User login
More evidence of better outcomes with 120–mm Hg BP target
Intensive lowering of blood pressure to a systolic target less than 120 mm Hg reduced cardiovascular events among individuals at high risk for cardiovascular disease, compared with standard treatment using a target less than 140 mm Hg in the ESPRIT trial.
a 39% lower cardiovascular mortality, and 21% lower all-cause mortality than the standard treatment targeting a systolic pressure below 140 mm Hg,” reported lead investigator, Jing Li, MD, PhD, director of the department of preventive medicine at the National Center for Cardiovascular Diseases in Beijing.
The trial included patients with diabetes and those with a history of stroke, two important groups that were excluded in the previous SPRINT trial of intensive BP lowering. Results suggested that the benefit of intensive BP lowering extends to these groups.
The results translate into the prevention of 14 major vascular events and 8 deaths for every 1,000 individuals are treated for 3 years to a target systolic pressure less than 120 mm Hg rather than less than 140 mm Hg, at the cost of an additional three patients experiencing the serious adverse event of syncope, Dr. Li said.
“Our study generates new evidence about benefit and safety of treatment targeting systolic blood pressure below 120 mm Hg among a diverse Asian population, which is generally consistent with those from other ethnicities. Implementing this intensive treatment strategy for high-risk adults has the potential to save more lives and reduce the public health burden of heart disease worldwide,” she concluded.
Dr. Li presented the ESPRIT trial at the annual scientific sessions of the American Heart Association.
The ESPRIT trial included 11,255 Chinese adults (average age, 64 years; 41% women) who had a baseline systolic BP measurement of 130-180 mm Hg (average was 147/83 mm Hg) and either established cardiovascular disease or at least two major risk factors for cardiovascular disease. Of those enrolled, 39% had diabetes, and 27% had a history of stroke.
They were randomly assigned to receive intensive BP treatment, with a systolic BP target less than 120 mm Hg, or standard treatment, with a target measurement less than 140 mm Hg, over a 3-year period. After 1 year, systolic pressure was lowered to 135.6 mm Hg in the standard care group and to 120.3 mm Hg in the intensive treatment group, with values remaining at around the same level for the remainder of the follow-up.
The primary outcome was a composite of myocardial infarction, coronary or noncoronary revascularization, hospitalization/ED visit for heart failure, stroke, or cardiovascular death.
After 3.4 years of follow-up, 624 primary outcome events had occurred in the standard arm (3.6%) versus 547 events in intensive arm (3.2%), a reduction of 12% (hazard ratio, 0.88; 95% confidence interval, 0.78-0.99). This gives a number needed to treat to prevent one event of 74.
Cardiovascular death occurred in 0.5% of the standard group versus 0.3% of the intensive group (HR 0.61; 95% CI, 0.44-0.84); and all-cause death occurred in 1.1% of the standard group versus 0.9% of the intensive group (HR, 0.79; 95% CI, 0.64-0.97).
The individual endpoints of MI, stroke, and heart failure showed positive trends to a reduction with intensive BP lowering, but these did not reach statistical significance.
In terms of serious adverse events, syncope was increased in the intensive group (0.4% vs 0.1%), but there were no significant differences in hypotension, electrolyte abnormality, falls resulting in an injury, acute kidney injury, or renal failure.
Should 120 mm Hg be new target?
Commenting on the study, Paul Whelton, MD, chair in global public health at Tulane University, New Orleans, said that the results were consistent with several other trials.
“When we look at meta-analysis of trials of different levels of blood pressure reduction, all the studies show the same thing – the lower the blood pressure, the better the outcome, with those starting at higher levels gaining the greatest the benefit of blood pressure reductions,” he noted.
“There are four trials that have looked at systolic targets of less than 120 mm Hg versus less than 140 mm Hg (SPRINT, ACCORD BP, RESPECT, and now ESPRIT), and when analyzed properly, they all show a similar benefit for cardiovascular outcomes with the lower 120 target,” said Dr. Whelton, who led the SPRINT trial.
“ESPRIT is a nicely done trial. It is reassuring because it is consistent with the other trials, in that it seems that the benefits are much greater than the risk of adverse effects,” he added.
Dr. Whelton pointed out that there are three more trials to come looking at this question, two in Brazil (one in individuals with diabetes and one in stroke survivors) and another trial in China in people with diabetes. “So, we will get more information from these.”
He said that guidelines committees will have to consider a lower systolic BP of 120 mm Hg as the optimal treatment target. In the United States, at present, the target is 130 mm Hg.
The current U.S. guidelines were based on the SPRINT trial, which showed a reduction in cardiovascular events in patients treated to a systolic target of 120 mm Hg versus 140 mm Hg.
Dr. Whelton, who was chair of the 2017 American College of Cardiology/American Heart Association hypertension guidelines committee, explained that, at the time the guidelines were written, there was only one trial, SPRINT, to base the evidence on.
“The committee could all comfortably agree on the 130 mm Hg target, but it was felt that there wasn’t enough evidence at the time to make a recommendation for 120 mm Hg,” he said. “But now we have four trials.”
He said that the trials included patients with high risk for cardiovascular disease, but they all brought some differences to the table, with ACCORD BP conducted in patients with diabetes; SPRINT having enrichment with African American patients, older adults, and patients with kidney disease; RESPECT was in stroke survivors; and ESPRIT had a mix of Chinese patients.
“I think we’ve got a nice mix of different participants and they’re all showing the same signal – that 120 mm Hg is better,” Dr. Whelton said.
But he stressed that although there is now good evidence in favor of lower BP targets, these findings were not being implemented in clinical practice.
“We are doing very badly in terms of implementation. There is a big gap between science and what’s happening in the real world.”
Dr. Whelton pointed out that only 30% of patients in high-income countries are controlled to the 140/90 target and that in low- and middle-income countries, only 8.8% get to that level, never mind lower targets. “The next job is to work on implementing these findings.”
He noted that several studies have shown better results in this regard using a team approach, with nonphysicians playing a major role in following up with patients.
A version of this article appeared on Medscape.com.
Intensive lowering of blood pressure to a systolic target less than 120 mm Hg reduced cardiovascular events among individuals at high risk for cardiovascular disease, compared with standard treatment using a target less than 140 mm Hg in the ESPRIT trial.
a 39% lower cardiovascular mortality, and 21% lower all-cause mortality than the standard treatment targeting a systolic pressure below 140 mm Hg,” reported lead investigator, Jing Li, MD, PhD, director of the department of preventive medicine at the National Center for Cardiovascular Diseases in Beijing.
The trial included patients with diabetes and those with a history of stroke, two important groups that were excluded in the previous SPRINT trial of intensive BP lowering. Results suggested that the benefit of intensive BP lowering extends to these groups.
The results translate into the prevention of 14 major vascular events and 8 deaths for every 1,000 individuals are treated for 3 years to a target systolic pressure less than 120 mm Hg rather than less than 140 mm Hg, at the cost of an additional three patients experiencing the serious adverse event of syncope, Dr. Li said.
“Our study generates new evidence about benefit and safety of treatment targeting systolic blood pressure below 120 mm Hg among a diverse Asian population, which is generally consistent with those from other ethnicities. Implementing this intensive treatment strategy for high-risk adults has the potential to save more lives and reduce the public health burden of heart disease worldwide,” she concluded.
Dr. Li presented the ESPRIT trial at the annual scientific sessions of the American Heart Association.
The ESPRIT trial included 11,255 Chinese adults (average age, 64 years; 41% women) who had a baseline systolic BP measurement of 130-180 mm Hg (average was 147/83 mm Hg) and either established cardiovascular disease or at least two major risk factors for cardiovascular disease. Of those enrolled, 39% had diabetes, and 27% had a history of stroke.
They were randomly assigned to receive intensive BP treatment, with a systolic BP target less than 120 mm Hg, or standard treatment, with a target measurement less than 140 mm Hg, over a 3-year period. After 1 year, systolic pressure was lowered to 135.6 mm Hg in the standard care group and to 120.3 mm Hg in the intensive treatment group, with values remaining at around the same level for the remainder of the follow-up.
The primary outcome was a composite of myocardial infarction, coronary or noncoronary revascularization, hospitalization/ED visit for heart failure, stroke, or cardiovascular death.
After 3.4 years of follow-up, 624 primary outcome events had occurred in the standard arm (3.6%) versus 547 events in intensive arm (3.2%), a reduction of 12% (hazard ratio, 0.88; 95% confidence interval, 0.78-0.99). This gives a number needed to treat to prevent one event of 74.
Cardiovascular death occurred in 0.5% of the standard group versus 0.3% of the intensive group (HR 0.61; 95% CI, 0.44-0.84); and all-cause death occurred in 1.1% of the standard group versus 0.9% of the intensive group (HR, 0.79; 95% CI, 0.64-0.97).
The individual endpoints of MI, stroke, and heart failure showed positive trends to a reduction with intensive BP lowering, but these did not reach statistical significance.
In terms of serious adverse events, syncope was increased in the intensive group (0.4% vs 0.1%), but there were no significant differences in hypotension, electrolyte abnormality, falls resulting in an injury, acute kidney injury, or renal failure.
Should 120 mm Hg be new target?
Commenting on the study, Paul Whelton, MD, chair in global public health at Tulane University, New Orleans, said that the results were consistent with several other trials.
“When we look at meta-analysis of trials of different levels of blood pressure reduction, all the studies show the same thing – the lower the blood pressure, the better the outcome, with those starting at higher levels gaining the greatest the benefit of blood pressure reductions,” he noted.
“There are four trials that have looked at systolic targets of less than 120 mm Hg versus less than 140 mm Hg (SPRINT, ACCORD BP, RESPECT, and now ESPRIT), and when analyzed properly, they all show a similar benefit for cardiovascular outcomes with the lower 120 target,” said Dr. Whelton, who led the SPRINT trial.
“ESPRIT is a nicely done trial. It is reassuring because it is consistent with the other trials, in that it seems that the benefits are much greater than the risk of adverse effects,” he added.
Dr. Whelton pointed out that there are three more trials to come looking at this question, two in Brazil (one in individuals with diabetes and one in stroke survivors) and another trial in China in people with diabetes. “So, we will get more information from these.”
He said that guidelines committees will have to consider a lower systolic BP of 120 mm Hg as the optimal treatment target. In the United States, at present, the target is 130 mm Hg.
The current U.S. guidelines were based on the SPRINT trial, which showed a reduction in cardiovascular events in patients treated to a systolic target of 120 mm Hg versus 140 mm Hg.
Dr. Whelton, who was chair of the 2017 American College of Cardiology/American Heart Association hypertension guidelines committee, explained that, at the time the guidelines were written, there was only one trial, SPRINT, to base the evidence on.
“The committee could all comfortably agree on the 130 mm Hg target, but it was felt that there wasn’t enough evidence at the time to make a recommendation for 120 mm Hg,” he said. “But now we have four trials.”
He said that the trials included patients with high risk for cardiovascular disease, but they all brought some differences to the table, with ACCORD BP conducted in patients with diabetes; SPRINT having enrichment with African American patients, older adults, and patients with kidney disease; RESPECT was in stroke survivors; and ESPRIT had a mix of Chinese patients.
“I think we’ve got a nice mix of different participants and they’re all showing the same signal – that 120 mm Hg is better,” Dr. Whelton said.
But he stressed that although there is now good evidence in favor of lower BP targets, these findings were not being implemented in clinical practice.
“We are doing very badly in terms of implementation. There is a big gap between science and what’s happening in the real world.”
Dr. Whelton pointed out that only 30% of patients in high-income countries are controlled to the 140/90 target and that in low- and middle-income countries, only 8.8% get to that level, never mind lower targets. “The next job is to work on implementing these findings.”
He noted that several studies have shown better results in this regard using a team approach, with nonphysicians playing a major role in following up with patients.
A version of this article appeared on Medscape.com.
