User login
MDedge conference coverage features onsite reporting of the latest study results and expert perspectives from leading researchers.
Triple Therapy Now Advised for Lupus Nephritis in Updated Guideline
WASHINGTON — A new guideline for management of lupus nephritis (LN) was unveiled at the annual meeting of the American College of Rheumatology (ACR), updating the 2012 LN guideline to recommend a more aggressive first-line approach to treating the disease.
“The biggest differences are that we are recommending what we’re calling triple therapy, where we incorporate the glucocorticoid therapy with baseline conventional immunosuppressants, usually mycophenolate with cyclophosphamide, and the addition of one of the newer agents more recently approved by the FDA [Food and Drug Administration] — belimumab, voclosporin, or another CNI [calcineurin inhibitor],” said Lisa Sammaritano, MD, director of the Rheumatology Reproductive Health Program of the Barbara Volcker Center for Women and Rheumatic Diseases at the Hospital for Special Surgery and professor of clinical medicine at Weill Cornell Medical College, both in New York City.

“This is a bit of a change from not only our previous guideline but some of the other guidelines out there, and it is based on the fact that we have very convincing evidence that starting with triple therapy yields to better long-term outcomes for our patients than starting with only two agents and waiting to see if they respond before escalating therapy,” she said. Other key updates include recommending use of pulse glucocorticoid therapy with a lower dose and more rapid steroid taper and treating patients with the recommended therapy for 3-5 years.
The guiding principles of the guideline are not only to preserve kidney function and minimize morbidity and mortality but also to ensure collaborative care with nephrology, to utilize shared decision-making that includes patients’ values and preferences, to reduce healthcare disparities, and to consider pediatric and geriatric populations. The guidelines are based on a quantitative synthesis of 105 studies that yielded 7 strong recommendations, 21 conditional recommendations, and 13 good practice statements — those commonly accepted as beneficial or practical advice even if there is little direct evidence to support them. The voting panel of 19 members included not only 3 nephrologists and 2 pediatric rheumatologists but also 2 patient representatives with LN.
The recommendations are just that, “a recommendation, not an order,” Sammaritano said, and strong recommendations are those “where we think, unequivocally, almost everybody should follow that recommendation. When we feel that we cannot make a strong recommendation, then we call our recommendation conditional, and it is conditional on looking at different things,” she said.
“Patients are different, especially lupus patients, and so one lupus nephritis patient may have different clinical characteristics, different thoughts about what therapy will work for them in their lives, or what therapy they really do not want to pursue,” Sammaritano said. “Maybe they can’t conceive of coming to the hospital once a month for intravenous therapy. Maybe they’re concerned about pill burden, which is something that our patient panel really emphasized to us. So, conditional recommendation means this voting panel thought that this was the best overall for most patients and most circumstances, recognizing there will still be a significant number of people, clinicians and patients, who may feel differently for that particular situation. So, that’s where you know the patient-clinician discussion can help with decision-making.”
What Are the Recommendations?
All patients with systemic lupus erythematosus (SLE) are strongly recommended to undergo proteinuria screening every 6-12 months or at the time of a flare. Those suspected of having LN should receive a prompt kidney biopsy and treatment with glucocorticoids while awaiting the biopsy and results. Two conditional recommendations for kidney biopsy include patients with SLE with unexplained impaired kidney function or a protein to creatinine ratio > 0.5 g/g, and patients with LN with a suspected flare after initial response or a lack of response or worsening after 6 months of therapy.
The guidelines include a strong recommendation for all patients with SLE to receive hydroxychloroquine and a conditional recommendation for all patients with elevated proteinuria (> 0.5 g/g) to receive renin-angiotensin-aldosterone system inhibitors (RAAS-I). Dosages in patients with LN with decreased glomerular filtration rate (GFR) should be adjusted as needed.
Sammaritano then reviewed the specifics on medication treatment. The glucocorticoid therapy in all patients with LN should begin with Pulse IV Therapy at 250-1000 mg/d for 1-3 days, followed by oral prednisone ≤ 0.5 mg/kg per day up to 40 mg/d, then tapered to a target dose > 5 mg/d within 6 months. The justification for this course comes from a 2024 systematic review finding pulse followed by oral glucocorticoids maximized complete renal response while minimizing toxicities, Sammaritano said.
“We have all become acutely aware of the very high risk of prolonged high dose of glucocorticoids for our patients,” she said, “and importantly, our patient panel participants strongly emphasized their preference for minimizing glucocorticoids dose.”
In addition to the recommendation of all patients receiving hydroxychloroquine and RAAS-I, first-line treatment of active, new-onset, or flaring LN should begin with triple therapy — glucocorticoids with two additional immunosuppressive agents. For patients with class III/IV LN, triple therapy includes the glucocorticoids course with a mycophenolic acid analog (MPAA) and either belimumab or a CNI. Conditional recommendations support MPAA with belimumab for significant extrarenal manifestations and MPAA with CNI for proteinuria ≥ 3 g/g.
An alternative triple therapy for class III/IV is glucocorticoids with low-dose cyclophosphamide and belimumab, but MPAA at 2-3 g/d is preferred over cyclophosphamide. The preferred regimen for cyclophosphamide is derived from the Euro-Lupus Nephritis Trial: Intravenous 500 mg every 2 weeks for six doses and then MPAA. Sammaritano noted that there are some limited data on using cyclophosphamide with belimumab, but “we do not specifically recommend cyclophosphamide with a CNI as one of our options because this combination has not been studied in randomized controlled trials.”
There are less data supporting class V recommendations, Sammaritano said, but for those with proteinuria of at least 1 g/g, the panel still recommends triple therapy with glucocorticoids, a MPAA, and a CNI. A CNI is preferred over belimumab because of its stabilizing effects on the podocyte cytoskeleton. Two alternative triple therapies for class V–only patients are glucocorticoids with belimumab and either low-dose cyclophosphamide or MPAA.
Dual therapy is only recommended if triple therapy is not available or not tolerated. The voting panel chose to recommend triple therapy over dual therapy with escalation for two reasons. First, the BLISS-LN and AURORA 1 trials showed improved outcomes with initial triple therapy over initial dual therapies.
Second, “nephron loss proceeds throughout a person’s lifetime even for those who do not have lupus nephritis, and every case of lupus nephritis or every period of time with uncontrolled lupus nephritis changes the course of that decline for the worse,” Sammaritano said. “So, we feel we can’t wait for nephron loss to implement what has been shown to be the most efficacious therapy. We want to gain rapid control of inflammation using the most effective regimen to prevent further damage and flare and maintain survival.”
Therapy is conditionally recommended for at least 3-5 years because “not only do we want to gain rapid control of disease activity [but we also] want to maintain control of disease activity until there’s sustained inactive disease,” Sammaritano said. “Repeat kidney biopsies show that immunologic activity persists in the kidneys for several years, and the withdrawal of immunosuppression when there is histologic activity predisposes patients to flare.” But immunosuppressive therapy can be tapered over time as determined by renal disease activity and medication tolerability.
For patients with refractory disease, consider additional factors that could be affecting the disease, such as adherence, the presence of other diagnoses, or advanced chronicity.
“If true refractory nephritis is present,” she said, “we recommend escalation to a more intensive regimen,” including the addition of anti-CD20 agents, combination therapy with three immunosuppressives, or referral for investigational therapy.
“We also emphasize the importance of other adjunctive therapies preventing comorbidities, such as cardiovascular disease, changes in bone health, or infection risk,” she said. In older patients, avoid polypharmacy as much as possible and be mindful of age-related GFR, she added.
A strong recommendation supported monitoring patients with LN and proteinuria at least every 3 months if they have not achieved complete renal response and every 3-6 months after sustained complete renal response.
Last, in patients with LN and end-stage kidney disease (ESKD), the voting panel strongly recommends transplant over dialysis and conditionally recommends proceeding to the transplant without requiring a complete clinical or serologic remission as long as no other organs are involved. In patients with LN at risk for ESKD, the guideline conditionally recommends consideration of a preemptive transplant, and patients on dialysis or post transplant are strongly recommended to regularly follow up with rheumatology.
Gabriel Kirsch, MD, a resident rheumatologist at the University of Florida College of Medicine, Jacksonville, said he found the guidelines helpful, “especially the guidance on the dichotomy between using belimumab and voclosporin and the clinical and patient preference that help you make that decision.”
Kirsch had hoped, however, to hear more about the impact of therapeutic drug monitoring of hydroxychloroquine on LN outcomes. He also noted a clinical scenario he’s come across that wasn’t addressed.
“When you’re checking GFR on these folks, a lot of our eGFR calculators are creatinine based, and creatinine at the extremes of muscle mass can be inaccurate,” such as getting artificially low creatinine readings from pediatric patients because of their low muscle mass or from patients with muscle atrophy caused by a lot of glucocorticoid exposure. “I was hoping for some more guidance on that,” he said.
Ellen Ginzler, MD, MPH, chief of rheumatology at SUNY Health Science Center in Brooklyn, New York, said the guidelines were pretty much what she expected them to be. She agreed with the panel’s advice that, when deciding between belimumab or voclosporin, “if it’s pure proteinuria, then you add voclosporin. If the patient has extra renal manifestations, you go with belimumab first.”
“They really made it quite clear that, despite the fact that people really want to reduce the amount of immunosuppression — and I agree you should taper steroids quickly — you really need to keep the immunosuppression for a prolonged period of time because all of the studies that have been done for years show that the longer you’re on immunosuppression after you achieve remission or a low disease activity state, the better your chance of not flaring,” Ginzler said. “Rapid tapering or discontinuation really increases the risk of flare.”
Sammaritano, Kirsch, and Ginzler had no disclosures. No external funding was used.
A version of this article appeared on Medscape.com.
WASHINGTON — A new guideline for management of lupus nephritis (LN) was unveiled at the annual meeting of the American College of Rheumatology (ACR), updating the 2012 LN guideline to recommend a more aggressive first-line approach to treating the disease.
“The biggest differences are that we are recommending what we’re calling triple therapy, where we incorporate the glucocorticoid therapy with baseline conventional immunosuppressants, usually mycophenolate with cyclophosphamide, and the addition of one of the newer agents more recently approved by the FDA [Food and Drug Administration] — belimumab, voclosporin, or another CNI [calcineurin inhibitor],” said Lisa Sammaritano, MD, director of the Rheumatology Reproductive Health Program of the Barbara Volcker Center for Women and Rheumatic Diseases at the Hospital for Special Surgery and professor of clinical medicine at Weill Cornell Medical College, both in New York City.
“This is a bit of a change from not only our previous guideline but some of the other guidelines out there, and it is based on the fact that we have very convincing evidence that starting with triple therapy yields to better long-term outcomes for our patients than starting with only two agents and waiting to see if they respond before escalating therapy,” she said. Other key updates include recommending use of pulse glucocorticoid therapy with a lower dose and more rapid steroid taper and treating patients with the recommended therapy for 3-5 years.
The guiding principles of the guideline are not only to preserve kidney function and minimize morbidity and mortality but also to ensure collaborative care with nephrology, to utilize shared decision-making that includes patients’ values and preferences, to reduce healthcare disparities, and to consider pediatric and geriatric populations. The guidelines are based on a quantitative synthesis of 105 studies that yielded 7 strong recommendations, 21 conditional recommendations, and 13 good practice statements — those commonly accepted as beneficial or practical advice even if there is little direct evidence to support them. The voting panel of 19 members included not only 3 nephrologists and 2 pediatric rheumatologists but also 2 patient representatives with LN.
The recommendations are just that, “a recommendation, not an order,” Sammaritano said, and strong recommendations are those “where we think, unequivocally, almost everybody should follow that recommendation. When we feel that we cannot make a strong recommendation, then we call our recommendation conditional, and it is conditional on looking at different things,” she said.
“Patients are different, especially lupus patients, and so one lupus nephritis patient may have different clinical characteristics, different thoughts about what therapy will work for them in their lives, or what therapy they really do not want to pursue,” Sammaritano said. “Maybe they can’t conceive of coming to the hospital once a month for intravenous therapy. Maybe they’re concerned about pill burden, which is something that our patient panel really emphasized to us. So, conditional recommendation means this voting panel thought that this was the best overall for most patients and most circumstances, recognizing there will still be a significant number of people, clinicians and patients, who may feel differently for that particular situation. So, that’s where you know the patient-clinician discussion can help with decision-making.”
What Are the Recommendations?
All patients with systemic lupus erythematosus (SLE) are strongly recommended to undergo proteinuria screening every 6-12 months or at the time of a flare. Those suspected of having LN should receive a prompt kidney biopsy and treatment with glucocorticoids while awaiting the biopsy and results. Two conditional recommendations for kidney biopsy include patients with SLE with unexplained impaired kidney function or a protein to creatinine ratio > 0.5 g/g, and patients with LN with a suspected flare after initial response or a lack of response or worsening after 6 months of therapy.
The guidelines include a strong recommendation for all patients with SLE to receive hydroxychloroquine and a conditional recommendation for all patients with elevated proteinuria (> 0.5 g/g) to receive renin-angiotensin-aldosterone system inhibitors (RAAS-I). Dosages in patients with LN with decreased glomerular filtration rate (GFR) should be adjusted as needed.
Sammaritano then reviewed the specifics on medication treatment. The glucocorticoid therapy in all patients with LN should begin with Pulse IV Therapy at 250-1000 mg/d for 1-3 days, followed by oral prednisone ≤ 0.5 mg/kg per day up to 40 mg/d, then tapered to a target dose > 5 mg/d within 6 months. The justification for this course comes from a 2024 systematic review finding pulse followed by oral glucocorticoids maximized complete renal response while minimizing toxicities, Sammaritano said.
“We have all become acutely aware of the very high risk of prolonged high dose of glucocorticoids for our patients,” she said, “and importantly, our patient panel participants strongly emphasized their preference for minimizing glucocorticoids dose.”
In addition to the recommendation of all patients receiving hydroxychloroquine and RAAS-I, first-line treatment of active, new-onset, or flaring LN should begin with triple therapy — glucocorticoids with two additional immunosuppressive agents. For patients with class III/IV LN, triple therapy includes the glucocorticoids course with a mycophenolic acid analog (MPAA) and either belimumab or a CNI. Conditional recommendations support MPAA with belimumab for significant extrarenal manifestations and MPAA with CNI for proteinuria ≥ 3 g/g.
An alternative triple therapy for class III/IV is glucocorticoids with low-dose cyclophosphamide and belimumab, but MPAA at 2-3 g/d is preferred over cyclophosphamide. The preferred regimen for cyclophosphamide is derived from the Euro-Lupus Nephritis Trial: Intravenous 500 mg every 2 weeks for six doses and then MPAA. Sammaritano noted that there are some limited data on using cyclophosphamide with belimumab, but “we do not specifically recommend cyclophosphamide with a CNI as one of our options because this combination has not been studied in randomized controlled trials.”
There are less data supporting class V recommendations, Sammaritano said, but for those with proteinuria of at least 1 g/g, the panel still recommends triple therapy with glucocorticoids, a MPAA, and a CNI. A CNI is preferred over belimumab because of its stabilizing effects on the podocyte cytoskeleton. Two alternative triple therapies for class V–only patients are glucocorticoids with belimumab and either low-dose cyclophosphamide or MPAA.
Dual therapy is only recommended if triple therapy is not available or not tolerated. The voting panel chose to recommend triple therapy over dual therapy with escalation for two reasons. First, the BLISS-LN and AURORA 1 trials showed improved outcomes with initial triple therapy over initial dual therapies.
Second, “nephron loss proceeds throughout a person’s lifetime even for those who do not have lupus nephritis, and every case of lupus nephritis or every period of time with uncontrolled lupus nephritis changes the course of that decline for the worse,” Sammaritano said. “So, we feel we can’t wait for nephron loss to implement what has been shown to be the most efficacious therapy. We want to gain rapid control of inflammation using the most effective regimen to prevent further damage and flare and maintain survival.”
Therapy is conditionally recommended for at least 3-5 years because “not only do we want to gain rapid control of disease activity [but we also] want to maintain control of disease activity until there’s sustained inactive disease,” Sammaritano said. “Repeat kidney biopsies show that immunologic activity persists in the kidneys for several years, and the withdrawal of immunosuppression when there is histologic activity predisposes patients to flare.” But immunosuppressive therapy can be tapered over time as determined by renal disease activity and medication tolerability.
For patients with refractory disease, consider additional factors that could be affecting the disease, such as adherence, the presence of other diagnoses, or advanced chronicity.
“If true refractory nephritis is present,” she said, “we recommend escalation to a more intensive regimen,” including the addition of anti-CD20 agents, combination therapy with three immunosuppressives, or referral for investigational therapy.
“We also emphasize the importance of other adjunctive therapies preventing comorbidities, such as cardiovascular disease, changes in bone health, or infection risk,” she said. In older patients, avoid polypharmacy as much as possible and be mindful of age-related GFR, she added.
A strong recommendation supported monitoring patients with LN and proteinuria at least every 3 months if they have not achieved complete renal response and every 3-6 months after sustained complete renal response.
Last, in patients with LN and end-stage kidney disease (ESKD), the voting panel strongly recommends transplant over dialysis and conditionally recommends proceeding to the transplant without requiring a complete clinical or serologic remission as long as no other organs are involved. In patients with LN at risk for ESKD, the guideline conditionally recommends consideration of a preemptive transplant, and patients on dialysis or post transplant are strongly recommended to regularly follow up with rheumatology.
Gabriel Kirsch, MD, a resident rheumatologist at the University of Florida College of Medicine, Jacksonville, said he found the guidelines helpful, “especially the guidance on the dichotomy between using belimumab and voclosporin and the clinical and patient preference that help you make that decision.”
Kirsch had hoped, however, to hear more about the impact of therapeutic drug monitoring of hydroxychloroquine on LN outcomes. He also noted a clinical scenario he’s come across that wasn’t addressed.
“When you’re checking GFR on these folks, a lot of our eGFR calculators are creatinine based, and creatinine at the extremes of muscle mass can be inaccurate,” such as getting artificially low creatinine readings from pediatric patients because of their low muscle mass or from patients with muscle atrophy caused by a lot of glucocorticoid exposure. “I was hoping for some more guidance on that,” he said.
Ellen Ginzler, MD, MPH, chief of rheumatology at SUNY Health Science Center in Brooklyn, New York, said the guidelines were pretty much what she expected them to be. She agreed with the panel’s advice that, when deciding between belimumab or voclosporin, “if it’s pure proteinuria, then you add voclosporin. If the patient has extra renal manifestations, you go with belimumab first.”
“They really made it quite clear that, despite the fact that people really want to reduce the amount of immunosuppression — and I agree you should taper steroids quickly — you really need to keep the immunosuppression for a prolonged period of time because all of the studies that have been done for years show that the longer you’re on immunosuppression after you achieve remission or a low disease activity state, the better your chance of not flaring,” Ginzler said. “Rapid tapering or discontinuation really increases the risk of flare.”
Sammaritano, Kirsch, and Ginzler had no disclosures. No external funding was used.
A version of this article appeared on Medscape.com.
WASHINGTON — A new guideline for management of lupus nephritis (LN) was unveiled at the annual meeting of the American College of Rheumatology (ACR), updating the 2012 LN guideline to recommend a more aggressive first-line approach to treating the disease.
“The biggest differences are that we are recommending what we’re calling triple therapy, where we incorporate the glucocorticoid therapy with baseline conventional immunosuppressants, usually mycophenolate with cyclophosphamide, and the addition of one of the newer agents more recently approved by the FDA [Food and Drug Administration] — belimumab, voclosporin, or another CNI [calcineurin inhibitor],” said Lisa Sammaritano, MD, director of the Rheumatology Reproductive Health Program of the Barbara Volcker Center for Women and Rheumatic Diseases at the Hospital for Special Surgery and professor of clinical medicine at Weill Cornell Medical College, both in New York City.
