User login
U.S. infant mortality continued slow decline in 2017
according to data released Aug. 1 by the National Center for Health Statistics, based on data from the National Vital Statistics System.
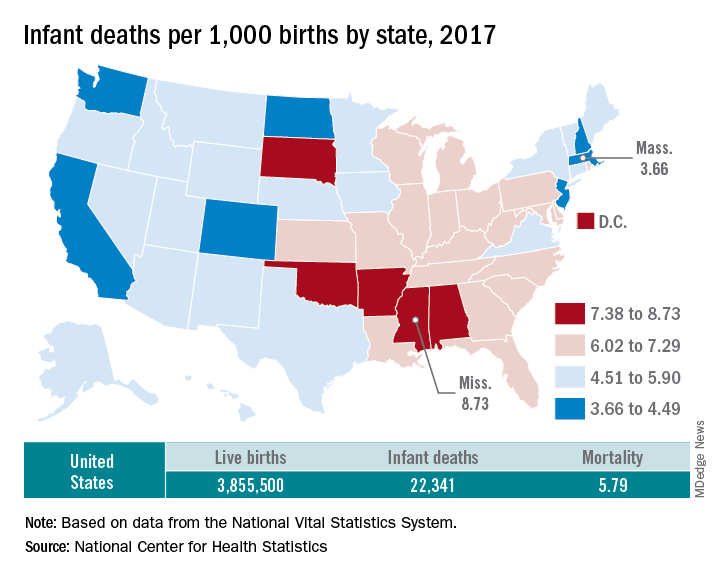
The rate for 2017 was 5.79 deaths per 1,000 live births, which was not statistically different from the rate of 5.87 in 2016, the National Center for Health Statistics said in a new report. Neonatal and postneonatal mortality – 3.85 and 1.94 per 1,000, respectively – both showed the same nonsignificant drop from 2016 to 2017.
About two-thirds of the infants who died in 2017 were children born preterm (less than 37 weeks’ gestation), the NCHS said, and “the mortality rate for infants born before 28 weeks of gestation [389.4 per 1,000] was 183 times the rate for term infants” born at 37-41 weeks.
Rates at the state level in 2017 ranged from a low of 3.66 deaths/1,000 live births in Massachusetts to a high of 8.73/1,000 in Mississippi. Washington (3.88) was the only other state with a rate below 4.0, while Arkansas (8.10) was the only other state above 8.0 (The District of Columbia had a rate of 8.16.). Infant mortality was significantly lower than the national rate in 11 states and significantly higher in 15 states and D.C., according to the report.
Overall, in 2017, 3,855,500 live births occurred, with 22,341 infants having died before the age of 1 year, data from the National Vital Statistics System’s linked birth/infant death file show. In 1995, the first year that the linked file was available, the corresponding numbers were 3,899,589 births and 29,505 deaths, for a rate of 7.57 deaths/1,000 live births.
according to data released Aug. 1 by the National Center for Health Statistics, based on data from the National Vital Statistics System.
The rate for 2017 was 5.79 deaths per 1,000 live births, which was not statistically different from the rate of 5.87 in 2016, the National Center for Health Statistics said in a new report. Neonatal and postneonatal mortality – 3.85 and 1.94 per 1,000, respectively – both showed the same nonsignificant drop from 2016 to 2017.
About two-thirds of the infants who died in 2017 were children born preterm (less than 37 weeks’ gestation), the NCHS said, and “the mortality rate for infants born before 28 weeks of gestation [389.4 per 1,000] was 183 times the rate for term infants” born at 37-41 weeks.
Rates at the state level in 2017 ranged from a low of 3.66 deaths/1,000 live births in Massachusetts to a high of 8.73/1,000 in Mississippi. Washington (3.88) was the only other state with a rate below 4.0, while Arkansas (8.10) was the only other state above 8.0 (The District of Columbia had a rate of 8.16.). Infant mortality was significantly lower than the national rate in 11 states and significantly higher in 15 states and D.C., according to the report.
Overall, in 2017, 3,855,500 live births occurred, with 22,341 infants having died before the age of 1 year, data from the National Vital Statistics System’s linked birth/infant death file show. In 1995, the first year that the linked file was available, the corresponding numbers were 3,899,589 births and 29,505 deaths, for a rate of 7.57 deaths/1,000 live births.
according to data released Aug. 1 by the National Center for Health Statistics, based on data from the National Vital Statistics System.
The rate for 2017 was 5.79 deaths per 1,000 live births, which was not statistically different from the rate of 5.87 in 2016, the National Center for Health Statistics said in a new report. Neonatal and postneonatal mortality – 3.85 and 1.94 per 1,000, respectively – both showed the same nonsignificant drop from 2016 to 2017.
About two-thirds of the infants who died in 2017 were children born preterm (less than 37 weeks’ gestation), the NCHS said, and “the mortality rate for infants born before 28 weeks of gestation [389.4 per 1,000] was 183 times the rate for term infants” born at 37-41 weeks.
Rates at the state level in 2017 ranged from a low of 3.66 deaths/1,000 live births in Massachusetts to a high of 8.73/1,000 in Mississippi. Washington (3.88) was the only other state with a rate below 4.0, while Arkansas (8.10) was the only other state above 8.0 (The District of Columbia had a rate of 8.16.). Infant mortality was significantly lower than the national rate in 11 states and significantly higher in 15 states and D.C., according to the report.
Overall, in 2017, 3,855,500 live births occurred, with 22,341 infants having died before the age of 1 year, data from the National Vital Statistics System’s linked birth/infant death file show. In 1995, the first year that the linked file was available, the corresponding numbers were 3,899,589 births and 29,505 deaths, for a rate of 7.57 deaths/1,000 live births.
HHS proposes pathways for drug importation
Officials at the U.S. Department of Health and Human Services have announced a new plan that they say would lay the foundation for safe importation of certain medications, with the aim of expanding drug access and lowering prescription costs for patients.
The action plan, unveiled July 31, outlines two pathways for drug importation from foreign markets. The first route would authorize states, wholesalers, or pharmacists to propose pilot demonstrations on how they would import drugs from Canada into the United States, provided these are versions of drugs already approved by the Food and Drug Administration. Similarly, a second pathway would allow manufacturers that sell in foreign countries the opportunity to import drugs that are versions of FDA-approved medications.
HHS Secretary Alex M. Azar II said the action plan is part of President Trump’s drug-pricing blueprint and is intended to combat the sky-high price tags on many prescription medications.
“President Trump has been clear: For too long American patients have been paying exorbitantly high prices for prescription drugs that are made available to other countries at lower prices,” Mr. Azar said in a statement. “[The] announcement outlines the pathways the administration intends to explore to allow safe importation of certain prescription drugs to lower prices and reduce out of pocket costs for American patients. This is the next important step in the administration’s work to end foreign freeloading and put American patients first.”

Under the first pathway, HHS would review plans submitted by states, pharmacists, or drugmakers that outline how the entities would import Health Canada–approved drugs that are in compliance with the federal Food, Drug, and Cosmetic Act. The importation would occur in a manner that assures the drug’s validity and meets the cost requirements of federal rule making, according to an HHS fact sheet.
Demonstration projects would be time-limited and require regular reporting to ensure safety and cost conditions are being met.