Intensive lowering of blood pressure to a systolic target less than 120 mm Hg reduced cardiovascular events among individuals at high risk for cardiovascular disease, compared with standard treatment using a target less than 140 mm Hg in the ESPRIT trial.
a 39% lower cardiovascular mortality, and 21% lower all-cause mortality than the standard treatment targeting a systolic pressure below 140 mm Hg,” reported lead investigator, Jing Li, MD, PhD, director of the department of preventive medicine at the National Center for Cardiovascular Diseases in Beijing.
The trial included patients with diabetes and those with a history of stroke, two important groups that were excluded in the previous SPRINT trial of intensive BP lowering. Results suggested that the benefit of intensive BP lowering extends to these groups.
The results translate into the prevention of 14 major vascular events and 8 deaths for every 1,000 individuals are treated for 3 years to a target systolic pressure less than 120 mm Hg rather than less than 140 mm Hg, at the cost of an additional three patients experiencing the serious adverse event of syncope, Dr. Li said.
“Our study generates new evidence about benefit and safety of treatment targeting systolic blood pressure below 120 mm Hg among a diverse Asian population, which is generally consistent with those from other ethnicities. Implementing this intensive treatment strategy for high-risk adults has the potential to save more lives and reduce the public health burden of heart disease worldwide,” she concluded.
Dr. Li presented the ESPRIT trial at the annual scientific sessions of the American Heart Association.
The ESPRIT trial included 11,255 Chinese adults (average age, 64 years; 41% women) who had a baseline systolic BP measurement of 130-180 mm Hg (average was 147/83 mm Hg) and either established cardiovascular disease or at least two major risk factors for cardiovascular disease. Of those enrolled, 39% had diabetes, and 27% had a history of stroke.
They were randomly assigned to receive intensive BP treatment, with a systolic BP target less than 120 mm Hg, or standard treatment, with a target measurement less than 140 mm Hg, over a 3-year period. After 1 year, systolic pressure was lowered to 135.6 mm Hg in the standard care group and to 120.3 mm Hg in the intensive treatment group, with values remaining at around the same level for the remainder of the follow-up.
The primary outcome was a composite of myocardial infarction, coronary or noncoronary revascularization, hospitalization/ED visit for heart failure, stroke, or cardiovascular death.
After 3.4 years of follow-up, 624 primary outcome events had occurred in the standard arm (3.6%) versus 547 events in intensive arm (3.2%), a reduction of 12% (hazard ratio, 0.88; 95% confidence interval, 0.78-0.99). This gives a number needed to treat to prevent one event of 74.
Cardiovascular death occurred in 0.5% of the standard group versus 0.3% of the intensive group (HR 0.61; 95% CI, 0.44-0.84); and all-cause death occurred in 1.1% of the standard group versus 0.9% of the intensive group (HR, 0.79; 95% CI, 0.64-0.97).
The individual endpoints of MI, stroke, and heart failure showed positive trends to a reduction with intensive BP lowering, but these did not reach statistical significance.
In terms of serious adverse events, syncope was increased in the intensive group (0.4% vs 0.1%), but there were no significant differences in hypotension, electrolyte abnormality, falls resulting in an injury, acute kidney injury, or renal failure.
Should 120 mm Hg be new target?
Commenting on the study, Paul Whelton, MD, chair in global public health at Tulane University, New Orleans, said that the results were consistent with several other trials.
“When we look at meta-analysis of trials of different levels of blood pressure reduction, all the studies show the same thing – the lower the blood pressure, the better the outcome, with those starting at higher levels gaining the greatest the benefit of blood pressure reductions,” he noted.
“There are four trials that have looked at systolic targets of less than 120 mm Hg versus less than 140 mm Hg (SPRINT, ACCORD BP, RESPECT, and now ESPRIT), and when analyzed properly, they all show a similar benefit for cardiovascular outcomes with the lower 120 target,” said Dr. Whelton, who led the SPRINT trial.
“ESPRIT is a nicely done trial. It is reassuring because it is consistent with the other trials, in that it seems that the benefits are much greater than the risk of adverse effects,” he added.
Dr. Whelton pointed out that there are three more trials to come looking at this question, two in Brazil (one in individuals with diabetes and one in stroke survivors) and another trial in China in people with diabetes. “So, we will get more information from these.”
He said that guidelines committees will have to consider a lower systolic BP of 120 mm Hg as the optimal treatment target. In the United States, at present, the target is 130 mm Hg.
The current U.S. guidelines were based on the SPRINT trial, which showed a reduction in cardiovascular events in patients treated to a systolic target of 120 mm Hg versus 140 mm Hg.
Dr. Whelton, who was chair of the 2017 American College of Cardiology/American Heart Association hypertension guidelines committee, explained that, at the time the guidelines were written, there was only one trial, SPRINT, to base the evidence on.
“The committee could all comfortably agree on the 130 mm Hg target, but it was felt that there wasn’t enough evidence at the time to make a recommendation for 120 mm Hg,” he said. “But now we have four trials.”
He said that the trials included patients with high risk for cardiovascular disease, but they all brought some differences to the table, with ACCORD BP conducted in patients with diabetes; SPRINT having enrichment with African American patients, older adults, and patients with kidney disease; RESPECT was in stroke survivors; and ESPRIT had a mix of Chinese patients.
“I think we’ve got a nice mix of different participants and they’re all showing the same signal – that 120 mm Hg is better,” Dr. Whelton said.
But he stressed that although there is now good evidence in favor of lower BP targets, these findings were not being implemented in clinical practice.
“We are doing very badly in terms of implementation. There is a big gap between science and what’s happening in the real world.”
Dr. Whelton pointed out that only 30% of patients in high-income countries are controlled to the 140/90 target and that in low- and middle-income countries, only 8.8% get to that level, never mind lower targets. “The next job is to work on implementing these findings.”
He noted that several studies have shown better results in this regard using a team approach, with nonphysicians playing a major role in following up with patients.
A version of this article appeared on Medscape.com.
FROM AHA 2023
Headache after drinking red wine? This could be why
This transcript has been edited for clarity.
Robert Louis Stevenson famously said, “Wine is bottled poetry.” And I think it works quite well. I’ve had wines that are simple, elegant, and unpretentious like Emily Dickinson, and passionate and mysterious like Pablo Neruda. And I’ve had wines that are more analogous to the limerick you might read scrawled on a rest-stop bathroom wall. Those ones give me headaches.
– and apparently it’s not just the alcohol.
Headaches are common, and headaches after drinking alcohol are particularly common. An interesting epidemiologic phenomenon, not yet adequately explained, is why red wine is associated with more headache than other forms of alcohol. There have been many studies fingering many suspects, from sulfites to tannins to various phenolic compounds, but none have really provided a concrete explanation for what might be going on.
A new hypothesis came to the fore on Nov. 20 in the journal Scientific Reports:
To understand the idea, first a reminder of what happens when you drink alcohol, physiologically.
Alcohol is metabolized by the enzyme alcohol dehydrogenase in the gut and then in the liver. That turns it into acetaldehyde, a toxic metabolite. In most of us, aldehyde dehydrogenase (ALDH) quickly metabolizes acetaldehyde to the inert acetate, which can be safely excreted.
I say “most of us” because some populations, particularly those with East Asian ancestry, have a mutation in the ALDH gene which can lead to accumulation of toxic acetaldehyde with alcohol consumption – leading to facial flushing, nausea, and headache.
We can also inhibit the enzyme medically. That’s what the drug disulfiram, also known as Antabuse, does. It doesn’t prevent you from wanting to drink; it makes the consequences of drinking incredibly aversive.
The researchers focused in on the aldehyde dehydrogenase enzyme and conducted a screening study. Are there any compounds in red wine that naturally inhibit ALDH?
The results pointed squarely at quercetin, and particularly its metabolite quercetin glucuronide, which, at 20 micromolar concentrations, inhibited about 80% of ALDH activity.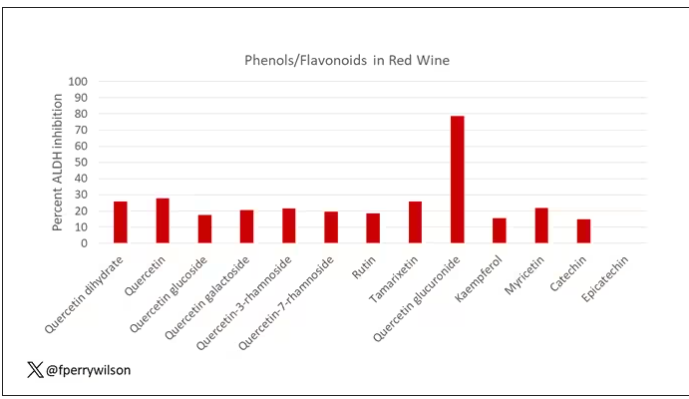
Quercetin is a flavonoid – a compound that gives color to a variety of vegetables and fruits, including grapes. In a test tube, it is an antioxidant, which is enough evidence to spawn a small quercetin-as-supplement industry, but there is no convincing evidence that it is medically useful. The authors then examined the concentration of quercetin glucuronide to achieve various inhibitions of ALDH, as you can see in this graph here.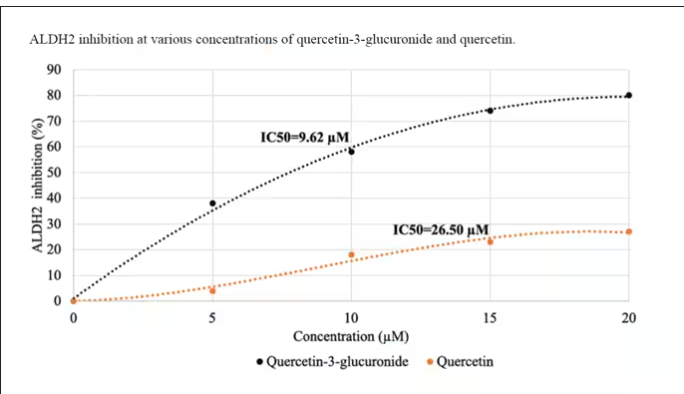
By about 10 micromolar, we see a decent amount of inhibition. Disulfiram is about 10 times more potent than that, but then again, you don’t drink three glasses of disulfiram with Thanksgiving dinner.
This is where this study stops. But it obviously tells us very little about what might be happening in the human body. For that, we need to ask the question: Can we get our quercetin levels to 10 micromolar? Is that remotely achievable?
Let’s start with how much quercetin there is in red wine. Like all things wine, it varies, but this study examining Australian wines found mean concentrations of 11 mg/L. The highest value I saw was close to 50 mg/L.
So let’s do some math. To make the numbers easy, let’s say you drank a liter of Australian wine, taking in 50 mg of quercetin glucuronide.
How much of that gets into your bloodstream? Some studies suggest a bioavailability of less than 1%, which basically means none and should probably put the quercetin hypothesis to bed. But there is some variation here too; it seems to depend on the form of quercetin you ingest.
Let’s say all 50 mg gets into your bloodstream. What blood concentration would that lead to? Well, I’ll keep the stoichiometry in the graphics and just say that if we assume that the volume of distribution of the compound is restricted to plasma alone, then you could achieve similar concentrations to what was done in petri dishes during this study.
Of course, if quercetin is really the culprit behind red wine headache, I have some questions: Why aren’t the Amazon reviews of quercetin supplements chock full of warnings not to take them with alcohol? And other foods have way higher quercetin concentration than wine, but you don’t hear people warning not to take your red onions with alcohol, or your capers, or lingonberries.
There’s some more work to be done here – most importantly, some human studies. Let’s give people wine with different amounts of quercetin and see what happens. Sign me up. Seriously.
As for Thanksgiving, it’s worth noting that cranberries have a lot of quercetin in them. So between the cranberry sauce, the Beaujolais, and your uncle ranting about the contrails again, the probability of headache is pretty darn high. Stay safe out there, and Happy Thanksgiving.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Robert Louis Stevenson famously said, “Wine is bottled poetry.” And I think it works quite well. I’ve had wines that are simple, elegant, and unpretentious like Emily Dickinson, and passionate and mysterious like Pablo Neruda. And I’ve had wines that are more analogous to the limerick you might read scrawled on a rest-stop bathroom wall. Those ones give me headaches.
