User login
Sleep apnea ABCs: Airway, breathing, circulation
Obstructive sleep apnea (OSA) is common and poorly recognized and, if untreated, leads to serious health consequences. This article discusses the epidemiology of OSA, describes common presenting signs and symptoms, and reviews diagnostic testing and treatment options. Adverse health effects related to untreated sleep apnea are also discussed.
COMMON, POORLY RECOGNIZED, AND COSTLY IF UNTREATED
OSA is very common in the general population and is associated with substantial morbidity and mortality. An estimated 17% of the general adult population has OSA, and the numbers are increasing with the obesity epidemic. Nearly 1 in 15 adults has at least moderate sleep apnea,1,2 and approximately 85% of cases are estimated to be undiagnosed.3 A 1999 study estimated that untreated OSA resulted in approximately $3.4 billion in additional medical costs per year in the United States,4 a figure that is likely to be higher now, given the rising prevalence of OSA. The prevalence of OSA in primary care and subspecialty clinics is even higher than in the community, as more than half of patients who have diabetes or hypertension and 30% to 40% of patients with coronary artery disease are estimated to have OSA.5–7
REPETITIVE UPPER-AIRWAY COLLAPSE
During sleep, parasympathetic activity is enhanced and the muscle tone of the upper airway is decreased, particularly in the pharyngeal dilator muscles. Still, even in the supine position, a healthy person maintains patency of the airway and adequate airflow during sleep.
OSA is characterized by repetitive complete or partial collapse of the upper airway during sleep, resulting in an apneic or hypopneic event, respectively, and often causing snoring from upper-airway tissue vibration.
People who are susceptible to OSA typically have a smaller, more collapsible airway that is often less distensible and has a higher critical closing pressure. Radiographic and physiologic data have shown that the airway dimensions of patients with OSA are smaller than in those without OSA. The shape of the airway of a patient with OSA is often elliptical, given the extrinsic compression of the lateral aspects of the airway by increased size of the parapharyngeal fat pads. OSA episodes are characterized by closure of the upper airway and by progressively increasing respiratory efforts driven by chemoreceptor and mechanoreceptor stimuli, culminating in an arousal from sleep and a reopening of the airway.
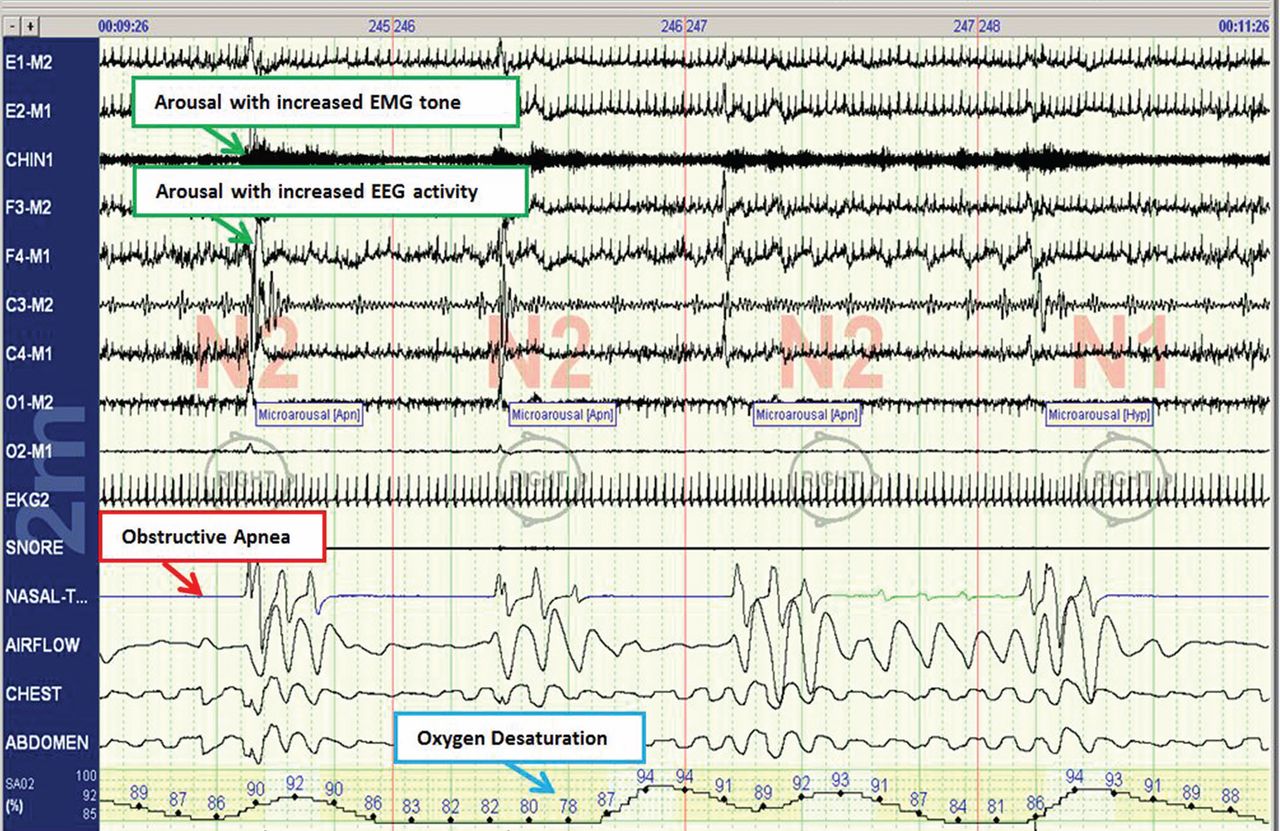
The disease-defining metric used for assessing OSA severity is the apnea-hypopnea index, ie, the number of apneas and hypopneas that occur per hour of sleep.8 An apneic or hypopneic event is identified during polysomnography by the complete cessation of airflow or by a reduction in airflow for 10 seconds or longer (Figure 1).
HEALTH CONSEQUENCES IF UNTREATED
Untreated sleep apnea causes numerous pathophysiologic perturbations, including chronic intermittent hypoxia, ventilatory overshoot hyperoxia, increased sympathetic nervous system activity, intrathoracic pressure swings, hypercapnea, sleep fragmentation, increased arousals, reduced sleep duration, and fragmentation of rapid-eye-movement sleep.
Intermittent hypoxia activates the sympathetic nervous system and causes pulmonary vasoconstriction, with increases in pulmonary arterial pressures and myocardial workload. Sympathetic activation, ascertained by peroneal microneurography, has been shown to be increased not only during sleep but also persisting during wakefulness in patients with untreated OSA vs those without OSA.9 Autonomic nervous system fluctuations accompany apneic episodes, resulting in enhanced parasympathetic tone and sympathetic activation associated with a rise in blood pressure and heart rate that occur after the respiratory event.
Intermediate pathways that link the negative pathophysiologic effects of OSA with adverse health outcomes include increased systemic inflammation, increased oxidative stress, metabolic dysfunction, insulin resistance, hypercoagulability, endothelial dysfunction, and autonomic dysfunction.
As a result, a variety of adverse clinical outcomes are associated with untreated OSA, including systemic hypertension, ischemic heart disease and atherosclerosis, diastolic dysfunction, congestive heart failure, cardiac arrhythmias, stroke, increased risk of death, and sudden death, as well as noncardiovascular outcomes such as gout, neurocognitive deficits, and mood disorders.10
Inflammatory and atherogenic effects
Increased levels of markers of systemic inflammation, prothrombosis, and oxidative stress have been observed in OSA and may be key pathophysiologic links between OSA and cardiovascular sequelae. OSA has been associated with up-regulation of a number of inflammatory mediators: interleukin (IL) 6, soluble IL-6 receptor, IL-8, tumor necrosis factor alpha, and C-reactive protein. Soluble IL-6 levels in particular are higher in people who have sleep-disordered breathing, as reflected by the apnea-hypopnea index independent of obesity, with relationships stronger in the morning than in the evening. This likely reflects the overnight OSA-related physiologic stress.11
Thrombotic potential is also enhanced, with higher levels of plasminogen activator inhibitor 1, fibrinogen, P-selectin, and vascular endothelial growth factor. Some of these factors normally have a diurnal cycle, with higher levels in the morning, but in OSA, increasing OSA severity is associated with increased prothrombotic potential in the morning hours. Of interest, levels of these substances showed a plateau effect, rising in people who had only mildly elevated apnea-hypopnea indices and then leveling off.12 Intermittent hypoxia followed by ventilatory overshoot hyperoxia, characteristic of sleep apnea, provides the ideal environment for augmentation of oxidative stress, with evidence of increased oxidation of serum proteins and lipids. Hypoxia and oxygen-derived free radicals may result in cardiac myocyte injury. Experimental data demonstrate that intermittent hypoxia combined with a high-fat diet results in synergistic acceleration of evidence of atherogenic lesions.
Patients with OSA also have evidence of endothelial dysfunction, insulin resistance, and dyslipidemia, all pathways that can facilitate the progression of atherosclerosis in OSA.13–15
Cardiac arrhythmias
In the Sleep Heart Health Study, a multicenter epidemiologic study designed to examine the relationships of OSA and cardiovascular outcomes, those who had moderate to severe OSA had a risk of ventricular and atrial arrhythmias two to four times higher than those without OSA, even after correction for the confounding influences of obesity and underlying cardiovascular risk.14 These findings were corroborated in subsequent work highlighting monotonic dose-response relationships with increasing OSA severity and increased odds of atrial and ventricular arrhythmia in a cohort of about 3,000 older men.11 Additional compelling evidence of a causal relationship is that the risk of discrete arrhythmic events is markedly increased after a respiratory disturbance in sleep.16
In patients who successfully underwent cardioversion for atrial fibrillation, those who had sleep apnea but were not treated with continuous positive airway pressure (CPAP) had a much higher rate of recurrence of atrial fibrillation during the subsequent year than those with CPAP-treated sleep apnea and than controls never diagnosed with sleep apnea. In the untreated patients with sleep apnea, the mean nocturnal fall in oxygen saturation was significantly greater in those who had recurrence of atrial fibrillation than in those who did not, suggesting hypoxia as an important mechanism contributing to atrial fibrillation.17
Since then, several other retrospective studies have shown similar findings after pulmonary vein antrum isolation and ablation in terms of reduction of atrial fibrillation recurrence with CPAP treatment in OSA.18
Walia et al19 described a patient with moderate sleep apnea who underwent a split-night study. During the baseline part of the study, the patient had about 18 ectopic beats per minute. During the second portion of the study while CPAP was applied, progressively fewer ectopic beats occurred as airway pressure was increased until a normal rhythm without ectopic beats was achieved at the goal treatment CPAP pressure setting.
Cardiovascular disease, stroke, and death
Marin et al20 followed about 1,500 men for 10 years, including some who had severe OSA, some with sleep apnea who were treated with CPAP, and controls. The risk of nonfatal and fatal cardiovascular disease events was nearly three times higher in those with severe disease than in healthy participants. Those treated with CPAP had a risk approximately the same as in the control group.
The Sleep Heart Study followed approximately 6,000 people with untreated sleep apnea for a median of nearly 9 years. It found a significant association between the apnea-hypopnea index and ischemic stroke, especially in men.21 Survival in patients with heart failure is also associated with the degree of OSA; patients with more severe disease (an apnea-hypopnea index ≥ 15) have a nearly three times greater risk of death than those with no disease or only mild disease (apnea-hypopnea index < 15).22
From the standpoint of health care utilization, findings that central sleep apnea predicts an increased risk of hospital readmission in heart failure are of particular interest.23
People with OSA are at increased risk of nocturnal sudden cardiac death.24 Sleep apnea is also associated with an increased overall death rate, and the higher the apnea-hypopnea index, the higher the death rate,25 even after adjusting for age, sex, body mass index, and underlying cardiovascular risk, with findings most pronounced in men under age 70.
Motor vehicle accidents
The need for caution during driving should be discussed with every patient, as motor vehicle accidents are an immediate danger to the patient and others. The association with motor vehicle accidents is independent of sleepiness, and drivers with sleep apnea often do not perceive performance impairment. Young et al26 found that men who snored were 3.4 times as likely to have an accident over a 5-year period, and that men and women with an apnea-hypopnea index greater than 15 were more than 7 times as likely to have multiple accidents over a 5-year period, highlighting the importance of discussing, documenting, and expeditiously diagnosing and treating OSA, particularly in those who report drowsiness while driving.
CLINICAL RISK FACTORS
Risk factors can be divided into nonmodifiable and modifiable ones.
Nonmodifiable factors
Age. Bimodal distributions in OSA prevalence have been observed; ie, that the pediatric population and people who are middle-aged have the highest prevalence of OSA. A linear relationship between sleep apnea prevalence and age until about age 65 was identified in data from the Sleep Heart Health Study.27 After that, the prevalence rates plateau; it is unclear if this is secondary to natural remission of the disease after a certain age or because patients with more severe disease have died by that age (ie, survivorship bias), blunting an increase in prevalence.
Sex. Men develop sleep apnea at a rate three to five times that of women. Several explanations have been proposed to account for this.28,29 Sex hormones are one factor; women with sleep apnea on hormone replacement therapy have a significantly less-severe sleep apnea burden than other postmenopausal women,30 suggesting a positive effect from estrogen. Sex-based differences in fat distribution, length and collapsibility of the upper airway, genioglossal activity, neurochemical control mechanisms, and arousal response may also contribute to prevalence differences between men and women.
As with coronary artery disease, the presentation of sleep apnea may be atypical in women, particularly around menopause. Sleep apnea should be considered in women who have snoring and daytime sleepiness.
Race. Whites, African Americans, and Asians have a similar prevalence of sleep apnea, but groups differ in obesity rates and craniofacial anatomy.31–34 Asians tend to have craniofacial skeletal restriction. African Americans are more likely to have upper-airway soft-tissue risk and to develop more severe OSA. Whites tend to have both craniofacial and soft-tissue risk. For those with craniofacial anatomy predisposing to OSA, even mild obesity can make it manifest.
Syndromes that predispose to OSA can include craniofacial structural abnormalities, connective tissue problems, or alterations in ventilatory control (eg, Marfan, Down, and Pierre Robin syndromes).
Modifiable risk factors
Obesity (body mass index ≥ 30 kg/m2) is a firmly established risk factor, but not all obese patients develop obstructive sleep apnea, and not all people with sleep apnea are obese.
Obesity increases risk by altering the geometry and function of the upper airway, increasing collapsibility. The changes are particularly pronounced in the lateral aspects of the pharynx.35
Obesity also affects respiratory drive, likely in part from leptin resistance. Load compensation is another contributing factor: the increased mass in the thorax and abdomen increases the work of breathing and reduces functional residual capacity, increasing oxygen demands and leading to atelectasis and ventilation-perfusion mismatch.
Although obesity is an important risk factor, it is important to recognize that obesity is not the only one to consider: most people with an apnea-hypopnea index of 5 or greater are not obese. The relationship between body mass index and sleep apnea is weaker in children and in the elderly, probably because other risk factors are more pronounced.36
Craniofacial structural abnormalities such as retrognathia (abnormal posterior position of the mandible) and micrognathia (undersized mandible) can increase the risk of OSA because of a resulting posteriorly displaced genioglossus muscle. Other conditions can alter chemosensitivity, affecting the pH and carbon dioxide level of the blood and therefore affecting ventilatory control mechanisms, making the person more prone to developing sleep apnea. Children and young adults may have tonsillar tissue that obstructs the airway.
The site of obstruction can be behind the palate (retropalatal), behind the tongue (retroglossal), or below the pharynx (hypopharyngeal). This helps explain why positive air way pressure—unlike surgery, which addresses a specific area—is often successful, as it serves to splint or treat all aspects of the airway.
FATIGUE, SLEEPINESS, SNORING, RESTLESS SLEEP

Sleep apnea can result in presentation of multiple signs and symptoms (Table 1).
Daytime sleepiness and fatigue are the most common symptoms. Although nonspecific, they are often quite pronounced. Two short questionnaires—the Epworth Sleepiness Scale37 and the Fatigue Severity Scale—can help distinguish between these two symptoms and assess their impact on a patient’s daily life. In the Epworth Sleepiness Scale, the patient rates his or her chance of dozing on a 4-point scale (0 = would never doze, to 3 = high chance of dozing) in eight situations:
- Sitting and reading
- Watching television
- Sitting inactive in a public place
- As a passenger in a car for an hour without a break
- Lying down to rest in the afternoon
- Sitting and talking to someone
- Sitting quietly after a lunch without alcohol
- In a car while stopped for a few minutes in traffic.
A score of 10 or more is consistent with significant subjective sleepiness.
The Fatigue Severity Scale assesses the impact of fatigue on daily living.
Snoring is a common and specific symptom of sleep apnea; however, not all patients who snore have OSA.
Restlessness during sleep is very common—patients may disturb their bed partner by moving around a lot during sleep or report that the sheets are “all over the place” by morning.
Nocturia can also be a sign of sleep apnea and can contribute to sleep fragmentation. A proposed mechanism of this symptom includes alterations of intrathoracic pressure resulting in atrial stretch, which release atrial natriuretic peptide, leading to nocturia. Treating with CPAP has been found to reduce levels of atrial natriuretic peptide, contributing to better sleep.38
Morning headache may occur and is likely related to increased CO2 levels, which appear to culminate in the morning hours. End-tidal or transcutaneous CO2 monitoring during polysomnography can help elucidate the presence of sleep-related hypoventilation.
Libido is often diminished and can actually be improved with CPAP. This is therefore an important point to discuss with patients, as improved libido can often serve as an incentive for adherence to OSA treatment.
Insomnia exists in about 15% of patients, primarily as a result of sleep apnea-related with treatment.
Sweating, particularly forehead sweating associated with sleep apnea, more commonly occurs in children.

The STOP-BANG questionnaire (Table 2)39 was primarily validated in preoperative anesthesia testing. However, because of its ease of use and favorable performance characteristics, it is increasingly used to predict the likelihood of finding OSA before polysomnography. A score of 3 or more has a sensitivity of 93%.
PHYSICAL EXAMINATION PROVIDES CLUES

Although the physical examination may be normal, certain findings indicate risk (Table 3). Obesity alone is not an accepted indication for polysomnography unless there are concomitant worrisome signs or symptoms. Of note, those who are morbidly obese (BMI > 40 kg/m2) have a prevalence of sleep apnea greater than 70%.
The classification by Friedman et al40 provides an indicator of risk. The patient is examined with the mouth opened wide and the tongue in a neutral natural position. Grades:
- I—Entire uvula and tonsils are visible
- II—Entire uvula is visible, but tonsils are not
- III—Soft palate is visible, but uvula is not
- IV—Only the hard palate is visible.
Especially in children and young adults, enlarged tonsils (or “kissing tonsils”) and a boggy edematous uvula set the stage for obstructive sleep apnea.
DIAGNOSIS REQUIRES SLEEP TESTING
A sleep study is the primary means of diagnosing OSA. Polysomnography includes electrooculography to determine when rapid-eye-movement sleep occurs; electromyography to measure muscle activity in the chin to help determine onset of sleep, with peripheral leads in the leg to measure leg movements; electroencephalography (EEG) to measure neural activity; electrocardiography; pulse oximetry to measure oxygen saturation; measurement of oronasal flow; and measurements of chest wall effort and body position using thoracic and abdominal belts that expand and contract with breathing; and audio recording to detect snoring.
Attended polysomnography requires the constant presence of a trained sleep technologist to monitor for technical issues and patient adherence.
End-tidal CO2 monitoring is a reasonable method to detect sleep-related hypoventilation but is not routinely performed in the United States. Transcutaneous CO2 monitoring is a different way to monitor CO2 used in the setting of positive airway pressure.
Polysomnography in a normal patient shows a regular pattern of increasing and decreasing airflow with inspiration and expiration while stable oxygen saturation is maintained.
In contrast, polysomnography of a patient with sleep apnea shows repetitive periods of no airflow, oxygen desaturation, and often evidence of thoracoabdominal paradox, punctuated by arousals on EEG associated with sympathetic activation (Figure 1). When the patient falls asleep, upper-airway muscle tone is reduced, causing an apneic event with hypoxia and pleural pressure swings. These prompt arousals with sympathetic activation that reestablish upper-airway muscle tone, allowing ventilation and reoxygenation to resume with a return to sleep.
Apnea-hypopnea index indicates severity
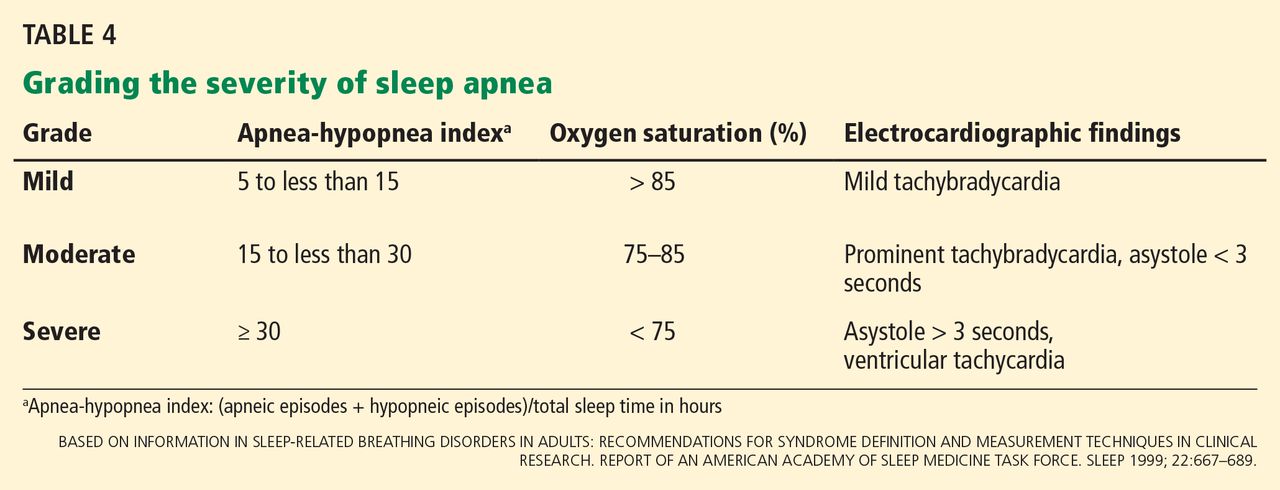
Sleep apnea severity is graded using the apnea-hypopnea index, ie, the number of apneic and hypopneic events per hour of sleep (Table 4).41 Events must last at least 10 seconds to be considered, ie, two consecutive missed breaths based on an average normal respiratory rate of about 12 breaths per minute for the typical adult.
The apnea-hypopnea index usually correlates with the severity of oxygen desaturation and with electrocardiographic abnormalities, including tachybradycardia and arrhythmias.
Although history, physical examination, and prediction tools are helpful in determining the likelihood that a patient has OSA, only polysomnography testing can establish the diagnosis. To diagnose OSA, 15 or more obstructive events per hour must be observed by polysomnography, or at least 5 events per hour with one of the following:
- Daytime sleepiness, sleep attacks, unrefreshing sleep, fatigue, or insomnia
- Waking with breath-holding, gasping, or choking
- Observer-reported loud snoring or breathing interruptions.41
Split-night study determines diagnosis and optimum treatment
The split-night study has two parts: the first is diagnostic polysomnography, followed by identification of the positive airway pressure that optimally treats the sleep apnea. The apnea-hypopnea index guides the need for the split-night study, with 40 being the established threshold according to the American Academy of Sleep Medicine.
A home sleep study is appropriate for some patients
Home sleep testing is typically more limited than standard polysomnography; it monitors airflow, effort, and oxygenation. The test is intended for adults with a high pretest probability of moderate to severe obstructive sleep apnea (STOP-BANG score ≥ 3). It is not intended for screening of asymptomatic patients or for those with coexisting sleep disorders (eg, central sleep apnea, sleep hypoventilation, periodic limb movements, insomnia, circadian rhythm disorders, parasomnias, narcolepsy) or medical disorders (eg, moderate to severe heart failure or other cardiac disease, symptomatic neurologic disease, moderate to severe pulmonary disease).42 Since March 2008, the Centers for Medicare and Medicaid Services has covered CPAP for obstructive sleep apnea based on diagnosis by home sleep study testing.43
TREATMENT OF SLEEP APNEA
Basic steps for reducing OSA are:
Weight loss. Even small weight changes can significantly affect the severity of sleep apnea, perhaps even leading to a reassessment of the degree of OSA and CPAP requirements. Longitudinal epidemiologic data demonstrate that a 10% weight loss correlates with a 26% reduction in the apnea-hypopnea index, and conversely, a 10% weight gain is associated with a 32% increase.44
Some studies have found that bariatric surgery cures OSA in 75% to 88% of cases, independent of approach.45,46 However, a trial in 60 obese patients with OSA who were randomized to either a low-calorie diet or bariatric surgery found no statistical difference in the apnea-hypopnea index between the two groups despite greater weight loss in the surgery group.47
Avoiding certain medications. Benzodiazepines, narcotics, and alcohol reduce upper airway muscle tone and should be avoided. No medications are associated with improvement of OSA, although acetazolamide may be used to treat central sleep apnea.
Positional therapy. Sleeping on the back exacerbates the problem. Supine-related OSA occurs as a result of several factors, including gravity, airway anatomy, airway critical closing pressures, and effects on upper-airway dilator muscle function.
Sleep hygiene. General recommendations to engage in behaviors to promote sleep are recommended, including keeping consistent sleep-wake times, not watching television in bed, and avoidance of caffeine intake, particularly within 4 to 6 hours of bedtime.
POSITIVE AIRWAY PRESSURE THERAPY
Nasal CPAP is the treatment of choice and is successful in 95% of patients when used consistently. It is not as costly as surgery, and results in improved long-term survival compared with uvulopalatopharyngoplasty. Another advantage is that the pressure can be retitrated as the patient’s condition changes, for example after a weight change or during pregnancy.
More than 15 randomized controlled trials have examined the effect of sleep apnea treatment with CPAP compared with either sham CPAP or another control. In a meta-analysis, CPAP was found to lead to an average systolic blood pressure reduction of about 2.5 mm Hg and a diastolic blood pressure reduction of 1.8 mm Hg. Although these reductions may seem negligible, benefits may be significant for cardiovascular outcomes.48,49
Challenges to treatment adherence
Adherence is the most commonly discussed problem with CPAP, but long-term adherence rates are comparable to medication compliance—about 60% to 70%. To optimize adherence, communication is important to ensure that problems are identified and addressed as they arise. Showing patients examples of apneic events and oxygen desaturation from their sleep study can enhance their understanding of OSA and its importance. Patients need to understand the serious nature of the disease and that CPAP therapy can significantly improve their quality of life and overall health, particularly from a cardiovascular perspective.
CPAP masks can be uncomfortable, posing a major barrier to compliance. But a number of mask designs are available, such as the nasal mask, the nasal pillow mask, and the oronasal mask. For patients with claustrophobia, the nasal pillow mask is an option, as it does not cover the face.
Some patients note symptoms of nasal congestion, although in many patients CPAP improves it. If congestion is a problem, the use of heated humidification with the machine, intranasal saline or gel, or nasal corticosteroids can help relieve it.
Pressure intolerance is a common problem. For those who feel that the pressure is too high, settings can be adjusted so that the pressure is gradually reduced between inspiration and expiration, ie, the use of expiratory pressure relief or consideration of the use of bilevel positive airway pressure.
Aerophagia (swallowing air) is a less common problem. It can also potentially be relieved with use of bilevel positive airway pressure.
Many patients develop skin irritation, which can be helped with moleskin, available at any pharmacy.
Social stigma can be a problem. Education regarding the importance of the treatment to health is essential.
Machine noise is less of a problem with the new machine models, but if it is a problem, a white-noise device or earplugs may help.
Other measures to improve compliance are keeping the regimen simple and ensuring that family support is adequate.
Medicare requires evidence of use and benefit
Medicare requires that clinical benefit be documented between the 31st and 91st day after initiating CPAP therapy. This requires face-to-face clinical reevaluation by the treating physician to document improved symptoms and objective evidence of adherence to use of the device. The devices can store usage patterns, and Medicare requires at least 4 hours per night on 70% of nights during a consecutive 30-day period in the first 3 months of use.
ALTERNATIVE THERAPIES
Alternative therapies may be options for some patients, in particular those who cannot use CPAP or who get no benefit from it. These include oral appliances for those with mild to moderate OSA50–53 and various surgical procedures, eg, uvulopalatopharyngoplasty,54,55 maxillomanibular advancement,56 tracheostomy (standard treatment before CPAP was identified as an effective treatment),57,58 and adenotonsillectomy (in children).59
Supplemental oxygen is not a first-line treatment for OSA and in general has not been found to be very effective, particularly in terms of intermediate cardiovascular outcomes,60–62 although a subset of patients with high loop gain may benefit from it.63 Loop gain is a measure of the tendency of the ventilatory control system to amplify respiration in response to a change, conferring less stable control of breathing.
Several novel alternative therapies are starting to be used. Although all of them have been shown to improve measures of OSA, none is as effective as CPAP in improving OSA severity. New therapies include the nasal expiratory positive airway pressure device,64 oral pressure therapy,65 and hypoglossal nerve stimulation.66
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 1993; 328:1230–1235.
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- Kapur VK, Redline S, Nieto FJ, Young TB, Newman AB, Henderson JA; Sleep Heart Health Research Group. The relationship between chronically disrupted sleep and healthcare use. Sleep 2002; 25:289–296.
- Kapur V, Blough DK, Sandblom RE, et al. The medical cost of undiagnosed sleep apnea. Sleep 1999; 22:749–755.
- Mooe T, Rabben T, Wiklund U, Franklin KA, Eriksson P. Sleep-disordered breathing in men with coronary artery disease. Chest 1996; 109:659–663.
- Schafer H, Koehler U, Ewig S, Hasper E, Tasci S, Luderitz B. Obstructive sleep apnea as a risk marker in coronary artery disease. Cardiology 1999; 92:79–84.
- Leung RS, Bradley TD. Sleep apnea and cardiovascular disease. Am J Respir Crit Care Med 2001; 164:2147–2165.
- American Academy of Sleep Medicine. International Classification of Sleep Disorders, Second Edition: Diagnostic and coding manual. Westchester, IL; American Academy of Sleep Medicine, 2005.
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995; 96:1897–1904.
- Mehra R. Sleep-disordered breathing and cardiovascular disease: exploring pathophysiology and existing data. Curr Resp Med Rev 2007; 3:258–269.
- Mehra R, Storfer-Isser A, Kirchner HL, et al. Soluble interleukin 6 receptor: a novel marker of moderate to severe sleep-related breathing disorder. Arch Intern Med 2006; 166:1725–1731.
- Mehra R, Xu F, Babineau DC, et al. Sleep-disordered breathing and prothrombotic biomarkers: cross-sectional results of the Cleveland Family Study. Am J Respir Crit Care Med 2010; 182:826–833.
- Mehra R, Storfer-Isser A, Tracy R, Jenny N, Redline S. Association of sleep disordered breathing and oxidized LDL [abstract]. Am J Respir Crit Care Med 2010; 181:A2474.
- Mehra R, Benjamin EJ, Shahar E, et al; Sleep Heart Health Study. Association of nocturnal arrhythmias with sleep-disordered breathing: the Sleep Heart Health Study. Am J Respir Crit Care Med 2006; 173:910–916.
- Mehra R, Xu F, Babineau DC, et al. Sleep-disordered breathing and prothrombotic biomarkers: cross-sectional results of the Cleveland Family Study. Am J Respir Crit Care Med 2010; 182:826–833.
- Monahan K, Storfer-Isser A, Mehra R, et al. Triggering of nocturnal arrhythmias by sleep-disordered breathing events. J Am Coll Cardiol 2009; 54:1797–1804.
- Kanagala R, Murali NS, Friedman PA, et al. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 2003; 107:2589–2594.
- Patel D, Mohanty P, Di Biase L, et al. Safety and efficacy of pulmonary vein antral isolation in patients with obstructive sleep apnea: the impact of continuous positive airway pressure. Circ Arrhythm Electrophysiol 2010; 3:445–451.
- Walia H, Strohl KP, Mehra R. Effect of continuous positive airway pressure on an atrial arrhythmia in a patient with mild obstructive sleep apnea. J Clin Sleep Med 2011; 7:397–398.
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365:1046–1053.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182:269–277.
- Wang H, Parker JD, Newton GE, et al. Influence of obstructive sleep apnea on mortality in patients with heart failure. J Am Coll Cardiol 2007; 49:1625–1631.
- Khayat R, Abraham W, Patt B, et al. Central sleep apnea is a predictor of cardiac readmission in hospitalized patients with systolic heart failure. J Card Fail 2012; 18:534–540.
- Gami AS, Howard DE, Olson EJ, Somers VK. Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med 2005; 352:1206–1214.
- Punjabi NM, Caffo BS, Goodwin JL, et al. Sleep-disordered breathing and mortality: a prospective cohort study. PLoS Med 2009; 6( 8):e1000132. doi: 10.1371/journal.pmed.1000132.
- Young T, Blustein J, Finn L, Palta M. Sleep-disordered breathing and motor vehicle accidents in a population-based sample of employed adults. Sleep 1997; 20:608–613.
- Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med 2002; 165:1217–1239.
- Lin CM, Davidson TM, Ancoli-Israel S. Gender differences in obstructive sleep apnea and treatment implications. Sleep Med Rev 2008; 12:481–496.
- Shaher E, Redline S, Young T, et al. Hormone replacement therapy and sleep-disordered breathing. Am J Respir Crit Care Med 2003; 167:1186–1192.
- Young T, Finn L, Austin D, Peterson A. Menopausal status and sleep-disordered breathing in the Wisconsin Sleep Cohort Study. Am J Respir Crit Care Med 2003; 167:1181–1185.
- Ancoli-Israel S, Klauber MR, Stepnowsky C, Estline E, Chinn A, Fell R. Sleep-disordered breathing in African-American elderly. Am J Respir Crit Care Med 1995; 152:1946–1949.
- Young T, Shahar E, Nieto FJ, et al; Sleep Heart Health Study Research Group. Predictors of sleep-disordered breathing in community-dwelling adults: the Sleep Heart Health Study. Arch Intern Med 2002; 162:893–900.
- Redline S, Tishler PV, Hans MG, Tosteson TD, Strohl KP, Spry K. Racial differences in sleep-disordered breathing in African-Americans and Caucasians. Am J Respir Crit Care Med 1997; 155:186–192. Erratum in: Am J Respir Crit Care Med 1997; 155:1820.
- Sutherland K, Lee RWW, Cistulli PA. Obesity and craniofacial structure as risk factors for obstructive sleep apnoea: impact of ethnicity. Respirology 2012; 17:213–222.
- Schwab RJ, Gupta KB, Gefter WB, Metzger LJ, Hoffman EA, Pack AI. Upper airway and soft tissue anatomy in normal subjects and patients with sleep-disordered breathing. Significance of the lateral pharyngeal walls. Am J Respir Crit Care Med 1995; 152:1673–1689.
- Nieto FJ, Young TB, Lind BK, et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 2000; 283:1829–1836.
- Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991; 14:540–545.
- Krieger J, Imbs J-L, Schmidt M, Kurtz D. Renal function in patients with obstructive sleep apnea. Effects of nasal continuous positive airway pressure. Arch Intern Med 1988; 148:1337–1340.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Friedman M, Ibrahim H, Bass L. Clinical staging for sleep-disordered breathing. Otolaryngal Head Neck Surg 2002; 127:13–21.
- Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. Report of an American Academy of Sleep Medicine Task Force. Sleep 1999; 22:667–689.
- Collop NA, Anderson WM, Boehlecke B, et al; Portable Monitoring Task Force of the American Academy of Sleep Medicine. Clinical guidelines for the use of unattended portable monitors in the diagnosis of obstructive sleep apnea in adult patients. Portable Monitoring Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2007; 3:737–747.
- Centers for Medicare & Medicaid Services (CMS). Continuous positive airway pressure (CPAP) therapy for obstructive sleep apnea (OSA). MLN Matters 2008. www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM6048.pdf. Accessed June 2, 2014.
- Peppard PE, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA 2000; 284:3015–3021.
- Guardiano SA, Scott JA, Ware JC, Schechner SA. The long-term results of gastric bypass on indexes of sleep apnea. Chest 2003; 124:1615–1619.
- Crooks PF. Surgical treatment of morbid obesity. Annu Rev Med 2006; 57:243–264.
- Dixon JB, Schachter LM, O’Brien PE, et al. Surgical vs conventional therapy for weight loss treatment of obstructive sleep apnea: a randomized controlled trial. JAMA 2012; 308:1142–1149.
- Bazzano LA, Khan Z, Reynolds K, He J. Effect of nocturnal nasal continuous positive airway pressure on blood pressure in obstructive sleep apnea. Hypertension 2007; 50:417–423.
- Logan AG, Perlikowski SM, Mente A, et al. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 2001; 19:2271–2277.
- Kushida CA, Morgenthaler TI, Littner MR, et al; American Academy of Sleep. Practice parameters for the treatment of snoring and obstructive sleep apnea with oral appliances: an update for 2005. Sleep 2006; 29:240–243.
- Otsuka R, Ribeiro de Almeida F, Lowe AA, Linden W, Ryan F. The effect of oral appliance therapy on blood pressure in patients with obstructive sleep apnea. Sleep Breath 2006; 10:29–36.
- Yoshida K. Effect on blood pressure of oral appliance therapy for sleep apnea syndrome. Int J Prosthodont 2006; 19:61–66.
- Inazawa T, Ayuse T, Kurata S, et al. Effect of mandibular position on upper airway collapsibility and resistance. J Dent Res 2005; 84:554–558.
- Fujita S, Conway W, Zorick F, Roth T. Surgical correction of anatomic abnormalities in obstructive sleep apnea syndrome: uvulopalatopharyngoplasty. Otolaryngol Head Neck Surg 1981; 89:923–934.
- Schwab RJ. Imaging for the snoring and sleep apnea patient. Dent Clin North Am 2001; 45:759–796.
- Prinsell JR. Maxillomandibular advancement surgery for obstructive sleep apnea syndrome. J Am Dent Assoc 2002; 133:1489–1497.
- Thatcher GW, Maisel RH. The long-term evaluation of tracheostomy in the management of severe obstructive sleep apnea. Laryngoscope 2003; 113:201–204.
- Conway WA, Victor LD, Magilligan DJ, Fujita S, Zorick FJ, Roth T. Adverse effects of tracheostomy for sleep apnea. JAMA 1981; 246:347–350.
- Marcus CL, Moore RH, Rosen CL, et al; Childhood Adenotonsillectomy Trial (CHAT). A randomized trial of adenotonsillectomy for childhood sleep apnea. N Engl J Med 2013; 368:2366–2376.
- Gottlieb DJ, Craig SE, Lorenzi-Filho G, et al. Sleep apnea cardiovascular clinical trials-current status and steps forward: The International Collaboration of Sleep Apnea Cardiovascular Trialists. Sleep 2013; 36:975–980.
- Loredo JS, Ancoli-Israel S, Kim EJ, Lim WJ, Dimsdale JE. Effect of continuous positive airway pressure versus supplemental oxygen on sleep quality in obstructive sleep apnea: a placebo-CPAP-controlled study. Sleep 2006; 29:564–571.
- Phillips BA, McConnell JW, Smith MD. The effects of hypoxemia on cardiac output. A dose-response curve. Chest 1988; 93:471–475.
- Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respire Physiol Neurobiol 2008; 162:144–151.
- Berry RB, Kryger MH, Massie CA. A novel nasal expiratory positive airway pressure (EPAP) device for the treatment of obstructive sleep apnea: a randomized controlled trial. Sleep 2011; 34:479–485.
- Colrain IM, Black J, Siegel LC, et al. A multicenter evaluation of oral pressure therapy for the treatment of obstructive sleep apnea. Sleep Med 2013; 14:830–837.
- Strollo PJ Jr, Soose RJ, Maurer JT, et al; STAR Trial Group. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med 2014; 370:139–149.
Obstructive sleep apnea (OSA) is common and poorly recognized and, if untreated, leads to serious health consequences. This article discusses the epidemiology of OSA, describes common presenting signs and symptoms, and reviews diagnostic testing and treatment options. Adverse health effects related to untreated sleep apnea are also discussed.
COMMON, POORLY RECOGNIZED, AND COSTLY IF UNTREATED
OSA is very common in the general population and is associated with substantial morbidity and mortality. An estimated 17% of the general adult population has OSA, and the numbers are increasing with the obesity epidemic. Nearly 1 in 15 adults has at least moderate sleep apnea,1,2 and approximately 85% of cases are estimated to be undiagnosed.3 A 1999 study estimated that untreated OSA resulted in approximately $3.4 billion in additional medical costs per year in the United States,4 a figure that is likely to be higher now, given the rising prevalence of OSA. The prevalence of OSA in primary care and subspecialty clinics is even higher than in the community, as more than half of patients who have diabetes or hypertension and 30% to 40% of patients with coronary artery disease are estimated to have OSA.5–7
REPETITIVE UPPER-AIRWAY COLLAPSE
During sleep, parasympathetic activity is enhanced and the muscle tone of the upper airway is decreased, particularly in the pharyngeal dilator muscles. Still, even in the supine position, a healthy person maintains patency of the airway and adequate airflow during sleep.
OSA is characterized by repetitive complete or partial collapse of the upper airway during sleep, resulting in an apneic or hypopneic event, respectively, and often causing snoring from upper-airway tissue vibration.
People who are susceptible to OSA typically have a smaller, more collapsible airway that is often less distensible and has a higher critical closing pressure. Radiographic and physiologic data have shown that the airway dimensions of patients with OSA are smaller than in those without OSA. The shape of the airway of a patient with OSA is often elliptical, given the extrinsic compression of the lateral aspects of the airway by increased size of the parapharyngeal fat pads. OSA episodes are characterized by closure of the upper airway and by progressively increasing respiratory efforts driven by chemoreceptor and mechanoreceptor stimuli, culminating in an arousal from sleep and a reopening of the airway.
The disease-defining metric used for assessing OSA severity is the apnea-hypopnea index, ie, the number of apneas and hypopneas that occur per hour of sleep.8 An apneic or hypopneic event is identified during polysomnography by the complete cessation of airflow or by a reduction in airflow for 10 seconds or longer (Figure 1).
HEALTH CONSEQUENCES IF UNTREATED
Untreated sleep apnea causes numerous pathophysiologic perturbations, including chronic intermittent hypoxia, ventilatory overshoot hyperoxia, increased sympathetic nervous system activity, intrathoracic pressure swings, hypercapnea, sleep fragmentation, increased arousals, reduced sleep duration, and fragmentation of rapid-eye-movement sleep.
Intermittent hypoxia activates the sympathetic nervous system and causes pulmonary vasoconstriction, with increases in pulmonary arterial pressures and myocardial workload. Sympathetic activation, ascertained by peroneal microneurography, has been shown to be increased not only during sleep but also persisting during wakefulness in patients with untreated OSA vs those without OSA.9 Autonomic nervous system fluctuations accompany apneic episodes, resulting in enhanced parasympathetic tone and sympathetic activation associated with a rise in blood pressure and heart rate that occur after the respiratory event.
Intermediate pathways that link the negative pathophysiologic effects of OSA with adverse health outcomes include increased systemic inflammation, increased oxidative stress, metabolic dysfunction, insulin resistance, hypercoagulability, endothelial dysfunction, and autonomic dysfunction.
As a result, a variety of adverse clinical outcomes are associated with untreated OSA, including systemic hypertension, ischemic heart disease and atherosclerosis, diastolic dysfunction, congestive heart failure, cardiac arrhythmias, stroke, increased risk of death, and sudden death, as well as noncardiovascular outcomes such as gout, neurocognitive deficits, and mood disorders.10
Inflammatory and atherogenic effects
Increased levels of markers of systemic inflammation, prothrombosis, and oxidative stress have been observed in OSA and may be key pathophysiologic links between OSA and cardiovascular sequelae. OSA has been associated with up-regulation of a number of inflammatory mediators: interleukin (IL) 6, soluble IL-6 receptor, IL-8, tumor necrosis factor alpha, and C-reactive protein. Soluble IL-6 levels in particular are higher in people who have sleep-disordered breathing, as reflected by the apnea-hypopnea index independent of obesity, with relationships stronger in the morning than in the evening. This likely reflects the overnight OSA-related physiologic stress.11
Thrombotic potential is also enhanced, with higher levels of plasminogen activator inhibitor 1, fibrinogen, P-selectin, and vascular endothelial growth factor. Some of these factors normally have a diurnal cycle, with higher levels in the morning, but in OSA, increasing OSA severity is associated with increased prothrombotic potential in the morning hours. Of interest, levels of these substances showed a plateau effect, rising in people who had only mildly elevated apnea-hypopnea indices and then leveling off.12 Intermittent hypoxia followed by ventilatory overshoot hyperoxia, characteristic of sleep apnea, provides the ideal environment for augmentation of oxidative stress, with evidence of increased oxidation of serum proteins and lipids. Hypoxia and oxygen-derived free radicals may result in cardiac myocyte injury. Experimental data demonstrate that intermittent hypoxia combined with a high-fat diet results in synergistic acceleration of evidence of atherogenic lesions.
Patients with OSA also have evidence of endothelial dysfunction, insulin resistance, and dyslipidemia, all pathways that can facilitate the progression of atherosclerosis in OSA.13–15
Cardiac arrhythmias
In the Sleep Heart Health Study, a multicenter epidemiologic study designed to examine the relationships of OSA and cardiovascular outcomes, those who had moderate to severe OSA had a risk of ventricular and atrial arrhythmias two to four times higher than those without OSA, even after correction for the confounding influences of obesity and underlying cardiovascular risk.14 These findings were corroborated in subsequent work highlighting monotonic dose-response relationships with increasing OSA severity and increased odds of atrial and ventricular arrhythmia in a cohort of about 3,000 older men.11 Additional compelling evidence of a causal relationship is that the risk of discrete arrhythmic events is markedly increased after a respiratory disturbance in sleep.16
In patients who successfully underwent cardioversion for atrial fibrillation, those who had sleep apnea but were not treated with continuous positive airway pressure (CPAP) had a much higher rate of recurrence of atrial fibrillation during the subsequent year than those with CPAP-treated sleep apnea and than controls never diagnosed with sleep apnea. In the untreated patients with sleep apnea, the mean nocturnal fall in oxygen saturation was significantly greater in those who had recurrence of atrial fibrillation than in those who did not, suggesting hypoxia as an important mechanism contributing to atrial fibrillation.17
Since then, several other retrospective studies have shown similar findings after pulmonary vein antrum isolation and ablation in terms of reduction of atrial fibrillation recurrence with CPAP treatment in OSA.18
Walia et al19 described a patient with moderate sleep apnea who underwent a split-night study. During the baseline part of the study, the patient had about 18 ectopic beats per minute. During the second portion of the study while CPAP was applied, progressively fewer ectopic beats occurred as airway pressure was increased until a normal rhythm without ectopic beats was achieved at the goal treatment CPAP pressure setting.
Cardiovascular disease, stroke, and death
Marin et al20 followed about 1,500 men for 10 years, including some who had severe OSA, some with sleep apnea who were treated with CPAP, and controls. The risk of nonfatal and fatal cardiovascular disease events was nearly three times higher in those with severe disease than in healthy participants. Those treated with CPAP had a risk approximately the same as in the control group.
The Sleep Heart Study followed approximately 6,000 people with untreated sleep apnea for a median of nearly 9 years. It found a significant association between the apnea-hypopnea index and ischemic stroke, especially in men.21 Survival in patients with heart failure is also associated with the degree of OSA; patients with more severe disease (an apnea-hypopnea index ≥ 15) have a nearly three times greater risk of death than those with no disease or only mild disease (apnea-hypopnea index < 15).22
From the standpoint of health care utilization, findings that central sleep apnea predicts an increased risk of hospital readmission in heart failure are of particular interest.23
People with OSA are at increased risk of nocturnal sudden cardiac death.24 Sleep apnea is also associated with an increased overall death rate, and the higher the apnea-hypopnea index, the higher the death rate,25 even after adjusting for age, sex, body mass index, and underlying cardiovascular risk, with findings most pronounced in men under age 70.
Motor vehicle accidents
The need for caution during driving should be discussed with every patient, as motor vehicle accidents are an immediate danger to the patient and others. The association with motor vehicle accidents is independent of sleepiness, and drivers with sleep apnea often do not perceive performance impairment. Young et al26 found that men who snored were 3.4 times as likely to have an accident over a 5-year period, and that men and women with an apnea-hypopnea index greater than 15 were more than 7 times as likely to have multiple accidents over a 5-year period, highlighting the importance of discussing, documenting, and expeditiously diagnosing and treating OSA, particularly in those who report drowsiness while driving.
CLINICAL RISK FACTORS
Risk factors can be divided into nonmodifiable and modifiable ones.
Nonmodifiable factors
Age. Bimodal distributions in OSA prevalence have been observed; ie, that the pediatric population and people who are middle-aged have the highest prevalence of OSA. A linear relationship between sleep apnea prevalence and age until about age 65 was identified in data from the Sleep Heart Health Study.27 After that, the prevalence rates plateau; it is unclear if this is secondary to natural remission of the disease after a certain age or because patients with more severe disease have died by that age (ie, survivorship bias), blunting an increase in prevalence.
Sex. Men develop sleep apnea at a rate three to five times that of women. Several explanations have been proposed to account for this.28,29 Sex hormones are one factor; women with sleep apnea on hormone replacement therapy have a significantly less-severe sleep apnea burden than other postmenopausal women,30 suggesting a positive effect from estrogen. Sex-based differences in fat distribution, length and collapsibility of the upper airway, genioglossal activity, neurochemical control mechanisms, and arousal response may also contribute to prevalence differences between men and women.
As with coronary artery disease, the presentation of sleep apnea may be atypical in women, particularly around menopause. Sleep apnea should be considered in women who have snoring and daytime sleepiness.
Race. Whites, African Americans, and Asians have a similar prevalence of sleep apnea, but groups differ in obesity rates and craniofacial anatomy.31–34 Asians tend to have craniofacial skeletal restriction. African Americans are more likely to have upper-airway soft-tissue risk and to develop more severe OSA. Whites tend to have both craniofacial and soft-tissue risk. For those with craniofacial anatomy predisposing to OSA, even mild obesity can make it manifest.
Syndromes that predispose to OSA can include craniofacial structural abnormalities, connective tissue problems, or alterations in ventilatory control (eg, Marfan, Down, and Pierre Robin syndromes).
Modifiable risk factors
Obesity (body mass index ≥ 30 kg/m2) is a firmly established risk factor, but not all obese patients develop obstructive sleep apnea, and not all people with sleep apnea are obese.
Obesity increases risk by altering the geometry and function of the upper airway, increasing collapsibility. The changes are particularly pronounced in the lateral aspects of the pharynx.35
Obesity also affects respiratory drive, likely in part from leptin resistance. Load compensation is another contributing factor: the increased mass in the thorax and abdomen increases the work of breathing and reduces functional residual capacity, increasing oxygen demands and leading to atelectasis and ventilation-perfusion mismatch.
Although obesity is an important risk factor, it is important to recognize that obesity is not the only one to consider: most people with an apnea-hypopnea index of 5 or greater are not obese. The relationship between body mass index and sleep apnea is weaker in children and in the elderly, probably because other risk factors are more pronounced.36
Craniofacial structural abnormalities such as retrognathia (abnormal posterior position of the mandible) and micrognathia (undersized mandible) can increase the risk of OSA because of a resulting posteriorly displaced genioglossus muscle. Other conditions can alter chemosensitivity, affecting the pH and carbon dioxide level of the blood and therefore affecting ventilatory control mechanisms, making the person more prone to developing sleep apnea. Children and young adults may have tonsillar tissue that obstructs the airway.
The site of obstruction can be behind the palate (retropalatal), behind the tongue (retroglossal), or below the pharynx (hypopharyngeal). This helps explain why positive air way pressure—unlike surgery, which addresses a specific area—is often successful, as it serves to splint or treat all aspects of the airway.
FATIGUE, SLEEPINESS, SNORING, RESTLESS SLEEP
Sleep apnea can result in presentation of multiple signs and symptoms (Table 1).
Daytime sleepiness and fatigue are the most common symptoms. Although nonspecific, they are often quite pronounced. Two short questionnaires—the Epworth Sleepiness Scale37 and the Fatigue Severity Scale—can help distinguish between these two symptoms and assess their impact on a patient’s daily life. In the Epworth Sleepiness Scale, the patient rates his or her chance of dozing on a 4-point scale (0 = would never doze, to 3 = high chance of dozing) in eight situations:
- Sitting and reading
- Watching television
- Sitting inactive in a public place
- As a passenger in a car for an hour without a break
- Lying down to rest in the afternoon
- Sitting and talking to someone
- Sitting quietly after a lunch without alcohol
- In a car while stopped for a few minutes in traffic.
A score of 10 or more is consistent with significant subjective sleepiness.
The Fatigue Severity Scale assesses the impact of fatigue on daily living.
Snoring is a common and specific symptom of sleep apnea; however, not all patients who snore have OSA.
Restlessness during sleep is very common—patients may disturb their bed partner by moving around a lot during sleep or report that the sheets are “all over the place” by morning.
Nocturia can also be a sign of sleep apnea and can contribute to sleep fragmentation. A proposed mechanism of this symptom includes alterations of intrathoracic pressure resulting in atrial stretch, which release atrial natriuretic peptide, leading to nocturia. Treating with CPAP has been found to reduce levels of atrial natriuretic peptide, contributing to better sleep.38
Morning headache may occur and is likely related to increased CO2 levels, which appear to culminate in the morning hours. End-tidal or transcutaneous CO2 monitoring during polysomnography can help elucidate the presence of sleep-related hypoventilation.
Libido is often diminished and can actually be improved with CPAP. This is therefore an important point to discuss with patients, as improved libido can often serve as an incentive for adherence to OSA treatment.
Insomnia exists in about 15% of patients, primarily as a result of sleep apnea-related with treatment.
Sweating, particularly forehead sweating associated with sleep apnea, more commonly occurs in children.
The STOP-BANG questionnaire (Table 2)39 was primarily validated in preoperative anesthesia testing. However, because of its ease of use and favorable performance characteristics, it is increasingly used to predict the likelihood of finding OSA before polysomnography. A score of 3 or more has a sensitivity of 93%.
PHYSICAL EXAMINATION PROVIDES CLUES
Although the physical examination may be normal, certain findings indicate risk (Table 3). Obesity alone is not an accepted indication for polysomnography unless there are concomitant worrisome signs or symptoms. Of note, those who are morbidly obese (BMI > 40 kg/m2) have a prevalence of sleep apnea greater than 70%.
The classification by Friedman et al40 provides an indicator of risk. The patient is examined with the mouth opened wide and the tongue in a neutral natural position. Grades:
- I—Entire uvula and tonsils are visible
- II—Entire uvula is visible, but tonsils are not
- III—Soft palate is visible, but uvula is not
- IV—Only the hard palate is visible.
Especially in children and young adults, enlarged tonsils (or “kissing tonsils”) and a boggy edematous uvula set the stage for obstructive sleep apnea.
DIAGNOSIS REQUIRES SLEEP TESTING
A sleep study is the primary means of diagnosing OSA. Polysomnography includes electrooculography to determine when rapid-eye-movement sleep occurs; electromyography to measure muscle activity in the chin to help determine onset of sleep, with peripheral leads in the leg to measure leg movements; electroencephalography (EEG) to measure neural activity; electrocardiography; pulse oximetry to measure oxygen saturation; measurement of oronasal flow; and measurements of chest wall effort and body position using thoracic and abdominal belts that expand and contract with breathing; and audio recording to detect snoring.
Attended polysomnography requires the constant presence of a trained sleep technologist to monitor for technical issues and patient adherence.
End-tidal CO2 monitoring is a reasonable method to detect sleep-related hypoventilation but is not routinely performed in the United States. Transcutaneous CO2 monitoring is a different way to monitor CO2 used in the setting of positive airway pressure.
Polysomnography in a normal patient shows a regular pattern of increasing and decreasing airflow with inspiration and expiration while stable oxygen saturation is maintained.
In contrast, polysomnography of a patient with sleep apnea shows repetitive periods of no airflow, oxygen desaturation, and often evidence of thoracoabdominal paradox, punctuated by arousals on EEG associated with sympathetic activation (Figure 1). When the patient falls asleep, upper-airway muscle tone is reduced, causing an apneic event with hypoxia and pleural pressure swings. These prompt arousals with sympathetic activation that reestablish upper-airway muscle tone, allowing ventilation and reoxygenation to resume with a return to sleep.
Apnea-hypopnea index indicates severity
Sleep apnea severity is graded using the apnea-hypopnea index, ie, the number of apneic and hypopneic events per hour of sleep (Table 4).41 Events must last at least 10 seconds to be considered, ie, two consecutive missed breaths based on an average normal respiratory rate of about 12 breaths per minute for the typical adult.
The apnea-hypopnea index usually correlates with the severity of oxygen desaturation and with electrocardiographic abnormalities, including tachybradycardia and arrhythmias.
Although history, physical examination, and prediction tools are helpful in determining the likelihood that a patient has OSA, only polysomnography testing can establish the diagnosis. To diagnose OSA, 15 or more obstructive events per hour must be observed by polysomnography, or at least 5 events per hour with one of the following:
- Daytime sleepiness, sleep attacks, unrefreshing sleep, fatigue, or insomnia
- Waking with breath-holding, gasping, or choking
- Observer-reported loud snoring or breathing interruptions.41
Split-night study determines diagnosis and optimum treatment
The split-night study has two parts: the first is diagnostic polysomnography, followed by identification of the positive airway pressure that optimally treats the sleep apnea. The apnea-hypopnea index guides the need for the split-night study, with 40 being the established threshold according to the American Academy of Sleep Medicine.
A home sleep study is appropriate for some patients
Home sleep testing is typically more limited than standard polysomnography; it monitors airflow, effort, and oxygenation. The test is intended for adults with a high pretest probability of moderate to severe obstructive sleep apnea (STOP-BANG score ≥ 3). It is not intended for screening of asymptomatic patients or for those with coexisting sleep disorders (eg, central sleep apnea, sleep hypoventilation, periodic limb movements, insomnia, circadian rhythm disorders, parasomnias, narcolepsy) or medical disorders (eg, moderate to severe heart failure or other cardiac disease, symptomatic neurologic disease, moderate to severe pulmonary disease).42 Since March 2008, the Centers for Medicare and Medicaid Services has covered CPAP for obstructive sleep apnea based on diagnosis by home sleep study testing.43
TREATMENT OF SLEEP APNEA
Basic steps for reducing OSA are:
Weight loss. Even small weight changes can significantly affect the severity of sleep apnea, perhaps even leading to a reassessment of the degree of OSA and CPAP requirements. Longitudinal epidemiologic data demonstrate that a 10% weight loss correlates with a 26% reduction in the apnea-hypopnea index, and conversely, a 10% weight gain is associated with a 32% increase.44
Some studies have found that bariatric surgery cures OSA in 75% to 88% of cases, independent of approach.45,46 However, a trial in 60 obese patients with OSA who were randomized to either a low-calorie diet or bariatric surgery found no statistical difference in the apnea-hypopnea index between the two groups despite greater weight loss in the surgery group.47
Avoiding certain medications. Benzodiazepines, narcotics, and alcohol reduce upper airway muscle tone and should be avoided. No medications are associated with improvement of OSA, although acetazolamide may be used to treat central sleep apnea.
Positional therapy. Sleeping on the back exacerbates the problem. Supine-related OSA occurs as a result of several factors, including gravity, airway anatomy, airway critical closing pressures, and effects on upper-airway dilator muscle function.
Sleep hygiene. General recommendations to engage in behaviors to promote sleep are recommended, including keeping consistent sleep-wake times, not watching television in bed, and avoidance of caffeine intake, particularly within 4 to 6 hours of bedtime.
POSITIVE AIRWAY PRESSURE THERAPY
Nasal CPAP is the treatment of choice and is successful in 95% of patients when used consistently. It is not as costly as surgery, and results in improved long-term survival compared with uvulopalatopharyngoplasty. Another advantage is that the pressure can be retitrated as the patient’s condition changes, for example after a weight change or during pregnancy.
More than 15 randomized controlled trials have examined the effect of sleep apnea treatment with CPAP compared with either sham CPAP or another control. In a meta-analysis, CPAP was found to lead to an average systolic blood pressure reduction of about 2.5 mm Hg and a diastolic blood pressure reduction of 1.8 mm Hg. Although these reductions may seem negligible, benefits may be significant for cardiovascular outcomes.48,49
Challenges to treatment adherence
Adherence is the most commonly discussed problem with CPAP, but long-term adherence rates are comparable to medication compliance—about 60% to 70%. To optimize adherence, communication is important to ensure that problems are identified and addressed as they arise. Showing patients examples of apneic events and oxygen desaturation from their sleep study can enhance their understanding of OSA and its importance. Patients need to understand the serious nature of the disease and that CPAP therapy can significantly improve their quality of life and overall health, particularly from a cardiovascular perspective.
CPAP masks can be uncomfortable, posing a major barrier to compliance. But a number of mask designs are available, such as the nasal mask, the nasal pillow mask, and the oronasal mask. For patients with claustrophobia, the nasal pillow mask is an option, as it does not cover the face.
Some patients note symptoms of nasal congestion, although in many patients CPAP improves it. If congestion is a problem, the use of heated humidification with the machine, intranasal saline or gel, or nasal corticosteroids can help relieve it.
Pressure intolerance is a common problem. For those who feel that the pressure is too high, settings can be adjusted so that the pressure is gradually reduced between inspiration and expiration, ie, the use of expiratory pressure relief or consideration of the use of bilevel positive airway pressure.
Aerophagia (swallowing air) is a less common problem. It can also potentially be relieved with use of bilevel positive airway pressure.
Many patients develop skin irritation, which can be helped with moleskin, available at any pharmacy.
Social stigma can be a problem. Education regarding the importance of the treatment to health is essential.
Machine noise is less of a problem with the new machine models, but if it is a problem, a white-noise device or earplugs may help.
Other measures to improve compliance are keeping the regimen simple and ensuring that family support is adequate.
Medicare requires evidence of use and benefit
Medicare requires that clinical benefit be documented between the 31st and 91st day after initiating CPAP therapy. This requires face-to-face clinical reevaluation by the treating physician to document improved symptoms and objective evidence of adherence to use of the device. The devices can store usage patterns, and Medicare requires at least 4 hours per night on 70% of nights during a consecutive 30-day period in the first 3 months of use.
ALTERNATIVE THERAPIES
Alternative therapies may be options for some patients, in particular those who cannot use CPAP or who get no benefit from it. These include oral appliances for those with mild to moderate OSA50–53 and various surgical procedures, eg, uvulopalatopharyngoplasty,54,55 maxillomanibular advancement,56 tracheostomy (standard treatment before CPAP was identified as an effective treatment),57,58 and adenotonsillectomy (in children).59
Supplemental oxygen is not a first-line treatment for OSA and in general has not been found to be very effective, particularly in terms of intermediate cardiovascular outcomes,60–62 although a subset of patients with high loop gain may benefit from it.63 Loop gain is a measure of the tendency of the ventilatory control system to amplify respiration in response to a change, conferring less stable control of breathing.
Several novel alternative therapies are starting to be used. Although all of them have been shown to improve measures of OSA, none is as effective as CPAP in improving OSA severity. New therapies include the nasal expiratory positive airway pressure device,64 oral pressure therapy,65 and hypoglossal nerve stimulation.66
Obstructive sleep apnea (OSA) is common and poorly recognized and, if untreated, leads to serious health consequences. This article discusses the epidemiology of OSA, describes common presenting signs and symptoms, and reviews diagnostic testing and treatment options. Adverse health effects related to untreated sleep apnea are also discussed.
COMMON, POORLY RECOGNIZED, AND COSTLY IF UNTREATED
OSA is very common in the general population and is associated with substantial morbidity and mortality. An estimated 17% of the general adult population has OSA, and the numbers are increasing with the obesity epidemic. Nearly 1 in 15 adults has at least moderate sleep apnea,1,2 and approximately 85% of cases are estimated to be undiagnosed.3 A 1999 study estimated that untreated OSA resulted in approximately $3.4 billion in additional medical costs per year in the United States,4 a figure that is likely to be higher now, given the rising prevalence of OSA. The prevalence of OSA in primary care and subspecialty clinics is even higher than in the community, as more than half of patients who have diabetes or hypertension and 30% to 40% of patients with coronary artery disease are estimated to have OSA.5–7
REPETITIVE UPPER-AIRWAY COLLAPSE
During sleep, parasympathetic activity is enhanced and the muscle tone of the upper airway is decreased, particularly in the pharyngeal dilator muscles. Still, even in the supine position, a healthy person maintains patency of the airway and adequate airflow during sleep.
OSA is characterized by repetitive complete or partial collapse of the upper airway during sleep, resulting in an apneic or hypopneic event, respectively, and often causing snoring from upper-airway tissue vibration.
People who are susceptible to OSA typically have a smaller, more collapsible airway that is often less distensible and has a higher critical closing pressure. Radiographic and physiologic data have shown that the airway dimensions of patients with OSA are smaller than in those without OSA. The shape of the airway of a patient with OSA is often elliptical, given the extrinsic compression of the lateral aspects of the airway by increased size of the parapharyngeal fat pads. OSA episodes are characterized by closure of the upper airway and by progressively increasing respiratory efforts driven by chemoreceptor and mechanoreceptor stimuli, culminating in an arousal from sleep and a reopening of the airway.
The disease-defining metric used for assessing OSA severity is the apnea-hypopnea index, ie, the number of apneas and hypopneas that occur per hour of sleep.8 An apneic or hypopneic event is identified during polysomnography by the complete cessation of airflow or by a reduction in airflow for 10 seconds or longer (Figure 1).
HEALTH CONSEQUENCES IF UNTREATED
Untreated sleep apnea causes numerous pathophysiologic perturbations, including chronic intermittent hypoxia, ventilatory overshoot hyperoxia, increased sympathetic nervous system activity, intrathoracic pressure swings, hypercapnea, sleep fragmentation, increased arousals, reduced sleep duration, and fragmentation of rapid-eye-movement sleep.
Intermittent hypoxia activates the sympathetic nervous system and causes pulmonary vasoconstriction, with increases in pulmonary arterial pressures and myocardial workload. Sympathetic activation, ascertained by peroneal microneurography, has been shown to be increased not only during sleep but also persisting during wakefulness in patients with untreated OSA vs those without OSA.9 Autonomic nervous system fluctuations accompany apneic episodes, resulting in enhanced parasympathetic tone and sympathetic activation associated with a rise in blood pressure and heart rate that occur after the respiratory event.
Intermediate pathways that link the negative pathophysiologic effects of OSA with adverse health outcomes include increased systemic inflammation, increased oxidative stress, metabolic dysfunction, insulin resistance, hypercoagulability, endothelial dysfunction, and autonomic dysfunction.
As a result, a variety of adverse clinical outcomes are associated with untreated OSA, including systemic hypertension, ischemic heart disease and atherosclerosis, diastolic dysfunction, congestive heart failure, cardiac arrhythmias, stroke, increased risk of death, and sudden death, as well as noncardiovascular outcomes such as gout, neurocognitive deficits, and mood disorders.10
Inflammatory and atherogenic effects
Increased levels of markers of systemic inflammation, prothrombosis, and oxidative stress have been observed in OSA and may be key pathophysiologic links between OSA and cardiovascular sequelae. OSA has been associated with up-regulation of a number of inflammatory mediators: interleukin (IL) 6, soluble IL-6 receptor, IL-8, tumor necrosis factor alpha, and C-reactive protein. Soluble IL-6 levels in particular are higher in people who have sleep-disordered breathing, as reflected by the apnea-hypopnea index independent of obesity, with relationships stronger in the morning than in the evening. This likely reflects the overnight OSA-related physiologic stress.11
Thrombotic potential is also enhanced, with higher levels of plasminogen activator inhibitor 1, fibrinogen, P-selectin, and vascular endothelial growth factor. Some of these factors normally have a diurnal cycle, with higher levels in the morning, but in OSA, increasing OSA severity is associated with increased prothrombotic potential in the morning hours. Of interest, levels of these substances showed a plateau effect, rising in people who had only mildly elevated apnea-hypopnea indices and then leveling off.12 Intermittent hypoxia followed by ventilatory overshoot hyperoxia, characteristic of sleep apnea, provides the ideal environment for augmentation of oxidative stress, with evidence of increased oxidation of serum proteins and lipids. Hypoxia and oxygen-derived free radicals may result in cardiac myocyte injury. Experimental data demonstrate that intermittent hypoxia combined with a high-fat diet results in synergistic acceleration of evidence of atherogenic lesions.
Patients with OSA also have evidence of endothelial dysfunction, insulin resistance, and dyslipidemia, all pathways that can facilitate the progression of atherosclerosis in OSA.13–15
Cardiac arrhythmias
In the Sleep Heart Health Study, a multicenter epidemiologic study designed to examine the relationships of OSA and cardiovascular outcomes, those who had moderate to severe OSA had a risk of ventricular and atrial arrhythmias two to four times higher than those without OSA, even after correction for the confounding influences of obesity and underlying cardiovascular risk.14 These findings were corroborated in subsequent work highlighting monotonic dose-response relationships with increasing OSA severity and increased odds of atrial and ventricular arrhythmia in a cohort of about 3,000 older men.11 Additional compelling evidence of a causal relationship is that the risk of discrete arrhythmic events is markedly increased after a respiratory disturbance in sleep.16
In patients who successfully underwent cardioversion for atrial fibrillation, those who had sleep apnea but were not treated with continuous positive airway pressure (CPAP) had a much higher rate of recurrence of atrial fibrillation during the subsequent year than those with CPAP-treated sleep apnea and than controls never diagnosed with sleep apnea. In the untreated patients with sleep apnea, the mean nocturnal fall in oxygen saturation was significantly greater in those who had recurrence of atrial fibrillation than in those who did not, suggesting hypoxia as an important mechanism contributing to atrial fibrillation.17
Since then, several other retrospective studies have shown similar findings after pulmonary vein antrum isolation and ablation in terms of reduction of atrial fibrillation recurrence with CPAP treatment in OSA.18
Walia et al19 described a patient with moderate sleep apnea who underwent a split-night study. During the baseline part of the study, the patient had about 18 ectopic beats per minute. During the second portion of the study while CPAP was applied, progressively fewer ectopic beats occurred as airway pressure was increased until a normal rhythm without ectopic beats was achieved at the goal treatment CPAP pressure setting.
Cardiovascular disease, stroke, and death
Marin et al20 followed about 1,500 men for 10 years, including some who had severe OSA, some with sleep apnea who were treated with CPAP, and controls. The risk of nonfatal and fatal cardiovascular disease events was nearly three times higher in those with severe disease than in healthy participants. Those treated with CPAP had a risk approximately the same as in the control group.
The Sleep Heart Study followed approximately 6,000 people with untreated sleep apnea for a median of nearly 9 years. It found a significant association between the apnea-hypopnea index and ischemic stroke, especially in men.21 Survival in patients with heart failure is also associated with the degree of OSA; patients with more severe disease (an apnea-hypopnea index ≥ 15) have a nearly three times greater risk of death than those with no disease or only mild disease (apnea-hypopnea index < 15).22
From the standpoint of health care utilization, findings that central sleep apnea predicts an increased risk of hospital readmission in heart failure are of particular interest.23
People with OSA are at increased risk of nocturnal sudden cardiac death.24 Sleep apnea is also associated with an increased overall death rate, and the higher the apnea-hypopnea index, the higher the death rate,25 even after adjusting for age, sex, body mass index, and underlying cardiovascular risk, with findings most pronounced in men under age 70.
Motor vehicle accidents
The need for caution during driving should be discussed with every patient, as motor vehicle accidents are an immediate danger to the patient and others. The association with motor vehicle accidents is independent of sleepiness, and drivers with sleep apnea often do not perceive performance impairment. Young et al26 found that men who snored were 3.4 times as likely to have an accident over a 5-year period, and that men and women with an apnea-hypopnea index greater than 15 were more than 7 times as likely to have multiple accidents over a 5-year period, highlighting the importance of discussing, documenting, and expeditiously diagnosing and treating OSA, particularly in those who report drowsiness while driving.
CLINICAL RISK FACTORS
Risk factors can be divided into nonmodifiable and modifiable ones.
Nonmodifiable factors
Age. Bimodal distributions in OSA prevalence have been observed; ie, that the pediatric population and people who are middle-aged have the highest prevalence of OSA. A linear relationship between sleep apnea prevalence and age until about age 65 was identified in data from the Sleep Heart Health Study.27 After that, the prevalence rates plateau; it is unclear if this is secondary to natural remission of the disease after a certain age or because patients with more severe disease have died by that age (ie, survivorship bias), blunting an increase in prevalence.
Sex. Men develop sleep apnea at a rate three to five times that of women. Several explanations have been proposed to account for this.28,29 Sex hormones are one factor; women with sleep apnea on hormone replacement therapy have a significantly less-severe sleep apnea burden than other postmenopausal women,30 suggesting a positive effect from estrogen. Sex-based differences in fat distribution, length and collapsibility of the upper airway, genioglossal activity, neurochemical control mechanisms, and arousal response may also contribute to prevalence differences between men and women.
As with coronary artery disease, the presentation of sleep apnea may be atypical in women, particularly around menopause. Sleep apnea should be considered in women who have snoring and daytime sleepiness.
Race. Whites, African Americans, and Asians have a similar prevalence of sleep apnea, but groups differ in obesity rates and craniofacial anatomy.31–34 Asians tend to have craniofacial skeletal restriction. African Americans are more likely to have upper-airway soft-tissue risk and to develop more severe OSA. Whites tend to have both craniofacial and soft-tissue risk. For those with craniofacial anatomy predisposing to OSA, even mild obesity can make it manifest.
Syndromes that predispose to OSA can include craniofacial structural abnormalities, connective tissue problems, or alterations in ventilatory control (eg, Marfan, Down, and Pierre Robin syndromes).
Modifiable risk factors
Obesity (body mass index ≥ 30 kg/m2) is a firmly established risk factor, but not all obese patients develop obstructive sleep apnea, and not all people with sleep apnea are obese.
Obesity increases risk by altering the geometry and function of the upper airway, increasing collapsibility. The changes are particularly pronounced in the lateral aspects of the pharynx.35
Obesity also affects respiratory drive, likely in part from leptin resistance. Load compensation is another contributing factor: the increased mass in the thorax and abdomen increases the work of breathing and reduces functional residual capacity, increasing oxygen demands and leading to atelectasis and ventilation-perfusion mismatch.
Although obesity is an important risk factor, it is important to recognize that obesity is not the only one to consider: most people with an apnea-hypopnea index of 5 or greater are not obese. The relationship between body mass index and sleep apnea is weaker in children and in the elderly, probably because other risk factors are more pronounced.36
Craniofacial structural abnormalities such as retrognathia (abnormal posterior position of the mandible) and micrognathia (undersized mandible) can increase the risk of OSA because of a resulting posteriorly displaced genioglossus muscle. Other conditions can alter chemosensitivity, affecting the pH and carbon dioxide level of the blood and therefore affecting ventilatory control mechanisms, making the person more prone to developing sleep apnea. Children and young adults may have tonsillar tissue that obstructs the airway.
The site of obstruction can be behind the palate (retropalatal), behind the tongue (retroglossal), or below the pharynx (hypopharyngeal). This helps explain why positive air way pressure—unlike surgery, which addresses a specific area—is often successful, as it serves to splint or treat all aspects of the airway.
FATIGUE, SLEEPINESS, SNORING, RESTLESS SLEEP
Sleep apnea can result in presentation of multiple signs and symptoms (Table 1).
Daytime sleepiness and fatigue are the most common symptoms. Although nonspecific, they are often quite pronounced. Two short questionnaires—the Epworth Sleepiness Scale37 and the Fatigue Severity Scale—can help distinguish between these two symptoms and assess their impact on a patient’s daily life. In the Epworth Sleepiness Scale, the patient rates his or her chance of dozing on a 4-point scale (0 = would never doze, to 3 = high chance of dozing) in eight situations:
- Sitting and reading
- Watching television
- Sitting inactive in a public place
- As a passenger in a car for an hour without a break
- Lying down to rest in the afternoon
- Sitting and talking to someone
- Sitting quietly after a lunch without alcohol
- In a car while stopped for a few minutes in traffic.
A score of 10 or more is consistent with significant subjective sleepiness.
The Fatigue Severity Scale assesses the impact of fatigue on daily living.
Snoring is a common and specific symptom of sleep apnea; however, not all patients who snore have OSA.
Restlessness during sleep is very common—patients may disturb their bed partner by moving around a lot during sleep or report that the sheets are “all over the place” by morning.
Nocturia can also be a sign of sleep apnea and can contribute to sleep fragmentation. A proposed mechanism of this symptom includes alterations of intrathoracic pressure resulting in atrial stretch, which release atrial natriuretic peptide, leading to nocturia. Treating with CPAP has been found to reduce levels of atrial natriuretic peptide, contributing to better sleep.38
Morning headache may occur and is likely related to increased CO2 levels, which appear to culminate in the morning hours. End-tidal or transcutaneous CO2 monitoring during polysomnography can help elucidate the presence of sleep-related hypoventilation.
Libido is often diminished and can actually be improved with CPAP. This is therefore an important point to discuss with patients, as improved libido can often serve as an incentive for adherence to OSA treatment.
Insomnia exists in about 15% of patients, primarily as a result of sleep apnea-related with treatment.
Sweating, particularly forehead sweating associated with sleep apnea, more commonly occurs in children.
The STOP-BANG questionnaire (Table 2)39 was primarily validated in preoperative anesthesia testing. However, because of its ease of use and favorable performance characteristics, it is increasingly used to predict the likelihood of finding OSA before polysomnography. A score of 3 or more has a sensitivity of 93%.
PHYSICAL EXAMINATION PROVIDES CLUES
Although the physical examination may be normal, certain findings indicate risk (Table 3). Obesity alone is not an accepted indication for polysomnography unless there are concomitant worrisome signs or symptoms. Of note, those who are morbidly obese (BMI > 40 kg/m2) have a prevalence of sleep apnea greater than 70%.
The classification by Friedman et al40 provides an indicator of risk. The patient is examined with the mouth opened wide and the tongue in a neutral natural position. Grades:
- I—Entire uvula and tonsils are visible
- II—Entire uvula is visible, but tonsils are not
- III—Soft palate is visible, but uvula is not
- IV—Only the hard palate is visible.
Especially in children and young adults, enlarged tonsils (or “kissing tonsils”) and a boggy edematous uvula set the stage for obstructive sleep apnea.
DIAGNOSIS REQUIRES SLEEP TESTING
A sleep study is the primary means of diagnosing OSA. Polysomnography includes electrooculography to determine when rapid-eye-movement sleep occurs; electromyography to measure muscle activity in the chin to help determine onset of sleep, with peripheral leads in the leg to measure leg movements; electroencephalography (EEG) to measure neural activity; electrocardiography; pulse oximetry to measure oxygen saturation; measurement of oronasal flow; and measurements of chest wall effort and body position using thoracic and abdominal belts that expand and contract with breathing; and audio recording to detect snoring.
Attended polysomnography requires the constant presence of a trained sleep technologist to monitor for technical issues and patient adherence.
End-tidal CO2 monitoring is a reasonable method to detect sleep-related hypoventilation but is not routinely performed in the United States. Transcutaneous CO2 monitoring is a different way to monitor CO2 used in the setting of positive airway pressure.
Polysomnography in a normal patient shows a regular pattern of increasing and decreasing airflow with inspiration and expiration while stable oxygen saturation is maintained.
In contrast, polysomnography of a patient with sleep apnea shows repetitive periods of no airflow, oxygen desaturation, and often evidence of thoracoabdominal paradox, punctuated by arousals on EEG associated with sympathetic activation (Figure 1). When the patient falls asleep, upper-airway muscle tone is reduced, causing an apneic event with hypoxia and pleural pressure swings. These prompt arousals with sympathetic activation that reestablish upper-airway muscle tone, allowing ventilation and reoxygenation to resume with a return to sleep.
Apnea-hypopnea index indicates severity
Sleep apnea severity is graded using the apnea-hypopnea index, ie, the number of apneic and hypopneic events per hour of sleep (Table 4).41 Events must last at least 10 seconds to be considered, ie, two consecutive missed breaths based on an average normal respiratory rate of about 12 breaths per minute for the typical adult.
The apnea-hypopnea index usually correlates with the severity of oxygen desaturation and with electrocardiographic abnormalities, including tachybradycardia and arrhythmias.
Although history, physical examination, and prediction tools are helpful in determining the likelihood that a patient has OSA, only polysomnography testing can establish the diagnosis. To diagnose OSA, 15 or more obstructive events per hour must be observed by polysomnography, or at least 5 events per hour with one of the following:
- Daytime sleepiness, sleep attacks, unrefreshing sleep, fatigue, or insomnia
- Waking with breath-holding, gasping, or choking
- Observer-reported loud snoring or breathing interruptions.41
Split-night study determines diagnosis and optimum treatment
The split-night study has two parts: the first is diagnostic polysomnography, followed by identification of the positive airway pressure that optimally treats the sleep apnea. The apnea-hypopnea index guides the need for the split-night study, with 40 being the established threshold according to the American Academy of Sleep Medicine.
A home sleep study is appropriate for some patients
Home sleep testing is typically more limited than standard polysomnography; it monitors airflow, effort, and oxygenation. The test is intended for adults with a high pretest probability of moderate to severe obstructive sleep apnea (STOP-BANG score ≥ 3). It is not intended for screening of asymptomatic patients or for those with coexisting sleep disorders (eg, central sleep apnea, sleep hypoventilation, periodic limb movements, insomnia, circadian rhythm disorders, parasomnias, narcolepsy) or medical disorders (eg, moderate to severe heart failure or other cardiac disease, symptomatic neurologic disease, moderate to severe pulmonary disease).42 Since March 2008, the Centers for Medicare and Medicaid Services has covered CPAP for obstructive sleep apnea based on diagnosis by home sleep study testing.43
TREATMENT OF SLEEP APNEA
Basic steps for reducing OSA are:
Weight loss. Even small weight changes can significantly affect the severity of sleep apnea, perhaps even leading to a reassessment of the degree of OSA and CPAP requirements. Longitudinal epidemiologic data demonstrate that a 10% weight loss correlates with a 26% reduction in the apnea-hypopnea index, and conversely, a 10% weight gain is associated with a 32% increase.44
Some studies have found that bariatric surgery cures OSA in 75% to 88% of cases, independent of approach.45,46 However, a trial in 60 obese patients with OSA who were randomized to either a low-calorie diet or bariatric surgery found no statistical difference in the apnea-hypopnea index between the two groups despite greater weight loss in the surgery group.47
Avoiding certain medications. Benzodiazepines, narcotics, and alcohol reduce upper airway muscle tone and should be avoided. No medications are associated with improvement of OSA, although acetazolamide may be used to treat central sleep apnea.
Positional therapy. Sleeping on the back exacerbates the problem. Supine-related OSA occurs as a result of several factors, including gravity, airway anatomy, airway critical closing pressures, and effects on upper-airway dilator muscle function.
Sleep hygiene. General recommendations to engage in behaviors to promote sleep are recommended, including keeping consistent sleep-wake times, not watching television in bed, and avoidance of caffeine intake, particularly within 4 to 6 hours of bedtime.
POSITIVE AIRWAY PRESSURE THERAPY
Nasal CPAP is the treatment of choice and is successful in 95% of patients when used consistently. It is not as costly as surgery, and results in improved long-term survival compared with uvulopalatopharyngoplasty. Another advantage is that the pressure can be retitrated as the patient’s condition changes, for example after a weight change or during pregnancy.
More than 15 randomized controlled trials have examined the effect of sleep apnea treatment with CPAP compared with either sham CPAP or another control. In a meta-analysis, CPAP was found to lead to an average systolic blood pressure reduction of about 2.5 mm Hg and a diastolic blood pressure reduction of 1.8 mm Hg. Although these reductions may seem negligible, benefits may be significant for cardiovascular outcomes.48,49
Challenges to treatment adherence
Adherence is the most commonly discussed problem with CPAP, but long-term adherence rates are comparable to medication compliance—about 60% to 70%. To optimize adherence, communication is important to ensure that problems are identified and addressed as they arise. Showing patients examples of apneic events and oxygen desaturation from their sleep study can enhance their understanding of OSA and its importance. Patients need to understand the serious nature of the disease and that CPAP therapy can significantly improve their quality of life and overall health, particularly from a cardiovascular perspective.
CPAP masks can be uncomfortable, posing a major barrier to compliance. But a number of mask designs are available, such as the nasal mask, the nasal pillow mask, and the oronasal mask. For patients with claustrophobia, the nasal pillow mask is an option, as it does not cover the face.
Some patients note symptoms of nasal congestion, although in many patients CPAP improves it. If congestion is a problem, the use of heated humidification with the machine, intranasal saline or gel, or nasal corticosteroids can help relieve it.
Pressure intolerance is a common problem. For those who feel that the pressure is too high, settings can be adjusted so that the pressure is gradually reduced between inspiration and expiration, ie, the use of expiratory pressure relief or consideration of the use of bilevel positive airway pressure.
Aerophagia (swallowing air) is a less common problem. It can also potentially be relieved with use of bilevel positive airway pressure.
Many patients develop skin irritation, which can be helped with moleskin, available at any pharmacy.
Social stigma can be a problem. Education regarding the importance of the treatment to health is essential.
Machine noise is less of a problem with the new machine models, but if it is a problem, a white-noise device or earplugs may help.
Other measures to improve compliance are keeping the regimen simple and ensuring that family support is adequate.
Medicare requires evidence of use and benefit
Medicare requires that clinical benefit be documented between the 31st and 91st day after initiating CPAP therapy. This requires face-to-face clinical reevaluation by the treating physician to document improved symptoms and objective evidence of adherence to use of the device. The devices can store usage patterns, and Medicare requires at least 4 hours per night on 70% of nights during a consecutive 30-day period in the first 3 months of use.
ALTERNATIVE THERAPIES
Alternative therapies may be options for some patients, in particular those who cannot use CPAP or who get no benefit from it. These include oral appliances for those with mild to moderate OSA50–53 and various surgical procedures, eg, uvulopalatopharyngoplasty,54,55 maxillomanibular advancement,56 tracheostomy (standard treatment before CPAP was identified as an effective treatment),57,58 and adenotonsillectomy (in children).59
Supplemental oxygen is not a first-line treatment for OSA and in general has not been found to be very effective, particularly in terms of intermediate cardiovascular outcomes,60–62 although a subset of patients with high loop gain may benefit from it.63 Loop gain is a measure of the tendency of the ventilatory control system to amplify respiration in response to a change, conferring less stable control of breathing.
Several novel alternative therapies are starting to be used. Although all of them have been shown to improve measures of OSA, none is as effective as CPAP in improving OSA severity. New therapies include the nasal expiratory positive airway pressure device,64 oral pressure therapy,65 and hypoglossal nerve stimulation.66
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 1993; 328:1230–1235.
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- Kapur VK, Redline S, Nieto FJ, Young TB, Newman AB, Henderson JA; Sleep Heart Health Research Group. The relationship between chronically disrupted sleep and healthcare use. Sleep 2002; 25:289–296.
- Kapur V, Blough DK, Sandblom RE, et al. The medical cost of undiagnosed sleep apnea. Sleep 1999; 22:749–755.
- Mooe T, Rabben T, Wiklund U, Franklin KA, Eriksson P. Sleep-disordered breathing in men with coronary artery disease. Chest 1996; 109:659–663.