Under the second pathway, manufacturers of FDA-approved drug products would be able to import versions of those drugs that they sell in foreign countries through a special process to be outlined by the agency. As part of the process, drugmakers would need to establish that the foreign version is the same as the U.S. version. The FDA would then allow the drug to be labeled for sale in the U.S. and imported, according to the fact sheet. HHS officials said they believe that manufacturers would use this pathway to offer U.S. patients lower-cost versions of their drugs and the medications affected could potentially include those used to treat diabetes, rheumatoid arthritis, cardiovascular disorders, and cancer.
“In recent years, multiple manufacturers have stated (either publicly or in statements to the Administration) that they wanted to offer lower cost versions but could not readily do so because they were locked into contracts with other parties in the supply chain,” HHS officials stated in the fact sheet. “This pathway would highlight an opportunity for manufacturers to use importation to offer lower-cost versions of their drugs.”
HHS plans to introduce its action plan through a formal notice of proposed rulemaking, which has not yet been finalized. Some elements of the final proposal may differ from its initial descriptions to reflect further consideration of the relevant issues, the agency noted.
Acting FDA Commissioner Ned Sharpless, MD, said the agency has a unique role to play in promoting competition that can help reduce drug prices and improve access to medicine for Americans.
“Driving down drug prices requires a comprehensive approach and we must continue to look at all innovative solutions to this challenge,” Dr. Sharpless said in a statement. “[The] proposal is the result of the hard work by the dedicated staff of the FDA, in close collaboration with HHS and the White House, to identify potential pathways we can pursue to support the safe importation of certain prescription drugs.”
Sen. Lamar Alexander (R-Tenn.), chair of the Health, Education, Labor and Pensions committee, said the administration’s proposal sounds promising as long as the plan ensures the safety and efficacy of imported medications.
“This is the first administration to take concrete steps to allow importation of prescription drugs to reduce their cost and I welcome it,” Sen. Alexander said in a statement. “The key for me is whether this plan preserves the Food and Drug Administration’s gold standard for safety and effectiveness. Millions of Americans every day buy prescription drugs relying on the FDA’s guarantee of quality.”
Officials at the U.S. Department of Health and Human Services have announced a new plan that they say would lay the foundation for safe importation of certain medications, with the aim of expanding drug access and lowering prescription costs for patients.
The action plan, unveiled July 31, outlines two pathways for drug importation from foreign markets. The first route would authorize states, wholesalers, or pharmacists to propose pilot demonstrations on how they would import drugs from Canada into the United States, provided these are versions of drugs already approved by the Food and Drug Administration. Similarly, a second pathway would allow manufacturers that sell in foreign countries the opportunity to import drugs that are versions of FDA-approved medications.
HHS Secretary Alex M. Azar II said the action plan is part of President Trump’s drug-pricing blueprint and is intended to combat the sky-high price tags on many prescription medications.
“President Trump has been clear: For too long American patients have been paying exorbitantly high prices for prescription drugs that are made available to other countries at lower prices,” Mr. Azar said in a statement. “[The] announcement outlines the pathways the administration intends to explore to allow safe importation of certain prescription drugs to lower prices and reduce out of pocket costs for American patients. This is the next important step in the administration’s work to end foreign freeloading and put American patients first.”
Under the first pathway, HHS would review plans submitted by states, pharmacists, or drugmakers that outline how the entities would import Health Canada–approved drugs that are in compliance with the federal Food, Drug, and Cosmetic Act. The importation would occur in a manner that assures the drug’s validity and meets the cost requirements of federal rule making, according to an HHS fact sheet.
Demonstration projects would be time-limited and require regular reporting to ensure safety and cost conditions are being met.
Under the second pathway, manufacturers of FDA-approved drug products would be able to import versions of those drugs that they sell in foreign countries through a special process to be outlined by the agency. As part of the process, drugmakers would need to establish that the foreign version is the same as the U.S. version. The FDA would then allow the drug to be labeled for sale in the U.S. and imported, according to the fact sheet. HHS officials said they believe that manufacturers would use this pathway to offer U.S. patients lower-cost versions of their drugs and the medications affected could potentially include those used to treat diabetes, rheumatoid arthritis, cardiovascular disorders, and cancer.
“In recent years, multiple manufacturers have stated (either publicly or in statements to the Administration) that they wanted to offer lower cost versions but could not readily do so because they were locked into contracts with other parties in the supply chain,” HHS officials stated in the fact sheet. “This pathway would highlight an opportunity for manufacturers to use importation to offer lower-cost versions of their drugs.”
HHS plans to introduce its action plan through a formal notice of proposed rulemaking, which has not yet been finalized. Some elements of the final proposal may differ from its initial descriptions to reflect further consideration of the relevant issues, the agency noted.
Acting FDA Commissioner Ned Sharpless, MD, said the agency has a unique role to play in promoting competition that can help reduce drug prices and improve access to medicine for Americans.
“Driving down drug prices requires a comprehensive approach and we must continue to look at all innovative solutions to this challenge,” Dr. Sharpless said in a statement. “[The] proposal is the result of the hard work by the dedicated staff of the FDA, in close collaboration with HHS and the White House, to identify potential pathways we can pursue to support the safe importation of certain prescription drugs.”
Sen. Lamar Alexander (R-Tenn.), chair of the Health, Education, Labor and Pensions committee, said the administration’s proposal sounds promising as long as the plan ensures the safety and efficacy of imported medications.
“This is the first administration to take concrete steps to allow importation of prescription drugs to reduce their cost and I welcome it,” Sen. Alexander said in a statement. “The key for me is whether this plan preserves the Food and Drug Administration’s gold standard for safety and effectiveness. Millions of Americans every day buy prescription drugs relying on the FDA’s guarantee of quality.”
Officials at the U.S. Department of Health and Human Services have announced a new plan that they say would lay the foundation for safe importation of certain medications, with the aim of expanding drug access and lowering prescription costs for patients.
The action plan, unveiled July 31, outlines two pathways for drug importation from foreign markets. The first route would authorize states, wholesalers, or pharmacists to propose pilot demonstrations on how they would import drugs from Canada into the United States, provided these are versions of drugs already approved by the Food and Drug Administration. Similarly, a second pathway would allow manufacturers that sell in foreign countries the opportunity to import drugs that are versions of FDA-approved medications.
HHS Secretary Alex M. Azar II said the action plan is part of President Trump’s drug-pricing blueprint and is intended to combat the sky-high price tags on many prescription medications.
“President Trump has been clear: For too long American patients have been paying exorbitantly high prices for prescription drugs that are made available to other countries at lower prices,” Mr. Azar said in a statement. “[The] announcement outlines the pathways the administration intends to explore to allow safe importation of certain prescription drugs to lower prices and reduce out of pocket costs for American patients. This is the next important step in the administration’s work to end foreign freeloading and put American patients first.”
Under the first pathway, HHS would review plans submitted by states, pharmacists, or drugmakers that outline how the entities would import Health Canada–approved drugs that are in compliance with the federal Food, Drug, and Cosmetic Act. The importation would occur in a manner that assures the drug’s validity and meets the cost requirements of federal rule making, according to an HHS fact sheet.
Demonstration projects would be time-limited and require regular reporting to ensure safety and cost conditions are being met.