– and apparently it’s not just the alcohol.
Headaches are common, and headaches after drinking alcohol are particularly common. An interesting epidemiologic phenomenon, not yet adequately explained, is why red wine is associated with more headache than other forms of alcohol. There have been many studies fingering many suspects, from sulfites to tannins to various phenolic compounds, but none have really provided a concrete explanation for what might be going on.
A new hypothesis came to the fore on Nov. 20 in the journal Scientific Reports:
To understand the idea, first a reminder of what happens when you drink alcohol, physiologically.
Alcohol is metabolized by the enzyme alcohol dehydrogenase in the gut and then in the liver. That turns it into acetaldehyde, a toxic metabolite. In most of us, aldehyde dehydrogenase (ALDH) quickly metabolizes acetaldehyde to the inert acetate, which can be safely excreted.
I say “most of us” because some populations, particularly those with East Asian ancestry, have a mutation in the ALDH gene which can lead to accumulation of toxic acetaldehyde with alcohol consumption – leading to facial flushing, nausea, and headache.
We can also inhibit the enzyme medically. That’s what the drug disulfiram, also known as Antabuse, does. It doesn’t prevent you from wanting to drink; it makes the consequences of drinking incredibly aversive.
The researchers focused in on the aldehyde dehydrogenase enzyme and conducted a screening study. Are there any compounds in red wine that naturally inhibit ALDH?
The results pointed squarely at quercetin, and particularly its metabolite quercetin glucuronide, which, at 20 micromolar concentrations, inhibited about 80% of ALDH activity.
Quercetin is a flavonoid – a compound that gives color to a variety of vegetables and fruits, including grapes. In a test tube, it is an antioxidant, which is enough evidence to spawn a small quercetin-as-supplement industry, but there is no convincing evidence that it is medically useful. The authors then examined the concentration of quercetin glucuronide to achieve various inhibitions of ALDH, as you can see in this graph here.
By about 10 micromolar, we see a decent amount of inhibition. Disulfiram is about 10 times more potent than that, but then again, you don’t drink three glasses of disulfiram with Thanksgiving dinner.
This is where this study stops. But it obviously tells us very little about what might be happening in the human body. For that, we need to ask the question: Can we get our quercetin levels to 10 micromolar? Is that remotely achievable?
Let’s start with how much quercetin there is in red wine. Like all things wine, it varies, but this study examining Australian wines found mean concentrations of 11 mg/L. The highest value I saw was close to 50 mg/L.
So let’s do some math. To make the numbers easy, let’s say you drank a liter of Australian wine, taking in 50 mg of quercetin glucuronide.
How much of that gets into your bloodstream? Some studies suggest a bioavailability of less than 1%, which basically means none and should probably put the quercetin hypothesis to bed. But there is some variation here too; it seems to depend on the form of quercetin you ingest.
Let’s say all 50 mg gets into your bloodstream. What blood concentration would that lead to? Well, I’ll keep the stoichiometry in the graphics and just say that if we assume that the volume of distribution of the compound is restricted to plasma alone, then you could achieve similar concentrations to what was done in petri dishes during this study.
Of course, if quercetin is really the culprit behind red wine headache, I have some questions: Why aren’t the Amazon reviews of quercetin supplements chock full of warnings not to take them with alcohol? And other foods have way higher quercetin concentration than wine, but you don’t hear people warning not to take your red onions with alcohol, or your capers, or lingonberries.
There’s some more work to be done here – most importantly, some human studies. Let’s give people wine with different amounts of quercetin and see what happens. Sign me up. Seriously.
As for Thanksgiving, it’s worth noting that cranberries have a lot of quercetin in them. So between the cranberry sauce, the Beaujolais, and your uncle ranting about the contrails again, the probability of headache is pretty darn high. Stay safe out there, and Happy Thanksgiving.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Robert Louis Stevenson famously said, “Wine is bottled poetry.” And I think it works quite well. I’ve had wines that are simple, elegant, and unpretentious like Emily Dickinson, and passionate and mysterious like Pablo Neruda. And I’ve had wines that are more analogous to the limerick you might read scrawled on a rest-stop bathroom wall. Those ones give me headaches.
– and apparently it’s not just the alcohol.
Headaches are common, and headaches after drinking alcohol are particularly common. An interesting epidemiologic phenomenon, not yet adequately explained, is why red wine is associated with more headache than other forms of alcohol. There have been many studies fingering many suspects, from sulfites to tannins to various phenolic compounds, but none have really provided a concrete explanation for what might be going on.
A new hypothesis came to the fore on Nov. 20 in the journal Scientific Reports:
To understand the idea, first a reminder of what happens when you drink alcohol, physiologically.
Alcohol is metabolized by the enzyme alcohol dehydrogenase in the gut and then in the liver. That turns it into acetaldehyde, a toxic metabolite. In most of us, aldehyde dehydrogenase (ALDH) quickly metabolizes acetaldehyde to the inert acetate, which can be safely excreted.
I say “most of us” because some populations, particularly those with East Asian ancestry, have a mutation in the ALDH gene which can lead to accumulation of toxic acetaldehyde with alcohol consumption – leading to facial flushing, nausea, and headache.
We can also inhibit the enzyme medically. That’s what the drug disulfiram, also known as Antabuse, does. It doesn’t prevent you from wanting to drink; it makes the consequences of drinking incredibly aversive.
The researchers focused in on the aldehyde dehydrogenase enzyme and conducted a screening study. Are there any compounds in red wine that naturally inhibit ALDH?
The results pointed squarely at quercetin, and particularly its metabolite quercetin glucuronide, which, at 20 micromolar concentrations, inhibited about 80% of ALDH activity.
Quercetin is a flavonoid – a compound that gives color to a variety of vegetables and fruits, including grapes. In a test tube, it is an antioxidant, which is enough evidence to spawn a small quercetin-as-supplement industry, but there is no convincing evidence that it is medically useful. The authors then examined the concentration of quercetin glucuronide to achieve various inhibitions of ALDH, as you can see in this graph here.
By about 10 micromolar, we see a decent amount of inhibition. Disulfiram is about 10 times more potent than that, but then again, you don’t drink three glasses of disulfiram with Thanksgiving dinner.
This is where this study stops. But it obviously tells us very little about what might be happening in the human body. For that, we need to ask the question: Can we get our quercetin levels to 10 micromolar? Is that remotely achievable?
Let’s start with how much quercetin there is in red wine. Like all things wine, it varies, but this study examining Australian wines found mean concentrations of 11 mg/L. The highest value I saw was close to 50 mg/L.
So let’s do some math. To make the numbers easy, let’s say you drank a liter of Australian wine, taking in 50 mg of quercetin glucuronide.
How much of that gets into your bloodstream? Some studies suggest a bioavailability of less than 1%, which basically means none and should probably put the quercetin hypothesis to bed. But there is some variation here too; it seems to depend on the form of quercetin you ingest.
Let’s say all 50 mg gets into your bloodstream. What blood concentration would that lead to? Well, I’ll keep the stoichiometry in the graphics and just say that if we assume that the volume of distribution of the compound is restricted to plasma alone, then you could achieve similar concentrations to what was done in petri dishes during this study.
Of course, if quercetin is really the culprit behind red wine headache, I have some questions: Why aren’t the Amazon reviews of quercetin supplements chock full of warnings not to take them with alcohol? And other foods have way higher quercetin concentration than wine, but you don’t hear people warning not to take your red onions with alcohol, or your capers, or lingonberries.
There’s some more work to be done here – most importantly, some human studies. Let’s give people wine with different amounts of quercetin and see what happens. Sign me up. Seriously.
As for Thanksgiving, it’s worth noting that cranberries have a lot of quercetin in them. So between the cranberry sauce, the Beaujolais, and your uncle ranting about the contrails again, the probability of headache is pretty darn high. Stay safe out there, and Happy Thanksgiving.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Surgery is falling out of favor in rectal cancer
TOPLINE:
METHODOLOGY:
- The National Comprehensive Cancer Network endorses watchful waiting, instead of surgery, when patients with rectal cancer have a complete clinical response to neoadjuvant therapy, but it’s unclear how often patients and providers opt for this organ preservation approach.
- To find out, investigators reviewed 175,545 adults in the National Cancer Database treated for rectal adenocarcinoma from 2006 to 2020.
- The research team assessed changes in the proportion of patients who were treated with chemotherapy and/or radiation without tumor resection, transanal local excision, or removal of the rectum.
- Patients had a mean age of 63 years, 39.7% were women, 17.4% had stage 1 disease, 24.7% had stage 2A-C disease, and 32.1% had stage 3A-C tumors; tumor stage was unknown in just over a quarter of patients.
TAKEAWAY:
- The absolute annual proportion of organ preservation increased by more than 50% from 18.4% in 2006 to 28.2% in 2020.
- In that time frame, organ preservation increased from 19.5% to 32.5% – a percent increase of about 67% – for patients with stage 2A-C disease, 16.2% to 29.1% – a percent increase of about 80% – for patients with stage 3A-C disease, and 16.5% to 26.6% – a percent increase of about 60% – for those with unknown stages.
- However, the rate of proctectomies increased by 6.1 percentage points, or by about 30%, among patients with stage I rectal cancer – from 20.3% to 26.4%.
- Among patients who did have surgery, the proportion who had complete pathologic responses to neoadjuvant therapy nearly tripled, increasing from 6.5% to 18.8%.
IN PRACTICE:
“This case series shows that rectal cancer is increasingly being managed medically, especially among patients whose treatment historically relied on proctectomy,” the authors concluded. However, protocols to standardize the approach are lacking, which is why “establishing quality standards for organ preservation is a pressing issue that should involve all relevant stakeholders, including patients.”
SOURCE:
The study, led by Anthony Loria, MD, MSCI, of the University of Rochester (N.Y.), was published online in JAMA Oncology.
LIMITATIONS:
The percentage of people who needed surgery for recurrence, patient and facility factors associated with organ preservation, and overall survival outcomes were not addressed.
DISCLOSURES:
No external funding was reported, and the investigators reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The National Comprehensive Cancer Network endorses watchful waiting, instead of surgery, when patients with rectal cancer have a complete clinical response to neoadjuvant therapy, but it’s unclear how often patients and providers opt for this organ preservation approach.
- To find out, investigators reviewed 175,545 adults in the National Cancer Database treated for rectal adenocarcinoma from 2006 to 2020.
- The research team assessed changes in the proportion of patients who were treated with chemotherapy and/or radiation without tumor resection, transanal local excision, or removal of the rectum.
- Patients had a mean age of 63 years, 39.7% were women, 17.4% had stage 1 disease, 24.7% had stage 2A-C disease, and 32.1% had stage 3A-C tumors; tumor stage was unknown in just over a quarter of patients.
TAKEAWAY:
- The absolute annual proportion of organ preservation increased by more than 50% from 18.4% in 2006 to 28.2% in 2020.
- In that time frame, organ preservation increased from 19.5% to 32.5% – a percent increase of about 67% – for patients with stage 2A-C disease, 16.2% to 29.1% – a percent increase of about 80% – for patients with stage 3A-C disease, and 16.5% to 26.6% – a percent increase of about 60% – for those with unknown stages.
- However, the rate of proctectomies increased by 6.1 percentage points, or by about 30%, among patients with stage I rectal cancer – from 20.3% to 26.4%.
- Among patients who did have surgery, the proportion who had complete pathologic responses to neoadjuvant therapy nearly tripled, increasing from 6.5% to 18.8%.
IN PRACTICE:
“This case series shows that rectal cancer is increasingly being managed medically, especially among patients whose treatment historically relied on proctectomy,” the authors concluded. However, protocols to standardize the approach are lacking, which is why “establishing quality standards for organ preservation is a pressing issue that should involve all relevant stakeholders, including patients.”
SOURCE:
The study, led by Anthony Loria, MD, MSCI, of the University of Rochester (N.Y.), was published online in JAMA Oncology.
LIMITATIONS:
The percentage of people who needed surgery for recurrence, patient and facility factors associated with organ preservation, and overall survival outcomes were not addressed.