“This is a bit of a change from not only our previous guideline but some of the other guidelines out there, and it is based on the fact that we have very convincing evidence that starting with triple therapy yields to better long-term outcomes for our patients than starting with only two agents and waiting to see if they respond before escalating therapy,” she said. Other key updates include recommending use of pulse glucocorticoid therapy with a lower dose and more rapid steroid taper and treating patients with the recommended therapy for 3-5 years.
The guiding principles of the guideline are not only to preserve kidney function and minimize morbidity and mortality but also to ensure collaborative care with nephrology, to utilize shared decision-making that includes patients’ values and preferences, to reduce healthcare disparities, and to consider pediatric and geriatric populations. The guidelines are based on a quantitative synthesis of 105 studies that yielded 7 strong recommendations, 21 conditional recommendations, and 13 good practice statements — those commonly accepted as beneficial or practical advice even if there is little direct evidence to support them. The voting panel of 19 members included not only 3 nephrologists and 2 pediatric rheumatologists but also 2 patient representatives with LN.
The recommendations are just that, “a recommendation, not an order,” Sammaritano said, and strong recommendations are those “where we think, unequivocally, almost everybody should follow that recommendation. When we feel that we cannot make a strong recommendation, then we call our recommendation conditional, and it is conditional on looking at different things,” she said.
“Patients are different, especially lupus patients, and so one lupus nephritis patient may have different clinical characteristics, different thoughts about what therapy will work for them in their lives, or what therapy they really do not want to pursue,” Sammaritano said. “Maybe they can’t conceive of coming to the hospital once a month for intravenous therapy. Maybe they’re concerned about pill burden, which is something that our patient panel really emphasized to us. So, conditional recommendation means this voting panel thought that this was the best overall for most patients and most circumstances, recognizing there will still be a significant number of people, clinicians and patients, who may feel differently for that particular situation. So, that’s where you know the patient-clinician discussion can help with decision-making.”
What Are the Recommendations?
All patients with systemic lupus erythematosus (SLE) are strongly recommended to undergo proteinuria screening every 6-12 months or at the time of a flare. Those suspected of having LN should receive a prompt kidney biopsy and treatment with glucocorticoids while awaiting the biopsy and results. Two conditional recommendations for kidney biopsy include patients with SLE with unexplained impaired kidney function or a protein to creatinine ratio > 0.5 g/g, and patients with LN with a suspected flare after initial response or a lack of response or worsening after 6 months of therapy.
The guidelines include a strong recommendation for all patients with SLE to receive hydroxychloroquine and a conditional recommendation for all patients with elevated proteinuria (> 0.5 g/g) to receive renin-angiotensin-aldosterone system inhibitors (RAAS-I). Dosages in patients with LN with decreased glomerular filtration rate (GFR) should be adjusted as needed.
Sammaritano then reviewed the specifics on medication treatment. The glucocorticoid therapy in all patients with LN should begin with Pulse IV Therapy at 250-1000 mg/d for 1-3 days, followed by oral prednisone ≤ 0.5 mg/kg per day up to 40 mg/d, then tapered to a target dose > 5 mg/d within 6 months. The justification for this course comes from a 2024 systematic review finding pulse followed by oral glucocorticoids maximized complete renal response while minimizing toxicities, Sammaritano said.
“We have all become acutely aware of the very high risk of prolonged high dose of glucocorticoids for our patients,” she said, “and importantly, our patient panel participants strongly emphasized their preference for minimizing glucocorticoids dose.”
In addition to the recommendation of all patients receiving hydroxychloroquine and RAAS-I, first-line treatment of active, new-onset, or flaring LN should begin with triple therapy — glucocorticoids with two additional immunosuppressive agents. For patients with class III/IV LN, triple therapy includes the glucocorticoids course with a mycophenolic acid analog (MPAA) and either belimumab or a CNI. Conditional recommendations support MPAA with belimumab for significant extrarenal manifestations and MPAA with CNI for proteinuria ≥ 3 g/g.
An alternative triple therapy for class III/IV is glucocorticoids with low-dose cyclophosphamide and belimumab, but MPAA at 2-3 g/d is preferred over cyclophosphamide. The preferred regimen for cyclophosphamide is derived from the Euro-Lupus Nephritis Trial: Intravenous 500 mg every 2 weeks for six doses and then MPAA. Sammaritano noted that there are some limited data on using cyclophosphamide with belimumab, but “we do not specifically recommend cyclophosphamide with a CNI as one of our options because this combination has not been studied in randomized controlled trials.”
There are less data supporting class V recommendations, Sammaritano said, but for those with proteinuria of at least 1 g/g, the panel still recommends triple therapy with glucocorticoids, a MPAA, and a CNI. A CNI is preferred over belimumab because of its stabilizing effects on the podocyte cytoskeleton. Two alternative triple therapies for class V–only patients are glucocorticoids with belimumab and either low-dose cyclophosphamide or MPAA.
Dual therapy is only recommended if triple therapy is not available or not tolerated. The voting panel chose to recommend triple therapy over dual therapy with escalation for two reasons. First, the BLISS-LN and AURORA 1 trials showed improved outcomes with initial triple therapy over initial dual therapies.
Second, “nephron loss proceeds throughout a person’s lifetime even for those who do not have lupus nephritis, and every case of lupus nephritis or every period of time with uncontrolled lupus nephritis changes the course of that decline for the worse,” Sammaritano said. “So, we feel we can’t wait for nephron loss to implement what has been shown to be the most efficacious therapy. We want to gain rapid control of inflammation using the most effective regimen to prevent further damage and flare and maintain survival.”
Therapy is conditionally recommended for at least 3-5 years because “not only do we want to gain rapid control of disease activity [but we also] want to maintain control of disease activity until there’s sustained inactive disease,” Sammaritano said. “Repeat kidney biopsies show that immunologic activity persists in the kidneys for several years, and the withdrawal of immunosuppression when there is histologic activity predisposes patients to flare.” But immunosuppressive therapy can be tapered over time as determined by renal disease activity and medication tolerability.
For patients with refractory disease, consider additional factors that could be affecting the disease, such as adherence, the presence of other diagnoses, or advanced chronicity.
“If true refractory nephritis is present,” she said, “we recommend escalation to a more intensive regimen,” including the addition of anti-CD20 agents, combination therapy with three immunosuppressives, or referral for investigational therapy.
“We also emphasize the importance of other adjunctive therapies preventing comorbidities, such as cardiovascular disease, changes in bone health, or infection risk,” she said. In older patients, avoid polypharmacy as much as possible and be mindful of age-related GFR, she added.
A strong recommendation supported monitoring patients with LN and proteinuria at least every 3 months if they have not achieved complete renal response and every 3-6 months after sustained complete renal response.
Last, in patients with LN and end-stage kidney disease (ESKD), the voting panel strongly recommends transplant over dialysis and conditionally recommends proceeding to the transplant without requiring a complete clinical or serologic remission as long as no other organs are involved. In patients with LN at risk for ESKD, the guideline conditionally recommends consideration of a preemptive transplant, and patients on dialysis or post transplant are strongly recommended to regularly follow up with rheumatology.
Gabriel Kirsch, MD, a resident rheumatologist at the University of Florida College of Medicine, Jacksonville, said he found the guidelines helpful, “especially the guidance on the dichotomy between using belimumab and voclosporin and the clinical and patient preference that help you make that decision.”
Kirsch had hoped, however, to hear more about the impact of therapeutic drug monitoring of hydroxychloroquine on LN outcomes. He also noted a clinical scenario he’s come across that wasn’t addressed.
“When you’re checking GFR on these folks, a lot of our eGFR calculators are creatinine based, and creatinine at the extremes of muscle mass can be inaccurate,” such as getting artificially low creatinine readings from pediatric patients because of their low muscle mass or from patients with muscle atrophy caused by a lot of glucocorticoid exposure. “I was hoping for some more guidance on that,” he said.
Ellen Ginzler, MD, MPH, chief of rheumatology at SUNY Health Science Center in Brooklyn, New York, said the guidelines were pretty much what she expected them to be. She agreed with the panel’s advice that, when deciding between belimumab or voclosporin, “if it’s pure proteinuria, then you add voclosporin. If the patient has extra renal manifestations, you go with belimumab first.”
“They really made it quite clear that, despite the fact that people really want to reduce the amount of immunosuppression — and I agree you should taper steroids quickly — you really need to keep the immunosuppression for a prolonged period of time because all of the studies that have been done for years show that the longer you’re on immunosuppression after you achieve remission or a low disease activity state, the better your chance of not flaring,” Ginzler said. “Rapid tapering or discontinuation really increases the risk of flare.”
Sammaritano, Kirsch, and Ginzler had no disclosures. No external funding was used.
A version of this article appeared on Medscape.com.
FROM ACR 2024
Treating Onychomycosis: Pearls from a Podiatrist
LAS VEGAS —
According to Tracey C. Vlahovic, DPM, a professor at the Samuel Merritt University College of Podiatric Medicine, Oakland, California, most cases of onychomycosis are caused by the dermatophytes Trichophyton rubrum and T mentagrophytes, although the cause can also be a mixed infection. “Dermatophytes are going to impact the nails first, and molds may come in and join the party later,” she said at the Society of Dermatology Physician Associates (SDPA) 22nd Annual Fall Dermatology Conference.

“The distal subungual onychomycosis (DSO) type is still the most common, but don’t forget that onychomycosis and nail psoriasis can happen at the same time. What we can’t lose sight of is that onychomycosis is a disease of the nail bed, which ultimately affects the nail plate; it’s not a disease of the nail plate first.”
Her diagnostic approach combines periodic acid-Schiff (PAS) staining with fungal culture “because I like to know the speciation,” she said. “PAS doesn’t give me the speciation; fungal cultures should. PCR can be expensive, but that can give me speciation.”
How Does This Happen?
Fungal DSO occurs because of exposure to a dermatophyte, which can be as simple as tinea pedis. “Perhaps it’s the environment in the shoe,” said Vlahovic, one of the authors of a textbook on onychomycosis. “That’s something I’m always concentrating on with the patient. What is your foot hygiene like? What’s your shoe and sock wear? What’s your level of physical activity? You can have trauma to the hyponychium, where the skin and the nail meet. Maybe they trim their nails too close to the skin, or maybe there’s another skin condition like psoriasis.”
The dermatophyte, she continued, enters and invades the nail at the hyponychium and uses the keratinase enzyme to digest keratin in the nail bed. Mild inflammation develops, and pH changes cause focal parakeratosis and subungual hyperkeratosis in the form of onycholysis and subungual debris. “Hyphae then invade the lamina of the nail plate, which causes brittle nails,” she said. “The compromised hyponychium creates a reservoir for molds and bacteria.”
Therapies approved by the Food and Drug Administration (FDA) for onychomycosis include the topical agents efinaconazole, tavaborole, and ciclopirox; the oral agents terbinafine and itraconazole; and laser therapy. Off-label, Vlahovic said that she sometimes uses oral fluconazole, pulsed dosing for terbinafine, and booster doses of terbinafine or any approved oral antifungal agent. Pulse dosing for itraconazole is FDA-approved for fingernails but not for toenails.
“We don’t have any oral antifungals that are approved for children, but we do have weight-based dosing,” she noted. Other off-label treatments for onychomycosis that patients may come across while browsing the internet but do not penetrate the nail plate, include products containing tolnaftate, tree oil, and undecylenic acid, “which is a very long-chain antifungal,” Vlahovic said. “It’s so huge that it can’t get through the nail plate. These products must get through the nail plate into the nail bed where the infection is.”
According to therapeutic recommendations for the treatment of toenail onychomycosis in the United States, published in 2021, terbinafine is the primary choice for oral treatment and efinaconazole 10% for topical treatment. There are no current treatment recommendations for pregnant or lactating patients. “I always defer to the obstetrician,” said Vlahovic, a coauthor of the recommendations. For pediatric patients, there are approved topical medications: Efinaconazole and tavaborole for ages 6 and up and ciclopirox for ages 12 years or older.
Treatment recommendations for adults vary based on clinical presentation and patient characteristics. Questions to consider: Are they older? Do they have diabetes? Are they able to reach their feet to apply medication? What other medications are they taking? Are there any kidney or liver issues that are cause for concern?
Another question to consider is whether they have concurrent nail psoriasis. “When I have those patients, I often treat the onychomycosis first and the nail psoriasis second,” she said.
Evidence for Lasers Weak
Though laser therapy is FDA approved for the temporary increase of clear nails in onychomycosis, Vlahovic is underwhelmed by the evidence of its use for onychomycosis. According to a systematic review of 261 studies, only 1 reported treatment success as 16.7%, and clinical cures ranged from 13% to 16%. “Many of the existing studies were so poorly done in terms of protocols; it was frustrating,” she said. “No study has reported complete cure. There’s a lack of standardization across laser companies and a lack of standardization across protocols.”
Before starting oral antifungal therapy, Vlahovic uses the Onychomycosis Severity Index to determine the number of nails involved and the proportion of nails that are affected. She also wants to know if the patient is taking any medication that might interfere with an oral antifungal and gets baseline liver function tests (LFTs) to document results in the chart. “You want to discuss the pros and cons of oral antifungal therapy, and you want to set realistic expectations,” she added. “These medications are not cosmetic products; they are meant to kill fungus. Sometimes patients lose sight of that.”
Vlahovic routinely offers pulse dosing of terbinafine, which is FDA approved at a dose of 250 mg/d for 90 days. Pulse dosing involves taking terbinafine 250 mg twice a day for 1 week, followed by a 3-week break. This cycle is repeated three or four times. A clinical trial found no significant difference in outcome between patients who received pulsed vs continuous terbinafine dosing for the treatment of dermatophyte onychomycosis.
What About Oral Antifungal Safety?
For patients who ask about the safety of oral antifungals, Vlahovic characterized them as “well tolerated and safe in an immunocompetent population.” In a meta-analysis of 122 studies of about 22,000 patients, the pooled risk for treatment discontinuation because of adverse events was 3.4% for terbinafine 250 mg/d and 4.21% for itraconazole 200 mg/d. The risk for liver injury requiring termination of treatment and the risk of having symptomatic elevation of LFTs were less than 2% for all regimens.
According to the best available published evidence, Vlahovic said, the onychomycosis recurrence rate ranges from 6% to 40%. “That’s a wild number. We really have no idea what the true recurrence rate is, and that’s a problem.”
Vlahovic disclosed having been a consultant to and an investigator for Ortho Dermatologics and Sagis Diagnostics.
A version of this article appeared on Medscape.com.
LAS VEGAS —
According to Tracey C. Vlahovic, DPM, a professor at the Samuel Merritt University College of Podiatric Medicine, Oakland, California, most cases of onychomycosis are caused by the dermatophytes Trichophyton rubrum and T mentagrophytes, although the cause can also be a mixed infection. “Dermatophytes are going to impact the nails first, and molds may come in and join the party later,” she said at the Society of Dermatology Physician Associates (SDPA) 22nd Annual Fall Dermatology Conference.
“The distal subungual onychomycosis (DSO) type is still the most common, but don’t forget that onychomycosis and nail psoriasis can happen at the same time. What we can’t lose sight of is that onychomycosis is a disease of the nail bed, which ultimately affects the nail plate; it’s not a disease of the nail plate first.”
Her diagnostic approach combines periodic acid-Schiff (PAS) staining with fungal culture “because I like to know the speciation,” she said. “PAS doesn’t give me the speciation; fungal cultures should. PCR can be expensive, but that can give me speciation.”
How Does This Happen?
Fungal DSO occurs because of exposure to a dermatophyte, which can be as simple as tinea pedis. “Perhaps it’s the environment in the shoe,” said Vlahovic, one of the authors of a textbook on onychomycosis. “That’s something I’m always concentrating on with the patient. What is your foot hygiene like? What’s your shoe and sock wear? What’s your level of physical activity? You can have trauma to the hyponychium, where the skin and the nail meet. Maybe they trim their nails too close to the skin, or maybe there’s another skin condition like psoriasis.”
The dermatophyte, she continued, enters and invades the nail at the hyponychium and uses the keratinase enzyme to digest keratin in the nail bed. Mild inflammation develops, and pH changes cause focal parakeratosis and subungual hyperkeratosis in the form of onycholysis and subungual debris. “Hyphae then invade the lamina of the nail plate, which causes brittle nails,” she said. “The compromised hyponychium creates a reservoir for molds and bacteria.”
Therapies approved by the Food and Drug Administration (FDA) for onychomycosis include the topical agents efinaconazole, tavaborole, and ciclopirox; the oral agents terbinafine and itraconazole; and laser therapy. Off-label, Vlahovic said that she sometimes uses oral fluconazole, pulsed dosing for terbinafine, and booster doses of terbinafine or any approved oral antifungal agent. Pulse dosing for itraconazole is FDA-approved for fingernails but not for toenails.
“We don’t have any oral antifungals that are approved for children, but we do have weight-based dosing,” she noted. Other off-label treatments for onychomycosis that patients may come across while browsing the internet but do not penetrate the nail plate, include products containing tolnaftate, tree oil, and undecylenic acid, “which is a very long-chain antifungal,” Vlahovic said. “It’s so huge that it can’t get through the nail plate. These products must get through the nail plate into the nail bed where the infection is.”
According to therapeutic recommendations for the treatment of toenail onychomycosis in the United States, published in 2021, terbinafine is the primary choice for oral treatment and efinaconazole 10% for topical treatment. There are no current treatment recommendations for pregnant or lactating patients. “I always defer to the obstetrician,” said Vlahovic, a coauthor of the recommendations. For pediatric patients, there are approved topical medications: Efinaconazole and tavaborole for ages 6 and up and ciclopirox for ages 12 years or older.
Treatment recommendations for adults vary based on clinical presentation and patient characteristics. Questions to consider: Are they older? Do they have diabetes? Are they able to reach their feet to apply medication? What other medications are they taking? Are there any kidney or liver issues that are cause for concern?
Another question to consider is whether they have concurrent nail psoriasis. “When I have those patients, I often treat the onychomycosis first and the nail psoriasis second,” she said.
Evidence for Lasers Weak
Though laser therapy is FDA approved for the temporary increase of clear nails in onychomycosis, Vlahovic is underwhelmed by the evidence of its use for onychomycosis. According to a systematic review of 261 studies, only 1 reported treatment success as 16.7%, and clinical cures ranged from 13% to 16%. “Many of the existing studies were so poorly done in terms of protocols; it was frustrating,” she said. “No study has reported complete cure. There’s a lack of standardization across laser companies and a lack of standardization across protocols.”
Before starting oral antifungal therapy, Vlahovic uses the Onychomycosis Severity Index to determine the number of nails involved and the proportion of nails that are affected. She also wants to know if the patient is taking any medication that might interfere with an oral antifungal and gets baseline liver function tests (LFTs) to document results in the chart. “You want to discuss the pros and cons of oral antifungal therapy, and you want to set realistic expectations,” she added. “These medications are not cosmetic products; they are meant to kill fungus. Sometimes patients lose sight of that.”
Vlahovic routinely offers pulse dosing of terbinafine, which is FDA approved at a dose of 250 mg/d for 90 days. Pulse dosing involves taking terbinafine 250 mg twice a day for 1 week, followed by a 3-week break. This cycle is repeated three or four times. A clinical trial found no significant difference in outcome between patients who received pulsed vs continuous terbinafine dosing for the treatment of dermatophyte onychomycosis.
What About Oral Antifungal Safety?
For patients who ask about the safety of oral antifungals, Vlahovic characterized them as “well tolerated and safe in an immunocompetent population.” In a meta-analysis of 122 studies of about 22,000 patients, the pooled risk for treatment discontinuation because of adverse events was 3.4% for terbinafine 250 mg/d and 4.21% for itraconazole 200 mg/d. The risk for liver injury requiring termination of treatment and the risk of having symptomatic elevation of LFTs were less than 2% for all regimens.
