User login
CDC warns of early uptick in respiratory disease
The Centers for Disease Control and Prevention is warning of an early surge in respiratory disease caused by multiple viruses. As influenza viruses, respiratory syncytial virus (RSV), SARS-CoV-2, and rhinovirus/enterovirus simultaneously circulate, the agency cautioned that this confluence of viral activity could strain the health care system, according to a CDC Health Network Alert advisory issued Nov. 4.
“This early increase in disease incidence highlights the importance of optimizing respiratory virus prevention and treatment measures, including prompt vaccination and antiviral treatment,” the alert stated.
The CDC reports that RSV activity is increasing nationally, but in some areas – such as the South and Mountain West – cases appear to be trending downward.
Influenza cases continue to climb, with the virus activity being the highest in the South, Mid-Atlantic, and the south-central West Coast, according to CDC data. “In fact, we’re seeing the highest influenza hospitalization rates going back a decade,” said José Romero, MD, director of the CDC’s National Center for Immunization and Respiratory Diseases, during a press briefing. The agency estimates that there have been 1.6 million illnesses, 13,000 hospitalizations, and 730 deaths from the flu so far this season. As of Nov. 4, there have been two pediatric deaths.
COVID-19 cases appear to have plateaued in the past three weeks, Dr. Romero said; however, the CDC expects that there will be “high-level circulation of SARS-CoV-2 this fall and winter,” the health alert stated.
The CDC advised that all eligible individuals aged 6-months or older should be vaccinated against COVID-19 and influenza. To protect against RSV-hospitalization, high-risk children should receive the monoclonal antibody drug palivizumab (Synagis). High-risk children include infants born before 29 weeks, children younger than age 2 with chronic lung disease or hemodynamically significant congenital heart disease, and children with suppressed immune systems or neuromuscular disorders.
Any patient with confirmed or suspected flu who is hospitalized, at higher risk for influenza complications, or who has a severe or progressive illness should be treated as early as possible with antivirals, such as oral oseltamivir (Tamiflu).
Patients with confirmed SARS-CoV-2 infection with increased risk of complications should also be treated with antivirals, such as nirmatrelvir and ritonavir (Paxlovid) or remdesivir (Veklury).
Patients should also be reminded to wash their hands frequently, cover coughs and sneezes, stay home when sick, and avoid close contact with people who are sick, the CDC advised.
“There’s no doubt that we will face some challenges this winter,” said Dawn O’Connell, HHS Assistant Secretary for Preparedness and Response, “but it’s important to remember that RSV and flu are not new, and we have safe and effective vaccines for COVID-19 and the flu.”
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention is warning of an early surge in respiratory disease caused by multiple viruses. As influenza viruses, respiratory syncytial virus (RSV), SARS-CoV-2, and rhinovirus/enterovirus simultaneously circulate, the agency cautioned that this confluence of viral activity could strain the health care system, according to a CDC Health Network Alert advisory issued Nov. 4.
“This early increase in disease incidence highlights the importance of optimizing respiratory virus prevention and treatment measures, including prompt vaccination and antiviral treatment,” the alert stated.
The CDC reports that RSV activity is increasing nationally, but in some areas – such as the South and Mountain West – cases appear to be trending downward.
Influenza cases continue to climb, with the virus activity being the highest in the South, Mid-Atlantic, and the south-central West Coast, according to CDC data. “In fact, we’re seeing the highest influenza hospitalization rates going back a decade,” said José Romero, MD, director of the CDC’s National Center for Immunization and Respiratory Diseases, during a press briefing. The agency estimates that there have been 1.6 million illnesses, 13,000 hospitalizations, and 730 deaths from the flu so far this season. As of Nov. 4, there have been two pediatric deaths.
COVID-19 cases appear to have plateaued in the past three weeks, Dr. Romero said; however, the CDC expects that there will be “high-level circulation of SARS-CoV-2 this fall and winter,” the health alert stated.
The CDC advised that all eligible individuals aged 6-months or older should be vaccinated against COVID-19 and influenza. To protect against RSV-hospitalization, high-risk children should receive the monoclonal antibody drug palivizumab (Synagis). High-risk children include infants born before 29 weeks, children younger than age 2 with chronic lung disease or hemodynamically significant congenital heart disease, and children with suppressed immune systems or neuromuscular disorders.
Any patient with confirmed or suspected flu who is hospitalized, at higher risk for influenza complications, or who has a severe or progressive illness should be treated as early as possible with antivirals, such as oral oseltamivir (Tamiflu).
Patients with confirmed SARS-CoV-2 infection with increased risk of complications should also be treated with antivirals, such as nirmatrelvir and ritonavir (Paxlovid) or remdesivir (Veklury).
Patients should also be reminded to wash their hands frequently, cover coughs and sneezes, stay home when sick, and avoid close contact with people who are sick, the CDC advised.
“There’s no doubt that we will face some challenges this winter,” said Dawn O’Connell, HHS Assistant Secretary for Preparedness and Response, “but it’s important to remember that RSV and flu are not new, and we have safe and effective vaccines for COVID-19 and the flu.”
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention is warning of an early surge in respiratory disease caused by multiple viruses. As influenza viruses, respiratory syncytial virus (RSV), SARS-CoV-2, and rhinovirus/enterovirus simultaneously circulate, the agency cautioned that this confluence of viral activity could strain the health care system, according to a CDC Health Network Alert advisory issued Nov. 4.
“This early increase in disease incidence highlights the importance of optimizing respiratory virus prevention and treatment measures, including prompt vaccination and antiviral treatment,” the alert stated.
The CDC reports that RSV activity is increasing nationally, but in some areas – such as the South and Mountain West – cases appear to be trending downward.
Influenza cases continue to climb, with the virus activity being the highest in the South, Mid-Atlantic, and the south-central West Coast, according to CDC data. “In fact, we’re seeing the highest influenza hospitalization rates going back a decade,” said José Romero, MD, director of the CDC’s National Center for Immunization and Respiratory Diseases, during a press briefing. The agency estimates that there have been 1.6 million illnesses, 13,000 hospitalizations, and 730 deaths from the flu so far this season. As of Nov. 4, there have been two pediatric deaths.
COVID-19 cases appear to have plateaued in the past three weeks, Dr. Romero said; however, the CDC expects that there will be “high-level circulation of SARS-CoV-2 this fall and winter,” the health alert stated.
The CDC advised that all eligible individuals aged 6-months or older should be vaccinated against COVID-19 and influenza. To protect against RSV-hospitalization, high-risk children should receive the monoclonal antibody drug palivizumab (Synagis). High-risk children include infants born before 29 weeks, children younger than age 2 with chronic lung disease or hemodynamically significant congenital heart disease, and children with suppressed immune systems or neuromuscular disorders.
Any patient with confirmed or suspected flu who is hospitalized, at higher risk for influenza complications, or who has a severe or progressive illness should be treated as early as possible with antivirals, such as oral oseltamivir (Tamiflu).
Patients with confirmed SARS-CoV-2 infection with increased risk of complications should also be treated with antivirals, such as nirmatrelvir and ritonavir (Paxlovid) or remdesivir (Veklury).
Patients should also be reminded to wash their hands frequently, cover coughs and sneezes, stay home when sick, and avoid close contact with people who are sick, the CDC advised.
“There’s no doubt that we will face some challenges this winter,” said Dawn O’Connell, HHS Assistant Secretary for Preparedness and Response, “but it’s important to remember that RSV and flu are not new, and we have safe and effective vaccines for COVID-19 and the flu.”
A version of this article first appeared on Medscape.com.
FDA expands tenofovir alafenamide (Vemlidy) use to adolescents with chronic HBV
the drug’s manufacturer has announced.
The approval in the pediatric patient population was supported by 24-week data from a phase 2 clinical trial comparing treatment with tenofovir alafenamide (25 mg once daily) with placebo in 70 treatment-naive and treatment-experienced patients aged 12-18 years weighing at least 35 kg.
The study met its primary endpoint of percentage of patients with HBV DNA levels less than 20 IU/mL at 24 weeks of therapy, Gilead Sciences said in a press release.
Overall, 10 of 47 (21%) patients treated with tenofovir alafenamide achieved HBV DNA less than 20 IU/mL at 24 weeks, compared with 0 of 23 (0%) treated with placebo.
The rates of serum ALT normalization were higher with tenofovir alafenamide than with placebo (44% vs 0%).
The mean percent changes in bone mineral density (BMD) from baseline to 24 weeks were numerically similar for tenofovir alafenamide– and placebo-treated patients (2.4% and 1.9% for lumbar spine, and 1.5% and 1.9% for whole body, respectively).
The mean changes from baseline BMD z scores were –0.03 and –0.09 for lumbar spine, and –0.05 and –0.01 for whole body, for tenofovir alafenamide and placebo groups, respectively.
The FDA initially approved the nucleoside analog reverse transcriptase inhibitor in 2016 for adults with chronic HBV.
The drug was approved in Europe in 2017 for chronic HBV infection in adults and adolescents aged 12 years and older weighing at least 35 kg.