Under the second pathway, manufacturers of FDA-approved drug products would be able to import versions of those drugs that they sell in foreign countries through a special process to be outlined by the agency. As part of the process, drugmakers would need to establish that the foreign version is the same as the U.S. version. The FDA would then allow the drug to be labeled for sale in the U.S. and imported, according to the fact sheet. HHS officials said they believe that manufacturers would use this pathway to offer U.S. patients lower-cost versions of their drugs and the medications affected could potentially include those used to treat diabetes, rheumatoid arthritis, cardiovascular disorders, and cancer.
“In recent years, multiple manufacturers have stated (either publicly or in statements to the Administration) that they wanted to offer lower cost versions but could not readily do so because they were locked into contracts with other parties in the supply chain,” HHS officials stated in the fact sheet. “This pathway would highlight an opportunity for manufacturers to use importation to offer lower-cost versions of their drugs.”
HHS plans to introduce its action plan through a formal notice of proposed rulemaking, which has not yet been finalized. Some elements of the final proposal may differ from its initial descriptions to reflect further consideration of the relevant issues, the agency noted.
Acting FDA Commissioner Ned Sharpless, MD, said the agency has a unique role to play in promoting competition that can help reduce drug prices and improve access to medicine for Americans.
“Driving down drug prices requires a comprehensive approach and we must continue to look at all innovative solutions to this challenge,” Dr. Sharpless said in a statement. “[The] proposal is the result of the hard work by the dedicated staff of the FDA, in close collaboration with HHS and the White House, to identify potential pathways we can pursue to support the safe importation of certain prescription drugs.”
Sen. Lamar Alexander (R-Tenn.), chair of the Health, Education, Labor and Pensions committee, said the administration’s proposal sounds promising as long as the plan ensures the safety and efficacy of imported medications.
“This is the first administration to take concrete steps to allow importation of prescription drugs to reduce their cost and I welcome it,” Sen. Alexander said in a statement. “The key for me is whether this plan preserves the Food and Drug Administration’s gold standard for safety and effectiveness. Millions of Americans every day buy prescription drugs relying on the FDA’s guarantee of quality.”
FDA approves darolutamide for nonmetastatic CRPC
The Food and Drug Administration has approved darolutamide for nonmetastatic, castration-resistant prostate cancer.

The approval was based on improved metastasis-free survival (MFS) in the randomized ARAMIS trial of 1,509 patients with nonmetastatic, castration-resistant prostate cancer.
Median MFS was 40.4 months (95% confidence interval, 34.3 months to not reached) for patients treated with darolutamide, compared with 18.4 months (95% CI, 15.5-22.3 months) for those receiving placebo (hazard ratio, 0.41; 95% CI, 0.34-0.50; P less than .0001), according to the FDA.
MFS is defined as the time from randomization to first evidence of distant metastasis or death from any cause within 33 weeks after the last evaluable scan, whichever occurred first.
In ARAMIS, patients were randomized 2:1 to receive either 600 mg darolutamide orally twice daily (n = 955) or matching placebo (n = 554). All patients received a gonadotropin-releasing hormone analog concurrently or had a previous bilateral orchiectomy. Twelve patients with previous seizure histories were treated on the darolutamide arm.
Overall survival data is not yet mature, the FDA said.
The most common adverse reactions in patients who received darolutamide were fatigue, extremity pain, and rash. Ischemic heart disease (4.3%) and heart failure (2.1%) were more common on the darolutamide arm, while seizure incidence was similar in the two arms (0.2%).
The recommended darolutamide dose is 600 mg (two 300-mg tablets) administered orally twice daily with food. Patients should also receive a gonadotropin-releasing hormone analog concurrently or should have had bilateral orchiectomy, the FDA said.
Darolutamide is marketed as Nubeqa by Bayer HealthCare Pharmaceuticals.
The Food and Drug Administration has approved darolutamide for nonmetastatic, castration-resistant prostate cancer.
The approval was based on improved metastasis-free survival (MFS) in the randomized ARAMIS trial of 1,509 patients with nonmetastatic, castration-resistant prostate cancer.
Median MFS was 40.4 months (95% confidence interval, 34.3 months to not reached) for patients treated with darolutamide, compared with 18.4 months (95% CI, 15.5-22.3 months) for those receiving placebo (hazard ratio, 0.41; 95% CI, 0.34-0.50; P less than .0001), according to the FDA.
MFS is defined as the time from randomization to first evidence of distant metastasis or death from any cause within 33 weeks after the last evaluable scan, whichever occurred first.
In ARAMIS, patients were randomized 2:1 to receive either 600 mg darolutamide orally twice daily (n = 955) or matching placebo (n = 554). All patients received a gonadotropin-releasing hormone analog concurrently or had a previous bilateral orchiectomy. Twelve patients with previous seizure histories were treated on the darolutamide arm.
Overall survival data is not yet mature, the FDA said.
The most common adverse reactions in patients who received darolutamide were fatigue, extremity pain, and rash. Ischemic heart disease (4.3%) and heart failure (2.1%) were more common on the darolutamide arm, while seizure incidence was similar in the two arms (0.2%).
The recommended darolutamide dose is 600 mg (two 300-mg tablets) administered orally twice daily with food. Patients should also receive a gonadotropin-releasing hormone analog concurrently or should have had bilateral orchiectomy, the FDA said.
Darolutamide is marketed as Nubeqa by Bayer HealthCare Pharmaceuticals.
The Food and Drug Administration has approved darolutamide for nonmetastatic, castration-resistant prostate cancer.
The approval was based on improved metastasis-free survival (MFS) in the randomized ARAMIS trial of 1,509 patients with nonmetastatic, castration-resistant prostate cancer.
Median MFS was 40.4 months (95% confidence interval, 34.3 months to not reached) for patients treated with darolutamide, compared with 18.4 months (95% CI, 15.5-22.3 months) for those receiving placebo (hazard ratio, 0.41; 95% CI, 0.34-0.50; P less than .0001), according to the FDA.
MFS is defined as the time from randomization to first evidence of distant metastasis or death from any cause within 33 weeks after the last evaluable scan, whichever occurred first.
In ARAMIS, patients were randomized 2:1 to receive either 600 mg darolutamide orally twice daily (n = 955) or matching placebo (n = 554). All patients received a gonadotropin-releasing hormone analog concurrently or had a previous bilateral orchiectomy. Twelve patients with previous seizure histories were treated on the darolutamide arm.
Overall survival data is not yet mature, the FDA said.
The most common adverse reactions in patients who received darolutamide were fatigue, extremity pain, and rash. Ischemic heart disease (4.3%) and heart failure (2.1%) were more common on the darolutamide arm, while seizure incidence was similar in the two arms (0.2%).
The recommended darolutamide dose is 600 mg (two 300-mg tablets) administered orally twice daily with food. Patients should also receive a gonadotropin-releasing hormone analog concurrently or should have had bilateral orchiectomy, the FDA said.
Darolutamide is marketed as Nubeqa by Bayer HealthCare Pharmaceuticals.
Hadlima approved as fourth adalimumab biosimilar in U.S.
The Food and Drug Administration has approved the Humira biosimilar Hadlima (adalimumab-bwwd), making it the fourth adalimumab biosimilar approved in the United States, the agency announced.

Hadlima is approved for seven of the reference product’s indications, which include rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, and ulcerative colitis.
