User login
Screening and Treating Hepatitis C in the VA: Achieving Excellence Using Lean and System Redesign
Hepatitis C virus (HCV) infection is a major public health problem in the US. Following the 2010 report of the Institute of Medicine/National Academies of Sciences, Engineering, and Medicine (NASEM) on hepatitis and liver cancer, the US Department of Health and Human Services (HHS) released the first National Viral Hepatitis Action Plan in 2011 with subsequent action plan updates for 2014-2016 and 2017-2020.1-3 A NASEM phase 2 report and the 2017-2020 HHS action plan outline a national strategy to prevent new viral hepatitis infections; reduce deaths and improve the health of people living with viral hepatitis; reduce viral hepatitis health disparities; and coordinate, monitor, and report on implementation of viral hepatitis activities.3,4 The Department of Veterans Affairs (VA) is the single largest HCV care provider in the US with about 165,000 veterans in care diagnosed with HCV in the beginning of 2014 and is a national leader in the testing and treatment of HCV.5,6
The VA’s recommendations for screening for HCV infection are in alignment with the United States Preventive Services Task Force (USPSTF) and Centers for Disease Control and Prevention (CDC) recommendations to test all veterans born between 1945 and 1965 and anyone with risk factors such as injection drug use.7-9 As of January 1, 2018, the VA had screened more than 80% of veterans in care within this highest risk birth cohort. As of January 1, 2018, more than 100,000 veterans in VA care have initiated treatment for HCV with direct-acting antivirals (DAAs) (Figure 1). 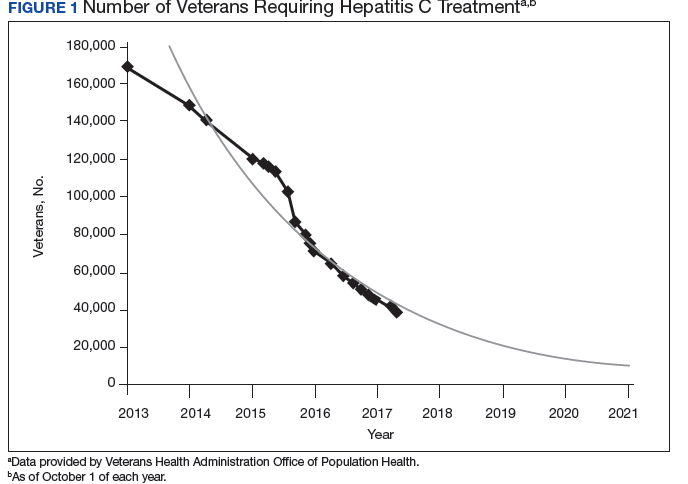
Several critical factors contributed to the VA success with HCV testing and treatment, including congressional appropriation of funding from fiscal year (FY) 2016 through FY 2018, unrestricted access to interferon-free DAA HCV treatments, and dedicated resources from the VA National Viral Hepatitis Program within the HIV, Hepatitis, and Related Conditions Programs (HHRC) in the Office of Specialty Care Services.5 In 2014, HHRC created and supported the Hepatitis Innovation Team (HIT) Collaborative, a VA process improvement initiative enabling
Veterans Integrated Service Network (VISN) -based, multidisciplinary teams to increase veterans’ access to HCV testing and treatment.
As the VA makes consistent progress toward eliminating HCV in veterans in VA care, it has become clear that achieving a cure is only a starting point in improving HCV care. Many patients with HCV infection also have advanced liver disease (ALD), or cirrhosis, which is a condition of permanent liver fibrosis that remains after the patient has been cured of HCV infection. In addition to hepatitis C, ALD also can be caused by excessive alcohol use, hepatitis B virus (HBV) infection, nonalcoholic fatty liver diseases, and several other inherited diseases. Advanced liver disease affects more than 80,000 veterans in VA care, and the HIT infrastructure provides an excellent framework to better understand and address facility-level and systemwide challenges in diagnosing, caring for, and treating veterans with ALD across the Veterans Health Administration (VHA) system.
This report will describe the elements that contributed to the success of the HIT Collaborative in redesigning care for patients affected by HCV in the VA and how these elements can be applied to improve the system of care for VHA ALD care.
Hepatitis Innovation Teams Collaborative Leadership
After the US Food and Drug Administration (FDA) approved new DAA medications to treat HCV, the VA recognized the need to mobilize the health care system quickly and allocate resources for these new, minimally toxic, and highly effective medications. Early in 2014, HHRC established the National Hepatitis C Resource Center (NHCRC), a successor program to the 4 regional hepatitis C resource centers that had addressed HCV care across the system.10 The NHCRC was charged with developing an operational strategy for VA to respond rapidly to the availability of DAAs. In collaboration with representatives from the Office of Strategic Integration | Veterans Engineering Resource Center (OSI|VERC), the NHCRC formed the HIT Collaborative Leadership Team (CLT).
The HIT CLT is responsible for executing the HIT Collaborative and uses a Lean process improvement framework focused on eliminating waste and maximizing value. Members of the CLT with expertise in facilitation, Lean process improvement, leadership, clinical knowledge, and population health management act as coaches for the VISN HITs. The CLT works to build and support the VISN HITs, identify opportunities for individual teams to improve and assist in finding the right local mix of “players” to be successful. The HIT CLT ensures all teams are functioning and working toward achieving their goals. The CLT obtains data from VA national databases, which are provided to the VISN HITs to inform and encourage continuous improvement of their strategies. Annual VA-wide aspirational goals are developed and disseminated to encourage a unified mission.
Catchment areas for each VISN include between 6 and 10 medical centers as well as outpatient and ambulatory care centers. Multidisciplinary HITs are composed of physicians, nurses, pharmacists, nurse practitioners, physician assistants, social workers, mental health and substance use providers, peer support specialists, administrators, information technology experts, and systems redesign professionals from medical centers within each VISN. Teams develop strong relationships across medical centers, implement context-specific strategies applicable to rural
and urban centers, and share expertise. In addition to intra-VISN process improvement, HITs collaborate monthly across VISNs via a virtual platform. They share strong practices, seek advice from one another, and compare outcomes on an established set of goals.
The HITs use process improvement tools to systematically assess the current steps involved in care. At the close of each year, the HITs analyze the current state of operations and set goals to improve over the following year guided by a target state map. Seed funding is provided to every VISN HIT annually to launch change initiatives. Many VISN HITs use these funds to support a VISN HIT coordinator, and HITs also use this financial support to conduct 2- to 3-day process improvement workshops and to purchase supplies, such as point-of-care testing kits. The HIT communication and work are predominantly executed virtually.
Each year, teams worked toward achieving goals set nationally. These included increasing HCV birth cohort testing and improving the percentage of patients who had SVR12 testing
(Table). 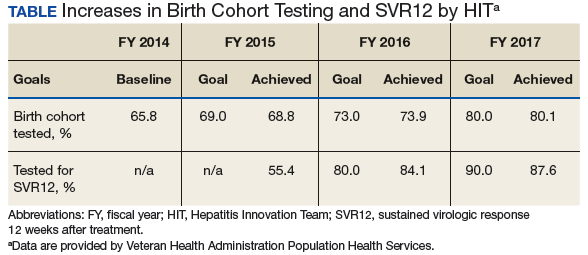
the percentage of patients who received SVR12 testing posttreatment completion was not included in the HIT Collaborative’s annual goals for the first year of the program. Recognizing this as a critical area for improvement, the HIT CLT set a goal to test 80% of all patients who completed treatment. The HITs applied Lean tools to identify and overcome gaps in the SVR12 testing process. By the end of the second year, 84% of all patients who completed treatment had been tested for SVR12.
The HITs also set specific local VISN and medical center goals, prioritizing projects that could have the greatest impact on local patient access and quality of care and build on existing strengths and address barriers. These projects encompass a wide range of areas that contribute to the overall national goals.
Focus on Lean
Lean process improvement is based on 2 key pillars: respect for people (those seeking service as customers and patients and those providing service as frontline staff and stakeholders) and continuous improvement. With Lean, personnel providing care should work to identify and eliminate waste in the system and to streamline care delivery to maximize process steps that are most valued by patients (eg, interaction with a clinical provider) and minimize those that are not valued (eg, time spent waiting to see a provider). With the knowledge that HHRC fully supports their work, HITs were encouraged to innovate based on local resources, context, and culture.
Teams receive basic training in Lean from the HIT CLT and local systems redesign specialists if available. The HITs apply the A3 structured approach to problem solving.11 The HITs follow prescribed problemsolving steps that help identify where to focus process improvement efforts, including analyzing the current state of care, outlining the target state, and prioritizing solution
approaches based on what will have the highest impact for patients. 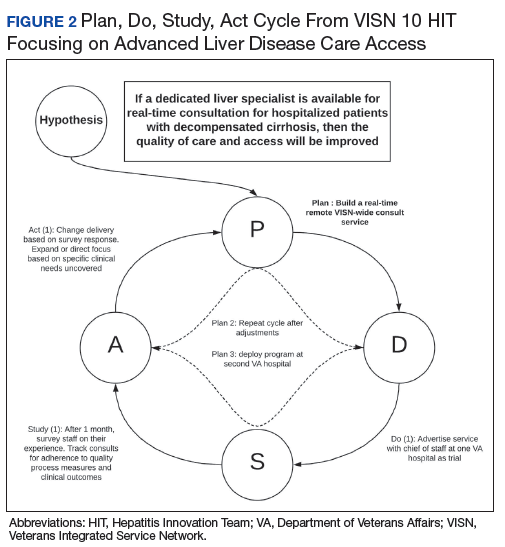
to accommodate the outcomes they observe (Figure 2).
Innovations
Over the course of the HIT Collaborative, numerous innovations have emerged to address and mitigate barriers to HCV screening and treatment. Examples of successful innovations include the following:
- To address transportation issues, several teams developed programs specific to patients with HCV in rural locations or with limited mobility. Mobile vans and units traditionally used as mobile cardiology clinics were transformed into HCV clinics, bringing testing and treatment services directly to veterans;
- Pharmacists and social workers developed outreach strategies to locate homeless veterans, provide point-of-care testing and utilize mobile technology to concurrently enroll and link veterans to care; and
- Many liver care teams partnered with inpatient and outpatient substance use treatment clinics to provide patient education and coordinate HCV treatment.
Inter-VISN working groups developed systemwide tools to address common needs. In the program’s first year, a few medical facilities across a handful of VISNs shared local population health management systems, programming, and best practices. Over time, this working group combined the virtual networking capacity of the HIT Collaborative with technical expertise to promote rapid dissemination and uptake of a population health management system. Providers at medical centers across VA use the tools to identify veterans who should be screened and treated for HCV with the ability to continuously update information, identifying patients who do not respond to treatment or patients overdue for SVR12 testing.
Providers with experience using telehepatology formed another inter-VISN working group. These subject matter experts provided guidance to care teams interested in implementing telehealth in areas where limited local resources or knowledge had prevented them from moving forward. The ability to build a strong coalition across content areas fostered a collaborative learning environment, adaptable to implementing new processes and technologies.
In 2017, the VA made significant efforts to reach out to veterans eligible for VA care who had not yet been screened or remained untreated. In May, Hepatitis Awareness Month, HITs held HCV testing and community outreach events and participated in veteran stand-downs and veteran service organization activities.
Evaluation
Since 2014, the VA has increased its HCV treatment and screening rates. To assess the components contributing to these achievements and the role of the HIT Collaborative in driving this success, a team of implementation scientists have been working with the CLT to conduct a HIT program evaluation. The goal of the evaluation is to establish the impact of the HIT Collaborative. The evaluation team catalogs the activities of the Collaborative and the HITs and assesses implementation strategies (use of specific techniques) to increase the uptake of evidence-based practices specifically related to HCV treatment.12
At the close of each FY, HCV providers and members of the HIT Collaborative are queried through an online survey to determine which strategies have been used to improve HCV care and how these strategies were associated with the HIT Collaborative. The use of more strategies was associated with more HCV treatment initiations.13 All utilized strategies were identified whether or not they were associated with treatment starts. These data are being used to understand which combinations of strategies are most effective at increasing treatment for HCV in the VA and to inform future initiatives.
Expanding the Scope
Inspired by the successful results of the HIT work in HCV and in the spirit of continuously improving health care delivery, HHRC expanded the scope of the HIT Collaborative in FY 2018 to include ALD. There are about 80,000 veterans in VA care with advanced scarring of the liver and between 10,000 to 15,000 new diagnoses each year. In addition to HCV as an etiology for ALD, cases of cirrhosis are projected to increase among veterans in care due to metabolic syndrome and alcohol use. A recent review of VA data from fiscal year 2016 found that 88.6% of ALD patients had been seen in primary care within the past 2 years, with about half (51%) seen in a gastroenterology (GI) or hepatology clinic (Personal communication, HIV, Hepatitis, and Related Conditions Program Office March 16, 2018). For patients in VA care with ALD, GI visits are associated with a lower 5-year mortality.14 Annual mortality for all ALD patients in VA is 6.2%, and of those with a hospital admission, mortality rises to 31%.15 In FY 2016, there were about 52,000 ALD-related discharges (more than 2 per patient). Of those discharges, 24% were readmitted within 30 days, with an average length of stay of 1.9 days and an estimated cost per patient of $47,000 over 3 years.16
Hepatologists from across the VA convened to identify critical opportunities for improvement for patients with ALD. Base on available evidence presented in the literature and their clinical expertise, these subject matter experts identified several areas for quality improvement, with the overarching goal to improve identification of patients with early cirrhosis and ensure appropriate linkage to care for all cirrhotic patients, thus improving quality of life and reducing mortality. Although not finalized, candidate improvement targets include consistent linkage to care and treatment for HCV and HBV, comprehensive case management, post-discharge patient follow-up, and adherence to evidence-based standards of care.
Conclusion
The VA has made great strides in nearly eliminating HCV among veterans in VA care. The national effort to redesign hepatitis care using Lean management strategies and develop local and regional teams and centralized support allowed VA to maximize available resources to achieve higher rates of HCV birth cohort testing and treatment of patients infected with HCV than has any other health care system in the US.
The HIT Collaborative has been a unique and innovative mechanism to promote directed, patient-outcome driven change in a large and dynamic health care system. It has allowed rural and urban providers to work together to develop and spread quality improvement innovations and as an integrated system to achieve national priorities. The focus of this foundational HIT structure is expanding to identifying, treat, and care for VA’s ALD population.
1. Colvin HM, Mitchell AE, eds; and the Committee on the Prevention and Control of Viral Hepatitis Infections Board on Population Health and Public Health Practice. Hepatitis and Liver Cancer: A National Strategy for Prevention and Control of Hepatitis B and C. Washington, DC: The National Academies Press; 2010.
2. US Department of Health and Human Services. Combating the silent epidemic of viral hepatitis: action plan for the prevention, care and treatment of viral hepatitis. https://www.hhs.gov/sites/default/files/action-plan-viral-hepatitis-2011.pdf. Accessed April 27, 2018.
3. Wolitski R. National viral hepatitis action plan: 2017-2020. https://www.hhs.gov/hepatitis/action-plan/national-viralhepatitis-action-plan-overview/index.html. Updated February
21, 2018. Accessed May 8, 2018.
4. National Academies of Sciences, Engineering, and Medicine. A National Strategy for the Elimination of Hepatitis B and C: Phase Two Report. Washington, DC: The National Academies Press; 2017.
5. Belperio PS, Chartier M, Ross DB, Alaigh P, Shulkin D. Curing hepatitis C infection: best practices from the Department of Veterans Affairs. Ann of Intern Med. 2017;167(7):499-504.
6. Kushner T, Serper M, Kaplan DE. Delta hepatitis within the Veterans Affairs medical system in the United States: prevalence, risk factors, and outcomes. J Hepatol. 2015;63(3):586-592.
7. US Department of Veterans Affairs, Veteran Health Administration. National Clinical Preventive Service Guidance Statements: Screening for Hepatitis C. http://www.prevention.va.gov/CPS/Screening_for_Hepatitis_C.asp. Published on June 20, 2017. [Nonpubic document; source not verified.]
8. Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
9. Smith BD, Morgan RL, Beckett GA, et al; Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945-1965. MMWR Recomm Rep. 2012;61(RR-4):1-32.
10. Garrard J, Choudary V, Groom H, et al. Organizational change in management of hepatitis C: evaluation of a CME program. J Contin Educ Health Prof. 2006;26(2):145-160.
11. Shook J. Managing to Learn: Using the A3 Management Process to Solve Problems, Gain Agreement, Mentor, and Lead. Cambridge, MA: Lean Enterprise Institute; 2010.
12. Powell BJ, Waltz TJ, Chinman MJ, et al. A refined compilation of implementation strategies: results from the Expert Recommendations for Implementing Change (ERIC) project. Implement Sci. 2015;10:21.
13. Rogal SS, Yakovchenko V, Waltz TJ, et al. The association between implementation strategy use and the uptake of hepatitis C treatment in a national sample. Implement Sci.
2017;12(1):60.
14. Mellinger JL, Moser S, Welsh DE, et al. Access to subspecialty care and survival among patients with liver disease. Am J Gastroenterol. 2016;111(6):838-844.
15. Beste LA, Leipertz SL, Green PK, Dominitz JA, Ross D, Ioannou GN. Trends in the burden of cirrhosis and hepatocellular carcinoma by underlying liver disease in US Veterans from 2001-2013. Gastroenterology. 2015;149(6):1471-1482.e5.
16. Kaplan DE, Chapko MK, Mehta R, et al; VOCAL Study Group. Healthcare costs related to treatment of hepatocellular carcinoma among veterans with cirrhosis in the United States. Clin Gastroenterol Hepatol. 2018;16(1):106-114.
Hepatitis C virus (HCV) infection is a major public health problem in the US. Following the 2010 report of the Institute of Medicine/National Academies of Sciences, Engineering, and Medicine (NASEM) on hepatitis and liver cancer, the US Department of Health and Human Services (HHS) released the first National Viral Hepatitis Action Plan in 2011 with subsequent action plan updates for 2014-2016 and 2017-2020.1-3 A NASEM phase 2 report and the 2017-2020 HHS action plan outline a national strategy to prevent new viral hepatitis infections; reduce deaths and improve the health of people living with viral hepatitis; reduce viral hepatitis health disparities; and coordinate, monitor, and report on implementation of viral hepatitis activities.3,4 The Department of Veterans Affairs (VA) is the single largest HCV care provider in the US with about 165,000 veterans in care diagnosed with HCV in the beginning of 2014 and is a national leader in the testing and treatment of HCV.5,6
The VA’s recommendations for screening for HCV infection are in alignment with the United States Preventive Services Task Force (USPSTF) and Centers for Disease Control and Prevention (CDC) recommendations to test all veterans born between 1945 and 1965 and anyone with risk factors such as injection drug use.7-9 As of January 1, 2018, the VA had screened more than 80% of veterans in care within this highest risk birth cohort. As of January 1, 2018, more than 100,000 veterans in VA care have initiated treatment for HCV with direct-acting antivirals (DAAs) (Figure 1).
Several critical factors contributed to the VA success with HCV testing and treatment, including congressional appropriation of funding from fiscal year (FY) 2016 through FY 2018, unrestricted access to interferon-free DAA HCV treatments, and dedicated resources from the VA National Viral Hepatitis Program within the HIV, Hepatitis, and Related Conditions Programs (HHRC) in the Office of Specialty Care Services.5 In 2014, HHRC created and supported the Hepatitis Innovation Team (HIT) Collaborative, a VA process improvement initiative enabling
Veterans Integrated Service Network (VISN) -based, multidisciplinary teams to increase veterans’ access to HCV testing and treatment.
As the VA makes consistent progress toward eliminating HCV in veterans in VA care, it has become clear that achieving a cure is only a starting point in improving HCV care. Many patients with HCV infection also have advanced liver disease (ALD), or cirrhosis, which is a condition of permanent liver fibrosis that remains after the patient has been cured of HCV infection. In addition to hepatitis C, ALD also can be caused by excessive alcohol use, hepatitis B virus (HBV) infection, nonalcoholic fatty liver diseases, and several other inherited diseases. Advanced liver disease affects more than 80,000 veterans in VA care, and the HIT infrastructure provides an excellent framework to better understand and address facility-level and systemwide challenges in diagnosing, caring for, and treating veterans with ALD across the Veterans Health Administration (VHA) system.
This report will describe the elements that contributed to the success of the HIT Collaborative in redesigning care for patients affected by HCV in the VA and how these elements can be applied to improve the system of care for VHA ALD care.
Hepatitis Innovation Teams Collaborative Leadership
After the US Food and Drug Administration (FDA) approved new DAA medications to treat HCV, the VA recognized the need to mobilize the health care system quickly and allocate resources for these new, minimally toxic, and highly effective medications. Early in 2014, HHRC established the National Hepatitis C Resource Center (NHCRC), a successor program to the 4 regional hepatitis C resource centers that had addressed HCV care across the system.10 The NHCRC was charged with developing an operational strategy for VA to respond rapidly to the availability of DAAs. In collaboration with representatives from the Office of Strategic Integration | Veterans Engineering Resource Center (OSI|VERC), the NHCRC formed the HIT Collaborative Leadership Team (CLT).
The HIT CLT is responsible for executing the HIT Collaborative and uses a Lean process improvement framework focused on eliminating waste and maximizing value. Members of the CLT with expertise in facilitation, Lean process improvement, leadership, clinical knowledge, and population health management act as coaches for the VISN HITs. The CLT works to build and support the VISN HITs, identify opportunities for individual teams to improve and assist in finding the right local mix of “players” to be successful. The HIT CLT ensures all teams are functioning and working toward achieving their goals. The CLT obtains data from VA national databases, which are provided to the VISN HITs to inform and encourage continuous improvement of their strategies. Annual VA-wide aspirational goals are developed and disseminated to encourage a unified mission.
Catchment areas for each VISN include between 6 and 10 medical centers as well as outpatient and ambulatory care centers. Multidisciplinary HITs are composed of physicians, nurses, pharmacists, nurse practitioners, physician assistants, social workers, mental health and substance use providers, peer support specialists, administrators, information technology experts, and systems redesign professionals from medical centers within each VISN. Teams develop strong relationships across medical centers, implement context-specific strategies applicable to rural
and urban centers, and share expertise. In addition to intra-VISN process improvement, HITs collaborate monthly across VISNs via a virtual platform. They share strong practices, seek advice from one another, and compare outcomes on an established set of goals.
The HITs use process improvement tools to systematically assess the current steps involved in care. At the close of each year, the HITs analyze the current state of operations and set goals to improve over the following year guided by a target state map. Seed funding is provided to every VISN HIT annually to launch change initiatives. Many VISN HITs use these funds to support a VISN HIT coordinator, and HITs also use this financial support to conduct 2- to 3-day process improvement workshops and to purchase supplies, such as point-of-care testing kits. The HIT communication and work are predominantly executed virtually.
Each year, teams worked toward achieving goals set nationally. These included increasing HCV birth cohort testing and improving the percentage of patients who had SVR12 testing
(Table).
the percentage of patients who received SVR12 testing posttreatment completion was not included in the HIT Collaborative’s annual goals for the first year of the program. Recognizing this as a critical area for improvement, the HIT CLT set a goal to test 80% of all patients who completed treatment. The HITs applied Lean tools to identify and overcome gaps in the SVR12 testing process. By the end of the second year, 84% of all patients who completed treatment had been tested for SVR12.
The HITs also set specific local VISN and medical center goals, prioritizing projects that could have the greatest impact on local patient access and quality of care and build on existing strengths and address barriers. These projects encompass a wide range of areas that contribute to the overall national goals.
Focus on Lean
Lean process improvement is based on 2 key pillars: respect for people (those seeking service as customers and patients and those providing service as frontline staff and stakeholders) and continuous improvement. With Lean, personnel providing care should work to identify and eliminate waste in the system and to streamline care delivery to maximize process steps that are most valued by patients (eg, interaction with a clinical provider) and minimize those that are not valued (eg, time spent waiting to see a provider). With the knowledge that HHRC fully supports their work, HITs were encouraged to innovate based on local resources, context, and culture.
Teams receive basic training in Lean from the HIT CLT and local systems redesign specialists if available. The HITs apply the A3 structured approach to problem solving.11 The HITs follow prescribed problemsolving steps that help identify where to focus process improvement efforts, including analyzing the current state of care, outlining the target state, and prioritizing solution
approaches based on what will have the highest impact for patients.
to accommodate the outcomes they observe (Figure 2).
Innovations
Over the course of the HIT Collaborative, numerous innovations have emerged to address and mitigate barriers to HCV screening and treatment. Examples of successful innovations include the following:
- To address transportation issues, several teams developed programs specific to patients with HCV in rural locations or with limited mobility. Mobile vans and units traditionally used as mobile cardiology clinics were transformed into HCV clinics, bringing testing and treatment services directly to veterans;
- Pharmacists and social workers developed outreach strategies to locate homeless veterans, provide point-of-care testing and utilize mobile technology to concurrently enroll and link veterans to care; and
- Many liver care teams partnered with inpatient and outpatient substance use treatment clinics to provide patient education and coordinate HCV treatment.
Inter-VISN working groups developed systemwide tools to address common needs. In the program’s first year, a few medical facilities across a handful of VISNs shared local population health management systems, programming, and best practices. Over time, this working group combined the virtual networking capacity of the HIT Collaborative with technical expertise to promote rapid dissemination and uptake of a population health management system. Providers at medical centers across VA use the tools to identify veterans who should be screened and treated for HCV with the ability to continuously update information, identifying patients who do not respond to treatment or patients overdue for SVR12 testing.
Providers with experience using telehepatology formed another inter-VISN working group. These subject matter experts provided guidance to care teams interested in implementing telehealth in areas where limited local resources or knowledge had prevented them from moving forward. The ability to build a strong coalition across content areas fostered a collaborative learning environment, adaptable to implementing new processes and technologies.
In 2017, the VA made significant efforts to reach out to veterans eligible for VA care who had not yet been screened or remained untreated. In May, Hepatitis Awareness Month, HITs held HCV testing and community outreach events and participated in veteran stand-downs and veteran service organization activities.
Evaluation
Since 2014, the VA has increased its HCV treatment and screening rates. To assess the components contributing to these achievements and the role of the HIT Collaborative in driving this success, a team of implementation scientists have been working with the CLT to conduct a HIT program evaluation. The goal of the evaluation is to establish the impact of the HIT Collaborative. The evaluation team catalogs the activities of the Collaborative and the HITs and assesses implementation strategies (use of specific techniques) to increase the uptake of evidence-based practices specifically related to HCV treatment.12
At the close of each FY, HCV providers and members of the HIT Collaborative are queried through an online survey to determine which strategies have been used to improve HCV care and how these strategies were associated with the HIT Collaborative. The use of more strategies was associated with more HCV treatment initiations.13 All utilized strategies were identified whether or not they were associated with treatment starts. These data are being used to understand which combinations of strategies are most effective at increasing treatment for HCV in the VA and to inform future initiatives.
Expanding the Scope
Inspired by the successful results of the HIT work in HCV and in the spirit of continuously improving health care delivery, HHRC expanded the scope of the HIT Collaborative in FY 2018 to include ALD. There are about 80,000 veterans in VA care with advanced scarring of the liver and between 10,000 to 15,000 new diagnoses each year. In addition to HCV as an etiology for ALD, cases of cirrhosis are projected to increase among veterans in care due to metabolic syndrome and alcohol use. A recent review of VA data from fiscal year 2016 found that 88.6% of ALD patients had been seen in primary care within the past 2 years, with about half (51%) seen in a gastroenterology (GI) or hepatology clinic (Personal communication, HIV, Hepatitis, and Related Conditions Program Office March 16, 2018). For patients in VA care with ALD, GI visits are associated with a lower 5-year mortality.14 Annual mortality for all ALD patients in VA is 6.2%, and of those with a hospital admission, mortality rises to 31%.15 In FY 2016, there were about 52,000 ALD-related discharges (more than 2 per patient). Of those discharges, 24% were readmitted within 30 days, with an average length of stay of 1.9 days and an estimated cost per patient of $47,000 over 3 years.16
Hepatologists from across the VA convened to identify critical opportunities for improvement for patients with ALD. Base on available evidence presented in the literature and their clinical expertise, these subject matter experts identified several areas for quality improvement, with the overarching goal to improve identification of patients with early cirrhosis and ensure appropriate linkage to care for all cirrhotic patients, thus improving quality of life and reducing mortality. Although not finalized, candidate improvement targets include consistent linkage to care and treatment for HCV and HBV, comprehensive case management, post-discharge patient follow-up, and adherence to evidence-based standards of care.
Conclusion
The VA has made great strides in nearly eliminating HCV among veterans in VA care. The national effort to redesign hepatitis care using Lean management strategies and develop local and regional teams and centralized support allowed VA to maximize available resources to achieve higher rates of HCV birth cohort testing and treatment of patients infected with HCV than has any other health care system in the US.
The HIT Collaborative has been a unique and innovative mechanism to promote directed, patient-outcome driven change in a large and dynamic health care system. It has allowed rural and urban providers to work together to develop and spread quality improvement innovations and as an integrated system to achieve national priorities. The focus of this foundational HIT structure is expanding to identifying, treat, and care for VA’s ALD population.
Hepatitis C virus (HCV) infection is a major public health problem in the US. Following the 2010 report of the Institute of Medicine/National Academies of Sciences, Engineering, and Medicine (NASEM) on hepatitis and liver cancer, the US Department of Health and Human Services (HHS) released the first National Viral Hepatitis Action Plan in 2011 with subsequent action plan updates for 2014-2016 and 2017-2020.1-3 A NASEM phase 2 report and the 2017-2020 HHS action plan outline a national strategy to prevent new viral hepatitis infections; reduce deaths and improve the health of people living with viral hepatitis; reduce viral hepatitis health disparities; and coordinate, monitor, and report on implementation of viral hepatitis activities.3,4 The Department of Veterans Affairs (VA) is the single largest HCV care provider in the US with about 165,000 veterans in care diagnosed with HCV in the beginning of 2014 and is a national leader in the testing and treatment of HCV.5,6
The VA’s recommendations for screening for HCV infection are in alignment with the United States Preventive Services Task Force (USPSTF) and Centers for Disease Control and Prevention (CDC) recommendations to test all veterans born between 1945 and 1965 and anyone with risk factors such as injection drug use.7-9 As of January 1, 2018, the VA had screened more than 80% of veterans in care within this highest risk birth cohort. As of January 1, 2018, more than 100,000 veterans in VA care have initiated treatment for HCV with direct-acting antivirals (DAAs) (Figure 1).
Several critical factors contributed to the VA success with HCV testing and treatment, including congressional appropriation of funding from fiscal year (FY) 2016 through FY 2018, unrestricted access to interferon-free DAA HCV treatments, and dedicated resources from the VA National Viral Hepatitis Program within the HIV, Hepatitis, and Related Conditions Programs (HHRC) in the Office of Specialty Care Services.5 In 2014, HHRC created and supported the Hepatitis Innovation Team (HIT) Collaborative, a VA process improvement initiative enabling
Veterans Integrated Service Network (VISN) -based, multidisciplinary teams to increase veterans’ access to HCV testing and treatment.
As the VA makes consistent progress toward eliminating HCV in veterans in VA care, it has become clear that achieving a cure is only a starting point in improving HCV care. Many patients with HCV infection also have advanced liver disease (ALD), or cirrhosis, which is a condition of permanent liver fibrosis that remains after the patient has been cured of HCV infection. In addition to hepatitis C, ALD also can be caused by excessive alcohol use, hepatitis B virus (HBV) infection, nonalcoholic fatty liver diseases, and several other inherited diseases. Advanced liver disease affects more than 80,000 veterans in VA care, and the HIT infrastructure provides an excellent framework to better understand and address facility-level and systemwide challenges in diagnosing, caring for, and treating veterans with ALD across the Veterans Health Administration (VHA) system.
This report will describe the elements that contributed to the success of the HIT Collaborative in redesigning care for patients affected by HCV in the VA and how these elements can be applied to improve the system of care for VHA ALD care.
Hepatitis Innovation Teams Collaborative Leadership
After the US Food and Drug Administration (FDA) approved new DAA medications to treat HCV, the VA recognized the need to mobilize the health care system quickly and allocate resources for these new, minimally toxic, and highly effective medications. Early in 2014, HHRC established the National Hepatitis C Resource Center (NHCRC), a successor program to the 4 regional hepatitis C resource centers that had addressed HCV care across the system.10 The NHCRC was charged with developing an operational strategy for VA to respond rapidly to the availability of DAAs. In collaboration with representatives from the Office of Strategic Integration | Veterans Engineering Resource Center (OSI|VERC), the NHCRC formed the HIT Collaborative Leadership Team (CLT).
The HIT CLT is responsible for executing the HIT Collaborative and uses a Lean process improvement framework focused on eliminating waste and maximizing value. Members of the CLT with expertise in facilitation, Lean process improvement, leadership, clinical knowledge, and population health management act as coaches for the VISN HITs. The CLT works to build and support the VISN HITs, identify opportunities for individual teams to improve and assist in finding the right local mix of “players” to be successful. The HIT CLT ensures all teams are functioning and working toward achieving their goals. The CLT obtains data from VA national databases, which are provided to the VISN HITs to inform and encourage continuous improvement of their strategies. Annual VA-wide aspirational goals are developed and disseminated to encourage a unified mission.
Catchment areas for each VISN include between 6 and 10 medical centers as well as outpatient and ambulatory care centers. Multidisciplinary HITs are composed of physicians, nurses, pharmacists, nurse practitioners, physician assistants, social workers, mental health and substance use providers, peer support specialists, administrators, information technology experts, and systems redesign professionals from medical centers within each VISN. Teams develop strong relationships across medical centers, implement context-specific strategies applicable to rural
and urban centers, and share expertise. In addition to intra-VISN process improvement, HITs collaborate monthly across VISNs via a virtual platform. They share strong practices, seek advice from one another, and compare outcomes on an established set of goals.
The HITs use process improvement tools to systematically assess the current steps involved in care. At the close of each year, the HITs analyze the current state of operations and set goals to improve over the following year guided by a target state map. Seed funding is provided to every VISN HIT annually to launch change initiatives. Many VISN HITs use these funds to support a VISN HIT coordinator, and HITs also use this financial support to conduct 2- to 3-day process improvement workshops and to purchase supplies, such as point-of-care testing kits. The HIT communication and work are predominantly executed virtually.
Each year, teams worked toward achieving goals set nationally. These included increasing HCV birth cohort testing and improving the percentage of patients who had SVR12 testing
(Table).
the percentage of patients who received SVR12 testing posttreatment completion was not included in the HIT Collaborative’s annual goals for the first year of the program. Recognizing this as a critical area for improvement, the HIT CLT set a goal to test 80% of all patients who completed treatment. The HITs applied Lean tools to identify and overcome gaps in the SVR12 testing process. By the end of the second year, 84% of all patients who completed treatment had been tested for SVR12.
The HITs also set specific local VISN and medical center goals, prioritizing projects that could have the greatest impact on local patient access and quality of care and build on existing strengths and address barriers. These projects encompass a wide range of areas that contribute to the overall national goals.
Focus on Lean
Lean process improvement is based on 2 key pillars: respect for people (those seeking service as customers and patients and those providing service as frontline staff and stakeholders) and continuous improvement. With Lean, personnel providing care should work to identify and eliminate waste in the system and to streamline care delivery to maximize process steps that are most valued by patients (eg, interaction with a clinical provider) and minimize those that are not valued (eg, time spent waiting to see a provider). With the knowledge that HHRC fully supports their work, HITs were encouraged to innovate based on local resources, context, and culture.
Teams receive basic training in Lean from the HIT CLT and local systems redesign specialists if available. The HITs apply the A3 structured approach to problem solving.11 The HITs follow prescribed problemsolving steps that help identify where to focus process improvement efforts, including analyzing the current state of care, outlining the target state, and prioritizing solution
approaches based on what will have the highest impact for patients.
to accommodate the outcomes they observe (Figure 2).
Innovations
Over the course of the HIT Collaborative, numerous innovations have emerged to address and mitigate barriers to HCV screening and treatment. Examples of successful innovations include the following:
- To address transportation issues, several teams developed programs specific to patients with HCV in rural locations or with limited mobility. Mobile vans and units traditionally used as mobile cardiology clinics were transformed into HCV clinics, bringing testing and treatment services directly to veterans;
- Pharmacists and social workers developed outreach strategies to locate homeless veterans, provide point-of-care testing and utilize mobile technology to concurrently enroll and link veterans to care; and
- Many liver care teams partnered with inpatient and outpatient substance use treatment clinics to provide patient education and coordinate HCV treatment.
Inter-VISN working groups developed systemwide tools to address common needs. In the program’s first year, a few medical facilities across a handful of VISNs shared local population health management systems, programming, and best practices. Over time, this working group combined the virtual networking capacity of the HIT Collaborative with technical expertise to promote rapid dissemination and uptake of a population health management system. Providers at medical centers across VA use the tools to identify veterans who should be screened and treated for HCV with the ability to continuously update information, identifying patients who do not respond to treatment or patients overdue for SVR12 testing.
Providers with experience using telehepatology formed another inter-VISN working group. These subject matter experts provided guidance to care teams interested in implementing telehealth in areas where limited local resources or knowledge had prevented them from moving forward. The ability to build a strong coalition across content areas fostered a collaborative learning environment, adaptable to implementing new processes and technologies.
In 2017, the VA made significant efforts to reach out to veterans eligible for VA care who had not yet been screened or remained untreated. In May, Hepatitis Awareness Month, HITs held HCV testing and community outreach events and participated in veteran stand-downs and veteran service organization activities.
Evaluation
Since 2014, the VA has increased its HCV treatment and screening rates. To assess the components contributing to these achievements and the role of the HIT Collaborative in driving this success, a team of implementation scientists have been working with the CLT to conduct a HIT program evaluation. The goal of the evaluation is to establish the impact of the HIT Collaborative. The evaluation team catalogs the activities of the Collaborative and the HITs and assesses implementation strategies (use of specific techniques) to increase the uptake of evidence-based practices specifically related to HCV treatment.12
At the close of each FY, HCV providers and members of the HIT Collaborative are queried through an online survey to determine which strategies have been used to improve HCV care and how these strategies were associated with the HIT Collaborative. The use of more strategies was associated with more HCV treatment initiations.13 All utilized strategies were identified whether or not they were associated with treatment starts. These data are being used to understand which combinations of strategies are most effective at increasing treatment for HCV in the VA and to inform future initiatives.
Expanding the Scope
Inspired by the successful results of the HIT work in HCV and in the spirit of continuously improving health care delivery, HHRC expanded the scope of the HIT Collaborative in FY 2018 to include ALD. There are about 80,000 veterans in VA care with advanced scarring of the liver and between 10,000 to 15,000 new diagnoses each year. In addition to HCV as an etiology for ALD, cases of cirrhosis are projected to increase among veterans in care due to metabolic syndrome and alcohol use. A recent review of VA data from fiscal year 2016 found that 88.6% of ALD patients had been seen in primary care within the past 2 years, with about half (51%) seen in a gastroenterology (GI) or hepatology clinic (Personal communication, HIV, Hepatitis, and Related Conditions Program Office March 16, 2018). For patients in VA care with ALD, GI visits are associated with a lower 5-year mortality.14 Annual mortality for all ALD patients in VA is 6.2%, and of those with a hospital admission, mortality rises to 31%.15 In FY 2016, there were about 52,000 ALD-related discharges (more than 2 per patient). Of those discharges, 24% were readmitted within 30 days, with an average length of stay of 1.9 days and an estimated cost per patient of $47,000 over 3 years.16
Hepatologists from across the VA convened to identify critical opportunities for improvement for patients with ALD. Base on available evidence presented in the literature and their clinical expertise, these subject matter experts identified several areas for quality improvement, with the overarching goal to improve identification of patients with early cirrhosis and ensure appropriate linkage to care for all cirrhotic patients, thus improving quality of life and reducing mortality. Although not finalized, candidate improvement targets include consistent linkage to care and treatment for HCV and HBV, comprehensive case management, post-discharge patient follow-up, and adherence to evidence-based standards of care.
Conclusion
The VA has made great strides in nearly eliminating HCV among veterans in VA care. The national effort to redesign hepatitis care using Lean management strategies and develop local and regional teams and centralized support allowed VA to maximize available resources to achieve higher rates of HCV birth cohort testing and treatment of patients infected with HCV than has any other health care system in the US.
The HIT Collaborative has been a unique and innovative mechanism to promote directed, patient-outcome driven change in a large and dynamic health care system. It has allowed rural and urban providers to work together to develop and spread quality improvement innovations and as an integrated system to achieve national priorities. The focus of this foundational HIT structure is expanding to identifying, treat, and care for VA’s ALD population.
1. Colvin HM, Mitchell AE, eds; and the Committee on the Prevention and Control of Viral Hepatitis Infections Board on Population Health and Public Health Practice. Hepatitis and Liver Cancer: A National Strategy for Prevention and Control of Hepatitis B and C. Washington, DC: The National Academies Press; 2010.
2. US Department of Health and Human Services. Combating the silent epidemic of viral hepatitis: action plan for the prevention, care and treatment of viral hepatitis. https://www.hhs.gov/sites/default/files/action-plan-viral-hepatitis-2011.pdf. Accessed April 27, 2018.
3. Wolitski R. National viral hepatitis action plan: 2017-2020. https://www.hhs.gov/hepatitis/action-plan/national-viralhepatitis-action-plan-overview/index.html. Updated February
21, 2018. Accessed May 8, 2018.
4. National Academies of Sciences, Engineering, and Medicine. A National Strategy for the Elimination of Hepatitis B and C: Phase Two Report. Washington, DC: The National Academies Press; 2017.
5. Belperio PS, Chartier M, Ross DB, Alaigh P, Shulkin D. Curing hepatitis C infection: best practices from the Department of Veterans Affairs. Ann of Intern Med. 2017;167(7):499-504.
6. Kushner T, Serper M, Kaplan DE. Delta hepatitis within the Veterans Affairs medical system in the United States: prevalence, risk factors, and outcomes. J Hepatol. 2015;63(3):586-592.
7. US Department of Veterans Affairs, Veteran Health Administration. National Clinical Preventive Service Guidance Statements: Screening for Hepatitis C. http://www.prevention.va.gov/CPS/Screening_for_Hepatitis_C.asp. Published on June 20, 2017. [Nonpubic document; source not verified.]
8. Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
9. Smith BD, Morgan RL, Beckett GA, et al; Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945-1965. MMWR Recomm Rep. 2012;61(RR-4):1-32.
10. Garrard J, Choudary V, Groom H, et al. Organizational change in management of hepatitis C: evaluation of a CME program. J Contin Educ Health Prof. 2006;26(2):145-160.
11. Shook J. Managing to Learn: Using the A3 Management Process to Solve Problems, Gain Agreement, Mentor, and Lead. Cambridge, MA: Lean Enterprise Institute; 2010.
12. Powell BJ, Waltz TJ, Chinman MJ, et al. A refined compilation of implementation strategies: results from the Expert Recommendations for Implementing Change (ERIC) project. Implement Sci. 2015;10:21.
13. Rogal SS, Yakovchenko V, Waltz TJ, et al. The association between implementation strategy use and the uptake of hepatitis C treatment in a national sample. Implement Sci.
2017;12(1):60.
14. Mellinger JL, Moser S, Welsh DE, et al. Access to subspecialty care and survival among patients with liver disease. Am J Gastroenterol. 2016;111(6):838-844.
15. Beste LA, Leipertz SL, Green PK, Dominitz JA, Ross D, Ioannou GN. Trends in the burden of cirrhosis and hepatocellular carcinoma by underlying liver disease in US Veterans from 2001-2013. Gastroenterology. 2015;149(6):1471-1482.e5.
16. Kaplan DE, Chapko MK, Mehta R, et al; VOCAL Study Group. Healthcare costs related to treatment of hepatocellular carcinoma among veterans with cirrhosis in the United States. Clin Gastroenterol Hepatol. 2018;16(1):106-114.
1. Colvin HM, Mitchell AE, eds; and the Committee on the Prevention and Control of Viral Hepatitis Infections Board on Population Health and Public Health Practice. Hepatitis and Liver Cancer: A National Strategy for Prevention and Control of Hepatitis B and C. Washington, DC: The National Academies Press; 2010.
2. US Department of Health and Human Services. Combating the silent epidemic of viral hepatitis: action plan for the prevention, care and treatment of viral hepatitis. https://www.hhs.gov/sites/default/files/action-plan-viral-hepatitis-2011.pdf. Accessed April 27, 2018.
3. Wolitski R. National viral hepatitis action plan: 2017-2020. https://www.hhs.gov/hepatitis/action-plan/national-viralhepatitis-action-plan-overview/index.html. Updated February
21, 2018. Accessed May 8, 2018.
4. National Academies of Sciences, Engineering, and Medicine. A National Strategy for the Elimination of Hepatitis B and C: Phase Two Report. Washington, DC: The National Academies Press; 2017.
5. Belperio PS, Chartier M, Ross DB, Alaigh P, Shulkin D. Curing hepatitis C infection: best practices from the Department of Veterans Affairs. Ann of Intern Med. 2017;167(7):499-504.
6. Kushner T, Serper M, Kaplan DE. Delta hepatitis within the Veterans Affairs medical system in the United States: prevalence, risk factors, and outcomes. J Hepatol. 2015;63(3):586-592.
7. US Department of Veterans Affairs, Veteran Health Administration. National Clinical Preventive Service Guidance Statements: Screening for Hepatitis C. http://www.prevention.va.gov/CPS/Screening_for_Hepatitis_C.asp. Published on June 20, 2017. [Nonpubic document; source not verified.]
8. Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
9. Smith BD, Morgan RL, Beckett GA, et al; Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945-1965. MMWR Recomm Rep. 2012;61(RR-4):1-32.
10. Garrard J, Choudary V, Groom H, et al. Organizational change in management of hepatitis C: evaluation of a CME program. J Contin Educ Health Prof. 2006;26(2):145-160.
11. Shook J. Managing to Learn: Using the A3 Management Process to Solve Problems, Gain Agreement, Mentor, and Lead. Cambridge, MA: Lean Enterprise Institute; 2010.
12. Powell BJ, Waltz TJ, Chinman MJ, et al. A refined compilation of implementation strategies: results from the Expert Recommendations for Implementing Change (ERIC) project. Implement Sci. 2015;10:21.
13. Rogal SS, Yakovchenko V, Waltz TJ, et al. The association between implementation strategy use and the uptake of hepatitis C treatment in a national sample. Implement Sci.
2017;12(1):60.
14. Mellinger JL, Moser S, Welsh DE, et al. Access to subspecialty care and survival among patients with liver disease. Am J Gastroenterol. 2016;111(6):838-844.
15. Beste LA, Leipertz SL, Green PK, Dominitz JA, Ross D, Ioannou GN. Trends in the burden of cirrhosis and hepatocellular carcinoma by underlying liver disease in US Veterans from 2001-2013. Gastroenterology. 2015;149(6):1471-1482.e5.
16. Kaplan DE, Chapko MK, Mehta R, et al; VOCAL Study Group. Healthcare costs related to treatment of hepatocellular carcinoma among veterans with cirrhosis in the United States. Clin Gastroenterol Hepatol. 2018;16(1):106-114.
Outcomes After Peripheral Nerve Block in Hip Arthroscopy
ABSTRACT
Pain control following hip arthroscopy presents a significant clinical challenge, with postoperative pain requiring considerable opioid use. Peripheral nerve blocks (PNBs) have emerged as one option to improve pain and limit the consequences of opioid use. The purpose of this study is to provide a comprehensive review of outcomes associated with PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications. A systematic review of PubMed, Medline, Scopus, and Embase databases was conducted through January 2015 for English-language articles reporting outcome data, with 2 reviewers independently reviewing studies for inclusion. When available, similar outcomes were combined to generate frequency-weighted means. Six studies met the inclusion criteria for this review, reporting on 710 patients undergoing hip arthroscopy. The mean ages were 37.0 and 37.7 years for the PNB and comparator groups, respectively, with a reported total of 281 (40.5%) male and 412 (59.5%) female patients. Postoperative post-anesthesia care unit (PACU) pain was consistently reduced in the PNB group, with the use of a lower morphine equivalent dose and lower rates of inpatient admission, compared with that in the control groups. Postoperative nausea and/or vomiting as well as PACU discharge time showed mixed results. High satisfaction and few complications were reported. In conclusion, PNB is associated with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions, though similar rates of nausea/vomiting and time to discharge were reported. Current PNB techniques are varied, and future research efforts should focus on examining which of these methods provides the optimal risk-benefit profile in hip arthroscopy.
Continue to: Hip arthroscopy has emerged...
Hip arthroscopy has emerged as a useful procedure in the diagnosis and treatment of hip pathology,1-8 experiencing a substantial rise in popularity in recent years, with the number of procedures growing by a factor of 18 from 1999 to 20099 and 25 from 2006 to 2013.10 Though hip arthroscopy is beneficial in many cases, marked postoperative pain has presented a substantial challenge, with patients requiring considerable doses of opiate-based medications in the post-anesthesia care unit (PACU).11,12 Increased narcotic use carries increased side effects, including postoperative nausea and vomiting,13 and poorly managed pain leads to increased unplanned admissions.14 Furthermore, patients with chronic hip pain and long-term opioid use may experience heightened and prolonged pain following the procedure, owing to medication tolerance and reduced opioid efficacy in this setting.15
Several pain control strategies have been employed in patients undergoing hip arthroscopy. General anesthesia16,17 and combined spinal epidural (CSE)18 are commonly used. However, such techniques rely heavily on opioids for postoperative pain control,11 and epidural anesthesia commonly requires adjunctive treatments (eg, neuromuscular blockade) to ensure muscle relaxation for joint distraction.19 One technique that has been employed recently is peripheral nerve block (PNB), which has been associated with a significant decrease in postoperative opioid use and nausea and vomiting.13,20 This method has proven successful in other fields of arthroscopy, including shoulder arthroscopy, in which it resulted in faster recovery, reduced opioid consumption,21 and demonstrated cost-effectiveness22 compared with general anesthesia and knee arthroscopy.23-26 As it is a relatively new field, little is known about the use of PNB in hip arthroscopy.
The goal of this systematic review was to comprehensively review the studies reporting on PNB in hip arthroscopy. We specifically focused on outcomes, including postoperative pain; analgesic use; nausea, vomiting, and antiemetic use; discharge time; inpatient admission; and patient satisfaction, as well as the complications associated with the use of PNB. Our knowledge of outcomes associated with PNB in hip arthroscopy is based on a few individual studies that have reported on small groups of patients using a variety of outcome measures and other findings. Furthermore, each of these studies commonly reflects the experience of an individual surgeon at a single institution and, when taken alone, may not be an accurate representation of the more general outcomes associated with PNB. A comprehensive review of such studies will provide surgeons, anesthesiologists, and patients with a better understanding of the anticipated outcomes of using PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications.
MATERIALS AND METHODS
A systematic review of outcomes associated with PNB in hip arthroscopy was performed using the available English-language literature in accordance with the guidelines laid out by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement and included studies retrieved from the PubMed, Medline, Scopus, and Embase computerized literature databases. Searches were executed comprising all years from database inception through January 2015. Articles were retrieved by an electronic search of medical subject headings and keyword terms and their respective combinations (Table 1). The inclusion criteria for studies in this systematic review were studies that (1) were written in the English language and (2) reported explicit outcome data. The exclusion criteria were (1) review articles, meta-analyses, case reports, abstracts/conference papers, comments/letters, or technique articles without reported patient data and (2) basic research, biomechanics, or animal/cadaveric studies without reported patient data.
Table 1. Search Terms Entered to Identify English-Language Studies Through January 2015
Database | Search terms |
PubMed, Scopus | Keyword: (hip AND arthroscopy) AND (pain control OR pain management OR pain regimen OR nerve block OR spinal anesthesia OR regional anesthesia OR general anesthesia) |
Medline | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “Anesthesia, General” OR “Anesthesia” OR “Anesthesia, Inhalation”, OR “Balanced Anesthesia” OR “Anesthesia, Local” OR “Anesthesia, Spinal” OR “Anesthesia, Conduction” OR “Nerve Block”) |
Embase | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “General Anesthesia” OR “Anesthesia” OR “Inhalation Anesthesia”, OR “Balanced Anesthesia” OR “Local Anesthesia” OR “Spinal Anesthesia” OR “Regional Anesthesia” OR “Nerve Block”) |
The literature search strategy is outlined in the Figure. The initial title search yielded a subset of possible articles that were then further included or excluded on the basis of the contents of the article’s abstract, wherein articles were again selected on the basis of the aforementioned inclusion and exclusion criteria. Articles selected in both the title and abstract phases underwent full-text review, during which the full text of each qualifying article was reviewed. In addition, the reference sections from articles undergoing full-text review were scanned to identify any additional studies that had not been identified in the original literature search. Appropriate studies for final inclusion were then selected at this stage. The title, abstract, and full-text selection process were performed by 2 of the study authors (Dr. Steinhaus and Dr. Lynch), with any discrepancies being discussed and resolved by mutual agreement.
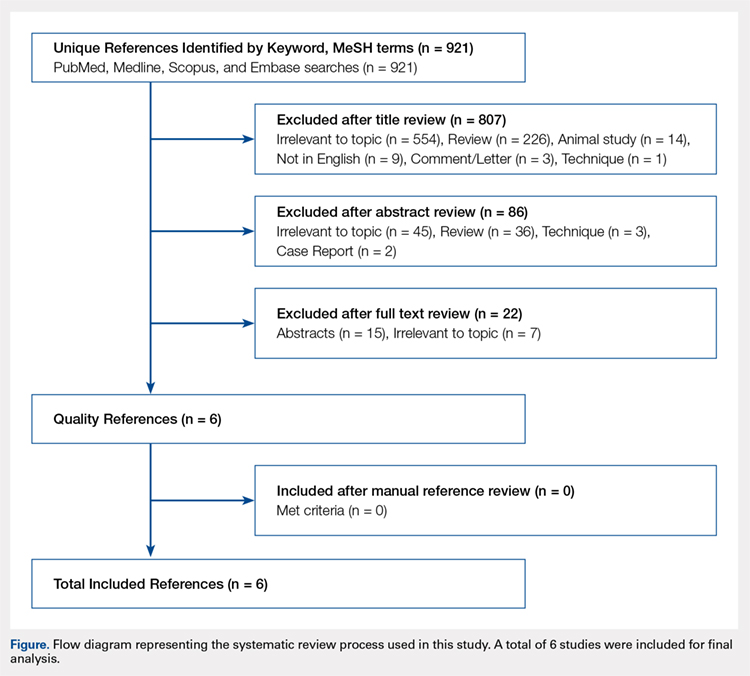
Continue to: For all 6 included studies...
For all 6 included studies,16-18,27-29 data were collected regarding the study specifics, patients included, and outcomes measured in the study. The journal of publication, type of study, level of evidence, and type of PNB, as well as the presence of a comparator group were noted (Table 2). Patient information included the number of patients at baseline and follow-up, mean age, gender, weight, height, body mass index, American Society of Anesthesiologists (ASA) status, and the specific procedures performed. In addition, data were collected on outcomes, including postoperative pain, as well as secondary outcomes and additional findings reported by the studies (Table 3). Where possible, weighted averages were calculated across all studies to obtain aggregate data.
RESULTS
STUDY INCLUSION
Six studies, all published between 2012 and 2014, were included in this systematic review (Table 2). Three studies involved lumbar plexus block, 2 studies involved femoral nerve block, and 1 study evaluated fascia iliaca block. Two studies used a control group of patients who received only general anesthesia (compared with the treatment group who received both general anesthesia and PNB); another study compared intravenous morphine with PNB; and 1 study compared CSE alone with PNB in addition to epidural.
DEMOGRAPHIC DATA
Demographic data from the included studies are presented in Table 2. In total, 710 and 549 patients were evaluated at baseline and final follow-up, respectively, which represents a follow-up rate of 77%. The frequency-weighted mean age of patients receiving PNB was 37.0 years compared with 37.7 years in the comparison groups, and the studies reported a total of 281 (40.5%) male and 412 (59.5%) female patients. The procedures performed were heterogeneously reported; therefore, totals were not tabulated, although the reported procedures included osteochondroplasty, labral débridement, labral and/or capsular repair, gluteus minimus repair, and synovectomy.
POSTOPERATIVE PAIN
Four studies reported on postoperative pain, and these data are presented in Table 3. In a retrospective study of patients receiving femoral nerve block in addition to general anesthesia, Dold and colleagues16 noted postoperative pain at 0, 15, 30, 45, and 60 minutes following arrival in the PACU, and discovered a statistically significantly lower level of pain at 60 minutes compared with inpatients receiving general anesthesia alone. YaDeau and colleagues18 found a significantly lower level of pain at rest in the PACU for those receiving CSE and lumbar plexus blockade compared with those receiving CSE alone. This significant difference did not persist at 24 hours or 6 months after the procedure, nor did it exist for pain with movement at any time point. Similarly, Schroeder and colleagues17 examined patients receiving general anesthesia and lumbar plexus block and found a significant reduction in pain immediately postoperatively in the PACU, though these effects disappeared the day following the procedure. Krych and colleagues27 also reported on postoperative pain in patients undergoing fascia iliaca blockade, although they did not include a comparator group. Outcome comparison between patients who received PNB and controls in the PACU and 1 day following the procedure are presented in Table 4.
ANALGESIC USE
Four studies reported on analgesic use after PNB, and these data are presented in Table 3. Dold and colleagues16 noted analgesic use intraoperatively, in the PACU, and in the surgical day care unit (SDCU). These authors found a significant reduction in morphine equivalent dose given in the operating room and in the PACU in the group receiving PNB, with a nonsignificant trend toward lower use of oxycodone in the SDCU. Schroeder and colleagues17 similarly reported significant reductions in morphine equivalent dose intraoperatively and in Phase I recovery for patients receiving PNB, and these differences disappeared in Phase II recovery as well as intraoperatively if the block dose was considered. In addition, these authors found a significant reduction in the use of fentanyl and hydromorphone in the operating room in the PNB group, as well as a significant reduction in the proportion of patients receiving ketorolac in the operating room or PACU. Finally, YaDeau and colleagues18 reported total analgesic usage in the PACU among PNB patients compared with those receiving CSE alone and showed a strong trend toward reduced use in the PNB group, although this difference was not significant (P = .051). PACU analgesic use is presented in Table 4.
Continue to: Postoperative nausea...
POSTOPERATIVE NAUSEA/VOMITING AND ANTIEMETIC USE
Five studies presented data on nausea, vomiting, or antiemetic use following PNB and are shown in Table 3. YaDeau and colleagues18 reported nausea among 34% of patients in the PNB group, compared with 20% in the control group, vomiting in 2% and 7%, respectively, and antiemetic use in 12% of both groups. Dold and colleagues16 identified a similar trend, with 41.1% of patients in the PNB group and 32.5% of patients in the control group experiencing postoperative nausea or vomiting, while Krych and colleagues27 noted only 10% of PNB patients with mild nausea and none requiring antiemetic use. In their study of patients receiving PNB, Schroeder and colleagues17 found a significant reduction in antiemetic use among PNB patients compared with those receiving general anesthesia alone. Similarly, Ward and colleagues29 noted a significant difference in postoperative nausea, with 10% of patients in the PNB group experiencing postoperative nausea compared with 75% of those in the comparator group who received intravenous morphine. The mean percentage of patients experiencing postoperative nausea and/or vomiting is shown in Table 4.
DISCHARGE TIME
Four studies presented data on discharge time from the PACU and are summarized in Table 3. Three of these studies included a comparator group. Both Dold and colleagues16 and YaDeau and colleagues18 reported an increase in the time to discharge for patients receiving PNB, although these differences were not significant. The study by Ward and colleagues,29 on the other hand, noted a significant reduction in the time to discharge for the PNB group. In addition to these studies, Krych and colleagues27 examined the time from skin closure to discharge for patients receiving PNB, noting a mean 199 minutes for the patients in their study. Mean times to discharge for the PNB and control groups are presented in Table 4.
INPATIENT ADMISSION
Four studies presented data on the proportion of study participants who were admitted as inpatients, and these data are shown in Table 3. Dold and colleagues16 reported no inpatient admissions in their PNB group compared with 5.0% for the control group (both cases of pain control), while YaDeau and colleagues18 found that 3 admissions occurred, with 2 in the control group (1 for oxygen desaturation and the other for intractable pain and nausea) and 1 from the PNB group (epidural spread and urinary retention). Two additional studies reported data on PNB groups alone. Krych and colleagues27 observed no overnight admissions in their study, while Nye and colleagues28 reported 1 readmission for bilateral leg numbness and weakness due to epidural spread, which resolved following discontinuation of the block. The mean proportion of inpatient admissions is presented in Table 4.
SATISFACTION
A total of 3 studies examined patient satisfaction, and these data are presented in Table 3. In their study, Ward and colleagues29 reported a significantly greater rate of satisfaction at 1 day postoperatively among the patients in the PNB group (90%) than among patients who received intravenous morphine (25%) (P < .0001). Similarly, YaDeau and colleagues18 noted greater satisfaction among the PNB group than among the control group, with PNB patients rating their satisfaction at a mean of 8.6 and control patients at a mean of 7.9 on a 10-point scale (0-10) 24 hours postoperatively, although this difference was not significant. Finally, Krych and colleagues27 found that 67% of patients were “very satisfied” and 33% were “satisfied”, based on a Likert scale.
COMPLICATIONS
Four studies presented data on complications, and these findings are summarized in Table 3. In their work, Nye and colleagues28 reported most extensively on complications associated with PNB. Overall, the authors found a rate of significant complications of 3.8%. In terms of specific complications, they noted local anesthetic systemic toxicity (0.9%), epidural spread (0.5%), sensory or motor deficits (9.4%), falls (0.5%), and catheter issues. In their study of patients receiving PNB and CSE, YaDeau and colleagues18 identified 1 patient in the PNB group with epidural spread and urinary retention, while they noted 1 case of oxygen desaturation and another case of intractable pain and nausea in the group receiving CSE alone, all 3 of which required inpatient admission. They found no permanent adverse events attributable to the PNB. In another study, Dold and colleagues16 observed no complications in patients receiving PNB compared with those in 2 admissions in the control group for inadequate pain control. Similarly, Krych and colleagues27 identified no complications in patients who received PNB in their study.
DISCUSSION
Hip arthroscopy has experienced a substantial gain in popularity in recent years, emerging as a beneficial technique for both the diagnosis and treatment of diverse hip pathologies in patients spanning a variety of demographics. Nevertheless, postoperative pain control, as well as medication side effects and unwanted patient admissions, present major challenges to the treating surgeon. As an adjuvant measure, peripheral nerve block represents one option to improve postoperative pain management, while at the same time addressing the adverse effects of considerable opioid use, which is commonly seen in these patients. Early experience with this method in hip arthroscopy was reported in a case series by Lee and colleagues.12 In an attempt to reduce postoperative pain, as well as limit the adverse effects and delay in discharge associated with considerable opioid use in the PACU, the authors used preoperative paravertebral blocks of L1 and L2 in 2 patients requiring hip arthroscopy with encouraging results. Since then, a number of studies have attempted the use of PNB in hip arthroscopy.16-18,27-29 However, we were unable to identify any prior reviews reporting on peripheral nerve blockade in hip arthroscopy, and thus this study is unique in providing a greater understanding of the outcomes associated with PNB use.
In general, we found that PNB was associated with improved outcomes. Based on the studies included in this review, there was a statistically significantly lower level of pain in the PACU for femoral nerve block (compared with general anesthesia alone)16 and lumbar plexus blockade (compared with general anesthesia17 and CSE18 alone). Nevertheless, these effects are likely short-lived, with differences disappearing the day following the procedure. In terms of analgesic use, 2 studies report significant reductions in analgesic use intraoperatively and in the PACU/Phase I recovery,16,17 with a third reporting a strong trend toward reduced analgesic use in the PACU (P = .051).18 Finally, we report fewer admissions for the PNB group, as well as high rates of satisfaction and few complications across these studies.
Continue to: Unlike these measures...
Unlike these measures, postoperative nausea, vomiting, and antiemetic use, as well as time to discharge, showed more mixed results. With regard to nausea/vomiting, 2 studies16,18 reported nonsignificantly increased rates in the PNB group, whereas others reported significant reductions in nausea/vomiting29 and in the proportion of patients receiving antiemetics.17 Similarly, mixed results were seen in terms of patient discharge time from the PACU. Two studies16,18 reported a nonsignificant increase in time to discharge for the PNB group, while another29 noted a significant reduction for the PNB group compared with those receiving intravenous morphine. These mixed results were surprising, as we expected reductions in opioid use to result in fewer instances of nausea/vomiting and a quicker time to discharge. The reasons underlying these findings are not clear, although it has been suggested that current discharge guidelines and clinical pathways limit the ability to take advantage of the accelerated timeline offered by regional anesthesia.16,30 As experience with PNB grows, our guidelines and pathways are likely to adapt to capitalize on these advantages, and future studies may show more reliable improvements in these measures.
While rare, the risk of bleeding requiring blood transfusion following hip arthroscopy is one of the most common complications of this procedure. Though the studies included in this review did not report on the need for transfusion, a recent study by Cvetanovich and colleagues10 used a national database and found that, of patients undergoing hip arthroscopy (n = 1338), 0.4% (n = 5) had bleeding requiring a transfusion, with 0.3% (n = 4) requiring return to the operating room, similar to an earlier study by Clarke and colleagues,31 who noted bleeding from the portal site in 0.4% of hip arthroscopy patients. In terms of risk factors, Cvetanovich and colleagues10 found that ASA class, older age, and prior cardiac surgery were significantly associated with minor and overall complications, whereas both regional anesthesia/monitored anesthesia care and alcohol consumption of >2 drinks a day were significantly associated with minor complications, including bleeding requiring transfusions. They noted, however, that these risk factors accounted for only 5% of the variance in complication rates, indicating that other unidentified variables better explained the variance in complication rates. These authors concluded that complications associated with hip arthroscopy are so rare that we may not be able to predict which risk factors or anesthesia types are more likely to cause them. Further characterization of bleeding following hip arthroscopy and its associated risk factors is a valuable area for future research.
LIMITATIONS
Our study contains a number of limitations. This review included studies whose level of evidence varied from I to IV; therefore, our study is limited by any bias or heterogeneity introduced in patient recruitment, selection, variability of technique, data collection, and analysis used in these studies. This heterogeneity is most apparent in the block types and comparator groups. Furthermore, several different outcome measures were reported across the 6 studies used in this review, which decreased the relevance of any one of these individual outcomes. Finally, given the limited data that currently exist for the use of PNB in hip arthroscopy, we are unable to note meaningful differences between various types of PNBs, such as differences in postoperative pain or other measures such as quadriceps weakness, which can accompany femoral nerve block.12 While it is important to read our work with these limitations in mind, this systematic review is, to our knowledge, the only comprehensive review to date of studies reporting on PNB in hip arthroscopy, providing clinicians and patients with a greater understanding of the associated outcomes across these studies.
CONCLUSION
This systematic review shows improved outcomes and few complications with PNB use in hip arthroscopy, with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions. Although opioid use was reduced in these studies, we found similar rates of postoperative nausea/vomiting as well as similar time to discharge from the PACU, which may reflect our continued reliance on outdated discharge guidelines and clinical pathways. Current attempts to provide peripheral nerve blockade are quite varied, with studies targeting femoral nerve, fascia iliaca, L1/L2 paravertebral, and lumbar plexus blockade. Future research efforts with a large prospective trial investigating these techniques should focus on which of these PNBs presents the optimal risk-benefit profile for hip arthroscopy patients and thus appropriately address the clinical questions at hand.
This paper will be judged for the Resident Writer’s Award.
- Baber YF, Robinson AH, Villar RN. Is diagnostic arthroscopy of the hip worthwhile? A prospective review of 328 adults investigated for hip pain. J Bone Joint Surg Br. 1999;81:600-603.
- Byrd JW, Jones KS. Arthroscopic management of femoroacetabular impingement: minimum 2-year follow-up. Arthroscopy. 2011;27:1379-1388.
- Larson CM, Giveans MR. Arthroscopic management of femoroacetabular impingement: early outcomes measures. Arthroscopy. 2008;24:540-546.
- O'Leary JA, Berend K, Vail TP. The relationship between diagnosis and outcome in arthroscopy of the hip. Arthroscopy. 2001;17:181-188.
- Philippon M, Schenker M, Briggs K, Kuppersmith D. Femoroacetabular impingement in 45 professional athletes: associated pathologies and return to sport following arthroscopic decompression. Knee Surg Sports Traumatol Arthrosc. 2007;15:908-914.
- Potter BK, Freedman BA, Andersen RC, Bojescul JA, Kuklo TR, Murphy KP. Correlation of Short Form-36 and disability status with outcomes of arthroscopic acetabular labral debridement. Am J Sports Med. 2005;33:864-870.
- Robertson WJ, Kadrmas WR, Kelly BT. Arthroscopic management of labral tears in the hip: a systematic review of the literature. Clin Orthop Relat Res. 2007;455:88-92.
- Yusaf MA, Hame SL. Arthroscopy of the hip. Curr Sports Med Rep. 2008;7:269-274.
- Colvin AC, Harrast J, Harner C. Trends in hip arthroscopy. J Bone Joint Surg Am. 2012;94:e23.
- Cvetanovich GL, Chalmers PN, Levy DM, et al. Hip arthroscopy surgical volume trends and 30-day postoperative complications. Arthroscopy. 2016 Apr 8. [Epub before print].
- Baker JF, Byrne DP, Hunter K, Mulhall KJ. Post-operative opiate requirements after hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2011;19:1399-1402.
- Lee EM, Murphy KP, Ben-David B. Postoperative analgesia for hip arthroscopy: combined L1 and L2 paravertebral blocks. J Clin Anesth. 2008;20:462-465.
- Ganesh A, Rose JB, Wells L, et al. Continuous peripheral nerve blockade for inpatient and outpatient postoperative analgesia in children. Anesth Analg. 2007;105:1234-1242.
- Williams BA, Kentor ML, Vogt MT, et al. Femoral-sciatic nerve blocks for complex outpatient knee surgery are associated with less postoperative pain before same-day discharge: a review of 1,200 consecutive cases from the period 1996-1999. Anesthesiology. 2003;98:1206-1213.
- Zywiel MG, Stroh DA, Lee SY, Bonutti PM, Mont MA. Chronic opioid use prior to total knee arthroplasty. J Bone Joint Surg Am. 2011;93:1988-1993.
- Dold AP, Murnaghan L, Xing J, Abdallah FW, Brull R, Whelan DB. Preoperative femoral nerve block in hip arthroscopic surgery: a retrospective review of 108 consecutive cases. Am J Sports Med. 2014;42:144-149.
- Schroeder KM, Donnelly MJ, Anderson BM, Ford MP, Keene JS. The analgesic impact of preoperative lumbar plexus blocks for hip arthroscopy. A retrospective review. Hip Int. 2013;23:93-98.
- YaDeau JT, Tedore T, Goytizolo EA, et al. Lumbar plexus blockade reduces pain after hip arthroscopy: a prospective randomized controlled trial. Anesth Analg. 2012;115:968-972.
- Smart LR, Oetgen M, Noonan B, Medvecky M. Beginning hip arthroscopy: indications, positioning, portals, basic techniques, and complications. Arthroscopy. 2007;23:1348-1353.
- Stevens M, Harrison G, McGrail M. A modified fascia iliaca compartment block has significant morphine-sparing effect after total hip arthroplasty. Anaesth Intensive Care. 2007;35:949-952.
- Lehmann LJ, Loosen G, Weiss C, Schmittner MD. Interscalene plexus block versus general anaesthesia for shoulder surgery: a randomized controlled study. Eur J Orthop Surg Traumatol. 2015;25:255-261.
- Gonano C, Kettner SC, Ernstbrunner M, Schebesta K, Chiari A, Marhofer P. Comparison of economical aspects of interscalene brachial plexus blockade and general anaesthesia for arthroscopic shoulder surgery. Br J Anaesth. 2009;103:428-433.
- Hadzic A, Karaca PE, Hobeika P, et al. Peripheral nerve blocks result in superior recovery profile compared with general anesthesia in outpatient knee arthroscopy. Anesth Analg. 2005;100:976-981.
- Hsu LP, Oh S, Nuber GW, et al. Nerve block of the infrapatellar branch of the saphenous nerve in knee arthroscopy: a prospective, double-blinded, randomized, placebo-controlled trial. J Bone Joint Surg Am. 2013;95:1465-1472.
- Montes FR, Zarate E, Grueso R, et al. Comparison of spinal anesthesia with combined sciatic-femoral nerve block for outpatient knee arthroscopy. J Clin Anesth. 2008;20:415-420.
- Wulf H, Lowe J, Gnutzmann KH, Steinfeldt T. Femoral nerve block with ropivacaine or bupivacaine in day case anterior crucial ligament reconstruction. Acta Anaesthesiol Scand. 2010;54:414-420.
- Krych AJ, Baran S, Kuzma SA, Smith HM, Johnson RL, Levy BA. Utility of multimodal analgesia with fascia iliaca blockade for acute pain management following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22:843-847.
- Nye ZB, Horn JL, Crittenden W, Abrahams MS, Aziz MF. Ambulatory continuous posterior lumbar plexus blocks following hip arthroscopy: a review of 213 cases. J Clin Anesth. 2013;25:268-274.
- Ward JP, Albert DB, Altman R, Goldstein RY, Cuff G, Youm T. Are femoral nerve blocks effective for early postoperative pain management after hip arthroscopy? Arthroscopy. 2012;28:1064-1069.
- Liu SS, Strodtbeck WM, Richman JM, Wu CL. A comparison of regional versus general anesthesia for ambulatory anesthesia: a meta-analysis of randomized controlled trials. Anesth Analg. 2005;101:1634-1642.
- Clarke MT, Arora A, Villar RN. Hip arthroscopy: complications in 1054 cases. Clin Orthop Relat Res. 2003;406:84-88.
ABSTRACT
Pain control following hip arthroscopy presents a significant clinical challenge, with postoperative pain requiring considerable opioid use. Peripheral nerve blocks (PNBs) have emerged as one option to improve pain and limit the consequences of opioid use. The purpose of this study is to provide a comprehensive review of outcomes associated with PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications. A systematic review of PubMed, Medline, Scopus, and Embase databases was conducted through January 2015 for English-language articles reporting outcome data, with 2 reviewers independently reviewing studies for inclusion. When available, similar outcomes were combined to generate frequency-weighted means. Six studies met the inclusion criteria for this review, reporting on 710 patients undergoing hip arthroscopy. The mean ages were 37.0 and 37.7 years for the PNB and comparator groups, respectively, with a reported total of 281 (40.5%) male and 412 (59.5%) female patients. Postoperative post-anesthesia care unit (PACU) pain was consistently reduced in the PNB group, with the use of a lower morphine equivalent dose and lower rates of inpatient admission, compared with that in the control groups. Postoperative nausea and/or vomiting as well as PACU discharge time showed mixed results. High satisfaction and few complications were reported. In conclusion, PNB is associated with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions, though similar rates of nausea/vomiting and time to discharge were reported. Current PNB techniques are varied, and future research efforts should focus on examining which of these methods provides the optimal risk-benefit profile in hip arthroscopy.
Continue to: Hip arthroscopy has emerged...
Hip arthroscopy has emerged as a useful procedure in the diagnosis and treatment of hip pathology,1-8 experiencing a substantial rise in popularity in recent years, with the number of procedures growing by a factor of 18 from 1999 to 20099 and 25 from 2006 to 2013.10 Though hip arthroscopy is beneficial in many cases, marked postoperative pain has presented a substantial challenge, with patients requiring considerable doses of opiate-based medications in the post-anesthesia care unit (PACU).11,12 Increased narcotic use carries increased side effects, including postoperative nausea and vomiting,13 and poorly managed pain leads to increased unplanned admissions.14 Furthermore, patients with chronic hip pain and long-term opioid use may experience heightened and prolonged pain following the procedure, owing to medication tolerance and reduced opioid efficacy in this setting.15
Several pain control strategies have been employed in patients undergoing hip arthroscopy. General anesthesia16,17 and combined spinal epidural (CSE)18 are commonly used. However, such techniques rely heavily on opioids for postoperative pain control,11 and epidural anesthesia commonly requires adjunctive treatments (eg, neuromuscular blockade) to ensure muscle relaxation for joint distraction.19 One technique that has been employed recently is peripheral nerve block (PNB), which has been associated with a significant decrease in postoperative opioid use and nausea and vomiting.13,20 This method has proven successful in other fields of arthroscopy, including shoulder arthroscopy, in which it resulted in faster recovery, reduced opioid consumption,21 and demonstrated cost-effectiveness22 compared with general anesthesia and knee arthroscopy.23-26 As it is a relatively new field, little is known about the use of PNB in hip arthroscopy.
The goal of this systematic review was to comprehensively review the studies reporting on PNB in hip arthroscopy. We specifically focused on outcomes, including postoperative pain; analgesic use; nausea, vomiting, and antiemetic use; discharge time; inpatient admission; and patient satisfaction, as well as the complications associated with the use of PNB. Our knowledge of outcomes associated with PNB in hip arthroscopy is based on a few individual studies that have reported on small groups of patients using a variety of outcome measures and other findings. Furthermore, each of these studies commonly reflects the experience of an individual surgeon at a single institution and, when taken alone, may not be an accurate representation of the more general outcomes associated with PNB. A comprehensive review of such studies will provide surgeons, anesthesiologists, and patients with a better understanding of the anticipated outcomes of using PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications.
MATERIALS AND METHODS
A systematic review of outcomes associated with PNB in hip arthroscopy was performed using the available English-language literature in accordance with the guidelines laid out by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement and included studies retrieved from the PubMed, Medline, Scopus, and Embase computerized literature databases. Searches were executed comprising all years from database inception through January 2015. Articles were retrieved by an electronic search of medical subject headings and keyword terms and their respective combinations (Table 1). The inclusion criteria for studies in this systematic review were studies that (1) were written in the English language and (2) reported explicit outcome data. The exclusion criteria were (1) review articles, meta-analyses, case reports, abstracts/conference papers, comments/letters, or technique articles without reported patient data and (2) basic research, biomechanics, or animal/cadaveric studies without reported patient data.
Table 1. Search Terms Entered to Identify English-Language Studies Through January 2015
Database | Search terms |
PubMed, Scopus | Keyword: (hip AND arthroscopy) AND (pain control OR pain management OR pain regimen OR nerve block OR spinal anesthesia OR regional anesthesia OR general anesthesia) |
Medline | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “Anesthesia, General” OR “Anesthesia” OR “Anesthesia, Inhalation”, OR “Balanced Anesthesia” OR “Anesthesia, Local” OR “Anesthesia, Spinal” OR “Anesthesia, Conduction” OR “Nerve Block”) |
Embase | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “General Anesthesia” OR “Anesthesia” OR “Inhalation Anesthesia”, OR “Balanced Anesthesia” OR “Local Anesthesia” OR “Spinal Anesthesia” OR “Regional Anesthesia” OR “Nerve Block”) |
The literature search strategy is outlined in the Figure. The initial title search yielded a subset of possible articles that were then further included or excluded on the basis of the contents of the article’s abstract, wherein articles were again selected on the basis of the aforementioned inclusion and exclusion criteria. Articles selected in both the title and abstract phases underwent full-text review, during which the full text of each qualifying article was reviewed. In addition, the reference sections from articles undergoing full-text review were scanned to identify any additional studies that had not been identified in the original literature search. Appropriate studies for final inclusion were then selected at this stage. The title, abstract, and full-text selection process were performed by 2 of the study authors (Dr. Steinhaus and Dr. Lynch), with any discrepancies being discussed and resolved by mutual agreement.
Continue to: For all 6 included studies...
For all 6 included studies,16-18,27-29 data were collected regarding the study specifics, patients included, and outcomes measured in the study. The journal of publication, type of study, level of evidence, and type of PNB, as well as the presence of a comparator group were noted (Table 2). Patient information included the number of patients at baseline and follow-up, mean age, gender, weight, height, body mass index, American Society of Anesthesiologists (ASA) status, and the specific procedures performed. In addition, data were collected on outcomes, including postoperative pain, as well as secondary outcomes and additional findings reported by the studies (Table 3). Where possible, weighted averages were calculated across all studies to obtain aggregate data.
RESULTS
STUDY INCLUSION
Six studies, all published between 2012 and 2014, were included in this systematic review (Table 2). Three studies involved lumbar plexus block, 2 studies involved femoral nerve block, and 1 study evaluated fascia iliaca block. Two studies used a control group of patients who received only general anesthesia (compared with the treatment group who received both general anesthesia and PNB); another study compared intravenous morphine with PNB; and 1 study compared CSE alone with PNB in addition to epidural.
DEMOGRAPHIC DATA
Demographic data from the included studies are presented in Table 2. In total, 710 and 549 patients were evaluated at baseline and final follow-up, respectively, which represents a follow-up rate of 77%. The frequency-weighted mean age of patients receiving PNB was 37.0 years compared with 37.7 years in the comparison groups, and the studies reported a total of 281 (40.5%) male and 412 (59.5%) female patients. The procedures performed were heterogeneously reported; therefore, totals were not tabulated, although the reported procedures included osteochondroplasty, labral débridement, labral and/or capsular repair, gluteus minimus repair, and synovectomy.