- Schafer H, Koehler U, Ewig S, Hasper E, Tasci S, Luderitz B. Obstructive sleep apnea as a risk marker in coronary artery disease. Cardiology 1999; 92:79–84.
- Leung RS, Bradley TD. Sleep apnea and cardiovascular disease. Am J Respir Crit Care Med 2001; 164:2147–2165.
- American Academy of Sleep Medicine. International Classification of Sleep Disorders, Second Edition: Diagnostic and coding manual. Westchester, IL; American Academy of Sleep Medicine, 2005.
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995; 96:1897–1904.
- Mehra R. Sleep-disordered breathing and cardiovascular disease: exploring pathophysiology and existing data. Curr Resp Med Rev 2007; 3:258–269.
- Mehra R, Storfer-Isser A, Kirchner HL, et al. Soluble interleukin 6 receptor: a novel marker of moderate to severe sleep-related breathing disorder. Arch Intern Med 2006; 166:1725–1731.
- Mehra R, Xu F, Babineau DC, et al. Sleep-disordered breathing and prothrombotic biomarkers: cross-sectional results of the Cleveland Family Study. Am J Respir Crit Care Med 2010; 182:826–833.
- Mehra R, Storfer-Isser A, Tracy R, Jenny N, Redline S. Association of sleep disordered breathing and oxidized LDL [abstract]. Am J Respir Crit Care Med 2010; 181:A2474.
- Mehra R, Benjamin EJ, Shahar E, et al; Sleep Heart Health Study. Association of nocturnal arrhythmias with sleep-disordered breathing: the Sleep Heart Health Study. Am J Respir Crit Care Med 2006; 173:910–916.
- Mehra R, Xu F, Babineau DC, et al. Sleep-disordered breathing and prothrombotic biomarkers: cross-sectional results of the Cleveland Family Study. Am J Respir Crit Care Med 2010; 182:826–833.
- Monahan K, Storfer-Isser A, Mehra R, et al. Triggering of nocturnal arrhythmias by sleep-disordered breathing events. J Am Coll Cardiol 2009; 54:1797–1804.
- Kanagala R, Murali NS, Friedman PA, et al. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 2003; 107:2589–2594.
- Patel D, Mohanty P, Di Biase L, et al. Safety and efficacy of pulmonary vein antral isolation in patients with obstructive sleep apnea: the impact of continuous positive airway pressure. Circ Arrhythm Electrophysiol 2010; 3:445–451.
- Walia H, Strohl KP, Mehra R. Effect of continuous positive airway pressure on an atrial arrhythmia in a patient with mild obstructive sleep apnea. J Clin Sleep Med 2011; 7:397–398.
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365:1046–1053.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182:269–277.
- Wang H, Parker JD, Newton GE, et al. Influence of obstructive sleep apnea on mortality in patients with heart failure. J Am Coll Cardiol 2007; 49:1625–1631.
- Khayat R, Abraham W, Patt B, et al. Central sleep apnea is a predictor of cardiac readmission in hospitalized patients with systolic heart failure. J Card Fail 2012; 18:534–540.
- Gami AS, Howard DE, Olson EJ, Somers VK. Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med 2005; 352:1206–1214.
- Punjabi NM, Caffo BS, Goodwin JL, et al. Sleep-disordered breathing and mortality: a prospective cohort study. PLoS Med 2009; 6( 8):e1000132. doi: 10.1371/journal.pmed.1000132.
- Young T, Blustein J, Finn L, Palta M. Sleep-disordered breathing and motor vehicle accidents in a population-based sample of employed adults. Sleep 1997; 20:608–613.
- Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med 2002; 165:1217–1239.
- Lin CM, Davidson TM, Ancoli-Israel S. Gender differences in obstructive sleep apnea and treatment implications. Sleep Med Rev 2008; 12:481–496.
- Shaher E, Redline S, Young T, et al. Hormone replacement therapy and sleep-disordered breathing. Am J Respir Crit Care Med 2003; 167:1186–1192.
- Young T, Finn L, Austin D, Peterson A. Menopausal status and sleep-disordered breathing in the Wisconsin Sleep Cohort Study. Am J Respir Crit Care Med 2003; 167:1181–1185.
- Ancoli-Israel S, Klauber MR, Stepnowsky C, Estline E, Chinn A, Fell R. Sleep-disordered breathing in African-American elderly. Am J Respir Crit Care Med 1995; 152:1946–1949.
- Young T, Shahar E, Nieto FJ, et al; Sleep Heart Health Study Research Group. Predictors of sleep-disordered breathing in community-dwelling adults: the Sleep Heart Health Study. Arch Intern Med 2002; 162:893–900.
- Redline S, Tishler PV, Hans MG, Tosteson TD, Strohl KP, Spry K. Racial differences in sleep-disordered breathing in African-Americans and Caucasians. Am J Respir Crit Care Med 1997; 155:186–192. Erratum in: Am J Respir Crit Care Med 1997; 155:1820.
- Sutherland K, Lee RWW, Cistulli PA. Obesity and craniofacial structure as risk factors for obstructive sleep apnoea: impact of ethnicity. Respirology 2012; 17:213–222.
- Schwab RJ, Gupta KB, Gefter WB, Metzger LJ, Hoffman EA, Pack AI. Upper airway and soft tissue anatomy in normal subjects and patients with sleep-disordered breathing. Significance of the lateral pharyngeal walls. Am J Respir Crit Care Med 1995; 152:1673–1689.
- Nieto FJ, Young TB, Lind BK, et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 2000; 283:1829–1836.
- Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991; 14:540–545.
- Krieger J, Imbs J-L, Schmidt M, Kurtz D. Renal function in patients with obstructive sleep apnea. Effects of nasal continuous positive airway pressure. Arch Intern Med 1988; 148:1337–1340.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Friedman M, Ibrahim H, Bass L. Clinical staging for sleep-disordered breathing. Otolaryngal Head Neck Surg 2002; 127:13–21.
- Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. Report of an American Academy of Sleep Medicine Task Force. Sleep 1999; 22:667–689.
- Collop NA, Anderson WM, Boehlecke B, et al; Portable Monitoring Task Force of the American Academy of Sleep Medicine. Clinical guidelines for the use of unattended portable monitors in the diagnosis of obstructive sleep apnea in adult patients. Portable Monitoring Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2007; 3:737–747.
- Centers for Medicare & Medicaid Services (CMS). Continuous positive airway pressure (CPAP) therapy for obstructive sleep apnea (OSA). MLN Matters 2008. www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM6048.pdf. Accessed June 2, 2014.
- Peppard PE, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA 2000; 284:3015–3021.
- Guardiano SA, Scott JA, Ware JC, Schechner SA. The long-term results of gastric bypass on indexes of sleep apnea. Chest 2003; 124:1615–1619.
- Crooks PF. Surgical treatment of morbid obesity. Annu Rev Med 2006; 57:243–264.
- Dixon JB, Schachter LM, O’Brien PE, et al. Surgical vs conventional therapy for weight loss treatment of obstructive sleep apnea: a randomized controlled trial. JAMA 2012; 308:1142–1149.
- Bazzano LA, Khan Z, Reynolds K, He J. Effect of nocturnal nasal continuous positive airway pressure on blood pressure in obstructive sleep apnea. Hypertension 2007; 50:417–423.
- Logan AG, Perlikowski SM, Mente A, et al. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 2001; 19:2271–2277.
- Kushida CA, Morgenthaler TI, Littner MR, et al; American Academy of Sleep. Practice parameters for the treatment of snoring and obstructive sleep apnea with oral appliances: an update for 2005. Sleep 2006; 29:240–243.
- Otsuka R, Ribeiro de Almeida F, Lowe AA, Linden W, Ryan F. The effect of oral appliance therapy on blood pressure in patients with obstructive sleep apnea. Sleep Breath 2006; 10:29–36.
- Yoshida K. Effect on blood pressure of oral appliance therapy for sleep apnea syndrome. Int J Prosthodont 2006; 19:61–66.
- Inazawa T, Ayuse T, Kurata S, et al. Effect of mandibular position on upper airway collapsibility and resistance. J Dent Res 2005; 84:554–558.
- Fujita S, Conway W, Zorick F, Roth T. Surgical correction of anatomic abnormalities in obstructive sleep apnea syndrome: uvulopalatopharyngoplasty. Otolaryngol Head Neck Surg 1981; 89:923–934.
- Schwab RJ. Imaging for the snoring and sleep apnea patient. Dent Clin North Am 2001; 45:759–796.
- Prinsell JR. Maxillomandibular advancement surgery for obstructive sleep apnea syndrome. J Am Dent Assoc 2002; 133:1489–1497.
- Thatcher GW, Maisel RH. The long-term evaluation of tracheostomy in the management of severe obstructive sleep apnea. Laryngoscope 2003; 113:201–204.
- Conway WA, Victor LD, Magilligan DJ, Fujita S, Zorick FJ, Roth T. Adverse effects of tracheostomy for sleep apnea. JAMA 1981; 246:347–350.
- Marcus CL, Moore RH, Rosen CL, et al; Childhood Adenotonsillectomy Trial (CHAT). A randomized trial of adenotonsillectomy for childhood sleep apnea. N Engl J Med 2013; 368:2366–2376.
- Gottlieb DJ, Craig SE, Lorenzi-Filho G, et al. Sleep apnea cardiovascular clinical trials-current status and steps forward: The International Collaboration of Sleep Apnea Cardiovascular Trialists. Sleep 2013; 36:975–980.
- Loredo JS, Ancoli-Israel S, Kim EJ, Lim WJ, Dimsdale JE. Effect of continuous positive airway pressure versus supplemental oxygen on sleep quality in obstructive sleep apnea: a placebo-CPAP-controlled study. Sleep 2006; 29:564–571.
- Phillips BA, McConnell JW, Smith MD. The effects of hypoxemia on cardiac output. A dose-response curve. Chest 1988; 93:471–475.
- Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respire Physiol Neurobiol 2008; 162:144–151.
- Berry RB, Kryger MH, Massie CA. A novel nasal expiratory positive airway pressure (EPAP) device for the treatment of obstructive sleep apnea: a randomized controlled trial. Sleep 2011; 34:479–485.
- Colrain IM, Black J, Siegel LC, et al. A multicenter evaluation of oral pressure therapy for the treatment of obstructive sleep apnea. Sleep Med 2013; 14:830–837.
- Strollo PJ Jr, Soose RJ, Maurer JT, et al; STAR Trial Group. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med 2014; 370:139–149.
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 1993; 328:1230–1235.
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- Kapur VK, Redline S, Nieto FJ, Young TB, Newman AB, Henderson JA; Sleep Heart Health Research Group. The relationship between chronically disrupted sleep and healthcare use. Sleep 2002; 25:289–296.
- Kapur V, Blough DK, Sandblom RE, et al. The medical cost of undiagnosed sleep apnea. Sleep 1999; 22:749–755.
- Mooe T, Rabben T, Wiklund U, Franklin KA, Eriksson P. Sleep-disordered breathing in men with coronary artery disease. Chest 1996; 109:659–663.
- Schafer H, Koehler U, Ewig S, Hasper E, Tasci S, Luderitz B. Obstructive sleep apnea as a risk marker in coronary artery disease. Cardiology 1999; 92:79–84.
- Leung RS, Bradley TD. Sleep apnea and cardiovascular disease. Am J Respir Crit Care Med 2001; 164:2147–2165.
- American Academy of Sleep Medicine. International Classification of Sleep Disorders, Second Edition: Diagnostic and coding manual. Westchester, IL; American Academy of Sleep Medicine, 2005.
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995; 96:1897–1904.
- Mehra R. Sleep-disordered breathing and cardiovascular disease: exploring pathophysiology and existing data. Curr Resp Med Rev 2007; 3:258–269.
- Mehra R, Storfer-Isser A, Kirchner HL, et al. Soluble interleukin 6 receptor: a novel marker of moderate to severe sleep-related breathing disorder. Arch Intern Med 2006; 166:1725–1731.
- Mehra R, Xu F, Babineau DC, et al. Sleep-disordered breathing and prothrombotic biomarkers: cross-sectional results of the Cleveland Family Study. Am J Respir Crit Care Med 2010; 182:826–833.
- Mehra R, Storfer-Isser A, Tracy R, Jenny N, Redline S. Association of sleep disordered breathing and oxidized LDL [abstract]. Am J Respir Crit Care Med 2010; 181:A2474.
- Mehra R, Benjamin EJ, Shahar E, et al; Sleep Heart Health Study. Association of nocturnal arrhythmias with sleep-disordered breathing: the Sleep Heart Health Study. Am J Respir Crit Care Med 2006; 173:910–916.
- Mehra R, Xu F, Babineau DC, et al. Sleep-disordered breathing and prothrombotic biomarkers: cross-sectional results of the Cleveland Family Study. Am J Respir Crit Care Med 2010; 182:826–833.
- Monahan K, Storfer-Isser A, Mehra R, et al. Triggering of nocturnal arrhythmias by sleep-disordered breathing events. J Am Coll Cardiol 2009; 54:1797–1804.
- Kanagala R, Murali NS, Friedman PA, et al. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 2003; 107:2589–2594.
- Patel D, Mohanty P, Di Biase L, et al. Safety and efficacy of pulmonary vein antral isolation in patients with obstructive sleep apnea: the impact of continuous positive airway pressure. Circ Arrhythm Electrophysiol 2010; 3:445–451.
- Walia H, Strohl KP, Mehra R. Effect of continuous positive airway pressure on an atrial arrhythmia in a patient with mild obstructive sleep apnea. J Clin Sleep Med 2011; 7:397–398.
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365:1046–1053.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182:269–277.
- Wang H, Parker JD, Newton GE, et al. Influence of obstructive sleep apnea on mortality in patients with heart failure. J Am Coll Cardiol 2007; 49:1625–1631.
- Khayat R, Abraham W, Patt B, et al. Central sleep apnea is a predictor of cardiac readmission in hospitalized patients with systolic heart failure. J Card Fail 2012; 18:534–540.
- Gami AS, Howard DE, Olson EJ, Somers VK. Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med 2005; 352:1206–1214.
- Punjabi NM, Caffo BS, Goodwin JL, et al. Sleep-disordered breathing and mortality: a prospective cohort study. PLoS Med 2009; 6( 8):e1000132. doi: 10.1371/journal.pmed.1000132.
- Young T, Blustein J, Finn L, Palta M. Sleep-disordered breathing and motor vehicle accidents in a population-based sample of employed adults. Sleep 1997; 20:608–613.
- Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med 2002; 165:1217–1239.
- Lin CM, Davidson TM, Ancoli-Israel S. Gender differences in obstructive sleep apnea and treatment implications. Sleep Med Rev 2008; 12:481–496.
- Shaher E, Redline S, Young T, et al. Hormone replacement therapy and sleep-disordered breathing. Am J Respir Crit Care Med 2003; 167:1186–1192.
- Young T, Finn L, Austin D, Peterson A. Menopausal status and sleep-disordered breathing in the Wisconsin Sleep Cohort Study. Am J Respir Crit Care Med 2003; 167:1181–1185.
- Ancoli-Israel S, Klauber MR, Stepnowsky C, Estline E, Chinn A, Fell R. Sleep-disordered breathing in African-American elderly. Am J Respir Crit Care Med 1995; 152:1946–1949.
- Young T, Shahar E, Nieto FJ, et al; Sleep Heart Health Study Research Group. Predictors of sleep-disordered breathing in community-dwelling adults: the Sleep Heart Health Study. Arch Intern Med 2002; 162:893–900.
- Redline S, Tishler PV, Hans MG, Tosteson TD, Strohl KP, Spry K. Racial differences in sleep-disordered breathing in African-Americans and Caucasians. Am J Respir Crit Care Med 1997; 155:186–192. Erratum in: Am J Respir Crit Care Med 1997; 155:1820.
- Sutherland K, Lee RWW, Cistulli PA. Obesity and craniofacial structure as risk factors for obstructive sleep apnoea: impact of ethnicity. Respirology 2012; 17:213–222.
- Schwab RJ, Gupta KB, Gefter WB, Metzger LJ, Hoffman EA, Pack AI. Upper airway and soft tissue anatomy in normal subjects and patients with sleep-disordered breathing. Significance of the lateral pharyngeal walls. Am J Respir Crit Care Med 1995; 152:1673–1689.
- Nieto FJ, Young TB, Lind BK, et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 2000; 283:1829–1836.
- Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991; 14:540–545.
- Krieger J, Imbs J-L, Schmidt M, Kurtz D. Renal function in patients with obstructive sleep apnea. Effects of nasal continuous positive airway pressure. Arch Intern Med 1988; 148:1337–1340.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Friedman M, Ibrahim H, Bass L. Clinical staging for sleep-disordered breathing. Otolaryngal Head Neck Surg 2002; 127:13–21.
- Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. Report of an American Academy of Sleep Medicine Task Force. Sleep 1999; 22:667–689.
- Collop NA, Anderson WM, Boehlecke B, et al; Portable Monitoring Task Force of the American Academy of Sleep Medicine. Clinical guidelines for the use of unattended portable monitors in the diagnosis of obstructive sleep apnea in adult patients. Portable Monitoring Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2007; 3:737–747.
- Centers for Medicare & Medicaid Services (CMS). Continuous positive airway pressure (CPAP) therapy for obstructive sleep apnea (OSA). MLN Matters 2008. www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM6048.pdf. Accessed June 2, 2014.
- Peppard PE, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA 2000; 284:3015–3021.
- Guardiano SA, Scott JA, Ware JC, Schechner SA. The long-term results of gastric bypass on indexes of sleep apnea. Chest 2003; 124:1615–1619.
- Crooks PF. Surgical treatment of morbid obesity. Annu Rev Med 2006; 57:243–264.
- Dixon JB, Schachter LM, O’Brien PE, et al. Surgical vs conventional therapy for weight loss treatment of obstructive sleep apnea: a randomized controlled trial. JAMA 2012; 308:1142–1149.
- Bazzano LA, Khan Z, Reynolds K, He J. Effect of nocturnal nasal continuous positive airway pressure on blood pressure in obstructive sleep apnea. Hypertension 2007; 50:417–423.
- Logan AG, Perlikowski SM, Mente A, et al. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 2001; 19:2271–2277.
- Kushida CA, Morgenthaler TI, Littner MR, et al; American Academy of Sleep. Practice parameters for the treatment of snoring and obstructive sleep apnea with oral appliances: an update for 2005. Sleep 2006; 29:240–243.
- Otsuka R, Ribeiro de Almeida F, Lowe AA, Linden W, Ryan F. The effect of oral appliance therapy on blood pressure in patients with obstructive sleep apnea. Sleep Breath 2006; 10:29–36.
- Yoshida K. Effect on blood pressure of oral appliance therapy for sleep apnea syndrome. Int J Prosthodont 2006; 19:61–66.
- Inazawa T, Ayuse T, Kurata S, et al. Effect of mandibular position on upper airway collapsibility and resistance. J Dent Res 2005; 84:554–558.
- Fujita S, Conway W, Zorick F, Roth T. Surgical correction of anatomic abnormalities in obstructive sleep apnea syndrome: uvulopalatopharyngoplasty. Otolaryngol Head Neck Surg 1981; 89:923–934.
- Schwab RJ. Imaging for the snoring and sleep apnea patient. Dent Clin North Am 2001; 45:759–796.
- Prinsell JR. Maxillomandibular advancement surgery for obstructive sleep apnea syndrome. J Am Dent Assoc 2002; 133:1489–1497.
- Thatcher GW, Maisel RH. The long-term evaluation of tracheostomy in the management of severe obstructive sleep apnea. Laryngoscope 2003; 113:201–204.
- Conway WA, Victor LD, Magilligan DJ, Fujita S, Zorick FJ, Roth T. Adverse effects of tracheostomy for sleep apnea. JAMA 1981; 246:347–350.
- Marcus CL, Moore RH, Rosen CL, et al; Childhood Adenotonsillectomy Trial (CHAT). A randomized trial of adenotonsillectomy for childhood sleep apnea. N Engl J Med 2013; 368:2366–2376.
- Gottlieb DJ, Craig SE, Lorenzi-Filho G, et al. Sleep apnea cardiovascular clinical trials-current status and steps forward: The International Collaboration of Sleep Apnea Cardiovascular Trialists. Sleep 2013; 36:975–980.
- Loredo JS, Ancoli-Israel S, Kim EJ, Lim WJ, Dimsdale JE. Effect of continuous positive airway pressure versus supplemental oxygen on sleep quality in obstructive sleep apnea: a placebo-CPAP-controlled study. Sleep 2006; 29:564–571.
- Phillips BA, McConnell JW, Smith MD. The effects of hypoxemia on cardiac output. A dose-response curve. Chest 1988; 93:471–475.
- Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respire Physiol Neurobiol 2008; 162:144–151.
- Berry RB, Kryger MH, Massie CA. A novel nasal expiratory positive airway pressure (EPAP) device for the treatment of obstructive sleep apnea: a randomized controlled trial. Sleep 2011; 34:479–485.
- Colrain IM, Black J, Siegel LC, et al. A multicenter evaluation of oral pressure therapy for the treatment of obstructive sleep apnea. Sleep Med 2013; 14:830–837.
- Strollo PJ Jr, Soose RJ, Maurer JT, et al; STAR Trial Group. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med 2014; 370:139–149.
KEY POINTS
- Although obesity and snoring are common features of OSA, they are not always present.
- Home sleep testing is appropriate for those highly likely to have sleep apnea and without other significant sleep or cardiovascular, respiratory, or neurologic disorders.
- Upper-airway surgery has a limited role—it is indicated primarily for those unable to tolerate CPAP.
- Risk of motor vehicle accidents is dramatically increased in untreated sleep apnea; patients should be counseled on the dangers of drowsy driving.
Managing acute coronary syndromes: Decades of progress
Most decisions for managing acute coronary syndromes can be based on ample data from large randomized trials with hard clinical end points, so there is little reason to provide care that is not evidence-based.
This article reviews some of the trials that provide guidance on diagnosing and managing acute coronary syndromes, including the timing of reperfusion and adjunctive therapies in different situations.
MOST ACUTE CORONARY SYNDROMES ARE NON-ST-ELEVATION CONDITIONS
Acute coronary syndromes range from unstable angina and non-ST-elevation myocardial infarction (NSTEMI) to ST-elevation MI (STEMI), reflecting a continuum of severity of coronary stenosis. The degree of coronary occlusion may ultimately determine whether a patient has unstable angina or MI with or without ST elevation.1
The substrate for all of these is vulnerable plaque. Angiographic studies have indicated that in many cases medium-size plaques (30%–40% stenosis) are more likely to rupture than larger, more obstructive ones. Moderate plaques may be vulnerable because they are less mature, with a large lipid core and a thin cap prone to rupture or erode, exposing the thrombogenic subendothelial components.2

Because the vulnerability of a coronary plaque may not correlate with the severity of stenosis before the plaque ruptures, stress tests and symptoms may not predict the risk of MI. The key role of thrombosis in the pathogenesis also highlights the importance of antithrombotic therapy in the acute phases of acute coronary syndromes, which can significantly reduce mortality and morbidity rates.
Perhaps because of the widespread use of aspirin and statins, most patients who currently present with an acute coronary syndrome have either unstable angina or NSTEMI: of about 1.57 million hospital admissions in 2004 for acute coronary syndromes, for example, only 330,000 (21%) were for STEMI.3
DIAGNOSING ACUTE CORONARY SYNDROME
Symptoms may not be classic
The classic symptoms of acute coronary syndromes are intense, oppressive chest pressure radiating to the left arm, but nearly any discomfort “between the nose and navel” (eg, including the jaw, arm, and epigastric and abdominal areas) may be an acute coronary syndrome. Associated symptoms may include chest heaviness or burning, radiation to the jaw, neck, shoulder, back, or arms, and dyspnea.
Particularly in older, female, postoperative, or diabetic patients, the presentation may be atypical or “silent,” including nausea or vomiting; breathlessness; sweating; arrhythmias; or light-headedness. Especially in these groups, symptoms may be mild or subtle, and acute coronary syndrome may manifest only as “not feeling well.”
The differential diagnosis of acute coronary syndromes is broad. Most important to immediately consider are pulmonary embolism and aortic dissection, as they are life-threatening and are treated differently from acute coronary syndromes. Otherwise, it is best to err on the side of caution and treat for an acute coronary syndrome until it is proven otherwise.
Electrocardiography is critical
Electrocardiography (ECG) gives valuable information about the location, extent, and prognosis of infarction, and it is critically important for distinguishing STEMI from NSTEMI, with ST elevation classically diagnostic of complete coronary occlusion. Q waves can occur early and do not necessarily signify completed infarction, as traditionally thought. ST depression or T inversion indicates that total coronary occlusion is unlikely unless they are in a pattern of circumflex infarct associated with an enlarging R wave in lead V1. An ST elevation in RV4 indicates right ventricular infarction.
The appearance on ECG may evolve over time, so a patient with atypical symptoms and a nonspecific electrocardiogram should be observed for 24 hours or until more specific criteria develop.
Biomarkers in NSTEMI
In MI, cardiac troponin levels begin to rise about 3 hours after the onset of chest pain, and elevations can last for up to 14 days. Levels can also be mildly elevated chronically in patients with renal dysfunction, so positive biomarker tests in that population should be interpreted cautiously.
For STEMI, the opportunity to reperfuse is lost if one waits for cardiac biomarkers to become elevated. But for NSTEMI, they are highly sensitive and specific for identifying patients at high risk and determining who should be treated aggressively. Patients who are biomarker-negative have a better prognosis than patients with identical symptoms and electrocardiograms who are biomarker-positive.
MI is currently defined as a rise in any biomarker (usually troponin) above the 99th percentile for a reference population, with at least one of the following:
- Ischemic symptoms
- New ST/T changes or left bundle branch block
- Pathologic Q waves
- Loss of myocardium or abnormal wall motion seen by imaging
- Intracoronary thrombus.
REPERFUSION FOR ACUTE STEMI
Because acute coronary syndromes have a common pathophysiology, for the most part, lessons from clinical trials in one syndrome are relevant to the others. However, important differences exist regarding the need for immediate reperfusion in STEMI, since in most cases these patients have total rather than partial occlusion.
Fibrinolysis has limitations
The standard of management for STEMI is immediate reperfusion. The goal is to interrupt the wave front of myocardial necrosis, salvage threatened myocardium, and ultimately improve survival.
Five placebo-controlled trials showed a 30% reduction in the death rate in patients who received fibrinolytic therapy within 6 to 12 hours of presentation.4
Patients with ST elevation or with new bundle branch block benefit most from fibrinolytic therapy. Those with ST depression, T inversion, or nonspecific changes on ECG do not benefit; they probably do not have complete coronary occlusion, so the prothrombotic or platelet-activating effects of fibrinolytic therapy may make them worse.5 Further, fibrinolytic therapy poses the risk of intracranial hemorrhage, which, although rare (occurring in up to 1% of cases depending on the drug regimen), is a devastating complication.
In general, absolute contraindications to fibrinolysis include intracranial abnormalities, hemorrhage, and head trauma. An important relative contraindication is uncontrolled blood pressure (> 180/110 mm Hg at any point during hospitalization, including during the immediate presentation). Studies show that even if blood pressure can be controlled, the risk of intracranial hemorrhage is substantially higher, although the risk may not outweigh the benefit of reperfusion, particularly for large infarctions when percutaneous coronary intervention (PCI) is not available as an alternative to fibrinolysis.
Prompt PCI is preferable to fibrinolysis
If PCI is available on site, there is nearly no role for fibrinolytic therapy. PCI is better than fibrinolytic therapy in terms of the degree of reperfusion, reocclusion, MI recurrence, and mortality rate, and it poses little or no risk of intracranial hemorrhage.6
For either fibrinolytic therapy or percutaneous therapy, “time is muscle”: the longer the ischemic time, the higher the mortality rate (relative risk = 1.075 for every 30 minutes of delay, P = .041).7
At centers that do not have PCI on site, studies (mainly from Europe) have shown that it is better to transport the patient for PCI than to give immediate fibrinolytic therapy.7,8 But because the centers studied tended to have short transport times (usually 40 minutes or less), it is uncertain whether the results are applicable throughout the United States.
The delay between symptom onset and presentation is also relevant. Reperfusion within the first 1 to 2 hours after the onset of symptoms provides the greatest degree of myocardial salvage and of reduction in the risk of death; the extent of benefit thereafter is substantially less. As a result, patients who present very early after symptom onset have the most to lose if their reperfusion is delayed by even a few more hours, whereas patients who have already experienced several hours of pain are affected less by additional delay.9 Thus, patients presenting within the “golden” 1 or 2 hours after symptoms begin should be considered for fibrinolytic therapy if transfer for PCI cannot be done expeditiously. It is important for hospitals without PCI available on site to have a system in place for rapid transport of patients when needed.
Guidelines advise that patients with STEMI should undergo PCI rather than receive fibrinolytic therapy as long as PCI is available within 90 minutes of first medical contact. Otherwise, fibrinolysis should be started within 30 minutes.10 For patients who present several hours after symptom onset, PCI may still be preferable even if the transport time is somewhat longer.
PCI after fibrinolytic therapy
In prior decades, PCI immediately after fibrinolytic therapy was associated with an increased risk of bleeding complications and reinfarction. That has changed with improvements in equipment and antithrombotic therapy.
Two large trials conclusively found that routinely transferring high-risk patients for PCI immediately after receiving fibrinolytic therapy (combined half-dose reteplase [Retavase] and abciximab [ReoPro]11 or full-dose tenecteplase [TNKase]12) resulted in much lower rates of ischemic end points without an increase in bleeding complications compared with transferring patients only for rescue PCI after fibrinolytic therapy.
Routine transfer is now the standard of care for high-risk patients after fibrinolytic therapy and probably is best for all patients after an MI.
MANAGING NSTEMI AND UNSTABLE ANGINA
For patients with NSTEMI, immediate reperfusion is usually not required, although initial triage for “early invasive” vs “initial conservative” management must be done early in the hospital course. Randomized trials have evaluated these two approaches, with most studies in the contemporary era reporting improved outcomes with an early invasive approach.
The TACTICS trial,13 the most important of these, enrolled more than 2,200 patients with unstable angina or NSTEMI and randomized them to an early invasive strategy or a conservative strategy. Overall, results were better with the early invasive strategy.
The ICTUS trial.14 Although several studies showed that an early invasive approach was better, the most recent study using the most modern practices—the ICTUS trial—did not find that it reduced death rates. Most patients eventually underwent angiography and revascularization, but not early on. However, all studies showed that rates of recurrent unstable angina and hospitalization were reduced by an early invasive approach, so revascularization does have a role in stabilizing the patient. But in situations of aggressive medical management with antithrombotic and other therapies, an early conservative approach may be an appropriate alternative for many patients.15
The selection of an invasive vs a conservative approach should include a consideration of risk, which can be estimated using a number of criteria, including the Thrombolysis in Myocardial Infarction (TIMI) or the GRACE risk score. When risk was stratified using the TIMI risk score,16 in the TACTICS trial, the higher the risk score, the more likely patients were to benefit from early revascularization.
When an invasive approach is chosen, it does not appear necessary to take patients to catheterization immediately (within 2–24 hours) compared with later during the hospital course.
The TIMACS trial,17 with more than 3,000 patients, tested the benefits of very early vs later revascularization for patients with NSTEMI and unstable angina. Early intervention did not significantly improve outcomes for the primary composite end point of death, MI, and stroke in the overall population enrolled in the trial, but when the secondary end point of refractory ischemia was added in, early intervention was found to be beneficial overall. Moreover, when stratified by risk, high-risk patients significantly benefited from early intervention for the primary end point.
Guidelines for NSTEMI and unstable angina continue to prefer an early invasive strategy, particularly for high-risk patients, although a conservative strategy is considered acceptable if patients receive intensive evidence-based medical therapy and remain clinically stable.18
ANTITHROMBOTIC THERAPIES
Once a revascularization strategy has been chosen, adjunctive therapies should be considered. The most important are the antithrombotic therapies.
Many drugs target platelet activity. Most important are the thromboxane inhibitor aspirin, the adenosine diphosphate (ADP) receptor antagonists clopidogrel (Plavix), prasugrel (Effient), and ticagrelor (Brilinta), and the glycoprotein (GP) IIb/IIIa antagonists abciximab and eptifibatide (Integrilin). Others, such as thrombin receptor antagonists, are under investigation.19
Aspirin for secondary prevention
Evidence is unequivocal for the benefit of aspirin therapy in patients with established or suspected vascular disease.
The ISIS-2 trial20 compared 35-day mortality rates in 16,000 patients with STEMI who were given aspirin, streptokinase, combined streptokinase and aspirin, or placebo. Mortality rates were reduced by aspirin compared with placebo by an extent similar to that achieved with streptokinase, with a further reduction when aspirin and streptokinase were given together.
Therefore, patients with STEMI should be given aspirin daily indefinitely unless they have true aspirin allergy. The dose is 165 to 325 mg initially and 75 to 162 mg daily thereafter.
For NSTEMI and even for secondary prevention in less-acute situations, a number of smaller trials also provide clear evidence of benefit from aspirin therapy.
The CURRENT-OASIS 7 trial21 showed that low maintenance dosages of aspirin (75–100 mg per day) resulted in the same incidence of ischemic end points (cardiovascular death, MI, or stroke) as higher dosages. Although rates of major bleeding events did not differ, a higher rate of gastrointestinal bleeding was evident at just 30 days in patients taking the higher doses. This large trial clearly established that there is no advantage to daily aspirin doses of more than 100 mg.
DUAL ANTIPLATELET THERAPY IS STANDARD
Standard practice now is to use aspirin plus another antiplatelet agent that acts by inhibiting either the ADP receptor (for which there is the most evidence) or the GP IIb/IIIa receptor (which is becoming less used). Dual therapy should begin early in patients with acute coronary syndrome.
Clopidogrel: Well studied with aspirin
The most commonly used ADP antagonist is clopidogrel, a thienopyridine. Much evidence exists for its benefit.
The CURE trial22 randomized more than 12,000 patients with NSTEMI or unstable angina to aspirin plus either clopidogrel or placebo. The incidence of the combined end point of MI, stroke, and cardiovascular death was 20% lower in the clopidogrel group than in the placebo group over 12 months of follow-up. The benefit of clopidogrel began to occur within the first 24 hours after randomization, with a 33% relative risk reduction in the combined end point of cardiovascular death, MI, stroke, and severe ischemia, demonstrating the importance of starting this agent early in the hospital course.
COMMIT23 found a benefit in adding clopidogrel to aspirin in patients with acute STEMI. Although it was only a 30-day trial, significant risk reduction was found in the dual-therapy group for combined death, stroke, or reinfarction. The results of this brief trial were less definitive, but the pathophysiology was similar to non-ST-elevation acute coronary syndromes, so it is reasonable to extrapolate the long-term findings to this setting.
The CURRENT-OASIS 7 trial21 randomized more than 25,000 patients to either clopidogrel in a double dosage (600 mg load, 150 mg/day for 6 days, then 75 mg/day) or standard dosage (300 mg load, 75 mg/day thereafter). Although no overall benefit was found for the higher dosage, a subgroup of more than 17,000 patients who underwent PCI after randomization had a lower risk of developing stent thrombosis. On the other hand, higher doses of clopidogrel caused more major bleeding events.
Ticagrelor and prasugrel: New alternatives to clopidogrel
The principal limitation of clopidogrel is its metabolism. It is a prodrug, ie, it is not active as taken and must be converted to its active state by cytochrome P450 enzymes in the liver. Patients who bear certain polymorphisms in the genes for these enzymes or who are taking other medications that affect this enzymatic pathway may derive less platelet inhibition from the drug, leading to considerable patient-to-patient variability in the degree of antiplatelet effect.
Alternatives to clopidogrel have been developed that inhibit platelets more intensely, are activated more rapidly, and have less interpatient variability. Available now are ticagrelor and prasugrel.24 Like clopidogrel, prasugrel is absorbed as an inactive prodrug, but it is efficiently metabolized by esterases to an active form, and then by a simpler step within the liver to its fully active metabolite.25 Ticagrelor is active as absorbed.26
Pharmacodynamically, the two drugs perform almost identically and much faster than clopidogrel, with equilibrium platelet inhibition reached in less than 1 hour. The degree of platelet inhibition is also more—sometimes twice as much—with the new drugs compared with clopidogrel, and the effect is much more consistent between patients.
Both clopidogrel and prasugrel permanently inhibit the platelet ADP receptor, and 3 to 7 days are therefore required for their antiplatelet effects to completely wear off. In contrast, ticagrelor is a reversible inhibitor and its effects wear off more rapidly. Despite achieving a much higher level of platelet inhibition than clopidogrel, ticagrelor’s activity falls below that of clopidogrel’s by 48 hours of discontinuing the drugs.
Trial of prasugrel vs clopidogrel
The TRITON-TIMI 38 trial27 enrolled more than 13,000 patients with acute coronary syndromes, randomized to receive, either prasugrel or clopidogrel, in addition to aspirin. The patients were all undergoing PCI, so the findings do not apply to patients treated medically with an early conservative approach. The study drug was given only after the decision was made to perform PCI in patients with non-ST-elevation acute coronary syndrome (but given immediately for patients with STEMI, because nearly all those patients undergo PCI).
Prasugrel was clearly beneficial, with a significant 20% lower rate of the combined end point of cardiovascular death, MI, and stroke at 15 months. However, bleeding risk was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% confidence interval 1.02–1.68, P = .03). Looking at individual end points, the advantages of prasugrel were primarily in reducing rates of stent thrombosis and nonfatal MI. Death rates with the two drugs were equivalent, possibly because of the higher risk of bleeding with prasugrel. Bleeding in the prasugrel group was particularly increased in patients who underwent bypass surgery; more patients also needed transfusion.
Subgroup analysis showed that patients with a history of stroke or transient ischemic attack had higher rates of ischemic and bleeding events with prasugrel than with clopidogrel, leading to these being labeled as absolute contraindications to prasugrel. Patients over age 75 or who weighed less than 60 kg experienced excess bleeding risk that closely matched the reduction in ischemic event rates and thus did not have a net benefit with prasugrel.
Trial of ticagrelor vs clopidogrel
The PLATO trial28 included 18,000 patients, of whom 65% underwent revascularization and 35% were treated medically. The drug—clopidogrel or ticagrelor—was given in addition to aspirin at randomization (within 24 hours of symptom onset); this more closely follows clinical practice, in which dual antiplatelet therapy is started as soon as possible. This difference makes the PLATO study more relevant to practice for patients with non-ST-elevation acute coronary syndrome. Also, because they gave the drugs to all patients regardless of whether they were to undergo PCI, this study likely had a higher-risk population, which may be refected in the higher mortality rate at 30 days (5.9% in the clopidogrel group in the PLATO study vs 3.2% in the clopidogrel group in the TRITON study).
Another important difference between the trials testing prasugrel and ticagrelor is that patients who had already received a thienopyridine were excluded from the prasugrel trial but not from the ticagrelor trial. Nearly half the patients in the ticagrelor group were already taking clopidogrel. The clinical implication is that for patients who arrive from another facility and already have been given clopidogrel, it is safe to give ticagrelor. There is limited information about whether that is also true for prasugrel, although there is no known reason why the safety of adding prasugrel to clopidogrel should be different from that of ticagrelor.
The rate of ischemic events was 20% lower in the ticagrelor group than in the clopidogrel group, importantly including reductions in the incidence of death, MI, and stent thrombosis. There was no increase with ticagrelor compared with clopidogrel in bleeding associated with coronary artery bypass graft surgery, likely because of the more rapid washout of the ticagrelor effect, or in the need for blood transfusions. However, the rate of bleeding unrelated to coronary artery bypass was about 20% higher with ticagrelor.
In summary, more intense platelet inhibition reduces the risk of ischemic events, but, particularly for the irreversible inhibitor prasugrel, at the cost of a higher risk of bleeding. In general, the net benefit of these agents in preventing the irreversible complications of MI and (in the case of ticagrelor) death favor the use of the more intense ADP inhibitors in appropriate patients. Ticagrelor is indicated in patients with acute coronary syndromes undergoing invasive or conservative management; prasugrel is indicated in patients undergoing PCI, but contraindicated in patients with a previous stroke or transient ischemic event. Neither drug is indicated in patients undergoing elective PCI outside the setting of acute coronary syndromes, although these agents may be appropriate in patients with intolerance or allergy to clopidogrel.
Glycoprotein IIb/IIIa antagonists for select cases only
GP IIb/IIIa antagonists such as abciximab were previously used more commonly than they are today. Now, with routine pretreatment using thienopyridines, their role in acute coronary syndromes is less clear. They still play a role when routine dual antiplatelet therapy is not used, when prasugrel or ticagrelor is not used, and when heparin rather than an alternative antithrombin agent is used.
A meta-analysis29 of 3,755 patients showed a clear reduction in ischemic complications with abciximab as an adjunct to primary PCI for STEMI in patients treated with heparin.
Kastrati et al30 found that patients with non-ST-elevation acute coronary syndromes benefited from abciximab at the time of PCI with heparin, even though they had been routinely pretreated with clopidogrel. However, benefits were seen only in high-risk patients who had presented with elevated troponins.
On the other hand, the role of GP IIb/IIIa blockade for “upstream” medical management in patients with acute coronary syndromes has been eroded by several studies.
The ACUITY trial31 randomized more than 9,000 patients to receive either routine treatment with a GP IIb/IIIa inhibitor before angiography or deferred selective use in the catheterization laboratory only for patients undergoing PCI. No significant differences were found in rates of MI and death.
The Early ACS trial32 compared early routine eptifibatide vs delayed, provisional eptifibatide in 9,492 patients with acute coronary syndromes without ST elevation and who were assigned to an invasive strategy. The early-eptifibatide group received two boluses and an infusion of eptifibatide before angiography; the others received a placebo infusion, with provisional eptifibatide after angiography if the patient underwent PCI and was deemed at high risk. No significant difference in rates of death or MI were noted, and the early-eptifibatide group had significantly higher rates of bleeding and need for transfusion.
The FINESSE trial33 also discredited “facilitating” PCI by giving GP IIb/IIIa antagonists in patients with STEMI before arrival in the catheterization laboratory, with no benefit to giving abciximab ahead of time vs in the catheterization laboratory, and with an increased risk of bleeding complications.
These studies have helped narrow the use of GP IIb/IIIa inhibitors to the catheterization laboratory in conjunction with heparin anticoagulation (as compared with bivalirudin [Angiomax]; see below) and only in select or high-risk cases. These drugs are indicated in the medical phase of management only if patients cannot be stabilized by aspirin or ADP inhibition.
NEWER ANTITHROMBOTICS: ADVANTAGES UNCLEAR
The complex coagulation cascade has a number of components, but only a few are targeted by drugs that are approved and recommended: fondaparinux (Arixtra) and oral factor Xa inhibitors affect the prothrombinase complex (including factor X); bivalirudin and oral factor IIa inhibitors affect thrombin; and heparin and the low-molecular-weight heparins inhibit both targets.
Low-molecular-weight heparins
The SYNERGY trial34 randomized nearly 10,000 patients with non-ST-elevation acute coronary syndromes at high risk for ischemic cardiac complications managed with an invasive approach to either the low-molecular-weight heparin enoxaparin (Lovenox) or intravenous unfractionated heparin immediately after enrollment. Most patients underwent catheterization and revascularization. No clinical advantage was found for enoxaparin, and bleeding complications were increased.
The EXTRACT-TIMI 25 trial35 randomized more than 20,000 patients with STEMI who were about to undergo fibrinolysis to receive either enoxaparin throughout hospitalization (average of 8 days) or unfractionated heparin for at least 48 hours. The enoxaparin group had a lower rate of recurrent MI, but it was unclear if the difference was in part attributable to the longer therapy time. The enoxaparin group also had more bleeding.
Fondaparinux
The OASIS-5 trial36,37 compared enoxaparin and fondaparinux, an exclusive factor Xa inhibitor, in more than 20,000 patients with unstable angina or NSTEMI. Fondaparinux was associated with a lower risk of death and reinfarction as well as fewer bleeding events. However, the benefits were almost exclusively in patients treated medically. In those undergoing PCI within the first 8 days, no benefit was found, although there was still a significant reduction in major bleeding events. Catheter thrombosis was also increased in patients taking fondaparinux, but only in those who did not receive adequate unfractionated heparin treatment before PCI.
Bivalirudin superior at time of catheterization
The most significant advance in antithrombotic therapy for patients with acute coronary syndromes is bivalirudin. This drug has a clear role only in the catheterization laboratory, where patients can be switched to it from heparin, low-molecular-weight heparin, or fondaparinux.
Three trials38–40 evaluated the drug in a total of more than 20,000 patients receiving invasive management of coronary artery disease undergoing PCI for elective indications, NSTEMI, or STEMI.
Results were remarkably similar across the three trials. Patients who were treated with bivalirudin alone had the same rate of ischemic end points at 30 days as those receiving heparin plus a GP IIb/IIIa inhibitor, but bivalirudin was associated with a consistent and significant 40% to 50% lower bleeding risk. For the highest-risk patients, those with STEMI, the bivalirudin group also had a significantly lower risk of death at 1 year.41
OTHER DRUGS: EARLY TREATMENT NO LONGER ROUTINE
Most data for the use of therapies aside from antithrombotics are from studies of patients with STEMI, but findings can logically be extrapolated to those with non-ST-elevation acute coronary syndromes.
Beta-blockers: Cardiogenic shock a risk
For beta-blockers, many historical trials were done in stable coronary disease, but there are no large trials in the setting of NSTEMI or unstable angina, and only recently have there been large trials for STEMI. Before the availability of recent evidence, standard practice was to treat STEMI routinely with intravenous metoprolol (Lopressor) and then oral metoprolol.
When large studies were finally conducted, the results were sobering.
COMMIT.42 Nearly 46,000 patients with suspected acute MI were randomized to receive either metoprolol (up to 15 mg intravenously, then 200 mg by mouth daily until discharge or for up to 4 weeks in the hospital) or placebo. Surprisingly, although rates of reinfarction and ventricular fibrillation were lower with metoprolol, a higher risk of cardiogenic shock with early beta-blockade offset these benefits and the net mortality rate was not reduced. This study led to a reduction in the early use of beta-blockers in patients with STEMI.
The standard of care has now shifted from beta-blockers in everyone as early as possible after MI to being more cautious in patients with contraindications, including signs of heart failure or a low-output state, or even in those of advanced age or with borderline low blood pressure or a high heart rate. Patients who present late and therefore may have a larger infarct are also at higher risk.
Although the goal should be to ultimately discharge patients on beta-blocker therapy after an MI, there should be no rush to start one early.
Carvedilol now preferred after STEMI
The CAPRICORN trial43 randomized nearly 2,000 patients following MI with left ventricular dysfunction (an ejection fraction of 40% or below) to either placebo or the beta-blocker carvedilol (Coreg). Patients taking the drug had a clear reduction in rates of death and reinfarction, leading to this drug becoming the beta-blocker of choice in patients with ventricular dysfunction after STEMI.
Angiotensin-converting enzyme inhibitors: Early risk of cardiogenic shock
The use of angiotensin-converting enzyme (ACE) inhibitors after MI is also supported by several studies.44 Two very large studies, one of nearly 60,000 patients and one of nearly 20,000, showed a clear reduction in the mortality rate in those who received an ACE inhibitor. Most of the benefit was in patients with an ejection fraction of less than 40%. On the basis of these trials, ACE inhibitors are indicated for all patients for the first 30 days after MI and then indefinitely for those with left ventricular dysfunction. However, the trial in which an ACE inhibitor was given intravenously early on had to be stopped prematurely because of worse outcomes owing to cardiogenic shock.
These studies highlight again that for patients who are unstable in the first few days of an acute coronary syndrome, it is best to wait until their condition stabilizes and to start these therapies before hospital discharge.
Intensive statin therapy
In the last 20 years, unequivocal evidence has emerged to support the beneficial role of statins for secondary prevention in patients with established coronary artery disease. More-recent trials have also shown that intensive statin therapy (a high dose of a potent statin) improves outcomes better than lower doses.
The PROVE-IT TIMI 22 trial45 randomized patients after an acute coronary syndrome to receive either standard therapy (pravastatin [Pravachol] 40 mg) or intensive therapy (atorvastatin [Lipitor] 80 mg). The intensive-therapy group had a significantly lower rate of major cardiovascular events, and the difference persisted and grew over 30 months of follow-up.
A number of studies confirmed this and broadened the patient population to those with unstable or stable coronary disease. Regardless of the risk profile, the effects were consistent and showed that high-dose statins were better in preventing coronary death and MI.46
Guidelines are evolving toward recommendation of highest doses of statins independently of the target level of low-density lipoprotein cholesterol.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction. Circulation 2004; 110:e82–e292. Erratum in: Circulation 2005; 111:2013–2014.
- Davies MJ. The pathophysiology of acute coronary syndromes. Heart 2000; 83:361–366.
- Rosamond W, Flegal K, Friday G, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007; 115:e69–e171.
- Granger CB, Califf RM, Topol EJ. Thrombolytic therapy for acute myocardial infarction. A review. Drugs 1992; 44:293–325.
- Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet 1994; 343:311–322.
- Keely EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20
- De Luca G, Suryapranata H, Ottervanger JP, Antman EM. Time delay to treatment and mortality in primary angioplasty for acute myocardial infarction: every minute of delay counts. Circulation 2004; 109:1223–1225.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Gersh BJ, Stone GW, White HD, Holmes DR Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA 2005; 293:979–986.
- Antman EM, Hand M, Armstron PW, et al; Canadian Cardiovascular Society; American Academy of Family Physicians; American College of Cardiology; American Heart Association. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab Reteplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Cannon CP, Weintraub WS, Demopoulos LA, et al; TACTICS (Treat Angina With Aggrastat and Determine Cost of Therapy With an Invasive or Conservative Strategy)–Thrombolysis in Myocardial Infarction 18 Investigators. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001; 344:1879–1887.
- Damman P, Hirsch A, Windhausen F, Tijssen JG, de Winter RJ; ICTUS Investigators. 5-year clinical outcomes in the ICTUS (Invasive versus Conservative Treatment in Unstable coronary Syndromes) trial a randomized comparison of an early invasive versus selective invasive management in patients with non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2010; 55:858–864.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic decision making. JAMA 2000; 284:835–842.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Yousef O, Bhatt DL. The evolution of antiplatelet therapy in cardiovascular disease. Nat Rev Cardiol 2011; 8:547–559.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Yusuf S, Mehta SR, Zhao F, et al; Clopidogrel in Unstable angina to prevent Recurrent Events Trial Investigators. Early and late effects of clopidogrel in patients with acute coronary syndromes. Circulation 2003; 107:966–972.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Schömig A. Ticagrelor—is there need for a new player in the antiplatelet-therapy field? N Engl J Med 2009; 361:1108–1111.
- Wiviott SD, Antman EM, Braunwald E. Prasugrel. Circulation 2010; 122:394–403.
- Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation 2009; 120:2577–2585.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- de Queiroz Fernandes Araujo JO, Veloso HH, Braga De Paiva JM, Fiho MW, Vincenzo De Paola AA. Efficacy and safety of abciximab on acute myocardial infarction treated with percutaneous coronary interventions: a meta-analysis of randomized, controlled trials. Am Heart J 2004; 148:937–943.
- Kastrati A, Mehilli J, Neuman FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Giugliano RP, White JA, Bode C, et al; Early ACS Investigators. Early vs delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med 2009; 360:2176–2190.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Fergusson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH; EXTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- The Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2007; 358:2218–2230.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Chen ZM, Pan HC, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1622–1632.
- Dargie JH. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 2001; 357:1385–1390.
- Hennekens CH, Albert CM, Godfried SL, Gaziano JM, Buring JE. Adjunctive drug therapy of acute myocardial infarction—evidence from clinical trials. N Engl J Med 1996; 335:1660–1667.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol 2006; 48:438–445.
Most decisions for managing acute coronary syndromes can be based on ample data from large randomized trials with hard clinical end points, so there is little reason to provide care that is not evidence-based.
This article reviews some of the trials that provide guidance on diagnosing and managing acute coronary syndromes, including the timing of reperfusion and adjunctive therapies in different situations.
MOST ACUTE CORONARY SYNDROMES ARE NON-ST-ELEVATION CONDITIONS
Acute coronary syndromes range from unstable angina and non-ST-elevation myocardial infarction (NSTEMI) to ST-elevation MI (STEMI), reflecting a continuum of severity of coronary stenosis. The degree of coronary occlusion may ultimately determine whether a patient has unstable angina or MI with or without ST elevation.1
The substrate for all of these is vulnerable plaque. Angiographic studies have indicated that in many cases medium-size plaques (30%–40% stenosis) are more likely to rupture than larger, more obstructive ones. Moderate plaques may be vulnerable because they are less mature, with a large lipid core and a thin cap prone to rupture or erode, exposing the thrombogenic subendothelial components.2
Because the vulnerability of a coronary plaque may not correlate with the severity of stenosis before the plaque ruptures, stress tests and symptoms may not predict the risk of MI. The key role of thrombosis in the pathogenesis also highlights the importance of antithrombotic therapy in the acute phases of acute coronary syndromes, which can significantly reduce mortality and morbidity rates.
Perhaps because of the widespread use of aspirin and statins, most patients who currently present with an acute coronary syndrome have either unstable angina or NSTEMI: of about 1.57 million hospital admissions in 2004 for acute coronary syndromes, for example, only 330,000 (21%) were for STEMI.3
DIAGNOSING ACUTE CORONARY SYNDROME
Symptoms may not be classic
The classic symptoms of acute coronary syndromes are intense, oppressive chest pressure radiating to the left arm, but nearly any discomfort “between the nose and navel” (eg, including the jaw, arm, and epigastric and abdominal areas) may be an acute coronary syndrome. Associated symptoms may include chest heaviness or burning, radiation to the jaw, neck, shoulder, back, or arms, and dyspnea.
Particularly in older, female, postoperative, or diabetic patients, the presentation may be atypical or “silent,” including nausea or vomiting; breathlessness; sweating; arrhythmias; or light-headedness. Especially in these groups, symptoms may be mild or subtle, and acute coronary syndrome may manifest only as “not feeling well.”
The differential diagnosis of acute coronary syndromes is broad. Most important to immediately consider are pulmonary embolism and aortic dissection, as they are life-threatening and are treated differently from acute coronary syndromes. Otherwise, it is best to err on the side of caution and treat for an acute coronary syndrome until it is proven otherwise.
Electrocardiography is critical
Electrocardiography (ECG) gives valuable information about the location, extent, and prognosis of infarction, and it is critically important for distinguishing STEMI from NSTEMI, with ST elevation classically diagnostic of complete coronary occlusion. Q waves can occur early and do not necessarily signify completed infarction, as traditionally thought. ST depression or T inversion indicates that total coronary occlusion is unlikely unless they are in a pattern of circumflex infarct associated with an enlarging R wave in lead V1. An ST elevation in RV4 indicates right ventricular infarction.
The appearance on ECG may evolve over time, so a patient with atypical symptoms and a nonspecific electrocardiogram should be observed for 24 hours or until more specific criteria develop.
Biomarkers in NSTEMI
In MI, cardiac troponin levels begin to rise about 3 hours after the onset of chest pain, and elevations can last for up to 14 days. Levels can also be mildly elevated chronically in patients with renal dysfunction, so positive biomarker tests in that population should be interpreted cautiously.
For STEMI, the opportunity to reperfuse is lost if one waits for cardiac biomarkers to become elevated. But for NSTEMI, they are highly sensitive and specific for identifying patients at high risk and determining who should be treated aggressively. Patients who are biomarker-negative have a better prognosis than patients with identical symptoms and electrocardiograms who are biomarker-positive.
MI is currently defined as a rise in any biomarker (usually troponin) above the 99th percentile for a reference population, with at least one of the following:
- Ischemic symptoms
- New ST/T changes or left bundle branch block
- Pathologic Q waves
- Loss of myocardium or abnormal wall motion seen by imaging
- Intracoronary thrombus.
REPERFUSION FOR ACUTE STEMI
Because acute coronary syndromes have a common pathophysiology, for the most part, lessons from clinical trials in one syndrome are relevant to the others. However, important differences exist regarding the need for immediate reperfusion in STEMI, since in most cases these patients have total rather than partial occlusion.
Fibrinolysis has limitations
The standard of management for STEMI is immediate reperfusion. The goal is to interrupt the wave front of myocardial necrosis, salvage threatened myocardium, and ultimately improve survival.
Five placebo-controlled trials showed a 30% reduction in the death rate in patients who received fibrinolytic therapy within 6 to 12 hours of presentation.4
Patients with ST elevation or with new bundle branch block benefit most from fibrinolytic therapy. Those with ST depression, T inversion, or nonspecific changes on ECG do not benefit; they probably do not have complete coronary occlusion, so the prothrombotic or platelet-activating effects of fibrinolytic therapy may make them worse.5 Further, fibrinolytic therapy poses the risk of intracranial hemorrhage, which, although rare (occurring in up to 1% of cases depending on the drug regimen), is a devastating complication.
In general, absolute contraindications to fibrinolysis include intracranial abnormalities, hemorrhage, and head trauma. An important relative contraindication is uncontrolled blood pressure (> 180/110 mm Hg at any point during hospitalization, including during the immediate presentation). Studies show that even if blood pressure can be controlled, the risk of intracranial hemorrhage is substantially higher, although the risk may not outweigh the benefit of reperfusion, particularly for large infarctions when percutaneous coronary intervention (PCI) is not available as an alternative to fibrinolysis.
Prompt PCI is preferable to fibrinolysis
If PCI is available on site, there is nearly no role for fibrinolytic therapy. PCI is better than fibrinolytic therapy in terms of the degree of reperfusion, reocclusion, MI recurrence, and mortality rate, and it poses little or no risk of intracranial hemorrhage.6
For either fibrinolytic therapy or percutaneous therapy, “time is muscle”: the longer the ischemic time, the higher the mortality rate (relative risk = 1.075 for every 30 minutes of delay, P = .041).7
At centers that do not have PCI on site, studies (mainly from Europe) have shown that it is better to transport the patient for PCI than to give immediate fibrinolytic therapy.7,8 But because the centers studied tended to have short transport times (usually 40 minutes or less), it is uncertain whether the results are applicable throughout the United States.
The delay between symptom onset and presentation is also relevant. Reperfusion within the first 1 to 2 hours after the onset of symptoms provides the greatest degree of myocardial salvage and of reduction in the risk of death; the extent of benefit thereafter is substantially less. As a result, patients who present very early after symptom onset have the most to lose if their reperfusion is delayed by even a few more hours, whereas patients who have already experienced several hours of pain are affected less by additional delay.9 Thus, patients presenting within the “golden” 1 or 2 hours after symptoms begin should be considered for fibrinolytic therapy if transfer for PCI cannot be done expeditiously. It is important for hospitals without PCI available on site to have a system in place for rapid transport of patients when needed.
Guidelines advise that patients with STEMI should undergo PCI rather than receive fibrinolytic therapy as long as PCI is available within 90 minutes of first medical contact. Otherwise, fibrinolysis should be started within 30 minutes.10 For patients who present several hours after symptom onset, PCI may still be preferable even if the transport time is somewhat longer.
PCI after fibrinolytic therapy
In prior decades, PCI immediately after fibrinolytic therapy was associated with an increased risk of bleeding complications and reinfarction. That has changed with improvements in equipment and antithrombotic therapy.
Two large trials conclusively found that routinely transferring high-risk patients for PCI immediately after receiving fibrinolytic therapy (combined half-dose reteplase [Retavase] and abciximab [ReoPro]11 or full-dose tenecteplase [TNKase]12) resulted in much lower rates of ischemic end points without an increase in bleeding complications compared with transferring patients only for rescue PCI after fibrinolytic therapy.
Routine transfer is now the standard of care for high-risk patients after fibrinolytic therapy and probably is best for all patients after an MI.
MANAGING NSTEMI AND UNSTABLE ANGINA
For patients with NSTEMI, immediate reperfusion is usually not required, although initial triage for “early invasive” vs “initial conservative” management must be done early in the hospital course. Randomized trials have evaluated these two approaches, with most studies in the contemporary era reporting improved outcomes with an early invasive approach.
The TACTICS trial,13 the most important of these, enrolled more than 2,200 patients with unstable angina or NSTEMI and randomized them to an early invasive strategy or a conservative strategy. Overall, results were better with the early invasive strategy.
The ICTUS trial.14 Although several studies showed that an early invasive approach was better, the most recent study using the most modern practices—the ICTUS trial—did not find that it reduced death rates. Most patients eventually underwent angiography and revascularization, but not early on. However, all studies showed that rates of recurrent unstable angina and hospitalization were reduced by an early invasive approach, so revascularization does have a role in stabilizing the patient. But in situations of aggressive medical management with antithrombotic and other therapies, an early conservative approach may be an appropriate alternative for many patients.15
The selection of an invasive vs a conservative approach should include a consideration of risk, which can be estimated using a number of criteria, including the Thrombolysis in Myocardial Infarction (TIMI) or the GRACE risk score. When risk was stratified using the TIMI risk score,16 in the TACTICS trial, the higher the risk score, the more likely patients were to benefit from early revascularization.
When an invasive approach is chosen, it does not appear necessary to take patients to catheterization immediately (within 2–24 hours) compared with later during the hospital course.
The TIMACS trial,17 with more than 3,000 patients, tested the benefits of very early vs later revascularization for patients with NSTEMI and unstable angina. Early intervention did not significantly improve outcomes for the primary composite end point of death, MI, and stroke in the overall population enrolled in the trial, but when the secondary end point of refractory ischemia was added in, early intervention was found to be beneficial overall. Moreover, when stratified by risk, high-risk patients significantly benefited from early intervention for the primary end point.
Guidelines for NSTEMI and unstable angina continue to prefer an early invasive strategy, particularly for high-risk patients, although a conservative strategy is considered acceptable if patients receive intensive evidence-based medical therapy and remain clinically stable.18
ANTITHROMBOTIC THERAPIES
Once a revascularization strategy has been chosen, adjunctive therapies should be considered. The most important are the antithrombotic therapies.
Many drugs target platelet activity. Most important are the thromboxane inhibitor aspirin, the adenosine diphosphate (ADP) receptor antagonists clopidogrel (Plavix), prasugrel (Effient), and ticagrelor (Brilinta), and the glycoprotein (GP) IIb/IIIa antagonists abciximab and eptifibatide (Integrilin). Others, such as thrombin receptor antagonists, are under investigation.19
Aspirin for secondary prevention
Evidence is unequivocal for the benefit of aspirin therapy in patients with established or suspected vascular disease.
The ISIS-2 trial20 compared 35-day mortality rates in 16,000 patients with STEMI who were given aspirin, streptokinase, combined streptokinase and aspirin, or placebo. Mortality rates were reduced by aspirin compared with placebo by an extent similar to that achieved with streptokinase, with a further reduction when aspirin and streptokinase were given together.
Therefore, patients with STEMI should be given aspirin daily indefinitely unless they have true aspirin allergy. The dose is 165 to 325 mg initially and 75 to 162 mg daily thereafter.
For NSTEMI and even for secondary prevention in less-acute situations, a number of smaller trials also provide clear evidence of benefit from aspirin therapy.
The CURRENT-OASIS 7 trial21 showed that low maintenance dosages of aspirin (75–100 mg per day) resulted in the same incidence of ischemic end points (cardiovascular death, MI, or stroke) as higher dosages. Although rates of major bleeding events did not differ, a higher rate of gastrointestinal bleeding was evident at just 30 days in patients taking the higher doses. This large trial clearly established that there is no advantage to daily aspirin doses of more than 100 mg.
DUAL ANTIPLATELET THERAPY IS STANDARD
Standard practice now is to use aspirin plus another antiplatelet agent that acts by inhibiting either the ADP receptor (for which there is the most evidence) or the GP IIb/IIIa receptor (which is becoming less used). Dual therapy should begin early in patients with acute coronary syndrome.
Clopidogrel: Well studied with aspirin
The most commonly used ADP antagonist is clopidogrel, a thienopyridine. Much evidence exists for its benefit.
The CURE trial22 randomized more than 12,000 patients with NSTEMI or unstable angina to aspirin plus either clopidogrel or placebo. The incidence of the combined end point of MI, stroke, and cardiovascular death was 20% lower in the clopidogrel group than in the placebo group over 12 months of follow-up. The benefit of clopidogrel began to occur within the first 24 hours after randomization, with a 33% relative risk reduction in the combined end point of cardiovascular death, MI, stroke, and severe ischemia, demonstrating the importance of starting this agent early in the hospital course.
COMMIT23 found a benefit in adding clopidogrel to aspirin in patients with acute STEMI. Although it was only a 30-day trial, significant risk reduction was found in the dual-therapy group for combined death, stroke, or reinfarction. The results of this brief trial were less definitive, but the pathophysiology was similar to non-ST-elevation acute coronary syndromes, so it is reasonable to extrapolate the long-term findings to this setting.
The CURRENT-OASIS 7 trial21 randomized more than 25,000 patients to either clopidogrel in a double dosage (600 mg load, 150 mg/day for 6 days, then 75 mg/day) or standard dosage (300 mg load, 75 mg/day thereafter). Although no overall benefit was found for the higher dosage, a subgroup of more than 17,000 patients who underwent PCI after randomization had a lower risk of developing stent thrombosis. On the other hand, higher doses of clopidogrel caused more major bleeding events.
Ticagrelor and prasugrel: New alternatives to clopidogrel
The principal limitation of clopidogrel is its metabolism. It is a prodrug, ie, it is not active as taken and must be converted to its active state by cytochrome P450 enzymes in the liver. Patients who bear certain polymorphisms in the genes for these enzymes or who are taking other medications that affect this enzymatic pathway may derive less platelet inhibition from the drug, leading to considerable patient-to-patient variability in the degree of antiplatelet effect.
Alternatives to clopidogrel have been developed that inhibit platelets more intensely, are activated more rapidly, and have less interpatient variability. Available now are ticagrelor and prasugrel.24 Like clopidogrel, prasugrel is absorbed as an inactive prodrug, but it is efficiently metabolized by esterases to an active form, and then by a simpler step within the liver to its fully active metabolite.25 Ticagrelor is active as absorbed.26
Pharmacodynamically, the two drugs perform almost identically and much faster than clopidogrel, with equilibrium platelet inhibition reached in less than 1 hour. The degree of platelet inhibition is also more—sometimes twice as much—with the new drugs compared with clopidogrel, and the effect is much more consistent between patients.
Both clopidogrel and prasugrel permanently inhibit the platelet ADP receptor, and 3 to 7 days are therefore required for their antiplatelet effects to completely wear off. In contrast, ticagrelor is a reversible inhibitor and its effects wear off more rapidly. Despite achieving a much higher level of platelet inhibition than clopidogrel, ticagrelor’s activity falls below that of clopidogrel’s by 48 hours of discontinuing the drugs.
Trial of prasugrel vs clopidogrel
The TRITON-TIMI 38 trial27 enrolled more than 13,000 patients with acute coronary syndromes, randomized to receive, either prasugrel or clopidogrel, in addition to aspirin. The patients were all undergoing PCI, so the findings do not apply to patients treated medically with an early conservative approach. The study drug was given only after the decision was made to perform PCI in patients with non-ST-elevation acute coronary syndrome (but given immediately for patients with STEMI, because nearly all those patients undergo PCI).
Prasugrel was clearly beneficial, with a significant 20% lower rate of the combined end point of cardiovascular death, MI, and stroke at 15 months. However, bleeding risk was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% confidence interval 1.02–1.68, P = .03). Looking at individual end points, the advantages of prasugrel were primarily in reducing rates of stent thrombosis and nonfatal MI. Death rates with the two drugs were equivalent, possibly because of the higher risk of bleeding with prasugrel. Bleeding in the prasugrel group was particularly increased in patients who underwent bypass surgery; more patients also needed transfusion.
Subgroup analysis showed that patients with a history of stroke or transient ischemic attack had higher rates of ischemic and bleeding events with prasugrel than with clopidogrel, leading to these being labeled as absolute contraindications to prasugrel. Patients over age 75 or who weighed less than 60 kg experienced excess bleeding risk that closely matched the reduction in ischemic event rates and thus did not have a net benefit with prasugrel.
Trial of ticagrelor vs clopidogrel
The PLATO trial28 included 18,000 patients, of whom 65% underwent revascularization and 35% were treated medically. The drug—clopidogrel or ticagrelor—was given in addition to aspirin at randomization (within 24 hours of symptom onset); this more closely follows clinical practice, in which dual antiplatelet therapy is started as soon as possible. This difference makes the PLATO study more relevant to practice for patients with non-ST-elevation acute coronary syndrome. Also, because they gave the drugs to all patients regardless of whether they were to undergo PCI, this study likely had a higher-risk population, which may be refected in the higher mortality rate at 30 days (5.9% in the clopidogrel group in the PLATO study vs 3.2% in the clopidogrel group in the TRITON study).
Another important difference between the trials testing prasugrel and ticagrelor is that patients who had already received a thienopyridine were excluded from the prasugrel trial but not from the ticagrelor trial. Nearly half the patients in the ticagrelor group were already taking clopidogrel. The clinical implication is that for patients who arrive from another facility and already have been given clopidogrel, it is safe to give ticagrelor. There is limited information about whether that is also true for prasugrel, although there is no known reason why the safety of adding prasugrel to clopidogrel should be different from that of ticagrelor.
The rate of ischemic events was 20% lower in the ticagrelor group than in the clopidogrel group, importantly including reductions in the incidence of death, MI, and stent thrombosis. There was no increase with ticagrelor compared with clopidogrel in bleeding associated with coronary artery bypass graft surgery, likely because of the more rapid washout of the ticagrelor effect, or in the need for blood transfusions. However, the rate of bleeding unrelated to coronary artery bypass was about 20% higher with ticagrelor.
In summary, more intense platelet inhibition reduces the risk of ischemic events, but, particularly for the irreversible inhibitor prasugrel, at the cost of a higher risk of bleeding. In general, the net benefit of these agents in preventing the irreversible complications of MI and (in the case of ticagrelor) death favor the use of the more intense ADP inhibitors in appropriate patients. Ticagrelor is indicated in patients with acute coronary syndromes undergoing invasive or conservative management; prasugrel is indicated in patients undergoing PCI, but contraindicated in patients with a previous stroke or transient ischemic event. Neither drug is indicated in patients undergoing elective PCI outside the setting of acute coronary syndromes, although these agents may be appropriate in patients with intolerance or allergy to clopidogrel.
Glycoprotein IIb/IIIa antagonists for select cases only
GP IIb/IIIa antagonists such as abciximab were previously used more commonly than they are today. Now, with routine pretreatment using thienopyridines, their role in acute coronary syndromes is less clear. They still play a role when routine dual antiplatelet therapy is not used, when prasugrel or ticagrelor is not used, and when heparin rather than an alternative antithrombin agent is used.
A meta-analysis29 of 3,755 patients showed a clear reduction in ischemic complications with abciximab as an adjunct to primary PCI for STEMI in patients treated with heparin.
Kastrati et al30 found that patients with non-ST-elevation acute coronary syndromes benefited from abciximab at the time of PCI with heparin, even though they had been routinely pretreated with clopidogrel. However, benefits were seen only in high-risk patients who had presented with elevated troponins.
On the other hand, the role of GP IIb/IIIa blockade for “upstream” medical management in patients with acute coronary syndromes has been eroded by several studies.
The ACUITY trial31 randomized more than 9,000 patients to receive either routine treatment with a GP IIb/IIIa inhibitor before angiography or deferred selective use in the catheterization laboratory only for patients undergoing PCI. No significant differences were found in rates of MI and death.
The Early ACS trial32 compared early routine eptifibatide vs delayed, provisional eptifibatide in 9,492 patients with acute coronary syndromes without ST elevation and who were assigned to an invasive strategy. The early-eptifibatide group received two boluses and an infusion of eptifibatide before angiography; the others received a placebo infusion, with provisional eptifibatide after angiography if the patient underwent PCI and was deemed at high risk. No significant difference in rates of death or MI were noted, and the early-eptifibatide group had significantly higher rates of bleeding and need for transfusion.
The FINESSE trial33 also discredited “facilitating” PCI by giving GP IIb/IIIa antagonists in patients with STEMI before arrival in the catheterization laboratory, with no benefit to giving abciximab ahead of time vs in the catheterization laboratory, and with an increased risk of bleeding complications.
These studies have helped narrow the use of GP IIb/IIIa inhibitors to the catheterization laboratory in conjunction with heparin anticoagulation (as compared with bivalirudin [Angiomax]; see below) and only in select or high-risk cases. These drugs are indicated in the medical phase of management only if patients cannot be stabilized by aspirin or ADP inhibition.
NEWER ANTITHROMBOTICS: ADVANTAGES UNCLEAR
The complex coagulation cascade has a number of components, but only a few are targeted by drugs that are approved and recommended: fondaparinux (Arixtra) and oral factor Xa inhibitors affect the prothrombinase complex (including factor X); bivalirudin and oral factor IIa inhibitors affect thrombin; and heparin and the low-molecular-weight heparins inhibit both targets.
Low-molecular-weight heparins
The SYNERGY trial34 randomized nearly 10,000 patients with non-ST-elevation acute coronary syndromes at high risk for ischemic cardiac complications managed with an invasive approach to either the low-molecular-weight heparin enoxaparin (Lovenox) or intravenous unfractionated heparin immediately after enrollment. Most patients underwent catheterization and revascularization. No clinical advantage was found for enoxaparin, and bleeding complications were increased.
The EXTRACT-TIMI 25 trial35 randomized more than 20,000 patients with STEMI who were about to undergo fibrinolysis to receive either enoxaparin throughout hospitalization (average of 8 days) or unfractionated heparin for at least 48 hours. The enoxaparin group had a lower rate of recurrent MI, but it was unclear if the difference was in part attributable to the longer therapy time. The enoxaparin group also had more bleeding.
Fondaparinux
The OASIS-5 trial36,37 compared enoxaparin and fondaparinux, an exclusive factor Xa inhibitor, in more than 20,000 patients with unstable angina or NSTEMI. Fondaparinux was associated with a lower risk of death and reinfarction as well as fewer bleeding events. However, the benefits were almost exclusively in patients treated medically. In those undergoing PCI within the first 8 days, no benefit was found, although there was still a significant reduction in major bleeding events. Catheter thrombosis was also increased in patients taking fondaparinux, but only in those who did not receive adequate unfractionated heparin treatment before PCI.
Bivalirudin superior at time of catheterization
The most significant advance in antithrombotic therapy for patients with acute coronary syndromes is bivalirudin. This drug has a clear role only in the catheterization laboratory, where patients can be switched to it from heparin, low-molecular-weight heparin, or fondaparinux.
Three trials38–40 evaluated the drug in a total of more than 20,000 patients receiving invasive management of coronary artery disease undergoing PCI for elective indications, NSTEMI, or STEMI.
Results were remarkably similar across the three trials. Patients who were treated with bivalirudin alone had the same rate of ischemic end points at 30 days as those receiving heparin plus a GP IIb/IIIa inhibitor, but bivalirudin was associated with a consistent and significant 40% to 50% lower bleeding risk. For the highest-risk patients, those with STEMI, the bivalirudin group also had a significantly lower risk of death at 1 year.41
OTHER DRUGS: EARLY TREATMENT NO LONGER ROUTINE
Most data for the use of therapies aside from antithrombotics are from studies of patients with STEMI, but findings can logically be extrapolated to those with non-ST-elevation acute coronary syndromes.
Beta-blockers: Cardiogenic shock a risk
For beta-blockers, many historical trials were done in stable coronary disease, but there are no large trials in the setting of NSTEMI or unstable angina, and only recently have there been large trials for STEMI. Before the availability of recent evidence, standard practice was to treat STEMI routinely with intravenous metoprolol (Lopressor) and then oral metoprolol.
When large studies were finally conducted, the results were sobering.
COMMIT.42 Nearly 46,000 patients with suspected acute MI were randomized to receive either metoprolol (up to 15 mg intravenously, then 200 mg by mouth daily until discharge or for up to 4 weeks in the hospital) or placebo. Surprisingly, although rates of reinfarction and ventricular fibrillation were lower with metoprolol, a higher risk of cardiogenic shock with early beta-blockade offset these benefits and the net mortality rate was not reduced. This study led to a reduction in the early use of beta-blockers in patients with STEMI.
The standard of care has now shifted from beta-blockers in everyone as early as possible after MI to being more cautious in patients with contraindications, including signs of heart failure or a low-output state, or even in those of advanced age or with borderline low blood pressure or a high heart rate. Patients who present late and therefore may have a larger infarct are also at higher risk.
Although the goal should be to ultimately discharge patients on beta-blocker therapy after an MI, there should be no rush to start one early.
Carvedilol now preferred after STEMI
The CAPRICORN trial43 randomized nearly 2,000 patients following MI with left ventricular dysfunction (an ejection fraction of 40% or below) to either placebo or the beta-blocker carvedilol (Coreg). Patients taking the drug had a clear reduction in rates of death and reinfarction, leading to this drug becoming the beta-blocker of choice in patients with ventricular dysfunction after STEMI.
Angiotensin-converting enzyme inhibitors: Early risk of cardiogenic shock
The use of angiotensin-converting enzyme (ACE) inhibitors after MI is also supported by several studies.44 Two very large studies, one of nearly 60,000 patients and one of nearly 20,000, showed a clear reduction in the mortality rate in those who received an ACE inhibitor. Most of the benefit was in patients with an ejection fraction of less than 40%. On the basis of these trials, ACE inhibitors are indicated for all patients for the first 30 days after MI and then indefinitely for those with left ventricular dysfunction. However, the trial in which an ACE inhibitor was given intravenously early on had to be stopped prematurely because of worse outcomes owing to cardiogenic shock.
These studies highlight again that for patients who are unstable in the first few days of an acute coronary syndrome, it is best to wait until their condition stabilizes and to start these therapies before hospital discharge.
Intensive statin therapy
In the last 20 years, unequivocal evidence has emerged to support the beneficial role of statins for secondary prevention in patients with established coronary artery disease. More-recent trials have also shown that intensive statin therapy (a high dose of a potent statin) improves outcomes better than lower doses.
The PROVE-IT TIMI 22 trial45 randomized patients after an acute coronary syndrome to receive either standard therapy (pravastatin [Pravachol] 40 mg) or intensive therapy (atorvastatin [Lipitor] 80 mg). The intensive-therapy group had a significantly lower rate of major cardiovascular events, and the difference persisted and grew over 30 months of follow-up.
A number of studies confirmed this and broadened the patient population to those with unstable or stable coronary disease. Regardless of the risk profile, the effects were consistent and showed that high-dose statins were better in preventing coronary death and MI.46
Guidelines are evolving toward recommendation of highest doses of statins independently of the target level of low-density lipoprotein cholesterol.
Most decisions for managing acute coronary syndromes can be based on ample data from large randomized trials with hard clinical end points, so there is little reason to provide care that is not evidence-based.
This article reviews some of the trials that provide guidance on diagnosing and managing acute coronary syndromes, including the timing of reperfusion and adjunctive therapies in different situations.
MOST ACUTE CORONARY SYNDROMES ARE NON-ST-ELEVATION CONDITIONS
Acute coronary syndromes range from unstable angina and non-ST-elevation myocardial infarction (NSTEMI) to ST-elevation MI (STEMI), reflecting a continuum of severity of coronary stenosis. The degree of coronary occlusion may ultimately determine whether a patient has unstable angina or MI with or without ST elevation.1
The substrate for all of these is vulnerable plaque. Angiographic studies have indicated that in many cases medium-size plaques (30%–40% stenosis) are more likely to rupture than larger, more obstructive ones. Moderate plaques may be vulnerable because they are less mature, with a large lipid core and a thin cap prone to rupture or erode, exposing the thrombogenic subendothelial components.2
Because the vulnerability of a coronary plaque may not correlate with the severity of stenosis before the plaque ruptures, stress tests and symptoms may not predict the risk of MI. The key role of thrombosis in the pathogenesis also highlights the importance of antithrombotic therapy in the acute phases of acute coronary syndromes, which can significantly reduce mortality and morbidity rates.
Perhaps because of the widespread use of aspirin and statins, most patients who currently present with an acute coronary syndrome have either unstable angina or NSTEMI: of about 1.57 million hospital admissions in 2004 for acute coronary syndromes, for example, only 330,000 (21%) were for STEMI.3
DIAGNOSING ACUTE CORONARY SYNDROME
Symptoms may not be classic
The classic symptoms of acute coronary syndromes are intense, oppressive chest pressure radiating to the left arm, but nearly any discomfort “between the nose and navel” (eg, including the jaw, arm, and epigastric and abdominal areas) may be an acute coronary syndrome. Associated symptoms may include chest heaviness or burning, radiation to the jaw, neck, shoulder, back, or arms, and dyspnea.
Particularly in older, female, postoperative, or diabetic patients, the presentation may be atypical or “silent,” including nausea or vomiting; breathlessness; sweating; arrhythmias; or light-headedness. Especially in these groups, symptoms may be mild or subtle, and acute coronary syndrome may manifest only as “not feeling well.”
The differential diagnosis of acute coronary syndromes is broad. Most important to immediately consider are pulmonary embolism and aortic dissection, as they are life-threatening and are treated differently from acute coronary syndromes. Otherwise, it is best to err on the side of caution and treat for an acute coronary syndrome until it is proven otherwise.
Electrocardiography is critical
Electrocardiography (ECG) gives valuable information about the location, extent, and prognosis of infarction, and it is critically important for distinguishing STEMI from NSTEMI, with ST elevation classically diagnostic of complete coronary occlusion. Q waves can occur early and do not necessarily signify completed infarction, as traditionally thought. ST depression or T inversion indicates that total coronary occlusion is unlikely unless they are in a pattern of circumflex infarct associated with an enlarging R wave in lead V1. An ST elevation in RV4 indicates right ventricular infarction.
The appearance on ECG may evolve over time, so a patient with atypical symptoms and a nonspecific electrocardiogram should be observed for 24 hours or until more specific criteria develop.
Biomarkers in NSTEMI
In MI, cardiac troponin levels begin to rise about 3 hours after the onset of chest pain, and elevations can last for up to 14 days. Levels can also be mildly elevated chronically in patients with renal dysfunction, so positive biomarker tests in that population should be interpreted cautiously.
For STEMI, the opportunity to reperfuse is lost if one waits for cardiac biomarkers to become elevated. But for NSTEMI, they are highly sensitive and specific for identifying patients at high risk and determining who should be treated aggressively. Patients who are biomarker-negative have a better prognosis than patients with identical symptoms and electrocardiograms who are biomarker-positive.
MI is currently defined as a rise in any biomarker (usually troponin) above the 99th percentile for a reference population, with at least one of the following:
- Ischemic symptoms
- New ST/T changes or left bundle branch block
- Pathologic Q waves
- Loss of myocardium or abnormal wall motion seen by imaging
- Intracoronary thrombus.
REPERFUSION FOR ACUTE STEMI
Because acute coronary syndromes have a common pathophysiology, for the most part, lessons from clinical trials in one syndrome are relevant to the others. However, important differences exist regarding the need for immediate reperfusion in STEMI, since in most cases these patients have total rather than partial occlusion.
Fibrinolysis has limitations
The standard of management for STEMI is immediate reperfusion. The goal is to interrupt the wave front of myocardial necrosis, salvage threatened myocardium, and ultimately improve survival.
Five placebo-controlled trials showed a 30% reduction in the death rate in patients who received fibrinolytic therapy within 6 to 12 hours of presentation.4
Patients with ST elevation or with new bundle branch block benefit most from fibrinolytic therapy. Those with ST depression, T inversion, or nonspecific changes on ECG do not benefit; they probably do not have complete coronary occlusion, so the prothrombotic or platelet-activating effects of fibrinolytic therapy may make them worse.5 Further, fibrinolytic therapy poses the risk of intracranial hemorrhage, which, although rare (occurring in up to 1% of cases depending on the drug regimen), is a devastating complication.
In general, absolute contraindications to fibrinolysis include intracranial abnormalities, hemorrhage, and head trauma. An important relative contraindication is uncontrolled blood pressure (> 180/110 mm Hg at any point during hospitalization, including during the immediate presentation). Studies show that even if blood pressure can be controlled, the risk of intracranial hemorrhage is substantially higher, although the risk may not outweigh the benefit of reperfusion, particularly for large infarctions when percutaneous coronary intervention (PCI) is not available as an alternative to fibrinolysis.
Prompt PCI is preferable to fibrinolysis
If PCI is available on site, there is nearly no role for fibrinolytic therapy. PCI is better than fibrinolytic therapy in terms of the degree of reperfusion, reocclusion, MI recurrence, and mortality rate, and it poses little or no risk of intracranial hemorrhage.6
For either fibrinolytic therapy or percutaneous therapy, “time is muscle”: the longer the ischemic time, the higher the mortality rate (relative risk = 1.075 for every 30 minutes of delay, P = .041).7
At centers that do not have PCI on site, studies (mainly from Europe) have shown that it is better to transport the patient for PCI than to give immediate fibrinolytic therapy.7,8 But because the centers studied tended to have short transport times (usually 40 minutes or less), it is uncertain whether the results are applicable throughout the United States.
The delay between symptom onset and presentation is also relevant. Reperfusion within the first 1 to 2 hours after the onset of symptoms provides the greatest degree of myocardial salvage and of reduction in the risk of death; the extent of benefit thereafter is substantially less. As a result, patients who present very early after symptom onset have the most to lose if their reperfusion is delayed by even a few more hours, whereas patients who have already experienced several hours of pain are affected less by additional delay.9 Thus, patients presenting within the “golden” 1 or 2 hours after symptoms begin should be considered for fibrinolytic therapy if transfer for PCI cannot be done expeditiously. It is important for hospitals without PCI available on site to have a system in place for rapid transport of patients when needed.
Guidelines advise that patients with STEMI should undergo PCI rather than receive fibrinolytic therapy as long as PCI is available within 90 minutes of first medical contact. Otherwise, fibrinolysis should be started within 30 minutes.10 For patients who present several hours after symptom onset, PCI may still be preferable even if the transport time is somewhat longer.
PCI after fibrinolytic therapy
In prior decades, PCI immediately after fibrinolytic therapy was associated with an increased risk of bleeding complications and reinfarction. That has changed with improvements in equipment and antithrombotic therapy.
Two large trials conclusively found that routinely transferring high-risk patients for PCI immediately after receiving fibrinolytic therapy (combined half-dose reteplase [Retavase] and abciximab [ReoPro]11 or full-dose tenecteplase [TNKase]12) resulted in much lower rates of ischemic end points without an increase in bleeding complications compared with transferring patients only for rescue PCI after fibrinolytic therapy.
Routine transfer is now the standard of care for high-risk patients after fibrinolytic therapy and probably is best for all patients after an MI.
MANAGING NSTEMI AND UNSTABLE ANGINA
For patients with NSTEMI, immediate reperfusion is usually not required, although initial triage for “early invasive” vs “initial conservative” management must be done early in the hospital course. Randomized trials have evaluated these two approaches, with most studies in the contemporary era reporting improved outcomes with an early invasive approach.
The TACTICS trial,13 the most important of these, enrolled more than 2,200 patients with unstable angina or NSTEMI and randomized them to an early invasive strategy or a conservative strategy. Overall, results were better with the early invasive strategy.