The product will launch in the United States on June 30, 2023. Other FDA-approved adalimumab biosimilars – Amjevita (adalimunab-atto), Cyltezo (adalimumab-adbm), Hyrimoz (adalimumab-adaz) – similarly will not reach the U.S. market until 2023.
Hadlima is developed by Samsung Bioepis and commercialized by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co.
Visit the AGA GI Patient Center for information to share with your patients about biologics and biosimilars at https://www.gastro.org/practice-guidance/gi-patient-center/topic/biosimilars.
The Food and Drug Administration has approved the Humira biosimilar Hadlima (adalimumab-bwwd), making it the fourth adalimumab biosimilar approved in the United States, the agency announced.
Hadlima is approved for seven of the reference product’s indications, which include rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, and ulcerative colitis.
The product will launch in the United States on June 30, 2023. Other FDA-approved adalimumab biosimilars – Amjevita (adalimunab-atto), Cyltezo (adalimumab-adbm), Hyrimoz (adalimumab-adaz) – similarly will not reach the U.S. market until 2023.
Hadlima is developed by Samsung Bioepis and commercialized by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co.
Visit the AGA GI Patient Center for information to share with your patients about biologics and biosimilars at https://www.gastro.org/practice-guidance/gi-patient-center/topic/biosimilars.
The Food and Drug Administration has approved the Humira biosimilar Hadlima (adalimumab-bwwd), making it the fourth adalimumab biosimilar approved in the United States, the agency announced.
Hadlima is approved for seven of the reference product’s indications, which include rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, and ulcerative colitis.
The product will launch in the United States on June 30, 2023. Other FDA-approved adalimumab biosimilars – Amjevita (adalimunab-atto), Cyltezo (adalimumab-adbm), Hyrimoz (adalimumab-adaz) – similarly will not reach the U.S. market until 2023.
Hadlima is developed by Samsung Bioepis and commercialized by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co.
Visit the AGA GI Patient Center for information to share with your patients about biologics and biosimilars at https://www.gastro.org/practice-guidance/gi-patient-center/topic/biosimilars.
FDA finds increased blood clot, death risk associated with Xeljanz
The Food and Drug Administration has issued a safety alert approving new boxed warnings about increased blood clot and mortality risk associated with the 10-mg, twice-daily dose of tofacitinib (Xeljanz), as well as a new limitation for patients with ulcerative colitis receiving the medication.

Tofacitinib, a Janus kinase inhibitor, was first approved by the FDA in 2012 for the treatment of rheumatoid arthritis (RA). An indication for psoriatic arthritis was added in 2017, and one for ulcerative colitis was added in 2018.
After the 2012 approval, the FDA commissioned a postmarketing trial in patients with RA on background methotrexate to evaluate safety and the risk of cancer, heart-related events, and infection. The 5- and 10-mg tofacitinib twice daily doses are being analyzed in an ongoing study in comparison with a tumor necrosis factor (TNF) inhibitor.
An interim analysis of the trial’s data, as of January 2019, found an increased risk of blood clots and death in patients receiving 10-mg tofacitinib twice daily, compared with the TNF inhibitor and the twice-daily, 5-mg dose. Overall, there were 19 cases of blood clots in the lung out of 3,884 patient-years of follow-up in patients who received tofacitinib 10 mg twice daily, compared with 3 cases out of 3,982 patient-years in patients who received TNF inhibitors. There were also 45 cases of death from all causes during follow-up for tofacitinib 10 mg twice daily, compared with 25 cases in patients who received TNF inhibitors.
Patients with symptoms of thrombosis also receiving tofacitinib should immediately discontinue the medication. Tofacitinib should not be given to patients with ulcerative colitis unless they are not treated effectively with a TNF inhibitor or do not tolerate TNF inhibitors; ulcerative colitis patients should receive the lowest effective dosage, and if the higher dosage is necessary, it should be limited to the shortest amount of time possible, the FDA noted.
The Food and Drug Administration has issued a safety alert approving new boxed warnings about increased blood clot and mortality risk associated with the 10-mg, twice-daily dose of tofacitinib (Xeljanz), as well as a new limitation for patients with ulcerative colitis receiving the medication.
Tofacitinib, a Janus kinase inhibitor, was first approved by the FDA in 2012 for the treatment of rheumatoid arthritis (RA). An indication for psoriatic arthritis was added in 2017, and one for ulcerative colitis was added in 2018.
After the 2012 approval, the FDA commissioned a postmarketing trial in patients with RA on background methotrexate to evaluate safety and the risk of cancer, heart-related events, and infection. The 5- and 10-mg tofacitinib twice daily doses are being analyzed in an ongoing study in comparison with a tumor necrosis factor (TNF) inhibitor.
An interim analysis of the trial’s data, as of January 2019, found an increased risk of blood clots and death in patients receiving 10-mg tofacitinib twice daily, compared with the TNF inhibitor and the twice-daily, 5-mg dose. Overall, there were 19 cases of blood clots in the lung out of 3,884 patient-years of follow-up in patients who received tofacitinib 10 mg twice daily, compared with 3 cases out of 3,982 patient-years in patients who received TNF inhibitors. There were also 45 cases of death from all causes during follow-up for tofacitinib 10 mg twice daily, compared with 25 cases in patients who received TNF inhibitors.
Patients with symptoms of thrombosis also receiving tofacitinib should immediately discontinue the medication. Tofacitinib should not be given to patients with ulcerative colitis unless they are not treated effectively with a TNF inhibitor or do not tolerate TNF inhibitors; ulcerative colitis patients should receive the lowest effective dosage, and if the higher dosage is necessary, it should be limited to the shortest amount of time possible, the FDA noted.
The Food and Drug Administration has issued a safety alert approving new boxed warnings about increased blood clot and mortality risk associated with the 10-mg, twice-daily dose of tofacitinib (Xeljanz), as well as a new limitation for patients with ulcerative colitis receiving the medication.
Tofacitinib, a Janus kinase inhibitor, was first approved by the FDA in 2012 for the treatment of rheumatoid arthritis (RA). An indication for psoriatic arthritis was added in 2017, and one for ulcerative colitis was added in 2018.
After the 2012 approval, the FDA commissioned a postmarketing trial in patients with RA on background methotrexate to evaluate safety and the risk of cancer, heart-related events, and infection. The 5- and 10-mg tofacitinib twice daily doses are being analyzed in an ongoing study in comparison with a tumor necrosis factor (TNF) inhibitor.
An interim analysis of the trial’s data, as of January 2019, found an increased risk of blood clots and death in patients receiving 10-mg tofacitinib twice daily, compared with the TNF inhibitor and the twice-daily, 5-mg dose. Overall, there were 19 cases of blood clots in the lung out of 3,884 patient-years of follow-up in patients who received tofacitinib 10 mg twice daily, compared with 3 cases out of 3,982 patient-years in patients who received TNF inhibitors. There were also 45 cases of death from all causes during follow-up for tofacitinib 10 mg twice daily, compared with 25 cases in patients who received TNF inhibitors.
Patients with symptoms of thrombosis also receiving tofacitinib should immediately discontinue the medication. Tofacitinib should not be given to patients with ulcerative colitis unless they are not treated effectively with a TNF inhibitor or do not tolerate TNF inhibitors; ulcerative colitis patients should receive the lowest effective dosage, and if the higher dosage is necessary, it should be limited to the shortest amount of time possible, the FDA noted.
