User login
New transfusion guidelines for thalassemia
Fresher blood products are not necessarily better for patients with beta thalassemia, according to a pair of experts.

Red blood cell units stored less than 2 weeks are ideal, but older units are acceptable, and phenotype matching should take priority over unit age, advised Ashutosh Lal, MD, and Elliott Vichinsky, MD, both of UCSF Benioff Children’s Hospital Oakland (Calif.). They discussed these and other recommendations for transfusing patients with thalassemia during a webinar hosted by the Centers for Disease Control and Prevention.
Indications for transfusion
Dr. Lal said patients with beta thalassemia major should be transfused if their hemoglobin is less than 7 g/dL on two occasions 2 weeks apart at baseline, or if their hemoglobin is greater than 7 g/dL and they have symptoms of anemia.
Patients with hemoglobin E beta thalassemia major should be transfused only if they have symptoms of anemia.
“The rationale is that, in beta thalassemia major, it is well established that, once the hemoglobin levels fall below 7 g/dL in young children, there is going to be massive bone marrow expansion, and there will be severe symptoms from anemia,” Dr. Lal said. “But the relationship of hemoglobin with symptoms in E beta thalassemia is less precise.”
The symptoms that should prompt transfusion include slowed growth, skeletal facial changes, splenomegaly, symptomatic or moderate to severe extramedullary hematopoiesis, cerebrovascular events, venous thromboembolism, pulmonary hypertension, osteoporotic fracture, and impaired quality of life in adults.
Dr. Lal said physicians should consider a 6-month trial of transfusions if the indication is unclear. He also noted that red cell antigen genotyping should be performed in all patients who may need transfusions.
Blood products
Dr. Lal said beta thalassemia patients should receive packed red blood cells that are leukoreduced prior to storage. The storage solution can be citrate-phosphate-dextrose solution with adenine (hematocrit 75%) or additive solution (hematocrit 60%).
“It’s important to note that the hematocrit of the two is quite different, and that needs to be inculcated into the decisions on how much volume to transfuse to younger children,” Dr. Lal said.
He noted that units should not be irradiated, as this damages the red cell membrane. And patients with severe allergic reactions should receive washed red blood cells because washing units removes residual donor plasma proteins.
Finally, units should be less than 2 weeks old if possible. Dr. Lal said using fresh units increases the survival of red blood cells post transfusion. However, he and Dr. Vichinsky both stressed that older units are acceptable, and phenotype matching is more important than the age of the unit.
Phenotype matching
Beta thalassemia patients who do not have preexisting alloantibodies or have transient autoantibodies should be matched to Rh and Kell, according to Dr. Lal.
Patients with preexisting alloantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and the specific alloantibody. Patients with persistent autoantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and any alloantibody.
Patients who start transfusions after 5 years of age should be matched to Rh, Kell, Duffy, Kidd, and S. Pregnant patients should be matched to Rh, Kell, Duffy, Kidd, and S, and units should be cytomegalovirus negative.
How to transfuse
Dr. Lal said the pretransfusion hemoglobin target is 10 g/dL, with a range of 9.5-10.5 g/dL in beta thalassemia major and a range of 9.0-10.5 g/dL for E beta thalassemia. A target of 10 g/dL is adequate for most individuals, Dr. Lal said, but he recommends individualization of hemoglobin target for patients with E beta thalassemia.
In general, patients should be transfused every 3 weeks, although 4 weeks is acceptable in younger children and those with hemoglobin E beta thalassemia.
As for the volume of a transfusion, children should receive 4 mL per kg of body weight, per gram increase in hemoglobin desired. Partial units can be used to avoid undertransfusion.
For adults, in general, those with pretransfusion hemoglobin less than 10 g/dL should receive three units, and those with pretransfusion hemoglobin of 10 g/dL or greater should receive two units.
The hemoglobin threshold should be adjusted based on fatigue or bone pain, Dr. Lal said. He also noted that patients with intact spleens have higher transfusion needs.
The rate of transfusion should be 5 mL/kg/hour in children and 200-300 mL/hour in adults, based on tolerance. Patients with impaired cardiac function should receive a reduced blood volume at a reduced rate.
Non–transfusion dependent thalassemia
Dr. Vichinsky discussed recommendations for non–transfusion dependent thalassemia (NTDT), noting that these patients may need transient transfusions to prevent morbidity.

Hemoglobin should not be the sole determinant of transfusion need in NTDT patients, he said. Their well-being – activity level, growth, and skeletal changes – is more important than hemoglobin levels. However, patients with hemoglobin levels less than 7 g/dL often have severe morbidity, and those with levels of 10 g/dL or greater are usually protected from severe morbidity.
Indications for transfusion in NTDT patients include:
- Growth failure.
- Hematopoietic tumors.
- Pulmonary hypertension.
- Silent brain infarcts.
- Skin ulcers.
- Severe bone pain.
- Poor quality of life.
- Frequent hemolytic crises.
- Marked and enlarging spleen.
- Failure of secondary sex development.
- Cosmetic and facial changes.
- Pregnancy.
“There is a risk to transfusing this population,” Dr. Vichinsky said. “They’re older, and when you transfuse them, they can get iron overloaded.”
He added that splenectomized NTDT patients have a high risk of alloimmunization, and the transfusion duration should be serially reevaluated in NTDT patients.
Alpha thalassemia major
For alpha thalassemia major, Dr. Vichinsky discussed the importance of prevention, screening, and fetal therapy. He said couples with a fetus at risk of alpha thalassemia major should be identified early and offered, in addition to termination, the option of early fetal transfusion.
Dr. Vichinsky recommended prenatal testing and monitoring of at-risk pregnancies with ultrasound. If the fetus requires a transfusion, monitoring hemoglobin Barts and hemoglobin A is necessary.
A fetus that requires a transfusion should receive packed red blood cells that are cytomegalovirus negative, are less than 7 days old, have been irradiated, have a hemoglobin mass greater than 75%, and have been optimally cross matched with the mother first.
“These babies appear, with serial transfusions, to survive and have a relatively normal neonatal period,” Dr. Vichinsky said.
He added, however, that postnatal management of alpha thalassemia major involves an aggressive transfusion protocol. These patients should be transfused to a higher hemoglobin level than patients with beta thalassemia – roughly 12 g/dL versus 10 g/dL.
These and Dr. Lal’s recommendations are based on information in the Standards of Care Guidelines for Thalassemia – Oakland 2011, the Thalassemia International Federation Guidelines – 2014, the Thalassemia Management Checklists: United States – 2018, the Thalassemia Western Consortium Consensus: US – 2019, and the International Collaboration for Transfusion Medicine Guidelines – 2019.
