User login
What’s new in treating older adults?
New clinical trials and observational studies are shedding light on ways to improve the health of elderly patients. Here is a brief summary of these trials and how they might influence your clinical practice.
EXERCISE HAS NEWLY DISCOVERED BENEFITS
According to government data,1 exercise has a dose-dependent effect on rates of all-cause mortality: the more hours one exercises per week, the lower the risk of death. The difference in risk is most pronounced as one goes from no exercise to about 3 hours of exercise per week; above 3 hours per week, the curve flattens out but continues to decline. Hence, we advise patients to engage in about 30 minutes of moderate-intensity exercise every day.
Lately, physical exercise has been found to have other, unexpected benefits.
Exercise helps cognition
ERICKSON KI, PRAKASH RS, VOSS MW, ET AL. AEROBIC FITNESS IS ASSOCIATED WITH HIPPOCAMPAL VOLUME IN ELDERLY HUMANS. HIPPOCAMPUS 2009; 19:1030–1039.
ETGEN T, SANDER D, HUNTGEBURTH U, POPPERT H, FÖRSTL H, BICKEL H. PHYSICAL ACTIVITY AND INCIDENT COGNITIVE IMPAIRMENT IN ELDERLY PERSONS: THE INVADE STUDY. ARCH INTERN MED 2010; 170:186–193.
The hippocampus is a structure deep in the brain that is involved in short-term memory. It atrophies with age, more so with dementia. Erickson2 found a correlation between aerobic fitness (as measured by maximum oxygen consumption), hippocampal volume, and spatial memory performance.
Etgen and colleagues3 studied nearly 4,000 older adults in Bavaria for 2 years. Among those reporting no physical activity, 21.4% had cognitive impairment at baseline, compared with 7.3% of those with high activity at baseline. Following those without cognitive impairment over a 2-year period, they found the incidence of new cognitive impairment was 13.9% in those with no physical activity at baseline, 6.7% in those with moderate activity, and 5.1% in those with high activity.
Exercise boosts the effect of influenza vaccine
WOODS JA, KEYLOCK KT, LOWDER T, ET AL. CARDIOVASCULAR EXERCISE TRAINING EXTENDS INFLUENZA VACCINE SEROPROTECTION IN SEDENTARY OLDER ADULTS: THE IMMUNE FUNCTION INTERVENTION TRIAL. J AM GERIATR SOC 2009; 57:2183–2191.
In a study in 144 sedentary but healthy older adults (ages 60 to 83), Woods et al4 randomized the participants to undergo either flexibility or cardiovascular training for 10 months, starting 4 months before their annual influenza shot. Exercise extended the duration of antibody protection, with more participants in the cardiovascular group than in the flexibility group showing protection at 24 weeks against all three strains covered by the vaccine: H1N1, H3N2, and influenza B.
PREVENTING FRACTURES
Each year, about 30% of people age 65 or older fall, sustaining serious injuries in 5% to 10% of cases. Unintentional falls are the main cause of hip fractures, which number 300,000 per year. They are also a common cause of death.
Vitamin D prevents fractures, but can there be too much of a good thing?
BISCHOFF-FERRARI HA, WILLETT WC, WONG JB, ET AL. PREVENTION OF NONVERTEBRAL FRACTURES WITH ORAL VITAMIN D AND DOSE DEPENDENCY: A META-ANALYSIS OF RANDOMIZED CONTROLLED TRIALS. ARCH INTERN MED 2009; 169:551–561.
SANDERS KM, STUART AL, WILLIAMSON EJ, ET AL. ANNUAL HIGH-DOSE ORAL VITAMIN D AND FALLS AND FRACTURES IN OLDER WOMEN: A RANDOMIZED CONTROLLED TRIAL. JAMA 2010; 303:1815–1822.
Bischoff-Ferrari5 performed a meta-analysis of 12 randomized controlled trials of oral supplemental vitamin D3 for preventing nonvertebral fractures in people age 65 and older, and eight trials for preventing hip fractures in the same age group. They found that the higher the daily dose of vitamin D, the lower the relative risk of hip fracture. The threshold dose at which supplementation significantly reduced the risk of falling was about 400 units per day. Higher doses of vitamin D reduced both falls and hip fractures by about 20%. The maximal effect was seen with studies using the maximum daily doses, ie, 770 to 800 units per day—not megadoses, but more than most Americans are taking. The threshold serum level of vitamin D of significance was 60 nmol/L (24 ng/mL).
Of interest, the effect on fractures was independent of calcium supplementation. This is important because calcium supplementation over and above ordinary dietary intake may increase the risk of cardiovascular events.6,7
Despite the benefits of vitamin D, too much may be too much of a good thing. Sanders et al8 performed a double-blind, placebo-controlled trial in 2,256 community-dwelling women, age 70 or older, who were considered to be at high risk for fractures. Half received a large oral dose (500,000 units) once a year for 3 to 5 years, and half got placebo. Their initial serum vitamin D level was 49 nmol/L; the level 30 days after a dose in the treatment group was 120 nmol/L.
Contrary to expectations, the incidence of falls was 15% higher in the vitamin D group than in the placebo group (P = .03), and the incidence of fractures was 26% higher (P = .047). The falls and fractures tended to cluster in the first 3 months after the dose in the active treatment group, when serum vitamin D levels were highest.
Comments. Unless future studies suggest a benefit to megadoses of vitamin D or prove calcium supplementation greater than 1,000 mg is safe, the optimal daily intake of vitamin D is likely 1,000 units, with approximately 200 units from diet and 800 units from supplements. A diet rich in low-fat dairy products may not require calcium supplementation. In those consuming a low-calcium diet, supplements of 500 to 1,000 mg/day are likely adequate.
Denosumab, a new drug for preventing fractures
CUMMINGS SR, SAN MARTIN J, MCCLUNG MR, ET AL; FREEDOM TRIAL. DENOSUMAB FOR PREVENTION OF FRACTURES IN POSTMENOPAUSAL WOMEN WITH OSTEOPOROSIS. N ENGL J MED 2009; 361:756–765.
SMITH MR, EGERDIE B, HERNÁNDEZ TORIZ N, ET AL; DENOSUMAB HALT PROSTATE CANCER STUDY GROUP. DENOSUMAB IN MEN RECEIVING ANDROGEN-DEPRIVATION THERAPY FOR PROSTATE CANCER. N ENGL J MED 2009; 361:745–755.
Denosumab (Prolia) is the first of a new class of drugs for the treatment of osteoporosis. It is a monoclonal antibody and member of the tumor necrosis factor superfamily that binds to the receptor activator nuclear factor kappa B (RANK) ligand. It has an antiresorptive effect, preventing osteoclast differentiation and activation. It is given by subcutaneous injection of 60 mg every 6 months; it is cleared by a nonrenal mechanism.
In a randomized controlled trial in 7,868 women between the ages of 60 and 90 who had osteoporosis, Cummings et al9 reported that denosumab reduced the 3-year incidence of vertebral fractures by 68% (P < .001), reduced the incidence of hip fractures by 40% (P = .01), and reduced the incidence of nonvertebral fractures by 20% (P = .01). In a trial in men receiving androgen deprivation therapy for prostate cancer, Smith et al10 reported that denosumab reduced the incidence of vertebral fracture by 62% (P = .006).
Comment. Denosumab was approved by the US Food and Drug Administration (FDA) on June 1, 2010, and is emerging in specialty clinics at the time of this publication. Its potential impact on clinical care is not yet known. It is costly—about $825 (average wholesale price) per injection—but since it is given by injection it may be easier than a yearly infusion of zoledronic acid (Reclast). It has the potential to suppress immune function, although this was not reported in the clinical trials. It may ultimately have a role in treating osteoporosis in men and women, prostate cancer following androgen deprivation, metastatic prostate cancer, metastatic breast cancer, osteoporosis with renal impairment, and other diseases.
DIALYSIS IN THE ELDERLY: A BLEAK STORY
KURELLA TAMURA M, COVINSKY KE, CHERTOW GM, YAFFE K, LANDEFELD CS, MCCOLLOCH CE. FUNCTIONAL STATUS OF ELDERLY ADULTS BEFORE AND AFTER INITIATION OF DIALYSIS. N ENGL J MED 2009; 361:1539–1547.
JASSAL SV, CHIU E, HLADUNEWITH M. LOSS OF INDEPENDENCE IN PATIENTS STARTING DIALYSIS AT 80 YEARS OF AGE OR OLDER (LETTER). N ENGL J MED 2009; 361:1612–1613.
Nursing home residents account for 4% of all patients in end-stage renal disease. However, the benefits of dialysis in older patients are uncertain. The mortality rate during the first year of dialysis is 35% in patients 70 years of age and older and 50% in patients 80 years and older.
Is dialysis helpful in the elderly, ie, does it improve survival and function?
Kurella Tamura et al11 retrospectively identified 3,702 nursing home residents starting dialysis in whom functional assessments had been done. The numbers told a bleak story. Initiation of dialysis was associated with a sharp decline in functional status, as reflected in an increase of 2.8 points on the 28-point Minimum Data Set–Activities of Daily Living (MDS-ADL) scale (the higher the score, the worse the function). MDS-ADL scores stabilized at a plateau for about 6 months and then continued to decline. Moreover, at 12 months, 58% of the patients had died.
The MDS-ADL score is based on seven components: eating, bed mobility, locomotion, transferring, toileting, hygiene, and dressing; function declined in all of these areas when patients started dialysis.
Patients were more likely to decline in activities of daily living after starting dialysis if they were older, were white, had cerebrovascular disease, had a diagnosis of dementia, were hospitalized at the start of dialysis, or had a serum albumin level lower than 3.5 g/dL.
The same thing happens to elders living in the community when they start dialysis. Jassal and colleagues12 reported that, of 97 community-dwelling patients (mean age 85), 46 (47%) were dead 2 years after starting dialysis. Although 76 (78%) had been living independently at the start of dialysis, only 11 (11%) were still doing so at 2 years.
Comment. These findings indicate that we do not know if hemodialysis improves survival. Hemodialysis may buy about 3 months of stable function, but it clearly does not restore function.
Is this the best we can do? Standard hemodialysis may have flaws, and nocturnal dialysis and peritoneal dialysis are used more in other countries. These dialysis techniques require more study in our older population. The lesson from these two publications on dialysis is that we should attend more carefully to slowing the decline in renal function before patients reach end-stage renal disease.
DABIGATRAN: AN ALTERNATIVE TO WARFARIN FOR ATRIAL FIBRILLATION
CONNOLLY SJ, EZEKOWITZ MD, YUSUF S, ET AL; RE-LY STEERING COMMITTEE AND INVESTIGATORS. DABIGATRAN VERSUS WARFARIN IN PATIENTS WITH ATRIAL FIBRILLATION. N ENGL J MED 2009; 361:1139–1151.
Atrial fibrillation is common, affecting 2.2 million adults. The median age of people who have atrial fibrillation is 75 years, and it is the most common arrhythmia in the elderly. Some 20% of ischemic strokes are attributed to it.13–15
Warfarin (Coumadin) is still the mainstay of treatment to prevent stroke in patients with atrial fibrillation. In an analysis of pooled data from five clinical trials,16 the relative risk reduction with warfarin was about 68% in the overall population (number needed to treat 32), 51% in people older than 75 years with no other risk factors (number needed to treat 56), and 85% in people older than 75 years with one or more risk factors (number needed to treat 15).
But warfarin carries a risk of bleeding, and its dose must be periodically adjusted on the basis of the international normalized ratio (INR) of the prothrombin time, so it carries a burden of laboratory monitoring. It is less safe in people who eat erratically, resulting in wide fluctuations in the INR.
Dabigatran (Pradaxa), a direct thrombin inhibitor, is expected to become an alternative to warfarin. It has been approved in Europe but not yet in the United States.
Connolly et al,17 in a randomized, double-blind trial, assigned 18,113 patients who had atrial fibrillation to receive either dabigatran 110 or 150 mg twice daily or adjusted-dose warfarin in an unblinded fashion. At 2 years, the rates of stroke and systemic embolism were about the same with dabigatran 110 mg as with warfarin but were lower with dabigatran 150 mg (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rate of major bleeding was lower with dabigatran 110 mg than with warfarin (2.71% per year vs 3.36% per year, P = .003), but it was similar with dabigatran 150 mg (3.11% per year). Rates of life-threatening bleeding were 1.80% with warfarin, 1.22% with dabigatran 110 mg (P < .05), and 1.45% with dabigatran 150 mg (P < .05).
Comment. I suspect that warfarin’s days are numbered. Dabigatran 110 or 150 mg was as safe and as effective as warfarin in clinical trials, and probably will be more effective than warfarin in clinical practice. It will also probably be safer than warfarin in clinical practice, particularly in challenging settings such as long-term care. On the other hand, it will likely be much more expensive than warfarin.
DEMENTIA
Adverse effects of cholinesterase inhibitors
GILL SS, ANDERSON GM, FISCHER HD, ET AL. SYNCOPE AND ITS CONSEQUENCES IN PATIENTS WITH DEMENTIA RECEIVING CHOLINESTERASE INHIBITORS: A POPULATION-BASED COHORT STUDY. ARCH INTERN MED 2009; 169:867–873.
Cholinesterase inhibitors, eg, donepezil (Aricept), galantamine (Razadyne), and rivastigmine (Exelon), are commonly used to treat Alzheimer disease. However, these drugs carry risks of serious adverse effects.
Gill et al18 retrospectively reviewed a database from Ontario, Canada, and identified about 20,000 community-dwelling elderly persons admitted to the hospital who had been prescribed cholinesterase inhibitors and about three times as many matched controls.
Several adverse events were more frequent in people receiving cholinesterase inhibitors. Findings (events per 1,000 person-years):
- Hospital visits for syncope: 31.5 vs 18.6, adjusted hazard ratio (HR) 1.76, 95% CI 1.57–1.98
- Hip fractures: 22.4 vs 19.8, HR 1.18, 85% CI 1.04–1.34
- Hospital visits for bradycardia: 6.9 vs 4.4, HR 1.69, 95% CI 1.32–2.15
- Permanent pacemaker insertion: 4.7 vs 3.3, HR 1.49, 95% CI 1.12–2.00.
Comment. This study adds to the concerns that cholinesterase inhibitors, which have only modest cognitive benefits, may increase the risk of falls, injury, and need for pacemaker placement in demented patients. A low threshold to stop medications in this class should be considered when a patient on a cholinesterase inhibitor presents with bradycardia, falls, and syncope.
The importance of ‘staging’ dementia
IVERSON DJ, GRONSETH GS, REGER MA, ET AL; STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. PRACTICE PARAMETER UPDATE: EVALUATION AND MANAGEMENT OF DRIVING RISK IN DEMENTIA: REPORT OF THE QUALITY STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. NEUROLOGY 2010; 74:1316–1324.
The Clinical Dementia Rating (CDR) is a simple scale that should be applied by clinicians to describe stage of dementia in patients with Alzheimer disease. This scale can be useful in a variety of settings, from prescribing antidementia drugs to determining whether a patient should still drive. Although research protocols utilize a survey or semistructured interview to derive the stage, the clinician can estimate the stage easily in the office, particularly if there is an informant who can comment on performance outside the office.
There are four stages to the CDR19:
- 0: No dementia
- 0.5: Mild memory deficit but intact function
- 1.0: Moderate memory loss with mild functional impairment
- 2.0: Severe memory loss, moderate functional impairment
- 3.0: Severe memory loss, no significant function outside of the house.
Comment. The first stage (0.5, mild memory deficit but intact function) corresponds to “mild cognitive impairment.” In the clinic, these patients tend to take more notes. They come to the appointment with a little book and they write everything down so they don’t forget. They do arrive at their appointments on time; they are not crashing the car; they are paying their bills.
Patients with CDR stage 1.0 dementia (moderate memory loss with mild functional impairment) may miss appointments, they may confuse their medications, and they may have problems driving. They are still taking care of their basic needs, and they show up for appointments acceptably washed and dressed. However, they are likely having trouble shopping and managing their finances.
Patients with severe memory loss and moderate functional impairment (CDR stage 2.0) may not realize they haven’t bathed for a week or have worn the same clothes repeatedly. They are having trouble with basic activities of daily living, such as bathing and toilet hygiene. However, if you were to encounter them socially and didn’t talk to them for too long, you might think they were normal.
Those with severe memory loss and no significant function outside the house (CDR stage 3.0) are the most severely disabled. Dementia in these individuals is recognizable at a glance, from across the room.
Alzheimer patients progress through the stages, from CDR stage 0.5 at about 1 year to stage 1 by about 2 years, to stage 2 by 5 years, and to stage 3 at 8 or 9 years.20
In prescribing antidementia medications. The CDR can help with prescribing antidementia drugs. No medications are approved by the FDA for stage 0 or 0.5. Cholinesterase inhibitors are approved for stages 1, 2, and 3; memantine (Namenda) is approved for stages 2 and 3.
Advising about driving. The CDR is the only risk predictor with a quality-of-evidence rating of A. More than half of people with stage 0.5 memory impairment are safe drivers; fewer than half of those with stage 1.0 are still safe drivers; and patients with stage 2.0 dementia should not be driving at all.21 An adverse rating by a caregiver carries a quality-of-evidence rating of B. Predictors of driving risk with a quality-of-evidence rating of C are decreased mileage due to self-restriction, agitation, or aggression; a crash in the past 1 to 5 years; a citation in the past 2 to 3 years; and a Folstein Mini-Mental State Examination score of 24 or less. Studies also show that a memory-impaired person’s self-rating of safe driving ability or of assurance that he or she avoids unsafe situations is not reliable.21
DELIRIUM
Delirium goes by a number of synonyms, eg, “sundowning,” acute confusional state, acute change in mental status, metabolic encephalopathy, toxic encephalopathy (psychosis), acute brain syndrome, and acute toxic psychosis.
Delirium is common in hospitalized elderly patients, occurring in 11% to 42% of elderly hospitalized patients overall, up to 53% of elderly surgical patients on regular hospital floors, 80% of elderly surgical patients in intensive care, and about half of elderly patients after undergoing coronary artery bypass grafting. Unfortunately, it is undiagnosed in 30% to 60% of cases.22–24
Many pathways can lead to delirium, including hypoxemia, metabolic derangement, drug effects, systemic inflammation, and infection.25
Outcomes can vary from full recovery to death. After 1 year, 50% of those who leave the hospital with some evidence of delirium have not regained their baseline function. Delirium also increases the cost of care and the risk of institutionalization.
Delirium can accelerate dementia
FONG TG, JONES RN, SHI P, ET AL. DELIRIUM ACCELERATES COGNITIVE DECLINE IN ALZHEIMER DISEASE. NEUROLOGY 2009; 72:1570–1575.
Delirium accelerates the course of dementia in patients who had some evidence of dementia before they entered the hospital. Often, the change is noticeable by the family.26
Preventing delirium
INOUYE SK BOGARDUS ST JR, CHARPENTIER PA, ET AL. A MULTICOMPONENT INTERVENTION TO PREVENT DELIRIUM IN HOSPITALIZED OLDER PATIENTS. N ENGL J MED 1999; 340:669–676.
LUNDSTRÖM M, OLOFSSON B, STENVALL M, ET AL. POSTOPERATIVE DELIRIUM IN OLD PATIENTS WITH FEMORAL NECK FRACTURE: A RANDOMIZED INTERVENTION STUDY. AGING CLIN EXP RES 2007; 19:178–186.
Delirium can often be prevented. In a report published in 1999, Inouye et al27 described the outcomes of a program to prevent delirium in hospitalized medically ill elderly patients. Interventions were aimed at optimizing cognitive function, preventing sleep deprivation, avoiding immobility, improving vision and hearing, and treating dehydration. The incidence of delirium was 9.9% in the intervention group vs 15% in the control group, a 40% reduction (P < .05).
Lundström et al28 implemented a similar program for elderly patients with hip fractures. Interventions included staff education and teamwork; active prevention, detection, and treatment of delirium; transfusions if hemoglobin levels were less than 10 g/dL; prompt removal of indwelling urinary catheters, with screening for urinary retention; active prevention and treatment of constipation; and protein-enriched meals. The incidence of delirium was 55% in the intervention group vs 75% in the control group, a 27% reduction.
Comment. Although we have long known that the risk of delirium in medical and surgical patients can be reduced, most hospitals do not have systematic programs to detect delirium and reduce its incidence. Hopefully, reduction in delirium risk will also reduce its adverse consequences, including worsening of dementia and increased mortality.
- Department of Health and Human Services. Physical activity guidelines for Americans. www.health.gov/paguidelines/reportG1_allcause.aspx
- Erickson KI, Prakash RS, Voss MW, et al. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus 2009; 19:1030–1039.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Woods JA, Keylock KT, Lowder T, et al. Cardiovascular exercise training extends influenza vaccine seroprotection in sedentary older adults: the immune function intervention trial. J Am Geriatr Soc 2009; 57:2183–2191.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ 2010; 341:c3691. doi:10.1136/bmj.c3691.
- Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ 2008; 336:262–266.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010; 303:1815–1822.
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McColloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med 2009; 361:1539–1547.
- Jassal SV, Chiu E, Hladunewich M. Loss of independence in patients starting dialysis at 80 years of age or older (letter). N Engl J Med 2009; 361:1612–1613.
- Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med 1995; 155:469–473.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. The Framingham Study. Stroke 1996; 27:1760–1764.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Gill SS, Anderson GM, Fischer HD, et al. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors: a population-based cohort study. Arch Intern Med 2009; 169:867–873.
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993; 43:2412–2414.
- Sloane PD. Advances in the treatment of Alzheimer’s disease. Am Fam Physician 1998; 58:1577–1586.
- Iverson DJ, Gronseth GS, Reger MA, et al; Standards Subcommittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Demeure MJ, Fain MJ. The elderly surgical patient and postoperative delirium. J Am Coll Surg 2006; 203:752–757.
- Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing 2006; 35:350–364.
- Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation 2009; 119:229–236.
- Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol 2009; 5:210–220.
- Fong TG, Jones RN, Shi P, et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology 2009; 72:1570–1575.
- Inouye SK, Bogardus ST, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med 1999; 340:669–676.
- Lundström M, Olofsson B, Stenvall M, et al. Postoperative delirium in old patients with femoral neck fracture: a randomized intervention study. Aging Clin Exp Res 2007; 19:178–186.
New clinical trials and observational studies are shedding light on ways to improve the health of elderly patients. Here is a brief summary of these trials and how they might influence your clinical practice.
EXERCISE HAS NEWLY DISCOVERED BENEFITS
According to government data,1 exercise has a dose-dependent effect on rates of all-cause mortality: the more hours one exercises per week, the lower the risk of death. The difference in risk is most pronounced as one goes from no exercise to about 3 hours of exercise per week; above 3 hours per week, the curve flattens out but continues to decline. Hence, we advise patients to engage in about 30 minutes of moderate-intensity exercise every day.
Lately, physical exercise has been found to have other, unexpected benefits.
Exercise helps cognition
ERICKSON KI, PRAKASH RS, VOSS MW, ET AL. AEROBIC FITNESS IS ASSOCIATED WITH HIPPOCAMPAL VOLUME IN ELDERLY HUMANS. HIPPOCAMPUS 2009; 19:1030–1039.
ETGEN T, SANDER D, HUNTGEBURTH U, POPPERT H, FÖRSTL H, BICKEL H. PHYSICAL ACTIVITY AND INCIDENT COGNITIVE IMPAIRMENT IN ELDERLY PERSONS: THE INVADE STUDY. ARCH INTERN MED 2010; 170:186–193.
The hippocampus is a structure deep in the brain that is involved in short-term memory. It atrophies with age, more so with dementia. Erickson2 found a correlation between aerobic fitness (as measured by maximum oxygen consumption), hippocampal volume, and spatial memory performance.
Etgen and colleagues3 studied nearly 4,000 older adults in Bavaria for 2 years. Among those reporting no physical activity, 21.4% had cognitive impairment at baseline, compared with 7.3% of those with high activity at baseline. Following those without cognitive impairment over a 2-year period, they found the incidence of new cognitive impairment was 13.9% in those with no physical activity at baseline, 6.7% in those with moderate activity, and 5.1% in those with high activity.
Exercise boosts the effect of influenza vaccine
WOODS JA, KEYLOCK KT, LOWDER T, ET AL. CARDIOVASCULAR EXERCISE TRAINING EXTENDS INFLUENZA VACCINE SEROPROTECTION IN SEDENTARY OLDER ADULTS: THE IMMUNE FUNCTION INTERVENTION TRIAL. J AM GERIATR SOC 2009; 57:2183–2191.
In a study in 144 sedentary but healthy older adults (ages 60 to 83), Woods et al4 randomized the participants to undergo either flexibility or cardiovascular training for 10 months, starting 4 months before their annual influenza shot. Exercise extended the duration of antibody protection, with more participants in the cardiovascular group than in the flexibility group showing protection at 24 weeks against all three strains covered by the vaccine: H1N1, H3N2, and influenza B.
PREVENTING FRACTURES
Each year, about 30% of people age 65 or older fall, sustaining serious injuries in 5% to 10% of cases. Unintentional falls are the main cause of hip fractures, which number 300,000 per year. They are also a common cause of death.
Vitamin D prevents fractures, but can there be too much of a good thing?
BISCHOFF-FERRARI HA, WILLETT WC, WONG JB, ET AL. PREVENTION OF NONVERTEBRAL FRACTURES WITH ORAL VITAMIN D AND DOSE DEPENDENCY: A META-ANALYSIS OF RANDOMIZED CONTROLLED TRIALS. ARCH INTERN MED 2009; 169:551–561.
SANDERS KM, STUART AL, WILLIAMSON EJ, ET AL. ANNUAL HIGH-DOSE ORAL VITAMIN D AND FALLS AND FRACTURES IN OLDER WOMEN: A RANDOMIZED CONTROLLED TRIAL. JAMA 2010; 303:1815–1822.
Bischoff-Ferrari5 performed a meta-analysis of 12 randomized controlled trials of oral supplemental vitamin D3 for preventing nonvertebral fractures in people age 65 and older, and eight trials for preventing hip fractures in the same age group. They found that the higher the daily dose of vitamin D, the lower the relative risk of hip fracture. The threshold dose at which supplementation significantly reduced the risk of falling was about 400 units per day. Higher doses of vitamin D reduced both falls and hip fractures by about 20%. The maximal effect was seen with studies using the maximum daily doses, ie, 770 to 800 units per day—not megadoses, but more than most Americans are taking. The threshold serum level of vitamin D of significance was 60 nmol/L (24 ng/mL).
Of interest, the effect on fractures was independent of calcium supplementation. This is important because calcium supplementation over and above ordinary dietary intake may increase the risk of cardiovascular events.6,7
Despite the benefits of vitamin D, too much may be too much of a good thing. Sanders et al8 performed a double-blind, placebo-controlled trial in 2,256 community-dwelling women, age 70 or older, who were considered to be at high risk for fractures. Half received a large oral dose (500,000 units) once a year for 3 to 5 years, and half got placebo. Their initial serum vitamin D level was 49 nmol/L; the level 30 days after a dose in the treatment group was 120 nmol/L.
Contrary to expectations, the incidence of falls was 15% higher in the vitamin D group than in the placebo group (P = .03), and the incidence of fractures was 26% higher (P = .047). The falls and fractures tended to cluster in the first 3 months after the dose in the active treatment group, when serum vitamin D levels were highest.
Comments. Unless future studies suggest a benefit to megadoses of vitamin D or prove calcium supplementation greater than 1,000 mg is safe, the optimal daily intake of vitamin D is likely 1,000 units, with approximately 200 units from diet and 800 units from supplements. A diet rich in low-fat dairy products may not require calcium supplementation. In those consuming a low-calcium diet, supplements of 500 to 1,000 mg/day are likely adequate.
Denosumab, a new drug for preventing fractures
CUMMINGS SR, SAN MARTIN J, MCCLUNG MR, ET AL; FREEDOM TRIAL. DENOSUMAB FOR PREVENTION OF FRACTURES IN POSTMENOPAUSAL WOMEN WITH OSTEOPOROSIS. N ENGL J MED 2009; 361:756–765.
SMITH MR, EGERDIE B, HERNÁNDEZ TORIZ N, ET AL; DENOSUMAB HALT PROSTATE CANCER STUDY GROUP. DENOSUMAB IN MEN RECEIVING ANDROGEN-DEPRIVATION THERAPY FOR PROSTATE CANCER. N ENGL J MED 2009; 361:745–755.
Denosumab (Prolia) is the first of a new class of drugs for the treatment of osteoporosis. It is a monoclonal antibody and member of the tumor necrosis factor superfamily that binds to the receptor activator nuclear factor kappa B (RANK) ligand. It has an antiresorptive effect, preventing osteoclast differentiation and activation. It is given by subcutaneous injection of 60 mg every 6 months; it is cleared by a nonrenal mechanism.
In a randomized controlled trial in 7,868 women between the ages of 60 and 90 who had osteoporosis, Cummings et al9 reported that denosumab reduced the 3-year incidence of vertebral fractures by 68% (P < .001), reduced the incidence of hip fractures by 40% (P = .01), and reduced the incidence of nonvertebral fractures by 20% (P = .01). In a trial in men receiving androgen deprivation therapy for prostate cancer, Smith et al10 reported that denosumab reduced the incidence of vertebral fracture by 62% (P = .006).
Comment. Denosumab was approved by the US Food and Drug Administration (FDA) on June 1, 2010, and is emerging in specialty clinics at the time of this publication. Its potential impact on clinical care is not yet known. It is costly—about $825 (average wholesale price) per injection—but since it is given by injection it may be easier than a yearly infusion of zoledronic acid (Reclast). It has the potential to suppress immune function, although this was not reported in the clinical trials. It may ultimately have a role in treating osteoporosis in men and women, prostate cancer following androgen deprivation, metastatic prostate cancer, metastatic breast cancer, osteoporosis with renal impairment, and other diseases.
DIALYSIS IN THE ELDERLY: A BLEAK STORY
KURELLA TAMURA M, COVINSKY KE, CHERTOW GM, YAFFE K, LANDEFELD CS, MCCOLLOCH CE. FUNCTIONAL STATUS OF ELDERLY ADULTS BEFORE AND AFTER INITIATION OF DIALYSIS. N ENGL J MED 2009; 361:1539–1547.
JASSAL SV, CHIU E, HLADUNEWITH M. LOSS OF INDEPENDENCE IN PATIENTS STARTING DIALYSIS AT 80 YEARS OF AGE OR OLDER (LETTER). N ENGL J MED 2009; 361:1612–1613.
Nursing home residents account for 4% of all patients in end-stage renal disease. However, the benefits of dialysis in older patients are uncertain. The mortality rate during the first year of dialysis is 35% in patients 70 years of age and older and 50% in patients 80 years and older.
Is dialysis helpful in the elderly, ie, does it improve survival and function?
Kurella Tamura et al11 retrospectively identified 3,702 nursing home residents starting dialysis in whom functional assessments had been done. The numbers told a bleak story. Initiation of dialysis was associated with a sharp decline in functional status, as reflected in an increase of 2.8 points on the 28-point Minimum Data Set–Activities of Daily Living (MDS-ADL) scale (the higher the score, the worse the function). MDS-ADL scores stabilized at a plateau for about 6 months and then continued to decline. Moreover, at 12 months, 58% of the patients had died.
The MDS-ADL score is based on seven components: eating, bed mobility, locomotion, transferring, toileting, hygiene, and dressing; function declined in all of these areas when patients started dialysis.
Patients were more likely to decline in activities of daily living after starting dialysis if they were older, were white, had cerebrovascular disease, had a diagnosis of dementia, were hospitalized at the start of dialysis, or had a serum albumin level lower than 3.5 g/dL.
The same thing happens to elders living in the community when they start dialysis. Jassal and colleagues12 reported that, of 97 community-dwelling patients (mean age 85), 46 (47%) were dead 2 years after starting dialysis. Although 76 (78%) had been living independently at the start of dialysis, only 11 (11%) were still doing so at 2 years.
Comment. These findings indicate that we do not know if hemodialysis improves survival. Hemodialysis may buy about 3 months of stable function, but it clearly does not restore function.
Is this the best we can do? Standard hemodialysis may have flaws, and nocturnal dialysis and peritoneal dialysis are used more in other countries. These dialysis techniques require more study in our older population. The lesson from these two publications on dialysis is that we should attend more carefully to slowing the decline in renal function before patients reach end-stage renal disease.
DABIGATRAN: AN ALTERNATIVE TO WARFARIN FOR ATRIAL FIBRILLATION
CONNOLLY SJ, EZEKOWITZ MD, YUSUF S, ET AL; RE-LY STEERING COMMITTEE AND INVESTIGATORS. DABIGATRAN VERSUS WARFARIN IN PATIENTS WITH ATRIAL FIBRILLATION. N ENGL J MED 2009; 361:1139–1151.
Atrial fibrillation is common, affecting 2.2 million adults. The median age of people who have atrial fibrillation is 75 years, and it is the most common arrhythmia in the elderly. Some 20% of ischemic strokes are attributed to it.13–15
Warfarin (Coumadin) is still the mainstay of treatment to prevent stroke in patients with atrial fibrillation. In an analysis of pooled data from five clinical trials,16 the relative risk reduction with warfarin was about 68% in the overall population (number needed to treat 32), 51% in people older than 75 years with no other risk factors (number needed to treat 56), and 85% in people older than 75 years with one or more risk factors (number needed to treat 15).
But warfarin carries a risk of bleeding, and its dose must be periodically adjusted on the basis of the international normalized ratio (INR) of the prothrombin time, so it carries a burden of laboratory monitoring. It is less safe in people who eat erratically, resulting in wide fluctuations in the INR.
Dabigatran (Pradaxa), a direct thrombin inhibitor, is expected to become an alternative to warfarin. It has been approved in Europe but not yet in the United States.
Connolly et al,17 in a randomized, double-blind trial, assigned 18,113 patients who had atrial fibrillation to receive either dabigatran 110 or 150 mg twice daily or adjusted-dose warfarin in an unblinded fashion. At 2 years, the rates of stroke and systemic embolism were about the same with dabigatran 110 mg as with warfarin but were lower with dabigatran 150 mg (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rate of major bleeding was lower with dabigatran 110 mg than with warfarin (2.71% per year vs 3.36% per year, P = .003), but it was similar with dabigatran 150 mg (3.11% per year). Rates of life-threatening bleeding were 1.80% with warfarin, 1.22% with dabigatran 110 mg (P < .05), and 1.45% with dabigatran 150 mg (P < .05).
Comment. I suspect that warfarin’s days are numbered. Dabigatran 110 or 150 mg was as safe and as effective as warfarin in clinical trials, and probably will be more effective than warfarin in clinical practice. It will also probably be safer than warfarin in clinical practice, particularly in challenging settings such as long-term care. On the other hand, it will likely be much more expensive than warfarin.
DEMENTIA
Adverse effects of cholinesterase inhibitors
GILL SS, ANDERSON GM, FISCHER HD, ET AL. SYNCOPE AND ITS CONSEQUENCES IN PATIENTS WITH DEMENTIA RECEIVING CHOLINESTERASE INHIBITORS: A POPULATION-BASED COHORT STUDY. ARCH INTERN MED 2009; 169:867–873.
Cholinesterase inhibitors, eg, donepezil (Aricept), galantamine (Razadyne), and rivastigmine (Exelon), are commonly used to treat Alzheimer disease. However, these drugs carry risks of serious adverse effects.
Gill et al18 retrospectively reviewed a database from Ontario, Canada, and identified about 20,000 community-dwelling elderly persons admitted to the hospital who had been prescribed cholinesterase inhibitors and about three times as many matched controls.
Several adverse events were more frequent in people receiving cholinesterase inhibitors. Findings (events per 1,000 person-years):
- Hospital visits for syncope: 31.5 vs 18.6, adjusted hazard ratio (HR) 1.76, 95% CI 1.57–1.98
- Hip fractures: 22.4 vs 19.8, HR 1.18, 85% CI 1.04–1.34
- Hospital visits for bradycardia: 6.9 vs 4.4, HR 1.69, 95% CI 1.32–2.15
- Permanent pacemaker insertion: 4.7 vs 3.3, HR 1.49, 95% CI 1.12–2.00.
Comment. This study adds to the concerns that cholinesterase inhibitors, which have only modest cognitive benefits, may increase the risk of falls, injury, and need for pacemaker placement in demented patients. A low threshold to stop medications in this class should be considered when a patient on a cholinesterase inhibitor presents with bradycardia, falls, and syncope.
The importance of ‘staging’ dementia
IVERSON DJ, GRONSETH GS, REGER MA, ET AL; STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. PRACTICE PARAMETER UPDATE: EVALUATION AND MANAGEMENT OF DRIVING RISK IN DEMENTIA: REPORT OF THE QUALITY STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. NEUROLOGY 2010; 74:1316–1324.
The Clinical Dementia Rating (CDR) is a simple scale that should be applied by clinicians to describe stage of dementia in patients with Alzheimer disease. This scale can be useful in a variety of settings, from prescribing antidementia drugs to determining whether a patient should still drive. Although research protocols utilize a survey or semistructured interview to derive the stage, the clinician can estimate the stage easily in the office, particularly if there is an informant who can comment on performance outside the office.
There are four stages to the CDR19:
- 0: No dementia
- 0.5: Mild memory deficit but intact function
- 1.0: Moderate memory loss with mild functional impairment
- 2.0: Severe memory loss, moderate functional impairment
- 3.0: Severe memory loss, no significant function outside of the house.
Comment. The first stage (0.5, mild memory deficit but intact function) corresponds to “mild cognitive impairment.” In the clinic, these patients tend to take more notes. They come to the appointment with a little book and they write everything down so they don’t forget. They do arrive at their appointments on time; they are not crashing the car; they are paying their bills.
Patients with CDR stage 1.0 dementia (moderate memory loss with mild functional impairment) may miss appointments, they may confuse their medications, and they may have problems driving. They are still taking care of their basic needs, and they show up for appointments acceptably washed and dressed. However, they are likely having trouble shopping and managing their finances.
Patients with severe memory loss and moderate functional impairment (CDR stage 2.0) may not realize they haven’t bathed for a week or have worn the same clothes repeatedly. They are having trouble with basic activities of daily living, such as bathing and toilet hygiene. However, if you were to encounter them socially and didn’t talk to them for too long, you might think they were normal.
Those with severe memory loss and no significant function outside the house (CDR stage 3.0) are the most severely disabled. Dementia in these individuals is recognizable at a glance, from across the room.
Alzheimer patients progress through the stages, from CDR stage 0.5 at about 1 year to stage 1 by about 2 years, to stage 2 by 5 years, and to stage 3 at 8 or 9 years.20
In prescribing antidementia medications. The CDR can help with prescribing antidementia drugs. No medications are approved by the FDA for stage 0 or 0.5. Cholinesterase inhibitors are approved for stages 1, 2, and 3; memantine (Namenda) is approved for stages 2 and 3.
Advising about driving. The CDR is the only risk predictor with a quality-of-evidence rating of A. More than half of people with stage 0.5 memory impairment are safe drivers; fewer than half of those with stage 1.0 are still safe drivers; and patients with stage 2.0 dementia should not be driving at all.21 An adverse rating by a caregiver carries a quality-of-evidence rating of B. Predictors of driving risk with a quality-of-evidence rating of C are decreased mileage due to self-restriction, agitation, or aggression; a crash in the past 1 to 5 years; a citation in the past 2 to 3 years; and a Folstein Mini-Mental State Examination score of 24 or less. Studies also show that a memory-impaired person’s self-rating of safe driving ability or of assurance that he or she avoids unsafe situations is not reliable.21
DELIRIUM
Delirium goes by a number of synonyms, eg, “sundowning,” acute confusional state, acute change in mental status, metabolic encephalopathy, toxic encephalopathy (psychosis), acute brain syndrome, and acute toxic psychosis.
Delirium is common in hospitalized elderly patients, occurring in 11% to 42% of elderly hospitalized patients overall, up to 53% of elderly surgical patients on regular hospital floors, 80% of elderly surgical patients in intensive care, and about half of elderly patients after undergoing coronary artery bypass grafting. Unfortunately, it is undiagnosed in 30% to 60% of cases.22–24
Many pathways can lead to delirium, including hypoxemia, metabolic derangement, drug effects, systemic inflammation, and infection.25
Outcomes can vary from full recovery to death. After 1 year, 50% of those who leave the hospital with some evidence of delirium have not regained their baseline function. Delirium also increases the cost of care and the risk of institutionalization.
Delirium can accelerate dementia
FONG TG, JONES RN, SHI P, ET AL. DELIRIUM ACCELERATES COGNITIVE DECLINE IN ALZHEIMER DISEASE. NEUROLOGY 2009; 72:1570–1575.
Delirium accelerates the course of dementia in patients who had some evidence of dementia before they entered the hospital. Often, the change is noticeable by the family.26
Preventing delirium
INOUYE SK BOGARDUS ST JR, CHARPENTIER PA, ET AL. A MULTICOMPONENT INTERVENTION TO PREVENT DELIRIUM IN HOSPITALIZED OLDER PATIENTS. N ENGL J MED 1999; 340:669–676.
LUNDSTRÖM M, OLOFSSON B, STENVALL M, ET AL. POSTOPERATIVE DELIRIUM IN OLD PATIENTS WITH FEMORAL NECK FRACTURE: A RANDOMIZED INTERVENTION STUDY. AGING CLIN EXP RES 2007; 19:178–186.