POSTOPERATIVE PAIN
Four studies reported on postoperative pain, and these data are presented in Table 3. In a retrospective study of patients receiving femoral nerve block in addition to general anesthesia, Dold and colleagues16 noted postoperative pain at 0, 15, 30, 45, and 60 minutes following arrival in the PACU, and discovered a statistically significantly lower level of pain at 60 minutes compared with inpatients receiving general anesthesia alone. YaDeau and colleagues18 found a significantly lower level of pain at rest in the PACU for those receiving CSE and lumbar plexus blockade compared with those receiving CSE alone. This significant difference did not persist at 24 hours or 6 months after the procedure, nor did it exist for pain with movement at any time point. Similarly, Schroeder and colleagues17 examined patients receiving general anesthesia and lumbar plexus block and found a significant reduction in pain immediately postoperatively in the PACU, though these effects disappeared the day following the procedure. Krych and colleagues27 also reported on postoperative pain in patients undergoing fascia iliaca blockade, although they did not include a comparator group. Outcome comparison between patients who received PNB and controls in the PACU and 1 day following the procedure are presented in Table 4.
ANALGESIC USE
Four studies reported on analgesic use after PNB, and these data are presented in Table 3. Dold and colleagues16 noted analgesic use intraoperatively, in the PACU, and in the surgical day care unit (SDCU). These authors found a significant reduction in morphine equivalent dose given in the operating room and in the PACU in the group receiving PNB, with a nonsignificant trend toward lower use of oxycodone in the SDCU. Schroeder and colleagues17 similarly reported significant reductions in morphine equivalent dose intraoperatively and in Phase I recovery for patients receiving PNB, and these differences disappeared in Phase II recovery as well as intraoperatively if the block dose was considered. In addition, these authors found a significant reduction in the use of fentanyl and hydromorphone in the operating room in the PNB group, as well as a significant reduction in the proportion of patients receiving ketorolac in the operating room or PACU. Finally, YaDeau and colleagues18 reported total analgesic usage in the PACU among PNB patients compared with those receiving CSE alone and showed a strong trend toward reduced use in the PNB group, although this difference was not significant (P = .051). PACU analgesic use is presented in Table 4.
Continue to: Postoperative nausea...
POSTOPERATIVE NAUSEA/VOMITING AND ANTIEMETIC USE
Five studies presented data on nausea, vomiting, or antiemetic use following PNB and are shown in Table 3. YaDeau and colleagues18 reported nausea among 34% of patients in the PNB group, compared with 20% in the control group, vomiting in 2% and 7%, respectively, and antiemetic use in 12% of both groups. Dold and colleagues16 identified a similar trend, with 41.1% of patients in the PNB group and 32.5% of patients in the control group experiencing postoperative nausea or vomiting, while Krych and colleagues27 noted only 10% of PNB patients with mild nausea and none requiring antiemetic use. In their study of patients receiving PNB, Schroeder and colleagues17 found a significant reduction in antiemetic use among PNB patients compared with those receiving general anesthesia alone. Similarly, Ward and colleagues29 noted a significant difference in postoperative nausea, with 10% of patients in the PNB group experiencing postoperative nausea compared with 75% of those in the comparator group who received intravenous morphine. The mean percentage of patients experiencing postoperative nausea and/or vomiting is shown in Table 4.
DISCHARGE TIME
Four studies presented data on discharge time from the PACU and are summarized in Table 3. Three of these studies included a comparator group. Both Dold and colleagues16 and YaDeau and colleagues18 reported an increase in the time to discharge for patients receiving PNB, although these differences were not significant. The study by Ward and colleagues,29 on the other hand, noted a significant reduction in the time to discharge for the PNB group. In addition to these studies, Krych and colleagues27 examined the time from skin closure to discharge for patients receiving PNB, noting a mean 199 minutes for the patients in their study. Mean times to discharge for the PNB and control groups are presented in Table 4.
INPATIENT ADMISSION
Four studies presented data on the proportion of study participants who were admitted as inpatients, and these data are shown in Table 3. Dold and colleagues16 reported no inpatient admissions in their PNB group compared with 5.0% for the control group (both cases of pain control), while YaDeau and colleagues18 found that 3 admissions occurred, with 2 in the control group (1 for oxygen desaturation and the other for intractable pain and nausea) and 1 from the PNB group (epidural spread and urinary retention). Two additional studies reported data on PNB groups alone. Krych and colleagues27 observed no overnight admissions in their study, while Nye and colleagues28 reported 1 readmission for bilateral leg numbness and weakness due to epidural spread, which resolved following discontinuation of the block. The mean proportion of inpatient admissions is presented in Table 4.
SATISFACTION
A total of 3 studies examined patient satisfaction, and these data are presented in Table 3. In their study, Ward and colleagues29 reported a significantly greater rate of satisfaction at 1 day postoperatively among the patients in the PNB group (90%) than among patients who received intravenous morphine (25%) (P < .0001). Similarly, YaDeau and colleagues18 noted greater satisfaction among the PNB group than among the control group, with PNB patients rating their satisfaction at a mean of 8.6 and control patients at a mean of 7.9 on a 10-point scale (0-10) 24 hours postoperatively, although this difference was not significant. Finally, Krych and colleagues27 found that 67% of patients were “very satisfied” and 33% were “satisfied”, based on a Likert scale.
COMPLICATIONS
Four studies presented data on complications, and these findings are summarized in Table 3. In their work, Nye and colleagues28 reported most extensively on complications associated with PNB. Overall, the authors found a rate of significant complications of 3.8%. In terms of specific complications, they noted local anesthetic systemic toxicity (0.9%), epidural spread (0.5%), sensory or motor deficits (9.4%), falls (0.5%), and catheter issues. In their study of patients receiving PNB and CSE, YaDeau and colleagues18 identified 1 patient in the PNB group with epidural spread and urinary retention, while they noted 1 case of oxygen desaturation and another case of intractable pain and nausea in the group receiving CSE alone, all 3 of which required inpatient admission. They found no permanent adverse events attributable to the PNB. In another study, Dold and colleagues16 observed no complications in patients receiving PNB compared with those in 2 admissions in the control group for inadequate pain control. Similarly, Krych and colleagues27 identified no complications in patients who received PNB in their study.
DISCUSSION
Hip arthroscopy has experienced a substantial gain in popularity in recent years, emerging as a beneficial technique for both the diagnosis and treatment of diverse hip pathologies in patients spanning a variety of demographics. Nevertheless, postoperative pain control, as well as medication side effects and unwanted patient admissions, present major challenges to the treating surgeon. As an adjuvant measure, peripheral nerve block represents one option to improve postoperative pain management, while at the same time addressing the adverse effects of considerable opioid use, which is commonly seen in these patients. Early experience with this method in hip arthroscopy was reported in a case series by Lee and colleagues.12 In an attempt to reduce postoperative pain, as well as limit the adverse effects and delay in discharge associated with considerable opioid use in the PACU, the authors used preoperative paravertebral blocks of L1 and L2 in 2 patients requiring hip arthroscopy with encouraging results. Since then, a number of studies have attempted the use of PNB in hip arthroscopy.16-18,27-29 However, we were unable to identify any prior reviews reporting on peripheral nerve blockade in hip arthroscopy, and thus this study is unique in providing a greater understanding of the outcomes associated with PNB use.
In general, we found that PNB was associated with improved outcomes. Based on the studies included in this review, there was a statistically significantly lower level of pain in the PACU for femoral nerve block (compared with general anesthesia alone)16 and lumbar plexus blockade (compared with general anesthesia17 and CSE18 alone). Nevertheless, these effects are likely short-lived, with differences disappearing the day following the procedure. In terms of analgesic use, 2 studies report significant reductions in analgesic use intraoperatively and in the PACU/Phase I recovery,16,17 with a third reporting a strong trend toward reduced analgesic use in the PACU (P = .051).18 Finally, we report fewer admissions for the PNB group, as well as high rates of satisfaction and few complications across these studies.
Continue to: Unlike these measures...
Unlike these measures, postoperative nausea, vomiting, and antiemetic use, as well as time to discharge, showed more mixed results. With regard to nausea/vomiting, 2 studies16,18 reported nonsignificantly increased rates in the PNB group, whereas others reported significant reductions in nausea/vomiting29 and in the proportion of patients receiving antiemetics.17 Similarly, mixed results were seen in terms of patient discharge time from the PACU. Two studies16,18 reported a nonsignificant increase in time to discharge for the PNB group, while another29 noted a significant reduction for the PNB group compared with those receiving intravenous morphine. These mixed results were surprising, as we expected reductions in opioid use to result in fewer instances of nausea/vomiting and a quicker time to discharge. The reasons underlying these findings are not clear, although it has been suggested that current discharge guidelines and clinical pathways limit the ability to take advantage of the accelerated timeline offered by regional anesthesia.16,30 As experience with PNB grows, our guidelines and pathways are likely to adapt to capitalize on these advantages, and future studies may show more reliable improvements in these measures.
While rare, the risk of bleeding requiring blood transfusion following hip arthroscopy is one of the most common complications of this procedure. Though the studies included in this review did not report on the need for transfusion, a recent study by Cvetanovich and colleagues10 used a national database and found that, of patients undergoing hip arthroscopy (n = 1338), 0.4% (n = 5) had bleeding requiring a transfusion, with 0.3% (n = 4) requiring return to the operating room, similar to an earlier study by Clarke and colleagues,31 who noted bleeding from the portal site in 0.4% of hip arthroscopy patients. In terms of risk factors, Cvetanovich and colleagues10 found that ASA class, older age, and prior cardiac surgery were significantly associated with minor and overall complications, whereas both regional anesthesia/monitored anesthesia care and alcohol consumption of >2 drinks a day were significantly associated with minor complications, including bleeding requiring transfusions. They noted, however, that these risk factors accounted for only 5% of the variance in complication rates, indicating that other unidentified variables better explained the variance in complication rates. These authors concluded that complications associated with hip arthroscopy are so rare that we may not be able to predict which risk factors or anesthesia types are more likely to cause them. Further characterization of bleeding following hip arthroscopy and its associated risk factors is a valuable area for future research.
LIMITATIONS
Our study contains a number of limitations. This review included studies whose level of evidence varied from I to IV; therefore, our study is limited by any bias or heterogeneity introduced in patient recruitment, selection, variability of technique, data collection, and analysis used in these studies. This heterogeneity is most apparent in the block types and comparator groups. Furthermore, several different outcome measures were reported across the 6 studies used in this review, which decreased the relevance of any one of these individual outcomes. Finally, given the limited data that currently exist for the use of PNB in hip arthroscopy, we are unable to note meaningful differences between various types of PNBs, such as differences in postoperative pain or other measures such as quadriceps weakness, which can accompany femoral nerve block.12 While it is important to read our work with these limitations in mind, this systematic review is, to our knowledge, the only comprehensive review to date of studies reporting on PNB in hip arthroscopy, providing clinicians and patients with a greater understanding of the associated outcomes across these studies.
CONCLUSION
This systematic review shows improved outcomes and few complications with PNB use in hip arthroscopy, with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions. Although opioid use was reduced in these studies, we found similar rates of postoperative nausea/vomiting as well as similar time to discharge from the PACU, which may reflect our continued reliance on outdated discharge guidelines and clinical pathways. Current attempts to provide peripheral nerve blockade are quite varied, with studies targeting femoral nerve, fascia iliaca, L1/L2 paravertebral, and lumbar plexus blockade. Future research efforts with a large prospective trial investigating these techniques should focus on which of these PNBs presents the optimal risk-benefit profile for hip arthroscopy patients and thus appropriately address the clinical questions at hand.
This paper will be judged for the Resident Writer’s Award.
ABSTRACT
Pain control following hip arthroscopy presents a significant clinical challenge, with postoperative pain requiring considerable opioid use. Peripheral nerve blocks (PNBs) have emerged as one option to improve pain and limit the consequences of opioid use. The purpose of this study is to provide a comprehensive review of outcomes associated with PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications. A systematic review of PubMed, Medline, Scopus, and Embase databases was conducted through January 2015 for English-language articles reporting outcome data, with 2 reviewers independently reviewing studies for inclusion. When available, similar outcomes were combined to generate frequency-weighted means. Six studies met the inclusion criteria for this review, reporting on 710 patients undergoing hip arthroscopy. The mean ages were 37.0 and 37.7 years for the PNB and comparator groups, respectively, with a reported total of 281 (40.5%) male and 412 (59.5%) female patients. Postoperative post-anesthesia care unit (PACU) pain was consistently reduced in the PNB group, with the use of a lower morphine equivalent dose and lower rates of inpatient admission, compared with that in the control groups. Postoperative nausea and/or vomiting as well as PACU discharge time showed mixed results. High satisfaction and few complications were reported. In conclusion, PNB is associated with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions, though similar rates of nausea/vomiting and time to discharge were reported. Current PNB techniques are varied, and future research efforts should focus on examining which of these methods provides the optimal risk-benefit profile in hip arthroscopy.
Continue to: Hip arthroscopy has emerged...
Hip arthroscopy has emerged as a useful procedure in the diagnosis and treatment of hip pathology,1-8 experiencing a substantial rise in popularity in recent years, with the number of procedures growing by a factor of 18 from 1999 to 20099 and 25 from 2006 to 2013.10 Though hip arthroscopy is beneficial in many cases, marked postoperative pain has presented a substantial challenge, with patients requiring considerable doses of opiate-based medications in the post-anesthesia care unit (PACU).11,12 Increased narcotic use carries increased side effects, including postoperative nausea and vomiting,13 and poorly managed pain leads to increased unplanned admissions.14 Furthermore, patients with chronic hip pain and long-term opioid use may experience heightened and prolonged pain following the procedure, owing to medication tolerance and reduced opioid efficacy in this setting.15
Several pain control strategies have been employed in patients undergoing hip arthroscopy. General anesthesia16,17 and combined spinal epidural (CSE)18 are commonly used. However, such techniques rely heavily on opioids for postoperative pain control,11 and epidural anesthesia commonly requires adjunctive treatments (eg, neuromuscular blockade) to ensure muscle relaxation for joint distraction.19 One technique that has been employed recently is peripheral nerve block (PNB), which has been associated with a significant decrease in postoperative opioid use and nausea and vomiting.13,20 This method has proven successful in other fields of arthroscopy, including shoulder arthroscopy, in which it resulted in faster recovery, reduced opioid consumption,21 and demonstrated cost-effectiveness22 compared with general anesthesia and knee arthroscopy.23-26 As it is a relatively new field, little is known about the use of PNB in hip arthroscopy.
The goal of this systematic review was to comprehensively review the studies reporting on PNB in hip arthroscopy. We specifically focused on outcomes, including postoperative pain; analgesic use; nausea, vomiting, and antiemetic use; discharge time; inpatient admission; and patient satisfaction, as well as the complications associated with the use of PNB. Our knowledge of outcomes associated with PNB in hip arthroscopy is based on a few individual studies that have reported on small groups of patients using a variety of outcome measures and other findings. Furthermore, each of these studies commonly reflects the experience of an individual surgeon at a single institution and, when taken alone, may not be an accurate representation of the more general outcomes associated with PNB. A comprehensive review of such studies will provide surgeons, anesthesiologists, and patients with a better understanding of the anticipated outcomes of using PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications.
MATERIALS AND METHODS
A systematic review of outcomes associated with PNB in hip arthroscopy was performed using the available English-language literature in accordance with the guidelines laid out by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement and included studies retrieved from the PubMed, Medline, Scopus, and Embase computerized literature databases. Searches were executed comprising all years from database inception through January 2015. Articles were retrieved by an electronic search of medical subject headings and keyword terms and their respective combinations (Table 1). The inclusion criteria for studies in this systematic review were studies that (1) were written in the English language and (2) reported explicit outcome data. The exclusion criteria were (1) review articles, meta-analyses, case reports, abstracts/conference papers, comments/letters, or technique articles without reported patient data and (2) basic research, biomechanics, or animal/cadaveric studies without reported patient data.
Table 1. Search Terms Entered to Identify English-Language Studies Through January 2015
Database | Search terms |
PubMed, Scopus | Keyword: (hip AND arthroscopy) AND (pain control OR pain management OR pain regimen OR nerve block OR spinal anesthesia OR regional anesthesia OR general anesthesia) |
Medline | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “Anesthesia, General” OR “Anesthesia” OR “Anesthesia, Inhalation”, OR “Balanced Anesthesia” OR “Anesthesia, Local” OR “Anesthesia, Spinal” OR “Anesthesia, Conduction” OR “Nerve Block”) |
Embase | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “General Anesthesia” OR “Anesthesia” OR “Inhalation Anesthesia”, OR “Balanced Anesthesia” OR “Local Anesthesia” OR “Spinal Anesthesia” OR “Regional Anesthesia” OR “Nerve Block”) |
The literature search strategy is outlined in the Figure. The initial title search yielded a subset of possible articles that were then further included or excluded on the basis of the contents of the article’s abstract, wherein articles were again selected on the basis of the aforementioned inclusion and exclusion criteria. Articles selected in both the title and abstract phases underwent full-text review, during which the full text of each qualifying article was reviewed. In addition, the reference sections from articles undergoing full-text review were scanned to identify any additional studies that had not been identified in the original literature search. Appropriate studies for final inclusion were then selected at this stage. The title, abstract, and full-text selection process were performed by 2 of the study authors (Dr. Steinhaus and Dr. Lynch), with any discrepancies being discussed and resolved by mutual agreement.
Continue to: For all 6 included studies...
For all 6 included studies,16-18,27-29 data were collected regarding the study specifics, patients included, and outcomes measured in the study. The journal of publication, type of study, level of evidence, and type of PNB, as well as the presence of a comparator group were noted (Table 2). Patient information included the number of patients at baseline and follow-up, mean age, gender, weight, height, body mass index, American Society of Anesthesiologists (ASA) status, and the specific procedures performed. In addition, data were collected on outcomes, including postoperative pain, as well as secondary outcomes and additional findings reported by the studies (Table 3). Where possible, weighted averages were calculated across all studies to obtain aggregate data.
RESULTS
STUDY INCLUSION
Six studies, all published between 2012 and 2014, were included in this systematic review (Table 2). Three studies involved lumbar plexus block, 2 studies involved femoral nerve block, and 1 study evaluated fascia iliaca block. Two studies used a control group of patients who received only general anesthesia (compared with the treatment group who received both general anesthesia and PNB); another study compared intravenous morphine with PNB; and 1 study compared CSE alone with PNB in addition to epidural.
DEMOGRAPHIC DATA
Demographic data from the included studies are presented in Table 2. In total, 710 and 549 patients were evaluated at baseline and final follow-up, respectively, which represents a follow-up rate of 77%. The frequency-weighted mean age of patients receiving PNB was 37.0 years compared with 37.7 years in the comparison groups, and the studies reported a total of 281 (40.5%) male and 412 (59.5%) female patients. The procedures performed were heterogeneously reported; therefore, totals were not tabulated, although the reported procedures included osteochondroplasty, labral débridement, labral and/or capsular repair, gluteus minimus repair, and synovectomy.
POSTOPERATIVE PAIN
Four studies reported on postoperative pain, and these data are presented in Table 3. In a retrospective study of patients receiving femoral nerve block in addition to general anesthesia, Dold and colleagues16 noted postoperative pain at 0, 15, 30, 45, and 60 minutes following arrival in the PACU, and discovered a statistically significantly lower level of pain at 60 minutes compared with inpatients receiving general anesthesia alone. YaDeau and colleagues18 found a significantly lower level of pain at rest in the PACU for those receiving CSE and lumbar plexus blockade compared with those receiving CSE alone. This significant difference did not persist at 24 hours or 6 months after the procedure, nor did it exist for pain with movement at any time point. Similarly, Schroeder and colleagues17 examined patients receiving general anesthesia and lumbar plexus block and found a significant reduction in pain immediately postoperatively in the PACU, though these effects disappeared the day following the procedure. Krych and colleagues27 also reported on postoperative pain in patients undergoing fascia iliaca blockade, although they did not include a comparator group. Outcome comparison between patients who received PNB and controls in the PACU and 1 day following the procedure are presented in Table 4.
ANALGESIC USE
Four studies reported on analgesic use after PNB, and these data are presented in Table 3. Dold and colleagues16 noted analgesic use intraoperatively, in the PACU, and in the surgical day care unit (SDCU). These authors found a significant reduction in morphine equivalent dose given in the operating room and in the PACU in the group receiving PNB, with a nonsignificant trend toward lower use of oxycodone in the SDCU. Schroeder and colleagues17 similarly reported significant reductions in morphine equivalent dose intraoperatively and in Phase I recovery for patients receiving PNB, and these differences disappeared in Phase II recovery as well as intraoperatively if the block dose was considered. In addition, these authors found a significant reduction in the use of fentanyl and hydromorphone in the operating room in the PNB group, as well as a significant reduction in the proportion of patients receiving ketorolac in the operating room or PACU. Finally, YaDeau and colleagues18 reported total analgesic usage in the PACU among PNB patients compared with those receiving CSE alone and showed a strong trend toward reduced use in the PNB group, although this difference was not significant (P = .051). PACU analgesic use is presented in Table 4.
Continue to: Postoperative nausea...
POSTOPERATIVE NAUSEA/VOMITING AND ANTIEMETIC USE
Five studies presented data on nausea, vomiting, or antiemetic use following PNB and are shown in Table 3. YaDeau and colleagues18 reported nausea among 34% of patients in the PNB group, compared with 20% in the control group, vomiting in 2% and 7%, respectively, and antiemetic use in 12% of both groups. Dold and colleagues16 identified a similar trend, with 41.1% of patients in the PNB group and 32.5% of patients in the control group experiencing postoperative nausea or vomiting, while Krych and colleagues27 noted only 10% of PNB patients with mild nausea and none requiring antiemetic use. In their study of patients receiving PNB, Schroeder and colleagues17 found a significant reduction in antiemetic use among PNB patients compared with those receiving general anesthesia alone. Similarly, Ward and colleagues29 noted a significant difference in postoperative nausea, with 10% of patients in the PNB group experiencing postoperative nausea compared with 75% of those in the comparator group who received intravenous morphine. The mean percentage of patients experiencing postoperative nausea and/or vomiting is shown in Table 4.
DISCHARGE TIME
Four studies presented data on discharge time from the PACU and are summarized in Table 3. Three of these studies included a comparator group. Both Dold and colleagues16 and YaDeau and colleagues18 reported an increase in the time to discharge for patients receiving PNB, although these differences were not significant. The study by Ward and colleagues,29 on the other hand, noted a significant reduction in the time to discharge for the PNB group. In addition to these studies, Krych and colleagues27 examined the time from skin closure to discharge for patients receiving PNB, noting a mean 199 minutes for the patients in their study. Mean times to discharge for the PNB and control groups are presented in Table 4.
INPATIENT ADMISSION
Four studies presented data on the proportion of study participants who were admitted as inpatients, and these data are shown in Table 3. Dold and colleagues16 reported no inpatient admissions in their PNB group compared with 5.0% for the control group (both cases of pain control), while YaDeau and colleagues18 found that 3 admissions occurred, with 2 in the control group (1 for oxygen desaturation and the other for intractable pain and nausea) and 1 from the PNB group (epidural spread and urinary retention). Two additional studies reported data on PNB groups alone. Krych and colleagues27 observed no overnight admissions in their study, while Nye and colleagues28 reported 1 readmission for bilateral leg numbness and weakness due to epidural spread, which resolved following discontinuation of the block. The mean proportion of inpatient admissions is presented in Table 4.
SATISFACTION
A total of 3 studies examined patient satisfaction, and these data are presented in Table 3. In their study, Ward and colleagues29 reported a significantly greater rate of satisfaction at 1 day postoperatively among the patients in the PNB group (90%) than among patients who received intravenous morphine (25%) (P < .0001). Similarly, YaDeau and colleagues18 noted greater satisfaction among the PNB group than among the control group, with PNB patients rating their satisfaction at a mean of 8.6 and control patients at a mean of 7.9 on a 10-point scale (0-10) 24 hours postoperatively, although this difference was not significant. Finally, Krych and colleagues27 found that 67% of patients were “very satisfied” and 33% were “satisfied”, based on a Likert scale.
COMPLICATIONS
Four studies presented data on complications, and these findings are summarized in Table 3. In their work, Nye and colleagues28 reported most extensively on complications associated with PNB. Overall, the authors found a rate of significant complications of 3.8%. In terms of specific complications, they noted local anesthetic systemic toxicity (0.9%), epidural spread (0.5%), sensory or motor deficits (9.4%), falls (0.5%), and catheter issues. In their study of patients receiving PNB and CSE, YaDeau and colleagues18 identified 1 patient in the PNB group with epidural spread and urinary retention, while they noted 1 case of oxygen desaturation and another case of intractable pain and nausea in the group receiving CSE alone, all 3 of which required inpatient admission. They found no permanent adverse events attributable to the PNB. In another study, Dold and colleagues16 observed no complications in patients receiving PNB compared with those in 2 admissions in the control group for inadequate pain control. Similarly, Krych and colleagues27 identified no complications in patients who received PNB in their study.
DISCUSSION
Hip arthroscopy has experienced a substantial gain in popularity in recent years, emerging as a beneficial technique for both the diagnosis and treatment of diverse hip pathologies in patients spanning a variety of demographics. Nevertheless, postoperative pain control, as well as medication side effects and unwanted patient admissions, present major challenges to the treating surgeon. As an adjuvant measure, peripheral nerve block represents one option to improve postoperative pain management, while at the same time addressing the adverse effects of considerable opioid use, which is commonly seen in these patients. Early experience with this method in hip arthroscopy was reported in a case series by Lee and colleagues.12 In an attempt to reduce postoperative pain, as well as limit the adverse effects and delay in discharge associated with considerable opioid use in the PACU, the authors used preoperative paravertebral blocks of L1 and L2 in 2 patients requiring hip arthroscopy with encouraging results. Since then, a number of studies have attempted the use of PNB in hip arthroscopy.16-18,27-29 However, we were unable to identify any prior reviews reporting on peripheral nerve blockade in hip arthroscopy, and thus this study is unique in providing a greater understanding of the outcomes associated with PNB use.
In general, we found that PNB was associated with improved outcomes. Based on the studies included in this review, there was a statistically significantly lower level of pain in the PACU for femoral nerve block (compared with general anesthesia alone)16 and lumbar plexus blockade (compared with general anesthesia17 and CSE18 alone). Nevertheless, these effects are likely short-lived, with differences disappearing the day following the procedure. In terms of analgesic use, 2 studies report significant reductions in analgesic use intraoperatively and in the PACU/Phase I recovery,16,17 with a third reporting a strong trend toward reduced analgesic use in the PACU (P = .051).18 Finally, we report fewer admissions for the PNB group, as well as high rates of satisfaction and few complications across these studies.
Continue to: Unlike these measures...
Unlike these measures, postoperative nausea, vomiting, and antiemetic use, as well as time to discharge, showed more mixed results. With regard to nausea/vomiting, 2 studies16,18 reported nonsignificantly increased rates in the PNB group, whereas others reported significant reductions in nausea/vomiting29 and in the proportion of patients receiving antiemetics.17 Similarly, mixed results were seen in terms of patient discharge time from the PACU. Two studies16,18 reported a nonsignificant increase in time to discharge for the PNB group, while another29 noted a significant reduction for the PNB group compared with those receiving intravenous morphine. These mixed results were surprising, as we expected reductions in opioid use to result in fewer instances of nausea/vomiting and a quicker time to discharge. The reasons underlying these findings are not clear, although it has been suggested that current discharge guidelines and clinical pathways limit the ability to take advantage of the accelerated timeline offered by regional anesthesia.16,30 As experience with PNB grows, our guidelines and pathways are likely to adapt to capitalize on these advantages, and future studies may show more reliable improvements in these measures.
While rare, the risk of bleeding requiring blood transfusion following hip arthroscopy is one of the most common complications of this procedure. Though the studies included in this review did not report on the need for transfusion, a recent study by Cvetanovich and colleagues10 used a national database and found that, of patients undergoing hip arthroscopy (n = 1338), 0.4% (n = 5) had bleeding requiring a transfusion, with 0.3% (n = 4) requiring return to the operating room, similar to an earlier study by Clarke and colleagues,31 who noted bleeding from the portal site in 0.4% of hip arthroscopy patients. In terms of risk factors, Cvetanovich and colleagues10 found that ASA class, older age, and prior cardiac surgery were significantly associated with minor and overall complications, whereas both regional anesthesia/monitored anesthesia care and alcohol consumption of >2 drinks a day were significantly associated with minor complications, including bleeding requiring transfusions. They noted, however, that these risk factors accounted for only 5% of the variance in complication rates, indicating that other unidentified variables better explained the variance in complication rates. These authors concluded that complications associated with hip arthroscopy are so rare that we may not be able to predict which risk factors or anesthesia types are more likely to cause them. Further characterization of bleeding following hip arthroscopy and its associated risk factors is a valuable area for future research.
LIMITATIONS
Our study contains a number of limitations. This review included studies whose level of evidence varied from I to IV; therefore, our study is limited by any bias or heterogeneity introduced in patient recruitment, selection, variability of technique, data collection, and analysis used in these studies. This heterogeneity is most apparent in the block types and comparator groups. Furthermore, several different outcome measures were reported across the 6 studies used in this review, which decreased the relevance of any one of these individual outcomes. Finally, given the limited data that currently exist for the use of PNB in hip arthroscopy, we are unable to note meaningful differences between various types of PNBs, such as differences in postoperative pain or other measures such as quadriceps weakness, which can accompany femoral nerve block.12 While it is important to read our work with these limitations in mind, this systematic review is, to our knowledge, the only comprehensive review to date of studies reporting on PNB in hip arthroscopy, providing clinicians and patients with a greater understanding of the associated outcomes across these studies.
CONCLUSION
This systematic review shows improved outcomes and few complications with PNB use in hip arthroscopy, with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions. Although opioid use was reduced in these studies, we found similar rates of postoperative nausea/vomiting as well as similar time to discharge from the PACU, which may reflect our continued reliance on outdated discharge guidelines and clinical pathways. Current attempts to provide peripheral nerve blockade are quite varied, with studies targeting femoral nerve, fascia iliaca, L1/L2 paravertebral, and lumbar plexus blockade. Future research efforts with a large prospective trial investigating these techniques should focus on which of these PNBs presents the optimal risk-benefit profile for hip arthroscopy patients and thus appropriately address the clinical questions at hand.
This paper will be judged for the Resident Writer’s Award.
- Baber YF, Robinson AH, Villar RN. Is diagnostic arthroscopy of the hip worthwhile? A prospective review of 328 adults investigated for hip pain. J Bone Joint Surg Br. 1999;81:600-603.
- Byrd JW, Jones KS. Arthroscopic management of femoroacetabular impingement: minimum 2-year follow-up. Arthroscopy. 2011;27:1379-1388.
- Larson CM, Giveans MR. Arthroscopic management of femoroacetabular impingement: early outcomes measures. Arthroscopy. 2008;24:540-546.
- O'Leary JA, Berend K, Vail TP. The relationship between diagnosis and outcome in arthroscopy of the hip. Arthroscopy. 2001;17:181-188.
- Philippon M, Schenker M, Briggs K, Kuppersmith D. Femoroacetabular impingement in 45 professional athletes: associated pathologies and return to sport following arthroscopic decompression. Knee Surg Sports Traumatol Arthrosc. 2007;15:908-914.
- Potter BK, Freedman BA, Andersen RC, Bojescul JA, Kuklo TR, Murphy KP. Correlation of Short Form-36 and disability status with outcomes of arthroscopic acetabular labral debridement. Am J Sports Med. 2005;33:864-870.
- Robertson WJ, Kadrmas WR, Kelly BT. Arthroscopic management of labral tears in the hip: a systematic review of the literature. Clin Orthop Relat Res. 2007;455:88-92.
- Yusaf MA, Hame SL. Arthroscopy of the hip. Curr Sports Med Rep. 2008;7:269-274.
- Colvin AC, Harrast J, Harner C. Trends in hip arthroscopy. J Bone Joint Surg Am. 2012;94:e23.
- Cvetanovich GL, Chalmers PN, Levy DM, et al. Hip arthroscopy surgical volume trends and 30-day postoperative complications. Arthroscopy. 2016 Apr 8. [Epub before print].
- Baker JF, Byrne DP, Hunter K, Mulhall KJ. Post-operative opiate requirements after hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2011;19:1399-1402.
- Lee EM, Murphy KP, Ben-David B. Postoperative analgesia for hip arthroscopy: combined L1 and L2 paravertebral blocks. J Clin Anesth. 2008;20:462-465.
- Ganesh A, Rose JB, Wells L, et al. Continuous peripheral nerve blockade for inpatient and outpatient postoperative analgesia in children. Anesth Analg. 2007;105:1234-1242.
- Williams BA, Kentor ML, Vogt MT, et al. Femoral-sciatic nerve blocks for complex outpatient knee surgery are associated with less postoperative pain before same-day discharge: a review of 1,200 consecutive cases from the period 1996-1999. Anesthesiology. 2003;98:1206-1213.
- Zywiel MG, Stroh DA, Lee SY, Bonutti PM, Mont MA. Chronic opioid use prior to total knee arthroplasty. J Bone Joint Surg Am. 2011;93:1988-1993.
- Dold AP, Murnaghan L, Xing J, Abdallah FW, Brull R, Whelan DB. Preoperative femoral nerve block in hip arthroscopic surgery: a retrospective review of 108 consecutive cases. Am J Sports Med. 2014;42:144-149.
- Schroeder KM, Donnelly MJ, Anderson BM, Ford MP, Keene JS. The analgesic impact of preoperative lumbar plexus blocks for hip arthroscopy. A retrospective review. Hip Int. 2013;23:93-98.
- YaDeau JT, Tedore T, Goytizolo EA, et al. Lumbar plexus blockade reduces pain after hip arthroscopy: a prospective randomized controlled trial. Anesth Analg. 2012;115:968-972.
- Smart LR, Oetgen M, Noonan B, Medvecky M. Beginning hip arthroscopy: indications, positioning, portals, basic techniques, and complications. Arthroscopy. 2007;23:1348-1353.
- Stevens M, Harrison G, McGrail M. A modified fascia iliaca compartment block has significant morphine-sparing effect after total hip arthroplasty. Anaesth Intensive Care. 2007;35:949-952.
- Lehmann LJ, Loosen G, Weiss C, Schmittner MD. Interscalene plexus block versus general anaesthesia for shoulder surgery: a randomized controlled study. Eur J Orthop Surg Traumatol. 2015;25:255-261.
- Gonano C, Kettner SC, Ernstbrunner M, Schebesta K, Chiari A, Marhofer P. Comparison of economical aspects of interscalene brachial plexus blockade and general anaesthesia for arthroscopic shoulder surgery. Br J Anaesth. 2009;103:428-433.
- Hadzic A, Karaca PE, Hobeika P, et al. Peripheral nerve blocks result in superior recovery profile compared with general anesthesia in outpatient knee arthroscopy. Anesth Analg. 2005;100:976-981.
- Hsu LP, Oh S, Nuber GW, et al. Nerve block of the infrapatellar branch of the saphenous nerve in knee arthroscopy: a prospective, double-blinded, randomized, placebo-controlled trial. J Bone Joint Surg Am. 2013;95:1465-1472.
- Montes FR, Zarate E, Grueso R, et al. Comparison of spinal anesthesia with combined sciatic-femoral nerve block for outpatient knee arthroscopy. J Clin Anesth. 2008;20:415-420.
- Wulf H, Lowe J, Gnutzmann KH, Steinfeldt T. Femoral nerve block with ropivacaine or bupivacaine in day case anterior crucial ligament reconstruction. Acta Anaesthesiol Scand. 2010;54:414-420.
- Krych AJ, Baran S, Kuzma SA, Smith HM, Johnson RL, Levy BA. Utility of multimodal analgesia with fascia iliaca blockade for acute pain management following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22:843-847.
- Nye ZB, Horn JL, Crittenden W, Abrahams MS, Aziz MF. Ambulatory continuous posterior lumbar plexus blocks following hip arthroscopy: a review of 213 cases. J Clin Anesth. 2013;25:268-274.
- Ward JP, Albert DB, Altman R, Goldstein RY, Cuff G, Youm T. Are femoral nerve blocks effective for early postoperative pain management after hip arthroscopy? Arthroscopy. 2012;28:1064-1069.
- Liu SS, Strodtbeck WM, Richman JM, Wu CL. A comparison of regional versus general anesthesia for ambulatory anesthesia: a meta-analysis of randomized controlled trials. Anesth Analg. 2005;101:1634-1642.
- Clarke MT, Arora A, Villar RN. Hip arthroscopy: complications in 1054 cases. Clin Orthop Relat Res. 2003;406:84-88.
- Baber YF, Robinson AH, Villar RN. Is diagnostic arthroscopy of the hip worthwhile? A prospective review of 328 adults investigated for hip pain. J Bone Joint Surg Br. 1999;81:600-603.
- Byrd JW, Jones KS. Arthroscopic management of femoroacetabular impingement: minimum 2-year follow-up. Arthroscopy. 2011;27:1379-1388.
- Larson CM, Giveans MR. Arthroscopic management of femoroacetabular impingement: early outcomes measures. Arthroscopy. 2008;24:540-546.
- O'Leary JA, Berend K, Vail TP. The relationship between diagnosis and outcome in arthroscopy of the hip. Arthroscopy. 2001;17:181-188.
- Philippon M, Schenker M, Briggs K, Kuppersmith D. Femoroacetabular impingement in 45 professional athletes: associated pathologies and return to sport following arthroscopic decompression. Knee Surg Sports Traumatol Arthrosc. 2007;15:908-914.
- Potter BK, Freedman BA, Andersen RC, Bojescul JA, Kuklo TR, Murphy KP. Correlation of Short Form-36 and disability status with outcomes of arthroscopic acetabular labral debridement. Am J Sports Med. 2005;33:864-870.
- Robertson WJ, Kadrmas WR, Kelly BT. Arthroscopic management of labral tears in the hip: a systematic review of the literature. Clin Orthop Relat Res. 2007;455:88-92.
- Yusaf MA, Hame SL. Arthroscopy of the hip. Curr Sports Med Rep. 2008;7:269-274.
- Colvin AC, Harrast J, Harner C. Trends in hip arthroscopy. J Bone Joint Surg Am. 2012;94:e23.
- Cvetanovich GL, Chalmers PN, Levy DM, et al. Hip arthroscopy surgical volume trends and 30-day postoperative complications. Arthroscopy. 2016 Apr 8. [Epub before print].
- Baker JF, Byrne DP, Hunter K, Mulhall KJ. Post-operative opiate requirements after hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2011;19:1399-1402.
- Lee EM, Murphy KP, Ben-David B. Postoperative analgesia for hip arthroscopy: combined L1 and L2 paravertebral blocks. J Clin Anesth. 2008;20:462-465.
- Ganesh A, Rose JB, Wells L, et al. Continuous peripheral nerve blockade for inpatient and outpatient postoperative analgesia in children. Anesth Analg. 2007;105:1234-1242.
- Williams BA, Kentor ML, Vogt MT, et al. Femoral-sciatic nerve blocks for complex outpatient knee surgery are associated with less postoperative pain before same-day discharge: a review of 1,200 consecutive cases from the period 1996-1999. Anesthesiology. 2003;98:1206-1213.
- Zywiel MG, Stroh DA, Lee SY, Bonutti PM, Mont MA. Chronic opioid use prior to total knee arthroplasty. J Bone Joint Surg Am. 2011;93:1988-1993.
- Dold AP, Murnaghan L, Xing J, Abdallah FW, Brull R, Whelan DB. Preoperative femoral nerve block in hip arthroscopic surgery: a retrospective review of 108 consecutive cases. Am J Sports Med. 2014;42:144-149.
- Schroeder KM, Donnelly MJ, Anderson BM, Ford MP, Keene JS. The analgesic impact of preoperative lumbar plexus blocks for hip arthroscopy. A retrospective review. Hip Int. 2013;23:93-98.
- YaDeau JT, Tedore T, Goytizolo EA, et al. Lumbar plexus blockade reduces pain after hip arthroscopy: a prospective randomized controlled trial. Anesth Analg. 2012;115:968-972.
- Smart LR, Oetgen M, Noonan B, Medvecky M. Beginning hip arthroscopy: indications, positioning, portals, basic techniques, and complications. Arthroscopy. 2007;23:1348-1353.
- Stevens M, Harrison G, McGrail M. A modified fascia iliaca compartment block has significant morphine-sparing effect after total hip arthroplasty. Anaesth Intensive Care. 2007;35:949-952.
- Lehmann LJ, Loosen G, Weiss C, Schmittner MD. Interscalene plexus block versus general anaesthesia for shoulder surgery: a randomized controlled study. Eur J Orthop Surg Traumatol. 2015;25:255-261.
- Gonano C, Kettner SC, Ernstbrunner M, Schebesta K, Chiari A, Marhofer P. Comparison of economical aspects of interscalene brachial plexus blockade and general anaesthesia for arthroscopic shoulder surgery. Br J Anaesth. 2009;103:428-433.
- Hadzic A, Karaca PE, Hobeika P, et al. Peripheral nerve blocks result in superior recovery profile compared with general anesthesia in outpatient knee arthroscopy. Anesth Analg. 2005;100:976-981.
- Hsu LP, Oh S, Nuber GW, et al. Nerve block of the infrapatellar branch of the saphenous nerve in knee arthroscopy: a prospective, double-blinded, randomized, placebo-controlled trial. J Bone Joint Surg Am. 2013;95:1465-1472.
- Montes FR, Zarate E, Grueso R, et al. Comparison of spinal anesthesia with combined sciatic-femoral nerve block for outpatient knee arthroscopy. J Clin Anesth. 2008;20:415-420.
- Wulf H, Lowe J, Gnutzmann KH, Steinfeldt T. Femoral nerve block with ropivacaine or bupivacaine in day case anterior crucial ligament reconstruction. Acta Anaesthesiol Scand. 2010;54:414-420.
- Krych AJ, Baran S, Kuzma SA, Smith HM, Johnson RL, Levy BA. Utility of multimodal analgesia with fascia iliaca blockade for acute pain management following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22:843-847.
- Nye ZB, Horn JL, Crittenden W, Abrahams MS, Aziz MF. Ambulatory continuous posterior lumbar plexus blocks following hip arthroscopy: a review of 213 cases. J Clin Anesth. 2013;25:268-274.
- Ward JP, Albert DB, Altman R, Goldstein RY, Cuff G, Youm T. Are femoral nerve blocks effective for early postoperative pain management after hip arthroscopy? Arthroscopy. 2012;28:1064-1069.
- Liu SS, Strodtbeck WM, Richman JM, Wu CL. A comparison of regional versus general anesthesia for ambulatory anesthesia: a meta-analysis of randomized controlled trials. Anesth Analg. 2005;101:1634-1642.
- Clarke MT, Arora A, Villar RN. Hip arthroscopy: complications in 1054 cases. Clin Orthop Relat Res. 2003;406:84-88.
TAKE-HOME POINTS
- Postoperative PACU pain was consistently reduced in the PNB group.
- Patients with PNBs had lower postoperative pain medication requirements and lower rates of inpatient admission compared with controls.
- Similar rates of nausea/vomiting and time to discharge were reported for PNB patients and controls.
- PNBs are associated with high rates of satisfaction and few complications.
- Future research should focus on comparing across PNB techniques.
Open vs Percutaneous vs Arthroscopic Surgical Treatment of Lateral Epicondylitis: An Updated Systematic Review
ABSTRACT
This study was performed to compare outcomes of open, arthroscopic, and percutaneous surgical techniques for lateral epicondylitis. We searched PubMed (MEDLINE) for literature published between January 1, 2004 and May 23, 2015 using these key words: lateral epicondylitis AND (surgery OR operative OR surgical OR open OR arthroscopic OR percutaneous). Meta-analyses were performed for outcomes reported in 3 studies using 2-sample and 2-proportion Z-tests. Thirty-five studies including 1640 elbows (1055 open, 401 arthroscopic, 184 percutaneous) met the inclusion criteria. There were no differences between groups regarding duration to return to work, complication rate, or patient satisfaction. A greater proportion of patients were pain free in the open group than in the arthroscopic group (70% vs 60%). Despite the absence of a difference among techniques regarding return to work and subjective function, we recommend open débridement as the technique most likely to achieve a pain-free outcome.
Continue to: Lateral epicondylitis affects...
Lateral epicondylitis affects 1% to 3% of adults each year. Although common, symptoms of lateral epicondylitis resolve spontaneously within a year of symptom onset in 80% of cases, and only 3% of patients who seek medical treatment ultimately require surgical intervention within 2 years of symptom onset.1 Despite a relatively low percentage of patients who require surgery, Sanders and colleagues1 noted a significant increase in the rate of surgical intervention from 1.1% to 3.2% of cases in the last 15 years. Surgical intervention is generally indicated when pain and functional disability persist after 6 to 12 months of nonsurgical treatment. Traditional surgical treatment involves open release/débridement of the extensor carpi radialis (ECRB) origin; however, with the increasing prevalence of surgical intervention, surgeons have demonstrated a rising interest in less invasive techniques like arthroscopic release/débridement and percutaneous tenotomy as alternatives to traditional open débridement. While favorable results have been reported for all 3 techniques, there is no current consensus regarding the optimal surgical technique. In 2007, Lo and Safran2 reported no difference in the results of open, percutaneous, and arthroscopic techniques regarding any outcome measure in a systematic review of 33 papers. We conducted a repeat systematic review of the current literature to update Lo and Safran’s2 review and to ascertain if more recent literature demonstrates superiority of 1 technique regarding pain relief, subjective questionnaire data, subjective satisfaction, restoration of strength, and return to work. We hypothesized that return to work would be accelerated, pain decreased, and function improved in the early postoperative period in the arthroscopic and percutaneous groups, but there would be no difference in ultimate pain, functional outcome, or subjective satisfaction.
METHODS
SEARCH STRATEGY AND STUDY SELECTION
We conducted a systematic review of the literature to update the topic of surgical intervention with lateral epicondylitis since the publication of the most recent review by Lo and Safran2 in 2007, which included all relevant studies published up to 2004. To include all relevant studies published since that time, we searched PubMed (MEDLINE) for all literature published from January 1, 2004 to May 23, 2015 using the following key words: lateral epicondylitis AND (surgery OR operative OR surgical OR open OR arthroscopic OR percutaneous). General search terms were utilized to avoid unintentional exclusion of relevant studies. Two authors reviewed the abstracts of all resultant citations. Table 1 outlines the inclusion and exclusion criteria for the search. References from all included studies were reviewed for applicable articles that were not captured by the initial broad search strategy. A Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) trial flow chart shows the study selection algorithm (Figure 1).
Table 1. Inclusion and Exclusion Criteria for the Analyzed Studies
Inclusion Criteria | Exclusion Criteria |
|
|
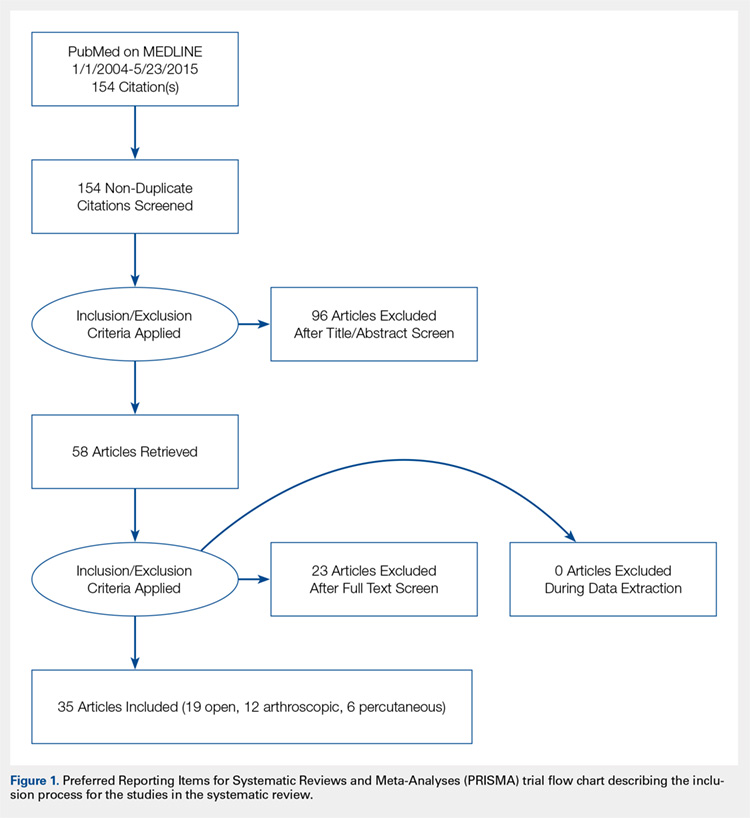
DATA EXTRACTION AND ANALYSIS
Data were extracted from the included studies by 2 reviewers using data abstraction forms. All study, subject, and surgery parameters were collected. The study and subject demographic parameters analyzed included year of publication, level of evidence, presence of study financial conflict of interest, number of subjects and elbows, gender, age, proportion in whom the dominant extremity was involved, proportion who were laborers, proportion who had a workman’s compensation claim, duration of symptoms prior to surgical intervention, and surgical technique employed (open, arthroscopic, or percutaneous). We recorded the following clinical outcomes: proportion of patients with complete pain relief, proportion who were partially or completely satisfied, proportion who were improved, duration to return to work, grip strength, Disabilities of the Arm, Shoulder, and Hand (DASH) score, visual analog scale (VAS) pain score, and complication rate.
Continue to: Statistical analysis...
STATISTICAL ANALYSIS
Data from all studies were pooled and descriptive statistics were reported as weighted mean ± weighted standard deviation for continuous variables and frequency with percentage for categorical variables. A meta-analysis was performed for all outcome measures that were reported in 3 or more studies within a specific treatment cohort. Data were analyzed using 2-sample and 2-proportion Z-tests. Results were considered statistically significant at P < .05.
RESULTS
LITERATURE RESEARCH
Using the aforementioned search strategy, 154 studies were identified. Following application of the inclusion and exclusion criteria, 35 studies were included in the analysis (Figure 1). One study compared open and percutaneous techniques, and another compared arthroscopic and percutaneous techniques, rendering a total of 19 studies examining open surgical techniques for treatment of lateral epicondylitis,3-21 12 studies examining arthroscopic techniques,14,22-32 and 6 studies reporting percutaneous surgical treatment of lateral epicondylitis29,33-37 (Table 2). There was1 level I study (3%), 6 level III studies (17%), and 28 level IV studies (80%).
Table 2. Study Demographic Data for Open, Arthroscopic, and Percutaneous Lateral Epicondylectomy
| Open | Arthroscopic | Percutaneous | Total |
Number of studies | 19 | 12 | 6 | 35 |
Level of evidence |
|
|
|
|
I | 1 (5%) | 0 | 0 | 1 (3%) |
II | 0 | 0 | 0 | 0 |
III | 3 (16%) | 4 (33%) | 1 (17%) | 6 (17%) |
IV | 15 (79%) | 8 (67%) | 5 (83%) | 28 (80%) |
US: International | 8:12 | 3:9 | 1:5 | 12:24 |
Journals of publication |
|
|
|
|
AJSM | 3 | 1 | 1 | 5 |
JSES | 2 | 2 | 1 | 5 |
Arthroscopy | 2 | 2 | 0 | 3 |
KSSTA | 1 | 2 | 0 | 3 |
CORR | 0 | 2 | 0 | 2 |
JHS | 0 | 1 | 0 | 1 |
JOS | 1 | 1 | 0 | 2 |
AJO | 2 | 0 | 0 | 2 |
Other | 8 | 1 | 4 | 12 |
Abbreviations: AJO, The American Journal of Orthopedics; AJSM, American Journal of Sports Medicine; Arthroscopy, The Journal of Arthroscopy and Related Surgery; CORR, Clinical Orthopaedics & Related Research; JHS, Journal of Hand Surgery; JOS, Journal of Orthopaedic Surgery; JSES, Journal of Shoulder and Elbow Surgery; KSSTA, Knee Surgery, Sports Traumatology, and Arthroscopy.
SUBJECT DEMOGRAPHICS
The 35 included studies comprised 1579 patients and 1640 elbows. Among these, 1055 (64%) elbows underwent open (O), 401 (25%) underwent arthroscopic (A), and 184 (11%) underwent percutaneous (P) treatment. The average age was 45.7 years, 47% of the patients were male, 43% were laborers, 31% had worker’s compensation claims, and the dominant extremity was involved in 62% of patients. The percutaneous cohort was older than the open cohort (P = 46.9, O = 45.4, A = 45.8; P = .036). The duration of symptoms was shorter in the percutaneous cohort than in the other 2 groups and shorter in the arthroscopic cohort than in the open cohort (P = 8 months, O = 23 months, A = 18 months; P < .001). There were no significant differences between groups regarding gender, occupation, worker’s compensation status, or involvement of the dominant extremity (Table 3).
Table 3. Subject Demographics for Open, Arthroscopic, and Percutaneous Groups
| Open | Arthroscopic | Percutaneous |
Subjects (N) | 999 | 397 | 183 |
Elbows (N) | 1055 | 401 | 184 |
Elbows with follow-up (%) | 915 (87%) | 350 (87%) | 181 (98%) |
Males (%) | 427 (47%) | 173 (49%) | 78 (43%) |
Females (%) | 488 (53%) | 177 (51%) | 103 (57%) |
Mean age (years) | 45.4 | 45.8 | 46.9 |
Dominant elbow (%) | 70% | 69% | 53% |
Laborer (%) | 56% | 53% | 48% |
Work comp (%) | 36% | 30% | NR |
Symptoms to operation (months) | 23 | 18 | 8 |
Min. symptoms to operation (months) | 6 | 6 | 3 |
Mean follow-up (months) | 60 | 44 | 11 |
MATA-ANALYSIS CLINICAL OUTCOMES
Clinical outcome results were pooled for all studies reporting the same outcome measure for the same technique (open, arthroscopic, or percutaneous). A meta-analysis was performed for all outcome measures that were reported in a minimum of 3 studies utilizing the same surgical technique (Table 4).
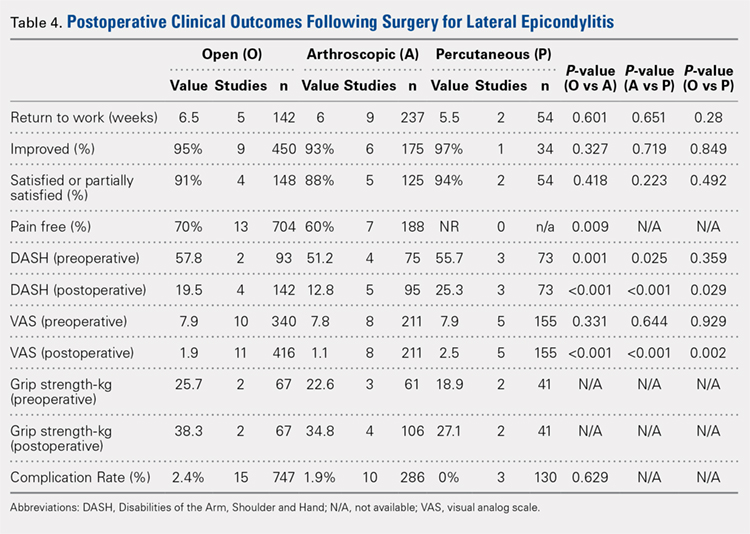
PAIN RELIEF
Thirteen open studies,3,5,7,8,11-16,18,19,21 7 arthroscopic studies14,22-24,26,27,31 and 0 percutaneous studies reported the proportion of patients who were pain free at final follow-up. The proportion of patients who were pain free following open débridement was greater than that in the arthroscopic cohort (O = 70%, A = 60%; P = .009) (Table 4).
Continue to: Subjective improvement and satisfaction...
SUBJECTIVE IMPROVEMENT AND SATISFACTION
Nine open studies, 6 arthroscopic studies, and 1 percutaneous study reported the proportion of patients who felt that their condition had been improved as a result of surgery. There was no difference in the proportion of patients who experienced improvement between the open and arthroscopic cohorts. Four open studies,3,11,12 5 arthroscopic studies,22,26,28,29,32 and 2 percutaneous studies29,36 reported the proportion of patients who were satisfied or partially satisfied with the results of the procedure. There was no difference between the open and arthroscopic groups in the proportion of patients who were satisfied or partially satisfied (Table 4).
RETURN TO WORK
The duration to return to work following surgery was reported in 5 open studies,4,5,10,13,14 9 arthroscopic studies,14,23-29,32 and 2 percutaneous studies.29,36 There was no statistically significant difference between the open and arthroscopic groups with regard to duration to return to work (O = 6.5 weeks, A = 6 weeks; P = .601). The percutaneous technique could not be included in the meta-analysis due to the presence of only 2 studies, but the pooled mean duration to return to work in these 2 studies was 5.5 weeks (Table 4).
GRIP STRENGTH
Postoperative grip strength was reported in 2 open studies,10,19 4 arthroscopic studies,28,30,32 and 2 percutaneous studies.35-36 A meta-analysis could not be performed on all the groups due to the presence of only 2 open and 2 percutaneous studies reporting grip strength. The pooled averages were O = 38.3 kg, A = 34.8 kg, and P = 27.1 kg (Table 4).
DASH SCORE
The postoperative DASH score was reported in 4 open studies,4,15,17,19,20 5 arthroscopic studies,28-31 and 3 percutaneous studies.29,33,36 At final follow-up, the mean DASH score was higher in the arthroscopic group than in the open and percutaneous groups (A = 12.8, O = 19.5, P = 25.3; P < .001 for both comparisons), and the mean DASH score was significantly higher in the open group than in the percutaneous group (P = .029). The reporting of DASH scores in the early postoperative period was not sufficiently consistent to allow us to test our hypothesis that there would be early differences in function between groups (Table 4).
VAS PAIN SCORE
Postoperative VAS pain scores were reported in 11 open studies,6,8-10,12,15,19-21 8 arthroscopic studies,24-26,29-32 and 5 percutaneous studies.29,33,35-37 At final follow-up, there was a lower mean VAS score in the arthroscopic group than in the open and percutaneous groups (A = 1.1, O = 1.9, and P = 2.5; P < .001 for both comparisons) and a lower mean VAS score in the open group than in the percutaneous group (P = .002) (Table 4). Reporting of VAS scores in the early postoperative period in the included studies wan not sufficiently consistent to allow us to test our hypothesis that there would be early differences in pain between groups.
COMPLICATIONS
The complication rate was reported in 15 open studies, 10 arthroscopic studies, and 3 percutaneous studies. There was no difference in the complication rate between the open and arthroscopic techniques (O = 2.4%, A = 1.9%; P = .629) (Table 4). Complications noted in the open cohort included superficial wound infection (6), hematoma (5), synovial fistula (2), seroma (2), and posterior interosseous nerve palsy (1). Complications noted in the arthroscopic cohort included superficial infection (3), hematoma (1), and transient paresthesia (1). Of note, there were no complications in the percutaneous group.
Continue to: Discussion...
DISCUSSION
The primary purpose of this review was to determine if definitive evidence suggests that any 1 of open, percutaneous, or arthroscopic surgical treatment is superior to the other 2 for relieving pain, improving functionality, restoring strength, or accelerating return to work. The most striking finding of this study was a significantly higher proportion of patients who were pain free at final follow-up in the open group than in the arthroscopic group (70% vs 60%, P = .009) (Table 4). At final follow-up, there were no significant differences between groups regarding duration to return to work, proportion who were improved, proportion who were satisfied or partially satisfied, and complication rate. Average VAS and DASH scores at final follow-up were lower in the arthroscopic group than in the open and percutaneous groups (Figure 2). However, although the difference between mean DASH scores in the arthroscopic and open groups (6.7 points) was statistically significant, it is likely not clinically significant, as the minimal clinically important difference (MCID) for the DASH score is 10 points, as demonstrated by Sorensen and colleagues.38 Although it has not been specifically defined for lateral epicondylitis, the MCID for VAS pain has been reported in the literature to range from 1.0 to 1.4.39-40 Therefore, as for the DASH score, the difference witnessed between the open and arthroscopic groups (0.8) is likely not clinically significant. Of note, the differences between values for arthroscopic and percutaneous techniques are greater than the MCID.

In light of a recent increase in the prevalence of surgical intervention for lateral epicondylitis, many authors have promoted arthroscopic and percutaneous techniques as alternatives to traditional open débridement with the goal of achieving the same results with decreased morbidity and accelerated return to work. Given the increased proportion of patients who were pain free at final follow-up in the open cohort, it is our contention that open release/débridement of the common extensor/ECRB origin allows the surgeon to fully appreciate the extent of tendinotic tissue that is contributing to the patient’s symptoms and to address the pathology in its entirety. Other authors have also questioned whether the full extent of extra-articular tendinosis can be accurately identified arthroscopically. Cummins41 demonstrated, in a series of 18 patients who underwent arthroscopic ECRB débridement, that 6 patients had residual tendinosis upon open evaluation and 10 had residual tendinosis on histologic assessment. Additionally, in the same series, residual tendinopathy was associated with poorer clinical outcomes.
The improved visualization associated with an open technique comes at minimal expense, as the incision was only 1.5 cm to 5 cm in 13 of 15 papers reporting incision length.3,4,6,8-11,13,15,18-20 This increased exposure may not translate into increased morbidity, as there was no increase in the duration to return to work nor the complication rate. As a result of the extensive instrumentation necessary for arthroscopic techniques, open techniques also appear to be less expensive. Analyses in the literature have suggested increased expenditures associated with arthroscopic treatment ranging from 23%42 to 100%43 greater than those of open treatment.
Although obvious, it should be noted that a percutaneous tenotomy does not permit assessment of the extent of pathologic tendinosis. As a result of an inability to visualize and débride pathologic tissue, percutaneous tenotomy rendered inferior outcomes to open and arthroscopic techniques in terms of both postoperative VAS pain score and DASH score. Nonetheless, it is a relatively rapid and simple technique and resulted in zero complications in 184 elbows. Overall, percutaneous tenotomy appears to be an inferior technique to open and arthroscopic techniques in terms of achieving complete pain relief and optimal functional recovery; however, it may be useful in those who wish to avoid a more invasive intervention.
LIMITATIONS
The most significant limitation of this study was the heterogeneity in the techniques utilized in each group. Among the 19 papers in the open cohort, 11 used techniques aimed at lengthening or release of the extensor origin, 7 performed débridement of tendinotic tissue at the ECRB origin, and 1 compared these approaches. Exposures ranged from 1.5 cm to 8 cm in length, 3 techniques added tendon repair following débridement, and 2 utilized a radiofrequency device.
Among the 12 papers in the arthroscopic cohort, 8 performed arthroscopic (inside-out) débridement of the tendinotic tissue at the ECRB origin, 3 performed arthroscopic release of the ECRB tendon, and 1 performed endoscopic ECRB release in an outside-in fashion. Four techniques added posterior synovial plica excision and 4 added decortication of the lateral epicondyle débridement or release. Some authors advocate for arthroscopic intervention on the grounds that it permits evaluation and correction of other intra-articular pathology. With this in mind, some authors have suggested that a synovial fold (plica) adjacent to the radiocapitellar joint may contribute to lateral elbow pain.27,44 Nevertheless, in the only comparative trial in the literature, Rhyou and Kim30 demonstrated that excision of posterior synovial fold failed to enhance pain relief or function in a retrospective cohort study comparing arthroscopic débridement with and without plica excision.
Continue to: Some authors advocate...
Some authors advocate decorticating the non-articular, lateral epicondyle with a shaver to stimulate bleeding and promote a healing response. However, 1 study in our review compared arthroscopic ECRB release with and without decortication and found that decortication significantly increased pain up to 4 weeks postoperatively, increased duration to return to work, and did not improve the ultimate clinical result.25 Of note, others have used a similar rationale to advocate drilling the lateral epicondyle when utilizing an open technique. However, Dunn and colleagues8 note that they have modified the Nirschl technique to eliminate drilling because they feel it increases postoperative pain and may damage the extensor digitorum communis origin.
Among the 6 papers in the percutaneous tenotomy cohort, 2 performed tenotomy with a hypodermic needle, 2 with a scalpel through a limited incision (0.5 cm-1 cm), 1 using a TX1 tissue removal system (Tenex Health), and 1 with a percutaneous radiofrequency probe. In 3 techniques, ultrasound was used to direct the tenotomy.
The quality of this review is also limited by the studies included for analysis, as with any systematic review. Because 28 of the 35 included studies were classified as evidence level IV, the likelihood of methodological bias is increased. The majority of studies contained ≥1 demonstrable biases, including selection, detection, attrition biases, or a combination. Selection bias is prevalent among predominantly level IV studies, in which the authors have selected their preferred surgical technique. There was heterogeneity in the reporting of preoperative variables and the outcome measures that were utilized. Scoring systems, such as the Nirschl Tennis Elbow Score and the Mayo Elbow Performance Index, would have been valuable in comparing the groups had they been more consistently reported. The heterogeneity in clinical outcome tools and the lack of reported outcome variance or standard deviations prevented a formal meta-analysis of some of these outcome measures. Due to inconsistent reporting, we were also unable to test our hypothesis that there would be less pain and improved function in the arthroscopic and/or percutaneous cohorts in the early postoperative period compared to the open cohort due to the less invasive techniques used. Although the differences in DASH and VAS scores at final follow-up likely did not meet the MCID threshold, these differences may have been greater and more clinically relevant in the early postoperative period.
CONCLUSION
We hypothesized that the arthroscopic and percutaneous groups would experience accelerated return to work and reduced pain in the early postoperative period but no difference in ultimate pain, functional outcome, or subjective satisfaction. There is no difference between open, arthroscopic, and percutaneous surgical treatment for lateral epicondylitis regarding return to work and subjective satisfaction; however, open treatment led to a greater percentage of patients being pain free at final follow-up. While arthroscopic treatment led to better pain and functional scores at final follow-up, the absolute differences were quite small and likely not clinically significant. In light of the available evidence, we recommend open débridement as the best means of minimizing cost and achieving a pain-free outcome in the long term. For future investigators, it would be useful to perform a randomized clinical study directly comparing open, arthroscopic, and percutaneous techniques, including assessment of pain and functional scores in the early postoperative period, and to further evaluate differences in cost among the various techniques.
This paper will be judged for the Resident Writer’s Award.
- Sanders TL Jr, Maradit Kremers H, Bryan AJ, Ransom JE, Smith J, Morrey BF. The epidemiology and health care burden of tennis elbow: a population-based study. Am J Sports Med. 2015;43(5):1066-1071. doi:10.1177/0363546514568087.
- Lo MY, Safran MR. Surgical treatment of lateral epicondylitis: a systematic review. Clin Orthop Relat Res. 2007;463:98-106. doi:10.1097/BLO.0b013e3181483dc4.
- Balk ML, Hagberg WC, Buterbaugh GA, Imbriglia JE. Outcome of surgery for lateral epicondylitis (tennis elbow): effect of worker’s compensation. Am J Orthop. 2005;34(3):122-126; discussion 126.
- Barth J, Mahieu P, Hollevoet N. Extensor tendon and fascia sectioning of extensors at the musculotendinous unit in lateral epicondylitis. Acta Orthop Belg. 2013;79(3):266-270.
- Bigorre N, Raimbeau G, Fouque PA, Cast YS, Rabarin F, Cesari B. Lateral epicondylitis treatment by extensor carpi radialis fasciotomy and radial nerve decompression: is outcome influenced by the occupational disease compensation aspect? Orthop Traumatol Surg Res. 2011;97(2):159-163. doi:10.1016/j.otsr.2010.11.007.
- Cho BK, Kim YM, Kim DS, et al. Mini-open muscle resection procedure under local anesthesia for lateral and medial epicondylitis. Clin Orthop Surg. 2009;1(3):123-127. doi:10.4055/cios.2009.1.3.123.
- Coleman B, Quinlan JF, Matheson JA. Surgical treatment for lateral epicondylitis: a long-term follow-up of results. J Shoulder Elbow Surg. 2010;19(3):363-367. doi:10.1016/j.jse.2009.09.008.
- Dunn JH, Kim JJ, Davis L, Nirschl RP. Ten- to 14-year follow-up of the Nirschl surgical technique for lateral epicondylitis. Am J Sports Med. 2008;36(2):261-266. doi:10.1177/0363546507308932.
- Manon-Matos Y, Oron A, Wolff TW. Combined common extensor and supinator aponeurotomy for the treatment of lateral epicondylitis. Tech Hand Up Extrem Surg. 2013;17(3):179-181. doi:10.1097/BTH.0b013e31829e0eeb.
- Meknas K, Odden-Miland A, Mercer JB, Castillejo M, Johansen O. Radiofrequency microtenotomy: a promising method for treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2008;36(10):1960-1965. doi:10.1177/0363546508318045.
- Pruzansky ME, Gantsoudes GD, Watters N. Late surgical results of reattachment to bone in repair of chronic lateral epicondylitis. Am J Orthop. 2009;38(6):295-299.
- Rayan F, Rao V Sr, Purushothamdas S, Mukundan C, Shafqat SO. Common extensor origin release in recalcitrant lateral epicondylitis – role justified? J Orthop Surg Res. 2010;5:31. doi:10.1186/1749-799X-5-31.
- Reddy VR, Satheesan KS, Bayliss N. Outcome of Boyd-McLeod procedure for recalcitrant lateral epicondylitis of elbow. Rheumatol Int. 2011;31(8):1081-1084. doi:10.1007/s00296-010-1450-1.
- Rubenthaler F, Wiese M, Senge A, Keller L, Wittenberg RH. Long-term follow-up of open and endoscopic Hohmann procedures for lateral epicondylitis. Arthroscopy. 2005;21(6):684-690. doi:10.1016/j.arthro.2005.03.017.
- Ruch DS, Orr SB, Richard MJ, Leversedge FJ, Mithani SK, Laino DK. A comparison of debridement with and without anconeus muscle flap for treatment of refractory lateral epicondylitis. J Shoulder Elbow Surg. 2015;24(2):236-241. doi:10.1016/j.jse.2014.09.035.
- Siddiqui MA, Koh J, Kua J, Cheung T, Chang P. Functional outcome assessment after open tennis elbow release: what are the predictor parameters? Singapore Med J. 2011;52(2):73-76.
- Solheim E, Hegna J, Øyen J. Extensor tendon release in tennis elbow: results and prognostic factors in 80 elbows. Knee Surg Sports Traumatol Arthrosc. 2011;19(6):1023-1027. doi:10.1007/s00167-011-1477-1.
- Svernlöv B, Adolfsson L. Outcome of release of the lateral extensor muscle origin for epicondylitis. Scand J Plast Reconstr Surg Hand Surg. 2006;40(3):161-165. doi:10.1080/02844310500491492.
- Tasto JP, Cummings J, Medlock V, Hardesty R, Amiel D. Microtenotomy using a radiofrequency probe to treat lateral epicondylitis. Arthroscopy. 2005;21(7):851-860. doi:10.1016/j.arthro.2005.03.019.
- Thornton SJ, Rogers JR, Prickett WD, Dunn WR, Allen AA, Hannafin JA. Treatment of recalcitrant lateral epicondylitis with suture anchor repair. Am J Sports Med. 2005;33(10):1558-1564. doi:10.1177/0363546505276758.
- Wang AW, Erak S. Fractional lengthening of forearm extensors for resistant lateral epicondylitis. ANZ J Surg. 2007;77(11):981-984. doi:10.1111/j.1445-2197.2007.04294.x.
- Baker CL Jr, Baker CL 3rd. Long-term follow-up of arthroscopic treatment of lateral epicondylitis. Am J Sports Med. 2008;36(2):254-260. doi:10.1177/0363546507311599.
- Grewal R, MacDermid JC, Shah P, King GJ. Functional outcome of arthroscopic extensor carpi radialis brevis tendon release in chronic lateral epicondylitis. J Hand Surg Am. 2009;34(5):849-857. doi:10.1016/j.jhsa.2009.02.006.
- Jerosch J, Schunck J. Arthroscopic treatment of lateral epicondylitis: indication, technique and early results. Knee Surg Sports Traumatol Arthrosc. 2006;14(4):379-382. doi:10.1007/s00167-005-0662-5.
- Kim JW, Chun CH, Shim DM, et al. Arthroscopic treatment of lateral epicondylitis: comparison of the outcome of ECRB release with and without decortication. Knee Surg Sports Traumatol Arthrosc. 2011;19(7):1178-1183. doi:10.1007/s00167-011-1507-z.
- Lattermann C, Romeo AA, Anbari A, et al. Arthroscopic debridement of the extensor carpi radialis brevis for recalcitrant lateral epicondylitis. J Shoulder Elbow Surg. 2010;19(5):651-656. doi:10.1016/j.jse.2010.02.008.
- Mullett H, Sprague M, Brown G, Hausman M. Arthroscopic treatment of lateral epicondylitis: clinical and cadaveric studies. Clin Orthop Relat Res. 2005;439:123-128. doi:10.1097/01.blo.0000176143.08886.fe.
- Oki G, Iba K, Sasaki K, Yamashita T, Wada T. Time to functional recovery after arthroscopic surgery for tennis elbow. J Shoulder Elbow Surg. 2014;23(10):1527-1531. doi:10.1016/j.jse.2014.05.010.
- Othman AM. Arthroscopic versus percutaneous release of common extensor origin for treatment of chronic tennis elbow. Arch Orthop Trauma Surg. 2011;131(3):383-388. doi:10.1007/s00402-011-1260-2.
- Rhyou IH, Kim KW. Is posterior synovial plica excision necessary for refractory lateral epicondylitis of the elbow? Clin Orthop Relat Res. 2013;471(1):284-290. doi:10.1007/s11999-012-2585-z.
- Wada T, Moriya T, Iba K, et al. Functional outcomes after arthroscopic treatment of lateral epicondylitis. J Orthop Sci. 2009;14(2):167-174. doi:10.1007/s00776-008-1304-9.
- Yoon JP, Chung SW, Yi JH, et al. Prognostic factors of arthroscopic extensor carpi radialis brevis release for lateral epicondylitis. Arthroscopy. 2015;31(7):1232-1237. doi:10.1016/j.arthro.2015.02.006.
- Barnes DE, Beckley JM, Smith J. Percutaneous ultrasonic tenotomy for chronic elbow tendinosis: a prospective study. J Shoulder Elbow Surg. 2015;24(1):67-73. doi:10.1016/j.jse.2014.07.017.
- Kaleli T, Ozturk C, Temiz A, Tirelioglu O. Surgical treatment of tennis elbow: percutaneous release of the common extensor origin. Acta Orthop Belg. 2004;70(2):131-133.
- Lin MT, Chou LW, Chen HS, Kao MJ. Percutaneous soft tissue release for treating chronic recurrent myofascial pain associated with lateral epicondylitis: 6 case studies. Evid Based Complement Alternat Med. 2012;2012:142941. doi:10.1155/2012/142941.
- Lin CL, Lee JS, Su WR, Kuo LC, Tai TW, Jou IM. Clinical and ultrasonographic results of ultrasonographically guided percutaneous radiofrequency lesioning in the treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2011;39(11):2429-2435. doi:10.1177/0363546511417096.
- Zhu J, Hu B, Xing C, Li J. Ultrasound-guided, minimally invasive, percutaneous needle puncture treatment for tennis elbow. Adv Ther. 2008;25(10):1031-1036. doi:10.1007/s12325-008-0099-6.
- Sorensen AA, Howard D, Tan WH, Ketchersid J, Calfee RP. Minimal clinically important differences of 3 patient-related outcomes instruments. J Hand Surg Am. 2013;38(4):641-649. doi:10.1016/j.jhsa.2012.12.032.
- Kelly AM. The minimum clinically significant difference in visual analogue scale pain score does not differ with severity of pain. Emerg Med J. 2001;18(3):205-207. doi:10.1136/emj.18.3.205.
- Tashjian RZ, Deloach J, Porucznik CA, Powell AP. Minimal clinically important differences (MCID) and patient acceptable symptomatic state (PASS) for visual analog scales (VAS) measuring pain in patients treated for rotator cuff disease. J Shoulder Elbow Surg. 2009;18(6):927-932. doi:10.1016/j.jse.2009.03.021.
- Cummins CA. Lateral epicondylitis: in vivo assessment of arthroscopic debridement and correlation with patient outcomes. Am J Sports Med. 2006;34(9):1486-1491. doi:10.1177/0363546506288016.
- Stapleton TR, Baker CL. Arthroscopic treatment of lateral epicondylitis: a clinical study. Arthroscopy. 1996;1:365-366.
- Hastings H. Open treatment for lateral tennis elbow good for certain indications. Orthop Today. 2009;2:1-2.
- Duparc F, Putz R, Michot C, Muller JM, Fréger P. The synovial fold of the humeroradial joint: anatomical and histological features, and clinical relevance in lateral epicondylalgia of the elbow. Surg Radiol Anat. 2002;24(5):302-307. doi:10.1007/s00276-002-0055-0.
ABSTRACT
This study was performed to compare outcomes of open, arthroscopic, and percutaneous surgical techniques for lateral epicondylitis. We searched PubMed (MEDLINE) for literature published between January 1, 2004 and May 23, 2015 using these key words: lateral epicondylitis AND (surgery OR operative OR surgical OR open OR arthroscopic OR percutaneous). Meta-analyses were performed for outcomes reported in 3 studies using 2-sample and 2-proportion Z-tests. Thirty-five studies including 1640 elbows (1055 open, 401 arthroscopic, 184 percutaneous) met the inclusion criteria. There were no differences between groups regarding duration to return to work, complication rate, or patient satisfaction. A greater proportion of patients were pain free in the open group than in the arthroscopic group (70% vs 60%). Despite the absence of a difference among techniques regarding return to work and subjective function, we recommend open débridement as the technique most likely to achieve a pain-free outcome.
Continue to: Lateral epicondylitis affects...
Lateral epicondylitis affects 1% to 3% of adults each year. Although common, symptoms of lateral epicondylitis resolve spontaneously within a year of symptom onset in 80% of cases, and only 3% of patients who seek medical treatment ultimately require surgical intervention within 2 years of symptom onset.1 Despite a relatively low percentage of patients who require surgery, Sanders and colleagues1 noted a significant increase in the rate of surgical intervention from 1.1% to 3.2% of cases in the last 15 years. Surgical intervention is generally indicated when pain and functional disability persist after 6 to 12 months of nonsurgical treatment. Traditional surgical treatment involves open release/débridement of the extensor carpi radialis (ECRB) origin; however, with the increasing prevalence of surgical intervention, surgeons have demonstrated a rising interest in less invasive techniques like arthroscopic release/débridement and percutaneous tenotomy as alternatives to traditional open débridement. While favorable results have been reported for all 3 techniques, there is no current consensus regarding the optimal surgical technique. In 2007, Lo and Safran2 reported no difference in the results of open, percutaneous, and arthroscopic techniques regarding any outcome measure in a systematic review of 33 papers. We conducted a repeat systematic review of the current literature to update Lo and Safran’s2 review and to ascertain if more recent literature demonstrates superiority of 1 technique regarding pain relief, subjective questionnaire data, subjective satisfaction, restoration of strength, and return to work. We hypothesized that return to work would be accelerated, pain decreased, and function improved in the early postoperative period in the arthroscopic and percutaneous groups, but there would be no difference in ultimate pain, functional outcome, or subjective satisfaction.
METHODS
SEARCH STRATEGY AND STUDY SELECTION
We conducted a systematic review of the literature to update the topic of surgical intervention with lateral epicondylitis since the publication of the most recent review by Lo and Safran2 in 2007, which included all relevant studies published up to 2004. To include all relevant studies published since that time, we searched PubMed (MEDLINE) for all literature published from January 1, 2004 to May 23, 2015 using the following key words: lateral epicondylitis AND (surgery OR operative OR surgical OR open OR arthroscopic OR percutaneous). General search terms were utilized to avoid unintentional exclusion of relevant studies. Two authors reviewed the abstracts of all resultant citations. Table 1 outlines the inclusion and exclusion criteria for the search. References from all included studies were reviewed for applicable articles that were not captured by the initial broad search strategy. A Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) trial flow chart shows the study selection algorithm (Figure 1).
Table 1. Inclusion and Exclusion Criteria for the Analyzed Studies
Inclusion Criteria | Exclusion Criteria |
|
|
DATA EXTRACTION AND ANALYSIS
Data were extracted from the included studies by 2 reviewers using data abstraction forms. All study, subject, and surgery parameters were collected. The study and subject demographic parameters analyzed included year of publication, level of evidence, presence of study financial conflict of interest, number of subjects and elbows, gender, age, proportion in whom the dominant extremity was involved, proportion who were laborers, proportion who had a workman’s compensation claim, duration of symptoms prior to surgical intervention, and surgical technique employed (open, arthroscopic, or percutaneous). We recorded the following clinical outcomes: proportion of patients with complete pain relief, proportion who were partially or completely satisfied, proportion who were improved, duration to return to work, grip strength, Disabilities of the Arm, Shoulder, and Hand (DASH) score, visual analog scale (VAS) pain score, and complication rate.
Continue to: Statistical analysis...