According to the best available published evidence, Vlahovic said, the onychomycosis recurrence rate ranges from 6% to 40%. “That’s a wild number. We really have no idea what the true recurrence rate is, and that’s a problem.”
Vlahovic disclosed having been a consultant to and an investigator for Ortho Dermatologics and Sagis Diagnostics.
A version of this article appeared on Medscape.com.
LAS VEGAS —
According to Tracey C. Vlahovic, DPM, a professor at the Samuel Merritt University College of Podiatric Medicine, Oakland, California, most cases of onychomycosis are caused by the dermatophytes Trichophyton rubrum and T mentagrophytes, although the cause can also be a mixed infection. “Dermatophytes are going to impact the nails first, and molds may come in and join the party later,” she said at the Society of Dermatology Physician Associates (SDPA) 22nd Annual Fall Dermatology Conference.
“The distal subungual onychomycosis (DSO) type is still the most common, but don’t forget that onychomycosis and nail psoriasis can happen at the same time. What we can’t lose sight of is that onychomycosis is a disease of the nail bed, which ultimately affects the nail plate; it’s not a disease of the nail plate first.”
Her diagnostic approach combines periodic acid-Schiff (PAS) staining with fungal culture “because I like to know the speciation,” she said. “PAS doesn’t give me the speciation; fungal cultures should. PCR can be expensive, but that can give me speciation.”
How Does This Happen?
Fungal DSO occurs because of exposure to a dermatophyte, which can be as simple as tinea pedis. “Perhaps it’s the environment in the shoe,” said Vlahovic, one of the authors of a textbook on onychomycosis. “That’s something I’m always concentrating on with the patient. What is your foot hygiene like? What’s your shoe and sock wear? What’s your level of physical activity? You can have trauma to the hyponychium, where the skin and the nail meet. Maybe they trim their nails too close to the skin, or maybe there’s another skin condition like psoriasis.”
The dermatophyte, she continued, enters and invades the nail at the hyponychium and uses the keratinase enzyme to digest keratin in the nail bed. Mild inflammation develops, and pH changes cause focal parakeratosis and subungual hyperkeratosis in the form of onycholysis and subungual debris. “Hyphae then invade the lamina of the nail plate, which causes brittle nails,” she said. “The compromised hyponychium creates a reservoir for molds and bacteria.”
Therapies approved by the Food and Drug Administration (FDA) for onychomycosis include the topical agents efinaconazole, tavaborole, and ciclopirox; the oral agents terbinafine and itraconazole; and laser therapy. Off-label, Vlahovic said that she sometimes uses oral fluconazole, pulsed dosing for terbinafine, and booster doses of terbinafine or any approved oral antifungal agent. Pulse dosing for itraconazole is FDA-approved for fingernails but not for toenails.
“We don’t have any oral antifungals that are approved for children, but we do have weight-based dosing,” she noted. Other off-label treatments for onychomycosis that patients may come across while browsing the internet but do not penetrate the nail plate, include products containing tolnaftate, tree oil, and undecylenic acid, “which is a very long-chain antifungal,” Vlahovic said. “It’s so huge that it can’t get through the nail plate. These products must get through the nail plate into the nail bed where the infection is.”
According to therapeutic recommendations for the treatment of toenail onychomycosis in the United States, published in 2021, terbinafine is the primary choice for oral treatment and efinaconazole 10% for topical treatment. There are no current treatment recommendations for pregnant or lactating patients. “I always defer to the obstetrician,” said Vlahovic, a coauthor of the recommendations. For pediatric patients, there are approved topical medications: Efinaconazole and tavaborole for ages 6 and up and ciclopirox for ages 12 years or older.
Treatment recommendations for adults vary based on clinical presentation and patient characteristics. Questions to consider: Are they older? Do they have diabetes? Are they able to reach their feet to apply medication? What other medications are they taking? Are there any kidney or liver issues that are cause for concern?
Another question to consider is whether they have concurrent nail psoriasis. “When I have those patients, I often treat the onychomycosis first and the nail psoriasis second,” she said.
Evidence for Lasers Weak
Though laser therapy is FDA approved for the temporary increase of clear nails in onychomycosis, Vlahovic is underwhelmed by the evidence of its use for onychomycosis. According to a systematic review of 261 studies, only 1 reported treatment success as 16.7%, and clinical cures ranged from 13% to 16%. “Many of the existing studies were so poorly done in terms of protocols; it was frustrating,” she said. “No study has reported complete cure. There’s a lack of standardization across laser companies and a lack of standardization across protocols.”
Before starting oral antifungal therapy, Vlahovic uses the Onychomycosis Severity Index to determine the number of nails involved and the proportion of nails that are affected. She also wants to know if the patient is taking any medication that might interfere with an oral antifungal and gets baseline liver function tests (LFTs) to document results in the chart. “You want to discuss the pros and cons of oral antifungal therapy, and you want to set realistic expectations,” she added. “These medications are not cosmetic products; they are meant to kill fungus. Sometimes patients lose sight of that.”
Vlahovic routinely offers pulse dosing of terbinafine, which is FDA approved at a dose of 250 mg/d for 90 days. Pulse dosing involves taking terbinafine 250 mg twice a day for 1 week, followed by a 3-week break. This cycle is repeated three or four times. A clinical trial found no significant difference in outcome between patients who received pulsed vs continuous terbinafine dosing for the treatment of dermatophyte onychomycosis.
What About Oral Antifungal Safety?
For patients who ask about the safety of oral antifungals, Vlahovic characterized them as “well tolerated and safe in an immunocompetent population.” In a meta-analysis of 122 studies of about 22,000 patients, the pooled risk for treatment discontinuation because of adverse events was 3.4% for terbinafine 250 mg/d and 4.21% for itraconazole 200 mg/d. The risk for liver injury requiring termination of treatment and the risk of having symptomatic elevation of LFTs were less than 2% for all regimens.
According to the best available published evidence, Vlahovic said, the onychomycosis recurrence rate ranges from 6% to 40%. “That’s a wild number. We really have no idea what the true recurrence rate is, and that’s a problem.”
Vlahovic disclosed having been a consultant to and an investigator for Ortho Dermatologics and Sagis Diagnostics.
A version of this article appeared on Medscape.com.
FROM SDPA 2024
Kidney, Cardiovascular Benefits Seen With GLP-1 RA Drugs in SLE, Lupus Nephritis
WASHINGTON — Glucagon-like peptide 1 receptor agonist (GLP-1 RA) medications appear beneficial for people with systemic lupus erythematosus (SLE) and lupus nephritis, two new studies suggest.
“The risk of cardiovascular disease is thought to be at least double that for people with lupus ... and we know the risk of progressing to end-stage renal disease [ESKD] for patients with lupus nephritis can be as high as 10%-30%, so there’s clearly a major unmet need for new treatments and approaches to improve these outcomes, perhaps with adjunctive treatment beyond our typical immunosuppressive therapy,” April Jorge, MD, of Massachusetts General Hospital, Boston, said at the annual meeting of the American College of Rheumatology (ACR).
The GLP-1 RAs are approved for the treatment of type 2 diabetes (T2D) and obesity. They also have proven cardiovascular benefit, along with emerging data suggesting kidney protection independent of glucose lowering. Jorge presented findings from a study using data from the US multicenter electronic health record database TriNetX, showing that, among patients who had both T2D and SLE, those using GLP-1 RAs had lower risks for major adverse cardiac events (MACE), venous thrombosis, kidney disease progression, and all-cause mortality, compared with those using a different class of T2D medication.
A second study using TriNetX, presented at the same ACR meeting session by Anna-Kay Palmer, MD, a third-year internal medicine resident at Jefferson Einstein Hospital, Philadelphia, Pennsylvania, showed that GLP-1 RAs reduced the risk of progression to ESKD in patients with lupus nephritis, possibly caused by reductions in pro-inflammatory mediators.
Asked to comment, session moderator Diane L. Kamen, MD, professor of medicine at the Medical University of South Carolina Division of Rheumatology, Charleston, said in an interview that she definitely supports the use of GLP-1 RAs for patients who have SLE and/or lupus nephritis and also a drug label indication, either T2D or obesity. “[The GLP-1 RA prescriber] will usually run it by rheumatology to make sure that it doesn’t conflict with any of their other medical treatment, and it’s very reassuring to know that they could actually get a win-win.”
But as far as prescribing off-label for those with SLE/lupus nephritis who don’t have other GLP-1 RA indications, Kamen said, “that’s a black hole at this point. We need to do those prospective studies. But if they have another indication, yes.”
Cardiovascular, Kidney Benefits of GLP-1 RAs
Jorge noted that patients with lupus were excluded from the randomized clinical trials of GLP-1 RAs, so the current study was designed to investigate the potential impact of these medications on cardiovascular and kidney outcomes in patients with SLE and lupus nephritis.
From TriNetX data for 46 healthcare organizations nationwide, a total of 96,511 patients with both SLE and T2D but not ESKD had initiated either a GLP-1 RA or another diabetes drug class, dipeptidyl peptidase 4 inhibitors (DPP4i), between October 2006 and August 2021. Of those, 29,177 had lupus nephritis.
Propensity score matching for factors such as demographics, lupus severity, comorbidities, and medication use was used to emulate a randomized trial. This yielded 25,838 with SLE and T2D, of whom 910 initiated a GLP-1 RA and 1004 started a DPP4i, and 12,387 with lupus nephritis and T2D, including 267 on a GLP-1 RA and 324 on a DPP4i. After matching, the mean age was 55 years, more than 90% were women, and just under half were White individuals. About one third had chronic kidney disease stages ≥ 3, and about 15% had heart failure.
Over an average follow-up time of 1.2-1.4 years among those with SLE, the hazard ratio (HR) for MACE (a composite of myocardial infarction, stroke, and heart failure) for those taking a GLP-1 RA vs a DPP4i was 0.66, a significant difference. And for venous thrombosis, the HR was also significant at 0.49.
Kidney disease progression, defined as an estimated glomerular filtration rate decline of 30% or more or new ESKD, was significantly less likely in the GLP-1 RA group, with a HR of 0.77. All-cause mortality also was dramatically reduced (HR, 0.26). As expected, there was no difference in control outcome, genital infections (HR, 1.02).
In the subgroup with lupus nephritis, there were also lower risks for both MACE (HR, 0.64) and for renal progression (HR, 0.70). “The findings suggest similar cardiac and kidney benefits among patients with SLE and lupus nephritis as have been observed in other populations,” Jorge concluded.
Kamen commented that the study design “was pretty brilliant, because you wouldn’t be able to do a placebo-controlled trial since the indication was diabetes ... but the fact is you do see that the GLP-1 RA gets the benefit whereas the other drug does not.”
Next steps, Jorge said, will be mechanistic studies to better understand the effects of GLP-1 RAs in lupus and other rheumatic diseases, prospective studies of GLP-1 RAs in SLE and lupus nephritis without diabetes, and clarification of ideal timing for GLP-1 RA use in SLE and lupus nephritis.
“Ideally, with our prospective studies with these patients we can try to isolate the effect on patients with lupus and also better understand whether there might be an impact on disease activity through the anti-inflammatory effects of these medications, rather than just the cardioprotective and nephroprotective benefits,” she said.
In Those With Lupus Nephritis, Kidney Protection Seen
In her presentation, Palmer noted that, despite immunosuppressive therapies for SLE, 10%-20% of patients who develop lupus nephritis will progress to ESKD within 5 years of diagnosis.
She added that GLP-1 RAs have been shown to reduce albuminuria in people with diabetes and have been hypothesized to reduce inflammation through multiple pathways, thereby potentially reducing kidney disease independently of the presence of diabetes or weight loss. These pathways include modulating immune cell signaling and reducing pro-inflammatory cytokines.
Based on all this, Palmer and colleagues used International Classification of Diseases – 10th edition diagnostic codes in TriNetX to identify 839 patients who had been diagnosed with lupus nephritis between 2014 and 2024 and who were prescribed liraglutide, dulaglutide, semaglutide, or exenatide for any time after the lupus nephritis diagnosis. Another 29,840 patients with lupus nephritis had not used GLP-1 RAs.
After 1:1 propensity score matching for age, sex, race, ethnicity, presence of hypertension, diabetes, use of immunosuppressive and diabetes medication, smoking, obesity, and statin use, there were 735 individuals in each group. About two thirds in each had diabetes, whereas the rest had been prescribed the GLP-1 RAs for other indications.
Patients who were not on GLP-1 RAs were twice as likely to develop ESKD or dialysis (8.88% vs 3.971%; odds ratio, 2.35; P = .001).
Kamen pointed out that not including the use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers was a study flaw. On the other hand, the fact that not everyone in this study had diabetes was an advantage.
Jorge received grant/research support from Bristol-Myers Squibb, Cabaletta Bio, and the Lupus Clinical Investigator Network. Kamen is an adviser/review panel member for Alpine Immune Sciences. Palmer had no disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — Glucagon-like peptide 1 receptor agonist (GLP-1 RA) medications appear beneficial for people with systemic lupus erythematosus (SLE) and lupus nephritis, two new studies suggest.
“The risk of cardiovascular disease is thought to be at least double that for people with lupus ... and we know the risk of progressing to end-stage renal disease [ESKD] for patients with lupus nephritis can be as high as 10%-30%, so there’s clearly a major unmet need for new treatments and approaches to improve these outcomes, perhaps with adjunctive treatment beyond our typical immunosuppressive therapy,” April Jorge, MD, of Massachusetts General Hospital, Boston, said at the annual meeting of the American College of Rheumatology (ACR).
The GLP-1 RAs are approved for the treatment of type 2 diabetes (T2D) and obesity. They also have proven cardiovascular benefit, along with emerging data suggesting kidney protection independent of glucose lowering. Jorge presented findings from a study using data from the US multicenter electronic health record database TriNetX, showing that, among patients who had both T2D and SLE, those using GLP-1 RAs had lower risks for major adverse cardiac events (MACE), venous thrombosis, kidney disease progression, and all-cause mortality, compared with those using a different class of T2D medication.
A second study using TriNetX, presented at the same ACR meeting session by Anna-Kay Palmer, MD, a third-year internal medicine resident at Jefferson Einstein Hospital, Philadelphia, Pennsylvania, showed that GLP-1 RAs reduced the risk of progression to ESKD in patients with lupus nephritis, possibly caused by reductions in pro-inflammatory mediators.
Asked to comment, session moderator Diane L. Kamen, MD, professor of medicine at the Medical University of South Carolina Division of Rheumatology, Charleston, said in an interview that she definitely supports the use of GLP-1 RAs for patients who have SLE and/or lupus nephritis and also a drug label indication, either T2D or obesity. “[The GLP-1 RA prescriber] will usually run it by rheumatology to make sure that it doesn’t conflict with any of their other medical treatment, and it’s very reassuring to know that they could actually get a win-win.”
But as far as prescribing off-label for those with SLE/lupus nephritis who don’t have other GLP-1 RA indications, Kamen said, “that’s a black hole at this point. We need to do those prospective studies. But if they have another indication, yes.”
Cardiovascular, Kidney Benefits of GLP-1 RAs
Jorge noted that patients with lupus were excluded from the randomized clinical trials of GLP-1 RAs, so the current study was designed to investigate the potential impact of these medications on cardiovascular and kidney outcomes in patients with SLE and lupus nephritis.
From TriNetX data for 46 healthcare organizations nationwide, a total of 96,511 patients with both SLE and T2D but not ESKD had initiated either a GLP-1 RA or another diabetes drug class, dipeptidyl peptidase 4 inhibitors (DPP4i), between October 2006 and August 2021. Of those, 29,177 had lupus nephritis.
Propensity score matching for factors such as demographics, lupus severity, comorbidities, and medication use was used to emulate a randomized trial. This yielded 25,838 with SLE and T2D, of whom 910 initiated a GLP-1 RA and 1004 started a DPP4i, and 12,387 with lupus nephritis and T2D, including 267 on a GLP-1 RA and 324 on a DPP4i. After matching, the mean age was 55 years, more than 90% were women, and just under half were White individuals. About one third had chronic kidney disease stages ≥ 3, and about 15% had heart failure.
Over an average follow-up time of 1.2-1.4 years among those with SLE, the hazard ratio (HR) for MACE (a composite of myocardial infarction, stroke, and heart failure) for those taking a GLP-1 RA vs a DPP4i was 0.66, a significant difference. And for venous thrombosis, the HR was also significant at 0.49.
Kidney disease progression, defined as an estimated glomerular filtration rate decline of 30% or more or new ESKD, was significantly less likely in the GLP-1 RA group, with a HR of 0.77. All-cause mortality also was dramatically reduced (HR, 0.26). As expected, there was no difference in control outcome, genital infections (HR, 1.02).
In the subgroup with lupus nephritis, there were also lower risks for both MACE (HR, 0.64) and for renal progression (HR, 0.70). “The findings suggest similar cardiac and kidney benefits among patients with SLE and lupus nephritis as have been observed in other populations,” Jorge concluded.
Kamen commented that the study design “was pretty brilliant, because you wouldn’t be able to do a placebo-controlled trial since the indication was diabetes ... but the fact is you do see that the GLP-1 RA gets the benefit whereas the other drug does not.”
Next steps, Jorge said, will be mechanistic studies to better understand the effects of GLP-1 RAs in lupus and other rheumatic diseases, prospective studies of GLP-1 RAs in SLE and lupus nephritis without diabetes, and clarification of ideal timing for GLP-1 RA use in SLE and lupus nephritis.
“Ideally, with our prospective studies with these patients we can try to isolate the effect on patients with lupus and also better understand whether there might be an impact on disease activity through the anti-inflammatory effects of these medications, rather than just the cardioprotective and nephroprotective benefits,” she said.
In Those With Lupus Nephritis, Kidney Protection Seen
In her presentation, Palmer noted that, despite immunosuppressive therapies for SLE, 10%-20% of patients who develop lupus nephritis will progress to ESKD within 5 years of diagnosis.
She added that GLP-1 RAs have been shown to reduce albuminuria in people with diabetes and have been hypothesized to reduce inflammation through multiple pathways, thereby potentially reducing kidney disease independently of the presence of diabetes or weight loss. These pathways include modulating immune cell signaling and reducing pro-inflammatory cytokines.
Based on all this, Palmer and colleagues used International Classification of Diseases – 10th edition diagnostic codes in TriNetX to identify 839 patients who had been diagnosed with lupus nephritis between 2014 and 2024 and who were prescribed liraglutide, dulaglutide, semaglutide, or exenatide for any time after the lupus nephritis diagnosis. Another 29,840 patients with lupus nephritis had not used GLP-1 RAs.
After 1:1 propensity score matching for age, sex, race, ethnicity, presence of hypertension, diabetes, use of immunosuppressive and diabetes medication, smoking, obesity, and statin use, there were 735 individuals in each group. About two thirds in each had diabetes, whereas the rest had been prescribed the GLP-1 RAs for other indications.
Patients who were not on GLP-1 RAs were twice as likely to develop ESKD or dialysis (8.88% vs 3.971%; odds ratio, 2.35; P = .001).
Kamen pointed out that not including the use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers was a study flaw. On the other hand, the fact that not everyone in this study had diabetes was an advantage.
Jorge received grant/research support from Bristol-Myers Squibb, Cabaletta Bio, and the Lupus Clinical Investigator Network. Kamen is an adviser/review panel member for Alpine Immune Sciences. Palmer had no disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — Glucagon-like peptide 1 receptor agonist (GLP-1 RA) medications appear beneficial for people with systemic lupus erythematosus (SLE) and lupus nephritis, two new studies suggest.
“The risk of cardiovascular disease is thought to be at least double that for people with lupus ... and we know the risk of progressing to end-stage renal disease [ESKD] for patients with lupus nephritis can be as high as 10%-30%, so there’s clearly a major unmet need for new treatments and approaches to improve these outcomes, perhaps with adjunctive treatment beyond our typical immunosuppressive therapy,” April Jorge, MD, of Massachusetts General Hospital, Boston, said at the annual meeting of the American College of Rheumatology (ACR).