DISCLOSURES:
No external funding was reported, and the investigators reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The National Comprehensive Cancer Network endorses watchful waiting, instead of surgery, when patients with rectal cancer have a complete clinical response to neoadjuvant therapy, but it’s unclear how often patients and providers opt for this organ preservation approach.
- To find out, investigators reviewed 175,545 adults in the National Cancer Database treated for rectal adenocarcinoma from 2006 to 2020.
- The research team assessed changes in the proportion of patients who were treated with chemotherapy and/or radiation without tumor resection, transanal local excision, or removal of the rectum.
- Patients had a mean age of 63 years, 39.7% were women, 17.4% had stage 1 disease, 24.7% had stage 2A-C disease, and 32.1% had stage 3A-C tumors; tumor stage was unknown in just over a quarter of patients.
TAKEAWAY:
- The absolute annual proportion of organ preservation increased by more than 50% from 18.4% in 2006 to 28.2% in 2020.
- In that time frame, organ preservation increased from 19.5% to 32.5% – a percent increase of about 67% – for patients with stage 2A-C disease, 16.2% to 29.1% – a percent increase of about 80% – for patients with stage 3A-C disease, and 16.5% to 26.6% – a percent increase of about 60% – for those with unknown stages.
- However, the rate of proctectomies increased by 6.1 percentage points, or by about 30%, among patients with stage I rectal cancer – from 20.3% to 26.4%.
- Among patients who did have surgery, the proportion who had complete pathologic responses to neoadjuvant therapy nearly tripled, increasing from 6.5% to 18.8%.
IN PRACTICE:
“This case series shows that rectal cancer is increasingly being managed medically, especially among patients whose treatment historically relied on proctectomy,” the authors concluded. However, protocols to standardize the approach are lacking, which is why “establishing quality standards for organ preservation is a pressing issue that should involve all relevant stakeholders, including patients.”
SOURCE:
The study, led by Anthony Loria, MD, MSCI, of the University of Rochester (N.Y.), was published online in JAMA Oncology.
LIMITATIONS:
The percentage of people who needed surgery for recurrence, patient and facility factors associated with organ preservation, and overall survival outcomes were not addressed.
DISCLOSURES:
No external funding was reported, and the investigators reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Single injection reduces blood pressure for 6 months: KARDIA-1
with what appeared to be an encouraging side-effect profile, in the phase 2 dose-ranging KARDIA-1 study.
“Our study demonstrates that either quarterly or biannual doses of zilebesiran can effectively and safely lower blood pressure in patients with uncontrolled hypertension,” said senior study investigator George Bakris, MD.
“Based on these results, zilebesiran has the potential to improve medication adherence, which will, in turn, reduce cardiovascular risk in people with hypertension,” added Dr. Bakris, who is professor of medicine and director of the Comprehensive Hypertension Center at the University of Chicago Medicine.
The KARDIA-1 study was presented at the American Heart Association scientific sessions.
Dr. Bakris noted that uncontrolled hypertension is a leading cause of morbidity and mortality, and despite availability of effective antihypertensives, many adults with hypertension are untreated, and up to 80% have uncontrolled disease, both globally and in the United States.
Zilebesiran is a subcutaneous RNA interference therapeutic that binds with high affinity to the hepatic asialoglycoprotein receptor, bringing about a reduction in the synthesis of angiotensinogen, the sole precursor of all angiotensin peptides. It is hoped that its hepatocyte-targeted delivery may allow extrahepatic angiotensinogen expression to be preserved, which could limit off-target effects in the kidney and other tissues.
The KARDIA-1 trial investigated the safety and efficacy of different doses of zilebesiran in patients with mild to moderate hypertension (systolic BP of 135-160 mm Hg), who are untreated or on stable therapy with up to two antihypertensive medications.
The study included 394 such patients (average baseline systolic BP was 142 mm Hg) who were randomly assigned to receive one of four different zilebesiran doses (150 mg, 300 mg, or 600 mg once every 6 months or 300 mg once every 2 months) or a placebo. The final analysis included 377 patients (56% men, 25% Black).
Results showed sustained reductions in serum angiotensinogen (between 88% and 98%) over the 6-month follow-up period.
Ambulatory systolic BP measured over 24 hours was significantly decreased with all zilebesiran regimens, with a mean reduction from baseline to month 6 of around 10 mm Hg in the three top doses studied and by around 14 mm Hg compared with placebo.
Patients receiving zilebesiran were more likely to achieve 24-hour average systolic BP measurements of 130 mm Hg or less at 6 months.
In addition, participants in all four zilebesiran groups consistently experienced significantly greater reductions in both daytime and nighttime systolic BP.
There were four nonserious adverse reactions leading to discontinuation in the zilebesiran groups: two instances of orthostatic hypotension, one of BP elevation, and one of injection site reaction.
Most hyperkalemia adverse events, which occurred in 6% of patients, were mild, did not require intervention, and generally resolved with repeat measurement; none were associated with acute kidney injury or led to study drug discontinuation. The incidence of hypotension events was low, and no clinically relevant changes in renal or hepatic function were observed, Dr. Bakris reported.
There was one death caused by cardiopulmonary arrest in a patient receiving zilebesiran 300 mg every 3 months, but this was not classified as drug related.
Zilebesiran is being further evaluated as an add-on therapy for treatment of hypertension in the ongoing KARDIA-2 phase 2 study.
Moderator of an AHA press conference at which the study was discussed, Sandra Taler, MD, professor of medicine at the Mayo Clinic, Rochester, Minn., said that “to have an injectable medicine that gives long-term blood pressure lowering is extremely exciting.”
Dr. Taler raised the point that some patients may not return for subsequent doses, but added that with subcutaneous dosing, administration at home may be a possibility.
Also commenting at the press conference, Keith Ferdinand, MD, professor of clinical medicine at Tulane University, New Orleans, said that this study “suggests we can now target the first step in the renin-angiotensin system – angiotensinogen – which then appears to lead to robust and continued blood pressure lowering for up to 6 months, which should improve adherence.”
Noting that only 50% of patients continue to take antihypertensive drugs after 1 year, Dr. Ferdinand added: “If we can increase adherence, we will increase efficacy and perhaps protect against some of the target organ damage.”
Designated discussant of the KARDIA-1 study at the AHA late-breaking clinical trial session, Anna Dominiczak, MD, University of Glasgow, noted that hypertension affects one in three adults worldwide, but only around 20% of people have it under control.
“An increase in the number of patients effectively treated for hypertension to levels observed in high-performing countries could prevent 76 million deaths, 120 million strokes, 79 million heart attacks, and 17 million cases of heart failure between now and 2050,” she said.
Dr. Bakris has received consulting fees from Alnylam Pharmaceuticals.
A version of this article first appeared on Medscape.com.
with what appeared to be an encouraging side-effect profile, in the phase 2 dose-ranging KARDIA-1 study.
“Our study demonstrates that either quarterly or biannual doses of zilebesiran can effectively and safely lower blood pressure in patients with uncontrolled hypertension,” said senior study investigator George Bakris, MD.
“Based on these results, zilebesiran has the potential to improve medication adherence, which will, in turn, reduce cardiovascular risk in people with hypertension,” added Dr. Bakris, who is professor of medicine and director of the Comprehensive Hypertension Center at the University of Chicago Medicine.
The KARDIA-1 study was presented at the American Heart Association scientific sessions.
Dr. Bakris noted that uncontrolled hypertension is a leading cause of morbidity and mortality, and despite availability of effective antihypertensives, many adults with hypertension are untreated, and up to 80% have uncontrolled disease, both globally and in the United States.
Zilebesiran is a subcutaneous RNA interference therapeutic that binds with high affinity to the hepatic asialoglycoprotein receptor, bringing about a reduction in the synthesis of angiotensinogen, the sole precursor of all angiotensin peptides. It is hoped that its hepatocyte-targeted delivery may allow extrahepatic angiotensinogen expression to be preserved, which could limit off-target effects in the kidney and other tissues.
The KARDIA-1 trial investigated the safety and efficacy of different doses of zilebesiran in patients with mild to moderate hypertension (systolic BP of 135-160 mm Hg), who are untreated or on stable therapy with up to two antihypertensive medications.
The study included 394 such patients (average baseline systolic BP was 142 mm Hg) who were randomly assigned to receive one of four different zilebesiran doses (150 mg, 300 mg, or 600 mg once every 6 months or 300 mg once every 2 months) or a placebo. The final analysis included 377 patients (56% men, 25% Black).
Results showed sustained reductions in serum angiotensinogen (between 88% and 98%) over the 6-month follow-up period.
Ambulatory systolic BP measured over 24 hours was significantly decreased with all zilebesiran regimens, with a mean reduction from baseline to month 6 of around 10 mm Hg in the three top doses studied and by around 14 mm Hg compared with placebo.
Patients receiving zilebesiran were more likely to achieve 24-hour average systolic BP measurements of 130 mm Hg or less at 6 months.
In addition, participants in all four zilebesiran groups consistently experienced significantly greater reductions in both daytime and nighttime systolic BP.
There were four nonserious adverse reactions leading to discontinuation in the zilebesiran groups: two instances of orthostatic hypotension, one of BP elevation, and one of injection site reaction.
Most hyperkalemia adverse events, which occurred in 6% of patients, were mild, did not require intervention, and generally resolved with repeat measurement; none were associated with acute kidney injury or led to study drug discontinuation. The incidence of hypotension events was low, and no clinically relevant changes in renal or hepatic function were observed, Dr. Bakris reported.
There was one death caused by cardiopulmonary arrest in a patient receiving zilebesiran 300 mg every 3 months, but this was not classified as drug related.
Zilebesiran is being further evaluated as an add-on therapy for treatment of hypertension in the ongoing KARDIA-2 phase 2 study.
Moderator of an AHA press conference at which the study was discussed, Sandra Taler, MD, professor of medicine at the Mayo Clinic, Rochester, Minn., said that “to have an injectable medicine that gives long-term blood pressure lowering is extremely exciting.”
Dr. Taler raised the point that some patients may not return for subsequent doses, but added that with subcutaneous dosing, administration at home may be a possibility.
Also commenting at the press conference, Keith Ferdinand, MD, professor of clinical medicine at Tulane University, New Orleans, said that this study “suggests we can now target the first step in the renin-angiotensin system – angiotensinogen – which then appears to lead to robust and continued blood pressure lowering for up to 6 months, which should improve adherence.”
Noting that only 50% of patients continue to take antihypertensive drugs after 1 year, Dr. Ferdinand added: “If we can increase adherence, we will increase efficacy and perhaps protect against some of the target organ damage.”
Designated discussant of the KARDIA-1 study at the AHA late-breaking clinical trial session, Anna Dominiczak, MD, University of Glasgow, noted that hypertension affects one in three adults worldwide, but only around 20% of people have it under control.
“An increase in the number of patients effectively treated for hypertension to levels observed in high-performing countries could prevent 76 million deaths, 120 million strokes, 79 million heart attacks, and 17 million cases of heart failure between now and 2050,” she said.
Dr. Bakris has received consulting fees from Alnylam Pharmaceuticals.
A version of this article first appeared on Medscape.com.
with what appeared to be an encouraging side-effect profile, in the phase 2 dose-ranging KARDIA-1 study.
“Our study demonstrates that either quarterly or biannual doses of zilebesiran can effectively and safely lower blood pressure in patients with uncontrolled hypertension,” said senior study investigator George Bakris, MD.
“Based on these results, zilebesiran has the potential to improve medication adherence, which will, in turn, reduce cardiovascular risk in people with hypertension,” added Dr. Bakris, who is professor of medicine and director of the Comprehensive Hypertension Center at the University of Chicago Medicine.
The KARDIA-1 study was presented at the American Heart Association scientific sessions.
Dr. Bakris noted that uncontrolled hypertension is a leading cause of morbidity and mortality, and despite availability of effective antihypertensives, many adults with hypertension are untreated, and up to 80% have uncontrolled disease, both globally and in the United States.
