User login
Semaglutide doesn’t improve fibrosis in NASH-related cirrhosis
according to a phase 2 trial.
However, the glucagonlike peptide–1 (GLP-1) receptor agonist led to improvements in liver enzymes, liver steatosis, weight, triglycerides, and very low-density lipoprotein (VLDL) cholesterol. Similar proportions of patients in each group reported adverse events, such as nausea, diarrhea, and vomiting.
“Previous studies in patients with NASH and stage 2 or 3 fibrosis have shown that semaglutide can improve NASH resolution over 72 weeks. However, there are limited data on whether any therapy is effective in patients with NASH cirrhosis,” lead author Rohit Loomba, MD, founding director of the NAFLD Research Center at the University of California, San Diego, said in an interview.
“Although semaglutide did not succeed in improving histological fibrosis, it had success in improving other clinically important parameters, such as cardiometabolic risk factors, liver enzymes, liver fat, and noninvasive biomarkers of fibrosis,” he said.
The study was published online in The Lancet Gastroenterology & Hepatology.
Analyzing safety and efficacy
Dr. Loomba and colleagues conducted a double-blind, placebo-controlled phase 2 trial that enrolled 71 patients at 38 centers in the United States and Europe between June 2019 and April 2021. Adults with biopsy-confirmed NASH-related cirrhosis and a body mass index (BMI) of at least 27 kg/m2 were randomly assigned 2:1 to receive either once-weekly subcutaneous semaglutide at 2.4 mg or a visually matching placebo.
Patients were randomly allocated through an interactive web system, which stratified participants on the basis of the presence or absence of type 2 diabetes. Patients, investigators, and outcomes analysts were masked to the treatment assignment.
The primary endpoint was the proportion of patients with an improvement in liver fibrosis of one stage or more without a worsening of NASH after 48 weeks, which was measured through biopsy in the intention-to-treat population. Safety was also assessed in all patients who received at least one dose of semaglutide.
Among the 71 patients, 47 were randomly assigned to the semaglutide group and 24 to the placebo group. About 90% completed treatment, and 63 had evaluable paired biopsies for primary endpoint assessment.
Between the groups, 49 participants (69%) were women and 22 were men. The average age was 59.5 years, and the average BMI was 34.9. About 75% of patients had diabetes at baseline, with an average hemoglobin A1c of 7.1%.
After 48 weeks, researchers found no statistically significant difference between the groups in the proportion of patients with an improvement in liver fibrosis of one stage or more without worsening of NASH. In the semaglutide group, five patients (11%) had an improvement, compared with seven patients (29%) in the placebo group (odds ratio, 0.28; 95% confidence interval, 0.06-1.24, P = .087).
There also wasn’t a significant difference between groups in the proportion of patients who achieved NASH resolution. In the semaglutide group, 16 patients (34%) had resolution, compared with 5 patients (21%) in the placebo group (OR, 1.97; 95% CI, 0.56-7.91; P = .29).
In addition, a lower proportion of patients achieved both NASH resolution and improvement in liver fibrosis with semaglutide versus placebo, although the difference wasn’t significant. In the semaglutide group, three patients (6%) achieved both, compared with three patients (13%) in the placebo group (OR, 0.48; 95% CI, 0.06-3.91; P = .4). A lower proportion of patients had an improvement in liver fibrosis stage with semaglutide versus placebo.
Some improvements seen
However, the semaglutide group had significantly greater improvements in liver steatosis (but not stiffness), liver fat volume, procollagen 3 peptide, and liver enzymes such as ALT, AST, and gamma-glutamyltransferase.
Body weight decreased by 8.83% in the semaglutide group, compared with 0.09% in the placebo group, which was a significant difference. BMI, waist circumference, triglycerides, and VLDL cholesterol were also significantly lower in the semaglutide group, but total cholesterol and blood pressure measurements weren’t significantly different. Among those with type 2 diabetes, A1c also decreased in the semaglutide group but did not in the placebo group.
Similar proportions of patients in each group reported adverse events. In the semaglutide group, 42 patients (89%) had an adverse event, compared with 19 patients (79%) in the placebo group. In addition, six patients (13%) in the semaglutide group and two patients (8%) in the placebo group reported serious adverse events.
The most common adverse events in the semaglutide and placebo groups were nausea (45% and 17%), diarrhea (19% and 8%), and vomiting (17% and none), which mainly occurred during treatment initiation or dose escalation. No patients withdrew from the trial because of adverse events, although five had a dose reduction. Hepatic and renal function remained stable after semaglutide treatment, and there were no decompensating events or deaths.