Delirium can often be prevented. In a report published in 1999, Inouye et al27 described the outcomes of a program to prevent delirium in hospitalized medically ill elderly patients. Interventions were aimed at optimizing cognitive function, preventing sleep deprivation, avoiding immobility, improving vision and hearing, and treating dehydration. The incidence of delirium was 9.9% in the intervention group vs 15% in the control group, a 40% reduction (P < .05).
Lundström et al28 implemented a similar program for elderly patients with hip fractures. Interventions included staff education and teamwork; active prevention, detection, and treatment of delirium; transfusions if hemoglobin levels were less than 10 g/dL; prompt removal of indwelling urinary catheters, with screening for urinary retention; active prevention and treatment of constipation; and protein-enriched meals. The incidence of delirium was 55% in the intervention group vs 75% in the control group, a 27% reduction.
Comment. Although we have long known that the risk of delirium in medical and surgical patients can be reduced, most hospitals do not have systematic programs to detect delirium and reduce its incidence. Hopefully, reduction in delirium risk will also reduce its adverse consequences, including worsening of dementia and increased mortality.
New clinical trials and observational studies are shedding light on ways to improve the health of elderly patients. Here is a brief summary of these trials and how they might influence your clinical practice.
EXERCISE HAS NEWLY DISCOVERED BENEFITS
According to government data,1 exercise has a dose-dependent effect on rates of all-cause mortality: the more hours one exercises per week, the lower the risk of death. The difference in risk is most pronounced as one goes from no exercise to about 3 hours of exercise per week; above 3 hours per week, the curve flattens out but continues to decline. Hence, we advise patients to engage in about 30 minutes of moderate-intensity exercise every day.
Lately, physical exercise has been found to have other, unexpected benefits.
Exercise helps cognition
ERICKSON KI, PRAKASH RS, VOSS MW, ET AL. AEROBIC FITNESS IS ASSOCIATED WITH HIPPOCAMPAL VOLUME IN ELDERLY HUMANS. HIPPOCAMPUS 2009; 19:1030–1039.
ETGEN T, SANDER D, HUNTGEBURTH U, POPPERT H, FÖRSTL H, BICKEL H. PHYSICAL ACTIVITY AND INCIDENT COGNITIVE IMPAIRMENT IN ELDERLY PERSONS: THE INVADE STUDY. ARCH INTERN MED 2010; 170:186–193.
The hippocampus is a structure deep in the brain that is involved in short-term memory. It atrophies with age, more so with dementia. Erickson2 found a correlation between aerobic fitness (as measured by maximum oxygen consumption), hippocampal volume, and spatial memory performance.
Etgen and colleagues3 studied nearly 4,000 older adults in Bavaria for 2 years. Among those reporting no physical activity, 21.4% had cognitive impairment at baseline, compared with 7.3% of those with high activity at baseline. Following those without cognitive impairment over a 2-year period, they found the incidence of new cognitive impairment was 13.9% in those with no physical activity at baseline, 6.7% in those with moderate activity, and 5.1% in those with high activity.
Exercise boosts the effect of influenza vaccine
WOODS JA, KEYLOCK KT, LOWDER T, ET AL. CARDIOVASCULAR EXERCISE TRAINING EXTENDS INFLUENZA VACCINE SEROPROTECTION IN SEDENTARY OLDER ADULTS: THE IMMUNE FUNCTION INTERVENTION TRIAL. J AM GERIATR SOC 2009; 57:2183–2191.
In a study in 144 sedentary but healthy older adults (ages 60 to 83), Woods et al4 randomized the participants to undergo either flexibility or cardiovascular training for 10 months, starting 4 months before their annual influenza shot. Exercise extended the duration of antibody protection, with more participants in the cardiovascular group than in the flexibility group showing protection at 24 weeks against all three strains covered by the vaccine: H1N1, H3N2, and influenza B.
PREVENTING FRACTURES
Each year, about 30% of people age 65 or older fall, sustaining serious injuries in 5% to 10% of cases. Unintentional falls are the main cause of hip fractures, which number 300,000 per year. They are also a common cause of death.
Vitamin D prevents fractures, but can there be too much of a good thing?
BISCHOFF-FERRARI HA, WILLETT WC, WONG JB, ET AL. PREVENTION OF NONVERTEBRAL FRACTURES WITH ORAL VITAMIN D AND DOSE DEPENDENCY: A META-ANALYSIS OF RANDOMIZED CONTROLLED TRIALS. ARCH INTERN MED 2009; 169:551–561.
SANDERS KM, STUART AL, WILLIAMSON EJ, ET AL. ANNUAL HIGH-DOSE ORAL VITAMIN D AND FALLS AND FRACTURES IN OLDER WOMEN: A RANDOMIZED CONTROLLED TRIAL. JAMA 2010; 303:1815–1822.
Bischoff-Ferrari5 performed a meta-analysis of 12 randomized controlled trials of oral supplemental vitamin D3 for preventing nonvertebral fractures in people age 65 and older, and eight trials for preventing hip fractures in the same age group. They found that the higher the daily dose of vitamin D, the lower the relative risk of hip fracture. The threshold dose at which supplementation significantly reduced the risk of falling was about 400 units per day. Higher doses of vitamin D reduced both falls and hip fractures by about 20%. The maximal effect was seen with studies using the maximum daily doses, ie, 770 to 800 units per day—not megadoses, but more than most Americans are taking. The threshold serum level of vitamin D of significance was 60 nmol/L (24 ng/mL).
Of interest, the effect on fractures was independent of calcium supplementation. This is important because calcium supplementation over and above ordinary dietary intake may increase the risk of cardiovascular events.6,7
Despite the benefits of vitamin D, too much may be too much of a good thing. Sanders et al8 performed a double-blind, placebo-controlled trial in 2,256 community-dwelling women, age 70 or older, who were considered to be at high risk for fractures. Half received a large oral dose (500,000 units) once a year for 3 to 5 years, and half got placebo. Their initial serum vitamin D level was 49 nmol/L; the level 30 days after a dose in the treatment group was 120 nmol/L.
Contrary to expectations, the incidence of falls was 15% higher in the vitamin D group than in the placebo group (P = .03), and the incidence of fractures was 26% higher (P = .047). The falls and fractures tended to cluster in the first 3 months after the dose in the active treatment group, when serum vitamin D levels were highest.
Comments. Unless future studies suggest a benefit to megadoses of vitamin D or prove calcium supplementation greater than 1,000 mg is safe, the optimal daily intake of vitamin D is likely 1,000 units, with approximately 200 units from diet and 800 units from supplements. A diet rich in low-fat dairy products may not require calcium supplementation. In those consuming a low-calcium diet, supplements of 500 to 1,000 mg/day are likely adequate.
Denosumab, a new drug for preventing fractures
CUMMINGS SR, SAN MARTIN J, MCCLUNG MR, ET AL; FREEDOM TRIAL. DENOSUMAB FOR PREVENTION OF FRACTURES IN POSTMENOPAUSAL WOMEN WITH OSTEOPOROSIS. N ENGL J MED 2009; 361:756–765.
SMITH MR, EGERDIE B, HERNÁNDEZ TORIZ N, ET AL; DENOSUMAB HALT PROSTATE CANCER STUDY GROUP. DENOSUMAB IN MEN RECEIVING ANDROGEN-DEPRIVATION THERAPY FOR PROSTATE CANCER. N ENGL J MED 2009; 361:745–755.
Denosumab (Prolia) is the first of a new class of drugs for the treatment of osteoporosis. It is a monoclonal antibody and member of the tumor necrosis factor superfamily that binds to the receptor activator nuclear factor kappa B (RANK) ligand. It has an antiresorptive effect, preventing osteoclast differentiation and activation. It is given by subcutaneous injection of 60 mg every 6 months; it is cleared by a nonrenal mechanism.
In a randomized controlled trial in 7,868 women between the ages of 60 and 90 who had osteoporosis, Cummings et al9 reported that denosumab reduced the 3-year incidence of vertebral fractures by 68% (P < .001), reduced the incidence of hip fractures by 40% (P = .01), and reduced the incidence of nonvertebral fractures by 20% (P = .01). In a trial in men receiving androgen deprivation therapy for prostate cancer, Smith et al10 reported that denosumab reduced the incidence of vertebral fracture by 62% (P = .006).
Comment. Denosumab was approved by the US Food and Drug Administration (FDA) on June 1, 2010, and is emerging in specialty clinics at the time of this publication. Its potential impact on clinical care is not yet known. It is costly—about $825 (average wholesale price) per injection—but since it is given by injection it may be easier than a yearly infusion of zoledronic acid (Reclast). It has the potential to suppress immune function, although this was not reported in the clinical trials. It may ultimately have a role in treating osteoporosis in men and women, prostate cancer following androgen deprivation, metastatic prostate cancer, metastatic breast cancer, osteoporosis with renal impairment, and other diseases.
DIALYSIS IN THE ELDERLY: A BLEAK STORY
KURELLA TAMURA M, COVINSKY KE, CHERTOW GM, YAFFE K, LANDEFELD CS, MCCOLLOCH CE. FUNCTIONAL STATUS OF ELDERLY ADULTS BEFORE AND AFTER INITIATION OF DIALYSIS. N ENGL J MED 2009; 361:1539–1547.
JASSAL SV, CHIU E, HLADUNEWITH M. LOSS OF INDEPENDENCE IN PATIENTS STARTING DIALYSIS AT 80 YEARS OF AGE OR OLDER (LETTER). N ENGL J MED 2009; 361:1612–1613.
Nursing home residents account for 4% of all patients in end-stage renal disease. However, the benefits of dialysis in older patients are uncertain. The mortality rate during the first year of dialysis is 35% in patients 70 years of age and older and 50% in patients 80 years and older.
Is dialysis helpful in the elderly, ie, does it improve survival and function?
Kurella Tamura et al11 retrospectively identified 3,702 nursing home residents starting dialysis in whom functional assessments had been done. The numbers told a bleak story. Initiation of dialysis was associated with a sharp decline in functional status, as reflected in an increase of 2.8 points on the 28-point Minimum Data Set–Activities of Daily Living (MDS-ADL) scale (the higher the score, the worse the function). MDS-ADL scores stabilized at a plateau for about 6 months and then continued to decline. Moreover, at 12 months, 58% of the patients had died.
The MDS-ADL score is based on seven components: eating, bed mobility, locomotion, transferring, toileting, hygiene, and dressing; function declined in all of these areas when patients started dialysis.
Patients were more likely to decline in activities of daily living after starting dialysis if they were older, were white, had cerebrovascular disease, had a diagnosis of dementia, were hospitalized at the start of dialysis, or had a serum albumin level lower than 3.5 g/dL.
The same thing happens to elders living in the community when they start dialysis. Jassal and colleagues12 reported that, of 97 community-dwelling patients (mean age 85), 46 (47%) were dead 2 years after starting dialysis. Although 76 (78%) had been living independently at the start of dialysis, only 11 (11%) were still doing so at 2 years.
Comment. These findings indicate that we do not know if hemodialysis improves survival. Hemodialysis may buy about 3 months of stable function, but it clearly does not restore function.
Is this the best we can do? Standard hemodialysis may have flaws, and nocturnal dialysis and peritoneal dialysis are used more in other countries. These dialysis techniques require more study in our older population. The lesson from these two publications on dialysis is that we should attend more carefully to slowing the decline in renal function before patients reach end-stage renal disease.
DABIGATRAN: AN ALTERNATIVE TO WARFARIN FOR ATRIAL FIBRILLATION
CONNOLLY SJ, EZEKOWITZ MD, YUSUF S, ET AL; RE-LY STEERING COMMITTEE AND INVESTIGATORS. DABIGATRAN VERSUS WARFARIN IN PATIENTS WITH ATRIAL FIBRILLATION. N ENGL J MED 2009; 361:1139–1151.
Atrial fibrillation is common, affecting 2.2 million adults. The median age of people who have atrial fibrillation is 75 years, and it is the most common arrhythmia in the elderly. Some 20% of ischemic strokes are attributed to it.13–15
Warfarin (Coumadin) is still the mainstay of treatment to prevent stroke in patients with atrial fibrillation. In an analysis of pooled data from five clinical trials,16 the relative risk reduction with warfarin was about 68% in the overall population (number needed to treat 32), 51% in people older than 75 years with no other risk factors (number needed to treat 56), and 85% in people older than 75 years with one or more risk factors (number needed to treat 15).
But warfarin carries a risk of bleeding, and its dose must be periodically adjusted on the basis of the international normalized ratio (INR) of the prothrombin time, so it carries a burden of laboratory monitoring. It is less safe in people who eat erratically, resulting in wide fluctuations in the INR.
Dabigatran (Pradaxa), a direct thrombin inhibitor, is expected to become an alternative to warfarin. It has been approved in Europe but not yet in the United States.
Connolly et al,17 in a randomized, double-blind trial, assigned 18,113 patients who had atrial fibrillation to receive either dabigatran 110 or 150 mg twice daily or adjusted-dose warfarin in an unblinded fashion. At 2 years, the rates of stroke and systemic embolism were about the same with dabigatran 110 mg as with warfarin but were lower with dabigatran 150 mg (relative risk 0.66, 95% confidence interval [CI] 0.53–0.82, P < .001). The rate of major bleeding was lower with dabigatran 110 mg than with warfarin (2.71% per year vs 3.36% per year, P = .003), but it was similar with dabigatran 150 mg (3.11% per year). Rates of life-threatening bleeding were 1.80% with warfarin, 1.22% with dabigatran 110 mg (P < .05), and 1.45% with dabigatran 150 mg (P < .05).
Comment. I suspect that warfarin’s days are numbered. Dabigatran 110 or 150 mg was as safe and as effective as warfarin in clinical trials, and probably will be more effective than warfarin in clinical practice. It will also probably be safer than warfarin in clinical practice, particularly in challenging settings such as long-term care. On the other hand, it will likely be much more expensive than warfarin.
DEMENTIA
Adverse effects of cholinesterase inhibitors
GILL SS, ANDERSON GM, FISCHER HD, ET AL. SYNCOPE AND ITS CONSEQUENCES IN PATIENTS WITH DEMENTIA RECEIVING CHOLINESTERASE INHIBITORS: A POPULATION-BASED COHORT STUDY. ARCH INTERN MED 2009; 169:867–873.
Cholinesterase inhibitors, eg, donepezil (Aricept), galantamine (Razadyne), and rivastigmine (Exelon), are commonly used to treat Alzheimer disease. However, these drugs carry risks of serious adverse effects.
Gill et al18 retrospectively reviewed a database from Ontario, Canada, and identified about 20,000 community-dwelling elderly persons admitted to the hospital who had been prescribed cholinesterase inhibitors and about three times as many matched controls.
Several adverse events were more frequent in people receiving cholinesterase inhibitors. Findings (events per 1,000 person-years):
- Hospital visits for syncope: 31.5 vs 18.6, adjusted hazard ratio (HR) 1.76, 95% CI 1.57–1.98
- Hip fractures: 22.4 vs 19.8, HR 1.18, 85% CI 1.04–1.34
- Hospital visits for bradycardia: 6.9 vs 4.4, HR 1.69, 95% CI 1.32–2.15
- Permanent pacemaker insertion: 4.7 vs 3.3, HR 1.49, 95% CI 1.12–2.00.
Comment. This study adds to the concerns that cholinesterase inhibitors, which have only modest cognitive benefits, may increase the risk of falls, injury, and need for pacemaker placement in demented patients. A low threshold to stop medications in this class should be considered when a patient on a cholinesterase inhibitor presents with bradycardia, falls, and syncope.
The importance of ‘staging’ dementia
IVERSON DJ, GRONSETH GS, REGER MA, ET AL; STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. PRACTICE PARAMETER UPDATE: EVALUATION AND MANAGEMENT OF DRIVING RISK IN DEMENTIA: REPORT OF THE QUALITY STANDARDS SUBCOMMITTEE OF THE AMERICAN ACADEMY OF NEUROLOGY. NEUROLOGY 2010; 74:1316–1324.
The Clinical Dementia Rating (CDR) is a simple scale that should be applied by clinicians to describe stage of dementia in patients with Alzheimer disease. This scale can be useful in a variety of settings, from prescribing antidementia drugs to determining whether a patient should still drive. Although research protocols utilize a survey or semistructured interview to derive the stage, the clinician can estimate the stage easily in the office, particularly if there is an informant who can comment on performance outside the office.
There are four stages to the CDR19:
- 0: No dementia
- 0.5: Mild memory deficit but intact function
- 1.0: Moderate memory loss with mild functional impairment
- 2.0: Severe memory loss, moderate functional impairment
- 3.0: Severe memory loss, no significant function outside of the house.
Comment. The first stage (0.5, mild memory deficit but intact function) corresponds to “mild cognitive impairment.” In the clinic, these patients tend to take more notes. They come to the appointment with a little book and they write everything down so they don’t forget. They do arrive at their appointments on time; they are not crashing the car; they are paying their bills.
Patients with CDR stage 1.0 dementia (moderate memory loss with mild functional impairment) may miss appointments, they may confuse their medications, and they may have problems driving. They are still taking care of their basic needs, and they show up for appointments acceptably washed and dressed. However, they are likely having trouble shopping and managing their finances.
Patients with severe memory loss and moderate functional impairment (CDR stage 2.0) may not realize they haven’t bathed for a week or have worn the same clothes repeatedly. They are having trouble with basic activities of daily living, such as bathing and toilet hygiene. However, if you were to encounter them socially and didn’t talk to them for too long, you might think they were normal.
Those with severe memory loss and no significant function outside the house (CDR stage 3.0) are the most severely disabled. Dementia in these individuals is recognizable at a glance, from across the room.
Alzheimer patients progress through the stages, from CDR stage 0.5 at about 1 year to stage 1 by about 2 years, to stage 2 by 5 years, and to stage 3 at 8 or 9 years.20
In prescribing antidementia medications. The CDR can help with prescribing antidementia drugs. No medications are approved by the FDA for stage 0 or 0.5. Cholinesterase inhibitors are approved for stages 1, 2, and 3; memantine (Namenda) is approved for stages 2 and 3.
Advising about driving. The CDR is the only risk predictor with a quality-of-evidence rating of A. More than half of people with stage 0.5 memory impairment are safe drivers; fewer than half of those with stage 1.0 are still safe drivers; and patients with stage 2.0 dementia should not be driving at all.21 An adverse rating by a caregiver carries a quality-of-evidence rating of B. Predictors of driving risk with a quality-of-evidence rating of C are decreased mileage due to self-restriction, agitation, or aggression; a crash in the past 1 to 5 years; a citation in the past 2 to 3 years; and a Folstein Mini-Mental State Examination score of 24 or less. Studies also show that a memory-impaired person’s self-rating of safe driving ability or of assurance that he or she avoids unsafe situations is not reliable.21
DELIRIUM
Delirium goes by a number of synonyms, eg, “sundowning,” acute confusional state, acute change in mental status, metabolic encephalopathy, toxic encephalopathy (psychosis), acute brain syndrome, and acute toxic psychosis.
Delirium is common in hospitalized elderly patients, occurring in 11% to 42% of elderly hospitalized patients overall, up to 53% of elderly surgical patients on regular hospital floors, 80% of elderly surgical patients in intensive care, and about half of elderly patients after undergoing coronary artery bypass grafting. Unfortunately, it is undiagnosed in 30% to 60% of cases.22–24
Many pathways can lead to delirium, including hypoxemia, metabolic derangement, drug effects, systemic inflammation, and infection.25
Outcomes can vary from full recovery to death. After 1 year, 50% of those who leave the hospital with some evidence of delirium have not regained their baseline function. Delirium also increases the cost of care and the risk of institutionalization.
Delirium can accelerate dementia
FONG TG, JONES RN, SHI P, ET AL. DELIRIUM ACCELERATES COGNITIVE DECLINE IN ALZHEIMER DISEASE. NEUROLOGY 2009; 72:1570–1575.
Delirium accelerates the course of dementia in patients who had some evidence of dementia before they entered the hospital. Often, the change is noticeable by the family.26
Preventing delirium
INOUYE SK BOGARDUS ST JR, CHARPENTIER PA, ET AL. A MULTICOMPONENT INTERVENTION TO PREVENT DELIRIUM IN HOSPITALIZED OLDER PATIENTS. N ENGL J MED 1999; 340:669–676.
LUNDSTRÖM M, OLOFSSON B, STENVALL M, ET AL. POSTOPERATIVE DELIRIUM IN OLD PATIENTS WITH FEMORAL NECK FRACTURE: A RANDOMIZED INTERVENTION STUDY. AGING CLIN EXP RES 2007; 19:178–186.
Delirium can often be prevented. In a report published in 1999, Inouye et al27 described the outcomes of a program to prevent delirium in hospitalized medically ill elderly patients. Interventions were aimed at optimizing cognitive function, preventing sleep deprivation, avoiding immobility, improving vision and hearing, and treating dehydration. The incidence of delirium was 9.9% in the intervention group vs 15% in the control group, a 40% reduction (P < .05).
Lundström et al28 implemented a similar program for elderly patients with hip fractures. Interventions included staff education and teamwork; active prevention, detection, and treatment of delirium; transfusions if hemoglobin levels were less than 10 g/dL; prompt removal of indwelling urinary catheters, with screening for urinary retention; active prevention and treatment of constipation; and protein-enriched meals. The incidence of delirium was 55% in the intervention group vs 75% in the control group, a 27% reduction.
Comment. Although we have long known that the risk of delirium in medical and surgical patients can be reduced, most hospitals do not have systematic programs to detect delirium and reduce its incidence. Hopefully, reduction in delirium risk will also reduce its adverse consequences, including worsening of dementia and increased mortality.
- Department of Health and Human Services. Physical activity guidelines for Americans. www.health.gov/paguidelines/reportG1_allcause.aspx
- Erickson KI, Prakash RS, Voss MW, et al. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus 2009; 19:1030–1039.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Woods JA, Keylock KT, Lowder T, et al. Cardiovascular exercise training extends influenza vaccine seroprotection in sedentary older adults: the immune function intervention trial. J Am Geriatr Soc 2009; 57:2183–2191.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ 2010; 341:c3691. doi:10.1136/bmj.c3691.
- Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ 2008; 336:262–266.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010; 303:1815–1822.
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McColloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med 2009; 361:1539–1547.
- Jassal SV, Chiu E, Hladunewich M. Loss of independence in patients starting dialysis at 80 years of age or older (letter). N Engl J Med 2009; 361:1612–1613.
- Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med 1995; 155:469–473.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. The Framingham Study. Stroke 1996; 27:1760–1764.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Gill SS, Anderson GM, Fischer HD, et al. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors: a population-based cohort study. Arch Intern Med 2009; 169:867–873.
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993; 43:2412–2414.
- Sloane PD. Advances in the treatment of Alzheimer’s disease. Am Fam Physician 1998; 58:1577–1586.
- Iverson DJ, Gronseth GS, Reger MA, et al; Standards Subcommittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Demeure MJ, Fain MJ. The elderly surgical patient and postoperative delirium. J Am Coll Surg 2006; 203:752–757.
- Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing 2006; 35:350–364.
- Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation 2009; 119:229–236.
- Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol 2009; 5:210–220.
- Fong TG, Jones RN, Shi P, et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology 2009; 72:1570–1575.
- Inouye SK, Bogardus ST, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med 1999; 340:669–676.
- Lundström M, Olofsson B, Stenvall M, et al. Postoperative delirium in old patients with femoral neck fracture: a randomized intervention study. Aging Clin Exp Res 2007; 19:178–186.
- Department of Health and Human Services. Physical activity guidelines for Americans. www.health.gov/paguidelines/reportG1_allcause.aspx
- Erickson KI, Prakash RS, Voss MW, et al. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus 2009; 19:1030–1039.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Woods JA, Keylock KT, Lowder T, et al. Cardiovascular exercise training extends influenza vaccine seroprotection in sedentary older adults: the immune function intervention trial. J Am Geriatr Soc 2009; 57:2183–2191.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ 2010; 341:c3691. doi:10.1136/bmj.c3691.
- Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ 2008; 336:262–266.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010; 303:1815–1822.
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McColloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med 2009; 361:1539–1547.
- Jassal SV, Chiu E, Hladunewich M. Loss of independence in patients starting dialysis at 80 years of age or older (letter). N Engl J Med 2009; 361:1612–1613.
- Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med 1995; 155:469–473.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. The Framingham Study. Stroke 1996; 27:1760–1764.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Gill SS, Anderson GM, Fischer HD, et al. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors: a population-based cohort study. Arch Intern Med 2009; 169:867–873.
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993; 43:2412–2414.
- Sloane PD. Advances in the treatment of Alzheimer’s disease. Am Fam Physician 1998; 58:1577–1586.
- Iverson DJ, Gronseth GS, Reger MA, et al; Standards Subcommittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Demeure MJ, Fain MJ. The elderly surgical patient and postoperative delirium. J Am Coll Surg 2006; 203:752–757.
- Siddiqi N, House AO, Holmes JD. Occurrence and outcome of delirium in medical in-patients: a systematic literature review. Age Ageing 2006; 35:350–364.
- Rudolph JL, Jones RN, Levkoff SE, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation 2009; 119:229–236.
- Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol 2009; 5:210–220.
- Fong TG, Jones RN, Shi P, et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology 2009; 72:1570–1575.
- Inouye SK, Bogardus ST, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med 1999; 340:669–676.
- Lundström M, Olofsson B, Stenvall M, et al. Postoperative delirium in old patients with femoral neck fracture: a randomized intervention study. Aging Clin Exp Res 2007; 19:178–186.
KEY POINTS
- Exercise has newly discovered benefits, such as preserving cognition and boosting the response to vaccination.
- Vitamin D supplementation has been found to prevent fractures, but yearly megadoses had the opposite effect.
- Denosumab (Prolia) has been approved for preventing fractures. It acts by inhibiting the receptor activator of nuclear factor kappa B (RANK) ligand.
- The outlook for elderly patients starting hemodialysis is bleak, with loss of function and a high risk of death.
- Dabigatran (Pradaxa), a direct thrombin inhibitor, may prove to be a safer alternative to warfarin (Coumadin).
- Cholinesterase inhibitors for Alzheimer disease are associated with higher risks of hospitalization for syncope, hip fractures, bradycardia, and pacemaker insertion.
- The Clinical Dementia Rating should be estimated when prescribing a cognitive enhancer and when advising a patient with memory impairment on driving safety.
- Delirium often accelerates dementia; interventions for hospitalized elderly patients may reduce its incidence.
Alzheimer disease prevention: Focus on cardiovascular risk, not amyloid?
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?

Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
Thiazolidinediones block microglial cell activation
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
- Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol 2006; 111:503–509.
- Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree? Pract Neurol 2006( 4):14–15.
- Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie? Neurobiol Aging 2002; 23:1101–1105.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 1996; 39:1392–1397.
- Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 2006; 63:1551–1555.
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006; 5:64–74.
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004; 53:474–481.
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 2004; 63:8–20.
- de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci 2002; 977:196–215.
- Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci 2002; 203–204:29–34.
- Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci 2007; 27:1973–1980.
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997; 277:813–817.
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996; 47:425–432.
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997; 48:626–632.
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004; 3:169–178.
- Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist 2002; 8:276–283.
- Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed 2006; 8:25.
- Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association 2006(2 suppl July):S148.
- Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 2010; 30:131–146.
- Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA 1998; 279:688–695.
- Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA 2002; 288:872–881.
- LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA 2001; 285:1489–1499.
- Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience 2000; 101:485–512.
- Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004; 291:2947–2958.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil 2004; 85:1694–1704.
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?
Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
Thiazolidinediones block microglial cell activation
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?
Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
Thiazolidinediones block microglial cell activation
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
- Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol 2006; 111:503–509.
- Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree? Pract Neurol 2006( 4):14–15.
- Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie? Neurobiol Aging 2002; 23:1101–1105.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 1996; 39:1392–1397.
- Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 2006; 63:1551–1555.
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006; 5:64–74.
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004; 53:474–481.
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 2004; 63:8–20.
- de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci 2002; 977:196–215.
- Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci 2002; 203–204:29–34.
- Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci 2007; 27:1973–1980.
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997; 277:813–817.
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996; 47:425–432.
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997; 48:626–632.
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004; 3:169–178.
- Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist 2002; 8:276–283.
- Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed 2006; 8:25.
- Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association 2006(2 suppl July):S148.
- Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 2010; 30:131–146.
- Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA 1998; 279:688–695.
- Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA 2002; 288:872–881.
- LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA 2001; 285:1489–1499.
- Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience 2000; 101:485–512.
- Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004; 291:2947–2958.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil 2004; 85:1694–1704.
- Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol 2006; 111:503–509.
- Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree? Pract Neurol 2006( 4):14–15.
- Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie? Neurobiol Aging 2002; 23:1101–1105.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 1996; 39:1392–1397.
- Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 2006; 63:1551–1555.
- Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006; 5:64–74.
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004; 53:474–481.
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 2004; 63:8–20.
- de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci 2002; 977:196–215.
- Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci 2002; 203–204:29–34.
- Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci 2007; 27:1973–1980.
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997; 277:813–817.
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996; 47:425–432.
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997; 48:626–632.
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004; 3:169–178.
- Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist 2002; 8:276–283.
- Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed 2006; 8:25.
- Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association 2006(2 suppl July):S148.
- Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 2010; 30:131–146.
- Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA 1998; 279:688–695.
- Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA 2002; 288:872–881.
- LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA 2001; 285:1489–1499.
- Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience 2000; 101:485–512.
- Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004; 291:2947–2958.
- Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol 2009; 66:1210–1215.
- Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med 2010; 170:186–193.
- Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil 2004; 85:1694–1704.
KEY POINTS
- Vascular risk factors clearly increase the risk of Alzheimer disease and can be addressed. However, controlled trials in patients with hypertension or with dyslipidemia have had negative results.
- Risk is lower with a diet high in antioxidants and polyunsaturated fatty acids.
- Estrogen therapy has had mixed results in observational studies, mostly hinting at lower risk. However, a randomized trial of hormone replacement therapy in late life indicated a higher risk of dementia with estrogen.
- Physical activity in midlife and in late life was associated with a lower risk of Alzheimer disease in observational studies. Controlled trials were not so positive, but the benefits of exercise may be slowly cumulative.
Controversies in non-ST-elevation acute coronary syndromes and percutaneous coronary interventions
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
KEY POINTS
- The data favor an aggressive strategy of routine catheterization, rather than a conservative strategy of catheterization only if a patient develops recurrent, spontaneous, or stress-induced ischemia.
- Early percutaneous intervention (within 24 hours) may be beneficial in patients at higher risk, but not necessarily in those at lower risk.
- Drug-eluting stents appear safe, assuming dual antiplatelet therapy is used. It is unclear how long this therapy needs to be continued.
- The choice of revascularization strategy—bypass surgery, bare-metal stent, or drug-eluting stent—should be individualized based on the risk of restenosis, thrombosis, and other factors.
Beyond office sphygmomanometry: Ways to better assess blood pressure
Hypertension is difficult to diagnose, and its treatment is difficult to monitor optimally on the basis of traditional office blood pressure measurements. To better protect our patients from the effects of undiagnosed or poorly controlled hypertension, we need to consider other options, such as ambulatory 24-hour blood pressure monitoring, automated measurement in the office, measurement in the patient’s home, and devices that analyze the peripheral pulse wave to estimate the central blood pressure and other indices of arterial stiffness.
MANUAL OFFICE MEASUREMENT HAS INHERENT LIMITATIONS
Office blood pressure measurements do provide enormous information about cardiovascular risk and the risk of death, as shown in epidemiologic studies. A meta-analysis1 of 61 prospective observational studies that included more than 1 million patients showed that office blood pressure levels clearly correlate with increased risk of death from cardiovascular disease and stroke.
But blood pressure is a dynamic measure with inherent minute-to-minute variability, and measurement will not be accurate if the correct technique is not followed. Traditional office sphygmomanometry is a snapshot and does not accurately reflect a patient’s blood pressure in the real world and in real time.
Recently, unique patterns of blood pressure have been identified that may not be detected in the physician’s office. It is clear from several clinical trials that some patients’ blood pressure is transiently elevated in the first few minutes during office measurements (the “white coat effect”). In addition, when office measurements are compared with out-of-office measurements, several patterns of hypertension emerge that have prognostic value. These patterns are white coat hypertension, masked hypertension, nocturnal hypertension, and failure of the blood pressure to dip during sleep.
WHITE COAT EFFECT
The white coat effect is described as a transient elevation in office blood pressure caused by an alerting reaction when the pressure is measured by a physician or a nurse. It may last for several minutes. The magnitude of blood pressure elevation has been noted to be higher when measured by a physician than when measured by a nurse. Multiple blood pressure measurements taken over 5 to 10 minutes help eliminate the white coat effect. In a recent study,2 36% of patients with hypertension demonstrated the white coat effect.
In a study by Mancia et al,3 46 patients underwent intra-arterial blood pressure monitoring for 2 days, during which time a physician or a nurse would check their blood pressure repeatedly over 10 minutes. This study found that most patients demonstrated the white coat effect: the blood pressure was higher in the first few measurements, but came down after 5 minutes. The white coat effect was as much as 22.6 ± 1.8 mm Hg when blood pressure was measured by a physician and was lower when measured by a nurse.
WHITE COAT HYPERTENSION
In contrast to the white coat effect, which is transient, white coat hypertension is defined as persistent elevation of office blood pressure measurements with normal blood pressure levels when measured outside the physician’s office. Depending on the population sampled, the prevalence of white coat hypertension ranges from 12% to 20%, but this is understandably difficult or almost impossible to detect with traditional office blood pressure measurements alone.4–7
MASKED HYPERTENSION
Patients with normal blood pressure in the physician’s office but high blood pressure during daily life were found to have a higher risk of cardiovascular events. This condition is called masked hypertension.8 For clinicians, the danger lies in underestimating the patient’s risk of cardiovascular events and, thus, undertreating the hypertension. Preliminary data on masked hypertension show that the rates of end-organ damage and cardiovascular events are slightly higher in patients with masked hypertension than in patients with sustained hypertension.
NOCTURNAL HYPERTENSION
Elevated nighttime blood pressure (>125/75 mm Hg) is considered nocturnal hypertension and is generally considered a subgroup of masked hypertension.9
In the African American Study of Kidney Disease and Hypertension (AASK),10,11 although most patients achieved their blood pressure goal during the trial, they were noted to have relentless progression of renal disease. On ambulatory 24-hour blood pressure monitoring during the cohort phase of the study,10 a high prevalence of elevated nighttime blood pressure (66%) was found. Further analysis showed that the elevated nighttime blood pressure was associated with worse hypertension-related end-organ damage. It is still unclear if lowering nighttime blood pressure improves clinical outcomes in this high-risk population.
DIPPING VS NONDIPPING
The mean blood pressure during sleep should normally decrease by 10% to 20% compared with daytime readings. “Nondipping,” ie, the lack of this nocturnal dip in blood pressure, carries a higher risk of death from cardiovascular causes, even if the person is otherwise normotensive.12,13 Nondipping is commonly noted in African Americans, patients with diabetes, and those with chronic kidney disease.
A study by Lurbe et al14 of patients with type 1 diabetes mellitus who underwent ambulatory 24-hour blood pressure monitoring found that the onset of the nondipping phenomenon preceded microalbuminuria (a risk factor for kidney disease). Data from our institution15 showed that nondipping was associated with a greater decline in glomerular filtration rate when compared with dipping.
The lack of reproducibility of a person’s dipping status has been a barrier in relying on this as a prognostic measure. White and Larocca16 found that only about half of the patients who appeared to be nondippers on one 24-hour recording still were nondippers on a second recording 4 to 8 weeks later. Compared with nondipping, nocturnal hypertension is a more stable blood pressure pattern that is being increasingly recognized in patients undergoing 24-hour blood pressure monitoring.
AUTOMATIC BLOOD PRESSURE DEVICES
An automated in-office blood pressure measurement device is one way to minimize the white coat effect and obtain a more accurate blood pressure assessment. Devices such as BpTRU (BpTRU Medical Devices Ltd, Coquitlam, BC, Canada) are programmed to take a series of automatic, oscillometric readings at regular intervals while the patient is left alone in a quiet room. BpTRU has been validated in several clinical trials and has been shown to overcome the white coat effect to some extent. Myers et al17 compared 24-hour blood pressure readings with those obtained by a family physician, by a research technician, and by the BpTRU device and found that the BpTRU readings were much closer to the average of awake blood pressure readings on 24-hour blood pressure monitoring.
AMBULATORY 24-HOUR BLOOD PRESSURE MONITORING
- Presence or absence of the nocturnal dip (the normal 10% to 20% drop in blood pressure at night during sleep)
- Morning surge (which in some studies was associated with higher incidence of stroke)
- Supine hypertension and sudden fluctuations in blood pressure seen in patients with autonomic failure.
Studies have shown that basing antihypertensive therapy on ambulatory 24-hour blood pressure monitoring results in better control of hypertension and lowers the rate of cardiovascular events.18,19
Perloff et al18 found that in patients whose hypertension was considered well controlled on the basis of office blood pressure measurements, those with higher blood pressures on ambulatory 24-hour monitoring had higher cardiovascular morbidity and mortality rates.
More recently, Clement et al19 showed that patients being treated for hypertension who have higher average ambulatory 24-hour blood pressures had a higher risk of cardiovascular events and cardiovascular death.
After following 790 patients for 3.7 years, Verdecchia et al20 concluded that controlling hypertension on the basis of ambulatory 24-hour blood pressure readings rather than traditional office measurements lowered the risk of cardiovascular disease.
‘Normal’ blood pressure on ambulatory 24-hour monitoring
It should be noted that the normal average blood pressure on ambulatory 24-hour monitoring tends to be lower than that on traditional office readings. According to the 2007 European guidelines,21 an average 24-hour blood pressure above the range of 125/80 to 130/80 mm Hg is considered diagnostic of hypertension.
The bottom line on ambulatory 24-hour monitoring: Not perfect, but helpful
Ambulatory 24-hour blood pressure monitoring is not perfect. It interferes with the patient’s activities and with sleep, and this can affect the readings. It is also expensive, and Medicare and Medicaid cover it only if the patient is diagnosed with white coat hypertension, based on stringent criteria that include three elevated clinic blood pressure measurements and two normal out-of-clinic blood pressure measurements and no evidence of end-organ damage. Despite these issues, almost all national guidelines for the management of hypertension recommend ambulatory 24-hour blood pressure monitoring to improve cardiovascular risk prediction and to measure the variability in blood pressure levels.
USING THE INTERNET IN MANAGING HYPERTENSION
Green et al22 studied a new model of care using home blood pressure monitoring via the Internet, and provided feedback and intervention to the patient via a pharmacist to achieve blood pressure goals. Patients measured their blood pressure at home on at least 2 days a week (two measurements each time), using an automatic oscillometric monitor (Omron Hem-705-CP, Kyoto, Japan), and entered the results in an electronic medical record on the Internet. In the intervention group, a pharmacist communicated with each patient by either phone or e-mail every 2 weeks, making changes to their antihypertensive regimens as needed.
Patients in the intervention group had an average reduction in blood pressure of 14 mm Hg from baseline, and their blood pressure was much better controlled compared with the control groups, who were being passively monitored or were receiving usual care based on office blood pressure readings.
MEASURING ARTERIAL STIFFNESS TO ASSES RISK OF END-ORGAN DAMAGE
Mean arterial blood pressure, derived from the extremes of systolic and diastolic pressure as measured with a traditional sphygmomanometer, is a product of cardiac output and total peripheral vascular resistance. In contrast, central aortic blood pressure, the central augmentation index, and pulse wave velocity are measures derived from brachial blood pressure as well as arterial pulse wave tracings. They provide additional information on arterial stiffness and help stratify patients at increased cardiovascular risk.
The art of evaluating the arterial pulse wave with the fingertips while examining a patient and diagnosing various ailments was well known and practiced by ancient Greek and Chinese physicians. Although this was less recognized in Western medicine, it was the pulse wave recording on a sphygmograph that was used to measure human blood pressure in the 19th century.23 In the early 20th century, this art was lost with the invention of the mercury sphygmomanometer.
Arterial stiffness indices—ie, central aortic blood pressure, the central augmentation index, and pulse wave velocity—can now be measured noninvasively and have been shown to correlate very well with measurements obtained via a central arterial catheter. In the past, the only way to measure central blood pressure was directly via a central arterial catheter. New devices now measure arterial stiffness indices indirectly by applanation tonometry and pulse wave analysis (reviewed by O’Rourke et al25).
Several trials have shown that these arterial indices have a better prognostic value than the mean arterial pressure or the brachial pulse pressure. For example, the Baltimore Longitudinal Study of Aging26 followed 100 normotensive individuals for 5 years and found that those with a higher pulse wave velocity had a greater chance of developing incident hypertension. Other studies showed that pulse wave velocity and other indices of arterial stiffness are associated with dysfunction of the microvasculature in the brain, with higher cardiovascular risk, and a higher risk of death.
A major limitation in measuring these arterial stiffness indices is that they are derived values and require measurement of brachial blood pressure in addition to the pulse wave tracing.
Recent hypertension guidelines21,27,28 released during the past 2 years in Europe, Latin America, and Japan have recommended measurement of arterial stiffness as part of a comprehensive evaluation of patients with hypertension.
EXCITING TIMES IN HYPERTENSION
These are exciting times in the field of hypertension. With advances in technology, we have new devices and techniques that provide a closer view of the hemodynamic changes and blood pressures experienced by vital organs. In addition, we can now go beyond the physician’s office and evaluate blood pressure changes that occur during the course of a usual day in a patient’s life. This enables us to make better decisions in the management of their hypertension, embodying Dr. Harvey Cushing’s teaching that the physician’s obligation is to “view the man in his world.”29
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Agespecific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- Culleton BF, McKay DW, Campbell NR. Performance of the automated BpTRU measurement device in the assessment of white-coat hypertension and white-coat effect. Blood Press Monit 2006; 11:37–42.
