User login
New and future therapies for lupus nephritis
Treatment for lupus nephritis has changed dramatically in recent years. Only 10 years ago, rheumatologists and nephrologists, whether specializing in adult or pediatric medicine, treated lupus nephritis with a similar regimen of monthly intravenous cyclophosphamide (Cytoxan) and glucocorticoids. Although the regimen is effective, side effects such as infection, hair loss, and infertility were extremely common.
Effective but very toxic therapy is common in autoimmune diseases. In the last decade, clinical trials have shown that less toxic drugs are as effective for treating lupus nephritis. This article will review new developments in therapy for lupus nephritis, which can be viewed as a prototype for other fields of medicine.
DEMOGRAPHICS ARE IMPORTANT
Although numerous factors have prognostic value in lupus nephritis (eg, serum creatinine, proteinuria, renal biopsy findings), the most important to consider when designing and interpreting studies are race and socioeconomic variables.
A retrospective study in Miami, FL,1 evaluated 213 patients with lupus nephritis, of whom 47% were Hispanic, 44% African American, and 20% white. At baseline, African Americans had higher blood pressure, higher serum creatinine levels, and lower household income. After 6 years, African Americans fared the worst in terms of doubling of serum creatinine, developing end-stage renal disease, and death; whites had the best outcomes, and Hispanics were in between. Low income was found to be a significant risk factor, independent of racial background.
In a similar retrospective study in New York City in 128 patients (43% white, 40% Hispanic, and 17% African American) with proliferative lupus nephritis,2 disease was much more likely to progress to renal failure over 10 years in patients living in a poor neighborhood, even after adjustment for race.
We need to keep in mind that racial and socioeconomic factors correlate with disease severity when we design and interpret studies of lupus nephritis. Study groups must be carefully balanced with patients of similar racial and socioeconomic profiles. Study findings must be interpreted with caution; for example, whether results from a study from China are applicable to an African American with lupus nephritis in New York City is unclear.
OLDER STANDARD THERAPY: EFFECTIVE BUT TOXIC
The last large National Institutes of Health study that involved only cyclophosphamide and a glucocorticoid was published in 2001,3 with 21 patients receiving cyclophosphamide alone and 20 patients receiving cyclophosphamide plus methylprednisolone. Although lupus nephritis improved, serious side effects occurred in one-third to one-half of patients in each group and included hypertension, hyperlipidemia, valvular heart disease, avascular necrosis, premature menopause, and major infections, including herpes zoster.
Less cyclophosphamide works just as well
The multicenter, prospective Euro-Lupus Nephritis Trial4 randomized 90 patients with proliferative lupus nephritis to receive either standard high-dose intravenous (IV) cyclophosphamide therapy (six monthly pulses and two quarterly pulses, with doses increasing according to the white blood cell count) or low-dose IV cyclophosphamide therapy (six pulses every 2 weeks at a fixed dose of 500 mg). Both regimens were followed by azathioprine (Imuran).
At 4 years, the two treatment groups were not significantly different in terms of treatment failure, remission rates, serum creatinine levels, 24-hour proteinuria, and freedom from renal flares. However, the rates of side effects were significantly different, with more patients in the low-dosage group free of severe infection.
One problem with this study is whether it is applicable to an American lupus nephritis population, since 84% of the patients were white. Since this study, others indicate that this regimen is probably also safe and effective for different racial groups in the United States.
At 10-year follow-up,5 both treatment groups still had identical excellent rates of freedom from end-stage renal disease. Serum creatinine and 24-hour proteinuria were also at excellent levels and identical in both groups. Nearly three quarters of patients still needed glucocorticoid therapy and more than half still needed immunosuppressive therapy, but the rates were not statistically significantly different between the treatment groups.
The cumulative dose of cyclophosphamide was 9.5 g in the standard-treatment group and 5.5 g in the low-dose group. This difference in exposure could make a tremendous difference to patients, not only for immediate side effects such as early menopause and infections, but for the risk of cancer in later decades.
This study showed clearly that low-dose cyclophosphamide is an option for induction therapy. Drawbacks of the study were that the population was mostly white and that patients had only moderately severe disease.
Low-dose cyclophosphamide has largely replaced the older National Institutes of Health regimen, although during the last decade drug therapy has undergone more changes.
MYCOPHENOLATE AND AZATHIOPRINE: ALTERNATIVES TO CYCLOPHOSPHAMIDE
In a Chinese study, mycophenolate was better than cyclophosphamide for induction
In a study in Hong Kong, Chan et al6 randomized 42 patients with severe lupus nephritis to receive either mycophenolate mofetil (available in the United States as CellCept; 2 g/day for 6 months, then 1 g/day for 6 months) or oral cyclophosphamide (2.5 mg/kg per day for 6 months) followed by azathioprine (1.5–2.0 mg/kg per day) for 6 months. Both groups also received prednisolone during the year.
At the end of the first year, the two groups were not significantly different in their rates of complete remission, partial remission, and relapse. The rate of infection, although not significantly different, was higher in the cyclophosphamide group (33% vs 19%). Two patients (10%) died in the cyclophosphamide group, but the difference in mortality rates was not statistically significant.
Nearly 5 years later,7 rates of chronic renal failure and relapse were still statistically the same in the two groups. Infections were fewer in the mycophenolate group (13% vs 40%, P = .013). The rate of amenorrhea was 36% in the cyclophosphamide group and only 4% in the mycophenolate group (P = .004). Four patients in the cyclophosphamide group and none in the mycophenolate group reached the composite end point of end-stage renal failure or death (P = .062).
This study appeared to offer a new option with equal efficacy and fewer side effects than standard therapy. However, its applicability to non-Chinese populations remained to be shown.
In a US study, mycophenolate or azathioprine was better than cyclophosphamide as maintenance
In a study in Miami,8 59 patients with lupus nephritis were given standard induction therapy with IV cyclophosphamide plus glucocorticoids for 6 months, then randomly assigned to one of three maintenance therapies for 1 to 3 years: IV injections of cyclophosphamide every 3 months (standard therapy), oral azathioprine, or oral mycophenolate. The population was 93% female, their average age was 33 years, and nearly half were African American, with many of the others being Hispanic. Patients tended to have severe disease, with nearly two-thirds having nephrotic syndrome.
After 6 years, there had been more deaths in the cyclophosphamide group than in the azathioprine group (P = .02) and in the mycophenolate group, although the latter difference was not statistically significant (P = .11). The combined rate of death and chronic renal failure was significantly higher with cyclophosphamide than with either of the oral agents. The cyclophosphamide group also had the highest relapse rate during the maintenance phase.
The differences in side effects were even more dramatic. Amenorrhea affected 32% of patients in the cyclophosphamide group, and only 7% and 6% in the azathioprine and mycophenolate groups, respectively. Rates of infections were 68% in the cyclophosphamide group and 28% and 21% in the azathioprine and mycophenolate groups, respectively. Patients given cyclophosphamide had 13 hospital days per patient per year, while the other groups each had only 1.
This study showed that maintenance therapy with oral azathioprine or mycophenolate was more effective and had fewer adverse effects than standard IV cyclophosphamide therapy. As a result of this study, oral agents for maintenance therapy became the new standard, but the question remained whether oral agents could safely be used for induction.
In a US study, mycophenolate was better than cyclophosphamide for induction
In a noninferiority study, Ginzler et al9 randomized 140 patients with severe lupus nephritis to receive either monthly IV cyclophosphamide or oral mycophenolate as induction therapy for 6 months. Adjunctive care with glucocorticoids was given in both groups. The study population was from 18 US academic centers and was predominantly female, and more than half were African American.
After 24 weeks, 22.5% of the mycophenolate patients were in complete remission by very strict criteria vs only 4% of those given cyclophosphamide (P = .005). The trend for partial remissions was also in favor of mycophenolate, although the difference was not statistically significant. The rate of complete and partial remissions, a prespecified end point, was significantly higher in the mycophenolate group. Although the study was trying to evaluate equivalency, it actually showed superiority for mycophenolate induction therapy.
Serum creatinine levels declined in both groups, but more in the mycophenolate group by 24 weeks. Urinary protein levels fell the same amount in both groups. At 3 years, the groups were statistically equivalent in terms of renal flares, renal failures, and deaths. However, the study groups were small, and the mycophenolate group did have a better trend for both renal failure (N = 4 vs 7) and deaths (N = 4 vs 8).
Mycophenolate also had fewer side effects, including infection, although again the numbers were too small to show statistical significance. The exception was diarrhea (N = 15 in the mycophenolate group vs 2 in the cyclophosphamide group).
A drawback of the study is that it was designed as a crossover study: a patient for whom therapy was failing after 3 months could switch to the other group, introducing potential confounding. Other problems involved the small population size and the question of whether results from patients in the United States were applicable to others worldwide.
In a worldwide study, mycophenolate was at least equivalent to cyclophosphamide for induction
The Aspreva Lupus Management Study (ALMS)10 used a similar design with 370 patients worldwide (United States, China, South America, and Europe) in one of the largest trials ever conducted in lupus nephritis. Patients were randomized to 6 months of induction therapy with either IV cyclophosphamide or oral mycophenolate but could not cross over.
At 6 months, response rates were identical between the two groups, with response defined as a combination of specific improvement in proteinuria, serum creatinine, and hematuria (50%–55%). In terms of individual renal and nonrenal variables, both groups appeared identical.
However, the side effect profiles differed between the two groups. As expected for mycophenolate, diarrhea was the most common side effect (occurring in 28% vs 12% in the cyclophosphamide group). Nausea and vomiting were more common with cyclophosphamide (45% and 37% respectively vs 14% and 13% in the mycophenolate group). Cyclophosphamide also caused hair loss in 35%, vs 10% in the mycophenolate group.
There were 14 deaths overall, which is a very low number considering the patients’ severity of illness, and it indicates the better results now achieved with therapy. The mortality rate was higher in the mycophenolate group (5% vs 3%), but the difference was not statistically significant. Six of the nine deaths with mycophenolate were from the same center in China, and none were from Europe or the United States. In summary, the study did not show that mycophenolate was superior to IV cyclophosphamide for induction therapy, but that they were equivalent in efficacy with different side effect profiles.
Membranous nephropathy: Mycophenolate vs cyclophosphamide
Less evidence is available about treatment for membranous disease, which is characterized by heavy proteinuria and the nephrotic syndrome but usually does not progress to renal failure. Radhakrishnan et al11 combined data from the trial by Ginzler et al9 and the ALMS trial10 and found 84 patients with pure membranous lupus, who were equally divided between the treatment groups receiving IV cyclophosphamide and mycophenolate. Consistent with the larger group’s data, mycophenolate and cyclophosphamide performed similarly in terms of efficacy, but there was a slightly higher rate of side effects with cyclophosphamide.
Maintenance therapy: Mycophenolate superior to azathioprine
The ALMS Maintenance Trial12 evaluated maintenance therapy in the same worldwide population that was studied for induction therapy. Of the 370 patients involved in the induction phase that compared IV cyclophosphamide and oral mycophenolate, 227 responded sufficiently to be rerandomized in a controlled, double-blinded trial of 36 months of maintenance therapy with corticosteroids and either mycophenolate (1 g twice daily) or azathioprine (2 mg/kg per day).
In intention-to-treat analysis, the time to treatment failure (ie, doubling of the serum creatinine level, progressing to renal failure, or death) was significantly shorter in the azathioprine group (P = .003). Every individual end point—end-stage renal disease, renal flares, doubling of serum creatinine, rescue immunosuppression required—was in favor of mycophenolate maintenance. At 3 years, the completion rate was 63% with mycophenolate and 49% with azathioprine. Serious adverse events and withdrawals because of adverse events were more common in the azathioprine group.
In summary, mycophenolate was superior to azathioprine in maintaining renal response and in preventing relapse in patients with active lupus nephritis who responded to induction therapy with either mycophenolate or IV cyclophosphamide. Mycophenolate was found to be superior regardless of initial induction treatment, race, or region and was confirmed by all key secondary end points.
Only one of the 227 patients died during the 3 years—from an auto accident. Again, this indicates the dramatically improved survival today compared with a decade ago.
RITUXIMAB: PROMISING BUT UNPROVEN
Rituximab (Rituxan) was originally approved to treat tumors, then rheumatoid arthritis, and most recently vasculitis. Evidence thus far is mixed regarding its use as a treatment for lupus nephritis. Although randomized clinical trials have not found it to be superior to standard regimens, there are many signs that it may be effective.
Rituximab in uncontrolled studies
Terrier et al13 analyzed prospective data from 136 patients with systemic lupus erythematosus, most of whom had renal disease, from the French Autoimmunity and Rituximab registry. Response occurred in 71% of patients using rituximab, with no difference found between patients receiving rituximab monotherapy and those concomitantly receiving immunosuppressive agents.
Melander et al14 retrospectively studied 19 women and 1 man who had been treated with rituximab for severe lupus nephritis and followed for at least 1 year. Three patients had concurrent therapy with cyclophosphamide, and 10 patients continued rituximab as maintenance therapy; 12 patients had lupus nephritis that had been refractory to standard treatment, and 6 had relapsing disease.
At a median follow-up of 22 months, 12 patients (60%) had achieved complete or partial renal remission.
Condon et al15 treated 21 patients who had severe lupus nephritis with two doses of rituximab and IV methylprednisolone 2 weeks apart, then maintenance therapy with mycophenolate without any oral steroids. At a mean follow-up of 35 months ( ± 14 months), 16 (76%) were in complete remission, with a mean time to remission of 12 months. Two (9.5%) achieved partial remission. The rate of toxicity was low.
Thus, rituximab appears promising in uncontrolled studies.
Placebo-controlled trials fail to prove rituximab effective
LUNAR trial. On the other hand, the largest placebo-controlled trial to evaluate rituximab in patients with proliferative lupus nephritis, the Lupus Nephritis Assessment With Rituximab (LUNAR) trial16 found differences in favor of rituximab, but none reached statistical significance. The trial randomized 140 patients to receive either mycophenolate plus periodic rituximab infusions or mycophenolate plus placebo infusions for 1 year. All patients received the same dosage of glucocorticoids, which was tapered over the year.
At the end of 1 year, the groups were not statistically different in terms of complete renal response and partial renal response. Rituximab appeared less likely to produce no response, but the difference was not statistically significant.
African Americans appeared to have a higher response rate to rituximab (70% in the rituximab group achieved a response vs 45% in the control group), but again, the difference did not reach statistical significance, and the total study population of African Americans was only 40.
Rituximab did have a statistically significant positive effect on two serologic markers at 1 year: levels of anti-dsDNA fell faster and complement rose faster. In addition, rates of adverse and serious adverse events were similar between the two groups, with no new or unexpected “safety signals.”
This study can be interpreted in a number of ways. The number of patients may have been too small to show significance and the follow-up may have been too short. On the other hand, it may simply not be effective to add rituximab to a full dose of mycophenolate and steroids, an already good treatment.
EXPLORER trial. Similarly, for patients with lupus without nephritis, the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial17 also tested rituximab against a background of an effective therapeutic regimen and found no additional benefit. This study had design problems similar to those of the LUNAR trial.
Rituximab as rescue therapy
The evidence so far indicates that rituximab may have a role as rescue therapy for refractory or relapsing disease. Rituximab must be used with other therapies, but maintenance corticosteroid therapy is not necessary. Its role as a first-line agent in induction therapy for lupus nephritis remains unclear, although it may have an important role for nonwhites. In general, it has been well tolerated. Until a large randomized trial indicates otherwise, it should not be used as a first-line therapy.
The US Food and Drug Administration (FDA) sent out a warning about the danger of progressive multifocal leukoencephalopathy as an adverse effect of rituximab and of mycophenolate, but this does not appear to be a major concern for most patients and is only likely to occur in those who have been over-immunosuppressed for many years.
MULTITARGET THERAPY
The concept of using multiple drugs simultaneously—such as mycophenolate, steroids, and rituximab—is increasingly being tried. Multi-target therapy appears to offer the advantages of combining different modes of action with better results, and it offers fewer side effects because dosages of each individual drug can be lower when combined with other immunosuppressives.
Bao et al18 in China randomly assigned 40 patients with diffuse proliferative and membranous nephritis to 6 to 9 months of induction treatment with either multitarget therapy (mycophenolate, tacrolimus [Prograf], and glucocorticoids) or IV cyclophosphamide. More complete remissions occurred in the multitarget therapy group, both at 6 months (50% vs 5%) and at 9 months (65% vs 15%). Most adverse events were less frequent in the multitarget therapy group, although three patients (15%) in the multitarget therapy group developed new-onset hypertension vs none in the cyclophosphamide group.
NEW MEDICATIONS
Entirely new classes of drugs are being developed with immunomodulatory effects, including tolerance molecules, cytokine blockers, inhibitors of human B lymphocyte stimulator, and costimulatory blockers.
Belimumab offers small improvement for lupus
Belimumab (Benlysta) is a human monoclonal antibody that inhibits the biologic activity of human B lymphocyte stimulator; it has recently been approved by the FDA for lupus nephritis. In a worldwide study,19 867 patients with systemic lupus erythematosus were randomized to receive either belimumab (1 mg/kg or 10 mg/kg) or placebo.
The primary end point was the reduction of disease activity by a scoring system (SELENA-SLEDAI) that incorporated multiple features of lupus, including arthritis, vasculitis, proteinuria, rash, and others. Patients in the belimumab group had better outcomes, but the results were not dramatic. Because the drug is so expensive (about $25,000 per year) and the improvement offered is only incremental, this drug will not likely change the treatment of lupus very much.
Moreover, patients with lupus nephritis were not included in the study, but a new study is being planned to do so. Improvement is harder to demonstrate in lupus nephritis than in rheumatoid arthritis and systemic lupus erythematosus: significant changes in creatinine levels and 24-hour urinary protein must be achieved, rather than more qualitative signs and symptoms of joint pain, rash, and feeling better. Although belimumab is still unproven for lupus nephritis, it might be worth trying for patients failing other therapy.
Laquinimod: A promising experimental drug
Laquinimod is an oral immunomodulatory drug with a number of effects, including down-regulating major histocompatability complex II, chemokines, and adhesion-related molecules related to inflammation. It has been studied in more than 2,500 patients with multiple sclerosis. Pilot studies are now being done for its use for lupus nephritis. If it shows promise, a large randomized, controlled trial will be conducted.
Abatacept is in clinical trials
Abatacept (Orencia), a costimulation blocker, is undergoing clinical trials in lupus nephritis. Results should be available shortly.
INDIVIDUALIZE THERAPY
This past decade has seen such an increase in options to treat lupus nephritis that therapy can now be individualized.
Choosing IV cyclophosphamide vs mycophenolate
As a result of recent trials, doctors in the United States are increasingly using mycophenolate as the first-line drug for lupus nephritis. In Europe, however, many are choosing the shorter regimen of IV cyclophosphamide because of the results of the Euro-Lupus study.
Nowadays, I tend to use IV cyclophosphamide as the first-line drug only for patients with severe crescenteric glomerulonephritis or a very high serum creatinine level. In such cases, there is more experience with cyclophosphamide, and such severe disease does not lend itself to the luxury of trying out different therapies sequentially. If such a severely ill patient insists that a future pregnancy is very important, an alternative therapy of mycophenolate plus rituximab should be considered. I prefer mycophenolate for induction and maintenance therapy in most patients.
Dosing and formulation considerations for mycophenolate
Large dosages of mycophenolate are much better tolerated when broken up throughout the day. A patient who cannot tolerate 1 g twice daily may be able to tolerate 500 mg four times a day. The formulation can also make a difference. Some patients tolerate sustained-release mycophenolate (Myfortic) better than CellCept, and vice versa.
For patients who cannot tolerate mycophenolate, azathioprine is an acceptable alternative. In addition, for a patient who is already doing well on azathioprine, there is no need to change to mycophenolate.
Long maintenance therapy now acceptable
The ALMS Maintenance Trial12 found 3 years of maintenance therapy to be safe and effective. Such a long maintenance period is increasingly viewed as important, especially for patients in their teens and 20s, as it allows them to live a normal life, ie, to finish their education, get married, and become settled socially. Whether 5 years of maintenance therapy or even 10 years is advisable is still unknown.
Treatment during pregnancy
Neither mycophenolate nor azathioprine is recommended during pregnancy, although their effects are unknown. Because we have much more renal transplant experience with azathioprine during pregnancy, I recommend either switching from mycophenolate to azathioprine or trying to stop medication altogether if the patient has been well controlled.
- Contreras G, Lenz O, Pardo V, et al. Outcomes in African Americans and Hispanics with lupus nephritis. Kidney Int 2006; 69:1846–1851.
- Barr RG, Seliger S, Appel GB, et al. Prognosis in proliferative lupus nephritis: the role of socio-economic status and race/ethnicity. Nephrol Dial Transplant 2003; 18:2039–2046.
- Illei GG, Austin HA, Crane M, et al. Combination therapy with pulse cyclophosphamide plus pulse methylprednisolone improves long-term renal outcome without adding toxicity in patients with lupus nephritis. Ann Intern Med 2001; 135:248–257.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum 2002; 46:2121–2131.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose and high-dose intravenous cyclophosphamide. Ann Rheum Dis 2010; 69:61–64.
- Chan TM, Li FK, Tang CS, et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong King-Guangzhou Nephrology Study Group. N Engl J Med 2000; 343:1156–1162.
- Chan TM, Tse KC, Tang CS, Mok MY, Li FK; Hong Kong Nephrology Study Group. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005; 16:1076–1084.
- Contreras G, Pardo V, Leclercq B, et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004; 350:971–980.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009; 20:1103–1112.
- Radhakrishnan J, Moutzouris DA, Ginzler EM, Solomons N, Siempos II, Appel GB. Mycophenolate mofetil and intravenous cyclophosphamide are similar as induction therapy for class V lupus nephritis. Kidney Int 2010; 77:152–160.
- Dooley MA, Jayne D, Ginzler EM, et al; for the ALMS Group. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N Engl J Med 2011; 365:1886–1895.
- Terrier B, Amoura Z, Ravaud P, et al; Club Rhumatismes et Inflammation. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum 2010; 62:2458–2466.
- Melander C, Sallée M, Troillet P, et al. Rituximab in severe lupus nephritis: early B-cell depletion affects long-term renal outcome. Clin J Am Soc Nephrol 2009; 4:579–587.
- Condon MB, Griffith M, Cook HT, Levy J, Lightstone L, Cairns T. Treatment of class IV lupus nephritis with rituximab & mycophenolate mofetil (MMF) with no oral steroids is effective and safe (abstract). J Am Soc Nephrol 2010; 21(suppl):625A–626A.
- Furie RA, Looney RJ, Rovin E, et al. Efficacy and safety of rituximab in subjects with active proliferative lupus nephritis (LN): results from the randomized, double-blind phase III LUNAR study (abstract). Arthritis Rheum 2009; 60(suppl 1):S429.
- Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 2010; 62:222–233.
- Bao H, Liu ZH, Zie HL, Hu WX, Zhang HT, Li LS. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J Am Soc Nephrol 2008; 19:2001–2010.
- Navarra SV, Guzmán RM, Gallacher AE, et al; BLISS-52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:721–731.
Treatment for lupus nephritis has changed dramatically in recent years. Only 10 years ago, rheumatologists and nephrologists, whether specializing in adult or pediatric medicine, treated lupus nephritis with a similar regimen of monthly intravenous cyclophosphamide (Cytoxan) and glucocorticoids. Although the regimen is effective, side effects such as infection, hair loss, and infertility were extremely common.
Effective but very toxic therapy is common in autoimmune diseases. In the last decade, clinical trials have shown that less toxic drugs are as effective for treating lupus nephritis. This article will review new developments in therapy for lupus nephritis, which can be viewed as a prototype for other fields of medicine.
DEMOGRAPHICS ARE IMPORTANT
Although numerous factors have prognostic value in lupus nephritis (eg, serum creatinine, proteinuria, renal biopsy findings), the most important to consider when designing and interpreting studies are race and socioeconomic variables.
A retrospective study in Miami, FL,1 evaluated 213 patients with lupus nephritis, of whom 47% were Hispanic, 44% African American, and 20% white. At baseline, African Americans had higher blood pressure, higher serum creatinine levels, and lower household income. After 6 years, African Americans fared the worst in terms of doubling of serum creatinine, developing end-stage renal disease, and death; whites had the best outcomes, and Hispanics were in between. Low income was found to be a significant risk factor, independent of racial background.
In a similar retrospective study in New York City in 128 patients (43% white, 40% Hispanic, and 17% African American) with proliferative lupus nephritis,2 disease was much more likely to progress to renal failure over 10 years in patients living in a poor neighborhood, even after adjustment for race.
We need to keep in mind that racial and socioeconomic factors correlate with disease severity when we design and interpret studies of lupus nephritis. Study groups must be carefully balanced with patients of similar racial and socioeconomic profiles. Study findings must be interpreted with caution; for example, whether results from a study from China are applicable to an African American with lupus nephritis in New York City is unclear.
OLDER STANDARD THERAPY: EFFECTIVE BUT TOXIC
The last large National Institutes of Health study that involved only cyclophosphamide and a glucocorticoid was published in 2001,3 with 21 patients receiving cyclophosphamide alone and 20 patients receiving cyclophosphamide plus methylprednisolone. Although lupus nephritis improved, serious side effects occurred in one-third to one-half of patients in each group and included hypertension, hyperlipidemia, valvular heart disease, avascular necrosis, premature menopause, and major infections, including herpes zoster.
Less cyclophosphamide works just as well
The multicenter, prospective Euro-Lupus Nephritis Trial4 randomized 90 patients with proliferative lupus nephritis to receive either standard high-dose intravenous (IV) cyclophosphamide therapy (six monthly pulses and two quarterly pulses, with doses increasing according to the white blood cell count) or low-dose IV cyclophosphamide therapy (six pulses every 2 weeks at a fixed dose of 500 mg). Both regimens were followed by azathioprine (Imuran).
At 4 years, the two treatment groups were not significantly different in terms of treatment failure, remission rates, serum creatinine levels, 24-hour proteinuria, and freedom from renal flares. However, the rates of side effects were significantly different, with more patients in the low-dosage group free of severe infection.
One problem with this study is whether it is applicable to an American lupus nephritis population, since 84% of the patients were white. Since this study, others indicate that this regimen is probably also safe and effective for different racial groups in the United States.
At 10-year follow-up,5 both treatment groups still had identical excellent rates of freedom from end-stage renal disease. Serum creatinine and 24-hour proteinuria were also at excellent levels and identical in both groups. Nearly three quarters of patients still needed glucocorticoid therapy and more than half still needed immunosuppressive therapy, but the rates were not statistically significantly different between the treatment groups.
The cumulative dose of cyclophosphamide was 9.5 g in the standard-treatment group and 5.5 g in the low-dose group. This difference in exposure could make a tremendous difference to patients, not only for immediate side effects such as early menopause and infections, but for the risk of cancer in later decades.
This study showed clearly that low-dose cyclophosphamide is an option for induction therapy. Drawbacks of the study were that the population was mostly white and that patients had only moderately severe disease.
Low-dose cyclophosphamide has largely replaced the older National Institutes of Health regimen, although during the last decade drug therapy has undergone more changes.
MYCOPHENOLATE AND AZATHIOPRINE: ALTERNATIVES TO CYCLOPHOSPHAMIDE
In a Chinese study, mycophenolate was better than cyclophosphamide for induction
In a study in Hong Kong, Chan et al6 randomized 42 patients with severe lupus nephritis to receive either mycophenolate mofetil (available in the United States as CellCept; 2 g/day for 6 months, then 1 g/day for 6 months) or oral cyclophosphamide (2.5 mg/kg per day for 6 months) followed by azathioprine (1.5–2.0 mg/kg per day) for 6 months. Both groups also received prednisolone during the year.
At the end of the first year, the two groups were not significantly different in their rates of complete remission, partial remission, and relapse. The rate of infection, although not significantly different, was higher in the cyclophosphamide group (33% vs 19%). Two patients (10%) died in the cyclophosphamide group, but the difference in mortality rates was not statistically significant.
Nearly 5 years later,7 rates of chronic renal failure and relapse were still statistically the same in the two groups. Infections were fewer in the mycophenolate group (13% vs 40%, P = .013). The rate of amenorrhea was 36% in the cyclophosphamide group and only 4% in the mycophenolate group (P = .004). Four patients in the cyclophosphamide group and none in the mycophenolate group reached the composite end point of end-stage renal failure or death (P = .062).
This study appeared to offer a new option with equal efficacy and fewer side effects than standard therapy. However, its applicability to non-Chinese populations remained to be shown.
In a US study, mycophenolate or azathioprine was better than cyclophosphamide as maintenance
In a study in Miami,8 59 patients with lupus nephritis were given standard induction therapy with IV cyclophosphamide plus glucocorticoids for 6 months, then randomly assigned to one of three maintenance therapies for 1 to 3 years: IV injections of cyclophosphamide every 3 months (standard therapy), oral azathioprine, or oral mycophenolate. The population was 93% female, their average age was 33 years, and nearly half were African American, with many of the others being Hispanic. Patients tended to have severe disease, with nearly two-thirds having nephrotic syndrome.
After 6 years, there had been more deaths in the cyclophosphamide group than in the azathioprine group (P = .02) and in the mycophenolate group, although the latter difference was not statistically significant (P = .11). The combined rate of death and chronic renal failure was significantly higher with cyclophosphamide than with either of the oral agents. The cyclophosphamide group also had the highest relapse rate during the maintenance phase.
The differences in side effects were even more dramatic. Amenorrhea affected 32% of patients in the cyclophosphamide group, and only 7% and 6% in the azathioprine and mycophenolate groups, respectively. Rates of infections were 68% in the cyclophosphamide group and 28% and 21% in the azathioprine and mycophenolate groups, respectively. Patients given cyclophosphamide had 13 hospital days per patient per year, while the other groups each had only 1.
This study showed that maintenance therapy with oral azathioprine or mycophenolate was more effective and had fewer adverse effects than standard IV cyclophosphamide therapy. As a result of this study, oral agents for maintenance therapy became the new standard, but the question remained whether oral agents could safely be used for induction.
In a US study, mycophenolate was better than cyclophosphamide for induction
In a noninferiority study, Ginzler et al9 randomized 140 patients with severe lupus nephritis to receive either monthly IV cyclophosphamide or oral mycophenolate as induction therapy for 6 months. Adjunctive care with glucocorticoids was given in both groups. The study population was from 18 US academic centers and was predominantly female, and more than half were African American.
After 24 weeks, 22.5% of the mycophenolate patients were in complete remission by very strict criteria vs only 4% of those given cyclophosphamide (P = .005). The trend for partial remissions was also in favor of mycophenolate, although the difference was not statistically significant. The rate of complete and partial remissions, a prespecified end point, was significantly higher in the mycophenolate group. Although the study was trying to evaluate equivalency, it actually showed superiority for mycophenolate induction therapy.
Serum creatinine levels declined in both groups, but more in the mycophenolate group by 24 weeks. Urinary protein levels fell the same amount in both groups. At 3 years, the groups were statistically equivalent in terms of renal flares, renal failures, and deaths. However, the study groups were small, and the mycophenolate group did have a better trend for both renal failure (N = 4 vs 7) and deaths (N = 4 vs 8).
Mycophenolate also had fewer side effects, including infection, although again the numbers were too small to show statistical significance. The exception was diarrhea (N = 15 in the mycophenolate group vs 2 in the cyclophosphamide group).
A drawback of the study is that it was designed as a crossover study: a patient for whom therapy was failing after 3 months could switch to the other group, introducing potential confounding. Other problems involved the small population size and the question of whether results from patients in the United States were applicable to others worldwide.
In a worldwide study, mycophenolate was at least equivalent to cyclophosphamide for induction
The Aspreva Lupus Management Study (ALMS)10 used a similar design with 370 patients worldwide (United States, China, South America, and Europe) in one of the largest trials ever conducted in lupus nephritis. Patients were randomized to 6 months of induction therapy with either IV cyclophosphamide or oral mycophenolate but could not cross over.
At 6 months, response rates were identical between the two groups, with response defined as a combination of specific improvement in proteinuria, serum creatinine, and hematuria (50%–55%). In terms of individual renal and nonrenal variables, both groups appeared identical.
However, the side effect profiles differed between the two groups. As expected for mycophenolate, diarrhea was the most common side effect (occurring in 28% vs 12% in the cyclophosphamide group). Nausea and vomiting were more common with cyclophosphamide (45% and 37% respectively vs 14% and 13% in the mycophenolate group). Cyclophosphamide also caused hair loss in 35%, vs 10% in the mycophenolate group.
There were 14 deaths overall, which is a very low number considering the patients’ severity of illness, and it indicates the better results now achieved with therapy. The mortality rate was higher in the mycophenolate group (5% vs 3%), but the difference was not statistically significant. Six of the nine deaths with mycophenolate were from the same center in China, and none were from Europe or the United States. In summary, the study did not show that mycophenolate was superior to IV cyclophosphamide for induction therapy, but that they were equivalent in efficacy with different side effect profiles.
Membranous nephropathy: Mycophenolate vs cyclophosphamide
Less evidence is available about treatment for membranous disease, which is characterized by heavy proteinuria and the nephrotic syndrome but usually does not progress to renal failure. Radhakrishnan et al11 combined data from the trial by Ginzler et al9 and the ALMS trial10 and found 84 patients with pure membranous lupus, who were equally divided between the treatment groups receiving IV cyclophosphamide and mycophenolate. Consistent with the larger group’s data, mycophenolate and cyclophosphamide performed similarly in terms of efficacy, but there was a slightly higher rate of side effects with cyclophosphamide.
Maintenance therapy: Mycophenolate superior to azathioprine
The ALMS Maintenance Trial12 evaluated maintenance therapy in the same worldwide population that was studied for induction therapy. Of the 370 patients involved in the induction phase that compared IV cyclophosphamide and oral mycophenolate, 227 responded sufficiently to be rerandomized in a controlled, double-blinded trial of 36 months of maintenance therapy with corticosteroids and either mycophenolate (1 g twice daily) or azathioprine (2 mg/kg per day).
In intention-to-treat analysis, the time to treatment failure (ie, doubling of the serum creatinine level, progressing to renal failure, or death) was significantly shorter in the azathioprine group (P = .003). Every individual end point—end-stage renal disease, renal flares, doubling of serum creatinine, rescue immunosuppression required—was in favor of mycophenolate maintenance. At 3 years, the completion rate was 63% with mycophenolate and 49% with azathioprine. Serious adverse events and withdrawals because of adverse events were more common in the azathioprine group.
In summary, mycophenolate was superior to azathioprine in maintaining renal response and in preventing relapse in patients with active lupus nephritis who responded to induction therapy with either mycophenolate or IV cyclophosphamide. Mycophenolate was found to be superior regardless of initial induction treatment, race, or region and was confirmed by all key secondary end points.
Only one of the 227 patients died during the 3 years—from an auto accident. Again, this indicates the dramatically improved survival today compared with a decade ago.
RITUXIMAB: PROMISING BUT UNPROVEN
Rituximab (Rituxan) was originally approved to treat tumors, then rheumatoid arthritis, and most recently vasculitis. Evidence thus far is mixed regarding its use as a treatment for lupus nephritis. Although randomized clinical trials have not found it to be superior to standard regimens, there are many signs that it may be effective.
Rituximab in uncontrolled studies
Terrier et al13 analyzed prospective data from 136 patients with systemic lupus erythematosus, most of whom had renal disease, from the French Autoimmunity and Rituximab registry. Response occurred in 71% of patients using rituximab, with no difference found between patients receiving rituximab monotherapy and those concomitantly receiving immunosuppressive agents.
Melander et al14 retrospectively studied 19 women and 1 man who had been treated with rituximab for severe lupus nephritis and followed for at least 1 year. Three patients had concurrent therapy with cyclophosphamide, and 10 patients continued rituximab as maintenance therapy; 12 patients had lupus nephritis that had been refractory to standard treatment, and 6 had relapsing disease.
At a median follow-up of 22 months, 12 patients (60%) had achieved complete or partial renal remission.
Condon et al15 treated 21 patients who had severe lupus nephritis with two doses of rituximab and IV methylprednisolone 2 weeks apart, then maintenance therapy with mycophenolate without any oral steroids. At a mean follow-up of 35 months ( ± 14 months), 16 (76%) were in complete remission, with a mean time to remission of 12 months. Two (9.5%) achieved partial remission. The rate of toxicity was low.
Thus, rituximab appears promising in uncontrolled studies.
Placebo-controlled trials fail to prove rituximab effective
LUNAR trial. On the other hand, the largest placebo-controlled trial to evaluate rituximab in patients with proliferative lupus nephritis, the Lupus Nephritis Assessment With Rituximab (LUNAR) trial16 found differences in favor of rituximab, but none reached statistical significance. The trial randomized 140 patients to receive either mycophenolate plus periodic rituximab infusions or mycophenolate plus placebo infusions for 1 year. All patients received the same dosage of glucocorticoids, which was tapered over the year.
At the end of 1 year, the groups were not statistically different in terms of complete renal response and partial renal response. Rituximab appeared less likely to produce no response, but the difference was not statistically significant.
African Americans appeared to have a higher response rate to rituximab (70% in the rituximab group achieved a response vs 45% in the control group), but again, the difference did not reach statistical significance, and the total study population of African Americans was only 40.
Rituximab did have a statistically significant positive effect on two serologic markers at 1 year: levels of anti-dsDNA fell faster and complement rose faster. In addition, rates of adverse and serious adverse events were similar between the two groups, with no new or unexpected “safety signals.”
This study can be interpreted in a number of ways. The number of patients may have been too small to show significance and the follow-up may have been too short. On the other hand, it may simply not be effective to add rituximab to a full dose of mycophenolate and steroids, an already good treatment.
EXPLORER trial. Similarly, for patients with lupus without nephritis, the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial17 also tested rituximab against a background of an effective therapeutic regimen and found no additional benefit. This study had design problems similar to those of the LUNAR trial.
Rituximab as rescue therapy
The evidence so far indicates that rituximab may have a role as rescue therapy for refractory or relapsing disease. Rituximab must be used with other therapies, but maintenance corticosteroid therapy is not necessary. Its role as a first-line agent in induction therapy for lupus nephritis remains unclear, although it may have an important role for nonwhites. In general, it has been well tolerated. Until a large randomized trial indicates otherwise, it should not be used as a first-line therapy.
The US Food and Drug Administration (FDA) sent out a warning about the danger of progressive multifocal leukoencephalopathy as an adverse effect of rituximab and of mycophenolate, but this does not appear to be a major concern for most patients and is only likely to occur in those who have been over-immunosuppressed for many years.
MULTITARGET THERAPY
The concept of using multiple drugs simultaneously—such as mycophenolate, steroids, and rituximab—is increasingly being tried. Multi-target therapy appears to offer the advantages of combining different modes of action with better results, and it offers fewer side effects because dosages of each individual drug can be lower when combined with other immunosuppressives.
Bao et al18 in China randomly assigned 40 patients with diffuse proliferative and membranous nephritis to 6 to 9 months of induction treatment with either multitarget therapy (mycophenolate, tacrolimus [Prograf], and glucocorticoids) or IV cyclophosphamide. More complete remissions occurred in the multitarget therapy group, both at 6 months (50% vs 5%) and at 9 months (65% vs 15%). Most adverse events were less frequent in the multitarget therapy group, although three patients (15%) in the multitarget therapy group developed new-onset hypertension vs none in the cyclophosphamide group.
NEW MEDICATIONS
Entirely new classes of drugs are being developed with immunomodulatory effects, including tolerance molecules, cytokine blockers, inhibitors of human B lymphocyte stimulator, and costimulatory blockers.
Belimumab offers small improvement for lupus
Belimumab (Benlysta) is a human monoclonal antibody that inhibits the biologic activity of human B lymphocyte stimulator; it has recently been approved by the FDA for lupus nephritis. In a worldwide study,19 867 patients with systemic lupus erythematosus were randomized to receive either belimumab (1 mg/kg or 10 mg/kg) or placebo.
The primary end point was the reduction of disease activity by a scoring system (SELENA-SLEDAI) that incorporated multiple features of lupus, including arthritis, vasculitis, proteinuria, rash, and others. Patients in the belimumab group had better outcomes, but the results were not dramatic. Because the drug is so expensive (about $25,000 per year) and the improvement offered is only incremental, this drug will not likely change the treatment of lupus very much.
Moreover, patients with lupus nephritis were not included in the study, but a new study is being planned to do so. Improvement is harder to demonstrate in lupus nephritis than in rheumatoid arthritis and systemic lupus erythematosus: significant changes in creatinine levels and 24-hour urinary protein must be achieved, rather than more qualitative signs and symptoms of joint pain, rash, and feeling better. Although belimumab is still unproven for lupus nephritis, it might be worth trying for patients failing other therapy.
Laquinimod: A promising experimental drug
Laquinimod is an oral immunomodulatory drug with a number of effects, including down-regulating major histocompatability complex II, chemokines, and adhesion-related molecules related to inflammation. It has been studied in more than 2,500 patients with multiple sclerosis. Pilot studies are now being done for its use for lupus nephritis. If it shows promise, a large randomized, controlled trial will be conducted.
Abatacept is in clinical trials
Abatacept (Orencia), a costimulation blocker, is undergoing clinical trials in lupus nephritis. Results should be available shortly.
INDIVIDUALIZE THERAPY
This past decade has seen such an increase in options to treat lupus nephritis that therapy can now be individualized.
Choosing IV cyclophosphamide vs mycophenolate
As a result of recent trials, doctors in the United States are increasingly using mycophenolate as the first-line drug for lupus nephritis. In Europe, however, many are choosing the shorter regimen of IV cyclophosphamide because of the results of the Euro-Lupus study.
Nowadays, I tend to use IV cyclophosphamide as the first-line drug only for patients with severe crescenteric glomerulonephritis or a very high serum creatinine level. In such cases, there is more experience with cyclophosphamide, and such severe disease does not lend itself to the luxury of trying out different therapies sequentially. If such a severely ill patient insists that a future pregnancy is very important, an alternative therapy of mycophenolate plus rituximab should be considered. I prefer mycophenolate for induction and maintenance therapy in most patients.
Dosing and formulation considerations for mycophenolate
Large dosages of mycophenolate are much better tolerated when broken up throughout the day. A patient who cannot tolerate 1 g twice daily may be able to tolerate 500 mg four times a day. The formulation can also make a difference. Some patients tolerate sustained-release mycophenolate (Myfortic) better than CellCept, and vice versa.
For patients who cannot tolerate mycophenolate, azathioprine is an acceptable alternative. In addition, for a patient who is already doing well on azathioprine, there is no need to change to mycophenolate.
Long maintenance therapy now acceptable
The ALMS Maintenance Trial12 found 3 years of maintenance therapy to be safe and effective. Such a long maintenance period is increasingly viewed as important, especially for patients in their teens and 20s, as it allows them to live a normal life, ie, to finish their education, get married, and become settled socially. Whether 5 years of maintenance therapy or even 10 years is advisable is still unknown.
Treatment during pregnancy
Neither mycophenolate nor azathioprine is recommended during pregnancy, although their effects are unknown. Because we have much more renal transplant experience with azathioprine during pregnancy, I recommend either switching from mycophenolate to azathioprine or trying to stop medication altogether if the patient has been well controlled.
Treatment for lupus nephritis has changed dramatically in recent years. Only 10 years ago, rheumatologists and nephrologists, whether specializing in adult or pediatric medicine, treated lupus nephritis with a similar regimen of monthly intravenous cyclophosphamide (Cytoxan) and glucocorticoids. Although the regimen is effective, side effects such as infection, hair loss, and infertility were extremely common.
Effective but very toxic therapy is common in autoimmune diseases. In the last decade, clinical trials have shown that less toxic drugs are as effective for treating lupus nephritis. This article will review new developments in therapy for lupus nephritis, which can be viewed as a prototype for other fields of medicine.
DEMOGRAPHICS ARE IMPORTANT
Although numerous factors have prognostic value in lupus nephritis (eg, serum creatinine, proteinuria, renal biopsy findings), the most important to consider when designing and interpreting studies are race and socioeconomic variables.
A retrospective study in Miami, FL,1 evaluated 213 patients with lupus nephritis, of whom 47% were Hispanic, 44% African American, and 20% white. At baseline, African Americans had higher blood pressure, higher serum creatinine levels, and lower household income. After 6 years, African Americans fared the worst in terms of doubling of serum creatinine, developing end-stage renal disease, and death; whites had the best outcomes, and Hispanics were in between. Low income was found to be a significant risk factor, independent of racial background.
In a similar retrospective study in New York City in 128 patients (43% white, 40% Hispanic, and 17% African American) with proliferative lupus nephritis,2 disease was much more likely to progress to renal failure over 10 years in patients living in a poor neighborhood, even after adjustment for race.
We need to keep in mind that racial and socioeconomic factors correlate with disease severity when we design and interpret studies of lupus nephritis. Study groups must be carefully balanced with patients of similar racial and socioeconomic profiles. Study findings must be interpreted with caution; for example, whether results from a study from China are applicable to an African American with lupus nephritis in New York City is unclear.
OLDER STANDARD THERAPY: EFFECTIVE BUT TOXIC
The last large National Institutes of Health study that involved only cyclophosphamide and a glucocorticoid was published in 2001,3 with 21 patients receiving cyclophosphamide alone and 20 patients receiving cyclophosphamide plus methylprednisolone. Although lupus nephritis improved, serious side effects occurred in one-third to one-half of patients in each group and included hypertension, hyperlipidemia, valvular heart disease, avascular necrosis, premature menopause, and major infections, including herpes zoster.
Less cyclophosphamide works just as well
The multicenter, prospective Euro-Lupus Nephritis Trial4 randomized 90 patients with proliferative lupus nephritis to receive either standard high-dose intravenous (IV) cyclophosphamide therapy (six monthly pulses and two quarterly pulses, with doses increasing according to the white blood cell count) or low-dose IV cyclophosphamide therapy (six pulses every 2 weeks at a fixed dose of 500 mg). Both regimens were followed by azathioprine (Imuran).
At 4 years, the two treatment groups were not significantly different in terms of treatment failure, remission rates, serum creatinine levels, 24-hour proteinuria, and freedom from renal flares. However, the rates of side effects were significantly different, with more patients in the low-dosage group free of severe infection.
One problem with this study is whether it is applicable to an American lupus nephritis population, since 84% of the patients were white. Since this study, others indicate that this regimen is probably also safe and effective for different racial groups in the United States.
At 10-year follow-up,5 both treatment groups still had identical excellent rates of freedom from end-stage renal disease. Serum creatinine and 24-hour proteinuria were also at excellent levels and identical in both groups. Nearly three quarters of patients still needed glucocorticoid therapy and more than half still needed immunosuppressive therapy, but the rates were not statistically significantly different between the treatment groups.
The cumulative dose of cyclophosphamide was 9.5 g in the standard-treatment group and 5.5 g in the low-dose group. This difference in exposure could make a tremendous difference to patients, not only for immediate side effects such as early menopause and infections, but for the risk of cancer in later decades.
This study showed clearly that low-dose cyclophosphamide is an option for induction therapy. Drawbacks of the study were that the population was mostly white and that patients had only moderately severe disease.
Low-dose cyclophosphamide has largely replaced the older National Institutes of Health regimen, although during the last decade drug therapy has undergone more changes.
MYCOPHENOLATE AND AZATHIOPRINE: ALTERNATIVES TO CYCLOPHOSPHAMIDE
In a Chinese study, mycophenolate was better than cyclophosphamide for induction
In a study in Hong Kong, Chan et al6 randomized 42 patients with severe lupus nephritis to receive either mycophenolate mofetil (available in the United States as CellCept; 2 g/day for 6 months, then 1 g/day for 6 months) or oral cyclophosphamide (2.5 mg/kg per day for 6 months) followed by azathioprine (1.5–2.0 mg/kg per day) for 6 months. Both groups also received prednisolone during the year.
At the end of the first year, the two groups were not significantly different in their rates of complete remission, partial remission, and relapse. The rate of infection, although not significantly different, was higher in the cyclophosphamide group (33% vs 19%). Two patients (10%) died in the cyclophosphamide group, but the difference in mortality rates was not statistically significant.
Nearly 5 years later,7 rates of chronic renal failure and relapse were still statistically the same in the two groups. Infections were fewer in the mycophenolate group (13% vs 40%, P = .013). The rate of amenorrhea was 36% in the cyclophosphamide group and only 4% in the mycophenolate group (P = .004). Four patients in the cyclophosphamide group and none in the mycophenolate group reached the composite end point of end-stage renal failure or death (P = .062).
This study appeared to offer a new option with equal efficacy and fewer side effects than standard therapy. However, its applicability to non-Chinese populations remained to be shown.