The GLP-1 RAs are approved for the treatment of type 2 diabetes (T2D) and obesity. They also have proven cardiovascular benefit, along with emerging data suggesting kidney protection independent of glucose lowering. Jorge presented findings from a study using data from the US multicenter electronic health record database TriNetX, showing that, among patients who had both T2D and SLE, those using GLP-1 RAs had lower risks for major adverse cardiac events (MACE), venous thrombosis, kidney disease progression, and all-cause mortality, compared with those using a different class of T2D medication.
A second study using TriNetX, presented at the same ACR meeting session by Anna-Kay Palmer, MD, a third-year internal medicine resident at Jefferson Einstein Hospital, Philadelphia, Pennsylvania, showed that GLP-1 RAs reduced the risk of progression to ESKD in patients with lupus nephritis, possibly caused by reductions in pro-inflammatory mediators.
Asked to comment, session moderator Diane L. Kamen, MD, professor of medicine at the Medical University of South Carolina Division of Rheumatology, Charleston, said in an interview that she definitely supports the use of GLP-1 RAs for patients who have SLE and/or lupus nephritis and also a drug label indication, either T2D or obesity. “[The GLP-1 RA prescriber] will usually run it by rheumatology to make sure that it doesn’t conflict with any of their other medical treatment, and it’s very reassuring to know that they could actually get a win-win.”
But as far as prescribing off-label for those with SLE/lupus nephritis who don’t have other GLP-1 RA indications, Kamen said, “that’s a black hole at this point. We need to do those prospective studies. But if they have another indication, yes.”
Cardiovascular, Kidney Benefits of GLP-1 RAs
Jorge noted that patients with lupus were excluded from the randomized clinical trials of GLP-1 RAs, so the current study was designed to investigate the potential impact of these medications on cardiovascular and kidney outcomes in patients with SLE and lupus nephritis.
From TriNetX data for 46 healthcare organizations nationwide, a total of 96,511 patients with both SLE and T2D but not ESKD had initiated either a GLP-1 RA or another diabetes drug class, dipeptidyl peptidase 4 inhibitors (DPP4i), between October 2006 and August 2021. Of those, 29,177 had lupus nephritis.
Propensity score matching for factors such as demographics, lupus severity, comorbidities, and medication use was used to emulate a randomized trial. This yielded 25,838 with SLE and T2D, of whom 910 initiated a GLP-1 RA and 1004 started a DPP4i, and 12,387 with lupus nephritis and T2D, including 267 on a GLP-1 RA and 324 on a DPP4i. After matching, the mean age was 55 years, more than 90% were women, and just under half were White individuals. About one third had chronic kidney disease stages ≥ 3, and about 15% had heart failure.
Over an average follow-up time of 1.2-1.4 years among those with SLE, the hazard ratio (HR) for MACE (a composite of myocardial infarction, stroke, and heart failure) for those taking a GLP-1 RA vs a DPP4i was 0.66, a significant difference. And for venous thrombosis, the HR was also significant at 0.49.
Kidney disease progression, defined as an estimated glomerular filtration rate decline of 30% or more or new ESKD, was significantly less likely in the GLP-1 RA group, with a HR of 0.77. All-cause mortality also was dramatically reduced (HR, 0.26). As expected, there was no difference in control outcome, genital infections (HR, 1.02).
In the subgroup with lupus nephritis, there were also lower risks for both MACE (HR, 0.64) and for renal progression (HR, 0.70). “The findings suggest similar cardiac and kidney benefits among patients with SLE and lupus nephritis as have been observed in other populations,” Jorge concluded.
Kamen commented that the study design “was pretty brilliant, because you wouldn’t be able to do a placebo-controlled trial since the indication was diabetes ... but the fact is you do see that the GLP-1 RA gets the benefit whereas the other drug does not.”
Next steps, Jorge said, will be mechanistic studies to better understand the effects of GLP-1 RAs in lupus and other rheumatic diseases, prospective studies of GLP-1 RAs in SLE and lupus nephritis without diabetes, and clarification of ideal timing for GLP-1 RA use in SLE and lupus nephritis.
“Ideally, with our prospective studies with these patients we can try to isolate the effect on patients with lupus and also better understand whether there might be an impact on disease activity through the anti-inflammatory effects of these medications, rather than just the cardioprotective and nephroprotective benefits,” she said.
In Those With Lupus Nephritis, Kidney Protection Seen
In her presentation, Palmer noted that, despite immunosuppressive therapies for SLE, 10%-20% of patients who develop lupus nephritis will progress to ESKD within 5 years of diagnosis.
She added that GLP-1 RAs have been shown to reduce albuminuria in people with diabetes and have been hypothesized to reduce inflammation through multiple pathways, thereby potentially reducing kidney disease independently of the presence of diabetes or weight loss. These pathways include modulating immune cell signaling and reducing pro-inflammatory cytokines.
Based on all this, Palmer and colleagues used International Classification of Diseases – 10th edition diagnostic codes in TriNetX to identify 839 patients who had been diagnosed with lupus nephritis between 2014 and 2024 and who were prescribed liraglutide, dulaglutide, semaglutide, or exenatide for any time after the lupus nephritis diagnosis. Another 29,840 patients with lupus nephritis had not used GLP-1 RAs.
After 1:1 propensity score matching for age, sex, race, ethnicity, presence of hypertension, diabetes, use of immunosuppressive and diabetes medication, smoking, obesity, and statin use, there were 735 individuals in each group. About two thirds in each had diabetes, whereas the rest had been prescribed the GLP-1 RAs for other indications.
Patients who were not on GLP-1 RAs were twice as likely to develop ESKD or dialysis (8.88% vs 3.971%; odds ratio, 2.35; P = .001).
Kamen pointed out that not including the use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers was a study flaw. On the other hand, the fact that not everyone in this study had diabetes was an advantage.
Jorge received grant/research support from Bristol-Myers Squibb, Cabaletta Bio, and the Lupus Clinical Investigator Network. Kamen is an adviser/review panel member for Alpine Immune Sciences. Palmer had no disclosures.
A version of this article appeared on Medscape.com.
FROM ACR 2024
RA Assessment Via Automated Ultrasound Scanner With AI Saves Time, Performs as Well as Rheumatologists
WASHINGTON — A fully automated ultrasound scanning system combined with artificial intelligence–based disease activity scoring performed as well as expert rheumatologists in hand joint assessment of patients with rheumatoid arthritis (RA), new research found.
The system, made by a Danish company called ROPCA, comprises an ultrasound scanner called ARTHUR (RA Ultrasound Robot) that interacts directly with the patient and scans 11 joints per hand and a neural network–based software system, DIANA (Diagnosis Aid Network for RA), that evaluates the images and monitors RA activity.
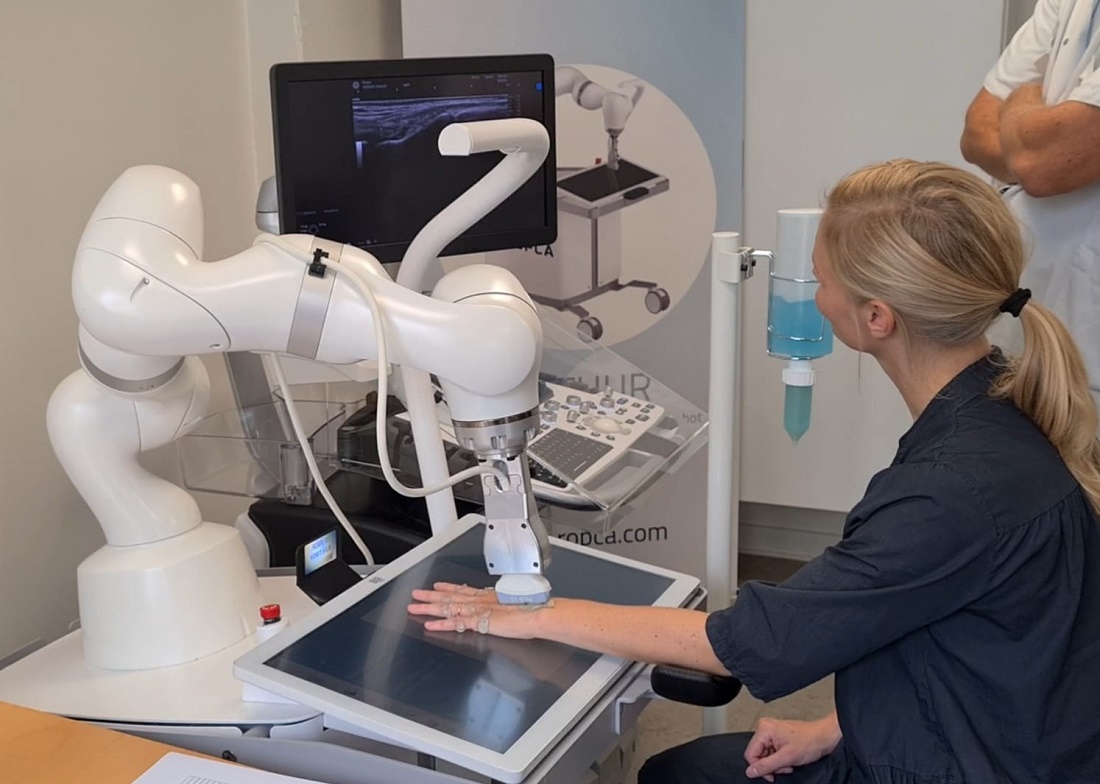
The combined system classifies the degree of RA according to the joint European Alliance of Associations for Rheumatology (EULAR)–Outcome Measures in Rheumatology (OMERACT) standards for RA diagnosis. It received a CE Mark in Europe in 2022 and is currently in use in six rheumatology clinics in Denmark, Germany, Switzerland, and Austria, with more to come, ROPCA Co-founder and Chief Medical Officer Søren A. Just, MD, said in an interview.
“Automated systems could help rheumatologists in the early detection and monitoring of arthritis diseases. Systems can be placed or move in areas with insufficient rheumatological expertise,” Just said during a special late-breaker session presentation at the annual meeting of the American College of Rheumatology (ACR).
He said in an interview: “Currently, there are so many people referred and few and fewer rheumatologists. So we need to think differently. We need good automated assistants.” As a screening tool, the system can determine whether a person with hand pain has RA or just osteoarthritis “and also can give the patient an immediate answer, instead of waiting sometimes up to 6 months to get the information.”
Just, who is also a senior physician in the Department of Internal Medicine at Odense University Hospital in Denmark, said that his department is also using the system to assess flares in patients with established RA. “They can have a blood sample taken. They’re scanned by the robot, and you can see if there is any disease activity. But I think that screening of patients with joint pain is the beginning.”
Asked to comment, session moderator Gregory C. Gardner, MD, Emeritus Professor in the Division of Rheumatology at the University of Washington, Seattle, and a member of the ACR conference program committee, said in an interview “one of the reasons we chose to feature this abstract is because we’re interested in science at the convergence. We really thought this was a potential way to move the field forward for rheumatologists.”
Gardner said it’s an advantage that the patient could potentially have an ARTHUR scan with a DIANA report and get blood tests done prior to a visit with the rheumatologist. “It’s really time-consuming for a human to do these studies, so if you automate it, that’s a step forward in terms of having the data available for the rheumatologist to view and use sequentially to follow how patients are doing.”
When introducing Just’s presentation, Gardner called it “the coolest abstract of the meeting.”
Both DIANA and ARTHUR Performed At Least as Well as Human Rheumatologists
In the study, 30 patients with RA underwent two scans by ARTHUR, followed by a scan from a rheumatologist specialist in musculoskeletal ultrasound. The scans were sent to DIANA, who graded the images according to the Global OMERACT-EULAR Synovitis Score, as did the human rheumatologist.
A “ground truth” was established by another human expert who evaluated both ARTHUR’s and the other rheumatologist’s images, blinded to the scanning method. The image with the highest disease activity was deemed “ground truth,” and agreement with that was assessed for the two individual methods.

Just showed a video of a patient being scanned by ARTHUR. The machine verbally guided her through removing her jewelry, applying the gel, and placing her hand on the screen under the scanner. ARTHUR’s arm moved around on the patient’s hand, locating the best angles to take grayscale images and Doppler images and Doppler video. The scan takes 15-20 minutes, and the images are stored, Just said.
The study patients had a mean age of 65 years, and 23 of the 30 were men. Their average disease duration was 11 years, and mean Disease Activity Score in 28 joints using C-reactive protein was 3.86, indicating moderate disease. A majority (73%) of patients were taking disease-modifying antirheumatic drugs, and about one third were taking biologics. ARTHUR scanned a total of 660 joints, and 564 scans were successful.
For repeatability between the two ARTHUR scans, percent exact agreement was 63% for synovial hypertrophy, 75% for Doppler activity, and 60% combined. Percent close (within a point) agreements were 93%, 94%, and 92%, respectively. Binary agreements as to whether the joint was healthy vs diseased were 88%, 91%, and 85%, respectively.
At the joint level, ARTHUR and DIANA’s percent exact agreement with ground truth was 49% for synovial hypertrophy, 63% for Doppler activity, and 48% combined. Binary agreements with disease vs healthy were 80%, 88%, and 78%, respectively.
The human rheumatologists scored very similarly. Percent exact agreement with ground truth was 51% for synovial hypertrophy, 64% for Doppler activity, and 50% combined. Percent close agreements were 94%, 94%, and 92%, respectively. And binary agreements with diseased vs healthy were 83%, 91%, and 80%, respectively.
At the patient level (all joints combined), ARTHUR and DIANA’s binary disease assessment of healthy vs disease showed agreement with the ground truth of 87% for synovial hypertrophy, 83% for Doppler activity, and 87% combined. Here, the rheumatologists scored lower, at 53%, 67%, and 60%, respectively.
“In this study, we think the precision of ARTHUR and DIANA was comparable to that of an experienced rheumatologist, at both the joint and patient level,” Just said.
Gardner pointed out another advantage of the system. “DIANA doesn’t get fatigued. ... With human reading, the precision may change based on the time of day or stress level. ... But with DIANA, you’re going to get consistent information.”
Just said that the Arthritis Foundation in Germany recently put ARTHUR and DIANA on a bus and took it to cities that lacked a rheumatologist. Patients lined up, answered a questionnaire, had blood drawn, and received their scans. A rheumatologist on the bus then interpreted the data and consulted with the individuals about their RA risk. “In the last trip, we screened 800 patients in 6 days. So there are definitely possibilities here.”
Just is co-owner of ROPCA. Gardner had no disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — A fully automated ultrasound scanning system combined with artificial intelligence–based disease activity scoring performed as well as expert rheumatologists in hand joint assessment of patients with rheumatoid arthritis (RA), new research found.
The system, made by a Danish company called ROPCA, comprises an ultrasound scanner called ARTHUR (RA Ultrasound Robot) that interacts directly with the patient and scans 11 joints per hand and a neural network–based software system, DIANA (Diagnosis Aid Network for RA), that evaluates the images and monitors RA activity.
The combined system classifies the degree of RA according to the joint European Alliance of Associations for Rheumatology (EULAR)–Outcome Measures in Rheumatology (OMERACT) standards for RA diagnosis. It received a CE Mark in Europe in 2022 and is currently in use in six rheumatology clinics in Denmark, Germany, Switzerland, and Austria, with more to come, ROPCA Co-founder and Chief Medical Officer Søren A. Just, MD, said in an interview.
“Automated systems could help rheumatologists in the early detection and monitoring of arthritis diseases. Systems can be placed or move in areas with insufficient rheumatological expertise,” Just said during a special late-breaker session presentation at the annual meeting of the American College of Rheumatology (ACR).
He said in an interview: “Currently, there are so many people referred and few and fewer rheumatologists. So we need to think differently. We need good automated assistants.” As a screening tool, the system can determine whether a person with hand pain has RA or just osteoarthritis “and also can give the patient an immediate answer, instead of waiting sometimes up to 6 months to get the information.”
Just, who is also a senior physician in the Department of Internal Medicine at Odense University Hospital in Denmark, said that his department is also using the system to assess flares in patients with established RA. “They can have a blood sample taken. They’re scanned by the robot, and you can see if there is any disease activity. But I think that screening of patients with joint pain is the beginning.”
Asked to comment, session moderator Gregory C. Gardner, MD, Emeritus Professor in the Division of Rheumatology at the University of Washington, Seattle, and a member of the ACR conference program committee, said in an interview “one of the reasons we chose to feature this abstract is because we’re interested in science at the convergence. We really thought this was a potential way to move the field forward for rheumatologists.”
Gardner said it’s an advantage that the patient could potentially have an ARTHUR scan with a DIANA report and get blood tests done prior to a visit with the rheumatologist. “It’s really time-consuming for a human to do these studies, so if you automate it, that’s a step forward in terms of having the data available for the rheumatologist to view and use sequentially to follow how patients are doing.”
When introducing Just’s presentation, Gardner called it “the coolest abstract of the meeting.”
Both DIANA and ARTHUR Performed At Least as Well as Human Rheumatologists
In the study, 30 patients with RA underwent two scans by ARTHUR, followed by a scan from a rheumatologist specialist in musculoskeletal ultrasound. The scans were sent to DIANA, who graded the images according to the Global OMERACT-EULAR Synovitis Score, as did the human rheumatologist.
A “ground truth” was established by another human expert who evaluated both ARTHUR’s and the other rheumatologist’s images, blinded to the scanning method. The image with the highest disease activity was deemed “ground truth,” and agreement with that was assessed for the two individual methods.
Just showed a video of a patient being scanned by ARTHUR. The machine verbally guided her through removing her jewelry, applying the gel, and placing her hand on the screen under the scanner. ARTHUR’s arm moved around on the patient’s hand, locating the best angles to take grayscale images and Doppler images and Doppler video. The scan takes 15-20 minutes, and the images are stored, Just said.
The study patients had a mean age of 65 years, and 23 of the 30 were men. Their average disease duration was 11 years, and mean Disease Activity Score in 28 joints using C-reactive protein was 3.86, indicating moderate disease. A majority (73%) of patients were taking disease-modifying antirheumatic drugs, and about one third were taking biologics. ARTHUR scanned a total of 660 joints, and 564 scans were successful.
For repeatability between the two ARTHUR scans, percent exact agreement was 63% for synovial hypertrophy, 75% for Doppler activity, and 60% combined. Percent close (within a point) agreements were 93%, 94%, and 92%, respectively. Binary agreements as to whether the joint was healthy vs diseased were 88%, 91%, and 85%, respectively.
At the joint level, ARTHUR and DIANA’s percent exact agreement with ground truth was 49% for synovial hypertrophy, 63% for Doppler activity, and 48% combined. Binary agreements with disease vs healthy were 80%, 88%, and 78%, respectively.
The human rheumatologists scored very similarly. Percent exact agreement with ground truth was 51% for synovial hypertrophy, 64% for Doppler activity, and 50% combined. Percent close agreements were 94%, 94%, and 92%, respectively. And binary agreements with diseased vs healthy were 83%, 91%, and 80%, respectively.
At the patient level (all joints combined), ARTHUR and DIANA’s binary disease assessment of healthy vs disease showed agreement with the ground truth of 87% for synovial hypertrophy, 83% for Doppler activity, and 87% combined. Here, the rheumatologists scored lower, at 53%, 67%, and 60%, respectively.
“In this study, we think the precision of ARTHUR and DIANA was comparable to that of an experienced rheumatologist, at both the joint and patient level,” Just said.
Gardner pointed out another advantage of the system. “DIANA doesn’t get fatigued. ... With human reading, the precision may change based on the time of day or stress level. ... But with DIANA, you’re going to get consistent information.”
