Smolle J, Kerl H. Multinucleate cell angiohistiocytoma: treatment with argon laser. Br J Dermatol. 1995;133:308-310.
- Väkevä L, Saksela O, Kariniemi AL. Multinucleate cell angiohistiocytoma: a report of four cases in Finland. Acta Derm Venereol. 2003;83:222-223.
Practice Points
- Multinucleate cell angiohistiocytoma (MCAH) is a rare underrecognized cutaneous tumor presenting as erythematous to violaceous papules.
- Although it clinically mimics Kaposi sarcoma, MCAH may be distinguished histopathologically by negative immunostaining for human herpesvirus 8.
- Surgical excision and laser therapies are definitive treatments for MCAH, which is a benign lesion.
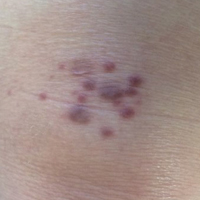
Pulmonary sarcomatoid carcinoma presenting as a necrotizing cavitary lung lesion: diagnostic dilemma
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear. The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsy of the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung, likely due to the underlying cavitary lesion (Figure 2B). Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic mass within the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2A) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3). Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine. Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was
recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade andnwith equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment. Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis and timely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high plateletderived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies. TSJ
Correspondence
Gaurang Nandkishor Vaidya, MD
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso. biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear. The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsy of the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung, likely due to the underlying cavitary lesion (Figure 2B). Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic mass within the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2A) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3). Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine. Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was
recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade andnwith equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment. Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis and timely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high plateletderived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies. TSJ
Correspondence
Gaurang Nandkishor Vaidya, MD
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso. biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
Pulmonary sarcomatoid carcinoma (PSC) is a rare histological subtype that has an aggressive course with average survival of 11-13 months.1 In clinical practice, the possible presentations of this rare cancer are not widely known, resulting in a misdiagnosis. That is what happened with our patient, who presented with necrotizing cavitary lung lesion and soft tissue necrotizing lymphadenitis. The clinical picture was reminiscent of tuberculosis or granulomatosis with polyangiitis and was further confounded by negative computed-tomography (CT)-guided biopsy and bronchoscopy findings, which added to the delay in diagnosis. With the currently available knowledge, the diagnosis of PSC depends largely on evaluation of the surgically resected specimen, which in most cases is avoided until there is a high suspicion of PSC. Biopsy is not useful due to extensive necrosis, as will be seen in our case. Consequently, most of the data in the literature is based on case series of autopsy specimen, and the clinical characteristics of PSC remain unclear. The rarity of PSC has prevented its characterization in literature. We report here a rare presentation of PSC with necrotizing lung lesion, to add to the paucity of the current data.
Case presentation and summary
A 58-year-old homeless man presented to the Upstate University Hospital, Syracuse, New York, with a 25-pound weight loss during the previous month and associated productive cough and hemoptysis for a week and a painful mass in the nape of his neck. He denied any fever, chest pain, sick contacts, or joint pain. He had a history of about 40 pack-years of smoking, and his brother had recently been diagnosed with lung cancer. A tender fluctuant mass was detected in the nape of his neck on examination (Figure 1).
The patient had presented 9 months earlier with persistent cough and hemoptysis, and at that visit was found to have a cavitary lesion in the right lung measuring 2 cm (0.8 in). He had undergone a computed-tomograpghy (CT)-guided biopsy of the lesion, which had shown acute and chronic inflammation with fibrosis, and he had negative bronchoscopy findings. The patient tested negative for tuberculosis during the first visit but he left the hospital against the medical advice of the physicians and he was lost to follow-up until his re-presentation.
On physical examination at his re-presentation, the patient seemed cachectic, with a blood pressure of 94/62 mm of Hg. The mass in the nape of his neck was about 3 cm (1.2 in) long, with erythema of the surrounding skin (Figure 1). Bronchial breath sounds were heard in the right upper lobe of the lung, likely due to the underlying cavitary lesion (Figure 2B). Relevant lab findings included a negative HIV test and repeat AFB (acid-fast bacilli) sputum cultures. A CT-guided biopsy with contrast of the thorax showed an interval increase in the size of the cavitary lesion in the patient’s right upper lobe, now measuring about 10 cm (4 in). Also seen were multiple nodules elsewhere in both lungs, with the largest measuring 8 mm (0.3 in). A CT scan of the neck showed 3 cm cystic mass within the posterior subcutaneous soft tissue of the C3 level, confirming the examination finding of the neck mass (Figure 2A) with peripheral enhancement and surrounding infiltrative changes, likely abscess or malignant lymph node versus necrotic infection. He underwent bronchoscopy, which again failed to reveal any endobronchial lesions. Bronchoalveolar lavage was sent for microbiological analysis, including AFB and fungus, but came back negative. Transbronchial biopsy cytology revealed fragments of tumor composed of large pleomorphic cells without glandular or squamous differentiation, within large areas of necrosis (Figure 3). Immunohistochemical studies showed strong reactivity with cytokeratin CAM5.2 (Figure 4), weak and focal reactivity with cytokeratin AE1/AE3 (Figure 5), and lack of reactivity with CD20, CD3, CD30, S-100, MART-1, TTF-1 and p63, all findings consistent with sarcomatoid carcinoma.