STATISTICAL ANALYSIS
Data from all studies were pooled and descriptive statistics were reported as weighted mean ± weighted standard deviation for continuous variables and frequency with percentage for categorical variables. A meta-analysis was performed for all outcome measures that were reported in 3 or more studies within a specific treatment cohort. Data were analyzed using 2-sample and 2-proportion Z-tests. Results were considered statistically significant at P < .05.
RESULTS
LITERATURE RESEARCH
Using the aforementioned search strategy, 154 studies were identified. Following application of the inclusion and exclusion criteria, 35 studies were included in the analysis (Figure 1). One study compared open and percutaneous techniques, and another compared arthroscopic and percutaneous techniques, rendering a total of 19 studies examining open surgical techniques for treatment of lateral epicondylitis,3-21 12 studies examining arthroscopic techniques,14,22-32 and 6 studies reporting percutaneous surgical treatment of lateral epicondylitis29,33-37 (Table 2). There was1 level I study (3%), 6 level III studies (17%), and 28 level IV studies (80%).
Table 2. Study Demographic Data for Open, Arthroscopic, and Percutaneous Lateral Epicondylectomy
| Open | Arthroscopic | Percutaneous | Total |
Number of studies | 19 | 12 | 6 | 35 |
Level of evidence |
|
|
|
|
I | 1 (5%) | 0 | 0 | 1 (3%) |
II | 0 | 0 | 0 | 0 |
III | 3 (16%) | 4 (33%) | 1 (17%) | 6 (17%) |
IV | 15 (79%) | 8 (67%) | 5 (83%) | 28 (80%) |
US: International | 8:12 | 3:9 | 1:5 | 12:24 |
Journals of publication |
|
|
|
|
AJSM | 3 | 1 | 1 | 5 |
JSES | 2 | 2 | 1 | 5 |
Arthroscopy | 2 | 2 | 0 | 3 |
KSSTA | 1 | 2 | 0 | 3 |
CORR | 0 | 2 | 0 | 2 |
JHS | 0 | 1 | 0 | 1 |
JOS | 1 | 1 | 0 | 2 |
AJO | 2 | 0 | 0 | 2 |
Other | 8 | 1 | 4 | 12 |
Abbreviations: AJO, The American Journal of Orthopedics; AJSM, American Journal of Sports Medicine; Arthroscopy, The Journal of Arthroscopy and Related Surgery; CORR, Clinical Orthopaedics & Related Research; JHS, Journal of Hand Surgery; JOS, Journal of Orthopaedic Surgery; JSES, Journal of Shoulder and Elbow Surgery; KSSTA, Knee Surgery, Sports Traumatology, and Arthroscopy.
SUBJECT DEMOGRAPHICS
The 35 included studies comprised 1579 patients and 1640 elbows. Among these, 1055 (64%) elbows underwent open (O), 401 (25%) underwent arthroscopic (A), and 184 (11%) underwent percutaneous (P) treatment. The average age was 45.7 years, 47% of the patients were male, 43% were laborers, 31% had worker’s compensation claims, and the dominant extremity was involved in 62% of patients. The percutaneous cohort was older than the open cohort (P = 46.9, O = 45.4, A = 45.8; P = .036). The duration of symptoms was shorter in the percutaneous cohort than in the other 2 groups and shorter in the arthroscopic cohort than in the open cohort (P = 8 months, O = 23 months, A = 18 months; P < .001). There were no significant differences between groups regarding gender, occupation, worker’s compensation status, or involvement of the dominant extremity (Table 3).
Table 3. Subject Demographics for Open, Arthroscopic, and Percutaneous Groups
| Open | Arthroscopic | Percutaneous |
Subjects (N) | 999 | 397 | 183 |
Elbows (N) | 1055 | 401 | 184 |
Elbows with follow-up (%) | 915 (87%) | 350 (87%) | 181 (98%) |
Males (%) | 427 (47%) | 173 (49%) | 78 (43%) |
Females (%) | 488 (53%) | 177 (51%) | 103 (57%) |
Mean age (years) | 45.4 | 45.8 | 46.9 |
Dominant elbow (%) | 70% | 69% | 53% |
Laborer (%) | 56% | 53% | 48% |
Work comp (%) | 36% | 30% | NR |
Symptoms to operation (months) | 23 | 18 | 8 |
Min. symptoms to operation (months) | 6 | 6 | 3 |
Mean follow-up (months) | 60 | 44 | 11 |
MATA-ANALYSIS CLINICAL OUTCOMES
Clinical outcome results were pooled for all studies reporting the same outcome measure for the same technique (open, arthroscopic, or percutaneous). A meta-analysis was performed for all outcome measures that were reported in a minimum of 3 studies utilizing the same surgical technique (Table 4).
PAIN RELIEF
Thirteen open studies,3,5,7,8,11-16,18,19,21 7 arthroscopic studies14,22-24,26,27,31 and 0 percutaneous studies reported the proportion of patients who were pain free at final follow-up. The proportion of patients who were pain free following open débridement was greater than that in the arthroscopic cohort (O = 70%, A = 60%; P = .009) (Table 4).
Continue to: Subjective improvement and satisfaction...
SUBJECTIVE IMPROVEMENT AND SATISFACTION
Nine open studies, 6 arthroscopic studies, and 1 percutaneous study reported the proportion of patients who felt that their condition had been improved as a result of surgery. There was no difference in the proportion of patients who experienced improvement between the open and arthroscopic cohorts. Four open studies,3,11,12 5 arthroscopic studies,22,26,28,29,32 and 2 percutaneous studies29,36 reported the proportion of patients who were satisfied or partially satisfied with the results of the procedure. There was no difference between the open and arthroscopic groups in the proportion of patients who were satisfied or partially satisfied (Table 4).
RETURN TO WORK
The duration to return to work following surgery was reported in 5 open studies,4,5,10,13,14 9 arthroscopic studies,14,23-29,32 and 2 percutaneous studies.29,36 There was no statistically significant difference between the open and arthroscopic groups with regard to duration to return to work (O = 6.5 weeks, A = 6 weeks; P = .601). The percutaneous technique could not be included in the meta-analysis due to the presence of only 2 studies, but the pooled mean duration to return to work in these 2 studies was 5.5 weeks (Table 4).
GRIP STRENGTH
Postoperative grip strength was reported in 2 open studies,10,19 4 arthroscopic studies,28,30,32 and 2 percutaneous studies.35-36 A meta-analysis could not be performed on all the groups due to the presence of only 2 open and 2 percutaneous studies reporting grip strength. The pooled averages were O = 38.3 kg, A = 34.8 kg, and P = 27.1 kg (Table 4).
DASH SCORE
The postoperative DASH score was reported in 4 open studies,4,15,17,19,20 5 arthroscopic studies,28-31 and 3 percutaneous studies.29,33,36 At final follow-up, the mean DASH score was higher in the arthroscopic group than in the open and percutaneous groups (A = 12.8, O = 19.5, P = 25.3; P < .001 for both comparisons), and the mean DASH score was significantly higher in the open group than in the percutaneous group (P = .029). The reporting of DASH scores in the early postoperative period was not sufficiently consistent to allow us to test our hypothesis that there would be early differences in function between groups (Table 4).
VAS PAIN SCORE
Postoperative VAS pain scores were reported in 11 open studies,6,8-10,12,15,19-21 8 arthroscopic studies,24-26,29-32 and 5 percutaneous studies.29,33,35-37 At final follow-up, there was a lower mean VAS score in the arthroscopic group than in the open and percutaneous groups (A = 1.1, O = 1.9, and P = 2.5; P < .001 for both comparisons) and a lower mean VAS score in the open group than in the percutaneous group (P = .002) (Table 4). Reporting of VAS scores in the early postoperative period in the included studies wan not sufficiently consistent to allow us to test our hypothesis that there would be early differences in pain between groups.
COMPLICATIONS
The complication rate was reported in 15 open studies, 10 arthroscopic studies, and 3 percutaneous studies. There was no difference in the complication rate between the open and arthroscopic techniques (O = 2.4%, A = 1.9%; P = .629) (Table 4). Complications noted in the open cohort included superficial wound infection (6), hematoma (5), synovial fistula (2), seroma (2), and posterior interosseous nerve palsy (1). Complications noted in the arthroscopic cohort included superficial infection (3), hematoma (1), and transient paresthesia (1). Of note, there were no complications in the percutaneous group.
Continue to: Discussion...
DISCUSSION
The primary purpose of this review was to determine if definitive evidence suggests that any 1 of open, percutaneous, or arthroscopic surgical treatment is superior to the other 2 for relieving pain, improving functionality, restoring strength, or accelerating return to work. The most striking finding of this study was a significantly higher proportion of patients who were pain free at final follow-up in the open group than in the arthroscopic group (70% vs 60%, P = .009) (Table 4). At final follow-up, there were no significant differences between groups regarding duration to return to work, proportion who were improved, proportion who were satisfied or partially satisfied, and complication rate. Average VAS and DASH scores at final follow-up were lower in the arthroscopic group than in the open and percutaneous groups (Figure 2). However, although the difference between mean DASH scores in the arthroscopic and open groups (6.7 points) was statistically significant, it is likely not clinically significant, as the minimal clinically important difference (MCID) for the DASH score is 10 points, as demonstrated by Sorensen and colleagues.38 Although it has not been specifically defined for lateral epicondylitis, the MCID for VAS pain has been reported in the literature to range from 1.0 to 1.4.39-40 Therefore, as for the DASH score, the difference witnessed between the open and arthroscopic groups (0.8) is likely not clinically significant. Of note, the differences between values for arthroscopic and percutaneous techniques are greater than the MCID.
In light of a recent increase in the prevalence of surgical intervention for lateral epicondylitis, many authors have promoted arthroscopic and percutaneous techniques as alternatives to traditional open débridement with the goal of achieving the same results with decreased morbidity and accelerated return to work. Given the increased proportion of patients who were pain free at final follow-up in the open cohort, it is our contention that open release/débridement of the common extensor/ECRB origin allows the surgeon to fully appreciate the extent of tendinotic tissue that is contributing to the patient’s symptoms and to address the pathology in its entirety. Other authors have also questioned whether the full extent of extra-articular tendinosis can be accurately identified arthroscopically. Cummins41 demonstrated, in a series of 18 patients who underwent arthroscopic ECRB débridement, that 6 patients had residual tendinosis upon open evaluation and 10 had residual tendinosis on histologic assessment. Additionally, in the same series, residual tendinopathy was associated with poorer clinical outcomes.
The improved visualization associated with an open technique comes at minimal expense, as the incision was only 1.5 cm to 5 cm in 13 of 15 papers reporting incision length.3,4,6,8-11,13,15,18-20 This increased exposure may not translate into increased morbidity, as there was no increase in the duration to return to work nor the complication rate. As a result of the extensive instrumentation necessary for arthroscopic techniques, open techniques also appear to be less expensive. Analyses in the literature have suggested increased expenditures associated with arthroscopic treatment ranging from 23%42 to 100%43 greater than those of open treatment.
Although obvious, it should be noted that a percutaneous tenotomy does not permit assessment of the extent of pathologic tendinosis. As a result of an inability to visualize and débride pathologic tissue, percutaneous tenotomy rendered inferior outcomes to open and arthroscopic techniques in terms of both postoperative VAS pain score and DASH score. Nonetheless, it is a relatively rapid and simple technique and resulted in zero complications in 184 elbows. Overall, percutaneous tenotomy appears to be an inferior technique to open and arthroscopic techniques in terms of achieving complete pain relief and optimal functional recovery; however, it may be useful in those who wish to avoid a more invasive intervention.
LIMITATIONS
The most significant limitation of this study was the heterogeneity in the techniques utilized in each group. Among the 19 papers in the open cohort, 11 used techniques aimed at lengthening or release of the extensor origin, 7 performed débridement of tendinotic tissue at the ECRB origin, and 1 compared these approaches. Exposures ranged from 1.5 cm to 8 cm in length, 3 techniques added tendon repair following débridement, and 2 utilized a radiofrequency device.
Among the 12 papers in the arthroscopic cohort, 8 performed arthroscopic (inside-out) débridement of the tendinotic tissue at the ECRB origin, 3 performed arthroscopic release of the ECRB tendon, and 1 performed endoscopic ECRB release in an outside-in fashion. Four techniques added posterior synovial plica excision and 4 added decortication of the lateral epicondyle débridement or release. Some authors advocate for arthroscopic intervention on the grounds that it permits evaluation and correction of other intra-articular pathology. With this in mind, some authors have suggested that a synovial fold (plica) adjacent to the radiocapitellar joint may contribute to lateral elbow pain.27,44 Nevertheless, in the only comparative trial in the literature, Rhyou and Kim30 demonstrated that excision of posterior synovial fold failed to enhance pain relief or function in a retrospective cohort study comparing arthroscopic débridement with and without plica excision.
Continue to: Some authors advocate...
Some authors advocate decorticating the non-articular, lateral epicondyle with a shaver to stimulate bleeding and promote a healing response. However, 1 study in our review compared arthroscopic ECRB release with and without decortication and found that decortication significantly increased pain up to 4 weeks postoperatively, increased duration to return to work, and did not improve the ultimate clinical result.25 Of note, others have used a similar rationale to advocate drilling the lateral epicondyle when utilizing an open technique. However, Dunn and colleagues8 note that they have modified the Nirschl technique to eliminate drilling because they feel it increases postoperative pain and may damage the extensor digitorum communis origin.
Among the 6 papers in the percutaneous tenotomy cohort, 2 performed tenotomy with a hypodermic needle, 2 with a scalpel through a limited incision (0.5 cm-1 cm), 1 using a TX1 tissue removal system (Tenex Health), and 1 with a percutaneous radiofrequency probe. In 3 techniques, ultrasound was used to direct the tenotomy.
The quality of this review is also limited by the studies included for analysis, as with any systematic review. Because 28 of the 35 included studies were classified as evidence level IV, the likelihood of methodological bias is increased. The majority of studies contained ≥1 demonstrable biases, including selection, detection, attrition biases, or a combination. Selection bias is prevalent among predominantly level IV studies, in which the authors have selected their preferred surgical technique. There was heterogeneity in the reporting of preoperative variables and the outcome measures that were utilized. Scoring systems, such as the Nirschl Tennis Elbow Score and the Mayo Elbow Performance Index, would have been valuable in comparing the groups had they been more consistently reported. The heterogeneity in clinical outcome tools and the lack of reported outcome variance or standard deviations prevented a formal meta-analysis of some of these outcome measures. Due to inconsistent reporting, we were also unable to test our hypothesis that there would be less pain and improved function in the arthroscopic and/or percutaneous cohorts in the early postoperative period compared to the open cohort due to the less invasive techniques used. Although the differences in DASH and VAS scores at final follow-up likely did not meet the MCID threshold, these differences may have been greater and more clinically relevant in the early postoperative period.
CONCLUSION
We hypothesized that the arthroscopic and percutaneous groups would experience accelerated return to work and reduced pain in the early postoperative period but no difference in ultimate pain, functional outcome, or subjective satisfaction. There is no difference between open, arthroscopic, and percutaneous surgical treatment for lateral epicondylitis regarding return to work and subjective satisfaction; however, open treatment led to a greater percentage of patients being pain free at final follow-up. While arthroscopic treatment led to better pain and functional scores at final follow-up, the absolute differences were quite small and likely not clinically significant. In light of the available evidence, we recommend open débridement as the best means of minimizing cost and achieving a pain-free outcome in the long term. For future investigators, it would be useful to perform a randomized clinical study directly comparing open, arthroscopic, and percutaneous techniques, including assessment of pain and functional scores in the early postoperative period, and to further evaluate differences in cost among the various techniques.
This paper will be judged for the Resident Writer’s Award.
ABSTRACT
This study was performed to compare outcomes of open, arthroscopic, and percutaneous surgical techniques for lateral epicondylitis. We searched PubMed (MEDLINE) for literature published between January 1, 2004 and May 23, 2015 using these key words: lateral epicondylitis AND (surgery OR operative OR surgical OR open OR arthroscopic OR percutaneous). Meta-analyses were performed for outcomes reported in 3 studies using 2-sample and 2-proportion Z-tests. Thirty-five studies including 1640 elbows (1055 open, 401 arthroscopic, 184 percutaneous) met the inclusion criteria. There were no differences between groups regarding duration to return to work, complication rate, or patient satisfaction. A greater proportion of patients were pain free in the open group than in the arthroscopic group (70% vs 60%). Despite the absence of a difference among techniques regarding return to work and subjective function, we recommend open débridement as the technique most likely to achieve a pain-free outcome.
Continue to: Lateral epicondylitis affects...
Lateral epicondylitis affects 1% to 3% of adults each year. Although common, symptoms of lateral epicondylitis resolve spontaneously within a year of symptom onset in 80% of cases, and only 3% of patients who seek medical treatment ultimately require surgical intervention within 2 years of symptom onset.1 Despite a relatively low percentage of patients who require surgery, Sanders and colleagues1 noted a significant increase in the rate of surgical intervention from 1.1% to 3.2% of cases in the last 15 years. Surgical intervention is generally indicated when pain and functional disability persist after 6 to 12 months of nonsurgical treatment. Traditional surgical treatment involves open release/débridement of the extensor carpi radialis (ECRB) origin; however, with the increasing prevalence of surgical intervention, surgeons have demonstrated a rising interest in less invasive techniques like arthroscopic release/débridement and percutaneous tenotomy as alternatives to traditional open débridement. While favorable results have been reported for all 3 techniques, there is no current consensus regarding the optimal surgical technique. In 2007, Lo and Safran2 reported no difference in the results of open, percutaneous, and arthroscopic techniques regarding any outcome measure in a systematic review of 33 papers. We conducted a repeat systematic review of the current literature to update Lo and Safran’s2 review and to ascertain if more recent literature demonstrates superiority of 1 technique regarding pain relief, subjective questionnaire data, subjective satisfaction, restoration of strength, and return to work. We hypothesized that return to work would be accelerated, pain decreased, and function improved in the early postoperative period in the arthroscopic and percutaneous groups, but there would be no difference in ultimate pain, functional outcome, or subjective satisfaction.
METHODS
SEARCH STRATEGY AND STUDY SELECTION
We conducted a systematic review of the literature to update the topic of surgical intervention with lateral epicondylitis since the publication of the most recent review by Lo and Safran2 in 2007, which included all relevant studies published up to 2004. To include all relevant studies published since that time, we searched PubMed (MEDLINE) for all literature published from January 1, 2004 to May 23, 2015 using the following key words: lateral epicondylitis AND (surgery OR operative OR surgical OR open OR arthroscopic OR percutaneous). General search terms were utilized to avoid unintentional exclusion of relevant studies. Two authors reviewed the abstracts of all resultant citations. Table 1 outlines the inclusion and exclusion criteria for the search. References from all included studies were reviewed for applicable articles that were not captured by the initial broad search strategy. A Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) trial flow chart shows the study selection algorithm (Figure 1).
Table 1. Inclusion and Exclusion Criteria for the Analyzed Studies
Inclusion Criteria | Exclusion Criteria |
|
|
DATA EXTRACTION AND ANALYSIS
Data were extracted from the included studies by 2 reviewers using data abstraction forms. All study, subject, and surgery parameters were collected. The study and subject demographic parameters analyzed included year of publication, level of evidence, presence of study financial conflict of interest, number of subjects and elbows, gender, age, proportion in whom the dominant extremity was involved, proportion who were laborers, proportion who had a workman’s compensation claim, duration of symptoms prior to surgical intervention, and surgical technique employed (open, arthroscopic, or percutaneous). We recorded the following clinical outcomes: proportion of patients with complete pain relief, proportion who were partially or completely satisfied, proportion who were improved, duration to return to work, grip strength, Disabilities of the Arm, Shoulder, and Hand (DASH) score, visual analog scale (VAS) pain score, and complication rate.
Continue to: Statistical analysis...
STATISTICAL ANALYSIS
Data from all studies were pooled and descriptive statistics were reported as weighted mean ± weighted standard deviation for continuous variables and frequency with percentage for categorical variables. A meta-analysis was performed for all outcome measures that were reported in 3 or more studies within a specific treatment cohort. Data were analyzed using 2-sample and 2-proportion Z-tests. Results were considered statistically significant at P < .05.
RESULTS
LITERATURE RESEARCH
Using the aforementioned search strategy, 154 studies were identified. Following application of the inclusion and exclusion criteria, 35 studies were included in the analysis (Figure 1). One study compared open and percutaneous techniques, and another compared arthroscopic and percutaneous techniques, rendering a total of 19 studies examining open surgical techniques for treatment of lateral epicondylitis,3-21 12 studies examining arthroscopic techniques,14,22-32 and 6 studies reporting percutaneous surgical treatment of lateral epicondylitis29,33-37 (Table 2). There was1 level I study (3%), 6 level III studies (17%), and 28 level IV studies (80%).
Table 2. Study Demographic Data for Open, Arthroscopic, and Percutaneous Lateral Epicondylectomy
| Open | Arthroscopic | Percutaneous | Total |
Number of studies | 19 | 12 | 6 | 35 |
Level of evidence |
|
|
|
|
I | 1 (5%) | 0 | 0 | 1 (3%) |
II | 0 | 0 | 0 | 0 |
III | 3 (16%) | 4 (33%) | 1 (17%) | 6 (17%) |
IV | 15 (79%) | 8 (67%) | 5 (83%) | 28 (80%) |
US: International | 8:12 | 3:9 | 1:5 | 12:24 |
Journals of publication |
|
|
|
|
AJSM | 3 | 1 | 1 | 5 |
JSES | 2 | 2 | 1 | 5 |
Arthroscopy | 2 | 2 | 0 | 3 |
KSSTA | 1 | 2 | 0 | 3 |
CORR | 0 | 2 | 0 | 2 |
JHS | 0 | 1 | 0 | 1 |
JOS | 1 | 1 | 0 | 2 |
AJO | 2 | 0 | 0 | 2 |
Other | 8 | 1 | 4 | 12 |
Abbreviations: AJO, The American Journal of Orthopedics; AJSM, American Journal of Sports Medicine; Arthroscopy, The Journal of Arthroscopy and Related Surgery; CORR, Clinical Orthopaedics & Related Research; JHS, Journal of Hand Surgery; JOS, Journal of Orthopaedic Surgery; JSES, Journal of Shoulder and Elbow Surgery; KSSTA, Knee Surgery, Sports Traumatology, and Arthroscopy.
SUBJECT DEMOGRAPHICS
The 35 included studies comprised 1579 patients and 1640 elbows. Among these, 1055 (64%) elbows underwent open (O), 401 (25%) underwent arthroscopic (A), and 184 (11%) underwent percutaneous (P) treatment. The average age was 45.7 years, 47% of the patients were male, 43% were laborers, 31% had worker’s compensation claims, and the dominant extremity was involved in 62% of patients. The percutaneous cohort was older than the open cohort (P = 46.9, O = 45.4, A = 45.8; P = .036). The duration of symptoms was shorter in the percutaneous cohort than in the other 2 groups and shorter in the arthroscopic cohort than in the open cohort (P = 8 months, O = 23 months, A = 18 months; P < .001). There were no significant differences between groups regarding gender, occupation, worker’s compensation status, or involvement of the dominant extremity (Table 3).
Table 3. Subject Demographics for Open, Arthroscopic, and Percutaneous Groups
| Open | Arthroscopic | Percutaneous |
Subjects (N) | 999 | 397 | 183 |
Elbows (N) | 1055 | 401 | 184 |
Elbows with follow-up (%) | 915 (87%) | 350 (87%) | 181 (98%) |
Males (%) | 427 (47%) | 173 (49%) | 78 (43%) |
Females (%) | 488 (53%) | 177 (51%) | 103 (57%) |
Mean age (years) | 45.4 | 45.8 | 46.9 |
Dominant elbow (%) | 70% | 69% | 53% |
Laborer (%) | 56% | 53% | 48% |
Work comp (%) | 36% | 30% | NR |
Symptoms to operation (months) | 23 | 18 | 8 |
Min. symptoms to operation (months) | 6 | 6 | 3 |
Mean follow-up (months) | 60 | 44 | 11 |
MATA-ANALYSIS CLINICAL OUTCOMES
Clinical outcome results were pooled for all studies reporting the same outcome measure for the same technique (open, arthroscopic, or percutaneous). A meta-analysis was performed for all outcome measures that were reported in a minimum of 3 studies utilizing the same surgical technique (Table 4).
PAIN RELIEF
Thirteen open studies,3,5,7,8,11-16,18,19,21 7 arthroscopic studies14,22-24,26,27,31 and 0 percutaneous studies reported the proportion of patients who were pain free at final follow-up. The proportion of patients who were pain free following open débridement was greater than that in the arthroscopic cohort (O = 70%, A = 60%; P = .009) (Table 4).
Continue to: Subjective improvement and satisfaction...
SUBJECTIVE IMPROVEMENT AND SATISFACTION
Nine open studies, 6 arthroscopic studies, and 1 percutaneous study reported the proportion of patients who felt that their condition had been improved as a result of surgery. There was no difference in the proportion of patients who experienced improvement between the open and arthroscopic cohorts. Four open studies,3,11,12 5 arthroscopic studies,22,26,28,29,32 and 2 percutaneous studies29,36 reported the proportion of patients who were satisfied or partially satisfied with the results of the procedure. There was no difference between the open and arthroscopic groups in the proportion of patients who were satisfied or partially satisfied (Table 4).
RETURN TO WORK
The duration to return to work following surgery was reported in 5 open studies,4,5,10,13,14 9 arthroscopic studies,14,23-29,32 and 2 percutaneous studies.29,36 There was no statistically significant difference between the open and arthroscopic groups with regard to duration to return to work (O = 6.5 weeks, A = 6 weeks; P = .601). The percutaneous technique could not be included in the meta-analysis due to the presence of only 2 studies, but the pooled mean duration to return to work in these 2 studies was 5.5 weeks (Table 4).
GRIP STRENGTH
Postoperative grip strength was reported in 2 open studies,10,19 4 arthroscopic studies,28,30,32 and 2 percutaneous studies.35-36 A meta-analysis could not be performed on all the groups due to the presence of only 2 open and 2 percutaneous studies reporting grip strength. The pooled averages were O = 38.3 kg, A = 34.8 kg, and P = 27.1 kg (Table 4).
DASH SCORE
The postoperative DASH score was reported in 4 open studies,4,15,17,19,20 5 arthroscopic studies,28-31 and 3 percutaneous studies.29,33,36 At final follow-up, the mean DASH score was higher in the arthroscopic group than in the open and percutaneous groups (A = 12.8, O = 19.5, P = 25.3; P < .001 for both comparisons), and the mean DASH score was significantly higher in the open group than in the percutaneous group (P = .029). The reporting of DASH scores in the early postoperative period was not sufficiently consistent to allow us to test our hypothesis that there would be early differences in function between groups (Table 4).
VAS PAIN SCORE
Postoperative VAS pain scores were reported in 11 open studies,6,8-10,12,15,19-21 8 arthroscopic studies,24-26,29-32 and 5 percutaneous studies.29,33,35-37 At final follow-up, there was a lower mean VAS score in the arthroscopic group than in the open and percutaneous groups (A = 1.1, O = 1.9, and P = 2.5; P < .001 for both comparisons) and a lower mean VAS score in the open group than in the percutaneous group (P = .002) (Table 4). Reporting of VAS scores in the early postoperative period in the included studies wan not sufficiently consistent to allow us to test our hypothesis that there would be early differences in pain between groups.
COMPLICATIONS
The complication rate was reported in 15 open studies, 10 arthroscopic studies, and 3 percutaneous studies. There was no difference in the complication rate between the open and arthroscopic techniques (O = 2.4%, A = 1.9%; P = .629) (Table 4). Complications noted in the open cohort included superficial wound infection (6), hematoma (5), synovial fistula (2), seroma (2), and posterior interosseous nerve palsy (1). Complications noted in the arthroscopic cohort included superficial infection (3), hematoma (1), and transient paresthesia (1). Of note, there were no complications in the percutaneous group.
Continue to: Discussion...
DISCUSSION
The primary purpose of this review was to determine if definitive evidence suggests that any 1 of open, percutaneous, or arthroscopic surgical treatment is superior to the other 2 for relieving pain, improving functionality, restoring strength, or accelerating return to work. The most striking finding of this study was a significantly higher proportion of patients who were pain free at final follow-up in the open group than in the arthroscopic group (70% vs 60%, P = .009) (Table 4). At final follow-up, there were no significant differences between groups regarding duration to return to work, proportion who were improved, proportion who were satisfied or partially satisfied, and complication rate. Average VAS and DASH scores at final follow-up were lower in the arthroscopic group than in the open and percutaneous groups (Figure 2). However, although the difference between mean DASH scores in the arthroscopic and open groups (6.7 points) was statistically significant, it is likely not clinically significant, as the minimal clinically important difference (MCID) for the DASH score is 10 points, as demonstrated by Sorensen and colleagues.38 Although it has not been specifically defined for lateral epicondylitis, the MCID for VAS pain has been reported in the literature to range from 1.0 to 1.4.39-40 Therefore, as for the DASH score, the difference witnessed between the open and arthroscopic groups (0.8) is likely not clinically significant. Of note, the differences between values for arthroscopic and percutaneous techniques are greater than the MCID.
In light of a recent increase in the prevalence of surgical intervention for lateral epicondylitis, many authors have promoted arthroscopic and percutaneous techniques as alternatives to traditional open débridement with the goal of achieving the same results with decreased morbidity and accelerated return to work. Given the increased proportion of patients who were pain free at final follow-up in the open cohort, it is our contention that open release/débridement of the common extensor/ECRB origin allows the surgeon to fully appreciate the extent of tendinotic tissue that is contributing to the patient’s symptoms and to address the pathology in its entirety. Other authors have also questioned whether the full extent of extra-articular tendinosis can be accurately identified arthroscopically. Cummins41 demonstrated, in a series of 18 patients who underwent arthroscopic ECRB débridement, that 6 patients had residual tendinosis upon open evaluation and 10 had residual tendinosis on histologic assessment. Additionally, in the same series, residual tendinopathy was associated with poorer clinical outcomes.
The improved visualization associated with an open technique comes at minimal expense, as the incision was only 1.5 cm to 5 cm in 13 of 15 papers reporting incision length.3,4,6,8-11,13,15,18-20 This increased exposure may not translate into increased morbidity, as there was no increase in the duration to return to work nor the complication rate. As a result of the extensive instrumentation necessary for arthroscopic techniques, open techniques also appear to be less expensive. Analyses in the literature have suggested increased expenditures associated with arthroscopic treatment ranging from 23%42 to 100%43 greater than those of open treatment.
Although obvious, it should be noted that a percutaneous tenotomy does not permit assessment of the extent of pathologic tendinosis. As a result of an inability to visualize and débride pathologic tissue, percutaneous tenotomy rendered inferior outcomes to open and arthroscopic techniques in terms of both postoperative VAS pain score and DASH score. Nonetheless, it is a relatively rapid and simple technique and resulted in zero complications in 184 elbows. Overall, percutaneous tenotomy appears to be an inferior technique to open and arthroscopic techniques in terms of achieving complete pain relief and optimal functional recovery; however, it may be useful in those who wish to avoid a more invasive intervention.
LIMITATIONS
The most significant limitation of this study was the heterogeneity in the techniques utilized in each group. Among the 19 papers in the open cohort, 11 used techniques aimed at lengthening or release of the extensor origin, 7 performed débridement of tendinotic tissue at the ECRB origin, and 1 compared these approaches. Exposures ranged from 1.5 cm to 8 cm in length, 3 techniques added tendon repair following débridement, and 2 utilized a radiofrequency device.
Among the 12 papers in the arthroscopic cohort, 8 performed arthroscopic (inside-out) débridement of the tendinotic tissue at the ECRB origin, 3 performed arthroscopic release of the ECRB tendon, and 1 performed endoscopic ECRB release in an outside-in fashion. Four techniques added posterior synovial plica excision and 4 added decortication of the lateral epicondyle débridement or release. Some authors advocate for arthroscopic intervention on the grounds that it permits evaluation and correction of other intra-articular pathology. With this in mind, some authors have suggested that a synovial fold (plica) adjacent to the radiocapitellar joint may contribute to lateral elbow pain.27,44 Nevertheless, in the only comparative trial in the literature, Rhyou and Kim30 demonstrated that excision of posterior synovial fold failed to enhance pain relief or function in a retrospective cohort study comparing arthroscopic débridement with and without plica excision.
Continue to: Some authors advocate...
Some authors advocate decorticating the non-articular, lateral epicondyle with a shaver to stimulate bleeding and promote a healing response. However, 1 study in our review compared arthroscopic ECRB release with and without decortication and found that decortication significantly increased pain up to 4 weeks postoperatively, increased duration to return to work, and did not improve the ultimate clinical result.25 Of note, others have used a similar rationale to advocate drilling the lateral epicondyle when utilizing an open technique. However, Dunn and colleagues8 note that they have modified the Nirschl technique to eliminate drilling because they feel it increases postoperative pain and may damage the extensor digitorum communis origin.
Among the 6 papers in the percutaneous tenotomy cohort, 2 performed tenotomy with a hypodermic needle, 2 with a scalpel through a limited incision (0.5 cm-1 cm), 1 using a TX1 tissue removal system (Tenex Health), and 1 with a percutaneous radiofrequency probe. In 3 techniques, ultrasound was used to direct the tenotomy.
The quality of this review is also limited by the studies included for analysis, as with any systematic review. Because 28 of the 35 included studies were classified as evidence level IV, the likelihood of methodological bias is increased. The majority of studies contained ≥1 demonstrable biases, including selection, detection, attrition biases, or a combination. Selection bias is prevalent among predominantly level IV studies, in which the authors have selected their preferred surgical technique. There was heterogeneity in the reporting of preoperative variables and the outcome measures that were utilized. Scoring systems, such as the Nirschl Tennis Elbow Score and the Mayo Elbow Performance Index, would have been valuable in comparing the groups had they been more consistently reported. The heterogeneity in clinical outcome tools and the lack of reported outcome variance or standard deviations prevented a formal meta-analysis of some of these outcome measures. Due to inconsistent reporting, we were also unable to test our hypothesis that there would be less pain and improved function in the arthroscopic and/or percutaneous cohorts in the early postoperative period compared to the open cohort due to the less invasive techniques used. Although the differences in DASH and VAS scores at final follow-up likely did not meet the MCID threshold, these differences may have been greater and more clinically relevant in the early postoperative period.
CONCLUSION
We hypothesized that the arthroscopic and percutaneous groups would experience accelerated return to work and reduced pain in the early postoperative period but no difference in ultimate pain, functional outcome, or subjective satisfaction. There is no difference between open, arthroscopic, and percutaneous surgical treatment for lateral epicondylitis regarding return to work and subjective satisfaction; however, open treatment led to a greater percentage of patients being pain free at final follow-up. While arthroscopic treatment led to better pain and functional scores at final follow-up, the absolute differences were quite small and likely not clinically significant. In light of the available evidence, we recommend open débridement as the best means of minimizing cost and achieving a pain-free outcome in the long term. For future investigators, it would be useful to perform a randomized clinical study directly comparing open, arthroscopic, and percutaneous techniques, including assessment of pain and functional scores in the early postoperative period, and to further evaluate differences in cost among the various techniques.
This paper will be judged for the Resident Writer’s Award.
- Sanders TL Jr, Maradit Kremers H, Bryan AJ, Ransom JE, Smith J, Morrey BF. The epidemiology and health care burden of tennis elbow: a population-based study. Am J Sports Med. 2015;43(5):1066-1071. doi:10.1177/0363546514568087.
- Lo MY, Safran MR. Surgical treatment of lateral epicondylitis: a systematic review. Clin Orthop Relat Res. 2007;463:98-106. doi:10.1097/BLO.0b013e3181483dc4.
- Balk ML, Hagberg WC, Buterbaugh GA, Imbriglia JE. Outcome of surgery for lateral epicondylitis (tennis elbow): effect of worker’s compensation. Am J Orthop. 2005;34(3):122-126; discussion 126.
- Barth J, Mahieu P, Hollevoet N. Extensor tendon and fascia sectioning of extensors at the musculotendinous unit in lateral epicondylitis. Acta Orthop Belg. 2013;79(3):266-270.
- Bigorre N, Raimbeau G, Fouque PA, Cast YS, Rabarin F, Cesari B. Lateral epicondylitis treatment by extensor carpi radialis fasciotomy and radial nerve decompression: is outcome influenced by the occupational disease compensation aspect? Orthop Traumatol Surg Res. 2011;97(2):159-163. doi:10.1016/j.otsr.2010.11.007.
- Cho BK, Kim YM, Kim DS, et al. Mini-open muscle resection procedure under local anesthesia for lateral and medial epicondylitis. Clin Orthop Surg. 2009;1(3):123-127. doi:10.4055/cios.2009.1.3.123.
- Coleman B, Quinlan JF, Matheson JA. Surgical treatment for lateral epicondylitis: a long-term follow-up of results. J Shoulder Elbow Surg. 2010;19(3):363-367. doi:10.1016/j.jse.2009.09.008.
- Dunn JH, Kim JJ, Davis L, Nirschl RP. Ten- to 14-year follow-up of the Nirschl surgical technique for lateral epicondylitis. Am J Sports Med. 2008;36(2):261-266. doi:10.1177/0363546507308932.
- Manon-Matos Y, Oron A, Wolff TW. Combined common extensor and supinator aponeurotomy for the treatment of lateral epicondylitis. Tech Hand Up Extrem Surg. 2013;17(3):179-181. doi:10.1097/BTH.0b013e31829e0eeb.
- Meknas K, Odden-Miland A, Mercer JB, Castillejo M, Johansen O. Radiofrequency microtenotomy: a promising method for treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2008;36(10):1960-1965. doi:10.1177/0363546508318045.
- Pruzansky ME, Gantsoudes GD, Watters N. Late surgical results of reattachment to bone in repair of chronic lateral epicondylitis. Am J Orthop. 2009;38(6):295-299.
- Rayan F, Rao V Sr, Purushothamdas S, Mukundan C, Shafqat SO. Common extensor origin release in recalcitrant lateral epicondylitis – role justified? J Orthop Surg Res. 2010;5:31. doi:10.1186/1749-799X-5-31.
- Reddy VR, Satheesan KS, Bayliss N. Outcome of Boyd-McLeod procedure for recalcitrant lateral epicondylitis of elbow. Rheumatol Int. 2011;31(8):1081-1084. doi:10.1007/s00296-010-1450-1.
- Rubenthaler F, Wiese M, Senge A, Keller L, Wittenberg RH. Long-term follow-up of open and endoscopic Hohmann procedures for lateral epicondylitis. Arthroscopy. 2005;21(6):684-690. doi:10.1016/j.arthro.2005.03.017.
- Ruch DS, Orr SB, Richard MJ, Leversedge FJ, Mithani SK, Laino DK. A comparison of debridement with and without anconeus muscle flap for treatment of refractory lateral epicondylitis. J Shoulder Elbow Surg. 2015;24(2):236-241. doi:10.1016/j.jse.2014.09.035.
- Siddiqui MA, Koh J, Kua J, Cheung T, Chang P. Functional outcome assessment after open tennis elbow release: what are the predictor parameters? Singapore Med J. 2011;52(2):73-76.
- Solheim E, Hegna J, Øyen J. Extensor tendon release in tennis elbow: results and prognostic factors in 80 elbows. Knee Surg Sports Traumatol Arthrosc. 2011;19(6):1023-1027. doi:10.1007/s00167-011-1477-1.
- Svernlöv B, Adolfsson L. Outcome of release of the lateral extensor muscle origin for epicondylitis. Scand J Plast Reconstr Surg Hand Surg. 2006;40(3):161-165. doi:10.1080/02844310500491492.
- Tasto JP, Cummings J, Medlock V, Hardesty R, Amiel D. Microtenotomy using a radiofrequency probe to treat lateral epicondylitis. Arthroscopy. 2005;21(7):851-860. doi:10.1016/j.arthro.2005.03.019.
- Thornton SJ, Rogers JR, Prickett WD, Dunn WR, Allen AA, Hannafin JA. Treatment of recalcitrant lateral epicondylitis with suture anchor repair. Am J Sports Med. 2005;33(10):1558-1564. doi:10.1177/0363546505276758.
- Wang AW, Erak S. Fractional lengthening of forearm extensors for resistant lateral epicondylitis. ANZ J Surg. 2007;77(11):981-984. doi:10.1111/j.1445-2197.2007.04294.x.
- Baker CL Jr, Baker CL 3rd. Long-term follow-up of arthroscopic treatment of lateral epicondylitis. Am J Sports Med. 2008;36(2):254-260. doi:10.1177/0363546507311599.
- Grewal R, MacDermid JC, Shah P, King GJ. Functional outcome of arthroscopic extensor carpi radialis brevis tendon release in chronic lateral epicondylitis. J Hand Surg Am. 2009;34(5):849-857. doi:10.1016/j.jhsa.2009.02.006.
- Jerosch J, Schunck J. Arthroscopic treatment of lateral epicondylitis: indication, technique and early results. Knee Surg Sports Traumatol Arthrosc. 2006;14(4):379-382. doi:10.1007/s00167-005-0662-5.
- Kim JW, Chun CH, Shim DM, et al. Arthroscopic treatment of lateral epicondylitis: comparison of the outcome of ECRB release with and without decortication. Knee Surg Sports Traumatol Arthrosc. 2011;19(7):1178-1183. doi:10.1007/s00167-011-1507-z.
- Lattermann C, Romeo AA, Anbari A, et al. Arthroscopic debridement of the extensor carpi radialis brevis for recalcitrant lateral epicondylitis. J Shoulder Elbow Surg. 2010;19(5):651-656. doi:10.1016/j.jse.2010.02.008.
- Mullett H, Sprague M, Brown G, Hausman M. Arthroscopic treatment of lateral epicondylitis: clinical and cadaveric studies. Clin Orthop Relat Res. 2005;439:123-128. doi:10.1097/01.blo.0000176143.08886.fe.
- Oki G, Iba K, Sasaki K, Yamashita T, Wada T. Time to functional recovery after arthroscopic surgery for tennis elbow. J Shoulder Elbow Surg. 2014;23(10):1527-1531. doi:10.1016/j.jse.2014.05.010.
- Othman AM. Arthroscopic versus percutaneous release of common extensor origin for treatment of chronic tennis elbow. Arch Orthop Trauma Surg. 2011;131(3):383-388. doi:10.1007/s00402-011-1260-2.
- Rhyou IH, Kim KW. Is posterior synovial plica excision necessary for refractory lateral epicondylitis of the elbow? Clin Orthop Relat Res. 2013;471(1):284-290. doi:10.1007/s11999-012-2585-z.
- Wada T, Moriya T, Iba K, et al. Functional outcomes after arthroscopic treatment of lateral epicondylitis. J Orthop Sci. 2009;14(2):167-174. doi:10.1007/s00776-008-1304-9.
- Yoon JP, Chung SW, Yi JH, et al. Prognostic factors of arthroscopic extensor carpi radialis brevis release for lateral epicondylitis. Arthroscopy. 2015;31(7):1232-1237. doi:10.1016/j.arthro.2015.02.006.
- Barnes DE, Beckley JM, Smith J. Percutaneous ultrasonic tenotomy for chronic elbow tendinosis: a prospective study. J Shoulder Elbow Surg. 2015;24(1):67-73. doi:10.1016/j.jse.2014.07.017.
- Kaleli T, Ozturk C, Temiz A, Tirelioglu O. Surgical treatment of tennis elbow: percutaneous release of the common extensor origin. Acta Orthop Belg. 2004;70(2):131-133.
- Lin MT, Chou LW, Chen HS, Kao MJ. Percutaneous soft tissue release for treating chronic recurrent myofascial pain associated with lateral epicondylitis: 6 case studies. Evid Based Complement Alternat Med. 2012;2012:142941. doi:10.1155/2012/142941.
- Lin CL, Lee JS, Su WR, Kuo LC, Tai TW, Jou IM. Clinical and ultrasonographic results of ultrasonographically guided percutaneous radiofrequency lesioning in the treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2011;39(11):2429-2435. doi:10.1177/0363546511417096.
- Zhu J, Hu B, Xing C, Li J. Ultrasound-guided, minimally invasive, percutaneous needle puncture treatment for tennis elbow. Adv Ther. 2008;25(10):1031-1036. doi:10.1007/s12325-008-0099-6.
- Sorensen AA, Howard D, Tan WH, Ketchersid J, Calfee RP. Minimal clinically important differences of 3 patient-related outcomes instruments. J Hand Surg Am. 2013;38(4):641-649. doi:10.1016/j.jhsa.2012.12.032.
- Kelly AM. The minimum clinically significant difference in visual analogue scale pain score does not differ with severity of pain. Emerg Med J. 2001;18(3):205-207. doi:10.1136/emj.18.3.205.
- Tashjian RZ, Deloach J, Porucznik CA, Powell AP. Minimal clinically important differences (MCID) and patient acceptable symptomatic state (PASS) for visual analog scales (VAS) measuring pain in patients treated for rotator cuff disease. J Shoulder Elbow Surg. 2009;18(6):927-932. doi:10.1016/j.jse.2009.03.021.
- Cummins CA. Lateral epicondylitis: in vivo assessment of arthroscopic debridement and correlation with patient outcomes. Am J Sports Med. 2006;34(9):1486-1491. doi:10.1177/0363546506288016.
- Stapleton TR, Baker CL. Arthroscopic treatment of lateral epicondylitis: a clinical study. Arthroscopy. 1996;1:365-366.
- Hastings H. Open treatment for lateral tennis elbow good for certain indications. Orthop Today. 2009;2:1-2.
- Duparc F, Putz R, Michot C, Muller JM, Fréger P. The synovial fold of the humeroradial joint: anatomical and histological features, and clinical relevance in lateral epicondylalgia of the elbow. Surg Radiol Anat. 2002;24(5):302-307. doi:10.1007/s00276-002-0055-0.
- Sanders TL Jr, Maradit Kremers H, Bryan AJ, Ransom JE, Smith J, Morrey BF. The epidemiology and health care burden of tennis elbow: a population-based study. Am J Sports Med. 2015;43(5):1066-1071. doi:10.1177/0363546514568087.
- Lo MY, Safran MR. Surgical treatment of lateral epicondylitis: a systematic review. Clin Orthop Relat Res. 2007;463:98-106. doi:10.1097/BLO.0b013e3181483dc4.
- Balk ML, Hagberg WC, Buterbaugh GA, Imbriglia JE. Outcome of surgery for lateral epicondylitis (tennis elbow): effect of worker’s compensation. Am J Orthop. 2005;34(3):122-126; discussion 126.
- Barth J, Mahieu P, Hollevoet N. Extensor tendon and fascia sectioning of extensors at the musculotendinous unit in lateral epicondylitis. Acta Orthop Belg. 2013;79(3):266-270.
- Bigorre N, Raimbeau G, Fouque PA, Cast YS, Rabarin F, Cesari B. Lateral epicondylitis treatment by extensor carpi radialis fasciotomy and radial nerve decompression: is outcome influenced by the occupational disease compensation aspect? Orthop Traumatol Surg Res. 2011;97(2):159-163. doi:10.1016/j.otsr.2010.11.007.
- Cho BK, Kim YM, Kim DS, et al. Mini-open muscle resection procedure under local anesthesia for lateral and medial epicondylitis. Clin Orthop Surg. 2009;1(3):123-127. doi:10.4055/cios.2009.1.3.123.
- Coleman B, Quinlan JF, Matheson JA. Surgical treatment for lateral epicondylitis: a long-term follow-up of results. J Shoulder Elbow Surg. 2010;19(3):363-367. doi:10.1016/j.jse.2009.09.008.
- Dunn JH, Kim JJ, Davis L, Nirschl RP. Ten- to 14-year follow-up of the Nirschl surgical technique for lateral epicondylitis. Am J Sports Med. 2008;36(2):261-266. doi:10.1177/0363546507308932.
- Manon-Matos Y, Oron A, Wolff TW. Combined common extensor and supinator aponeurotomy for the treatment of lateral epicondylitis. Tech Hand Up Extrem Surg. 2013;17(3):179-181. doi:10.1097/BTH.0b013e31829e0eeb.
- Meknas K, Odden-Miland A, Mercer JB, Castillejo M, Johansen O. Radiofrequency microtenotomy: a promising method for treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2008;36(10):1960-1965. doi:10.1177/0363546508318045.
- Pruzansky ME, Gantsoudes GD, Watters N. Late surgical results of reattachment to bone in repair of chronic lateral epicondylitis. Am J Orthop. 2009;38(6):295-299.
- Rayan F, Rao V Sr, Purushothamdas S, Mukundan C, Shafqat SO. Common extensor origin release in recalcitrant lateral epicondylitis – role justified? J Orthop Surg Res. 2010;5:31. doi:10.1186/1749-799X-5-31.
- Reddy VR, Satheesan KS, Bayliss N. Outcome of Boyd-McLeod procedure for recalcitrant lateral epicondylitis of elbow. Rheumatol Int. 2011;31(8):1081-1084. doi:10.1007/s00296-010-1450-1.
- Rubenthaler F, Wiese M, Senge A, Keller L, Wittenberg RH. Long-term follow-up of open and endoscopic Hohmann procedures for lateral epicondylitis. Arthroscopy. 2005;21(6):684-690. doi:10.1016/j.arthro.2005.03.017.
- Ruch DS, Orr SB, Richard MJ, Leversedge FJ, Mithani SK, Laino DK. A comparison of debridement with and without anconeus muscle flap for treatment of refractory lateral epicondylitis. J Shoulder Elbow Surg. 2015;24(2):236-241. doi:10.1016/j.jse.2014.09.035.
- Siddiqui MA, Koh J, Kua J, Cheung T, Chang P. Functional outcome assessment after open tennis elbow release: what are the predictor parameters? Singapore Med J. 2011;52(2):73-76.
- Solheim E, Hegna J, Øyen J. Extensor tendon release in tennis elbow: results and prognostic factors in 80 elbows. Knee Surg Sports Traumatol Arthrosc. 2011;19(6):1023-1027. doi:10.1007/s00167-011-1477-1.
- Svernlöv B, Adolfsson L. Outcome of release of the lateral extensor muscle origin for epicondylitis. Scand J Plast Reconstr Surg Hand Surg. 2006;40(3):161-165. doi:10.1080/02844310500491492.
- Tasto JP, Cummings J, Medlock V, Hardesty R, Amiel D. Microtenotomy using a radiofrequency probe to treat lateral epicondylitis. Arthroscopy. 2005;21(7):851-860. doi:10.1016/j.arthro.2005.03.019.
- Thornton SJ, Rogers JR, Prickett WD, Dunn WR, Allen AA, Hannafin JA. Treatment of recalcitrant lateral epicondylitis with suture anchor repair. Am J Sports Med. 2005;33(10):1558-1564. doi:10.1177/0363546505276758.
- Wang AW, Erak S. Fractional lengthening of forearm extensors for resistant lateral epicondylitis. ANZ J Surg. 2007;77(11):981-984. doi:10.1111/j.1445-2197.2007.04294.x.
- Baker CL Jr, Baker CL 3rd. Long-term follow-up of arthroscopic treatment of lateral epicondylitis. Am J Sports Med. 2008;36(2):254-260. doi:10.1177/0363546507311599.
- Grewal R, MacDermid JC, Shah P, King GJ. Functional outcome of arthroscopic extensor carpi radialis brevis tendon release in chronic lateral epicondylitis. J Hand Surg Am. 2009;34(5):849-857. doi:10.1016/j.jhsa.2009.02.006.
- Jerosch J, Schunck J. Arthroscopic treatment of lateral epicondylitis: indication, technique and early results. Knee Surg Sports Traumatol Arthrosc. 2006;14(4):379-382. doi:10.1007/s00167-005-0662-5.
- Kim JW, Chun CH, Shim DM, et al. Arthroscopic treatment of lateral epicondylitis: comparison of the outcome of ECRB release with and without decortication. Knee Surg Sports Traumatol Arthrosc. 2011;19(7):1178-1183. doi:10.1007/s00167-011-1507-z.
- Lattermann C, Romeo AA, Anbari A, et al. Arthroscopic debridement of the extensor carpi radialis brevis for recalcitrant lateral epicondylitis. J Shoulder Elbow Surg. 2010;19(5):651-656. doi:10.1016/j.jse.2010.02.008.
- Mullett H, Sprague M, Brown G, Hausman M. Arthroscopic treatment of lateral epicondylitis: clinical and cadaveric studies. Clin Orthop Relat Res. 2005;439:123-128. doi:10.1097/01.blo.0000176143.08886.fe.
- Oki G, Iba K, Sasaki K, Yamashita T, Wada T. Time to functional recovery after arthroscopic surgery for tennis elbow. J Shoulder Elbow Surg. 2014;23(10):1527-1531. doi:10.1016/j.jse.2014.05.010.
- Othman AM. Arthroscopic versus percutaneous release of common extensor origin for treatment of chronic tennis elbow. Arch Orthop Trauma Surg. 2011;131(3):383-388. doi:10.1007/s00402-011-1260-2.
- Rhyou IH, Kim KW. Is posterior synovial plica excision necessary for refractory lateral epicondylitis of the elbow? Clin Orthop Relat Res. 2013;471(1):284-290. doi:10.1007/s11999-012-2585-z.
- Wada T, Moriya T, Iba K, et al. Functional outcomes after arthroscopic treatment of lateral epicondylitis. J Orthop Sci. 2009;14(2):167-174. doi:10.1007/s00776-008-1304-9.
- Yoon JP, Chung SW, Yi JH, et al. Prognostic factors of arthroscopic extensor carpi radialis brevis release for lateral epicondylitis. Arthroscopy. 2015;31(7):1232-1237. doi:10.1016/j.arthro.2015.02.006.
- Barnes DE, Beckley JM, Smith J. Percutaneous ultrasonic tenotomy for chronic elbow tendinosis: a prospective study. J Shoulder Elbow Surg. 2015;24(1):67-73. doi:10.1016/j.jse.2014.07.017.
- Kaleli T, Ozturk C, Temiz A, Tirelioglu O. Surgical treatment of tennis elbow: percutaneous release of the common extensor origin. Acta Orthop Belg. 2004;70(2):131-133.
- Lin MT, Chou LW, Chen HS, Kao MJ. Percutaneous soft tissue release for treating chronic recurrent myofascial pain associated with lateral epicondylitis: 6 case studies. Evid Based Complement Alternat Med. 2012;2012:142941. doi:10.1155/2012/142941.
- Lin CL, Lee JS, Su WR, Kuo LC, Tai TW, Jou IM. Clinical and ultrasonographic results of ultrasonographically guided percutaneous radiofrequency lesioning in the treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2011;39(11):2429-2435. doi:10.1177/0363546511417096.
- Zhu J, Hu B, Xing C, Li J. Ultrasound-guided, minimally invasive, percutaneous needle puncture treatment for tennis elbow. Adv Ther. 2008;25(10):1031-1036. doi:10.1007/s12325-008-0099-6.
- Sorensen AA, Howard D, Tan WH, Ketchersid J, Calfee RP. Minimal clinically important differences of 3 patient-related outcomes instruments. J Hand Surg Am. 2013;38(4):641-649. doi:10.1016/j.jhsa.2012.12.032.
- Kelly AM. The minimum clinically significant difference in visual analogue scale pain score does not differ with severity of pain. Emerg Med J. 2001;18(3):205-207. doi:10.1136/emj.18.3.205.
- Tashjian RZ, Deloach J, Porucznik CA, Powell AP. Minimal clinically important differences (MCID) and patient acceptable symptomatic state (PASS) for visual analog scales (VAS) measuring pain in patients treated for rotator cuff disease. J Shoulder Elbow Surg. 2009;18(6):927-932. doi:10.1016/j.jse.2009.03.021.
- Cummins CA. Lateral epicondylitis: in vivo assessment of arthroscopic debridement and correlation with patient outcomes. Am J Sports Med. 2006;34(9):1486-1491. doi:10.1177/0363546506288016.
- Stapleton TR, Baker CL. Arthroscopic treatment of lateral epicondylitis: a clinical study. Arthroscopy. 1996;1:365-366.
- Hastings H. Open treatment for lateral tennis elbow good for certain indications. Orthop Today. 2009;2:1-2.
- Duparc F, Putz R, Michot C, Muller JM, Fréger P. The synovial fold of the humeroradial joint: anatomical and histological features, and clinical relevance in lateral epicondylalgia of the elbow. Surg Radiol Anat. 2002;24(5):302-307. doi:10.1007/s00276-002-0055-0.
TAKE-HOME POINTS
- While favorable results have been reported for open, arthroscopic, and percutaneous surgical techniques, there is no current consensus regarding the optimal technique for lateral epicondylitis.
- There is no difference between open, arthroscopic, and percutaneous surgical treatment for lateral epicondylitis regarding return to work and subjective satisfaction.
- Open treatment led to a greater percentage of patients being pain free at final follow-up.
- While arthroscopic treatment led to better pain and functional scores at final follow-up, the absolute differences were quite small and likely not clinically significant.
- We recommend open débridement as the best means of minimizing cost and achieving a pain-free outcome in the long-term.
Prevention of Central Line–Associated Bloodstream Infections
Division of Infectious Diseases, Department of Internal Medicine, VA Ann Arbor Healthcare System and University of Michigan Health System, Ann Arbor, MI.
Abstract
- Objective: To review prevention of central line–associated bloodstream infection (CLABSI).
- Method: Review of the literature.
- Results: Evidence-based prevention practices include ensuring hand hygiene before the procedure, using maximal sterile barrier precautions, cleaning the skin with alcoholic chlorhexidine before central line insertion, avoiding the femoral site for insertion, and removing unneeded catheters.
- Conclusion: For continued success in CLABSI prevention, best practices should be followed and patient safety should be emphasized.
Health care–associated infections (HAIs) are a preventable cause of morbidity and mortality in the United States and internationally. A Centers for Disease Control and Prevention (CDC) report estimates that in acute care hospitals, 1 in 25 patients end up with at least one HAI during their hospital stay [1]. HAIs can also be costly; in the United States, the indirect and direct cost has been estimated to be between $96 to $147 billion dollars [2]. National initiatives to prevent these types of infections have included efforts from the Department of Health and Human Services (HHS), the Institute of Medicine (IOM), the Institute for Healthcare Improvement (IHI) and the Centers for Medicare and Medicaid Services (CMS). This work has led to particular success in preventing central line–associated bloodstream infection (CLABSI).
CLABSI can lead to considerable mortality, morbidity, and cost. An estimated 250,000 CLABSIs occur in patients yearly, and about 80,000 of those are estimated to occur in the intensive care unit (ICU) setting [3]. Since central venous catheters (CVCs), or central lines, are most often used in the ICU setting, much of the work on prevention and management of CLABSI has been within the ICU population [4,5]. The increased use of peripherally inserted central catheters (PICCs) in the non-ICU setting and recognition of CLABSI in non-ICU settings has led to new efforts to understand the best way to prevent CLABSI in the non-ICU setting [4,6]. Regardless of setting, the annual cost of these infections has been estimated to be as high as $2.3 billion [7]. One episode is estimated to cost a hospital up to $46,485 per episode with components of excess length of stay, antibiotic cost, and cost of care [8]. In this review, selected best practices in CLABSI prevention are identified and described.
Elements of CLABSI Prevention
One of the key papers in the CLABSI literature was the Keystone ICU project in Michigan [9]. This state-wide effort grew out of a successful pilot patient-safety program that was trialed at Johns Hopkins Medical Institutions to reduce CLABSI in the ICU setting. In 2003, the Agency for Healthcare Research and Quality (AHRQ) funded a study to examine the intervention in ICUs in the state of Michigan. A total of 108 ICUs from 67 individual hospitals participated in the pre-intervention/post-intervention study [9]. A combination of technical and socio-adaptive interventions to prevent CLABSI included clinician education on best practices in insertion of central lines, having a central-line cart in each ICU, an insertion checklist of best practices, empowering nursing staff to stop the procedure if best practices were not being followed, discussing removal of catheters daily, and providing feedback to units regarding rates of CLABSI [10]. Executive administration of each hospital was also involved and there were monthly phone calls for hospital teams to share successes and barriers.
In the pre-intervention phase, the median catheter- related bloodstream infection rate was 2.7 infections per 1000 catheter days for the sum of hospitals. After the interventions were put in place, the median rate of catheter related bloodstream infections was down to 0.34 at 18 months. The study showed that results from a relatively inexpensive and straightforward intervention could be effective and could last in the long term. This study led to many other single center and multicenter studies, nationally and internationally, to replicate results in efforts to decrease CLABSI in ICU populations [5]. The CDC and AHRQ have continued to partner with regional, state and national efforts to focus on CLABSI prevention.
The Bundle Approach
A number of interventions have been proven to be effective at preventing CLABSI. Combining more than one intervention can often have additive effects. This effect has been recognized in numerous quality improvement studies on CLABSI and has been termed using the “bundle” approach. 
Hand Hygiene
Poor hand hygiene by health care workers is generally thought to be the most common cause of HAIs [12]. Guidelines recommend an alcohol-based waterless product or antiseptic soap and water prior to catheter insertion [13]. The most common underlying etiology of CLABSI is through microorganisms introduced at time of insertion of catheter. This can be extraluminally mediated via skin flora of the patient, or due to lack of hand washing on the inserter’s part and can lead to CLABSI [14]. While a randomized controlled trial would be unethical, several studies have shown when targeted hand hygiene campaigns are held, CLABSI rates tend to decrease [15–17].
Maximal Barrier Precautions
The use of maximal sterile barrier precautions has been associated with less mortality, decreasing catheter colonization, incidence of HAI and cost savings [18–20]. Like most components of the bundle, maximal sterile barrier precautions have rarely been studied alone, but are often a part of a “bundle” or number of interventions [21]. Like hand hygiene, while regularly a part of many hospital’s checklist or bundle process, compliance with this key part of infection prevention can be deficient; one study noted measured maximal sterile barriers compliance to be 44% [22].
Chlorhexidine Skin Antisepsis
Chlorhexidine skin preparation decreases bacterial burden at site of insertion and is thought to reduce infection from this mechanism. Chlorhexidine-alcohol skin preparation has been proven in randomized controlled trials to outperform povidone iodine-alcohol in preventing CLABSI [23,24]. Chlorhexidine skin preparation is considered a technical element of checklists and is thought to be a straightforward and easily implementable action [25]. If a hospital supplies only alcoholic chlorhexidine and doesn’t provide povidone-iodine for skin preparation, then clinicians can be “nudged” towards performing this part of the bundle.
Optimal Catheter Site Selection
For all sites of insertion of CVC, the risk of mechanical and infectious complications depends on the skill and proficiency of operators, the clinical situation, and the availability of ultrasound to help guide placement. These factors are important in determining which anatomical site is best for each patient [26]. The femoral site has been associated with a greater risk of catheter-related infection and catheter-related thrombosis and is not recommended as the initial choice for non-emergent CVC insertion according to national guidelines [13,27]. The internal jugular vein site is associated with a lower risk of severe mechanical complications such as pneumothorax when compared to subclavian vein site [27]. The subclavian vein site is associated with a lower risk of catheter-related blood stream infection and lower rate of thrombosis, but this greatly depends on experience of operator. Experts have proposed that the subclavian site has a lower burden of colonization by bacteria than other sites and is anatomically more protected by catheter dressing; also the subcutaneous course of the central line itself is longer for the subclavian site than other sites and these reasons could contribute to the lower risk of infection [28]. The subclavian site is, however, associated with a higher risk of mechanical complications that can be serious for ICU patients. In general, the femoral vein site should be avoided in non-emergent line placement situations, particularly if the patient is an obese adult [13]. Using ultrasound as a guidance for catheter insertion has also been shown to reduce risk of CLABSI and other mechanical complications and is recommended [29,30].
Daily Review of Line Necessity
Removing unnecessary catheters as soon as possible decreases catheter dwell time and risk of infection. Few studies have concentrated on this step alone in CLABSI prevention, but the studies that have focused on catheter removal usually implement electronic reminders or multidisciplinary catheter rounds (where need for catheter is incorporated into daily rounds or discussed separately by a multidisciplinary group) [5,31].
Additional Considerations
Other basic practices that all hospitals should adopt include the above strategies and providing all inclusive catheter carts or kits, disinfecting hubs in maintenance care of catheters, covering the CVC site with sterile dressings, having recurrent educational interventions and using checklists to assure adherence to the evidence-based bundle (Table) [4,13]. As prevalence of non-ICU central lines has also grown, maintenance care is particularly important in reducing CLABSI. Maintenance bundles that highlight best practices such as aseptic technique, correct hand hygiene, chlorhexidine skin disinfection scrub, antimicrobial bandage application, and catheter hub disinfection have been used with success [32]. Specialized CVC insertion teams with trained personnel have also been recommended [4]. When these basic evidence-based practices are still unable to bring down CLABSI rates for select populations or during an outbreak, supplemental strategies can be tried to reduce CLABSI. These include antimicrobial-impregnated catheters, chlorhexidine-impregnated dressings, and chlorhexidine bathing, which is increasingly being used in the ICU setting [5,13,33].
Epidemiology/Risk Factors
At-risk Populations
ICU patients are at risk for CLABSI because of frequent use of multiple catheters, and the comorbidities and acuity of care that these patients have. ICU patients also tend to have lots of manipulation of their catheters and often these catheters are placed in emergent situations [13]. Patients in the non-ICU and outpatient setting are also at risk for CLABSI when they have a central venous catheter. Long courses of antibiotics for disease states such as osteomyelitis and endocarditis often entail central venous catheters. Recent work has shown that PICCs carry as high of a CLABSI risk as short-term CVCs in hospitalized patients [34]. Patients with end-stage renal disease, especially those undergoing maintenance hemodialysis via a tunneled dialysis catheter are particularly vulnerable to CLABSI [13,35].
Risk Factors for CLABSI
A number of studies have reviewed risk factors and epidemiology of CLABSI in the adult and pediatric population. Factors that have been associated with risk of CLABSI in more than one study include prolonged hospitalization before placement of the central line, prolonged duration of the central line, heavy microbial colonization at the site of insertion, heavy microbial colonization of the catheter hub, multiple lumens, internal jugular site catheterization, femoral vein site catheterization, neutropenia of the patient, a reduced nurse to patient ratio in the ICU setting, presence of total parenteral nutrition, and poor maintenance care of the catheter [4,13,36–40]. One study [41] that calculated a score to help predict risk of PICC-CLABSI found that previous CLABSI (within 3 months of PICC insertion) significantly increases risk of repeat CLABSI.
Conclusion
CLABSI is an important cause of morbidity, mortality and cost. There has been remarkable success in prevention of these infections in recent years due to focused efforts on patient safety. As efforts have multiplied to put into place interventions to decrease CLABSI nationally, the CDC published a Vital Signs report discussing the impact of these efforts [42]. It was estimated that over one decade, infection prevention efforts had avoided 25,000 CLABSIs in U.S. ICUs, a 58% reduction in this infection [42]. CLABSI has served as the best example of using evidence-based interventions through an infection prevention bundle or framework to reduce HAIs. Similar approaches are being used to try to reduce catheter-associated urinary tract infection, Clostridium difficile infection, surgical site infection, and ventilator-associated pneumonia, but there have been less distinct successes nationally and internationally for these other HAIs.
The literature emphasizes that there are several evidence-based measures that can prevent CLABSI. These include hand hygiene, using alcoholic chlorhexidine for skin preparation prior to insertion, maximal sterile barrier precautions, avoiding the femoral site for CVC insertion, and removing unnecessary catheters as soon as possible. Support from administration in emphasizing patient safety and HAI prevention along with following evidence-based practice could lead to long-term improvement in CLABSI prevention across hospital systems.
Corresponding author: Payal K. Patel, MD, MPH, Div of Infectious Diseases, Dept of Internal Medicine, VA Ann Arbor Healthcare System, 2215 Fuller Rd, Ann Arbor, MI 48105, [email protected].
Financial disclosures: None.
1. Magill SS, Edwards JR, Bamberg W, et al; Emerging Infections Program Healthcare-Associated Infections and Antimicrobial Use Prevalence Survey Team. Multistate point-prevalence survey of health care-associated infections. N Engl J Med 2014;370:1198–208.
2. Marchetti A, Rossiter R. Economic burden of healthcare-associated infection in US acute care hospitals: societal perspective. J Med Econ 2013;16:1399–404.
3. O’Neil C, Ball K, Wood H, et al. A central line care maintenance bundle for the prevention of central line-associated bloodstream infection in non-intensive care unit settings. Infect Control Hosp Epidemiol 2016;37:1–7.
4. Shekelle PG, Wachter RM, Pronovost PJ, et al. Making health care safer II: an updated critical analysis of the evidence for patient safety practices. Evid Rep Technol Assess (Full Rep) 2013:1–945.
5. Patel PK, Gupta A, Vaughn VM, Mann JD, Ameling JM, Meddings J. Review of strategies to reduce central line-associated bloodstream infection (CLABSI) and catheter-associated urinary tract infection (CAUTI) in adult ICUs. J Hosp Med 2018;13:105–16.
6. Chopra V, Ratz D, Kuhn L, et al. PICC-associated bloodstream infections: prevalence, patterns, and predictors. Am J Med 2014;127:319–28.
7. Sagana R, Hyzy RC. Achieving zero central line-associated bloodstream infection rates in your intensive care unit. Crit Care Clin 2013;29:1–9.
8. Nelson RE, Angelovic AW, Nelson SD, Gleed JR, Drews FA. An economic analysis of adherence engineering to improve use of best practices during central line maintenance procedures. Infect Control Hosp Epidemiol 2015;36:550–6.
9. Pronovost P, Needham D, Berenholtz S, et al. An intervention to decrease catheter-related bloodstream infections in the ICU. N Engl J Med 2006;355:2725–32.
10. Dumyati G, Concannon C, van Wijngaarden E, et al. Sustained reduction of central line-associated bloodstream infections outside the intensive care unit with a multimodal intervention focusing on central line maintenance. Am J Infect Control 2014;42:723–30.
11. Sacks GD, Diggs BS, Hadjizacharia P, et al. Reducing the rate of catheter-associated bloodstream infections in a surgical intensive care unit using the Institute for Healthcare Improvement Central Line Bundle. Am J Surg 2014;207:817–23.
12. Boyce JM, Pittet D; Healthcare Infection Control Practices Advisory Committee; HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Guideline for hand hygiene in health-care settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Society for Healthcare Epidemiology of America/Association for Professionals in Infection Control/Infectious Diseases Society of America. MMWR Recomm Rep 2002; 51:1-45, quiz CE1–4.
13. Marschall J, Mermel LA, et al; Society for Healthcare Epidemiology of America. Strategies to prevent central line-associated bloodstream infections in acute care hospitals: 2014 update. Infect Control Hosp Epidemiol 2014;35:753–71.
14. Safdar N, Maki DG. The pathogenesis of catheter-related bloodstream infection with noncuffed short-term central venous catheters. Intensive Care Med 2004;30:62–7.
15. Shabot MM, Chassin MR, France AC, et al. Using the targeted solutions tool(R) to improve hand hygiene compliance is associated with decreased health care-associated infections. Jt Comm J Qual Patient Saf 2016;42:6–17.
16. Johnson L, Grueber S, Schlotzhauer C, et al. A multifactorial action plan improves hand hygiene adherence and significantly reduces central line-associated bloodstream infections. Am J Infect Control 2014;42:1146–51.
17. Barrera L, Zingg W, Mendez F, Pittet D. Effectiveness of a hand hygiene promotion strategy using alcohol-based handrub in 6 intensive care units in Colombia. Am J Infect Control 2011;39:633–9.
18. Hu KK, Lipsky BA, Veenstra DL, Saint S. Using maximal sterile barriers to prevent central venous catheter-related infection: a systematic evidence-based review. Am J Infect Control 2004;32:142–6.
19. Hu KK, Veenstra DL, Lipsky BA, Saint S. Use of maximal sterile barriers during central venous catheter insertion: clinical and economic outcomes. Clin Infect Dis 2004;39:1441–5.
20. Raad II, Hohn DC, Gilbreath BJ, et al. Prevention of central venous catheter-related infections by using maximal sterile barrier precautions during insertion. Infect Control Hosp Epidemiol 1994;15:231–8.
21. Furuya EY, Dick AW, Herzig CT, Pogorzelska-Maziarz M, Larson EL, Stone PW. Central line-associated bloodstream infection reduction and bundle compliance in intensive care units: a national study. Infect Control Hosp Epidemiol 2016;37:805–10.
22. Sherertz RJ, Ely EW, Westbrook DM, et al. Education of physicians-in-training can decrease the risk for vascular catheter infection. Ann Intern Med 2000;132:641–8.
23. Mimoz O, Lucet JC, Kerforne T, Pascal J, et al. Skin antisepsis with chlorhexidine-alcohol versus povidone iodine-alcohol, with and without skin scrubbing, for prevention of intravascular-catheter-related infection (CLEAN): an open-label, multicentre, randomised, controlled, two-by-two factorial trial. Lancet 2015;386:2069–77.
24. Lai NM, Lai NA, O’Riordan E, et al. Skin antisepsis for reducing central venous catheter-related infections. Cochrane Database Syst Rev 2016;7:CD010140.
25. Chopra V, Shojania KG. Recipes for checklists and bundles: one part active ingredient, two parts measurement. BMJ Qual Saf 2013;22:93–6.
26. Marik PE, Flemmer M, Harrison W. The risk of catheter-related bloodstream infection with femoral venous catheters as compared to subclavian and internal jugular venous catheters: a systematic review of the literature and meta-analysis. Crit Care Med 2012;40:2479–85.
27. Timsit JF. What is the best site for central venous catheter insertion in critically ill patients? Crit Care 2003;7:397–99.
28. Parienti JJ, Mongardon N, Megarbane B, et al; 3SITES Study Group. Intravascular complications of central venous catheterization by insertion site. N Engl J Med 2015;373:1220–9.
29. Hind D, Calvert N, McWilliams R, et al. Ultrasonic locating devices for central venous cannulation: meta-analysis. BMJ 2003;327:361.
30. Fragou M, Gravvanis A, Dimitriou V, et al. Real-time ultrasound-guided subclavian vein cannulation versus the landmark method in critical care patients: a prospective randomized study. Crit Care Med 2011;39:1607–12.
31. Pageler NM, Longhurst CA, Wood M, et al. Use of electronic medical record-enhanced checklist and electronic dashboard to decrease CLABSIs. Pediatrics 2014;133:e738–46.
32. Drews FA, Bakdash JZ, Gleed JR. Improving central line maintenance to reduce central line-associated bloodstream infections. Am J Infect Control 2017;45:1224–30.
33. Frost SA, Alogso MC, Metcalfe L, et al. Chlorhexidine bathing and health care-associated infections among adult intensive care patients: a systematic review and meta-analysis. Crit Care 2016;20:379.
34. Chopra V, O’Horo JC, Rogers MA, et al. The risk of bloodstream infection associated with peripherally inserted central catheters compared with central venous catheters in adults: a systematic review and meta-analysis. Infect Control Hosp Epidemiol 2013;34:908–18.
35. Xue H, Ix JH, Wang W, et al. Hemodialysis access usage patterns in the incident dialysis year and associated catheter-related complications. Am J Kidney Dis 2013;61:123–30.
36. Almuneef MA, Memish ZA, Balkhy HH, et al. Rate, risk factors and outcomes of catheter-related bloodstream infection in a paediatric intensive care unit in Saudi Arabia. J Hosp Infect 2006;62:207–13.
37. Alonso-Echanove J, Edwards JR, Richards MJ, et al. Effect of nurse staffing and antimicrobial-impregnated central venous catheters on the risk for bloodstream infections in intensive care units. Infect Control Hosp Epidemiol 2003;24:916–25.
38. Lorente L, Henry C, Martin MM, et al. Central venous catheter-related infection in a prospective and observational study of 2,595 catheters. Crit Care 2005;9:R631–5.
39. Rey C, Alvarez F, De-La-Rua V, et al. Intervention to reduce catheter-related bloodstream infections in a pediatric intensive care unit. Intensive Care Med 2011;37:678–85.
40. O’Brien J, Paquet F, Lindsay R, Valenti D. Insertion of PICCs with minimum number of lumens reduces complications and costs. J Am Coll Radiol. 2013;10:864–8.
41. Herc E, Patel P, Washer LL, et al. A model to predict central-line-associated bloodstream infection among patients with peripherally inserted central catheters: the MPC score. Infect Control Hosp Epidemiol 2017;38:1155–66.
42. Centers for Disease Control and Prevention. Vital signs: central line-associated blood stream infections--United States, 2001, 2008, and 2009. MMWR Morb Mortal Wkly Rep 2011;60:243–8.
Division of Infectious Diseases, Department of Internal Medicine, VA Ann Arbor Healthcare System and University of Michigan Health System, Ann Arbor, MI.
Abstract
- Objective: To review prevention of central line–associated bloodstream infection (CLABSI).
- Method: Review of the literature.
- Results: Evidence-based prevention practices include ensuring hand hygiene before the procedure, using maximal sterile barrier precautions, cleaning the skin with alcoholic chlorhexidine before central line insertion, avoiding the femoral site for insertion, and removing unneeded catheters.
- Conclusion: For continued success in CLABSI prevention, best practices should be followed and patient safety should be emphasized.
Health care–associated infections (HAIs) are a preventable cause of morbidity and mortality in the United States and internationally. A Centers for Disease Control and Prevention (CDC) report estimates that in acute care hospitals, 1 in 25 patients end up with at least one HAI during their hospital stay [1]. HAIs can also be costly; in the United States, the indirect and direct cost has been estimated to be between $96 to $147 billion dollars [2]. National initiatives to prevent these types of infections have included efforts from the Department of Health and Human Services (HHS), the Institute of Medicine (IOM), the Institute for Healthcare Improvement (IHI) and the Centers for Medicare and Medicaid Services (CMS). This work has led to particular success in preventing central line–associated bloodstream infection (CLABSI).
CLABSI can lead to considerable mortality, morbidity, and cost. An estimated 250,000 CLABSIs occur in patients yearly, and about 80,000 of those are estimated to occur in the intensive care unit (ICU) setting [3]. Since central venous catheters (CVCs), or central lines, are most often used in the ICU setting, much of the work on prevention and management of CLABSI has been within the ICU population [4,5]. The increased use of peripherally inserted central catheters (PICCs) in the non-ICU setting and recognition of CLABSI in non-ICU settings has led to new efforts to understand the best way to prevent CLABSI in the non-ICU setting [4,6]. Regardless of setting, the annual cost of these infections has been estimated to be as high as $2.3 billion [7]. One episode is estimated to cost a hospital up to $46,485 per episode with components of excess length of stay, antibiotic cost, and cost of care [8]. In this review, selected best practices in CLABSI prevention are identified and described.
Elements of CLABSI Prevention
One of the key papers in the CLABSI literature was the Keystone ICU project in Michigan [9]. This state-wide effort grew out of a successful pilot patient-safety program that was trialed at Johns Hopkins Medical Institutions to reduce CLABSI in the ICU setting. In 2003, the Agency for Healthcare Research and Quality (AHRQ) funded a study to examine the intervention in ICUs in the state of Michigan. A total of 108 ICUs from 67 individual hospitals participated in the pre-intervention/post-intervention study [9]. A combination of technical and socio-adaptive interventions to prevent CLABSI included clinician education on best practices in insertion of central lines, having a central-line cart in each ICU, an insertion checklist of best practices, empowering nursing staff to stop the procedure if best practices were not being followed, discussing removal of catheters daily, and providing feedback to units regarding rates of CLABSI [10]. Executive administration of each hospital was also involved and there were monthly phone calls for hospital teams to share successes and barriers.
In the pre-intervention phase, the median catheter- related bloodstream infection rate was 2.7 infections per 1000 catheter days for the sum of hospitals. After the interventions were put in place, the median rate of catheter related bloodstream infections was down to 0.34 at 18 months. The study showed that results from a relatively inexpensive and straightforward intervention could be effective and could last in the long term. This study led to many other single center and multicenter studies, nationally and internationally, to replicate results in efforts to decrease CLABSI in ICU populations [5]. The CDC and AHRQ have continued to partner with regional, state and national efforts to focus on CLABSI prevention.
The Bundle Approach
A number of interventions have been proven to be effective at preventing CLABSI. Combining more than one intervention can often have additive effects. This effect has been recognized in numerous quality improvement studies on CLABSI and has been termed using the “bundle” approach.
Hand Hygiene
Poor hand hygiene by health care workers is generally thought to be the most common cause of HAIs [12]. Guidelines recommend an alcohol-based waterless product or antiseptic soap and water prior to catheter insertion [13]. The most common underlying etiology of CLABSI is through microorganisms introduced at time of insertion of catheter. This can be extraluminally mediated via skin flora of the patient, or due to lack of hand washing on the inserter’s part and can lead to CLABSI [14]. While a randomized controlled trial would be unethical, several studies have shown when targeted hand hygiene campaigns are held, CLABSI rates tend to decrease [15–17].
Maximal Barrier Precautions
The use of maximal sterile barrier precautions has been associated with less mortality, decreasing catheter colonization, incidence of HAI and cost savings [18–20]. Like most components of the bundle, maximal sterile barrier precautions have rarely been studied alone, but are often a part of a “bundle” or number of interventions [21]. Like hand hygiene, while regularly a part of many hospital’s checklist or bundle process, compliance with this key part of infection prevention can be deficient; one study noted measured maximal sterile barriers compliance to be 44% [22].
