User login
Atezolizumab TNBC indication ‘in jeopardy’ because of phase 3 results
The FDA said the phase 3 IMpassion131 trial showed that atezolizumab plus paclitaxel did not significantly reduce the risk of cancer progression and death, when compared with paclitaxel plus placebo, in programmed death-ligand 1 (PD-L1)–positive patients.
“Additionally, interim overall survival results favored paclitaxel plus placebo over paclitaxel plus atezolizumab in both the PD-L1-positive population and total population,” the FDA noted.
As a result, “health care professionals should not replace paclitaxel protein-bound (Abraxane) with paclitaxel in clinical practice,” the FDA advised.
Atezolizumab is approved for use in combination with protein-bound paclitaxel, also known as nanoparticle albumin–bound paclitaxel (nab-paclitaxel), to treat patients with unresectable, locally advanced, or metastatic TNBC whose tumors express PD-L1, as detected by an FDA-approved test. The combination was granted accelerated approval for this indication last year.
Atezolizumab plus nab-paclitaxel is the combination most often used in PD-L1-positive TNBC, as opposed to atezolizumab and unbound paclitaxel, said Melinda L. Telli, MD, an associate professor of medicine and director of the Stanford Cancer Institute Breast Cancer Program at Stanford (Calif.) University.
However, as the FDA noted, “continued approval of atezolizumab in combination with [nab-paclitaxel] may be contingent on proven benefit of the treatment in additional trials.”
Dr. Telli explained that atezolizumab was granted accelerated approval for the treatment of PD-L1-positive TNBC based on results of the phase 3 IMpassion130 trial, which compared nab-paclitaxel plus atezolizumab with nab-paclitaxel plus placebo.
“Additional data from IMpassion131 was hoped to be used to support the conversion of the accelerated approval to a full approval. Since IMpassion131 was negative, it unfortunately places the status of atezolizumab in [TNBC] in jeopardy as the benefits were not corroborated. The FDA may move to revoke the approval of atezolizumab for [TNBC],” Dr. Telli said.
In its alert, the FDA stated that it “will review the findings of IMpassion131 and will communicate new information regarding the IMpassion131 results and any potential changes to prescribing information.”
“We need to wait for full presentation and publication of the study results, but, in my assessment, the negative results in IMpassion131 are most likely due to differences in patient selection [from IMpassion130],” Dr. Telli said.
Results from IMpassion131 are scheduled to be presented at the ESMO Virtual Congress 2020.
The IMpassion trials were funded by Roche, maker of atezolizumab. Dr. Telli disclosed relationships with AbbVie, AstraZeneca, Merck, PharmaMar, Pfizer, and Tesaro.
The FDA said the phase 3 IMpassion131 trial showed that atezolizumab plus paclitaxel did not significantly reduce the risk of cancer progression and death, when compared with paclitaxel plus placebo, in programmed death-ligand 1 (PD-L1)–positive patients.
“Additionally, interim overall survival results favored paclitaxel plus placebo over paclitaxel plus atezolizumab in both the PD-L1-positive population and total population,” the FDA noted.
As a result, “health care professionals should not replace paclitaxel protein-bound (Abraxane) with paclitaxel in clinical practice,” the FDA advised.
Atezolizumab is approved for use in combination with protein-bound paclitaxel, also known as nanoparticle albumin–bound paclitaxel (nab-paclitaxel), to treat patients with unresectable, locally advanced, or metastatic TNBC whose tumors express PD-L1, as detected by an FDA-approved test. The combination was granted accelerated approval for this indication last year.
Atezolizumab plus nab-paclitaxel is the combination most often used in PD-L1-positive TNBC, as opposed to atezolizumab and unbound paclitaxel, said Melinda L. Telli, MD, an associate professor of medicine and director of the Stanford Cancer Institute Breast Cancer Program at Stanford (Calif.) University.
However, as the FDA noted, “continued approval of atezolizumab in combination with [nab-paclitaxel] may be contingent on proven benefit of the treatment in additional trials.”
Dr. Telli explained that atezolizumab was granted accelerated approval for the treatment of PD-L1-positive TNBC based on results of the phase 3 IMpassion130 trial, which compared nab-paclitaxel plus atezolizumab with nab-paclitaxel plus placebo.
“Additional data from IMpassion131 was hoped to be used to support the conversion of the accelerated approval to a full approval. Since IMpassion131 was negative, it unfortunately places the status of atezolizumab in [TNBC] in jeopardy as the benefits were not corroborated. The FDA may move to revoke the approval of atezolizumab for [TNBC],” Dr. Telli said.
In its alert, the FDA stated that it “will review the findings of IMpassion131 and will communicate new information regarding the IMpassion131 results and any potential changes to prescribing information.”
“We need to wait for full presentation and publication of the study results, but, in my assessment, the negative results in IMpassion131 are most likely due to differences in patient selection [from IMpassion130],” Dr. Telli said.
Results from IMpassion131 are scheduled to be presented at the ESMO Virtual Congress 2020.
The IMpassion trials were funded by Roche, maker of atezolizumab. Dr. Telli disclosed relationships with AbbVie, AstraZeneca, Merck, PharmaMar, Pfizer, and Tesaro.
The FDA said the phase 3 IMpassion131 trial showed that atezolizumab plus paclitaxel did not significantly reduce the risk of cancer progression and death, when compared with paclitaxel plus placebo, in programmed death-ligand 1 (PD-L1)–positive patients.
“Additionally, interim overall survival results favored paclitaxel plus placebo over paclitaxel plus atezolizumab in both the PD-L1-positive population and total population,” the FDA noted.
As a result, “health care professionals should not replace paclitaxel protein-bound (Abraxane) with paclitaxel in clinical practice,” the FDA advised.
Atezolizumab is approved for use in combination with protein-bound paclitaxel, also known as nanoparticle albumin–bound paclitaxel (nab-paclitaxel), to treat patients with unresectable, locally advanced, or metastatic TNBC whose tumors express PD-L1, as detected by an FDA-approved test. The combination was granted accelerated approval for this indication last year.
Atezolizumab plus nab-paclitaxel is the combination most often used in PD-L1-positive TNBC, as opposed to atezolizumab and unbound paclitaxel, said Melinda L. Telli, MD, an associate professor of medicine and director of the Stanford Cancer Institute Breast Cancer Program at Stanford (Calif.) University.
However, as the FDA noted, “continued approval of atezolizumab in combination with [nab-paclitaxel] may be contingent on proven benefit of the treatment in additional trials.”
Dr. Telli explained that atezolizumab was granted accelerated approval for the treatment of PD-L1-positive TNBC based on results of the phase 3 IMpassion130 trial, which compared nab-paclitaxel plus atezolizumab with nab-paclitaxel plus placebo.
“Additional data from IMpassion131 was hoped to be used to support the conversion of the accelerated approval to a full approval. Since IMpassion131 was negative, it unfortunately places the status of atezolizumab in [TNBC] in jeopardy as the benefits were not corroborated. The FDA may move to revoke the approval of atezolizumab for [TNBC],” Dr. Telli said.
In its alert, the FDA stated that it “will review the findings of IMpassion131 and will communicate new information regarding the IMpassion131 results and any potential changes to prescribing information.”
“We need to wait for full presentation and publication of the study results, but, in my assessment, the negative results in IMpassion131 are most likely due to differences in patient selection [from IMpassion130],” Dr. Telli said.
Results from IMpassion131 are scheduled to be presented at the ESMO Virtual Congress 2020.
The IMpassion trials were funded by Roche, maker of atezolizumab. Dr. Telli disclosed relationships with AbbVie, AstraZeneca, Merck, PharmaMar, Pfizer, and Tesaro.
FDA grants approval to weekly growth hormone for adults
The human growth hormone formulation somapacitan for adults with growth hormone deficiency was approved by the Food and Drug Administration on Sept. 1. .
Somapacitan contains an albumin-binding element attached to the growth hormone, causing the reversible binding to albumin proteins in the body. This reduces clearance and increases the half-life of the hormone. The formulation has previous demonstrated safety and efficacy in children with growth hormone deficiency (J Clin Endocrinol Metab. 2020 Apr 1. doi: 10.1210/clinem/dgz310).
Growth hormone treatment can counter abdominal obesity, reduced lean body mass, fatigue, osteopenia, cardiovascular risks, and other manifestations of growth hormone deficiency in adults, but daily injections can be burdensome for patients. That makes long-acting versions attractive, but the lifelong nature of the treatment makes it important to characterize safety and tolerability.
The approval comes on the strength of a randomized, placebo-controlled phase 3 trial (REAL 1) of 300 adult patients in 17 countries with growth hormone deficiency (J Clin Endocrinol Metab. 2020 Apr 1. doi: 10.1210/clinem/dgaa049). Participants had either never received growth hormone treatment, or had stopped taking one at least 6 months before starting the trial. Subjects received once-weekly somapacitan, once-weekly placebo, or daily somatropin, which is FDA approved.