The ICTUS trial.14 Although several studies showed that an early invasive approach was better, the most recent study using the most modern practices—the ICTUS trial—did not find that it reduced death rates. Most patients eventually underwent angiography and revascularization, but not early on. However, all studies showed that rates of recurrent unstable angina and hospitalization were reduced by an early invasive approach, so revascularization does have a role in stabilizing the patient. But in situations of aggressive medical management with antithrombotic and other therapies, an early conservative approach may be an appropriate alternative for many patients.15
The selection of an invasive vs a conservative approach should include a consideration of risk, which can be estimated using a number of criteria, including the Thrombolysis in Myocardial Infarction (TIMI) or the GRACE risk score. When risk was stratified using the TIMI risk score,16 in the TACTICS trial, the higher the risk score, the more likely patients were to benefit from early revascularization.
When an invasive approach is chosen, it does not appear necessary to take patients to catheterization immediately (within 2–24 hours) compared with later during the hospital course.
The TIMACS trial,17 with more than 3,000 patients, tested the benefits of very early vs later revascularization for patients with NSTEMI and unstable angina. Early intervention did not significantly improve outcomes for the primary composite end point of death, MI, and stroke in the overall population enrolled in the trial, but when the secondary end point of refractory ischemia was added in, early intervention was found to be beneficial overall. Moreover, when stratified by risk, high-risk patients significantly benefited from early intervention for the primary end point.
Guidelines for NSTEMI and unstable angina continue to prefer an early invasive strategy, particularly for high-risk patients, although a conservative strategy is considered acceptable if patients receive intensive evidence-based medical therapy and remain clinically stable.18
ANTITHROMBOTIC THERAPIES
Once a revascularization strategy has been chosen, adjunctive therapies should be considered. The most important are the antithrombotic therapies.
Many drugs target platelet activity. Most important are the thromboxane inhibitor aspirin, the adenosine diphosphate (ADP) receptor antagonists clopidogrel (Plavix), prasugrel (Effient), and ticagrelor (Brilinta), and the glycoprotein (GP) IIb/IIIa antagonists abciximab and eptifibatide (Integrilin). Others, such as thrombin receptor antagonists, are under investigation.19
Aspirin for secondary prevention
Evidence is unequivocal for the benefit of aspirin therapy in patients with established or suspected vascular disease.
The ISIS-2 trial20 compared 35-day mortality rates in 16,000 patients with STEMI who were given aspirin, streptokinase, combined streptokinase and aspirin, or placebo. Mortality rates were reduced by aspirin compared with placebo by an extent similar to that achieved with streptokinase, with a further reduction when aspirin and streptokinase were given together.
Therefore, patients with STEMI should be given aspirin daily indefinitely unless they have true aspirin allergy. The dose is 165 to 325 mg initially and 75 to 162 mg daily thereafter.
For NSTEMI and even for secondary prevention in less-acute situations, a number of smaller trials also provide clear evidence of benefit from aspirin therapy.
The CURRENT-OASIS 7 trial21 showed that low maintenance dosages of aspirin (75–100 mg per day) resulted in the same incidence of ischemic end points (cardiovascular death, MI, or stroke) as higher dosages. Although rates of major bleeding events did not differ, a higher rate of gastrointestinal bleeding was evident at just 30 days in patients taking the higher doses. This large trial clearly established that there is no advantage to daily aspirin doses of more than 100 mg.
DUAL ANTIPLATELET THERAPY IS STANDARD
Standard practice now is to use aspirin plus another antiplatelet agent that acts by inhibiting either the ADP receptor (for which there is the most evidence) or the GP IIb/IIIa receptor (which is becoming less used). Dual therapy should begin early in patients with acute coronary syndrome.
Clopidogrel: Well studied with aspirin
The most commonly used ADP antagonist is clopidogrel, a thienopyridine. Much evidence exists for its benefit.
The CURE trial22 randomized more than 12,000 patients with NSTEMI or unstable angina to aspirin plus either clopidogrel or placebo. The incidence of the combined end point of MI, stroke, and cardiovascular death was 20% lower in the clopidogrel group than in the placebo group over 12 months of follow-up. The benefit of clopidogrel began to occur within the first 24 hours after randomization, with a 33% relative risk reduction in the combined end point of cardiovascular death, MI, stroke, and severe ischemia, demonstrating the importance of starting this agent early in the hospital course.
COMMIT23 found a benefit in adding clopidogrel to aspirin in patients with acute STEMI. Although it was only a 30-day trial, significant risk reduction was found in the dual-therapy group for combined death, stroke, or reinfarction. The results of this brief trial were less definitive, but the pathophysiology was similar to non-ST-elevation acute coronary syndromes, so it is reasonable to extrapolate the long-term findings to this setting.
The CURRENT-OASIS 7 trial21 randomized more than 25,000 patients to either clopidogrel in a double dosage (600 mg load, 150 mg/day for 6 days, then 75 mg/day) or standard dosage (300 mg load, 75 mg/day thereafter). Although no overall benefit was found for the higher dosage, a subgroup of more than 17,000 patients who underwent PCI after randomization had a lower risk of developing stent thrombosis. On the other hand, higher doses of clopidogrel caused more major bleeding events.
Ticagrelor and prasugrel: New alternatives to clopidogrel
The principal limitation of clopidogrel is its metabolism. It is a prodrug, ie, it is not active as taken and must be converted to its active state by cytochrome P450 enzymes in the liver. Patients who bear certain polymorphisms in the genes for these enzymes or who are taking other medications that affect this enzymatic pathway may derive less platelet inhibition from the drug, leading to considerable patient-to-patient variability in the degree of antiplatelet effect.
Alternatives to clopidogrel have been developed that inhibit platelets more intensely, are activated more rapidly, and have less interpatient variability. Available now are ticagrelor and prasugrel.24 Like clopidogrel, prasugrel is absorbed as an inactive prodrug, but it is efficiently metabolized by esterases to an active form, and then by a simpler step within the liver to its fully active metabolite.25 Ticagrelor is active as absorbed.26
Pharmacodynamically, the two drugs perform almost identically and much faster than clopidogrel, with equilibrium platelet inhibition reached in less than 1 hour. The degree of platelet inhibition is also more—sometimes twice as much—with the new drugs compared with clopidogrel, and the effect is much more consistent between patients.
Both clopidogrel and prasugrel permanently inhibit the platelet ADP receptor, and 3 to 7 days are therefore required for their antiplatelet effects to completely wear off. In contrast, ticagrelor is a reversible inhibitor and its effects wear off more rapidly. Despite achieving a much higher level of platelet inhibition than clopidogrel, ticagrelor’s activity falls below that of clopidogrel’s by 48 hours of discontinuing the drugs.
Trial of prasugrel vs clopidogrel
The TRITON-TIMI 38 trial27 enrolled more than 13,000 patients with acute coronary syndromes, randomized to receive, either prasugrel or clopidogrel, in addition to aspirin. The patients were all undergoing PCI, so the findings do not apply to patients treated medically with an early conservative approach. The study drug was given only after the decision was made to perform PCI in patients with non-ST-elevation acute coronary syndrome (but given immediately for patients with STEMI, because nearly all those patients undergo PCI).
Prasugrel was clearly beneficial, with a significant 20% lower rate of the combined end point of cardiovascular death, MI, and stroke at 15 months. However, bleeding risk was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% confidence interval 1.02–1.68, P = .03). Looking at individual end points, the advantages of prasugrel were primarily in reducing rates of stent thrombosis and nonfatal MI. Death rates with the two drugs were equivalent, possibly because of the higher risk of bleeding with prasugrel. Bleeding in the prasugrel group was particularly increased in patients who underwent bypass surgery; more patients also needed transfusion.
Subgroup analysis showed that patients with a history of stroke or transient ischemic attack had higher rates of ischemic and bleeding events with prasugrel than with clopidogrel, leading to these being labeled as absolute contraindications to prasugrel. Patients over age 75 or who weighed less than 60 kg experienced excess bleeding risk that closely matched the reduction in ischemic event rates and thus did not have a net benefit with prasugrel.
Trial of ticagrelor vs clopidogrel
The PLATO trial28 included 18,000 patients, of whom 65% underwent revascularization and 35% were treated medically. The drug—clopidogrel or ticagrelor—was given in addition to aspirin at randomization (within 24 hours of symptom onset); this more closely follows clinical practice, in which dual antiplatelet therapy is started as soon as possible. This difference makes the PLATO study more relevant to practice for patients with non-ST-elevation acute coronary syndrome. Also, because they gave the drugs to all patients regardless of whether they were to undergo PCI, this study likely had a higher-risk population, which may be refected in the higher mortality rate at 30 days (5.9% in the clopidogrel group in the PLATO study vs 3.2% in the clopidogrel group in the TRITON study).
Another important difference between the trials testing prasugrel and ticagrelor is that patients who had already received a thienopyridine were excluded from the prasugrel trial but not from the ticagrelor trial. Nearly half the patients in the ticagrelor group were already taking clopidogrel. The clinical implication is that for patients who arrive from another facility and already have been given clopidogrel, it is safe to give ticagrelor. There is limited information about whether that is also true for prasugrel, although there is no known reason why the safety of adding prasugrel to clopidogrel should be different from that of ticagrelor.
The rate of ischemic events was 20% lower in the ticagrelor group than in the clopidogrel group, importantly including reductions in the incidence of death, MI, and stent thrombosis. There was no increase with ticagrelor compared with clopidogrel in bleeding associated with coronary artery bypass graft surgery, likely because of the more rapid washout of the ticagrelor effect, or in the need for blood transfusions. However, the rate of bleeding unrelated to coronary artery bypass was about 20% higher with ticagrelor.
In summary, more intense platelet inhibition reduces the risk of ischemic events, but, particularly for the irreversible inhibitor prasugrel, at the cost of a higher risk of bleeding. In general, the net benefit of these agents in preventing the irreversible complications of MI and (in the case of ticagrelor) death favor the use of the more intense ADP inhibitors in appropriate patients. Ticagrelor is indicated in patients with acute coronary syndromes undergoing invasive or conservative management; prasugrel is indicated in patients undergoing PCI, but contraindicated in patients with a previous stroke or transient ischemic event. Neither drug is indicated in patients undergoing elective PCI outside the setting of acute coronary syndromes, although these agents may be appropriate in patients with intolerance or allergy to clopidogrel.
Glycoprotein IIb/IIIa antagonists for select cases only
GP IIb/IIIa antagonists such as abciximab were previously used more commonly than they are today. Now, with routine pretreatment using thienopyridines, their role in acute coronary syndromes is less clear. They still play a role when routine dual antiplatelet therapy is not used, when prasugrel or ticagrelor is not used, and when heparin rather than an alternative antithrombin agent is used.
A meta-analysis29 of 3,755 patients showed a clear reduction in ischemic complications with abciximab as an adjunct to primary PCI for STEMI in patients treated with heparin.
Kastrati et al30 found that patients with non-ST-elevation acute coronary syndromes benefited from abciximab at the time of PCI with heparin, even though they had been routinely pretreated with clopidogrel. However, benefits were seen only in high-risk patients who had presented with elevated troponins.
On the other hand, the role of GP IIb/IIIa blockade for “upstream” medical management in patients with acute coronary syndromes has been eroded by several studies.
The ACUITY trial31 randomized more than 9,000 patients to receive either routine treatment with a GP IIb/IIIa inhibitor before angiography or deferred selective use in the catheterization laboratory only for patients undergoing PCI. No significant differences were found in rates of MI and death.
The Early ACS trial32 compared early routine eptifibatide vs delayed, provisional eptifibatide in 9,492 patients with acute coronary syndromes without ST elevation and who were assigned to an invasive strategy. The early-eptifibatide group received two boluses and an infusion of eptifibatide before angiography; the others received a placebo infusion, with provisional eptifibatide after angiography if the patient underwent PCI and was deemed at high risk. No significant difference in rates of death or MI were noted, and the early-eptifibatide group had significantly higher rates of bleeding and need for transfusion.
The FINESSE trial33 also discredited “facilitating” PCI by giving GP IIb/IIIa antagonists in patients with STEMI before arrival in the catheterization laboratory, with no benefit to giving abciximab ahead of time vs in the catheterization laboratory, and with an increased risk of bleeding complications.
These studies have helped narrow the use of GP IIb/IIIa inhibitors to the catheterization laboratory in conjunction with heparin anticoagulation (as compared with bivalirudin [Angiomax]; see below) and only in select or high-risk cases. These drugs are indicated in the medical phase of management only if patients cannot be stabilized by aspirin or ADP inhibition.
NEWER ANTITHROMBOTICS: ADVANTAGES UNCLEAR
The complex coagulation cascade has a number of components, but only a few are targeted by drugs that are approved and recommended: fondaparinux (Arixtra) and oral factor Xa inhibitors affect the prothrombinase complex (including factor X); bivalirudin and oral factor IIa inhibitors affect thrombin; and heparin and the low-molecular-weight heparins inhibit both targets.
Low-molecular-weight heparins
The SYNERGY trial34 randomized nearly 10,000 patients with non-ST-elevation acute coronary syndromes at high risk for ischemic cardiac complications managed with an invasive approach to either the low-molecular-weight heparin enoxaparin (Lovenox) or intravenous unfractionated heparin immediately after enrollment. Most patients underwent catheterization and revascularization. No clinical advantage was found for enoxaparin, and bleeding complications were increased.
The EXTRACT-TIMI 25 trial35 randomized more than 20,000 patients with STEMI who were about to undergo fibrinolysis to receive either enoxaparin throughout hospitalization (average of 8 days) or unfractionated heparin for at least 48 hours. The enoxaparin group had a lower rate of recurrent MI, but it was unclear if the difference was in part attributable to the longer therapy time. The enoxaparin group also had more bleeding.
Fondaparinux
The OASIS-5 trial36,37 compared enoxaparin and fondaparinux, an exclusive factor Xa inhibitor, in more than 20,000 patients with unstable angina or NSTEMI. Fondaparinux was associated with a lower risk of death and reinfarction as well as fewer bleeding events. However, the benefits were almost exclusively in patients treated medically. In those undergoing PCI within the first 8 days, no benefit was found, although there was still a significant reduction in major bleeding events. Catheter thrombosis was also increased in patients taking fondaparinux, but only in those who did not receive adequate unfractionated heparin treatment before PCI.
Bivalirudin superior at time of catheterization
The most significant advance in antithrombotic therapy for patients with acute coronary syndromes is bivalirudin. This drug has a clear role only in the catheterization laboratory, where patients can be switched to it from heparin, low-molecular-weight heparin, or fondaparinux.
Three trials38–40 evaluated the drug in a total of more than 20,000 patients receiving invasive management of coronary artery disease undergoing PCI for elective indications, NSTEMI, or STEMI.
Results were remarkably similar across the three trials. Patients who were treated with bivalirudin alone had the same rate of ischemic end points at 30 days as those receiving heparin plus a GP IIb/IIIa inhibitor, but bivalirudin was associated with a consistent and significant 40% to 50% lower bleeding risk. For the highest-risk patients, those with STEMI, the bivalirudin group also had a significantly lower risk of death at 1 year.41
OTHER DRUGS: EARLY TREATMENT NO LONGER ROUTINE
Most data for the use of therapies aside from antithrombotics are from studies of patients with STEMI, but findings can logically be extrapolated to those with non-ST-elevation acute coronary syndromes.
Beta-blockers: Cardiogenic shock a risk
For beta-blockers, many historical trials were done in stable coronary disease, but there are no large trials in the setting of NSTEMI or unstable angina, and only recently have there been large trials for STEMI. Before the availability of recent evidence, standard practice was to treat STEMI routinely with intravenous metoprolol (Lopressor) and then oral metoprolol.
When large studies were finally conducted, the results were sobering.
COMMIT.42 Nearly 46,000 patients with suspected acute MI were randomized to receive either metoprolol (up to 15 mg intravenously, then 200 mg by mouth daily until discharge or for up to 4 weeks in the hospital) or placebo. Surprisingly, although rates of reinfarction and ventricular fibrillation were lower with metoprolol, a higher risk of cardiogenic shock with early beta-blockade offset these benefits and the net mortality rate was not reduced. This study led to a reduction in the early use of beta-blockers in patients with STEMI.
The standard of care has now shifted from beta-blockers in everyone as early as possible after MI to being more cautious in patients with contraindications, including signs of heart failure or a low-output state, or even in those of advanced age or with borderline low blood pressure or a high heart rate. Patients who present late and therefore may have a larger infarct are also at higher risk.
Although the goal should be to ultimately discharge patients on beta-blocker therapy after an MI, there should be no rush to start one early.
Carvedilol now preferred after STEMI
The CAPRICORN trial43 randomized nearly 2,000 patients following MI with left ventricular dysfunction (an ejection fraction of 40% or below) to either placebo or the beta-blocker carvedilol (Coreg). Patients taking the drug had a clear reduction in rates of death and reinfarction, leading to this drug becoming the beta-blocker of choice in patients with ventricular dysfunction after STEMI.
Angiotensin-converting enzyme inhibitors: Early risk of cardiogenic shock
The use of angiotensin-converting enzyme (ACE) inhibitors after MI is also supported by several studies.44 Two very large studies, one of nearly 60,000 patients and one of nearly 20,000, showed a clear reduction in the mortality rate in those who received an ACE inhibitor. Most of the benefit was in patients with an ejection fraction of less than 40%. On the basis of these trials, ACE inhibitors are indicated for all patients for the first 30 days after MI and then indefinitely for those with left ventricular dysfunction. However, the trial in which an ACE inhibitor was given intravenously early on had to be stopped prematurely because of worse outcomes owing to cardiogenic shock.
These studies highlight again that for patients who are unstable in the first few days of an acute coronary syndrome, it is best to wait until their condition stabilizes and to start these therapies before hospital discharge.
Intensive statin therapy
In the last 20 years, unequivocal evidence has emerged to support the beneficial role of statins for secondary prevention in patients with established coronary artery disease. More-recent trials have also shown that intensive statin therapy (a high dose of a potent statin) improves outcomes better than lower doses.
The PROVE-IT TIMI 22 trial45 randomized patients after an acute coronary syndrome to receive either standard therapy (pravastatin [Pravachol] 40 mg) or intensive therapy (atorvastatin [Lipitor] 80 mg). The intensive-therapy group had a significantly lower rate of major cardiovascular events, and the difference persisted and grew over 30 months of follow-up.
A number of studies confirmed this and broadened the patient population to those with unstable or stable coronary disease. Regardless of the risk profile, the effects were consistent and showed that high-dose statins were better in preventing coronary death and MI.46
Guidelines are evolving toward recommendation of highest doses of statins independently of the target level of low-density lipoprotein cholesterol.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction. Circulation 2004; 110:e82–e292. Erratum in: Circulation 2005; 111:2013–2014.
- Davies MJ. The pathophysiology of acute coronary syndromes. Heart 2000; 83:361–366.
- Rosamond W, Flegal K, Friday G, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007; 115:e69–e171.
- Granger CB, Califf RM, Topol EJ. Thrombolytic therapy for acute myocardial infarction. A review. Drugs 1992; 44:293–325.
- Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet 1994; 343:311–322.
- Keely EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20
- De Luca G, Suryapranata H, Ottervanger JP, Antman EM. Time delay to treatment and mortality in primary angioplasty for acute myocardial infarction: every minute of delay counts. Circulation 2004; 109:1223–1225.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Gersh BJ, Stone GW, White HD, Holmes DR Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA 2005; 293:979–986.
- Antman EM, Hand M, Armstron PW, et al; Canadian Cardiovascular Society; American Academy of Family Physicians; American College of Cardiology; American Heart Association. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab Reteplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Cannon CP, Weintraub WS, Demopoulos LA, et al; TACTICS (Treat Angina With Aggrastat and Determine Cost of Therapy With an Invasive or Conservative Strategy)–Thrombolysis in Myocardial Infarction 18 Investigators. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001; 344:1879–1887.
- Damman P, Hirsch A, Windhausen F, Tijssen JG, de Winter RJ; ICTUS Investigators. 5-year clinical outcomes in the ICTUS (Invasive versus Conservative Treatment in Unstable coronary Syndromes) trial a randomized comparison of an early invasive versus selective invasive management in patients with non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2010; 55:858–864.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic decision making. JAMA 2000; 284:835–842.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Yousef O, Bhatt DL. The evolution of antiplatelet therapy in cardiovascular disease. Nat Rev Cardiol 2011; 8:547–559.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Yusuf S, Mehta SR, Zhao F, et al; Clopidogrel in Unstable angina to prevent Recurrent Events Trial Investigators. Early and late effects of clopidogrel in patients with acute coronary syndromes. Circulation 2003; 107:966–972.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Schömig A. Ticagrelor—is there need for a new player in the antiplatelet-therapy field? N Engl J Med 2009; 361:1108–1111.
- Wiviott SD, Antman EM, Braunwald E. Prasugrel. Circulation 2010; 122:394–403.
- Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation 2009; 120:2577–2585.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- de Queiroz Fernandes Araujo JO, Veloso HH, Braga De Paiva JM, Fiho MW, Vincenzo De Paola AA. Efficacy and safety of abciximab on acute myocardial infarction treated with percutaneous coronary interventions: a meta-analysis of randomized, controlled trials. Am Heart J 2004; 148:937–943.
- Kastrati A, Mehilli J, Neuman FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Giugliano RP, White JA, Bode C, et al; Early ACS Investigators. Early vs delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med 2009; 360:2176–2190.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Fergusson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH; EXTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- The Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2007; 358:2218–2230.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Chen ZM, Pan HC, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1622–1632.
- Dargie JH. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 2001; 357:1385–1390.
- Hennekens CH, Albert CM, Godfried SL, Gaziano JM, Buring JE. Adjunctive drug therapy of acute myocardial infarction—evidence from clinical trials. N Engl J Med 1996; 335:1660–1667.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol 2006; 48:438–445.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction. Circulation 2004; 110:e82–e292. Erratum in: Circulation 2005; 111:2013–2014.
- Davies MJ. The pathophysiology of acute coronary syndromes. Heart 2000; 83:361–366.
- Rosamond W, Flegal K, Friday G, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007; 115:e69–e171.
- Granger CB, Califf RM, Topol EJ. Thrombolytic therapy for acute myocardial infarction. A review. Drugs 1992; 44:293–325.
- Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet 1994; 343:311–322.
- Keely EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20
- De Luca G, Suryapranata H, Ottervanger JP, Antman EM. Time delay to treatment and mortality in primary angioplasty for acute myocardial infarction: every minute of delay counts. Circulation 2004; 109:1223–1225.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Gersh BJ, Stone GW, White HD, Holmes DR Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA 2005; 293:979–986.
- Antman EM, Hand M, Armstron PW, et al; Canadian Cardiovascular Society; American Academy of Family Physicians; American College of Cardiology; American Heart Association. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab Reteplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Cannon CP, Weintraub WS, Demopoulos LA, et al; TACTICS (Treat Angina With Aggrastat and Determine Cost of Therapy With an Invasive or Conservative Strategy)–Thrombolysis in Myocardial Infarction 18 Investigators. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001; 344:1879–1887.
- Damman P, Hirsch A, Windhausen F, Tijssen JG, de Winter RJ; ICTUS Investigators. 5-year clinical outcomes in the ICTUS (Invasive versus Conservative Treatment in Unstable coronary Syndromes) trial a randomized comparison of an early invasive versus selective invasive management in patients with non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2010; 55:858–864.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic decision making. JAMA 2000; 284:835–842.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Yousef O, Bhatt DL. The evolution of antiplatelet therapy in cardiovascular disease. Nat Rev Cardiol 2011; 8:547–559.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Yusuf S, Mehta SR, Zhao F, et al; Clopidogrel in Unstable angina to prevent Recurrent Events Trial Investigators. Early and late effects of clopidogrel in patients with acute coronary syndromes. Circulation 2003; 107:966–972.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Schömig A. Ticagrelor—is there need for a new player in the antiplatelet-therapy field? N Engl J Med 2009; 361:1108–1111.
- Wiviott SD, Antman EM, Braunwald E. Prasugrel. Circulation 2010; 122:394–403.
- Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation 2009; 120:2577–2585.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- de Queiroz Fernandes Araujo JO, Veloso HH, Braga De Paiva JM, Fiho MW, Vincenzo De Paola AA. Efficacy and safety of abciximab on acute myocardial infarction treated with percutaneous coronary interventions: a meta-analysis of randomized, controlled trials. Am Heart J 2004; 148:937–943.
- Kastrati A, Mehilli J, Neuman FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Giugliano RP, White JA, Bode C, et al; Early ACS Investigators. Early vs delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med 2009; 360:2176–2190.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Fergusson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH; EXTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- The Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2007; 358:2218–2230.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Chen ZM, Pan HC, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1622–1632.
- Dargie JH. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 2001; 357:1385–1390.
- Hennekens CH, Albert CM, Godfried SL, Gaziano JM, Buring JE. Adjunctive drug therapy of acute myocardial infarction—evidence from clinical trials. N Engl J Med 1996; 335:1660–1667.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol 2006; 48:438–445.
KEY POINTS
- For acute ST-elevation myocardial infarction, primary percutaneous coronary intervention is preferred over fibrinolytic therapy if it is available within 90 minutes of first medical contact.
- For non-ST-elevation acute coronary syndromes, either an early invasive or conservative strategy is recommended depending on patient risk and whether intensive medical therapy is available and appropriate.
- Daily aspirin therapy is indicated for all patients with acute coronary syndromes unless they have a true aspirin allergy.
- Adenosine diphosphate receptor inhibitors—clopidogrel, prasugrel, and ticagrelor—reduce ischemic events but increase bleeding risk and should be used only for patients with no history of stroke or transient ischemic attack.
Acute and critical limb ischemia: When time is limb
In many ways, vascular disease in the leg is similar to that in the heart. The risk factors, underlying conditions, and pathogenetic processes are the same, and in many cases, patients have both conditions. And just as cardiologists and emergency physicians have learned that in acute myocardial infarction “time is muscle,” we are coming to appreciate that in many cases of limb ischemia, “time is limb.”
Most physicians well understand the clinical spectrum of coronary artery disease, which ranges from stable angina to ST-elevation myocardial infarction. In the leg, the same situation exists: at the more benign end of the spectrum, patients experience no symptoms, but often that is because they lead a sedentary lifestyle, modifying their activity level to avoid pain. As the disease worsens, they can develop claudication and critical leg ischemia, comparable to non-ST-elevation myocardial infarction. The most severe condition is acute limb ischemia, analagous to ST-elevation myocardial infarction.
Distinguishing acute from critical limb ischemia is essential in patients who present with leg problems, whether it be leg pain or ulcers. The farther along the clinical spectrum the patient’s condition is, the more important it is to be aggressive in diagnosis and treatment. The history and physical examination are the most important first steps, focusing on the onset of symptoms, history, risk factors, and past interventions.
Peripheral artery disease is increasingly becoming a worldwide problem that is now being emphasized by the World Health Organization. Unfortunately, not enough attention is paid to the problem, not only in less-developed countries but also in the United States. Patients with peripheral artery disease tend to be elderly, in the lowest economic classes, and uninsured, and they often do not understand the impact of the disease on their health.
LEG ULCERS: CAUSES AND COSTS

Finding the underlying cause of leg ulcers is important, and the differential diagnosis is large (Table 1). However, knowing the cause does not necessarily lead to healing; it is still essential to assess perfusion, infection, and wound care, and to arrest edema.
Causes of leg and foot ulcers include venous insufficiency (with an estimated 2.5 million cases annually),1,2 diabetes (nearly 1 million cases),3 and pressure (ie, bedsores, occurring in up to 28% of patients in extended care),4 all at a cost in the billions of dollars.5–7
In general, peripheral artery disease itself does not cause ulcers; it is an inciting factor. It is important to find what started the process. Ill-fitting shoes, poor sensation because of diabetes, or a cut when trimming toenails can all contribute to a wound, and peripheral artery disease makes it unable to heal. The healing process requires more nutrients and oxygen than poor circulation can provide.
ACUTE LIMB ISCHEMIA
Acute limb ischemia is defined as any sudden decrease in limb perfusion causing a potential threat to limb viability.8 Although it comes on suddenly, it does not imply that the patient has not had long-standing peripheral artery disease. It is important to determine what suddenly changed to cause the onset of symptoms.
History and physical examination: The six Ps
A good history includes a thorough evaluation of the present illness, including the pain’s time of onset, abruptness, location, intensity, and change over time, and whether it is present at rest. The medical history should focus on claudication, diabetes, smoking, heart disease, palpitations, atrial fibrillation, and previous ischemic symptoms.8
The physical examination should focus on the “six Ps”:
- Pain
- Pulselessness
- Paresthesia (numbness occurs in about half of patients)
- Pallor (obstruction is typically one joint above the level of demarcation of pallor)
- Paralysis (a bad sign, particularly if the calf is tight)
- Poikilothermia (inability to regulate temperature).
A good pulse examination includes measuring the ankle-brachial index and a Doppler examination of both legs. A neurologic examination focusing on sensory and motor function is critical for determining the level of ischemia and the urgency of intervention.
Classification of acute limb ischemia

If it is determined that a patient has acute leg ischemia, it is important to categorize the condition using the classification system devised by the Society of Vascular Surgery and International Society of Cardiovascular Surgery (Table 2).9 The category establishes the type and urgency of treatment. This classification system is simple and depends on factors that can be assessed easily by nonspecialists:
- Pulses—arterial and venous pulses assessed by Doppler ultrasonography
- Sensation—the patient closes the eyes and answers if he or she can feel the examiner’s touch
- Motor function—can the patient move his or her toes?
Venous pulses can be difficult to assess. However, if the arterial pulse is present, the venous pulse should be next to it. Knowing the other criteria can determine the category, so not being certain of the venous pulse should not deter a clinician from assessing the other factors.
Category I is “viable.” Patients have intact sensory and motor functions and audible pulses. Patients in this category should be admitted and possibly started on anticoagulation therapy and referred to a vascular specialist within hours.
Category IIa is “threatened.” Sensation is starting to be lost but motor function is still present. These patients are considered to have reversible ischemia, analogous to myocardial infarction of the leg, and they require immediate attention.
Category IIb is similar and it also requires immediate attention.
Category III is usually irreversible, with loss of motor function and sensation.
CAUSES OF ACUTE LIMB ISCHEMIA
Thrombosis accounts for about 50% of cases. Underlying causes of the thrombosis are artherosclerosis (native or bypass), aneurysm, trauma, vasculitis (eg, in a rheumatologic disease such as lupus), and hypercoagulable states (particularly in patients with cancer).
Embolism accounts for about 30% of cases. Emboli usually arise from plaque rupture in atherosclerotic arteries or a clot breaking off from an aneurysm or from within the heart in patients with atrial fibrillation or another underlying heart disease. Paradoxical embolism, caused by an embolism crossing the heart through an opening such as a patent foramen ovale, is rare.
Uncommon causes include arterial dissection following trauma, adventitial cystic disease, popliteal artery entrapment, ergotism (from consuming fungus-contaminated grains), and human immunodeficiency virus arteriopathy.
The physical examination provides clues to the origin: livedo reticularis (purple discoloration in a mottled pattern) and blue nail beds indicate that an embolus is likely. Tests, including electrocardiography, echocardiography, and computed tomography of the chest and abdomen to look for an aneurysm, can help identify the cause. Ultrasonography of the popliteal arteries should also be considered to search for an aneurysm.
CRITICAL LIMB ISCHEMIA
Critical limb ischemia is more likely than acute limb ischemia to be seen in a general practice. Many aspects need to be addressed simultaneously, by different specialists: vascular and endocrine systems, infection, and wound care. The most successful management strategy is a dynamic approach using every piece of information.10
The Rutherford classification of peripheral artery disease has six categories based on the clinical presentation, with categories I through III being mild to severe claudication. We discuss here only the more severe categories: IV (pain at rest), V (tissue loss), and VI (gangrene).
Strong indicators of pain at rest are that the patient has to get up at night to dangle the leg over the bed or walk a few steps, or sleeps in a chair, or refuses to elevate the leg because of pain. The affected leg tends to appear red when the patient is standing (dependent rubor), but pale when the foot is elevated (elevation pallor).
Confirming that a patient has dependent rubor can be challenging, especially in people with dark skin. Classically, redness is seen when the leg is down and disappears with elevation, but in cellulitis, redness can also be reduced by elevating the leg. A foot that is hot to the touch is an indication of infection and not lack of perfusion alone.
The hemodynamic definition of critical limb ischemia is11:
- Ankle-brachial pressure index less than 0.4
- Reduced toebrachial pressure index, ie, less than 0.7
- Reduced transcutaneous pressure of oxygen (Tcpo2), ie, less than 40 mm Hg.
From 15% to 20% of patients with claudication will progress to critical ischemia over their lifetime, and in patients with claudication who also have diabetes, the risk is nearly 10 times higher. Without revascularization, the risk of amputation within 1 year is 73% for patients in Rutherford class IV and 95% for patients in class V or VI.
Revascularization and limb preservation
Preserving the limb is a prime goal. For patients who have an amputation, the mortality rate is 40% within 2 years.8 These patients tend to be elderly, and after an amputation, most will not learn to use a prosthesis and resume their previous level of activity. Other treatment objectives are to relieve pain, reduce cardiovascular risk, and minimize procedural complications.
Although limb preservation is not a controversial goal, best practices to preserve limbs are not universally available. Goodney et al12 studied variation in the United States in the use of lower-extremity vascular procedures for critical limb ischemia. They defined “low-intensity” to “high-intensity” regions of the country depending on the proportion of patients who underwent a vascular procedure in the year before amputation. They found considerable variation, but even in the region of highest intensity, more than 40% of patients did not have a vascular procedure in the year before amputation.
Similarly, Jones et al13 mapped amputation rates by US state and found significant variation even after adjusting for risk factors such as tobacco use and obesity.
Controversy surrounds the specifics of revascularization treatment, as in many fields in vascular medicine. However, most experts agree that improved perfusion is the goal.
The Trans-Atlantic Inter-Society Consensus for the Management of Peripheral Artery Disease recommends revascularization as the best treatment for patients with critical limb ischemia.8 In addition, the American College of Cardiology and American Heart Association Guidelines for the Management of Patients With Peripheral Arterial Disease (Lower Extremity, Renal, Mesenteric, and Abdominal Aortic) state that the tibial or pedal artery that is capable of providing continuous and uncompromised outflow to the foot should be used as the site of distal anastomosis.14 These guidelines do not yet mention endovascular therapy.
Angiosomes guide revascularization
In the past few years, the ability to facilitate healing of foot ulcers has improved. Angiosomes—regions of vascularization supplied by specific arteries—can be mapped on the skin, similar to the way dermatomes are mapped for neural innervation (Figure 1). The foot and lower leg region has six angiosomes perfused by three arteries that branch off the popliteal artery after it passes behind the knee:
- The anterior tibial artery supplies the dorsum of the foot and the front of the lower limb.
- The posterior tibial artery supplies the plantar surface of the foot via three branches—the medial plantar, lateral plantar, and calcaneal branches.
- The peroneal artery supplies the lateral part of the foot with collaterals to the anterior and posterior tibial arteries if they are compromised.
Studies have compared angiosome-based treatment vs revascularizing the best available artery (thus depending on collateral flow to compensate to surrounding areas). They have found that regardless of whether an endovascular or bypass method of revascularization was used, an angiosome-based approach led to significantly higher amputation-free survival rates.15–17
Patients typically do not have blockage of only a single tibial artery. Graziani et al18 assessed the vascular lesions in 417 patients with critical limb ischemia and found that multiple below-knee arteries were frequently involved. This makes it difficult to decide where to target revascularization efforts, and the angiosome concept helps with that.
ASSESSING WOUND PERFUSION
Ankle- and toe-brachial indices assess perfusion
The ankle-brachial index19 is a good superficial assessment of perfusion. Multiple epidemiologic studies have shown the prognostic value of the ankle brachial index beyond the traditional risk factors and even the Framingham risk score.19 Values:
- Normal 1.1–1.30 (> 1.31 is abnormal and consistent with calcified vessels, and is an unreliable measure)
- Low normal 0.91–1.00
- Mild disease 0.71–0.90
- Moderate disease 0.41–0.70
- Severe disease ≤ 0.40.
However, the ankle-brachial index assesses perfusion only to the ankle, and many patients have ulcers in the toes and distal foot. The toe-brachial index must be specifically ordered in most institutions (if the first toe has an ulcer, the second toe should be assessed). The toe-brachial index is also important if the ankle-brachial index cannot be obtained because of calcified, noncompressible arteries in the ankle. A normal toe-brachial index is greater than 0.7.
The segmental blood pressure examination compares blood pressure measurements at multiple sites in the lower extremity. A drop of more than 20 mm Hg between segments indicates obstruction at that location. The test is simple and noninvasive and often can replace computed tomography.20
Transcutaneous oximetry
Transcutaneous oximetry measures the Tcpo2 from 1 to 2 mm deep in the skin from local capillaries. Measured adjacent to an ulcer, it is useful to predict wound healing and to assess the response to hyperbaric oxygen therapy.21 The values are:
- Normal > 70 mm Hg
- Impaired wound healing < 40 mm Hg
- Critical limb ischemia < 30 mm Hg.
Although most agree that a Tcpo2 below 40 mm Hg requires revascularization, low values can arise from many causes other than peripheral artery disease, including high altitude, pulmonary disease, heart failure, edema, inflammation, callus, and skin diseases such as scleroderma.
Skin perfusion pressure better predicts healing
Skin perfusion pressure is a measure of the capillary opening pressure after occlusion and is another way to assess perfusion. This test is not routinely done and must be specially requested.
The test is performed by inflating a blood pressure cuff on the leg until blood flow is occluded, then using laser Doppler to determine reactive hyperemia, ie, the gradual return of blood flow during controlled pressure release. The pressure at which movement is detected is the skin perfusion pressure.22
The laser Doppler probe emits and detects light scattered in the tissue. Light hitting moving blood cells undergoes a change in frequency, ie, a Doppler shift. An algorithm converts the optical information in the skin perfusion pressure by capturing the onset of capillary flow return and determining the pressure at which flow returns. Categories of results:
- > 50 mm Hg—normal
- 40–50 mm Hg—mild ischemia (wound healing probable)
- 30–40 mm Hg—moderate ischemia (wound healing uncertain)
- < 30 mm Hg—critical limb ischemia (wound healing unlikely).
Skin perfusion pressure testing has the advantages of not being affected by vessel calcification, thickened skin, or edema. It can be used on the plantar aspect of the foot and on digits. Recent small studies indicate that it is more sensitive for predicting wound healing than Tcpo2 measures.
On the other hand, skin perfusion pressure testing is not useful for predicting response to hyperbaric oxygen therapy. Also, blood flow occlusion by the cuff may be painful.
Intraoperative fluorescence angiography
Intraoperative fluorescence angiography is used to assess flap viability during reconstructive surgery and is being studied to determine its usefulness for assessing tissue viability in limb ischemia.
The test provides real-time assessment of capillary perfusion, determining surface tissue viability. The imaging head contains a digital camera, a laser light source, and a distance sensor. The test requires intravenous administration of indocyanine green, which binds to plasma proteins and is cleared through the liver, making it safe for patients with renal dysfunction. It cannot be used in patients with allergies to iodine contrast, penicillin, or sulfa.23
PREVENTION TARGETS CARDIOVASCULAR RISK FACTORS
Preventive measures are the same as for cardiovascular disease, ie, aggressive risk-factor modification: quitting smoking, lowering low-density lipoprotein cholesterol, reducing blood pressure, controlling diabetes, and managing heart failure.
Dual antiplatelet therapy should be instituted with aspirin and clopidogrel (Plavix) in patients undergoing revascularization. One can also consider cilostazol (Pletal); however, the role of this agent in patients with critical limb ischemia is less defined.
BYPASS OR ANGIOPLASTY?
The Bypass Versus Angioplasty in Severe Ischaemia of the Leg (BASIL) trial24 randomly assigned 452 patients with severe limb ischemia due to infrainguinal atherosclerosis to receive either surgery-first or angioplasty-first care and followed them for 5.5 years.
No significant differences between the two groups were found in amputation-free survival, deaths, or health-related quality of life. However, hospital costs associated with the surgery-first strategy were about one-third higher. As expected, more patients in the surgery group developed a wound infection, and more patients in the angioplasty group required bypass surgery at some point.
The conclusion that can be reached from this study is that patients presenting with severe limb ischemia due to infrainguinal atherosclerotic occlusive disease who are suitable for both surgical and interventional procedures can be treated with either method. However, most experts consider endovascular therapy as the first option in many patients. The National Institutes of Health recently funded a study to compare contemporary endovascular therapy vs surgery in patients with critical limb ischemia.
TAKE-HOME POINTS
In the last decade, significant endovascular advances have been made. New devices and techniques have enhanced our ability to treat high-risk patients who have critical limb ischemia. The combination of risk factor modification, accurate diagnosis, and aggressive revascularization should prevent limb loss in many of these patients. For the primary care physician, a low threshold for assessing perfusion in patients with critical limb ischemia is important using a screening ankle-brachial index and toe-brachial index. These patients should promptly be referred to a vascular specialist for further evaluation and treatment.
- Phillips T, Stanton B, Provan A, Lew R. A study of the impact of leg ulcers on quality of life: financial, social, and psychologic implications. J Am Acad Dermatol 1994; 31:49–53.
- Brem H, Kirsner RS, Falanga V. Protocol for the successful treatment of venous ulcers. Am J Surg 2004; 188(1A suppl):1–8.
- Ramsey SD, Newton K, Blough D, et al. Incidence, outcomes, and cost of foot ulcers in patients with diabetes. Diabetes Care 1999; 22:382–387.
- Cuddigan J, Berlowitz DR, Ayello E; National Pressure Ulcer Advisory Panel. Pressure ulcers in America: Prevalence, incidence, and implications for the future: an executive summary of the National Pressure Ulcer Advisory Panel monograph. Adv Skin Wound Care 2001; 14:208–215.
- Olin JW, Beusterien KM, Childs MB, Seavey C, McHugh L, Griffiths RI. Medical costs of treating venous stasis ulcers: evidence from a retrospective cohort study. Vasc Med 1999; 4:1–7.
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26:1790–1795.
- Kumar RN, Gupchup GV, Dodd MA, et al. Direct health care costs of 4 common skin ulcers in New Mexico Medicaid fee-for-service patients. Adv Skin Wound Care 2004; 17:143–149.
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG; TASC II Working Group. Inter-society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg 2007; 45(suppl):S5–S67.
- Rutherford RB, Baker JD, Ernst C, et al. Recommended standards for reports dealing with lower extremity ischemia: revised version. J Vasc Surg 1997; 26:517–538. Erratum in J Vasc Surg 2001; 33:805.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304. Erratum in N Engl J Med 2010; 363:1092.
- Dormandy JA, Rutherford RB. Management of peripheral arterial disease (PAD). TASC Working Group. TransAtlantic Inter-Society Consensus (TASC). J Vasc Surg 2000; 31:S1–S296.
- Goodney PP, Travis LL, Nallamothu BK, et al. Variation in the use of lower extremity vascular procedures for critical limb ischemia. Circ Cardiovasc Qual Outcomes 2012; 5:94–102.
- Jones WS, Patel MR, Dai D, et al. Temporal trends and geographic variation of lower-extremity amputation in patients with peripheral artery disease: results from U.S. Medicare 2000–2008. J Am Coll Cardiol 2012; 60:2230–2236.
- Hirsch AT, Haskal ZJ, Mertzer NR, et al. ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Alexandrescu VA, Hubermont G, Philips Y, et al. Selective primary angioplasty following an angiosome model of reperfusion in the treatment of Wagner 1–4 diabetic foot lesions: practice in a multidisciplinary diabetic limb service. J Endovasc Ther 2008; 15:580–593.
- Neville RF, Attinger CE, Bulan EJ, Ducic I, Thomassen M, Sidawy AN. Revascularization of a specific angiosome for limb salvage: does the target artery matter? Ann Vasc Surg 2009; 23:367–373.
- Iida O, Soga Y, Hirano K, et al. Long-term results of direct and indirect endovascular revascularization based on the angiosome concept in patients with critical limb ischemia presenting with isolated below-the-knee lesions. J Vasc Surg 2012; 55:363–370.
- Graziani L, Silvestro A, Bertone V, et al. Vascular involvement in diabetic subjects with ischemic foot ulcer: a new morphologic categorization of disease severity. Eur J Vasc Endovasc Surg 2007; 33:453–460.
- Newman AB, Siscovick DS, Manolio TA, et al., Cardiovascular Heart Study (CHS) Collaborative Research Group. Ankle-arm index as a marker of atherosclerosis in the Cardiovascular Health Study. Circulation 1993; 88:837–845.
- Cronenwett JL, Johnston KW. Rutherford’s Vascular Surgery. 7th ed. Philadelphia, PA: Saunders Elsevier; 2010.
- Fife CE, Smart DR, Sheffield PJ, Hopf HW, Hawkins G, Clarke D. Transcutaneous oximetry in clinical practice: consensus statements from an expert panel based on evidence. Undersea Hyperb Med 2009; 36:43–53.
- Lo T, Sample R, Moore P, Gold P. Prediction of wound healing outcome using skin perfusion pressure and transcutaneous oximetry. Wounds 2009; 21:310–316.
- Perry D, Bharara M, Armstrong DG, Mills J. Intraoperative fluorescence vascular angiography: during tibial bypass. J Diabetes Sci Technol 2012; 6:204–208.
- Adam DJ, Beard JD, Cleveland T, et al; BASIL trial participants. Bypass versus angioplasty in severe ischaemia of the leg (BASIL): multicentre, randomised controlled trial. Lancet 2005; 366:1925–1934.
In many ways, vascular disease in the leg is similar to that in the heart. The risk factors, underlying conditions, and pathogenetic processes are the same, and in many cases, patients have both conditions. And just as cardiologists and emergency physicians have learned that in acute myocardial infarction “time is muscle,” we are coming to appreciate that in many cases of limb ischemia, “time is limb.”
Most physicians well understand the clinical spectrum of coronary artery disease, which ranges from stable angina to ST-elevation myocardial infarction. In the leg, the same situation exists: at the more benign end of the spectrum, patients experience no symptoms, but often that is because they lead a sedentary lifestyle, modifying their activity level to avoid pain. As the disease worsens, they can develop claudication and critical leg ischemia, comparable to non-ST-elevation myocardial infarction. The most severe condition is acute limb ischemia, analagous to ST-elevation myocardial infarction.
Distinguishing acute from critical limb ischemia is essential in patients who present with leg problems, whether it be leg pain or ulcers. The farther along the clinical spectrum the patient’s condition is, the more important it is to be aggressive in diagnosis and treatment. The history and physical examination are the most important first steps, focusing on the onset of symptoms, history, risk factors, and past interventions.
Peripheral artery disease is increasingly becoming a worldwide problem that is now being emphasized by the World Health Organization. Unfortunately, not enough attention is paid to the problem, not only in less-developed countries but also in the United States. Patients with peripheral artery disease tend to be elderly, in the lowest economic classes, and uninsured, and they often do not understand the impact of the disease on their health.
LEG ULCERS: CAUSES AND COSTS
Finding the underlying cause of leg ulcers is important, and the differential diagnosis is large (Table 1). However, knowing the cause does not necessarily lead to healing; it is still essential to assess perfusion, infection, and wound care, and to arrest edema.
Causes of leg and foot ulcers include venous insufficiency (with an estimated 2.5 million cases annually),1,2 diabetes (nearly 1 million cases),3 and pressure (ie, bedsores, occurring in up to 28% of patients in extended care),4 all at a cost in the billions of dollars.5–7
In general, peripheral artery disease itself does not cause ulcers; it is an inciting factor. It is important to find what started the process. Ill-fitting shoes, poor sensation because of diabetes, or a cut when trimming toenails can all contribute to a wound, and peripheral artery disease makes it unable to heal. The healing process requires more nutrients and oxygen than poor circulation can provide.
ACUTE LIMB ISCHEMIA
Acute limb ischemia is defined as any sudden decrease in limb perfusion causing a potential threat to limb viability.8 Although it comes on suddenly, it does not imply that the patient has not had long-standing peripheral artery disease. It is important to determine what suddenly changed to cause the onset of symptoms.
History and physical examination: The six Ps
A good history includes a thorough evaluation of the present illness, including the pain’s time of onset, abruptness, location, intensity, and change over time, and whether it is present at rest. The medical history should focus on claudication, diabetes, smoking, heart disease, palpitations, atrial fibrillation, and previous ischemic symptoms.8
The physical examination should focus on the “six Ps”:
- Pain
- Pulselessness
- Paresthesia (numbness occurs in about half of patients)
- Pallor (obstruction is typically one joint above the level of demarcation of pallor)
- Paralysis (a bad sign, particularly if the calf is tight)
- Poikilothermia (inability to regulate temperature).
A good pulse examination includes measuring the ankle-brachial index and a Doppler examination of both legs. A neurologic examination focusing on sensory and motor function is critical for determining the level of ischemia and the urgency of intervention.
Classification of acute limb ischemia
If it is determined that a patient has acute leg ischemia, it is important to categorize the condition using the classification system devised by the Society of Vascular Surgery and International Society of Cardiovascular Surgery (Table 2).9 The category establishes the type and urgency of treatment. This classification system is simple and depends on factors that can be assessed easily by nonspecialists:
- Pulses—arterial and venous pulses assessed by Doppler ultrasonography
- Sensation—the patient closes the eyes and answers if he or she can feel the examiner’s touch
- Motor function—can the patient move his or her toes?
Venous pulses can be difficult to assess. However, if the arterial pulse is present, the venous pulse should be next to it. Knowing the other criteria can determine the category, so not being certain of the venous pulse should not deter a clinician from assessing the other factors.
Category I is “viable.” Patients have intact sensory and motor functions and audible pulses. Patients in this category should be admitted and possibly started on anticoagulation therapy and referred to a vascular specialist within hours.
Category IIa is “threatened.” Sensation is starting to be lost but motor function is still present. These patients are considered to have reversible ischemia, analogous to myocardial infarction of the leg, and they require immediate attention.
Category IIb is similar and it also requires immediate attention.
Category III is usually irreversible, with loss of motor function and sensation.
CAUSES OF ACUTE LIMB ISCHEMIA
Thrombosis accounts for about 50% of cases. Underlying causes of the thrombosis are artherosclerosis (native or bypass), aneurysm, trauma, vasculitis (eg, in a rheumatologic disease such as lupus), and hypercoagulable states (particularly in patients with cancer).
Embolism accounts for about 30% of cases. Emboli usually arise from plaque rupture in atherosclerotic arteries or a clot breaking off from an aneurysm or from within the heart in patients with atrial fibrillation or another underlying heart disease. Paradoxical embolism, caused by an embolism crossing the heart through an opening such as a patent foramen ovale, is rare.
Uncommon causes include arterial dissection following trauma, adventitial cystic disease, popliteal artery entrapment, ergotism (from consuming fungus-contaminated grains), and human immunodeficiency virus arteriopathy.
The physical examination provides clues to the origin: livedo reticularis (purple discoloration in a mottled pattern) and blue nail beds indicate that an embolus is likely. Tests, including electrocardiography, echocardiography, and computed tomography of the chest and abdomen to look for an aneurysm, can help identify the cause. Ultrasonography of the popliteal arteries should also be considered to search for an aneurysm.
CRITICAL LIMB ISCHEMIA
Critical limb ischemia is more likely than acute limb ischemia to be seen in a general practice. Many aspects need to be addressed simultaneously, by different specialists: vascular and endocrine systems, infection, and wound care. The most successful management strategy is a dynamic approach using every piece of information.10
The Rutherford classification of peripheral artery disease has six categories based on the clinical presentation, with categories I through III being mild to severe claudication. We discuss here only the more severe categories: IV (pain at rest), V (tissue loss), and VI (gangrene).
Strong indicators of pain at rest are that the patient has to get up at night to dangle the leg over the bed or walk a few steps, or sleeps in a chair, or refuses to elevate the leg because of pain. The affected leg tends to appear red when the patient is standing (dependent rubor), but pale when the foot is elevated (elevation pallor).
Confirming that a patient has dependent rubor can be challenging, especially in people with dark skin. Classically, redness is seen when the leg is down and disappears with elevation, but in cellulitis, redness can also be reduced by elevating the leg. A foot that is hot to the touch is an indication of infection and not lack of perfusion alone.
The hemodynamic definition of critical limb ischemia is11:
- Ankle-brachial pressure index less than 0.4
- Reduced toebrachial pressure index, ie, less than 0.7
- Reduced transcutaneous pressure of oxygen (Tcpo2), ie, less than 40 mm Hg.
From 15% to 20% of patients with claudication will progress to critical ischemia over their lifetime, and in patients with claudication who also have diabetes, the risk is nearly 10 times higher. Without revascularization, the risk of amputation within 1 year is 73% for patients in Rutherford class IV and 95% for patients in class V or VI.
Revascularization and limb preservation
Preserving the limb is a prime goal. For patients who have an amputation, the mortality rate is 40% within 2 years.8 These patients tend to be elderly, and after an amputation, most will not learn to use a prosthesis and resume their previous level of activity. Other treatment objectives are to relieve pain, reduce cardiovascular risk, and minimize procedural complications.
Although limb preservation is not a controversial goal, best practices to preserve limbs are not universally available. Goodney et al12 studied variation in the United States in the use of lower-extremity vascular procedures for critical limb ischemia. They defined “low-intensity” to “high-intensity” regions of the country depending on the proportion of patients who underwent a vascular procedure in the year before amputation. They found considerable variation, but even in the region of highest intensity, more than 40% of patients did not have a vascular procedure in the year before amputation.
Similarly, Jones et al13 mapped amputation rates by US state and found significant variation even after adjusting for risk factors such as tobacco use and obesity.
Controversy surrounds the specifics of revascularization treatment, as in many fields in vascular medicine. However, most experts agree that improved perfusion is the goal.
The Trans-Atlantic Inter-Society Consensus for the Management of Peripheral Artery Disease recommends revascularization as the best treatment for patients with critical limb ischemia.8 In addition, the American College of Cardiology and American Heart Association Guidelines for the Management of Patients With Peripheral Arterial Disease (Lower Extremity, Renal, Mesenteric, and Abdominal Aortic) state that the tibial or pedal artery that is capable of providing continuous and uncompromised outflow to the foot should be used as the site of distal anastomosis.14 These guidelines do not yet mention endovascular therapy.
Angiosomes guide revascularization
In the past few years, the ability to facilitate healing of foot ulcers has improved. Angiosomes—regions of vascularization supplied by specific arteries—can be mapped on the skin, similar to the way dermatomes are mapped for neural innervation (Figure 1). The foot and lower leg region has six angiosomes perfused by three arteries that branch off the popliteal artery after it passes behind the knee:
- The anterior tibial artery supplies the dorsum of the foot and the front of the lower limb.
- The posterior tibial artery supplies the plantar surface of the foot via three branches—the medial plantar, lateral plantar, and calcaneal branches.
- The peroneal artery supplies the lateral part of the foot with collaterals to the anterior and posterior tibial arteries if they are compromised.
Studies have compared angiosome-based treatment vs revascularizing the best available artery (thus depending on collateral flow to compensate to surrounding areas). They have found that regardless of whether an endovascular or bypass method of revascularization was used, an angiosome-based approach led to significantly higher amputation-free survival rates.15–17
Patients typically do not have blockage of only a single tibial artery. Graziani et al18 assessed the vascular lesions in 417 patients with critical limb ischemia and found that multiple below-knee arteries were frequently involved. This makes it difficult to decide where to target revascularization efforts, and the angiosome concept helps with that.
ASSESSING WOUND PERFUSION
Ankle- and toe-brachial indices assess perfusion
The ankle-brachial index19 is a good superficial assessment of perfusion. Multiple epidemiologic studies have shown the prognostic value of the ankle brachial index beyond the traditional risk factors and even the Framingham risk score.19 Values:
- Normal 1.1–1.30 (> 1.31 is abnormal and consistent with calcified vessels, and is an unreliable measure)
- Low normal 0.91–1.00
- Mild disease 0.71–0.90
- Moderate disease 0.41–0.70
- Severe disease ≤ 0.40.
However, the ankle-brachial index assesses perfusion only to the ankle, and many patients have ulcers in the toes and distal foot. The toe-brachial index must be specifically ordered in most institutions (if the first toe has an ulcer, the second toe should be assessed). The toe-brachial index is also important if the ankle-brachial index cannot be obtained because of calcified, noncompressible arteries in the ankle. A normal toe-brachial index is greater than 0.7.
The segmental blood pressure examination compares blood pressure measurements at multiple sites in the lower extremity. A drop of more than 20 mm Hg between segments indicates obstruction at that location. The test is simple and noninvasive and often can replace computed tomography.20
Transcutaneous oximetry
Transcutaneous oximetry measures the Tcpo2 from 1 to 2 mm deep in the skin from local capillaries. Measured adjacent to an ulcer, it is useful to predict wound healing and to assess the response to hyperbaric oxygen therapy.21 The values are:
- Normal > 70 mm Hg
- Impaired wound healing < 40 mm Hg
- Critical limb ischemia < 30 mm Hg.
Although most agree that a Tcpo2 below 40 mm Hg requires revascularization, low values can arise from many causes other than peripheral artery disease, including high altitude, pulmonary disease, heart failure, edema, inflammation, callus, and skin diseases such as scleroderma.
Skin perfusion pressure better predicts healing
Skin perfusion pressure is a measure of the capillary opening pressure after occlusion and is another way to assess perfusion. This test is not routinely done and must be specially requested.
The test is performed by inflating a blood pressure cuff on the leg until blood flow is occluded, then using laser Doppler to determine reactive hyperemia, ie, the gradual return of blood flow during controlled pressure release. The pressure at which movement is detected is the skin perfusion pressure.22
The laser Doppler probe emits and detects light scattered in the tissue. Light hitting moving blood cells undergoes a change in frequency, ie, a Doppler shift. An algorithm converts the optical information in the skin perfusion pressure by capturing the onset of capillary flow return and determining the pressure at which flow returns. Categories of results:
- > 50 mm Hg—normal
- 40–50 mm Hg—mild ischemia (wound healing probable)
- 30–40 mm Hg—moderate ischemia (wound healing uncertain)
- < 30 mm Hg—critical limb ischemia (wound healing unlikely).
Skin perfusion pressure testing has the advantages of not being affected by vessel calcification, thickened skin, or edema. It can be used on the plantar aspect of the foot and on digits. Recent small studies indicate that it is more sensitive for predicting wound healing than Tcpo2 measures.
On the other hand, skin perfusion pressure testing is not useful for predicting response to hyperbaric oxygen therapy. Also, blood flow occlusion by the cuff may be painful.
Intraoperative fluorescence angiography
Intraoperative fluorescence angiography is used to assess flap viability during reconstructive surgery and is being studied to determine its usefulness for assessing tissue viability in limb ischemia.
The test provides real-time assessment of capillary perfusion, determining surface tissue viability. The imaging head contains a digital camera, a laser light source, and a distance sensor. The test requires intravenous administration of indocyanine green, which binds to plasma proteins and is cleared through the liver, making it safe for patients with renal dysfunction. It cannot be used in patients with allergies to iodine contrast, penicillin, or sulfa.23
PREVENTION TARGETS CARDIOVASCULAR RISK FACTORS
Preventive measures are the same as for cardiovascular disease, ie, aggressive risk-factor modification: quitting smoking, lowering low-density lipoprotein cholesterol, reducing blood pressure, controlling diabetes, and managing heart failure.
Dual antiplatelet therapy should be instituted with aspirin and clopidogrel (Plavix) in patients undergoing revascularization. One can also consider cilostazol (Pletal); however, the role of this agent in patients with critical limb ischemia is less defined.
BYPASS OR ANGIOPLASTY?
The Bypass Versus Angioplasty in Severe Ischaemia of the Leg (BASIL) trial24 randomly assigned 452 patients with severe limb ischemia due to infrainguinal atherosclerosis to receive either surgery-first or angioplasty-first care and followed them for 5.5 years.
No significant differences between the two groups were found in amputation-free survival, deaths, or health-related quality of life. However, hospital costs associated with the surgery-first strategy were about one-third higher. As expected, more patients in the surgery group developed a wound infection, and more patients in the angioplasty group required bypass surgery at some point.
The conclusion that can be reached from this study is that patients presenting with severe limb ischemia due to infrainguinal atherosclerotic occlusive disease who are suitable for both surgical and interventional procedures can be treated with either method. However, most experts consider endovascular therapy as the first option in many patients. The National Institutes of Health recently funded a study to compare contemporary endovascular therapy vs surgery in patients with critical limb ischemia.
TAKE-HOME POINTS
In the last decade, significant endovascular advances have been made. New devices and techniques have enhanced our ability to treat high-risk patients who have critical limb ischemia. The combination of risk factor modification, accurate diagnosis, and aggressive revascularization should prevent limb loss in many of these patients. For the primary care physician, a low threshold for assessing perfusion in patients with critical limb ischemia is important using a screening ankle-brachial index and toe-brachial index. These patients should promptly be referred to a vascular specialist for further evaluation and treatment.
In many ways, vascular disease in the leg is similar to that in the heart. The risk factors, underlying conditions, and pathogenetic processes are the same, and in many cases, patients have both conditions. And just as cardiologists and emergency physicians have learned that in acute myocardial infarction “time is muscle,” we are coming to appreciate that in many cases of limb ischemia, “time is limb.”
Most physicians well understand the clinical spectrum of coronary artery disease, which ranges from stable angina to ST-elevation myocardial infarction. In the leg, the same situation exists: at the more benign end of the spectrum, patients experience no symptoms, but often that is because they lead a sedentary lifestyle, modifying their activity level to avoid pain. As the disease worsens, they can develop claudication and critical leg ischemia, comparable to non-ST-elevation myocardial infarction. The most severe condition is acute limb ischemia, analagous to ST-elevation myocardial infarction.
Distinguishing acute from critical limb ischemia is essential in patients who present with leg problems, whether it be leg pain or ulcers. The farther along the clinical spectrum the patient’s condition is, the more important it is to be aggressive in diagnosis and treatment. The history and physical examination are the most important first steps, focusing on the onset of symptoms, history, risk factors, and past interventions.
Peripheral artery disease is increasingly becoming a worldwide problem that is now being emphasized by the World Health Organization. Unfortunately, not enough attention is paid to the problem, not only in less-developed countries but also in the United States. Patients with peripheral artery disease tend to be elderly, in the lowest economic classes, and uninsured, and they often do not understand the impact of the disease on their health.
LEG ULCERS: CAUSES AND COSTS
Finding the underlying cause of leg ulcers is important, and the differential diagnosis is large (Table 1). However, knowing the cause does not necessarily lead to healing; it is still essential to assess perfusion, infection, and wound care, and to arrest edema.
Causes of leg and foot ulcers include venous insufficiency (with an estimated 2.5 million cases annually),1,2 diabetes (nearly 1 million cases),3 and pressure (ie, bedsores, occurring in up to 28% of patients in extended care),4 all at a cost in the billions of dollars.5–7
In general, peripheral artery disease itself does not cause ulcers; it is an inciting factor. It is important to find what started the process. Ill-fitting shoes, poor sensation because of diabetes, or a cut when trimming toenails can all contribute to a wound, and peripheral artery disease makes it unable to heal. The healing process requires more nutrients and oxygen than poor circulation can provide.
ACUTE LIMB ISCHEMIA
Acute limb ischemia is defined as any sudden decrease in limb perfusion causing a potential threat to limb viability.8 Although it comes on suddenly, it does not imply that the patient has not had long-standing peripheral artery disease. It is important to determine what suddenly changed to cause the onset of symptoms.
History and physical examination: The six Ps
A good history includes a thorough evaluation of the present illness, including the pain’s time of onset, abruptness, location, intensity, and change over time, and whether it is present at rest. The medical history should focus on claudication, diabetes, smoking, heart disease, palpitations, atrial fibrillation, and previous ischemic symptoms.8
The physical examination should focus on the “six Ps”:
- Pain
- Pulselessness
- Paresthesia (numbness occurs in about half of patients)
- Pallor (obstruction is typically one joint above the level of demarcation of pallor)
- Paralysis (a bad sign, particularly if the calf is tight)
- Poikilothermia (inability to regulate temperature).
A good pulse examination includes measuring the ankle-brachial index and a Doppler examination of both legs. A neurologic examination focusing on sensory and motor function is critical for determining the level of ischemia and the urgency of intervention.
Classification of acute limb ischemia
If it is determined that a patient has acute leg ischemia, it is important to categorize the condition using the classification system devised by the Society of Vascular Surgery and International Society of Cardiovascular Surgery (Table 2).9 The category establishes the type and urgency of treatment. This classification system is simple and depends on factors that can be assessed easily by nonspecialists:
- Pulses—arterial and venous pulses assessed by Doppler ultrasonography
- Sensation—the patient closes the eyes and answers if he or she can feel the examiner’s touch
- Motor function—can the patient move his or her toes?
Venous pulses can be difficult to assess. However, if the arterial pulse is present, the venous pulse should be next to it. Knowing the other criteria can determine the category, so not being certain of the venous pulse should not deter a clinician from assessing the other factors.
Category I is “viable.” Patients have intact sensory and motor functions and audible pulses. Patients in this category should be admitted and possibly started on anticoagulation therapy and referred to a vascular specialist within hours.
Category IIa is “threatened.” Sensation is starting to be lost but motor function is still present. These patients are considered to have reversible ischemia, analogous to myocardial infarction of the leg, and they require immediate attention.
Category IIb is similar and it also requires immediate attention.
Category III is usually irreversible, with loss of motor function and sensation.
CAUSES OF ACUTE LIMB ISCHEMIA
Thrombosis accounts for about 50% of cases. Underlying causes of the thrombosis are artherosclerosis (native or bypass), aneurysm, trauma, vasculitis (eg, in a rheumatologic disease such as lupus), and hypercoagulable states (particularly in patients with cancer).
Embolism accounts for about 30% of cases. Emboli usually arise from plaque rupture in atherosclerotic arteries or a clot breaking off from an aneurysm or from within the heart in patients with atrial fibrillation or another underlying heart disease. Paradoxical embolism, caused by an embolism crossing the heart through an opening such as a patent foramen ovale, is rare.
Uncommon causes include arterial dissection following trauma, adventitial cystic disease, popliteal artery entrapment, ergotism (from consuming fungus-contaminated grains), and human immunodeficiency virus arteriopathy.
The physical examination provides clues to the origin: livedo reticularis (purple discoloration in a mottled pattern) and blue nail beds indicate that an embolus is likely. Tests, including electrocardiography, echocardiography, and computed tomography of the chest and abdomen to look for an aneurysm, can help identify the cause. Ultrasonography of the popliteal arteries should also be considered to search for an aneurysm.
CRITICAL LIMB ISCHEMIA
Critical limb ischemia is more likely than acute limb ischemia to be seen in a general practice. Many aspects need to be addressed simultaneously, by different specialists: vascular and endocrine systems, infection, and wound care. The most successful management strategy is a dynamic approach using every piece of information.10
The Rutherford classification of peripheral artery disease has six categories based on the clinical presentation, with categories I through III being mild to severe claudication. We discuss here only the more severe categories: IV (pain at rest), V (tissue loss), and VI (gangrene).
Strong indicators of pain at rest are that the patient has to get up at night to dangle the leg over the bed or walk a few steps, or sleeps in a chair, or refuses to elevate the leg because of pain. The affected leg tends to appear red when the patient is standing (dependent rubor), but pale when the foot is elevated (elevation pallor).
Confirming that a patient has dependent rubor can be challenging, especially in people with dark skin. Classically, redness is seen when the leg is down and disappears with elevation, but in cellulitis, redness can also be reduced by elevating the leg. A foot that is hot to the touch is an indication of infection and not lack of perfusion alone.
The hemodynamic definition of critical limb ischemia is11:
- Ankle-brachial pressure index less than 0.4
- Reduced toebrachial pressure index, ie, less than 0.7
- Reduced transcutaneous pressure of oxygen (Tcpo2), ie, less than 40 mm Hg.
From 15% to 20% of patients with claudication will progress to critical ischemia over their lifetime, and in patients with claudication who also have diabetes, the risk is nearly 10 times higher. Without revascularization, the risk of amputation within 1 year is 73% for patients in Rutherford class IV and 95% for patients in class V or VI.
Revascularization and limb preservation
Preserving the limb is a prime goal. For patients who have an amputation, the mortality rate is 40% within 2 years.8 These patients tend to be elderly, and after an amputation, most will not learn to use a prosthesis and resume their previous level of activity. Other treatment objectives are to relieve pain, reduce cardiovascular risk, and minimize procedural complications.
Although limb preservation is not a controversial goal, best practices to preserve limbs are not universally available. Goodney et al12 studied variation in the United States in the use of lower-extremity vascular procedures for critical limb ischemia. They defined “low-intensity” to “high-intensity” regions of the country depending on the proportion of patients who underwent a vascular procedure in the year before amputation. They found considerable variation, but even in the region of highest intensity, more than 40% of patients did not have a vascular procedure in the year before amputation.
Similarly, Jones et al13 mapped amputation rates by US state and found significant variation even after adjusting for risk factors such as tobacco use and obesity.
Controversy surrounds the specifics of revascularization treatment, as in many fields in vascular medicine. However, most experts agree that improved perfusion is the goal.
The Trans-Atlantic Inter-Society Consensus for the Management of Peripheral Artery Disease recommends revascularization as the best treatment for patients with critical limb ischemia.8 In addition, the American College of Cardiology and American Heart Association Guidelines for the Management of Patients With Peripheral Arterial Disease (Lower Extremity, Renal, Mesenteric, and Abdominal Aortic) state that the tibial or pedal artery that is capable of providing continuous and uncompromised outflow to the foot should be used as the site of distal anastomosis.14 These guidelines do not yet mention endovascular therapy.
Angiosomes guide revascularization
In the past few years, the ability to facilitate healing of foot ulcers has improved. Angiosomes—regions of vascularization supplied by specific arteries—can be mapped on the skin, similar to the way dermatomes are mapped for neural innervation (Figure 1). The foot and lower leg region has six angiosomes perfused by three arteries that branch off the popliteal artery after it passes behind the knee:
- The anterior tibial artery supplies the dorsum of the foot and the front of the lower limb.
- The posterior tibial artery supplies the plantar surface of the foot via three branches—the medial plantar, lateral plantar, and calcaneal branches.
- The peroneal artery supplies the lateral part of the foot with collaterals to the anterior and posterior tibial arteries if they are compromised.
Studies have compared angiosome-based treatment vs revascularizing the best available artery (thus depending on collateral flow to compensate to surrounding areas). They have found that regardless of whether an endovascular or bypass method of revascularization was used, an angiosome-based approach led to significantly higher amputation-free survival rates.15–17
Patients typically do not have blockage of only a single tibial artery. Graziani et al18 assessed the vascular lesions in 417 patients with critical limb ischemia and found that multiple below-knee arteries were frequently involved. This makes it difficult to decide where to target revascularization efforts, and the angiosome concept helps with that.
ASSESSING WOUND PERFUSION
Ankle- and toe-brachial indices assess perfusion
The ankle-brachial index19 is a good superficial assessment of perfusion. Multiple epidemiologic studies have shown the prognostic value of the ankle brachial index beyond the traditional risk factors and even the Framingham risk score.19 Values:
- Normal 1.1–1.30 (> 1.31 is abnormal and consistent with calcified vessels, and is an unreliable measure)
- Low normal 0.91–1.00
- Mild disease 0.71–0.90
- Moderate disease 0.41–0.70
- Severe disease ≤ 0.40.
However, the ankle-brachial index assesses perfusion only to the ankle, and many patients have ulcers in the toes and distal foot. The toe-brachial index must be specifically ordered in most institutions (if the first toe has an ulcer, the second toe should be assessed). The toe-brachial index is also important if the ankle-brachial index cannot be obtained because of calcified, noncompressible arteries in the ankle. A normal toe-brachial index is greater than 0.7.
The segmental blood pressure examination compares blood pressure measurements at multiple sites in the lower extremity. A drop of more than 20 mm Hg between segments indicates obstruction at that location. The test is simple and noninvasive and often can replace computed tomography.20
Transcutaneous oximetry
Transcutaneous oximetry measures the Tcpo2 from 1 to 2 mm deep in the skin from local capillaries. Measured adjacent to an ulcer, it is useful to predict wound healing and to assess the response to hyperbaric oxygen therapy.21 The values are:
- Normal > 70 mm Hg
- Impaired wound healing < 40 mm Hg
- Critical limb ischemia < 30 mm Hg.
Although most agree that a Tcpo2 below 40 mm Hg requires revascularization, low values can arise from many causes other than peripheral artery disease, including high altitude, pulmonary disease, heart failure, edema, inflammation, callus, and skin diseases such as scleroderma.
Skin perfusion pressure better predicts healing
Skin perfusion pressure is a measure of the capillary opening pressure after occlusion and is another way to assess perfusion. This test is not routinely done and must be specially requested.
The test is performed by inflating a blood pressure cuff on the leg until blood flow is occluded, then using laser Doppler to determine reactive hyperemia, ie, the gradual return of blood flow during controlled pressure release. The pressure at which movement is detected is the skin perfusion pressure.22
The laser Doppler probe emits and detects light scattered in the tissue. Light hitting moving blood cells undergoes a change in frequency, ie, a Doppler shift. An algorithm converts the optical information in the skin perfusion pressure by capturing the onset of capillary flow return and determining the pressure at which flow returns. Categories of results:
- > 50 mm Hg—normal
- 40–50 mm Hg—mild ischemia (wound healing probable)
- 30–40 mm Hg—moderate ischemia (wound healing uncertain)
- < 30 mm Hg—critical limb ischemia (wound healing unlikely).
Skin perfusion pressure testing has the advantages of not being affected by vessel calcification, thickened skin, or edema. It can be used on the plantar aspect of the foot and on digits. Recent small studies indicate that it is more sensitive for predicting wound healing than Tcpo2 measures.
On the other hand, skin perfusion pressure testing is not useful for predicting response to hyperbaric oxygen therapy. Also, blood flow occlusion by the cuff may be painful.
Intraoperative fluorescence angiography
Intraoperative fluorescence angiography is used to assess flap viability during reconstructive surgery and is being studied to determine its usefulness for assessing tissue viability in limb ischemia.
The test provides real-time assessment of capillary perfusion, determining surface tissue viability. The imaging head contains a digital camera, a laser light source, and a distance sensor. The test requires intravenous administration of indocyanine green, which binds to plasma proteins and is cleared through the liver, making it safe for patients with renal dysfunction. It cannot be used in patients with allergies to iodine contrast, penicillin, or sulfa.23
PREVENTION TARGETS CARDIOVASCULAR RISK FACTORS
Preventive measures are the same as for cardiovascular disease, ie, aggressive risk-factor modification: quitting smoking, lowering low-density lipoprotein cholesterol, reducing blood pressure, controlling diabetes, and managing heart failure.
Dual antiplatelet therapy should be instituted with aspirin and clopidogrel (Plavix) in patients undergoing revascularization. One can also consider cilostazol (Pletal); however, the role of this agent in patients with critical limb ischemia is less defined.
BYPASS OR ANGIOPLASTY?
The Bypass Versus Angioplasty in Severe Ischaemia of the Leg (BASIL) trial24 randomly assigned 452 patients with severe limb ischemia due to infrainguinal atherosclerosis to receive either surgery-first or angioplasty-first care and followed them for 5.5 years.
No significant differences between the two groups were found in amputation-free survival, deaths, or health-related quality of life. However, hospital costs associated with the surgery-first strategy were about one-third higher. As expected, more patients in the surgery group developed a wound infection, and more patients in the angioplasty group required bypass surgery at some point.
The conclusion that can be reached from this study is that patients presenting with severe limb ischemia due to infrainguinal atherosclerotic occlusive disease who are suitable for both surgical and interventional procedures can be treated with either method. However, most experts consider endovascular therapy as the first option in many patients. The National Institutes of Health recently funded a study to compare contemporary endovascular therapy vs surgery in patients with critical limb ischemia.
TAKE-HOME POINTS
In the last decade, significant endovascular advances have been made. New devices and techniques have enhanced our ability to treat high-risk patients who have critical limb ischemia. The combination of risk factor modification, accurate diagnosis, and aggressive revascularization should prevent limb loss in many of these patients. For the primary care physician, a low threshold for assessing perfusion in patients with critical limb ischemia is important using a screening ankle-brachial index and toe-brachial index. These patients should promptly be referred to a vascular specialist for further evaluation and treatment.
- Phillips T, Stanton B, Provan A, Lew R. A study of the impact of leg ulcers on quality of life: financial, social, and psychologic implications. J Am Acad Dermatol 1994; 31:49–53.
- Brem H, Kirsner RS, Falanga V. Protocol for the successful treatment of venous ulcers. Am J Surg 2004; 188(1A suppl):1–8.
- Ramsey SD, Newton K, Blough D, et al. Incidence, outcomes, and cost of foot ulcers in patients with diabetes. Diabetes Care 1999; 22:382–387.
- Cuddigan J, Berlowitz DR, Ayello E; National Pressure Ulcer Advisory Panel. Pressure ulcers in America: Prevalence, incidence, and implications for the future: an executive summary of the National Pressure Ulcer Advisory Panel monograph. Adv Skin Wound Care 2001; 14:208–215.
- Olin JW, Beusterien KM, Childs MB, Seavey C, McHugh L, Griffiths RI. Medical costs of treating venous stasis ulcers: evidence from a retrospective cohort study. Vasc Med 1999; 4:1–7.
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26:1790–1795.
- Kumar RN, Gupchup GV, Dodd MA, et al. Direct health care costs of 4 common skin ulcers in New Mexico Medicaid fee-for-service patients. Adv Skin Wound Care 2004; 17:143–149.
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG; TASC II Working Group. Inter-society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg 2007; 45(suppl):S5–S67.
- Rutherford RB, Baker JD, Ernst C, et al. Recommended standards for reports dealing with lower extremity ischemia: revised version. J Vasc Surg 1997; 26:517–538. Erratum in J Vasc Surg 2001; 33:805.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304. Erratum in N Engl J Med 2010; 363:1092.
- Dormandy JA, Rutherford RB. Management of peripheral arterial disease (PAD). TASC Working Group. TransAtlantic Inter-Society Consensus (TASC). J Vasc Surg 2000; 31:S1–S296.
- Goodney PP, Travis LL, Nallamothu BK, et al. Variation in the use of lower extremity vascular procedures for critical limb ischemia. Circ Cardiovasc Qual Outcomes 2012; 5:94–102.
- Jones WS, Patel MR, Dai D, et al. Temporal trends and geographic variation of lower-extremity amputation in patients with peripheral artery disease: results from U.S. Medicare 2000–2008. J Am Coll Cardiol 2012; 60:2230–2236.
- Hirsch AT, Haskal ZJ, Mertzer NR, et al. ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Alexandrescu VA, Hubermont G, Philips Y, et al. Selective primary angioplasty following an angiosome model of reperfusion in the treatment of Wagner 1–4 diabetic foot lesions: practice in a multidisciplinary diabetic limb service. J Endovasc Ther 2008; 15:580–593.
- Neville RF, Attinger CE, Bulan EJ, Ducic I, Thomassen M, Sidawy AN. Revascularization of a specific angiosome for limb salvage: does the target artery matter? Ann Vasc Surg 2009; 23:367–373.
- Iida O, Soga Y, Hirano K, et al. Long-term results of direct and indirect endovascular revascularization based on the angiosome concept in patients with critical limb ischemia presenting with isolated below-the-knee lesions. J Vasc Surg 2012; 55:363–370.
- Graziani L, Silvestro A, Bertone V, et al. Vascular involvement in diabetic subjects with ischemic foot ulcer: a new morphologic categorization of disease severity. Eur J Vasc Endovasc Surg 2007; 33:453–460.
- Newman AB, Siscovick DS, Manolio TA, et al., Cardiovascular Heart Study (CHS) Collaborative Research Group. Ankle-arm index as a marker of atherosclerosis in the Cardiovascular Health Study. Circulation 1993; 88:837–845.
- Cronenwett JL, Johnston KW. Rutherford’s Vascular Surgery. 7th ed. Philadelphia, PA: Saunders Elsevier; 2010.
- Fife CE, Smart DR, Sheffield PJ, Hopf HW, Hawkins G, Clarke D. Transcutaneous oximetry in clinical practice: consensus statements from an expert panel based on evidence. Undersea Hyperb Med 2009; 36:43–53.
- Lo T, Sample R, Moore P, Gold P. Prediction of wound healing outcome using skin perfusion pressure and transcutaneous oximetry. Wounds 2009; 21:310–316.
- Perry D, Bharara M, Armstrong DG, Mills J. Intraoperative fluorescence vascular angiography: during tibial bypass. J Diabetes Sci Technol 2012; 6:204–208.
- Adam DJ, Beard JD, Cleveland T, et al; BASIL trial participants. Bypass versus angioplasty in severe ischaemia of the leg (BASIL): multicentre, randomised controlled trial. Lancet 2005; 366:1925–1934.
- Phillips T, Stanton B, Provan A, Lew R. A study of the impact of leg ulcers on quality of life: financial, social, and psychologic implications. J Am Acad Dermatol 1994; 31:49–53.
- Brem H, Kirsner RS, Falanga V. Protocol for the successful treatment of venous ulcers. Am J Surg 2004; 188(1A suppl):1–8.
- Ramsey SD, Newton K, Blough D, et al. Incidence, outcomes, and cost of foot ulcers in patients with diabetes. Diabetes Care 1999; 22:382–387.
- Cuddigan J, Berlowitz DR, Ayello E; National Pressure Ulcer Advisory Panel. Pressure ulcers in America: Prevalence, incidence, and implications for the future: an executive summary of the National Pressure Ulcer Advisory Panel monograph. Adv Skin Wound Care 2001; 14:208–215.
- Olin JW, Beusterien KM, Childs MB, Seavey C, McHugh L, Griffiths RI. Medical costs of treating venous stasis ulcers: evidence from a retrospective cohort study. Vasc Med 1999; 4:1–7.
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26:1790–1795.
- Kumar RN, Gupchup GV, Dodd MA, et al. Direct health care costs of 4 common skin ulcers in New Mexico Medicaid fee-for-service patients. Adv Skin Wound Care 2004; 17:143–149.
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG; TASC II Working Group. Inter-society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg 2007; 45(suppl):S5–S67.
- Rutherford RB, Baker JD, Ernst C, et al. Recommended standards for reports dealing with lower extremity ischemia: revised version. J Vasc Surg 1997; 26:517–538. Erratum in J Vasc Surg 2001; 33:805.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304. Erratum in N Engl J Med 2010; 363:1092.
- Dormandy JA, Rutherford RB. Management of peripheral arterial disease (PAD). TASC Working Group. TransAtlantic Inter-Society Consensus (TASC). J Vasc Surg 2000; 31:S1–S296.
- Goodney PP, Travis LL, Nallamothu BK, et al. Variation in the use of lower extremity vascular procedures for critical limb ischemia. Circ Cardiovasc Qual Outcomes 2012; 5:94–102.
- Jones WS, Patel MR, Dai D, et al. Temporal trends and geographic variation of lower-extremity amputation in patients with peripheral artery disease: results from U.S. Medicare 2000–2008. J Am Coll Cardiol 2012; 60:2230–2236.
- Hirsch AT, Haskal ZJ, Mertzer NR, et al. ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Alexandrescu VA, Hubermont G, Philips Y, et al. Selective primary angioplasty following an angiosome model of reperfusion in the treatment of Wagner 1–4 diabetic foot lesions: practice in a multidisciplinary diabetic limb service. J Endovasc Ther 2008; 15:580–593.
- Neville RF, Attinger CE, Bulan EJ, Ducic I, Thomassen M, Sidawy AN. Revascularization of a specific angiosome for limb salvage: does the target artery matter? Ann Vasc Surg 2009; 23:367–373.
- Iida O, Soga Y, Hirano K, et al. Long-term results of direct and indirect endovascular revascularization based on the angiosome concept in patients with critical limb ischemia presenting with isolated below-the-knee lesions. J Vasc Surg 2012; 55:363–370.
- Graziani L, Silvestro A, Bertone V, et al. Vascular involvement in diabetic subjects with ischemic foot ulcer: a new morphologic categorization of disease severity. Eur J Vasc Endovasc Surg 2007; 33:453–460.
- Newman AB, Siscovick DS, Manolio TA, et al., Cardiovascular Heart Study (CHS) Collaborative Research Group. Ankle-arm index as a marker of atherosclerosis in the Cardiovascular Health Study. Circulation 1993; 88:837–845.
- Cronenwett JL, Johnston KW. Rutherford’s Vascular Surgery. 7th ed. Philadelphia, PA: Saunders Elsevier; 2010.
- Fife CE, Smart DR, Sheffield PJ, Hopf HW, Hawkins G, Clarke D. Transcutaneous oximetry in clinical practice: consensus statements from an expert panel based on evidence. Undersea Hyperb Med 2009; 36:43–53.
- Lo T, Sample R, Moore P, Gold P. Prediction of wound healing outcome using skin perfusion pressure and transcutaneous oximetry. Wounds 2009; 21:310–316.
- Perry D, Bharara M, Armstrong DG, Mills J. Intraoperative fluorescence vascular angiography: during tibial bypass. J Diabetes Sci Technol 2012; 6:204–208.
- Adam DJ, Beard JD, Cleveland T, et al; BASIL trial participants. Bypass versus angioplasty in severe ischaemia of the leg (BASIL): multicentre, randomised controlled trial. Lancet 2005; 366:1925–1934.
KEY POINTS
- In assessing peripheral artery disease, perform a thorough history and physical examination, paying close attention to the onset and characteristics of pain, activity level, history, and pulses, and the condition of the feet.
- Acute limb ischemia is a sudden decrease in limb perfusion, potentially threatening limb viability. Patients who have acute cessation of blood flow, sensation, or motor function need immediate revascularization to avoid amputation.
- Critical limb ischemia ranges from rest pain to gangrene and must be addressed with a multidisciplinary approach.
- The ankle-brachial index is a noninvasive, inexpensive test that can be done in the office with a hand-held Doppler device to assess the presence and severity of peripheral artery disease.
Managing severe acute pancreatitis
Severe acute pancreatitis has been known since the time of Rembrandt, with Nicolaes Tulp—the physician credited as first describing it—immortalized in the famous painting, The Anatomy Lesson. However, progress in managing this disease has been disappointing. Treatment is mainly supportive, and we lack any true disease-modifying therapy. But we are learning to recognize the disease and treat it supportively better than in the past.
The early hours of severe acute pancreatitis are critical for instituting appropriate intervention. Prompt fluid resuscitation is key to preventing immediate and later morbidity and death. This article focuses on identifying and managing the most severe form of acute pancreatitis—necrotizing disease—and its complications.
NECROTIZING DISEASE ACCOUNTS FOR MOST PANCREATITIS DEATHS
The classification and definitions of acute pancreatitis were recently revised from the 1992 Atlanta system and published early in 2013.1 In addition, the American Pancreatic Association and the International Association of Pancreatology met in 2012 to develop evidence-based guidelines on managing severe pancreatitis.
An estimated 210,000 new cases of acute pancreatitis occur each year in the United States. About 20% of cases of severe acute pancreatitis are necrotizing disease, which accounts for nearly all the morbidity and death associated with acute pancreatitis.
The clinical spectrum of acute pancreatitis ranges from mild to life-threatening, reflecting interstitial (death rate < 1%) to necrotizing histology (the latter associated with a 25% risk of death if the pancreatitis becomes infected and a 10% risk if it is sterile). When death occurs early in the disease course, it tends to be from multiorgan failure; when death occurs later in the course, it tends to be from infection. Appropriate early treatment may prevent death in both categories.
