User login
Scaly Pink Patches: Differentiating Psoriasis From Basal Cell Carcinoma
Dermoscopy increases diagnostic accuracy in the analysis of skin growths.1,2 Recently the use of dermoscopy has broadened to include inflammatory dermatoses and skin infections.3 To substantiate the value of dermoscopy in assessing psoriasis, we performed a systematic review of the literature and briefly reviewed 31 articles. We also report a case that highlights the differences between psoriasis and basal cell carcinoma (BCC) under dermoscopic examination, and we discuss the literature on the dermoscopic findings of psoriasis with an emphasis on the relative sensitivities and specificities of dermoscopic findings for psoriasis and for BCC.
Case Report
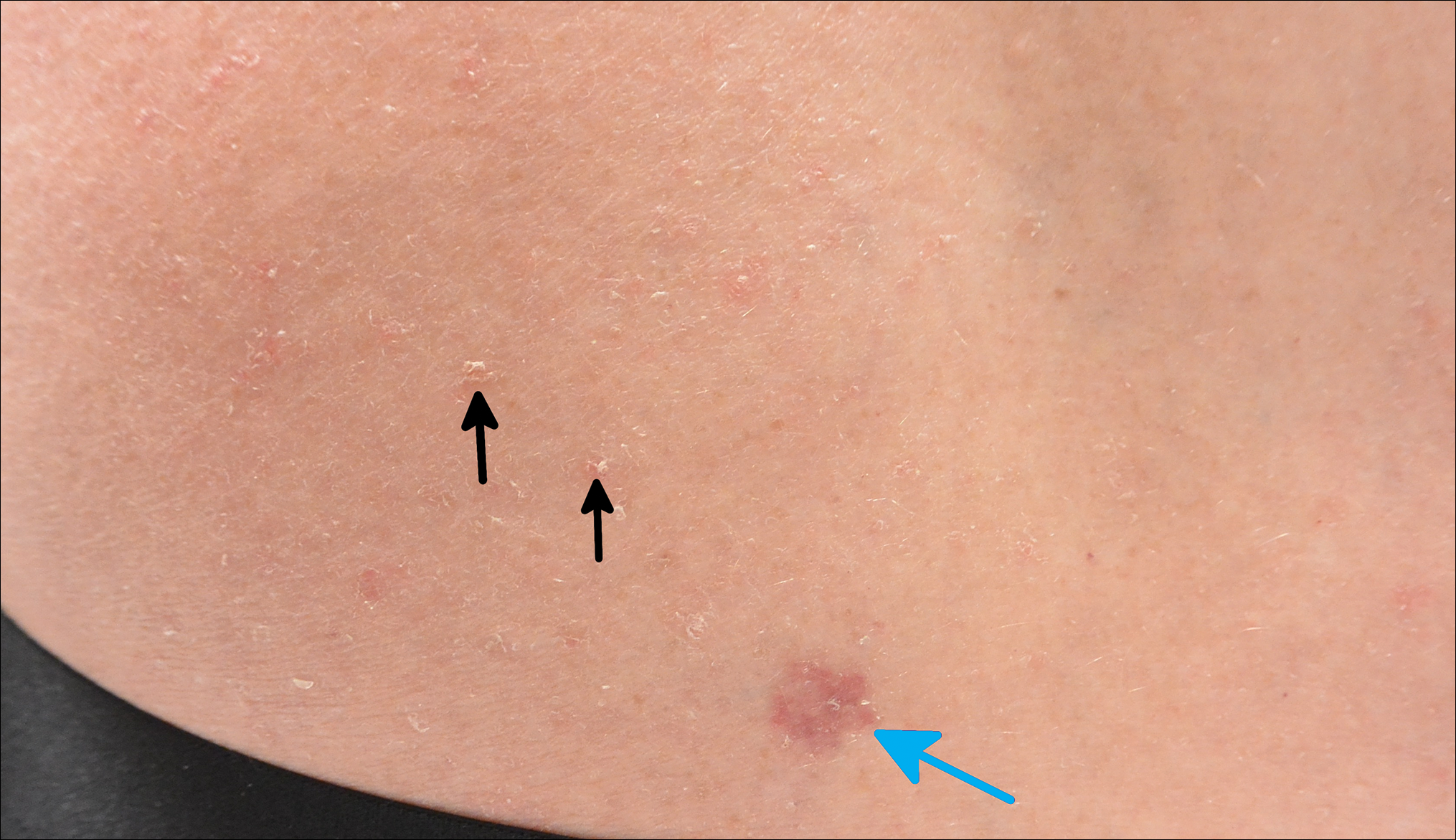
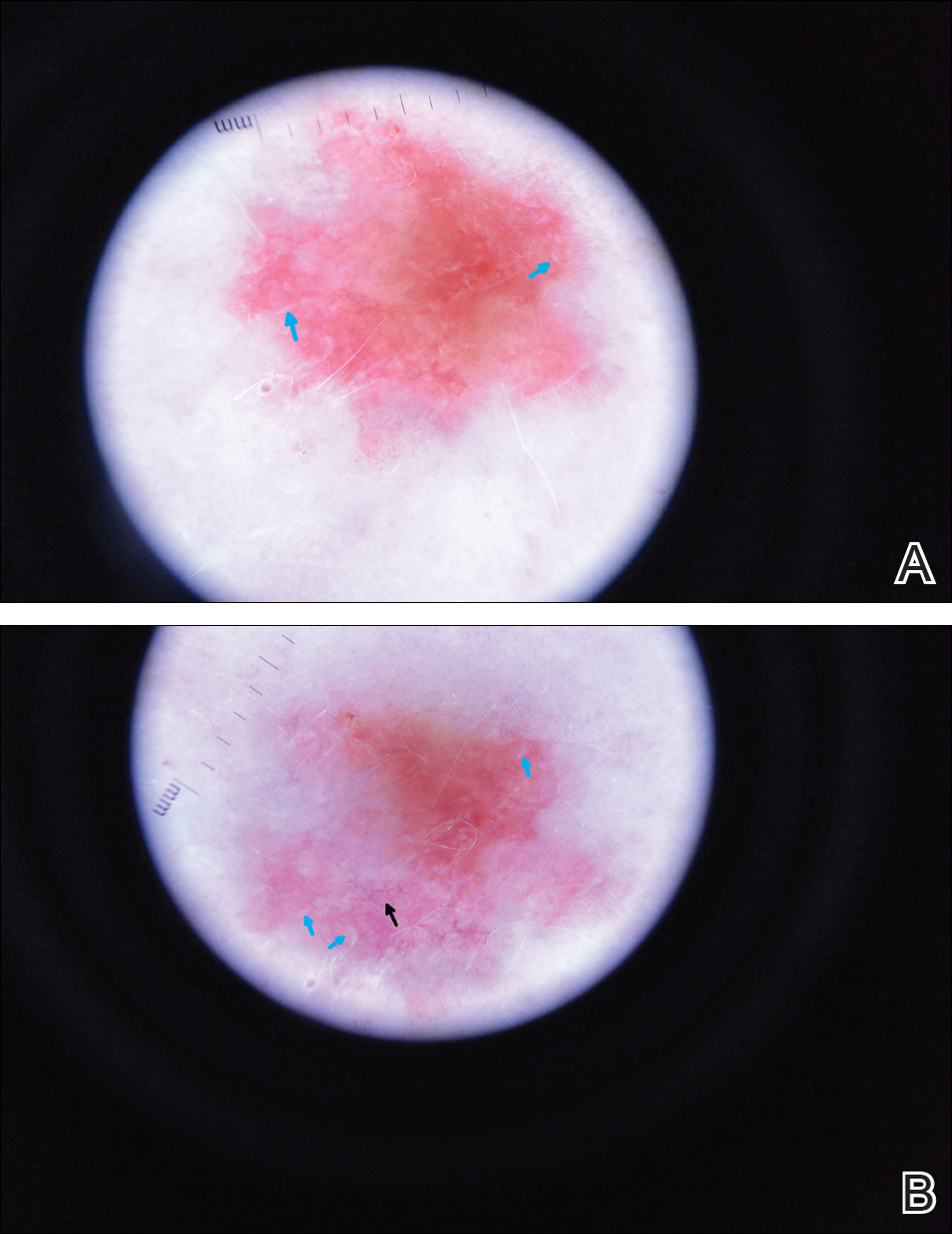
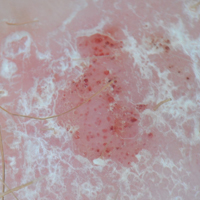
A 63-year-old man with psoriasis and a history of BCC presented for follow-up of psoriasis, which was well-controlled on etanercept. The physical examination was remarkable for scaly pink papules scattered on the trunk and extremities. A new larger red-pink patch was located on the left lower back (Figure 1). Dermoscopic evaluation of the new patch revealed shiny white lines and branching blood vessels (Figure 2).
Comment
The clinical morphology of psoriasis and BCC can be similar, and dermoscopy can help in differentiating between the 2 conditions.
Literature Search on Dermoscopy and Psoriasis
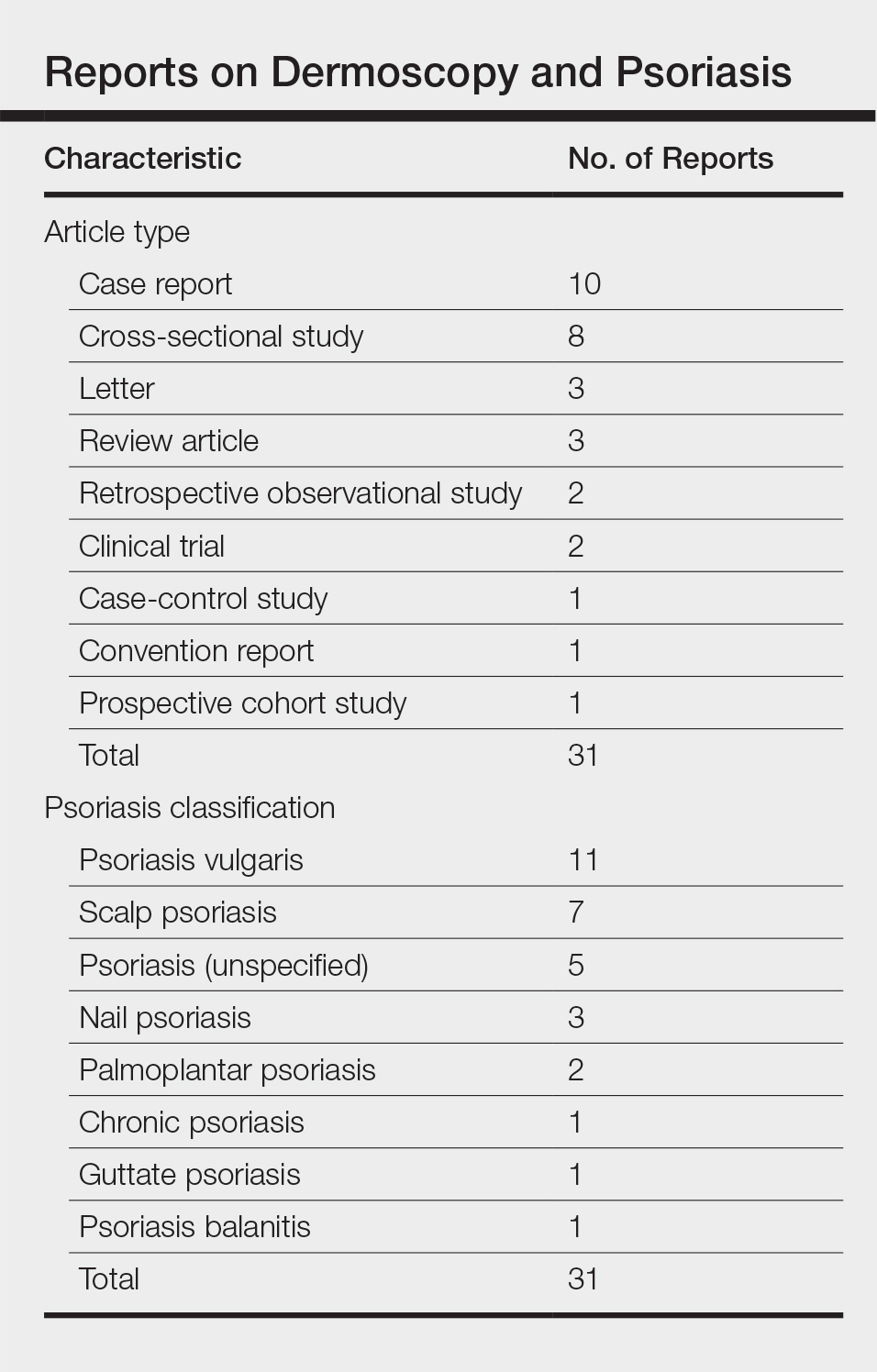
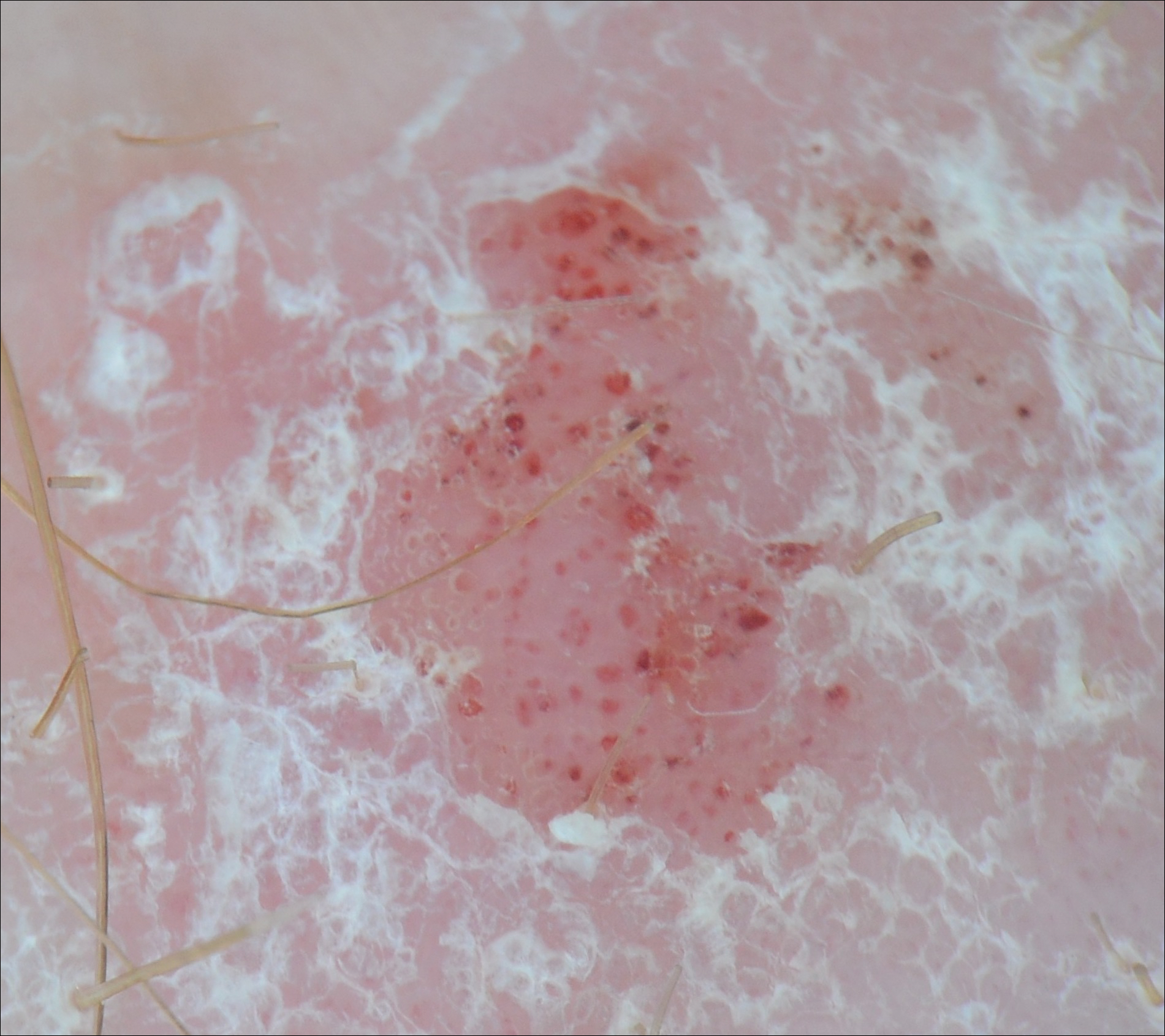
We performed a PubMed search of articles indexed for MEDLINE to review the published literature on dermoscopy and psoriasis. Two reviewers (C.H. and L.C.) searched for psoriasis paired with the terms dermoscopy or dermatoscopy or epiluminescence microscopy. Only English-language articles published between 1996 and 2016 were included in the search. Articles that focused solely on confocal microscopy were excluded. Article titles and abstracts were evaluated and articles that omitted mention of dermoscopy and psoriasis were excluded, yielding a total of 31 articles. Of these articles, only 2 discussed the specificity or sensitivity of the dermoscopic findings of psoriasis.4,5 Most of the articles were case reports and descriptive cross-sectional studies. The reports addressed multiple subtypes of psoriasis, but reports on psoriasis vulgaris and scalp psoriasis were most common (Table). Lallas et al6 provided a comprehensive descriptive review of the main findings on dermoscopy for psoriasis and other inflammatory skin conditions, but it lacked a comparison between psoriasis and BCC or data on the sensitivity and specificity of the findings. Two studies reported sensitivity and specificity values for the dermoscopic findings of psoriasis.4,5 Pan et al5 reported a 98% diagnostic probability of psoriasis if red dots, homogeneous vascular pattern, and a light red background are all present. Additionally, they reported that the presence of 4 of 6 criteria for BCC—scattered vascular pattern, arborizing microvessels, telangiectatic or atypical vessels, milky-pink background, and brown dots⁄globules—yielded a diagnostic probability of 99%.5 Similarly, Lallas et al6 demonstrated that the presence of dotted vessels alone is not sufficient to presume a diagnosis of psoriasis, as this finding can be seen in other inflammatory skin conditions. However, “the combination of regularly distributed dotted vessels over a light red background associated with diffuse white scales was highly predictive of [plaque psoriasis] and allowed a correct diagnosis with 88.0% specificity and 84.9% sensitivity.”4 Figure 3 shows a dermoscopic image of plaque psoriasis that demonstrates these findings. The remaining literature corroborated this evidence, with the most commonly reported dermoscopic findings of psoriasis being red dots, red globules, glomerular vessels (also known as twisted capillary loops), red globular ring
Dermoscopy and BCC
Much has been published on the dermoscopic findings of BCC.5,13-15 The dermoscopic findings of BCC include large blue-gray ovoid nests, leaflike areas, spoke-wheel–like areas, arborizing vessels (telangiectasia), and ulceration.15 Superficial BCC is characterized by short fine or arborizing telangiectasia, shallow erosions, and shiny white areas.15 The positive predictive value of dermoscopy in BCC is as high as 97%.16 Additionally, multiple studies report a sensitivity of 95% to 99%5,13,14 and a specificity of 79% to 99% in the use of dermoscopy for identifying BCC. According to Pan et al,5 the most sensitive finding for BCC is a scattered vascular pattern (97%), while the most specific finding is arborizing microvessels (99%).
Utility of Dermoscopy
Our case of a 63-year-old man with a history of psoriasis and BCC highlights the usefulness of dermoscopy in accurately determining the features of each condition. Additionally, dermoscopy aids in differentiating between psoriasis and squamous cell carcinoma. In contrast to the dotted vessels seen in psoriasis, squamous cell carcinomas often have peripheral hairpin (glomerular) vessels.17
If future reports confirm dermoscopy’s utility in accurately diagnosing psoriasis, fewer biopsies may be needed when evaluating patients with new rashes. Furthermore, dermoscopy may expedite treatment of psoriasis (as it can for malignant conditions) by obviating the wait for pathology results currently needed to initiate systemic treatment. For patients with psoriasis who also have sun-damaged skin, dermoscopy may assist in differentiating pink patches and plaques of psoriasis from skin cancer, such as superficial BCCs, which often have shiny white lines not seen in psoriasis.15
- Kittler H, Pehamberger H, Wolff K, et al. Diagnostic accuracy of dermoscopy. Lancet Oncol. 2002;3:159-165.
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
- Lallas A, Giacomel J, Argenziano G, et al. Dermoscopy in general dermatology: practical tips for the clinician. Br J Dermatol. 2014;170:514-526.
- Lallas A, Kyrgidis A, Tzellos TG, et al. Accuracy of dermoscopic criteria for the diagnosis of psoriasis, dermatitis, lichen planus and pityriasis rosea. Br J Dermatol. 2012;166:1198-1205.
- Pan Y, Chamberlain AJ, Bailey M, et al. Dermatoscopy aids in the diagnosis of the solitary red scaly patch or plaque–features distinguishing superficial basal cell carcinoma, intraepidermal carcinoma, and psoriasis. J Am Acad Dermatol. 2008;59:268-274.
- Lallas A, Apalla Z, Argenziano G, et al. Dermoscopic pattern of psoriatic lesions on specific body sites. Dermatology. 2014;228:250-254.
- Almeida MC, Romiti R, Doche I, et al. Psoriatic scarring alopecia. An Bras Dermatol. 2013;88:29-31.
- Zalaudek I, Argenziano G. Dermoscopy subpatterns of inflammatory skin disorders. Arch Dermatol. 2006;142:808.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
- Vázquez-López F, Zaballos P, Fueyo-Casado A, et al. A dermoscopy subpattern of plaque-type psoriasis: red globular rings. Arch Dermatol. 2007;143:1612.
- Lacarrubba F, Nasca MR, Micali G. Videodermatoscopy enhances diagnostic capability in psoriatic balanitis. J Am Acad Dermatol. 2009;61:1084-1086.
- Liebman TN, Wang SQ. Detection of early basal cell carcinoma with dermoscopy in a patient with psoriasis. Dermatol Online J. 2011;17:12.
- Menzies SW, Westerhoff K, Rabinovitz H, et al. Surface microscopy of pigmented basal cell carcinoma. Arch Dermatol. 2000;136:1012-1016.
- Altamura D, Menzies SW, Argenziano G, et al. Dermatoscopy of basal cell carcinoma: morphologic variability of global and local features and accuracy of diagnosis. J Am Acad Dermatol. 2010;62:67-75.
- Marghoob AA, Malvehy J, Braun RP, eds. An Atlas of Dermoscopy. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Nelson SA, Scope A, Rishpon A, et al. Accuracy and confidence in the clinical diagnosis of basal cell cancer using dermoscopy and reflex confocal microscopy. Int J Dermatol. 2016;55:1351-1356.
- Zalaudek I, Kreusch J, Giacomel J, et al. How to diagnose nonpigmented skin tumors: a review of vascular structures seen with dermoscopy: part I. melanocytic skin tumors. J Am Acad Dermatol. 2010;63:361-374.
Dermoscopy increases diagnostic accuracy in the analysis of skin growths.1,2 Recently the use of dermoscopy has broadened to include inflammatory dermatoses and skin infections.3 To substantiate the value of dermoscopy in assessing psoriasis, we performed a systematic review of the literature and briefly reviewed 31 articles. We also report a case that highlights the differences between psoriasis and basal cell carcinoma (BCC) under dermoscopic examination, and we discuss the literature on the dermoscopic findings of psoriasis with an emphasis on the relative sensitivities and specificities of dermoscopic findings for psoriasis and for BCC.
Case Report
A 63-year-old man with psoriasis and a history of BCC presented for follow-up of psoriasis, which was well-controlled on etanercept. The physical examination was remarkable for scaly pink papules scattered on the trunk and extremities. A new larger red-pink patch was located on the left lower back (Figure 1). Dermoscopic evaluation of the new patch revealed shiny white lines and branching blood vessels (Figure 2).
Comment
The clinical morphology of psoriasis and BCC can be similar, and dermoscopy can help in differentiating between the 2 conditions.
Literature Search on Dermoscopy and Psoriasis
We performed a PubMed search of articles indexed for MEDLINE to review the published literature on dermoscopy and psoriasis. Two reviewers (C.H. and L.C.) searched for psoriasis paired with the terms dermoscopy or dermatoscopy or epiluminescence microscopy. Only English-language articles published between 1996 and 2016 were included in the search. Articles that focused solely on confocal microscopy were excluded. Article titles and abstracts were evaluated and articles that omitted mention of dermoscopy and psoriasis were excluded, yielding a total of 31 articles. Of these articles, only 2 discussed the specificity or sensitivity of the dermoscopic findings of psoriasis.4,5 Most of the articles were case reports and descriptive cross-sectional studies. The reports addressed multiple subtypes of psoriasis, but reports on psoriasis vulgaris and scalp psoriasis were most common (Table). Lallas et al6 provided a comprehensive descriptive review of the main findings on dermoscopy for psoriasis and other inflammatory skin conditions, but it lacked a comparison between psoriasis and BCC or data on the sensitivity and specificity of the findings. Two studies reported sensitivity and specificity values for the dermoscopic findings of psoriasis.4,5 Pan et al5 reported a 98% diagnostic probability of psoriasis if red dots, homogeneous vascular pattern, and a light red background are all present. Additionally, they reported that the presence of 4 of 6 criteria for BCC—scattered vascular pattern, arborizing microvessels, telangiectatic or atypical vessels, milky-pink background, and brown dots⁄globules—yielded a diagnostic probability of 99%.5 Similarly, Lallas et al6 demonstrated that the presence of dotted vessels alone is not sufficient to presume a diagnosis of psoriasis, as this finding can be seen in other inflammatory skin conditions. However, “the combination of regularly distributed dotted vessels over a light red background associated with diffuse white scales was highly predictive of [plaque psoriasis] and allowed a correct diagnosis with 88.0% specificity and 84.9% sensitivity.”4 Figure 3 shows a dermoscopic image of plaque psoriasis that demonstrates these findings. The remaining literature corroborated this evidence, with the most commonly reported dermoscopic findings of psoriasis being red dots, red globules, glomerular vessels (also known as twisted capillary loops), red globular ring
Dermoscopy and BCC
Much has been published on the dermoscopic findings of BCC.5,13-15 The dermoscopic findings of BCC include large blue-gray ovoid nests, leaflike areas, spoke-wheel–like areas, arborizing vessels (telangiectasia), and ulceration.15 Superficial BCC is characterized by short fine or arborizing telangiectasia, shallow erosions, and shiny white areas.15 The positive predictive value of dermoscopy in BCC is as high as 97%.16 Additionally, multiple studies report a sensitivity of 95% to 99%5,13,14 and a specificity of 79% to 99% in the use of dermoscopy for identifying BCC. According to Pan et al,5 the most sensitive finding for BCC is a scattered vascular pattern (97%), while the most specific finding is arborizing microvessels (99%).
Utility of Dermoscopy
Our case of a 63-year-old man with a history of psoriasis and BCC highlights the usefulness of dermoscopy in accurately determining the features of each condition. Additionally, dermoscopy aids in differentiating between psoriasis and squamous cell carcinoma. In contrast to the dotted vessels seen in psoriasis, squamous cell carcinomas often have peripheral hairpin (glomerular) vessels.17
If future reports confirm dermoscopy’s utility in accurately diagnosing psoriasis, fewer biopsies may be needed when evaluating patients with new rashes. Furthermore, dermoscopy may expedite treatment of psoriasis (as it can for malignant conditions) by obviating the wait for pathology results currently needed to initiate systemic treatment. For patients with psoriasis who also have sun-damaged skin, dermoscopy may assist in differentiating pink patches and plaques of psoriasis from skin cancer, such as superficial BCCs, which often have shiny white lines not seen in psoriasis.15
Dermoscopy increases diagnostic accuracy in the analysis of skin growths.1,2 Recently the use of dermoscopy has broadened to include inflammatory dermatoses and skin infections.3 To substantiate the value of dermoscopy in assessing psoriasis, we performed a systematic review of the literature and briefly reviewed 31 articles. We also report a case that highlights the differences between psoriasis and basal cell carcinoma (BCC) under dermoscopic examination, and we discuss the literature on the dermoscopic findings of psoriasis with an emphasis on the relative sensitivities and specificities of dermoscopic findings for psoriasis and for BCC.
Case Report
A 63-year-old man with psoriasis and a history of BCC presented for follow-up of psoriasis, which was well-controlled on etanercept. The physical examination was remarkable for scaly pink papules scattered on the trunk and extremities. A new larger red-pink patch was located on the left lower back (Figure 1). Dermoscopic evaluation of the new patch revealed shiny white lines and branching blood vessels (Figure 2).
Comment
The clinical morphology of psoriasis and BCC can be similar, and dermoscopy can help in differentiating between the 2 conditions.
Literature Search on Dermoscopy and Psoriasis
We performed a PubMed search of articles indexed for MEDLINE to review the published literature on dermoscopy and psoriasis. Two reviewers (C.H. and L.C.) searched for psoriasis paired with the terms dermoscopy or dermatoscopy or epiluminescence microscopy. Only English-language articles published between 1996 and 2016 were included in the search. Articles that focused solely on confocal microscopy were excluded. Article titles and abstracts were evaluated and articles that omitted mention of dermoscopy and psoriasis were excluded, yielding a total of 31 articles. Of these articles, only 2 discussed the specificity or sensitivity of the dermoscopic findings of psoriasis.4,5 Most of the articles were case reports and descriptive cross-sectional studies. The reports addressed multiple subtypes of psoriasis, but reports on psoriasis vulgaris and scalp psoriasis were most common (Table). Lallas et al6 provided a comprehensive descriptive review of the main findings on dermoscopy for psoriasis and other inflammatory skin conditions, but it lacked a comparison between psoriasis and BCC or data on the sensitivity and specificity of the findings. Two studies reported sensitivity and specificity values for the dermoscopic findings of psoriasis.4,5 Pan et al5 reported a 98% diagnostic probability of psoriasis if red dots, homogeneous vascular pattern, and a light red background are all present. Additionally, they reported that the presence of 4 of 6 criteria for BCC—scattered vascular pattern, arborizing microvessels, telangiectatic or atypical vessels, milky-pink background, and brown dots⁄globules—yielded a diagnostic probability of 99%.5 Similarly, Lallas et al6 demonstrated that the presence of dotted vessels alone is not sufficient to presume a diagnosis of psoriasis, as this finding can be seen in other inflammatory skin conditions. However, “the combination of regularly distributed dotted vessels over a light red background associated with diffuse white scales was highly predictive of [plaque psoriasis] and allowed a correct diagnosis with 88.0% specificity and 84.9% sensitivity.”4 Figure 3 shows a dermoscopic image of plaque psoriasis that demonstrates these findings. The remaining literature corroborated this evidence, with the most commonly reported dermoscopic findings of psoriasis being red dots, red globules, glomerular vessels (also known as twisted capillary loops), red globular ring
Dermoscopy and BCC
Much has been published on the dermoscopic findings of BCC.5,13-15 The dermoscopic findings of BCC include large blue-gray ovoid nests, leaflike areas, spoke-wheel–like areas, arborizing vessels (telangiectasia), and ulceration.15 Superficial BCC is characterized by short fine or arborizing telangiectasia, shallow erosions, and shiny white areas.15 The positive predictive value of dermoscopy in BCC is as high as 97%.16 Additionally, multiple studies report a sensitivity of 95% to 99%5,13,14 and a specificity of 79% to 99% in the use of dermoscopy for identifying BCC. According to Pan et al,5 the most sensitive finding for BCC is a scattered vascular pattern (97%), while the most specific finding is arborizing microvessels (99%).
Utility of Dermoscopy
Our case of a 63-year-old man with a history of psoriasis and BCC highlights the usefulness of dermoscopy in accurately determining the features of each condition. Additionally, dermoscopy aids in differentiating between psoriasis and squamous cell carcinoma. In contrast to the dotted vessels seen in psoriasis, squamous cell carcinomas often have peripheral hairpin (glomerular) vessels.17
If future reports confirm dermoscopy’s utility in accurately diagnosing psoriasis, fewer biopsies may be needed when evaluating patients with new rashes. Furthermore, dermoscopy may expedite treatment of psoriasis (as it can for malignant conditions) by obviating the wait for pathology results currently needed to initiate systemic treatment. For patients with psoriasis who also have sun-damaged skin, dermoscopy may assist in differentiating pink patches and plaques of psoriasis from skin cancer, such as superficial BCCs, which often have shiny white lines not seen in psoriasis.15
- Kittler H, Pehamberger H, Wolff K, et al. Diagnostic accuracy of dermoscopy. Lancet Oncol. 2002;3:159-165.
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
- Lallas A, Giacomel J, Argenziano G, et al. Dermoscopy in general dermatology: practical tips for the clinician. Br J Dermatol. 2014;170:514-526.
- Lallas A, Kyrgidis A, Tzellos TG, et al. Accuracy of dermoscopic criteria for the diagnosis of psoriasis, dermatitis, lichen planus and pityriasis rosea. Br J Dermatol. 2012;166:1198-1205.
- Pan Y, Chamberlain AJ, Bailey M, et al. Dermatoscopy aids in the diagnosis of the solitary red scaly patch or plaque–features distinguishing superficial basal cell carcinoma, intraepidermal carcinoma, and psoriasis. J Am Acad Dermatol. 2008;59:268-274.
- Lallas A, Apalla Z, Argenziano G, et al. Dermoscopic pattern of psoriatic lesions on specific body sites. Dermatology. 2014;228:250-254.
- Almeida MC, Romiti R, Doche I, et al. Psoriatic scarring alopecia. An Bras Dermatol. 2013;88:29-31.
- Zalaudek I, Argenziano G. Dermoscopy subpatterns of inflammatory skin disorders. Arch Dermatol. 2006;142:808.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
- Vázquez-López F, Zaballos P, Fueyo-Casado A, et al. A dermoscopy subpattern of plaque-type psoriasis: red globular rings. Arch Dermatol. 2007;143:1612.
- Lacarrubba F, Nasca MR, Micali G. Videodermatoscopy enhances diagnostic capability in psoriatic balanitis. J Am Acad Dermatol. 2009;61:1084-1086.
- Liebman TN, Wang SQ. Detection of early basal cell carcinoma with dermoscopy in a patient with psoriasis. Dermatol Online J. 2011;17:12.
- Menzies SW, Westerhoff K, Rabinovitz H, et al. Surface microscopy of pigmented basal cell carcinoma. Arch Dermatol. 2000;136:1012-1016.
- Altamura D, Menzies SW, Argenziano G, et al. Dermatoscopy of basal cell carcinoma: morphologic variability of global and local features and accuracy of diagnosis. J Am Acad Dermatol. 2010;62:67-75.
- Marghoob AA, Malvehy J, Braun RP, eds. An Atlas of Dermoscopy. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Nelson SA, Scope A, Rishpon A, et al. Accuracy and confidence in the clinical diagnosis of basal cell cancer using dermoscopy and reflex confocal microscopy. Int J Dermatol. 2016;55:1351-1356.
- Zalaudek I, Kreusch J, Giacomel J, et al. How to diagnose nonpigmented skin tumors: a review of vascular structures seen with dermoscopy: part I. melanocytic skin tumors. J Am Acad Dermatol. 2010;63:361-374.
- Kittler H, Pehamberger H, Wolff K, et al. Diagnostic accuracy of dermoscopy. Lancet Oncol. 2002;3:159-165.
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
- Lallas A, Giacomel J, Argenziano G, et al. Dermoscopy in general dermatology: practical tips for the clinician. Br J Dermatol. 2014;170:514-526.
- Lallas A, Kyrgidis A, Tzellos TG, et al. Accuracy of dermoscopic criteria for the diagnosis of psoriasis, dermatitis, lichen planus and pityriasis rosea. Br J Dermatol. 2012;166:1198-1205.
- Pan Y, Chamberlain AJ, Bailey M, et al. Dermatoscopy aids in the diagnosis of the solitary red scaly patch or plaque–features distinguishing superficial basal cell carcinoma, intraepidermal carcinoma, and psoriasis. J Am Acad Dermatol. 2008;59:268-274.
- Lallas A, Apalla Z, Argenziano G, et al. Dermoscopic pattern of psoriatic lesions on specific body sites. Dermatology. 2014;228:250-254.
- Almeida MC, Romiti R, Doche I, et al. Psoriatic scarring alopecia. An Bras Dermatol. 2013;88:29-31.
- Zalaudek I, Argenziano G. Dermoscopy subpatterns of inflammatory skin disorders. Arch Dermatol. 2006;142:808.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
- Vázquez-López F, Zaballos P, Fueyo-Casado A, et al. A dermoscopy subpattern of plaque-type psoriasis: red globular rings. Arch Dermatol. 2007;143:1612.
- Lacarrubba F, Nasca MR, Micali G. Videodermatoscopy enhances diagnostic capability in psoriatic balanitis. J Am Acad Dermatol. 2009;61:1084-1086.
- Liebman TN, Wang SQ. Detection of early basal cell carcinoma with dermoscopy in a patient with psoriasis. Dermatol Online J. 2011;17:12.
- Menzies SW, Westerhoff K, Rabinovitz H, et al. Surface microscopy of pigmented basal cell carcinoma. Arch Dermatol. 2000;136:1012-1016.
- Altamura D, Menzies SW, Argenziano G, et al. Dermatoscopy of basal cell carcinoma: morphologic variability of global and local features and accuracy of diagnosis. J Am Acad Dermatol. 2010;62:67-75.
- Marghoob AA, Malvehy J, Braun RP, eds. An Atlas of Dermoscopy. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Nelson SA, Scope A, Rishpon A, et al. Accuracy and confidence in the clinical diagnosis of basal cell cancer using dermoscopy and reflex confocal microscopy. Int J Dermatol. 2016;55:1351-1356.
- Zalaudek I, Kreusch J, Giacomel J, et al. How to diagnose nonpigmented skin tumors: a review of vascular structures seen with dermoscopy: part I. melanocytic skin tumors. J Am Acad Dermatol. 2010;63:361-374.
Practice Points
- Dermoscopy has been largely utilized for the evaluation of malignant lesions. It also is gaining traction in the evaluation of inflammatory dermatoses.
- Early distinction between basal cell carcinoma and psoriasis is important for both treatment options and health care costs.
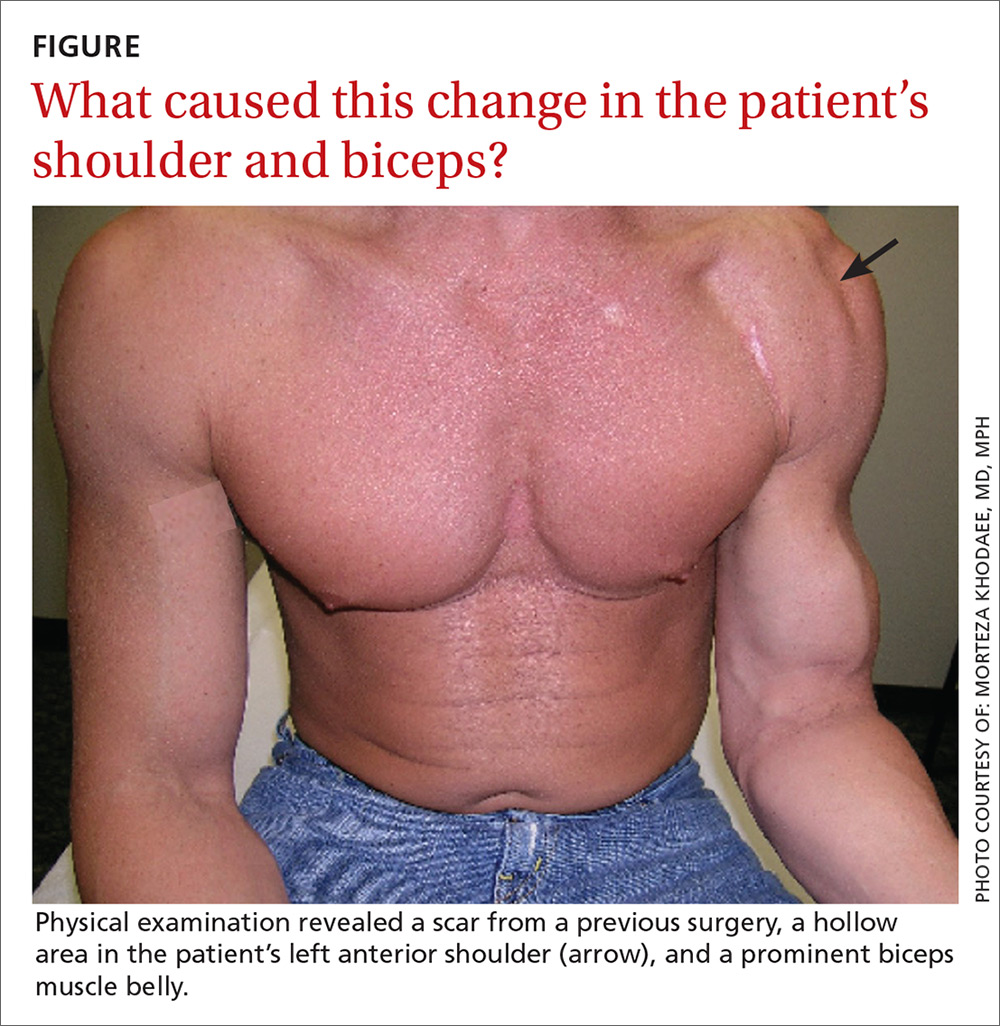
Weakness with left elbow flexion • left anterior shoulder pain • Dx?