The primary endpoint was percentage change of truncal fat, which is regulated by growth hormone, and can lead to medical problems. After 34 weeks, subjects in the somapacitan group experienced a 1.06% decrease in truncal fat, compared with a 0.47% increase in the placebo group (P = .009) and a 2.23% decrease in the daily somatropin group.
After 34 weeks, a 52-week extension trial began. The somapacitan group continued on the drug and the placebo group was offered somapacitan. Patients on daily somatropin were randomized to continue daily treatment with somatropin or to switch to somapacitan.
At the end of the extension trial, those taking somapacitan for the full 86-week duration had an average reduction of 1.52% in truncal fat. After 86 weeks, the somapacitan and daily somatropin groups had similar values for percentage change in visceral fat, lean body mass, or appendicular skeletal muscle mass.
Common side effects of somapacitan were back pain, joint paint, indigestion, a sleep disorder, dizziness, tonsillitis, swelling in the arms or lower legs, vomiting, adrenal insufficiency, hypertension, increase in blood creatine phosphokinase, weight increase, and anemia.
Somapacitan, marketed as Sogroya by Novo Nordisk, is contraindicated in patients with an allergy to the drug, as well as those with an active malignancy, diabetic eye disease where increases in blood sugars could lead to retinal damage, acute critical illness, or acute respiratory failure.
The FDA recommends that providers perform an eye examination before drug initiation, as well as periodically while the patient is taking the drug, to rule out preexisting papilledema. This could be a sign of intracranial hypertension, which could be caused or worsened by growth hormones.
The human growth hormone formulation somapacitan for adults with growth hormone deficiency was approved by the Food and Drug Administration on Sept. 1. .
Somapacitan contains an albumin-binding element attached to the growth hormone, causing the reversible binding to albumin proteins in the body. This reduces clearance and increases the half-life of the hormone. The formulation has previous demonstrated safety and efficacy in children with growth hormone deficiency (J Clin Endocrinol Metab. 2020 Apr 1. doi: 10.1210/clinem/dgz310).
Growth hormone treatment can counter abdominal obesity, reduced lean body mass, fatigue, osteopenia, cardiovascular risks, and other manifestations of growth hormone deficiency in adults, but daily injections can be burdensome for patients. That makes long-acting versions attractive, but the lifelong nature of the treatment makes it important to characterize safety and tolerability.
The approval comes on the strength of a randomized, placebo-controlled phase 3 trial (REAL 1) of 300 adult patients in 17 countries with growth hormone deficiency (J Clin Endocrinol Metab. 2020 Apr 1. doi: 10.1210/clinem/dgaa049). Participants had either never received growth hormone treatment, or had stopped taking one at least 6 months before starting the trial. Subjects received once-weekly somapacitan, once-weekly placebo, or daily somatropin, which is FDA approved.
The primary endpoint was percentage change of truncal fat, which is regulated by growth hormone, and can lead to medical problems. After 34 weeks, subjects in the somapacitan group experienced a 1.06% decrease in truncal fat, compared with a 0.47% increase in the placebo group (P = .009) and a 2.23% decrease in the daily somatropin group.
After 34 weeks, a 52-week extension trial began. The somapacitan group continued on the drug and the placebo group was offered somapacitan. Patients on daily somatropin were randomized to continue daily treatment with somatropin or to switch to somapacitan.
At the end of the extension trial, those taking somapacitan for the full 86-week duration had an average reduction of 1.52% in truncal fat. After 86 weeks, the somapacitan and daily somatropin groups had similar values for percentage change in visceral fat, lean body mass, or appendicular skeletal muscle mass.
Common side effects of somapacitan were back pain, joint paint, indigestion, a sleep disorder, dizziness, tonsillitis, swelling in the arms or lower legs, vomiting, adrenal insufficiency, hypertension, increase in blood creatine phosphokinase, weight increase, and anemia.
Somapacitan, marketed as Sogroya by Novo Nordisk, is contraindicated in patients with an allergy to the drug, as well as those with an active malignancy, diabetic eye disease where increases in blood sugars could lead to retinal damage, acute critical illness, or acute respiratory failure.
The FDA recommends that providers perform an eye examination before drug initiation, as well as periodically while the patient is taking the drug, to rule out preexisting papilledema. This could be a sign of intracranial hypertension, which could be caused or worsened by growth hormones.
The human growth hormone formulation somapacitan for adults with growth hormone deficiency was approved by the Food and Drug Administration on Sept. 1. .
Somapacitan contains an albumin-binding element attached to the growth hormone, causing the reversible binding to albumin proteins in the body. This reduces clearance and increases the half-life of the hormone. The formulation has previous demonstrated safety and efficacy in children with growth hormone deficiency (J Clin Endocrinol Metab. 2020 Apr 1. doi: 10.1210/clinem/dgz310).
Growth hormone treatment can counter abdominal obesity, reduced lean body mass, fatigue, osteopenia, cardiovascular risks, and other manifestations of growth hormone deficiency in adults, but daily injections can be burdensome for patients. That makes long-acting versions attractive, but the lifelong nature of the treatment makes it important to characterize safety and tolerability.
The approval comes on the strength of a randomized, placebo-controlled phase 3 trial (REAL 1) of 300 adult patients in 17 countries with growth hormone deficiency (J Clin Endocrinol Metab. 2020 Apr 1. doi: 10.1210/clinem/dgaa049). Participants had either never received growth hormone treatment, or had stopped taking one at least 6 months before starting the trial. Subjects received once-weekly somapacitan, once-weekly placebo, or daily somatropin, which is FDA approved.
The primary endpoint was percentage change of truncal fat, which is regulated by growth hormone, and can lead to medical problems. After 34 weeks, subjects in the somapacitan group experienced a 1.06% decrease in truncal fat, compared with a 0.47% increase in the placebo group (P = .009) and a 2.23% decrease in the daily somatropin group.
After 34 weeks, a 52-week extension trial began. The somapacitan group continued on the drug and the placebo group was offered somapacitan. Patients on daily somatropin were randomized to continue daily treatment with somatropin or to switch to somapacitan.
At the end of the extension trial, those taking somapacitan for the full 86-week duration had an average reduction of 1.52% in truncal fat. After 86 weeks, the somapacitan and daily somatropin groups had similar values for percentage change in visceral fat, lean body mass, or appendicular skeletal muscle mass.
Common side effects of somapacitan were back pain, joint paint, indigestion, a sleep disorder, dizziness, tonsillitis, swelling in the arms or lower legs, vomiting, adrenal insufficiency, hypertension, increase in blood creatine phosphokinase, weight increase, and anemia.
Somapacitan, marketed as Sogroya by Novo Nordisk, is contraindicated in patients with an allergy to the drug, as well as those with an active malignancy, diabetic eye disease where increases in blood sugars could lead to retinal damage, acute critical illness, or acute respiratory failure.
The FDA recommends that providers perform an eye examination before drug initiation, as well as periodically while the patient is taking the drug, to rule out preexisting papilledema. This could be a sign of intracranial hypertension, which could be caused or worsened by growth hormones.
Pralsetinib: Second drug for RET+ NSCLC approved in U.S.
A second drug is now available in the United States for use in the treatment of patients with metastatic non–small cell lung cancer (NSCLC) that tests positive for rearranged during transfection (RET) fusions.
The new drug is pralsetinib (Gavreto). The Food and Drug Administration granted it an accelerated approval for this indication on the basis of response rate data. Continued approval for this indication depends on clinical benefit in a confirmatory trial.
Pralsetinib joins selpercatinib (Retevmo), which was approved in the United States in May 2020 as the first RET-targeted therapy. Selpercatinib is also indicated for use in RET+ NSCLC and was approved for use in RET+ medullary thyroid cancer and RET+ thyroid cancer.
Pralsetinib is still awaiting approval for these thyroid cancer indications.
Both drugs are taken orally; pralsetinib is taken once daily, and selpercatinib is taken twice daily.
For both drugs, before treatment is initiated, laboratory testing is needed to show that a RET gene alteration is present in the tumor.
RET fusions are found in approximately 1%-2% of patients with NSCLC.
They are the latest of a number of tumor-specific gene alterations found in NSCLC that are targeted with an approved drug.
“Targeted therapies have dramatically improved care for patients with non–small cell lung cancer driven by oncogenes, including EGFR and ALK, and the approval of the selective RET inhibitor pralsetinib, or Gavreto, marks another milestone in a paradigm shift toward precision medicine,” Vivek Subbiah, MD, of the University of Texas MD Anderson Cancer Center, Houston, said in a press release.
Dr. Subbiah was an investigator of the phase 1/2 clinical trial known as ARROW, which provided the data on which the accelerated approval was based. In this trial, patients with RET+ NSCLC were found by testing with next-generation sequencing, FISH (fluorescence in situ hybridization), or other methods.