In a US study, mycophenolate or azathioprine was better than cyclophosphamide as maintenance
In a study in Miami,8 59 patients with lupus nephritis were given standard induction therapy with IV cyclophosphamide plus glucocorticoids for 6 months, then randomly assigned to one of three maintenance therapies for 1 to 3 years: IV injections of cyclophosphamide every 3 months (standard therapy), oral azathioprine, or oral mycophenolate. The population was 93% female, their average age was 33 years, and nearly half were African American, with many of the others being Hispanic. Patients tended to have severe disease, with nearly two-thirds having nephrotic syndrome.
After 6 years, there had been more deaths in the cyclophosphamide group than in the azathioprine group (P = .02) and in the mycophenolate group, although the latter difference was not statistically significant (P = .11). The combined rate of death and chronic renal failure was significantly higher with cyclophosphamide than with either of the oral agents. The cyclophosphamide group also had the highest relapse rate during the maintenance phase.
The differences in side effects were even more dramatic. Amenorrhea affected 32% of patients in the cyclophosphamide group, and only 7% and 6% in the azathioprine and mycophenolate groups, respectively. Rates of infections were 68% in the cyclophosphamide group and 28% and 21% in the azathioprine and mycophenolate groups, respectively. Patients given cyclophosphamide had 13 hospital days per patient per year, while the other groups each had only 1.
This study showed that maintenance therapy with oral azathioprine or mycophenolate was more effective and had fewer adverse effects than standard IV cyclophosphamide therapy. As a result of this study, oral agents for maintenance therapy became the new standard, but the question remained whether oral agents could safely be used for induction.
In a US study, mycophenolate was better than cyclophosphamide for induction
In a noninferiority study, Ginzler et al9 randomized 140 patients with severe lupus nephritis to receive either monthly IV cyclophosphamide or oral mycophenolate as induction therapy for 6 months. Adjunctive care with glucocorticoids was given in both groups. The study population was from 18 US academic centers and was predominantly female, and more than half were African American.
After 24 weeks, 22.5% of the mycophenolate patients were in complete remission by very strict criteria vs only 4% of those given cyclophosphamide (P = .005). The trend for partial remissions was also in favor of mycophenolate, although the difference was not statistically significant. The rate of complete and partial remissions, a prespecified end point, was significantly higher in the mycophenolate group. Although the study was trying to evaluate equivalency, it actually showed superiority for mycophenolate induction therapy.
Serum creatinine levels declined in both groups, but more in the mycophenolate group by 24 weeks. Urinary protein levels fell the same amount in both groups. At 3 years, the groups were statistically equivalent in terms of renal flares, renal failures, and deaths. However, the study groups were small, and the mycophenolate group did have a better trend for both renal failure (N = 4 vs 7) and deaths (N = 4 vs 8).
Mycophenolate also had fewer side effects, including infection, although again the numbers were too small to show statistical significance. The exception was diarrhea (N = 15 in the mycophenolate group vs 2 in the cyclophosphamide group).
A drawback of the study is that it was designed as a crossover study: a patient for whom therapy was failing after 3 months could switch to the other group, introducing potential confounding. Other problems involved the small population size and the question of whether results from patients in the United States were applicable to others worldwide.
In a worldwide study, mycophenolate was at least equivalent to cyclophosphamide for induction
The Aspreva Lupus Management Study (ALMS)10 used a similar design with 370 patients worldwide (United States, China, South America, and Europe) in one of the largest trials ever conducted in lupus nephritis. Patients were randomized to 6 months of induction therapy with either IV cyclophosphamide or oral mycophenolate but could not cross over.
At 6 months, response rates were identical between the two groups, with response defined as a combination of specific improvement in proteinuria, serum creatinine, and hematuria (50%–55%). In terms of individual renal and nonrenal variables, both groups appeared identical.
However, the side effect profiles differed between the two groups. As expected for mycophenolate, diarrhea was the most common side effect (occurring in 28% vs 12% in the cyclophosphamide group). Nausea and vomiting were more common with cyclophosphamide (45% and 37% respectively vs 14% and 13% in the mycophenolate group). Cyclophosphamide also caused hair loss in 35%, vs 10% in the mycophenolate group.
There were 14 deaths overall, which is a very low number considering the patients’ severity of illness, and it indicates the better results now achieved with therapy. The mortality rate was higher in the mycophenolate group (5% vs 3%), but the difference was not statistically significant. Six of the nine deaths with mycophenolate were from the same center in China, and none were from Europe or the United States. In summary, the study did not show that mycophenolate was superior to IV cyclophosphamide for induction therapy, but that they were equivalent in efficacy with different side effect profiles.
Membranous nephropathy: Mycophenolate vs cyclophosphamide
Less evidence is available about treatment for membranous disease, which is characterized by heavy proteinuria and the nephrotic syndrome but usually does not progress to renal failure. Radhakrishnan et al11 combined data from the trial by Ginzler et al9 and the ALMS trial10 and found 84 patients with pure membranous lupus, who were equally divided between the treatment groups receiving IV cyclophosphamide and mycophenolate. Consistent with the larger group’s data, mycophenolate and cyclophosphamide performed similarly in terms of efficacy, but there was a slightly higher rate of side effects with cyclophosphamide.
Maintenance therapy: Mycophenolate superior to azathioprine
The ALMS Maintenance Trial12 evaluated maintenance therapy in the same worldwide population that was studied for induction therapy. Of the 370 patients involved in the induction phase that compared IV cyclophosphamide and oral mycophenolate, 227 responded sufficiently to be rerandomized in a controlled, double-blinded trial of 36 months of maintenance therapy with corticosteroids and either mycophenolate (1 g twice daily) or azathioprine (2 mg/kg per day).
In intention-to-treat analysis, the time to treatment failure (ie, doubling of the serum creatinine level, progressing to renal failure, or death) was significantly shorter in the azathioprine group (P = .003). Every individual end point—end-stage renal disease, renal flares, doubling of serum creatinine, rescue immunosuppression required—was in favor of mycophenolate maintenance. At 3 years, the completion rate was 63% with mycophenolate and 49% with azathioprine. Serious adverse events and withdrawals because of adverse events were more common in the azathioprine group.
In summary, mycophenolate was superior to azathioprine in maintaining renal response and in preventing relapse in patients with active lupus nephritis who responded to induction therapy with either mycophenolate or IV cyclophosphamide. Mycophenolate was found to be superior regardless of initial induction treatment, race, or region and was confirmed by all key secondary end points.
Only one of the 227 patients died during the 3 years—from an auto accident. Again, this indicates the dramatically improved survival today compared with a decade ago.
RITUXIMAB: PROMISING BUT UNPROVEN
Rituximab (Rituxan) was originally approved to treat tumors, then rheumatoid arthritis, and most recently vasculitis. Evidence thus far is mixed regarding its use as a treatment for lupus nephritis. Although randomized clinical trials have not found it to be superior to standard regimens, there are many signs that it may be effective.
Rituximab in uncontrolled studies
Terrier et al13 analyzed prospective data from 136 patients with systemic lupus erythematosus, most of whom had renal disease, from the French Autoimmunity and Rituximab registry. Response occurred in 71% of patients using rituximab, with no difference found between patients receiving rituximab monotherapy and those concomitantly receiving immunosuppressive agents.
Melander et al14 retrospectively studied 19 women and 1 man who had been treated with rituximab for severe lupus nephritis and followed for at least 1 year. Three patients had concurrent therapy with cyclophosphamide, and 10 patients continued rituximab as maintenance therapy; 12 patients had lupus nephritis that had been refractory to standard treatment, and 6 had relapsing disease.
At a median follow-up of 22 months, 12 patients (60%) had achieved complete or partial renal remission.
Condon et al15 treated 21 patients who had severe lupus nephritis with two doses of rituximab and IV methylprednisolone 2 weeks apart, then maintenance therapy with mycophenolate without any oral steroids. At a mean follow-up of 35 months ( ± 14 months), 16 (76%) were in complete remission, with a mean time to remission of 12 months. Two (9.5%) achieved partial remission. The rate of toxicity was low.
Thus, rituximab appears promising in uncontrolled studies.
Placebo-controlled trials fail to prove rituximab effective
LUNAR trial. On the other hand, the largest placebo-controlled trial to evaluate rituximab in patients with proliferative lupus nephritis, the Lupus Nephritis Assessment With Rituximab (LUNAR) trial16 found differences in favor of rituximab, but none reached statistical significance. The trial randomized 140 patients to receive either mycophenolate plus periodic rituximab infusions or mycophenolate plus placebo infusions for 1 year. All patients received the same dosage of glucocorticoids, which was tapered over the year.
At the end of 1 year, the groups were not statistically different in terms of complete renal response and partial renal response. Rituximab appeared less likely to produce no response, but the difference was not statistically significant.
African Americans appeared to have a higher response rate to rituximab (70% in the rituximab group achieved a response vs 45% in the control group), but again, the difference did not reach statistical significance, and the total study population of African Americans was only 40.
Rituximab did have a statistically significant positive effect on two serologic markers at 1 year: levels of anti-dsDNA fell faster and complement rose faster. In addition, rates of adverse and serious adverse events were similar between the two groups, with no new or unexpected “safety signals.”
This study can be interpreted in a number of ways. The number of patients may have been too small to show significance and the follow-up may have been too short. On the other hand, it may simply not be effective to add rituximab to a full dose of mycophenolate and steroids, an already good treatment.
EXPLORER trial. Similarly, for patients with lupus without nephritis, the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial17 also tested rituximab against a background of an effective therapeutic regimen and found no additional benefit. This study had design problems similar to those of the LUNAR trial.
Rituximab as rescue therapy
The evidence so far indicates that rituximab may have a role as rescue therapy for refractory or relapsing disease. Rituximab must be used with other therapies, but maintenance corticosteroid therapy is not necessary. Its role as a first-line agent in induction therapy for lupus nephritis remains unclear, although it may have an important role for nonwhites. In general, it has been well tolerated. Until a large randomized trial indicates otherwise, it should not be used as a first-line therapy.
The US Food and Drug Administration (FDA) sent out a warning about the danger of progressive multifocal leukoencephalopathy as an adverse effect of rituximab and of mycophenolate, but this does not appear to be a major concern for most patients and is only likely to occur in those who have been over-immunosuppressed for many years.
MULTITARGET THERAPY
The concept of using multiple drugs simultaneously—such as mycophenolate, steroids, and rituximab—is increasingly being tried. Multi-target therapy appears to offer the advantages of combining different modes of action with better results, and it offers fewer side effects because dosages of each individual drug can be lower when combined with other immunosuppressives.
Bao et al18 in China randomly assigned 40 patients with diffuse proliferative and membranous nephritis to 6 to 9 months of induction treatment with either multitarget therapy (mycophenolate, tacrolimus [Prograf], and glucocorticoids) or IV cyclophosphamide. More complete remissions occurred in the multitarget therapy group, both at 6 months (50% vs 5%) and at 9 months (65% vs 15%). Most adverse events were less frequent in the multitarget therapy group, although three patients (15%) in the multitarget therapy group developed new-onset hypertension vs none in the cyclophosphamide group.
NEW MEDICATIONS
Entirely new classes of drugs are being developed with immunomodulatory effects, including tolerance molecules, cytokine blockers, inhibitors of human B lymphocyte stimulator, and costimulatory blockers.
Belimumab offers small improvement for lupus
Belimumab (Benlysta) is a human monoclonal antibody that inhibits the biologic activity of human B lymphocyte stimulator; it has recently been approved by the FDA for lupus nephritis. In a worldwide study,19 867 patients with systemic lupus erythematosus were randomized to receive either belimumab (1 mg/kg or 10 mg/kg) or placebo.
The primary end point was the reduction of disease activity by a scoring system (SELENA-SLEDAI) that incorporated multiple features of lupus, including arthritis, vasculitis, proteinuria, rash, and others. Patients in the belimumab group had better outcomes, but the results were not dramatic. Because the drug is so expensive (about $25,000 per year) and the improvement offered is only incremental, this drug will not likely change the treatment of lupus very much.
Moreover, patients with lupus nephritis were not included in the study, but a new study is being planned to do so. Improvement is harder to demonstrate in lupus nephritis than in rheumatoid arthritis and systemic lupus erythematosus: significant changes in creatinine levels and 24-hour urinary protein must be achieved, rather than more qualitative signs and symptoms of joint pain, rash, and feeling better. Although belimumab is still unproven for lupus nephritis, it might be worth trying for patients failing other therapy.
Laquinimod: A promising experimental drug
Laquinimod is an oral immunomodulatory drug with a number of effects, including down-regulating major histocompatability complex II, chemokines, and adhesion-related molecules related to inflammation. It has been studied in more than 2,500 patients with multiple sclerosis. Pilot studies are now being done for its use for lupus nephritis. If it shows promise, a large randomized, controlled trial will be conducted.
Abatacept is in clinical trials
Abatacept (Orencia), a costimulation blocker, is undergoing clinical trials in lupus nephritis. Results should be available shortly.
INDIVIDUALIZE THERAPY
This past decade has seen such an increase in options to treat lupus nephritis that therapy can now be individualized.
Choosing IV cyclophosphamide vs mycophenolate
As a result of recent trials, doctors in the United States are increasingly using mycophenolate as the first-line drug for lupus nephritis. In Europe, however, many are choosing the shorter regimen of IV cyclophosphamide because of the results of the Euro-Lupus study.
Nowadays, I tend to use IV cyclophosphamide as the first-line drug only for patients with severe crescenteric glomerulonephritis or a very high serum creatinine level. In such cases, there is more experience with cyclophosphamide, and such severe disease does not lend itself to the luxury of trying out different therapies sequentially. If such a severely ill patient insists that a future pregnancy is very important, an alternative therapy of mycophenolate plus rituximab should be considered. I prefer mycophenolate for induction and maintenance therapy in most patients.
Dosing and formulation considerations for mycophenolate
Large dosages of mycophenolate are much better tolerated when broken up throughout the day. A patient who cannot tolerate 1 g twice daily may be able to tolerate 500 mg four times a day. The formulation can also make a difference. Some patients tolerate sustained-release mycophenolate (Myfortic) better than CellCept, and vice versa.
For patients who cannot tolerate mycophenolate, azathioprine is an acceptable alternative. In addition, for a patient who is already doing well on azathioprine, there is no need to change to mycophenolate.
Long maintenance therapy now acceptable
The ALMS Maintenance Trial12 found 3 years of maintenance therapy to be safe and effective. Such a long maintenance period is increasingly viewed as important, especially for patients in their teens and 20s, as it allows them to live a normal life, ie, to finish their education, get married, and become settled socially. Whether 5 years of maintenance therapy or even 10 years is advisable is still unknown.
Treatment during pregnancy
Neither mycophenolate nor azathioprine is recommended during pregnancy, although their effects are unknown. Because we have much more renal transplant experience with azathioprine during pregnancy, I recommend either switching from mycophenolate to azathioprine or trying to stop medication altogether if the patient has been well controlled.
- Contreras G, Lenz O, Pardo V, et al. Outcomes in African Americans and Hispanics with lupus nephritis. Kidney Int 2006; 69:1846–1851.
- Barr RG, Seliger S, Appel GB, et al. Prognosis in proliferative lupus nephritis: the role of socio-economic status and race/ethnicity. Nephrol Dial Transplant 2003; 18:2039–2046.
- Illei GG, Austin HA, Crane M, et al. Combination therapy with pulse cyclophosphamide plus pulse methylprednisolone improves long-term renal outcome without adding toxicity in patients with lupus nephritis. Ann Intern Med 2001; 135:248–257.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum 2002; 46:2121–2131.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose and high-dose intravenous cyclophosphamide. Ann Rheum Dis 2010; 69:61–64.
- Chan TM, Li FK, Tang CS, et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong King-Guangzhou Nephrology Study Group. N Engl J Med 2000; 343:1156–1162.
- Chan TM, Tse KC, Tang CS, Mok MY, Li FK; Hong Kong Nephrology Study Group. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005; 16:1076–1084.
- Contreras G, Pardo V, Leclercq B, et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004; 350:971–980.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009; 20:1103–1112.
- Radhakrishnan J, Moutzouris DA, Ginzler EM, Solomons N, Siempos II, Appel GB. Mycophenolate mofetil and intravenous cyclophosphamide are similar as induction therapy for class V lupus nephritis. Kidney Int 2010; 77:152–160.
- Dooley MA, Jayne D, Ginzler EM, et al; for the ALMS Group. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N Engl J Med 2011; 365:1886–1895.
- Terrier B, Amoura Z, Ravaud P, et al; Club Rhumatismes et Inflammation. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum 2010; 62:2458–2466.
- Melander C, Sallée M, Troillet P, et al. Rituximab in severe lupus nephritis: early B-cell depletion affects long-term renal outcome. Clin J Am Soc Nephrol 2009; 4:579–587.
- Condon MB, Griffith M, Cook HT, Levy J, Lightstone L, Cairns T. Treatment of class IV lupus nephritis with rituximab & mycophenolate mofetil (MMF) with no oral steroids is effective and safe (abstract). J Am Soc Nephrol 2010; 21(suppl):625A–626A.
- Furie RA, Looney RJ, Rovin E, et al. Efficacy and safety of rituximab in subjects with active proliferative lupus nephritis (LN): results from the randomized, double-blind phase III LUNAR study (abstract). Arthritis Rheum 2009; 60(suppl 1):S429.
- Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 2010; 62:222–233.
- Bao H, Liu ZH, Zie HL, Hu WX, Zhang HT, Li LS. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J Am Soc Nephrol 2008; 19:2001–2010.
- Navarra SV, Guzmán RM, Gallacher AE, et al; BLISS-52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:721–731.
- Contreras G, Lenz O, Pardo V, et al. Outcomes in African Americans and Hispanics with lupus nephritis. Kidney Int 2006; 69:1846–1851.
- Barr RG, Seliger S, Appel GB, et al. Prognosis in proliferative lupus nephritis: the role of socio-economic status and race/ethnicity. Nephrol Dial Transplant 2003; 18:2039–2046.
- Illei GG, Austin HA, Crane M, et al. Combination therapy with pulse cyclophosphamide plus pulse methylprednisolone improves long-term renal outcome without adding toxicity in patients with lupus nephritis. Ann Intern Med 2001; 135:248–257.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum 2002; 46:2121–2131.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose and high-dose intravenous cyclophosphamide. Ann Rheum Dis 2010; 69:61–64.
- Chan TM, Li FK, Tang CS, et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong King-Guangzhou Nephrology Study Group. N Engl J Med 2000; 343:1156–1162.
- Chan TM, Tse KC, Tang CS, Mok MY, Li FK; Hong Kong Nephrology Study Group. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005; 16:1076–1084.
- Contreras G, Pardo V, Leclercq B, et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004; 350:971–980.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009; 20:1103–1112.
- Radhakrishnan J, Moutzouris DA, Ginzler EM, Solomons N, Siempos II, Appel GB. Mycophenolate mofetil and intravenous cyclophosphamide are similar as induction therapy for class V lupus nephritis. Kidney Int 2010; 77:152–160.
- Dooley MA, Jayne D, Ginzler EM, et al; for the ALMS Group. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N Engl J Med 2011; 365:1886–1895.
- Terrier B, Amoura Z, Ravaud P, et al; Club Rhumatismes et Inflammation. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum 2010; 62:2458–2466.
- Melander C, Sallée M, Troillet P, et al. Rituximab in severe lupus nephritis: early B-cell depletion affects long-term renal outcome. Clin J Am Soc Nephrol 2009; 4:579–587.
- Condon MB, Griffith M, Cook HT, Levy J, Lightstone L, Cairns T. Treatment of class IV lupus nephritis with rituximab & mycophenolate mofetil (MMF) with no oral steroids is effective and safe (abstract). J Am Soc Nephrol 2010; 21(suppl):625A–626A.
- Furie RA, Looney RJ, Rovin E, et al. Efficacy and safety of rituximab in subjects with active proliferative lupus nephritis (LN): results from the randomized, double-blind phase III LUNAR study (abstract). Arthritis Rheum 2009; 60(suppl 1):S429.
- Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 2010; 62:222–233.
- Bao H, Liu ZH, Zie HL, Hu WX, Zhang HT, Li LS. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J Am Soc Nephrol 2008; 19:2001–2010.
- Navarra SV, Guzmán RM, Gallacher AE, et al; BLISS-52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:721–731.
KEY POINTS
- Mycophenolate is at least equivalent to intravenous cyclophosphamide for induction and maintenance treatment of severe lupus nephritis.
- The role of rituximab is unclear, and for now it should only be used in relapsing patients or patients whose disease is resistant to standard therapy.
- Using combination therapies for induction treatment and maintenance is becoming increasingly common.
- Three-year maintenance therapy is now considered advisable in most patients.
- Entirely new drugs under study include costimulatory blockers, inhibitors of human B lymphocyte stimulator, tolerance molecules, and cytokine blockers.
Deep brain stimulation: What can patients expect from it?
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
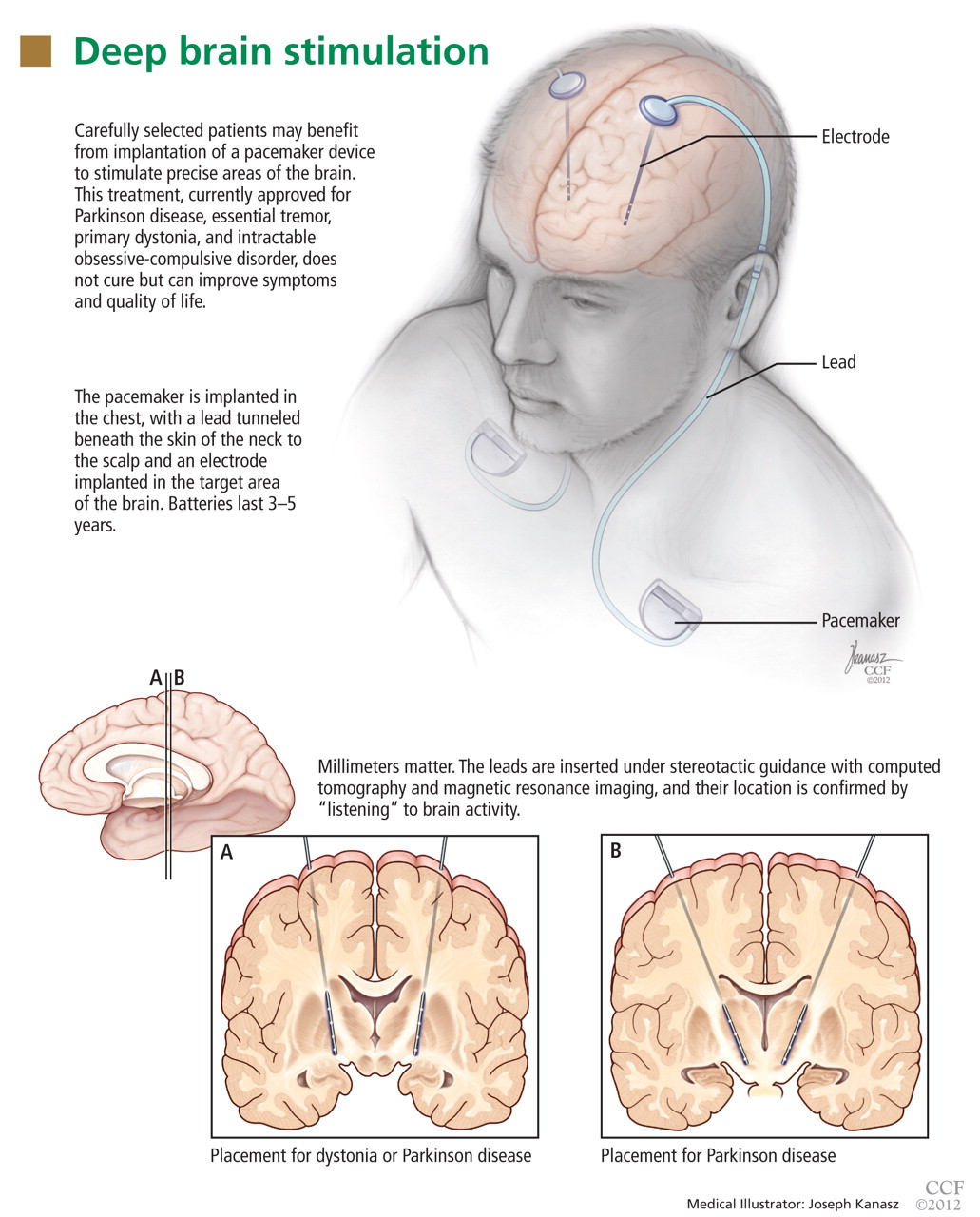
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
KEY POINTS
- Compared with ablative procedures, deep brain stimulation has the advantage of being reversible and adjustable. It is considered safer than ablative surgery, in particular for bilateral procedures, which are often needed for patients with advanced Parkinson disease and other movement disorders.
- For Parkinson disease, deep brain stimulation improves the cardinal motor symptoms, extends medication “on” time, and reduces motor fluctuations during the day.
- In general, patients with Parkinson disease are likely to benefit from this therapy if they show a clear response to levodopa. Patients are therefore asked to stop their Parkinson medications overnight to permit a formal evaluation of their motor response before and after a dose of levodopa.
- Candidates require a thorough evaluation to assess whether they are likely to benefit from deep brain stimulation and if they can comply with the maintenance often required for a successful outcome.
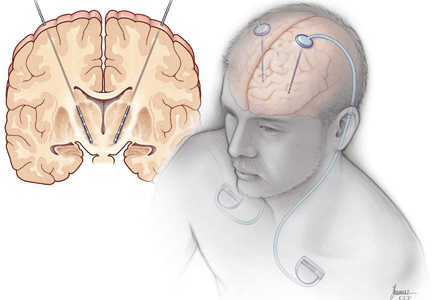
Updates in the medical management of Parkinson disease
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING

Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
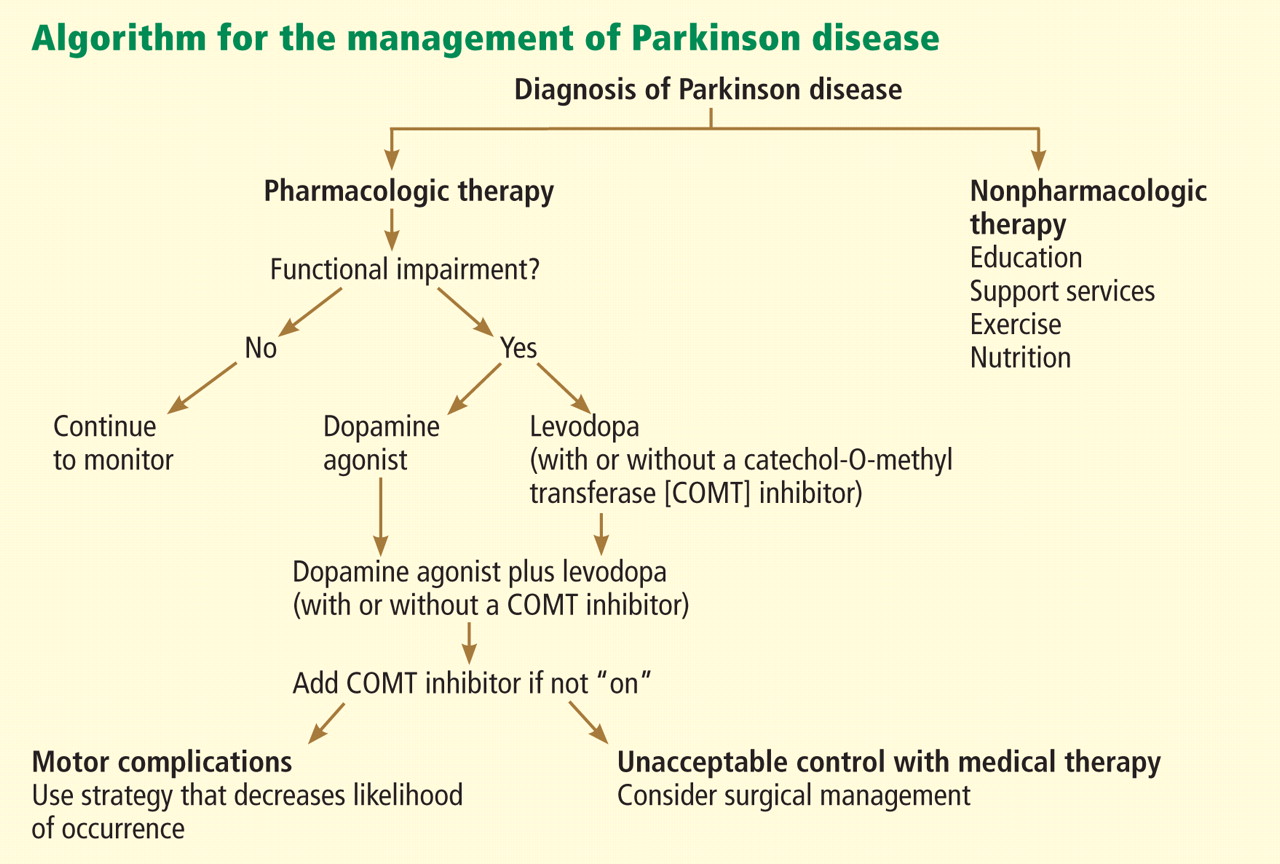
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING
Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING
Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
KEY POINTS
- Parkinson disease can usually be diagnosed on the basis of clinical features: slow movement, resting tremor, rigidity, and asymmetrical presentation, as well as alleviation of symptoms with dopaminergic therapy.
- Early disease can be treated with levodopa, dopamine agonists, anticholinergics, and monoamine oxidase-B inhibitors.
- Advanced Parkinson disease may require a catechol-O-methyltransferase (COMT) inhibitor, apomorphine, and amantadine (Symmetrel). Side effects include motor fluctuations, dyskinesias, and cognitive problems.
Accountable care organizations, the patient-centered medical home, and health care reform: What does it all mean?
The US health care system cannot continue with “business as usual.” The current model is broken: it does not deliver the kind of care we want for our patients, ourselves, our families, and our communities. It is our role as professionals to help drive change and make medical care more cost-effective and of higher quality, with better satisfaction for patients as well as for providers.
Central to efforts to reform the system are two concepts. One is the “patient-centered medical home,” in which a single provider is responsible for coordinating care for individual patients. The other is “accountable care organizations,” a new way of organizing care along a continuum from doctor to hospital, mandated by the new health care reform law (technically known as the Patient Protection and Affordable Care Act).
CURRENT STATE OF HEALTH CARE: HIGH COST AND POOR QUALITY
Since health care reform was initially proposed in the 1990s, trends in the United States have grown steadily worse. Escalating health care costs have outstripped inflation, consuming an increasing percentage of the gross domestic product (GDP) at an unsustainable rate. Despite increased spending, quality outcomes are suboptimal. In addition, with the emergence of specialization and technology, care is increasingly fragmented and poorly coordinated, with multiple providers and poorly managed resources.
Over the last 15 years, the United States has far surpassed most countries in the developed world for total health care expenditures per capita.1,2 In 2009, we spent 17.4% of our GDP on health care, translating to $7,960 per capita, while Japan spent only 8.5% of its GDP, averaging $2,878 per capita.2 At the current rate, health care spending in the United States will increase from $2.5 trillion in 2009 to over $4.6 trillion in 2020.3
Paradoxically, costlier care is often of poorer quality. Many countries that spend far less per capita on health care achieve far better outcomes. Even within the United States, greater Medicare spending on a state and regional basis tends to correlate with poorer quality of care.4 Spending among Medicare beneficiaries is not standardized and varies widely throughout the country.5 The amount of care a patient receives also varies dramatically by region. The number of specialists involved in care during the last year of life is steadily increasing in many regions of the country, indicating poor care coordination.6
PATIENT-CENTERED MEDICAL HOMES: A POSITIVE TREND
The problems of high cost, poor quality, and poor coordination of care have led to the emergence of the concept of the patient-centered medical home. Originally proposed in 1967 by the American Academy of Pediatrics in response to the need for care coordination by a single physician, the idea did not really take root until the early 1990s. In 2002, the American Academy of Family Medicine embraced the concept and moved it forward.
According to the National Committee for Quality Assurance (NCQA), a nonprofit organization that provides voluntary certification for medical organizations, the patient-centered medical home is a model of care in which “patients have a direct relationship with a provider who coordinates a cooperative team of healthcare professionals, takes collective responsibility for the care provided to the patient, and arranges for appropriate care with other qualified providers as needed.”7
Patient-centered medical homes are supposed to improve quality outcomes and lower costs. In addition, they can compete for public or private incentives that reward this model of care and, as we will see later, are at the heart of ACO readiness.
Medical homes meet certification standards
NCQA first formally licensed patient-centered medical homes in 2008, based on nine standards and six key elements. A scoring system was used to rank the level of certification from level 1 (the lowest) to level 3. From 2008 to the end of 2010, the number of certified homes grew from 28 to 1,506. New York has the largest number of medical homes.

The new elements in the NCQA program align more closely with federal programs that are designed to drive quality, including the Centers for Medicare and Medicaid Services program to encourage the use of the electronic medical record, and with federal rule-making this last spring designed to implement accountable care organizations (ACOs).
Same-day access is now emphasized, as is managing patient populations—rather than just individual patients—with certain chronic diseases, such as diabetes and congestive heart failure. The requirements for tracking and coordinating care have profound implications about how resources are allocated. Ideally, coordinators of chronic disease management are embedded within practices to help manage high-risk patients, although the current reimbursement mechanism does not support this model. Population management may not be feasible for institutions that still rely on paper-based medical records.
Medical homes lower costs, improve quality
Integrated delivery system models such as patient-centered medical homes have demonstrated cost-savings while improving quality of care.8,9 Reducing hospital admissions and visits to the emergency department shows the greatest cost-savings in these models. Several projects have shown significant cost-savings10:
The Group Health Cooperative of Puget Sound reduced total costs by $10 per member per month (from $498 to $488, P = 0.76), with a 16% reduction in hospital admissions (P < .001) and a 29% reduction in emergency department visits (P < .001).
The Geisinger Health System Proven-Health Navigator in Pennsylvania reduced readmissions by 18% (P < .01). They also had a 7% reduction in total costs per member per month relative to a matched control group also in the Geisinger system but not in a medical home, although this difference did not reach statistical significance. Private payer demonstration projects of patient-centered medical homes have also shown cost-savings.
Blue Cross Blue Shield of South Carolina randomized patients to participate in either a patient-centered medical home or their standard system. The patient-centered medical home group had 36% fewer hospital days, 12.4% fewer emergency department visits, and a 6.5% reduction in total medical and pharmacy costs compared with controls.
Finally, the use of chronic care coordinators in a patient-centered medical home has been shown to be cost-effective and can lower the overall cost of care despite the investment to hire them. Johns Hopkins Guided Care program demonstrated a 24% reduction in hospital days, 15% fewer emergency department visits, and a 37% reduction in days in a skilled nursing facility. The annual net Medicare savings was $75,000 per coordinator nurse hired.
ACCOUNTABLE CARE ORGANIZATIONS: A NEW SYSTEM OF HEALTH CARE DELIVERY
While the patient-centered medical home is designed to improve the coordination of care among physicians, ACOs have the broader goal of coordinating care across the entire continuum of health care, from physicians to hospitals to other clinicians. The concept of ACOs was spawned in 2006 by Elliott S. Fisher, MD, MPH, of the Dartmouth Institute for Health Policy and Clinical Practice. The idea is that, by improving care coordination within an ACO and reducing fragmented care, costs can be controlled and outcomes improved. Of course, the devil is in the details.
As part of its health care reform initiative, the state of Massachusetts’ Special Commission on the Health Care Payment System defined ACOs as health care delivery systems composed of hospitals, physicians, and other clinician and nonclinician providers that manage care across the entire spectrum of care. An ACO could be a real (incorporated) or virtual (contractually networked) organization, for example, a large physician organization that would contract with one or more hospitals and ancillary providers.11
In a 2009 report to Congress, the Medicare Payment Advisory Committee (MedPac) similarly defined ACOs for the Medicare population. But MedPac also introduced the concept of financial risk: providers in the ACO would share in efficiency gains from improved care coordination and could be subjected to financial penalties for poor performance, depending on the structure of the ACO.12
But what has placed ACOs at center stage is the new health care reform law, which encourages the formation of ACOs. On March 31, 2011, the Centers for Medicare and Medicaid Services published proposed rules to implement ACOs for Medicare patients (they appeared in the Federal Register on April 7, 2011).13,14 Comments on the 129-page proposed rules were due by June 6, 2011. Final rules are supposed to be published later this year.
The proposed new rule has a three-part aim:
- Better care for individuals, as described by all six dimensions of quality in the Institute of Medicine report “Crossing the Quality Chasm”15: safety, effectiveness, patient-centeredness, timeliness, efficiency, and equity
- Better health for populations, with respect to educating beneficiaries about the major causes of ill health—poor nutrition, physical inactivity, substance abuse, and poverty—as well as about the importance of preventive services such as an annual physical examination and annual influenza vaccination
- Lower growth in expenditures by eliminating waste and inefficiencies while not withholding any needed care that helps beneficiaries.
DETAILS OF THE PROPOSED ACO RULE
Here are some of the highlights of the proposed ACO rule.
Two shared-savings options
Although the program could start as soon as January 1, 2012, the application process is formidable, so this timeline may not be realistic. Moreover, a final rule is pending.
The proposed rule requires at least a 3-year contract, and primary care physicians must be included. Shared savings will be available and will depend on an ACO’s ability to manage costs and to achieve quality target performances. Two shared-savings options will be available: one with no risk until the third year and the other with risk during all 3 years but greater potential benefit. In the one-sided model with no risk until year 3, an ACO would begin to accrue shared savings at a rate of 50% after an initial 2% of savings compared with a risk-adjusted per capita benchmark based on performance during the previous 3 years. In the second plan, an ACO would immediately realize shared savings at a rate of 60% as long as savings were achieved compared with prior benchmark performance. However, in this second model, the ACO would be at risk to repay a share of all losses that were more than 2% higher than the benchmark expenditures, with loss caps of 5%, 7.5%, and 10% above benchmark in years 1, 2, and 3, respectively.
Structure of an ACO
Under the proposed rule, the minimum population size of Medicare beneficiaries is 5,000 patients, with some exceptions in rural or other shortage areas, or areas with critical access hospitals. ACO founders can be primary care physicians, primary care independent practice associations, or employee groups. Participants may include hospitals, critical access hospitals, specialists, and other providers. The ACO must be a legal entity with its own tax identification number and its own governance and management structure.
Concerns have been expressed that, in some markets, certain groups may come together and achieve market dominance with more than half of the population. Proposed ACOs with less than 30% of the market share will be exempt from antitrust concerns, and those with greater than 50% of market share will undergo detailed review.
Patient assignment
Patients will be assigned to an ACO retrospectively, at the end of the 3 years. The Centers for Medicare and Medicaid Services argues that retrospective assignment will encourage the ACO to design a system to help all patients, not just those assigned to the ACO.
Patients may not opt out of being counted against ACO performance measures. Although Medicare will share beneficiaries’ data with the ACO retrospectively so that it can learn more about costs per patient, patients may opt out of this data-sharing. Patients also retain unrestricted choice to see other providers, with attribution of costs incurred to the ACO.
Quality and reporting
The proposed rule has 65 equally weighted quality measures, many of which are not presently reported by most health care organizations. The measures fall within five broad categories: patient and caregiver experience, care coordination, patient safety, preventive health, and managing at-risk populations, including the frail elderly. Bonus payments for cost-savings will be adjusted based on meeting the quality measures.
Governance and management
Under the proposed rule, an ACO must meet stringent governance requirements. It must be a distinct legal entity as governed by state law. There must be proportional representation of all participants (eg, hospitals, community organizations, providers), comprising at least 75% of its Board of Trustees. These members must have authority to execute statutory functions of the ACO. Medicare beneficiaries and community stakeholder organizations must also be represented on the Board.
ACO operations must be managed by an executive director, manager, or general partner, who may or may not be a physician. A board-certified physician who is licensed in the state in which the ACO is domiciled must serve on location as the full-time, senior-level medical director, overseeing and managing clinical operations. A leadership team must be able to influence clinical practice, and a physician-directed process-improvement and quality-assurance committee is required.
Infrastructure and policies
The proposed rule outlines a number of infrastructure and policy requirements that must be addressed in the application process. These include:
- Written performance standards for quality and efficiency
- Evidence-based practice guidelines
- Tools to collect, evaluate, and share data to influence decision-making at the point of care
- Processes to identify and correct poor performance
- Description of how shared savings will be used to further improve care.

CONCERNS ABOUT THE PROPOSED NEW ACO RULE
While there is broad consensus in the health care community that the current system of care delivery fails to achieve the desired outcomes and is financially unsustainable and in need of reform, many concerns have been expressed about the proposed new ACO rule.
The regulations are too detailed. The regulations are highly prescriptive with detailed application, reporting, and regulatory requirements that create significant administrative burdens. Small medical groups are unlikely to have the administrative infrastructure to become involved.
Potential savings are inadequate. The shared savings concept has modest upside gain when modeled with holdback.16 Moreover, a recent analysis from the University Health System Consortium suggested that 50% of ACOs with 5,000 or more attributed lives would sustain unwarranted penalties as a result of random fluctuation of expenditures in the population.17
Participation involves a big investment. Participation requires significant resource investment, such as hiring chronic-disease managers and, in some practices, creating a whole new concept of managing wellness and continuity of care.
Retrospective beneficiary assignment is unpopular. Groups would generally prefer to know beforehand for whom they are responsible financially. A prospective assignment model was considered for the proposed rule but was ultimately rejected.
The patient assignment system is too risky. The plurality rule requires only a single visit with the ACO in order to be responsible for a patient for the entire year. In addition, the fact that the patient has the freedom to choose care elsewhere with expense assigned to the ACO confers significant financial risk.
There are too many quality measures. The high number of quality metrics—65—required to be measured and reported is onerous for most organizations.
Advertising is micromanaged. All marketing materials that are sent to patients about the ACO and any subsequent revisions must first be approved by Medicare, a potentially burdensome and time-consuming requirement.
Specialists are excluded. Using only generalists could actually be less cost-effective for some patients, such as those with human immunodeficiency virus, end-stage renal disease, certain malignancies, or advanced congestive heart failure.
Provider replacement is prohibited. Providers cannot be replaced over the 3 years of the demonstration, but the departing physician’s patients are still the responsibility of the plan. This would be especially problematic for small practices.
PREDICTING ACO READINESS
I believe there are five core competencies that are required to be an ACO:
- Operational excellence in care delivery
- Ability to deliver care across the continuum
- Cultural alignment among participating organizations
- Technical and informatics support to manage individual and population data
- Physician alignment around the concept of the ACO.
Certain strategies will increase the chances of success of an ACO:
Reduce emergency department usage and hospitalization. Cost-savings in patient-centered medical homes have been greatest by reducing hospitalizations, rehospitalizations, and emergency department visits.
Develop a high-quality, efficient primary care network. Have enough of a share in the primary care physician network to deliver effective primary care. Make sure there is good access to care and effective communication between patients and the primary care network. Deliver comprehensive services and have good care coordination. Aggressively manage communication, care coordination, and “hand-offs” across the care continuum and with specialists.
Create an effective patient-centered medical home. The current reimbursement climate fails to incentivize all of the necessary elements, which ultimately need to include chronic-care coordinators for medically complex patients, pharmacy support for patient medication management, adequate support staff to optimize efficiency, and a culture of wellness and necessary resources to support wellness.
PHYSICIANS NEED TO DRIVE SOLUTIONS
Soaring health care costs in the United States, poor quality outcomes, and increasing fragmentation of care are the major drivers of health care reform. The Patient Centered Medical Home is a key component to the solution and has already been shown to improve outcomes and lower costs. Further refinement of this concept and implementation should be priorities for primary care physicians and health care organizations.
The ACO concept attempts to further improve quality and lower costs. The proposed ACO rule released by the Centers for Medicare and Medicaid Services on March 31, 2011, has generated significant controversy in the health care community. In its current form, few health care systems are likely to participate. A revised rule is awaited in the coming months. In the meantime, the Centers for Medicare and Medicaid Services has released a request for application for a Pioneer ACO model, which offers up to 30 organizations the opportunity to participate in an ACO pilot that allows for prospective patient assignment and greater shared savings.
Whether ACOs as proposed achieve widespread implementation remains to be seen. However, the current system of health care delivery in this country is broken. Physicians and health care systems need to drive solutions to the challenges we face about quality, cost, access, care coordination, and outcomes.
- The Concord Coalition. Escalating Health Care Costs and the Federal Budget. April 2, 2009. http://www.concordcoalition.org/files/uploaded_for_nodes/docs/Iowa_Handout_final.pdf. Accessed August 8, 2011.
- The Henry J. Kaiser Family Foundation. Snapshots: Health Care Costs. Health Care Spending in the United States and OECD Countries. April 2011. http://www.kff.org/insurance/snapshot/OECD042111.cfm. Accessed August 8, 2011.
- Centers for Medicare and Medicaid Services. National health expenditure projections 2010–2020. http://www.cms.gov/NationalHealthExpendData/downloads/proj2010.pdf. Accessed August 8, 2011.
- The Commonwealth Fund. Performance snapshots, 2006. http://www.cmwf.org/snapshots. Accessed August 8, 2011.
- Fisher E, Goodman D, Skinner J, Bronner K. Health care spending, quality, and outcomes. More isn’t always better. The Dartmouth Atlas of Health Care. The Dartmouth Institute for Health Policy and Clinical Practice, 2009. http://www.dartmouthatlas.org/downloads/reports/Spending_Brief_022709.pdf. Accessed August 8, 2011.
- Goodman DC, Esty AR, Fisher ES, Chang C-H. Trends and variation in end-of-life care for Medicare beneficiaries with severe chronic illness. The Dartmouth Atlas of Health Care. The Dartmouth Institute for Health Policy and Clinical Practice, 2011. http://www.dartmouthatlas.org/downloads/reports/EOL_Trend_Report_0411.pdf. Accessed August 8, 2011.
- National Committee for Quality Assurance (NCQA). Leveraging health IT to achieve ambulatory quality: the patient-centered medical home (PCMH). www.ncqa.org/Portals/0/Public%20Policy/HIMSS_NCQA_PCMH_Factsheet.pdf. Accessed August 8, 2011.
- Bodenheimer T. Lessons from the trenches—a high-functioning primary care clinic. N Eng J Med 2011; 365:5–8.
- Gabbay RA, Bailit MH, Mauger DT, Wagner EH, Siminerio L. Multipayer patient-centered medical home implementation guided by the chronic care model. Jt Comm J Qual Patient Saf 2011; 37:265–273.
- Grumbach K, Grundy P. Outcomes of implementing Patient Centered Medical Home interventions: a review of the evidence from prospective evaluation studies in the United States. Patient-Centered Primary Care Collaborative. November 16, 2010. http://www.pcpcc.net/files/evidence_outcomes_in_pcmh.pdf. Accessed August 8, 2011.
- Kirwan LA, Iselin S. Recommendations of the Special Commission on the Health Care Payment System. Commonwealth of Massachusetts, July 16, 2009. http://www.mass.gov/Eeohhs2/docs/dhcfp/pc/Final_Report/Final_Report.pdf. Accessed August 8, 2011.
- Medicare Payment Advisory Commission. Report to the Congress. Improving incentives in the Medicare Program. http://www.medpac.gov/documents/jun09_entirereport.pdf. Accessed August 8, 2011.
- National Archives and Records Administration. Federal Register Volume 76, Number 67, Thursday, April 7, 2011. http://edocket.access.gpo.gov/2011/pdf/2011-7880.pdf. Accessed August 8, 2011.
- Berwick DM. Launching accountable care organizations—the proposed rule for the Medicare Shared Savings Program. N Engl J Med 2011; 364:e32.
- Institute of Medicine. Crossing the Quality Chasm. Washington, DC: National Academy Press; 2001.
- Fitch K, Mirkin D, Murphy-Barron C, Parke R, Pyenson B. A first look at ACOs’ risky business: quality is not enough. Seattle, WA: Millman, Inc; 2011. http://publications.milliman.com/publications/healthreform/pdfs/at-first-lookacos.pdf. Accessed August 10, 2011.
- University HealthSystem Consortium. Accountable care organizations: a measured view for academic medical centers. May 2011.
The US health care system cannot continue with “business as usual.” The current model is broken: it does not deliver the kind of care we want for our patients, ourselves, our families, and our communities. It is our role as professionals to help drive change and make medical care more cost-effective and of higher quality, with better satisfaction for patients as well as for providers.