Tenofovir alafenamide carries a boxed warning citing risks for lactic acidosis/severe hepatomegaly with steatosis and posttreatment severe acute exacerbation of HBV.
A version of this article first appeared on Medscape.com.
the drug’s manufacturer has announced.
The approval in the pediatric patient population was supported by 24-week data from a phase 2 clinical trial comparing treatment with tenofovir alafenamide (25 mg once daily) with placebo in 70 treatment-naive and treatment-experienced patients aged 12-18 years weighing at least 35 kg.
The study met its primary endpoint of percentage of patients with HBV DNA levels less than 20 IU/mL at 24 weeks of therapy, Gilead Sciences said in a press release.
Overall, 10 of 47 (21%) patients treated with tenofovir alafenamide achieved HBV DNA less than 20 IU/mL at 24 weeks, compared with 0 of 23 (0%) treated with placebo.
The rates of serum ALT normalization were higher with tenofovir alafenamide than with placebo (44% vs 0%).
The mean percent changes in bone mineral density (BMD) from baseline to 24 weeks were numerically similar for tenofovir alafenamide– and placebo-treated patients (2.4% and 1.9% for lumbar spine, and 1.5% and 1.9% for whole body, respectively).
The mean changes from baseline BMD z scores were –0.03 and –0.09 for lumbar spine, and –0.05 and –0.01 for whole body, for tenofovir alafenamide and placebo groups, respectively.
The FDA initially approved the nucleoside analog reverse transcriptase inhibitor in 2016 for adults with chronic HBV.
The drug was approved in Europe in 2017 for chronic HBV infection in adults and adolescents aged 12 years and older weighing at least 35 kg.
Tenofovir alafenamide carries a boxed warning citing risks for lactic acidosis/severe hepatomegaly with steatosis and posttreatment severe acute exacerbation of HBV.
A version of this article first appeared on Medscape.com.
the drug’s manufacturer has announced.
The approval in the pediatric patient population was supported by 24-week data from a phase 2 clinical trial comparing treatment with tenofovir alafenamide (25 mg once daily) with placebo in 70 treatment-naive and treatment-experienced patients aged 12-18 years weighing at least 35 kg.
The study met its primary endpoint of percentage of patients with HBV DNA levels less than 20 IU/mL at 24 weeks of therapy, Gilead Sciences said in a press release.
Overall, 10 of 47 (21%) patients treated with tenofovir alafenamide achieved HBV DNA less than 20 IU/mL at 24 weeks, compared with 0 of 23 (0%) treated with placebo.
The rates of serum ALT normalization were higher with tenofovir alafenamide than with placebo (44% vs 0%).
The mean percent changes in bone mineral density (BMD) from baseline to 24 weeks were numerically similar for tenofovir alafenamide– and placebo-treated patients (2.4% and 1.9% for lumbar spine, and 1.5% and 1.9% for whole body, respectively).
The mean changes from baseline BMD z scores were –0.03 and –0.09 for lumbar spine, and –0.05 and –0.01 for whole body, for tenofovir alafenamide and placebo groups, respectively.
The FDA initially approved the nucleoside analog reverse transcriptase inhibitor in 2016 for adults with chronic HBV.
The drug was approved in Europe in 2017 for chronic HBV infection in adults and adolescents aged 12 years and older weighing at least 35 kg.
Tenofovir alafenamide carries a boxed warning citing risks for lactic acidosis/severe hepatomegaly with steatosis and posttreatment severe acute exacerbation of HBV.
A version of this article first appeared on Medscape.com.
Mid-October flulike illness cases higher than past 5 years
Outpatient visits for influenzalike illness (ILI), which includes influenza, SARS-CoV-2, and RSV, were higher after 3 weeks than for any of the previous five flu seasons: 3.3% of visits reported through the CDC’s Outpatient Influenza-like Illness Surveillance Network involved ILI as of Oct. 22. The highest comparable rate in the previous 5 years was the 1.9% recorded in late October of 2021, shortly after the definition of ILI was changed to also include illnesses other than influenza.
This season’s higher flu activity is in contrast to the previous two, which were unusually mild. The change, however, is not unexpected, as William Schaffner, MD, an infectious disease expert and professor of preventive medicine at Vanderbilt University, recently told CNN.
“Here we are in the middle of October – not the middle of November – we’re already seeing scattered influenza cases, even hospitalized influenza cases, around the country,” he said. “So we know that this virus is now spreading out in the community already. It’s gathering speed already. It looks to me to be about a month early.”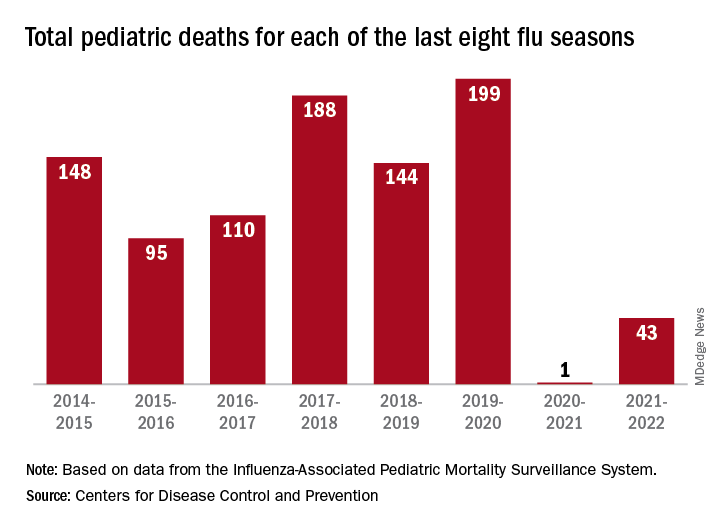
One indication of the mildness of the previous two flu seasons was the number of deaths, both pediatric and overall. Influenza-associated pediatric deaths had averaged about 110 per season over the previous eight seasons, compared with just 1 for 2020-2021 and 43 in 2021-2022. Overall flu deaths never reached 1% of all weekly deaths for either season, well below baseline levels for the flu, which range from 5.5% to 6.8%, CDC data show.
Other indicators of early severity
This season’s early rise in viral activity also can be seen in hospitalizations. The cumulative rate of flu-related admissions was 1.5 per 100,000 population as of Oct. 22, higher than the rate observed in the comparable week of previous seasons going back to 2010-2011, according to the CDC’s Influenza Hospitalization Surveillance Network.
A look at state reports of ILI outpatient visit rates shows that the District of Columbia and South Carolina are already in the very high range of the CDC’s severity scale, while 11 states are in the high range. Again going back to 2010-2011, no jurisdiction has ever been in the very high range this early in the season, based on data from the Outpatient Influenza-like Illness Surveillance Network.
Outpatient visits for influenzalike illness (ILI), which includes influenza, SARS-CoV-2, and RSV, were higher after 3 weeks than for any of the previous five flu seasons: 3.3% of visits reported through the CDC’s Outpatient Influenza-like Illness Surveillance Network involved ILI as of Oct. 22. The highest comparable rate in the previous 5 years was the 1.9% recorded in late October of 2021, shortly after the definition of ILI was changed to also include illnesses other than influenza.
This season’s higher flu activity is in contrast to the previous two, which were unusually mild. The change, however, is not unexpected, as William Schaffner, MD, an infectious disease expert and professor of preventive medicine at Vanderbilt University, recently told CNN.
“Here we are in the middle of October – not the middle of November – we’re already seeing scattered influenza cases, even hospitalized influenza cases, around the country,” he said. “So we know that this virus is now spreading out in the community already. It’s gathering speed already. It looks to me to be about a month early.”
One indication of the mildness of the previous two flu seasons was the number of deaths, both pediatric and overall. Influenza-associated pediatric deaths had averaged about 110 per season over the previous eight seasons, compared with just 1 for 2020-2021 and 43 in 2021-2022. Overall flu deaths never reached 1% of all weekly deaths for either season, well below baseline levels for the flu, which range from 5.5% to 6.8%, CDC data show.
Other indicators of early severity
This season’s early rise in viral activity also can be seen in hospitalizations. The cumulative rate of flu-related admissions was 1.5 per 100,000 population as of Oct. 22, higher than the rate observed in the comparable week of previous seasons going back to 2010-2011, according to the CDC’s Influenza Hospitalization Surveillance Network.
A look at state reports of ILI outpatient visit rates shows that the District of Columbia and South Carolina are already in the very high range of the CDC’s severity scale, while 11 states are in the high range. Again going back to 2010-2011, no jurisdiction has ever been in the very high range this early in the season, based on data from the Outpatient Influenza-like Illness Surveillance Network.
Outpatient visits for influenzalike illness (ILI), which includes influenza, SARS-CoV-2, and RSV, were higher after 3 weeks than for any of the previous five flu seasons: 3.3% of visits reported through the CDC’s Outpatient Influenza-like Illness Surveillance Network involved ILI as of Oct. 22. The highest comparable rate in the previous 5 years was the 1.9% recorded in late October of 2021, shortly after the definition of ILI was changed to also include illnesses other than influenza.