- Mancia G, Parati G, Pomidossi G, Grassi G, Casadei R, Zanchetti A. Alerting reaction and rise in blood pressure during measurement by physician and nurse. Hypertension 1987; 9:209–215.
- Mancia G, Sega R, Bravi C, et al. Ambulatory blood pressure normality: results from the PAMELA study. J Hypertens 1995; 13:1377–1390.
- Ohkubo T, Kikuya M, Metoki H, et al. Prognosis of “masked” hypertension and “white-coat” hypertension detected by 24-h ambulatory blood pressure monitoring 10-year follow-up from the Ohasama study. J Am Coll Cardiol 2005; 46:508–515.
- Kotsis V, Stabouli S, Toumanidis S, et al. Target organ damage in “white coat hypertension” and “masked hypertension.” Am J Hypertens 2008; 21:393–399.
- Obara T, Ohkubo T, Funahashi J, et al. Isolated uncontrolled hypertension at home and in the office among treated hypertensive patients from the J-HOME study. J Hypertens 2005; 23:1653–1660.
- Pickering TG DK, Rafey MA, Schwartz J, Gerin W. Masked hypertension: are those with normal office but elevated ambulatory blood pressure at risk? J Hypertens 2002; 20( suppl 4):176.
- Pickering TG, Hall JE, Appel LJ. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation 2005; 111:697–716.
- Pogue V, Rahman M, Lipkowitz M, et al. Disparate estimates of hypertension control from ambulatory and clinic blood pressure measurements in hypertensive kidney disease. Hypertension 2009; 53:20–27.
- Agodoa LY, Appel L, Bakris GL, et al. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Ohkubo T, Hozawa A, Yamaguchi J, et al. Prognostic significance of the nocturnal decline in blood pressure in individuals with and without high 24-h blood pressure: the Ohasama study. J Hypertens 2002; 20:2183–2189.
- Brotman DJ, Davidson MB, Boumitri M, Vidt DG. Impaired diurnal blood pressure variation and all-cause mortality. Am J Hypertens 2008; 21:92–97.
- Lurbe E, Redon J, Kesani A, et al. Increase in nocturnal blood pressure and progression to microalbuminuria in type 1 diabetes. N Engl J Med 2002; 347:797–805.
- Davidson MB, Hix JK, Vidt DG, Brotman DJ. Association of impaired diurnal blood pressure variation with a subsequent decline in glo-merular filtration rate. Arch Intern Med 2006; 166:846–852.
- White WB, Larocca GM. Improving the utility of the nocturnal hypertension definition by using absolute sleep blood pressure rather than the “dipping” proportion. Am J Cardiol 2003; 92:1439–1441.
- Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce the white coat response. J Hypertens 2009; 27:280–286.
- Perloff D, Sokolow M, Cowan R. The prognostic value of ambulatory blood pressures. JAMA 1983; 249:2792–2798.
- Clement DL, De Buyzere ML, De Bacquer DA, et al. Prognostic value of ambulatory blood-pressure recordings in patients with treated hypertension. N Engl J Med 2003; 348:2407–2415.
- Verdecchia P, Reboldi G, Porcellati C, et al. Risk of cardiovascular disease in relation to achieved office and ambulatory blood pressure control in treated hypertensive subjects. J Am Coll Cardiol 2002; 39:878–885.
- Mansia G, De Backer G, Dominiczak A, et al. 2007 ESH-ESC Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Blood Press 2007; 16:135–232.
- Green BB, Cook AJ, Ralston JD, et al. Effectiveness of home blood pressure monitoring, Web communication, and pharmacist care on hypertension control: a randomized controlled trial. JAMA 2008; 299:2857–2867.
- Mohamed F. On chronic Bright’s disease, and its essential symptoms. Lancet 1879; 1:399–401.
- Liew Y, Rafey MA, Allam S, Arrigain S, Butler R, Schreiber M. Blood pressure goals and arterial stiffness in chronic kidney disease. J Clin Hypertens (Greenwich) 2009; 11:201–206.
- O’Rourke MF, Pauca A, Jiang XJ. Pulse wave analysis. Br J Clin Pharmacol 2001; 51:507–522.
- Najjar SS, Scuteri A, Shetty V, et al. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol 2008; 51:1377–1383.
- Sanchez RA, Ayala M, Baglivo H, et al. Latin American guidelines on hypertension. J Hypertens 2009; 27:905–922.
- Japanese Society of Hypertension. The Japanese Society of Hypertension Committee for Guidelines for the Management of Hypertension: Measurement and clinical evaluation of blood pressure. Hypertens Res 2009; 32:11–23.
- Dubos RJ. Man Adapting. New Haven, CT: Yale University Press, 1980.
Hypertension is difficult to diagnose, and its treatment is difficult to monitor optimally on the basis of traditional office blood pressure measurements. To better protect our patients from the effects of undiagnosed or poorly controlled hypertension, we need to consider other options, such as ambulatory 24-hour blood pressure monitoring, automated measurement in the office, measurement in the patient’s home, and devices that analyze the peripheral pulse wave to estimate the central blood pressure and other indices of arterial stiffness.
MANUAL OFFICE MEASUREMENT HAS INHERENT LIMITATIONS
Office blood pressure measurements do provide enormous information about cardiovascular risk and the risk of death, as shown in epidemiologic studies. A meta-analysis1 of 61 prospective observational studies that included more than 1 million patients showed that office blood pressure levels clearly correlate with increased risk of death from cardiovascular disease and stroke.
But blood pressure is a dynamic measure with inherent minute-to-minute variability, and measurement will not be accurate if the correct technique is not followed. Traditional office sphygmomanometry is a snapshot and does not accurately reflect a patient’s blood pressure in the real world and in real time.
Recently, unique patterns of blood pressure have been identified that may not be detected in the physician’s office. It is clear from several clinical trials that some patients’ blood pressure is transiently elevated in the first few minutes during office measurements (the “white coat effect”). In addition, when office measurements are compared with out-of-office measurements, several patterns of hypertension emerge that have prognostic value. These patterns are white coat hypertension, masked hypertension, nocturnal hypertension, and failure of the blood pressure to dip during sleep.
WHITE COAT EFFECT
The white coat effect is described as a transient elevation in office blood pressure caused by an alerting reaction when the pressure is measured by a physician or a nurse. It may last for several minutes. The magnitude of blood pressure elevation has been noted to be higher when measured by a physician than when measured by a nurse. Multiple blood pressure measurements taken over 5 to 10 minutes help eliminate the white coat effect. In a recent study,2 36% of patients with hypertension demonstrated the white coat effect.
In a study by Mancia et al,3 46 patients underwent intra-arterial blood pressure monitoring for 2 days, during which time a physician or a nurse would check their blood pressure repeatedly over 10 minutes. This study found that most patients demonstrated the white coat effect: the blood pressure was higher in the first few measurements, but came down after 5 minutes. The white coat effect was as much as 22.6 ± 1.8 mm Hg when blood pressure was measured by a physician and was lower when measured by a nurse.
WHITE COAT HYPERTENSION
In contrast to the white coat effect, which is transient, white coat hypertension is defined as persistent elevation of office blood pressure measurements with normal blood pressure levels when measured outside the physician’s office. Depending on the population sampled, the prevalence of white coat hypertension ranges from 12% to 20%, but this is understandably difficult or almost impossible to detect with traditional office blood pressure measurements alone.4–7
MASKED HYPERTENSION
Patients with normal blood pressure in the physician’s office but high blood pressure during daily life were found to have a higher risk of cardiovascular events. This condition is called masked hypertension.8 For clinicians, the danger lies in underestimating the patient’s risk of cardiovascular events and, thus, undertreating the hypertension. Preliminary data on masked hypertension show that the rates of end-organ damage and cardiovascular events are slightly higher in patients with masked hypertension than in patients with sustained hypertension.
NOCTURNAL HYPERTENSION
Elevated nighttime blood pressure (>125/75 mm Hg) is considered nocturnal hypertension and is generally considered a subgroup of masked hypertension.9
In the African American Study of Kidney Disease and Hypertension (AASK),10,11 although most patients achieved their blood pressure goal during the trial, they were noted to have relentless progression of renal disease. On ambulatory 24-hour blood pressure monitoring during the cohort phase of the study,10 a high prevalence of elevated nighttime blood pressure (66%) was found. Further analysis showed that the elevated nighttime blood pressure was associated with worse hypertension-related end-organ damage. It is still unclear if lowering nighttime blood pressure improves clinical outcomes in this high-risk population.
DIPPING VS NONDIPPING
The mean blood pressure during sleep should normally decrease by 10% to 20% compared with daytime readings. “Nondipping,” ie, the lack of this nocturnal dip in blood pressure, carries a higher risk of death from cardiovascular causes, even if the person is otherwise normotensive.12,13 Nondipping is commonly noted in African Americans, patients with diabetes, and those with chronic kidney disease.
A study by Lurbe et al14 of patients with type 1 diabetes mellitus who underwent ambulatory 24-hour blood pressure monitoring found that the onset of the nondipping phenomenon preceded microalbuminuria (a risk factor for kidney disease). Data from our institution15 showed that nondipping was associated with a greater decline in glomerular filtration rate when compared with dipping.
The lack of reproducibility of a person’s dipping status has been a barrier in relying on this as a prognostic measure. White and Larocca16 found that only about half of the patients who appeared to be nondippers on one 24-hour recording still were nondippers on a second recording 4 to 8 weeks later. Compared with nondipping, nocturnal hypertension is a more stable blood pressure pattern that is being increasingly recognized in patients undergoing 24-hour blood pressure monitoring.
AUTOMATIC BLOOD PRESSURE DEVICES
An automated in-office blood pressure measurement device is one way to minimize the white coat effect and obtain a more accurate blood pressure assessment. Devices such as BpTRU (BpTRU Medical Devices Ltd, Coquitlam, BC, Canada) are programmed to take a series of automatic, oscillometric readings at regular intervals while the patient is left alone in a quiet room. BpTRU has been validated in several clinical trials and has been shown to overcome the white coat effect to some extent. Myers et al17 compared 24-hour blood pressure readings with those obtained by a family physician, by a research technician, and by the BpTRU device and found that the BpTRU readings were much closer to the average of awake blood pressure readings on 24-hour blood pressure monitoring.
AMBULATORY 24-HOUR BLOOD PRESSURE MONITORING
- Presence or absence of the nocturnal dip (the normal 10% to 20% drop in blood pressure at night during sleep)
- Morning surge (which in some studies was associated with higher incidence of stroke)
- Supine hypertension and sudden fluctuations in blood pressure seen in patients with autonomic failure.
Studies have shown that basing antihypertensive therapy on ambulatory 24-hour blood pressure monitoring results in better control of hypertension and lowers the rate of cardiovascular events.18,19
Perloff et al18 found that in patients whose hypertension was considered well controlled on the basis of office blood pressure measurements, those with higher blood pressures on ambulatory 24-hour monitoring had higher cardiovascular morbidity and mortality rates.
More recently, Clement et al19 showed that patients being treated for hypertension who have higher average ambulatory 24-hour blood pressures had a higher risk of cardiovascular events and cardiovascular death.
After following 790 patients for 3.7 years, Verdecchia et al20 concluded that controlling hypertension on the basis of ambulatory 24-hour blood pressure readings rather than traditional office measurements lowered the risk of cardiovascular disease.
‘Normal’ blood pressure on ambulatory 24-hour monitoring
It should be noted that the normal average blood pressure on ambulatory 24-hour monitoring tends to be lower than that on traditional office readings. According to the 2007 European guidelines,21 an average 24-hour blood pressure above the range of 125/80 to 130/80 mm Hg is considered diagnostic of hypertension.
The bottom line on ambulatory 24-hour monitoring: Not perfect, but helpful
Ambulatory 24-hour blood pressure monitoring is not perfect. It interferes with the patient’s activities and with sleep, and this can affect the readings. It is also expensive, and Medicare and Medicaid cover it only if the patient is diagnosed with white coat hypertension, based on stringent criteria that include three elevated clinic blood pressure measurements and two normal out-of-clinic blood pressure measurements and no evidence of end-organ damage. Despite these issues, almost all national guidelines for the management of hypertension recommend ambulatory 24-hour blood pressure monitoring to improve cardiovascular risk prediction and to measure the variability in blood pressure levels.
USING THE INTERNET IN MANAGING HYPERTENSION
Green et al22 studied a new model of care using home blood pressure monitoring via the Internet, and provided feedback and intervention to the patient via a pharmacist to achieve blood pressure goals. Patients measured their blood pressure at home on at least 2 days a week (two measurements each time), using an automatic oscillometric monitor (Omron Hem-705-CP, Kyoto, Japan), and entered the results in an electronic medical record on the Internet. In the intervention group, a pharmacist communicated with each patient by either phone or e-mail every 2 weeks, making changes to their antihypertensive regimens as needed.
Patients in the intervention group had an average reduction in blood pressure of 14 mm Hg from baseline, and their blood pressure was much better controlled compared with the control groups, who were being passively monitored or were receiving usual care based on office blood pressure readings.
MEASURING ARTERIAL STIFFNESS TO ASSES RISK OF END-ORGAN DAMAGE
Mean arterial blood pressure, derived from the extremes of systolic and diastolic pressure as measured with a traditional sphygmomanometer, is a product of cardiac output and total peripheral vascular resistance. In contrast, central aortic blood pressure, the central augmentation index, and pulse wave velocity are measures derived from brachial blood pressure as well as arterial pulse wave tracings. They provide additional information on arterial stiffness and help stratify patients at increased cardiovascular risk.
The art of evaluating the arterial pulse wave with the fingertips while examining a patient and diagnosing various ailments was well known and practiced by ancient Greek and Chinese physicians. Although this was less recognized in Western medicine, it was the pulse wave recording on a sphygmograph that was used to measure human blood pressure in the 19th century.23 In the early 20th century, this art was lost with the invention of the mercury sphygmomanometer.
Arterial stiffness indices—ie, central aortic blood pressure, the central augmentation index, and pulse wave velocity—can now be measured noninvasively and have been shown to correlate very well with measurements obtained via a central arterial catheter. In the past, the only way to measure central blood pressure was directly via a central arterial catheter. New devices now measure arterial stiffness indices indirectly by applanation tonometry and pulse wave analysis (reviewed by O’Rourke et al25).
Several trials have shown that these arterial indices have a better prognostic value than the mean arterial pressure or the brachial pulse pressure. For example, the Baltimore Longitudinal Study of Aging26 followed 100 normotensive individuals for 5 years and found that those with a higher pulse wave velocity had a greater chance of developing incident hypertension. Other studies showed that pulse wave velocity and other indices of arterial stiffness are associated with dysfunction of the microvasculature in the brain, with higher cardiovascular risk, and a higher risk of death.
A major limitation in measuring these arterial stiffness indices is that they are derived values and require measurement of brachial blood pressure in addition to the pulse wave tracing.
Recent hypertension guidelines21,27,28 released during the past 2 years in Europe, Latin America, and Japan have recommended measurement of arterial stiffness as part of a comprehensive evaluation of patients with hypertension.
EXCITING TIMES IN HYPERTENSION
These are exciting times in the field of hypertension. With advances in technology, we have new devices and techniques that provide a closer view of the hemodynamic changes and blood pressures experienced by vital organs. In addition, we can now go beyond the physician’s office and evaluate blood pressure changes that occur during the course of a usual day in a patient’s life. This enables us to make better decisions in the management of their hypertension, embodying Dr. Harvey Cushing’s teaching that the physician’s obligation is to “view the man in his world.”29
Hypertension is difficult to diagnose, and its treatment is difficult to monitor optimally on the basis of traditional office blood pressure measurements. To better protect our patients from the effects of undiagnosed or poorly controlled hypertension, we need to consider other options, such as ambulatory 24-hour blood pressure monitoring, automated measurement in the office, measurement in the patient’s home, and devices that analyze the peripheral pulse wave to estimate the central blood pressure and other indices of arterial stiffness.
MANUAL OFFICE MEASUREMENT HAS INHERENT LIMITATIONS
Office blood pressure measurements do provide enormous information about cardiovascular risk and the risk of death, as shown in epidemiologic studies. A meta-analysis1 of 61 prospective observational studies that included more than 1 million patients showed that office blood pressure levels clearly correlate with increased risk of death from cardiovascular disease and stroke.
But blood pressure is a dynamic measure with inherent minute-to-minute variability, and measurement will not be accurate if the correct technique is not followed. Traditional office sphygmomanometry is a snapshot and does not accurately reflect a patient’s blood pressure in the real world and in real time.
Recently, unique patterns of blood pressure have been identified that may not be detected in the physician’s office. It is clear from several clinical trials that some patients’ blood pressure is transiently elevated in the first few minutes during office measurements (the “white coat effect”). In addition, when office measurements are compared with out-of-office measurements, several patterns of hypertension emerge that have prognostic value. These patterns are white coat hypertension, masked hypertension, nocturnal hypertension, and failure of the blood pressure to dip during sleep.
WHITE COAT EFFECT
The white coat effect is described as a transient elevation in office blood pressure caused by an alerting reaction when the pressure is measured by a physician or a nurse. It may last for several minutes. The magnitude of blood pressure elevation has been noted to be higher when measured by a physician than when measured by a nurse. Multiple blood pressure measurements taken over 5 to 10 minutes help eliminate the white coat effect. In a recent study,2 36% of patients with hypertension demonstrated the white coat effect.
In a study by Mancia et al,3 46 patients underwent intra-arterial blood pressure monitoring for 2 days, during which time a physician or a nurse would check their blood pressure repeatedly over 10 minutes. This study found that most patients demonstrated the white coat effect: the blood pressure was higher in the first few measurements, but came down after 5 minutes. The white coat effect was as much as 22.6 ± 1.8 mm Hg when blood pressure was measured by a physician and was lower when measured by a nurse.
WHITE COAT HYPERTENSION
In contrast to the white coat effect, which is transient, white coat hypertension is defined as persistent elevation of office blood pressure measurements with normal blood pressure levels when measured outside the physician’s office. Depending on the population sampled, the prevalence of white coat hypertension ranges from 12% to 20%, but this is understandably difficult or almost impossible to detect with traditional office blood pressure measurements alone.4–7
MASKED HYPERTENSION
Patients with normal blood pressure in the physician’s office but high blood pressure during daily life were found to have a higher risk of cardiovascular events. This condition is called masked hypertension.8 For clinicians, the danger lies in underestimating the patient’s risk of cardiovascular events and, thus, undertreating the hypertension. Preliminary data on masked hypertension show that the rates of end-organ damage and cardiovascular events are slightly higher in patients with masked hypertension than in patients with sustained hypertension.
NOCTURNAL HYPERTENSION
Elevated nighttime blood pressure (>125/75 mm Hg) is considered nocturnal hypertension and is generally considered a subgroup of masked hypertension.9
In the African American Study of Kidney Disease and Hypertension (AASK),10,11 although most patients achieved their blood pressure goal during the trial, they were noted to have relentless progression of renal disease. On ambulatory 24-hour blood pressure monitoring during the cohort phase of the study,10 a high prevalence of elevated nighttime blood pressure (66%) was found. Further analysis showed that the elevated nighttime blood pressure was associated with worse hypertension-related end-organ damage. It is still unclear if lowering nighttime blood pressure improves clinical outcomes in this high-risk population.
DIPPING VS NONDIPPING
The mean blood pressure during sleep should normally decrease by 10% to 20% compared with daytime readings. “Nondipping,” ie, the lack of this nocturnal dip in blood pressure, carries a higher risk of death from cardiovascular causes, even if the person is otherwise normotensive.12,13 Nondipping is commonly noted in African Americans, patients with diabetes, and those with chronic kidney disease.
A study by Lurbe et al14 of patients with type 1 diabetes mellitus who underwent ambulatory 24-hour blood pressure monitoring found that the onset of the nondipping phenomenon preceded microalbuminuria (a risk factor for kidney disease). Data from our institution15 showed that nondipping was associated with a greater decline in glomerular filtration rate when compared with dipping.
The lack of reproducibility of a person’s dipping status has been a barrier in relying on this as a prognostic measure. White and Larocca16 found that only about half of the patients who appeared to be nondippers on one 24-hour recording still were nondippers on a second recording 4 to 8 weeks later. Compared with nondipping, nocturnal hypertension is a more stable blood pressure pattern that is being increasingly recognized in patients undergoing 24-hour blood pressure monitoring.
AUTOMATIC BLOOD PRESSURE DEVICES
An automated in-office blood pressure measurement device is one way to minimize the white coat effect and obtain a more accurate blood pressure assessment. Devices such as BpTRU (BpTRU Medical Devices Ltd, Coquitlam, BC, Canada) are programmed to take a series of automatic, oscillometric readings at regular intervals while the patient is left alone in a quiet room. BpTRU has been validated in several clinical trials and has been shown to overcome the white coat effect to some extent. Myers et al17 compared 24-hour blood pressure readings with those obtained by a family physician, by a research technician, and by the BpTRU device and found that the BpTRU readings were much closer to the average of awake blood pressure readings on 24-hour blood pressure monitoring.
AMBULATORY 24-HOUR BLOOD PRESSURE MONITORING
- Presence or absence of the nocturnal dip (the normal 10% to 20% drop in blood pressure at night during sleep)
- Morning surge (which in some studies was associated with higher incidence of stroke)
- Supine hypertension and sudden fluctuations in blood pressure seen in patients with autonomic failure.
Studies have shown that basing antihypertensive therapy on ambulatory 24-hour blood pressure monitoring results in better control of hypertension and lowers the rate of cardiovascular events.18,19
Perloff et al18 found that in patients whose hypertension was considered well controlled on the basis of office blood pressure measurements, those with higher blood pressures on ambulatory 24-hour monitoring had higher cardiovascular morbidity and mortality rates.
More recently, Clement et al19 showed that patients being treated for hypertension who have higher average ambulatory 24-hour blood pressures had a higher risk of cardiovascular events and cardiovascular death.
After following 790 patients for 3.7 years, Verdecchia et al20 concluded that controlling hypertension on the basis of ambulatory 24-hour blood pressure readings rather than traditional office measurements lowered the risk of cardiovascular disease.
‘Normal’ blood pressure on ambulatory 24-hour monitoring
It should be noted that the normal average blood pressure on ambulatory 24-hour monitoring tends to be lower than that on traditional office readings. According to the 2007 European guidelines,21 an average 24-hour blood pressure above the range of 125/80 to 130/80 mm Hg is considered diagnostic of hypertension.
The bottom line on ambulatory 24-hour monitoring: Not perfect, but helpful
Ambulatory 24-hour blood pressure monitoring is not perfect. It interferes with the patient’s activities and with sleep, and this can affect the readings. It is also expensive, and Medicare and Medicaid cover it only if the patient is diagnosed with white coat hypertension, based on stringent criteria that include three elevated clinic blood pressure measurements and two normal out-of-clinic blood pressure measurements and no evidence of end-organ damage. Despite these issues, almost all national guidelines for the management of hypertension recommend ambulatory 24-hour blood pressure monitoring to improve cardiovascular risk prediction and to measure the variability in blood pressure levels.
USING THE INTERNET IN MANAGING HYPERTENSION
Green et al22 studied a new model of care using home blood pressure monitoring via the Internet, and provided feedback and intervention to the patient via a pharmacist to achieve blood pressure goals. Patients measured their blood pressure at home on at least 2 days a week (two measurements each time), using an automatic oscillometric monitor (Omron Hem-705-CP, Kyoto, Japan), and entered the results in an electronic medical record on the Internet. In the intervention group, a pharmacist communicated with each patient by either phone or e-mail every 2 weeks, making changes to their antihypertensive regimens as needed.
Patients in the intervention group had an average reduction in blood pressure of 14 mm Hg from baseline, and their blood pressure was much better controlled compared with the control groups, who were being passively monitored or were receiving usual care based on office blood pressure readings.
MEASURING ARTERIAL STIFFNESS TO ASSES RISK OF END-ORGAN DAMAGE
Mean arterial blood pressure, derived from the extremes of systolic and diastolic pressure as measured with a traditional sphygmomanometer, is a product of cardiac output and total peripheral vascular resistance. In contrast, central aortic blood pressure, the central augmentation index, and pulse wave velocity are measures derived from brachial blood pressure as well as arterial pulse wave tracings. They provide additional information on arterial stiffness and help stratify patients at increased cardiovascular risk.
The art of evaluating the arterial pulse wave with the fingertips while examining a patient and diagnosing various ailments was well known and practiced by ancient Greek and Chinese physicians. Although this was less recognized in Western medicine, it was the pulse wave recording on a sphygmograph that was used to measure human blood pressure in the 19th century.23 In the early 20th century, this art was lost with the invention of the mercury sphygmomanometer.
Arterial stiffness indices—ie, central aortic blood pressure, the central augmentation index, and pulse wave velocity—can now be measured noninvasively and have been shown to correlate very well with measurements obtained via a central arterial catheter. In the past, the only way to measure central blood pressure was directly via a central arterial catheter. New devices now measure arterial stiffness indices indirectly by applanation tonometry and pulse wave analysis (reviewed by O’Rourke et al25).
Several trials have shown that these arterial indices have a better prognostic value than the mean arterial pressure or the brachial pulse pressure. For example, the Baltimore Longitudinal Study of Aging26 followed 100 normotensive individuals for 5 years and found that those with a higher pulse wave velocity had a greater chance of developing incident hypertension. Other studies showed that pulse wave velocity and other indices of arterial stiffness are associated with dysfunction of the microvasculature in the brain, with higher cardiovascular risk, and a higher risk of death.
A major limitation in measuring these arterial stiffness indices is that they are derived values and require measurement of brachial blood pressure in addition to the pulse wave tracing.
Recent hypertension guidelines21,27,28 released during the past 2 years in Europe, Latin America, and Japan have recommended measurement of arterial stiffness as part of a comprehensive evaluation of patients with hypertension.
EXCITING TIMES IN HYPERTENSION
These are exciting times in the field of hypertension. With advances in technology, we have new devices and techniques that provide a closer view of the hemodynamic changes and blood pressures experienced by vital organs. In addition, we can now go beyond the physician’s office and evaluate blood pressure changes that occur during the course of a usual day in a patient’s life. This enables us to make better decisions in the management of their hypertension, embodying Dr. Harvey Cushing’s teaching that the physician’s obligation is to “view the man in his world.”29
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Agespecific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- Culleton BF, McKay DW, Campbell NR. Performance of the automated BpTRU measurement device in the assessment of white-coat hypertension and white-coat effect. Blood Press Monit 2006; 11:37–42.
- Mancia G, Parati G, Pomidossi G, Grassi G, Casadei R, Zanchetti A. Alerting reaction and rise in blood pressure during measurement by physician and nurse. Hypertension 1987; 9:209–215.
- Mancia G, Sega R, Bravi C, et al. Ambulatory blood pressure normality: results from the PAMELA study. J Hypertens 1995; 13:1377–1390.
- Ohkubo T, Kikuya M, Metoki H, et al. Prognosis of “masked” hypertension and “white-coat” hypertension detected by 24-h ambulatory blood pressure monitoring 10-year follow-up from the Ohasama study. J Am Coll Cardiol 2005; 46:508–515.
- Kotsis V, Stabouli S, Toumanidis S, et al. Target organ damage in “white coat hypertension” and “masked hypertension.” Am J Hypertens 2008; 21:393–399.
- Obara T, Ohkubo T, Funahashi J, et al. Isolated uncontrolled hypertension at home and in the office among treated hypertensive patients from the J-HOME study. J Hypertens 2005; 23:1653–1660.
- Pickering TG DK, Rafey MA, Schwartz J, Gerin W. Masked hypertension: are those with normal office but elevated ambulatory blood pressure at risk? J Hypertens 2002; 20( suppl 4):176.
- Pickering TG, Hall JE, Appel LJ. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation 2005; 111:697–716.
- Pogue V, Rahman M, Lipkowitz M, et al. Disparate estimates of hypertension control from ambulatory and clinic blood pressure measurements in hypertensive kidney disease. Hypertension 2009; 53:20–27.
- Agodoa LY, Appel L, Bakris GL, et al. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Ohkubo T, Hozawa A, Yamaguchi J, et al. Prognostic significance of the nocturnal decline in blood pressure in individuals with and without high 24-h blood pressure: the Ohasama study. J Hypertens 2002; 20:2183–2189.
- Brotman DJ, Davidson MB, Boumitri M, Vidt DG. Impaired diurnal blood pressure variation and all-cause mortality. Am J Hypertens 2008; 21:92–97.
- Lurbe E, Redon J, Kesani A, et al. Increase in nocturnal blood pressure and progression to microalbuminuria in type 1 diabetes. N Engl J Med 2002; 347:797–805.
- Davidson MB, Hix JK, Vidt DG, Brotman DJ. Association of impaired diurnal blood pressure variation with a subsequent decline in glo-merular filtration rate. Arch Intern Med 2006; 166:846–852.
- White WB, Larocca GM. Improving the utility of the nocturnal hypertension definition by using absolute sleep blood pressure rather than the “dipping” proportion. Am J Cardiol 2003; 92:1439–1441.
- Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce the white coat response. J Hypertens 2009; 27:280–286.
- Perloff D, Sokolow M, Cowan R. The prognostic value of ambulatory blood pressures. JAMA 1983; 249:2792–2798.
- Clement DL, De Buyzere ML, De Bacquer DA, et al. Prognostic value of ambulatory blood-pressure recordings in patients with treated hypertension. N Engl J Med 2003; 348:2407–2415.
- Verdecchia P, Reboldi G, Porcellati C, et al. Risk of cardiovascular disease in relation to achieved office and ambulatory blood pressure control in treated hypertensive subjects. J Am Coll Cardiol 2002; 39:878–885.
- Mansia G, De Backer G, Dominiczak A, et al. 2007 ESH-ESC Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Blood Press 2007; 16:135–232.
- Green BB, Cook AJ, Ralston JD, et al. Effectiveness of home blood pressure monitoring, Web communication, and pharmacist care on hypertension control: a randomized controlled trial. JAMA 2008; 299:2857–2867.
- Mohamed F. On chronic Bright’s disease, and its essential symptoms. Lancet 1879; 1:399–401.
- Liew Y, Rafey MA, Allam S, Arrigain S, Butler R, Schreiber M. Blood pressure goals and arterial stiffness in chronic kidney disease. J Clin Hypertens (Greenwich) 2009; 11:201–206.
- O’Rourke MF, Pauca A, Jiang XJ. Pulse wave analysis. Br J Clin Pharmacol 2001; 51:507–522.
- Najjar SS, Scuteri A, Shetty V, et al. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol 2008; 51:1377–1383.
- Sanchez RA, Ayala M, Baglivo H, et al. Latin American guidelines on hypertension. J Hypertens 2009; 27:905–922.
- Japanese Society of Hypertension. The Japanese Society of Hypertension Committee for Guidelines for the Management of Hypertension: Measurement and clinical evaluation of blood pressure. Hypertens Res 2009; 32:11–23.
- Dubos RJ. Man Adapting. New Haven, CT: Yale University Press, 1980.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Agespecific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- Culleton BF, McKay DW, Campbell NR. Performance of the automated BpTRU measurement device in the assessment of white-coat hypertension and white-coat effect. Blood Press Monit 2006; 11:37–42.
- Mancia G, Parati G, Pomidossi G, Grassi G, Casadei R, Zanchetti A. Alerting reaction and rise in blood pressure during measurement by physician and nurse. Hypertension 1987; 9:209–215.
- Mancia G, Sega R, Bravi C, et al. Ambulatory blood pressure normality: results from the PAMELA study. J Hypertens 1995; 13:1377–1390.
- Ohkubo T, Kikuya M, Metoki H, et al. Prognosis of “masked” hypertension and “white-coat” hypertension detected by 24-h ambulatory blood pressure monitoring 10-year follow-up from the Ohasama study. J Am Coll Cardiol 2005; 46:508–515.
- Kotsis V, Stabouli S, Toumanidis S, et al. Target organ damage in “white coat hypertension” and “masked hypertension.” Am J Hypertens 2008; 21:393–399.
- Obara T, Ohkubo T, Funahashi J, et al. Isolated uncontrolled hypertension at home and in the office among treated hypertensive patients from the J-HOME study. J Hypertens 2005; 23:1653–1660.
- Pickering TG DK, Rafey MA, Schwartz J, Gerin W. Masked hypertension: are those with normal office but elevated ambulatory blood pressure at risk? J Hypertens 2002; 20( suppl 4):176.
- Pickering TG, Hall JE, Appel LJ. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation 2005; 111:697–716.
- Pogue V, Rahman M, Lipkowitz M, et al. Disparate estimates of hypertension control from ambulatory and clinic blood pressure measurements in hypertensive kidney disease. Hypertension 2009; 53:20–27.
- Agodoa LY, Appel L, Bakris GL, et al. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Ohkubo T, Hozawa A, Yamaguchi J, et al. Prognostic significance of the nocturnal decline in blood pressure in individuals with and without high 24-h blood pressure: the Ohasama study. J Hypertens 2002; 20:2183–2189.
- Brotman DJ, Davidson MB, Boumitri M, Vidt DG. Impaired diurnal blood pressure variation and all-cause mortality. Am J Hypertens 2008; 21:92–97.
- Lurbe E, Redon J, Kesani A, et al. Increase in nocturnal blood pressure and progression to microalbuminuria in type 1 diabetes. N Engl J Med 2002; 347:797–805.
- Davidson MB, Hix JK, Vidt DG, Brotman DJ. Association of impaired diurnal blood pressure variation with a subsequent decline in glo-merular filtration rate. Arch Intern Med 2006; 166:846–852.
- White WB, Larocca GM. Improving the utility of the nocturnal hypertension definition by using absolute sleep blood pressure rather than the “dipping” proportion. Am J Cardiol 2003; 92:1439–1441.
- Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce the white coat response. J Hypertens 2009; 27:280–286.
- Perloff D, Sokolow M, Cowan R. The prognostic value of ambulatory blood pressures. JAMA 1983; 249:2792–2798.
- Clement DL, De Buyzere ML, De Bacquer DA, et al. Prognostic value of ambulatory blood-pressure recordings in patients with treated hypertension. N Engl J Med 2003; 348:2407–2415.
- Verdecchia P, Reboldi G, Porcellati C, et al. Risk of cardiovascular disease in relation to achieved office and ambulatory blood pressure control in treated hypertensive subjects. J Am Coll Cardiol 2002; 39:878–885.
- Mansia G, De Backer G, Dominiczak A, et al. 2007 ESH-ESC Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Blood Press 2007; 16:135–232.
- Green BB, Cook AJ, Ralston JD, et al. Effectiveness of home blood pressure monitoring, Web communication, and pharmacist care on hypertension control: a randomized controlled trial. JAMA 2008; 299:2857–2867.
- Mohamed F. On chronic Bright’s disease, and its essential symptoms. Lancet 1879; 1:399–401.
- Liew Y, Rafey MA, Allam S, Arrigain S, Butler R, Schreiber M. Blood pressure goals and arterial stiffness in chronic kidney disease. J Clin Hypertens (Greenwich) 2009; 11:201–206.
- O’Rourke MF, Pauca A, Jiang XJ. Pulse wave analysis. Br J Clin Pharmacol 2001; 51:507–522.
- Najjar SS, Scuteri A, Shetty V, et al. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol 2008; 51:1377–1383.
- Sanchez RA, Ayala M, Baglivo H, et al. Latin American guidelines on hypertension. J Hypertens 2009; 27:905–922.
- Japanese Society of Hypertension. The Japanese Society of Hypertension Committee for Guidelines for the Management of Hypertension: Measurement and clinical evaluation of blood pressure. Hypertens Res 2009; 32:11–23.
- Dubos RJ. Man Adapting. New Haven, CT: Yale University Press, 1980.
KEY POINTS
- Traditional office blood pressure measurements have diagnostic limitations, since they are only snapshots of a very dynamic variable.
- Ambulatory 24-hour blood pressure monitoring is a useful and proven tool and can reveal nocturnal hypertension, a possible new marker of risk.
- Automatic devices can be used in the clinician’s office to minimize the “white coat effect” and measure blood pressure accurately.
- Pulse-wave analysis provides physiologic data on central blood pressure and arterial stiffness. This information may help in the early identification and management of patients at risk for end-organ damage.

Update on 2009 pandemic influenza A (H1N1) virus
A 69-year-old ohio man with leukemia was treated in another state in late June. During the car trip back to Ohio, he developed a sore throat, fever, cough, and nasal congestion. He was admitted to Cleveland Clinic with a presumed diagnosis of neutropenic fever; his absolute neutrophil count was 0.4 × 109/L (reference range 1.8–7.7). His chest radiograph was normal. He was treated with empiric broad-spectrum antimicrobials. On his second day in the hospital, he was tested for influenza by a polymerase chain reaction (PCR) test, which was positive for influenza A. He was moved to a private room and started on oseltamivir (Tamiflu) and rimantadine (Flumadine). The patient’s previous roommate subsequently tested positive for influenza A, as did two health care workers working on the ward. All patients on the floor received prophylactic oseltamivir.
The patient’s condition worsened, and he subsequently went into respiratory distress with diffuse pulmonary infiltrates. He was transferred to the intensive care unit, where he was intubated. Influenza A was isolated from a bronchoscopic specimen. He subsequently recovered after a prolonged course and was discharged on hospital day 50. Testing by the Ohio Department of Health confirmed that this was the 2009 pandemic influenza A (H1N1) virus.
THE CHALLENGES WE FACE
We are now in the midst of an influenza pandemic of the 2009 influenza A (H1N1) virus, with pandemic defined as “worldwide sustained community transmission.” The circulation of seasonal and 2009 pandemic influenza A (H1N1) strains will make this flu season both interesting and challenging.
The approaches to vaccination, prophylaxis, and treatment will be more complex. As of this writing (mid-September 2009), it is clear that we will be giving two influenza vaccines this season: a trivalent vaccine for seasonal influenza, and a monovalent vaccine for pandemic H1N1. It appears the monovalent vaccine may require only one dose to provide protective immunity.1 Fortunately, the vast majority of cases of pandemic H1N1 are relatively mild and uncomplicated. Still, some people are at higher risk of complications, including young patients, pregnant women, and people with immune deficiency or concomitant health conditions that put them at higher risk of flu-associated complications. Thus, clinicians will need to be educated about whom to test, who needs prophylaxis, and who should not be treated.
As our case demonstrates, unsuspected cases of influenza in hospitalized patients or health care workers working with influenza pose the greatest threat for transmission of influenza within the hospital. Adults hospitalized with influenza tend to present late (more than 48 hours after the onset of symptoms) and tend to have prolonged illness.2 Ambulatory adults shed virus for 3 to 6 days; virus shedding is more prolonged for hospitalized patients. Antiviral agents started within 4 days of illness enhance viral clearance and are associated with a shorter stay.3 Therefore, we should have a low threshold for testing for influenza and for isolating all suspected cases.
This is also creating a paradigm shift for health care workers, who are notorious for working through an illness. If you are sick, stay home! This applies whether you have pandemic H1N1 or something else.
EPIDEMIOLOGY OF PANDEMIC 2009 INFLUENZA A (H1N1) VIRUS
The location of cases can now be found on Google Maps; the US Centers for Disease Control and Prevention (CDC) provides weekly influenza reports at www.cdc.gov/flu/weekly/fluactivity.htm.
Pandemic H1N1 appeared in the spring of 2009, and cases continued to mount all summer in the United States (when influenza is normally absent) and around the world. In Mexico in March and April 2009, 2,155 cases of pneumonia, 821 hospitalizations, and 100 deaths were reported.4
In contrast with seasonal influenza, children and younger adults were hit the hardest in Mexico. The age group 5 through 59 years accounted for 87% of the deaths (usually, they account for about 17%) and 71% of the cases of severe pneumonia (usually, they account for 32%). These observations may be explained in part by the possibility that people who were alive during the 1957 pandemic (which was an H1N1 strain) have some immunity to the new virus. However, the case-fatality rate was highest in people age 65 and older.4
As of July 2009, there were more than 43,000 confirmed cases of pandemic H1N1 in the United States, and actual cases probably exceed 1 million, with more than 400 deaths. An underlying risk factor was identified in more than half of the fatal cases.5 Ten percent of the women who died were pregnant.
Pandemic H1N1 has several distinctive epidemiologic features:
- The distribution of cases is similar across multiple geographic areas.
- The distribution of cases by age group is markedly different than that of seasonal influenza, with more cases in school children and fewer cases in older adults.
- Fewer cases have been reported in older adults, but this group has the highest case-fatality rate.
2009 PANDEMIC H1N1 IS A MONGREL
There are three types of influenza viruses, designated A, B, and C. Type A undergoes antigenic shift (rapid changes) and antigenic drift (gradual changes) from year to year, and so it is the type associated with pandemics. In contrast, type B undergoes antigenic drift only, and type C is relatively stable.
Influenza virus is subtyped on the basis of surface glycoproteins: 16 hemagglutinins and nine neuraminidases. The circulating subtypes change every year; the current circulating human subtypes are a seasonal subtype of H1N1 that is different than the pandemic H1N1 subtype, and H3N2.
The 2009 pandemic H1N1 is a new virus never seen before in North America.6 Genetically, it is a mongrel, coming from three recognized sources (pigs, birds, and humans) which were combined in pigs.7 It is similar to subtypes that circulated in the 1920s through the 1940s.