Central to efforts to reform the system are two concepts. One is the “patient-centered medical home,” in which a single provider is responsible for coordinating care for individual patients. The other is “accountable care organizations,” a new way of organizing care along a continuum from doctor to hospital, mandated by the new health care reform law (technically known as the Patient Protection and Affordable Care Act).
CURRENT STATE OF HEALTH CARE: HIGH COST AND POOR QUALITY
Since health care reform was initially proposed in the 1990s, trends in the United States have grown steadily worse. Escalating health care costs have outstripped inflation, consuming an increasing percentage of the gross domestic product (GDP) at an unsustainable rate. Despite increased spending, quality outcomes are suboptimal. In addition, with the emergence of specialization and technology, care is increasingly fragmented and poorly coordinated, with multiple providers and poorly managed resources.
Over the last 15 years, the United States has far surpassed most countries in the developed world for total health care expenditures per capita.1,2 In 2009, we spent 17.4% of our GDP on health care, translating to $7,960 per capita, while Japan spent only 8.5% of its GDP, averaging $2,878 per capita.2 At the current rate, health care spending in the United States will increase from $2.5 trillion in 2009 to over $4.6 trillion in 2020.3
Paradoxically, costlier care is often of poorer quality. Many countries that spend far less per capita on health care achieve far better outcomes. Even within the United States, greater Medicare spending on a state and regional basis tends to correlate with poorer quality of care.4 Spending among Medicare beneficiaries is not standardized and varies widely throughout the country.5 The amount of care a patient receives also varies dramatically by region. The number of specialists involved in care during the last year of life is steadily increasing in many regions of the country, indicating poor care coordination.6
PATIENT-CENTERED MEDICAL HOMES: A POSITIVE TREND
The problems of high cost, poor quality, and poor coordination of care have led to the emergence of the concept of the patient-centered medical home. Originally proposed in 1967 by the American Academy of Pediatrics in response to the need for care coordination by a single physician, the idea did not really take root until the early 1990s. In 2002, the American Academy of Family Medicine embraced the concept and moved it forward.
According to the National Committee for Quality Assurance (NCQA), a nonprofit organization that provides voluntary certification for medical organizations, the patient-centered medical home is a model of care in which “patients have a direct relationship with a provider who coordinates a cooperative team of healthcare professionals, takes collective responsibility for the care provided to the patient, and arranges for appropriate care with other qualified providers as needed.”7
Patient-centered medical homes are supposed to improve quality outcomes and lower costs. In addition, they can compete for public or private incentives that reward this model of care and, as we will see later, are at the heart of ACO readiness.
Medical homes meet certification standards
NCQA first formally licensed patient-centered medical homes in 2008, based on nine standards and six key elements. A scoring system was used to rank the level of certification from level 1 (the lowest) to level 3. From 2008 to the end of 2010, the number of certified homes grew from 28 to 1,506. New York has the largest number of medical homes.
The new elements in the NCQA program align more closely with federal programs that are designed to drive quality, including the Centers for Medicare and Medicaid Services program to encourage the use of the electronic medical record, and with federal rule-making this last spring designed to implement accountable care organizations (ACOs).
Same-day access is now emphasized, as is managing patient populations—rather than just individual patients—with certain chronic diseases, such as diabetes and congestive heart failure. The requirements for tracking and coordinating care have profound implications about how resources are allocated. Ideally, coordinators of chronic disease management are embedded within practices to help manage high-risk patients, although the current reimbursement mechanism does not support this model. Population management may not be feasible for institutions that still rely on paper-based medical records.
Medical homes lower costs, improve quality
Integrated delivery system models such as patient-centered medical homes have demonstrated cost-savings while improving quality of care.8,9 Reducing hospital admissions and visits to the emergency department shows the greatest cost-savings in these models. Several projects have shown significant cost-savings10:
The Group Health Cooperative of Puget Sound reduced total costs by $10 per member per month (from $498 to $488, P = 0.76), with a 16% reduction in hospital admissions (P < .001) and a 29% reduction in emergency department visits (P < .001).
The Geisinger Health System Proven-Health Navigator in Pennsylvania reduced readmissions by 18% (P < .01). They also had a 7% reduction in total costs per member per month relative to a matched control group also in the Geisinger system but not in a medical home, although this difference did not reach statistical significance. Private payer demonstration projects of patient-centered medical homes have also shown cost-savings.
Blue Cross Blue Shield of South Carolina randomized patients to participate in either a patient-centered medical home or their standard system. The patient-centered medical home group had 36% fewer hospital days, 12.4% fewer emergency department visits, and a 6.5% reduction in total medical and pharmacy costs compared with controls.
Finally, the use of chronic care coordinators in a patient-centered medical home has been shown to be cost-effective and can lower the overall cost of care despite the investment to hire them. Johns Hopkins Guided Care program demonstrated a 24% reduction in hospital days, 15% fewer emergency department visits, and a 37% reduction in days in a skilled nursing facility. The annual net Medicare savings was $75,000 per coordinator nurse hired.
ACCOUNTABLE CARE ORGANIZATIONS: A NEW SYSTEM OF HEALTH CARE DELIVERY
While the patient-centered medical home is designed to improve the coordination of care among physicians, ACOs have the broader goal of coordinating care across the entire continuum of health care, from physicians to hospitals to other clinicians. The concept of ACOs was spawned in 2006 by Elliott S. Fisher, MD, MPH, of the Dartmouth Institute for Health Policy and Clinical Practice. The idea is that, by improving care coordination within an ACO and reducing fragmented care, costs can be controlled and outcomes improved. Of course, the devil is in the details.
As part of its health care reform initiative, the state of Massachusetts’ Special Commission on the Health Care Payment System defined ACOs as health care delivery systems composed of hospitals, physicians, and other clinician and nonclinician providers that manage care across the entire spectrum of care. An ACO could be a real (incorporated) or virtual (contractually networked) organization, for example, a large physician organization that would contract with one or more hospitals and ancillary providers.11
In a 2009 report to Congress, the Medicare Payment Advisory Committee (MedPac) similarly defined ACOs for the Medicare population. But MedPac also introduced the concept of financial risk: providers in the ACO would share in efficiency gains from improved care coordination and could be subjected to financial penalties for poor performance, depending on the structure of the ACO.12
But what has placed ACOs at center stage is the new health care reform law, which encourages the formation of ACOs. On March 31, 2011, the Centers for Medicare and Medicaid Services published proposed rules to implement ACOs for Medicare patients (they appeared in the Federal Register on April 7, 2011).13,14 Comments on the 129-page proposed rules were due by June 6, 2011. Final rules are supposed to be published later this year.
The proposed new rule has a three-part aim:
- Better care for individuals, as described by all six dimensions of quality in the Institute of Medicine report “Crossing the Quality Chasm”15: safety, effectiveness, patient-centeredness, timeliness, efficiency, and equity
- Better health for populations, with respect to educating beneficiaries about the major causes of ill health—poor nutrition, physical inactivity, substance abuse, and poverty—as well as about the importance of preventive services such as an annual physical examination and annual influenza vaccination
- Lower growth in expenditures by eliminating waste and inefficiencies while not withholding any needed care that helps beneficiaries.
DETAILS OF THE PROPOSED ACO RULE
Here are some of the highlights of the proposed ACO rule.
Two shared-savings options
Although the program could start as soon as January 1, 2012, the application process is formidable, so this timeline may not be realistic. Moreover, a final rule is pending.
The proposed rule requires at least a 3-year contract, and primary care physicians must be included. Shared savings will be available and will depend on an ACO’s ability to manage costs and to achieve quality target performances. Two shared-savings options will be available: one with no risk until the third year and the other with risk during all 3 years but greater potential benefit. In the one-sided model with no risk until year 3, an ACO would begin to accrue shared savings at a rate of 50% after an initial 2% of savings compared with a risk-adjusted per capita benchmark based on performance during the previous 3 years. In the second plan, an ACO would immediately realize shared savings at a rate of 60% as long as savings were achieved compared with prior benchmark performance. However, in this second model, the ACO would be at risk to repay a share of all losses that were more than 2% higher than the benchmark expenditures, with loss caps of 5%, 7.5%, and 10% above benchmark in years 1, 2, and 3, respectively.
Structure of an ACO
Under the proposed rule, the minimum population size of Medicare beneficiaries is 5,000 patients, with some exceptions in rural or other shortage areas, or areas with critical access hospitals. ACO founders can be primary care physicians, primary care independent practice associations, or employee groups. Participants may include hospitals, critical access hospitals, specialists, and other providers. The ACO must be a legal entity with its own tax identification number and its own governance and management structure.
Concerns have been expressed that, in some markets, certain groups may come together and achieve market dominance with more than half of the population. Proposed ACOs with less than 30% of the market share will be exempt from antitrust concerns, and those with greater than 50% of market share will undergo detailed review.
Patient assignment
Patients will be assigned to an ACO retrospectively, at the end of the 3 years. The Centers for Medicare and Medicaid Services argues that retrospective assignment will encourage the ACO to design a system to help all patients, not just those assigned to the ACO.
Patients may not opt out of being counted against ACO performance measures. Although Medicare will share beneficiaries’ data with the ACO retrospectively so that it can learn more about costs per patient, patients may opt out of this data-sharing. Patients also retain unrestricted choice to see other providers, with attribution of costs incurred to the ACO.
Quality and reporting
The proposed rule has 65 equally weighted quality measures, many of which are not presently reported by most health care organizations. The measures fall within five broad categories: patient and caregiver experience, care coordination, patient safety, preventive health, and managing at-risk populations, including the frail elderly. Bonus payments for cost-savings will be adjusted based on meeting the quality measures.
Governance and management
Under the proposed rule, an ACO must meet stringent governance requirements. It must be a distinct legal entity as governed by state law. There must be proportional representation of all participants (eg, hospitals, community organizations, providers), comprising at least 75% of its Board of Trustees. These members must have authority to execute statutory functions of the ACO. Medicare beneficiaries and community stakeholder organizations must also be represented on the Board.
ACO operations must be managed by an executive director, manager, or general partner, who may or may not be a physician. A board-certified physician who is licensed in the state in which the ACO is domiciled must serve on location as the full-time, senior-level medical director, overseeing and managing clinical operations. A leadership team must be able to influence clinical practice, and a physician-directed process-improvement and quality-assurance committee is required.
Infrastructure and policies
The proposed rule outlines a number of infrastructure and policy requirements that must be addressed in the application process. These include:
- Written performance standards for quality and efficiency
- Evidence-based practice guidelines
- Tools to collect, evaluate, and share data to influence decision-making at the point of care
- Processes to identify and correct poor performance
- Description of how shared savings will be used to further improve care.
CONCERNS ABOUT THE PROPOSED NEW ACO RULE
While there is broad consensus in the health care community that the current system of care delivery fails to achieve the desired outcomes and is financially unsustainable and in need of reform, many concerns have been expressed about the proposed new ACO rule.
The regulations are too detailed. The regulations are highly prescriptive with detailed application, reporting, and regulatory requirements that create significant administrative burdens. Small medical groups are unlikely to have the administrative infrastructure to become involved.
Potential savings are inadequate. The shared savings concept has modest upside gain when modeled with holdback.16 Moreover, a recent analysis from the University Health System Consortium suggested that 50% of ACOs with 5,000 or more attributed lives would sustain unwarranted penalties as a result of random fluctuation of expenditures in the population.17
Participation involves a big investment. Participation requires significant resource investment, such as hiring chronic-disease managers and, in some practices, creating a whole new concept of managing wellness and continuity of care.
Retrospective beneficiary assignment is unpopular. Groups would generally prefer to know beforehand for whom they are responsible financially. A prospective assignment model was considered for the proposed rule but was ultimately rejected.
The patient assignment system is too risky. The plurality rule requires only a single visit with the ACO in order to be responsible for a patient for the entire year. In addition, the fact that the patient has the freedom to choose care elsewhere with expense assigned to the ACO confers significant financial risk.
There are too many quality measures. The high number of quality metrics—65—required to be measured and reported is onerous for most organizations.
Advertising is micromanaged. All marketing materials that are sent to patients about the ACO and any subsequent revisions must first be approved by Medicare, a potentially burdensome and time-consuming requirement.
Specialists are excluded. Using only generalists could actually be less cost-effective for some patients, such as those with human immunodeficiency virus, end-stage renal disease, certain malignancies, or advanced congestive heart failure.
Provider replacement is prohibited. Providers cannot be replaced over the 3 years of the demonstration, but the departing physician’s patients are still the responsibility of the plan. This would be especially problematic for small practices.
PREDICTING ACO READINESS
I believe there are five core competencies that are required to be an ACO:
- Operational excellence in care delivery
- Ability to deliver care across the continuum
- Cultural alignment among participating organizations
- Technical and informatics support to manage individual and population data
- Physician alignment around the concept of the ACO.
Certain strategies will increase the chances of success of an ACO:
Reduce emergency department usage and hospitalization. Cost-savings in patient-centered medical homes have been greatest by reducing hospitalizations, rehospitalizations, and emergency department visits.
Develop a high-quality, efficient primary care network. Have enough of a share in the primary care physician network to deliver effective primary care. Make sure there is good access to care and effective communication between patients and the primary care network. Deliver comprehensive services and have good care coordination. Aggressively manage communication, care coordination, and “hand-offs” across the care continuum and with specialists.
Create an effective patient-centered medical home. The current reimbursement climate fails to incentivize all of the necessary elements, which ultimately need to include chronic-care coordinators for medically complex patients, pharmacy support for patient medication management, adequate support staff to optimize efficiency, and a culture of wellness and necessary resources to support wellness.
PHYSICIANS NEED TO DRIVE SOLUTIONS
Soaring health care costs in the United States, poor quality outcomes, and increasing fragmentation of care are the major drivers of health care reform. The Patient Centered Medical Home is a key component to the solution and has already been shown to improve outcomes and lower costs. Further refinement of this concept and implementation should be priorities for primary care physicians and health care organizations.
The ACO concept attempts to further improve quality and lower costs. The proposed ACO rule released by the Centers for Medicare and Medicaid Services on March 31, 2011, has generated significant controversy in the health care community. In its current form, few health care systems are likely to participate. A revised rule is awaited in the coming months. In the meantime, the Centers for Medicare and Medicaid Services has released a request for application for a Pioneer ACO model, which offers up to 30 organizations the opportunity to participate in an ACO pilot that allows for prospective patient assignment and greater shared savings.
Whether ACOs as proposed achieve widespread implementation remains to be seen. However, the current system of health care delivery in this country is broken. Physicians and health care systems need to drive solutions to the challenges we face about quality, cost, access, care coordination, and outcomes.
The US health care system cannot continue with “business as usual.” The current model is broken: it does not deliver the kind of care we want for our patients, ourselves, our families, and our communities. It is our role as professionals to help drive change and make medical care more cost-effective and of higher quality, with better satisfaction for patients as well as for providers.
Central to efforts to reform the system are two concepts. One is the “patient-centered medical home,” in which a single provider is responsible for coordinating care for individual patients. The other is “accountable care organizations,” a new way of organizing care along a continuum from doctor to hospital, mandated by the new health care reform law (technically known as the Patient Protection and Affordable Care Act).
CURRENT STATE OF HEALTH CARE: HIGH COST AND POOR QUALITY
Since health care reform was initially proposed in the 1990s, trends in the United States have grown steadily worse. Escalating health care costs have outstripped inflation, consuming an increasing percentage of the gross domestic product (GDP) at an unsustainable rate. Despite increased spending, quality outcomes are suboptimal. In addition, with the emergence of specialization and technology, care is increasingly fragmented and poorly coordinated, with multiple providers and poorly managed resources.
Over the last 15 years, the United States has far surpassed most countries in the developed world for total health care expenditures per capita.1,2 In 2009, we spent 17.4% of our GDP on health care, translating to $7,960 per capita, while Japan spent only 8.5% of its GDP, averaging $2,878 per capita.2 At the current rate, health care spending in the United States will increase from $2.5 trillion in 2009 to over $4.6 trillion in 2020.3
Paradoxically, costlier care is often of poorer quality. Many countries that spend far less per capita on health care achieve far better outcomes. Even within the United States, greater Medicare spending on a state and regional basis tends to correlate with poorer quality of care.4 Spending among Medicare beneficiaries is not standardized and varies widely throughout the country.5 The amount of care a patient receives also varies dramatically by region. The number of specialists involved in care during the last year of life is steadily increasing in many regions of the country, indicating poor care coordination.6
PATIENT-CENTERED MEDICAL HOMES: A POSITIVE TREND
The problems of high cost, poor quality, and poor coordination of care have led to the emergence of the concept of the patient-centered medical home. Originally proposed in 1967 by the American Academy of Pediatrics in response to the need for care coordination by a single physician, the idea did not really take root until the early 1990s. In 2002, the American Academy of Family Medicine embraced the concept and moved it forward.
According to the National Committee for Quality Assurance (NCQA), a nonprofit organization that provides voluntary certification for medical organizations, the patient-centered medical home is a model of care in which “patients have a direct relationship with a provider who coordinates a cooperative team of healthcare professionals, takes collective responsibility for the care provided to the patient, and arranges for appropriate care with other qualified providers as needed.”7
Patient-centered medical homes are supposed to improve quality outcomes and lower costs. In addition, they can compete for public or private incentives that reward this model of care and, as we will see later, are at the heart of ACO readiness.
Medical homes meet certification standards
NCQA first formally licensed patient-centered medical homes in 2008, based on nine standards and six key elements. A scoring system was used to rank the level of certification from level 1 (the lowest) to level 3. From 2008 to the end of 2010, the number of certified homes grew from 28 to 1,506. New York has the largest number of medical homes.
The new elements in the NCQA program align more closely with federal programs that are designed to drive quality, including the Centers for Medicare and Medicaid Services program to encourage the use of the electronic medical record, and with federal rule-making this last spring designed to implement accountable care organizations (ACOs).
Same-day access is now emphasized, as is managing patient populations—rather than just individual patients—with certain chronic diseases, such as diabetes and congestive heart failure. The requirements for tracking and coordinating care have profound implications about how resources are allocated. Ideally, coordinators of chronic disease management are embedded within practices to help manage high-risk patients, although the current reimbursement mechanism does not support this model. Population management may not be feasible for institutions that still rely on paper-based medical records.
Medical homes lower costs, improve quality
Integrated delivery system models such as patient-centered medical homes have demonstrated cost-savings while improving quality of care.8,9 Reducing hospital admissions and visits to the emergency department shows the greatest cost-savings in these models. Several projects have shown significant cost-savings10:
The Group Health Cooperative of Puget Sound reduced total costs by $10 per member per month (from $498 to $488, P = 0.76), with a 16% reduction in hospital admissions (P < .001) and a 29% reduction in emergency department visits (P < .001).
The Geisinger Health System Proven-Health Navigator in Pennsylvania reduced readmissions by 18% (P < .01). They also had a 7% reduction in total costs per member per month relative to a matched control group also in the Geisinger system but not in a medical home, although this difference did not reach statistical significance. Private payer demonstration projects of patient-centered medical homes have also shown cost-savings.
Blue Cross Blue Shield of South Carolina randomized patients to participate in either a patient-centered medical home or their standard system. The patient-centered medical home group had 36% fewer hospital days, 12.4% fewer emergency department visits, and a 6.5% reduction in total medical and pharmacy costs compared with controls.
Finally, the use of chronic care coordinators in a patient-centered medical home has been shown to be cost-effective and can lower the overall cost of care despite the investment to hire them. Johns Hopkins Guided Care program demonstrated a 24% reduction in hospital days, 15% fewer emergency department visits, and a 37% reduction in days in a skilled nursing facility. The annual net Medicare savings was $75,000 per coordinator nurse hired.
ACCOUNTABLE CARE ORGANIZATIONS: A NEW SYSTEM OF HEALTH CARE DELIVERY
While the patient-centered medical home is designed to improve the coordination of care among physicians, ACOs have the broader goal of coordinating care across the entire continuum of health care, from physicians to hospitals to other clinicians. The concept of ACOs was spawned in 2006 by Elliott S. Fisher, MD, MPH, of the Dartmouth Institute for Health Policy and Clinical Practice. The idea is that, by improving care coordination within an ACO and reducing fragmented care, costs can be controlled and outcomes improved. Of course, the devil is in the details.
As part of its health care reform initiative, the state of Massachusetts’ Special Commission on the Health Care Payment System defined ACOs as health care delivery systems composed of hospitals, physicians, and other clinician and nonclinician providers that manage care across the entire spectrum of care. An ACO could be a real (incorporated) or virtual (contractually networked) organization, for example, a large physician organization that would contract with one or more hospitals and ancillary providers.11
In a 2009 report to Congress, the Medicare Payment Advisory Committee (MedPac) similarly defined ACOs for the Medicare population. But MedPac also introduced the concept of financial risk: providers in the ACO would share in efficiency gains from improved care coordination and could be subjected to financial penalties for poor performance, depending on the structure of the ACO.12
But what has placed ACOs at center stage is the new health care reform law, which encourages the formation of ACOs. On March 31, 2011, the Centers for Medicare and Medicaid Services published proposed rules to implement ACOs for Medicare patients (they appeared in the Federal Register on April 7, 2011).13,14 Comments on the 129-page proposed rules were due by June 6, 2011. Final rules are supposed to be published later this year.
The proposed new rule has a three-part aim:
- Better care for individuals, as described by all six dimensions of quality in the Institute of Medicine report “Crossing the Quality Chasm”15: safety, effectiveness, patient-centeredness, timeliness, efficiency, and equity
- Better health for populations, with respect to educating beneficiaries about the major causes of ill health—poor nutrition, physical inactivity, substance abuse, and poverty—as well as about the importance of preventive services such as an annual physical examination and annual influenza vaccination
- Lower growth in expenditures by eliminating waste and inefficiencies while not withholding any needed care that helps beneficiaries.
DETAILS OF THE PROPOSED ACO RULE
Here are some of the highlights of the proposed ACO rule.
Two shared-savings options
Although the program could start as soon as January 1, 2012, the application process is formidable, so this timeline may not be realistic. Moreover, a final rule is pending.
The proposed rule requires at least a 3-year contract, and primary care physicians must be included. Shared savings will be available and will depend on an ACO’s ability to manage costs and to achieve quality target performances. Two shared-savings options will be available: one with no risk until the third year and the other with risk during all 3 years but greater potential benefit. In the one-sided model with no risk until year 3, an ACO would begin to accrue shared savings at a rate of 50% after an initial 2% of savings compared with a risk-adjusted per capita benchmark based on performance during the previous 3 years. In the second plan, an ACO would immediately realize shared savings at a rate of 60% as long as savings were achieved compared with prior benchmark performance. However, in this second model, the ACO would be at risk to repay a share of all losses that were more than 2% higher than the benchmark expenditures, with loss caps of 5%, 7.5%, and 10% above benchmark in years 1, 2, and 3, respectively.
Structure of an ACO
Under the proposed rule, the minimum population size of Medicare beneficiaries is 5,000 patients, with some exceptions in rural or other shortage areas, or areas with critical access hospitals. ACO founders can be primary care physicians, primary care independent practice associations, or employee groups. Participants may include hospitals, critical access hospitals, specialists, and other providers. The ACO must be a legal entity with its own tax identification number and its own governance and management structure.
Concerns have been expressed that, in some markets, certain groups may come together and achieve market dominance with more than half of the population. Proposed ACOs with less than 30% of the market share will be exempt from antitrust concerns, and those with greater than 50% of market share will undergo detailed review.
Patient assignment
Patients will be assigned to an ACO retrospectively, at the end of the 3 years. The Centers for Medicare and Medicaid Services argues that retrospective assignment will encourage the ACO to design a system to help all patients, not just those assigned to the ACO.
Patients may not opt out of being counted against ACO performance measures. Although Medicare will share beneficiaries’ data with the ACO retrospectively so that it can learn more about costs per patient, patients may opt out of this data-sharing. Patients also retain unrestricted choice to see other providers, with attribution of costs incurred to the ACO.
Quality and reporting
The proposed rule has 65 equally weighted quality measures, many of which are not presently reported by most health care organizations. The measures fall within five broad categories: patient and caregiver experience, care coordination, patient safety, preventive health, and managing at-risk populations, including the frail elderly. Bonus payments for cost-savings will be adjusted based on meeting the quality measures.
Governance and management
Under the proposed rule, an ACO must meet stringent governance requirements. It must be a distinct legal entity as governed by state law. There must be proportional representation of all participants (eg, hospitals, community organizations, providers), comprising at least 75% of its Board of Trustees. These members must have authority to execute statutory functions of the ACO. Medicare beneficiaries and community stakeholder organizations must also be represented on the Board.
ACO operations must be managed by an executive director, manager, or general partner, who may or may not be a physician. A board-certified physician who is licensed in the state in which the ACO is domiciled must serve on location as the full-time, senior-level medical director, overseeing and managing clinical operations. A leadership team must be able to influence clinical practice, and a physician-directed process-improvement and quality-assurance committee is required.
Infrastructure and policies
The proposed rule outlines a number of infrastructure and policy requirements that must be addressed in the application process. These include:
- Written performance standards for quality and efficiency
- Evidence-based practice guidelines
- Tools to collect, evaluate, and share data to influence decision-making at the point of care
- Processes to identify and correct poor performance
- Description of how shared savings will be used to further improve care.
CONCERNS ABOUT THE PROPOSED NEW ACO RULE
While there is broad consensus in the health care community that the current system of care delivery fails to achieve the desired outcomes and is financially unsustainable and in need of reform, many concerns have been expressed about the proposed new ACO rule.
The regulations are too detailed. The regulations are highly prescriptive with detailed application, reporting, and regulatory requirements that create significant administrative burdens. Small medical groups are unlikely to have the administrative infrastructure to become involved.
Potential savings are inadequate. The shared savings concept has modest upside gain when modeled with holdback.16 Moreover, a recent analysis from the University Health System Consortium suggested that 50% of ACOs with 5,000 or more attributed lives would sustain unwarranted penalties as a result of random fluctuation of expenditures in the population.17
Participation involves a big investment. Participation requires significant resource investment, such as hiring chronic-disease managers and, in some practices, creating a whole new concept of managing wellness and continuity of care.
Retrospective beneficiary assignment is unpopular. Groups would generally prefer to know beforehand for whom they are responsible financially. A prospective assignment model was considered for the proposed rule but was ultimately rejected.
The patient assignment system is too risky. The plurality rule requires only a single visit with the ACO in order to be responsible for a patient for the entire year. In addition, the fact that the patient has the freedom to choose care elsewhere with expense assigned to the ACO confers significant financial risk.
There are too many quality measures. The high number of quality metrics—65—required to be measured and reported is onerous for most organizations.
Advertising is micromanaged. All marketing materials that are sent to patients about the ACO and any subsequent revisions must first be approved by Medicare, a potentially burdensome and time-consuming requirement.
Specialists are excluded. Using only generalists could actually be less cost-effective for some patients, such as those with human immunodeficiency virus, end-stage renal disease, certain malignancies, or advanced congestive heart failure.
Provider replacement is prohibited. Providers cannot be replaced over the 3 years of the demonstration, but the departing physician’s patients are still the responsibility of the plan. This would be especially problematic for small practices.
PREDICTING ACO READINESS
I believe there are five core competencies that are required to be an ACO:
- Operational excellence in care delivery
- Ability to deliver care across the continuum
- Cultural alignment among participating organizations
- Technical and informatics support to manage individual and population data
- Physician alignment around the concept of the ACO.
Certain strategies will increase the chances of success of an ACO:
Reduce emergency department usage and hospitalization. Cost-savings in patient-centered medical homes have been greatest by reducing hospitalizations, rehospitalizations, and emergency department visits.
Develop a high-quality, efficient primary care network. Have enough of a share in the primary care physician network to deliver effective primary care. Make sure there is good access to care and effective communication between patients and the primary care network. Deliver comprehensive services and have good care coordination. Aggressively manage communication, care coordination, and “hand-offs” across the care continuum and with specialists.
Create an effective patient-centered medical home. The current reimbursement climate fails to incentivize all of the necessary elements, which ultimately need to include chronic-care coordinators for medically complex patients, pharmacy support for patient medication management, adequate support staff to optimize efficiency, and a culture of wellness and necessary resources to support wellness.
PHYSICIANS NEED TO DRIVE SOLUTIONS
Soaring health care costs in the United States, poor quality outcomes, and increasing fragmentation of care are the major drivers of health care reform. The Patient Centered Medical Home is a key component to the solution and has already been shown to improve outcomes and lower costs. Further refinement of this concept and implementation should be priorities for primary care physicians and health care organizations.
The ACO concept attempts to further improve quality and lower costs. The proposed ACO rule released by the Centers for Medicare and Medicaid Services on March 31, 2011, has generated significant controversy in the health care community. In its current form, few health care systems are likely to participate. A revised rule is awaited in the coming months. In the meantime, the Centers for Medicare and Medicaid Services has released a request for application for a Pioneer ACO model, which offers up to 30 organizations the opportunity to participate in an ACO pilot that allows for prospective patient assignment and greater shared savings.
Whether ACOs as proposed achieve widespread implementation remains to be seen. However, the current system of health care delivery in this country is broken. Physicians and health care systems need to drive solutions to the challenges we face about quality, cost, access, care coordination, and outcomes.
- The Concord Coalition. Escalating Health Care Costs and the Federal Budget. April 2, 2009. http://www.concordcoalition.org/files/uploaded_for_nodes/docs/Iowa_Handout_final.pdf. Accessed August 8, 2011.
- The Henry J. Kaiser Family Foundation. Snapshots: Health Care Costs. Health Care Spending in the United States and OECD Countries. April 2011. http://www.kff.org/insurance/snapshot/OECD042111.cfm. Accessed August 8, 2011.
- Centers for Medicare and Medicaid Services. National health expenditure projections 2010–2020. http://www.cms.gov/NationalHealthExpendData/downloads/proj2010.pdf. Accessed August 8, 2011.
- The Commonwealth Fund. Performance snapshots, 2006. http://www.cmwf.org/snapshots. Accessed August 8, 2011.
- Fisher E, Goodman D, Skinner J, Bronner K. Health care spending, quality, and outcomes. More isn’t always better. The Dartmouth Atlas of Health Care. The Dartmouth Institute for Health Policy and Clinical Practice, 2009. http://www.dartmouthatlas.org/downloads/reports/Spending_Brief_022709.pdf. Accessed August 8, 2011.
- Goodman DC, Esty AR, Fisher ES, Chang C-H. Trends and variation in end-of-life care for Medicare beneficiaries with severe chronic illness. The Dartmouth Atlas of Health Care. The Dartmouth Institute for Health Policy and Clinical Practice, 2011. http://www.dartmouthatlas.org/downloads/reports/EOL_Trend_Report_0411.pdf. Accessed August 8, 2011.
- National Committee for Quality Assurance (NCQA). Leveraging health IT to achieve ambulatory quality: the patient-centered medical home (PCMH). www.ncqa.org/Portals/0/Public%20Policy/HIMSS_NCQA_PCMH_Factsheet.pdf. Accessed August 8, 2011.
- Bodenheimer T. Lessons from the trenches—a high-functioning primary care clinic. N Eng J Med 2011; 365:5–8.
- Gabbay RA, Bailit MH, Mauger DT, Wagner EH, Siminerio L. Multipayer patient-centered medical home implementation guided by the chronic care model. Jt Comm J Qual Patient Saf 2011; 37:265–273.
- Grumbach K, Grundy P. Outcomes of implementing Patient Centered Medical Home interventions: a review of the evidence from prospective evaluation studies in the United States. Patient-Centered Primary Care Collaborative. November 16, 2010. http://www.pcpcc.net/files/evidence_outcomes_in_pcmh.pdf. Accessed August 8, 2011.
- Kirwan LA, Iselin S. Recommendations of the Special Commission on the Health Care Payment System. Commonwealth of Massachusetts, July 16, 2009. http://www.mass.gov/Eeohhs2/docs/dhcfp/pc/Final_Report/Final_Report.pdf. Accessed August 8, 2011.
- Medicare Payment Advisory Commission. Report to the Congress. Improving incentives in the Medicare Program. http://www.medpac.gov/documents/jun09_entirereport.pdf. Accessed August 8, 2011.
- National Archives and Records Administration. Federal Register Volume 76, Number 67, Thursday, April 7, 2011. http://edocket.access.gpo.gov/2011/pdf/2011-7880.pdf. Accessed August 8, 2011.
- Berwick DM. Launching accountable care organizations—the proposed rule for the Medicare Shared Savings Program. N Engl J Med 2011; 364:e32.
- Institute of Medicine. Crossing the Quality Chasm. Washington, DC: National Academy Press; 2001.
- Fitch K, Mirkin D, Murphy-Barron C, Parke R, Pyenson B. A first look at ACOs’ risky business: quality is not enough. Seattle, WA: Millman, Inc; 2011. http://publications.milliman.com/publications/healthreform/pdfs/at-first-lookacos.pdf. Accessed August 10, 2011.
- University HealthSystem Consortium. Accountable care organizations: a measured view for academic medical centers. May 2011.
- The Concord Coalition. Escalating Health Care Costs and the Federal Budget. April 2, 2009. http://www.concordcoalition.org/files/uploaded_for_nodes/docs/Iowa_Handout_final.pdf. Accessed August 8, 2011.
- The Henry J. Kaiser Family Foundation. Snapshots: Health Care Costs. Health Care Spending in the United States and OECD Countries. April 2011. http://www.kff.org/insurance/snapshot/OECD042111.cfm. Accessed August 8, 2011.
- Centers for Medicare and Medicaid Services. National health expenditure projections 2010–2020. http://www.cms.gov/NationalHealthExpendData/downloads/proj2010.pdf. Accessed August 8, 2011.
- The Commonwealth Fund. Performance snapshots, 2006. http://www.cmwf.org/snapshots. Accessed August 8, 2011.
- Fisher E, Goodman D, Skinner J, Bronner K. Health care spending, quality, and outcomes. More isn’t always better. The Dartmouth Atlas of Health Care. The Dartmouth Institute for Health Policy and Clinical Practice, 2009. http://www.dartmouthatlas.org/downloads/reports/Spending_Brief_022709.pdf. Accessed August 8, 2011.
- Goodman DC, Esty AR, Fisher ES, Chang C-H. Trends and variation in end-of-life care for Medicare beneficiaries with severe chronic illness. The Dartmouth Atlas of Health Care. The Dartmouth Institute for Health Policy and Clinical Practice, 2011. http://www.dartmouthatlas.org/downloads/reports/EOL_Trend_Report_0411.pdf. Accessed August 8, 2011.
- National Committee for Quality Assurance (NCQA). Leveraging health IT to achieve ambulatory quality: the patient-centered medical home (PCMH). www.ncqa.org/Portals/0/Public%20Policy/HIMSS_NCQA_PCMH_Factsheet.pdf. Accessed August 8, 2011.
- Bodenheimer T. Lessons from the trenches—a high-functioning primary care clinic. N Eng J Med 2011; 365:5–8.
- Gabbay RA, Bailit MH, Mauger DT, Wagner EH, Siminerio L. Multipayer patient-centered medical home implementation guided by the chronic care model. Jt Comm J Qual Patient Saf 2011; 37:265–273.
- Grumbach K, Grundy P. Outcomes of implementing Patient Centered Medical Home interventions: a review of the evidence from prospective evaluation studies in the United States. Patient-Centered Primary Care Collaborative. November 16, 2010. http://www.pcpcc.net/files/evidence_outcomes_in_pcmh.pdf. Accessed August 8, 2011.
- Kirwan LA, Iselin S. Recommendations of the Special Commission on the Health Care Payment System. Commonwealth of Massachusetts, July 16, 2009. http://www.mass.gov/Eeohhs2/docs/dhcfp/pc/Final_Report/Final_Report.pdf. Accessed August 8, 2011.
- Medicare Payment Advisory Commission. Report to the Congress. Improving incentives in the Medicare Program. http://www.medpac.gov/documents/jun09_entirereport.pdf. Accessed August 8, 2011.
- National Archives and Records Administration. Federal Register Volume 76, Number 67, Thursday, April 7, 2011. http://edocket.access.gpo.gov/2011/pdf/2011-7880.pdf. Accessed August 8, 2011.
- Berwick DM. Launching accountable care organizations—the proposed rule for the Medicare Shared Savings Program. N Engl J Med 2011; 364:e32.
- Institute of Medicine. Crossing the Quality Chasm. Washington, DC: National Academy Press; 2001.
- Fitch K, Mirkin D, Murphy-Barron C, Parke R, Pyenson B. A first look at ACOs’ risky business: quality is not enough. Seattle, WA: Millman, Inc; 2011. http://publications.milliman.com/publications/healthreform/pdfs/at-first-lookacos.pdf. Accessed August 10, 2011.
- University HealthSystem Consortium. Accountable care organizations: a measured view for academic medical centers. May 2011.
KEY POINTS
- Compared with other developed countries, health care in the United States is among the costliest and has poor quality measures.
- The patient-centered medical home is an increasingly popular model that emphasizes continuous coordinated patient care. It has been shown to lower costs while improving health care outcomes.
- Patient-centered medical homes are at the heart of ACOs, which establish a team approach to health care delivery systems that includes doctors and hospitals.
- Applications are now being accepted for participation in the Centers for Medicare and Medicaid Services’ ACO Proposed Rule. The 3-year minimum contract specifies numerous details regarding structure, governance, and management, and may or may not involve risk—as well as savings—according to the plan chosen.
Update in hospital medicine: Studies likely to affect inpatient practice in 2011
A number of studies published in the last few years will likely affect the way we practice medicine in the hospital. Here, we will use a hypothetical case scenario to focus on the issues of anticoagulants, patient safety, quality improvement, critical care, transitions of care, and perioperative medicine.
AN ELDERLY MAN WITH NEW-ONSET ATRIAL FIBRILLATION
P.G. is an 80-year-old man with a history of hypertension and type 2 diabetes mellitus who is admitted with new-onset atrial fibrillation. In the hospital, his heart rate is brought under control with intravenous metoprolol (Lopressor). On discharge, he will be followed by his primary care physician (PCP). He does not have access to an anticoagulation clinic.
1. What are this patient’s options for stroke prevention?
- Aspirin 81 mg daily and clopidogrel (Plavix) 75 mg daily
- Warfarin (Coumadin) with a target international normalized ratio (INR) of 2.0 to 3.0
- Aspirin mg daily by itself
- Dabigatran (Pradaxa) 150 mg daily
A new oral anticoagulant agent
In deciding what type of anticoagulation to give to a patient with atrial fibrillation, it is useful to look at the CHADS2 score (1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack. This patient has a CHADS2 score of 3, indicating that he should receive warfarin. An alternative is dabigatran, the first new anticoagulant agent in more than 50 years.
In a multicenter, international trial, Connolly et al1 randomized 18,113 patients (mean age 71, 64% men) to receive dabigatran 110 mg twice daily, dabigatran 150 mg twice daily, or warfarin with a target INR of 2.0 to 3.0. In this noninferiority trial, dabigatran was given in a blinded manner, but the use of warfarin was open-label. Patients were eligible if they had atrial fibrillation at screening or within the previous 6 months and were at risk of stroke—ie, if they had at least one of the following: a history of stroke or transient ischemic attack, a left ventricular ejection fraction of less than 40%, symptoms of congestive heart failure (New York Heart Association class II or higher), and an age of 75 or older or an age of 65 to 74 with diabetes mellitus, hypertension, or coronary artery disease.
At a mean follow-up of 2 years, the rate of stroke or systolic embolism was 1.69% per year in the warfarin group compared with 1.1% in the higher-dose dabigatran group (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rates of major hemorrhage were similar between these two groups. Comparing lower-dose dabigatran and warfarin, the rates of stroke or systolic embolism were not significantly different, but the rate of major bleeding was significantly lower with lower-dose dabigatran.
In a trial in patients with acute venous thromboembolism, Schulman et al2 found that dabigatran was not inferior to warfarin in preventing venous thromboembolism.
Guidelines from the American College of Cardiology Foundation and the American Heart Association now endorse dabigatran as an alternative to warfarin for patients with atrial fibrillation.3 However, the guidelines state that it should be reserved for those patients who:
- Do not have a prosthetic heart valve or hemodynamically significant valve disease
- Have good kidney function (dabigatran is cleared by the kidney; the creatinine clearance rate should be greater than 30 mL/min for patients to receive dabigatran 150 mg twice a day, and at least 15 mL/min to receive 75 mg twice a day)
- Do not have severe hepatic dysfunction (which would impair baseline clotting function).
They note that other factors to consider are whether the patient:
- Can comply with the twice-daily dosing required
- Can afford the drug
- Has access to an anticoagulation management program (which would argue in favor of using warfarin).
Dabigatran is not yet approved to prevent venous thromboembolism.
CASE CONTINUED: HE GETS AN INFECTION
P.G. is started on dabigatran 150 mg by mouth twice a day.
While in the hospital he develops shortness of breath and needs intravenous furosemide (Lasix). Because he has bad veins, a percutaneous intravenous central catheter (PICC) line is placed. However, 2 days later, his temperature is 101.5°F, and his systolic blood pressure is 70 mm Hg. He is transferred to the medical intensive care unit (ICU) for treatment of sepsis. The anticoagulant is held, the PICC line is removed, and a new central catheter is inserted.
2. Which of the following directions is incorrect?
- Wash your hands before inserting the catheter. The accompanying nurse is required to directly observe this procedure or, if this step is not observed, to confirm that the physician did it.
- Before inserting the catheter, clean the patient’s skin with chlorhexidine antiseptic.
- Place sterile drapes over the entire patient.
- Wear any mask, hat, gown, and gloves available.
- Put a sterile dressing over the catheter.
A checklist can prevent infections when inserting central catheters
A checklist developed at Johns Hopkins Hospital consists of the five statements above, except for the second to last one—you should wear a sterile mask, hat, gown and gloves. This is important to ensure that sterility is not broken at any point during the procedure.
Pronovost et al4 launched a multicenter initiative at 90 ICUs, predominantly in the state of Michigan, to implement interventions to improve staff culture and teamwork and to translate research into practice by increasing the extent to which these five evidence-based recommendations were applied. The mean rate of catheter-related blood stream infections at baseline was 7.7%; this dropped to 2.8% during the implementation period, 2.3% in the first 3 months after implementation, 1.3% in months 16 through 18, and 1.1% in months 34 through 36, demonstrating that the gains from this quality-improvement project were sustainable.
If this intervention and collaborative model were implemented in all ICUs across the United States and if similar success rates were achieved, substantial and sustained reductions could be made in the 82,000 infections, 28,000 deaths, and $2.3 billion in costs attributed to these infections annually.
CASE CONTINUED: HE IS RESUSCITATED
P.G. is started on a 1-L fluid bolus but he remains hypotensive, necessitating a norepinephrine drip. He does well for about 6 hours, but in the middle of the night he develops ventricular tachycardia and ventricular fibrillation, and a code is called. He is successfully resuscitated, but the family is looking for prognostic information.
3. What are P.G.’s chances of surviving and leaving the hospital?
- 5%
- 8%
- 15%
- 23%
A registry of cardiopulmonary resuscitation
Tian et al5 evaluated outcomes in the largest registry of cardiopulmonary resuscitation to date. In this analysis, 49,656 adult patients with a first cardiopulmonary arrest occurring in an ICU between January 1, 2000, and August 26, 2008, were evaluated for their outcomes on pressors vs those not on pressors.
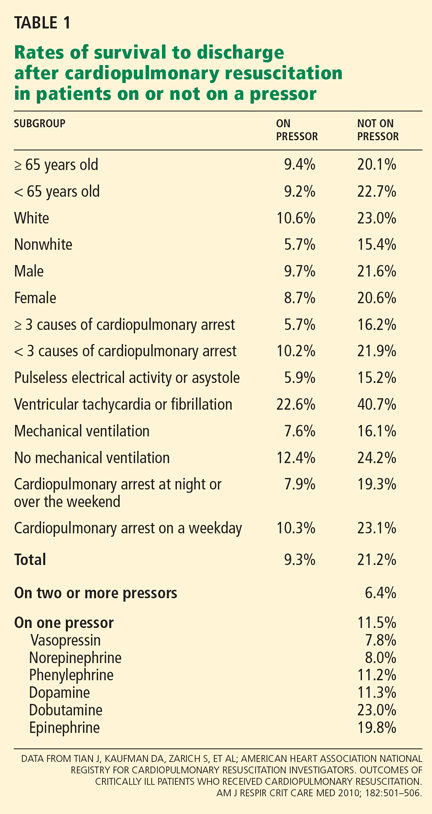
Other independent predictors of a lower survival rate were nonwhite race, mechanical ventilation, having three or more immediate causes of cardiopulmonary arrest, age 65 years or older, and cardiopulmonary arrest occurring at night or over the weekend.
Fortunately, for our patient, survival rates were higher for patients with ventricular tachycardia or fibrillation than with other causes of cardiopulmonary arrest: 22.6% for those on pressors (like our patient) and 40.7% for those on no pressors.
CASE CONTINUED: HE RECOVERS AND GOES HOME
P.G. makes a remarkable recovery and is now ready to go home. It is the weekend, and you are unable to schedule a follow-up appointment before his discharge, so you ask him to make an appointment with his PCP.
4. What is the likelihood that P.G. will be readmitted within 1 month?
- 5%
- 12%
- 20%
- 25%
- 30%
The importance of follow-up with a primary care physician
Misky et al,6 in a small study, attempted to identify the characteristics and outcomes of discharged patients who lack timely follow-up with a PCP. They prospectively enrolled 65 patients admitted to University of Colorado Hospital, an urban 425-bed tertiary care center, collecting information about patient demographics, diagnosis, payer source, and PCPs. After discharge, they called the patients to determine their PCP follow-up and readmission status. Thirty-day readmission rates and hospital length of stay were compared in patients with and without timely PCP follow-up (ie, within 4 weeks).
Patients lacking timely PCP follow-up were 10 times more likely to be readmitted (odds ratio [OR] = 9.9, P = .04): the rate was 21% in patients lacking timely PCP follow-up vs 3% in patients with timely PCP follow-up, P = .03. Lack of insurance was associated with lower rates of timely PCP follow-up: 29% vs 56% (P = .06), but did not independently increase the readmission rate or length of stay (OR = 1.0, P = .96). Index hospital length of stay was longer in patients lacking timely PCP follow-up: 4.4 days vs 6.3 days, P = 0.11.
Comment. Nearly half of the patients in this study, who were discharged from a large urban academic center, lacked timely follow-up with a PCP, resulting in higher rates of readmission and a nonsignificant trend toward longer length of stay. Timely follow-up is necessary for vulnerable patients.
Since the lack of timely PCP follow-up results in higher readmission rates and possibly a longer length of stay, a PCP appointment at discharge should perhaps be considered a core quality measure. This would be problematic in our American health care system, in which many patients lack health insurance and do not have a PCP.
A MAN UNDERGOING GASTRIC BYPASS SURGERY
A 55-year-old morbidly obese man (body mass index 45 kg/m2) with a history of type 2 diabetes mellitus, chronic renal insufficiency (serum creatinine level 2.1 mg/dL), hypercholesterolemia, and previous stroke is scheduled for gastric bypass surgery. His functional capacity is low, but he is able to do his activities of daily living. He reports having dyspnea on exertion and intermittently at rest, but no chest pain. His medications include insulin, atorvastatin (Lipitor), aspirin, and atenolol (Tenormin). He is afebrile; his blood pressure is 130/80 mm Hg, pulse 75, and oxygen saturation 97% on room air. His baseline electrocardiogram shows no Q waves.
5. Which of the following is an appropriate next step before proceeding to surgery?
- Echocardiography
- Cardiac catheterization
- Dobutamine stress echocardiography or adenosine thallium scanning
- No cardiac testing is necessary before surgery
Is cardiac testing necessary before noncardiac surgery?
Wijeysundera et al7 performed a retrospective cohort study of patients who underwent elective surgery at acute care hospitals in Ontario, Canada, in the years 1994 through 2004. The aim was to determine the association of noninvasive cardiac stress testing before surgery with survival rates and length of hospital stay. Included were 271,082 patients, of whom 23,991 (8.9%) underwent stress testing less than 6 months before surgery. These patients were matched with 46,120 who did not undergo testing.
One year after surgery, fewer patients who underwent stress testing had died: 1,622 (7.0%) vs 1,738 (7.5%); hazard ratio 0.92, 95% CI 0.86–0.99, P = .03. The number needed to treat (ie, to be tested) to prevent one death was 221. The tested patients also had a shorter mean hospital stay: 8.72 vs 8.96 days, a difference of 0.24 days (95% CI −0.07 to −0.43; P < .001).