This season’s higher flu activity is in contrast to the previous two, which were unusually mild. The change, however, is not unexpected, as William Schaffner, MD, an infectious disease expert and professor of preventive medicine at Vanderbilt University, recently told CNN.
“Here we are in the middle of October – not the middle of November – we’re already seeing scattered influenza cases, even hospitalized influenza cases, around the country,” he said. “So we know that this virus is now spreading out in the community already. It’s gathering speed already. It looks to me to be about a month early.”
One indication of the mildness of the previous two flu seasons was the number of deaths, both pediatric and overall. Influenza-associated pediatric deaths had averaged about 110 per season over the previous eight seasons, compared with just 1 for 2020-2021 and 43 in 2021-2022. Overall flu deaths never reached 1% of all weekly deaths for either season, well below baseline levels for the flu, which range from 5.5% to 6.8%, CDC data show.
Other indicators of early severity
This season’s early rise in viral activity also can be seen in hospitalizations. The cumulative rate of flu-related admissions was 1.5 per 100,000 population as of Oct. 22, higher than the rate observed in the comparable week of previous seasons going back to 2010-2011, according to the CDC’s Influenza Hospitalization Surveillance Network.
A look at state reports of ILI outpatient visit rates shows that the District of Columbia and South Carolina are already in the very high range of the CDC’s severity scale, while 11 states are in the high range. Again going back to 2010-2011, no jurisdiction has ever been in the very high range this early in the season, based on data from the Outpatient Influenza-like Illness Surveillance Network.
Children and COVID: Weekly cases can’t sustain downward trend
New COVID-19 cases in children inched up in late October, just 1 week after dipping to their lowest level in more than a year, and some measures of pediatric emergency visits and hospital admissions rose as well.
There was an 8% increase in the number of cases for the week of Oct. 21-27, compared with the previous week, but this week’s total was still below 25,000, and the overall trend since the beginning of September is still one of decline, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.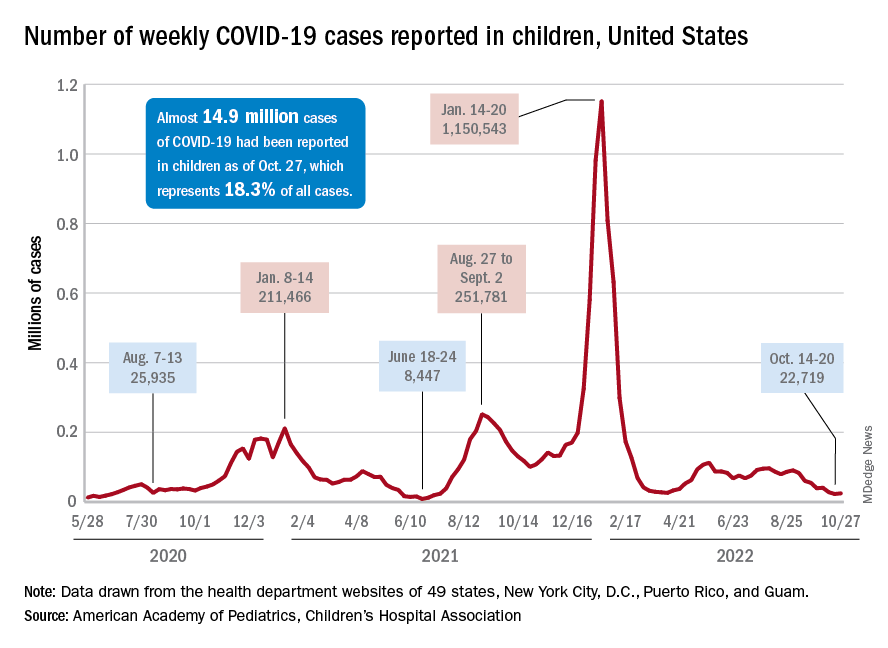
A similar increase can be seen for hospitalizations with confirmed COVID. The rate for children aged 0-17 years fell from 0.44 admissions per 100,000 population at the end of August to 0.16 per 100,000 on Oct. 23. Hospitalizations have since ticked up to 0.17 per 100,000, according to the Centers for Disease Control and Prevention.
Emergency department visits with diagnosed COVID among children aged 16-17 years, as a percentage of all ED visits, rose from 0.6% on Oct. 21 to 0.8% on Oct. 26. ED visits for 12- to 15-year-olds rose from 0.6% to 0.7% at about the same time, with both increases coming after declines that started in late August. No such increase has occurred yet among children aged 0-11 years, the CDC reported on its COVID Data Tracker.
One small milestone reached in the past week involved the proportion of all COVID cases that have occurred in children. The total number of child cases as of Oct. 27 was almost 14.9 million, which represents 18.3% of cases in all Americans, according to the AAP and CHA. That figure had been sitting at 18.4% since mid-August after reaching as high as 19.0% during the spring.
The CDC puts total COVID-related hospital admissions for children aged 0-17 at 163,588 since Aug. 1, 2020, which is 3.0% of all U.S. admissions. Total pediatric deaths number 1,843, or just about 0.2% of all COVID-related fatalities since the start of the pandemic, the CDC data show.
The latest vaccination figures show that 71.3% of children aged 12-17 years have received at least one dose, as have 38.8% of 5- to 11-year-olds, 8.4% of 2- to 4-year-olds, and 5.5% of those under age 2. Full vaccination by age group looks like this: 60.9% (12-17 years), 31.7% (5-11 years), 3.7% (2-4 years), and 2.1% (<2 years), the CDC reported. Almost 30% of children aged 12-17 have gotten a first booster dose, as have 16% of 5- to 11-year-olds.
New COVID-19 cases in children inched up in late October, just 1 week after dipping to their lowest level in more than a year, and some measures of pediatric emergency visits and hospital admissions rose as well.
There was an 8% increase in the number of cases for the week of Oct. 21-27, compared with the previous week, but this week’s total was still below 25,000, and the overall trend since the beginning of September is still one of decline, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
A similar increase can be seen for hospitalizations with confirmed COVID. The rate for children aged 0-17 years fell from 0.44 admissions per 100,000 population at the end of August to 0.16 per 100,000 on Oct. 23. Hospitalizations have since ticked up to 0.17 per 100,000, according to the Centers for Disease Control and Prevention.
Emergency department visits with diagnosed COVID among children aged 16-17 years, as a percentage of all ED visits, rose from 0.6% on Oct. 21 to 0.8% on Oct. 26. ED visits for 12- to 15-year-olds rose from 0.6% to 0.7% at about the same time, with both increases coming after declines that started in late August. No such increase has occurred yet among children aged 0-11 years, the CDC reported on its COVID Data Tracker.
One small milestone reached in the past week involved the proportion of all COVID cases that have occurred in children. The total number of child cases as of Oct. 27 was almost 14.9 million, which represents 18.3% of cases in all Americans, according to the AAP and CHA. That figure had been sitting at 18.4% since mid-August after reaching as high as 19.0% during the spring.
The CDC puts total COVID-related hospital admissions for children aged 0-17 at 163,588 since Aug. 1, 2020, which is 3.0% of all U.S. admissions. Total pediatric deaths number 1,843, or just about 0.2% of all COVID-related fatalities since the start of the pandemic, the CDC data show.
The latest vaccination figures show that 71.3% of children aged 12-17 years have received at least one dose, as have 38.8% of 5- to 11-year-olds, 8.4% of 2- to 4-year-olds, and 5.5% of those under age 2. Full vaccination by age group looks like this: 60.9% (12-17 years), 31.7% (5-11 years), 3.7% (2-4 years), and 2.1% (<2 years), the CDC reported. Almost 30% of children aged 12-17 have gotten a first booster dose, as have 16% of 5- to 11-year-olds.
New COVID-19 cases in children inched up in late October, just 1 week after dipping to their lowest level in more than a year, and some measures of pediatric emergency visits and hospital admissions rose as well.
There was an 8% increase in the number of cases for the week of Oct. 21-27, compared with the previous week, but this week’s total was still below 25,000, and the overall trend since the beginning of September is still one of decline, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
A similar increase can be seen for hospitalizations with confirmed COVID. The rate for children aged 0-17 years fell from 0.44 admissions per 100,000 population at the end of August to 0.16 per 100,000 on Oct. 23. Hospitalizations have since ticked up to 0.17 per 100,000, according to the Centers for Disease Control and Prevention.
Emergency department visits with diagnosed COVID among children aged 16-17 years, as a percentage of all ED visits, rose from 0.6% on Oct. 21 to 0.8% on Oct. 26. ED visits for 12- to 15-year-olds rose from 0.6% to 0.7% at about the same time, with both increases coming after declines that started in late August. No such increase has occurred yet among children aged 0-11 years, the CDC reported on its COVID Data Tracker.
One small milestone reached in the past week involved the proportion of all COVID cases that have occurred in children. The total number of child cases as of Oct. 27 was almost 14.9 million, which represents 18.3% of cases in all Americans, according to the AAP and CHA. That figure had been sitting at 18.4% since mid-August after reaching as high as 19.0% during the spring.