FDA advisors recommend nintedanib for SSc interstitial lung disease
The Food and Drug Administration Arthritis Advisory Committee recommended approval of nintedanib for the treatment of interstitial lung disease in patients with systemic sclerosis by a 10-7 vote on July 25, 2019. If the FDA acts in accord with the panel’s recommendation, it would make nintedanib (Ofev) the first drug to receive marketing approval for this indication.

Nintedanib has had FDA approval for treating idiopathic pulmonary fibrosis since 2014, and the manufacturer, Boehringer Ingelheim, designed the current pivotal trial with 576 patients to broaden the indication to patients with a different but similar fibrotic lung disease, interstitial lung disease (ILD), that is a common and eventually lethal complication of systemic sclerosis. The results of the pivotal study, the SENSCIS (Safety and Efficacy of Nintedanib in Systemic Sclerosis) trial, recently appeared in print and showed that patients randomized to receive 150 mg of nintedanib orally twice daily had an average 41-mL cut in the rate of loss of forced vital capacity (FVC) during 52 weeks on treatment, compared with those randomized to placebo. This was a 44% relative reduction in rate of FVC loss that was statistically significant for the study’s primary endpoint (N Engl J Med. 2019 June 27;380[26]:2518-28).
Votes in favor of FDA approval for many on the panel seemed to stem from a combination of the fact that nintedanib met the pivotal trial’s primary endpoint; which had been developed in consultation with the FDA, as well as the absence of any new safety signals when compared with prior experience using the drug; the lack of any treatment specifically recognized as beneficial to systemic sclerosis patients who develop the terminal complication of ILD; and the challenge of running a second trial in an orphan disease with an estimated U.S. prevalence of no more than 100,000 patients. Several committee members who voted in favor of nintedanib’s approval also voiced concern that the case in favor of its benefit/risk balance was not open and shut.![]()
“I have a fair amount of apprehension,” admitted the committee’s chair, Daniel H. Solomon, MD, a rheumatologist and professor of medicine at Harvard Medical School, Boston. “I support the needs of patients, but we don’t want to give them false hope. We need to be able to say who will benefit, and the single study [SENSCIS] results don’t tell us how to use the drug. I want to understand which patient subgroups benefit.” He suggested that the FDA mandate further data collection through postmarketing studies.
Comments from panel members who voted against recommending approval generally focused on what was generally agreed to be a very modest treatment effect with a 41-mL average difference in FVC decline that has marginal clinical meaningfulness. Although the SENSCIS results met the study’s primary endpoint it was neutral for all prespecified secondary endpoints, including a measure of quality of life, although many on the panel agreed that a good measure of quality of life in the target patient population is lacking. Some sensitivity analyses run by FDA staffers also failed to confirm the primary result. Fewer questions arose about safety, although some panelists expressed concern about gastrointestinal effects, especially diarrhea, that seemed to link with treatment, as well as a signal for an increased incidence of pneumonia among patients on nintedanib. The data also showed a possible signal of reduced efficacy among patients who also received treatment with the immunosuppressive agent mycophenolate mofetil, often used off label to treat systemic sclerosis patients with ILD. However, a statistician involved in the discussion warned against overinterpreting this or other subgroup analyses.
Dr. Solomon has received research support from AbbVie, Amgen, Bristol-Myers Squibb, Genentech, Janssen, and Pfizer.
The Food and Drug Administration Arthritis Advisory Committee recommended approval of nintedanib for the treatment of interstitial lung disease in patients with systemic sclerosis by a 10-7 vote on July 25, 2019. If the FDA acts in accord with the panel’s recommendation, it would make nintedanib (Ofev) the first drug to receive marketing approval for this indication.
Nintedanib has had FDA approval for treating idiopathic pulmonary fibrosis since 2014, and the manufacturer, Boehringer Ingelheim, designed the current pivotal trial with 576 patients to broaden the indication to patients with a different but similar fibrotic lung disease, interstitial lung disease (ILD), that is a common and eventually lethal complication of systemic sclerosis. The results of the pivotal study, the SENSCIS (Safety and Efficacy of Nintedanib in Systemic Sclerosis) trial, recently appeared in print and showed that patients randomized to receive 150 mg of nintedanib orally twice daily had an average 41-mL cut in the rate of loss of forced vital capacity (FVC) during 52 weeks on treatment, compared with those randomized to placebo. This was a 44% relative reduction in rate of FVC loss that was statistically significant for the study’s primary endpoint (N Engl J Med. 2019 June 27;380[26]:2518-28).
Votes in favor of FDA approval for many on the panel seemed to stem from a combination of the fact that nintedanib met the pivotal trial’s primary endpoint; which had been developed in consultation with the FDA, as well as the absence of any new safety signals when compared with prior experience using the drug; the lack of any treatment specifically recognized as beneficial to systemic sclerosis patients who develop the terminal complication of ILD; and the challenge of running a second trial in an orphan disease with an estimated U.S. prevalence of no more than 100,000 patients. Several committee members who voted in favor of nintedanib’s approval also voiced concern that the case in favor of its benefit/risk balance was not open and shut.![]()
“I have a fair amount of apprehension,” admitted the committee’s chair, Daniel H. Solomon, MD, a rheumatologist and professor of medicine at Harvard Medical School, Boston. “I support the needs of patients, but we don’t want to give them false hope. We need to be able to say who will benefit, and the single study [SENSCIS] results don’t tell us how to use the drug. I want to understand which patient subgroups benefit.” He suggested that the FDA mandate further data collection through postmarketing studies.
Comments from panel members who voted against recommending approval generally focused on what was generally agreed to be a very modest treatment effect with a 41-mL average difference in FVC decline that has marginal clinical meaningfulness. Although the SENSCIS results met the study’s primary endpoint it was neutral for all prespecified secondary endpoints, including a measure of quality of life, although many on the panel agreed that a good measure of quality of life in the target patient population is lacking. Some sensitivity analyses run by FDA staffers also failed to confirm the primary result. Fewer questions arose about safety, although some panelists expressed concern about gastrointestinal effects, especially diarrhea, that seemed to link with treatment, as well as a signal for an increased incidence of pneumonia among patients on nintedanib. The data also showed a possible signal of reduced efficacy among patients who also received treatment with the immunosuppressive agent mycophenolate mofetil, often used off label to treat systemic sclerosis patients with ILD. However, a statistician involved in the discussion warned against overinterpreting this or other subgroup analyses.
Dr. Solomon has received research support from AbbVie, Amgen, Bristol-Myers Squibb, Genentech, Janssen, and Pfizer.
The Food and Drug Administration Arthritis Advisory Committee recommended approval of nintedanib for the treatment of interstitial lung disease in patients with systemic sclerosis by a 10-7 vote on July 25, 2019. If the FDA acts in accord with the panel’s recommendation, it would make nintedanib (Ofev) the first drug to receive marketing approval for this indication.
Nintedanib has had FDA approval for treating idiopathic pulmonary fibrosis since 2014, and the manufacturer, Boehringer Ingelheim, designed the current pivotal trial with 576 patients to broaden the indication to patients with a different but similar fibrotic lung disease, interstitial lung disease (ILD), that is a common and eventually lethal complication of systemic sclerosis. The results of the pivotal study, the SENSCIS (Safety and Efficacy of Nintedanib in Systemic Sclerosis) trial, recently appeared in print and showed that patients randomized to receive 150 mg of nintedanib orally twice daily had an average 41-mL cut in the rate of loss of forced vital capacity (FVC) during 52 weeks on treatment, compared with those randomized to placebo. This was a 44% relative reduction in rate of FVC loss that was statistically significant for the study’s primary endpoint (N Engl J Med. 2019 June 27;380[26]:2518-28).