However, the elderly patients (ie, older than 66 years) who underwent testing were more likely to be on beta-blockers and statins than those who did not undergo testing, which may be a confounding factor.
Furthermore, the benefit was all in the patients at intermediate or high risk. The authors performed a subgroup analysis, dividing the patients on the basis of their Revised Cardiac Risk Index (RCRI; 1 point each for ischemic heart disease, congestive heart failure, cerebrovascular disease, diabetes, renal insufficiency, and high-risk surgery).8 Patients with an RCRI of 0 points (indicating low risk) actually had a higher risk of death with testing than without testing: hazard ratio 1.35 (95% CI 1.03–1.74), number needed to harm 179—ie, for every 179 low-risk patients tested, one excess death occurred. Those with an RCRI of 1 or 2 points (indicating intermediate risk) had a hazard ratio of 0.92 with testing (95% CI 085–0.99), and those with an RCRI of 3 to 6 points (indicating high risk) had a hazard ratio of 0.80 with testing (95% CI 0.67- 0.97; number needed to treat = 38).
Comment. These findings indicate that cardiac stress testing should be done selectively before noncardiac surgery, and primarily for patients at high risk (with an RCRI of 3 or higher) and in some patients at intermediate risk, but not in patients at low risk, in whom it may be harmful. Stress testing may change patient management because a positive stress test allows one to start a beta-blocker or a statin, use more aggressive intraoperative and postoperative care, and identify patients who have indications for revascularization.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Wann LS, Curtis AB, Ellenbogen KA, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Pronovost PJ, Goeschel CA, Colantuoni E, et al. Sustaining reductions in catheter-related bloodstream infections in Michigan intensive care units: observational study. BMJ 2010; 340:c309.
- Tian J, Kaufman DA, Zarich S, et al; American Heart Association National Registry for Cardiopulmonary Resuscitation Investigators. Outcomes of critically ill patients who received cardiopulmonary resuscitation. Am J Respir Crit Care Med 2010; 182:501–506.
- Misky GJ, Wald HL, Coleman EA. Post-hospitalization transitions: examining the effects of timing of primary care provider follow-up. J Hosp Med 2010; 5:392–397.
- Wijeysundera DN, Beattie WS, Austin PC, Hux JE, Laupacis A. Non-invasive cardiac stress testing before elective major non-cardiac surgery: population based cohort study. BMJ 2010; 340:b5526.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
A number of studies published in the last few years will likely affect the way we practice medicine in the hospital. Here, we will use a hypothetical case scenario to focus on the issues of anticoagulants, patient safety, quality improvement, critical care, transitions of care, and perioperative medicine.
AN ELDERLY MAN WITH NEW-ONSET ATRIAL FIBRILLATION
P.G. is an 80-year-old man with a history of hypertension and type 2 diabetes mellitus who is admitted with new-onset atrial fibrillation. In the hospital, his heart rate is brought under control with intravenous metoprolol (Lopressor). On discharge, he will be followed by his primary care physician (PCP). He does not have access to an anticoagulation clinic.
1. What are this patient’s options for stroke prevention?
- Aspirin 81 mg daily and clopidogrel (Plavix) 75 mg daily
- Warfarin (Coumadin) with a target international normalized ratio (INR) of 2.0 to 3.0
- Aspirin mg daily by itself
- Dabigatran (Pradaxa) 150 mg daily
A new oral anticoagulant agent
In deciding what type of anticoagulation to give to a patient with atrial fibrillation, it is useful to look at the CHADS2 score (1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack. This patient has a CHADS2 score of 3, indicating that he should receive warfarin. An alternative is dabigatran, the first new anticoagulant agent in more than 50 years.
In a multicenter, international trial, Connolly et al1 randomized 18,113 patients (mean age 71, 64% men) to receive dabigatran 110 mg twice daily, dabigatran 150 mg twice daily, or warfarin with a target INR of 2.0 to 3.0. In this noninferiority trial, dabigatran was given in a blinded manner, but the use of warfarin was open-label. Patients were eligible if they had atrial fibrillation at screening or within the previous 6 months and were at risk of stroke—ie, if they had at least one of the following: a history of stroke or transient ischemic attack, a left ventricular ejection fraction of less than 40%, symptoms of congestive heart failure (New York Heart Association class II or higher), and an age of 75 or older or an age of 65 to 74 with diabetes mellitus, hypertension, or coronary artery disease.
At a mean follow-up of 2 years, the rate of stroke or systolic embolism was 1.69% per year in the warfarin group compared with 1.1% in the higher-dose dabigatran group (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rates of major hemorrhage were similar between these two groups. Comparing lower-dose dabigatran and warfarin, the rates of stroke or systolic embolism were not significantly different, but the rate of major bleeding was significantly lower with lower-dose dabigatran.
In a trial in patients with acute venous thromboembolism, Schulman et al2 found that dabigatran was not inferior to warfarin in preventing venous thromboembolism.
Guidelines from the American College of Cardiology Foundation and the American Heart Association now endorse dabigatran as an alternative to warfarin for patients with atrial fibrillation.3 However, the guidelines state that it should be reserved for those patients who:
- Do not have a prosthetic heart valve or hemodynamically significant valve disease
- Have good kidney function (dabigatran is cleared by the kidney; the creatinine clearance rate should be greater than 30 mL/min for patients to receive dabigatran 150 mg twice a day, and at least 15 mL/min to receive 75 mg twice a day)
- Do not have severe hepatic dysfunction (which would impair baseline clotting function).
They note that other factors to consider are whether the patient:
- Can comply with the twice-daily dosing required
- Can afford the drug
- Has access to an anticoagulation management program (which would argue in favor of using warfarin).
Dabigatran is not yet approved to prevent venous thromboembolism.
CASE CONTINUED: HE GETS AN INFECTION
P.G. is started on dabigatran 150 mg by mouth twice a day.
While in the hospital he develops shortness of breath and needs intravenous furosemide (Lasix). Because he has bad veins, a percutaneous intravenous central catheter (PICC) line is placed. However, 2 days later, his temperature is 101.5°F, and his systolic blood pressure is 70 mm Hg. He is transferred to the medical intensive care unit (ICU) for treatment of sepsis. The anticoagulant is held, the PICC line is removed, and a new central catheter is inserted.
2. Which of the following directions is incorrect?
- Wash your hands before inserting the catheter. The accompanying nurse is required to directly observe this procedure or, if this step is not observed, to confirm that the physician did it.
- Before inserting the catheter, clean the patient’s skin with chlorhexidine antiseptic.
- Place sterile drapes over the entire patient.
- Wear any mask, hat, gown, and gloves available.
- Put a sterile dressing over the catheter.
A checklist can prevent infections when inserting central catheters
A checklist developed at Johns Hopkins Hospital consists of the five statements above, except for the second to last one—you should wear a sterile mask, hat, gown and gloves. This is important to ensure that sterility is not broken at any point during the procedure.
Pronovost et al4 launched a multicenter initiative at 90 ICUs, predominantly in the state of Michigan, to implement interventions to improve staff culture and teamwork and to translate research into practice by increasing the extent to which these five evidence-based recommendations were applied. The mean rate of catheter-related blood stream infections at baseline was 7.7%; this dropped to 2.8% during the implementation period, 2.3% in the first 3 months after implementation, 1.3% in months 16 through 18, and 1.1% in months 34 through 36, demonstrating that the gains from this quality-improvement project were sustainable.
If this intervention and collaborative model were implemented in all ICUs across the United States and if similar success rates were achieved, substantial and sustained reductions could be made in the 82,000 infections, 28,000 deaths, and $2.3 billion in costs attributed to these infections annually.
CASE CONTINUED: HE IS RESUSCITATED
P.G. is started on a 1-L fluid bolus but he remains hypotensive, necessitating a norepinephrine drip. He does well for about 6 hours, but in the middle of the night he develops ventricular tachycardia and ventricular fibrillation, and a code is called. He is successfully resuscitated, but the family is looking for prognostic information.
3. What are P.G.’s chances of surviving and leaving the hospital?
- 5%
- 8%
- 15%
- 23%
A registry of cardiopulmonary resuscitation
Tian et al5 evaluated outcomes in the largest registry of cardiopulmonary resuscitation to date. In this analysis, 49,656 adult patients with a first cardiopulmonary arrest occurring in an ICU between January 1, 2000, and August 26, 2008, were evaluated for their outcomes on pressors vs those not on pressors.
Other independent predictors of a lower survival rate were nonwhite race, mechanical ventilation, having three or more immediate causes of cardiopulmonary arrest, age 65 years or older, and cardiopulmonary arrest occurring at night or over the weekend.
Fortunately, for our patient, survival rates were higher for patients with ventricular tachycardia or fibrillation than with other causes of cardiopulmonary arrest: 22.6% for those on pressors (like our patient) and 40.7% for those on no pressors.
CASE CONTINUED: HE RECOVERS AND GOES HOME
P.G. makes a remarkable recovery and is now ready to go home. It is the weekend, and you are unable to schedule a follow-up appointment before his discharge, so you ask him to make an appointment with his PCP.
4. What is the likelihood that P.G. will be readmitted within 1 month?
- 5%
- 12%
- 20%
- 25%
- 30%
The importance of follow-up with a primary care physician
Misky et al,6 in a small study, attempted to identify the characteristics and outcomes of discharged patients who lack timely follow-up with a PCP. They prospectively enrolled 65 patients admitted to University of Colorado Hospital, an urban 425-bed tertiary care center, collecting information about patient demographics, diagnosis, payer source, and PCPs. After discharge, they called the patients to determine their PCP follow-up and readmission status. Thirty-day readmission rates and hospital length of stay were compared in patients with and without timely PCP follow-up (ie, within 4 weeks).
Patients lacking timely PCP follow-up were 10 times more likely to be readmitted (odds ratio [OR] = 9.9, P = .04): the rate was 21% in patients lacking timely PCP follow-up vs 3% in patients with timely PCP follow-up, P = .03. Lack of insurance was associated with lower rates of timely PCP follow-up: 29% vs 56% (P = .06), but did not independently increase the readmission rate or length of stay (OR = 1.0, P = .96). Index hospital length of stay was longer in patients lacking timely PCP follow-up: 4.4 days vs 6.3 days, P = 0.11.
Comment. Nearly half of the patients in this study, who were discharged from a large urban academic center, lacked timely follow-up with a PCP, resulting in higher rates of readmission and a nonsignificant trend toward longer length of stay. Timely follow-up is necessary for vulnerable patients.
Since the lack of timely PCP follow-up results in higher readmission rates and possibly a longer length of stay, a PCP appointment at discharge should perhaps be considered a core quality measure. This would be problematic in our American health care system, in which many patients lack health insurance and do not have a PCP.
A MAN UNDERGOING GASTRIC BYPASS SURGERY
A 55-year-old morbidly obese man (body mass index 45 kg/m2) with a history of type 2 diabetes mellitus, chronic renal insufficiency (serum creatinine level 2.1 mg/dL), hypercholesterolemia, and previous stroke is scheduled for gastric bypass surgery. His functional capacity is low, but he is able to do his activities of daily living. He reports having dyspnea on exertion and intermittently at rest, but no chest pain. His medications include insulin, atorvastatin (Lipitor), aspirin, and atenolol (Tenormin). He is afebrile; his blood pressure is 130/80 mm Hg, pulse 75, and oxygen saturation 97% on room air. His baseline electrocardiogram shows no Q waves.
5. Which of the following is an appropriate next step before proceeding to surgery?
- Echocardiography
- Cardiac catheterization
- Dobutamine stress echocardiography or adenosine thallium scanning
- No cardiac testing is necessary before surgery
Is cardiac testing necessary before noncardiac surgery?
Wijeysundera et al7 performed a retrospective cohort study of patients who underwent elective surgery at acute care hospitals in Ontario, Canada, in the years 1994 through 2004. The aim was to determine the association of noninvasive cardiac stress testing before surgery with survival rates and length of hospital stay. Included were 271,082 patients, of whom 23,991 (8.9%) underwent stress testing less than 6 months before surgery. These patients were matched with 46,120 who did not undergo testing.
One year after surgery, fewer patients who underwent stress testing had died: 1,622 (7.0%) vs 1,738 (7.5%); hazard ratio 0.92, 95% CI 0.86–0.99, P = .03. The number needed to treat (ie, to be tested) to prevent one death was 221. The tested patients also had a shorter mean hospital stay: 8.72 vs 8.96 days, a difference of 0.24 days (95% CI −0.07 to −0.43; P < .001).
However, the elderly patients (ie, older than 66 years) who underwent testing were more likely to be on beta-blockers and statins than those who did not undergo testing, which may be a confounding factor.
Furthermore, the benefit was all in the patients at intermediate or high risk. The authors performed a subgroup analysis, dividing the patients on the basis of their Revised Cardiac Risk Index (RCRI; 1 point each for ischemic heart disease, congestive heart failure, cerebrovascular disease, diabetes, renal insufficiency, and high-risk surgery).8 Patients with an RCRI of 0 points (indicating low risk) actually had a higher risk of death with testing than without testing: hazard ratio 1.35 (95% CI 1.03–1.74), number needed to harm 179—ie, for every 179 low-risk patients tested, one excess death occurred. Those with an RCRI of 1 or 2 points (indicating intermediate risk) had a hazard ratio of 0.92 with testing (95% CI 085–0.99), and those with an RCRI of 3 to 6 points (indicating high risk) had a hazard ratio of 0.80 with testing (95% CI 0.67- 0.97; number needed to treat = 38).
Comment. These findings indicate that cardiac stress testing should be done selectively before noncardiac surgery, and primarily for patients at high risk (with an RCRI of 3 or higher) and in some patients at intermediate risk, but not in patients at low risk, in whom it may be harmful. Stress testing may change patient management because a positive stress test allows one to start a beta-blocker or a statin, use more aggressive intraoperative and postoperative care, and identify patients who have indications for revascularization.
A number of studies published in the last few years will likely affect the way we practice medicine in the hospital. Here, we will use a hypothetical case scenario to focus on the issues of anticoagulants, patient safety, quality improvement, critical care, transitions of care, and perioperative medicine.
AN ELDERLY MAN WITH NEW-ONSET ATRIAL FIBRILLATION
P.G. is an 80-year-old man with a history of hypertension and type 2 diabetes mellitus who is admitted with new-onset atrial fibrillation. In the hospital, his heart rate is brought under control with intravenous metoprolol (Lopressor). On discharge, he will be followed by his primary care physician (PCP). He does not have access to an anticoagulation clinic.
1. What are this patient’s options for stroke prevention?
- Aspirin 81 mg daily and clopidogrel (Plavix) 75 mg daily
- Warfarin (Coumadin) with a target international normalized ratio (INR) of 2.0 to 3.0
- Aspirin mg daily by itself
- Dabigatran (Pradaxa) 150 mg daily
A new oral anticoagulant agent
In deciding what type of anticoagulation to give to a patient with atrial fibrillation, it is useful to look at the CHADS2 score (1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack. This patient has a CHADS2 score of 3, indicating that he should receive warfarin. An alternative is dabigatran, the first new anticoagulant agent in more than 50 years.
In a multicenter, international trial, Connolly et al1 randomized 18,113 patients (mean age 71, 64% men) to receive dabigatran 110 mg twice daily, dabigatran 150 mg twice daily, or warfarin with a target INR of 2.0 to 3.0. In this noninferiority trial, dabigatran was given in a blinded manner, but the use of warfarin was open-label. Patients were eligible if they had atrial fibrillation at screening or within the previous 6 months and were at risk of stroke—ie, if they had at least one of the following: a history of stroke or transient ischemic attack, a left ventricular ejection fraction of less than 40%, symptoms of congestive heart failure (New York Heart Association class II or higher), and an age of 75 or older or an age of 65 to 74 with diabetes mellitus, hypertension, or coronary artery disease.
At a mean follow-up of 2 years, the rate of stroke or systolic embolism was 1.69% per year in the warfarin group compared with 1.1% in the higher-dose dabigatran group (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rates of major hemorrhage were similar between these two groups. Comparing lower-dose dabigatran and warfarin, the rates of stroke or systolic embolism were not significantly different, but the rate of major bleeding was significantly lower with lower-dose dabigatran.
In a trial in patients with acute venous thromboembolism, Schulman et al2 found that dabigatran was not inferior to warfarin in preventing venous thromboembolism.
Guidelines from the American College of Cardiology Foundation and the American Heart Association now endorse dabigatran as an alternative to warfarin for patients with atrial fibrillation.3 However, the guidelines state that it should be reserved for those patients who:
- Do not have a prosthetic heart valve or hemodynamically significant valve disease
- Have good kidney function (dabigatran is cleared by the kidney; the creatinine clearance rate should be greater than 30 mL/min for patients to receive dabigatran 150 mg twice a day, and at least 15 mL/min to receive 75 mg twice a day)
- Do not have severe hepatic dysfunction (which would impair baseline clotting function).
They note that other factors to consider are whether the patient:
- Can comply with the twice-daily dosing required
- Can afford the drug
- Has access to an anticoagulation management program (which would argue in favor of using warfarin).
Dabigatran is not yet approved to prevent venous thromboembolism.
CASE CONTINUED: HE GETS AN INFECTION
P.G. is started on dabigatran 150 mg by mouth twice a day.
While in the hospital he develops shortness of breath and needs intravenous furosemide (Lasix). Because he has bad veins, a percutaneous intravenous central catheter (PICC) line is placed. However, 2 days later, his temperature is 101.5°F, and his systolic blood pressure is 70 mm Hg. He is transferred to the medical intensive care unit (ICU) for treatment of sepsis. The anticoagulant is held, the PICC line is removed, and a new central catheter is inserted.
2. Which of the following directions is incorrect?
- Wash your hands before inserting the catheter. The accompanying nurse is required to directly observe this procedure or, if this step is not observed, to confirm that the physician did it.
- Before inserting the catheter, clean the patient’s skin with chlorhexidine antiseptic.
- Place sterile drapes over the entire patient.
- Wear any mask, hat, gown, and gloves available.
- Put a sterile dressing over the catheter.
A checklist can prevent infections when inserting central catheters
A checklist developed at Johns Hopkins Hospital consists of the five statements above, except for the second to last one—you should wear a sterile mask, hat, gown and gloves. This is important to ensure that sterility is not broken at any point during the procedure.
Pronovost et al4 launched a multicenter initiative at 90 ICUs, predominantly in the state of Michigan, to implement interventions to improve staff culture and teamwork and to translate research into practice by increasing the extent to which these five evidence-based recommendations were applied. The mean rate of catheter-related blood stream infections at baseline was 7.7%; this dropped to 2.8% during the implementation period, 2.3% in the first 3 months after implementation, 1.3% in months 16 through 18, and 1.1% in months 34 through 36, demonstrating that the gains from this quality-improvement project were sustainable.
If this intervention and collaborative model were implemented in all ICUs across the United States and if similar success rates were achieved, substantial and sustained reductions could be made in the 82,000 infections, 28,000 deaths, and $2.3 billion in costs attributed to these infections annually.
CASE CONTINUED: HE IS RESUSCITATED
P.G. is started on a 1-L fluid bolus but he remains hypotensive, necessitating a norepinephrine drip. He does well for about 6 hours, but in the middle of the night he develops ventricular tachycardia and ventricular fibrillation, and a code is called. He is successfully resuscitated, but the family is looking for prognostic information.
3. What are P.G.’s chances of surviving and leaving the hospital?
- 5%
- 8%
- 15%
- 23%
A registry of cardiopulmonary resuscitation
Tian et al5 evaluated outcomes in the largest registry of cardiopulmonary resuscitation to date. In this analysis, 49,656 adult patients with a first cardiopulmonary arrest occurring in an ICU between January 1, 2000, and August 26, 2008, were evaluated for their outcomes on pressors vs those not on pressors.
Other independent predictors of a lower survival rate were nonwhite race, mechanical ventilation, having three or more immediate causes of cardiopulmonary arrest, age 65 years or older, and cardiopulmonary arrest occurring at night or over the weekend.
Fortunately, for our patient, survival rates were higher for patients with ventricular tachycardia or fibrillation than with other causes of cardiopulmonary arrest: 22.6% for those on pressors (like our patient) and 40.7% for those on no pressors.
CASE CONTINUED: HE RECOVERS AND GOES HOME
P.G. makes a remarkable recovery and is now ready to go home. It is the weekend, and you are unable to schedule a follow-up appointment before his discharge, so you ask him to make an appointment with his PCP.
4. What is the likelihood that P.G. will be readmitted within 1 month?
- 5%
- 12%
- 20%
- 25%
- 30%
The importance of follow-up with a primary care physician
Misky et al,6 in a small study, attempted to identify the characteristics and outcomes of discharged patients who lack timely follow-up with a PCP. They prospectively enrolled 65 patients admitted to University of Colorado Hospital, an urban 425-bed tertiary care center, collecting information about patient demographics, diagnosis, payer source, and PCPs. After discharge, they called the patients to determine their PCP follow-up and readmission status. Thirty-day readmission rates and hospital length of stay were compared in patients with and without timely PCP follow-up (ie, within 4 weeks).
Patients lacking timely PCP follow-up were 10 times more likely to be readmitted (odds ratio [OR] = 9.9, P = .04): the rate was 21% in patients lacking timely PCP follow-up vs 3% in patients with timely PCP follow-up, P = .03. Lack of insurance was associated with lower rates of timely PCP follow-up: 29% vs 56% (P = .06), but did not independently increase the readmission rate or length of stay (OR = 1.0, P = .96). Index hospital length of stay was longer in patients lacking timely PCP follow-up: 4.4 days vs 6.3 days, P = 0.11.
Comment. Nearly half of the patients in this study, who were discharged from a large urban academic center, lacked timely follow-up with a PCP, resulting in higher rates of readmission and a nonsignificant trend toward longer length of stay. Timely follow-up is necessary for vulnerable patients.
Since the lack of timely PCP follow-up results in higher readmission rates and possibly a longer length of stay, a PCP appointment at discharge should perhaps be considered a core quality measure. This would be problematic in our American health care system, in which many patients lack health insurance and do not have a PCP.
A MAN UNDERGOING GASTRIC BYPASS SURGERY
A 55-year-old morbidly obese man (body mass index 45 kg/m2) with a history of type 2 diabetes mellitus, chronic renal insufficiency (serum creatinine level 2.1 mg/dL), hypercholesterolemia, and previous stroke is scheduled for gastric bypass surgery. His functional capacity is low, but he is able to do his activities of daily living. He reports having dyspnea on exertion and intermittently at rest, but no chest pain. His medications include insulin, atorvastatin (Lipitor), aspirin, and atenolol (Tenormin). He is afebrile; his blood pressure is 130/80 mm Hg, pulse 75, and oxygen saturation 97% on room air. His baseline electrocardiogram shows no Q waves.
5. Which of the following is an appropriate next step before proceeding to surgery?
- Echocardiography
- Cardiac catheterization
- Dobutamine stress echocardiography or adenosine thallium scanning
- No cardiac testing is necessary before surgery
Is cardiac testing necessary before noncardiac surgery?
Wijeysundera et al7 performed a retrospective cohort study of patients who underwent elective surgery at acute care hospitals in Ontario, Canada, in the years 1994 through 2004. The aim was to determine the association of noninvasive cardiac stress testing before surgery with survival rates and length of hospital stay. Included were 271,082 patients, of whom 23,991 (8.9%) underwent stress testing less than 6 months before surgery. These patients were matched with 46,120 who did not undergo testing.
One year after surgery, fewer patients who underwent stress testing had died: 1,622 (7.0%) vs 1,738 (7.5%); hazard ratio 0.92, 95% CI 0.86–0.99, P = .03. The number needed to treat (ie, to be tested) to prevent one death was 221. The tested patients also had a shorter mean hospital stay: 8.72 vs 8.96 days, a difference of 0.24 days (95% CI −0.07 to −0.43; P < .001).
However, the elderly patients (ie, older than 66 years) who underwent testing were more likely to be on beta-blockers and statins than those who did not undergo testing, which may be a confounding factor.
Furthermore, the benefit was all in the patients at intermediate or high risk. The authors performed a subgroup analysis, dividing the patients on the basis of their Revised Cardiac Risk Index (RCRI; 1 point each for ischemic heart disease, congestive heart failure, cerebrovascular disease, diabetes, renal insufficiency, and high-risk surgery).8 Patients with an RCRI of 0 points (indicating low risk) actually had a higher risk of death with testing than without testing: hazard ratio 1.35 (95% CI 1.03–1.74), number needed to harm 179—ie, for every 179 low-risk patients tested, one excess death occurred. Those with an RCRI of 1 or 2 points (indicating intermediate risk) had a hazard ratio of 0.92 with testing (95% CI 085–0.99), and those with an RCRI of 3 to 6 points (indicating high risk) had a hazard ratio of 0.80 with testing (95% CI 0.67- 0.97; number needed to treat = 38).
Comment. These findings indicate that cardiac stress testing should be done selectively before noncardiac surgery, and primarily for patients at high risk (with an RCRI of 3 or higher) and in some patients at intermediate risk, but not in patients at low risk, in whom it may be harmful. Stress testing may change patient management because a positive stress test allows one to start a beta-blocker or a statin, use more aggressive intraoperative and postoperative care, and identify patients who have indications for revascularization.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Wann LS, Curtis AB, Ellenbogen KA, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Pronovost PJ, Goeschel CA, Colantuoni E, et al. Sustaining reductions in catheter-related bloodstream infections in Michigan intensive care units: observational study. BMJ 2010; 340:c309.
- Tian J, Kaufman DA, Zarich S, et al; American Heart Association National Registry for Cardiopulmonary Resuscitation Investigators. Outcomes of critically ill patients who received cardiopulmonary resuscitation. Am J Respir Crit Care Med 2010; 182:501–506.
- Misky GJ, Wald HL, Coleman EA. Post-hospitalization transitions: examining the effects of timing of primary care provider follow-up. J Hosp Med 2010; 5:392–397.
- Wijeysundera DN, Beattie WS, Austin PC, Hux JE, Laupacis A. Non-invasive cardiac stress testing before elective major non-cardiac surgery: population based cohort study. BMJ 2010; 340:b5526.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Wann LS, Curtis AB, Ellenbogen KA, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Pronovost PJ, Goeschel CA, Colantuoni E, et al. Sustaining reductions in catheter-related bloodstream infections in Michigan intensive care units: observational study. BMJ 2010; 340:c309.
- Tian J, Kaufman DA, Zarich S, et al; American Heart Association National Registry for Cardiopulmonary Resuscitation Investigators. Outcomes of critically ill patients who received cardiopulmonary resuscitation. Am J Respir Crit Care Med 2010; 182:501–506.
- Misky GJ, Wald HL, Coleman EA. Post-hospitalization transitions: examining the effects of timing of primary care provider follow-up. J Hosp Med 2010; 5:392–397.
- Wijeysundera DN, Beattie WS, Austin PC, Hux JE, Laupacis A. Non-invasive cardiac stress testing before elective major non-cardiac surgery: population based cohort study. BMJ 2010; 340:b5526.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
KEY POINTS
- Dabigatran (Pradaxa) will likely start to replace warfarin (Coumadin) both to prevent stroke in patients with atrial fibrillation and to prevent recurrent venous thromboembolism.
- Using a checklist during insertion of central venous catheters can decrease the rate of catheter-related bloodstream infections in the intensive care unit.
- The overall survival rate of patients who undergo cardiopulmonary resuscitation in the intensive care unit is approximately 16%; the rate is lower in patients who are receiving pressor drugs and higher in those with ventricular tachycardia or ventricular fibrillation.
- Patients lacking follow-up with a primary care physician within 30 days of discharge are at high risk of readmission and have a trend for longer length of hospital stay.
- Preoperative stress testing for patients undergoing noncardiac surgery should be done selectively, ie, in patients at high risk.
How to manage type 2 diabetes in medical and surgical patients in the hospital
Hyperglycemia and diabetes mellitus are very common in hospitalized patients. Although more data are available on the prevalence of this problem and on how to manage it in the intensive care unit (ICU) than on regular hospital floors, the situation is changing. Information is emerging on the prevalence and impact of hyperglycemia and diabetes in the non-ICU setting, which is the focus of this paper.
HYPERGLYCEMIA IS COMMON AND PREDICTS POOR OUTCOMES
Cook et al,1 in a survey of 126 US hospitals, found that the prevalence of hyperglycemia (blood glucose > 180 mg/dL) was 46% in the ICU and 32% in regular wards.
Kosiborod et al2 reported that hyperglycemia (blood glucose > 140 mg/dL) was present in 78% of diabetic patients hospitalized with acute coronary syndrome and 26% of similar hospitalized nondiabetic patients.
Hyperglycemia is a common comorbidity in medical-surgical patients in community hospitals. Our group3 found that, in our hospital, 62% of patients were normoglycemic (ie, had a fasting blood glucose < 126 mg/dL or a random blood glucose < 200 mg/dL on two occasions), 26% had known diabetes, and 12% had new hyperglycemia. Further, new hyperglycemia was associated with a higher in-hospital death rate than the other two conditions.
Failure to identify diabetes is a predictor of rehospitalization. Robbins and Webb4 reported that 30.6% of those who had diabetes that was missed during hospitalization were readmitted within 30 days, compared with 9.4% of patients with diabetes first diagnosed during hospitalization.
WHAT DIAGNOSTIC CRITERIA SHOULD WE USE?
Blood glucose greater than 140 mg/dL
A consensus statement from the American Association of Clinical Endocrinologists (ACE) and the American Diabetes Association (ADA)5 defines in-hospital hyperglycemia as a blood glucose level greater than 140 mg/dL on admission or in the hospital. If the blood glucose is higher than this, the question arises as to whether the patient has preexisting diabetes or has stress hyperglycemia.
Hemoglobin A1c of 6.5% or higher
In view of the uncertainty as to whether a patient with an elevated blood glucose level has preexisting diabetes or stress hyperglycemia, upcoming guidelines will recommend measuring the hemoglobin A1c level if the blood glucose level is higher than 140 mg/dL.
A patient with an elevated blood glucose level (>140 mg/dL) whose hemoglobin A1c level is 6.5% or higher can be identified as having diabetes that preceded the hospitalization. Hemoglobin A1c testing can also be useful to assess glycemic control before admission and in designing an optional regimen at the time of discharge. In patients with newly recognized hyperglycemia, a hemoglobin A1c measurement can help differentiate patients with previously undiagnosed diabetes from those with stress-induced hyperglycemia.
Clinicians should keep in mind that a hemoglobin A1c cutoff of 6.5% identifies fewer cases of undiagnosed diabetes than does a high fasting glucose concentration, and that a level less than 6.5% does not rule out the diagnosis of diabetes. Several epidemiologic studies6 have reported a low sensitivity (44% to 66%) but a high specificity (76% to 99%) for hemoglobin A1c values higher than 6.5% in an outpatient population. The high specificity therefore supports the use of hemoglobin A1c to confirm the diagnosis of diabetes in patients with hyperglycemia, but the low sensitivity indicates that this test should not be used for universal screening in the hospital.
Many factors can influence the hemoglobin A1c level, such as anemia, iron deficiency, blood transfusions, hemolytic anemia, and renal failure.
Until now, if patients had hyperglycemia but no prior diagnosis of diabetes, the recommendation was for an oral 2-hour glucose tolerance test shortly after discharge to confirm the diagnosis of diabetes. Norhammar et al7 performed oral glucose tolerance tests in patients admitted with acute myocardial infarction, and Matz et al8 performed glucose tolerance tests in patients with acute stroke. They found that impaired glucose tolerance and undiagnosed type 2 diabetes were very common in these two groups. However, physicians rarely order oral glucose tolerance tests. We believe that hemoglobin A1c will be a better tool than an oral glucose tolerance test to confirm diabetes in hyperglycemic patients in the hospital setting.

WHAT IS THE ASSOCIATION BETWEEN HYPERGLYCEMIA AND OUTCOMES?
In 2,471 patients admitted to the hospital with community-acquired pneumonia, McAlister et al10 found that the rates of hospital complications and of death rose with blood glucose levels.
Falguera et al11 found that, in 660 episodes of community-acquired pneumonia, the rates of hospitalization, death, pleural effusion, and concomitant illnesses were all significantly higher in diabetic patients than in nondiabetic patients.
Noordzij et al12 performed a case-control study of 108,593 patients who underwent noncardiac surgery. The odds ratio for perioperative death was 1.19 (95% confidence interval [CI] 1.1–1.3) for every 1-mmol/L increase in the glucose level.
Frisch et al,13 in patients undergoing noncardiac surgery, found that the 30-day rates of death and of in-hospital complications were all higher in patients with diabetes than without diabetes.
Our group3 identified hyperglycemia as an independent marker of in-hospital death in patients with undiagnosed diabetes. The rates of death were 1.7% in those with normoglycemia, 3.0% in those with known diabetes, and 16.0% (P < .01) in those with new hyperglycemia.
The ACE/ADA consensus panel14 set the following glucose targets for patients in the non-ICU setting:
- Pre-meal blood glucose < 140 mg/dL
- Random blood glucose < 180 mg/dL.
On the other hand, hypoglycemia is also associated with adverse outcomes. Therefore, to avoid hypoglycemia, the insulin regimen should be reassessed if blood glucose levels fall below 100 mg/dL. New guidelines will suggest keeping the blood glucose between 100 and 140 mg/dL.
HOW SHOULD WE MANAGE HYPERGLYCEMIA IN THE NON-ICU SETTING?
The ACE/ADA guidelines recommend subcutaneous insulin therapy for most medical-surgical patients with diabetes, reserving intravenous insulin therapy for hyperglycemic crises and uncontrolled hyperglycemia.14
Oral antidiabetic agents are not generally recommended, as we have no data to support their use in the hospital. Another argument against using noninsulin therapies in the hospital is that sulfonylureas, especially glyburide (Diabeta, Micronase) are a major cause of hypoglycemia. Metformin (Glucophage) is contraindicated in decreased renal function, in hemodynamic instability, in surgical patients, and with the use of iodinated contrast dye. Thiazolidinediones are associated with edema and congestive heart failure, and they take up to 12 weeks to lower blood glucose levels. Alpha-glucosidase inhibitors are weak glucose-lowering agents. Also, therapies directed at glucagon-like-protein 1 can cause nausea and have a greater effect on postprandial glucose.14
The two main options for managing hyperglycemia and diabetes in the non-ICU setting are short-acting insulin on a sliding scale and basal-bolus therapy, the latter with either NPH plus regular insulin or long-acting plus rapid-acting insulin analogues.
Basal-bolus vs sliding scale insulin: The RABBIT-2 trial
In the RABBIT 2 trial (Randomized Basal Bolus Versus Sliding Scale Regular Insulin in Patients With Type 2 Diabetes Mellitus),15 our group compared the efficacy and safety of a basal-bolus regimen and a sliding-scale regimen in 130 hospitalized patients with type 2 diabetes treated with diet, with oral hypoglycemic agents, or with both. Oral antidiabetic drugs were discontinued on admission, and patients were randomized to one of the treatment groups.
In the basal-bolus group, the starting total daily dose was 0.4 U/kg/day if the blood glucose level on admission was between 140 and 200 mg/dL, or 0.5 U/kg/day if the glucose level was between 201 and 400 mg/dL. Half of the total daily dose was given as insulin glargine (Lantus) once daily, and the other half was given as insulin glulisine (Apidra) before meals. These doses were adjusted if the patient’s fasting or pre-meal blood glucose levels rose above 140 mg/dL or fell below 70 mg/dL.
The sliding-scale group received regular insulin four times daily (before meals and at bedtime) for glucose levels higher than 140 mg/dL; the higher the level, the more they got.
The basal-bolus regimen was better than sliding-scale regular insulin. At admission, the mean glucose values and hemoglobin A1c values were similar in both groups, but the mean glucose level on therapy was significantly lower in the basal-bolus group than in the sliding-scale group, 166 ± 32 mg/dL vs 193 ± 54 mg/dL, P < .001). About two-thirds of the basal-bolus group achieved a blood glucose target of less than 140 mg/dL, compared with only about one-third of the sliding-scale group. The basal-bolus group received more insulin, a mean of 42 units per day vs 12.5 units per day in the sliding-scale group. Yet the incidence of hypoglycemia was 3% in both groups.
NPH plus regular vs detemir plus aspart: The DEAN trial
Several long-acting insulin analogues are available and have a longer duration of action than NPH. Similarly, several newer rapid-acting analogues act more rapidly than regular insulin. Do these pharmacokinetic advantages matter? And do they justify the higher costs of the newer agents?
In the randomized Insulin Detemir Versus NPH Insulin in Hospitalized Patients With Diabetes (DEAN) trial,16 we compared two regimens: detemir plus aspart in a basal-bolus regimen, and NPH plus regular insulin in two divided doses, two-thirds of the total daily dose in the morning before breakfast and one-third before dinner, both doses in a ratio of two-thirds NPH and one-third regular, mixed in the same syringe. We recruited 130 patients with type 2 diabetes mellitus who were on oral hypoglycemic agents or insulin therapy.
NPH plus regular was just as good as detemir plus aspart in improving glycemic control. Blood glucose levels fell during the first day of therapy and were similar in both groups throughout the trial, as measured before breakfast, lunch, and dinner and at bedtime. The mean total daily insulin dose was not significantly different between treatment groups: 56 ± 45 units in the basal-bolus detemir-aspart group and 45 ± 32 units in the NPH-regular group. However, the basal-bolus group received significantly more short-acting insulin: 27 ± 20 units a day of aspart vs 18 ± 14 units of regular.
Somewhat fewer patients in the NPH-regular group had episodes of hypoglycemia, although the difference between groups was not statistically significant.
In a univariate analysis of the RABBIT-2 and DEAN trials,17 factors that predicted a blood glucose level less than 60 mg/dL were older age, lower body weight, higher serum creatinine level, and previous insulin therapy. Factors that were not predictive were the hemoglobin A1c level and the enrollment blood glucose level. Based on these data, we believe that to reduce the rate of hypoglycemia, lower insulin doses are needed in elderly patients and patients with renal impairment, and that if patients have been taking insulin before they come to the hospital, the dose should be cut back by about 25% while they are hospitalized.
Basal-bolus vs sliding-scale insulin for surgical patients: The RABBIT 2 Surgery trial
Does better glucose control in surgical patients affect outcomes in patients undergoing general surgery? To find out, we performed a prospective, multicenter, randomized, open-label trial in general surgery patients not in the ICU.18 We recruited and randomized 211 patients with type 2 diabetes who were on diet therapy or oral hypoglycemic agents or insulin in low doses (< 0.4 U/kg/day).
Oral drugs were discontinued on admission, and patients were randomized to receive either a basal-bolus regimen of glargine plus glulisine or regular insulin on a sliding scale. The basal-bolus group got 0.5 U/kg/day, half of it as glargine once daily and half as glulisine before meals. The total daily dose was reduced to 0.3 U/kg/day in patients age 70 and older or who had a serum creatinine level of 2.0 mg/dL or higher.
The goal was to maintain fasting and pre-meal glucose concentrations between 100 and 140 mg/dL. The total daily dose was raised by 10% (mostly in the glargine dose) if the blood glucose level was in the range of 141 to 180 mg/dL, and by 20% if the glucose level was higher than 181 mg/dL. The dose was decreased by 10% for glucose levels between 70 and 99 mg/dL, was decreased by 20% if the glucose level was between 40 and 69, and was held if the glucose level was lower than 40 mg/dL. If a patient was not able to eat, insulin glulisine was held until meals were resumed.
The sliding-scale group received regular insulin four times a day for blood glucose levels higher than 140 mg/dL.
The primary outcomes measured were the difference between groups in mean daily blood glucose concentration and a composite of hospital complications including postoperative wound infection, pneumonia, respiratory failure, acute renal failure, and bacteremia. Secondary outcomes were differences between groups in mean fasting and pre-meal blood glucose, number of hypoglycemic episodes (blood glucose < 70 mg/dL), hyperglycemic episodes (blood glucose > 200 mg/dL), length of hospital stay, need for intensive care, and rate of complications including wound infection, pneumonia, acute renal failure, and death.
Blood glucose levels were significantly lower in the basal-bolus group through the first 7 days after randomization, as measured before breakfast, lunch, and dinner, and at bedtime, and then they converged.
More patients in the sliding-scale group had hospital complications, 26 vs 9, P = .003. On the other hand, more patients in the basal-bolus group had episodes of hypoglycemia: 24 (23%) vs 5 (4.7%) had episodes of less than 70 mg/dL (P < .001), 12 (12%) vs 2 (1.9%) had episodes of less than 60 mg/dL (P = .005), and 4 (3.8%) vs 0 had episodes of less than 40 mg/dL (P = .057). The mean total daily dose of insulin was 33.4 units in the basal-bolus group and 12.3 units in the sliding-scale group.
WHAT HAVE WE LEARNED?
Don’t use a sliding-scale regimen as a single agent in patients with diabetes. Glycemic control is better with a basal-bolus regimen than with a sliding-scale regimen, and a basal-bolus insulin regimen is preferred for most patients with hyperglycemia.
The old human insulins (ie, regular and NPH) are still good and improve glycemic control as well as the new basal insulin analogues (detemir and aspart) do.
Improved control may reduce the rate of hospital complications, according to preliminary evidence. More studies are under way.
One size does not fit all. Those who are elderly or who have impaired renal function should receive lower doses of insulin, eg, 0.3 U/kg/day instead of 0.5 U/kg/day. Those who are on insulin should have their dose decreased when they are admitted to the hospital. Perhaps lean patients with type 2 diabetes should also have a lower dose.
Most hospitalized patients with diabetes and elevated blood glucose values (or hyperglycemia) should receive subcutaneous insulin treatment with a basal-bolus regimen or a multidose combination of NPH plus regular insulin. Selected patients with severe insulin resistance and persistent hyperglycemia despite subcutaneous insulin may benefit from continuous intravenous insulin infusion.
Patients treated with insulin at home should continue to receive insulin therapy in the hospital. However, the insulin dosage should be reduced by about 25% to allow for lower food intake.
QUESTIONS FOR FURTHER STUDY
Should we modify the standard basal-bolus regimen?
In a typical basal-bolus regimen, patients get 50% of their total daily insulin dose in the form of a basal injection and 50% in the form of rapid-acting boluses before meals. However, for a variety of reasons, hospitalized patients do not eat very much. Thus, a 50-50 basal-bolus regimen may not be ideal for patients with poor oral intake.
In the Basal-PLUS trial, currently under way, we are comparing the safety and efficacy of a daily dose of basal insulin (glargine) plus correction doses of a rapid-acting insulin analogue (glulisine) on a sliding scale and a standard basal-bolus regimen in medical and surgical patients.
Does one glycemic target fit all patients?
Falciglia et al19 found an association between hyperglycemia and death in patients with unstable angina, arrhythmias, stroke, pneumonia, gastrointestinal bleeding, respiratory failure, sepsis, acute renal failure, and congestive heart failure. However, they found no such association in patients with chronic obstructive pulmonary disease, liver failure, diabetic ketoacidosis, gastrointestinal neoplasm, musculoskeletal disease, peripheral vascular disease with bypass, hip fracture, amputation due to peripheral vascular disease, or prostate surgery. Should patients in this second group be treated with a less-intensive insulin regimen?
What is the best regimen after hospital discharge?
We are conducting a prospective clinical trial to assess the impact of insulin after hospital discharge. Our current practice when a patient is discharged from the hospital is as follows:
- If the admission hemoglobin A1c level is less than 7%, we restart the previous outpatient treatment regimen of oral antidiabetic agents, or insulin, or both.
- If the admission hemoglobin A1c is between 7% and 9%, we restart the outpatient oral agents and continue glargine once daily at 50% to 80% of the hospital dose.
- If the hemoglobin A1c level is higher than 9%, we discharge the patient on a basal-bolus regimen at the same dosage as in the hospital. As an alternative, we could restart the oral agents and add glargine once daily at 80% of the hospital dose.
- Cook CB, Kongable GL, Potter DJ, Abad VJ, Leija DE, Anderson M. Inpatient glucose control: a glycemic survey of 126 U.S. hospitals. J Hosp Med 2009; 4:E7–E14.
- Kosiborod M, Inzucchi S, Clark B, et al. National patterns of glucose control among patients hospitalized with acute myocardial infarction [abstract]. J Am Coll Cardiol 2007; 49:283A–284A.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Robbins JM, Webb DA. Diagnosing diabetes and preventing rehospitalizations: the urban diabetes study. Med Care 2006; 44:292–296.
- Moghissi ES, Korytkowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Saudek D, Herman WH, Sacks DB, Bergenstal RM, Edelman D, Davidson MB. A new look at screening and diagnosing diabetes mellitus. J Clin Endocrinol Metab 2008; 93:2447–2453.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study. Lancet 2002; 359:2140–2144.
- Matz K, Keresztes K, Tatschl C, et al. Disorders of glucose metabolism in acute stroke patients: an underrecognized problem. Diabetes Care 2006; 29:792–797.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010; 33(suppl 1):S62–S69.
- McAlister FA, Majumdar SR, Blitz S, Rowe BH, Romney J, Marrie TJ. The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community-acquired pneumonia. Diabetes Care 2005; 28:810–815.
- Falguera M, Pifarre R, Martin A, Sheikh A, Moreno A. Etiology and outcome of community-acquired pneumonia in patients with diabetes mellitus. Chest 2005; 128:3233–3239.
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol 2007; 156:137–142.
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care 2010; 33:1783–1788.
- Moghissi ES, Korythowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Endocrine Pract 2009; 15:1–17.
- Umpierrrez GE, Smiley D, Zisman A, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial). Diabetes Care 2007; 30:2181–2186.
- Umpierrez GE, Hor T, Smiley D, et al. Comparison of inpatient insulin regimens with detemir plus aspart versus neutral protamine Hagedorn plus regular in medical patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:564–569.
- Umpierrez GE, Smiley D, Umpierrez D, Ceron M, Temponi A. Hypoglycemic events during subcutaneous insulin therapy in type 2 diabetes (abstract). Presented at American Diabetes Association 69th Scientific Sessions, New Orleans, LA, June 5–9, 2009.
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care 2011; 34:256–261.
- Falciglia M, Freyberg RW, Almenoff PL, D’Alessio DA, Render ML. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med 2009; 37:3001–3009.
Hyperglycemia and diabetes mellitus are very common in hospitalized patients. Although more data are available on the prevalence of this problem and on how to manage it in the intensive care unit (ICU) than on regular hospital floors, the situation is changing. Information is emerging on the prevalence and impact of hyperglycemia and diabetes in the non-ICU setting, which is the focus of this paper.
HYPERGLYCEMIA IS COMMON AND PREDICTS POOR OUTCOMES
Cook et al,1 in a survey of 126 US hospitals, found that the prevalence of hyperglycemia (blood glucose > 180 mg/dL) was 46% in the ICU and 32% in regular wards.
Kosiborod et al2 reported that hyperglycemia (blood glucose > 140 mg/dL) was present in 78% of diabetic patients hospitalized with acute coronary syndrome and 26% of similar hospitalized nondiabetic patients.
Hyperglycemia is a common comorbidity in medical-surgical patients in community hospitals. Our group3 found that, in our hospital, 62% of patients were normoglycemic (ie, had a fasting blood glucose < 126 mg/dL or a random blood glucose < 200 mg/dL on two occasions), 26% had known diabetes, and 12% had new hyperglycemia. Further, new hyperglycemia was associated with a higher in-hospital death rate than the other two conditions.
Failure to identify diabetes is a predictor of rehospitalization. Robbins and Webb4 reported that 30.6% of those who had diabetes that was missed during hospitalization were readmitted within 30 days, compared with 9.4% of patients with diabetes first diagnosed during hospitalization.
WHAT DIAGNOSTIC CRITERIA SHOULD WE USE?
Blood glucose greater than 140 mg/dL
A consensus statement from the American Association of Clinical Endocrinologists (ACE) and the American Diabetes Association (ADA)5 defines in-hospital hyperglycemia as a blood glucose level greater than 140 mg/dL on admission or in the hospital. If the blood glucose is higher than this, the question arises as to whether the patient has preexisting diabetes or has stress hyperglycemia.
Hemoglobin A1c of 6.5% or higher
In view of the uncertainty as to whether a patient with an elevated blood glucose level has preexisting diabetes or stress hyperglycemia, upcoming guidelines will recommend measuring the hemoglobin A1c level if the blood glucose level is higher than 140 mg/dL.
A patient with an elevated blood glucose level (>140 mg/dL) whose hemoglobin A1c level is 6.5% or higher can be identified as having diabetes that preceded the hospitalization. Hemoglobin A1c testing can also be useful to assess glycemic control before admission and in designing an optional regimen at the time of discharge. In patients with newly recognized hyperglycemia, a hemoglobin A1c measurement can help differentiate patients with previously undiagnosed diabetes from those with stress-induced hyperglycemia.