THE CASE
A 41-year-old, right-hand dominant man sought care at our facility one day after trying to pull his boat out of the water. He’d tried to lift the boat with his hands while his forearms were fully supinated and his elbows were flexed to about 90°. He then felt a sharp burning sensation in his left anterior shoulder and was unable to lift the boat. The patient denied feeling a popping sensation at the time of the injury. He had mild pain at night, but was able to sleep. He said that he had mild diminished strength with elbow flexion, but denied having any numbness, tingling, or discoloration of his skin.
The patient said he did weightlifting and strength training of his upper and lower extremities 4 times/week. He was in good general health, was not taking any medications or supplements, and denied smoking or using illicit drugs. His surgical history was significant for a Bankart repair 8 years ago.
On physical examination, the patient had a scar from the previous surgery, a hollow area in his left anterior shoulder, and a prominent biceps muscle belly (FIGURE). His shoulder range of motion was normal. Left shoulder Neer, Hawkins-Kennedy, drop-arm, cross-arm, empty can, and apprehension tests were negative. A left Speed’s test (resisted elbow flexion when elbow is flexed 20° to 30° with the forearm in supination and the arm in about 60° of flexion) was positive for mild anterior shoulder pain. So, too, was a Yergason’s test (resisted forearm supination and elbow flexion when forearm is pronated and elbow is flexed to 90°). The patient’s elbow flexion strength was 4 out of 5, and his supination strength was 5 out of 5. Neurovascular and sensory examinations of his upper extremities, including radial and ulnar pulses, were normal.
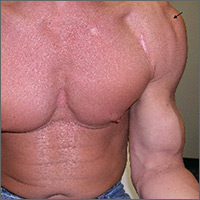
THE DIAGNOSIS
A diagnostic musculoskeletal ultrasound revealed an empty tendon sheath of the long head of the biceps in the bicipital groove and a retracted echogenic stump with associated hematoma at the proximal musculotendinous junction. Based on the patient’s history, physical examination, and ultrasound, a diagnosis of an acute rupture of the left long head of the biceps brachii tendon was made.
DISCUSSION
Diagnosis of acute rupture is often made clinically based on a visually apparent defect proximally and a bulbous mass distally (“Popeye deformity”).1 Ultrasound and magnetic resonance imaging (MRI) may aid in the diagnosis by demonstrating an absence of the long head in the bicipital groove or at its insertion.
The biceps brachii tendon functions in flexion and supination of the forearm. The long head of the biceps also plays a stabilizing role in the glenohumeral joint during elbow flexion and supination.2 Injury to the biceps most often occurs in middle-aged men following a traumatic sudden eccentric bicipital contraction event, during which most patients describe a snapping or popping sensation.3,4
Rupture of the proximal biceps tendon represents about 90% of all biceps ruptures, which almost exclusively involve the long head of the biceps.3,5,6 Risk factors for tendon rupture include obesity, smoking, steroid injection in or around the tendon, and previous tendinopathy.7-10
Functional limitations. It is generally thought that functional limitations following a proximal biceps rupture are relatively minimal, due to the work of other flexors and supinators, including the brachialis and brachioradialis. However, because strength and endurance of the muscle can decrease by about 25%, physical laborers and high-demand athletes may notice a degree of residual weakness with supination and elbow flexion.11,12
Surgery is suitable for some, but not all
Surgical repair is recommended for acute ruptures in patients with high physical demands and for whom a slight loss of flexion and supination strength would not be well tolerated.13 Tenotomy and tenodesis are the main techniques used to surgically repair a rupture of the long head of the biceps brachii tendon. Although there is no consensus on which technique is superior, it seems that there is less cosmetic deformity and better post-surgery biomechanical strength with tenodesis compared with tenotomy.14 However, tenodesis is associated with a higher likelihood of bicipital pain,14 and recent case reports have suggested it is associated with an increased risk of humeral fracture.15 Therefore, each patient should be treated on an individual case basis, taking into account age, activity level, and physical demand.14
For most patients, treatment remains conservative with typically excellent outcomes. Nonoperative management includes gentle range-of-motion exercises for the prevention of contractures of the elbow and shoulder. Such exercises can be started almost immediately after injury. In one study, nonoperative management was recommended for patients with sedentary work, injury in the non-dominant arm, and acceptable cosmetic deformity. Researchers noted that patients who opt for a nonsurgical treatment generally do well with a home exercise program and rarely have stiffness.1
If the patient is a young athlete, if cosmetic deformity is unacceptable, or if the injury is in the dominant arm of a laborer, then the patient may want to consider tenodesis.1 Tangari et al found that in high-demand athletes, biceps tenodesis resulted in excellent functional and cosmetic results with no clinically significant decrease in strength after an average follow-up of 7.6 years.13 In a case s
Our patient elected to proceed with a tenodesis procedure. Two months after the surgery, he had fully recovered.
THE TAKEAWAY
Rupture of the biceps brachii tendon is relatively uncommon. In the vast majority of cases, it happens in the long head of the dominant arm of middle-aged men. Diagnosis is mainly clinical; however, ultrasound and MRI can confirm the diagnosis when there is doubt. Nonoperative management is appropriate for the majority of patients. Young athletes, patients who are concerned with cosmetic appearance, and labor workers with injury to their dominant arm should be referred to an orthopedic surgeon for possible surgery.
1. Geaney LE, Mazzocca AD. Biceps brachii tendon ruptures: a review of diagnosis and treatment of proximal and distal biceps tendon ruptures. Phys Sportsmed. 2010;38:117-125.
2. Payne LZ, Deng XH, Craig EV, et al. The combined dynamic and static contributions to subacromial impingement. A biomechanical analysis. Am J Sports Med. 1997;25:801-808.
3. Jayamoorthy T, Field JR, Costi JJ, et al. Biceps tenodesis: a biomechanical study of fixation methods. J Shoulder Elbow Surg. 2004;13:160-164.
4. Mazzocca AD, Spang JT, Arciero RA. Distal biceps rupture. Orthop Clin North Am. 2008;39:237-249, vii.
5. Carter AN, Erickson SM. Proximal biceps tendon rupture: primarily an injury of middle age. Phys Sportsmed. 1999;27:95-101.
6. Elser F, Braun S, Dewing CB, et al. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27:581-592.
7. Kelly MP, Perkinson SG, Ablove RH, et al. Distal biceps tendon ruptures: an epidemiological analysis using a large population database. Am J Sports Med. 2015;43:2012-2017.
8. Schneider A, Bennett JM, O’Connor DP, et al. Bilateral ruptures of the distal biceps brachii tendon. J Shoulder Elbow Surg. 2009;18:804-807.
9. Sethi N, Wright R, Yamaguchi K. Disorders of the long head of the biceps tendon. J Shoulder Elbow Surg. 1999;8:644-654.
10. The Physician and Sportsmedicine. Complete rupture of large tendons. Risk factors, signs, and definitive treatment. Available at: https://orthony.com/directory/uploads/flik_complete-rupture-of-large-tendons.pdf. Accessed December 8, 2017.
11. Pearl ML, Bessos K, Wong K. Strength deficits related to distal biceps tendon rupture and repair. A case report. Am J Sports Med. 1998;26:295-296.
12. Deutch SR, Gelineck J, Johannsen HV, et al. Permanent disabilities in the displaced muscle from rupture of the long head tendon of the biceps. Scand J Med Sci Sports. 2005;15:159-162.
13. Tangari M, Carbone S, Gallo M, et al. Long head of the biceps tendon rupture in professional wrestlers: treatment with a mini-open tenodesis. J Shoulder Elbow Surg. 2011;20:409-413.
14. Hsu AR, Ghodadra NS, Provencher MT, et al. Biceps tenotomy versus tenodesis: a review of clinical outcomes and biomechanical results. J Shoulder Elbow Surg. 2011;20:326-332.
15. Sears BW, Spencer EE, Getz CL. Humeral fracture following subpectoral biceps tenodesis in 2 active, healthy patients. J Shoulder Elbow Surg. 2011;20:e7-e11.
THE CASE
A 41-year-old, right-hand dominant man sought care at our facility one day after trying to pull his boat out of the water. He’d tried to lift the boat with his hands while his forearms were fully supinated and his elbows were flexed to about 90°. He then felt a sharp burning sensation in his left anterior shoulder and was unable to lift the boat. The patient denied feeling a popping sensation at the time of the injury. He had mild pain at night, but was able to sleep. He said that he had mild diminished strength with elbow flexion, but denied having any numbness, tingling, or discoloration of his skin.
The patient said he did weightlifting and strength training of his upper and lower extremities 4 times/week. He was in good general health, was not taking any medications or supplements, and denied smoking or using illicit drugs. His surgical history was significant for a Bankart repair 8 years ago.
On physical examination, the patient had a scar from the previous surgery, a hollow area in his left anterior shoulder, and a prominent biceps muscle belly (FIGURE). His shoulder range of motion was normal. Left shoulder Neer, Hawkins-Kennedy, drop-arm, cross-arm, empty can, and apprehension tests were negative. A left Speed’s test (resisted elbow flexion when elbow is flexed 20° to 30° with the forearm in supination and the arm in about 60° of flexion) was positive for mild anterior shoulder pain. So, too, was a Yergason’s test (resisted forearm supination and elbow flexion when forearm is pronated and elbow is flexed to 90°). The patient’s elbow flexion strength was 4 out of 5, and his supination strength was 5 out of 5. Neurovascular and sensory examinations of his upper extremities, including radial and ulnar pulses, were normal.
THE DIAGNOSIS
A diagnostic musculoskeletal ultrasound revealed an empty tendon sheath of the long head of the biceps in the bicipital groove and a retracted echogenic stump with associated hematoma at the proximal musculotendinous junction. Based on the patient’s history, physical examination, and ultrasound, a diagnosis of an acute rupture of the left long head of the biceps brachii tendon was made.
DISCUSSION
Diagnosis of acute rupture is often made clinically based on a visually apparent defect proximally and a bulbous mass distally (“Popeye deformity”).1 Ultrasound and magnetic resonance imaging (MRI) may aid in the diagnosis by demonstrating an absence of the long head in the bicipital groove or at its insertion.
The biceps brachii tendon functions in flexion and supination of the forearm. The long head of the biceps also plays a stabilizing role in the glenohumeral joint during elbow flexion and supination.2 Injury to the biceps most often occurs in middle-aged men following a traumatic sudden eccentric bicipital contraction event, during which most patients describe a snapping or popping sensation.3,4
Rupture of the proximal biceps tendon represents about 90% of all biceps ruptures, which almost exclusively involve the long head of the biceps.3,5,6 Risk factors for tendon rupture include obesity, smoking, steroid injection in or around the tendon, and previous tendinopathy.7-10
Functional limitations. It is generally thought that functional limitations following a proximal biceps rupture are relatively minimal, due to the work of other flexors and supinators, including the brachialis and brachioradialis. However, because strength and endurance of the muscle can decrease by about 25%, physical laborers and high-demand athletes may notice a degree of residual weakness with supination and elbow flexion.11,12
Surgery is suitable for some, but not all
Surgical repair is recommended for acute ruptures in patients with high physical demands and for whom a slight loss of flexion and supination strength would not be well tolerated.13 Tenotomy and tenodesis are the main techniques used to surgically repair a rupture of the long head of the biceps brachii tendon. Although there is no consensus on which technique is superior, it seems that there is less cosmetic deformity and better post-surgery biomechanical strength with tenodesis compared with tenotomy.14 However, tenodesis is associated with a higher likelihood of bicipital pain,14 and recent case reports have suggested it is associated with an increased risk of humeral fracture.15 Therefore, each patient should be treated on an individual case basis, taking into account age, activity level, and physical demand.14
For most patients, treatment remains conservative with typically excellent outcomes. Nonoperative management includes gentle range-of-motion exercises for the prevention of contractures of the elbow and shoulder. Such exercises can be started almost immediately after injury. In one study, nonoperative management was recommended for patients with sedentary work, injury in the non-dominant arm, and acceptable cosmetic deformity. Researchers noted that patients who opt for a nonsurgical treatment generally do well with a home exercise program and rarely have stiffness.1
If the patient is a young athlete, if cosmetic deformity is unacceptable, or if the injury is in the dominant arm of a laborer, then the patient may want to consider tenodesis.1 Tangari et al found that in high-demand athletes, biceps tenodesis resulted in excellent functional and cosmetic results with no clinically significant decrease in strength after an average follow-up of 7.6 years.13 In a case s
Our patient elected to proceed with a tenodesis procedure. Two months after the surgery, he had fully recovered.
THE TAKEAWAY
Rupture of the biceps brachii tendon is relatively uncommon. In the vast majority of cases, it happens in the long head of the dominant arm of middle-aged men. Diagnosis is mainly clinical; however, ultrasound and MRI can confirm the diagnosis when there is doubt. Nonoperative management is appropriate for the majority of patients. Young athletes, patients who are concerned with cosmetic appearance, and labor workers with injury to their dominant arm should be referred to an orthopedic surgeon for possible surgery.
THE CASE
A 41-year-old, right-hand dominant man sought care at our facility one day after trying to pull his boat out of the water. He’d tried to lift the boat with his hands while his forearms were fully supinated and his elbows were flexed to about 90°. He then felt a sharp burning sensation in his left anterior shoulder and was unable to lift the boat. The patient denied feeling a popping sensation at the time of the injury. He had mild pain at night, but was able to sleep. He said that he had mild diminished strength with elbow flexion, but denied having any numbness, tingling, or discoloration of his skin.
The patient said he did weightlifting and strength training of his upper and lower extremities 4 times/week. He was in good general health, was not taking any medications or supplements, and denied smoking or using illicit drugs. His surgical history was significant for a Bankart repair 8 years ago.
On physical examination, the patient had a scar from the previous surgery, a hollow area in his left anterior shoulder, and a prominent biceps muscle belly (FIGURE). His shoulder range of motion was normal. Left shoulder Neer, Hawkins-Kennedy, drop-arm, cross-arm, empty can, and apprehension tests were negative. A left Speed’s test (resisted elbow flexion when elbow is flexed 20° to 30° with the forearm in supination and the arm in about 60° of flexion) was positive for mild anterior shoulder pain. So, too, was a Yergason’s test (resisted forearm supination and elbow flexion when forearm is pronated and elbow is flexed to 90°). The patient’s elbow flexion strength was 4 out of 5, and his supination strength was 5 out of 5. Neurovascular and sensory examinations of his upper extremities, including radial and ulnar pulses, were normal.
THE DIAGNOSIS
A diagnostic musculoskeletal ultrasound revealed an empty tendon sheath of the long head of the biceps in the bicipital groove and a retracted echogenic stump with associated hematoma at the proximal musculotendinous junction. Based on the patient’s history, physical examination, and ultrasound, a diagnosis of an acute rupture of the left long head of the biceps brachii tendon was made.
DISCUSSION
Diagnosis of acute rupture is often made clinically based on a visually apparent defect proximally and a bulbous mass distally (“Popeye deformity”).1 Ultrasound and magnetic resonance imaging (MRI) may aid in the diagnosis by demonstrating an absence of the long head in the bicipital groove or at its insertion.
The biceps brachii tendon functions in flexion and supination of the forearm. The long head of the biceps also plays a stabilizing role in the glenohumeral joint during elbow flexion and supination.2 Injury to the biceps most often occurs in middle-aged men following a traumatic sudden eccentric bicipital contraction event, during which most patients describe a snapping or popping sensation.3,4
Rupture of the proximal biceps tendon represents about 90% of all biceps ruptures, which almost exclusively involve the long head of the biceps.3,5,6 Risk factors for tendon rupture include obesity, smoking, steroid injection in or around the tendon, and previous tendinopathy.7-10
Functional limitations. It is generally thought that functional limitations following a proximal biceps rupture are relatively minimal, due to the work of other flexors and supinators, including the brachialis and brachioradialis. However, because strength and endurance of the muscle can decrease by about 25%, physical laborers and high-demand athletes may notice a degree of residual weakness with supination and elbow flexion.11,12
Surgery is suitable for some, but not all
Surgical repair is recommended for acute ruptures in patients with high physical demands and for whom a slight loss of flexion and supination strength would not be well tolerated.13 Tenotomy and tenodesis are the main techniques used to surgically repair a rupture of the long head of the biceps brachii tendon. Although there is no consensus on which technique is superior, it seems that there is less cosmetic deformity and better post-surgery biomechanical strength with tenodesis compared with tenotomy.14 However, tenodesis is associated with a higher likelihood of bicipital pain,14 and recent case reports have suggested it is associated with an increased risk of humeral fracture.15 Therefore, each patient should be treated on an individual case basis, taking into account age, activity level, and physical demand.14
For most patients, treatment remains conservative with typically excellent outcomes. Nonoperative management includes gentle range-of-motion exercises for the prevention of contractures of the elbow and shoulder. Such exercises can be started almost immediately after injury. In one study, nonoperative management was recommended for patients with sedentary work, injury in the non-dominant arm, and acceptable cosmetic deformity. Researchers noted that patients who opt for a nonsurgical treatment generally do well with a home exercise program and rarely have stiffness.1
If the patient is a young athlete, if cosmetic deformity is unacceptable, or if the injury is in the dominant arm of a laborer, then the patient may want to consider tenodesis.1 Tangari et al found that in high-demand athletes, biceps tenodesis resulted in excellent functional and cosmetic results with no clinically significant decrease in strength after an average follow-up of 7.6 years.13 In a case s
Our patient elected to proceed with a tenodesis procedure. Two months after the surgery, he had fully recovered.
THE TAKEAWAY
Rupture of the biceps brachii tendon is relatively uncommon. In the vast majority of cases, it happens in the long head of the dominant arm of middle-aged men. Diagnosis is mainly clinical; however, ultrasound and MRI can confirm the diagnosis when there is doubt. Nonoperative management is appropriate for the majority of patients. Young athletes, patients who are concerned with cosmetic appearance, and labor workers with injury to their dominant arm should be referred to an orthopedic surgeon for possible surgery.
1. Geaney LE, Mazzocca AD. Biceps brachii tendon ruptures: a review of diagnosis and treatment of proximal and distal biceps tendon ruptures. Phys Sportsmed. 2010;38:117-125.
2. Payne LZ, Deng XH, Craig EV, et al. The combined dynamic and static contributions to subacromial impingement. A biomechanical analysis. Am J Sports Med. 1997;25:801-808.
3. Jayamoorthy T, Field JR, Costi JJ, et al. Biceps tenodesis: a biomechanical study of fixation methods. J Shoulder Elbow Surg. 2004;13:160-164.
4. Mazzocca AD, Spang JT, Arciero RA. Distal biceps rupture. Orthop Clin North Am. 2008;39:237-249, vii.
5. Carter AN, Erickson SM. Proximal biceps tendon rupture: primarily an injury of middle age. Phys Sportsmed. 1999;27:95-101.
6. Elser F, Braun S, Dewing CB, et al. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27:581-592.
7. Kelly MP, Perkinson SG, Ablove RH, et al. Distal biceps tendon ruptures: an epidemiological analysis using a large population database. Am J Sports Med. 2015;43:2012-2017.
8. Schneider A, Bennett JM, O’Connor DP, et al. Bilateral ruptures of the distal biceps brachii tendon. J Shoulder Elbow Surg. 2009;18:804-807.
9. Sethi N, Wright R, Yamaguchi K. Disorders of the long head of the biceps tendon. J Shoulder Elbow Surg. 1999;8:644-654.
10. The Physician and Sportsmedicine. Complete rupture of large tendons. Risk factors, signs, and definitive treatment. Available at: https://orthony.com/directory/uploads/flik_complete-rupture-of-large-tendons.pdf. Accessed December 8, 2017.
11. Pearl ML, Bessos K, Wong K. Strength deficits related to distal biceps tendon rupture and repair. A case report. Am J Sports Med. 1998;26:295-296.
12. Deutch SR, Gelineck J, Johannsen HV, et al. Permanent disabilities in the displaced muscle from rupture of the long head tendon of the biceps. Scand J Med Sci Sports. 2005;15:159-162.
13. Tangari M, Carbone S, Gallo M, et al. Long head of the biceps tendon rupture in professional wrestlers: treatment with a mini-open tenodesis. J Shoulder Elbow Surg. 2011;20:409-413.
14. Hsu AR, Ghodadra NS, Provencher MT, et al. Biceps tenotomy versus tenodesis: a review of clinical outcomes and biomechanical results. J Shoulder Elbow Surg. 2011;20:326-332.
15. Sears BW, Spencer EE, Getz CL. Humeral fracture following subpectoral biceps tenodesis in 2 active, healthy patients. J Shoulder Elbow Surg. 2011;20:e7-e11.
1. Geaney LE, Mazzocca AD. Biceps brachii tendon ruptures: a review of diagnosis and treatment of proximal and distal biceps tendon ruptures. Phys Sportsmed. 2010;38:117-125.
2. Payne LZ, Deng XH, Craig EV, et al. The combined dynamic and static contributions to subacromial impingement. A biomechanical analysis. Am J Sports Med. 1997;25:801-808.
3. Jayamoorthy T, Field JR, Costi JJ, et al. Biceps tenodesis: a biomechanical study of fixation methods. J Shoulder Elbow Surg. 2004;13:160-164.
4. Mazzocca AD, Spang JT, Arciero RA. Distal biceps rupture. Orthop Clin North Am. 2008;39:237-249, vii.
5. Carter AN, Erickson SM. Proximal biceps tendon rupture: primarily an injury of middle age. Phys Sportsmed. 1999;27:95-101.
6. Elser F, Braun S, Dewing CB, et al. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27:581-592.
7. Kelly MP, Perkinson SG, Ablove RH, et al. Distal biceps tendon ruptures: an epidemiological analysis using a large population database. Am J Sports Med. 2015;43:2012-2017.
8. Schneider A, Bennett JM, O’Connor DP, et al. Bilateral ruptures of the distal biceps brachii tendon. J Shoulder Elbow Surg. 2009;18:804-807.
9. Sethi N, Wright R, Yamaguchi K. Disorders of the long head of the biceps tendon. J Shoulder Elbow Surg. 1999;8:644-654.
10. The Physician and Sportsmedicine. Complete rupture of large tendons. Risk factors, signs, and definitive treatment. Available at: https://orthony.com/directory/uploads/flik_complete-rupture-of-large-tendons.pdf. Accessed December 8, 2017.
11. Pearl ML, Bessos K, Wong K. Strength deficits related to distal biceps tendon rupture and repair. A case report. Am J Sports Med. 1998;26:295-296.
12. Deutch SR, Gelineck J, Johannsen HV, et al. Permanent disabilities in the displaced muscle from rupture of the long head tendon of the biceps. Scand J Med Sci Sports. 2005;15:159-162.
13. Tangari M, Carbone S, Gallo M, et al. Long head of the biceps tendon rupture in professional wrestlers: treatment with a mini-open tenodesis. J Shoulder Elbow Surg. 2011;20:409-413.
14. Hsu AR, Ghodadra NS, Provencher MT, et al. Biceps tenotomy versus tenodesis: a review of clinical outcomes and biomechanical results. J Shoulder Elbow Surg. 2011;20:326-332.
15. Sears BW, Spencer EE, Getz CL. Humeral fracture following subpectoral biceps tenodesis in 2 active, healthy patients. J Shoulder Elbow Surg. 2011;20:e7-e11.
She’s Not My Mother: A 24-Year-Old Man With Capgras Delusion
Many patients admitted to inpatient psychiatric hospitals present with delusions; however, the Capgras delusion is a rare type that often appears as a sequela of certain medical and neurologic conditions.1 The Capgras delusion is a condition in which a person believes that either an individual or a group of people has been replaced by doubles or imposters.
In 1923, French psychiatrist Joseph Capgras first described the delusion. He and Jean Reboul-Lachaux coauthored a paper on a 53-year-old woman. The patient was a paranoid megalomaniac who “transformed everyone in her entourage, even those closest to her, such as her husband and daughter, into various and numerous doubles.”2 She believed she was famous, wealthy, and of royal lineage. Although 3 of her children had died, she believed that they were abducted, and that her only surviving child was replaced by a look-alike.2,3 Although the prevalence of such delusions in the general population has not been fully studied, a psychiatric hospital in Turkey found a 1.3% prevalence (1.8% women and 0.9% men) in 920 admissions over 5 years.4
The Capgras delusion is one of many delusions related to the misidentification of people, places, or objects; these delusions collectively are known as delusional misidentification syndrome (DMS).5,6 The Fregoli delusion involves the belief that several different people are the same person in disguise. Intermetamorphosis is the belief that an individual has been transformed internally and externally to another person. Subjective doubles is the belief that a doppelganger of the afflicted person exists, living and functioning independently in the world. Reduplicative paramnesia is the belief that a person, place, or object has been duplicated. A rarer example of DMS is the Cotard delusion, which is the belief that the patient himself or herself is dead, putrefying, exsanguinating, or lacking internal organs.
The most common of the DMS is the Capgras delusion. One common presentation of Capgras delusion involves the spouse of the patient, who believes that an imposter of the same sex as their spouse has taken over his or her body. Rarer delusions are those in which a person misidentifies him or herself as the imposter.3,5,6
Case Presentation
This case involved a 24-year-old male veteran who had received a wide range of mental health diagnoses in the past, including major depressive disorder (MDD) with psychotic features, generalized anxiety disorder, cannabis use disorder, adjustment disorder, and borderline personality disorder. He also had a medical history related to a motor vehicle accident with subsequent intestinal rupture and colostomy placement that had occurred a year and a half prior to presentation. He had no history of brain trauma.
The patient voluntarily presented to the hospital for increased suicidal thoughts and was admitted voluntarily for stabilization and self-harm prevention. He stated that “I feel everything is unreal. I feel suicidal and guilt” and endorsed a plan to either walk into traffic or shoot himself in the head due to increasingly distressing thoughts and memories. According to the patient, he had reported to the police that he raped his ex-girlfriend a year previously, although she denied the claim to the police.
The patient further disclosed that he did not believe his mother was real. “Last year my sister told me it was not 2016, but it was 2022,” he said. “She told me that I have hurt my mother with a padlock—that you could no longer identify her face. I don’t remember having done this. I have lived with her since that time, so I don’t think it’s really [my mother].” He believed that his mother was replaced by “government employees” who were sent to elicit confessions for his behavior while in the military. He expressed guilt over several actions he had performed while in military service, such as punching a wall during boot camp, stealing “soak-up” pads, and napping during work hours. His mother was contacted by a staff psychiatrist in the inpatient unit and denied that any assault had taken place.
The patient’s psychiatric review of systems was positive for visual hallucinations (specifically “blurs” next to his bed in the morning that disappeared as he tried to touch them), depressed mood, anxiety, hopelessness, and insomnia. Pertinent negatives of the review of systems included a denial of manic symptoms and auditory hallucinations. For additional details of his past psychiatric history, the patient admitted that his motor vehicle accident, intestinal rupture, and colostomy were the result of his 1 suicide attempt a year and a half prior after a verbal dispute with the same ex-girlfriend that he believed he had raped. After undergoing extensive medical and surgical treatment, he began seeing an outpatient psychiatrist as well as attending substance use counseling to curtail his marijuana use. He was prescribed a combination of duloxetine and risperidone as an outpatient, which he was taking with intermittent adherence.
Regarding substance use, the patient admitted to using marijuana regularly in the past but quit completely 1 month prior and denied any other drug use or alcohol use. He reported a family history of a sister who was undergoing treatment for bipolar disorder. In his social history, the patient disclosed that he was raised by both parents and described a good childhood with a life absent of abuse in any form. He was single with no children. Although he was unemployed, he lived off the funds from an insurance settlement from his motor vehicle accident. He was living in a trailer with his brother and mother. He also denied having access to firearms.
The patient was overweight, neatly groomed, had good eye contact, and was calm and cooperative. He seemed anxious as evidenced by his continuous shaking of his feet; although speech was normal in rate and tone. He reported his mood as “depressed and anxious” with congruent and tearful affect. His thought process was concrete, although his thought content contained delusions, suicidal ideation, and paranoia. He denied any homicidal thoughts or thoughts of harming others. He did not present with any auditory or visual hallucinations. Insight and judgment were poor. The mental status examination revealed no notable deficits in cognition.
The patient’s differential diagnosis included schizophreniform disorder, exacerbation of MDD with psychotic features, and the psychotic component of cannabis use disorder. His outpatient risperidone and duloxetine were not restarted. Aripiprazole 15 mg daily was prescribed for his delusions, paranoia, and visual hallucinations. The patient also received a prescription for hydroxyzine 50 mg every 6 hours as needed for anxiety.
Because of the nature of his delusions, comorbid medical and neurologic conditions were considered. Neurology consultation recommended a noncontrast head computer tomography (CT) scan and an electroencephalogram (EEG). Laboratory workup included HIV antibody, thyroid panel, chemistry panel, complete blood count, hepatitis B serum antigen, urine drug screen, hepatitis C virus, and rapid plasma reagin. All laboratory results were benign and unremarkable, and the urine drug screen was negative. The noncontrast CT revealed no acute findings, and the EEG revealed no recorded epileptiform abnormalities or seizures.
Throughout his hospital course, the patient remained cooperative with treatment. Three days into the hospitalization, he stated that he believed the entire family had been replaced by imposters. He began to distrust members of his family and was reticent to communicate with them when they attempted to contact him. He also experienced fragmented sleep during his hospital stay, and trazodone 50 mg at bedtime was added.
After aripiprazole was increased to 20 mg daily on hospital day 2 and then to 30 mg daily on hospital day 3 due to the patient’s delusions, he began to doubt the validity of his beliefs. After showing gradual improvement over 6 days, the patient reported that he no longer believed that those memories were real. His sleep, depressed mood, anxiety, and paranoia had markedly improved toward the end of the hospitalization and suicidal ideation/intent resolved. The patient was discharged home to his mother and brother after 6 days of hospitalization with aripiprazole 30 mg daily and trazodone 50 mg at bedtime.
Discussion
The Capgras delusion can present in several different contexts. A psychiatric differential diagnosis includes disorders in the schizophrenia spectrum (brief psychotic disorder, schizophreniform disorder, and schizophrenia), schizoaffective disorder, delusional disorder, and substance-induced psychotic disorder. In addition to psychiatric disorders, the Capgras delusion has been shown to occur in several medical conditions, which include stroke, central nervous system tumors, subarachnoid hemorrhage, vitamin B12 deficiency, hepatic encephalopathy, hypothyroidism, hyperparathyroidism, epilepsy, and dementia.1,2,4,7
A 2007 retrospective study by Josephs examined 47 patients diagnosed with the Capgras delusion from several tertiary care centers. Of those patients, 38 (81%) had a neurodegenerative disease, most commonly Lewy body dementia (LBD).1 In his review of the Josephs study, Devinsky proposed that the loss of striatal D2 receptors in LBD may be implicated in the manifestation of Capgras delusions.2 The data suggest multiple brain regions may be involved, including the frontal lobes, right temporal lobe, right parietal lobe, parahippocampus, and amygdala.1,2 Most patients in the Josephs study demonstrated global atrophy on imaging studies. One hypothesis is that it is the disconnection of the frontal lobe to other brain regions that may be implicated.1,2,4 This results in intact recognition of facial features of familiar people, impaired emotional recognition, and impaired self-correction due to executive dysfunction.
Methamphetamine also has been implicated in a small number of cases of Capgras; the proposed mechanism involves dopaminergic neuronal impairment/loss.1,2 Additionally, Capgras delusions have been described in cases of patients treated with antimalarial medications, such as chloroquine.8 Younger patients with the Capgras delusion were more likely to have purely psychiatric comorbidities—such as schizophrenia, substance-induced psychosis, or schizoaffective disorder—as opposed to underlying medical conditions.1 In the case presented here, the Capgras delusion was thought to be due to a disorder in the schizophrenia spectrum, specifically schizophreniform disorder.
Because an increasing amount of evidence indicates that the Capgras delusion is associated with certain medical conditions, a workup should be performed to rule out underlying medical etiology. Of note, no official guidelines for the workup have been produced for the Capgras delusion. However, the workup may include brain imaging, such as magnetic resonance imaging and/or CT scan to rule out mass lesions, vascular malformations, stroke, or neuro-infectious processes; laboratory tests, such as vitamin B12, liver panel, HIV, rapid plasma reagin, hepatitis B and C viruses, parathyroid hormone levels, urine drug screen, and thyroid panel can be ordered to rule out other medical causes.1,2,6,7,9
Consultations with internal medicine and neurology departments may be beneficial. Although treatment of the underlying condition may lead to an improvement in the symptoms, full remission in all cases has not been consistently demonstrated in the current literature.5,7,9,10 Patients with the Capgras delusion are challenging to treat, because their delusions have been shown to be refractory to antipsychotic therapy. However, antipsychotics are currently the mainstay of treatment. Some case studies have shown efficacy with pimozide, tricyclic antidepressants, and mirtazapine.6,9
One case study in 2014 in India of a 45-year-old woman who believed her husband and son were replaced by imposters out to kill her, showed a 40% to 50% reduction of paranoia, irritability, and suspicious scanning behaviors with a combination of risperidone and trihexyphenidyl. Despite the improvements, the woman continued to have delusions.7
A notable feature associated with those experiencing the Capgras delusion is the increased risk of violent behaviors, often because of suspiciousness and paranoia. A 2004 review suggested the risk of violence and homicidality is much higher in male patients compared with that of female patients with the Capgras delusion.9 This is despite evidence suggesting that the prevalence of the Capgras delusion seems to be greater in women.6,9 Moreover, patients often demonstrated social withdrawal and self-isolation prior to violent acts. The victims often were family members or those who live with the patient, which is consistent with the evidence that those most familiar to patients are more likely to be misidentified.1,2,7,9,10
A 1989 case series that examined 8 cases of the Capgras delusion listed the following violent behaviors: shot and killed father, pointed knife at mother, held knife to mother’s throat, punched parents, threatened to stab husband with scissors, nonspecifically threatened physical harm to family, injured mother with axe, and threatened to stab son with knife and burn him. Seven of the 8 patients lived with the misidentified persons, and 5 of the 8 patients were treatment resistant. The study posited that chronicity of the delusion, content of the delusion, and accessibility of misidentified persons seemed to increase the risk of violent behaviors. These authors went on to suggest that despite the appearance of stability, patients may react violently to minute changes.10 Overall literature seems to suggest the importance of performing a violence and homicidality assessment with special attention to assessment for themes of hostility toward misidentified individuals.9,10
Conclusion
The Capgras delusion is an uncommon symptom associated with varied psychiatric, medical, iatrogenic, and neurologic conditions. Treatment of underlying medical conditions may improve or resolve the delusions. However, in this case, the patient did not seem to have any underlying medical conditions, and it was thought that he may have been experiencing a prodrome within the schizophrenia spectrum. This is consistent with the literature, which suggests that those with the delusions at younger ages may have a psychiatric etiology.
Although this patient was responsive to aripiprazole, the Capgras delusion has been known to be resistant to antipsychotic therapy. It is worth considering a medical and neurologic workup with the addition of a psychiatry referral. Further, while the patient in the presented case had the delusion that he had assaulted his mother, whom he misidentified as an imposter, the patient did not demonstrate any hostility and denied thoughts of harming her. However, given the increased risk of violence in patients with the Capgras delusion, a homicidality and violence assessment should be performed. While further recommendations are outside the scope of this article, the provider should be cognizant of local duty-to-warn and duty-to-protect laws regarding potentially homicidal patients.
1. Josephs KA. The Capgras delusion and its relationship to neurodegenerative disease. Arch Neurol. 2007;64(12):1762-1766.
2. Devinsky O. Behavioral neurology. The neurology of the Capgras delusion. Rev Neurol Dis. 2008;5(2):97-100.
3. Sadock BJ, Sadock VA. Kaplan & Sadock’s Synopsis of Psychiatry: Behavioral Sciences/Clinical Psychiatry. 10th ed. Philadelphia, PA: Wolters Kluwer; 2007.
4. Tamam L, Karatas G, Zeren T, Ozpyraz N. The prevalence of Capgras syndrome in a university hospital setting. Acta Neuropsychiatr. 2003;15(5):290-295.
5. Klein CA, Hirchan S. The masks of identities: who’s who? Delusional misidentification syndromes. J Am Acad Psychiatry Law. 2014;42(3):369-378.
6. Atta K. Forlenza N, Gujski M, Hashmi S, Isaac G. Delusional misidentification syndromes: separate disorders or unusual presentations of existing DSM-IV categories? Psychiatry (Edgemont). 2006;3(9):56-61.
7. Sathe H, Karia S, De Sousa A, Shah N. Capgras syndrome: a case report. Paripex Indian J Res. 2014;3(8):134-135. 8. Bhatia MS, Singhal PK, Agrawal P, Malik SC. Capgras’ syndrome in chloroquine induced psychosis. Indian J Psychiatry. 1988;30(3):311-313.
9. Bourget D, Whitehurst L. Capgras syndrome: a review of the neurophysiological correlates and presenting clinical features in cases involving physical violence. Can J Psychiatry. 2004;49(11):719-725.
10. Silva JA, Leong GB, Weinstock R, Boyer CL. Capgras syndrome and dangerousness. Bull Am Acad Psychiatry Law. 1989;17(1):5-14.
Many patients admitted to inpatient psychiatric hospitals present with delusions; however, the Capgras delusion is a rare type that often appears as a sequela of certain medical and neurologic conditions.1 The Capgras delusion is a condition in which a person believes that either an individual or a group of people has been replaced by doubles or imposters.
In 1923, French psychiatrist Joseph Capgras first described the delusion. He and Jean Reboul-Lachaux coauthored a paper on a 53-year-old woman. The patient was a paranoid megalomaniac who “transformed everyone in her entourage, even those closest to her, such as her husband and daughter, into various and numerous doubles.”2 She believed she was famous, wealthy, and of royal lineage. Although 3 of her children had died, she believed that they were abducted, and that her only surviving child was replaced by a look-alike.2,3 Although the prevalence of such delusions in the general population has not been fully studied, a psychiatric hospital in Turkey found a 1.3% prevalence (1.8% women and 0.9% men) in 920 admissions over 5 years.4
The Capgras delusion is one of many delusions related to the misidentification of people, places, or objects; these delusions collectively are known as delusional misidentification syndrome (DMS).5,6 The Fregoli delusion involves the belief that several different people are the same person in disguise. Intermetamorphosis is the belief that an individual has been transformed internally and externally to another person. Subjective doubles is the belief that a doppelganger of the afflicted person exists, living and functioning independently in the world. Reduplicative paramnesia is the belief that a person, place, or object has been duplicated. A rarer example of DMS is the Cotard delusion, which is the belief that the patient himself or herself is dead, putrefying, exsanguinating, or lacking internal organs.