Dr. Lal and Dr. Vichinsky did not disclose any conflicts of interest.
Fresher blood products are not necessarily better for patients with beta thalassemia, according to a pair of experts.
Red blood cell units stored less than 2 weeks are ideal, but older units are acceptable, and phenotype matching should take priority over unit age, advised Ashutosh Lal, MD, and Elliott Vichinsky, MD, both of UCSF Benioff Children’s Hospital Oakland (Calif.). They discussed these and other recommendations for transfusing patients with thalassemia during a webinar hosted by the Centers for Disease Control and Prevention.
Indications for transfusion
Dr. Lal said patients with beta thalassemia major should be transfused if their hemoglobin is less than 7 g/dL on two occasions 2 weeks apart at baseline, or if their hemoglobin is greater than 7 g/dL and they have symptoms of anemia.
Patients with hemoglobin E beta thalassemia major should be transfused only if they have symptoms of anemia.
“The rationale is that, in beta thalassemia major, it is well established that, once the hemoglobin levels fall below 7 g/dL in young children, there is going to be massive bone marrow expansion, and there will be severe symptoms from anemia,” Dr. Lal said. “But the relationship of hemoglobin with symptoms in E beta thalassemia is less precise.”
The symptoms that should prompt transfusion include slowed growth, skeletal facial changes, splenomegaly, symptomatic or moderate to severe extramedullary hematopoiesis, cerebrovascular events, venous thromboembolism, pulmonary hypertension, osteoporotic fracture, and impaired quality of life in adults.
Dr. Lal said physicians should consider a 6-month trial of transfusions if the indication is unclear. He also noted that red cell antigen genotyping should be performed in all patients who may need transfusions.
Blood products
Dr. Lal said beta thalassemia patients should receive packed red blood cells that are leukoreduced prior to storage. The storage solution can be citrate-phosphate-dextrose solution with adenine (hematocrit 75%) or additive solution (hematocrit 60%).
“It’s important to note that the hematocrit of the two is quite different, and that needs to be inculcated into the decisions on how much volume to transfuse to younger children,” Dr. Lal said.
He noted that units should not be irradiated, as this damages the red cell membrane. And patients with severe allergic reactions should receive washed red blood cells because washing units removes residual donor plasma proteins.
Finally, units should be less than 2 weeks old if possible. Dr. Lal said using fresh units increases the survival of red blood cells post transfusion. However, he and Dr. Vichinsky both stressed that older units are acceptable, and phenotype matching is more important than the age of the unit.
Phenotype matching
Beta thalassemia patients who do not have preexisting alloantibodies or have transient autoantibodies should be matched to Rh and Kell, according to Dr. Lal.
Patients with preexisting alloantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and the specific alloantibody. Patients with persistent autoantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and any alloantibody.
Patients who start transfusions after 5 years of age should be matched to Rh, Kell, Duffy, Kidd, and S. Pregnant patients should be matched to Rh, Kell, Duffy, Kidd, and S, and units should be cytomegalovirus negative.
How to transfuse
Dr. Lal said the pretransfusion hemoglobin target is 10 g/dL, with a range of 9.5-10.5 g/dL in beta thalassemia major and a range of 9.0-10.5 g/dL for E beta thalassemia. A target of 10 g/dL is adequate for most individuals, Dr. Lal said, but he recommends individualization of hemoglobin target for patients with E beta thalassemia.
In general, patients should be transfused every 3 weeks, although 4 weeks is acceptable in younger children and those with hemoglobin E beta thalassemia.
As for the volume of a transfusion, children should receive 4 mL per kg of body weight, per gram increase in hemoglobin desired. Partial units can be used to avoid undertransfusion.
For adults, in general, those with pretransfusion hemoglobin less than 10 g/dL should receive three units, and those with pretransfusion hemoglobin of 10 g/dL or greater should receive two units.
The hemoglobin threshold should be adjusted based on fatigue or bone pain, Dr. Lal said. He also noted that patients with intact spleens have higher transfusion needs.
The rate of transfusion should be 5 mL/kg/hour in children and 200-300 mL/hour in adults, based on tolerance. Patients with impaired cardiac function should receive a reduced blood volume at a reduced rate.
Non–transfusion dependent thalassemia
Dr. Vichinsky discussed recommendations for non–transfusion dependent thalassemia (NTDT), noting that these patients may need transient transfusions to prevent morbidity.
Hemoglobin should not be the sole determinant of transfusion need in NTDT patients, he said. Their well-being – activity level, growth, and skeletal changes – is more important than hemoglobin levels. However, patients with hemoglobin levels less than 7 g/dL often have severe morbidity, and those with levels of 10 g/dL or greater are usually protected from severe morbidity.
Indications for transfusion in NTDT patients include:
- Growth failure.
- Hematopoietic tumors.
- Pulmonary hypertension.
- Silent brain infarcts.
- Skin ulcers.
- Severe bone pain.
- Poor quality of life.
- Frequent hemolytic crises.
- Marked and enlarging spleen.
- Failure of secondary sex development.
- Cosmetic and facial changes.
- Pregnancy.
“There is a risk to transfusing this population,” Dr. Vichinsky said. “They’re older, and when you transfuse them, they can get iron overloaded.”
He added that splenectomized NTDT patients have a high risk of alloimmunization, and the transfusion duration should be serially reevaluated in NTDT patients.
Alpha thalassemia major
For alpha thalassemia major, Dr. Vichinsky discussed the importance of prevention, screening, and fetal therapy. He said couples with a fetus at risk of alpha thalassemia major should be identified early and offered, in addition to termination, the option of early fetal transfusion.
Dr. Vichinsky recommended prenatal testing and monitoring of at-risk pregnancies with ultrasound. If the fetus requires a transfusion, monitoring hemoglobin Barts and hemoglobin A is necessary.
A fetus that requires a transfusion should receive packed red blood cells that are cytomegalovirus negative, are less than 7 days old, have been irradiated, have a hemoglobin mass greater than 75%, and have been optimally cross matched with the mother first.
“These babies appear, with serial transfusions, to survive and have a relatively normal neonatal period,” Dr. Vichinsky said.
He added, however, that postnatal management of alpha thalassemia major involves an aggressive transfusion protocol. These patients should be transfused to a higher hemoglobin level than patients with beta thalassemia – roughly 12 g/dL versus 10 g/dL.