Chlorhexidine Skin Antisepsis
Chlorhexidine skin preparation decreases bacterial burden at site of insertion and is thought to reduce infection from this mechanism. Chlorhexidine-alcohol skin preparation has been proven in randomized controlled trials to outperform povidone iodine-alcohol in preventing CLABSI [23,24]. Chlorhexidine skin preparation is considered a technical element of checklists and is thought to be a straightforward and easily implementable action [25]. If a hospital supplies only alcoholic chlorhexidine and doesn’t provide povidone-iodine for skin preparation, then clinicians can be “nudged” towards performing this part of the bundle.
Optimal Catheter Site Selection
For all sites of insertion of CVC, the risk of mechanical and infectious complications depends on the skill and proficiency of operators, the clinical situation, and the availability of ultrasound to help guide placement. These factors are important in determining which anatomical site is best for each patient [26]. The femoral site has been associated with a greater risk of catheter-related infection and catheter-related thrombosis and is not recommended as the initial choice for non-emergent CVC insertion according to national guidelines [13,27]. The internal jugular vein site is associated with a lower risk of severe mechanical complications such as pneumothorax when compared to subclavian vein site [27]. The subclavian vein site is associated with a lower risk of catheter-related blood stream infection and lower rate of thrombosis, but this greatly depends on experience of operator. Experts have proposed that the subclavian site has a lower burden of colonization by bacteria than other sites and is anatomically more protected by catheter dressing; also the subcutaneous course of the central line itself is longer for the subclavian site than other sites and these reasons could contribute to the lower risk of infection [28]. The subclavian site is, however, associated with a higher risk of mechanical complications that can be serious for ICU patients. In general, the femoral vein site should be avoided in non-emergent line placement situations, particularly if the patient is an obese adult [13]. Using ultrasound as a guidance for catheter insertion has also been shown to reduce risk of CLABSI and other mechanical complications and is recommended [29,30].
Daily Review of Line Necessity
Removing unnecessary catheters as soon as possible decreases catheter dwell time and risk of infection. Few studies have concentrated on this step alone in CLABSI prevention, but the studies that have focused on catheter removal usually implement electronic reminders or multidisciplinary catheter rounds (where need for catheter is incorporated into daily rounds or discussed separately by a multidisciplinary group) [5,31].
Additional Considerations
Other basic practices that all hospitals should adopt include the above strategies and providing all inclusive catheter carts or kits, disinfecting hubs in maintenance care of catheters, covering the CVC site with sterile dressings, having recurrent educational interventions and using checklists to assure adherence to the evidence-based bundle (Table) [4,13]. As prevalence of non-ICU central lines has also grown, maintenance care is particularly important in reducing CLABSI. Maintenance bundles that highlight best practices such as aseptic technique, correct hand hygiene, chlorhexidine skin disinfection scrub, antimicrobial bandage application, and catheter hub disinfection have been used with success [32]. Specialized CVC insertion teams with trained personnel have also been recommended [4]. When these basic evidence-based practices are still unable to bring down CLABSI rates for select populations or during an outbreak, supplemental strategies can be tried to reduce CLABSI. These include antimicrobial-impregnated catheters, chlorhexidine-impregnated dressings, and chlorhexidine bathing, which is increasingly being used in the ICU setting [5,13,33].
Epidemiology/Risk Factors
At-risk Populations
ICU patients are at risk for CLABSI because of frequent use of multiple catheters, and the comorbidities and acuity of care that these patients have. ICU patients also tend to have lots of manipulation of their catheters and often these catheters are placed in emergent situations [13]. Patients in the non-ICU and outpatient setting are also at risk for CLABSI when they have a central venous catheter. Long courses of antibiotics for disease states such as osteomyelitis and endocarditis often entail central venous catheters. Recent work has shown that PICCs carry as high of a CLABSI risk as short-term CVCs in hospitalized patients [34]. Patients with end-stage renal disease, especially those undergoing maintenance hemodialysis via a tunneled dialysis catheter are particularly vulnerable to CLABSI [13,35].
Risk Factors for CLABSI
A number of studies have reviewed risk factors and epidemiology of CLABSI in the adult and pediatric population. Factors that have been associated with risk of CLABSI in more than one study include prolonged hospitalization before placement of the central line, prolonged duration of the central line, heavy microbial colonization at the site of insertion, heavy microbial colonization of the catheter hub, multiple lumens, internal jugular site catheterization, femoral vein site catheterization, neutropenia of the patient, a reduced nurse to patient ratio in the ICU setting, presence of total parenteral nutrition, and poor maintenance care of the catheter [4,13,36–40]. One study [41] that calculated a score to help predict risk of PICC-CLABSI found that previous CLABSI (within 3 months of PICC insertion) significantly increases risk of repeat CLABSI.
Conclusion
CLABSI is an important cause of morbidity, mortality and cost. There has been remarkable success in prevention of these infections in recent years due to focused efforts on patient safety. As efforts have multiplied to put into place interventions to decrease CLABSI nationally, the CDC published a Vital Signs report discussing the impact of these efforts [42]. It was estimated that over one decade, infection prevention efforts had avoided 25,000 CLABSIs in U.S. ICUs, a 58% reduction in this infection [42]. CLABSI has served as the best example of using evidence-based interventions through an infection prevention bundle or framework to reduce HAIs. Similar approaches are being used to try to reduce catheter-associated urinary tract infection, Clostridium difficile infection, surgical site infection, and ventilator-associated pneumonia, but there have been less distinct successes nationally and internationally for these other HAIs.
The literature emphasizes that there are several evidence-based measures that can prevent CLABSI. These include hand hygiene, using alcoholic chlorhexidine for skin preparation prior to insertion, maximal sterile barrier precautions, avoiding the femoral site for CVC insertion, and removing unnecessary catheters as soon as possible. Support from administration in emphasizing patient safety and HAI prevention along with following evidence-based practice could lead to long-term improvement in CLABSI prevention across hospital systems.
Corresponding author: Payal K. Patel, MD, MPH, Div of Infectious Diseases, Dept of Internal Medicine, VA Ann Arbor Healthcare System, 2215 Fuller Rd, Ann Arbor, MI 48105, [email protected].
Financial disclosures: None.
Division of Infectious Diseases, Department of Internal Medicine, VA Ann Arbor Healthcare System and University of Michigan Health System, Ann Arbor, MI.
Abstract
- Objective: To review prevention of central line–associated bloodstream infection (CLABSI).
- Method: Review of the literature.
- Results: Evidence-based prevention practices include ensuring hand hygiene before the procedure, using maximal sterile barrier precautions, cleaning the skin with alcoholic chlorhexidine before central line insertion, avoiding the femoral site for insertion, and removing unneeded catheters.
- Conclusion: For continued success in CLABSI prevention, best practices should be followed and patient safety should be emphasized.
Health care–associated infections (HAIs) are a preventable cause of morbidity and mortality in the United States and internationally. A Centers for Disease Control and Prevention (CDC) report estimates that in acute care hospitals, 1 in 25 patients end up with at least one HAI during their hospital stay [1]. HAIs can also be costly; in the United States, the indirect and direct cost has been estimated to be between $96 to $147 billion dollars [2]. National initiatives to prevent these types of infections have included efforts from the Department of Health and Human Services (HHS), the Institute of Medicine (IOM), the Institute for Healthcare Improvement (IHI) and the Centers for Medicare and Medicaid Services (CMS). This work has led to particular success in preventing central line–associated bloodstream infection (CLABSI).
CLABSI can lead to considerable mortality, morbidity, and cost. An estimated 250,000 CLABSIs occur in patients yearly, and about 80,000 of those are estimated to occur in the intensive care unit (ICU) setting [3]. Since central venous catheters (CVCs), or central lines, are most often used in the ICU setting, much of the work on prevention and management of CLABSI has been within the ICU population [4,5]. The increased use of peripherally inserted central catheters (PICCs) in the non-ICU setting and recognition of CLABSI in non-ICU settings has led to new efforts to understand the best way to prevent CLABSI in the non-ICU setting [4,6]. Regardless of setting, the annual cost of these infections has been estimated to be as high as $2.3 billion [7]. One episode is estimated to cost a hospital up to $46,485 per episode with components of excess length of stay, antibiotic cost, and cost of care [8]. In this review, selected best practices in CLABSI prevention are identified and described.
Elements of CLABSI Prevention
One of the key papers in the CLABSI literature was the Keystone ICU project in Michigan [9]. This state-wide effort grew out of a successful pilot patient-safety program that was trialed at Johns Hopkins Medical Institutions to reduce CLABSI in the ICU setting. In 2003, the Agency for Healthcare Research and Quality (AHRQ) funded a study to examine the intervention in ICUs in the state of Michigan. A total of 108 ICUs from 67 individual hospitals participated in the pre-intervention/post-intervention study [9]. A combination of technical and socio-adaptive interventions to prevent CLABSI included clinician education on best practices in insertion of central lines, having a central-line cart in each ICU, an insertion checklist of best practices, empowering nursing staff to stop the procedure if best practices were not being followed, discussing removal of catheters daily, and providing feedback to units regarding rates of CLABSI [10]. Executive administration of each hospital was also involved and there were monthly phone calls for hospital teams to share successes and barriers.
In the pre-intervention phase, the median catheter- related bloodstream infection rate was 2.7 infections per 1000 catheter days for the sum of hospitals. After the interventions were put in place, the median rate of catheter related bloodstream infections was down to 0.34 at 18 months. The study showed that results from a relatively inexpensive and straightforward intervention could be effective and could last in the long term. This study led to many other single center and multicenter studies, nationally and internationally, to replicate results in efforts to decrease CLABSI in ICU populations [5]. The CDC and AHRQ have continued to partner with regional, state and national efforts to focus on CLABSI prevention.
The Bundle Approach
A number of interventions have been proven to be effective at preventing CLABSI. Combining more than one intervention can often have additive effects. This effect has been recognized in numerous quality improvement studies on CLABSI and has been termed using the “bundle” approach.
Hand Hygiene
Poor hand hygiene by health care workers is generally thought to be the most common cause of HAIs [12]. Guidelines recommend an alcohol-based waterless product or antiseptic soap and water prior to catheter insertion [13]. The most common underlying etiology of CLABSI is through microorganisms introduced at time of insertion of catheter. This can be extraluminally mediated via skin flora of the patient, or due to lack of hand washing on the inserter’s part and can lead to CLABSI [14]. While a randomized controlled trial would be unethical, several studies have shown when targeted hand hygiene campaigns are held, CLABSI rates tend to decrease [15–17].
Maximal Barrier Precautions
The use of maximal sterile barrier precautions has been associated with less mortality, decreasing catheter colonization, incidence of HAI and cost savings [18–20]. Like most components of the bundle, maximal sterile barrier precautions have rarely been studied alone, but are often a part of a “bundle” or number of interventions [21]. Like hand hygiene, while regularly a part of many hospital’s checklist or bundle process, compliance with this key part of infection prevention can be deficient; one study noted measured maximal sterile barriers compliance to be 44% [22].
Chlorhexidine Skin Antisepsis
Chlorhexidine skin preparation decreases bacterial burden at site of insertion and is thought to reduce infection from this mechanism. Chlorhexidine-alcohol skin preparation has been proven in randomized controlled trials to outperform povidone iodine-alcohol in preventing CLABSI [23,24]. Chlorhexidine skin preparation is considered a technical element of checklists and is thought to be a straightforward and easily implementable action [25]. If a hospital supplies only alcoholic chlorhexidine and doesn’t provide povidone-iodine for skin preparation, then clinicians can be “nudged” towards performing this part of the bundle.
Optimal Catheter Site Selection
For all sites of insertion of CVC, the risk of mechanical and infectious complications depends on the skill and proficiency of operators, the clinical situation, and the availability of ultrasound to help guide placement. These factors are important in determining which anatomical site is best for each patient [26]. The femoral site has been associated with a greater risk of catheter-related infection and catheter-related thrombosis and is not recommended as the initial choice for non-emergent CVC insertion according to national guidelines [13,27]. The internal jugular vein site is associated with a lower risk of severe mechanical complications such as pneumothorax when compared to subclavian vein site [27]. The subclavian vein site is associated with a lower risk of catheter-related blood stream infection and lower rate of thrombosis, but this greatly depends on experience of operator. Experts have proposed that the subclavian site has a lower burden of colonization by bacteria than other sites and is anatomically more protected by catheter dressing; also the subcutaneous course of the central line itself is longer for the subclavian site than other sites and these reasons could contribute to the lower risk of infection [28]. The subclavian site is, however, associated with a higher risk of mechanical complications that can be serious for ICU patients. In general, the femoral vein site should be avoided in non-emergent line placement situations, particularly if the patient is an obese adult [13]. Using ultrasound as a guidance for catheter insertion has also been shown to reduce risk of CLABSI and other mechanical complications and is recommended [29,30].
Daily Review of Line Necessity
Removing unnecessary catheters as soon as possible decreases catheter dwell time and risk of infection. Few studies have concentrated on this step alone in CLABSI prevention, but the studies that have focused on catheter removal usually implement electronic reminders or multidisciplinary catheter rounds (where need for catheter is incorporated into daily rounds or discussed separately by a multidisciplinary group) [5,31].
Additional Considerations
Other basic practices that all hospitals should adopt include the above strategies and providing all inclusive catheter carts or kits, disinfecting hubs in maintenance care of catheters, covering the CVC site with sterile dressings, having recurrent educational interventions and using checklists to assure adherence to the evidence-based bundle (Table) [4,13]. As prevalence of non-ICU central lines has also grown, maintenance care is particularly important in reducing CLABSI. Maintenance bundles that highlight best practices such as aseptic technique, correct hand hygiene, chlorhexidine skin disinfection scrub, antimicrobial bandage application, and catheter hub disinfection have been used with success [32]. Specialized CVC insertion teams with trained personnel have also been recommended [4]. When these basic evidence-based practices are still unable to bring down CLABSI rates for select populations or during an outbreak, supplemental strategies can be tried to reduce CLABSI. These include antimicrobial-impregnated catheters, chlorhexidine-impregnated dressings, and chlorhexidine bathing, which is increasingly being used in the ICU setting [5,13,33].
Epidemiology/Risk Factors
At-risk Populations
ICU patients are at risk for CLABSI because of frequent use of multiple catheters, and the comorbidities and acuity of care that these patients have. ICU patients also tend to have lots of manipulation of their catheters and often these catheters are placed in emergent situations [13]. Patients in the non-ICU and outpatient setting are also at risk for CLABSI when they have a central venous catheter. Long courses of antibiotics for disease states such as osteomyelitis and endocarditis often entail central venous catheters. Recent work has shown that PICCs carry as high of a CLABSI risk as short-term CVCs in hospitalized patients [34]. Patients with end-stage renal disease, especially those undergoing maintenance hemodialysis via a tunneled dialysis catheter are particularly vulnerable to CLABSI [13,35].
Risk Factors for CLABSI
A number of studies have reviewed risk factors and epidemiology of CLABSI in the adult and pediatric population. Factors that have been associated with risk of CLABSI in more than one study include prolonged hospitalization before placement of the central line, prolonged duration of the central line, heavy microbial colonization at the site of insertion, heavy microbial colonization of the catheter hub, multiple lumens, internal jugular site catheterization, femoral vein site catheterization, neutropenia of the patient, a reduced nurse to patient ratio in the ICU setting, presence of total parenteral nutrition, and poor maintenance care of the catheter [4,13,36–40]. One study [41] that calculated a score to help predict risk of PICC-CLABSI found that previous CLABSI (within 3 months of PICC insertion) significantly increases risk of repeat CLABSI.
Conclusion
CLABSI is an important cause of morbidity, mortality and cost. There has been remarkable success in prevention of these infections in recent years due to focused efforts on patient safety. As efforts have multiplied to put into place interventions to decrease CLABSI nationally, the CDC published a Vital Signs report discussing the impact of these efforts [42]. It was estimated that over one decade, infection prevention efforts had avoided 25,000 CLABSIs in U.S. ICUs, a 58% reduction in this infection [42]. CLABSI has served as the best example of using evidence-based interventions through an infection prevention bundle or framework to reduce HAIs. Similar approaches are being used to try to reduce catheter-associated urinary tract infection, Clostridium difficile infection, surgical site infection, and ventilator-associated pneumonia, but there have been less distinct successes nationally and internationally for these other HAIs.
The literature emphasizes that there are several evidence-based measures that can prevent CLABSI. These include hand hygiene, using alcoholic chlorhexidine for skin preparation prior to insertion, maximal sterile barrier precautions, avoiding the femoral site for CVC insertion, and removing unnecessary catheters as soon as possible. Support from administration in emphasizing patient safety and HAI prevention along with following evidence-based practice could lead to long-term improvement in CLABSI prevention across hospital systems.
Corresponding author: Payal K. Patel, MD, MPH, Div of Infectious Diseases, Dept of Internal Medicine, VA Ann Arbor Healthcare System, 2215 Fuller Rd, Ann Arbor, MI 48105, [email protected].
Financial disclosures: None.
1. Magill SS, Edwards JR, Bamberg W, et al; Emerging Infections Program Healthcare-Associated Infections and Antimicrobial Use Prevalence Survey Team. Multistate point-prevalence survey of health care-associated infections. N Engl J Med 2014;370:1198–208.
2. Marchetti A, Rossiter R. Economic burden of healthcare-associated infection in US acute care hospitals: societal perspective. J Med Econ 2013;16:1399–404.
3. O’Neil C, Ball K, Wood H, et al. A central line care maintenance bundle for the prevention of central line-associated bloodstream infection in non-intensive care unit settings. Infect Control Hosp Epidemiol 2016;37:1–7.
4. Shekelle PG, Wachter RM, Pronovost PJ, et al. Making health care safer II: an updated critical analysis of the evidence for patient safety practices. Evid Rep Technol Assess (Full Rep) 2013:1–945.
5. Patel PK, Gupta A, Vaughn VM, Mann JD, Ameling JM, Meddings J. Review of strategies to reduce central line-associated bloodstream infection (CLABSI) and catheter-associated urinary tract infection (CAUTI) in adult ICUs. J Hosp Med 2018;13:105–16.
6. Chopra V, Ratz D, Kuhn L, et al. PICC-associated bloodstream infections: prevalence, patterns, and predictors. Am J Med 2014;127:319–28.
7. Sagana R, Hyzy RC. Achieving zero central line-associated bloodstream infection rates in your intensive care unit. Crit Care Clin 2013;29:1–9.
8. Nelson RE, Angelovic AW, Nelson SD, Gleed JR, Drews FA. An economic analysis of adherence engineering to improve use of best practices during central line maintenance procedures. Infect Control Hosp Epidemiol 2015;36:550–6.
9. Pronovost P, Needham D, Berenholtz S, et al. An intervention to decrease catheter-related bloodstream infections in the ICU. N Engl J Med 2006;355:2725–32.
10. Dumyati G, Concannon C, van Wijngaarden E, et al. Sustained reduction of central line-associated bloodstream infections outside the intensive care unit with a multimodal intervention focusing on central line maintenance. Am J Infect Control 2014;42:723–30.
11. Sacks GD, Diggs BS, Hadjizacharia P, et al. Reducing the rate of catheter-associated bloodstream infections in a surgical intensive care unit using the Institute for Healthcare Improvement Central Line Bundle. Am J Surg 2014;207:817–23.
12. Boyce JM, Pittet D; Healthcare Infection Control Practices Advisory Committee; HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Guideline for hand hygiene in health-care settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Society for Healthcare Epidemiology of America/Association for Professionals in Infection Control/Infectious Diseases Society of America. MMWR Recomm Rep 2002; 51:1-45, quiz CE1–4.
13. Marschall J, Mermel LA, et al; Society for Healthcare Epidemiology of America. Strategies to prevent central line-associated bloodstream infections in acute care hospitals: 2014 update. Infect Control Hosp Epidemiol 2014;35:753–71.
14. Safdar N, Maki DG. The pathogenesis of catheter-related bloodstream infection with noncuffed short-term central venous catheters. Intensive Care Med 2004;30:62–7.
15. Shabot MM, Chassin MR, France AC, et al. Using the targeted solutions tool(R) to improve hand hygiene compliance is associated with decreased health care-associated infections. Jt Comm J Qual Patient Saf 2016;42:6–17.
16. Johnson L, Grueber S, Schlotzhauer C, et al. A multifactorial action plan improves hand hygiene adherence and significantly reduces central line-associated bloodstream infections. Am J Infect Control 2014;42:1146–51.
17. Barrera L, Zingg W, Mendez F, Pittet D. Effectiveness of a hand hygiene promotion strategy using alcohol-based handrub in 6 intensive care units in Colombia. Am J Infect Control 2011;39:633–9.
18. Hu KK, Lipsky BA, Veenstra DL, Saint S. Using maximal sterile barriers to prevent central venous catheter-related infection: a systematic evidence-based review. Am J Infect Control 2004;32:142–6.
19. Hu KK, Veenstra DL, Lipsky BA, Saint S. Use of maximal sterile barriers during central venous catheter insertion: clinical and economic outcomes. Clin Infect Dis 2004;39:1441–5.
20. Raad II, Hohn DC, Gilbreath BJ, et al. Prevention of central venous catheter-related infections by using maximal sterile barrier precautions during insertion. Infect Control Hosp Epidemiol 1994;15:231–8.
21. Furuya EY, Dick AW, Herzig CT, Pogorzelska-Maziarz M, Larson EL, Stone PW. Central line-associated bloodstream infection reduction and bundle compliance in intensive care units: a national study. Infect Control Hosp Epidemiol 2016;37:805–10.
22. Sherertz RJ, Ely EW, Westbrook DM, et al. Education of physicians-in-training can decrease the risk for vascular catheter infection. Ann Intern Med 2000;132:641–8.
23. Mimoz O, Lucet JC, Kerforne T, Pascal J, et al. Skin antisepsis with chlorhexidine-alcohol versus povidone iodine-alcohol, with and without skin scrubbing, for prevention of intravascular-catheter-related infection (CLEAN): an open-label, multicentre, randomised, controlled, two-by-two factorial trial. Lancet 2015;386:2069–77.
24. Lai NM, Lai NA, O’Riordan E, et al. Skin antisepsis for reducing central venous catheter-related infections. Cochrane Database Syst Rev 2016;7:CD010140.
25. Chopra V, Shojania KG. Recipes for checklists and bundles: one part active ingredient, two parts measurement. BMJ Qual Saf 2013;22:93–6.
26. Marik PE, Flemmer M, Harrison W. The risk of catheter-related bloodstream infection with femoral venous catheters as compared to subclavian and internal jugular venous catheters: a systematic review of the literature and meta-analysis. Crit Care Med 2012;40:2479–85.
27. Timsit JF. What is the best site for central venous catheter insertion in critically ill patients? Crit Care 2003;7:397–99.
28. Parienti JJ, Mongardon N, Megarbane B, et al; 3SITES Study Group. Intravascular complications of central venous catheterization by insertion site. N Engl J Med 2015;373:1220–9.
29. Hind D, Calvert N, McWilliams R, et al. Ultrasonic locating devices for central venous cannulation: meta-analysis. BMJ 2003;327:361.
30. Fragou M, Gravvanis A, Dimitriou V, et al. Real-time ultrasound-guided subclavian vein cannulation versus the landmark method in critical care patients: a prospective randomized study. Crit Care Med 2011;39:1607–12.
31. Pageler NM, Longhurst CA, Wood M, et al. Use of electronic medical record-enhanced checklist and electronic dashboard to decrease CLABSIs. Pediatrics 2014;133:e738–46.
32. Drews FA, Bakdash JZ, Gleed JR. Improving central line maintenance to reduce central line-associated bloodstream infections. Am J Infect Control 2017;45:1224–30.
33. Frost SA, Alogso MC, Metcalfe L, et al. Chlorhexidine bathing and health care-associated infections among adult intensive care patients: a systematic review and meta-analysis. Crit Care 2016;20:379.
34. Chopra V, O’Horo JC, Rogers MA, et al. The risk of bloodstream infection associated with peripherally inserted central catheters compared with central venous catheters in adults: a systematic review and meta-analysis. Infect Control Hosp Epidemiol 2013;34:908–18.
35. Xue H, Ix JH, Wang W, et al. Hemodialysis access usage patterns in the incident dialysis year and associated catheter-related complications. Am J Kidney Dis 2013;61:123–30.
36. Almuneef MA, Memish ZA, Balkhy HH, et al. Rate, risk factors and outcomes of catheter-related bloodstream infection in a paediatric intensive care unit in Saudi Arabia. J Hosp Infect 2006;62:207–13.
37. Alonso-Echanove J, Edwards JR, Richards MJ, et al. Effect of nurse staffing and antimicrobial-impregnated central venous catheters on the risk for bloodstream infections in intensive care units. Infect Control Hosp Epidemiol 2003;24:916–25.
38. Lorente L, Henry C, Martin MM, et al. Central venous catheter-related infection in a prospective and observational study of 2,595 catheters. Crit Care 2005;9:R631–5.
39. Rey C, Alvarez F, De-La-Rua V, et al. Intervention to reduce catheter-related bloodstream infections in a pediatric intensive care unit. Intensive Care Med 2011;37:678–85.
40. O’Brien J, Paquet F, Lindsay R, Valenti D. Insertion of PICCs with minimum number of lumens reduces complications and costs. J Am Coll Radiol. 2013;10:864–8.
41. Herc E, Patel P, Washer LL, et al. A model to predict central-line-associated bloodstream infection among patients with peripherally inserted central catheters: the MPC score. Infect Control Hosp Epidemiol 2017;38:1155–66.
42. Centers for Disease Control and Prevention. Vital signs: central line-associated blood stream infections--United States, 2001, 2008, and 2009. MMWR Morb Mortal Wkly Rep 2011;60:243–8.
1. Magill SS, Edwards JR, Bamberg W, et al; Emerging Infections Program Healthcare-Associated Infections and Antimicrobial Use Prevalence Survey Team. Multistate point-prevalence survey of health care-associated infections. N Engl J Med 2014;370:1198–208.
2. Marchetti A, Rossiter R. Economic burden of healthcare-associated infection in US acute care hospitals: societal perspective. J Med Econ 2013;16:1399–404.
3. O’Neil C, Ball K, Wood H, et al. A central line care maintenance bundle for the prevention of central line-associated bloodstream infection in non-intensive care unit settings. Infect Control Hosp Epidemiol 2016;37:1–7.
4. Shekelle PG, Wachter RM, Pronovost PJ, et al. Making health care safer II: an updated critical analysis of the evidence for patient safety practices. Evid Rep Technol Assess (Full Rep) 2013:1–945.
5. Patel PK, Gupta A, Vaughn VM, Mann JD, Ameling JM, Meddings J. Review of strategies to reduce central line-associated bloodstream infection (CLABSI) and catheter-associated urinary tract infection (CAUTI) in adult ICUs. J Hosp Med 2018;13:105–16.
6. Chopra V, Ratz D, Kuhn L, et al. PICC-associated bloodstream infections: prevalence, patterns, and predictors. Am J Med 2014;127:319–28.
7. Sagana R, Hyzy RC. Achieving zero central line-associated bloodstream infection rates in your intensive care unit. Crit Care Clin 2013;29:1–9.
8. Nelson RE, Angelovic AW, Nelson SD, Gleed JR, Drews FA. An economic analysis of adherence engineering to improve use of best practices during central line maintenance procedures. Infect Control Hosp Epidemiol 2015;36:550–6.
9. Pronovost P, Needham D, Berenholtz S, et al. An intervention to decrease catheter-related bloodstream infections in the ICU. N Engl J Med 2006;355:2725–32.
10. Dumyati G, Concannon C, van Wijngaarden E, et al. Sustained reduction of central line-associated bloodstream infections outside the intensive care unit with a multimodal intervention focusing on central line maintenance. Am J Infect Control 2014;42:723–30.
11. Sacks GD, Diggs BS, Hadjizacharia P, et al. Reducing the rate of catheter-associated bloodstream infections in a surgical intensive care unit using the Institute for Healthcare Improvement Central Line Bundle. Am J Surg 2014;207:817–23.
12. Boyce JM, Pittet D; Healthcare Infection Control Practices Advisory Committee; HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Guideline for hand hygiene in health-care settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force. Society for Healthcare Epidemiology of America/Association for Professionals in Infection Control/Infectious Diseases Society of America. MMWR Recomm Rep 2002; 51:1-45, quiz CE1–4.
13. Marschall J, Mermel LA, et al; Society for Healthcare Epidemiology of America. Strategies to prevent central line-associated bloodstream infections in acute care hospitals: 2014 update. Infect Control Hosp Epidemiol 2014;35:753–71.
14. Safdar N, Maki DG. The pathogenesis of catheter-related bloodstream infection with noncuffed short-term central venous catheters. Intensive Care Med 2004;30:62–7.
15. Shabot MM, Chassin MR, France AC, et al. Using the targeted solutions tool(R) to improve hand hygiene compliance is associated with decreased health care-associated infections. Jt Comm J Qual Patient Saf 2016;42:6–17.
16. Johnson L, Grueber S, Schlotzhauer C, et al. A multifactorial action plan improves hand hygiene adherence and significantly reduces central line-associated bloodstream infections. Am J Infect Control 2014;42:1146–51.
17. Barrera L, Zingg W, Mendez F, Pittet D. Effectiveness of a hand hygiene promotion strategy using alcohol-based handrub in 6 intensive care units in Colombia. Am J Infect Control 2011;39:633–9.
18. Hu KK, Lipsky BA, Veenstra DL, Saint S. Using maximal sterile barriers to prevent central venous catheter-related infection: a systematic evidence-based review. Am J Infect Control 2004;32:142–6.
19. Hu KK, Veenstra DL, Lipsky BA, Saint S. Use of maximal sterile barriers during central venous catheter insertion: clinical and economic outcomes. Clin Infect Dis 2004;39:1441–5.
20. Raad II, Hohn DC, Gilbreath BJ, et al. Prevention of central venous catheter-related infections by using maximal sterile barrier precautions during insertion. Infect Control Hosp Epidemiol 1994;15:231–8.
21. Furuya EY, Dick AW, Herzig CT, Pogorzelska-Maziarz M, Larson EL, Stone PW. Central line-associated bloodstream infection reduction and bundle compliance in intensive care units: a national study. Infect Control Hosp Epidemiol 2016;37:805–10.
22. Sherertz RJ, Ely EW, Westbrook DM, et al. Education of physicians-in-training can decrease the risk for vascular catheter infection. Ann Intern Med 2000;132:641–8.
23. Mimoz O, Lucet JC, Kerforne T, Pascal J, et al. Skin antisepsis with chlorhexidine-alcohol versus povidone iodine-alcohol, with and without skin scrubbing, for prevention of intravascular-catheter-related infection (CLEAN): an open-label, multicentre, randomised, controlled, two-by-two factorial trial. Lancet 2015;386:2069–77.
24. Lai NM, Lai NA, O’Riordan E, et al. Skin antisepsis for reducing central venous catheter-related infections. Cochrane Database Syst Rev 2016;7:CD010140.
25. Chopra V, Shojania KG. Recipes for checklists and bundles: one part active ingredient, two parts measurement. BMJ Qual Saf 2013;22:93–6.
26. Marik PE, Flemmer M, Harrison W. The risk of catheter-related bloodstream infection with femoral venous catheters as compared to subclavian and internal jugular venous catheters: a systematic review of the literature and meta-analysis. Crit Care Med 2012;40:2479–85.
27. Timsit JF. What is the best site for central venous catheter insertion in critically ill patients? Crit Care 2003;7:397–99.
28. Parienti JJ, Mongardon N, Megarbane B, et al; 3SITES Study Group. Intravascular complications of central venous catheterization by insertion site. N Engl J Med 2015;373:1220–9.
29. Hind D, Calvert N, McWilliams R, et al. Ultrasonic locating devices for central venous cannulation: meta-analysis. BMJ 2003;327:361.
30. Fragou M, Gravvanis A, Dimitriou V, et al. Real-time ultrasound-guided subclavian vein cannulation versus the landmark method in critical care patients: a prospective randomized study. Crit Care Med 2011;39:1607–12.
31. Pageler NM, Longhurst CA, Wood M, et al. Use of electronic medical record-enhanced checklist and electronic dashboard to decrease CLABSIs. Pediatrics 2014;133:e738–46.
32. Drews FA, Bakdash JZ, Gleed JR. Improving central line maintenance to reduce central line-associated bloodstream infections. Am J Infect Control 2017;45:1224–30.
33. Frost SA, Alogso MC, Metcalfe L, et al. Chlorhexidine bathing and health care-associated infections among adult intensive care patients: a systematic review and meta-analysis. Crit Care 2016;20:379.
34. Chopra V, O’Horo JC, Rogers MA, et al. The risk of bloodstream infection associated with peripherally inserted central catheters compared with central venous catheters in adults: a systematic review and meta-analysis. Infect Control Hosp Epidemiol 2013;34:908–18.
35. Xue H, Ix JH, Wang W, et al. Hemodialysis access usage patterns in the incident dialysis year and associated catheter-related complications. Am J Kidney Dis 2013;61:123–30.
36. Almuneef MA, Memish ZA, Balkhy HH, et al. Rate, risk factors and outcomes of catheter-related bloodstream infection in a paediatric intensive care unit in Saudi Arabia. J Hosp Infect 2006;62:207–13.
37. Alonso-Echanove J, Edwards JR, Richards MJ, et al. Effect of nurse staffing and antimicrobial-impregnated central venous catheters on the risk for bloodstream infections in intensive care units. Infect Control Hosp Epidemiol 2003;24:916–25.
38. Lorente L, Henry C, Martin MM, et al. Central venous catheter-related infection in a prospective and observational study of 2,595 catheters. Crit Care 2005;9:R631–5.
39. Rey C, Alvarez F, De-La-Rua V, et al. Intervention to reduce catheter-related bloodstream infections in a pediatric intensive care unit. Intensive Care Med 2011;37:678–85.
40. O’Brien J, Paquet F, Lindsay R, Valenti D. Insertion of PICCs with minimum number of lumens reduces complications and costs. J Am Coll Radiol. 2013;10:864–8.
41. Herc E, Patel P, Washer LL, et al. A model to predict central-line-associated bloodstream infection among patients with peripherally inserted central catheters: the MPC score. Infect Control Hosp Epidemiol 2017;38:1155–66.
42. Centers for Disease Control and Prevention. Vital signs: central line-associated blood stream infections--United States, 2001, 2008, and 2009. MMWR Morb Mortal Wkly Rep 2011;60:243–8.
Current Controversies Regarding Nutrition Therapy in the ICU
From the Center for Nursing Science & Clinical Inquiry (Dr. McCarthy), and Nutrition Care Division, (Ms. Phipps), Madigan Army Medical Center, Tacoma, WA.
Abstract
- Background: Many controversies exist in the field of nutrition support today, particularly in the critical care environment where nutrition plays a more primary rather than adjunctive role.
- Objective: To provide a brief review of current controversies regarding nutrition therapy in the ICU focusing on the choices regarding the nutrition regimen and the safe, consistent delivery of nutrition as measured by clinical outcomes.
- Methods: Selected areas of controversy are discussed detailing the strengths and weaknesses of the research behind opposing opinions.
- Results: ICU nutrition support controversies include enteral vs parenteral nutrition, use of supplmental parenteral nutrition, protein quantity and quality, and polymeric vs immune-modulating nutrients. Issues surrounding the safety of nutrition support therapy include gastric vs small bowel feeding and trophic vs full feeding. Evidence-based recommendations published by professional societies are presented.
- Conclusion: Understanding a patient’s risk for disease and predicting their response to treatment will assist clinicians with selecting those nutrition interventions that will achieve the best possible outcomes.
According to the National Library of Medicine’s translation of the Hippocratic oath, nowhere does it explicitly say “First, do no harm.” What is written is this: “I will use those dietary regimens which will benefit my patients according to my greatest ability and judgement, and I will do no harm or injustice to them” [1]. In another renowned text, one can find this observation regarding diet by a noted scholar, clinician, and the founder of modern nursing, Florence Nightingale: “Every careful observer of the sick will agree in this that thousands of patients are annually starved in the midst of plenty, from want of attention to the ways which alone make it possible for them to take food” [2,]. While Nightingale was alluding to malnutrition of hospitalized patients, it seems that her real concern may have been the iatrogenic malnutrition that inevitably accompanies hospitalization, even today [3].
From these philosophic texts, we have two ongoing controversies in modern day nutrition therapy identified: (1) what evidence do we have to support the choice of dietary regimens (ie, enteral vs parenteral therapy, timing of supplemental parenteral nutrition, standard vs high protein formula, polymeric vs immune-modulating nutrients) that best serve critically ill patients, and (2) how do we ensure that ICU patients are fed in a safe, consistent, and effective manner (gastric vs small bowel tube placement, gastric residual monitoring or not, trophic vs full feeding) as measured by clinically relevant outcomes? Many controversies exist in the field of nutrition support today [4–7] and a comprehensive discussion of all of them is beyond the scope of this paper. In this paper we will provide a brief review of current controversies focusing on those mentioned above which have only recently been challenged by new rigorous randomized clinical trials (RCTs), and in some cases, subsequent systematic reviews and meta-analyses [8–11].
The Path to Modern Day Nutrition Support Therapy
The field of nutrition support, in general, has expanded greatly over the last 4 decades, but perhaps the most notable advancements have occurred in the critical care environment where efforts have been directed at advancing our understanding of the molecular and biological effects of nutrients in maintaining homeostasis in the critically ill [6]. In recent years, specialized nutrition, delivered by the enteral or parenteral route, was finally recognized for its contribution to important clinical outcomes in the critically ill population [12]. Critical care clinicians have been educated about the advances in nutrition therapy designed to address the unique needs of a vulnerable population where survival is threatened by poor nutritional status upon admission, compromised immune function, weakened respiratory muscles with decreased ventilation capacity, and gastrointestinal (GI) dysfunction [6]. The rapid deterioration seen in these patients is exaggerated by the all too common ICU conditions of systemic inflammatory response syndrome (SIRS), sepsis, hemodynamic instability, respiratory failure, coagulation disorders, and acute kidney injury [13,14].
Beginning in the early 1990s, formulations of enteral nutrition (EN) contained active nutrients that reportedly reduced oxidative damage to cells and tissues, modulated inflammation, and improved feeding tolerance. These benefits are now referred to as the non-nutritive benefits of enteral feeding [15]. For the next 20 years, scientific publications released new results from studies examining the role of omega-3 fatty acids, antioxidant vitamins, minerals such as selenium and zinc, ribonucleotides, and conditionally essential amino acids like glutamine and arginine, in healing and recovery from critical illness. The excitement was summarized succinctly by Hegazi and Wischmeyer in 2011 when they remarked that the modern ICU clinician now has scientific data to guide specialized nutrition therapy, for example, choosing formulas supplemented with anti-inflammatory, immune-modulating, or tolerance-promoting nutrients that have the potential to enhance natural recovery processes and prevent complications [16].
The improvements in nutritional formulas were accompanied by numerous technological advances including bedside devices (electromagnetic enteral access system, real-time image-guided disposable feeding tube, smart feeding pumps with water flush technology) that quickly and safely establish access for small bowel feedings, which help minimize risk of gastric aspiration and ventilator-associated pneumonia, promote tolerance, decrease radiologic exposure, and may reduce nursing time consumed by tube placements, GI dysfunction, and patient discomfort [17–20]. Nasogastric feeding remains the most common first approach, with local practices, contraindications, and ease of placement usually determining the location of the feeding tube [5]. The advancements helped to overcome the many barriers to initiating and maintaining feedings and thus, efforts to feed critically ill patients early and effectively became more routine, along with nurse, patient, and family satisfaction. In conjunction with the innovative approaches to establishing nutrition therapy, practice guidelines published by United States, European, and Canadian nutrition societies became widely available in the past decade with graded evidence-based recommendations for who, when, what, and how to feed, and unique considerations for various critically ill populations [12,21,22]. The tireless efforts by the nutrition societies to provide much needed guidelines for clinicians were appreciated, yet there was a wide range in the grade of the recommendations, with many based on expert opinion alone. In some cases, the research conducted lacked rigor or had missing data with obvious limits to the generalizability of results. Nevertheless, for the 7 years between the publication of the old and newly revised Society of Critical Care Medicine (SCCM)/ American Society of Parenteral and Enteral Nutrition (ASPEN) Guidelines (2016), [12,23] nutrition therapy was a high-priority intervention in most ICUs. The goal was to initiate feeding within 48 hours, select an immune-modulating or other metabolic support formula, and aggressively advance the rate to 80% to 100% of goal to avoid caloric deficit, impaired intestinal integrity, nitrogen losses, and functional impairments [9,24,25]. Nutrition support evolved from adjunctive care to its rightful place in the ABCD mnemonic of early priorities of ICU care: Airway, Breathing, Circulation, Diet.
The 2016 joint publication of the SCCM/ASPEN guidelines includes primarily randomized controlled trial (RCT) data, along with some observational trial data, indexed in any major publication database through December 2013. In these guidelines there were 98 recommendations, of which only 5 were a Level 1A; most of the recommendations were categorized as “expert consensus” [12]. The results of several important clinical trials in the United States and Europe that were underway at the time have since been published and compared to the SCCM/ASPEN relevant recommendations [7]. The results have forced nutrition support clinicians to take a step back and re-examine their practice. For many seasoned clinicians who comprised the nutrition support teams of the 1980s and 1990s, it feels like a return to the basics. Until biology-driven personalized medicine is commonplace and genotype data is readily available to guide nutrition therapy for each critically ill patient, standard enteral feeding that begins slow and proceeds carefully over 5 to 7 days towards 80% of goal caloric intake under judicious monitoring of biochemical and metabolic indices may be the “best practice” today, without risk of harm [15,26]. As in all aspects of clinical care, this practice is not without controversy.
ICU Nutrition Support Controversies Today
Enteral vs Parenteral Nutrition
There is universal consensus that EN is the preferred route for nutrition therapy due to the superior physiological response and both nutritional and non-nutritional benefits [24]. Changes in gut permeability tend to occur as illness progresses and consequences include increased bacterial challenge, risk for multiple organ dysfunction syndrome, and systemic infection. It is best to intervene with nutrition early, defined as within the first 48 hours of ICU admission, while the likelihood of success and opportunity to impact the disease process is greater [12]. Early initiation of feeding provides the necessary nutrients to support gut-associated lymphoid tissue (GALT), mucosal-associated lymphoid tissue (MALT), and preserve gut integrity and microbial diversity [27]. The intestine is an effective barrier against bacteria and intraluminal toxins due to the high rate of enterocyte turnover, the mucus secreted by the goblet cells, and the large amount of protective immunological tissue; 80% of the immunoglobulins are synthesized in the GI tract [28]. Fasting states for procedures or delays in feeding longer than 3 days for any reason may contribute to disruption of intestinal integrity through atrophy and derangements in the physical structure and function of the microvilli and crypts [29]. Intestinal dysfunction leads to increased intestinal permeability and the possibility of bacterial translocation. Intestinal ischemia resulting from shock and sepsis may produce hypoxia and reperfusion injuries further affecting intestinal wall permeability [29]. In surgical patients, early enteral feeding has been found to reduce inflammation, oxidative stress, and the catabolic response to anesthesia and surgical-induced stress, help restore intestinal motility, reverse enteric mucosal atrophy, and improve wound healing [26].
We did not have sufficient data to refute the benefits of EN over PN until the paper by Harvey et al (2014), which reported no difference in mortality or infectious complications in ICU patients receiving EN or PN within 36 hours of admission and for up to 5 days [30]. This was the largest published pragmatic RCT, referred to as the CALORIES trial, which analyzed 2388 patients from 33 ICUs and resulted in controversy over what was an unchallenged approach up until this time. It was only a matter of time before other investigators would set out to confirm or negate this finding, which is what Elke and coworkers (2016) did a few years later [31]. They performed an updated systematic review and meta-analysis to evaluate the overall effect of the route of nutrition (EN versus PN) on clinical outcomes in adult critically ill patients. Similar to the Harvey et al report, they found no difference in mortality between the two routes of nutrition. However, unlike the earlier report, patients receiving EN compared to PN had a significant reduction in the number of infectious complications and ICU length of stay. No significant effect was found for hospital length of stay or days requiring mechanical ventilation. The authors suggest that EN delivery of macronutrients below predefined targets may be responsible as PN is more likely to meet or exceed these targets and overwhelm metabolic capacity in the early days of critical illness [31].
The most recent trial prompting even more discussion about early PN versus early EN in mechanically ventilated ICU patients in shock is the Reignier et al (2018) NUTRIREA-2 trial involving 2410 patients from 44 ICUs in France [32]. The investigators hypothesized that outcomes would be better with early exclusive EN compared to early exclusive PN; their hypothesis was not supported by the results, which found no difference in 28-day mortality or ICU-acquired infections. Also unexpected was the higher cumulative incidence of gastrointestinal complications including vomiting, diarrhea, bowel ischemia, and acute colonic obstruction in the EN group. The trial was stopped early after an interim analysis determined that additional enrollment was not likely to significantly change the results of the trial. Given the similarities between the CALORIES trial and this NUTRIREA-2 trial, clinicians now have mounting evidence that equivalent options for nutrition therapy exist and an appropriate selection should be made based on patient-specific indications and treatment goals. In summary, EN remains preferable to PN for the majority of adult critically ill patients due to crucial support of gut integrity, but the optimal dose or rate of delivery to favorably influence clinical outcomes in the first few days following admission remains unknown.
Use of Supplemental Parenteral Nutrition
Both the nutrition support and bench science communities have learned a great deal about PN over the 4 decades it has been in existence, with the most compelling data coming from more recent trials [31–38]. This is because it has taken many years to recover from the days of hyperalimentation or overfeeding ICU patients by providing excessive calories to meet the elevated energy demands and to reverse the hypercatabolism of critical illness. This approach contributed to the complications of hyperglycemia, hyperlipidemia, increased infectious complications, and liver steatosis, all of which gave PN a negative reputation [37]. We now have adjusted the caloric distribution and the actual formulation of PN using the recent FDA-approved lipid emulsion (Soy, Medium-chain triglycerides, Olive oil, and Fish oil; SMOF) and created protocols for administering it based on specific indications, such as loss of GI integrity or demonstrated intolerance. In general, the advances in PN have led to a safer more therapeutic formulation that has its place in critical illness. Manzanares et al [40] reported a trend toward a decrease in ventilation requirement and mortality when a fish oil–containing lipid emulsion was administered to patients who were receiving nutrition support either enterally or parenterally. The meta-analysis combined all soybean oil–sparing lipid emulsions for comparison with soybean oil and was able to show the trend for improved clinical outcomes with soybean oil–sparing lipid emulsions. The main findings of this meta-analysis were that fish oil–containing lipid emulsions may reduce infections and may be associated with a tendency toward fewer mechanical ventilation days, although not mortality, when compared with soybean oil-based strategies or administration of other alternative lipid emulsions in ICU patients [40]. Recent trial results do not change the recommendation for selecting EN first but do suggest lowering the threshold for using PN when EN alone is insufficient to meet nutrition goals. A systematic review reported no benefit of early supplemental PN over late supplemental PN and cautioned that our continued inability to explain greater infectious morbidity and unresolved organ failure limits any justification for early PN [35].
Protein Quantity and Quality
The practice of providing protein in the range of 1.2–2.0 g/kg actual body weight early in critical illness is suggested by expert consensus in the 2016 SCCM/ASPEN guidelines [12]; however, the evidence for efficacy remains controversial. It is argued that the evidence for benefit comes from observational studies, not from prospective RCTs, and that the patients at high risk are often excluded from study protocols. It has also been suggested that in patients with limited vital organ function increased protein delivery may lead to organ compromise. In a recent (2017) paper, Rooyackers et al discussed the post-hoc analyses of data from the EPANIC Trial stating the statistical correlation between protein intake and outcomes indicate that protein was associated with unfavorable outcomes, possibly by inhibiting autophagy [41].
The nutrition support community may have widely varying approaches to feeding critically ill patients but most experts agree that protein may be the most important macronutrient delivered during critical illness. There is consensus that the hypercatabolism associated with stress induces proteolysis and the loss of lean muscle mass, which may affect clinical and functional outcomes beyond the ICU stay. Using multiple assessment modalities, Puthucheary et al (2013) demonstrated a reduction in the rectus femoris muscle of 12.5% over the first week of hospitalization in the ICU and up to 17.7% by day 10. These numbers imply that sufficient protein of at least 1.2 g/kg/day should be provided to minimize these losses, even if the effect on overall outcome remains unknown [42]. Evidence is lacking for whether or not we can prevent the muscle wasting that occurs in critical illness with increased protein dosing. We also need to better identify the possible risks involved with a high-protein intake at the level of the individual patient. A secondary analysis done by Heyland et al (2013) determined that no specific dose or type of macronutrient was found to be associated with improved outcome [43]. It is clear that more large-scale RCTs of protein/amino acid interventions are needed to prove that these nutrition interventions have favorable effects on clinically important outcomes, including long-term physical function.
Polymeric vs Immune-Modulating Nutrients
The Marik and Zaloga (2008) systematic review on immunonutrition in the critically ill convinced most clinicians that while fish-oil based immunonutrition improves the outcome of medical ICU patients, diets supplemented with arginine, with or without glutamine or fish oils, do not demonstrate an advantage over standard enteral products in general ICU, trauma, or burn patients[44]. What followed these trials examining early formulations of immunonutrition was decades of well-intentioned research dedicated to elucidating the mechanism of action for individual immune-modulating nutrients for various populations, including those with acute lung injury/acute respiratory distress syndrome (ARDS) [45–47], sepsis/systemic inflammatory response syndrome [48–50], head and neck cancer [51], upper and lower GI cancer [52–55], and severe acute pancreatitis [56]. Our understanding of immunonutrition and the administration of this formulation in specific disease conditions has grown considerably yet clinicians are still asking exactly what is the role of immunonutrition and who stands to benefit the most from immune-modulating nutrition therapy. The enteral formulations currently available have a proprietary composition and dosage of individual nutrients which yield unpredictable physiologic effects. In addition, the pervasive problem of underfeeding during hospitalization prevents adequate delivery of physiologic doses of nutrients thus contributing to the widely variable research results.
Prevailing expert opinion today is that the majority of critically ill patients will benefit from nutrition support initiated early and delivered consistently; the standard polymeric formula will suffice for the majority of patients with surgical ICU patients potentially deriving benefit from immunonutrition that supports a reduction in infectious complications [57]. In the recent multiple-treatment meta-analysis performed by Mazaki et al (2015) involving 74 studies and 7572 patients, immunonutrition was ranked first for reducing the incidence of 7 complications according to the surface under the cumulative ranking curve; these were as follows: any infection, 0.86; overall complication, 0.88; mortality, 0.81; wound infection, 0.79; intra-abdominal abscess, 0.98; anastomotic leak, 0.79; and sepsis, 0.92. immunonutrition was ranked second for reducing ventilator-associated pneumonia and catheter-associated urinary tract infection (CAUTI), behind immune-modulating PN. The authors stated that immunonutrition was efficacious for reducing the incidence of complications in GI surgery unrelated to the timing of administration [57]. The 2014 publication of results from the MetaPlus Trial [58]challenged the published recommendations for the use of immunonutrition in the medical critically ill population. This trial used high-protein immunonutrition or standard formula for 301 adult critically ill patients in 14 European ICUs with diagnoses such as pneumonia or infections of the urinary tract, bloodstream, central nervous system, eye, ear, nose or throat, and the skin and soft tissue. Even with higher than average target energy intakes of 70% for the high protein immunonutrition group and 80% for the high protein standard group, there were no statistically significant differences in the primary outcome of new infections, or the secondary outcomes of days on mechanical ventilation, Sequential Organ Failure Assessment scores, or ICU and hospital length of stay. However, the 6-month mortality rate of 28% was higher in the medical subgroup [58]. Using these results, as well as existing publications of negative outcomes in medical ICU patients [44,46], the SCCM/ASPEN Guidelines Committee updated its position in 2016 to suggest that immunonutrition formulations or disease-specific formulations should no longer be used routinely in medical ICU patients, including those with acute lung injury/ARDS [12]. The Committee did suggest that these formulations should be reserved for patients with traumatic brain injury and for surgical ICU patients. The benefit for ICU postoperative patients has been linked to the synergistic effect of fish oil and arginine, which must both be present to achieve outcome benefits. A meta-analysis comprised of 35 trials was conducted by Drover et al [58], who reported that administering an arginine and fish oil-containing formula postoperatively reduced infection (RR 0.78; 95% CI, 0.64 to 0.95; P = 0.01) and hospital length of stay (WMD –2.23, 95% CI, –3.80 to –0.65; P = 0.006) but not mortality, when compared to use of a standard formula. Similar results were reported in a second meta-analysis [56], thus providing supportive evidence for the current SCCM/ASPEN recommendation to use an immune-modulating formula (containing both arginine and fish oils) in the SICU for the postoperative patient who requires EN therapy [12].
Safe, Consistent, and Effective Nutrition Support Therapy
Gastric vs Small Bowel Feeding
There is a large group of critically ill patients in whom impaired gastric emptying presents challenges to feeding; 50% of mechanically ventilated patients demonstrate delayed gastric emptying, and 80% of patients with increased intracranial pressure following head injury [60]. In one prospective RCT, Huang et al (2012) showed that severely ill patients (defined by an APACHE II score > 20) fed by the nasoduodenal route experienced significantly shortened hospital LOS, fewer complications, and improved nutrient delivery compared to similar patients fed by the nasogastric route. Less severely ill patients (APACHE II < 20) showed no differences between nasogastric and nasoduodenal feeding for the same outcomes [61]. A recent systematic review [17] pooled data from 14 trials of 1109 participants who received either gastric or small bowel feeding. Moderate quality evidence suggested that post-pyloric feeding was associated with lower rates of pneumonia compared with gastric feeding (RR 0.65, 95% CI, 0.51 to 0.84). Low-quality evidence showed an increase in the percentage of total nutrient delivered to the patient by post-pyloric feeding (mean difference 7.8%, 95% CI, 1.43 to 14.18). Overall, the authors found a 30% lower rate of pneumonia associated with post-pyloric feeding. There is insufficient evidence to show that other clinically important outcomes such as duration of mechanical ventilation, mortality, or LOS were affected by the site of feeding. The American Thoracic Society and ASPEN, as well as the Infectious Diseases Society of America, have published guidelines in support of small bowel feeding in the ICU setting due to its association with reduced incidence of health care–associated infections, specifically ventilator-associated pneumonia [62]. The experts who developed the SCCM/ASPEN and Canadian guidelines stress that critically ill patients at high risk for aspiration or feeding intolerance should be fed using small bowel access [12,21]. The reality in ICU clinical practice is that many centers will begin with gastric feeding, barring absolute contraindications, and carefully monitor the patient for signs of intolerance before moving the feeding tube tip into a post-pyloric location. This follows the general recommendation by experts saying in most critically ill patients, it is acceptable to initiate EN in the stomach [12,21]. Protocols that guide management of risk prevention and intolerance typically recommend head of bed elevation, prokinetic agents, and frequent abdominal assessments [63,64].
Once the decision is made to use a post-pyloric feeding tube for nutrition therapy, the next decision is how to safely place the tube, ensure the tip is in an acceptable small bowel location, and minimize delays in feeding. Challenges related to feeding tube insertion may preclude timely advancement to nutrition goals. Placement of feeding tubes into the post-pyloric position is often done at the bedside by trained nursing or medical staff without endoscopic or fluoroscopic guidance; however, the blind bedside approach is not without risks. Success rates of this approach vary greatly depending on the patient population and provider expertise. Placement using endoscopic or fluoroscopic guidance is a safe alternative but usually requires coordinating a transport to the radiologic suite, posing safety risks and possible feeding delays for the patient [65]. Bedside use of an electromagnetic placement device (EMPD), such as Cortrak, provides yet another alternative with reports in the literature of 98% success rates for initial placement in less than 20 minutes. In a multicenter prospective study by Powers et al (2011), only one of 194 patients enrolled had data showing a discrepancy between the original EMPD verification and the final radiograph interpretation, demonstrating a 99.5% agreement between the two readings [20]. Median placement time was 12 minutes and no patient experienced an adverse event related to tube insertion using this device. The ability to monitor the location of the feeding tube tip in real time provides a desirable safety feature for the clinician performing bedside insertions. Nurses should consider incorporating the EMPD into the unit feeding protocol, as this would reduce the time to initiation of feedings with early and accurate tube insertion. Ongoing staff education and experience with the procedure are necessary elements to achieve the high rates of success often reported in the literature [66,67]. Procedural complications from placement of nasoenteral feeding tubes by all methods can be as high as 10%, with complication rates of 1% to 3% for inadvertent placement of the feeding tube in the airway alone [65]. Radiographic confirmation of tube placement is advised prior to initiating feeding, thus eliminating any possibility of misplacement and administration of formula into the lungs.
Gastric Residual Volume Monitoring
A number of factors impede the delivery of EN in the critical care setting; these include gastrointestinal intolerance, under-prescribing to meet daily requirements, frequent interruptions for procedures, and technical issues with tube placement and maintaining patency [68]. Monitoring gastric residual volumes (GRV) contributes to these factors, yet volumes do not correlate well with incidence of pneumonia [69], measures of gastric emptying, or to the incidence of regurgitation and aspiration [70,71]. However, few studies have highlighted the difficulty of obtaining an accurate GRV due to feeding tube tip location, patient position, and type of tube [69]. Several high quality studies have demonstrated that raising the cutoff value for GRV from a lower number of 50–150 mL to a higher number of 250–500 mL does not increase risk for regurgitation, aspiration, or pneumonia [70,71]. A lower cutoff value for GRV does not protect the patient from complications, often leads to inappropriate cessation, and may adversely affect outcome through reduced volume of EN infused [72]. Gastric residual volumes in the range of 200–500 mL should raise concern and lead to the implementation of measures to reduce risk of aspiration, but automatic cessation of feeding should not occur for GRV < 500 mL in the absence of other signs of intolerance [12,69]. Metheny et al (2012) conducted a survey in which more than 97% of nurses responded that they assessed intolerance by measuring GRV; the most frequently cited threshold levels for interrupting feedings were 200 mL and 250 mL [73]. While threshold levels varied widely, only 12.6% of the nurse respondents reported allowing GRV up to 500 mL before interrupting feedings. While monitoring GRV is unnecessary with small bowel feeding, the location of the feeding tube tip should be questioned if gastric contents are obtained from a small bowel tube. The use of GRV as a parameter for trending may also yield important information regarding tolerance of feeding when the patient is unable to communicate abdominal discomfort. Other objective measures to use in the assessment of tolerance include an abdominal exam with documentation of changes in bowel sounds, expanding girth, tenderness or firmness on palpation, increasing nasogastric output, and vomiting [12,68]. If there are indications of intolerance, it is appropriate to divert the tip of the feeding tube into the distal small bowel as discussed previously.
Trophic vs Full Feeding
For the patient with low nutrition risk, there is a lack of convincing data to support an aggressive approach to feeding, either EN or PN, in the first week of critical illness [7]. In recent years, results of several trials suggest early goal-directed feeding in this population may cause net harm with increased morbidity and mortality. When discussing recent controversies in critical care nutrition, one must mention the two schools of thought when it comes to full versus limited energy provision in the first week following ICU admission. Studies in animals and humans have shown a trophic effect of enteral nutrients on the integrity of the gut mucosa, a finding that has provided the rationale for instituting enteral nutrition early during critical illness [15]. However, the inability to provide enteral nutrition early may be a marker of the severity of illness (ie, patients who can be fed enterally are less ill than those who cannot) rather than a mediator of complications and poor outcomes. Compher et al (2017) stated that greater nutritional intake is associated with lower mortality and faster time to discharge alive in high-risk, chronic patients but does not seem to be significant in nutritionally low-risk patients [74]. The findings of the EPaNIC and EDEN trials raised concern that targeting goals that meet full energy needs early in critical illness does not provide benefit and may cause harm in some populations or settings [32,75]. The EDEN trial [32] left us believing that trophic feeding at 10–20 mL/hr may be just as effective as any feeding in the first few days of critical illness striving for 15% to 20% of daily goal calories. After establishing tolerance, advancing daily intake to > 50% to 65% of goal calories, and up to 80% for the highest risk patients, may be required to prevent intestinal permeability and achieve positive clinical outcomes [33].
The systematic review and meta-analysis performed by Al-Dorzi et al (2016) adds further evidence for judicious advancement of EN for critically ill patients [76]. The authors reported finding no association between the dose of caloric intake and hospital mortality. Furthermore, a lower caloric intake resulted in lower risk of bloodstream infections and the need for renal replacement therapy (in 5 of the 21 trials only). As with many other meta-analyses, the authors reported that their results are most assuredly impacted by the heterogeneity in design, feeding route, and dose prescribed and delivered [16,76,77]. Other recent trials such as Arabi et al (2015) that enrolled 894 patients with different feeding targets further confirmed that there is no difference in outcome between groups when it comes to moderate (40% to 60% of goal) vs high (70% to 100% of goal) energy intake, infection rates, or 90-day mortality. The authors summarized their findings saying feeding closer to target is associated with better outcomes compared with severe underfeeding [78]. This adds to the controversy when considering the findings of still other RCTs or meta-analyses that evaluated minimal or trophic feeding versus standard feeding rates [9,46,77]. The meta-analysis performed by Marik and Hooper concluded that there were no differences in the risk of acquired infections, hospital mortality, ICU LOS, or ventilator-free days whether patients received intentional hypocaloric or normocaloric nutrition support [9]. Similarly, there was no significant difference in overall mortality between the underfeeding and full-feeding groups (OR, 0.94; 95% CI, 0.74–1.19; I2 = 26.6%; P = 0.61) in the meta-analysis done by Choi et al (2015), although only 4 trials were included to ensure homogeneity of the population and the intervention [77]. Furthermore, the hospital LOS and ICU LOS did not differ between the 2 groups, nor did any other secondary clinical outcome, leading the authors to conclude that calorie intake of the initial EN support for ICU patients had no bearing on relevant outcomes.
Recent studies have attempted to correlate caloric intake and patient outcomes without success; achieving 100% of caloric goal has not favorably impacted morbidity and mortality. Evidence suggests that intake greater than 65% to 70% of daily caloric requirement in the first 7 to 10 days of ICU stay may be associated with poorer outcomes, particularly when parenteral nutrition is used to supplement intake to achieve the caloric target [33–35].
Conclusion
In this review we described current ICU controversies surrounding nutrition therapy and briefly discussed the data that support more than one course of action. A summary of key points covered is presented in the Table. 
Disclaimer: The views expressed in this paper are those of the authors and do not reflect the official policy of the Department of the Army, the Department of Defense, or the U.S. government.
Corresponding author: Mary S. McCarthy, PhD, RN, CNSC, 1611 Nisqually St, Steilacoom, WA 98388.
Financial disclosures: None.
1. Hippocratic Oath. Translated by Michael North, National Library of Medicine, 2002.
2. Nightingale F. Notes on Nursing. What it is and what it is not. Radford, VA: Wilder Publications, LLC;2007
3. White JV, Guenter P, Jensen G, et al; the Academy Malnutrition Work Group; the ASPEN Malnutrition Task Force; and the ASPEN Board of Directors. Consensus statement: Academy of Nutrition and Dietetics and American Society for Parenteral and Enteral Nutrition: characteristics recommended for the identification and documentation of adult malnutrition (undernutrition). JPEN J Parenter Enteral Nutr 2012;36:275–83.
4. Hooper MH, Marik PE. Controversies and misconceptions in Intensive Care Unit nutrition. Clin Chest Med 2015;36:409–18.
5. Patel JJ, Codner P. Controversies in critical care nutrition support. Crit Care Clin 2016;32:173–89.
6. Rosenthal MD, Vanzant EL, Martindale RG, Moore FA. Evolving paradigms in the nutritional support of critically ill surgical patients. Curr Probl Surg 2015;52:147–82.
7. McCarthy MS, Warren M, Roberts PR. Recent critical care nutrition trials and the revised guidelines: do they reconcile? Nutr Clin Pract 2016;31:150–4.
8. Barker LA, Gray C, Wilson L, et al. Preoperative immunonutrition and its effect on postoperative outcomes in well-nourished and malnourished gastrointestinal surgery patients: a randomised controlled trial. Eur J Clin Nutr 2013;67: 802–807.
9. Marik PE, Hooper MH. Normocaloric versus hypocaloric feeding on the outcomes of ICU patients: a systematic review and meta-analysis. Intensive Care Med 2016;42:316–323.
10. Patkova A, Joskova V, Havel E, et al. Energy, protein, carbohydrate, and lipid intakes and their effects on morbidity and mortality in critically ill adult patients: a systematic review. Adv Nutr 2017;8:624–34.
11. Wong CS, Aly EH. The effects of enteral immunonutrition in upper gastrointestinal surgery: a systematic review and meta-analysis. Int J Surg 2016;29:137–50.
12. McClave SA, Taylor BE, Martindale RG, et al; Society of Critical Care Medicine; American Society for Parenteral and Enteral Nutrition. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society of Parenteral and Enteral Nutrition (ASPEN). JPEN J Parenter Enteral Nutr 2016; 40:159–211.
13. Ammori BJ. Importance of the early increase in intestinal permeability in critically ill patients. Eur J Surg 2002;168:660–1.
14. Vazquez-Sandoval A, Ghamande S, Surani S. Critically ill patients and gut motility: are we addressing it? World J Gastrointest Pharmacol Ther 2017;8:174–9.
15. Patel JJ, Martindale RG, McClave SA. Controversies surrounding critical care nutrition: an appraisal of permissive underfeeding, protein, and outcomes. JPEN J Parenter Enteral Nutr 2017; 148607117721908.
16. Hegazi RA, Hustead DS, Evans DC. Preoperative standard oral nutrition supplements vs immunonutrition: results of a systematic review and meta-analysis. J Am Coll Surg 2014;219:1078–87.
17. Alkhawaja S, Martin C, Butler RJ, Gwadry-Sridhar F. Post-pyloric versus gastric tube feeding for preventing pneumonia and improving nutritional outcomes in critically ill adults. Cochrane Database of Syst Rev 2015; CD008875.
18. Davies AR, Morrison SS, Bailey MJ, et al ; ENTERIC Study Investigators; ANZICS Clinical Trials Group. A multi-center randomized controlled trial comparing early nasojejunal with nasogastric nutrition in critical illness. Crit Care Med 2012;40:2342–8.
19. Hsu CW, Sun SF, Lin SL, et al. Duodenal versus gastric feeding in medical intensive care unit patients: a prospective, randomized, clinical study. Crit Care Med 2009;37:1866–72.
20. Powers J, Luebbehusen M, Spitzer T, et al. Verification of an electromagnetic placement device compared with abdominal radiograph to predict accuracy of feeding tube placement. JPEN J Parenter Enteral Nutr 2011;35:535–9.
21. Dhaliwal R, Cahill N, Lemieux M, Heyland DK. The Canadian critical care nutrition guidelines in 2013: an update on current recommendations and implementation strategies. Nutr Clin Pract 2014; 29:29–43.22. Kreymann K, Berger M, Deutz N. et al; DGEM (German Society for Nutritional Medicine); ESPEN (European Society for Parenteral and Enteral Nutrition). ESPEN guidelines on enteral nutrition: intensive care. Clin Nutr 2006;25:210–23.
23. McClave SA, Martindale RG, Vanek VW, et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (ASPEN). JPEN J Parenter Ent Nutr 2009;33:277–316.
24. McClave SA, Martindale RG, Rice TW, Heyland DK. Feeding the critically ill patient. Crit Care Med 2014;42:2600–10.
25. Tian F, Gao X, Wu C, et al. Initial energy supplementation in critically ill patients receiving enteral nutrition: a systematic review and meta-analysis of randomized controlled trials. Asia Pac J Clin Nutr 2017;26:11–9.
26. Martindale RG, Warren M. Should enteral nutrition be started in the first week of critical illness? Curr Opin Clin Nutr Metab Care 2015;18:202–6.
27. McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract 2009;24:305–15.
28. Kang W, Kudsk KA. Is there evidence that the gut contributes to mucosal immunity in humans? JPEN J Parenter Enteral Nutr 2007;31:461–82.
29. Seron-Arbeloa C, Zamora-Elson M, Labarta-Monzon L, Mallor-Bonet T. Enteral nutrition in critical care. J Clin Med Res 2013;5:1-11.
30. Harvey SE, Parrott F, Harrison DA, et al; CALORIES Trial Investigators. Trial of the route of early nutritional support in critically ill adults. N Engl J Med 2014;371:1673–84.
31. Elke G, van Zanten AR, Lemieux M, et al. Enteral versus parenteral nutrition in critically ill patients: an updated systematic review and meta-analysis of randomized controlled trials. Crit Care 2016;20:117.
32. Reignier J, Boisramé-Helms J, Brisard L, et al. Enteral versus parenteral nutrition in ventilated adults with shock: a randomized, controlled, multicenter, open-label, parallel-group study (NUTRIREA-2). Lancet 2018;391:133–43.
33. Rice TW , Wheeler AP, Thompson BT et al;National Heart, Lung, and Blood Institute, Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Initial trophic vs full enteral feeding in patients with acute lung injury: the EDEN randomized trial. JAMA 2012;307:795–803.
34. Heyland DK, Dhaliwal R, Jiang X, Day AG. Identifying critically ill patients who benefit the most from nutrition therapy: the development and initial validation of a novel risk assessment tool. Crit Care 2011;15:R258.
35. Bost RB, Tjan DH, van Zanten AR. Timing of (supplemental) parenteral nutrition in critically ill patients: a systematic review. Ann Intensive Care 2014;4:31.
36. Casaer MP, Mesotten D, Hermans G, et al. Early verus late parenteral nutrition in critically ill adults. N Eng J Med 2011;365:506–17.
37. Harvey SE, Parrott F, Harrison DA, et al; CALORIES Trial Investigators. Trial of the route of early nutritional support in critically ill adults. N Eng J Med 2014;371:1673–84.
38. Manzanares W, Dhaliwal R, Jurewitsch B, et al. Parenteral fish oil lipid emulsions in the critically ill: A systematic review and meta-analysis. JPEN J Parenter Enteral Nutr 2014;38:20–8.
39. Oshima T, Heidegger CP, Pichard C. Supplemental parenteral nutrition is the key to prevent energy deficits in critically ill patients. Nutr Clin Prac 2016;31:432–7.
40. Manzanares W, Langlois PL, Dhaliwal R, Lemieux M, Heyland DK. Intravenous fish oil lipid emulsions in critically ill patients: an updated systematic review and meta-analysis. Crit Care 2015;19:167.
41. Rooyackers O, Sundström Rehal M, Liebau F, et al. High protein intake without concerns? Crit Care 2017;21:106.
42. Puthucheary ZA, Rawal J, McPhail M, et al. Acute skeletal muscle wasting in critical illness. JAMA 2013;310:1591–600.
43. Heyland D, Muscedere J, Wischmeyer PE, et al; Canadian Critical Care Trials Group. A randomized trial of glutamine and antioxidants in critically ill patients. N Engl J Med 2013;368:1489–97.
44. Marik PE, Zaloga GP. Immunonutrition in critically ill patients: a systematic review and analysis of the literature. Intensive Care Med 2008;34:1980–90.
45. Gadek JE, DeMichele SJ, Karlstad MD, et al; Enteral Nutrition in ARDS Study Group. Effect of enteral feeding with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome. Crit Care Med 1999;27:1409–20.
46. Rice TW, Wheeler AP, Thompson BT, et al; NIH NHLBI Acute Respiratory Distress Syndrome Network of Investigators. Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA 2011;306:1574–81.
47. Singer P, Theilla M, Fisher H, et al. Benefit of an enteral diet enriched with eicosapentaenoic acid and gamma-linolenic acid in ventilated patients with acute lung injury. Crit Care Med 2006;34:1033–38.
48. Atkinson S, Sieffert E, Bihari D. A prospective, randomized, double-blind, controlled clinical trial of enteral immunonutrition in the critically ill. Guy’s Hospital Intensive Care Group. Crit Care Med 1998;26:1164–72.
49. Galbán C, Montejo JC, Mesejo A, et al. An immune-enhancing enteral diet reduces mortality rate and episodes of bacteremia in septic intensive care unit patients. Crit Care Med 2000;28:643–8.
50. Weimann A, Bastian L, Bischoff WE, et al. Influence of arginine, omega-3 fatty acids and nucleotide-supplemented enteral support on systemic inflammatory response syndrome and multiple organ failure in patients after severe trauma. Nutrition 1998;14:165–72.
51. van Bokhorst-De Van Der Schueren MA, Quak JJ, von Blomberg-van der Flier BM, et al. Effect of perioperative nutrition with and without arginine supplementation, on nutritional status, immune function, postoperative morbidity, and survival in severely malnourished head and neck cancer patients. Am J Clin Nutr 2001;73:323–32.
52. Cerantola Y, Hübner M, Grass F, et al. Immunonutrition in gastrointestinal surgery. Br J Surg 2011;98:37–48.
53. Marik PE, Zaloga GP. Immunonutrition in high-risk surgical patients: a systematic review and analysis of the literature. JPEN J Parenter Enteral Nutr 2010;34:378–86.
54. Sultan J, Griffin SM, Di Franco F, et al. Randomized clinical trial of omega-3 fatty acid–supplemented enteral nutrition vs. standard enteral nutrition in patients undergoing oesophagogastric cancer surgery. Br J Surg 2012;99:346–55.
55. Waitzberg DL, Saito H, Plank LD, et al. Postsurgical infections are reduced with specialized nutrition support. World J Surg 2006;30:1592–604.
56. Pearce CB, Sadek SA, Walters AM, et al. A double-blind, randomised, controlled trial to study the effects of an enteral feed supplemented with glutamine, arginine, and omega-3 fatty acid in predicted acute severe pancreatitis. JOP 2006;7:361–71.
57. Mazaki T, Ishii Y, Murai I. Immunoenhancing enteral and parenteral nutrition for gastrointestinal surgery: a multiple treatments meta-analysis. Ann Surg 2015;261:662–9.
58. van Zanten ARH, Sztark F, Kaisers UX, et al. High-protein enteral nutrition enriched with immune-modulating nutrients vs standard high protein enteral nutrition and nosocomial infections in the ICU. JAMA 2014;312:514–24.
59. Drover JW, Dhaliwal R, Weitzel L, et al. Perioperative use of arginine supplemented diets: a systematic review of the evidence. J Am Coll Surg 2011;212:385–99.
60. Stupak D, Abdelsayed GG, Soloway GN. Motility disorders of the upper gastrointestinal tract in the intensive care unit: pathophysiology and contemporary management. J Clin Gastroenterol 2012;46:449–56.
61. Huang HH, Chang SJ, Hsu CW, et al. Severity of illness influences the efficacy of enteral feeding route on clinical outcomes in patients with critical illness. J Acad Nutr Diet 2012;112:1138–46.
62. American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005;171:388–416.
63. Heyland DK, Cahill NE, Dhaliwal R, et al. Impact of enteral feeding protocols on enteral nutrition delivery: results of a multicenter observational study. JPEN J Parenter Enteral Nutr 2010;34:675–84.
64. Landzinski J, Kiser TH, Fish DN, et al. Gastric motility function in critically ill patients tolerant vs intolerant to gastric nutrition. JPEN J Parenter Enteral Nutr 2008;32:45–50.
65. de Aguilar-Nascimento JE, Kudsk KA. Use of small bore feeding tubes: successes and failures. Curr Opin Clin Nutr Metab Care 2007;10:291–6.
66. Boyer N, McCarthy MS, Mount CA. Analysis of an electromagnetic tube placement device vs a self-advancing nasal jejunal device for postpyloric feeding tube placement . J Hosp Med 2014;9:23–8.
67. Metheny NA, Meert KL. Effectiveness of an electromagnetic feeding tube placement device in detecting inadvertent respiratory placement. Am J Crit Care 2014;23:240–8.
68. Montejo JC, Miñambres E, Bordejé L, et al. Gastric residual volume during enteral nutrition in ICU patients: the REGANE study. Intensive Care Med 2010;36:1386–93.
69. Hurt RT, McClave SA. Gastric residual volumes in critical illness: what do they really mean? Crit Care Clin 2010;26:481–90.
70. Poulard F, Dimet J, Martin-Lefevre L, et al. Impact of not measuring residual gastric volume in mechanically ventilated patients receiving early enteral feeding: a prospective before-after study. JPEN J Parenter Enteral Nutr 2010;34:125–30.
71. Reignier J, Mercier E, Gouge AL, et al; Clinical Research in Intensive Care and Sepsis (CRICS) Group. Effect of not monitoring residual gastric volume on risk of ventilator-associated pneumonia in adults receiving mechanical ventilation and early enteral feeding: a randomized controlled trial. JAMA 2013;309:249–56.
72. Williams TA, Leslie GD, Leen T, et al. Reducing interruptions to continuous enteral nutrition in the intensive care unit: a comparative study. J Clin Nurs 2013;22:2838-2848.
73. Metheny NA, Stewart BJ, Mills AC. Blind insertion of feeding tubes in intensive care units: a national survey. Am J Crit Care 2012;21:352–360.
74. Compher C, Chittams J, Sammarco T, et al. Greater protein and energy intake may be associated with improved mortality in higher risk critically ill patients: A multicenter, multinational observational study. Crit Care Med 2017;45:156–163.
75. Casaer MP, Wilmer A, Hermans G, et al. Role of disease and macronutrient dose in the randomized controlled EPANIC Trial: a post hoc analysis. Am J Resp Crit Care Med 2013;187:247–55.
76. Al-Dorzi HM, Albarrak A, Ferwana M, et al. Lower versus higher dose of enteral caloric intake in adult critically ill patients: a systematic review and meta-analysis. Crit Care 2016;20:358.
77. Choi EY, Park DA, Park J. Calorie intake of enteral nutrition and clinical outcomes in acutely critically ill patients: a meta-analysis of randomized controlled trials. JPEN J Parenter Enteral Nutr 2015;39:291–300.
78. Arabi YM, Aldawood AS, Haddad SH, et al. Permissive underfeeding or standard enteral feeding in critically ill adults. N Engl J Med 2015;372:2398–408.
From the Center for Nursing Science & Clinical Inquiry (Dr. McCarthy), and Nutrition Care Division, (Ms. Phipps), Madigan Army Medical Center, Tacoma, WA.
Abstract
- Background: Many controversies exist in the field of nutrition support today, particularly in the critical care environment where nutrition plays a more primary rather than adjunctive role.
- Objective: To provide a brief review of current controversies regarding nutrition therapy in the ICU focusing on the choices regarding the nutrition regimen and the safe, consistent delivery of nutrition as measured by clinical outcomes.
- Methods: Selected areas of controversy are discussed detailing the strengths and weaknesses of the research behind opposing opinions.
- Results: ICU nutrition support controversies include enteral vs parenteral nutrition, use of supplmental parenteral nutrition, protein quantity and quality, and polymeric vs immune-modulating nutrients. Issues surrounding the safety of nutrition support therapy include gastric vs small bowel feeding and trophic vs full feeding. Evidence-based recommendations published by professional societies are presented.
- Conclusion: Understanding a patient’s risk for disease and predicting their response to treatment will assist clinicians with selecting those nutrition interventions that will achieve the best possible outcomes.
According to the National Library of Medicine’s translation of the Hippocratic oath, nowhere does it explicitly say “First, do no harm.” What is written is this: “I will use those dietary regimens which will benefit my patients according to my greatest ability and judgement, and I will do no harm or injustice to them” [1]. In another renowned text, one can find this observation regarding diet by a noted scholar, clinician, and the founder of modern nursing, Florence Nightingale: “Every careful observer of the sick will agree in this that thousands of patients are annually starved in the midst of plenty, from want of attention to the ways which alone make it possible for them to take food” [2,]. While Nightingale was alluding to malnutrition of hospitalized patients, it seems that her real concern may have been the iatrogenic malnutrition that inevitably accompanies hospitalization, even today [3].
From these philosophic texts, we have two ongoing controversies in modern day nutrition therapy identified: (1) what evidence do we have to support the choice of dietary regimens (ie, enteral vs parenteral therapy, timing of supplemental parenteral nutrition, standard vs high protein formula, polymeric vs immune-modulating nutrients) that best serve critically ill patients, and (2) how do we ensure that ICU patients are fed in a safe, consistent, and effective manner (gastric vs small bowel tube placement, gastric residual monitoring or not, trophic vs full feeding) as measured by clinically relevant outcomes? Many controversies exist in the field of nutrition support today [4–7] and a comprehensive discussion of all of them is beyond the scope of this paper. In this paper we will provide a brief review of current controversies focusing on those mentioned above which have only recently been challenged by new rigorous randomized clinical trials (RCTs), and in some cases, subsequent systematic reviews and meta-analyses [8–11].
The Path to Modern Day Nutrition Support Therapy
The field of nutrition support, in general, has expanded greatly over the last 4 decades, but perhaps the most notable advancements have occurred in the critical care environment where efforts have been directed at advancing our understanding of the molecular and biological effects of nutrients in maintaining homeostasis in the critically ill [6]. In recent years, specialized nutrition, delivered by the enteral or parenteral route, was finally recognized for its contribution to important clinical outcomes in the critically ill population [12]. Critical care clinicians have been educated about the advances in nutrition therapy designed to address the unique needs of a vulnerable population where survival is threatened by poor nutritional status upon admission, compromised immune function, weakened respiratory muscles with decreased ventilation capacity, and gastrointestinal (GI) dysfunction [6]. The rapid deterioration seen in these patients is exaggerated by the all too common ICU conditions of systemic inflammatory response syndrome (SIRS), sepsis, hemodynamic instability, respiratory failure, coagulation disorders, and acute kidney injury [13,14].