Most influenza in the Western world comes from Asia every fall, and its arrival is probably facilitated by air travel. The spread is usually unidirectional and is unlikely to contribute to long-term viral evolution.8 It appears that 2009 H1N1 virus is the predominant strain circulating in the current influenza season in the Southern Hemisphere. Virologic studies indicate that the H1N1 virus strain has remained antigenically stable since it appeared in April 2009. Thus, it appears likely that the strain selected by the United States for vaccine manufacturing will match the currently circulating seasonal and pandemic H1N1 strains.
VACCINATION IS THE FIRST LINE OF DEFENSE
In addition to the trivalent vaccine against seasonal influenza, a monovalent vaccine for pandemic H1N1 virus is being produced. The CDC has indicated that 45 million doses of pandemic influenza vaccine are expected in October 2009, with an average of 20 million doses each week thereafter. It is anticipated that half of these will be in multidose vials, that 20% will be in prefilled syringes for children over 5 years old and for pregnant women, and that 20% will be in the form of live-attenuated influenza vaccine (nasal spray). The inhaled vaccine should not be given to children under 2 years old, to children under 5 years old who have recurrent wheezing, or to anyone with severe asthma. Neither vaccine should be given to people allergic to hen eggs, from which the vaccine is produced.
An ample supply of the seasonal trivalent vaccine should be available. Once the CDC has more information about specific product availability of the pandemic H1N1 vaccine, that vaccine will be distributed. It can be given concurrently with seasonal influenza vaccine.
Several definitions should be kept in mind when discussing vaccination strategies. Supply is the number of vaccine doses available for distribution. Availability is the ability of a person recommended to be vaccinated to do so in a local venue. Prioritization is the recommendation to vaccination venues to selectively use vaccine for certain population groups first. Targeting is the recommendation that immunization programs encourage and promote vaccination for certain population groups.
The Advisory Committee on Immunization Practices and the CDC recommend both seasonal and H1N1 vaccinations for anyone 6 months of age or older who is at risk of becoming ill or of transmitting the viruses to others. Based on a review of epidemiologic data, the recommendation is for targeting the following five groups for H1N1 vaccination: children and young adults aged 6 months through 24 years; pregnant women; health care workers and emergency medical service workers; people ages 25 through 64 years who have certain health conditions (eg, diabetes, heart disease, lung disease); and people who live with or care for children younger than 6 months of age. This represents approximately 159 million people in the United States.
If the estimates for the vaccine supply are met, and if pandemic H1N1 vaccine requires only a single injection, there should be no need for prioritization of vaccine. If the supply of pandemic H1N1 vaccine is inadequate, then those groups who are targeted would also receive the first doses of the pandemic H1N1 vaccine. It should be used only with caution after consideration of potential benefits and risks in people who have had Guillain-Barré syndrome during the previous 6 weeks, in people with altered immunocompetence, or in people with medical conditions predisposing to influenza complications.
A mass vaccination campaign involving two separate flu vaccines can pose challenges in execution and messaging for public health officials and politicians. In 1976, an aggressive vaccination program turned into a disaster, as there was no pandemic and the vaccine was associated with adverse effects such as Guillain-Barré syndrome. The government and the medical profession need to prepare for a vaccine controversy and to communicate and continue to explain the plan to the public. As pointed out in a recent op-ed piece,9 we would hope that all expectant women in the fall flu season will get the flu vaccines. We also know that, normally, one in seven pregnancies would be expected to miscarry. The challenge for public health officials and physicians will be to explain to these patients that there may be an association rather than a causal relationship.
In health care workers, the average vaccination rate is only 37%. We should be doing much better. Cleveland Clinic previously increased the rate of vaccination among its employees via a program in which all workers must either be vaccinated or formally declare (on an internal Web site) that they decline to be vaccinated.10 This season, even more resources are being directed at decreasing the barriers to flu vaccinations for our health care workers with the support from hospital leadership.
INFECTION CONTROL IN THE HOSPITAL AND IN THE COMMUNITY
Influenza is very contagious and is spread in droplets via sneezing and coughing (within a 3-foot radius), or via unwashed hands—thus the infection-control campaigns urging you to cover your cough and wash your hands.
As noted, for patients being admitted or transferred to the hospital, we need to have a low threshold for testing for influenza and for isolating patients suspected of having influenza. For patients with suspected or proven seasonal influenza, transmission precautions are those recommended by the CDC for droplet precautions (www.cdc.gov/ncidod/dhqp/gl_isolation_droplet.html). A face mask is deemed adequate to protect transmission when coming within 3 feet of an infected person. CDC guidelines for pandemic H1N1 recommends airborne-transmission-based precautions for health care workers who are in close contact with patients with proven or possible H1N1 (www.cdc.gov/ncidod/dhqp/gl_isolation_airborne.html). This recommendation implies the use of fit-tested N95 respirators and negative air pressure rooms (if available).
The recent Institute of Medicine report, Respiratory Protection for Healthcare Workers in the Workplace Against Novel H1N1 Influenza A (www.iom.edu/CMS/3740/71769/72967/72970.aspx) endorses the current CDC guidelines and recommends following these guidelines until we have evidence that other forms of protection or guidelines are equally or more effective.
Personally, I am against this requirement because it creates a terrible administrative burden with no proven benefit. Requiring a respirator means requiring fit-testing, and this will negatively affect our ability to deliver patient care. Recent studies have shown that surgical masks may not be as effective11 but are probably sufficient. Lim et al12 reported that 79 (37%) of 212 workers who responded to a survey experienced headaches while wearing N95 masks. This remains a controversial issue.
Besides getting the flu shot, what can one do to avoid getting influenza or transmitting to others?
- Cover your cough (cough etiquette) and sneeze.
- Practice good hand hygiene.
- Avoid close contact with people who are sick.
- Do not go to school or work if sick.
A recent study of influenza in households suggested that having the person with flu and household contacts wear face masks and practice hand hygiene within the first 36 hours decreased transmission of flu within the household.13
The United States does have a national influenza pandemic plan that outlines specific roles in the event of a pandemic, and I urge you to peruse it at www.hhs.gov/pandemicflu/plan/.
RECOGNIZING AND DIAGNOSING INFLUENZA
The familiar signs and symptoms of influenza—fever, cough, muscle aches, and headache—are nonspecific. Call et al14 analyzed the diagnostic accuracy of symptoms and signs of influenza and found that fever and cough during an epidemic suggest but do not confirm influenza, and that sneezing in those over age 60 argues against influenza. They concluded that signs and symptoms can tell us whether a patient has an influenza-like illness, but do not confirm or exclude the diagnosis of influenza: “Clinicians need to consider whether influenza is circulating in their communities, and then either treat patients with influenza-like illness empirically or obtain a rapid influenza test.”14
The signs and symptoms of pandemic 2009 H1N1 are the same as for seasonal flu, except that about 25% of patients with pandemic flu develop gastrointestinal symptoms. It has not been more virulent than seasonal influenza to date.
Should you order a test for influenza?
Most people with influenza are neither tested nor treated. Before ordering a test for influenza, ask, “Does this patient actually have influenza?” Patients diagnosed with “influenza” may have a range of infectious and noninfectious causes, such as vasculitis, endocarditis, or any other condition that can cause a fever and cough.
If I truly suspect influenza, I would still only order a test if the results would change how I manage the patient—for example, a patient being admitted to the hospital where isolation would be required.
Pandemic H1N1 will be detected only as influenza A in our current PCR screen for human influenza. The test does not differentiate between seasonal strains of influenza A (which is resistant to oseltamivir) and pandemic H1N1 (which is susceptible to oseltamivir). This means if you intend to treat, you will have to address further complexity.
Testing for influenza
The clinician should be familiar with the types of tests available. Each test has advantages and disadvantages15:
Rapid antigen assay is a point-of-care test that can give results in 15 minutes but unfortunately is only 20% to 30% sensitive, so a negative result does not exclude the diagnosis. The positive predictive value is high, meaning a positive test means the patient does have the flu.
Direct fluorescent antibody testing takes about 2.5 hours to complete and requires special training for technicians. It has a sensitivity of 47%, a positive predictive value of 95%, and a negative predictive value of 92%.
PCR testing takes about 6 hours and has a sensitivity of 98%, a positive predictive value of 100%, and a negative predictive value of 98%. This is probably the best test, in view of its all-around performance, but it is not a point-of-care test.
Culture takes 2 to 3 days, has a sensitivity of 89%, a positive predictive value of 100%, and a negative predictive value of 88%.
These tests can determine that the patient has influenza A, but a confirmatory test is always required to confirm pandemic H1N1. This confirmatory testing can be done by the CDC, by state public health laboratories, and by commercial reference laboratories.
ANTIVIRAL TREATMENT
Since influenza test results do not specify whether the patient has seasonal or pandemic influenza, treatment decisions are a sticky wicket. Most patients with pandemic H1N1 do not need to be tested or treated.
Several drugs are approved for treating influenza and shorten the duration of symptoms by about 1 day. The earlier the treatment is started, the better: the time of antiviral initiation affects influenza viral load and the duration of viral shedding.3
The neuraminidase inhibitors oseltamivir and zanamivir (Relenza) block release of virus from the cell. Resistance to oseltamivir is emerging in seasonal influenza A, while most pandemic H1N1 strains are susceptible.
Oseltamivir resistance in pandemic H1N1
A total of 11 cases of oseltamivir-resistant pandemic H1N1 have been confirmed worldwide, including 3 in the United States (2 in immunosuppressed patients in Seattle, WA). Ten of the 11 cases occurred with oseltamivir exposure. All involved a histidine-to-tyrosine substitution at position 275 (H275Y) of the neuraminidase gene. Most were susceptible to zanamivir.
Supplies of oseltamivir and zanamivir are limited, so they should be used only in those who will benefit the most, ie, those at higher risk of influenza complications. These include children under 5 years old, adults age 65 and older, children and adolescents on long-term aspirin therapy, pregnant women, patients who have chronic conditions or who are immunosuppressed, and residents of long-term care facilities.
- Greenberg MA, Lai MH, Hartel GF. Response after one dose of a monovalent influenza A (H1N1) 2009 vaccine—preliminary report. N Engl J Med 2009;361doi:10.1056/NEJMoa0907413 [published online ahead of print].
- Ison M. Influenza in hospitalized adults: gaining insight into a significant problem. J Infect Dis 2009; 200:485–488.
- Lee N, Chan PKS, Hui DSC, et al. Viral loads and duration of viral shedding in adult patients hospitalized with influenza. J Infect Dis 2009; 200:492–500.
- Chowell G, Bertozzi SM, Colchero MA, et al. Severe respiratory disease concurrent with the circulation of H1N1 influenza. N Engl J Med 2009; 361:674–679.
- Vaillant L, La Ruche G, Tarantola A, Barboza P; for the Epidemic Intelligence Team at InVS. Epidemiology of fatal cases associated with pandemic H1N1 influenza 2009. Euro Surveill 2009; 14(33):1–6. Available online at www.eurosurveillance.org/ViewArticle.aspx?ArticleID=19309.
- Zimmer SM, Burke DS. Historical perspective—emergence of influenza A (H1N1) viruses. N Engl J Med 2009; 361:279–285.
- Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009; 325:197–201.
- Russell CA, Jones TC, Barr IG, et al. The global circulation of seasonal influenza A (H3N2) viruses. Science 2008; 320:340–346.
- Allen A. Prepare for a vaccine controversy. New York Times. 9/1/2009.
- Bertin M, Scarpelli M, Proctor AW, et al. Novel use of the intranet to document health care personnel participation in a mandatory influenza vaccination reporting program. Am J Infect Control 2007; 35:33–37.
- Johnson DF, Druce JD, Birch C, Grayson ML. A quantitative assessment of the efficacy of surgical and N95 masks to filter influenza virus in patients with acute influenza infection. Clin Infect Dis 2009; 49:275–277.
- Lim EC, Seet RC, Lee KH, Wilder-Smith EP, Chuah BY, Ong BK. Headaches and the N95 face-mask amongst healthcare providers. Acta Neurol Scand 2006; 113:199–202.
- Cowling BJ, Chan KH, Fang VJ, et al. Facemasks and hand hygiene to prevent influenza transmission in households: a randomized trial. Ann Intern Med 2009; 151(6 Oct) [published online ahead of print].
- Call SA, Vollenweider MA, Hornung CA, Simel DL, McKinney WP. Does this patient have influenza? JAMA 2005; 293:987–997.
- Ginocchio CC, Zhang F, Manji R, et al. Evaluation of multiple test methods for the detection of the novel 2009 influenza A (H1N1) during the New York City outbreak. J Clin Virol 2009; 45:191–195.
- US Centers for Disease Control and Prevention. Oseltamivir-resistant novel influenza A (H1N1) virus infection in two immunosuppressed patients—Seattle, Washington, 2009. MMWR 2009; 58:893–896.
A 69-year-old ohio man with leukemia was treated in another state in late June. During the car trip back to Ohio, he developed a sore throat, fever, cough, and nasal congestion. He was admitted to Cleveland Clinic with a presumed diagnosis of neutropenic fever; his absolute neutrophil count was 0.4 × 109/L (reference range 1.8–7.7). His chest radiograph was normal. He was treated with empiric broad-spectrum antimicrobials. On his second day in the hospital, he was tested for influenza by a polymerase chain reaction (PCR) test, which was positive for influenza A. He was moved to a private room and started on oseltamivir (Tamiflu) and rimantadine (Flumadine). The patient’s previous roommate subsequently tested positive for influenza A, as did two health care workers working on the ward. All patients on the floor received prophylactic oseltamivir.
The patient’s condition worsened, and he subsequently went into respiratory distress with diffuse pulmonary infiltrates. He was transferred to the intensive care unit, where he was intubated. Influenza A was isolated from a bronchoscopic specimen. He subsequently recovered after a prolonged course and was discharged on hospital day 50. Testing by the Ohio Department of Health confirmed that this was the 2009 pandemic influenza A (H1N1) virus.
THE CHALLENGES WE FACE
We are now in the midst of an influenza pandemic of the 2009 influenza A (H1N1) virus, with pandemic defined as “worldwide sustained community transmission.” The circulation of seasonal and 2009 pandemic influenza A (H1N1) strains will make this flu season both interesting and challenging.
The approaches to vaccination, prophylaxis, and treatment will be more complex. As of this writing (mid-September 2009), it is clear that we will be giving two influenza vaccines this season: a trivalent vaccine for seasonal influenza, and a monovalent vaccine for pandemic H1N1. It appears the monovalent vaccine may require only one dose to provide protective immunity.1 Fortunately, the vast majority of cases of pandemic H1N1 are relatively mild and uncomplicated. Still, some people are at higher risk of complications, including young patients, pregnant women, and people with immune deficiency or concomitant health conditions that put them at higher risk of flu-associated complications. Thus, clinicians will need to be educated about whom to test, who needs prophylaxis, and who should not be treated.
As our case demonstrates, unsuspected cases of influenza in hospitalized patients or health care workers working with influenza pose the greatest threat for transmission of influenza within the hospital. Adults hospitalized with influenza tend to present late (more than 48 hours after the onset of symptoms) and tend to have prolonged illness.2 Ambulatory adults shed virus for 3 to 6 days; virus shedding is more prolonged for hospitalized patients. Antiviral agents started within 4 days of illness enhance viral clearance and are associated with a shorter stay.3 Therefore, we should have a low threshold for testing for influenza and for isolating all suspected cases.
This is also creating a paradigm shift for health care workers, who are notorious for working through an illness. If you are sick, stay home! This applies whether you have pandemic H1N1 or something else.
EPIDEMIOLOGY OF PANDEMIC 2009 INFLUENZA A (H1N1) VIRUS
The location of cases can now be found on Google Maps; the US Centers for Disease Control and Prevention (CDC) provides weekly influenza reports at www.cdc.gov/flu/weekly/fluactivity.htm.
Pandemic H1N1 appeared in the spring of 2009, and cases continued to mount all summer in the United States (when influenza is normally absent) and around the world. In Mexico in March and April 2009, 2,155 cases of pneumonia, 821 hospitalizations, and 100 deaths were reported.4
In contrast with seasonal influenza, children and younger adults were hit the hardest in Mexico. The age group 5 through 59 years accounted for 87% of the deaths (usually, they account for about 17%) and 71% of the cases of severe pneumonia (usually, they account for 32%). These observations may be explained in part by the possibility that people who were alive during the 1957 pandemic (which was an H1N1 strain) have some immunity to the new virus. However, the case-fatality rate was highest in people age 65 and older.4
As of July 2009, there were more than 43,000 confirmed cases of pandemic H1N1 in the United States, and actual cases probably exceed 1 million, with more than 400 deaths. An underlying risk factor was identified in more than half of the fatal cases.5 Ten percent of the women who died were pregnant.
Pandemic H1N1 has several distinctive epidemiologic features:
- The distribution of cases is similar across multiple geographic areas.
- The distribution of cases by age group is markedly different than that of seasonal influenza, with more cases in school children and fewer cases in older adults.
- Fewer cases have been reported in older adults, but this group has the highest case-fatality rate.
2009 PANDEMIC H1N1 IS A MONGREL
There are three types of influenza viruses, designated A, B, and C. Type A undergoes antigenic shift (rapid changes) and antigenic drift (gradual changes) from year to year, and so it is the type associated with pandemics. In contrast, type B undergoes antigenic drift only, and type C is relatively stable.
Influenza virus is subtyped on the basis of surface glycoproteins: 16 hemagglutinins and nine neuraminidases. The circulating subtypes change every year; the current circulating human subtypes are a seasonal subtype of H1N1 that is different than the pandemic H1N1 subtype, and H3N2.
The 2009 pandemic H1N1 is a new virus never seen before in North America.6 Genetically, it is a mongrel, coming from three recognized sources (pigs, birds, and humans) which were combined in pigs.7 It is similar to subtypes that circulated in the 1920s through the 1940s.
Most influenza in the Western world comes from Asia every fall, and its arrival is probably facilitated by air travel. The spread is usually unidirectional and is unlikely to contribute to long-term viral evolution.8 It appears that 2009 H1N1 virus is the predominant strain circulating in the current influenza season in the Southern Hemisphere. Virologic studies indicate that the H1N1 virus strain has remained antigenically stable since it appeared in April 2009. Thus, it appears likely that the strain selected by the United States for vaccine manufacturing will match the currently circulating seasonal and pandemic H1N1 strains.
VACCINATION IS THE FIRST LINE OF DEFENSE
In addition to the trivalent vaccine against seasonal influenza, a monovalent vaccine for pandemic H1N1 virus is being produced. The CDC has indicated that 45 million doses of pandemic influenza vaccine are expected in October 2009, with an average of 20 million doses each week thereafter. It is anticipated that half of these will be in multidose vials, that 20% will be in prefilled syringes for children over 5 years old and for pregnant women, and that 20% will be in the form of live-attenuated influenza vaccine (nasal spray). The inhaled vaccine should not be given to children under 2 years old, to children under 5 years old who have recurrent wheezing, or to anyone with severe asthma. Neither vaccine should be given to people allergic to hen eggs, from which the vaccine is produced.
An ample supply of the seasonal trivalent vaccine should be available. Once the CDC has more information about specific product availability of the pandemic H1N1 vaccine, that vaccine will be distributed. It can be given concurrently with seasonal influenza vaccine.
Several definitions should be kept in mind when discussing vaccination strategies. Supply is the number of vaccine doses available for distribution. Availability is the ability of a person recommended to be vaccinated to do so in a local venue. Prioritization is the recommendation to vaccination venues to selectively use vaccine for certain population groups first. Targeting is the recommendation that immunization programs encourage and promote vaccination for certain population groups.
The Advisory Committee on Immunization Practices and the CDC recommend both seasonal and H1N1 vaccinations for anyone 6 months of age or older who is at risk of becoming ill or of transmitting the viruses to others. Based on a review of epidemiologic data, the recommendation is for targeting the following five groups for H1N1 vaccination: children and young adults aged 6 months through 24 years; pregnant women; health care workers and emergency medical service workers; people ages 25 through 64 years who have certain health conditions (eg, diabetes, heart disease, lung disease); and people who live with or care for children younger than 6 months of age. This represents approximately 159 million people in the United States.
If the estimates for the vaccine supply are met, and if pandemic H1N1 vaccine requires only a single injection, there should be no need for prioritization of vaccine. If the supply of pandemic H1N1 vaccine is inadequate, then those groups who are targeted would also receive the first doses of the pandemic H1N1 vaccine. It should be used only with caution after consideration of potential benefits and risks in people who have had Guillain-Barré syndrome during the previous 6 weeks, in people with altered immunocompetence, or in people with medical conditions predisposing to influenza complications.
A mass vaccination campaign involving two separate flu vaccines can pose challenges in execution and messaging for public health officials and politicians. In 1976, an aggressive vaccination program turned into a disaster, as there was no pandemic and the vaccine was associated with adverse effects such as Guillain-Barré syndrome. The government and the medical profession need to prepare for a vaccine controversy and to communicate and continue to explain the plan to the public. As pointed out in a recent op-ed piece,9 we would hope that all expectant women in the fall flu season will get the flu vaccines. We also know that, normally, one in seven pregnancies would be expected to miscarry. The challenge for public health officials and physicians will be to explain to these patients that there may be an association rather than a causal relationship.
In health care workers, the average vaccination rate is only 37%. We should be doing much better. Cleveland Clinic previously increased the rate of vaccination among its employees via a program in which all workers must either be vaccinated or formally declare (on an internal Web site) that they decline to be vaccinated.10 This season, even more resources are being directed at decreasing the barriers to flu vaccinations for our health care workers with the support from hospital leadership.
INFECTION CONTROL IN THE HOSPITAL AND IN THE COMMUNITY
Influenza is very contagious and is spread in droplets via sneezing and coughing (within a 3-foot radius), or via unwashed hands—thus the infection-control campaigns urging you to cover your cough and wash your hands.
As noted, for patients being admitted or transferred to the hospital, we need to have a low threshold for testing for influenza and for isolating patients suspected of having influenza. For patients with suspected or proven seasonal influenza, transmission precautions are those recommended by the CDC for droplet precautions (www.cdc.gov/ncidod/dhqp/gl_isolation_droplet.html). A face mask is deemed adequate to protect transmission when coming within 3 feet of an infected person. CDC guidelines for pandemic H1N1 recommends airborne-transmission-based precautions for health care workers who are in close contact with patients with proven or possible H1N1 (www.cdc.gov/ncidod/dhqp/gl_isolation_airborne.html). This recommendation implies the use of fit-tested N95 respirators and negative air pressure rooms (if available).
The recent Institute of Medicine report, Respiratory Protection for Healthcare Workers in the Workplace Against Novel H1N1 Influenza A (www.iom.edu/CMS/3740/71769/72967/72970.aspx) endorses the current CDC guidelines and recommends following these guidelines until we have evidence that other forms of protection or guidelines are equally or more effective.
Personally, I am against this requirement because it creates a terrible administrative burden with no proven benefit. Requiring a respirator means requiring fit-testing, and this will negatively affect our ability to deliver patient care. Recent studies have shown that surgical masks may not be as effective11 but are probably sufficient. Lim et al12 reported that 79 (37%) of 212 workers who responded to a survey experienced headaches while wearing N95 masks. This remains a controversial issue.
Besides getting the flu shot, what can one do to avoid getting influenza or transmitting to others?
- Cover your cough (cough etiquette) and sneeze.
- Practice good hand hygiene.
- Avoid close contact with people who are sick.
- Do not go to school or work if sick.
A recent study of influenza in households suggested that having the person with flu and household contacts wear face masks and practice hand hygiene within the first 36 hours decreased transmission of flu within the household.13
The United States does have a national influenza pandemic plan that outlines specific roles in the event of a pandemic, and I urge you to peruse it at www.hhs.gov/pandemicflu/plan/.
RECOGNIZING AND DIAGNOSING INFLUENZA
The familiar signs and symptoms of influenza—fever, cough, muscle aches, and headache—are nonspecific. Call et al14 analyzed the diagnostic accuracy of symptoms and signs of influenza and found that fever and cough during an epidemic suggest but do not confirm influenza, and that sneezing in those over age 60 argues against influenza. They concluded that signs and symptoms can tell us whether a patient has an influenza-like illness, but do not confirm or exclude the diagnosis of influenza: “Clinicians need to consider whether influenza is circulating in their communities, and then either treat patients with influenza-like illness empirically or obtain a rapid influenza test.”14
The signs and symptoms of pandemic 2009 H1N1 are the same as for seasonal flu, except that about 25% of patients with pandemic flu develop gastrointestinal symptoms. It has not been more virulent than seasonal influenza to date.
Should you order a test for influenza?
Most people with influenza are neither tested nor treated. Before ordering a test for influenza, ask, “Does this patient actually have influenza?” Patients diagnosed with “influenza” may have a range of infectious and noninfectious causes, such as vasculitis, endocarditis, or any other condition that can cause a fever and cough.
If I truly suspect influenza, I would still only order a test if the results would change how I manage the patient—for example, a patient being admitted to the hospital where isolation would be required.
Pandemic H1N1 will be detected only as influenza A in our current PCR screen for human influenza. The test does not differentiate between seasonal strains of influenza A (which is resistant to oseltamivir) and pandemic H1N1 (which is susceptible to oseltamivir). This means if you intend to treat, you will have to address further complexity.
Testing for influenza
The clinician should be familiar with the types of tests available. Each test has advantages and disadvantages15:
Rapid antigen assay is a point-of-care test that can give results in 15 minutes but unfortunately is only 20% to 30% sensitive, so a negative result does not exclude the diagnosis. The positive predictive value is high, meaning a positive test means the patient does have the flu.
Direct fluorescent antibody testing takes about 2.5 hours to complete and requires special training for technicians. It has a sensitivity of 47%, a positive predictive value of 95%, and a negative predictive value of 92%.
PCR testing takes about 6 hours and has a sensitivity of 98%, a positive predictive value of 100%, and a negative predictive value of 98%. This is probably the best test, in view of its all-around performance, but it is not a point-of-care test.
Culture takes 2 to 3 days, has a sensitivity of 89%, a positive predictive value of 100%, and a negative predictive value of 88%.
These tests can determine that the patient has influenza A, but a confirmatory test is always required to confirm pandemic H1N1. This confirmatory testing can be done by the CDC, by state public health laboratories, and by commercial reference laboratories.
ANTIVIRAL TREATMENT
Since influenza test results do not specify whether the patient has seasonal or pandemic influenza, treatment decisions are a sticky wicket. Most patients with pandemic H1N1 do not need to be tested or treated.
Several drugs are approved for treating influenza and shorten the duration of symptoms by about 1 day. The earlier the treatment is started, the better: the time of antiviral initiation affects influenza viral load and the duration of viral shedding.3
The neuraminidase inhibitors oseltamivir and zanamivir (Relenza) block release of virus from the cell. Resistance to oseltamivir is emerging in seasonal influenza A, while most pandemic H1N1 strains are susceptible.
Oseltamivir resistance in pandemic H1N1
A total of 11 cases of oseltamivir-resistant pandemic H1N1 have been confirmed worldwide, including 3 in the United States (2 in immunosuppressed patients in Seattle, WA). Ten of the 11 cases occurred with oseltamivir exposure. All involved a histidine-to-tyrosine substitution at position 275 (H275Y) of the neuraminidase gene. Most were susceptible to zanamivir.
Supplies of oseltamivir and zanamivir are limited, so they should be used only in those who will benefit the most, ie, those at higher risk of influenza complications. These include children under 5 years old, adults age 65 and older, children and adolescents on long-term aspirin therapy, pregnant women, patients who have chronic conditions or who are immunosuppressed, and residents of long-term care facilities.
A 69-year-old ohio man with leukemia was treated in another state in late June. During the car trip back to Ohio, he developed a sore throat, fever, cough, and nasal congestion. He was admitted to Cleveland Clinic with a presumed diagnosis of neutropenic fever; his absolute neutrophil count was 0.4 × 109/L (reference range 1.8–7.7). His chest radiograph was normal. He was treated with empiric broad-spectrum antimicrobials. On his second day in the hospital, he was tested for influenza by a polymerase chain reaction (PCR) test, which was positive for influenza A. He was moved to a private room and started on oseltamivir (Tamiflu) and rimantadine (Flumadine). The patient’s previous roommate subsequently tested positive for influenza A, as did two health care workers working on the ward. All patients on the floor received prophylactic oseltamivir.
The patient’s condition worsened, and he subsequently went into respiratory distress with diffuse pulmonary infiltrates. He was transferred to the intensive care unit, where he was intubated. Influenza A was isolated from a bronchoscopic specimen. He subsequently recovered after a prolonged course and was discharged on hospital day 50. Testing by the Ohio Department of Health confirmed that this was the 2009 pandemic influenza A (H1N1) virus.
THE CHALLENGES WE FACE
We are now in the midst of an influenza pandemic of the 2009 influenza A (H1N1) virus, with pandemic defined as “worldwide sustained community transmission.” The circulation of seasonal and 2009 pandemic influenza A (H1N1) strains will make this flu season both interesting and challenging.
The approaches to vaccination, prophylaxis, and treatment will be more complex. As of this writing (mid-September 2009), it is clear that we will be giving two influenza vaccines this season: a trivalent vaccine for seasonal influenza, and a monovalent vaccine for pandemic H1N1. It appears the monovalent vaccine may require only one dose to provide protective immunity.1 Fortunately, the vast majority of cases of pandemic H1N1 are relatively mild and uncomplicated. Still, some people are at higher risk of complications, including young patients, pregnant women, and people with immune deficiency or concomitant health conditions that put them at higher risk of flu-associated complications. Thus, clinicians will need to be educated about whom to test, who needs prophylaxis, and who should not be treated.
As our case demonstrates, unsuspected cases of influenza in hospitalized patients or health care workers working with influenza pose the greatest threat for transmission of influenza within the hospital. Adults hospitalized with influenza tend to present late (more than 48 hours after the onset of symptoms) and tend to have prolonged illness.2 Ambulatory adults shed virus for 3 to 6 days; virus shedding is more prolonged for hospitalized patients. Antiviral agents started within 4 days of illness enhance viral clearance and are associated with a shorter stay.3 Therefore, we should have a low threshold for testing for influenza and for isolating all suspected cases.
This is also creating a paradigm shift for health care workers, who are notorious for working through an illness. If you are sick, stay home! This applies whether you have pandemic H1N1 or something else.
EPIDEMIOLOGY OF PANDEMIC 2009 INFLUENZA A (H1N1) VIRUS
The location of cases can now be found on Google Maps; the US Centers for Disease Control and Prevention (CDC) provides weekly influenza reports at www.cdc.gov/flu/weekly/fluactivity.htm.
Pandemic H1N1 appeared in the spring of 2009, and cases continued to mount all summer in the United States (when influenza is normally absent) and around the world. In Mexico in March and April 2009, 2,155 cases of pneumonia, 821 hospitalizations, and 100 deaths were reported.4
In contrast with seasonal influenza, children and younger adults were hit the hardest in Mexico. The age group 5 through 59 years accounted for 87% of the deaths (usually, they account for about 17%) and 71% of the cases of severe pneumonia (usually, they account for 32%). These observations may be explained in part by the possibility that people who were alive during the 1957 pandemic (which was an H1N1 strain) have some immunity to the new virus. However, the case-fatality rate was highest in people age 65 and older.4
As of July 2009, there were more than 43,000 confirmed cases of pandemic H1N1 in the United States, and actual cases probably exceed 1 million, with more than 400 deaths. An underlying risk factor was identified in more than half of the fatal cases.5 Ten percent of the women who died were pregnant.
Pandemic H1N1 has several distinctive epidemiologic features:
- The distribution of cases is similar across multiple geographic areas.
- The distribution of cases by age group is markedly different than that of seasonal influenza, with more cases in school children and fewer cases in older adults.
- Fewer cases have been reported in older adults, but this group has the highest case-fatality rate.
2009 PANDEMIC H1N1 IS A MONGREL
There are three types of influenza viruses, designated A, B, and C. Type A undergoes antigenic shift (rapid changes) and antigenic drift (gradual changes) from year to year, and so it is the type associated with pandemics. In contrast, type B undergoes antigenic drift only, and type C is relatively stable.
Influenza virus is subtyped on the basis of surface glycoproteins: 16 hemagglutinins and nine neuraminidases. The circulating subtypes change every year; the current circulating human subtypes are a seasonal subtype of H1N1 that is different than the pandemic H1N1 subtype, and H3N2.
The 2009 pandemic H1N1 is a new virus never seen before in North America.6 Genetically, it is a mongrel, coming from three recognized sources (pigs, birds, and humans) which were combined in pigs.7 It is similar to subtypes that circulated in the 1920s through the 1940s.
Most influenza in the Western world comes from Asia every fall, and its arrival is probably facilitated by air travel. The spread is usually unidirectional and is unlikely to contribute to long-term viral evolution.8 It appears that 2009 H1N1 virus is the predominant strain circulating in the current influenza season in the Southern Hemisphere. Virologic studies indicate that the H1N1 virus strain has remained antigenically stable since it appeared in April 2009. Thus, it appears likely that the strain selected by the United States for vaccine manufacturing will match the currently circulating seasonal and pandemic H1N1 strains.
VACCINATION IS THE FIRST LINE OF DEFENSE
In addition to the trivalent vaccine against seasonal influenza, a monovalent vaccine for pandemic H1N1 virus is being produced. The CDC has indicated that 45 million doses of pandemic influenza vaccine are expected in October 2009, with an average of 20 million doses each week thereafter. It is anticipated that half of these will be in multidose vials, that 20% will be in prefilled syringes for children over 5 years old and for pregnant women, and that 20% will be in the form of live-attenuated influenza vaccine (nasal spray). The inhaled vaccine should not be given to children under 2 years old, to children under 5 years old who have recurrent wheezing, or to anyone with severe asthma. Neither vaccine should be given to people allergic to hen eggs, from which the vaccine is produced.
An ample supply of the seasonal trivalent vaccine should be available. Once the CDC has more information about specific product availability of the pandemic H1N1 vaccine, that vaccine will be distributed. It can be given concurrently with seasonal influenza vaccine.
Several definitions should be kept in mind when discussing vaccination strategies. Supply is the number of vaccine doses available for distribution. Availability is the ability of a person recommended to be vaccinated to do so in a local venue. Prioritization is the recommendation to vaccination venues to selectively use vaccine for certain population groups first. Targeting is the recommendation that immunization programs encourage and promote vaccination for certain population groups.
The Advisory Committee on Immunization Practices and the CDC recommend both seasonal and H1N1 vaccinations for anyone 6 months of age or older who is at risk of becoming ill or of transmitting the viruses to others. Based on a review of epidemiologic data, the recommendation is for targeting the following five groups for H1N1 vaccination: children and young adults aged 6 months through 24 years; pregnant women; health care workers and emergency medical service workers; people ages 25 through 64 years who have certain health conditions (eg, diabetes, heart disease, lung disease); and people who live with or care for children younger than 6 months of age. This represents approximately 159 million people in the United States.
If the estimates for the vaccine supply are met, and if pandemic H1N1 vaccine requires only a single injection, there should be no need for prioritization of vaccine. If the supply of pandemic H1N1 vaccine is inadequate, then those groups who are targeted would also receive the first doses of the pandemic H1N1 vaccine. It should be used only with caution after consideration of potential benefits and risks in people who have had Guillain-Barré syndrome during the previous 6 weeks, in people with altered immunocompetence, or in people with medical conditions predisposing to influenza complications.
A mass vaccination campaign involving two separate flu vaccines can pose challenges in execution and messaging for public health officials and politicians. In 1976, an aggressive vaccination program turned into a disaster, as there was no pandemic and the vaccine was associated with adverse effects such as Guillain-Barré syndrome. The government and the medical profession need to prepare for a vaccine controversy and to communicate and continue to explain the plan to the public. As pointed out in a recent op-ed piece,9 we would hope that all expectant women in the fall flu season will get the flu vaccines. We also know that, normally, one in seven pregnancies would be expected to miscarry. The challenge for public health officials and physicians will be to explain to these patients that there may be an association rather than a causal relationship.
In health care workers, the average vaccination rate is only 37%. We should be doing much better. Cleveland Clinic previously increased the rate of vaccination among its employees via a program in which all workers must either be vaccinated or formally declare (on an internal Web site) that they decline to be vaccinated.10 This season, even more resources are being directed at decreasing the barriers to flu vaccinations for our health care workers with the support from hospital leadership.
INFECTION CONTROL IN THE HOSPITAL AND IN THE COMMUNITY
Influenza is very contagious and is spread in droplets via sneezing and coughing (within a 3-foot radius), or via unwashed hands—thus the infection-control campaigns urging you to cover your cough and wash your hands.
As noted, for patients being admitted or transferred to the hospital, we need to have a low threshold for testing for influenza and for isolating patients suspected of having influenza. For patients with suspected or proven seasonal influenza, transmission precautions are those recommended by the CDC for droplet precautions (www.cdc.gov/ncidod/dhqp/gl_isolation_droplet.html). A face mask is deemed adequate to protect transmission when coming within 3 feet of an infected person. CDC guidelines for pandemic H1N1 recommends airborne-transmission-based precautions for health care workers who are in close contact with patients with proven or possible H1N1 (www.cdc.gov/ncidod/dhqp/gl_isolation_airborne.html). This recommendation implies the use of fit-tested N95 respirators and negative air pressure rooms (if available).
The recent Institute of Medicine report, Respiratory Protection for Healthcare Workers in the Workplace Against Novel H1N1 Influenza A (www.iom.edu/CMS/3740/71769/72967/72970.aspx) endorses the current CDC guidelines and recommends following these guidelines until we have evidence that other forms of protection or guidelines are equally or more effective.
Personally, I am against this requirement because it creates a terrible administrative burden with no proven benefit. Requiring a respirator means requiring fit-testing, and this will negatively affect our ability to deliver patient care. Recent studies have shown that surgical masks may not be as effective11 but are probably sufficient. Lim et al12 reported that 79 (37%) of 212 workers who responded to a survey experienced headaches while wearing N95 masks. This remains a controversial issue.
Besides getting the flu shot, what can one do to avoid getting influenza or transmitting to others?
- Cover your cough (cough etiquette) and sneeze.
- Practice good hand hygiene.
- Avoid close contact with people who are sick.
- Do not go to school or work if sick.
A recent study of influenza in households suggested that having the person with flu and household contacts wear face masks and practice hand hygiene within the first 36 hours decreased transmission of flu within the household.13
The United States does have a national influenza pandemic plan that outlines specific roles in the event of a pandemic, and I urge you to peruse it at www.hhs.gov/pandemicflu/plan/.
RECOGNIZING AND DIAGNOSING INFLUENZA
The familiar signs and symptoms of influenza—fever, cough, muscle aches, and headache—are nonspecific. Call et al14 analyzed the diagnostic accuracy of symptoms and signs of influenza and found that fever and cough during an epidemic suggest but do not confirm influenza, and that sneezing in those over age 60 argues against influenza. They concluded that signs and symptoms can tell us whether a patient has an influenza-like illness, but do not confirm or exclude the diagnosis of influenza: “Clinicians need to consider whether influenza is circulating in their communities, and then either treat patients with influenza-like illness empirically or obtain a rapid influenza test.”14
The signs and symptoms of pandemic 2009 H1N1 are the same as for seasonal flu, except that about 25% of patients with pandemic flu develop gastrointestinal symptoms. It has not been more virulent than seasonal influenza to date.
Should you order a test for influenza?
Most people with influenza are neither tested nor treated. Before ordering a test for influenza, ask, “Does this patient actually have influenza?” Patients diagnosed with “influenza” may have a range of infectious and noninfectious causes, such as vasculitis, endocarditis, or any other condition that can cause a fever and cough.
If I truly suspect influenza, I would still only order a test if the results would change how I manage the patient—for example, a patient being admitted to the hospital where isolation would be required.
Pandemic H1N1 will be detected only as influenza A in our current PCR screen for human influenza. The test does not differentiate between seasonal strains of influenza A (which is resistant to oseltamivir) and pandemic H1N1 (which is susceptible to oseltamivir). This means if you intend to treat, you will have to address further complexity.
Testing for influenza
The clinician should be familiar with the types of tests available. Each test has advantages and disadvantages15:
Rapid antigen assay is a point-of-care test that can give results in 15 minutes but unfortunately is only 20% to 30% sensitive, so a negative result does not exclude the diagnosis. The positive predictive value is high, meaning a positive test means the patient does have the flu.
Direct fluorescent antibody testing takes about 2.5 hours to complete and requires special training for technicians. It has a sensitivity of 47%, a positive predictive value of 95%, and a negative predictive value of 92%.
PCR testing takes about 6 hours and has a sensitivity of 98%, a positive predictive value of 100%, and a negative predictive value of 98%. This is probably the best test, in view of its all-around performance, but it is not a point-of-care test.
Culture takes 2 to 3 days, has a sensitivity of 89%, a positive predictive value of 100%, and a negative predictive value of 88%.
These tests can determine that the patient has influenza A, but a confirmatory test is always required to confirm pandemic H1N1. This confirmatory testing can be done by the CDC, by state public health laboratories, and by commercial reference laboratories.
ANTIVIRAL TREATMENT
Since influenza test results do not specify whether the patient has seasonal or pandemic influenza, treatment decisions are a sticky wicket. Most patients with pandemic H1N1 do not need to be tested or treated.
Several drugs are approved for treating influenza and shorten the duration of symptoms by about 1 day. The earlier the treatment is started, the better: the time of antiviral initiation affects influenza viral load and the duration of viral shedding.3
The neuraminidase inhibitors oseltamivir and zanamivir (Relenza) block release of virus from the cell. Resistance to oseltamivir is emerging in seasonal influenza A, while most pandemic H1N1 strains are susceptible.