Just said that the Arthritis Foundation in Germany recently put ARTHUR and DIANA on a bus and took it to cities that lacked a rheumatologist. Patients lined up, answered a questionnaire, had blood drawn, and received their scans. A rheumatologist on the bus then interpreted the data and consulted with the individuals about their RA risk. “In the last trip, we screened 800 patients in 6 days. So there are definitely possibilities here.”
Just is co-owner of ROPCA. Gardner had no disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — A fully automated ultrasound scanning system combined with artificial intelligence–based disease activity scoring performed as well as expert rheumatologists in hand joint assessment of patients with rheumatoid arthritis (RA), new research found.
The system, made by a Danish company called ROPCA, comprises an ultrasound scanner called ARTHUR (RA Ultrasound Robot) that interacts directly with the patient and scans 11 joints per hand and a neural network–based software system, DIANA (Diagnosis Aid Network for RA), that evaluates the images and monitors RA activity.
The combined system classifies the degree of RA according to the joint European Alliance of Associations for Rheumatology (EULAR)–Outcome Measures in Rheumatology (OMERACT) standards for RA diagnosis. It received a CE Mark in Europe in 2022 and is currently in use in six rheumatology clinics in Denmark, Germany, Switzerland, and Austria, with more to come, ROPCA Co-founder and Chief Medical Officer Søren A. Just, MD, said in an interview.
“Automated systems could help rheumatologists in the early detection and monitoring of arthritis diseases. Systems can be placed or move in areas with insufficient rheumatological expertise,” Just said during a special late-breaker session presentation at the annual meeting of the American College of Rheumatology (ACR).
He said in an interview: “Currently, there are so many people referred and few and fewer rheumatologists. So we need to think differently. We need good automated assistants.” As a screening tool, the system can determine whether a person with hand pain has RA or just osteoarthritis “and also can give the patient an immediate answer, instead of waiting sometimes up to 6 months to get the information.”
Just, who is also a senior physician in the Department of Internal Medicine at Odense University Hospital in Denmark, said that his department is also using the system to assess flares in patients with established RA. “They can have a blood sample taken. They’re scanned by the robot, and you can see if there is any disease activity. But I think that screening of patients with joint pain is the beginning.”
Asked to comment, session moderator Gregory C. Gardner, MD, Emeritus Professor in the Division of Rheumatology at the University of Washington, Seattle, and a member of the ACR conference program committee, said in an interview “one of the reasons we chose to feature this abstract is because we’re interested in science at the convergence. We really thought this was a potential way to move the field forward for rheumatologists.”
Gardner said it’s an advantage that the patient could potentially have an ARTHUR scan with a DIANA report and get blood tests done prior to a visit with the rheumatologist. “It’s really time-consuming for a human to do these studies, so if you automate it, that’s a step forward in terms of having the data available for the rheumatologist to view and use sequentially to follow how patients are doing.”
When introducing Just’s presentation, Gardner called it “the coolest abstract of the meeting.”
Both DIANA and ARTHUR Performed At Least as Well as Human Rheumatologists
In the study, 30 patients with RA underwent two scans by ARTHUR, followed by a scan from a rheumatologist specialist in musculoskeletal ultrasound. The scans were sent to DIANA, who graded the images according to the Global OMERACT-EULAR Synovitis Score, as did the human rheumatologist.
A “ground truth” was established by another human expert who evaluated both ARTHUR’s and the other rheumatologist’s images, blinded to the scanning method. The image with the highest disease activity was deemed “ground truth,” and agreement with that was assessed for the two individual methods.
Just showed a video of a patient being scanned by ARTHUR. The machine verbally guided her through removing her jewelry, applying the gel, and placing her hand on the screen under the scanner. ARTHUR’s arm moved around on the patient’s hand, locating the best angles to take grayscale images and Doppler images and Doppler video. The scan takes 15-20 minutes, and the images are stored, Just said.
The study patients had a mean age of 65 years, and 23 of the 30 were men. Their average disease duration was 11 years, and mean Disease Activity Score in 28 joints using C-reactive protein was 3.86, indicating moderate disease. A majority (73%) of patients were taking disease-modifying antirheumatic drugs, and about one third were taking biologics. ARTHUR scanned a total of 660 joints, and 564 scans were successful.
For repeatability between the two ARTHUR scans, percent exact agreement was 63% for synovial hypertrophy, 75% for Doppler activity, and 60% combined. Percent close (within a point) agreements were 93%, 94%, and 92%, respectively. Binary agreements as to whether the joint was healthy vs diseased were 88%, 91%, and 85%, respectively.
At the joint level, ARTHUR and DIANA’s percent exact agreement with ground truth was 49% for synovial hypertrophy, 63% for Doppler activity, and 48% combined. Binary agreements with disease vs healthy were 80%, 88%, and 78%, respectively.
The human rheumatologists scored very similarly. Percent exact agreement with ground truth was 51% for synovial hypertrophy, 64% for Doppler activity, and 50% combined. Percent close agreements were 94%, 94%, and 92%, respectively. And binary agreements with diseased vs healthy were 83%, 91%, and 80%, respectively.
At the patient level (all joints combined), ARTHUR and DIANA’s binary disease assessment of healthy vs disease showed agreement with the ground truth of 87% for synovial hypertrophy, 83% for Doppler activity, and 87% combined. Here, the rheumatologists scored lower, at 53%, 67%, and 60%, respectively.
“In this study, we think the precision of ARTHUR and DIANA was comparable to that of an experienced rheumatologist, at both the joint and patient level,” Just said.
Gardner pointed out another advantage of the system. “DIANA doesn’t get fatigued. ... With human reading, the precision may change based on the time of day or stress level. ... But with DIANA, you’re going to get consistent information.”
Just said that the Arthritis Foundation in Germany recently put ARTHUR and DIANA on a bus and took it to cities that lacked a rheumatologist. Patients lined up, answered a questionnaire, had blood drawn, and received their scans. A rheumatologist on the bus then interpreted the data and consulted with the individuals about their RA risk. “In the last trip, we screened 800 patients in 6 days. So there are definitely possibilities here.”
Just is co-owner of ROPCA. Gardner had no disclosures.
A version of this article appeared on Medscape.com.
FROM ACR 2024
Uric Acid Levels, Gout Symptoms Improved With Plant-Based Diet in Pilot Trial
A Mediterranean-inspired plant-based diet improved self-reported measures of gout as well as uric acid levels, a pilot study has found.
There hasn’t been a lot of research on diet in gout, according to Anna Kretova, RD, who presented the study at the annual research symposium of the Gout Hyperuricemia and Crystal-Associated Disease Network. She noted that a 2019 systematic review of low-calorie diets, low-purine diets, and Mediterranean diets found that uric acid levels below 0.6 mmol/L were achieved only in those on the Mediterranean diet (Nutrients. 2019 Dec 4;11[12]:2955). A 2020 study compared a low-fat, high-carbohydrate, plant-based diet vs an animal-based, ketogenic diet in healthy individuals. After 2 weeks, uric acid levels increased in those on the animal-based, low-carb diet and decreased in those on the plant-based diet.
Some foods are considered to be proinflammatory and generally come from animal origins, including saturated fats and animal protein in addition to ultraprocessed foods. Foods that have anti-inflammatory properties are mostly plant based and unprocessed and often rich in fiber. “From recent interventional studies, we also know that the whole-foods plant-based diet has shown to be effective as treatments of the main comorbidities of gout, such as obesity, cardiovascular disease, or [osteoarthritis],” said Kretova, who is a registered dietitian and a researcher at the Reade Rehabilitation and Rheumatology Center, Amsterdam, the Netherlands.
Those findings led the researchers to develop a whole-foods, plant-based diet and test its effect on serum uric acid in patients with gout, as well as gout disease activity and cardiovascular disease risk. Participants could not eat meat, fish, eggs, or dairy.
The trial included 33 individuals with gout who were randomized to a 16-week intervention with five consultations with a registered dietitian (n = 18) or a wait-list control group (n = 15) who received standard care. The mean age overall was 52 years, and 91% were men. The mean body mass index (BMI) was 32.6 kg/m2, and the median uric acid level was 0.50 mmol/L (8.4 mg/dL).
Among gout-related outcomes, the researchers noted improvements in gout severity as measured by visual analog scale (VAS; between group difference, –2.0; P =.01), pain as measured by VAS (between group difference, –2.0; P =.04), and uric acid levels after adjustment for age, sex, and BMI (between group difference, –0.05 mmol/L, P =.004). There were also improvements in the intervention group in weight loss (between group difference, –5.3 kg; P <.0001), BMI (between group difference, –1.7; P < .0001), waist circumference (between group difference, –3.9 cm; P = .004), and low-density lipoprotein (LDL) cholesterol (between group difference, –0.5; P = .007).
At 16 weeks, “we concluded that a Mediterranean-inspired whole-foods, plant-based diet significantly lowers serum uric acid in patients with gout and abdominal obesity, and additionally, the diet reduces gout-related pain and disease activity, promotes substantial weight loss, decreases weight circumference, and improves LDL cholesterol levels, and thus decreases [cardiovascular disease] risk in these patients,” Kretova said.
She added that some might question whether a uric acid reduction of –0.05 mmol/L is clinically relevant. “We would argue it is because of the strong decrease in disease activity and pain in the intervention group,” Kretova said.
The study is limited by its small size, the fact that it was not blinded, and the 4-month duration, which might be too short to capture potential indirect effects of diet on hyperuricemia and chronic inflammation, Kretova said. The group is planning to follow participants out to 12 months in an extension study.
During the Q&A session after the presentation, an audience member asked if the participants were vegetarians before they entered the study, and whether the dietary change could be sustained. “It’s a very good proof-of-concept study, but whether an intervention based entirely on plant-based therapy will be something that patients will be able to adhere to long term [is uncertain],” Kretova said.
She was optimistic, even though the participants generally enjoyed food and ate a lot of red meat. “I think there will be a gradation of people who can sustain and who cannot sustain [the diet]. From what we saw, people actually found it easier to follow than they expected, and a lot of participants changed their diet permanently for the better. Not everyone became [entirely] plant-based, but they became much more plant-based than they expected from themselves. So, it is definitely feasible,” she said.
Kretova reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
A Mediterranean-inspired plant-based diet improved self-reported measures of gout as well as uric acid levels, a pilot study has found.
There hasn’t been a lot of research on diet in gout, according to Anna Kretova, RD, who presented the study at the annual research symposium of the Gout Hyperuricemia and Crystal-Associated Disease Network. She noted that a 2019 systematic review of low-calorie diets, low-purine diets, and Mediterranean diets found that uric acid levels below 0.6 mmol/L were achieved only in those on the Mediterranean diet (Nutrients. 2019 Dec 4;11[12]:2955). A 2020 study compared a low-fat, high-carbohydrate, plant-based diet vs an animal-based, ketogenic diet in healthy individuals. After 2 weeks, uric acid levels increased in those on the animal-based, low-carb diet and decreased in those on the plant-based diet.
Some foods are considered to be proinflammatory and generally come from animal origins, including saturated fats and animal protein in addition to ultraprocessed foods. Foods that have anti-inflammatory properties are mostly plant based and unprocessed and often rich in fiber. “From recent interventional studies, we also know that the whole-foods plant-based diet has shown to be effective as treatments of the main comorbidities of gout, such as obesity, cardiovascular disease, or [osteoarthritis],” said Kretova, who is a registered dietitian and a researcher at the Reade Rehabilitation and Rheumatology Center, Amsterdam, the Netherlands.
Those findings led the researchers to develop a whole-foods, plant-based diet and test its effect on serum uric acid in patients with gout, as well as gout disease activity and cardiovascular disease risk. Participants could not eat meat, fish, eggs, or dairy.
The trial included 33 individuals with gout who were randomized to a 16-week intervention with five consultations with a registered dietitian (n = 18) or a wait-list control group (n = 15) who received standard care. The mean age overall was 52 years, and 91% were men. The mean body mass index (BMI) was 32.6 kg/m2, and the median uric acid level was 0.50 mmol/L (8.4 mg/dL).
Among gout-related outcomes, the researchers noted improvements in gout severity as measured by visual analog scale (VAS; between group difference, –2.0; P =.01), pain as measured by VAS (between group difference, –2.0; P =.04), and uric acid levels after adjustment for age, sex, and BMI (between group difference, –0.05 mmol/L, P =.004). There were also improvements in the intervention group in weight loss (between group difference, –5.3 kg; P <.0001), BMI (between group difference, –1.7; P < .0001), waist circumference (between group difference, –3.9 cm; P = .004), and low-density lipoprotein (LDL) cholesterol (between group difference, –0.5; P = .007).
At 16 weeks, “we concluded that a Mediterranean-inspired whole-foods, plant-based diet significantly lowers serum uric acid in patients with gout and abdominal obesity, and additionally, the diet reduces gout-related pain and disease activity, promotes substantial weight loss, decreases weight circumference, and improves LDL cholesterol levels, and thus decreases [cardiovascular disease] risk in these patients,” Kretova said.
She added that some might question whether a uric acid reduction of –0.05 mmol/L is clinically relevant. “We would argue it is because of the strong decrease in disease activity and pain in the intervention group,” Kretova said.
The study is limited by its small size, the fact that it was not blinded, and the 4-month duration, which might be too short to capture potential indirect effects of diet on hyperuricemia and chronic inflammation, Kretova said. The group is planning to follow participants out to 12 months in an extension study.
During the Q&A session after the presentation, an audience member asked if the participants were vegetarians before they entered the study, and whether the dietary change could be sustained. “It’s a very good proof-of-concept study, but whether an intervention based entirely on plant-based therapy will be something that patients will be able to adhere to long term [is uncertain],” Kretova said.
She was optimistic, even though the participants generally enjoyed food and ate a lot of red meat. “I think there will be a gradation of people who can sustain and who cannot sustain [the diet]. From what we saw, people actually found it easier to follow than they expected, and a lot of participants changed their diet permanently for the better. Not everyone became [entirely] plant-based, but they became much more plant-based than they expected from themselves. So, it is definitely feasible,” she said.
Kretova reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
A Mediterranean-inspired plant-based diet improved self-reported measures of gout as well as uric acid levels, a pilot study has found.
There hasn’t been a lot of research on diet in gout, according to Anna Kretova, RD, who presented the study at the annual research symposium of the Gout Hyperuricemia and Crystal-Associated Disease Network. She noted that a 2019 systematic review of low-calorie diets, low-purine diets, and Mediterranean diets found that uric acid levels below 0.6 mmol/L were achieved only in those on the Mediterranean diet (Nutrients. 2019 Dec 4;11[12]:2955). A 2020 study compared a low-fat, high-carbohydrate, plant-based diet vs an animal-based, ketogenic diet in healthy individuals. After 2 weeks, uric acid levels increased in those on the animal-based, low-carb diet and decreased in those on the plant-based diet.
Some foods are considered to be proinflammatory and generally come from animal origins, including saturated fats and animal protein in addition to ultraprocessed foods. Foods that have anti-inflammatory properties are mostly plant based and unprocessed and often rich in fiber. “From recent interventional studies, we also know that the whole-foods plant-based diet has shown to be effective as treatments of the main comorbidities of gout, such as obesity, cardiovascular disease, or [osteoarthritis],” said Kretova, who is a registered dietitian and a researcher at the Reade Rehabilitation and Rheumatology Center, Amsterdam, the Netherlands.
Those findings led the researchers to develop a whole-foods, plant-based diet and test its effect on serum uric acid in patients with gout, as well as gout disease activity and cardiovascular disease risk. Participants could not eat meat, fish, eggs, or dairy.
The trial included 33 individuals with gout who were randomized to a 16-week intervention with five consultations with a registered dietitian (n = 18) or a wait-list control group (n = 15) who received standard care. The mean age overall was 52 years, and 91% were men. The mean body mass index (BMI) was 32.6 kg/m2, and the median uric acid level was 0.50 mmol/L (8.4 mg/dL).
Among gout-related outcomes, the researchers noted improvements in gout severity as measured by visual analog scale (VAS; between group difference, –2.0; P =.01), pain as measured by VAS (between group difference, –2.0; P =.04), and uric acid levels after adjustment for age, sex, and BMI (between group difference, –0.05 mmol/L, P =.004). There were also improvements in the intervention group in weight loss (between group difference, –5.3 kg; P <.0001), BMI (between group difference, –1.7; P < .0001), waist circumference (between group difference, –3.9 cm; P = .004), and low-density lipoprotein (LDL) cholesterol (between group difference, –0.5; P = .007).
At 16 weeks, “we concluded that a Mediterranean-inspired whole-foods, plant-based diet significantly lowers serum uric acid in patients with gout and abdominal obesity, and additionally, the diet reduces gout-related pain and disease activity, promotes substantial weight loss, decreases weight circumference, and improves LDL cholesterol levels, and thus decreases [cardiovascular disease] risk in these patients,” Kretova said.
She added that some might question whether a uric acid reduction of –0.05 mmol/L is clinically relevant. “We would argue it is because of the strong decrease in disease activity and pain in the intervention group,” Kretova said.
The study is limited by its small size, the fact that it was not blinded, and the 4-month duration, which might be too short to capture potential indirect effects of diet on hyperuricemia and chronic inflammation, Kretova said. The group is planning to follow participants out to 12 months in an extension study.
During the Q&A session after the presentation, an audience member asked if the participants were vegetarians before they entered the study, and whether the dietary change could be sustained. “It’s a very good proof-of-concept study, but whether an intervention based entirely on plant-based therapy will be something that patients will be able to adhere to long term [is uncertain],” Kretova said.
She was optimistic, even though the participants generally enjoyed food and ate a lot of red meat. “I think there will be a gradation of people who can sustain and who cannot sustain [the diet]. From what we saw, people actually found it easier to follow than they expected, and a lot of participants changed their diet permanently for the better. Not everyone became [entirely] plant-based, but they became much more plant-based than they expected from themselves. So, it is definitely feasible,” she said.
Kretova reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
FROM G-CAN 2024
Phase 3 Lupus Trial Shows Promising Results for Dapirolizumab Pegol
WASHINGTON — The investigational anti-CD40 ligand agent dapirolizumab pegol (DZP) outperformed placebo in improving disease activity and reducing high-dose corticosteroid use in patients with systemic lupus erythematosus (SLE) in the phase 3 PHOENYCS GO trial.
“We really think that dapirolizumab pegol may represent a novel treatment for lupus, particularly given its broad immune modulatory effects,” said Megan Clowse, MD, MPH, associate professor of medicine and chief of the Division of Rheumatology and Immunology at Duke University School of Medicine in Durham, North Carolina. She presented the study in a late-breaking poster session at the American College of Rheumatology (ACR) 2024 Annual Meeting.
There is a “huge unmet need” for drugs for lupus, Clowse told this news organization. Patients with SLE continue to have high disease burden, including ongoing symptoms often driven by inflammation. Corticosteroids are often the best medications to control disease activity, she said, but they can result in long-term toxicity.
What Makes DZP Unique?
Through CD40 ligand signaling, DZP has been shown to reduce B- and T-cell activation and to downregulate interferon pathways. Previous antibodies targeting the CD40 ligand have been associated with an increased risk for thromboembolic events. However, DZP lacks the Fc portion of the antibody, which can bind to platelets and cause clotting. Data from phase 1, 2, and 3 trials thus far do not show an elevated risk for these events, Clowse explained. In fact, safety signals were strong enough that patients with antiphospholipid antibodies — a key driver for blood clots in patients with SLE — were included in the trial.
In PHOENYCS GO, investigators enrolled 321 patients with moderate to severe SLE with persistently active or frequently flaring/relapsing-remitting disease activity despite stable standard of care (SOC) medications such as antimalarials, corticosteroids, and immunosuppressants.
Patients were randomized 2:1 to receive intravenous DZP (24 mg/kg) plus SOC or intravenous placebo plus SOC every 4 weeks, with patients and investigators blinded to treatment assignments.