The CDC puts total COVID-related hospital admissions for children aged 0-17 at 163,588 since Aug. 1, 2020, which is 3.0% of all U.S. admissions. Total pediatric deaths number 1,843, or just about 0.2% of all COVID-related fatalities since the start of the pandemic, the CDC data show.
The latest vaccination figures show that 71.3% of children aged 12-17 years have received at least one dose, as have 38.8% of 5- to 11-year-olds, 8.4% of 2- to 4-year-olds, and 5.5% of those under age 2. Full vaccination by age group looks like this: 60.9% (12-17 years), 31.7% (5-11 years), 3.7% (2-4 years), and 2.1% (<2 years), the CDC reported. Almost 30% of children aged 12-17 have gotten a first booster dose, as have 16% of 5- to 11-year-olds.
Children and COVID: Weekly cases fall to lowest level in over a year
With the third autumn of the COVID era now upon us, the discussion has turned again to a possible influenza/COVID twindemic, as well as the new-for-2022 influenza/COVID/respiratory syncytial virus tripledemic. It appears, however, that COVID may have missed the memo.
For the sixth time in the last 7 weeks, the number of new COVID cases in children fell, with just under 23,000 reported during the week of Oct. 14-20, according to the American Academy of Pediatrics and the Children’s Hospital Association. That is the lowest weekly count so far this year, and the lowest since early July of 2021, just as the Delta surge was starting. New pediatric cases had dipped to 8,500, the lowest for any week during the pandemic, a couple of weeks before that, the AAP/CHA data show.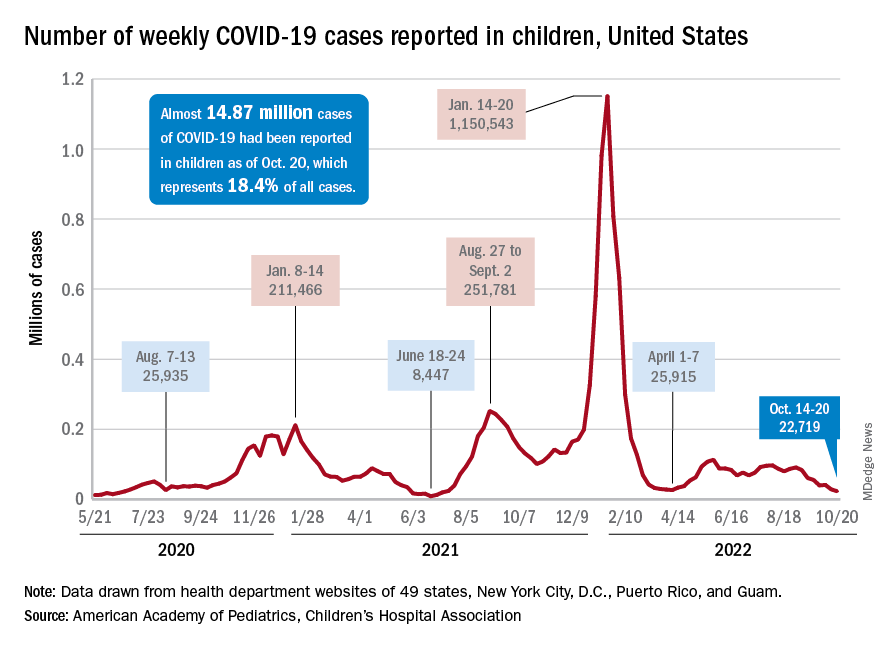
Weekly cases have fallen by almost 75% since over 90,000 were reported for the week of Aug. 26 to Sept. 1, even as children have returned to school and vaccine uptake remains slow in the youngest age groups. Rates of emergency department visits with diagnosed COVID also have continued to drop, as have new admissions, and both are nearing their 2021 lows, according to the Centers for Disease Control and Prevention.
New vaccinations in children under age 5 years were up slightly for the most recent week (Oct. 13-19), but total uptake for that age group is only 7.1% for an initial dose and 2.9% for full vaccination. Among children aged 5-11 years, 38.7% have received at least one dose and 31.6% have completed the primary series, with corresponding figures of 71.2% and 60.9% for those aged 12-17, the CDC said on its COVID Data Tracker.
Despite the low overall numbers, though, the youngest children are, in one respect, punching above their weight when it comes to vaccinations. In the 2 weeks from Oct. 6 to Oct. 19, children under 5 years of age, who represent 5.9% of the U.S. population, received 9.2% of the initial vaccine doses administered. Children aged 5-11 years, who represent 8.7% of the total population, got just 4.2% of all first doses over those same 2 weeks, while 12- to 17-year-olds, who make up 7.6% of the population, got 3.4% of the vaccine doses, the CDC reported.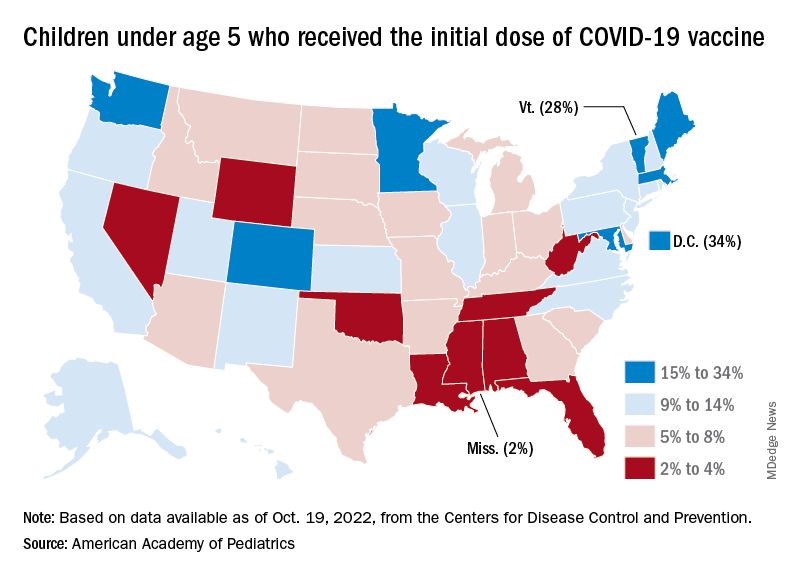
On the vaccine-approval front, the Food and Drug Administration recently announced that the new bivalent COVID-19 vaccines are now included in the emergency use authorizations for children who have completed primary or booster vaccination. The Moderna vaccine is authorized as a single-dose booster for children as young as 6 years and the Pfizer-BioNTech vaccine can be given as a single booster dose in children as young as 5 years, the FDA said.
“These bivalent COVID-19 vaccines include an mRNA component of the original strain to provide an immune response that is broadly protective against COVID-19 and an mRNA component in common between the omicron variant BA.4 and BA.5 lineages,” the FDA said.
With the third autumn of the COVID era now upon us, the discussion has turned again to a possible influenza/COVID twindemic, as well as the new-for-2022 influenza/COVID/respiratory syncytial virus tripledemic. It appears, however, that COVID may have missed the memo.
For the sixth time in the last 7 weeks, the number of new COVID cases in children fell, with just under 23,000 reported during the week of Oct. 14-20, according to the American Academy of Pediatrics and the Children’s Hospital Association. That is the lowest weekly count so far this year, and the lowest since early July of 2021, just as the Delta surge was starting. New pediatric cases had dipped to 8,500, the lowest for any week during the pandemic, a couple of weeks before that, the AAP/CHA data show.
Weekly cases have fallen by almost 75% since over 90,000 were reported for the week of Aug. 26 to Sept. 1, even as children have returned to school and vaccine uptake remains slow in the youngest age groups. Rates of emergency department visits with diagnosed COVID also have continued to drop, as have new admissions, and both are nearing their 2021 lows, according to the Centers for Disease Control and Prevention.
New vaccinations in children under age 5 years were up slightly for the most recent week (Oct. 13-19), but total uptake for that age group is only 7.1% for an initial dose and 2.9% for full vaccination. Among children aged 5-11 years, 38.7% have received at least one dose and 31.6% have completed the primary series, with corresponding figures of 71.2% and 60.9% for those aged 12-17, the CDC said on its COVID Data Tracker.
Despite the low overall numbers, though, the youngest children are, in one respect, punching above their weight when it comes to vaccinations. In the 2 weeks from Oct. 6 to Oct. 19, children under 5 years of age, who represent 5.9% of the U.S. population, received 9.2% of the initial vaccine doses administered. Children aged 5-11 years, who represent 8.7% of the total population, got just 4.2% of all first doses over those same 2 weeks, while 12- to 17-year-olds, who make up 7.6% of the population, got 3.4% of the vaccine doses, the CDC reported.
On the vaccine-approval front, the Food and Drug Administration recently announced that the new bivalent COVID-19 vaccines are now included in the emergency use authorizations for children who have completed primary or booster vaccination. The Moderna vaccine is authorized as a single-dose booster for children as young as 6 years and the Pfizer-BioNTech vaccine can be given as a single booster dose in children as young as 5 years, the FDA said.
“These bivalent COVID-19 vaccines include an mRNA component of the original strain to provide an immune response that is broadly protective against COVID-19 and an mRNA component in common between the omicron variant BA.4 and BA.5 lineages,” the FDA said.