The ARROW trial involved one cohort of 87 patients who had previously been treated with platinum-based chemotherapy. In these patients, the overall response rate (ORR) was 57%, the complete response (CR) rate was 5.7%, and the median duration of response (DOR) was not estimable, according to the manufacturer, Blueprint Medicines.
The trial also involved 27 treatment-naive patients who were ineligible for platinum-based chemotherapy per the study protocol. In this group, the ORR was 70%, and the CR rate was 11%. The median DOR was 9.0 months.
“Patients treated with [pralsetinib] had durable clinical responses, with a subset achieving complete responses characterized by the resolution of all target lesions, an uncommon outcome in metastatic lung cancer,” Dr. Subbiah commented.
“We observed this activity with or without prior therapy and regardless of RET fusion partner or the presence of brain metastases. This approval represents an important advance with the potential to change standards of care for patients with RET fusion-positive NSCLC, who have historically had limited treatment options,” Dr. Subbiah added.
Product information for pralsetinib has warnings and precautions of interstitial lung disease/pneumonitis, hypertension, hepatotoxicity, hemorrhagic events, risk for impaired wound healing, and risk for embryo-fetal toxicity.
This article first appeared on Medscape.com.
A second drug is now available in the United States for use in the treatment of patients with metastatic non–small cell lung cancer (NSCLC) that tests positive for rearranged during transfection (RET) fusions.
The new drug is pralsetinib (Gavreto). The Food and Drug Administration granted it an accelerated approval for this indication on the basis of response rate data. Continued approval for this indication depends on clinical benefit in a confirmatory trial.
Pralsetinib joins selpercatinib (Retevmo), which was approved in the United States in May 2020 as the first RET-targeted therapy. Selpercatinib is also indicated for use in RET+ NSCLC and was approved for use in RET+ medullary thyroid cancer and RET+ thyroid cancer.
Pralsetinib is still awaiting approval for these thyroid cancer indications.
Both drugs are taken orally; pralsetinib is taken once daily, and selpercatinib is taken twice daily.
For both drugs, before treatment is initiated, laboratory testing is needed to show that a RET gene alteration is present in the tumor.
RET fusions are found in approximately 1%-2% of patients with NSCLC.
They are the latest of a number of tumor-specific gene alterations found in NSCLC that are targeted with an approved drug.
“Targeted therapies have dramatically improved care for patients with non–small cell lung cancer driven by oncogenes, including EGFR and ALK, and the approval of the selective RET inhibitor pralsetinib, or Gavreto, marks another milestone in a paradigm shift toward precision medicine,” Vivek Subbiah, MD, of the University of Texas MD Anderson Cancer Center, Houston, said in a press release.
Dr. Subbiah was an investigator of the phase 1/2 clinical trial known as ARROW, which provided the data on which the accelerated approval was based. In this trial, patients with RET+ NSCLC were found by testing with next-generation sequencing, FISH (fluorescence in situ hybridization), or other methods.
The ARROW trial involved one cohort of 87 patients who had previously been treated with platinum-based chemotherapy. In these patients, the overall response rate (ORR) was 57%, the complete response (CR) rate was 5.7%, and the median duration of response (DOR) was not estimable, according to the manufacturer, Blueprint Medicines.
The trial also involved 27 treatment-naive patients who were ineligible for platinum-based chemotherapy per the study protocol. In this group, the ORR was 70%, and the CR rate was 11%. The median DOR was 9.0 months.
“Patients treated with [pralsetinib] had durable clinical responses, with a subset achieving complete responses characterized by the resolution of all target lesions, an uncommon outcome in metastatic lung cancer,” Dr. Subbiah commented.
“We observed this activity with or without prior therapy and regardless of RET fusion partner or the presence of brain metastases. This approval represents an important advance with the potential to change standards of care for patients with RET fusion-positive NSCLC, who have historically had limited treatment options,” Dr. Subbiah added.
Product information for pralsetinib has warnings and precautions of interstitial lung disease/pneumonitis, hypertension, hepatotoxicity, hemorrhagic events, risk for impaired wound healing, and risk for embryo-fetal toxicity.
This article first appeared on Medscape.com.
A second drug is now available in the United States for use in the treatment of patients with metastatic non–small cell lung cancer (NSCLC) that tests positive for rearranged during transfection (RET) fusions.
The new drug is pralsetinib (Gavreto). The Food and Drug Administration granted it an accelerated approval for this indication on the basis of response rate data. Continued approval for this indication depends on clinical benefit in a confirmatory trial.
Pralsetinib joins selpercatinib (Retevmo), which was approved in the United States in May 2020 as the first RET-targeted therapy. Selpercatinib is also indicated for use in RET+ NSCLC and was approved for use in RET+ medullary thyroid cancer and RET+ thyroid cancer.
Pralsetinib is still awaiting approval for these thyroid cancer indications.
Both drugs are taken orally; pralsetinib is taken once daily, and selpercatinib is taken twice daily.
For both drugs, before treatment is initiated, laboratory testing is needed to show that a RET gene alteration is present in the tumor.
RET fusions are found in approximately 1%-2% of patients with NSCLC.
They are the latest of a number of tumor-specific gene alterations found in NSCLC that are targeted with an approved drug.
“Targeted therapies have dramatically improved care for patients with non–small cell lung cancer driven by oncogenes, including EGFR and ALK, and the approval of the selective RET inhibitor pralsetinib, or Gavreto, marks another milestone in a paradigm shift toward precision medicine,” Vivek Subbiah, MD, of the University of Texas MD Anderson Cancer Center, Houston, said in a press release.
Dr. Subbiah was an investigator of the phase 1/2 clinical trial known as ARROW, which provided the data on which the accelerated approval was based. In this trial, patients with RET+ NSCLC were found by testing with next-generation sequencing, FISH (fluorescence in situ hybridization), or other methods.
The ARROW trial involved one cohort of 87 patients who had previously been treated with platinum-based chemotherapy. In these patients, the overall response rate (ORR) was 57%, the complete response (CR) rate was 5.7%, and the median duration of response (DOR) was not estimable, according to the manufacturer, Blueprint Medicines.
The trial also involved 27 treatment-naive patients who were ineligible for platinum-based chemotherapy per the study protocol. In this group, the ORR was 70%, and the CR rate was 11%. The median DOR was 9.0 months.
“Patients treated with [pralsetinib] had durable clinical responses, with a subset achieving complete responses characterized by the resolution of all target lesions, an uncommon outcome in metastatic lung cancer,” Dr. Subbiah commented.
“We observed this activity with or without prior therapy and regardless of RET fusion partner or the presence of brain metastases. This approval represents an important advance with the potential to change standards of care for patients with RET fusion-positive NSCLC, who have historically had limited treatment options,” Dr. Subbiah added.
Product information for pralsetinib has warnings and precautions of interstitial lung disease/pneumonitis, hypertension, hepatotoxicity, hemorrhagic events, risk for impaired wound healing, and risk for embryo-fetal toxicity.
This article first appeared on Medscape.com.
FDA approves first maintenance therapy for AML
The Food and Drug Administration has approved an oral form of azacitidine (Onureg) for use as maintenance therapy for patients with acute myeloid leukemia (AML) who have achieved a first complete remission.
The approval extends to patients who have achieved complete remission with incomplete blood count recovery following intensive induction chemotherapy and who are unable to complete intensive curative therapy.
The approval was based on data from the QUAZAR AML-001 trial, which showed that oral azacitidine significantly improved overall survival when compared with placebo.
“It’s not too hard to get these patients into remission,” Harry P. Erba, MD, PhD, director of the Leukemia Program at the Duke Cancer Institute, Durham, N.C., said in an interview last year when these results were first presented at the 2019 annual meeting of the American Society of Hematology. “The problem comes in keeping them in remission.”
Despite various attempts, there has been no success over the past 30 years in defining maintenance treatment for these patients, Andrew H. Wei, MBBS, PhD, from the Alfred Hospital in Melbourne, Australia, said.
“Oral azacitidine represents a new therapeutic standard for patients with AML in remission,” he said.
Azacitidine is a hypomethylating agent that incorporates into DNA and RNA. It has long been used as an injectable therapy for the treatment of myelodysplastic syndromes.
The approval of the new oral formulation for the new indication of AML “is the culmination of over a decade of research and 13 preclinical and clinical trials,” said Giovanni Caforio, M.D., chairman and chief executive officer of Bristol-Myers Squibb, in a statement.
QUAZAR results
The QUAZAR AML-001 trial was a phase 3, international study involving 472 patients with AML who were within achieving a first complete remission or remission with incomplete blood recovery. All patients had received intensive induction chemotherapy with or without consolidation treatment per investigator preference prior to study entry and were not candidates for hematopoietic stem cell transplant at the time of screening.
Patients were randomly assigned to receive either oral azacitidine 200 mg daily on days 1-14 of a repeat 28-day cycle (n = 278) or matching placebo (n = 274). Treatment was continued indefinitely until blast count was more than 15% or patients experienced unacceptable toxicity or underwent transplant.