Clinicians should keep in mind that a hemoglobin A1c cutoff of 6.5% identifies fewer cases of undiagnosed diabetes than does a high fasting glucose concentration, and that a level less than 6.5% does not rule out the diagnosis of diabetes. Several epidemiologic studies6 have reported a low sensitivity (44% to 66%) but a high specificity (76% to 99%) for hemoglobin A1c values higher than 6.5% in an outpatient population. The high specificity therefore supports the use of hemoglobin A1c to confirm the diagnosis of diabetes in patients with hyperglycemia, but the low sensitivity indicates that this test should not be used for universal screening in the hospital.
Many factors can influence the hemoglobin A1c level, such as anemia, iron deficiency, blood transfusions, hemolytic anemia, and renal failure.
Until now, if patients had hyperglycemia but no prior diagnosis of diabetes, the recommendation was for an oral 2-hour glucose tolerance test shortly after discharge to confirm the diagnosis of diabetes. Norhammar et al7 performed oral glucose tolerance tests in patients admitted with acute myocardial infarction, and Matz et al8 performed glucose tolerance tests in patients with acute stroke. They found that impaired glucose tolerance and undiagnosed type 2 diabetes were very common in these two groups. However, physicians rarely order oral glucose tolerance tests. We believe that hemoglobin A1c will be a better tool than an oral glucose tolerance test to confirm diabetes in hyperglycemic patients in the hospital setting.
WHAT IS THE ASSOCIATION BETWEEN HYPERGLYCEMIA AND OUTCOMES?
In 2,471 patients admitted to the hospital with community-acquired pneumonia, McAlister et al10 found that the rates of hospital complications and of death rose with blood glucose levels.
Falguera et al11 found that, in 660 episodes of community-acquired pneumonia, the rates of hospitalization, death, pleural effusion, and concomitant illnesses were all significantly higher in diabetic patients than in nondiabetic patients.
Noordzij et al12 performed a case-control study of 108,593 patients who underwent noncardiac surgery. The odds ratio for perioperative death was 1.19 (95% confidence interval [CI] 1.1–1.3) for every 1-mmol/L increase in the glucose level.
Frisch et al,13 in patients undergoing noncardiac surgery, found that the 30-day rates of death and of in-hospital complications were all higher in patients with diabetes than without diabetes.
Our group3 identified hyperglycemia as an independent marker of in-hospital death in patients with undiagnosed diabetes. The rates of death were 1.7% in those with normoglycemia, 3.0% in those with known diabetes, and 16.0% (P < .01) in those with new hyperglycemia.
The ACE/ADA consensus panel14 set the following glucose targets for patients in the non-ICU setting:
- Pre-meal blood glucose < 140 mg/dL
- Random blood glucose < 180 mg/dL.
On the other hand, hypoglycemia is also associated with adverse outcomes. Therefore, to avoid hypoglycemia, the insulin regimen should be reassessed if blood glucose levels fall below 100 mg/dL. New guidelines will suggest keeping the blood glucose between 100 and 140 mg/dL.
HOW SHOULD WE MANAGE HYPERGLYCEMIA IN THE NON-ICU SETTING?
The ACE/ADA guidelines recommend subcutaneous insulin therapy for most medical-surgical patients with diabetes, reserving intravenous insulin therapy for hyperglycemic crises and uncontrolled hyperglycemia.14
Oral antidiabetic agents are not generally recommended, as we have no data to support their use in the hospital. Another argument against using noninsulin therapies in the hospital is that sulfonylureas, especially glyburide (Diabeta, Micronase) are a major cause of hypoglycemia. Metformin (Glucophage) is contraindicated in decreased renal function, in hemodynamic instability, in surgical patients, and with the use of iodinated contrast dye. Thiazolidinediones are associated with edema and congestive heart failure, and they take up to 12 weeks to lower blood glucose levels. Alpha-glucosidase inhibitors are weak glucose-lowering agents. Also, therapies directed at glucagon-like-protein 1 can cause nausea and have a greater effect on postprandial glucose.14
The two main options for managing hyperglycemia and diabetes in the non-ICU setting are short-acting insulin on a sliding scale and basal-bolus therapy, the latter with either NPH plus regular insulin or long-acting plus rapid-acting insulin analogues.
Basal-bolus vs sliding scale insulin: The RABBIT-2 trial
In the RABBIT 2 trial (Randomized Basal Bolus Versus Sliding Scale Regular Insulin in Patients With Type 2 Diabetes Mellitus),15 our group compared the efficacy and safety of a basal-bolus regimen and a sliding-scale regimen in 130 hospitalized patients with type 2 diabetes treated with diet, with oral hypoglycemic agents, or with both. Oral antidiabetic drugs were discontinued on admission, and patients were randomized to one of the treatment groups.
In the basal-bolus group, the starting total daily dose was 0.4 U/kg/day if the blood glucose level on admission was between 140 and 200 mg/dL, or 0.5 U/kg/day if the glucose level was between 201 and 400 mg/dL. Half of the total daily dose was given as insulin glargine (Lantus) once daily, and the other half was given as insulin glulisine (Apidra) before meals. These doses were adjusted if the patient’s fasting or pre-meal blood glucose levels rose above 140 mg/dL or fell below 70 mg/dL.
The sliding-scale group received regular insulin four times daily (before meals and at bedtime) for glucose levels higher than 140 mg/dL; the higher the level, the more they got.
The basal-bolus regimen was better than sliding-scale regular insulin. At admission, the mean glucose values and hemoglobin A1c values were similar in both groups, but the mean glucose level on therapy was significantly lower in the basal-bolus group than in the sliding-scale group, 166 ± 32 mg/dL vs 193 ± 54 mg/dL, P < .001). About two-thirds of the basal-bolus group achieved a blood glucose target of less than 140 mg/dL, compared with only about one-third of the sliding-scale group. The basal-bolus group received more insulin, a mean of 42 units per day vs 12.5 units per day in the sliding-scale group. Yet the incidence of hypoglycemia was 3% in both groups.
NPH plus regular vs detemir plus aspart: The DEAN trial
Several long-acting insulin analogues are available and have a longer duration of action than NPH. Similarly, several newer rapid-acting analogues act more rapidly than regular insulin. Do these pharmacokinetic advantages matter? And do they justify the higher costs of the newer agents?
In the randomized Insulin Detemir Versus NPH Insulin in Hospitalized Patients With Diabetes (DEAN) trial,16 we compared two regimens: detemir plus aspart in a basal-bolus regimen, and NPH plus regular insulin in two divided doses, two-thirds of the total daily dose in the morning before breakfast and one-third before dinner, both doses in a ratio of two-thirds NPH and one-third regular, mixed in the same syringe. We recruited 130 patients with type 2 diabetes mellitus who were on oral hypoglycemic agents or insulin therapy.
NPH plus regular was just as good as detemir plus aspart in improving glycemic control. Blood glucose levels fell during the first day of therapy and were similar in both groups throughout the trial, as measured before breakfast, lunch, and dinner and at bedtime. The mean total daily insulin dose was not significantly different between treatment groups: 56 ± 45 units in the basal-bolus detemir-aspart group and 45 ± 32 units in the NPH-regular group. However, the basal-bolus group received significantly more short-acting insulin: 27 ± 20 units a day of aspart vs 18 ± 14 units of regular.
Somewhat fewer patients in the NPH-regular group had episodes of hypoglycemia, although the difference between groups was not statistically significant.
In a univariate analysis of the RABBIT-2 and DEAN trials,17 factors that predicted a blood glucose level less than 60 mg/dL were older age, lower body weight, higher serum creatinine level, and previous insulin therapy. Factors that were not predictive were the hemoglobin A1c level and the enrollment blood glucose level. Based on these data, we believe that to reduce the rate of hypoglycemia, lower insulin doses are needed in elderly patients and patients with renal impairment, and that if patients have been taking insulin before they come to the hospital, the dose should be cut back by about 25% while they are hospitalized.
Basal-bolus vs sliding-scale insulin for surgical patients: The RABBIT 2 Surgery trial
Does better glucose control in surgical patients affect outcomes in patients undergoing general surgery? To find out, we performed a prospective, multicenter, randomized, open-label trial in general surgery patients not in the ICU.18 We recruited and randomized 211 patients with type 2 diabetes who were on diet therapy or oral hypoglycemic agents or insulin in low doses (< 0.4 U/kg/day).
Oral drugs were discontinued on admission, and patients were randomized to receive either a basal-bolus regimen of glargine plus glulisine or regular insulin on a sliding scale. The basal-bolus group got 0.5 U/kg/day, half of it as glargine once daily and half as glulisine before meals. The total daily dose was reduced to 0.3 U/kg/day in patients age 70 and older or who had a serum creatinine level of 2.0 mg/dL or higher.
The goal was to maintain fasting and pre-meal glucose concentrations between 100 and 140 mg/dL. The total daily dose was raised by 10% (mostly in the glargine dose) if the blood glucose level was in the range of 141 to 180 mg/dL, and by 20% if the glucose level was higher than 181 mg/dL. The dose was decreased by 10% for glucose levels between 70 and 99 mg/dL, was decreased by 20% if the glucose level was between 40 and 69, and was held if the glucose level was lower than 40 mg/dL. If a patient was not able to eat, insulin glulisine was held until meals were resumed.
The sliding-scale group received regular insulin four times a day for blood glucose levels higher than 140 mg/dL.
The primary outcomes measured were the difference between groups in mean daily blood glucose concentration and a composite of hospital complications including postoperative wound infection, pneumonia, respiratory failure, acute renal failure, and bacteremia. Secondary outcomes were differences between groups in mean fasting and pre-meal blood glucose, number of hypoglycemic episodes (blood glucose < 70 mg/dL), hyperglycemic episodes (blood glucose > 200 mg/dL), length of hospital stay, need for intensive care, and rate of complications including wound infection, pneumonia, acute renal failure, and death.
Blood glucose levels were significantly lower in the basal-bolus group through the first 7 days after randomization, as measured before breakfast, lunch, and dinner, and at bedtime, and then they converged.
More patients in the sliding-scale group had hospital complications, 26 vs 9, P = .003. On the other hand, more patients in the basal-bolus group had episodes of hypoglycemia: 24 (23%) vs 5 (4.7%) had episodes of less than 70 mg/dL (P < .001), 12 (12%) vs 2 (1.9%) had episodes of less than 60 mg/dL (P = .005), and 4 (3.8%) vs 0 had episodes of less than 40 mg/dL (P = .057). The mean total daily dose of insulin was 33.4 units in the basal-bolus group and 12.3 units in the sliding-scale group.
WHAT HAVE WE LEARNED?
Don’t use a sliding-scale regimen as a single agent in patients with diabetes. Glycemic control is better with a basal-bolus regimen than with a sliding-scale regimen, and a basal-bolus insulin regimen is preferred for most patients with hyperglycemia.
The old human insulins (ie, regular and NPH) are still good and improve glycemic control as well as the new basal insulin analogues (detemir and aspart) do.
Improved control may reduce the rate of hospital complications, according to preliminary evidence. More studies are under way.
One size does not fit all. Those who are elderly or who have impaired renal function should receive lower doses of insulin, eg, 0.3 U/kg/day instead of 0.5 U/kg/day. Those who are on insulin should have their dose decreased when they are admitted to the hospital. Perhaps lean patients with type 2 diabetes should also have a lower dose.
Most hospitalized patients with diabetes and elevated blood glucose values (or hyperglycemia) should receive subcutaneous insulin treatment with a basal-bolus regimen or a multidose combination of NPH plus regular insulin. Selected patients with severe insulin resistance and persistent hyperglycemia despite subcutaneous insulin may benefit from continuous intravenous insulin infusion.
Patients treated with insulin at home should continue to receive insulin therapy in the hospital. However, the insulin dosage should be reduced by about 25% to allow for lower food intake.
QUESTIONS FOR FURTHER STUDY
Should we modify the standard basal-bolus regimen?
In a typical basal-bolus regimen, patients get 50% of their total daily insulin dose in the form of a basal injection and 50% in the form of rapid-acting boluses before meals. However, for a variety of reasons, hospitalized patients do not eat very much. Thus, a 50-50 basal-bolus regimen may not be ideal for patients with poor oral intake.
In the Basal-PLUS trial, currently under way, we are comparing the safety and efficacy of a daily dose of basal insulin (glargine) plus correction doses of a rapid-acting insulin analogue (glulisine) on a sliding scale and a standard basal-bolus regimen in medical and surgical patients.
Does one glycemic target fit all patients?
Falciglia et al19 found an association between hyperglycemia and death in patients with unstable angina, arrhythmias, stroke, pneumonia, gastrointestinal bleeding, respiratory failure, sepsis, acute renal failure, and congestive heart failure. However, they found no such association in patients with chronic obstructive pulmonary disease, liver failure, diabetic ketoacidosis, gastrointestinal neoplasm, musculoskeletal disease, peripheral vascular disease with bypass, hip fracture, amputation due to peripheral vascular disease, or prostate surgery. Should patients in this second group be treated with a less-intensive insulin regimen?
What is the best regimen after hospital discharge?
We are conducting a prospective clinical trial to assess the impact of insulin after hospital discharge. Our current practice when a patient is discharged from the hospital is as follows:
- If the admission hemoglobin A1c level is less than 7%, we restart the previous outpatient treatment regimen of oral antidiabetic agents, or insulin, or both.
- If the admission hemoglobin A1c is between 7% and 9%, we restart the outpatient oral agents and continue glargine once daily at 50% to 80% of the hospital dose.
- If the hemoglobin A1c level is higher than 9%, we discharge the patient on a basal-bolus regimen at the same dosage as in the hospital. As an alternative, we could restart the oral agents and add glargine once daily at 80% of the hospital dose.
Hyperglycemia and diabetes mellitus are very common in hospitalized patients. Although more data are available on the prevalence of this problem and on how to manage it in the intensive care unit (ICU) than on regular hospital floors, the situation is changing. Information is emerging on the prevalence and impact of hyperglycemia and diabetes in the non-ICU setting, which is the focus of this paper.
HYPERGLYCEMIA IS COMMON AND PREDICTS POOR OUTCOMES
Cook et al,1 in a survey of 126 US hospitals, found that the prevalence of hyperglycemia (blood glucose > 180 mg/dL) was 46% in the ICU and 32% in regular wards.
Kosiborod et al2 reported that hyperglycemia (blood glucose > 140 mg/dL) was present in 78% of diabetic patients hospitalized with acute coronary syndrome and 26% of similar hospitalized nondiabetic patients.
Hyperglycemia is a common comorbidity in medical-surgical patients in community hospitals. Our group3 found that, in our hospital, 62% of patients were normoglycemic (ie, had a fasting blood glucose < 126 mg/dL or a random blood glucose < 200 mg/dL on two occasions), 26% had known diabetes, and 12% had new hyperglycemia. Further, new hyperglycemia was associated with a higher in-hospital death rate than the other two conditions.
Failure to identify diabetes is a predictor of rehospitalization. Robbins and Webb4 reported that 30.6% of those who had diabetes that was missed during hospitalization were readmitted within 30 days, compared with 9.4% of patients with diabetes first diagnosed during hospitalization.
WHAT DIAGNOSTIC CRITERIA SHOULD WE USE?
Blood glucose greater than 140 mg/dL
A consensus statement from the American Association of Clinical Endocrinologists (ACE) and the American Diabetes Association (ADA)5 defines in-hospital hyperglycemia as a blood glucose level greater than 140 mg/dL on admission or in the hospital. If the blood glucose is higher than this, the question arises as to whether the patient has preexisting diabetes or has stress hyperglycemia.
Hemoglobin A1c of 6.5% or higher
In view of the uncertainty as to whether a patient with an elevated blood glucose level has preexisting diabetes or stress hyperglycemia, upcoming guidelines will recommend measuring the hemoglobin A1c level if the blood glucose level is higher than 140 mg/dL.
A patient with an elevated blood glucose level (>140 mg/dL) whose hemoglobin A1c level is 6.5% or higher can be identified as having diabetes that preceded the hospitalization. Hemoglobin A1c testing can also be useful to assess glycemic control before admission and in designing an optional regimen at the time of discharge. In patients with newly recognized hyperglycemia, a hemoglobin A1c measurement can help differentiate patients with previously undiagnosed diabetes from those with stress-induced hyperglycemia.
Clinicians should keep in mind that a hemoglobin A1c cutoff of 6.5% identifies fewer cases of undiagnosed diabetes than does a high fasting glucose concentration, and that a level less than 6.5% does not rule out the diagnosis of diabetes. Several epidemiologic studies6 have reported a low sensitivity (44% to 66%) but a high specificity (76% to 99%) for hemoglobin A1c values higher than 6.5% in an outpatient population. The high specificity therefore supports the use of hemoglobin A1c to confirm the diagnosis of diabetes in patients with hyperglycemia, but the low sensitivity indicates that this test should not be used for universal screening in the hospital.
Many factors can influence the hemoglobin A1c level, such as anemia, iron deficiency, blood transfusions, hemolytic anemia, and renal failure.
Until now, if patients had hyperglycemia but no prior diagnosis of diabetes, the recommendation was for an oral 2-hour glucose tolerance test shortly after discharge to confirm the diagnosis of diabetes. Norhammar et al7 performed oral glucose tolerance tests in patients admitted with acute myocardial infarction, and Matz et al8 performed glucose tolerance tests in patients with acute stroke. They found that impaired glucose tolerance and undiagnosed type 2 diabetes were very common in these two groups. However, physicians rarely order oral glucose tolerance tests. We believe that hemoglobin A1c will be a better tool than an oral glucose tolerance test to confirm diabetes in hyperglycemic patients in the hospital setting.
WHAT IS THE ASSOCIATION BETWEEN HYPERGLYCEMIA AND OUTCOMES?
In 2,471 patients admitted to the hospital with community-acquired pneumonia, McAlister et al10 found that the rates of hospital complications and of death rose with blood glucose levels.
Falguera et al11 found that, in 660 episodes of community-acquired pneumonia, the rates of hospitalization, death, pleural effusion, and concomitant illnesses were all significantly higher in diabetic patients than in nondiabetic patients.
Noordzij et al12 performed a case-control study of 108,593 patients who underwent noncardiac surgery. The odds ratio for perioperative death was 1.19 (95% confidence interval [CI] 1.1–1.3) for every 1-mmol/L increase in the glucose level.
Frisch et al,13 in patients undergoing noncardiac surgery, found that the 30-day rates of death and of in-hospital complications were all higher in patients with diabetes than without diabetes.
Our group3 identified hyperglycemia as an independent marker of in-hospital death in patients with undiagnosed diabetes. The rates of death were 1.7% in those with normoglycemia, 3.0% in those with known diabetes, and 16.0% (P < .01) in those with new hyperglycemia.
The ACE/ADA consensus panel14 set the following glucose targets for patients in the non-ICU setting:
- Pre-meal blood glucose < 140 mg/dL
- Random blood glucose < 180 mg/dL.
On the other hand, hypoglycemia is also associated with adverse outcomes. Therefore, to avoid hypoglycemia, the insulin regimen should be reassessed if blood glucose levels fall below 100 mg/dL. New guidelines will suggest keeping the blood glucose between 100 and 140 mg/dL.
HOW SHOULD WE MANAGE HYPERGLYCEMIA IN THE NON-ICU SETTING?
The ACE/ADA guidelines recommend subcutaneous insulin therapy for most medical-surgical patients with diabetes, reserving intravenous insulin therapy for hyperglycemic crises and uncontrolled hyperglycemia.14
Oral antidiabetic agents are not generally recommended, as we have no data to support their use in the hospital. Another argument against using noninsulin therapies in the hospital is that sulfonylureas, especially glyburide (Diabeta, Micronase) are a major cause of hypoglycemia. Metformin (Glucophage) is contraindicated in decreased renal function, in hemodynamic instability, in surgical patients, and with the use of iodinated contrast dye. Thiazolidinediones are associated with edema and congestive heart failure, and they take up to 12 weeks to lower blood glucose levels. Alpha-glucosidase inhibitors are weak glucose-lowering agents. Also, therapies directed at glucagon-like-protein 1 can cause nausea and have a greater effect on postprandial glucose.14
The two main options for managing hyperglycemia and diabetes in the non-ICU setting are short-acting insulin on a sliding scale and basal-bolus therapy, the latter with either NPH plus regular insulin or long-acting plus rapid-acting insulin analogues.
Basal-bolus vs sliding scale insulin: The RABBIT-2 trial
In the RABBIT 2 trial (Randomized Basal Bolus Versus Sliding Scale Regular Insulin in Patients With Type 2 Diabetes Mellitus),15 our group compared the efficacy and safety of a basal-bolus regimen and a sliding-scale regimen in 130 hospitalized patients with type 2 diabetes treated with diet, with oral hypoglycemic agents, or with both. Oral antidiabetic drugs were discontinued on admission, and patients were randomized to one of the treatment groups.
In the basal-bolus group, the starting total daily dose was 0.4 U/kg/day if the blood glucose level on admission was between 140 and 200 mg/dL, or 0.5 U/kg/day if the glucose level was between 201 and 400 mg/dL. Half of the total daily dose was given as insulin glargine (Lantus) once daily, and the other half was given as insulin glulisine (Apidra) before meals. These doses were adjusted if the patient’s fasting or pre-meal blood glucose levels rose above 140 mg/dL or fell below 70 mg/dL.
The sliding-scale group received regular insulin four times daily (before meals and at bedtime) for glucose levels higher than 140 mg/dL; the higher the level, the more they got.
The basal-bolus regimen was better than sliding-scale regular insulin. At admission, the mean glucose values and hemoglobin A1c values were similar in both groups, but the mean glucose level on therapy was significantly lower in the basal-bolus group than in the sliding-scale group, 166 ± 32 mg/dL vs 193 ± 54 mg/dL, P < .001). About two-thirds of the basal-bolus group achieved a blood glucose target of less than 140 mg/dL, compared with only about one-third of the sliding-scale group. The basal-bolus group received more insulin, a mean of 42 units per day vs 12.5 units per day in the sliding-scale group. Yet the incidence of hypoglycemia was 3% in both groups.
NPH plus regular vs detemir plus aspart: The DEAN trial
Several long-acting insulin analogues are available and have a longer duration of action than NPH. Similarly, several newer rapid-acting analogues act more rapidly than regular insulin. Do these pharmacokinetic advantages matter? And do they justify the higher costs of the newer agents?
In the randomized Insulin Detemir Versus NPH Insulin in Hospitalized Patients With Diabetes (DEAN) trial,16 we compared two regimens: detemir plus aspart in a basal-bolus regimen, and NPH plus regular insulin in two divided doses, two-thirds of the total daily dose in the morning before breakfast and one-third before dinner, both doses in a ratio of two-thirds NPH and one-third regular, mixed in the same syringe. We recruited 130 patients with type 2 diabetes mellitus who were on oral hypoglycemic agents or insulin therapy.
NPH plus regular was just as good as detemir plus aspart in improving glycemic control. Blood glucose levels fell during the first day of therapy and were similar in both groups throughout the trial, as measured before breakfast, lunch, and dinner and at bedtime. The mean total daily insulin dose was not significantly different between treatment groups: 56 ± 45 units in the basal-bolus detemir-aspart group and 45 ± 32 units in the NPH-regular group. However, the basal-bolus group received significantly more short-acting insulin: 27 ± 20 units a day of aspart vs 18 ± 14 units of regular.
Somewhat fewer patients in the NPH-regular group had episodes of hypoglycemia, although the difference between groups was not statistically significant.
In a univariate analysis of the RABBIT-2 and DEAN trials,17 factors that predicted a blood glucose level less than 60 mg/dL were older age, lower body weight, higher serum creatinine level, and previous insulin therapy. Factors that were not predictive were the hemoglobin A1c level and the enrollment blood glucose level. Based on these data, we believe that to reduce the rate of hypoglycemia, lower insulin doses are needed in elderly patients and patients with renal impairment, and that if patients have been taking insulin before they come to the hospital, the dose should be cut back by about 25% while they are hospitalized.
Basal-bolus vs sliding-scale insulin for surgical patients: The RABBIT 2 Surgery trial
Does better glucose control in surgical patients affect outcomes in patients undergoing general surgery? To find out, we performed a prospective, multicenter, randomized, open-label trial in general surgery patients not in the ICU.18 We recruited and randomized 211 patients with type 2 diabetes who were on diet therapy or oral hypoglycemic agents or insulin in low doses (< 0.4 U/kg/day).
Oral drugs were discontinued on admission, and patients were randomized to receive either a basal-bolus regimen of glargine plus glulisine or regular insulin on a sliding scale. The basal-bolus group got 0.5 U/kg/day, half of it as glargine once daily and half as glulisine before meals. The total daily dose was reduced to 0.3 U/kg/day in patients age 70 and older or who had a serum creatinine level of 2.0 mg/dL or higher.
The goal was to maintain fasting and pre-meal glucose concentrations between 100 and 140 mg/dL. The total daily dose was raised by 10% (mostly in the glargine dose) if the blood glucose level was in the range of 141 to 180 mg/dL, and by 20% if the glucose level was higher than 181 mg/dL. The dose was decreased by 10% for glucose levels between 70 and 99 mg/dL, was decreased by 20% if the glucose level was between 40 and 69, and was held if the glucose level was lower than 40 mg/dL. If a patient was not able to eat, insulin glulisine was held until meals were resumed.
The sliding-scale group received regular insulin four times a day for blood glucose levels higher than 140 mg/dL.
The primary outcomes measured were the difference between groups in mean daily blood glucose concentration and a composite of hospital complications including postoperative wound infection, pneumonia, respiratory failure, acute renal failure, and bacteremia. Secondary outcomes were differences between groups in mean fasting and pre-meal blood glucose, number of hypoglycemic episodes (blood glucose < 70 mg/dL), hyperglycemic episodes (blood glucose > 200 mg/dL), length of hospital stay, need for intensive care, and rate of complications including wound infection, pneumonia, acute renal failure, and death.
Blood glucose levels were significantly lower in the basal-bolus group through the first 7 days after randomization, as measured before breakfast, lunch, and dinner, and at bedtime, and then they converged.
More patients in the sliding-scale group had hospital complications, 26 vs 9, P = .003. On the other hand, more patients in the basal-bolus group had episodes of hypoglycemia: 24 (23%) vs 5 (4.7%) had episodes of less than 70 mg/dL (P < .001), 12 (12%) vs 2 (1.9%) had episodes of less than 60 mg/dL (P = .005), and 4 (3.8%) vs 0 had episodes of less than 40 mg/dL (P = .057). The mean total daily dose of insulin was 33.4 units in the basal-bolus group and 12.3 units in the sliding-scale group.
WHAT HAVE WE LEARNED?
Don’t use a sliding-scale regimen as a single agent in patients with diabetes. Glycemic control is better with a basal-bolus regimen than with a sliding-scale regimen, and a basal-bolus insulin regimen is preferred for most patients with hyperglycemia.
The old human insulins (ie, regular and NPH) are still good and improve glycemic control as well as the new basal insulin analogues (detemir and aspart) do.
Improved control may reduce the rate of hospital complications, according to preliminary evidence. More studies are under way.
One size does not fit all. Those who are elderly or who have impaired renal function should receive lower doses of insulin, eg, 0.3 U/kg/day instead of 0.5 U/kg/day. Those who are on insulin should have their dose decreased when they are admitted to the hospital. Perhaps lean patients with type 2 diabetes should also have a lower dose.
Most hospitalized patients with diabetes and elevated blood glucose values (or hyperglycemia) should receive subcutaneous insulin treatment with a basal-bolus regimen or a multidose combination of NPH plus regular insulin. Selected patients with severe insulin resistance and persistent hyperglycemia despite subcutaneous insulin may benefit from continuous intravenous insulin infusion.
Patients treated with insulin at home should continue to receive insulin therapy in the hospital. However, the insulin dosage should be reduced by about 25% to allow for lower food intake.
QUESTIONS FOR FURTHER STUDY
Should we modify the standard basal-bolus regimen?
In a typical basal-bolus regimen, patients get 50% of their total daily insulin dose in the form of a basal injection and 50% in the form of rapid-acting boluses before meals. However, for a variety of reasons, hospitalized patients do not eat very much. Thus, a 50-50 basal-bolus regimen may not be ideal for patients with poor oral intake.
In the Basal-PLUS trial, currently under way, we are comparing the safety and efficacy of a daily dose of basal insulin (glargine) plus correction doses of a rapid-acting insulin analogue (glulisine) on a sliding scale and a standard basal-bolus regimen in medical and surgical patients.
Does one glycemic target fit all patients?
Falciglia et al19 found an association between hyperglycemia and death in patients with unstable angina, arrhythmias, stroke, pneumonia, gastrointestinal bleeding, respiratory failure, sepsis, acute renal failure, and congestive heart failure. However, they found no such association in patients with chronic obstructive pulmonary disease, liver failure, diabetic ketoacidosis, gastrointestinal neoplasm, musculoskeletal disease, peripheral vascular disease with bypass, hip fracture, amputation due to peripheral vascular disease, or prostate surgery. Should patients in this second group be treated with a less-intensive insulin regimen?
What is the best regimen after hospital discharge?
We are conducting a prospective clinical trial to assess the impact of insulin after hospital discharge. Our current practice when a patient is discharged from the hospital is as follows:
- If the admission hemoglobin A1c level is less than 7%, we restart the previous outpatient treatment regimen of oral antidiabetic agents, or insulin, or both.
- If the admission hemoglobin A1c is between 7% and 9%, we restart the outpatient oral agents and continue glargine once daily at 50% to 80% of the hospital dose.
- If the hemoglobin A1c level is higher than 9%, we discharge the patient on a basal-bolus regimen at the same dosage as in the hospital. As an alternative, we could restart the oral agents and add glargine once daily at 80% of the hospital dose.
- Cook CB, Kongable GL, Potter DJ, Abad VJ, Leija DE, Anderson M. Inpatient glucose control: a glycemic survey of 126 U.S. hospitals. J Hosp Med 2009; 4:E7–E14.
- Kosiborod M, Inzucchi S, Clark B, et al. National patterns of glucose control among patients hospitalized with acute myocardial infarction [abstract]. J Am Coll Cardiol 2007; 49:283A–284A.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Robbins JM, Webb DA. Diagnosing diabetes and preventing rehospitalizations: the urban diabetes study. Med Care 2006; 44:292–296.
- Moghissi ES, Korytkowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Saudek D, Herman WH, Sacks DB, Bergenstal RM, Edelman D, Davidson MB. A new look at screening and diagnosing diabetes mellitus. J Clin Endocrinol Metab 2008; 93:2447–2453.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study. Lancet 2002; 359:2140–2144.
- Matz K, Keresztes K, Tatschl C, et al. Disorders of glucose metabolism in acute stroke patients: an underrecognized problem. Diabetes Care 2006; 29:792–797.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010; 33(suppl 1):S62–S69.
- McAlister FA, Majumdar SR, Blitz S, Rowe BH, Romney J, Marrie TJ. The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community-acquired pneumonia. Diabetes Care 2005; 28:810–815.
- Falguera M, Pifarre R, Martin A, Sheikh A, Moreno A. Etiology and outcome of community-acquired pneumonia in patients with diabetes mellitus. Chest 2005; 128:3233–3239.
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol 2007; 156:137–142.
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care 2010; 33:1783–1788.
- Moghissi ES, Korythowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Endocrine Pract 2009; 15:1–17.
- Umpierrrez GE, Smiley D, Zisman A, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial). Diabetes Care 2007; 30:2181–2186.
- Umpierrez GE, Hor T, Smiley D, et al. Comparison of inpatient insulin regimens with detemir plus aspart versus neutral protamine Hagedorn plus regular in medical patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:564–569.
- Umpierrez GE, Smiley D, Umpierrez D, Ceron M, Temponi A. Hypoglycemic events during subcutaneous insulin therapy in type 2 diabetes (abstract). Presented at American Diabetes Association 69th Scientific Sessions, New Orleans, LA, June 5–9, 2009.
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care 2011; 34:256–261.
- Falciglia M, Freyberg RW, Almenoff PL, D’Alessio DA, Render ML. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med 2009; 37:3001–3009.
- Cook CB, Kongable GL, Potter DJ, Abad VJ, Leija DE, Anderson M. Inpatient glucose control: a glycemic survey of 126 U.S. hospitals. J Hosp Med 2009; 4:E7–E14.
- Kosiborod M, Inzucchi S, Clark B, et al. National patterns of glucose control among patients hospitalized with acute myocardial infarction [abstract]. J Am Coll Cardiol 2007; 49:283A–284A.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Robbins JM, Webb DA. Diagnosing diabetes and preventing rehospitalizations: the urban diabetes study. Med Care 2006; 44:292–296.
- Moghissi ES, Korytkowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Saudek D, Herman WH, Sacks DB, Bergenstal RM, Edelman D, Davidson MB. A new look at screening and diagnosing diabetes mellitus. J Clin Endocrinol Metab 2008; 93:2447–2453.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study. Lancet 2002; 359:2140–2144.
- Matz K, Keresztes K, Tatschl C, et al. Disorders of glucose metabolism in acute stroke patients: an underrecognized problem. Diabetes Care 2006; 29:792–797.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010; 33(suppl 1):S62–S69.
- McAlister FA, Majumdar SR, Blitz S, Rowe BH, Romney J, Marrie TJ. The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community-acquired pneumonia. Diabetes Care 2005; 28:810–815.
- Falguera M, Pifarre R, Martin A, Sheikh A, Moreno A. Etiology and outcome of community-acquired pneumonia in patients with diabetes mellitus. Chest 2005; 128:3233–3239.
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol 2007; 156:137–142.
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care 2010; 33:1783–1788.
- Moghissi ES, Korythowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Endocrine Pract 2009; 15:1–17.
- Umpierrrez GE, Smiley D, Zisman A, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial). Diabetes Care 2007; 30:2181–2186.
- Umpierrez GE, Hor T, Smiley D, et al. Comparison of inpatient insulin regimens with detemir plus aspart versus neutral protamine Hagedorn plus regular in medical patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:564–569.
- Umpierrez GE, Smiley D, Umpierrez D, Ceron M, Temponi A. Hypoglycemic events during subcutaneous insulin therapy in type 2 diabetes (abstract). Presented at American Diabetes Association 69th Scientific Sessions, New Orleans, LA, June 5–9, 2009.
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care 2011; 34:256–261.
- Falciglia M, Freyberg RW, Almenoff PL, D’Alessio DA, Render ML. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med 2009; 37:3001–3009.
KEY POINTS
- Hyperglycemia and undiagnosed diabetes are very common in hospitalized patients and are associated with poorer outcomes.
- Hospitalized patients should be screened for diabetes with a blood glucose measurement. Those who have a value of 140 mg/dL or higher should be tested for hemoglobin A1c. A value higher than 6.5% is very specific for diabetes, although not very sensitive for it.
- Most hospitalized patients with diabetes and elevated blood glucose values (or hyperglycemia) should receive subcutaneous insulin treatment with a basal-bolus regimen or a multidose combination of neutral protamine Hagedorn (NPH) plus regular insulin. Selected patients with severe insulin resistance and persistent hyperglycemia despite subcutaneous insulin may benefit from continuous intravenous insulin infusion.
- Sliding-scale insulin as a single form of therapy in patients with diabetes is undesirable.
Immune thrombocytopenia: No longer ‘idiopathic’
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.

FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.
FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.
FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
KEY POINTS
- ITP is defined as an isolated platelet count of less than 100 × 109/L (100,000/μL) and usually presents without symptoms.
- Patients without symptoms who have a platelet count above 30 × 109/L should generally not be treated unless they have an increased risk of bleeding.
- Recent studies suggest that viruses and other pathogens play an important role in secondary ITP.
- Initially, corticosteroids are usually given as prednisone (1–2 mg/kg/day, then tapered), though recent studies suggest that dexamethasone pulses (40 mg/day for 4 days) may provide more durable responses when used in this setting.
- Thrombopoietic agents are important new treatments, although their place in the overall therapy of ITP has not been established.
Ending LGBT invisibility in health care: The first step in ensuring equitable care
In speaking about lesbian, gay, bisexual, and transgender (LGBT) health, it is not uncommon for me to be asked what is so unique about the health care needs of lesbians, gay men, bisexuals, and transgender individuals that it warrants focused attention in the training of health professionals and while providing care.1 Although it is true that most health issues affecting LGBT individuals parallel those of the general population, people who are LGBT have been shown to have unique health needs and to experience disparities in care.
There is a growing if limited number of good studies of LGBT health. The Institute of Medicine2 reported on lesbian health in 1999, concluding that enough evidence of disparities exists to support more research and to develop better methods of conducting the research. Healthy People 2020 actually recognizes significant health care disparities.3 Finally, the Institute of Medicine recently formed a committee on LGBT health issues to identify gaps in our knowledge and priorities for research. Their findings were expected to be published in late March 2011, after this article went to press.
MAKING A DIFFERENCE
While this article will not attempt to discuss all the disparities, the focus will be on how physicians can take the first critical step to helping LGBT individuals feel comfortable seeking care, ie, by being proactive in taking a history that includes discussion of sexual orientation and gender identity. Only by knowing this about patients will clinicians appropriately care for specific health needs, and will patients feel comfortable discussing their concerns in clinical settings.
While some feel this is relevant only in select areas of the country, recent data show that the LGBT population is both spread throughout the country and diverse in how they might present themselves in clinical settings.1,4 In the United States, 1.4% to 4.1% of people identify themselves as lesbian, gay, or bisexual.5 About 3% of women and 4% of men say they have had a same-sex sexual contact in the last year, and 4% to 11% of women and 6% to 9% of men report having ever had one.
Everyone who practices clinical medicine needs to understand whether patients are LGBT and how to engage in conversation about sexual orientation and gender identity.
GETTING TO KNOW LGBT PATIENTS
What questions should a clinician ask to get this information? In thinking about what to ask, it helps to realize that patients generally do not mind being questioned about personal matters if the provider approaches the topic and the patient with genuine respect, empathy, and even curiosity.
On the other hand, providers often feel ill-prepared to discuss intimate issues, or feel uncomfortable doing so. Successfully achieving a change in clinical practice involves learning an approach to doing so and becoming comfortable with discussions that may follow. One question to consider is how you will feel and how you will follow up if a patient tells you that he or she is LGBT.
The core comprehensive history for LGBT patients is the same as for all patients, keeping in mind the unique LGBT health risks and issues. Clinicians may begin by getting to know each patient as a person (eg, ask about partners, children, and jobs). I like to begin a session with a patient who is otherwise in good health with an open-ended question such as “Tell me a bit about yourself.” This provides an opportunity for patients to raise a range of issues without any additional focused questions being asked. In this context, if a patient brings up issues regarding sexual orientation or gender identity, ask permission to include this information in the medical record and assure the patient of its importance and that it will be confidential.
If these issues do not come up in response to general questions, they can be embedded in the sexual history, which should be more than a history of risk behaviors and should include a discussion of sexual health, sexual orientation (including identity, behavior, and desire), and gender identity. One can start by simply asking, “Do you have any concerns or questions about your sexuality, sexual orientation, or sexual desires?”
When it is necessary to ask more directed questions, it helps to provide some context so patients do not wonder why you are asking questions they may never have been asked by a physician before. It is best to explain that these are questions you ask all patients, as the information can be important in providing quality care. Patients should be told that discussion of sexual identity, behavior, and desire, as well as gender identity, is routine and confidential. For example, you might say: “I am going to ask you some questions about your sexual health and sexuality that I ask all my patients. The answers to these questions are important for me to know to help keep you healthy. Like the rest of this visit, this information is strictly confidential.”
One usually need not be too probing to get answers; people are often very forthcoming. During such conversations, patients often tell me that it is the first time a doctor has shown any interest in talking about these topics.
In having these conversations, initially it is best to use gender-neutral terms and pronouns when referring to partners until you know which to use: for example, “Do you have a partner or a spouse?” “Are you currently in a relationship?” “What do you call your partner?” Even if you make an incorrect assumption, and the patient corrects you, you can always apologize if a mistake is made and ask which term the patient prefers. Once you know it, use the pronoun that matches a person’s gender identity.
In order to get more information from the patient, the physician can engage in a series of questions, such as:
- Are you sexually active?
- When was the last time you had sex?
- When you have sex, do you do so with men, women, or both?
- How many sexual partners have you had during the last year?
- Do you have any desires regarding sexual intimacy that you would like to discuss?
In general, it is best to mirror the patient’s language. If patients use the term “gay” or “lesbian” to describe themselves, it would be off-putting to the patient to use a more clinical term, such as homosexual, in response. Some patients may use terms such as “queer” to indicate that they do not choose to identify as gay or straight. If terms like this are unclear to you, you may simply ask what this term means to the patient.
ASSESS SEXUAL BEHAVIOR TO DETERMINE RISK
In taking a history, it is important to distinguish sexual identity from sexual behavior. Physicians need to discuss sexual behavior with patients regardless of their sexual identity in order to do a risk-assessment, ascertaining what activities they engage in and to learn what they do to prevent transmission of sexually transmitted disease. In a 2006 study of more than 4,000 men in New York City,4 9.4% of those who identified themselves as straight had had sex with a man in the previous year. These men were more likely to be either foreign-born or from minority racial and ethnic groups with lower socioeconomic status. They were also less likely to have used a condom. A study of lesbians reported that 77% to 91% had at least one prior sexual experience with men, and 8% reported having had sex with a man in the previous year.6
Once you understand more about a patient’s sexual behavior, it is important to ask how patients protect themselves from human immunodeficiency virus (HIV) and other sexually transmitted diseases. If they use condoms or latex dams, they should be asked whether they do so consistently. Many patients have the misconception that they are practicing safe sex by only engaging in oral sex and do not realize that although it is probably protective against HIV infection, it does not protect against gonorrhea, syphilis, and other sexually transmitted diseases. Although most sexually transmitted diseases are treatable, their presence increases the risk of transmission of HIV.
Counseling on safer sex should include behavioral risk-reduction approaches. Depending on what behaviors a patient already engages in and what counseling he or she would be willing to accept, one could counsel abstinence, monogamy with an uninfected partner, reducing the number of partners, low-risk sexual practices, consistent and correct use of barrier methods, ceasing to engage in at least one high-risk activity, and avoiding excessive substance abuse. Physicians should advise patients to have a proactive plan to protect themselves and their partners. Patients should also be counseled on the correct use of barrier protection and on what is available for prophylaxis in case of high-risk HIV exposure (eg, a condom breaking or postcoital HIV disclosure). Another important question is, “Do you use alcohol or drugs when you have sex, and does your partner?” because these behaviors are often associated with unsafe sexual practices.
A new dimension of care will be biomedical prevention. While there are many ongoing studies of vaginal and anal microbicides to prevent HIV infection, there are also ongoing studies of antiretroviral therapies to do so.
One important new study demonstrated the effectiveness a biomedical intervention using antiretroviral therapy to prevent HIV infection in high-risk individuals.7 The study showed that men who were assigned to take a combination antiretroviral medication orally on a daily basis decreased their HIV risk by almost half compared with those assigned to take a placebo. The therapy was given along with intensive behavioral counseling. While this study was done in men who have sex with men, it is a major breakthrough and suggests there will be many new approaches to preventing HIV in the future.
A guide for clinicians has not been published by any government agency at this point, but guidance for clinicians is available from the Fenway Institute at www.fenwayhealth.org.
ASSESS GENDER-IDENTITY ISSUES
One should also routinely ask about whether patients are transgender or have gender-identity concerns. Psychologists start the conversation with the following example, which can also be used by general clinicians:
“Because so many people are impacted by gender issues, I have begun to ask everyone if they have any concerns about their gender. Anything you say about gender issues will be kept confidential. If this topic isn’t relevant to you, tell me and I’ll move on.”8
It is important to open the door to conversation, because many transgender people see a doctor for years and the topic never comes up. When they realize that they want to change their life, no one has ever helped them deal with the issues.
If appropriate, one can also say:
“Out of respect for my clients’ right to self-identify, I ask all clients what gender pronoun they’d prefer I use for them. What pronoun would you like me to use for you?”
Once these issues have been raised, it is important to support transgender people and help them explore a number of choices, including whether they wish to undergo hormone treatment, cosmetic surgery, and genital surgery. This may not be easy for many clinicians, so it will be important to learn about resources to care for transgender individuals in your community. Resources that can be very helpful for primary care clinicians include the following:
- The World Professional Association for Transgender Health (www.wpath.org) is the oldest and most traditional source for establishing standards of care.
- Vancouver Coastal Health published a series of monographs online (http://transhealth.vch.ca) that were developed by the University of British Columbia so that transgender people could be cared for in the community by primary care clinicians.
- The Endocrine Society in the United States published guidelines in 2009.9
PROVIDE SUPPORT FOR ‘COMING OUT’
We should also be understanding of people’s desires and support those who are “coming out.” The desire to reveal sexual orientation to others can happen at any age, including in childhood and among those who appear to have a traditional life because they are married and have children. Sometimes people do not know how to come out and would like to discuss such issues with their doctor.
MENTAL HEALTH CONCERNS
Given the marginalization and stigma that LGBT people face throughout their lives, it is not surprising that mental health problems are more prevalent in this population than in the general population. Gay and bisexual men have more depression, panic attacks, suicidal ideation, psychological distress, and body image and eating disorders than do heterosexual men. Lesbian and bisexual women are at greater risk of generalized anxiety disorder, depression, antidepressant use, and psychological distress.10 Care providers should screen for mental health disorders, assess comfort with sexual identity, and ask about social support.
FAMILY LIFE
Gays and lesbians increasingly want to discuss commitment, marriage, having children, parenting, and legal issues. A lot of research is being conducted on the sexual orientation of children raised by gay parents, and evidence shows that they are not more likely to be gay or lesbian than children raised by straight parents.
Elderly same-sex couples face special difficulties. They are less likely to feel comfortable “out of the closet” than are younger people. Fewer family and community supports are available to them, and they are often unable to live together in an assisted living facility. They particularly need to have advanced directives because they do not have the legal protections of other couples.
JUST A BEGINNING
While the points made above are relatively straightforward, they will open the door for many patients to have more meaningful conversations about their lives with their health care providers. It may only be a first step, but it can make a world of difference helping LGBT people feel comfortable accessing health care and receiving appropriate preventive care and treatment. Beyond the interaction with clinicians, health care providers should consider their overall environment and ensure that it is welcoming to LGBT individuals who come there for care.11
RESOURCES
Family Acceptance Project. familyproject.sfsu.edu
Gay & Lesbian Medical Association. www.glma.org
Human Rights Campaign. HRC.org
Parents, Families and Friends of Lesbians and Gays. PFLAG.org
World Professional Association for Transgender Health. www.wpath.org
Youth Resource (website by and for LGBT youth). Youthresource.com
- Makadon HJ. Improving health care for the lesbian and gay communities. N Engl J Med 2006; 354:895–897.
- Solarz AL, editor. Committee on Lesbian Health Research Priorities, Institute of Medicine. Lesbian Health: Current Assessment and Directions for the Future. Washington, DC: National Academy Press; 1999.
- Healthy People 2020. Lesbian, gay, bisexuaal, and transgender health. http://www.healthypeople.gov/2020/topicsobjectives2020/overview.aspx?topicid=25. Accessed 3/10/2011.
- Pathela P, Hajat A, Schillinger J, Blank S, Sell R, Mostashari F. Discordance between sexual behavior and self-reported sexual identity: a population-based survey of New York City men. Ann Intern Med 2006; 145:416–425. Erratum in: Ann Intern Med 2006; 145:936.
- Mosher WD, Chandra A, Jones J. Sexual behavior and selected health measures: men and women 15–44 years of age, United States, 2002. Adv Data 2005; 362:1–55.
- O’Hanlan KA, Robertson PA, Cabaj R, Schatz B, Nemrow P. A review of the medical consequences of homophobia with suggestions for resolution. Journal of the Gay and Lesbian Medical Association 1997; 1( 1):25–39.
- Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 2010; 363:2587–2599.
- Feldman J, Goldberg JM. Transgender Primary Medical Care: Suggested Guidelines for Clinicians in British Columbia. Vancouver, BC: Vancouver Coastal Health Authority, 2006.
- Hembree WC, Cohen-Kettenis P, Delemarre-van de Waal HA, et al; Endocrine Society. Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2009; 94:3132–3154.
- Cochran SD, Mays VM, Sullivan JG. Prevalence of mental disorders, psychological distress, and mental health services use among lesbian, gay, and bisexual adults in the United States. J Consult Clin Psychol 2003; 71:53–61.
- Human Rights Campaign Foundation. Healthcare equality index 2010. www.hrc.org/hei.