With the third autumn of the COVID era now upon us, the discussion has turned again to a possible influenza/COVID twindemic, as well as the new-for-2022 influenza/COVID/respiratory syncytial virus tripledemic. It appears, however, that COVID may have missed the memo.
For the sixth time in the last 7 weeks, the number of new COVID cases in children fell, with just under 23,000 reported during the week of Oct. 14-20, according to the American Academy of Pediatrics and the Children’s Hospital Association. That is the lowest weekly count so far this year, and the lowest since early July of 2021, just as the Delta surge was starting. New pediatric cases had dipped to 8,500, the lowest for any week during the pandemic, a couple of weeks before that, the AAP/CHA data show.
Weekly cases have fallen by almost 75% since over 90,000 were reported for the week of Aug. 26 to Sept. 1, even as children have returned to school and vaccine uptake remains slow in the youngest age groups. Rates of emergency department visits with diagnosed COVID also have continued to drop, as have new admissions, and both are nearing their 2021 lows, according to the Centers for Disease Control and Prevention.
New vaccinations in children under age 5 years were up slightly for the most recent week (Oct. 13-19), but total uptake for that age group is only 7.1% for an initial dose and 2.9% for full vaccination. Among children aged 5-11 years, 38.7% have received at least one dose and 31.6% have completed the primary series, with corresponding figures of 71.2% and 60.9% for those aged 12-17, the CDC said on its COVID Data Tracker.
Despite the low overall numbers, though, the youngest children are, in one respect, punching above their weight when it comes to vaccinations. In the 2 weeks from Oct. 6 to Oct. 19, children under 5 years of age, who represent 5.9% of the U.S. population, received 9.2% of the initial vaccine doses administered. Children aged 5-11 years, who represent 8.7% of the total population, got just 4.2% of all first doses over those same 2 weeks, while 12- to 17-year-olds, who make up 7.6% of the population, got 3.4% of the vaccine doses, the CDC reported.
On the vaccine-approval front, the Food and Drug Administration recently announced that the new bivalent COVID-19 vaccines are now included in the emergency use authorizations for children who have completed primary or booster vaccination. The Moderna vaccine is authorized as a single-dose booster for children as young as 6 years and the Pfizer-BioNTech vaccine can be given as a single booster dose in children as young as 5 years, the FDA said.
“These bivalent COVID-19 vaccines include an mRNA component of the original strain to provide an immune response that is broadly protective against COVID-19 and an mRNA component in common between the omicron variant BA.4 and BA.5 lineages,” the FDA said.
FDA approves upadacitinib (Rinvoq) for sixth indication
The United States Food and Drug Administration has approved the Janus kinase (JAK) inhibitor upadacitinib (Rinvoq) for adults with nonradiographic axial spondyloarthritis (nr-axSpA) who have objective signs of inflammation and who have had an inadequate response to or are intolerant of one or more tumor necrosis factor (TNF) inhibitors, according to an announcement from the manufacturer, AbbVie.
The indication is the sixth in the United States for the JAK inhibitor. Upadacitinib 15 mg once daily is already approved in the United States for adults with moderately to severely active rheumatoid arthritis, active psoriatic arthritis (PsA), and active ankylosing spondylitis (AS). All these indications are for patients who have had an inadequate response to or are intolerant of one or more TNF inhibitors.
Upadacitinib is now the only JAK inhibitor that has been approved for both nr-axSpA and AS.
“Many patients living with nr-axSpA continue to experience symptoms and are unable to control disease with current treatments. In the SELECT-AXIS 2 trials, Rinvoq demonstrated efficacy in both nr-axSpA and AS with safety that was consistent across indications,” Atul Deodhar, MD, lead investigator of the trial, said in the announcement. “Today’s FDA approval offers an important new therapeutic option for patients and their caregivers to help take control of their symptoms and disease.”
Upadacitinib is also approved at a dose of 15 mg once daily for adults and children 12 years of age and older who weigh at least 40 kg and who have refractory, moderate to severe atopic dermatitis that is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
It is approved as well at 45 mg once daily for 8 weeks as an induction therapy for adult patients with moderately to severely active ulcerative colitis who have had an inadequate response to or are intolerant of one or more TNF blockers. Following induction therapy for patients with ulcerative colitis, the recommended dose for maintenance treatment is 15 mg once daily, but a dose of 30 mg once daily may be considered for patients with refractory, severe, or extensive disease.
The FDA’s decision is supported by data from the phase 3 SELECT-AXIS 2 clinical trial, which assessed the efficacy, safety, and tolerability of upadacitinib in adults with active nr-axSpA.
Nearly half of patients treated with upadacitinib had achieved 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS 40), the primary endpoint, at week 14, compared with placebo (44.9% vs. 22.3%). These responses were observed as early as 2 weeks after initiation of therapy. The safety profile was consistent with what’s known in patients with RA, PsA, and AS.
Upadacitinib can lower the ability to fight infections. Serious infections, some fatal, have occurred, including tuberculosis and infections caused by bacteria, fungi, or viruses. It is associated with an increased risk of death and major cardiovascular events in people aged 50 and older who have at least one heart disease risk factor, and it is associated with an increased risk of some cancers, including lymphoma and skin cancers.
A version of this article first appeared on Medscape.com.
The United States Food and Drug Administration has approved the Janus kinase (JAK) inhibitor upadacitinib (Rinvoq) for adults with nonradiographic axial spondyloarthritis (nr-axSpA) who have objective signs of inflammation and who have had an inadequate response to or are intolerant of one or more tumor necrosis factor (TNF) inhibitors, according to an announcement from the manufacturer, AbbVie.
The indication is the sixth in the United States for the JAK inhibitor. Upadacitinib 15 mg once daily is already approved in the United States for adults with moderately to severely active rheumatoid arthritis, active psoriatic arthritis (PsA), and active ankylosing spondylitis (AS). All these indications are for patients who have had an inadequate response to or are intolerant of one or more TNF inhibitors.
Upadacitinib is now the only JAK inhibitor that has been approved for both nr-axSpA and AS.
“Many patients living with nr-axSpA continue to experience symptoms and are unable to control disease with current treatments. In the SELECT-AXIS 2 trials, Rinvoq demonstrated efficacy in both nr-axSpA and AS with safety that was consistent across indications,” Atul Deodhar, MD, lead investigator of the trial, said in the announcement. “Today’s FDA approval offers an important new therapeutic option for patients and their caregivers to help take control of their symptoms and disease.”
Upadacitinib is also approved at a dose of 15 mg once daily for adults and children 12 years of age and older who weigh at least 40 kg and who have refractory, moderate to severe atopic dermatitis that is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
It is approved as well at 45 mg once daily for 8 weeks as an induction therapy for adult patients with moderately to severely active ulcerative colitis who have had an inadequate response to or are intolerant of one or more TNF blockers. Following induction therapy for patients with ulcerative colitis, the recommended dose for maintenance treatment is 15 mg once daily, but a dose of 30 mg once daily may be considered for patients with refractory, severe, or extensive disease.
The FDA’s decision is supported by data from the phase 3 SELECT-AXIS 2 clinical trial, which assessed the efficacy, safety, and tolerability of upadacitinib in adults with active nr-axSpA.
Nearly half of patients treated with upadacitinib had achieved 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS 40), the primary endpoint, at week 14, compared with placebo (44.9% vs. 22.3%). These responses were observed as early as 2 weeks after initiation of therapy. The safety profile was consistent with what’s known in patients with RA, PsA, and AS.
Upadacitinib can lower the ability to fight infections. Serious infections, some fatal, have occurred, including tuberculosis and infections caused by bacteria, fungi, or viruses. It is associated with an increased risk of death and major cardiovascular events in people aged 50 and older who have at least one heart disease risk factor, and it is associated with an increased risk of some cancers, including lymphoma and skin cancers.
A version of this article first appeared on Medscape.com.
The United States Food and Drug Administration has approved the Janus kinase (JAK) inhibitor upadacitinib (Rinvoq) for adults with nonradiographic axial spondyloarthritis (nr-axSpA) who have objective signs of inflammation and who have had an inadequate response to or are intolerant of one or more tumor necrosis factor (TNF) inhibitors, according to an announcement from the manufacturer, AbbVie.
The indication is the sixth in the United States for the JAK inhibitor. Upadacitinib 15 mg once daily is already approved in the United States for adults with moderately to severely active rheumatoid arthritis, active psoriatic arthritis (PsA), and active ankylosing spondylitis (AS). All these indications are for patients who have had an inadequate response to or are intolerant of one or more TNF inhibitors.
Upadacitinib is now the only JAK inhibitor that has been approved for both nr-axSpA and AS.
“Many patients living with nr-axSpA continue to experience symptoms and are unable to control disease with current treatments. In the SELECT-AXIS 2 trials, Rinvoq demonstrated efficacy in both nr-axSpA and AS with safety that was consistent across indications,” Atul Deodhar, MD, lead investigator of the trial, said in the announcement. “Today’s FDA approval offers an important new therapeutic option for patients and their caregivers to help take control of their symptoms and disease.”