These and Dr. Lal’s recommendations are based on information in the Standards of Care Guidelines for Thalassemia – Oakland 2011, the Thalassemia International Federation Guidelines – 2014, the Thalassemia Management Checklists: United States – 2018, the Thalassemia Western Consortium Consensus: US – 2019, and the International Collaboration for Transfusion Medicine Guidelines – 2019.
Dr. Lal and Dr. Vichinsky did not disclose any conflicts of interest.
Fresher blood products are not necessarily better for patients with beta thalassemia, according to a pair of experts.
Red blood cell units stored less than 2 weeks are ideal, but older units are acceptable, and phenotype matching should take priority over unit age, advised Ashutosh Lal, MD, and Elliott Vichinsky, MD, both of UCSF Benioff Children’s Hospital Oakland (Calif.). They discussed these and other recommendations for transfusing patients with thalassemia during a webinar hosted by the Centers for Disease Control and Prevention.
Indications for transfusion
Dr. Lal said patients with beta thalassemia major should be transfused if their hemoglobin is less than 7 g/dL on two occasions 2 weeks apart at baseline, or if their hemoglobin is greater than 7 g/dL and they have symptoms of anemia.
Patients with hemoglobin E beta thalassemia major should be transfused only if they have symptoms of anemia.
“The rationale is that, in beta thalassemia major, it is well established that, once the hemoglobin levels fall below 7 g/dL in young children, there is going to be massive bone marrow expansion, and there will be severe symptoms from anemia,” Dr. Lal said. “But the relationship of hemoglobin with symptoms in E beta thalassemia is less precise.”
The symptoms that should prompt transfusion include slowed growth, skeletal facial changes, splenomegaly, symptomatic or moderate to severe extramedullary hematopoiesis, cerebrovascular events, venous thromboembolism, pulmonary hypertension, osteoporotic fracture, and impaired quality of life in adults.
Dr. Lal said physicians should consider a 6-month trial of transfusions if the indication is unclear. He also noted that red cell antigen genotyping should be performed in all patients who may need transfusions.
Blood products
Dr. Lal said beta thalassemia patients should receive packed red blood cells that are leukoreduced prior to storage. The storage solution can be citrate-phosphate-dextrose solution with adenine (hematocrit 75%) or additive solution (hematocrit 60%).
“It’s important to note that the hematocrit of the two is quite different, and that needs to be inculcated into the decisions on how much volume to transfuse to younger children,” Dr. Lal said.
He noted that units should not be irradiated, as this damages the red cell membrane. And patients with severe allergic reactions should receive washed red blood cells because washing units removes residual donor plasma proteins.
Finally, units should be less than 2 weeks old if possible. Dr. Lal said using fresh units increases the survival of red blood cells post transfusion. However, he and Dr. Vichinsky both stressed that older units are acceptable, and phenotype matching is more important than the age of the unit.
Phenotype matching
Beta thalassemia patients who do not have preexisting alloantibodies or have transient autoantibodies should be matched to Rh and Kell, according to Dr. Lal.
Patients with preexisting alloantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and the specific alloantibody. Patients with persistent autoantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and any alloantibody.
Patients who start transfusions after 5 years of age should be matched to Rh, Kell, Duffy, Kidd, and S. Pregnant patients should be matched to Rh, Kell, Duffy, Kidd, and S, and units should be cytomegalovirus negative.
How to transfuse
Dr. Lal said the pretransfusion hemoglobin target is 10 g/dL, with a range of 9.5-10.5 g/dL in beta thalassemia major and a range of 9.0-10.5 g/dL for E beta thalassemia. A target of 10 g/dL is adequate for most individuals, Dr. Lal said, but he recommends individualization of hemoglobin target for patients with E beta thalassemia.
In general, patients should be transfused every 3 weeks, although 4 weeks is acceptable in younger children and those with hemoglobin E beta thalassemia.
As for the volume of a transfusion, children should receive 4 mL per kg of body weight, per gram increase in hemoglobin desired. Partial units can be used to avoid undertransfusion.
For adults, in general, those with pretransfusion hemoglobin less than 10 g/dL should receive three units, and those with pretransfusion hemoglobin of 10 g/dL or greater should receive two units.
The hemoglobin threshold should be adjusted based on fatigue or bone pain, Dr. Lal said. He also noted that patients with intact spleens have higher transfusion needs.
The rate of transfusion should be 5 mL/kg/hour in children and 200-300 mL/hour in adults, based on tolerance. Patients with impaired cardiac function should receive a reduced blood volume at a reduced rate.
Non–transfusion dependent thalassemia
Dr. Vichinsky discussed recommendations for non–transfusion dependent thalassemia (NTDT), noting that these patients may need transient transfusions to prevent morbidity.
Hemoglobin should not be the sole determinant of transfusion need in NTDT patients, he said. Their well-being – activity level, growth, and skeletal changes – is more important than hemoglobin levels. However, patients with hemoglobin levels less than 7 g/dL often have severe morbidity, and those with levels of 10 g/dL or greater are usually protected from severe morbidity.
Indications for transfusion in NTDT patients include:
- Growth failure.
- Hematopoietic tumors.
- Pulmonary hypertension.
- Silent brain infarcts.
- Skin ulcers.
- Severe bone pain.
- Poor quality of life.
- Frequent hemolytic crises.
- Marked and enlarging spleen.
- Failure of secondary sex development.
- Cosmetic and facial changes.
- Pregnancy.
“There is a risk to transfusing this population,” Dr. Vichinsky said. “They’re older, and when you transfuse them, they can get iron overloaded.”
He added that splenectomized NTDT patients have a high risk of alloimmunization, and the transfusion duration should be serially reevaluated in NTDT patients.
Alpha thalassemia major
For alpha thalassemia major, Dr. Vichinsky discussed the importance of prevention, screening, and fetal therapy. He said couples with a fetus at risk of alpha thalassemia major should be identified early and offered, in addition to termination, the option of early fetal transfusion.
Dr. Vichinsky recommended prenatal testing and monitoring of at-risk pregnancies with ultrasound. If the fetus requires a transfusion, monitoring hemoglobin Barts and hemoglobin A is necessary.
A fetus that requires a transfusion should receive packed red blood cells that are cytomegalovirus negative, are less than 7 days old, have been irradiated, have a hemoglobin mass greater than 75%, and have been optimally cross matched with the mother first.
“These babies appear, with serial transfusions, to survive and have a relatively normal neonatal period,” Dr. Vichinsky said.