Votes in favor of FDA approval for many on the panel seemed to stem from a combination of the fact that nintedanib met the pivotal trial’s primary endpoint; which had been developed in consultation with the FDA, as well as the absence of any new safety signals when compared with prior experience using the drug; the lack of any treatment specifically recognized as beneficial to systemic sclerosis patients who develop the terminal complication of ILD; and the challenge of running a second trial in an orphan disease with an estimated U.S. prevalence of no more than 100,000 patients. Several committee members who voted in favor of nintedanib’s approval also voiced concern that the case in favor of its benefit/risk balance was not open and shut.![]()
“I have a fair amount of apprehension,” admitted the committee’s chair, Daniel H. Solomon, MD, a rheumatologist and professor of medicine at Harvard Medical School, Boston. “I support the needs of patients, but we don’t want to give them false hope. We need to be able to say who will benefit, and the single study [SENSCIS] results don’t tell us how to use the drug. I want to understand which patient subgroups benefit.” He suggested that the FDA mandate further data collection through postmarketing studies.
Comments from panel members who voted against recommending approval generally focused on what was generally agreed to be a very modest treatment effect with a 41-mL average difference in FVC decline that has marginal clinical meaningfulness. Although the SENSCIS results met the study’s primary endpoint it was neutral for all prespecified secondary endpoints, including a measure of quality of life, although many on the panel agreed that a good measure of quality of life in the target patient population is lacking. Some sensitivity analyses run by FDA staffers also failed to confirm the primary result. Fewer questions arose about safety, although some panelists expressed concern about gastrointestinal effects, especially diarrhea, that seemed to link with treatment, as well as a signal for an increased incidence of pneumonia among patients on nintedanib. The data also showed a possible signal of reduced efficacy among patients who also received treatment with the immunosuppressive agent mycophenolate mofetil, often used off label to treat systemic sclerosis patients with ILD. However, a statistician involved in the discussion warned against overinterpreting this or other subgroup analyses.
Dr. Solomon has received research support from AbbVie, Amgen, Bristol-Myers Squibb, Genentech, Janssen, and Pfizer.
Lymphoma risk prompts FDA recall of Allergan’s textured breast implants
The Food and Drug Administration requested on July 24 that Allergan pull six brands of textured breast implants and breast expanders from the U.S. market, an action the agency took because of new data that substantially increased the number of women who developed a rare cancer – anaplastic large-cell lymphoma – in association with receiving these textured breast devices.

This is the first product recall the FDA has made to address the issue of breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL), a complication that first came to national attention with a 2011 FDA report that had tallied 60 identified BIA-ALCL cases worldwide. By the end of September 2018, the number of reported worldwide BIA-ALCL cases had jumped to 457 cases reported to the agency via medical device reporting. In July 2019, the FDA cited a total of 573 unique, global case reports for BIA-ALCL sent to the agency through July 6, including 33 episodes that led to death.
It was inclusion of these additional 116 cases since September 2018 and 24 additional deaths that led FDA researchers to conclude that “the risk of BIA-ALCL with Allergan BIOCELL textured implants is approximately six-times the risk of BIA-ALCL with textured implants from other manufacturers marketing in the U.S.,” according to a statement from the agency.
The FDA is not recommending that patients who received one of the six products covered by the recall have the material removed if symptoms have not appeared because of the potential risk from explantation.
The agency also stressed that its investigation of the risk posed by placement of other brands of textured breast implants is ongoing and that overall less than 5% of all breast implants performed in current U.S. practice involve the macrotextured implants of the type specified in the Allergan recall.
This U.S. recall follows similar actions taken in France (and the rest of the European Union), Canada, and Australia, and it contrasts with the agency’s prior decision in May 2019 not to start a recall or ban of textured implants following a advisory committee meeting that discussed BIA-ALCL.
The six products that Allergan agreed to recall from marketing at the FDA’s request are four textured breast implants (Natrelle Saline-Filled Breast Implants, Natrelle Silicone-Filled Breast Implants, Natrelle Inspira Silicone-Filled breast Implants, and Natrelle 410 Highly Cohesive Anatomically Shaped Silicone-Filled Breast Implants) and two tissue expanders used prior to a breast implant (Natrelle 133 Plus Tissue Expander and the Natrelle 133 Tissue Expander with Suture Tabs).

FDA officials said they are considering recommendations for changes to the labeling of breast implant products, including a possible boxed warning and beefed up patient information.
“The recall of these textured implants is a big deal in protecting women from the potential risks of developing, and dying from, this rare type of aggressive lymphoma,” Joshua Brody, MD, a medical oncologist and director of the lymphoma immunotherapy program at Mount Sinai Medical Center in New York said in a statement. “While case reports have suggested a potential link between some types of breast implants and this disease – anaplastic lymphoma – for over 20 years, it has taken time to gain sufficient evidence to suggest, and understand, the causality. Some types of implants induce inflammation, which can both increase the chance of developing cancer, and also help to ‘hide’ developing cancers from the immune system. By preventing further use of these implants, the FDA is helping women to protect themselves from the medically serious and emotionally exhausting effects of these risks.”
Dr. Brody reported having no relevant disclosures.
The Food and Drug Administration requested on July 24 that Allergan pull six brands of textured breast implants and breast expanders from the U.S. market, an action the agency took because of new data that substantially increased the number of women who developed a rare cancer – anaplastic large-cell lymphoma – in association with receiving these textured breast devices.
This is the first product recall the FDA has made to address the issue of breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL), a complication that first came to national attention with a 2011 FDA report that had tallied 60 identified BIA-ALCL cases worldwide. By the end of September 2018, the number of reported worldwide BIA-ALCL cases had jumped to 457 cases reported to the agency via medical device reporting. In July 2019, the FDA cited a total of 573 unique, global case reports for BIA-ALCL sent to the agency through July 6, including 33 episodes that led to death.
It was inclusion of these additional 116 cases since September 2018 and 24 additional deaths that led FDA researchers to conclude that “the risk of BIA-ALCL with Allergan BIOCELL textured implants is approximately six-times the risk of BIA-ALCL with textured implants from other manufacturers marketing in the U.S.,” according to a statement from the agency.
The FDA is not recommending that patients who received one of the six products covered by the recall have the material removed if symptoms have not appeared because of the potential risk from explantation.
The agency also stressed that its investigation of the risk posed by placement of other brands of textured breast implants is ongoing and that overall less than 5% of all breast implants performed in current U.S. practice involve the macrotextured implants of the type specified in the Allergan recall.
This U.S. recall follows similar actions taken in France (and the rest of the European Union), Canada, and Australia, and it contrasts with the agency’s prior decision in May 2019 not to start a recall or ban of textured implants following a advisory committee meeting that discussed BIA-ALCL.