The most common of the DMS is the Capgras delusion. One common presentation of Capgras delusion involves the spouse of the patient, who believes that an imposter of the same sex as their spouse has taken over his or her body. Rarer delusions are those in which a person misidentifies him or herself as the imposter.3,5,6
Case Presentation
This case involved a 24-year-old male veteran who had received a wide range of mental health diagnoses in the past, including major depressive disorder (MDD) with psychotic features, generalized anxiety disorder, cannabis use disorder, adjustment disorder, and borderline personality disorder. He also had a medical history related to a motor vehicle accident with subsequent intestinal rupture and colostomy placement that had occurred a year and a half prior to presentation. He had no history of brain trauma.
The patient voluntarily presented to the hospital for increased suicidal thoughts and was admitted voluntarily for stabilization and self-harm prevention. He stated that “I feel everything is unreal. I feel suicidal and guilt” and endorsed a plan to either walk into traffic or shoot himself in the head due to increasingly distressing thoughts and memories. According to the patient, he had reported to the police that he raped his ex-girlfriend a year previously, although she denied the claim to the police.
The patient further disclosed that he did not believe his mother was real. “Last year my sister told me it was not 2016, but it was 2022,” he said. “She told me that I have hurt my mother with a padlock—that you could no longer identify her face. I don’t remember having done this. I have lived with her since that time, so I don’t think it’s really [my mother].” He believed that his mother was replaced by “government employees” who were sent to elicit confessions for his behavior while in the military. He expressed guilt over several actions he had performed while in military service, such as punching a wall during boot camp, stealing “soak-up” pads, and napping during work hours. His mother was contacted by a staff psychiatrist in the inpatient unit and denied that any assault had taken place.
The patient’s psychiatric review of systems was positive for visual hallucinations (specifically “blurs” next to his bed in the morning that disappeared as he tried to touch them), depressed mood, anxiety, hopelessness, and insomnia. Pertinent negatives of the review of systems included a denial of manic symptoms and auditory hallucinations. For additional details of his past psychiatric history, the patient admitted that his motor vehicle accident, intestinal rupture, and colostomy were the result of his 1 suicide attempt a year and a half prior after a verbal dispute with the same ex-girlfriend that he believed he had raped. After undergoing extensive medical and surgical treatment, he began seeing an outpatient psychiatrist as well as attending substance use counseling to curtail his marijuana use. He was prescribed a combination of duloxetine and risperidone as an outpatient, which he was taking with intermittent adherence.
Regarding substance use, the patient admitted to using marijuana regularly in the past but quit completely 1 month prior and denied any other drug use or alcohol use. He reported a family history of a sister who was undergoing treatment for bipolar disorder. In his social history, the patient disclosed that he was raised by both parents and described a good childhood with a life absent of abuse in any form. He was single with no children. Although he was unemployed, he lived off the funds from an insurance settlement from his motor vehicle accident. He was living in a trailer with his brother and mother. He also denied having access to firearms.
The patient was overweight, neatly groomed, had good eye contact, and was calm and cooperative. He seemed anxious as evidenced by his continuous shaking of his feet; although speech was normal in rate and tone. He reported his mood as “depressed and anxious” with congruent and tearful affect. His thought process was concrete, although his thought content contained delusions, suicidal ideation, and paranoia. He denied any homicidal thoughts or thoughts of harming others. He did not present with any auditory or visual hallucinations. Insight and judgment were poor. The mental status examination revealed no notable deficits in cognition.
The patient’s differential diagnosis included schizophreniform disorder, exacerbation of MDD with psychotic features, and the psychotic component of cannabis use disorder. His outpatient risperidone and duloxetine were not restarted. Aripiprazole 15 mg daily was prescribed for his delusions, paranoia, and visual hallucinations. The patient also received a prescription for hydroxyzine 50 mg every 6 hours as needed for anxiety.
Because of the nature of his delusions, comorbid medical and neurologic conditions were considered. Neurology consultation recommended a noncontrast head computer tomography (CT) scan and an electroencephalogram (EEG). Laboratory workup included HIV antibody, thyroid panel, chemistry panel, complete blood count, hepatitis B serum antigen, urine drug screen, hepatitis C virus, and rapid plasma reagin. All laboratory results were benign and unremarkable, and the urine drug screen was negative. The noncontrast CT revealed no acute findings, and the EEG revealed no recorded epileptiform abnormalities or seizures.
Throughout his hospital course, the patient remained cooperative with treatment. Three days into the hospitalization, he stated that he believed the entire family had been replaced by imposters. He began to distrust members of his family and was reticent to communicate with them when they attempted to contact him. He also experienced fragmented sleep during his hospital stay, and trazodone 50 mg at bedtime was added.
After aripiprazole was increased to 20 mg daily on hospital day 2 and then to 30 mg daily on hospital day 3 due to the patient’s delusions, he began to doubt the validity of his beliefs. After showing gradual improvement over 6 days, the patient reported that he no longer believed that those memories were real. His sleep, depressed mood, anxiety, and paranoia had markedly improved toward the end of the hospitalization and suicidal ideation/intent resolved. The patient was discharged home to his mother and brother after 6 days of hospitalization with aripiprazole 30 mg daily and trazodone 50 mg at bedtime.
Discussion
The Capgras delusion can present in several different contexts. A psychiatric differential diagnosis includes disorders in the schizophrenia spectrum (brief psychotic disorder, schizophreniform disorder, and schizophrenia), schizoaffective disorder, delusional disorder, and substance-induced psychotic disorder. In addition to psychiatric disorders, the Capgras delusion has been shown to occur in several medical conditions, which include stroke, central nervous system tumors, subarachnoid hemorrhage, vitamin B12 deficiency, hepatic encephalopathy, hypothyroidism, hyperparathyroidism, epilepsy, and dementia.1,2,4,7
A 2007 retrospective study by Josephs examined 47 patients diagnosed with the Capgras delusion from several tertiary care centers. Of those patients, 38 (81%) had a neurodegenerative disease, most commonly Lewy body dementia (LBD).1 In his review of the Josephs study, Devinsky proposed that the loss of striatal D2 receptors in LBD may be implicated in the manifestation of Capgras delusions.2 The data suggest multiple brain regions may be involved, including the frontal lobes, right temporal lobe, right parietal lobe, parahippocampus, and amygdala.1,2 Most patients in the Josephs study demonstrated global atrophy on imaging studies. One hypothesis is that it is the disconnection of the frontal lobe to other brain regions that may be implicated.1,2,4 This results in intact recognition of facial features of familiar people, impaired emotional recognition, and impaired self-correction due to executive dysfunction.
Methamphetamine also has been implicated in a small number of cases of Capgras; the proposed mechanism involves dopaminergic neuronal impairment/loss.1,2 Additionally, Capgras delusions have been described in cases of patients treated with antimalarial medications, such as chloroquine.8 Younger patients with the Capgras delusion were more likely to have purely psychiatric comorbidities—such as schizophrenia, substance-induced psychosis, or schizoaffective disorder—as opposed to underlying medical conditions.1 In the case presented here, the Capgras delusion was thought to be due to a disorder in the schizophrenia spectrum, specifically schizophreniform disorder.
Because an increasing amount of evidence indicates that the Capgras delusion is associated with certain medical conditions, a workup should be performed to rule out underlying medical etiology. Of note, no official guidelines for the workup have been produced for the Capgras delusion. However, the workup may include brain imaging, such as magnetic resonance imaging and/or CT scan to rule out mass lesions, vascular malformations, stroke, or neuro-infectious processes; laboratory tests, such as vitamin B12, liver panel, HIV, rapid plasma reagin, hepatitis B and C viruses, parathyroid hormone levels, urine drug screen, and thyroid panel can be ordered to rule out other medical causes.1,2,6,7,9
Consultations with internal medicine and neurology departments may be beneficial. Although treatment of the underlying condition may lead to an improvement in the symptoms, full remission in all cases has not been consistently demonstrated in the current literature.5,7,9,10 Patients with the Capgras delusion are challenging to treat, because their delusions have been shown to be refractory to antipsychotic therapy. However, antipsychotics are currently the mainstay of treatment. Some case studies have shown efficacy with pimozide, tricyclic antidepressants, and mirtazapine.6,9
One case study in 2014 in India of a 45-year-old woman who believed her husband and son were replaced by imposters out to kill her, showed a 40% to 50% reduction of paranoia, irritability, and suspicious scanning behaviors with a combination of risperidone and trihexyphenidyl. Despite the improvements, the woman continued to have delusions.7
A notable feature associated with those experiencing the Capgras delusion is the increased risk of violent behaviors, often because of suspiciousness and paranoia. A 2004 review suggested the risk of violence and homicidality is much higher in male patients compared with that of female patients with the Capgras delusion.9 This is despite evidence suggesting that the prevalence of the Capgras delusion seems to be greater in women.6,9 Moreover, patients often demonstrated social withdrawal and self-isolation prior to violent acts. The victims often were family members or those who live with the patient, which is consistent with the evidence that those most familiar to patients are more likely to be misidentified.1,2,7,9,10
A 1989 case series that examined 8 cases of the Capgras delusion listed the following violent behaviors: shot and killed father, pointed knife at mother, held knife to mother’s throat, punched parents, threatened to stab husband with scissors, nonspecifically threatened physical harm to family, injured mother with axe, and threatened to stab son with knife and burn him. Seven of the 8 patients lived with the misidentified persons, and 5 of the 8 patients were treatment resistant. The study posited that chronicity of the delusion, content of the delusion, and accessibility of misidentified persons seemed to increase the risk of violent behaviors. These authors went on to suggest that despite the appearance of stability, patients may react violently to minute changes.10 Overall literature seems to suggest the importance of performing a violence and homicidality assessment with special attention to assessment for themes of hostility toward misidentified individuals.9,10
Conclusion
The Capgras delusion is an uncommon symptom associated with varied psychiatric, medical, iatrogenic, and neurologic conditions. Treatment of underlying medical conditions may improve or resolve the delusions. However, in this case, the patient did not seem to have any underlying medical conditions, and it was thought that he may have been experiencing a prodrome within the schizophrenia spectrum. This is consistent with the literature, which suggests that those with the delusions at younger ages may have a psychiatric etiology.
Although this patient was responsive to aripiprazole, the Capgras delusion has been known to be resistant to antipsychotic therapy. It is worth considering a medical and neurologic workup with the addition of a psychiatry referral. Further, while the patient in the presented case had the delusion that he had assaulted his mother, whom he misidentified as an imposter, the patient did not demonstrate any hostility and denied thoughts of harming her. However, given the increased risk of violence in patients with the Capgras delusion, a homicidality and violence assessment should be performed. While further recommendations are outside the scope of this article, the provider should be cognizant of local duty-to-warn and duty-to-protect laws regarding potentially homicidal patients.
Many patients admitted to inpatient psychiatric hospitals present with delusions; however, the Capgras delusion is a rare type that often appears as a sequela of certain medical and neurologic conditions.1 The Capgras delusion is a condition in which a person believes that either an individual or a group of people has been replaced by doubles or imposters.
In 1923, French psychiatrist Joseph Capgras first described the delusion. He and Jean Reboul-Lachaux coauthored a paper on a 53-year-old woman. The patient was a paranoid megalomaniac who “transformed everyone in her entourage, even those closest to her, such as her husband and daughter, into various and numerous doubles.”2 She believed she was famous, wealthy, and of royal lineage. Although 3 of her children had died, she believed that they were abducted, and that her only surviving child was replaced by a look-alike.2,3 Although the prevalence of such delusions in the general population has not been fully studied, a psychiatric hospital in Turkey found a 1.3% prevalence (1.8% women and 0.9% men) in 920 admissions over 5 years.4
The Capgras delusion is one of many delusions related to the misidentification of people, places, or objects; these delusions collectively are known as delusional misidentification syndrome (DMS).5,6 The Fregoli delusion involves the belief that several different people are the same person in disguise. Intermetamorphosis is the belief that an individual has been transformed internally and externally to another person. Subjective doubles is the belief that a doppelganger of the afflicted person exists, living and functioning independently in the world. Reduplicative paramnesia is the belief that a person, place, or object has been duplicated. A rarer example of DMS is the Cotard delusion, which is the belief that the patient himself or herself is dead, putrefying, exsanguinating, or lacking internal organs.
The most common of the DMS is the Capgras delusion. One common presentation of Capgras delusion involves the spouse of the patient, who believes that an imposter of the same sex as their spouse has taken over his or her body. Rarer delusions are those in which a person misidentifies him or herself as the imposter.3,5,6
Case Presentation
This case involved a 24-year-old male veteran who had received a wide range of mental health diagnoses in the past, including major depressive disorder (MDD) with psychotic features, generalized anxiety disorder, cannabis use disorder, adjustment disorder, and borderline personality disorder. He also had a medical history related to a motor vehicle accident with subsequent intestinal rupture and colostomy placement that had occurred a year and a half prior to presentation. He had no history of brain trauma.
The patient voluntarily presented to the hospital for increased suicidal thoughts and was admitted voluntarily for stabilization and self-harm prevention. He stated that “I feel everything is unreal. I feel suicidal and guilt” and endorsed a plan to either walk into traffic or shoot himself in the head due to increasingly distressing thoughts and memories. According to the patient, he had reported to the police that he raped his ex-girlfriend a year previously, although she denied the claim to the police.
The patient further disclosed that he did not believe his mother was real. “Last year my sister told me it was not 2016, but it was 2022,” he said. “She told me that I have hurt my mother with a padlock—that you could no longer identify her face. I don’t remember having done this. I have lived with her since that time, so I don’t think it’s really [my mother].” He believed that his mother was replaced by “government employees” who were sent to elicit confessions for his behavior while in the military. He expressed guilt over several actions he had performed while in military service, such as punching a wall during boot camp, stealing “soak-up” pads, and napping during work hours. His mother was contacted by a staff psychiatrist in the inpatient unit and denied that any assault had taken place.
The patient’s psychiatric review of systems was positive for visual hallucinations (specifically “blurs” next to his bed in the morning that disappeared as he tried to touch them), depressed mood, anxiety, hopelessness, and insomnia. Pertinent negatives of the review of systems included a denial of manic symptoms and auditory hallucinations. For additional details of his past psychiatric history, the patient admitted that his motor vehicle accident, intestinal rupture, and colostomy were the result of his 1 suicide attempt a year and a half prior after a verbal dispute with the same ex-girlfriend that he believed he had raped. After undergoing extensive medical and surgical treatment, he began seeing an outpatient psychiatrist as well as attending substance use counseling to curtail his marijuana use. He was prescribed a combination of duloxetine and risperidone as an outpatient, which he was taking with intermittent adherence.
Regarding substance use, the patient admitted to using marijuana regularly in the past but quit completely 1 month prior and denied any other drug use or alcohol use. He reported a family history of a sister who was undergoing treatment for bipolar disorder. In his social history, the patient disclosed that he was raised by both parents and described a good childhood with a life absent of abuse in any form. He was single with no children. Although he was unemployed, he lived off the funds from an insurance settlement from his motor vehicle accident. He was living in a trailer with his brother and mother. He also denied having access to firearms.
The patient was overweight, neatly groomed, had good eye contact, and was calm and cooperative. He seemed anxious as evidenced by his continuous shaking of his feet; although speech was normal in rate and tone. He reported his mood as “depressed and anxious” with congruent and tearful affect. His thought process was concrete, although his thought content contained delusions, suicidal ideation, and paranoia. He denied any homicidal thoughts or thoughts of harming others. He did not present with any auditory or visual hallucinations. Insight and judgment were poor. The mental status examination revealed no notable deficits in cognition.
The patient’s differential diagnosis included schizophreniform disorder, exacerbation of MDD with psychotic features, and the psychotic component of cannabis use disorder. His outpatient risperidone and duloxetine were not restarted. Aripiprazole 15 mg daily was prescribed for his delusions, paranoia, and visual hallucinations. The patient also received a prescription for hydroxyzine 50 mg every 6 hours as needed for anxiety.
Because of the nature of his delusions, comorbid medical and neurologic conditions were considered. Neurology consultation recommended a noncontrast head computer tomography (CT) scan and an electroencephalogram (EEG). Laboratory workup included HIV antibody, thyroid panel, chemistry panel, complete blood count, hepatitis B serum antigen, urine drug screen, hepatitis C virus, and rapid plasma reagin. All laboratory results were benign and unremarkable, and the urine drug screen was negative. The noncontrast CT revealed no acute findings, and the EEG revealed no recorded epileptiform abnormalities or seizures.
Throughout his hospital course, the patient remained cooperative with treatment. Three days into the hospitalization, he stated that he believed the entire family had been replaced by imposters. He began to distrust members of his family and was reticent to communicate with them when they attempted to contact him. He also experienced fragmented sleep during his hospital stay, and trazodone 50 mg at bedtime was added.
After aripiprazole was increased to 20 mg daily on hospital day 2 and then to 30 mg daily on hospital day 3 due to the patient’s delusions, he began to doubt the validity of his beliefs. After showing gradual improvement over 6 days, the patient reported that he no longer believed that those memories were real. His sleep, depressed mood, anxiety, and paranoia had markedly improved toward the end of the hospitalization and suicidal ideation/intent resolved. The patient was discharged home to his mother and brother after 6 days of hospitalization with aripiprazole 30 mg daily and trazodone 50 mg at bedtime.
Discussion
The Capgras delusion can present in several different contexts. A psychiatric differential diagnosis includes disorders in the schizophrenia spectrum (brief psychotic disorder, schizophreniform disorder, and schizophrenia), schizoaffective disorder, delusional disorder, and substance-induced psychotic disorder. In addition to psychiatric disorders, the Capgras delusion has been shown to occur in several medical conditions, which include stroke, central nervous system tumors, subarachnoid hemorrhage, vitamin B12 deficiency, hepatic encephalopathy, hypothyroidism, hyperparathyroidism, epilepsy, and dementia.1,2,4,7
A 2007 retrospective study by Josephs examined 47 patients diagnosed with the Capgras delusion from several tertiary care centers. Of those patients, 38 (81%) had a neurodegenerative disease, most commonly Lewy body dementia (LBD).1 In his review of the Josephs study, Devinsky proposed that the loss of striatal D2 receptors in LBD may be implicated in the manifestation of Capgras delusions.2 The data suggest multiple brain regions may be involved, including the frontal lobes, right temporal lobe, right parietal lobe, parahippocampus, and amygdala.1,2 Most patients in the Josephs study demonstrated global atrophy on imaging studies. One hypothesis is that it is the disconnection of the frontal lobe to other brain regions that may be implicated.1,2,4 This results in intact recognition of facial features of familiar people, impaired emotional recognition, and impaired self-correction due to executive dysfunction.
Methamphetamine also has been implicated in a small number of cases of Capgras; the proposed mechanism involves dopaminergic neuronal impairment/loss.1,2 Additionally, Capgras delusions have been described in cases of patients treated with antimalarial medications, such as chloroquine.8 Younger patients with the Capgras delusion were more likely to have purely psychiatric comorbidities—such as schizophrenia, substance-induced psychosis, or schizoaffective disorder—as opposed to underlying medical conditions.1 In the case presented here, the Capgras delusion was thought to be due to a disorder in the schizophrenia spectrum, specifically schizophreniform disorder.
Because an increasing amount of evidence indicates that the Capgras delusion is associated with certain medical conditions, a workup should be performed to rule out underlying medical etiology. Of note, no official guidelines for the workup have been produced for the Capgras delusion. However, the workup may include brain imaging, such as magnetic resonance imaging and/or CT scan to rule out mass lesions, vascular malformations, stroke, or neuro-infectious processes; laboratory tests, such as vitamin B12, liver panel, HIV, rapid plasma reagin, hepatitis B and C viruses, parathyroid hormone levels, urine drug screen, and thyroid panel can be ordered to rule out other medical causes.1,2,6,7,9
Consultations with internal medicine and neurology departments may be beneficial. Although treatment of the underlying condition may lead to an improvement in the symptoms, full remission in all cases has not been consistently demonstrated in the current literature.5,7,9,10 Patients with the Capgras delusion are challenging to treat, because their delusions have been shown to be refractory to antipsychotic therapy. However, antipsychotics are currently the mainstay of treatment. Some case studies have shown efficacy with pimozide, tricyclic antidepressants, and mirtazapine.6,9
One case study in 2014 in India of a 45-year-old woman who believed her husband and son were replaced by imposters out to kill her, showed a 40% to 50% reduction of paranoia, irritability, and suspicious scanning behaviors with a combination of risperidone and trihexyphenidyl. Despite the improvements, the woman continued to have delusions.7
A notable feature associated with those experiencing the Capgras delusion is the increased risk of violent behaviors, often because of suspiciousness and paranoia. A 2004 review suggested the risk of violence and homicidality is much higher in male patients compared with that of female patients with the Capgras delusion.9 This is despite evidence suggesting that the prevalence of the Capgras delusion seems to be greater in women.6,9 Moreover, patients often demonstrated social withdrawal and self-isolation prior to violent acts. The victims often were family members or those who live with the patient, which is consistent with the evidence that those most familiar to patients are more likely to be misidentified.1,2,7,9,10
A 1989 case series that examined 8 cases of the Capgras delusion listed the following violent behaviors: shot and killed father, pointed knife at mother, held knife to mother’s throat, punched parents, threatened to stab husband with scissors, nonspecifically threatened physical harm to family, injured mother with axe, and threatened to stab son with knife and burn him. Seven of the 8 patients lived with the misidentified persons, and 5 of the 8 patients were treatment resistant. The study posited that chronicity of the delusion, content of the delusion, and accessibility of misidentified persons seemed to increase the risk of violent behaviors. These authors went on to suggest that despite the appearance of stability, patients may react violently to minute changes.10 Overall literature seems to suggest the importance of performing a violence and homicidality assessment with special attention to assessment for themes of hostility toward misidentified individuals.9,10
Conclusion
The Capgras delusion is an uncommon symptom associated with varied psychiatric, medical, iatrogenic, and neurologic conditions. Treatment of underlying medical conditions may improve or resolve the delusions. However, in this case, the patient did not seem to have any underlying medical conditions, and it was thought that he may have been experiencing a prodrome within the schizophrenia spectrum. This is consistent with the literature, which suggests that those with the delusions at younger ages may have a psychiatric etiology.
Although this patient was responsive to aripiprazole, the Capgras delusion has been known to be resistant to antipsychotic therapy. It is worth considering a medical and neurologic workup with the addition of a psychiatry referral. Further, while the patient in the presented case had the delusion that he had assaulted his mother, whom he misidentified as an imposter, the patient did not demonstrate any hostility and denied thoughts of harming her. However, given the increased risk of violence in patients with the Capgras delusion, a homicidality and violence assessment should be performed. While further recommendations are outside the scope of this article, the provider should be cognizant of local duty-to-warn and duty-to-protect laws regarding potentially homicidal patients.
1. Josephs KA. The Capgras delusion and its relationship to neurodegenerative disease. Arch Neurol. 2007;64(12):1762-1766.
2. Devinsky O. Behavioral neurology. The neurology of the Capgras delusion. Rev Neurol Dis. 2008;5(2):97-100.
3. Sadock BJ, Sadock VA. Kaplan & Sadock’s Synopsis of Psychiatry: Behavioral Sciences/Clinical Psychiatry. 10th ed. Philadelphia, PA: Wolters Kluwer; 2007.
4. Tamam L, Karatas G, Zeren T, Ozpyraz N. The prevalence of Capgras syndrome in a university hospital setting. Acta Neuropsychiatr. 2003;15(5):290-295.
5. Klein CA, Hirchan S. The masks of identities: who’s who? Delusional misidentification syndromes. J Am Acad Psychiatry Law. 2014;42(3):369-378.
6. Atta K. Forlenza N, Gujski M, Hashmi S, Isaac G. Delusional misidentification syndromes: separate disorders or unusual presentations of existing DSM-IV categories? Psychiatry (Edgemont). 2006;3(9):56-61.
7. Sathe H, Karia S, De Sousa A, Shah N. Capgras syndrome: a case report. Paripex Indian J Res. 2014;3(8):134-135. 8. Bhatia MS, Singhal PK, Agrawal P, Malik SC. Capgras’ syndrome in chloroquine induced psychosis. Indian J Psychiatry. 1988;30(3):311-313.
9. Bourget D, Whitehurst L. Capgras syndrome: a review of the neurophysiological correlates and presenting clinical features in cases involving physical violence. Can J Psychiatry. 2004;49(11):719-725.
10. Silva JA, Leong GB, Weinstock R, Boyer CL. Capgras syndrome and dangerousness. Bull Am Acad Psychiatry Law. 1989;17(1):5-14.
1. Josephs KA. The Capgras delusion and its relationship to neurodegenerative disease. Arch Neurol. 2007;64(12):1762-1766.
2. Devinsky O. Behavioral neurology. The neurology of the Capgras delusion. Rev Neurol Dis. 2008;5(2):97-100.
3. Sadock BJ, Sadock VA. Kaplan & Sadock’s Synopsis of Psychiatry: Behavioral Sciences/Clinical Psychiatry. 10th ed. Philadelphia, PA: Wolters Kluwer; 2007.
4. Tamam L, Karatas G, Zeren T, Ozpyraz N. The prevalence of Capgras syndrome in a university hospital setting. Acta Neuropsychiatr. 2003;15(5):290-295.
5. Klein CA, Hirchan S. The masks of identities: who’s who? Delusional misidentification syndromes. J Am Acad Psychiatry Law. 2014;42(3):369-378.
6. Atta K. Forlenza N, Gujski M, Hashmi S, Isaac G. Delusional misidentification syndromes: separate disorders or unusual presentations of existing DSM-IV categories? Psychiatry (Edgemont). 2006;3(9):56-61.
7. Sathe H, Karia S, De Sousa A, Shah N. Capgras syndrome: a case report. Paripex Indian J Res. 2014;3(8):134-135. 8. Bhatia MS, Singhal PK, Agrawal P, Malik SC. Capgras’ syndrome in chloroquine induced psychosis. Indian J Psychiatry. 1988;30(3):311-313.
9. Bourget D, Whitehurst L. Capgras syndrome: a review of the neurophysiological correlates and presenting clinical features in cases involving physical violence. Can J Psychiatry. 2004;49(11):719-725.
10. Silva JA, Leong GB, Weinstock R, Boyer CL. Capgras syndrome and dangerousness. Bull Am Acad Psychiatry Law. 1989;17(1):5-14.
Transformation of Benign Giant Cell Tumor of Bone Into Epithelioid Angiosarcoma
Take-Home Points
- Malignant transformation of a benign GCT is extremely rare.
- It is difficult to distinguish between an early malignant transformation and an overlooked malignancy.
- The most common clinical presentation of transformation of GCT into malignancy is pain, often with swelling.
- Interval monitoring of GCTs may be necessary in patients with symptoms concerning for malignant transformation.
- Clinicians should maintain a high clinical suspicion for malignant transformation or late recurrence of GCT in a patient with new pain at the wound site.
Giant cell tumors (GCTs) of bone account for about 5% of all primary bone tumors in adults, with a predominance in the third decade in life.1 Clinically, GCT of bone often presents with pain, pathologic fracture, and/or soft- tissue expansion in the epiphysis of long bones. However, GCT of bone also has been reported in non-long bones, such as the talus and the calcaneus.2,3 Histologically, GCT of bone consists of neoplastic stromal cells, mononuclear histiocytic cells, and multinucleated giant cells that resemble osteoclasts.4 The radiologic appearance of GCT is often described as a lytic, eccentrically located bony lesion that extends near the articular surface in patients with closed physes. Many GCTs have aggressive radiologic features with possible extensive bony destruction and soft-tissue extension.
Although categorized as a benign lesion, GCT can be locally aggressive, with a variable local recurrence rate of 0% to 65%, depending on treatment modality and skeletal location. Given the aggressiveness of GCT of bone, recommendations for operative intervention include intralesional curettage with adjuvant therapy (eg, cryotherapy, phenol, argon beam, electrocautery) and placement of bone void fillers (eg, bone graft polymethylmethacrylate). Wide resection is recommended when the articular surface is no longer viable for reconstruction secondary to extensive destruction. Some authors have reported that surgical margin is the only risk factor in local recurrence,5,6 and thus complete resection may be needed for tumor eradication. In addition, about 3% of GCTs demonstrate benign pulmonary implants, which have been cited as cause of death in 16% to 25% of reported cases of pulmonary spread.7,8
The literature includes few reports of primary or secondary malignant transformation of GCT. Hutter and colleagues9 defined primary malignant GCT as GCT with sarcomatous tissue juxtaposed with zones of typical benign GCT cells. Secondary malignant GCT is a sarcomatous lesion at the site of a previously documented benign GCT. Secondary malignant GCT of bone histologically has been classified as a fibrosarcoma, malignant fibrous histiocytoma, or osteosarcoma transformation.10
Most malignant transformations of GCT of bone have been attributed to previous irradiation of the lesion.11,12 However, there are some case reports of benign bone GCT malignant transformation in situ without any other medical intervention. It was reported that non-radiation-induced secondary transformations occur relatively early after GCT treatment.13 During the early stages of tumor recurrence, however, it is difficult to distinguish between malignant transformation and primary disease overlooked as a result of sampling error.
We report a case of secondary malignant transformation of GCT of bone 11 years after surgical curettage, cryotherapy, and cementation without adjuvant radiation therapy. To our knowledge, this case report is the first to describe transformation of a nonirradiated benign GCT into an aggressive, high-grade epithelioid angiosarcoma, a very rare vascular bone tumor. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
In July 2003, a 46-year-old woman presented with left heel pain of several months’ duration. Plain radiographs showed a nonaggressive-appearing lytic lesion of the superior aspect of the posterior calcaneal tuberosity with a small cortical incongruity along the superior margin of the lesion (Figures 1A-1D).
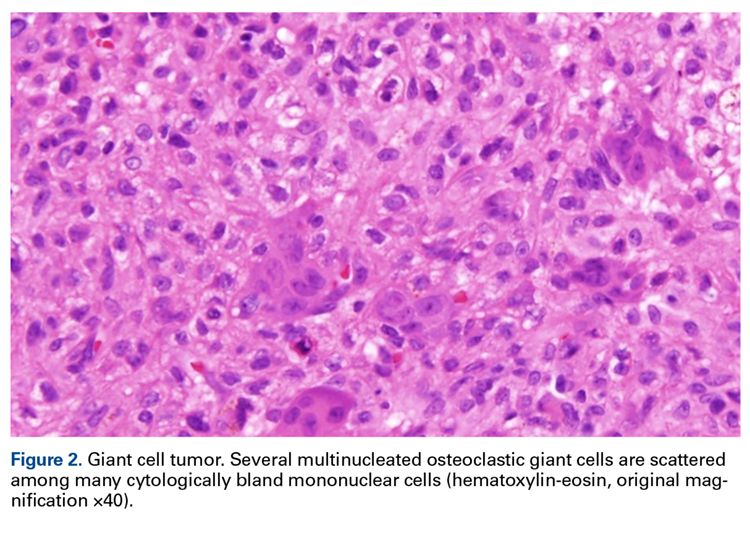
A postoperative splint was placed, and weight-bearing progressed over 6 weeks. The patient was followed at 2- to 3-month intervals over the first 5 postoperative years. She was able to work and perform activities of daily living, but her postoperative course was complicated by significant chronic pain in multiple extremities and long-term treatment by the chronic pain service. At no time did postoperative imaging—magnetic resonance imaging (MRI) at 6 years, whole-body bone scan at 7 years, plain radiographs at 10 years—show evidence of recurrence.
Radiographs showed stable postoperative changes with a small radiolucent area (with sclerotic rim) surrounding the cement-bone interface. Given its proximity to the Achilles tendon and more motion than usual at the wound site, the radiolucency likely was caused by small movements of the interface. The radiolucent area remained stable over a 15-month period.
Whole-body bone scan showed a small area of osteoblastic activity in the left calcaneus, consistent with inflammation surrounding the bone- cement interface, but the uptake was minor relative to other areas of signal, and there were no significant inflammatory reactive changes on MRI (Figures 3A, 3B).
Over 11 years, regular 6- to 12-month follow-up examinations revealed no significant changes in the left foot or in plain radiographs of the chest. In addition, physical examinations revealed no evidence of a palpable mass of the left foot.
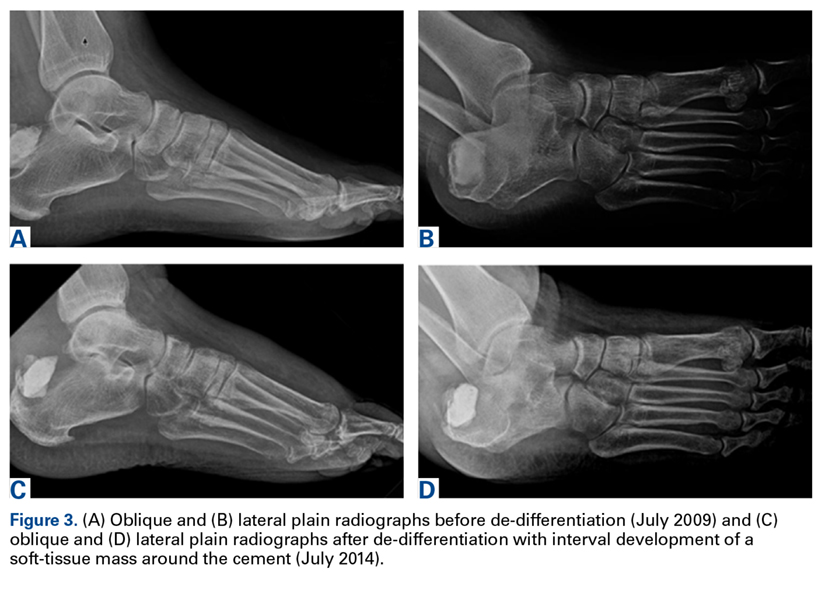
In July 2014 (11 years after curettage and cementation), the patient presented to her pain clinic appointment with severe left foot pain. She said that, over a few weeks, she experienced a significant increase in pain and developed posterolateral foot swelling, which limited her ability to ambulate. Plain radiographs showed a significant soft-tissue prominence around the posterior calcaneus, increased lucency around the bone-cement interface in the calcaneus with elevation, and a cortical break of the superior margin of the posterior calcaneus (Figures 3C, 3D). MRI showed a large lobular mass in the calcaneus and surrounding soft tissue with T1 and T2 signal heterogeneity and enhancement after administration of gadolinium (Figures 4A-4D). There was a large extraosseous extension of the calcaneus-based mass laterally and superiorly with edema in the surrounding hindfoot region (Figure 4).
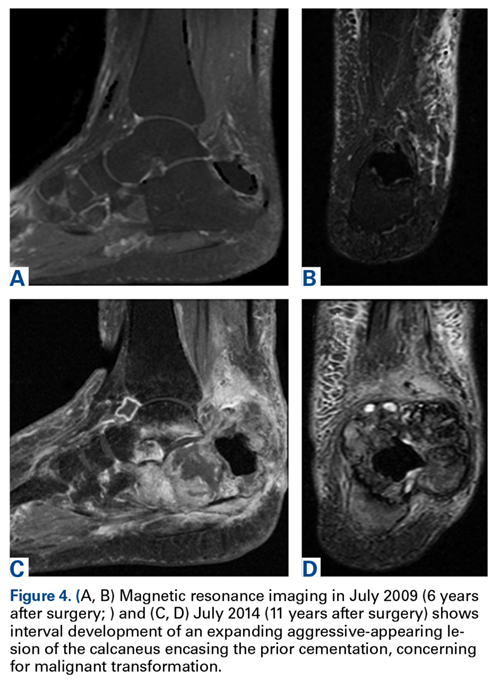
Physical examination revealed exquisite tenderness along the lateral and posterior aspects of the left hindfoot. The patient was unable to bear weight and had soft-tissue swelling throughout the foot and mid calf as well as a palpable mass in the posterior heel. She was otherwise neurovascularly intact through all distributions of the left lower extremity. It was unclear if the GCT of the calcaneus had recurred or if there was a new, secondary tumor. Given her severe pain and morbidity, the patient decided to proceed with open biopsy and a pathology-pending plan for possible amputation in the near future.
In August 2014, an open biopsy with intraoperative frozen evaluation yielded a diagnosis of malignant neoplasm not otherwise specified. Permanent sections showed a proliferation of malignant epithelioid cells with extensive necrosis, hemorrhage, and hemosiderin deposition but no multinucleated giant cells.
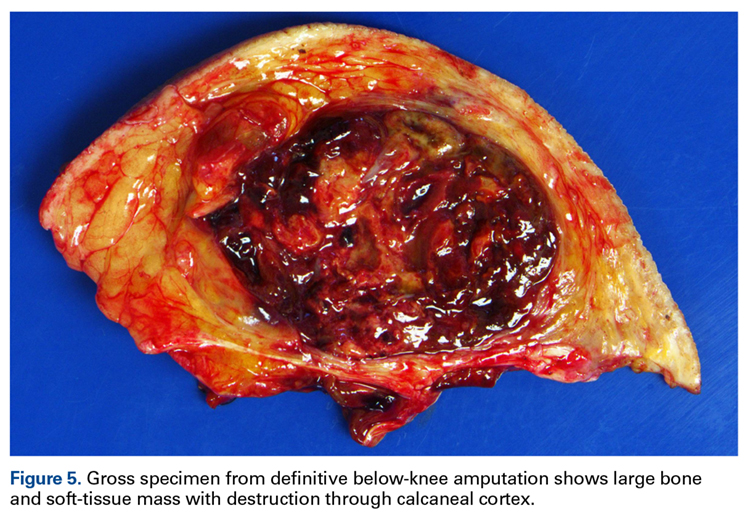
Transformation of the GCT into a high-grade epithelioid angiosarcoma prompted presentation of the patient’s case to a multidisciplinary board of physicians with a focused clinical practice in sarcoma management. The board included board-certified specialists in orthopedic oncology, pathology, musculoskeletal radiology, medical oncology, and radiation oncology. Although discussion included pre-resection use of neoadjuvant chemotherapy to evaluate for disease response, the patient’s severe pain led her to forgo this treatment and proceed directly to below-knee amputation.
Amputation revealed a 7.7-cm hemorrhagic necrotic mass composed of a highly cellular spindle and epithelioid malignancy with abundant hemosiderin deposition (Figure 5). In addition, several atypical mitotic figures and malignant multinucleated tumor giant cells were randomly scattered throughout the neoplasm.
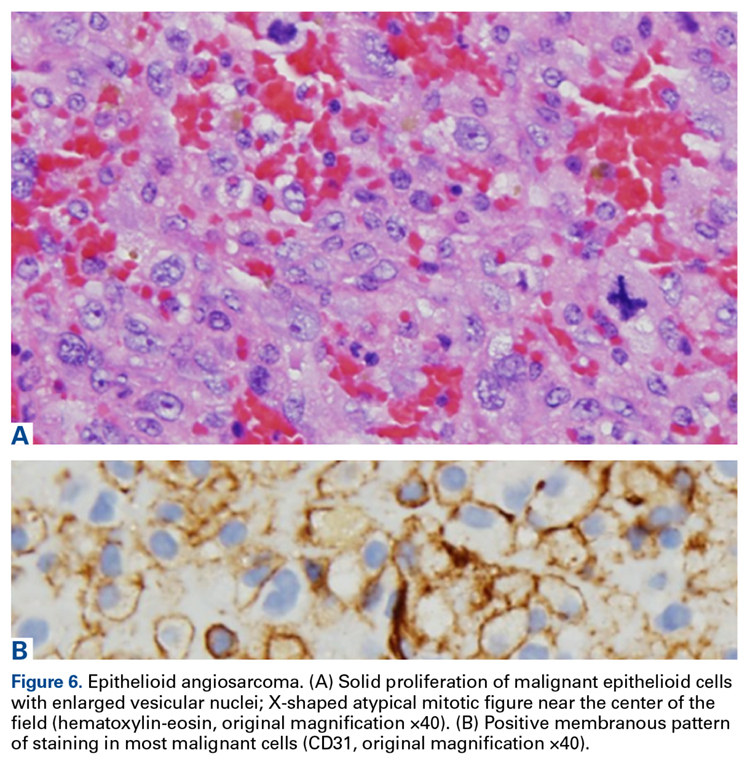
At first follow-up, the patient reported significant pain relief and asked to begin titrating off her chronic pain medicine. Clinical staging, which involved performing whole-body positron emission tomography/computed tomography, revealed nothing concerning for metastases. When this report was being written, the patient was being monitored for recurrent disease in accordance with National Comprehensive Cancer Network guidelines. In the absence of residual sarcoma, our medical oncology team discussed adjuvant chemotherapy options with her. Subsequently, however, she proceeded only with observation and periodic imaging.