DIAGNOSING ACUTE PANCREATITIS AND PREDICTING ITS SEVERITY
The diagnosis of acute pancreatitis requires two of the following three criteria:
- Clinical presentation—epigastric pain, nausea, vomiting
- Biochemical—amylase level more than three times the upper limit of normal, or lipase more than three times the upper limit of normal
- Evidence from computed tomography (CT), ultrasonography, or magnetic resonance imaging.
Although the biochemical criteria are variably sensitive for detecting acute pancreatitis (55%–100%), the specificity is very high (93% to 99%).
Recently, urinary trypsinogen-2, measured by dipstick, has also been used to aid diagnosis. It has a reasonable sensitivity (53%–96%) and specificity (85%) if positive (> 50 ng/mL).
Speed is critical
Over the years, many clinical prediction rules have been used for predicting the severity of acute pancreatitis. The Ranson criteria,2 from 1974, and the Acute Physiology and Chronic Health Evaluation (APACHE) II system3 are cumbersome and require waiting up to 48 hours after the onset of acute pancreatitis to obtain a complete score. The Imrie-Glasgow score is another predictor.
The systemic inflammatory response syndrome (SIRS) is currently the most important indicator of prognosis.4 Originally adopted for predicting the development of organ failure with sepsis, it requires at least two of the following criteria:
- Heart rate > 90 beats/min
- Core temperature < 36°C or > 38°C
- White blood cells < 4,000 or > 12,000/mm3
- Respirations > 20/min.
The advantages of this system are that it identifies risk very early in the course of the disease and can be assessed quickly in the emergency department.
The Bedside Index for Severity of Acute Pancreatitis (BISAP) score is another simple, easy-to-perform prognostic index,5,6 calculated by assigning 1 point for each of the following if present within the first 24 hours of presentation:
- Blood urea nitrogen > 25 mg/dL
- Abnormal mental status (Glasgow coma score < 15)
- Evidence of systemic inflammatory response syndrome
- Age > 60 years
- Pleural effusion seen on imaging study.
A score of 3 points is associated with a 5.3% rate of hospital death, 4 points with 12.7%, and 5 points with 22.5%.
At its most basic, severe acute pancreatitis is defined by organ failure (at least one organ from the respiratory, renal, or cardiovascular system) lasting for more than 48 hours. Failure for each organ is defined by the Marshall scoring system.1
EARLY MANAGEMENT IS KEY TO OUTCOME
The window of opportunity to make a significant difference in outcome is within the first 12 to 24 hours of presentation. Volume resuscitation is the cornerstone of early management. By the time of presentation for severe acute pancreatitis, the pancreas is already necrotic, so the aim is to minimize the systemic inflammatory response syndrome with the goals of reducing rates of organ failure, morbidity, and death. Necrotizing pancreatitis is essentially an ischemic event, and the goal of volume resuscitation is to maintain pancreatic and intestinal microcirculation to prevent intestinal ischemia and subsequent bacterial translocation.7
Early resuscitation with lactated Ringer’s solution recommended
The evidence supporting a specific protocol for fluid resuscitation in severe acute pancreatitis is not strong, but a few studies provide guidance.
Wu et al8 randomized 40 patients with acute pancreatitis to one of four arms: “goal-directed fluid resuscitation” with either lactated Ringer’s solution or normal saline, or standard therapy (by physician discretion) with either lactated Ringer’s solution or normal saline. Goal-directed therapy involved a bolus of 20 mL/kg given over 30 to 45 minutes at presentation followed by infusion with rates dependent on an algorithm based on change in blood urea nitrogen level at set times. Patients receiving either goal-directed or standard therapy had significantly lower rates of systemic inflammatory response syndrome at 24 hours than at admission. Most striking was that treatment with lactated Ringer’s solution was associated with dramatically improved rates, whereas normal saline showed no improvement.
In a retrospective study of patients with acute pancreatitis, Warndorf et al9 identified 340 patients who received early resuscitation (more than one-third of the total 72-hour fluid volume within 24 hours of presentation) and 90 patients who received late resuscitation (less than one-third of the total 72-hour fluid volume within 24 hours of presentation). Patients who received early resuscitation developed less systemic inflammatory response syndrome and organ failure, and required fewer interventions.
Monitoring for optimum fluid resuscitation
Fluid resuscitation should be carefully managed to avoid administering either inadequate or excessive amounts of fluid. Inadequate fluid resuscitation can result in renal failure, progression of necrosis, and possibly infectious complications. Excessive resuscitation—defined as more than 4 L in the first 24 hours—is associated with respiratory failure, pancreatic fluid collections, and abdominal compartment syndrome.
Optimum resuscitation is controlled fluid expansion averaging 5 to 10 mL/kg per hour, with 2,500 to 4,000 mL given in the first 24 hours.
Adequate volume resuscitation can be evaluated clinically with the following goals:
- Heart rate < 120 beats per minute
- Mean arterial pressure 65–85 mm Hg
- Urinary output > 1 mL/kg per hour
- Hematocrit 35%–44%.
EARLY CT IS JUSTIFIED ONLY IF DIAGNOSIS IS UNCLEAR
The normal pancreas takes up contrast in the same way as do the liver and spleen, so its enhancement on CT is similar. If there is interstitial pancreatitis, CT shows the pancreas with normal contrast uptake, but the organ appears “boggy” with indistinct outlines. With necrotizing pancreatitis, only small areas of tissue with normal contrast may be apparent.
Peripancreatic fat necrosis may also be visible on CT. Obese patients tend to have a worse clinical course of necrotizing pancreatitis, probably because of the associated peripancreatic fat that is incorporated into the pancreatic necrosis.
For clear-cut cases of acute pancreatitis, time is wasted waiting to obtain CT images, and this could delay fluid resuscitation. Results from immediate CT almost never change the clinical management during the first week of acute pancreatitis, and obtaining CT images is usually not recommended if the diagnosis of acute pancreatitis is clear. CT’s sensitivity for detecting necrosis is only 70% in the first 48 hours of presentation, so it is easy to be fooled by a false-negative scan: frequently, a scan does not show necrotizing pancreatitis until after 72 hours. In addition, evidence from animal studies indicates that contrast agents might worsen pancreatic necrosis.
Immediate CT is justified if the diagnosis is in doubt at presentation, such as to evaluate for other intra-abdominal conditions such as intestinal ischemia or a perforated duodenal ulcer.
Contrast-enhanced CT is recommended 72 to 96 hours after presentation, or earlier if the patient is worsening despite treatment. Specific CT protocols will be included in new management guidelines, expected to be published soon.
PREVENTING INFECTIOUS COMPLICATIONS
Risk of infection is associated with the degree of pancreatic necrosis. Patients with less than 30% necrosis have a 22.5% chance of infection, whereas those with more than 50% necrosis have a 46.5% risk of infection.10
Infection can develop from a variety of sources:
Bacterial translocation from the colon and small bowel is thought to be one of the major sources of infection in necrotic pancreatitis. Volume resuscitation and maintaining gut integrity with early enteral nutrition are believed to minimize the risk of bacterial translocation.
Hematogenous spread of bacteria is another suspected source of infection into the pancreas. Again, enteral nutrition also reduces the risk by minimizing the need for central catheters.
Biliary sources may also play a role. Bile duct stones or gall bladder infection can lead to infected pancreatic necrosis.
ANTIBIOTICS NOT ROUTINELY RECOMMENDED
Treating acute pancreatitis with antibiotics has fallen in and out of favor over the past decades. From being standard practice in the 1970s, it dropped off in the 1980s and 1990s and then became more common again.
Current recommendations from the American Pancreatic Association and the International Association of Pancreatology are not to routinely use intravenous antibiotics to prevent infection in necrotizing pancreatitis because of lack of evidence that it changes overall outcome. Antibiotic usage may be associated with more bacterial resistance and the introduction of fungal infections into the pancreas.
Selective gut decontamination, involving oral and rectal administration of neomycin and other antibiotics, was shown in a single randomized trial to reduce the incidence of infection, but it is very cumbersome and is not recommended for acute pancreatitis.
Treatment with probiotics is also not recommended and was shown in one study to lead to a worse outcome.11
ENTERAL BETTER THAN TOTAL PARENTERAL NUTRITION
Enteral tube feeding with either an elemental diet or a polymeric enteral formulation is the first-line therapy for necrotizing pancreatitis. Compared with total parenteral nutrition, it reduces infection, organ failure, hospital length of stay, the need for surgical intervention, and the risk of death. Total parenteral nutrition should be considered only for patients who do not tolerate enteral feeding because of severe ileus.
Conventional thinking for many years was to provide enteral feeding with a tube passed beyond the ligament of Treitz, thinking that it reduced stimulation to the pancreas. However, recent studies indicate that nasogastric feeding is equivalent to nasojejunal feeding in terms of nutrition, maintaining gut integrity, and outcome.
INTRA-ABDOMINAL HYPERTENSION AND ABDOMINAL COMPARTMENT SYNDROME
Movement of fluid into the intracellular space (“third-spacing”) occurs in acute pancreatitis and is exacerbated by fluid resuscitation. Intra-abdominal hypertension is associated with poor outcomes in patients with severe acute pancreatitis. Especially for patients with severe pancreatitis who are on mechanical ventilation, pressure should be monitored with transvesicular bladder measurements.
Intra-abdominal hypertension is defined as a sustained intra-abdominal pressure of more than 12 mm Hg, with the following grades:
- Grade 1: 12–15 mm Hg
- Grade 2: 16–20 mm Hg
- Grade 3: 21–25 mm Hg
- Grade 4: > 25 mm Hg.
Abdominal compartment syndrome is defined as a sustained intra-abdominal pressure of more than 20 mm Hg. It is associated with new organ dysfunction or failure. It should first be managed with ultrafiltration or diuretics to try to reduce the amount of fluid in the abdomen. Lumenal decompression can be tried with nasogastric or rectal tubes for the stomach and bowels. Ascites or retroperitoneal fluid can be drained percutaneously. In addition, analgesia and sedation to reduce abdominal muscle tone can help the patient become better ventilated. Neuromuscular blockade can also relax the abdomen.
Open abdominal decompression is the treatment of last resort to relieve abdominal compartment syndrome. The abdominal wall is not closed surgically but is allowed to heal by secondary intention (it “granulates in”).12
IDENTIFYING INFECTION
Fine-needle aspiration if clinical and imaging signs are not clear
Untreated infected pancreatitis is associated with a much higher risk of death than sterile pancreatic necrosis. Unfortunately, it can be difficult to determine if a patient with necrotizing pancreatitis has an infection because fever, tachycardia, and leukocytosis are usually present regardless. It is important to determine because mechanically intervening for sterile necrosis does not improve outcome.
Fine-needle aspiration, either guided by CT or done at the bedside with ultrasonography, with evaluation with Gram stain and culture, was widely used in the 1990s in cases of necrotizing pancreatitis to determine if infection was present. There has been a shift away from this because, although it can confirm the presence of infection, the false-negative rate is 15%. Clinical and imaging signs can be relied on in most cases to determine the presence of infection, and it is now recognized that fineneedle aspiration should be used only for select cases. Clinical studies have not shown that fine-needle aspiration improves outcomes.
Clinical scenarios typical of infected pancreatic necrosis include patients who have obvious signs of infection with no identifiable source, such as those who stabilize after acute severe acute pancreatitis, and then 10 to 14 days later become worse, with a dramatically higher white blood cell count and tachycardia. Such a patient likely needs an intervention regardless of the results of fine-needle aspiration.
On the other hand, a patient with a continually up-and-down course that never stabilizes over 3 weeks, with no identifiable source of infection, and with no peripancreatic gas apparent on imaging would be a good candidate for fine-needle aspiration.
If peripancreatic gas is seen on imaging, fine-needle aspiration is unnecessary. Peripancreatic gas is traditionally attributed to gasforming bacteria within the pancreas, but in my experience, it is usually from a fistula from the necrosis to the duodenum or the colon, the fistula being caused as the necrosis erodes at the hepatic flexure, the transverse colon, or the splenic flexure.
MECHANICAL INTERVENTIONS FOR INFECTIVE NECROSIS
Late, minimally invasive procedures preferred
Conventional management has shifted away from removing the necrosis with early surgical debridement of the pancreas. Experience with myocardial infarction shows that it is not necessary to remove a sterile necrotic organ, and studies with sterile pancreatic necrosis have found that surgical intervention is associated with a higher risk of death than medical management.
Documented infection has traditionally been considered a definite indication for debridement, but even that is being called into question as more studies are emerging of infected necrosis treated successfully with antibiotics alone.
Sterile necrosis with a fulminant course is a controversial indication for surgery. It was traditionally felt that surgery was worth trying for such patients, but this is no longer common practice.
For cases in which debridement was deemed advisable, surgery was done more frequently in the past. Now, a minimally invasive approach such as with endoscopy or percutaneous catheter is also used. Waiting until at least 4 weeks after the onset of acute pancreatitis is associated with a better outcome than intervening early.
WALLED-OFF NECROSIS
Watchful waiting or minimally invasive intervention
Patients who survive multiorgan failure but are still ill more than 4 weeks after the onset of pancreatitis should be suspected of having walled-off necrosis, formerly referred to as a pancreatic phlegmon. This term was abandoned after the 1992 Atlanta symposium.13 In the mid to late 1990s, the process was referred to as organized pancreatic necrosis. It is characterized by a mature, encapsulated collection of pancreatic or peripancreatic necrosis that contains variable amounts of amylase-rich fluid from pancreatic duct disruption.
Walled-off pancreatic necrosis (WOPN) is often confused with pancreatic pseudocyst; these may appear similar on CT, and higherdensity solid debris may be visible in walled-off necrosis within an otherwise homogenous-appearing collection. Magnetic resonance imaging defines liquid and solid much better than CT.
The best way to distinguish WOPN from pseudocyst is by clinical history: a patient with a preceding history of clinically severe acute pancreatitis almost always has necrotizing pancreatitis that evolves to walled-off necrosis, usually over 3 to 4 weeks.
Endoscopic removal and other minimally invasive approaches, such as aggressive percutaneous interventions, have replaced open necrosectomy for treatment, which was associated with high morbidity and mortality rates.14–16
Intervening for sterile walled-off necrosis is still a controversial topic: although systemically ill, the patient is no longer having life-threatening consequences, and watchful waiting might be just as expedient as intervention. Evidence to support either view is lacking. Most experts believe that intervention should be done if the patient has gastric outlet obstruction and intractable pain and is unable to eat 4 to 6 weeks after the onset of pancreatitis with WOPN. Infected WOPN is considered an indication for drainage.
- Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62:102–111.
- Ranson JH, Rifkind KM, Roses DF, Fink SD, Eng K, Spencer FC. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974; 139:69–81.
- Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med 1985; 13:818–829.
- American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 1992; 20:864–874.
- Wu BU, Johannes RS, Sun X, Tabak Y, Conwell DL, Banks PA. The early prediction of mortality in acute pancreatitis: a large population-based study. Gut 2008; 57:1698–1703.
- Singh VK, Wu BU, Bollen TL, et al. A prospective evaluation of the bedside index for severity in acute pancreatitis score in assessing mortality and intermediate markers of severity in acute pancreatitis. Am J Gastroenterol 2009; 104:966–971.
- Fisher JM, Gardner TB. The “golden hours” of management in acute pancreatitis. Am J Gastroenterol 2012; 107:1146–1150.
- Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol 2011; 9:710–717.
- Warndorf MG, Kurtzman JT, Bartel MJ, et al. Early fluid resuscitation reduces morbidity among patients with acute pancreatitis. Clin Gastroenterol Hepatol 2011; 9:705–709.
- Beger HG, Rau BM. Severe acute pancreatitis: clinical course and management. World J Gastroenterol 2007; 13:5043–5051.
- Besselink MG, van Santvoort HC, Buskens E, et al; Dutch Acute Pancreatitis Study Group. Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet 2008; 371:651–659.
- Fitzgerald JE, Gupta S, Masterson S, Sigurdsson HH. Laparostomy management using the ABThera open abdomen negative pressure therapy system in a grade IV open abdomen secondary to acute pancreatitis. Int Wound J 2012. doi: 1111/j.1742-481X2012.00953.x. [epub ahead of print]
- Bradley EL. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11–13, 1992. Arch Surg 1993; 128:586–590.
- Baron TH, Thaggard WG, Morgan DE, Stanley RJ. Endoscopic therapy for organized pancreatic necrosis. Gastroenterology 1996; 111:755–764.
- van Santvoort HC, Besselink MG, Bakker OJ, et al; Dutch Pancreatitis Study Group. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med 2010; 362:1491–1502.
- Bakker OJ, van Santvoort HC, van Brunschot S, et al; Dutch Pancreatitis Study Group. Endoscopic transgastric vs surgical necrosectomy for infected necrotizing pancreatitis: a randomized trial. JAMA 2012; 307:1053–1061.
Severe acute pancreatitis has been known since the time of Rembrandt, with Nicolaes Tulp—the physician credited as first describing it—immortalized in the famous painting, The Anatomy Lesson. However, progress in managing this disease has been disappointing. Treatment is mainly supportive, and we lack any true disease-modifying therapy. But we are learning to recognize the disease and treat it supportively better than in the past.
The early hours of severe acute pancreatitis are critical for instituting appropriate intervention. Prompt fluid resuscitation is key to preventing immediate and later morbidity and death. This article focuses on identifying and managing the most severe form of acute pancreatitis—necrotizing disease—and its complications.
NECROTIZING DISEASE ACCOUNTS FOR MOST PANCREATITIS DEATHS
The classification and definitions of acute pancreatitis were recently revised from the 1992 Atlanta system and published early in 2013.1 In addition, the American Pancreatic Association and the International Association of Pancreatology met in 2012 to develop evidence-based guidelines on managing severe pancreatitis.
An estimated 210,000 new cases of acute pancreatitis occur each year in the United States. About 20% of cases of severe acute pancreatitis are necrotizing disease, which accounts for nearly all the morbidity and death associated with acute pancreatitis.
The clinical spectrum of acute pancreatitis ranges from mild to life-threatening, reflecting interstitial (death rate < 1%) to necrotizing histology (the latter associated with a 25% risk of death if the pancreatitis becomes infected and a 10% risk if it is sterile). When death occurs early in the disease course, it tends to be from multiorgan failure; when death occurs later in the course, it tends to be from infection. Appropriate early treatment may prevent death in both categories.
DIAGNOSING ACUTE PANCREATITIS AND PREDICTING ITS SEVERITY
The diagnosis of acute pancreatitis requires two of the following three criteria:
- Clinical presentation—epigastric pain, nausea, vomiting
- Biochemical—amylase level more than three times the upper limit of normal, or lipase more than three times the upper limit of normal
- Evidence from computed tomography (CT), ultrasonography, or magnetic resonance imaging.
Although the biochemical criteria are variably sensitive for detecting acute pancreatitis (55%–100%), the specificity is very high (93% to 99%).
Recently, urinary trypsinogen-2, measured by dipstick, has also been used to aid diagnosis. It has a reasonable sensitivity (53%–96%) and specificity (85%) if positive (> 50 ng/mL).
Speed is critical
Over the years, many clinical prediction rules have been used for predicting the severity of acute pancreatitis. The Ranson criteria,2 from 1974, and the Acute Physiology and Chronic Health Evaluation (APACHE) II system3 are cumbersome and require waiting up to 48 hours after the onset of acute pancreatitis to obtain a complete score. The Imrie-Glasgow score is another predictor.
The systemic inflammatory response syndrome (SIRS) is currently the most important indicator of prognosis.4 Originally adopted for predicting the development of organ failure with sepsis, it requires at least two of the following criteria:
- Heart rate > 90 beats/min
- Core temperature < 36°C or > 38°C
- White blood cells < 4,000 or > 12,000/mm3
- Respirations > 20/min.
The advantages of this system are that it identifies risk very early in the course of the disease and can be assessed quickly in the emergency department.
The Bedside Index for Severity of Acute Pancreatitis (BISAP) score is another simple, easy-to-perform prognostic index,5,6 calculated by assigning 1 point for each of the following if present within the first 24 hours of presentation:
- Blood urea nitrogen > 25 mg/dL
- Abnormal mental status (Glasgow coma score < 15)
- Evidence of systemic inflammatory response syndrome
- Age > 60 years
- Pleural effusion seen on imaging study.
A score of 3 points is associated with a 5.3% rate of hospital death, 4 points with 12.7%, and 5 points with 22.5%.
At its most basic, severe acute pancreatitis is defined by organ failure (at least one organ from the respiratory, renal, or cardiovascular system) lasting for more than 48 hours. Failure for each organ is defined by the Marshall scoring system.1
EARLY MANAGEMENT IS KEY TO OUTCOME
The window of opportunity to make a significant difference in outcome is within the first 12 to 24 hours of presentation. Volume resuscitation is the cornerstone of early management. By the time of presentation for severe acute pancreatitis, the pancreas is already necrotic, so the aim is to minimize the systemic inflammatory response syndrome with the goals of reducing rates of organ failure, morbidity, and death. Necrotizing pancreatitis is essentially an ischemic event, and the goal of volume resuscitation is to maintain pancreatic and intestinal microcirculation to prevent intestinal ischemia and subsequent bacterial translocation.7
Early resuscitation with lactated Ringer’s solution recommended
The evidence supporting a specific protocol for fluid resuscitation in severe acute pancreatitis is not strong, but a few studies provide guidance.
Wu et al8 randomized 40 patients with acute pancreatitis to one of four arms: “goal-directed fluid resuscitation” with either lactated Ringer’s solution or normal saline, or standard therapy (by physician discretion) with either lactated Ringer’s solution or normal saline. Goal-directed therapy involved a bolus of 20 mL/kg given over 30 to 45 minutes at presentation followed by infusion with rates dependent on an algorithm based on change in blood urea nitrogen level at set times. Patients receiving either goal-directed or standard therapy had significantly lower rates of systemic inflammatory response syndrome at 24 hours than at admission. Most striking was that treatment with lactated Ringer’s solution was associated with dramatically improved rates, whereas normal saline showed no improvement.
In a retrospective study of patients with acute pancreatitis, Warndorf et al9 identified 340 patients who received early resuscitation (more than one-third of the total 72-hour fluid volume within 24 hours of presentation) and 90 patients who received late resuscitation (less than one-third of the total 72-hour fluid volume within 24 hours of presentation). Patients who received early resuscitation developed less systemic inflammatory response syndrome and organ failure, and required fewer interventions.
Monitoring for optimum fluid resuscitation
Fluid resuscitation should be carefully managed to avoid administering either inadequate or excessive amounts of fluid. Inadequate fluid resuscitation can result in renal failure, progression of necrosis, and possibly infectious complications. Excessive resuscitation—defined as more than 4 L in the first 24 hours—is associated with respiratory failure, pancreatic fluid collections, and abdominal compartment syndrome.
Optimum resuscitation is controlled fluid expansion averaging 5 to 10 mL/kg per hour, with 2,500 to 4,000 mL given in the first 24 hours.
Adequate volume resuscitation can be evaluated clinically with the following goals:
- Heart rate < 120 beats per minute
- Mean arterial pressure 65–85 mm Hg
- Urinary output > 1 mL/kg per hour
- Hematocrit 35%–44%.
EARLY CT IS JUSTIFIED ONLY IF DIAGNOSIS IS UNCLEAR
The normal pancreas takes up contrast in the same way as do the liver and spleen, so its enhancement on CT is similar. If there is interstitial pancreatitis, CT shows the pancreas with normal contrast uptake, but the organ appears “boggy” with indistinct outlines. With necrotizing pancreatitis, only small areas of tissue with normal contrast may be apparent.
Peripancreatic fat necrosis may also be visible on CT. Obese patients tend to have a worse clinical course of necrotizing pancreatitis, probably because of the associated peripancreatic fat that is incorporated into the pancreatic necrosis.
For clear-cut cases of acute pancreatitis, time is wasted waiting to obtain CT images, and this could delay fluid resuscitation. Results from immediate CT almost never change the clinical management during the first week of acute pancreatitis, and obtaining CT images is usually not recommended if the diagnosis of acute pancreatitis is clear. CT’s sensitivity for detecting necrosis is only 70% in the first 48 hours of presentation, so it is easy to be fooled by a false-negative scan: frequently, a scan does not show necrotizing pancreatitis until after 72 hours. In addition, evidence from animal studies indicates that contrast agents might worsen pancreatic necrosis.
Immediate CT is justified if the diagnosis is in doubt at presentation, such as to evaluate for other intra-abdominal conditions such as intestinal ischemia or a perforated duodenal ulcer.
Contrast-enhanced CT is recommended 72 to 96 hours after presentation, or earlier if the patient is worsening despite treatment. Specific CT protocols will be included in new management guidelines, expected to be published soon.
PREVENTING INFECTIOUS COMPLICATIONS
Risk of infection is associated with the degree of pancreatic necrosis. Patients with less than 30% necrosis have a 22.5% chance of infection, whereas those with more than 50% necrosis have a 46.5% risk of infection.10
Infection can develop from a variety of sources:
Bacterial translocation from the colon and small bowel is thought to be one of the major sources of infection in necrotic pancreatitis. Volume resuscitation and maintaining gut integrity with early enteral nutrition are believed to minimize the risk of bacterial translocation.
Hematogenous spread of bacteria is another suspected source of infection into the pancreas. Again, enteral nutrition also reduces the risk by minimizing the need for central catheters.
Biliary sources may also play a role. Bile duct stones or gall bladder infection can lead to infected pancreatic necrosis.
ANTIBIOTICS NOT ROUTINELY RECOMMENDED
Treating acute pancreatitis with antibiotics has fallen in and out of favor over the past decades. From being standard practice in the 1970s, it dropped off in the 1980s and 1990s and then became more common again.
Current recommendations from the American Pancreatic Association and the International Association of Pancreatology are not to routinely use intravenous antibiotics to prevent infection in necrotizing pancreatitis because of lack of evidence that it changes overall outcome. Antibiotic usage may be associated with more bacterial resistance and the introduction of fungal infections into the pancreas.
Selective gut decontamination, involving oral and rectal administration of neomycin and other antibiotics, was shown in a single randomized trial to reduce the incidence of infection, but it is very cumbersome and is not recommended for acute pancreatitis.
Treatment with probiotics is also not recommended and was shown in one study to lead to a worse outcome.11
ENTERAL BETTER THAN TOTAL PARENTERAL NUTRITION
Enteral tube feeding with either an elemental diet or a polymeric enteral formulation is the first-line therapy for necrotizing pancreatitis. Compared with total parenteral nutrition, it reduces infection, organ failure, hospital length of stay, the need for surgical intervention, and the risk of death. Total parenteral nutrition should be considered only for patients who do not tolerate enteral feeding because of severe ileus.
Conventional thinking for many years was to provide enteral feeding with a tube passed beyond the ligament of Treitz, thinking that it reduced stimulation to the pancreas. However, recent studies indicate that nasogastric feeding is equivalent to nasojejunal feeding in terms of nutrition, maintaining gut integrity, and outcome.
INTRA-ABDOMINAL HYPERTENSION AND ABDOMINAL COMPARTMENT SYNDROME
Movement of fluid into the intracellular space (“third-spacing”) occurs in acute pancreatitis and is exacerbated by fluid resuscitation. Intra-abdominal hypertension is associated with poor outcomes in patients with severe acute pancreatitis. Especially for patients with severe pancreatitis who are on mechanical ventilation, pressure should be monitored with transvesicular bladder measurements.
Intra-abdominal hypertension is defined as a sustained intra-abdominal pressure of more than 12 mm Hg, with the following grades:
- Grade 1: 12–15 mm Hg
- Grade 2: 16–20 mm Hg
- Grade 3: 21–25 mm Hg
- Grade 4: > 25 mm Hg.
Abdominal compartment syndrome is defined as a sustained intra-abdominal pressure of more than 20 mm Hg. It is associated with new organ dysfunction or failure. It should first be managed with ultrafiltration or diuretics to try to reduce the amount of fluid in the abdomen. Lumenal decompression can be tried with nasogastric or rectal tubes for the stomach and bowels. Ascites or retroperitoneal fluid can be drained percutaneously. In addition, analgesia and sedation to reduce abdominal muscle tone can help the patient become better ventilated. Neuromuscular blockade can also relax the abdomen.
Open abdominal decompression is the treatment of last resort to relieve abdominal compartment syndrome. The abdominal wall is not closed surgically but is allowed to heal by secondary intention (it “granulates in”).12
IDENTIFYING INFECTION
Fine-needle aspiration if clinical and imaging signs are not clear
Untreated infected pancreatitis is associated with a much higher risk of death than sterile pancreatic necrosis. Unfortunately, it can be difficult to determine if a patient with necrotizing pancreatitis has an infection because fever, tachycardia, and leukocytosis are usually present regardless. It is important to determine because mechanically intervening for sterile necrosis does not improve outcome.
Fine-needle aspiration, either guided by CT or done at the bedside with ultrasonography, with evaluation with Gram stain and culture, was widely used in the 1990s in cases of necrotizing pancreatitis to determine if infection was present. There has been a shift away from this because, although it can confirm the presence of infection, the false-negative rate is 15%. Clinical and imaging signs can be relied on in most cases to determine the presence of infection, and it is now recognized that fineneedle aspiration should be used only for select cases. Clinical studies have not shown that fine-needle aspiration improves outcomes.
Clinical scenarios typical of infected pancreatic necrosis include patients who have obvious signs of infection with no identifiable source, such as those who stabilize after acute severe acute pancreatitis, and then 10 to 14 days later become worse, with a dramatically higher white blood cell count and tachycardia. Such a patient likely needs an intervention regardless of the results of fine-needle aspiration.
On the other hand, a patient with a continually up-and-down course that never stabilizes over 3 weeks, with no identifiable source of infection, and with no peripancreatic gas apparent on imaging would be a good candidate for fine-needle aspiration.
If peripancreatic gas is seen on imaging, fine-needle aspiration is unnecessary. Peripancreatic gas is traditionally attributed to gasforming bacteria within the pancreas, but in my experience, it is usually from a fistula from the necrosis to the duodenum or the colon, the fistula being caused as the necrosis erodes at the hepatic flexure, the transverse colon, or the splenic flexure.
MECHANICAL INTERVENTIONS FOR INFECTIVE NECROSIS
Late, minimally invasive procedures preferred
Conventional management has shifted away from removing the necrosis with early surgical debridement of the pancreas. Experience with myocardial infarction shows that it is not necessary to remove a sterile necrotic organ, and studies with sterile pancreatic necrosis have found that surgical intervention is associated with a higher risk of death than medical management.
Documented infection has traditionally been considered a definite indication for debridement, but even that is being called into question as more studies are emerging of infected necrosis treated successfully with antibiotics alone.
Sterile necrosis with a fulminant course is a controversial indication for surgery. It was traditionally felt that surgery was worth trying for such patients, but this is no longer common practice.
For cases in which debridement was deemed advisable, surgery was done more frequently in the past. Now, a minimally invasive approach such as with endoscopy or percutaneous catheter is also used. Waiting until at least 4 weeks after the onset of acute pancreatitis is associated with a better outcome than intervening early.
WALLED-OFF NECROSIS
Watchful waiting or minimally invasive intervention
Patients who survive multiorgan failure but are still ill more than 4 weeks after the onset of pancreatitis should be suspected of having walled-off necrosis, formerly referred to as a pancreatic phlegmon. This term was abandoned after the 1992 Atlanta symposium.13 In the mid to late 1990s, the process was referred to as organized pancreatic necrosis. It is characterized by a mature, encapsulated collection of pancreatic or peripancreatic necrosis that contains variable amounts of amylase-rich fluid from pancreatic duct disruption.
Walled-off pancreatic necrosis (WOPN) is often confused with pancreatic pseudocyst; these may appear similar on CT, and higherdensity solid debris may be visible in walled-off necrosis within an otherwise homogenous-appearing collection. Magnetic resonance imaging defines liquid and solid much better than CT.
The best way to distinguish WOPN from pseudocyst is by clinical history: a patient with a preceding history of clinically severe acute pancreatitis almost always has necrotizing pancreatitis that evolves to walled-off necrosis, usually over 3 to 4 weeks.
Endoscopic removal and other minimally invasive approaches, such as aggressive percutaneous interventions, have replaced open necrosectomy for treatment, which was associated with high morbidity and mortality rates.14–16
Intervening for sterile walled-off necrosis is still a controversial topic: although systemically ill, the patient is no longer having life-threatening consequences, and watchful waiting might be just as expedient as intervention. Evidence to support either view is lacking. Most experts believe that intervention should be done if the patient has gastric outlet obstruction and intractable pain and is unable to eat 4 to 6 weeks after the onset of pancreatitis with WOPN. Infected WOPN is considered an indication for drainage.
Severe acute pancreatitis has been known since the time of Rembrandt, with Nicolaes Tulp—the physician credited as first describing it—immortalized in the famous painting, The Anatomy Lesson. However, progress in managing this disease has been disappointing. Treatment is mainly supportive, and we lack any true disease-modifying therapy. But we are learning to recognize the disease and treat it supportively better than in the past.
The early hours of severe acute pancreatitis are critical for instituting appropriate intervention. Prompt fluid resuscitation is key to preventing immediate and later morbidity and death. This article focuses on identifying and managing the most severe form of acute pancreatitis—necrotizing disease—and its complications.
NECROTIZING DISEASE ACCOUNTS FOR MOST PANCREATITIS DEATHS
The classification and definitions of acute pancreatitis were recently revised from the 1992 Atlanta system and published early in 2013.1 In addition, the American Pancreatic Association and the International Association of Pancreatology met in 2012 to develop evidence-based guidelines on managing severe pancreatitis.
An estimated 210,000 new cases of acute pancreatitis occur each year in the United States. About 20% of cases of severe acute pancreatitis are necrotizing disease, which accounts for nearly all the morbidity and death associated with acute pancreatitis.
The clinical spectrum of acute pancreatitis ranges from mild to life-threatening, reflecting interstitial (death rate < 1%) to necrotizing histology (the latter associated with a 25% risk of death if the pancreatitis becomes infected and a 10% risk if it is sterile). When death occurs early in the disease course, it tends to be from multiorgan failure; when death occurs later in the course, it tends to be from infection. Appropriate early treatment may prevent death in both categories.
DIAGNOSING ACUTE PANCREATITIS AND PREDICTING ITS SEVERITY
The diagnosis of acute pancreatitis requires two of the following three criteria:
- Clinical presentation—epigastric pain, nausea, vomiting
- Biochemical—amylase level more than three times the upper limit of normal, or lipase more than three times the upper limit of normal
- Evidence from computed tomography (CT), ultrasonography, or magnetic resonance imaging.
Although the biochemical criteria are variably sensitive for detecting acute pancreatitis (55%–100%), the specificity is very high (93% to 99%).
Recently, urinary trypsinogen-2, measured by dipstick, has also been used to aid diagnosis. It has a reasonable sensitivity (53%–96%) and specificity (85%) if positive (> 50 ng/mL).
Speed is critical
Over the years, many clinical prediction rules have been used for predicting the severity of acute pancreatitis. The Ranson criteria,2 from 1974, and the Acute Physiology and Chronic Health Evaluation (APACHE) II system3 are cumbersome and require waiting up to 48 hours after the onset of acute pancreatitis to obtain a complete score. The Imrie-Glasgow score is another predictor.
The systemic inflammatory response syndrome (SIRS) is currently the most important indicator of prognosis.4 Originally adopted for predicting the development of organ failure with sepsis, it requires at least two of the following criteria:
- Heart rate > 90 beats/min
- Core temperature < 36°C or > 38°C
- White blood cells < 4,000 or > 12,000/mm3
- Respirations > 20/min.
The advantages of this system are that it identifies risk very early in the course of the disease and can be assessed quickly in the emergency department.
The Bedside Index for Severity of Acute Pancreatitis (BISAP) score is another simple, easy-to-perform prognostic index,5,6 calculated by assigning 1 point for each of the following if present within the first 24 hours of presentation:
- Blood urea nitrogen > 25 mg/dL
- Abnormal mental status (Glasgow coma score < 15)
- Evidence of systemic inflammatory response syndrome
- Age > 60 years
- Pleural effusion seen on imaging study.
A score of 3 points is associated with a 5.3% rate of hospital death, 4 points with 12.7%, and 5 points with 22.5%.
At its most basic, severe acute pancreatitis is defined by organ failure (at least one organ from the respiratory, renal, or cardiovascular system) lasting for more than 48 hours. Failure for each organ is defined by the Marshall scoring system.1
EARLY MANAGEMENT IS KEY TO OUTCOME
The window of opportunity to make a significant difference in outcome is within the first 12 to 24 hours of presentation. Volume resuscitation is the cornerstone of early management. By the time of presentation for severe acute pancreatitis, the pancreas is already necrotic, so the aim is to minimize the systemic inflammatory response syndrome with the goals of reducing rates of organ failure, morbidity, and death. Necrotizing pancreatitis is essentially an ischemic event, and the goal of volume resuscitation is to maintain pancreatic and intestinal microcirculation to prevent intestinal ischemia and subsequent bacterial translocation.7
Early resuscitation with lactated Ringer’s solution recommended
The evidence supporting a specific protocol for fluid resuscitation in severe acute pancreatitis is not strong, but a few studies provide guidance.
Wu et al8 randomized 40 patients with acute pancreatitis to one of four arms: “goal-directed fluid resuscitation” with either lactated Ringer’s solution or normal saline, or standard therapy (by physician discretion) with either lactated Ringer’s solution or normal saline. Goal-directed therapy involved a bolus of 20 mL/kg given over 30 to 45 minutes at presentation followed by infusion with rates dependent on an algorithm based on change in blood urea nitrogen level at set times. Patients receiving either goal-directed or standard therapy had significantly lower rates of systemic inflammatory response syndrome at 24 hours than at admission. Most striking was that treatment with lactated Ringer’s solution was associated with dramatically improved rates, whereas normal saline showed no improvement.
In a retrospective study of patients with acute pancreatitis, Warndorf et al9 identified 340 patients who received early resuscitation (more than one-third of the total 72-hour fluid volume within 24 hours of presentation) and 90 patients who received late resuscitation (less than one-third of the total 72-hour fluid volume within 24 hours of presentation). Patients who received early resuscitation developed less systemic inflammatory response syndrome and organ failure, and required fewer interventions.
Monitoring for optimum fluid resuscitation
Fluid resuscitation should be carefully managed to avoid administering either inadequate or excessive amounts of fluid. Inadequate fluid resuscitation can result in renal failure, progression of necrosis, and possibly infectious complications. Excessive resuscitation—defined as more than 4 L in the first 24 hours—is associated with respiratory failure, pancreatic fluid collections, and abdominal compartment syndrome.
Optimum resuscitation is controlled fluid expansion averaging 5 to 10 mL/kg per hour, with 2,500 to 4,000 mL given in the first 24 hours.
Adequate volume resuscitation can be evaluated clinically with the following goals:
- Heart rate < 120 beats per minute
- Mean arterial pressure 65–85 mm Hg
- Urinary output > 1 mL/kg per hour
- Hematocrit 35%–44%.
EARLY CT IS JUSTIFIED ONLY IF DIAGNOSIS IS UNCLEAR
The normal pancreas takes up contrast in the same way as do the liver and spleen, so its enhancement on CT is similar. If there is interstitial pancreatitis, CT shows the pancreas with normal contrast uptake, but the organ appears “boggy” with indistinct outlines. With necrotizing pancreatitis, only small areas of tissue with normal contrast may be apparent.
Peripancreatic fat necrosis may also be visible on CT. Obese patients tend to have a worse clinical course of necrotizing pancreatitis, probably because of the associated peripancreatic fat that is incorporated into the pancreatic necrosis.
For clear-cut cases of acute pancreatitis, time is wasted waiting to obtain CT images, and this could delay fluid resuscitation. Results from immediate CT almost never change the clinical management during the first week of acute pancreatitis, and obtaining CT images is usually not recommended if the diagnosis of acute pancreatitis is clear. CT’s sensitivity for detecting necrosis is only 70% in the first 48 hours of presentation, so it is easy to be fooled by a false-negative scan: frequently, a scan does not show necrotizing pancreatitis until after 72 hours. In addition, evidence from animal studies indicates that contrast agents might worsen pancreatic necrosis.
Immediate CT is justified if the diagnosis is in doubt at presentation, such as to evaluate for other intra-abdominal conditions such as intestinal ischemia or a perforated duodenal ulcer.
Contrast-enhanced CT is recommended 72 to 96 hours after presentation, or earlier if the patient is worsening despite treatment. Specific CT protocols will be included in new management guidelines, expected to be published soon.
PREVENTING INFECTIOUS COMPLICATIONS
Risk of infection is associated with the degree of pancreatic necrosis. Patients with less than 30% necrosis have a 22.5% chance of infection, whereas those with more than 50% necrosis have a 46.5% risk of infection.10
Infection can develop from a variety of sources:
Bacterial translocation from the colon and small bowel is thought to be one of the major sources of infection in necrotic pancreatitis. Volume resuscitation and maintaining gut integrity with early enteral nutrition are believed to minimize the risk of bacterial translocation.
Hematogenous spread of bacteria is another suspected source of infection into the pancreas. Again, enteral nutrition also reduces the risk by minimizing the need for central catheters.
Biliary sources may also play a role. Bile duct stones or gall bladder infection can lead to infected pancreatic necrosis.
ANTIBIOTICS NOT ROUTINELY RECOMMENDED
Treating acute pancreatitis with antibiotics has fallen in and out of favor over the past decades. From being standard practice in the 1970s, it dropped off in the 1980s and 1990s and then became more common again.
Current recommendations from the American Pancreatic Association and the International Association of Pancreatology are not to routinely use intravenous antibiotics to prevent infection in necrotizing pancreatitis because of lack of evidence that it changes overall outcome. Antibiotic usage may be associated with more bacterial resistance and the introduction of fungal infections into the pancreas.
Selective gut decontamination, involving oral and rectal administration of neomycin and other antibiotics, was shown in a single randomized trial to reduce the incidence of infection, but it is very cumbersome and is not recommended for acute pancreatitis.
Treatment with probiotics is also not recommended and was shown in one study to lead to a worse outcome.11
ENTERAL BETTER THAN TOTAL PARENTERAL NUTRITION
Enteral tube feeding with either an elemental diet or a polymeric enteral formulation is the first-line therapy for necrotizing pancreatitis. Compared with total parenteral nutrition, it reduces infection, organ failure, hospital length of stay, the need for surgical intervention, and the risk of death. Total parenteral nutrition should be considered only for patients who do not tolerate enteral feeding because of severe ileus.
Conventional thinking for many years was to provide enteral feeding with a tube passed beyond the ligament of Treitz, thinking that it reduced stimulation to the pancreas. However, recent studies indicate that nasogastric feeding is equivalent to nasojejunal feeding in terms of nutrition, maintaining gut integrity, and outcome.
INTRA-ABDOMINAL HYPERTENSION AND ABDOMINAL COMPARTMENT SYNDROME
Movement of fluid into the intracellular space (“third-spacing”) occurs in acute pancreatitis and is exacerbated by fluid resuscitation. Intra-abdominal hypertension is associated with poor outcomes in patients with severe acute pancreatitis. Especially for patients with severe pancreatitis who are on mechanical ventilation, pressure should be monitored with transvesicular bladder measurements.
Intra-abdominal hypertension is defined as a sustained intra-abdominal pressure of more than 12 mm Hg, with the following grades:
- Grade 1: 12–15 mm Hg
- Grade 2: 16–20 mm Hg
- Grade 3: 21–25 mm Hg
- Grade 4: > 25 mm Hg.
Abdominal compartment syndrome is defined as a sustained intra-abdominal pressure of more than 20 mm Hg. It is associated with new organ dysfunction or failure. It should first be managed with ultrafiltration or diuretics to try to reduce the amount of fluid in the abdomen. Lumenal decompression can be tried with nasogastric or rectal tubes for the stomach and bowels. Ascites or retroperitoneal fluid can be drained percutaneously. In addition, analgesia and sedation to reduce abdominal muscle tone can help the patient become better ventilated. Neuromuscular blockade can also relax the abdomen.
Open abdominal decompression is the treatment of last resort to relieve abdominal compartment syndrome. The abdominal wall is not closed surgically but is allowed to heal by secondary intention (it “granulates in”).12
IDENTIFYING INFECTION
Fine-needle aspiration if clinical and imaging signs are not clear
Untreated infected pancreatitis is associated with a much higher risk of death than sterile pancreatic necrosis. Unfortunately, it can be difficult to determine if a patient with necrotizing pancreatitis has an infection because fever, tachycardia, and leukocytosis are usually present regardless. It is important to determine because mechanically intervening for sterile necrosis does not improve outcome.
Fine-needle aspiration, either guided by CT or done at the bedside with ultrasonography, with evaluation with Gram stain and culture, was widely used in the 1990s in cases of necrotizing pancreatitis to determine if infection was present. There has been a shift away from this because, although it can confirm the presence of infection, the false-negative rate is 15%. Clinical and imaging signs can be relied on in most cases to determine the presence of infection, and it is now recognized that fineneedle aspiration should be used only for select cases. Clinical studies have not shown that fine-needle aspiration improves outcomes.
Clinical scenarios typical of infected pancreatic necrosis include patients who have obvious signs of infection with no identifiable source, such as those who stabilize after acute severe acute pancreatitis, and then 10 to 14 days later become worse, with a dramatically higher white blood cell count and tachycardia. Such a patient likely needs an intervention regardless of the results of fine-needle aspiration.
On the other hand, a patient with a continually up-and-down course that never stabilizes over 3 weeks, with no identifiable source of infection, and with no peripancreatic gas apparent on imaging would be a good candidate for fine-needle aspiration.
If peripancreatic gas is seen on imaging, fine-needle aspiration is unnecessary. Peripancreatic gas is traditionally attributed to gasforming bacteria within the pancreas, but in my experience, it is usually from a fistula from the necrosis to the duodenum or the colon, the fistula being caused as the necrosis erodes at the hepatic flexure, the transverse colon, or the splenic flexure.
MECHANICAL INTERVENTIONS FOR INFECTIVE NECROSIS
Late, minimally invasive procedures preferred
Conventional management has shifted away from removing the necrosis with early surgical debridement of the pancreas. Experience with myocardial infarction shows that it is not necessary to remove a sterile necrotic organ, and studies with sterile pancreatic necrosis have found that surgical intervention is associated with a higher risk of death than medical management.
Documented infection has traditionally been considered a definite indication for debridement, but even that is being called into question as more studies are emerging of infected necrosis treated successfully with antibiotics alone.
Sterile necrosis with a fulminant course is a controversial indication for surgery. It was traditionally felt that surgery was worth trying for such patients, but this is no longer common practice.
For cases in which debridement was deemed advisable, surgery was done more frequently in the past. Now, a minimally invasive approach such as with endoscopy or percutaneous catheter is also used. Waiting until at least 4 weeks after the onset of acute pancreatitis is associated with a better outcome than intervening early.
WALLED-OFF NECROSIS
Watchful waiting or minimally invasive intervention
Patients who survive multiorgan failure but are still ill more than 4 weeks after the onset of pancreatitis should be suspected of having walled-off necrosis, formerly referred to as a pancreatic phlegmon. This term was abandoned after the 1992 Atlanta symposium.13 In the mid to late 1990s, the process was referred to as organized pancreatic necrosis. It is characterized by a mature, encapsulated collection of pancreatic or peripancreatic necrosis that contains variable amounts of amylase-rich fluid from pancreatic duct disruption.
Walled-off pancreatic necrosis (WOPN) is often confused with pancreatic pseudocyst; these may appear similar on CT, and higherdensity solid debris may be visible in walled-off necrosis within an otherwise homogenous-appearing collection. Magnetic resonance imaging defines liquid and solid much better than CT.
The best way to distinguish WOPN from pseudocyst is by clinical history: a patient with a preceding history of clinically severe acute pancreatitis almost always has necrotizing pancreatitis that evolves to walled-off necrosis, usually over 3 to 4 weeks.
Endoscopic removal and other minimally invasive approaches, such as aggressive percutaneous interventions, have replaced open necrosectomy for treatment, which was associated with high morbidity and mortality rates.14–16
Intervening for sterile walled-off necrosis is still a controversial topic: although systemically ill, the patient is no longer having life-threatening consequences, and watchful waiting might be just as expedient as intervention. Evidence to support either view is lacking. Most experts believe that intervention should be done if the patient has gastric outlet obstruction and intractable pain and is unable to eat 4 to 6 weeks after the onset of pancreatitis with WOPN. Infected WOPN is considered an indication for drainage.
- Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62:102–111.
- Ranson JH, Rifkind KM, Roses DF, Fink SD, Eng K, Spencer FC. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974; 139:69–81.
- Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med 1985; 13:818–829.
- American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 1992; 20:864–874.
- Wu BU, Johannes RS, Sun X, Tabak Y, Conwell DL, Banks PA. The early prediction of mortality in acute pancreatitis: a large population-based study. Gut 2008; 57:1698–1703.
- Singh VK, Wu BU, Bollen TL, et al. A prospective evaluation of the bedside index for severity in acute pancreatitis score in assessing mortality and intermediate markers of severity in acute pancreatitis. Am J Gastroenterol 2009; 104:966–971.
- Fisher JM, Gardner TB. The “golden hours” of management in acute pancreatitis. Am J Gastroenterol 2012; 107:1146–1150.
- Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol 2011; 9:710–717.
- Warndorf MG, Kurtzman JT, Bartel MJ, et al. Early fluid resuscitation reduces morbidity among patients with acute pancreatitis. Clin Gastroenterol Hepatol 2011; 9:705–709.
- Beger HG, Rau BM. Severe acute pancreatitis: clinical course and management. World J Gastroenterol 2007; 13:5043–5051.
- Besselink MG, van Santvoort HC, Buskens E, et al; Dutch Acute Pancreatitis Study Group. Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet 2008; 371:651–659.
- Fitzgerald JE, Gupta S, Masterson S, Sigurdsson HH. Laparostomy management using the ABThera open abdomen negative pressure therapy system in a grade IV open abdomen secondary to acute pancreatitis. Int Wound J 2012. doi: 1111/j.1742-481X2012.00953.x. [epub ahead of print]
- Bradley EL. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11–13, 1992. Arch Surg 1993; 128:586–590.
- Baron TH, Thaggard WG, Morgan DE, Stanley RJ. Endoscopic therapy for organized pancreatic necrosis. Gastroenterology 1996; 111:755–764.
- van Santvoort HC, Besselink MG, Bakker OJ, et al; Dutch Pancreatitis Study Group. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med 2010; 362:1491–1502.
- Bakker OJ, van Santvoort HC, van Brunschot S, et al; Dutch Pancreatitis Study Group. Endoscopic transgastric vs surgical necrosectomy for infected necrotizing pancreatitis: a randomized trial. JAMA 2012; 307:1053–1061.
- Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62:102–111.
- Ranson JH, Rifkind KM, Roses DF, Fink SD, Eng K, Spencer FC. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974; 139:69–81.
- Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med 1985; 13:818–829.
- American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 1992; 20:864–874.
- Wu BU, Johannes RS, Sun X, Tabak Y, Conwell DL, Banks PA. The early prediction of mortality in acute pancreatitis: a large population-based study. Gut 2008; 57:1698–1703.
- Singh VK, Wu BU, Bollen TL, et al. A prospective evaluation of the bedside index for severity in acute pancreatitis score in assessing mortality and intermediate markers of severity in acute pancreatitis. Am J Gastroenterol 2009; 104:966–971.
- Fisher JM, Gardner TB. The “golden hours” of management in acute pancreatitis. Am J Gastroenterol 2012; 107:1146–1150.
- Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol 2011; 9:710–717.
- Warndorf MG, Kurtzman JT, Bartel MJ, et al. Early fluid resuscitation reduces morbidity among patients with acute pancreatitis. Clin Gastroenterol Hepatol 2011; 9:705–709.
- Beger HG, Rau BM. Severe acute pancreatitis: clinical course and management. World J Gastroenterol 2007; 13:5043–5051.
- Besselink MG, van Santvoort HC, Buskens E, et al; Dutch Acute Pancreatitis Study Group. Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet 2008; 371:651–659.
- Fitzgerald JE, Gupta S, Masterson S, Sigurdsson HH. Laparostomy management using the ABThera open abdomen negative pressure therapy system in a grade IV open abdomen secondary to acute pancreatitis. Int Wound J 2012. doi: 1111/j.1742-481X2012.00953.x. [epub ahead of print]
- Bradley EL. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11–13, 1992. Arch Surg 1993; 128:586–590.
- Baron TH, Thaggard WG, Morgan DE, Stanley RJ. Endoscopic therapy for organized pancreatic necrosis. Gastroenterology 1996; 111:755–764.
- van Santvoort HC, Besselink MG, Bakker OJ, et al; Dutch Pancreatitis Study Group. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med 2010; 362:1491–1502.
- Bakker OJ, van Santvoort HC, van Brunschot S, et al; Dutch Pancreatitis Study Group. Endoscopic transgastric vs surgical necrosectomy for infected necrotizing pancreatitis: a randomized trial. JAMA 2012; 307:1053–1061.
KEY POINTS
- Routine early computed tomography to evaluate patients with severe acute pancreatitis wastes time and is necessary only if the diagnosis at presentation is not clearly consistent with acute pancreatitis.
- Optimum fluid resuscitation is now recommended, using lactated Ringer’s solution at a rate of 5 to 10 mL/kg per hour, with 2,500 to 4,000 mL given in the first 24 hours.
- Enteral feeding with either an elemental diet or a polymeric enteral formulation is first-line nutritional therapy.
- Antibiotics are no longer routinely used to prevent infection.
- Relief of abdominal compartment syndrome should be attempted by multiple means before resorting to open abdominal decompression.
Detecting and controlling diabetic nephropathy: What do we know?
Diabetes is on the rise, and so is diabetic nephropathy. In view of this epidemic, physicians should consider strategies to detect and control kidney disease in their diabetic patients.
This article will focus on kidney disease in adult-onset type 2 diabetes. Although it has different pathogenetic mechanisms than type 1 diabetes, the clinical course of the two conditions is very similar in terms of the prevalence of proteinuria after diagnosis, the progression to renal failure after the onset of proteinuria, and treatment options.1
DIABETES AND DIABETIC KIDNEY DISEASE ARE ON THE RISE
The incidence of diabetes increases with age, and with the aging of the baby boomers, its prevalence is growing dramatically. The 2005– 2008 National Health and Nutrition Examination Survey estimated the prevalence as 3.7% in adults age 20 to 44, 13.7% at age 45 to 64, and 26.9% in people age 65 and older. The obesity epidemic is also contributing to the increase in diabetes in all age groups.
Diabetic kidney disease has increased in the United States from about 4 million cases 20 years ago to about 7 million in 2005–2008.2 Diabetes is the major cause of end-stage renal disease in the developed world, accounting for 40% to 50% of cases. Other major causes are hypertension (27%) and glomerulonephritis (13%).3
Physicians in nearly every field of medicine now care for patients with diabetic nephropathy. The classic presentation—a patient who has impaired vision, fluid retention with edema, and hypertension—is commonly seen in dialysis units and ophthalmology and cardiovascular clinics.
CLINICAL PROGRESSION
Early in the course of diabetic nephropathy, blood pressure is normal and microalbuminuria is not evident, but many patients have a high glomerular filtration rate (GFR), indicating temporarily “enhanced” renal function or hyperfiltration. The next stage is characterized by microalbuminuria, correlating with glomerular mesangial expansion: the GFR falls back into the normal range and blood pressure starts to increase. Finally, macroalbuminuria occurs, accompanied by rising blood pressure and a declining GFR, correlating with the histologic appearance of glomerulosclerosis and Kimmelstiel-Wilson nodules.4
Hypertension develops in 5% of patients by 10 years after type 1 diabetes is diagnosed, 33% by 20 years, and 70% by 40 years. In contrast, 40% of patients with type 2 diabetes have high blood pressure at diagnosis.
Unfortunately, in most cases, this progression is a one-way street, so it is critical to intervene to try to slow the progression early in the course of the disease process.
SCREENING FOR DIABETIC NEPHROPATHY
Nephropathy screening guidelines for patients with diabetes are provided in Table 1.5

Blood pressure should be monitored at each office visit (Table 1). The goal for adults with diabetes should be to reduce blood pressure to 130/80 mm Hg. Reduction beyond this level may be associated with an increased mortality rate.6 Very high blood pressure (> 180 mm Hg systolic) should be lowered slowly. Lowering blood pressure delays the progression from microalbuminuria (30–299 mg/day or 20–199 μg/min) to macroalbuminuria (> 300 mg/day or > 200 μg/min) and slows the progression to renal failure.
Urinary albumin. Proteinuria takes 5 to 10 years to develop after the onset of diabetes. Because it is possible for patients with type 2 diabetes to have had the disease for some time before being diagnosed, urinary albumin screening should be performed at diagnosis and annually thereafter. Patients with type 1 are usually diagnosed with diabetes at or near onset of disease; therefore, annual screening for urinary albumin can begin 5 years after diagnosis.5
Proteinuria can be measured in different ways (Table 2). The basic screening test for clinical proteinuria is the urine dipstick, which is very sensitive to albumin and relatively insensitive to other proteins. “Trace-positive” results are common in healthy people, so proteinuria is not confirmed unless a patient has repeatedly positive results.
Microalbuminuria is important to measure, especially if it helps determine therapy. It is not detectable by the urinary dipstick, but can be measured in the following ways:
- Measurement of the albumin-creatinine ratio in a random spot collection
- 24-hour collection (creatinine should simultaneously be measured and creatinine clearance calculated)
- Timed collection (4 hours or overnight).
The first method is preferred, and any positive test result must be confirmed by repeat analyses of urinary albumin before a patient is diagnosed with microalbuminuria.
Occasionally a patient presenting with proteinuria but normal blood sugar and hemoglobin A1c will have a biopsy that reveals morphologic changes of classic diabetic nephropathy. Most such patients have a history of hyperglycemia, indicating that they actually have been diabetic.
Proteinuria—the best marker of disease progression
Proteinuria is the strongest predictor of renal outcomes. The Reduction in End Points in Noninsulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) study was a randomized, placebo-controlled trial in more than 1,500 patients with type 2 diabetes to test the effects of losartan on renal outcome. Those with high albuminuria (> 3.0 g albumin/g creatinine) at baseline were five times more likely to reach a renal end point and were eight times more likely to have progression to end-stage renal disease than patients with low albuminuria (< 1.5 g/g).7 The degree of albuminuria after 6 months of treatment showed similar predictive trends, indicating that monitoring and treating proteinuria are extremely important goals.
STRATEGY 1 TO LIMIT RENAL INJURY: REDUCE BLOOD PRESSURE
Blood pressure control improves renal and cardiovascular function.
As early as 1983, Parving et al,8 in a study of only 10 insulin-dependent diabetic patients, showed strong evidence that early aggressive antihypertensive treatment improved the course of diabetic nephropathy. During the mean pretreatment period of 29 months, the GFR decreased significantly and the urinary albumin excretion rate and arterial blood pressure rose significantly. During the mean 39-month period of antihypertensive treatment with metoprolol, hydralazine, and furosemide or a thiazide, mean arterial blood pressure fell from 144/97 to 128/84 mm Hg and urinary albumin excretion from 977 to 433 μg/ min. The rate of decline in GFR slowed from 0.91 mL/min/month before treatment to 0.39 mL/min/month during treatment.
The Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) trial9 enrolled more than 11,000 patients internationally with type 2 diabetes at high risk for cardiovascular events. In addition to standard therapy, blood pressure was intensively controlled in one group with a combination of the angiotensin-converting enzyme (ACE) inhibitor perindopril and the diuretic indapamide. The intensive-therapy group achieved blood pressures less than 140/80 mm Hg and had a mean reduction of systolic blood pressure of 5.6 mm Hg and diastolic blood pressure of 2.2 mm Hg vs controls. Despite these apparently modest reductions, the intensively controlled group had a significant 9% reduction of the primary outcome of combined macrovascular events (cardiovascular death, myocardial infarction, and stroke) and microvascular events (new or worsening nephropathy, or retinopathy).10
A meta-analysis of studies of patients with type 2 diabetes found reduced nephropathy with systolic blood pressure control to less than 130 mm Hg.11
The United Kingdom Prospective Diabetes Study (UKPDS) is a series of studies of diabetes. The original study in 1998 enrolled 5,102 patients with newly diagnosed type 2 diabetes.12 The more than 1,000 patients with hypertension were randomized to either tight blood pressure control or regular care. The intensive treatment group had a mean blood pressure reduction of 9 mm Hg systolic and 3 mm Hg diastolic, along with major reductions in all diabetes end points, diabetes deaths, microvascular disease, and stroke over a median follow-up of 8.4 years.
Continuous blood pressure control is critical
Tight blood pressure control must be maintained to have continued benefit. During the 10 years following the UKPDS, no attempts were made to maintain the previously assigned therapies. A follow-up study13 of 884 UKPDS patients found that blood pressures were the same again between the two groups 2 years after the trial was stopped, and no beneficial legacy effect from previous blood pressure control was evident on end points.
Control below 120 mm Hg systolic not needed
Blood pressure control slows kidney disease and prevents major macrovascular disease, but there is no evidence that lowering systolic blood pressure below 120 mm Hg provides additional benefit. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,14 more than 10,000 patients with type 2 diabetes and existing cardiovascular disease or additional cardiovascular risk factors were randomized to a goal of systolic blood pressure less than 120 mm Hg or less than 140 mm Hg (actual mean systolic pressures were 119 vs 134 mm Hg, respectively). Over nearly 5 years, there was no difference in cardiovascular events or deaths between the two groups.15

Since 1997, six international organizations have revised their recommended blood pressure goals in diabetes mellitus and renal diseases. Randomized clinical trials and observational studies have demonstrated the importance of blood pressure control to the level of 125/75 to 140/80 mm Hg. The National Kidney Foundation, the American Diabetes Association, and the Canadian Hypertension Society have developed consensus guidelines for blood pressure control to less than 130/80 mm Hg.16–21 Table 3 summarizes blood pressure goals for patients with diabetes.
STRATEGY 2: CONTROL BLOOD SUGAR
Recommendations for blood sugar goals are more controversial.
The Diabetes Control and Complications Trial22 provided early evidence that tight blood sugar control slows the development of microalbuminuria and macroalbuminuria. The study randomized more than 1,400 patients with type 1 diabetes to either standard therapy (1 or 2 daily insulin injections) or intensive therapy (an external insulin pump or 3 or more insulin injections guided by frequent blood glucose monitoring) to keep blood glucose levels close to normal. About half the patients had mild retinopathy at baseline and the others had no retinopathy. After 6.5 years, intensive therapy was found to significantly delay the onset and slow the progression of diabetic retinopathy and nephropathy.
The Kumamoto Study23 randomized 110 patients with type 2 diabetes and either no retinopathy (primary prevention cohort) or simple retinopathy (secondary prevention cohort) to receive either multiple insulin injections or conventional insulin therapy over 8 years. Intensive therapy led to lower rates of retinopathy (7.7% vs 32% in primary prevention and 19% vs 44% in secondary prevention) and progressive nephropathy (7% vs 28% in primary prevention at 6 years and 11% vs 32% in secondary prevention).
In addition to studying the effects of blood pressure control, the UKPDS also studied the effects of intensive blood glucose control.24,25 Nearly 4,000 patients with newly diagnosed type 2 diabetes were randomized to intensive treatment with a sulfonylurea or insulin, or to conventional treatment with diet. Over 10 years, the mean hemoglobin A1c was reduced to 7.0% in the intensive group and 7.9% in the conventional group. The risk of any diabetes-related end point was 12% lower in the intensive group, 10% lower for diabetes-related death, and 6% lower for all-cause mortality. There was also a 25% reduction in microvascular disease (retinopathy and nephropathy). However, the intensive group had more hypoglycemic episodes than the conventional group and a tendency to some increase in macrovascular events. A legacy effect was evident: patients who had intensive treatment had less microvascular disease progression years after stopping therapy.
Tight glycemic control reduces nephropathy, but does it increase cardiovascular risk?
Earlier trials provided strong evidence that blood glucose control prevents or slows retinopathy and nephropathy. The critical question is, “At what expense?” Although diabetes is the most common cause of kidney failure in the United States, most people with diabetes do not die of kidney failure, but of cardiovascular disease. Two recent large trials had different results regarding glycemic control below hemoglobin A1c of 7.0% and macrovascular risk, creating a controversy about what recommendations are best.
The ADVANCE trial, enrolling 11,140 patients with type 2 diabetes, was largely conducted in Australia and used the sulfonylurea glipizide for glycemic control. Compared with the group that received standard therapy (n=5,569), the intensive-treatment group (n=5,571) achieved mean hemoglobin A1c levels of 6.5% compared with 7.3% in the standard group, and had less nephropathy, less microalbuminuria, less doubling of creatinine, and a lower rate of end-stage renal disease (4% vs 5% in the standard therapy group). No difference between the two groups was found in retinopathy. Rates of all-cause mortality did not differ between the groups.9
The ACCORD trial had more than 10,000 subjects with type 2 diabetes and took place mostly in the United States. Using mainly rosiglitazone for intensive therapy, the intensive group achieved hemoglobin A1c levels of 6.4% vs 7.5% in the standard-therapy group. The trial was stopped early, at 3.7 years, because of a higher risk of death and cardiovascular events in the group with intensive glycemic control. However, the intensive-therapy group did have a significant decrease in microvascular renal outcomes and a reduction in the progression of retinopathy.14,26
In summary, tighter glycemic control improves microvascular complications—both retinopathy and nephropathy—in patients with type 2 diabetes. The benefit of intensive therapy on macrovascular complications (stroke, myocardial infarction) in long-standing diabetes has not been convincingly demonstrated in randomized trials. The UKPDS suggested that maintaining a hemoglobin A1c of 7% in patients newly diagnosed with type 2 diabetes confers long-term cardiovascular benefits. The target hemoglobin A1c for type 2 diabetes should be tailored to the patient: 7% is a reasonable goal for most patients, but the goal should be higher for the elderly and frail. Reducing the risk of cardiovascular death is still best done by controlling blood pressure, reducing lipids, quitting smoking, and losing weight.
STRATEGY 3: INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE AXIS
Components of the renin-angiotensin-aldo-sterone system are present not only in the circulation but also in many tissues, including the heart, brain, kidney, blood vessels, and adrenal glands. The role of renin-angiotensin-aldosterone system blockers in treating and preventing diabetic nephropathy has become controversial in recent years with findings from new studies.
The renin-angiotensin-aldosterone system is important in the development or maintenance of high blood pressure and the resultant damage to the brain, heart, and kidney. Drug development has focused on inhibiting steps in the biochemical pathway. ACE inhibitors block the formation of angiotensin II—the most biologically potent angiotensin peptide—and are among the most commonly used drugs to treat hypertension and concomitant conditions, such as renal insufficiency, proteinuria, and heart failure. Angiotensin receptor blockers (ARBs) interact with the angiotensin AT1 receptor and block most of its actions. They are approved by the US Food and Drug Administration (FDA) for the treatment of hypertension, and they help prevent left ventricular hypertrophy and mesangial sclerosis. Large studies have shown that ACE inhibitors and ARBs offer similar cardiovascular benefit.
The glomerulus has the only capillary bed with a blood supply that drains into an efferent arteriole instead of a venule, providing high resistance to aid filtration. Efferent arterioles are rich in AT1 receptors. In the presence of angiotensin II they constrict, increasing pressure in the glomerulus, which can lead to proteinuria and glomerulosclerosis. ACE inhibitors and ARBs relax the efferent arteriole, allowing increased blood flow through the glomerulus. This reduction in intraglomerular pressure is associated with less proteinuria and less glomerulosclerosis.
Diabetes promotes renal disease in many ways. Glucose and advanced glycation end products can lead to increased blood flow and increased pressure in the glomerulus. Through a variety of pathways, hyperglycemia, acting on angiotensin II, leads to NF-kapa beta production, profibrotic cytokines, increased matrix, and eventual fibrosis. ACE inhibitors and ARBs counteract many of these.
ACE inhibitors and ARBs slow nephropathy progression beyond blood pressure control
Several major clinical trials27–32 examined the effects of either ACE inhibitors or ARBs in slowing the progression of diabetic nephropathy and have had consistently positive results.
The Collaborative Study Group30 was a 3-year randomized trial in 419 patients with type 1 diabetes, using the ACE inhibitor captopril vs placebo. Captopril was associated with less decline in kidney function and a 50% reduction in the risk of the combined end points of death, dialysis, and transplantation that was independent of the small difference in blood pressures between the two groups.
The Irbesartan Diabetic Nephropathy Trial (IDNT)31 studied the effect of the ARB irbesartan vs the calcium channel blocker amlodipine vs placebo over 2.6 years in 1,715 patients with type 2 diabetes. Irbesartan was found to be significantly more effective in protecting against the progression of nephropathy, independent of reduction in blood pressure.
The RENAAL trial,32 published in 2001, was a 3-year, randomized, double-blind study comparing the ARB losartan at increasing dosages with placebo (both taken in addition to conventional antihypertensive treatment) in 1,513 patients with type 2 diabetes and nephropathy. The blood pressure goal was 140/90 mm Hg in both groups, but the losartan group had a lower rate of doubling of serum creatinine, end-stage renal disease, and combined end-stage renal disease or death.
‘Aldosterone escape’ motivates the search for new therapies
An important reason for developing more ways to block the renin-angiotensin-aldosterone system is because of “aldosterone escape,” the phenomenon of angiotensin II or aldosterone returning to pretreatment levels despite continued ACE inhibition.
Biollaz et al,33 in a 1982 study of 19 patients with hypertension, showed that despite reducing blood pressure and keeping the blood level of ACE very low with twice-daily enalapril 20 mg, blood and urine levels of angiotensin II steadily rose back to baseline levels within a few months.
A growing body of evidence suggests that despite effective inhibition of angiotensin II activity, non-ACE synthetic pathways still permit angiotensin II generation via serine proteases such as chymase, cathepsin G, and tissue plasminogen activator.
Thus, efforts have been made to block the renin-angiotensin system in other places. In addition to ACE inhibitors and ARBs, two aldosterone receptor antagonists are available, spironolactone and eplerenone, both used to treat heart failure. A direct renin inhibitor, aliskiren, is also available.
Combination therapy—less proteinuria, but…
A number of studies have shown that combination treatment with agents having different targets in the renin-angiotensin-aldosterone system leads to larger reductions in albuminuria than does single-agent therapy.
Mogensen et al34 studied the effect of the ACE inhibitor lisinopril (20 mg per day) plus the ARB candesartan (16 mg per day) in subjects with microalbuminuria, hypertension, and type 2 diabetes. Combined treatment was more effective in reducing proteinuria.
Epstein et al35 studied the effects of the ACE inhibitor enalapril (20 mg/day) combined with either of two doses of the selective aldosterone receptor antagonist eplerenone (50 or 100 mg/day) or placebo. Both eplerenone dosages, when added to the enalapril treatment, significantly reduced albuminuria from baseline as early as week 4 (P < .001), but placebo treatment added to the enalapril did not result in any significant decrease in urinary albumin excretion. Systolic blood pressure decreased significantly in all treatment groups and by about the same amount.
The Aliskiren Combined With Losartan in Type 2 Diabetes and Nephropathy (AVOID) trial36 randomized more than 600 patients with type 2 diabetes and nephropathy to aliskiren (a renin inhibitor) or placebo added to the ARB losartan. Again, combination treatment was more renoprotective, independent of blood pressure lowering.
Worse outcomes with combination therapy?
More recent studies have indicated that although combination therapy reduces proteinuria to a greater extent than monotherapy, overall it worsens major renal and cardiovascular outcomes. The multicenter Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)37 randomized more than 25,000 patients age 55 and older with established atherosclerotic vascular disease or with diabetes and end-organ damage to receive either the ARB telmisartan 80 mg daily, the ACE inhibitor ramipril 10 mg daily, or both. Mean follow-up was 56 months. The combination-treatment group had higher rates of death and renal disease than the single-therapy groups (which did not differ from one another).
Why the combination therapy had poorer outcomes is under debate. Patients may get sudden drops in blood pressure that are not detected with only periodic monitoring. Renal failure was mostly acute rather than chronic, and the estimated GFR declined more in the combined therapy group than in the single-therapy groups.
The Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints (ALTITUDE) was designed to test the effect of the direct renin inhibitor aliskiren or placebo, both arms combined with either an ACE inhibitor or an ARB in patients with type 2 diabetes at high risk for cardiovascular and renal events. The trial was terminated early because of more strokes and deaths in the combination therapy arms. The results led the FDA to issue black box warnings against using aliskiren with these other classes of agents, and all studies testing similar combinations have been stopped. (In one study that was stopped and has not yet been published, 100 patients with proteinuria were treated with either aliskiren, the ARB losartan, or both, to evaluate the effects of aldosterone escape. Results showed no differences: about one-third of each group had this phenomenon.)
My personal recommendation is as follows: for younger patients with proteinuria, at lower risk for cardiovascular events and with disease due not to diabetes but to immunoglobulin A nephropathy or another proteinuric kidney disease, treat with both an ACE inhibitor and ARB. But the combination should not be used for patients at high risk of cardiovascular disease, which includes almost all patients with diabetes.
If more aggressive renin-angiotensin system blockade is needed against diabetic nephropathy, adding a diuretic increases the impact of blocking the renin-angiotensin-aldosterone system on both proteinuria and progression of renal disease. The aldosterone blocker spironolactone 25 mg can be added if potassium levels are carefully monitored.
ACE inhibitor plus calcium channel blocker is safer than ACE inhibitor plus diuretic
The Avoiding Cardiovascular Events Through Combination Therapy in Patients Living With Systolic Hypertension (ACCOMPLISH) trial38 randomized more than 11,000 high-risk patients with hypertension to receive an ACE inhibitor (benazepril) plus either a calcium channel blocker (amlodipine) or thiazide diuretic (hydrochlorothiazide). Blood pressures were identical between the two groups, but the trial was terminated early, at 36 months, because of a higher risk of the combined end point of cardiovascular death, myocardial infarction, stroke, and other major cardiac events in the ACE inhibitor-thiazide group.
Although some experts believe this study is definitive and indicates that high blood pressure should never be treated with an ACE inhibitor-thiazide combination, I believe that caution is needed in interpreting these findings. This regimen should be avoided in older patients with diabetes at high risk for cardiovascular disease, but otherwise, getting blood pressure under control is critical, and this combination can be used if it works and the patient is tolerating it well.
In summary, the choice of blood pressure-lowering medications is based on reducing cardiovascular events and slowing the progression of kidney disease. Either an ACE inhibitor or an ARB is the first choice for patients with diabetes, hypertension, and any degree of proteinuria. Many experts recommend beginning one of these agents even if proteinuria is not present. However, the combination of an ACE inhibitor and ARB should not be used in diabetic patients, especially if they have cardiovascular disease, until further data clarify the results of the ONTARGET and ALTITUDE trials.
STRATEGY 4: METABOLIC MANIPULATION WITH NOVEL AGENTS
Several new agents have recently been studied for the treatment of diabetic nephropathy, including aminoguanidine, which reduces levels of advanced glycation end-products, and sulodexide, which blocks basement membrane permeability. Neither agent has been shown to be safe and effective in diabetic nephropathy. The newest agent is bardoxolone methyl. It induces the Keap1–Nrf2 pathway, which up-regulates cytoprotective factors, suppressing inflammatory and other cytokines that are major mediators of progression of chronic kidney disease.39
Pergola et al,40 in a phase 2, double-blind trial, randomized 227 adults with diabetic kidney disease and a low estimated GFR (20–45 mL/min/1.73 m2) to receive placebo or bardoxolone 25, 75, or 150 mg daily. Drug treatment was associated with improvement in the estimated GFR, a finding that persisted throughout the 52 weeks of treatment. Surprisingly, proteinuria did not decrease with drug treatment.
As of this writing, a large multicenter controlled randomized trial has been halted because of concerns by the data safety monitoring board, which found increased rates of death and fluid retention with the drug. A number of recent trials have shown a beneficial effect of sodium bicarbonate therapy in patients with late-stage chronic kidney disease. They have shown slowing of the progression of GFR decline in a number of renal diseases, including diabetes.
- Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med 1999; 341:1127–1133.
- de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011; 305:2532–2539.
- United States Renal Data System (USRDS) 2000 Annual Data Report. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases – Division of Kidney, Urologic and Hematologic Diseases. USRDS Coordinating Center operated by the Minneapolis Medical Research Foundation. www.usrds.org
- Macisaac RJ, Jerums G. Diabetic kidney disease with and without albuminuria. Curr Opin Nephrol Hypertens 2011; 20:246–257.
- Molitch ME, DeFronzo RA, Franz MJ, et al; American Diabetes Association. Nephropathy in diabetes. Diabetes Care 2004; 27(suppl 1):S79–S83.
- Vamos EP, Harris M, Millett C, et al. Association of systolic and diastolic blood pressure and all cause mortality in people with newly diagnosed type 2 diabetes: retrospective cohort study. BMJ 2012; 345:e5567.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Parving HH, Andersen AR, Smidt UM, Svendsen PA. Early aggressive antihypertensive treatment reduces rate of decline in kidney function in diabetic nephropathy. Lancet 1983; 1:1175–1179.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet 2007; 370:829–840.
- Bangalore S, Kumar S, Lobach I, Messerli FH. Blood pressure targets in subjects with type 2 diabetes mellitus/impaired fasting glucose: observations from traditional and bayesian random-effects meta-analyses of randomized trials. Circulation 2011; 123:2799–2810.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63. (Erratum in: Diabetes Care 2012; 35:660.)
- Bakris GL, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 2000; 36:646–661.
- Ramsay L, Williams B, Johnston G, et al. Guidelines for management of hypertension: report of the third working party of the British Hypertension Society. J Hum Hypertens 1999; 13:569–592.
- Feldman RD, Campbell N, Larochelle P, et al. 1999 Canadian recommendations for the management of hypertension. Task Force for the Development of the 1999 Canadian Recommendations for the Management of Hypertension. CMAJ 1999; 161(suppl):12:S1–S17.
- Chalmers J, MacMahon S, Mancia G, et al. 1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension. Guidelines Sub-committee of the World Health Organization. Clin Exp Hypertens 1999; 21:1009–1060.
- The seventh report of the Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23(suppl 2):B21–B29.
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Ismail-Beigi F, Craven T, Banerji MA, et al; ACCORD trial group. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010; 376:419–430. Erratum in: Lancet 2010; 376:1466.
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000: 355:253–259. Erratum in: Lancet2000; 356:860.
- Parving HH, Lehnert H, Bröchner-Mortensen J, et al; Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001; 345:870–878.
- Viberti G, Wheeldon NM; MicroAlbuminuria Reduction With VALsartan (MARVAL) Study Investigators. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: a blood pressure-independent effect. Circulation 2002; 106:672–678.
- Lewis EJ, Hunsicker LG, Bain R P, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Biollaz J, Brunner HR, Gavras I, Waeber B, Gavras H. Antihypertensive therapy with MK 421: angiotensin II--renin relationships to evaluate efficacy of converting enzyme blockade. J Cardiovasc Pharmacol 1982; 4:966–972.
- Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ 2000; 321:1440–1444.
- Epstein M, Williams GH, Weinberger M, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol 2006; 1:940–951.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK; AVOID Study Investigators. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008; 358:2433–2446.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Jamerson K, Weber MA, Bakris GL, et al; ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 2010; 298:F662–F671.
- Pergola PE, Raskin P, Toto RD, et al; BEAM Study Investigators Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 2011; 365:327–336.
Diabetes is on the rise, and so is diabetic nephropathy. In view of this epidemic, physicians should consider strategies to detect and control kidney disease in their diabetic patients.
This article will focus on kidney disease in adult-onset type 2 diabetes. Although it has different pathogenetic mechanisms than type 1 diabetes, the clinical course of the two conditions is very similar in terms of the prevalence of proteinuria after diagnosis, the progression to renal failure after the onset of proteinuria, and treatment options.1
DIABETES AND DIABETIC KIDNEY DISEASE ARE ON THE RISE
The incidence of diabetes increases with age, and with the aging of the baby boomers, its prevalence is growing dramatically. The 2005– 2008 National Health and Nutrition Examination Survey estimated the prevalence as 3.7% in adults age 20 to 44, 13.7% at age 45 to 64, and 26.9% in people age 65 and older. The obesity epidemic is also contributing to the increase in diabetes in all age groups.
Diabetic kidney disease has increased in the United States from about 4 million cases 20 years ago to about 7 million in 2005–2008.2 Diabetes is the major cause of end-stage renal disease in the developed world, accounting for 40% to 50% of cases. Other major causes are hypertension (27%) and glomerulonephritis (13%).3
Physicians in nearly every field of medicine now care for patients with diabetic nephropathy. The classic presentation—a patient who has impaired vision, fluid retention with edema, and hypertension—is commonly seen in dialysis units and ophthalmology and cardiovascular clinics.
CLINICAL PROGRESSION
Early in the course of diabetic nephropathy, blood pressure is normal and microalbuminuria is not evident, but many patients have a high glomerular filtration rate (GFR), indicating temporarily “enhanced” renal function or hyperfiltration. The next stage is characterized by microalbuminuria, correlating with glomerular mesangial expansion: the GFR falls back into the normal range and blood pressure starts to increase. Finally, macroalbuminuria occurs, accompanied by rising blood pressure and a declining GFR, correlating with the histologic appearance of glomerulosclerosis and Kimmelstiel-Wilson nodules.4
Hypertension develops in 5% of patients by 10 years after type 1 diabetes is diagnosed, 33% by 20 years, and 70% by 40 years. In contrast, 40% of patients with type 2 diabetes have high blood pressure at diagnosis.
Unfortunately, in most cases, this progression is a one-way street, so it is critical to intervene to try to slow the progression early in the course of the disease process.
SCREENING FOR DIABETIC NEPHROPATHY
Nephropathy screening guidelines for patients with diabetes are provided in Table 1.5
Blood pressure should be monitored at each office visit (Table 1). The goal for adults with diabetes should be to reduce blood pressure to 130/80 mm Hg. Reduction beyond this level may be associated with an increased mortality rate.6 Very high blood pressure (> 180 mm Hg systolic) should be lowered slowly. Lowering blood pressure delays the progression from microalbuminuria (30–299 mg/day or 20–199 μg/min) to macroalbuminuria (> 300 mg/day or > 200 μg/min) and slows the progression to renal failure.
Urinary albumin. Proteinuria takes 5 to 10 years to develop after the onset of diabetes. Because it is possible for patients with type 2 diabetes to have had the disease for some time before being diagnosed, urinary albumin screening should be performed at diagnosis and annually thereafter. Patients with type 1 are usually diagnosed with diabetes at or near onset of disease; therefore, annual screening for urinary albumin can begin 5 years after diagnosis.5
Proteinuria can be measured in different ways (Table 2). The basic screening test for clinical proteinuria is the urine dipstick, which is very sensitive to albumin and relatively insensitive to other proteins. “Trace-positive” results are common in healthy people, so proteinuria is not confirmed unless a patient has repeatedly positive results.
Microalbuminuria is important to measure, especially if it helps determine therapy. It is not detectable by the urinary dipstick, but can be measured in the following ways:
- Measurement of the albumin-creatinine ratio in a random spot collection
- 24-hour collection (creatinine should simultaneously be measured and creatinine clearance calculated)
- Timed collection (4 hours or overnight).
The first method is preferred, and any positive test result must be confirmed by repeat analyses of urinary albumin before a patient is diagnosed with microalbuminuria.
Occasionally a patient presenting with proteinuria but normal blood sugar and hemoglobin A1c will have a biopsy that reveals morphologic changes of classic diabetic nephropathy. Most such patients have a history of hyperglycemia, indicating that they actually have been diabetic.
Proteinuria—the best marker of disease progression
Proteinuria is the strongest predictor of renal outcomes. The Reduction in End Points in Noninsulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) study was a randomized, placebo-controlled trial in more than 1,500 patients with type 2 diabetes to test the effects of losartan on renal outcome. Those with high albuminuria (> 3.0 g albumin/g creatinine) at baseline were five times more likely to reach a renal end point and were eight times more likely to have progression to end-stage renal disease than patients with low albuminuria (< 1.5 g/g).7 The degree of albuminuria after 6 months of treatment showed similar predictive trends, indicating that monitoring and treating proteinuria are extremely important goals.
STRATEGY 1 TO LIMIT RENAL INJURY: REDUCE BLOOD PRESSURE
Blood pressure control improves renal and cardiovascular function.
As early as 1983, Parving et al,8 in a study of only 10 insulin-dependent diabetic patients, showed strong evidence that early aggressive antihypertensive treatment improved the course of diabetic nephropathy. During the mean pretreatment period of 29 months, the GFR decreased significantly and the urinary albumin excretion rate and arterial blood pressure rose significantly. During the mean 39-month period of antihypertensive treatment with metoprolol, hydralazine, and furosemide or a thiazide, mean arterial blood pressure fell from 144/97 to 128/84 mm Hg and urinary albumin excretion from 977 to 433 μg/ min. The rate of decline in GFR slowed from 0.91 mL/min/month before treatment to 0.39 mL/min/month during treatment.
The Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) trial9 enrolled more than 11,000 patients internationally with type 2 diabetes at high risk for cardiovascular events. In addition to standard therapy, blood pressure was intensively controlled in one group with a combination of the angiotensin-converting enzyme (ACE) inhibitor perindopril and the diuretic indapamide. The intensive-therapy group achieved blood pressures less than 140/80 mm Hg and had a mean reduction of systolic blood pressure of 5.6 mm Hg and diastolic blood pressure of 2.2 mm Hg vs controls. Despite these apparently modest reductions, the intensively controlled group had a significant 9% reduction of the primary outcome of combined macrovascular events (cardiovascular death, myocardial infarction, and stroke) and microvascular events (new or worsening nephropathy, or retinopathy).10
A meta-analysis of studies of patients with type 2 diabetes found reduced nephropathy with systolic blood pressure control to less than 130 mm Hg.11
The United Kingdom Prospective Diabetes Study (UKPDS) is a series of studies of diabetes. The original study in 1998 enrolled 5,102 patients with newly diagnosed type 2 diabetes.12 The more than 1,000 patients with hypertension were randomized to either tight blood pressure control or regular care. The intensive treatment group had a mean blood pressure reduction of 9 mm Hg systolic and 3 mm Hg diastolic, along with major reductions in all diabetes end points, diabetes deaths, microvascular disease, and stroke over a median follow-up of 8.4 years.
Continuous blood pressure control is critical
Tight blood pressure control must be maintained to have continued benefit. During the 10 years following the UKPDS, no attempts were made to maintain the previously assigned therapies. A follow-up study13 of 884 UKPDS patients found that blood pressures were the same again between the two groups 2 years after the trial was stopped, and no beneficial legacy effect from previous blood pressure control was evident on end points.
Control below 120 mm Hg systolic not needed
Blood pressure control slows kidney disease and prevents major macrovascular disease, but there is no evidence that lowering systolic blood pressure below 120 mm Hg provides additional benefit. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,14 more than 10,000 patients with type 2 diabetes and existing cardiovascular disease or additional cardiovascular risk factors were randomized to a goal of systolic blood pressure less than 120 mm Hg or less than 140 mm Hg (actual mean systolic pressures were 119 vs 134 mm Hg, respectively). Over nearly 5 years, there was no difference in cardiovascular events or deaths between the two groups.15
Since 1997, six international organizations have revised their recommended blood pressure goals in diabetes mellitus and renal diseases. Randomized clinical trials and observational studies have demonstrated the importance of blood pressure control to the level of 125/75 to 140/80 mm Hg. The National Kidney Foundation, the American Diabetes Association, and the Canadian Hypertension Society have developed consensus guidelines for blood pressure control to less than 130/80 mm Hg.16–21 Table 3 summarizes blood pressure goals for patients with diabetes.