Upadacitinib is also approved at a dose of 15 mg once daily for adults and children 12 years of age and older who weigh at least 40 kg and who have refractory, moderate to severe atopic dermatitis that is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
It is approved as well at 45 mg once daily for 8 weeks as an induction therapy for adult patients with moderately to severely active ulcerative colitis who have had an inadequate response to or are intolerant of one or more TNF blockers. Following induction therapy for patients with ulcerative colitis, the recommended dose for maintenance treatment is 15 mg once daily, but a dose of 30 mg once daily may be considered for patients with refractory, severe, or extensive disease.
The FDA’s decision is supported by data from the phase 3 SELECT-AXIS 2 clinical trial, which assessed the efficacy, safety, and tolerability of upadacitinib in adults with active nr-axSpA.
Nearly half of patients treated with upadacitinib had achieved 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS 40), the primary endpoint, at week 14, compared with placebo (44.9% vs. 22.3%). These responses were observed as early as 2 weeks after initiation of therapy. The safety profile was consistent with what’s known in patients with RA, PsA, and AS.
Upadacitinib can lower the ability to fight infections. Serious infections, some fatal, have occurred, including tuberculosis and infections caused by bacteria, fungi, or viruses. It is associated with an increased risk of death and major cardiovascular events in people aged 50 and older who have at least one heart disease risk factor, and it is associated with an increased risk of some cancers, including lymphoma and skin cancers.
A version of this article first appeared on Medscape.com.
People of color more likely to be hospitalized for influenza, CDC report finds
Black Americans are 80% more likely to be hospitalized for the flu, compared with White Americans, according to new federal data.
Black, Hispanic, and American Indian/Alaska Native (AI/AN) adults in the United States also have had lower influenza vaccination rates, compared with their White counterparts, since 2010, researchers at the Centers for Disease Control and Prevention (CDC) revealed in a report.
The inequalities are the result of barriers to care, distrust of the medical system, and misinformation, the report said.
“We have many of the tools we need to address inequities and flu vaccination coverage and outcomes,” said CDC Acting Principal Deputy Director Debra Houry, MD, MPH, in a press call; “however, we must acknowledge that inequities in access to care continue to exist. To improve vaccine uptake, we must address the root causes of these ongoing disparities.”
The CDC has already reported early increases in flu activity in the United States, with the highest activity in the southeastern and south-central parts of the country. Experts also warn of a potentially more severe influenza season than in the previous 2 years. CDC officials emphasized that vaccination is the best protection against severe illness, hospitalization, and death from the flu. “Everyone should get vaccinated against flu today and encourage others and their community to get a flu vaccine for the best protection against flu this fall and winter,” Dr. Houry said.
In the recent report on disparities by community published October 18 in CDC Vital Signs, researchers looked at hospitalization rates from 2009 to 2022 and vaccination rates from 2010 to 2022 based on race and ethnicity using two national databases, the Influenza-Associated Hospitalization Surveillance Network and the Behavioral Risk Factor Surveillance System. All individuals included in the analysis were aged 18 years or older, and the 2020-2021 flu season was excluded from the analysis because of insufficient data.
Compared with those for White adults, hospitalization rates were 80% higher for Black adults, 30% higher for Hispanic adults, and 20% higher for AI/AN adults. While flu vaccination rates were similar in White and Asian adults (about 54%), coverage was lower in Black (42%), Hispanic (38%), AI/AN (41%), and other/multiracial (43%) adults. This disparity persisted even among individuals who had medical insurance, a personal health care provider, and a routine checkup within the last year.
Carla Black, PhD, MPH, an epidemiologist at the CDC’s Immunization Services Division, said during the press call. While flu vaccines may not always prevent infection, people who do get sick after being vaccinated tend to have better outcomes, she added. The report noted that building trust, increasing access to vaccination services, and combating misinformation are important steps to increasing vaccine coverage in minority groups.
While social distancing measures such as masking have made it difficult for the flu to spread, the relaxation of these safety measures could also lead to higher case counts. “We’ve had two mild flu seasons, and this means we might be ripe for a severe season,” Dr. Black said. “People haven’t had natural disease in 2 years, so there’s less natural immunity out there. People are going back to work. People are traveling again. All of these factors could contribute to us having a more severe flu season.”
A version of this article first appeared on Medscape.com.
Black Americans are 80% more likely to be hospitalized for the flu, compared with White Americans, according to new federal data.
Black, Hispanic, and American Indian/Alaska Native (AI/AN) adults in the United States also have had lower influenza vaccination rates, compared with their White counterparts, since 2010, researchers at the Centers for Disease Control and Prevention (CDC) revealed in a report.
The inequalities are the result of barriers to care, distrust of the medical system, and misinformation, the report said.
“We have many of the tools we need to address inequities and flu vaccination coverage and outcomes,” said CDC Acting Principal Deputy Director Debra Houry, MD, MPH, in a press call; “however, we must acknowledge that inequities in access to care continue to exist. To improve vaccine uptake, we must address the root causes of these ongoing disparities.”
The CDC has already reported early increases in flu activity in the United States, with the highest activity in the southeastern and south-central parts of the country. Experts also warn of a potentially more severe influenza season than in the previous 2 years. CDC officials emphasized that vaccination is the best protection against severe illness, hospitalization, and death from the flu. “Everyone should get vaccinated against flu today and encourage others and their community to get a flu vaccine for the best protection against flu this fall and winter,” Dr. Houry said.
In the recent report on disparities by community published October 18 in CDC Vital Signs, researchers looked at hospitalization rates from 2009 to 2022 and vaccination rates from 2010 to 2022 based on race and ethnicity using two national databases, the Influenza-Associated Hospitalization Surveillance Network and the Behavioral Risk Factor Surveillance System. All individuals included in the analysis were aged 18 years or older, and the 2020-2021 flu season was excluded from the analysis because of insufficient data.
Compared with those for White adults, hospitalization rates were 80% higher for Black adults, 30% higher for Hispanic adults, and 20% higher for AI/AN adults. While flu vaccination rates were similar in White and Asian adults (about 54%), coverage was lower in Black (42%), Hispanic (38%), AI/AN (41%), and other/multiracial (43%) adults. This disparity persisted even among individuals who had medical insurance, a personal health care provider, and a routine checkup within the last year.
Carla Black, PhD, MPH, an epidemiologist at the CDC’s Immunization Services Division, said during the press call. While flu vaccines may not always prevent infection, people who do get sick after being vaccinated tend to have better outcomes, she added. The report noted that building trust, increasing access to vaccination services, and combating misinformation are important steps to increasing vaccine coverage in minority groups.
While social distancing measures such as masking have made it difficult for the flu to spread, the relaxation of these safety measures could also lead to higher case counts. “We’ve had two mild flu seasons, and this means we might be ripe for a severe season,” Dr. Black said. “People haven’t had natural disease in 2 years, so there’s less natural immunity out there. People are going back to work. People are traveling again. All of these factors could contribute to us having a more severe flu season.”
A version of this article first appeared on Medscape.com.
Black Americans are 80% more likely to be hospitalized for the flu, compared with White Americans, according to new federal data.
Black, Hispanic, and American Indian/Alaska Native (AI/AN) adults in the United States also have had lower influenza vaccination rates, compared with their White counterparts, since 2010, researchers at the Centers for Disease Control and Prevention (CDC) revealed in a report.
The inequalities are the result of barriers to care, distrust of the medical system, and misinformation, the report said.
“We have many of the tools we need to address inequities and flu vaccination coverage and outcomes,” said CDC Acting Principal Deputy Director Debra Houry, MD, MPH, in a press call; “however, we must acknowledge that inequities in access to care continue to exist. To improve vaccine uptake, we must address the root causes of these ongoing disparities.”
The CDC has already reported early increases in flu activity in the United States, with the highest activity in the southeastern and south-central parts of the country. Experts also warn of a potentially more severe influenza season than in the previous 2 years. CDC officials emphasized that vaccination is the best protection against severe illness, hospitalization, and death from the flu. “Everyone should get vaccinated against flu today and encourage others and their community to get a flu vaccine for the best protection against flu this fall and winter,” Dr. Houry said.
In the recent report on disparities by community published October 18 in CDC Vital Signs, researchers looked at hospitalization rates from 2009 to 2022 and vaccination rates from 2010 to 2022 based on race and ethnicity using two national databases, the Influenza-Associated Hospitalization Surveillance Network and the Behavioral Risk Factor Surveillance System. All individuals included in the analysis were aged 18 years or older, and the 2020-2021 flu season was excluded from the analysis because of insufficient data.
Compared with those for White adults, hospitalization rates were 80% higher for Black adults, 30% higher for Hispanic adults, and 20% higher for AI/AN adults. While flu vaccination rates were similar in White and Asian adults (about 54%), coverage was lower in Black (42%), Hispanic (38%), AI/AN (41%), and other/multiracial (43%) adults. This disparity persisted even among individuals who had medical insurance, a personal health care provider, and a routine checkup within the last year.