At a median follow-up of more than 41.2 months, the median overall survival was significantly longer for patients who received oral azacitidine at 24.7 months versus 14.8 months for those who received placebo (hazard ratio, 0.69; P < .0009).
Relapse-free survival was also significantly prolonged to 10.2 months for patients who received oral azacitidine vs. 4.8 months for those who received placebo (HR, 0.65; P < .0001).
Serious adverse reactions occurred in 15% of patients who received azacitidine. Events that occurred in 2% of patients or more include pneumonia (8%) and febrile neutropenia (7%). There was one fatal event.
The most common adverse reactions were nausea (65% vs. 24%), vomiting (60% vs. 10%), diarrhea (50% vs. 21%), fatigue/asthenia (44% vs. 25%), constipation (39% vs. 24%), pneumonia (27% vs. 17%), abdominal pain (22% vs. 13%) arthralgia (14% vs. 10%), decreased appetite (13% vs. 6%), febrile neutropenia (12% vs. 8%), dizziness (11% vs. 9%), and pain in extremity (11% vs. 5%). Permanent discontinuation because of an adverse reaction occurred in 8% of patients.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved an oral form of azacitidine (Onureg) for use as maintenance therapy for patients with acute myeloid leukemia (AML) who have achieved a first complete remission.
The approval extends to patients who have achieved complete remission with incomplete blood count recovery following intensive induction chemotherapy and who are unable to complete intensive curative therapy.
The approval was based on data from the QUAZAR AML-001 trial, which showed that oral azacitidine significantly improved overall survival when compared with placebo.
“It’s not too hard to get these patients into remission,” Harry P. Erba, MD, PhD, director of the Leukemia Program at the Duke Cancer Institute, Durham, N.C., said in an interview last year when these results were first presented at the 2019 annual meeting of the American Society of Hematology. “The problem comes in keeping them in remission.”
Despite various attempts, there has been no success over the past 30 years in defining maintenance treatment for these patients, Andrew H. Wei, MBBS, PhD, from the Alfred Hospital in Melbourne, Australia, said.
“Oral azacitidine represents a new therapeutic standard for patients with AML in remission,” he said.
Azacitidine is a hypomethylating agent that incorporates into DNA and RNA. It has long been used as an injectable therapy for the treatment of myelodysplastic syndromes.
The approval of the new oral formulation for the new indication of AML “is the culmination of over a decade of research and 13 preclinical and clinical trials,” said Giovanni Caforio, M.D., chairman and chief executive officer of Bristol-Myers Squibb, in a statement.
QUAZAR results
The QUAZAR AML-001 trial was a phase 3, international study involving 472 patients with AML who were within achieving a first complete remission or remission with incomplete blood recovery. All patients had received intensive induction chemotherapy with or without consolidation treatment per investigator preference prior to study entry and were not candidates for hematopoietic stem cell transplant at the time of screening.
Patients were randomly assigned to receive either oral azacitidine 200 mg daily on days 1-14 of a repeat 28-day cycle (n = 278) or matching placebo (n = 274). Treatment was continued indefinitely until blast count was more than 15% or patients experienced unacceptable toxicity or underwent transplant.
At a median follow-up of more than 41.2 months, the median overall survival was significantly longer for patients who received oral azacitidine at 24.7 months versus 14.8 months for those who received placebo (hazard ratio, 0.69; P < .0009).
Relapse-free survival was also significantly prolonged to 10.2 months for patients who received oral azacitidine vs. 4.8 months for those who received placebo (HR, 0.65; P < .0001).
Serious adverse reactions occurred in 15% of patients who received azacitidine. Events that occurred in 2% of patients or more include pneumonia (8%) and febrile neutropenia (7%). There was one fatal event.
The most common adverse reactions were nausea (65% vs. 24%), vomiting (60% vs. 10%), diarrhea (50% vs. 21%), fatigue/asthenia (44% vs. 25%), constipation (39% vs. 24%), pneumonia (27% vs. 17%), abdominal pain (22% vs. 13%) arthralgia (14% vs. 10%), decreased appetite (13% vs. 6%), febrile neutropenia (12% vs. 8%), dizziness (11% vs. 9%), and pain in extremity (11% vs. 5%). Permanent discontinuation because of an adverse reaction occurred in 8% of patients.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved an oral form of azacitidine (Onureg) for use as maintenance therapy for patients with acute myeloid leukemia (AML) who have achieved a first complete remission.
The approval extends to patients who have achieved complete remission with incomplete blood count recovery following intensive induction chemotherapy and who are unable to complete intensive curative therapy.
The approval was based on data from the QUAZAR AML-001 trial, which showed that oral azacitidine significantly improved overall survival when compared with placebo.
“It’s not too hard to get these patients into remission,” Harry P. Erba, MD, PhD, director of the Leukemia Program at the Duke Cancer Institute, Durham, N.C., said in an interview last year when these results were first presented at the 2019 annual meeting of the American Society of Hematology. “The problem comes in keeping them in remission.”
Despite various attempts, there has been no success over the past 30 years in defining maintenance treatment for these patients, Andrew H. Wei, MBBS, PhD, from the Alfred Hospital in Melbourne, Australia, said.
“Oral azacitidine represents a new therapeutic standard for patients with AML in remission,” he said.
Azacitidine is a hypomethylating agent that incorporates into DNA and RNA. It has long been used as an injectable therapy for the treatment of myelodysplastic syndromes.
The approval of the new oral formulation for the new indication of AML “is the culmination of over a decade of research and 13 preclinical and clinical trials,” said Giovanni Caforio, M.D., chairman and chief executive officer of Bristol-Myers Squibb, in a statement.
QUAZAR results
The QUAZAR AML-001 trial was a phase 3, international study involving 472 patients with AML who were within achieving a first complete remission or remission with incomplete blood recovery. All patients had received intensive induction chemotherapy with or without consolidation treatment per investigator preference prior to study entry and were not candidates for hematopoietic stem cell transplant at the time of screening.
Patients were randomly assigned to receive either oral azacitidine 200 mg daily on days 1-14 of a repeat 28-day cycle (n = 278) or matching placebo (n = 274). Treatment was continued indefinitely until blast count was more than 15% or patients experienced unacceptable toxicity or underwent transplant.
At a median follow-up of more than 41.2 months, the median overall survival was significantly longer for patients who received oral azacitidine at 24.7 months versus 14.8 months for those who received placebo (hazard ratio, 0.69; P < .0009).
Relapse-free survival was also significantly prolonged to 10.2 months for patients who received oral azacitidine vs. 4.8 months for those who received placebo (HR, 0.65; P < .0001).
Serious adverse reactions occurred in 15% of patients who received azacitidine. Events that occurred in 2% of patients or more include pneumonia (8%) and febrile neutropenia (7%). There was one fatal event.
The most common adverse reactions were nausea (65% vs. 24%), vomiting (60% vs. 10%), diarrhea (50% vs. 21%), fatigue/asthenia (44% vs. 25%), constipation (39% vs. 24%), pneumonia (27% vs. 17%), abdominal pain (22% vs. 13%) arthralgia (14% vs. 10%), decreased appetite (13% vs. 6%), febrile neutropenia (12% vs. 8%), dizziness (11% vs. 9%), and pain in extremity (11% vs. 5%). Permanent discontinuation because of an adverse reaction occurred in 8% of patients.
A version of this article originally appeared on Medscape.com.
FDA OKs new ‘artificial pancreas’ Medtronic 770G
The Food and Drug Administration has approved the MiniMed 770G (Medtronic) automated insulin delivery system for children aged 2-6 years.
The 770G system adds Bluetooth smartphone connectivity to the SmartGuard technology that is present in the hybrid closed-loop MiniMed 670G system, which has been available in the United States since 2016 for individuals aged 14 years and older who have type 1 diabetes. It has been available since 2018 for children aged 7 years.
The 770G will also be available to older children and adults once it has been launched.
As with other so-called artificial pancreas systems, the 770G is made up of an insulin pump and continuous glucose monitor that are connected via software that allows the pump to deliver or withhold insulin on the basis of glucose readings.
It is a “hybrid closed-loop” system in that users or caregivers must still manually signal carbohydrate consumption.
The 770G includes a “share” feature that allows health care providers, users, and caregivers to follow the user’s glucose levels remotely via smartphones. In-app notices indicate when glucose levels are out of range. The data can be uploaded prior to telehealth visits.
The approval was based on a 3-month study of 151 children aged 2-6 years who showed improvement in outcomes comparable with those seen in 124 older adolescents and adults with the 770G system as compared to patients who used manual (nonlooped) mode over a 2-week period. There were no episodes of severe hypoglycemia or diabetic ketoacidosis and no serious device-related adverse events while in hybrid closed-loop mode.
The FDA will require Medtronic to conduct a postmarketing study to evaluate the 770G in real-world settings. It is not approved for use in children younger than 2 years nor in any patient who requires less than 8 units of insulin per day.