The six products that Allergan agreed to recall from marketing at the FDA’s request are four textured breast implants (Natrelle Saline-Filled Breast Implants, Natrelle Silicone-Filled Breast Implants, Natrelle Inspira Silicone-Filled breast Implants, and Natrelle 410 Highly Cohesive Anatomically Shaped Silicone-Filled Breast Implants) and two tissue expanders used prior to a breast implant (Natrelle 133 Plus Tissue Expander and the Natrelle 133 Tissue Expander with Suture Tabs).
FDA officials said they are considering recommendations for changes to the labeling of breast implant products, including a possible boxed warning and beefed up patient information.
“The recall of these textured implants is a big deal in protecting women from the potential risks of developing, and dying from, this rare type of aggressive lymphoma,” Joshua Brody, MD, a medical oncologist and director of the lymphoma immunotherapy program at Mount Sinai Medical Center in New York said in a statement. “While case reports have suggested a potential link between some types of breast implants and this disease – anaplastic lymphoma – for over 20 years, it has taken time to gain sufficient evidence to suggest, and understand, the causality. Some types of implants induce inflammation, which can both increase the chance of developing cancer, and also help to ‘hide’ developing cancers from the immune system. By preventing further use of these implants, the FDA is helping women to protect themselves from the medically serious and emotionally exhausting effects of these risks.”
Dr. Brody reported having no relevant disclosures.
The Food and Drug Administration requested on July 24 that Allergan pull six brands of textured breast implants and breast expanders from the U.S. market, an action the agency took because of new data that substantially increased the number of women who developed a rare cancer – anaplastic large-cell lymphoma – in association with receiving these textured breast devices.
This is the first product recall the FDA has made to address the issue of breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL), a complication that first came to national attention with a 2011 FDA report that had tallied 60 identified BIA-ALCL cases worldwide. By the end of September 2018, the number of reported worldwide BIA-ALCL cases had jumped to 457 cases reported to the agency via medical device reporting. In July 2019, the FDA cited a total of 573 unique, global case reports for BIA-ALCL sent to the agency through July 6, including 33 episodes that led to death.
It was inclusion of these additional 116 cases since September 2018 and 24 additional deaths that led FDA researchers to conclude that “the risk of BIA-ALCL with Allergan BIOCELL textured implants is approximately six-times the risk of BIA-ALCL with textured implants from other manufacturers marketing in the U.S.,” according to a statement from the agency.
The FDA is not recommending that patients who received one of the six products covered by the recall have the material removed if symptoms have not appeared because of the potential risk from explantation.
The agency also stressed that its investigation of the risk posed by placement of other brands of textured breast implants is ongoing and that overall less than 5% of all breast implants performed in current U.S. practice involve the macrotextured implants of the type specified in the Allergan recall.
This U.S. recall follows similar actions taken in France (and the rest of the European Union), Canada, and Australia, and it contrasts with the agency’s prior decision in May 2019 not to start a recall or ban of textured implants following a advisory committee meeting that discussed BIA-ALCL.
The six products that Allergan agreed to recall from marketing at the FDA’s request are four textured breast implants (Natrelle Saline-Filled Breast Implants, Natrelle Silicone-Filled Breast Implants, Natrelle Inspira Silicone-Filled breast Implants, and Natrelle 410 Highly Cohesive Anatomically Shaped Silicone-Filled Breast Implants) and two tissue expanders used prior to a breast implant (Natrelle 133 Plus Tissue Expander and the Natrelle 133 Tissue Expander with Suture Tabs).
FDA officials said they are considering recommendations for changes to the labeling of breast implant products, including a possible boxed warning and beefed up patient information.
“The recall of these textured implants is a big deal in protecting women from the potential risks of developing, and dying from, this rare type of aggressive lymphoma,” Joshua Brody, MD, a medical oncologist and director of the lymphoma immunotherapy program at Mount Sinai Medical Center in New York said in a statement. “While case reports have suggested a potential link between some types of breast implants and this disease – anaplastic lymphoma – for over 20 years, it has taken time to gain sufficient evidence to suggest, and understand, the causality. Some types of implants induce inflammation, which can both increase the chance of developing cancer, and also help to ‘hide’ developing cancers from the immune system. By preventing further use of these implants, the FDA is helping women to protect themselves from the medically serious and emotionally exhausting effects of these risks.”
Dr. Brody reported having no relevant disclosures.
Hadlima approved as fourth adalimumab biosimilar in U.S.
The Food and Drug Administration has approved the Humira biosimilar Hadlima (adalimumab-bwwd), making it the fourth adalimumab biosimilar approved in the United States, the agency announced.
Hadlima is approved for seven of the reference product’s indications, which include rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, and ulcerative colitis.
The product will launch in the United States on June 30, 2023. Other FDA-approved adalimumab biosimilars – Amjevita (adalimunab-atto), Cyltezo (adalimumab-adbm), Hyrimoz (adalimumab-adaz) – similarly will not reach the U.S. market until 2023.
Hadlima is developed by Samsung Bioepis and commercialized by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co.
*This article was updated on July 24, 2019.
The Food and Drug Administration has approved the Humira biosimilar Hadlima (adalimumab-bwwd), making it the fourth adalimumab biosimilar approved in the United States, the agency announced.
Hadlima is approved for seven of the reference product’s indications, which include rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, and ulcerative colitis.
The product will launch in the United States on June 30, 2023. Other FDA-approved adalimumab biosimilars – Amjevita (adalimunab-atto), Cyltezo (adalimumab-adbm), Hyrimoz (adalimumab-adaz) – similarly will not reach the U.S. market until 2023.
Hadlima is developed by Samsung Bioepis and commercialized by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co.
*This article was updated on July 24, 2019.
The Food and Drug Administration has approved the Humira biosimilar Hadlima (adalimumab-bwwd), making it the fourth adalimumab biosimilar approved in the United States, the agency announced.
Hadlima is approved for seven of the reference product’s indications, which include rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, and ulcerative colitis.
The product will launch in the United States on June 30, 2023. Other FDA-approved adalimumab biosimilars – Amjevita (adalimunab-atto), Cyltezo (adalimumab-adbm), Hyrimoz (adalimumab-adaz) – similarly will not reach the U.S. market until 2023.
Hadlima is developed by Samsung Bioepis and commercialized by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co.
*This article was updated on July 24, 2019.
FDA approves rituximab biosimilar for cancer, autoimmune disorders
The Food and Drug Administration has approved rituximab-pvvr (Ruxience) for adults with non-Hodgkin lymphoma, chronic lymphocytic leukemia (CLL), and granulomatosis with polyangiitis and microscopic polyangiitis. It is the first biosimilar approved to treat these two rare autoimmune conditions.
Specifically, the biosimilar product is approved as single-agent therapy for relapsed or refractory, low grade or follicular, CD20-positive B-cell non-Hodgkin lymphoma; in combination with chemotherapy for other types of previously untreated CD20-positive B-cell non-Hodgkin lymphoma; and as a single agent for nonprogressing, low-grade, CD20-positive B-cell non-Hodgkin lymphoma after first-line chemotherapy treatment. It is also approved for both previously untreated and previously treated CD20-positive CLL in combination with chemotherapy. And it is approved for granulomatosis with polyangiitis and microscopic polyangiitis in combination with glucocorticoids.