STRATEGY 2: CONTROL BLOOD SUGAR
Recommendations for blood sugar goals are more controversial.
The Diabetes Control and Complications Trial22 provided early evidence that tight blood sugar control slows the development of microalbuminuria and macroalbuminuria. The study randomized more than 1,400 patients with type 1 diabetes to either standard therapy (1 or 2 daily insulin injections) or intensive therapy (an external insulin pump or 3 or more insulin injections guided by frequent blood glucose monitoring) to keep blood glucose levels close to normal. About half the patients had mild retinopathy at baseline and the others had no retinopathy. After 6.5 years, intensive therapy was found to significantly delay the onset and slow the progression of diabetic retinopathy and nephropathy.
The Kumamoto Study23 randomized 110 patients with type 2 diabetes and either no retinopathy (primary prevention cohort) or simple retinopathy (secondary prevention cohort) to receive either multiple insulin injections or conventional insulin therapy over 8 years. Intensive therapy led to lower rates of retinopathy (7.7% vs 32% in primary prevention and 19% vs 44% in secondary prevention) and progressive nephropathy (7% vs 28% in primary prevention at 6 years and 11% vs 32% in secondary prevention).
In addition to studying the effects of blood pressure control, the UKPDS also studied the effects of intensive blood glucose control.24,25 Nearly 4,000 patients with newly diagnosed type 2 diabetes were randomized to intensive treatment with a sulfonylurea or insulin, or to conventional treatment with diet. Over 10 years, the mean hemoglobin A1c was reduced to 7.0% in the intensive group and 7.9% in the conventional group. The risk of any diabetes-related end point was 12% lower in the intensive group, 10% lower for diabetes-related death, and 6% lower for all-cause mortality. There was also a 25% reduction in microvascular disease (retinopathy and nephropathy). However, the intensive group had more hypoglycemic episodes than the conventional group and a tendency to some increase in macrovascular events. A legacy effect was evident: patients who had intensive treatment had less microvascular disease progression years after stopping therapy.
Tight glycemic control reduces nephropathy, but does it increase cardiovascular risk?
Earlier trials provided strong evidence that blood glucose control prevents or slows retinopathy and nephropathy. The critical question is, “At what expense?” Although diabetes is the most common cause of kidney failure in the United States, most people with diabetes do not die of kidney failure, but of cardiovascular disease. Two recent large trials had different results regarding glycemic control below hemoglobin A1c of 7.0% and macrovascular risk, creating a controversy about what recommendations are best.
The ADVANCE trial, enrolling 11,140 patients with type 2 diabetes, was largely conducted in Australia and used the sulfonylurea glipizide for glycemic control. Compared with the group that received standard therapy (n=5,569), the intensive-treatment group (n=5,571) achieved mean hemoglobin A1c levels of 6.5% compared with 7.3% in the standard group, and had less nephropathy, less microalbuminuria, less doubling of creatinine, and a lower rate of end-stage renal disease (4% vs 5% in the standard therapy group). No difference between the two groups was found in retinopathy. Rates of all-cause mortality did not differ between the groups.9
The ACCORD trial had more than 10,000 subjects with type 2 diabetes and took place mostly in the United States. Using mainly rosiglitazone for intensive therapy, the intensive group achieved hemoglobin A1c levels of 6.4% vs 7.5% in the standard-therapy group. The trial was stopped early, at 3.7 years, because of a higher risk of death and cardiovascular events in the group with intensive glycemic control. However, the intensive-therapy group did have a significant decrease in microvascular renal outcomes and a reduction in the progression of retinopathy.14,26
In summary, tighter glycemic control improves microvascular complications—both retinopathy and nephropathy—in patients with type 2 diabetes. The benefit of intensive therapy on macrovascular complications (stroke, myocardial infarction) in long-standing diabetes has not been convincingly demonstrated in randomized trials. The UKPDS suggested that maintaining a hemoglobin A1c of 7% in patients newly diagnosed with type 2 diabetes confers long-term cardiovascular benefits. The target hemoglobin A1c for type 2 diabetes should be tailored to the patient: 7% is a reasonable goal for most patients, but the goal should be higher for the elderly and frail. Reducing the risk of cardiovascular death is still best done by controlling blood pressure, reducing lipids, quitting smoking, and losing weight.
STRATEGY 3: INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE AXIS
Components of the renin-angiotensin-aldo-sterone system are present not only in the circulation but also in many tissues, including the heart, brain, kidney, blood vessels, and adrenal glands. The role of renin-angiotensin-aldosterone system blockers in treating and preventing diabetic nephropathy has become controversial in recent years with findings from new studies.
The renin-angiotensin-aldosterone system is important in the development or maintenance of high blood pressure and the resultant damage to the brain, heart, and kidney. Drug development has focused on inhibiting steps in the biochemical pathway. ACE inhibitors block the formation of angiotensin II—the most biologically potent angiotensin peptide—and are among the most commonly used drugs to treat hypertension and concomitant conditions, such as renal insufficiency, proteinuria, and heart failure. Angiotensin receptor blockers (ARBs) interact with the angiotensin AT1 receptor and block most of its actions. They are approved by the US Food and Drug Administration (FDA) for the treatment of hypertension, and they help prevent left ventricular hypertrophy and mesangial sclerosis. Large studies have shown that ACE inhibitors and ARBs offer similar cardiovascular benefit.
The glomerulus has the only capillary bed with a blood supply that drains into an efferent arteriole instead of a venule, providing high resistance to aid filtration. Efferent arterioles are rich in AT1 receptors. In the presence of angiotensin II they constrict, increasing pressure in the glomerulus, which can lead to proteinuria and glomerulosclerosis. ACE inhibitors and ARBs relax the efferent arteriole, allowing increased blood flow through the glomerulus. This reduction in intraglomerular pressure is associated with less proteinuria and less glomerulosclerosis.
Diabetes promotes renal disease in many ways. Glucose and advanced glycation end products can lead to increased blood flow and increased pressure in the glomerulus. Through a variety of pathways, hyperglycemia, acting on angiotensin II, leads to NF-kapa beta production, profibrotic cytokines, increased matrix, and eventual fibrosis. ACE inhibitors and ARBs counteract many of these.
ACE inhibitors and ARBs slow nephropathy progression beyond blood pressure control
Several major clinical trials27–32 examined the effects of either ACE inhibitors or ARBs in slowing the progression of diabetic nephropathy and have had consistently positive results.
The Collaborative Study Group30 was a 3-year randomized trial in 419 patients with type 1 diabetes, using the ACE inhibitor captopril vs placebo. Captopril was associated with less decline in kidney function and a 50% reduction in the risk of the combined end points of death, dialysis, and transplantation that was independent of the small difference in blood pressures between the two groups.
The Irbesartan Diabetic Nephropathy Trial (IDNT)31 studied the effect of the ARB irbesartan vs the calcium channel blocker amlodipine vs placebo over 2.6 years in 1,715 patients with type 2 diabetes. Irbesartan was found to be significantly more effective in protecting against the progression of nephropathy, independent of reduction in blood pressure.
The RENAAL trial,32 published in 2001, was a 3-year, randomized, double-blind study comparing the ARB losartan at increasing dosages with placebo (both taken in addition to conventional antihypertensive treatment) in 1,513 patients with type 2 diabetes and nephropathy. The blood pressure goal was 140/90 mm Hg in both groups, but the losartan group had a lower rate of doubling of serum creatinine, end-stage renal disease, and combined end-stage renal disease or death.
‘Aldosterone escape’ motivates the search for new therapies
An important reason for developing more ways to block the renin-angiotensin-aldosterone system is because of “aldosterone escape,” the phenomenon of angiotensin II or aldosterone returning to pretreatment levels despite continued ACE inhibition.
Biollaz et al,33 in a 1982 study of 19 patients with hypertension, showed that despite reducing blood pressure and keeping the blood level of ACE very low with twice-daily enalapril 20 mg, blood and urine levels of angiotensin II steadily rose back to baseline levels within a few months.
A growing body of evidence suggests that despite effective inhibition of angiotensin II activity, non-ACE synthetic pathways still permit angiotensin II generation via serine proteases such as chymase, cathepsin G, and tissue plasminogen activator.
Thus, efforts have been made to block the renin-angiotensin system in other places. In addition to ACE inhibitors and ARBs, two aldosterone receptor antagonists are available, spironolactone and eplerenone, both used to treat heart failure. A direct renin inhibitor, aliskiren, is also available.
Combination therapy—less proteinuria, but…
A number of studies have shown that combination treatment with agents having different targets in the renin-angiotensin-aldosterone system leads to larger reductions in albuminuria than does single-agent therapy.
Mogensen et al34 studied the effect of the ACE inhibitor lisinopril (20 mg per day) plus the ARB candesartan (16 mg per day) in subjects with microalbuminuria, hypertension, and type 2 diabetes. Combined treatment was more effective in reducing proteinuria.
Epstein et al35 studied the effects of the ACE inhibitor enalapril (20 mg/day) combined with either of two doses of the selective aldosterone receptor antagonist eplerenone (50 or 100 mg/day) or placebo. Both eplerenone dosages, when added to the enalapril treatment, significantly reduced albuminuria from baseline as early as week 4 (P < .001), but placebo treatment added to the enalapril did not result in any significant decrease in urinary albumin excretion. Systolic blood pressure decreased significantly in all treatment groups and by about the same amount.
The Aliskiren Combined With Losartan in Type 2 Diabetes and Nephropathy (AVOID) trial36 randomized more than 600 patients with type 2 diabetes and nephropathy to aliskiren (a renin inhibitor) or placebo added to the ARB losartan. Again, combination treatment was more renoprotective, independent of blood pressure lowering.
Worse outcomes with combination therapy?
More recent studies have indicated that although combination therapy reduces proteinuria to a greater extent than monotherapy, overall it worsens major renal and cardiovascular outcomes. The multicenter Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)37 randomized more than 25,000 patients age 55 and older with established atherosclerotic vascular disease or with diabetes and end-organ damage to receive either the ARB telmisartan 80 mg daily, the ACE inhibitor ramipril 10 mg daily, or both. Mean follow-up was 56 months. The combination-treatment group had higher rates of death and renal disease than the single-therapy groups (which did not differ from one another).
Why the combination therapy had poorer outcomes is under debate. Patients may get sudden drops in blood pressure that are not detected with only periodic monitoring. Renal failure was mostly acute rather than chronic, and the estimated GFR declined more in the combined therapy group than in the single-therapy groups.
The Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints (ALTITUDE) was designed to test the effect of the direct renin inhibitor aliskiren or placebo, both arms combined with either an ACE inhibitor or an ARB in patients with type 2 diabetes at high risk for cardiovascular and renal events. The trial was terminated early because of more strokes and deaths in the combination therapy arms. The results led the FDA to issue black box warnings against using aliskiren with these other classes of agents, and all studies testing similar combinations have been stopped. (In one study that was stopped and has not yet been published, 100 patients with proteinuria were treated with either aliskiren, the ARB losartan, or both, to evaluate the effects of aldosterone escape. Results showed no differences: about one-third of each group had this phenomenon.)
My personal recommendation is as follows: for younger patients with proteinuria, at lower risk for cardiovascular events and with disease due not to diabetes but to immunoglobulin A nephropathy or another proteinuric kidney disease, treat with both an ACE inhibitor and ARB. But the combination should not be used for patients at high risk of cardiovascular disease, which includes almost all patients with diabetes.
If more aggressive renin-angiotensin system blockade is needed against diabetic nephropathy, adding a diuretic increases the impact of blocking the renin-angiotensin-aldosterone system on both proteinuria and progression of renal disease. The aldosterone blocker spironolactone 25 mg can be added if potassium levels are carefully monitored.
ACE inhibitor plus calcium channel blocker is safer than ACE inhibitor plus diuretic
The Avoiding Cardiovascular Events Through Combination Therapy in Patients Living With Systolic Hypertension (ACCOMPLISH) trial38 randomized more than 11,000 high-risk patients with hypertension to receive an ACE inhibitor (benazepril) plus either a calcium channel blocker (amlodipine) or thiazide diuretic (hydrochlorothiazide). Blood pressures were identical between the two groups, but the trial was terminated early, at 36 months, because of a higher risk of the combined end point of cardiovascular death, myocardial infarction, stroke, and other major cardiac events in the ACE inhibitor-thiazide group.
Although some experts believe this study is definitive and indicates that high blood pressure should never be treated with an ACE inhibitor-thiazide combination, I believe that caution is needed in interpreting these findings. This regimen should be avoided in older patients with diabetes at high risk for cardiovascular disease, but otherwise, getting blood pressure under control is critical, and this combination can be used if it works and the patient is tolerating it well.
In summary, the choice of blood pressure-lowering medications is based on reducing cardiovascular events and slowing the progression of kidney disease. Either an ACE inhibitor or an ARB is the first choice for patients with diabetes, hypertension, and any degree of proteinuria. Many experts recommend beginning one of these agents even if proteinuria is not present. However, the combination of an ACE inhibitor and ARB should not be used in diabetic patients, especially if they have cardiovascular disease, until further data clarify the results of the ONTARGET and ALTITUDE trials.
STRATEGY 4: METABOLIC MANIPULATION WITH NOVEL AGENTS
Several new agents have recently been studied for the treatment of diabetic nephropathy, including aminoguanidine, which reduces levels of advanced glycation end-products, and sulodexide, which blocks basement membrane permeability. Neither agent has been shown to be safe and effective in diabetic nephropathy. The newest agent is bardoxolone methyl. It induces the Keap1–Nrf2 pathway, which up-regulates cytoprotective factors, suppressing inflammatory and other cytokines that are major mediators of progression of chronic kidney disease.39
Pergola et al,40 in a phase 2, double-blind trial, randomized 227 adults with diabetic kidney disease and a low estimated GFR (20–45 mL/min/1.73 m2) to receive placebo or bardoxolone 25, 75, or 150 mg daily. Drug treatment was associated with improvement in the estimated GFR, a finding that persisted throughout the 52 weeks of treatment. Surprisingly, proteinuria did not decrease with drug treatment.
As of this writing, a large multicenter controlled randomized trial has been halted because of concerns by the data safety monitoring board, which found increased rates of death and fluid retention with the drug. A number of recent trials have shown a beneficial effect of sodium bicarbonate therapy in patients with late-stage chronic kidney disease. They have shown slowing of the progression of GFR decline in a number of renal diseases, including diabetes.
Diabetes is on the rise, and so is diabetic nephropathy. In view of this epidemic, physicians should consider strategies to detect and control kidney disease in their diabetic patients.
This article will focus on kidney disease in adult-onset type 2 diabetes. Although it has different pathogenetic mechanisms than type 1 diabetes, the clinical course of the two conditions is very similar in terms of the prevalence of proteinuria after diagnosis, the progression to renal failure after the onset of proteinuria, and treatment options.1
DIABETES AND DIABETIC KIDNEY DISEASE ARE ON THE RISE
The incidence of diabetes increases with age, and with the aging of the baby boomers, its prevalence is growing dramatically. The 2005– 2008 National Health and Nutrition Examination Survey estimated the prevalence as 3.7% in adults age 20 to 44, 13.7% at age 45 to 64, and 26.9% in people age 65 and older. The obesity epidemic is also contributing to the increase in diabetes in all age groups.
Diabetic kidney disease has increased in the United States from about 4 million cases 20 years ago to about 7 million in 2005–2008.2 Diabetes is the major cause of end-stage renal disease in the developed world, accounting for 40% to 50% of cases. Other major causes are hypertension (27%) and glomerulonephritis (13%).3
Physicians in nearly every field of medicine now care for patients with diabetic nephropathy. The classic presentation—a patient who has impaired vision, fluid retention with edema, and hypertension—is commonly seen in dialysis units and ophthalmology and cardiovascular clinics.
CLINICAL PROGRESSION
Early in the course of diabetic nephropathy, blood pressure is normal and microalbuminuria is not evident, but many patients have a high glomerular filtration rate (GFR), indicating temporarily “enhanced” renal function or hyperfiltration. The next stage is characterized by microalbuminuria, correlating with glomerular mesangial expansion: the GFR falls back into the normal range and blood pressure starts to increase. Finally, macroalbuminuria occurs, accompanied by rising blood pressure and a declining GFR, correlating with the histologic appearance of glomerulosclerosis and Kimmelstiel-Wilson nodules.4
Hypertension develops in 5% of patients by 10 years after type 1 diabetes is diagnosed, 33% by 20 years, and 70% by 40 years. In contrast, 40% of patients with type 2 diabetes have high blood pressure at diagnosis.
Unfortunately, in most cases, this progression is a one-way street, so it is critical to intervene to try to slow the progression early in the course of the disease process.
SCREENING FOR DIABETIC NEPHROPATHY
Nephropathy screening guidelines for patients with diabetes are provided in Table 1.5
Blood pressure should be monitored at each office visit (Table 1). The goal for adults with diabetes should be to reduce blood pressure to 130/80 mm Hg. Reduction beyond this level may be associated with an increased mortality rate.6 Very high blood pressure (> 180 mm Hg systolic) should be lowered slowly. Lowering blood pressure delays the progression from microalbuminuria (30–299 mg/day or 20–199 μg/min) to macroalbuminuria (> 300 mg/day or > 200 μg/min) and slows the progression to renal failure.
Urinary albumin. Proteinuria takes 5 to 10 years to develop after the onset of diabetes. Because it is possible for patients with type 2 diabetes to have had the disease for some time before being diagnosed, urinary albumin screening should be performed at diagnosis and annually thereafter. Patients with type 1 are usually diagnosed with diabetes at or near onset of disease; therefore, annual screening for urinary albumin can begin 5 years after diagnosis.5
Proteinuria can be measured in different ways (Table 2). The basic screening test for clinical proteinuria is the urine dipstick, which is very sensitive to albumin and relatively insensitive to other proteins. “Trace-positive” results are common in healthy people, so proteinuria is not confirmed unless a patient has repeatedly positive results.
Microalbuminuria is important to measure, especially if it helps determine therapy. It is not detectable by the urinary dipstick, but can be measured in the following ways:
- Measurement of the albumin-creatinine ratio in a random spot collection
- 24-hour collection (creatinine should simultaneously be measured and creatinine clearance calculated)
- Timed collection (4 hours or overnight).
The first method is preferred, and any positive test result must be confirmed by repeat analyses of urinary albumin before a patient is diagnosed with microalbuminuria.
Occasionally a patient presenting with proteinuria but normal blood sugar and hemoglobin A1c will have a biopsy that reveals morphologic changes of classic diabetic nephropathy. Most such patients have a history of hyperglycemia, indicating that they actually have been diabetic.
Proteinuria—the best marker of disease progression
Proteinuria is the strongest predictor of renal outcomes. The Reduction in End Points in Noninsulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) study was a randomized, placebo-controlled trial in more than 1,500 patients with type 2 diabetes to test the effects of losartan on renal outcome. Those with high albuminuria (> 3.0 g albumin/g creatinine) at baseline were five times more likely to reach a renal end point and were eight times more likely to have progression to end-stage renal disease than patients with low albuminuria (< 1.5 g/g).7 The degree of albuminuria after 6 months of treatment showed similar predictive trends, indicating that monitoring and treating proteinuria are extremely important goals.
STRATEGY 1 TO LIMIT RENAL INJURY: REDUCE BLOOD PRESSURE
Blood pressure control improves renal and cardiovascular function.
As early as 1983, Parving et al,8 in a study of only 10 insulin-dependent diabetic patients, showed strong evidence that early aggressive antihypertensive treatment improved the course of diabetic nephropathy. During the mean pretreatment period of 29 months, the GFR decreased significantly and the urinary albumin excretion rate and arterial blood pressure rose significantly. During the mean 39-month period of antihypertensive treatment with metoprolol, hydralazine, and furosemide or a thiazide, mean arterial blood pressure fell from 144/97 to 128/84 mm Hg and urinary albumin excretion from 977 to 433 μg/ min. The rate of decline in GFR slowed from 0.91 mL/min/month before treatment to 0.39 mL/min/month during treatment.
The Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) trial9 enrolled more than 11,000 patients internationally with type 2 diabetes at high risk for cardiovascular events. In addition to standard therapy, blood pressure was intensively controlled in one group with a combination of the angiotensin-converting enzyme (ACE) inhibitor perindopril and the diuretic indapamide. The intensive-therapy group achieved blood pressures less than 140/80 mm Hg and had a mean reduction of systolic blood pressure of 5.6 mm Hg and diastolic blood pressure of 2.2 mm Hg vs controls. Despite these apparently modest reductions, the intensively controlled group had a significant 9% reduction of the primary outcome of combined macrovascular events (cardiovascular death, myocardial infarction, and stroke) and microvascular events (new or worsening nephropathy, or retinopathy).10
A meta-analysis of studies of patients with type 2 diabetes found reduced nephropathy with systolic blood pressure control to less than 130 mm Hg.11
The United Kingdom Prospective Diabetes Study (UKPDS) is a series of studies of diabetes. The original study in 1998 enrolled 5,102 patients with newly diagnosed type 2 diabetes.12 The more than 1,000 patients with hypertension were randomized to either tight blood pressure control or regular care. The intensive treatment group had a mean blood pressure reduction of 9 mm Hg systolic and 3 mm Hg diastolic, along with major reductions in all diabetes end points, diabetes deaths, microvascular disease, and stroke over a median follow-up of 8.4 years.
Continuous blood pressure control is critical
Tight blood pressure control must be maintained to have continued benefit. During the 10 years following the UKPDS, no attempts were made to maintain the previously assigned therapies. A follow-up study13 of 884 UKPDS patients found that blood pressures were the same again between the two groups 2 years after the trial was stopped, and no beneficial legacy effect from previous blood pressure control was evident on end points.
Control below 120 mm Hg systolic not needed
Blood pressure control slows kidney disease and prevents major macrovascular disease, but there is no evidence that lowering systolic blood pressure below 120 mm Hg provides additional benefit. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,14 more than 10,000 patients with type 2 diabetes and existing cardiovascular disease or additional cardiovascular risk factors were randomized to a goal of systolic blood pressure less than 120 mm Hg or less than 140 mm Hg (actual mean systolic pressures were 119 vs 134 mm Hg, respectively). Over nearly 5 years, there was no difference in cardiovascular events or deaths between the two groups.15
Since 1997, six international organizations have revised their recommended blood pressure goals in diabetes mellitus and renal diseases. Randomized clinical trials and observational studies have demonstrated the importance of blood pressure control to the level of 125/75 to 140/80 mm Hg. The National Kidney Foundation, the American Diabetes Association, and the Canadian Hypertension Society have developed consensus guidelines for blood pressure control to less than 130/80 mm Hg.16–21 Table 3 summarizes blood pressure goals for patients with diabetes.
STRATEGY 2: CONTROL BLOOD SUGAR
Recommendations for blood sugar goals are more controversial.
The Diabetes Control and Complications Trial22 provided early evidence that tight blood sugar control slows the development of microalbuminuria and macroalbuminuria. The study randomized more than 1,400 patients with type 1 diabetes to either standard therapy (1 or 2 daily insulin injections) or intensive therapy (an external insulin pump or 3 or more insulin injections guided by frequent blood glucose monitoring) to keep blood glucose levels close to normal. About half the patients had mild retinopathy at baseline and the others had no retinopathy. After 6.5 years, intensive therapy was found to significantly delay the onset and slow the progression of diabetic retinopathy and nephropathy.
The Kumamoto Study23 randomized 110 patients with type 2 diabetes and either no retinopathy (primary prevention cohort) or simple retinopathy (secondary prevention cohort) to receive either multiple insulin injections or conventional insulin therapy over 8 years. Intensive therapy led to lower rates of retinopathy (7.7% vs 32% in primary prevention and 19% vs 44% in secondary prevention) and progressive nephropathy (7% vs 28% in primary prevention at 6 years and 11% vs 32% in secondary prevention).
In addition to studying the effects of blood pressure control, the UKPDS also studied the effects of intensive blood glucose control.24,25 Nearly 4,000 patients with newly diagnosed type 2 diabetes were randomized to intensive treatment with a sulfonylurea or insulin, or to conventional treatment with diet. Over 10 years, the mean hemoglobin A1c was reduced to 7.0% in the intensive group and 7.9% in the conventional group. The risk of any diabetes-related end point was 12% lower in the intensive group, 10% lower for diabetes-related death, and 6% lower for all-cause mortality. There was also a 25% reduction in microvascular disease (retinopathy and nephropathy). However, the intensive group had more hypoglycemic episodes than the conventional group and a tendency to some increase in macrovascular events. A legacy effect was evident: patients who had intensive treatment had less microvascular disease progression years after stopping therapy.
Tight glycemic control reduces nephropathy, but does it increase cardiovascular risk?
Earlier trials provided strong evidence that blood glucose control prevents or slows retinopathy and nephropathy. The critical question is, “At what expense?” Although diabetes is the most common cause of kidney failure in the United States, most people with diabetes do not die of kidney failure, but of cardiovascular disease. Two recent large trials had different results regarding glycemic control below hemoglobin A1c of 7.0% and macrovascular risk, creating a controversy about what recommendations are best.
The ADVANCE trial, enrolling 11,140 patients with type 2 diabetes, was largely conducted in Australia and used the sulfonylurea glipizide for glycemic control. Compared with the group that received standard therapy (n=5,569), the intensive-treatment group (n=5,571) achieved mean hemoglobin A1c levels of 6.5% compared with 7.3% in the standard group, and had less nephropathy, less microalbuminuria, less doubling of creatinine, and a lower rate of end-stage renal disease (4% vs 5% in the standard therapy group). No difference between the two groups was found in retinopathy. Rates of all-cause mortality did not differ between the groups.9
The ACCORD trial had more than 10,000 subjects with type 2 diabetes and took place mostly in the United States. Using mainly rosiglitazone for intensive therapy, the intensive group achieved hemoglobin A1c levels of 6.4% vs 7.5% in the standard-therapy group. The trial was stopped early, at 3.7 years, because of a higher risk of death and cardiovascular events in the group with intensive glycemic control. However, the intensive-therapy group did have a significant decrease in microvascular renal outcomes and a reduction in the progression of retinopathy.14,26
In summary, tighter glycemic control improves microvascular complications—both retinopathy and nephropathy—in patients with type 2 diabetes. The benefit of intensive therapy on macrovascular complications (stroke, myocardial infarction) in long-standing diabetes has not been convincingly demonstrated in randomized trials. The UKPDS suggested that maintaining a hemoglobin A1c of 7% in patients newly diagnosed with type 2 diabetes confers long-term cardiovascular benefits. The target hemoglobin A1c for type 2 diabetes should be tailored to the patient: 7% is a reasonable goal for most patients, but the goal should be higher for the elderly and frail. Reducing the risk of cardiovascular death is still best done by controlling blood pressure, reducing lipids, quitting smoking, and losing weight.
STRATEGY 3: INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE AXIS
Components of the renin-angiotensin-aldo-sterone system are present not only in the circulation but also in many tissues, including the heart, brain, kidney, blood vessels, and adrenal glands. The role of renin-angiotensin-aldosterone system blockers in treating and preventing diabetic nephropathy has become controversial in recent years with findings from new studies.
The renin-angiotensin-aldosterone system is important in the development or maintenance of high blood pressure and the resultant damage to the brain, heart, and kidney. Drug development has focused on inhibiting steps in the biochemical pathway. ACE inhibitors block the formation of angiotensin II—the most biologically potent angiotensin peptide—and are among the most commonly used drugs to treat hypertension and concomitant conditions, such as renal insufficiency, proteinuria, and heart failure. Angiotensin receptor blockers (ARBs) interact with the angiotensin AT1 receptor and block most of its actions. They are approved by the US Food and Drug Administration (FDA) for the treatment of hypertension, and they help prevent left ventricular hypertrophy and mesangial sclerosis. Large studies have shown that ACE inhibitors and ARBs offer similar cardiovascular benefit.
The glomerulus has the only capillary bed with a blood supply that drains into an efferent arteriole instead of a venule, providing high resistance to aid filtration. Efferent arterioles are rich in AT1 receptors. In the presence of angiotensin II they constrict, increasing pressure in the glomerulus, which can lead to proteinuria and glomerulosclerosis. ACE inhibitors and ARBs relax the efferent arteriole, allowing increased blood flow through the glomerulus. This reduction in intraglomerular pressure is associated with less proteinuria and less glomerulosclerosis.
Diabetes promotes renal disease in many ways. Glucose and advanced glycation end products can lead to increased blood flow and increased pressure in the glomerulus. Through a variety of pathways, hyperglycemia, acting on angiotensin II, leads to NF-kapa beta production, profibrotic cytokines, increased matrix, and eventual fibrosis. ACE inhibitors and ARBs counteract many of these.
ACE inhibitors and ARBs slow nephropathy progression beyond blood pressure control
Several major clinical trials27–32 examined the effects of either ACE inhibitors or ARBs in slowing the progression of diabetic nephropathy and have had consistently positive results.
The Collaborative Study Group30 was a 3-year randomized trial in 419 patients with type 1 diabetes, using the ACE inhibitor captopril vs placebo. Captopril was associated with less decline in kidney function and a 50% reduction in the risk of the combined end points of death, dialysis, and transplantation that was independent of the small difference in blood pressures between the two groups.
The Irbesartan Diabetic Nephropathy Trial (IDNT)31 studied the effect of the ARB irbesartan vs the calcium channel blocker amlodipine vs placebo over 2.6 years in 1,715 patients with type 2 diabetes. Irbesartan was found to be significantly more effective in protecting against the progression of nephropathy, independent of reduction in blood pressure.
The RENAAL trial,32 published in 2001, was a 3-year, randomized, double-blind study comparing the ARB losartan at increasing dosages with placebo (both taken in addition to conventional antihypertensive treatment) in 1,513 patients with type 2 diabetes and nephropathy. The blood pressure goal was 140/90 mm Hg in both groups, but the losartan group had a lower rate of doubling of serum creatinine, end-stage renal disease, and combined end-stage renal disease or death.
‘Aldosterone escape’ motivates the search for new therapies
An important reason for developing more ways to block the renin-angiotensin-aldosterone system is because of “aldosterone escape,” the phenomenon of angiotensin II or aldosterone returning to pretreatment levels despite continued ACE inhibition.
Biollaz et al,33 in a 1982 study of 19 patients with hypertension, showed that despite reducing blood pressure and keeping the blood level of ACE very low with twice-daily enalapril 20 mg, blood and urine levels of angiotensin II steadily rose back to baseline levels within a few months.
A growing body of evidence suggests that despite effective inhibition of angiotensin II activity, non-ACE synthetic pathways still permit angiotensin II generation via serine proteases such as chymase, cathepsin G, and tissue plasminogen activator.
Thus, efforts have been made to block the renin-angiotensin system in other places. In addition to ACE inhibitors and ARBs, two aldosterone receptor antagonists are available, spironolactone and eplerenone, both used to treat heart failure. A direct renin inhibitor, aliskiren, is also available.
Combination therapy—less proteinuria, but…
A number of studies have shown that combination treatment with agents having different targets in the renin-angiotensin-aldosterone system leads to larger reductions in albuminuria than does single-agent therapy.
Mogensen et al34 studied the effect of the ACE inhibitor lisinopril (20 mg per day) plus the ARB candesartan (16 mg per day) in subjects with microalbuminuria, hypertension, and type 2 diabetes. Combined treatment was more effective in reducing proteinuria.
Epstein et al35 studied the effects of the ACE inhibitor enalapril (20 mg/day) combined with either of two doses of the selective aldosterone receptor antagonist eplerenone (50 or 100 mg/day) or placebo. Both eplerenone dosages, when added to the enalapril treatment, significantly reduced albuminuria from baseline as early as week 4 (P < .001), but placebo treatment added to the enalapril did not result in any significant decrease in urinary albumin excretion. Systolic blood pressure decreased significantly in all treatment groups and by about the same amount.
The Aliskiren Combined With Losartan in Type 2 Diabetes and Nephropathy (AVOID) trial36 randomized more than 600 patients with type 2 diabetes and nephropathy to aliskiren (a renin inhibitor) or placebo added to the ARB losartan. Again, combination treatment was more renoprotective, independent of blood pressure lowering.
Worse outcomes with combination therapy?
More recent studies have indicated that although combination therapy reduces proteinuria to a greater extent than monotherapy, overall it worsens major renal and cardiovascular outcomes. The multicenter Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)37 randomized more than 25,000 patients age 55 and older with established atherosclerotic vascular disease or with diabetes and end-organ damage to receive either the ARB telmisartan 80 mg daily, the ACE inhibitor ramipril 10 mg daily, or both. Mean follow-up was 56 months. The combination-treatment group had higher rates of death and renal disease than the single-therapy groups (which did not differ from one another).
Why the combination therapy had poorer outcomes is under debate. Patients may get sudden drops in blood pressure that are not detected with only periodic monitoring. Renal failure was mostly acute rather than chronic, and the estimated GFR declined more in the combined therapy group than in the single-therapy groups.
The Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints (ALTITUDE) was designed to test the effect of the direct renin inhibitor aliskiren or placebo, both arms combined with either an ACE inhibitor or an ARB in patients with type 2 diabetes at high risk for cardiovascular and renal events. The trial was terminated early because of more strokes and deaths in the combination therapy arms. The results led the FDA to issue black box warnings against using aliskiren with these other classes of agents, and all studies testing similar combinations have been stopped. (In one study that was stopped and has not yet been published, 100 patients with proteinuria were treated with either aliskiren, the ARB losartan, or both, to evaluate the effects of aldosterone escape. Results showed no differences: about one-third of each group had this phenomenon.)
My personal recommendation is as follows: for younger patients with proteinuria, at lower risk for cardiovascular events and with disease due not to diabetes but to immunoglobulin A nephropathy or another proteinuric kidney disease, treat with both an ACE inhibitor and ARB. But the combination should not be used for patients at high risk of cardiovascular disease, which includes almost all patients with diabetes.
If more aggressive renin-angiotensin system blockade is needed against diabetic nephropathy, adding a diuretic increases the impact of blocking the renin-angiotensin-aldosterone system on both proteinuria and progression of renal disease. The aldosterone blocker spironolactone 25 mg can be added if potassium levels are carefully monitored.
ACE inhibitor plus calcium channel blocker is safer than ACE inhibitor plus diuretic
The Avoiding Cardiovascular Events Through Combination Therapy in Patients Living With Systolic Hypertension (ACCOMPLISH) trial38 randomized more than 11,000 high-risk patients with hypertension to receive an ACE inhibitor (benazepril) plus either a calcium channel blocker (amlodipine) or thiazide diuretic (hydrochlorothiazide). Blood pressures were identical between the two groups, but the trial was terminated early, at 36 months, because of a higher risk of the combined end point of cardiovascular death, myocardial infarction, stroke, and other major cardiac events in the ACE inhibitor-thiazide group.
Although some experts believe this study is definitive and indicates that high blood pressure should never be treated with an ACE inhibitor-thiazide combination, I believe that caution is needed in interpreting these findings. This regimen should be avoided in older patients with diabetes at high risk for cardiovascular disease, but otherwise, getting blood pressure under control is critical, and this combination can be used if it works and the patient is tolerating it well.
In summary, the choice of blood pressure-lowering medications is based on reducing cardiovascular events and slowing the progression of kidney disease. Either an ACE inhibitor or an ARB is the first choice for patients with diabetes, hypertension, and any degree of proteinuria. Many experts recommend beginning one of these agents even if proteinuria is not present. However, the combination of an ACE inhibitor and ARB should not be used in diabetic patients, especially if they have cardiovascular disease, until further data clarify the results of the ONTARGET and ALTITUDE trials.
STRATEGY 4: METABOLIC MANIPULATION WITH NOVEL AGENTS
Several new agents have recently been studied for the treatment of diabetic nephropathy, including aminoguanidine, which reduces levels of advanced glycation end-products, and sulodexide, which blocks basement membrane permeability. Neither agent has been shown to be safe and effective in diabetic nephropathy. The newest agent is bardoxolone methyl. It induces the Keap1–Nrf2 pathway, which up-regulates cytoprotective factors, suppressing inflammatory and other cytokines that are major mediators of progression of chronic kidney disease.39
Pergola et al,40 in a phase 2, double-blind trial, randomized 227 adults with diabetic kidney disease and a low estimated GFR (20–45 mL/min/1.73 m2) to receive placebo or bardoxolone 25, 75, or 150 mg daily. Drug treatment was associated with improvement in the estimated GFR, a finding that persisted throughout the 52 weeks of treatment. Surprisingly, proteinuria did not decrease with drug treatment.
As of this writing, a large multicenter controlled randomized trial has been halted because of concerns by the data safety monitoring board, which found increased rates of death and fluid retention with the drug. A number of recent trials have shown a beneficial effect of sodium bicarbonate therapy in patients with late-stage chronic kidney disease. They have shown slowing of the progression of GFR decline in a number of renal diseases, including diabetes.
- Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med 1999; 341:1127–1133.
- de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011; 305:2532–2539.
- United States Renal Data System (USRDS) 2000 Annual Data Report. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases – Division of Kidney, Urologic and Hematologic Diseases. USRDS Coordinating Center operated by the Minneapolis Medical Research Foundation. www.usrds.org
- Macisaac RJ, Jerums G. Diabetic kidney disease with and without albuminuria. Curr Opin Nephrol Hypertens 2011; 20:246–257.
- Molitch ME, DeFronzo RA, Franz MJ, et al; American Diabetes Association. Nephropathy in diabetes. Diabetes Care 2004; 27(suppl 1):S79–S83.
- Vamos EP, Harris M, Millett C, et al. Association of systolic and diastolic blood pressure and all cause mortality in people with newly diagnosed type 2 diabetes: retrospective cohort study. BMJ 2012; 345:e5567.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Parving HH, Andersen AR, Smidt UM, Svendsen PA. Early aggressive antihypertensive treatment reduces rate of decline in kidney function in diabetic nephropathy. Lancet 1983; 1:1175–1179.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet 2007; 370:829–840.
- Bangalore S, Kumar S, Lobach I, Messerli FH. Blood pressure targets in subjects with type 2 diabetes mellitus/impaired fasting glucose: observations from traditional and bayesian random-effects meta-analyses of randomized trials. Circulation 2011; 123:2799–2810.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63. (Erratum in: Diabetes Care 2012; 35:660.)
- Bakris GL, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 2000; 36:646–661.
- Ramsay L, Williams B, Johnston G, et al. Guidelines for management of hypertension: report of the third working party of the British Hypertension Society. J Hum Hypertens 1999; 13:569–592.
- Feldman RD, Campbell N, Larochelle P, et al. 1999 Canadian recommendations for the management of hypertension. Task Force for the Development of the 1999 Canadian Recommendations for the Management of Hypertension. CMAJ 1999; 161(suppl):12:S1–S17.
- Chalmers J, MacMahon S, Mancia G, et al. 1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension. Guidelines Sub-committee of the World Health Organization. Clin Exp Hypertens 1999; 21:1009–1060.
- The seventh report of the Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23(suppl 2):B21–B29.
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Ismail-Beigi F, Craven T, Banerji MA, et al; ACCORD trial group. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010; 376:419–430. Erratum in: Lancet 2010; 376:1466.
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000: 355:253–259. Erratum in: Lancet2000; 356:860.
- Parving HH, Lehnert H, Bröchner-Mortensen J, et al; Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001; 345:870–878.
- Viberti G, Wheeldon NM; MicroAlbuminuria Reduction With VALsartan (MARVAL) Study Investigators. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: a blood pressure-independent effect. Circulation 2002; 106:672–678.
- Lewis EJ, Hunsicker LG, Bain R P, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Biollaz J, Brunner HR, Gavras I, Waeber B, Gavras H. Antihypertensive therapy with MK 421: angiotensin II--renin relationships to evaluate efficacy of converting enzyme blockade. J Cardiovasc Pharmacol 1982; 4:966–972.
- Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ 2000; 321:1440–1444.
- Epstein M, Williams GH, Weinberger M, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol 2006; 1:940–951.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK; AVOID Study Investigators. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008; 358:2433–2446.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Jamerson K, Weber MA, Bakris GL, et al; ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 2010; 298:F662–F671.
- Pergola PE, Raskin P, Toto RD, et al; BEAM Study Investigators Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 2011; 365:327–336.
- Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med 1999; 341:1127–1133.
- de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011; 305:2532–2539.
- United States Renal Data System (USRDS) 2000 Annual Data Report. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases – Division of Kidney, Urologic and Hematologic Diseases. USRDS Coordinating Center operated by the Minneapolis Medical Research Foundation. www.usrds.org
- Macisaac RJ, Jerums G. Diabetic kidney disease with and without albuminuria. Curr Opin Nephrol Hypertens 2011; 20:246–257.
- Molitch ME, DeFronzo RA, Franz MJ, et al; American Diabetes Association. Nephropathy in diabetes. Diabetes Care 2004; 27(suppl 1):S79–S83.
- Vamos EP, Harris M, Millett C, et al. Association of systolic and diastolic blood pressure and all cause mortality in people with newly diagnosed type 2 diabetes: retrospective cohort study. BMJ 2012; 345:e5567.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Parving HH, Andersen AR, Smidt UM, Svendsen PA. Early aggressive antihypertensive treatment reduces rate of decline in kidney function in diabetic nephropathy. Lancet 1983; 1:1175–1179.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet 2007; 370:829–840.
- Bangalore S, Kumar S, Lobach I, Messerli FH. Blood pressure targets in subjects with type 2 diabetes mellitus/impaired fasting glucose: observations from traditional and bayesian random-effects meta-analyses of randomized trials. Circulation 2011; 123:2799–2810.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63. (Erratum in: Diabetes Care 2012; 35:660.)
- Bakris GL, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 2000; 36:646–661.
- Ramsay L, Williams B, Johnston G, et al. Guidelines for management of hypertension: report of the third working party of the British Hypertension Society. J Hum Hypertens 1999; 13:569–592.
- Feldman RD, Campbell N, Larochelle P, et al. 1999 Canadian recommendations for the management of hypertension. Task Force for the Development of the 1999 Canadian Recommendations for the Management of Hypertension. CMAJ 1999; 161(suppl):12:S1–S17.
- Chalmers J, MacMahon S, Mancia G, et al. 1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension. Guidelines Sub-committee of the World Health Organization. Clin Exp Hypertens 1999; 21:1009–1060.
- The seventh report of the Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23(suppl 2):B21–B29.
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Ismail-Beigi F, Craven T, Banerji MA, et al; ACCORD trial group. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010; 376:419–430. Erratum in: Lancet 2010; 376:1466.
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000: 355:253–259. Erratum in: Lancet2000; 356:860.
- Parving HH, Lehnert H, Bröchner-Mortensen J, et al; Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001; 345:870–878.
- Viberti G, Wheeldon NM; MicroAlbuminuria Reduction With VALsartan (MARVAL) Study Investigators. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: a blood pressure-independent effect. Circulation 2002; 106:672–678.
- Lewis EJ, Hunsicker LG, Bain R P, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Biollaz J, Brunner HR, Gavras I, Waeber B, Gavras H. Antihypertensive therapy with MK 421: angiotensin II--renin relationships to evaluate efficacy of converting enzyme blockade. J Cardiovasc Pharmacol 1982; 4:966–972.
- Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ 2000; 321:1440–1444.
- Epstein M, Williams GH, Weinberger M, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol 2006; 1:940–951.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK; AVOID Study Investigators. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008; 358:2433–2446.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Jamerson K, Weber MA, Bakris GL, et al; ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 2010; 298:F662–F671.
- Pergola PE, Raskin P, Toto RD, et al; BEAM Study Investigators Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 2011; 365:327–336.
KEY POINTS
- The progression from no proteinuria to microalbuminuria to clinical proteinuria parallels glomerular changes of thickening of the basement membrane, mesangial expansion, and the development of Kimmelstiel-Wilson nodules and sclerosis.
- Blood pressure control to 130/80 mm Hg slows microvascular and macrovascular disease, but the goal should not be lower in older patients with diabetes.
- Glycemic control slows microvascular disease: the goal for most patients for hemoglobin A1c is 7.0%. Tighter control may increase cardiovascular risk.
- Either an angiotensin-converting enzyme inhibitor or an angiotensin receptor blocker is the first-line treatment for diabetic nephropathy; combining the two is no longer recommended.
- If more aggressive treatment is needed, a diuretic or spironolactone (with potassium monitoring) can be added.
- The role of sodium bicarbonate and new agents such as blockers of transcription factors is still emerging.
Resistant hypertension: Diagnostic strategies and management
Poor control of blood pressure is one of the most common risk factors for death worldwide, responsible for 62% of cases of cerebral vascular disease and 49% of cases of ischemic heart disease as well as 7.1 million deaths annually. As our population ages and the prevalence of obesity, diabetes, and chronic kidney disease increases, resistant hypertension will be seen more often in general practice.
Using a case study, this article will provide a strategy for diagnosing and treating resistant hypertension.
CASE: A WOMAN WITH LONG-STANDING HIGH BLOOD PRESSURE
A 37-year-old woman was referred for help with managing difficult-to-control hypertension. She had been diagnosed with hypertension at age 32, and it was well controlled until about 2 years ago. Various combinations of antihypertensive drugs had been tried, and a search for a cause of secondary hypertension revealed no clues.
On examination, her blood pressure averaged 212/124 mm Hg, and her heart rate was 109 beats per minute. Her medications were:
- Amlodipine (Norvasc), a calcium channel blocker, 10 mg once daily
- Valsartan (Diovan), an angiotensin II receptor antagonist, 160 mg once daily
- Carvedilol (Coreg), a beta-blocker, 25 mg twice daily
- Labetalol (Normodyne), a beta-blocker, 400 mg three times daily
- Clonidine (Catapres), a sympatholytic agent, 0.05 mg three times daily
- Doxazosin (Cardura), a peripheral alpha-blocker, 16 mg once daily
- Xylometazoline (Xylomet), an alpha agonist nasal spray for nasal congestion.
She had previously been taking spironolactone (Aldactone), hydralazine (Apresoline), and hydrochlorothiazide, but they were discontinued because of adverse effects.
Does this patient have resistant hypertension? How should her condition be managed?
RESISTANT HYPERTENSION DEFINED
The seventh Joint National Committee and the American Heart Association define resistant hypertension as an office blood pressure above the appropriate goal of therapy (< 140/90 mm Hg for most patients, and < 130/80 mm Hg for those with ischemic heart disease, diabetes, or renal insufficiency) despite the use of three or more antihypertensive drugs from different classes at full dosages, one of which is a diuretic.1,2
In this definition, the number of antihypertensive drugs required is arbitrary. More importantly, the concept of resistant hypertension is focused on identifying patients who may have a reversible cause of hypertension, as well as those who could benefit from special diagnostic or therapeutic intervention because of persistently high blood pressure.
This definition does not apply to patients who have recently been diagnosed with hypertension.
Resistant hypertension is not synonymous with uncontrolled hypertension, which includes all cases of hypertension that is not optimally controlled despite treatment, including apparent resistance (ie, pseudoresistance) and true resistance (defined below).
COMMON, BUT ITS PREVALENCE IS HARD TO PINPOINT
The prevalence of resistant hypertension is unknown because of inadequate sample sizes in published studies. However, it is common and is likely to become more common with the aging of the population and with the increasing prevalence of obesity, diabetes mellitus, and chronic kidney disease.
In small studies, the prevalence of resistance in hypertensive patients ranged from 5% in general medical practice to more than 50% in nephrology clinics. In the National Health and Nutrition Examination Survey in 2003 to 2004, only 58% of people being treated for hypertension had achieved blood pressure levels lower than 140/90 mm Hg,3 and the control rate in those with diabetes mellitus or chronic kidney disease was less than 40%.4
Isolated systolic hypertension—elevated systolic pressure with normal diastolic pressure—increases in prevalence with age in those with treated, uncontrolled hypertension. It accounted for 29.1% of cases of treated, uncontrolled hypertension in patients ages 25 to 44, 66.1% of cases in patients ages 45 to 64, and 87.6% of cases in patients age 65 and older.5
Even in clinical trials, in which one would expect excellent control of hypertension, rates of control ranged from 45% to 82%.6–10
APPARENT RESISTANCE VS TRUE RESISTANCE
Resistant hypertension can be divided arbitrarily into two broad categories: apparent resistance and true resistance, with the prevalence of apparent resistance being considerably higher. Each broad category has a long list of possible causes; most are readily identifiable in the course of a thorough history and physical examination and routine laboratory testing. If resistance to therapy persists, referral to a hypertension specialist is a logical next step.
Detecting pseudoresistance
Causes of apparent resistance include improper technique in measuring blood pressure, such as not having the patient rest before measurement, allowing the patient to have coffee or to smoke just before measurement, or not positioning the patient’s arm at the level of the heart during measurement.
Many elderly patients have calcified arteries that are hard to compress, leading to erroneously high systolic blood pressure measurements, a situation called pseudohypertension and a cause of pseudoresistance. The only way to measure blood pressure accurately in such cases is intra-arterially. These patients often do not have target-organ disease, which would be expected with high systolic pressure.
The white-coat phenomenon is another common cause of apparent resistance. It is defined as persistently elevated clinic or office blood pressure (> 140/90 mm Hg), together with normal daytime ambulatory blood pressure (the “white-coat effect” is the difference between those blood pressures).
Finally, poor patient adherence to treatment is estimated to account for 40% of cases of resistant hypertension.4,5,11 Poor adherence is difficult to prove because patients often claim they are compliant, but certain clues are indicative. For example, patients taking a diuretic should have increased uric acid levels, so normal uric acid levels in a patient on a diuretic could be a clue that he or she is not taking the medication. If poor adherence is suspected, patients should be admitted to the hospital to take the medications under close observation.
Many factors can contribute to true resistance
Many cases of resistant hypertension are drug-induced, particularly in patients taking a nonsteroidal anti-inflammatory drug or a cyclooxygenase II inhibitor. Use of ginseng, ma huang, and bitter lemon should also be suspected. Drugs or herbal preparations contributing to high blood pressure should be discontinued or minimized.
Alcohol intake in excess of two drinks (1 oz of alcohol) per day for men and half that amount for women can also contribute to hypertension.
Volume overload is common and has many causes, including a compensatory response to vasodilators, excessive salt intake, or an undetected reduction in the glomerular filtration rate causing retention of salt and water.
Drug considerations
A common cause of apparent resistant hypertension is physicians not following blood pressure treatment guidelines by not increasing the dosage when needed or by prescribing inappropriate drug combinations.
We commonly see furosemide (Lasix) being misused, ie, being prescribed once daily for hypertension. (It has a shorter duration of action than thiazide diuretics, the usual class of diuretics used for hypertension.)
For a patient who is already on many medications but whose hypertension is not responding, the first step should be to give a diuretic of an appropriate class in an appropriate dosage.
Diuretics are often inappropriately stopped if a patient develops hypokalemia. Potassium supplementation should always be an adjunct to diuretic therapy. Potassium itself is a potent vasodilator and, given as a supplement, has been shown to reduce stroke risk in rats.
The combination of an angiotensin receptor blocker and an angiotensin-converting enzyme inhibitor should not be used for patients with true resistant hypertension. The direct renin inhibitor aliskiren (Tekturna) should not be used in combination with these drugs, and the combination of aliskiren and valsartan (Valturna) has now been taken off the market.
Spironolactone (Aldactone) is sometimes used for resistant hypertension in the belief that in some cases primary aldosteronism is the underlying cause. A study in 1,400 participants confirms that it lowers blood pressure,9 but the reason is unclear: the blood pressure response was unrelated to levels of renin, angiotensin, or the plasma aldosterone-to-renin ratio.
Identify secondary causes of hypertension
Patients should be evaluated for kidney disease, which is the most common secondary medical reason for resistant hypertension. For patients with poor renal function (estimated glomerular filtration rate < 50 mL/minute), hydrochlorothiazide is not effective against hypertension, but chlorthalidone is. In addition, patients with poor renal function should be given loop diuretics such as furosemide two or three times daily, or the long-acting drug torsemide (Demadex) should be used instead.
Genetic variation can cause different rates of metabolism of drugs, contributing to resistant hypertension. Certain people metabolize hydralazine very fast, making it less effective. The same is true for some beta-blockers.
Obesity and diabetes can also contribute to resistant hypertension.
Ancillary neurohumoral studies are occasionally indicated to rule out identifiable causes of secondary hypertension that may be correctable. There are many identifiable causes of hypertension, but detailing each is beyond the scope of this article.

Patients should be tested for thyroid disease. Hypothyroidism can cause high blood pressure, although usually diastolic rather than systolic hypertension. Hyperthyroidism can cause marked systolic hypertension.
Table 1 provides a step-by-step guide for evaluating and managing patients with resistant hypertension.
EXPERIMENTAL DRUG THERAPY
Endothelin receptor antagonists are currently under investigation for the treatment of resistant hypertension. The protein endothelin-1 (ET-1) is a potent vasoconstrictor (30–50 times more potent than angiotensin II and norepinephrine) and has a long duration of action. ET-1 binds to two receptors with opposing effects: ET-A promotes vasoconstriction, and ET-B promotes vasodilation and clears ET-1.
Darusentan, a selective blocker of ET-A, was tested in the phase III DORADO trial, which was discontinued because the initial results did not meet primary outcome measures. Initial findings had indicated that it might not be as useful as hoped. Side effects included headache, flushing, and edema.
EXPERIMENTAL NONPHARMACOLOGIC THERAPIES
Electrical stimulation of carotid sinus baroreceptors is being tried under the assumption that a high sympathoexcitatory state contributes to resistant hypertension. Devices are placed around the carotid artery bifurcation, and stimulation is believed to increase the depressor influences that modulate blood pressure. Large-scale trials are under way, but it is too early to tell if the approach will be useful. Patients complain of neck pain from the device.
Renal denervation is another experimental approach.12 The kidney has a central role in blood pressure regulation: efferent nerves regulate renal vascular resistance, renal blood flow, and renin release from the juxtaglomerular apparatus; afferent nerves modulate sympathetic output from the central nervous system. The results of the Renal Denervation in Patients With Uncontrolled Hypertension (Symplicity HTN) trials 1 and 2 have been encouraging. The Symplicity HTN-3 trial will begin soon in the United States.
OUR PATIENT UNDERGOES ADDITIONAL STUDIES
To rule out the white-coat effect in our patient, we measured her blood pressure with an automated device that takes several readings without the clinician in the room. (This topic has been reviewed by Vidt et al in this journal13). The average of the automated readings was 183/113 mm Hg, and her average pulse was 109 beats per minute, arguing against a white-coat effect.
Her blood pressure was also markedly elevated (average 198/129) during 24-hour ambulatory blood pressure monitoring.
Findings on physical examination were unremarkable except for grade III hypertensive retinopathy. She had no carotid or abdominal bruits. Her peripheral pulses were strong and synchronous bilaterally.
Laboratory testing found the patient had normal serum electrolyte levels and good renal function but relatively low urinary sodium, 90 mmol/day (normal 40–220), and very low renin activity, 0.7 μg/L/h (normal up-right 0.8–5.8 μg/L/h, supine 0.5–1.8 μg/L/h), calling into question the wisdom of treatment with an angiotensin receptor blocker.
Hemodynamic studies were performed using impedance cardiography and found very high systemic vascular resistance with normal cardiac output, indicating that the patient had a high preload, which could be from hypervolemia or intense venous constriction. It is especially interesting that her vascular resistance was high despite her treatment regimen that included an angiotensin receptor blocker and a vasodilator, perhaps an indication of nonadherence with her medications.
Diuresis reduces her blood pressure
The patient was admitted to the hospital, and because her laboratory results indicated that plasma renin activity was suppressed, the angiotensin receptor blocker valsartan was discontinued.
On day 1, her weight was 162 lb and average blood pressure was 194/128 mm Hg. After 4 days of diuresis with escalating doses of furosemide, her weight was 153 lb and blood pressures ranged from 140 to 158 over 82 to 98 mm Hg. Her heart rate was 90 beats per minute. The hospital stay showed that volume overload was one of the factors maintaining her hypertension. She was discharged on metoprolol succinate (Toprol-XL) 100 mg twice daily and furosemide 80 mg twice daily.
Her blood pressure fluctuates widely after discharge
Over the next 5 days after discharge, the patient’s blood pressure rose steadily to 180/122 mm Hg, her heart rate was in excess of 100 beats per minute, and her weight increased to 158 lb. Blood screening found that the level of metoprolol was undetectable, and a diuretic screen showed no furosemide in the urine. Both the patient and her husband were adamant that she was taking her medications.
Hydrochlorothiazide 25 mg daily was added, and nadolol (Corgard) 80 mg once daily was started in place of metoprolol. On a return visit, her blood pressure and heart rate were finally good at 138/86 mm Hg and 60 beats per minute (sitting) and 134/92 and 63 (standing).
On 24-hour monitoring, some fluctuations of elevated blood pressure were still evident, with an average of 142/91 mm Hg, so nifedipine (Procardia) 60 mg daily was added.
Her final list of medications is hydrochlorothiazide 25 mg, nadolol 80 mg, and nifedipine XL 60 mg, all taken once daily.
Volume overload complicated by nonadherence
In summary, the main pathogenetic mechanism that sustained this patient’s hypertension was volume overload. Her urinary sodium level indicated that she was not taking excessive amounts of sodium. The volume overload may have been a compensatory response to the concomitant use of peripheral vasodilators plus sympatholytic agents.
In addition, she was not adherent to her antihypertensive regimen. The fact that her heart rate was 109 beats per minute despite having a drug regimen that included five sympathetic blocking agents was a strong clue. She eventually admitted that she did not like taking diuretics because they made her skin wrinkle.
In general, in a case like this, I try to minimize the number of drugs and give a diuretic as well as different classes of appropriate drugs.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2007; 51:1403–1419.
- Ong KL, Cheung BM, Man YB, Lau CP, Lam KS. Prevalence, awareness, treatment and control of hypertension among United States adults 1999–2004. Hypertension 2007; 49:69–75.
- Sarafidis PA, Li S, Chen SC, et al. Hypertension awareness, treatment, and control in chronic kidney disease. Am J Med 2008; 121:332–340.
- Sarafidis PA, Bakris GL. State of hypertension management in the United States: confluence of risk factors and the prevalence of resistant hypertension. J Clin Hypertens (Greenwich) 2008; 10:130–139.
- Jamerson K, Bakris GL, Dahlöf B, et al; for the ACCOMPLISH Investigators. Exceptional early blood pressure control rates: the ACCOMPLISH trial. Blood Pressure 2007; 16:80–86.
- Dahlöf B, Devereux RB, Kjeldsen S, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–003.
- Cushman WC, Ford CE, Cutler JA, et al; for the ALLHAT Collaborative Research Group. Success and predictors of blood pressure control in diverse North American settings: the Antihypertensive and Lipid-Lowering and Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens (Greenwich) 2002; 4:393–404.
- Chapman N, Dobson J, Wilson S, et al; on behalf of the Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension 2007; 49:839–845.
- Pepine CJ, Handberg EM, Cooper-DeHoff RM, et al; INVEST Investigators. A calcium antagonist vs a noncalcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Calhoun DA, Jones D, Textor S, et al. AHA Scientific Statement. Resistant hypertension: diagnosis, evaluation, and treatment. Circulation 2008; 17:e510–e526.
- Thomas G, Shishehbor MH, Bravo EL, Nally JV. Renal denervation to treat resistant hypertension: guarded optimism. Cleve Clin J Med 2012; 79:501–510.
- Vidt DG, Lang RS, Seballos RJ, Misra-Hebert A, Campbell J, Bena JF. Taking blood pressure: too important to trust to humans? Cleve Clin J Med 2010; 77:683–688.
Poor control of blood pressure is one of the most common risk factors for death worldwide, responsible for 62% of cases of cerebral vascular disease and 49% of cases of ischemic heart disease as well as 7.1 million deaths annually. As our population ages and the prevalence of obesity, diabetes, and chronic kidney disease increases, resistant hypertension will be seen more often in general practice.
Using a case study, this article will provide a strategy for diagnosing and treating resistant hypertension.
CASE: A WOMAN WITH LONG-STANDING HIGH BLOOD PRESSURE
A 37-year-old woman was referred for help with managing difficult-to-control hypertension. She had been diagnosed with hypertension at age 32, and it was well controlled until about 2 years ago. Various combinations of antihypertensive drugs had been tried, and a search for a cause of secondary hypertension revealed no clues.
On examination, her blood pressure averaged 212/124 mm Hg, and her heart rate was 109 beats per minute. Her medications were:
- Amlodipine (Norvasc), a calcium channel blocker, 10 mg once daily
- Valsartan (Diovan), an angiotensin II receptor antagonist, 160 mg once daily
- Carvedilol (Coreg), a beta-blocker, 25 mg twice daily
- Labetalol (Normodyne), a beta-blocker, 400 mg three times daily
- Clonidine (Catapres), a sympatholytic agent, 0.05 mg three times daily
- Doxazosin (Cardura), a peripheral alpha-blocker, 16 mg once daily
- Xylometazoline (Xylomet), an alpha agonist nasal spray for nasal congestion.
She had previously been taking spironolactone (Aldactone), hydralazine (Apresoline), and hydrochlorothiazide, but they were discontinued because of adverse effects.
Does this patient have resistant hypertension? How should her condition be managed?
RESISTANT HYPERTENSION DEFINED
The seventh Joint National Committee and the American Heart Association define resistant hypertension as an office blood pressure above the appropriate goal of therapy (< 140/90 mm Hg for most patients, and < 130/80 mm Hg for those with ischemic heart disease, diabetes, or renal insufficiency) despite the use of three or more antihypertensive drugs from different classes at full dosages, one of which is a diuretic.1,2
In this definition, the number of antihypertensive drugs required is arbitrary. More importantly, the concept of resistant hypertension is focused on identifying patients who may have a reversible cause of hypertension, as well as those who could benefit from special diagnostic or therapeutic intervention because of persistently high blood pressure.
This definition does not apply to patients who have recently been diagnosed with hypertension.
Resistant hypertension is not synonymous with uncontrolled hypertension, which includes all cases of hypertension that is not optimally controlled despite treatment, including apparent resistance (ie, pseudoresistance) and true resistance (defined below).
COMMON, BUT ITS PREVALENCE IS HARD TO PINPOINT
The prevalence of resistant hypertension is unknown because of inadequate sample sizes in published studies. However, it is common and is likely to become more common with the aging of the population and with the increasing prevalence of obesity, diabetes mellitus, and chronic kidney disease.
In small studies, the prevalence of resistance in hypertensive patients ranged from 5% in general medical practice to more than 50% in nephrology clinics. In the National Health and Nutrition Examination Survey in 2003 to 2004, only 58% of people being treated for hypertension had achieved blood pressure levels lower than 140/90 mm Hg,3 and the control rate in those with diabetes mellitus or chronic kidney disease was less than 40%.4
Isolated systolic hypertension—elevated systolic pressure with normal diastolic pressure—increases in prevalence with age in those with treated, uncontrolled hypertension. It accounted for 29.1% of cases of treated, uncontrolled hypertension in patients ages 25 to 44, 66.1% of cases in patients ages 45 to 64, and 87.6% of cases in patients age 65 and older.5
Even in clinical trials, in which one would expect excellent control of hypertension, rates of control ranged from 45% to 82%.6–10
APPARENT RESISTANCE VS TRUE RESISTANCE
Resistant hypertension can be divided arbitrarily into two broad categories: apparent resistance and true resistance, with the prevalence of apparent resistance being considerably higher. Each broad category has a long list of possible causes; most are readily identifiable in the course of a thorough history and physical examination and routine laboratory testing. If resistance to therapy persists, referral to a hypertension specialist is a logical next step.
Detecting pseudoresistance
Causes of apparent resistance include improper technique in measuring blood pressure, such as not having the patient rest before measurement, allowing the patient to have coffee or to smoke just before measurement, or not positioning the patient’s arm at the level of the heart during measurement.
Many elderly patients have calcified arteries that are hard to compress, leading to erroneously high systolic blood pressure measurements, a situation called pseudohypertension and a cause of pseudoresistance. The only way to measure blood pressure accurately in such cases is intra-arterially. These patients often do not have target-organ disease, which would be expected with high systolic pressure.
The white-coat phenomenon is another common cause of apparent resistance. It is defined as persistently elevated clinic or office blood pressure (> 140/90 mm Hg), together with normal daytime ambulatory blood pressure (the “white-coat effect” is the difference between those blood pressures).
Finally, poor patient adherence to treatment is estimated to account for 40% of cases of resistant hypertension.4,5,11 Poor adherence is difficult to prove because patients often claim they are compliant, but certain clues are indicative. For example, patients taking a diuretic should have increased uric acid levels, so normal uric acid levels in a patient on a diuretic could be a clue that he or she is not taking the medication. If poor adherence is suspected, patients should be admitted to the hospital to take the medications under close observation.
Many factors can contribute to true resistance
Many cases of resistant hypertension are drug-induced, particularly in patients taking a nonsteroidal anti-inflammatory drug or a cyclooxygenase II inhibitor. Use of ginseng, ma huang, and bitter lemon should also be suspected. Drugs or herbal preparations contributing to high blood pressure should be discontinued or minimized.
Alcohol intake in excess of two drinks (1 oz of alcohol) per day for men and half that amount for women can also contribute to hypertension.
Volume overload is common and has many causes, including a compensatory response to vasodilators, excessive salt intake, or an undetected reduction in the glomerular filtration rate causing retention of salt and water.
Drug considerations
A common cause of apparent resistant hypertension is physicians not following blood pressure treatment guidelines by not increasing the dosage when needed or by prescribing inappropriate drug combinations.
We commonly see furosemide (Lasix) being misused, ie, being prescribed once daily for hypertension. (It has a shorter duration of action than thiazide diuretics, the usual class of diuretics used for hypertension.)
For a patient who is already on many medications but whose hypertension is not responding, the first step should be to give a diuretic of an appropriate class in an appropriate dosage.
Diuretics are often inappropriately stopped if a patient develops hypokalemia. Potassium supplementation should always be an adjunct to diuretic therapy. Potassium itself is a potent vasodilator and, given as a supplement, has been shown to reduce stroke risk in rats.
The combination of an angiotensin receptor blocker and an angiotensin-converting enzyme inhibitor should not be used for patients with true resistant hypertension. The direct renin inhibitor aliskiren (Tekturna) should not be used in combination with these drugs, and the combination of aliskiren and valsartan (Valturna) has now been taken off the market.
Spironolactone (Aldactone) is sometimes used for resistant hypertension in the belief that in some cases primary aldosteronism is the underlying cause. A study in 1,400 participants confirms that it lowers blood pressure,9 but the reason is unclear: the blood pressure response was unrelated to levels of renin, angiotensin, or the plasma aldosterone-to-renin ratio.
Identify secondary causes of hypertension
Patients should be evaluated for kidney disease, which is the most common secondary medical reason for resistant hypertension. For patients with poor renal function (estimated glomerular filtration rate < 50 mL/minute), hydrochlorothiazide is not effective against hypertension, but chlorthalidone is. In addition, patients with poor renal function should be given loop diuretics such as furosemide two or three times daily, or the long-acting drug torsemide (Demadex) should be used instead.
Genetic variation can cause different rates of metabolism of drugs, contributing to resistant hypertension. Certain people metabolize hydralazine very fast, making it less effective. The same is true for some beta-blockers.
Obesity and diabetes can also contribute to resistant hypertension.
Ancillary neurohumoral studies are occasionally indicated to rule out identifiable causes of secondary hypertension that may be correctable. There are many identifiable causes of hypertension, but detailing each is beyond the scope of this article.
Patients should be tested for thyroid disease. Hypothyroidism can cause high blood pressure, although usually diastolic rather than systolic hypertension. Hyperthyroidism can cause marked systolic hypertension.
Table 1 provides a step-by-step guide for evaluating and managing patients with resistant hypertension.
EXPERIMENTAL DRUG THERAPY
Endothelin receptor antagonists are currently under investigation for the treatment of resistant hypertension. The protein endothelin-1 (ET-1) is a potent vasoconstrictor (30–50 times more potent than angiotensin II and norepinephrine) and has a long duration of action. ET-1 binds to two receptors with opposing effects: ET-A promotes vasoconstriction, and ET-B promotes vasodilation and clears ET-1.
Darusentan, a selective blocker of ET-A, was tested in the phase III DORADO trial, which was discontinued because the initial results did not meet primary outcome measures. Initial findings had indicated that it might not be as useful as hoped. Side effects included headache, flushing, and edema.
EXPERIMENTAL NONPHARMACOLOGIC THERAPIES
Electrical stimulation of carotid sinus baroreceptors is being tried under the assumption that a high sympathoexcitatory state contributes to resistant hypertension. Devices are placed around the carotid artery bifurcation, and stimulation is believed to increase the depressor influences that modulate blood pressure. Large-scale trials are under way, but it is too early to tell if the approach will be useful. Patients complain of neck pain from the device.
Renal denervation is another experimental approach.12 The kidney has a central role in blood pressure regulation: efferent nerves regulate renal vascular resistance, renal blood flow, and renin release from the juxtaglomerular apparatus; afferent nerves modulate sympathetic output from the central nervous system. The results of the Renal Denervation in Patients With Uncontrolled Hypertension (Symplicity HTN) trials 1 and 2 have been encouraging. The Symplicity HTN-3 trial will begin soon in the United States.
OUR PATIENT UNDERGOES ADDITIONAL STUDIES
To rule out the white-coat effect in our patient, we measured her blood pressure with an automated device that takes several readings without the clinician in the room. (This topic has been reviewed by Vidt et al in this journal13). The average of the automated readings was 183/113 mm Hg, and her average pulse was 109 beats per minute, arguing against a white-coat effect.
Her blood pressure was also markedly elevated (average 198/129) during 24-hour ambulatory blood pressure monitoring.
Findings on physical examination were unremarkable except for grade III hypertensive retinopathy. She had no carotid or abdominal bruits. Her peripheral pulses were strong and synchronous bilaterally.
Laboratory testing found the patient had normal serum electrolyte levels and good renal function but relatively low urinary sodium, 90 mmol/day (normal 40–220), and very low renin activity, 0.7 μg/L/h (normal up-right 0.8–5.8 μg/L/h, supine 0.5–1.8 μg/L/h), calling into question the wisdom of treatment with an angiotensin receptor blocker.
Hemodynamic studies were performed using impedance cardiography and found very high systemic vascular resistance with normal cardiac output, indicating that the patient had a high preload, which could be from hypervolemia or intense venous constriction. It is especially interesting that her vascular resistance was high despite her treatment regimen that included an angiotensin receptor blocker and a vasodilator, perhaps an indication of nonadherence with her medications.
Diuresis reduces her blood pressure
The patient was admitted to the hospital, and because her laboratory results indicated that plasma renin activity was suppressed, the angiotensin receptor blocker valsartan was discontinued.
On day 1, her weight was 162 lb and average blood pressure was 194/128 mm Hg. After 4 days of diuresis with escalating doses of furosemide, her weight was 153 lb and blood pressures ranged from 140 to 158 over 82 to 98 mm Hg. Her heart rate was 90 beats per minute. The hospital stay showed that volume overload was one of the factors maintaining her hypertension. She was discharged on metoprolol succinate (Toprol-XL) 100 mg twice daily and furosemide 80 mg twice daily.
Her blood pressure fluctuates widely after discharge
Over the next 5 days after discharge, the patient’s blood pressure rose steadily to 180/122 mm Hg, her heart rate was in excess of 100 beats per minute, and her weight increased to 158 lb. Blood screening found that the level of metoprolol was undetectable, and a diuretic screen showed no furosemide in the urine. Both the patient and her husband were adamant that she was taking her medications.
Hydrochlorothiazide 25 mg daily was added, and nadolol (Corgard) 80 mg once daily was started in place of metoprolol. On a return visit, her blood pressure and heart rate were finally good at 138/86 mm Hg and 60 beats per minute (sitting) and 134/92 and 63 (standing).
On 24-hour monitoring, some fluctuations of elevated blood pressure were still evident, with an average of 142/91 mm Hg, so nifedipine (Procardia) 60 mg daily was added.
Her final list of medications is hydrochlorothiazide 25 mg, nadolol 80 mg, and nifedipine XL 60 mg, all taken once daily.
Volume overload complicated by nonadherence
In summary, the main pathogenetic mechanism that sustained this patient’s hypertension was volume overload. Her urinary sodium level indicated that she was not taking excessive amounts of sodium. The volume overload may have been a compensatory response to the concomitant use of peripheral vasodilators plus sympatholytic agents.
In addition, she was not adherent to her antihypertensive regimen. The fact that her heart rate was 109 beats per minute despite having a drug regimen that included five sympathetic blocking agents was a strong clue. She eventually admitted that she did not like taking diuretics because they made her skin wrinkle.
In general, in a case like this, I try to minimize the number of drugs and give a diuretic as well as different classes of appropriate drugs.
Poor control of blood pressure is one of the most common risk factors for death worldwide, responsible for 62% of cases of cerebral vascular disease and 49% of cases of ischemic heart disease as well as 7.1 million deaths annually. As our population ages and the prevalence of obesity, diabetes, and chronic kidney disease increases, resistant hypertension will be seen more often in general practice.
Using a case study, this article will provide a strategy for diagnosing and treating resistant hypertension.
CASE: A WOMAN WITH LONG-STANDING HIGH BLOOD PRESSURE
A 37-year-old woman was referred for help with managing difficult-to-control hypertension. She had been diagnosed with hypertension at age 32, and it was well controlled until about 2 years ago. Various combinations of antihypertensive drugs had been tried, and a search for a cause of secondary hypertension revealed no clues.
On examination, her blood pressure averaged 212/124 mm Hg, and her heart rate was 109 beats per minute. Her medications were:
- Amlodipine (Norvasc), a calcium channel blocker, 10 mg once daily
- Valsartan (Diovan), an angiotensin II receptor antagonist, 160 mg once daily
- Carvedilol (Coreg), a beta-blocker, 25 mg twice daily
- Labetalol (Normodyne), a beta-blocker, 400 mg three times daily
- Clonidine (Catapres), a sympatholytic agent, 0.05 mg three times daily
- Doxazosin (Cardura), a peripheral alpha-blocker, 16 mg once daily
- Xylometazoline (Xylomet), an alpha agonist nasal spray for nasal congestion.
She had previously been taking spironolactone (Aldactone), hydralazine (Apresoline), and hydrochlorothiazide, but they were discontinued because of adverse effects.
Does this patient have resistant hypertension? How should her condition be managed?
RESISTANT HYPERTENSION DEFINED
The seventh Joint National Committee and the American Heart Association define resistant hypertension as an office blood pressure above the appropriate goal of therapy (< 140/90 mm Hg for most patients, and < 130/80 mm Hg for those with ischemic heart disease, diabetes, or renal insufficiency) despite the use of three or more antihypertensive drugs from different classes at full dosages, one of which is a diuretic.1,2
In this definition, the number of antihypertensive drugs required is arbitrary. More importantly, the concept of resistant hypertension is focused on identifying patients who may have a reversible cause of hypertension, as well as those who could benefit from special diagnostic or therapeutic intervention because of persistently high blood pressure.
This definition does not apply to patients who have recently been diagnosed with hypertension.
Resistant hypertension is not synonymous with uncontrolled hypertension, which includes all cases of hypertension that is not optimally controlled despite treatment, including apparent resistance (ie, pseudoresistance) and true resistance (defined below).
COMMON, BUT ITS PREVALENCE IS HARD TO PINPOINT
The prevalence of resistant hypertension is unknown because of inadequate sample sizes in published studies. However, it is common and is likely to become more common with the aging of the population and with the increasing prevalence of obesity, diabetes mellitus, and chronic kidney disease.
In small studies, the prevalence of resistance in hypertensive patients ranged from 5% in general medical practice to more than 50% in nephrology clinics. In the National Health and Nutrition Examination Survey in 2003 to 2004, only 58% of people being treated for hypertension had achieved blood pressure levels lower than 140/90 mm Hg,3 and the control rate in those with diabetes mellitus or chronic kidney disease was less than 40%.4
Isolated systolic hypertension—elevated systolic pressure with normal diastolic pressure—increases in prevalence with age in those with treated, uncontrolled hypertension. It accounted for 29.1% of cases of treated, uncontrolled hypertension in patients ages 25 to 44, 66.1% of cases in patients ages 45 to 64, and 87.6% of cases in patients age 65 and older.5
Even in clinical trials, in which one would expect excellent control of hypertension, rates of control ranged from 45% to 82%.6–10
APPARENT RESISTANCE VS TRUE RESISTANCE
Resistant hypertension can be divided arbitrarily into two broad categories: apparent resistance and true resistance, with the prevalence of apparent resistance being considerably higher. Each broad category has a long list of possible causes; most are readily identifiable in the course of a thorough history and physical examination and routine laboratory testing. If resistance to therapy persists, referral to a hypertension specialist is a logical next step.
Detecting pseudoresistance
Causes of apparent resistance include improper technique in measuring blood pressure, such as not having the patient rest before measurement, allowing the patient to have coffee or to smoke just before measurement, or not positioning the patient’s arm at the level of the heart during measurement.
Many elderly patients have calcified arteries that are hard to compress, leading to erroneously high systolic blood pressure measurements, a situation called pseudohypertension and a cause of pseudoresistance. The only way to measure blood pressure accurately in such cases is intra-arterially. These patients often do not have target-organ disease, which would be expected with high systolic pressure.
The white-coat phenomenon is another common cause of apparent resistance. It is defined as persistently elevated clinic or office blood pressure (> 140/90 mm Hg), together with normal daytime ambulatory blood pressure (the “white-coat effect” is the difference between those blood pressures).
Finally, poor patient adherence to treatment is estimated to account for 40% of cases of resistant hypertension.4,5,11 Poor adherence is difficult to prove because patients often claim they are compliant, but certain clues are indicative. For example, patients taking a diuretic should have increased uric acid levels, so normal uric acid levels in a patient on a diuretic could be a clue that he or she is not taking the medication. If poor adherence is suspected, patients should be admitted to the hospital to take the medications under close observation.
Many factors can contribute to true resistance
Many cases of resistant hypertension are drug-induced, particularly in patients taking a nonsteroidal anti-inflammatory drug or a cyclooxygenase II inhibitor. Use of ginseng, ma huang, and bitter lemon should also be suspected. Drugs or herbal preparations contributing to high blood pressure should be discontinued or minimized.
Alcohol intake in excess of two drinks (1 oz of alcohol) per day for men and half that amount for women can also contribute to hypertension.
Volume overload is common and has many causes, including a compensatory response to vasodilators, excessive salt intake, or an undetected reduction in the glomerular filtration rate causing retention of salt and water.
Drug considerations
A common cause of apparent resistant hypertension is physicians not following blood pressure treatment guidelines by not increasing the dosage when needed or by prescribing inappropriate drug combinations.
We commonly see furosemide (Lasix) being misused, ie, being prescribed once daily for hypertension. (It has a shorter duration of action than thiazide diuretics, the usual class of diuretics used for hypertension.)
For a patient who is already on many medications but whose hypertension is not responding, the first step should be to give a diuretic of an appropriate class in an appropriate dosage.
Diuretics are often inappropriately stopped if a patient develops hypokalemia. Potassium supplementation should always be an adjunct to diuretic therapy. Potassium itself is a potent vasodilator and, given as a supplement, has been shown to reduce stroke risk in rats.
The combination of an angiotensin receptor blocker and an angiotensin-converting enzyme inhibitor should not be used for patients with true resistant hypertension. The direct renin inhibitor aliskiren (Tekturna) should not be used in combination with these drugs, and the combination of aliskiren and valsartan (Valturna) has now been taken off the market.
Spironolactone (Aldactone) is sometimes used for resistant hypertension in the belief that in some cases primary aldosteronism is the underlying cause. A study in 1,400 participants confirms that it lowers blood pressure,9 but the reason is unclear: the blood pressure response was unrelated to levels of renin, angiotensin, or the plasma aldosterone-to-renin ratio.
Identify secondary causes of hypertension
Patients should be evaluated for kidney disease, which is the most common secondary medical reason for resistant hypertension. For patients with poor renal function (estimated glomerular filtration rate < 50 mL/minute), hydrochlorothiazide is not effective against hypertension, but chlorthalidone is. In addition, patients with poor renal function should be given loop diuretics such as furosemide two or three times daily, or the long-acting drug torsemide (Demadex) should be used instead.
Genetic variation can cause different rates of metabolism of drugs, contributing to resistant hypertension. Certain people metabolize hydralazine very fast, making it less effective. The same is true for some beta-blockers.
Obesity and diabetes can also contribute to resistant hypertension.
Ancillary neurohumoral studies are occasionally indicated to rule out identifiable causes of secondary hypertension that may be correctable. There are many identifiable causes of hypertension, but detailing each is beyond the scope of this article.
Patients should be tested for thyroid disease. Hypothyroidism can cause high blood pressure, although usually diastolic rather than systolic hypertension. Hyperthyroidism can cause marked systolic hypertension.
Table 1 provides a step-by-step guide for evaluating and managing patients with resistant hypertension.
EXPERIMENTAL DRUG THERAPY
Endothelin receptor antagonists are currently under investigation for the treatment of resistant hypertension. The protein endothelin-1 (ET-1) is a potent vasoconstrictor (30–50 times more potent than angiotensin II and norepinephrine) and has a long duration of action. ET-1 binds to two receptors with opposing effects: ET-A promotes vasoconstriction, and ET-B promotes vasodilation and clears ET-1.
Darusentan, a selective blocker of ET-A, was tested in the phase III DORADO trial, which was discontinued because the initial results did not meet primary outcome measures. Initial findings had indicated that it might not be as useful as hoped. Side effects included headache, flushing, and edema.
EXPERIMENTAL NONPHARMACOLOGIC THERAPIES
Electrical stimulation of carotid sinus baroreceptors is being tried under the assumption that a high sympathoexcitatory state contributes to resistant hypertension. Devices are placed around the carotid artery bifurcation, and stimulation is believed to increase the depressor influences that modulate blood pressure. Large-scale trials are under way, but it is too early to tell if the approach will be useful. Patients complain of neck pain from the device.
Renal denervation is another experimental approach.12 The kidney has a central role in blood pressure regulation: efferent nerves regulate renal vascular resistance, renal blood flow, and renin release from the juxtaglomerular apparatus; afferent nerves modulate sympathetic output from the central nervous system. The results of the Renal Denervation in Patients With Uncontrolled Hypertension (Symplicity HTN) trials 1 and 2 have been encouraging. The Symplicity HTN-3 trial will begin soon in the United States.
OUR PATIENT UNDERGOES ADDITIONAL STUDIES
To rule out the white-coat effect in our patient, we measured her blood pressure with an automated device that takes several readings without the clinician in the room. (This topic has been reviewed by Vidt et al in this journal13). The average of the automated readings was 183/113 mm Hg, and her average pulse was 109 beats per minute, arguing against a white-coat effect.
Her blood pressure was also markedly elevated (average 198/129) during 24-hour ambulatory blood pressure monitoring.
Findings on physical examination were unremarkable except for grade III hypertensive retinopathy. She had no carotid or abdominal bruits. Her peripheral pulses were strong and synchronous bilaterally.
Laboratory testing found the patient had normal serum electrolyte levels and good renal function but relatively low urinary sodium, 90 mmol/day (normal 40–220), and very low renin activity, 0.7 μg/L/h (normal up-right 0.8–5.8 μg/L/h, supine 0.5–1.8 μg/L/h), calling into question the wisdom of treatment with an angiotensin receptor blocker.
Hemodynamic studies were performed using impedance cardiography and found very high systemic vascular resistance with normal cardiac output, indicating that the patient had a high preload, which could be from hypervolemia or intense venous constriction. It is especially interesting that her vascular resistance was high despite her treatment regimen that included an angiotensin receptor blocker and a vasodilator, perhaps an indication of nonadherence with her medications.
Diuresis reduces her blood pressure
The patient was admitted to the hospital, and because her laboratory results indicated that plasma renin activity was suppressed, the angiotensin receptor blocker valsartan was discontinued.
On day 1, her weight was 162 lb and average blood pressure was 194/128 mm Hg. After 4 days of diuresis with escalating doses of furosemide, her weight was 153 lb and blood pressures ranged from 140 to 158 over 82 to 98 mm Hg. Her heart rate was 90 beats per minute. The hospital stay showed that volume overload was one of the factors maintaining her hypertension. She was discharged on metoprolol succinate (Toprol-XL) 100 mg twice daily and furosemide 80 mg twice daily.
Her blood pressure fluctuates widely after discharge
Over the next 5 days after discharge, the patient’s blood pressure rose steadily to 180/122 mm Hg, her heart rate was in excess of 100 beats per minute, and her weight increased to 158 lb. Blood screening found that the level of metoprolol was undetectable, and a diuretic screen showed no furosemide in the urine. Both the patient and her husband were adamant that she was taking her medications.
Hydrochlorothiazide 25 mg daily was added, and nadolol (Corgard) 80 mg once daily was started in place of metoprolol. On a return visit, her blood pressure and heart rate were finally good at 138/86 mm Hg and 60 beats per minute (sitting) and 134/92 and 63 (standing).
On 24-hour monitoring, some fluctuations of elevated blood pressure were still evident, with an average of 142/91 mm Hg, so nifedipine (Procardia) 60 mg daily was added.
Her final list of medications is hydrochlorothiazide 25 mg, nadolol 80 mg, and nifedipine XL 60 mg, all taken once daily.
Volume overload complicated by nonadherence
In summary, the main pathogenetic mechanism that sustained this patient’s hypertension was volume overload. Her urinary sodium level indicated that she was not taking excessive amounts of sodium. The volume overload may have been a compensatory response to the concomitant use of peripheral vasodilators plus sympatholytic agents.
In addition, she was not adherent to her antihypertensive regimen. The fact that her heart rate was 109 beats per minute despite having a drug regimen that included five sympathetic blocking agents was a strong clue. She eventually admitted that she did not like taking diuretics because they made her skin wrinkle.
In general, in a case like this, I try to minimize the number of drugs and give a diuretic as well as different classes of appropriate drugs.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2007; 51:1403–1419.
- Ong KL, Cheung BM, Man YB, Lau CP, Lam KS. Prevalence, awareness, treatment and control of hypertension among United States adults 1999–2004. Hypertension 2007; 49:69–75.
- Sarafidis PA, Li S, Chen SC, et al. Hypertension awareness, treatment, and control in chronic kidney disease. Am J Med 2008; 121:332–340.
- Sarafidis PA, Bakris GL. State of hypertension management in the United States: confluence of risk factors and the prevalence of resistant hypertension. J Clin Hypertens (Greenwich) 2008; 10:130–139.
- Jamerson K, Bakris GL, Dahlöf B, et al; for the ACCOMPLISH Investigators. Exceptional early blood pressure control rates: the ACCOMPLISH trial. Blood Pressure 2007; 16:80–86.
- Dahlöf B, Devereux RB, Kjeldsen S, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–003.
- Cushman WC, Ford CE, Cutler JA, et al; for the ALLHAT Collaborative Research Group. Success and predictors of blood pressure control in diverse North American settings: the Antihypertensive and Lipid-Lowering and Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens (Greenwich) 2002; 4:393–404.
- Chapman N, Dobson J, Wilson S, et al; on behalf of the Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension 2007; 49:839–845.
- Pepine CJ, Handberg EM, Cooper-DeHoff RM, et al; INVEST Investigators. A calcium antagonist vs a noncalcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Calhoun DA, Jones D, Textor S, et al. AHA Scientific Statement. Resistant hypertension: diagnosis, evaluation, and treatment. Circulation 2008; 17:e510–e526.
- Thomas G, Shishehbor MH, Bravo EL, Nally JV. Renal denervation to treat resistant hypertension: guarded optimism. Cleve Clin J Med 2012; 79:501–510.