Zilebesiran is a subcutaneous RNA interference therapeutic that binds with high affinity to the hepatic asialoglycoprotein receptor, bringing about a reduction in the synthesis of angiotensinogen, the sole precursor of all angiotensin peptides. It is hoped that its hepatocyte-targeted delivery may allow extrahepatic angiotensinogen expression to be preserved, which could limit off-target effects in the kidney and other tissues.
The KARDIA-1 trial investigated the safety and efficacy of different doses of zilebesiran in patients with mild to moderate hypertension (systolic BP of 135-160 mm Hg), who are untreated or on stable therapy with up to two antihypertensive medications.
The study included 394 such patients (average baseline systolic BP was 142 mm Hg) who were randomly assigned to receive one of four different zilebesiran doses (150 mg, 300 mg, or 600 mg once every 6 months or 300 mg once every 2 months) or a placebo. The final analysis included 377 patients (56% men, 25% Black).
Results showed sustained reductions in serum angiotensinogen (between 88% and 98%) over the 6-month follow-up period.
Ambulatory systolic BP measured over 24 hours was significantly decreased with all zilebesiran regimens, with a mean reduction from baseline to month 6 of around 10 mm Hg in the three top doses studied and by around 14 mm Hg compared with placebo.
Patients receiving zilebesiran were more likely to achieve 24-hour average systolic BP measurements of 130 mm Hg or less at 6 months.
In addition, participants in all four zilebesiran groups consistently experienced significantly greater reductions in both daytime and nighttime systolic BP.
There were four nonserious adverse reactions leading to discontinuation in the zilebesiran groups: two instances of orthostatic hypotension, one of BP elevation, and one of injection site reaction.
Most hyperkalemia adverse events, which occurred in 6% of patients, were mild, did not require intervention, and generally resolved with repeat measurement; none were associated with acute kidney injury or led to study drug discontinuation. The incidence of hypotension events was low, and no clinically relevant changes in renal or hepatic function were observed, Dr. Bakris reported.
There was one death caused by cardiopulmonary arrest in a patient receiving zilebesiran 300 mg every 3 months, but this was not classified as drug related.
Zilebesiran is being further evaluated as an add-on therapy for treatment of hypertension in the ongoing KARDIA-2 phase 2 study.
Moderator of an AHA press conference at which the study was discussed, Sandra Taler, MD, professor of medicine at the Mayo Clinic, Rochester, Minn., said that “to have an injectable medicine that gives long-term blood pressure lowering is extremely exciting.”
Dr. Taler raised the point that some patients may not return for subsequent doses, but added that with subcutaneous dosing, administration at home may be a possibility.
Also commenting at the press conference, Keith Ferdinand, MD, professor of clinical medicine at Tulane University, New Orleans, said that this study “suggests we can now target the first step in the renin-angiotensin system – angiotensinogen – which then appears to lead to robust and continued blood pressure lowering for up to 6 months, which should improve adherence.”
Noting that only 50% of patients continue to take antihypertensive drugs after 1 year, Dr. Ferdinand added: “If we can increase adherence, we will increase efficacy and perhaps protect against some of the target organ damage.”
Designated discussant of the KARDIA-1 study at the AHA late-breaking clinical trial session, Anna Dominiczak, MD, University of Glasgow, noted that hypertension affects one in three adults worldwide, but only around 20% of people have it under control.
“An increase in the number of patients effectively treated for hypertension to levels observed in high-performing countries could prevent 76 million deaths, 120 million strokes, 79 million heart attacks, and 17 million cases of heart failure between now and 2050,” she said.
Dr. Bakris has received consulting fees from Alnylam Pharmaceuticals.
A version of this article first appeared on Medscape.com.
FROM AHA 2023
Infographic: Careers that tempt doctors to leave medicine
In a recently published Medscape report, 26% of American physicians said they were considering a career away from practicing medicine, for various reasons. Becoming a teacher was one of the nonclinical careers that most enthused them. What were the others?
For more details, check out the Medscape Physicians and Nonclinical Careers Report 2023.

A version of this article first appeared on Medscape.com.
In a recently published Medscape report, 26% of American physicians said they were considering a career away from practicing medicine, for various reasons. Becoming a teacher was one of the nonclinical careers that most enthused them. What were the others?
For more details, check out the Medscape Physicians and Nonclinical Careers Report 2023.
A version of this article first appeared on Medscape.com.
In a recently published Medscape report, 26% of American physicians said they were considering a career away from practicing medicine, for various reasons. Becoming a teacher was one of the nonclinical careers that most enthused them. What were the others?
For more details, check out the Medscape Physicians and Nonclinical Careers Report 2023.
A version of this article first appeared on Medscape.com.
WHO: Smoking cessation reduces risk of type 2 diabetes up to 40%
TOPLINE:
, and quitting even after one has developed type 2 diabetes is important in preventing a worsening of the disease’s many serious comorbidities, according to a new policy brief jointly issued by the World Health Organization, the International Diabetes Federation (IDF), and the University of Newcastle, Callaghan, Australia.
With type 2 diabetes representing one of the most prevalent chronic diseases worldwide and the ninth cause of death globally, the potential to reduce the risk and worsening of the disease by quitting smoking adds to the urgency of smoking cessation as a public health interest.
METHODOLOGY:
- The policy brief summarizes the evidence on the health impacts of type 2 diabetes, tobacco smoking, and the pathophysiology of tobacco use and its role in the development of type 2 diabetes.
- The brief also describes the latest data on newer products that target smokers or potential smokers, including smokeless tobacco, new nicotine and tobacco products, and their relationship with type 2 diabetes. For instance, evidence suggests that even with smokeless tobacco, heavy use or high consumption increases the risk of developing type 2 diabetes, as the products often contain nicotine, known to contribute to the development of type 2 diabetes and related health conditions.
- Evidence on the effectiveness of tobacco control interventions among those with type 2 diabetes is also summarized, including discussion of a systematic review of six studies suggesting that interventions focusing on education and the involvement of health care professionals and pharmacists can be beneficial for people with type 2 diabetes.
TAKEAWAY:
- Smoking exacerbates the known serious complications of diabetic neuropathy and foot ulcers with type 2 diabetes, while further impeding wound healing.
- Smoking also causes damage to retinal blood vessels already at risk with type 2 diabetes, increasing the risk of diabetic retinopathy and vision loss.
- Quitting tobacco use can help prevent those and other major health complications already linked to diabetes, including kidney failure and cardiovascular events.
- Studies show that key misconceptions among smokers with type 2 diabetes that can prevent cessation include concerns about post-cessation weight gain, the influence of peers who smoke, and the psychological aspect of addiction.
- Clinicians are urged to provide advice on how to stop smoking to all tobacco users during the course of a routine consultation or interaction, which can be accomplished in only a few minutes.
IN PRACTICE:
“Health professionals play a vital role in motivating and guiding individuals with type 2 diabetes in their journey to quit tobacco,” Ruediger Krech, MD, director of the Department of Health Promotion at the World Health Organization in Geneva, Switzerland, said in a press statement on the policy brief.
“Simultaneously, governments must take the crucial step of ensuring all indoor public places, workplaces, and public transport are completely smoke-free. These interventions are essential safeguards against the onset and progression of this and many other chronic diseases,” he emphasized.
SOURCE:
The policy brief was jointly developed by the World Health Organization, the International Diabetes Federation, and the University of Newcastle.
The detailed policy brief can be downloaded on the IDF website.
LIMITATIONS:
Research remains limited on some issues, including the effectiveness of tobacco control interventions and smoking cessation methods for people with type 2 diabetes.
Likewise, specific guidelines for smoking cessation in the type 2 diabetes population are lacking. However, the general approaches of building patient motivation, behavioral interventions, and pharmacological treatments are advised.
“These interventions should be at least as intensive as those for the general population, while considering the unique characteristics of the disease and the individual,” the authors asserted.
DISCLOSURES:
The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
, and quitting even after one has developed type 2 diabetes is important in preventing a worsening of the disease’s many serious comorbidities, according to a new policy brief jointly issued by the World Health Organization, the International Diabetes Federation (IDF), and the University of Newcastle, Callaghan, Australia.
With type 2 diabetes representing one of the most prevalent chronic diseases worldwide and the ninth cause of death globally, the potential to reduce the risk and worsening of the disease by quitting smoking adds to the urgency of smoking cessation as a public health interest.
METHODOLOGY:
- The policy brief summarizes the evidence on the health impacts of type 2 diabetes, tobacco smoking, and the pathophysiology of tobacco use and its role in the development of type 2 diabetes.
- The brief also describes the latest data on newer products that target smokers or potential smokers, including smokeless tobacco, new nicotine and tobacco products, and their relationship with type 2 diabetes. For instance, evidence suggests that even with smokeless tobacco, heavy use or high consumption increases the risk of developing type 2 diabetes, as the products often contain nicotine, known to contribute to the development of type 2 diabetes and related health conditions.
- Evidence on the effectiveness of tobacco control interventions among those with type 2 diabetes is also summarized, including discussion of a systematic review of six studies suggesting that interventions focusing on education and the involvement of health care professionals and pharmacists can be beneficial for people with type 2 diabetes.
TAKEAWAY:
- Smoking exacerbates the known serious complications of diabetic neuropathy and foot ulcers with type 2 diabetes, while further impeding wound healing.
- Smoking also causes damage to retinal blood vessels already at risk with type 2 diabetes, increasing the risk of diabetic retinopathy and vision loss.
- Quitting tobacco use can help prevent those and other major health complications already linked to diabetes, including kidney failure and cardiovascular events.
- Studies show that key misconceptions among smokers with type 2 diabetes that can prevent cessation include concerns about post-cessation weight gain, the influence of peers who smoke, and the psychological aspect of addiction.
- Clinicians are urged to provide advice on how to stop smoking to all tobacco users during the course of a routine consultation or interaction, which can be accomplished in only a few minutes.
IN PRACTICE:
“Health professionals play a vital role in motivating and guiding individuals with type 2 diabetes in their journey to quit tobacco,” Ruediger Krech, MD, director of the Department of Health Promotion at the World Health Organization in Geneva, Switzerland, said in a press statement on the policy brief.
“Simultaneously, governments must take the crucial step of ensuring all indoor public places, workplaces, and public transport are completely smoke-free. These interventions are essential safeguards against the onset and progression of this and many other chronic diseases,” he emphasized.
SOURCE:
The policy brief was jointly developed by the World Health Organization, the International Diabetes Federation, and the University of Newcastle.
The detailed policy brief can be downloaded on the IDF website.
LIMITATIONS:
Research remains limited on some issues, including the effectiveness of tobacco control interventions and smoking cessation methods for people with type 2 diabetes.
Likewise, specific guidelines for smoking cessation in the type 2 diabetes population are lacking. However, the general approaches of building patient motivation, behavioral interventions, and pharmacological treatments are advised.
“These interventions should be at least as intensive as those for the general population, while considering the unique characteristics of the disease and the individual,” the authors asserted.
DISCLOSURES:
The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
, and quitting even after one has developed type 2 diabetes is important in preventing a worsening of the disease’s many serious comorbidities, according to a new policy brief jointly issued by the World Health Organization, the International Diabetes Federation (IDF), and the University of Newcastle, Callaghan, Australia.
With type 2 diabetes representing one of the most prevalent chronic diseases worldwide and the ninth cause of death globally, the potential to reduce the risk and worsening of the disease by quitting smoking adds to the urgency of smoking cessation as a public health interest.
METHODOLOGY:
- The policy brief summarizes the evidence on the health impacts of type 2 diabetes, tobacco smoking, and the pathophysiology of tobacco use and its role in the development of type 2 diabetes.
- The brief also describes the latest data on newer products that target smokers or potential smokers, including smokeless tobacco, new nicotine and tobacco products, and their relationship with type 2 diabetes. For instance, evidence suggests that even with smokeless tobacco, heavy use or high consumption increases the risk of developing type 2 diabetes, as the products often contain nicotine, known to contribute to the development of type 2 diabetes and related health conditions.