He added, however, that postnatal management of alpha thalassemia major involves an aggressive transfusion protocol. These patients should be transfused to a higher hemoglobin level than patients with beta thalassemia – roughly 12 g/dL versus 10 g/dL.
These and Dr. Lal’s recommendations are based on information in the Standards of Care Guidelines for Thalassemia – Oakland 2011, the Thalassemia International Federation Guidelines – 2014, the Thalassemia Management Checklists: United States – 2018, the Thalassemia Western Consortium Consensus: US – 2019, and the International Collaboration for Transfusion Medicine Guidelines – 2019.
Dr. Lal and Dr. Vichinsky did not disclose any conflicts of interest.
FDA approves upadacitinib for rheumatoid arthritis
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.

The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.
The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.
The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
Prescription drug use varies between U.S. and Canada
The United States and Canada deliver health care in different ways, and patterns of prescription drug use also vary between the two countries, according to the National Center for Health Statistics.
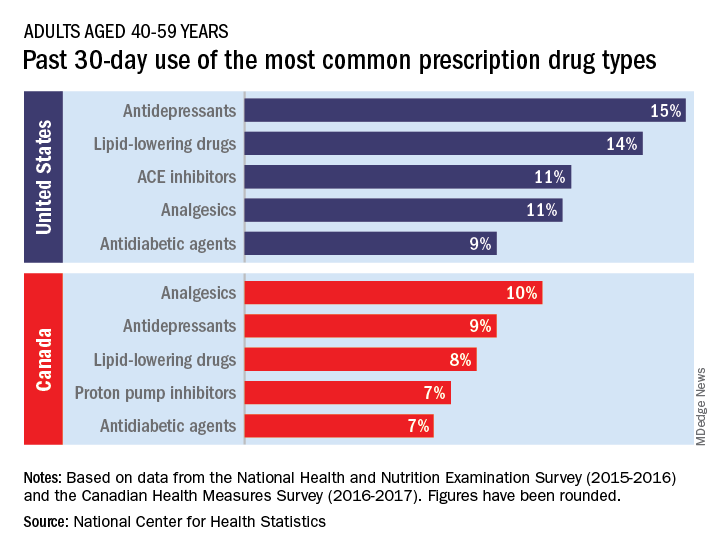
The populations of the two countries, however, have similar age distributions – adults aged 40-79 years made up 44% of the population in the United States and 48% in Canada in 2016 – so “monitoring the use of prescription drugs provides insights into the health and health care of U.S. and Canadian adults,” the NCHS investigators wrote.
Data from the 2015-2016 National Health and Nutritional Examination Survey show that 15% of Americans aged 40-59 years had used antidepressants in the past 30 days, putting them ahead of lipid-lowering drugs (14%) and ACE inhibitors (11%), the NCHS said in a recent Data Brief.
Analgesics were the leading drug type in Canada, with 10% of adults aged 40-59 years reporting use in the past month, although that’s still lower than in the United States (11%), where they were fourth in popularity. The American top two were second and third among Canadians, while proton pump inhibitors were fourth in Canada but did not crack the top five in the United States, the NCHS reported based on 2016-2017 data from the Canadian Health Measures Survey.
Older adults (60-79 years) in the two countries managed to share some common ground: Lipid-lowering drugs were the most commonly used prescription medication both north and south of the border, although past 30-day use was considerably higher in the United States (45% vs. 34%), the NCHS investigators said.
There were differences to be found, however, in the older age group. Analgesics were the second most commonly used drug type in Canada but did not even reach the top five in the United States, while beta-blockers were third among Americans but missed the Canadian top five, they noted.
(60% vs. 53%) and to have used at least five such drugs (15% vs. 10%). The differences among adults aged 60-79 were not significant, although American use was higher for at least one drug (84% vs. 83%) and for at least five (35% vs. 31%), according to the report.
The United States and Canada deliver health care in different ways, and patterns of prescription drug use also vary between the two countries, according to the National Center for Health Statistics.
The populations of the two countries, however, have similar age distributions – adults aged 40-79 years made up 44% of the population in the United States and 48% in Canada in 2016 – so “monitoring the use of prescription drugs provides insights into the health and health care of U.S. and Canadian adults,” the NCHS investigators wrote.
Data from the 2015-2016 National Health and Nutritional Examination Survey show that 15% of Americans aged 40-59 years had used antidepressants in the past 30 days, putting them ahead of lipid-lowering drugs (14%) and ACE inhibitors (11%), the NCHS said in a recent Data Brief.
Analgesics were the leading drug type in Canada, with 10% of adults aged 40-59 years reporting use in the past month, although that’s still lower than in the United States (11%), where they were fourth in popularity. The American top two were second and third among Canadians, while proton pump inhibitors were fourth in Canada but did not crack the top five in the United States, the NCHS reported based on 2016-2017 data from the Canadian Health Measures Survey.
Older adults (60-79 years) in the two countries managed to share some common ground: Lipid-lowering drugs were the most commonly used prescription medication both north and south of the border, although past 30-day use was considerably higher in the United States (45% vs. 34%), the NCHS investigators said.
There were differences to be found, however, in the older age group. Analgesics were the second most commonly used drug type in Canada but did not even reach the top five in the United States, while beta-blockers were third among Americans but missed the Canadian top five, they noted.
(60% vs. 53%) and to have used at least five such drugs (15% vs. 10%). The differences among adults aged 60-79 were not significant, although American use was higher for at least one drug (84% vs. 83%) and for at least five (35% vs. 31%), according to the report.
The United States and Canada deliver health care in different ways, and patterns of prescription drug use also vary between the two countries, according to the National Center for Health Statistics.
The populations of the two countries, however, have similar age distributions – adults aged 40-79 years made up 44% of the population in the United States and 48% in Canada in 2016 – so “monitoring the use of prescription drugs provides insights into the health and health care of U.S. and Canadian adults,” the NCHS investigators wrote.
Data from the 2015-2016 National Health and Nutritional Examination Survey show that 15% of Americans aged 40-59 years had used antidepressants in the past 30 days, putting them ahead of lipid-lowering drugs (14%) and ACE inhibitors (11%), the NCHS said in a recent Data Brief.
Analgesics were the leading drug type in Canada, with 10% of adults aged 40-59 years reporting use in the past month, although that’s still lower than in the United States (11%), where they were fourth in popularity. The American top two were second and third among Canadians, while proton pump inhibitors were fourth in Canada but did not crack the top five in the United States, the NCHS reported based on 2016-2017 data from the Canadian Health Measures Survey.