- Vidt DG, Lang RS, Seballos RJ, Misra-Hebert A, Campbell J, Bena JF. Taking blood pressure: too important to trust to humans? Cleve Clin J Med 2010; 77:683–688.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2007; 51:1403–1419.
- Ong KL, Cheung BM, Man YB, Lau CP, Lam KS. Prevalence, awareness, treatment and control of hypertension among United States adults 1999–2004. Hypertension 2007; 49:69–75.
- Sarafidis PA, Li S, Chen SC, et al. Hypertension awareness, treatment, and control in chronic kidney disease. Am J Med 2008; 121:332–340.
- Sarafidis PA, Bakris GL. State of hypertension management in the United States: confluence of risk factors and the prevalence of resistant hypertension. J Clin Hypertens (Greenwich) 2008; 10:130–139.
- Jamerson K, Bakris GL, Dahlöf B, et al; for the ACCOMPLISH Investigators. Exceptional early blood pressure control rates: the ACCOMPLISH trial. Blood Pressure 2007; 16:80–86.
- Dahlöf B, Devereux RB, Kjeldsen S, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–003.
- Cushman WC, Ford CE, Cutler JA, et al; for the ALLHAT Collaborative Research Group. Success and predictors of blood pressure control in diverse North American settings: the Antihypertensive and Lipid-Lowering and Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens (Greenwich) 2002; 4:393–404.
- Chapman N, Dobson J, Wilson S, et al; on behalf of the Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension 2007; 49:839–845.
- Pepine CJ, Handberg EM, Cooper-DeHoff RM, et al; INVEST Investigators. A calcium antagonist vs a noncalcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Calhoun DA, Jones D, Textor S, et al. AHA Scientific Statement. Resistant hypertension: diagnosis, evaluation, and treatment. Circulation 2008; 17:e510–e526.
- Thomas G, Shishehbor MH, Bravo EL, Nally JV. Renal denervation to treat resistant hypertension: guarded optimism. Cleve Clin J Med 2012; 79:501–510.
- Vidt DG, Lang RS, Seballos RJ, Misra-Hebert A, Campbell J, Bena JF. Taking blood pressure: too important to trust to humans? Cleve Clin J Med 2010; 77:683–688.
KEY POINTS
- Resistant hypertension is arbitrarily divided into two categories: apparent resistance (pseudoresistant hypertension) and true resistance. Apparent resistance is much more common.
- Common causes of true resistant hypertension are volume overload, excessive alcohol use, some drugs (eg, nonsteroidal anti-inflammatory drugs), and some over-the-counter supplements.
- Volume overload commonly results from excess sodium intake, kidney disease, or a counterregulatory response to arterial vasodilation.
- To address volume overload, an appropriate diuretic at an adequate dosage is a cornerstone of therapy, along with potassium supplementation.
- Hospitalization may be needed to monitor drug intake if poor compliance is suspected.
Advanced heart failure: Transplantation, LVADs, and beyond
Patients with advanced heart failure far outnumber the hearts available for transplantation. Partly as a consequence of this shortage, left-ventricular assist devices (LVADs) are being used more widely.
This article is an update on options for managing severe, advanced heart failure, with special attention to new developments and continuing challenges in heart transplantation and LVADs.
THE PREVALENCE OF HEART FAILURE
About 2.6% of the US population age 20 and older have heart failure—some 5.8 million people. Of these, about half have systolic heart failure.1 Patients with systolic heart failure can be classified by degree of severity under two systems:
The New York Heart Association (NYHA) classifies patients by their functional status, from I (no limitation in activities) to IV (symptoms at rest). NYHA class III (symptoms with minimal exertion) is sometimes further broken down into IIIa and IIIb, with the latter defined as having a recent history of dyspnea at rest.
The joint American College of Cardiology and American Heart Association (ACC/AHA) classification uses four stages, from A (high risk of developing heart failure, ie, having risk factors such as family history of heart disease, hypertension, or diabetes) to D (advanced heart disease despite treatment). Patients in stage D tend to be recurrently hospitalized despite cardiac resynchronization therapy and drug therapy, and they cannot be safely discharged without specialized interventions. The options for these patients are limited: either end-of-life care or extraordinary measures such as heart transplantation, long-term treatment with inotropic drugs, permanent mechanical circulatory support, or experimental therapies.2
The estimated number of people in ACC/AHA stage D or NYHA class IV is 15,600 to 156,000. The approximate number of heart transplants performed in the United States each year is 2,100.3
WHICH AMBULATORY PATIENTS ARE MOST AT RISK?
The range for the estimated number of patients with advanced heart failure (NYHA class IIIb or IV) is wide (see above) because these patients may be hard to recognize. The most debilitated patients are obvious: they tend to be in the intensive care unit with end-organ failure. However, it is a challenge to recognize patients at extremely high risk who are still ambulatory.
The European Society of Cardiology4 developed a definition of advanced chronic heart failure that can help identify patients who are candidates for the transplant list and for an LVAD. All the following features must be present despite optimal therapy that includes diuretics, inhibitors of the renin-angiotensin-aldosterone system, and beta-blockers, unless these are poorly tolerated or contraindicated, and cardiac resynchronization therapy if indicated:
- Severe symptoms, with dyspnea or fatigue at rest or with minimal exertion (NYHA class III or IV)
- Episodes of fluid retention (pulmonary or systemic congestion, peripheral edema) or of reduced cardiac output at rest (peripheral hypoperfusion)
- Objective evidence of severe cardiac dysfunction (at least one of the following): left ventricular ejection fraction less than 30%, pseudonormal or restrictive mitral inflow pattern on Doppler echocardiography, high left or right ventricular filling pressure (or both left and right filling pressures), and elevated B-type natriuretic peptides
- Severely impaired functional capacity demonstrated by one of the following: inability to exercise, 6-minute walk test distance less than 300 m (or less in women or patients who are age 75 and older), or peak oxygen intake less than 12 to 14 mL/kg/min
- One or more hospitalizations for heart failure in the past 6 months.
Treadmill exercise time is an easily performed test. Hsich et al5 found that the longer patients can walk, the lower their risk of death, and that this variable is about as predictive of survival in patients with systolic left ventricular dysfunction as peak oxygen consumption, which is much more cumbersome to measure.
The Seattle Heart Failure Model gives an estimate of prognosis for ambulatory patients with advanced heart failure. Available at http://depts.washington.edu/shfm/, it is based on age, sex, NYHA class, weight, ejection fraction, blood pressure, medications, a few laboratory values, and other clinical information. The model has been validated in numerous cohorts,6 but it may underestimate risk and is currently being tested in clinical trials (REVIVE-IT and ROADMAP; see at www.clinicaltrials.gov).
Recurrent hospitalization is a simple predictor of risk. A study of about 7,000 patients worldwide found that after hospitalization with acute decompensated heart failure, the strongest predictor of death within 6 months was readmission for any reason within 30 days of the index hospitalization (Starling RC, unpublished observation, 2011). Any patient with heart failure who is repeatedly hospitalized should have a consultation with a heart failure specialist.
INOTROPIC THERAPY FOR BRIDGING
Inotropic drugs, which include intravenous dobutamine (Dobutrex) and milrinone (Primacor), are used to help maintain end-organ function until a patient can obtain a heart transplant or LVAD.
Inotropic therapy should not be viewed as an alternative to heart transplantation or device implantation. We inform patients that inotropic therapy is purely palliative and may actually increase the risk of death, which is about 50% at 6 months and nearly 100% at 1 year. A patient on inotropic therapy who is not a candidate for a transplant or for an assist device should be referred to a hospice program.7
CARDIAC TRANSPLANTATION: SUCCESSES, CHALLENGES
Survival rates after heart transplantation are now excellent. The 1-year survival rate is about 90%, the 5-year rate is about 70%, but only about 20% survive 20 years or longer.8,9 The prognosis is not as good as for combined heart-lung transplantation patients.
Age is an important factor and is a contentious issue: some medical centers will not offer transplantation to patients over age 65. Others regard age as just another risk factor, like renal dysfunction or diabetes.
Quality of life after heart transplantation is excellent: patients are usually able to return to work, regardless of their profession.
The leading cause of death after heart transplantation is malignancy, followed by coronary artery vasculopathy, then by graft failure. Some patients develop left ventricular dysfunction and heart failure of unknown cause. Others develop antibody-mediated rejection; in recent years this has been more promptly recognized, but treatment remains a challenge.
Acute rejection, which used to be one of the main causes of death, now has an extremely low incidence because of modern drug therapies. In a US observational study currently being conducted in about 200 patients receiving a heart transplant (details on CTOT-05 at www.clinicaltrials.gov), the incidence of moderate rejection during the first year is less than 10% (Starling RC, unpublished observation). But several concerns remain.
Adverse effects of immunosuppressive drugs continue to be problematic. These include infection, malignancy, osteoporosis, chronic kidney toxicity, hypertension, and neuropathy.
Renal dysfunction is one of the largest issues. About 10% of heart transplant recipients develop stage 4 kidney disease (with a glomerular filtration rate < 30 mL/min) and need kidney transplantation or renal replacement therapy because of the use of calcineurin inhibitors for immunosuppression.10
Coronary artery vasculopathy was the largest problem when heart transplantation began and continues to be a major concern and focus of research.11,12 Case 1 (below) is an example of the problem.
Case 1: Poor outcome despite an ideal scenario
A 57-year-old businessman had dilated cardiomyopathy and progressive heart failure for 10 years. He was listed for transplantation in 2008 and was given an LVAD (HeartMate II, Thoratec Corp, Pleasanton, CA) as a bridge until a donor heart became available.
In 2009, he received a heart transplant under ideal conditions: the donor was a large 30-year-old man who died of a gunshot wound to the head in the same city in which the patient and transplant hospital were located. Cardiopulmonary resuscitation was not performed, and the cold ischemic time was just a little more than 3 hours. Immune indicators were ideal with a negative prospective cross-match.
Laboratory results after transplantation included creatinine 1.7 mg/dL (normal 0.6–1.2 mg/dL), low-density lipoprotein cholesterol 75 mg/dL, high-density lipoprotein cholesterol 64 mg/dL, and triglycerides 90 mg/dL.
The patient was given immunosuppressive therapy with cyclosporine (Neoral), mycophenolate (CellCept), and prednisone. Because his creatinine level was high, he was also perioperatively given basiliximab (Simulect), a monoclonal antibody to the alpha chain (CD25) of the interleukin-2 receptor. (In a patient who has poor renal function, basilixumab may help by enabling us to delay the use of calcineurin inhibitors.) He also received simvastatin (Zocor) 10 mg.
Per Cleveland Clinic protocol, he underwent 13 biopsy procedures during his first year, and each was normal (grade 0 or 1R). Evaluation by cardiac catheterization at 1 year showed some irregularities in the left anterior descending artery, but a stent was not deemed necessary. Also, per protocol, he underwent intravascular ultrasonography, which revealed abnormal thickness in the intima and media, indicating that coronary artery disease was developing, although it was nonobstructive.
Two months after this checkup, the patient collapsed and suddenly died while shopping. At autopsy, his left anterior descending artery was found to be severely obstructed.
Coronary artery vasculopathy is still a major problem
This case shows that coronary artery vasculopathy may develop despite an ideal transplantation scenario. It remains a large concern and a focus of research.
Coronary vasculopathy develops in 30% to 40% of heart transplant recipients within 5 years, and the incidence has not been reduced by much over the years. However, probably fewer than 5% of these patients die or even need bypass surgery or stenting, and the problem is managed the same as native atherosclerosis. We perform routine annual cardiac catheterizations or stress tests, or both, and place stents in severely blocked arteries.
THE DONOR SHORTAGE: CHANGING HOW HEARTS ARE ALLOCATED
The number of patients receiving a heart transplant in the United States—about 2,000 per year—has not increased in the past decade. The European Union also has great difficulty obtaining hearts for people in need, and almost every transplant candidate there gets mechanical support for some time. The gap between those listed for transplant and the number transplanted each year continues to widen in both the United States and Europe.
All transplant candidates are assigned a status by the United Network of Organ Sharing (UNOS) based on their medical condition. The highest status, 1A, goes to patients who are seriously ill, in the hospital, on high doses of inotropic drugs (specific dosages are defined) and mechanical circulatory support such as an LVAD, and expected to live less than 1 month without a transplant. Status 1B patients are stable on lower-dose inotropic therapy or on mechanical support, and can be in the hospital or at home. Status 2 patients are stable and ambulatory and are not on inotropic drugs.
In July 2006, UNOS changed the rules on how patients are prioritized for obtaining an organ. The new rules are based both on severity of illness (see above) and geographic proximity to the donor heart—local, within 500 miles (“zone A”) or within 500 to 1,000 miles (“zone B”). The order of priority for donor hearts is:
- Local, status 1A
- Local, status 1B
- Zone A, status 1A
- Zone A, status 1B
- Local, status 2
- Zone B, status 1A
- Zone B, status 1B
- Zone A, status 2.
As a result of the change, donor hearts that become available in a particular hospital do not necessarily go to a patient in that state. Another result is that status 2 patients, who were previously the most common transplant recipients, now have much less access because all status 1 patients within 500 miles are given higher priority. Since the change, only 8% of hearts nationwide go to status 2 patients, which is 67% fewer than before. At the same time, organs allocated to status 1A patients have increased by 26%, and their death rates have fallen.3
The new allocation system is a positive change for the sickest patients, providing quicker access and a reduction in waiting-list mortality.13 The drawback is that status 2 patients who are less ill are less likely to ever receive an organ until their condition worsens.
Heart transplant outcomes are publicly reported
The Scientific Registry of Transplant Recipients publicly reports heart transplant outcomes (www.srtr.org). For any transplant center, the public can learn the number of patients waiting for a transplant, the death rate on the waiting list, the number of transplants performed in the previous 12 months, the waiting time in months, and observed and risk-adjusted expected survival rates. A center that deviates from the expected survival rates by 10% or more may be audited and could lose its certification.
Also listed on the Web site is the percentage of patients who receive a support device before receiving a transplant. This can vary widely between institutions and may reflect the organ availability at the transplant center (waiting times) or the preferences and expertise of the transplantation team. We believe that the mortality rate on the waiting list will be reduced with appropriate use of LVADs as a bridge to transplantation when indicated. We have now transitioned to the use of the improved continuous-flow LVADs and rarely maintain patients on continuous inotropic therapy at home to await a donor organ.
MECHANICAL CIRCULATORY SUPPORT: BRIDGE OR DESTINATION?
Mechanical circulatory support devices are increasingly being used to sustain patients with advanced heart failure. Currently at Cleveland Clinic, more LVADs are implanted than hearts are transplanted.
Mechanical circulatory support is indicated for patients who are listed for transplant to keep them functioning as well as possible while they are waiting (bridge to transplant). For others it is “destination therapy”: they are not candidates for a transplant, but a device may improve and prolong the rest of their life.
Case 2: A good outcome despite a poor prognosis
A 71-year-old man was rejected for transplantation by his local hospital because of his age and also because he had pulmonary artery hypertension (78/42 mm Hg; reference range 15–30/5–15 mm Hg) and creatinine elevation (3.0 mg/dL; reference range 0.6–1.5 mg/dL). Nevertheless, he did well on a mechanical device and was accepted for transplantation by Cleveland Clinic. He received a transplant and is still alive and active 14 years later.
Comment. Determining that a patient is not a good transplantation candidate is often impossible. Putting the patient on mechanical support for a period of time can often help clarify whether transplantation is advisable. Probably most patients who receive mechanical support do so as a bridge to decision: most are acutely ill and many have organ dysfunction, pulmonary hypertension, and renal insufficiency. After a period of support, they can be assessed for suitability for transplantation.
LVADs continue to improve
Many devices are available for mechanical circulatory support.14 In addition to LVADs, there are right-ventricular assist devices (RVADs), and devices that simultaneously support both ventricles (BiVADs). Total artificial hearts are also available, as are acute temporary percutaneous devices. These temporary devices—TandemHeart (CardiacAssist, Pittsburgh, PA) and Impella (Abiomed, Danvers, MD)—can be used before a long-term mechanical device can be surgically implanted.
LVADs are of three types:
- Pulsatile volume-displacement pumps, which mimic the pumping action of the natural heart. These early devices were large and placed in the abdomen.
- Continuous axial-flow pumps, which do not have a “heartbeat.” These are quieter and lighter than the early pumps, and use a turbine that spins at 8,000 to 10,000 rpm.
- Continuous centrifugal-flow pumps. These have a rotor spinning at 2,000 to 3,000 rpm, and most of them are magnetically powered and suspended.
The superiority of LVADs over medical therapy was clearly shown even in early studies that used pulsatile LVADs.15 The results of such studies and the increased durability of the devices have led to their rapidly expanded use.
The newer continuous-flow pumps offer significant improvements over the old pulsatile-flow pumps, being smaller, lighter, quieter, and more durable (Table 1). A 2007 study of 133 patients on a continuous axial-flow LVAD (HeartMate II) found that 76% were still alive after 6 months, and patients had significant improvement in functional status and quality of life.16 In a postapproval study based on registry data, HeartMate II was found superior to pulsatile pumps in terms of survival up to 12 months, percentage of patients reaching transplant, and cardiac recovery. Adverse event rates were similar or lower for HeartMate II.17
Another study compared a continuousflow with a pulsatile-flow LVAD for patients who were ineligible for transplantation. Survival at 2 years was 58% with the continuousflow device vs 24% with the pulsatile-flow device (P = .008).18 Since then, postmarket data of patients who received an LVAD showed that 85% are still alive at 1 year.19 This study can be viewed as supporting the use of LVADs as destination therapy.
Quality of life for patients receiving an LVAD has been excellent. When biventricular pacemakers for resynchronization therapy first became available, distances on the 6-minute walk test improved by 39 m, which was deemed a big improvement. LVAD devices have increased the 6-minute walk distance by 156 m.20
Adverse events with LVADs have improved, but continue to be of concern
Infections can arise in the blood stream, in the device pocket, or especially where the driveline exits the skin. As devices have become smaller, driveline diameters have become smaller as well, allowing for a better seal at the skin and making this less of a problem. Some centers report the incidence of driveline infections as less than 20%, but they continue to be a focus of concern.18
Stroke rates continue to improve, although patients still require intensive lifelong anticoagulation. The target international normalized ratio varies by device manufacturer, ranging from 1.7 to 2.5.
Bleeding. Acquired von Willebrand syndrome can develop in patients who have an LVAD, with the gastrointestinal system being the most frequent site of bleeding.21
Device thrombosis occurs very rarely (2%–3%) but is very serious and may require pump exchange.
Mechanical malfunction. As duration of therapy lengthens, problems are arising with aging devices, such as broken wires or short circuits. New-generation pumps have markedly improved durability and reliability.
Good data are kept on device outcomes
The Interagency for Mechanically Assisted Circulatory Support (INTERMACS) maintains a national registry of patients with a mechanical circulatory support device to treat advanced heart failure. It was jointly established in 2006 by the National Heart, Lung, and Blood Institute, Centers for Medicare and Medicaid Services (CMS), the US Food and Drug Administration, and others. Reporting to INTERMACS is required for CMS reimbursement.
The INTERMACS database now has about 4,500 patients at 126 medical centers and is yielding useful information that is published in annual reports.22 The 2011 report focused on the experience with mechanical circulatory support as destination therapy and showed that patients who receive continuousflow pumps have significantly better survival rates than those with pulsatile-flow pumps.23 An earlier report showed that the level of illness at the time of implantation predicts survival24; this information now drives cardiologists to try to improve patient status with a temporary support device or intra-aortic balloon pump before implanting a durable device. The sickest patients (INTERMACS level 1) have the poorest outcomes, and centers now do fewer implantations in patients in this category. We have learned this important lesson from the INTERMACS registry.
The new devices have received a lot of media attention, and patient accrual has increased steadily as the devices have been approved.
On November 20, 2012, the US Food and Drug Administration approved the HeartWare Ventricular Assist System (HeartWare, Framingham, MA) for heart failure patients awaiting a transplant.
FUTURE DIRECTIONS
PROCEED II is an ongoing global clinical trial comparing the outcomes with donor hearts transported in standard cold storage to those transported in an experimental transport device that pumps the heart under physiologic conditions. If proven effective, this device could allow long-distance transport of donor hearts and expand the donor population.
A prospective, randomized study is now enrolling patients to evaluate induction therapy with rituximab (Rituxan) plus conventional immunosuppression (tacrolimus [Prograf], mycophenolate, steroid taper) vs placebo induction plus conventional immunosuppression. The study will enroll 400 patients (200 to each treatment arm) at 25 sites and will have a 36-month accrual period with 12-month follow-up (see http://clinicaltrials.gov/show/NCT01278745). The study is based on data in primates that found that eliminating B cells with an anti-CD20 drug before transplantation markedly reduced the incidence of coronary artery vasculopathy.
- Lloyd-Jones D, Adams RJ, Brown TM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 2010; 1221:e46–e215.
- Jessup M, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:1977–2016.
- 2009 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1999–2008. U.S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation, Rockville, MD.
- Metra M, Ponikowski P, Dickstein K, et al; Heart Failure Association of the European Society of Cardiology. Advanced chronic heart failure: a position statement from the Study Group on Advanced Heart Failure of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2007; 9:684–694.
- Hsich E, Gorodeski EZ, Starling RC, Blackstone EH, Ishwaran H, Lauer MS. Importance of treadmill exercise time as an initial prognostic screening tool in patients with systolic left ventricular dysfunction. Circulation 2009; 119:3189–3197.
- Gorodeski EZ, Chu EC, Chow CH, Levy WC, Hsich E, Starling RC. Application of the Seattle Heart Failure Model in ambulatory patients presented to an advanced heart failure therapeutics committee. Circ Heart Fail 2010; 3:706–714.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Taylor DO, Stehlik J, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth official adult heart transplant report—2009. J Heart Lung Transplant 2009; 28:1007–1022.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult heart transplant report—2010. J Heart Lung Transplant 2010; 29:1089–1103.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med 1999; 340:272–277. Erratum in: N Engl J Med 1999; 340:976.
- Eisen HJ, Tuzcu EM, Dorent R, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiactransplant recipients. N Engl J Med 2003; 349:847–858.
- Singh TP, Almond CS, Taylor DO, Graham DA. Decline in heart transplant wait list mortality in the United States following broader regional sharing of donor hearts. Circ Heart Fail 2012; 5:249–258.
- Baughman KL, Jarcho JA. Bridge to life—cardiac mechanical support. N Engl J Med 2007; 357:846–849.
- Rose EA, Gelijns AC, Moskowitz AJ, et al; Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med 2001; 345:1435–1443.
- Miller LW, Pagani FD, Russell SD, et al; HeartMate II Clinical Investigators. Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med 2007; 357:885–896.
- Starling RC, Naka Y, Boyle AJ, et al. Results of the post-U.S. Food and Drug Administration-approval study with a continuous flow left ventricular assist device as a bridge to heart transplantation: a prospective study using the INTERMACS (Interagency Registry for Mechanically Assisted Circulatory Support). J Am Coll Cardiol 2011; 57:1890–1898.
- Slaughter MS, Rogers JG, Milano CA, et al; HeartMate II Investigators. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009; 361:2241–2251.
- John R, Naka Y, Smedira NG, et al. Continuous flow left ventricular assist device outcomes in commercial use compared with the prior clinical trial. Ann Thorac Surg 2011; 92:1406–1413.
- Starling RC. Improved quantity and quality of life: a winning combination to treat advanced heart failure. J Am Coll Cardiol 2010; 55:1835–1836.
- Uriel N, Pak SW, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010; 56:1207–1213.
- Kirklin JK, Naftel DC, Kormos RL, et al. The fourth INTERMACS annual report: 4,000 implants and counting. J Heart Lung Transplant 2012; 31:117–126.
- Kirklin JK, Naftel DC, Kormos RL, et al. Third INTERMACS Annual Report: the evolution of destination therapy in the United States. J Heart Lung Transplant 2011; 30:115–123.
- Kirklin JK, Naftel DC, Kormos RL, et al. Second INTERMACS annual report: more than 1,000 primary left ventricular assist device implants. J Heart Lung Transplant 2010; 29:1–10.
SUGGESTED READING
Costanzo MR, Dipchand A, Starling R, et al; International Society of Heart and Lung Transplantation Guidelines. The International Society of Heart and Lung Transplantation guidelines for the care of heart transplant recipients. J Heart Lung Transplant 2010; 29:914–956.
Mehra MR, Kobashigawa J, Starling R, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates–2006. J Heart Lung Transplant 2006; 25:1024–1042.
Slaughter MS, Pagani FD, Rogers JG, et al; HeartMate II Clinical Investigators. Clinical management of continuous flow left ventricular assist devices in advanced heart failure. J Heart Lung Transplant 2010; 29(4 suppl):S1–S39.
Patients with advanced heart failure far outnumber the hearts available for transplantation. Partly as a consequence of this shortage, left-ventricular assist devices (LVADs) are being used more widely.
This article is an update on options for managing severe, advanced heart failure, with special attention to new developments and continuing challenges in heart transplantation and LVADs.
THE PREVALENCE OF HEART FAILURE
About 2.6% of the US population age 20 and older have heart failure—some 5.8 million people. Of these, about half have systolic heart failure.1 Patients with systolic heart failure can be classified by degree of severity under two systems:
The New York Heart Association (NYHA) classifies patients by their functional status, from I (no limitation in activities) to IV (symptoms at rest). NYHA class III (symptoms with minimal exertion) is sometimes further broken down into IIIa and IIIb, with the latter defined as having a recent history of dyspnea at rest.
The joint American College of Cardiology and American Heart Association (ACC/AHA) classification uses four stages, from A (high risk of developing heart failure, ie, having risk factors such as family history of heart disease, hypertension, or diabetes) to D (advanced heart disease despite treatment). Patients in stage D tend to be recurrently hospitalized despite cardiac resynchronization therapy and drug therapy, and they cannot be safely discharged without specialized interventions. The options for these patients are limited: either end-of-life care or extraordinary measures such as heart transplantation, long-term treatment with inotropic drugs, permanent mechanical circulatory support, or experimental therapies.2
The estimated number of people in ACC/AHA stage D or NYHA class IV is 15,600 to 156,000. The approximate number of heart transplants performed in the United States each year is 2,100.3
WHICH AMBULATORY PATIENTS ARE MOST AT RISK?
The range for the estimated number of patients with advanced heart failure (NYHA class IIIb or IV) is wide (see above) because these patients may be hard to recognize. The most debilitated patients are obvious: they tend to be in the intensive care unit with end-organ failure. However, it is a challenge to recognize patients at extremely high risk who are still ambulatory.
The European Society of Cardiology4 developed a definition of advanced chronic heart failure that can help identify patients who are candidates for the transplant list and for an LVAD. All the following features must be present despite optimal therapy that includes diuretics, inhibitors of the renin-angiotensin-aldosterone system, and beta-blockers, unless these are poorly tolerated or contraindicated, and cardiac resynchronization therapy if indicated:
- Severe symptoms, with dyspnea or fatigue at rest or with minimal exertion (NYHA class III or IV)
- Episodes of fluid retention (pulmonary or systemic congestion, peripheral edema) or of reduced cardiac output at rest (peripheral hypoperfusion)
- Objective evidence of severe cardiac dysfunction (at least one of the following): left ventricular ejection fraction less than 30%, pseudonormal or restrictive mitral inflow pattern on Doppler echocardiography, high left or right ventricular filling pressure (or both left and right filling pressures), and elevated B-type natriuretic peptides
- Severely impaired functional capacity demonstrated by one of the following: inability to exercise, 6-minute walk test distance less than 300 m (or less in women or patients who are age 75 and older), or peak oxygen intake less than 12 to 14 mL/kg/min
- One or more hospitalizations for heart failure in the past 6 months.
Treadmill exercise time is an easily performed test. Hsich et al5 found that the longer patients can walk, the lower their risk of death, and that this variable is about as predictive of survival in patients with systolic left ventricular dysfunction as peak oxygen consumption, which is much more cumbersome to measure.
The Seattle Heart Failure Model gives an estimate of prognosis for ambulatory patients with advanced heart failure. Available at http://depts.washington.edu/shfm/, it is based on age, sex, NYHA class, weight, ejection fraction, blood pressure, medications, a few laboratory values, and other clinical information. The model has been validated in numerous cohorts,6 but it may underestimate risk and is currently being tested in clinical trials (REVIVE-IT and ROADMAP; see at www.clinicaltrials.gov).
Recurrent hospitalization is a simple predictor of risk. A study of about 7,000 patients worldwide found that after hospitalization with acute decompensated heart failure, the strongest predictor of death within 6 months was readmission for any reason within 30 days of the index hospitalization (Starling RC, unpublished observation, 2011). Any patient with heart failure who is repeatedly hospitalized should have a consultation with a heart failure specialist.
INOTROPIC THERAPY FOR BRIDGING
Inotropic drugs, which include intravenous dobutamine (Dobutrex) and milrinone (Primacor), are used to help maintain end-organ function until a patient can obtain a heart transplant or LVAD.
Inotropic therapy should not be viewed as an alternative to heart transplantation or device implantation. We inform patients that inotropic therapy is purely palliative and may actually increase the risk of death, which is about 50% at 6 months and nearly 100% at 1 year. A patient on inotropic therapy who is not a candidate for a transplant or for an assist device should be referred to a hospice program.7
CARDIAC TRANSPLANTATION: SUCCESSES, CHALLENGES
Survival rates after heart transplantation are now excellent. The 1-year survival rate is about 90%, the 5-year rate is about 70%, but only about 20% survive 20 years or longer.8,9 The prognosis is not as good as for combined heart-lung transplantation patients.
Age is an important factor and is a contentious issue: some medical centers will not offer transplantation to patients over age 65. Others regard age as just another risk factor, like renal dysfunction or diabetes.
Quality of life after heart transplantation is excellent: patients are usually able to return to work, regardless of their profession.
The leading cause of death after heart transplantation is malignancy, followed by coronary artery vasculopathy, then by graft failure. Some patients develop left ventricular dysfunction and heart failure of unknown cause. Others develop antibody-mediated rejection; in recent years this has been more promptly recognized, but treatment remains a challenge.
Acute rejection, which used to be one of the main causes of death, now has an extremely low incidence because of modern drug therapies. In a US observational study currently being conducted in about 200 patients receiving a heart transplant (details on CTOT-05 at www.clinicaltrials.gov), the incidence of moderate rejection during the first year is less than 10% (Starling RC, unpublished observation). But several concerns remain.
Adverse effects of immunosuppressive drugs continue to be problematic. These include infection, malignancy, osteoporosis, chronic kidney toxicity, hypertension, and neuropathy.
Renal dysfunction is one of the largest issues. About 10% of heart transplant recipients develop stage 4 kidney disease (with a glomerular filtration rate < 30 mL/min) and need kidney transplantation or renal replacement therapy because of the use of calcineurin inhibitors for immunosuppression.10
Coronary artery vasculopathy was the largest problem when heart transplantation began and continues to be a major concern and focus of research.11,12 Case 1 (below) is an example of the problem.
Case 1: Poor outcome despite an ideal scenario
A 57-year-old businessman had dilated cardiomyopathy and progressive heart failure for 10 years. He was listed for transplantation in 2008 and was given an LVAD (HeartMate II, Thoratec Corp, Pleasanton, CA) as a bridge until a donor heart became available.
In 2009, he received a heart transplant under ideal conditions: the donor was a large 30-year-old man who died of a gunshot wound to the head in the same city in which the patient and transplant hospital were located. Cardiopulmonary resuscitation was not performed, and the cold ischemic time was just a little more than 3 hours. Immune indicators were ideal with a negative prospective cross-match.
Laboratory results after transplantation included creatinine 1.7 mg/dL (normal 0.6–1.2 mg/dL), low-density lipoprotein cholesterol 75 mg/dL, high-density lipoprotein cholesterol 64 mg/dL, and triglycerides 90 mg/dL.
The patient was given immunosuppressive therapy with cyclosporine (Neoral), mycophenolate (CellCept), and prednisone. Because his creatinine level was high, he was also perioperatively given basiliximab (Simulect), a monoclonal antibody to the alpha chain (CD25) of the interleukin-2 receptor. (In a patient who has poor renal function, basilixumab may help by enabling us to delay the use of calcineurin inhibitors.) He also received simvastatin (Zocor) 10 mg.
Per Cleveland Clinic protocol, he underwent 13 biopsy procedures during his first year, and each was normal (grade 0 or 1R). Evaluation by cardiac catheterization at 1 year showed some irregularities in the left anterior descending artery, but a stent was not deemed necessary. Also, per protocol, he underwent intravascular ultrasonography, which revealed abnormal thickness in the intima and media, indicating that coronary artery disease was developing, although it was nonobstructive.
Two months after this checkup, the patient collapsed and suddenly died while shopping. At autopsy, his left anterior descending artery was found to be severely obstructed.
Coronary artery vasculopathy is still a major problem
This case shows that coronary artery vasculopathy may develop despite an ideal transplantation scenario. It remains a large concern and a focus of research.
Coronary vasculopathy develops in 30% to 40% of heart transplant recipients within 5 years, and the incidence has not been reduced by much over the years. However, probably fewer than 5% of these patients die or even need bypass surgery or stenting, and the problem is managed the same as native atherosclerosis. We perform routine annual cardiac catheterizations or stress tests, or both, and place stents in severely blocked arteries.
THE DONOR SHORTAGE: CHANGING HOW HEARTS ARE ALLOCATED
The number of patients receiving a heart transplant in the United States—about 2,000 per year—has not increased in the past decade. The European Union also has great difficulty obtaining hearts for people in need, and almost every transplant candidate there gets mechanical support for some time. The gap between those listed for transplant and the number transplanted each year continues to widen in both the United States and Europe.
All transplant candidates are assigned a status by the United Network of Organ Sharing (UNOS) based on their medical condition. The highest status, 1A, goes to patients who are seriously ill, in the hospital, on high doses of inotropic drugs (specific dosages are defined) and mechanical circulatory support such as an LVAD, and expected to live less than 1 month without a transplant. Status 1B patients are stable on lower-dose inotropic therapy or on mechanical support, and can be in the hospital or at home. Status 2 patients are stable and ambulatory and are not on inotropic drugs.
In July 2006, UNOS changed the rules on how patients are prioritized for obtaining an organ. The new rules are based both on severity of illness (see above) and geographic proximity to the donor heart—local, within 500 miles (“zone A”) or within 500 to 1,000 miles (“zone B”). The order of priority for donor hearts is:
- Local, status 1A
- Local, status 1B
- Zone A, status 1A
- Zone A, status 1B
- Local, status 2
- Zone B, status 1A
- Zone B, status 1B
- Zone A, status 2.
As a result of the change, donor hearts that become available in a particular hospital do not necessarily go to a patient in that state. Another result is that status 2 patients, who were previously the most common transplant recipients, now have much less access because all status 1 patients within 500 miles are given higher priority. Since the change, only 8% of hearts nationwide go to status 2 patients, which is 67% fewer than before. At the same time, organs allocated to status 1A patients have increased by 26%, and their death rates have fallen.3
The new allocation system is a positive change for the sickest patients, providing quicker access and a reduction in waiting-list mortality.13 The drawback is that status 2 patients who are less ill are less likely to ever receive an organ until their condition worsens.
Heart transplant outcomes are publicly reported
The Scientific Registry of Transplant Recipients publicly reports heart transplant outcomes (www.srtr.org). For any transplant center, the public can learn the number of patients waiting for a transplant, the death rate on the waiting list, the number of transplants performed in the previous 12 months, the waiting time in months, and observed and risk-adjusted expected survival rates. A center that deviates from the expected survival rates by 10% or more may be audited and could lose its certification.
Also listed on the Web site is the percentage of patients who receive a support device before receiving a transplant. This can vary widely between institutions and may reflect the organ availability at the transplant center (waiting times) or the preferences and expertise of the transplantation team. We believe that the mortality rate on the waiting list will be reduced with appropriate use of LVADs as a bridge to transplantation when indicated. We have now transitioned to the use of the improved continuous-flow LVADs and rarely maintain patients on continuous inotropic therapy at home to await a donor organ.
MECHANICAL CIRCULATORY SUPPORT: BRIDGE OR DESTINATION?
Mechanical circulatory support devices are increasingly being used to sustain patients with advanced heart failure. Currently at Cleveland Clinic, more LVADs are implanted than hearts are transplanted.
Mechanical circulatory support is indicated for patients who are listed for transplant to keep them functioning as well as possible while they are waiting (bridge to transplant). For others it is “destination therapy”: they are not candidates for a transplant, but a device may improve and prolong the rest of their life.
Case 2: A good outcome despite a poor prognosis
A 71-year-old man was rejected for transplantation by his local hospital because of his age and also because he had pulmonary artery hypertension (78/42 mm Hg; reference range 15–30/5–15 mm Hg) and creatinine elevation (3.0 mg/dL; reference range 0.6–1.5 mg/dL). Nevertheless, he did well on a mechanical device and was accepted for transplantation by Cleveland Clinic. He received a transplant and is still alive and active 14 years later.
Comment. Determining that a patient is not a good transplantation candidate is often impossible. Putting the patient on mechanical support for a period of time can often help clarify whether transplantation is advisable. Probably most patients who receive mechanical support do so as a bridge to decision: most are acutely ill and many have organ dysfunction, pulmonary hypertension, and renal insufficiency. After a period of support, they can be assessed for suitability for transplantation.
LVADs continue to improve
Many devices are available for mechanical circulatory support.14 In addition to LVADs, there are right-ventricular assist devices (RVADs), and devices that simultaneously support both ventricles (BiVADs). Total artificial hearts are also available, as are acute temporary percutaneous devices. These temporary devices—TandemHeart (CardiacAssist, Pittsburgh, PA) and Impella (Abiomed, Danvers, MD)—can be used before a long-term mechanical device can be surgically implanted.
LVADs are of three types:
- Pulsatile volume-displacement pumps, which mimic the pumping action of the natural heart. These early devices were large and placed in the abdomen.
- Continuous axial-flow pumps, which do not have a “heartbeat.” These are quieter and lighter than the early pumps, and use a turbine that spins at 8,000 to 10,000 rpm.
- Continuous centrifugal-flow pumps. These have a rotor spinning at 2,000 to 3,000 rpm, and most of them are magnetically powered and suspended.
The superiority of LVADs over medical therapy was clearly shown even in early studies that used pulsatile LVADs.15 The results of such studies and the increased durability of the devices have led to their rapidly expanded use.
The newer continuous-flow pumps offer significant improvements over the old pulsatile-flow pumps, being smaller, lighter, quieter, and more durable (Table 1). A 2007 study of 133 patients on a continuous axial-flow LVAD (HeartMate II) found that 76% were still alive after 6 months, and patients had significant improvement in functional status and quality of life.16 In a postapproval study based on registry data, HeartMate II was found superior to pulsatile pumps in terms of survival up to 12 months, percentage of patients reaching transplant, and cardiac recovery. Adverse event rates were similar or lower for HeartMate II.17
Another study compared a continuousflow with a pulsatile-flow LVAD for patients who were ineligible for transplantation. Survival at 2 years was 58% with the continuousflow device vs 24% with the pulsatile-flow device (P = .008).18 Since then, postmarket data of patients who received an LVAD showed that 85% are still alive at 1 year.19 This study can be viewed as supporting the use of LVADs as destination therapy.
Quality of life for patients receiving an LVAD has been excellent. When biventricular pacemakers for resynchronization therapy first became available, distances on the 6-minute walk test improved by 39 m, which was deemed a big improvement. LVAD devices have increased the 6-minute walk distance by 156 m.20
Adverse events with LVADs have improved, but continue to be of concern
Infections can arise in the blood stream, in the device pocket, or especially where the driveline exits the skin. As devices have become smaller, driveline diameters have become smaller as well, allowing for a better seal at the skin and making this less of a problem. Some centers report the incidence of driveline infections as less than 20%, but they continue to be a focus of concern.18
Stroke rates continue to improve, although patients still require intensive lifelong anticoagulation. The target international normalized ratio varies by device manufacturer, ranging from 1.7 to 2.5.
Bleeding. Acquired von Willebrand syndrome can develop in patients who have an LVAD, with the gastrointestinal system being the most frequent site of bleeding.21
Device thrombosis occurs very rarely (2%–3%) but is very serious and may require pump exchange.
Mechanical malfunction. As duration of therapy lengthens, problems are arising with aging devices, such as broken wires or short circuits. New-generation pumps have markedly improved durability and reliability.
Good data are kept on device outcomes
The Interagency for Mechanically Assisted Circulatory Support (INTERMACS) maintains a national registry of patients with a mechanical circulatory support device to treat advanced heart failure. It was jointly established in 2006 by the National Heart, Lung, and Blood Institute, Centers for Medicare and Medicaid Services (CMS), the US Food and Drug Administration, and others. Reporting to INTERMACS is required for CMS reimbursement.
The INTERMACS database now has about 4,500 patients at 126 medical centers and is yielding useful information that is published in annual reports.22 The 2011 report focused on the experience with mechanical circulatory support as destination therapy and showed that patients who receive continuousflow pumps have significantly better survival rates than those with pulsatile-flow pumps.23 An earlier report showed that the level of illness at the time of implantation predicts survival24; this information now drives cardiologists to try to improve patient status with a temporary support device or intra-aortic balloon pump before implanting a durable device. The sickest patients (INTERMACS level 1) have the poorest outcomes, and centers now do fewer implantations in patients in this category. We have learned this important lesson from the INTERMACS registry.
The new devices have received a lot of media attention, and patient accrual has increased steadily as the devices have been approved.
On November 20, 2012, the US Food and Drug Administration approved the HeartWare Ventricular Assist System (HeartWare, Framingham, MA) for heart failure patients awaiting a transplant.
FUTURE DIRECTIONS
PROCEED II is an ongoing global clinical trial comparing the outcomes with donor hearts transported in standard cold storage to those transported in an experimental transport device that pumps the heart under physiologic conditions. If proven effective, this device could allow long-distance transport of donor hearts and expand the donor population.
A prospective, randomized study is now enrolling patients to evaluate induction therapy with rituximab (Rituxan) plus conventional immunosuppression (tacrolimus [Prograf], mycophenolate, steroid taper) vs placebo induction plus conventional immunosuppression. The study will enroll 400 patients (200 to each treatment arm) at 25 sites and will have a 36-month accrual period with 12-month follow-up (see http://clinicaltrials.gov/show/NCT01278745). The study is based on data in primates that found that eliminating B cells with an anti-CD20 drug before transplantation markedly reduced the incidence of coronary artery vasculopathy.
Patients with advanced heart failure far outnumber the hearts available for transplantation. Partly as a consequence of this shortage, left-ventricular assist devices (LVADs) are being used more widely.
This article is an update on options for managing severe, advanced heart failure, with special attention to new developments and continuing challenges in heart transplantation and LVADs.
THE PREVALENCE OF HEART FAILURE
About 2.6% of the US population age 20 and older have heart failure—some 5.8 million people. Of these, about half have systolic heart failure.1 Patients with systolic heart failure can be classified by degree of severity under two systems:
The New York Heart Association (NYHA) classifies patients by their functional status, from I (no limitation in activities) to IV (symptoms at rest). NYHA class III (symptoms with minimal exertion) is sometimes further broken down into IIIa and IIIb, with the latter defined as having a recent history of dyspnea at rest.
The joint American College of Cardiology and American Heart Association (ACC/AHA) classification uses four stages, from A (high risk of developing heart failure, ie, having risk factors such as family history of heart disease, hypertension, or diabetes) to D (advanced heart disease despite treatment). Patients in stage D tend to be recurrently hospitalized despite cardiac resynchronization therapy and drug therapy, and they cannot be safely discharged without specialized interventions. The options for these patients are limited: either end-of-life care or extraordinary measures such as heart transplantation, long-term treatment with inotropic drugs, permanent mechanical circulatory support, or experimental therapies.2
The estimated number of people in ACC/AHA stage D or NYHA class IV is 15,600 to 156,000. The approximate number of heart transplants performed in the United States each year is 2,100.3
WHICH AMBULATORY PATIENTS ARE MOST AT RISK?
The range for the estimated number of patients with advanced heart failure (NYHA class IIIb or IV) is wide (see above) because these patients may be hard to recognize. The most debilitated patients are obvious: they tend to be in the intensive care unit with end-organ failure. However, it is a challenge to recognize patients at extremely high risk who are still ambulatory.
The European Society of Cardiology4 developed a definition of advanced chronic heart failure that can help identify patients who are candidates for the transplant list and for an LVAD. All the following features must be present despite optimal therapy that includes diuretics, inhibitors of the renin-angiotensin-aldosterone system, and beta-blockers, unless these are poorly tolerated or contraindicated, and cardiac resynchronization therapy if indicated:
- Severe symptoms, with dyspnea or fatigue at rest or with minimal exertion (NYHA class III or IV)
- Episodes of fluid retention (pulmonary or systemic congestion, peripheral edema) or of reduced cardiac output at rest (peripheral hypoperfusion)
- Objective evidence of severe cardiac dysfunction (at least one of the following): left ventricular ejection fraction less than 30%, pseudonormal or restrictive mitral inflow pattern on Doppler echocardiography, high left or right ventricular filling pressure (or both left and right filling pressures), and elevated B-type natriuretic peptides
- Severely impaired functional capacity demonstrated by one of the following: inability to exercise, 6-minute walk test distance less than 300 m (or less in women or patients who are age 75 and older), or peak oxygen intake less than 12 to 14 mL/kg/min
- One or more hospitalizations for heart failure in the past 6 months.
Treadmill exercise time is an easily performed test. Hsich et al5 found that the longer patients can walk, the lower their risk of death, and that this variable is about as predictive of survival in patients with systolic left ventricular dysfunction as peak oxygen consumption, which is much more cumbersome to measure.
The Seattle Heart Failure Model gives an estimate of prognosis for ambulatory patients with advanced heart failure. Available at http://depts.washington.edu/shfm/, it is based on age, sex, NYHA class, weight, ejection fraction, blood pressure, medications, a few laboratory values, and other clinical information. The model has been validated in numerous cohorts,6 but it may underestimate risk and is currently being tested in clinical trials (REVIVE-IT and ROADMAP; see at www.clinicaltrials.gov).
Recurrent hospitalization is a simple predictor of risk. A study of about 7,000 patients worldwide found that after hospitalization with acute decompensated heart failure, the strongest predictor of death within 6 months was readmission for any reason within 30 days of the index hospitalization (Starling RC, unpublished observation, 2011). Any patient with heart failure who is repeatedly hospitalized should have a consultation with a heart failure specialist.
INOTROPIC THERAPY FOR BRIDGING
Inotropic drugs, which include intravenous dobutamine (Dobutrex) and milrinone (Primacor), are used to help maintain end-organ function until a patient can obtain a heart transplant or LVAD.
Inotropic therapy should not be viewed as an alternative to heart transplantation or device implantation. We inform patients that inotropic therapy is purely palliative and may actually increase the risk of death, which is about 50% at 6 months and nearly 100% at 1 year. A patient on inotropic therapy who is not a candidate for a transplant or for an assist device should be referred to a hospice program.7
CARDIAC TRANSPLANTATION: SUCCESSES, CHALLENGES
Survival rates after heart transplantation are now excellent. The 1-year survival rate is about 90%, the 5-year rate is about 70%, but only about 20% survive 20 years or longer.8,9 The prognosis is not as good as for combined heart-lung transplantation patients.
Age is an important factor and is a contentious issue: some medical centers will not offer transplantation to patients over age 65. Others regard age as just another risk factor, like renal dysfunction or diabetes.
Quality of life after heart transplantation is excellent: patients are usually able to return to work, regardless of their profession.
The leading cause of death after heart transplantation is malignancy, followed by coronary artery vasculopathy, then by graft failure. Some patients develop left ventricular dysfunction and heart failure of unknown cause. Others develop antibody-mediated rejection; in recent years this has been more promptly recognized, but treatment remains a challenge.
Acute rejection, which used to be one of the main causes of death, now has an extremely low incidence because of modern drug therapies. In a US observational study currently being conducted in about 200 patients receiving a heart transplant (details on CTOT-05 at www.clinicaltrials.gov), the incidence of moderate rejection during the first year is less than 10% (Starling RC, unpublished observation). But several concerns remain.
Adverse effects of immunosuppressive drugs continue to be problematic. These include infection, malignancy, osteoporosis, chronic kidney toxicity, hypertension, and neuropathy.
Renal dysfunction is one of the largest issues. About 10% of heart transplant recipients develop stage 4 kidney disease (with a glomerular filtration rate < 30 mL/min) and need kidney transplantation or renal replacement therapy because of the use of calcineurin inhibitors for immunosuppression.10
Coronary artery vasculopathy was the largest problem when heart transplantation began and continues to be a major concern and focus of research.11,12 Case 1 (below) is an example of the problem.
Case 1: Poor outcome despite an ideal scenario
A 57-year-old businessman had dilated cardiomyopathy and progressive heart failure for 10 years. He was listed for transplantation in 2008 and was given an LVAD (HeartMate II, Thoratec Corp, Pleasanton, CA) as a bridge until a donor heart became available.
In 2009, he received a heart transplant under ideal conditions: the donor was a large 30-year-old man who died of a gunshot wound to the head in the same city in which the patient and transplant hospital were located. Cardiopulmonary resuscitation was not performed, and the cold ischemic time was just a little more than 3 hours. Immune indicators were ideal with a negative prospective cross-match.
Laboratory results after transplantation included creatinine 1.7 mg/dL (normal 0.6–1.2 mg/dL), low-density lipoprotein cholesterol 75 mg/dL, high-density lipoprotein cholesterol 64 mg/dL, and triglycerides 90 mg/dL.
The patient was given immunosuppressive therapy with cyclosporine (Neoral), mycophenolate (CellCept), and prednisone. Because his creatinine level was high, he was also perioperatively given basiliximab (Simulect), a monoclonal antibody to the alpha chain (CD25) of the interleukin-2 receptor. (In a patient who has poor renal function, basilixumab may help by enabling us to delay the use of calcineurin inhibitors.) He also received simvastatin (Zocor) 10 mg.
Per Cleveland Clinic protocol, he underwent 13 biopsy procedures during his first year, and each was normal (grade 0 or 1R). Evaluation by cardiac catheterization at 1 year showed some irregularities in the left anterior descending artery, but a stent was not deemed necessary. Also, per protocol, he underwent intravascular ultrasonography, which revealed abnormal thickness in the intima and media, indicating that coronary artery disease was developing, although it was nonobstructive.
Two months after this checkup, the patient collapsed and suddenly died while shopping. At autopsy, his left anterior descending artery was found to be severely obstructed.
Coronary artery vasculopathy is still a major problem
This case shows that coronary artery vasculopathy may develop despite an ideal transplantation scenario. It remains a large concern and a focus of research.
Coronary vasculopathy develops in 30% to 40% of heart transplant recipients within 5 years, and the incidence has not been reduced by much over the years. However, probably fewer than 5% of these patients die or even need bypass surgery or stenting, and the problem is managed the same as native atherosclerosis. We perform routine annual cardiac catheterizations or stress tests, or both, and place stents in severely blocked arteries.
THE DONOR SHORTAGE: CHANGING HOW HEARTS ARE ALLOCATED
The number of patients receiving a heart transplant in the United States—about 2,000 per year—has not increased in the past decade. The European Union also has great difficulty obtaining hearts for people in need, and almost every transplant candidate there gets mechanical support for some time. The gap between those listed for transplant and the number transplanted each year continues to widen in both the United States and Europe.
All transplant candidates are assigned a status by the United Network of Organ Sharing (UNOS) based on their medical condition. The highest status, 1A, goes to patients who are seriously ill, in the hospital, on high doses of inotropic drugs (specific dosages are defined) and mechanical circulatory support such as an LVAD, and expected to live less than 1 month without a transplant. Status 1B patients are stable on lower-dose inotropic therapy or on mechanical support, and can be in the hospital or at home. Status 2 patients are stable and ambulatory and are not on inotropic drugs.
In July 2006, UNOS changed the rules on how patients are prioritized for obtaining an organ. The new rules are based both on severity of illness (see above) and geographic proximity to the donor heart—local, within 500 miles (“zone A”) or within 500 to 1,000 miles (“zone B”). The order of priority for donor hearts is:
- Local, status 1A
- Local, status 1B
- Zone A, status 1A
- Zone A, status 1B
- Local, status 2
- Zone B, status 1A
- Zone B, status 1B
- Zone A, status 2.
As a result of the change, donor hearts that become available in a particular hospital do not necessarily go to a patient in that state. Another result is that status 2 patients, who were previously the most common transplant recipients, now have much less access because all status 1 patients within 500 miles are given higher priority. Since the change, only 8% of hearts nationwide go to status 2 patients, which is 67% fewer than before. At the same time, organs allocated to status 1A patients have increased by 26%, and their death rates have fallen.3
The new allocation system is a positive change for the sickest patients, providing quicker access and a reduction in waiting-list mortality.13 The drawback is that status 2 patients who are less ill are less likely to ever receive an organ until their condition worsens.
Heart transplant outcomes are publicly reported
The Scientific Registry of Transplant Recipients publicly reports heart transplant outcomes (www.srtr.org). For any transplant center, the public can learn the number of patients waiting for a transplant, the death rate on the waiting list, the number of transplants performed in the previous 12 months, the waiting time in months, and observed and risk-adjusted expected survival rates. A center that deviates from the expected survival rates by 10% or more may be audited and could lose its certification.
Also listed on the Web site is the percentage of patients who receive a support device before receiving a transplant. This can vary widely between institutions and may reflect the organ availability at the transplant center (waiting times) or the preferences and expertise of the transplantation team. We believe that the mortality rate on the waiting list will be reduced with appropriate use of LVADs as a bridge to transplantation when indicated. We have now transitioned to the use of the improved continuous-flow LVADs and rarely maintain patients on continuous inotropic therapy at home to await a donor organ.
MECHANICAL CIRCULATORY SUPPORT: BRIDGE OR DESTINATION?
Mechanical circulatory support devices are increasingly being used to sustain patients with advanced heart failure. Currently at Cleveland Clinic, more LVADs are implanted than hearts are transplanted.
Mechanical circulatory support is indicated for patients who are listed for transplant to keep them functioning as well as possible while they are waiting (bridge to transplant). For others it is “destination therapy”: they are not candidates for a transplant, but a device may improve and prolong the rest of their life.
Case 2: A good outcome despite a poor prognosis
A 71-year-old man was rejected for transplantation by his local hospital because of his age and also because he had pulmonary artery hypertension (78/42 mm Hg; reference range 15–30/5–15 mm Hg) and creatinine elevation (3.0 mg/dL; reference range 0.6–1.5 mg/dL). Nevertheless, he did well on a mechanical device and was accepted for transplantation by Cleveland Clinic. He received a transplant and is still alive and active 14 years later.
Comment. Determining that a patient is not a good transplantation candidate is often impossible. Putting the patient on mechanical support for a period of time can often help clarify whether transplantation is advisable. Probably most patients who receive mechanical support do so as a bridge to decision: most are acutely ill and many have organ dysfunction, pulmonary hypertension, and renal insufficiency. After a period of support, they can be assessed for suitability for transplantation.
LVADs continue to improve
Many devices are available for mechanical circulatory support.14 In addition to LVADs, there are right-ventricular assist devices (RVADs), and devices that simultaneously support both ventricles (BiVADs). Total artificial hearts are also available, as are acute temporary percutaneous devices. These temporary devices—TandemHeart (CardiacAssist, Pittsburgh, PA) and Impella (Abiomed, Danvers, MD)—can be used before a long-term mechanical device can be surgically implanted.
LVADs are of three types:
- Pulsatile volume-displacement pumps, which mimic the pumping action of the natural heart. These early devices were large and placed in the abdomen.
- Continuous axial-flow pumps, which do not have a “heartbeat.” These are quieter and lighter than the early pumps, and use a turbine that spins at 8,000 to 10,000 rpm.
- Continuous centrifugal-flow pumps. These have a rotor spinning at 2,000 to 3,000 rpm, and most of them are magnetically powered and suspended.
The superiority of LVADs over medical therapy was clearly shown even in early studies that used pulsatile LVADs.15 The results of such studies and the increased durability of the devices have led to their rapidly expanded use.
The newer continuous-flow pumps offer significant improvements over the old pulsatile-flow pumps, being smaller, lighter, quieter, and more durable (Table 1). A 2007 study of 133 patients on a continuous axial-flow LVAD (HeartMate II) found that 76% were still alive after 6 months, and patients had significant improvement in functional status and quality of life.16 In a postapproval study based on registry data, HeartMate II was found superior to pulsatile pumps in terms of survival up to 12 months, percentage of patients reaching transplant, and cardiac recovery. Adverse event rates were similar or lower for HeartMate II.17
Another study compared a continuousflow with a pulsatile-flow LVAD for patients who were ineligible for transplantation. Survival at 2 years was 58% with the continuousflow device vs 24% with the pulsatile-flow device (P = .008).18 Since then, postmarket data of patients who received an LVAD showed that 85% are still alive at 1 year.19 This study can be viewed as supporting the use of LVADs as destination therapy.
Quality of life for patients receiving an LVAD has been excellent. When biventricular pacemakers for resynchronization therapy first became available, distances on the 6-minute walk test improved by 39 m, which was deemed a big improvement. LVAD devices have increased the 6-minute walk distance by 156 m.20
Adverse events with LVADs have improved, but continue to be of concern
Infections can arise in the blood stream, in the device pocket, or especially where the driveline exits the skin. As devices have become smaller, driveline diameters have become smaller as well, allowing for a better seal at the skin and making this less of a problem. Some centers report the incidence of driveline infections as less than 20%, but they continue to be a focus of concern.18
Stroke rates continue to improve, although patients still require intensive lifelong anticoagulation. The target international normalized ratio varies by device manufacturer, ranging from 1.7 to 2.5.
Bleeding. Acquired von Willebrand syndrome can develop in patients who have an LVAD, with the gastrointestinal system being the most frequent site of bleeding.21
Device thrombosis occurs very rarely (2%–3%) but is very serious and may require pump exchange.
Mechanical malfunction. As duration of therapy lengthens, problems are arising with aging devices, such as broken wires or short circuits. New-generation pumps have markedly improved durability and reliability.
Good data are kept on device outcomes
The Interagency for Mechanically Assisted Circulatory Support (INTERMACS) maintains a national registry of patients with a mechanical circulatory support device to treat advanced heart failure. It was jointly established in 2006 by the National Heart, Lung, and Blood Institute, Centers for Medicare and Medicaid Services (CMS), the US Food and Drug Administration, and others. Reporting to INTERMACS is required for CMS reimbursement.
The INTERMACS database now has about 4,500 patients at 126 medical centers and is yielding useful information that is published in annual reports.22 The 2011 report focused on the experience with mechanical circulatory support as destination therapy and showed that patients who receive continuousflow pumps have significantly better survival rates than those with pulsatile-flow pumps.23 An earlier report showed that the level of illness at the time of implantation predicts survival24; this information now drives cardiologists to try to improve patient status with a temporary support device or intra-aortic balloon pump before implanting a durable device. The sickest patients (INTERMACS level 1) have the poorest outcomes, and centers now do fewer implantations in patients in this category. We have learned this important lesson from the INTERMACS registry.
The new devices have received a lot of media attention, and patient accrual has increased steadily as the devices have been approved.
On November 20, 2012, the US Food and Drug Administration approved the HeartWare Ventricular Assist System (HeartWare, Framingham, MA) for heart failure patients awaiting a transplant.
FUTURE DIRECTIONS
PROCEED II is an ongoing global clinical trial comparing the outcomes with donor hearts transported in standard cold storage to those transported in an experimental transport device that pumps the heart under physiologic conditions. If proven effective, this device could allow long-distance transport of donor hearts and expand the donor population.
A prospective, randomized study is now enrolling patients to evaluate induction therapy with rituximab (Rituxan) plus conventional immunosuppression (tacrolimus [Prograf], mycophenolate, steroid taper) vs placebo induction plus conventional immunosuppression. The study will enroll 400 patients (200 to each treatment arm) at 25 sites and will have a 36-month accrual period with 12-month follow-up (see http://clinicaltrials.gov/show/NCT01278745). The study is based on data in primates that found that eliminating B cells with an anti-CD20 drug before transplantation markedly reduced the incidence of coronary artery vasculopathy.
- Lloyd-Jones D, Adams RJ, Brown TM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 2010; 1221:e46–e215.
- Jessup M, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:1977–2016.
- 2009 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1999–2008. U.S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation, Rockville, MD.
- Metra M, Ponikowski P, Dickstein K, et al; Heart Failure Association of the European Society of Cardiology. Advanced chronic heart failure: a position statement from the Study Group on Advanced Heart Failure of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2007; 9:684–694.
- Hsich E, Gorodeski EZ, Starling RC, Blackstone EH, Ishwaran H, Lauer MS. Importance of treadmill exercise time as an initial prognostic screening tool in patients with systolic left ventricular dysfunction. Circulation 2009; 119:3189–3197.
- Gorodeski EZ, Chu EC, Chow CH, Levy WC, Hsich E, Starling RC. Application of the Seattle Heart Failure Model in ambulatory patients presented to an advanced heart failure therapeutics committee. Circ Heart Fail 2010; 3:706–714.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Taylor DO, Stehlik J, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth official adult heart transplant report—2009. J Heart Lung Transplant 2009; 28:1007–1022.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult heart transplant report—2010. J Heart Lung Transplant 2010; 29:1089–1103.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med 1999; 340:272–277. Erratum in: N Engl J Med 1999; 340:976.
- Eisen HJ, Tuzcu EM, Dorent R, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiactransplant recipients. N Engl J Med 2003; 349:847–858.
- Singh TP, Almond CS, Taylor DO, Graham DA. Decline in heart transplant wait list mortality in the United States following broader regional sharing of donor hearts. Circ Heart Fail 2012; 5:249–258.
- Baughman KL, Jarcho JA. Bridge to life—cardiac mechanical support. N Engl J Med 2007; 357:846–849.
- Rose EA, Gelijns AC, Moskowitz AJ, et al; Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med 2001; 345:1435–1443.
- Miller LW, Pagani FD, Russell SD, et al; HeartMate II Clinical Investigators. Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med 2007; 357:885–896.
- Starling RC, Naka Y, Boyle AJ, et al. Results of the post-U.S. Food and Drug Administration-approval study with a continuous flow left ventricular assist device as a bridge to heart transplantation: a prospective study using the INTERMACS (Interagency Registry for Mechanically Assisted Circulatory Support). J Am Coll Cardiol 2011; 57:1890–1898.
- Slaughter MS, Rogers JG, Milano CA, et al; HeartMate II Investigators. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009; 361:2241–2251.
- John R, Naka Y, Smedira NG, et al. Continuous flow left ventricular assist device outcomes in commercial use compared with the prior clinical trial. Ann Thorac Surg 2011; 92:1406–1413.
- Starling RC. Improved quantity and quality of life: a winning combination to treat advanced heart failure. J Am Coll Cardiol 2010; 55:1835–1836.
- Uriel N, Pak SW, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010; 56:1207–1213.
- Kirklin JK, Naftel DC, Kormos RL, et al. The fourth INTERMACS annual report: 4,000 implants and counting. J Heart Lung Transplant 2012; 31:117–126.
- Kirklin JK, Naftel DC, Kormos RL, et al. Third INTERMACS Annual Report: the evolution of destination therapy in the United States. J Heart Lung Transplant 2011; 30:115–123.
- Kirklin JK, Naftel DC, Kormos RL, et al. Second INTERMACS annual report: more than 1,000 primary left ventricular assist device implants. J Heart Lung Transplant 2010; 29:1–10.
SUGGESTED READING
Costanzo MR, Dipchand A, Starling R, et al; International Society of Heart and Lung Transplantation Guidelines. The International Society of Heart and Lung Transplantation guidelines for the care of heart transplant recipients. J Heart Lung Transplant 2010; 29:914–956.
Mehra MR, Kobashigawa J, Starling R, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates–2006. J Heart Lung Transplant 2006; 25:1024–1042.
Slaughter MS, Pagani FD, Rogers JG, et al; HeartMate II Clinical Investigators. Clinical management of continuous flow left ventricular assist devices in advanced heart failure. J Heart Lung Transplant 2010; 29(4 suppl):S1–S39.
- Lloyd-Jones D, Adams RJ, Brown TM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 2010; 1221:e46–e215.
- Jessup M, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:1977–2016.
- 2009 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1999–2008. U.S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation, Rockville, MD.
- Metra M, Ponikowski P, Dickstein K, et al; Heart Failure Association of the European Society of Cardiology. Advanced chronic heart failure: a position statement from the Study Group on Advanced Heart Failure of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2007; 9:684–694.
- Hsich E, Gorodeski EZ, Starling RC, Blackstone EH, Ishwaran H, Lauer MS. Importance of treadmill exercise time as an initial prognostic screening tool in patients with systolic left ventricular dysfunction. Circulation 2009; 119:3189–3197.
- Gorodeski EZ, Chu EC, Chow CH, Levy WC, Hsich E, Starling RC. Application of the Seattle Heart Failure Model in ambulatory patients presented to an advanced heart failure therapeutics committee. Circ Heart Fail 2010; 3:706–714.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Taylor DO, Stehlik J, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth official adult heart transplant report—2009. J Heart Lung Transplant 2009; 28:1007–1022.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult heart transplant report—2010. J Heart Lung Transplant 2010; 29:1089–1103.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med 1999; 340:272–277. Erratum in: N Engl J Med 1999; 340:976.
- Eisen HJ, Tuzcu EM, Dorent R, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiactransplant recipients. N Engl J Med 2003; 349:847–858.
- Singh TP, Almond CS, Taylor DO, Graham DA. Decline in heart transplant wait list mortality in the United States following broader regional sharing of donor hearts. Circ Heart Fail 2012; 5:249–258.
- Baughman KL, Jarcho JA. Bridge to life—cardiac mechanical support. N Engl J Med 2007; 357:846–849.
- Rose EA, Gelijns AC, Moskowitz AJ, et al; Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med 2001; 345:1435–1443.
- Miller LW, Pagani FD, Russell SD, et al; HeartMate II Clinical Investigators. Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med 2007; 357:885–896.
- Starling RC, Naka Y, Boyle AJ, et al. Results of the post-U.S. Food and Drug Administration-approval study with a continuous flow left ventricular assist device as a bridge to heart transplantation: a prospective study using the INTERMACS (Interagency Registry for Mechanically Assisted Circulatory Support). J Am Coll Cardiol 2011; 57:1890–1898.
- Slaughter MS, Rogers JG, Milano CA, et al; HeartMate II Investigators. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009; 361:2241–2251.
- John R, Naka Y, Smedira NG, et al. Continuous flow left ventricular assist device outcomes in commercial use compared with the prior clinical trial. Ann Thorac Surg 2011; 92:1406–1413.
- Starling RC. Improved quantity and quality of life: a winning combination to treat advanced heart failure. J Am Coll Cardiol 2010; 55:1835–1836.
- Uriel N, Pak SW, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010; 56:1207–1213.
- Kirklin JK, Naftel DC, Kormos RL, et al. The fourth INTERMACS annual report: 4,000 implants and counting. J Heart Lung Transplant 2012; 31:117–126.
- Kirklin JK, Naftel DC, Kormos RL, et al. Third INTERMACS Annual Report: the evolution of destination therapy in the United States. J Heart Lung Transplant 2011; 30:115–123.
- Kirklin JK, Naftel DC, Kormos RL, et al. Second INTERMACS annual report: more than 1,000 primary left ventricular assist device implants. J Heart Lung Transplant 2010; 29:1–10.
SUGGESTED READING
Costanzo MR, Dipchand A, Starling R, et al; International Society of Heart and Lung Transplantation Guidelines. The International Society of Heart and Lung Transplantation guidelines for the care of heart transplant recipients. J Heart Lung Transplant 2010; 29:914–956.
Mehra MR, Kobashigawa J, Starling R, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates–2006. J Heart Lung Transplant 2006; 25:1024–1042.
Slaughter MS, Pagani FD, Rogers JG, et al; HeartMate II Clinical Investigators. Clinical management of continuous flow left ventricular assist devices in advanced heart failure. J Heart Lung Transplant 2010; 29(4 suppl):S1–S39.
KEY POINTS
- After heart transplantation, survival rates are high and quality of life is excellent, although coronary artery disease, renal dysfunction, and the need for immunosuppressive drugs are ongoing challenges.
- Changes in donor heart allocation made in 2006 more strongly favor the sickest patients and have reduced the rate of mortality on the waiting list.
- Continuous-flow left-ventricular assist devices offer many advantages over the older pulsatile-flow devices, including improved outcomes, smaller size, less noise, and greater durability.
- Inotropic therapy is purely palliative and should not be viewed as an alternative to heart transplantation or device implantation.
Atrial fibrillation: New drugs, devices, and procedures
Although many developments have occurred in the last decade for managing atrial fibrillation, challenges remain. New and emerging alternatives to warfarin (Coumadin) for anticoagulation therapy prevent stroke marginally better and pose slightly less risk of hemorrhage, but they have important drawbacks.
The antiarrhythmic drug dronedarone (Multaq) has been found to offer only temporary benefit for persistent atrial fibrillation, and significant risks have emerged.
Radiofrequency ablation is gaining prominence, but repeat procedures are sometimes necessary.
An investigational device can be implanted via percutaneous catheter in the left atrial appendage to prevent embolization. It is too soon to know its eventual role in clinical practice.
This article reviews the results of clinical trials of these new treatments and discusses their role in clinical practice.
CHALLENGES OF ANTICOAGULATION
The main focus of managing atrial fibrillation is on alleviating symptoms, by either rate control or rhythm control. The other focus is on preventing stroke—a devastating outcome—with anticoagulation therapy.
For deciding whether to give warfarin to patients with atrial fibrillation, the six-point CHADS2 score is a crude but effective way of assessing the risk of stroke based on the following risk factors: congestive heart failure, hypertension, age 75 years or older, and diabetes (1 point each); or a history of stroke or transient ischemic attack (2 points).1 Warfarin is given if patients have a score of at least 2 points.
Warfarin has a narrow therapeutic window, with a higher risk of ischemic stroke if the international normalized ratio (INR) is less than 2.0,2 and a higher risk of intracranial hemorrhage if the INR is more than 3.0.3 Keeping the INR in the therapeutic range is difficult because of variations in diet, concurrent medications, and other factors.
The percent of time that the INR is within the therapeutic range predicts the risk of adverse events. Connolly et al4 showed that the cumulative risk of stroke, myocardial infarction, systemic embolism, or vascular death was no better with warfarin than with clopidogrel (Plavix) plus aspirin if the INR was in the therapeutic range less than 65% of the time, but the risk was significantly less if the INR was in the therapeutic range more than 65% of the time.
Also, comparing warfarin with the combination of aspirin and clopidogrel, Verheugt5 found that the rates of stroke of any kind, of disabling and fatal stroke, and of stroke per major bleed were lower in patients taking warfarin. Although many physicians prefer aspirin plus clopidogrel because of concerns about bleeding with warfarin, the rates of major bleeding were about the same in the two groups.
In a trial in patients for whom warfarin was “unsuitable,”6 the combination of aspirin plus clopidogrel was associated with a lower rate of stroke than aspirin alone (2.4% per year vs 3.3% per year, relative risk 0.762) but a higher rate of major bleeding events (2.0% per year vs 1.3% per year, relative risk 1.57).
NEW ALTERNATIVES TO WARFARIN

Because of the problems with warfarin, alternatives have been sought for many years. Several new oral anticoagulants are available or are being developed,7 including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis) and the direct factor II (thrombin) inhibitor dabigatran (Pradaxa) (Table 1).
Dabigatran’s advantages and drawbacks
Dabigatran has been on the market for more than a year and has gained rapid acceptance. The dosage is 150 mg twice a day, or 75 mg twice a day if renal function is impaired. Cleared by the kidneys, it has a half-life of 12 to 17 hours; 75% is cleared within 24 hours. For a patient who needs surgery that poses a low risk of bleeding, the general recommendation is to stop dabigatran the night before the surgical procedure. For operations with a greater risk of bleeding, many surgeons recommend stopping the drug 3 or 4 days before.
Advantages of dabigatran include that it is not influenced by diet and that the onset of therapeutic benefit is within 1 hour. Although some drugs affect dabigatran, drug interactions are more troublesome with warfarin.
A serious concern about dabigatran and the other new agents is that if a bleeding problem arises, the effects of these drugs are not reversible by administration of fresh frozen plasma. Dabigatran is reversible by dialysis; however, if a patient is also hypotensive, dialysis is not an option, and simply waiting for the drug to clear is the only choice.
Another drawback is that therapeutic levels cannot be monitored. If a patient taking warfarin requires cardioversion, the INR is carefully monitored for several weeks beforehand to reduce the risk of stroke. With dabigatran, there is no way to know if a patient is actually taking the drug as prescribed.
Clinical trials show that alternatives are marginally better than warfarin
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,8 dabigatran was associated with a significantly lower incidence of intracranial hemorrhage, combined strokes, and systemic embolization than warfarin. The incidence of major bleeds was slightly lower with dabigatran. Although dabigatran performed better, the differences were small and would not require patients to change from warfarin if they are already doing well.
Apixaban and rivaroxaban are other alternatives to warfarin, with different mechanisms of action and metabolism. Although rivaroxaban’s half-life is similar to that of apixaban and dabigatran, it is being marketed as allowing once-daily dosing instead of twice-daily.
Recent randomized controlled clinical trials of the new drugs include:
- The Apixaban Versus Acetylsalicylic Acid (ASA) to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment (AVERROES) trial,9 which compared apixaban and aspirin
- The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial comparing apixaban and warfarin10
- The Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF),11 comparing rivaroxaban and warfarin
- RE-LY,8 comparing dabigatran and warfarin.
In the ARISTOTLE,10 ROCKET AF,11 and RE-LY trials,8 the time that the warfarin patients’ INRs were in the therapeutic range varied from 55% to 68%. This seems low and is a problem when trying to compare therapies, but is probably about as high as one can expect in the real world.
In AVERROES,9 the combined rate of stroke and embolism was higher with aspirin than with apixaban. In the other trials, the rates were slightly better with the new drugs than with warfarin, and the rates of major hemorrhage and hemorrhagic stroke were only slightly higher with warfarin than with the new drugs. Because the differences in benefits and risks are so small, the main advantage of the newer drugs will probably be for patients who have difficulty staying in the therapeutic INR range on warfarin.
RATE CONTROL VS RESTORATION OF SINUS RHYTHM
Evidence is insufficient to determine the risk of very-long-term asymptomatic atrial fibrillation in patients on appropriate anticoagulation. Rate control is an option for asymptomatic patients but provides no change in quality of life and no definitive reduction in the risk of stroke. The main argument for restoring normal sinus rhythm in patients with mild to moderate symptoms is that it improves exercise capacity. The need for anticoagulation persists when patients are converted to sinus rhythm because the risk of recurrent atrial fibrillation remains high.
For patients with symptomatic atrial fibrillation, rate control is sometimes achieved with beta-blockers or calcium channel blockers. Rate control may be augmented with the addition of digoxin, but when used alone digoxin generally does not control the rate of atrial fibrillation. However, in many cases of atrial fibrillation, symptoms are not rate-related, and cardioversion to normal sinus rhythm should be attempted. In such cases, the symptoms may be attributable to a loss of atrial transport function.
Patients with the following risk factors should be admitted to the hospital to start antiarrhythmic drugs:
- Borderline or a long QTc interval at baseline (> 450 msec)
- Treatment with dofetilide (Tikosyn) because of its effects on the QT interval
- Heart failure or poor left-ventricular function
- Sinus node dysfunction
- Significant atrioventricular conduction disease.
Selecting an antiarrhythmic drug
Any of the antiarrhythmic drugs listed in Table 2 can be used for a patient with lone atrial fibrillation (ie, not caused by underlying heart disease). The choice of drug should be determined by whether coronary artery disease or renal failure is present as well. Liver disease or chronic obstructive pulmonary disease also may affect this decision.
Benefits of dronedarone are mixed
In a randomized trial of dronedarone vs placebo in patients with atrial fibrillation, the rate of death and the rate of first hospitalization due to a cardiovascular event at 21 months were significantly lower with dronedarone.12 No difference was found between the two groups in the rate of death from all causes, but fewer people died of cardiovascular causes in the dronedarone group. More patients taking dronedarone developed bradycardia, QT-interval prolongation, nausea, diarrhea, rash, or a higher serum creatinine level. Gastrointestinal side effects are often a problem with dronedarone: 20% to 30% of patients cannot tolerate the drug.
Dronedarone may cause a small rise in creatinine, and although this effect should be monitored, by itself it should not be interpreted as impairment of renal function. In a study in healthy people,13 dronedarone caused a 10% to 15% increase in serum creatinine, but the glomerular filtration rate was unchanged, as were renal plasma flow and anion secretion.
Another trial, in patients with severe heart failure, found that patients taking dronedarone had higher rates of hospitalization and overall mortality, raising serious concern about the safety of this drug in patients with advanced heart failure.14
Singh et al15 pooled the data from two multicenter, randomized trials that compared dronedarone with placebo for maintaining sinus rhythm in patients with atrial fibrillation or flutter. The mean time to the recurrence of atrial fibrillation was 116 days with dronedarone and 53 days with placebo. Other trials also showed longer times to recurrence and lower recurrence rates with dronedarone. Although the differences were statistically significant, they may not be clinically meaningful for patients.
Dronedarone is structurally similar to amiodarone (Cordarone), but the two drugs work differently. A meta-analysis of clinical trials16 found that amiodarone recipients had a lower rate of recurrence of atrial fibrillation than did those receiving dronedarone.
Two safety warnings for dronedarone
In January 2011, the US Food and Drug Administration (FDA) issued an alert about cases of rare but severe liver injury in patients treated with dronedarone, including two cases of acute liver failure leading to liver transplantation.17
The Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS)18 compared dronedarone and placebo in patients with permanent atrial fibrillation. More people died or had serious cardiovascular adverse events in the dronedarone group. The study was stopped early after data monitoring showed that rates of death, stroke, and hospitalization for heart failure were two times higher in patients receiving dronedarone. This prompted the FDA to issue another safety alert in July 2011.
Interestingly, the PALLAS study did not set out to determine whether dronedarone controls atrial fibrillation, as the study patients had long-standing, persistent atrial fibrillation. The study was designed only to determine if the drug reduces the rate of adverse events; it clearly does not, and the study shows that dronedarone should not be used to control the heart rate in patients with persistent atrial fibrillation. Instead, its use is best restricted to patients with paroxysmal atrial fibrillation without significant cardiovascular disease.
ABLATION OF ATRIAL FIBRILLATION
Another way to try to restore sinus rhythm is to destroy or isolate the area that is generating the abnormal beats via a catheter-based procedure.
Radiofrequency ablation is generally tried in patients in whom one or two drugs have failed to control atrial fibrillation. Direct comparisons show that ablation is superior to drug therapy and is effective in about 75% of patients with paroxysmal atrial fibrillation vs 20% to 40% of patients on drug therapy. Ablation plus drug therapy is often more effective than either treatment alone.
Mechanisms of atrial fibrillation and ablation
In many cases, atrial fibrillation is stimulated by vagal and sympathetic inputs to the atrium that enter around the pulmonary veins and trigger electrical activations in the area, generating spiraling, reentering circuits. Focal atrial fibrillation also originates predominantly in the pulmonary veins. Ablation of tissue widely circumscribing the mouth of the pulmonary veins prevents the electrical signal from exiting into the atrium.
In about 11% to 37% of cases, atrial fibrillation originates elsewhere, eg, in the left atrium, in the superior vena cava, or in the vein of Marshall. Techniques have evolved to also ablate these regions.
Anticoagulation therapy is recommended before the procedure, and patients at low risk should continue it for a minimum of 2 months afterward. Patients with a higher CHADS2 score should receive anticoagulation therapy for at least 1 year. The consensus statement by the Heart Rhythm Society19 recommends that patients remain on warfarin or one of the newer anticoagulants if their CHADS2 score is 2 or higher. This is because patients have a significant risk of recurrence of atrial fibrillation after radiofrequency ablation, so if their stroke risk is high they should remain on anticoagulant therapy.
Ablation is usually effective, but it carries rare but serious risks
The efficacy of a single radiofrequency ablation procedure is in the range of 60% to 80% for paroxysmal atrial fibrillation and 40% to 60% for persistent atrial fibrillation. The Second International Ablation Registry20 shows a success rate of about 75% in patients with paroxysmal atrial fibrillation and about 65% in patients with persistent and permanent atrial fibrillation. Registry data are often more favorable because reporting is optional, but these results are consistent with those from experienced medical centers. Rates of suppression of atrial fibrillation are higher in patients who also take antiarrhythmic drugs, making a “hybrid” approach useful when ablation alone fails.
According to a worldwide survey, the risk of serious complications is 4.5%. These include stroke (0.23%), tamponade (1.3%), and pulmonary vein stenosis (< 0.29%). The esophagus lies just behind the right atrium, and burning through and creating a fistula between them occurs in about 0.04% of cases and is almost uniformly fatal.20
A second ablation procedure is sometimes indicated for the recurrence of atrial fibrillation, which is almost always caused by recovery of the pulmonary veins. Bhargava et al21 found that the success rate at Cleveland Clinic for a single procedure for paroxysmal atrial fibrillation was 77%, and that it was 92% after a repeat procedure. For persistent atrial fibrillation, success rates were 76% after the first procedure and 90% after the second. Even for long-standing persistent atrial fibrillation (ie, lasting more than 1 year), 80% success was achieved after two procedures. Patients who are less likely to have a successful ablation procedure are those with long-standing atrial fibrillation and coexisting heart disease, including severe valvular disease, although mitral regurgitation sometimes improves if sinus rhythm can be maintained.
The need for a second procedure
After ablation, patients should be closely monitored for a recurrence of atrial fibrillation. Continuous monitoring with implantable cardiac monitor loop recorders can detect unrecognized episodes of arrhythmia. Long-term follow-up is also required to track outcomes and quality of life.
According to the Heart Rhythm Society Task Force on Catheter and Surgical Ablation of Atrial Fibrillation,19 atrial fibrillation recurs after ablation in about 35% to 60% of patients in the first 3 months, with recurrence rates after 1 year ranging from 5% to 16%. The rate of success is determined by the skill of the surgeon, underlying heart disease, attention to follow-up, and how success is defined.
Freedom from recurrence early on is a good predictor that late recurrence is unlikely. Patients who only have a very early recurrence (within the first 4 weeks) are more likely to have long-term freedom from atrial fibrillation tha those who have recurrences after that time.22
In a study of 831 patients, Hussein et al23 found recurrence rates of 24% between months 3 to 13 following ablation and 9% after 12 months. At 55 months, 79% were free from atrial fibrillation without drugs, 11% were free of atrial fibrillation with medications, and 5% had refractory atrial fibrillation.
Recurrence—whether early or late—was more likely to occur in people with persistent vs paroxysmal atrial fibrillation. Other risk factors for late recurrence included older age and larger left atrial size (which is also a risk factor for recurrence on drug therapy). Although recurrent arrhythmia was most often atrial fibrillation, atrial flutter also occurred frequently (in 27% of patients with late recurrence). Three patients (4% of patients with late recurrence) developed atrial tachycardia.23
In patients with early recurrence, 81% underwent repeat ablation, all of whom had recovery of one or more pulmonary veins. After the second ablation, 21% had recurrence, 65% of whom were controlled by medications.23
Whether a patient should undergo subsequent ablation procedures depends on the severity of symptoms, the likelihood of success (based on an educated guess), and the patient’s willingness to undergo another procedure.
ATRIAL APPENDAGE OCCLUSION DEVICE UNDER INVESTIGATION
New devices are being investigated that occlude the left atrial appendage to try to prevent embolization.
The Watchman device, resembling an umbrella, is implanted via a percutaneous catheter in the left atrial appendage, closing it off to preclude a thrombus from forming in the appendage and embolizing to the body. Clinical trials showed that patients who received a device had a slightly lower risk of stroke than otherwise seen in clinical practice.24 Safety and efficacy are still being determined.
The device cannot be deployed in a patient with an existing thrombus because of the danger of dislodging the thrombus, allowing it to embolize.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the national Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Hylek EM, Skates SJ, Sheehan MA, Singer DE. An analysis of the lowest effective intensity of prophylactic anticoagulation for patients with nonrheumatic atrial fibrillation. N Engl J Med 1996; 335:540–546.
- Hylek EM, Singer DE. Risk factors for intracranial hemorrhage in outpatients taking warfarin. Ann Intern Med 1994; 120:897–902.
- Connolly SJ, Pogue J, Eikelboom J, et al; ACTIVE W Investigators. Benefit of oral anticoagulant over antiplatelet therapy in atrial fibrillation depends on the quality of international normalized ratio control achieved by centers and countries as measured by time in therapeutic range. Circulation 2008; 118:2029–2037.
- Verheugt FWA. Who is ineligible for warfarin in atrial fibrillation? Lancet 2009; 374:510–511.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Harenberg J. New anticoagulants in atrial fibrillation. Semin Thromb Hemost 2009; 35:574–585.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Connolly SJ, Eikelboom J, Joyner C, et al; AVERROES Steering Committee and Investigators. Apixaban in patients with atrial fibrillation. N Engl J Med 2011; 364:806–817.
- Granger CB, Alexander JH, McMurray JJV, et al; for the ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med August 28, 2011; 10.1056/nejmoa1107039.
- Patel MR, Mahaffey KW, Garg J, et al; the ROCKET AF Steering Committee. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Hohnloser SH, Crijns HJ, van Eickels M, et al; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:688–678.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
- Kóber L, Torp-Pederson C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Piccini JP, Hasselblad V, Peterson ED, Washam JB, Califf RM. Comparative efficacy of dronedarone and amiodarone for the maintenance of sinus rhythm in patients with atrial fibrillation. J Am Coll Cardiol 2009; 54:1089–1095.
- US Food and Drug Administration. FDA drug safety communication: severe liver injury associated with the use of dronedarone (marketed as Multaq). http://www.fda.gov/drugs/drugsafety/ucm240011.htm. Accessed July 5, 2012.
- Connolly SJ, Camm AJ, Halperin JL, et al; for the PALLAS Investigators. Dronedarone in high-risk permanent atrial fibrillation. N Engl J Med 2011; 365:2268–2276.
- HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for personnel, policy, procedures and follow-up. Heart Rhythm 2007; 4:1–46.
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3:32–38.
- Bhargava M, Di Biase L, Mohanty P, et al. Impact of type of atrial fibrillation and repeat catheter ablation on long-term freedom from atrial fibrillation: results from a multicenter study. Heart Rhythm 2009; 6:1403–1412.
- Themistoclakis S, Schweikert RA, Sliba WI, et al. Clinical predictors and relationship between early and late atrial tachyarrhythmias after pulmonary vein antrum isolation. Heart Rhythm 2008; 5:679–685.
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4:271–278.
- Holmes DR, Reddy VY, Turi ZG, et al; for the PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
Although many developments have occurred in the last decade for managing atrial fibrillation, challenges remain. New and emerging alternatives to warfarin (Coumadin) for anticoagulation therapy prevent stroke marginally better and pose slightly less risk of hemorrhage, but they have important drawbacks.
The antiarrhythmic drug dronedarone (Multaq) has been found to offer only temporary benefit for persistent atrial fibrillation, and significant risks have emerged.
Radiofrequency ablation is gaining prominence, but repeat procedures are sometimes necessary.
An investigational device can be implanted via percutaneous catheter in the left atrial appendage to prevent embolization. It is too soon to know its eventual role in clinical practice.