Discussion
Malignant transformation of a benign GCT is extremely rare, especially in cases in which the tumor bed has not previously undergone radiation therapy. Although the literature includes historical case reports, primary and secondary malignant GCTs comprise <9% of all GCTs.11,13,14 Primary bone epithelioid angiosarcoma is also extremely rare, especially in the calcaneus; only 1 case is described in the literature.15 In this article, we report on a benign GCT of bone that transformed into an epithelioid angiosarcoma more than a decade after the GCT was treated with curettage and cementation.
The fact that the malignant areas of a previous tumor may have been missed because of sampling error is important for benign GCT of bone in the early postoperative period, as distinguishing between early malignant transformation and an overlooked malignancy may not be possible. However, transformation is more likely the case when a benign GCT becomes a high-grade malignancy after a long disease-free interval. Several authors have indicated that a benign GCT tumor recurring with a secondary malignancy 2 to 5 years after initial GCT treatment suggests malignant transformation.16 Grote and colleagues10 compiled reports of malignant transformation of GCT of bone and described the clinicopathologic features of secondary malignant transformation of GCTs. The data they compiled and data from several other studies indicate a poor prognosis after malignant transformation of GCT; 4 years after diagnosis, mean survival is 40% to 50%.10,16 The most common clinical presentation of transformation of GCT into malignancy is pain, often with coincident swelling of the native wound bed. However, a few cases have been identified with radiologic imaging alone and without a period of clinical symptoms.16
To our knowledge, this case report is the first to describe a longitudinal assessment of the transformation of a benign GCT of bone into an epithelioid angiosarcoma. Whereas an earlier reported GCT of bone transformed into epithelioid angiosarcoma after irradiation,12 our patient’s GCT of bone transformed without irradiation. GCTs of bone are locally aggressive benign tumors and are relatively rare. Malignant transformation of a benign bone tumor a decade after initial, definitive treatment is concerning, especially given the poor prognosis after malignant transformation in this clinical scenario. Current adjuvant treatments have not changed the prognosis. The literature includes a wide variety of histologic transformations, including high-grade sarcomas, after a long disease-free interval. Although malignant transformation of benign GCTs is rare, clinicians should be aware of the potential. Interval monitoring of GCTs may be necessary in patients with symptoms concerning for malignant transformation—pain or swelling in the wound bed—and patients should know to immediately inform their physician of any changes in pain level or local wound bed. Clinicians should maintain a high clinical suspicion for malignant transformation or late recurrence of GCT in a patient with new pain at the site of a previously treated GCT of bone with a disease-free interval of several years.
1. Unni KK. Dahlin’s Bone Tumors: General Aspects and Data on 11,087 Cases. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
2. Errani C, Ruggieri P, Asenzio MA, et al. Giant cell tumor of the extremity: a review of 349 cases from a single institution. Cancer Treat Rev. 2010;36(1):1-7.
3. Campanacci M, Baldini N, Boriani S, Sudanese A. Giant-cell tumor of bone. J Bone Joint Surg Am. 1987;69(1):106-114.
4. Werner M. Giant cell tumour of bone: morphological, biological and histogenetical aspects. Int Orthop. 2006;30(6):484-489.
5 Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Recurrent giant cell tumor of long bones: analysis of surgical management. Clin Orthop Relat Res. 2011;469(4):1181-1187.
6. McDonald DJ, Sim FH, McLeod RA, Dahlin DC. Giant-cell tumor of bone. J Bone Joint Surg Am. 1986;68(2):235-242.
7. Kay RM, Eckardt JJ, Seeger LL, Mirra JM, Hak DJ. Pulmonary metastasis of benign giant cell tumor of bone. Six histologically confirmed cases, including one of spontaneous regression. Clin Orthop Relat Res. 1994;(302):219-230.
8. Maloney WJ, Vaughan LM, Jones HH, Ross J, Nagel DA. Benign metastasizing giant-cell tumor of bone. Report of three cases and review of the literature. Clin Orthop Relat Res. 1989;(243):208-215.
9. Hutter RV, Worcester JN Jr, Francis KC, Foote FW Jr, Stewart FW. Benign and malignant giant cell tumors of bone. A clinicopathological analysis of the natural history of the disease. Cancer. 1962;15:653-690.
10. Grote HJ, Braun M, Kalinski T, et al. Spontaneous malignant transformation of conventional giant cell tumor. Skeletal Radiol. 2004;33(3):169-175.
11. Rock MG, Sim FH, Unni KK, et al. Secondary malignant giant-cell tumor of bone. Clinicopathological assessment of nineteen patients. J Bone Joint Surg Am. 1986;68(7):1073-1079.
12. Mittal S, Goswami C, Kanoria N, Bhattacharya A. Post-irradiation angiosarcoma of bone. J Cancer Res Ther. 2007;3(2):96-99.
13. Bertoni F, Bacchini P, Staals EL. Malignancy in giant cell tumor of bone. Cancer. 2003;97(10):2520-2529.
14. Dahlin DC, Cupps RE, Johnson EW Jr. Giant-cell tumor: a study of 195 cases. Cancer. 1970;25(5):1061-1070.
15. Balaji GG, Arockiaraj JS, Roy AC, Deepak B. Primary epithelioid angiosarcoma of the calcaneum: a diagnostic dilemma. J Foot Ankle Surg. 2014;53(2):239-242.
16. Anract P, De Pinieux G, Cottias P, Pouillart P, Forest M, Tomeno B. Malignant giant-cell tumours of bone. Clinico-pathological types and prognosis: a review of 29 cases. Int Orthop. 1998;22(1):19-26.
Take-Home Points
- Malignant transformation of a benign GCT is extremely rare.
- It is difficult to distinguish between an early malignant transformation and an overlooked malignancy.
- The most common clinical presentation of transformation of GCT into malignancy is pain, often with swelling.
- Interval monitoring of GCTs may be necessary in patients with symptoms concerning for malignant transformation.
- Clinicians should maintain a high clinical suspicion for malignant transformation or late recurrence of GCT in a patient with new pain at the wound site.
Giant cell tumors (GCTs) of bone account for about 5% of all primary bone tumors in adults, with a predominance in the third decade in life.1 Clinically, GCT of bone often presents with pain, pathologic fracture, and/or soft- tissue expansion in the epiphysis of long bones. However, GCT of bone also has been reported in non-long bones, such as the talus and the calcaneus.2,3 Histologically, GCT of bone consists of neoplastic stromal cells, mononuclear histiocytic cells, and multinucleated giant cells that resemble osteoclasts.4 The radiologic appearance of GCT is often described as a lytic, eccentrically located bony lesion that extends near the articular surface in patients with closed physes. Many GCTs have aggressive radiologic features with possible extensive bony destruction and soft-tissue extension.
Although categorized as a benign lesion, GCT can be locally aggressive, with a variable local recurrence rate of 0% to 65%, depending on treatment modality and skeletal location. Given the aggressiveness of GCT of bone, recommendations for operative intervention include intralesional curettage with adjuvant therapy (eg, cryotherapy, phenol, argon beam, electrocautery) and placement of bone void fillers (eg, bone graft polymethylmethacrylate). Wide resection is recommended when the articular surface is no longer viable for reconstruction secondary to extensive destruction. Some authors have reported that surgical margin is the only risk factor in local recurrence,5,6 and thus complete resection may be needed for tumor eradication. In addition, about 3% of GCTs demonstrate benign pulmonary implants, which have been cited as cause of death in 16% to 25% of reported cases of pulmonary spread.7,8
The literature includes few reports of primary or secondary malignant transformation of GCT. Hutter and colleagues9 defined primary malignant GCT as GCT with sarcomatous tissue juxtaposed with zones of typical benign GCT cells. Secondary malignant GCT is a sarcomatous lesion at the site of a previously documented benign GCT. Secondary malignant GCT of bone histologically has been classified as a fibrosarcoma, malignant fibrous histiocytoma, or osteosarcoma transformation.10
Most malignant transformations of GCT of bone have been attributed to previous irradiation of the lesion.11,12 However, there are some case reports of benign bone GCT malignant transformation in situ without any other medical intervention. It was reported that non-radiation-induced secondary transformations occur relatively early after GCT treatment.13 During the early stages of tumor recurrence, however, it is difficult to distinguish between malignant transformation and primary disease overlooked as a result of sampling error.
We report a case of secondary malignant transformation of GCT of bone 11 years after surgical curettage, cryotherapy, and cementation without adjuvant radiation therapy. To our knowledge, this case report is the first to describe transformation of a nonirradiated benign GCT into an aggressive, high-grade epithelioid angiosarcoma, a very rare vascular bone tumor. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
In July 2003, a 46-year-old woman presented with left heel pain of several months’ duration. Plain radiographs showed a nonaggressive-appearing lytic lesion of the superior aspect of the posterior calcaneal tuberosity with a small cortical incongruity along the superior margin of the lesion (Figures 1A-1D).
A postoperative splint was placed, and weight-bearing progressed over 6 weeks. The patient was followed at 2- to 3-month intervals over the first 5 postoperative years. She was able to work and perform activities of daily living, but her postoperative course was complicated by significant chronic pain in multiple extremities and long-term treatment by the chronic pain service. At no time did postoperative imaging—magnetic resonance imaging (MRI) at 6 years, whole-body bone scan at 7 years, plain radiographs at 10 years—show evidence of recurrence.
Radiographs showed stable postoperative changes with a small radiolucent area (with sclerotic rim) surrounding the cement-bone interface. Given its proximity to the Achilles tendon and more motion than usual at the wound site, the radiolucency likely was caused by small movements of the interface. The radiolucent area remained stable over a 15-month period.
Whole-body bone scan showed a small area of osteoblastic activity in the left calcaneus, consistent with inflammation surrounding the bone- cement interface, but the uptake was minor relative to other areas of signal, and there were no significant inflammatory reactive changes on MRI (Figures 3A, 3B).
Over 11 years, regular 6- to 12-month follow-up examinations revealed no significant changes in the left foot or in plain radiographs of the chest. In addition, physical examinations revealed no evidence of a palpable mass of the left foot.
In July 2014 (11 years after curettage and cementation), the patient presented to her pain clinic appointment with severe left foot pain. She said that, over a few weeks, she experienced a significant increase in pain and developed posterolateral foot swelling, which limited her ability to ambulate. Plain radiographs showed a significant soft-tissue prominence around the posterior calcaneus, increased lucency around the bone-cement interface in the calcaneus with elevation, and a cortical break of the superior margin of the posterior calcaneus (Figures 3C, 3D). MRI showed a large lobular mass in the calcaneus and surrounding soft tissue with T1 and T2 signal heterogeneity and enhancement after administration of gadolinium (Figures 4A-4D). There was a large extraosseous extension of the calcaneus-based mass laterally and superiorly with edema in the surrounding hindfoot region (Figure 4).
Physical examination revealed exquisite tenderness along the lateral and posterior aspects of the left hindfoot. The patient was unable to bear weight and had soft-tissue swelling throughout the foot and mid calf as well as a palpable mass in the posterior heel. She was otherwise neurovascularly intact through all distributions of the left lower extremity. It was unclear if the GCT of the calcaneus had recurred or if there was a new, secondary tumor. Given her severe pain and morbidity, the patient decided to proceed with open biopsy and a pathology-pending plan for possible amputation in the near future.
In August 2014, an open biopsy with intraoperative frozen evaluation yielded a diagnosis of malignant neoplasm not otherwise specified. Permanent sections showed a proliferation of malignant epithelioid cells with extensive necrosis, hemorrhage, and hemosiderin deposition but no multinucleated giant cells.
Transformation of the GCT into a high-grade epithelioid angiosarcoma prompted presentation of the patient’s case to a multidisciplinary board of physicians with a focused clinical practice in sarcoma management. The board included board-certified specialists in orthopedic oncology, pathology, musculoskeletal radiology, medical oncology, and radiation oncology. Although discussion included pre-resection use of neoadjuvant chemotherapy to evaluate for disease response, the patient’s severe pain led her to forgo this treatment and proceed directly to below-knee amputation.
Amputation revealed a 7.7-cm hemorrhagic necrotic mass composed of a highly cellular spindle and epithelioid malignancy with abundant hemosiderin deposition (Figure 5). In addition, several atypical mitotic figures and malignant multinucleated tumor giant cells were randomly scattered throughout the neoplasm.
At first follow-up, the patient reported significant pain relief and asked to begin titrating off her chronic pain medicine. Clinical staging, which involved performing whole-body positron emission tomography/computed tomography, revealed nothing concerning for metastases. When this report was being written, the patient was being monitored for recurrent disease in accordance with National Comprehensive Cancer Network guidelines. In the absence of residual sarcoma, our medical oncology team discussed adjuvant chemotherapy options with her. Subsequently, however, she proceeded only with observation and periodic imaging.
Discussion
Malignant transformation of a benign GCT is extremely rare, especially in cases in which the tumor bed has not previously undergone radiation therapy. Although the literature includes historical case reports, primary and secondary malignant GCTs comprise <9% of all GCTs.11,13,14 Primary bone epithelioid angiosarcoma is also extremely rare, especially in the calcaneus; only 1 case is described in the literature.15 In this article, we report on a benign GCT of bone that transformed into an epithelioid angiosarcoma more than a decade after the GCT was treated with curettage and cementation.
The fact that the malignant areas of a previous tumor may have been missed because of sampling error is important for benign GCT of bone in the early postoperative period, as distinguishing between early malignant transformation and an overlooked malignancy may not be possible. However, transformation is more likely the case when a benign GCT becomes a high-grade malignancy after a long disease-free interval. Several authors have indicated that a benign GCT tumor recurring with a secondary malignancy 2 to 5 years after initial GCT treatment suggests malignant transformation.16 Grote and colleagues10 compiled reports of malignant transformation of GCT of bone and described the clinicopathologic features of secondary malignant transformation of GCTs. The data they compiled and data from several other studies indicate a poor prognosis after malignant transformation of GCT; 4 years after diagnosis, mean survival is 40% to 50%.10,16 The most common clinical presentation of transformation of GCT into malignancy is pain, often with coincident swelling of the native wound bed. However, a few cases have been identified with radiologic imaging alone and without a period of clinical symptoms.16
To our knowledge, this case report is the first to describe a longitudinal assessment of the transformation of a benign GCT of bone into an epithelioid angiosarcoma. Whereas an earlier reported GCT of bone transformed into epithelioid angiosarcoma after irradiation,12 our patient’s GCT of bone transformed without irradiation. GCTs of bone are locally aggressive benign tumors and are relatively rare. Malignant transformation of a benign bone tumor a decade after initial, definitive treatment is concerning, especially given the poor prognosis after malignant transformation in this clinical scenario. Current adjuvant treatments have not changed the prognosis. The literature includes a wide variety of histologic transformations, including high-grade sarcomas, after a long disease-free interval. Although malignant transformation of benign GCTs is rare, clinicians should be aware of the potential. Interval monitoring of GCTs may be necessary in patients with symptoms concerning for malignant transformation—pain or swelling in the wound bed—and patients should know to immediately inform their physician of any changes in pain level or local wound bed. Clinicians should maintain a high clinical suspicion for malignant transformation or late recurrence of GCT in a patient with new pain at the site of a previously treated GCT of bone with a disease-free interval of several years.
Take-Home Points
- Malignant transformation of a benign GCT is extremely rare.
- It is difficult to distinguish between an early malignant transformation and an overlooked malignancy.
- The most common clinical presentation of transformation of GCT into malignancy is pain, often with swelling.
- Interval monitoring of GCTs may be necessary in patients with symptoms concerning for malignant transformation.
- Clinicians should maintain a high clinical suspicion for malignant transformation or late recurrence of GCT in a patient with new pain at the wound site.
Giant cell tumors (GCTs) of bone account for about 5% of all primary bone tumors in adults, with a predominance in the third decade in life.1 Clinically, GCT of bone often presents with pain, pathologic fracture, and/or soft- tissue expansion in the epiphysis of long bones. However, GCT of bone also has been reported in non-long bones, such as the talus and the calcaneus.2,3 Histologically, GCT of bone consists of neoplastic stromal cells, mononuclear histiocytic cells, and multinucleated giant cells that resemble osteoclasts.4 The radiologic appearance of GCT is often described as a lytic, eccentrically located bony lesion that extends near the articular surface in patients with closed physes. Many GCTs have aggressive radiologic features with possible extensive bony destruction and soft-tissue extension.
Although categorized as a benign lesion, GCT can be locally aggressive, with a variable local recurrence rate of 0% to 65%, depending on treatment modality and skeletal location. Given the aggressiveness of GCT of bone, recommendations for operative intervention include intralesional curettage with adjuvant therapy (eg, cryotherapy, phenol, argon beam, electrocautery) and placement of bone void fillers (eg, bone graft polymethylmethacrylate). Wide resection is recommended when the articular surface is no longer viable for reconstruction secondary to extensive destruction. Some authors have reported that surgical margin is the only risk factor in local recurrence,5,6 and thus complete resection may be needed for tumor eradication. In addition, about 3% of GCTs demonstrate benign pulmonary implants, which have been cited as cause of death in 16% to 25% of reported cases of pulmonary spread.7,8
The literature includes few reports of primary or secondary malignant transformation of GCT. Hutter and colleagues9 defined primary malignant GCT as GCT with sarcomatous tissue juxtaposed with zones of typical benign GCT cells. Secondary malignant GCT is a sarcomatous lesion at the site of a previously documented benign GCT. Secondary malignant GCT of bone histologically has been classified as a fibrosarcoma, malignant fibrous histiocytoma, or osteosarcoma transformation.10
Most malignant transformations of GCT of bone have been attributed to previous irradiation of the lesion.11,12 However, there are some case reports of benign bone GCT malignant transformation in situ without any other medical intervention. It was reported that non-radiation-induced secondary transformations occur relatively early after GCT treatment.13 During the early stages of tumor recurrence, however, it is difficult to distinguish between malignant transformation and primary disease overlooked as a result of sampling error.
We report a case of secondary malignant transformation of GCT of bone 11 years after surgical curettage, cryotherapy, and cementation without adjuvant radiation therapy. To our knowledge, this case report is the first to describe transformation of a nonirradiated benign GCT into an aggressive, high-grade epithelioid angiosarcoma, a very rare vascular bone tumor. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
In July 2003, a 46-year-old woman presented with left heel pain of several months’ duration. Plain radiographs showed a nonaggressive-appearing lytic lesion of the superior aspect of the posterior calcaneal tuberosity with a small cortical incongruity along the superior margin of the lesion (Figures 1A-1D).
A postoperative splint was placed, and weight-bearing progressed over 6 weeks. The patient was followed at 2- to 3-month intervals over the first 5 postoperative years. She was able to work and perform activities of daily living, but her postoperative course was complicated by significant chronic pain in multiple extremities and long-term treatment by the chronic pain service. At no time did postoperative imaging—magnetic resonance imaging (MRI) at 6 years, whole-body bone scan at 7 years, plain radiographs at 10 years—show evidence of recurrence.
Radiographs showed stable postoperative changes with a small radiolucent area (with sclerotic rim) surrounding the cement-bone interface. Given its proximity to the Achilles tendon and more motion than usual at the wound site, the radiolucency likely was caused by small movements of the interface. The radiolucent area remained stable over a 15-month period.
Whole-body bone scan showed a small area of osteoblastic activity in the left calcaneus, consistent with inflammation surrounding the bone- cement interface, but the uptake was minor relative to other areas of signal, and there were no significant inflammatory reactive changes on MRI (Figures 3A, 3B).
Over 11 years, regular 6- to 12-month follow-up examinations revealed no significant changes in the left foot or in plain radiographs of the chest. In addition, physical examinations revealed no evidence of a palpable mass of the left foot.
In July 2014 (11 years after curettage and cementation), the patient presented to her pain clinic appointment with severe left foot pain. She said that, over a few weeks, she experienced a significant increase in pain and developed posterolateral foot swelling, which limited her ability to ambulate. Plain radiographs showed a significant soft-tissue prominence around the posterior calcaneus, increased lucency around the bone-cement interface in the calcaneus with elevation, and a cortical break of the superior margin of the posterior calcaneus (Figures 3C, 3D). MRI showed a large lobular mass in the calcaneus and surrounding soft tissue with T1 and T2 signal heterogeneity and enhancement after administration of gadolinium (Figures 4A-4D). There was a large extraosseous extension of the calcaneus-based mass laterally and superiorly with edema in the surrounding hindfoot region (Figure 4).
Physical examination revealed exquisite tenderness along the lateral and posterior aspects of the left hindfoot. The patient was unable to bear weight and had soft-tissue swelling throughout the foot and mid calf as well as a palpable mass in the posterior heel. She was otherwise neurovascularly intact through all distributions of the left lower extremity. It was unclear if the GCT of the calcaneus had recurred or if there was a new, secondary tumor. Given her severe pain and morbidity, the patient decided to proceed with open biopsy and a pathology-pending plan for possible amputation in the near future.
In August 2014, an open biopsy with intraoperative frozen evaluation yielded a diagnosis of malignant neoplasm not otherwise specified. Permanent sections showed a proliferation of malignant epithelioid cells with extensive necrosis, hemorrhage, and hemosiderin deposition but no multinucleated giant cells.
Transformation of the GCT into a high-grade epithelioid angiosarcoma prompted presentation of the patient’s case to a multidisciplinary board of physicians with a focused clinical practice in sarcoma management. The board included board-certified specialists in orthopedic oncology, pathology, musculoskeletal radiology, medical oncology, and radiation oncology. Although discussion included pre-resection use of neoadjuvant chemotherapy to evaluate for disease response, the patient’s severe pain led her to forgo this treatment and proceed directly to below-knee amputation.
Amputation revealed a 7.7-cm hemorrhagic necrotic mass composed of a highly cellular spindle and epithelioid malignancy with abundant hemosiderin deposition (Figure 5). In addition, several atypical mitotic figures and malignant multinucleated tumor giant cells were randomly scattered throughout the neoplasm.
At first follow-up, the patient reported significant pain relief and asked to begin titrating off her chronic pain medicine. Clinical staging, which involved performing whole-body positron emission tomography/computed tomography, revealed nothing concerning for metastases. When this report was being written, the patient was being monitored for recurrent disease in accordance with National Comprehensive Cancer Network guidelines. In the absence of residual sarcoma, our medical oncology team discussed adjuvant chemotherapy options with her. Subsequently, however, she proceeded only with observation and periodic imaging.
Discussion
Malignant transformation of a benign GCT is extremely rare, especially in cases in which the tumor bed has not previously undergone radiation therapy. Although the literature includes historical case reports, primary and secondary malignant GCTs comprise <9% of all GCTs.11,13,14 Primary bone epithelioid angiosarcoma is also extremely rare, especially in the calcaneus; only 1 case is described in the literature.15 In this article, we report on a benign GCT of bone that transformed into an epithelioid angiosarcoma more than a decade after the GCT was treated with curettage and cementation.
The fact that the malignant areas of a previous tumor may have been missed because of sampling error is important for benign GCT of bone in the early postoperative period, as distinguishing between early malignant transformation and an overlooked malignancy may not be possible. However, transformation is more likely the case when a benign GCT becomes a high-grade malignancy after a long disease-free interval. Several authors have indicated that a benign GCT tumor recurring with a secondary malignancy 2 to 5 years after initial GCT treatment suggests malignant transformation.16 Grote and colleagues10 compiled reports of malignant transformation of GCT of bone and described the clinicopathologic features of secondary malignant transformation of GCTs. The data they compiled and data from several other studies indicate a poor prognosis after malignant transformation of GCT; 4 years after diagnosis, mean survival is 40% to 50%.10,16 The most common clinical presentation of transformation of GCT into malignancy is pain, often with coincident swelling of the native wound bed. However, a few cases have been identified with radiologic imaging alone and without a period of clinical symptoms.16
To our knowledge, this case report is the first to describe a longitudinal assessment of the transformation of a benign GCT of bone into an epithelioid angiosarcoma. Whereas an earlier reported GCT of bone transformed into epithelioid angiosarcoma after irradiation,12 our patient’s GCT of bone transformed without irradiation. GCTs of bone are locally aggressive benign tumors and are relatively rare. Malignant transformation of a benign bone tumor a decade after initial, definitive treatment is concerning, especially given the poor prognosis after malignant transformation in this clinical scenario. Current adjuvant treatments have not changed the prognosis. The literature includes a wide variety of histologic transformations, including high-grade sarcomas, after a long disease-free interval. Although malignant transformation of benign GCTs is rare, clinicians should be aware of the potential. Interval monitoring of GCTs may be necessary in patients with symptoms concerning for malignant transformation—pain or swelling in the wound bed—and patients should know to immediately inform their physician of any changes in pain level or local wound bed. Clinicians should maintain a high clinical suspicion for malignant transformation or late recurrence of GCT in a patient with new pain at the site of a previously treated GCT of bone with a disease-free interval of several years.
1. Unni KK. Dahlin’s Bone Tumors: General Aspects and Data on 11,087 Cases. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
2. Errani C, Ruggieri P, Asenzio MA, et al. Giant cell tumor of the extremity: a review of 349 cases from a single institution. Cancer Treat Rev. 2010;36(1):1-7.
3. Campanacci M, Baldini N, Boriani S, Sudanese A. Giant-cell tumor of bone. J Bone Joint Surg Am. 1987;69(1):106-114.
4. Werner M. Giant cell tumour of bone: morphological, biological and histogenetical aspects. Int Orthop. 2006;30(6):484-489.
5 Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Recurrent giant cell tumor of long bones: analysis of surgical management. Clin Orthop Relat Res. 2011;469(4):1181-1187.
6. McDonald DJ, Sim FH, McLeod RA, Dahlin DC. Giant-cell tumor of bone. J Bone Joint Surg Am. 1986;68(2):235-242.
7. Kay RM, Eckardt JJ, Seeger LL, Mirra JM, Hak DJ. Pulmonary metastasis of benign giant cell tumor of bone. Six histologically confirmed cases, including one of spontaneous regression. Clin Orthop Relat Res. 1994;(302):219-230.
8. Maloney WJ, Vaughan LM, Jones HH, Ross J, Nagel DA. Benign metastasizing giant-cell tumor of bone. Report of three cases and review of the literature. Clin Orthop Relat Res. 1989;(243):208-215.
9. Hutter RV, Worcester JN Jr, Francis KC, Foote FW Jr, Stewart FW. Benign and malignant giant cell tumors of bone. A clinicopathological analysis of the natural history of the disease. Cancer. 1962;15:653-690.
10. Grote HJ, Braun M, Kalinski T, et al. Spontaneous malignant transformation of conventional giant cell tumor. Skeletal Radiol. 2004;33(3):169-175.
11. Rock MG, Sim FH, Unni KK, et al. Secondary malignant giant-cell tumor of bone. Clinicopathological assessment of nineteen patients. J Bone Joint Surg Am. 1986;68(7):1073-1079.
12. Mittal S, Goswami C, Kanoria N, Bhattacharya A. Post-irradiation angiosarcoma of bone. J Cancer Res Ther. 2007;3(2):96-99.
13. Bertoni F, Bacchini P, Staals EL. Malignancy in giant cell tumor of bone. Cancer. 2003;97(10):2520-2529.
14. Dahlin DC, Cupps RE, Johnson EW Jr. Giant-cell tumor: a study of 195 cases. Cancer. 1970;25(5):1061-1070.
15. Balaji GG, Arockiaraj JS, Roy AC, Deepak B. Primary epithelioid angiosarcoma of the calcaneum: a diagnostic dilemma. J Foot Ankle Surg. 2014;53(2):239-242.
16. Anract P, De Pinieux G, Cottias P, Pouillart P, Forest M, Tomeno B. Malignant giant-cell tumours of bone. Clinico-pathological types and prognosis: a review of 29 cases. Int Orthop. 1998;22(1):19-26.
1. Unni KK. Dahlin’s Bone Tumors: General Aspects and Data on 11,087 Cases. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
2. Errani C, Ruggieri P, Asenzio MA, et al. Giant cell tumor of the extremity: a review of 349 cases from a single institution. Cancer Treat Rev. 2010;36(1):1-7.
3. Campanacci M, Baldini N, Boriani S, Sudanese A. Giant-cell tumor of bone. J Bone Joint Surg Am. 1987;69(1):106-114.
4. Werner M. Giant cell tumour of bone: morphological, biological and histogenetical aspects. Int Orthop. 2006;30(6):484-489.
5 Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Recurrent giant cell tumor of long bones: analysis of surgical management. Clin Orthop Relat Res. 2011;469(4):1181-1187.
6. McDonald DJ, Sim FH, McLeod RA, Dahlin DC. Giant-cell tumor of bone. J Bone Joint Surg Am. 1986;68(2):235-242.
7. Kay RM, Eckardt JJ, Seeger LL, Mirra JM, Hak DJ. Pulmonary metastasis of benign giant cell tumor of bone. Six histologically confirmed cases, including one of spontaneous regression. Clin Orthop Relat Res. 1994;(302):219-230.
8. Maloney WJ, Vaughan LM, Jones HH, Ross J, Nagel DA. Benign metastasizing giant-cell tumor of bone. Report of three cases and review of the literature. Clin Orthop Relat Res. 1989;(243):208-215.
9. Hutter RV, Worcester JN Jr, Francis KC, Foote FW Jr, Stewart FW. Benign and malignant giant cell tumors of bone. A clinicopathological analysis of the natural history of the disease. Cancer. 1962;15:653-690.
10. Grote HJ, Braun M, Kalinski T, et al. Spontaneous malignant transformation of conventional giant cell tumor. Skeletal Radiol. 2004;33(3):169-175.
11. Rock MG, Sim FH, Unni KK, et al. Secondary malignant giant-cell tumor of bone. Clinicopathological assessment of nineteen patients. J Bone Joint Surg Am. 1986;68(7):1073-1079.
12. Mittal S, Goswami C, Kanoria N, Bhattacharya A. Post-irradiation angiosarcoma of bone. J Cancer Res Ther. 2007;3(2):96-99.
13. Bertoni F, Bacchini P, Staals EL. Malignancy in giant cell tumor of bone. Cancer. 2003;97(10):2520-2529.
14. Dahlin DC, Cupps RE, Johnson EW Jr. Giant-cell tumor: a study of 195 cases. Cancer. 1970;25(5):1061-1070.
15. Balaji GG, Arockiaraj JS, Roy AC, Deepak B. Primary epithelioid angiosarcoma of the calcaneum: a diagnostic dilemma. J Foot Ankle Surg. 2014;53(2):239-242.
16. Anract P, De Pinieux G, Cottias P, Pouillart P, Forest M, Tomeno B. Malignant giant-cell tumours of bone. Clinico-pathological types and prognosis: a review of 29 cases. Int Orthop. 1998;22(1):19-26.
Pseudomyogenic Hemangioendothelioma
Pseudomyogenic hemangioendothelioma (PMHE), also referred to as epithelioid sarcoma–like hemangioendothelioma,1 is a rare soft tissue tumor that was described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. It predominantly affects males between the second and fifth decades of life and most commonly presents as multiple nodules that may involve either the superficial or deep soft tissues of the legs and less often the arms. It also can arise on the trunk. We present a case of PMHE occurring in a young man and briefly review the literature on clinical presentation and histologic differentiation of this unique tumor, comparing these findings to its mimickers.
Case Report
A 20-year-old man presented with skin lesions on the left leg that had been present for 1 year. The patient described the lesions as tender pimples that would drain yellow discharge on occasion but had now transformed into large brown plaques. Physical examination showed 4 verrucous plaques ranging in size from 1 to 3 cm with hyperpigmentation and a central crust (Figure 1). Initially, the patient thought the lesions appeared due to shaving his legs for sports. He presented to the emergency department multiple times over the past year; pain control was provided and local skin care was recommended. Culture of the discharge had been performed 6 months prior to biopsy with negative results. No biopsy was performed on initial presentation and the lesions were diagnosed in the emergency department clinically as boils.
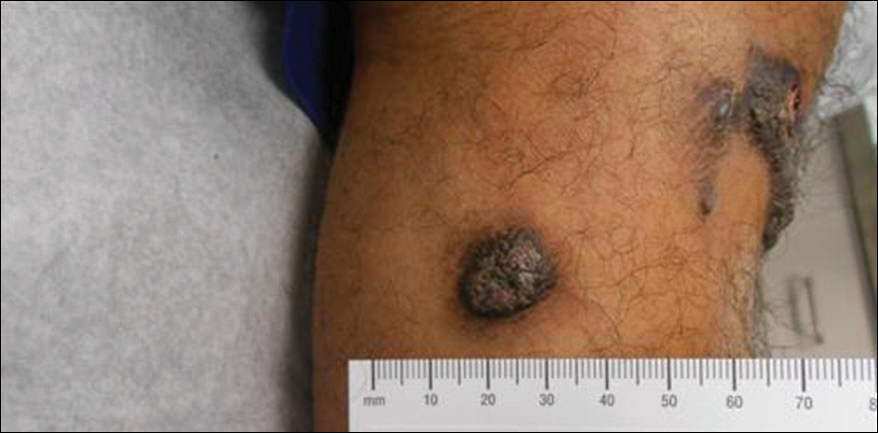
After failing to improve, the patient was seen by an outside dermatologist and the clinical differential diagnosis included deep fungal infection, atypical mycobacterial infection, and keloids. A 4-mm punch biopsy was taken from the periphery of one of the lesions and demonstrated hyperkeratosis, papillomatosis, and acanthosis (Figure 2). Within the superficial and deep dermis and focally extending into the subcutaneous tissue, there were sheets of spindled to epithelioid-appearing cells with moderate cytologic atypia (Figure 3). The tumor showed infiltrative margins. There was moderate cellularity. The individual cells had a rhabdoid appearance with large eccentric vesicular nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm (Figure 4). No definitive evidence of glandular, squamous, or vascular differentiation was present. There was an associated moderate inflammatory host response composed of neutrophils and lymphocytes. Occasional extravasated red blood cells were present. Immunohistochemistry staining was performed and the atypical cells demonstrated diffuse positive staining for friend leukemia integration 1 transcription factor (FLI-1), erythroblastosis virus E26 transforming sequence-related gene (ERG)(Figure 5), CD31, and CD68. There was patchy positive staining for cytokeratin AE1/AE3, CD10, and factor VIII. There was no remarkable staining for human herpesvirus 8, epithelial membrane antigen, S-100, CD34, cytokeratin 903, and desmin. Overall, the histologic features in conjunction with the immunohistochemistry staining were consistent with a diagnosis of PMHE.

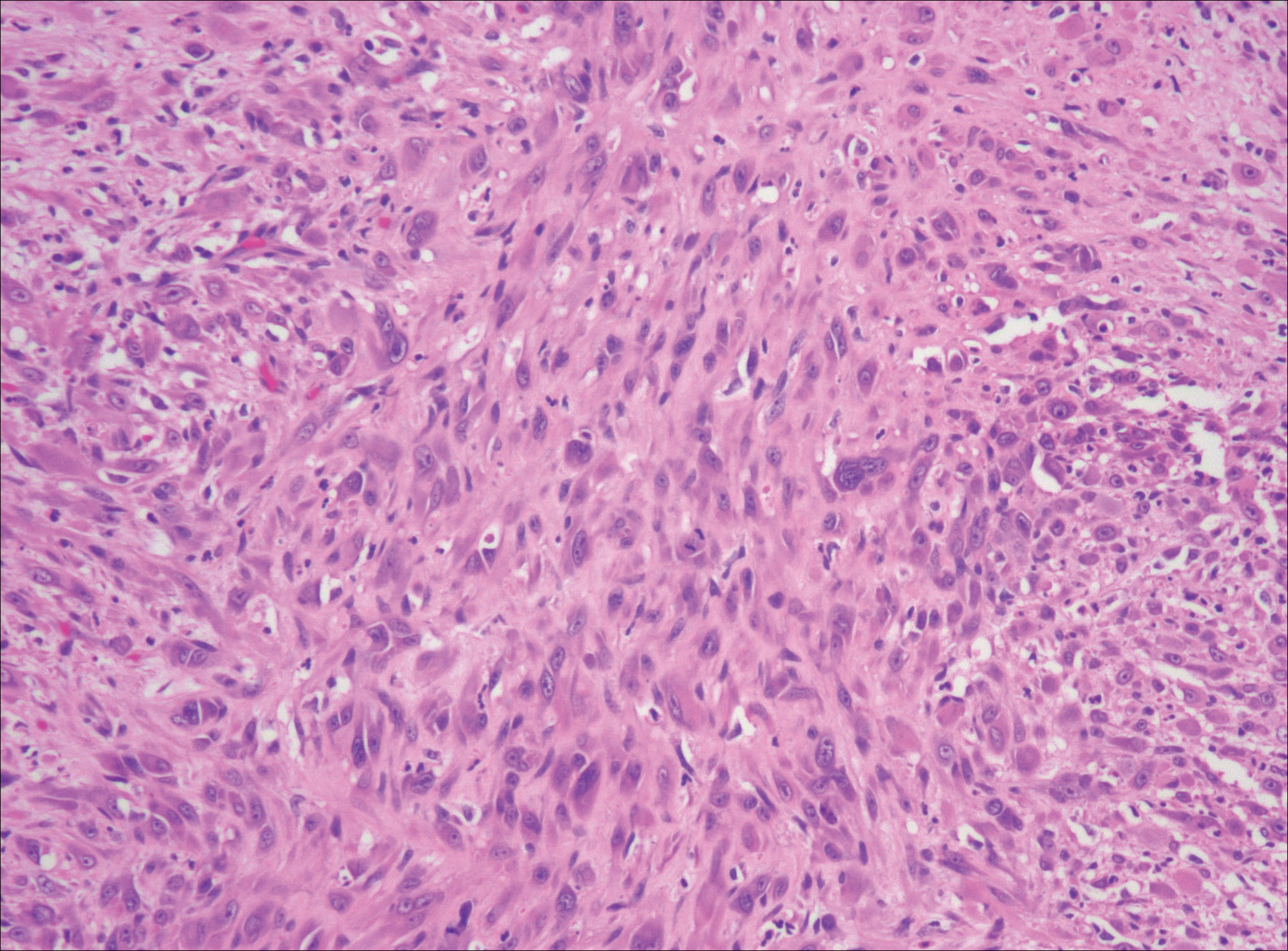
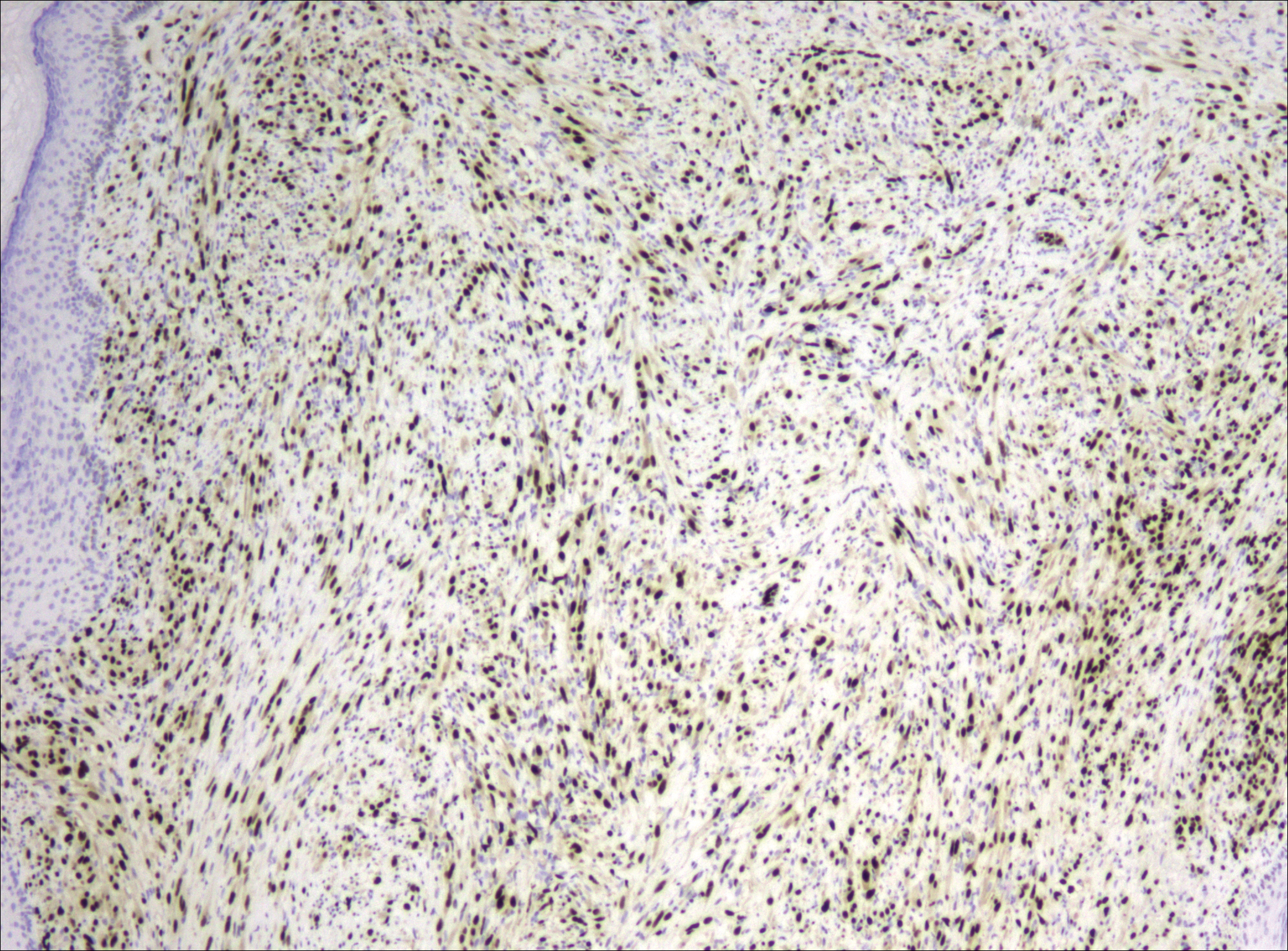
Magnetic resonance imaging was then performed to evaluate the depth and extent of the lesions for surgical excision planning (Figure 6), which showed 5 nodular lesions within the dermis and subcutis adjacent to the proximal aspect of the left tibia and medial aspect of the left knee. An additional lesion was noted between the sartorius and semimembranosus muscles, which was thought to represent either a lymph node or an additional neoplastic lesion. Chest computed tomography also displayed indeterminate lesions in the lungs.