Patients taking a corticosteroid dose > 7.5 mg/day began a mandatory steroid taper by week 8 of the trial, with the goal of reducing that to < 7.5 mg/day. The tapering regimen was at the discretion of providers and was adapted to each patient’s individual disease activity.
The primary endpoint was British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA) response at week 48.
Patients in the DZP and placebo groups were on average 43.5 and 41.5 years old, respectively. More than 90% of patients were women, all on concomitant SLE medications. About half of the participants took a daily corticosteroid dose > 7.5 mg.
At 48 weeks, half of the DZP group (49.5%) achieved BICLA response compared with 34.6% in the placebo group (P = .0110). A higher proportion of patients taking DZP achieved SLE Responder Index-4 response than those taking placebo (60.1% vs 41.1%, respectively; P = .0014), and the rate of severe British Isles Lupus Assessment Group flares in the DZP group was half that of the placebo group (11.6% vs 23.4%; P = .0257). In the subgroup of patients who underwent corticosteroid tapering, 72.4% receiving DZP and 52.9% taking placebo reduced their dose to < 7.5 mg/day by 48 weeks (P = .0404).
DZP was generally well tolerated. Over 48 weeks, 82.6% of the DZP group and 75% of the placebo group reported treatment-emergent adverse events, but serious occurrences were more common in the placebo group (14.8%) than in the DZP group (9.9%). Herpes viral infections were higher in the placebo group, although there were three ophthalmic herpes cases in the DZP group. There was one case of acute myocardial infarction and one death linked to gangrene-related sepsis in patients receiving DZP.
A ‘Mild to Moderate’ Response
Although these are definitely positive results, they show a “mild to moderate response” to DZP, commented Gregory Gardner, MD, an emeritus professor in the Division of Rheumatology at the University of Washington, Seattle, and chair of the American College of Rheumatology’s annual meeting planning committee. He moderated the session where the research was presented. Although DZP showed efficacy among some patients, he noted, “there were still 51% patients that it didn’t work for.”
The drug uses an alternative pathway to current lupus drugs, Gardner added, and more research is needed to understand how best to use this medication in practice.
Clowse noted that DZP could be particularly beneficial for patients with SLE who want to get pregnant. Many drugs used to treat the disease are teratogenic; however, “because of the lack of Fc portion on this drug, it very likely does not cross the placenta in any kind of significant amount,” she said. Although there are not yet any reproductive safety data on DZP, she added, “that is a great potential niche.”
Biogen and UCB, which are jointly developing DZP, aim to start a second phase 3 trial of DZP in patients with SLE, called PHOENYCS FLY, in 2024.
The trial was sponsored by UCB. Clowse is a consultant and has received grant/research support from GSK and UCB. Gardner had no relevant disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — The investigational anti-CD40 ligand agent dapirolizumab pegol (DZP) outperformed placebo in improving disease activity and reducing high-dose corticosteroid use in patients with systemic lupus erythematosus (SLE) in the phase 3 PHOENYCS GO trial.
“We really think that dapirolizumab pegol may represent a novel treatment for lupus, particularly given its broad immune modulatory effects,” said Megan Clowse, MD, MPH, associate professor of medicine and chief of the Division of Rheumatology and Immunology at Duke University School of Medicine in Durham, North Carolina. She presented the study in a late-breaking poster session at the American College of Rheumatology (ACR) 2024 Annual Meeting.
There is a “huge unmet need” for drugs for lupus, Clowse told this news organization. Patients with SLE continue to have high disease burden, including ongoing symptoms often driven by inflammation. Corticosteroids are often the best medications to control disease activity, she said, but they can result in long-term toxicity.
What Makes DZP Unique?
Through CD40 ligand signaling, DZP has been shown to reduce B- and T-cell activation and to downregulate interferon pathways. Previous antibodies targeting the CD40 ligand have been associated with an increased risk for thromboembolic events. However, DZP lacks the Fc portion of the antibody, which can bind to platelets and cause clotting. Data from phase 1, 2, and 3 trials thus far do not show an elevated risk for these events, Clowse explained. In fact, safety signals were strong enough that patients with antiphospholipid antibodies — a key driver for blood clots in patients with SLE — were included in the trial.
In PHOENYCS GO, investigators enrolled 321 patients with moderate to severe SLE with persistently active or frequently flaring/relapsing-remitting disease activity despite stable standard of care (SOC) medications such as antimalarials, corticosteroids, and immunosuppressants.
Patients were randomized 2:1 to receive intravenous DZP (24 mg/kg) plus SOC or intravenous placebo plus SOC every 4 weeks, with patients and investigators blinded to treatment assignments.
Patients taking a corticosteroid dose > 7.5 mg/day began a mandatory steroid taper by week 8 of the trial, with the goal of reducing that to < 7.5 mg/day. The tapering regimen was at the discretion of providers and was adapted to each patient’s individual disease activity.
The primary endpoint was British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA) response at week 48.
Patients in the DZP and placebo groups were on average 43.5 and 41.5 years old, respectively. More than 90% of patients were women, all on concomitant SLE medications. About half of the participants took a daily corticosteroid dose > 7.5 mg.
At 48 weeks, half of the DZP group (49.5%) achieved BICLA response compared with 34.6% in the placebo group (P = .0110). A higher proportion of patients taking DZP achieved SLE Responder Index-4 response than those taking placebo (60.1% vs 41.1%, respectively; P = .0014), and the rate of severe British Isles Lupus Assessment Group flares in the DZP group was half that of the placebo group (11.6% vs 23.4%; P = .0257). In the subgroup of patients who underwent corticosteroid tapering, 72.4% receiving DZP and 52.9% taking placebo reduced their dose to < 7.5 mg/day by 48 weeks (P = .0404).
DZP was generally well tolerated. Over 48 weeks, 82.6% of the DZP group and 75% of the placebo group reported treatment-emergent adverse events, but serious occurrences were more common in the placebo group (14.8%) than in the DZP group (9.9%). Herpes viral infections were higher in the placebo group, although there were three ophthalmic herpes cases in the DZP group. There was one case of acute myocardial infarction and one death linked to gangrene-related sepsis in patients receiving DZP.
A ‘Mild to Moderate’ Response
Although these are definitely positive results, they show a “mild to moderate response” to DZP, commented Gregory Gardner, MD, an emeritus professor in the Division of Rheumatology at the University of Washington, Seattle, and chair of the American College of Rheumatology’s annual meeting planning committee. He moderated the session where the research was presented. Although DZP showed efficacy among some patients, he noted, “there were still 51% patients that it didn’t work for.”
The drug uses an alternative pathway to current lupus drugs, Gardner added, and more research is needed to understand how best to use this medication in practice.
Clowse noted that DZP could be particularly beneficial for patients with SLE who want to get pregnant. Many drugs used to treat the disease are teratogenic; however, “because of the lack of Fc portion on this drug, it very likely does not cross the placenta in any kind of significant amount,” she said. Although there are not yet any reproductive safety data on DZP, she added, “that is a great potential niche.”
Biogen and UCB, which are jointly developing DZP, aim to start a second phase 3 trial of DZP in patients with SLE, called PHOENYCS FLY, in 2024.
The trial was sponsored by UCB. Clowse is a consultant and has received grant/research support from GSK and UCB. Gardner had no relevant disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — The investigational anti-CD40 ligand agent dapirolizumab pegol (DZP) outperformed placebo in improving disease activity and reducing high-dose corticosteroid use in patients with systemic lupus erythematosus (SLE) in the phase 3 PHOENYCS GO trial.
“We really think that dapirolizumab pegol may represent a novel treatment for lupus, particularly given its broad immune modulatory effects,” said Megan Clowse, MD, MPH, associate professor of medicine and chief of the Division of Rheumatology and Immunology at Duke University School of Medicine in Durham, North Carolina. She presented the study in a late-breaking poster session at the American College of Rheumatology (ACR) 2024 Annual Meeting.
There is a “huge unmet need” for drugs for lupus, Clowse told this news organization. Patients with SLE continue to have high disease burden, including ongoing symptoms often driven by inflammation. Corticosteroids are often the best medications to control disease activity, she said, but they can result in long-term toxicity.
What Makes DZP Unique?
Through CD40 ligand signaling, DZP has been shown to reduce B- and T-cell activation and to downregulate interferon pathways. Previous antibodies targeting the CD40 ligand have been associated with an increased risk for thromboembolic events. However, DZP lacks the Fc portion of the antibody, which can bind to platelets and cause clotting. Data from phase 1, 2, and 3 trials thus far do not show an elevated risk for these events, Clowse explained. In fact, safety signals were strong enough that patients with antiphospholipid antibodies — a key driver for blood clots in patients with SLE — were included in the trial.
In PHOENYCS GO, investigators enrolled 321 patients with moderate to severe SLE with persistently active or frequently flaring/relapsing-remitting disease activity despite stable standard of care (SOC) medications such as antimalarials, corticosteroids, and immunosuppressants.
Patients were randomized 2:1 to receive intravenous DZP (24 mg/kg) plus SOC or intravenous placebo plus SOC every 4 weeks, with patients and investigators blinded to treatment assignments.
Patients taking a corticosteroid dose > 7.5 mg/day began a mandatory steroid taper by week 8 of the trial, with the goal of reducing that to < 7.5 mg/day. The tapering regimen was at the discretion of providers and was adapted to each patient’s individual disease activity.
The primary endpoint was British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA) response at week 48.
Patients in the DZP and placebo groups were on average 43.5 and 41.5 years old, respectively. More than 90% of patients were women, all on concomitant SLE medications. About half of the participants took a daily corticosteroid dose > 7.5 mg.
At 48 weeks, half of the DZP group (49.5%) achieved BICLA response compared with 34.6% in the placebo group (P = .0110). A higher proportion of patients taking DZP achieved SLE Responder Index-4 response than those taking placebo (60.1% vs 41.1%, respectively; P = .0014), and the rate of severe British Isles Lupus Assessment Group flares in the DZP group was half that of the placebo group (11.6% vs 23.4%; P = .0257). In the subgroup of patients who underwent corticosteroid tapering, 72.4% receiving DZP and 52.9% taking placebo reduced their dose to < 7.5 mg/day by 48 weeks (P = .0404).
DZP was generally well tolerated. Over 48 weeks, 82.6% of the DZP group and 75% of the placebo group reported treatment-emergent adverse events, but serious occurrences were more common in the placebo group (14.8%) than in the DZP group (9.9%). Herpes viral infections were higher in the placebo group, although there were three ophthalmic herpes cases in the DZP group. There was one case of acute myocardial infarction and one death linked to gangrene-related sepsis in patients receiving DZP.
A ‘Mild to Moderate’ Response
Although these are definitely positive results, they show a “mild to moderate response” to DZP, commented Gregory Gardner, MD, an emeritus professor in the Division of Rheumatology at the University of Washington, Seattle, and chair of the American College of Rheumatology’s annual meeting planning committee. He moderated the session where the research was presented. Although DZP showed efficacy among some patients, he noted, “there were still 51% patients that it didn’t work for.”
The drug uses an alternative pathway to current lupus drugs, Gardner added, and more research is needed to understand how best to use this medication in practice.
Clowse noted that DZP could be particularly beneficial for patients with SLE who want to get pregnant. Many drugs used to treat the disease are teratogenic; however, “because of the lack of Fc portion on this drug, it very likely does not cross the placenta in any kind of significant amount,” she said. Although there are not yet any reproductive safety data on DZP, she added, “that is a great potential niche.”
Biogen and UCB, which are jointly developing DZP, aim to start a second phase 3 trial of DZP in patients with SLE, called PHOENYCS FLY, in 2024.
The trial was sponsored by UCB. Clowse is a consultant and has received grant/research support from GSK and UCB. Gardner had no relevant disclosures.
A version of this article appeared on Medscape.com.
FROM ACR 2024
New Gout Remission Criteria Approved
In a nearly unanimous vote at the annual research symposium of the Gout Hyperuricemia and Crystal-Associated Disease Network (G-CAN), members approved a revision to gout remission criteria first established in 2016. The new version simplifies the definition in response to patient comments that the earlier version was redundant in some areas.
The previous version was developed following deliberations by 49 clinicians and researchers with experience in gout. They settled on a definition of gout remission that included five criteria:
- Serum urate levels lower than 0.36 mmol/L measured at least twice over 12 months, with no intervening values of 0.36 mmol/L or higher
- No gout flares over 12 months
- No tophi
- Pain score due to gout < 2 at least twice over 12 months on a 10-point Likert scale or 10-cm visual analog scale, with no intervening values ≥ 2
- Patient global assessment of gout disease activity < 2 on a 10-point Likert scale or 10-cm visual analog scale, with no intervening values of ≥ 2.
Some participants reported that patients sometimes misattributed pain from other sources while using patient-reported outcomes (PROs). The argument for keeping PROs was that they are validated measures and endorsed by Outcome Measures in Rheumatology. Nevertheless, there was no direct patient involvement in the development of the 2016 criteria.
Researchers later interviewed 20 individuals with well-controlled gout to get their feedback on the 2016 criteria. Those individuals endorsed the existing criteria and did not suggest any new ones, but they suggested that the pain due to gout and the absence of gout flares were redundant measures. One said: “If you have no flare-ups, you’ve got no pain; it sort of answers itself.”
“That was a bit challenging for us because it wasn’t quite what we expected, but I think it did make us look again at the definition and think about whether we could simplify the definition further,” Nicola Dalbeth, MBChB, said during a presentation at G-CAN. Dalbeth is an academic rheumatologist at the University of Auckland, in New Zealand, who was also the lead author of the original criteria.
Simplified Version Created With Only Three Criteria
In response to these points, researchers produced a revised version with only three criteria, including the serum urate, absence of gout flares, and absence of subcutaneous tophi at the time of assessment.
To determine if the simplified criteria performed well, they compared the original and revised remission criteria in the context of the CARES trial, the Nottingham nurse-led trial, and randomized controlled trials in patients with gout that were conducted in New Zealand (here and here).
Dansoa Tabi-Amponsah, a PhD candidate at the University of Auckland, presented results of a study comparing the two versions in the Nottingham trial, which included 517 participants who received nurse-led or usual general practitioner care. The nurse-led care included education, regular follow-up and serum urate testing, individualized advice on gout flare management, and escalation of urate-lowering therapy with a treat-to-target strategy.
Both definitions demonstrated a link between the nurse-led strategy and increased rates of remission at year 1 and year 2, although the simplified definition found that more patients were in remission (17.6% vs 9.9% at year 1 and 42.7% vs 28.4% at year 2, both P < .001). “This is something we’ve seen across all of our analyses,” said Tabi-Amponsah.
Both criteria also found significant differences in remission rates between the nurse-led group in year 2 vs year 1 but not in the usual care group.
Participants who achieved remission had better gout impact scale scores in areas like worrying that a gout attack will occur, fears of worsening gout, and concerns about the impact of gout on future activities. “This is important because during that qualitative study, a key aspect of being in remission was no longer being worried about their gout, no longer feeling anxious about having constant gout flares, and having control over their gout. So, it’s important to note that despite the absence of PROs in that simplified definition, it’s still able to align with the patients’ perspectives of their disease state,” said Tabi-Amponsah.
During the Q&A period after her talk, an audience member asked whether the higher rate of remission found by the simplified criteria is actually a good thing. “If I compare that to rheumatoid arthritis, when you use DAS28 you have a lot more remission, but still progression. So, are we missing some people? Are we including people in remission that still have disease?” she asked.
Tabi-Amponsah responded that the pain and patient global assessment domains seem to be quite difficult to achieve. In a separate analysis, the researchers examined tender and swollen joint counts and found that those achieving remission no longer had tender or swollen joints. “So, we don’t think the simplified definition is heavily misclassifying anyone as being in remission,” she said.
During the Q&A following Dalbeth’s talk, an audience member asked about patients with what he described as “mountains of tophi,” despite responding well to uricase therapy. “They may take months or even a year to really resolve that burden. They may be doing very well, yet they’re not going to be in remission because they’ve still got visible tophi. So, are we underselling them, and do we need a different definition for them doing well that this doesn’t capture?” he asked.
Dalbeth suggested that patients with large amounts of tophi aren’t really in remission. “I think we do need to be thinking about the disease, not just in terms of just crystals or just inflammation, but actually trying to integrate both of those, and I think this is where these composite measures might work quite well. I think we need to be aiming for holistic disease control, which is essentially what this is,” she said.
Tabi-Amponsah and Dalbeth did not disclose any financial relationships.
A version of this article appeared on Medscape.com.
In a nearly unanimous vote at the annual research symposium of the Gout Hyperuricemia and Crystal-Associated Disease Network (G-CAN), members approved a revision to gout remission criteria first established in 2016. The new version simplifies the definition in response to patient comments that the earlier version was redundant in some areas.
The previous version was developed following deliberations by 49 clinicians and researchers with experience in gout. They settled on a definition of gout remission that included five criteria:
- Serum urate levels lower than 0.36 mmol/L measured at least twice over 12 months, with no intervening values of 0.36 mmol/L or higher
- No gout flares over 12 months
- No tophi
- Pain score due to gout < 2 at least twice over 12 months on a 10-point Likert scale or 10-cm visual analog scale, with no intervening values ≥ 2
- Patient global assessment of gout disease activity < 2 on a 10-point Likert scale or 10-cm visual analog scale, with no intervening values of ≥ 2.
Some participants reported that patients sometimes misattributed pain from other sources while using patient-reported outcomes (PROs). The argument for keeping PROs was that they are validated measures and endorsed by Outcome Measures in Rheumatology. Nevertheless, there was no direct patient involvement in the development of the 2016 criteria.
Researchers later interviewed 20 individuals with well-controlled gout to get their feedback on the 2016 criteria. Those individuals endorsed the existing criteria and did not suggest any new ones, but they suggested that the pain due to gout and the absence of gout flares were redundant measures. One said: “If you have no flare-ups, you’ve got no pain; it sort of answers itself.”
“That was a bit challenging for us because it wasn’t quite what we expected, but I think it did make us look again at the definition and think about whether we could simplify the definition further,” Nicola Dalbeth, MBChB, said during a presentation at G-CAN. Dalbeth is an academic rheumatologist at the University of Auckland, in New Zealand, who was also the lead author of the original criteria.
Simplified Version Created With Only Three Criteria
In response to these points, researchers produced a revised version with only three criteria, including the serum urate, absence of gout flares, and absence of subcutaneous tophi at the time of assessment.
To determine if the simplified criteria performed well, they compared the original and revised remission criteria in the context of the CARES trial, the Nottingham nurse-led trial, and randomized controlled trials in patients with gout that were conducted in New Zealand (here and here).
Dansoa Tabi-Amponsah, a PhD candidate at the University of Auckland, presented results of a study comparing the two versions in the Nottingham trial, which included 517 participants who received nurse-led or usual general practitioner care. The nurse-led care included education, regular follow-up and serum urate testing, individualized advice on gout flare management, and escalation of urate-lowering therapy with a treat-to-target strategy.
Both definitions demonstrated a link between the nurse-led strategy and increased rates of remission at year 1 and year 2, although the simplified definition found that more patients were in remission (17.6% vs 9.9% at year 1 and 42.7% vs 28.4% at year 2, both P < .001). “This is something we’ve seen across all of our analyses,” said Tabi-Amponsah.
Both criteria also found significant differences in remission rates between the nurse-led group in year 2 vs year 1 but not in the usual care group.
Participants who achieved remission had better gout impact scale scores in areas like worrying that a gout attack will occur, fears of worsening gout, and concerns about the impact of gout on future activities. “This is important because during that qualitative study, a key aspect of being in remission was no longer being worried about their gout, no longer feeling anxious about having constant gout flares, and having control over their gout. So, it’s important to note that despite the absence of PROs in that simplified definition, it’s still able to align with the patients’ perspectives of their disease state,” said Tabi-Amponsah.