The next-generation Medtronic closed-loop system, the 780G, has already been approved in Europe. It improves on the technology by delivering automated bolus correction doses in addition to basal insulin every 5 minutes. The company is preparing to submit the 780G for approval in the United States.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved the MiniMed 770G (Medtronic) automated insulin delivery system for children aged 2-6 years.
The 770G system adds Bluetooth smartphone connectivity to the SmartGuard technology that is present in the hybrid closed-loop MiniMed 670G system, which has been available in the United States since 2016 for individuals aged 14 years and older who have type 1 diabetes. It has been available since 2018 for children aged 7 years.
The 770G will also be available to older children and adults once it has been launched.
As with other so-called artificial pancreas systems, the 770G is made up of an insulin pump and continuous glucose monitor that are connected via software that allows the pump to deliver or withhold insulin on the basis of glucose readings.
It is a “hybrid closed-loop” system in that users or caregivers must still manually signal carbohydrate consumption.
The 770G includes a “share” feature that allows health care providers, users, and caregivers to follow the user’s glucose levels remotely via smartphones. In-app notices indicate when glucose levels are out of range. The data can be uploaded prior to telehealth visits.
The approval was based on a 3-month study of 151 children aged 2-6 years who showed improvement in outcomes comparable with those seen in 124 older adolescents and adults with the 770G system as compared to patients who used manual (nonlooped) mode over a 2-week period. There were no episodes of severe hypoglycemia or diabetic ketoacidosis and no serious device-related adverse events while in hybrid closed-loop mode.
The FDA will require Medtronic to conduct a postmarketing study to evaluate the 770G in real-world settings. It is not approved for use in children younger than 2 years nor in any patient who requires less than 8 units of insulin per day.
The next-generation Medtronic closed-loop system, the 780G, has already been approved in Europe. It improves on the technology by delivering automated bolus correction doses in addition to basal insulin every 5 minutes. The company is preparing to submit the 780G for approval in the United States.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved the MiniMed 770G (Medtronic) automated insulin delivery system for children aged 2-6 years.
The 770G system adds Bluetooth smartphone connectivity to the SmartGuard technology that is present in the hybrid closed-loop MiniMed 670G system, which has been available in the United States since 2016 for individuals aged 14 years and older who have type 1 diabetes. It has been available since 2018 for children aged 7 years.
The 770G will also be available to older children and adults once it has been launched.
As with other so-called artificial pancreas systems, the 770G is made up of an insulin pump and continuous glucose monitor that are connected via software that allows the pump to deliver or withhold insulin on the basis of glucose readings.
It is a “hybrid closed-loop” system in that users or caregivers must still manually signal carbohydrate consumption.
The 770G includes a “share” feature that allows health care providers, users, and caregivers to follow the user’s glucose levels remotely via smartphones. In-app notices indicate when glucose levels are out of range. The data can be uploaded prior to telehealth visits.
The approval was based on a 3-month study of 151 children aged 2-6 years who showed improvement in outcomes comparable with those seen in 124 older adolescents and adults with the 770G system as compared to patients who used manual (nonlooped) mode over a 2-week period. There were no episodes of severe hypoglycemia or diabetic ketoacidosis and no serious device-related adverse events while in hybrid closed-loop mode.
The FDA will require Medtronic to conduct a postmarketing study to evaluate the 770G in real-world settings. It is not approved for use in children younger than 2 years nor in any patient who requires less than 8 units of insulin per day.
The next-generation Medtronic closed-loop system, the 780G, has already been approved in Europe. It improves on the technology by delivering automated bolus correction doses in addition to basal insulin every 5 minutes. The company is preparing to submit the 780G for approval in the United States.
A version of this article originally appeared on Medscape.com.
Two PR employees at FDA fired after plasma therapy controversy
The US Food and Drug Administration has removed two senior public relations employees, one of whom advised the agency against unbridled promotion of convalescent blood plasma as a treatment for people with COVID-19, multiple media outlets reported Aug. 28.
Officials claim the dismissals are coincidental and are not related to a controversy about whether claims regarding convalescent plasma therapy that were put forth by President Donald Trump and FDA Commissioner Stephen M. Hahn, MD, were exaggerated, according to reports from The New York Times , CNN, and elsewhere.
One of the PR employees, Emily Miller, was on the job less than 2 weeks. The White House named her FDA chief spokeswoman 11 days ago, but Hahn removed her from that post Aug. 28.
On Aug. 27, the US Department of Health and Human Services terminated the contract for Wayne L. Pines, a PR consultant to the FDA. Pines reportedly advised Hahn to apologize for making misleading claims about the therapeutic benefits of convalescent plasma therapy for COVID-19.
The FDA did not respond to multiple requests for comment.
The controversy stems from comments Hahn made about the announcement of the emergency use authorization for convalescent plasma for patients with COVID-19. He said that plasma had been found to save the lives of 35 out of every 100 people who were treated. That statement was later found to be erroneous because he presented a relative risk reduction as an absolute decrease in risk. He later apologized via Twitter.
Researchers running clinical trials to evaluate the efficacy of convalescent plasma for COVID-19 are concerned that the emergency use authorization could thwart efforts to recruit participants for their studies.
This article first appeared on Medscape.com.
The US Food and Drug Administration has removed two senior public relations employees, one of whom advised the agency against unbridled promotion of convalescent blood plasma as a treatment for people with COVID-19, multiple media outlets reported Aug. 28.
Officials claim the dismissals are coincidental and are not related to a controversy about whether claims regarding convalescent plasma therapy that were put forth by President Donald Trump and FDA Commissioner Stephen M. Hahn, MD, were exaggerated, according to reports from The New York Times , CNN, and elsewhere.
One of the PR employees, Emily Miller, was on the job less than 2 weeks. The White House named her FDA chief spokeswoman 11 days ago, but Hahn removed her from that post Aug. 28.
On Aug. 27, the US Department of Health and Human Services terminated the contract for Wayne L. Pines, a PR consultant to the FDA. Pines reportedly advised Hahn to apologize for making misleading claims about the therapeutic benefits of convalescent plasma therapy for COVID-19.
The FDA did not respond to multiple requests for comment.
The controversy stems from comments Hahn made about the announcement of the emergency use authorization for convalescent plasma for patients with COVID-19. He said that plasma had been found to save the lives of 35 out of every 100 people who were treated. That statement was later found to be erroneous because he presented a relative risk reduction as an absolute decrease in risk. He later apologized via Twitter.
Researchers running clinical trials to evaluate the efficacy of convalescent plasma for COVID-19 are concerned that the emergency use authorization could thwart efforts to recruit participants for their studies.
This article first appeared on Medscape.com.
The US Food and Drug Administration has removed two senior public relations employees, one of whom advised the agency against unbridled promotion of convalescent blood plasma as a treatment for people with COVID-19, multiple media outlets reported Aug. 28.
Officials claim the dismissals are coincidental and are not related to a controversy about whether claims regarding convalescent plasma therapy that were put forth by President Donald Trump and FDA Commissioner Stephen M. Hahn, MD, were exaggerated, according to reports from The New York Times , CNN, and elsewhere.
One of the PR employees, Emily Miller, was on the job less than 2 weeks. The White House named her FDA chief spokeswoman 11 days ago, but Hahn removed her from that post Aug. 28.
On Aug. 27, the US Department of Health and Human Services terminated the contract for Wayne L. Pines, a PR consultant to the FDA. Pines reportedly advised Hahn to apologize for making misleading claims about the therapeutic benefits of convalescent plasma therapy for COVID-19.
The FDA did not respond to multiple requests for comment.
The controversy stems from comments Hahn made about the announcement of the emergency use authorization for convalescent plasma for patients with COVID-19. He said that plasma had been found to save the lives of 35 out of every 100 people who were treated. That statement was later found to be erroneous because he presented a relative risk reduction as an absolute decrease in risk. He later apologized via Twitter.
Researchers running clinical trials to evaluate the efficacy of convalescent plasma for COVID-19 are concerned that the emergency use authorization could thwart efforts to recruit participants for their studies.
This article first appeared on Medscape.com.
Attempted suicide in high school America, 2019
according to newly released data from the 2019 Youth Risk Behavior Survey.
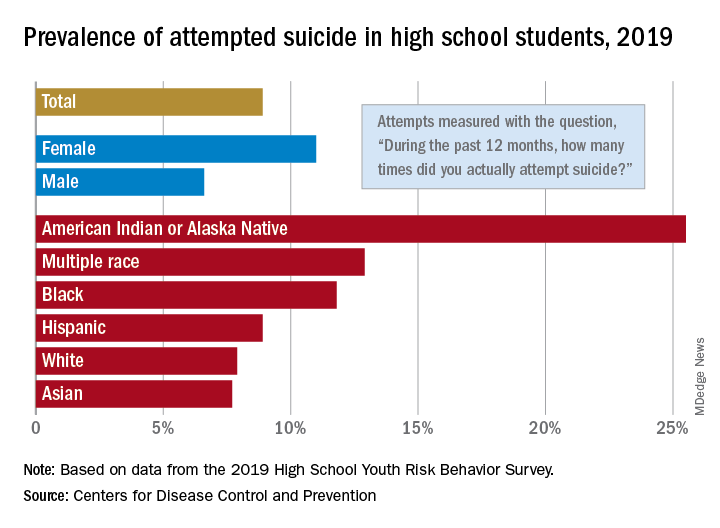
The prevalence of attempted suicide during the previous 12 months was 8.9% among the 13,677 students in grades 9-12 who took the survey last year, but the rate was 25.5% for American Indian/Alaska Native (AI/AN) respondents, almost 2.9 times higher, the YRBS data show.