Beginning in the early 1990s, formulations of enteral nutrition (EN) contained active nutrients that reportedly reduced oxidative damage to cells and tissues, modulated inflammation, and improved feeding tolerance. These benefits are now referred to as the non-nutritive benefits of enteral feeding [15]. For the next 20 years, scientific publications released new results from studies examining the role of omega-3 fatty acids, antioxidant vitamins, minerals such as selenium and zinc, ribonucleotides, and conditionally essential amino acids like glutamine and arginine, in healing and recovery from critical illness. The excitement was summarized succinctly by Hegazi and Wischmeyer in 2011 when they remarked that the modern ICU clinician now has scientific data to guide specialized nutrition therapy, for example, choosing formulas supplemented with anti-inflammatory, immune-modulating, or tolerance-promoting nutrients that have the potential to enhance natural recovery processes and prevent complications [16].
The improvements in nutritional formulas were accompanied by numerous technological advances including bedside devices (electromagnetic enteral access system, real-time image-guided disposable feeding tube, smart feeding pumps with water flush technology) that quickly and safely establish access for small bowel feedings, which help minimize risk of gastric aspiration and ventilator-associated pneumonia, promote tolerance, decrease radiologic exposure, and may reduce nursing time consumed by tube placements, GI dysfunction, and patient discomfort [17–20]. Nasogastric feeding remains the most common first approach, with local practices, contraindications, and ease of placement usually determining the location of the feeding tube [5]. The advancements helped to overcome the many barriers to initiating and maintaining feedings and thus, efforts to feed critically ill patients early and effectively became more routine, along with nurse, patient, and family satisfaction. In conjunction with the innovative approaches to establishing nutrition therapy, practice guidelines published by United States, European, and Canadian nutrition societies became widely available in the past decade with graded evidence-based recommendations for who, when, what, and how to feed, and unique considerations for various critically ill populations [12,21,22]. The tireless efforts by the nutrition societies to provide much needed guidelines for clinicians were appreciated, yet there was a wide range in the grade of the recommendations, with many based on expert opinion alone. In some cases, the research conducted lacked rigor or had missing data with obvious limits to the generalizability of results. Nevertheless, for the 7 years between the publication of the old and newly revised Society of Critical Care Medicine (SCCM)/ American Society of Parenteral and Enteral Nutrition (ASPEN) Guidelines (2016), [12,23] nutrition therapy was a high-priority intervention in most ICUs. The goal was to initiate feeding within 48 hours, select an immune-modulating or other metabolic support formula, and aggressively advance the rate to 80% to 100% of goal to avoid caloric deficit, impaired intestinal integrity, nitrogen losses, and functional impairments [9,24,25]. Nutrition support evolved from adjunctive care to its rightful place in the ABCD mnemonic of early priorities of ICU care: Airway, Breathing, Circulation, Diet.
The 2016 joint publication of the SCCM/ASPEN guidelines includes primarily randomized controlled trial (RCT) data, along with some observational trial data, indexed in any major publication database through December 2013. In these guidelines there were 98 recommendations, of which only 5 were a Level 1A; most of the recommendations were categorized as “expert consensus” [12]. The results of several important clinical trials in the United States and Europe that were underway at the time have since been published and compared to the SCCM/ASPEN relevant recommendations [7]. The results have forced nutrition support clinicians to take a step back and re-examine their practice. For many seasoned clinicians who comprised the nutrition support teams of the 1980s and 1990s, it feels like a return to the basics. Until biology-driven personalized medicine is commonplace and genotype data is readily available to guide nutrition therapy for each critically ill patient, standard enteral feeding that begins slow and proceeds carefully over 5 to 7 days towards 80% of goal caloric intake under judicious monitoring of biochemical and metabolic indices may be the “best practice” today, without risk of harm [15,26]. As in all aspects of clinical care, this practice is not without controversy.
ICU Nutrition Support Controversies Today
Enteral vs Parenteral Nutrition
There is universal consensus that EN is the preferred route for nutrition therapy due to the superior physiological response and both nutritional and non-nutritional benefits [24]. Changes in gut permeability tend to occur as illness progresses and consequences include increased bacterial challenge, risk for multiple organ dysfunction syndrome, and systemic infection. It is best to intervene with nutrition early, defined as within the first 48 hours of ICU admission, while the likelihood of success and opportunity to impact the disease process is greater [12]. Early initiation of feeding provides the necessary nutrients to support gut-associated lymphoid tissue (GALT), mucosal-associated lymphoid tissue (MALT), and preserve gut integrity and microbial diversity [27]. The intestine is an effective barrier against bacteria and intraluminal toxins due to the high rate of enterocyte turnover, the mucus secreted by the goblet cells, and the large amount of protective immunological tissue; 80% of the immunoglobulins are synthesized in the GI tract [28]. Fasting states for procedures or delays in feeding longer than 3 days for any reason may contribute to disruption of intestinal integrity through atrophy and derangements in the physical structure and function of the microvilli and crypts [29]. Intestinal dysfunction leads to increased intestinal permeability and the possibility of bacterial translocation. Intestinal ischemia resulting from shock and sepsis may produce hypoxia and reperfusion injuries further affecting intestinal wall permeability [29]. In surgical patients, early enteral feeding has been found to reduce inflammation, oxidative stress, and the catabolic response to anesthesia and surgical-induced stress, help restore intestinal motility, reverse enteric mucosal atrophy, and improve wound healing [26].
We did not have sufficient data to refute the benefits of EN over PN until the paper by Harvey et al (2014), which reported no difference in mortality or infectious complications in ICU patients receiving EN or PN within 36 hours of admission and for up to 5 days [30]. This was the largest published pragmatic RCT, referred to as the CALORIES trial, which analyzed 2388 patients from 33 ICUs and resulted in controversy over what was an unchallenged approach up until this time. It was only a matter of time before other investigators would set out to confirm or negate this finding, which is what Elke and coworkers (2016) did a few years later [31]. They performed an updated systematic review and meta-analysis to evaluate the overall effect of the route of nutrition (EN versus PN) on clinical outcomes in adult critically ill patients. Similar to the Harvey et al report, they found no difference in mortality between the two routes of nutrition. However, unlike the earlier report, patients receiving EN compared to PN had a significant reduction in the number of infectious complications and ICU length of stay. No significant effect was found for hospital length of stay or days requiring mechanical ventilation. The authors suggest that EN delivery of macronutrients below predefined targets may be responsible as PN is more likely to meet or exceed these targets and overwhelm metabolic capacity in the early days of critical illness [31].
The most recent trial prompting even more discussion about early PN versus early EN in mechanically ventilated ICU patients in shock is the Reignier et al (2018) NUTRIREA-2 trial involving 2410 patients from 44 ICUs in France [32]. The investigators hypothesized that outcomes would be better with early exclusive EN compared to early exclusive PN; their hypothesis was not supported by the results, which found no difference in 28-day mortality or ICU-acquired infections. Also unexpected was the higher cumulative incidence of gastrointestinal complications including vomiting, diarrhea, bowel ischemia, and acute colonic obstruction in the EN group. The trial was stopped early after an interim analysis determined that additional enrollment was not likely to significantly change the results of the trial. Given the similarities between the CALORIES trial and this NUTRIREA-2 trial, clinicians now have mounting evidence that equivalent options for nutrition therapy exist and an appropriate selection should be made based on patient-specific indications and treatment goals. In summary, EN remains preferable to PN for the majority of adult critically ill patients due to crucial support of gut integrity, but the optimal dose or rate of delivery to favorably influence clinical outcomes in the first few days following admission remains unknown.
Use of Supplemental Parenteral Nutrition
Both the nutrition support and bench science communities have learned a great deal about PN over the 4 decades it has been in existence, with the most compelling data coming from more recent trials [31–38]. This is because it has taken many years to recover from the days of hyperalimentation or overfeeding ICU patients by providing excessive calories to meet the elevated energy demands and to reverse the hypercatabolism of critical illness. This approach contributed to the complications of hyperglycemia, hyperlipidemia, increased infectious complications, and liver steatosis, all of which gave PN a negative reputation [37]. We now have adjusted the caloric distribution and the actual formulation of PN using the recent FDA-approved lipid emulsion (Soy, Medium-chain triglycerides, Olive oil, and Fish oil; SMOF) and created protocols for administering it based on specific indications, such as loss of GI integrity or demonstrated intolerance. In general, the advances in PN have led to a safer more therapeutic formulation that has its place in critical illness. Manzanares et al [40] reported a trend toward a decrease in ventilation requirement and mortality when a fish oil–containing lipid emulsion was administered to patients who were receiving nutrition support either enterally or parenterally. The meta-analysis combined all soybean oil–sparing lipid emulsions for comparison with soybean oil and was able to show the trend for improved clinical outcomes with soybean oil–sparing lipid emulsions. The main findings of this meta-analysis were that fish oil–containing lipid emulsions may reduce infections and may be associated with a tendency toward fewer mechanical ventilation days, although not mortality, when compared with soybean oil-based strategies or administration of other alternative lipid emulsions in ICU patients [40]. Recent trial results do not change the recommendation for selecting EN first but do suggest lowering the threshold for using PN when EN alone is insufficient to meet nutrition goals. A systematic review reported no benefit of early supplemental PN over late supplemental PN and cautioned that our continued inability to explain greater infectious morbidity and unresolved organ failure limits any justification for early PN [35].
Protein Quantity and Quality
The practice of providing protein in the range of 1.2–2.0 g/kg actual body weight early in critical illness is suggested by expert consensus in the 2016 SCCM/ASPEN guidelines [12]; however, the evidence for efficacy remains controversial. It is argued that the evidence for benefit comes from observational studies, not from prospective RCTs, and that the patients at high risk are often excluded from study protocols. It has also been suggested that in patients with limited vital organ function increased protein delivery may lead to organ compromise. In a recent (2017) paper, Rooyackers et al discussed the post-hoc analyses of data from the EPANIC Trial stating the statistical correlation between protein intake and outcomes indicate that protein was associated with unfavorable outcomes, possibly by inhibiting autophagy [41].
The nutrition support community may have widely varying approaches to feeding critically ill patients but most experts agree that protein may be the most important macronutrient delivered during critical illness. There is consensus that the hypercatabolism associated with stress induces proteolysis and the loss of lean muscle mass, which may affect clinical and functional outcomes beyond the ICU stay. Using multiple assessment modalities, Puthucheary et al (2013) demonstrated a reduction in the rectus femoris muscle of 12.5% over the first week of hospitalization in the ICU and up to 17.7% by day 10. These numbers imply that sufficient protein of at least 1.2 g/kg/day should be provided to minimize these losses, even if the effect on overall outcome remains unknown [42]. Evidence is lacking for whether or not we can prevent the muscle wasting that occurs in critical illness with increased protein dosing. We also need to better identify the possible risks involved with a high-protein intake at the level of the individual patient. A secondary analysis done by Heyland et al (2013) determined that no specific dose or type of macronutrient was found to be associated with improved outcome [43]. It is clear that more large-scale RCTs of protein/amino acid interventions are needed to prove that these nutrition interventions have favorable effects on clinically important outcomes, including long-term physical function.
Polymeric vs Immune-Modulating Nutrients
The Marik and Zaloga (2008) systematic review on immunonutrition in the critically ill convinced most clinicians that while fish-oil based immunonutrition improves the outcome of medical ICU patients, diets supplemented with arginine, with or without glutamine or fish oils, do not demonstrate an advantage over standard enteral products in general ICU, trauma, or burn patients[44]. What followed these trials examining early formulations of immunonutrition was decades of well-intentioned research dedicated to elucidating the mechanism of action for individual immune-modulating nutrients for various populations, including those with acute lung injury/acute respiratory distress syndrome (ARDS) [45–47], sepsis/systemic inflammatory response syndrome [48–50], head and neck cancer [51], upper and lower GI cancer [52–55], and severe acute pancreatitis [56]. Our understanding of immunonutrition and the administration of this formulation in specific disease conditions has grown considerably yet clinicians are still asking exactly what is the role of immunonutrition and who stands to benefit the most from immune-modulating nutrition therapy. The enteral formulations currently available have a proprietary composition and dosage of individual nutrients which yield unpredictable physiologic effects. In addition, the pervasive problem of underfeeding during hospitalization prevents adequate delivery of physiologic doses of nutrients thus contributing to the widely variable research results.
Prevailing expert opinion today is that the majority of critically ill patients will benefit from nutrition support initiated early and delivered consistently; the standard polymeric formula will suffice for the majority of patients with surgical ICU patients potentially deriving benefit from immunonutrition that supports a reduction in infectious complications [57]. In the recent multiple-treatment meta-analysis performed by Mazaki et al (2015) involving 74 studies and 7572 patients, immunonutrition was ranked first for reducing the incidence of 7 complications according to the surface under the cumulative ranking curve; these were as follows: any infection, 0.86; overall complication, 0.88; mortality, 0.81; wound infection, 0.79; intra-abdominal abscess, 0.98; anastomotic leak, 0.79; and sepsis, 0.92. immunonutrition was ranked second for reducing ventilator-associated pneumonia and catheter-associated urinary tract infection (CAUTI), behind immune-modulating PN. The authors stated that immunonutrition was efficacious for reducing the incidence of complications in GI surgery unrelated to the timing of administration [57]. The 2014 publication of results from the MetaPlus Trial [58]challenged the published recommendations for the use of immunonutrition in the medical critically ill population. This trial used high-protein immunonutrition or standard formula for 301 adult critically ill patients in 14 European ICUs with diagnoses such as pneumonia or infections of the urinary tract, bloodstream, central nervous system, eye, ear, nose or throat, and the skin and soft tissue. Even with higher than average target energy intakes of 70% for the high protein immunonutrition group and 80% for the high protein standard group, there were no statistically significant differences in the primary outcome of new infections, or the secondary outcomes of days on mechanical ventilation, Sequential Organ Failure Assessment scores, or ICU and hospital length of stay. However, the 6-month mortality rate of 28% was higher in the medical subgroup [58]. Using these results, as well as existing publications of negative outcomes in medical ICU patients [44,46], the SCCM/ASPEN Guidelines Committee updated its position in 2016 to suggest that immunonutrition formulations or disease-specific formulations should no longer be used routinely in medical ICU patients, including those with acute lung injury/ARDS [12]. The Committee did suggest that these formulations should be reserved for patients with traumatic brain injury and for surgical ICU patients. The benefit for ICU postoperative patients has been linked to the synergistic effect of fish oil and arginine, which must both be present to achieve outcome benefits. A meta-analysis comprised of 35 trials was conducted by Drover et al [58], who reported that administering an arginine and fish oil-containing formula postoperatively reduced infection (RR 0.78; 95% CI, 0.64 to 0.95; P = 0.01) and hospital length of stay (WMD –2.23, 95% CI, –3.80 to –0.65; P = 0.006) but not mortality, when compared to use of a standard formula. Similar results were reported in a second meta-analysis [56], thus providing supportive evidence for the current SCCM/ASPEN recommendation to use an immune-modulating formula (containing both arginine and fish oils) in the SICU for the postoperative patient who requires EN therapy [12].
Safe, Consistent, and Effective Nutrition Support Therapy
Gastric vs Small Bowel Feeding
There is a large group of critically ill patients in whom impaired gastric emptying presents challenges to feeding; 50% of mechanically ventilated patients demonstrate delayed gastric emptying, and 80% of patients with increased intracranial pressure following head injury [60]. In one prospective RCT, Huang et al (2012) showed that severely ill patients (defined by an APACHE II score > 20) fed by the nasoduodenal route experienced significantly shortened hospital LOS, fewer complications, and improved nutrient delivery compared to similar patients fed by the nasogastric route. Less severely ill patients (APACHE II < 20) showed no differences between nasogastric and nasoduodenal feeding for the same outcomes [61]. A recent systematic review [17] pooled data from 14 trials of 1109 participants who received either gastric or small bowel feeding. Moderate quality evidence suggested that post-pyloric feeding was associated with lower rates of pneumonia compared with gastric feeding (RR 0.65, 95% CI, 0.51 to 0.84). Low-quality evidence showed an increase in the percentage of total nutrient delivered to the patient by post-pyloric feeding (mean difference 7.8%, 95% CI, 1.43 to 14.18). Overall, the authors found a 30% lower rate of pneumonia associated with post-pyloric feeding. There is insufficient evidence to show that other clinically important outcomes such as duration of mechanical ventilation, mortality, or LOS were affected by the site of feeding. The American Thoracic Society and ASPEN, as well as the Infectious Diseases Society of America, have published guidelines in support of small bowel feeding in the ICU setting due to its association with reduced incidence of health care–associated infections, specifically ventilator-associated pneumonia [62]. The experts who developed the SCCM/ASPEN and Canadian guidelines stress that critically ill patients at high risk for aspiration or feeding intolerance should be fed using small bowel access [12,21]. The reality in ICU clinical practice is that many centers will begin with gastric feeding, barring absolute contraindications, and carefully monitor the patient for signs of intolerance before moving the feeding tube tip into a post-pyloric location. This follows the general recommendation by experts saying in most critically ill patients, it is acceptable to initiate EN in the stomach [12,21]. Protocols that guide management of risk prevention and intolerance typically recommend head of bed elevation, prokinetic agents, and frequent abdominal assessments [63,64].
Once the decision is made to use a post-pyloric feeding tube for nutrition therapy, the next decision is how to safely place the tube, ensure the tip is in an acceptable small bowel location, and minimize delays in feeding. Challenges related to feeding tube insertion may preclude timely advancement to nutrition goals. Placement of feeding tubes into the post-pyloric position is often done at the bedside by trained nursing or medical staff without endoscopic or fluoroscopic guidance; however, the blind bedside approach is not without risks. Success rates of this approach vary greatly depending on the patient population and provider expertise. Placement using endoscopic or fluoroscopic guidance is a safe alternative but usually requires coordinating a transport to the radiologic suite, posing safety risks and possible feeding delays for the patient [65]. Bedside use of an electromagnetic placement device (EMPD), such as Cortrak, provides yet another alternative with reports in the literature of 98% success rates for initial placement in less than 20 minutes. In a multicenter prospective study by Powers et al (2011), only one of 194 patients enrolled had data showing a discrepancy between the original EMPD verification and the final radiograph interpretation, demonstrating a 99.5% agreement between the two readings [20]. Median placement time was 12 minutes and no patient experienced an adverse event related to tube insertion using this device. The ability to monitor the location of the feeding tube tip in real time provides a desirable safety feature for the clinician performing bedside insertions. Nurses should consider incorporating the EMPD into the unit feeding protocol, as this would reduce the time to initiation of feedings with early and accurate tube insertion. Ongoing staff education and experience with the procedure are necessary elements to achieve the high rates of success often reported in the literature [66,67]. Procedural complications from placement of nasoenteral feeding tubes by all methods can be as high as 10%, with complication rates of 1% to 3% for inadvertent placement of the feeding tube in the airway alone [65]. Radiographic confirmation of tube placement is advised prior to initiating feeding, thus eliminating any possibility of misplacement and administration of formula into the lungs.
Gastric Residual Volume Monitoring
A number of factors impede the delivery of EN in the critical care setting; these include gastrointestinal intolerance, under-prescribing to meet daily requirements, frequent interruptions for procedures, and technical issues with tube placement and maintaining patency [68]. Monitoring gastric residual volumes (GRV) contributes to these factors, yet volumes do not correlate well with incidence of pneumonia [69], measures of gastric emptying, or to the incidence of regurgitation and aspiration [70,71]. However, few studies have highlighted the difficulty of obtaining an accurate GRV due to feeding tube tip location, patient position, and type of tube [69]. Several high quality studies have demonstrated that raising the cutoff value for GRV from a lower number of 50–150 mL to a higher number of 250–500 mL does not increase risk for regurgitation, aspiration, or pneumonia [70,71]. A lower cutoff value for GRV does not protect the patient from complications, often leads to inappropriate cessation, and may adversely affect outcome through reduced volume of EN infused [72]. Gastric residual volumes in the range of 200–500 mL should raise concern and lead to the implementation of measures to reduce risk of aspiration, but automatic cessation of feeding should not occur for GRV < 500 mL in the absence of other signs of intolerance [12,69]. Metheny et al (2012) conducted a survey in which more than 97% of nurses responded that they assessed intolerance by measuring GRV; the most frequently cited threshold levels for interrupting feedings were 200 mL and 250 mL [73]. While threshold levels varied widely, only 12.6% of the nurse respondents reported allowing GRV up to 500 mL before interrupting feedings. While monitoring GRV is unnecessary with small bowel feeding, the location of the feeding tube tip should be questioned if gastric contents are obtained from a small bowel tube. The use of GRV as a parameter for trending may also yield important information regarding tolerance of feeding when the patient is unable to communicate abdominal discomfort. Other objective measures to use in the assessment of tolerance include an abdominal exam with documentation of changes in bowel sounds, expanding girth, tenderness or firmness on palpation, increasing nasogastric output, and vomiting [12,68]. If there are indications of intolerance, it is appropriate to divert the tip of the feeding tube into the distal small bowel as discussed previously.
Trophic vs Full Feeding
For the patient with low nutrition risk, there is a lack of convincing data to support an aggressive approach to feeding, either EN or PN, in the first week of critical illness [7]. In recent years, results of several trials suggest early goal-directed feeding in this population may cause net harm with increased morbidity and mortality. When discussing recent controversies in critical care nutrition, one must mention the two schools of thought when it comes to full versus limited energy provision in the first week following ICU admission. Studies in animals and humans have shown a trophic effect of enteral nutrients on the integrity of the gut mucosa, a finding that has provided the rationale for instituting enteral nutrition early during critical illness [15]. However, the inability to provide enteral nutrition early may be a marker of the severity of illness (ie, patients who can be fed enterally are less ill than those who cannot) rather than a mediator of complications and poor outcomes. Compher et al (2017) stated that greater nutritional intake is associated with lower mortality and faster time to discharge alive in high-risk, chronic patients but does not seem to be significant in nutritionally low-risk patients [74]. The findings of the EPaNIC and EDEN trials raised concern that targeting goals that meet full energy needs early in critical illness does not provide benefit and may cause harm in some populations or settings [32,75]. The EDEN trial [32] left us believing that trophic feeding at 10–20 mL/hr may be just as effective as any feeding in the first few days of critical illness striving for 15% to 20% of daily goal calories. After establishing tolerance, advancing daily intake to > 50% to 65% of goal calories, and up to 80% for the highest risk patients, may be required to prevent intestinal permeability and achieve positive clinical outcomes [33].
The systematic review and meta-analysis performed by Al-Dorzi et al (2016) adds further evidence for judicious advancement of EN for critically ill patients [76]. The authors reported finding no association between the dose of caloric intake and hospital mortality. Furthermore, a lower caloric intake resulted in lower risk of bloodstream infections and the need for renal replacement therapy (in 5 of the 21 trials only). As with many other meta-analyses, the authors reported that their results are most assuredly impacted by the heterogeneity in design, feeding route, and dose prescribed and delivered [16,76,77]. Other recent trials such as Arabi et al (2015) that enrolled 894 patients with different feeding targets further confirmed that there is no difference in outcome between groups when it comes to moderate (40% to 60% of goal) vs high (70% to 100% of goal) energy intake, infection rates, or 90-day mortality. The authors summarized their findings saying feeding closer to target is associated with better outcomes compared with severe underfeeding [78]. This adds to the controversy when considering the findings of still other RCTs or meta-analyses that evaluated minimal or trophic feeding versus standard feeding rates [9,46,77]. The meta-analysis performed by Marik and Hooper concluded that there were no differences in the risk of acquired infections, hospital mortality, ICU LOS, or ventilator-free days whether patients received intentional hypocaloric or normocaloric nutrition support [9]. Similarly, there was no significant difference in overall mortality between the underfeeding and full-feeding groups (OR, 0.94; 95% CI, 0.74–1.19; I2 = 26.6%; P = 0.61) in the meta-analysis done by Choi et al (2015), although only 4 trials were included to ensure homogeneity of the population and the intervention [77]. Furthermore, the hospital LOS and ICU LOS did not differ between the 2 groups, nor did any other secondary clinical outcome, leading the authors to conclude that calorie intake of the initial EN support for ICU patients had no bearing on relevant outcomes.
Recent studies have attempted to correlate caloric intake and patient outcomes without success; achieving 100% of caloric goal has not favorably impacted morbidity and mortality. Evidence suggests that intake greater than 65% to 70% of daily caloric requirement in the first 7 to 10 days of ICU stay may be associated with poorer outcomes, particularly when parenteral nutrition is used to supplement intake to achieve the caloric target [33–35].
Conclusion
In this review we described current ICU controversies surrounding nutrition therapy and briefly discussed the data that support more than one course of action. A summary of key points covered is presented in the Table.
Disclaimer: The views expressed in this paper are those of the authors and do not reflect the official policy of the Department of the Army, the Department of Defense, or the U.S. government.
Corresponding author: Mary S. McCarthy, PhD, RN, CNSC, 1611 Nisqually St, Steilacoom, WA 98388.
Financial disclosures: None.
From the Center for Nursing Science & Clinical Inquiry (Dr. McCarthy), and Nutrition Care Division, (Ms. Phipps), Madigan Army Medical Center, Tacoma, WA.
Abstract
- Background: Many controversies exist in the field of nutrition support today, particularly in the critical care environment where nutrition plays a more primary rather than adjunctive role.
- Objective: To provide a brief review of current controversies regarding nutrition therapy in the ICU focusing on the choices regarding the nutrition regimen and the safe, consistent delivery of nutrition as measured by clinical outcomes.
- Methods: Selected areas of controversy are discussed detailing the strengths and weaknesses of the research behind opposing opinions.
- Results: ICU nutrition support controversies include enteral vs parenteral nutrition, use of supplmental parenteral nutrition, protein quantity and quality, and polymeric vs immune-modulating nutrients. Issues surrounding the safety of nutrition support therapy include gastric vs small bowel feeding and trophic vs full feeding. Evidence-based recommendations published by professional societies are presented.
- Conclusion: Understanding a patient’s risk for disease and predicting their response to treatment will assist clinicians with selecting those nutrition interventions that will achieve the best possible outcomes.
According to the National Library of Medicine’s translation of the Hippocratic oath, nowhere does it explicitly say “First, do no harm.” What is written is this: “I will use those dietary regimens which will benefit my patients according to my greatest ability and judgement, and I will do no harm or injustice to them” [1]. In another renowned text, one can find this observation regarding diet by a noted scholar, clinician, and the founder of modern nursing, Florence Nightingale: “Every careful observer of the sick will agree in this that thousands of patients are annually starved in the midst of plenty, from want of attention to the ways which alone make it possible for them to take food” [2,]. While Nightingale was alluding to malnutrition of hospitalized patients, it seems that her real concern may have been the iatrogenic malnutrition that inevitably accompanies hospitalization, even today [3].
From these philosophic texts, we have two ongoing controversies in modern day nutrition therapy identified: (1) what evidence do we have to support the choice of dietary regimens (ie, enteral vs parenteral therapy, timing of supplemental parenteral nutrition, standard vs high protein formula, polymeric vs immune-modulating nutrients) that best serve critically ill patients, and (2) how do we ensure that ICU patients are fed in a safe, consistent, and effective manner (gastric vs small bowel tube placement, gastric residual monitoring or not, trophic vs full feeding) as measured by clinically relevant outcomes? Many controversies exist in the field of nutrition support today [4–7] and a comprehensive discussion of all of them is beyond the scope of this paper. In this paper we will provide a brief review of current controversies focusing on those mentioned above which have only recently been challenged by new rigorous randomized clinical trials (RCTs), and in some cases, subsequent systematic reviews and meta-analyses [8–11].
The Path to Modern Day Nutrition Support Therapy
The field of nutrition support, in general, has expanded greatly over the last 4 decades, but perhaps the most notable advancements have occurred in the critical care environment where efforts have been directed at advancing our understanding of the molecular and biological effects of nutrients in maintaining homeostasis in the critically ill [6]. In recent years, specialized nutrition, delivered by the enteral or parenteral route, was finally recognized for its contribution to important clinical outcomes in the critically ill population [12]. Critical care clinicians have been educated about the advances in nutrition therapy designed to address the unique needs of a vulnerable population where survival is threatened by poor nutritional status upon admission, compromised immune function, weakened respiratory muscles with decreased ventilation capacity, and gastrointestinal (GI) dysfunction [6]. The rapid deterioration seen in these patients is exaggerated by the all too common ICU conditions of systemic inflammatory response syndrome (SIRS), sepsis, hemodynamic instability, respiratory failure, coagulation disorders, and acute kidney injury [13,14].
Beginning in the early 1990s, formulations of enteral nutrition (EN) contained active nutrients that reportedly reduced oxidative damage to cells and tissues, modulated inflammation, and improved feeding tolerance. These benefits are now referred to as the non-nutritive benefits of enteral feeding [15]. For the next 20 years, scientific publications released new results from studies examining the role of omega-3 fatty acids, antioxidant vitamins, minerals such as selenium and zinc, ribonucleotides, and conditionally essential amino acids like glutamine and arginine, in healing and recovery from critical illness. The excitement was summarized succinctly by Hegazi and Wischmeyer in 2011 when they remarked that the modern ICU clinician now has scientific data to guide specialized nutrition therapy, for example, choosing formulas supplemented with anti-inflammatory, immune-modulating, or tolerance-promoting nutrients that have the potential to enhance natural recovery processes and prevent complications [16].
The improvements in nutritional formulas were accompanied by numerous technological advances including bedside devices (electromagnetic enteral access system, real-time image-guided disposable feeding tube, smart feeding pumps with water flush technology) that quickly and safely establish access for small bowel feedings, which help minimize risk of gastric aspiration and ventilator-associated pneumonia, promote tolerance, decrease radiologic exposure, and may reduce nursing time consumed by tube placements, GI dysfunction, and patient discomfort [17–20]. Nasogastric feeding remains the most common first approach, with local practices, contraindications, and ease of placement usually determining the location of the feeding tube [5]. The advancements helped to overcome the many barriers to initiating and maintaining feedings and thus, efforts to feed critically ill patients early and effectively became more routine, along with nurse, patient, and family satisfaction. In conjunction with the innovative approaches to establishing nutrition therapy, practice guidelines published by United States, European, and Canadian nutrition societies became widely available in the past decade with graded evidence-based recommendations for who, when, what, and how to feed, and unique considerations for various critically ill populations [12,21,22]. The tireless efforts by the nutrition societies to provide much needed guidelines for clinicians were appreciated, yet there was a wide range in the grade of the recommendations, with many based on expert opinion alone. In some cases, the research conducted lacked rigor or had missing data with obvious limits to the generalizability of results. Nevertheless, for the 7 years between the publication of the old and newly revised Society of Critical Care Medicine (SCCM)/ American Society of Parenteral and Enteral Nutrition (ASPEN) Guidelines (2016), [12,23] nutrition therapy was a high-priority intervention in most ICUs. The goal was to initiate feeding within 48 hours, select an immune-modulating or other metabolic support formula, and aggressively advance the rate to 80% to 100% of goal to avoid caloric deficit, impaired intestinal integrity, nitrogen losses, and functional impairments [9,24,25]. Nutrition support evolved from adjunctive care to its rightful place in the ABCD mnemonic of early priorities of ICU care: Airway, Breathing, Circulation, Diet.
The 2016 joint publication of the SCCM/ASPEN guidelines includes primarily randomized controlled trial (RCT) data, along with some observational trial data, indexed in any major publication database through December 2013. In these guidelines there were 98 recommendations, of which only 5 were a Level 1A; most of the recommendations were categorized as “expert consensus” [12]. The results of several important clinical trials in the United States and Europe that were underway at the time have since been published and compared to the SCCM/ASPEN relevant recommendations [7]. The results have forced nutrition support clinicians to take a step back and re-examine their practice. For many seasoned clinicians who comprised the nutrition support teams of the 1980s and 1990s, it feels like a return to the basics. Until biology-driven personalized medicine is commonplace and genotype data is readily available to guide nutrition therapy for each critically ill patient, standard enteral feeding that begins slow and proceeds carefully over 5 to 7 days towards 80% of goal caloric intake under judicious monitoring of biochemical and metabolic indices may be the “best practice” today, without risk of harm [15,26]. As in all aspects of clinical care, this practice is not without controversy.
ICU Nutrition Support Controversies Today
Enteral vs Parenteral Nutrition
There is universal consensus that EN is the preferred route for nutrition therapy due to the superior physiological response and both nutritional and non-nutritional benefits [24]. Changes in gut permeability tend to occur as illness progresses and consequences include increased bacterial challenge, risk for multiple organ dysfunction syndrome, and systemic infection. It is best to intervene with nutrition early, defined as within the first 48 hours of ICU admission, while the likelihood of success and opportunity to impact the disease process is greater [12]. Early initiation of feeding provides the necessary nutrients to support gut-associated lymphoid tissue (GALT), mucosal-associated lymphoid tissue (MALT), and preserve gut integrity and microbial diversity [27]. The intestine is an effective barrier against bacteria and intraluminal toxins due to the high rate of enterocyte turnover, the mucus secreted by the goblet cells, and the large amount of protective immunological tissue; 80% of the immunoglobulins are synthesized in the GI tract [28]. Fasting states for procedures or delays in feeding longer than 3 days for any reason may contribute to disruption of intestinal integrity through atrophy and derangements in the physical structure and function of the microvilli and crypts [29]. Intestinal dysfunction leads to increased intestinal permeability and the possibility of bacterial translocation. Intestinal ischemia resulting from shock and sepsis may produce hypoxia and reperfusion injuries further affecting intestinal wall permeability [29]. In surgical patients, early enteral feeding has been found to reduce inflammation, oxidative stress, and the catabolic response to anesthesia and surgical-induced stress, help restore intestinal motility, reverse enteric mucosal atrophy, and improve wound healing [26].
We did not have sufficient data to refute the benefits of EN over PN until the paper by Harvey et al (2014), which reported no difference in mortality or infectious complications in ICU patients receiving EN or PN within 36 hours of admission and for up to 5 days [30]. This was the largest published pragmatic RCT, referred to as the CALORIES trial, which analyzed 2388 patients from 33 ICUs and resulted in controversy over what was an unchallenged approach up until this time. It was only a matter of time before other investigators would set out to confirm or negate this finding, which is what Elke and coworkers (2016) did a few years later [31]. They performed an updated systematic review and meta-analysis to evaluate the overall effect of the route of nutrition (EN versus PN) on clinical outcomes in adult critically ill patients. Similar to the Harvey et al report, they found no difference in mortality between the two routes of nutrition. However, unlike the earlier report, patients receiving EN compared to PN had a significant reduction in the number of infectious complications and ICU length of stay. No significant effect was found for hospital length of stay or days requiring mechanical ventilation. The authors suggest that EN delivery of macronutrients below predefined targets may be responsible as PN is more likely to meet or exceed these targets and overwhelm metabolic capacity in the early days of critical illness [31].
The most recent trial prompting even more discussion about early PN versus early EN in mechanically ventilated ICU patients in shock is the Reignier et al (2018) NUTRIREA-2 trial involving 2410 patients from 44 ICUs in France [32]. The investigators hypothesized that outcomes would be better with early exclusive EN compared to early exclusive PN; their hypothesis was not supported by the results, which found no difference in 28-day mortality or ICU-acquired infections. Also unexpected was the higher cumulative incidence of gastrointestinal complications including vomiting, diarrhea, bowel ischemia, and acute colonic obstruction in the EN group. The trial was stopped early after an interim analysis determined that additional enrollment was not likely to significantly change the results of the trial. Given the similarities between the CALORIES trial and this NUTRIREA-2 trial, clinicians now have mounting evidence that equivalent options for nutrition therapy exist and an appropriate selection should be made based on patient-specific indications and treatment goals. In summary, EN remains preferable to PN for the majority of adult critically ill patients due to crucial support of gut integrity, but the optimal dose or rate of delivery to favorably influence clinical outcomes in the first few days following admission remains unknown.
Use of Supplemental Parenteral Nutrition
Both the nutrition support and bench science communities have learned a great deal about PN over the 4 decades it has been in existence, with the most compelling data coming from more recent trials [31–38]. This is because it has taken many years to recover from the days of hyperalimentation or overfeeding ICU patients by providing excessive calories to meet the elevated energy demands and to reverse the hypercatabolism of critical illness. This approach contributed to the complications of hyperglycemia, hyperlipidemia, increased infectious complications, and liver steatosis, all of which gave PN a negative reputation [37]. We now have adjusted the caloric distribution and the actual formulation of PN using the recent FDA-approved lipid emulsion (Soy, Medium-chain triglycerides, Olive oil, and Fish oil; SMOF) and created protocols for administering it based on specific indications, such as loss of GI integrity or demonstrated intolerance. In general, the advances in PN have led to a safer more therapeutic formulation that has its place in critical illness. Manzanares et al [40] reported a trend toward a decrease in ventilation requirement and mortality when a fish oil–containing lipid emulsion was administered to patients who were receiving nutrition support either enterally or parenterally. The meta-analysis combined all soybean oil–sparing lipid emulsions for comparison with soybean oil and was able to show the trend for improved clinical outcomes with soybean oil–sparing lipid emulsions. The main findings of this meta-analysis were that fish oil–containing lipid emulsions may reduce infections and may be associated with a tendency toward fewer mechanical ventilation days, although not mortality, when compared with soybean oil-based strategies or administration of other alternative lipid emulsions in ICU patients [40]. Recent trial results do not change the recommendation for selecting EN first but do suggest lowering the threshold for using PN when EN alone is insufficient to meet nutrition goals. A systematic review reported no benefit of early supplemental PN over late supplemental PN and cautioned that our continued inability to explain greater infectious morbidity and unresolved organ failure limits any justification for early PN [35].
Protein Quantity and Quality
The practice of providing protein in the range of 1.2–2.0 g/kg actual body weight early in critical illness is suggested by expert consensus in the 2016 SCCM/ASPEN guidelines [12]; however, the evidence for efficacy remains controversial. It is argued that the evidence for benefit comes from observational studies, not from prospective RCTs, and that the patients at high risk are often excluded from study protocols. It has also been suggested that in patients with limited vital organ function increased protein delivery may lead to organ compromise. In a recent (2017) paper, Rooyackers et al discussed the post-hoc analyses of data from the EPANIC Trial stating the statistical correlation between protein intake and outcomes indicate that protein was associated with unfavorable outcomes, possibly by inhibiting autophagy [41].
The nutrition support community may have widely varying approaches to feeding critically ill patients but most experts agree that protein may be the most important macronutrient delivered during critical illness. There is consensus that the hypercatabolism associated with stress induces proteolysis and the loss of lean muscle mass, which may affect clinical and functional outcomes beyond the ICU stay. Using multiple assessment modalities, Puthucheary et al (2013) demonstrated a reduction in the rectus femoris muscle of 12.5% over the first week of hospitalization in the ICU and up to 17.7% by day 10. These numbers imply that sufficient protein of at least 1.2 g/kg/day should be provided to minimize these losses, even if the effect on overall outcome remains unknown [42]. Evidence is lacking for whether or not we can prevent the muscle wasting that occurs in critical illness with increased protein dosing. We also need to better identify the possible risks involved with a high-protein intake at the level of the individual patient. A secondary analysis done by Heyland et al (2013) determined that no specific dose or type of macronutrient was found to be associated with improved outcome [43]. It is clear that more large-scale RCTs of protein/amino acid interventions are needed to prove that these nutrition interventions have favorable effects on clinically important outcomes, including long-term physical function.
Polymeric vs Immune-Modulating Nutrients
The Marik and Zaloga (2008) systematic review on immunonutrition in the critically ill convinced most clinicians that while fish-oil based immunonutrition improves the outcome of medical ICU patients, diets supplemented with arginine, with or without glutamine or fish oils, do not demonstrate an advantage over standard enteral products in general ICU, trauma, or burn patients[44]. What followed these trials examining early formulations of immunonutrition was decades of well-intentioned research dedicated to elucidating the mechanism of action for individual immune-modulating nutrients for various populations, including those with acute lung injury/acute respiratory distress syndrome (ARDS) [45–47], sepsis/systemic inflammatory response syndrome [48–50], head and neck cancer [51], upper and lower GI cancer [52–55], and severe acute pancreatitis [56]. Our understanding of immunonutrition and the administration of this formulation in specific disease conditions has grown considerably yet clinicians are still asking exactly what is the role of immunonutrition and who stands to benefit the most from immune-modulating nutrition therapy. The enteral formulations currently available have a proprietary composition and dosage of individual nutrients which yield unpredictable physiologic effects. In addition, the pervasive problem of underfeeding during hospitalization prevents adequate delivery of physiologic doses of nutrients thus contributing to the widely variable research results.
Prevailing expert opinion today is that the majority of critically ill patients will benefit from nutrition support initiated early and delivered consistently; the standard polymeric formula will suffice for the majority of patients with surgical ICU patients potentially deriving benefit from immunonutrition that supports a reduction in infectious complications [57]. In the recent multiple-treatment meta-analysis performed by Mazaki et al (2015) involving 74 studies and 7572 patients, immunonutrition was ranked first for reducing the incidence of 7 complications according to the surface under the cumulative ranking curve; these were as follows: any infection, 0.86; overall complication, 0.88; mortality, 0.81; wound infection, 0.79; intra-abdominal abscess, 0.98; anastomotic leak, 0.79; and sepsis, 0.92. immunonutrition was ranked second for reducing ventilator-associated pneumonia and catheter-associated urinary tract infection (CAUTI), behind immune-modulating PN. The authors stated that immunonutrition was efficacious for reducing the incidence of complications in GI surgery unrelated to the timing of administration [57]. The 2014 publication of results from the MetaPlus Trial [58]challenged the published recommendations for the use of immunonutrition in the medical critically ill population. This trial used high-protein immunonutrition or standard formula for 301 adult critically ill patients in 14 European ICUs with diagnoses such as pneumonia or infections of the urinary tract, bloodstream, central nervous system, eye, ear, nose or throat, and the skin and soft tissue. Even with higher than average target energy intakes of 70% for the high protein immunonutrition group and 80% for the high protein standard group, there were no statistically significant differences in the primary outcome of new infections, or the secondary outcomes of days on mechanical ventilation, Sequential Organ Failure Assessment scores, or ICU and hospital length of stay. However, the 6-month mortality rate of 28% was higher in the medical subgroup [58]. Using these results, as well as existing publications of negative outcomes in medical ICU patients [44,46], the SCCM/ASPEN Guidelines Committee updated its position in 2016 to suggest that immunonutrition formulations or disease-specific formulations should no longer be used routinely in medical ICU patients, including those with acute lung injury/ARDS [12]. The Committee did suggest that these formulations should be reserved for patients with traumatic brain injury and for surgical ICU patients. The benefit for ICU postoperative patients has been linked to the synergistic effect of fish oil and arginine, which must both be present to achieve outcome benefits. A meta-analysis comprised of 35 trials was conducted by Drover et al [58], who reported that administering an arginine and fish oil-containing formula postoperatively reduced infection (RR 0.78; 95% CI, 0.64 to 0.95; P = 0.01) and hospital length of stay (WMD –2.23, 95% CI, –3.80 to –0.65; P = 0.006) but not mortality, when compared to use of a standard formula. Similar results were reported in a second meta-analysis [56], thus providing supportive evidence for the current SCCM/ASPEN recommendation to use an immune-modulating formula (containing both arginine and fish oils) in the SICU for the postoperative patient who requires EN therapy [12].
Safe, Consistent, and Effective Nutrition Support Therapy
Gastric vs Small Bowel Feeding
There is a large group of critically ill patients in whom impaired gastric emptying presents challenges to feeding; 50% of mechanically ventilated patients demonstrate delayed gastric emptying, and 80% of patients with increased intracranial pressure following head injury [60]. In one prospective RCT, Huang et al (2012) showed that severely ill patients (defined by an APACHE II score > 20) fed by the nasoduodenal route experienced significantly shortened hospital LOS, fewer complications, and improved nutrient delivery compared to similar patients fed by the nasogastric route. Less severely ill patients (APACHE II < 20) showed no differences between nasogastric and nasoduodenal feeding for the same outcomes [61]. A recent systematic review [17] pooled data from 14 trials of 1109 participants who received either gastric or small bowel feeding. Moderate quality evidence suggested that post-pyloric feeding was associated with lower rates of pneumonia compared with gastric feeding (RR 0.65, 95% CI, 0.51 to 0.84). Low-quality evidence showed an increase in the percentage of total nutrient delivered to the patient by post-pyloric feeding (mean difference 7.8%, 95% CI, 1.43 to 14.18). Overall, the authors found a 30% lower rate of pneumonia associated with post-pyloric feeding. There is insufficient evidence to show that other clinically important outcomes such as duration of mechanical ventilation, mortality, or LOS were affected by the site of feeding. The American Thoracic Society and ASPEN, as well as the Infectious Diseases Society of America, have published guidelines in support of small bowel feeding in the ICU setting due to its association with reduced incidence of health care–associated infections, specifically ventilator-associated pneumonia [62]. The experts who developed the SCCM/ASPEN and Canadian guidelines stress that critically ill patients at high risk for aspiration or feeding intolerance should be fed using small bowel access [12,21]. The reality in ICU clinical practice is that many centers will begin with gastric feeding, barring absolute contraindications, and carefully monitor the patient for signs of intolerance before moving the feeding tube tip into a post-pyloric location. This follows the general recommendation by experts saying in most critically ill patients, it is acceptable to initiate EN in the stomach [12,21]. Protocols that guide management of risk prevention and intolerance typically recommend head of bed elevation, prokinetic agents, and frequent abdominal assessments [63,64].
Once the decision is made to use a post-pyloric feeding tube for nutrition therapy, the next decision is how to safely place the tube, ensure the tip is in an acceptable small bowel location, and minimize delays in feeding. Challenges related to feeding tube insertion may preclude timely advancement to nutrition goals. Placement of feeding tubes into the post-pyloric position is often done at the bedside by trained nursing or medical staff without endoscopic or fluoroscopic guidance; however, the blind bedside approach is not without risks. Success rates of this approach vary greatly depending on the patient population and provider expertise. Placement using endoscopic or fluoroscopic guidance is a safe alternative but usually requires coordinating a transport to the radiologic suite, posing safety risks and possible feeding delays for the patient [65]. Bedside use of an electromagnetic placement device (EMPD), such as Cortrak, provides yet another alternative with reports in the literature of 98% success rates for initial placement in less than 20 minutes. In a multicenter prospective study by Powers et al (2011), only one of 194 patients enrolled had data showing a discrepancy between the original EMPD verification and the final radiograph interpretation, demonstrating a 99.5% agreement between the two readings [20]. Median placement time was 12 minutes and no patient experienced an adverse event related to tube insertion using this device. The ability to monitor the location of the feeding tube tip in real time provides a desirable safety feature for the clinician performing bedside insertions. Nurses should consider incorporating the EMPD into the unit feeding protocol, as this would reduce the time to initiation of feedings with early and accurate tube insertion. Ongoing staff education and experience with the procedure are necessary elements to achieve the high rates of success often reported in the literature [66,67]. Procedural complications from placement of nasoenteral feeding tubes by all methods can be as high as 10%, with complication rates of 1% to 3% for inadvertent placement of the feeding tube in the airway alone [65]. Radiographic confirmation of tube placement is advised prior to initiating feeding, thus eliminating any possibility of misplacement and administration of formula into the lungs.
Gastric Residual Volume Monitoring
A number of factors impede the delivery of EN in the critical care setting; these include gastrointestinal intolerance, under-prescribing to meet daily requirements, frequent interruptions for procedures, and technical issues with tube placement and maintaining patency [68]. Monitoring gastric residual volumes (GRV) contributes to these factors, yet volumes do not correlate well with incidence of pneumonia [69], measures of gastric emptying, or to the incidence of regurgitation and aspiration [70,71]. However, few studies have highlighted the difficulty of obtaining an accurate GRV due to feeding tube tip location, patient position, and type of tube [69]. Several high quality studies have demonstrated that raising the cutoff value for GRV from a lower number of 50–150 mL to a higher number of 250–500 mL does not increase risk for regurgitation, aspiration, or pneumonia [70,71]. A lower cutoff value for GRV does not protect the patient from complications, often leads to inappropriate cessation, and may adversely affect outcome through reduced volume of EN infused [72]. Gastric residual volumes in the range of 200–500 mL should raise concern and lead to the implementation of measures to reduce risk of aspiration, but automatic cessation of feeding should not occur for GRV < 500 mL in the absence of other signs of intolerance [12,69]. Metheny et al (2012) conducted a survey in which more than 97% of nurses responded that they assessed intolerance by measuring GRV; the most frequently cited threshold levels for interrupting feedings were 200 mL and 250 mL [73]. While threshold levels varied widely, only 12.6% of the nurse respondents reported allowing GRV up to 500 mL before interrupting feedings. While monitoring GRV is unnecessary with small bowel feeding, the location of the feeding tube tip should be questioned if gastric contents are obtained from a small bowel tube. The use of GRV as a parameter for trending may also yield important information regarding tolerance of feeding when the patient is unable to communicate abdominal discomfort. Other objective measures to use in the assessment of tolerance include an abdominal exam with documentation of changes in bowel sounds, expanding girth, tenderness or firmness on palpation, increasing nasogastric output, and vomiting [12,68]. If there are indications of intolerance, it is appropriate to divert the tip of the feeding tube into the distal small bowel as discussed previously.
Trophic vs Full Feeding
For the patient with low nutrition risk, there is a lack of convincing data to support an aggressive approach to feeding, either EN or PN, in the first week of critical illness [7]. In recent years, results of several trials suggest early goal-directed feeding in this population may cause net harm with increased morbidity and mortality. When discussing recent controversies in critical care nutrition, one must mention the two schools of thought when it comes to full versus limited energy provision in the first week following ICU admission. Studies in animals and humans have shown a trophic effect of enteral nutrients on the integrity of the gut mucosa, a finding that has provided the rationale for instituting enteral nutrition early during critical illness [15]. However, the inability to provide enteral nutrition early may be a marker of the severity of illness (ie, patients who can be fed enterally are less ill than those who cannot) rather than a mediator of complications and poor outcomes. Compher et al (2017) stated that greater nutritional intake is associated with lower mortality and faster time to discharge alive in high-risk, chronic patients but does not seem to be significant in nutritionally low-risk patients [74]. The findings of the EPaNIC and EDEN trials raised concern that targeting goals that meet full energy needs early in critical illness does not provide benefit and may cause harm in some populations or settings [32,75]. The EDEN trial [32] left us believing that trophic feeding at 10–20 mL/hr may be just as effective as any feeding in the first few days of critical illness striving for 15% to 20% of daily goal calories. After establishing tolerance, advancing daily intake to > 50% to 65% of goal calories, and up to 80% for the highest risk patients, may be required to prevent intestinal permeability and achieve positive clinical outcomes [33].
The systematic review and meta-analysis performed by Al-Dorzi et al (2016) adds further evidence for judicious advancement of EN for critically ill patients [76]. The authors reported finding no association between the dose of caloric intake and hospital mortality. Furthermore, a lower caloric intake resulted in lower risk of bloodstream infections and the need for renal replacement therapy (in 5 of the 21 trials only). As with many other meta-analyses, the authors reported that their results are most assuredly impacted by the heterogeneity in design, feeding route, and dose prescribed and delivered [16,76,77]. Other recent trials such as Arabi et al (2015) that enrolled 894 patients with different feeding targets further confirmed that there is no difference in outcome between groups when it comes to moderate (40% to 60% of goal) vs high (70% to 100% of goal) energy intake, infection rates, or 90-day mortality. The authors summarized their findings saying feeding closer to target is associated with better outcomes compared with severe underfeeding [78]. This adds to the controversy when considering the findings of still other RCTs or meta-analyses that evaluated minimal or trophic feeding versus standard feeding rates [9,46,77]. The meta-analysis performed by Marik and Hooper concluded that there were no differences in the risk of acquired infections, hospital mortality, ICU LOS, or ventilator-free days whether patients received intentional hypocaloric or normocaloric nutrition support [9]. Similarly, there was no significant difference in overall mortality between the underfeeding and full-feeding groups (OR, 0.94; 95% CI, 0.74–1.19; I2 = 26.6%; P = 0.61) in the meta-analysis done by Choi et al (2015), although only 4 trials were included to ensure homogeneity of the population and the intervention [77]. Furthermore, the hospital LOS and ICU LOS did not differ between the 2 groups, nor did any other secondary clinical outcome, leading the authors to conclude that calorie intake of the initial EN support for ICU patients had no bearing on relevant outcomes.
Recent studies have attempted to correlate caloric intake and patient outcomes without success; achieving 100% of caloric goal has not favorably impacted morbidity and mortality. Evidence suggests that intake greater than 65% to 70% of daily caloric requirement in the first 7 to 10 days of ICU stay may be associated with poorer outcomes, particularly when parenteral nutrition is used to supplement intake to achieve the caloric target [33–35].
Conclusion
In this review we described current ICU controversies surrounding nutrition therapy and briefly discussed the data that support more than one course of action. A summary of key points covered is presented in the Table.
Disclaimer: The views expressed in this paper are those of the authors and do not reflect the official policy of the Department of the Army, the Department of Defense, or the U.S. government.
Corresponding author: Mary S. McCarthy, PhD, RN, CNSC, 1611 Nisqually St, Steilacoom, WA 98388.
Financial disclosures: None.
1. Hippocratic Oath. Translated by Michael North, National Library of Medicine, 2002.
2. Nightingale F. Notes on Nursing. What it is and what it is not. Radford, VA: Wilder Publications, LLC;2007
3. White JV, Guenter P, Jensen G, et al; the Academy Malnutrition Work Group; the ASPEN Malnutrition Task Force; and the ASPEN Board of Directors. Consensus statement: Academy of Nutrition and Dietetics and American Society for Parenteral and Enteral Nutrition: characteristics recommended for the identification and documentation of adult malnutrition (undernutrition). JPEN J Parenter Enteral Nutr 2012;36:275–83.
4. Hooper MH, Marik PE. Controversies and misconceptions in Intensive Care Unit nutrition. Clin Chest Med 2015;36:409–18.
5. Patel JJ, Codner P. Controversies in critical care nutrition support. Crit Care Clin 2016;32:173–89.
6. Rosenthal MD, Vanzant EL, Martindale RG, Moore FA. Evolving paradigms in the nutritional support of critically ill surgical patients. Curr Probl Surg 2015;52:147–82.
7. McCarthy MS, Warren M, Roberts PR. Recent critical care nutrition trials and the revised guidelines: do they reconcile? Nutr Clin Pract 2016;31:150–4.
8. Barker LA, Gray C, Wilson L, et al. Preoperative immunonutrition and its effect on postoperative outcomes in well-nourished and malnourished gastrointestinal surgery patients: a randomised controlled trial. Eur J Clin Nutr 2013;67: 802–807.
9. Marik PE, Hooper MH. Normocaloric versus hypocaloric feeding on the outcomes of ICU patients: a systematic review and meta-analysis. Intensive Care Med 2016;42:316–323.
10. Patkova A, Joskova V, Havel E, et al. Energy, protein, carbohydrate, and lipid intakes and their effects on morbidity and mortality in critically ill adult patients: a systematic review. Adv Nutr 2017;8:624–34.
11. Wong CS, Aly EH. The effects of enteral immunonutrition in upper gastrointestinal surgery: a systematic review and meta-analysis. Int J Surg 2016;29:137–50.
12. McClave SA, Taylor BE, Martindale RG, et al; Society of Critical Care Medicine; American Society for Parenteral and Enteral Nutrition. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society of Parenteral and Enteral Nutrition (ASPEN). JPEN J Parenter Enteral Nutr 2016; 40:159–211.
13. Ammori BJ. Importance of the early increase in intestinal permeability in critically ill patients. Eur J Surg 2002;168:660–1.
14. Vazquez-Sandoval A, Ghamande S, Surani S. Critically ill patients and gut motility: are we addressing it? World J Gastrointest Pharmacol Ther 2017;8:174–9.
15. Patel JJ, Martindale RG, McClave SA. Controversies surrounding critical care nutrition: an appraisal of permissive underfeeding, protein, and outcomes. JPEN J Parenter Enteral Nutr 2017; 148607117721908.
16. Hegazi RA, Hustead DS, Evans DC. Preoperative standard oral nutrition supplements vs immunonutrition: results of a systematic review and meta-analysis. J Am Coll Surg 2014;219:1078–87.
17. Alkhawaja S, Martin C, Butler RJ, Gwadry-Sridhar F. Post-pyloric versus gastric tube feeding for preventing pneumonia and improving nutritional outcomes in critically ill adults. Cochrane Database of Syst Rev 2015; CD008875.
18. Davies AR, Morrison SS, Bailey MJ, et al ; ENTERIC Study Investigators; ANZICS Clinical Trials Group. A multi-center randomized controlled trial comparing early nasojejunal with nasogastric nutrition in critical illness. Crit Care Med 2012;40:2342–8.
19. Hsu CW, Sun SF, Lin SL, et al. Duodenal versus gastric feeding in medical intensive care unit patients: a prospective, randomized, clinical study. Crit Care Med 2009;37:1866–72.
20. Powers J, Luebbehusen M, Spitzer T, et al. Verification of an electromagnetic placement device compared with abdominal radiograph to predict accuracy of feeding tube placement. JPEN J Parenter Enteral Nutr 2011;35:535–9.
21. Dhaliwal R, Cahill N, Lemieux M, Heyland DK. The Canadian critical care nutrition guidelines in 2013: an update on current recommendations and implementation strategies. Nutr Clin Pract 2014; 29:29–43.22. Kreymann K, Berger M, Deutz N. et al; DGEM (German Society for Nutritional Medicine); ESPEN (European Society for Parenteral and Enteral Nutrition). ESPEN guidelines on enteral nutrition: intensive care. Clin Nutr 2006;25:210–23.
23. McClave SA, Martindale RG, Vanek VW, et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (ASPEN). JPEN J Parenter Ent Nutr 2009;33:277–316.
24. McClave SA, Martindale RG, Rice TW, Heyland DK. Feeding the critically ill patient. Crit Care Med 2014;42:2600–10.
25. Tian F, Gao X, Wu C, et al. Initial energy supplementation in critically ill patients receiving enteral nutrition: a systematic review and meta-analysis of randomized controlled trials. Asia Pac J Clin Nutr 2017;26:11–9.
26. Martindale RG, Warren M. Should enteral nutrition be started in the first week of critical illness? Curr Opin Clin Nutr Metab Care 2015;18:202–6.
27. McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract 2009;24:305–15.
28. Kang W, Kudsk KA. Is there evidence that the gut contributes to mucosal immunity in humans? JPEN J Parenter Enteral Nutr 2007;31:461–82.
29. Seron-Arbeloa C, Zamora-Elson M, Labarta-Monzon L, Mallor-Bonet T. Enteral nutrition in critical care. J Clin Med Res 2013;5:1-11.
30. Harvey SE, Parrott F, Harrison DA, et al; CALORIES Trial Investigators. Trial of the route of early nutritional support in critically ill adults. N Engl J Med 2014;371:1673–84.
31. Elke G, van Zanten AR, Lemieux M, et al. Enteral versus parenteral nutrition in critically ill patients: an updated systematic review and meta-analysis of randomized controlled trials. Crit Care 2016;20:117.
32. Reignier J, Boisramé-Helms J, Brisard L, et al. Enteral versus parenteral nutrition in ventilated adults with shock: a randomized, controlled, multicenter, open-label, parallel-group study (NUTRIREA-2). Lancet 2018;391:133–43.
33. Rice TW , Wheeler AP, Thompson BT et al;National Heart, Lung, and Blood Institute, Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Initial trophic vs full enteral feeding in patients with acute lung injury: the EDEN randomized trial. JAMA 2012;307:795–803.
34. Heyland DK, Dhaliwal R, Jiang X, Day AG. Identifying critically ill patients who benefit the most from nutrition therapy: the development and initial validation of a novel risk assessment tool. Crit Care 2011;15:R258.
35. Bost RB, Tjan DH, van Zanten AR. Timing of (supplemental) parenteral nutrition in critically ill patients: a systematic review. Ann Intensive Care 2014;4:31.
36. Casaer MP, Mesotten D, Hermans G, et al. Early verus late parenteral nutrition in critically ill adults. N Eng J Med 2011;365:506–17.
37. Harvey SE, Parrott F, Harrison DA, et al; CALORIES Trial Investigators. Trial of the route of early nutritional support in critically ill adults. N Eng J Med 2014;371:1673–84.
38. Manzanares W, Dhaliwal R, Jurewitsch B, et al. Parenteral fish oil lipid emulsions in the critically ill: A systematic review and meta-analysis. JPEN J Parenter Enteral Nutr 2014;38:20–8.
39. Oshima T, Heidegger CP, Pichard C. Supplemental parenteral nutrition is the key to prevent energy deficits in critically ill patients. Nutr Clin Prac 2016;31:432–7.
40. Manzanares W, Langlois PL, Dhaliwal R, Lemieux M, Heyland DK. Intravenous fish oil lipid emulsions in critically ill patients: an updated systematic review and meta-analysis. Crit Care 2015;19:167.
41. Rooyackers O, Sundström Rehal M, Liebau F, et al. High protein intake without concerns? Crit Care 2017;21:106.
42. Puthucheary ZA, Rawal J, McPhail M, et al. Acute skeletal muscle wasting in critical illness. JAMA 2013;310:1591–600.
43. Heyland D, Muscedere J, Wischmeyer PE, et al; Canadian Critical Care Trials Group. A randomized trial of glutamine and antioxidants in critically ill patients. N Engl J Med 2013;368:1489–97.
44. Marik PE, Zaloga GP. Immunonutrition in critically ill patients: a systematic review and analysis of the literature. Intensive Care Med 2008;34:1980–90.
45. Gadek JE, DeMichele SJ, Karlstad MD, et al; Enteral Nutrition in ARDS Study Group. Effect of enteral feeding with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome. Crit Care Med 1999;27:1409–20.
46. Rice TW, Wheeler AP, Thompson BT, et al; NIH NHLBI Acute Respiratory Distress Syndrome Network of Investigators. Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA 2011;306:1574–81.
47. Singer P, Theilla M, Fisher H, et al. Benefit of an enteral diet enriched with eicosapentaenoic acid and gamma-linolenic acid in ventilated patients with acute lung injury. Crit Care Med 2006;34:1033–38.
48. Atkinson S, Sieffert E, Bihari D. A prospective, randomized, double-blind, controlled clinical trial of enteral immunonutrition in the critically ill. Guy’s Hospital Intensive Care Group. Crit Care Med 1998;26:1164–72.
49. Galbán C, Montejo JC, Mesejo A, et al. An immune-enhancing enteral diet reduces mortality rate and episodes of bacteremia in septic intensive care unit patients. Crit Care Med 2000;28:643–8.
50. Weimann A, Bastian L, Bischoff WE, et al. Influence of arginine, omega-3 fatty acids and nucleotide-supplemented enteral support on systemic inflammatory response syndrome and multiple organ failure in patients after severe trauma. Nutrition 1998;14:165–72.
51. van Bokhorst-De Van Der Schueren MA, Quak JJ, von Blomberg-van der Flier BM, et al. Effect of perioperative nutrition with and without arginine supplementation, on nutritional status, immune function, postoperative morbidity, and survival in severely malnourished head and neck cancer patients. Am J Clin Nutr 2001;73:323–32.
52. Cerantola Y, Hübner M, Grass F, et al. Immunonutrition in gastrointestinal surgery. Br J Surg 2011;98:37–48.
53. Marik PE, Zaloga GP. Immunonutrition in high-risk surgical patients: a systematic review and analysis of the literature. JPEN J Parenter Enteral Nutr 2010;34:378–86.
54. Sultan J, Griffin SM, Di Franco F, et al. Randomized clinical trial of omega-3 fatty acid–supplemented enteral nutrition vs. standard enteral nutrition in patients undergoing oesophagogastric cancer surgery. Br J Surg 2012;99:346–55.
55. Waitzberg DL, Saito H, Plank LD, et al. Postsurgical infections are reduced with specialized nutrition support. World J Surg 2006;30:1592–604.
56. Pearce CB, Sadek SA, Walters AM, et al. A double-blind, randomised, controlled trial to study the effects of an enteral feed supplemented with glutamine, arginine, and omega-3 fatty acid in predicted acute severe pancreatitis. JOP 2006;7:361–71.
57. Mazaki T, Ishii Y, Murai I. Immunoenhancing enteral and parenteral nutrition for gastrointestinal surgery: a multiple treatments meta-analysis. Ann Surg 2015;261:662–9.
58. van Zanten ARH, Sztark F, Kaisers UX, et al. High-protein enteral nutrition enriched with immune-modulating nutrients vs standard high protein enteral nutrition and nosocomial infections in the ICU. JAMA 2014;312:514–24.
59. Drover JW, Dhaliwal R, Weitzel L, et al. Perioperative use of arginine supplemented diets: a systematic review of the evidence. J Am Coll Surg 2011;212:385–99.
60. Stupak D, Abdelsayed GG, Soloway GN. Motility disorders of the upper gastrointestinal tract in the intensive care unit: pathophysiology and contemporary management. J Clin Gastroenterol 2012;46:449–56.
61. Huang HH, Chang SJ, Hsu CW, et al. Severity of illness influences the efficacy of enteral feeding route on clinical outcomes in patients with critical illness. J Acad Nutr Diet 2012;112:1138–46.
62. American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005;171:388–416.
63. Heyland DK, Cahill NE, Dhaliwal R, et al. Impact of enteral feeding protocols on enteral nutrition delivery: results of a multicenter observational study. JPEN J Parenter Enteral Nutr 2010;34:675–84.
64. Landzinski J, Kiser TH, Fish DN, et al. Gastric motility function in critically ill patients tolerant vs intolerant to gastric nutrition. JPEN J Parenter Enteral Nutr 2008;32:45–50.
65. de Aguilar-Nascimento JE, Kudsk KA. Use of small bore feeding tubes: successes and failures. Curr Opin Clin Nutr Metab Care 2007;10:291–6.
66. Boyer N, McCarthy MS, Mount CA. Analysis of an electromagnetic tube placement device vs a self-advancing nasal jejunal device for postpyloric feeding tube placement . J Hosp Med 2014;9:23–8.
67. Metheny NA, Meert KL. Effectiveness of an electromagnetic feeding tube placement device in detecting inadvertent respiratory placement. Am J Crit Care 2014;23:240–8.
68. Montejo JC, Miñambres E, Bordejé L, et al. Gastric residual volume during enteral nutrition in ICU patients: the REGANE study. Intensive Care Med 2010;36:1386–93.
69. Hurt RT, McClave SA. Gastric residual volumes in critical illness: what do they really mean? Crit Care Clin 2010;26:481–90.
70. Poulard F, Dimet J, Martin-Lefevre L, et al. Impact of not measuring residual gastric volume in mechanically ventilated patients receiving early enteral feeding: a prospective before-after study. JPEN J Parenter Enteral Nutr 2010;34:125–30.
71. Reignier J, Mercier E, Gouge AL, et al; Clinical Research in Intensive Care and Sepsis (CRICS) Group. Effect of not monitoring residual gastric volume on risk of ventilator-associated pneumonia in adults receiving mechanical ventilation and early enteral feeding: a randomized controlled trial. JAMA 2013;309:249–56.
72. Williams TA, Leslie GD, Leen T, et al. Reducing interruptions to continuous enteral nutrition in the intensive care unit: a comparative study. J Clin Nurs 2013;22:2838-2848.
73. Metheny NA, Stewart BJ, Mills AC. Blind insertion of feeding tubes in intensive care units: a national survey. Am J Crit Care 2012;21:352–360.
74. Compher C, Chittams J, Sammarco T, et al. Greater protein and energy intake may be associated with improved mortality in higher risk critically ill patients: A multicenter, multinational observational study. Crit Care Med 2017;45:156–163.
75. Casaer MP, Wilmer A, Hermans G, et al. Role of disease and macronutrient dose in the randomized controlled EPANIC Trial: a post hoc analysis. Am J Resp Crit Care Med 2013;187:247–55.
76. Al-Dorzi HM, Albarrak A, Ferwana M, et al. Lower versus higher dose of enteral caloric intake in adult critically ill patients: a systematic review and meta-analysis. Crit Care 2016;20:358.
77. Choi EY, Park DA, Park J. Calorie intake of enteral nutrition and clinical outcomes in acutely critically ill patients: a meta-analysis of randomized controlled trials. JPEN J Parenter Enteral Nutr 2015;39:291–300.
78. Arabi YM, Aldawood AS, Haddad SH, et al. Permissive underfeeding or standard enteral feeding in critically ill adults. N Engl J Med 2015;372:2398–408.
1. Hippocratic Oath. Translated by Michael North, National Library of Medicine, 2002.
2. Nightingale F. Notes on Nursing. What it is and what it is not. Radford, VA: Wilder Publications, LLC;2007
3. White JV, Guenter P, Jensen G, et al; the Academy Malnutrition Work Group; the ASPEN Malnutrition Task Force; and the ASPEN Board of Directors. Consensus statement: Academy of Nutrition and Dietetics and American Society for Parenteral and Enteral Nutrition: characteristics recommended for the identification and documentation of adult malnutrition (undernutrition). JPEN J Parenter Enteral Nutr 2012;36:275–83.
4. Hooper MH, Marik PE. Controversies and misconceptions in Intensive Care Unit nutrition. Clin Chest Med 2015;36:409–18.
5. Patel JJ, Codner P. Controversies in critical care nutrition support. Crit Care Clin 2016;32:173–89.
6. Rosenthal MD, Vanzant EL, Martindale RG, Moore FA. Evolving paradigms in the nutritional support of critically ill surgical patients. Curr Probl Surg 2015;52:147–82.
7. McCarthy MS, Warren M, Roberts PR. Recent critical care nutrition trials and the revised guidelines: do they reconcile? Nutr Clin Pract 2016;31:150–4.
8. Barker LA, Gray C, Wilson L, et al. Preoperative immunonutrition and its effect on postoperative outcomes in well-nourished and malnourished gastrointestinal surgery patients: a randomised controlled trial. Eur J Clin Nutr 2013;67: 802–807.
9. Marik PE, Hooper MH. Normocaloric versus hypocaloric feeding on the outcomes of ICU patients: a systematic review and meta-analysis. Intensive Care Med 2016;42:316–323.
10. Patkova A, Joskova V, Havel E, et al. Energy, protein, carbohydrate, and lipid intakes and their effects on morbidity and mortality in critically ill adult patients: a systematic review. Adv Nutr 2017;8:624–34.
11. Wong CS, Aly EH. The effects of enteral immunonutrition in upper gastrointestinal surgery: a systematic review and meta-analysis. Int J Surg 2016;29:137–50.
12. McClave SA, Taylor BE, Martindale RG, et al; Society of Critical Care Medicine; American Society for Parenteral and Enteral Nutrition. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society of Parenteral and Enteral Nutrition (ASPEN). JPEN J Parenter Enteral Nutr 2016; 40:159–211.
13. Ammori BJ. Importance of the early increase in intestinal permeability in critically ill patients. Eur J Surg 2002;168:660–1.
14. Vazquez-Sandoval A, Ghamande S, Surani S. Critically ill patients and gut motility: are we addressing it? World J Gastrointest Pharmacol Ther 2017;8:174–9.
15. Patel JJ, Martindale RG, McClave SA. Controversies surrounding critical care nutrition: an appraisal of permissive underfeeding, protein, and outcomes. JPEN J Parenter Enteral Nutr 2017; 148607117721908.
16. Hegazi RA, Hustead DS, Evans DC. Preoperative standard oral nutrition supplements vs immunonutrition: results of a systematic review and meta-analysis. J Am Coll Surg 2014;219:1078–87.
17. Alkhawaja S, Martin C, Butler RJ, Gwadry-Sridhar F. Post-pyloric versus gastric tube feeding for preventing pneumonia and improving nutritional outcomes in critically ill adults. Cochrane Database of Syst Rev 2015; CD008875.
18. Davies AR, Morrison SS, Bailey MJ, et al ; ENTERIC Study Investigators; ANZICS Clinical Trials Group. A multi-center randomized controlled trial comparing early nasojejunal with nasogastric nutrition in critical illness. Crit Care Med 2012;40:2342–8.
19. Hsu CW, Sun SF, Lin SL, et al. Duodenal versus gastric feeding in medical intensive care unit patients: a prospective, randomized, clinical study. Crit Care Med 2009;37:1866–72.
20. Powers J, Luebbehusen M, Spitzer T, et al. Verification of an electromagnetic placement device compared with abdominal radiograph to predict accuracy of feeding tube placement. JPEN J Parenter Enteral Nutr 2011;35:535–9.
21. Dhaliwal R, Cahill N, Lemieux M, Heyland DK. The Canadian critical care nutrition guidelines in 2013: an update on current recommendations and implementation strategies. Nutr Clin Pract 2014; 29:29–43.22. Kreymann K, Berger M, Deutz N. et al; DGEM (German Society for Nutritional Medicine); ESPEN (European Society for Parenteral and Enteral Nutrition). ESPEN guidelines on enteral nutrition: intensive care. Clin Nutr 2006;25:210–23.
23. McClave SA, Martindale RG, Vanek VW, et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (ASPEN). JPEN J Parenter Ent Nutr 2009;33:277–316.
24. McClave SA, Martindale RG, Rice TW, Heyland DK. Feeding the critically ill patient. Crit Care Med 2014;42:2600–10.
25. Tian F, Gao X, Wu C, et al. Initial energy supplementation in critically ill patients receiving enteral nutrition: a systematic review and meta-analysis of randomized controlled trials. Asia Pac J Clin Nutr 2017;26:11–9.
26. Martindale RG, Warren M. Should enteral nutrition be started in the first week of critical illness? Curr Opin Clin Nutr Metab Care 2015;18:202–6.
27. McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract 2009;24:305–15.
28. Kang W, Kudsk KA. Is there evidence that the gut contributes to mucosal immunity in humans? JPEN J Parenter Enteral Nutr 2007;31:461–82.
29. Seron-Arbeloa C, Zamora-Elson M, Labarta-Monzon L, Mallor-Bonet T. Enteral nutrition in critical care. J Clin Med Res 2013;5:1-11.
30. Harvey SE, Parrott F, Harrison DA, et al; CALORIES Trial Investigators. Trial of the route of early nutritional support in critically ill adults. N Engl J Med 2014;371:1673–84.
31. Elke G, van Zanten AR, Lemieux M, et al. Enteral versus parenteral nutrition in critically ill patients: an updated systematic review and meta-analysis of randomized controlled trials. Crit Care 2016;20:117.
32. Reignier J, Boisramé-Helms J, Brisard L, et al. Enteral versus parenteral nutrition in ventilated adults with shock: a randomized, controlled, multicenter, open-label, parallel-group study (NUTRIREA-2). Lancet 2018;391:133–43.
33. Rice TW , Wheeler AP, Thompson BT et al;National Heart, Lung, and Blood Institute, Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Initial trophic vs full enteral feeding in patients with acute lung injury: the EDEN randomized trial. JAMA 2012;307:795–803.
34. Heyland DK, Dhaliwal R, Jiang X, Day AG. Identifying critically ill patients who benefit the most from nutrition therapy: the development and initial validation of a novel risk assessment tool. Crit Care 2011;15:R258.
35. Bost RB, Tjan DH, van Zanten AR. Timing of (supplemental) parenteral nutrition in critically ill patients: a systematic review. Ann Intensive Care 2014;4:31.
36. Casaer MP, Mesotten D, Hermans G, et al. Early verus late parenteral nutrition in critically ill adults. N Eng J Med 2011;365:506–17.
37. Harvey SE, Parrott F, Harrison DA, et al; CALORIES Trial Investigators. Trial of the route of early nutritional support in critically ill adults. N Eng J Med 2014;371:1673–84.
38. Manzanares W, Dhaliwal R, Jurewitsch B, et al. Parenteral fish oil lipid emulsions in the critically ill: A systematic review and meta-analysis. JPEN J Parenter Enteral Nutr 2014;38:20–8.
39. Oshima T, Heidegger CP, Pichard C. Supplemental parenteral nutrition is the key to prevent energy deficits in critically ill patients. Nutr Clin Prac 2016;31:432–7.
40. Manzanares W, Langlois PL, Dhaliwal R, Lemieux M, Heyland DK. Intravenous fish oil lipid emulsions in critically ill patients: an updated systematic review and meta-analysis. Crit Care 2015;19:167.
41. Rooyackers O, Sundström Rehal M, Liebau F, et al. High protein intake without concerns? Crit Care 2017;21:106.
42. Puthucheary ZA, Rawal J, McPhail M, et al. Acute skeletal muscle wasting in critical illness. JAMA 2013;310:1591–600.
43. Heyland D, Muscedere J, Wischmeyer PE, et al; Canadian Critical Care Trials Group. A randomized trial of glutamine and antioxidants in critically ill patients. N Engl J Med 2013;368:1489–97.
44. Marik PE, Zaloga GP. Immunonutrition in critically ill patients: a systematic review and analysis of the literature. Intensive Care Med 2008;34:1980–90.
45. Gadek JE, DeMichele SJ, Karlstad MD, et al; Enteral Nutrition in ARDS Study Group. Effect of enteral feeding with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome. Crit Care Med 1999;27:1409–20.
46. Rice TW, Wheeler AP, Thompson BT, et al; NIH NHLBI Acute Respiratory Distress Syndrome Network of Investigators. Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA 2011;306:1574–81.
47. Singer P, Theilla M, Fisher H, et al. Benefit of an enteral diet enriched with eicosapentaenoic acid and gamma-linolenic acid in ventilated patients with acute lung injury. Crit Care Med 2006;34:1033–38.
48. Atkinson S, Sieffert E, Bihari D. A prospective, randomized, double-blind, controlled clinical trial of enteral immunonutrition in the critically ill. Guy’s Hospital Intensive Care Group. Crit Care Med 1998;26:1164–72.
49. Galbán C, Montejo JC, Mesejo A, et al. An immune-enhancing enteral diet reduces mortality rate and episodes of bacteremia in septic intensive care unit patients. Crit Care Med 2000;28:643–8.
50. Weimann A, Bastian L, Bischoff WE, et al. Influence of arginine, omega-3 fatty acids and nucleotide-supplemented enteral support on systemic inflammatory response syndrome and multiple organ failure in patients after severe trauma. Nutrition 1998;14:165–72.
51. van Bokhorst-De Van Der Schueren MA, Quak JJ, von Blomberg-van der Flier BM, et al. Effect of perioperative nutrition with and without arginine supplementation, on nutritional status, immune function, postoperative morbidity, and survival in severely malnourished head and neck cancer patients. Am J Clin Nutr 2001;73:323–32.
52. Cerantola Y, Hübner M, Grass F, et al. Immunonutrition in gastrointestinal surgery. Br J Surg 2011;98:37–48.
53. Marik PE, Zaloga GP. Immunonutrition in high-risk surgical patients: a systematic review and analysis of the literature. JPEN J Parenter Enteral Nutr 2010;34:378–86.
54. Sultan J, Griffin SM, Di Franco F, et al. Randomized clinical trial of omega-3 fatty acid–supplemented enteral nutrition vs. standard enteral nutrition in patients undergoing oesophagogastric cancer surgery. Br J Surg 2012;99:346–55.
55. Waitzberg DL, Saito H, Plank LD, et al. Postsurgical infections are reduced with specialized nutrition support. World J Surg 2006;30:1592–604.
56. Pearce CB, Sadek SA, Walters AM, et al. A double-blind, randomised, controlled trial to study the effects of an enteral feed supplemented with glutamine, arginine, and omega-3 fatty acid in predicted acute severe pancreatitis. JOP 2006;7:361–71.
57. Mazaki T, Ishii Y, Murai I. Immunoenhancing enteral and parenteral nutrition for gastrointestinal surgery: a multiple treatments meta-analysis. Ann Surg 2015;261:662–9.
58. van Zanten ARH, Sztark F, Kaisers UX, et al. High-protein enteral nutrition enriched with immune-modulating nutrients vs standard high protein enteral nutrition and nosocomial infections in the ICU. JAMA 2014;312:514–24.
59. Drover JW, Dhaliwal R, Weitzel L, et al. Perioperative use of arginine supplemented diets: a systematic review of the evidence. J Am Coll Surg 2011;212:385–99.
60. Stupak D, Abdelsayed GG, Soloway GN. Motility disorders of the upper gastrointestinal tract in the intensive care unit: pathophysiology and contemporary management. J Clin Gastroenterol 2012;46:449–56.
61. Huang HH, Chang SJ, Hsu CW, et al. Severity of illness influences the efficacy of enteral feeding route on clinical outcomes in patients with critical illness. J Acad Nutr Diet 2012;112:1138–46.
62. American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005;171:388–416.
63. Heyland DK, Cahill NE, Dhaliwal R, et al. Impact of enteral feeding protocols on enteral nutrition delivery: results of a multicenter observational study. JPEN J Parenter Enteral Nutr 2010;34:675–84.