Oseltamivir resistance in pandemic H1N1
A total of 11 cases of oseltamivir-resistant pandemic H1N1 have been confirmed worldwide, including 3 in the United States (2 in immunosuppressed patients in Seattle, WA). Ten of the 11 cases occurred with oseltamivir exposure. All involved a histidine-to-tyrosine substitution at position 275 (H275Y) of the neuraminidase gene. Most were susceptible to zanamivir.
Supplies of oseltamivir and zanamivir are limited, so they should be used only in those who will benefit the most, ie, those at higher risk of influenza complications. These include children under 5 years old, adults age 65 and older, children and adolescents on long-term aspirin therapy, pregnant women, patients who have chronic conditions or who are immunosuppressed, and residents of long-term care facilities.
- Greenberg MA, Lai MH, Hartel GF. Response after one dose of a monovalent influenza A (H1N1) 2009 vaccine—preliminary report. N Engl J Med 2009;361doi:10.1056/NEJMoa0907413 [published online ahead of print].
- Ison M. Influenza in hospitalized adults: gaining insight into a significant problem. J Infect Dis 2009; 200:485–488.
- Lee N, Chan PKS, Hui DSC, et al. Viral loads and duration of viral shedding in adult patients hospitalized with influenza. J Infect Dis 2009; 200:492–500.
- Chowell G, Bertozzi SM, Colchero MA, et al. Severe respiratory disease concurrent with the circulation of H1N1 influenza. N Engl J Med 2009; 361:674–679.
- Vaillant L, La Ruche G, Tarantola A, Barboza P; for the Epidemic Intelligence Team at InVS. Epidemiology of fatal cases associated with pandemic H1N1 influenza 2009. Euro Surveill 2009; 14(33):1–6. Available online at www.eurosurveillance.org/ViewArticle.aspx?ArticleID=19309.
- Zimmer SM, Burke DS. Historical perspective—emergence of influenza A (H1N1) viruses. N Engl J Med 2009; 361:279–285.
- Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009; 325:197–201.
- Russell CA, Jones TC, Barr IG, et al. The global circulation of seasonal influenza A (H3N2) viruses. Science 2008; 320:340–346.
- Allen A. Prepare for a vaccine controversy. New York Times. 9/1/2009.
- Bertin M, Scarpelli M, Proctor AW, et al. Novel use of the intranet to document health care personnel participation in a mandatory influenza vaccination reporting program. Am J Infect Control 2007; 35:33–37.
- Johnson DF, Druce JD, Birch C, Grayson ML. A quantitative assessment of the efficacy of surgical and N95 masks to filter influenza virus in patients with acute influenza infection. Clin Infect Dis 2009; 49:275–277.
- Lim EC, Seet RC, Lee KH, Wilder-Smith EP, Chuah BY, Ong BK. Headaches and the N95 face-mask amongst healthcare providers. Acta Neurol Scand 2006; 113:199–202.
- Cowling BJ, Chan KH, Fang VJ, et al. Facemasks and hand hygiene to prevent influenza transmission in households: a randomized trial. Ann Intern Med 2009; 151(6 Oct) [published online ahead of print].
- Call SA, Vollenweider MA, Hornung CA, Simel DL, McKinney WP. Does this patient have influenza? JAMA 2005; 293:987–997.
- Ginocchio CC, Zhang F, Manji R, et al. Evaluation of multiple test methods for the detection of the novel 2009 influenza A (H1N1) during the New York City outbreak. J Clin Virol 2009; 45:191–195.
- US Centers for Disease Control and Prevention. Oseltamivir-resistant novel influenza A (H1N1) virus infection in two immunosuppressed patients—Seattle, Washington, 2009. MMWR 2009; 58:893–896.
- Greenberg MA, Lai MH, Hartel GF. Response after one dose of a monovalent influenza A (H1N1) 2009 vaccine—preliminary report. N Engl J Med 2009;361doi:10.1056/NEJMoa0907413 [published online ahead of print].
- Ison M. Influenza in hospitalized adults: gaining insight into a significant problem. J Infect Dis 2009; 200:485–488.
- Lee N, Chan PKS, Hui DSC, et al. Viral loads and duration of viral shedding in adult patients hospitalized with influenza. J Infect Dis 2009; 200:492–500.
- Chowell G, Bertozzi SM, Colchero MA, et al. Severe respiratory disease concurrent with the circulation of H1N1 influenza. N Engl J Med 2009; 361:674–679.
- Vaillant L, La Ruche G, Tarantola A, Barboza P; for the Epidemic Intelligence Team at InVS. Epidemiology of fatal cases associated with pandemic H1N1 influenza 2009. Euro Surveill 2009; 14(33):1–6. Available online at www.eurosurveillance.org/ViewArticle.aspx?ArticleID=19309.
- Zimmer SM, Burke DS. Historical perspective—emergence of influenza A (H1N1) viruses. N Engl J Med 2009; 361:279–285.
- Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009; 325:197–201.
- Russell CA, Jones TC, Barr IG, et al. The global circulation of seasonal influenza A (H3N2) viruses. Science 2008; 320:340–346.
- Allen A. Prepare for a vaccine controversy. New York Times. 9/1/2009.
- Bertin M, Scarpelli M, Proctor AW, et al. Novel use of the intranet to document health care personnel participation in a mandatory influenza vaccination reporting program. Am J Infect Control 2007; 35:33–37.
- Johnson DF, Druce JD, Birch C, Grayson ML. A quantitative assessment of the efficacy of surgical and N95 masks to filter influenza virus in patients with acute influenza infection. Clin Infect Dis 2009; 49:275–277.
- Lim EC, Seet RC, Lee KH, Wilder-Smith EP, Chuah BY, Ong BK. Headaches and the N95 face-mask amongst healthcare providers. Acta Neurol Scand 2006; 113:199–202.
- Cowling BJ, Chan KH, Fang VJ, et al. Facemasks and hand hygiene to prevent influenza transmission in households: a randomized trial. Ann Intern Med 2009; 151(6 Oct) [published online ahead of print].
- Call SA, Vollenweider MA, Hornung CA, Simel DL, McKinney WP. Does this patient have influenza? JAMA 2005; 293:987–997.
- Ginocchio CC, Zhang F, Manji R, et al. Evaluation of multiple test methods for the detection of the novel 2009 influenza A (H1N1) during the New York City outbreak. J Clin Virol 2009; 45:191–195.
- US Centers for Disease Control and Prevention. Oseltamivir-resistant novel influenza A (H1N1) virus infection in two immunosuppressed patients—Seattle, Washington, 2009. MMWR 2009; 58:893–896.
KEY POINTS
- Vaccination this season will require two vaccines: a trivalent vaccine for seasonal influenza and a monovalent vaccine for 2009 pandemic influenza A (H1N1).
- Recent studies indicate that the monovalent vaccine for 2009 pandemic influenza A (H1N1) may require only one injection.
- To date, 2009 pandemic influenza A (H1N1) virus has not been exceptionally virulent and differs from conventional influenza in that it seems to disproportionately affect children and young adults. Pregnant women are at a higher risk of complications.
- Most people with 2009 pandemic influenza A (H1N1) do not need to be tested, treated, or seen by a clinician.
- Antiviral drugs should be reserved only for those at high risk of influenza complications.
What’s new in prostate cancer screening and prevention?
In spite of some recent studies, or perhaps because of them, we still are unsure about how best to screen for and prevent prostate cancer. Two large trials of screening with prostate-specific antigen (PSA) measurements came to seemingly opposite conclusions.1,2 Furthermore, a large trial of selenium and vitamin E found that these agents have no value as preventive agents.3
Nevertheless, negative studies also advance science, and steady progress is being made in prostate cancer research. In this paper I briefly summarize and comment on some of the recent findings.
TO SCREEN OR NOT TO SCREEN?
All cases of prostate cancer are clinically relevant in that they can cause anxiety or can lead to treatment-related morbidity. The challenge is to detect the minority of cases of cancer that are biologically significant, ie, those that will cause serious illness or death.
Many men have prostate cancer
In the United States, the lifetime probability of developing prostate cancer is 1 in 6, and the probability increases with age. Prostate cancer is primarily a disease of the Western world, but it is becoming more common in other areas as well.
Risk factors for prostate cancer are age, race, and family history. Clinically apparent disease is very rare in men younger than 40 years; until recently, most guidelines suggested that screening for it should begin at age 50. African American men have the highest risk of developing and dying of prostate cancer, for reasons that are not clear. In the past, this finding was attributed to disparities in access and less aggressive therapy in black men, but recent studies suggest the differences persist even in the absence of these factors, suggesting there is a biological difference in cancers between blacks and whites. Having a father or brother who had prostate cancer increases one’s risk twofold (threefold if the father or brother was affected before the age of 60); having a father and a brother with prostate cancer increases one’s risk fourfold, and true hereditary cancer raises the risk fivefold.4
But relatively few men die of it
The Scandinavian Prostate Cancer Group5 randomized 695 men with early prostate cancer (mostly discovered by digital rectal examination or by symptoms) to undergo either radical prostatectomy or a program of watchful waiting. In 8.2 years of follow-up, 8.6% of the men in the surgery group and 14.4% of those in the watchful waiting group died of prostate cancer. Thus, we can conclude that surgery is beneficial in this situation.
Despite these calculations, in contemporary practice in the United States, about 90% of men with newly diagnosed low-grade prostate cancer choose to be treated.6 This high level of intervention reflects our current inability to predict which cancers will remain indolent vs which will progress and the lack of validated markers that tell us when to intervene in patients who are managed expectantly and not lose the chance for cure. Most often, patients and their physicians, who are paid to intervene, deal with this uncertainty by choosing the high likelihood of cure with early intervention despite treatment-related morbidity.
What PSA has wrought
When PSA screening was introduced in the late 1980s and early 1990s, it brought about several changes in the epidemiology and clinical profile of this disease that led us to believe that it was making a meaningful difference.
A spike in the apparent incidence of prostate cancer occurred in the late 1980s and early 1990s with the introduction of PSA screening. The spike was temporary, representing detection of preexisting cases. Now, the incidence may have leveled off.7
A shift in the stages of cancers detected. In 1982, half of men with newly diagnosed prostate cancer had incurable disease.8 Five years after the introduction of PSA testing, 95% had curable disease.9
An increase in the rate of cure after radical prostatectomy was seen.
A decrease in the death rate from prostate cancer since the early 1990s has been noted, which is likely due not only to earlier detection but also to earlier and better treatment.
Limitations of PSA screening
PSA screening has low specificity. PSA is more sensitive than digital rectal examination, but most men with “elevated” PSA do not have prostate cancer. Nevertheless, although it is not a perfect screening test, it is still the best cancer marker that we have.
In the Prostate Cancer Prevention Trial (PCPT),10 finasteride (Proscar) decreased the incidence of prostate cancer by about 25% over 7 years. But there were also lessons to be learned from the placebo group, which underwent PSA testing every year and prostate biopsy at the end of the study.
We used to think the cutoff PSA level that had high sensitivity and specificity for finding cancer was 4 ng/mL. However, in the PCPT, 6.6% of men with PSA levels below 0.5 ng/mL were found to have cancer, and 12.5% of those cancers were high-grade. Of those with PSA levels of 3.1 to 4.0 ng/mL, 26.9% had cancer, and 25.0% of the cancers were high-grade. These data demonstrate that there is no PSA level below which risk of cancer is zero, and that there is no PSA cutoff with sufficient sensitivity and specificity to be clinically useful.
The PCPT risk calculator (http://deb.uthscsa.edu/URORiskCalc/Pages/uroriskcalc.jsp) is a wonderful tool that came out of that study. It uses seven variables—race, age, PSA level, family history of prostate cancer, findings on digital rectal examination, whether the patient has ever undergone a prostate biopsy, and whether the patient is taking finasteride—and calculates the patient’s risk of harboring prostate cancer and, more important, the risk of having high-grade prostate cancer. This tool allows estimation of individual risk and helps identify who is at risk of cancer that may require therapy.
Other factors can affect PSA levels. Men with a higher body mass index have lower PSA levels. The reason is not clear; it may be a hormonal effect, or heavier men may simply have higher blood volume, which may dilute the PSA. Furthermore, there are genetic differences that make some men secrete more PSA, but this effect is probably not clinically important. And a study by Hamilton et al11 suggested that statin drugs lower PSA levels. As these findings are confirmed, in the future it may be necessary to adjust PSA levels to account for their effects before deciding on the need for biopsy.
Two new, conflicting studies
Two large trials of PSA screening, published simultaneously in March 2009, came to opposite conclusions.
The European Randomized Study of Screening for Prostate Cancer2 randomized 162,243 men between the ages of 55 and 69 to undergo PSA screening at an average of once every 4 years or to a control group. Most of the participating centers used a PSA level of 3.0 ng/mL as an indication for biopsy. At an average follow-up time of 8.8 years, 214 men had died of prostate cancer in the screening group, compared with 326 in the control group, for an adjusted rate ratio of 0.80 (95% confidence interval [CI] 0.65–0.98, P = .04). In other words, screening decreased the risk of death from prostate cancer by 20%.
The Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial,1 conducted in the United States, came to the opposite conclusion, ie, that there is no benefit from PSA screening. This study was smaller, with 76,693 men between ages 55 and 74 randomly assigned to receive PSA testing every year for 6 years and digital rectal examination for 4 years, or usual care. A PSA level of more than 4.0 ng/mL was considered to be positive for prostate cancer. At 7 years, of those who reported undergoing no more than one PSA test at baseline, 48 men had died of prostate cancer in the screening group, compared with 41 in the control group (rate ratio 1.16, 95% CI 0.76–1.76).
Why were the findings different? The PLCO investigators offered several possible explanations for their negative results. The PSA threshold of 4 ng/mL that was used in that study may not be effective. More than half the men in the control group actually had a PSA test in the first 6 years of the study, potentially diluting any effect of testing. (This was the most worrisome flaw in the study, in my opinion.) About 44% of the men in the study had already had one or more PSA tests at baseline, which would have eliminated cancers detectable on screening from the study, and not all men who were advised to undergo biopsy actually did so. The follow-up time may not yet be long enough for the benefit to be apparent. Most important, in their opinion, treatment for prostate cancer improved during the time of the trial, so that fewer men than expected died of prostate cancer in both groups.
Improvements to PSA screening
Derivatives of PSA have been used in an attempt to improve its performance characteristics for detecting cancer.
PSA density, defined as serum PSA divided by prostate volume, has some predictive power but requires performance of transrectal ultrasonography. It is therefore not a good screening test in the primary care setting.
PSA velocity or doubling time, based on the rate of change over time, is predictive of prostate cancer, but is highly dependent on the absolute value of PSA and does not add independent information to the variables defined in the PCPT risk calculator or other standard predictive variables.12
A PSA level between the ages of 44 and 50 may predict the lifetime risk of prostate cancer, according to a study by Lilja et al13 in Sweden. This finding suggests that we should measure PSA early in life and screen men who have higher values more frequently or with better strategies. This recommendation has been adopted by the American Urological Association, which released updated screening guidelines in April 2009 (available at www.auanet.org/content/guidelines-and-quality-care/clinical-guidelines/main-reports/psa09.pdf).
New markers under study
A number of new biological markers probably will improve our ability to detect prostate cancer, although they are not yet ready for widespread use.
Urinary PCA3. Prostate cancer gene 3 (PCA3) codes for a messenger RNA that is highly overexpressed in the urine of men with prostate cancer. Urine is collected after prostate massage. Marks et al14 reported that PCA3 scores predicted biopsy outcomes in men with serum PSA levels of 2.5 ng/mL or higher.
Serum EPCA-2 (early prostate cancer antigen 2) is another candidate marker undergoing study.
Gene fusions, specifically of TRMPSS2 and the ETS gene family, are detectable in high levels in the urine of some men with prostate cancer, and appear to be very promising markers for detection.
Metabolomics is a technique that uses mass spectroscopy to detect the metabolic signature of cancer. Sreekumar et al15 identified sarcosine as a potential marker of prostate cancer using this technique.
Genetic tests: Not yet
Some data suggest that we can use genetic tests to screen for prostate cancer, but the tests are not yet as good as we would like.
Zheng et al16 reported that 16 singlenucleotide polymorphisms (SNPs) in five chromosomal regions plus a family history of prostate cancer have a cumulative association with prostate cancer: men who had any five or more of these SNPs had a risk of prostate cancer nearly 10 times as high as men without any of them. However, the number of men who actually fall into this category is so low that routine use in the general population is not cost-effective; it may, however, be useful in men with a family history of prostate cancer.
Other SNPs have been linked to prostate cancer (reviewed by Witte17). Having any one of these loci increases one’s risk only modestly, however. Only about 2% of the population has five or more of these SNPS, and the sensitivity is about only about 16%.
A commercially available DNA test (Decode Genetics, Reykjavik, Iceland) can detect eight variants that, according to the company, account for about half of all cases of prostate cancer.
Prostate cancer screening: My interpretation
I believe the two new studies of PSA screening suggest there is a modest benefit from screening in terms of preventing deaths from prostate cancer. But I also believe we should be more judicious in recommending treatment for men whom we know have biologically indolent tumors, although we cannot yet identify them perfectly.
In the past, we used an arbitrary PSA cutoff to detect prostate cancer of any grade, and men with high levels were advised to have a biopsy. Currently, we use continuous-risk models to look for any cancer and biologically significant cancers. These involve nomograms, a risk calculator, and new markers.
We use the PCPT risk calculator routinely in our practice. I recommend—completely arbitrarily—that a man undergo biopsy if he has a 10% or higher risk of high-grade cancer, but not if the risk is less. I believe this is more accurate than a simple PSA cutoff value.
CAN WE PREVENT PROSTATE CANCER?
Prostate cancer is a significant public health risk, with 186,000 new cases and 26,000 deaths yearly. Its risk factors (age, race, and genes) are not modifiable. The benefit of screening in terms of preventing deaths is not as good as we would like, and therapy is associated with morbidity. That leaves prevention as a potential way to reduce the morbidity and perhaps mortality of prostate cancer and its therapy.
Epidemiologic studies suggest that certain lifestyle factors may increase the risk, ie, consumption of fat, red meat, fried foods, and dairy; high calcium intake; smoking; total calories; and body size. Other factors may decrease the risk: plant-based foods and vegetables, especially lycopene-containing foods such as tomatoes, cruciferous vegetables, soy, and legumes, specific nutrients such as carotenoids, lycopene, total antioxidants, fish oil (omega-3 fatty acids), and moderate to vigorous exercise. However, there have been few randomized trials to determine if any of these agents are beneficial.
Findings of trials of prevention
Selenium and vitamin E do not prevent prostate cancer, lung cancer, colorectal cancer, other primary cancers, or deaths. The Selenium and Vitamin E Cancer Prevention Trial (SELECT)3 involved 35,533 men 55 years of age or older (or 50 and older if they were African American). They were randomized to receive one of four treatments: selenium 200 μg/day plus vitamin E placebo, vitamin E 400 IU/day plus selenium placebo, selenium plus vitamin E, or double placebo. At a median follow-up of 5.46 years, compared with the placebo group, the hazard ratio for prostate cancer was 1.04 in the selenium-only group, 1.13 in the vitamin E-only group, and 1.05 in the selenium-plus-vitamin E group. None of the differences was statistically significant.
The Physician’s Health Study18 also found that vitamin E at the same dose given every other day does not prevent prostate cancer.
Finasteride prevents prostate cancer. The PCPT19 included 18,882 men, 55 years of age or older, who had PSA levels of 3.0 ng/mL or less and normal findings on digital rectal examination. Treatment was with finasteride 5 mg/day or placebo. At 7 years, prostate cancer had been discovered in 18.4% of the finasteride group vs 24.4% of the placebo group, a 24.8% reduction (95% CI 18.6–30.6, P < .001). Sexual side effects were more common in the men who received finasteride, while urinary symptoms were more common in the placebo group.
At the time of the original PCPT report in 2003,19 tumors of Gleason grade 7 or higher were more common in the finasteride group, accounting for 37.0% of the tumors discovered, than in the placebo group (22.2%), creating concern that finasteride might somehow cause the tumors that occurred to be more aggressive. However, a subsequent analysis20 found the opposite to be true, ie, that finasteride decreases the risk of high-grade cancers. A companion quality-of-life study showed that chronic use of finasteride had clinically insignificant effects on sexual function, and the PCPT and other studies have shown benefits of finasteride in reducing lower urinary tract symptoms due to benign prostatic hyperplasia (BPH), reducing the risk of acute urinary retention and the need for surgical intervention for BPH, and reducing the risk of prostatitis.
Dutasteride also prevents prostate cancer. A large-scale trial of another 5-alpha reductase inhibitor, dutasteride (Avodart), was reported by Andriole at the annual meeting of the American Urological Association in April 2009.21 The Reduction by Dutasteride of Prostate Events (REDUCE) trial included men who were 50 to 75 years old, inclusively, and who had PSA levels between 2.5 and 10 ng/mL, prostate volume less than 80 cc, and one prior negative prostate biopsy within 6 months of enrollment, representing a group at high risk for cancer on a subsequent biopsy. The trial accrued 8,231 men. At 4 years, prostate cancer had occurred in 659 men in the dutasteride group vs 857 in the placebo group, a 23% reduction (P < .0001). Interestingly, no significant increase in Gleason grade 8 to grade 10 tumors was observed in the study.
Preliminary analyses also suggest that dutasteride enhanced the utility of PSA as a diagnostic test for prostate cancer, had beneficial effects on BPH, and was generally well tolerated. The fact that the results of REDUCE were congruent with those of the PCPT with respect to the magnitude of risk reduction, beneficial effects on benign prostatic hypertrophy, minimal toxicity, and no issues related to tumor grade suggests a class effect for 5-alpha reductase inhibitors, and suggests that these agents should be used more liberally for the prevention of prostate cancer.
There is current debate about whether 5-alpha reductase inhibitors should be used by all men at risk of prostate cancer or only by those at high risk. However, the American Urological Association and the American Society of Clinical Oncology have issued guidelines stating that men at risk should consider this intervention.22
- Andriole GL, Grubb RL, Buys SS, et al; PLCO Project Team. Mortality results from a randomized prostate cancer screening trial. N Engl J Med 2009; 360:1310–1319.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate cancer mortality in a randomized European study. N Engl J Med 2009; 360:1351–1354.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Bratt O. Hereditary prostate cancer: clinical aspects. J Urol 2002; 168:906–913.
- Bill-Axelson A, Holmberg L, Ruutu M, et al; Scandinavian Prostate Cancer Group Study No. 4. Radical prostatectomy versus watchful waiting in early prostate cancer. N Engl J Med 2005; 352:1977–1984.
- Cooperberg MR, Broering JM, Kantoff PW, Carroll PR. Contemporary trends in low risk prostate cancer: risk assessment and treatment. J Urol 2007; 178:S14–S19.
- Horner MJ, Ries LAG, Krapcho M, et al, editors. SEER Cancer Statistics Review, 1975–2006, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER web site, 2009. Accessed 6/28/2009.
- Murphy GP, Natarajan N, Pontes JE, et al. The national survey of prostate cancer in the United States by the American College of Surgeons. J Urol 1982; 127:928–934.
- Catalona WJ, Smith DS, Ratliff TL, Basler JW. Detection of organconfined prostate cancer is increased through prostate-specific antigen-based screening. JAMA 1993; 270:948–954.
- Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level < or = 4.0 ng per milliliter. N Engl J Med 2004; 350:2239–2246.
- Hamilton RJ, Goldberg KC, Platz EA, Freedland SJ. The influence of statin medications on prostate-specific antigen levels. N Natl Cancer Inst 2008; 100:1487–1488.
- Vickers AJ, Savage C, O’Brien MF, Lilja H. Systematic review of pretreatment prostate-specific antigen velocity and doubling time as predictors for prostate cancer. J Clin Oncol 2009; 27:398–403.
- Lilja H, Ulmert D, Vickers AJ. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat Rev Cancer 2008; 8:268–278.
- Marks LS, Fradet Y, Deras IL, et al. PCA molecular urine assay for prostate cancer in men undergoing repeat biopsy. Urology 2007; 69:532–535.
- Sreekumar A, Poisson LM, Thekkelnaycke M, et al. Metabolomic profile delineates potential role for sarcosine in prostate cancer progression. Nature 2009; 457:910–914.
- Zheng SL, Sun J, Wiklund F, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med 2008; 358:910–919.
- Witte JS. Prostate cancer genomics: toward a new understanding. Nat Rev Genet 2009; 10:77–82.
- Gaziano JM, Glynn RJ, Christen WG, et al. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA 2009; 301:52–62.
- Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med 2003; 349:215–224.
- Lucia MS, Darke AK, Goodman PJ, et al. Pathologic characteristics of cancers detected in the Prostate Cancer Prevention Trial: implications for prostate cancer detection and chemoprevention. Cancer Prev Res (Phila PA) 2008; 1:167–173.
- Andriole G, Bostwick D, Brawley O, et al. Further analyses from the REDUCE prostate cancer risk reduction trial [abstract]. J Urol 2009; 181:( suppl):555.
- Kramer BS, Hagerty KL, Justman S, et al; American Society of Clinical Oncology/American Urological Association. Use of 5-alpha-reductase inhibitors for prostate cancer chemoprevention: American Society of Clinical Oncology/American Urological Association 2008 Clinical Practice Guideline. J Urol 2009; 181:1642–1657.
In spite of some recent studies, or perhaps because of them, we still are unsure about how best to screen for and prevent prostate cancer. Two large trials of screening with prostate-specific antigen (PSA) measurements came to seemingly opposite conclusions.1,2 Furthermore, a large trial of selenium and vitamin E found that these agents have no value as preventive agents.3
Nevertheless, negative studies also advance science, and steady progress is being made in prostate cancer research. In this paper I briefly summarize and comment on some of the recent findings.
TO SCREEN OR NOT TO SCREEN?
All cases of prostate cancer are clinically relevant in that they can cause anxiety or can lead to treatment-related morbidity. The challenge is to detect the minority of cases of cancer that are biologically significant, ie, those that will cause serious illness or death.
Many men have prostate cancer
In the United States, the lifetime probability of developing prostate cancer is 1 in 6, and the probability increases with age. Prostate cancer is primarily a disease of the Western world, but it is becoming more common in other areas as well.
Risk factors for prostate cancer are age, race, and family history. Clinically apparent disease is very rare in men younger than 40 years; until recently, most guidelines suggested that screening for it should begin at age 50. African American men have the highest risk of developing and dying of prostate cancer, for reasons that are not clear. In the past, this finding was attributed to disparities in access and less aggressive therapy in black men, but recent studies suggest the differences persist even in the absence of these factors, suggesting there is a biological difference in cancers between blacks and whites. Having a father or brother who had prostate cancer increases one’s risk twofold (threefold if the father or brother was affected before the age of 60); having a father and a brother with prostate cancer increases one’s risk fourfold, and true hereditary cancer raises the risk fivefold.4
But relatively few men die of it
The Scandinavian Prostate Cancer Group5 randomized 695 men with early prostate cancer (mostly discovered by digital rectal examination or by symptoms) to undergo either radical prostatectomy or a program of watchful waiting. In 8.2 years of follow-up, 8.6% of the men in the surgery group and 14.4% of those in the watchful waiting group died of prostate cancer. Thus, we can conclude that surgery is beneficial in this situation.
Despite these calculations, in contemporary practice in the United States, about 90% of men with newly diagnosed low-grade prostate cancer choose to be treated.6 This high level of intervention reflects our current inability to predict which cancers will remain indolent vs which will progress and the lack of validated markers that tell us when to intervene in patients who are managed expectantly and not lose the chance for cure. Most often, patients and their physicians, who are paid to intervene, deal with this uncertainty by choosing the high likelihood of cure with early intervention despite treatment-related morbidity.
What PSA has wrought
When PSA screening was introduced in the late 1980s and early 1990s, it brought about several changes in the epidemiology and clinical profile of this disease that led us to believe that it was making a meaningful difference.
A spike in the apparent incidence of prostate cancer occurred in the late 1980s and early 1990s with the introduction of PSA screening. The spike was temporary, representing detection of preexisting cases. Now, the incidence may have leveled off.7
A shift in the stages of cancers detected. In 1982, half of men with newly diagnosed prostate cancer had incurable disease.8 Five years after the introduction of PSA testing, 95% had curable disease.9
An increase in the rate of cure after radical prostatectomy was seen.
A decrease in the death rate from prostate cancer since the early 1990s has been noted, which is likely due not only to earlier detection but also to earlier and better treatment.
Limitations of PSA screening
PSA screening has low specificity. PSA is more sensitive than digital rectal examination, but most men with “elevated” PSA do not have prostate cancer. Nevertheless, although it is not a perfect screening test, it is still the best cancer marker that we have.
In the Prostate Cancer Prevention Trial (PCPT),10 finasteride (Proscar) decreased the incidence of prostate cancer by about 25% over 7 years. But there were also lessons to be learned from the placebo group, which underwent PSA testing every year and prostate biopsy at the end of the study.
We used to think the cutoff PSA level that had high sensitivity and specificity for finding cancer was 4 ng/mL. However, in the PCPT, 6.6% of men with PSA levels below 0.5 ng/mL were found to have cancer, and 12.5% of those cancers were high-grade. Of those with PSA levels of 3.1 to 4.0 ng/mL, 26.9% had cancer, and 25.0% of the cancers were high-grade. These data demonstrate that there is no PSA level below which risk of cancer is zero, and that there is no PSA cutoff with sufficient sensitivity and specificity to be clinically useful.
The PCPT risk calculator (http://deb.uthscsa.edu/URORiskCalc/Pages/uroriskcalc.jsp) is a wonderful tool that came out of that study. It uses seven variables—race, age, PSA level, family history of prostate cancer, findings on digital rectal examination, whether the patient has ever undergone a prostate biopsy, and whether the patient is taking finasteride—and calculates the patient’s risk of harboring prostate cancer and, more important, the risk of having high-grade prostate cancer. This tool allows estimation of individual risk and helps identify who is at risk of cancer that may require therapy.
Other factors can affect PSA levels. Men with a higher body mass index have lower PSA levels. The reason is not clear; it may be a hormonal effect, or heavier men may simply have higher blood volume, which may dilute the PSA. Furthermore, there are genetic differences that make some men secrete more PSA, but this effect is probably not clinically important. And a study by Hamilton et al11 suggested that statin drugs lower PSA levels. As these findings are confirmed, in the future it may be necessary to adjust PSA levels to account for their effects before deciding on the need for biopsy.
Two new, conflicting studies
Two large trials of PSA screening, published simultaneously in March 2009, came to opposite conclusions.
The European Randomized Study of Screening for Prostate Cancer2 randomized 162,243 men between the ages of 55 and 69 to undergo PSA screening at an average of once every 4 years or to a control group. Most of the participating centers used a PSA level of 3.0 ng/mL as an indication for biopsy. At an average follow-up time of 8.8 years, 214 men had died of prostate cancer in the screening group, compared with 326 in the control group, for an adjusted rate ratio of 0.80 (95% confidence interval [CI] 0.65–0.98, P = .04). In other words, screening decreased the risk of death from prostate cancer by 20%.
The Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial,1 conducted in the United States, came to the opposite conclusion, ie, that there is no benefit from PSA screening. This study was smaller, with 76,693 men between ages 55 and 74 randomly assigned to receive PSA testing every year for 6 years and digital rectal examination for 4 years, or usual care. A PSA level of more than 4.0 ng/mL was considered to be positive for prostate cancer. At 7 years, of those who reported undergoing no more than one PSA test at baseline, 48 men had died of prostate cancer in the screening group, compared with 41 in the control group (rate ratio 1.16, 95% CI 0.76–1.76).
Why were the findings different? The PLCO investigators offered several possible explanations for their negative results. The PSA threshold of 4 ng/mL that was used in that study may not be effective. More than half the men in the control group actually had a PSA test in the first 6 years of the study, potentially diluting any effect of testing. (This was the most worrisome flaw in the study, in my opinion.) About 44% of the men in the study had already had one or more PSA tests at baseline, which would have eliminated cancers detectable on screening from the study, and not all men who were advised to undergo biopsy actually did so. The follow-up time may not yet be long enough for the benefit to be apparent. Most important, in their opinion, treatment for prostate cancer improved during the time of the trial, so that fewer men than expected died of prostate cancer in both groups.
Improvements to PSA screening
Derivatives of PSA have been used in an attempt to improve its performance characteristics for detecting cancer.
PSA density, defined as serum PSA divided by prostate volume, has some predictive power but requires performance of transrectal ultrasonography. It is therefore not a good screening test in the primary care setting.
PSA velocity or doubling time, based on the rate of change over time, is predictive of prostate cancer, but is highly dependent on the absolute value of PSA and does not add independent information to the variables defined in the PCPT risk calculator or other standard predictive variables.12
A PSA level between the ages of 44 and 50 may predict the lifetime risk of prostate cancer, according to a study by Lilja et al13 in Sweden. This finding suggests that we should measure PSA early in life and screen men who have higher values more frequently or with better strategies. This recommendation has been adopted by the American Urological Association, which released updated screening guidelines in April 2009 (available at www.auanet.org/content/guidelines-and-quality-care/clinical-guidelines/main-reports/psa09.pdf).
New markers under study
A number of new biological markers probably will improve our ability to detect prostate cancer, although they are not yet ready for widespread use.
Urinary PCA3. Prostate cancer gene 3 (PCA3) codes for a messenger RNA that is highly overexpressed in the urine of men with prostate cancer. Urine is collected after prostate massage. Marks et al14 reported that PCA3 scores predicted biopsy outcomes in men with serum PSA levels of 2.5 ng/mL or higher.
Serum EPCA-2 (early prostate cancer antigen 2) is another candidate marker undergoing study.
Gene fusions, specifically of TRMPSS2 and the ETS gene family, are detectable in high levels in the urine of some men with prostate cancer, and appear to be very promising markers for detection.
Metabolomics is a technique that uses mass spectroscopy to detect the metabolic signature of cancer. Sreekumar et al15 identified sarcosine as a potential marker of prostate cancer using this technique.
Genetic tests: Not yet
Some data suggest that we can use genetic tests to screen for prostate cancer, but the tests are not yet as good as we would like.
Zheng et al16 reported that 16 singlenucleotide polymorphisms (SNPs) in five chromosomal regions plus a family history of prostate cancer have a cumulative association with prostate cancer: men who had any five or more of these SNPs had a risk of prostate cancer nearly 10 times as high as men without any of them. However, the number of men who actually fall into this category is so low that routine use in the general population is not cost-effective; it may, however, be useful in men with a family history of prostate cancer.
Other SNPs have been linked to prostate cancer (reviewed by Witte17). Having any one of these loci increases one’s risk only modestly, however. Only about 2% of the population has five or more of these SNPS, and the sensitivity is about only about 16%.
A commercially available DNA test (Decode Genetics, Reykjavik, Iceland) can detect eight variants that, according to the company, account for about half of all cases of prostate cancer.
Prostate cancer screening: My interpretation
I believe the two new studies of PSA screening suggest there is a modest benefit from screening in terms of preventing deaths from prostate cancer. But I also believe we should be more judicious in recommending treatment for men whom we know have biologically indolent tumors, although we cannot yet identify them perfectly.
In the past, we used an arbitrary PSA cutoff to detect prostate cancer of any grade, and men with high levels were advised to have a biopsy. Currently, we use continuous-risk models to look for any cancer and biologically significant cancers. These involve nomograms, a risk calculator, and new markers.
We use the PCPT risk calculator routinely in our practice. I recommend—completely arbitrarily—that a man undergo biopsy if he has a 10% or higher risk of high-grade cancer, but not if the risk is less. I believe this is more accurate than a simple PSA cutoff value.
CAN WE PREVENT PROSTATE CANCER?
Prostate cancer is a significant public health risk, with 186,000 new cases and 26,000 deaths yearly. Its risk factors (age, race, and genes) are not modifiable. The benefit of screening in terms of preventing deaths is not as good as we would like, and therapy is associated with morbidity. That leaves prevention as a potential way to reduce the morbidity and perhaps mortality of prostate cancer and its therapy.
Epidemiologic studies suggest that certain lifestyle factors may increase the risk, ie, consumption of fat, red meat, fried foods, and dairy; high calcium intake; smoking; total calories; and body size. Other factors may decrease the risk: plant-based foods and vegetables, especially lycopene-containing foods such as tomatoes, cruciferous vegetables, soy, and legumes, specific nutrients such as carotenoids, lycopene, total antioxidants, fish oil (omega-3 fatty acids), and moderate to vigorous exercise. However, there have been few randomized trials to determine if any of these agents are beneficial.
Findings of trials of prevention
Selenium and vitamin E do not prevent prostate cancer, lung cancer, colorectal cancer, other primary cancers, or deaths. The Selenium and Vitamin E Cancer Prevention Trial (SELECT)3 involved 35,533 men 55 years of age or older (or 50 and older if they were African American). They were randomized to receive one of four treatments: selenium 200 μg/day plus vitamin E placebo, vitamin E 400 IU/day plus selenium placebo, selenium plus vitamin E, or double placebo. At a median follow-up of 5.46 years, compared with the placebo group, the hazard ratio for prostate cancer was 1.04 in the selenium-only group, 1.13 in the vitamin E-only group, and 1.05 in the selenium-plus-vitamin E group. None of the differences was statistically significant.
The Physician’s Health Study18 also found that vitamin E at the same dose given every other day does not prevent prostate cancer.
Finasteride prevents prostate cancer. The PCPT19 included 18,882 men, 55 years of age or older, who had PSA levels of 3.0 ng/mL or less and normal findings on digital rectal examination. Treatment was with finasteride 5 mg/day or placebo. At 7 years, prostate cancer had been discovered in 18.4% of the finasteride group vs 24.4% of the placebo group, a 24.8% reduction (95% CI 18.6–30.6, P < .001). Sexual side effects were more common in the men who received finasteride, while urinary symptoms were more common in the placebo group.
At the time of the original PCPT report in 2003,19 tumors of Gleason grade 7 or higher were more common in the finasteride group, accounting for 37.0% of the tumors discovered, than in the placebo group (22.2%), creating concern that finasteride might somehow cause the tumors that occurred to be more aggressive. However, a subsequent analysis20 found the opposite to be true, ie, that finasteride decreases the risk of high-grade cancers. A companion quality-of-life study showed that chronic use of finasteride had clinically insignificant effects on sexual function, and the PCPT and other studies have shown benefits of finasteride in reducing lower urinary tract symptoms due to benign prostatic hyperplasia (BPH), reducing the risk of acute urinary retention and the need for surgical intervention for BPH, and reducing the risk of prostatitis.
Dutasteride also prevents prostate cancer. A large-scale trial of another 5-alpha reductase inhibitor, dutasteride (Avodart), was reported by Andriole at the annual meeting of the American Urological Association in April 2009.21 The Reduction by Dutasteride of Prostate Events (REDUCE) trial included men who were 50 to 75 years old, inclusively, and who had PSA levels between 2.5 and 10 ng/mL, prostate volume less than 80 cc, and one prior negative prostate biopsy within 6 months of enrollment, representing a group at high risk for cancer on a subsequent biopsy. The trial accrued 8,231 men. At 4 years, prostate cancer had occurred in 659 men in the dutasteride group vs 857 in the placebo group, a 23% reduction (P < .0001). Interestingly, no significant increase in Gleason grade 8 to grade 10 tumors was observed in the study.
Preliminary analyses also suggest that dutasteride enhanced the utility of PSA as a diagnostic test for prostate cancer, had beneficial effects on BPH, and was generally well tolerated. The fact that the results of REDUCE were congruent with those of the PCPT with respect to the magnitude of risk reduction, beneficial effects on benign prostatic hypertrophy, minimal toxicity, and no issues related to tumor grade suggests a class effect for 5-alpha reductase inhibitors, and suggests that these agents should be used more liberally for the prevention of prostate cancer.
There is current debate about whether 5-alpha reductase inhibitors should be used by all men at risk of prostate cancer or only by those at high risk. However, the American Urological Association and the American Society of Clinical Oncology have issued guidelines stating that men at risk should consider this intervention.22
In spite of some recent studies, or perhaps because of them, we still are unsure about how best to screen for and prevent prostate cancer. Two large trials of screening with prostate-specific antigen (PSA) measurements came to seemingly opposite conclusions.1,2 Furthermore, a large trial of selenium and vitamin E found that these agents have no value as preventive agents.3
Nevertheless, negative studies also advance science, and steady progress is being made in prostate cancer research. In this paper I briefly summarize and comment on some of the recent findings.
TO SCREEN OR NOT TO SCREEN?
All cases of prostate cancer are clinically relevant in that they can cause anxiety or can lead to treatment-related morbidity. The challenge is to detect the minority of cases of cancer that are biologically significant, ie, those that will cause serious illness or death.
Many men have prostate cancer
In the United States, the lifetime probability of developing prostate cancer is 1 in 6, and the probability increases with age. Prostate cancer is primarily a disease of the Western world, but it is becoming more common in other areas as well.
Risk factors for prostate cancer are age, race, and family history. Clinically apparent disease is very rare in men younger than 40 years; until recently, most guidelines suggested that screening for it should begin at age 50. African American men have the highest risk of developing and dying of prostate cancer, for reasons that are not clear. In the past, this finding was attributed to disparities in access and less aggressive therapy in black men, but recent studies suggest the differences persist even in the absence of these factors, suggesting there is a biological difference in cancers between blacks and whites. Having a father or brother who had prostate cancer increases one’s risk twofold (threefold if the father or brother was affected before the age of 60); having a father and a brother with prostate cancer increases one’s risk fourfold, and true hereditary cancer raises the risk fivefold.4
But relatively few men die of it
The Scandinavian Prostate Cancer Group5 randomized 695 men with early prostate cancer (mostly discovered by digital rectal examination or by symptoms) to undergo either radical prostatectomy or a program of watchful waiting. In 8.2 years of follow-up, 8.6% of the men in the surgery group and 14.4% of those in the watchful waiting group died of prostate cancer. Thus, we can conclude that surgery is beneficial in this situation.