Respondents with multiple races in their backgrounds, at 12.9%, and African Americans, with a prevalence of 11.8%, also were above the high school average for suicide attempts, while Whites (7.9%) and Asians (7.7%) were under it and Hispanics equaled it, the Centers for Disease Control and Prevention reported.
The number of AI/AN students was insufficient to examine differences by sex, but females in all of the other racial/ethnic groups were more likely than males to have attempted suicide: multiple race (17.8% vs. 7.3%), African American (15.2% vs. 8.5%), Hispanic (11.9% vs. 5.5%), White (9.4% vs. 6.4%), and Asian (8.4% vs. 7.1%), the CDC’s Division of Adolescent and School Health said.
Among all respondents, 11.0% of females had attempted suicide in the 12 months before the survey, a figure that is significantly higher than the 6.6% prevalence in males. Females also were significantly more likely than males to make a plan about how they would attempt suicide (19.9% vs. 11.3%) and to seriously consider an attempt (24.1% vs. 13.3%), CDC investigators said in a separate report.
Significant differences also were seen when looking at sexual identity. Suicide attempts were reported by 6.4% of heterosexuals, 16.1% of those who weren’t sure, and 23.4% of lesbians/gays/bisexuals (LGBs). For serious consideration of suicide, the respective numbers were 14.5%, 30.4%, and 46.8%, they reported (MMWR Supp. 2020 Aug 21;69[1]:47-55).
For nonheterosexuals, however, males were slightly more likely (23.8%) than females (23.6%) to have attempted suicide, but females were more likely to seriously consider it (49.0% vs. 40.4%) and to make a plan (42.4% vs. 33.0%), according to the YRBS data.
“Adolescence … represents a time for expanded identity development, with sexual identity development representing a complex, multidimensional, and often stressful process for youths,” the CDC investigators said in the MMWR. “To address the health differences in suicidal ideation and behaviors observed by student demographics and to decrease these outcomes overall, a comprehensive approach to suicide prevention, including programs, practices, and policies based on the best available evidence, is needed.”
according to newly released data from the 2019 Youth Risk Behavior Survey.
The prevalence of attempted suicide during the previous 12 months was 8.9% among the 13,677 students in grades 9-12 who took the survey last year, but the rate was 25.5% for American Indian/Alaska Native (AI/AN) respondents, almost 2.9 times higher, the YRBS data show.
Respondents with multiple races in their backgrounds, at 12.9%, and African Americans, with a prevalence of 11.8%, also were above the high school average for suicide attempts, while Whites (7.9%) and Asians (7.7%) were under it and Hispanics equaled it, the Centers for Disease Control and Prevention reported.
The number of AI/AN students was insufficient to examine differences by sex, but females in all of the other racial/ethnic groups were more likely than males to have attempted suicide: multiple race (17.8% vs. 7.3%), African American (15.2% vs. 8.5%), Hispanic (11.9% vs. 5.5%), White (9.4% vs. 6.4%), and Asian (8.4% vs. 7.1%), the CDC’s Division of Adolescent and School Health said.
Among all respondents, 11.0% of females had attempted suicide in the 12 months before the survey, a figure that is significantly higher than the 6.6% prevalence in males. Females also were significantly more likely than males to make a plan about how they would attempt suicide (19.9% vs. 11.3%) and to seriously consider an attempt (24.1% vs. 13.3%), CDC investigators said in a separate report.
Significant differences also were seen when looking at sexual identity. Suicide attempts were reported by 6.4% of heterosexuals, 16.1% of those who weren’t sure, and 23.4% of lesbians/gays/bisexuals (LGBs). For serious consideration of suicide, the respective numbers were 14.5%, 30.4%, and 46.8%, they reported (MMWR Supp. 2020 Aug 21;69[1]:47-55).
For nonheterosexuals, however, males were slightly more likely (23.8%) than females (23.6%) to have attempted suicide, but females were more likely to seriously consider it (49.0% vs. 40.4%) and to make a plan (42.4% vs. 33.0%), according to the YRBS data.
“Adolescence … represents a time for expanded identity development, with sexual identity development representing a complex, multidimensional, and often stressful process for youths,” the CDC investigators said in the MMWR. “To address the health differences in suicidal ideation and behaviors observed by student demographics and to decrease these outcomes overall, a comprehensive approach to suicide prevention, including programs, practices, and policies based on the best available evidence, is needed.”
according to newly released data from the 2019 Youth Risk Behavior Survey.
The prevalence of attempted suicide during the previous 12 months was 8.9% among the 13,677 students in grades 9-12 who took the survey last year, but the rate was 25.5% for American Indian/Alaska Native (AI/AN) respondents, almost 2.9 times higher, the YRBS data show.
Respondents with multiple races in their backgrounds, at 12.9%, and African Americans, with a prevalence of 11.8%, also were above the high school average for suicide attempts, while Whites (7.9%) and Asians (7.7%) were under it and Hispanics equaled it, the Centers for Disease Control and Prevention reported.
The number of AI/AN students was insufficient to examine differences by sex, but females in all of the other racial/ethnic groups were more likely than males to have attempted suicide: multiple race (17.8% vs. 7.3%), African American (15.2% vs. 8.5%), Hispanic (11.9% vs. 5.5%), White (9.4% vs. 6.4%), and Asian (8.4% vs. 7.1%), the CDC’s Division of Adolescent and School Health said.
Among all respondents, 11.0% of females had attempted suicide in the 12 months before the survey, a figure that is significantly higher than the 6.6% prevalence in males. Females also were significantly more likely than males to make a plan about how they would attempt suicide (19.9% vs. 11.3%) and to seriously consider an attempt (24.1% vs. 13.3%), CDC investigators said in a separate report.
Significant differences also were seen when looking at sexual identity. Suicide attempts were reported by 6.4% of heterosexuals, 16.1% of those who weren’t sure, and 23.4% of lesbians/gays/bisexuals (LGBs). For serious consideration of suicide, the respective numbers were 14.5%, 30.4%, and 46.8%, they reported (MMWR Supp. 2020 Aug 21;69[1]:47-55).
For nonheterosexuals, however, males were slightly more likely (23.8%) than females (23.6%) to have attempted suicide, but females were more likely to seriously consider it (49.0% vs. 40.4%) and to make a plan (42.4% vs. 33.0%), according to the YRBS data.
“Adolescence … represents a time for expanded identity development, with sexual identity development representing a complex, multidimensional, and often stressful process for youths,” the CDC investigators said in the MMWR. “To address the health differences in suicidal ideation and behaviors observed by student demographics and to decrease these outcomes overall, a comprehensive approach to suicide prevention, including programs, practices, and policies based on the best available evidence, is needed.”
FDA clears first brain stimulation device to help smokers quit
The Food and Drug Administration has granted marketing approval for the BrainsWay deep transcranial magnetic stimulation (TMS) system to help adult smokers kick tobacco.

in a press release.
As previously reported, the system has already been approved by the FDA as a treatment for patients suffering from obsessive-compulsive disorder and major depressive disorder.
The BrainsWay deep TMS system with H4-coil is designed to target addiction-related brain circuits.
It was evaluated as an aid to short-term smoking cessation in a prospective, double-blind, randomized, sham-controlled, multicenter study that involved 262 adults who had a history of smoking an average of more than 26 years and had attempted to quit multiple times but failed.
Active and sham treatments were performed daily 5 days a week for 3 weeks, followed by an additional three sessions once weekly for 3 weeks, for a total of 18 sessions over 6 weeks.
In the full intention-to-treat population (all 262 participants), the 4-week continuous quit rate (CQR, the primary endpoint) was higher in the active deep TMS group than in the sham TMS group (17.1% vs. 7.9%; P = .0238).
Among participants who completed the study, that is, those who underwent treatment for 4 weeks, who kept daily records, and for whom confirmatory urine samples were available, the CQR was 28.4% in the active deep TMS group, compared with 11.7% in the sham treatment group (P = .0063).
The average number of cigarettes smoked per day, as determined on the basis of daily records (secondary endpoint), was statistically significantly lower in the active deep TMS group, compared with the sham treatment group (P = .0311).