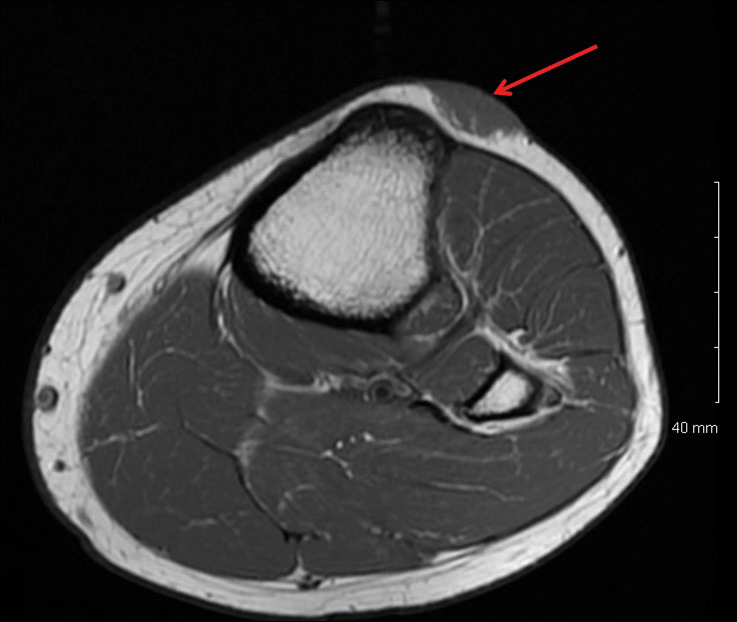
Excision of the superficial lesions was performed. All of the lesions demonstrated similar histologic changes to the previously described biopsy specimen. The tumor was limited to the dermis and subcutaneous tissue. The patient was lost to follow-up and the etiology of the lung lesions was unknown.
Comment
Nomenclature
Pseudomyogenic hemangioendothelioma is a relatively new type of vascular tumor that has been included in the updated 2013 edition of the World Health Organization classification as an intermediate malignant tumor that rarely metastasizes.3 It typically involves multiple tissue planes, most notably the dermis and subcutaneous layers but also muscle and bone.4 The term pseudomyogenic refers to the histologic resemblance of some of the cells to rhabdomyoblasts; however, these tumors are negative for all immunohistochemical muscle markers, most notably myogenin, desmin, and α-smooth muscle actin.5
Clinical Presentation
Gross features of PMHE typically include multiple firm nodules with ill-defined margins. The tumor was initially described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. In 2003, a series of 7 cases of PMHE was reported by Billings et al6 under the term epithelioid sarcomalike hemangioendothelioma. Other than the predominance of an epithelioid morphology, the cases reported as epithelioid sarcomalike hemangioendothelioma had similar clinical features and immunophenotype to what has been reported as PMHE.
Based on a PubMed search of articles indexed for MEDLINE using the term pseudomyogenic hemangioendothelioma, the 2 largest case series were reported by Pradhan et al7 (N=8) in 2017 and Hornick and Fletcher4 (N=50) in 2011. Hornick and Fletcher4 reported a male (41/50 [82.0%]) to female (9/50 [18.0%]) ratio of 4.6 to 1, and an average age at presentation of 31 years with 82% (41/50) of patients 40 years or younger. Pradhan et al7 also reported a male predominance (7/8 [87.5%]) with a similar average age at presentation of 29 years (age range, 9–62 years). The size of individual tumors ranged from 0.3 to 5.5 cm (mean size, 1.9 cm) in the series by Hornick and Fletcher4 and 0.3 to 6.0 com in the series by Pradhan et al.7 Hornick and Fletcher4 reported the most common site of involvement was the leg (27/50 [54.0%]), followed by the arm (12/50 [24.0%]), trunk (9/50 [18.0%]), and head and neck (2/50 [4.0%]). The leg (6/8 [75.0%]) also was the most common site of involvement in the series by Pradhan et al,7 with 2 cases occurring on the arm. In the series by Hornick and Fletcher,4 the tumors typically involved the dermis and subcutaneous tissue (26/50 [52%]) with a smaller number involving skeletal muscle (17/50 [34%]) and bone (7/50 [14%]). They reported 66% of their patients (33/50) had multifocal disease at presentation.4 Pradhan et al7 also reported 2 (25.0%) cases being limited to the superficial soft tissue, 2 (25.0%) being limited to the deep soft tissue, and 4 (50.0%) involving the bone; 5 (62.5%) patients had multifocal disease at presentation. The presentation of our patient in regards to gender, age, and tumor characteristics is consistent with other published cases.5-10
Histopathology
Microscopic features of PMHE include sheets of spindled to epithelioid-appearing cells with mild to moderate nuclear atypia and eosinophilic cytoplasm. The tumor has an infiltrative growth pattern. Some of the cells may resemble rhabdomyoblastlike cells, hence the moniker pseudomyogenic. There is no recapitulation of vascular structures or remarkable cytoplasmic vacuolization. Mitotic rate is low and there is no tumor necrosis.4 The tumor cells do not appear to arise from a vessel or display an angiocentric growth pattern. Many cases report the presence of an inflammatory infiltrate containing neutrophils interspersed within the tumor.4,5,7 The overlying epidermis will commonly show hyperkeratosis, epidermal hyperplasia, and acanthosis.4,11
Differential Diagnosis
The histopathologic differential diagnosis would include epithelioid sarcoma, epithelioid hemangioendothelioma, and to a lesser extent dermatofibrosarcoma protuberans (DFSP) and rhabdomyosarcoma. Dermatofibrosarcoma protuberans is the most commonly encountered of these tumors. Histologically, DFSP is characterized by a cellular proliferation of small spindle cells with plump nuclei arranged in a storiform or cartwheel pattern. Dermatofibrosarcoma protuberans tends to be limited to the dermis and subcutaneous tissue and only rarely involves underlying skeletal muscle. The presence of the storiform growth pattern in conjunction with the lack of rhabdoid changes would favor a diagnosis of DFSP. Another characteristic histologic finding typically only associated with DFSP is the interdigitating growth pattern of the spindle cells within the lobules of the subcutaneous tissue, creating a lacelike or honeycomb appearance.
Immunohistochemistry staining is necessary to help differentiate PMHE from other tumors in the differential diagnosis. Pseudomyogenic hemangioendothelioma stains positive for cytokeratin AE1/AE3; integrase interactor 1; and vascular markers FLI-1, CD31, and ERG, and negative for CD34.4,6,12-15 In contrast to epithelioid hemangioendothelioma, DFSP, and to a lesser extent epithelioid sarcoma, all of which are positive for CD34, epithelioid sarcoma is negative for both CD31 and integrase interactor 1. Dermatofibrosarcoma protuberans is negative for cytokeratin AE1/AE3. Rhabdomyosarcomas are positive for myogenic markers such as MyoD1 and myogenin, unlike any of the other tumors mentioned. Histologically, epithelioid sarcomas will tend to have a granulomalike growth pattern with central necrosis, unlike PMHE.12 Epithelioid hemangioendothelioma often will have a cordlike growth pattern in a myxochondroid background. Unlike PMHE, these tumors often will appear to be arising from vessels, and intracytoplasmic vacuoles are common. Three cases of PMHE have been reported to have a t(7;19)(q22;q13) chromosomal anomaly, which is not consistent with every case.16
Treatment Options
Standard treatment typically includes wide excision of the lesions, as was done in our case. Because of the substantial risk of local recurrence, which was up to 58% in the series by Hornick and Fletcher,4 adjuvant therapy may be considered if positive margins are found on excision. Metastasis to lymph nodes and the lungs has been reported but is rare.2,4 Most cases have been shown to have a favorable prognosis; however, local recurrence seems to be common. Rarely, amputation of the limb may be required.5 In contrast, epithelioid sarcomas have been found to spread to lymph nodes and the lungs in up to 50% of cases with a 5-year survival rate of 10% to 30%.13
Conclusion
In summary, we describe a case of PMHE involving the lower leg in a 20-year-old man. These tumors often are multinodular and multiplanar, with the dermis and subcutaneous tissues being the most common areas affected. It has a high rate of local recurrence but rarely has distant metastasis. Pseudomyogenic hemangioendothelioma, similar to other soft tissue tumors, can be difficult to diagnose on shave biopsy or superficial punch biopsy not extending into subcutaneous tissue. Deep incisional or punch biopsies are required to more definitively diagnose these types of tumors. The diagnosis of PMHE versus other soft tissue tumors requires correlation of histology and immunohistochemistry staining with clinical information and radiographic findings.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma (pseudomyogenic hemangioendothelioma). Am J Surg Pathol. 2011;35:1088; author reply 1088-1089.
- Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
- Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014;46:95-104.
- Hornick JL, Fletcher CD. Pseudomyogenic hemangioendothelioma: a distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol. 2011;35:190-201.
- Sheng W, Pan Y, Wang J. Pseudomyogenic hemangioendothelioma: report of an additional case with aggressive clinical course. Am J Dermatopathol. 2013;35:597-600.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol. 2003;27:48-57.
- Pradhan D, Schoedel K, McGough RL, et al. Pseudomyogenic hemangioendothelioma of skin, bone and soft tissue—a clinicopathological, immunohistochemical and fluorescence in situ hybridization study [published online November 2, 2017]. Hum Pathol. 2017. doi:0.1016/j.humpath.2017.10.023.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- McGinity M, Bartanusz V, Dengler B, et al. Pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma, fibroma-like variant of epithelioid sarcoma) of the thoracic spine. Eur Spine J. 2013;22(suppl 3):S506-S511.
- Stuart LN, Gardner JM, Lauer SR, et al. Epithelioid sarcoma-like (pseudomyogenic) hemangioendothelioma, clinically mimicking dermatofibroma, diagnosed by skin biopsy in a 30-year-old man. J Cutan Pathol. 2013;40:909-913.
- Amary MF, O’Donnell P, Berisha F, et al. Pseudomyogenic (epithelioid sarcoma-like) hemangioendothelioma: characterization of five cases. Skeletal Radiol. 2013;42:947-957.
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542-550.
- Chbani L, Guillou L, Terrier P, et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French Sarcoma Group. Am J Clin Pathol. 2009;131:222-227.
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- Trombetta D, Magnusson L, von Steyern FV, et al. Translocation t(7;19)(q22;q13)—a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet. 2011;204:211-215.
Pseudomyogenic hemangioendothelioma (PMHE), also referred to as epithelioid sarcoma–like hemangioendothelioma,1 is a rare soft tissue tumor that was described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. It predominantly affects males between the second and fifth decades of life and most commonly presents as multiple nodules that may involve either the superficial or deep soft tissues of the legs and less often the arms. It also can arise on the trunk. We present a case of PMHE occurring in a young man and briefly review the literature on clinical presentation and histologic differentiation of this unique tumor, comparing these findings to its mimickers.
Case Report
A 20-year-old man presented with skin lesions on the left leg that had been present for 1 year. The patient described the lesions as tender pimples that would drain yellow discharge on occasion but had now transformed into large brown plaques. Physical examination showed 4 verrucous plaques ranging in size from 1 to 3 cm with hyperpigmentation and a central crust (Figure 1). Initially, the patient thought the lesions appeared due to shaving his legs for sports. He presented to the emergency department multiple times over the past year; pain control was provided and local skin care was recommended. Culture of the discharge had been performed 6 months prior to biopsy with negative results. No biopsy was performed on initial presentation and the lesions were diagnosed in the emergency department clinically as boils.
After failing to improve, the patient was seen by an outside dermatologist and the clinical differential diagnosis included deep fungal infection, atypical mycobacterial infection, and keloids. A 4-mm punch biopsy was taken from the periphery of one of the lesions and demonstrated hyperkeratosis, papillomatosis, and acanthosis (Figure 2). Within the superficial and deep dermis and focally extending into the subcutaneous tissue, there were sheets of spindled to epithelioid-appearing cells with moderate cytologic atypia (Figure 3). The tumor showed infiltrative margins. There was moderate cellularity. The individual cells had a rhabdoid appearance with large eccentric vesicular nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm (Figure 4). No definitive evidence of glandular, squamous, or vascular differentiation was present. There was an associated moderate inflammatory host response composed of neutrophils and lymphocytes. Occasional extravasated red blood cells were present. Immunohistochemistry staining was performed and the atypical cells demonstrated diffuse positive staining for friend leukemia integration 1 transcription factor (FLI-1), erythroblastosis virus E26 transforming sequence-related gene (ERG)(Figure 5), CD31, and CD68. There was patchy positive staining for cytokeratin AE1/AE3, CD10, and factor VIII. There was no remarkable staining for human herpesvirus 8, epithelial membrane antigen, S-100, CD34, cytokeratin 903, and desmin. Overall, the histologic features in conjunction with the immunohistochemistry staining were consistent with a diagnosis of PMHE.
Magnetic resonance imaging was then performed to evaluate the depth and extent of the lesions for surgical excision planning (Figure 6), which showed 5 nodular lesions within the dermis and subcutis adjacent to the proximal aspect of the left tibia and medial aspect of the left knee. An additional lesion was noted between the sartorius and semimembranosus muscles, which was thought to represent either a lymph node or an additional neoplastic lesion. Chest computed tomography also displayed indeterminate lesions in the lungs.
Excision of the superficial lesions was performed. All of the lesions demonstrated similar histologic changes to the previously described biopsy specimen. The tumor was limited to the dermis and subcutaneous tissue. The patient was lost to follow-up and the etiology of the lung lesions was unknown.
Comment
Nomenclature
Pseudomyogenic hemangioendothelioma is a relatively new type of vascular tumor that has been included in the updated 2013 edition of the World Health Organization classification as an intermediate malignant tumor that rarely metastasizes.3 It typically involves multiple tissue planes, most notably the dermis and subcutaneous layers but also muscle and bone.4 The term pseudomyogenic refers to the histologic resemblance of some of the cells to rhabdomyoblasts; however, these tumors are negative for all immunohistochemical muscle markers, most notably myogenin, desmin, and α-smooth muscle actin.5
Clinical Presentation
Gross features of PMHE typically include multiple firm nodules with ill-defined margins. The tumor was initially described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. In 2003, a series of 7 cases of PMHE was reported by Billings et al6 under the term epithelioid sarcomalike hemangioendothelioma. Other than the predominance of an epithelioid morphology, the cases reported as epithelioid sarcomalike hemangioendothelioma had similar clinical features and immunophenotype to what has been reported as PMHE.
Based on a PubMed search of articles indexed for MEDLINE using the term pseudomyogenic hemangioendothelioma, the 2 largest case series were reported by Pradhan et al7 (N=8) in 2017 and Hornick and Fletcher4 (N=50) in 2011. Hornick and Fletcher4 reported a male (41/50 [82.0%]) to female (9/50 [18.0%]) ratio of 4.6 to 1, and an average age at presentation of 31 years with 82% (41/50) of patients 40 years or younger. Pradhan et al7 also reported a male predominance (7/8 [87.5%]) with a similar average age at presentation of 29 years (age range, 9–62 years). The size of individual tumors ranged from 0.3 to 5.5 cm (mean size, 1.9 cm) in the series by Hornick and Fletcher4 and 0.3 to 6.0 com in the series by Pradhan et al.7 Hornick and Fletcher4 reported the most common site of involvement was the leg (27/50 [54.0%]), followed by the arm (12/50 [24.0%]), trunk (9/50 [18.0%]), and head and neck (2/50 [4.0%]). The leg (6/8 [75.0%]) also was the most common site of involvement in the series by Pradhan et al,7 with 2 cases occurring on the arm. In the series by Hornick and Fletcher,4 the tumors typically involved the dermis and subcutaneous tissue (26/50 [52%]) with a smaller number involving skeletal muscle (17/50 [34%]) and bone (7/50 [14%]). They reported 66% of their patients (33/50) had multifocal disease at presentation.4 Pradhan et al7 also reported 2 (25.0%) cases being limited to the superficial soft tissue, 2 (25.0%) being limited to the deep soft tissue, and 4 (50.0%) involving the bone; 5 (62.5%) patients had multifocal disease at presentation. The presentation of our patient in regards to gender, age, and tumor characteristics is consistent with other published cases.5-10
Histopathology
Microscopic features of PMHE include sheets of spindled to epithelioid-appearing cells with mild to moderate nuclear atypia and eosinophilic cytoplasm. The tumor has an infiltrative growth pattern. Some of the cells may resemble rhabdomyoblastlike cells, hence the moniker pseudomyogenic. There is no recapitulation of vascular structures or remarkable cytoplasmic vacuolization. Mitotic rate is low and there is no tumor necrosis.4 The tumor cells do not appear to arise from a vessel or display an angiocentric growth pattern. Many cases report the presence of an inflammatory infiltrate containing neutrophils interspersed within the tumor.4,5,7 The overlying epidermis will commonly show hyperkeratosis, epidermal hyperplasia, and acanthosis.4,11
Differential Diagnosis
The histopathologic differential diagnosis would include epithelioid sarcoma, epithelioid hemangioendothelioma, and to a lesser extent dermatofibrosarcoma protuberans (DFSP) and rhabdomyosarcoma. Dermatofibrosarcoma protuberans is the most commonly encountered of these tumors. Histologically, DFSP is characterized by a cellular proliferation of small spindle cells with plump nuclei arranged in a storiform or cartwheel pattern. Dermatofibrosarcoma protuberans tends to be limited to the dermis and subcutaneous tissue and only rarely involves underlying skeletal muscle. The presence of the storiform growth pattern in conjunction with the lack of rhabdoid changes would favor a diagnosis of DFSP. Another characteristic histologic finding typically only associated with DFSP is the interdigitating growth pattern of the spindle cells within the lobules of the subcutaneous tissue, creating a lacelike or honeycomb appearance.
Immunohistochemistry staining is necessary to help differentiate PMHE from other tumors in the differential diagnosis. Pseudomyogenic hemangioendothelioma stains positive for cytokeratin AE1/AE3; integrase interactor 1; and vascular markers FLI-1, CD31, and ERG, and negative for CD34.4,6,12-15 In contrast to epithelioid hemangioendothelioma, DFSP, and to a lesser extent epithelioid sarcoma, all of which are positive for CD34, epithelioid sarcoma is negative for both CD31 and integrase interactor 1. Dermatofibrosarcoma protuberans is negative for cytokeratin AE1/AE3. Rhabdomyosarcomas are positive for myogenic markers such as MyoD1 and myogenin, unlike any of the other tumors mentioned. Histologically, epithelioid sarcomas will tend to have a granulomalike growth pattern with central necrosis, unlike PMHE.12 Epithelioid hemangioendothelioma often will have a cordlike growth pattern in a myxochondroid background. Unlike PMHE, these tumors often will appear to be arising from vessels, and intracytoplasmic vacuoles are common. Three cases of PMHE have been reported to have a t(7;19)(q22;q13) chromosomal anomaly, which is not consistent with every case.16
Treatment Options
Standard treatment typically includes wide excision of the lesions, as was done in our case. Because of the substantial risk of local recurrence, which was up to 58% in the series by Hornick and Fletcher,4 adjuvant therapy may be considered if positive margins are found on excision. Metastasis to lymph nodes and the lungs has been reported but is rare.2,4 Most cases have been shown to have a favorable prognosis; however, local recurrence seems to be common. Rarely, amputation of the limb may be required.5 In contrast, epithelioid sarcomas have been found to spread to lymph nodes and the lungs in up to 50% of cases with a 5-year survival rate of 10% to 30%.13
Conclusion
In summary, we describe a case of PMHE involving the lower leg in a 20-year-old man. These tumors often are multinodular and multiplanar, with the dermis and subcutaneous tissues being the most common areas affected. It has a high rate of local recurrence but rarely has distant metastasis. Pseudomyogenic hemangioendothelioma, similar to other soft tissue tumors, can be difficult to diagnose on shave biopsy or superficial punch biopsy not extending into subcutaneous tissue. Deep incisional or punch biopsies are required to more definitively diagnose these types of tumors. The diagnosis of PMHE versus other soft tissue tumors requires correlation of histology and immunohistochemistry staining with clinical information and radiographic findings.
Pseudomyogenic hemangioendothelioma (PMHE), also referred to as epithelioid sarcoma–like hemangioendothelioma,1 is a rare soft tissue tumor that was described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. It predominantly affects males between the second and fifth decades of life and most commonly presents as multiple nodules that may involve either the superficial or deep soft tissues of the legs and less often the arms. It also can arise on the trunk. We present a case of PMHE occurring in a young man and briefly review the literature on clinical presentation and histologic differentiation of this unique tumor, comparing these findings to its mimickers.
Case Report
A 20-year-old man presented with skin lesions on the left leg that had been present for 1 year. The patient described the lesions as tender pimples that would drain yellow discharge on occasion but had now transformed into large brown plaques. Physical examination showed 4 verrucous plaques ranging in size from 1 to 3 cm with hyperpigmentation and a central crust (Figure 1). Initially, the patient thought the lesions appeared due to shaving his legs for sports. He presented to the emergency department multiple times over the past year; pain control was provided and local skin care was recommended. Culture of the discharge had been performed 6 months prior to biopsy with negative results. No biopsy was performed on initial presentation and the lesions were diagnosed in the emergency department clinically as boils.
After failing to improve, the patient was seen by an outside dermatologist and the clinical differential diagnosis included deep fungal infection, atypical mycobacterial infection, and keloids. A 4-mm punch biopsy was taken from the periphery of one of the lesions and demonstrated hyperkeratosis, papillomatosis, and acanthosis (Figure 2). Within the superficial and deep dermis and focally extending into the subcutaneous tissue, there were sheets of spindled to epithelioid-appearing cells with moderate cytologic atypia (Figure 3). The tumor showed infiltrative margins. There was moderate cellularity. The individual cells had a rhabdoid appearance with large eccentric vesicular nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm (Figure 4). No definitive evidence of glandular, squamous, or vascular differentiation was present. There was an associated moderate inflammatory host response composed of neutrophils and lymphocytes. Occasional extravasated red blood cells were present. Immunohistochemistry staining was performed and the atypical cells demonstrated diffuse positive staining for friend leukemia integration 1 transcription factor (FLI-1), erythroblastosis virus E26 transforming sequence-related gene (ERG)(Figure 5), CD31, and CD68. There was patchy positive staining for cytokeratin AE1/AE3, CD10, and factor VIII. There was no remarkable staining for human herpesvirus 8, epithelial membrane antigen, S-100, CD34, cytokeratin 903, and desmin. Overall, the histologic features in conjunction with the immunohistochemistry staining were consistent with a diagnosis of PMHE.
Magnetic resonance imaging was then performed to evaluate the depth and extent of the lesions for surgical excision planning (Figure 6), which showed 5 nodular lesions within the dermis and subcutis adjacent to the proximal aspect of the left tibia and medial aspect of the left knee. An additional lesion was noted between the sartorius and semimembranosus muscles, which was thought to represent either a lymph node or an additional neoplastic lesion. Chest computed tomography also displayed indeterminate lesions in the lungs.
Excision of the superficial lesions was performed. All of the lesions demonstrated similar histologic changes to the previously described biopsy specimen. The tumor was limited to the dermis and subcutaneous tissue. The patient was lost to follow-up and the etiology of the lung lesions was unknown.
Comment
Nomenclature
Pseudomyogenic hemangioendothelioma is a relatively new type of vascular tumor that has been included in the updated 2013 edition of the World Health Organization classification as an intermediate malignant tumor that rarely metastasizes.3 It typically involves multiple tissue planes, most notably the dermis and subcutaneous layers but also muscle and bone.4 The term pseudomyogenic refers to the histologic resemblance of some of the cells to rhabdomyoblasts; however, these tumors are negative for all immunohistochemical muscle markers, most notably myogenin, desmin, and α-smooth muscle actin.5
Clinical Presentation
Gross features of PMHE typically include multiple firm nodules with ill-defined margins. The tumor was initially described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. In 2003, a series of 7 cases of PMHE was reported by Billings et al6 under the term epithelioid sarcomalike hemangioendothelioma. Other than the predominance of an epithelioid morphology, the cases reported as epithelioid sarcomalike hemangioendothelioma had similar clinical features and immunophenotype to what has been reported as PMHE.
Based on a PubMed search of articles indexed for MEDLINE using the term pseudomyogenic hemangioendothelioma, the 2 largest case series were reported by Pradhan et al7 (N=8) in 2017 and Hornick and Fletcher4 (N=50) in 2011. Hornick and Fletcher4 reported a male (41/50 [82.0%]) to female (9/50 [18.0%]) ratio of 4.6 to 1, and an average age at presentation of 31 years with 82% (41/50) of patients 40 years or younger. Pradhan et al7 also reported a male predominance (7/8 [87.5%]) with a similar average age at presentation of 29 years (age range, 9–62 years). The size of individual tumors ranged from 0.3 to 5.5 cm (mean size, 1.9 cm) in the series by Hornick and Fletcher4 and 0.3 to 6.0 com in the series by Pradhan et al.7 Hornick and Fletcher4 reported the most common site of involvement was the leg (27/50 [54.0%]), followed by the arm (12/50 [24.0%]), trunk (9/50 [18.0%]), and head and neck (2/50 [4.0%]). The leg (6/8 [75.0%]) also was the most common site of involvement in the series by Pradhan et al,7 with 2 cases occurring on the arm. In the series by Hornick and Fletcher,4 the tumors typically involved the dermis and subcutaneous tissue (26/50 [52%]) with a smaller number involving skeletal muscle (17/50 [34%]) and bone (7/50 [14%]). They reported 66% of their patients (33/50) had multifocal disease at presentation.4 Pradhan et al7 also reported 2 (25.0%) cases being limited to the superficial soft tissue, 2 (25.0%) being limited to the deep soft tissue, and 4 (50.0%) involving the bone; 5 (62.5%) patients had multifocal disease at presentation. The presentation of our patient in regards to gender, age, and tumor characteristics is consistent with other published cases.5-10
Histopathology
Microscopic features of PMHE include sheets of spindled to epithelioid-appearing cells with mild to moderate nuclear atypia and eosinophilic cytoplasm. The tumor has an infiltrative growth pattern. Some of the cells may resemble rhabdomyoblastlike cells, hence the moniker pseudomyogenic. There is no recapitulation of vascular structures or remarkable cytoplasmic vacuolization. Mitotic rate is low and there is no tumor necrosis.4 The tumor cells do not appear to arise from a vessel or display an angiocentric growth pattern. Many cases report the presence of an inflammatory infiltrate containing neutrophils interspersed within the tumor.4,5,7 The overlying epidermis will commonly show hyperkeratosis, epidermal hyperplasia, and acanthosis.4,11
Differential Diagnosis
The histopathologic differential diagnosis would include epithelioid sarcoma, epithelioid hemangioendothelioma, and to a lesser extent dermatofibrosarcoma protuberans (DFSP) and rhabdomyosarcoma. Dermatofibrosarcoma protuberans is the most commonly encountered of these tumors. Histologically, DFSP is characterized by a cellular proliferation of small spindle cells with plump nuclei arranged in a storiform or cartwheel pattern. Dermatofibrosarcoma protuberans tends to be limited to the dermis and subcutaneous tissue and only rarely involves underlying skeletal muscle. The presence of the storiform growth pattern in conjunction with the lack of rhabdoid changes would favor a diagnosis of DFSP. Another characteristic histologic finding typically only associated with DFSP is the interdigitating growth pattern of the spindle cells within the lobules of the subcutaneous tissue, creating a lacelike or honeycomb appearance.
Immunohistochemistry staining is necessary to help differentiate PMHE from other tumors in the differential diagnosis. Pseudomyogenic hemangioendothelioma stains positive for cytokeratin AE1/AE3; integrase interactor 1; and vascular markers FLI-1, CD31, and ERG, and negative for CD34.4,6,12-15 In contrast to epithelioid hemangioendothelioma, DFSP, and to a lesser extent epithelioid sarcoma, all of which are positive for CD34, epithelioid sarcoma is negative for both CD31 and integrase interactor 1. Dermatofibrosarcoma protuberans is negative for cytokeratin AE1/AE3. Rhabdomyosarcomas are positive for myogenic markers such as MyoD1 and myogenin, unlike any of the other tumors mentioned. Histologically, epithelioid sarcomas will tend to have a granulomalike growth pattern with central necrosis, unlike PMHE.12 Epithelioid hemangioendothelioma often will have a cordlike growth pattern in a myxochondroid background. Unlike PMHE, these tumors often will appear to be arising from vessels, and intracytoplasmic vacuoles are common. Three cases of PMHE have been reported to have a t(7;19)(q22;q13) chromosomal anomaly, which is not consistent with every case.16
Treatment Options
Standard treatment typically includes wide excision of the lesions, as was done in our case. Because of the substantial risk of local recurrence, which was up to 58% in the series by Hornick and Fletcher,4 adjuvant therapy may be considered if positive margins are found on excision. Metastasis to lymph nodes and the lungs has been reported but is rare.2,4 Most cases have been shown to have a favorable prognosis; however, local recurrence seems to be common. Rarely, amputation of the limb may be required.5 In contrast, epithelioid sarcomas have been found to spread to lymph nodes and the lungs in up to 50% of cases with a 5-year survival rate of 10% to 30%.13
Conclusion
In summary, we describe a case of PMHE involving the lower leg in a 20-year-old man. These tumors often are multinodular and multiplanar, with the dermis and subcutaneous tissues being the most common areas affected. It has a high rate of local recurrence but rarely has distant metastasis. Pseudomyogenic hemangioendothelioma, similar to other soft tissue tumors, can be difficult to diagnose on shave biopsy or superficial punch biopsy not extending into subcutaneous tissue. Deep incisional or punch biopsies are required to more definitively diagnose these types of tumors. The diagnosis of PMHE versus other soft tissue tumors requires correlation of histology and immunohistochemistry staining with clinical information and radiographic findings.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma (pseudomyogenic hemangioendothelioma). Am J Surg Pathol. 2011;35:1088; author reply 1088-1089.
- Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
- Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014;46:95-104.
- Hornick JL, Fletcher CD. Pseudomyogenic hemangioendothelioma: a distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol. 2011;35:190-201.
- Sheng W, Pan Y, Wang J. Pseudomyogenic hemangioendothelioma: report of an additional case with aggressive clinical course. Am J Dermatopathol. 2013;35:597-600.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol. 2003;27:48-57.
- Pradhan D, Schoedel K, McGough RL, et al. Pseudomyogenic hemangioendothelioma of skin, bone and soft tissue—a clinicopathological, immunohistochemical and fluorescence in situ hybridization study [published online November 2, 2017]. Hum Pathol. 2017. doi:0.1016/j.humpath.2017.10.023.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- McGinity M, Bartanusz V, Dengler B, et al. Pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma, fibroma-like variant of epithelioid sarcoma) of the thoracic spine. Eur Spine J. 2013;22(suppl 3):S506-S511.
- Stuart LN, Gardner JM, Lauer SR, et al. Epithelioid sarcoma-like (pseudomyogenic) hemangioendothelioma, clinically mimicking dermatofibroma, diagnosed by skin biopsy in a 30-year-old man. J Cutan Pathol. 2013;40:909-913.
- Amary MF, O’Donnell P, Berisha F, et al. Pseudomyogenic (epithelioid sarcoma-like) hemangioendothelioma: characterization of five cases. Skeletal Radiol. 2013;42:947-957.
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542-550.
- Chbani L, Guillou L, Terrier P, et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French Sarcoma Group. Am J Clin Pathol. 2009;131:222-227.
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- Trombetta D, Magnusson L, von Steyern FV, et al. Translocation t(7;19)(q22;q13)—a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet. 2011;204:211-215.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma (pseudomyogenic hemangioendothelioma). Am J Surg Pathol. 2011;35:1088; author reply 1088-1089.
- Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
- Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014;46:95-104.
- Hornick JL, Fletcher CD. Pseudomyogenic hemangioendothelioma: a distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol. 2011;35:190-201.
- Sheng W, Pan Y, Wang J. Pseudomyogenic hemangioendothelioma: report of an additional case with aggressive clinical course. Am J Dermatopathol. 2013;35:597-600.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol. 2003;27:48-57.
- Pradhan D, Schoedel K, McGough RL, et al. Pseudomyogenic hemangioendothelioma of skin, bone and soft tissue—a clinicopathological, immunohistochemical and fluorescence in situ hybridization study [published online November 2, 2017]. Hum Pathol. 2017. doi:0.1016/j.humpath.2017.10.023.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- McGinity M, Bartanusz V, Dengler B, et al. Pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma, fibroma-like variant of epithelioid sarcoma) of the thoracic spine. Eur Spine J. 2013;22(suppl 3):S506-S511.
- Stuart LN, Gardner JM, Lauer SR, et al. Epithelioid sarcoma-like (pseudomyogenic) hemangioendothelioma, clinically mimicking dermatofibroma, diagnosed by skin biopsy in a 30-year-old man. J Cutan Pathol. 2013;40:909-913.
- Amary MF, O’Donnell P, Berisha F, et al. Pseudomyogenic (epithelioid sarcoma-like) hemangioendothelioma: characterization of five cases. Skeletal Radiol. 2013;42:947-957.
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542-550.
- Chbani L, Guillou L, Terrier P, et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French Sarcoma Group. Am J Clin Pathol. 2009;131:222-227.
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- Trombetta D, Magnusson L, von Steyern FV, et al. Translocation t(7;19)(q22;q13)—a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet. 2011;204:211-215.
Practice Points
- Pseudomyogenic hemangioendothelioma (PMHE) is an uncommon vascular tumor that most often presents as multiple distinct nodules on the legs in young men.
- Pseudomyogenic hemangioendothelioma has an unusual immunohistochemistry staining pattern, with positive staining for cytokeratin AE1/AE3, CD31, and ERG but negative for CD34.
- Although local reoccurrence is common, PMHE metastasis is very uncommon.
Medication-Induced Pruritus From Direct Oral Anticoagulants
Pruritus is a subjective report of itching, which can be caused by dermatologic or systemic conditions. Pruritus accounts for about 5% of all skin adverse drug reactions (ADRs) after administration.1 Mechanisms by which medication-induced pruritus occurs are still unknown and have been understudied. Treatment modalities also have been understudied; however, the understood method for treatment is cessation of the causative agent.2
Anticoagulants commonly are used in several conditions, including prevention of ischemic cerebrovascular accident (CVA) in patients with atrial fibrillation (AF) as well as for the treatment and prevention of deep vein thrombosis (DVT) and pulmonary embolism (PE).3 Traditionally, warfarin was the gold standard anticoagulant. With the relatively recent approval of several direct oral anticoagulants (DOACs), such as rivaroxaban, apixaban, and dabigatran, the landscape of anticoagulation is changing. One benefit of using DOACs as opposed to warfarin is that they often require less frequent laboratory monitoring. However, rare ADRs not detected during clinical trials have appeared following drug approval.4
In a VA anticoagulation clinic that managed more than 60 patients taking DOACs, the authors identified 2 cases of pruritus, possibly associated with DOAC agents. These 2 cases point to a higher incidence rate than the rate reported in the rivaroxaban package insert (2%).5 Of note, pruritus is not mentioned in the apixaban package insert.6
Patient 1 Case Presentation
Patient 1 was a 69-year-old male with AF who was switched to rivaroxaban after 5 years of warfarin therapy. Past medical history included iron deficiency anemia, hypertension, type 2 diabetes mellitus, systolic heart failure, hyperlipidemia, hepatic steatosis, benign prostatic hyperplasia, and gastroesophageal reflux disease. The patient reported “itching all over” soon after initiation of rivaroxaban and that the itching was intolerable and always began 90 to 120 minutes after each dose of rivaroxaban with no associated rash.
After about 6 months of treatment with rivaroxaban, the patient was switched to apixaban; however, the pruritus persisted even after the switch. The onset of itching had similar timing with regard to the apixaban doses. When apixaban was initiated, the patient also was started on amiodarone and tamsulosin. A full pharmacotherapy review of the patient’s medication list for the incidence of pruritus was conducted. Regarding amiodarone and tamsulosin, incidence of pruritus was < 1%.7,8 Neither agent had yet been started during the rivaroxaban therapy; therefore, it was unlikely that either of these 2 medications were the causative agent of the pruritic ADR.
In response to the itching, the patient was given diphenhydramine 25 mg twice daily, taken with each dose of apixaban. Shortly thereafter, the patient reported that diphenhydramine lessened the severity of the pruritus. He was switched to loratadine 10 mg twice daily with each dose of apixaban, to avoid drowsiness as well as the increased anticholinergic ADRs of first-generation antihistamines. The patient reported that the itching was tolerable.
Patient 2 Case Presentation
Patient 2 was a 63-year-old male with AF and hypertension who was initially started on rivaroxaban and reported pruritus after about 1 month. Despite the uncomfortable itch, the patient elected to continue therapy and began diphenhydramine 25 mg daily with each dose of rivaroxaban. Diphenhydramine seemed to improve the pruritus but did not completely alleviate it. While on rivaroxaban, the patient experienced an acute drop in hemoglobin; however, no source of bleeding was found. Although the pruritus was largely resolved, he was switched to apixaban due to its favorable bleeding profile.9 The patient continued to have pruritus on apixaban; however, he reported that pruritus was less severe than it had been while taking rivaroxaban. The patient restarted on diphenhydramine twice daily with each dose of apixaban and reported cessation of pruritus.
Discussion
After observing both cases in relation to the timing between the administration of a DOAC and onset of pruritus, it seemed likely that the causative agent could be the DOAC. A Naranjo Nomogram was used to determine the likeliness of each drug to be the causative agent of the ADR.10 This nomogram is scaled from a low score of -4 to a high score of 13. Patients 1 and 2 had a score of 4, which is reflective of a possible ADR (score 1-4). Using the nomogram as well as the subjective information provided by the patients, it is reasonable to conclude that the pruritus was possibly associated with the use of the DOACs. Nonadherence to anticoagulants may lead to potentially fatal adverse outcomes, such as stroke. Medication-associated pruritus could lead to medication nonadherence if left unaddressed. It is notable that prescribing an antihistamine that is taken at the time of the anticoagulant dose allowed these patients with possible DOAC-associated pruritus to continue therapy with the selected anticoagulant. Further research on this topic is needed.