The patient underwent fine-needle aspiration and drainage of the neck lesion and the culture grew mixed organisms The results of a bone scan, which was done within a week, showed multiple foci of uptake in the ribs and cervical spine. Given the patient’s advanced disease, he was started on palliative radiotherapy with radiosensitizing chemotherapy with carboplatin (target AUC 6) and paclitaxel (135 mg/m2 over 24 hours). His symptoms of hemoptysis improved transiently after the first cycle, but he became hypotensive and drowsy during the second cycle of therapy, and the family decided to make the patient comfort care and withdraw all further treatment. He was discharged to hospice.
Discussion
PSC is a rare variant of non-small-cell carcinoma lung cancer, accounting for up to 0.4% of lung malignancy.1 It was
recently subtyped by the World Health Organization as a non-small cell lung carcinoma with certain amount of differentiation resembling sarcoma or containing elements of sarcoma.2-4 It is not known why both elements co-exist in the tumor, but Franks and colleagues some theories have been postulated in the literature, including possible origin from a single, aberrant stem cell with progenies differentiating in two separate pathways.3
Sarcomatoid carcinoma consists of spectrum of tumors including pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and blastoma.3,4 It usually shows male preponderance, and association with smoking.3 The diagnosis commonly occurs in the sixth decade of life, except for pulmonary blastoma, which is more common in the fourth decade andnwith equal gender distribution.4
The presenting symptoms can be variable and nonspecific, but predominantly include chest pain, cough, hemoptysis, and/or weight loss.5 Radiologically, pulmonary sarcomatoid cancer presenting as a necrotizing cavitary lesion in the lung is a rare finding, seldom reported in the past.6,7 The presentation in our case, with necrotizing lymphadenitis, was reminiscent of an infectious or autoimmune etiology such as tuberculosis or granulomatosis with polyangiitis. The presence of extensive necrosis in the lesion and the characteristic heterogeneity of the tumor had resulted in inconclusive biopsy findings during the previous presentation. In clinical practice, there is over-reliance on biopsy findings to make the distinction between cancer and other mimicking conditions. This is especially true for rare tumors such as PSC, which often results in misdiagnosis and a delay in administering the proper treatment. Transbronchial biopsy in cases such as the present case, carries little benefit because the diagnosis depends on the site from which the biopsy is taken and whether the biopsied tissue is representative of the entire mass. The diagnosis can be suspected based on the clinical and radiological findings but confirmation requires a surgical resection to delineate the accurate cytology and architecture.5,6,8 Huang and colleagues showed a misdiagnosis rate of PSC of >70% preoperatively.4 Resective surgery is feasible only in patients with high index of suspicion for a malignancy, which in most cases requires previous confirmation with a biopsy. The rarity of this cancer, its unusual presentations, and the lack of specific testing preclude early diagnosis and timely treatment of this fatal condition.
Initial treatment options for localized or with limited spread disease is resective surgery. The role of chemo- or radiation therapy is not known, but they have not previously shown promising results,6,8 except in some cases when they are used as postoperative adjuvant chemotherapy4 or in bulky, locally invasive tumors.1 The recurrence rate after surgery is very high, resulting in a poor 5-year survival rate.1,8 Experimental therapies, such as antibodies that target epidermal growth factor receptor mutations, have not shown much success either.8 In conclusion, the outlook for patients with PSC with the current available knowledge and treatment protocols, is dismal.
Most of the current knowledge and data in the literature is based on cases from autopsy or early-stage surgical resections rather than on patients with advanced cancer.5 Moreover, the role of surgical resection in PSC is questionable, given the high recurrence rate. Subsequently, the clinical and pathological manifestations have yet to be well characterized.4 There has been advance with the publication of more studies recently. Cytokeratin markers such as CAM 5.2 and AE1/AE3 are commonly useful to support the diagnosis when suspected.3 Other markers, including the carcinoembryonic antigen, CD15, and thyroid transcription factor-1 may be variably positive, based on the differentiation of the cancer. Other exciting prospects in the study of PSC include the suggestion of a modified vimentin histologic score for better characterization of the cancer and the discovery of high plateletderived growth factor receptor beta immunohistochemistry expression in PSC as a potential target for future therapy.
Conclusion
Pulmonary sarcomatoid lung cancer can present with a predominant necrotizing picture that mimics diseases such as tuberculosis. In such case, transbronchial biopsy carries little benefit because the diagnosis depends on whether the biopsied tissue is representative of the entire mass, often confounded by the extensive necrosis. More data is needed to determine prognostic factors and appropriate therapeutic strategies. TSJ
Correspondence
Gaurang Nandkishor Vaidya, MD
References
1. Martin LW, Correa AM, Ordonez NG, et al. Sarcomatoid carcinoma of the lung: a predictor of poor prognosis. Ann Thorac Surg. 2007;84(3):973-980.
2. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059-1068.
3. Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134(1):49-54.
4. Huang SY, Shen SJ, Li XY. Pulmonary sarcomatoid carcinoma: a clinicopathologic study and prognostic analysis of 51 cases. http://wjso. biomedcentral.com/articles/10.1186/1477-7819-11-252. Published 2013. Accessed March 12, 2017.
5. Travis WD. Sarcomatoid neoplasms of the lung and pleura. Arch Pathol Lab Med. 2010;134(11):1645-1658.
6. Pelosi G, Sonzogni A, De Pas T, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18(2):103-120.
7. Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91-97.
8. Park JS, Lee Y, Han J, et al. Clinicopathologic outcomes of curative resection for sarcomatoid carcinoma of the lung. Oncology. 2011;81(3-4):206-213.
This article was originally published in the Journal of Community and Supportive Oncology (JCSO 2017;15(2):103-105). doi: https://doi.org/10.12788/jcso.0259. It is reproduced here with permission of the copyright owner. Further reproduction is prohibited without permission.