Carla Black, PhD, MPH, an epidemiologist at the CDC’s Immunization Services Division, said during the press call. While flu vaccines may not always prevent infection, people who do get sick after being vaccinated tend to have better outcomes, she added. The report noted that building trust, increasing access to vaccination services, and combating misinformation are important steps to increasing vaccine coverage in minority groups.
While social distancing measures such as masking have made it difficult for the flu to spread, the relaxation of these safety measures could also lead to higher case counts. “We’ve had two mild flu seasons, and this means we might be ripe for a severe season,” Dr. Black said. “People haven’t had natural disease in 2 years, so there’s less natural immunity out there. People are going back to work. People are traveling again. All of these factors could contribute to us having a more severe flu season.”
A version of this article first appeared on Medscape.com.
FDA confirms nationwide Adderall shortage
The U.S. Food and Drug Administration which are approved for treating attention deficit hyperactivity disorder and narcolepsy.
The FDA announcement follows weeks of reports of a shortage of the drug by pharmacy chains and Adderall users.
The agency said it is in “frequent” contact with all manufacturers of Adderall – and reported that one of those companies, Teva, is experiencing ongoing intermittent manufacturing delays.
Other manufacturers continue to produce amphetamine mixed salts, but there is not enough supply to continue to meet U.S. market demand through those producers, the FDA noted.
“Until supply is restored, there are alternative therapies, including the extended-release version of amphetamine mixed salts, available to health care professionals and their patients for amphetamine mixed salts’ approved indications,” the agency said.
Patients should work with their health care provider to determine their best treatment option, it added.
The organization is continuing to monitor the supply of Adderall and to help manufacturers resolve the shortage.
Its Drug Shortage webpage has additional information about the situation and is updated regularly.
“We continue to use all the tools we have available to help keep supply available for patients and will provide public updates regarding the Adderall shortage,” the FDA said.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration which are approved for treating attention deficit hyperactivity disorder and narcolepsy.
The FDA announcement follows weeks of reports of a shortage of the drug by pharmacy chains and Adderall users.
The agency said it is in “frequent” contact with all manufacturers of Adderall – and reported that one of those companies, Teva, is experiencing ongoing intermittent manufacturing delays.
Other manufacturers continue to produce amphetamine mixed salts, but there is not enough supply to continue to meet U.S. market demand through those producers, the FDA noted.
“Until supply is restored, there are alternative therapies, including the extended-release version of amphetamine mixed salts, available to health care professionals and their patients for amphetamine mixed salts’ approved indications,” the agency said.
Patients should work with their health care provider to determine their best treatment option, it added.
The organization is continuing to monitor the supply of Adderall and to help manufacturers resolve the shortage.
Its Drug Shortage webpage has additional information about the situation and is updated regularly.
“We continue to use all the tools we have available to help keep supply available for patients and will provide public updates regarding the Adderall shortage,” the FDA said.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration which are approved for treating attention deficit hyperactivity disorder and narcolepsy.
The FDA announcement follows weeks of reports of a shortage of the drug by pharmacy chains and Adderall users.
The agency said it is in “frequent” contact with all manufacturers of Adderall – and reported that one of those companies, Teva, is experiencing ongoing intermittent manufacturing delays.
Other manufacturers continue to produce amphetamine mixed salts, but there is not enough supply to continue to meet U.S. market demand through those producers, the FDA noted.
“Until supply is restored, there are alternative therapies, including the extended-release version of amphetamine mixed salts, available to health care professionals and their patients for amphetamine mixed salts’ approved indications,” the agency said.
Patients should work with their health care provider to determine their best treatment option, it added.
The organization is continuing to monitor the supply of Adderall and to help manufacturers resolve the shortage.
Its Drug Shortage webpage has additional information about the situation and is updated regularly.
“We continue to use all the tools we have available to help keep supply available for patients and will provide public updates regarding the Adderall shortage,” the FDA said.
A version of this article first appeared on Medscape.com.
Children and COVID: Downward trend reverses with small increase in new cases
A small increase in new cases brought COVID-19’s latest losing streak to an end at 4 weeks, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
The 40,656 new cases reported bring the U.S. cumulative count of child COVID-19 cases to over 14.8 million since the pandemic began, which represents 18.4% of all cases, the AAP and CHA said in their weekly report based on state-level data.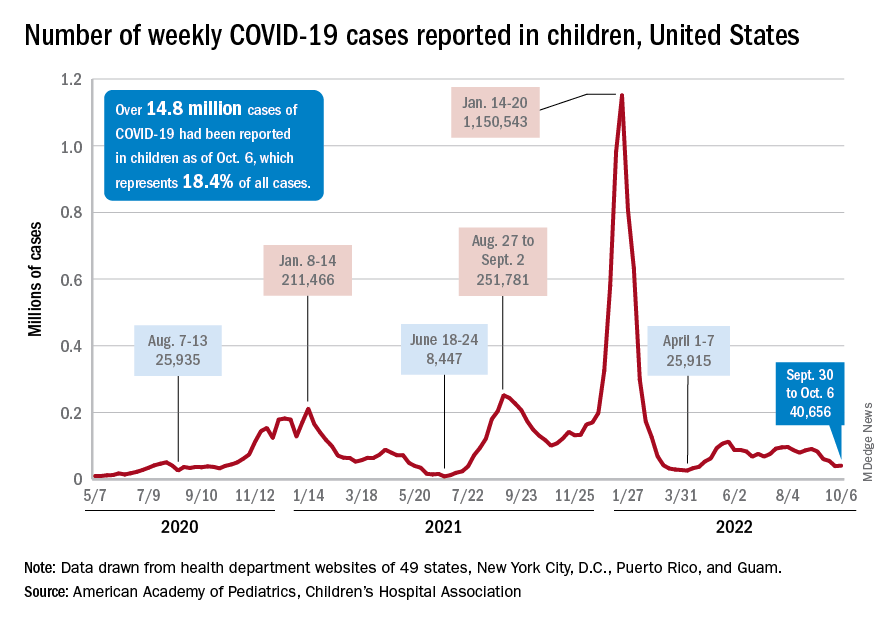
The increase in new cases was not reflected in emergency department visits or hospital admissions, which both continued sustained declines that started in August. In the week from Sept. 27 to Oct. 4, the 7-day averages for ED visits with diagnosed COVID were down by 21.5% (age 0-11), 27.3% (12-15), and 18.2% (16-17), the Centers for Disease Control and Prevention said, while the most recent 7-day average for new admissions – 127 per day for Oct. 2-8 – among children aged 0-17 years with confirmed COVID was down from 161 per day the previous week, a drop of over 21%.
The state-level data that are currently available (several states are no longer reporting) show Alaska (25.5%) and Vermont (25.4%) have the highest proportions of cumulative cases in children, and Florida (12.3%) and Utah (13.5%) have the lowest. Rhode Island has the highest rate of COVID-19 per 100,000 children at 40,427, while Missouri has the lowest at 14,252. The national average is 19,687 per 100,000, the AAP and CHA reported.
Taking a look at vaccination
Vaccinations were up slightly in children aged 12-17 years, as 20,000 initial doses were given during the week of Sept. 29 to Oct. 5, compared with 17,000 and 18,000 the previous 2 weeks. Initial vaccinations in younger children, however, continued declines dating back to August, the AAP said in its weekly vaccination trends report.
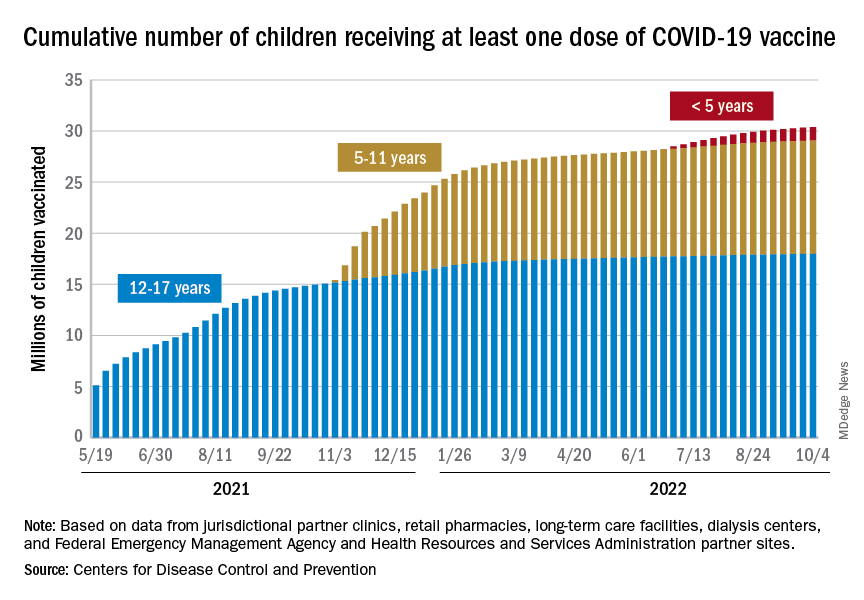
The District of Columbia and Massachusetts have the most highly vaccinated groups of 12- to 17-year-olds, as 100% and 95%, respectively, have received initial doses, while Wyoming (39%) and Idaho (42%) have the lowest. D.C. (73%) and Vermont (68%) have the highest proportions of vaccinated 5- to 11-year-olds, and Alabama (17%) and Mississippi (18%) have the lowest. For children under age 5 years, those in D.C. (33%) and Vermont (26%) are the most likely to have received an initial COVID vaccination, while Alabama, Louisiana, and Mississippi share national-low rates of 2%, the AAP said its report, which is based on CDC data.