Older adults (60-79 years) in the two countries managed to share some common ground: Lipid-lowering drugs were the most commonly used prescription medication both north and south of the border, although past 30-day use was considerably higher in the United States (45% vs. 34%), the NCHS investigators said.
There were differences to be found, however, in the older age group. Analgesics were the second most commonly used drug type in Canada but did not even reach the top five in the United States, while beta-blockers were third among Americans but missed the Canadian top five, they noted.
(60% vs. 53%) and to have used at least five such drugs (15% vs. 10%). The differences among adults aged 60-79 were not significant, although American use was higher for at least one drug (84% vs. 83%) and for at least five (35% vs. 31%), according to the report.
FDA approves fedratinib for myelofibrosis
The Food and Drug Administration has approved fedratinib (Inrebic), an oral JAK2/FLT3 inhibitor, to treat myelofibrosis.
Fedratinib is approved to treat adults with intermediate-2 or high-risk primary or secondary (post–polycythemia vera or post–essential thrombocythemia) myelofibrosis.
The prescribing information for fedratinib includes a boxed warning detailing the risk of serious and fatal encephalopathy, including Wernicke’s.
The encephalopathy risk prompted Sanofi to stop developing fedratinib in 2013. The FDA placed a clinical hold on all trials of fedratinib after potential cases of Wernicke’s encephalopathy were observed in eight patients.
The FDA lifted the clinical hold in 2017, and Celgene Corporation decided to develop fedratinib when the company acquired Impact Biomedicines in 2018.
In the phase 3 JAKARTA trial, fedratinib significantly reduced splenomegaly and symptom burden in patients with primary or secondary myelofibrosis (JAMA Oncol. 2015 Aug;1[5]:643-51). In the phase 2 JAKARTA2 trial, fedratinib produced responses in myelofibrosis patients previously treated with ruxolitinib (Lancet Haematol. 2017 Jul;4[7]:e317-e324).
Fedratinib received orphan drug designation from the FDA, and the application for fedratinib received priority review.
The FDA granted approval of fedratinib to Impact Biomedicines, a wholly owned subsidiary of Celgene.
The Food and Drug Administration has approved fedratinib (Inrebic), an oral JAK2/FLT3 inhibitor, to treat myelofibrosis.
Fedratinib is approved to treat adults with intermediate-2 or high-risk primary or secondary (post–polycythemia vera or post–essential thrombocythemia) myelofibrosis.
The prescribing information for fedratinib includes a boxed warning detailing the risk of serious and fatal encephalopathy, including Wernicke’s.
The encephalopathy risk prompted Sanofi to stop developing fedratinib in 2013. The FDA placed a clinical hold on all trials of fedratinib after potential cases of Wernicke’s encephalopathy were observed in eight patients.
The FDA lifted the clinical hold in 2017, and Celgene Corporation decided to develop fedratinib when the company acquired Impact Biomedicines in 2018.
In the phase 3 JAKARTA trial, fedratinib significantly reduced splenomegaly and symptom burden in patients with primary or secondary myelofibrosis (JAMA Oncol. 2015 Aug;1[5]:643-51). In the phase 2 JAKARTA2 trial, fedratinib produced responses in myelofibrosis patients previously treated with ruxolitinib (Lancet Haematol. 2017 Jul;4[7]:e317-e324).
Fedratinib received orphan drug designation from the FDA, and the application for fedratinib received priority review.
The FDA granted approval of fedratinib to Impact Biomedicines, a wholly owned subsidiary of Celgene.
The Food and Drug Administration has approved fedratinib (Inrebic), an oral JAK2/FLT3 inhibitor, to treat myelofibrosis.
Fedratinib is approved to treat adults with intermediate-2 or high-risk primary or secondary (post–polycythemia vera or post–essential thrombocythemia) myelofibrosis.
The prescribing information for fedratinib includes a boxed warning detailing the risk of serious and fatal encephalopathy, including Wernicke’s.
The encephalopathy risk prompted Sanofi to stop developing fedratinib in 2013. The FDA placed a clinical hold on all trials of fedratinib after potential cases of Wernicke’s encephalopathy were observed in eight patients.
The FDA lifted the clinical hold in 2017, and Celgene Corporation decided to develop fedratinib when the company acquired Impact Biomedicines in 2018.
In the phase 3 JAKARTA trial, fedratinib significantly reduced splenomegaly and symptom burden in patients with primary or secondary myelofibrosis (JAMA Oncol. 2015 Aug;1[5]:643-51). In the phase 2 JAKARTA2 trial, fedratinib produced responses in myelofibrosis patients previously treated with ruxolitinib (Lancet Haematol. 2017 Jul;4[7]:e317-e324).
Fedratinib received orphan drug designation from the FDA, and the application for fedratinib received priority review.
The FDA granted approval of fedratinib to Impact Biomedicines, a wholly owned subsidiary of Celgene.
CDC updates recommendation for serologic Lyme disease detection
, according to CDC investigators.
![]()
At the 1994 Second National Conference on Serologic Diagnosis of Lyme Disease, several groups and organizations convened, recommending a two-test methodology for Lyme disease detection. First, an enzyme immunoassay (EIA) or immunofluorescence assay should be used, followed by a western immunoblot assay for specimens yielding positive or equivocal results. The guideline advised that all future tests should be evaluated against a challenge panel, and that new assays should only move forward if their specificity, sensitivity, and precision equaled or surpassed the performance of tests used in the recommended two-test procedure.
On July 29, 2019, the Food and Drug Administration approved several Lyme disease serologic assays with new indications for use based on a modified two-test methodology, with a second EIA replacing the western immunoblot assay.
“Clearance by FDA of the new Lyme disease assays indicates that test performance has been evaluated and is ‘substantially equivalent to or better than’ a legally marketed predicate test,” the CDC investigators noted (MMWR Morb Mortal Wkly Rep. 2019 Aug 15;68(32):703).
The recommendation advises that FDA-cleared “serologic assays that utilize EIA rather than western immunoblot assay in a two-test format are acceptable alternatives for the laboratory diagnosis of Lyme disease.”
, according to CDC investigators.
![]()
At the 1994 Second National Conference on Serologic Diagnosis of Lyme Disease, several groups and organizations convened, recommending a two-test methodology for Lyme disease detection. First, an enzyme immunoassay (EIA) or immunofluorescence assay should be used, followed by a western immunoblot assay for specimens yielding positive or equivocal results. The guideline advised that all future tests should be evaluated against a challenge panel, and that new assays should only move forward if their specificity, sensitivity, and precision equaled or surpassed the performance of tests used in the recommended two-test procedure.