This article reviews the results of clinical trials of these new treatments and discusses their role in clinical practice.
CHALLENGES OF ANTICOAGULATION
The main focus of managing atrial fibrillation is on alleviating symptoms, by either rate control or rhythm control. The other focus is on preventing stroke—a devastating outcome—with anticoagulation therapy.
For deciding whether to give warfarin to patients with atrial fibrillation, the six-point CHADS2 score is a crude but effective way of assessing the risk of stroke based on the following risk factors: congestive heart failure, hypertension, age 75 years or older, and diabetes (1 point each); or a history of stroke or transient ischemic attack (2 points).1 Warfarin is given if patients have a score of at least 2 points.
Warfarin has a narrow therapeutic window, with a higher risk of ischemic stroke if the international normalized ratio (INR) is less than 2.0,2 and a higher risk of intracranial hemorrhage if the INR is more than 3.0.3 Keeping the INR in the therapeutic range is difficult because of variations in diet, concurrent medications, and other factors.
The percent of time that the INR is within the therapeutic range predicts the risk of adverse events. Connolly et al4 showed that the cumulative risk of stroke, myocardial infarction, systemic embolism, or vascular death was no better with warfarin than with clopidogrel (Plavix) plus aspirin if the INR was in the therapeutic range less than 65% of the time, but the risk was significantly less if the INR was in the therapeutic range more than 65% of the time.
Also, comparing warfarin with the combination of aspirin and clopidogrel, Verheugt5 found that the rates of stroke of any kind, of disabling and fatal stroke, and of stroke per major bleed were lower in patients taking warfarin. Although many physicians prefer aspirin plus clopidogrel because of concerns about bleeding with warfarin, the rates of major bleeding were about the same in the two groups.
In a trial in patients for whom warfarin was “unsuitable,”6 the combination of aspirin plus clopidogrel was associated with a lower rate of stroke than aspirin alone (2.4% per year vs 3.3% per year, relative risk 0.762) but a higher rate of major bleeding events (2.0% per year vs 1.3% per year, relative risk 1.57).
NEW ALTERNATIVES TO WARFARIN
Because of the problems with warfarin, alternatives have been sought for many years. Several new oral anticoagulants are available or are being developed,7 including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis) and the direct factor II (thrombin) inhibitor dabigatran (Pradaxa) (Table 1).
Dabigatran’s advantages and drawbacks
Dabigatran has been on the market for more than a year and has gained rapid acceptance. The dosage is 150 mg twice a day, or 75 mg twice a day if renal function is impaired. Cleared by the kidneys, it has a half-life of 12 to 17 hours; 75% is cleared within 24 hours. For a patient who needs surgery that poses a low risk of bleeding, the general recommendation is to stop dabigatran the night before the surgical procedure. For operations with a greater risk of bleeding, many surgeons recommend stopping the drug 3 or 4 days before.
Advantages of dabigatran include that it is not influenced by diet and that the onset of therapeutic benefit is within 1 hour. Although some drugs affect dabigatran, drug interactions are more troublesome with warfarin.
A serious concern about dabigatran and the other new agents is that if a bleeding problem arises, the effects of these drugs are not reversible by administration of fresh frozen plasma. Dabigatran is reversible by dialysis; however, if a patient is also hypotensive, dialysis is not an option, and simply waiting for the drug to clear is the only choice.
Another drawback is that therapeutic levels cannot be monitored. If a patient taking warfarin requires cardioversion, the INR is carefully monitored for several weeks beforehand to reduce the risk of stroke. With dabigatran, there is no way to know if a patient is actually taking the drug as prescribed.
Clinical trials show that alternatives are marginally better than warfarin
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,8 dabigatran was associated with a significantly lower incidence of intracranial hemorrhage, combined strokes, and systemic embolization than warfarin. The incidence of major bleeds was slightly lower with dabigatran. Although dabigatran performed better, the differences were small and would not require patients to change from warfarin if they are already doing well.
Apixaban and rivaroxaban are other alternatives to warfarin, with different mechanisms of action and metabolism. Although rivaroxaban’s half-life is similar to that of apixaban and dabigatran, it is being marketed as allowing once-daily dosing instead of twice-daily.
Recent randomized controlled clinical trials of the new drugs include:
- The Apixaban Versus Acetylsalicylic Acid (ASA) to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment (AVERROES) trial,9 which compared apixaban and aspirin
- The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial comparing apixaban and warfarin10
- The Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF),11 comparing rivaroxaban and warfarin
- RE-LY,8 comparing dabigatran and warfarin.
In the ARISTOTLE,10 ROCKET AF,11 and RE-LY trials,8 the time that the warfarin patients’ INRs were in the therapeutic range varied from 55% to 68%. This seems low and is a problem when trying to compare therapies, but is probably about as high as one can expect in the real world.
In AVERROES,9 the combined rate of stroke and embolism was higher with aspirin than with apixaban. In the other trials, the rates were slightly better with the new drugs than with warfarin, and the rates of major hemorrhage and hemorrhagic stroke were only slightly higher with warfarin than with the new drugs. Because the differences in benefits and risks are so small, the main advantage of the newer drugs will probably be for patients who have difficulty staying in the therapeutic INR range on warfarin.
RATE CONTROL VS RESTORATION OF SINUS RHYTHM
Evidence is insufficient to determine the risk of very-long-term asymptomatic atrial fibrillation in patients on appropriate anticoagulation. Rate control is an option for asymptomatic patients but provides no change in quality of life and no definitive reduction in the risk of stroke. The main argument for restoring normal sinus rhythm in patients with mild to moderate symptoms is that it improves exercise capacity. The need for anticoagulation persists when patients are converted to sinus rhythm because the risk of recurrent atrial fibrillation remains high.
For patients with symptomatic atrial fibrillation, rate control is sometimes achieved with beta-blockers or calcium channel blockers. Rate control may be augmented with the addition of digoxin, but when used alone digoxin generally does not control the rate of atrial fibrillation. However, in many cases of atrial fibrillation, symptoms are not rate-related, and cardioversion to normal sinus rhythm should be attempted. In such cases, the symptoms may be attributable to a loss of atrial transport function.
Patients with the following risk factors should be admitted to the hospital to start antiarrhythmic drugs:
- Borderline or a long QTc interval at baseline (> 450 msec)
- Treatment with dofetilide (Tikosyn) because of its effects on the QT interval
- Heart failure or poor left-ventricular function
- Sinus node dysfunction
- Significant atrioventricular conduction disease.
Selecting an antiarrhythmic drug
Any of the antiarrhythmic drugs listed in Table 2 can be used for a patient with lone atrial fibrillation (ie, not caused by underlying heart disease). The choice of drug should be determined by whether coronary artery disease or renal failure is present as well. Liver disease or chronic obstructive pulmonary disease also may affect this decision.
Benefits of dronedarone are mixed
In a randomized trial of dronedarone vs placebo in patients with atrial fibrillation, the rate of death and the rate of first hospitalization due to a cardiovascular event at 21 months were significantly lower with dronedarone.12 No difference was found between the two groups in the rate of death from all causes, but fewer people died of cardiovascular causes in the dronedarone group. More patients taking dronedarone developed bradycardia, QT-interval prolongation, nausea, diarrhea, rash, or a higher serum creatinine level. Gastrointestinal side effects are often a problem with dronedarone: 20% to 30% of patients cannot tolerate the drug.
Dronedarone may cause a small rise in creatinine, and although this effect should be monitored, by itself it should not be interpreted as impairment of renal function. In a study in healthy people,13 dronedarone caused a 10% to 15% increase in serum creatinine, but the glomerular filtration rate was unchanged, as were renal plasma flow and anion secretion.
Another trial, in patients with severe heart failure, found that patients taking dronedarone had higher rates of hospitalization and overall mortality, raising serious concern about the safety of this drug in patients with advanced heart failure.14
Singh et al15 pooled the data from two multicenter, randomized trials that compared dronedarone with placebo for maintaining sinus rhythm in patients with atrial fibrillation or flutter. The mean time to the recurrence of atrial fibrillation was 116 days with dronedarone and 53 days with placebo. Other trials also showed longer times to recurrence and lower recurrence rates with dronedarone. Although the differences were statistically significant, they may not be clinically meaningful for patients.
Dronedarone is structurally similar to amiodarone (Cordarone), but the two drugs work differently. A meta-analysis of clinical trials16 found that amiodarone recipients had a lower rate of recurrence of atrial fibrillation than did those receiving dronedarone.
Two safety warnings for dronedarone
In January 2011, the US Food and Drug Administration (FDA) issued an alert about cases of rare but severe liver injury in patients treated with dronedarone, including two cases of acute liver failure leading to liver transplantation.17
The Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS)18 compared dronedarone and placebo in patients with permanent atrial fibrillation. More people died or had serious cardiovascular adverse events in the dronedarone group. The study was stopped early after data monitoring showed that rates of death, stroke, and hospitalization for heart failure were two times higher in patients receiving dronedarone. This prompted the FDA to issue another safety alert in July 2011.
Interestingly, the PALLAS study did not set out to determine whether dronedarone controls atrial fibrillation, as the study patients had long-standing, persistent atrial fibrillation. The study was designed only to determine if the drug reduces the rate of adverse events; it clearly does not, and the study shows that dronedarone should not be used to control the heart rate in patients with persistent atrial fibrillation. Instead, its use is best restricted to patients with paroxysmal atrial fibrillation without significant cardiovascular disease.
ABLATION OF ATRIAL FIBRILLATION
Another way to try to restore sinus rhythm is to destroy or isolate the area that is generating the abnormal beats via a catheter-based procedure.
Radiofrequency ablation is generally tried in patients in whom one or two drugs have failed to control atrial fibrillation. Direct comparisons show that ablation is superior to drug therapy and is effective in about 75% of patients with paroxysmal atrial fibrillation vs 20% to 40% of patients on drug therapy. Ablation plus drug therapy is often more effective than either treatment alone.
Mechanisms of atrial fibrillation and ablation
In many cases, atrial fibrillation is stimulated by vagal and sympathetic inputs to the atrium that enter around the pulmonary veins and trigger electrical activations in the area, generating spiraling, reentering circuits. Focal atrial fibrillation also originates predominantly in the pulmonary veins. Ablation of tissue widely circumscribing the mouth of the pulmonary veins prevents the electrical signal from exiting into the atrium.
In about 11% to 37% of cases, atrial fibrillation originates elsewhere, eg, in the left atrium, in the superior vena cava, or in the vein of Marshall. Techniques have evolved to also ablate these regions.
Anticoagulation therapy is recommended before the procedure, and patients at low risk should continue it for a minimum of 2 months afterward. Patients with a higher CHADS2 score should receive anticoagulation therapy for at least 1 year. The consensus statement by the Heart Rhythm Society19 recommends that patients remain on warfarin or one of the newer anticoagulants if their CHADS2 score is 2 or higher. This is because patients have a significant risk of recurrence of atrial fibrillation after radiofrequency ablation, so if their stroke risk is high they should remain on anticoagulant therapy.
Ablation is usually effective, but it carries rare but serious risks
The efficacy of a single radiofrequency ablation procedure is in the range of 60% to 80% for paroxysmal atrial fibrillation and 40% to 60% for persistent atrial fibrillation. The Second International Ablation Registry20 shows a success rate of about 75% in patients with paroxysmal atrial fibrillation and about 65% in patients with persistent and permanent atrial fibrillation. Registry data are often more favorable because reporting is optional, but these results are consistent with those from experienced medical centers. Rates of suppression of atrial fibrillation are higher in patients who also take antiarrhythmic drugs, making a “hybrid” approach useful when ablation alone fails.
According to a worldwide survey, the risk of serious complications is 4.5%. These include stroke (0.23%), tamponade (1.3%), and pulmonary vein stenosis (< 0.29%). The esophagus lies just behind the right atrium, and burning through and creating a fistula between them occurs in about 0.04% of cases and is almost uniformly fatal.20
A second ablation procedure is sometimes indicated for the recurrence of atrial fibrillation, which is almost always caused by recovery of the pulmonary veins. Bhargava et al21 found that the success rate at Cleveland Clinic for a single procedure for paroxysmal atrial fibrillation was 77%, and that it was 92% after a repeat procedure. For persistent atrial fibrillation, success rates were 76% after the first procedure and 90% after the second. Even for long-standing persistent atrial fibrillation (ie, lasting more than 1 year), 80% success was achieved after two procedures. Patients who are less likely to have a successful ablation procedure are those with long-standing atrial fibrillation and coexisting heart disease, including severe valvular disease, although mitral regurgitation sometimes improves if sinus rhythm can be maintained.
The need for a second procedure
After ablation, patients should be closely monitored for a recurrence of atrial fibrillation. Continuous monitoring with implantable cardiac monitor loop recorders can detect unrecognized episodes of arrhythmia. Long-term follow-up is also required to track outcomes and quality of life.
According to the Heart Rhythm Society Task Force on Catheter and Surgical Ablation of Atrial Fibrillation,19 atrial fibrillation recurs after ablation in about 35% to 60% of patients in the first 3 months, with recurrence rates after 1 year ranging from 5% to 16%. The rate of success is determined by the skill of the surgeon, underlying heart disease, attention to follow-up, and how success is defined.
Freedom from recurrence early on is a good predictor that late recurrence is unlikely. Patients who only have a very early recurrence (within the first 4 weeks) are more likely to have long-term freedom from atrial fibrillation tha those who have recurrences after that time.22
In a study of 831 patients, Hussein et al23 found recurrence rates of 24% between months 3 to 13 following ablation and 9% after 12 months. At 55 months, 79% were free from atrial fibrillation without drugs, 11% were free of atrial fibrillation with medications, and 5% had refractory atrial fibrillation.
Recurrence—whether early or late—was more likely to occur in people with persistent vs paroxysmal atrial fibrillation. Other risk factors for late recurrence included older age and larger left atrial size (which is also a risk factor for recurrence on drug therapy). Although recurrent arrhythmia was most often atrial fibrillation, atrial flutter also occurred frequently (in 27% of patients with late recurrence). Three patients (4% of patients with late recurrence) developed atrial tachycardia.23
In patients with early recurrence, 81% underwent repeat ablation, all of whom had recovery of one or more pulmonary veins. After the second ablation, 21% had recurrence, 65% of whom were controlled by medications.23
Whether a patient should undergo subsequent ablation procedures depends on the severity of symptoms, the likelihood of success (based on an educated guess), and the patient’s willingness to undergo another procedure.
ATRIAL APPENDAGE OCCLUSION DEVICE UNDER INVESTIGATION
New devices are being investigated that occlude the left atrial appendage to try to prevent embolization.
The Watchman device, resembling an umbrella, is implanted via a percutaneous catheter in the left atrial appendage, closing it off to preclude a thrombus from forming in the appendage and embolizing to the body. Clinical trials showed that patients who received a device had a slightly lower risk of stroke than otherwise seen in clinical practice.24 Safety and efficacy are still being determined.
The device cannot be deployed in a patient with an existing thrombus because of the danger of dislodging the thrombus, allowing it to embolize.
Although many developments have occurred in the last decade for managing atrial fibrillation, challenges remain. New and emerging alternatives to warfarin (Coumadin) for anticoagulation therapy prevent stroke marginally better and pose slightly less risk of hemorrhage, but they have important drawbacks.
The antiarrhythmic drug dronedarone (Multaq) has been found to offer only temporary benefit for persistent atrial fibrillation, and significant risks have emerged.
Radiofrequency ablation is gaining prominence, but repeat procedures are sometimes necessary.
An investigational device can be implanted via percutaneous catheter in the left atrial appendage to prevent embolization. It is too soon to know its eventual role in clinical practice.
This article reviews the results of clinical trials of these new treatments and discusses their role in clinical practice.
CHALLENGES OF ANTICOAGULATION
The main focus of managing atrial fibrillation is on alleviating symptoms, by either rate control or rhythm control. The other focus is on preventing stroke—a devastating outcome—with anticoagulation therapy.
For deciding whether to give warfarin to patients with atrial fibrillation, the six-point CHADS2 score is a crude but effective way of assessing the risk of stroke based on the following risk factors: congestive heart failure, hypertension, age 75 years or older, and diabetes (1 point each); or a history of stroke or transient ischemic attack (2 points).1 Warfarin is given if patients have a score of at least 2 points.
Warfarin has a narrow therapeutic window, with a higher risk of ischemic stroke if the international normalized ratio (INR) is less than 2.0,2 and a higher risk of intracranial hemorrhage if the INR is more than 3.0.3 Keeping the INR in the therapeutic range is difficult because of variations in diet, concurrent medications, and other factors.
The percent of time that the INR is within the therapeutic range predicts the risk of adverse events. Connolly et al4 showed that the cumulative risk of stroke, myocardial infarction, systemic embolism, or vascular death was no better with warfarin than with clopidogrel (Plavix) plus aspirin if the INR was in the therapeutic range less than 65% of the time, but the risk was significantly less if the INR was in the therapeutic range more than 65% of the time.
Also, comparing warfarin with the combination of aspirin and clopidogrel, Verheugt5 found that the rates of stroke of any kind, of disabling and fatal stroke, and of stroke per major bleed were lower in patients taking warfarin. Although many physicians prefer aspirin plus clopidogrel because of concerns about bleeding with warfarin, the rates of major bleeding were about the same in the two groups.
In a trial in patients for whom warfarin was “unsuitable,”6 the combination of aspirin plus clopidogrel was associated with a lower rate of stroke than aspirin alone (2.4% per year vs 3.3% per year, relative risk 0.762) but a higher rate of major bleeding events (2.0% per year vs 1.3% per year, relative risk 1.57).
NEW ALTERNATIVES TO WARFARIN
Because of the problems with warfarin, alternatives have been sought for many years. Several new oral anticoagulants are available or are being developed,7 including the factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis) and the direct factor II (thrombin) inhibitor dabigatran (Pradaxa) (Table 1).
Dabigatran’s advantages and drawbacks
Dabigatran has been on the market for more than a year and has gained rapid acceptance. The dosage is 150 mg twice a day, or 75 mg twice a day if renal function is impaired. Cleared by the kidneys, it has a half-life of 12 to 17 hours; 75% is cleared within 24 hours. For a patient who needs surgery that poses a low risk of bleeding, the general recommendation is to stop dabigatran the night before the surgical procedure. For operations with a greater risk of bleeding, many surgeons recommend stopping the drug 3 or 4 days before.
Advantages of dabigatran include that it is not influenced by diet and that the onset of therapeutic benefit is within 1 hour. Although some drugs affect dabigatran, drug interactions are more troublesome with warfarin.
A serious concern about dabigatran and the other new agents is that if a bleeding problem arises, the effects of these drugs are not reversible by administration of fresh frozen plasma. Dabigatran is reversible by dialysis; however, if a patient is also hypotensive, dialysis is not an option, and simply waiting for the drug to clear is the only choice.
Another drawback is that therapeutic levels cannot be monitored. If a patient taking warfarin requires cardioversion, the INR is carefully monitored for several weeks beforehand to reduce the risk of stroke. With dabigatran, there is no way to know if a patient is actually taking the drug as prescribed.
Clinical trials show that alternatives are marginally better than warfarin
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial,8 dabigatran was associated with a significantly lower incidence of intracranial hemorrhage, combined strokes, and systemic embolization than warfarin. The incidence of major bleeds was slightly lower with dabigatran. Although dabigatran performed better, the differences were small and would not require patients to change from warfarin if they are already doing well.
Apixaban and rivaroxaban are other alternatives to warfarin, with different mechanisms of action and metabolism. Although rivaroxaban’s half-life is similar to that of apixaban and dabigatran, it is being marketed as allowing once-daily dosing instead of twice-daily.
Recent randomized controlled clinical trials of the new drugs include:
- The Apixaban Versus Acetylsalicylic Acid (ASA) to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment (AVERROES) trial,9 which compared apixaban and aspirin
- The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial comparing apixaban and warfarin10
- The Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF),11 comparing rivaroxaban and warfarin
- RE-LY,8 comparing dabigatran and warfarin.
In the ARISTOTLE,10 ROCKET AF,11 and RE-LY trials,8 the time that the warfarin patients’ INRs were in the therapeutic range varied from 55% to 68%. This seems low and is a problem when trying to compare therapies, but is probably about as high as one can expect in the real world.
In AVERROES,9 the combined rate of stroke and embolism was higher with aspirin than with apixaban. In the other trials, the rates were slightly better with the new drugs than with warfarin, and the rates of major hemorrhage and hemorrhagic stroke were only slightly higher with warfarin than with the new drugs. Because the differences in benefits and risks are so small, the main advantage of the newer drugs will probably be for patients who have difficulty staying in the therapeutic INR range on warfarin.
RATE CONTROL VS RESTORATION OF SINUS RHYTHM
Evidence is insufficient to determine the risk of very-long-term asymptomatic atrial fibrillation in patients on appropriate anticoagulation. Rate control is an option for asymptomatic patients but provides no change in quality of life and no definitive reduction in the risk of stroke. The main argument for restoring normal sinus rhythm in patients with mild to moderate symptoms is that it improves exercise capacity. The need for anticoagulation persists when patients are converted to sinus rhythm because the risk of recurrent atrial fibrillation remains high.
For patients with symptomatic atrial fibrillation, rate control is sometimes achieved with beta-blockers or calcium channel blockers. Rate control may be augmented with the addition of digoxin, but when used alone digoxin generally does not control the rate of atrial fibrillation. However, in many cases of atrial fibrillation, symptoms are not rate-related, and cardioversion to normal sinus rhythm should be attempted. In such cases, the symptoms may be attributable to a loss of atrial transport function.
Patients with the following risk factors should be admitted to the hospital to start antiarrhythmic drugs:
- Borderline or a long QTc interval at baseline (> 450 msec)
- Treatment with dofetilide (Tikosyn) because of its effects on the QT interval
- Heart failure or poor left-ventricular function
- Sinus node dysfunction
- Significant atrioventricular conduction disease.
Selecting an antiarrhythmic drug
Any of the antiarrhythmic drugs listed in Table 2 can be used for a patient with lone atrial fibrillation (ie, not caused by underlying heart disease). The choice of drug should be determined by whether coronary artery disease or renal failure is present as well. Liver disease or chronic obstructive pulmonary disease also may affect this decision.
Benefits of dronedarone are mixed
In a randomized trial of dronedarone vs placebo in patients with atrial fibrillation, the rate of death and the rate of first hospitalization due to a cardiovascular event at 21 months were significantly lower with dronedarone.12 No difference was found between the two groups in the rate of death from all causes, but fewer people died of cardiovascular causes in the dronedarone group. More patients taking dronedarone developed bradycardia, QT-interval prolongation, nausea, diarrhea, rash, or a higher serum creatinine level. Gastrointestinal side effects are often a problem with dronedarone: 20% to 30% of patients cannot tolerate the drug.
Dronedarone may cause a small rise in creatinine, and although this effect should be monitored, by itself it should not be interpreted as impairment of renal function. In a study in healthy people,13 dronedarone caused a 10% to 15% increase in serum creatinine, but the glomerular filtration rate was unchanged, as were renal plasma flow and anion secretion.
Another trial, in patients with severe heart failure, found that patients taking dronedarone had higher rates of hospitalization and overall mortality, raising serious concern about the safety of this drug in patients with advanced heart failure.14
Singh et al15 pooled the data from two multicenter, randomized trials that compared dronedarone with placebo for maintaining sinus rhythm in patients with atrial fibrillation or flutter. The mean time to the recurrence of atrial fibrillation was 116 days with dronedarone and 53 days with placebo. Other trials also showed longer times to recurrence and lower recurrence rates with dronedarone. Although the differences were statistically significant, they may not be clinically meaningful for patients.
Dronedarone is structurally similar to amiodarone (Cordarone), but the two drugs work differently. A meta-analysis of clinical trials16 found that amiodarone recipients had a lower rate of recurrence of atrial fibrillation than did those receiving dronedarone.
Two safety warnings for dronedarone
In January 2011, the US Food and Drug Administration (FDA) issued an alert about cases of rare but severe liver injury in patients treated with dronedarone, including two cases of acute liver failure leading to liver transplantation.17
The Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS)18 compared dronedarone and placebo in patients with permanent atrial fibrillation. More people died or had serious cardiovascular adverse events in the dronedarone group. The study was stopped early after data monitoring showed that rates of death, stroke, and hospitalization for heart failure were two times higher in patients receiving dronedarone. This prompted the FDA to issue another safety alert in July 2011.
Interestingly, the PALLAS study did not set out to determine whether dronedarone controls atrial fibrillation, as the study patients had long-standing, persistent atrial fibrillation. The study was designed only to determine if the drug reduces the rate of adverse events; it clearly does not, and the study shows that dronedarone should not be used to control the heart rate in patients with persistent atrial fibrillation. Instead, its use is best restricted to patients with paroxysmal atrial fibrillation without significant cardiovascular disease.
ABLATION OF ATRIAL FIBRILLATION
Another way to try to restore sinus rhythm is to destroy or isolate the area that is generating the abnormal beats via a catheter-based procedure.
Radiofrequency ablation is generally tried in patients in whom one or two drugs have failed to control atrial fibrillation. Direct comparisons show that ablation is superior to drug therapy and is effective in about 75% of patients with paroxysmal atrial fibrillation vs 20% to 40% of patients on drug therapy. Ablation plus drug therapy is often more effective than either treatment alone.
Mechanisms of atrial fibrillation and ablation
In many cases, atrial fibrillation is stimulated by vagal and sympathetic inputs to the atrium that enter around the pulmonary veins and trigger electrical activations in the area, generating spiraling, reentering circuits. Focal atrial fibrillation also originates predominantly in the pulmonary veins. Ablation of tissue widely circumscribing the mouth of the pulmonary veins prevents the electrical signal from exiting into the atrium.
In about 11% to 37% of cases, atrial fibrillation originates elsewhere, eg, in the left atrium, in the superior vena cava, or in the vein of Marshall. Techniques have evolved to also ablate these regions.
Anticoagulation therapy is recommended before the procedure, and patients at low risk should continue it for a minimum of 2 months afterward. Patients with a higher CHADS2 score should receive anticoagulation therapy for at least 1 year. The consensus statement by the Heart Rhythm Society19 recommends that patients remain on warfarin or one of the newer anticoagulants if their CHADS2 score is 2 or higher. This is because patients have a significant risk of recurrence of atrial fibrillation after radiofrequency ablation, so if their stroke risk is high they should remain on anticoagulant therapy.
Ablation is usually effective, but it carries rare but serious risks
The efficacy of a single radiofrequency ablation procedure is in the range of 60% to 80% for paroxysmal atrial fibrillation and 40% to 60% for persistent atrial fibrillation. The Second International Ablation Registry20 shows a success rate of about 75% in patients with paroxysmal atrial fibrillation and about 65% in patients with persistent and permanent atrial fibrillation. Registry data are often more favorable because reporting is optional, but these results are consistent with those from experienced medical centers. Rates of suppression of atrial fibrillation are higher in patients who also take antiarrhythmic drugs, making a “hybrid” approach useful when ablation alone fails.
According to a worldwide survey, the risk of serious complications is 4.5%. These include stroke (0.23%), tamponade (1.3%), and pulmonary vein stenosis (< 0.29%). The esophagus lies just behind the right atrium, and burning through and creating a fistula between them occurs in about 0.04% of cases and is almost uniformly fatal.20
A second ablation procedure is sometimes indicated for the recurrence of atrial fibrillation, which is almost always caused by recovery of the pulmonary veins. Bhargava et al21 found that the success rate at Cleveland Clinic for a single procedure for paroxysmal atrial fibrillation was 77%, and that it was 92% after a repeat procedure. For persistent atrial fibrillation, success rates were 76% after the first procedure and 90% after the second. Even for long-standing persistent atrial fibrillation (ie, lasting more than 1 year), 80% success was achieved after two procedures. Patients who are less likely to have a successful ablation procedure are those with long-standing atrial fibrillation and coexisting heart disease, including severe valvular disease, although mitral regurgitation sometimes improves if sinus rhythm can be maintained.
The need for a second procedure
After ablation, patients should be closely monitored for a recurrence of atrial fibrillation. Continuous monitoring with implantable cardiac monitor loop recorders can detect unrecognized episodes of arrhythmia. Long-term follow-up is also required to track outcomes and quality of life.
According to the Heart Rhythm Society Task Force on Catheter and Surgical Ablation of Atrial Fibrillation,19 atrial fibrillation recurs after ablation in about 35% to 60% of patients in the first 3 months, with recurrence rates after 1 year ranging from 5% to 16%. The rate of success is determined by the skill of the surgeon, underlying heart disease, attention to follow-up, and how success is defined.
Freedom from recurrence early on is a good predictor that late recurrence is unlikely. Patients who only have a very early recurrence (within the first 4 weeks) are more likely to have long-term freedom from atrial fibrillation tha those who have recurrences after that time.22
In a study of 831 patients, Hussein et al23 found recurrence rates of 24% between months 3 to 13 following ablation and 9% after 12 months. At 55 months, 79% were free from atrial fibrillation without drugs, 11% were free of atrial fibrillation with medications, and 5% had refractory atrial fibrillation.
Recurrence—whether early or late—was more likely to occur in people with persistent vs paroxysmal atrial fibrillation. Other risk factors for late recurrence included older age and larger left atrial size (which is also a risk factor for recurrence on drug therapy). Although recurrent arrhythmia was most often atrial fibrillation, atrial flutter also occurred frequently (in 27% of patients with late recurrence). Three patients (4% of patients with late recurrence) developed atrial tachycardia.23
In patients with early recurrence, 81% underwent repeat ablation, all of whom had recovery of one or more pulmonary veins. After the second ablation, 21% had recurrence, 65% of whom were controlled by medications.23
Whether a patient should undergo subsequent ablation procedures depends on the severity of symptoms, the likelihood of success (based on an educated guess), and the patient’s willingness to undergo another procedure.
ATRIAL APPENDAGE OCCLUSION DEVICE UNDER INVESTIGATION
New devices are being investigated that occlude the left atrial appendage to try to prevent embolization.
The Watchman device, resembling an umbrella, is implanted via a percutaneous catheter in the left atrial appendage, closing it off to preclude a thrombus from forming in the appendage and embolizing to the body. Clinical trials showed that patients who received a device had a slightly lower risk of stroke than otherwise seen in clinical practice.24 Safety and efficacy are still being determined.
The device cannot be deployed in a patient with an existing thrombus because of the danger of dislodging the thrombus, allowing it to embolize.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the national Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Hylek EM, Skates SJ, Sheehan MA, Singer DE. An analysis of the lowest effective intensity of prophylactic anticoagulation for patients with nonrheumatic atrial fibrillation. N Engl J Med 1996; 335:540–546.
- Hylek EM, Singer DE. Risk factors for intracranial hemorrhage in outpatients taking warfarin. Ann Intern Med 1994; 120:897–902.
- Connolly SJ, Pogue J, Eikelboom J, et al; ACTIVE W Investigators. Benefit of oral anticoagulant over antiplatelet therapy in atrial fibrillation depends on the quality of international normalized ratio control achieved by centers and countries as measured by time in therapeutic range. Circulation 2008; 118:2029–2037.
- Verheugt FWA. Who is ineligible for warfarin in atrial fibrillation? Lancet 2009; 374:510–511.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Harenberg J. New anticoagulants in atrial fibrillation. Semin Thromb Hemost 2009; 35:574–585.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Connolly SJ, Eikelboom J, Joyner C, et al; AVERROES Steering Committee and Investigators. Apixaban in patients with atrial fibrillation. N Engl J Med 2011; 364:806–817.
- Granger CB, Alexander JH, McMurray JJV, et al; for the ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med August 28, 2011; 10.1056/nejmoa1107039.
- Patel MR, Mahaffey KW, Garg J, et al; the ROCKET AF Steering Committee. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Hohnloser SH, Crijns HJ, van Eickels M, et al; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:688–678.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
- Kóber L, Torp-Pederson C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Piccini JP, Hasselblad V, Peterson ED, Washam JB, Califf RM. Comparative efficacy of dronedarone and amiodarone for the maintenance of sinus rhythm in patients with atrial fibrillation. J Am Coll Cardiol 2009; 54:1089–1095.
- US Food and Drug Administration. FDA drug safety communication: severe liver injury associated with the use of dronedarone (marketed as Multaq). http://www.fda.gov/drugs/drugsafety/ucm240011.htm. Accessed July 5, 2012.
- Connolly SJ, Camm AJ, Halperin JL, et al; for the PALLAS Investigators. Dronedarone in high-risk permanent atrial fibrillation. N Engl J Med 2011; 365:2268–2276.
- HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for personnel, policy, procedures and follow-up. Heart Rhythm 2007; 4:1–46.
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3:32–38.
- Bhargava M, Di Biase L, Mohanty P, et al. Impact of type of atrial fibrillation and repeat catheter ablation on long-term freedom from atrial fibrillation: results from a multicenter study. Heart Rhythm 2009; 6:1403–1412.
- Themistoclakis S, Schweikert RA, Sliba WI, et al. Clinical predictors and relationship between early and late atrial tachyarrhythmias after pulmonary vein antrum isolation. Heart Rhythm 2008; 5:679–685.
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4:271–278.
- Holmes DR, Reddy VY, Turi ZG, et al; for the PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the national Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Hylek EM, Skates SJ, Sheehan MA, Singer DE. An analysis of the lowest effective intensity of prophylactic anticoagulation for patients with nonrheumatic atrial fibrillation. N Engl J Med 1996; 335:540–546.
- Hylek EM, Singer DE. Risk factors for intracranial hemorrhage in outpatients taking warfarin. Ann Intern Med 1994; 120:897–902.
- Connolly SJ, Pogue J, Eikelboom J, et al; ACTIVE W Investigators. Benefit of oral anticoagulant over antiplatelet therapy in atrial fibrillation depends on the quality of international normalized ratio control achieved by centers and countries as measured by time in therapeutic range. Circulation 2008; 118:2029–2037.
- Verheugt FWA. Who is ineligible for warfarin in atrial fibrillation? Lancet 2009; 374:510–511.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Harenberg J. New anticoagulants in atrial fibrillation. Semin Thromb Hemost 2009; 35:574–585.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Connolly SJ, Eikelboom J, Joyner C, et al; AVERROES Steering Committee and Investigators. Apixaban in patients with atrial fibrillation. N Engl J Med 2011; 364:806–817.
- Granger CB, Alexander JH, McMurray JJV, et al; for the ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med August 28, 2011; 10.1056/nejmoa1107039.
- Patel MR, Mahaffey KW, Garg J, et al; the ROCKET AF Steering Committee. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Hohnloser SH, Crijns HJ, van Eickels M, et al; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009; 360:688–678.
- Tschuppert Y, Buclin T, Rothuizen LE, et al. Effect of dronedarone on renal function in healthy subjects. Br J Clin Pharmacol 2007; 64:785–791.
- Kóber L, Torp-Pederson C, McMurray JJ, et al; Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008; 358:2678–2687.
- Singh BN, Connolly SJ, Crijns HJ, et al; EURDIS and ADONIS Investigators. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007; 357:987–999.
- Piccini JP, Hasselblad V, Peterson ED, Washam JB, Califf RM. Comparative efficacy of dronedarone and amiodarone for the maintenance of sinus rhythm in patients with atrial fibrillation. J Am Coll Cardiol 2009; 54:1089–1095.
- US Food and Drug Administration. FDA drug safety communication: severe liver injury associated with the use of dronedarone (marketed as Multaq). http://www.fda.gov/drugs/drugsafety/ucm240011.htm. Accessed July 5, 2012.
- Connolly SJ, Camm AJ, Halperin JL, et al; for the PALLAS Investigators. Dronedarone in high-risk permanent atrial fibrillation. N Engl J Med 2011; 365:2268–2276.
- HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for personnel, policy, procedures and follow-up. Heart Rhythm 2007; 4:1–46.
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3:32–38.
- Bhargava M, Di Biase L, Mohanty P, et al. Impact of type of atrial fibrillation and repeat catheter ablation on long-term freedom from atrial fibrillation: results from a multicenter study. Heart Rhythm 2009; 6:1403–1412.
- Themistoclakis S, Schweikert RA, Sliba WI, et al. Clinical predictors and relationship between early and late atrial tachyarrhythmias after pulmonary vein antrum isolation. Heart Rhythm 2008; 5:679–685.
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4:271–278.
- Holmes DR, Reddy VY, Turi ZG, et al; for the PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
KEY POINTS
- Warfarin is as safe as—and more effective than—the combination of aspirin and clopidogrel (Plavix) if the international normalized ratio is in the therapeutic range 65% of the time or more.
- New anticoagulants are promising alternatives to warfarin, but they also pose risks. Patients who are doing well on warfarin need not change.
- Several antiarrhythmic drugs are available to control symptomatic atrial fibrillation. Dronedarone (Multaq) should only be considered for patients with paroxysmal atrial fibrillation without significant cardiovascular disease.
- Ablation is often effective in controlling atrial fibrillation, but recurrence is common. Early recurrence often subsides, but late recurrence often requires a repeat procedure.
Acute community-acquired bacterial meningitis in adults: An evidence-based review
Although the incidence and rates of morbidity and death from acute community-acquired bacterial meningitis have dramatically declined, probably as a result of vaccination and better antimicrobial and adjuvant therapy, the disease still has a high toll. From 10% to 20% of people who contract it in the United States still die of it.1,2
In the United States, meningitis from all causes accounts for about 72,000 hospitalizations and up to $1.2 billion in hospital costs annually.3 However, the incidence of bacterial meningitis has declined from 3 to 5 per 100,000 per year a few decades ago to 1.3 to 2 per 100,000 per year currently.2 In less-developed countries, rates are much higher.
In the early 1900s in the United States, the death rate from bacterial meningitis was 80% to 100%. The use of intrathecal equine meningococcal antiserum during the first decades of the 1900s dramatically reduced the rate of death from meningococcal meningitis. With the advent of antimicrobial drugs in the 1930s and 1940s, the death rate from bacterial meningitis further declined.
The organisms that cause community-acquired bacterial meningitis differ somewhat by geographic region and by age. In a recent paper based on surveillance data, in the United States, from 1998 to 2007, the most common cause of bacterial meningitis among adults was Streptococcus pneumoniae. Among young adults, Neisseria meningitidis is nearly as common as S pneumoniae. The incidence of Listeria infections increases with age in adults.2
The epidemiologic features of bacterial meningitis have changed dramatically over the past decades with the advent of the Haemophilus influenzae vaccine. In 1986, about half the cases of acute bacterial meningitis were caused by H influenzae, but a decade later the incidence of H influenzae meningitis had been reduced by 94%.4
Meningitis is inflammation of the pia and arachnoid (the inner two layers of the meninges). Acute community-acquired meningitis can develop within hours to days and can be viral or bacterial. Viral meningitis usually has a good prognosis, whereas bacterial meningitis is associated with significant rates of morbidity and death, so it is critical to recognize and differentiate them promptly.
PATHOGENESIS
Most cases of community-acquired bacterial meningitis begin with colonization of the nasopharyngeal mucosa. In certain individuals this leads to mucosal invasion and bacteremia. Not all organisms that cause bacteremia are capable of breaching the blood-cerebrospinal fluid barrier to enter the subarachnoid space to cause meningitis. Very few organisms have this capacity, but N meningitidis and S pneumoniae do.5
Some patients are at higher risk of meningitis because of an abnormal communication between the nasopharynx and the subarachnoid space due either to trauma or a congenital anatomic abnormality. The organisms in these instances can directly spread from the nasopharynx to the meninges. Patients without a spleen or with an immunoglobulin deficiency are also more prone to infections from encapsulated organisms such as pneumococci and meningococci. The opsonizing immunoglobulins coat the capsule, helping phagocytes in the spleen to remove them from the bloodstream. A patient presenting with multiple episodes of bacterial meningitis merits evaluation for these conditions.
In contrast, Listeria spp and, rarely, gramnegative bacteria enter the bloodstream through the gastrointestinal tract and then spread to the meninges.
Once in the subarachnoid space, bacteria elicit a profuse inflammatory response, which can be damaging.5 The inflammation in the subarachnoid space can extend along the Virchow-Robin spaces surrounding the blood vessels deep into the brain parenchyma. This perivascular inflammation can cause thrombosis in both the arterial and venous circulation.
Thus, the inflammation can lead to intracranial complications such as cerebral edema, hydrocephalus, and stroke. The complications of bacterial meningitis can be remembered by the acronym HACTIVE: hydrocephalus, abscess, cerebritis and cranial nerve lesions, thrombosis, infarct, ventriculitis and vasculopathy, and extra-axial collection.5,6
MICROBIOLOGY: WHEN TO SUSPECT DIFFERENT ORGANISMS
S pneumoniae: The most common cause in adults
Patients without a spleen and patients with either a primary or secondary immunoglobulin deficiency, including patients with multiple myeloma or human immunodeficiency virus infection, are at a higher risk of infection with this organism.
N meningitidis: More common in young adults
N meningitidis is easily transmitted and is associated with crowding, as in school dormitories and military barracks. People with congenital deficiencies of components of terminal complement are at greater risk for both meningococcal and gonococcal infections. Patients with recurrent episodes of Neisseria infection should be evaluated for complement deficiency.
Meningococcal infection is more commonly associated with a rash. The most common rash of meningococcal meningitis is a very transient, maculopapular rash that appears early in the course of the disease. More pathognomonic is a petechial rash (Figure 1) with thrombocytopenia, which can very rapidly progress to purpura, ecchymosis, and disseminated intravascular coagulation. The petechial rash is evident in 60% of adults and up to 90% of children,7 and it is most likely to appear in dependent areas (such as the back of a patient lying down) and in areas of pressure, such as under the elastic band of underwear or stockings.
Listeria
Listeria infection is usually acquired through contaminated food such as raw vegetables, unpasteurized milk and cheese, and deli meats. From the gastrointestinal tract, it spreads to the bloodstream and then to the meninges.
Listeria is an intracellular pathogen; thus, people at greater risk are those with poor cell-mediated immunity due to immunosuppressant medications such as steroids or tumor necrosis factor inhibitors.
The rate of Listeria meningitis starts to increase with age, especially after age 50, probably due to immune senescence or decreased immunity with age.
Aerobic gram-negative bacilli
Gram-negative enteric bacilli usually cause meningitis after head trauma or neurosurgery and are very uncommon causes of community-acquired meningitis. Disseminated strongyloidiasis, also known as hyperinfection syndrome, should be suspected in any patient with community-acquired meningitis caused by enteric gram-negative bacilli.
Strongyloides stercoralis is a parasitic intestinal roundworm that is found in the tropics, in the subtropics, and in certain parts of the United States and Europe. The adult worm lives in the intestines and lays eggs, which hatch in the mucosa; the larvae are excreted in the stool. A small percentage of larvae penetrate the perianal skin and gut mucosa to cause an autoinfection. People may asymptomatically harbor the parasite for decades, then develop the hyperinfection syndrome when treated with immunosuppressive drugs such as steroids. In the hyperinfection syndrome a significant proportion of the larvae penetrate the gut mucosa to enter the bloodstream and travel throughout the body, including into the brain, carrying gram-negative bacteria with them.
The mortality rate of untreated hyperinfection syndrome can sometimes reach 100%.8 Thus, it is important to identify and treat the hyperinfection syndrome in the context of gram-negative bacillary meningitis.
SUSPECTED MENINGITIS: CLINICAL SCENARIO
A 36-year-old man presents to the emergency department with high fever, headache, and lethargy that developed over the past 24 hours. His temperature is 104°F (40°C), pulse 120 beats/min, respiratory rate 30/min, and blood pressure 130/70 mm Hg. He is oriented only to person and has nuchal rigidity. His white blood cell count is 30 × 109/L, with 20% bands.
The clinical questions that arise with such a patient are:
- Does the patient have bacterial or viral meningitis?
- Can we reliably rule out meningitis based on a history and physical examination?
- Is a lumbar puncture for cerebrospinal fluid (CSF) analysis needed? How should these studies be interpreted?
- Should computed tomography of the head be done before lumbar puncture?
- Which antimicrobial drugs should be started empirically at the outset?
- What is the role of steroids in treatment?
CLINICAL SIGNS AND SYMPTOMS
The classic triad of meningitis is fever, neck stiffness, and altered mental status. Other signs and symptoms that have been described are photophobia, headache, nausea, vomiting, focal neurologic symptoms, altered mental status, the Kernig sign (inability to allow full knee extension when the hip is flexed to a 90° angle), and the Brudzinski sign (spontaneous flexion of the hips during attempted passive flexion of the neck).
Can meningitis be ruled out if the patient does not have this classic presentation?
Unfortunately, only a few high-quality studies of the diagnostic accuracy of signs and symptoms of bacterial meningitis have been done. Fourteen retrospective studies examined this issue, but they were heterogeneous with respect to patient age, immunosuppression status, and clinical presentation, as well as to how meningitis was diagnosed (via culture or cerebrospinal fluid analysis), making the results difficult to interpret.9 Retrospective studies are more prone to bias, as they lack a control group, and examiner bias is more likely. Based on retrospective data, the combination of fever, neck stiffness, and altered mental status has a sensitivity of only 0.46.9
Two prospective studies examined symptoms and signs. Thomas et al10 evaluated 297 patients with “clinically suspected meningitis.” Unfortunately, in this study the physical examination was not standardized. In a study by Uchihara and Tsukagoshi,11 the measurement was more reliable, as they used a single examiner to evaluate patients presenting with fever and headache, but only 54 patients were studied.
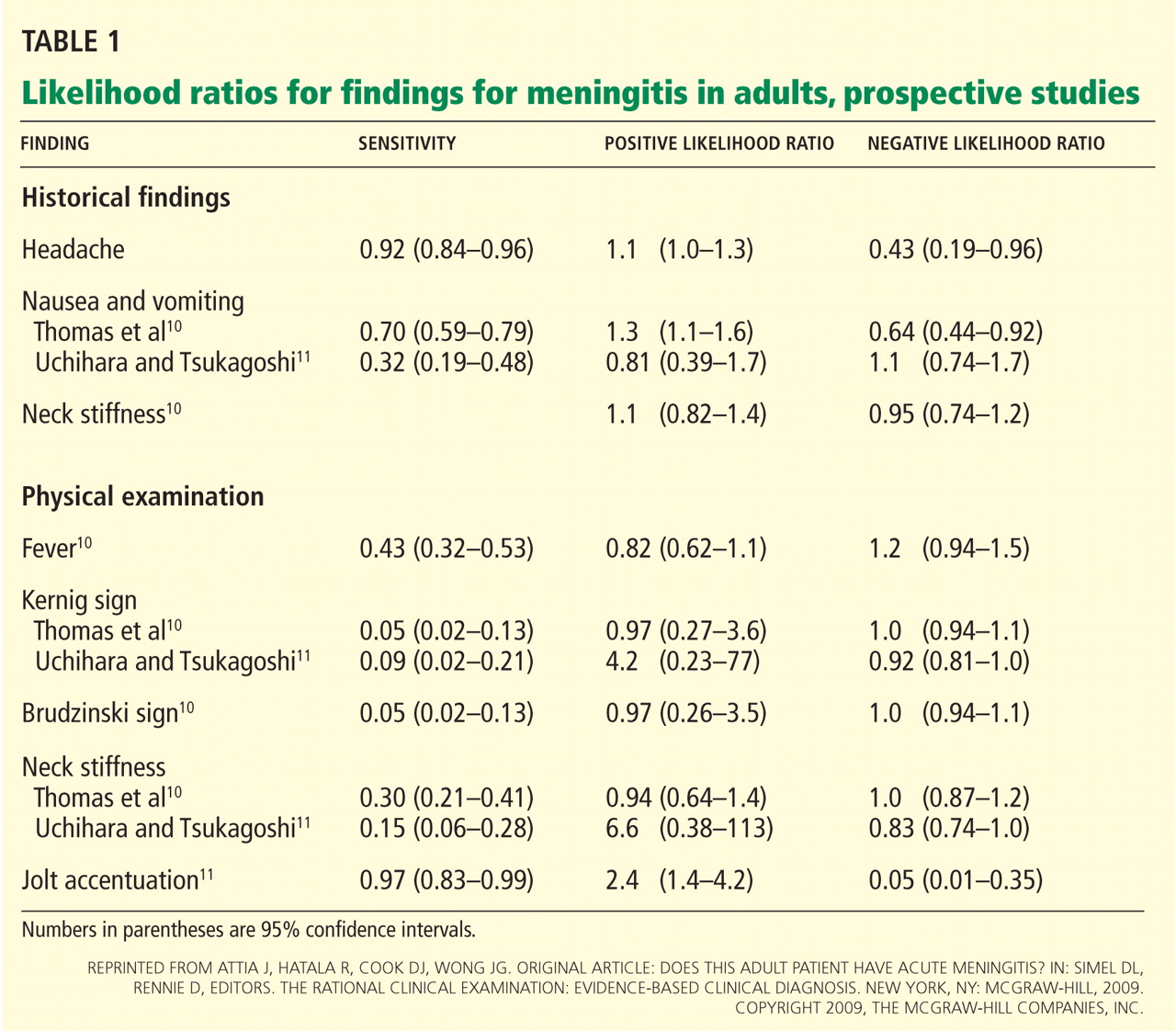
Based on these prospective studies, the presence of nausea and vomiting, headache, or neck stiffness does not reliably rule in meningitis (Table 1).9 Similarly, the absence of these does not rule it out. The 95% confidence intervals (CIs) of the positive and negative likelihood ratios include the value 1. (A simple interpretation of that would be that the likelihood of finding these features is the same in patients with meningitis when compared with those without meningitis.9)
For the physical examination, the presence or absence of fever, the Kernig sign, or the Brudzinski sign were also inconclusive. The CIs of the positive and negative likelihood ratios, like those of the symptoms, included the value 1. Only one test done on physical examination looked promising in having diagnostic utility to rule out meningitis: the jolt accentuation test (performed by asking a patient with a headache to quickly move his or her head twice horizontally; the result is positive if the headache worsens). If the result is negative, meningitis is unlikely (negative likelihood ratio 0.05, 95% CI 0.01–0.35).9 However, a positive test is not useful in making the diagnosis. A caveat is that this is based on a single study.
In summary, the history and physical examination are not sufficient to determine whether a patient has meningitis. If a patient is suspected of having meningitis, a lumbar puncture is needed.
WORKUP AND DIAGNOSTIC TESTS
Which tests are needed?
Blood cultures should be drawn before antimicrobial treatment is started.12–14 Although positive only 19% to 70% of the time, they can help identify the pathogen.15–17
Lumbar puncture with CSF study is essential to make the diagnosis and to identify the organism and its susceptibility to various antibiotics. If lumbar puncture can be performed immediately, it should be done before starting antibiotics, to maximize the yield of cultures. Pediatric studies show that after starting antibiotics, complete sterilization of the cerebrospinal fluid can occur within 2 hours for N meningitides and within 4 hours for S pneumoniae.14 However, starting antimicrobials should not be delayed if a lumbar puncture cannot be done expeditiously.
Is computed tomography of the brain necessary before a lumbar puncture?
The rationale behind performing CT before lumbar puncture is to determine if the patient has elevated intracranial pressure, which would increase the risk of brain herniation due to lowering of the lumbar CSF pressure during lumbar puncture. For ethical and practical reasons, it would be difficult to evaluate this in a randomized clinical trial.
Hasbun et al18 performed a study to evaluate if any features on clinical presentation can predict abnormal findings on CT of the head suggestive of elevated intracranial pressure and thus the risk of herniation. The study included 301 adults with suspected meningitis. It found that abnormal findings on CT were unlikely if all of the following features were absent at baseline:
- Immunocompromised state
- History of central nervous system disease (mass lesion, stroke, or a focal infection)
- New onset of seizure (≤ 1 week from presentation)
- Specific abnormal neurologic findings (eg, an abnormal level of consciousness, inability to answer two consecutive questions correctly or to follow two consecutive commands, gaze palsy, abnormal visual fields, facial palsy, arm drift, leg drift, abnormal language).
Absence of these baseline features made it unlikely that CT would be abnormal (negative likelihood ratio 0.1, 95% CI 0.03–0.31).
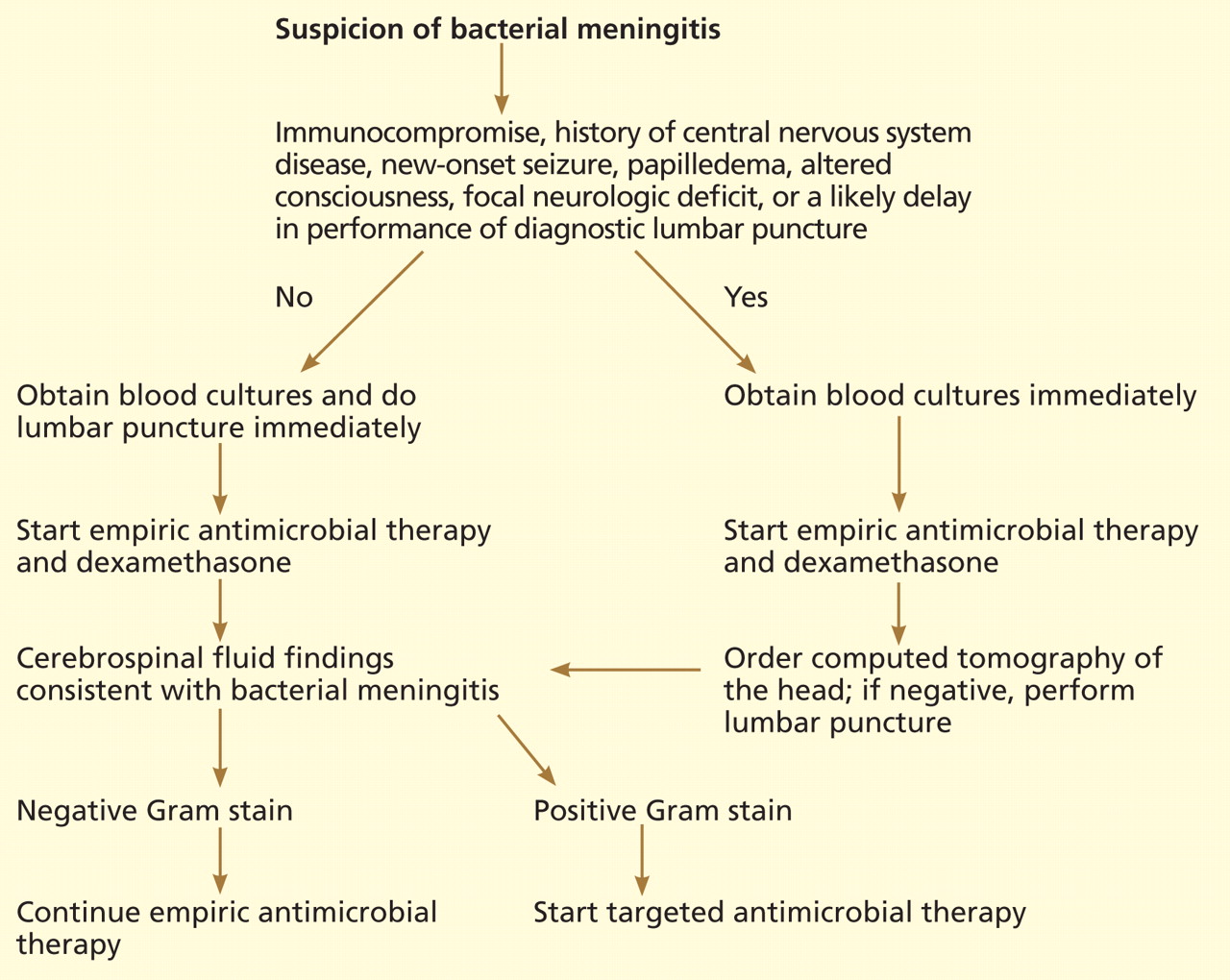
According to the guidelines from the Infectious Diseases Society of America (IDSA),19 if none of those features is present, blood cultures and a lumbar puncture should be done immediately, followed by empiric antimicrobial therapy. If any of the features is present, blood cultures should be obtained first, then empiric antimicrobial therapy started, followed by CT of the brain to look for contraindications to a lumbar puncture (Figure 2).
What can lumbar puncture tell us?
Results of lumbar puncture studies can help determine whether meningitis is present and, if so, whether the cause is likely bacterial or viral.20
The opening pressure is elevated (usually > 180 mm H2O) in acute bacterial meningitis. The CSF white blood cell count is usually more than 1.0 × 109/L, consisting predominantly of neutrophils, in acute bacterial meningitis. In viral meningitis, it is usually less than 0.1 × 109/L, mostly lymphocytes.
Protein shows a mild to marked elevation in bacterial meningitis but is normal to elevated in viral meningitis.
The CSF glucose level is lower in bacterial meningitis than in viral meningitis.
The ratio of CSF glucose to blood glucose. Because the glucose levels in the CSF and the blood equilibrate, the ratio of CSF glucose to serum glucose has better diagnostic accuracy than the CSF glucose level alone. The equilibration takes place within a few hours, so the serum glucose level should be ordered at the same time lumbar puncture is done. The CSF glucose-blood glucose ratio is a better predictor of bacterial meningitis than the CSF white blood cell count. Bacterial meningitis is likely if the ratio is lower than 0.4.
Lactate levels are not usually measured, but a lactate level greater than 31.5 mg/dL (3.5 mmol/L) is predictive of meningitis, and a lower level makes the diagnosis unlikely.
The diagnostic accuracies (likelihood ratios) of the CSF tests were analyzed by Straus et al.21 The positive likelihood ratios for the CSF white blood cell count and for the CSF glucose-blood glucose ratio are greater than 10, but these tests have negative likelihood ratios of more than 0.1. (It is generally thought that a test with a positive likelihood ratio of more than 10 is considered good for ruling in a diagnosis, whereas one with a negative likelihood ratio of less than 0.1 is good for ruling out a diagnosis.) Thus, these tests are good to rule in bacterial meningitis, but not as good to rule it out. There are some data to show that CSF lactate and procalcitonin might be more sensitive in ruling out bacterial meningitis, but more studies are needed.22
Gram stain of the cerebrospinal fluid can be done quickly. If no bacteria are seen, the information is not helpful in ruling out bacterial meningitis (negative likelihood ratio 0.14, 95% CI 0.08–0.27). If it is positive, it is almost 100% specific for meningitis due to the organism seen (positive likelihood ratio 735, 95% CI 230–2,295).21
MANAGEMENT
Empiric antimicrobial therapy must be started as soon as feasible
Most studies of the timing of antimicrobial drugs were retrospective and included a very heterogeneous population. They were thus more prone to bias and confounding.23,24 Proulx et al,23 in a retrospective study, found that if antibiotics were given within 6 hours of the time the patient presented to the emergency department, the case fatality rate was only 5% to 6%. If treatment started 6 to 8 hours after presentation, the death rate was 45%, and if it started from 8 to 10 hours after presentation, the death rate was 75%. Most physicians would agree that starting antimicrobials early would be beneficial.
CSF concentrations of most antimicrobial drugs are considerably less than in the serum due to poor penetration of the blood-CSF barrier. Thus, the dose for treating meningitis is usually higher than the regular dose. For example, for the treatment of pneumococcal pneumonia, ceftriaxone (Rocephin) is used at a dose of 1 g every 24 hours, but for pneumococcal meningitis the dose is 2 g every 12 hours.
Empiric treatment of community-acquired bacterial meningitis in immunocompetent adults up to 50 years of age consists of a third-generation cephalosporin such as cefotaxime (Claforan) 2 g intravenously every 4 hours or ceftriaxone 2 g intravenously every 12 hours, which covers most S pneumoniae and N meningitides strains.19 The IDSA guidelines recommend adding vancomycin (Vancocin) empirically in suspected S pneumoniae meningitis due to concerns about drug-resistant pneumococcal strains.19 For vancomycin, 45 to 60 mg/kg intravenously per day divided into every-6-hour or every-8-hour doses would achieve better CSF concentrations.25
In patients over age 50 or those with a cell-mediated immunodeficiency, empiric therapy should also include ampicillin 2 g intravenously every 4 hours to cover Listeria.
It is important to tailor therapy to the results of Gram stain, culture, and susceptibility as they become available.
Role of corticosteroids
Glucocorticoids, especially dexamethasone (Decadron), have been well studied as adjunctive therapies in bacterial meningitis. The rationale behind their use is that the profuse inflammatory response to the bacterial components in the CSF by itself has deleterious effects, and steroids can reduce that.
In 2004, a Cochrane meta-analysis26 of five randomized clinical trials, including 623 adults with bacterial meningitis (234 with pneumococcal meningitis and 232 with meningococcal meningitis), found a significant reduction in the death rate for patients who received steroids: the death rate was 12% in patients who received steroids vs 22% in those who did not (odds ratio 0.6; 95% CI 0.40–0.81). This led to an IDSA practice guideline recommendation that in adults with suspected or proven pneumococcal meningitis, dexamethasone would be beneficial.19
But since then, many more studies have emerged from Europe, South America, Malawi, and Vietnam, and another Cochrane metaanalysis27 incorporated the new studies. Twenty-four studies involving 4,041 participants were included. Similar numbers of participants died in the corticosteroid and placebo groups (18% vs 20%; risk ratio [RR] 0.92, 95% CI 0.82–1.04, P = .18). A trend towards a lower mortality rate was noticed in adults receiving corticosteroids (RR 0.74, 95% CI 0.53–1.05, P = .09). In adults, corticosteroids were associated with lower rates of hearing loss (RR 0.74, 95% CI 0.56–0.98), and there was a trend towards fewer neurologic sequelae (RR 0.72, 95% CI 0.51–1.01). The benefits were shown in studies in adults in high-income countries, but the studies from low-income countries showed neither harm nor benefit. Based on these findings, the authors recommended the use of steroids in high-income countries, though the strength of the evidence was not optimal. The recommended steroid was dexamethasone 0.15 mg/kg intravenously every 6 hours for 4 days.
- Swartz MN. Bacterial meningitis—a view of the past 90 years. N Engl J Med 2004; 351:1826–1828.
- Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med 2011; 364:2010–2025.
- Holmquist L, Russo CA, Elixhauser A. Meningitis-related hospitalizations in the United States, 2006. Statistical Brief #57. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville, MD, 2008. www.hcup-us.ahrq.gov/reports/statbriefs/sb57.jsp. Accessed May 4, 2012.
- Schuchat A, Robinson K, Wenger JD, et al. Bacterial meningitis in the United States in 1995. N Engl J Med 1997; 337:970–976.
- Koedel U, Scheld WM, Pfister H-W. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis 2002; 2:721–736.
- Hughes DC, Raghavan A, Mordekar SR, Griffiths PD, Connolly DJ. Role of imaging in the diagnosis of acute bacterial meningitis and its complications. Postgrad Med J 2010; 86:478–485.
- Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev 2010; 23:467–492.
- Maguire JH. Intestinal nematodes (roundworms). In:Mandell G, Bennett J, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia: Elsevier, 2009:3577–3586.
- Attia J, Hatala R, Cook DJ, Wong JG. Original article: does this adult patient have acute meningitis?In:Simel DL, Rennie D, editors. The Rational Clinical Examinatino: Evidence-Based Clinical Diagnosis. New York, NY: McGraw-Hill; 2009.
- Thomas KE, Hasbun R, Jekel J, Quagliarello VJ. The diagnostic accuracy of Kernig’s sign, Brudzinski’s sign, and nuchal rigidity in adults with suspected meningitis. Clin Infect Dis 2002; 35:46–52.
- Uchihara T, Tsukagoshi H. Jolt accenulation of headache: the most sensitive sign of CSF pleocytosis. Headache 1991; 31:167–171.
- Geiseler PJ, Nelson KE, Levin S, Reddi KT, Moses VK. Community-acquired purulent meningitis: a review of 1,316 cases during the antibiotic era, 1954–1976. Rev Infect Dis 1980; 2:725–745.
- Talan DA, Hoffman JR, Yoshikawa TT, Overturf GD. Role of empiric parenteral antibiotics prior to lumbar puncture in suspected bacterial meningitis: state of the art. Rev Infect Dis 1988; 10:365–376.
- Kanegaye JT, Soliemanzadeh P, Bradley JS. Lumbar puncture in pediatric bacterial meningitis: defining the time interval for recovery of cerebrospinal fluid pathogens after parenteral antibiotic pretreatment. Pediatrics 2001; 108:1169–1174.
- Sigurdardóttir B, Björnsson OM, Jónsdóttir KE, Erlendsdóttir H, Gudmundsson S. Acute bacterial meningitis in adults. A 20-year overview. Arch Intern Med 1997; 157:425–430.
- Aronin SI, Peduzzi P, Quagliarello VJ. Community-acquired bacterial meningitis: risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med 1998; 129:862–869.
- Andersen J, Backer V, Voldsgaard P, Skinhój P, Wandall JH. Acute meningococcal meningitis: analysis of features of the disease according to the age of 255 patients. Copenhagen Meningitis Study Group. J Infect 1997; 34:227–235.
- Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med 2001; 345:1727–1733.
- Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004; 39:1267–1284.
- Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician 2003; 68:1103–1108.
- Straus SE, Thorpe KE, Holroyd-Leduc J. How do I perform a lumbar puncture and analyze the results to diagnose bacterial meningitis? JAMA 2006; 296:2010–2022.
- Viallon A, Desseigne N, Marjollet O, et al. Meningitis in adult patients with a negative direct cerebrospinal fluid examination: value of cytochemical markers for differential diagnosis. Crit Care 2011; 15:R136.
- Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM 2005; 98:291–298.
- Radetsky M. Duration of symptoms and outcome in bacterial meningitis: an analysis of causation and the implications of a delay in diagnosis. Pediatr Infect Dis J 1992; 11:694–698.
- Ricard JD, Wolff M, Lacherade JC, et al. Levels of vancomycin in cerebrospinal fluid of adult patients receiving adjunctive corticosteroids to treat pneumococcal meningitis: a prospective multicenter observational study. Clin Infect Dis 2007; 44:250–255.
- van de Beek D, de Gans J, McIntyre P, Prasad K. Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis 2004; 4:139–143.
- van de Beek D, Farrar JJ, de Gans J, et al. Adjunctive dexamethasone in bacterial meningitis: a meta-analysis of individual patient data. Lancet Neurol 2010; 9:254–263.
Although the incidence and rates of morbidity and death from acute community-acquired bacterial meningitis have dramatically declined, probably as a result of vaccination and better antimicrobial and adjuvant therapy, the disease still has a high toll. From 10% to 20% of people who contract it in the United States still die of it.1,2
In the United States, meningitis from all causes accounts for about 72,000 hospitalizations and up to $1.2 billion in hospital costs annually.3 However, the incidence of bacterial meningitis has declined from 3 to 5 per 100,000 per year a few decades ago to 1.3 to 2 per 100,000 per year currently.2 In less-developed countries, rates are much higher.
In the early 1900s in the United States, the death rate from bacterial meningitis was 80% to 100%. The use of intrathecal equine meningococcal antiserum during the first decades of the 1900s dramatically reduced the rate of death from meningococcal meningitis. With the advent of antimicrobial drugs in the 1930s and 1940s, the death rate from bacterial meningitis further declined.
The organisms that cause community-acquired bacterial meningitis differ somewhat by geographic region and by age. In a recent paper based on surveillance data, in the United States, from 1998 to 2007, the most common cause of bacterial meningitis among adults was Streptococcus pneumoniae. Among young adults, Neisseria meningitidis is nearly as common as S pneumoniae. The incidence of Listeria infections increases with age in adults.2
The epidemiologic features of bacterial meningitis have changed dramatically over the past decades with the advent of the Haemophilus influenzae vaccine. In 1986, about half the cases of acute bacterial meningitis were caused by H influenzae, but a decade later the incidence of H influenzae meningitis had been reduced by 94%.4
Meningitis is inflammation of the pia and arachnoid (the inner two layers of the meninges). Acute community-acquired meningitis can develop within hours to days and can be viral or bacterial. Viral meningitis usually has a good prognosis, whereas bacterial meningitis is associated with significant rates of morbidity and death, so it is critical to recognize and differentiate them promptly.
PATHOGENESIS
Most cases of community-acquired bacterial meningitis begin with colonization of the nasopharyngeal mucosa. In certain individuals this leads to mucosal invasion and bacteremia. Not all organisms that cause bacteremia are capable of breaching the blood-cerebrospinal fluid barrier to enter the subarachnoid space to cause meningitis. Very few organisms have this capacity, but N meningitidis and S pneumoniae do.5
Some patients are at higher risk of meningitis because of an abnormal communication between the nasopharynx and the subarachnoid space due either to trauma or a congenital anatomic abnormality. The organisms in these instances can directly spread from the nasopharynx to the meninges. Patients without a spleen or with an immunoglobulin deficiency are also more prone to infections from encapsulated organisms such as pneumococci and meningococci. The opsonizing immunoglobulins coat the capsule, helping phagocytes in the spleen to remove them from the bloodstream. A patient presenting with multiple episodes of bacterial meningitis merits evaluation for these conditions.
In contrast, Listeria spp and, rarely, gramnegative bacteria enter the bloodstream through the gastrointestinal tract and then spread to the meninges.
Once in the subarachnoid space, bacteria elicit a profuse inflammatory response, which can be damaging.5 The inflammation in the subarachnoid space can extend along the Virchow-Robin spaces surrounding the blood vessels deep into the brain parenchyma. This perivascular inflammation can cause thrombosis in both the arterial and venous circulation.
Thus, the inflammation can lead to intracranial complications such as cerebral edema, hydrocephalus, and stroke. The complications of bacterial meningitis can be remembered by the acronym HACTIVE: hydrocephalus, abscess, cerebritis and cranial nerve lesions, thrombosis, infarct, ventriculitis and vasculopathy, and extra-axial collection.5,6
MICROBIOLOGY: WHEN TO SUSPECT DIFFERENT ORGANISMS
S pneumoniae: The most common cause in adults
Patients without a spleen and patients with either a primary or secondary immunoglobulin deficiency, including patients with multiple myeloma or human immunodeficiency virus infection, are at a higher risk of infection with this organism.
N meningitidis: More common in young adults
N meningitidis is easily transmitted and is associated with crowding, as in school dormitories and military barracks. People with congenital deficiencies of components of terminal complement are at greater risk for both meningococcal and gonococcal infections. Patients with recurrent episodes of Neisseria infection should be evaluated for complement deficiency.
Meningococcal infection is more commonly associated with a rash. The most common rash of meningococcal meningitis is a very transient, maculopapular rash that appears early in the course of the disease. More pathognomonic is a petechial rash (Figure 1) with thrombocytopenia, which can very rapidly progress to purpura, ecchymosis, and disseminated intravascular coagulation. The petechial rash is evident in 60% of adults and up to 90% of children,7 and it is most likely to appear in dependent areas (such as the back of a patient lying down) and in areas of pressure, such as under the elastic band of underwear or stockings.
Listeria
Listeria infection is usually acquired through contaminated food such as raw vegetables, unpasteurized milk and cheese, and deli meats. From the gastrointestinal tract, it spreads to the bloodstream and then to the meninges.
Listeria is an intracellular pathogen; thus, people at greater risk are those with poor cell-mediated immunity due to immunosuppressant medications such as steroids or tumor necrosis factor inhibitors.
The rate of Listeria meningitis starts to increase with age, especially after age 50, probably due to immune senescence or decreased immunity with age.
Aerobic gram-negative bacilli
Gram-negative enteric bacilli usually cause meningitis after head trauma or neurosurgery and are very uncommon causes of community-acquired meningitis. Disseminated strongyloidiasis, also known as hyperinfection syndrome, should be suspected in any patient with community-acquired meningitis caused by enteric gram-negative bacilli.
Strongyloides stercoralis is a parasitic intestinal roundworm that is found in the tropics, in the subtropics, and in certain parts of the United States and Europe. The adult worm lives in the intestines and lays eggs, which hatch in the mucosa; the larvae are excreted in the stool. A small percentage of larvae penetrate the perianal skin and gut mucosa to cause an autoinfection. People may asymptomatically harbor the parasite for decades, then develop the hyperinfection syndrome when treated with immunosuppressive drugs such as steroids. In the hyperinfection syndrome a significant proportion of the larvae penetrate the gut mucosa to enter the bloodstream and travel throughout the body, including into the brain, carrying gram-negative bacteria with them.
The mortality rate of untreated hyperinfection syndrome can sometimes reach 100%.8 Thus, it is important to identify and treat the hyperinfection syndrome in the context of gram-negative bacillary meningitis.
SUSPECTED MENINGITIS: CLINICAL SCENARIO
A 36-year-old man presents to the emergency department with high fever, headache, and lethargy that developed over the past 24 hours. His temperature is 104°F (40°C), pulse 120 beats/min, respiratory rate 30/min, and blood pressure 130/70 mm Hg. He is oriented only to person and has nuchal rigidity. His white blood cell count is 30 × 109/L, with 20% bands.
The clinical questions that arise with such a patient are:
- Does the patient have bacterial or viral meningitis?
- Can we reliably rule out meningitis based on a history and physical examination?
- Is a lumbar puncture for cerebrospinal fluid (CSF) analysis needed? How should these studies be interpreted?
- Should computed tomography of the head be done before lumbar puncture?
- Which antimicrobial drugs should be started empirically at the outset?
- What is the role of steroids in treatment?
CLINICAL SIGNS AND SYMPTOMS
The classic triad of meningitis is fever, neck stiffness, and altered mental status. Other signs and symptoms that have been described are photophobia, headache, nausea, vomiting, focal neurologic symptoms, altered mental status, the Kernig sign (inability to allow full knee extension when the hip is flexed to a 90° angle), and the Brudzinski sign (spontaneous flexion of the hips during attempted passive flexion of the neck).
Can meningitis be ruled out if the patient does not have this classic presentation?
Unfortunately, only a few high-quality studies of the diagnostic accuracy of signs and symptoms of bacterial meningitis have been done. Fourteen retrospective studies examined this issue, but they were heterogeneous with respect to patient age, immunosuppression status, and clinical presentation, as well as to how meningitis was diagnosed (via culture or cerebrospinal fluid analysis), making the results difficult to interpret.9 Retrospective studies are more prone to bias, as they lack a control group, and examiner bias is more likely. Based on retrospective data, the combination of fever, neck stiffness, and altered mental status has a sensitivity of only 0.46.9
Two prospective studies examined symptoms and signs. Thomas et al10 evaluated 297 patients with “clinically suspected meningitis.” Unfortunately, in this study the physical examination was not standardized. In a study by Uchihara and Tsukagoshi,11 the measurement was more reliable, as they used a single examiner to evaluate patients presenting with fever and headache, but only 54 patients were studied.
Based on these prospective studies, the presence of nausea and vomiting, headache, or neck stiffness does not reliably rule in meningitis (Table 1).9 Similarly, the absence of these does not rule it out. The 95% confidence intervals (CIs) of the positive and negative likelihood ratios include the value 1. (A simple interpretation of that would be that the likelihood of finding these features is the same in patients with meningitis when compared with those without meningitis.9)
For the physical examination, the presence or absence of fever, the Kernig sign, or the Brudzinski sign were also inconclusive. The CIs of the positive and negative likelihood ratios, like those of the symptoms, included the value 1. Only one test done on physical examination looked promising in having diagnostic utility to rule out meningitis: the jolt accentuation test (performed by asking a patient with a headache to quickly move his or her head twice horizontally; the result is positive if the headache worsens). If the result is negative, meningitis is unlikely (negative likelihood ratio 0.05, 95% CI 0.01–0.35).9 However, a positive test is not useful in making the diagnosis. A caveat is that this is based on a single study.
In summary, the history and physical examination are not sufficient to determine whether a patient has meningitis. If a patient is suspected of having meningitis, a lumbar puncture is needed.
WORKUP AND DIAGNOSTIC TESTS
Which tests are needed?
Blood cultures should be drawn before antimicrobial treatment is started.12–14 Although positive only 19% to 70% of the time, they can help identify the pathogen.15–17
Lumbar puncture with CSF study is essential to make the diagnosis and to identify the organism and its susceptibility to various antibiotics. If lumbar puncture can be performed immediately, it should be done before starting antibiotics, to maximize the yield of cultures. Pediatric studies show that after starting antibiotics, complete sterilization of the cerebrospinal fluid can occur within 2 hours for N meningitides and within 4 hours for S pneumoniae.14 However, starting antimicrobials should not be delayed if a lumbar puncture cannot be done expeditiously.
Is computed tomography of the brain necessary before a lumbar puncture?
The rationale behind performing CT before lumbar puncture is to determine if the patient has elevated intracranial pressure, which would increase the risk of brain herniation due to lowering of the lumbar CSF pressure during lumbar puncture. For ethical and practical reasons, it would be difficult to evaluate this in a randomized clinical trial.
Hasbun et al18 performed a study to evaluate if any features on clinical presentation can predict abnormal findings on CT of the head suggestive of elevated intracranial pressure and thus the risk of herniation. The study included 301 adults with suspected meningitis. It found that abnormal findings on CT were unlikely if all of the following features were absent at baseline:
- Immunocompromised state
- History of central nervous system disease (mass lesion, stroke, or a focal infection)
- New onset of seizure (≤ 1 week from presentation)
- Specific abnormal neurologic findings (eg, an abnormal level of consciousness, inability to answer two consecutive questions correctly or to follow two consecutive commands, gaze palsy, abnormal visual fields, facial palsy, arm drift, leg drift, abnormal language).
Absence of these baseline features made it unlikely that CT would be abnormal (negative likelihood ratio 0.1, 95% CI 0.03–0.31).
According to the guidelines from the Infectious Diseases Society of America (IDSA),19 if none of those features is present, blood cultures and a lumbar puncture should be done immediately, followed by empiric antimicrobial therapy. If any of the features is present, blood cultures should be obtained first, then empiric antimicrobial therapy started, followed by CT of the brain to look for contraindications to a lumbar puncture (Figure 2).
What can lumbar puncture tell us?
Results of lumbar puncture studies can help determine whether meningitis is present and, if so, whether the cause is likely bacterial or viral.20
The opening pressure is elevated (usually > 180 mm H2O) in acute bacterial meningitis. The CSF white blood cell count is usually more than 1.0 × 109/L, consisting predominantly of neutrophils, in acute bacterial meningitis. In viral meningitis, it is usually less than 0.1 × 109/L, mostly lymphocytes.
Protein shows a mild to marked elevation in bacterial meningitis but is normal to elevated in viral meningitis.
The CSF glucose level is lower in bacterial meningitis than in viral meningitis.
The ratio of CSF glucose to blood glucose. Because the glucose levels in the CSF and the blood equilibrate, the ratio of CSF glucose to serum glucose has better diagnostic accuracy than the CSF glucose level alone. The equilibration takes place within a few hours, so the serum glucose level should be ordered at the same time lumbar puncture is done. The CSF glucose-blood glucose ratio is a better predictor of bacterial meningitis than the CSF white blood cell count. Bacterial meningitis is likely if the ratio is lower than 0.4.
Lactate levels are not usually measured, but a lactate level greater than 31.5 mg/dL (3.5 mmol/L) is predictive of meningitis, and a lower level makes the diagnosis unlikely.
The diagnostic accuracies (likelihood ratios) of the CSF tests were analyzed by Straus et al.21 The positive likelihood ratios for the CSF white blood cell count and for the CSF glucose-blood glucose ratio are greater than 10, but these tests have negative likelihood ratios of more than 0.1. (It is generally thought that a test with a positive likelihood ratio of more than 10 is considered good for ruling in a diagnosis, whereas one with a negative likelihood ratio of less than 0.1 is good for ruling out a diagnosis.) Thus, these tests are good to rule in bacterial meningitis, but not as good to rule it out. There are some data to show that CSF lactate and procalcitonin might be more sensitive in ruling out bacterial meningitis, but more studies are needed.22
Gram stain of the cerebrospinal fluid can be done quickly. If no bacteria are seen, the information is not helpful in ruling out bacterial meningitis (negative likelihood ratio 0.14, 95% CI 0.08–0.27). If it is positive, it is almost 100% specific for meningitis due to the organism seen (positive likelihood ratio 735, 95% CI 230–2,295).21
MANAGEMENT
Empiric antimicrobial therapy must be started as soon as feasible
Most studies of the timing of antimicrobial drugs were retrospective and included a very heterogeneous population. They were thus more prone to bias and confounding.23,24 Proulx et al,23 in a retrospective study, found that if antibiotics were given within 6 hours of the time the patient presented to the emergency department, the case fatality rate was only 5% to 6%. If treatment started 6 to 8 hours after presentation, the death rate was 45%, and if it started from 8 to 10 hours after presentation, the death rate was 75%. Most physicians would agree that starting antimicrobials early would be beneficial.
CSF concentrations of most antimicrobial drugs are considerably less than in the serum due to poor penetration of the blood-CSF barrier. Thus, the dose for treating meningitis is usually higher than the regular dose. For example, for the treatment of pneumococcal pneumonia, ceftriaxone (Rocephin) is used at a dose of 1 g every 24 hours, but for pneumococcal meningitis the dose is 2 g every 12 hours.
Empiric treatment of community-acquired bacterial meningitis in immunocompetent adults up to 50 years of age consists of a third-generation cephalosporin such as cefotaxime (Claforan) 2 g intravenously every 4 hours or ceftriaxone 2 g intravenously every 12 hours, which covers most S pneumoniae and N meningitides strains.19 The IDSA guidelines recommend adding vancomycin (Vancocin) empirically in suspected S pneumoniae meningitis due to concerns about drug-resistant pneumococcal strains.19 For vancomycin, 45 to 60 mg/kg intravenously per day divided into every-6-hour or every-8-hour doses would achieve better CSF concentrations.25
In patients over age 50 or those with a cell-mediated immunodeficiency, empiric therapy should also include ampicillin 2 g intravenously every 4 hours to cover Listeria.
It is important to tailor therapy to the results of Gram stain, culture, and susceptibility as they become available.
Role of corticosteroids
Glucocorticoids, especially dexamethasone (Decadron), have been well studied as adjunctive therapies in bacterial meningitis. The rationale behind their use is that the profuse inflammatory response to the bacterial components in the CSF by itself has deleterious effects, and steroids can reduce that.
In 2004, a Cochrane meta-analysis26 of five randomized clinical trials, including 623 adults with bacterial meningitis (234 with pneumococcal meningitis and 232 with meningococcal meningitis), found a significant reduction in the death rate for patients who received steroids: the death rate was 12% in patients who received steroids vs 22% in those who did not (odds ratio 0.6; 95% CI 0.40–0.81). This led to an IDSA practice guideline recommendation that in adults with suspected or proven pneumococcal meningitis, dexamethasone would be beneficial.19
But since then, many more studies have emerged from Europe, South America, Malawi, and Vietnam, and another Cochrane metaanalysis27 incorporated the new studies. Twenty-four studies involving 4,041 participants were included. Similar numbers of participants died in the corticosteroid and placebo groups (18% vs 20%; risk ratio [RR] 0.92, 95% CI 0.82–1.04, P = .18). A trend towards a lower mortality rate was noticed in adults receiving corticosteroids (RR 0.74, 95% CI 0.53–1.05, P = .09). In adults, corticosteroids were associated with lower rates of hearing loss (RR 0.74, 95% CI 0.56–0.98), and there was a trend towards fewer neurologic sequelae (RR 0.72, 95% CI 0.51–1.01). The benefits were shown in studies in adults in high-income countries, but the studies from low-income countries showed neither harm nor benefit. Based on these findings, the authors recommended the use of steroids in high-income countries, though the strength of the evidence was not optimal. The recommended steroid was dexamethasone 0.15 mg/kg intravenously every 6 hours for 4 days.
Although the incidence and rates of morbidity and death from acute community-acquired bacterial meningitis have dramatically declined, probably as a result of vaccination and better antimicrobial and adjuvant therapy, the disease still has a high toll. From 10% to 20% of people who contract it in the United States still die of it.1,2
In the United States, meningitis from all causes accounts for about 72,000 hospitalizations and up to $1.2 billion in hospital costs annually.3 However, the incidence of bacterial meningitis has declined from 3 to 5 per 100,000 per year a few decades ago to 1.3 to 2 per 100,000 per year currently.2 In less-developed countries, rates are much higher.
In the early 1900s in the United States, the death rate from bacterial meningitis was 80% to 100%. The use of intrathecal equine meningococcal antiserum during the first decades of the 1900s dramatically reduced the rate of death from meningococcal meningitis. With the advent of antimicrobial drugs in the 1930s and 1940s, the death rate from bacterial meningitis further declined.
The organisms that cause community-acquired bacterial meningitis differ somewhat by geographic region and by age. In a recent paper based on surveillance data, in the United States, from 1998 to 2007, the most common cause of bacterial meningitis among adults was Streptococcus pneumoniae. Among young adults, Neisseria meningitidis is nearly as common as S pneumoniae. The incidence of Listeria infections increases with age in adults.2
The epidemiologic features of bacterial meningitis have changed dramatically over the past decades with the advent of the Haemophilus influenzae vaccine. In 1986, about half the cases of acute bacterial meningitis were caused by H influenzae, but a decade later the incidence of H influenzae meningitis had been reduced by 94%.4
Meningitis is inflammation of the pia and arachnoid (the inner two layers of the meninges). Acute community-acquired meningitis can develop within hours to days and can be viral or bacterial. Viral meningitis usually has a good prognosis, whereas bacterial meningitis is associated with significant rates of morbidity and death, so it is critical to recognize and differentiate them promptly.
PATHOGENESIS
Most cases of community-acquired bacterial meningitis begin with colonization of the nasopharyngeal mucosa. In certain individuals this leads to mucosal invasion and bacteremia. Not all organisms that cause bacteremia are capable of breaching the blood-cerebrospinal fluid barrier to enter the subarachnoid space to cause meningitis. Very few organisms have this capacity, but N meningitidis and S pneumoniae do.5
Some patients are at higher risk of meningitis because of an abnormal communication between the nasopharynx and the subarachnoid space due either to trauma or a congenital anatomic abnormality. The organisms in these instances can directly spread from the nasopharynx to the meninges. Patients without a spleen or with an immunoglobulin deficiency are also more prone to infections from encapsulated organisms such as pneumococci and meningococci. The opsonizing immunoglobulins coat the capsule, helping phagocytes in the spleen to remove them from the bloodstream. A patient presenting with multiple episodes of bacterial meningitis merits evaluation for these conditions.
In contrast, Listeria spp and, rarely, gramnegative bacteria enter the bloodstream through the gastrointestinal tract and then spread to the meninges.
Once in the subarachnoid space, bacteria elicit a profuse inflammatory response, which can be damaging.5 The inflammation in the subarachnoid space can extend along the Virchow-Robin spaces surrounding the blood vessels deep into the brain parenchyma. This perivascular inflammation can cause thrombosis in both the arterial and venous circulation.
Thus, the inflammation can lead to intracranial complications such as cerebral edema, hydrocephalus, and stroke. The complications of bacterial meningitis can be remembered by the acronym HACTIVE: hydrocephalus, abscess, cerebritis and cranial nerve lesions, thrombosis, infarct, ventriculitis and vasculopathy, and extra-axial collection.5,6
MICROBIOLOGY: WHEN TO SUSPECT DIFFERENT ORGANISMS
S pneumoniae: The most common cause in adults
Patients without a spleen and patients with either a primary or secondary immunoglobulin deficiency, including patients with multiple myeloma or human immunodeficiency virus infection, are at a higher risk of infection with this organism.
N meningitidis: More common in young adults
N meningitidis is easily transmitted and is associated with crowding, as in school dormitories and military barracks. People with congenital deficiencies of components of terminal complement are at greater risk for both meningococcal and gonococcal infections. Patients with recurrent episodes of Neisseria infection should be evaluated for complement deficiency.