No patient suffered a seizure. The most common adverse event was headache, for which there was no statistical difference between the active and sham treatment groups. Other side effects included application site discomfort, back pain, muscle twitching, and discomfort.
“This FDA clearance represents a significant milestone for BrainsWay and our deep TMS platform technology,” Christopher von Jako, PhD, president and CEO of the company, said in the release.
“While other therapies are currently available, a substantial medical need continues to exist for treatments that can increase the continuous quit rate among smokers,” Dr. von Jako noted.
“Based on the compelling data from our large, randomized pivotal study of 262 subjects, we are confident that our deep TMS technology can play an important role in treating cigarette smokers who seek to quit,” he added.
The company plans a “controlled” U.S. market release of the system for this indication early next year.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has granted marketing approval for the BrainsWay deep transcranial magnetic stimulation (TMS) system to help adult smokers kick tobacco.
in a press release.
As previously reported, the system has already been approved by the FDA as a treatment for patients suffering from obsessive-compulsive disorder and major depressive disorder.
The BrainsWay deep TMS system with H4-coil is designed to target addiction-related brain circuits.
It was evaluated as an aid to short-term smoking cessation in a prospective, double-blind, randomized, sham-controlled, multicenter study that involved 262 adults who had a history of smoking an average of more than 26 years and had attempted to quit multiple times but failed.
Active and sham treatments were performed daily 5 days a week for 3 weeks, followed by an additional three sessions once weekly for 3 weeks, for a total of 18 sessions over 6 weeks.
In the full intention-to-treat population (all 262 participants), the 4-week continuous quit rate (CQR, the primary endpoint) was higher in the active deep TMS group than in the sham TMS group (17.1% vs. 7.9%; P = .0238).
Among participants who completed the study, that is, those who underwent treatment for 4 weeks, who kept daily records, and for whom confirmatory urine samples were available, the CQR was 28.4% in the active deep TMS group, compared with 11.7% in the sham treatment group (P = .0063).
The average number of cigarettes smoked per day, as determined on the basis of daily records (secondary endpoint), was statistically significantly lower in the active deep TMS group, compared with the sham treatment group (P = .0311).
No patient suffered a seizure. The most common adverse event was headache, for which there was no statistical difference between the active and sham treatment groups. Other side effects included application site discomfort, back pain, muscle twitching, and discomfort.
“This FDA clearance represents a significant milestone for BrainsWay and our deep TMS platform technology,” Christopher von Jako, PhD, president and CEO of the company, said in the release.
“While other therapies are currently available, a substantial medical need continues to exist for treatments that can increase the continuous quit rate among smokers,” Dr. von Jako noted.
“Based on the compelling data from our large, randomized pivotal study of 262 subjects, we are confident that our deep TMS technology can play an important role in treating cigarette smokers who seek to quit,” he added.
The company plans a “controlled” U.S. market release of the system for this indication early next year.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has granted marketing approval for the BrainsWay deep transcranial magnetic stimulation (TMS) system to help adult smokers kick tobacco.
in a press release.
As previously reported, the system has already been approved by the FDA as a treatment for patients suffering from obsessive-compulsive disorder and major depressive disorder.
The BrainsWay deep TMS system with H4-coil is designed to target addiction-related brain circuits.
It was evaluated as an aid to short-term smoking cessation in a prospective, double-blind, randomized, sham-controlled, multicenter study that involved 262 adults who had a history of smoking an average of more than 26 years and had attempted to quit multiple times but failed.
Active and sham treatments were performed daily 5 days a week for 3 weeks, followed by an additional three sessions once weekly for 3 weeks, for a total of 18 sessions over 6 weeks.
In the full intention-to-treat population (all 262 participants), the 4-week continuous quit rate (CQR, the primary endpoint) was higher in the active deep TMS group than in the sham TMS group (17.1% vs. 7.9%; P = .0238).
Among participants who completed the study, that is, those who underwent treatment for 4 weeks, who kept daily records, and for whom confirmatory urine samples were available, the CQR was 28.4% in the active deep TMS group, compared with 11.7% in the sham treatment group (P = .0063).
The average number of cigarettes smoked per day, as determined on the basis of daily records (secondary endpoint), was statistically significantly lower in the active deep TMS group, compared with the sham treatment group (P = .0311).
No patient suffered a seizure. The most common adverse event was headache, for which there was no statistical difference between the active and sham treatment groups. Other side effects included application site discomfort, back pain, muscle twitching, and discomfort.
“This FDA clearance represents a significant milestone for BrainsWay and our deep TMS platform technology,” Christopher von Jako, PhD, president and CEO of the company, said in the release.
“While other therapies are currently available, a substantial medical need continues to exist for treatments that can increase the continuous quit rate among smokers,” Dr. von Jako noted.
“Based on the compelling data from our large, randomized pivotal study of 262 subjects, we are confident that our deep TMS technology can play an important role in treating cigarette smokers who seek to quit,” he added.
The company plans a “controlled” U.S. market release of the system for this indication early next year.
A version of this article originally appeared on Medscape.com.
FDA authorizes convalescent plasma for COVID-19
Convalescent plasma contains antibodies from the blood of recovered COVID-19 patients, which can be used to treat people with severe infections. Convalescent plasma has been used to treat patients for other infectious diseases. The authorization allows the plasma to be distributed in the United States and administered by health care providers.
“COVID-19 convalescent plasma is safe and shows promising efficacy,” Stephen Hahn, MD, commissioner of the FDA, said during a press briefing with President Donald Trump.
In April, the FDA approved a program to test convalescent plasma in COVID-19 patients at the Mayo Clinic, followed by other institutions. More than 90,000 patients have enrolled in the program, and 70,000 have received the treatment, Dr. Hahn said.
The data indicate that the plasma can reduce mortality in patients by 35%, particularly if patients are treated within 3 days of being diagnosed. Those who have benefited the most were under age 80 and not on artificial respiration, Alex Azar, the secretary for the Department of Health & Human Services, said during the briefing.
“We dream, in drug development, of something like a 35% mortality reduction,” he said.
But top scientists pushed back against the announcement.
Eric Topol, MD, director of the Scripps Research Translational Institute, professor of molecular medicine, and executive vice president of Scripps Research, said the data the FDA are relying on did not come from the rigorous randomized, double-blind placebo trials that best determine if a treatment is successful.
Still, convalescent plasma is “one more tool added to the arsenal” of combating COVID-19, Mr. Azar said. The FDA will continue to study convalescent plasma as a COVID-19 treatment, Dr. Hahn added.
“We’re waiting for more data. We’re going to continue to gather data,” Dr. Hahn said during the briefing, but the current results meet FDA criteria for issuing an emergency use authorization.
Convalescent plasma “may be effective in lessening the severity or shortening the length of COVID-19 illness in some hospitalized patients,” according to the FDA announcement. Potential side effects include allergic reactions, transfusion-transmitted infections, and transfusion-associated lung injury.
“We’ve seen a great deal of demand for this from doctors around the country,” Dr. Hahn said during the briefing. “The EUA … allows us to continue that and meet that demand.”
Dr. Topol, however, said it appears Trump and the FDA are playing politics with science.
“There’s no evidence to support any survival benefit,” Dr. Topol said on Twitter. “Two days ago [the] FDA’s website stated there was no evidence for an EUA.”
The American Red Cross and other blood centers put out a national call for blood donors in July, especially for patients who have recovered from COVID-19. Mr. Azar and Dr. Hahn emphasized the need for blood donors during the press briefing.
“If you donate plasma, you could save a life,” Mr. Azar said.
The study has not been peer reviewed and did not include a placebo group for comparison, STAT reported.
Last week several health officials warned that the scientific data were too weak to warrant an emergency authorization, the New York Times reported.
A version of this originally appeared on WebMD.com.
Convalescent plasma contains antibodies from the blood of recovered COVID-19 patients, which can be used to treat people with severe infections. Convalescent plasma has been used to treat patients for other infectious diseases. The authorization allows the plasma to be distributed in the United States and administered by health care providers.
“COVID-19 convalescent plasma is safe and shows promising efficacy,” Stephen Hahn, MD, commissioner of the FDA, said during a press briefing with President Donald Trump.
In April, the FDA approved a program to test convalescent plasma in COVID-19 patients at the Mayo Clinic, followed by other institutions. More than 90,000 patients have enrolled in the program, and 70,000 have received the treatment, Dr. Hahn said.
The data indicate that the plasma can reduce mortality in patients by 35%, particularly if patients are treated within 3 days of being diagnosed. Those who have benefited the most were under age 80 and not on artificial respiration, Alex Azar, the secretary for the Department of Health & Human Services, said during the briefing.
“We dream, in drug development, of something like a 35% mortality reduction,” he said.
But top scientists pushed back against the announcement.
Eric Topol, MD, director of the Scripps Research Translational Institute, professor of molecular medicine, and executive vice president of Scripps Research, said the data the FDA are relying on did not come from the rigorous randomized, double-blind placebo trials that best determine if a treatment is successful.