On July 29, 2019, the Food and Drug Administration approved several Lyme disease serologic assays with new indications for use based on a modified two-test methodology, with a second EIA replacing the western immunoblot assay.
“Clearance by FDA of the new Lyme disease assays indicates that test performance has been evaluated and is ‘substantially equivalent to or better than’ a legally marketed predicate test,” the CDC investigators noted (MMWR Morb Mortal Wkly Rep. 2019 Aug 15;68(32):703).
The recommendation advises that FDA-cleared “serologic assays that utilize EIA rather than western immunoblot assay in a two-test format are acceptable alternatives for the laboratory diagnosis of Lyme disease.”
, according to CDC investigators.
![]()
At the 1994 Second National Conference on Serologic Diagnosis of Lyme Disease, several groups and organizations convened, recommending a two-test methodology for Lyme disease detection. First, an enzyme immunoassay (EIA) or immunofluorescence assay should be used, followed by a western immunoblot assay for specimens yielding positive or equivocal results. The guideline advised that all future tests should be evaluated against a challenge panel, and that new assays should only move forward if their specificity, sensitivity, and precision equaled or surpassed the performance of tests used in the recommended two-test procedure.
On July 29, 2019, the Food and Drug Administration approved several Lyme disease serologic assays with new indications for use based on a modified two-test methodology, with a second EIA replacing the western immunoblot assay.
“Clearance by FDA of the new Lyme disease assays indicates that test performance has been evaluated and is ‘substantially equivalent to or better than’ a legally marketed predicate test,” the CDC investigators noted (MMWR Morb Mortal Wkly Rep. 2019 Aug 15;68(32):703).
The recommendation advises that FDA-cleared “serologic assays that utilize EIA rather than western immunoblot assay in a two-test format are acceptable alternatives for the laboratory diagnosis of Lyme disease.”
FROM THE MMWR
FDA approves drug combo to treat highly resistant TB
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.
The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.
The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.
The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
NEWS FROM THE FDA
FDA update: Higher late mortality with paclitaxel-coated devices
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.
This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.
This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.
This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
CDC finds that too little naloxone is dispensed
Although the CDC recommends that clinicians consider prescribing naloxone, which can reverse the effects of an opioid overdose, to patients who receive high-dose opioid prescriptions, one naloxone prescription was dispensed in 2018 for every 69 such patients, according to a Vital Signs investigation published Aug. 6 in the Morbidity and Mortality Weekly Report.
Approximately 9 million more naloxone prescriptions could have been dispensed in 2018 if every patient with a high-dose opioid prescription were offered the drug, according to the agency. In addition, the rate at which naloxone is dispensed varies significantly according to region.
“Thousands of Americans are alive today thanks to the use of naloxone,” said Alex M. Azar, secretary of Health and Human Services, in a press release. “Giving people a chance to survive an opioid overdose and safely enter recovery is one of the five key pillars of our HHS strategy for ending the overdose epidemic. With help from Congress, the private sector, state, and local governments and communities, targeted access to naloxone has expanded dramatically over the last several years, but today’s CDC report is a reminder that there is much more all of us need to do to save lives.”
Investigators examined retail pharmacy data
In 2017, 47,600 (67.8%) drug overdose deaths in the United States involved opioids. For decades, emergency medical service providers have administered naloxone to patients with suspected drug overdose. A major focus of public health initiatives intended to address the opioid overdose crisis has been to increase access to naloxone through clinician prescribing and pharmacy dispensing. The CDC recommends considering prescribing naloxone to patients with a history of overdose or substance use disorder, those receiving opioid dosages of 50 morphine milligram equivalents per day or greater (that is, high-dose prescriptions), and those who are using benzodiazepines concurrently.
Investigators at the CDC examined retail pharmacy data from IQVIA, a company that maintains information on prescriptions from approximately 50,400 retail pharmacies. They extracted data from 2012 through 2018 to analyze naloxone dispensing by region, urban versus rural status, prescriber specialty, and recipient characteristics (for example, age group, sex, out-of-pocket costs, and method of payment).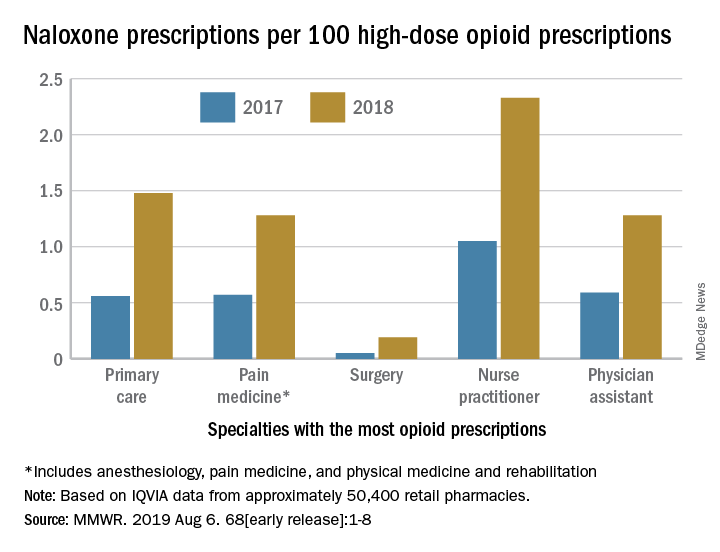
Dispensations doubled from 2017 to 2018
Naloxone dispensing from retail pharmacies increased from 0.4 prescriptions per 100,000 in 2012 to 170.2 prescriptions per 100,000 in 2018. From 2017 to 2018 alone, the number of prescriptions dispensed increased by 106%.
Despite consistency among state laws, naloxone dispensation varied by region. The average rate of naloxone prescriptions per 100 high-dose opioid prescriptions ranged from 0.2 in the lowest quartile to 2.9 in the highest quartile. In 2018, the rate of naloxone prescriptions per 100 high-dose opioid prescriptions ranged from 1.5 in metropolitan counties and 1.6 in the Northeast to 1.2 in rural counties and 1.3 in the Midwest. Rural counties were nearly three times more likely to be low-dispensing counties, compared with metropolitan counties.
The rate of naloxone prescriptions per 100 high-dose opioid prescriptions also varied by provider specialty. This rate was lowest among surgeons (0.2) and highest among psychiatrists (12.9).
Most naloxone prescriptions entailed out-of-pocket costs. About 71% of prescriptions paid for by Medicare entailed out-of-pocket costs, compared with 43.8% of prescriptions paid for by Medicaid, and 41.5% of prescriptions paid for by commercial insurance.