Despite these calculations, in contemporary practice in the United States, about 90% of men with newly diagnosed low-grade prostate cancer choose to be treated.6 This high level of intervention reflects our current inability to predict which cancers will remain indolent vs which will progress and the lack of validated markers that tell us when to intervene in patients who are managed expectantly and not lose the chance for cure. Most often, patients and their physicians, who are paid to intervene, deal with this uncertainty by choosing the high likelihood of cure with early intervention despite treatment-related morbidity.
What PSA has wrought
When PSA screening was introduced in the late 1980s and early 1990s, it brought about several changes in the epidemiology and clinical profile of this disease that led us to believe that it was making a meaningful difference.
A spike in the apparent incidence of prostate cancer occurred in the late 1980s and early 1990s with the introduction of PSA screening. The spike was temporary, representing detection of preexisting cases. Now, the incidence may have leveled off.7
A shift in the stages of cancers detected. In 1982, half of men with newly diagnosed prostate cancer had incurable disease.8 Five years after the introduction of PSA testing, 95% had curable disease.9
An increase in the rate of cure after radical prostatectomy was seen.
A decrease in the death rate from prostate cancer since the early 1990s has been noted, which is likely due not only to earlier detection but also to earlier and better treatment.
Limitations of PSA screening
PSA screening has low specificity. PSA is more sensitive than digital rectal examination, but most men with “elevated” PSA do not have prostate cancer. Nevertheless, although it is not a perfect screening test, it is still the best cancer marker that we have.
In the Prostate Cancer Prevention Trial (PCPT),10 finasteride (Proscar) decreased the incidence of prostate cancer by about 25% over 7 years. But there were also lessons to be learned from the placebo group, which underwent PSA testing every year and prostate biopsy at the end of the study.
We used to think the cutoff PSA level that had high sensitivity and specificity for finding cancer was 4 ng/mL. However, in the PCPT, 6.6% of men with PSA levels below 0.5 ng/mL were found to have cancer, and 12.5% of those cancers were high-grade. Of those with PSA levels of 3.1 to 4.0 ng/mL, 26.9% had cancer, and 25.0% of the cancers were high-grade. These data demonstrate that there is no PSA level below which risk of cancer is zero, and that there is no PSA cutoff with sufficient sensitivity and specificity to be clinically useful.
The PCPT risk calculator (http://deb.uthscsa.edu/URORiskCalc/Pages/uroriskcalc.jsp) is a wonderful tool that came out of that study. It uses seven variables—race, age, PSA level, family history of prostate cancer, findings on digital rectal examination, whether the patient has ever undergone a prostate biopsy, and whether the patient is taking finasteride—and calculates the patient’s risk of harboring prostate cancer and, more important, the risk of having high-grade prostate cancer. This tool allows estimation of individual risk and helps identify who is at risk of cancer that may require therapy.
Other factors can affect PSA levels. Men with a higher body mass index have lower PSA levels. The reason is not clear; it may be a hormonal effect, or heavier men may simply have higher blood volume, which may dilute the PSA. Furthermore, there are genetic differences that make some men secrete more PSA, but this effect is probably not clinically important. And a study by Hamilton et al11 suggested that statin drugs lower PSA levels. As these findings are confirmed, in the future it may be necessary to adjust PSA levels to account for their effects before deciding on the need for biopsy.
Two new, conflicting studies
Two large trials of PSA screening, published simultaneously in March 2009, came to opposite conclusions.
The European Randomized Study of Screening for Prostate Cancer2 randomized 162,243 men between the ages of 55 and 69 to undergo PSA screening at an average of once every 4 years or to a control group. Most of the participating centers used a PSA level of 3.0 ng/mL as an indication for biopsy. At an average follow-up time of 8.8 years, 214 men had died of prostate cancer in the screening group, compared with 326 in the control group, for an adjusted rate ratio of 0.80 (95% confidence interval [CI] 0.65–0.98, P = .04). In other words, screening decreased the risk of death from prostate cancer by 20%.
The Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial,1 conducted in the United States, came to the opposite conclusion, ie, that there is no benefit from PSA screening. This study was smaller, with 76,693 men between ages 55 and 74 randomly assigned to receive PSA testing every year for 6 years and digital rectal examination for 4 years, or usual care. A PSA level of more than 4.0 ng/mL was considered to be positive for prostate cancer. At 7 years, of those who reported undergoing no more than one PSA test at baseline, 48 men had died of prostate cancer in the screening group, compared with 41 in the control group (rate ratio 1.16, 95% CI 0.76–1.76).
Why were the findings different? The PLCO investigators offered several possible explanations for their negative results. The PSA threshold of 4 ng/mL that was used in that study may not be effective. More than half the men in the control group actually had a PSA test in the first 6 years of the study, potentially diluting any effect of testing. (This was the most worrisome flaw in the study, in my opinion.) About 44% of the men in the study had already had one or more PSA tests at baseline, which would have eliminated cancers detectable on screening from the study, and not all men who were advised to undergo biopsy actually did so. The follow-up time may not yet be long enough for the benefit to be apparent. Most important, in their opinion, treatment for prostate cancer improved during the time of the trial, so that fewer men than expected died of prostate cancer in both groups.
Improvements to PSA screening
Derivatives of PSA have been used in an attempt to improve its performance characteristics for detecting cancer.
PSA density, defined as serum PSA divided by prostate volume, has some predictive power but requires performance of transrectal ultrasonography. It is therefore not a good screening test in the primary care setting.
PSA velocity or doubling time, based on the rate of change over time, is predictive of prostate cancer, but is highly dependent on the absolute value of PSA and does not add independent information to the variables defined in the PCPT risk calculator or other standard predictive variables.12
A PSA level between the ages of 44 and 50 may predict the lifetime risk of prostate cancer, according to a study by Lilja et al13 in Sweden. This finding suggests that we should measure PSA early in life and screen men who have higher values more frequently or with better strategies. This recommendation has been adopted by the American Urological Association, which released updated screening guidelines in April 2009 (available at www.auanet.org/content/guidelines-and-quality-care/clinical-guidelines/main-reports/psa09.pdf).
New markers under study
A number of new biological markers probably will improve our ability to detect prostate cancer, although they are not yet ready for widespread use.
Urinary PCA3. Prostate cancer gene 3 (PCA3) codes for a messenger RNA that is highly overexpressed in the urine of men with prostate cancer. Urine is collected after prostate massage. Marks et al14 reported that PCA3 scores predicted biopsy outcomes in men with serum PSA levels of 2.5 ng/mL or higher.
Serum EPCA-2 (early prostate cancer antigen 2) is another candidate marker undergoing study.
Gene fusions, specifically of TRMPSS2 and the ETS gene family, are detectable in high levels in the urine of some men with prostate cancer, and appear to be very promising markers for detection.
Metabolomics is a technique that uses mass spectroscopy to detect the metabolic signature of cancer. Sreekumar et al15 identified sarcosine as a potential marker of prostate cancer using this technique.
Genetic tests: Not yet
Some data suggest that we can use genetic tests to screen for prostate cancer, but the tests are not yet as good as we would like.
Zheng et al16 reported that 16 singlenucleotide polymorphisms (SNPs) in five chromosomal regions plus a family history of prostate cancer have a cumulative association with prostate cancer: men who had any five or more of these SNPs had a risk of prostate cancer nearly 10 times as high as men without any of them. However, the number of men who actually fall into this category is so low that routine use in the general population is not cost-effective; it may, however, be useful in men with a family history of prostate cancer.
Other SNPs have been linked to prostate cancer (reviewed by Witte17). Having any one of these loci increases one’s risk only modestly, however. Only about 2% of the population has five or more of these SNPS, and the sensitivity is about only about 16%.
A commercially available DNA test (Decode Genetics, Reykjavik, Iceland) can detect eight variants that, according to the company, account for about half of all cases of prostate cancer.
Prostate cancer screening: My interpretation
I believe the two new studies of PSA screening suggest there is a modest benefit from screening in terms of preventing deaths from prostate cancer. But I also believe we should be more judicious in recommending treatment for men whom we know have biologically indolent tumors, although we cannot yet identify them perfectly.
In the past, we used an arbitrary PSA cutoff to detect prostate cancer of any grade, and men with high levels were advised to have a biopsy. Currently, we use continuous-risk models to look for any cancer and biologically significant cancers. These involve nomograms, a risk calculator, and new markers.
We use the PCPT risk calculator routinely in our practice. I recommend—completely arbitrarily—that a man undergo biopsy if he has a 10% or higher risk of high-grade cancer, but not if the risk is less. I believe this is more accurate than a simple PSA cutoff value.
CAN WE PREVENT PROSTATE CANCER?
Prostate cancer is a significant public health risk, with 186,000 new cases and 26,000 deaths yearly. Its risk factors (age, race, and genes) are not modifiable. The benefit of screening in terms of preventing deaths is not as good as we would like, and therapy is associated with morbidity. That leaves prevention as a potential way to reduce the morbidity and perhaps mortality of prostate cancer and its therapy.
Epidemiologic studies suggest that certain lifestyle factors may increase the risk, ie, consumption of fat, red meat, fried foods, and dairy; high calcium intake; smoking; total calories; and body size. Other factors may decrease the risk: plant-based foods and vegetables, especially lycopene-containing foods such as tomatoes, cruciferous vegetables, soy, and legumes, specific nutrients such as carotenoids, lycopene, total antioxidants, fish oil (omega-3 fatty acids), and moderate to vigorous exercise. However, there have been few randomized trials to determine if any of these agents are beneficial.
Findings of trials of prevention
Selenium and vitamin E do not prevent prostate cancer, lung cancer, colorectal cancer, other primary cancers, or deaths. The Selenium and Vitamin E Cancer Prevention Trial (SELECT)3 involved 35,533 men 55 years of age or older (or 50 and older if they were African American). They were randomized to receive one of four treatments: selenium 200 μg/day plus vitamin E placebo, vitamin E 400 IU/day plus selenium placebo, selenium plus vitamin E, or double placebo. At a median follow-up of 5.46 years, compared with the placebo group, the hazard ratio for prostate cancer was 1.04 in the selenium-only group, 1.13 in the vitamin E-only group, and 1.05 in the selenium-plus-vitamin E group. None of the differences was statistically significant.
The Physician’s Health Study18 also found that vitamin E at the same dose given every other day does not prevent prostate cancer.
Finasteride prevents prostate cancer. The PCPT19 included 18,882 men, 55 years of age or older, who had PSA levels of 3.0 ng/mL or less and normal findings on digital rectal examination. Treatment was with finasteride 5 mg/day or placebo. At 7 years, prostate cancer had been discovered in 18.4% of the finasteride group vs 24.4% of the placebo group, a 24.8% reduction (95% CI 18.6–30.6, P < .001). Sexual side effects were more common in the men who received finasteride, while urinary symptoms were more common in the placebo group.
At the time of the original PCPT report in 2003,19 tumors of Gleason grade 7 or higher were more common in the finasteride group, accounting for 37.0% of the tumors discovered, than in the placebo group (22.2%), creating concern that finasteride might somehow cause the tumors that occurred to be more aggressive. However, a subsequent analysis20 found the opposite to be true, ie, that finasteride decreases the risk of high-grade cancers. A companion quality-of-life study showed that chronic use of finasteride had clinically insignificant effects on sexual function, and the PCPT and other studies have shown benefits of finasteride in reducing lower urinary tract symptoms due to benign prostatic hyperplasia (BPH), reducing the risk of acute urinary retention and the need for surgical intervention for BPH, and reducing the risk of prostatitis.
Dutasteride also prevents prostate cancer. A large-scale trial of another 5-alpha reductase inhibitor, dutasteride (Avodart), was reported by Andriole at the annual meeting of the American Urological Association in April 2009.21 The Reduction by Dutasteride of Prostate Events (REDUCE) trial included men who were 50 to 75 years old, inclusively, and who had PSA levels between 2.5 and 10 ng/mL, prostate volume less than 80 cc, and one prior negative prostate biopsy within 6 months of enrollment, representing a group at high risk for cancer on a subsequent biopsy. The trial accrued 8,231 men. At 4 years, prostate cancer had occurred in 659 men in the dutasteride group vs 857 in the placebo group, a 23% reduction (P < .0001). Interestingly, no significant increase in Gleason grade 8 to grade 10 tumors was observed in the study.
Preliminary analyses also suggest that dutasteride enhanced the utility of PSA as a diagnostic test for prostate cancer, had beneficial effects on BPH, and was generally well tolerated. The fact that the results of REDUCE were congruent with those of the PCPT with respect to the magnitude of risk reduction, beneficial effects on benign prostatic hypertrophy, minimal toxicity, and no issues related to tumor grade suggests a class effect for 5-alpha reductase inhibitors, and suggests that these agents should be used more liberally for the prevention of prostate cancer.
There is current debate about whether 5-alpha reductase inhibitors should be used by all men at risk of prostate cancer or only by those at high risk. However, the American Urological Association and the American Society of Clinical Oncology have issued guidelines stating that men at risk should consider this intervention.22
- Andriole GL, Grubb RL, Buys SS, et al; PLCO Project Team. Mortality results from a randomized prostate cancer screening trial. N Engl J Med 2009; 360:1310–1319.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate cancer mortality in a randomized European study. N Engl J Med 2009; 360:1351–1354.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Bratt O. Hereditary prostate cancer: clinical aspects. J Urol 2002; 168:906–913.
- Bill-Axelson A, Holmberg L, Ruutu M, et al; Scandinavian Prostate Cancer Group Study No. 4. Radical prostatectomy versus watchful waiting in early prostate cancer. N Engl J Med 2005; 352:1977–1984.
- Cooperberg MR, Broering JM, Kantoff PW, Carroll PR. Contemporary trends in low risk prostate cancer: risk assessment and treatment. J Urol 2007; 178:S14–S19.
- Horner MJ, Ries LAG, Krapcho M, et al, editors. SEER Cancer Statistics Review, 1975–2006, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER web site, 2009. Accessed 6/28/2009.
- Murphy GP, Natarajan N, Pontes JE, et al. The national survey of prostate cancer in the United States by the American College of Surgeons. J Urol 1982; 127:928–934.
- Catalona WJ, Smith DS, Ratliff TL, Basler JW. Detection of organconfined prostate cancer is increased through prostate-specific antigen-based screening. JAMA 1993; 270:948–954.
- Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level < or = 4.0 ng per milliliter. N Engl J Med 2004; 350:2239–2246.
- Hamilton RJ, Goldberg KC, Platz EA, Freedland SJ. The influence of statin medications on prostate-specific antigen levels. N Natl Cancer Inst 2008; 100:1487–1488.
- Vickers AJ, Savage C, O’Brien MF, Lilja H. Systematic review of pretreatment prostate-specific antigen velocity and doubling time as predictors for prostate cancer. J Clin Oncol 2009; 27:398–403.
- Lilja H, Ulmert D, Vickers AJ. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat Rev Cancer 2008; 8:268–278.
- Marks LS, Fradet Y, Deras IL, et al. PCA molecular urine assay for prostate cancer in men undergoing repeat biopsy. Urology 2007; 69:532–535.
- Sreekumar A, Poisson LM, Thekkelnaycke M, et al. Metabolomic profile delineates potential role for sarcosine in prostate cancer progression. Nature 2009; 457:910–914.
- Zheng SL, Sun J, Wiklund F, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med 2008; 358:910–919.
- Witte JS. Prostate cancer genomics: toward a new understanding. Nat Rev Genet 2009; 10:77–82.
- Gaziano JM, Glynn RJ, Christen WG, et al. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA 2009; 301:52–62.
- Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med 2003; 349:215–224.
- Lucia MS, Darke AK, Goodman PJ, et al. Pathologic characteristics of cancers detected in the Prostate Cancer Prevention Trial: implications for prostate cancer detection and chemoprevention. Cancer Prev Res (Phila PA) 2008; 1:167–173.
- Andriole G, Bostwick D, Brawley O, et al. Further analyses from the REDUCE prostate cancer risk reduction trial [abstract]. J Urol 2009; 181:( suppl):555.
- Kramer BS, Hagerty KL, Justman S, et al; American Society of Clinical Oncology/American Urological Association. Use of 5-alpha-reductase inhibitors for prostate cancer chemoprevention: American Society of Clinical Oncology/American Urological Association 2008 Clinical Practice Guideline. J Urol 2009; 181:1642–1657.
- Andriole GL, Grubb RL, Buys SS, et al; PLCO Project Team. Mortality results from a randomized prostate cancer screening trial. N Engl J Med 2009; 360:1310–1319.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate cancer mortality in a randomized European study. N Engl J Med 2009; 360:1351–1354.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Bratt O. Hereditary prostate cancer: clinical aspects. J Urol 2002; 168:906–913.
- Bill-Axelson A, Holmberg L, Ruutu M, et al; Scandinavian Prostate Cancer Group Study No. 4. Radical prostatectomy versus watchful waiting in early prostate cancer. N Engl J Med 2005; 352:1977–1984.
- Cooperberg MR, Broering JM, Kantoff PW, Carroll PR. Contemporary trends in low risk prostate cancer: risk assessment and treatment. J Urol 2007; 178:S14–S19.
- Horner MJ, Ries LAG, Krapcho M, et al, editors. SEER Cancer Statistics Review, 1975–2006, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER web site, 2009. Accessed 6/28/2009.
- Murphy GP, Natarajan N, Pontes JE, et al. The national survey of prostate cancer in the United States by the American College of Surgeons. J Urol 1982; 127:928–934.
- Catalona WJ, Smith DS, Ratliff TL, Basler JW. Detection of organconfined prostate cancer is increased through prostate-specific antigen-based screening. JAMA 1993; 270:948–954.
- Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level < or = 4.0 ng per milliliter. N Engl J Med 2004; 350:2239–2246.
- Hamilton RJ, Goldberg KC, Platz EA, Freedland SJ. The influence of statin medications on prostate-specific antigen levels. N Natl Cancer Inst 2008; 100:1487–1488.
- Vickers AJ, Savage C, O’Brien MF, Lilja H. Systematic review of pretreatment prostate-specific antigen velocity and doubling time as predictors for prostate cancer. J Clin Oncol 2009; 27:398–403.
- Lilja H, Ulmert D, Vickers AJ. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat Rev Cancer 2008; 8:268–278.
- Marks LS, Fradet Y, Deras IL, et al. PCA molecular urine assay for prostate cancer in men undergoing repeat biopsy. Urology 2007; 69:532–535.
- Sreekumar A, Poisson LM, Thekkelnaycke M, et al. Metabolomic profile delineates potential role for sarcosine in prostate cancer progression. Nature 2009; 457:910–914.
- Zheng SL, Sun J, Wiklund F, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med 2008; 358:910–919.
- Witte JS. Prostate cancer genomics: toward a new understanding. Nat Rev Genet 2009; 10:77–82.
- Gaziano JM, Glynn RJ, Christen WG, et al. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA 2009; 301:52–62.
- Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med 2003; 349:215–224.
- Lucia MS, Darke AK, Goodman PJ, et al. Pathologic characteristics of cancers detected in the Prostate Cancer Prevention Trial: implications for prostate cancer detection and chemoprevention. Cancer Prev Res (Phila PA) 2008; 1:167–173.
- Andriole G, Bostwick D, Brawley O, et al. Further analyses from the REDUCE prostate cancer risk reduction trial [abstract]. J Urol 2009; 181:( suppl):555.
- Kramer BS, Hagerty KL, Justman S, et al; American Society of Clinical Oncology/American Urological Association. Use of 5-alpha-reductase inhibitors for prostate cancer chemoprevention: American Society of Clinical Oncology/American Urological Association 2008 Clinical Practice Guideline. J Urol 2009; 181:1642–1657.
KEY POINTS
- An elevated PSA level lacks specificity as a test for prostate cancer, but PSA measurements can be useful in combination with clinical risk factors or to measure changes in PSA over time.
- Rather than relying on PSA screening alone, we should stratify the risk of prostate cancer on the basis of race, age, PSA level, family history, findings on digital rectal examination, whether the patient has ever undergone a prostate biopsy, and whether the patient is taking finasteride (Proscar). A simple online tool is available to do this.
- There is no PSA level below which the risk of cancer is zero.
- Finasteride has been found in a randomized trial to decrease the risk of prostate cancer, but vitamin E and selenium supplements have failed to show a benefit.
Lupus update: Perspective and clinical pearls
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
KEY POINTS
- Lupus is often misdiagnosed. A person may be given a diagnosis based on a positive antinuclear antibody (ANA) test, a finding that alone is not sufficient to establish the diagnosis. In contrast, some patients with lupus may go several years and see numerous physicians before the proper diagnosis is made.
- One of the major mechanisms for lupus involves defective clearance of apoptotic cells, which act as a source of self-antigens. Because sun exposure can result in massive cell death of keratinocytes (skin cells), protection from the damaging effects of ultraviolet light plays an important role in the management of lupus.
- Patients at any age with SLE should have their modifiable cardiovascular risk factors managed.
- Drugs on the horizon for treating SLE inactivate B cells or their actions.
Autosomal dominant polycystic kidney disease: Emerging concepts of pathogenesis and new treatments
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
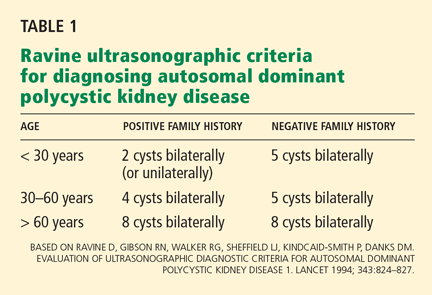
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
KEY POINTS
- In ADPKD the expanding cysts destroy normally functioning kidney tissue, causing hypertension, pain, and other complications, but renal function remains relatively stable until kidney volumes reach a critical size.
- Testing for genetic defects that cause ADPKD is available. The specific mutation involved (PKD1 or PKD2) affects the age of onset and therefore the rate of disease progression as well as the likelihood of cardiovascular complications. Other factors include somatic mutations (“second hits”) of the normal paired chromosome.
- Intracranial aneurysms are a key noncystic feature and may present with a very severe (“sentinel” or “thunderclap”) headache requiring immediate medical attention. Their occurrence is strongly influenced by family history.
- Basic research indicates that patients may be advised to increase their water intake, limit their sodium intake, and avoid caffeine and methylxanthine derivatives.
Update in infectious disease treatment
Studies published during the past year provide information that could influence how we treat several infectious diseases in daily practice. Here is a brief overview of these “impact” studies.
VANCOMYCIN BEATS METRONIDAZOLE FOR SEVERE C DIFFICILE DIARRHEA
Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-assoicated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
Clostridium difficile is the most common infectious cause of nosocomial diarrhea. Furthermore, a unique and highly virulent strain has emerged.
Which drug should be the treatment of choice: metronidazole (Flagyl) or oral vancomycin (Vancocin)? Over time, some infectious disease practitioners have believed that oral vancomycin is superior to oral metronidazole for the treatment of severe C difficile-associated diarrhea. Indeed, in a recently published survey, more than 25% of infectious disease practitioners said they used vancomycin as initial therapy for C difficile-associated diarrhea.1 Until recently, there has been no evidence to support this preference.
Ever since the first description of C difficile-associated diarrhea in the late 1970s, only two head-to-head studies have compared the efficacy of metronidazole vs vancomycin for the treatment of this disorder. Both studies were underpowered and neither was blinded. In 1983, Teasley et al2 treated 101 patients with metronidazole or vancomycin in a non-blinded, nonrandomized study and found no difference in efficacy. In 1996, Wenisch et al,3 in a prospective, randomized, but nonblinded study in 119 patients, compared vancomycin, metronidazole, fusidic acid, and teicoplanin (Targocid) and also found no significant difference in efficacy.
The study. Zar et al,4 in a prospective, double-blind trial at a single institution over an 8-year period, randomized 172 patients with C difficile-associated diarrhea to receive either oral metronidazole 250 mg four times a day or oral vancomycin 125 mg four times a day, both for 10 days. (The appropriate dosage of vancomycin has been debated over the years. In 1989, Fekety et al5 treated patients who had antibiotic-associated C difficile colitis with either 125 or 500 mg of vancomycin, four times a day, and found that the low dosage was as effective as the high dosage.) Both groups also received an oral placebo in addition to the study drug.
In the study of Zar et al, criteria for inclusion were diarrhea (defined as having more than two nonformed stools per 24 hours) and the finding of either toxin A in the stool or pseudomembranes on endoscopic examination. Patients were excluded if they were pregnant, had suspected or proven life-threatening intra-abdominal complications, were allergic to either study drug, had taken one of the study drugs during the last 14 days, or had previously had C difficile-associated diarrhea that did not respond to either study drug.
Patients were followed for up to 21 days. The primary end points were cure, treatment failure, or relapse. Cure was defined as the resolution of diarrhea and no C difficile toxin A detected on stool assay at days 6 and 10.
Disease severity was classified as either mild or severe based on a point system: patients received a single point each for being older than 60 years, being febrile, having an albumin level of less than 2.5 mg/dL, or having a white blood cell count of more than 15 × 109/L. Patients were classified as having severe disease if they had two or more points. They received two points (ie, they were automatically classified as having severe disease) if they had pseudomembranous colitis or if they developed C difficile infection that required treatment in an intensive care unit.
Findings. The overall cure rate in patients receiving vancomycin was 97%, compared with 84% for those on metronidazole (P = .006). This difference was attributable to the group of patients with severe disease; no difference in treatment outcome was found in patients with mild disease. The relapse rates did not differ significantly between treatment groups in patients with either mild or severe disease.
Comments. The study was limited in that it was done at a single center and was done before the current highly virulent strain emerged. Whether these data can be extrapolated to today’s epidemic is unclear. Moreover, the investigators did not test for antimicrobial susceptibility (although metronidazole resistance is still uncommon). Finally, the development of colonization with vancomycin-resistant enterococci, one of the reasons that oral vancomycin is often not recommended, was not assessed.
Despite the study’s limitations, it shows that for severely ill patients with C difficile-associated diarrhea, oral vancomycin should be the treatment of choice.
IS CEFEPIME SAFE?
Yahav D, Paul M, Fraser A, Sarid N, Leibovici L. Efficacy and safety of cefepime: a systematic review and meta-analysis. Lancet Infect Dis 2007; 7:338 348.
Cefepime (Maxipime) is a broad-spectrum, fourth-generation cephalosporin. It is widely used for its approved indications: pneumonia; bacteremia; urinary tract, abdominal, skin, and soft-tissue infections; and febrile neutropenia.
In 2006, Paul et al6 reviewed 33 controlled trials of empiric cefepime monotherapy for febrile neutropenia and found a higher death rate with cefepime than with other beta-lactam antibiotics. That preliminary study spawned the following more comprehensive review by the same group.
The study. Yahav et al7 performed a meta-analysis of randomized trials that compared cefepime with another beta-lactam antibiotic alone or combined with a non-beta-lactam drug given in both treatment groups. Two reviewers independently identified studies from a number of databases and extracted data.
The primary end point was the rate of death from all causes at 30 days. Secondary end points were clinical failure (defined as unresolved infection, treatment modification, or death from infection), failure to eradicate the causative pathogens, superinfection with different bacterial, fungal, or viral organisms, and adverse events.
More than 8,000 patients were involved in 57 trials: 20 trials evaluated therapy for neutropenic fever, 18 for pneumonia, 5 for urogenital infections, 2 for meningitis, and 10 for mixed infections.
Comparison drugs for febrile neutropenia were ceftazidime (Ceptaz, Fortaz, Tazicef); im-ipenem-cilastatin (Primaxin) or meropenem (Merrem); piperacillin-tazobactam (Zosyn); and ceftriaxone (Rocephin). Aminoglycosides were added to both treatment groups in six trials and vancomycin was added in one trial.
For pneumonia, comparison drugs were ceftazidime, ceftriaxone, cefotaxime (Claforan), and cefoperazone-sulbactam.
Adequate allocation concealment and allocation-sequence generation were described in 30 studies. Scores for baseline patient risk factors did not differ significantly between study populations.
Findings. The death rate from all causes was higher in patients taking cefepime than with other beta-lactam antibiotics (risk ratio [RR] 1.26, 95% confidence interval [CI] 1.08–1.49, P = .005). The rate was lower with each of the alternative antibiotics, but the difference was statistically significant only for cefepime vs piperacillin-tazobactam (RR 2.14, 95% CI 1.17–3.89, P = .05).
The rate of death from all causes was higher for cefepime in all types of infections (except urinary tract infection, in which no deaths occurred in any of the treatment arms), although the difference was statistically significant only for febrile neutropenia (RR 1.42, 95% CI 1.09–1.84, P = .009). No differences were found in secondary outcomes, either by disease or by drug used.
Comments. This meta-analysis supports previous findings that more patients die when cefepime is used. The mechanism, however, is unclear. The authors call for reconsideration of the use of cefepime for febrile neutropenia, community-acquired pneumonia, and health-care associated pneumonia. In November 2007, the US Food and Drug Administration (FDA) launched an investigation into the risk of cefepime but has not yet made recommendations. Practitioners should be aware of these data when considering antimicrobial options for treatment in these settings. Knowledge of local antimicrobial susceptibility data of key pathogens is essential in determining optimal empiric and pathogen-specific therapy.
AN ANTIBIOTIC AND A NASAL STEROID ARE INEFFECTIVE IN ACUTE SINUSITIS
Williamson IG, Rumsby K, Benge S, et al. Antibiotics and topical nasal steroid for treatment of acute maxillary sinusitis: a randomized controlled trial. JAMA 2007; 298:2487 2496.
In the United States and Europe 1% to 2% of all primary care office visits are for acute sinusitis. Studies indicate that 67% to nearly 100% of patients with symptoms of sinusitis receive an antibiotic for it, even though the evidence of efficacy is weak and guidelines do not support this practice. Cochrane reviews8,9 have suggested that topical corticosteroids, penicillin, and amoxicillin have marginal benefit in acute sinusitis, but the studies on which the analyses were based were flawed.
The Berg and Carenfelt criteria were developed to help diagnose bacterial sinusitis.10 At the time they were developed, computed tomography was not routinely done to search for sinusitis, so plain film diagnosis was compared with clinical criteria. The Berg and Carenfelt criteria include three symptoms and one sign: a history of purulent unilateral nasal discharge, unilateral facial pain, or bilateral purulent discharge and pus in the nares on inspection. The presence of two criteria has reasonable sensitivity (81%), specificity (89%), and positive predictive value (86%) for detecting acute bacterial or maxillary sinusitis in the office setting.
The study. Williamson et al11 conducted a double-blind, randomized, placebo-controlled trial of antibiotic and topical nasal steroid use in patients with suspected acute maxillary sinusitis. The trial included 240 patients who were seen in 58 family practices over 4 years in the United Kingdom and who had acute nonrecurrent sinusitis based on Berg and Carenfelt criteria. Patients were at least 16 years old; the average age was 44. Three-quarters were women. Few had fever, and 70% met only two Berg and Carenfelt criteria; the remaining 30% met three or all four criteria. Patients were excluded who had at least two sinusitis attacks per year, underlying nasal pathology, significant comorbidities, or a history of penicillin allergy, or if they had been treated with antibiotics or steroids during the past month.
Patients were randomized to receive one of four treatments:
- Amoxicillin 500 mg three times a day for 7 days plus budesonide (Rhinocort) 200 μg in each nostril once a day for 10 days
- Placebo amoxicillin plus real budesonide
- Amoxicillin plus placebo budesonide
- Placebo amoxicillin plus placebo budes-onide.
The groups were well matched. Outcomes were based on a questionnaire and a patient diary that assessed the duration and severity of 11 symptoms.
Findings. No difference was found between the treatment groups in overall outcome, in the proportion of those with symptoms at 10 days, or in daily symptom severity. The secondary analysis suggested that nasal steroids were marginally more effective in patients with less severe symptoms.
The authors concluded that neither an antibiotic nor a nasal steroid, alone or in combination, is effective for acute maxillary sinusitis in the primary care setting, and they recommended against their routine use.
Comments. This study had limitations. Some cases of viral disease may have been included: no objective reference standard (ie, computed tomography of the sinuses or sinus aspiration) was used, and although the Berg and Carenfelt criteria have been validated in secondary care settings, they have not been validated in primary care settings. In addition, fever was absent in most patients, and mild symptoms were poorly defined. Moreover, recruitment of patients was slow, raising questions of bias and generalizability. The study also did not address patients with comorbidities.
Nevertheless, the study shows that outpatients with symptoms of sinusitis without fever or significant comorbidities should not be treated with oral antibiotics or nasal steroids. Otherwise, antibiotic therapy may still be appropriate in certain patients at high risk and in those with fever.
PREDNISOLONE IS BENEFICIAL IN ACUTE BELL PALSY, ACYCLOVIR IS NOT
Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell palsy. N Engl J Med 2007; 357:1598 1607.
Bell palsy accounts for about two-thirds of cases of acute unilateral facial nerve palsy in the United States. Virologic studies from patients undergoing surgery for facial nerve decompression have suggested a possible association with herpes simplex virus. Other causes of acute unilateral facial nerve palsy include Lyme disease, sarcoidosis, Sjögren syndrome, trauma, carotid tumors, and diabetes. Bell palsy occurs most often during middle age, peaking between ages 30 and 45. As many as 30% of patients are left with significant neurologic residua. Corticosteroids and antiviral medications are commonly used to treat Bell palsy, but evidence for their efficacy is weak.
The study. Sullivan et al12 conducted a double-blind, placebo-controlled, randomized trial over 2 years in Scotland with 551 patients, age 16 years or older, recruited within 72 hours of the onset of symptoms. Patients who were pregnant or breastfeeding or who had uncontrolled diabetes, peptic ulcer disease, suppurative otitis, zoster, multiple sclerosis, sarcoidosis, or systemic infection were excluded. They were randomized to treatment for 10 days with either acyclovir (Zovirax) 400 mg five times daily or prednisolone 25 mg twice daily, both agents, or placebo.
The primary outcome was recovery of facial function based on the House-Brackmann grading system. Digital photographs of patients at 3 and 9 months of treatment were evaluated independently by three experts who were unaware of study group assignment or stage of assessment. These included a neurologist, an otorhinolaryn-gologist, and a plastic surgeon. The secondary outcomes were quality of life, facial appearance, and pain, as assessed by the patients.
Findings. At 3 months, 83% of the prednisolone recipients had no facial asymmetry, increasing to 94% at 9 months. In comparison, the numbers were 64% and 82% in those who did not receive prednisolone, and these differences were statistically significant. Acyclovir was found to be of no benefit at either 3 or 9 months.
The authors concluded that early treatment of Bell palsy with prednisolone improves the chance of complete recovery, and that acyclovir alone or in combination with steroids confers no benefit.
Comments. At about the same time that this study was published, Hato et al13 evaluated valacyclovir (Valtrex) plus prednisolone vs placebo plus prednisolone and found that patients with severe Bell palsy (defined as complete facial nerve paralysis) benefited from antiviral therapy.
Corticosteroids are indicated for acute Bell palsy. In patients with complete facial nerve paralysis, valacyclovir should be considered.
POSACONAZOLE AS PROPHYLAXIS IN FEBRILE NEUTROPENIA
Cornely OA, Maertens J, Winston DJ, et al. Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med 2007; 356:348 359.
For many years, amphotericin B was the only drug available for antifungal prophylaxis and therapy. Then, in the early 1990s, a number of studies suggested that the triazoles, notably fluconazole (Diflucan), were effective in a variety of clinical settings for both prophylaxis and therapy of serious fungal infections. In 1992 and 1995, two studies found that fluconazole prophylaxis was as effective as amphotericin B in preventing fungal infections in patients undergoing hematopoietic stem cell transplantation.14,15 Based on these studies, clinical practice changed, not only for patients undergoing hematopoietic stem cell transplantation, but also for empiric antifungal prophylaxis in patients receiving myeloablative chemotherapy to treat hematologic malignancies.
Fluconazole is not active against invasive molds, and newer drugs—itraconazole (Sporanox), voriconazole (Vfend), and most recently posaconazole (Noxafil)—were developed with expanded clinical activity. Studies in the 1990s found that itraconazole and voriconazole performed better than fluconazole but did not provide complete prophylaxis.
The study. Cornely et al16 compared posaconazole with fluconazole or itraconazole in 602 patients undergoing chemotherapy for acute myelogenous leukemia or myelodysplasia. Although patients were randomized to either the posaconazole group or the fluconazole-or-itraconazole group, investigators could choose either fluconazole or itraconazole for patients randomized to that group. Most patients in the latter group (240 of 298) received fluconazole.
Patients were at least 13 years old, were able to take oral medications, had newly diagnosed disease or were having a first relapse, and had or were anticipated to have neutropenia for at least 7 days. The study excluded patients with invasive fungal infection within 30 days, significant liver or kidney dysfunction, an abnormal QT interval corrected for heart rate, an Eastern Cooperative Oncology Group performance status score of more than 2 (in bed more than half of the day), or allergy or a contraindication to azoles.
The trial treatment was started with each cycle of chemotherapy and was continued until recovery from neutropenia and complete remission, until invasive fungal infection developed, or for up to 12 weeks, whichever came first.
The primary end point was the incidence of proven or probable invasive fungal infection during the treatment phase. Secondary end points included death from any cause and time to death.
Findings. Posaconazole recipients fared significantly better than patients in the other treatment group with respect to the incidence of proven or probable invasive fungal infection, invasive aspergillosis, probability of death, death at 100 days, and death secondary to fungal infection. Treatment-related severe adverse events were a bit more common with posaconazole.
The authors suggest that posaconazole prophylaxis may have a place in prophylaxis in patients undergoing chemotherapy for acute myelogenous leukemia or myelodysplasia.
Comments. It is not surprising that posaconazole performed better, because the standard treatment arm contained an agent (fluconazole) that did not cover Aspergillus, the most frequently identified source of invasive fungal infection during the treatment phase of the study.
In an editorial accompanying the article, De Pauw and Donnelly17 pointed out that whether posaconazole prophylaxis would be appropriate in a given case depends upon how likely infection is with Aspergillus. An institution with very few Aspergillus infections would have a much higher number needed to treat with posaconazole to prevent one case of aspergillosis than in this study, in which the number needed to treat was 16. Thus, knowledge of local epidemiology and incidence of invasive mold infections should guide selection of the optimal antifungal agent for prophylaxis in patients undergoing myeloablative chemotherapy for acute myelogenous leukemia or myelodysplasia.
ANIDULAFUNGIN VS FLUCONAZOLE FOR INVASIVE CANDIDIASIS
Reboli AC, Rotstein C, Pappas PG, et al; Anidulafungin Study Group. Anidulafungin versus fluconazole for invasive candidiasis. N Engl J Med 2007; 356:2472 2482.
In 2002, caspofungin (Cancidas) was the first of a new class of drugs, the echinocandins, to be approved by the FDA. The echinocandins have been shown to be as effective as amphotericin B for the treatment of invasive candidiasis, but how they compare with azoles is an ongoing debate. Currently approved treatments for candidiasis, an important cause of disease and death in hospitalized patients, include fluconazole, voriconazole, caspofungin, and amphotericin B. Anidulafungin is the newest echinocandin and has been shown in a phase 2 study to be effective against invasive candidiasis.
The study. Reboli et al18 performed a randomized, double-blind, noninferiority trial comparing anidulafungin and fluconazole to treat candidemia and other forms of candidiasis. The trial was conducted in multiple centers over 15 months and involved 245 patients at least 16 years old who had a single blood culture or culture from a normally sterile site that was positive for Candida species, and who also had one or more of the following: fever, hypothermia, hypotension, local signs and symptoms, or radiographic findings of candidiasis. Patients were excluded if they had had more than 48 hours of systemic therapy with either of these agents or another antifungal drug, if they had had prophylaxis with an azole for more than 7 of the previous 30 days, or if they had refractory candidal infection, elevated liver function test results, Candida krusei infection, meningitis, endocarditis, or osteomyelitis. Removal of central venous catheters was recommended for all patients with candidemia.
Patients were initially stratified by severity of illness based on the Acute Physiology and Chronic Health Evaluation (APACHE II) score (= 20 or > 20, with higher scores indicating more severe disease) and the presence or absence of neutropenia at enrollment. They were then randomly assigned to receive either intravenous anidulafungin (200 mg on day 1 and then 100 mg daily) or intravenous fluconazole (800 mg on day 1 and then 400 mg daily, with the dose adjusted according to creatinine clearance) for at least 14 days after a negative blood culture and improved clinical state and for up to 42 days in total. After 10 days of intravenous therapy, all patients could receive oral fluconazole 400 mg daily at the investigators’ discretion if clinical improvement criteria were met.
The primary end point was global response at the end of intravenous therapy, defined as clinical and microbiologic improvement. A number of secondary end points were also studied. Response failure was defined as no significant clinical improvement, death due to candidiasis, persistent or recurrent candidiasis or a new Candida infection, or an indeterminate response (eg, loss to follow-up or death not attributed to candidiasis).
Of the 245 patients in the primary analysis, 89% had candidemia alone, and nearly two-thirds of those cases were caused by Candida albicans. Only 3% of patients had neutropenia at baseline. Fluconazole resistance was monitored and was rare.