64. Landzinski J, Kiser TH, Fish DN, et al. Gastric motility function in critically ill patients tolerant vs intolerant to gastric nutrition. JPEN J Parenter Enteral Nutr 2008;32:45–50.
65. de Aguilar-Nascimento JE, Kudsk KA. Use of small bore feeding tubes: successes and failures. Curr Opin Clin Nutr Metab Care 2007;10:291–6.
66. Boyer N, McCarthy MS, Mount CA. Analysis of an electromagnetic tube placement device vs a self-advancing nasal jejunal device for postpyloric feeding tube placement . J Hosp Med 2014;9:23–8.
67. Metheny NA, Meert KL. Effectiveness of an electromagnetic feeding tube placement device in detecting inadvertent respiratory placement. Am J Crit Care 2014;23:240–8.
68. Montejo JC, Miñambres E, Bordejé L, et al. Gastric residual volume during enteral nutrition in ICU patients: the REGANE study. Intensive Care Med 2010;36:1386–93.
69. Hurt RT, McClave SA. Gastric residual volumes in critical illness: what do they really mean? Crit Care Clin 2010;26:481–90.
70. Poulard F, Dimet J, Martin-Lefevre L, et al. Impact of not measuring residual gastric volume in mechanically ventilated patients receiving early enteral feeding: a prospective before-after study. JPEN J Parenter Enteral Nutr 2010;34:125–30.
71. Reignier J, Mercier E, Gouge AL, et al; Clinical Research in Intensive Care and Sepsis (CRICS) Group. Effect of not monitoring residual gastric volume on risk of ventilator-associated pneumonia in adults receiving mechanical ventilation and early enteral feeding: a randomized controlled trial. JAMA 2013;309:249–56.
72. Williams TA, Leslie GD, Leen T, et al. Reducing interruptions to continuous enteral nutrition in the intensive care unit: a comparative study. J Clin Nurs 2013;22:2838-2848.
73. Metheny NA, Stewart BJ, Mills AC. Blind insertion of feeding tubes in intensive care units: a national survey. Am J Crit Care 2012;21:352–360.
74. Compher C, Chittams J, Sammarco T, et al. Greater protein and energy intake may be associated with improved mortality in higher risk critically ill patients: A multicenter, multinational observational study. Crit Care Med 2017;45:156–163.
75. Casaer MP, Wilmer A, Hermans G, et al. Role of disease and macronutrient dose in the randomized controlled EPANIC Trial: a post hoc analysis. Am J Resp Crit Care Med 2013;187:247–55.
76. Al-Dorzi HM, Albarrak A, Ferwana M, et al. Lower versus higher dose of enteral caloric intake in adult critically ill patients: a systematic review and meta-analysis. Crit Care 2016;20:358.
77. Choi EY, Park DA, Park J. Calorie intake of enteral nutrition and clinical outcomes in acutely critically ill patients: a meta-analysis of randomized controlled trials. JPEN J Parenter Enteral Nutr 2015;39:291–300.
78. Arabi YM, Aldawood AS, Haddad SH, et al. Permissive underfeeding or standard enteral feeding in critically ill adults. N Engl J Med 2015;372:2398–408.
Pancreatic Adenocarcinoma: Management of Advanced Unresectable and Metastatic Disease
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
Aggressive B-Cell Non-Hodgkin Lymphoma
Introduction
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features.1 While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women.2 NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides,3 immunosuppression associated with HIV infection,4 autoimmune disorders,5 iatrogenically induced immune suppression in the post-transplant and other settings,6 family history of NHL,7 and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL.8 In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma.9 Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas.10 Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes,11–13 including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Work-Up
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1. Both hepatitis B and C have been associated with increased risk of NHL. In addition, there is a risk of hepatitis B reactivation following certain NHL therapies. A contrast-enhanced computed tomography (CT) scan in addition to positron emission tomography (PET) is useful to define the extent of disease in situations needing greater definition (eg, lymphadenopathy close to the bowel, cervical and supraclavicular nodal involvement, and lymphadenopathy causing thrombosis or compression of nearby structures).14 In cases where it is apparent that the patient has advanced stage disease (Ann Arbor stage III/IV) based on imaging, bone marrow biopsy is unlikely to alter the treatment plan. For such patients, if the complete blood count is unremarkable, deferral of bone marrow biopsy may be reasonable. For new cases of DLBCL, assessment for MYC translocation by fluorescence in situ hybridization (FISH) is recommended. If a MYC translocation is identified, then testing for BCL2 and BCL6 translocations by FISH should be performed.
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL.14 This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases.2 It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes.15
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races.16 There is a slight male predominance (55%), median age at diagnosis is 65 years,16,17 and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue.18 Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis.19
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease.20 The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement,21 this feature does not carry the same poor prognosis associated with transformed disease22 or DLBCL involvement of the bone marrow.23
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein,24 and about 70% express the BCL-6 protein.25 C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL.26
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2). Subsequently, modifications were made to adjust for age and stage, with the latest iteration being the NCCN (National Comprehensive Cancer Network) IPI.27 This tool uses age, performance status, LDH ratio (relative to the upper limit of normal), a more precise definition for presence of extranodal sites of disease (defined as lymphomatous involvement in the bone marrow, CNS, liver/GI tract, or lung), and Ann Arbor stage to stratify patients into 4 risk groups with significantly different 5-year OS, ranging from 38% to 96% based on the subgroup. Importantly, the NCCN-IPI was derived in a cohort of patients treated with rituximab-based therapy.
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP.28 The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis.29,30 Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma.1 In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL.31,32 Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy
DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years.33 The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS.34,35
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues36 studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms,37 with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT.36
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed.38 Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]);39 dose-dense therapy (R-CHOP-14);replacement of rituximab with obinutuzuimab;40 addition of novel agents such as bortezomib,41 lenalidomide,42 or ibrutinib43,44 to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide,45 enzastaurin,46 and everolimus.47 Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit.48 More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however.49 As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%).50,51 Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes,52 with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission.53
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively.54 A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF).55 In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months.56 For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series.57
Relapsed/Refractory Disease
Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype.58 However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone.59 The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort.60
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT.61 Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained.62 However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL.64 PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL.20 It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious.64 Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease,65 with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL.66 PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis.67 This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT.68–70 The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78%71 when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases.72,73 For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80%.74–76 In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease.77 This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe.78 MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease.79 Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases.80 In rare cases, CD5 or cyclin D1 may be negative.80 Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression.81 Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome.82 In cyclin D1–negative cases, SOX11 expression may help with diagnosis.83 A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range.84 In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range.85–87 Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004,88 and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010.89 The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients.90 The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement.91
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course,92 but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy
For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission.87,93,94 Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial.93 At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy.95 However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach.96 Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP.97,98
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years.99 In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib,100,101 the BTK inhibitors ibrutinib102,103 and acalabrutinib,104 and the immunomodulatory agent lenalidomide.105
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission.95 Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease.95,106 A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma.107 The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90%.107 Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients.108 In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT.109
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70%.110,111 High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse.112 However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease.113
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients.114
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.
Introduction
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features.1 While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women.2 NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides,3 immunosuppression associated with HIV infection,4 autoimmune disorders,5 iatrogenically induced immune suppression in the post-transplant and other settings,6 family history of NHL,7 and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL.8 In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma.9 Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas.10 Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes,11–13 including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Work-Up
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1. Both hepatitis B and C have been associated with increased risk of NHL. In addition, there is a risk of hepatitis B reactivation following certain NHL therapies. A contrast-enhanced computed tomography (CT) scan in addition to positron emission tomography (PET) is useful to define the extent of disease in situations needing greater definition (eg, lymphadenopathy close to the bowel, cervical and supraclavicular nodal involvement, and lymphadenopathy causing thrombosis or compression of nearby structures).14 In cases where it is apparent that the patient has advanced stage disease (Ann Arbor stage III/IV) based on imaging, bone marrow biopsy is unlikely to alter the treatment plan. For such patients, if the complete blood count is unremarkable, deferral of bone marrow biopsy may be reasonable. For new cases of DLBCL, assessment for MYC translocation by fluorescence in situ hybridization (FISH) is recommended. If a MYC translocation is identified, then testing for BCL2 and BCL6 translocations by FISH should be performed.
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL.14 This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases.2 It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes.15
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races.16 There is a slight male predominance (55%), median age at diagnosis is 65 years,16,17 and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue.18 Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis.19
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease.20 The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement,21 this feature does not carry the same poor prognosis associated with transformed disease22 or DLBCL involvement of the bone marrow.23
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein,24 and about 70% express the BCL-6 protein.25 C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL.26
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2). Subsequently, modifications were made to adjust for age and stage, with the latest iteration being the NCCN (National Comprehensive Cancer Network) IPI.27 This tool uses age, performance status, LDH ratio (relative to the upper limit of normal), a more precise definition for presence of extranodal sites of disease (defined as lymphomatous involvement in the bone marrow, CNS, liver/GI tract, or lung), and Ann Arbor stage to stratify patients into 4 risk groups with significantly different 5-year OS, ranging from 38% to 96% based on the subgroup. Importantly, the NCCN-IPI was derived in a cohort of patients treated with rituximab-based therapy.
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP.28 The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis.29,30 Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma.1 In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL.31,32 Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy
DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years.33 The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS.34,35
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues36 studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms,37 with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT.36
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed.38 Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]);39 dose-dense therapy (R-CHOP-14);replacement of rituximab with obinutuzuimab;40 addition of novel agents such as bortezomib,41 lenalidomide,42 or ibrutinib43,44 to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide,45 enzastaurin,46 and everolimus.47 Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit.48 More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however.49 As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%).50,51 Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes,52 with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission.53
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively.54 A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF).55 In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months.56 For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series.57
Relapsed/Refractory Disease
Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype.58 However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone.59 The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort.60
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT.61 Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained.62 However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL.64 PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL.20 It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious.64 Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease,65 with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL.66 PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis.67 This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT.68–70 The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78%71 when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases.72,73 For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80%.74–76 In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease.77 This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe.78 MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease.79 Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases.80 In rare cases, CD5 or cyclin D1 may be negative.80 Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression.81 Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome.82 In cyclin D1–negative cases, SOX11 expression may help with diagnosis.83 A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range.84 In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range.85–87 Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004,88 and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010.89 The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients.90 The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement.91
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course,92 but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy
For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission.87,93,94 Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial.93 At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy.95 However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach.96 Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP.97,98
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years.99 In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib,100,101 the BTK inhibitors ibrutinib102,103 and acalabrutinib,104 and the immunomodulatory agent lenalidomide.105
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission.95 Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease.95,106 A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma.107 The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90%.107 Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients.108 In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT.109
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70%.110,111 High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse.112 However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease.113
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients.114
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
Introduction
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features.1 While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women.2 NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides,3 immunosuppression associated with HIV infection,4 autoimmune disorders,5 iatrogenically induced immune suppression in the post-transplant and other settings,6 family history of NHL,7 and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL.8 In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma.9 Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas.10 Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes,11–13 including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Work-Up
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1. Both hepatitis B and C have been associated with increased risk of NHL. In addition, there is a risk of hepatitis B reactivation following certain NHL therapies. A contrast-enhanced computed tomography (CT) scan in addition to positron emission tomography (PET) is useful to define the extent of disease in situations needing greater definition (eg, lymphadenopathy close to the bowel, cervical and supraclavicular nodal involvement, and lymphadenopathy causing thrombosis or compression of nearby structures).14 In cases where it is apparent that the patient has advanced stage disease (Ann Arbor stage III/IV) based on imaging, bone marrow biopsy is unlikely to alter the treatment plan. For such patients, if the complete blood count is unremarkable, deferral of bone marrow biopsy may be reasonable. For new cases of DLBCL, assessment for MYC translocation by fluorescence in situ hybridization (FISH) is recommended. If a MYC translocation is identified, then testing for BCL2 and BCL6 translocations by FISH should be performed.
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL.14 This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases.2 It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes.15
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races.16 There is a slight male predominance (55%), median age at diagnosis is 65 years,16,17 and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue.18 Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis.19
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease.20 The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement,21 this feature does not carry the same poor prognosis associated with transformed disease22 or DLBCL involvement of the bone marrow.23
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein,24 and about 70% express the BCL-6 protein.25 C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL.26
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2). Subsequently, modifications were made to adjust for age and stage, with the latest iteration being the NCCN (National Comprehensive Cancer Network) IPI.27 This tool uses age, performance status, LDH ratio (relative to the upper limit of normal), a more precise definition for presence of extranodal sites of disease (defined as lymphomatous involvement in the bone marrow, CNS, liver/GI tract, or lung), and Ann Arbor stage to stratify patients into 4 risk groups with significantly different 5-year OS, ranging from 38% to 96% based on the subgroup. Importantly, the NCCN-IPI was derived in a cohort of patients treated with rituximab-based therapy.
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP.28 The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis.29,30 Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma.1 In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL.31,32 Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy
DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years.33 The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS.34,35
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues36 studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms,37 with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT.36
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed.38 Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]);39 dose-dense therapy (R-CHOP-14);replacement of rituximab with obinutuzuimab;40 addition of novel agents such as bortezomib,41 lenalidomide,42 or ibrutinib43,44 to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide,45 enzastaurin,46 and everolimus.47 Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit.48 More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however.49 As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%).50,51 Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes,52 with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission.53
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively.54 A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF).55 In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months.56 For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series.57
Relapsed/Refractory Disease
Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype.58 However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone.59 The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort.60
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT.61 Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained.62 However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL.64 PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL.20 It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious.64 Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease,65 with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL.66 PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis.67 This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT.68–70 The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78%71 when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases.72,73 For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80%.74–76 In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease.77 This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe.78 MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease.79 Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases.80 In rare cases, CD5 or cyclin D1 may be negative.80 Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression.81 Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome.82 In cyclin D1–negative cases, SOX11 expression may help with diagnosis.83 A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range.84 In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range.85–87 Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004,88 and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010.89 The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients.90 The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement.91
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course,92 but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy
For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission.87,93,94 Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial.93 At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy.95 However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach.96 Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP.97,98
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years.99 In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib,100,101 the BTK inhibitors ibrutinib102,103 and acalabrutinib,104 and the immunomodulatory agent lenalidomide.105
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission.95 Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease.95,106 A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma.107 The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90%.107 Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients.108 In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT.109
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70%.110,111 High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse.112 However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease.113
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients.114
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.
HER2-Positive Breast Cancer: Current Management
Introduction
Breast cancer is the second leading cause of cancer deaths among women in the United States, according to the SEER database. It is estimated that 1 in 8 women will be diagnosed with breast cancer at some point during their lifetime (12.4% lifetime risk).1,2 Because breast tumors are clinically and histopathologically heterogeneous, different diagnostic and therapeutic approaches are required for each subtype. Among the subtypes, tumors that are positive for human epidermal growth factor receptor 2 (HER2) account for approximately 15% to 20% of all newly diagnosed localized and metastatic invasive breast tumors.3,4 Historically, this subset of tumors has been considered the most aggressive due to a higher propensity to relapse and metastasize, translating into poorer prognosis compared with other subtypes.5–7 However, with the advent of HER2-targeted therapy in the late 1990s, prognosis has significantly improved for both early- and late-stage HER2-positive tumors.8
Pathogenesis
The HER2 proto-oncogene belongs to a family of human epidermal growth factor receptors that includes 4 transmembrane tyrosine kinase receptors: HER1 (also commonly known as epidermal growth factor receptor, EGFR), HER2, HER3, and HER4. Another commonly used nomenclature for this family of receptors is ERBB1 to ERBB4. Each of the receptors has a similar structure consisting of a growth factor–binding extracellular domain, a single transmembrane segment, an intracellular protein-tyrosine kinase catalytic domain, and a tyrosine-containing cytoplasmic tail. Activation of the extracellular domain leads to conformational changes that initiate a cascade of reactions resulting in protein kinase activation. ERBB tyrosine receptor kinases subsequently activate several intracellular pathways that are critical for cellular function and survival, including the PI3K-AKT, RAS-MAPK, and mTOR pathways. Hyperactivation or overexpression of these receptors leads to uncontrolled cell growth and proliferation, and eventually cancerogenesis.9,10
HER2 gene amplification can cause activation of the receptor’s extramembranous domain by way of either dimerization of two HER2 receptors or heterodimerization with other ERBB family receptors, leading to ligand-independent activation of cell signaling (ie, activation in the absence of external growth factors). Besides breast cancer, HER2 protein is overexpressed in several other tumor types, including esophageal and gastric adenocarcinomas, colon and gynecological malignancies, and to a lesser extent in other malignancies.
Biomarker Testing
All patients with newly diagnosed breast cancer should have their tumor tissue submitted for biomarker testing for estrogen receptors (ER), progesterone receptors (PR), and HER2 overexpression, as the result this testing dictates therapy choices. The purpose of HER2 testing is to investigate whether the HER2 gene, located on chromosome 17, is overexpressed or amplified. HER2 status provides the basis for treatment selection, which impacts long-term outcome measures such as recurrence and survival. Routine testing of carcinoma in situ for HER2 expression/amplification is not recommended and has no implication on choice of therapy at this time.
In 2013, the American Society of Clinical Oncology and the College of American Pathologists (ASCO/CAP) updated their clinical guideline recommendations for HER2 testing in breast cancer to improve its accuracy and its utility as a predictive marker.11 There are currently 2 approved modalities for HER2 testing: detection of HER2 protein overexpression by i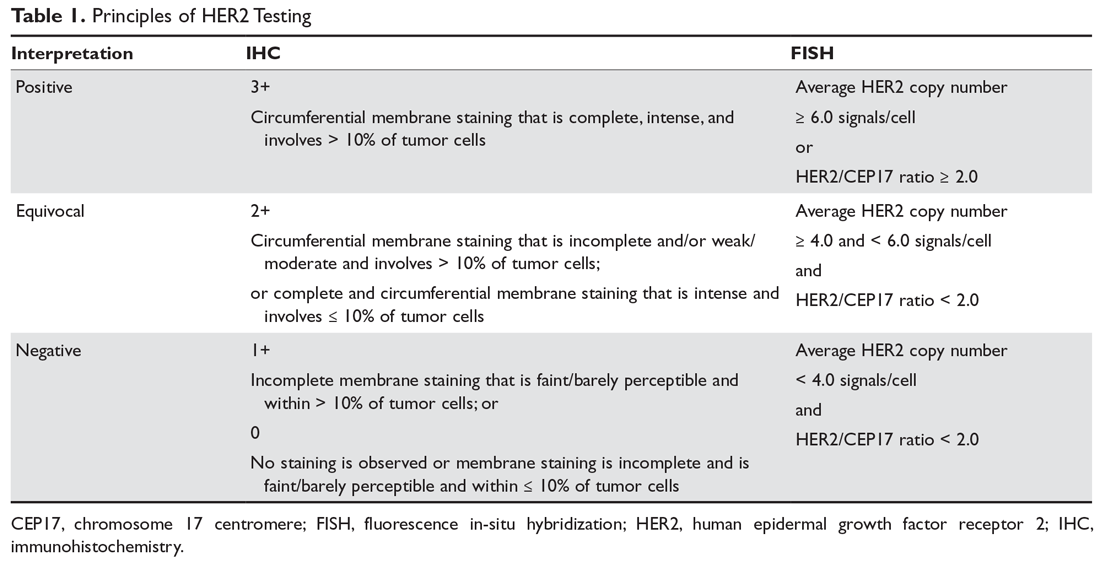
Fluorescence in-situ hybridization (FISH) testing assesses for HER2 amplification by determining the number of HER2 signals and 
Neoadjuvant and Adjuvant Therapy for Locoregional Disease
Case Patient 1
A 56-year-old woman undergoes ultrasound-guided biopsy of a self-palpated breast lump. Pathology shows invasive ductal carcinoma that is ER-positive, PR-negative, and HER2 equivocal by IHC (2+ staining). Follow-up FISH testing shows a HER2/CEP17 ratio of 2.5. The tumor is estimated to be 2 cm in diameter by imaging and exam with no clinically palpable axillary lymphadenopathy. The patient exhibits no constitutional or localized symptoms concerning for metastases.
- What is the recommended management approach for this patient?
According to the ASCO/CAP guidelines, this patient’s tumor qualifies as HER2-positive based upon testing results showing amplification of the gene. This result has important implications for management since nearly all patients with macroscopically invasive HER2-positive tumors should be considered for adjuvant chemotherapy in combination with anti-HER2 therapy. The patient should proceed with upfront tumor resection and sentinel lymph node biopsy. Systemic staging imaging (ie, computed tomography [CT] or bone scan) is not indicated in early stage breast cancer.12,13 Systemic staging scans are indicated when (1) any anatomical stage III disease is suspected (eg, with involvement of the skin or chest wall, the presence of enlarged matted or fixed axillary lymph nodes, and involvement of nodal stations other than in the axilla), and (2) when symptoms or abnormal laboratory values raise suspicion for distant metastases (eg, unexplained bone pain, unintentional weight loss, elevated serum alkaline phosphatase, and transaminitis).
Case 1 Continued
The patient presents to discuss treatment options after undergoing a lumpectomy and sentinel node biopsy procedure. The pathology report notes a single focus of carcinoma measuring 2 cm with negative sentinel lymph nodes.
- What agents are used for adjuvant therapy in HER2-postive breast cancer?
Nearly all patients with macroscopically invasive (> 1 mm) breast carcinoma should be considered for adjuvant therapy using a regimen that contains a taxane and trastuzumab. However, the benefit may be small for patients with tumors ≤ 5 mm (T1a, N0), so it is important to carefully weigh the risk against the benefit. Among the agents that targeting HER2, only trastuzumab has been shown to improve overall survival (OS) in the adjuvant setting; long-term follow-up data are awaited for other agents.8 A trastuzumab biosimilar, trastuzumab-dkst, was recently approved by the US Food and Drug Administration (FDA) for the same indications as trastuzumab.14 The regimens most commonly used in the adjuvant and neoadjuvant settings for nonmetastatic breast cancer are summarized in Table 2.
Patients with small (≤ 3 cm), node-negative tumors can generally be considered for a reduced-intensity regimen that includes weekly paclitaxel plus trastuzumab. This combination proved efficacious in a single-group, multicenter study that enrolled 406 patients.15 Paclitaxel and trastuzumab were given once weekly for 12 weeks, followed by trastuzumab, either weekly or every 3 weeks, to complete 1 year of therapy.After a median follow-up of more than 6 years, the rates of distant and locoregional recurrence were 1% and 1.2%, respectively.16
A combination of docetaxel, carboplatin, and trastuzumab is a nonanthracycline regimen that is also appropriate in this setting, based on the results of the Breast Cancer International Research Group 006 (BCIRG-006) trial.17 This phase 3 randomized trial enrolled 3222 women with HER2-positive, invasive, high-risk adenocarcinoma. Eligible patients had a T1–3 tumor and either lymph node–negative or –positive disease and were randomly assigned to receive 1 of 3 regimens: group 1 received doxorubicin and cyclophosphamide every 3 weeks for 4 cycles followed by docetaxel every 3 weeks for 4 cycles (AC-T); group 2 received the AC-T regimen in combination with trastuzumab; and group 3 received docetaxel, carboplatin, and trastuzumab once every 3 weeks for 6 cycles (TCH). Groups 2 and 3 also received trastuzumab for an additional 34 weeks to complete 1 year of therapy. Trastuzumab-containing regimens were found to offer superior disease-free survival (DFS) and OS. When comparing the 2 trastuzumab arms after more than 10 years of follow-up, no statistically significant advantage of an anthracycline regimen over a nonanthracycline regimen was found.18 Furthermore, the anthracycline arm had a fivefold higher incidence of symptomatic congestive heart failure (grades 3 and 4), and the nonanthracycline regimen was associated with a lower incidence of treatment-related leukemia, a clinically significant finding despite not reaching statistical significance due to low overall numbers.
BCIRG-006, NSABP B-31, NCCTG N9831, and HERA are all large randomized trials with consistent results confirming trastuzumab’s role in reducing recurrence and improving survival in HER2-positive breast cancer in the adjuvant settings. The estimated overall benefit from addition of this agent was a 34% to 41% improvement in survival and a 33% to 52% improvement in DFS.8,17–20
Dual anti-HER2 therapy containing both trastuzumab and pertuzumab should be strongly considered for patients with macroscopic lymph node involvement based on the results of the APHINITY trial.21 In this study, the addition of pertuzumab to standard trastuzumab-based therapy led to a significant improvement in invasive-disease-free survival at 3 years. In subgroup analysis, the benefit was restricted to the node-positive group (3-year invasive-disease-free survival rates of 92% in the pertuzumab group versus 90.2% in the placebo group, P = 0.02). Patients with hormone receptor–negative disease derived greater benefit from the addition of pertuzumab. Regimens used in the APHINITY trial included the anti-HER2 agents trastuzumab and pertuzumab in combination with 1 of the following chemotherapy regimens: sequential cyclophosphamide plus either doxorubicin or epirubicin, followed by either 4 cycles of docetaxel or 12 weekly doses of paclitaxel; sequential fluorouracil plus either epirubicin or doxorubicin plus cyclophosphamide (3 or 4 cycles), followed by 3 or 4 cycles of docetaxel or 12 weekly cycles of paclitaxel; or 6 cycles of concurrent docetaxel plus carboplatin.
One-year therapy with neratinib, an oral tyrosine kinase inhibitor of HER2, is now approved by the FDA after completion of trastuzumab in the adjuvant setting, based on the results of the ExteNET trial.22 In this study, patients who had completed trastuzumab within the preceding 12 months, without evidence of recurrence, were randomly assigned to receive either oral neratinib or placebo daily for 1 year. The 2-year DFS rate was 93.9% and 91.6% for the neratinib and placebo groups, respectively. The most common adverse effect of neratinib was diarrhea, with approximately 40% of patients experiencing grade 3 diarrhea. In subgroup analyses, hormone receptor–positive patients derived the most benefit, while hormone receptor–negative patients derived no or marginal benefit.22 OS benefit has not yet been established.23
Trastuzumab therapy (with pertuzumab if indicated) should be offered for an optimal duration of 12 months (17 cycles, including those given with chemotherapy backbone). A shorter duration of therapy, 6 months, has been shown to be inferior,24 while a longer duration, 24 months, has been shown to provide no additional benefit.25
Finally, sequential addition of anti-estrogen endocrine therapy is indicated for hormone-positive tumors. Endocrine therapy is usually added after completion of the chemotherapy backbone of the regimen, but may be given concurrently with anti-HER2 therapy. If radiation is being administered, endocrine therapy can be given concurrently or started after radiation therapy is completed.
Case 1 Conclusion
The patient can be offered 1 of 2 adjuvant treatment regimens, either TH or TCH (Table 2). Since the patient had lumpectomy, she is an appropriate candidate for adjuvant radiation, which would be started after completion of the chemotherapy backbone (taxane/platinum). Endocrine therapy for at least 5 years should be offered sequentially or concurrently with radiation. Her long-term prognosis is very favorable.
Case Patient 2
A 43-year-old woman presents with a 4-cm breast mass, a separate skin nodule, and palpable matted axillary lymphadenopathy. Biopsies of the breast mass and subcutaneous nodule reveal invasive ductal carcinoma that is ER-negative, PR-negative, and HER2-positive by IHC (3+ staining). Based on clinical findings, the patient is staged as T4b (separate tumor nodule), N2 (matted axillary lymph nodes). Systemic staging with CT scan of the chest, abdomen, and pelvis shows no evidence of distant metastases.
- What is the recommended approach to management for this patient?
Recommendations for neoadjuvant therapy, given before definitive surgery, follow the same path as with other subtypes of breast cancer. Patients with suspected anatomical stage III disease are strongly encouraged to undergo upfront (neoadjuvant) chemotherapy in combination with HER2-targeted agents. In addition, all HER2-positive patients with clinically node-positive disease can be offered neoadjuvant therapy using chemotherapy plus dual anti-HER2 therapy (trastuzumab and pertuzumab), with complete pathological response expected in more than 60% of patients.26,27 Because this patient has locally advanced disease, especially skin involvement and matted axillary nodes, she should undergo neoadjuvant therapy. Preferred regimens contain both trastuzumab and pertuzumab in combination with cytotoxic chemotherapy. The latter may be given concurrently (nonanthracycline regimens, such as docetaxel plus carboplatin) or sequentially (anthracycline-based regimens), as outlined in Table 2. Administration of anthracyclines and trastuzumab simultaneously is contraindicated due to increased risk of cardiomyopathy.28
Endocrine therapy is not indicated for this patient per the current standard of care because the tumor was ER- and PR-negative. Had the tumor been hormone receptor–positive, endocrine therapy for a minimum of 5 years would have been indicated. Likewise, in the case of hormone receptor–positive disease, 12 months of neratinib therapy after completion of trastuzumab may add further benefit, as shown in the ExteNET trial.22,23 Neratinib seems to have a propensity to prevent or delay trastuzumab-induced overexpression of estrogen receptors. This is mainly due to hormone receptor/HER2 crosstalk, a potential mechanism of resistance to trastuzumab.29,30
In addition to the medical therapy options discussed here, this patient would be expected to benefit from adjuvant radiation to the breast and regional lymph nodes, given the presence of T4 disease and bulky adenopathy in the axilla.31
Case 2 Conclusion
The patient undergoes neoadjuvant treatment (docetaxel, carboplatin, trastuzumab, and pertuzumab every 21 days for a total of 6 cycles), followed by surgical resection (modified radical mastectomy) that reveals complete pathological response (no residual invasive carcinoma). Subsequently, she receives radiation therapy to the primary tumor site and regional lymph nodes while continuing trastuzumab and pertuzumab for 11 more cycles (17 total). Despite presenting with locally advanced disease, the patient has a favorable overall prognosis due to an excellent pathological response.
- What is the approach to follow-up after completion of primary therapy?
Patients may follow up every 3 to 6 months for clinical evaluation in the first 5 years after completing primary adjuvant therapy. An annual screening mammogram is recommended as well. Body imaging can be done if dictated by symptoms. However, routine CT, positron emission tomography, or bone scans are not recommended as part of follow-up in the absence of symptoms, mainly because of a lack of evidence that such surveillance improves survival.32
Metastatic HER2-Positive Breast Cancer
Metastatic breast cancer most commonly presents as a distant recurrence of previously treated local disease. However, 6% to 18% of patients have no prior history of breast cancer and present with de novo metastatic disease.33,34 The most commonly involved distant organs are the skeletal bones, liver, lung, distant lymph node stations, and brain. Compared to other subtypes, HER2-positive tumors have an increased tendency to involve the central nervous system.35–38 Although metastatic HER2-positive breast cancer is not considered curable, significant improvement in survival has been achieved, and patients with metastatic disease have median survival approaching 5 years.39
Case Presentation 3
A 69-year-old woman with a history of breast cancer 4 years ago presents with new-onset back pain and unintentional weight loss. On exam, she is found to have palpable axillary adenopathy on the same side as her previous cancer. Her initial disease was stage IIB ER-positive and HER2-positive and was treated with chemotherapy, mastectomy, and anastrozole, which the patient is still taking. She undergoes CT scan of the chest, abdomen, and pelvis and radionucleotide bone scan, which show multiple liver and bony lesions suspicious for metastatic disease. Axillary lymph node biopsy confirms recurrent invasive carcinoma that is ER-positive and HER2-positive by IHC (3+).
- What is the approach to management of a patient who presents with symptoms of recurrent HER2-positive disease?
This patient likely has metastatic breast cancer based on the imaging findings. In such cases, a biopsy of the recurrent disease should always be considered, if feasible, to confirm the diagnosis and rule out other etiologies such as different malignances and benign conditions. Hormone-receptor and HER2 testing should also be performed on recurrent disease, since a change in HER2 status can be seen in 15% to 33% of cases.40–42
Based on data from the phase 3 CLEOPATRA trial, first-line systemic regimens for patients with metastatic breast cancer that is positive for HER2 should consist of a combination of docetaxel, trastuzumab, and pertuzumab. Compared to placebo, adding pertuzumab yielded superior progression-free survival of 18.4 months (versus 12.4 months for placebo) and an unprecedented OS of 56.5 months (versus 40.8 for placebo).39 Weekly paclitaxel can replace docetaxel with comparable efficacy (Table 3).43
Patients can develop significant neuropathy as well as skin and nail changes after multiple cycles of taxane-based chemotherapy. Therefore, the taxane backbone may be dropped after 6 to 8 cycles, while patients continue the trastuzumab and pertuzumab combination until disease progression or unacceptable toxicity. Some patients may enjoy remarkable long-term survival on “maintenance” anti-HER2 therapy.44 Despite lack of high-level evidence, such as from large randomized trials, some experts recommend the addition of a hormone blocker after discontinuation of the taxane in ER-positive tumors.45
Premenopausal and perimenopausal women with hormone receptor–positive metastatic disease should be considered for simultaneous ovarian suppression. Ovarian suppression can be accomplished medically using a gonadotropin-releasing hormone agonist (goserelin) or surgically via salpingo-oophorectomy.46–48
Case 3 Conclusion
The patient receives 6 cycles of docetaxel, trastuzumab, and pertuzumab, after which the docetaxel is discontinued due to neuropathy while she continues the other 2 agents. After 26 months of disease control, the patient is found to have new liver metastatic lesions, indicating progression of disease.
- What therapeutic options are available for this patient?
Patients whose disease progresses after receiving taxane- and trastuzumab-containing regimens are candidates to receive the novel antibody-drug conjugate ado-trastuzumab emtansine (T-DM1). Early progressors (ie, patients with early stage disease who have progression of disease while receiving adjuvant trastuzumab or within 6 months of completion of adjuvant trastuzumab) are also candidates for T-DM1. Treatment usually fits in the second line or beyond based on data from the EMILIA trial, which randomly assigned patients to receive either capecitabine plus lapatinib or T-DM1.49,50 Progression-free survival in the T-DM1 group was 9.6 months versus 6.4 months for the comparator. Improvement of 4 months in OS persisted with longer follow-up despite a crossover rate of 27%. Furthermore, a significantly higher objective response rate and fewer adverse effects were reported in the T-DM1 patients. Most patients included in the EMILIA trial were pertuzumab-naive. However, the benefit of T-DM1 appears to persist, albeit to a lesser extent, for pertuzumab-pretreated patients.51,52
Patients in whom treatment fails with 2 or more lines of therapy containing taxane-trastuzumab (with or without pertuzumab) and T-DM1 are candidates to receive a combination of capecitabine and lapatinib, a TKI, in the third line and beyond. Similarly, the combination of capecitabine with trastuzumab in the same settings appears to have equal efficacy.53,54 Trastuzumab may be continued beyond progression while changing the single-agent chemotherapy drug for subsequent lines of therapy, per ASCO guidelines,55 although improvement in OS has not been demonstrated beyond the third line in a large randomized trial (Table 3).
Approved HER2-Targeted Drugs
HER2-directed therapy is implemented in the management of nearly all stages of HER2-positive invasive breast cancer, including early and late stages (Table 4).
Trastuzumab
Trastuzumab was the first anti-HER2 agent to be approved by the FDA in 1998. It is a humanized monoclonal antibody directed against the extracellular domain of the HER2 receptor (domain IV). Trastuzumab functions by interrupting HER2 signal transduction and by flagging tumor cells for immune destruction.56 Cardiotoxicity, usually manifested as left ventricular systolic dysfunction, is the most noteworthy adverse effect of trastuzumab. The most prominent risk factors for cardiomyopathy in patients receiving trastuzumab are low baseline ejection fraction (< 55%), age > 50 years, co-administration and higher cumulative dose of anthracyclines, and increased body mass index and obesity.57–59 Whether patients receive therapy in the neoadjuvant, adjuvant, or metastatic settings, baseline cardiac function assessment with echocardiogram or multiple-gated acquisition scan is required. While well-designed randomized trials validating the value and frequency of monitoring are lacking, repeated cardiac testing every 3 months is generally recommended for patients undergoing adjuvant therapy. Patients with metastatic disease who are receiving treatment with palliative intent may be monitored less frequently.60,61
An asymptomatic drop in ejection fraction is the most common manifestation of cardiac toxicity. Other cardiac manifestations have also been reported with much less frequency, including arrhythmias, severe congestive heart failure, ventricular thrombus formation, and even cardiac death. Until monitoring and dose-adjustment guidelines are issued, the guidance provided in the FDA-approved prescribing information should be followed, which recommends holding trastuzumab when there is ≥ 16% absolute reduction in left ventricular ejection fraction (LVEF) from the baseline value; or if the LVEF value is below the institutional lower limit of normal and the drop is ≥ 10%. After holding the drug, cardiac function can be re-evaluated every 4 weeks. In most patients, trastuzumab-induced cardiotoxicity can be reversed by withholding trastuzumab and initiating cardioprotective therapy, although the latter remains controversial. Re-challenging after recovery of ejection fraction is possible and toxicity does not appear to be proportional to cumulative dose. Cardiomyopathy due to trastuzumab therapy is potentially reversible within 6 months in more than 80% of cases.28,57,60–63
Other notable adverse effects of trastuzumab include pulmonary toxicity (such as interstitial lung disease) and infusion reactions (usually during or within 24 hours of first dose).
Pertuzumab
Pertuzumab is another humanized monoclonal antibody directed to a different extracellular domain of the HER2 receptor, the dimerization domain (domain II), which is responsible for heterodimerization of HER2 with other HER receptors, especially HER3. This agent should always be co-administered with trastuzumab as the 2 drugs produce synergistic anti-tumor effect, without competition for the receptor. Activation of HER3, via dimerization with HER2, produces an alternative mechanism of downstream signaling, even in the presence of trastuzumab and in the absence of growth factors (Figure 2). 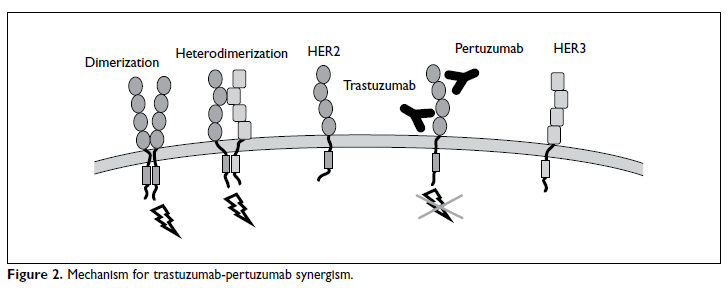
Ado-Trastuzumab Emtansine
Ado-trastuzumab emtansine (T-DM1) is an antibody-drug conjugate that combines the monoclonal antibody trastuzumab with the cytotoxic agent DM1 (emtansine), a potent microtubule inhibitor and a derivative of maytansine, in a single structure (Figure 3). 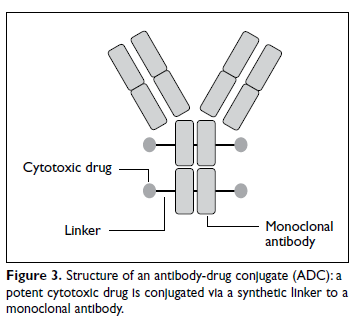
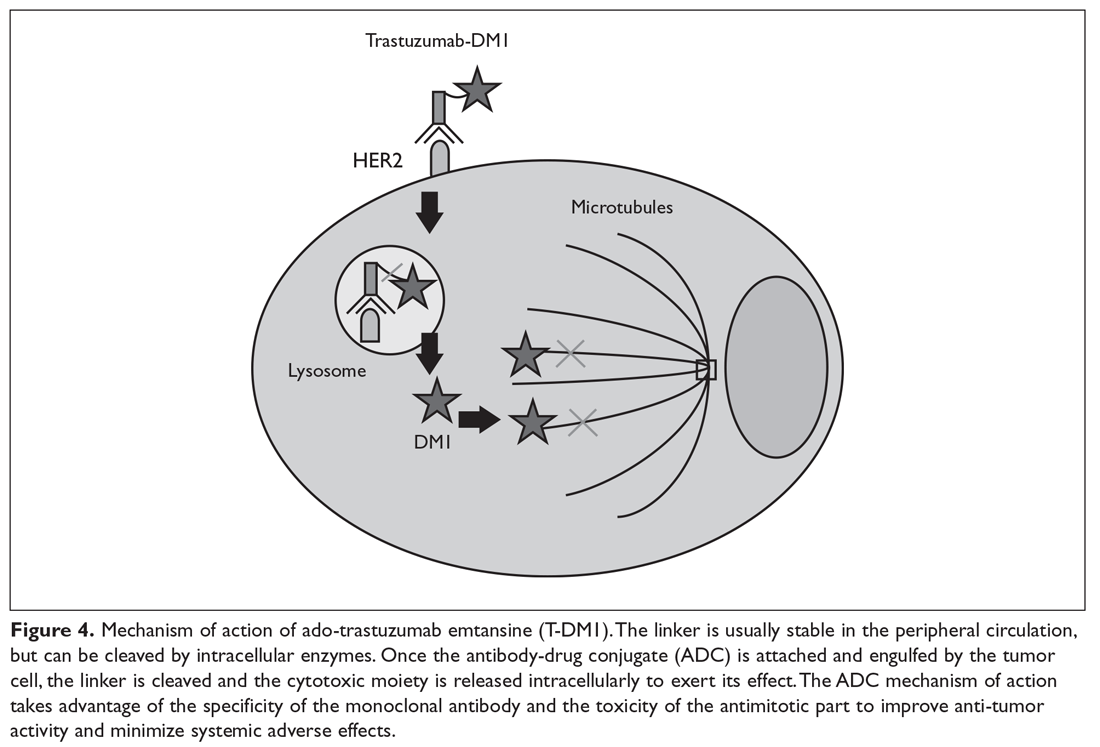
Lapatinib
Lapatinib is an oral small-molecule tyrosine kinase inhibitor of EGFR (HER1) and HER2 receptors. It is approved in combination with capecitabine for patients with HER2-expressing metastatic breast cancer who previously received trastuzumab, an anthracycline, and a taxane chemotherapy or T-DM1. Lapatinib is also approved in combination with letrozole in postmenopausal women with HER2-positive, hormone receptor–positive metastatic disease, although it is unclear where this regimen would fit in the current schema. It may be considered for patients with hormone receptor–positive disease who are not candidates for therapy with taxane-trastuzumab and T-DM1 or who decline this therapy. Diarrhea is seen in most patients treated with lapatinib and may be severe in 20% of cases when lapatinib is combined with capecitabine. Decreases in LVEF have been reported and cardiac function monitoring at baseline and periodically may be considered.69,70 Lapatinib is not approved for use in adjuvant settings.
Neratinib
Neratinib is an oral small-molecule irreversible tyrosine kinase inhibitor of HER1, HER2, and HER4. It is currently approved only for extended adjuvant therapy after completion of 1 year of standard trastuzumab therapy. It is given orally every day for 1 year. The main side effect, expected in nearly all patients, is diarrhea, which can be severe in up to 40% of patients and may lead to dehydration and electrolyte imbalance. Diarrhea usually starts early in the course of therapy and can be most intense during the first cycle. Therefore, prophylactic antidiarrheal therapy is recommended to reduce the intensity of diarrhea. Loperamide prophylaxis may be initiated simultaneously for all patients using a tapering schedule. Drug interruption or dose reduction may be required if diarrhea is severe or refractory.21,71 Neratinib is not FDA-approved in the metastatic settings.
Conclusion
HER2-positive tumors represent a distinct subset(s) of breast tumors with unique pathological and clinical characteristics. Treatment with a combination of cytotoxic chemotherapy and HER2-targeted agents has led to a dramatic improvement in survival for patients with locoregional and advanced disease. Trastuzumab is an integral part of adjuvant therapy for HER2-positive invasive disease. Pertuzumab should be added to trastuzumab in node-positive disease. Neratinib may be considered after completion of trastuzumab therapy in patients with hormone receptor–positive disease. For metastatic HER2-positive breast cancer, a regimen consisting of docetaxel plus trastuzumab and pertuzumab is the standard first-line therapy. Ado-trastuzumab is an ideal next line option for patients whose disease progresses on trastuzumab and taxanes.
1. Yedjou CG, Tchounwou PB, Payton M, et al. Assessing the racial and ethnic disparities in breast cancer mortality in the United States. Int J Environ Res Public Health 2017;14(5).
2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016;66:271–89.
3. Huang HJ, Neven P, Drijkoningen M, et al. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. J Clin Pathol 2005;58:611–6.
4. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
5. Cronin KA, Harlan LC, Dodd KW, et al. Population-based estimate of the prevalence of HER-2 positive breast cancer tumors for early stage patients in the US. Cancer Invest 2010;28:963–-8.
6. Huang HJ, Neven P, Drijkoningen M, et al. Hormone receptors do not predict the HER2/neu status in all age groups of women with an operable breast cancer. Ann Oncol 2005;16:1755–61.
7. Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006;295:2492–502.
8. Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol 2014;32:3744–52.
9. Brennan PJ, Kumagai T, Berezov A, et al. HER2/neu: mechanisms of dimerization/oligomerization. Oncogene 2000;19:6093–101.
10. Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun 2004;319:1–11.
11. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013.
12. Ravaioli A, Pasini G, Polselli A, et al. Staging of breast cancer: new recommended standard procedure. Breast Cancer Res Treat 2002;72:53–60.
13. Puglisi F, Follador A, Minisini AM, et al. Baseline staging tests after a new diagnosis of breast cancer: further evidence of their limited indications. Ann Oncol 2005;16:263–6.
14. FDA approves trastuzumab biosimilar. Cancer Discov 2018;8:130.
15. Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med 2015;372:134–41.
16. Tolaney SM, Barry WT, Guo H, Dillon D, et al. Seven-year (yr) follow-up of adjuvant paclitaxel (T) and trastuzumab (H) (APT trial) for node-negative, HER2-positive breast cancer (BC) [ASCO abstract]. J Clin Oncol. 2017;35(suppl):511.
17. Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 2011;365:1273–83.
18. Slamon DJ, Eiermann W, Robert NJ, et al. Ten year follow-up of BCIRG-006 comparing doxorubicin plus cyclophosphamide followed by docetaxel (AC -> T) with doxorubicin plus cyclophosphamide followed by docetaxel and trastuzumab (AC -> TH) with docetaxel, carboplatin and trastuzumab (TCH) in HER2+early breast cancer [SABC abstract]. Cancer Res 2016;76(4 supplement):S5-04.
19. Jahanzeb M. Adjuvant trastuzumab therapy for HER2-positive breast cancer. Clin Breast Cancer 2008;8:324–33.
20. Cameron D, Piccart-Gebhart MJ, Gelber RD, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017;389:1195–205.
21. von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med 2017;377:122–31.
22. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17:367–77.
23. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1688–700.
24. Pivot X, Romieu G, Debled M, et al. 6 months versus 12 months of adjuvant trastuzumab for patients with HER2-positive early breast cancer (PHARE): a randomised phase 3 trial. Lancet Oncol 2013;14:741–8.
25. Goldhirsch A, Gelber RD, Piccart-Gebhart MJ, et al. 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet 2013;382:1021–8.
26. Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol 2013;24:2278–84.
27. Schneeweiss A, Chia S, Hickish T, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: Evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. Eur J Cancer 2018;89:27–35
28. de Azambuja E, Procter MJ, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac events at 8 years of median follow-up in the Herceptin Adjuvant trial (BIG 1-01). J Clin Oncol 2014;32:2159–65.
29. Dowsett M, Harper-Wynne C, Boeddinghaus I, et al. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res 2001;61:8452–8.
30. Nahta R, O’Regan RM. Therapeutic implications of estrogen receptor signaling in HER2-positive breast cancers. Breast Cancer Res Treat 2012;135:39–48.
31. Recht A, Comen EA, Fine RE, et al. Postmastectomy radiotherapy: An American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology focused guideline update. Pract Radiat Oncol 2016;6:e219-e34.
32. Runowicz CD, Leach CR, Henry NL, et al. American Cancer Society/American Society of Clinical Oncology breast cancer survivorship care guideline. J Clin Oncol 2016;34:611–35.
33. Zeichner SB, Herna S, Mani A, et al. Survival of patients with de-novo metastatic breast cancer: analysis of data from a large breast cancer-specific private practice, a university-based cancer center and review of the literature. Breast Cancer Res Treat 2015;153:617–24.
34. Dawood S, Broglio K, Ensor J, et al. Survival differences among women with de novo stage IV and relapsed breast cancer. Ann Oncol 2010;21:2169–74.
35. Savci-Heijink CD, Halfwerk H, Hooijer GK, et al. Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat 2015;150:547–57.
36. Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol 2015;35:85–95.
37. Bendell JC, Domchek SM, Burstein HJ, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 2003;97:2972–7.
38. Burstein HJ, Lieberman G, Slamon DJ, et al. Isolated central nervous system metastases in patients with HER2-overexpressing advanced breast cancer treated with first-line trastuzumab-based therapy. Ann Oncol 2005;16:1772–7.
39. Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372:724–34.
40. Lindstrom LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol 2012;30:2601–8.
41. Guarneri V, Giovannelli S, Ficarra G, et al. Comparison of HER-2 and hormone receptor expression in primary breast cancers and asynchronous paired metastases: impact on patient management. Oncologist 2008;13:838–44.
42. Salkeni MA, Hall SJ. Metastatic breast cancer: Endocrine therapy landscape reshaped. Avicenna J Med 2017;7:144–52.
43. Dang C, Iyengar N, Datko F, et al. Phase II study of paclitaxel given once per week along with trastuzumab and pertuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:442–7.
44. Cantini L, Pistelli M, Savini A, et al. Long-responders to anti-HER2 therapies: A case report and review of the literature. Mol Clin Oncol 2018;8:147–52.
45. Sutherland S, Miles D, Makris A. Use of maintenance endocrine therapy after chemotherapy in metastatic breast cancer. Eur J Cancer 2016;69:216–22.
46. Falkson G, Holcroft C, Gelman RS, et al. Ten-year follow-up study of premenopausal women with metastatic breast cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol 1995;13:1453–8.
47. Boccardo F, Rubagotti A, Perrotta A, et al. Ovarian ablation versus goserelin with or without tamoxifen in pre-perimenopausal patients with advanced breast cancer: results of a multicentric Italian study. Ann Oncol 1994;5:337–42.
48 Taylor CW, Green S, Dalton WS, et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: an intergroup study. J Clin Oncol 1998;16:994–9.
49. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783–91.
50. Dieras V, Miles D, Verma S, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:732–42.
51. Dzimitrowicz H, Berger M, Vargo C, et al. T-DM1 Activity in metastatic human epidermal growth factor receptor 2-positive breast cancers that received prior therapy with trastuzumab and pertuzumab. J Clin Oncol 2016;34:3511–7.
52. Fabi A, Giannarelli D, Moscetti L, et al. Ado-trastuzumab emtansine (T-DM1) in HER2+ advanced breast cancer patients: does pretreatment with pertuzumab matter? Future Oncol 2017;13:2791–7.
53. Madden R, Kosari S, Peterson GM, et al. Lapatinib plus capecitabine in patients with HER2-positive metastatic breast cancer: A systematic review. Int J Clin Pharmacol Ther 2018;56:72–80.
54. Pivot X, Manikhas A, Zurawski B, et al. CEREBEL (EGF111438): A phase III, randomized, open-label study of lapatinib plus capecitabine versus trastuzumab plus capecitabine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:1564–73.
55. Giordano SH, Temin S, Kirshner JJ, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014;32:2078–99.
56. Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007;357:39–51.
57. Russell SD, Blackwell KL, Lawrence J, et al. Independent adjudication of symptomatic heart failure with the use of doxorubicin and cyclophosphamide followed by trastuzumab adjuvant therapy: a combined review of cardiac data from the National Surgical Adjuvant breast and Bowel Project B-31 and the North Central Cancer Treatment Group N9831 clinical trials. J Clin Oncol 2010;28:3416–21.
58. Ewer SM, Ewer MS. Cardiotoxicity profile of trastuzumab. Drug Saf 2008;31:459–67.
59. Guenancia C, Lefebvre A, Cardinale D, et al. Obesity as a risk factor for anthracyclines and trastuzumab cardiotoxicity in breast cancer: a systematic review and meta-analysis. J Clin Oncol 2016;34:3157–65.
60. Dang CT, Yu AF, Jones LW, et al. Cardiac surveillance guidelines for trastuzumab-containing therapy in early-stage breast cancer: getting to the heart of the matter. J Clin Oncol 2016;34:1030–3.
61. Brann AM, Cobleigh MA, Okwuosa TM. Cardiovascular monitoring with trastuzumab therapy: how frequent is too frequent? JAMA Oncol 2016;2:1123–4.
62. Suter TM, Procter M, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol 2007;25:3859–65.
63. Procter M, Suter TM, de Azambuja E, et al. Longer-term assessment of trastuzumab-related cardiac adverse events in the Herceptin Adjuvant (HERA) trial. J Clin Oncol 2010;28:3422–8.
64. Yamashita-Kashima Y, Shu S, Yorozu K, et al. Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncol Lett 2017;14:4197–205.
65. Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer 2017;117:1736–42.
66. Girish S, Gupta M, Wang B, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol 2012;69:1229–40.
67. Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res 2015;21:123–33.
68. Yan H, Endo Y, Shen Y, et al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther 2016;15:480–90.
69. Spector NL, Xia W, Burris H 3rd, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 2005;23:2502–12.
70. Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 2009;27:5538–46.
71. Neratinib (Nerlynx) for HER2-positive breast cancer. Med Lett Drugs Ther 2018;60(1539):23.
Introduction
Breast cancer is the second leading cause of cancer deaths among women in the United States, according to the SEER database. It is estimated that 1 in 8 women will be diagnosed with breast cancer at some point during their lifetime (12.4% lifetime risk).1,2 Because breast tumors are clinically and histopathologically heterogeneous, different diagnostic and therapeutic approaches are required for each subtype. Among the subtypes, tumors that are positive for human epidermal growth factor receptor 2 (HER2) account for approximately 15% to 20% of all newly diagnosed localized and metastatic invasive breast tumors.3,4 Historically, this subset of tumors has been considered the most aggressive due to a higher propensity to relapse and metastasize, translating into poorer prognosis compared with other subtypes.5–7 However, with the advent of HER2-targeted therapy in the late 1990s, prognosis has significantly improved for both early- and late-stage HER2-positive tumors.8
Pathogenesis
The HER2 proto-oncogene belongs to a family of human epidermal growth factor receptors that includes 4 transmembrane tyrosine kinase receptors: HER1 (also commonly known as epidermal growth factor receptor, EGFR), HER2, HER3, and HER4. Another commonly used nomenclature for this family of receptors is ERBB1 to ERBB4. Each of the receptors has a similar structure consisting of a growth factor–binding extracellular domain, a single transmembrane segment, an intracellular protein-tyrosine kinase catalytic domain, and a tyrosine-containing cytoplasmic tail. Activation of the extracellular domain leads to conformational changes that initiate a cascade of reactions resulting in protein kinase activation. ERBB tyrosine receptor kinases subsequently activate several intracellular pathways that are critical for cellular function and survival, including the PI3K-AKT, RAS-MAPK, and mTOR pathways. Hyperactivation or overexpression of these receptors leads to uncontrolled cell growth and proliferation, and eventually cancerogenesis.9,10
HER2 gene amplification can cause activation of the receptor’s extramembranous domain by way of either dimerization of two HER2 receptors or heterodimerization with other ERBB family receptors, leading to ligand-independent activation of cell signaling (ie, activation in the absence of external growth factors). Besides breast cancer, HER2 protein is overexpressed in several other tumor types, including esophageal and gastric adenocarcinomas, colon and gynecological malignancies, and to a lesser extent in other malignancies.
Biomarker Testing
All patients with newly diagnosed breast cancer should have their tumor tissue submitted for biomarker testing for estrogen receptors (ER), progesterone receptors (PR), and HER2 overexpression, as the result this testing dictates therapy choices. The purpose of HER2 testing is to investigate whether the HER2 gene, located on chromosome 17, is overexpressed or amplified. HER2 status provides the basis for treatment selection, which impacts long-term outcome measures such as recurrence and survival. Routine testing of carcinoma in situ for HER2 expression/amplification is not recommended and has no implication on choice of therapy at this time.
In 2013, the American Society of Clinical Oncology and the College of American Pathologists (ASCO/CAP) updated their clinical guideline recommendations for HER2 testing in breast cancer to improve its accuracy and its utility as a predictive marker.11 There are currently 2 approved modalities for HER2 testing: detection of HER2 protein overexpression by i
Fluorescence in-situ hybridization (FISH) testing assesses for HER2 amplification by determining the number of HER2 signals and
Neoadjuvant and Adjuvant Therapy for Locoregional Disease
Case Patient 1
A 56-year-old woman undergoes ultrasound-guided biopsy of a self-palpated breast lump. Pathology shows invasive ductal carcinoma that is ER-positive, PR-negative, and HER2 equivocal by IHC (2+ staining). Follow-up FISH testing shows a HER2/CEP17 ratio of 2.5. The tumor is estimated to be 2 cm in diameter by imaging and exam with no clinically palpable axillary lymphadenopathy. The patient exhibits no constitutional or localized symptoms concerning for metastases.
- What is the recommended management approach for this patient?
According to the ASCO/CAP guidelines, this patient’s tumor qualifies as HER2-positive based upon testing results showing amplification of the gene. This result has important implications for management since nearly all patients with macroscopically invasive HER2-positive tumors should be considered for adjuvant chemotherapy in combination with anti-HER2 therapy. The patient should proceed with upfront tumor resection and sentinel lymph node biopsy. Systemic staging imaging (ie, computed tomography [CT] or bone scan) is not indicated in early stage breast cancer.12,13 Systemic staging scans are indicated when (1) any anatomical stage III disease is suspected (eg, with involvement of the skin or chest wall, the presence of enlarged matted or fixed axillary lymph nodes, and involvement of nodal stations other than in the axilla), and (2) when symptoms or abnormal laboratory values raise suspicion for distant metastases (eg, unexplained bone pain, unintentional weight loss, elevated serum alkaline phosphatase, and transaminitis).
Case 1 Continued
The patient presents to discuss treatment options after undergoing a lumpectomy and sentinel node biopsy procedure. The pathology report notes a single focus of carcinoma measuring 2 cm with negative sentinel lymph nodes.
- What agents are used for adjuvant therapy in HER2-postive breast cancer?
Nearly all patients with macroscopically invasive (> 1 mm) breast carcinoma should be considered for adjuvant therapy using a regimen that contains a taxane and trastuzumab. However, the benefit may be small for patients with tumors ≤ 5 mm (T1a, N0), so it is important to carefully weigh the risk against the benefit. Among the agents that targeting HER2, only trastuzumab has been shown to improve overall survival (OS) in the adjuvant setting; long-term follow-up data are awaited for other agents.8 A trastuzumab biosimilar, trastuzumab-dkst, was recently approved by the US Food and Drug Administration (FDA) for the same indications as trastuzumab.14 The regimens most commonly used in the adjuvant and neoadjuvant settings for nonmetastatic breast cancer are summarized in Table 2.
Patients with small (≤ 3 cm), node-negative tumors can generally be considered for a reduced-intensity regimen that includes weekly paclitaxel plus trastuzumab. This combination proved efficacious in a single-group, multicenter study that enrolled 406 patients.15 Paclitaxel and trastuzumab were given once weekly for 12 weeks, followed by trastuzumab, either weekly or every 3 weeks, to complete 1 year of therapy.After a median follow-up of more than 6 years, the rates of distant and locoregional recurrence were 1% and 1.2%, respectively.16
A combination of docetaxel, carboplatin, and trastuzumab is a nonanthracycline regimen that is also appropriate in this setting, based on the results of the Breast Cancer International Research Group 006 (BCIRG-006) trial.17 This phase 3 randomized trial enrolled 3222 women with HER2-positive, invasive, high-risk adenocarcinoma. Eligible patients had a T1–3 tumor and either lymph node–negative or –positive disease and were randomly assigned to receive 1 of 3 regimens: group 1 received doxorubicin and cyclophosphamide every 3 weeks for 4 cycles followed by docetaxel every 3 weeks for 4 cycles (AC-T); group 2 received the AC-T regimen in combination with trastuzumab; and group 3 received docetaxel, carboplatin, and trastuzumab once every 3 weeks for 6 cycles (TCH). Groups 2 and 3 also received trastuzumab for an additional 34 weeks to complete 1 year of therapy. Trastuzumab-containing regimens were found to offer superior disease-free survival (DFS) and OS. When comparing the 2 trastuzumab arms after more than 10 years of follow-up, no statistically significant advantage of an anthracycline regimen over a nonanthracycline regimen was found.18 Furthermore, the anthracycline arm had a fivefold higher incidence of symptomatic congestive heart failure (grades 3 and 4), and the nonanthracycline regimen was associated with a lower incidence of treatment-related leukemia, a clinically significant finding despite not reaching statistical significance due to low overall numbers.
BCIRG-006, NSABP B-31, NCCTG N9831, and HERA are all large randomized trials with consistent results confirming trastuzumab’s role in reducing recurrence and improving survival in HER2-positive breast cancer in the adjuvant settings. The estimated overall benefit from addition of this agent was a 34% to 41% improvement in survival and a 33% to 52% improvement in DFS.8,17–20
Dual anti-HER2 therapy containing both trastuzumab and pertuzumab should be strongly considered for patients with macroscopic lymph node involvement based on the results of the APHINITY trial.21 In this study, the addition of pertuzumab to standard trastuzumab-based therapy led to a significant improvement in invasive-disease-free survival at 3 years. In subgroup analysis, the benefit was restricted to the node-positive group (3-year invasive-disease-free survival rates of 92% in the pertuzumab group versus 90.2% in the placebo group, P = 0.02). Patients with hormone receptor–negative disease derived greater benefit from the addition of pertuzumab. Regimens used in the APHINITY trial included the anti-HER2 agents trastuzumab and pertuzumab in combination with 1 of the following chemotherapy regimens: sequential cyclophosphamide plus either doxorubicin or epirubicin, followed by either 4 cycles of docetaxel or 12 weekly doses of paclitaxel; sequential fluorouracil plus either epirubicin or doxorubicin plus cyclophosphamide (3 or 4 cycles), followed by 3 or 4 cycles of docetaxel or 12 weekly cycles of paclitaxel; or 6 cycles of concurrent docetaxel plus carboplatin.
One-year therapy with neratinib, an oral tyrosine kinase inhibitor of HER2, is now approved by the FDA after completion of trastuzumab in the adjuvant setting, based on the results of the ExteNET trial.22 In this study, patients who had completed trastuzumab within the preceding 12 months, without evidence of recurrence, were randomly assigned to receive either oral neratinib or placebo daily for 1 year. The 2-year DFS rate was 93.9% and 91.6% for the neratinib and placebo groups, respectively. The most common adverse effect of neratinib was diarrhea, with approximately 40% of patients experiencing grade 3 diarrhea. In subgroup analyses, hormone receptor–positive patients derived the most benefit, while hormone receptor–negative patients derived no or marginal benefit.22 OS benefit has not yet been established.23
Trastuzumab therapy (with pertuzumab if indicated) should be offered for an optimal duration of 12 months (17 cycles, including those given with chemotherapy backbone). A shorter duration of therapy, 6 months, has been shown to be inferior,24 while a longer duration, 24 months, has been shown to provide no additional benefit.25
Finally, sequential addition of anti-estrogen endocrine therapy is indicated for hormone-positive tumors. Endocrine therapy is usually added after completion of the chemotherapy backbone of the regimen, but may be given concurrently with anti-HER2 therapy. If radiation is being administered, endocrine therapy can be given concurrently or started after radiation therapy is completed.
Case 1 Conclusion
The patient can be offered 1 of 2 adjuvant treatment regimens, either TH or TCH (Table 2). Since the patient had lumpectomy, she is an appropriate candidate for adjuvant radiation, which would be started after completion of the chemotherapy backbone (taxane/platinum). Endocrine therapy for at least 5 years should be offered sequentially or concurrently with radiation. Her long-term prognosis is very favorable.
Case Patient 2
A 43-year-old woman presents with a 4-cm breast mass, a separate skin nodule, and palpable matted axillary lymphadenopathy. Biopsies of the breast mass and subcutaneous nodule reveal invasive ductal carcinoma that is ER-negative, PR-negative, and HER2-positive by IHC (3+ staining). Based on clinical findings, the patient is staged as T4b (separate tumor nodule), N2 (matted axillary lymph nodes). Systemic staging with CT scan of the chest, abdomen, and pelvis shows no evidence of distant metastases.
- What is the recommended approach to management for this patient?
Recommendations for neoadjuvant therapy, given before definitive surgery, follow the same path as with other subtypes of breast cancer. Patients with suspected anatomical stage III disease are strongly encouraged to undergo upfront (neoadjuvant) chemotherapy in combination with HER2-targeted agents. In addition, all HER2-positive patients with clinically node-positive disease can be offered neoadjuvant therapy using chemotherapy plus dual anti-HER2 therapy (trastuzumab and pertuzumab), with complete pathological response expected in more than 60% of patients.26,27 Because this patient has locally advanced disease, especially skin involvement and matted axillary nodes, she should undergo neoadjuvant therapy. Preferred regimens contain both trastuzumab and pertuzumab in combination with cytotoxic chemotherapy. The latter may be given concurrently (nonanthracycline regimens, such as docetaxel plus carboplatin) or sequentially (anthracycline-based regimens), as outlined in Table 2. Administration of anthracyclines and trastuzumab simultaneously is contraindicated due to increased risk of cardiomyopathy.28
Endocrine therapy is not indicated for this patient per the current standard of care because the tumor was ER- and PR-negative. Had the tumor been hormone receptor–positive, endocrine therapy for a minimum of 5 years would have been indicated. Likewise, in the case of hormone receptor–positive disease, 12 months of neratinib therapy after completion of trastuzumab may add further benefit, as shown in the ExteNET trial.22,23 Neratinib seems to have a propensity to prevent or delay trastuzumab-induced overexpression of estrogen receptors. This is mainly due to hormone receptor/HER2 crosstalk, a potential mechanism of resistance to trastuzumab.29,30
In addition to the medical therapy options discussed here, this patient would be expected to benefit from adjuvant radiation to the breast and regional lymph nodes, given the presence of T4 disease and bulky adenopathy in the axilla.31
Case 2 Conclusion
The patient undergoes neoadjuvant treatment (docetaxel, carboplatin, trastuzumab, and pertuzumab every 21 days for a total of 6 cycles), followed by surgical resection (modified radical mastectomy) that reveals complete pathological response (no residual invasive carcinoma). Subsequently, she receives radiation therapy to the primary tumor site and regional lymph nodes while continuing trastuzumab and pertuzumab for 11 more cycles (17 total). Despite presenting with locally advanced disease, the patient has a favorable overall prognosis due to an excellent pathological response.
- What is the approach to follow-up after completion of primary therapy?
Patients may follow up every 3 to 6 months for clinical evaluation in the first 5 years after completing primary adjuvant therapy. An annual screening mammogram is recommended as well. Body imaging can be done if dictated by symptoms. However, routine CT, positron emission tomography, or bone scans are not recommended as part of follow-up in the absence of symptoms, mainly because of a lack of evidence that such surveillance improves survival.32
Metastatic HER2-Positive Breast Cancer
Metastatic breast cancer most commonly presents as a distant recurrence of previously treated local disease. However, 6% to 18% of patients have no prior history of breast cancer and present with de novo metastatic disease.33,34 The most commonly involved distant organs are the skeletal bones, liver, lung, distant lymph node stations, and brain. Compared to other subtypes, HER2-positive tumors have an increased tendency to involve the central nervous system.35–38 Although metastatic HER2-positive breast cancer is not considered curable, significant improvement in survival has been achieved, and patients with metastatic disease have median survival approaching 5 years.39
Case Presentation 3
A 69-year-old woman with a history of breast cancer 4 years ago presents with new-onset back pain and unintentional weight loss. On exam, she is found to have palpable axillary adenopathy on the same side as her previous cancer. Her initial disease was stage IIB ER-positive and HER2-positive and was treated with chemotherapy, mastectomy, and anastrozole, which the patient is still taking. She undergoes CT scan of the chest, abdomen, and pelvis and radionucleotide bone scan, which show multiple liver and bony lesions suspicious for metastatic disease. Axillary lymph node biopsy confirms recurrent invasive carcinoma that is ER-positive and HER2-positive by IHC (3+).
- What is the approach to management of a patient who presents with symptoms of recurrent HER2-positive disease?
This patient likely has metastatic breast cancer based on the imaging findings. In such cases, a biopsy of the recurrent disease should always be considered, if feasible, to confirm the diagnosis and rule out other etiologies such as different malignances and benign conditions. Hormone-receptor and HER2 testing should also be performed on recurrent disease, since a change in HER2 status can be seen in 15% to 33% of cases.40–42
Based on data from the phase 3 CLEOPATRA trial, first-line systemic regimens for patients with metastatic breast cancer that is positive for HER2 should consist of a combination of docetaxel, trastuzumab, and pertuzumab. Compared to placebo, adding pertuzumab yielded superior progression-free survival of 18.4 months (versus 12.4 months for placebo) and an unprecedented OS of 56.5 months (versus 40.8 for placebo).39 Weekly paclitaxel can replace docetaxel with comparable efficacy (Table 3).43
Patients can develop significant neuropathy as well as skin and nail changes after multiple cycles of taxane-based chemotherapy. Therefore, the taxane backbone may be dropped after 6 to 8 cycles, while patients continue the trastuzumab and pertuzumab combination until disease progression or unacceptable toxicity. Some patients may enjoy remarkable long-term survival on “maintenance” anti-HER2 therapy.44 Despite lack of high-level evidence, such as from large randomized trials, some experts recommend the addition of a hormone blocker after discontinuation of the taxane in ER-positive tumors.45
Premenopausal and perimenopausal women with hormone receptor–positive metastatic disease should be considered for simultaneous ovarian suppression. Ovarian suppression can be accomplished medically using a gonadotropin-releasing hormone agonist (goserelin) or surgically via salpingo-oophorectomy.46–48
Case 3 Conclusion
The patient receives 6 cycles of docetaxel, trastuzumab, and pertuzumab, after which the docetaxel is discontinued due to neuropathy while she continues the other 2 agents. After 26 months of disease control, the patient is found to have new liver metastatic lesions, indicating progression of disease.
- What therapeutic options are available for this patient?
Patients whose disease progresses after receiving taxane- and trastuzumab-containing regimens are candidates to receive the novel antibody-drug conjugate ado-trastuzumab emtansine (T-DM1). Early progressors (ie, patients with early stage disease who have progression of disease while receiving adjuvant trastuzumab or within 6 months of completion of adjuvant trastuzumab) are also candidates for T-DM1. Treatment usually fits in the second line or beyond based on data from the EMILIA trial, which randomly assigned patients to receive either capecitabine plus lapatinib or T-DM1.49,50 Progression-free survival in the T-DM1 group was 9.6 months versus 6.4 months for the comparator. Improvement of 4 months in OS persisted with longer follow-up despite a crossover rate of 27%. Furthermore, a significantly higher objective response rate and fewer adverse effects were reported in the T-DM1 patients. Most patients included in the EMILIA trial were pertuzumab-naive. However, the benefit of T-DM1 appears to persist, albeit to a lesser extent, for pertuzumab-pretreated patients.51,52
Patients in whom treatment fails with 2 or more lines of therapy containing taxane-trastuzumab (with or without pertuzumab) and T-DM1 are candidates to receive a combination of capecitabine and lapatinib, a TKI, in the third line and beyond. Similarly, the combination of capecitabine with trastuzumab in the same settings appears to have equal efficacy.53,54 Trastuzumab may be continued beyond progression while changing the single-agent chemotherapy drug for subsequent lines of therapy, per ASCO guidelines,55 although improvement in OS has not been demonstrated beyond the third line in a large randomized trial (Table 3).
Approved HER2-Targeted Drugs
HER2-directed therapy is implemented in the management of nearly all stages of HER2-positive invasive breast cancer, including early and late stages (Table 4).
Trastuzumab
Trastuzumab was the first anti-HER2 agent to be approved by the FDA in 1998. It is a humanized monoclonal antibody directed against the extracellular domain of the HER2 receptor (domain IV). Trastuzumab functions by interrupting HER2 signal transduction and by flagging tumor cells for immune destruction.56 Cardiotoxicity, usually manifested as left ventricular systolic dysfunction, is the most noteworthy adverse effect of trastuzumab. The most prominent risk factors for cardiomyopathy in patients receiving trastuzumab are low baseline ejection fraction (< 55%), age > 50 years, co-administration and higher cumulative dose of anthracyclines, and increased body mass index and obesity.57–59 Whether patients receive therapy in the neoadjuvant, adjuvant, or metastatic settings, baseline cardiac function assessment with echocardiogram or multiple-gated acquisition scan is required. While well-designed randomized trials validating the value and frequency of monitoring are lacking, repeated cardiac testing every 3 months is generally recommended for patients undergoing adjuvant therapy. Patients with metastatic disease who are receiving treatment with palliative intent may be monitored less frequently.60,61
An asymptomatic drop in ejection fraction is the most common manifestation of cardiac toxicity. Other cardiac manifestations have also been reported with much less frequency, including arrhythmias, severe congestive heart failure, ventricular thrombus formation, and even cardiac death. Until monitoring and dose-adjustment guidelines are issued, the guidance provided in the FDA-approved prescribing information should be followed, which recommends holding trastuzumab when there is ≥ 16% absolute reduction in left ventricular ejection fraction (LVEF) from the baseline value; or if the LVEF value is below the institutional lower limit of normal and the drop is ≥ 10%. After holding the drug, cardiac function can be re-evaluated every 4 weeks. In most patients, trastuzumab-induced cardiotoxicity can be reversed by withholding trastuzumab and initiating cardioprotective therapy, although the latter remains controversial. Re-challenging after recovery of ejection fraction is possible and toxicity does not appear to be proportional to cumulative dose. Cardiomyopathy due to trastuzumab therapy is potentially reversible within 6 months in more than 80% of cases.28,57,60–63
Other notable adverse effects of trastuzumab include pulmonary toxicity (such as interstitial lung disease) and infusion reactions (usually during or within 24 hours of first dose).
Pertuzumab
Pertuzumab is another humanized monoclonal antibody directed to a different extracellular domain of the HER2 receptor, the dimerization domain (domain II), which is responsible for heterodimerization of HER2 with other HER receptors, especially HER3. This agent should always be co-administered with trastuzumab as the 2 drugs produce synergistic anti-tumor effect, without competition for the receptor. Activation of HER3, via dimerization with HER2, produces an alternative mechanism of downstream signaling, even in the presence of trastuzumab and in the absence of growth factors (Figure 2).
Ado-Trastuzumab Emtansine
Ado-trastuzumab emtansine (T-DM1) is an antibody-drug conjugate that combines the monoclonal antibody trastuzumab with the cytotoxic agent DM1 (emtansine), a potent microtubule inhibitor and a derivative of maytansine, in a single structure (Figure 3).
Lapatinib
Lapatinib is an oral small-molecule tyrosine kinase inhibitor of EGFR (HER1) and HER2 receptors. It is approved in combination with capecitabine for patients with HER2-expressing metastatic breast cancer who previously received trastuzumab, an anthracycline, and a taxane chemotherapy or T-DM1. Lapatinib is also approved in combination with letrozole in postmenopausal women with HER2-positive, hormone receptor–positive metastatic disease, although it is unclear where this regimen would fit in the current schema. It may be considered for patients with hormone receptor–positive disease who are not candidates for therapy with taxane-trastuzumab and T-DM1 or who decline this therapy. Diarrhea is seen in most patients treated with lapatinib and may be severe in 20% of cases when lapatinib is combined with capecitabine. Decreases in LVEF have been reported and cardiac function monitoring at baseline and periodically may be considered.69,70 Lapatinib is not approved for use in adjuvant settings.
Neratinib
Neratinib is an oral small-molecule irreversible tyrosine kinase inhibitor of HER1, HER2, and HER4. It is currently approved only for extended adjuvant therapy after completion of 1 year of standard trastuzumab therapy. It is given orally every day for 1 year. The main side effect, expected in nearly all patients, is diarrhea, which can be severe in up to 40% of patients and may lead to dehydration and electrolyte imbalance. Diarrhea usually starts early in the course of therapy and can be most intense during the first cycle. Therefore, prophylactic antidiarrheal therapy is recommended to reduce the intensity of diarrhea. Loperamide prophylaxis may be initiated simultaneously for all patients using a tapering schedule. Drug interruption or dose reduction may be required if diarrhea is severe or refractory.21,71 Neratinib is not FDA-approved in the metastatic settings.
Conclusion
HER2-positive tumors represent a distinct subset(s) of breast tumors with unique pathological and clinical characteristics. Treatment with a combination of cytotoxic chemotherapy and HER2-targeted agents has led to a dramatic improvement in survival for patients with locoregional and advanced disease. Trastuzumab is an integral part of adjuvant therapy for HER2-positive invasive disease. Pertuzumab should be added to trastuzumab in node-positive disease. Neratinib may be considered after completion of trastuzumab therapy in patients with hormone receptor–positive disease. For metastatic HER2-positive breast cancer, a regimen consisting of docetaxel plus trastuzumab and pertuzumab is the standard first-line therapy. Ado-trastuzumab is an ideal next line option for patients whose disease progresses on trastuzumab and taxanes.
Introduction
Breast cancer is the second leading cause of cancer deaths among women in the United States, according to the SEER database. It is estimated that 1 in 8 women will be diagnosed with breast cancer at some point during their lifetime (12.4% lifetime risk).1,2 Because breast tumors are clinically and histopathologically heterogeneous, different diagnostic and therapeutic approaches are required for each subtype. Among the subtypes, tumors that are positive for human epidermal growth factor receptor 2 (HER2) account for approximately 15% to 20% of all newly diagnosed localized and metastatic invasive breast tumors.3,4 Historically, this subset of tumors has been considered the most aggressive due to a higher propensity to relapse and metastasize, translating into poorer prognosis compared with other subtypes.5–7 However, with the advent of HER2-targeted therapy in the late 1990s, prognosis has significantly improved for both early- and late-stage HER2-positive tumors.8
Pathogenesis
The HER2 proto-oncogene belongs to a family of human epidermal growth factor receptors that includes 4 transmembrane tyrosine kinase receptors: HER1 (also commonly known as epidermal growth factor receptor, EGFR), HER2, HER3, and HER4. Another commonly used nomenclature for this family of receptors is ERBB1 to ERBB4. Each of the receptors has a similar structure consisting of a growth factor–binding extracellular domain, a single transmembrane segment, an intracellular protein-tyrosine kinase catalytic domain, and a tyrosine-containing cytoplasmic tail. Activation of the extracellular domain leads to conformational changes that initiate a cascade of reactions resulting in protein kinase activation. ERBB tyrosine receptor kinases subsequently activate several intracellular pathways that are critical for cellular function and survival, including the PI3K-AKT, RAS-MAPK, and mTOR pathways. Hyperactivation or overexpression of these receptors leads to uncontrolled cell growth and proliferation, and eventually cancerogenesis.9,10
HER2 gene amplification can cause activation of the receptor’s extramembranous domain by way of either dimerization of two HER2 receptors or heterodimerization with other ERBB family receptors, leading to ligand-independent activation of cell signaling (ie, activation in the absence of external growth factors). Besides breast cancer, HER2 protein is overexpressed in several other tumor types, including esophageal and gastric adenocarcinomas, colon and gynecological malignancies, and to a lesser extent in other malignancies.
Biomarker Testing
All patients with newly diagnosed breast cancer should have their tumor tissue submitted for biomarker testing for estrogen receptors (ER), progesterone receptors (PR), and HER2 overexpression, as the result this testing dictates therapy choices. The purpose of HER2 testing is to investigate whether the HER2 gene, located on chromosome 17, is overexpressed or amplified. HER2 status provides the basis for treatment selection, which impacts long-term outcome measures such as recurrence and survival. Routine testing of carcinoma in situ for HER2 expression/amplification is not recommended and has no implication on choice of therapy at this time.
In 2013, the American Society of Clinical Oncology and the College of American Pathologists (ASCO/CAP) updated their clinical guideline recommendations for HER2 testing in breast cancer to improve its accuracy and its utility as a predictive marker.11 There are currently 2 approved modalities for HER2 testing: detection of HER2 protein overexpression by i
Fluorescence in-situ hybridization (FISH) testing assesses for HER2 amplification by determining the number of HER2 signals and
Neoadjuvant and Adjuvant Therapy for Locoregional Disease
Case Patient 1
A 56-year-old woman undergoes ultrasound-guided biopsy of a self-palpated breast lump. Pathology shows invasive ductal carcinoma that is ER-positive, PR-negative, and HER2 equivocal by IHC (2+ staining). Follow-up FISH testing shows a HER2/CEP17 ratio of 2.5. The tumor is estimated to be 2 cm in diameter by imaging and exam with no clinically palpable axillary lymphadenopathy. The patient exhibits no constitutional or localized symptoms concerning for metastases.
- What is the recommended management approach for this patient?