In speaking about lesbian, gay, bisexual, and transgender (LGBT) health, it is not uncommon for me to be asked what is so unique about the health care needs of lesbians, gay men, bisexuals, and transgender individuals that it warrants focused attention in the training of health professionals and while providing care.1 Although it is true that most health issues affecting LGBT individuals parallel those of the general population, people who are LGBT have been shown to have unique health needs and to experience disparities in care.
There is a growing if limited number of good studies of LGBT health. The Institute of Medicine2 reported on lesbian health in 1999, concluding that enough evidence of disparities exists to support more research and to develop better methods of conducting the research. Healthy People 2020 actually recognizes significant health care disparities.3 Finally, the Institute of Medicine recently formed a committee on LGBT health issues to identify gaps in our knowledge and priorities for research. Their findings were expected to be published in late March 2011, after this article went to press.
MAKING A DIFFERENCE
While this article will not attempt to discuss all the disparities, the focus will be on how physicians can take the first critical step to helping LGBT individuals feel comfortable seeking care, ie, by being proactive in taking a history that includes discussion of sexual orientation and gender identity. Only by knowing this about patients will clinicians appropriately care for specific health needs, and will patients feel comfortable discussing their concerns in clinical settings.
While some feel this is relevant only in select areas of the country, recent data show that the LGBT population is both spread throughout the country and diverse in how they might present themselves in clinical settings.1,4 In the United States, 1.4% to 4.1% of people identify themselves as lesbian, gay, or bisexual.5 About 3% of women and 4% of men say they have had a same-sex sexual contact in the last year, and 4% to 11% of women and 6% to 9% of men report having ever had one.
Everyone who practices clinical medicine needs to understand whether patients are LGBT and how to engage in conversation about sexual orientation and gender identity.
GETTING TO KNOW LGBT PATIENTS
What questions should a clinician ask to get this information? In thinking about what to ask, it helps to realize that patients generally do not mind being questioned about personal matters if the provider approaches the topic and the patient with genuine respect, empathy, and even curiosity.
On the other hand, providers often feel ill-prepared to discuss intimate issues, or feel uncomfortable doing so. Successfully achieving a change in clinical practice involves learning an approach to doing so and becoming comfortable with discussions that may follow. One question to consider is how you will feel and how you will follow up if a patient tells you that he or she is LGBT.
The core comprehensive history for LGBT patients is the same as for all patients, keeping in mind the unique LGBT health risks and issues. Clinicians may begin by getting to know each patient as a person (eg, ask about partners, children, and jobs). I like to begin a session with a patient who is otherwise in good health with an open-ended question such as “Tell me a bit about yourself.” This provides an opportunity for patients to raise a range of issues without any additional focused questions being asked. In this context, if a patient brings up issues regarding sexual orientation or gender identity, ask permission to include this information in the medical record and assure the patient of its importance and that it will be confidential.
If these issues do not come up in response to general questions, they can be embedded in the sexual history, which should be more than a history of risk behaviors and should include a discussion of sexual health, sexual orientation (including identity, behavior, and desire), and gender identity. One can start by simply asking, “Do you have any concerns or questions about your sexuality, sexual orientation, or sexual desires?”
When it is necessary to ask more directed questions, it helps to provide some context so patients do not wonder why you are asking questions they may never have been asked by a physician before. It is best to explain that these are questions you ask all patients, as the information can be important in providing quality care. Patients should be told that discussion of sexual identity, behavior, and desire, as well as gender identity, is routine and confidential. For example, you might say: “I am going to ask you some questions about your sexual health and sexuality that I ask all my patients. The answers to these questions are important for me to know to help keep you healthy. Like the rest of this visit, this information is strictly confidential.”
One usually need not be too probing to get answers; people are often very forthcoming. During such conversations, patients often tell me that it is the first time a doctor has shown any interest in talking about these topics.
In having these conversations, initially it is best to use gender-neutral terms and pronouns when referring to partners until you know which to use: for example, “Do you have a partner or a spouse?” “Are you currently in a relationship?” “What do you call your partner?” Even if you make an incorrect assumption, and the patient corrects you, you can always apologize if a mistake is made and ask which term the patient prefers. Once you know it, use the pronoun that matches a person’s gender identity.
In order to get more information from the patient, the physician can engage in a series of questions, such as:
- Are you sexually active?
- When was the last time you had sex?
- When you have sex, do you do so with men, women, or both?
- How many sexual partners have you had during the last year?
- Do you have any desires regarding sexual intimacy that you would like to discuss?
In general, it is best to mirror the patient’s language. If patients use the term “gay” or “lesbian” to describe themselves, it would be off-putting to the patient to use a more clinical term, such as homosexual, in response. Some patients may use terms such as “queer” to indicate that they do not choose to identify as gay or straight. If terms like this are unclear to you, you may simply ask what this term means to the patient.
ASSESS SEXUAL BEHAVIOR TO DETERMINE RISK
In taking a history, it is important to distinguish sexual identity from sexual behavior. Physicians need to discuss sexual behavior with patients regardless of their sexual identity in order to do a risk-assessment, ascertaining what activities they engage in and to learn what they do to prevent transmission of sexually transmitted disease. In a 2006 study of more than 4,000 men in New York City,4 9.4% of those who identified themselves as straight had had sex with a man in the previous year. These men were more likely to be either foreign-born or from minority racial and ethnic groups with lower socioeconomic status. They were also less likely to have used a condom. A study of lesbians reported that 77% to 91% had at least one prior sexual experience with men, and 8% reported having had sex with a man in the previous year.6
Once you understand more about a patient’s sexual behavior, it is important to ask how patients protect themselves from human immunodeficiency virus (HIV) and other sexually transmitted diseases. If they use condoms or latex dams, they should be asked whether they do so consistently. Many patients have the misconception that they are practicing safe sex by only engaging in oral sex and do not realize that although it is probably protective against HIV infection, it does not protect against gonorrhea, syphilis, and other sexually transmitted diseases. Although most sexually transmitted diseases are treatable, their presence increases the risk of transmission of HIV.
Counseling on safer sex should include behavioral risk-reduction approaches. Depending on what behaviors a patient already engages in and what counseling he or she would be willing to accept, one could counsel abstinence, monogamy with an uninfected partner, reducing the number of partners, low-risk sexual practices, consistent and correct use of barrier methods, ceasing to engage in at least one high-risk activity, and avoiding excessive substance abuse. Physicians should advise patients to have a proactive plan to protect themselves and their partners. Patients should also be counseled on the correct use of barrier protection and on what is available for prophylaxis in case of high-risk HIV exposure (eg, a condom breaking or postcoital HIV disclosure). Another important question is, “Do you use alcohol or drugs when you have sex, and does your partner?” because these behaviors are often associated with unsafe sexual practices.
A new dimension of care will be biomedical prevention. While there are many ongoing studies of vaginal and anal microbicides to prevent HIV infection, there are also ongoing studies of antiretroviral therapies to do so.
One important new study demonstrated the effectiveness a biomedical intervention using antiretroviral therapy to prevent HIV infection in high-risk individuals.7 The study showed that men who were assigned to take a combination antiretroviral medication orally on a daily basis decreased their HIV risk by almost half compared with those assigned to take a placebo. The therapy was given along with intensive behavioral counseling. While this study was done in men who have sex with men, it is a major breakthrough and suggests there will be many new approaches to preventing HIV in the future.
A guide for clinicians has not been published by any government agency at this point, but guidance for clinicians is available from the Fenway Institute at www.fenwayhealth.org.
ASSESS GENDER-IDENTITY ISSUES
One should also routinely ask about whether patients are transgender or have gender-identity concerns. Psychologists start the conversation with the following example, which can also be used by general clinicians:
“Because so many people are impacted by gender issues, I have begun to ask everyone if they have any concerns about their gender. Anything you say about gender issues will be kept confidential. If this topic isn’t relevant to you, tell me and I’ll move on.”8
It is important to open the door to conversation, because many transgender people see a doctor for years and the topic never comes up. When they realize that they want to change their life, no one has ever helped them deal with the issues.
If appropriate, one can also say:
“Out of respect for my clients’ right to self-identify, I ask all clients what gender pronoun they’d prefer I use for them. What pronoun would you like me to use for you?”
Once these issues have been raised, it is important to support transgender people and help them explore a number of choices, including whether they wish to undergo hormone treatment, cosmetic surgery, and genital surgery. This may not be easy for many clinicians, so it will be important to learn about resources to care for transgender individuals in your community. Resources that can be very helpful for primary care clinicians include the following:
- The World Professional Association for Transgender Health (www.wpath.org) is the oldest and most traditional source for establishing standards of care.
- Vancouver Coastal Health published a series of monographs online (http://transhealth.vch.ca) that were developed by the University of British Columbia so that transgender people could be cared for in the community by primary care clinicians.
- The Endocrine Society in the United States published guidelines in 2009.9
PROVIDE SUPPORT FOR ‘COMING OUT’
We should also be understanding of people’s desires and support those who are “coming out.” The desire to reveal sexual orientation to others can happen at any age, including in childhood and among those who appear to have a traditional life because they are married and have children. Sometimes people do not know how to come out and would like to discuss such issues with their doctor.
MENTAL HEALTH CONCERNS
Given the marginalization and stigma that LGBT people face throughout their lives, it is not surprising that mental health problems are more prevalent in this population than in the general population. Gay and bisexual men have more depression, panic attacks, suicidal ideation, psychological distress, and body image and eating disorders than do heterosexual men. Lesbian and bisexual women are at greater risk of generalized anxiety disorder, depression, antidepressant use, and psychological distress.10 Care providers should screen for mental health disorders, assess comfort with sexual identity, and ask about social support.
FAMILY LIFE
Gays and lesbians increasingly want to discuss commitment, marriage, having children, parenting, and legal issues. A lot of research is being conducted on the sexual orientation of children raised by gay parents, and evidence shows that they are not more likely to be gay or lesbian than children raised by straight parents.
Elderly same-sex couples face special difficulties. They are less likely to feel comfortable “out of the closet” than are younger people. Fewer family and community supports are available to them, and they are often unable to live together in an assisted living facility. They particularly need to have advanced directives because they do not have the legal protections of other couples.
JUST A BEGINNING
While the points made above are relatively straightforward, they will open the door for many patients to have more meaningful conversations about their lives with their health care providers. It may only be a first step, but it can make a world of difference helping LGBT people feel comfortable accessing health care and receiving appropriate preventive care and treatment. Beyond the interaction with clinicians, health care providers should consider their overall environment and ensure that it is welcoming to LGBT individuals who come there for care.11
RESOURCES
Family Acceptance Project. familyproject.sfsu.edu
Gay & Lesbian Medical Association. www.glma.org
Human Rights Campaign. HRC.org
Parents, Families and Friends of Lesbians and Gays. PFLAG.org
World Professional Association for Transgender Health. www.wpath.org
Youth Resource (website by and for LGBT youth). Youthresource.com
In speaking about lesbian, gay, bisexual, and transgender (LGBT) health, it is not uncommon for me to be asked what is so unique about the health care needs of lesbians, gay men, bisexuals, and transgender individuals that it warrants focused attention in the training of health professionals and while providing care.1 Although it is true that most health issues affecting LGBT individuals parallel those of the general population, people who are LGBT have been shown to have unique health needs and to experience disparities in care.
There is a growing if limited number of good studies of LGBT health. The Institute of Medicine2 reported on lesbian health in 1999, concluding that enough evidence of disparities exists to support more research and to develop better methods of conducting the research. Healthy People 2020 actually recognizes significant health care disparities.3 Finally, the Institute of Medicine recently formed a committee on LGBT health issues to identify gaps in our knowledge and priorities for research. Their findings were expected to be published in late March 2011, after this article went to press.
MAKING A DIFFERENCE
While this article will not attempt to discuss all the disparities, the focus will be on how physicians can take the first critical step to helping LGBT individuals feel comfortable seeking care, ie, by being proactive in taking a history that includes discussion of sexual orientation and gender identity. Only by knowing this about patients will clinicians appropriately care for specific health needs, and will patients feel comfortable discussing their concerns in clinical settings.
While some feel this is relevant only in select areas of the country, recent data show that the LGBT population is both spread throughout the country and diverse in how they might present themselves in clinical settings.1,4 In the United States, 1.4% to 4.1% of people identify themselves as lesbian, gay, or bisexual.5 About 3% of women and 4% of men say they have had a same-sex sexual contact in the last year, and 4% to 11% of women and 6% to 9% of men report having ever had one.
Everyone who practices clinical medicine needs to understand whether patients are LGBT and how to engage in conversation about sexual orientation and gender identity.
GETTING TO KNOW LGBT PATIENTS
What questions should a clinician ask to get this information? In thinking about what to ask, it helps to realize that patients generally do not mind being questioned about personal matters if the provider approaches the topic and the patient with genuine respect, empathy, and even curiosity.
On the other hand, providers often feel ill-prepared to discuss intimate issues, or feel uncomfortable doing so. Successfully achieving a change in clinical practice involves learning an approach to doing so and becoming comfortable with discussions that may follow. One question to consider is how you will feel and how you will follow up if a patient tells you that he or she is LGBT.
The core comprehensive history for LGBT patients is the same as for all patients, keeping in mind the unique LGBT health risks and issues. Clinicians may begin by getting to know each patient as a person (eg, ask about partners, children, and jobs). I like to begin a session with a patient who is otherwise in good health with an open-ended question such as “Tell me a bit about yourself.” This provides an opportunity for patients to raise a range of issues without any additional focused questions being asked. In this context, if a patient brings up issues regarding sexual orientation or gender identity, ask permission to include this information in the medical record and assure the patient of its importance and that it will be confidential.
If these issues do not come up in response to general questions, they can be embedded in the sexual history, which should be more than a history of risk behaviors and should include a discussion of sexual health, sexual orientation (including identity, behavior, and desire), and gender identity. One can start by simply asking, “Do you have any concerns or questions about your sexuality, sexual orientation, or sexual desires?”
When it is necessary to ask more directed questions, it helps to provide some context so patients do not wonder why you are asking questions they may never have been asked by a physician before. It is best to explain that these are questions you ask all patients, as the information can be important in providing quality care. Patients should be told that discussion of sexual identity, behavior, and desire, as well as gender identity, is routine and confidential. For example, you might say: “I am going to ask you some questions about your sexual health and sexuality that I ask all my patients. The answers to these questions are important for me to know to help keep you healthy. Like the rest of this visit, this information is strictly confidential.”
One usually need not be too probing to get answers; people are often very forthcoming. During such conversations, patients often tell me that it is the first time a doctor has shown any interest in talking about these topics.
In having these conversations, initially it is best to use gender-neutral terms and pronouns when referring to partners until you know which to use: for example, “Do you have a partner or a spouse?” “Are you currently in a relationship?” “What do you call your partner?” Even if you make an incorrect assumption, and the patient corrects you, you can always apologize if a mistake is made and ask which term the patient prefers. Once you know it, use the pronoun that matches a person’s gender identity.
In order to get more information from the patient, the physician can engage in a series of questions, such as:
- Are you sexually active?
- When was the last time you had sex?
- When you have sex, do you do so with men, women, or both?
- How many sexual partners have you had during the last year?
- Do you have any desires regarding sexual intimacy that you would like to discuss?
In general, it is best to mirror the patient’s language. If patients use the term “gay” or “lesbian” to describe themselves, it would be off-putting to the patient to use a more clinical term, such as homosexual, in response. Some patients may use terms such as “queer” to indicate that they do not choose to identify as gay or straight. If terms like this are unclear to you, you may simply ask what this term means to the patient.
ASSESS SEXUAL BEHAVIOR TO DETERMINE RISK
In taking a history, it is important to distinguish sexual identity from sexual behavior. Physicians need to discuss sexual behavior with patients regardless of their sexual identity in order to do a risk-assessment, ascertaining what activities they engage in and to learn what they do to prevent transmission of sexually transmitted disease. In a 2006 study of more than 4,000 men in New York City,4 9.4% of those who identified themselves as straight had had sex with a man in the previous year. These men were more likely to be either foreign-born or from minority racial and ethnic groups with lower socioeconomic status. They were also less likely to have used a condom. A study of lesbians reported that 77% to 91% had at least one prior sexual experience with men, and 8% reported having had sex with a man in the previous year.6
Once you understand more about a patient’s sexual behavior, it is important to ask how patients protect themselves from human immunodeficiency virus (HIV) and other sexually transmitted diseases. If they use condoms or latex dams, they should be asked whether they do so consistently. Many patients have the misconception that they are practicing safe sex by only engaging in oral sex and do not realize that although it is probably protective against HIV infection, it does not protect against gonorrhea, syphilis, and other sexually transmitted diseases. Although most sexually transmitted diseases are treatable, their presence increases the risk of transmission of HIV.
Counseling on safer sex should include behavioral risk-reduction approaches. Depending on what behaviors a patient already engages in and what counseling he or she would be willing to accept, one could counsel abstinence, monogamy with an uninfected partner, reducing the number of partners, low-risk sexual practices, consistent and correct use of barrier methods, ceasing to engage in at least one high-risk activity, and avoiding excessive substance abuse. Physicians should advise patients to have a proactive plan to protect themselves and their partners. Patients should also be counseled on the correct use of barrier protection and on what is available for prophylaxis in case of high-risk HIV exposure (eg, a condom breaking or postcoital HIV disclosure). Another important question is, “Do you use alcohol or drugs when you have sex, and does your partner?” because these behaviors are often associated with unsafe sexual practices.
A new dimension of care will be biomedical prevention. While there are many ongoing studies of vaginal and anal microbicides to prevent HIV infection, there are also ongoing studies of antiretroviral therapies to do so.
One important new study demonstrated the effectiveness a biomedical intervention using antiretroviral therapy to prevent HIV infection in high-risk individuals.7 The study showed that men who were assigned to take a combination antiretroviral medication orally on a daily basis decreased their HIV risk by almost half compared with those assigned to take a placebo. The therapy was given along with intensive behavioral counseling. While this study was done in men who have sex with men, it is a major breakthrough and suggests there will be many new approaches to preventing HIV in the future.
A guide for clinicians has not been published by any government agency at this point, but guidance for clinicians is available from the Fenway Institute at www.fenwayhealth.org.
ASSESS GENDER-IDENTITY ISSUES
One should also routinely ask about whether patients are transgender or have gender-identity concerns. Psychologists start the conversation with the following example, which can also be used by general clinicians:
“Because so many people are impacted by gender issues, I have begun to ask everyone if they have any concerns about their gender. Anything you say about gender issues will be kept confidential. If this topic isn’t relevant to you, tell me and I’ll move on.”8
It is important to open the door to conversation, because many transgender people see a doctor for years and the topic never comes up. When they realize that they want to change their life, no one has ever helped them deal with the issues.
If appropriate, one can also say:
“Out of respect for my clients’ right to self-identify, I ask all clients what gender pronoun they’d prefer I use for them. What pronoun would you like me to use for you?”
Once these issues have been raised, it is important to support transgender people and help them explore a number of choices, including whether they wish to undergo hormone treatment, cosmetic surgery, and genital surgery. This may not be easy for many clinicians, so it will be important to learn about resources to care for transgender individuals in your community. Resources that can be very helpful for primary care clinicians include the following:
- The World Professional Association for Transgender Health (www.wpath.org) is the oldest and most traditional source for establishing standards of care.
- Vancouver Coastal Health published a series of monographs online (http://transhealth.vch.ca) that were developed by the University of British Columbia so that transgender people could be cared for in the community by primary care clinicians.
- The Endocrine Society in the United States published guidelines in 2009.9
PROVIDE SUPPORT FOR ‘COMING OUT’
We should also be understanding of people’s desires and support those who are “coming out.” The desire to reveal sexual orientation to others can happen at any age, including in childhood and among those who appear to have a traditional life because they are married and have children. Sometimes people do not know how to come out and would like to discuss such issues with their doctor.
MENTAL HEALTH CONCERNS
Given the marginalization and stigma that LGBT people face throughout their lives, it is not surprising that mental health problems are more prevalent in this population than in the general population. Gay and bisexual men have more depression, panic attacks, suicidal ideation, psychological distress, and body image and eating disorders than do heterosexual men. Lesbian and bisexual women are at greater risk of generalized anxiety disorder, depression, antidepressant use, and psychological distress.10 Care providers should screen for mental health disorders, assess comfort with sexual identity, and ask about social support.
FAMILY LIFE
Gays and lesbians increasingly want to discuss commitment, marriage, having children, parenting, and legal issues. A lot of research is being conducted on the sexual orientation of children raised by gay parents, and evidence shows that they are not more likely to be gay or lesbian than children raised by straight parents.
Elderly same-sex couples face special difficulties. They are less likely to feel comfortable “out of the closet” than are younger people. Fewer family and community supports are available to them, and they are often unable to live together in an assisted living facility. They particularly need to have advanced directives because they do not have the legal protections of other couples.
JUST A BEGINNING
While the points made above are relatively straightforward, they will open the door for many patients to have more meaningful conversations about their lives with their health care providers. It may only be a first step, but it can make a world of difference helping LGBT people feel comfortable accessing health care and receiving appropriate preventive care and treatment. Beyond the interaction with clinicians, health care providers should consider their overall environment and ensure that it is welcoming to LGBT individuals who come there for care.11
RESOURCES
Family Acceptance Project. familyproject.sfsu.edu
Gay & Lesbian Medical Association. www.glma.org
Human Rights Campaign. HRC.org
Parents, Families and Friends of Lesbians and Gays. PFLAG.org
World Professional Association for Transgender Health. www.wpath.org
Youth Resource (website by and for LGBT youth). Youthresource.com
- Makadon HJ. Improving health care for the lesbian and gay communities. N Engl J Med 2006; 354:895–897.
- Solarz AL, editor. Committee on Lesbian Health Research Priorities, Institute of Medicine. Lesbian Health: Current Assessment and Directions for the Future. Washington, DC: National Academy Press; 1999.
- Healthy People 2020. Lesbian, gay, bisexuaal, and transgender health. http://www.healthypeople.gov/2020/topicsobjectives2020/overview.aspx?topicid=25. Accessed 3/10/2011.
- Pathela P, Hajat A, Schillinger J, Blank S, Sell R, Mostashari F. Discordance between sexual behavior and self-reported sexual identity: a population-based survey of New York City men. Ann Intern Med 2006; 145:416–425. Erratum in: Ann Intern Med 2006; 145:936.
- Mosher WD, Chandra A, Jones J. Sexual behavior and selected health measures: men and women 15–44 years of age, United States, 2002. Adv Data 2005; 362:1–55.
- O’Hanlan KA, Robertson PA, Cabaj R, Schatz B, Nemrow P. A review of the medical consequences of homophobia with suggestions for resolution. Journal of the Gay and Lesbian Medical Association 1997; 1( 1):25–39.
- Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 2010; 363:2587–2599.
- Feldman J, Goldberg JM. Transgender Primary Medical Care: Suggested Guidelines for Clinicians in British Columbia. Vancouver, BC: Vancouver Coastal Health Authority, 2006.
- Hembree WC, Cohen-Kettenis P, Delemarre-van de Waal HA, et al; Endocrine Society. Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2009; 94:3132–3154.
- Cochran SD, Mays VM, Sullivan JG. Prevalence of mental disorders, psychological distress, and mental health services use among lesbian, gay, and bisexual adults in the United States. J Consult Clin Psychol 2003; 71:53–61.
- Human Rights Campaign Foundation. Healthcare equality index 2010. www.hrc.org/hei.
- Makadon HJ. Improving health care for the lesbian and gay communities. N Engl J Med 2006; 354:895–897.
- Solarz AL, editor. Committee on Lesbian Health Research Priorities, Institute of Medicine. Lesbian Health: Current Assessment and Directions for the Future. Washington, DC: National Academy Press; 1999.
- Healthy People 2020. Lesbian, gay, bisexuaal, and transgender health. http://www.healthypeople.gov/2020/topicsobjectives2020/overview.aspx?topicid=25. Accessed 3/10/2011.
- Pathela P, Hajat A, Schillinger J, Blank S, Sell R, Mostashari F. Discordance between sexual behavior and self-reported sexual identity: a population-based survey of New York City men. Ann Intern Med 2006; 145:416–425. Erratum in: Ann Intern Med 2006; 145:936.
- Mosher WD, Chandra A, Jones J. Sexual behavior and selected health measures: men and women 15–44 years of age, United States, 2002. Adv Data 2005; 362:1–55.
- O’Hanlan KA, Robertson PA, Cabaj R, Schatz B, Nemrow P. A review of the medical consequences of homophobia with suggestions for resolution. Journal of the Gay and Lesbian Medical Association 1997; 1( 1):25–39.
- Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 2010; 363:2587–2599.
- Feldman J, Goldberg JM. Transgender Primary Medical Care: Suggested Guidelines for Clinicians in British Columbia. Vancouver, BC: Vancouver Coastal Health Authority, 2006.
- Hembree WC, Cohen-Kettenis P, Delemarre-van de Waal HA, et al; Endocrine Society. Endocrine treatment of transsexual persons: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2009; 94:3132–3154.
- Cochran SD, Mays VM, Sullivan JG. Prevalence of mental disorders, psychological distress, and mental health services use among lesbian, gay, and bisexual adults in the United States. J Consult Clin Psychol 2003; 71:53–61.
- Human Rights Campaign Foundation. Healthcare equality index 2010. www.hrc.org/hei.
KEY POINTS
- LGBT people are represented in most medical practices, and their health issues, including sexually transmitted diseases such as human immunodeficiency virus, can generally be managed in traditional health care settings rather than in special clinics.
- Physicians need to become more comfortable asking patients about sexual health, identity, and behavior, and make such queries more routine.
- Sexual behavior is not always congruent with routine understanding of sexual identity. For example, many men who do not identify themselves as gay occasionally have sex with men, as do many self-identified lesbians. It is important to know this to provide appropriate preventive screening and care.
Seek and treat: HIV update 2011
With early treatment of human immunodeficiency virus (HIV) infection, we can now expect patients to live a much longer life and, in some situations, have a near-normal lifespan.1 Unfortunately, in screening for HIV infection, the United States lags behind many regions of the world, and infection is often not diagnosed until patients present with advanced disease, ie, the acquired immunodeficiency syndrome (AIDS). In this country there is a critical need to make HIV screening a routine part of medical care in all health settings in order to give patients their best chance for a healthy life, to prevent mother-to-child transmission, and to reduce the spread of HIV in the community.
HIV infection meets the criteria that justify routine screening, as laid out by the World Health Organization2:
- It is a serious health disorder that can be detected before symptoms develop
- Treatment is more beneficial if begun before symptoms develop
- Reliable, inexpensive, and acceptable screening tests exist
- The costs of screening are reasonable in relation to the anticipated benefits.
This article will review the epidemiology of the HIV epidemic, present the benefits of early treatment, and make the case for widely expanding screening for HIV infection in the US health care system.
HIV INFECTION CONTINUES TO BE A LARGE BURDEN
In 2008, an estimated 33.4 million people worldwide were HIV-positive. The vast majority of infected people—more than 22 million—live in sub-Saharan Africa.3
The United States has approximately 1.2 million cases.4 Although this is a small proportion of cases worldwide, it still represents a significant health care burden. In this country, the number of AIDS cases peaked in 1993, and the rate of deaths from AIDS began to decrease over the ensuing years as adequate therapy for HIV was developed. Standard therapy then and now consists of at least three drugs from two different classes.
Unfortunately, we have made little progress on the incidence of this disease. The estimated number of new HIV infections in the United States in 2008 was 56,000 and had remained about the same over the previous 15 years.5,6 Because of improved rates of survival, the prevalence has risen steadily since the mid-1990s to the current estimate of 1.2 million persons living with HIV/AIDS in the US.
About 25% of people infected with HIV are unaware of it. This group accounts for more than half of all new infections annually, which highlights the importance of enhanced screening. Once people know they are infected, they tend to change their behavior and are less likely to spread the disease.7
HIV disproportionately affects minority populations and gay men
Cases of HIV infection are reported among all age groups, although most patients tend to have been infected as young adults. Currently, the largest age group living with HIV is middle-aged. As this cohort grows older, an increasing burden of comorbidities due to aging can be expected. In 5 years, about half of the people with HIV in this country are expected to be 50 years of age or older. Although survival rates have steadily increased due to better treatment, survival tends to be shorter for older people newly diagnosed with HIV.
Worldwide, about an equal number of men and women are infected with HIV, but in the United States infected men outnumber women. In this country, about half the cases of HIV transmission among adults are by male-to-male sexual contact, about 30% are by high-risk heterosexual contact (ie, with a partner known to be HIV-infected or at high risk for being infected), and about 10% are by injection drug use.
In the United States, AIDS is predominantly and disproportionately a disease of minorities and those who live in poverty. African Americans account for the largest number of cases, followed by whites and then by Hispanics. Combined, African Americans and Hispanics account for two-thirds to three-fourths of all new cases, although they make up less than one-fourth of the US population. The incidence rate is nearly 137 per 100,000 for African Americans, 56 per 100,000 for Hispanics, and 19 per 100,000 for whites. The incidence is highest in New York and in the southeast, the geographic areas where the greatest number of minorities and people living in poverty reside. These groups also often lack access to health care.
HIV TREATMENT IS MORE EFFECTIVE IF STARTED EARLY
Treatment guidelines from the US Department of Health and Human Services (DHHS) have changed over the years. When effective medications were first introduced in the 1990s, the trend was to treat everyone as soon as they were diagnosed. As the burden of therapy began to unfold (side effects, cost, adherence, and drug resistance), the consensus was to wait until the CD4 T-cell count dropped to a lower level. As the medications have improved and have become better tolerated, the pendulum has swung back to treating earlier in the course of the disease. Currently, the DHHS recommends that therapy be started at CD4 counts of 350 cells/mL or lower (level of evidence: A1).8 It also recommends therapy for CD4 counts between 350 and 500 cells/mL, but the level of evidence is lower.8
The CD4 T cell is the prime target of the HIV virus and also an important marker of the health of the immune system. The lower the CD4 count at the start of therapy, the more challenging it is to normalize.9 If HIV infection is diagnosed early and therapy is started early, the likelihood is higher of normalizing the CD4 count and preserving immune function.
Progress is being made toward diagnosing HIV earlier. The CD4 count at presentation is increasing, but patients in the United States still present for care later than in other countries. In 1997, the median CD4 count at presentation was 234 cells/mL; in 2007, it was 327 (normal is about 550–1,000). Although this is a significant improvement, more than 50% of patients still have fewer than 350 cells/mL at presentation, which is the current threshold for beginning therapy, according to the most recent guidelines.10
Before triple therapy was available, almost all HIV-infected patients died of AIDS-related diseases. Now, about half of treated HIV-infected patients in Europe and North America die of other causes.11 However, many diseases not previously attributed to AIDS are now also known to be exacerbated by HIV infection.
Cancer risk increases with lower CD4 counts
The cumulative incidence of AIDS-defining cancers (Kaposi sarcoma, non-Hodgkin lymphoma, cervical carcinoma) has decreased steadily from 8.7% in the 1980s to 6.4% during the years 1990 to 1995, and to 2.1% between 1996 and 2006. This is attributable to improved immune function as a result of treatment success with antiviral therapy.12
But the incidence of non-AIDS-defining cancers (Hodgkin disease, anal cancer, oral and respiratory cancers) has increased.11 As therapy has regenerated the immune system, patients are surviving longer and are developing the more common cancers but with higher rates than in the general population.
Higher cancer risk is attributed to reduced immune surveillance. Many of these cancers are associated with viruses, such as human papillomavirus (anal and oral or pharyngeal cancers) and Epstein-Barr virus (Hodgkin disease), which can usually be controlled by a fully functioning immune system. The lower the CD4 count, the higher the risk of cancer, which highlights the need to diagnose HIV and start treatment early.13
Cardiovascular disease increases with lower CD4 counts
Associations have recently been identified between coronary disease and HIV as well as with HIV medications. Protease inhibitors tend to raise the levels of triglycerides, low-density lipoprotein cholesterol, and total cholesterol and increase the risk of heart attack.14
Regardless of therapy, HIV appears to be an independent risk factor for coronary disease. Arterial stiffness, as measured by carotid femoral pulse-wave velocity, was found to be increased among a sample of 80 HIV-infected men. This was associated with the usual risk factors of increasing age, blood pressure, and diabetes, as well as with lower nadir CD4 count.15
Fractures and neurocognitive disorders increase with HIV
Osteoporotic fractures are also more common in patients with HIV than in the general population. Risk factors include the traditional risks of older age, hepatitis C infection, diabetes, and substance abuse, but also nadir CD4 count less than 200.16
The risk of neurocognitive disorders is also associated with lower nadir CD4 counts. The lower the CD4 count, the higher the risk of developing neurocognitive deficits.17 The potential benefits of earlier diagnosis and treatment are obvious based upon the multiple recent findings outlined above.
CLINICAL PRESENTATION OF PRIMARY HIV INFECTION
During primary HIV infection, when patients are first infected, 50% to 90% are symptomatic. Symptoms usually appear in the first 6 weeks. The viral load tends to be highest at this time. Higher viral loads appear directly correlated with the degree of infectivity, highlighting the urgency of finding and treating new infections promptly to help avoid transmission to others.18
The clinical picture during primary infection is similar to that of acute mononucleosis. Signs and symptoms include fever, fatigue, rash, headache, lymphadenopathy, sore throat, and muscle aches. Although this presentation is common to many viral infections, questioning the patient about high-risk behavior (unprotected sex, multiple partners, intravenous drug use) will lead the astute physician to the correct testing and diagnosis.
Other early manifestations include mucocutaneous signs, such as seborrheic dermatitis, psoriasis, folliculitis, and thrush. Laboratory test results demonstrating leukopenia, thrombocytopenia, elevated total protein levels, proteinuria, and transaminitis are also suggestive of HIV infection.
THE CASE FOR INCREASED TESTING AND TREATMENT
The estimated prevalence of HIV in the United States is approximately 0.3%. However, its prevalence in Washington, DC, is 3%, which rivals rates in some areas of the developing world. From 2004 to 2008, health officials made a concerted effort in Washington, DC, to screen more people, particularly those at high risk. The number of publicly funded HIV tests performed increased by a factor of 3.7, and the number of newly reported cases increased by 17%. There was also a significant increase in the median CD4 count at the time of HIV diagnosis and a significant delay in time to progression to AIDS after HIV diagnosis.19
A study in British Columbia expanded access to highly active antiretroviral therapy during 2004 through 2009. High-risk individuals were targeted for increased screening. All those diagnosed with HIV were provided free medication. This resulted in a 50% reduction in new diagnoses of HIV infection throughout the community, especially among injectable drug users, a usually marginalized population. The proportion of patients with HIV-1 RNA levels above 1,500 copies/mL fell from about 50% to about 20%, indicating that the viral load—a measure of infectivity throughout the community—was reduced. Interestingly, this trend occurred during a time of increased rates of gonorrhea, syphilis, and other sexually transmitted diseases known to be associated with enhanced HIV transmission.20
In Africa, antiretroviral therapy was offered to discordant couples (one partner was infected with HIV and the other was not). Among those who chose therapy, the rate of HIV transmission was 92% lower than in those not receiving antiretroviral drugs,21 once again demonstrating that control of HIV by treatment can lead to decreased transmission.
US HIV testing is inadequate
The current state of HIV testing in the United States needs to be improved. Testing is not performed routinely, leading to delayed diagnosis when patients present with symptomatic, advanced disease. Patients who are tested late (within 12 months before being diagnosed with AIDS) tend to be younger and less educated and are more likely to be heterosexual and either African American or Hispanic than patients who are tested earlier.22 When retrospectively evaluated, these patients often have been in the health care system but not tested. Routine universal screening and targeted testing could lead to a much earlier diagnosis and potential better long-term outcomes.
A 1996 survey of 95 academic emergency departments found that for patients with suspected sexually transmitted infections, 93% of physicians said they screen for gonorrhea, 88% for Chlamydia infection, 58% for syphilis, but only 3% for HIV.23 Sexually transmitted infections and HIV are often transmitted together.
A similar 2002 survey of 154 emergency department providers who saw an average of 13 patients with sexually transmitted infections per week found that only 10% always recommend HIV testing to these patients. Reasons given for not testing were concern about follow-up (51%), not having a “certified” counselor (45%), HIV testing being too time-consuming (19%), and HIV testing being unavailable (27%).24
Although most HIV tests are given by private doctors and health maintenance organizations, the likelihood of finding patients with HIV is greatest in hospitals, emergency departments, outpatient clinics, and public community clinics.
The Advancing HIV Prevention initiative of the US Centers for Disease Control and Prevention (CDC) has four priorities:
- To make voluntary HIV testing a routine part of medical care
- To implement new models for diagnosing HIV infection outside medical settings
- To prevent HIV infection by working with patients with HIV and their partners
- To further decrease the rate of perinatal HIV transmission.
Rapid tests for HIV are available
There is a public health need to have rapid HIV testing available in all health care settings. With standard HIV tests, which can take 48 to 72 hours to run, about one-third of patients do not return for results. Subsequently locating them can be a huge challenge and is sometimes impossible. The ability to have rapid test results can improve this situation. It is especially important in prenatal care settings, where the mother can be immediately treated to reduce the risk of transmission to the child. Rapid testing increases the feasibility of testing in multiple venues, particularly acute-care settings with almost immediate results and linkage to care.
Several rapid tests are available and can be performed on whole blood, serum, plasma, and oral fluid. The tests provide reliable results in minutes, with 99% sensitivity and specificity. Positive results must be confirmed by subsequent two-stage laboratory testing, enzyme-linked immunosorbent assay, and Western blot. Patients who have negative or have indeterminate results on Western blot testing should be tested again after 4 weeks.
The cost-effectiveness of routine screening for HIV, even in populations with a low prevalence, is similar to that of commonly accepted interventions.25 In populations with a 1% prevalence of HIV, the cost is $15,078 per quality-adjusted life-year.26 Even if the prevalence is less than 0.05%, the cost is less than $50,000 per quality-adjusted life-year, which is normally the cutoff for acceptability for screening tests.25,26
‘OPT-OUT’ TESTING
In the past, patients were asked if they would like to have HIV testing (“opt-in” testing). It is now recommended that physicians request testing to be performed (“opt-out” testing). This still allows the patient to decline but also conveys a “matter of fact” nonjudgmental message, indicative of a routine procedure no different than other screening tests. When testing was done on an opt-in basis, only 35% of pregnant women agreed to be tested. Some women felt that accepting an HIV test indicated that they engage in high-risk behavior. When testing was instead offered as routine but with an opportunity to decline, 88% accepted testing, and they were significantly less anxious about testing.27
CDC RECOMMENDATIONS
The CDC now recommends that routine, voluntary HIV screening be done for all persons ages 13 to 64 in health care settings, regardless of risk.28 Screening should be repeated at least annually in persons with known risk. Screening should be done on an opt-out basis, with the opportunity to ask questions and the option to decline. Consent for HIV testing should be included with general consent for care. A separate signed informed consent is not recommended, and verbal consent can merely be documented in the medical record. Prevention counseling in conjunction with HIV screening in health care settings is not required.
Testing should be done in all health care settings, including primary care settings, inpatient services, emergency departments, urgent care clinics, and sexually transmitted disease clinics. Test results should be communicated in the same manner as other diagnostic and screening care. Clinical HIV care should be available onsite or reliable referral to qualified providers should be established.
For pregnant women, the CDC recommends universal opt-out HIV screening, with HIV testing as part of the routine panel of prenatal screening tests. The consent for prenatal care includes HIV testing, with notification and the option to decline. Women should be tested again in the third trimester if they are known to be at risk for HIV, and in areas and health care facilities in which the prevalence of HIV is high.
In women whose HIV status is undocumented in labor and delivery, opt-out rapid testing should be performed, and antiretroviral prophylaxis should be given on the basis of the rapid test result. Rapid testing of the newborn is recommended if the mother’s status is unknown at delivery, and antiretroviral prophylaxis should be started within 12 hours of birth on the basis of the rapid test result.
Widespread routine screening and earlier treatment could significantly reduce the incidence and improve the outcomes of HIV in this country. Health care providers are encouraged to adopt these practices.
- Van Sighem A, Gras L, Reiss P, Brinkman K, de Wolf F, and ATHENA Natl Observational Cohort Study. Life expectancy of recently diagnosed asymptomatic HIV-infected patients approaches that of uninfected individuals. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 526.
- World Health Organization. Principles and Practice of Screening for Disease. WHO Public Health Paper, 1968.
- Joint United Nations Programme on HIV/AIDS (UNAIDS) and World Health Organization (WHO). Global Facts & Figures 09. http://data.unaids.org/pub/FactSheet/2009/20091124_FS_global_en.pdf. Accessed 1/4/2011.
- World Health Organization. Epidemiological Fact Sheet on HIV and AIDS. Core data on epidemiology and response. United States of America. 2008 Update. http://apps.who.int/globalatlas/predefinedReports/EFS2008/full/EFS2008_US.pdf. Accessed 1/4/2011.
- US Centers for Disease Control and Prevention. HIV Surveillance Report, 2008; vol. 20. http://www.cdc.gov/hiv/topics/surveillance/resources/reports/. Published June 2010. Accessed 8/7/2010.
- Hall HI, Song R, Rhodes P, et al; HIV Incidence Surveillance Group. Estimation of HIV incidence in the United States. JAMA 2008; 300:520–529.
- Marks G, Crepaz N, Janssen RS. Estimated sexual transmission of HIV from persons aware and unaware that they are infected with the virus in the USA. AIDS 2006; 20:1447–1450.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. December 1, 2009;1–161. http://www.aidsinfo.nih.gov/ContentFiles/AdultsandAdolescentGL.pdf. Accessed 1/4/2011.
- Palella F, Armon C, Buchacz , et al; the HOPS Investigators. CD4 at HAART initiation predicts long term CD4 responses and mortality from AIDS and non-AIDS causes in the HIV Outpatient Study (HOPS). Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 983.
- Althoff K, Gange S, Klein M, et al; the North American-AIDS Cohort Collaboration on Res and Design. Late presentation for HIV care in the United States and Canada. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 982.
- Antiretroviral Therapy Cohort Collaboration. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996–2006: collaborative analysis of 13 HIV cohort studies. Clin Infect Dis 2010; 50:1387–1396.
- Simard E, Pfeiffer R, Engels E. Cancer incidence and cancer-attributable mortality among persons with AIDS in the United States. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 27.
- Silverberg M, Xu L, Chao C, et al. Immunodeficiency, HIV RNA levels, and risk of non-AIDS-defining cancers. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 28.
- DAD Study Group, Friis-Møller N, Reiss P, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007; 356:1723–1735.
- Ho J, Deeks S, Hecht F, et al. Earlier initiation of antiretroviral therapy in HIV-infected individuals is associated with reduced arterial stiffness. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 707.
- Dao C, Young B, Buchacz K, Baker R, Brooks J, and the HIV Outpatient Study Investigators. Higher and increasing rates of fracture among HIV-infected persons in the HIV Outpatient Study (HOPS) compared to the general US population 1994 to 2008. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 128.
- Ellis R, Heaton R, Letendre S, et al; the CHARTER Group. Higher CD4 nadir is associated with reduced rates of HIV-associated neurocognitive disorders in the CHARTER study: potential implications for early treatment initiation. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 429.
- Schacker T, Collier AC, Hughes J, Shea T, Corey L. Clinical and epidemiologic features of primary HIV infection. Ann Intern Med 1996; 125:257–264.
- Castel A, Samala R, Griffin A, et al. Monitoring the impact of expanded HIV testing in the District of Columbia using population-based HIV/AIDS surveillance data. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 34.
- Montaner J, Wood E, Kerr T, et al. Association of expanded HAART coverage with a decrease in new HIV diagnoses, particularly mong injection drug users in British Columbia, Canada. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 88LB.
- Donnell D, Kiarie J, Thomas K, et al. ART and risk of heterosexual HIV-1 transmissin in HIV-1 serodiscordant African couples: a multinational prospective study. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 136.
- Centers for Disease Control and Prevention. Late versus early testing of HIV—16 sites, United States, 2000–2003. MMWR Morb Mortal Wkly Rep 2003; Jun 27; 52( 25):581–586.
- Wilson SR, Mitchell C, Bradbury DR, Chavez J. Testing for HIV: current practies in the academic ED. Am J Emerg Med 1999; 17:346–356.
- Fincher-Mergi M, Cartone KJ, Mischler J, Pasieka P, Lerner EB, Billittier AJ. Assessment of emergency department heatlh care professionals’ behaviors regaridng HIV testing and referral for patients with STDs. AIDS Patient Care STDs 2002; 16:549–553.
- Paltiel AD, Weinstein MC, Kimmel AD, et al. Expanded screening for HIV in the United States—an analysis of cost-effectiveness. N Engl J Med 2005; 352:586–595.
- Sanders GD, Gayoumi AM, Sundaram V, et al. Cost-effectiveness of screening for HIV in the era of highly active antiretroviral therapy. N Engl J Med 2005; 352:570–585.
- Simpson WM, Johnstone FD, Goldberg DJ, Gormley SM, Hart GJ. Antenatal HIV testing: assessment of a routine voluntary approach. BMJ 1999; 318:1660–1661.
- Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention. Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep 2006; 55(RR-14):1–17.
With early treatment of human immunodeficiency virus (HIV) infection, we can now expect patients to live a much longer life and, in some situations, have a near-normal lifespan.1 Unfortunately, in screening for HIV infection, the United States lags behind many regions of the world, and infection is often not diagnosed until patients present with advanced disease, ie, the acquired immunodeficiency syndrome (AIDS). In this country there is a critical need to make HIV screening a routine part of medical care in all health settings in order to give patients their best chance for a healthy life, to prevent mother-to-child transmission, and to reduce the spread of HIV in the community.
HIV infection meets the criteria that justify routine screening, as laid out by the World Health Organization2:
- It is a serious health disorder that can be detected before symptoms develop
- Treatment is more beneficial if begun before symptoms develop
- Reliable, inexpensive, and acceptable screening tests exist
- The costs of screening are reasonable in relation to the anticipated benefits.
This article will review the epidemiology of the HIV epidemic, present the benefits of early treatment, and make the case for widely expanding screening for HIV infection in the US health care system.
HIV INFECTION CONTINUES TO BE A LARGE BURDEN
In 2008, an estimated 33.4 million people worldwide were HIV-positive. The vast majority of infected people—more than 22 million—live in sub-Saharan Africa.3
The United States has approximately 1.2 million cases.4 Although this is a small proportion of cases worldwide, it still represents a significant health care burden. In this country, the number of AIDS cases peaked in 1993, and the rate of deaths from AIDS began to decrease over the ensuing years as adequate therapy for HIV was developed. Standard therapy then and now consists of at least three drugs from two different classes.
Unfortunately, we have made little progress on the incidence of this disease. The estimated number of new HIV infections in the United States in 2008 was 56,000 and had remained about the same over the previous 15 years.5,6 Because of improved rates of survival, the prevalence has risen steadily since the mid-1990s to the current estimate of 1.2 million persons living with HIV/AIDS in the US.
About 25% of people infected with HIV are unaware of it. This group accounts for more than half of all new infections annually, which highlights the importance of enhanced screening. Once people know they are infected, they tend to change their behavior and are less likely to spread the disease.7
HIV disproportionately affects minority populations and gay men
Cases of HIV infection are reported among all age groups, although most patients tend to have been infected as young adults. Currently, the largest age group living with HIV is middle-aged. As this cohort grows older, an increasing burden of comorbidities due to aging can be expected. In 5 years, about half of the people with HIV in this country are expected to be 50 years of age or older. Although survival rates have steadily increased due to better treatment, survival tends to be shorter for older people newly diagnosed with HIV.
Worldwide, about an equal number of men and women are infected with HIV, but in the United States infected men outnumber women. In this country, about half the cases of HIV transmission among adults are by male-to-male sexual contact, about 30% are by high-risk heterosexual contact (ie, with a partner known to be HIV-infected or at high risk for being infected), and about 10% are by injection drug use.
In the United States, AIDS is predominantly and disproportionately a disease of minorities and those who live in poverty. African Americans account for the largest number of cases, followed by whites and then by Hispanics. Combined, African Americans and Hispanics account for two-thirds to three-fourths of all new cases, although they make up less than one-fourth of the US population. The incidence rate is nearly 137 per 100,000 for African Americans, 56 per 100,000 for Hispanics, and 19 per 100,000 for whites. The incidence is highest in New York and in the southeast, the geographic areas where the greatest number of minorities and people living in poverty reside. These groups also often lack access to health care.