When all states and territories are combined, 71% of children aged 12-17 have received at least one dose of vaccine, as have 38.6% of all children 5-11 years old and 6.7% of those under age 5. Almost 61% of the nation’s 16- to 17-year-olds have been fully vaccinated, along with 31.5% of those aged 5-11 and 2.4% of children younger than 5 years, the CDC said on its COVID Data Tracker.
About 42 million children – 58% of the population under the age of 18 years – have not received any vaccine yet, the AAP noted. Meanwhile, CDC data indicate that 36 children died of COVID in the last week, with pediatric deaths now totaling 1,781 over the course of the pandemic.
A small increase in new cases brought COVID-19’s latest losing streak to an end at 4 weeks, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
The 40,656 new cases reported bring the U.S. cumulative count of child COVID-19 cases to over 14.8 million since the pandemic began, which represents 18.4% of all cases, the AAP and CHA said in their weekly report based on state-level data.
The increase in new cases was not reflected in emergency department visits or hospital admissions, which both continued sustained declines that started in August. In the week from Sept. 27 to Oct. 4, the 7-day averages for ED visits with diagnosed COVID were down by 21.5% (age 0-11), 27.3% (12-15), and 18.2% (16-17), the Centers for Disease Control and Prevention said, while the most recent 7-day average for new admissions – 127 per day for Oct. 2-8 – among children aged 0-17 years with confirmed COVID was down from 161 per day the previous week, a drop of over 21%.
The state-level data that are currently available (several states are no longer reporting) show Alaska (25.5%) and Vermont (25.4%) have the highest proportions of cumulative cases in children, and Florida (12.3%) and Utah (13.5%) have the lowest. Rhode Island has the highest rate of COVID-19 per 100,000 children at 40,427, while Missouri has the lowest at 14,252. The national average is 19,687 per 100,000, the AAP and CHA reported.
Taking a look at vaccination
Vaccinations were up slightly in children aged 12-17 years, as 20,000 initial doses were given during the week of Sept. 29 to Oct. 5, compared with 17,000 and 18,000 the previous 2 weeks. Initial vaccinations in younger children, however, continued declines dating back to August, the AAP said in its weekly vaccination trends report.
The District of Columbia and Massachusetts have the most highly vaccinated groups of 12- to 17-year-olds, as 100% and 95%, respectively, have received initial doses, while Wyoming (39%) and Idaho (42%) have the lowest. D.C. (73%) and Vermont (68%) have the highest proportions of vaccinated 5- to 11-year-olds, and Alabama (17%) and Mississippi (18%) have the lowest. For children under age 5 years, those in D.C. (33%) and Vermont (26%) are the most likely to have received an initial COVID vaccination, while Alabama, Louisiana, and Mississippi share national-low rates of 2%, the AAP said its report, which is based on CDC data.
When all states and territories are combined, 71% of children aged 12-17 have received at least one dose of vaccine, as have 38.6% of all children 5-11 years old and 6.7% of those under age 5. Almost 61% of the nation’s 16- to 17-year-olds have been fully vaccinated, along with 31.5% of those aged 5-11 and 2.4% of children younger than 5 years, the CDC said on its COVID Data Tracker.
About 42 million children – 58% of the population under the age of 18 years – have not received any vaccine yet, the AAP noted. Meanwhile, CDC data indicate that 36 children died of COVID in the last week, with pediatric deaths now totaling 1,781 over the course of the pandemic.
A small increase in new cases brought COVID-19’s latest losing streak to an end at 4 weeks, based on data from the American Academy of Pediatrics and the Children’s Hospital Association.
The 40,656 new cases reported bring the U.S. cumulative count of child COVID-19 cases to over 14.8 million since the pandemic began, which represents 18.4% of all cases, the AAP and CHA said in their weekly report based on state-level data.
The increase in new cases was not reflected in emergency department visits or hospital admissions, which both continued sustained declines that started in August. In the week from Sept. 27 to Oct. 4, the 7-day averages for ED visits with diagnosed COVID were down by 21.5% (age 0-11), 27.3% (12-15), and 18.2% (16-17), the Centers for Disease Control and Prevention said, while the most recent 7-day average for new admissions – 127 per day for Oct. 2-8 – among children aged 0-17 years with confirmed COVID was down from 161 per day the previous week, a drop of over 21%.
The state-level data that are currently available (several states are no longer reporting) show Alaska (25.5%) and Vermont (25.4%) have the highest proportions of cumulative cases in children, and Florida (12.3%) and Utah (13.5%) have the lowest. Rhode Island has the highest rate of COVID-19 per 100,000 children at 40,427, while Missouri has the lowest at 14,252. The national average is 19,687 per 100,000, the AAP and CHA reported.
Taking a look at vaccination
Vaccinations were up slightly in children aged 12-17 years, as 20,000 initial doses were given during the week of Sept. 29 to Oct. 5, compared with 17,000 and 18,000 the previous 2 weeks. Initial vaccinations in younger children, however, continued declines dating back to August, the AAP said in its weekly vaccination trends report.
The District of Columbia and Massachusetts have the most highly vaccinated groups of 12- to 17-year-olds, as 100% and 95%, respectively, have received initial doses, while Wyoming (39%) and Idaho (42%) have the lowest. D.C. (73%) and Vermont (68%) have the highest proportions of vaccinated 5- to 11-year-olds, and Alabama (17%) and Mississippi (18%) have the lowest. For children under age 5 years, those in D.C. (33%) and Vermont (26%) are the most likely to have received an initial COVID vaccination, while Alabama, Louisiana, and Mississippi share national-low rates of 2%, the AAP said its report, which is based on CDC data.
When all states and territories are combined, 71% of children aged 12-17 have received at least one dose of vaccine, as have 38.6% of all children 5-11 years old and 6.7% of those under age 5. Almost 61% of the nation’s 16- to 17-year-olds have been fully vaccinated, along with 31.5% of those aged 5-11 and 2.4% of children younger than 5 years, the CDC said on its COVID Data Tracker.
About 42 million children – 58% of the population under the age of 18 years – have not received any vaccine yet, the AAP noted. Meanwhile, CDC data indicate that 36 children died of COVID in the last week, with pediatric deaths now totaling 1,781 over the course of the pandemic.
FDA: Newborns protected by whooping cough vaccine
The Food and Drug Administration has approved a whooping cough vaccine that protects newborns under 2 months of age.
The vaccine, manufactured by GlaxoSmithKline, was previously approved among pregnant people for their own protection.
“Infants younger than 2 months of age are too young to be protected by the childhood pertussis vaccine series,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in a press release. “This is the first vaccine approved specifically for use during pregnancy to prevent a disease in young infants whose mothers are vaccinated during pregnancy.”
Pertussis is a highly contagious respiratory tract infection caused by the bacterium Bordetella pertussis. Most cases that result in hospitalizations and death are among infants within 2 months of birth.
The FDA said its decision was based on data from observational studies, which included 108 cases of pertussis in infants younger than 2 months old. According to data evaluated by the agency, the vaccine was 78% effective in preventing whooping cough.
Boostrix is administered as a single 0.5-mL dose.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved a whooping cough vaccine that protects newborns under 2 months of age.
The vaccine, manufactured by GlaxoSmithKline, was previously approved among pregnant people for their own protection.
“Infants younger than 2 months of age are too young to be protected by the childhood pertussis vaccine series,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in a press release. “This is the first vaccine approved specifically for use during pregnancy to prevent a disease in young infants whose mothers are vaccinated during pregnancy.”
Pertussis is a highly contagious respiratory tract infection caused by the bacterium Bordetella pertussis. Most cases that result in hospitalizations and death are among infants within 2 months of birth.
The FDA said its decision was based on data from observational studies, which included 108 cases of pertussis in infants younger than 2 months old. According to data evaluated by the agency, the vaccine was 78% effective in preventing whooping cough.
Boostrix is administered as a single 0.5-mL dose.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved a whooping cough vaccine that protects newborns under 2 months of age.
The vaccine, manufactured by GlaxoSmithKline, was previously approved among pregnant people for their own protection.
“Infants younger than 2 months of age are too young to be protected by the childhood pertussis vaccine series,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in a press release. “This is the first vaccine approved specifically for use during pregnancy to prevent a disease in young infants whose mothers are vaccinated during pregnancy.”
Pertussis is a highly contagious respiratory tract infection caused by the bacterium Bordetella pertussis. Most cases that result in hospitalizations and death are among infants within 2 months of birth.
The FDA said its decision was based on data from observational studies, which included 108 cases of pertussis in infants younger than 2 months old. According to data evaluated by the agency, the vaccine was 78% effective in preventing whooping cough.
Boostrix is administered as a single 0.5-mL dose.
A version of this article first appeared on Medscape.com.