During the Q&A period after her talk, an audience member asked whether the higher rate of remission found by the simplified criteria is actually a good thing. “If I compare that to rheumatoid arthritis, when you use DAS28 you have a lot more remission, but still progression. So, are we missing some people? Are we including people in remission that still have disease?” she asked.
Tabi-Amponsah responded that the pain and patient global assessment domains seem to be quite difficult to achieve. In a separate analysis, the researchers examined tender and swollen joint counts and found that those achieving remission no longer had tender or swollen joints. “So, we don’t think the simplified definition is heavily misclassifying anyone as being in remission,” she said.
During the Q&A following Dalbeth’s talk, an audience member asked about patients with what he described as “mountains of tophi,” despite responding well to uricase therapy. “They may take months or even a year to really resolve that burden. They may be doing very well, yet they’re not going to be in remission because they’ve still got visible tophi. So, are we underselling them, and do we need a different definition for them doing well that this doesn’t capture?” he asked.
Dalbeth suggested that patients with large amounts of tophi aren’t really in remission. “I think we do need to be thinking about the disease, not just in terms of just crystals or just inflammation, but actually trying to integrate both of those, and I think this is where these composite measures might work quite well. I think we need to be aiming for holistic disease control, which is essentially what this is,” she said.
Tabi-Amponsah and Dalbeth did not disclose any financial relationships.
A version of this article appeared on Medscape.com.
In a nearly unanimous vote at the annual research symposium of the Gout Hyperuricemia and Crystal-Associated Disease Network (G-CAN), members approved a revision to gout remission criteria first established in 2016. The new version simplifies the definition in response to patient comments that the earlier version was redundant in some areas.
The previous version was developed following deliberations by 49 clinicians and researchers with experience in gout. They settled on a definition of gout remission that included five criteria:
- Serum urate levels lower than 0.36 mmol/L measured at least twice over 12 months, with no intervening values of 0.36 mmol/L or higher
- No gout flares over 12 months
- No tophi
- Pain score due to gout < 2 at least twice over 12 months on a 10-point Likert scale or 10-cm visual analog scale, with no intervening values ≥ 2
- Patient global assessment of gout disease activity < 2 on a 10-point Likert scale or 10-cm visual analog scale, with no intervening values of ≥ 2.
Some participants reported that patients sometimes misattributed pain from other sources while using patient-reported outcomes (PROs). The argument for keeping PROs was that they are validated measures and endorsed by Outcome Measures in Rheumatology. Nevertheless, there was no direct patient involvement in the development of the 2016 criteria.
Researchers later interviewed 20 individuals with well-controlled gout to get their feedback on the 2016 criteria. Those individuals endorsed the existing criteria and did not suggest any new ones, but they suggested that the pain due to gout and the absence of gout flares were redundant measures. One said: “If you have no flare-ups, you’ve got no pain; it sort of answers itself.”
“That was a bit challenging for us because it wasn’t quite what we expected, but I think it did make us look again at the definition and think about whether we could simplify the definition further,” Nicola Dalbeth, MBChB, said during a presentation at G-CAN. Dalbeth is an academic rheumatologist at the University of Auckland, in New Zealand, who was also the lead author of the original criteria.
Simplified Version Created With Only Three Criteria
In response to these points, researchers produced a revised version with only three criteria, including the serum urate, absence of gout flares, and absence of subcutaneous tophi at the time of assessment.
To determine if the simplified criteria performed well, they compared the original and revised remission criteria in the context of the CARES trial, the Nottingham nurse-led trial, and randomized controlled trials in patients with gout that were conducted in New Zealand (here and here).
Dansoa Tabi-Amponsah, a PhD candidate at the University of Auckland, presented results of a study comparing the two versions in the Nottingham trial, which included 517 participants who received nurse-led or usual general practitioner care. The nurse-led care included education, regular follow-up and serum urate testing, individualized advice on gout flare management, and escalation of urate-lowering therapy with a treat-to-target strategy.
Both definitions demonstrated a link between the nurse-led strategy and increased rates of remission at year 1 and year 2, although the simplified definition found that more patients were in remission (17.6% vs 9.9% at year 1 and 42.7% vs 28.4% at year 2, both P < .001). “This is something we’ve seen across all of our analyses,” said Tabi-Amponsah.
Both criteria also found significant differences in remission rates between the nurse-led group in year 2 vs year 1 but not in the usual care group.
Participants who achieved remission had better gout impact scale scores in areas like worrying that a gout attack will occur, fears of worsening gout, and concerns about the impact of gout on future activities. “This is important because during that qualitative study, a key aspect of being in remission was no longer being worried about their gout, no longer feeling anxious about having constant gout flares, and having control over their gout. So, it’s important to note that despite the absence of PROs in that simplified definition, it’s still able to align with the patients’ perspectives of their disease state,” said Tabi-Amponsah.
During the Q&A period after her talk, an audience member asked whether the higher rate of remission found by the simplified criteria is actually a good thing. “If I compare that to rheumatoid arthritis, when you use DAS28 you have a lot more remission, but still progression. So, are we missing some people? Are we including people in remission that still have disease?” she asked.
Tabi-Amponsah responded that the pain and patient global assessment domains seem to be quite difficult to achieve. In a separate analysis, the researchers examined tender and swollen joint counts and found that those achieving remission no longer had tender or swollen joints. “So, we don’t think the simplified definition is heavily misclassifying anyone as being in remission,” she said.
During the Q&A following Dalbeth’s talk, an audience member asked about patients with what he described as “mountains of tophi,” despite responding well to uricase therapy. “They may take months or even a year to really resolve that burden. They may be doing very well, yet they’re not going to be in remission because they’ve still got visible tophi. So, are we underselling them, and do we need a different definition for them doing well that this doesn’t capture?” he asked.
Dalbeth suggested that patients with large amounts of tophi aren’t really in remission. “I think we do need to be thinking about the disease, not just in terms of just crystals or just inflammation, but actually trying to integrate both of those, and I think this is where these composite measures might work quite well. I think we need to be aiming for holistic disease control, which is essentially what this is,” she said.
Tabi-Amponsah and Dalbeth did not disclose any financial relationships.
A version of this article appeared on Medscape.com.
FROM G-CAN 2024
Family Doctors Can Intervene to Treat and Prevent STIs
VANCOUVER, BRITISH COLUMBIA — Family physicians should familiarize themselves with doxycycline post-exposure prophylaxis (doxy PEP) as a prescription for patients who frequently have unprotected sex (including oral, anal, and vaginal) with multiple partners, according to a presentation at the Family Medicine Forum (FMF) 2024. Doxy PEP decreases the risk for bacterial sexually transmitted infections (STIs).
Many family physicians may be unaware of doxy PEP as a means of avoiding STIs. “Doxy PEP is an incredible tool that can be used within 72 hours of unprotected sex to reduce bacterial STI risk,” said James Owen, MD, assistant professor of family and community medicine at the University of Toronto in Ontario, Canada.
The US Centers for Disease Control and Prevention point to data from three randomized, controlled trials that demonstrate the ability of doxy PEP to decrease syphilis and chlamydia infections by more than 70% and decrease gonococcal infections by about 50%, said Jordan Goodridge, MD, assistant professor of family and community medicine at the University of Toronto.
Optimizing Doxy PEP
Doxy PEP (200 mg) is most effective when administered in the first 24 hours after unprotected sex, explained Goodridge. It is recommended that patients repeat the 200-mg dose if they are sexually active again within 24 hours.
To ensure optimal absorption of doxy PEP, the drug should not be taken with antacids or multivitamins, said Goodridge. Antacids “can bind to doxycycline and prevent it from being absorbed,” he said.
A key concern about doxy PEP is the development of antimicrobial resistance, noted Goodridge. “We don’t have long-term studies to give us an idea about what this risk is, to quantify it. The studies we have are relatively short, generally less than a year, and didn’t suggest that there was a huge risk [for antimicrobial resistance].”
Moreover, doxycycline is teratogenic, and its use is contraindicated in pregnancy, said Goodridge. If a pregnant patient is being treated for syphilis, then penicillin is the treatment of choice. For pregnant patients with penicillin allergy, Public Health Agency of Canada guidelines call for penicillin desensitization followed by penicillin.
The rate of syphilis has been rising in Canadian women of reproductive age, thus increasing the potential for congenital syphilis, noted Goodridge.
Benefits of HPV Vaccine
The 9-valent HPV vaccine is recommended in Canada for patients aged 9-26 years, but those aged 27 years or older at ongoing risk for HPV exposure may receive the 9-valent HPV vaccine after discussion with their healthcare providers, noted Owen.
High-risk patients can benefit from the vaccine even though they have likely had exposure to HPV, he added. “If someone has multiple sexual partners, they have probably been exposed to HPV at some point,” said Owen. “You still could reduce a patient’s risk for being exposed to certain oncogenic strains [of HPV]. Certainly, within our practices, we’re often giving it [that is, the HPV vaccine] to higher-risk individuals, including men having sex with men.”
HIV Prevention
“Condoms are still a mainstay of HIV and STI prevention, but condom use is going down,” said Owen.
In a national survey commissioned by the Toronto-based organization LetsStopAIDS and including more than 1100 Canadians aged 18-24 years, 24% of respondents said they use condoms “all the time.” In 2020, by contrast, more than half (53%) of respondents reported that they use condoms all the time.
Updated Canadian guidelines on the use of HIV pre-exposure prophylaxis (PrEP) are expected to be released in 2025 and will address questions about how frequently individuals taking HIV PrEP should be tested to ensure a negative HIV result. The guidelines will also provide guidance as to whether HIV serology or HIV viral load should be captured, said Owen. Patients in Canada who take HIV PrEP are now generally screened for HIV every 3 months.
A new HIV PrEP tool that has become available to Canadian clinicians is the injectable drug cabotegravir, which is dosed every 2 months.
Owen and Goodridge reported having no relevant financial relationships.
A version of this article appeared on Medscape.com.
VANCOUVER, BRITISH COLUMBIA — Family physicians should familiarize themselves with doxycycline post-exposure prophylaxis (doxy PEP) as a prescription for patients who frequently have unprotected sex (including oral, anal, and vaginal) with multiple partners, according to a presentation at the Family Medicine Forum (FMF) 2024. Doxy PEP decreases the risk for bacterial sexually transmitted infections (STIs).
Many family physicians may be unaware of doxy PEP as a means of avoiding STIs. “Doxy PEP is an incredible tool that can be used within 72 hours of unprotected sex to reduce bacterial STI risk,” said James Owen, MD, assistant professor of family and community medicine at the University of Toronto in Ontario, Canada.
The US Centers for Disease Control and Prevention point to data from three randomized, controlled trials that demonstrate the ability of doxy PEP to decrease syphilis and chlamydia infections by more than 70% and decrease gonococcal infections by about 50%, said Jordan Goodridge, MD, assistant professor of family and community medicine at the University of Toronto.
Optimizing Doxy PEP
Doxy PEP (200 mg) is most effective when administered in the first 24 hours after unprotected sex, explained Goodridge. It is recommended that patients repeat the 200-mg dose if they are sexually active again within 24 hours.
To ensure optimal absorption of doxy PEP, the drug should not be taken with antacids or multivitamins, said Goodridge. Antacids “can bind to doxycycline and prevent it from being absorbed,” he said.
A key concern about doxy PEP is the development of antimicrobial resistance, noted Goodridge. “We don’t have long-term studies to give us an idea about what this risk is, to quantify it. The studies we have are relatively short, generally less than a year, and didn’t suggest that there was a huge risk [for antimicrobial resistance].”
Moreover, doxycycline is teratogenic, and its use is contraindicated in pregnancy, said Goodridge. If a pregnant patient is being treated for syphilis, then penicillin is the treatment of choice. For pregnant patients with penicillin allergy, Public Health Agency of Canada guidelines call for penicillin desensitization followed by penicillin.
The rate of syphilis has been rising in Canadian women of reproductive age, thus increasing the potential for congenital syphilis, noted Goodridge.
Benefits of HPV Vaccine
The 9-valent HPV vaccine is recommended in Canada for patients aged 9-26 years, but those aged 27 years or older at ongoing risk for HPV exposure may receive the 9-valent HPV vaccine after discussion with their healthcare providers, noted Owen.
High-risk patients can benefit from the vaccine even though they have likely had exposure to HPV, he added. “If someone has multiple sexual partners, they have probably been exposed to HPV at some point,” said Owen. “You still could reduce a patient’s risk for being exposed to certain oncogenic strains [of HPV]. Certainly, within our practices, we’re often giving it [that is, the HPV vaccine] to higher-risk individuals, including men having sex with men.”
HIV Prevention
“Condoms are still a mainstay of HIV and STI prevention, but condom use is going down,” said Owen.
In a national survey commissioned by the Toronto-based organization LetsStopAIDS and including more than 1100 Canadians aged 18-24 years, 24% of respondents said they use condoms “all the time.” In 2020, by contrast, more than half (53%) of respondents reported that they use condoms all the time.
Updated Canadian guidelines on the use of HIV pre-exposure prophylaxis (PrEP) are expected to be released in 2025 and will address questions about how frequently individuals taking HIV PrEP should be tested to ensure a negative HIV result. The guidelines will also provide guidance as to whether HIV serology or HIV viral load should be captured, said Owen. Patients in Canada who take HIV PrEP are now generally screened for HIV every 3 months.
A new HIV PrEP tool that has become available to Canadian clinicians is the injectable drug cabotegravir, which is dosed every 2 months.
Owen and Goodridge reported having no relevant financial relationships.
A version of this article appeared on Medscape.com.
VANCOUVER, BRITISH COLUMBIA — Family physicians should familiarize themselves with doxycycline post-exposure prophylaxis (doxy PEP) as a prescription for patients who frequently have unprotected sex (including oral, anal, and vaginal) with multiple partners, according to a presentation at the Family Medicine Forum (FMF) 2024. Doxy PEP decreases the risk for bacterial sexually transmitted infections (STIs).
Many family physicians may be unaware of doxy PEP as a means of avoiding STIs. “Doxy PEP is an incredible tool that can be used within 72 hours of unprotected sex to reduce bacterial STI risk,” said James Owen, MD, assistant professor of family and community medicine at the University of Toronto in Ontario, Canada.
The US Centers for Disease Control and Prevention point to data from three randomized, controlled trials that demonstrate the ability of doxy PEP to decrease syphilis and chlamydia infections by more than 70% and decrease gonococcal infections by about 50%, said Jordan Goodridge, MD, assistant professor of family and community medicine at the University of Toronto.
Optimizing Doxy PEP
Doxy PEP (200 mg) is most effective when administered in the first 24 hours after unprotected sex, explained Goodridge. It is recommended that patients repeat the 200-mg dose if they are sexually active again within 24 hours.
To ensure optimal absorption of doxy PEP, the drug should not be taken with antacids or multivitamins, said Goodridge. Antacids “can bind to doxycycline and prevent it from being absorbed,” he said.
A key concern about doxy PEP is the development of antimicrobial resistance, noted Goodridge. “We don’t have long-term studies to give us an idea about what this risk is, to quantify it. The studies we have are relatively short, generally less than a year, and didn’t suggest that there was a huge risk [for antimicrobial resistance].”
Moreover, doxycycline is teratogenic, and its use is contraindicated in pregnancy, said Goodridge. If a pregnant patient is being treated for syphilis, then penicillin is the treatment of choice. For pregnant patients with penicillin allergy, Public Health Agency of Canada guidelines call for penicillin desensitization followed by penicillin.
The rate of syphilis has been rising in Canadian women of reproductive age, thus increasing the potential for congenital syphilis, noted Goodridge.
Benefits of HPV Vaccine
The 9-valent HPV vaccine is recommended in Canada for patients aged 9-26 years, but those aged 27 years or older at ongoing risk for HPV exposure may receive the 9-valent HPV vaccine after discussion with their healthcare providers, noted Owen.
High-risk patients can benefit from the vaccine even though they have likely had exposure to HPV, he added. “If someone has multiple sexual partners, they have probably been exposed to HPV at some point,” said Owen. “You still could reduce a patient’s risk for being exposed to certain oncogenic strains [of HPV]. Certainly, within our practices, we’re often giving it [that is, the HPV vaccine] to higher-risk individuals, including men having sex with men.”
HIV Prevention
“Condoms are still a mainstay of HIV and STI prevention, but condom use is going down,” said Owen.
In a national survey commissioned by the Toronto-based organization LetsStopAIDS and including more than 1100 Canadians aged 18-24 years, 24% of respondents said they use condoms “all the time.” In 2020, by contrast, more than half (53%) of respondents reported that they use condoms all the time.
Updated Canadian guidelines on the use of HIV pre-exposure prophylaxis (PrEP) are expected to be released in 2025 and will address questions about how frequently individuals taking HIV PrEP should be tested to ensure a negative HIV result. The guidelines will also provide guidance as to whether HIV serology or HIV viral load should be captured, said Owen. Patients in Canada who take HIV PrEP are now generally screened for HIV every 3 months.
A new HIV PrEP tool that has become available to Canadian clinicians is the injectable drug cabotegravir, which is dosed every 2 months.
Owen and Goodridge reported having no relevant financial relationships.
A version of this article appeared on Medscape.com.
FROM FMF 2024
AI Tool Identifies Undiagnosed Early-Stage MASLD
SAN DIEGO — according to new research.
Among the patients identified by the algorithm as meeting the criteria for MASLD, only a small percentage had an MASLD-associated diagnostic code.
“A significant portion of patients who meet criteria for MASLD go undiagnosed, which can lead to delays in care and progression to advanced liver disease,” said lead author Ariana Stuart, MD, an internal medicine resident at the University of Washington, Seattle, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
“However, people shouldn’t interpret our findings as a lack of primary care training or management,” she said. “Instead, this study indicates that AI can complement physician workflow and address the limitations of traditional clinical practice.”
Developing an MASLD Algorithm
Typically, the identification of MASLD has relied on clinician recognition and descriptions in chart notes, Stuart said. Early-stage disease often goes unnoticed, particularly if patients remain asymptomatic, until cirrhosis develops.
To address this, Stuart and colleagues created a machine learning, natural language processing AI algorithm on the basis of MASLD criteria from AASLD: Hepatic steatosis on imaging and at least one metabolic factor (elevated body mass index, hypertension, prediabetes or diabetes, or dyslipidemia). The model was validated by two physicians, who manually reviewed monthly cohorts generated by the algorithm.
Between December 2023 and May 2024, the researchers used the algorithm to analyze an MASLD cohort from medical centers in the Seattle area. The mean age was 51 years, 44% were women, and 68% were White. Those with alcohol-associated liver disease, metastatic malignancy, and autoimmune, genetic, and infectious causes of liver disease were excluded.
The algorithm identified 957 patients with imaging that matched MASLD criteria.
Among those, 137 patients (17%) identified by the algorithm had an MASLD-associated diagnostic code. For these patients, the mean time from initial imaging with steatosis to diagnosis was 33 days, according to patient records.
An additional 26 patients received an MASLD diagnosis during the study period, with a mean time to diagnosis of 56.2 days.
In terms of patient management, 245 patients (26%) had contact with a gastroenterologist or hepatologist based on documentation of a letter, phone call, or office visit. In addition, 546 patients (57%) were screened for hepatitis C.
After adjusting for an over-inclusion error rate of 12.8% and an overdiagnosis rate of 0.02%, the research team found 697 patients (83%) lacked a relevant diagnosis. After multiple iterations, the algorithm achieved an accuracy of about 88%, Stuart said.
Considering Future AI Use
Stuart and colleagues are now testing the algorithm in larger groups and across longer periods.
After that, they intend to implement a quality improvement program to increase awareness for clinicians and primary care providers, as well as train users on how to interpret and move forward with findings of hepatic steatosis in patient records.
For instance, future AI models could flag patients for additional testing, improve chart review, and aid in research efforts around cardiometabolic comorbidities associated with MASLD, she said.