- Evidence on the effectiveness of tobacco control interventions among those with type 2 diabetes is also summarized, including discussion of a systematic review of six studies suggesting that interventions focusing on education and the involvement of health care professionals and pharmacists can be beneficial for people with type 2 diabetes.
TAKEAWAY:
- Smoking exacerbates the known serious complications of diabetic neuropathy and foot ulcers with type 2 diabetes, while further impeding wound healing.
- Smoking also causes damage to retinal blood vessels already at risk with type 2 diabetes, increasing the risk of diabetic retinopathy and vision loss.
- Quitting tobacco use can help prevent those and other major health complications already linked to diabetes, including kidney failure and cardiovascular events.
- Studies show that key misconceptions among smokers with type 2 diabetes that can prevent cessation include concerns about post-cessation weight gain, the influence of peers who smoke, and the psychological aspect of addiction.
- Clinicians are urged to provide advice on how to stop smoking to all tobacco users during the course of a routine consultation or interaction, which can be accomplished in only a few minutes.
IN PRACTICE:
“Health professionals play a vital role in motivating and guiding individuals with type 2 diabetes in their journey to quit tobacco,” Ruediger Krech, MD, director of the Department of Health Promotion at the World Health Organization in Geneva, Switzerland, said in a press statement on the policy brief.
“Simultaneously, governments must take the crucial step of ensuring all indoor public places, workplaces, and public transport are completely smoke-free. These interventions are essential safeguards against the onset and progression of this and many other chronic diseases,” he emphasized.
SOURCE:
The policy brief was jointly developed by the World Health Organization, the International Diabetes Federation, and the University of Newcastle.
The detailed policy brief can be downloaded on the IDF website.
LIMITATIONS:
Research remains limited on some issues, including the effectiveness of tobacco control interventions and smoking cessation methods for people with type 2 diabetes.
Likewise, specific guidelines for smoking cessation in the type 2 diabetes population are lacking. However, the general approaches of building patient motivation, behavioral interventions, and pharmacological treatments are advised.
“These interventions should be at least as intensive as those for the general population, while considering the unique characteristics of the disease and the individual,” the authors asserted.
DISCLOSURES:
The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Breast milk liquid biopsy under study for early-stage breast cancer detection
Breast cancer has a worse prognosis when diagnosed during pregnancy or postpartum. Methods for early detection are needed, as evidenced every day in the multidisciplinary unit for treating pregnancy-associated breast cancer, which operates within the breast unit at the Vall d’Hebron University Hospital in Barcelona.
The team working in this field is led by Cristina Saura, PhD, who is also head of the Breast Cancer Group at the Vall d’Hebron Institute of Oncology (VHIO). The results of a study recently published in Cancer Discovery show, for the first time, that breast milk from breast cancer patients contains circulating tumor DNA that can be detected by a liquid biopsy of the milk.
Dr. Saura explained in an interview why they began to pursue this research, which, in one sense, fell into their laps. “In this case, it arose from the concerns of a breast cancer patient who was diagnosed while pregnant with her third daughter. She was actually the one who came up with the idea for the project. She was worried that she had transmitted the tumor through her breast milk to her second daughter while breastfeeding. She had been breastfeeding for a long time and had stretched it out until shortly before she was diagnosed with breast cancer. So she brought us a sample of breast milk that she had stored in her freezer.
“So, thanks to her, that’s where our project started. Though we knew that breast cancer is not transmitted through breast milk, we decided to test the sample and look for markers that could help our research. In the end, when we analyzed the patient’s breast milk, we found DNA with the same mutation that was present in her tumor,” explained Dr. Saura. She noted that the breast milk they analyzed had been frozen for more than a year before the patient’s cancer diagnosis.
In terms of methodology, Ana Vivancos, PhD, head of the VHIO cancer genomics group and also one of the authors of the study, explained that they used two techniques to analyze the breast milk and blood samples: next-generation sequencing and droplet digital polymerase chain reaction. These methods confirmed the presence of ctDNA in the breast milk.
High-sensitivity genomic panel
“We were able to detect tumor mutations in milk samples from 13 of the 15 patients with breast cancer who were tested, while circulating tumor DNA was detected in only one of all the blood samples that were collected at the same time,” said Dr. Vivancos. “The samples from the two patients for whom no mutation was detected were discovered to be colostrum that had been collected during the first few hours of lactation.”
As a next step to make this finding practically useful, the research team designed a genomic panel using next-generation sequencing as a potential method for early detection of breast cancer. “We’ve developed a panel that uses hybrid capture chemistry and unique molecular identifiers that ensure better sensitivity during next-generation sequencing. The panel has been calibrated, based on the existing literature, to detect the genes that are most frequently mutated in breast cancer in young women under 45 years old.”
According to Dr. Vivancos, the sensitivity of this panel exceeds 70%. This means that for all the patient samples analyzed using this panel, 7 out of 10 cases are detected with 100% specificity.
“In practice, the panel design allows us to detect mutations in more than 95% of breast cancer cases in women under 45 years old. noted Dr. Vivancos.
As for this unresolved need, Dr. Saura explained that there is currently no system or tool available to allow early suspicion of breast tumors in pregnant women prior to diagnosis. “That’s exactly the goal of this research: to screen for breast cancer in women who have just given birth. Now, it needs to be validated in a larger group of women in a clinical trial.”
More direct contact with tumor cells
In Dr. Saura’s opinion, in Spain, just like taking a small blood sample from newborns in a heel-prick test to rule out metabolic diseases, milk samples could be taken from women who give birth to rule out or diagnose breast cancer.
As to the potential advantages that breast milk liquid biopsy could have over similar techniques like blood liquid biopsy, Dr. Vivancos pointed to the results of her study: “We have seen that breast milk liquid biopsy was positive for the presence of circulating tumor DNA in 87% of cases, whereas blood only revealed the presence of this marker in 8% of cases. This difference indicates that breast milk is a biofluid that is in more direct contact with tumor cells and therefore will be more informative in earlier stages.”
Dr. Saura explained that the data does not lie when it comes to these tumors in pregnant or postpartum women. “In general, they tend to have a worse prognosis because, in most cases, they are diagnosed in advanced stages. Furthermore, it is typically assumed that the physiological changes in the breasts during gestation and lactation, which are considered to be normal, may hide a developing tumor. The fact is that postpartum breast cancer, understood to be the 10 years after delivery, accounts for 40%-45% of breast cancer cases diagnosed before age 45.”
The researchers plan to continue this project. “Our next step to confirm the usefulness of breast milk as a new tool for liquid biopsy for early detection of breast cancer during the postpartum period is to perform this noninvasive test in thousands of women,” said Dr. Saura.
Goal: Standardize the test as a screening method
“Based on the results we’ve published, we’re starting a study aimed at collecting breast milk samples from 5,000 healthy women around the world who became pregnant at age 40 or older, or who got pregnant at any age and carry mutations that increase their risk of breast cancer,” Dr. Saura added.
When asked when they expect to have preliminary results from this new study, Dr. Saura stated that it’s not yet possible to say exactly when. “We’re still waiting for funding to continue this project, but we continue performing analyses on a case-by-case basis. Of course, if we detect any abnormalities in these women, we will follow the established protocol to confirm diagnosis and start treatment if necessary.”
When asked whether it is reasonable to expect breast milk liquid biopsy to become normalized as a screening method for women of childbearing age who have a history or risk factors for developing breast cancer, Dr. Vivancos said, “That’s the scenario we see in the future and what we wish to contribute toward by providing scientific evidence to make it a reality.”
“For now, our goal is to validate whether circulating tumor DNA can be detected by breast milk liquid biopsy even before breast cancer can be diagnosed using conventional imaging techniques. If we can validate these preliminary results, we will be able to detect breast cancer early using a noninvasive test like breast milk liquid biopsy,” explained Saura.
Lastly, and in view of the issues that are still unresolved when it comes to the detection and treatment of breast cancer during pregnancy, Dr. Saura highlighted the emotional impact that a diagnosis of pregnancy-related cancer has on women and on those close to them. “But the first thing they need to know is that diagnosis is not necessarily synonymous with termination of the pregnancy. On the contrary, this tumor can be treated during pregnancy, since surgery can be performed at any time, and chemotherapy can be started in the second trimester. Proof of this is the 72 children who have been born under these circumstances in the past 20 years at the Vall d’Hebrón University Hospital. This hospital is a pioneer in Spain thanks to its multidisciplinary program for education and specific follow-up with women who have been diagnosed with a breast tumor during pregnancy.”
Dr. Saura and Dr. Vivancos reported no relevant financial relationships.
This article was translated from the Medscape Spanish edition. A version of this article appeared on Medscape.com.
Breast cancer has a worse prognosis when diagnosed during pregnancy or postpartum. Methods for early detection are needed, as evidenced every day in the multidisciplinary unit for treating pregnancy-associated breast cancer, which operates within the breast unit at the Vall d’Hebron University Hospital in Barcelona.
The team working in this field is led by Cristina Saura, PhD, who is also head of the Breast Cancer Group at the Vall d’Hebron Institute of Oncology (VHIO). The results of a study recently published in Cancer Discovery show, for the first time, that breast milk from breast cancer patients contains circulating tumor DNA that can be detected by a liquid biopsy of the milk.
Dr. Saura explained in an interview why they began to pursue this research, which, in one sense, fell into their laps. “In this case, it arose from the concerns of a breast cancer patient who was diagnosed while pregnant with her third daughter. She was actually the one who came up with the idea for the project. She was worried that she had transmitted the tumor through her breast milk to her second daughter while breastfeeding. She had been breastfeeding for a long time and had stretched it out until shortly before she was diagnosed with breast cancer. So she brought us a sample of breast milk that she had stored in her freezer.
“So, thanks to her, that’s where our project started. Though we knew that breast cancer is not transmitted through breast milk, we decided to test the sample and look for markers that could help our research. In the end, when we analyzed the patient’s breast milk, we found DNA with the same mutation that was present in her tumor,” explained Dr. Saura. She noted that the breast milk they analyzed had been frozen for more than a year before the patient’s cancer diagnosis.
In terms of methodology, Ana Vivancos, PhD, head of the VHIO cancer genomics group and also one of the authors of the study, explained that they used two techniques to analyze the breast milk and blood samples: next-generation sequencing and droplet digital polymerase chain reaction. These methods confirmed the presence of ctDNA in the breast milk.
High-sensitivity genomic panel
“We were able to detect tumor mutations in milk samples from 13 of the 15 patients with breast cancer who were tested, while circulating tumor DNA was detected in only one of all the blood samples that were collected at the same time,” said Dr. Vivancos. “The samples from the two patients for whom no mutation was detected were discovered to be colostrum that had been collected during the first few hours of lactation.”
As a next step to make this finding practically useful, the research team designed a genomic panel using next-generation sequencing as a potential method for early detection of breast cancer. “We’ve developed a panel that uses hybrid capture chemistry and unique molecular identifiers that ensure better sensitivity during next-generation sequencing. The panel has been calibrated, based on the existing literature, to detect the genes that are most frequently mutated in breast cancer in young women under 45 years old.”
According to Dr. Vivancos, the sensitivity of this panel exceeds 70%. This means that for all the patient samples analyzed using this panel, 7 out of 10 cases are detected with 100% specificity.
“In practice, the panel design allows us to detect mutations in more than 95% of breast cancer cases in women under 45 years old. noted Dr. Vivancos.
As for this unresolved need, Dr. Saura explained that there is currently no system or tool available to allow early suspicion of breast tumors in pregnant women prior to diagnosis. “That’s exactly the goal of this research: to screen for breast cancer in women who have just given birth. Now, it needs to be validated in a larger group of women in a clinical trial.”
More direct contact with tumor cells
In Dr. Saura’s opinion, in Spain, just like taking a small blood sample from newborns in a heel-prick test to rule out metabolic diseases, milk samples could be taken from women who give birth to rule out or diagnose breast cancer.