1. Reich A, Ständer S, Szepietowski C. Drug-induced pruritus: a review. Acta Derm Venereol. 2009;89(3):236-244.
2. Ebata T. Drug-induced itch management. Curr Probl Dermatol. 2016;50:155-163.
3. Burnett AE, Mahan CE, Vazquez SR, Oertel LB, Garcia DA, Ansell J. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis. 2016;41(1):206-232.
4. U.S. Department of Health and Human Services, U.S. Food & Drug Administration. FDA Adverse Events Reporting System (FAERS) public dashboard. Apixaban. https://fis.fda.gov/sense/app/777e9f4d-0cf8-448e-8068-f564c31baa25/sheet/45beeb74-30ab-46be-8267-5756582633b4/state/analysis. Updated August 31, 2017. Accessed November 8, 2017.
5. Xarelto [package insert]. Titusville, NJ: Janssen Pharmaceuticals Inc; 2011.
6. Eliquis [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; New York, NY: Pfizer Inc; 2016.
7. Pacerone [package insert]. Minneapolis, MN: Upsher-Smith Laboratories Inc; 2008. Minneapolis, MN.
8. Flomax [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc; 2016.
9. Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patient with atrial fibrillation. N Engl J Med. 2011;365(11):981-992.
10. Michel DJ, Knodel LC. Comparison of three algorithms used to evaluate adverse drug reactions. Am J Hosp Pharm. 1986;43(7):1709-1714.
Pruritus is a subjective report of itching, which can be caused by dermatologic or systemic conditions. Pruritus accounts for about 5% of all skin adverse drug reactions (ADRs) after administration.1 Mechanisms by which medication-induced pruritus occurs are still unknown and have been understudied. Treatment modalities also have been understudied; however, the understood method for treatment is cessation of the causative agent.2
Anticoagulants commonly are used in several conditions, including prevention of ischemic cerebrovascular accident (CVA) in patients with atrial fibrillation (AF) as well as for the treatment and prevention of deep vein thrombosis (DVT) and pulmonary embolism (PE).3 Traditionally, warfarin was the gold standard anticoagulant. With the relatively recent approval of several direct oral anticoagulants (DOACs), such as rivaroxaban, apixaban, and dabigatran, the landscape of anticoagulation is changing. One benefit of using DOACs as opposed to warfarin is that they often require less frequent laboratory monitoring. However, rare ADRs not detected during clinical trials have appeared following drug approval.4
In a VA anticoagulation clinic that managed more than 60 patients taking DOACs, the authors identified 2 cases of pruritus, possibly associated with DOAC agents. These 2 cases point to a higher incidence rate than the rate reported in the rivaroxaban package insert (2%).5 Of note, pruritus is not mentioned in the apixaban package insert.6
Patient 1 Case Presentation
Patient 1 was a 69-year-old male with AF who was switched to rivaroxaban after 5 years of warfarin therapy. Past medical history included iron deficiency anemia, hypertension, type 2 diabetes mellitus, systolic heart failure, hyperlipidemia, hepatic steatosis, benign prostatic hyperplasia, and gastroesophageal reflux disease. The patient reported “itching all over” soon after initiation of rivaroxaban and that the itching was intolerable and always began 90 to 120 minutes after each dose of rivaroxaban with no associated rash.
After about 6 months of treatment with rivaroxaban, the patient was switched to apixaban; however, the pruritus persisted even after the switch. The onset of itching had similar timing with regard to the apixaban doses. When apixaban was initiated, the patient also was started on amiodarone and tamsulosin. A full pharmacotherapy review of the patient’s medication list for the incidence of pruritus was conducted. Regarding amiodarone and tamsulosin, incidence of pruritus was < 1%.7,8 Neither agent had yet been started during the rivaroxaban therapy; therefore, it was unlikely that either of these 2 medications were the causative agent of the pruritic ADR.
In response to the itching, the patient was given diphenhydramine 25 mg twice daily, taken with each dose of apixaban. Shortly thereafter, the patient reported that diphenhydramine lessened the severity of the pruritus. He was switched to loratadine 10 mg twice daily with each dose of apixaban, to avoid drowsiness as well as the increased anticholinergic ADRs of first-generation antihistamines. The patient reported that the itching was tolerable.
Patient 2 Case Presentation
Patient 2 was a 63-year-old male with AF and hypertension who was initially started on rivaroxaban and reported pruritus after about 1 month. Despite the uncomfortable itch, the patient elected to continue therapy and began diphenhydramine 25 mg daily with each dose of rivaroxaban. Diphenhydramine seemed to improve the pruritus but did not completely alleviate it. While on rivaroxaban, the patient experienced an acute drop in hemoglobin; however, no source of bleeding was found. Although the pruritus was largely resolved, he was switched to apixaban due to its favorable bleeding profile.9 The patient continued to have pruritus on apixaban; however, he reported that pruritus was less severe than it had been while taking rivaroxaban. The patient restarted on diphenhydramine twice daily with each dose of apixaban and reported cessation of pruritus.
Discussion
After observing both cases in relation to the timing between the administration of a DOAC and onset of pruritus, it seemed likely that the causative agent could be the DOAC. A Naranjo Nomogram was used to determine the likeliness of each drug to be the causative agent of the ADR.10 This nomogram is scaled from a low score of -4 to a high score of 13. Patients 1 and 2 had a score of 4, which is reflective of a possible ADR (score 1-4). Using the nomogram as well as the subjective information provided by the patients, it is reasonable to conclude that the pruritus was possibly associated with the use of the DOACs. Nonadherence to anticoagulants may lead to potentially fatal adverse outcomes, such as stroke. Medication-associated pruritus could lead to medication nonadherence if left unaddressed. It is notable that prescribing an antihistamine that is taken at the time of the anticoagulant dose allowed these patients with possible DOAC-associated pruritus to continue therapy with the selected anticoagulant. Further research on this topic is needed.
Pruritus is a subjective report of itching, which can be caused by dermatologic or systemic conditions. Pruritus accounts for about 5% of all skin adverse drug reactions (ADRs) after administration.1 Mechanisms by which medication-induced pruritus occurs are still unknown and have been understudied. Treatment modalities also have been understudied; however, the understood method for treatment is cessation of the causative agent.2
Anticoagulants commonly are used in several conditions, including prevention of ischemic cerebrovascular accident (CVA) in patients with atrial fibrillation (AF) as well as for the treatment and prevention of deep vein thrombosis (DVT) and pulmonary embolism (PE).3 Traditionally, warfarin was the gold standard anticoagulant. With the relatively recent approval of several direct oral anticoagulants (DOACs), such as rivaroxaban, apixaban, and dabigatran, the landscape of anticoagulation is changing. One benefit of using DOACs as opposed to warfarin is that they often require less frequent laboratory monitoring. However, rare ADRs not detected during clinical trials have appeared following drug approval.4
In a VA anticoagulation clinic that managed more than 60 patients taking DOACs, the authors identified 2 cases of pruritus, possibly associated with DOAC agents. These 2 cases point to a higher incidence rate than the rate reported in the rivaroxaban package insert (2%).5 Of note, pruritus is not mentioned in the apixaban package insert.6
Patient 1 Case Presentation
Patient 1 was a 69-year-old male with AF who was switched to rivaroxaban after 5 years of warfarin therapy. Past medical history included iron deficiency anemia, hypertension, type 2 diabetes mellitus, systolic heart failure, hyperlipidemia, hepatic steatosis, benign prostatic hyperplasia, and gastroesophageal reflux disease. The patient reported “itching all over” soon after initiation of rivaroxaban and that the itching was intolerable and always began 90 to 120 minutes after each dose of rivaroxaban with no associated rash.
After about 6 months of treatment with rivaroxaban, the patient was switched to apixaban; however, the pruritus persisted even after the switch. The onset of itching had similar timing with regard to the apixaban doses. When apixaban was initiated, the patient also was started on amiodarone and tamsulosin. A full pharmacotherapy review of the patient’s medication list for the incidence of pruritus was conducted. Regarding amiodarone and tamsulosin, incidence of pruritus was < 1%.7,8 Neither agent had yet been started during the rivaroxaban therapy; therefore, it was unlikely that either of these 2 medications were the causative agent of the pruritic ADR.
In response to the itching, the patient was given diphenhydramine 25 mg twice daily, taken with each dose of apixaban. Shortly thereafter, the patient reported that diphenhydramine lessened the severity of the pruritus. He was switched to loratadine 10 mg twice daily with each dose of apixaban, to avoid drowsiness as well as the increased anticholinergic ADRs of first-generation antihistamines. The patient reported that the itching was tolerable.
Patient 2 Case Presentation
Patient 2 was a 63-year-old male with AF and hypertension who was initially started on rivaroxaban and reported pruritus after about 1 month. Despite the uncomfortable itch, the patient elected to continue therapy and began diphenhydramine 25 mg daily with each dose of rivaroxaban. Diphenhydramine seemed to improve the pruritus but did not completely alleviate it. While on rivaroxaban, the patient experienced an acute drop in hemoglobin; however, no source of bleeding was found. Although the pruritus was largely resolved, he was switched to apixaban due to its favorable bleeding profile.9 The patient continued to have pruritus on apixaban; however, he reported that pruritus was less severe than it had been while taking rivaroxaban. The patient restarted on diphenhydramine twice daily with each dose of apixaban and reported cessation of pruritus.
Discussion
After observing both cases in relation to the timing between the administration of a DOAC and onset of pruritus, it seemed likely that the causative agent could be the DOAC. A Naranjo Nomogram was used to determine the likeliness of each drug to be the causative agent of the ADR.10 This nomogram is scaled from a low score of -4 to a high score of 13. Patients 1 and 2 had a score of 4, which is reflective of a possible ADR (score 1-4). Using the nomogram as well as the subjective information provided by the patients, it is reasonable to conclude that the pruritus was possibly associated with the use of the DOACs. Nonadherence to anticoagulants may lead to potentially fatal adverse outcomes, such as stroke. Medication-associated pruritus could lead to medication nonadherence if left unaddressed. It is notable that prescribing an antihistamine that is taken at the time of the anticoagulant dose allowed these patients with possible DOAC-associated pruritus to continue therapy with the selected anticoagulant. Further research on this topic is needed.
1. Reich A, Ständer S, Szepietowski C. Drug-induced pruritus: a review. Acta Derm Venereol. 2009;89(3):236-244.
2. Ebata T. Drug-induced itch management. Curr Probl Dermatol. 2016;50:155-163.
3. Burnett AE, Mahan CE, Vazquez SR, Oertel LB, Garcia DA, Ansell J. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis. 2016;41(1):206-232.
4. U.S. Department of Health and Human Services, U.S. Food & Drug Administration. FDA Adverse Events Reporting System (FAERS) public dashboard. Apixaban. https://fis.fda.gov/sense/app/777e9f4d-0cf8-448e-8068-f564c31baa25/sheet/45beeb74-30ab-46be-8267-5756582633b4/state/analysis. Updated August 31, 2017. Accessed November 8, 2017.
5. Xarelto [package insert]. Titusville, NJ: Janssen Pharmaceuticals Inc; 2011.
6. Eliquis [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; New York, NY: Pfizer Inc; 2016.
7. Pacerone [package insert]. Minneapolis, MN: Upsher-Smith Laboratories Inc; 2008. Minneapolis, MN.
8. Flomax [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc; 2016.
9. Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patient with atrial fibrillation. N Engl J Med. 2011;365(11):981-992.
10. Michel DJ, Knodel LC. Comparison of three algorithms used to evaluate adverse drug reactions. Am J Hosp Pharm. 1986;43(7):1709-1714.
1. Reich A, Ständer S, Szepietowski C. Drug-induced pruritus: a review. Acta Derm Venereol. 2009;89(3):236-244.
2. Ebata T. Drug-induced itch management. Curr Probl Dermatol. 2016;50:155-163.
3. Burnett AE, Mahan CE, Vazquez SR, Oertel LB, Garcia DA, Ansell J. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis. 2016;41(1):206-232.
4. U.S. Department of Health and Human Services, U.S. Food & Drug Administration. FDA Adverse Events Reporting System (FAERS) public dashboard. Apixaban. https://fis.fda.gov/sense/app/777e9f4d-0cf8-448e-8068-f564c31baa25/sheet/45beeb74-30ab-46be-8267-5756582633b4/state/analysis. Updated August 31, 2017. Accessed November 8, 2017.
5. Xarelto [package insert]. Titusville, NJ: Janssen Pharmaceuticals Inc; 2011.
6. Eliquis [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; New York, NY: Pfizer Inc; 2016.
7. Pacerone [package insert]. Minneapolis, MN: Upsher-Smith Laboratories Inc; 2008. Minneapolis, MN.
8. Flomax [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc; 2016.
9. Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patient with atrial fibrillation. N Engl J Med. 2011;365(11):981-992.
10. Michel DJ, Knodel LC. Comparison of three algorithms used to evaluate adverse drug reactions. Am J Hosp Pharm. 1986;43(7):1709-1714.
Translunate, Transradial, Transtriquetral, Transtrapezoid Perilunate Dislocation With Multiple Metacarpal Neck Fractures
Take-Home Points
- Emergency physicians should be aware of radiological markers to avoid missing perilunate injuries.
- They should have a low threshold to refer a suspected perilunate injury for urgent specialist assessment.
- Although majority of the injuries demonstrate the classical pattern, one should be aware of atypical injuries.
- The principles of early anatomic reduction and stable fixation remain the same.
- Salvage procedures are only indicated in extensive irreparable injuries.
Perilunate fracture-dislocations, rare injuries representing <10% of wrist injuries,1 are part of a wide spectrum of high-energy trauma injuries. The typical mechanism of injury is a fall on a dorsiflexed and ulnar-deviated wrist with forces progressively traversing the scapholunate, lunocapitate, and lunotriquetral ligaments.2
In this article, we report a very unusual case of translunate, transradial, transtriquetral, transtrapezoid perilunate dislocation with multiple metacarpal neck fractures. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A fit and healthy 30-year-old male software professional fell down stairs, landed on his nondominant right hand, and sustained a high-energy wrist injury. The patient also sustained a concussion, without focal neurologic deficit, and was unable to recall the exact mechanism of the wrist injury (there were no other witnesses). Radiographs of the right wrist in the emergency department showed only a nondisplaced fracture of the neck of the second, third, fourth, and fifth metacarpals and a nondisplaced fracture of the radial styloid.
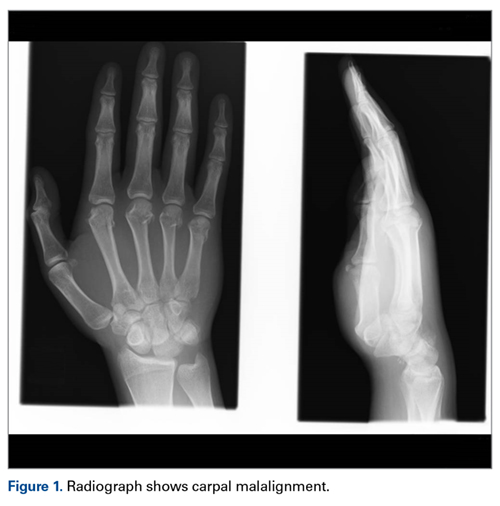
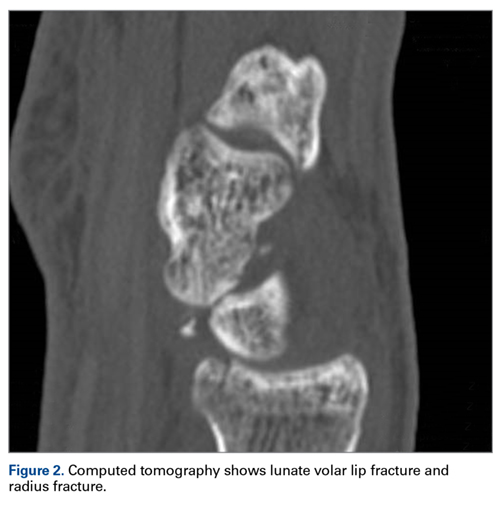
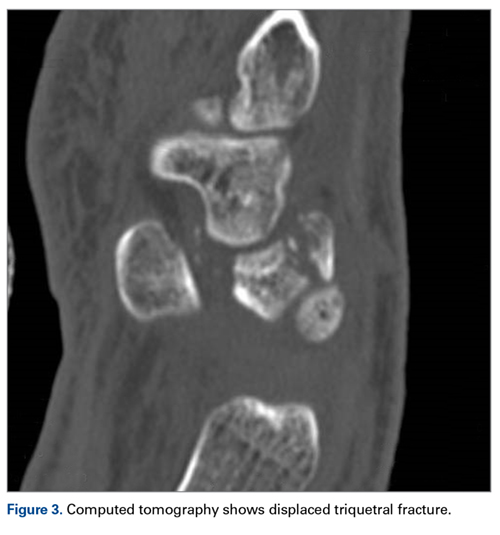
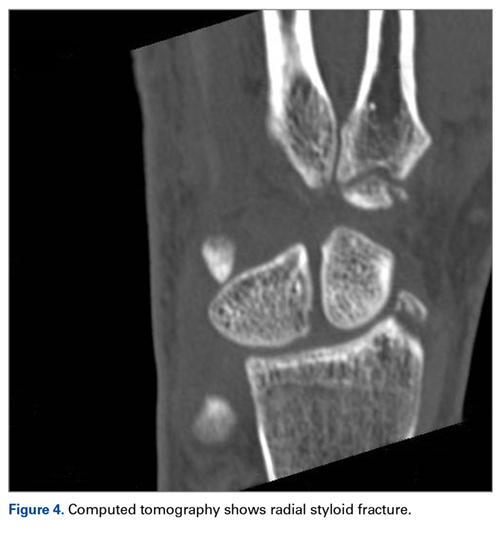
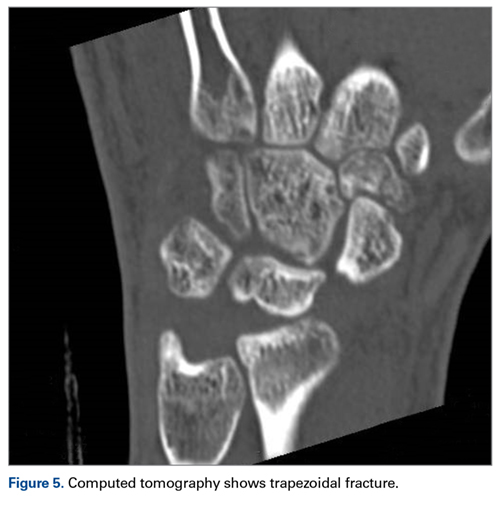
The next day, with the patient under general anesthesia, an attempt to reduce the perilunate dislocation by manipulation was unsuccessful. Open reduction and internal fixation (ORIF) were performed through a dorsal approach; the perilunate dislocation was reduced and stabilized with lunocapitate 1.2-mm Kirschner wire (K-wire). The scapholunate and lunotriquetral ligaments were found to be intact, and the significantly displaced triquetral fracture was treated with internal fixation involving 2 minifragment screws (Figure 6).
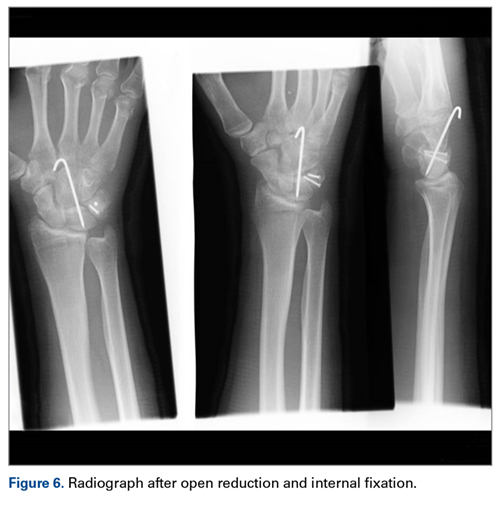
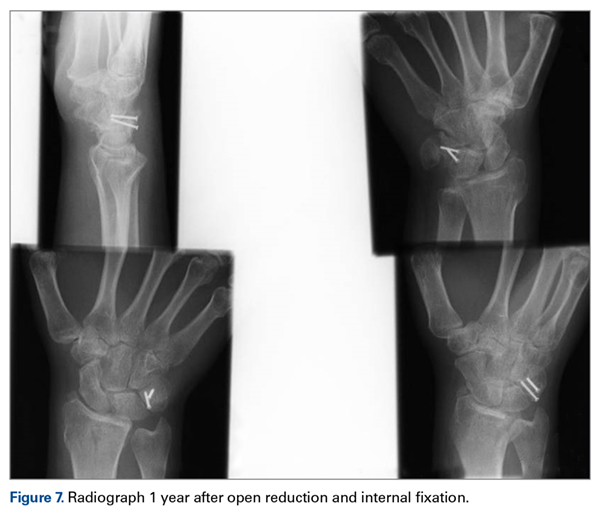
Discussion
Perilunate injuries are classified as lesser arc injuries (purely ligamentous) or greater arc injuries (osseoligamentous). Greater arc injuries involve fracture of one or more carpal bones with associated ligamentous injuries.3 The greater or lesser arc injuries described by Mayfield and colleagues2 imply a specific pattern of force transmission with axial loading in a dorsiflexed and ulnar-deviated wrist with intercarpal supination. Graham4 introduced a concept of inferior arc injury with the forces passing through the radiocarpal joint with fracture of the radial styloid or juxta-articular margin. Similarly, lunate fracture in perilunate dislocations was explained by Bain and colleagues5 in the translunate arc concept in which forces pass through the lunate bone. A study involving a literature review of translunate perilunate dislocations noted associated transradial, trans-scaphoid, transcapitate, and transtriquetral fractures in order of decreasing frequency.6 To our knowledge, no case of translunate perilunate dislocation with multiple carpal and metacarpal fractures with radial styloid fracture has been reported in the literature.
Our patient’s associated multiple metacarpal neck fractures can be explained by the peculiar double-impact injury with initial axial loading across the hyperextended metacarpophalangeal joint, followed by axial loading across the hyperextended and ulnar-deviated wrist, causing greater arc perilunate fracture-dislocation. The mechanism of lunate injury in this case seems to be longitudinal impaction of the capitate shearing against the volar lunate in the axial plane causing a volar lip fracture (Teisen type I), and this may be accentuated by tension in the volar radiolunate ligament.6,7 Associated triquetral fracture in perilunate dislocation is well described in the literature.6 However, the trapezoid fracture in our case implies a very atypical pattern of force transmission with the arc probably passing more distally through the trapezoid laterally and the triquetrum medially.
This case, which represents a very rare fracture pattern associated with perilunate dislocation, may have been caused by the variable position of the wrist and the pattern of load transmission at time of impact. Although the majority of cases demonstrate the classical pattern described in the literature, it may not be unusual to find atypical fracture patterns, especially those associated with high-energy trauma.
Perilunate injuries have been missed in busy emergency departments and orthopedic practices. An estimated 25% of such injuries can be missed on initial presentation.8 In the present case, fracture of the radial styloid provided a clue to possible more complex carpal injuries involving the scaphoid, lunate, or scapholunate ligament, as Graham4 suggested with the concept of the “transverse pattern” of force transmission. In this case as well, the injury was initially missed, and its extent became evident only with CT. Therefore, emergency teams should have a very low threshold for suspecting and evaluating high-energy wrist injuries.
The goal in the treatment of perilunate dislocation with multiple carpal fractures is anatomical reduction and restoration of carpal alignment—which frequently require ORIF, though acute salvage procedures like proximal row carpectomy may be considered in irreparable fractures with extensive ligament injuries.9 For open reduction, the approach can be dorsal, volar, or a combination. The approach in our patient’s case was dorsal. His triquetral fracture, his only displaced fracture, was treated with internal fixation. All other fractures were nondisplaced, stable, and did not warrant internal fixation.
A high index of suspicion and urgent specialist consultation are essential in suspected perilunate injuries. The injury and fracture pattern may be atypical, but the principles of early anatomical reduction and stable fixation remain the same.
1. Youssef B, Deshmukh SC. Volar perilunate dislocation: a case report and review of the literature. Open Orthop J. 2008;2:57-58.
2. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
3. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
4. Graham TJ. The inferior arc injury: an addition to the family of complex carpal fracture-dislocation patterns. Am J Orthop. 2003;32(9 suppl):10-19.
5. Bain GI, McLean JM, Turner PC, Sood A, Pourgiezis N. Translunate fracture with associated perilunate injury: 3 case reports with introduction of the translunate arc concept. J Hand Surg Am. 2008;33(10):1770-1776.
6. Bain GI, Pallapati S, Eng K. Translunate perilunate injuries—a spectrum of this uncommon injury. J Wrist Surg. 2013;2(1):63-68.
7. Teisen H, Hjarbaek J. Classification of fresh fractures of the lunate. J Hand Surg Br. 1988;13(4):458-462.
8. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
9. Huish EG Jr, Vitale MA, Shin AY. Acute proximal row carpectomy to treat a transscaphoid, transtriquetral perilunate fracture dislocation: case report and review of the literature. Hand
(N Y). 2013;8(1):105-109.
Take-Home Points
- Emergency physicians should be aware of radiological markers to avoid missing perilunate injuries.
- They should have a low threshold to refer a suspected perilunate injury for urgent specialist assessment.
- Although majority of the injuries demonstrate the classical pattern, one should be aware of atypical injuries.
- The principles of early anatomic reduction and stable fixation remain the same.
- Salvage procedures are only indicated in extensive irreparable injuries.
Perilunate fracture-dislocations, rare injuries representing <10% of wrist injuries,1 are part of a wide spectrum of high-energy trauma injuries. The typical mechanism of injury is a fall on a dorsiflexed and ulnar-deviated wrist with forces progressively traversing the scapholunate, lunocapitate, and lunotriquetral ligaments.2
In this article, we report a very unusual case of translunate, transradial, transtriquetral, transtrapezoid perilunate dislocation with multiple metacarpal neck fractures. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A fit and healthy 30-year-old male software professional fell down stairs, landed on his nondominant right hand, and sustained a high-energy wrist injury. The patient also sustained a concussion, without focal neurologic deficit, and was unable to recall the exact mechanism of the wrist injury (there were no other witnesses). Radiographs of the right wrist in the emergency department showed only a nondisplaced fracture of the neck of the second, third, fourth, and fifth metacarpals and a nondisplaced fracture of the radial styloid.
The next day, with the patient under general anesthesia, an attempt to reduce the perilunate dislocation by manipulation was unsuccessful. Open reduction and internal fixation (ORIF) were performed through a dorsal approach; the perilunate dislocation was reduced and stabilized with lunocapitate 1.2-mm Kirschner wire (K-wire). The scapholunate and lunotriquetral ligaments were found to be intact, and the significantly displaced triquetral fracture was treated with internal fixation involving 2 minifragment screws (Figure 6).
Discussion
Perilunate injuries are classified as lesser arc injuries (purely ligamentous) or greater arc injuries (osseoligamentous). Greater arc injuries involve fracture of one or more carpal bones with associated ligamentous injuries.3 The greater or lesser arc injuries described by Mayfield and colleagues2 imply a specific pattern of force transmission with axial loading in a dorsiflexed and ulnar-deviated wrist with intercarpal supination. Graham4 introduced a concept of inferior arc injury with the forces passing through the radiocarpal joint with fracture of the radial styloid or juxta-articular margin. Similarly, lunate fracture in perilunate dislocations was explained by Bain and colleagues5 in the translunate arc concept in which forces pass through the lunate bone. A study involving a literature review of translunate perilunate dislocations noted associated transradial, trans-scaphoid, transcapitate, and transtriquetral fractures in order of decreasing frequency.6 To our knowledge, no case of translunate perilunate dislocation with multiple carpal and metacarpal fractures with radial styloid fracture has been reported in the literature.
Our patient’s associated multiple metacarpal neck fractures can be explained by the peculiar double-impact injury with initial axial loading across the hyperextended metacarpophalangeal joint, followed by axial loading across the hyperextended and ulnar-deviated wrist, causing greater arc perilunate fracture-dislocation. The mechanism of lunate injury in this case seems to be longitudinal impaction of the capitate shearing against the volar lunate in the axial plane causing a volar lip fracture (Teisen type I), and this may be accentuated by tension in the volar radiolunate ligament.6,7 Associated triquetral fracture in perilunate dislocation is well described in the literature.6 However, the trapezoid fracture in our case implies a very atypical pattern of force transmission with the arc probably passing more distally through the trapezoid laterally and the triquetrum medially.
This case, which represents a very rare fracture pattern associated with perilunate dislocation, may have been caused by the variable position of the wrist and the pattern of load transmission at time of impact. Although the majority of cases demonstrate the classical pattern described in the literature, it may not be unusual to find atypical fracture patterns, especially those associated with high-energy trauma.
Perilunate injuries have been missed in busy emergency departments and orthopedic practices. An estimated 25% of such injuries can be missed on initial presentation.8 In the present case, fracture of the radial styloid provided a clue to possible more complex carpal injuries involving the scaphoid, lunate, or scapholunate ligament, as Graham4 suggested with the concept of the “transverse pattern” of force transmission. In this case as well, the injury was initially missed, and its extent became evident only with CT. Therefore, emergency teams should have a very low threshold for suspecting and evaluating high-energy wrist injuries.
The goal in the treatment of perilunate dislocation with multiple carpal fractures is anatomical reduction and restoration of carpal alignment—which frequently require ORIF, though acute salvage procedures like proximal row carpectomy may be considered in irreparable fractures with extensive ligament injuries.9 For open reduction, the approach can be dorsal, volar, or a combination. The approach in our patient’s case was dorsal. His triquetral fracture, his only displaced fracture, was treated with internal fixation. All other fractures were nondisplaced, stable, and did not warrant internal fixation.
A high index of suspicion and urgent specialist consultation are essential in suspected perilunate injuries. The injury and fracture pattern may be atypical, but the principles of early anatomical reduction and stable fixation remain the same.
Take-Home Points
- Emergency physicians should be aware of radiological markers to avoid missing perilunate injuries.
- They should have a low threshold to refer a suspected perilunate injury for urgent specialist assessment.
- Although majority of the injuries demonstrate the classical pattern, one should be aware of atypical injuries.
- The principles of early anatomic reduction and stable fixation remain the same.
- Salvage procedures are only indicated in extensive irreparable injuries.
Perilunate fracture-dislocations, rare injuries representing <10% of wrist injuries,1 are part of a wide spectrum of high-energy trauma injuries. The typical mechanism of injury is a fall on a dorsiflexed and ulnar-deviated wrist with forces progressively traversing the scapholunate, lunocapitate, and lunotriquetral ligaments.2
In this article, we report a very unusual case of translunate, transradial, transtriquetral, transtrapezoid perilunate dislocation with multiple metacarpal neck fractures. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A fit and healthy 30-year-old male software professional fell down stairs, landed on his nondominant right hand, and sustained a high-energy wrist injury. The patient also sustained a concussion, without focal neurologic deficit, and was unable to recall the exact mechanism of the wrist injury (there were no other witnesses). Radiographs of the right wrist in the emergency department showed only a nondisplaced fracture of the neck of the second, third, fourth, and fifth metacarpals and a nondisplaced fracture of the radial styloid.
The next day, with the patient under general anesthesia, an attempt to reduce the perilunate dislocation by manipulation was unsuccessful. Open reduction and internal fixation (ORIF) were performed through a dorsal approach; the perilunate dislocation was reduced and stabilized with lunocapitate 1.2-mm Kirschner wire (K-wire). The scapholunate and lunotriquetral ligaments were found to be intact, and the significantly displaced triquetral fracture was treated with internal fixation involving 2 minifragment screws (Figure 6).
Discussion
Perilunate injuries are classified as lesser arc injuries (purely ligamentous) or greater arc injuries (osseoligamentous). Greater arc injuries involve fracture of one or more carpal bones with associated ligamentous injuries.3 The greater or lesser arc injuries described by Mayfield and colleagues2 imply a specific pattern of force transmission with axial loading in a dorsiflexed and ulnar-deviated wrist with intercarpal supination. Graham4 introduced a concept of inferior arc injury with the forces passing through the radiocarpal joint with fracture of the radial styloid or juxta-articular margin. Similarly, lunate fracture in perilunate dislocations was explained by Bain and colleagues5 in the translunate arc concept in which forces pass through the lunate bone. A study involving a literature review of translunate perilunate dislocations noted associated transradial, trans-scaphoid, transcapitate, and transtriquetral fractures in order of decreasing frequency.6 To our knowledge, no case of translunate perilunate dislocation with multiple carpal and metacarpal fractures with radial styloid fracture has been reported in the literature.
Our patient’s associated multiple metacarpal neck fractures can be explained by the peculiar double-impact injury with initial axial loading across the hyperextended metacarpophalangeal joint, followed by axial loading across the hyperextended and ulnar-deviated wrist, causing greater arc perilunate fracture-dislocation. The mechanism of lunate injury in this case seems to be longitudinal impaction of the capitate shearing against the volar lunate in the axial plane causing a volar lip fracture (Teisen type I), and this may be accentuated by tension in the volar radiolunate ligament.6,7 Associated triquetral fracture in perilunate dislocation is well described in the literature.6 However, the trapezoid fracture in our case implies a very atypical pattern of force transmission with the arc probably passing more distally through the trapezoid laterally and the triquetrum medially.
This case, which represents a very rare fracture pattern associated with perilunate dislocation, may have been caused by the variable position of the wrist and the pattern of load transmission at time of impact. Although the majority of cases demonstrate the classical pattern described in the literature, it may not be unusual to find atypical fracture patterns, especially those associated with high-energy trauma.
Perilunate injuries have been missed in busy emergency departments and orthopedic practices. An estimated 25% of such injuries can be missed on initial presentation.8 In the present case, fracture of the radial styloid provided a clue to possible more complex carpal injuries involving the scaphoid, lunate, or scapholunate ligament, as Graham4 suggested with the concept of the “transverse pattern” of force transmission. In this case as well, the injury was initially missed, and its extent became evident only with CT. Therefore, emergency teams should have a very low threshold for suspecting and evaluating high-energy wrist injuries.
The goal in the treatment of perilunate dislocation with multiple carpal fractures is anatomical reduction and restoration of carpal alignment—which frequently require ORIF, though acute salvage procedures like proximal row carpectomy may be considered in irreparable fractures with extensive ligament injuries.9 For open reduction, the approach can be dorsal, volar, or a combination. The approach in our patient’s case was dorsal. His triquetral fracture, his only displaced fracture, was treated with internal fixation. All other fractures were nondisplaced, stable, and did not warrant internal fixation.
A high index of suspicion and urgent specialist consultation are essential in suspected perilunate injuries. The injury and fracture pattern may be atypical, but the principles of early anatomical reduction and stable fixation remain the same.
1. Youssef B, Deshmukh SC. Volar perilunate dislocation: a case report and review of the literature. Open Orthop J. 2008;2:57-58.
2. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
3. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
4. Graham TJ. The inferior arc injury: an addition to the family of complex carpal fracture-dislocation patterns. Am J Orthop. 2003;32(9 suppl):10-19.
5. Bain GI, McLean JM, Turner PC, Sood A, Pourgiezis N. Translunate fracture with associated perilunate injury: 3 case reports with introduction of the translunate arc concept. J Hand Surg Am. 2008;33(10):1770-1776.
6. Bain GI, Pallapati S, Eng K. Translunate perilunate injuries—a spectrum of this uncommon injury. J Wrist Surg. 2013;2(1):63-68.
7. Teisen H, Hjarbaek J. Classification of fresh fractures of the lunate. J Hand Surg Br. 1988;13(4):458-462.
8. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
9. Huish EG Jr, Vitale MA, Shin AY. Acute proximal row carpectomy to treat a transscaphoid, transtriquetral perilunate fracture dislocation: case report and review of the literature. Hand
(N Y). 2013;8(1):105-109.
1. Youssef B, Deshmukh SC. Volar perilunate dislocation: a case report and review of the literature. Open Orthop J. 2008;2:57-58.
2. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
3. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
4. Graham TJ. The inferior arc injury: an addition to the family of complex carpal fracture-dislocation patterns. Am J Orthop. 2003;32(9 suppl):10-19.
5. Bain GI, McLean JM, Turner PC, Sood A, Pourgiezis N. Translunate fracture with associated perilunate injury: 3 case reports with introduction of the translunate arc concept. J Hand Surg Am. 2008;33(10):1770-1776.
6. Bain GI, Pallapati S, Eng K. Translunate perilunate injuries—a spectrum of this uncommon injury. J Wrist Surg. 2013;2(1):63-68.
7. Teisen H, Hjarbaek J. Classification of fresh fractures of the lunate. J Hand Surg Br. 1988;13(4):458-462.
8. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
9. Huish EG Jr, Vitale MA, Shin AY. Acute proximal row carpectomy to treat a transscaphoid, transtriquetral perilunate fracture dislocation: case report and review of the literature. Hand
(N Y). 2013;8(1):105-109.
Metastatic eccrine carcinoma with stomach and pericardial involvement
Skin adnexal tumors (SAT) are rare tumors that make up about 1%-2% of all cutaneous malignancies. They represent a various group of benign and malignant tumors that arise from skin adnexal epithelial structures: hair follicle, pilosebaceous unit, and apocrine or eccrine sweat glands. Although this derivation provides a practical basis for classification, some tumors may exhibit a mixed or more than one line of differentiation, rendering precise classification of those neoplasms difficult, and such cases should be categorized according to prevailing phenotype. In this report, we present a patient with metastatic eccrine carcinoma. Clinical experience for metastatic disease treatment is derived from a few reports, and there are no universal treatment guidelines. Given the few reported cases and the absence of randomized clinical trials for these patients, it is important to collect clinical experiences.
Case presentation and summary
A 56-year-old African man presented with a 5-week history of multiple nontender subcutaneous skin nodules all over his body except for his palms and soles, and associated with generalized itching. He had a mass in the sole of his right foot 35 years previously in another country. The mass had recurred 15 years later and was excised again. The exact etiology of the mass was unknown to the patient. He had no other medical problems. He was on no medications and did not smoke, drink, or use recreational drugs.
His vital signs on admission were normal. Examination was significant for innumerable superficial skin nodules in the scalp, back, torso, and abdomen. The largest was in the neck and measured 4 x 2 cm. A firm right inguinal mass of 7 x 4 cm was palpable. An abdominal exam revealed large ascites but no organomegaly.
The results of laboratory tests were significant for hyponatremia 126 mEq/L (normal, 135-145), hypercalcemia of 12.2 mg/dL (8.5-10.5), with normal phosphorous of 2.5 mg/dL (2.5-4.5), parathyroid of 11.5 pg/ml (6-65), and low vitamin D level of <7 ng/ml (30-100). Other test results were: carcinoembryonic antigen (CEA), 4.36 ng/ml (0.00-2.99); alpha fetoprotein, 2.39 IU/ml (0.00-9.0); calcium 11.6 mg/dL (8.5-10.2); lactate dehydrogenase, 325 U/L (85-210); aspartate aminotransferase, 59 U/L (0-40); alanine aminotransferase 43 U/L (5-35); alkaline phosphatase, 65 u/L (50-120); albumin, 2.7 g/dL (3.8-5.2); white blood cell count, 14.1 k/uL (4.4-10.6); h
A chest and abdomen computed-tomography scan on presentation showed presence of innumerable subcutaneous and intramuscular nodules throughout the chest, abdomen, and pelvis (Figure 1).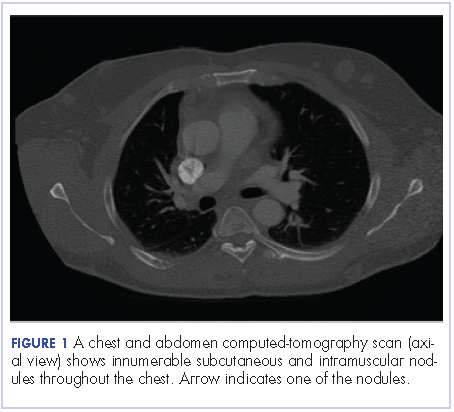
Extensive peritoneal carcinomatosis in addition to moderate ascites and perivascular lymphadenopathy were evident in the abdomen cuts. Remarkably, multiple lytic, osseous metastases were seen with subacute pathologic fracture of right fourth rib in addition to mediastinal lymphadenopathy with small pericardial effusion in the chest cuts. The right thigh mass was described as a large lobulated solid and cystic mass. Ascitic fluid analysis was negative for malignant cells. Biopsy of one the skin nodules in the upper back showed carcinoma involving the skin with focal tubular differentiation (Figure 2).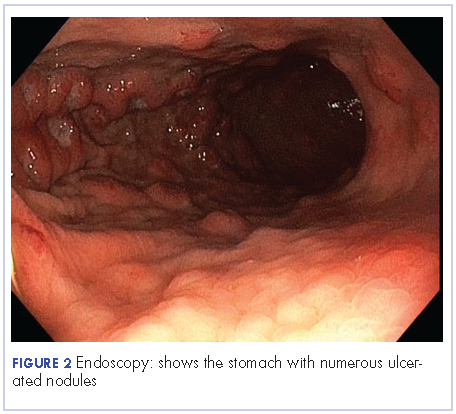
Immunohistochemical stains were positive for p63, epithelial membrane antigen, high molecular weight keratin, and p40. The lesional cells were negative for CEA, bcl-2, Ber-Ep4, CK7, and CK20. The profile was compatible with a skin adnexal carcinoma of sweat gland origin. The groin lymph node showed eccrine acrospiroma.