Looking ahead, AI tools such as these represent what’s possible for advancements in research, patient care, and clinical workflows, said Ashley Spann, MD, assistant professor and transplant hepatologist at Vanderbilt University, Nashville, Tennessee, and director of clinical research informatics for Vanderbilt’s Gastroenterology Division.
“AI, in my view, is actually augmented intelligence,” she added. “We need to think about the people and processes involved.”
Spann, who spoke about the use of AI tools in medicine in general, stressed the need for transparency in AI use, careful validation of input-output data, frameworks for machine learning models in medicine, and standardization across institutions.
“What we ultimately need is an infrastructure that supports the simultaneous deployment and evaluation of these models,” she said. “We all need to be on the same page and make sure our models work in multiple settings and make adjustments based on algorithmovigilance afterward.”
Stuart reported no relevant disclosures. Spann serves on Epic’s hepatology steering board, which has focused on how to use AI tools in electronic medical records.
A version of this article appeared on Medscape.com.
SAN DIEGO — according to new research.
Among the patients identified by the algorithm as meeting the criteria for MASLD, only a small percentage had an MASLD-associated diagnostic code.
“A significant portion of patients who meet criteria for MASLD go undiagnosed, which can lead to delays in care and progression to advanced liver disease,” said lead author Ariana Stuart, MD, an internal medicine resident at the University of Washington, Seattle, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
“However, people shouldn’t interpret our findings as a lack of primary care training or management,” she said. “Instead, this study indicates that AI can complement physician workflow and address the limitations of traditional clinical practice.”
Developing an MASLD Algorithm
Typically, the identification of MASLD has relied on clinician recognition and descriptions in chart notes, Stuart said. Early-stage disease often goes unnoticed, particularly if patients remain asymptomatic, until cirrhosis develops.
To address this, Stuart and colleagues created a machine learning, natural language processing AI algorithm on the basis of MASLD criteria from AASLD: Hepatic steatosis on imaging and at least one metabolic factor (elevated body mass index, hypertension, prediabetes or diabetes, or dyslipidemia). The model was validated by two physicians, who manually reviewed monthly cohorts generated by the algorithm.
Between December 2023 and May 2024, the researchers used the algorithm to analyze an MASLD cohort from medical centers in the Seattle area. The mean age was 51 years, 44% were women, and 68% were White. Those with alcohol-associated liver disease, metastatic malignancy, and autoimmune, genetic, and infectious causes of liver disease were excluded.
The algorithm identified 957 patients with imaging that matched MASLD criteria.
Among those, 137 patients (17%) identified by the algorithm had an MASLD-associated diagnostic code. For these patients, the mean time from initial imaging with steatosis to diagnosis was 33 days, according to patient records.
An additional 26 patients received an MASLD diagnosis during the study period, with a mean time to diagnosis of 56.2 days.
In terms of patient management, 245 patients (26%) had contact with a gastroenterologist or hepatologist based on documentation of a letter, phone call, or office visit. In addition, 546 patients (57%) were screened for hepatitis C.
After adjusting for an over-inclusion error rate of 12.8% and an overdiagnosis rate of 0.02%, the research team found 697 patients (83%) lacked a relevant diagnosis. After multiple iterations, the algorithm achieved an accuracy of about 88%, Stuart said.
Considering Future AI Use
Stuart and colleagues are now testing the algorithm in larger groups and across longer periods.
After that, they intend to implement a quality improvement program to increase awareness for clinicians and primary care providers, as well as train users on how to interpret and move forward with findings of hepatic steatosis in patient records.
For instance, future AI models could flag patients for additional testing, improve chart review, and aid in research efforts around cardiometabolic comorbidities associated with MASLD, she said.
Looking ahead, AI tools such as these represent what’s possible for advancements in research, patient care, and clinical workflows, said Ashley Spann, MD, assistant professor and transplant hepatologist at Vanderbilt University, Nashville, Tennessee, and director of clinical research informatics for Vanderbilt’s Gastroenterology Division.
“AI, in my view, is actually augmented intelligence,” she added. “We need to think about the people and processes involved.”
Spann, who spoke about the use of AI tools in medicine in general, stressed the need for transparency in AI use, careful validation of input-output data, frameworks for machine learning models in medicine, and standardization across institutions.
“What we ultimately need is an infrastructure that supports the simultaneous deployment and evaluation of these models,” she said. “We all need to be on the same page and make sure our models work in multiple settings and make adjustments based on algorithmovigilance afterward.”
Stuart reported no relevant disclosures. Spann serves on Epic’s hepatology steering board, which has focused on how to use AI tools in electronic medical records.
A version of this article appeared on Medscape.com.
SAN DIEGO — according to new research.
Among the patients identified by the algorithm as meeting the criteria for MASLD, only a small percentage had an MASLD-associated diagnostic code.
“A significant portion of patients who meet criteria for MASLD go undiagnosed, which can lead to delays in care and progression to advanced liver disease,” said lead author Ariana Stuart, MD, an internal medicine resident at the University of Washington, Seattle, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
“However, people shouldn’t interpret our findings as a lack of primary care training or management,” she said. “Instead, this study indicates that AI can complement physician workflow and address the limitations of traditional clinical practice.”
Developing an MASLD Algorithm
Typically, the identification of MASLD has relied on clinician recognition and descriptions in chart notes, Stuart said. Early-stage disease often goes unnoticed, particularly if patients remain asymptomatic, until cirrhosis develops.
To address this, Stuart and colleagues created a machine learning, natural language processing AI algorithm on the basis of MASLD criteria from AASLD: Hepatic steatosis on imaging and at least one metabolic factor (elevated body mass index, hypertension, prediabetes or diabetes, or dyslipidemia). The model was validated by two physicians, who manually reviewed monthly cohorts generated by the algorithm.
Between December 2023 and May 2024, the researchers used the algorithm to analyze an MASLD cohort from medical centers in the Seattle area. The mean age was 51 years, 44% were women, and 68% were White. Those with alcohol-associated liver disease, metastatic malignancy, and autoimmune, genetic, and infectious causes of liver disease were excluded.
The algorithm identified 957 patients with imaging that matched MASLD criteria.
Among those, 137 patients (17%) identified by the algorithm had an MASLD-associated diagnostic code. For these patients, the mean time from initial imaging with steatosis to diagnosis was 33 days, according to patient records.
An additional 26 patients received an MASLD diagnosis during the study period, with a mean time to diagnosis of 56.2 days.
In terms of patient management, 245 patients (26%) had contact with a gastroenterologist or hepatologist based on documentation of a letter, phone call, or office visit. In addition, 546 patients (57%) were screened for hepatitis C.
After adjusting for an over-inclusion error rate of 12.8% and an overdiagnosis rate of 0.02%, the research team found 697 patients (83%) lacked a relevant diagnosis. After multiple iterations, the algorithm achieved an accuracy of about 88%, Stuart said.
Considering Future AI Use
Stuart and colleagues are now testing the algorithm in larger groups and across longer periods.
After that, they intend to implement a quality improvement program to increase awareness for clinicians and primary care providers, as well as train users on how to interpret and move forward with findings of hepatic steatosis in patient records.
For instance, future AI models could flag patients for additional testing, improve chart review, and aid in research efforts around cardiometabolic comorbidities associated with MASLD, she said.
Looking ahead, AI tools such as these represent what’s possible for advancements in research, patient care, and clinical workflows, said Ashley Spann, MD, assistant professor and transplant hepatologist at Vanderbilt University, Nashville, Tennessee, and director of clinical research informatics for Vanderbilt’s Gastroenterology Division.
“AI, in my view, is actually augmented intelligence,” she added. “We need to think about the people and processes involved.”
Spann, who spoke about the use of AI tools in medicine in general, stressed the need for transparency in AI use, careful validation of input-output data, frameworks for machine learning models in medicine, and standardization across institutions.
“What we ultimately need is an infrastructure that supports the simultaneous deployment and evaluation of these models,” she said. “We all need to be on the same page and make sure our models work in multiple settings and make adjustments based on algorithmovigilance afterward.”
Stuart reported no relevant disclosures. Spann serves on Epic’s hepatology steering board, which has focused on how to use AI tools in electronic medical records.
A version of this article appeared on Medscape.com.
FROM AASLD 24
MELD 3.0 Reduces Sex-Based Liver Transplant Disparities
SAN DIEGO — , according to new research.
In particular, women are now more likely to be added to the waitlist for a liver transplant, more likely to receive a transplant, and less likely to fall off the waitlist because of death.
“MELD 3.0 improved access to transplantation for women, and now waitlist mortality and transplant rates for women more closely approximate the rates for men,” said lead author Allison Kwong, MD, assistant professor of medicine and transplant hepatologist at Stanford Medicine in California.
“Overall transplant outcomes have also improved year over year,” said Kwong, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
Changes in MELD and Transplant Numbers
MELD, which estimates liver failure severity and short-term survival in patients with chronic liver disease, has been used since 2002 to determine organ allocation priority for patients in the United States awaiting liver transplantation. Originally, the score incorporated three variables: creatinine, bilirubin, and the international normalized ratio (INR). MELDNa1, or MELD 2.0, was adopted in 2016 to add sodium.
“Under this system, however, there have been sex-based disparities” with women receiving lower priority scores despite similar disease severity, said Kwong.
“This has been attributed to several factors, such as the creatinine term in the MELD score underestimating renal dysfunction in women, height and body size differences, and differences in disease etiology, and how we’ve assigned exception points historically,” she reported.
Men have had a lower pretransplant mortality rate and higher deceased donor transplant rates, she added.
MELD 3.0 was developed to address these gender differences and other determinants of waitlist outcomes. The updated equation added 1.33 points for women, as well as adding other variables, such as albumin, interactions between bilirubin and sodium, and interactions between albumin and creatinine, to increase prediction accuracy.
To observe the effects of the new system, Kwong and colleagues analyzed OPTN data for patients aged 12 years or older, focusing on the records of more than 20,300 newly registered liver transplant candidates, and about 18,700 transplant recipients, during the 12 months before and 12 months after MELD 3.0 was implemented.
After the switch, 43.7% of newly registered liver transplant candidates were women, compared with 40.4% before the switch. At registration, the median age was 55, both before and after the change in policy, and the median MELD score changed from 23 to 22 after implementation.
In addition, 42.1% of transplants occurred among women after MELD 3.0 implementation, as compared with 37.3% before. Overall, deceased donor transplant rates were similar for men and women after MELD 3.0 implementation.
The 90-day waitlist dropout rate — patients who died or became too sick to receive a transplant — decreased from 13.5% to 9.1% among women, which may be partially attributable to MELD 3.0, said Kwong.
However, waitlist dropout rates also decreased among men, from 9.8% to 7.4%, probably because of improvements in technology, such as machine perfusion, which have increased the number of available livers, she added.
Disparities Continue to Exist
Some disparities still exist. Although the total median MELD score at transplant decreased from 29 to 27, women still had a higher median score of 29 at transplant, compared with a median score of 27 among men.
“This indicates that there may still be differences in transplant access between the sexes,” Kwong said. “There are still body size differences that can affect the probability of transplant, and this would not be addressed by MELD 3.0.”
Additional transplant disparities exist related to other patient characteristics, such as age, race, and ethnicity.
Future versions of MELD could potentially consider these factors, said session moderator Aleksander Krag, MD, PhD, MBA, professor of clinical medicine at the University of Southern Denmark, Odense, and secretary general of the European Association for the Study of the Liver, 2023-2025.
“There are infinite versions of MELD that can be made,” Kwong said. “It’s still early to see how MELD 3.0 will serve the system, but so far, so good.”
In a comment, Tamar Taddei, MD, professor of medicine in digestive diseases at Yale School of Medicine, New Haven, Connecticut, who comoderated the session, noted the importance of using a MELD score that considers sex-based differences.
This study brings MELD 3.0 to its fruition by reducing the disparities experienced by women who were underserved by the previous scoring systems, she said.
It was lovely to see that MELD 3.0 reduced the disparities with transplants, and also that the waitlist dropout was reduced — for both men and women,” Taddei said. “This change is a no-brainer.”
Kwong, Krag, and Taddei reported no relevant disclosures.
A version of this article appeared on Medscape.com.
SAN DIEGO — , according to new research.
In particular, women are now more likely to be added to the waitlist for a liver transplant, more likely to receive a transplant, and less likely to fall off the waitlist because of death.
“MELD 3.0 improved access to transplantation for women, and now waitlist mortality and transplant rates for women more closely approximate the rates for men,” said lead author Allison Kwong, MD, assistant professor of medicine and transplant hepatologist at Stanford Medicine in California.
“Overall transplant outcomes have also improved year over year,” said Kwong, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
Changes in MELD and Transplant Numbers
MELD, which estimates liver failure severity and short-term survival in patients with chronic liver disease, has been used since 2002 to determine organ allocation priority for patients in the United States awaiting liver transplantation. Originally, the score incorporated three variables: creatinine, bilirubin, and the international normalized ratio (INR). MELDNa1, or MELD 2.0, was adopted in 2016 to add sodium.
“Under this system, however, there have been sex-based disparities” with women receiving lower priority scores despite similar disease severity, said Kwong.
“This has been attributed to several factors, such as the creatinine term in the MELD score underestimating renal dysfunction in women, height and body size differences, and differences in disease etiology, and how we’ve assigned exception points historically,” she reported.
Men have had a lower pretransplant mortality rate and higher deceased donor transplant rates, she added.
MELD 3.0 was developed to address these gender differences and other determinants of waitlist outcomes. The updated equation added 1.33 points for women, as well as adding other variables, such as albumin, interactions between bilirubin and sodium, and interactions between albumin and creatinine, to increase prediction accuracy.
To observe the effects of the new system, Kwong and colleagues analyzed OPTN data for patients aged 12 years or older, focusing on the records of more than 20,300 newly registered liver transplant candidates, and about 18,700 transplant recipients, during the 12 months before and 12 months after MELD 3.0 was implemented.
After the switch, 43.7% of newly registered liver transplant candidates were women, compared with 40.4% before the switch. At registration, the median age was 55, both before and after the change in policy, and the median MELD score changed from 23 to 22 after implementation.
In addition, 42.1% of transplants occurred among women after MELD 3.0 implementation, as compared with 37.3% before. Overall, deceased donor transplant rates were similar for men and women after MELD 3.0 implementation.
The 90-day waitlist dropout rate — patients who died or became too sick to receive a transplant — decreased from 13.5% to 9.1% among women, which may be partially attributable to MELD 3.0, said Kwong.
However, waitlist dropout rates also decreased among men, from 9.8% to 7.4%, probably because of improvements in technology, such as machine perfusion, which have increased the number of available livers, she added.
Disparities Continue to Exist
Some disparities still exist. Although the total median MELD score at transplant decreased from 29 to 27, women still had a higher median score of 29 at transplant, compared with a median score of 27 among men.
“This indicates that there may still be differences in transplant access between the sexes,” Kwong said. “There are still body size differences that can affect the probability of transplant, and this would not be addressed by MELD 3.0.”
Additional transplant disparities exist related to other patient characteristics, such as age, race, and ethnicity.
Future versions of MELD could potentially consider these factors, said session moderator Aleksander Krag, MD, PhD, MBA, professor of clinical medicine at the University of Southern Denmark, Odense, and secretary general of the European Association for the Study of the Liver, 2023-2025.
“There are infinite versions of MELD that can be made,” Kwong said. “It’s still early to see how MELD 3.0 will serve the system, but so far, so good.”
In a comment, Tamar Taddei, MD, professor of medicine in digestive diseases at Yale School of Medicine, New Haven, Connecticut, who comoderated the session, noted the importance of using a MELD score that considers sex-based differences.
This study brings MELD 3.0 to its fruition by reducing the disparities experienced by women who were underserved by the previous scoring systems, she said.
It was lovely to see that MELD 3.0 reduced the disparities with transplants, and also that the waitlist dropout was reduced — for both men and women,” Taddei said. “This change is a no-brainer.”
Kwong, Krag, and Taddei reported no relevant disclosures.
A version of this article appeared on Medscape.com.
SAN DIEGO — , according to new research.
In particular, women are now more likely to be added to the waitlist for a liver transplant, more likely to receive a transplant, and less likely to fall off the waitlist because of death.
“MELD 3.0 improved access to transplantation for women, and now waitlist mortality and transplant rates for women more closely approximate the rates for men,” said lead author Allison Kwong, MD, assistant professor of medicine and transplant hepatologist at Stanford Medicine in California.
“Overall transplant outcomes have also improved year over year,” said Kwong, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
Changes in MELD and Transplant Numbers
MELD, which estimates liver failure severity and short-term survival in patients with chronic liver disease, has been used since 2002 to determine organ allocation priority for patients in the United States awaiting liver transplantation. Originally, the score incorporated three variables: creatinine, bilirubin, and the international normalized ratio (INR). MELDNa1, or MELD 2.0, was adopted in 2016 to add sodium.
“Under this system, however, there have been sex-based disparities” with women receiving lower priority scores despite similar disease severity, said Kwong.
“This has been attributed to several factors, such as the creatinine term in the MELD score underestimating renal dysfunction in women, height and body size differences, and differences in disease etiology, and how we’ve assigned exception points historically,” she reported.
Men have had a lower pretransplant mortality rate and higher deceased donor transplant rates, she added.
MELD 3.0 was developed to address these gender differences and other determinants of waitlist outcomes. The updated equation added 1.33 points for women, as well as adding other variables, such as albumin, interactions between bilirubin and sodium, and interactions between albumin and creatinine, to increase prediction accuracy.
To observe the effects of the new system, Kwong and colleagues analyzed OPTN data for patients aged 12 years or older, focusing on the records of more than 20,300 newly registered liver transplant candidates, and about 18,700 transplant recipients, during the 12 months before and 12 months after MELD 3.0 was implemented.
After the switch, 43.7% of newly registered liver transplant candidates were women, compared with 40.4% before the switch. At registration, the median age was 55, both before and after the change in policy, and the median MELD score changed from 23 to 22 after implementation.
In addition, 42.1% of transplants occurred among women after MELD 3.0 implementation, as compared with 37.3% before. Overall, deceased donor transplant rates were similar for men and women after MELD 3.0 implementation.
The 90-day waitlist dropout rate — patients who died or became too sick to receive a transplant — decreased from 13.5% to 9.1% among women, which may be partially attributable to MELD 3.0, said Kwong.
However, waitlist dropout rates also decreased among men, from 9.8% to 7.4%, probably because of improvements in technology, such as machine perfusion, which have increased the number of available livers, she added.
Disparities Continue to Exist
Some disparities still exist. Although the total median MELD score at transplant decreased from 29 to 27, women still had a higher median score of 29 at transplant, compared with a median score of 27 among men.
“This indicates that there may still be differences in transplant access between the sexes,” Kwong said. “There are still body size differences that can affect the probability of transplant, and this would not be addressed by MELD 3.0.”
Additional transplant disparities exist related to other patient characteristics, such as age, race, and ethnicity.
Future versions of MELD could potentially consider these factors, said session moderator Aleksander Krag, MD, PhD, MBA, professor of clinical medicine at the University of Southern Denmark, Odense, and secretary general of the European Association for the Study of the Liver, 2023-2025.
“There are infinite versions of MELD that can be made,” Kwong said. “It’s still early to see how MELD 3.0 will serve the system, but so far, so good.”
In a comment, Tamar Taddei, MD, professor of medicine in digestive diseases at Yale School of Medicine, New Haven, Connecticut, who comoderated the session, noted the importance of using a MELD score that considers sex-based differences.
This study brings MELD 3.0 to its fruition by reducing the disparities experienced by women who were underserved by the previous scoring systems, she said.
It was lovely to see that MELD 3.0 reduced the disparities with transplants, and also that the waitlist dropout was reduced — for both men and women,” Taddei said. “This change is a no-brainer.”
Kwong, Krag, and Taddei reported no relevant disclosures.
A version of this article appeared on Medscape.com.
FROM AASLD 24