Findings. Intravenous therapy was successful in 76% of patients receiving anidulafungin and in 60% of fluconazole recipients, a difference of 15.4 percentage points (95% CI 3.9–27.0). Results were similar for other efficacy end points. The rate of death from all causes was 31% in the fluconazole group and 23% in the anidulafungin group (P = .13). The frequency and types of adverse events were similar in the two groups. The authors concluded that anidulafungin was not inferior to fluconazole in the treatment of invasive candidiasis.
Comments. Does this study prove that anidulafungin is the treatment of choice for invasive candidiasis? Although the study noted trends in favor of anidulafungin, the differences did not achieve statistical significance for superiority. In addition, the study included so few patients with neutropenia that the results are not applicable to those patients. Finally, anidulafungin is several times more expensive than fluconazole.
Fluconazole has stood the test of time and is probably still the treatment of choice in patients who have suspected or proven candidemia or invasive candidiasis, unless they have already been treated with azoles or are critically ill. In those settings, echinocandins may be the preferred treatment.
- Nielsen ND, Layton BA, McDonald LC, Gerding DN, Liedtke LA, Strausbaugh LJ Infectious Diseases Society of America Emerging Infections Network. Changing epidemiology of Clostridium difficile-associated disease. Infect Dis Clin Pract 2006; 14:296–302.
- Teasley DG, Gerding DN, Olson MM, et al. Prospective randomised trial of metronidazole versus vancomycin for Clostridium-difficile-associated diarrhoea and colitis. Lancet 1983; 2:1043–1046.
- Wenisch C, Parschalk B, Hasenhündl M, Hirschl AM, Graninger W. Comparison of vancomycin, teicoplanin, metronidazole, and fusidic acid for the treatment of Clostridium difficile-associated diarrhea. Clin Infect Dis 1996; 22:813–818. Erratum in: Clin Infect Dis 1996; 23:423.
- Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
- Fekety R, Silva J, Kauffman C, Buggy B, Deery HG. Treatment of antibiotic-associated Clostridium difficile colitis with oral vancomycin: comparison of two dosage regimens. Am J Med 1989; 86:15–19.
- Paul M, Yahav D, Fraser A, Leibovici L. Empirical antibiotic monotherapy for febrile neutropenia: systematic review and meta-analysis of randomized controlled trials. J Antimicrob Chemother 2006; 57:176–189.
- Yahav D, Paul M, Fraser A, Sarid N, Leibovici L. Efficacy and safety of cefepime: a systematic review and meta-analysis. Lancet Infect Dis 2007; 7:338–348.
- Williams JW, Aguilar C, Cornell J, et al. Antibiotics for acute maxillary sinusitis. Cochrane Database Syst Rev 2003; 2:CD000243.
- Zalmanovici A, Yaphe J. Steroids for acute sinusitis. Cochrane Database Syst Rev 2007; 2:CD005149.
- Berg O, Carenfelt C. Analysis of symptoms and clinical signs in the maxillary sinus empyema. Acta Otolaryngol 1988; 105:343–349.
- Williamson IG, Rumsby K, Benge S, et al. Antibiotics and topical nasal steroid for treatment of acute maxillary sinusitis: a randomized controlled trial. JAMA 2007; 298:2487–2496.
- Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med 2007; 357:1598–1607.
- Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol 2007; 28:408–413.
- Goodman JL, Winston DJ, Greenfield RA, et al. A controlled trial of fluconazole to prevent fungal infections in patients undergoing bone marrow transplantation. N Engl J Med 1992; 326:845–851.
- Slavin MA, Osborne B, Adams R, et al. Efficacy and safety of fluconazole prophylaxis for fungal infections after bone marrow transplantation—a prospective, randomized, double-blind study. J Infect Dis 1995; 171:1545–1552.
- Cornely OA, Maertens J, Winston DJ, et al. Posaconazole vs fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med 2007; 356:348–359.
- De Pauw BE, Donnelly JP. Prophylaxis and aspergillosis—Has the principle been proven? N Engl J Med 2007; 356:409–411.
- Reboli AC, Rotstein C, Pappas PG, et al Anidulafungin Study Group. Anidulafungin versus fluconazole for invasive candidiasis. N Engl J Med 2007; 356:2472–2482.
Studies published during the past year provide information that could influence how we treat several infectious diseases in daily practice. Here is a brief overview of these “impact” studies.
VANCOMYCIN BEATS METRONIDAZOLE FOR SEVERE C DIFFICILE DIARRHEA
Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-assoicated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
Clostridium difficile is the most common infectious cause of nosocomial diarrhea. Furthermore, a unique and highly virulent strain has emerged.
Which drug should be the treatment of choice: metronidazole (Flagyl) or oral vancomycin (Vancocin)? Over time, some infectious disease practitioners have believed that oral vancomycin is superior to oral metronidazole for the treatment of severe C difficile-associated diarrhea. Indeed, in a recently published survey, more than 25% of infectious disease practitioners said they used vancomycin as initial therapy for C difficile-associated diarrhea.1 Until recently, there has been no evidence to support this preference.
Ever since the first description of C difficile-associated diarrhea in the late 1970s, only two head-to-head studies have compared the efficacy of metronidazole vs vancomycin for the treatment of this disorder. Both studies were underpowered and neither was blinded. In 1983, Teasley et al2 treated 101 patients with metronidazole or vancomycin in a non-blinded, nonrandomized study and found no difference in efficacy. In 1996, Wenisch et al,3 in a prospective, randomized, but nonblinded study in 119 patients, compared vancomycin, metronidazole, fusidic acid, and teicoplanin (Targocid) and also found no significant difference in efficacy.
The study. Zar et al,4 in a prospective, double-blind trial at a single institution over an 8-year period, randomized 172 patients with C difficile-associated diarrhea to receive either oral metronidazole 250 mg four times a day or oral vancomycin 125 mg four times a day, both for 10 days. (The appropriate dosage of vancomycin has been debated over the years. In 1989, Fekety et al5 treated patients who had antibiotic-associated C difficile colitis with either 125 or 500 mg of vancomycin, four times a day, and found that the low dosage was as effective as the high dosage.) Both groups also received an oral placebo in addition to the study drug.
In the study of Zar et al, criteria for inclusion were diarrhea (defined as having more than two nonformed stools per 24 hours) and the finding of either toxin A in the stool or pseudomembranes on endoscopic examination. Patients were excluded if they were pregnant, had suspected or proven life-threatening intra-abdominal complications, were allergic to either study drug, had taken one of the study drugs during the last 14 days, or had previously had C difficile-associated diarrhea that did not respond to either study drug.
Patients were followed for up to 21 days. The primary end points were cure, treatment failure, or relapse. Cure was defined as the resolution of diarrhea and no C difficile toxin A detected on stool assay at days 6 and 10.
Disease severity was classified as either mild or severe based on a point system: patients received a single point each for being older than 60 years, being febrile, having an albumin level of less than 2.5 mg/dL, or having a white blood cell count of more than 15 × 109/L. Patients were classified as having severe disease if they had two or more points. They received two points (ie, they were automatically classified as having severe disease) if they had pseudomembranous colitis or if they developed C difficile infection that required treatment in an intensive care unit.
Findings. The overall cure rate in patients receiving vancomycin was 97%, compared with 84% for those on metronidazole (P = .006). This difference was attributable to the group of patients with severe disease; no difference in treatment outcome was found in patients with mild disease. The relapse rates did not differ significantly between treatment groups in patients with either mild or severe disease.
Comments. The study was limited in that it was done at a single center and was done before the current highly virulent strain emerged. Whether these data can be extrapolated to today’s epidemic is unclear. Moreover, the investigators did not test for antimicrobial susceptibility (although metronidazole resistance is still uncommon). Finally, the development of colonization with vancomycin-resistant enterococci, one of the reasons that oral vancomycin is often not recommended, was not assessed.
Despite the study’s limitations, it shows that for severely ill patients with C difficile-associated diarrhea, oral vancomycin should be the treatment of choice.
IS CEFEPIME SAFE?
Yahav D, Paul M, Fraser A, Sarid N, Leibovici L. Efficacy and safety of cefepime: a systematic review and meta-analysis. Lancet Infect Dis 2007; 7:338 348.
Cefepime (Maxipime) is a broad-spectrum, fourth-generation cephalosporin. It is widely used for its approved indications: pneumonia; bacteremia; urinary tract, abdominal, skin, and soft-tissue infections; and febrile neutropenia.
In 2006, Paul et al6 reviewed 33 controlled trials of empiric cefepime monotherapy for febrile neutropenia and found a higher death rate with cefepime than with other beta-lactam antibiotics. That preliminary study spawned the following more comprehensive review by the same group.
The study. Yahav et al7 performed a meta-analysis of randomized trials that compared cefepime with another beta-lactam antibiotic alone or combined with a non-beta-lactam drug given in both treatment groups. Two reviewers independently identified studies from a number of databases and extracted data.
The primary end point was the rate of death from all causes at 30 days. Secondary end points were clinical failure (defined as unresolved infection, treatment modification, or death from infection), failure to eradicate the causative pathogens, superinfection with different bacterial, fungal, or viral organisms, and adverse events.
More than 8,000 patients were involved in 57 trials: 20 trials evaluated therapy for neutropenic fever, 18 for pneumonia, 5 for urogenital infections, 2 for meningitis, and 10 for mixed infections.
Comparison drugs for febrile neutropenia were ceftazidime (Ceptaz, Fortaz, Tazicef); im-ipenem-cilastatin (Primaxin) or meropenem (Merrem); piperacillin-tazobactam (Zosyn); and ceftriaxone (Rocephin). Aminoglycosides were added to both treatment groups in six trials and vancomycin was added in one trial.
For pneumonia, comparison drugs were ceftazidime, ceftriaxone, cefotaxime (Claforan), and cefoperazone-sulbactam.
Adequate allocation concealment and allocation-sequence generation were described in 30 studies. Scores for baseline patient risk factors did not differ significantly between study populations.
Findings. The death rate from all causes was higher in patients taking cefepime than with other beta-lactam antibiotics (risk ratio [RR] 1.26, 95% confidence interval [CI] 1.08–1.49, P = .005). The rate was lower with each of the alternative antibiotics, but the difference was statistically significant only for cefepime vs piperacillin-tazobactam (RR 2.14, 95% CI 1.17–3.89, P = .05).
The rate of death from all causes was higher for cefepime in all types of infections (except urinary tract infection, in which no deaths occurred in any of the treatment arms), although the difference was statistically significant only for febrile neutropenia (RR 1.42, 95% CI 1.09–1.84, P = .009). No differences were found in secondary outcomes, either by disease or by drug used.
Comments. This meta-analysis supports previous findings that more patients die when cefepime is used. The mechanism, however, is unclear. The authors call for reconsideration of the use of cefepime for febrile neutropenia, community-acquired pneumonia, and health-care associated pneumonia. In November 2007, the US Food and Drug Administration (FDA) launched an investigation into the risk of cefepime but has not yet made recommendations. Practitioners should be aware of these data when considering antimicrobial options for treatment in these settings. Knowledge of local antimicrobial susceptibility data of key pathogens is essential in determining optimal empiric and pathogen-specific therapy.
AN ANTIBIOTIC AND A NASAL STEROID ARE INEFFECTIVE IN ACUTE SINUSITIS
Williamson IG, Rumsby K, Benge S, et al. Antibiotics and topical nasal steroid for treatment of acute maxillary sinusitis: a randomized controlled trial. JAMA 2007; 298:2487 2496.
In the United States and Europe 1% to 2% of all primary care office visits are for acute sinusitis. Studies indicate that 67% to nearly 100% of patients with symptoms of sinusitis receive an antibiotic for it, even though the evidence of efficacy is weak and guidelines do not support this practice. Cochrane reviews8,9 have suggested that topical corticosteroids, penicillin, and amoxicillin have marginal benefit in acute sinusitis, but the studies on which the analyses were based were flawed.
The Berg and Carenfelt criteria were developed to help diagnose bacterial sinusitis.10 At the time they were developed, computed tomography was not routinely done to search for sinusitis, so plain film diagnosis was compared with clinical criteria. The Berg and Carenfelt criteria include three symptoms and one sign: a history of purulent unilateral nasal discharge, unilateral facial pain, or bilateral purulent discharge and pus in the nares on inspection. The presence of two criteria has reasonable sensitivity (81%), specificity (89%), and positive predictive value (86%) for detecting acute bacterial or maxillary sinusitis in the office setting.
The study. Williamson et al11 conducted a double-blind, randomized, placebo-controlled trial of antibiotic and topical nasal steroid use in patients with suspected acute maxillary sinusitis. The trial included 240 patients who were seen in 58 family practices over 4 years in the United Kingdom and who had acute nonrecurrent sinusitis based on Berg and Carenfelt criteria. Patients were at least 16 years old; the average age was 44. Three-quarters were women. Few had fever, and 70% met only two Berg and Carenfelt criteria; the remaining 30% met three or all four criteria. Patients were excluded who had at least two sinusitis attacks per year, underlying nasal pathology, significant comorbidities, or a history of penicillin allergy, or if they had been treated with antibiotics or steroids during the past month.
Patients were randomized to receive one of four treatments:
- Amoxicillin 500 mg three times a day for 7 days plus budesonide (Rhinocort) 200 μg in each nostril once a day for 10 days
- Placebo amoxicillin plus real budesonide
- Amoxicillin plus placebo budesonide
- Placebo amoxicillin plus placebo budes-onide.
The groups were well matched. Outcomes were based on a questionnaire and a patient diary that assessed the duration and severity of 11 symptoms.
Findings. No difference was found between the treatment groups in overall outcome, in the proportion of those with symptoms at 10 days, or in daily symptom severity. The secondary analysis suggested that nasal steroids were marginally more effective in patients with less severe symptoms.
The authors concluded that neither an antibiotic nor a nasal steroid, alone or in combination, is effective for acute maxillary sinusitis in the primary care setting, and they recommended against their routine use.
Comments. This study had limitations. Some cases of viral disease may have been included: no objective reference standard (ie, computed tomography of the sinuses or sinus aspiration) was used, and although the Berg and Carenfelt criteria have been validated in secondary care settings, they have not been validated in primary care settings. In addition, fever was absent in most patients, and mild symptoms were poorly defined. Moreover, recruitment of patients was slow, raising questions of bias and generalizability. The study also did not address patients with comorbidities.
Nevertheless, the study shows that outpatients with symptoms of sinusitis without fever or significant comorbidities should not be treated with oral antibiotics or nasal steroids. Otherwise, antibiotic therapy may still be appropriate in certain patients at high risk and in those with fever.
PREDNISOLONE IS BENEFICIAL IN ACUTE BELL PALSY, ACYCLOVIR IS NOT
Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell palsy. N Engl J Med 2007; 357:1598 1607.
Bell palsy accounts for about two-thirds of cases of acute unilateral facial nerve palsy in the United States. Virologic studies from patients undergoing surgery for facial nerve decompression have suggested a possible association with herpes simplex virus. Other causes of acute unilateral facial nerve palsy include Lyme disease, sarcoidosis, Sjögren syndrome, trauma, carotid tumors, and diabetes. Bell palsy occurs most often during middle age, peaking between ages 30 and 45. As many as 30% of patients are left with significant neurologic residua. Corticosteroids and antiviral medications are commonly used to treat Bell palsy, but evidence for their efficacy is weak.
The study. Sullivan et al12 conducted a double-blind, placebo-controlled, randomized trial over 2 years in Scotland with 551 patients, age 16 years or older, recruited within 72 hours of the onset of symptoms. Patients who were pregnant or breastfeeding or who had uncontrolled diabetes, peptic ulcer disease, suppurative otitis, zoster, multiple sclerosis, sarcoidosis, or systemic infection were excluded. They were randomized to treatment for 10 days with either acyclovir (Zovirax) 400 mg five times daily or prednisolone 25 mg twice daily, both agents, or placebo.
The primary outcome was recovery of facial function based on the House-Brackmann grading system. Digital photographs of patients at 3 and 9 months of treatment were evaluated independently by three experts who were unaware of study group assignment or stage of assessment. These included a neurologist, an otorhinolaryn-gologist, and a plastic surgeon. The secondary outcomes were quality of life, facial appearance, and pain, as assessed by the patients.
Findings. At 3 months, 83% of the prednisolone recipients had no facial asymmetry, increasing to 94% at 9 months. In comparison, the numbers were 64% and 82% in those who did not receive prednisolone, and these differences were statistically significant. Acyclovir was found to be of no benefit at either 3 or 9 months.
The authors concluded that early treatment of Bell palsy with prednisolone improves the chance of complete recovery, and that acyclovir alone or in combination with steroids confers no benefit.
Comments. At about the same time that this study was published, Hato et al13 evaluated valacyclovir (Valtrex) plus prednisolone vs placebo plus prednisolone and found that patients with severe Bell palsy (defined as complete facial nerve paralysis) benefited from antiviral therapy.
Corticosteroids are indicated for acute Bell palsy. In patients with complete facial nerve paralysis, valacyclovir should be considered.
POSACONAZOLE AS PROPHYLAXIS IN FEBRILE NEUTROPENIA
Cornely OA, Maertens J, Winston DJ, et al. Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med 2007; 356:348 359.
For many years, amphotericin B was the only drug available for antifungal prophylaxis and therapy. Then, in the early 1990s, a number of studies suggested that the triazoles, notably fluconazole (Diflucan), were effective in a variety of clinical settings for both prophylaxis and therapy of serious fungal infections. In 1992 and 1995, two studies found that fluconazole prophylaxis was as effective as amphotericin B in preventing fungal infections in patients undergoing hematopoietic stem cell transplantation.14,15 Based on these studies, clinical practice changed, not only for patients undergoing hematopoietic stem cell transplantation, but also for empiric antifungal prophylaxis in patients receiving myeloablative chemotherapy to treat hematologic malignancies.
Fluconazole is not active against invasive molds, and newer drugs—itraconazole (Sporanox), voriconazole (Vfend), and most recently posaconazole (Noxafil)—were developed with expanded clinical activity. Studies in the 1990s found that itraconazole and voriconazole performed better than fluconazole but did not provide complete prophylaxis.
The study. Cornely et al16 compared posaconazole with fluconazole or itraconazole in 602 patients undergoing chemotherapy for acute myelogenous leukemia or myelodysplasia. Although patients were randomized to either the posaconazole group or the fluconazole-or-itraconazole group, investigators could choose either fluconazole or itraconazole for patients randomized to that group. Most patients in the latter group (240 of 298) received fluconazole.
Patients were at least 13 years old, were able to take oral medications, had newly diagnosed disease or were having a first relapse, and had or were anticipated to have neutropenia for at least 7 days. The study excluded patients with invasive fungal infection within 30 days, significant liver or kidney dysfunction, an abnormal QT interval corrected for heart rate, an Eastern Cooperative Oncology Group performance status score of more than 2 (in bed more than half of the day), or allergy or a contraindication to azoles.
The trial treatment was started with each cycle of chemotherapy and was continued until recovery from neutropenia and complete remission, until invasive fungal infection developed, or for up to 12 weeks, whichever came first.
The primary end point was the incidence of proven or probable invasive fungal infection during the treatment phase. Secondary end points included death from any cause and time to death.
Findings. Posaconazole recipients fared significantly better than patients in the other treatment group with respect to the incidence of proven or probable invasive fungal infection, invasive aspergillosis, probability of death, death at 100 days, and death secondary to fungal infection. Treatment-related severe adverse events were a bit more common with posaconazole.
The authors suggest that posaconazole prophylaxis may have a place in prophylaxis in patients undergoing chemotherapy for acute myelogenous leukemia or myelodysplasia.
Comments. It is not surprising that posaconazole performed better, because the standard treatment arm contained an agent (fluconazole) that did not cover Aspergillus, the most frequently identified source of invasive fungal infection during the treatment phase of the study.
In an editorial accompanying the article, De Pauw and Donnelly17 pointed out that whether posaconazole prophylaxis would be appropriate in a given case depends upon how likely infection is with Aspergillus. An institution with very few Aspergillus infections would have a much higher number needed to treat with posaconazole to prevent one case of aspergillosis than in this study, in which the number needed to treat was 16. Thus, knowledge of local epidemiology and incidence of invasive mold infections should guide selection of the optimal antifungal agent for prophylaxis in patients undergoing myeloablative chemotherapy for acute myelogenous leukemia or myelodysplasia.
ANIDULAFUNGIN VS FLUCONAZOLE FOR INVASIVE CANDIDIASIS
Reboli AC, Rotstein C, Pappas PG, et al; Anidulafungin Study Group. Anidulafungin versus fluconazole for invasive candidiasis. N Engl J Med 2007; 356:2472 2482.
In 2002, caspofungin (Cancidas) was the first of a new class of drugs, the echinocandins, to be approved by the FDA. The echinocandins have been shown to be as effective as amphotericin B for the treatment of invasive candidiasis, but how they compare with azoles is an ongoing debate. Currently approved treatments for candidiasis, an important cause of disease and death in hospitalized patients, include fluconazole, voriconazole, caspofungin, and amphotericin B. Anidulafungin is the newest echinocandin and has been shown in a phase 2 study to be effective against invasive candidiasis.
The study. Reboli et al18 performed a randomized, double-blind, noninferiority trial comparing anidulafungin and fluconazole to treat candidemia and other forms of candidiasis. The trial was conducted in multiple centers over 15 months and involved 245 patients at least 16 years old who had a single blood culture or culture from a normally sterile site that was positive for Candida species, and who also had one or more of the following: fever, hypothermia, hypotension, local signs and symptoms, or radiographic findings of candidiasis. Patients were excluded if they had had more than 48 hours of systemic therapy with either of these agents or another antifungal drug, if they had had prophylaxis with an azole for more than 7 of the previous 30 days, or if they had refractory candidal infection, elevated liver function test results, Candida krusei infection, meningitis, endocarditis, or osteomyelitis. Removal of central venous catheters was recommended for all patients with candidemia.
Patients were initially stratified by severity of illness based on the Acute Physiology and Chronic Health Evaluation (APACHE II) score (= 20 or > 20, with higher scores indicating more severe disease) and the presence or absence of neutropenia at enrollment. They were then randomly assigned to receive either intravenous anidulafungin (200 mg on day 1 and then 100 mg daily) or intravenous fluconazole (800 mg on day 1 and then 400 mg daily, with the dose adjusted according to creatinine clearance) for at least 14 days after a negative blood culture and improved clinical state and for up to 42 days in total. After 10 days of intravenous therapy, all patients could receive oral fluconazole 400 mg daily at the investigators’ discretion if clinical improvement criteria were met.
The primary end point was global response at the end of intravenous therapy, defined as clinical and microbiologic improvement. A number of secondary end points were also studied. Response failure was defined as no significant clinical improvement, death due to candidiasis, persistent or recurrent candidiasis or a new Candida infection, or an indeterminate response (eg, loss to follow-up or death not attributed to candidiasis).
Of the 245 patients in the primary analysis, 89% had candidemia alone, and nearly two-thirds of those cases were caused by Candida albicans. Only 3% of patients had neutropenia at baseline. Fluconazole resistance was monitored and was rare.
Findings. Intravenous therapy was successful in 76% of patients receiving anidulafungin and in 60% of fluconazole recipients, a difference of 15.4 percentage points (95% CI 3.9–27.0). Results were similar for other efficacy end points. The rate of death from all causes was 31% in the fluconazole group and 23% in the anidulafungin group (P = .13). The frequency and types of adverse events were similar in the two groups. The authors concluded that anidulafungin was not inferior to fluconazole in the treatment of invasive candidiasis.
Comments. Does this study prove that anidulafungin is the treatment of choice for invasive candidiasis? Although the study noted trends in favor of anidulafungin, the differences did not achieve statistical significance for superiority. In addition, the study included so few patients with neutropenia that the results are not applicable to those patients. Finally, anidulafungin is several times more expensive than fluconazole.
Fluconazole has stood the test of time and is probably still the treatment of choice in patients who have suspected or proven candidemia or invasive candidiasis, unless they have already been treated with azoles or are critically ill. In those settings, echinocandins may be the preferred treatment.
Studies published during the past year provide information that could influence how we treat several infectious diseases in daily practice. Here is a brief overview of these “impact” studies.
VANCOMYCIN BEATS METRONIDAZOLE FOR SEVERE C DIFFICILE DIARRHEA
Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-assoicated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
Clostridium difficile is the most common infectious cause of nosocomial diarrhea. Furthermore, a unique and highly virulent strain has emerged.
Which drug should be the treatment of choice: metronidazole (Flagyl) or oral vancomycin (Vancocin)? Over time, some infectious disease practitioners have believed that oral vancomycin is superior to oral metronidazole for the treatment of severe C difficile-associated diarrhea. Indeed, in a recently published survey, more than 25% of infectious disease practitioners said they used vancomycin as initial therapy for C difficile-associated diarrhea.1 Until recently, there has been no evidence to support this preference.
Ever since the first description of C difficile-associated diarrhea in the late 1970s, only two head-to-head studies have compared the efficacy of metronidazole vs vancomycin for the treatment of this disorder. Both studies were underpowered and neither was blinded. In 1983, Teasley et al2 treated 101 patients with metronidazole or vancomycin in a non-blinded, nonrandomized study and found no difference in efficacy. In 1996, Wenisch et al,3 in a prospective, randomized, but nonblinded study in 119 patients, compared vancomycin, metronidazole, fusidic acid, and teicoplanin (Targocid) and also found no significant difference in efficacy.
The study. Zar et al,4 in a prospective, double-blind trial at a single institution over an 8-year period, randomized 172 patients with C difficile-associated diarrhea to receive either oral metronidazole 250 mg four times a day or oral vancomycin 125 mg four times a day, both for 10 days. (The appropriate dosage of vancomycin has been debated over the years. In 1989, Fekety et al5 treated patients who had antibiotic-associated C difficile colitis with either 125 or 500 mg of vancomycin, four times a day, and found that the low dosage was as effective as the high dosage.) Both groups also received an oral placebo in addition to the study drug.
In the study of Zar et al, criteria for inclusion were diarrhea (defined as having more than two nonformed stools per 24 hours) and the finding of either toxin A in the stool or pseudomembranes on endoscopic examination. Patients were excluded if they were pregnant, had suspected or proven life-threatening intra-abdominal complications, were allergic to either study drug, had taken one of the study drugs during the last 14 days, or had previously had C difficile-associated diarrhea that did not respond to either study drug.
Patients were followed for up to 21 days. The primary end points were cure, treatment failure, or relapse. Cure was defined as the resolution of diarrhea and no C difficile toxin A detected on stool assay at days 6 and 10.
Disease severity was classified as either mild or severe based on a point system: patients received a single point each for being older than 60 years, being febrile, having an albumin level of less than 2.5 mg/dL, or having a white blood cell count of more than 15 × 109/L. Patients were classified as having severe disease if they had two or more points. They received two points (ie, they were automatically classified as having severe disease) if they had pseudomembranous colitis or if they developed C difficile infection that required treatment in an intensive care unit.
Findings. The overall cure rate in patients receiving vancomycin was 97%, compared with 84% for those on metronidazole (P = .006). This difference was attributable to the group of patients with severe disease; no difference in treatment outcome was found in patients with mild disease. The relapse rates did not differ significantly between treatment groups in patients with either mild or severe disease.
Comments. The study was limited in that it was done at a single center and was done before the current highly virulent strain emerged. Whether these data can be extrapolated to today’s epidemic is unclear. Moreover, the investigators did not test for antimicrobial susceptibility (although metronidazole resistance is still uncommon). Finally, the development of colonization with vancomycin-resistant enterococci, one of the reasons that oral vancomycin is often not recommended, was not assessed.
Despite the study’s limitations, it shows that for severely ill patients with C difficile-associated diarrhea, oral vancomycin should be the treatment of choice.
IS CEFEPIME SAFE?
Yahav D, Paul M, Fraser A, Sarid N, Leibovici L. Efficacy and safety of cefepime: a systematic review and meta-analysis. Lancet Infect Dis 2007; 7:338 348.
Cefepime (Maxipime) is a broad-spectrum, fourth-generation cephalosporin. It is widely used for its approved indications: pneumonia; bacteremia; urinary tract, abdominal, skin, and soft-tissue infections; and febrile neutropenia.
In 2006, Paul et al6 reviewed 33 controlled trials of empiric cefepime monotherapy for febrile neutropenia and found a higher death rate with cefepime than with other beta-lactam antibiotics. That preliminary study spawned the following more comprehensive review by the same group.
The study. Yahav et al7 performed a meta-analysis of randomized trials that compared cefepime with another beta-lactam antibiotic alone or combined with a non-beta-lactam drug given in both treatment groups. Two reviewers independently identified studies from a number of databases and extracted data.
The primary end point was the rate of death from all causes at 30 days. Secondary end points were clinical failure (defined as unresolved infection, treatment modification, or death from infection), failure to eradicate the causative pathogens, superinfection with different bacterial, fungal, or viral organisms, and adverse events.
More than 8,000 patients were involved in 57 trials: 20 trials evaluated therapy for neutropenic fever, 18 for pneumonia, 5 for urogenital infections, 2 for meningitis, and 10 for mixed infections.
Comparison drugs for febrile neutropenia were ceftazidime (Ceptaz, Fortaz, Tazicef); im-ipenem-cilastatin (Primaxin) or meropenem (Merrem); piperacillin-tazobactam (Zosyn); and ceftriaxone (Rocephin). Aminoglycosides were added to both treatment groups in six trials and vancomycin was added in one trial.
For pneumonia, comparison drugs were ceftazidime, ceftriaxone, cefotaxime (Claforan), and cefoperazone-sulbactam.
Adequate allocation concealment and allocation-sequence generation were described in 30 studies. Scores for baseline patient risk factors did not differ significantly between study populations.
Findings. The death rate from all causes was higher in patients taking cefepime than with other beta-lactam antibiotics (risk ratio [RR] 1.26, 95% confidence interval [CI] 1.08–1.49, P = .005). The rate was lower with each of the alternative antibiotics, but the difference was statistically significant only for cefepime vs piperacillin-tazobactam (RR 2.14, 95% CI 1.17–3.89, P = .05).
The rate of death from all causes was higher for cefepime in all types of infections (except urinary tract infection, in which no deaths occurred in any of the treatment arms), although the difference was statistically significant only for febrile neutropenia (RR 1.42, 95% CI 1.09–1.84, P = .009). No differences were found in secondary outcomes, either by disease or by drug used.
Comments. This meta-analysis supports previous findings that more patients die when cefepime is used. The mechanism, however, is unclear. The authors call for reconsideration of the use of cefepime for febrile neutropenia, community-acquired pneumonia, and health-care associated pneumonia. In November 2007, the US Food and Drug Administration (FDA) launched an investigation into the risk of cefepime but has not yet made recommendations. Practitioners should be aware of these data when considering antimicrobial options for treatment in these settings. Knowledge of local antimicrobial susceptibility data of key pathogens is essential in determining optimal empiric and pathogen-specific therapy.
AN ANTIBIOTIC AND A NASAL STEROID ARE INEFFECTIVE IN ACUTE SINUSITIS
Williamson IG, Rumsby K, Benge S, et al. Antibiotics and topical nasal steroid for treatment of acute maxillary sinusitis: a randomized controlled trial. JAMA 2007; 298:2487 2496.
In the United States and Europe 1% to 2% of all primary care office visits are for acute sinusitis. Studies indicate that 67% to nearly 100% of patients with symptoms of sinusitis receive an antibiotic for it, even though the evidence of efficacy is weak and guidelines do not support this practice. Cochrane reviews8,9 have suggested that topical corticosteroids, penicillin, and amoxicillin have marginal benefit in acute sinusitis, but the studies on which the analyses were based were flawed.
The Berg and Carenfelt criteria were developed to help diagnose bacterial sinusitis.10 At the time they were developed, computed tomography was not routinely done to search for sinusitis, so plain film diagnosis was compared with clinical criteria. The Berg and Carenfelt criteria include three symptoms and one sign: a history of purulent unilateral nasal discharge, unilateral facial pain, or bilateral purulent discharge and pus in the nares on inspection. The presence of two criteria has reasonable sensitivity (81%), specificity (89%), and positive predictive value (86%) for detecting acute bacterial or maxillary sinusitis in the office setting.
The study. Williamson et al11 conducted a double-blind, randomized, placebo-controlled trial of antibiotic and topical nasal steroid use in patients with suspected acute maxillary sinusitis. The trial included 240 patients who were seen in 58 family practices over 4 years in the United Kingdom and who had acute nonrecurrent sinusitis based on Berg and Carenfelt criteria. Patients were at least 16 years old; the average age was 44. Three-quarters were women. Few had fever, and 70% met only two Berg and Carenfelt criteria; the remaining 30% met three or all four criteria. Patients were excluded who had at least two sinusitis attacks per year, underlying nasal pathology, significant comorbidities, or a history of penicillin allergy, or if they had been treated with antibiotics or steroids during the past month.
Patients were randomized to receive one of four treatments:
- Amoxicillin 500 mg three times a day for 7 days plus budesonide (Rhinocort) 200 μg in each nostril once a day for 10 days
- Placebo amoxicillin plus real budesonide
- Amoxicillin plus placebo budesonide
- Placebo amoxicillin plus placebo budes-onide.
The groups were well matched. Outcomes were based on a questionnaire and a patient diary that assessed the duration and severity of 11 symptoms.
Findings. No difference was found between the treatment groups in overall outcome, in the proportion of those with symptoms at 10 days, or in daily symptom severity. The secondary analysis suggested that nasal steroids were marginally more effective in patients with less severe symptoms.
The authors concluded that neither an antibiotic nor a nasal steroid, alone or in combination, is effective for acute maxillary sinusitis in the primary care setting, and they recommended against their routine use.
Comments. This study had limitations. Some cases of viral disease may have been included: no objective reference standard (ie, computed tomography of the sinuses or sinus aspiration) was used, and although the Berg and Carenfelt criteria have been validated in secondary care settings, they have not been validated in primary care settings. In addition, fever was absent in most patients, and mild symptoms were poorly defined. Moreover, recruitment of patients was slow, raising questions of bias and generalizability. The study also did not address patients with comorbidities.
Nevertheless, the study shows that outpatients with symptoms of sinusitis without fever or significant comorbidities should not be treated with oral antibiotics or nasal steroids. Otherwise, antibiotic therapy may still be appropriate in certain patients at high risk and in those with fever.
PREDNISOLONE IS BENEFICIAL IN ACUTE BELL PALSY, ACYCLOVIR IS NOT
Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell palsy. N Engl J Med 2007; 357:1598 1607.
Bell palsy accounts for about two-thirds of cases of acute unilateral facial nerve palsy in the United States. Virologic studies from patients undergoing surgery for facial nerve decompression have suggested a possible association with herpes simplex virus. Other causes of acute unilateral facial nerve palsy include Lyme disease, sarcoidosis, Sjögren syndrome, trauma, carotid tumors, and diabetes. Bell palsy occurs most often during middle age, peaking between ages 30 and 45. As many as 30% of patients are left with significant neurologic residua. Corticosteroids and antiviral medications are commonly used to treat Bell palsy, but evidence for their efficacy is weak.
The study. Sullivan et al12 conducted a double-blind, placebo-controlled, randomized trial over 2 years in Scotland with 551 patients, age 16 years or older, recruited within 72 hours of the onset of symptoms. Patients who were pregnant or breastfeeding or who had uncontrolled diabetes, peptic ulcer disease, suppurative otitis, zoster, multiple sclerosis, sarcoidosis, or systemic infection were excluded. They were randomized to treatment for 10 days with either acyclovir (Zovirax) 400 mg five times daily or prednisolone 25 mg twice daily, both agents, or placebo.
The primary outcome was recovery of facial function based on the House-Brackmann grading system. Digital photographs of patients at 3 and 9 months of treatment were evaluated independently by three experts who were unaware of study group assignment or stage of assessment. These included a neurologist, an otorhinolaryn-gologist, and a plastic surgeon. The secondary outcomes were quality of life, facial appearance, and pain, as assessed by the patients.
Findings. At 3 months, 83% of the prednisolone recipients had no facial asymmetry, increasing to 94% at 9 months. In comparison, the numbers were 64% and 82% in those who did not receive prednisolone, and these differences were statistically significant. Acyclovir was found to be of no benefit at either 3 or 9 months.
The authors concluded that early treatment of Bell palsy with prednisolone improves the chance of complete recovery, and that acyclovir alone or in combination with steroids confers no benefit.
Comments. At about the same time that this study was published, Hato et al13 evaluated valacyclovir (Valtrex) plus prednisolone vs placebo plus prednisolone and found that patients with severe Bell palsy (defined as complete facial nerve paralysis) benefited from antiviral therapy.
Corticosteroids are indicated for acute Bell palsy. In patients with complete facial nerve paralysis, valacyclovir should be considered.
POSACONAZOLE AS PROPHYLAXIS IN FEBRILE NEUTROPENIA
Cornely OA, Maertens J, Winston DJ, et al. Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med 2007; 356:348 359.
For many years, amphotericin B was the only drug available for antifungal prophylaxis and therapy. Then, in the early 1990s, a number of studies suggested that the triazoles, notably fluconazole (Diflucan), were effective in a variety of clinical settings for both prophylaxis and therapy of serious fungal infections. In 1992 and 1995, two studies found that fluconazole prophylaxis was as effective as amphotericin B in preventing fungal infections in patients undergoing hematopoietic stem cell transplantation.14,15 Based on these studies, clinical practice changed, not only for patients undergoing hematopoietic stem cell transplantation, but also for empiric antifungal prophylaxis in patients receiving myeloablative chemotherapy to treat hematologic malignancies.
Fluconazole is not active against invasive molds, and newer drugs—itraconazole (Sporanox), voriconazole (Vfend), and most recently posaconazole (Noxafil)—were developed with expanded clinical activity. Studies in the 1990s found that itraconazole and voriconazole performed better than fluconazole but did not provide complete prophylaxis.
The study. Cornely et al16 compared posaconazole with fluconazole or itraconazole in 602 patients undergoing chemotherapy for acute myelogenous leukemia or myelodysplasia. Although patients were randomized to either the posaconazole group or the fluconazole-or-itraconazole group, investigators could choose either fluconazole or itraconazole for patients randomized to that group. Most patients in the latter group (240 of 298) received fluconazole.
Patients were at least 13 years old, were able to take oral medications, had newly diagnosed disease or were having a first relapse, and had or were anticipated to have neutropenia for at least 7 days. The study excluded patients with invasive fungal infection within 30 days, significant liver or kidney dysfunction, an abnormal QT interval corrected for heart rate, an Eastern Cooperative Oncology Group performance status score of more than 2 (in bed more than half of the day), or allergy or a contraindication to azoles.
The trial treatment was started with each cycle of chemotherapy and was continued until recovery from neutropenia and complete remission, until invasive fungal infection developed, or for up to 12 weeks, whichever came first.
The primary end point was the incidence of proven or probable invasive fungal infection during the treatment phase. Secondary end points included death from any cause and time to death.
Findings. Posaconazole recipients fared significantly better than patients in the other treatment group with respect to the incidence of proven or probable invasive fungal infection, invasive aspergillosis, probability of death, death at 100 days, and death secondary to fungal infection. Treatment-related severe adverse events were a bit more common with posaconazole.
The authors suggest that posaconazole prophylaxis may have a place in prophylaxis in patients undergoing chemotherapy for acute myelogenous leukemia or myelodysplasia.
Comments. It is not surprising that posaconazole performed better, because the standard treatment arm contained an agent (fluconazole) that did not cover Aspergillus, the most frequently identified source of invasive fungal infection during the treatment phase of the study.
In an editorial accompanying the article, De Pauw and Donnelly17 pointed out that whether posaconazole prophylaxis would be appropriate in a given case depends upon how likely infection is with Aspergillus. An institution with very few Aspergillus infections would have a much higher number needed to treat with posaconazole to prevent one case of aspergillosis than in this study, in which the number needed to treat was 16. Thus, knowledge of local epidemiology and incidence of invasive mold infections should guide selection of the optimal antifungal agent for prophylaxis in patients undergoing myeloablative chemotherapy for acute myelogenous leukemia or myelodysplasia.
ANIDULAFUNGIN VS FLUCONAZOLE FOR INVASIVE CANDIDIASIS
Reboli AC, Rotstein C, Pappas PG, et al; Anidulafungin Study Group. Anidulafungin versus fluconazole for invasive candidiasis. N Engl J Med 2007; 356:2472 2482.
In 2002, caspofungin (Cancidas) was the first of a new class of drugs, the echinocandins, to be approved by the FDA. The echinocandins have been shown to be as effective as amphotericin B for the treatment of invasive candidiasis, but how they compare with azoles is an ongoing debate. Currently approved treatments for candidiasis, an important cause of disease and death in hospitalized patients, include fluconazole, voriconazole, caspofungin, and amphotericin B. Anidulafungin is the newest echinocandin and has been shown in a phase 2 study to be effective against invasive candidiasis.
The study. Reboli et al18 performed a randomized, double-blind, noninferiority trial comparing anidulafungin and fluconazole to treat candidemia and other forms of candidiasis. The trial was conducted in multiple centers over 15 months and involved 245 patients at least 16 years old who had a single blood culture or culture from a normally sterile site that was positive for Candida species, and who also had one or more of the following: fever, hypothermia, hypotension, local signs and symptoms, or radiographic findings of candidiasis. Patients were excluded if they had had more than 48 hours of systemic therapy with either of these agents or another antifungal drug, if they had had prophylaxis with an azole for more than 7 of the previous 30 days, or if they had refractory candidal infection, elevated liver function test results, Candida krusei infection, meningitis, endocarditis, or osteomyelitis. Removal of central venous catheters was recommended for all patients with candidemia.