The patient underwent an upper endoscopy to assess for recurrent vomiting and it revealed diffuse areas of large erythematous ulcerated nodules noted in the cardia, fundus, and body of the stomach (Figure 3). A biopsy of the gastric nodules revealed gastric mucosa with metastatic carcinoma.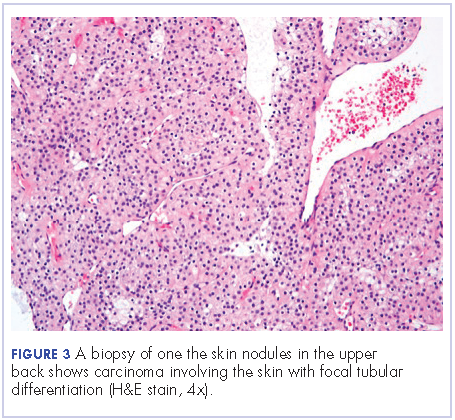
After a thorough review of the literature, he was started on palliative chemotherapy 13 days after initial presentation with docetaxel 75 mg/m2, carboplatin AUC 5 (470 mg), and 5-FU (5-fluorouracil, 750 mg/m2) over 24 hours on days 1 through 5. However, on day 2 of the chemotherapy, he became hypotensive and was found to have cardiac tamponade. He underwent an emergent pericardial window procedure. Analysis of the pericardial fluid was consistent with metastatic carcinoma (Figure 4). Chemotherapy was discontinued while he remained hypotensive requiring multiple vasopressors. His clinical condition did not improve and he passed away 27 days from initial presentation.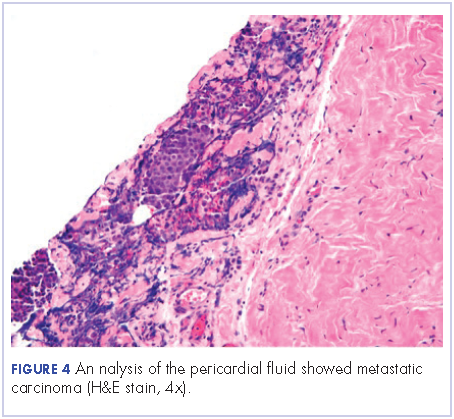
Discussion
Sweat gland carcinomas are very rare malignant tumors of the adnexal epithelial structures of the skin, sebaceous, hair follicle, apocrine or eccrine glands that were first described by Cornil in 1865.1 They occur primarily in adult patients, with a peak incidence in fifth and sixth decades of life.2,3 The etiology is unknown, but some cases have been reported to be a consequence of radiation therapy.4 They are almost always an incidental histologic diagnosis.2,5 The tumors usually appear as single nodule, and multinodularity usually associated with both local and metastatic disease.6 There are no characteristic findings to suggest that a particular nodule may represent sweat gland carcinoma, and even if sweat gland tumor is suspected, benign counterparts are more common.
Eccrine carcinoma is the most aggressive among skin adnexal tumors. They can arise on the lower limbs, trunk, head and neck, scalp and ears, upper extremities, abdomen, and genital sites.7
The cells of eccrine sweat glands express low molecular weight keratin, epithelial membrane antigen, carcinoembryonic antigen, as well as S100 protein, smooth muscle actin, p63, calponin, cytokeratin 14, and bcl-2.8 Skin tumors with eccrine differentiation may stain for estrogen and progesterone, which has important clinical implications because those patients can be treated with hormonal therapy.9 Positivity for estrogen receptors does not differentiate cutaneous eccrine tumors from cutaneous metastases of breast cancers.8,9 Androgen receptor evaluation in these cases can help distinguish between the two.10 Human epidermal growth factor receptor 2 (HER-2) is expressed in 3.5% of skin adnexal tumors.11
The molecular pathogenesis of malignant adnexal tumors is not clear, but overexpression of tumor suppressor protein p16 has been described as a common feature in eccrine carcinomas.12
Prognostic factors for sweat gland carcinoma are difficult to identify, because of the small number of reported cases. The likely prognostic factors include size, histological type, lymph node involvement, and presence of distant metastasis. Absent of lymph node involvement correlates with 10-year disease-free survival rate of 56%, which falls to 9% if nodes are involved.13
There are no uniform guidelines for the treatment sweat gland carcinomas, and the clinical experience described in the literature is the only source of available information.
The treatment of choice of all subtypes of localized sweat gland carcinomas is wide surgical excision with broad tumor margins, given the propensity for local recurrences along with regional lymph node dissection in the presence of clinically positive nodes. Prophylactic lymph node resection does not seem to improve survival or decrease recurrence rates.7 The use of adjuvant radiotherapy to prevent local recurrence also is not well established. One report suggested radiosensitivity of these tumors, and adjuvant radiation was therefore recommended in high-risk cases (ie, large tumors of 5 cm and positive surgical margins of 1 cm) and moderate to poorly differentiated tumors with lymphovascular invasion.14 Adjuvant radiation to the involved lymph node basin is suggested in the setting of extranodal extension or extensive involvement, that is, 4 lymph nodes.15 The role of lymphadenectomy has not been adequately addressed in the literature.
The role of chemotherapy in metastatic disease is not clear, but sweat gland carcinomas are considered chemoresistant (Table). Several combinations have been used with short-term responses. In one case treated with doxorubicin, mitomycin, vincristine, and 5-FU followed by maintenance therapy, the patient achieved a complete response that lasted for 16 months.16 In another report, the treatment response was 2 years with treatment consisted of anthracyclin, cyclophosphamide, vincristine, and bloemycin.17 Other combinations used in the literature include carboplatin and paclitaxel, which led to prolonged remission.14 Cisplatin and 5-FU, or cisplatin plus cetuximab have been reported but with discouraging results.18 Results to taxanes showed conflicting results.19,20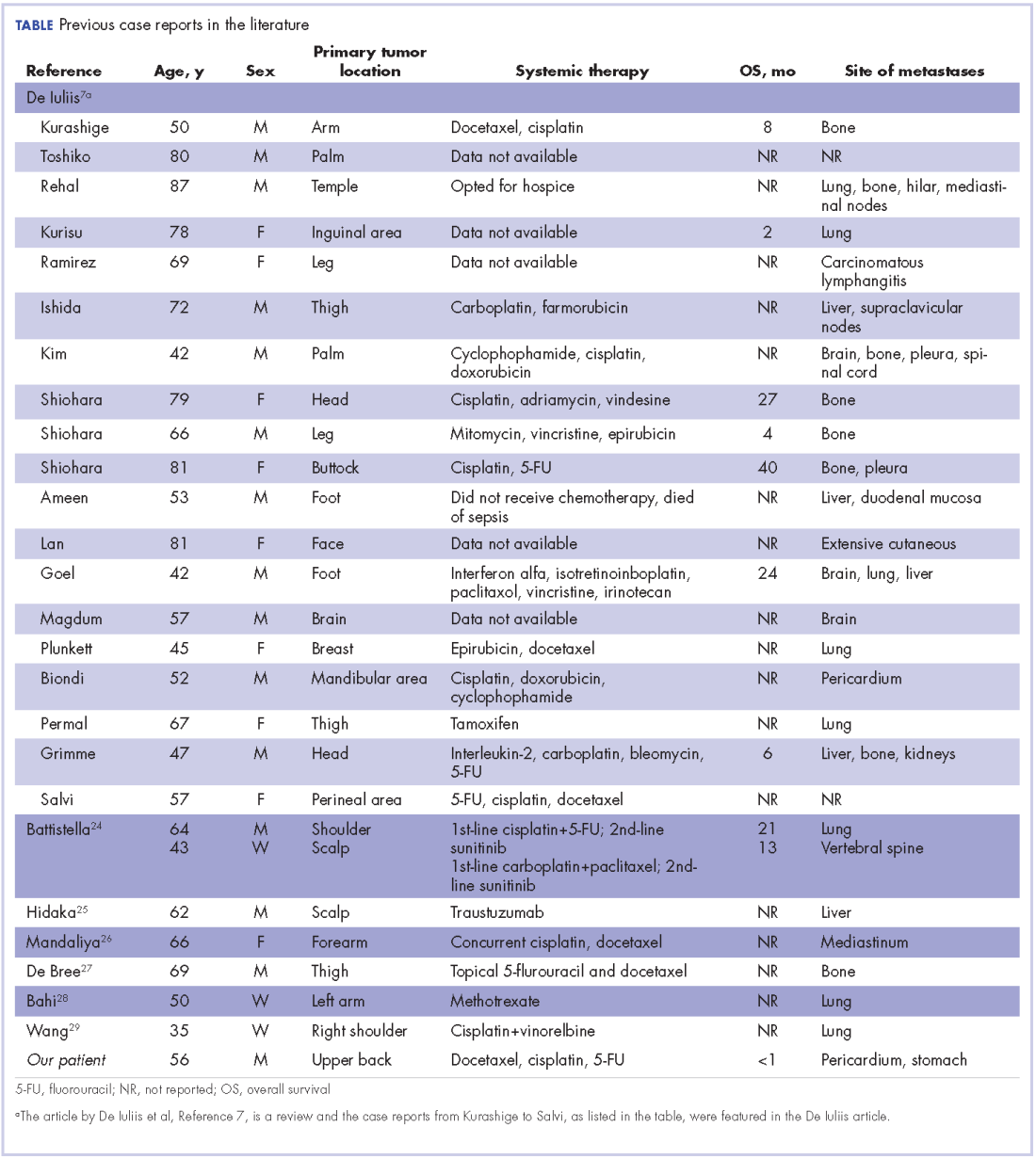
Hormonal therapy can be effective in cases in which estrogen and progesterone receptors are expressed, which can range from 19%-30% of eccrine sweat gland carcinomas.21,22 Two cases have reported complete regression of lymph nodes in patients with metastatic disease, and in 1 patient relief from pain caused by bone metastases with durable response of around 3 years.23,24 a
Experience with targeted therapy is very limited. Sunitinib has been reported to have some activity in metastatic adnexal tumors as a second-line therapy in 2 patients, with disease control for 8 and 10 months respectively.25 Trastuzumab has been reported as having activity in 1 patient with strong HER2 expression (IHC score of 3+, denoting HER2 positivity), with complete regression of metastatic tumor. Upon progression in the same patient, a combination of lapatinib and capecitabine also showed positive response.26
In conclusion, metastatic sweat gland tumors treatment has not been standardized because of a dearth of reports in the literatures. Its early identification and complete excision gives the best chance of a cure. Neither chemotherapy nor radiation therapy has been proven to be of clinical benefit in treating metastatic disease.
1. Gates O, Warren S, Warvi WN. Tumors of sweat glands. Am J Pathol. 1943;19(4):591-631.
2. Mitts DL, Smith MT, Russell L, Bannayan GA, Cruz AB. Sweat gland carcinoma: a clinico-pathological reappraisal. J Surg Oncol. 1976;8(1):23-29.
3. Panoussopoulos D, Darom A, Lazaris AC, Misthos P, Papadimitriou K, Androulakis G. Sweat gland carcinoma with multiple local recurrences: a case report. Adv Clin Path. 1999;3(3):63-68.
4. Marone U, Caracò C, Anniciello AM, et al. Metastatic eccrine porocarcinoma : report of a case and review of the literature. World J Surg Oncol. 2011;9:32.
5. Yildirim S, Aköz T, Akan M, Ege GA. De novo malignant eccrine spiradenoma with an interesting and unusual location. Dermatol Surg. 2001;27(4):417-420.
6. Shaw M, McKee PH, Lowe D, Black MM. Malignant eccrine poroma: a study of twenty-seven cases. Br J Dermatol. 1982;107(6):675-680.
7. De Iuliis F, Amoroso L, Taglieri L, et al. Chemotherapy of rare skin adnexal tumors: a review of literature. Anticancer Res. 2014;34(10):5263-5268.
8. Alsaad KO, Obaidat NA, Ghazarian D. Skin adnexal neoplasms – part 1: an approach to tumours of the pilosebaceous unit. J Clin Pathol. 2007;60(2):129-144.
9. Serhrouchni KI, Harmouch T, Chbani L, et al. Eccrine carcinoma : a rare cutaneous neoplasm. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3570399/. Published online February 4, 2013. Accessed October 11, 2017.
10. Shidham VB, Komorowski RA, Machhi JK. Androgen receptor expression in metastatic adenocarcinoma in females favors a breast primary. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1601970/. Published online October 4, 2006. Accessed October 11, 2017.
11. Hiatt KM, Pillow JL, Smoller BR. Her-2 expression in cutaneous eccrine and apocrine neoplasms. Mod Pathol. 2004;17(1):28-32.
12. Gu L-H, Ichiki Y, Kitajima Y. Aberrant expression of p16 and RB protein in eccrine porocarcinoma. J Cutan Pathol. 2002;29(8):473-479.
13. el-Domeiri AA, Brasfield RD, Huvos AG, Strong EW. Sweat gland carcinoma: a clinico-pathologic study of 83 patients. Ann Surg. 1971;173(2):270-274.
14. Tlemcani K, Levine D, Smith R V, et al. Metastatic apocrine carcinoma of the scalp: prolonged response to systemic chemotherapy. J Clin Oncol. 2010;28(24):e412-e414.
15. Chamberlain RS, Huber K, White JC, Travaglino-Parda R. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22(2):131-135.
16. Gutermuth J, Audring H, Voit C, Trefzer U, Haas N. Antitumour activity of paclitaxel and interferon-alpha in a case of metastatic eccrine porocarcinoma. J Eur Acad Dermatol Venereol. 2004;18(4):477-479.
17. Mezger J, Remberger K, Schalhorn A, Wohlrab A, Wilmanns W. Treatment of metastatic sweat gland carcinoma by a four drug combination chemotherapy: response in two cases. Med Oncol Tumor Pharmacother. 1986;3(1):29-34.
18. Aaribi I, Mohtaram A, Ben Ameur El Youbi M, et al. Successful management of metastatic eccrine porocarcinoma. https://www.hindawi.com/journals/crionm/2013/282536/. Published 2013. Accessed October 10, 2017.
19. Shiohara J, Koga H, Uhara H, Takata M, Saida T. Eccrine porocarcinoma: clinical and pathological studies of 12 cases. J Dermatol. 2007;34(8):516-522.
20. Swanson PE, Mazoujian G, Mills SE, Campbell RJ, Wick MR. Immunoreactivity for estrogen receptor protein in sweat gland tumors. Am J Surg Pathol. 1991;15(9):835-841.
21. Busam KJ, Tan LK, Granter SR, et al. Epidermal growth factor, estrogen, and progesterone receptor expression in primary sweat gland carcinomas and primary and metastatic mammary carcinomas. Mod Pathol. 1999;12(8):786-793.
22. Sridhar KS, Benedetto P, Otrakji CL, Charyulu KK. Response of eccrine adenocarcinoma to tamoxifen. Cancer. 1989;64(2):366-370.
23. Daniel SJ, Nader R, Kost K, Hüttner I. Facial sweat gland carcinoma metastasizing to neck nodes: a diagnostic and therapeutic challenge. Arch Otolaryngol Head Neck Surg. 2001;127(12):1495-1498.
24. Battistella M, Mateus C, Lassau N, et al. Sunitinib efficacy in the treatment of metastatic skin adnexal carcinomas: report of two patients with hidradenocarcinoma and trichoblastic carcinoma. J Eur Acad Dermatol Venereol. 2010;24(2):199-203.
25. Hidaka T, Fujimura T, Watabe A, et al. Successful treatment of HER-2-positive metastatic apocrine carcinoma of the skin with lapatinib and capecitabine. Acta Derm Venereol. 2012;92(6):654-655.
26. Mandaliya H, Nordman I. Metastatic eccrine porocarcinoma: a rare case of successful treatment. Case Rep Oncol. 2016;9(2):454-456.
27. de Bree E, Volalakis E, Tsetis D, et al. Treatment of advanced malignant eccrine poroma with locoregional chemotherapy. Br J Dermatol. 2005;152(5):1051-1055.
28. Bahl A, Sharma DN, Julka PK, Das A, Rath GK. Sweat gland carcinoma with lung metastases. J Cancer Res Ther. 2(4):209-211.
29. Wang X-X, Wang H-Y, Zheng J-N, Sui J-C. Primary cutaneous sweat gland carcinoma. J Cancer Res Ther. 10(2):390-392.
Skin adnexal tumors (SAT) are rare tumors that make up about 1%-2% of all cutaneous malignancies. They represent a various group of benign and malignant tumors that arise from skin adnexal epithelial structures: hair follicle, pilosebaceous unit, and apocrine or eccrine sweat glands. Although this derivation provides a practical basis for classification, some tumors may exhibit a mixed or more than one line of differentiation, rendering precise classification of those neoplasms difficult, and such cases should be categorized according to prevailing phenotype. In this report, we present a patient with metastatic eccrine carcinoma. Clinical experience for metastatic disease treatment is derived from a few reports, and there are no universal treatment guidelines. Given the few reported cases and the absence of randomized clinical trials for these patients, it is important to collect clinical experiences.
Case presentation and summary
A 56-year-old African man presented with a 5-week history of multiple nontender subcutaneous skin nodules all over his body except for his palms and soles, and associated with generalized itching. He had a mass in the sole of his right foot 35 years previously in another country. The mass had recurred 15 years later and was excised again. The exact etiology of the mass was unknown to the patient. He had no other medical problems. He was on no medications and did not smoke, drink, or use recreational drugs.
His vital signs on admission were normal. Examination was significant for innumerable superficial skin nodules in the scalp, back, torso, and abdomen. The largest was in the neck and measured 4 x 2 cm. A firm right inguinal mass of 7 x 4 cm was palpable. An abdominal exam revealed large ascites but no organomegaly.
The results of laboratory tests were significant for hyponatremia 126 mEq/L (normal, 135-145), hypercalcemia of 12.2 mg/dL (8.5-10.5), with normal phosphorous of 2.5 mg/dL (2.5-4.5), parathyroid of 11.5 pg/ml (6-65), and low vitamin D level of <7 ng/ml (30-100). Other test results were: carcinoembryonic antigen (CEA), 4.36 ng/ml (0.00-2.99); alpha fetoprotein, 2.39 IU/ml (0.00-9.0); calcium 11.6 mg/dL (8.5-10.2); lactate dehydrogenase, 325 U/L (85-210); aspartate aminotransferase, 59 U/L (0-40); alanine aminotransferase 43 U/L (5-35); alkaline phosphatase, 65 u/L (50-120); albumin, 2.7 g/dL (3.8-5.2); white blood cell count, 14.1 k/uL (4.4-10.6); h
A chest and abdomen computed-tomography scan on presentation showed presence of innumerable subcutaneous and intramuscular nodules throughout the chest, abdomen, and pelvis (Figure 1).
Extensive peritoneal carcinomatosis in addition to moderate ascites and perivascular lymphadenopathy were evident in the abdomen cuts. Remarkably, multiple lytic, osseous metastases were seen with subacute pathologic fracture of right fourth rib in addition to mediastinal lymphadenopathy with small pericardial effusion in the chest cuts. The right thigh mass was described as a large lobulated solid and cystic mass. Ascitic fluid analysis was negative for malignant cells. Biopsy of one the skin nodules in the upper back showed carcinoma involving the skin with focal tubular differentiation (Figure 2).
Immunohistochemical stains were positive for p63, epithelial membrane antigen, high molecular weight keratin, and p40. The lesional cells were negative for CEA, bcl-2, Ber-Ep4, CK7, and CK20. The profile was compatible with a skin adnexal carcinoma of sweat gland origin. The groin lymph node showed eccrine acrospiroma.
The patient underwent an upper endoscopy to assess for recurrent vomiting and it revealed diffuse areas of large erythematous ulcerated nodules noted in the cardia, fundus, and body of the stomach (Figure 3). A biopsy of the gastric nodules revealed gastric mucosa with metastatic carcinoma.
After a thorough review of the literature, he was started on palliative chemotherapy 13 days after initial presentation with docetaxel 75 mg/m2, carboplatin AUC 5 (470 mg), and 5-FU (5-fluorouracil, 750 mg/m2) over 24 hours on days 1 through 5. However, on day 2 of the chemotherapy, he became hypotensive and was found to have cardiac tamponade. He underwent an emergent pericardial window procedure. Analysis of the pericardial fluid was consistent with metastatic carcinoma (Figure 4). Chemotherapy was discontinued while he remained hypotensive requiring multiple vasopressors. His clinical condition did not improve and he passed away 27 days from initial presentation.
Discussion
Sweat gland carcinomas are very rare malignant tumors of the adnexal epithelial structures of the skin, sebaceous, hair follicle, apocrine or eccrine glands that were first described by Cornil in 1865.1 They occur primarily in adult patients, with a peak incidence in fifth and sixth decades of life.2,3 The etiology is unknown, but some cases have been reported to be a consequence of radiation therapy.4 They are almost always an incidental histologic diagnosis.2,5 The tumors usually appear as single nodule, and multinodularity usually associated with both local and metastatic disease.6 There are no characteristic findings to suggest that a particular nodule may represent sweat gland carcinoma, and even if sweat gland tumor is suspected, benign counterparts are more common.
Eccrine carcinoma is the most aggressive among skin adnexal tumors. They can arise on the lower limbs, trunk, head and neck, scalp and ears, upper extremities, abdomen, and genital sites.7
The cells of eccrine sweat glands express low molecular weight keratin, epithelial membrane antigen, carcinoembryonic antigen, as well as S100 protein, smooth muscle actin, p63, calponin, cytokeratin 14, and bcl-2.8 Skin tumors with eccrine differentiation may stain for estrogen and progesterone, which has important clinical implications because those patients can be treated with hormonal therapy.9 Positivity for estrogen receptors does not differentiate cutaneous eccrine tumors from cutaneous metastases of breast cancers.8,9 Androgen receptor evaluation in these cases can help distinguish between the two.10 Human epidermal growth factor receptor 2 (HER-2) is expressed in 3.5% of skin adnexal tumors.11
The molecular pathogenesis of malignant adnexal tumors is not clear, but overexpression of tumor suppressor protein p16 has been described as a common feature in eccrine carcinomas.12
Prognostic factors for sweat gland carcinoma are difficult to identify, because of the small number of reported cases. The likely prognostic factors include size, histological type, lymph node involvement, and presence of distant metastasis. Absent of lymph node involvement correlates with 10-year disease-free survival rate of 56%, which falls to 9% if nodes are involved.13
There are no uniform guidelines for the treatment sweat gland carcinomas, and the clinical experience described in the literature is the only source of available information.
The treatment of choice of all subtypes of localized sweat gland carcinomas is wide surgical excision with broad tumor margins, given the propensity for local recurrences along with regional lymph node dissection in the presence of clinically positive nodes. Prophylactic lymph node resection does not seem to improve survival or decrease recurrence rates.7 The use of adjuvant radiotherapy to prevent local recurrence also is not well established. One report suggested radiosensitivity of these tumors, and adjuvant radiation was therefore recommended in high-risk cases (ie, large tumors of 5 cm and positive surgical margins of 1 cm) and moderate to poorly differentiated tumors with lymphovascular invasion.14 Adjuvant radiation to the involved lymph node basin is suggested in the setting of extranodal extension or extensive involvement, that is, 4 lymph nodes.15 The role of lymphadenectomy has not been adequately addressed in the literature.
The role of chemotherapy in metastatic disease is not clear, but sweat gland carcinomas are considered chemoresistant (Table). Several combinations have been used with short-term responses. In one case treated with doxorubicin, mitomycin, vincristine, and 5-FU followed by maintenance therapy, the patient achieved a complete response that lasted for 16 months.16 In another report, the treatment response was 2 years with treatment consisted of anthracyclin, cyclophosphamide, vincristine, and bloemycin.17 Other combinations used in the literature include carboplatin and paclitaxel, which led to prolonged remission.14 Cisplatin and 5-FU, or cisplatin plus cetuximab have been reported but with discouraging results.18 Results to taxanes showed conflicting results.19,20
Hormonal therapy can be effective in cases in which estrogen and progesterone receptors are expressed, which can range from 19%-30% of eccrine sweat gland carcinomas.21,22 Two cases have reported complete regression of lymph nodes in patients with metastatic disease, and in 1 patient relief from pain caused by bone metastases with durable response of around 3 years.23,24 a
Experience with targeted therapy is very limited. Sunitinib has been reported to have some activity in metastatic adnexal tumors as a second-line therapy in 2 patients, with disease control for 8 and 10 months respectively.25 Trastuzumab has been reported as having activity in 1 patient with strong HER2 expression (IHC score of 3+, denoting HER2 positivity), with complete regression of metastatic tumor. Upon progression in the same patient, a combination of lapatinib and capecitabine also showed positive response.26
In conclusion, metastatic sweat gland tumors treatment has not been standardized because of a dearth of reports in the literatures. Its early identification and complete excision gives the best chance of a cure. Neither chemotherapy nor radiation therapy has been proven to be of clinical benefit in treating metastatic disease.
Skin adnexal tumors (SAT) are rare tumors that make up about 1%-2% of all cutaneous malignancies. They represent a various group of benign and malignant tumors that arise from skin adnexal epithelial structures: hair follicle, pilosebaceous unit, and apocrine or eccrine sweat glands. Although this derivation provides a practical basis for classification, some tumors may exhibit a mixed or more than one line of differentiation, rendering precise classification of those neoplasms difficult, and such cases should be categorized according to prevailing phenotype. In this report, we present a patient with metastatic eccrine carcinoma. Clinical experience for metastatic disease treatment is derived from a few reports, and there are no universal treatment guidelines. Given the few reported cases and the absence of randomized clinical trials for these patients, it is important to collect clinical experiences.
Case presentation and summary
A 56-year-old African man presented with a 5-week history of multiple nontender subcutaneous skin nodules all over his body except for his palms and soles, and associated with generalized itching. He had a mass in the sole of his right foot 35 years previously in another country. The mass had recurred 15 years later and was excised again. The exact etiology of the mass was unknown to the patient. He had no other medical problems. He was on no medications and did not smoke, drink, or use recreational drugs.
His vital signs on admission were normal. Examination was significant for innumerable superficial skin nodules in the scalp, back, torso, and abdomen. The largest was in the neck and measured 4 x 2 cm. A firm right inguinal mass of 7 x 4 cm was palpable. An abdominal exam revealed large ascites but no organomegaly.
The results of laboratory tests were significant for hyponatremia 126 mEq/L (normal, 135-145), hypercalcemia of 12.2 mg/dL (8.5-10.5), with normal phosphorous of 2.5 mg/dL (2.5-4.5), parathyroid of 11.5 pg/ml (6-65), and low vitamin D level of <7 ng/ml (30-100). Other test results were: carcinoembryonic antigen (CEA), 4.36 ng/ml (0.00-2.99); alpha fetoprotein, 2.39 IU/ml (0.00-9.0); calcium 11.6 mg/dL (8.5-10.2); lactate dehydrogenase, 325 U/L (85-210); aspartate aminotransferase, 59 U/L (0-40); alanine aminotransferase 43 U/L (5-35); alkaline phosphatase, 65 u/L (50-120); albumin, 2.7 g/dL (3.8-5.2); white blood cell count, 14.1 k/uL (4.4-10.6); h
A chest and abdomen computed-tomography scan on presentation showed presence of innumerable subcutaneous and intramuscular nodules throughout the chest, abdomen, and pelvis (Figure 1).
Extensive peritoneal carcinomatosis in addition to moderate ascites and perivascular lymphadenopathy were evident in the abdomen cuts. Remarkably, multiple lytic, osseous metastases were seen with subacute pathologic fracture of right fourth rib in addition to mediastinal lymphadenopathy with small pericardial effusion in the chest cuts. The right thigh mass was described as a large lobulated solid and cystic mass. Ascitic fluid analysis was negative for malignant cells. Biopsy of one the skin nodules in the upper back showed carcinoma involving the skin with focal tubular differentiation (Figure 2).
Immunohistochemical stains were positive for p63, epithelial membrane antigen, high molecular weight keratin, and p40. The lesional cells were negative for CEA, bcl-2, Ber-Ep4, CK7, and CK20. The profile was compatible with a skin adnexal carcinoma of sweat gland origin. The groin lymph node showed eccrine acrospiroma.
The patient underwent an upper endoscopy to assess for recurrent vomiting and it revealed diffuse areas of large erythematous ulcerated nodules noted in the cardia, fundus, and body of the stomach (Figure 3). A biopsy of the gastric nodules revealed gastric mucosa with metastatic carcinoma.
After a thorough review of the literature, he was started on palliative chemotherapy 13 days after initial presentation with docetaxel 75 mg/m2, carboplatin AUC 5 (470 mg), and 5-FU (5-fluorouracil, 750 mg/m2) over 24 hours on days 1 through 5. However, on day 2 of the chemotherapy, he became hypotensive and was found to have cardiac tamponade. He underwent an emergent pericardial window procedure. Analysis of the pericardial fluid was consistent with metastatic carcinoma (Figure 4). Chemotherapy was discontinued while he remained hypotensive requiring multiple vasopressors. His clinical condition did not improve and he passed away 27 days from initial presentation.
Discussion
Sweat gland carcinomas are very rare malignant tumors of the adnexal epithelial structures of the skin, sebaceous, hair follicle, apocrine or eccrine glands that were first described by Cornil in 1865.1 They occur primarily in adult patients, with a peak incidence in fifth and sixth decades of life.2,3 The etiology is unknown, but some cases have been reported to be a consequence of radiation therapy.4 They are almost always an incidental histologic diagnosis.2,5 The tumors usually appear as single nodule, and multinodularity usually associated with both local and metastatic disease.6 There are no characteristic findings to suggest that a particular nodule may represent sweat gland carcinoma, and even if sweat gland tumor is suspected, benign counterparts are more common.
Eccrine carcinoma is the most aggressive among skin adnexal tumors. They can arise on the lower limbs, trunk, head and neck, scalp and ears, upper extremities, abdomen, and genital sites.7
The cells of eccrine sweat glands express low molecular weight keratin, epithelial membrane antigen, carcinoembryonic antigen, as well as S100 protein, smooth muscle actin, p63, calponin, cytokeratin 14, and bcl-2.8 Skin tumors with eccrine differentiation may stain for estrogen and progesterone, which has important clinical implications because those patients can be treated with hormonal therapy.9 Positivity for estrogen receptors does not differentiate cutaneous eccrine tumors from cutaneous metastases of breast cancers.8,9 Androgen receptor evaluation in these cases can help distinguish between the two.10 Human epidermal growth factor receptor 2 (HER-2) is expressed in 3.5% of skin adnexal tumors.11
The molecular pathogenesis of malignant adnexal tumors is not clear, but overexpression of tumor suppressor protein p16 has been described as a common feature in eccrine carcinomas.12
Prognostic factors for sweat gland carcinoma are difficult to identify, because of the small number of reported cases. The likely prognostic factors include size, histological type, lymph node involvement, and presence of distant metastasis. Absent of lymph node involvement correlates with 10-year disease-free survival rate of 56%, which falls to 9% if nodes are involved.13
There are no uniform guidelines for the treatment sweat gland carcinomas, and the clinical experience described in the literature is the only source of available information.
The treatment of choice of all subtypes of localized sweat gland carcinomas is wide surgical excision with broad tumor margins, given the propensity for local recurrences along with regional lymph node dissection in the presence of clinically positive nodes. Prophylactic lymph node resection does not seem to improve survival or decrease recurrence rates.7 The use of adjuvant radiotherapy to prevent local recurrence also is not well established. One report suggested radiosensitivity of these tumors, and adjuvant radiation was therefore recommended in high-risk cases (ie, large tumors of 5 cm and positive surgical margins of 1 cm) and moderate to poorly differentiated tumors with lymphovascular invasion.14 Adjuvant radiation to the involved lymph node basin is suggested in the setting of extranodal extension or extensive involvement, that is, 4 lymph nodes.15 The role of lymphadenectomy has not been adequately addressed in the literature.
The role of chemotherapy in metastatic disease is not clear, but sweat gland carcinomas are considered chemoresistant (Table). Several combinations have been used with short-term responses. In one case treated with doxorubicin, mitomycin, vincristine, and 5-FU followed by maintenance therapy, the patient achieved a complete response that lasted for 16 months.16 In another report, the treatment response was 2 years with treatment consisted of anthracyclin, cyclophosphamide, vincristine, and bloemycin.17 Other combinations used in the literature include carboplatin and paclitaxel, which led to prolonged remission.14 Cisplatin and 5-FU, or cisplatin plus cetuximab have been reported but with discouraging results.18 Results to taxanes showed conflicting results.19,20
Hormonal therapy can be effective in cases in which estrogen and progesterone receptors are expressed, which can range from 19%-30% of eccrine sweat gland carcinomas.21,22 Two cases have reported complete regression of lymph nodes in patients with metastatic disease, and in 1 patient relief from pain caused by bone metastases with durable response of around 3 years.23,24 a
Experience with targeted therapy is very limited. Sunitinib has been reported to have some activity in metastatic adnexal tumors as a second-line therapy in 2 patients, with disease control for 8 and 10 months respectively.25 Trastuzumab has been reported as having activity in 1 patient with strong HER2 expression (IHC score of 3+, denoting HER2 positivity), with complete regression of metastatic tumor. Upon progression in the same patient, a combination of lapatinib and capecitabine also showed positive response.26
In conclusion, metastatic sweat gland tumors treatment has not been standardized because of a dearth of reports in the literatures. Its early identification and complete excision gives the best chance of a cure. Neither chemotherapy nor radiation therapy has been proven to be of clinical benefit in treating metastatic disease.
1. Gates O, Warren S, Warvi WN. Tumors of sweat glands. Am J Pathol. 1943;19(4):591-631.
2. Mitts DL, Smith MT, Russell L, Bannayan GA, Cruz AB. Sweat gland carcinoma: a clinico-pathological reappraisal. J Surg Oncol. 1976;8(1):23-29.
3. Panoussopoulos D, Darom A, Lazaris AC, Misthos P, Papadimitriou K, Androulakis G. Sweat gland carcinoma with multiple local recurrences: a case report. Adv Clin Path. 1999;3(3):63-68.
4. Marone U, Caracò C, Anniciello AM, et al. Metastatic eccrine porocarcinoma : report of a case and review of the literature. World J Surg Oncol. 2011;9:32.
5. Yildirim S, Aköz T, Akan M, Ege GA. De novo malignant eccrine spiradenoma with an interesting and unusual location. Dermatol Surg. 2001;27(4):417-420.
6. Shaw M, McKee PH, Lowe D, Black MM. Malignant eccrine poroma: a study of twenty-seven cases. Br J Dermatol. 1982;107(6):675-680.
7. De Iuliis F, Amoroso L, Taglieri L, et al. Chemotherapy of rare skin adnexal tumors: a review of literature. Anticancer Res. 2014;34(10):5263-5268.
8. Alsaad KO, Obaidat NA, Ghazarian D. Skin adnexal neoplasms – part 1: an approach to tumours of the pilosebaceous unit. J Clin Pathol. 2007;60(2):129-144.
9. Serhrouchni KI, Harmouch T, Chbani L, et al. Eccrine carcinoma : a rare cutaneous neoplasm. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3570399/. Published online February 4, 2013. Accessed October 11, 2017.
10. Shidham VB, Komorowski RA, Machhi JK. Androgen receptor expression in metastatic adenocarcinoma in females favors a breast primary. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1601970/. Published online October 4, 2006. Accessed October 11, 2017.
11. Hiatt KM, Pillow JL, Smoller BR. Her-2 expression in cutaneous eccrine and apocrine neoplasms. Mod Pathol. 2004;17(1):28-32.
12. Gu L-H, Ichiki Y, Kitajima Y. Aberrant expression of p16 and RB protein in eccrine porocarcinoma. J Cutan Pathol. 2002;29(8):473-479.
13. el-Domeiri AA, Brasfield RD, Huvos AG, Strong EW. Sweat gland carcinoma: a clinico-pathologic study of 83 patients. Ann Surg. 1971;173(2):270-274.
14. Tlemcani K, Levine D, Smith R V, et al. Metastatic apocrine carcinoma of the scalp: prolonged response to systemic chemotherapy. J Clin Oncol. 2010;28(24):e412-e414.
15. Chamberlain RS, Huber K, White JC, Travaglino-Parda R. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22(2):131-135.
16. Gutermuth J, Audring H, Voit C, Trefzer U, Haas N. Antitumour activity of paclitaxel and interferon-alpha in a case of metastatic eccrine porocarcinoma. J Eur Acad Dermatol Venereol. 2004;18(4):477-479.
17. Mezger J, Remberger K, Schalhorn A, Wohlrab A, Wilmanns W. Treatment of metastatic sweat gland carcinoma by a four drug combination chemotherapy: response in two cases. Med Oncol Tumor Pharmacother. 1986;3(1):29-34.
18. Aaribi I, Mohtaram A, Ben Ameur El Youbi M, et al. Successful management of metastatic eccrine porocarcinoma. https://www.hindawi.com/journals/crionm/2013/282536/. Published 2013. Accessed October 10, 2017.
19. Shiohara J, Koga H, Uhara H, Takata M, Saida T. Eccrine porocarcinoma: clinical and pathological studies of 12 cases. J Dermatol. 2007;34(8):516-522.
20. Swanson PE, Mazoujian G, Mills SE, Campbell RJ, Wick MR. Immunoreactivity for estrogen receptor protein in sweat gland tumors. Am J Surg Pathol. 1991;15(9):835-841.
21. Busam KJ, Tan LK, Granter SR, et al. Epidermal growth factor, estrogen, and progesterone receptor expression in primary sweat gland carcinomas and primary and metastatic mammary carcinomas. Mod Pathol. 1999;12(8):786-793.
22. Sridhar KS, Benedetto P, Otrakji CL, Charyulu KK. Response of eccrine adenocarcinoma to tamoxifen. Cancer. 1989;64(2):366-370.
23. Daniel SJ, Nader R, Kost K, Hüttner I. Facial sweat gland carcinoma metastasizing to neck nodes: a diagnostic and therapeutic challenge. Arch Otolaryngol Head Neck Surg. 2001;127(12):1495-1498.
24. Battistella M, Mateus C, Lassau N, et al. Sunitinib efficacy in the treatment of metastatic skin adnexal carcinomas: report of two patients with hidradenocarcinoma and trichoblastic carcinoma. J Eur Acad Dermatol Venereol. 2010;24(2):199-203.
25. Hidaka T, Fujimura T, Watabe A, et al. Successful treatment of HER-2-positive metastatic apocrine carcinoma of the skin with lapatinib and capecitabine. Acta Derm Venereol. 2012;92(6):654-655.
26. Mandaliya H, Nordman I. Metastatic eccrine porocarcinoma: a rare case of successful treatment. Case Rep Oncol. 2016;9(2):454-456.