As to the potential advantages that breast milk liquid biopsy could have over similar techniques like blood liquid biopsy, Dr. Vivancos pointed to the results of her study: “We have seen that breast milk liquid biopsy was positive for the presence of circulating tumor DNA in 87% of cases, whereas blood only revealed the presence of this marker in 8% of cases. This difference indicates that breast milk is a biofluid that is in more direct contact with tumor cells and therefore will be more informative in earlier stages.”
Dr. Saura explained that the data does not lie when it comes to these tumors in pregnant or postpartum women. “In general, they tend to have a worse prognosis because, in most cases, they are diagnosed in advanced stages. Furthermore, it is typically assumed that the physiological changes in the breasts during gestation and lactation, which are considered to be normal, may hide a developing tumor. The fact is that postpartum breast cancer, understood to be the 10 years after delivery, accounts for 40%-45% of breast cancer cases diagnosed before age 45.”
The researchers plan to continue this project. “Our next step to confirm the usefulness of breast milk as a new tool for liquid biopsy for early detection of breast cancer during the postpartum period is to perform this noninvasive test in thousands of women,” said Dr. Saura.
Goal: Standardize the test as a screening method
“Based on the results we’ve published, we’re starting a study aimed at collecting breast milk samples from 5,000 healthy women around the world who became pregnant at age 40 or older, or who got pregnant at any age and carry mutations that increase their risk of breast cancer,” Dr. Saura added.
When asked when they expect to have preliminary results from this new study, Dr. Saura stated that it’s not yet possible to say exactly when. “We’re still waiting for funding to continue this project, but we continue performing analyses on a case-by-case basis. Of course, if we detect any abnormalities in these women, we will follow the established protocol to confirm diagnosis and start treatment if necessary.”
When asked whether it is reasonable to expect breast milk liquid biopsy to become normalized as a screening method for women of childbearing age who have a history or risk factors for developing breast cancer, Dr. Vivancos said, “That’s the scenario we see in the future and what we wish to contribute toward by providing scientific evidence to make it a reality.”
“For now, our goal is to validate whether circulating tumor DNA can be detected by breast milk liquid biopsy even before breast cancer can be diagnosed using conventional imaging techniques. If we can validate these preliminary results, we will be able to detect breast cancer early using a noninvasive test like breast milk liquid biopsy,” explained Saura.
Lastly, and in view of the issues that are still unresolved when it comes to the detection and treatment of breast cancer during pregnancy, Dr. Saura highlighted the emotional impact that a diagnosis of pregnancy-related cancer has on women and on those close to them. “But the first thing they need to know is that diagnosis is not necessarily synonymous with termination of the pregnancy. On the contrary, this tumor can be treated during pregnancy, since surgery can be performed at any time, and chemotherapy can be started in the second trimester. Proof of this is the 72 children who have been born under these circumstances in the past 20 years at the Vall d’Hebrón University Hospital. This hospital is a pioneer in Spain thanks to its multidisciplinary program for education and specific follow-up with women who have been diagnosed with a breast tumor during pregnancy.”
Dr. Saura and Dr. Vivancos reported no relevant financial relationships.
This article was translated from the Medscape Spanish edition. A version of this article appeared on Medscape.com.
Breast cancer has a worse prognosis when diagnosed during pregnancy or postpartum. Methods for early detection are needed, as evidenced every day in the multidisciplinary unit for treating pregnancy-associated breast cancer, which operates within the breast unit at the Vall d’Hebron University Hospital in Barcelona.
The team working in this field is led by Cristina Saura, PhD, who is also head of the Breast Cancer Group at the Vall d’Hebron Institute of Oncology (VHIO). The results of a study recently published in Cancer Discovery show, for the first time, that breast milk from breast cancer patients contains circulating tumor DNA that can be detected by a liquid biopsy of the milk.
Dr. Saura explained in an interview why they began to pursue this research, which, in one sense, fell into their laps. “In this case, it arose from the concerns of a breast cancer patient who was diagnosed while pregnant with her third daughter. She was actually the one who came up with the idea for the project. She was worried that she had transmitted the tumor through her breast milk to her second daughter while breastfeeding. She had been breastfeeding for a long time and had stretched it out until shortly before she was diagnosed with breast cancer. So she brought us a sample of breast milk that she had stored in her freezer.
“So, thanks to her, that’s where our project started. Though we knew that breast cancer is not transmitted through breast milk, we decided to test the sample and look for markers that could help our research. In the end, when we analyzed the patient’s breast milk, we found DNA with the same mutation that was present in her tumor,” explained Dr. Saura. She noted that the breast milk they analyzed had been frozen for more than a year before the patient’s cancer diagnosis.
In terms of methodology, Ana Vivancos, PhD, head of the VHIO cancer genomics group and also one of the authors of the study, explained that they used two techniques to analyze the breast milk and blood samples: next-generation sequencing and droplet digital polymerase chain reaction. These methods confirmed the presence of ctDNA in the breast milk.
High-sensitivity genomic panel
“We were able to detect tumor mutations in milk samples from 13 of the 15 patients with breast cancer who were tested, while circulating tumor DNA was detected in only one of all the blood samples that were collected at the same time,” said Dr. Vivancos. “The samples from the two patients for whom no mutation was detected were discovered to be colostrum that had been collected during the first few hours of lactation.”
As a next step to make this finding practically useful, the research team designed a genomic panel using next-generation sequencing as a potential method for early detection of breast cancer. “We’ve developed a panel that uses hybrid capture chemistry and unique molecular identifiers that ensure better sensitivity during next-generation sequencing. The panel has been calibrated, based on the existing literature, to detect the genes that are most frequently mutated in breast cancer in young women under 45 years old.”
According to Dr. Vivancos, the sensitivity of this panel exceeds 70%. This means that for all the patient samples analyzed using this panel, 7 out of 10 cases are detected with 100% specificity.
“In practice, the panel design allows us to detect mutations in more than 95% of breast cancer cases in women under 45 years old. noted Dr. Vivancos.
As for this unresolved need, Dr. Saura explained that there is currently no system or tool available to allow early suspicion of breast tumors in pregnant women prior to diagnosis. “That’s exactly the goal of this research: to screen for breast cancer in women who have just given birth. Now, it needs to be validated in a larger group of women in a clinical trial.”
More direct contact with tumor cells
In Dr. Saura’s opinion, in Spain, just like taking a small blood sample from newborns in a heel-prick test to rule out metabolic diseases, milk samples could be taken from women who give birth to rule out or diagnose breast cancer.
As to the potential advantages that breast milk liquid biopsy could have over similar techniques like blood liquid biopsy, Dr. Vivancos pointed to the results of her study: “We have seen that breast milk liquid biopsy was positive for the presence of circulating tumor DNA in 87% of cases, whereas blood only revealed the presence of this marker in 8% of cases. This difference indicates that breast milk is a biofluid that is in more direct contact with tumor cells and therefore will be more informative in earlier stages.”
Dr. Saura explained that the data does not lie when it comes to these tumors in pregnant or postpartum women. “In general, they tend to have a worse prognosis because, in most cases, they are diagnosed in advanced stages. Furthermore, it is typically assumed that the physiological changes in the breasts during gestation and lactation, which are considered to be normal, may hide a developing tumor. The fact is that postpartum breast cancer, understood to be the 10 years after delivery, accounts for 40%-45% of breast cancer cases diagnosed before age 45.”
The researchers plan to continue this project. “Our next step to confirm the usefulness of breast milk as a new tool for liquid biopsy for early detection of breast cancer during the postpartum period is to perform this noninvasive test in thousands of women,” said Dr. Saura.
Goal: Standardize the test as a screening method
“Based on the results we’ve published, we’re starting a study aimed at collecting breast milk samples from 5,000 healthy women around the world who became pregnant at age 40 or older, or who got pregnant at any age and carry mutations that increase their risk of breast cancer,” Dr. Saura added.
When asked when they expect to have preliminary results from this new study, Dr. Saura stated that it’s not yet possible to say exactly when. “We’re still waiting for funding to continue this project, but we continue performing analyses on a case-by-case basis. Of course, if we detect any abnormalities in these women, we will follow the established protocol to confirm diagnosis and start treatment if necessary.”
When asked whether it is reasonable to expect breast milk liquid biopsy to become normalized as a screening method for women of childbearing age who have a history or risk factors for developing breast cancer, Dr. Vivancos said, “That’s the scenario we see in the future and what we wish to contribute toward by providing scientific evidence to make it a reality.”
“For now, our goal is to validate whether circulating tumor DNA can be detected by breast milk liquid biopsy even before breast cancer can be diagnosed using conventional imaging techniques. If we can validate these preliminary results, we will be able to detect breast cancer early using a noninvasive test like breast milk liquid biopsy,” explained Saura.
Lastly, and in view of the issues that are still unresolved when it comes to the detection and treatment of breast cancer during pregnancy, Dr. Saura highlighted the emotional impact that a diagnosis of pregnancy-related cancer has on women and on those close to them. “But the first thing they need to know is that diagnosis is not necessarily synonymous with termination of the pregnancy. On the contrary, this tumor can be treated during pregnancy, since surgery can be performed at any time, and chemotherapy can be started in the second trimester. Proof of this is the 72 children who have been born under these circumstances in the past 20 years at the Vall d’Hebrón University Hospital. This hospital is a pioneer in Spain thanks to its multidisciplinary program for education and specific follow-up with women who have been diagnosed with a breast tumor during pregnancy.”
Dr. Saura and Dr. Vivancos reported no relevant financial relationships.
This article was translated from the Medscape Spanish edition. A version of this article appeared on Medscape.com.
FROM CANCER DISCOVERY
Can a Mediterranean diet reduce breast cancer recurrence?
TOPLINE:
However, women at high risk for recurrence who made the greatest improvements in their diet quality demonstrated a 41% lower risk for recurrence, compared with peers who made the fewest improvements.
METHODOLOGY:
- A growing body of evidence suggests that a better dietary quality may improve survival among patients with breast cancer, but whether diet impacts breast cancer–specific mortality remains controversial.
- To better understand the relationship between diet and breast cancer outcomes, investigators recruited 1,542 women with breast cancer who had undergone surgical resection in the past 5 years and were considered high risk for recurrence.
- All women received general recommendations for cancer prevention, while the intervention group received active support to adhere to a macro–Mediterranean-style diet, which encourages mainly consuming whole grains, legumes, and high-fiber vegetables and discourages eating foods high in saturated and trans fats, processed meats, and foods and beverages high in sugar.
- Diet was assessed at baseline, 1 year, and every few months in subsequent years via food frequency diaries. Compliance with dietary recommendations for the whole cohort was assessed using a dietary index developed for the trial.
- In addition to diet, women in the diet intervention group were encouraged to maintain moderate to intense physical activity – 30 minutes, on average, each day – and received pedometers to track steps, aiming for 10,000 per day.
TAKEAWAY:
- Over 5 years of follow-up, the rate of breast cancer recurrence did not differ between women in the diet intervention group and those in the control group. Overall, 95 of 769 women in the intervention group and 98 of 773 in the control group had a breast cancer recurrence (hazard ratio, 0.99).
- When evaluating outcomes in the entire cohort, looking at everyone’s level of compliance with dietary recommendations, women who adhered the most to the dietary guidelines had a 41% lower recurrence risk compared with women who adhered the least (HR, 0.59).
- The greatest protective effect among women who demonstrated high compliance occurred in those with ER-positive cancers (HR, 0.42) and those with ER-positive cancers who received tamoxifen (HR, 0.30).
IN PRACTICE:
This intervention trial “did not confirm the hypothesis that a comprehensive dietary modification reduces breast cancer recurrence and metastases,” but when looking at compliance to the Mediterranean diet overall, the analysis did find “a significantly better prognosis” for women with the best adherence.
SOURCE:
The study, with first author Franco Berrino, MD, PhD, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, was published online in Clinical Cancer Research.
LIMITATIONS:
The study relied on self-reported dietary data. No dietary instrument was used to estimate nutrient intake and the dietary index developed for the trial remains unvalidated.