HIV TREATMENT IS MORE EFFECTIVE IF STARTED EARLY
Treatment guidelines from the US Department of Health and Human Services (DHHS) have changed over the years. When effective medications were first introduced in the 1990s, the trend was to treat everyone as soon as they were diagnosed. As the burden of therapy began to unfold (side effects, cost, adherence, and drug resistance), the consensus was to wait until the CD4 T-cell count dropped to a lower level. As the medications have improved and have become better tolerated, the pendulum has swung back to treating earlier in the course of the disease. Currently, the DHHS recommends that therapy be started at CD4 counts of 350 cells/mL or lower (level of evidence: A1).8 It also recommends therapy for CD4 counts between 350 and 500 cells/mL, but the level of evidence is lower.8
The CD4 T cell is the prime target of the HIV virus and also an important marker of the health of the immune system. The lower the CD4 count at the start of therapy, the more challenging it is to normalize.9 If HIV infection is diagnosed early and therapy is started early, the likelihood is higher of normalizing the CD4 count and preserving immune function.
Progress is being made toward diagnosing HIV earlier. The CD4 count at presentation is increasing, but patients in the United States still present for care later than in other countries. In 1997, the median CD4 count at presentation was 234 cells/mL; in 2007, it was 327 (normal is about 550–1,000). Although this is a significant improvement, more than 50% of patients still have fewer than 350 cells/mL at presentation, which is the current threshold for beginning therapy, according to the most recent guidelines.10
Before triple therapy was available, almost all HIV-infected patients died of AIDS-related diseases. Now, about half of treated HIV-infected patients in Europe and North America die of other causes.11 However, many diseases not previously attributed to AIDS are now also known to be exacerbated by HIV infection.
Cancer risk increases with lower CD4 counts
The cumulative incidence of AIDS-defining cancers (Kaposi sarcoma, non-Hodgkin lymphoma, cervical carcinoma) has decreased steadily from 8.7% in the 1980s to 6.4% during the years 1990 to 1995, and to 2.1% between 1996 and 2006. This is attributable to improved immune function as a result of treatment success with antiviral therapy.12
But the incidence of non-AIDS-defining cancers (Hodgkin disease, anal cancer, oral and respiratory cancers) has increased.11 As therapy has regenerated the immune system, patients are surviving longer and are developing the more common cancers but with higher rates than in the general population.
Higher cancer risk is attributed to reduced immune surveillance. Many of these cancers are associated with viruses, such as human papillomavirus (anal and oral or pharyngeal cancers) and Epstein-Barr virus (Hodgkin disease), which can usually be controlled by a fully functioning immune system. The lower the CD4 count, the higher the risk of cancer, which highlights the need to diagnose HIV and start treatment early.13
Cardiovascular disease increases with lower CD4 counts
Associations have recently been identified between coronary disease and HIV as well as with HIV medications. Protease inhibitors tend to raise the levels of triglycerides, low-density lipoprotein cholesterol, and total cholesterol and increase the risk of heart attack.14
Regardless of therapy, HIV appears to be an independent risk factor for coronary disease. Arterial stiffness, as measured by carotid femoral pulse-wave velocity, was found to be increased among a sample of 80 HIV-infected men. This was associated with the usual risk factors of increasing age, blood pressure, and diabetes, as well as with lower nadir CD4 count.15
Fractures and neurocognitive disorders increase with HIV
Osteoporotic fractures are also more common in patients with HIV than in the general population. Risk factors include the traditional risks of older age, hepatitis C infection, diabetes, and substance abuse, but also nadir CD4 count less than 200.16
The risk of neurocognitive disorders is also associated with lower nadir CD4 counts. The lower the CD4 count, the higher the risk of developing neurocognitive deficits.17 The potential benefits of earlier diagnosis and treatment are obvious based upon the multiple recent findings outlined above.
CLINICAL PRESENTATION OF PRIMARY HIV INFECTION
During primary HIV infection, when patients are first infected, 50% to 90% are symptomatic. Symptoms usually appear in the first 6 weeks. The viral load tends to be highest at this time. Higher viral loads appear directly correlated with the degree of infectivity, highlighting the urgency of finding and treating new infections promptly to help avoid transmission to others.18
The clinical picture during primary infection is similar to that of acute mononucleosis. Signs and symptoms include fever, fatigue, rash, headache, lymphadenopathy, sore throat, and muscle aches. Although this presentation is common to many viral infections, questioning the patient about high-risk behavior (unprotected sex, multiple partners, intravenous drug use) will lead the astute physician to the correct testing and diagnosis.
Other early manifestations include mucocutaneous signs, such as seborrheic dermatitis, psoriasis, folliculitis, and thrush. Laboratory test results demonstrating leukopenia, thrombocytopenia, elevated total protein levels, proteinuria, and transaminitis are also suggestive of HIV infection.
THE CASE FOR INCREASED TESTING AND TREATMENT
The estimated prevalence of HIV in the United States is approximately 0.3%. However, its prevalence in Washington, DC, is 3%, which rivals rates in some areas of the developing world. From 2004 to 2008, health officials made a concerted effort in Washington, DC, to screen more people, particularly those at high risk. The number of publicly funded HIV tests performed increased by a factor of 3.7, and the number of newly reported cases increased by 17%. There was also a significant increase in the median CD4 count at the time of HIV diagnosis and a significant delay in time to progression to AIDS after HIV diagnosis.19
A study in British Columbia expanded access to highly active antiretroviral therapy during 2004 through 2009. High-risk individuals were targeted for increased screening. All those diagnosed with HIV were provided free medication. This resulted in a 50% reduction in new diagnoses of HIV infection throughout the community, especially among injectable drug users, a usually marginalized population. The proportion of patients with HIV-1 RNA levels above 1,500 copies/mL fell from about 50% to about 20%, indicating that the viral load—a measure of infectivity throughout the community—was reduced. Interestingly, this trend occurred during a time of increased rates of gonorrhea, syphilis, and other sexually transmitted diseases known to be associated with enhanced HIV transmission.20
In Africa, antiretroviral therapy was offered to discordant couples (one partner was infected with HIV and the other was not). Among those who chose therapy, the rate of HIV transmission was 92% lower than in those not receiving antiretroviral drugs,21 once again demonstrating that control of HIV by treatment can lead to decreased transmission.
US HIV testing is inadequate
The current state of HIV testing in the United States needs to be improved. Testing is not performed routinely, leading to delayed diagnosis when patients present with symptomatic, advanced disease. Patients who are tested late (within 12 months before being diagnosed with AIDS) tend to be younger and less educated and are more likely to be heterosexual and either African American or Hispanic than patients who are tested earlier.22 When retrospectively evaluated, these patients often have been in the health care system but not tested. Routine universal screening and targeted testing could lead to a much earlier diagnosis and potential better long-term outcomes.
A 1996 survey of 95 academic emergency departments found that for patients with suspected sexually transmitted infections, 93% of physicians said they screen for gonorrhea, 88% for Chlamydia infection, 58% for syphilis, but only 3% for HIV.23 Sexually transmitted infections and HIV are often transmitted together.
A similar 2002 survey of 154 emergency department providers who saw an average of 13 patients with sexually transmitted infections per week found that only 10% always recommend HIV testing to these patients. Reasons given for not testing were concern about follow-up (51%), not having a “certified” counselor (45%), HIV testing being too time-consuming (19%), and HIV testing being unavailable (27%).24
Although most HIV tests are given by private doctors and health maintenance organizations, the likelihood of finding patients with HIV is greatest in hospitals, emergency departments, outpatient clinics, and public community clinics.
The Advancing HIV Prevention initiative of the US Centers for Disease Control and Prevention (CDC) has four priorities:
- To make voluntary HIV testing a routine part of medical care
- To implement new models for diagnosing HIV infection outside medical settings
- To prevent HIV infection by working with patients with HIV and their partners
- To further decrease the rate of perinatal HIV transmission.
Rapid tests for HIV are available
There is a public health need to have rapid HIV testing available in all health care settings. With standard HIV tests, which can take 48 to 72 hours to run, about one-third of patients do not return for results. Subsequently locating them can be a huge challenge and is sometimes impossible. The ability to have rapid test results can improve this situation. It is especially important in prenatal care settings, where the mother can be immediately treated to reduce the risk of transmission to the child. Rapid testing increases the feasibility of testing in multiple venues, particularly acute-care settings with almost immediate results and linkage to care.
Several rapid tests are available and can be performed on whole blood, serum, plasma, and oral fluid. The tests provide reliable results in minutes, with 99% sensitivity and specificity. Positive results must be confirmed by subsequent two-stage laboratory testing, enzyme-linked immunosorbent assay, and Western blot. Patients who have negative or have indeterminate results on Western blot testing should be tested again after 4 weeks.
The cost-effectiveness of routine screening for HIV, even in populations with a low prevalence, is similar to that of commonly accepted interventions.25 In populations with a 1% prevalence of HIV, the cost is $15,078 per quality-adjusted life-year.26 Even if the prevalence is less than 0.05%, the cost is less than $50,000 per quality-adjusted life-year, which is normally the cutoff for acceptability for screening tests.25,26
‘OPT-OUT’ TESTING
In the past, patients were asked if they would like to have HIV testing (“opt-in” testing). It is now recommended that physicians request testing to be performed (“opt-out” testing). This still allows the patient to decline but also conveys a “matter of fact” nonjudgmental message, indicative of a routine procedure no different than other screening tests. When testing was done on an opt-in basis, only 35% of pregnant women agreed to be tested. Some women felt that accepting an HIV test indicated that they engage in high-risk behavior. When testing was instead offered as routine but with an opportunity to decline, 88% accepted testing, and they were significantly less anxious about testing.27
CDC RECOMMENDATIONS
The CDC now recommends that routine, voluntary HIV screening be done for all persons ages 13 to 64 in health care settings, regardless of risk.28 Screening should be repeated at least annually in persons with known risk. Screening should be done on an opt-out basis, with the opportunity to ask questions and the option to decline. Consent for HIV testing should be included with general consent for care. A separate signed informed consent is not recommended, and verbal consent can merely be documented in the medical record. Prevention counseling in conjunction with HIV screening in health care settings is not required.
Testing should be done in all health care settings, including primary care settings, inpatient services, emergency departments, urgent care clinics, and sexually transmitted disease clinics. Test results should be communicated in the same manner as other diagnostic and screening care. Clinical HIV care should be available onsite or reliable referral to qualified providers should be established.
For pregnant women, the CDC recommends universal opt-out HIV screening, with HIV testing as part of the routine panel of prenatal screening tests. The consent for prenatal care includes HIV testing, with notification and the option to decline. Women should be tested again in the third trimester if they are known to be at risk for HIV, and in areas and health care facilities in which the prevalence of HIV is high.
In women whose HIV status is undocumented in labor and delivery, opt-out rapid testing should be performed, and antiretroviral prophylaxis should be given on the basis of the rapid test result. Rapid testing of the newborn is recommended if the mother’s status is unknown at delivery, and antiretroviral prophylaxis should be started within 12 hours of birth on the basis of the rapid test result.
Widespread routine screening and earlier treatment could significantly reduce the incidence and improve the outcomes of HIV in this country. Health care providers are encouraged to adopt these practices.
With early treatment of human immunodeficiency virus (HIV) infection, we can now expect patients to live a much longer life and, in some situations, have a near-normal lifespan.1 Unfortunately, in screening for HIV infection, the United States lags behind many regions of the world, and infection is often not diagnosed until patients present with advanced disease, ie, the acquired immunodeficiency syndrome (AIDS). In this country there is a critical need to make HIV screening a routine part of medical care in all health settings in order to give patients their best chance for a healthy life, to prevent mother-to-child transmission, and to reduce the spread of HIV in the community.
HIV infection meets the criteria that justify routine screening, as laid out by the World Health Organization2:
- It is a serious health disorder that can be detected before symptoms develop
- Treatment is more beneficial if begun before symptoms develop
- Reliable, inexpensive, and acceptable screening tests exist
- The costs of screening are reasonable in relation to the anticipated benefits.
This article will review the epidemiology of the HIV epidemic, present the benefits of early treatment, and make the case for widely expanding screening for HIV infection in the US health care system.
HIV INFECTION CONTINUES TO BE A LARGE BURDEN
In 2008, an estimated 33.4 million people worldwide were HIV-positive. The vast majority of infected people—more than 22 million—live in sub-Saharan Africa.3
The United States has approximately 1.2 million cases.4 Although this is a small proportion of cases worldwide, it still represents a significant health care burden. In this country, the number of AIDS cases peaked in 1993, and the rate of deaths from AIDS began to decrease over the ensuing years as adequate therapy for HIV was developed. Standard therapy then and now consists of at least three drugs from two different classes.
Unfortunately, we have made little progress on the incidence of this disease. The estimated number of new HIV infections in the United States in 2008 was 56,000 and had remained about the same over the previous 15 years.5,6 Because of improved rates of survival, the prevalence has risen steadily since the mid-1990s to the current estimate of 1.2 million persons living with HIV/AIDS in the US.
About 25% of people infected with HIV are unaware of it. This group accounts for more than half of all new infections annually, which highlights the importance of enhanced screening. Once people know they are infected, they tend to change their behavior and are less likely to spread the disease.7
HIV disproportionately affects minority populations and gay men
Cases of HIV infection are reported among all age groups, although most patients tend to have been infected as young adults. Currently, the largest age group living with HIV is middle-aged. As this cohort grows older, an increasing burden of comorbidities due to aging can be expected. In 5 years, about half of the people with HIV in this country are expected to be 50 years of age or older. Although survival rates have steadily increased due to better treatment, survival tends to be shorter for older people newly diagnosed with HIV.
Worldwide, about an equal number of men and women are infected with HIV, but in the United States infected men outnumber women. In this country, about half the cases of HIV transmission among adults are by male-to-male sexual contact, about 30% are by high-risk heterosexual contact (ie, with a partner known to be HIV-infected or at high risk for being infected), and about 10% are by injection drug use.
In the United States, AIDS is predominantly and disproportionately a disease of minorities and those who live in poverty. African Americans account for the largest number of cases, followed by whites and then by Hispanics. Combined, African Americans and Hispanics account for two-thirds to three-fourths of all new cases, although they make up less than one-fourth of the US population. The incidence rate is nearly 137 per 100,000 for African Americans, 56 per 100,000 for Hispanics, and 19 per 100,000 for whites. The incidence is highest in New York and in the southeast, the geographic areas where the greatest number of minorities and people living in poverty reside. These groups also often lack access to health care.
HIV TREATMENT IS MORE EFFECTIVE IF STARTED EARLY
Treatment guidelines from the US Department of Health and Human Services (DHHS) have changed over the years. When effective medications were first introduced in the 1990s, the trend was to treat everyone as soon as they were diagnosed. As the burden of therapy began to unfold (side effects, cost, adherence, and drug resistance), the consensus was to wait until the CD4 T-cell count dropped to a lower level. As the medications have improved and have become better tolerated, the pendulum has swung back to treating earlier in the course of the disease. Currently, the DHHS recommends that therapy be started at CD4 counts of 350 cells/mL or lower (level of evidence: A1).8 It also recommends therapy for CD4 counts between 350 and 500 cells/mL, but the level of evidence is lower.8
The CD4 T cell is the prime target of the HIV virus and also an important marker of the health of the immune system. The lower the CD4 count at the start of therapy, the more challenging it is to normalize.9 If HIV infection is diagnosed early and therapy is started early, the likelihood is higher of normalizing the CD4 count and preserving immune function.
Progress is being made toward diagnosing HIV earlier. The CD4 count at presentation is increasing, but patients in the United States still present for care later than in other countries. In 1997, the median CD4 count at presentation was 234 cells/mL; in 2007, it was 327 (normal is about 550–1,000). Although this is a significant improvement, more than 50% of patients still have fewer than 350 cells/mL at presentation, which is the current threshold for beginning therapy, according to the most recent guidelines.10
Before triple therapy was available, almost all HIV-infected patients died of AIDS-related diseases. Now, about half of treated HIV-infected patients in Europe and North America die of other causes.11 However, many diseases not previously attributed to AIDS are now also known to be exacerbated by HIV infection.
Cancer risk increases with lower CD4 counts
The cumulative incidence of AIDS-defining cancers (Kaposi sarcoma, non-Hodgkin lymphoma, cervical carcinoma) has decreased steadily from 8.7% in the 1980s to 6.4% during the years 1990 to 1995, and to 2.1% between 1996 and 2006. This is attributable to improved immune function as a result of treatment success with antiviral therapy.12
But the incidence of non-AIDS-defining cancers (Hodgkin disease, anal cancer, oral and respiratory cancers) has increased.11 As therapy has regenerated the immune system, patients are surviving longer and are developing the more common cancers but with higher rates than in the general population.
Higher cancer risk is attributed to reduced immune surveillance. Many of these cancers are associated with viruses, such as human papillomavirus (anal and oral or pharyngeal cancers) and Epstein-Barr virus (Hodgkin disease), which can usually be controlled by a fully functioning immune system. The lower the CD4 count, the higher the risk of cancer, which highlights the need to diagnose HIV and start treatment early.13
Cardiovascular disease increases with lower CD4 counts
Associations have recently been identified between coronary disease and HIV as well as with HIV medications. Protease inhibitors tend to raise the levels of triglycerides, low-density lipoprotein cholesterol, and total cholesterol and increase the risk of heart attack.14
Regardless of therapy, HIV appears to be an independent risk factor for coronary disease. Arterial stiffness, as measured by carotid femoral pulse-wave velocity, was found to be increased among a sample of 80 HIV-infected men. This was associated with the usual risk factors of increasing age, blood pressure, and diabetes, as well as with lower nadir CD4 count.15
Fractures and neurocognitive disorders increase with HIV
Osteoporotic fractures are also more common in patients with HIV than in the general population. Risk factors include the traditional risks of older age, hepatitis C infection, diabetes, and substance abuse, but also nadir CD4 count less than 200.16
The risk of neurocognitive disorders is also associated with lower nadir CD4 counts. The lower the CD4 count, the higher the risk of developing neurocognitive deficits.17 The potential benefits of earlier diagnosis and treatment are obvious based upon the multiple recent findings outlined above.
CLINICAL PRESENTATION OF PRIMARY HIV INFECTION
During primary HIV infection, when patients are first infected, 50% to 90% are symptomatic. Symptoms usually appear in the first 6 weeks. The viral load tends to be highest at this time. Higher viral loads appear directly correlated with the degree of infectivity, highlighting the urgency of finding and treating new infections promptly to help avoid transmission to others.18
The clinical picture during primary infection is similar to that of acute mononucleosis. Signs and symptoms include fever, fatigue, rash, headache, lymphadenopathy, sore throat, and muscle aches. Although this presentation is common to many viral infections, questioning the patient about high-risk behavior (unprotected sex, multiple partners, intravenous drug use) will lead the astute physician to the correct testing and diagnosis.
Other early manifestations include mucocutaneous signs, such as seborrheic dermatitis, psoriasis, folliculitis, and thrush. Laboratory test results demonstrating leukopenia, thrombocytopenia, elevated total protein levels, proteinuria, and transaminitis are also suggestive of HIV infection.
THE CASE FOR INCREASED TESTING AND TREATMENT
The estimated prevalence of HIV in the United States is approximately 0.3%. However, its prevalence in Washington, DC, is 3%, which rivals rates in some areas of the developing world. From 2004 to 2008, health officials made a concerted effort in Washington, DC, to screen more people, particularly those at high risk. The number of publicly funded HIV tests performed increased by a factor of 3.7, and the number of newly reported cases increased by 17%. There was also a significant increase in the median CD4 count at the time of HIV diagnosis and a significant delay in time to progression to AIDS after HIV diagnosis.19
A study in British Columbia expanded access to highly active antiretroviral therapy during 2004 through 2009. High-risk individuals were targeted for increased screening. All those diagnosed with HIV were provided free medication. This resulted in a 50% reduction in new diagnoses of HIV infection throughout the community, especially among injectable drug users, a usually marginalized population. The proportion of patients with HIV-1 RNA levels above 1,500 copies/mL fell from about 50% to about 20%, indicating that the viral load—a measure of infectivity throughout the community—was reduced. Interestingly, this trend occurred during a time of increased rates of gonorrhea, syphilis, and other sexually transmitted diseases known to be associated with enhanced HIV transmission.20
In Africa, antiretroviral therapy was offered to discordant couples (one partner was infected with HIV and the other was not). Among those who chose therapy, the rate of HIV transmission was 92% lower than in those not receiving antiretroviral drugs,21 once again demonstrating that control of HIV by treatment can lead to decreased transmission.
US HIV testing is inadequate
The current state of HIV testing in the United States needs to be improved. Testing is not performed routinely, leading to delayed diagnosis when patients present with symptomatic, advanced disease. Patients who are tested late (within 12 months before being diagnosed with AIDS) tend to be younger and less educated and are more likely to be heterosexual and either African American or Hispanic than patients who are tested earlier.22 When retrospectively evaluated, these patients often have been in the health care system but not tested. Routine universal screening and targeted testing could lead to a much earlier diagnosis and potential better long-term outcomes.
A 1996 survey of 95 academic emergency departments found that for patients with suspected sexually transmitted infections, 93% of physicians said they screen for gonorrhea, 88% for Chlamydia infection, 58% for syphilis, but only 3% for HIV.23 Sexually transmitted infections and HIV are often transmitted together.
A similar 2002 survey of 154 emergency department providers who saw an average of 13 patients with sexually transmitted infections per week found that only 10% always recommend HIV testing to these patients. Reasons given for not testing were concern about follow-up (51%), not having a “certified” counselor (45%), HIV testing being too time-consuming (19%), and HIV testing being unavailable (27%).24
Although most HIV tests are given by private doctors and health maintenance organizations, the likelihood of finding patients with HIV is greatest in hospitals, emergency departments, outpatient clinics, and public community clinics.
The Advancing HIV Prevention initiative of the US Centers for Disease Control and Prevention (CDC) has four priorities:
- To make voluntary HIV testing a routine part of medical care
- To implement new models for diagnosing HIV infection outside medical settings
- To prevent HIV infection by working with patients with HIV and their partners
- To further decrease the rate of perinatal HIV transmission.
Rapid tests for HIV are available
There is a public health need to have rapid HIV testing available in all health care settings. With standard HIV tests, which can take 48 to 72 hours to run, about one-third of patients do not return for results. Subsequently locating them can be a huge challenge and is sometimes impossible. The ability to have rapid test results can improve this situation. It is especially important in prenatal care settings, where the mother can be immediately treated to reduce the risk of transmission to the child. Rapid testing increases the feasibility of testing in multiple venues, particularly acute-care settings with almost immediate results and linkage to care.
Several rapid tests are available and can be performed on whole blood, serum, plasma, and oral fluid. The tests provide reliable results in minutes, with 99% sensitivity and specificity. Positive results must be confirmed by subsequent two-stage laboratory testing, enzyme-linked immunosorbent assay, and Western blot. Patients who have negative or have indeterminate results on Western blot testing should be tested again after 4 weeks.
The cost-effectiveness of routine screening for HIV, even in populations with a low prevalence, is similar to that of commonly accepted interventions.25 In populations with a 1% prevalence of HIV, the cost is $15,078 per quality-adjusted life-year.26 Even if the prevalence is less than 0.05%, the cost is less than $50,000 per quality-adjusted life-year, which is normally the cutoff for acceptability for screening tests.25,26
‘OPT-OUT’ TESTING
In the past, patients were asked if they would like to have HIV testing (“opt-in” testing). It is now recommended that physicians request testing to be performed (“opt-out” testing). This still allows the patient to decline but also conveys a “matter of fact” nonjudgmental message, indicative of a routine procedure no different than other screening tests. When testing was done on an opt-in basis, only 35% of pregnant women agreed to be tested. Some women felt that accepting an HIV test indicated that they engage in high-risk behavior. When testing was instead offered as routine but with an opportunity to decline, 88% accepted testing, and they were significantly less anxious about testing.27
CDC RECOMMENDATIONS
The CDC now recommends that routine, voluntary HIV screening be done for all persons ages 13 to 64 in health care settings, regardless of risk.28 Screening should be repeated at least annually in persons with known risk. Screening should be done on an opt-out basis, with the opportunity to ask questions and the option to decline. Consent for HIV testing should be included with general consent for care. A separate signed informed consent is not recommended, and verbal consent can merely be documented in the medical record. Prevention counseling in conjunction with HIV screening in health care settings is not required.
Testing should be done in all health care settings, including primary care settings, inpatient services, emergency departments, urgent care clinics, and sexually transmitted disease clinics. Test results should be communicated in the same manner as other diagnostic and screening care. Clinical HIV care should be available onsite or reliable referral to qualified providers should be established.
For pregnant women, the CDC recommends universal opt-out HIV screening, with HIV testing as part of the routine panel of prenatal screening tests. The consent for prenatal care includes HIV testing, with notification and the option to decline. Women should be tested again in the third trimester if they are known to be at risk for HIV, and in areas and health care facilities in which the prevalence of HIV is high.
In women whose HIV status is undocumented in labor and delivery, opt-out rapid testing should be performed, and antiretroviral prophylaxis should be given on the basis of the rapid test result. Rapid testing of the newborn is recommended if the mother’s status is unknown at delivery, and antiretroviral prophylaxis should be started within 12 hours of birth on the basis of the rapid test result.
Widespread routine screening and earlier treatment could significantly reduce the incidence and improve the outcomes of HIV in this country. Health care providers are encouraged to adopt these practices.
- Van Sighem A, Gras L, Reiss P, Brinkman K, de Wolf F, and ATHENA Natl Observational Cohort Study. Life expectancy of recently diagnosed asymptomatic HIV-infected patients approaches that of uninfected individuals. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 526.
- World Health Organization. Principles and Practice of Screening for Disease. WHO Public Health Paper, 1968.
- Joint United Nations Programme on HIV/AIDS (UNAIDS) and World Health Organization (WHO). Global Facts & Figures 09. http://data.unaids.org/pub/FactSheet/2009/20091124_FS_global_en.pdf. Accessed 1/4/2011.
- World Health Organization. Epidemiological Fact Sheet on HIV and AIDS. Core data on epidemiology and response. United States of America. 2008 Update. http://apps.who.int/globalatlas/predefinedReports/EFS2008/full/EFS2008_US.pdf. Accessed 1/4/2011.
- US Centers for Disease Control and Prevention. HIV Surveillance Report, 2008; vol. 20. http://www.cdc.gov/hiv/topics/surveillance/resources/reports/. Published June 2010. Accessed 8/7/2010.
- Hall HI, Song R, Rhodes P, et al; HIV Incidence Surveillance Group. Estimation of HIV incidence in the United States. JAMA 2008; 300:520–529.
- Marks G, Crepaz N, Janssen RS. Estimated sexual transmission of HIV from persons aware and unaware that they are infected with the virus in the USA. AIDS 2006; 20:1447–1450.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. December 1, 2009;1–161. http://www.aidsinfo.nih.gov/ContentFiles/AdultsandAdolescentGL.pdf. Accessed 1/4/2011.
- Palella F, Armon C, Buchacz , et al; the HOPS Investigators. CD4 at HAART initiation predicts long term CD4 responses and mortality from AIDS and non-AIDS causes in the HIV Outpatient Study (HOPS). Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 983.
- Althoff K, Gange S, Klein M, et al; the North American-AIDS Cohort Collaboration on Res and Design. Late presentation for HIV care in the United States and Canada. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 982.
- Antiretroviral Therapy Cohort Collaboration. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996–2006: collaborative analysis of 13 HIV cohort studies. Clin Infect Dis 2010; 50:1387–1396.
- Simard E, Pfeiffer R, Engels E. Cancer incidence and cancer-attributable mortality among persons with AIDS in the United States. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 27.
- Silverberg M, Xu L, Chao C, et al. Immunodeficiency, HIV RNA levels, and risk of non-AIDS-defining cancers. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 28.
- DAD Study Group, Friis-Møller N, Reiss P, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007; 356:1723–1735.
- Ho J, Deeks S, Hecht F, et al. Earlier initiation of antiretroviral therapy in HIV-infected individuals is associated with reduced arterial stiffness. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 707.
- Dao C, Young B, Buchacz K, Baker R, Brooks J, and the HIV Outpatient Study Investigators. Higher and increasing rates of fracture among HIV-infected persons in the HIV Outpatient Study (HOPS) compared to the general US population 1994 to 2008. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 128.
- Ellis R, Heaton R, Letendre S, et al; the CHARTER Group. Higher CD4 nadir is associated with reduced rates of HIV-associated neurocognitive disorders in the CHARTER study: potential implications for early treatment initiation. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 429.
- Schacker T, Collier AC, Hughes J, Shea T, Corey L. Clinical and epidemiologic features of primary HIV infection. Ann Intern Med 1996; 125:257–264.
- Castel A, Samala R, Griffin A, et al. Monitoring the impact of expanded HIV testing in the District of Columbia using population-based HIV/AIDS surveillance data. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 34.
- Montaner J, Wood E, Kerr T, et al. Association of expanded HAART coverage with a decrease in new HIV diagnoses, particularly mong injection drug users in British Columbia, Canada. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 88LB.
- Donnell D, Kiarie J, Thomas K, et al. ART and risk of heterosexual HIV-1 transmissin in HIV-1 serodiscordant African couples: a multinational prospective study. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 136.
- Centers for Disease Control and Prevention. Late versus early testing of HIV—16 sites, United States, 2000–2003. MMWR Morb Mortal Wkly Rep 2003; Jun 27; 52( 25):581–586.
- Wilson SR, Mitchell C, Bradbury DR, Chavez J. Testing for HIV: current practies in the academic ED. Am J Emerg Med 1999; 17:346–356.
- Fincher-Mergi M, Cartone KJ, Mischler J, Pasieka P, Lerner EB, Billittier AJ. Assessment of emergency department heatlh care professionals’ behaviors regaridng HIV testing and referral for patients with STDs. AIDS Patient Care STDs 2002; 16:549–553.
- Paltiel AD, Weinstein MC, Kimmel AD, et al. Expanded screening for HIV in the United States—an analysis of cost-effectiveness. N Engl J Med 2005; 352:586–595.
- Sanders GD, Gayoumi AM, Sundaram V, et al. Cost-effectiveness of screening for HIV in the era of highly active antiretroviral therapy. N Engl J Med 2005; 352:570–585.
- Simpson WM, Johnstone FD, Goldberg DJ, Gormley SM, Hart GJ. Antenatal HIV testing: assessment of a routine voluntary approach. BMJ 1999; 318:1660–1661.
- Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention. Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep 2006; 55(RR-14):1–17.
- Van Sighem A, Gras L, Reiss P, Brinkman K, de Wolf F, and ATHENA Natl Observational Cohort Study. Life expectancy of recently diagnosed asymptomatic HIV-infected patients approaches that of uninfected individuals. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 526.
- World Health Organization. Principles and Practice of Screening for Disease. WHO Public Health Paper, 1968.
- Joint United Nations Programme on HIV/AIDS (UNAIDS) and World Health Organization (WHO). Global Facts & Figures 09. http://data.unaids.org/pub/FactSheet/2009/20091124_FS_global_en.pdf. Accessed 1/4/2011.
- World Health Organization. Epidemiological Fact Sheet on HIV and AIDS. Core data on epidemiology and response. United States of America. 2008 Update. http://apps.who.int/globalatlas/predefinedReports/EFS2008/full/EFS2008_US.pdf. Accessed 1/4/2011.
- US Centers for Disease Control and Prevention. HIV Surveillance Report, 2008; vol. 20. http://www.cdc.gov/hiv/topics/surveillance/resources/reports/. Published June 2010. Accessed 8/7/2010.
- Hall HI, Song R, Rhodes P, et al; HIV Incidence Surveillance Group. Estimation of HIV incidence in the United States. JAMA 2008; 300:520–529.
- Marks G, Crepaz N, Janssen RS. Estimated sexual transmission of HIV from persons aware and unaware that they are infected with the virus in the USA. AIDS 2006; 20:1447–1450.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. December 1, 2009;1–161. http://www.aidsinfo.nih.gov/ContentFiles/AdultsandAdolescentGL.pdf. Accessed 1/4/2011.
- Palella F, Armon C, Buchacz , et al; the HOPS Investigators. CD4 at HAART initiation predicts long term CD4 responses and mortality from AIDS and non-AIDS causes in the HIV Outpatient Study (HOPS). Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 983.
- Althoff K, Gange S, Klein M, et al; the North American-AIDS Cohort Collaboration on Res and Design. Late presentation for HIV care in the United States and Canada. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 982.
- Antiretroviral Therapy Cohort Collaboration. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996–2006: collaborative analysis of 13 HIV cohort studies. Clin Infect Dis 2010; 50:1387–1396.
- Simard E, Pfeiffer R, Engels E. Cancer incidence and cancer-attributable mortality among persons with AIDS in the United States. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 27.
- Silverberg M, Xu L, Chao C, et al. Immunodeficiency, HIV RNA levels, and risk of non-AIDS-defining cancers. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 28.
- DAD Study Group, Friis-Møller N, Reiss P, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007; 356:1723–1735.
- Ho J, Deeks S, Hecht F, et al. Earlier initiation of antiretroviral therapy in HIV-infected individuals is associated with reduced arterial stiffness. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 707.
- Dao C, Young B, Buchacz K, Baker R, Brooks J, and the HIV Outpatient Study Investigators. Higher and increasing rates of fracture among HIV-infected persons in the HIV Outpatient Study (HOPS) compared to the general US population 1994 to 2008. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 128.
- Ellis R, Heaton R, Letendre S, et al; the CHARTER Group. Higher CD4 nadir is associated with reduced rates of HIV-associated neurocognitive disorders in the CHARTER study: potential implications for early treatment initiation. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 429.
- Schacker T, Collier AC, Hughes J, Shea T, Corey L. Clinical and epidemiologic features of primary HIV infection. Ann Intern Med 1996; 125:257–264.
- Castel A, Samala R, Griffin A, et al. Monitoring the impact of expanded HIV testing in the District of Columbia using population-based HIV/AIDS surveillance data. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 34.
- Montaner J, Wood E, Kerr T, et al. Association of expanded HAART coverage with a decrease in new HIV diagnoses, particularly mong injection drug users in British Columbia, Canada. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 88LB.
- Donnell D, Kiarie J, Thomas K, et al. ART and risk of heterosexual HIV-1 transmissin in HIV-1 serodiscordant African couples: a multinational prospective study. Presented at the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA, February 16–19, 2010. Abstract 136.
- Centers for Disease Control and Prevention. Late versus early testing of HIV—16 sites, United States, 2000–2003. MMWR Morb Mortal Wkly Rep 2003; Jun 27; 52( 25):581–586.
- Wilson SR, Mitchell C, Bradbury DR, Chavez J. Testing for HIV: current practies in the academic ED. Am J Emerg Med 1999; 17:346–356.
- Fincher-Mergi M, Cartone KJ, Mischler J, Pasieka P, Lerner EB, Billittier AJ. Assessment of emergency department heatlh care professionals’ behaviors regaridng HIV testing and referral for patients with STDs. AIDS Patient Care STDs 2002; 16:549–553.
- Paltiel AD, Weinstein MC, Kimmel AD, et al. Expanded screening for HIV in the United States—an analysis of cost-effectiveness. N Engl J Med 2005; 352:586–595.
- Sanders GD, Gayoumi AM, Sundaram V, et al. Cost-effectiveness of screening for HIV in the era of highly active antiretroviral therapy. N Engl J Med 2005; 352:570–585.
- Simpson WM, Johnstone FD, Goldberg DJ, Gormley SM, Hart GJ. Antenatal HIV testing: assessment of a routine voluntary approach. BMJ 1999; 318:1660–1661.
- Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention. Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep 2006; 55(RR-14):1–17.
KEY POINTS
- Recommendations from the US Centers for Disease Control and Prevention call for routine HIV screening for all people ages 13 to 64 at least once regardless of their risk profile, and annual testing for people with known risk factors for acquiring HIV.
- Early treatment of HIV infection may reduce the risk of cancer, cardiovascular disease, neurocognitive disorders, and osteoporotic fractures and improve the rate of survival compared with patients treated late in the course of HIV infection.
- Finding and treating patients early in the course of infection has the potential to reduce infectivity in the community.
- Reliable rapid testing is now available to screen for HIV in community settings, emergency departments, and public health clinics, and during labor for those not tested in the prenatal period. It is also useful when follow-up is uncertain.
Vitamin D and the heart: Why we need large-scale clinical trials
Vitamin D is viewed as a promising supplement by the medical, public health, and lay communities, potentially offering many health benefits. But enthusiasm for a new intervention too often gets far ahead of the evidence, as was the case with beta-carotene, selenium, folic acid, and vitamins C and E.
Despite the enthusiasm for vitamin D, there have been no large-scale primary prevention trials that have had either cardiovascular disease or cancer as a prespecified primary outcome. Previous randomized trials of vitamin D have focused primarily on osteoporosis, fracture, falls, and physical function. Although the investigators often reported their findings on vitamin D and cardiovascular disease or cancer, these outcomes were generally secondary or tertiary end points that were not prespecified. These studies should be viewed as hypothesis-generating rather than hypothesis-testing. The increasing prevalence of use of vitamin D supplements underscores the need for rigorous and conclusive evidence from randomized clinical trials that have cardiovascular disease and cancer as primary outcomes.
This article will explain the rationale for a large-scale, randomized clinical trial to evaluate the role of vitamin D in the prevention of cardiovascular disease and cancer. It will also describe the biological mechanisms and currently available evidence relating vitamin D to potential health benefits. Finally, the design, dosage considerations, and logistics of the Vitamin D and Omega-3 Trial (VITAL) will be presented.

EVIDENCE IS MOUNTING FOR VITAMIN D’S BIOLOGICAL IMPORTANCE
Vitamin D is undoubtedly important to health: not only is it produced endogenously, but at least 500 genes have been identified with vitamin D response elements. The vitamin D receptor is found in nearly all cells in the body, and the 1-alpha-hydroxylase enzyme is present in many tissues. Some studies suggest that almost 10% of the human genome may be at least partially regulated by vitamin D.
Although we know vitamin D is important, what our optimal intake and our blood level of 25-hydroxyvitamin D3 should be are key unknowns.
RECOMMENDATIONS FOR VITAMIN D INTAKE
During winter, late fall, and early spring, people who live above the 37th parallel (geographically, about one-half of the contiguous United States) do not get enough ultraviolet B energy from the sun to make all the vitamin D they need, even if they spend several hours outside every day. In addition, dark skin pigmentation serves as a sun block, as do sunscreens.
The Institute of Medicine (IOM) provided guidelines for vitamin D intake in 1997 and, most recently, in 2010. However, these guidelines are based on the amount of vitamin D required for bone health and do not address the amount that may be of benefit for prevention of cancer and cardiovascular disease. The latter outcomes are not addressed because the IOM committee believed that evidence was insufficient to determine the role of vitamin D in the prevention of cardiovascular disease, cancer, and other chronic diseases. Thus, current IOM guidelines, which generally recommend less than 1,000 IU of vitamin D daily, are relevant to bone health but not necessarily to other health outcomes. More research is needed to understand whether the guidelines should be modified for the prevention of other chronic diseases.
Moreover, whether or not everyone should be screened for 25-hydroxyvitamin D3 blood levels is controversial. Most experts agree that a level less than 20 ng/mL is deficient or insufficient. Conversely, potentially harmful are levels 150 ng/mL or more (> 375 nmol/L), which entail the risk of hypercalcemia, vascular soft tissue calcification, and hyperphosphatemia.
People do not reach toxic levels with ultraviolet light exposure because the amount of 25-hydroxyvitamin D3 synthesis is well regulated. Dietary supplements, however, can bring about toxic levels, and patients taking high doses need to be monitored carefully. The level that should be considered optimal is controversial and requires further study.
RISK FACTORS FOR LOW VITAMIN D LEVELS
Risk factors for low vitamin D levels include older age, living in northern latitudes, sun avoidance, dark skin pigmentation, obesity, low dietary intake, and various medical conditions, especially malabsorption syndromes. Some of these are also risk factors for cardiovascular disease, cancer, and other chronic diseases, and potentially confound outcomes in many studies. Older age, which is usually adjusted for in multivariate models, is important to recognize as a major risk factor for vitamin D deficiency, owing to reduced absorption and synthesis, less time outdoors, and low dietary intake.
Wearing sunscreen decreases the synthesis of vitamin D in the skin, but because ultra-violet light has been clearly classified as a carcinogen, it is a not advisable to increase sun exposure for the sake of increasing vitamin D levels. That is a poor trade-off, given the high incidence rate of skin cancer and the adverse effects of solar radiation on skin aging.
Obesity is a risk factor for vitamin D deficiency because vitamin D is fat-soluble and becomes sequestered in fat tissue. Vitamin D may also play a role in the differentiation of adipocytes and may affect their function. In observational studies, it is very important for researchers to adjust for body mass index, physical activity (which may be correlated with more time outdoors), and other potential confounders in their analyses.
HOW VITAMIN D MAY LOWER CANCER RISK
Because of the important effect of vitamin D in regulating cell differentiation and cell growth, there are multiple ways that it may affect cancer risk. Laboratory, cell culture, and animal studies suggest that vitamin D may lower cancer risk by inhibiting cell proliferation, angiogenesis, metastasis, and inflammation and inducing apoptosis and cellular differentiation. Several of these mechanisms are also relevant to atherosclerosis and cardiovascular disease. Although VITAL is addressing the role of vitamin D in preventing both cancer and cardiovascular disease, the remainder of this article will focus on cardiovascular outcomes.
HOW VITAMIN D MAY REDUCE CARDIOVASCULAR RISK
Vitamin D may lower cardiovascular risk via several mechanisms:
Inhibiting inflammation. Vitamin D has a powerful immunomodulatory effect: laboratory studies show that it inhibits prostaglandin and cyclooxygenase 2 pathways, reduces matrix metalloproteinase 9 and several proinflammatory cytokines, and increases interleukin 10, all of which result in suppressed inflammation.1
Inhibiting vascular muscle proliferation and vascular calcification. Animal studies indicate that in moderate doses vitamin D decreases calcium cellular influx and increases matrix Gla protein, which inhibits vascular smooth muscle proliferation and vascular calcification. These protective effects contrast with the hypercalcemia associated with a high intake of vitamin D, especially in the context of renal failure or other risk factors, which may lead to increased vascular calcification.1
Regulates blood pressure. Vitamin D decreases renin gene expression and the synthesis of renin, which reduces activity of the renin-angiotensin-aldosterone system, leading to a reduction of blood pressure and a favorable effect on volume homeostasis.1
Regulates glucose metabolism. Limited evidence shows that vitamin D may increase insulin sensitivity and regulate glucose metabolism.1
Vitamin D and cardiac hypertrophy
The vitamin D receptor is present in virtually all tissues, including cardiac myocytes and endothelial cells. Animals with vitamin D deficiency have higher blood pressures, and animals genetically altered to have no vitamin D receptors (knock-out models) develop left ventricular hypertrophy and heart failure.
Animals genetically altered to have no 1-alpha-hydroxylase (so that the most active form of vitamin D is not made) also develop left ventricular hypertrophy. They can be rescued by the administration of 1,25-dihydroxy vitamin D3.1
These findings are consistent with what is observed in patients with end-stage renal disease, who produce very little 1,25-dihydroxyvitamin D3: they often develop left ventricular hypertrophy, diastolic heart failure, atherosclerosis, and vascular calcification.
EVIDENCE FOR CARDIOVASCULAR DISEASE REDUCTION
Wang et al1 recently reviewed available prospective cohort and randomized clinical trials from 1966 to 2009 that examined vitamin D or calcium supplementation and cardiovascular disease. Comparing people with the lowest to the highest levels of serum 25-hydroxyvitamin D3 indicated that a low level is a risk factor for coronary artery disease and cardiovascular death. Unfortunately, most studies were not designed to assess primary effects on cardiovascular outcomes, and so have many potential confounders.
Prospective observational studies
Observational studies suggest that vitamin D deficiency is associated with an increased risk of cardiovascular disease. Some examples:
The Framingham Offspring Study2 followed 1,739 men and women with a mean age of 59 for 5.4 years. The study compared the incidence of cardiovascular events in those with a serum 25-dihydroxyvitamin D level of at least 37.5 nmol/L vs those with lower levels. The risk of cardiovascular disease was 1.62 times higher in those with the lowest levels of vitamin D, a statistically significant difference. However, a threshold effect was apparent (discussed below).
The Health Professionals Follow-up Study3 prospectively evaluated more than 18,000 men ages 40 to 75 for 10 years. The study compared men with a low serum level of vitamin D (< 37.5 nmol/L) to those with a more optimal level (> 75 nmol/L). The incidence of cardiovascular events was 2.09 times higher in men with low levels of vitamin D, a difference that was statistically significant.
The Third National Health and Nutrition Examination Survey (NHANES III) included data for more than 13,300 men and women age 20 years and older. Using a cohort that was followed for 8.7 years, Melamed et al4 compared the quartile with the lowest serum vitamin D level (< 44.4 nmol/L) against the quartile with the highest level (> 80.1 nmol/L). The associations were modest: those with low levels had a 1.20-times higher rate of death from cardiovascular disease and a statistically significant 1.26-times higher rate of death from all causes.
Randomized clinical trials
A meta-analysis of 18 randomized trials5 of vitamin D supplementation (300–2,000 IU/day, mean 528 IU/day vs placebo), including 57,311 participants, evaluated the rate of death from all causes and found a modest but significant reduction in risk (relative risk 0.93, 95% confidence interval [CI] 0.87–0.99). These were generally trials looking at fracture rates or physical performance, and a dose-response relationship was not evident. A recent systematic review of randomized controlled trials of vitamin D1 that included cardiovascular disease as a secondary outcome found a pooled relative risk for cardiovascular disease of 0.90 (95% CI 0.77–1.05) for vitamin D supplementation compared with placebo and 1.04 (95% CI 0.92–1.18) for combination vitamin D plus calcium supplementation vs placebo.1 Two individual trials are discussed below.
Trivedi et al6 randomized 2,686 British men and women to vitamin D3 100,000 IU given every 4 months over 5 years (equivalent to 800 IU/day) or placebo. The relative risk of cardiovascular events was 0.90 (95% CI 0.77–1.06) and of cardiovascular deaths 0.84 (95% CI 0.65–1.10). Although the results were promising, the trial was designed to assess fracture risk and was not large enough for the differences in cardiovascular outcomes to reach statistical significance.
The Women’s Health Initiative,7,8 which included 36,282 postmenopausal women aged 50 to 79, tested combined vitamin D3 (400 IU/day) with calcium (1,000 mg/day) vs placebo. No benefit was seen for preventing coronary events or stroke, which may be due to the low dosage of vitamin D. The hazard ratio for coronary disease was 1.04 (0.92–1.18). Regarding mortality, the hazard ratio for cardiovascular death was 0.92, for cerebrovascular death 0.89, for cancer death 0.89, and for other deaths 0.95. None of these hazard ratios reached statistical significance.
MORE MAY NOT BE BETTER
As is probably true for everything in biological systems, there apparently is an optimal level of intake to meet vitamin D needs.
The Framingham Offspring Study,2 which found a higher risk with vitamin D deficiency, also found a suggestion of a threshold. Participants who had levels of 50 to 65 nmol/L had the lowest risk. Higher levels did not confer lower risk and even suggested a slight upturn.
Evidence from the Women’s Health Initiative8 also indicates that high dosages may not be better than moderate dosages. The meta-analysis of vitamin D and all-cause mortality5 found a relative risk of 0.93, but one of the largest studies in that meta-analysis tested only 400 IU a day and found a similar relative risk of 0.91 (95% confidence interval, 0.83–1.01).
Moreover, the NHANES study found that with increasing serum 25-hydroxyvitamin D3 levels, the risk of all-cause mortality fell until about 100 nmol/L, but then plateaued and even increased with higher serum levels.4
VITAL: STUDY DESIGN AND LOGISTICS
In VITAL, the investigators aim to recruit 20,000 healthy men (age 60 and older) and women (65 and older) who are representative of the US population (www.vitalstudy.org). Because it is a primary prevention trial, people with a known history of cardiovascular disease or cancer will be excluded. Participants will be randomized to receive either 2,000 IU of vitamin D3 per day or placebo. Each group will be further randomized to receive either 1 g per day of fish oil (combined eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) or placebo. The mean treatment period will be 5 years. Recruitment began in early 2010.
Blood will be collected in about 80% (ideally 100%) of participants, with follow-up blood collection in at least 2,000.
Primary aims of the trial are to test whether vitamin D3 and the omega-3 fatty acids reduce the risk of total cancer and major cardiovascular events (a composite of myocardial infarction, stroke, and death due to cardiovascular events).