According to the ASCO/CAP guidelines, this patient’s tumor qualifies as HER2-positive based upon testing results showing amplification of the gene. This result has important implications for management since nearly all patients with macroscopically invasive HER2-positive tumors should be considered for adjuvant chemotherapy in combination with anti-HER2 therapy. The patient should proceed with upfront tumor resection and sentinel lymph node biopsy. Systemic staging imaging (ie, computed tomography [CT] or bone scan) is not indicated in early stage breast cancer.12,13 Systemic staging scans are indicated when (1) any anatomical stage III disease is suspected (eg, with involvement of the skin or chest wall, the presence of enlarged matted or fixed axillary lymph nodes, and involvement of nodal stations other than in the axilla), and (2) when symptoms or abnormal laboratory values raise suspicion for distant metastases (eg, unexplained bone pain, unintentional weight loss, elevated serum alkaline phosphatase, and transaminitis).
Case 1 Continued
The patient presents to discuss treatment options after undergoing a lumpectomy and sentinel node biopsy procedure. The pathology report notes a single focus of carcinoma measuring 2 cm with negative sentinel lymph nodes.
- What agents are used for adjuvant therapy in HER2-postive breast cancer?
Nearly all patients with macroscopically invasive (> 1 mm) breast carcinoma should be considered for adjuvant therapy using a regimen that contains a taxane and trastuzumab. However, the benefit may be small for patients with tumors ≤ 5 mm (T1a, N0), so it is important to carefully weigh the risk against the benefit. Among the agents that targeting HER2, only trastuzumab has been shown to improve overall survival (OS) in the adjuvant setting; long-term follow-up data are awaited for other agents.8 A trastuzumab biosimilar, trastuzumab-dkst, was recently approved by the US Food and Drug Administration (FDA) for the same indications as trastuzumab.14 The regimens most commonly used in the adjuvant and neoadjuvant settings for nonmetastatic breast cancer are summarized in Table 2.
Patients with small (≤ 3 cm), node-negative tumors can generally be considered for a reduced-intensity regimen that includes weekly paclitaxel plus trastuzumab. This combination proved efficacious in a single-group, multicenter study that enrolled 406 patients.15 Paclitaxel and trastuzumab were given once weekly for 12 weeks, followed by trastuzumab, either weekly or every 3 weeks, to complete 1 year of therapy.After a median follow-up of more than 6 years, the rates of distant and locoregional recurrence were 1% and 1.2%, respectively.16
A combination of docetaxel, carboplatin, and trastuzumab is a nonanthracycline regimen that is also appropriate in this setting, based on the results of the Breast Cancer International Research Group 006 (BCIRG-006) trial.17 This phase 3 randomized trial enrolled 3222 women with HER2-positive, invasive, high-risk adenocarcinoma. Eligible patients had a T1–3 tumor and either lymph node–negative or –positive disease and were randomly assigned to receive 1 of 3 regimens: group 1 received doxorubicin and cyclophosphamide every 3 weeks for 4 cycles followed by docetaxel every 3 weeks for 4 cycles (AC-T); group 2 received the AC-T regimen in combination with trastuzumab; and group 3 received docetaxel, carboplatin, and trastuzumab once every 3 weeks for 6 cycles (TCH). Groups 2 and 3 also received trastuzumab for an additional 34 weeks to complete 1 year of therapy. Trastuzumab-containing regimens were found to offer superior disease-free survival (DFS) and OS. When comparing the 2 trastuzumab arms after more than 10 years of follow-up, no statistically significant advantage of an anthracycline regimen over a nonanthracycline regimen was found.18 Furthermore, the anthracycline arm had a fivefold higher incidence of symptomatic congestive heart failure (grades 3 and 4), and the nonanthracycline regimen was associated with a lower incidence of treatment-related leukemia, a clinically significant finding despite not reaching statistical significance due to low overall numbers.
BCIRG-006, NSABP B-31, NCCTG N9831, and HERA are all large randomized trials with consistent results confirming trastuzumab’s role in reducing recurrence and improving survival in HER2-positive breast cancer in the adjuvant settings. The estimated overall benefit from addition of this agent was a 34% to 41% improvement in survival and a 33% to 52% improvement in DFS.8,17–20
Dual anti-HER2 therapy containing both trastuzumab and pertuzumab should be strongly considered for patients with macroscopic lymph node involvement based on the results of the APHINITY trial.21 In this study, the addition of pertuzumab to standard trastuzumab-based therapy led to a significant improvement in invasive-disease-free survival at 3 years. In subgroup analysis, the benefit was restricted to the node-positive group (3-year invasive-disease-free survival rates of 92% in the pertuzumab group versus 90.2% in the placebo group, P = 0.02). Patients with hormone receptor–negative disease derived greater benefit from the addition of pertuzumab. Regimens used in the APHINITY trial included the anti-HER2 agents trastuzumab and pertuzumab in combination with 1 of the following chemotherapy regimens: sequential cyclophosphamide plus either doxorubicin or epirubicin, followed by either 4 cycles of docetaxel or 12 weekly doses of paclitaxel; sequential fluorouracil plus either epirubicin or doxorubicin plus cyclophosphamide (3 or 4 cycles), followed by 3 or 4 cycles of docetaxel or 12 weekly cycles of paclitaxel; or 6 cycles of concurrent docetaxel plus carboplatin.
One-year therapy with neratinib, an oral tyrosine kinase inhibitor of HER2, is now approved by the FDA after completion of trastuzumab in the adjuvant setting, based on the results of the ExteNET trial.22 In this study, patients who had completed trastuzumab within the preceding 12 months, without evidence of recurrence, were randomly assigned to receive either oral neratinib or placebo daily for 1 year. The 2-year DFS rate was 93.9% and 91.6% for the neratinib and placebo groups, respectively. The most common adverse effect of neratinib was diarrhea, with approximately 40% of patients experiencing grade 3 diarrhea. In subgroup analyses, hormone receptor–positive patients derived the most benefit, while hormone receptor–negative patients derived no or marginal benefit.22 OS benefit has not yet been established.23
Trastuzumab therapy (with pertuzumab if indicated) should be offered for an optimal duration of 12 months (17 cycles, including those given with chemotherapy backbone). A shorter duration of therapy, 6 months, has been shown to be inferior,24 while a longer duration, 24 months, has been shown to provide no additional benefit.25
Finally, sequential addition of anti-estrogen endocrine therapy is indicated for hormone-positive tumors. Endocrine therapy is usually added after completion of the chemotherapy backbone of the regimen, but may be given concurrently with anti-HER2 therapy. If radiation is being administered, endocrine therapy can be given concurrently or started after radiation therapy is completed.
Case 1 Conclusion
The patient can be offered 1 of 2 adjuvant treatment regimens, either TH or TCH (Table 2). Since the patient had lumpectomy, she is an appropriate candidate for adjuvant radiation, which would be started after completion of the chemotherapy backbone (taxane/platinum). Endocrine therapy for at least 5 years should be offered sequentially or concurrently with radiation. Her long-term prognosis is very favorable.
Case Patient 2
A 43-year-old woman presents with a 4-cm breast mass, a separate skin nodule, and palpable matted axillary lymphadenopathy. Biopsies of the breast mass and subcutaneous nodule reveal invasive ductal carcinoma that is ER-negative, PR-negative, and HER2-positive by IHC (3+ staining). Based on clinical findings, the patient is staged as T4b (separate tumor nodule), N2 (matted axillary lymph nodes). Systemic staging with CT scan of the chest, abdomen, and pelvis shows no evidence of distant metastases.
- What is the recommended approach to management for this patient?
Recommendations for neoadjuvant therapy, given before definitive surgery, follow the same path as with other subtypes of breast cancer. Patients with suspected anatomical stage III disease are strongly encouraged to undergo upfront (neoadjuvant) chemotherapy in combination with HER2-targeted agents. In addition, all HER2-positive patients with clinically node-positive disease can be offered neoadjuvant therapy using chemotherapy plus dual anti-HER2 therapy (trastuzumab and pertuzumab), with complete pathological response expected in more than 60% of patients.26,27 Because this patient has locally advanced disease, especially skin involvement and matted axillary nodes, she should undergo neoadjuvant therapy. Preferred regimens contain both trastuzumab and pertuzumab in combination with cytotoxic chemotherapy. The latter may be given concurrently (nonanthracycline regimens, such as docetaxel plus carboplatin) or sequentially (anthracycline-based regimens), as outlined in Table 2. Administration of anthracyclines and trastuzumab simultaneously is contraindicated due to increased risk of cardiomyopathy.28
Endocrine therapy is not indicated for this patient per the current standard of care because the tumor was ER- and PR-negative. Had the tumor been hormone receptor–positive, endocrine therapy for a minimum of 5 years would have been indicated. Likewise, in the case of hormone receptor–positive disease, 12 months of neratinib therapy after completion of trastuzumab may add further benefit, as shown in the ExteNET trial.22,23 Neratinib seems to have a propensity to prevent or delay trastuzumab-induced overexpression of estrogen receptors. This is mainly due to hormone receptor/HER2 crosstalk, a potential mechanism of resistance to trastuzumab.29,30
In addition to the medical therapy options discussed here, this patient would be expected to benefit from adjuvant radiation to the breast and regional lymph nodes, given the presence of T4 disease and bulky adenopathy in the axilla.31
Case 2 Conclusion
The patient undergoes neoadjuvant treatment (docetaxel, carboplatin, trastuzumab, and pertuzumab every 21 days for a total of 6 cycles), followed by surgical resection (modified radical mastectomy) that reveals complete pathological response (no residual invasive carcinoma). Subsequently, she receives radiation therapy to the primary tumor site and regional lymph nodes while continuing trastuzumab and pertuzumab for 11 more cycles (17 total). Despite presenting with locally advanced disease, the patient has a favorable overall prognosis due to an excellent pathological response.
- What is the approach to follow-up after completion of primary therapy?
Patients may follow up every 3 to 6 months for clinical evaluation in the first 5 years after completing primary adjuvant therapy. An annual screening mammogram is recommended as well. Body imaging can be done if dictated by symptoms. However, routine CT, positron emission tomography, or bone scans are not recommended as part of follow-up in the absence of symptoms, mainly because of a lack of evidence that such surveillance improves survival.32
Metastatic HER2-Positive Breast Cancer
Metastatic breast cancer most commonly presents as a distant recurrence of previously treated local disease. However, 6% to 18% of patients have no prior history of breast cancer and present with de novo metastatic disease.33,34 The most commonly involved distant organs are the skeletal bones, liver, lung, distant lymph node stations, and brain. Compared to other subtypes, HER2-positive tumors have an increased tendency to involve the central nervous system.35–38 Although metastatic HER2-positive breast cancer is not considered curable, significant improvement in survival has been achieved, and patients with metastatic disease have median survival approaching 5 years.39
Case Presentation 3
A 69-year-old woman with a history of breast cancer 4 years ago presents with new-onset back pain and unintentional weight loss. On exam, she is found to have palpable axillary adenopathy on the same side as her previous cancer. Her initial disease was stage IIB ER-positive and HER2-positive and was treated with chemotherapy, mastectomy, and anastrozole, which the patient is still taking. She undergoes CT scan of the chest, abdomen, and pelvis and radionucleotide bone scan, which show multiple liver and bony lesions suspicious for metastatic disease. Axillary lymph node biopsy confirms recurrent invasive carcinoma that is ER-positive and HER2-positive by IHC (3+).
- What is the approach to management of a patient who presents with symptoms of recurrent HER2-positive disease?
This patient likely has metastatic breast cancer based on the imaging findings. In such cases, a biopsy of the recurrent disease should always be considered, if feasible, to confirm the diagnosis and rule out other etiologies such as different malignances and benign conditions. Hormone-receptor and HER2 testing should also be performed on recurrent disease, since a change in HER2 status can be seen in 15% to 33% of cases.40–42
Based on data from the phase 3 CLEOPATRA trial, first-line systemic regimens for patients with metastatic breast cancer that is positive for HER2 should consist of a combination of docetaxel, trastuzumab, and pertuzumab. Compared to placebo, adding pertuzumab yielded superior progression-free survival of 18.4 months (versus 12.4 months for placebo) and an unprecedented OS of 56.5 months (versus 40.8 for placebo).39 Weekly paclitaxel can replace docetaxel with comparable efficacy (Table 3).43
Patients can develop significant neuropathy as well as skin and nail changes after multiple cycles of taxane-based chemotherapy. Therefore, the taxane backbone may be dropped after 6 to 8 cycles, while patients continue the trastuzumab and pertuzumab combination until disease progression or unacceptable toxicity. Some patients may enjoy remarkable long-term survival on “maintenance” anti-HER2 therapy.44 Despite lack of high-level evidence, such as from large randomized trials, some experts recommend the addition of a hormone blocker after discontinuation of the taxane in ER-positive tumors.45
Premenopausal and perimenopausal women with hormone receptor–positive metastatic disease should be considered for simultaneous ovarian suppression. Ovarian suppression can be accomplished medically using a gonadotropin-releasing hormone agonist (goserelin) or surgically via salpingo-oophorectomy.46–48
Case 3 Conclusion
The patient receives 6 cycles of docetaxel, trastuzumab, and pertuzumab, after which the docetaxel is discontinued due to neuropathy while she continues the other 2 agents. After 26 months of disease control, the patient is found to have new liver metastatic lesions, indicating progression of disease.
- What therapeutic options are available for this patient?
Patients whose disease progresses after receiving taxane- and trastuzumab-containing regimens are candidates to receive the novel antibody-drug conjugate ado-trastuzumab emtansine (T-DM1). Early progressors (ie, patients with early stage disease who have progression of disease while receiving adjuvant trastuzumab or within 6 months of completion of adjuvant trastuzumab) are also candidates for T-DM1. Treatment usually fits in the second line or beyond based on data from the EMILIA trial, which randomly assigned patients to receive either capecitabine plus lapatinib or T-DM1.49,50 Progression-free survival in the T-DM1 group was 9.6 months versus 6.4 months for the comparator. Improvement of 4 months in OS persisted with longer follow-up despite a crossover rate of 27%. Furthermore, a significantly higher objective response rate and fewer adverse effects were reported in the T-DM1 patients. Most patients included in the EMILIA trial were pertuzumab-naive. However, the benefit of T-DM1 appears to persist, albeit to a lesser extent, for pertuzumab-pretreated patients.51,52
Patients in whom treatment fails with 2 or more lines of therapy containing taxane-trastuzumab (with or without pertuzumab) and T-DM1 are candidates to receive a combination of capecitabine and lapatinib, a TKI, in the third line and beyond. Similarly, the combination of capecitabine with trastuzumab in the same settings appears to have equal efficacy.53,54 Trastuzumab may be continued beyond progression while changing the single-agent chemotherapy drug for subsequent lines of therapy, per ASCO guidelines,55 although improvement in OS has not been demonstrated beyond the third line in a large randomized trial (Table 3).
Approved HER2-Targeted Drugs
HER2-directed therapy is implemented in the management of nearly all stages of HER2-positive invasive breast cancer, including early and late stages (Table 4).
Trastuzumab
Trastuzumab was the first anti-HER2 agent to be approved by the FDA in 1998. It is a humanized monoclonal antibody directed against the extracellular domain of the HER2 receptor (domain IV). Trastuzumab functions by interrupting HER2 signal transduction and by flagging tumor cells for immune destruction.56 Cardiotoxicity, usually manifested as left ventricular systolic dysfunction, is the most noteworthy adverse effect of trastuzumab. The most prominent risk factors for cardiomyopathy in patients receiving trastuzumab are low baseline ejection fraction (< 55%), age > 50 years, co-administration and higher cumulative dose of anthracyclines, and increased body mass index and obesity.57–59 Whether patients receive therapy in the neoadjuvant, adjuvant, or metastatic settings, baseline cardiac function assessment with echocardiogram or multiple-gated acquisition scan is required. While well-designed randomized trials validating the value and frequency of monitoring are lacking, repeated cardiac testing every 3 months is generally recommended for patients undergoing adjuvant therapy. Patients with metastatic disease who are receiving treatment with palliative intent may be monitored less frequently.60,61
An asymptomatic drop in ejection fraction is the most common manifestation of cardiac toxicity. Other cardiac manifestations have also been reported with much less frequency, including arrhythmias, severe congestive heart failure, ventricular thrombus formation, and even cardiac death. Until monitoring and dose-adjustment guidelines are issued, the guidance provided in the FDA-approved prescribing information should be followed, which recommends holding trastuzumab when there is ≥ 16% absolute reduction in left ventricular ejection fraction (LVEF) from the baseline value; or if the LVEF value is below the institutional lower limit of normal and the drop is ≥ 10%. After holding the drug, cardiac function can be re-evaluated every 4 weeks. In most patients, trastuzumab-induced cardiotoxicity can be reversed by withholding trastuzumab and initiating cardioprotective therapy, although the latter remains controversial. Re-challenging after recovery of ejection fraction is possible and toxicity does not appear to be proportional to cumulative dose. Cardiomyopathy due to trastuzumab therapy is potentially reversible within 6 months in more than 80% of cases.28,57,60–63
Other notable adverse effects of trastuzumab include pulmonary toxicity (such as interstitial lung disease) and infusion reactions (usually during or within 24 hours of first dose).
Pertuzumab
Pertuzumab is another humanized monoclonal antibody directed to a different extracellular domain of the HER2 receptor, the dimerization domain (domain II), which is responsible for heterodimerization of HER2 with other HER receptors, especially HER3. This agent should always be co-administered with trastuzumab as the 2 drugs produce synergistic anti-tumor effect, without competition for the receptor. Activation of HER3, via dimerization with HER2, produces an alternative mechanism of downstream signaling, even in the presence of trastuzumab and in the absence of growth factors (Figure 2).
Ado-Trastuzumab Emtansine
Ado-trastuzumab emtansine (T-DM1) is an antibody-drug conjugate that combines the monoclonal antibody trastuzumab with the cytotoxic agent DM1 (emtansine), a potent microtubule inhibitor and a derivative of maytansine, in a single structure (Figure 3).
Lapatinib
Lapatinib is an oral small-molecule tyrosine kinase inhibitor of EGFR (HER1) and HER2 receptors. It is approved in combination with capecitabine for patients with HER2-expressing metastatic breast cancer who previously received trastuzumab, an anthracycline, and a taxane chemotherapy or T-DM1. Lapatinib is also approved in combination with letrozole in postmenopausal women with HER2-positive, hormone receptor–positive metastatic disease, although it is unclear where this regimen would fit in the current schema. It may be considered for patients with hormone receptor–positive disease who are not candidates for therapy with taxane-trastuzumab and T-DM1 or who decline this therapy. Diarrhea is seen in most patients treated with lapatinib and may be severe in 20% of cases when lapatinib is combined with capecitabine. Decreases in LVEF have been reported and cardiac function monitoring at baseline and periodically may be considered.69,70 Lapatinib is not approved for use in adjuvant settings.
Neratinib
Neratinib is an oral small-molecule irreversible tyrosine kinase inhibitor of HER1, HER2, and HER4. It is currently approved only for extended adjuvant therapy after completion of 1 year of standard trastuzumab therapy. It is given orally every day for 1 year. The main side effect, expected in nearly all patients, is diarrhea, which can be severe in up to 40% of patients and may lead to dehydration and electrolyte imbalance. Diarrhea usually starts early in the course of therapy and can be most intense during the first cycle. Therefore, prophylactic antidiarrheal therapy is recommended to reduce the intensity of diarrhea. Loperamide prophylaxis may be initiated simultaneously for all patients using a tapering schedule. Drug interruption or dose reduction may be required if diarrhea is severe or refractory.21,71 Neratinib is not FDA-approved in the metastatic settings.
Conclusion
HER2-positive tumors represent a distinct subset(s) of breast tumors with unique pathological and clinical characteristics. Treatment with a combination of cytotoxic chemotherapy and HER2-targeted agents has led to a dramatic improvement in survival for patients with locoregional and advanced disease. Trastuzumab is an integral part of adjuvant therapy for HER2-positive invasive disease. Pertuzumab should be added to trastuzumab in node-positive disease. Neratinib may be considered after completion of trastuzumab therapy in patients with hormone receptor–positive disease. For metastatic HER2-positive breast cancer, a regimen consisting of docetaxel plus trastuzumab and pertuzumab is the standard first-line therapy. Ado-trastuzumab is an ideal next line option for patients whose disease progresses on trastuzumab and taxanes.
1. Yedjou CG, Tchounwou PB, Payton M, et al. Assessing the racial and ethnic disparities in breast cancer mortality in the United States. Int J Environ Res Public Health 2017;14(5).
2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016;66:271–89.
3. Huang HJ, Neven P, Drijkoningen M, et al. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. J Clin Pathol 2005;58:611–6.
4. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
5. Cronin KA, Harlan LC, Dodd KW, et al. Population-based estimate of the prevalence of HER-2 positive breast cancer tumors for early stage patients in the US. Cancer Invest 2010;28:963–-8.
6. Huang HJ, Neven P, Drijkoningen M, et al. Hormone receptors do not predict the HER2/neu status in all age groups of women with an operable breast cancer. Ann Oncol 2005;16:1755–61.
7. Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006;295:2492–502.
8. Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol 2014;32:3744–52.
9. Brennan PJ, Kumagai T, Berezov A, et al. HER2/neu: mechanisms of dimerization/oligomerization. Oncogene 2000;19:6093–101.
10. Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun 2004;319:1–11.
11. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013.
12. Ravaioli A, Pasini G, Polselli A, et al. Staging of breast cancer: new recommended standard procedure. Breast Cancer Res Treat 2002;72:53–60.
13. Puglisi F, Follador A, Minisini AM, et al. Baseline staging tests after a new diagnosis of breast cancer: further evidence of their limited indications. Ann Oncol 2005;16:263–6.
14. FDA approves trastuzumab biosimilar. Cancer Discov 2018;8:130.
15. Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med 2015;372:134–41.
16. Tolaney SM, Barry WT, Guo H, Dillon D, et al. Seven-year (yr) follow-up of adjuvant paclitaxel (T) and trastuzumab (H) (APT trial) for node-negative, HER2-positive breast cancer (BC) [ASCO abstract]. J Clin Oncol. 2017;35(suppl):511.
17. Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 2011;365:1273–83.
18. Slamon DJ, Eiermann W, Robert NJ, et al. Ten year follow-up of BCIRG-006 comparing doxorubicin plus cyclophosphamide followed by docetaxel (AC -> T) with doxorubicin plus cyclophosphamide followed by docetaxel and trastuzumab (AC -> TH) with docetaxel, carboplatin and trastuzumab (TCH) in HER2+early breast cancer [SABC abstract]. Cancer Res 2016;76(4 supplement):S5-04.
19. Jahanzeb M. Adjuvant trastuzumab therapy for HER2-positive breast cancer. Clin Breast Cancer 2008;8:324–33.
20. Cameron D, Piccart-Gebhart MJ, Gelber RD, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017;389:1195–205.
21. von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med 2017;377:122–31.
22. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17:367–77.
23. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1688–700.
24. Pivot X, Romieu G, Debled M, et al. 6 months versus 12 months of adjuvant trastuzumab for patients with HER2-positive early breast cancer (PHARE): a randomised phase 3 trial. Lancet Oncol 2013;14:741–8.
25. Goldhirsch A, Gelber RD, Piccart-Gebhart MJ, et al. 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet 2013;382:1021–8.
26. Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol 2013;24:2278–84.
27. Schneeweiss A, Chia S, Hickish T, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: Evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. Eur J Cancer 2018;89:27–35
28. de Azambuja E, Procter MJ, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac events at 8 years of median follow-up in the Herceptin Adjuvant trial (BIG 1-01). J Clin Oncol 2014;32:2159–65.
29. Dowsett M, Harper-Wynne C, Boeddinghaus I, et al. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res 2001;61:8452–8.
30. Nahta R, O’Regan RM. Therapeutic implications of estrogen receptor signaling in HER2-positive breast cancers. Breast Cancer Res Treat 2012;135:39–48.
31. Recht A, Comen EA, Fine RE, et al. Postmastectomy radiotherapy: An American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology focused guideline update. Pract Radiat Oncol 2016;6:e219-e34.
32. Runowicz CD, Leach CR, Henry NL, et al. American Cancer Society/American Society of Clinical Oncology breast cancer survivorship care guideline. J Clin Oncol 2016;34:611–35.
33. Zeichner SB, Herna S, Mani A, et al. Survival of patients with de-novo metastatic breast cancer: analysis of data from a large breast cancer-specific private practice, a university-based cancer center and review of the literature. Breast Cancer Res Treat 2015;153:617–24.
34. Dawood S, Broglio K, Ensor J, et al. Survival differences among women with de novo stage IV and relapsed breast cancer. Ann Oncol 2010;21:2169–74.
35. Savci-Heijink CD, Halfwerk H, Hooijer GK, et al. Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat 2015;150:547–57.
36. Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol 2015;35:85–95.
37. Bendell JC, Domchek SM, Burstein HJ, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 2003;97:2972–7.
38. Burstein HJ, Lieberman G, Slamon DJ, et al. Isolated central nervous system metastases in patients with HER2-overexpressing advanced breast cancer treated with first-line trastuzumab-based therapy. Ann Oncol 2005;16:1772–7.
39. Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372:724–34.
40. Lindstrom LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol 2012;30:2601–8.
41. Guarneri V, Giovannelli S, Ficarra G, et al. Comparison of HER-2 and hormone receptor expression in primary breast cancers and asynchronous paired metastases: impact on patient management. Oncologist 2008;13:838–44.
42. Salkeni MA, Hall SJ. Metastatic breast cancer: Endocrine therapy landscape reshaped. Avicenna J Med 2017;7:144–52.
43. Dang C, Iyengar N, Datko F, et al. Phase II study of paclitaxel given once per week along with trastuzumab and pertuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:442–7.
44. Cantini L, Pistelli M, Savini A, et al. Long-responders to anti-HER2 therapies: A case report and review of the literature. Mol Clin Oncol 2018;8:147–52.
45. Sutherland S, Miles D, Makris A. Use of maintenance endocrine therapy after chemotherapy in metastatic breast cancer. Eur J Cancer 2016;69:216–22.
46. Falkson G, Holcroft C, Gelman RS, et al. Ten-year follow-up study of premenopausal women with metastatic breast cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol 1995;13:1453–8.
47. Boccardo F, Rubagotti A, Perrotta A, et al. Ovarian ablation versus goserelin with or without tamoxifen in pre-perimenopausal patients with advanced breast cancer: results of a multicentric Italian study. Ann Oncol 1994;5:337–42.
48 Taylor CW, Green S, Dalton WS, et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: an intergroup study. J Clin Oncol 1998;16:994–9.
49. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783–91.
50. Dieras V, Miles D, Verma S, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:732–42.
51. Dzimitrowicz H, Berger M, Vargo C, et al. T-DM1 Activity in metastatic human epidermal growth factor receptor 2-positive breast cancers that received prior therapy with trastuzumab and pertuzumab. J Clin Oncol 2016;34:3511–7.
52. Fabi A, Giannarelli D, Moscetti L, et al. Ado-trastuzumab emtansine (T-DM1) in HER2+ advanced breast cancer patients: does pretreatment with pertuzumab matter? Future Oncol 2017;13:2791–7.
53. Madden R, Kosari S, Peterson GM, et al. Lapatinib plus capecitabine in patients with HER2-positive metastatic breast cancer: A systematic review. Int J Clin Pharmacol Ther 2018;56:72–80.
54. Pivot X, Manikhas A, Zurawski B, et al. CEREBEL (EGF111438): A phase III, randomized, open-label study of lapatinib plus capecitabine versus trastuzumab plus capecitabine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:1564–73.
55. Giordano SH, Temin S, Kirshner JJ, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014;32:2078–99.
56. Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007;357:39–51.
57. Russell SD, Blackwell KL, Lawrence J, et al. Independent adjudication of symptomatic heart failure with the use of doxorubicin and cyclophosphamide followed by trastuzumab adjuvant therapy: a combined review of cardiac data from the National Surgical Adjuvant breast and Bowel Project B-31 and the North Central Cancer Treatment Group N9831 clinical trials. J Clin Oncol 2010;28:3416–21.
58. Ewer SM, Ewer MS. Cardiotoxicity profile of trastuzumab. Drug Saf 2008;31:459–67.
59. Guenancia C, Lefebvre A, Cardinale D, et al. Obesity as a risk factor for anthracyclines and trastuzumab cardiotoxicity in breast cancer: a systematic review and meta-analysis. J Clin Oncol 2016;34:3157–65.
60. Dang CT, Yu AF, Jones LW, et al. Cardiac surveillance guidelines for trastuzumab-containing therapy in early-stage breast cancer: getting to the heart of the matter. J Clin Oncol 2016;34:1030–3.
61. Brann AM, Cobleigh MA, Okwuosa TM. Cardiovascular monitoring with trastuzumab therapy: how frequent is too frequent? JAMA Oncol 2016;2:1123–4.
62. Suter TM, Procter M, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol 2007;25:3859–65.
63. Procter M, Suter TM, de Azambuja E, et al. Longer-term assessment of trastuzumab-related cardiac adverse events in the Herceptin Adjuvant (HERA) trial. J Clin Oncol 2010;28:3422–8.
64. Yamashita-Kashima Y, Shu S, Yorozu K, et al. Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncol Lett 2017;14:4197–205.
65. Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer 2017;117:1736–42.
66. Girish S, Gupta M, Wang B, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol 2012;69:1229–40.
67. Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res 2015;21:123–33.
68. Yan H, Endo Y, Shen Y, et al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther 2016;15:480–90.
69. Spector NL, Xia W, Burris H 3rd, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 2005;23:2502–12.
70. Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 2009;27:5538–46.
71. Neratinib (Nerlynx) for HER2-positive breast cancer. Med Lett Drugs Ther 2018;60(1539):23.
1. Yedjou CG, Tchounwou PB, Payton M, et al. Assessing the racial and ethnic disparities in breast cancer mortality in the United States. Int J Environ Res Public Health 2017;14(5).
2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016;66:271–89.
3. Huang HJ, Neven P, Drijkoningen M, et al. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. J Clin Pathol 2005;58:611–6.
4. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
5. Cronin KA, Harlan LC, Dodd KW, et al. Population-based estimate of the prevalence of HER-2 positive breast cancer tumors for early stage patients in the US. Cancer Invest 2010;28:963–-8.
6. Huang HJ, Neven P, Drijkoningen M, et al. Hormone receptors do not predict the HER2/neu status in all age groups of women with an operable breast cancer. Ann Oncol 2005;16:1755–61.
7. Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006;295:2492–502.
8. Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol 2014;32:3744–52.
9. Brennan PJ, Kumagai T, Berezov A, et al. HER2/neu: mechanisms of dimerization/oligomerization. Oncogene 2000;19:6093–101.
10. Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun 2004;319:1–11.
11. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013.
12. Ravaioli A, Pasini G, Polselli A, et al. Staging of breast cancer: new recommended standard procedure. Breast Cancer Res Treat 2002;72:53–60.
13. Puglisi F, Follador A, Minisini AM, et al. Baseline staging tests after a new diagnosis of breast cancer: further evidence of their limited indications. Ann Oncol 2005;16:263–6.
14. FDA approves trastuzumab biosimilar. Cancer Discov 2018;8:130.
15. Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med 2015;372:134–41.
16. Tolaney SM, Barry WT, Guo H, Dillon D, et al. Seven-year (yr) follow-up of adjuvant paclitaxel (T) and trastuzumab (H) (APT trial) for node-negative, HER2-positive breast cancer (BC) [ASCO abstract]. J Clin Oncol. 2017;35(suppl):511.
17. Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 2011;365:1273–83.
18. Slamon DJ, Eiermann W, Robert NJ, et al. Ten year follow-up of BCIRG-006 comparing doxorubicin plus cyclophosphamide followed by docetaxel (AC -> T) with doxorubicin plus cyclophosphamide followed by docetaxel and trastuzumab (AC -> TH) with docetaxel, carboplatin and trastuzumab (TCH) in HER2+early breast cancer [SABC abstract]. Cancer Res 2016;76(4 supplement):S5-04.
19. Jahanzeb M. Adjuvant trastuzumab therapy for HER2-positive breast cancer. Clin Breast Cancer 2008;8:324–33.
20. Cameron D, Piccart-Gebhart MJ, Gelber RD, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017;389:1195–205.
21. von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med 2017;377:122–31.
22. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17:367–77.
23. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1688–700.
24. Pivot X, Romieu G, Debled M, et al. 6 months versus 12 months of adjuvant trastuzumab for patients with HER2-positive early breast cancer (PHARE): a randomised phase 3 trial. Lancet Oncol 2013;14:741–8.
25. Goldhirsch A, Gelber RD, Piccart-Gebhart MJ, et al. 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet 2013;382:1021–8.
26. Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol 2013;24:2278–84.
27. Schneeweiss A, Chia S, Hickish T, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: Evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. Eur J Cancer 2018;89:27–35
28. de Azambuja E, Procter MJ, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac events at 8 years of median follow-up in the Herceptin Adjuvant trial (BIG 1-01). J Clin Oncol 2014;32:2159–65.
29. Dowsett M, Harper-Wynne C, Boeddinghaus I, et al. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res 2001;61:8452–8.
30. Nahta R, O’Regan RM. Therapeutic implications of estrogen receptor signaling in HER2-positive breast cancers. Breast Cancer Res Treat 2012;135:39–48.
31. Recht A, Comen EA, Fine RE, et al. Postmastectomy radiotherapy: An American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology focused guideline update. Pract Radiat Oncol 2016;6:e219-e34.
32. Runowicz CD, Leach CR, Henry NL, et al. American Cancer Society/American Society of Clinical Oncology breast cancer survivorship care guideline. J Clin Oncol 2016;34:611–35.
33. Zeichner SB, Herna S, Mani A, et al. Survival of patients with de-novo metastatic breast cancer: analysis of data from a large breast cancer-specific private practice, a university-based cancer center and review of the literature. Breast Cancer Res Treat 2015;153:617–24.
34. Dawood S, Broglio K, Ensor J, et al. Survival differences among women with de novo stage IV and relapsed breast cancer. Ann Oncol 2010;21:2169–74.
35. Savci-Heijink CD, Halfwerk H, Hooijer GK, et al. Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat 2015;150:547–57.
36. Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol 2015;35:85–95.
37. Bendell JC, Domchek SM, Burstein HJ, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 2003;97:2972–7.
38. Burstein HJ, Lieberman G, Slamon DJ, et al. Isolated central nervous system metastases in patients with HER2-overexpressing advanced breast cancer treated with first-line trastuzumab-based therapy. Ann Oncol 2005;16:1772–7.
39. Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372:724–34.
40. Lindstrom LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol 2012;30:2601–8.
41. Guarneri V, Giovannelli S, Ficarra G, et al. Comparison of HER-2 and hormone receptor expression in primary breast cancers and asynchronous paired metastases: impact on patient management. Oncologist 2008;13:838–44.
42. Salkeni MA, Hall SJ. Metastatic breast cancer: Endocrine therapy landscape reshaped. Avicenna J Med 2017;7:144–52.
43. Dang C, Iyengar N, Datko F, et al. Phase II study of paclitaxel given once per week along with trastuzumab and pertuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:442–7.
44. Cantini L, Pistelli M, Savini A, et al. Long-responders to anti-HER2 therapies: A case report and review of the literature. Mol Clin Oncol 2018;8:147–52.
45. Sutherland S, Miles D, Makris A. Use of maintenance endocrine therapy after chemotherapy in metastatic breast cancer. Eur J Cancer 2016;69:216–22.
46. Falkson G, Holcroft C, Gelman RS, et al. Ten-year follow-up study of premenopausal women with metastatic breast cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol 1995;13:1453–8.
47. Boccardo F, Rubagotti A, Perrotta A, et al. Ovarian ablation versus goserelin with or without tamoxifen in pre-perimenopausal patients with advanced breast cancer: results of a multicentric Italian study. Ann Oncol 1994;5:337–42.
48 Taylor CW, Green S, Dalton WS, et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: an intergroup study. J Clin Oncol 1998;16:994–9.
49. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783–91.
50. Dieras V, Miles D, Verma S, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:732–42.
51. Dzimitrowicz H, Berger M, Vargo C, et al. T-DM1 Activity in metastatic human epidermal growth factor receptor 2-positive breast cancers that received prior therapy with trastuzumab and pertuzumab. J Clin Oncol 2016;34:3511–7.
52. Fabi A, Giannarelli D, Moscetti L, et al. Ado-trastuzumab emtansine (T-DM1) in HER2+ advanced breast cancer patients: does pretreatment with pertuzumab matter? Future Oncol 2017;13:2791–7.
53. Madden R, Kosari S, Peterson GM, et al. Lapatinib plus capecitabine in patients with HER2-positive metastatic breast cancer: A systematic review. Int J Clin Pharmacol Ther 2018;56:72–80.
54. Pivot X, Manikhas A, Zurawski B, et al. CEREBEL (EGF111438): A phase III, randomized, open-label study of lapatinib plus capecitabine versus trastuzumab plus capecitabine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:1564–73.
55. Giordano SH, Temin S, Kirshner JJ, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014;32:2078–99.
56. Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007;357:39–51.
57. Russell SD, Blackwell KL, Lawrence J, et al. Independent adjudication of symptomatic heart failure with the use of doxorubicin and cyclophosphamide followed by trastuzumab adjuvant therapy: a combined review of cardiac data from the National Surgical Adjuvant breast and Bowel Project B-31 and the North Central Cancer Treatment Group N9831 clinical trials. J Clin Oncol 2010;28:3416–21.
58. Ewer SM, Ewer MS. Cardiotoxicity profile of trastuzumab. Drug Saf 2008;31:459–67.
59. Guenancia C, Lefebvre A, Cardinale D, et al. Obesity as a risk factor for anthracyclines and trastuzumab cardiotoxicity in breast cancer: a systematic review and meta-analysis. J Clin Oncol 2016;34:3157–65.
60. Dang CT, Yu AF, Jones LW, et al. Cardiac surveillance guidelines for trastuzumab-containing therapy in early-stage breast cancer: getting to the heart of the matter. J Clin Oncol 2016;34:1030–3.
61. Brann AM, Cobleigh MA, Okwuosa TM. Cardiovascular monitoring with trastuzumab therapy: how frequent is too frequent? JAMA Oncol 2016;2:1123–4.
62. Suter TM, Procter M, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol 2007;25:3859–65.
63. Procter M, Suter TM, de Azambuja E, et al. Longer-term assessment of trastuzumab-related cardiac adverse events in the Herceptin Adjuvant (HERA) trial. J Clin Oncol 2010;28:3422–8.
64. Yamashita-Kashima Y, Shu S, Yorozu K, et al. Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncol Lett 2017;14:4197–205.
65. Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer 2017;117:1736–42.
66. Girish S, Gupta M, Wang B, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol 2012;69:1229–40.
67. Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res 2015;21:123–33.
68. Yan H, Endo Y, Shen Y, et al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther 2016;15:480–90.
69. Spector NL, Xia W, Burris H 3rd, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 2005;23:2502–12.
70. Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 2009;27:5538–46.
71. Neratinib (Nerlynx) for HER2-positive breast cancer. Med Lett Drugs Ther 2018;60(1539):23.
Open Clinical Trials for Patients With HIV and/or Hepatitis B and C (FULL)
Web-Based Intervention to Reduce Alcohol Use in Veterans With Hepatitis C
The primary objective of this study is to implement and evaluate a web-based brief alcohol intervention (BAI) for treating veterans with hepatitis C virus (HCV) and seeking care at 2 VA HCV clinics–VAPAHCS and SFVAMC. This study will have 3 aims: First, the investigators plan to assess patient, provider, and system factors that may impact the initial adoption of this intervention in 2 VA HCV clinics. These data will result in the development of a protocol for the initial implementation of the web-based BAI at the investigators’ 2 study sites. A secondary aim will involve obtaining patient and provider feedback on an existing web-based BAI (see www.bmi-aft.org, VA intranet only) to help inform its redesign for use with this population. Second, the investigators will implement and examine the effectiveness of a web-based BAI in 2 HCV clinics to reduce alcohol consumption in veterans with HCV at 3- and 6-months post-treatment. Third, the investigators will conduct a budget impact analysis to estimate the short-term costs (1-3 years) of adoption and diffusion of the web-based BAI and the trajectory of health care spending for study participants.
ID: NCT01707030
Sponsor: VA Office of Research and Development
Location (contact): VA Palo Alto Health Care System, California (Keith Humphreys); San Francisco VAMC, California (Alex Monto)
Pilot Study of the Effect of Rifaximin On B-Cell Dysregulation in Cirrhosis
Hepatitis C is the leading cause of chronic liver disease and cirrhosis in U.S. veterans. Cirrhosis is associated with impaired antibody responses and increased risk of bacterial infections. We have recently identified that cirrhosis is associated with abnormalities of memory B-cells, cells that make antibodies and help protect against bacterial infections. We have identified that chemicals associated with gut bacteria might play a role in causing these B-cell abnormalities. It is well known that gut bacteria have increased access to the blood in individuals with cirrhosis, a process called bacterial translocation. We hypothesize that reducing bacteria counts in the gut by using poorly-absorbed antibiotics (also known as selective gut decontamination) will partially reverse losses of memory B-cells in cirrhosis by reducing bacterial translocation.
ID: NCT01951209
Sponsor: David E. Kaplan, MD MS
Location (contact): Corporal Michael J. Crescenz VAMC, Philadelphia, Pennsylvania (David Kaplan)
Long-Term Study of Liver Disease in People With Hepatitis B and/or Hepatitis C With or Without HIV Infection
The primary objective of the proposed study is to characterize viral liver disease and factors affecting the natural history of viral liver disease in persons with and without HIV with an emphasis on those living in the Washington DC metropolitan area. There are few longitudinal research cohorts of participants with viral hepatitis and HIV coinfection, especially at integrated medical care centers. The study, including a participant questionnaire for HCV infected participants only and phlebotomy, will be administered on-site at clinical facilities in the District of Columbia and at the National Institutes of Health. The cohort will be designed to study research questions with respect to liver disease, disease pathogenesis using genomics, proteomics, and immunologic disease models. Secondary objectives include study of the immunopathogenesis of hepatitis B virus (HBV) and HCV disease progression in HIV-infected subjects. In addition, this is an invaluable opportunity to determine the prevalence and risk factors associated with the development of hepatocellular carcinoma, the long-term effects of HCV clearance with direct-acting antivirals (DAAs), along with biomarker profile(s) for diagnosis and outcome. Moreover, this will serve as a catchment protocol to select appropriate participants for novel HBV and HCV therapeutic trials.
ID: NCT01350648
Sponsor: National Institutes of Health Clinical Center
Location: Washington DC VAMC; National Institutes of Health Clinical Center, Bethesda, Maryland
Evaluating the Use of Pitavastatin to Reduce the Risk of Cardiovascular Disease in HIV-Infected Adults (REPRIEVE)
Currently, there are few strategies to prevent cardiovascular disease (CVD) in HIV-infected people, even though they are at high risk for developing CVD. Statin medications are used to lower cholesterol and may be effective at reducing the risk of CVD in people infected with HIV. The purpose of this study is to evaluate the use of pitavastatin to reduce the risk of CVD in adults infected with HIV who are on antiretroviral therapy (ART). This study will enroll adults infected with HIV who are on any ART regimen (ART is not provided by the study) for at least 6 months before study entry considered low-to-moderate risk using the 2013 American College of Cardiology/American Heart Association guideline thresholds forecommended statin initiation. Total study duration will be approximately 72 months from the time the first participant is enrolled.
ID: NCT02344290
Sponsor: National Institute of Allergy and Infectious Diseases (NIAID)
Location (contact): VA West Los Angeles Medical Center CRS, California (Wendy Rossen); VA Connecticut Healthcare System CRS, West Haven (Laurie Andrews); Washington DC VAMC; Malcom Randall VA Medical Center CRS, Gainesville, Florida (Evan Waters); VA New York Harbor Healthcare System (Christine Reel-Brander); Dallas VAMC, Texas (Ashley Liggion-Turk); Michael E. DeBakey VAMC, Houston, Texas (Mahwish Mushtaq)
Development of a City-Wide Cohort of HIVInfected Persons in Care in the District of Columbia: The DC Cohort
All major community and academic clinics treating HIVinfected persons in the District of Columbia (DC) will initially be included in the development of a city-wide “DC Cohort” of HIV-infected persons in care, with consideration to be given subsequently to the inclusion of large private physician practices. Socio-demographics, risk factors, treatments, diagnoses, labs and procedures documented in outpatient medical record systems will be included in the DC Cohort database. Routine reports will be generated every 6 months for sites comparing their participants’ socio-demographics, clinical status, treatments, and outcomes to all other data in the DC Cohort database, and other comparisons specifically requested by sites. All sites will be provided analytic support in research areas of interest.
ID: NCT01206920
Sponsor: George Washington University
Location: Washington DC VAMC
Study of Immune Responses Induced by a HIV Vaccine
The primary purpose of this study is to define in HIV-uninfected volunteers the innate, cell-mediated and humoral responses induced by AIDSVAX B/E in the systemic and mucosal compartments and to characterize B cell functional specificities in peripheral blood, bone marrow and sigmoid compartments.
ID: NCT01933685
Sponsor: U.S. Army Medical Research and Materiel Command
Strength Training and Endurance Exercise for LIFE (STEEL)
The objective of this study is to determine the effect of exercise training on the central (cardiovascular) and peripheral (muscular) impairments underlying poor physical function by comparing older HIV-infected veterans randomized to combine aerobic and resistance exercise training versus usual care. The study hypothesis is that a progressive aerobic and resistance rehabilitation program will increase aerobic capacity and muscle strength, which will be mediated by improved diastolic function, increased muscle mass, and decreased systemic inflammation. To test this hypothesis, investigators will conduct a randomized 16-week trial of progressive aerobic and resistance training versus usual care control in 40 sedentary older (50+ years) HIVinfected veterans. The study will determine the effects of exercise training on aerobic capacity and diastolic function, and their relationship to changes in biomarkers of systemic inflammation and cardiac fibrosis (AIM 1). The study also will determine the effect of exercise training on strength and muscle mass, and their relationship to changes in biomarkers of systemic inflammation (AIM 2).
ID: NCT02101060
Sponsor: VA Office of Research and Development
Location (contact): Salem VA Medical Center, Virginia (Carolyn Jones, Tracy A Hicks)
Click here to read the digital edition.
Web-Based Intervention to Reduce Alcohol Use in Veterans With Hepatitis C
The primary objective of this study is to implement and evaluate a web-based brief alcohol intervention (BAI) for treating veterans with hepatitis C virus (HCV) and seeking care at 2 VA HCV clinics–VAPAHCS and SFVAMC. This study will have 3 aims: First, the investigators plan to assess patient, provider, and system factors that may impact the initial adoption of this intervention in 2 VA HCV clinics. These data will result in the development of a protocol for the initial implementation of the web-based BAI at the investigators’ 2 study sites. A secondary aim will involve obtaining patient and provider feedback on an existing web-based BAI (see www.bmi-aft.org, VA intranet only) to help inform its redesign for use with this population. Second, the investigators will implement and examine the effectiveness of a web-based BAI in 2 HCV clinics to reduce alcohol consumption in veterans with HCV at 3- and 6-months post-treatment. Third, the investigators will conduct a budget impact analysis to estimate the short-term costs (1-3 years) of adoption and diffusion of the web-based BAI and the trajectory of health care spending for study participants.
ID: NCT01707030
Sponsor: VA Office of Research and Development
Location (contact): VA Palo Alto Health Care System, California (Keith Humphreys); San Francisco VAMC, California (Alex Monto)
Pilot Study of the Effect of Rifaximin On B-Cell Dysregulation in Cirrhosis
Hepatitis C is the leading cause of chronic liver disease and cirrhosis in U.S. veterans. Cirrhosis is associated with impaired antibody responses and increased risk of bacterial infections. We have recently identified that cirrhosis is associated with abnormalities of memory B-cells, cells that make antibodies and help protect against bacterial infections. We have identified that chemicals associated with gut bacteria might play a role in causing these B-cell abnormalities. It is well known that gut bacteria have increased access to the blood in individuals with cirrhosis, a process called bacterial translocation. We hypothesize that reducing bacteria counts in the gut by using poorly-absorbed antibiotics (also known as selective gut decontamination) will partially reverse losses of memory B-cells in cirrhosis by reducing bacterial translocation.
ID: NCT01951209
Sponsor: David E. Kaplan, MD MS
Location (contact): Corporal Michael J. Crescenz VAMC, Philadelphia, Pennsylvania (David Kaplan)
Long-Term Study of Liver Disease in People With Hepatitis B and/or Hepatitis C With or Without HIV Infection
The primary objective of the proposed study is to characterize viral liver disease and factors affecting the natural history of viral liver disease in persons with and without HIV with an emphasis on those living in the Washington DC metropolitan area. There are few longitudinal research cohorts of participants with viral hepatitis and HIV coinfection, especially at integrated medical care centers. The study, including a participant questionnaire for HCV infected participants only and phlebotomy, will be administered on-site at clinical facilities in the District of Columbia and at the National Institutes of Health. The cohort will be designed to study research questions with respect to liver disease, disease pathogenesis using genomics, proteomics, and immunologic disease models. Secondary objectives include study of the immunopathogenesis of hepatitis B virus (HBV) and HCV disease progression in HIV-infected subjects. In addition, this is an invaluable opportunity to determine the prevalence and risk factors associated with the development of hepatocellular carcinoma, the long-term effects of HCV clearance with direct-acting antivirals (DAAs), along with biomarker profile(s) for diagnosis and outcome. Moreover, this will serve as a catchment protocol to select appropriate participants for novel HBV and HCV therapeutic trials.
ID: NCT01350648
Sponsor: National Institutes of Health Clinical Center
Location: Washington DC VAMC; National Institutes of Health Clinical Center, Bethesda, Maryland
Evaluating the Use of Pitavastatin to Reduce the Risk of Cardiovascular Disease in HIV-Infected Adults (REPRIEVE)
Currently, there are few strategies to prevent cardiovascular disease (CVD) in HIV-infected people, even though they are at high risk for developing CVD. Statin medications are used to lower cholesterol and may be effective at reducing the risk of CVD in people infected with HIV. The purpose of this study is to evaluate the use of pitavastatin to reduce the risk of CVD in adults infected with HIV who are on antiretroviral therapy (ART). This study will enroll adults infected with HIV who are on any ART regimen (ART is not provided by the study) for at least 6 months before study entry considered low-to-moderate risk using the 2013 American College of Cardiology/American Heart Association guideline thresholds forecommended statin initiation. Total study duration will be approximately 72 months from the time the first participant is enrolled.
ID: NCT02344290
Sponsor: National Institute of Allergy and Infectious Diseases (NIAID)
Location (contact): VA West Los Angeles Medical Center CRS, California (Wendy Rossen); VA Connecticut Healthcare System CRS, West Haven (Laurie Andrews); Washington DC VAMC; Malcom Randall VA Medical Center CRS, Gainesville, Florida (Evan Waters); VA New York Harbor Healthcare System (Christine Reel-Brander); Dallas VAMC, Texas (Ashley Liggion-Turk); Michael E. DeBakey VAMC, Houston, Texas (Mahwish Mushtaq)
Development of a City-Wide Cohort of HIVInfected Persons in Care in the District of Columbia: The DC Cohort
All major community and academic clinics treating HIVinfected persons in the District of Columbia (DC) will initially be included in the development of a city-wide “DC Cohort” of HIV-infected persons in care, with consideration to be given subsequently to the inclusion of large private physician practices. Socio-demographics, risk factors, treatments, diagnoses, labs and procedures documented in outpatient medical record systems will be included in the DC Cohort database. Routine reports will be generated every 6 months for sites comparing their participants’ socio-demographics, clinical status, treatments, and outcomes to all other data in the DC Cohort database, and other comparisons specifically requested by sites. All sites will be provided analytic support in research areas of interest.
ID: NCT01206920
Sponsor: George Washington University
Location: Washington DC VAMC
Study of Immune Responses Induced by a HIV Vaccine
The primary purpose of this study is to define in HIV-uninfected volunteers the innate, cell-mediated and humoral responses induced by AIDSVAX B/E in the systemic and mucosal compartments and to characterize B cell functional specificities in peripheral blood, bone marrow and sigmoid compartments.
ID: NCT01933685
Sponsor: U.S. Army Medical Research and Materiel Command
Strength Training and Endurance Exercise for LIFE (STEEL)
The objective of this study is to determine the effect of exercise training on the central (cardiovascular) and peripheral (muscular) impairments underlying poor physical function by comparing older HIV-infected veterans randomized to combine aerobic and resistance exercise training versus usual care. The study hypothesis is that a progressive aerobic and resistance rehabilitation program will increase aerobic capacity and muscle strength, which will be mediated by improved diastolic function, increased muscle mass, and decreased systemic inflammation. To test this hypothesis, investigators will conduct a randomized 16-week trial of progressive aerobic and resistance training versus usual care control in 40 sedentary older (50+ years) HIVinfected veterans. The study will determine the effects of exercise training on aerobic capacity and diastolic function, and their relationship to changes in biomarkers of systemic inflammation and cardiac fibrosis (AIM 1). The study also will determine the effect of exercise training on strength and muscle mass, and their relationship to changes in biomarkers of systemic inflammation (AIM 2).
ID: NCT02101060
Sponsor: VA Office of Research and Development
Location (contact): Salem VA Medical Center, Virginia (Carolyn Jones, Tracy A Hicks)
Click here to read the digital edition.
Web-Based Intervention to Reduce Alcohol Use in Veterans With Hepatitis C
The primary objective of this study is to implement and evaluate a web-based brief alcohol intervention (BAI) for treating veterans with hepatitis C virus (HCV) and seeking care at 2 VA HCV clinics–VAPAHCS and SFVAMC. This study will have 3 aims: First, the investigators plan to assess patient, provider, and system factors that may impact the initial adoption of this intervention in 2 VA HCV clinics. These data will result in the development of a protocol for the initial implementation of the web-based BAI at the investigators’ 2 study sites. A secondary aim will involve obtaining patient and provider feedback on an existing web-based BAI (see www.bmi-aft.org, VA intranet only) to help inform its redesign for use with this population. Second, the investigators will implement and examine the effectiveness of a web-based BAI in 2 HCV clinics to reduce alcohol consumption in veterans with HCV at 3- and 6-months post-treatment. Third, the investigators will conduct a budget impact analysis to estimate the short-term costs (1-3 years) of adoption and diffusion of the web-based BAI and the trajectory of health care spending for study participants.
ID: NCT01707030
Sponsor: VA Office of Research and Development
Location (contact): VA Palo Alto Health Care System, California (Keith Humphreys); San Francisco VAMC, California (Alex Monto)
Pilot Study of the Effect of Rifaximin On B-Cell Dysregulation in Cirrhosis
Hepatitis C is the leading cause of chronic liver disease and cirrhosis in U.S. veterans. Cirrhosis is associated with impaired antibody responses and increased risk of bacterial infections. We have recently identified that cirrhosis is associated with abnormalities of memory B-cells, cells that make antibodies and help protect against bacterial infections. We have identified that chemicals associated with gut bacteria might play a role in causing these B-cell abnormalities. It is well known that gut bacteria have increased access to the blood in individuals with cirrhosis, a process called bacterial translocation. We hypothesize that reducing bacteria counts in the gut by using poorly-absorbed antibiotics (also known as selective gut decontamination) will partially reverse losses of memory B-cells in cirrhosis by reducing bacterial translocation.
ID: NCT01951209
Sponsor: David E. Kaplan, MD MS
Location (contact): Corporal Michael J. Crescenz VAMC, Philadelphia, Pennsylvania (David Kaplan)
Long-Term Study of Liver Disease in People With Hepatitis B and/or Hepatitis C With or Without HIV Infection
The primary objective of the proposed study is to characterize viral liver disease and factors affecting the natural history of viral liver disease in persons with and without HIV with an emphasis on those living in the Washington DC metropolitan area. There are few longitudinal research cohorts of participants with viral hepatitis and HIV coinfection, especially at integrated medical care centers. The study, including a participant questionnaire for HCV infected participants only and phlebotomy, will be administered on-site at clinical facilities in the District of Columbia and at the National Institutes of Health. The cohort will be designed to study research questions with respect to liver disease, disease pathogenesis using genomics, proteomics, and immunologic disease models. Secondary objectives include study of the immunopathogenesis of hepatitis B virus (HBV) and HCV disease progression in HIV-infected subjects. In addition, this is an invaluable opportunity to determine the prevalence and risk factors associated with the development of hepatocellular carcinoma, the long-term effects of HCV clearance with direct-acting antivirals (DAAs), along with biomarker profile(s) for diagnosis and outcome. Moreover, this will serve as a catchment protocol to select appropriate participants for novel HBV and HCV therapeutic trials.
ID: NCT01350648
Sponsor: National Institutes of Health Clinical Center
Location: Washington DC VAMC; National Institutes of Health Clinical Center, Bethesda, Maryland
Evaluating the Use of Pitavastatin to Reduce the Risk of Cardiovascular Disease in HIV-Infected Adults (REPRIEVE)
Currently, there are few strategies to prevent cardiovascular disease (CVD) in HIV-infected people, even though they are at high risk for developing CVD. Statin medications are used to lower cholesterol and may be effective at reducing the risk of CVD in people infected with HIV. The purpose of this study is to evaluate the use of pitavastatin to reduce the risk of CVD in adults infected with HIV who are on antiretroviral therapy (ART). This study will enroll adults infected with HIV who are on any ART regimen (ART is not provided by the study) for at least 6 months before study entry considered low-to-moderate risk using the 2013 American College of Cardiology/American Heart Association guideline thresholds forecommended statin initiation. Total study duration will be approximately 72 months from the time the first participant is enrolled.
ID: NCT02344290
Sponsor: National Institute of Allergy and Infectious Diseases (NIAID)
Location (contact): VA West Los Angeles Medical Center CRS, California (Wendy Rossen); VA Connecticut Healthcare System CRS, West Haven (Laurie Andrews); Washington DC VAMC; Malcom Randall VA Medical Center CRS, Gainesville, Florida (Evan Waters); VA New York Harbor Healthcare System (Christine Reel-Brander); Dallas VAMC, Texas (Ashley Liggion-Turk); Michael E. DeBakey VAMC, Houston, Texas (Mahwish Mushtaq)
Development of a City-Wide Cohort of HIVInfected Persons in Care in the District of Columbia: The DC Cohort
All major community and academic clinics treating HIVinfected persons in the District of Columbia (DC) will initially be included in the development of a city-wide “DC Cohort” of HIV-infected persons in care, with consideration to be given subsequently to the inclusion of large private physician practices. Socio-demographics, risk factors, treatments, diagnoses, labs and procedures documented in outpatient medical record systems will be included in the DC Cohort database. Routine reports will be generated every 6 months for sites comparing their participants’ socio-demographics, clinical status, treatments, and outcomes to all other data in the DC Cohort database, and other comparisons specifically requested by sites. All sites will be provided analytic support in research areas of interest.
ID: NCT01206920
Sponsor: George Washington University
Location: Washington DC VAMC
Study of Immune Responses Induced by a HIV Vaccine
The primary purpose of this study is to define in HIV-uninfected volunteers the innate, cell-mediated and humoral responses induced by AIDSVAX B/E in the systemic and mucosal compartments and to characterize B cell functional specificities in peripheral blood, bone marrow and sigmoid compartments.
ID: NCT01933685
Sponsor: U.S. Army Medical Research and Materiel Command
Strength Training and Endurance Exercise for LIFE (STEEL)
The objective of this study is to determine the effect of exercise training on the central (cardiovascular) and peripheral (muscular) impairments underlying poor physical function by comparing older HIV-infected veterans randomized to combine aerobic and resistance exercise training versus usual care. The study hypothesis is that a progressive aerobic and resistance rehabilitation program will increase aerobic capacity and muscle strength, which will be mediated by improved diastolic function, increased muscle mass, and decreased systemic inflammation. To test this hypothesis, investigators will conduct a randomized 16-week trial of progressive aerobic and resistance training versus usual care control in 40 sedentary older (50+ years) HIVinfected veterans. The study will determine the effects of exercise training on aerobic capacity and diastolic function, and their relationship to changes in biomarkers of systemic inflammation and cardiac fibrosis (AIM 1). The study also will determine the effect of exercise training on strength and muscle mass, and their relationship to changes in biomarkers of systemic inflammation (AIM 2).
ID: NCT02101060
Sponsor: VA Office of Research and Development
Location (contact): Salem VA Medical Center, Virginia (Carolyn Jones, Tracy A Hicks)
Click here to read the digital edition.
The Evidence for Herbal and Botanical Remedies, Part 2
More than a third of American adults use complementary and alternative medicine.1 Unfortunately, the public’s enthusiasm for herbal products is not always consistent with the scientific evidence supporting their use. In part one of this series, we discussed the studies that have been done on capsaicin, butterbur, green tea, and peppermint. In this installment, we outline the research on 5 additional remedies: turmeric/curcumin, which may be of benefit in ulcerative colitis; chamomile, which appears to offer relief to patients with anxiety; rosemary, which may help treat alopecia; as well as coffee and cocoa, which may have some cardiovascular benefits (TABLE).
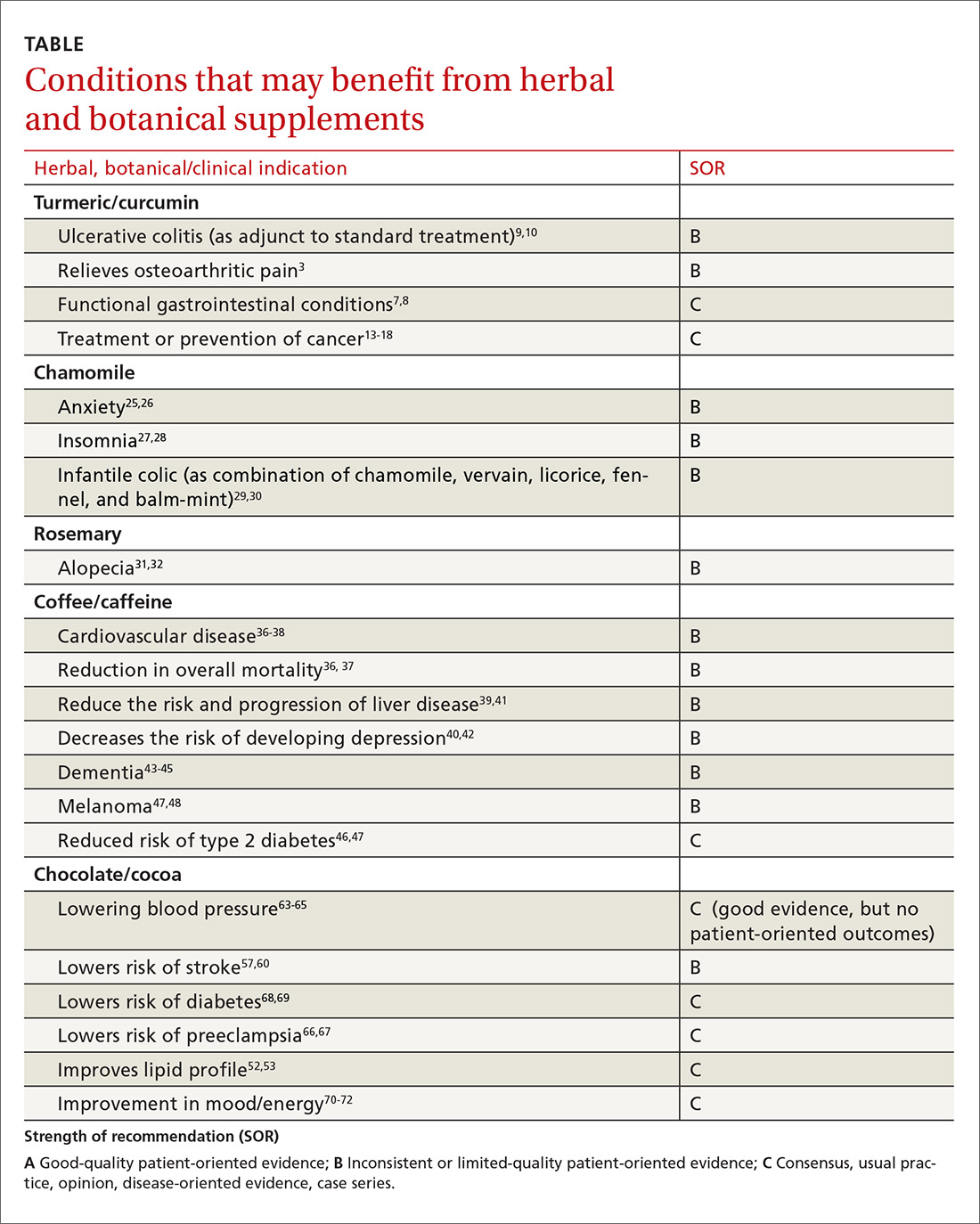
Turmeric/curcumin
Overview
Turmeric (Curcuma longa), a relative of ginger, has been used for 4000 years to treat a variety of conditions.2,3 Curcumin is the yellow pigment isolated from the rhizomes of Curcuma longa, commonly known as turmeric.3 Turmeric powder contains 5% curcumin, which is the main biologically active compound. Although it grows in many tropical locations, most turmeric is grown in India, where it is used as a main ingredient in curry. The roots and bulbs of turmeric that are used in medicine are generally boiled and dried, which results in a yellow powder.
Turmeric has been used in both Ayurvedic and Chinese medicine for its anti-inflammatory properties, in the treatment of digestive and liver problems, to fight infections, and to help heal skin diseases and wounds.3-7
Functional GI disorders. A recent review noted that curcumin has been shown in several preclinical studies and uncontrolled clinical trials to have effects on gut inflammation, gut permeability, and the brain-gut axis, especially in functional GI disorders.7 A double-blind, placebo-controlled study from 1989 found that turmeric reduced symptoms of bloating and gas in subjects suffering from undifferentiated dyspepsia.8
Ulcerative colitis (UC). A 2012 Cochrane review noted that curcumin appears to be a safe and effective therapy for maintenance of remission in quiescent UC when given as adjunctive therapy along with mesalamine or sulfasalazine.9 In a 2015 randomized controlled trial (RCT), the addition of curcumin to mesalamine therapy was superior to the combination of placebo and mesalamine in inducing clinical and endoscopic remission in patients with mild-to-moderate active UC, producing no apparent adverse effects.10
Osteoarthritis (OA). Because of turmeric’s ability to reduce inflammation, it may help relieve OA pain.3 Clinical evidence is scant for the anti-arthritic efficacy of turmeric dietary supplements, although animal studies indicate that turmeric prevents inflammation through regulation of NF-kappaB-regulated genes that regulate the immune and inflammatory response.6 Inflammatory cell influx, joint levels of prostaglandin E2, and periarticular osteoclast formation were also inhibited by turmeric extract treatment.6
A 2013 review of turmeric for OA concluded that observational studies and in vitro results are promising for the use of curcumin for OA, but well-designed clinical studies were lacking and are needed to support the efficacy of curcumin in OA patients.11 However, in a 2014 randomized trial of 367 patients, turmeric appeared to be similar in efficacy to ibuprofen for the treatment of pain and disability in adults with knee OA.12 The curcumin (turmeric) group also had fewer adverse effects.12
Cancer. There has been a great deal of research on turmeric’s anti-cancer properties, but clinical evidence is lacking. In vitro evidence, animal studies, and small clinical trials suggest that curcumin may help prevent or treat several types of cancers, but the overall evidence is poor. Nonetheless, curcumin and turmeric have been or are currently being evaluated for the treatment or prevention of prostate, liver, breast, skin, gynecologic, hematologic, pulmonary, thymic, bone, brain, and colon cancer.13-18
Oral submucous fibrosis. A small randomized trial found improvement in oral function with curcumin lozenges, when compared to placebo, indicating that turmeric may hold promise as a treatment of oral submucous fibrosis.19
Uveitis. A small pilot study of 32 patients suggested that oral curcumin may be as effective as corticosteroids for uveitis.20
Heart disease. Curcumin may have a cardiovascular protective role, as it has been shown to reduce atherosclerosis, but a reduction in myocardial infarction or stroke has not been documented.21
Alzheimer’s dementia. Animal studies have shown a reduction in amyloid plaque formation with curcumin.22
Adverse effects (and precautions)
Turmeric in food is considered safe. A variety of animal and human studies have also indicated that curcumin is safe and well tolerated, even at very high doses.13 However, taking large amounts of turmeric for long periods of time could cause stomach upset and gastric ulcers. In addition, patients with gallstones or bile obstruction should use it with caution due to increased bile production.7
Because turmeric may lower blood sugar levels, patients with diabetes should monitor for hypoglycemia when using turmeric in combination with diabetic medications. Similarly, those with bleeding disorders taking blood thinners should use turmeric and curcumin with caution, because it can inhibit platelet aggregation.23
Although it is safe to eat foods with turmeric during pregnancy, pregnant and breastfeeding women should not take turmeric supplements, as the safety of large doses in pregnancy is unknown.
The bottom line
Turmeric/curcumin has anti-inflammatory properties and may be useful as an adjunct for ulcerative colitis and to improve the symptoms of OA. It may also have anti-carcinogenic properties, although definitive data are lacking. Those with a history of gastrointestinal conditions such as gastric ulcer, patients taking blood thinners, and patients with diabetes who are prone to low blood sugar levels should use turmeric/curcumin with caution.
Chamomile
Overview
Chamomile, a member of the Asteraceae/Compositae family, is one of the oldest herbal medicines. It has been used for hay fever, inflammation, muscle spasms, menstrual disorders, insomnia, ulcers, wounds, gastrointestinal disorders, rheumatic pain, and hemorrhoids. Essential oils of chamomile are used extensively in cosmetics and aromatherapy. Many different preparations have been developed, the most popular being herbal tea.24
Individuals with a hypersensitivity to plants of the Asteraceae (Compositae) family such as ragweed (Ambrosia spp.), marigold flower (Calendula officinalis), and chrysanthemum (Chrysanthemum spp.) may show a similar reaction to chamomile.25
Anxiety. A controlled clinical trial of chamomile extract for generalized anxiety disorder (GAD) suggested that it may have modest anxiolytic activity in patients with mild to moderate GAD.26 Another randomized, double-blind, placebo-controlled trial found oral chamomile extract was efficacious and well-tolerated in patients experiencing mild to moderate GAD and may provide an alternative therapeutic anxiolytic for patients with mild GAD.25 In addition to its anxiolytic activity, chamomile may also provide clinically meaningful antidepressant activity.26
Insomnia. Chamomile may have some impact on sleep diary measures (total sleep time, sleep efficiency, sleep latency, wake after sleep onset, sleep quality, and number of awakenings) relative to placebo in adults with chronic primary insomnia, according to a small randomized, double-blind, placebo-controlled pilot trial involving 34 patients.27 However, a systematic review found no statistically significant difference between any herbal medicine (including chamomile) and placebo, for clinical efficacy in patients with insomnia. A similar, or smaller, number of adverse events per person were reported with chamomile compared with placebo, suggesting safe use.28
Infantile colic. A small prospective double-blind study on the use of chamomile-containing tea on infantile colic showed statistically significant symptom improvement in tea-treated infants. The study did note, however, that prolonged ingestion of herbal teas may lead to decreased milk intake.29,30
Adverse effects
As noted earlier, a systematic review found that the number of adverse events per person reported with chamomile was comparable to the number associated with placebo, suggesting that it is safe.28
The bottom line
Chamomile appears to be safe with minimal adverse effects and may be effective for the treatment of anxiety, insomnia, and infantile colic.
Rosemary
Overview
Rosemary, officially known as Rosmarinus officinalis, is a medicinal evergreen plant native to the Mediterranean area that appears to increase microcapillary perfusion.31
Alopecia. A randomized double-blind controlled trial found that essential oils including rosemary oil (as well as thyme, lavender, and cedarwood) massaged into the scalp improved hair growth in almost half of patients with alopecia areata after 7 months.32 Another randomized trial comparing rosemary oil to minoxidil 2% for androgenetic alopecia showed a significant increase in hair count at the 6-month endpoint compared with the baseline, but no significant difference was found between the study groups regarding hair count either at Month 3 or Month 6 (P >.05). 31
Adverse effects
In the randomized trial described above comparing rosemary oil to minoxidil 2%, adverse effects appeared to be rare for topical rosemary oil. Scalp itching was more frequent in the minoxidil group.31
The bottom line
Topical rosemary oil may be useful in the treatment of alopecia with minimal adverse effects.
Coffee/caffeine
Overview
Coffee is one of the most widely used botanicals with approximately 3.5 billion cups of coffee consumed per day worldwide. It is a popular beverage because of its unique aromatic taste and its use as a central nervous system stimulant. The coffee tree (genus coffea) is found throughout Latin America, Africa, and eastern Asia. Two of the most common commercially grown species are Coffea arabica (Arabicas) and Coffea canephora (Robusta). Processing and roasting methods may differ and produce variations in flavor and aroma. The degree of roasting also affects the caffeine content.
Coffee consumption leads to increased alertness and can boost mental performance. Based on the literature and US Food and Drug Administration recommendations, four 8-oz cups of coffee (about 400 mg of caffeine) daily is an acceptable average amount of caffeine. More than 500 mg/d is considered excessive use of coffee.33,34
Overall mortality. A 2008 study showed that regular coffee was not associated with increased or decreased mortality in both men and women.35 However, more recent studies show an inverse relationship between mortality and coffee consumption.
Specifically, a 2014 meta-analysis found an inverse relationship between coffee and mortality.36 A large prospective cohort study from 2015 that included 79,234 women and 76,704 men found that drinking coffee was inversely associated with overall mortality.37 In this cohort study, an inverse association were observed for deaths from heart disease, respiratory disease, diabetes, and self-harm.37 While mechanisms were not analyzed, coffee may reduce mortality risk by affecting inflammation, lung function, insulin sensitivity, and depression.
Cardiovascular disease. Coffee consumption may modestly reduce the risk of stroke, according to a prospective cohort study of 83,076 women from the Nurses’ Health Study who were followed for 24 years.38 Reduced cardiovascular mortality was also found in a large prospective cohort study, as noted in the mortality discussion above.37 A 2014 meta-analysis concluded that coffee consumption is inversely associated with cardiovascular mortality. Drinking 3 or 4 cups a day appears to be the amount that may decrease one’s risk of death when compared to those who do not drink coffee at all.36
Liver disease. Friedrich et al performed a study involving 379 patients with end stage liver disease, and found that coffee consumption delayed the progression of disease in patients with both alcoholic liver disease and primary sclerosing cholangitis.39 Coffee consumption also increased long-term survival after liver transplantation.39 However, the study found that coffee did not have any effect on patients with chronic viral hepatitis.
In a 2016 meta-analysis, caffeinated coffee consumption reduced hepatic fibrosis of nonalcoholic fatty liver disease, although caffeine consumption did not reduce the prevalence of nonalcoholic fatty liver disease.40 Another meta-analysis, including 16 studies, also found caffeine reduced the risk for hepatic fibrosis and cirrhosis.41
Depression. Based on 2 different systematic reviews and meta-analyses from 2016, coffee consumption appears to have a significant protective effect, decreasing the risk of developing depression.40,42
Alzheimer’s disease/dementia. Coffee, tea, and caffeine consumption show promise in reducing the risk of cognitive decline and dementia. Individuals who consume one to 2 cups of coffee per day had a decreased incidence of mild cognitive impairment compared to non-drinkers.43 A 2015 Japanese study also found an inverse association between coffee consumption and dementia among women, nonsmokers, and those who do not drink alcohol.44 Most recently, a 2016 study, the Women’s Health Initiative Memory Study, looked at incident dementia rates in women >65 years of age with high vs low caffeine intake. Women with higher caffeine intake were less likely to develop dementia or any cognitive impairment compared with those consuming <64 mg/day.45
Type 2 diabetes. A 2009 prospective cohort study, which included 40,011 participants followed for more than 10 years, found that drinking at least 3 cups of coffee or tea was associated with a lowered risk of type 2 diabetes.46 A 2009 systematic review of 20 cohort studies showed that high intakes of coffee, decaffeinated coffee, and tea are associated with a reduced risk of diabetes.47
Melanoma.
Adverse effects
Despite the many potential benefits of coffee, caffeine is a potent drug that should be used with caution.49 People with underlying heart problems should avoid caffeine due to concern that it may cause palpitations from tachycardia. It may worsen anxiety problems or depression. Coffee may increase the production of stomach acids, which can worsen acid reflux or stomach ulcers.
Caffeine is a potent diuretic and may decrease absorption of calcium and cause OA. Caffeine may cause dependence and withdrawal symptoms. Some of the symptoms of withdrawal include drowsiness, headaches, irritability, nausea, and vomiting. It may disrupt sleeping patterns by causing jitters and sleeplessness.49 Additionally, large amounts of caffeine may cause overdose and death.
The bottom line
Regular coffee intake is associated with a lower risk of mortality, reduced cardiovascular events, and a reduction in liver disease progression. Coffee may also have some utility for improving cognitive function and reducing the risk of type 2 diabetes. Caffeinated coffee should be limited to no more than 32 oz per day, due to the risk of insomnia, palpitations, anxiety, and gastritis.
Chocolate/cocoa
Overview
Few natural products have been claimed to successfully treat as many disorders as chocolate. The modern concept of chocolate as food has overshadowed its traditional medicinal use, although recent trials have looked at evidence for some of its traditional uses. Chocolate is processed from the pod of the cacao plant. The earliest evidence for its medical use is in Mayan civilizations, and for most of its approximately 4000-year history, chocolate was consumed as a bitter drink referred to as the “drink of the Gods.” The traditional drink was mixed with water, vanilla, honey, chili peppers, and other spices. Important components in chocolate include flavonoids (antioxidants), cocoa butter, caffeine, theobromine, and phenylethylamine.
Chocolate has stimulating, anti-inflammatory, neuroprotective, and cardioprotective effects, and improves the bioavailability of nitric oxide, which can improve blood pressure and platelet function.50 Epicatechin (an antioxidant) in cocoa is primarily responsible for its favorable impact on vascular endothelium via its effect on both acute and chronic upregulation of nitric oxide production. Other cardiovascular effects are mediated by the anti-inflammatory effects of cocoa polyphenols, and modulated through the activity of NF-kappaB.51
Dark chocolate appears to have the greatest benefit, as milk binds to antioxidants in chocolate, making them unavailable. Therefore, milk chocolate is not a good antioxidant source. There is no specific amount of chocolate that is known to be ideal, but an average of one to 2 ounces per day is often used in studies.
Cardiovascular effects. Chocolate does contain saturated fat, but a comparative, double-blind study found that short-term use of cocoa powder lowered plasma low-density lipoprotein (LDL) cholesterol, oxidized LDL, and apo B concentrations, and the plasma high-density lipoprotein (HDL) cholesterol concentration increased, relative to baseline in the low-, middle-, and high-cocoa groups.52 A small randomized crossover trial without clinical outcomes indicated that chocolate may increase HDL cholesterol without increasing weight.53
A meta-analysis of short-term (2-12 weeks) treatment with dark chocolate/cocoa products showed reductions in LDL and total cholesterol, but no changes in HDL or triglycerides.54 Another meta-analysis of RCTs, however, showed no short-term effect of cocoa/chocolate on lipid concentrations.55 A randomized, placebo-controlled double-blind study of 62 patients with diabetes and hypertension showed that high polyphenol chocolate improved triglyceride levels.56
Multiple studies have shown that chocolate is associated with a reduction in cardiovascular risk.57-59 A best case scenario analysis using a Markov model to predict the long-term effectiveness and cost effectiveness of daily dark chocolate consumption in a population with metabolic syndrome at high risk of cardiovascular disease concluded that daily consumption of dark chocolate can reduce cardiovascular events by 85 per 10,000 population treated over 10 years. The study concluded that $42 could be cost effectively spent per person per year on prevention strategies using dark chocolate.59
In addition, a meta-analysis of 7 observational studies showed that high levels of chocolate consumption (any type) were associated with a 29% reduction in stroke compared with the lowest levels of chocolate intake.57 Results of a similar meta-analysis from Neurology in 2012 also suggested that moderate chocolate consumption (any type) may lower the risk of stroke.60
That said, 2 systematic reviews specifically relating to the risk of coronary heart disease and chocolate intake were inconclusive.61-62
Blood pressure (BP). An RCT published in JAMA indicates that inclusion of small amounts of polyphenol-rich dark chocolate as part of a usual diet efficiently reduced BP and improved the formation of vasodilative nitric oxide.63 A meta-analysis of 10 RCTs also showed mean BP change in the active cocoa treatment arms across all trials was -4.5 mm Hg (95% confidence interval (CI), -5.9 to -3.2; P<.001) for systolic BP and -2.5 mm Hg (95% CI, -3.9 to -1.2; P<.001) for diastolic BP.64
A Cochrane Review meta-analysis of 20 studies revealed a statistically significant BP-reducing effect of flavanol-rich cocoa products compared with control in short-term trials of 2 to 18 weeks' duration.65 Because studies have shown improvement in BP with chocolate intake, investigations into a role of chocolate in the prevention of preeclampsia have been undertaken. In some studies, chocolate intake was associated with reduced odds of preeclampsia and gestational hypertension.66,67
Diabetes. Chocolate may exert significant vascular protection because of its antioxidant properties and possible increase of nitric oxide bioavailability, which can influence glucose uptake. A small trial comparing the effects of either dark or white chocolate bars (which do not contain the polyphenols) showed improved BP and glucose and insulin responses to an oral glucose tolerance test in healthy subjects on dark chocolate, but not white chocolate.68 A comparison of chocolate consumption and risk of diabetes in the Physicians’ Health Study showed an inverse relationship between chocolate intake with incident disease, but this association appeared only to apply in younger and normal-body weight men after controlling for comprehensive lifestyles, including total energy consumption.69
Fatigue. The effect of chocolate on a person’s energy level has been noted for centuries.70 A small randomized trial showed improved energy levels in those treated with higher chocolate intakes. In a double-blind, randomized, clinical pilot crossover study, high cocoa liquor/polyphenol rich chocolate, reduced fatigue in subjects with chronic fatigue syndrome.71
Anxiety. A small randomized trial showed chocolate decreased anxiety in high-anxiety trait subjects and improved the anxiety level and the energy levels of low-anxiety trait participants.72
Eye effects. The literature presents conflicting evidence regarding the effect of flavonoids on patients with glaucoma and ocular hypertension. However, a meta-analysis showed that flavonoids have a promising role in improving visual function in patients with glaucoma and ocular hypertension, and appear to play a part in both improving and slowing the progression of visual field loss.73
Cognitive decline. Chocolate intake (any type) was associated with a lower risk of cognitive decline (RR = 0.59; 95% CI, 0.38-0.92) with the greatest benefit noted in those who averaged more than one chocolate bar or one tablespoon of cocoa powder per week. This protective effect was observed only among subjects with an average daily consumption of caffeine <75 mg (69% of the participants; RR = 0.50; 95% CI, 0.31-0.82).74
The bottom line
Chocolate with high cocoa content (dark chocolate) appears to be safe and beneficial as part of a healthy diet and lifestyle that includes exercise and stress reduction to decrease cardiovascular risk and may improve energy levels.