Meningococcal infection is more commonly associated with a rash. The most common rash of meningococcal meningitis is a very transient, maculopapular rash that appears early in the course of the disease. More pathognomonic is a petechial rash (Figure 1) with thrombocytopenia, which can very rapidly progress to purpura, ecchymosis, and disseminated intravascular coagulation. The petechial rash is evident in 60% of adults and up to 90% of children,7 and it is most likely to appear in dependent areas (such as the back of a patient lying down) and in areas of pressure, such as under the elastic band of underwear or stockings.
Listeria
Listeria infection is usually acquired through contaminated food such as raw vegetables, unpasteurized milk and cheese, and deli meats. From the gastrointestinal tract, it spreads to the bloodstream and then to the meninges.
Listeria is an intracellular pathogen; thus, people at greater risk are those with poor cell-mediated immunity due to immunosuppressant medications such as steroids or tumor necrosis factor inhibitors.
The rate of Listeria meningitis starts to increase with age, especially after age 50, probably due to immune senescence or decreased immunity with age.
Aerobic gram-negative bacilli
Gram-negative enteric bacilli usually cause meningitis after head trauma or neurosurgery and are very uncommon causes of community-acquired meningitis. Disseminated strongyloidiasis, also known as hyperinfection syndrome, should be suspected in any patient with community-acquired meningitis caused by enteric gram-negative bacilli.
Strongyloides stercoralis is a parasitic intestinal roundworm that is found in the tropics, in the subtropics, and in certain parts of the United States and Europe. The adult worm lives in the intestines and lays eggs, which hatch in the mucosa; the larvae are excreted in the stool. A small percentage of larvae penetrate the perianal skin and gut mucosa to cause an autoinfection. People may asymptomatically harbor the parasite for decades, then develop the hyperinfection syndrome when treated with immunosuppressive drugs such as steroids. In the hyperinfection syndrome a significant proportion of the larvae penetrate the gut mucosa to enter the bloodstream and travel throughout the body, including into the brain, carrying gram-negative bacteria with them.
The mortality rate of untreated hyperinfection syndrome can sometimes reach 100%.8 Thus, it is important to identify and treat the hyperinfection syndrome in the context of gram-negative bacillary meningitis.
SUSPECTED MENINGITIS: CLINICAL SCENARIO
A 36-year-old man presents to the emergency department with high fever, headache, and lethargy that developed over the past 24 hours. His temperature is 104°F (40°C), pulse 120 beats/min, respiratory rate 30/min, and blood pressure 130/70 mm Hg. He is oriented only to person and has nuchal rigidity. His white blood cell count is 30 × 109/L, with 20% bands.
The clinical questions that arise with such a patient are:
- Does the patient have bacterial or viral meningitis?
- Can we reliably rule out meningitis based on a history and physical examination?
- Is a lumbar puncture for cerebrospinal fluid (CSF) analysis needed? How should these studies be interpreted?
- Should computed tomography of the head be done before lumbar puncture?
- Which antimicrobial drugs should be started empirically at the outset?
- What is the role of steroids in treatment?
CLINICAL SIGNS AND SYMPTOMS
The classic triad of meningitis is fever, neck stiffness, and altered mental status. Other signs and symptoms that have been described are photophobia, headache, nausea, vomiting, focal neurologic symptoms, altered mental status, the Kernig sign (inability to allow full knee extension when the hip is flexed to a 90° angle), and the Brudzinski sign (spontaneous flexion of the hips during attempted passive flexion of the neck).
Can meningitis be ruled out if the patient does not have this classic presentation?
Unfortunately, only a few high-quality studies of the diagnostic accuracy of signs and symptoms of bacterial meningitis have been done. Fourteen retrospective studies examined this issue, but they were heterogeneous with respect to patient age, immunosuppression status, and clinical presentation, as well as to how meningitis was diagnosed (via culture or cerebrospinal fluid analysis), making the results difficult to interpret.9 Retrospective studies are more prone to bias, as they lack a control group, and examiner bias is more likely. Based on retrospective data, the combination of fever, neck stiffness, and altered mental status has a sensitivity of only 0.46.9
Two prospective studies examined symptoms and signs. Thomas et al10 evaluated 297 patients with “clinically suspected meningitis.” Unfortunately, in this study the physical examination was not standardized. In a study by Uchihara and Tsukagoshi,11 the measurement was more reliable, as they used a single examiner to evaluate patients presenting with fever and headache, but only 54 patients were studied.
Based on these prospective studies, the presence of nausea and vomiting, headache, or neck stiffness does not reliably rule in meningitis (Table 1).9 Similarly, the absence of these does not rule it out. The 95% confidence intervals (CIs) of the positive and negative likelihood ratios include the value 1. (A simple interpretation of that would be that the likelihood of finding these features is the same in patients with meningitis when compared with those without meningitis.9)
For the physical examination, the presence or absence of fever, the Kernig sign, or the Brudzinski sign were also inconclusive. The CIs of the positive and negative likelihood ratios, like those of the symptoms, included the value 1. Only one test done on physical examination looked promising in having diagnostic utility to rule out meningitis: the jolt accentuation test (performed by asking a patient with a headache to quickly move his or her head twice horizontally; the result is positive if the headache worsens). If the result is negative, meningitis is unlikely (negative likelihood ratio 0.05, 95% CI 0.01–0.35).9 However, a positive test is not useful in making the diagnosis. A caveat is that this is based on a single study.
In summary, the history and physical examination are not sufficient to determine whether a patient has meningitis. If a patient is suspected of having meningitis, a lumbar puncture is needed.
WORKUP AND DIAGNOSTIC TESTS
Which tests are needed?
Blood cultures should be drawn before antimicrobial treatment is started.12–14 Although positive only 19% to 70% of the time, they can help identify the pathogen.15–17
Lumbar puncture with CSF study is essential to make the diagnosis and to identify the organism and its susceptibility to various antibiotics. If lumbar puncture can be performed immediately, it should be done before starting antibiotics, to maximize the yield of cultures. Pediatric studies show that after starting antibiotics, complete sterilization of the cerebrospinal fluid can occur within 2 hours for N meningitides and within 4 hours for S pneumoniae.14 However, starting antimicrobials should not be delayed if a lumbar puncture cannot be done expeditiously.
Is computed tomography of the brain necessary before a lumbar puncture?
The rationale behind performing CT before lumbar puncture is to determine if the patient has elevated intracranial pressure, which would increase the risk of brain herniation due to lowering of the lumbar CSF pressure during lumbar puncture. For ethical and practical reasons, it would be difficult to evaluate this in a randomized clinical trial.
Hasbun et al18 performed a study to evaluate if any features on clinical presentation can predict abnormal findings on CT of the head suggestive of elevated intracranial pressure and thus the risk of herniation. The study included 301 adults with suspected meningitis. It found that abnormal findings on CT were unlikely if all of the following features were absent at baseline:
- Immunocompromised state
- History of central nervous system disease (mass lesion, stroke, or a focal infection)
- New onset of seizure (≤ 1 week from presentation)
- Specific abnormal neurologic findings (eg, an abnormal level of consciousness, inability to answer two consecutive questions correctly or to follow two consecutive commands, gaze palsy, abnormal visual fields, facial palsy, arm drift, leg drift, abnormal language).
Absence of these baseline features made it unlikely that CT would be abnormal (negative likelihood ratio 0.1, 95% CI 0.03–0.31).
According to the guidelines from the Infectious Diseases Society of America (IDSA),19 if none of those features is present, blood cultures and a lumbar puncture should be done immediately, followed by empiric antimicrobial therapy. If any of the features is present, blood cultures should be obtained first, then empiric antimicrobial therapy started, followed by CT of the brain to look for contraindications to a lumbar puncture (Figure 2).
What can lumbar puncture tell us?
Results of lumbar puncture studies can help determine whether meningitis is present and, if so, whether the cause is likely bacterial or viral.20
The opening pressure is elevated (usually > 180 mm H2O) in acute bacterial meningitis. The CSF white blood cell count is usually more than 1.0 × 109/L, consisting predominantly of neutrophils, in acute bacterial meningitis. In viral meningitis, it is usually less than 0.1 × 109/L, mostly lymphocytes.
Protein shows a mild to marked elevation in bacterial meningitis but is normal to elevated in viral meningitis.
The CSF glucose level is lower in bacterial meningitis than in viral meningitis.
The ratio of CSF glucose to blood glucose. Because the glucose levels in the CSF and the blood equilibrate, the ratio of CSF glucose to serum glucose has better diagnostic accuracy than the CSF glucose level alone. The equilibration takes place within a few hours, so the serum glucose level should be ordered at the same time lumbar puncture is done. The CSF glucose-blood glucose ratio is a better predictor of bacterial meningitis than the CSF white blood cell count. Bacterial meningitis is likely if the ratio is lower than 0.4.
Lactate levels are not usually measured, but a lactate level greater than 31.5 mg/dL (3.5 mmol/L) is predictive of meningitis, and a lower level makes the diagnosis unlikely.
The diagnostic accuracies (likelihood ratios) of the CSF tests were analyzed by Straus et al.21 The positive likelihood ratios for the CSF white blood cell count and for the CSF glucose-blood glucose ratio are greater than 10, but these tests have negative likelihood ratios of more than 0.1. (It is generally thought that a test with a positive likelihood ratio of more than 10 is considered good for ruling in a diagnosis, whereas one with a negative likelihood ratio of less than 0.1 is good for ruling out a diagnosis.) Thus, these tests are good to rule in bacterial meningitis, but not as good to rule it out. There are some data to show that CSF lactate and procalcitonin might be more sensitive in ruling out bacterial meningitis, but more studies are needed.22
Gram stain of the cerebrospinal fluid can be done quickly. If no bacteria are seen, the information is not helpful in ruling out bacterial meningitis (negative likelihood ratio 0.14, 95% CI 0.08–0.27). If it is positive, it is almost 100% specific for meningitis due to the organism seen (positive likelihood ratio 735, 95% CI 230–2,295).21
MANAGEMENT
Empiric antimicrobial therapy must be started as soon as feasible
Most studies of the timing of antimicrobial drugs were retrospective and included a very heterogeneous population. They were thus more prone to bias and confounding.23,24 Proulx et al,23 in a retrospective study, found that if antibiotics were given within 6 hours of the time the patient presented to the emergency department, the case fatality rate was only 5% to 6%. If treatment started 6 to 8 hours after presentation, the death rate was 45%, and if it started from 8 to 10 hours after presentation, the death rate was 75%. Most physicians would agree that starting antimicrobials early would be beneficial.
CSF concentrations of most antimicrobial drugs are considerably less than in the serum due to poor penetration of the blood-CSF barrier. Thus, the dose for treating meningitis is usually higher than the regular dose. For example, for the treatment of pneumococcal pneumonia, ceftriaxone (Rocephin) is used at a dose of 1 g every 24 hours, but for pneumococcal meningitis the dose is 2 g every 12 hours.
Empiric treatment of community-acquired bacterial meningitis in immunocompetent adults up to 50 years of age consists of a third-generation cephalosporin such as cefotaxime (Claforan) 2 g intravenously every 4 hours or ceftriaxone 2 g intravenously every 12 hours, which covers most S pneumoniae and N meningitides strains.19 The IDSA guidelines recommend adding vancomycin (Vancocin) empirically in suspected S pneumoniae meningitis due to concerns about drug-resistant pneumococcal strains.19 For vancomycin, 45 to 60 mg/kg intravenously per day divided into every-6-hour or every-8-hour doses would achieve better CSF concentrations.25
In patients over age 50 or those with a cell-mediated immunodeficiency, empiric therapy should also include ampicillin 2 g intravenously every 4 hours to cover Listeria.
It is important to tailor therapy to the results of Gram stain, culture, and susceptibility as they become available.
Role of corticosteroids
Glucocorticoids, especially dexamethasone (Decadron), have been well studied as adjunctive therapies in bacterial meningitis. The rationale behind their use is that the profuse inflammatory response to the bacterial components in the CSF by itself has deleterious effects, and steroids can reduce that.
In 2004, a Cochrane meta-analysis26 of five randomized clinical trials, including 623 adults with bacterial meningitis (234 with pneumococcal meningitis and 232 with meningococcal meningitis), found a significant reduction in the death rate for patients who received steroids: the death rate was 12% in patients who received steroids vs 22% in those who did not (odds ratio 0.6; 95% CI 0.40–0.81). This led to an IDSA practice guideline recommendation that in adults with suspected or proven pneumococcal meningitis, dexamethasone would be beneficial.19
But since then, many more studies have emerged from Europe, South America, Malawi, and Vietnam, and another Cochrane metaanalysis27 incorporated the new studies. Twenty-four studies involving 4,041 participants were included. Similar numbers of participants died in the corticosteroid and placebo groups (18% vs 20%; risk ratio [RR] 0.92, 95% CI 0.82–1.04, P = .18). A trend towards a lower mortality rate was noticed in adults receiving corticosteroids (RR 0.74, 95% CI 0.53–1.05, P = .09). In adults, corticosteroids were associated with lower rates of hearing loss (RR 0.74, 95% CI 0.56–0.98), and there was a trend towards fewer neurologic sequelae (RR 0.72, 95% CI 0.51–1.01). The benefits were shown in studies in adults in high-income countries, but the studies from low-income countries showed neither harm nor benefit. Based on these findings, the authors recommended the use of steroids in high-income countries, though the strength of the evidence was not optimal. The recommended steroid was dexamethasone 0.15 mg/kg intravenously every 6 hours for 4 days.
- Swartz MN. Bacterial meningitis—a view of the past 90 years. N Engl J Med 2004; 351:1826–1828.
- Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med 2011; 364:2010–2025.
- Holmquist L, Russo CA, Elixhauser A. Meningitis-related hospitalizations in the United States, 2006. Statistical Brief #57. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville, MD, 2008. www.hcup-us.ahrq.gov/reports/statbriefs/sb57.jsp. Accessed May 4, 2012.
- Schuchat A, Robinson K, Wenger JD, et al. Bacterial meningitis in the United States in 1995. N Engl J Med 1997; 337:970–976.
- Koedel U, Scheld WM, Pfister H-W. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis 2002; 2:721–736.
- Hughes DC, Raghavan A, Mordekar SR, Griffiths PD, Connolly DJ. Role of imaging in the diagnosis of acute bacterial meningitis and its complications. Postgrad Med J 2010; 86:478–485.
- Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev 2010; 23:467–492.
- Maguire JH. Intestinal nematodes (roundworms). In:Mandell G, Bennett J, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia: Elsevier, 2009:3577–3586.
- Attia J, Hatala R, Cook DJ, Wong JG. Original article: does this adult patient have acute meningitis?In:Simel DL, Rennie D, editors. The Rational Clinical Examinatino: Evidence-Based Clinical Diagnosis. New York, NY: McGraw-Hill; 2009.
- Thomas KE, Hasbun R, Jekel J, Quagliarello VJ. The diagnostic accuracy of Kernig’s sign, Brudzinski’s sign, and nuchal rigidity in adults with suspected meningitis. Clin Infect Dis 2002; 35:46–52.
- Uchihara T, Tsukagoshi H. Jolt accenulation of headache: the most sensitive sign of CSF pleocytosis. Headache 1991; 31:167–171.
- Geiseler PJ, Nelson KE, Levin S, Reddi KT, Moses VK. Community-acquired purulent meningitis: a review of 1,316 cases during the antibiotic era, 1954–1976. Rev Infect Dis 1980; 2:725–745.
- Talan DA, Hoffman JR, Yoshikawa TT, Overturf GD. Role of empiric parenteral antibiotics prior to lumbar puncture in suspected bacterial meningitis: state of the art. Rev Infect Dis 1988; 10:365–376.
- Kanegaye JT, Soliemanzadeh P, Bradley JS. Lumbar puncture in pediatric bacterial meningitis: defining the time interval for recovery of cerebrospinal fluid pathogens after parenteral antibiotic pretreatment. Pediatrics 2001; 108:1169–1174.
- Sigurdardóttir B, Björnsson OM, Jónsdóttir KE, Erlendsdóttir H, Gudmundsson S. Acute bacterial meningitis in adults. A 20-year overview. Arch Intern Med 1997; 157:425–430.
- Aronin SI, Peduzzi P, Quagliarello VJ. Community-acquired bacterial meningitis: risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med 1998; 129:862–869.
- Andersen J, Backer V, Voldsgaard P, Skinhój P, Wandall JH. Acute meningococcal meningitis: analysis of features of the disease according to the age of 255 patients. Copenhagen Meningitis Study Group. J Infect 1997; 34:227–235.
- Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med 2001; 345:1727–1733.
- Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004; 39:1267–1284.
- Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician 2003; 68:1103–1108.
- Straus SE, Thorpe KE, Holroyd-Leduc J. How do I perform a lumbar puncture and analyze the results to diagnose bacterial meningitis? JAMA 2006; 296:2010–2022.
- Viallon A, Desseigne N, Marjollet O, et al. Meningitis in adult patients with a negative direct cerebrospinal fluid examination: value of cytochemical markers for differential diagnosis. Crit Care 2011; 15:R136.
- Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM 2005; 98:291–298.
- Radetsky M. Duration of symptoms and outcome in bacterial meningitis: an analysis of causation and the implications of a delay in diagnosis. Pediatr Infect Dis J 1992; 11:694–698.
- Ricard JD, Wolff M, Lacherade JC, et al. Levels of vancomycin in cerebrospinal fluid of adult patients receiving adjunctive corticosteroids to treat pneumococcal meningitis: a prospective multicenter observational study. Clin Infect Dis 2007; 44:250–255.
- van de Beek D, de Gans J, McIntyre P, Prasad K. Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis 2004; 4:139–143.
- van de Beek D, Farrar JJ, de Gans J, et al. Adjunctive dexamethasone in bacterial meningitis: a meta-analysis of individual patient data. Lancet Neurol 2010; 9:254–263.
- Swartz MN. Bacterial meningitis—a view of the past 90 years. N Engl J Med 2004; 351:1826–1828.
- Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med 2011; 364:2010–2025.
- Holmquist L, Russo CA, Elixhauser A. Meningitis-related hospitalizations in the United States, 2006. Statistical Brief #57. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville, MD, 2008. www.hcup-us.ahrq.gov/reports/statbriefs/sb57.jsp. Accessed May 4, 2012.
- Schuchat A, Robinson K, Wenger JD, et al. Bacterial meningitis in the United States in 1995. N Engl J Med 1997; 337:970–976.
- Koedel U, Scheld WM, Pfister H-W. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis 2002; 2:721–736.
- Hughes DC, Raghavan A, Mordekar SR, Griffiths PD, Connolly DJ. Role of imaging in the diagnosis of acute bacterial meningitis and its complications. Postgrad Med J 2010; 86:478–485.
- Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev 2010; 23:467–492.
- Maguire JH. Intestinal nematodes (roundworms). In:Mandell G, Bennett J, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia: Elsevier, 2009:3577–3586.
- Attia J, Hatala R, Cook DJ, Wong JG. Original article: does this adult patient have acute meningitis?In:Simel DL, Rennie D, editors. The Rational Clinical Examinatino: Evidence-Based Clinical Diagnosis. New York, NY: McGraw-Hill; 2009.
- Thomas KE, Hasbun R, Jekel J, Quagliarello VJ. The diagnostic accuracy of Kernig’s sign, Brudzinski’s sign, and nuchal rigidity in adults with suspected meningitis. Clin Infect Dis 2002; 35:46–52.
- Uchihara T, Tsukagoshi H. Jolt accenulation of headache: the most sensitive sign of CSF pleocytosis. Headache 1991; 31:167–171.
- Geiseler PJ, Nelson KE, Levin S, Reddi KT, Moses VK. Community-acquired purulent meningitis: a review of 1,316 cases during the antibiotic era, 1954–1976. Rev Infect Dis 1980; 2:725–745.
- Talan DA, Hoffman JR, Yoshikawa TT, Overturf GD. Role of empiric parenteral antibiotics prior to lumbar puncture in suspected bacterial meningitis: state of the art. Rev Infect Dis 1988; 10:365–376.
- Kanegaye JT, Soliemanzadeh P, Bradley JS. Lumbar puncture in pediatric bacterial meningitis: defining the time interval for recovery of cerebrospinal fluid pathogens after parenteral antibiotic pretreatment. Pediatrics 2001; 108:1169–1174.
- Sigurdardóttir B, Björnsson OM, Jónsdóttir KE, Erlendsdóttir H, Gudmundsson S. Acute bacterial meningitis in adults. A 20-year overview. Arch Intern Med 1997; 157:425–430.
- Aronin SI, Peduzzi P, Quagliarello VJ. Community-acquired bacterial meningitis: risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med 1998; 129:862–869.
- Andersen J, Backer V, Voldsgaard P, Skinhój P, Wandall JH. Acute meningococcal meningitis: analysis of features of the disease according to the age of 255 patients. Copenhagen Meningitis Study Group. J Infect 1997; 34:227–235.
- Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med 2001; 345:1727–1733.
- Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004; 39:1267–1284.
- Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician 2003; 68:1103–1108.
- Straus SE, Thorpe KE, Holroyd-Leduc J. How do I perform a lumbar puncture and analyze the results to diagnose bacterial meningitis? JAMA 2006; 296:2010–2022.
- Viallon A, Desseigne N, Marjollet O, et al. Meningitis in adult patients with a negative direct cerebrospinal fluid examination: value of cytochemical markers for differential diagnosis. Crit Care 2011; 15:R136.
- Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM 2005; 98:291–298.
- Radetsky M. Duration of symptoms and outcome in bacterial meningitis: an analysis of causation and the implications of a delay in diagnosis. Pediatr Infect Dis J 1992; 11:694–698.
- Ricard JD, Wolff M, Lacherade JC, et al. Levels of vancomycin in cerebrospinal fluid of adult patients receiving adjunctive corticosteroids to treat pneumococcal meningitis: a prospective multicenter observational study. Clin Infect Dis 2007; 44:250–255.
- van de Beek D, de Gans J, McIntyre P, Prasad K. Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis 2004; 4:139–143.
- van de Beek D, Farrar JJ, de Gans J, et al. Adjunctive dexamethasone in bacterial meningitis: a meta-analysis of individual patient data. Lancet Neurol 2010; 9:254–263.
KEY POINTS
- The most common organisms that cause community-acquired bacterial meningitis are Streptococcus pneumoniae and Neisseria meningitidis. The incidence of Listeria infection increases in patients over age 50 and in those with compromised cell-mediated immunity.
- Symptoms and signs are not sensitive or specific enough to diagnose community-acquired bacterial meningitis. A lumbar puncture for cerebrospinal fluid studies is needed to reach the diagnosis, to identify the organism, and to determine antimicrobial susceptibilities.
- Gram stain of cerebrospinal fluid may quickly identify the causative organism. It is not very sensitive, but it is specific.
- Lumbar puncture should be performed as soon as possible. Computed tomography of the head is not necessary in all patients, only in immunocompromised patients and those who have features suggestive of or who are at risk of increased intracranial pressure.
- Try to obtain blood and cerebrospinal fluid cultures before staring antimicrobial therapy, but do not delay therapy if obtaining them is not feasible.
Geriatrics update 2012: What parts of our practice to change, what to ‘think about’
A number of new studies and guidelines published over the last few years are changing the way we treat older patients. This article summarizes these recent developments in a variety of areas—from prevention of falls to targets for hypertension therapy—relevant to the treatment of geriatric patients.
A MULTICOMPONENT APPROACH TO PREVENTING FALS
The American Geriatrics Society and British Geriatrics Society’s 2010 Clinical Practice Guideline for Prevention of Falls in Older Persons1 has added an important new element since the 2001 guideline: in addition to asking older patients about a fall, clinicians should also ask whether a gait or balance problem has developed.
A complete falls evaluation and multicomponent intervention is indicated for patients who in the past year or since the previous visit have had one fall with an injury or more than one fall, or for patients who report or have been diagnosed with a gait or balance problem. A falls risk assessment is not indicated for a patient with no gait or balance problem and who has had only one noninjurious fall in the previous year that did not require medical attention.
The multicomponent evaluation detailed in the guideline is very thorough and comprises more elements than can be done in a follow-up office visit. In addition to the relevant medical history, physical examination, and cognitive and functional assessment, the fall-risk evaluation includes a falls history, medication review, visual acuity testing, gait and balance assessment, postural and heart-rate evaluation, examination of the feet and footwear, and, if appropriate, a referral for home assessment of environmental hazards.
Intervention consists of many aspects
Of the interventions, exercise has the strongest correlation with falls prevention, and a prescription should include exercises for balance, gait, and strength. Tai chi is specifically recommended.
Medications should be reduced or withdrawn. The previous guideline recommended reducing medications for patients taking four or more medications, but the current guideline applies to everyone.
First cataract removal is associated with reducing the risk of falls.
Postural hypotension should be treated if present.
Vitamin D at 800 U per day is recommended for all elderly people at risk. For elderly people in long-term care, giving vitamin D for proven or suspected deficiency is by itself correlated with risk reduction.
Interventions that by themselves are not associated with risk reduction include education (eg, providing a handout on preventing falls) and having vision checked. For adults who are cognitively impaired, there is insufficient evidence that even the multicomponent intervention helps prevent falls.
CALCIUM AND VITAMIN D MAY NOT BE HARMLESS

Calcium supplements: A cause of heart attack?
Questions have arisen in recent studies about the potential risks of calcium supplementation.
A meta-analysis of 11 trials with nearly 12,000 participants found that the risk of myocardial infarction was significantly higher in people taking calcium supplementation (relative risk 1.27; 95% confidence interval [CI] 1.01–1.59, P = .038).2 Patients were predominantly postmenopausal women and were followed for a mean of 4 years. The incidence of stroke and death were also higher in people who took calcium, but the differences did not reach statistical significance. The dosages were primarily 1,000 mg per day (range 600 mg to 2 g). Risk was independent of age, sex, and type of supplement.
The authors concluded (somewhat provocatively, because only the risk of myocardial infarction reached statistical significance) that if 1,000 people were treated with calcium supplementation for 5 years, 26 fractures would be prevented but 14 myocardial infarctions, 10 strokes, and 13 deaths would be caused.

Comments. These data suggest that physicians may wish to prescribe calcium to supplement (not replace) dietary calcium to help patients reach but not exceed current guidelines for total calcium intake for age and sex. They may also want to advise the patient to take the calcium supplement separately from medications, as indicated in Table 2.
Benefits of vitamin D may depend on dosing
Studies show that the risk of hip fracture can be reduced with modest daily vitamin D supplementation, up to 800 U daily, regardless of calcium intake.3 Some vitamin D dosing regimens, however, may also entail risk.
Sanders et al4 randomized women age 70 and older to receive an annual injection of a high dose of vitamin D (500,000 U) or placebo for 3 to 5 years. Women in the vitamin D group had 15% more falls and 25% more fractures than those in the placebo group. The once-yearly dose of 500,000 U equates to 1,370 U/day, which is not much higher than the recommended daily dosage. The median baseline serum level was 49 nmol/L and reached 120 nmol/L at 30 days in the treatment group, which was not in the toxic range.
Comments. This study cautions physicians against giving large doses of vitamin D at long intervals. Future studies should focus on long-term clinical outcomes of falls and fractures for dosing regimens currently in practice, such as 50,000 units weekly or monthly.
BISPHOSPHONATES AND NONTRAUMATIC THICK BONE FRACTURES
Bisphosphonates have been regarded as the best drugs for preventing hip fracture. But in 2010, the US Food and Drug Administration (FDA) issued a warning that bisphosphonates have been associated with “atypical” femoral fractures. The atypical fracture pattern is a clean break through the thick bone of the shaft that occurs after minimal or no trauma.5 This pattern contrasts with the splintering “typical” fracture in the proximal femur in osteoporotic bone, usually after a fall.
Another characteristic of the atypical fractures is a higher incidence of postoperative complications requiring revision surgery. In more than 14,000 women in secondary analyses of three large randomized bisphosphonate trials, 12 fractures in 10 patients were found that were classified as atypical, averaging to an incidence of 2.3 per 10,000 patient-years.6
A population-based, nested case-control study7 using Canadian pharmacy records evaluated more than 200,000 women at least 68 years old who received bisphosphonate therapy. Of these, 716 (0.35%) sustained an atypical femoral fracture and 9,723 (4.7%) had a typical osteoporotic femoral fracture. Comparing the duration of bisphosphonate use between the two groups, the authors found that the risk of an atypical fracture increased with years of usage (at 5 years or more, the adjusted odds ratio was 2.74, 95% CI 1.25–6.02), but the risk of a typical fracture decreased (at 5 years or more, the adjusted odds ratio was 0.76, 95% CI 0.63–0.93). The study suggests that for every 100 hip fractures that bisphosphonate therapy prevents, it causes one atypical hip fracture.
Comments. These studies have caused some experts to advocate periodic bisphosphonate “vacations,”8 but for how long remains an open question because the risk of a typical fracture will increase. It is possible that a biomarker can help establish the best course, but that has yet to be determined.
DENOSUMAB: A NEW DRUG FOR OSTEOPOROSIS WITH A BIG PRICE TAG
Denosumab (Prolia, Xgeva), a newly available injectable drug, is a monoclonal antibody member of the tumor necrosis factor super-family.9 It is FDA-approved for osteoporosis in postmenopausal women at a dosage of 60 mg every 6 months and for skeletal metastases from solid tumors (120 mg every 4 weeks). It is also being used off-label for skeletal protection in women taking aromatase inhibitors and for men with androgen deficiency.
This drug is expensive, costing $850 per 60-mg dose wholesale, and no data are yet available on its long-term effects.
Since the drug is not cleared via renal mechanisms, there is some hope that it can be used to treat osteoporosis in patients with advanced chronic kidney disease, since bisphosphonates are contraindicated in those with an estimated glomerular filtration rate (GFR) less than 30 to 35 mL/min. However, the major study of denosumab to date, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) study, had no patients with stage 5 chronic kidney disease (GFR < 15 mL/min/1.73 m2 or on dialysis), and too few with stage 4 chronic kidney disease (GFR 15–29) to demonstrate either the safety or efficacy of denosumab in patients with advanced chronic kidney disease.10
HYPERTENSION TREATMENT
A secondary analysis of a recent large hypertension study confirmed the benefits of antihypertensive therapy in very old adults and suggested new targets for systolic and diastolic blood pressures.11,12
The Systolic Hypertension in the Elderly Program (SHEP) trial,13 the Systolic Hypertension in Europe (Syst-Eur) trial,14 and the Hypertension in the Very Elderly Trial (HYVET)15 are the major, randomized, placebo-controlled antihypertensive trials in older adults. They all showed a reduction in the risk of stroke and cardiovascular events. The diuretic studies (SHEP and HYVET)13,15 also showed a lower risk of heart failure and death.
Most recently, secondary analysis of the International Verapamil-Trandolapril (INVEST) study11,12 showed that adults in the oldest groups (age 70–79 and 80 and older), experienced a greater risk of adverse cardiovascular outcomes if systolic blood pressure was lowered to below about 130 mm Hg. As diastolic blood pressure was lowered to about the 65–70 mm Hg range, all age groups in the study experienced an increased risk of cardiovascular events. These results confirm the findings of a secondary analysis of the SHEP trial,16 showing an increased risk of cardiovascular events when diastolic pressure was lowered to below approximately 65 mm Hg.
These studies have been incorporated into 75 pages of the 2011 Expert Consensus Document on Hypertension in the Elderly issued by the American College of Cardiology Foundation and the American Heart Association.17 In a nutshell, the guidelines suggest that older adults less than 80 years of age be treated comparably to middle-aged adults. However, for adults age 80 and older:
- A target for systolic blood pressure of 140 to 145 mm Hg “can be acceptable.”
- Initiating treatment with monotherapy (with a low-dose thiazide, calcium channel blocker, or renin-angiotensin-aldosterone system drug) is reasonable. A second drug may be added if needed.
- Patients should be monitored for “excessive” orthostasis.
- Systolic blood pressure lower than 130 mm Hg and diastolic blood pressure lower than 65 mm Hg should be avoided.
TRANSCATHETER AORTIC VALVE IMPLANTATION APPROVED BY THE FDA
An estimated 2% to 9% of the elderly have aortic stenosis. Aortic valve replacement reduces mortality rates and improves function in all age groups, including octogenarians. Those with asymptomatic aortic stenosis tend to decline very quickly once they develop heart failure, syncope, or angina. Aortic valve replacement has been shown to put people back on the course they were on before they became symptomatic.
Transcatheter self-expanding transaortic valve implantation was approved by the FDA in November 2011. The procedure does not require open surgery and involves angioplasty of the old valve, with the new valve being passed into place through a catheter and expanded. Access is either transfemoral or transapical.
Transaortic valve implantation has been rapidly adopted in Europe since 2002 without any randomized control trials. The Placement of Aortic Transcatheter Valves (PARTNER) trial18 in 2011 was the first randomized trial of this therapy. It was conducted at 25 centers, with nearly 700 patients with severe aortic stenosis randomized to undergo either transcatheter aortic valve replacement with a balloon-expandable valve (244 via the transfemoral and 104 via the transapical approach) or surgical replacement. The mean age of the patients was 84 years, and the Society of Thoracic Surgeons mean score was 12%, indicating high perioperative risk.
At 30 days after the procedure, the rates of death were 3.4% with transcatheter implantation and 6.5% with surgical replacement (P = .07). At 1 year, the rates were 24.2% and 26.8%, respectively (P = 0.44, and P = .001 for noninferiority). However, the rate of major stroke was higher in the transcatheter implantation group: 3.8% vs 2.1% in the surgical group (P = .20) at 1 month and 5.1% vs 2.4% (P = .07) at 1 year. Vascular complications were significantly more frequent in the transcatheter implantation group, and the new onset of atrial fibrillation and major bleeding were significantly higher in the surgical group.
Patients in the transcatheter implantation group had a significantly shorter length of stay in the intensive care unit and a shorter index hospitalization. At 30 days, the transcatheter group also had a significant improvement in New York Heart Association functional status and a better 6-minute walk performance, although at 1 year, these measures were similar between the two groups and were greatly improved over baseline. Quality of life, measured using the Kansas City Cardiomyopathy Questionnaire, was higher both at 6 months and at 1 year in the transcatheter implantation group compared with those who underwent the open surgical procedure.19
Comments. The higher risk of stroke with the transcatheter implantation procedure remains a concern. More evaluation is also needed with respect to function and cognition in the very elderly, and of efficacy and safety in higher- and lower-risk patients.
DEPRESSION CAN BE EFFECTIVELY TREATED WITH MEDICATION
Many placebo-controlled trials have demonstrated the effectiveness of treating depression with medications in elderly people who are cognitively intact and living in the community. A Cochrane Review20 found that in placebo-controlled trials, the number needed to treat to produce one recovery with tricyclic antidepressants, selective serotonin reuptake inhibitors, and monoamine oxidase inhibitors was less than 10 for each of the drug classes.
Since the newer drugs appear to be safer and to have fewer adverse effects than the older drugs, more older adults have been treated with antidepressants, including patients with comorbidities such as dementia that were exclusion criteria in early studies. For example, the number of older adults treated with antidepressants has increased 25% since 1992; at the same time the number being referred for cognitive-based therapies has been reduced by 43%.21 Similar trends are apparent in elderly people in long-term care. In 1999, about one-third of people in long-term care were diagnosed with depression; in 2007 more than one-half were.22
Treating depression is less effective when dementia is present
Up to half of adults age 85 and older living in the community may have dementia. In long-term care facilities, most residents likely have some cognitive impairment or are diagnosed with dementia. Many of these are also taking antidepressive agents.
A review of studies in the Medline and Cochrane registries found seven trials that treated 330 patients with antidepressants for combined depression and dementia. Efficacy was not confirmed.23
After this study was published, Banerjee et al24 treated 218 patients who had depression and dementia in nine centers in the United Kingdom. Patients received sertraline (Zoloft), mirtazapine (Remeron), or placebo. Reductions in depression scores at 13 weeks and at 39 weeks did not differ between the groups, and adverse events were more frequent in the treatment groups than in the placebo groups.
Comments. The poor performance of antidepressants in patients with dementia may be due to misdiagnosis, such as mistaking apathy for depression.25 It is also possible that better criteria than we have now are needed to diagnose depression in patients with dementia, or that current outcome measures are not sensitive for depression when dementia is present.
It may also be unsafe to treat older adults long-term with antidepressive agents. For example, although selective serotonin reuptake inhibitors, the most commonly prescribed antidepressive agents, are considered safe, their side effects are numerous and include sexual dysfunction, bleeding (due to platelet dysfunction), hyponatremia, early weight loss, tremor (mostly with paroxetine [Paxil]), sedation, apathy (especially with high doses), loose stools (with sertraline), urinary incontinence, falls, bone loss, and QTc prolongation.
Citalopram: Maximum dosage in elderly
In August 2011, an FDA Safety Communication was issued for citalopram (Celexa), stating that the daily dose should not exceed 40 mg in the general population and should not exceed 20 mg in patients age 60 and older. The dose should also not exceed 20 mg for a patient at any age who has hepatic impairment, who is known to be a poor metabolizer of CYP 2C19, or who takes cimetidine (Tagamet), since that drug inhibits the metabolism of citalopram at the CYP 2C19 enzyme site.
Although the FDA warning specifically mentions only cimetidine, physicians may have concerns about other drugs that inhibit CYP 2C19, such as proton pump inhibitors (eg, omeprazole [Prilosec]) when taken concomitantly with citalopram. Also, escitalopram (Lexapro) and sertraline are quite similar to citalopram; although they were not mentioned in the FDA Safety Communication, higher doses of these drugs may put patients at similar risk.
ALZHEIMER DISEASE: NEED TO BETTER IDENTIFY PEOPLE AT RISK
The definition of dementia is essentially the presence of a cognitive problem that affects the ability to function. For people with Alzheimer disease, impairment of cognitive performance precedes functional decline. Those with a cognitive deficit who still function well have, by definition, mild cognitive impairment (MCI). Although MCI could be caused by a variety of vascular and other neurologic processes, the most common cause of MCI in the United States is Alzheimer disease.
Unfortunately, the population with MCI currently enrolled in clinical trials to reduce the risk of progression to Alzheimer disease is heterogeneous. Many study participants may never get dementia, and others may have had the pathology present for decades and are progressing rapidly. Imaging and biomarkers are emerging as good indicators that predict progression and could help to better define populations for clinical trials.26
Studies now indicate that people with MCI that is ultimately due to Alzheimer disease are likely to have amyloid beta peptide 42 evident in the cerebrospinal fluid 10 to 20 years before symptoms arise. At the same time, amyloid is also likely to be evident in the brain with amyloid-imaging positron emission tomography (PET). Some time later, abnormalities in metabolism are also evident on fluorodeoxyglucose (FDG) PET, as are changes such as reduced hippocampal volume on magnetic resonance imaging (MRI).
The 1984 criteria for diagnosing MCI due to Alzheimer disease were recently revised to incorporate the evolving availability of biomarkers.27,28 The diagnosis of MCI itself is still based on clinical ascertainment including history, physical examination, and cognitive testing. It requires diagnosis of a cognitive decline from a prior level but maintenance of activities of daily living with no or minimal assistance. This diagnosis is certainly challenging since it requires ascertainment of a prior level of function and corroboration, when feasible, with an informant. Blood tests and imaging, which are readily available, constitute an important part of the assessment.
Attributing the MCI to Alzheimer disease requires consistency of the disease course—a gradual decline in Alzheimer disease, rather than a stroke, head injury, neurologic disease such as Parkinson disease, or mixed causes.
Knowledge of genetic factors, such as the presence of a mutation in APP, PS1, or PS2, can be predictive with young patients. The presence of one or two 34 alleles in the apolipoprotein E (APOE) gene is the only genetic variant broadly accepted as increasing the risk for late-onset Alzheimer dementia, whereas the 32 allele decreases risk.
Refining the risk attribution to Alzheimer disease requires biomarkers, currently available only in research settings:
- High likelihood—amyloid beta peptide detected by PET or cerebrospinal fluid analysis and evidence of neuronal degeneration or injury (elevated tau in the cerebrospinal fluid, decreased FDG uptake on PET, and atrophy evident by structural MRI)
- Intermediate likelihood—presence of amyloid beta peptide or evidence of neuronal degeneration or injury
- Unlikely—biomarkers tested and negative
- No comment—biomarkers not tested or reporting is indeterminate.
Comments. There is significant potential for misunderstanding the new definition for MCI. Patients who are concerned about their memory may request biomarker testing in an effort to determine if they currently have or will acquire Alzheimer disease. Doctors may be tempted to refer patients for biomarker testing (via imaging or lumbar puncture) to “screen” for MCI or Alzheimer disease.
It should be emphasized that MCI itself is still a clinical diagnosis, with the challenges noted above of determining whether there has been a cognitive decline from a prior level of function but preservation of activities of daily living. The biomarkers are not proposed to diagnose MCI, but only to help identify the subset of MCI patients most likely to progress rapidly to Alzheimer disease.
At present, the best use of biomarker testing is to aid research by identifying high-risk people among those with MCI who enroll in prospective trials for testing interventions to reduce the progression of Alzheimer disease.
- Panel on Prevention of Falls in Older Persons, American Geriatrics Society and British Geriatrics Society. J Am Geriatr Soc 2011; 59:148–157.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: metaanalysis. BMJ 2010; 341:c3691.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women. A randomized controlled trial. JAMA 2010; 303:1815–1822.
- Kuehn BM. Prolonged bisphosphonate use linked to rare fractures, esophageal cancer. JAMA 2010; 304:2114–2115.
- Black DM, Kelly MP, Genant HK, et al; Fracture Intervention Trial Steering Committee; HORIZON Pivotal Fracture Trial Steering Committee. Bisphosphonates and fractures of the subtrochanteric or diaphyseal femur. N Engl J Med 2010; 362:1761–1771.
- Park-Wyllie LY, Mamdani MM, Juurlink DN, et al. Bisphosphonate use and the risk of subtrochanteric or femoral shaft fractures in older women. JAMA 2011; 305:783–789.
- Ott SM. What is the optimal duration of bisphosphonate therapy? Cleve Clin J Med 2011; 78:619–630.
- Cummings SR, San Martin J, McClung MR, et al; for the FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Pepine CJ, Handberg EM, Cooper-Dehoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Denardo SJ, Gong Y, Nichols WW, et al. Blood pressure and outcomes in very old hypertensive coronary artery disease patients: an INVEST substudy. Am J Med 2010; 123:719–726.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al; for the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension (erratum published in Lancet 1997; 350:1636). Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; for the HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Somes G, Pahor M, Shorr R, Cushman WC, Applegate WB. The role of diastolic blood pressure when treating isolated systolic hypertension. Arch Intern Med 1999; 159:2004–2009.
- Aronow WS, Fleg JL, Pepine CJ, et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly. J Am Coll Cardiol 2011; 57:2037–2114.
- Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
- Reynolds MR, Magnuson EA, Lei Y, et al; Placement of Aortic Transcatheter Valves (PARTNER) Investigators. Health-related quality of life after transcatheter aortic valve replacement in inoperable patients with severe aortic stenosis. Circulation 2011; 124:1964–1972.
- Wilson K, Mottram P, Sivanranthan A, Nightingale A. Antidepressant versus placebo for depressed elderly. Cochrane Database Syst Rev 2001;(2):CD000561.
- Akincigil A, Olfson M, Walkup JT, et al. Diagnosis and treatment of depression in older community-dwelling adults: 1992–2005. J Am Geriatr Soc 2011; 59:1042–1051.
- Gaboda D, Lucas J, Siegel M, Kalay E, Crystal S. No longer undertreated? Depression diagnosis and antidepressant therapy in elderly long-stay nursing home residents, 1999 to 2007. J Am Geriatr Soc 2011; 59:673–680.
- Nelson JC, Devanand DP. A systematic review and meta-analysis of placebo-controlled antidepressant studies in peoloe with depression and dementia. J Am Geriatr Soc 2011; 59:577–585.
- Banerjee S, Hellier J, Dewey M, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet 2011; 378:403–411.
- Landes AM, Sperry SD, Strauss ME, Geldmacher DS. Apathy in Alzheimer’s disease. J Am Geriatr Soc 2001; 49:1700–1707.
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010; 9:1118–1127.
- Daviglus ML, Bell CC, Berrettini W, et al. National Institutes of Health State-of-the-Science Conference statement: preventing Alzheimer disease and cognitive decline. Ann Intern Med 2010; 153:176–181.
- McKhann GM, Knopman DS, Chertkow H, et al The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269.
A number of new studies and guidelines published over the last few years are changing the way we treat older patients. This article summarizes these recent developments in a variety of areas—from prevention of falls to targets for hypertension therapy—relevant to the treatment of geriatric patients.
A MULTICOMPONENT APPROACH TO PREVENTING FALS
The American Geriatrics Society and British Geriatrics Society’s 2010 Clinical Practice Guideline for Prevention of Falls in Older Persons1 has added an important new element since the 2001 guideline: in addition to asking older patients about a fall, clinicians should also ask whether a gait or balance problem has developed.
A complete falls evaluation and multicomponent intervention is indicated for patients who in the past year or since the previous visit have had one fall with an injury or more than one fall, or for patients who report or have been diagnosed with a gait or balance problem. A falls risk assessment is not indicated for a patient with no gait or balance problem and who has had only one noninjurious fall in the previous year that did not require medical attention.
The multicomponent evaluation detailed in the guideline is very thorough and comprises more elements than can be done in a follow-up office visit. In addition to the relevant medical history, physical examination, and cognitive and functional assessment, the fall-risk evaluation includes a falls history, medication review, visual acuity testing, gait and balance assessment, postural and heart-rate evaluation, examination of the feet and footwear, and, if appropriate, a referral for home assessment of environmental hazards.
Intervention consists of many aspects
Of the interventions, exercise has the strongest correlation with falls prevention, and a prescription should include exercises for balance, gait, and strength. Tai chi is specifically recommended.
Medications should be reduced or withdrawn. The previous guideline recommended reducing medications for patients taking four or more medications, but the current guideline applies to everyone.
First cataract removal is associated with reducing the risk of falls.
Postural hypotension should be treated if present.
Vitamin D at 800 U per day is recommended for all elderly people at risk. For elderly people in long-term care, giving vitamin D for proven or suspected deficiency is by itself correlated with risk reduction.
Interventions that by themselves are not associated with risk reduction include education (eg, providing a handout on preventing falls) and having vision checked. For adults who are cognitively impaired, there is insufficient evidence that even the multicomponent intervention helps prevent falls.
CALCIUM AND VITAMIN D MAY NOT BE HARMLESS
Calcium supplements: A cause of heart attack?
Questions have arisen in recent studies about the potential risks of calcium supplementation.
A meta-analysis of 11 trials with nearly 12,000 participants found that the risk of myocardial infarction was significantly higher in people taking calcium supplementation (relative risk 1.27; 95% confidence interval [CI] 1.01–1.59, P = .038).2 Patients were predominantly postmenopausal women and were followed for a mean of 4 years. The incidence of stroke and death were also higher in people who took calcium, but the differences did not reach statistical significance. The dosages were primarily 1,000 mg per day (range 600 mg to 2 g). Risk was independent of age, sex, and type of supplement.
The authors concluded (somewhat provocatively, because only the risk of myocardial infarction reached statistical significance) that if 1,000 people were treated with calcium supplementation for 5 years, 26 fractures would be prevented but 14 myocardial infarctions, 10 strokes, and 13 deaths would be caused.
Comments. These data suggest that physicians may wish to prescribe calcium to supplement (not replace) dietary calcium to help patients reach but not exceed current guidelines for total calcium intake for age and sex. They may also want to advise the patient to take the calcium supplement separately from medications, as indicated in Table 2.
Benefits of vitamin D may depend on dosing
Studies show that the risk of hip fracture can be reduced with modest daily vitamin D supplementation, up to 800 U daily, regardless of calcium intake.3 Some vitamin D dosing regimens, however, may also entail risk.
Sanders et al4 randomized women age 70 and older to receive an annual injection of a high dose of vitamin D (500,000 U) or placebo for 3 to 5 years. Women in the vitamin D group had 15% more falls and 25% more fractures than those in the placebo group. The once-yearly dose of 500,000 U equates to 1,370 U/day, which is not much higher than the recommended daily dosage. The median baseline serum level was 49 nmol/L and reached 120 nmol/L at 30 days in the treatment group, which was not in the toxic range.
Comments. This study cautions physicians against giving large doses of vitamin D at long intervals. Future studies should focus on long-term clinical outcomes of falls and fractures for dosing regimens currently in practice, such as 50,000 units weekly or monthly.
BISPHOSPHONATES AND NONTRAUMATIC THICK BONE FRACTURES
Bisphosphonates have been regarded as the best drugs for preventing hip fracture. But in 2010, the US Food and Drug Administration (FDA) issued a warning that bisphosphonates have been associated with “atypical” femoral fractures. The atypical fracture pattern is a clean break through the thick bone of the shaft that occurs after minimal or no trauma.5 This pattern contrasts with the splintering “typical” fracture in the proximal femur in osteoporotic bone, usually after a fall.
Another characteristic of the atypical fractures is a higher incidence of postoperative complications requiring revision surgery. In more than 14,000 women in secondary analyses of three large randomized bisphosphonate trials, 12 fractures in 10 patients were found that were classified as atypical, averaging to an incidence of 2.3 per 10,000 patient-years.6
A population-based, nested case-control study7 using Canadian pharmacy records evaluated more than 200,000 women at least 68 years old who received bisphosphonate therapy. Of these, 716 (0.35%) sustained an atypical femoral fracture and 9,723 (4.7%) had a typical osteoporotic femoral fracture. Comparing the duration of bisphosphonate use between the two groups, the authors found that the risk of an atypical fracture increased with years of usage (at 5 years or more, the adjusted odds ratio was 2.74, 95% CI 1.25–6.02), but the risk of a typical fracture decreased (at 5 years or more, the adjusted odds ratio was 0.76, 95% CI 0.63–0.93). The study suggests that for every 100 hip fractures that bisphosphonate therapy prevents, it causes one atypical hip fracture.
Comments. These studies have caused some experts to advocate periodic bisphosphonate “vacations,”8 but for how long remains an open question because the risk of a typical fracture will increase. It is possible that a biomarker can help establish the best course, but that has yet to be determined.
DENOSUMAB: A NEW DRUG FOR OSTEOPOROSIS WITH A BIG PRICE TAG
Denosumab (Prolia, Xgeva), a newly available injectable drug, is a monoclonal antibody member of the tumor necrosis factor super-family.9 It is FDA-approved for osteoporosis in postmenopausal women at a dosage of 60 mg every 6 months and for skeletal metastases from solid tumors (120 mg every 4 weeks). It is also being used off-label for skeletal protection in women taking aromatase inhibitors and for men with androgen deficiency.
This drug is expensive, costing $850 per 60-mg dose wholesale, and no data are yet available on its long-term effects.
Since the drug is not cleared via renal mechanisms, there is some hope that it can be used to treat osteoporosis in patients with advanced chronic kidney disease, since bisphosphonates are contraindicated in those with an estimated glomerular filtration rate (GFR) less than 30 to 35 mL/min. However, the major study of denosumab to date, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) study, had no patients with stage 5 chronic kidney disease (GFR < 15 mL/min/1.73 m2 or on dialysis), and too few with stage 4 chronic kidney disease (GFR 15–29) to demonstrate either the safety or efficacy of denosumab in patients with advanced chronic kidney disease.10
HYPERTENSION TREATMENT
A secondary analysis of a recent large hypertension study confirmed the benefits of antihypertensive therapy in very old adults and suggested new targets for systolic and diastolic blood pressures.11,12
The Systolic Hypertension in the Elderly Program (SHEP) trial,13 the Systolic Hypertension in Europe (Syst-Eur) trial,14 and the Hypertension in the Very Elderly Trial (HYVET)15 are the major, randomized, placebo-controlled antihypertensive trials in older adults. They all showed a reduction in the risk of stroke and cardiovascular events. The diuretic studies (SHEP and HYVET)13,15 also showed a lower risk of heart failure and death.
Most recently, secondary analysis of the International Verapamil-Trandolapril (INVEST) study11,12 showed that adults in the oldest groups (age 70–79 and 80 and older), experienced a greater risk of adverse cardiovascular outcomes if systolic blood pressure was lowered to below about 130 mm Hg. As diastolic blood pressure was lowered to about the 65–70 mm Hg range, all age groups in the study experienced an increased risk of cardiovascular events. These results confirm the findings of a secondary analysis of the SHEP trial,16 showing an increased risk of cardiovascular events when diastolic pressure was lowered to below approximately 65 mm Hg.
These studies have been incorporated into 75 pages of the 2011 Expert Consensus Document on Hypertension in the Elderly issued by the American College of Cardiology Foundation and the American Heart Association.17 In a nutshell, the guidelines suggest that older adults less than 80 years of age be treated comparably to middle-aged adults. However, for adults age 80 and older:
- A target for systolic blood pressure of 140 to 145 mm Hg “can be acceptable.”
- Initiating treatment with monotherapy (with a low-dose thiazide, calcium channel blocker, or renin-angiotensin-aldosterone system drug) is reasonable. A second drug may be added if needed.
- Patients should be monitored for “excessive” orthostasis.
- Systolic blood pressure lower than 130 mm Hg and diastolic blood pressure lower than 65 mm Hg should be avoided.
TRANSCATHETER AORTIC VALVE IMPLANTATION APPROVED BY THE FDA
An estimated 2% to 9% of the elderly have aortic stenosis. Aortic valve replacement reduces mortality rates and improves function in all age groups, including octogenarians. Those with asymptomatic aortic stenosis tend to decline very quickly once they develop heart failure, syncope, or angina. Aortic valve replacement has been shown to put people back on the course they were on before they became symptomatic.
Transcatheter self-expanding transaortic valve implantation was approved by the FDA in November 2011. The procedure does not require open surgery and involves angioplasty of the old valve, with the new valve being passed into place through a catheter and expanded. Access is either transfemoral or transapical.
Transaortic valve implantation has been rapidly adopted in Europe since 2002 without any randomized control trials. The Placement of Aortic Transcatheter Valves (PARTNER) trial18 in 2011 was the first randomized trial of this therapy. It was conducted at 25 centers, with nearly 700 patients with severe aortic stenosis randomized to undergo either transcatheter aortic valve replacement with a balloon-expandable valve (244 via the transfemoral and 104 via the transapical approach) or surgical replacement. The mean age of the patients was 84 years, and the Society of Thoracic Surgeons mean score was 12%, indicating high perioperative risk.
At 30 days after the procedure, the rates of death were 3.4% with transcatheter implantation and 6.5% with surgical replacement (P = .07). At 1 year, the rates were 24.2% and 26.8%, respectively (P = 0.44, and P = .001 for noninferiority). However, the rate of major stroke was higher in the transcatheter implantation group: 3.8% vs 2.1% in the surgical group (P = .20) at 1 month and 5.1% vs 2.4% (P = .07) at 1 year. Vascular complications were significantly more frequent in the transcatheter implantation group, and the new onset of atrial fibrillation and major bleeding were significantly higher in the surgical group.
Patients in the transcatheter implantation group had a significantly shorter length of stay in the intensive care unit and a shorter index hospitalization. At 30 days, the transcatheter group also had a significant improvement in New York Heart Association functional status and a better 6-minute walk performance, although at 1 year, these measures were similar between the two groups and were greatly improved over baseline. Quality of life, measured using the Kansas City Cardiomyopathy Questionnaire, was higher both at 6 months and at 1 year in the transcatheter implantation group compared with those who underwent the open surgical procedure.19
Comments. The higher risk of stroke with the transcatheter implantation procedure remains a concern. More evaluation is also needed with respect to function and cognition in the very elderly, and of efficacy and safety in higher- and lower-risk patients.
DEPRESSION CAN BE EFFECTIVELY TREATED WITH MEDICATION
Many placebo-controlled trials have demonstrated the effectiveness of treating depression with medications in elderly people who are cognitively intact and living in the community. A Cochrane Review20 found that in placebo-controlled trials, the number needed to treat to produce one recovery with tricyclic antidepressants, selective serotonin reuptake inhibitors, and monoamine oxidase inhibitors was less than 10 for each of the drug classes.
Since the newer drugs appear to be safer and to have fewer adverse effects than the older drugs, more older adults have been treated with antidepressants, including patients with comorbidities such as dementia that were exclusion criteria in early studies. For example, the number of older adults treated with antidepressants has increased 25% since 1992; at the same time the number being referred for cognitive-based therapies has been reduced by 43%.21 Similar trends are apparent in elderly people in long-term care. In 1999, about one-third of people in long-term care were diagnosed with depression; in 2007 more than one-half were.22
Treating depression is less effective when dementia is present
Up to half of adults age 85 and older living in the community may have dementia. In long-term care facilities, most residents likely have some cognitive impairment or are diagnosed with dementia. Many of these are also taking antidepressive agents.
A review of studies in the Medline and Cochrane registries found seven trials that treated 330 patients with antidepressants for combined depression and dementia. Efficacy was not confirmed.23
After this study was published, Banerjee et al24 treated 218 patients who had depression and dementia in nine centers in the United Kingdom. Patients received sertraline (Zoloft), mirtazapine (Remeron), or placebo. Reductions in depression scores at 13 weeks and at 39 weeks did not differ between the groups, and adverse events were more frequent in the treatment groups than in the placebo groups.
Comments. The poor performance of antidepressants in patients with dementia may be due to misdiagnosis, such as mistaking apathy for depression.25 It is also possible that better criteria than we have now are needed to diagnose depression in patients with dementia, or that current outcome measures are not sensitive for depression when dementia is present.
It may also be unsafe to treat older adults long-term with antidepressive agents. For example, although selective serotonin reuptake inhibitors, the most commonly prescribed antidepressive agents, are considered safe, their side effects are numerous and include sexual dysfunction, bleeding (due to platelet dysfunction), hyponatremia, early weight loss, tremor (mostly with paroxetine [Paxil]), sedation, apathy (especially with high doses), loose stools (with sertraline), urinary incontinence, falls, bone loss, and QTc prolongation.
Citalopram: Maximum dosage in elderly
In August 2011, an FDA Safety Communication was issued for citalopram (Celexa), stating that the daily dose should not exceed 40 mg in the general population and should not exceed 20 mg in patients age 60 and older. The dose should also not exceed 20 mg for a patient at any age who has hepatic impairment, who is known to be a poor metabolizer of CYP 2C19, or who takes cimetidine (Tagamet), since that drug inhibits the metabolism of citalopram at the CYP 2C19 enzyme site.
Although the FDA warning specifically mentions only cimetidine, physicians may have concerns about other drugs that inhibit CYP 2C19, such as proton pump inhibitors (eg, omeprazole [Prilosec]) when taken concomitantly with citalopram. Also, escitalopram (Lexapro) and sertraline are quite similar to citalopram; although they were not mentioned in the FDA Safety Communication, higher doses of these drugs may put patients at similar risk.
ALZHEIMER DISEASE: NEED TO BETTER IDENTIFY PEOPLE AT RISK
The definition of dementia is essentially the presence of a cognitive problem that affects the ability to function. For people with Alzheimer disease, impairment of cognitive performance precedes functional decline. Those with a cognitive deficit who still function well have, by definition, mild cognitive impairment (MCI). Although MCI could be caused by a variety of vascular and other neurologic processes, the most common cause of MCI in the United States is Alzheimer disease.
Unfortunately, the population with MCI currently enrolled in clinical trials to reduce the risk of progression to Alzheimer disease is heterogeneous. Many study participants may never get dementia, and others may have had the pathology present for decades and are progressing rapidly. Imaging and biomarkers are emerging as good indicators that predict progression and could help to better define populations for clinical trials.26
Studies now indicate that people with MCI that is ultimately due to Alzheimer disease are likely to have amyloid beta peptide 42 evident in the cerebrospinal fluid 10 to 20 years before symptoms arise. At the same time, amyloid is also likely to be evident in the brain with amyloid-imaging positron emission tomography (PET). Some time later, abnormalities in metabolism are also evident on fluorodeoxyglucose (FDG) PET, as are changes such as reduced hippocampal volume on magnetic resonance imaging (MRI).
The 1984 criteria for diagnosing MCI due to Alzheimer disease were recently revised to incorporate the evolving availability of biomarkers.27,28 The diagnosis of MCI itself is still based on clinical ascertainment including history, physical examination, and cognitive testing. It requires diagnosis of a cognitive decline from a prior level but maintenance of activities of daily living with no or minimal assistance. This diagnosis is certainly challenging since it requires ascertainment of a prior level of function and corroboration, when feasible, with an informant. Blood tests and imaging, which are readily available, constitute an important part of the assessment.
Attributing the MCI to Alzheimer disease requires consistency of the disease course—a gradual decline in Alzheimer disease, rather than a stroke, head injury, neurologic disease such as Parkinson disease, or mixed causes.
Knowledge of genetic factors, such as the presence of a mutation in APP, PS1, or PS2, can be predictive with young patients. The presence of one or two 34 alleles in the apolipoprotein E (APOE) gene is the only genetic variant broadly accepted as increasing the risk for late-onset Alzheimer dementia, whereas the 32 allele decreases risk.
Refining the risk attribution to Alzheimer disease requires biomarkers, currently available only in research settings:
- High likelihood—amyloid beta peptide detected by PET or cerebrospinal fluid analysis and evidence of neuronal degeneration or injury (elevated tau in the cerebrospinal fluid, decreased FDG uptake on PET, and atrophy evident by structural MRI)
- Intermediate likelihood—presence of amyloid beta peptide or evidence of neuronal degeneration or injury
- Unlikely—biomarkers tested and negative
- No comment—biomarkers not tested or reporting is indeterminate.
Comments. There is significant potential for misunderstanding the new definition for MCI. Patients who are concerned about their memory may request biomarker testing in an effort to determine if they currently have or will acquire Alzheimer disease. Doctors may be tempted to refer patients for biomarker testing (via imaging or lumbar puncture) to “screen” for MCI or Alzheimer disease.
It should be emphasized that MCI itself is still a clinical diagnosis, with the challenges noted above of determining whether there has been a cognitive decline from a prior level of function but preservation of activities of daily living. The biomarkers are not proposed to diagnose MCI, but only to help identify the subset of MCI patients most likely to progress rapidly to Alzheimer disease.
At present, the best use of biomarker testing is to aid research by identifying high-risk people among those with MCI who enroll in prospective trials for testing interventions to reduce the progression of Alzheimer disease.
A number of new studies and guidelines published over the last few years are changing the way we treat older patients. This article summarizes these recent developments in a variety of areas—from prevention of falls to targets for hypertension therapy—relevant to the treatment of geriatric patients.
A MULTICOMPONENT APPROACH TO PREVENTING FALS
The American Geriatrics Society and British Geriatrics Society’s 2010 Clinical Practice Guideline for Prevention of Falls in Older Persons1 has added an important new element since the 2001 guideline: in addition to asking older patients about a fall, clinicians should also ask whether a gait or balance problem has developed.
A complete falls evaluation and multicomponent intervention is indicated for patients who in the past year or since the previous visit have had one fall with an injury or more than one fall, or for patients who report or have been diagnosed with a gait or balance problem. A falls risk assessment is not indicated for a patient with no gait or balance problem and who has had only one noninjurious fall in the previous year that did not require medical attention.
The multicomponent evaluation detailed in the guideline is very thorough and comprises more elements than can be done in a follow-up office visit. In addition to the relevant medical history, physical examination, and cognitive and functional assessment, the fall-risk evaluation includes a falls history, medication review, visual acuity testing, gait and balance assessment, postural and heart-rate evaluation, examination of the feet and footwear, and, if appropriate, a referral for home assessment of environmental hazards.
Intervention consists of many aspects
Of the interventions, exercise has the strongest correlation with falls prevention, and a prescription should include exercises for balance, gait, and strength. Tai chi is specifically recommended.
Medications should be reduced or withdrawn. The previous guideline recommended reducing medications for patients taking four or more medications, but the current guideline applies to everyone.
First cataract removal is associated with reducing the risk of falls.
Postural hypotension should be treated if present.
Vitamin D at 800 U per day is recommended for all elderly people at risk. For elderly people in long-term care, giving vitamin D for proven or suspected deficiency is by itself correlated with risk reduction.
Interventions that by themselves are not associated with risk reduction include education (eg, providing a handout on preventing falls) and having vision checked. For adults who are cognitively impaired, there is insufficient evidence that even the multicomponent intervention helps prevent falls.
CALCIUM AND VITAMIN D MAY NOT BE HARMLESS
Calcium supplements: A cause of heart attack?
Questions have arisen in recent studies about the potential risks of calcium supplementation.
A meta-analysis of 11 trials with nearly 12,000 participants found that the risk of myocardial infarction was significantly higher in people taking calcium supplementation (relative risk 1.27; 95% confidence interval [CI] 1.01–1.59, P = .038).2 Patients were predominantly postmenopausal women and were followed for a mean of 4 years. The incidence of stroke and death were also higher in people who took calcium, but the differences did not reach statistical significance. The dosages were primarily 1,000 mg per day (range 600 mg to 2 g). Risk was independent of age, sex, and type of supplement.
The authors concluded (somewhat provocatively, because only the risk of myocardial infarction reached statistical significance) that if 1,000 people were treated with calcium supplementation for 5 years, 26 fractures would be prevented but 14 myocardial infarctions, 10 strokes, and 13 deaths would be caused.
Comments. These data suggest that physicians may wish to prescribe calcium to supplement (not replace) dietary calcium to help patients reach but not exceed current guidelines for total calcium intake for age and sex. They may also want to advise the patient to take the calcium supplement separately from medications, as indicated in Table 2.
Benefits of vitamin D may depend on dosing
Studies show that the risk of hip fracture can be reduced with modest daily vitamin D supplementation, up to 800 U daily, regardless of calcium intake.3 Some vitamin D dosing regimens, however, may also entail risk.
Sanders et al4 randomized women age 70 and older to receive an annual injection of a high dose of vitamin D (500,000 U) or placebo for 3 to 5 years. Women in the vitamin D group had 15% more falls and 25% more fractures than those in the placebo group. The once-yearly dose of 500,000 U equates to 1,370 U/day, which is not much higher than the recommended daily dosage. The median baseline serum level was 49 nmol/L and reached 120 nmol/L at 30 days in the treatment group, which was not in the toxic range.
Comments. This study cautions physicians against giving large doses of vitamin D at long intervals. Future studies should focus on long-term clinical outcomes of falls and fractures for dosing regimens currently in practice, such as 50,000 units weekly or monthly.
BISPHOSPHONATES AND NONTRAUMATIC THICK BONE FRACTURES
Bisphosphonates have been regarded as the best drugs for preventing hip fracture. But in 2010, the US Food and Drug Administration (FDA) issued a warning that bisphosphonates have been associated with “atypical” femoral fractures. The atypical fracture pattern is a clean break through the thick bone of the shaft that occurs after minimal or no trauma.5 This pattern contrasts with the splintering “typical” fracture in the proximal femur in osteoporotic bone, usually after a fall.
Another characteristic of the atypical fractures is a higher incidence of postoperative complications requiring revision surgery. In more than 14,000 women in secondary analyses of three large randomized bisphosphonate trials, 12 fractures in 10 patients were found that were classified as atypical, averaging to an incidence of 2.3 per 10,000 patient-years.6
A population-based, nested case-control study7 using Canadian pharmacy records evaluated more than 200,000 women at least 68 years old who received bisphosphonate therapy. Of these, 716 (0.35%) sustained an atypical femoral fracture and 9,723 (4.7%) had a typical osteoporotic femoral fracture. Comparing the duration of bisphosphonate use between the two groups, the authors found that the risk of an atypical fracture increased with years of usage (at 5 years or more, the adjusted odds ratio was 2.74, 95% CI 1.25–6.02), but the risk of a typical fracture decreased (at 5 years or more, the adjusted odds ratio was 0.76, 95% CI 0.63–0.93). The study suggests that for every 100 hip fractures that bisphosphonate therapy prevents, it causes one atypical hip fracture.
Comments. These studies have caused some experts to advocate periodic bisphosphonate “vacations,”8 but for how long remains an open question because the risk of a typical fracture will increase. It is possible that a biomarker can help establish the best course, but that has yet to be determined.
DENOSUMAB: A NEW DRUG FOR OSTEOPOROSIS WITH A BIG PRICE TAG
Denosumab (Prolia, Xgeva), a newly available injectable drug, is a monoclonal antibody member of the tumor necrosis factor super-family.9 It is FDA-approved for osteoporosis in postmenopausal women at a dosage of 60 mg every 6 months and for skeletal metastases from solid tumors (120 mg every 4 weeks). It is also being used off-label for skeletal protection in women taking aromatase inhibitors and for men with androgen deficiency.
This drug is expensive, costing $850 per 60-mg dose wholesale, and no data are yet available on its long-term effects.
Since the drug is not cleared via renal mechanisms, there is some hope that it can be used to treat osteoporosis in patients with advanced chronic kidney disease, since bisphosphonates are contraindicated in those with an estimated glomerular filtration rate (GFR) less than 30 to 35 mL/min. However, the major study of denosumab to date, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) study, had no patients with stage 5 chronic kidney disease (GFR < 15 mL/min/1.73 m2 or on dialysis), and too few with stage 4 chronic kidney disease (GFR 15–29) to demonstrate either the safety or efficacy of denosumab in patients with advanced chronic kidney disease.10
HYPERTENSION TREATMENT
A secondary analysis of a recent large hypertension study confirmed the benefits of antihypertensive therapy in very old adults and suggested new targets for systolic and diastolic blood pressures.11,12
The Systolic Hypertension in the Elderly Program (SHEP) trial,13 the Systolic Hypertension in Europe (Syst-Eur) trial,14 and the Hypertension in the Very Elderly Trial (HYVET)15 are the major, randomized, placebo-controlled antihypertensive trials in older adults. They all showed a reduction in the risk of stroke and cardiovascular events. The diuretic studies (SHEP and HYVET)13,15 also showed a lower risk of heart failure and death.
Most recently, secondary analysis of the International Verapamil-Trandolapril (INVEST) study11,12 showed that adults in the oldest groups (age 70–79 and 80 and older), experienced a greater risk of adverse cardiovascular outcomes if systolic blood pressure was lowered to below about 130 mm Hg. As diastolic blood pressure was lowered to about the 65–70 mm Hg range, all age groups in the study experienced an increased risk of cardiovascular events. These results confirm the findings of a secondary analysis of the SHEP trial,16 showing an increased risk of cardiovascular events when diastolic pressure was lowered to below approximately 65 mm Hg.
These studies have been incorporated into 75 pages of the 2011 Expert Consensus Document on Hypertension in the Elderly issued by the American College of Cardiology Foundation and the American Heart Association.17 In a nutshell, the guidelines suggest that older adults less than 80 years of age be treated comparably to middle-aged adults. However, for adults age 80 and older:
- A target for systolic blood pressure of 140 to 145 mm Hg “can be acceptable.”
- Initiating treatment with monotherapy (with a low-dose thiazide, calcium channel blocker, or renin-angiotensin-aldosterone system drug) is reasonable. A second drug may be added if needed.
- Patients should be monitored for “excessive” orthostasis.
- Systolic blood pressure lower than 130 mm Hg and diastolic blood pressure lower than 65 mm Hg should be avoided.
TRANSCATHETER AORTIC VALVE IMPLANTATION APPROVED BY THE FDA
An estimated 2% to 9% of the elderly have aortic stenosis. Aortic valve replacement reduces mortality rates and improves function in all age groups, including octogenarians. Those with asymptomatic aortic stenosis tend to decline very quickly once they develop heart failure, syncope, or angina. Aortic valve replacement has been shown to put people back on the course they were on before they became symptomatic.
Transcatheter self-expanding transaortic valve implantation was approved by the FDA in November 2011. The procedure does not require open surgery and involves angioplasty of the old valve, with the new valve being passed into place through a catheter and expanded. Access is either transfemoral or transapical.
Transaortic valve implantation has been rapidly adopted in Europe since 2002 without any randomized control trials. The Placement of Aortic Transcatheter Valves (PARTNER) trial18 in 2011 was the first randomized trial of this therapy. It was conducted at 25 centers, with nearly 700 patients with severe aortic stenosis randomized to undergo either transcatheter aortic valve replacement with a balloon-expandable valve (244 via the transfemoral and 104 via the transapical approach) or surgical replacement. The mean age of the patients was 84 years, and the Society of Thoracic Surgeons mean score was 12%, indicating high perioperative risk.
At 30 days after the procedure, the rates of death were 3.4% with transcatheter implantation and 6.5% with surgical replacement (P = .07). At 1 year, the rates were 24.2% and 26.8%, respectively (P = 0.44, and P = .001 for noninferiority). However, the rate of major stroke was higher in the transcatheter implantation group: 3.8% vs 2.1% in the surgical group (P = .20) at 1 month and 5.1% vs 2.4% (P = .07) at 1 year. Vascular complications were significantly more frequent in the transcatheter implantation group, and the new onset of atrial fibrillation and major bleeding were significantly higher in the surgical group.
Patients in the transcatheter implantation group had a significantly shorter length of stay in the intensive care unit and a shorter index hospitalization. At 30 days, the transcatheter group also had a significant improvement in New York Heart Association functional status and a better 6-minute walk performance, although at 1 year, these measures were similar between the two groups and were greatly improved over baseline. Quality of life, measured using the Kansas City Cardiomyopathy Questionnaire, was higher both at 6 months and at 1 year in the transcatheter implantation group compared with those who underwent the open surgical procedure.19
Comments. The higher risk of stroke with the transcatheter implantation procedure remains a concern. More evaluation is also needed with respect to function and cognition in the very elderly, and of efficacy and safety in higher- and lower-risk patients.
DEPRESSION CAN BE EFFECTIVELY TREATED WITH MEDICATION
Many placebo-controlled trials have demonstrated the effectiveness of treating depression with medications in elderly people who are cognitively intact and living in the community. A Cochrane Review20 found that in placebo-controlled trials, the number needed to treat to produce one recovery with tricyclic antidepressants, selective serotonin reuptake inhibitors, and monoamine oxidase inhibitors was less than 10 for each of the drug classes.
Since the newer drugs appear to be safer and to have fewer adverse effects than the older drugs, more older adults have been treated with antidepressants, including patients with comorbidities such as dementia that were exclusion criteria in early studies. For example, the number of older adults treated with antidepressants has increased 25% since 1992; at the same time the number being referred for cognitive-based therapies has been reduced by 43%.21 Similar trends are apparent in elderly people in long-term care. In 1999, about one-third of people in long-term care were diagnosed with depression; in 2007 more than one-half were.22
Treating depression is less effective when dementia is present
Up to half of adults age 85 and older living in the community may have dementia. In long-term care facilities, most residents likely have some cognitive impairment or are diagnosed with dementia. Many of these are also taking antidepressive agents.
A review of studies in the Medline and Cochrane registries found seven trials that treated 330 patients with antidepressants for combined depression and dementia. Efficacy was not confirmed.23
After this study was published, Banerjee et al24 treated 218 patients who had depression and dementia in nine centers in the United Kingdom. Patients received sertraline (Zoloft), mirtazapine (Remeron), or placebo. Reductions in depression scores at 13 weeks and at 39 weeks did not differ between the groups, and adverse events were more frequent in the treatment groups than in the placebo groups.
Comments. The poor performance of antidepressants in patients with dementia may be due to misdiagnosis, such as mistaking apathy for depression.25 It is also possible that better criteria than we have now are needed to diagnose depression in patients with dementia, or that current outcome measures are not sensitive for depression when dementia is present.
It may also be unsafe to treat older adults long-term with antidepressive agents. For example, although selective serotonin reuptake inhibitors, the most commonly prescribed antidepressive agents, are considered safe, their side effects are numerous and include sexual dysfunction, bleeding (due to platelet dysfunction), hyponatremia, early weight loss, tremor (mostly with paroxetine [Paxil]), sedation, apathy (especially with high doses), loose stools (with sertraline), urinary incontinence, falls, bone loss, and QTc prolongation.
Citalopram: Maximum dosage in elderly
In August 2011, an FDA Safety Communication was issued for citalopram (Celexa), stating that the daily dose should not exceed 40 mg in the general population and should not exceed 20 mg in patients age 60 and older. The dose should also not exceed 20 mg for a patient at any age who has hepatic impairment, who is known to be a poor metabolizer of CYP 2C19, or who takes cimetidine (Tagamet), since that drug inhibits the metabolism of citalopram at the CYP 2C19 enzyme site.
Although the FDA warning specifically mentions only cimetidine, physicians may have concerns about other drugs that inhibit CYP 2C19, such as proton pump inhibitors (eg, omeprazole [Prilosec]) when taken concomitantly with citalopram. Also, escitalopram (Lexapro) and sertraline are quite similar to citalopram; although they were not mentioned in the FDA Safety Communication, higher doses of these drugs may put patients at similar risk.
ALZHEIMER DISEASE: NEED TO BETTER IDENTIFY PEOPLE AT RISK
The definition of dementia is essentially the presence of a cognitive problem that affects the ability to function. For people with Alzheimer disease, impairment of cognitive performance precedes functional decline. Those with a cognitive deficit who still function well have, by definition, mild cognitive impairment (MCI). Although MCI could be caused by a variety of vascular and other neurologic processes, the most common cause of MCI in the United States is Alzheimer disease.
Unfortunately, the population with MCI currently enrolled in clinical trials to reduce the risk of progression to Alzheimer disease is heterogeneous. Many study participants may never get dementia, and others may have had the pathology present for decades and are progressing rapidly. Imaging and biomarkers are emerging as good indicators that predict progression and could help to better define populations for clinical trials.26
Studies now indicate that people with MCI that is ultimately due to Alzheimer disease are likely to have amyloid beta peptide 42 evident in the cerebrospinal fluid 10 to 20 years before symptoms arise. At the same time, amyloid is also likely to be evident in the brain with amyloid-imaging positron emission tomography (PET). Some time later, abnormalities in metabolism are also evident on fluorodeoxyglucose (FDG) PET, as are changes such as reduced hippocampal volume on magnetic resonance imaging (MRI).
The 1984 criteria for diagnosing MCI due to Alzheimer disease were recently revised to incorporate the evolving availability of biomarkers.27,28 The diagnosis of MCI itself is still based on clinical ascertainment including history, physical examination, and cognitive testing. It requires diagnosis of a cognitive decline from a prior level but maintenance of activities of daily living with no or minimal assistance. This diagnosis is certainly challenging since it requires ascertainment of a prior level of function and corroboration, when feasible, with an informant. Blood tests and imaging, which are readily available, constitute an important part of the assessment.
Attributing the MCI to Alzheimer disease requires consistency of the disease course—a gradual decline in Alzheimer disease, rather than a stroke, head injury, neurologic disease such as Parkinson disease, or mixed causes.
Knowledge of genetic factors, such as the presence of a mutation in APP, PS1, or PS2, can be predictive with young patients. The presence of one or two 34 alleles in the apolipoprotein E (APOE) gene is the only genetic variant broadly accepted as increasing the risk for late-onset Alzheimer dementia, whereas the 32 allele decreases risk.
Refining the risk attribution to Alzheimer disease requires biomarkers, currently available only in research settings:
- High likelihood—amyloid beta peptide detected by PET or cerebrospinal fluid analysis and evidence of neuronal degeneration or injury (elevated tau in the cerebrospinal fluid, decreased FDG uptake on PET, and atrophy evident by structural MRI)
- Intermediate likelihood—presence of amyloid beta peptide or evidence of neuronal degeneration or injury
- Unlikely—biomarkers tested and negative
- No comment—biomarkers not tested or reporting is indeterminate.
Comments. There is significant potential for misunderstanding the new definition for MCI. Patients who are concerned about their memory may request biomarker testing in an effort to determine if they currently have or will acquire Alzheimer disease. Doctors may be tempted to refer patients for biomarker testing (via imaging or lumbar puncture) to “screen” for MCI or Alzheimer disease.
It should be emphasized that MCI itself is still a clinical diagnosis, with the challenges noted above of determining whether there has been a cognitive decline from a prior level of function but preservation of activities of daily living. The biomarkers are not proposed to diagnose MCI, but only to help identify the subset of MCI patients most likely to progress rapidly to Alzheimer disease.
At present, the best use of biomarker testing is to aid research by identifying high-risk people among those with MCI who enroll in prospective trials for testing interventions to reduce the progression of Alzheimer disease.
- Panel on Prevention of Falls in Older Persons, American Geriatrics Society and British Geriatrics Society. J Am Geriatr Soc 2011; 59:148–157.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: metaanalysis. BMJ 2010; 341:c3691.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women. A randomized controlled trial. JAMA 2010; 303:1815–1822.
- Kuehn BM. Prolonged bisphosphonate use linked to rare fractures, esophageal cancer. JAMA 2010; 304:2114–2115.
- Black DM, Kelly MP, Genant HK, et al; Fracture Intervention Trial Steering Committee; HORIZON Pivotal Fracture Trial Steering Committee. Bisphosphonates and fractures of the subtrochanteric or diaphyseal femur. N Engl J Med 2010; 362:1761–1771.
- Park-Wyllie LY, Mamdani MM, Juurlink DN, et al. Bisphosphonate use and the risk of subtrochanteric or femoral shaft fractures in older women. JAMA 2011; 305:783–789.
- Ott SM. What is the optimal duration of bisphosphonate therapy? Cleve Clin J Med 2011; 78:619–630.
- Cummings SR, San Martin J, McClung MR, et al; for the FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Pepine CJ, Handberg EM, Cooper-Dehoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Denardo SJ, Gong Y, Nichols WW, et al. Blood pressure and outcomes in very old hypertensive coronary artery disease patients: an INVEST substudy. Am J Med 2010; 123:719–726.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al; for the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension (erratum published in Lancet 1997; 350:1636). Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; for the HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Somes G, Pahor M, Shorr R, Cushman WC, Applegate WB. The role of diastolic blood pressure when treating isolated systolic hypertension. Arch Intern Med 1999; 159:2004–2009.
- Aronow WS, Fleg JL, Pepine CJ, et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly. J Am Coll Cardiol 2011; 57:2037–2114.
- Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
- Reynolds MR, Magnuson EA, Lei Y, et al; Placement of Aortic Transcatheter Valves (PARTNER) Investigators. Health-related quality of life after transcatheter aortic valve replacement in inoperable patients with severe aortic stenosis. Circulation 2011; 124:1964–1972.
- Wilson K, Mottram P, Sivanranthan A, Nightingale A. Antidepressant versus placebo for depressed elderly. Cochrane Database Syst Rev 2001;(2):CD000561.
- Akincigil A, Olfson M, Walkup JT, et al. Diagnosis and treatment of depression in older community-dwelling adults: 1992–2005. J Am Geriatr Soc 2011; 59:1042–1051.
- Gaboda D, Lucas J, Siegel M, Kalay E, Crystal S. No longer undertreated? Depression diagnosis and antidepressant therapy in elderly long-stay nursing home residents, 1999 to 2007. J Am Geriatr Soc 2011; 59:673–680.
- Nelson JC, Devanand DP. A systematic review and meta-analysis of placebo-controlled antidepressant studies in peoloe with depression and dementia. J Am Geriatr Soc 2011; 59:577–585.
- Banerjee S, Hellier J, Dewey M, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet 2011; 378:403–411.
- Landes AM, Sperry SD, Strauss ME, Geldmacher DS. Apathy in Alzheimer’s disease. J Am Geriatr Soc 2001; 49:1700–1707.
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010; 9:1118–1127.
- Daviglus ML, Bell CC, Berrettini W, et al. National Institutes of Health State-of-the-Science Conference statement: preventing Alzheimer disease and cognitive decline. Ann Intern Med 2010; 153:176–181.
- McKhann GM, Knopman DS, Chertkow H, et al The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269.
- Panel on Prevention of Falls in Older Persons, American Geriatrics Society and British Geriatrics Society. J Am Geriatr Soc 2011; 59:148–157.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: metaanalysis. BMJ 2010; 341:c3691.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women. A randomized controlled trial. JAMA 2010; 303:1815–1822.
- Kuehn BM. Prolonged bisphosphonate use linked to rare fractures, esophageal cancer. JAMA 2010; 304:2114–2115.
- Black DM, Kelly MP, Genant HK, et al; Fracture Intervention Trial Steering Committee; HORIZON Pivotal Fracture Trial Steering Committee. Bisphosphonates and fractures of the subtrochanteric or diaphyseal femur. N Engl J Med 2010; 362:1761–1771.
- Park-Wyllie LY, Mamdani MM, Juurlink DN, et al. Bisphosphonate use and the risk of subtrochanteric or femoral shaft fractures in older women. JAMA 2011; 305:783–789.
- Ott SM. What is the optimal duration of bisphosphonate therapy? Cleve Clin J Med 2011; 78:619–630.
- Cummings SR, San Martin J, McClung MR, et al; for the FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Pepine CJ, Handberg EM, Cooper-Dehoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Denardo SJ, Gong Y, Nichols WW, et al. Blood pressure and outcomes in very old hypertensive coronary artery disease patients: an INVEST substudy. Am J Med 2010; 123:719–726.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al; for the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension (erratum published in Lancet 1997; 350:1636). Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; for the HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Somes G, Pahor M, Shorr R, Cushman WC, Applegate WB. The role of diastolic blood pressure when treating isolated systolic hypertension. Arch Intern Med 1999; 159:2004–2009.
- Aronow WS, Fleg JL, Pepine CJ, et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly. J Am Coll Cardiol 2011; 57:2037–2114.
- Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
- Reynolds MR, Magnuson EA, Lei Y, et al; Placement of Aortic Transcatheter Valves (PARTNER) Investigators. Health-related quality of life after transcatheter aortic valve replacement in inoperable patients with severe aortic stenosis. Circulation 2011; 124:1964–1972.
- Wilson K, Mottram P, Sivanranthan A, Nightingale A. Antidepressant versus placebo for depressed elderly. Cochrane Database Syst Rev 2001;(2):CD000561.
- Akincigil A, Olfson M, Walkup JT, et al. Diagnosis and treatment of depression in older community-dwelling adults: 1992–2005. J Am Geriatr Soc 2011; 59:1042–1051.
- Gaboda D, Lucas J, Siegel M, Kalay E, Crystal S. No longer undertreated? Depression diagnosis and antidepressant therapy in elderly long-stay nursing home residents, 1999 to 2007. J Am Geriatr Soc 2011; 59:673–680.
- Nelson JC, Devanand DP. A systematic review and meta-analysis of placebo-controlled antidepressant studies in peoloe with depression and dementia. J Am Geriatr Soc 2011; 59:577–585.
- Banerjee S, Hellier J, Dewey M, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet 2011; 378:403–411.
- Landes AM, Sperry SD, Strauss ME, Geldmacher DS. Apathy in Alzheimer’s disease. J Am Geriatr Soc 2001; 49:1700–1707.
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010; 9:1118–1127.
- Daviglus ML, Bell CC, Berrettini W, et al. National Institutes of Health State-of-the-Science Conference statement: preventing Alzheimer disease and cognitive decline. Ann Intern Med 2010; 153:176–181.
- McKhann GM, Knopman DS, Chertkow H, et al The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269.
KEY POINTS
- To prevent falls, patients should be asked not only about recent falls but about balance. Referral for a multicomponent falls evaluation should be considered.
- For patients age 80 and older, a target systolic blood pressure of 140 to 145 mm Hg is acceptable, and blood pressure below 130 mm Hg systolic and 65 mm Hg diastolic should be avoided.
- The dosage of the antidepressant citalopram (Celexa) should not exceed 40 mg per day in the general population and 20 mg in patients age 60 and older.
- Calcium supplementation may increase the risk of myocardial infarction and stroke. A large annual dose of vitamin D appears harmful, raising questions about the long-term safety of large doses given weekly or monthly.