Secondary aims are to test whether these agents lower the risk of:
- Site-specific cancer, including colorectal, breast, and prostate cancer, and the total cancer mortality rate
- An expanded composite outcome including myocardial infarction, stroke, cardiovascular death, coronary artery bypass grafting, percutaneous coronary intervention, and its individual components.
Tertiary aims are to explore whether vitamin D3 and omega-3 fatty acids have additive effects on the primary and secondary end points. The trial will also explore whether the effects of vitamin D3 and omega-3 fatty acids on cancer and cardiovascular disease vary by baseline blood levels of these nutrients, and whether race, skin pigmentation, or body mass index modify the effects of vitamin D3.
Ancillary studies will assess the effect of the interventions on risk of diabetes, hypertension, cognitive decline, depression, fracture, infections, respiratory disorders, and autoimmune diseases. The primary sponsor of this trial is the National Cancer Institute, and the secondary sponsor is the National Heart, Lung and Blood Institute. Other institutes and agencies also are cosponsors of the study.
The timing of VITAL is optimal
There is a limited window of opportunity for conducting a randomized clinical trial: the evidence must be strong enough to justify mounting a very large trial with enough power to look at cardiovascular events and cancer, but the evidence must not be so strong that it would be unethical to have a placebo group. Thus, there must be a state of equipoise. Our trial allows the study population to have a background intake of vitamin D that is currently recommended by national guidelines. Therefore, even the placebo group should have adequate intake of vitamin D.
The growing use of vitamin D supplementation by the public underscores the need for conclusive evidence of its benefits and risks. No previous large-scale randomized clinical trial has tested moderate to high doses of vitamin D for the primary prevention of cancer and cardiovascular disease.
Setting the dosage
VITAL set the vitamin D3 dosage at 2,000 IU per day (50 μg/day), which is designed to provide the best balance of efficacy and safety. As a general rule, each microgram of vitamin D3 is expected to raise the serum 25-hydroxyvitamin D3 level about 1 nmol/L, although the response is not linear: if baseline levels are lower, the increase is greater. In the United States, people commonly have a baseline level of about 40 nmol/L, so we expect that levels of people treated in the study will reach about 90 nmol/L (range 75–100 nmol/L), about 35 to 50 nmol/L higher than in the placebo group.
The target range of 75 to 100 nmol/L is the level at which greatest efficacy has been suggested in observational studies. Previous randomized trials of vitamin D have not tested high enough doses to achieve this level of 25-hydroxyvitamin D3. VITAL will test whether reaching this serum level lowers the risk of cardiovascular disease, cancer, and other chronic diseases. This level may be associated with benefit and has minimal risk of hypercalcemia. Risk of hypercalcemia may be present in participants with an occult chronic granulomatous condition such as sarcoidosis or Wegener granulomatosis, in which activated macrophages synthesize 1,25-dihydroxyvitamin D3. These conditions are very rare, however, and the risk of hypercalcemia in the trial is exceedingly low.
VITAL participants will also be randomized to take placebo or 1 g per day of combined EPA and DHA, about 5 to 10 times more than most Americans consume.
Nationwide recruitment among senior citizens
We aim to recruit 20,000 people (10,000 men and 10,000 women) nationwide who are willing, eligible, and compliant (ie, who take more than two-thirds of study pills during a 3-month placebo “run-in” phase of the trial). The trial aims to enroll 40,000 in the run-in period, and 20,000 will be randomized. To get this many participants, we will send invitational mailings and screening questionnaires to at least 2.5 million people around the United States, with mailing lists selected by age—ie, members of the American Association of Retired Persons, health professionals, teachers, and subscription lists for selected magazines. A pilot study in 5,000 people has indicated that recruiting and randomizing 20,000 participants via large mailings should be possible.
The trial is expected to be extremely cost-effective because it will be conducted largely by mail. Medication will be mailed in calendar blister packs. Participants report outcomes, which are then confirmed by medical record review. The Centers for Medicare and Medicaid Services and the National Death Index will also be used to ascertain outcomes.
We hope to recruit a more racially diverse study population than is typically seen in US trials: 63% (12,620) whites, 25% (5,000) African Americans, 7% (1,400) Hispanics, 2.5% (500) Asians, 2% (400) American Indians and Alaska natives, and 0.4% (80) native Hawaiian and Pacific Islanders.
Eligibility criteria ensure primary prevention is tested
To enter the study, men must be at least 60 years old and women at least 65. At a minimum, a high school education is required due to the detailed forms and questionnaires to be completed. Because this is a primary prevention trial, anyone with a history of cancer (except nonmelanoma skin cancer) or cardiovascular disease (including myocardial infarction, stroke, or coronary revascularization) will be excluded, as will anyone with a history of kidney stones, renal failure or dialysis, hypercalcemia, hypoparathyroidism or hyperparathyroidism, severe liver disease (eg, cirrhosis), sarcoidosis, tuberculosis, or other granulomatous disease. People with an allergy to fish will also be excluded.
We do not expect that those in the placebo group will develop vitamin D deficiency due to their participation in the study. The trial will allow a background intake in the study population of up to 800 IU of vitamin D and 1,200 mg of calcium per day in supplements. Assuming they also get about 200 IU of vitamin D in the diet, the background intake in the placebo group may be close to 1,000 IU of vitamin D. Assuming that the active treatment group has a similar background intake, their total intake will be about 3,000 IU per day (about 1,000 IU/day from background intake plus 2,000 IU/day from the intervention).
Cohort power sufficient to see effect in 5 years
The trial is expected to have sufficient power to evaluate cardiovascular disease and cancer end points as primary outcomes during 5 years of follow-up. The trial is designed to have a power of 91% to 92% to detect a relative risk of 0.85 for the primary cancer end point of total cancer incidence and 0.80 for the cardiovascular disease end point of myocardial infarction, stroke, and cardiovascular mortality. Power will be even greater for the expanded composite outcome for cardiovascular disease.
Ancillary studies
Ancillary studies include evaluating the interventions’ role in preventing diabetes and glucose intolerance, hypertension, heart failure, atrial fibrillation, cognitive decline, mood disorders, osteoporosis and fractures, asthma and respiratory diseases, infections, macular degeneration, rheumatoid arthritis, systemic lupus erythematosus, and a composite of autoimmune diseases. Imaging studies also are planned, including dual energy x-ray absorptiometry, mammographic density, and non-invasive vascular imaging (carotid intima medial thickness, coronary calcium measurements, and two-dimensional echocardiography to assess cardiac function).
Several biomarker and genetic studies will also be carried out. We intend to perform genetic studies on most of the study population to evaluate gene variants in the vitamin D receptor, vitamin D binding protein, and other vitamin-D-related genes that may contribute to lower baseline levels of 25-hydroxyvitamin D3 or different responses to the interventions.
Clinical and Translational Science Center visits are planned to provide more detailed assessments of 1,000 participants, including blood pressure measurements, height, weight, waist circumference, other anthropometric measurements, a 2-hour glucose tolerance test, a fasting blood collection, hemoglobin A1c measurements, spirometry, and assessment of physical performance, strength, frailty, cognitive function, mood, and depression. Dual-energy x-ray absorptiometry and noninvasive vascular imaging studies are also planned for those visits.
- Wang L, Manson JE, Song Y, Sesso HD. Systematic review: vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med 2010; 152:315–323.
- Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008; 117:503–511.
- Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med 2008; 168:1174–1180.
- Melamed ML, Michos ED, Post W, Astor B. 25-Hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med 2008; 168:1629–1637.
- Autier P, Gandini S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med 2007; 167:1730–1737.
- Trivedi DP, Doll R, Khaw KT. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ 2003; 326:469.
- Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation 2007; 115:846–854.
- LaCroix AZ, Kotchen J, Anderson G, et al. Calcium plus vitamin D supplementation and mortality in postmenopausal women: the Women’s Health Initiative calcium-vitamin D randomized controlled trial. J Gerontol A Biol Sci Med Sci 2009; 64:559–567.
Vitamin D is viewed as a promising supplement by the medical, public health, and lay communities, potentially offering many health benefits. But enthusiasm for a new intervention too often gets far ahead of the evidence, as was the case with beta-carotene, selenium, folic acid, and vitamins C and E.
Despite the enthusiasm for vitamin D, there have been no large-scale primary prevention trials that have had either cardiovascular disease or cancer as a prespecified primary outcome. Previous randomized trials of vitamin D have focused primarily on osteoporosis, fracture, falls, and physical function. Although the investigators often reported their findings on vitamin D and cardiovascular disease or cancer, these outcomes were generally secondary or tertiary end points that were not prespecified. These studies should be viewed as hypothesis-generating rather than hypothesis-testing. The increasing prevalence of use of vitamin D supplements underscores the need for rigorous and conclusive evidence from randomized clinical trials that have cardiovascular disease and cancer as primary outcomes.
This article will explain the rationale for a large-scale, randomized clinical trial to evaluate the role of vitamin D in the prevention of cardiovascular disease and cancer. It will also describe the biological mechanisms and currently available evidence relating vitamin D to potential health benefits. Finally, the design, dosage considerations, and logistics of the Vitamin D and Omega-3 Trial (VITAL) will be presented.
EVIDENCE IS MOUNTING FOR VITAMIN D’S BIOLOGICAL IMPORTANCE
Vitamin D is undoubtedly important to health: not only is it produced endogenously, but at least 500 genes have been identified with vitamin D response elements. The vitamin D receptor is found in nearly all cells in the body, and the 1-alpha-hydroxylase enzyme is present in many tissues. Some studies suggest that almost 10% of the human genome may be at least partially regulated by vitamin D.
Although we know vitamin D is important, what our optimal intake and our blood level of 25-hydroxyvitamin D3 should be are key unknowns.
RECOMMENDATIONS FOR VITAMIN D INTAKE
During winter, late fall, and early spring, people who live above the 37th parallel (geographically, about one-half of the contiguous United States) do not get enough ultraviolet B energy from the sun to make all the vitamin D they need, even if they spend several hours outside every day. In addition, dark skin pigmentation serves as a sun block, as do sunscreens.
The Institute of Medicine (IOM) provided guidelines for vitamin D intake in 1997 and, most recently, in 2010. However, these guidelines are based on the amount of vitamin D required for bone health and do not address the amount that may be of benefit for prevention of cancer and cardiovascular disease. The latter outcomes are not addressed because the IOM committee believed that evidence was insufficient to determine the role of vitamin D in the prevention of cardiovascular disease, cancer, and other chronic diseases. Thus, current IOM guidelines, which generally recommend less than 1,000 IU of vitamin D daily, are relevant to bone health but not necessarily to other health outcomes. More research is needed to understand whether the guidelines should be modified for the prevention of other chronic diseases.
Moreover, whether or not everyone should be screened for 25-hydroxyvitamin D3 blood levels is controversial. Most experts agree that a level less than 20 ng/mL is deficient or insufficient. Conversely, potentially harmful are levels 150 ng/mL or more (> 375 nmol/L), which entail the risk of hypercalcemia, vascular soft tissue calcification, and hyperphosphatemia.
People do not reach toxic levels with ultraviolet light exposure because the amount of 25-hydroxyvitamin D3 synthesis is well regulated. Dietary supplements, however, can bring about toxic levels, and patients taking high doses need to be monitored carefully. The level that should be considered optimal is controversial and requires further study.
RISK FACTORS FOR LOW VITAMIN D LEVELS
Risk factors for low vitamin D levels include older age, living in northern latitudes, sun avoidance, dark skin pigmentation, obesity, low dietary intake, and various medical conditions, especially malabsorption syndromes. Some of these are also risk factors for cardiovascular disease, cancer, and other chronic diseases, and potentially confound outcomes in many studies. Older age, which is usually adjusted for in multivariate models, is important to recognize as a major risk factor for vitamin D deficiency, owing to reduced absorption and synthesis, less time outdoors, and low dietary intake.
Wearing sunscreen decreases the synthesis of vitamin D in the skin, but because ultra-violet light has been clearly classified as a carcinogen, it is a not advisable to increase sun exposure for the sake of increasing vitamin D levels. That is a poor trade-off, given the high incidence rate of skin cancer and the adverse effects of solar radiation on skin aging.
Obesity is a risk factor for vitamin D deficiency because vitamin D is fat-soluble and becomes sequestered in fat tissue. Vitamin D may also play a role in the differentiation of adipocytes and may affect their function. In observational studies, it is very important for researchers to adjust for body mass index, physical activity (which may be correlated with more time outdoors), and other potential confounders in their analyses.
HOW VITAMIN D MAY LOWER CANCER RISK
Because of the important effect of vitamin D in regulating cell differentiation and cell growth, there are multiple ways that it may affect cancer risk. Laboratory, cell culture, and animal studies suggest that vitamin D may lower cancer risk by inhibiting cell proliferation, angiogenesis, metastasis, and inflammation and inducing apoptosis and cellular differentiation. Several of these mechanisms are also relevant to atherosclerosis and cardiovascular disease. Although VITAL is addressing the role of vitamin D in preventing both cancer and cardiovascular disease, the remainder of this article will focus on cardiovascular outcomes.
HOW VITAMIN D MAY REDUCE CARDIOVASCULAR RISK
Vitamin D may lower cardiovascular risk via several mechanisms:
Inhibiting inflammation. Vitamin D has a powerful immunomodulatory effect: laboratory studies show that it inhibits prostaglandin and cyclooxygenase 2 pathways, reduces matrix metalloproteinase 9 and several proinflammatory cytokines, and increases interleukin 10, all of which result in suppressed inflammation.1
Inhibiting vascular muscle proliferation and vascular calcification. Animal studies indicate that in moderate doses vitamin D decreases calcium cellular influx and increases matrix Gla protein, which inhibits vascular smooth muscle proliferation and vascular calcification. These protective effects contrast with the hypercalcemia associated with a high intake of vitamin D, especially in the context of renal failure or other risk factors, which may lead to increased vascular calcification.1
Regulates blood pressure. Vitamin D decreases renin gene expression and the synthesis of renin, which reduces activity of the renin-angiotensin-aldosterone system, leading to a reduction of blood pressure and a favorable effect on volume homeostasis.1
Regulates glucose metabolism. Limited evidence shows that vitamin D may increase insulin sensitivity and regulate glucose metabolism.1
Vitamin D and cardiac hypertrophy
The vitamin D receptor is present in virtually all tissues, including cardiac myocytes and endothelial cells. Animals with vitamin D deficiency have higher blood pressures, and animals genetically altered to have no vitamin D receptors (knock-out models) develop left ventricular hypertrophy and heart failure.
Animals genetically altered to have no 1-alpha-hydroxylase (so that the most active form of vitamin D is not made) also develop left ventricular hypertrophy. They can be rescued by the administration of 1,25-dihydroxy vitamin D3.1
These findings are consistent with what is observed in patients with end-stage renal disease, who produce very little 1,25-dihydroxyvitamin D3: they often develop left ventricular hypertrophy, diastolic heart failure, atherosclerosis, and vascular calcification.
EVIDENCE FOR CARDIOVASCULAR DISEASE REDUCTION
Wang et al1 recently reviewed available prospective cohort and randomized clinical trials from 1966 to 2009 that examined vitamin D or calcium supplementation and cardiovascular disease. Comparing people with the lowest to the highest levels of serum 25-hydroxyvitamin D3 indicated that a low level is a risk factor for coronary artery disease and cardiovascular death. Unfortunately, most studies were not designed to assess primary effects on cardiovascular outcomes, and so have many potential confounders.
Prospective observational studies
Observational studies suggest that vitamin D deficiency is associated with an increased risk of cardiovascular disease. Some examples:
The Framingham Offspring Study2 followed 1,739 men and women with a mean age of 59 for 5.4 years. The study compared the incidence of cardiovascular events in those with a serum 25-dihydroxyvitamin D level of at least 37.5 nmol/L vs those with lower levels. The risk of cardiovascular disease was 1.62 times higher in those with the lowest levels of vitamin D, a statistically significant difference. However, a threshold effect was apparent (discussed below).
The Health Professionals Follow-up Study3 prospectively evaluated more than 18,000 men ages 40 to 75 for 10 years. The study compared men with a low serum level of vitamin D (< 37.5 nmol/L) to those with a more optimal level (> 75 nmol/L). The incidence of cardiovascular events was 2.09 times higher in men with low levels of vitamin D, a difference that was statistically significant.
The Third National Health and Nutrition Examination Survey (NHANES III) included data for more than 13,300 men and women age 20 years and older. Using a cohort that was followed for 8.7 years, Melamed et al4 compared the quartile with the lowest serum vitamin D level (< 44.4 nmol/L) against the quartile with the highest level (> 80.1 nmol/L). The associations were modest: those with low levels had a 1.20-times higher rate of death from cardiovascular disease and a statistically significant 1.26-times higher rate of death from all causes.
Randomized clinical trials
A meta-analysis of 18 randomized trials5 of vitamin D supplementation (300–2,000 IU/day, mean 528 IU/day vs placebo), including 57,311 participants, evaluated the rate of death from all causes and found a modest but significant reduction in risk (relative risk 0.93, 95% confidence interval [CI] 0.87–0.99). These were generally trials looking at fracture rates or physical performance, and a dose-response relationship was not evident. A recent systematic review of randomized controlled trials of vitamin D1 that included cardiovascular disease as a secondary outcome found a pooled relative risk for cardiovascular disease of 0.90 (95% CI 0.77–1.05) for vitamin D supplementation compared with placebo and 1.04 (95% CI 0.92–1.18) for combination vitamin D plus calcium supplementation vs placebo.1 Two individual trials are discussed below.
Trivedi et al6 randomized 2,686 British men and women to vitamin D3 100,000 IU given every 4 months over 5 years (equivalent to 800 IU/day) or placebo. The relative risk of cardiovascular events was 0.90 (95% CI 0.77–1.06) and of cardiovascular deaths 0.84 (95% CI 0.65–1.10). Although the results were promising, the trial was designed to assess fracture risk and was not large enough for the differences in cardiovascular outcomes to reach statistical significance.
The Women’s Health Initiative,7,8 which included 36,282 postmenopausal women aged 50 to 79, tested combined vitamin D3 (400 IU/day) with calcium (1,000 mg/day) vs placebo. No benefit was seen for preventing coronary events or stroke, which may be due to the low dosage of vitamin D. The hazard ratio for coronary disease was 1.04 (0.92–1.18). Regarding mortality, the hazard ratio for cardiovascular death was 0.92, for cerebrovascular death 0.89, for cancer death 0.89, and for other deaths 0.95. None of these hazard ratios reached statistical significance.
MORE MAY NOT BE BETTER
As is probably true for everything in biological systems, there apparently is an optimal level of intake to meet vitamin D needs.
The Framingham Offspring Study,2 which found a higher risk with vitamin D deficiency, also found a suggestion of a threshold. Participants who had levels of 50 to 65 nmol/L had the lowest risk. Higher levels did not confer lower risk and even suggested a slight upturn.
Evidence from the Women’s Health Initiative8 also indicates that high dosages may not be better than moderate dosages. The meta-analysis of vitamin D and all-cause mortality5 found a relative risk of 0.93, but one of the largest studies in that meta-analysis tested only 400 IU a day and found a similar relative risk of 0.91 (95% confidence interval, 0.83–1.01).
Moreover, the NHANES study found that with increasing serum 25-hydroxyvitamin D3 levels, the risk of all-cause mortality fell until about 100 nmol/L, but then plateaued and even increased with higher serum levels.4
VITAL: STUDY DESIGN AND LOGISTICS
In VITAL, the investigators aim to recruit 20,000 healthy men (age 60 and older) and women (65 and older) who are representative of the US population (www.vitalstudy.org). Because it is a primary prevention trial, people with a known history of cardiovascular disease or cancer will be excluded. Participants will be randomized to receive either 2,000 IU of vitamin D3 per day or placebo. Each group will be further randomized to receive either 1 g per day of fish oil (combined eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) or placebo. The mean treatment period will be 5 years. Recruitment began in early 2010.
Blood will be collected in about 80% (ideally 100%) of participants, with follow-up blood collection in at least 2,000.
Primary aims of the trial are to test whether vitamin D3 and the omega-3 fatty acids reduce the risk of total cancer and major cardiovascular events (a composite of myocardial infarction, stroke, and death due to cardiovascular events).
Secondary aims are to test whether these agents lower the risk of:
- Site-specific cancer, including colorectal, breast, and prostate cancer, and the total cancer mortality rate
- An expanded composite outcome including myocardial infarction, stroke, cardiovascular death, coronary artery bypass grafting, percutaneous coronary intervention, and its individual components.
Tertiary aims are to explore whether vitamin D3 and omega-3 fatty acids have additive effects on the primary and secondary end points. The trial will also explore whether the effects of vitamin D3 and omega-3 fatty acids on cancer and cardiovascular disease vary by baseline blood levels of these nutrients, and whether race, skin pigmentation, or body mass index modify the effects of vitamin D3.
Ancillary studies will assess the effect of the interventions on risk of diabetes, hypertension, cognitive decline, depression, fracture, infections, respiratory disorders, and autoimmune diseases. The primary sponsor of this trial is the National Cancer Institute, and the secondary sponsor is the National Heart, Lung and Blood Institute. Other institutes and agencies also are cosponsors of the study.
The timing of VITAL is optimal
There is a limited window of opportunity for conducting a randomized clinical trial: the evidence must be strong enough to justify mounting a very large trial with enough power to look at cardiovascular events and cancer, but the evidence must not be so strong that it would be unethical to have a placebo group. Thus, there must be a state of equipoise. Our trial allows the study population to have a background intake of vitamin D that is currently recommended by national guidelines. Therefore, even the placebo group should have adequate intake of vitamin D.
The growing use of vitamin D supplementation by the public underscores the need for conclusive evidence of its benefits and risks. No previous large-scale randomized clinical trial has tested moderate to high doses of vitamin D for the primary prevention of cancer and cardiovascular disease.
Setting the dosage
VITAL set the vitamin D3 dosage at 2,000 IU per day (50 μg/day), which is designed to provide the best balance of efficacy and safety. As a general rule, each microgram of vitamin D3 is expected to raise the serum 25-hydroxyvitamin D3 level about 1 nmol/L, although the response is not linear: if baseline levels are lower, the increase is greater. In the United States, people commonly have a baseline level of about 40 nmol/L, so we expect that levels of people treated in the study will reach about 90 nmol/L (range 75–100 nmol/L), about 35 to 50 nmol/L higher than in the placebo group.
The target range of 75 to 100 nmol/L is the level at which greatest efficacy has been suggested in observational studies. Previous randomized trials of vitamin D have not tested high enough doses to achieve this level of 25-hydroxyvitamin D3. VITAL will test whether reaching this serum level lowers the risk of cardiovascular disease, cancer, and other chronic diseases. This level may be associated with benefit and has minimal risk of hypercalcemia. Risk of hypercalcemia may be present in participants with an occult chronic granulomatous condition such as sarcoidosis or Wegener granulomatosis, in which activated macrophages synthesize 1,25-dihydroxyvitamin D3. These conditions are very rare, however, and the risk of hypercalcemia in the trial is exceedingly low.
VITAL participants will also be randomized to take placebo or 1 g per day of combined EPA and DHA, about 5 to 10 times more than most Americans consume.
Nationwide recruitment among senior citizens
We aim to recruit 20,000 people (10,000 men and 10,000 women) nationwide who are willing, eligible, and compliant (ie, who take more than two-thirds of study pills during a 3-month placebo “run-in” phase of the trial). The trial aims to enroll 40,000 in the run-in period, and 20,000 will be randomized. To get this many participants, we will send invitational mailings and screening questionnaires to at least 2.5 million people around the United States, with mailing lists selected by age—ie, members of the American Association of Retired Persons, health professionals, teachers, and subscription lists for selected magazines. A pilot study in 5,000 people has indicated that recruiting and randomizing 20,000 participants via large mailings should be possible.
The trial is expected to be extremely cost-effective because it will be conducted largely by mail. Medication will be mailed in calendar blister packs. Participants report outcomes, which are then confirmed by medical record review. The Centers for Medicare and Medicaid Services and the National Death Index will also be used to ascertain outcomes.
We hope to recruit a more racially diverse study population than is typically seen in US trials: 63% (12,620) whites, 25% (5,000) African Americans, 7% (1,400) Hispanics, 2.5% (500) Asians, 2% (400) American Indians and Alaska natives, and 0.4% (80) native Hawaiian and Pacific Islanders.
Eligibility criteria ensure primary prevention is tested
To enter the study, men must be at least 60 years old and women at least 65. At a minimum, a high school education is required due to the detailed forms and questionnaires to be completed. Because this is a primary prevention trial, anyone with a history of cancer (except nonmelanoma skin cancer) or cardiovascular disease (including myocardial infarction, stroke, or coronary revascularization) will be excluded, as will anyone with a history of kidney stones, renal failure or dialysis, hypercalcemia, hypoparathyroidism or hyperparathyroidism, severe liver disease (eg, cirrhosis), sarcoidosis, tuberculosis, or other granulomatous disease. People with an allergy to fish will also be excluded.
We do not expect that those in the placebo group will develop vitamin D deficiency due to their participation in the study. The trial will allow a background intake in the study population of up to 800 IU of vitamin D and 1,200 mg of calcium per day in supplements. Assuming they also get about 200 IU of vitamin D in the diet, the background intake in the placebo group may be close to 1,000 IU of vitamin D. Assuming that the active treatment group has a similar background intake, their total intake will be about 3,000 IU per day (about 1,000 IU/day from background intake plus 2,000 IU/day from the intervention).
Cohort power sufficient to see effect in 5 years
The trial is expected to have sufficient power to evaluate cardiovascular disease and cancer end points as primary outcomes during 5 years of follow-up. The trial is designed to have a power of 91% to 92% to detect a relative risk of 0.85 for the primary cancer end point of total cancer incidence and 0.80 for the cardiovascular disease end point of myocardial infarction, stroke, and cardiovascular mortality. Power will be even greater for the expanded composite outcome for cardiovascular disease.
Ancillary studies
Ancillary studies include evaluating the interventions’ role in preventing diabetes and glucose intolerance, hypertension, heart failure, atrial fibrillation, cognitive decline, mood disorders, osteoporosis and fractures, asthma and respiratory diseases, infections, macular degeneration, rheumatoid arthritis, systemic lupus erythematosus, and a composite of autoimmune diseases. Imaging studies also are planned, including dual energy x-ray absorptiometry, mammographic density, and non-invasive vascular imaging (carotid intima medial thickness, coronary calcium measurements, and two-dimensional echocardiography to assess cardiac function).
Several biomarker and genetic studies will also be carried out. We intend to perform genetic studies on most of the study population to evaluate gene variants in the vitamin D receptor, vitamin D binding protein, and other vitamin-D-related genes that may contribute to lower baseline levels of 25-hydroxyvitamin D3 or different responses to the interventions.
Clinical and Translational Science Center visits are planned to provide more detailed assessments of 1,000 participants, including blood pressure measurements, height, weight, waist circumference, other anthropometric measurements, a 2-hour glucose tolerance test, a fasting blood collection, hemoglobin A1c measurements, spirometry, and assessment of physical performance, strength, frailty, cognitive function, mood, and depression. Dual-energy x-ray absorptiometry and noninvasive vascular imaging studies are also planned for those visits.
Vitamin D is viewed as a promising supplement by the medical, public health, and lay communities, potentially offering many health benefits. But enthusiasm for a new intervention too often gets far ahead of the evidence, as was the case with beta-carotene, selenium, folic acid, and vitamins C and E.
Despite the enthusiasm for vitamin D, there have been no large-scale primary prevention trials that have had either cardiovascular disease or cancer as a prespecified primary outcome. Previous randomized trials of vitamin D have focused primarily on osteoporosis, fracture, falls, and physical function. Although the investigators often reported their findings on vitamin D and cardiovascular disease or cancer, these outcomes were generally secondary or tertiary end points that were not prespecified. These studies should be viewed as hypothesis-generating rather than hypothesis-testing. The increasing prevalence of use of vitamin D supplements underscores the need for rigorous and conclusive evidence from randomized clinical trials that have cardiovascular disease and cancer as primary outcomes.
This article will explain the rationale for a large-scale, randomized clinical trial to evaluate the role of vitamin D in the prevention of cardiovascular disease and cancer. It will also describe the biological mechanisms and currently available evidence relating vitamin D to potential health benefits. Finally, the design, dosage considerations, and logistics of the Vitamin D and Omega-3 Trial (VITAL) will be presented.
EVIDENCE IS MOUNTING FOR VITAMIN D’S BIOLOGICAL IMPORTANCE
Vitamin D is undoubtedly important to health: not only is it produced endogenously, but at least 500 genes have been identified with vitamin D response elements. The vitamin D receptor is found in nearly all cells in the body, and the 1-alpha-hydroxylase enzyme is present in many tissues. Some studies suggest that almost 10% of the human genome may be at least partially regulated by vitamin D.
Although we know vitamin D is important, what our optimal intake and our blood level of 25-hydroxyvitamin D3 should be are key unknowns.
RECOMMENDATIONS FOR VITAMIN D INTAKE
During winter, late fall, and early spring, people who live above the 37th parallel (geographically, about one-half of the contiguous United States) do not get enough ultraviolet B energy from the sun to make all the vitamin D they need, even if they spend several hours outside every day. In addition, dark skin pigmentation serves as a sun block, as do sunscreens.
The Institute of Medicine (IOM) provided guidelines for vitamin D intake in 1997 and, most recently, in 2010. However, these guidelines are based on the amount of vitamin D required for bone health and do not address the amount that may be of benefit for prevention of cancer and cardiovascular disease. The latter outcomes are not addressed because the IOM committee believed that evidence was insufficient to determine the role of vitamin D in the prevention of cardiovascular disease, cancer, and other chronic diseases. Thus, current IOM guidelines, which generally recommend less than 1,000 IU of vitamin D daily, are relevant to bone health but not necessarily to other health outcomes. More research is needed to understand whether the guidelines should be modified for the prevention of other chronic diseases.
Moreover, whether or not everyone should be screened for 25-hydroxyvitamin D3 blood levels is controversial. Most experts agree that a level less than 20 ng/mL is deficient or insufficient. Conversely, potentially harmful are levels 150 ng/mL or more (> 375 nmol/L), which entail the risk of hypercalcemia, vascular soft tissue calcification, and hyperphosphatemia.
People do not reach toxic levels with ultraviolet light exposure because the amount of 25-hydroxyvitamin D3 synthesis is well regulated. Dietary supplements, however, can bring about toxic levels, and patients taking high doses need to be monitored carefully. The level that should be considered optimal is controversial and requires further study.
RISK FACTORS FOR LOW VITAMIN D LEVELS
Risk factors for low vitamin D levels include older age, living in northern latitudes, sun avoidance, dark skin pigmentation, obesity, low dietary intake, and various medical conditions, especially malabsorption syndromes. Some of these are also risk factors for cardiovascular disease, cancer, and other chronic diseases, and potentially confound outcomes in many studies. Older age, which is usually adjusted for in multivariate models, is important to recognize as a major risk factor for vitamin D deficiency, owing to reduced absorption and synthesis, less time outdoors, and low dietary intake.
Wearing sunscreen decreases the synthesis of vitamin D in the skin, but because ultra-violet light has been clearly classified as a carcinogen, it is a not advisable to increase sun exposure for the sake of increasing vitamin D levels. That is a poor trade-off, given the high incidence rate of skin cancer and the adverse effects of solar radiation on skin aging.
Obesity is a risk factor for vitamin D deficiency because vitamin D is fat-soluble and becomes sequestered in fat tissue. Vitamin D may also play a role in the differentiation of adipocytes and may affect their function. In observational studies, it is very important for researchers to adjust for body mass index, physical activity (which may be correlated with more time outdoors), and other potential confounders in their analyses.
HOW VITAMIN D MAY LOWER CANCER RISK
Because of the important effect of vitamin D in regulating cell differentiation and cell growth, there are multiple ways that it may affect cancer risk. Laboratory, cell culture, and animal studies suggest that vitamin D may lower cancer risk by inhibiting cell proliferation, angiogenesis, metastasis, and inflammation and inducing apoptosis and cellular differentiation. Several of these mechanisms are also relevant to atherosclerosis and cardiovascular disease. Although VITAL is addressing the role of vitamin D in preventing both cancer and cardiovascular disease, the remainder of this article will focus on cardiovascular outcomes.
HOW VITAMIN D MAY REDUCE CARDIOVASCULAR RISK
Vitamin D may lower cardiovascular risk via several mechanisms:
Inhibiting inflammation. Vitamin D has a powerful immunomodulatory effect: laboratory studies show that it inhibits prostaglandin and cyclooxygenase 2 pathways, reduces matrix metalloproteinase 9 and several proinflammatory cytokines, and increases interleukin 10, all of which result in suppressed inflammation.1
Inhibiting vascular muscle proliferation and vascular calcification. Animal studies indicate that in moderate doses vitamin D decreases calcium cellular influx and increases matrix Gla protein, which inhibits vascular smooth muscle proliferation and vascular calcification. These protective effects contrast with the hypercalcemia associated with a high intake of vitamin D, especially in the context of renal failure or other risk factors, which may lead to increased vascular calcification.1
Regulates blood pressure. Vitamin D decreases renin gene expression and the synthesis of renin, which reduces activity of the renin-angiotensin-aldosterone system, leading to a reduction of blood pressure and a favorable effect on volume homeostasis.1
Regulates glucose metabolism. Limited evidence shows that vitamin D may increase insulin sensitivity and regulate glucose metabolism.1
Vitamin D and cardiac hypertrophy
The vitamin D receptor is present in virtually all tissues, including cardiac myocytes and endothelial cells. Animals with vitamin D deficiency have higher blood pressures, and animals genetically altered to have no vitamin D receptors (knock-out models) develop left ventricular hypertrophy and heart failure.
Animals genetically altered to have no 1-alpha-hydroxylase (so that the most active form of vitamin D is not made) also develop left ventricular hypertrophy. They can be rescued by the administration of 1,25-dihydroxy vitamin D3.1
These findings are consistent with what is observed in patients with end-stage renal disease, who produce very little 1,25-dihydroxyvitamin D3: they often develop left ventricular hypertrophy, diastolic heart failure, atherosclerosis, and vascular calcification.
EVIDENCE FOR CARDIOVASCULAR DISEASE REDUCTION
Wang et al1 recently reviewed available prospective cohort and randomized clinical trials from 1966 to 2009 that examined vitamin D or calcium supplementation and cardiovascular disease. Comparing people with the lowest to the highest levels of serum 25-hydroxyvitamin D3 indicated that a low level is a risk factor for coronary artery disease and cardiovascular death. Unfortunately, most studies were not designed to assess primary effects on cardiovascular outcomes, and so have many potential confounders.
Prospective observational studies
Observational studies suggest that vitamin D deficiency is associated with an increased risk of cardiovascular disease. Some examples:
The Framingham Offspring Study2 followed 1,739 men and women with a mean age of 59 for 5.4 years. The study compared the incidence of cardiovascular events in those with a serum 25-dihydroxyvitamin D level of at least 37.5 nmol/L vs those with lower levels. The risk of cardiovascular disease was 1.62 times higher in those with the lowest levels of vitamin D, a statistically significant difference. However, a threshold effect was apparent (discussed below).
The Health Professionals Follow-up Study3 prospectively evaluated more than 18,000 men ages 40 to 75 for 10 years. The study compared men with a low serum level of vitamin D (< 37.5 nmol/L) to those with a more optimal level (> 75 nmol/L). The incidence of cardiovascular events was 2.09 times higher in men with low levels of vitamin D, a difference that was statistically significant.
The Third National Health and Nutrition Examination Survey (NHANES III) included data for more than 13,300 men and women age 20 years and older. Using a cohort that was followed for 8.7 years, Melamed et al4 compared the quartile with the lowest serum vitamin D level (< 44.4 nmol/L) against the quartile with the highest level (> 80.1 nmol/L). The associations were modest: those with low levels had a 1.20-times higher rate of death from cardiovascular disease and a statistically significant 1.26-times higher rate of death from all causes.
Randomized clinical trials
A meta-analysis of 18 randomized trials5 of vitamin D supplementation (300–2,000 IU/day, mean 528 IU/day vs placebo), including 57,311 participants, evaluated the rate of death from all causes and found a modest but significant reduction in risk (relative risk 0.93, 95% confidence interval [CI] 0.87–0.99). These were generally trials looking at fracture rates or physical performance, and a dose-response relationship was not evident. A recent systematic review of randomized controlled trials of vitamin D1 that included cardiovascular disease as a secondary outcome found a pooled relative risk for cardiovascular disease of 0.90 (95% CI 0.77–1.05) for vitamin D supplementation compared with placebo and 1.04 (95% CI 0.92–1.18) for combination vitamin D plus calcium supplementation vs placebo.1 Two individual trials are discussed below.
Trivedi et al6 randomized 2,686 British men and women to vitamin D3 100,000 IU given every 4 months over 5 years (equivalent to 800 IU/day) or placebo. The relative risk of cardiovascular events was 0.90 (95% CI 0.77–1.06) and of cardiovascular deaths 0.84 (95% CI 0.65–1.10). Although the results were promising, the trial was designed to assess fracture risk and was not large enough for the differences in cardiovascular outcomes to reach statistical significance.
The Women’s Health Initiative,7,8 which included 36,282 postmenopausal women aged 50 to 79, tested combined vitamin D3 (400 IU/day) with calcium (1,000 mg/day) vs placebo. No benefit was seen for preventing coronary events or stroke, which may be due to the low dosage of vitamin D. The hazard ratio for coronary disease was 1.04 (0.92–1.18). Regarding mortality, the hazard ratio for cardiovascular death was 0.92, for cerebrovascular death 0.89, for cancer death 0.89, and for other deaths 0.95. None of these hazard ratios reached statistical significance.
MORE MAY NOT BE BETTER
As is probably true for everything in biological systems, there apparently is an optimal level of intake to meet vitamin D needs.
The Framingham Offspring Study,2 which found a higher risk with vitamin D deficiency, also found a suggestion of a threshold. Participants who had levels of 50 to 65 nmol/L had the lowest risk. Higher levels did not confer lower risk and even suggested a slight upturn.
Evidence from the Women’s Health Initiative8 also indicates that high dosages may not be better than moderate dosages. The meta-analysis of vitamin D and all-cause mortality5 found a relative risk of 0.93, but one of the largest studies in that meta-analysis tested only 400 IU a day and found a similar relative risk of 0.91 (95% confidence interval, 0.83–1.01).
Moreover, the NHANES study found that with increasing serum 25-hydroxyvitamin D3 levels, the risk of all-cause mortality fell until about 100 nmol/L, but then plateaued and even increased with higher serum levels.4
VITAL: STUDY DESIGN AND LOGISTICS
In VITAL, the investigators aim to recruit 20,000 healthy men (age 60 and older) and women (65 and older) who are representative of the US population (www.vitalstudy.org). Because it is a primary prevention trial, people with a known history of cardiovascular disease or cancer will be excluded. Participants will be randomized to receive either 2,000 IU of vitamin D3 per day or placebo. Each group will be further randomized to receive either 1 g per day of fish oil (combined eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) or placebo. The mean treatment period will be 5 years. Recruitment began in early 2010.
Blood will be collected in about 80% (ideally 100%) of participants, with follow-up blood collection in at least 2,000.
Primary aims of the trial are to test whether vitamin D3 and the omega-3 fatty acids reduce the risk of total cancer and major cardiovascular events (a composite of myocardial infarction, stroke, and death due to cardiovascular events).
Secondary aims are to test whether these agents lower the risk of:
- Site-specific cancer, including colorectal, breast, and prostate cancer, and the total cancer mortality rate
- An expanded composite outcome including myocardial infarction, stroke, cardiovascular death, coronary artery bypass grafting, percutaneous coronary intervention, and its individual components.
Tertiary aims are to explore whether vitamin D3 and omega-3 fatty acids have additive effects on the primary and secondary end points. The trial will also explore whether the effects of vitamin D3 and omega-3 fatty acids on cancer and cardiovascular disease vary by baseline blood levels of these nutrients, and whether race, skin pigmentation, or body mass index modify the effects of vitamin D3.
Ancillary studies will assess the effect of the interventions on risk of diabetes, hypertension, cognitive decline, depression, fracture, infections, respiratory disorders, and autoimmune diseases. The primary sponsor of this trial is the National Cancer Institute, and the secondary sponsor is the National Heart, Lung and Blood Institute. Other institutes and agencies also are cosponsors of the study.
The timing of VITAL is optimal
There is a limited window of opportunity for conducting a randomized clinical trial: the evidence must be strong enough to justify mounting a very large trial with enough power to look at cardiovascular events and cancer, but the evidence must not be so strong that it would be unethical to have a placebo group. Thus, there must be a state of equipoise. Our trial allows the study population to have a background intake of vitamin D that is currently recommended by national guidelines. Therefore, even the placebo group should have adequate intake of vitamin D.
The growing use of vitamin D supplementation by the public underscores the need for conclusive evidence of its benefits and risks. No previous large-scale randomized clinical trial has tested moderate to high doses of vitamin D for the primary prevention of cancer and cardiovascular disease.
Setting the dosage
VITAL set the vitamin D3 dosage at 2,000 IU per day (50 μg/day), which is designed to provide the best balance of efficacy and safety. As a general rule, each microgram of vitamin D3 is expected to raise the serum 25-hydroxyvitamin D3 level about 1 nmol/L, although the response is not linear: if baseline levels are lower, the increase is greater. In the United States, people commonly have a baseline level of about 40 nmol/L, so we expect that levels of people treated in the study will reach about 90 nmol/L (range 75–100 nmol/L), about 35 to 50 nmol/L higher than in the placebo group.
The target range of 75 to 100 nmol/L is the level at which greatest efficacy has been suggested in observational studies. Previous randomized trials of vitamin D have not tested high enough doses to achieve this level of 25-hydroxyvitamin D3. VITAL will test whether reaching this serum level lowers the risk of cardiovascular disease, cancer, and other chronic diseases. This level may be associated with benefit and has minimal risk of hypercalcemia. Risk of hypercalcemia may be present in participants with an occult chronic granulomatous condition such as sarcoidosis or Wegener granulomatosis, in which activated macrophages synthesize 1,25-dihydroxyvitamin D3. These conditions are very rare, however, and the risk of hypercalcemia in the trial is exceedingly low.
VITAL participants will also be randomized to take placebo or 1 g per day of combined EPA and DHA, about 5 to 10 times more than most Americans consume.
Nationwide recruitment among senior citizens
We aim to recruit 20,000 people (10,000 men and 10,000 women) nationwide who are willing, eligible, and compliant (ie, who take more than two-thirds of study pills during a 3-month placebo “run-in” phase of the trial). The trial aims to enroll 40,000 in the run-in period, and 20,000 will be randomized. To get this many participants, we will send invitational mailings and screening questionnaires to at least 2.5 million people around the United States, with mailing lists selected by age—ie, members of the American Association of Retired Persons, health professionals, teachers, and subscription lists for selected magazines. A pilot study in 5,000 people has indicated that recruiting and randomizing 20,000 participants via large mailings should be possible.
The trial is expected to be extremely cost-effective because it will be conducted largely by mail. Medication will be mailed in calendar blister packs. Participants report outcomes, which are then confirmed by medical record review. The Centers for Medicare and Medicaid Services and the National Death Index will also be used to ascertain outcomes.
We hope to recruit a more racially diverse study population than is typically seen in US trials: 63% (12,620) whites, 25% (5,000) African Americans, 7% (1,400) Hispanics, 2.5% (500) Asians, 2% (400) American Indians and Alaska natives, and 0.4% (80) native Hawaiian and Pacific Islanders.
Eligibility criteria ensure primary prevention is tested
To enter the study, men must be at least 60 years old and women at least 65. At a minimum, a high school education is required due to the detailed forms and questionnaires to be completed. Because this is a primary prevention trial, anyone with a history of cancer (except nonmelanoma skin cancer) or cardiovascular disease (including myocardial infarction, stroke, or coronary revascularization) will be excluded, as will anyone with a history of kidney stones, renal failure or dialysis, hypercalcemia, hypoparathyroidism or hyperparathyroidism, severe liver disease (eg, cirrhosis), sarcoidosis, tuberculosis, or other granulomatous disease. People with an allergy to fish will also be excluded.
We do not expect that those in the placebo group will develop vitamin D deficiency due to their participation in the study. The trial will allow a background intake in the study population of up to 800 IU of vitamin D and 1,200 mg of calcium per day in supplements. Assuming they also get about 200 IU of vitamin D in the diet, the background intake in the placebo group may be close to 1,000 IU of vitamin D. Assuming that the active treatment group has a similar background intake, their total intake will be about 3,000 IU per day (about 1,000 IU/day from background intake plus 2,000 IU/day from the intervention).
Cohort power sufficient to see effect in 5 years
The trial is expected to have sufficient power to evaluate cardiovascular disease and cancer end points as primary outcomes during 5 years of follow-up. The trial is designed to have a power of 91% to 92% to detect a relative risk of 0.85 for the primary cancer end point of total cancer incidence and 0.80 for the cardiovascular disease end point of myocardial infarction, stroke, and cardiovascular mortality. Power will be even greater for the expanded composite outcome for cardiovascular disease.
Ancillary studies
Ancillary studies include evaluating the interventions’ role in preventing diabetes and glucose intolerance, hypertension, heart failure, atrial fibrillation, cognitive decline, mood disorders, osteoporosis and fractures, asthma and respiratory diseases, infections, macular degeneration, rheumatoid arthritis, systemic lupus erythematosus, and a composite of autoimmune diseases. Imaging studies also are planned, including dual energy x-ray absorptiometry, mammographic density, and non-invasive vascular imaging (carotid intima medial thickness, coronary calcium measurements, and two-dimensional echocardiography to assess cardiac function).
Several biomarker and genetic studies will also be carried out. We intend to perform genetic studies on most of the study population to evaluate gene variants in the vitamin D receptor, vitamin D binding protein, and other vitamin-D-related genes that may contribute to lower baseline levels of 25-hydroxyvitamin D3 or different responses to the interventions.
Clinical and Translational Science Center visits are planned to provide more detailed assessments of 1,000 participants, including blood pressure measurements, height, weight, waist circumference, other anthropometric measurements, a 2-hour glucose tolerance test, a fasting blood collection, hemoglobin A1c measurements, spirometry, and assessment of physical performance, strength, frailty, cognitive function, mood, and depression. Dual-energy x-ray absorptiometry and noninvasive vascular imaging studies are also planned for those visits.
- Wang L, Manson JE, Song Y, Sesso HD. Systematic review: vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med 2010; 152:315–323.
- Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008; 117:503–511.
- Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med 2008; 168:1174–1180.
- Melamed ML, Michos ED, Post W, Astor B. 25-Hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med 2008; 168:1629–1637.
- Autier P, Gandini S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med 2007; 167:1730–1737.
- Trivedi DP, Doll R, Khaw KT. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ 2003; 326:469.
- Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation 2007; 115:846–854.
- LaCroix AZ, Kotchen J, Anderson G, et al. Calcium plus vitamin D supplementation and mortality in postmenopausal women: the Women’s Health Initiative calcium-vitamin D randomized controlled trial. J Gerontol A Biol Sci Med Sci 2009; 64:559–567.
- Wang L, Manson JE, Song Y, Sesso HD. Systematic review: vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med 2010; 152:315–323.
- Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008; 117:503–511.
- Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med 2008; 168:1174–1180.
- Melamed ML, Michos ED, Post W, Astor B. 25-Hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med 2008; 168:1629–1637.
- Autier P, Gandini S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med 2007; 167:1730–1737.
- Trivedi DP, Doll R, Khaw KT. Effect of four monthly oral vitamin D3 (cholecalciferol) supplementation on fractures and mortality in men and women living in the community: randomised double blind controlled trial. BMJ 2003; 326:469.
- Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation 2007; 115:846–854.
- LaCroix AZ, Kotchen J, Anderson G, et al. Calcium plus vitamin D supplementation and mortality in postmenopausal women: the Women’s Health Initiative calcium-vitamin D randomized controlled trial. J Gerontol A Biol Sci Med Sci 2009; 64:559–567.
KEY POINTS
- Laboratory evidence suggests that vitamin D may lower cancer risk by inhibiting cell proliferation, angiogenesis, metastasis, and inflammation.
- Vitamin D may also reduce cardiovascular risk by inhibiting vascular smooth muscle proliferation, regulating blood pressure and glucose metabolism, and reducing inflammation.
- Some observational studies indicate there may be a threshold for vitamin D intake above which there is no increase in benefit and which may increase risk.
- The VITAL trial is currently randomizing 20,000 healthy older men and women throughout the United States to receive either 2,000 IU of vitamin D3 (cholecalciferol) per day or placebo, as well as 1 g of marine omega-3 fatty acids per day or placebo, for 5 years.