Still, convalescent plasma is “one more tool added to the arsenal” of combating COVID-19, Mr. Azar said. The FDA will continue to study convalescent plasma as a COVID-19 treatment, Dr. Hahn added.
“We’re waiting for more data. We’re going to continue to gather data,” Dr. Hahn said during the briefing, but the current results meet FDA criteria for issuing an emergency use authorization.
Convalescent plasma “may be effective in lessening the severity or shortening the length of COVID-19 illness in some hospitalized patients,” according to the FDA announcement. Potential side effects include allergic reactions, transfusion-transmitted infections, and transfusion-associated lung injury.
“We’ve seen a great deal of demand for this from doctors around the country,” Dr. Hahn said during the briefing. “The EUA … allows us to continue that and meet that demand.”
Dr. Topol, however, said it appears Trump and the FDA are playing politics with science.
“There’s no evidence to support any survival benefit,” Dr. Topol said on Twitter. “Two days ago [the] FDA’s website stated there was no evidence for an EUA.”
The American Red Cross and other blood centers put out a national call for blood donors in July, especially for patients who have recovered from COVID-19. Mr. Azar and Dr. Hahn emphasized the need for blood donors during the press briefing.
“If you donate plasma, you could save a life,” Mr. Azar said.
The study has not been peer reviewed and did not include a placebo group for comparison, STAT reported.
Last week several health officials warned that the scientific data were too weak to warrant an emergency authorization, the New York Times reported.
A version of this originally appeared on WebMD.com.
Convalescent plasma contains antibodies from the blood of recovered COVID-19 patients, which can be used to treat people with severe infections. Convalescent plasma has been used to treat patients for other infectious diseases. The authorization allows the plasma to be distributed in the United States and administered by health care providers.
“COVID-19 convalescent plasma is safe and shows promising efficacy,” Stephen Hahn, MD, commissioner of the FDA, said during a press briefing with President Donald Trump.
In April, the FDA approved a program to test convalescent plasma in COVID-19 patients at the Mayo Clinic, followed by other institutions. More than 90,000 patients have enrolled in the program, and 70,000 have received the treatment, Dr. Hahn said.
The data indicate that the plasma can reduce mortality in patients by 35%, particularly if patients are treated within 3 days of being diagnosed. Those who have benefited the most were under age 80 and not on artificial respiration, Alex Azar, the secretary for the Department of Health & Human Services, said during the briefing.
“We dream, in drug development, of something like a 35% mortality reduction,” he said.
But top scientists pushed back against the announcement.
Eric Topol, MD, director of the Scripps Research Translational Institute, professor of molecular medicine, and executive vice president of Scripps Research, said the data the FDA are relying on did not come from the rigorous randomized, double-blind placebo trials that best determine if a treatment is successful.
Still, convalescent plasma is “one more tool added to the arsenal” of combating COVID-19, Mr. Azar said. The FDA will continue to study convalescent plasma as a COVID-19 treatment, Dr. Hahn added.
“We’re waiting for more data. We’re going to continue to gather data,” Dr. Hahn said during the briefing, but the current results meet FDA criteria for issuing an emergency use authorization.
Convalescent plasma “may be effective in lessening the severity or shortening the length of COVID-19 illness in some hospitalized patients,” according to the FDA announcement. Potential side effects include allergic reactions, transfusion-transmitted infections, and transfusion-associated lung injury.
“We’ve seen a great deal of demand for this from doctors around the country,” Dr. Hahn said during the briefing. “The EUA … allows us to continue that and meet that demand.”
Dr. Topol, however, said it appears Trump and the FDA are playing politics with science.
“There’s no evidence to support any survival benefit,” Dr. Topol said on Twitter. “Two days ago [the] FDA’s website stated there was no evidence for an EUA.”
The American Red Cross and other blood centers put out a national call for blood donors in July, especially for patients who have recovered from COVID-19. Mr. Azar and Dr. Hahn emphasized the need for blood donors during the press briefing.
“If you donate plasma, you could save a life,” Mr. Azar said.
The study has not been peer reviewed and did not include a placebo group for comparison, STAT reported.
Last week several health officials warned that the scientific data were too weak to warrant an emergency authorization, the New York Times reported.
A version of this originally appeared on WebMD.com.
FDA updates hydrochlorothiazide label to include nonmelanoma skin cancer risk
and undergo regular skin cancer screening, according to updates to the medication’s label.
The skin cancer risk is small, however, and patients should continue taking HCTZ, a commonly used diuretic and antihypertensive drug, unless their doctor says otherwise, according to a U.S. Food and Drug Administration announcement about the labeling changes, which the agency approved on Aug. 20.
HCTZ, first approved in 1959, is associated with photosensitivity. Researchers identified a relationship between HCTZ and nonmelanoma skin cancer in postmarketing studies. Investigators have described dose-response patterns for basal cell carcinoma and squamous cell carcinoma (SCC).
An FDA analysis found that the risk mostly was increased for SCC. The drug was associated with approximately one additional case of SCC per 16,000 patients per year. For white patients who received a cumulative dose of 50,000 mg or more, the risk was greater. In this patient population, HCTZ was associated with about one additional case of SCC per 6,700 patients per year, according to the label.
Reliably estimating the frequency of nonmelanoma skin cancer and establishing a causal relationship to drug exposure is not possible with the available postmarketing data, the label notes
“Treatment for nonmelanoma skin cancer is typically local and successful, with very low rates of death,” the FDA said. “Meanwhile, the risks of uncontrolled blood pressure can be severe and include life-threatening heart attacks or stroke. Given this information, patients should continue to use HCTZ and take protective skin care measures to reduce their risk of nonmelanoma skin cancer, unless directed otherwise from their health care provider.”
Patients can reduce sun exposure by using broad-spectrum sunscreens with a sun protection factor value of at least 15, limiting time in the sun, and wearing protective clothing, the agency advised.
and undergo regular skin cancer screening, according to updates to the medication’s label.
The skin cancer risk is small, however, and patients should continue taking HCTZ, a commonly used diuretic and antihypertensive drug, unless their doctor says otherwise, according to a U.S. Food and Drug Administration announcement about the labeling changes, which the agency approved on Aug. 20.
HCTZ, first approved in 1959, is associated with photosensitivity. Researchers identified a relationship between HCTZ and nonmelanoma skin cancer in postmarketing studies. Investigators have described dose-response patterns for basal cell carcinoma and squamous cell carcinoma (SCC).
An FDA analysis found that the risk mostly was increased for SCC. The drug was associated with approximately one additional case of SCC per 16,000 patients per year. For white patients who received a cumulative dose of 50,000 mg or more, the risk was greater. In this patient population, HCTZ was associated with about one additional case of SCC per 6,700 patients per year, according to the label.
Reliably estimating the frequency of nonmelanoma skin cancer and establishing a causal relationship to drug exposure is not possible with the available postmarketing data, the label notes
“Treatment for nonmelanoma skin cancer is typically local and successful, with very low rates of death,” the FDA said. “Meanwhile, the risks of uncontrolled blood pressure can be severe and include life-threatening heart attacks or stroke. Given this information, patients should continue to use HCTZ and take protective skin care measures to reduce their risk of nonmelanoma skin cancer, unless directed otherwise from their health care provider.”
Patients can reduce sun exposure by using broad-spectrum sunscreens with a sun protection factor value of at least 15, limiting time in the sun, and wearing protective clothing, the agency advised.
and undergo regular skin cancer screening, according to updates to the medication’s label.
The skin cancer risk is small, however, and patients should continue taking HCTZ, a commonly used diuretic and antihypertensive drug, unless their doctor says otherwise, according to a U.S. Food and Drug Administration announcement about the labeling changes, which the agency approved on Aug. 20.
HCTZ, first approved in 1959, is associated with photosensitivity. Researchers identified a relationship between HCTZ and nonmelanoma skin cancer in postmarketing studies. Investigators have described dose-response patterns for basal cell carcinoma and squamous cell carcinoma (SCC).
An FDA analysis found that the risk mostly was increased for SCC. The drug was associated with approximately one additional case of SCC per 16,000 patients per year. For white patients who received a cumulative dose of 50,000 mg or more, the risk was greater. In this patient population, HCTZ was associated with about one additional case of SCC per 6,700 patients per year, according to the label.
Reliably estimating the frequency of nonmelanoma skin cancer and establishing a causal relationship to drug exposure is not possible with the available postmarketing data, the label notes
“Treatment for nonmelanoma skin cancer is typically local and successful, with very low rates of death,” the FDA said. “Meanwhile, the risks of uncontrolled blood pressure can be severe and include life-threatening heart attacks or stroke. Given this information, patients should continue to use HCTZ and take protective skin care measures to reduce their risk of nonmelanoma skin cancer, unless directed otherwise from their health care provider.”
Patients can reduce sun exposure by using broad-spectrum sunscreens with a sun protection factor value of at least 15, limiting time in the sun, and wearing protective clothing, the agency advised.