CORRESPONDENCE
Michael Malone, MD, Family and Community Medicine, Penn State College of Medicine, 500 University Drive, Hershey, PA 17033; [email protected].
1. National Center for Complementary and Integrative Health. The use of complementary and alternative medicine in the United States. Available at: https://nccih.nih.gov/research/statistics/2007/camsurvey_fs1.htm. Accessed Nov 28, 2017.
2. Aggarwal BB. Curcumin-free turmeric exhibits anti-inflammatory and anticancer activities: identification of novel components of turmeric. Mol Nutr Food Res. 2013;57:1529-1542.
3. Henrotin Y, Clutterbuck AL, Allaway D, et al. Biological actions of curcumin on articular chondrocytes. Osteoarthritis Cartilage. 2010;18:141-149.
4. Asher GN, Spelman K. Clinical utility of curcumin extract. Altern Ther Health Med. 2013;19:20-22.
5. Phan TT, See P, Lee ST, et al. Protective effects of curcumin against oxidative damage on skin cells in vitro: its implication for wound healing. J Trauma. 2001;51:927-931.
6. Funk JL, Frye JB, Oyarzo JN, et al. Efficacy and mechanism of action of turmeric supplements in the treatment of experimental arthritis. Arthritis Rheum. 2006;54:3452-3464.
7. Patcharatrakul T, Gonlachanvit S. Chili peppers, curcumins, and prebiotics in gastrointestinal health and disease. Curr Gastroenterol Rep. 2016;18:19.
8. Thamlikitkul V, Bunyapraphatsara N, Dechatiwongse T, et al. Randomized double blind study of Curcuma domestica Val. for dyspepsia. J Med Assoc Thai. 1989;72:613-620.
9. Kumar S, Ahuja V, Sankar MJ, et al. Curcumin for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2012;10:CD008424.
10. Lang A, Salomon N, Wu JC, et al. Curcumin in combination with mesalamine induces remission in patients with mild-to-moderate ulcerative colitis in a randomized controlled trial. Clin Gastroenterol Hepatol. 2015;13:1444-1449.e1.
11. Henrotin Y, Priem F, Mobasheri A. Curcumin: a new paradigm and therapeutic opportunity for the treatment of osteoarthritis: curcumin for osteoarthritis management. Springerplus. 2013;2:56.
12. Kuptniratsaikul V, Dajpratham P, Taechaarpornkul W, et al. Efficacy and safety of Curcuma domestica extracts compared with ibuprofen in patients with knee osteoarthritis: a multicenter study. Clin Interv Aging. 2014;9:451-458.
13. Shehzad A, Lee J, Lee YS. Curcumin in various cancers. Biofactors. 2013;39:56-68.
14. Sordillo LA, Sordillo PP, Helson L. Curcumin for the treatment of glioblastoma. Anticancer Res. 2015;35:6373-6378.
15. Darvesh AS, Aggarwal BB, Bishayee A. Curcumin and liver cancer: a review. Curr Pharm Biotechnol. 2012;13:218-228.
16. Nagaraju GP, Aliya S, Zafar SF, et al. The impact of curcumin on breast cancer. Integr Biol (Camb). 2012;4:996-1007.
17. Johnson JJ, Mukhtar H. Curcumin for chemoprevention of colon cancer. Cancer Lett. 2007;255:170-181.
18. Dorai T, Cao YC, Dorai B, et al. Therapeutic potential of curcumin in human prostate cancer. III. Curcumin inhibits proliferation, induces apoptosis, and inhibits angiogenesis of LNCaP prostate cancer cells in vivo. Prostate. 2001;47:293-303.
19. Hazarey VK, Sakrikar AR, Ganvir SM. Efficacy of curcumin in the treatment for oral submucous fibrosis - a randomized clinical trial. J Oral Maxillofac Pathol. 2015;19:145-152.
20. Lal B, Kapoor AK, Asthana OP, et al. Efficacy of curcumin in the management of chronic anterior uveitis. Phytother Res. 1999;13:318-322.
21. Kapakos G, Youreva V, Srivastava AK. Cardiovascular protection by curcumin: molecular aspects. Indian J Biochem Biophys. 2012;49:306-315.
22. Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892-5901.
23. Heck AM, DeWitt BA, Lukes AL. Potential interactions between alternative therapies and warfarin. Am J Health Syst Pharm. 2000;57:1221-1227.
24. Srivastava JK, Shankar E, Gupta S. Chamomile: a herbal medicine of the past with bright future. Mol Med Rep. 2010;3:895-901.
25. Ross SM. Generalized anxiety disorder (GAD): efficacy of standardized matricaria recutita (german chamomile) extract in the treatment of generalized anxiety disorder. Holistic Nursing Practice. 2013;27:366- 368.
26. Amsterdam JD, Li Y, Soeller I, et al. A randomized, double-blind, placebo-controlled trial of oral Matricaria recutita (chamomile) extract therapy for generalized anxiety disorder. J Clin Psychopharmacol. 2009;29:378-382.
27. Zick SM, Wright BD, Sen A, et al. Preliminary examination of the efficacy and safety of a standardized chamomile extract for chronic primary insomnia: a randomized placebo-controlled pilot study. BMC Complement Altern Med. 2011;11:78.
28. Leach MJ, Page AT. Herbal medicine for insomnia: a systematic review and meta-analysis. Sleep Med Rev. 2015;24:1-12.
29. Weizman Z, Alkrinawi S, Goldfarb D, et al. Efficacy of herbal tea preparation in infantile colic. J Pediatr. 1993;122:650.
30. Crotteau CA, Wright ST, Eglash A. Clinical inquiries. What is the best treatment for infants with colic? J Fam Pract. 2006;55:634-636.
31. Panahi Y, Taghizadeh M, Marzony ET, et al. Rosemary oil vs minoxidil 2% for the treatment of androgenetic alopecia: a randomized comparative trial. Skinmed. 2015;13:15-21.
32. Hay IC, Jamieson M, Ormerod AD. Randomized trial of aromatherapy. Successful treatment for alopecia areata. Arch Dermatol. 1998;134:1349-1352.
33. Caffeine and kids: FDA takes a closer look. Available at: https://www.fda.gov/ForConsumers/ConsumerUpdates/ucm350570.htm. Accessed: November 1, 2017.
34. Torpy JM, Livingston EH. Energy Drinks. JAMA. 2013;309:297.
35. Lopez-Garcia E, van Dam RM, Li TY, et al. The relationship of coffee consumption with mortality. Ann Intern Med. 2008;148:904-914.
36. Crippa A, Discacciati A, Larsson SC, et al. Coffee consumption and mortality from all causes, cardiovascular disease, and cancer: a dose-response meta-analysis. Am J Epidemiol. 2014;180:763-775.
37. Loftfield E, Freedman ND, Graubard BI, et al. Association of coffee consumption with overall and cause-specific mortality in a large US prospective cohort study. Am J Epidemiol. 2015;182:1010-1022.
38. Lopez-Garcia E, Rodriguez-Artalejo F, Rexrode KM, et al. Coffee consumption and risk of stroke in women. Circulation. 2009;119:1116-1123.
39. Friedrich K, Smit M, Wannhoff A, et al. Coffee consumption protects against progression in liver cirrhosis and increases long-term survival after liver transplantation. J Gastroenterol Hepatol. 2016;31:1470-1475.
40. Wang L, Shen X, Wu Y, et al. Coffee and caffeine consumption and depression: a meta-analysis of observational studies. Aust N Z J Psychiatry. 2016;50:228-242.
41. Liu F, Wang X, Wu G, et al. Coffee consumption decreases risks for hepatic fibrosis and cirrhosis: a meta-analysis. PLoS One. 2015;10:e0142457.
42. Grosso G, Micek A, Castellano S, et al. Coffee, tea, caffeine and risk of depression: a systematic review and dose-response meta-analysis of observational studies. Mol Nutr Food Res. 2016;60:223-234.
43. Solfrizzi V, Panza F, Imbimbo BP, et al. Italian longitudinal study on aging working group. Coffee consumption habits and the risk of mild cognitive impairment: The Italian Longitudinal Study on Aging. J Alzheimers Dis. 2015;47:889-899.
44. Sugiyama K, Tomata Y, Kaiho Y, et al. Association between coffee consumption and incident risk of disabling dementia in elderly japanese: The Ohsaki Cohort 2006 Study. J Alzheimers Dis. 2015;50:491-500.
45. Driscoll I, Shumaker SA, Snively BM, et al. Relationships between caffeine intake and risk for probable dementia or global cognitive impairment: The Women’s Health Initiative Memory Study. J Gerontol A Biol Sci Med Sci. 2016;71:1596-1602.
46. van Dieren S, Uiterwaal CS, van der Schouw YT, et al. Coffee and tea consumption and risk of type 2 diabetes. Diabetologia. 2009;52:2561-2569.
47. Wang J, Li X, Zhang D. Coffee consumption and the risk of cutaneous melanoma: a meta-analysis. Eur J Nutr. 2016;55:1317-1329.
48. Liu J, Shen B, Shi M, et al. Higher caffeinated coffee intake is associated with reduced malignant melanoma risk: a meta-analysis study. PLoS One. 2016;11:e0147056.
49. Wikoff D, Welsh BT, Henderson R, et al. Systematic review of the potential adverse effects of caffeine consumption in healthy adults, pregnant women, adolescents, and children. Food Chem Toxical. 2017;109(Pt 1):585-648.
50. Verna R. The history and science of chocolate. Malays J Pathol. 2013;35:111-121.
51. Katz DL, Doughty K, Ali A. Cocoa and chocolate in human health and disease. Antioxid Redox Signal. 2011;15:2779-2811.
52. Baba S, Natsume M, Yasuda A, et al. Plasma LDL and HDL cholesterol and oxidized LDL concentrations are altered in normo- and hypercholesterolemic humans after intake of different levels of cocoa powder. J Nutr. 2007;137:1436-1441.
53. Mellor DD, Sathyapalan T, Kilpatrick ES, et al. High-cocoa polyphenol-rich chocolate improves HDL cholesterol in type 2 diabetes patients. Diabet Med. 2010;27:1318-1321.
54. Tokede OA, Gaziano JM, Djoussé L. Effects of cocoa products/dark chocolate on serum lipids: a meta-analysis. Eur J Clin Nutr. 2011;65:879-886.
55. Jia L, Liu X, Bai YY, et al. Short-term effect of cocoa product consumption on lipid profile: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2010;92:218-225.
56. Rostami A, Khalili M, Haghighat N, et al. High-cocoa polyphenol-rich chocolate improves blood pressure in patients with diabetes and hypertension. ARYA Atheroscler. 2015;11:21-29.
57. Buitrago-Lopez A, Sanderson J, Johnson L, et al. Chocolate consumption and cardiometabolic disorders: systematic review and meta-analysis. BMJ. 2011;26;343:d4488.
58. Wang X, Ouyang YY, Liu J, et al. Flavonoid intake and risk of CVD: a systematic review and meta-analysis of prospective cohort studies. Br J Nutr. 2014;111:1-11.
59. Zomer E, Owen A, Magliano DJ, et al. The effectiveness and cost effectiveness of dark chocolate consumption as prevention therapy in people at high risk of cardiovascular disease: best case scenario analysis using a Markov model. BMJ. 2012;344:e3657.
60. Larsson SC, Virtamo J, Wolk A. Chocolate consumption and risk of stroke: a prospective cohort of men and meta-analysis. Neurology. 2012;79:1223-1229.
61. Khawaja O, Gaziano JM, Djoussé L. Chocolate and coronary heart disease: a systematic review. Curr Atheroscler Rep. 2011;13:447-452.
62. Jacques PF, Cassidy A, Rogers G, et al. Dietary flavonoid intakes and CVD incidence in the Framingham Offspring Cohort. Br J Nutr. 2015;114:1496-1503.
63. Taubert D, Roesen R, Lehmann C, et al. Effects of low habitual cocoa intake on blood pressure and bioactive nitric oxide: a randomized controlled trial. JAMA. 2007;298:49-60.
64. Desch S, Schmidt J, Kobler D, et al. Effect of cocoa products on blood pressure: systematic review and meta-analysis. Am J Hypertens. 2010;23:97-103.
65. Ried K, Sullivan TR, Fakler P, et al. Effect of cocoa on blood pressure. Cochrane Database Syst Rev. 2012;8:CD008893.
66. Saftlas AF, Triche EW, Beydoun H, et al. Does chocolate intake during pregnancy reduce the risks of preeclampsia and gestational hypertension? Ann Epidemiol. 2010;20:584-591.
67. Triche EW, Grosso LM, Belanger K, et al. Chocolate consumption in pregnancy and reduced likelihood of preeclampsia. Epidemiology. 2008;19:459-464.
68. Grassi D, Lippi C, Necozione S, et al. Short-term administration of dark chocolate is followed by a significant increase in insulin sensitivity and a decrease in blood pressure in healthy persons. Am J Clin Nutr. 2005;81:611-614.
69. Matsumoto C, Petrone AB, Sesso HD, et al. Chocolate consumption and risk of diabetes mellitus in the Physicians’ Health Study. Am J Clin Nutr. 2015;101:362-367.
70. Lippi D. Chocolate in history: food, medicine, medi-food. Nutrients. 2013;5:1573-1584.
71. Sathyapalan T, Beckett S, Rigby AS, et al. High cocoa polyphenol rich chocolate may reduce the burden of the symptoms in chronic fatigue syndrome. Nutr J. 2010;9:55.
72. Martin FP, Antille N, Rezzi S, et al. Everyday eating experiences of chocolate and non-chocolate snacks impact postprandial anxiety, energy and emotional states. Nutrients. 2012;4:554-567.
73. Patel S, Mathan JJ, Vaghefi E, et al. The effect of flavonoids on visual function in patients with glaucoma or ocular hypertension: a systematic review and meta-analysis. Graefes Arch Clin Exp Ophthalmol. 2015;253:1841-1850.
74. Moreira A, Diógenes MJ, de Mendonça A, et al. Chocolate consumption is associated with a lower risk of cognitive decline. J Alzheimers Dis. 2016;53:85-93.
More than a third of American adults use complementary and alternative medicine.1 Unfortunately, the public’s enthusiasm for herbal products is not always consistent with the scientific evidence supporting their use. In part one of this series, we discussed the studies that have been done on capsaicin, butterbur, green tea, and peppermint. In this installment, we outline the research on 5 additional remedies: turmeric/curcumin, which may be of benefit in ulcerative colitis; chamomile, which appears to offer relief to patients with anxiety; rosemary, which may help treat alopecia; as well as coffee and cocoa, which may have some cardiovascular benefits (TABLE).
Turmeric/curcumin
Overview
Turmeric (Curcuma longa), a relative of ginger, has been used for 4000 years to treat a variety of conditions.2,3 Curcumin is the yellow pigment isolated from the rhizomes of Curcuma longa, commonly known as turmeric.3 Turmeric powder contains 5% curcumin, which is the main biologically active compound. Although it grows in many tropical locations, most turmeric is grown in India, where it is used as a main ingredient in curry. The roots and bulbs of turmeric that are used in medicine are generally boiled and dried, which results in a yellow powder.
Turmeric has been used in both Ayurvedic and Chinese medicine for its anti-inflammatory properties, in the treatment of digestive and liver problems, to fight infections, and to help heal skin diseases and wounds.3-7
Functional GI disorders. A recent review noted that curcumin has been shown in several preclinical studies and uncontrolled clinical trials to have effects on gut inflammation, gut permeability, and the brain-gut axis, especially in functional GI disorders.7 A double-blind, placebo-controlled study from 1989 found that turmeric reduced symptoms of bloating and gas in subjects suffering from undifferentiated dyspepsia.8
Ulcerative colitis (UC). A 2012 Cochrane review noted that curcumin appears to be a safe and effective therapy for maintenance of remission in quiescent UC when given as adjunctive therapy along with mesalamine or sulfasalazine.9 In a 2015 randomized controlled trial (RCT), the addition of curcumin to mesalamine therapy was superior to the combination of placebo and mesalamine in inducing clinical and endoscopic remission in patients with mild-to-moderate active UC, producing no apparent adverse effects.10
Osteoarthritis (OA). Because of turmeric’s ability to reduce inflammation, it may help relieve OA pain.3 Clinical evidence is scant for the anti-arthritic efficacy of turmeric dietary supplements, although animal studies indicate that turmeric prevents inflammation through regulation of NF-kappaB-regulated genes that regulate the immune and inflammatory response.6 Inflammatory cell influx, joint levels of prostaglandin E2, and periarticular osteoclast formation were also inhibited by turmeric extract treatment.6
A 2013 review of turmeric for OA concluded that observational studies and in vitro results are promising for the use of curcumin for OA, but well-designed clinical studies were lacking and are needed to support the efficacy of curcumin in OA patients.11 However, in a 2014 randomized trial of 367 patients, turmeric appeared to be similar in efficacy to ibuprofen for the treatment of pain and disability in adults with knee OA.12 The curcumin (turmeric) group also had fewer adverse effects.12
Cancer. There has been a great deal of research on turmeric’s anti-cancer properties, but clinical evidence is lacking. In vitro evidence, animal studies, and small clinical trials suggest that curcumin may help prevent or treat several types of cancers, but the overall evidence is poor. Nonetheless, curcumin and turmeric have been or are currently being evaluated for the treatment or prevention of prostate, liver, breast, skin, gynecologic, hematologic, pulmonary, thymic, bone, brain, and colon cancer.13-18
Oral submucous fibrosis. A small randomized trial found improvement in oral function with curcumin lozenges, when compared to placebo, indicating that turmeric may hold promise as a treatment of oral submucous fibrosis.19
Uveitis. A small pilot study of 32 patients suggested that oral curcumin may be as effective as corticosteroids for uveitis.20
Heart disease. Curcumin may have a cardiovascular protective role, as it has been shown to reduce atherosclerosis, but a reduction in myocardial infarction or stroke has not been documented.21
Alzheimer’s dementia. Animal studies have shown a reduction in amyloid plaque formation with curcumin.22
Adverse effects (and precautions)
Turmeric in food is considered safe. A variety of animal and human studies have also indicated that curcumin is safe and well tolerated, even at very high doses.13 However, taking large amounts of turmeric for long periods of time could cause stomach upset and gastric ulcers. In addition, patients with gallstones or bile obstruction should use it with caution due to increased bile production.7
Because turmeric may lower blood sugar levels, patients with diabetes should monitor for hypoglycemia when using turmeric in combination with diabetic medications. Similarly, those with bleeding disorders taking blood thinners should use turmeric and curcumin with caution, because it can inhibit platelet aggregation.23
Although it is safe to eat foods with turmeric during pregnancy, pregnant and breastfeeding women should not take turmeric supplements, as the safety of large doses in pregnancy is unknown.
The bottom line
Turmeric/curcumin has anti-inflammatory properties and may be useful as an adjunct for ulcerative colitis and to improve the symptoms of OA. It may also have anti-carcinogenic properties, although definitive data are lacking. Those with a history of gastrointestinal conditions such as gastric ulcer, patients taking blood thinners, and patients with diabetes who are prone to low blood sugar levels should use turmeric/curcumin with caution.
Chamomile
Overview
Chamomile, a member of the Asteraceae/Compositae family, is one of the oldest herbal medicines. It has been used for hay fever, inflammation, muscle spasms, menstrual disorders, insomnia, ulcers, wounds, gastrointestinal disorders, rheumatic pain, and hemorrhoids. Essential oils of chamomile are used extensively in cosmetics and aromatherapy. Many different preparations have been developed, the most popular being herbal tea.24
Individuals with a hypersensitivity to plants of the Asteraceae (Compositae) family such as ragweed (Ambrosia spp.), marigold flower (Calendula officinalis), and chrysanthemum (Chrysanthemum spp.) may show a similar reaction to chamomile.25
Anxiety. A controlled clinical trial of chamomile extract for generalized anxiety disorder (GAD) suggested that it may have modest anxiolytic activity in patients with mild to moderate GAD.26 Another randomized, double-blind, placebo-controlled trial found oral chamomile extract was efficacious and well-tolerated in patients experiencing mild to moderate GAD and may provide an alternative therapeutic anxiolytic for patients with mild GAD.25 In addition to its anxiolytic activity, chamomile may also provide clinically meaningful antidepressant activity.26
Insomnia. Chamomile may have some impact on sleep diary measures (total sleep time, sleep efficiency, sleep latency, wake after sleep onset, sleep quality, and number of awakenings) relative to placebo in adults with chronic primary insomnia, according to a small randomized, double-blind, placebo-controlled pilot trial involving 34 patients.27 However, a systematic review found no statistically significant difference between any herbal medicine (including chamomile) and placebo, for clinical efficacy in patients with insomnia. A similar, or smaller, number of adverse events per person were reported with chamomile compared with placebo, suggesting safe use.28
Infantile colic. A small prospective double-blind study on the use of chamomile-containing tea on infantile colic showed statistically significant symptom improvement in tea-treated infants. The study did note, however, that prolonged ingestion of herbal teas may lead to decreased milk intake.29,30
Adverse effects
As noted earlier, a systematic review found that the number of adverse events per person reported with chamomile was comparable to the number associated with placebo, suggesting that it is safe.28
The bottom line
Chamomile appears to be safe with minimal adverse effects and may be effective for the treatment of anxiety, insomnia, and infantile colic.
Rosemary
Overview
Rosemary, officially known as Rosmarinus officinalis, is a medicinal evergreen plant native to the Mediterranean area that appears to increase microcapillary perfusion.31
Alopecia. A randomized double-blind controlled trial found that essential oils including rosemary oil (as well as thyme, lavender, and cedarwood) massaged into the scalp improved hair growth in almost half of patients with alopecia areata after 7 months.32 Another randomized trial comparing rosemary oil to minoxidil 2% for androgenetic alopecia showed a significant increase in hair count at the 6-month endpoint compared with the baseline, but no significant difference was found between the study groups regarding hair count either at Month 3 or Month 6 (P >.05). 31
Adverse effects
In the randomized trial described above comparing rosemary oil to minoxidil 2%, adverse effects appeared to be rare for topical rosemary oil. Scalp itching was more frequent in the minoxidil group.31
The bottom line
Topical rosemary oil may be useful in the treatment of alopecia with minimal adverse effects.
Coffee/caffeine
Overview
Coffee is one of the most widely used botanicals with approximately 3.5 billion cups of coffee consumed per day worldwide. It is a popular beverage because of its unique aromatic taste and its use as a central nervous system stimulant. The coffee tree (genus coffea) is found throughout Latin America, Africa, and eastern Asia. Two of the most common commercially grown species are Coffea arabica (Arabicas) and Coffea canephora (Robusta). Processing and roasting methods may differ and produce variations in flavor and aroma. The degree of roasting also affects the caffeine content.
Coffee consumption leads to increased alertness and can boost mental performance. Based on the literature and US Food and Drug Administration recommendations, four 8-oz cups of coffee (about 400 mg of caffeine) daily is an acceptable average amount of caffeine. More than 500 mg/d is considered excessive use of coffee.33,34
Overall mortality. A 2008 study showed that regular coffee was not associated with increased or decreased mortality in both men and women.35 However, more recent studies show an inverse relationship between mortality and coffee consumption.
Specifically, a 2014 meta-analysis found an inverse relationship between coffee and mortality.36 A large prospective cohort study from 2015 that included 79,234 women and 76,704 men found that drinking coffee was inversely associated with overall mortality.37 In this cohort study, an inverse association were observed for deaths from heart disease, respiratory disease, diabetes, and self-harm.37 While mechanisms were not analyzed, coffee may reduce mortality risk by affecting inflammation, lung function, insulin sensitivity, and depression.
Cardiovascular disease. Coffee consumption may modestly reduce the risk of stroke, according to a prospective cohort study of 83,076 women from the Nurses’ Health Study who were followed for 24 years.38 Reduced cardiovascular mortality was also found in a large prospective cohort study, as noted in the mortality discussion above.37 A 2014 meta-analysis concluded that coffee consumption is inversely associated with cardiovascular mortality. Drinking 3 or 4 cups a day appears to be the amount that may decrease one’s risk of death when compared to those who do not drink coffee at all.36
Liver disease. Friedrich et al performed a study involving 379 patients with end stage liver disease, and found that coffee consumption delayed the progression of disease in patients with both alcoholic liver disease and primary sclerosing cholangitis.39 Coffee consumption also increased long-term survival after liver transplantation.39 However, the study found that coffee did not have any effect on patients with chronic viral hepatitis.
In a 2016 meta-analysis, caffeinated coffee consumption reduced hepatic fibrosis of nonalcoholic fatty liver disease, although caffeine consumption did not reduce the prevalence of nonalcoholic fatty liver disease.40 Another meta-analysis, including 16 studies, also found caffeine reduced the risk for hepatic fibrosis and cirrhosis.41
Depression. Based on 2 different systematic reviews and meta-analyses from 2016, coffee consumption appears to have a significant protective effect, decreasing the risk of developing depression.40,42
Alzheimer’s disease/dementia. Coffee, tea, and caffeine consumption show promise in reducing the risk of cognitive decline and dementia. Individuals who consume one to 2 cups of coffee per day had a decreased incidence of mild cognitive impairment compared to non-drinkers.43 A 2015 Japanese study also found an inverse association between coffee consumption and dementia among women, nonsmokers, and those who do not drink alcohol.44 Most recently, a 2016 study, the Women’s Health Initiative Memory Study, looked at incident dementia rates in women >65 years of age with high vs low caffeine intake. Women with higher caffeine intake were less likely to develop dementia or any cognitive impairment compared with those consuming <64 mg/day.45
Type 2 diabetes. A 2009 prospective cohort study, which included 40,011 participants followed for more than 10 years, found that drinking at least 3 cups of coffee or tea was associated with a lowered risk of type 2 diabetes.46 A 2009 systematic review of 20 cohort studies showed that high intakes of coffee, decaffeinated coffee, and tea are associated with a reduced risk of diabetes.47
Melanoma.
Adverse effects
Despite the many potential benefits of coffee, caffeine is a potent drug that should be used with caution.49 People with underlying heart problems should avoid caffeine due to concern that it may cause palpitations from tachycardia. It may worsen anxiety problems or depression. Coffee may increase the production of stomach acids, which can worsen acid reflux or stomach ulcers.
Caffeine is a potent diuretic and may decrease absorption of calcium and cause OA. Caffeine may cause dependence and withdrawal symptoms. Some of the symptoms of withdrawal include drowsiness, headaches, irritability, nausea, and vomiting. It may disrupt sleeping patterns by causing jitters and sleeplessness.49 Additionally, large amounts of caffeine may cause overdose and death.
The bottom line
Regular coffee intake is associated with a lower risk of mortality, reduced cardiovascular events, and a reduction in liver disease progression. Coffee may also have some utility for improving cognitive function and reducing the risk of type 2 diabetes. Caffeinated coffee should be limited to no more than 32 oz per day, due to the risk of insomnia, palpitations, anxiety, and gastritis.
Chocolate/cocoa
Overview
Few natural products have been claimed to successfully treat as many disorders as chocolate. The modern concept of chocolate as food has overshadowed its traditional medicinal use, although recent trials have looked at evidence for some of its traditional uses. Chocolate is processed from the pod of the cacao plant. The earliest evidence for its medical use is in Mayan civilizations, and for most of its approximately 4000-year history, chocolate was consumed as a bitter drink referred to as the “drink of the Gods.” The traditional drink was mixed with water, vanilla, honey, chili peppers, and other spices. Important components in chocolate include flavonoids (antioxidants), cocoa butter, caffeine, theobromine, and phenylethylamine.
Chocolate has stimulating, anti-inflammatory, neuroprotective, and cardioprotective effects, and improves the bioavailability of nitric oxide, which can improve blood pressure and platelet function.50 Epicatechin (an antioxidant) in cocoa is primarily responsible for its favorable impact on vascular endothelium via its effect on both acute and chronic upregulation of nitric oxide production. Other cardiovascular effects are mediated by the anti-inflammatory effects of cocoa polyphenols, and modulated through the activity of NF-kappaB.51
Dark chocolate appears to have the greatest benefit, as milk binds to antioxidants in chocolate, making them unavailable. Therefore, milk chocolate is not a good antioxidant source. There is no specific amount of chocolate that is known to be ideal, but an average of one to 2 ounces per day is often used in studies.
Cardiovascular effects. Chocolate does contain saturated fat, but a comparative, double-blind study found that short-term use of cocoa powder lowered plasma low-density lipoprotein (LDL) cholesterol, oxidized LDL, and apo B concentrations, and the plasma high-density lipoprotein (HDL) cholesterol concentration increased, relative to baseline in the low-, middle-, and high-cocoa groups.52 A small randomized crossover trial without clinical outcomes indicated that chocolate may increase HDL cholesterol without increasing weight.53
A meta-analysis of short-term (2-12 weeks) treatment with dark chocolate/cocoa products showed reductions in LDL and total cholesterol, but no changes in HDL or triglycerides.54 Another meta-analysis of RCTs, however, showed no short-term effect of cocoa/chocolate on lipid concentrations.55 A randomized, placebo-controlled double-blind study of 62 patients with diabetes and hypertension showed that high polyphenol chocolate improved triglyceride levels.56
Multiple studies have shown that chocolate is associated with a reduction in cardiovascular risk.57-59 A best case scenario analysis using a Markov model to predict the long-term effectiveness and cost effectiveness of daily dark chocolate consumption in a population with metabolic syndrome at high risk of cardiovascular disease concluded that daily consumption of dark chocolate can reduce cardiovascular events by 85 per 10,000 population treated over 10 years. The study concluded that $42 could be cost effectively spent per person per year on prevention strategies using dark chocolate.59
In addition, a meta-analysis of 7 observational studies showed that high levels of chocolate consumption (any type) were associated with a 29% reduction in stroke compared with the lowest levels of chocolate intake.57 Results of a similar meta-analysis from Neurology in 2012 also suggested that moderate chocolate consumption (any type) may lower the risk of stroke.60
That said, 2 systematic reviews specifically relating to the risk of coronary heart disease and chocolate intake were inconclusive.61-62
Blood pressure (BP). An RCT published in JAMA indicates that inclusion of small amounts of polyphenol-rich dark chocolate as part of a usual diet efficiently reduced BP and improved the formation of vasodilative nitric oxide.63 A meta-analysis of 10 RCTs also showed mean BP change in the active cocoa treatment arms across all trials was -4.5 mm Hg (95% confidence interval (CI), -5.9 to -3.2; P<.001) for systolic BP and -2.5 mm Hg (95% CI, -3.9 to -1.2; P<.001) for diastolic BP.64
A Cochrane Review meta-analysis of 20 studies revealed a statistically significant BP-reducing effect of flavanol-rich cocoa products compared with control in short-term trials of 2 to 18 weeks' duration.65 Because studies have shown improvement in BP with chocolate intake, investigations into a role of chocolate in the prevention of preeclampsia have been undertaken. In some studies, chocolate intake was associated with reduced odds of preeclampsia and gestational hypertension.66,67
Diabetes. Chocolate may exert significant vascular protection because of its antioxidant properties and possible increase of nitric oxide bioavailability, which can influence glucose uptake. A small trial comparing the effects of either dark or white chocolate bars (which do not contain the polyphenols) showed improved BP and glucose and insulin responses to an oral glucose tolerance test in healthy subjects on dark chocolate, but not white chocolate.68 A comparison of chocolate consumption and risk of diabetes in the Physicians’ Health Study showed an inverse relationship between chocolate intake with incident disease, but this association appeared only to apply in younger and normal-body weight men after controlling for comprehensive lifestyles, including total energy consumption.69
Fatigue. The effect of chocolate on a person’s energy level has been noted for centuries.70 A small randomized trial showed improved energy levels in those treated with higher chocolate intakes. In a double-blind, randomized, clinical pilot crossover study, high cocoa liquor/polyphenol rich chocolate, reduced fatigue in subjects with chronic fatigue syndrome.71
Anxiety. A small randomized trial showed chocolate decreased anxiety in high-anxiety trait subjects and improved the anxiety level and the energy levels of low-anxiety trait participants.72
Eye effects. The literature presents conflicting evidence regarding the effect of flavonoids on patients with glaucoma and ocular hypertension. However, a meta-analysis showed that flavonoids have a promising role in improving visual function in patients with glaucoma and ocular hypertension, and appear to play a part in both improving and slowing the progression of visual field loss.73
Cognitive decline. Chocolate intake (any type) was associated with a lower risk of cognitive decline (RR = 0.59; 95% CI, 0.38-0.92) with the greatest benefit noted in those who averaged more than one chocolate bar or one tablespoon of cocoa powder per week. This protective effect was observed only among subjects with an average daily consumption of caffeine <75 mg (69% of the participants; RR = 0.50; 95% CI, 0.31-0.82).74
The bottom line
Chocolate with high cocoa content (dark chocolate) appears to be safe and beneficial as part of a healthy diet and lifestyle that includes exercise and stress reduction to decrease cardiovascular risk and may improve energy levels.
CORRESPONDENCE
Michael Malone, MD, Family and Community Medicine, Penn State College of Medicine, 500 University Drive, Hershey, PA 17033; [email protected].
More than a third of American adults use complementary and alternative medicine.1 Unfortunately, the public’s enthusiasm for herbal products is not always consistent with the scientific evidence supporting their use. In part one of this series, we discussed the studies that have been done on capsaicin, butterbur, green tea, and peppermint. In this installment, we outline the research on 5 additional remedies: turmeric/curcumin, which may be of benefit in ulcerative colitis; chamomile, which appears to offer relief to patients with anxiety; rosemary, which may help treat alopecia; as well as coffee and cocoa, which may have some cardiovascular benefits (TABLE).
Turmeric/curcumin
Overview
Turmeric (Curcuma longa), a relative of ginger, has been used for 4000 years to treat a variety of conditions.2,3 Curcumin is the yellow pigment isolated from the rhizomes of Curcuma longa, commonly known as turmeric.3 Turmeric powder contains 5% curcumin, which is the main biologically active compound. Although it grows in many tropical locations, most turmeric is grown in India, where it is used as a main ingredient in curry. The roots and bulbs of turmeric that are used in medicine are generally boiled and dried, which results in a yellow powder.
Turmeric has been used in both Ayurvedic and Chinese medicine for its anti-inflammatory properties, in the treatment of digestive and liver problems, to fight infections, and to help heal skin diseases and wounds.3-7
Functional GI disorders. A recent review noted that curcumin has been shown in several preclinical studies and uncontrolled clinical trials to have effects on gut inflammation, gut permeability, and the brain-gut axis, especially in functional GI disorders.7 A double-blind, placebo-controlled study from 1989 found that turmeric reduced symptoms of bloating and gas in subjects suffering from undifferentiated dyspepsia.8
Ulcerative colitis (UC). A 2012 Cochrane review noted that curcumin appears to be a safe and effective therapy for maintenance of remission in quiescent UC when given as adjunctive therapy along with mesalamine or sulfasalazine.9 In a 2015 randomized controlled trial (RCT), the addition of curcumin to mesalamine therapy was superior to the combination of placebo and mesalamine in inducing clinical and endoscopic remission in patients with mild-to-moderate active UC, producing no apparent adverse effects.10
Osteoarthritis (OA). Because of turmeric’s ability to reduce inflammation, it may help relieve OA pain.3 Clinical evidence is scant for the anti-arthritic efficacy of turmeric dietary supplements, although animal studies indicate that turmeric prevents inflammation through regulation of NF-kappaB-regulated genes that regulate the immune and inflammatory response.6 Inflammatory cell influx, joint levels of prostaglandin E2, and periarticular osteoclast formation were also inhibited by turmeric extract treatment.6
A 2013 review of turmeric for OA concluded that observational studies and in vitro results are promising for the use of curcumin for OA, but well-designed clinical studies were lacking and are needed to support the efficacy of curcumin in OA patients.11 However, in a 2014 randomized trial of 367 patients, turmeric appeared to be similar in efficacy to ibuprofen for the treatment of pain and disability in adults with knee OA.12 The curcumin (turmeric) group also had fewer adverse effects.12
Cancer. There has been a great deal of research on turmeric’s anti-cancer properties, but clinical evidence is lacking. In vitro evidence, animal studies, and small clinical trials suggest that curcumin may help prevent or treat several types of cancers, but the overall evidence is poor. Nonetheless, curcumin and turmeric have been or are currently being evaluated for the treatment or prevention of prostate, liver, breast, skin, gynecologic, hematologic, pulmonary, thymic, bone, brain, and colon cancer.13-18
Oral submucous fibrosis. A small randomized trial found improvement in oral function with curcumin lozenges, when compared to placebo, indicating that turmeric may hold promise as a treatment of oral submucous fibrosis.19
Uveitis. A small pilot study of 32 patients suggested that oral curcumin may be as effective as corticosteroids for uveitis.20
Heart disease. Curcumin may have a cardiovascular protective role, as it has been shown to reduce atherosclerosis, but a reduction in myocardial infarction or stroke has not been documented.21
Alzheimer’s dementia. Animal studies have shown a reduction in amyloid plaque formation with curcumin.22
Adverse effects (and precautions)
Turmeric in food is considered safe. A variety of animal and human studies have also indicated that curcumin is safe and well tolerated, even at very high doses.13 However, taking large amounts of turmeric for long periods of time could cause stomach upset and gastric ulcers. In addition, patients with gallstones or bile obstruction should use it with caution due to increased bile production.7
Because turmeric may lower blood sugar levels, patients with diabetes should monitor for hypoglycemia when using turmeric in combination with diabetic medications. Similarly, those with bleeding disorders taking blood thinners should use turmeric and curcumin with caution, because it can inhibit platelet aggregation.23
Although it is safe to eat foods with turmeric during pregnancy, pregnant and breastfeeding women should not take turmeric supplements, as the safety of large doses in pregnancy is unknown.
The bottom line
Turmeric/curcumin has anti-inflammatory properties and may be useful as an adjunct for ulcerative colitis and to improve the symptoms of OA. It may also have anti-carcinogenic properties, although definitive data are lacking. Those with a history of gastrointestinal conditions such as gastric ulcer, patients taking blood thinners, and patients with diabetes who are prone to low blood sugar levels should use turmeric/curcumin with caution.
Chamomile
Overview
Chamomile, a member of the Asteraceae/Compositae family, is one of the oldest herbal medicines. It has been used for hay fever, inflammation, muscle spasms, menstrual disorders, insomnia, ulcers, wounds, gastrointestinal disorders, rheumatic pain, and hemorrhoids. Essential oils of chamomile are used extensively in cosmetics and aromatherapy. Many different preparations have been developed, the most popular being herbal tea.24
Individuals with a hypersensitivity to plants of the Asteraceae (Compositae) family such as ragweed (Ambrosia spp.), marigold flower (Calendula officinalis), and chrysanthemum (Chrysanthemum spp.) may show a similar reaction to chamomile.25
Anxiety. A controlled clinical trial of chamomile extract for generalized anxiety disorder (GAD) suggested that it may have modest anxiolytic activity in patients with mild to moderate GAD.26 Another randomized, double-blind, placebo-controlled trial found oral chamomile extract was efficacious and well-tolerated in patients experiencing mild to moderate GAD and may provide an alternative therapeutic anxiolytic for patients with mild GAD.25 In addition to its anxiolytic activity, chamomile may also provide clinically meaningful antidepressant activity.26
Insomnia. Chamomile may have some impact on sleep diary measures (total sleep time, sleep efficiency, sleep latency, wake after sleep onset, sleep quality, and number of awakenings) relative to placebo in adults with chronic primary insomnia, according to a small randomized, double-blind, placebo-controlled pilot trial involving 34 patients.27 However, a systematic review found no statistically significant difference between any herbal medicine (including chamomile) and placebo, for clinical efficacy in patients with insomnia. A similar, or smaller, number of adverse events per person were reported with chamomile compared with placebo, suggesting safe use.28
Infantile colic. A small prospective double-blind study on the use of chamomile-containing tea on infantile colic showed statistically significant symptom improvement in tea-treated infants. The study did note, however, that prolonged ingestion of herbal teas may lead to decreased milk intake.29,30
Adverse effects
As noted earlier, a systematic review found that the number of adverse events per person reported with chamomile was comparable to the number associated with placebo, suggesting that it is safe.28
The bottom line
Chamomile appears to be safe with minimal adverse effects and may be effective for the treatment of anxiety, insomnia, and infantile colic.
Rosemary
Overview
Rosemary, officially known as Rosmarinus officinalis, is a medicinal evergreen plant native to the Mediterranean area that appears to increase microcapillary perfusion.31
Alopecia. A randomized double-blind controlled trial found that essential oils including rosemary oil (as well as thyme, lavender, and cedarwood) massaged into the scalp improved hair growth in almost half of patients with alopecia areata after 7 months.32 Another randomized trial comparing rosemary oil to minoxidil 2% for androgenetic alopecia showed a significant increase in hair count at the 6-month endpoint compared with the baseline, but no significant difference was found between the study groups regarding hair count either at Month 3 or Month 6 (P >.05). 31
Adverse effects
In the randomized trial described above comparing rosemary oil to minoxidil 2%, adverse effects appeared to be rare for topical rosemary oil. Scalp itching was more frequent in the minoxidil group.31
The bottom line
Topical rosemary oil may be useful in the treatment of alopecia with minimal adverse effects.
Coffee/caffeine
Overview
Coffee is one of the most widely used botanicals with approximately 3.5 billion cups of coffee consumed per day worldwide. It is a popular beverage because of its unique aromatic taste and its use as a central nervous system stimulant. The coffee tree (genus coffea) is found throughout Latin America, Africa, and eastern Asia. Two of the most common commercially grown species are Coffea arabica (Arabicas) and Coffea canephora (Robusta). Processing and roasting methods may differ and produce variations in flavor and aroma. The degree of roasting also affects the caffeine content.
Coffee consumption leads to increased alertness and can boost mental performance. Based on the literature and US Food and Drug Administration recommendations, four 8-oz cups of coffee (about 400 mg of caffeine) daily is an acceptable average amount of caffeine. More than 500 mg/d is considered excessive use of coffee.33,34
Overall mortality. A 2008 study showed that regular coffee was not associated with increased or decreased mortality in both men and women.35 However, more recent studies show an inverse relationship between mortality and coffee consumption.
Specifically, a 2014 meta-analysis found an inverse relationship between coffee and mortality.36 A large prospective cohort study from 2015 that included 79,234 women and 76,704 men found that drinking coffee was inversely associated with overall mortality.37 In this cohort study, an inverse association were observed for deaths from heart disease, respiratory disease, diabetes, and self-harm.37 While mechanisms were not analyzed, coffee may reduce mortality risk by affecting inflammation, lung function, insulin sensitivity, and depression.
Cardiovascular disease. Coffee consumption may modestly reduce the risk of stroke, according to a prospective cohort study of 83,076 women from the Nurses’ Health Study who were followed for 24 years.38 Reduced cardiovascular mortality was also found in a large prospective cohort study, as noted in the mortality discussion above.37 A 2014 meta-analysis concluded that coffee consumption is inversely associated with cardiovascular mortality. Drinking 3 or 4 cups a day appears to be the amount that may decrease one’s risk of death when compared to those who do not drink coffee at all.36
Liver disease. Friedrich et al performed a study involving 379 patients with end stage liver disease, and found that coffee consumption delayed the progression of disease in patients with both alcoholic liver disease and primary sclerosing cholangitis.39 Coffee consumption also increased long-term survival after liver transplantation.39 However, the study found that coffee did not have any effect on patients with chronic viral hepatitis.
In a 2016 meta-analysis, caffeinated coffee consumption reduced hepatic fibrosis of nonalcoholic fatty liver disease, although caffeine consumption did not reduce the prevalence of nonalcoholic fatty liver disease.40 Another meta-analysis, including 16 studies, also found caffeine reduced the risk for hepatic fibrosis and cirrhosis.41
Depression. Based on 2 different systematic reviews and meta-analyses from 2016, coffee consumption appears to have a significant protective effect, decreasing the risk of developing depression.40,42
Alzheimer’s disease/dementia. Coffee, tea, and caffeine consumption show promise in reducing the risk of cognitive decline and dementia. Individuals who consume one to 2 cups of coffee per day had a decreased incidence of mild cognitive impairment compared to non-drinkers.43 A 2015 Japanese study also found an inverse association between coffee consumption and dementia among women, nonsmokers, and those who do not drink alcohol.44 Most recently, a 2016 study, the Women’s Health Initiative Memory Study, looked at incident dementia rates in women >65 years of age with high vs low caffeine intake. Women with higher caffeine intake were less likely to develop dementia or any cognitive impairment compared with those consuming <64 mg/day.45
Type 2 diabetes. A 2009 prospective cohort study, which included 40,011 participants followed for more than 10 years, found that drinking at least 3 cups of coffee or tea was associated with a lowered risk of type 2 diabetes.46 A 2009 systematic review of 20 cohort studies showed that high intakes of coffee, decaffeinated coffee, and tea are associated with a reduced risk of diabetes.47
Melanoma.
Adverse effects
Despite the many potential benefits of coffee, caffeine is a potent drug that should be used with caution.49 People with underlying heart problems should avoid caffeine due to concern that it may cause palpitations from tachycardia. It may worsen anxiety problems or depression. Coffee may increase the production of stomach acids, which can worsen acid reflux or stomach ulcers.
Caffeine is a potent diuretic and may decrease absorption of calcium and cause OA. Caffeine may cause dependence and withdrawal symptoms. Some of the symptoms of withdrawal include drowsiness, headaches, irritability, nausea, and vomiting. It may disrupt sleeping patterns by causing jitters and sleeplessness.49 Additionally, large amounts of caffeine may cause overdose and death.
The bottom line
Regular coffee intake is associated with a lower risk of mortality, reduced cardiovascular events, and a reduction in liver disease progression. Coffee may also have some utility for improving cognitive function and reducing the risk of type 2 diabetes. Caffeinated coffee should be limited to no more than 32 oz per day, due to the risk of insomnia, palpitations, anxiety, and gastritis.
Chocolate/cocoa
Overview
Few natural products have been claimed to successfully treat as many disorders as chocolate. The modern concept of chocolate as food has overshadowed its traditional medicinal use, although recent trials have looked at evidence for some of its traditional uses. Chocolate is processed from the pod of the cacao plant. The earliest evidence for its medical use is in Mayan civilizations, and for most of its approximately 4000-year history, chocolate was consumed as a bitter drink referred to as the “drink of the Gods.” The traditional drink was mixed with water, vanilla, honey, chili peppers, and other spices. Important components in chocolate include flavonoids (antioxidants), cocoa butter, caffeine, theobromine, and phenylethylamine.
Chocolate has stimulating, anti-inflammatory, neuroprotective, and cardioprotective effects, and improves the bioavailability of nitric oxide, which can improve blood pressure and platelet function.50 Epicatechin (an antioxidant) in cocoa is primarily responsible for its favorable impact on vascular endothelium via its effect on both acute and chronic upregulation of nitric oxide production. Other cardiovascular effects are mediated by the anti-inflammatory effects of cocoa polyphenols, and modulated through the activity of NF-kappaB.51
Dark chocolate appears to have the greatest benefit, as milk binds to antioxidants in chocolate, making them unavailable. Therefore, milk chocolate is not a good antioxidant source. There is no specific amount of chocolate that is known to be ideal, but an average of one to 2 ounces per day is often used in studies.
Cardiovascular effects. Chocolate does contain saturated fat, but a comparative, double-blind study found that short-term use of cocoa powder lowered plasma low-density lipoprotein (LDL) cholesterol, oxidized LDL, and apo B concentrations, and the plasma high-density lipoprotein (HDL) cholesterol concentration increased, relative to baseline in the low-, middle-, and high-cocoa groups.52 A small randomized crossover trial without clinical outcomes indicated that chocolate may increase HDL cholesterol without increasing weight.53
A meta-analysis of short-term (2-12 weeks) treatment with dark chocolate/cocoa products showed reductions in LDL and total cholesterol, but no changes in HDL or triglycerides.54 Another meta-analysis of RCTs, however, showed no short-term effect of cocoa/chocolate on lipid concentrations.55 A randomized, placebo-controlled double-blind study of 62 patients with diabetes and hypertension showed that high polyphenol chocolate improved triglyceride levels.56
Multiple studies have shown that chocolate is associated with a reduction in cardiovascular risk.57-59 A best case scenario analysis using a Markov model to predict the long-term effectiveness and cost effectiveness of daily dark chocolate consumption in a population with metabolic syndrome at high risk of cardiovascular disease concluded that daily consumption of dark chocolate can reduce cardiovascular events by 85 per 10,000 population treated over 10 years. The study concluded that $42 could be cost effectively spent per person per year on prevention strategies using dark chocolate.59
In addition, a meta-analysis of 7 observational studies showed that high levels of chocolate consumption (any type) were associated with a 29% reduction in stroke compared with the lowest levels of chocolate intake.57 Results of a similar meta-analysis from Neurology in 2012 also suggested that moderate chocolate consumption (any type) may lower the risk of stroke.60
That said, 2 systematic reviews specifically relating to the risk of coronary heart disease and chocolate intake were inconclusive.61-62
Blood pressure (BP). An RCT published in JAMA indicates that inclusion of small amounts of polyphenol-rich dark chocolate as part of a usual diet efficiently reduced BP and improved the formation of vasodilative nitric oxide.63 A meta-analysis of 10 RCTs also showed mean BP change in the active cocoa treatment arms across all trials was -4.5 mm Hg (95% confidence interval (CI), -5.9 to -3.2; P<.001) for systolic BP and -2.5 mm Hg (95% CI, -3.9 to -1.2; P<.001) for diastolic BP.64
A Cochrane Review meta-analysis of 20 studies revealed a statistically significant BP-reducing effect of flavanol-rich cocoa products compared with control in short-term trials of 2 to 18 weeks' duration.65 Because studies have shown improvement in BP with chocolate intake, investigations into a role of chocolate in the prevention of preeclampsia have been undertaken. In some studies, chocolate intake was associated with reduced odds of preeclampsia and gestational hypertension.66,67
Diabetes. Chocolate may exert significant vascular protection because of its antioxidant properties and possible increase of nitric oxide bioavailability, which can influence glucose uptake. A small trial comparing the effects of either dark or white chocolate bars (which do not contain the polyphenols) showed improved BP and glucose and insulin responses to an oral glucose tolerance test in healthy subjects on dark chocolate, but not white chocolate.68 A comparison of chocolate consumption and risk of diabetes in the Physicians’ Health Study showed an inverse relationship between chocolate intake with incident disease, but this association appeared only to apply in younger and normal-body weight men after controlling for comprehensive lifestyles, including total energy consumption.69
Fatigue. The effect of chocolate on a person’s energy level has been noted for centuries.70 A small randomized trial showed improved energy levels in those treated with higher chocolate intakes. In a double-blind, randomized, clinical pilot crossover study, high cocoa liquor/polyphenol rich chocolate, reduced fatigue in subjects with chronic fatigue syndrome.71
Anxiety. A small randomized trial showed chocolate decreased anxiety in high-anxiety trait subjects and improved the anxiety level and the energy levels of low-anxiety trait participants.72
Eye effects. The literature presents conflicting evidence regarding the effect of flavonoids on patients with glaucoma and ocular hypertension. However, a meta-analysis showed that flavonoids have a promising role in improving visual function in patients with glaucoma and ocular hypertension, and appear to play a part in both improving and slowing the progression of visual field loss.73
Cognitive decline. Chocolate intake (any type) was associated with a lower risk of cognitive decline (RR = 0.59; 95% CI, 0.38-0.92) with the greatest benefit noted in those who averaged more than one chocolate bar or one tablespoon of cocoa powder per week. This protective effect was observed only among subjects with an average daily consumption of caffeine <75 mg (69% of the participants; RR = 0.50; 95% CI, 0.31-0.82).74
The bottom line
Chocolate with high cocoa content (dark chocolate) appears to be safe and beneficial as part of a healthy diet and lifestyle that includes exercise and stress reduction to decrease cardiovascular risk and may improve energy levels.
CORRESPONDENCE
Michael Malone, MD, Family and Community Medicine, Penn State College of Medicine, 500 University Drive, Hershey, PA 17033; [email protected].
1. National Center for Complementary and Integrative Health. The use of complementary and alternative medicine in the United States. Available at: https://nccih.nih.gov/research/statistics/2007/camsurvey_fs1.htm. Accessed Nov 28, 2017.
2. Aggarwal BB. Curcumin-free turmeric exhibits anti-inflammatory and anticancer activities: identification of novel components of turmeric. Mol Nutr Food Res. 2013;57:1529-1542.
3. Henrotin Y, Clutterbuck AL, Allaway D, et al. Biological actions of curcumin on articular chondrocytes. Osteoarthritis Cartilage. 2010;18:141-149.
4. Asher GN, Spelman K. Clinical utility of curcumin extract. Altern Ther Health Med. 2013;19:20-22.
5. Phan TT, See P, Lee ST, et al. Protective effects of curcumin against oxidative damage on skin cells in vitro: its implication for wound healing. J Trauma. 2001;51:927-931.
6. Funk JL, Frye JB, Oyarzo JN, et al. Efficacy and mechanism of action of turmeric supplements in the treatment of experimental arthritis. Arthritis Rheum. 2006;54:3452-3464.
7. Patcharatrakul T, Gonlachanvit S. Chili peppers, curcumins, and prebiotics in gastrointestinal health and disease. Curr Gastroenterol Rep. 2016;18:19.
8. Thamlikitkul V, Bunyapraphatsara N, Dechatiwongse T, et al. Randomized double blind study of Curcuma domestica Val. for dyspepsia. J Med Assoc Thai. 1989;72:613-620.
9. Kumar S, Ahuja V, Sankar MJ, et al. Curcumin for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2012;10:CD008424.
10. Lang A, Salomon N, Wu JC, et al. Curcumin in combination with mesalamine induces remission in patients with mild-to-moderate ulcerative colitis in a randomized controlled trial. Clin Gastroenterol Hepatol. 2015;13:1444-1449.e1.
11. Henrotin Y, Priem F, Mobasheri A. Curcumin: a new paradigm and therapeutic opportunity for the treatment of osteoarthritis: curcumin for osteoarthritis management. Springerplus. 2013;2:56.
12. Kuptniratsaikul V, Dajpratham P, Taechaarpornkul W, et al. Efficacy and safety of Curcuma domestica extracts compared with ibuprofen in patients with knee osteoarthritis: a multicenter study. Clin Interv Aging. 2014;9:451-458.
13. Shehzad A, Lee J, Lee YS. Curcumin in various cancers. Biofactors. 2013;39:56-68.
14. Sordillo LA, Sordillo PP, Helson L. Curcumin for the treatment of glioblastoma. Anticancer Res. 2015;35:6373-6378.
15. Darvesh AS, Aggarwal BB, Bishayee A. Curcumin and liver cancer: a review. Curr Pharm Biotechnol. 2012;13:218-228.
16. Nagaraju GP, Aliya S, Zafar SF, et al. The impact of curcumin on breast cancer. Integr Biol (Camb). 2012;4:996-1007.
17. Johnson JJ, Mukhtar H. Curcumin for chemoprevention of colon cancer. Cancer Lett. 2007;255:170-181.
18. Dorai T, Cao YC, Dorai B, et al. Therapeutic potential of curcumin in human prostate cancer. III. Curcumin inhibits proliferation, induces apoptosis, and inhibits angiogenesis of LNCaP prostate cancer cells in vivo. Prostate. 2001;47:293-303.
19. Hazarey VK, Sakrikar AR, Ganvir SM. Efficacy of curcumin in the treatment for oral submucous fibrosis - a randomized clinical trial. J Oral Maxillofac Pathol. 2015;19:145-152.
20. Lal B, Kapoor AK, Asthana OP, et al. Efficacy of curcumin in the management of chronic anterior uveitis. Phytother Res. 1999;13:318-322.
21. Kapakos G, Youreva V, Srivastava AK. Cardiovascular protection by curcumin: molecular aspects. Indian J Biochem Biophys. 2012;49:306-315.
22. Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892-5901.
23. Heck AM, DeWitt BA, Lukes AL. Potential interactions between alternative therapies and warfarin. Am J Health Syst Pharm. 2000;57:1221-1227.
24. Srivastava JK, Shankar E, Gupta S. Chamomile: a herbal medicine of the past with bright future. Mol Med Rep. 2010;3:895-901.
25. Ross SM. Generalized anxiety disorder (GAD): efficacy of standardized matricaria recutita (german chamomile) extract in the treatment of generalized anxiety disorder. Holistic Nursing Practice. 2013;27:366- 368.
26. Amsterdam JD, Li Y, Soeller I, et al. A randomized, double-blind, placebo-controlled trial of oral Matricaria recutita (chamomile) extract therapy for generalized anxiety disorder. J Clin Psychopharmacol. 2009;29:378-382.
27. Zick SM, Wright BD, Sen A, et al. Preliminary examination of the efficacy and safety of a standardized chamomile extract for chronic primary insomnia: a randomized placebo-controlled pilot study. BMC Complement Altern Med. 2011;11:78.
28. Leach MJ, Page AT. Herbal medicine for insomnia: a systematic review and meta-analysis. Sleep Med Rev. 2015;24:1-12.
29. Weizman Z, Alkrinawi S, Goldfarb D, et al. Efficacy of herbal tea preparation in infantile colic. J Pediatr. 1993;122:650.
30. Crotteau CA, Wright ST, Eglash A. Clinical inquiries. What is the best treatment for infants with colic? J Fam Pract. 2006;55:634-636.
31. Panahi Y, Taghizadeh M, Marzony ET, et al. Rosemary oil vs minoxidil 2% for the treatment of androgenetic alopecia: a randomized comparative trial. Skinmed. 2015;13:15-21.
32. Hay IC, Jamieson M, Ormerod AD. Randomized trial of aromatherapy. Successful treatment for alopecia areata. Arch Dermatol. 1998;134:1349-1352.
33. Caffeine and kids: FDA takes a closer look. Available at: https://www.fda.gov/ForConsumers/ConsumerUpdates/ucm350570.htm. Accessed: November 1, 2017.
34. Torpy JM, Livingston EH. Energy Drinks. JAMA. 2013;309:297.
35. Lopez-Garcia E, van Dam RM, Li TY, et al. The relationship of coffee consumption with mortality. Ann Intern Med. 2008;148:904-914.
36. Crippa A, Discacciati A, Larsson SC, et al. Coffee consumption and mortality from all causes, cardiovascular disease, and cancer: a dose-response meta-analysis. Am J Epidemiol. 2014;180:763-775.
37. Loftfield E, Freedman ND, Graubard BI, et al. Association of coffee consumption with overall and cause-specific mortality in a large US prospective cohort study. Am J Epidemiol. 2015;182:1010-1022.
38. Lopez-Garcia E, Rodriguez-Artalejo F, Rexrode KM, et al. Coffee consumption and risk of stroke in women. Circulation. 2009;119:1116-1123.
39. Friedrich K, Smit M, Wannhoff A, et al. Coffee consumption protects against progression in liver cirrhosis and increases long-term survival after liver transplantation. J Gastroenterol Hepatol. 2016;31:1470-1475.
40. Wang L, Shen X, Wu Y, et al. Coffee and caffeine consumption and depression: a meta-analysis of observational studies. Aust N Z J Psychiatry. 2016;50:228-242.
41. Liu F, Wang X, Wu G, et al. Coffee consumption decreases risks for hepatic fibrosis and cirrhosis: a meta-analysis. PLoS One. 2015;10:e0142457.
42. Grosso G, Micek A, Castellano S, et al. Coffee, tea, caffeine and risk of depression: a systematic review and dose-response meta-analysis of observational studies. Mol Nutr Food Res. 2016;60:223-234.
43. Solfrizzi V, Panza F, Imbimbo BP, et al. Italian longitudinal study on aging working group. Coffee consumption habits and the risk of mild cognitive impairment: The Italian Longitudinal Study on Aging. J Alzheimers Dis. 2015;47:889-899.
44. Sugiyama K, Tomata Y, Kaiho Y, et al. Association between coffee consumption and incident risk of disabling dementia in elderly japanese: The Ohsaki Cohort 2006 Study. J Alzheimers Dis. 2015;50:491-500.
45. Driscoll I, Shumaker SA, Snively BM, et al. Relationships between caffeine intake and risk for probable dementia or global cognitive impairment: The Women’s Health Initiative Memory Study. J Gerontol A Biol Sci Med Sci. 2016;71:1596-1602.
46. van Dieren S, Uiterwaal CS, van der Schouw YT, et al. Coffee and tea consumption and risk of type 2 diabetes. Diabetologia. 2009;52:2561-2569.
47. Wang J, Li X, Zhang D. Coffee consumption and the risk of cutaneous melanoma: a meta-analysis. Eur J Nutr. 2016;55:1317-1329.
48. Liu J, Shen B, Shi M, et al. Higher caffeinated coffee intake is associated with reduced malignant melanoma risk: a meta-analysis study. PLoS One. 2016;11:e0147056.
49. Wikoff D, Welsh BT, Henderson R, et al. Systematic review of the potential adverse effects of caffeine consumption in healthy adults, pregnant women, adolescents, and children. Food Chem Toxical. 2017;109(Pt 1):585-648.
50. Verna R. The history and science of chocolate. Malays J Pathol. 2013;35:111-121.
51. Katz DL, Doughty K, Ali A. Cocoa and chocolate in human health and disease. Antioxid Redox Signal. 2011;15:2779-2811.
52. Baba S, Natsume M, Yasuda A, et al. Plasma LDL and HDL cholesterol and oxidized LDL concentrations are altered in normo- and hypercholesterolemic humans after intake of different levels of cocoa powder. J Nutr. 2007;137:1436-1441.
53. Mellor DD, Sathyapalan T, Kilpatrick ES, et al. High-cocoa polyphenol-rich chocolate improves HDL cholesterol in type 2 diabetes patients. Diabet Med. 2010;27:1318-1321.
54. Tokede OA, Gaziano JM, Djoussé L. Effects of cocoa products/dark chocolate on serum lipids: a meta-analysis. Eur J Clin Nutr. 2011;65:879-886.
55. Jia L, Liu X, Bai YY, et al. Short-term effect of cocoa product consumption on lipid profile: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2010;92:218-225.
56. Rostami A, Khalili M, Haghighat N, et al. High-cocoa polyphenol-rich chocolate improves blood pressure in patients with diabetes and hypertension. ARYA Atheroscler. 2015;11:21-29.
57. Buitrago-Lopez A, Sanderson J, Johnson L, et al. Chocolate consumption and cardiometabolic disorders: systematic review and meta-analysis. BMJ. 2011;26;343:d4488.
58. Wang X, Ouyang YY, Liu J, et al. Flavonoid intake and risk of CVD: a systematic review and meta-analysis of prospective cohort studies. Br J Nutr. 2014;111:1-11.
59. Zomer E, Owen A, Magliano DJ, et al. The effectiveness and cost effectiveness of dark chocolate consumption as prevention therapy in people at high risk of cardiovascular disease: best case scenario analysis using a Markov model. BMJ. 2012;344:e3657.
60. Larsson SC, Virtamo J, Wolk A. Chocolate consumption and risk of stroke: a prospective cohort of men and meta-analysis. Neurology. 2012;79:1223-1229.
61. Khawaja O, Gaziano JM, Djoussé L. Chocolate and coronary heart disease: a systematic review. Curr Atheroscler Rep. 2011;13:447-452.
62. Jacques PF, Cassidy A, Rogers G, et al. Dietary flavonoid intakes and CVD incidence in the Framingham Offspring Cohort. Br J Nutr. 2015;114:1496-1503.
63. Taubert D, Roesen R, Lehmann C, et al. Effects of low habitual cocoa intake on blood pressure and bioactive nitric oxide: a randomized controlled trial. JAMA. 2007;298:49-60.
64. Desch S, Schmidt J, Kobler D, et al. Effect of cocoa products on blood pressure: systematic review and meta-analysis. Am J Hypertens. 2010;23:97-103.
65. Ried K, Sullivan TR, Fakler P, et al. Effect of cocoa on blood pressure. Cochrane Database Syst Rev. 2012;8:CD008893.
66. Saftlas AF, Triche EW, Beydoun H, et al. Does chocolate intake during pregnancy reduce the risks of preeclampsia and gestational hypertension? Ann Epidemiol. 2010;20:584-591.
67. Triche EW, Grosso LM, Belanger K, et al. Chocolate consumption in pregnancy and reduced likelihood of preeclampsia. Epidemiology. 2008;19:459-464.
68. Grassi D, Lippi C, Necozione S, et al. Short-term administration of dark chocolate is followed by a significant increase in insulin sensitivity and a decrease in blood pressure in healthy persons. Am J Clin Nutr. 2005;81:611-614.
69. Matsumoto C, Petrone AB, Sesso HD, et al. Chocolate consumption and risk of diabetes mellitus in the Physicians’ Health Study. Am J Clin Nutr. 2015;101:362-367.
70. Lippi D. Chocolate in history: food, medicine, medi-food. Nutrients. 2013;5:1573-1584.
71. Sathyapalan T, Beckett S, Rigby AS, et al. High cocoa polyphenol rich chocolate may reduce the burden of the symptoms in chronic fatigue syndrome. Nutr J. 2010;9:55.
72. Martin FP, Antille N, Rezzi S, et al. Everyday eating experiences of chocolate and non-chocolate snacks impact postprandial anxiety, energy and emotional states. Nutrients. 2012;4:554-567.
73. Patel S, Mathan JJ, Vaghefi E, et al. The effect of flavonoids on visual function in patients with glaucoma or ocular hypertension: a systematic review and meta-analysis. Graefes Arch Clin Exp Ophthalmol. 2015;253:1841-1850.
74. Moreira A, Diógenes MJ, de Mendonça A, et al. Chocolate consumption is associated with a lower risk of cognitive decline. J Alzheimers Dis. 2016;53:85-93.
1. National Center for Complementary and Integrative Health. The use of complementary and alternative medicine in the United States. Available at: https://nccih.nih.gov/research/statistics/2007/camsurvey_fs1.htm. Accessed Nov 28, 2017.
2. Aggarwal BB. Curcumin-free turmeric exhibits anti-inflammatory and anticancer activities: identification of novel components of turmeric. Mol Nutr Food Res. 2013;57:1529-1542.
3. Henrotin Y, Clutterbuck AL, Allaway D, et al. Biological actions of curcumin on articular chondrocytes. Osteoarthritis Cartilage. 2010;18:141-149.
4. Asher GN, Spelman K. Clinical utility of curcumin extract. Altern Ther Health Med. 2013;19:20-22.
5. Phan TT, See P, Lee ST, et al. Protective effects of curcumin against oxidative damage on skin cells in vitro: its implication for wound healing. J Trauma. 2001;51:927-931.
6. Funk JL, Frye JB, Oyarzo JN, et al. Efficacy and mechanism of action of turmeric supplements in the treatment of experimental arthritis. Arthritis Rheum. 2006;54:3452-3464.
7. Patcharatrakul T, Gonlachanvit S. Chili peppers, curcumins, and prebiotics in gastrointestinal health and disease. Curr Gastroenterol Rep. 2016;18:19.
8. Thamlikitkul V, Bunyapraphatsara N, Dechatiwongse T, et al. Randomized double blind study of Curcuma domestica Val. for dyspepsia. J Med Assoc Thai. 1989;72:613-620.
9. Kumar S, Ahuja V, Sankar MJ, et al. Curcumin for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2012;10:CD008424.
10. Lang A, Salomon N, Wu JC, et al. Curcumin in combination with mesalamine induces remission in patients with mild-to-moderate ulcerative colitis in a randomized controlled trial. Clin Gastroenterol Hepatol. 2015;13:1444-1449.e1.
11. Henrotin Y, Priem F, Mobasheri A. Curcumin: a new paradigm and therapeutic opportunity for the treatment of osteoarthritis: curcumin for osteoarthritis management. Springerplus. 2013;2:56.
12. Kuptniratsaikul V, Dajpratham P, Taechaarpornkul W, et al. Efficacy and safety of Curcuma domestica extracts compared with ibuprofen in patients with knee osteoarthritis: a multicenter study. Clin Interv Aging. 2014;9:451-458.
13. Shehzad A, Lee J, Lee YS. Curcumin in various cancers. Biofactors. 2013;39:56-68.
14. Sordillo LA, Sordillo PP, Helson L. Curcumin for the treatment of glioblastoma. Anticancer Res. 2015;35:6373-6378.
15. Darvesh AS, Aggarwal BB, Bishayee A. Curcumin and liver cancer: a review. Curr Pharm Biotechnol. 2012;13:218-228.
16. Nagaraju GP, Aliya S, Zafar SF, et al. The impact of curcumin on breast cancer. Integr Biol (Camb). 2012;4:996-1007.
17. Johnson JJ, Mukhtar H. Curcumin for chemoprevention of colon cancer. Cancer Lett. 2007;255:170-181.
18. Dorai T, Cao YC, Dorai B, et al. Therapeutic potential of curcumin in human prostate cancer. III. Curcumin inhibits proliferation, induces apoptosis, and inhibits angiogenesis of LNCaP prostate cancer cells in vivo. Prostate. 2001;47:293-303.
19. Hazarey VK, Sakrikar AR, Ganvir SM. Efficacy of curcumin in the treatment for oral submucous fibrosis - a randomized clinical trial. J Oral Maxillofac Pathol. 2015;19:145-152.
20. Lal B, Kapoor AK, Asthana OP, et al. Efficacy of curcumin in the management of chronic anterior uveitis. Phytother Res. 1999;13:318-322.
21. Kapakos G, Youreva V, Srivastava AK. Cardiovascular protection by curcumin: molecular aspects. Indian J Biochem Biophys. 2012;49:306-315.
22. Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892-5901.
23. Heck AM, DeWitt BA, Lukes AL. Potential interactions between alternative therapies and warfarin. Am J Health Syst Pharm. 2000;57:1221-1227.
24. Srivastava JK, Shankar E, Gupta S. Chamomile: a herbal medicine of the past with bright future. Mol Med Rep. 2010;3:895-901.
25. Ross SM. Generalized anxiety disorder (GAD): efficacy of standardized matricaria recutita (german chamomile) extract in the treatment of generalized anxiety disorder. Holistic Nursing Practice. 2013;27:366- 368.
26. Amsterdam JD, Li Y, Soeller I, et al. A randomized, double-blind, placebo-controlled trial of oral Matricaria recutita (chamomile) extract therapy for generalized anxiety disorder. J Clin Psychopharmacol. 2009;29:378-382.
27. Zick SM, Wright BD, Sen A, et al. Preliminary examination of the efficacy and safety of a standardized chamomile extract for chronic primary insomnia: a randomized placebo-controlled pilot study. BMC Complement Altern Med. 2011;11:78.
28. Leach MJ, Page AT. Herbal medicine for insomnia: a systematic review and meta-analysis. Sleep Med Rev. 2015;24:1-12.
29. Weizman Z, Alkrinawi S, Goldfarb D, et al. Efficacy of herbal tea preparation in infantile colic. J Pediatr. 1993;122:650.
30. Crotteau CA, Wright ST, Eglash A. Clinical inquiries. What is the best treatment for infants with colic? J Fam Pract. 2006;55:634-636.
31. Panahi Y, Taghizadeh M, Marzony ET, et al. Rosemary oil vs minoxidil 2% for the treatment of androgenetic alopecia: a randomized comparative trial. Skinmed. 2015;13:15-21.
32. Hay IC, Jamieson M, Ormerod AD. Randomized trial of aromatherapy. Successful treatment for alopecia areata. Arch Dermatol. 1998;134:1349-1352.
33. Caffeine and kids: FDA takes a closer look. Available at: https://www.fda.gov/ForConsumers/ConsumerUpdates/ucm350570.htm. Accessed: November 1, 2017.
34. Torpy JM, Livingston EH. Energy Drinks. JAMA. 2013;309:297.
35. Lopez-Garcia E, van Dam RM, Li TY, et al. The relationship of coffee consumption with mortality. Ann Intern Med. 2008;148:904-914.
36. Crippa A, Discacciati A, Larsson SC, et al. Coffee consumption and mortality from all causes, cardiovascular disease, and cancer: a dose-response meta-analysis. Am J Epidemiol. 2014;180:763-775.
37. Loftfield E, Freedman ND, Graubard BI, et al. Association of coffee consumption with overall and cause-specific mortality in a large US prospective cohort study. Am J Epidemiol. 2015;182:1010-1022.
38. Lopez-Garcia E, Rodriguez-Artalejo F, Rexrode KM, et al. Coffee consumption and risk of stroke in women. Circulation. 2009;119:1116-1123.
39. Friedrich K, Smit M, Wannhoff A, et al. Coffee consumption protects against progression in liver cirrhosis and increases long-term survival after liver transplantation. J Gastroenterol Hepatol. 2016;31:1470-1475.
40. Wang L, Shen X, Wu Y, et al. Coffee and caffeine consumption and depression: a meta-analysis of observational studies. Aust N Z J Psychiatry. 2016;50:228-242.
41. Liu F, Wang X, Wu G, et al. Coffee consumption decreases risks for hepatic fibrosis and cirrhosis: a meta-analysis. PLoS One. 2015;10:e0142457.
42. Grosso G, Micek A, Castellano S, et al. Coffee, tea, caffeine and risk of depression: a systematic review and dose-response meta-analysis of observational studies. Mol Nutr Food Res. 2016;60:223-234.
43. Solfrizzi V, Panza F, Imbimbo BP, et al. Italian longitudinal study on aging working group. Coffee consumption habits and the risk of mild cognitive impairment: The Italian Longitudinal Study on Aging. J Alzheimers Dis. 2015;47:889-899.
44. Sugiyama K, Tomata Y, Kaiho Y, et al. Association between coffee consumption and incident risk of disabling dementia in elderly japanese: The Ohsaki Cohort 2006 Study. J Alzheimers Dis. 2015;50:491-500.
45. Driscoll I, Shumaker SA, Snively BM, et al. Relationships between caffeine intake and risk for probable dementia or global cognitive impairment: The Women’s Health Initiative Memory Study. J Gerontol A Biol Sci Med Sci. 2016;71:1596-1602.
46. van Dieren S, Uiterwaal CS, van der Schouw YT, et al. Coffee and tea consumption and risk of type 2 diabetes. Diabetologia. 2009;52:2561-2569.
47. Wang J, Li X, Zhang D. Coffee consumption and the risk of cutaneous melanoma: a meta-analysis. Eur J Nutr. 2016;55:1317-1329.
48. Liu J, Shen B, Shi M, et al. Higher caffeinated coffee intake is associated with reduced malignant melanoma risk: a meta-analysis study. PLoS One. 2016;11:e0147056.
49. Wikoff D, Welsh BT, Henderson R, et al. Systematic review of the potential adverse effects of caffeine consumption in healthy adults, pregnant women, adolescents, and children. Food Chem Toxical. 2017;109(Pt 1):585-648.
50. Verna R. The history and science of chocolate. Malays J Pathol. 2013;35:111-121.
51. Katz DL, Doughty K, Ali A. Cocoa and chocolate in human health and disease. Antioxid Redox Signal. 2011;15:2779-2811.
52. Baba S, Natsume M, Yasuda A, et al. Plasma LDL and HDL cholesterol and oxidized LDL concentrations are altered in normo- and hypercholesterolemic humans after intake of different levels of cocoa powder. J Nutr. 2007;137:1436-1441.
53. Mellor DD, Sathyapalan T, Kilpatrick ES, et al. High-cocoa polyphenol-rich chocolate improves HDL cholesterol in type 2 diabetes patients. Diabet Med. 2010;27:1318-1321.
54. Tokede OA, Gaziano JM, Djoussé L. Effects of cocoa products/dark chocolate on serum lipids: a meta-analysis. Eur J Clin Nutr. 2011;65:879-886.
55. Jia L, Liu X, Bai YY, et al. Short-term effect of cocoa product consumption on lipid profile: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2010;92:218-225.
56. Rostami A, Khalili M, Haghighat N, et al. High-cocoa polyphenol-rich chocolate improves blood pressure in patients with diabetes and hypertension. ARYA Atheroscler. 2015;11:21-29.
57. Buitrago-Lopez A, Sanderson J, Johnson L, et al. Chocolate consumption and cardiometabolic disorders: systematic review and meta-analysis. BMJ. 2011;26;343:d4488.
58. Wang X, Ouyang YY, Liu J, et al. Flavonoid intake and risk of CVD: a systematic review and meta-analysis of prospective cohort studies. Br J Nutr. 2014;111:1-11.
59. Zomer E, Owen A, Magliano DJ, et al. The effectiveness and cost effectiveness of dark chocolate consumption as prevention therapy in people at high risk of cardiovascular disease: best case scenario analysis using a Markov model. BMJ. 2012;344:e3657.
60. Larsson SC, Virtamo J, Wolk A. Chocolate consumption and risk of stroke: a prospective cohort of men and meta-analysis. Neurology. 2012;79:1223-1229.
61. Khawaja O, Gaziano JM, Djoussé L. Chocolate and coronary heart disease: a systematic review. Curr Atheroscler Rep. 2011;13:447-452.
62. Jacques PF, Cassidy A, Rogers G, et al. Dietary flavonoid intakes and CVD incidence in the Framingham Offspring Cohort. Br J Nutr. 2015;114:1496-1503.
63. Taubert D, Roesen R, Lehmann C, et al. Effects of low habitual cocoa intake on blood pressure and bioactive nitric oxide: a randomized controlled trial. JAMA. 2007;298:49-60.
64. Desch S, Schmidt J, Kobler D, et al. Effect of cocoa products on blood pressure: systematic review and meta-analysis. Am J Hypertens. 2010;23:97-103.
65. Ried K, Sullivan TR, Fakler P, et al. Effect of cocoa on blood pressure. Cochrane Database Syst Rev. 2012;8:CD008893.
66. Saftlas AF, Triche EW, Beydoun H, et al. Does chocolate intake during pregnancy reduce the risks of preeclampsia and gestational hypertension? Ann Epidemiol. 2010;20:584-591.
67. Triche EW, Grosso LM, Belanger K, et al. Chocolate consumption in pregnancy and reduced likelihood of preeclampsia. Epidemiology. 2008;19:459-464.
68. Grassi D, Lippi C, Necozione S, et al. Short-term administration of dark chocolate is followed by a significant increase in insulin sensitivity and a decrease in blood pressure in healthy persons. Am J Clin Nutr. 2005;81:611-614.
69. Matsumoto C, Petrone AB, Sesso HD, et al. Chocolate consumption and risk of diabetes mellitus in the Physicians’ Health Study. Am J Clin Nutr. 2015;101:362-367.
70. Lippi D. Chocolate in history: food, medicine, medi-food. Nutrients. 2013;5:1573-1584.
71. Sathyapalan T, Beckett S, Rigby AS, et al. High cocoa polyphenol rich chocolate may reduce the burden of the symptoms in chronic fatigue syndrome. Nutr J. 2010;9:55.
72. Martin FP, Antille N, Rezzi S, et al. Everyday eating experiences of chocolate and non-chocolate snacks impact postprandial anxiety, energy and emotional states. Nutrients. 2012;4:554-567.
73. Patel S, Mathan JJ, Vaghefi E, et al. The effect of flavonoids on visual function in patients with glaucoma or ocular hypertension: a systematic review and meta-analysis. Graefes Arch Clin Exp Ophthalmol. 2015;253:1841-1850.
74. Moreira A, Diógenes MJ, de Mendonça A, et al. Chocolate consumption is associated with a lower risk of cognitive decline. J Alzheimers Dis. 2016;53:85-93.
PRACTICE RECOMMENDATIONS
› Inform patients that curcumin appears to be a safe and effective adjunctive therapy for ulcerative colitis when used along with mesalamine or sulfasalazine. B
› Recommend chamomile extract to patients experiencing mild to moderate generalized anxiety disorder. B
› Tell patients that coffee is associated with a lower risk of mortality, reduced cardiovascular events, and a reduction in liver disease progression (in patients with end-stage liver disease). B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