 Centers for Disease Control and Prevention
Centers for Disease Control and Prevention
More can be done
“It is clear from the data that there is still much needed education around the important role naloxone plays in reducing overdose deaths,” said Robert R. Redfield, MD, director of the CDC, in a press release. “The time is now to ensure all individuals who are prescribed high-dose opioids also receive naloxone as a potential life-saving intervention. As we aggressively confront what is the public health crisis of our time, CDC will continue to stress with health care providers the benefit of making this overdose-reversing medicine available to patients.”
“While we’ve seen these important increases [in naloxone prescriptions], we are not as far along as we’d like to be,” said Anne Schuchat, MD, principal deputy director of the CDC, during a press conference. “Cost is one of the issues, but I think awareness is another.” These data should prompt pharmacies to make sure that they stock naloxone and remind clinicians to consider naloxone when they prescribe opioids, she added. Patients and their family members should be aware of naloxone and ask their health care providers about it. “We’d really like to see the increase [in naloxone prescriptions] move much more rapidly,” she concluded.
The investigators disclosed no potential conflicts of interest.
SOURCE: Guy GP et al. MMWR Morb Mortal Wkly Rep. 2019 Aug 6.
Although the CDC recommends that clinicians consider prescribing naloxone, which can reverse the effects of an opioid overdose, to patients who receive high-dose opioid prescriptions, one naloxone prescription was dispensed in 2018 for every 69 such patients, according to a Vital Signs investigation published Aug. 6 in the Morbidity and Mortality Weekly Report.
Approximately 9 million more naloxone prescriptions could have been dispensed in 2018 if every patient with a high-dose opioid prescription were offered the drug, according to the agency. In addition, the rate at which naloxone is dispensed varies significantly according to region.
“Thousands of Americans are alive today thanks to the use of naloxone,” said Alex M. Azar, secretary of Health and Human Services, in a press release. “Giving people a chance to survive an opioid overdose and safely enter recovery is one of the five key pillars of our HHS strategy for ending the overdose epidemic. With help from Congress, the private sector, state, and local governments and communities, targeted access to naloxone has expanded dramatically over the last several years, but today’s CDC report is a reminder that there is much more all of us need to do to save lives.”
Investigators examined retail pharmacy data
In 2017, 47,600 (67.8%) drug overdose deaths in the United States involved opioids. For decades, emergency medical service providers have administered naloxone to patients with suspected drug overdose. A major focus of public health initiatives intended to address the opioid overdose crisis has been to increase access to naloxone through clinician prescribing and pharmacy dispensing. The CDC recommends considering prescribing naloxone to patients with a history of overdose or substance use disorder, those receiving opioid dosages of 50 morphine milligram equivalents per day or greater (that is, high-dose prescriptions), and those who are using benzodiazepines concurrently.
Investigators at the CDC examined retail pharmacy data from IQVIA, a company that maintains information on prescriptions from approximately 50,400 retail pharmacies. They extracted data from 2012 through 2018 to analyze naloxone dispensing by region, urban versus rural status, prescriber specialty, and recipient characteristics (for example, age group, sex, out-of-pocket costs, and method of payment).
Dispensations doubled from 2017 to 2018
Naloxone dispensing from retail pharmacies increased from 0.4 prescriptions per 100,000 in 2012 to 170.2 prescriptions per 100,000 in 2018. From 2017 to 2018 alone, the number of prescriptions dispensed increased by 106%.
Despite consistency among state laws, naloxone dispensation varied by region. The average rate of naloxone prescriptions per 100 high-dose opioid prescriptions ranged from 0.2 in the lowest quartile to 2.9 in the highest quartile. In 2018, the rate of naloxone prescriptions per 100 high-dose opioid prescriptions ranged from 1.5 in metropolitan counties and 1.6 in the Northeast to 1.2 in rural counties and 1.3 in the Midwest. Rural counties were nearly three times more likely to be low-dispensing counties, compared with metropolitan counties.
The rate of naloxone prescriptions per 100 high-dose opioid prescriptions also varied by provider specialty. This rate was lowest among surgeons (0.2) and highest among psychiatrists (12.9).
Most naloxone prescriptions entailed out-of-pocket costs. About 71% of prescriptions paid for by Medicare entailed out-of-pocket costs, compared with 43.8% of prescriptions paid for by Medicaid, and 41.5% of prescriptions paid for by commercial insurance.
Centers for Disease Control and Prevention
More can be done
“It is clear from the data that there is still much needed education around the important role naloxone plays in reducing overdose deaths,” said Robert R. Redfield, MD, director of the CDC, in a press release. “The time is now to ensure all individuals who are prescribed high-dose opioids also receive naloxone as a potential life-saving intervention. As we aggressively confront what is the public health crisis of our time, CDC will continue to stress with health care providers the benefit of making this overdose-reversing medicine available to patients.”
“While we’ve seen these important increases [in naloxone prescriptions], we are not as far along as we’d like to be,” said Anne Schuchat, MD, principal deputy director of the CDC, during a press conference. “Cost is one of the issues, but I think awareness is another.” These data should prompt pharmacies to make sure that they stock naloxone and remind clinicians to consider naloxone when they prescribe opioids, she added. Patients and their family members should be aware of naloxone and ask their health care providers about it. “We’d really like to see the increase [in naloxone prescriptions] move much more rapidly,” she concluded.
The investigators disclosed no potential conflicts of interest.
SOURCE: Guy GP et al. MMWR Morb Mortal Wkly Rep. 2019 Aug 6.
Although the CDC recommends that clinicians consider prescribing naloxone, which can reverse the effects of an opioid overdose, to patients who receive high-dose opioid prescriptions, one naloxone prescription was dispensed in 2018 for every 69 such patients, according to a Vital Signs investigation published Aug. 6 in the Morbidity and Mortality Weekly Report.
Approximately 9 million more naloxone prescriptions could have been dispensed in 2018 if every patient with a high-dose opioid prescription were offered the drug, according to the agency. In addition, the rate at which naloxone is dispensed varies significantly according to region.
“Thousands of Americans are alive today thanks to the use of naloxone,” said Alex M. Azar, secretary of Health and Human Services, in a press release. “Giving people a chance to survive an opioid overdose and safely enter recovery is one of the five key pillars of our HHS strategy for ending the overdose epidemic. With help from Congress, the private sector, state, and local governments and communities, targeted access to naloxone has expanded dramatically over the last several years, but today’s CDC report is a reminder that there is much more all of us need to do to save lives.”