Patients were initially stratified by severity of illness based on the Acute Physiology and Chronic Health Evaluation (APACHE II) score (= 20 or > 20, with higher scores indicating more severe disease) and the presence or absence of neutropenia at enrollment. They were then randomly assigned to receive either intravenous anidulafungin (200 mg on day 1 and then 100 mg daily) or intravenous fluconazole (800 mg on day 1 and then 400 mg daily, with the dose adjusted according to creatinine clearance) for at least 14 days after a negative blood culture and improved clinical state and for up to 42 days in total. After 10 days of intravenous therapy, all patients could receive oral fluconazole 400 mg daily at the investigators’ discretion if clinical improvement criteria were met.
The primary end point was global response at the end of intravenous therapy, defined as clinical and microbiologic improvement. A number of secondary end points were also studied. Response failure was defined as no significant clinical improvement, death due to candidiasis, persistent or recurrent candidiasis or a new Candida infection, or an indeterminate response (eg, loss to follow-up or death not attributed to candidiasis).
Of the 245 patients in the primary analysis, 89% had candidemia alone, and nearly two-thirds of those cases were caused by Candida albicans. Only 3% of patients had neutropenia at baseline. Fluconazole resistance was monitored and was rare.
Findings. Intravenous therapy was successful in 76% of patients receiving anidulafungin and in 60% of fluconazole recipients, a difference of 15.4 percentage points (95% CI 3.9–27.0). Results were similar for other efficacy end points. The rate of death from all causes was 31% in the fluconazole group and 23% in the anidulafungin group (P = .13). The frequency and types of adverse events were similar in the two groups. The authors concluded that anidulafungin was not inferior to fluconazole in the treatment of invasive candidiasis.
Comments. Does this study prove that anidulafungin is the treatment of choice for invasive candidiasis? Although the study noted trends in favor of anidulafungin, the differences did not achieve statistical significance for superiority. In addition, the study included so few patients with neutropenia that the results are not applicable to those patients. Finally, anidulafungin is several times more expensive than fluconazole.
Fluconazole has stood the test of time and is probably still the treatment of choice in patients who have suspected or proven candidemia or invasive candidiasis, unless they have already been treated with azoles or are critically ill. In those settings, echinocandins may be the preferred treatment.
- Nielsen ND, Layton BA, McDonald LC, Gerding DN, Liedtke LA, Strausbaugh LJ Infectious Diseases Society of America Emerging Infections Network. Changing epidemiology of Clostridium difficile-associated disease. Infect Dis Clin Pract 2006; 14:296–302.
- Teasley DG, Gerding DN, Olson MM, et al. Prospective randomised trial of metronidazole versus vancomycin for Clostridium-difficile-associated diarrhoea and colitis. Lancet 1983; 2:1043–1046.
- Wenisch C, Parschalk B, Hasenhündl M, Hirschl AM, Graninger W. Comparison of vancomycin, teicoplanin, metronidazole, and fusidic acid for the treatment of Clostridium difficile-associated diarrhea. Clin Infect Dis 1996; 22:813–818. Erratum in: Clin Infect Dis 1996; 23:423.
- Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
- Fekety R, Silva J, Kauffman C, Buggy B, Deery HG. Treatment of antibiotic-associated Clostridium difficile colitis with oral vancomycin: comparison of two dosage regimens. Am J Med 1989; 86:15–19.
- Paul M, Yahav D, Fraser A, Leibovici L. Empirical antibiotic monotherapy for febrile neutropenia: systematic review and meta-analysis of randomized controlled trials. J Antimicrob Chemother 2006; 57:176–189.
- Yahav D, Paul M, Fraser A, Sarid N, Leibovici L. Efficacy and safety of cefepime: a systematic review and meta-analysis. Lancet Infect Dis 2007; 7:338–348.
- Williams JW, Aguilar C, Cornell J, et al. Antibiotics for acute maxillary sinusitis. Cochrane Database Syst Rev 2003; 2:CD000243.
- Zalmanovici A, Yaphe J. Steroids for acute sinusitis. Cochrane Database Syst Rev 2007; 2:CD005149.
- Berg O, Carenfelt C. Analysis of symptoms and clinical signs in the maxillary sinus empyema. Acta Otolaryngol 1988; 105:343–349.
- Williamson IG, Rumsby K, Benge S, et al. Antibiotics and topical nasal steroid for treatment of acute maxillary sinusitis: a randomized controlled trial. JAMA 2007; 298:2487–2496.
- Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med 2007; 357:1598–1607.
- Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol 2007; 28:408–413.
- Goodman JL, Winston DJ, Greenfield RA, et al. A controlled trial of fluconazole to prevent fungal infections in patients undergoing bone marrow transplantation. N Engl J Med 1992; 326:845–851.
- Slavin MA, Osborne B, Adams R, et al. Efficacy and safety of fluconazole prophylaxis for fungal infections after bone marrow transplantation—a prospective, randomized, double-blind study. J Infect Dis 1995; 171:1545–1552.
- Cornely OA, Maertens J, Winston DJ, et al. Posaconazole vs fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med 2007; 356:348–359.
- De Pauw BE, Donnelly JP. Prophylaxis and aspergillosis—Has the principle been proven? N Engl J Med 2007; 356:409–411.
- Reboli AC, Rotstein C, Pappas PG, et al Anidulafungin Study Group. Anidulafungin versus fluconazole for invasive candidiasis. N Engl J Med 2007; 356:2472–2482.
- Nielsen ND, Layton BA, McDonald LC, Gerding DN, Liedtke LA, Strausbaugh LJ Infectious Diseases Society of America Emerging Infections Network. Changing epidemiology of Clostridium difficile-associated disease. Infect Dis Clin Pract 2006; 14:296–302.
- Teasley DG, Gerding DN, Olson MM, et al. Prospective randomised trial of metronidazole versus vancomycin for Clostridium-difficile-associated diarrhoea and colitis. Lancet 1983; 2:1043–1046.
- Wenisch C, Parschalk B, Hasenhündl M, Hirschl AM, Graninger W. Comparison of vancomycin, teicoplanin, metronidazole, and fusidic acid for the treatment of Clostridium difficile-associated diarrhea. Clin Infect Dis 1996; 22:813–818. Erratum in: Clin Infect Dis 1996; 23:423.
- Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
- Fekety R, Silva J, Kauffman C, Buggy B, Deery HG. Treatment of antibiotic-associated Clostridium difficile colitis with oral vancomycin: comparison of two dosage regimens. Am J Med 1989; 86:15–19.
- Paul M, Yahav D, Fraser A, Leibovici L. Empirical antibiotic monotherapy for febrile neutropenia: systematic review and meta-analysis of randomized controlled trials. J Antimicrob Chemother 2006; 57:176–189.
- Yahav D, Paul M, Fraser A, Sarid N, Leibovici L. Efficacy and safety of cefepime: a systematic review and meta-analysis. Lancet Infect Dis 2007; 7:338–348.
- Williams JW, Aguilar C, Cornell J, et al. Antibiotics for acute maxillary sinusitis. Cochrane Database Syst Rev 2003; 2:CD000243.
- Zalmanovici A, Yaphe J. Steroids for acute sinusitis. Cochrane Database Syst Rev 2007; 2:CD005149.
- Berg O, Carenfelt C. Analysis of symptoms and clinical signs in the maxillary sinus empyema. Acta Otolaryngol 1988; 105:343–349.
- Williamson IG, Rumsby K, Benge S, et al. Antibiotics and topical nasal steroid for treatment of acute maxillary sinusitis: a randomized controlled trial. JAMA 2007; 298:2487–2496.
- Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med 2007; 357:1598–1607.
- Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol 2007; 28:408–413.
- Goodman JL, Winston DJ, Greenfield RA, et al. A controlled trial of fluconazole to prevent fungal infections in patients undergoing bone marrow transplantation. N Engl J Med 1992; 326:845–851.
- Slavin MA, Osborne B, Adams R, et al. Efficacy and safety of fluconazole prophylaxis for fungal infections after bone marrow transplantation—a prospective, randomized, double-blind study. J Infect Dis 1995; 171:1545–1552.
- Cornely OA, Maertens J, Winston DJ, et al. Posaconazole vs fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med 2007; 356:348–359.
- De Pauw BE, Donnelly JP. Prophylaxis and aspergillosis—Has the principle been proven? N Engl J Med 2007; 356:409–411.
- Reboli AC, Rotstein C, Pappas PG, et al Anidulafungin Study Group. Anidulafungin versus fluconazole for invasive candidiasis. N Engl J Med 2007; 356:2472–2482.
Update on infectious disease prevention: Human papillomavirus, hepatitis A
How we prevent human papillomavirus (HPV) infection, and how we prevent hepatitis A following exposure to an index case have changed, based on the results of several key clinical trials published during the past year. The results of these studies should influence the measures we take in our daily practice to prevent these diseases. Here is a brief overview of these “impact” studies.
QUADRIVALENT HPV VACCINE PREVENTS CERVICAL LESIONS
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Cervical cancer is the second most common type of cancer in women and is the leading cause of cancer-related deaths in developing countries. More than 500,000 new cases of cervical cancer are reported worldwide each year, and about 250,000 women die of it.1
Nearly all cases of cervical cancer are caused by HPVs, and the oncogenic types HPV-16 and HPV-18 together account for about 70%. These two types also cause vulvo-vaginal cancer, which accounts for about 6% of all gynecologic malignancies.2 Two other HPV types, HPV-6 and HPV-11, cause genital warts and, less often, cervical intraepithelial neoplasia and cervical invasive cancers.
Two HPV vaccines have been developed. One, sold as Cervarix, is directed against HPV-16 and HPV-18; it is not yet available in the United States. The other, sold as Gardasil, is directed against four HPV types: 6, 11, 16, and 18, and it is currently available (reviewed by Widdice and Kahn3).
The study. The Females United to Unilaterally Reduce Endo/Ectocervical Cancer (FUTURE) II study4 assessed the ability of the quadrivalent vaccine to prevent high-grade cervical lesions. Between June 2002 and September 2003, more than 12,000 women ages 15 to 26 were enrolled at 90 sites in 13 countries. Eligible women were not pregnant, had no abnormal Papanicolaou (Pap) smear, had had four or fewer lifetime sexual partners, and agreed to use effective contraception throughout the course of the study.
In a randomized, double-blind fashion, patients received vaccine or a placebo injection at day 1 and again 2 and 6 months later. They returned for follow-up 1, 6, 24, 36, and 48 months after the third injection, with Pap smears and colposcopy of cervical lesions.
The primary composite end point was the development of grade 2 or 3 cervical intraepithelial neoplasia, adenocarcinoma in situ, or invasive cervical carcinoma, with detection of HPV-16 or HPV-18 or both in one or more of the adjacent sections of the same lesion.
In all, 6,087 patients received vaccine and 6,080 received placebo; the two groups were well matched. About 23% had serologic evidence of exposure to either HPV-16 or HPV-18 at enrollment.
Findings. In the analysis of the data, the patients were divided into three overlapping subgroups. The first comprised women who had no serologic evidence of HPV-16 or HPV-18 infection at enrollment, who received all three injections, who remained DNA-negative at month 7, and who had no protocol violations. In this “per-protocol susceptible population,” at an average of 3 years of follow-up, lesions associated with HPV-16 or HPV-18 had developed in 42 of 5,260 women who received placebo, compared with only 1 of 5,305 who received the vaccine. The vaccine efficacy was calculated at 98% (95% confidence interval [CI] 86–100).
The second subgroup were women who had no evidence of HPV-16 or HPV-18 infection at baseline, but whose compliance with the protocol was considered imperfect. In this “unrestricted susceptible population,” the vaccine efficacy was 95% (95% CI 85–99).
The third group included all comers, regardless of whether they were already infected at baseline. In this “intention-to-treat population,” the vaccine efficacy was 44% (95% CI 26–58).
The authors concluded that in young women not previously infected with HPV-16 or HPV-18, vaccine recipients had a significantly lower occurrence of high-grade cervical intraepithelial neoplasia related to these two oncogenic HPV types.
QUADRIVALENT HPV VACCINE PREVENTS ANOGENITAL DISEASE
Garland SM, Hernandez-Avila M, Wheeler CM, et al; Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. J Engl J Med 2007; 356:1928–1943.
The study. This double-blind, placebo-controlled study5 tested the usefulness of the quadrivalent HPV vaccine to prevent anogenital disease. It included 5,400 women ages 16 to 24 and was conducted over 14 months in 2002 and 2003 at 62 sites in 16 countries. Women received vaccine or placebo at day 1 and again 2 and 6 months later, and then underwent anogenital and gynecologic examinations at intervals for up to 4 years.
The co-primary composite end points were the incidence of genital warts, vulvar or vaginal intraepithelial neoplasia or cancer, cervical intraepithelial neoplasia, cervical adenocarcinoma in situ, or cervical cancer associated with HPV types 6, 11, 16, or 18.
Findings. In all, 2,700 women were assigned to receive vaccine and 2,700 to receive placebo, and they were followed for an average of 3 years. Twenty percent had pre-existing serologic evidence of infection with one of these four HPV types. In the per-protocol population who were seronegative at day 1 and were compliant, the vaccine efficacy was 100%. In the intention-to-treat group, vaccine reduced the rate of vulvar or vaginal perianal lesions regardless of HPV type by 34%, and reduced the rate of cervical lesions regardless of type by 20%.
HPV VACCINE LIKELY COST-EFFECTIVE IN GIRLS, BUT NOT BOYS
Newall AT, Beutels P, Wood JG, Edmunds WJ, MacIntyre CR. Cost-effectiveness analyses of human papillomavirus vaccination. Lancet Infect Dis 2007; 7:289–296.
The study. In a review, Newall et al6 looked at four studies that examined the cost-effectiveness of the HPV vaccine. These studies were not perfect and had methodologic limitations because of uncertainty about vaccine efficacy, duration of protection, and the contribution of herd immunity. The studies nevertheless suggested that immunization of young girls but not young boys may be cost-effective, though they suggested the need for further research.
Findings. Three of the studies showed an incremental cost-effectiveness ratio of $14,000 to $24,000 per quality-adjusted year of life gained, which is well within the range for many preventive strategies that we employ in this country.
One of the studies examined the cost-effectiveness of immunizing males, and in that study it was found not to be cost-effective.
TAKE-HOME POINTS ON HPV VACCINATION
Quadrivalent vaccine does indeed reduce the incidence of HPV-associated cervical intra-epithelial neoplasia, vulvar and vaginal intra-epithelial neoplasia, and anogenital diseases in young women, and it is likely cost-effective.
The vaccine works only against HPV types 6, 11, 16, and 18, and 30% of cervical cancers are due to types other than HPV-16 and HPV-18. Also, vaccination is much more effective in patients not yet exposed to HPV, so it would be best to vaccinate them before they become sexually active.
The Advisory Committee on Immunization Practices voted to recommend that girls ages 11 to 12 in this country should receive vaccine.
Regrettably, many third-party payers do not yet pay for the vaccine, and the cost (around $375) must be paid out of pocket. Also, this issue remains politically charged and controversial. Some states have mandated vaccination and another 15 are presently considering legislation mandating vaccination. Such legislation has been defeated in four states.
My own practice is to offer the vaccine to 11- and 12-year old girls, and to older girls and young women (not to boys), especially if the health insurance plan covers it or if the patient or the patient’s family can afford it.
HEPATITIS A VACCINE IS AS GOOD AS IMMUNE GLOBULIN AFTER EXPOSURE
Victor JC, Monto AS, Surdina TY, et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med 2007; 357:1685–1694.
Before 1995, when the first hepatitis A vaccine was introduced, about 30,000 cases of hepatitis A were reported each year in the United States. This was thought to be the tip of the iceberg: since this infection is often subclinical, estimates of up to 300,000 cases per year were given.
At first, immunization against hepatitis A in this country was confined to children over age 2 in states in which hepatitis A occurred more often than the norm. In 2005, after it had become clear that the vaccine was highly effective, the Advisory Committee on Immunization Practices revised its recommendations to include immunization of children between the ages of 12 and 23 months,7 so that they would complete this two-stage vaccination procedure by the time they reached the age of 2 years. With that strategy, the annual occurrence of hepatitis A in the United States fell dramatically, to about 4,000 cases per year in 2005, the lowest number of cases reported in the last 40 years. At present, most hepatitis A infections in this country are not from casual idiosyncratic transmission but rather are food-borne.
Still, hepatitis A remains a major problem in many parts of the world. Moreover, the availability of immune globulin, the traditional recommended agent for postexposure pro-phylaxis, has been limited because only one company manufactures it and the price has steadily escalated.
The study. Investigators at the University of Michigan and in Kazakhstan compared conventional doses of immune globulin vs hepatitis A vaccine as postexposure prophylaxis, given within 14 days of exposure to index cases of hepatitis A.8 Excluded were persons under the age of 2 years or over the age of 40, those with a history of hepatitis A or vaccination, those with liver disease, and those with other contraindications. The primary end point was the development of symptomatic, laboratory-confirmed hepatitis A, defined as a positive test for immunoglobulin M antibodies to hepatitis A; transaminase levels greater than two times the upper limit of normal; and symptoms consistent with hepatitis A in the absence of another identifiable disease that occurred within 15 to 56 days of exposure to the index case.
Findings. Of 4,524 contacts randomized, only 1,414 (31%) were susceptible to hepatitis A, suggesting that the prevalence of hepatitis A in Kazakhstan was high at that time. Of these, 1,090 completed the immunization and follow-up protocol and were eligible for the final analysis. Of these, 568 received vaccine and 522 received globulin. The average age was 12 years, the average time to vaccination after exposure was 10 days; 16% of the exposures occurred in the day-care setting, and 84% of the exposures occurred from household contacts.
Symptomatic hepatitis A occurred in 4.4% of vaccine recipients vs 3.3% of immunoglobulin recipients. The authors concluded that hepatitis A vaccine met the test of noninferiority, that both strategies were highly protective, but that immunoglobulin was modestly better. Thus, in June 2007, the Advisory Committee on Immunization Practices recommended hepatitis A vaccine as the preferred regimen for postexposure prophylaxis.9
This approach has several advantages:
- Hepatitis A vaccine confers immunity and long-term protection, which globulin does not
- The supply of vaccine is abundant
- Vaccine is relatively cheap
- Vaccine is easy to give.
This study, however, does not apply to people younger than 2 years or older than 40, those who are immunocompromised, or those who have chronic liver disease. In these groups, the recommendation is still to use immunoglobulin in postexposure prophylaxis.
- CancerMondial. International Agency for Research on Cancer. www-dep.iarc.fr/. Accessed May 12, 2008.
- Munoz N, Bosch FX, de Sanjose S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003; 348:518–527.
- Widdice LE, Kahn JA. Using the new HPV vaccines in clinical practice. Cleve Clin J Med 2006; 73:929–935.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Garland SM, Hernandez-Avila M, Wheeler CM, et al Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) I Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med 2007; 356:1928–1943.
- Newall AT, Beutels P, Wood JG, Edmunds WJ, MacIntyre CR. Cost-effectiveness analyses of human papillomavirus vaccination. Lancet Infect Dis 2007; 7:289–296.
- Advisory Committee on Immunization Practices (ACIP)Fiore AE, Wasley A, Bell BP. Prevention of hepatitis A through active or passive immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55( RR–7):1–23.
- Victor JC, Monto AS, Surdina TY, et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med 2007; 357:1685–1694.
- Advisory Committee on Immunization Practices, US Centers for Disease Control and Prevention. Update: prevention of hepatitis A after exposure to hepatitis A virus and in international travelers. Updated recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2007; 56:1080–1084.
How we prevent human papillomavirus (HPV) infection, and how we prevent hepatitis A following exposure to an index case have changed, based on the results of several key clinical trials published during the past year. The results of these studies should influence the measures we take in our daily practice to prevent these diseases. Here is a brief overview of these “impact” studies.
QUADRIVALENT HPV VACCINE PREVENTS CERVICAL LESIONS
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Cervical cancer is the second most common type of cancer in women and is the leading cause of cancer-related deaths in developing countries. More than 500,000 new cases of cervical cancer are reported worldwide each year, and about 250,000 women die of it.1
Nearly all cases of cervical cancer are caused by HPVs, and the oncogenic types HPV-16 and HPV-18 together account for about 70%. These two types also cause vulvo-vaginal cancer, which accounts for about 6% of all gynecologic malignancies.2 Two other HPV types, HPV-6 and HPV-11, cause genital warts and, less often, cervical intraepithelial neoplasia and cervical invasive cancers.
Two HPV vaccines have been developed. One, sold as Cervarix, is directed against HPV-16 and HPV-18; it is not yet available in the United States. The other, sold as Gardasil, is directed against four HPV types: 6, 11, 16, and 18, and it is currently available (reviewed by Widdice and Kahn3).
The study. The Females United to Unilaterally Reduce Endo/Ectocervical Cancer (FUTURE) II study4 assessed the ability of the quadrivalent vaccine to prevent high-grade cervical lesions. Between June 2002 and September 2003, more than 12,000 women ages 15 to 26 were enrolled at 90 sites in 13 countries. Eligible women were not pregnant, had no abnormal Papanicolaou (Pap) smear, had had four or fewer lifetime sexual partners, and agreed to use effective contraception throughout the course of the study.
In a randomized, double-blind fashion, patients received vaccine or a placebo injection at day 1 and again 2 and 6 months later. They returned for follow-up 1, 6, 24, 36, and 48 months after the third injection, with Pap smears and colposcopy of cervical lesions.
The primary composite end point was the development of grade 2 or 3 cervical intraepithelial neoplasia, adenocarcinoma in situ, or invasive cervical carcinoma, with detection of HPV-16 or HPV-18 or both in one or more of the adjacent sections of the same lesion.
In all, 6,087 patients received vaccine and 6,080 received placebo; the two groups were well matched. About 23% had serologic evidence of exposure to either HPV-16 or HPV-18 at enrollment.
Findings. In the analysis of the data, the patients were divided into three overlapping subgroups. The first comprised women who had no serologic evidence of HPV-16 or HPV-18 infection at enrollment, who received all three injections, who remained DNA-negative at month 7, and who had no protocol violations. In this “per-protocol susceptible population,” at an average of 3 years of follow-up, lesions associated with HPV-16 or HPV-18 had developed in 42 of 5,260 women who received placebo, compared with only 1 of 5,305 who received the vaccine. The vaccine efficacy was calculated at 98% (95% confidence interval [CI] 86–100).
The second subgroup were women who had no evidence of HPV-16 or HPV-18 infection at baseline, but whose compliance with the protocol was considered imperfect. In this “unrestricted susceptible population,” the vaccine efficacy was 95% (95% CI 85–99).
The third group included all comers, regardless of whether they were already infected at baseline. In this “intention-to-treat population,” the vaccine efficacy was 44% (95% CI 26–58).
The authors concluded that in young women not previously infected with HPV-16 or HPV-18, vaccine recipients had a significantly lower occurrence of high-grade cervical intraepithelial neoplasia related to these two oncogenic HPV types.
QUADRIVALENT HPV VACCINE PREVENTS ANOGENITAL DISEASE
Garland SM, Hernandez-Avila M, Wheeler CM, et al; Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. J Engl J Med 2007; 356:1928–1943.
The study. This double-blind, placebo-controlled study5 tested the usefulness of the quadrivalent HPV vaccine to prevent anogenital disease. It included 5,400 women ages 16 to 24 and was conducted over 14 months in 2002 and 2003 at 62 sites in 16 countries. Women received vaccine or placebo at day 1 and again 2 and 6 months later, and then underwent anogenital and gynecologic examinations at intervals for up to 4 years.
The co-primary composite end points were the incidence of genital warts, vulvar or vaginal intraepithelial neoplasia or cancer, cervical intraepithelial neoplasia, cervical adenocarcinoma in situ, or cervical cancer associated with HPV types 6, 11, 16, or 18.
Findings. In all, 2,700 women were assigned to receive vaccine and 2,700 to receive placebo, and they were followed for an average of 3 years. Twenty percent had pre-existing serologic evidence of infection with one of these four HPV types. In the per-protocol population who were seronegative at day 1 and were compliant, the vaccine efficacy was 100%. In the intention-to-treat group, vaccine reduced the rate of vulvar or vaginal perianal lesions regardless of HPV type by 34%, and reduced the rate of cervical lesions regardless of type by 20%.
HPV VACCINE LIKELY COST-EFFECTIVE IN GIRLS, BUT NOT BOYS
Newall AT, Beutels P, Wood JG, Edmunds WJ, MacIntyre CR. Cost-effectiveness analyses of human papillomavirus vaccination. Lancet Infect Dis 2007; 7:289–296.
The study. In a review, Newall et al6 looked at four studies that examined the cost-effectiveness of the HPV vaccine. These studies were not perfect and had methodologic limitations because of uncertainty about vaccine efficacy, duration of protection, and the contribution of herd immunity. The studies nevertheless suggested that immunization of young girls but not young boys may be cost-effective, though they suggested the need for further research.
Findings. Three of the studies showed an incremental cost-effectiveness ratio of $14,000 to $24,000 per quality-adjusted year of life gained, which is well within the range for many preventive strategies that we employ in this country.
One of the studies examined the cost-effectiveness of immunizing males, and in that study it was found not to be cost-effective.
TAKE-HOME POINTS ON HPV VACCINATION
Quadrivalent vaccine does indeed reduce the incidence of HPV-associated cervical intra-epithelial neoplasia, vulvar and vaginal intra-epithelial neoplasia, and anogenital diseases in young women, and it is likely cost-effective.
The vaccine works only against HPV types 6, 11, 16, and 18, and 30% of cervical cancers are due to types other than HPV-16 and HPV-18. Also, vaccination is much more effective in patients not yet exposed to HPV, so it would be best to vaccinate them before they become sexually active.
The Advisory Committee on Immunization Practices voted to recommend that girls ages 11 to 12 in this country should receive vaccine.
Regrettably, many third-party payers do not yet pay for the vaccine, and the cost (around $375) must be paid out of pocket. Also, this issue remains politically charged and controversial. Some states have mandated vaccination and another 15 are presently considering legislation mandating vaccination. Such legislation has been defeated in four states.
My own practice is to offer the vaccine to 11- and 12-year old girls, and to older girls and young women (not to boys), especially if the health insurance plan covers it or if the patient or the patient’s family can afford it.
HEPATITIS A VACCINE IS AS GOOD AS IMMUNE GLOBULIN AFTER EXPOSURE
Victor JC, Monto AS, Surdina TY, et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med 2007; 357:1685–1694.
Before 1995, when the first hepatitis A vaccine was introduced, about 30,000 cases of hepatitis A were reported each year in the United States. This was thought to be the tip of the iceberg: since this infection is often subclinical, estimates of up to 300,000 cases per year were given.
At first, immunization against hepatitis A in this country was confined to children over age 2 in states in which hepatitis A occurred more often than the norm. In 2005, after it had become clear that the vaccine was highly effective, the Advisory Committee on Immunization Practices revised its recommendations to include immunization of children between the ages of 12 and 23 months,7 so that they would complete this two-stage vaccination procedure by the time they reached the age of 2 years. With that strategy, the annual occurrence of hepatitis A in the United States fell dramatically, to about 4,000 cases per year in 2005, the lowest number of cases reported in the last 40 years. At present, most hepatitis A infections in this country are not from casual idiosyncratic transmission but rather are food-borne.
Still, hepatitis A remains a major problem in many parts of the world. Moreover, the availability of immune globulin, the traditional recommended agent for postexposure pro-phylaxis, has been limited because only one company manufactures it and the price has steadily escalated.
The study. Investigators at the University of Michigan and in Kazakhstan compared conventional doses of immune globulin vs hepatitis A vaccine as postexposure prophylaxis, given within 14 days of exposure to index cases of hepatitis A.8 Excluded were persons under the age of 2 years or over the age of 40, those with a history of hepatitis A or vaccination, those with liver disease, and those with other contraindications. The primary end point was the development of symptomatic, laboratory-confirmed hepatitis A, defined as a positive test for immunoglobulin M antibodies to hepatitis A; transaminase levels greater than two times the upper limit of normal; and symptoms consistent with hepatitis A in the absence of another identifiable disease that occurred within 15 to 56 days of exposure to the index case.
Findings. Of 4,524 contacts randomized, only 1,414 (31%) were susceptible to hepatitis A, suggesting that the prevalence of hepatitis A in Kazakhstan was high at that time. Of these, 1,090 completed the immunization and follow-up protocol and were eligible for the final analysis. Of these, 568 received vaccine and 522 received globulin. The average age was 12 years, the average time to vaccination after exposure was 10 days; 16% of the exposures occurred in the day-care setting, and 84% of the exposures occurred from household contacts.
Symptomatic hepatitis A occurred in 4.4% of vaccine recipients vs 3.3% of immunoglobulin recipients. The authors concluded that hepatitis A vaccine met the test of noninferiority, that both strategies were highly protective, but that immunoglobulin was modestly better. Thus, in June 2007, the Advisory Committee on Immunization Practices recommended hepatitis A vaccine as the preferred regimen for postexposure prophylaxis.9
This approach has several advantages:
- Hepatitis A vaccine confers immunity and long-term protection, which globulin does not
- The supply of vaccine is abundant
- Vaccine is relatively cheap
- Vaccine is easy to give.
This study, however, does not apply to people younger than 2 years or older than 40, those who are immunocompromised, or those who have chronic liver disease. In these groups, the recommendation is still to use immunoglobulin in postexposure prophylaxis.
How we prevent human papillomavirus (HPV) infection, and how we prevent hepatitis A following exposure to an index case have changed, based on the results of several key clinical trials published during the past year. The results of these studies should influence the measures we take in our daily practice to prevent these diseases. Here is a brief overview of these “impact” studies.
QUADRIVALENT HPV VACCINE PREVENTS CERVICAL LESIONS
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Cervical cancer is the second most common type of cancer in women and is the leading cause of cancer-related deaths in developing countries. More than 500,000 new cases of cervical cancer are reported worldwide each year, and about 250,000 women die of it.1
Nearly all cases of cervical cancer are caused by HPVs, and the oncogenic types HPV-16 and HPV-18 together account for about 70%. These two types also cause vulvo-vaginal cancer, which accounts for about 6% of all gynecologic malignancies.2 Two other HPV types, HPV-6 and HPV-11, cause genital warts and, less often, cervical intraepithelial neoplasia and cervical invasive cancers.
Two HPV vaccines have been developed. One, sold as Cervarix, is directed against HPV-16 and HPV-18; it is not yet available in the United States. The other, sold as Gardasil, is directed against four HPV types: 6, 11, 16, and 18, and it is currently available (reviewed by Widdice and Kahn3).
The study. The Females United to Unilaterally Reduce Endo/Ectocervical Cancer (FUTURE) II study4 assessed the ability of the quadrivalent vaccine to prevent high-grade cervical lesions. Between June 2002 and September 2003, more than 12,000 women ages 15 to 26 were enrolled at 90 sites in 13 countries. Eligible women were not pregnant, had no abnormal Papanicolaou (Pap) smear, had had four or fewer lifetime sexual partners, and agreed to use effective contraception throughout the course of the study.
In a randomized, double-blind fashion, patients received vaccine or a placebo injection at day 1 and again 2 and 6 months later. They returned for follow-up 1, 6, 24, 36, and 48 months after the third injection, with Pap smears and colposcopy of cervical lesions.
The primary composite end point was the development of grade 2 or 3 cervical intraepithelial neoplasia, adenocarcinoma in situ, or invasive cervical carcinoma, with detection of HPV-16 or HPV-18 or both in one or more of the adjacent sections of the same lesion.
In all, 6,087 patients received vaccine and 6,080 received placebo; the two groups were well matched. About 23% had serologic evidence of exposure to either HPV-16 or HPV-18 at enrollment.
Findings. In the analysis of the data, the patients were divided into three overlapping subgroups. The first comprised women who had no serologic evidence of HPV-16 or HPV-18 infection at enrollment, who received all three injections, who remained DNA-negative at month 7, and who had no protocol violations. In this “per-protocol susceptible population,” at an average of 3 years of follow-up, lesions associated with HPV-16 or HPV-18 had developed in 42 of 5,260 women who received placebo, compared with only 1 of 5,305 who received the vaccine. The vaccine efficacy was calculated at 98% (95% confidence interval [CI] 86–100).
The second subgroup were women who had no evidence of HPV-16 or HPV-18 infection at baseline, but whose compliance with the protocol was considered imperfect. In this “unrestricted susceptible population,” the vaccine efficacy was 95% (95% CI 85–99).
The third group included all comers, regardless of whether they were already infected at baseline. In this “intention-to-treat population,” the vaccine efficacy was 44% (95% CI 26–58).
The authors concluded that in young women not previously infected with HPV-16 or HPV-18, vaccine recipients had a significantly lower occurrence of high-grade cervical intraepithelial neoplasia related to these two oncogenic HPV types.
QUADRIVALENT HPV VACCINE PREVENTS ANOGENITAL DISEASE
Garland SM, Hernandez-Avila M, Wheeler CM, et al; Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. J Engl J Med 2007; 356:1928–1943.
The study. This double-blind, placebo-controlled study5 tested the usefulness of the quadrivalent HPV vaccine to prevent anogenital disease. It included 5,400 women ages 16 to 24 and was conducted over 14 months in 2002 and 2003 at 62 sites in 16 countries. Women received vaccine or placebo at day 1 and again 2 and 6 months later, and then underwent anogenital and gynecologic examinations at intervals for up to 4 years.
The co-primary composite end points were the incidence of genital warts, vulvar or vaginal intraepithelial neoplasia or cancer, cervical intraepithelial neoplasia, cervical adenocarcinoma in situ, or cervical cancer associated with HPV types 6, 11, 16, or 18.
Findings. In all, 2,700 women were assigned to receive vaccine and 2,700 to receive placebo, and they were followed for an average of 3 years. Twenty percent had pre-existing serologic evidence of infection with one of these four HPV types. In the per-protocol population who were seronegative at day 1 and were compliant, the vaccine efficacy was 100%. In the intention-to-treat group, vaccine reduced the rate of vulvar or vaginal perianal lesions regardless of HPV type by 34%, and reduced the rate of cervical lesions regardless of type by 20%.
HPV VACCINE LIKELY COST-EFFECTIVE IN GIRLS, BUT NOT BOYS
Newall AT, Beutels P, Wood JG, Edmunds WJ, MacIntyre CR. Cost-effectiveness analyses of human papillomavirus vaccination. Lancet Infect Dis 2007; 7:289–296.
The study. In a review, Newall et al6 looked at four studies that examined the cost-effectiveness of the HPV vaccine. These studies were not perfect and had methodologic limitations because of uncertainty about vaccine efficacy, duration of protection, and the contribution of herd immunity. The studies nevertheless suggested that immunization of young girls but not young boys may be cost-effective, though they suggested the need for further research.
Findings. Three of the studies showed an incremental cost-effectiveness ratio of $14,000 to $24,000 per quality-adjusted year of life gained, which is well within the range for many preventive strategies that we employ in this country.
One of the studies examined the cost-effectiveness of immunizing males, and in that study it was found not to be cost-effective.
TAKE-HOME POINTS ON HPV VACCINATION
Quadrivalent vaccine does indeed reduce the incidence of HPV-associated cervical intra-epithelial neoplasia, vulvar and vaginal intra-epithelial neoplasia, and anogenital diseases in young women, and it is likely cost-effective.
The vaccine works only against HPV types 6, 11, 16, and 18, and 30% of cervical cancers are due to types other than HPV-16 and HPV-18. Also, vaccination is much more effective in patients not yet exposed to HPV, so it would be best to vaccinate them before they become sexually active.
The Advisory Committee on Immunization Practices voted to recommend that girls ages 11 to 12 in this country should receive vaccine.
Regrettably, many third-party payers do not yet pay for the vaccine, and the cost (around $375) must be paid out of pocket. Also, this issue remains politically charged and controversial. Some states have mandated vaccination and another 15 are presently considering legislation mandating vaccination. Such legislation has been defeated in four states.
My own practice is to offer the vaccine to 11- and 12-year old girls, and to older girls and young women (not to boys), especially if the health insurance plan covers it or if the patient or the patient’s family can afford it.
HEPATITIS A VACCINE IS AS GOOD AS IMMUNE GLOBULIN AFTER EXPOSURE
Victor JC, Monto AS, Surdina TY, et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med 2007; 357:1685–1694.
Before 1995, when the first hepatitis A vaccine was introduced, about 30,000 cases of hepatitis A were reported each year in the United States. This was thought to be the tip of the iceberg: since this infection is often subclinical, estimates of up to 300,000 cases per year were given.
At first, immunization against hepatitis A in this country was confined to children over age 2 in states in which hepatitis A occurred more often than the norm. In 2005, after it had become clear that the vaccine was highly effective, the Advisory Committee on Immunization Practices revised its recommendations to include immunization of children between the ages of 12 and 23 months,7 so that they would complete this two-stage vaccination procedure by the time they reached the age of 2 years. With that strategy, the annual occurrence of hepatitis A in the United States fell dramatically, to about 4,000 cases per year in 2005, the lowest number of cases reported in the last 40 years. At present, most hepatitis A infections in this country are not from casual idiosyncratic transmission but rather are food-borne.
Still, hepatitis A remains a major problem in many parts of the world. Moreover, the availability of immune globulin, the traditional recommended agent for postexposure pro-phylaxis, has been limited because only one company manufactures it and the price has steadily escalated.
The study. Investigators at the University of Michigan and in Kazakhstan compared conventional doses of immune globulin vs hepatitis A vaccine as postexposure prophylaxis, given within 14 days of exposure to index cases of hepatitis A.8 Excluded were persons under the age of 2 years or over the age of 40, those with a history of hepatitis A or vaccination, those with liver disease, and those with other contraindications. The primary end point was the development of symptomatic, laboratory-confirmed hepatitis A, defined as a positive test for immunoglobulin M antibodies to hepatitis A; transaminase levels greater than two times the upper limit of normal; and symptoms consistent with hepatitis A in the absence of another identifiable disease that occurred within 15 to 56 days of exposure to the index case.
Findings. Of 4,524 contacts randomized, only 1,414 (31%) were susceptible to hepatitis A, suggesting that the prevalence of hepatitis A in Kazakhstan was high at that time. Of these, 1,090 completed the immunization and follow-up protocol and were eligible for the final analysis. Of these, 568 received vaccine and 522 received globulin. The average age was 12 years, the average time to vaccination after exposure was 10 days; 16% of the exposures occurred in the day-care setting, and 84% of the exposures occurred from household contacts.
Symptomatic hepatitis A occurred in 4.4% of vaccine recipients vs 3.3% of immunoglobulin recipients. The authors concluded that hepatitis A vaccine met the test of noninferiority, that both strategies were highly protective, but that immunoglobulin was modestly better. Thus, in June 2007, the Advisory Committee on Immunization Practices recommended hepatitis A vaccine as the preferred regimen for postexposure prophylaxis.9
This approach has several advantages:
- Hepatitis A vaccine confers immunity and long-term protection, which globulin does not
- The supply of vaccine is abundant
- Vaccine is relatively cheap
- Vaccine is easy to give.
This study, however, does not apply to people younger than 2 years or older than 40, those who are immunocompromised, or those who have chronic liver disease. In these groups, the recommendation is still to use immunoglobulin in postexposure prophylaxis.
- CancerMondial. International Agency for Research on Cancer. www-dep.iarc.fr/. Accessed May 12, 2008.
- Munoz N, Bosch FX, de Sanjose S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003; 348:518–527.
- Widdice LE, Kahn JA. Using the new HPV vaccines in clinical practice. Cleve Clin J Med 2006; 73:929–935.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Garland SM, Hernandez-Avila M, Wheeler CM, et al Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) I Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med 2007; 356:1928–1943.
- Newall AT, Beutels P, Wood JG, Edmunds WJ, MacIntyre CR. Cost-effectiveness analyses of human papillomavirus vaccination. Lancet Infect Dis 2007; 7:289–296.
- Advisory Committee on Immunization Practices (ACIP)Fiore AE, Wasley A, Bell BP. Prevention of hepatitis A through active or passive immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55( RR–7):1–23.
- Victor JC, Monto AS, Surdina TY, et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med 2007; 357:1685–1694.
- Advisory Committee on Immunization Practices, US Centers for Disease Control and Prevention. Update: prevention of hepatitis A after exposure to hepatitis A virus and in international travelers. Updated recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2007; 56:1080–1084.
- CancerMondial. International Agency for Research on Cancer. www-dep.iarc.fr/. Accessed May 12, 2008.
- Munoz N, Bosch FX, de Sanjose S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003; 348:518–527.
- Widdice LE, Kahn JA. Using the new HPV vaccines in clinical practice. Cleve Clin J Med 2006; 73:929–935.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Garland SM, Hernandez-Avila M, Wheeler CM, et al Females United to Unilaterally Reduce Endo/Ectocervical Disease (FUTURE) I Investigators. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med 2007; 356:1928–1943.
- Newall AT, Beutels P, Wood JG, Edmunds WJ, MacIntyre CR. Cost-effectiveness analyses of human papillomavirus vaccination. Lancet Infect Dis 2007; 7:289–296.
- Advisory Committee on Immunization Practices (ACIP)Fiore AE, Wasley A, Bell BP. Prevention of hepatitis A through active or passive immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2006; 55( RR–7):1–23.
- Victor JC, Monto AS, Surdina TY, et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med 2007; 357:1685–1694.
- Advisory Committee on Immunization Practices, US Centers for Disease Control and Prevention. Update: prevention of hepatitis A after exposure to hepatitis A virus and in international travelers. Updated recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2007; 56:1080–1084.