The approval is based on demonstration that rituximab-pvvr had no clinically meaningful differences in safety or efficacy when compared with the reference drug, rituximab (Rituxan), according to a release from the biosimilar’s developer. As with rituximab, rituximab-pvvr’s label comes with an FDA boxed warning. In the biosimilar’s case, it warns against fatal infusion-related reactions, severe mucocutaneous reactions, hepatitis B virus reactivation, and progressive multifocal leukoencephalopathy. Other adverse reactions include fever, headache, neutropenia, and lymphopenia.
The Food and Drug Administration has approved rituximab-pvvr (Ruxience) for adults with non-Hodgkin lymphoma, chronic lymphocytic leukemia (CLL), and granulomatosis with polyangiitis and microscopic polyangiitis. It is the first biosimilar approved to treat these two rare autoimmune conditions.
Specifically, the biosimilar product is approved as single-agent therapy for relapsed or refractory, low grade or follicular, CD20-positive B-cell non-Hodgkin lymphoma; in combination with chemotherapy for other types of previously untreated CD20-positive B-cell non-Hodgkin lymphoma; and as a single agent for nonprogressing, low-grade, CD20-positive B-cell non-Hodgkin lymphoma after first-line chemotherapy treatment. It is also approved for both previously untreated and previously treated CD20-positive CLL in combination with chemotherapy. And it is approved for granulomatosis with polyangiitis and microscopic polyangiitis in combination with glucocorticoids.
The approval is based on demonstration that rituximab-pvvr had no clinically meaningful differences in safety or efficacy when compared with the reference drug, rituximab (Rituxan), according to a release from the biosimilar’s developer. As with rituximab, rituximab-pvvr’s label comes with an FDA boxed warning. In the biosimilar’s case, it warns against fatal infusion-related reactions, severe mucocutaneous reactions, hepatitis B virus reactivation, and progressive multifocal leukoencephalopathy. Other adverse reactions include fever, headache, neutropenia, and lymphopenia.
The Food and Drug Administration has approved rituximab-pvvr (Ruxience) for adults with non-Hodgkin lymphoma, chronic lymphocytic leukemia (CLL), and granulomatosis with polyangiitis and microscopic polyangiitis. It is the first biosimilar approved to treat these two rare autoimmune conditions.
Specifically, the biosimilar product is approved as single-agent therapy for relapsed or refractory, low grade or follicular, CD20-positive B-cell non-Hodgkin lymphoma; in combination with chemotherapy for other types of previously untreated CD20-positive B-cell non-Hodgkin lymphoma; and as a single agent for nonprogressing, low-grade, CD20-positive B-cell non-Hodgkin lymphoma after first-line chemotherapy treatment. It is also approved for both previously untreated and previously treated CD20-positive CLL in combination with chemotherapy. And it is approved for granulomatosis with polyangiitis and microscopic polyangiitis in combination with glucocorticoids.
The approval is based on demonstration that rituximab-pvvr had no clinically meaningful differences in safety or efficacy when compared with the reference drug, rituximab (Rituxan), according to a release from the biosimilar’s developer. As with rituximab, rituximab-pvvr’s label comes with an FDA boxed warning. In the biosimilar’s case, it warns against fatal infusion-related reactions, severe mucocutaneous reactions, hepatitis B virus reactivation, and progressive multifocal leukoencephalopathy. Other adverse reactions include fever, headache, neutropenia, and lymphopenia.
FDA approves Otezla for treatment of Behçet’s-associated oral ulcers
The Food and Drug Administration has expanded the indication for apremilast (Otezla) to include the treatment of oral ulcers associated with Behçet’s disease in adults, according to an announcement from the manufacturer, Celgene.
FDA approval was based on results of the randomized, placebo-controlled, double-blind, phase 3 RELIEF trial, in which 207 patients with Behçet’s disease with active ulcers underwent treatment for 12 weeks with 30 mg apremilast or placebo. When measured on a visual analog scale, the reduction in pain from oral ulcers after 12 weeks in patients receiving apremilast was 42.7 points, compared with 18.7 points in the placebo group. Just over 50% of apremilast patients achieved complete response by week 12, compared with 22.3% in the placebo group.
The most common adverse events associated with apremilast during RELIEF were diarrhea, nausea, headache, and upper respiratory infection. This was consistent with apremilast’s known safety profile.
Apremilast is also indicated for treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy, and for patients with active psoriatic arthritis.
“Oral ulcers are a recurring and debilitating manifestation that affects nearly everyone living with Behçet’s disease and have an important negative impact on the quality of life for these patients. In the clinical trial, Otezla demonstrated improvements in measures of oral ulcers at week 12. Otezla has the potential to be a needed treatment option for U.S. patients and their physicians, who previously had limited options available,” Yusuf Yazici, MD, clinical associate professor in the department of medicine at New York University, said in the announcement.
The Food and Drug Administration has expanded the indication for apremilast (Otezla) to include the treatment of oral ulcers associated with Behçet’s disease in adults, according to an announcement from the manufacturer, Celgene.
FDA approval was based on results of the randomized, placebo-controlled, double-blind, phase 3 RELIEF trial, in which 207 patients with Behçet’s disease with active ulcers underwent treatment for 12 weeks with 30 mg apremilast or placebo. When measured on a visual analog scale, the reduction in pain from oral ulcers after 12 weeks in patients receiving apremilast was 42.7 points, compared with 18.7 points in the placebo group. Just over 50% of apremilast patients achieved complete response by week 12, compared with 22.3% in the placebo group.
The most common adverse events associated with apremilast during RELIEF were diarrhea, nausea, headache, and upper respiratory infection. This was consistent with apremilast’s known safety profile.
Apremilast is also indicated for treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy, and for patients with active psoriatic arthritis.
“Oral ulcers are a recurring and debilitating manifestation that affects nearly everyone living with Behçet’s disease and have an important negative impact on the quality of life for these patients. In the clinical trial, Otezla demonstrated improvements in measures of oral ulcers at week 12. Otezla has the potential to be a needed treatment option for U.S. patients and their physicians, who previously had limited options available,” Yusuf Yazici, MD, clinical associate professor in the department of medicine at New York University, said in the announcement.
The Food and Drug Administration has expanded the indication for apremilast (Otezla) to include the treatment of oral ulcers associated with Behçet’s disease in adults, according to an announcement from the manufacturer, Celgene.
FDA approval was based on results of the randomized, placebo-controlled, double-blind, phase 3 RELIEF trial, in which 207 patients with Behçet’s disease with active ulcers underwent treatment for 12 weeks with 30 mg apremilast or placebo. When measured on a visual analog scale, the reduction in pain from oral ulcers after 12 weeks in patients receiving apremilast was 42.7 points, compared with 18.7 points in the placebo group. Just over 50% of apremilast patients achieved complete response by week 12, compared with 22.3% in the placebo group.
The most common adverse events associated with apremilast during RELIEF were diarrhea, nausea, headache, and upper respiratory infection. This was consistent with apremilast’s known safety profile.
Apremilast is also indicated for treatment of patients with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy, and for patients with active psoriatic arthritis.
“Oral ulcers are a recurring and debilitating manifestation that affects nearly everyone living with Behçet’s disease and have an important negative impact on the quality of life for these patients. In the clinical trial, Otezla demonstrated improvements in measures of oral ulcers at week 12. Otezla has the potential to be a needed treatment option for U.S. patients and their physicians, who previously had limited options available,” Yusuf Yazici, MD, clinical associate professor in the department of medicine at New York University, said in the announcement.