Investigators examined retail pharmacy data
In 2017, 47,600 (67.8%) drug overdose deaths in the United States involved opioids. For decades, emergency medical service providers have administered naloxone to patients with suspected drug overdose. A major focus of public health initiatives intended to address the opioid overdose crisis has been to increase access to naloxone through clinician prescribing and pharmacy dispensing. The CDC recommends considering prescribing naloxone to patients with a history of overdose or substance use disorder, those receiving opioid dosages of 50 morphine milligram equivalents per day or greater (that is, high-dose prescriptions), and those who are using benzodiazepines concurrently.
Investigators at the CDC examined retail pharmacy data from IQVIA, a company that maintains information on prescriptions from approximately 50,400 retail pharmacies. They extracted data from 2012 through 2018 to analyze naloxone dispensing by region, urban versus rural status, prescriber specialty, and recipient characteristics (for example, age group, sex, out-of-pocket costs, and method of payment).
Dispensations doubled from 2017 to 2018
Naloxone dispensing from retail pharmacies increased from 0.4 prescriptions per 100,000 in 2012 to 170.2 prescriptions per 100,000 in 2018. From 2017 to 2018 alone, the number of prescriptions dispensed increased by 106%.
Despite consistency among state laws, naloxone dispensation varied by region. The average rate of naloxone prescriptions per 100 high-dose opioid prescriptions ranged from 0.2 in the lowest quartile to 2.9 in the highest quartile. In 2018, the rate of naloxone prescriptions per 100 high-dose opioid prescriptions ranged from 1.5 in metropolitan counties and 1.6 in the Northeast to 1.2 in rural counties and 1.3 in the Midwest. Rural counties were nearly three times more likely to be low-dispensing counties, compared with metropolitan counties.
The rate of naloxone prescriptions per 100 high-dose opioid prescriptions also varied by provider specialty. This rate was lowest among surgeons (0.2) and highest among psychiatrists (12.9).
Most naloxone prescriptions entailed out-of-pocket costs. About 71% of prescriptions paid for by Medicare entailed out-of-pocket costs, compared with 43.8% of prescriptions paid for by Medicaid, and 41.5% of prescriptions paid for by commercial insurance.
Centers for Disease Control and Prevention
More can be done
“It is clear from the data that there is still much needed education around the important role naloxone plays in reducing overdose deaths,” said Robert R. Redfield, MD, director of the CDC, in a press release. “The time is now to ensure all individuals who are prescribed high-dose opioids also receive naloxone as a potential life-saving intervention. As we aggressively confront what is the public health crisis of our time, CDC will continue to stress with health care providers the benefit of making this overdose-reversing medicine available to patients.”
“While we’ve seen these important increases [in naloxone prescriptions], we are not as far along as we’d like to be,” said Anne Schuchat, MD, principal deputy director of the CDC, during a press conference. “Cost is one of the issues, but I think awareness is another.” These data should prompt pharmacies to make sure that they stock naloxone and remind clinicians to consider naloxone when they prescribe opioids, she added. Patients and their family members should be aware of naloxone and ask their health care providers about it. “We’d really like to see the increase [in naloxone prescriptions] move much more rapidly,” she concluded.
The investigators disclosed no potential conflicts of interest.
SOURCE: Guy GP et al. MMWR Morb Mortal Wkly Rep. 2019 Aug 6.
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
Abbott issues recall on Ellipse ICDs
Abbott Laboratories has issued a recall of all Ellipse Implantable Cardioverter Defibrillators manufactured between April 5, 2019, and May 29, 2019, because of exposed aluminum wires within the device, potentially preventing defibrillation.
The Ellipse Implantable Cardioverter Defibrillators, formerly manufactured by St. Jude Medical (now a wholly-owned subsidiary of Abbott), provide pacing for patients with bradycardia and electric shock or pacing for patients with tachycardia. The device is implanted under the skin in the upper chest area with leads running into the heart.
The recall has been issued because electrical failures have occurred; Abbott has determined that these failures are caused by a faulty manufacturing process that left some aluminum wires in the leads partially exposed. Wires without proper insulation are likely to short, leaving the device without the ability to provide high voltage therapy.
Abbott is aware of no related reports of electrical failure in any of the devices that have already been implanted, the Food and Drug Administration announced, and no reports of patient harm, adverse events, or death have occurred. All affected devices that were implanted have either been replaced or are scheduled to be replaced, the agency said.
Abbott Laboratories has issued a recall of all Ellipse Implantable Cardioverter Defibrillators manufactured between April 5, 2019, and May 29, 2019, because of exposed aluminum wires within the device, potentially preventing defibrillation.
The Ellipse Implantable Cardioverter Defibrillators, formerly manufactured by St. Jude Medical (now a wholly-owned subsidiary of Abbott), provide pacing for patients with bradycardia and electric shock or pacing for patients with tachycardia. The device is implanted under the skin in the upper chest area with leads running into the heart.
The recall has been issued because electrical failures have occurred; Abbott has determined that these failures are caused by a faulty manufacturing process that left some aluminum wires in the leads partially exposed. Wires without proper insulation are likely to short, leaving the device without the ability to provide high voltage therapy.
Abbott is aware of no related reports of electrical failure in any of the devices that have already been implanted, the Food and Drug Administration announced, and no reports of patient harm, adverse events, or death have occurred. All affected devices that were implanted have either been replaced or are scheduled to be replaced, the agency said.
Abbott Laboratories has issued a recall of all Ellipse Implantable Cardioverter Defibrillators manufactured between April 5, 2019, and May 29, 2019, because of exposed aluminum wires within the device, potentially preventing defibrillation.
The Ellipse Implantable Cardioverter Defibrillators, formerly manufactured by St. Jude Medical (now a wholly-owned subsidiary of Abbott), provide pacing for patients with bradycardia and electric shock or pacing for patients with tachycardia. The device is implanted under the skin in the upper chest area with leads running into the heart.
The recall has been issued because electrical failures have occurred; Abbott has determined that these failures are caused by a faulty manufacturing process that left some aluminum wires in the leads partially exposed. Wires without proper insulation are likely to short, leaving the device without the ability to provide high voltage therapy.
Abbott is aware of no related reports of electrical failure in any of the devices that have already been implanted, the Food and Drug Administration announced, and no reports of patient harm, adverse events, or death have occurred. All affected devices that were implanted have either been replaced or are scheduled to be replaced, the agency said.
FDA approves Turalio for symptomatic tenosynovial giant cell tumor
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.

Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.