DISCLOSURES:
The study was supported by the Italian Department of Health, the Associazione Italiana per la Ricerca sul Cancro, and the Vita e Salute Foundation. The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
However, women at high risk for recurrence who made the greatest improvements in their diet quality demonstrated a 41% lower risk for recurrence, compared with peers who made the fewest improvements.
METHODOLOGY:
- A growing body of evidence suggests that a better dietary quality may improve survival among patients with breast cancer, but whether diet impacts breast cancer–specific mortality remains controversial.
- To better understand the relationship between diet and breast cancer outcomes, investigators recruited 1,542 women with breast cancer who had undergone surgical resection in the past 5 years and were considered high risk for recurrence.
- All women received general recommendations for cancer prevention, while the intervention group received active support to adhere to a macro–Mediterranean-style diet, which encourages mainly consuming whole grains, legumes, and high-fiber vegetables and discourages eating foods high in saturated and trans fats, processed meats, and foods and beverages high in sugar.
- Diet was assessed at baseline, 1 year, and every few months in subsequent years via food frequency diaries. Compliance with dietary recommendations for the whole cohort was assessed using a dietary index developed for the trial.
- In addition to diet, women in the diet intervention group were encouraged to maintain moderate to intense physical activity – 30 minutes, on average, each day – and received pedometers to track steps, aiming for 10,000 per day.
TAKEAWAY:
- Over 5 years of follow-up, the rate of breast cancer recurrence did not differ between women in the diet intervention group and those in the control group. Overall, 95 of 769 women in the intervention group and 98 of 773 in the control group had a breast cancer recurrence (hazard ratio, 0.99).
- When evaluating outcomes in the entire cohort, looking at everyone’s level of compliance with dietary recommendations, women who adhered the most to the dietary guidelines had a 41% lower recurrence risk compared with women who adhered the least (HR, 0.59).
- The greatest protective effect among women who demonstrated high compliance occurred in those with ER-positive cancers (HR, 0.42) and those with ER-positive cancers who received tamoxifen (HR, 0.30).
IN PRACTICE:
This intervention trial “did not confirm the hypothesis that a comprehensive dietary modification reduces breast cancer recurrence and metastases,” but when looking at compliance to the Mediterranean diet overall, the analysis did find “a significantly better prognosis” for women with the best adherence.
SOURCE:
The study, with first author Franco Berrino, MD, PhD, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, was published online in Clinical Cancer Research.
LIMITATIONS:
The study relied on self-reported dietary data. No dietary instrument was used to estimate nutrient intake and the dietary index developed for the trial remains unvalidated.
DISCLOSURES:
The study was supported by the Italian Department of Health, the Associazione Italiana per la Ricerca sul Cancro, and the Vita e Salute Foundation. The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
However, women at high risk for recurrence who made the greatest improvements in their diet quality demonstrated a 41% lower risk for recurrence, compared with peers who made the fewest improvements.
METHODOLOGY:
- A growing body of evidence suggests that a better dietary quality may improve survival among patients with breast cancer, but whether diet impacts breast cancer–specific mortality remains controversial.
- To better understand the relationship between diet and breast cancer outcomes, investigators recruited 1,542 women with breast cancer who had undergone surgical resection in the past 5 years and were considered high risk for recurrence.
- All women received general recommendations for cancer prevention, while the intervention group received active support to adhere to a macro–Mediterranean-style diet, which encourages mainly consuming whole grains, legumes, and high-fiber vegetables and discourages eating foods high in saturated and trans fats, processed meats, and foods and beverages high in sugar.
- Diet was assessed at baseline, 1 year, and every few months in subsequent years via food frequency diaries. Compliance with dietary recommendations for the whole cohort was assessed using a dietary index developed for the trial.
- In addition to diet, women in the diet intervention group were encouraged to maintain moderate to intense physical activity – 30 minutes, on average, each day – and received pedometers to track steps, aiming for 10,000 per day.
TAKEAWAY:
- Over 5 years of follow-up, the rate of breast cancer recurrence did not differ between women in the diet intervention group and those in the control group. Overall, 95 of 769 women in the intervention group and 98 of 773 in the control group had a breast cancer recurrence (hazard ratio, 0.99).
- When evaluating outcomes in the entire cohort, looking at everyone’s level of compliance with dietary recommendations, women who adhered the most to the dietary guidelines had a 41% lower recurrence risk compared with women who adhered the least (HR, 0.59).
- The greatest protective effect among women who demonstrated high compliance occurred in those with ER-positive cancers (HR, 0.42) and those with ER-positive cancers who received tamoxifen (HR, 0.30).
IN PRACTICE:
This intervention trial “did not confirm the hypothesis that a comprehensive dietary modification reduces breast cancer recurrence and metastases,” but when looking at compliance to the Mediterranean diet overall, the analysis did find “a significantly better prognosis” for women with the best adherence.
SOURCE:
The study, with first author Franco Berrino, MD, PhD, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, was published online in Clinical Cancer Research.
LIMITATIONS:
The study relied on self-reported dietary data. No dietary instrument was used to estimate nutrient intake and the dietary index developed for the trial remains unvalidated.
DISCLOSURES:
The study was supported by the Italian Department of Health, the Associazione Italiana per la Ricerca sul Cancro, and the Vita e Salute Foundation. The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FDA OKs capivasertib for certain advanced breast cancers
Specifically, the first-in-class AKT kinase inhibitor approval is for patients with one or more PIK3CA/AKT1/PTEN alterations, as detected by an FDA-approved test, whose metastatic disease progressed on at least one endocrine-based regimen or who experienced recurrence on or within 12 months of completing adjuvant therapy, according to the FDA approval announcement.
The FDA also approved a companion diagnostic device, the FoundationOne CDx assay, to identify patients who are eligible for treatment with capivasertib.
Approval of capivasertib was based on findings from the randomized, placebo-controlled, phase 3 CAPItello-291 trial, which involved 708 patients with locally advanced or metastatic HR-positive, HER2-negative breast cancer, including 289 whose tumors had PIK3CA/AKT1/PTEN alterations. All had progressed on aromatase inhibitor–based treatment and may have received up to two prior lines of endocrine therapy and up to one line of chemotherapy.
Patients were randomized to either 400 mg of oral capivasertib or placebo twice daily for 4 days, followed by 3 days off treatment each week over a 28-day treatment cycle. Patients in both arms received 500 mg intramuscular fulvestrant on cycle 1 days 1 and 15, and then every 28 days thereafter. Treatment continued until disease progression or unacceptable toxicity.
In the 289 patients with PIK3CA/AKT1/PTEN–altered tumors, median progression-free survival (PFS) in the capivasertib arm was 7.3 months versus 3.1 months in the placebo group (hazard ratio, 0.50).
An exploratory analysis of PFS in the 313 (44%) patients whose tumors did not have a PIK3CA/AKT1/PTEN alteration demonstrated a less notable benefit to the combination (HR, 0.79; 95% confidence interval, 0.61-1.02), indicating that “the difference in the overall population was primarily attributed to the results seen in the population of patients whose tumors have PIK3CA/AKT1/PTEN alteration,” the FDA explained.
Adverse reactions occurring in at least 20% of patients included decreased lymphocytes, leukocytes, hemoglobin, and neutrophils; increased fasting glucose, creatinine, and triglycerides; and diarrhea, nausea, fatigue, vomiting, and stomatitis.
The recommended capivasertib dose is 400 mg orally twice daily, given about 12 hours apart with or without food, for 4 days followed by 3 off days until disease progression or unacceptable toxicity, according to the prescribing information.
A version of this article first appeared on Medscape.com.
Specifically, the first-in-class AKT kinase inhibitor approval is for patients with one or more PIK3CA/AKT1/PTEN alterations, as detected by an FDA-approved test, whose metastatic disease progressed on at least one endocrine-based regimen or who experienced recurrence on or within 12 months of completing adjuvant therapy, according to the FDA approval announcement.
The FDA also approved a companion diagnostic device, the FoundationOne CDx assay, to identify patients who are eligible for treatment with capivasertib.
Approval of capivasertib was based on findings from the randomized, placebo-controlled, phase 3 CAPItello-291 trial, which involved 708 patients with locally advanced or metastatic HR-positive, HER2-negative breast cancer, including 289 whose tumors had PIK3CA/AKT1/PTEN alterations. All had progressed on aromatase inhibitor–based treatment and may have received up to two prior lines of endocrine therapy and up to one line of chemotherapy.
Patients were randomized to either 400 mg of oral capivasertib or placebo twice daily for 4 days, followed by 3 days off treatment each week over a 28-day treatment cycle. Patients in both arms received 500 mg intramuscular fulvestrant on cycle 1 days 1 and 15, and then every 28 days thereafter. Treatment continued until disease progression or unacceptable toxicity.
In the 289 patients with PIK3CA/AKT1/PTEN–altered tumors, median progression-free survival (PFS) in the capivasertib arm was 7.3 months versus 3.1 months in the placebo group (hazard ratio, 0.50).
An exploratory analysis of PFS in the 313 (44%) patients whose tumors did not have a PIK3CA/AKT1/PTEN alteration demonstrated a less notable benefit to the combination (HR, 0.79; 95% confidence interval, 0.61-1.02), indicating that “the difference in the overall population was primarily attributed to the results seen in the population of patients whose tumors have PIK3CA/AKT1/PTEN alteration,” the FDA explained.
Adverse reactions occurring in at least 20% of patients included decreased lymphocytes, leukocytes, hemoglobin, and neutrophils; increased fasting glucose, creatinine, and triglycerides; and diarrhea, nausea, fatigue, vomiting, and stomatitis.
The recommended capivasertib dose is 400 mg orally twice daily, given about 12 hours apart with or without food, for 4 days followed by 3 off days until disease progression or unacceptable toxicity, according to the prescribing information.
A version of this article first appeared on Medscape.com.
Specifically, the first-in-class AKT kinase inhibitor approval is for patients with one or more PIK3CA/AKT1/PTEN alterations, as detected by an FDA-approved test, whose metastatic disease progressed on at least one endocrine-based regimen or who experienced recurrence on or within 12 months of completing adjuvant therapy, according to the FDA approval announcement.
The FDA also approved a companion diagnostic device, the FoundationOne CDx assay, to identify patients who are eligible for treatment with capivasertib.
Approval of capivasertib was based on findings from the randomized, placebo-controlled, phase 3 CAPItello-291 trial, which involved 708 patients with locally advanced or metastatic HR-positive, HER2-negative breast cancer, including 289 whose tumors had PIK3CA/AKT1/PTEN alterations. All had progressed on aromatase inhibitor–based treatment and may have received up to two prior lines of endocrine therapy and up to one line of chemotherapy.
Patients were randomized to either 400 mg of oral capivasertib or placebo twice daily for 4 days, followed by 3 days off treatment each week over a 28-day treatment cycle. Patients in both arms received 500 mg intramuscular fulvestrant on cycle 1 days 1 and 15, and then every 28 days thereafter. Treatment continued until disease progression or unacceptable toxicity.
In the 289 patients with PIK3CA/AKT1/PTEN–altered tumors, median progression-free survival (PFS) in the capivasertib arm was 7.3 months versus 3.1 months in the placebo group (hazard ratio, 0.50).
An exploratory analysis of PFS in the 313 (44%) patients whose tumors did not have a PIK3CA/AKT1/PTEN alteration demonstrated a less notable benefit to the combination (HR, 0.79; 95% confidence interval, 0.61-1.02), indicating that “the difference in the overall population was primarily attributed to the results seen in the population of patients whose tumors have PIK3CA/AKT1/PTEN alteration,” the FDA explained.
Adverse reactions occurring in at least 20% of patients included decreased lymphocytes, leukocytes, hemoglobin, and neutrophils; increased fasting glucose, creatinine, and triglycerides; and diarrhea, nausea, fatigue, vomiting, and stomatitis.
The recommended capivasertib dose is 400 mg orally twice daily, given about 12 hours apart with or without food, for 4 days followed by 3 off days until disease progression or unacceptable toxicity, according to the prescribing information.
A version of this article first appeared on Medscape.com.
FDA panel voices concerns over 2 lymphoma accelerated approvals
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”