27. de Bree E, Volalakis E, Tsetis D, et al. Treatment of advanced malignant eccrine poroma with locoregional chemotherapy. Br J Dermatol. 2005;152(5):1051-1055.
28. Bahl A, Sharma DN, Julka PK, Das A, Rath GK. Sweat gland carcinoma with lung metastases. J Cancer Res Ther. 2(4):209-211.
29. Wang X-X, Wang H-Y, Zheng J-N, Sui J-C. Primary cutaneous sweat gland carcinoma. J Cancer Res Ther. 10(2):390-392.
1. Gates O, Warren S, Warvi WN. Tumors of sweat glands. Am J Pathol. 1943;19(4):591-631.
2. Mitts DL, Smith MT, Russell L, Bannayan GA, Cruz AB. Sweat gland carcinoma: a clinico-pathological reappraisal. J Surg Oncol. 1976;8(1):23-29.
3. Panoussopoulos D, Darom A, Lazaris AC, Misthos P, Papadimitriou K, Androulakis G. Sweat gland carcinoma with multiple local recurrences: a case report. Adv Clin Path. 1999;3(3):63-68.
4. Marone U, Caracò C, Anniciello AM, et al. Metastatic eccrine porocarcinoma : report of a case and review of the literature. World J Surg Oncol. 2011;9:32.
5. Yildirim S, Aköz T, Akan M, Ege GA. De novo malignant eccrine spiradenoma with an interesting and unusual location. Dermatol Surg. 2001;27(4):417-420.
6. Shaw M, McKee PH, Lowe D, Black MM. Malignant eccrine poroma: a study of twenty-seven cases. Br J Dermatol. 1982;107(6):675-680.
7. De Iuliis F, Amoroso L, Taglieri L, et al. Chemotherapy of rare skin adnexal tumors: a review of literature. Anticancer Res. 2014;34(10):5263-5268.
8. Alsaad KO, Obaidat NA, Ghazarian D. Skin adnexal neoplasms – part 1: an approach to tumours of the pilosebaceous unit. J Clin Pathol. 2007;60(2):129-144.
9. Serhrouchni KI, Harmouch T, Chbani L, et al. Eccrine carcinoma : a rare cutaneous neoplasm. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3570399/. Published online February 4, 2013. Accessed October 11, 2017.
10. Shidham VB, Komorowski RA, Machhi JK. Androgen receptor expression in metastatic adenocarcinoma in females favors a breast primary. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1601970/. Published online October 4, 2006. Accessed October 11, 2017.
11. Hiatt KM, Pillow JL, Smoller BR. Her-2 expression in cutaneous eccrine and apocrine neoplasms. Mod Pathol. 2004;17(1):28-32.
12. Gu L-H, Ichiki Y, Kitajima Y. Aberrant expression of p16 and RB protein in eccrine porocarcinoma. J Cutan Pathol. 2002;29(8):473-479.
13. el-Domeiri AA, Brasfield RD, Huvos AG, Strong EW. Sweat gland carcinoma: a clinico-pathologic study of 83 patients. Ann Surg. 1971;173(2):270-274.
14. Tlemcani K, Levine D, Smith R V, et al. Metastatic apocrine carcinoma of the scalp: prolonged response to systemic chemotherapy. J Clin Oncol. 2010;28(24):e412-e414.
15. Chamberlain RS, Huber K, White JC, Travaglino-Parda R. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22(2):131-135.
16. Gutermuth J, Audring H, Voit C, Trefzer U, Haas N. Antitumour activity of paclitaxel and interferon-alpha in a case of metastatic eccrine porocarcinoma. J Eur Acad Dermatol Venereol. 2004;18(4):477-479.
17. Mezger J, Remberger K, Schalhorn A, Wohlrab A, Wilmanns W. Treatment of metastatic sweat gland carcinoma by a four drug combination chemotherapy: response in two cases. Med Oncol Tumor Pharmacother. 1986;3(1):29-34.
18. Aaribi I, Mohtaram A, Ben Ameur El Youbi M, et al. Successful management of metastatic eccrine porocarcinoma. https://www.hindawi.com/journals/crionm/2013/282536/. Published 2013. Accessed October 10, 2017.
19. Shiohara J, Koga H, Uhara H, Takata M, Saida T. Eccrine porocarcinoma: clinical and pathological studies of 12 cases. J Dermatol. 2007;34(8):516-522.
20. Swanson PE, Mazoujian G, Mills SE, Campbell RJ, Wick MR. Immunoreactivity for estrogen receptor protein in sweat gland tumors. Am J Surg Pathol. 1991;15(9):835-841.
21. Busam KJ, Tan LK, Granter SR, et al. Epidermal growth factor, estrogen, and progesterone receptor expression in primary sweat gland carcinomas and primary and metastatic mammary carcinomas. Mod Pathol. 1999;12(8):786-793.
22. Sridhar KS, Benedetto P, Otrakji CL, Charyulu KK. Response of eccrine adenocarcinoma to tamoxifen. Cancer. 1989;64(2):366-370.
23. Daniel SJ, Nader R, Kost K, Hüttner I. Facial sweat gland carcinoma metastasizing to neck nodes: a diagnostic and therapeutic challenge. Arch Otolaryngol Head Neck Surg. 2001;127(12):1495-1498.
24. Battistella M, Mateus C, Lassau N, et al. Sunitinib efficacy in the treatment of metastatic skin adnexal carcinomas: report of two patients with hidradenocarcinoma and trichoblastic carcinoma. J Eur Acad Dermatol Venereol. 2010;24(2):199-203.
25. Hidaka T, Fujimura T, Watabe A, et al. Successful treatment of HER-2-positive metastatic apocrine carcinoma of the skin with lapatinib and capecitabine. Acta Derm Venereol. 2012;92(6):654-655.
26. Mandaliya H, Nordman I. Metastatic eccrine porocarcinoma: a rare case of successful treatment. Case Rep Oncol. 2016;9(2):454-456.
27. de Bree E, Volalakis E, Tsetis D, et al. Treatment of advanced malignant eccrine poroma with locoregional chemotherapy. Br J Dermatol. 2005;152(5):1051-1055.
28. Bahl A, Sharma DN, Julka PK, Das A, Rath GK. Sweat gland carcinoma with lung metastases. J Cancer Res Ther. 2(4):209-211.
29. Wang X-X, Wang H-Y, Zheng J-N, Sui J-C. Primary cutaneous sweat gland carcinoma. J Cancer Res Ther. 10(2):390-392.
Cold hemolytic anemia: a rare complication of influenza A
Autoimmune hemolytic anemia (AIHA) is characterized by the temperature at which the auto-antibody has the greatest avidity for the target red cell antigen, either warm or cold forms. It is detected by a positive direct antiglobulin test (DAT) also known as the direct Coombs test. DAT is used to determine if red cells have been coated in vivo with immunoglobulin, complement, or both.1 Some causes of a positive DAT include hemolytic transfusion reactions, hemolytic disease of the fetus and newborn, AIHA, and drug-induced immune hemolysis.
Case presentation and summary
A 58-year-old woman from Brazil with past medical history only significant for cholecystectomy and cesarean section had been visiting in United States for 2 months when she presented to an outside hospital with fever, shortness of breath, and syncope that had resulted in a foot injury. She reported she had been feeling short of breath and had a nonproductive cough and malaise for about 2 weeks before presentation with sick contacts at home. On admission it was noted that she had a hemoglobin level of 7.7 g/dL (normal, 12.0-15.5 g/dL; MCV, 94 fL), total bilirubin of 2.14 mg/dL (normal, 0.2-1.0 mg/dL), and lactate dehydrogenase of 523 U/L (normal, 81-234 U/L). There were no signs of bleeding on her examination. Her DAT was positive and moderate red blood cell agglutination was reported. During the first admission at the outside hospital she was diagnosed with influenza A and completed a full course of oseltamivir (75 mg po twice daily for 5 days). A chest X-ray was negative for infiltrates and showed that the patient’s lung fields were clear. She was transfused 2 units of packed red blood cells with response in hemoglobin up to 9.8 g/dL. The patient was treated with dexamethasone (4 mg IV Q8) as an inpatient and was discharged on a prednisone taper (40 mg, with taper by 10 mg every 3 days) with hemoglobin of 8.1 g/dL.
The patient continued to have nonproductive cough, dyspnea, fevers, chills, and generalized weakness, when she returned to the same outside hospital’s emergency department 2 days after her discharge. At that time, it was noted that she had leucocytosis (white blood cell count, 34.6 x 109 per L), a hemoglobin level of 6.8 g/dL, and her total bilirubin level was 6.9 mg/dL. Her hemodynamics were unstable and she was admitted to their intensive care unit. The results of a chest X-ray revealed right lung consolidation.
The day after this admission, her hemoglobin level fell to 4.7 g/dL, and she was transfused 2 units of packed red blood cells before being transferred to our hospital. A chest X-ray at our hospital confirmed a right lung infiltrate. Vancomycin (1,250 mg IV Q12), levaquin (750 mg IV Q24), and maxipime (1 g IV Q12) were initiated for pneumonia and the patient was transferred to our hospital’s intensive care unit. She was afebrile at 98.3°F, her pulse rate was 84 beats per minute, she was tachypneic with respiratory rate of 26 breaths per minute, her blood pressure was 98/51 mmHg, and she had an oxygen saturation of 99% on 2L oxygen via nasal cannula.
On physical examination she was noted to have scleral icterus and was in mild respiratory distress. A chest X-ray revealed a patchy opacity in the right mid to lower lung. Her initial complete blood panel revealed anemia, with hemoglobin, 6.3 g/dL; white blood cell count, 27 x 109 per L; and platelets, 533 x 109 per L. The patient was then transfused another 2 units of packed red blood cells. She was given intravenous hydration, acetaminophen, and albuterol nebulizer treatments as supportive care. She was provided with blankets to keep warm. In addition to her antibiotics, she was also given prednisone 70 mg for her respiratory symptoms.
Further tests revealed haptoglobin, <30 mg/dL (normal, 36-195 mg/dL); lactate dehydrogenase, 371 U/L (normal, 98-192 U/L); and complements C3, 90 mg/dL (normal, 79-152 mg/dL) and C4, <8 mg/dL (normal, 18-55 mg/dL). Her DAT was positive, and agglutination was seen on peripheral smear (Figure 1). This was her second positive DAT as she had positive one at the outside hospital initially. Her tests for mycoplasma pneumonia, the PCR and IgM, were negative, as were the Monospot for mononucleosis and the ANA for autoimmune disorders. Her cold agglutinin titer was 1:256 (normal, no agglutination <1:64). The patient’s repeat respiratory viral panel was negative given recent full treatment for her influenza A at the previous hospital. Her blood and urine cultures were negative.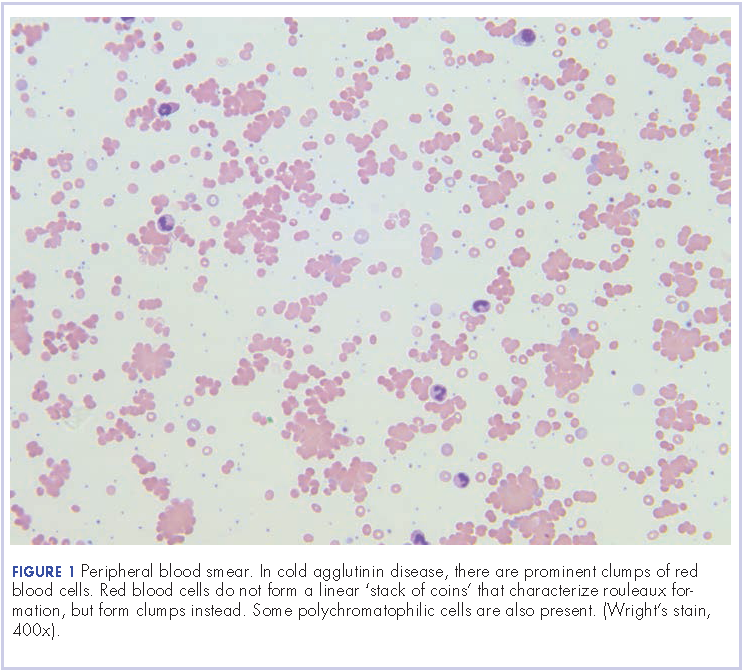
The patient was given antibiotics (vancomycin 1,250 mg IV Q12, cefepime 2 g IV Q8, and azithromycin 500 mg daily) for her pneumonia. Her respiratory status improved, and she was transferred to general medical floors after the first day of her admission. Her total bilirubin trended down to 1.9 mg/dL. She remained on prednisone 70 mg daily.
The patient remained in the hospital for an additional 6 days before being discharged home on prednisone. She wanted to return to her home country of Brazil as soon as she was able to and said she would seek outpatient follow-up there with a hematologist. At the time of her discharge, her hemoglobin was 6.6 g/dL and her reticulocyte count, 6.0%. Figures 2 and 3 illustrate her hemoglobin and reticulocyte trend during her admission at our hospital.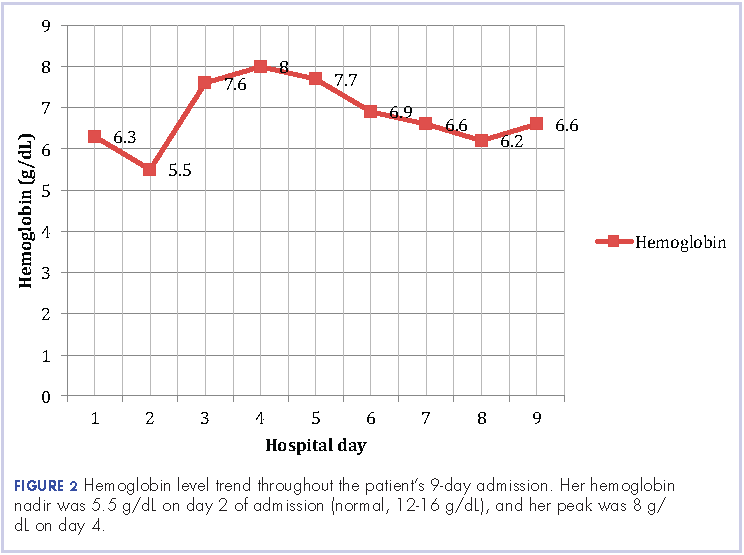
Discussion
The incidence of cold AIHA or cold agglutinin disease (CAD) occurs about 4 per 1 million people and commonly affects women more often than men.2 The cause of CAD can be subdivided into primary, idiopathic, or secondary causes, which can include infections, malignancies, or benign diseases.3,4 Primary CAD is a chronic disorder that is generally seen in older women. Secondary CAD can be associated with B-cell lymphoproliferative disorders, such as Waldenstrom macroglobulinemia or chronic lymphocytic leukemia, and infectious agents such as Mycoplasma pneumoniae and mononucleosis caused by Epstein-Barr virus.
Mild hemolysis or acrocyanosis may occur with exposure to cold. The blood smear in CAD demonstrates red blood cell agglutination or clumping, polychromasia, and an absence of spherocytosis. In general, most cases require no treatment, but cytotoxic agents or rituximab can be used to treat more severe cases. Appropriate treatment for infectious causes of CAD includes supportive care aimed at the underlying disease process. In addition, it is helpful to keep the patient warm. There is no role for steroid therapy in CAD unlike in warm AIHA. However, our patient was symptomatic from her pneumonia, so we added steroids to help with her pulmonary insult.
The patient had a cold agglutinin titer of 1:256. Titers of 1:32 or higher are considered elevated by this technique. Elevated titers are generally rarely seen except in primary atypical pneumonia due to either M. pneumoniae, influenza A, influenza B, parainfluenza, and adenovirus, and in certain hemolytic anemias. Low titers of cold agglutinins have been demonstrated in malaria, peripheral vascular disease, and common respiratory diseases.
Warm AIHA is caused by IgG antibody activities at body temperature or at 98.6°F. They may or may not bind complement and are removed from circulation by the spleen. Cold AIHA is due to IgM antibodies coating red cells at lower temperatures. They bind complement and lead to red blood cell destruction of agglutinated cells. If the antibody is active at temperatures approaching 98.6°F, clinically significant intravascular and sometimes extravascular complement-mediated hemolysis occur in the liver.5
The incidence of warm AIHA occurs about 10 per 1 million people and affects women twice often as men.2 It can be primary or idiopathic, or associated with various underlying conditions, including autoimmune disorders, immunodeficiency syndromes, lymphoproliferative disorders, other malignancies, and certain drugs. In more severe cases, jaundice and splenomegaly may occur. The blood smear in warm AIHA demonstrates variable spherocytosis, polychromasia, and rare erythrophagocytosis. Treatment usually includes steroids, cytotoxic agents, and splenectomy in severe cases.
There have been few case reports describing influenza as a cause of cold agglutinin hemolytic anemia. Chen and colleagues reported a case of influenza A infection in a 22-month-old boy.6Schoindre and colleagues reported the case of a 60-year-old woman infected with influenza A H1N1 virus who died from CAD.7 Shizuma reported the case of a 67-year-old man with alcoholic cirrhosis who developed a mixed hemolytic anemia and was positive for influenza A.8Our patient presented with influenza A, which had been diagnosed by respiratory virus panel at a different hospital, and she was anemic at the time of presentation to the outside hospital, with a positive DAT test. She was treated for influenza A with a full course of osltamivir and then returned with complaints of worsening fatigue and was again noted to be anemic with the development of patchy opacities on chest X-ray. The patient was subsequently transferred to our hospital and remained anemic during the course of her treatment. She received supportive care for her underlying influenza A and had symptomatic improvement. She ultimately decided the she would like to pursue further treatment in her native country and was discharged.
In conclusion, this case represents a rare complication of a common illness. Few cases of influenza causing hemolytic anemia have been reported in the literature. There have been reports of oseltamivir causing hemolytic anemia, but our patient presented with evidence of hemolytic anemia before initiation of the medication. In all the aforementioned cases, the patients died as a result of comorbid conditions. Our patient was stable enough to be discharged from the hospital after treatment of her comorbid conditions.
Acknowledgment
The authors thank David Henry, MD, at Pennsylvania Hospital, Philadelphia, for sharing this case and for his guidance during this patient’s treatment.
1. Roback JD, Grossman BJ, Harris T, Hillyer CD. Technical manual [17th ed]. Bethesda, MD; American Association of Blood Banks; 2011.
2. Jaffee ES, Harris NL, Vardiman JW, Campo E, Arber DA. Hematopathology. St. Louis, MO; Elsevier Saunders, 2011.
3. Feizi T. Monotypic cold agglutinins in infection by Mycoplasma pneumoniae. Nature. 1967;215(5100):540-542.
4. Horwitz CA, Moulds J, Henle W, et al. Cold agglutinins in infectious mononucleosis and heterophil-antibody-negative mononucleosis-like syndromes. Blood. 1977;50(2):195-202.
5. Hsi ED, editor. Hematopathology [3rd ed]. Philadelphia, PA; Elsevier Saunders; 2012.
6. Chen H, Jia XL, Gao HM, Qian SY. Comorbid presentation of severe novel influenza A (H1N1) and Evans syndrome: a case report. Chin Med J. 2011;124(11):1743-1746.
7. Schoindre Y, Bollée G, Dumont MD, Lesavre P, Servais A. Cold agglutinin syndrome associated with a 2009 influenza A H1N1 infection. http://www.amjmed.com/article/S0002-9343(10)00482-1/fulltext. Published February 2011. Accessed October 10, 2017.
8. [Article in Japanese] Shizuma T. [A case of autoimmune hemolytic anemia caused by type A influenza infection in a patient with alcoholic liver cirrhosis]. Kansenshogaku Zasshi. 2010;84(3):296-299.
Autoimmune hemolytic anemia (AIHA) is characterized by the temperature at which the auto-antibody has the greatest avidity for the target red cell antigen, either warm or cold forms. It is detected by a positive direct antiglobulin test (DAT) also known as the direct Coombs test. DAT is used to determine if red cells have been coated in vivo with immunoglobulin, complement, or both.1 Some causes of a positive DAT include hemolytic transfusion reactions, hemolytic disease of the fetus and newborn, AIHA, and drug-induced immune hemolysis.
Case presentation and summary
A 58-year-old woman from Brazil with past medical history only significant for cholecystectomy and cesarean section had been visiting in United States for 2 months when she presented to an outside hospital with fever, shortness of breath, and syncope that had resulted in a foot injury. She reported she had been feeling short of breath and had a nonproductive cough and malaise for about 2 weeks before presentation with sick contacts at home. On admission it was noted that she had a hemoglobin level of 7.7 g/dL (normal, 12.0-15.5 g/dL; MCV, 94 fL), total bilirubin of 2.14 mg/dL (normal, 0.2-1.0 mg/dL), and lactate dehydrogenase of 523 U/L (normal, 81-234 U/L). There were no signs of bleeding on her examination. Her DAT was positive and moderate red blood cell agglutination was reported. During the first admission at the outside hospital she was diagnosed with influenza A and completed a full course of oseltamivir (75 mg po twice daily for 5 days). A chest X-ray was negative for infiltrates and showed that the patient’s lung fields were clear. She was transfused 2 units of packed red blood cells with response in hemoglobin up to 9.8 g/dL. The patient was treated with dexamethasone (4 mg IV Q8) as an inpatient and was discharged on a prednisone taper (40 mg, with taper by 10 mg every 3 days) with hemoglobin of 8.1 g/dL.
The patient continued to have nonproductive cough, dyspnea, fevers, chills, and generalized weakness, when she returned to the same outside hospital’s emergency department 2 days after her discharge. At that time, it was noted that she had leucocytosis (white blood cell count, 34.6 x 109 per L), a hemoglobin level of 6.8 g/dL, and her total bilirubin level was 6.9 mg/dL. Her hemodynamics were unstable and she was admitted to their intensive care unit. The results of a chest X-ray revealed right lung consolidation.
The day after this admission, her hemoglobin level fell to 4.7 g/dL, and she was transfused 2 units of packed red blood cells before being transferred to our hospital. A chest X-ray at our hospital confirmed a right lung infiltrate. Vancomycin (1,250 mg IV Q12), levaquin (750 mg IV Q24), and maxipime (1 g IV Q12) were initiated for pneumonia and the patient was transferred to our hospital’s intensive care unit. She was afebrile at 98.3°F, her pulse rate was 84 beats per minute, she was tachypneic with respiratory rate of 26 breaths per minute, her blood pressure was 98/51 mmHg, and she had an oxygen saturation of 99% on 2L oxygen via nasal cannula.
On physical examination she was noted to have scleral icterus and was in mild respiratory distress. A chest X-ray revealed a patchy opacity in the right mid to lower lung. Her initial complete blood panel revealed anemia, with hemoglobin, 6.3 g/dL; white blood cell count, 27 x 109 per L; and platelets, 533 x 109 per L. The patient was then transfused another 2 units of packed red blood cells. She was given intravenous hydration, acetaminophen, and albuterol nebulizer treatments as supportive care. She was provided with blankets to keep warm. In addition to her antibiotics, she was also given prednisone 70 mg for her respiratory symptoms.
Further tests revealed haptoglobin, <30 mg/dL (normal, 36-195 mg/dL); lactate dehydrogenase, 371 U/L (normal, 98-192 U/L); and complements C3, 90 mg/dL (normal, 79-152 mg/dL) and C4, <8 mg/dL (normal, 18-55 mg/dL). Her DAT was positive, and agglutination was seen on peripheral smear (Figure 1). This was her second positive DAT as she had positive one at the outside hospital initially. Her tests for mycoplasma pneumonia, the PCR and IgM, were negative, as were the Monospot for mononucleosis and the ANA for autoimmune disorders. Her cold agglutinin titer was 1:256 (normal, no agglutination <1:64). The patient’s repeat respiratory viral panel was negative given recent full treatment for her influenza A at the previous hospital. Her blood and urine cultures were negative.
The patient was given antibiotics (vancomycin 1,250 mg IV Q12, cefepime 2 g IV Q8, and azithromycin 500 mg daily) for her pneumonia. Her respiratory status improved, and she was transferred to general medical floors after the first day of her admission. Her total bilirubin trended down to 1.9 mg/dL. She remained on prednisone 70 mg daily.
The patient remained in the hospital for an additional 6 days before being discharged home on prednisone. She wanted to return to her home country of Brazil as soon as she was able to and said she would seek outpatient follow-up there with a hematologist. At the time of her discharge, her hemoglobin was 6.6 g/dL and her reticulocyte count, 6.0%. Figures 2 and 3 illustrate her hemoglobin and reticulocyte trend during her admission at our hospital.
Discussion
The incidence of cold AIHA or cold agglutinin disease (CAD) occurs about 4 per 1 million people and commonly affects women more often than men.2 The cause of CAD can be subdivided into primary, idiopathic, or secondary causes, which can include infections, malignancies, or benign diseases.3,4 Primary CAD is a chronic disorder that is generally seen in older women. Secondary CAD can be associated with B-cell lymphoproliferative disorders, such as Waldenstrom macroglobulinemia or chronic lymphocytic leukemia, and infectious agents such as Mycoplasma pneumoniae and mononucleosis caused by Epstein-Barr virus.
Mild hemolysis or acrocyanosis may occur with exposure to cold. The blood smear in CAD demonstrates red blood cell agglutination or clumping, polychromasia, and an absence of spherocytosis. In general, most cases require no treatment, but cytotoxic agents or rituximab can be used to treat more severe cases. Appropriate treatment for infectious causes of CAD includes supportive care aimed at the underlying disease process. In addition, it is helpful to keep the patient warm. There is no role for steroid therapy in CAD unlike in warm AIHA. However, our patient was symptomatic from her pneumonia, so we added steroids to help with her pulmonary insult.
The patient had a cold agglutinin titer of 1:256. Titers of 1:32 or higher are considered elevated by this technique. Elevated titers are generally rarely seen except in primary atypical pneumonia due to either M. pneumoniae, influenza A, influenza B, parainfluenza, and adenovirus, and in certain hemolytic anemias. Low titers of cold agglutinins have been demonstrated in malaria, peripheral vascular disease, and common respiratory diseases.
Warm AIHA is caused by IgG antibody activities at body temperature or at 98.6°F. They may or may not bind complement and are removed from circulation by the spleen. Cold AIHA is due to IgM antibodies coating red cells at lower temperatures. They bind complement and lead to red blood cell destruction of agglutinated cells. If the antibody is active at temperatures approaching 98.6°F, clinically significant intravascular and sometimes extravascular complement-mediated hemolysis occur in the liver.5
The incidence of warm AIHA occurs about 10 per 1 million people and affects women twice often as men.2 It can be primary or idiopathic, or associated with various underlying conditions, including autoimmune disorders, immunodeficiency syndromes, lymphoproliferative disorders, other malignancies, and certain drugs. In more severe cases, jaundice and splenomegaly may occur. The blood smear in warm AIHA demonstrates variable spherocytosis, polychromasia, and rare erythrophagocytosis. Treatment usually includes steroids, cytotoxic agents, and splenectomy in severe cases.
There have been few case reports describing influenza as a cause of cold agglutinin hemolytic anemia. Chen and colleagues reported a case of influenza A infection in a 22-month-old boy.6Schoindre and colleagues reported the case of a 60-year-old woman infected with influenza A H1N1 virus who died from CAD.7 Shizuma reported the case of a 67-year-old man with alcoholic cirrhosis who developed a mixed hemolytic anemia and was positive for influenza A.8Our patient presented with influenza A, which had been diagnosed by respiratory virus panel at a different hospital, and she was anemic at the time of presentation to the outside hospital, with a positive DAT test. She was treated for influenza A with a full course of osltamivir and then returned with complaints of worsening fatigue and was again noted to be anemic with the development of patchy opacities on chest X-ray. The patient was subsequently transferred to our hospital and remained anemic during the course of her treatment. She received supportive care for her underlying influenza A and had symptomatic improvement. She ultimately decided the she would like to pursue further treatment in her native country and was discharged.
In conclusion, this case represents a rare complication of a common illness. Few cases of influenza causing hemolytic anemia have been reported in the literature. There have been reports of oseltamivir causing hemolytic anemia, but our patient presented with evidence of hemolytic anemia before initiation of the medication. In all the aforementioned cases, the patients died as a result of comorbid conditions. Our patient was stable enough to be discharged from the hospital after treatment of her comorbid conditions.
Acknowledgment
The authors thank David Henry, MD, at Pennsylvania Hospital, Philadelphia, for sharing this case and for his guidance during this patient’s treatment.
Autoimmune hemolytic anemia (AIHA) is characterized by the temperature at which the auto-antibody has the greatest avidity for the target red cell antigen, either warm or cold forms. It is detected by a positive direct antiglobulin test (DAT) also known as the direct Coombs test. DAT is used to determine if red cells have been coated in vivo with immunoglobulin, complement, or both.1 Some causes of a positive DAT include hemolytic transfusion reactions, hemolytic disease of the fetus and newborn, AIHA, and drug-induced immune hemolysis.
Case presentation and summary
A 58-year-old woman from Brazil with past medical history only significant for cholecystectomy and cesarean section had been visiting in United States for 2 months when she presented to an outside hospital with fever, shortness of breath, and syncope that had resulted in a foot injury. She reported she had been feeling short of breath and had a nonproductive cough and malaise for about 2 weeks before presentation with sick contacts at home. On admission it was noted that she had a hemoglobin level of 7.7 g/dL (normal, 12.0-15.5 g/dL; MCV, 94 fL), total bilirubin of 2.14 mg/dL (normal, 0.2-1.0 mg/dL), and lactate dehydrogenase of 523 U/L (normal, 81-234 U/L). There were no signs of bleeding on her examination. Her DAT was positive and moderate red blood cell agglutination was reported. During the first admission at the outside hospital she was diagnosed with influenza A and completed a full course of oseltamivir (75 mg po twice daily for 5 days). A chest X-ray was negative for infiltrates and showed that the patient’s lung fields were clear. She was transfused 2 units of packed red blood cells with response in hemoglobin up to 9.8 g/dL. The patient was treated with dexamethasone (4 mg IV Q8) as an inpatient and was discharged on a prednisone taper (40 mg, with taper by 10 mg every 3 days) with hemoglobin of 8.1 g/dL.
The patient continued to have nonproductive cough, dyspnea, fevers, chills, and generalized weakness, when she returned to the same outside hospital’s emergency department 2 days after her discharge. At that time, it was noted that she had leucocytosis (white blood cell count, 34.6 x 109 per L), a hemoglobin level of 6.8 g/dL, and her total bilirubin level was 6.9 mg/dL. Her hemodynamics were unstable and she was admitted to their intensive care unit. The results of a chest X-ray revealed right lung consolidation.
The day after this admission, her hemoglobin level fell to 4.7 g/dL, and she was transfused 2 units of packed red blood cells before being transferred to our hospital. A chest X-ray at our hospital confirmed a right lung infiltrate. Vancomycin (1,250 mg IV Q12), levaquin (750 mg IV Q24), and maxipime (1 g IV Q12) were initiated for pneumonia and the patient was transferred to our hospital’s intensive care unit. She was afebrile at 98.3°F, her pulse rate was 84 beats per minute, she was tachypneic with respiratory rate of 26 breaths per minute, her blood pressure was 98/51 mmHg, and she had an oxygen saturation of 99% on 2L oxygen via nasal cannula.
On physical examination she was noted to have scleral icterus and was in mild respiratory distress. A chest X-ray revealed a patchy opacity in the right mid to lower lung. Her initial complete blood panel revealed anemia, with hemoglobin, 6.3 g/dL; white blood cell count, 27 x 109 per L; and platelets, 533 x 109 per L. The patient was then transfused another 2 units of packed red blood cells. She was given intravenous hydration, acetaminophen, and albuterol nebulizer treatments as supportive care. She was provided with blankets to keep warm. In addition to her antibiotics, she was also given prednisone 70 mg for her respiratory symptoms.
Further tests revealed haptoglobin, <30 mg/dL (normal, 36-195 mg/dL); lactate dehydrogenase, 371 U/L (normal, 98-192 U/L); and complements C3, 90 mg/dL (normal, 79-152 mg/dL) and C4, <8 mg/dL (normal, 18-55 mg/dL). Her DAT was positive, and agglutination was seen on peripheral smear (Figure 1). This was her second positive DAT as she had positive one at the outside hospital initially. Her tests for mycoplasma pneumonia, the PCR and IgM, were negative, as were the Monospot for mononucleosis and the ANA for autoimmune disorders. Her cold agglutinin titer was 1:256 (normal, no agglutination <1:64). The patient’s repeat respiratory viral panel was negative given recent full treatment for her influenza A at the previous hospital. Her blood and urine cultures were negative.
The patient was given antibiotics (vancomycin 1,250 mg IV Q12, cefepime 2 g IV Q8, and azithromycin 500 mg daily) for her pneumonia. Her respiratory status improved, and she was transferred to general medical floors after the first day of her admission. Her total bilirubin trended down to 1.9 mg/dL. She remained on prednisone 70 mg daily.
The patient remained in the hospital for an additional 6 days before being discharged home on prednisone. She wanted to return to her home country of Brazil as soon as she was able to and said she would seek outpatient follow-up there with a hematologist. At the time of her discharge, her hemoglobin was 6.6 g/dL and her reticulocyte count, 6.0%. Figures 2 and 3 illustrate her hemoglobin and reticulocyte trend during her admission at our hospital.
Discussion
The incidence of cold AIHA or cold agglutinin disease (CAD) occurs about 4 per 1 million people and commonly affects women more often than men.2 The cause of CAD can be subdivided into primary, idiopathic, or secondary causes, which can include infections, malignancies, or benign diseases.3,4 Primary CAD is a chronic disorder that is generally seen in older women. Secondary CAD can be associated with B-cell lymphoproliferative disorders, such as Waldenstrom macroglobulinemia or chronic lymphocytic leukemia, and infectious agents such as Mycoplasma pneumoniae and mononucleosis caused by Epstein-Barr virus.
Mild hemolysis or acrocyanosis may occur with exposure to cold. The blood smear in CAD demonstrates red blood cell agglutination or clumping, polychromasia, and an absence of spherocytosis. In general, most cases require no treatment, but cytotoxic agents or rituximab can be used to treat more severe cases. Appropriate treatment for infectious causes of CAD includes supportive care aimed at the underlying disease process. In addition, it is helpful to keep the patient warm. There is no role for steroid therapy in CAD unlike in warm AIHA. However, our patient was symptomatic from her pneumonia, so we added steroids to help with her pulmonary insult.
The patient had a cold agglutinin titer of 1:256. Titers of 1:32 or higher are considered elevated by this technique. Elevated titers are generally rarely seen except in primary atypical pneumonia due to either M. pneumoniae, influenza A, influenza B, parainfluenza, and adenovirus, and in certain hemolytic anemias. Low titers of cold agglutinins have been demonstrated in malaria, peripheral vascular disease, and common respiratory diseases.
Warm AIHA is caused by IgG antibody activities at body temperature or at 98.6°F. They may or may not bind complement and are removed from circulation by the spleen. Cold AIHA is due to IgM antibodies coating red cells at lower temperatures. They bind complement and lead to red blood cell destruction of agglutinated cells. If the antibody is active at temperatures approaching 98.6°F, clinically significant intravascular and sometimes extravascular complement-mediated hemolysis occur in the liver.5
The incidence of warm AIHA occurs about 10 per 1 million people and affects women twice often as men.2 It can be primary or idiopathic, or associated with various underlying conditions, including autoimmune disorders, immunodeficiency syndromes, lymphoproliferative disorders, other malignancies, and certain drugs. In more severe cases, jaundice and splenomegaly may occur. The blood smear in warm AIHA demonstrates variable spherocytosis, polychromasia, and rare erythrophagocytosis. Treatment usually includes steroids, cytotoxic agents, and splenectomy in severe cases.
There have been few case reports describing influenza as a cause of cold agglutinin hemolytic anemia. Chen and colleagues reported a case of influenza A infection in a 22-month-old boy.6Schoindre and colleagues reported the case of a 60-year-old woman infected with influenza A H1N1 virus who died from CAD.7 Shizuma reported the case of a 67-year-old man with alcoholic cirrhosis who developed a mixed hemolytic anemia and was positive for influenza A.8Our patient presented with influenza A, which had been diagnosed by respiratory virus panel at a different hospital, and she was anemic at the time of presentation to the outside hospital, with a positive DAT test. She was treated for influenza A with a full course of osltamivir and then returned with complaints of worsening fatigue and was again noted to be anemic with the development of patchy opacities on chest X-ray. The patient was subsequently transferred to our hospital and remained anemic during the course of her treatment. She received supportive care for her underlying influenza A and had symptomatic improvement. She ultimately decided the she would like to pursue further treatment in her native country and was discharged.
In conclusion, this case represents a rare complication of a common illness. Few cases of influenza causing hemolytic anemia have been reported in the literature. There have been reports of oseltamivir causing hemolytic anemia, but our patient presented with evidence of hemolytic anemia before initiation of the medication. In all the aforementioned cases, the patients died as a result of comorbid conditions. Our patient was stable enough to be discharged from the hospital after treatment of her comorbid conditions.
Acknowledgment
The authors thank David Henry, MD, at Pennsylvania Hospital, Philadelphia, for sharing this case and for his guidance during this patient’s treatment.
1. Roback JD, Grossman BJ, Harris T, Hillyer CD. Technical manual [17th ed]. Bethesda, MD; American Association of Blood Banks; 2011.
2. Jaffee ES, Harris NL, Vardiman JW, Campo E, Arber DA. Hematopathology. St. Louis, MO; Elsevier Saunders, 2011.
3. Feizi T. Monotypic cold agglutinins in infection by Mycoplasma pneumoniae. Nature. 1967;215(5100):540-542.
4. Horwitz CA, Moulds J, Henle W, et al. Cold agglutinins in infectious mononucleosis and heterophil-antibody-negative mononucleosis-like syndromes. Blood. 1977;50(2):195-202.
5. Hsi ED, editor. Hematopathology [3rd ed]. Philadelphia, PA; Elsevier Saunders; 2012.
6. Chen H, Jia XL, Gao HM, Qian SY. Comorbid presentation of severe novel influenza A (H1N1) and Evans syndrome: a case report. Chin Med J. 2011;124(11):1743-1746.
7. Schoindre Y, Bollée G, Dumont MD, Lesavre P, Servais A. Cold agglutinin syndrome associated with a 2009 influenza A H1N1 infection. http://www.amjmed.com/article/S0002-9343(10)00482-1/fulltext. Published February 2011. Accessed October 10, 2017.
8. [Article in Japanese] Shizuma T. [A case of autoimmune hemolytic anemia caused by type A influenza infection in a patient with alcoholic liver cirrhosis]. Kansenshogaku Zasshi. 2010;84(3):296-299.
1. Roback JD, Grossman BJ, Harris T, Hillyer CD. Technical manual [17th ed]. Bethesda, MD; American Association of Blood Banks; 2011.
2. Jaffee ES, Harris NL, Vardiman JW, Campo E, Arber DA. Hematopathology. St. Louis, MO; Elsevier Saunders, 2011.
3. Feizi T. Monotypic cold agglutinins in infection by Mycoplasma pneumoniae. Nature. 1967;215(5100):540-542.
4. Horwitz CA, Moulds J, Henle W, et al. Cold agglutinins in infectious mononucleosis and heterophil-antibody-negative mononucleosis-like syndromes. Blood. 1977;50(2):195-202.
5. Hsi ED, editor. Hematopathology [3rd ed]. Philadelphia, PA; Elsevier Saunders; 2012.
6. Chen H, Jia XL, Gao HM, Qian SY. Comorbid presentation of severe novel influenza A (H1N1) and Evans syndrome: a case report. Chin Med J. 2011;124(11):1743-1746.
7. Schoindre Y, Bollée G, Dumont MD, Lesavre P, Servais A. Cold agglutinin syndrome associated with a 2009 influenza A H1N1 infection. http://www.amjmed.com/article/S0002-9343(10)00482-1/fulltext. Published February 2011. Accessed October 10, 2017.
8. [Article in Japanese] Shizuma T. [A case of autoimmune hemolytic anemia caused by type A influenza infection in a patient with alcoholic liver cirrhosis]. Kansenshogaku Zasshi. 2010;84(3):296-299.