“GLP-1 analogue exposure – among patients with compensated cirrhosis who suffer from morbid obesity and type 2 diabetes – for the treatment of diabetes appears to be well-tolerated and may be safe,” Dr. Loomba said. “Further studies are needed in this study population.”
Considering next steps
Dr. Loomba and colleagues are continuing research around risk factors linked to advanced fibrosis, such as type 2 diabetes, a family history of cirrhosis, and the presence of key genetic risk alleles. Gut dysbiosis also appears to increase the risk for advanced fatty liver disease, he said.
Future clinical trials could focus on therapeutic options for patients with advanced fibrosis, particularly those with cirrhosis who face increased risks for liver-related complications and mortality.
“As these patients are oftentimes excluded from initial randomized controlled trials, we have significantly less information on how to address obesity, type 2 diabetes, and NASH in these patients,” Fernando Bril, MD, a physician-scientist focused on NASH-related research at the University of Alabama at Birmingham, said in an interview.
Dr. Bril, who wasn’t involved with this study, wrote an accompanying editorial in The Lancet Gastroenterology & Hepatology.
Patients with NASH-related cirrhosis may have progressed to a point of the disease where fibrosis regression may be more difficult to achieve, he said.
“This emphasizes that early diagnosis of patients with NASH is crucial,” he said.
“Therefore, primary care providers, endocrinologists, and diabetologists need to have a low threshold to suspect liver disease in patients with overweight, obesity, and/or type 2 diabetes. Only this will allow for early initiation of therapy, which may delay the progression of liver disease.”
In further research, investigators may want to consider the lack of NASH resolution, a result that could be caused by this study being underpowered, Dr. Bril noted. The trend in resolution in this study appeared similar to improvements seen in NASH patients without cirrhosis in other studies, he said. The weight reduction and improved diabetes control in this group also shows promise.
“While a purist may be adamant that this was a negative study for histological outcomes, it is essential to take note of the positive results in many secondary outcomes,” he said. “Improving cardiometabolic risk in these patients is essential because many still die of cardiovascular disease and not liver-related complications.”
At the same time, it’s important to note that NASH can’t be oversimplified as “a matter of weight,” Dr. Bril said. Significant weight loss in the study didn’t result in histologic improvement, which means other strategies are needed to treat the disease.
“Negative results from this study emphasize that monotherapy may not be enough to improve NASH and liver fibrosis,” he said. “In a similar way we treat type 2 diabetes and hypertension with combination therapy, we need to consider a similar approach for patients with NASH.”
The study was sponsored by Novo Nordisk, which manufactures semaglutide. The authors declared grant funding, speaker fees, and consultant roles with numerous pharmaceutical companies. Dr. Bril had no relevant disclosures.
A version of this article first appeared on Medscape.com.
according to a phase 2 trial.
However, the glucagonlike peptide–1 (GLP-1) receptor agonist led to improvements in liver enzymes, liver steatosis, weight, triglycerides, and very low-density lipoprotein (VLDL) cholesterol. Similar proportions of patients in each group reported adverse events, such as nausea, diarrhea, and vomiting.
“Previous studies in patients with NASH and stage 2 or 3 fibrosis have shown that semaglutide can improve NASH resolution over 72 weeks. However, there are limited data on whether any therapy is effective in patients with NASH cirrhosis,” lead author Rohit Loomba, MD, founding director of the NAFLD Research Center at the University of California, San Diego, said in an interview.
“Although semaglutide did not succeed in improving histological fibrosis, it had success in improving other clinically important parameters, such as cardiometabolic risk factors, liver enzymes, liver fat, and noninvasive biomarkers of fibrosis,” he said.
The study was published online in The Lancet Gastroenterology & Hepatology.
Analyzing safety and efficacy
Dr. Loomba and colleagues conducted a double-blind, placebo-controlled phase 2 trial that enrolled 71 patients at 38 centers in the United States and Europe between June 2019 and April 2021. Adults with biopsy-confirmed NASH-related cirrhosis and a body mass index (BMI) of at least 27 kg/m2 were randomly assigned 2:1 to receive either once-weekly subcutaneous semaglutide at 2.4 mg or a visually matching placebo.
Patients were randomly allocated through an interactive web system, which stratified participants on the basis of the presence or absence of type 2 diabetes. Patients, investigators, and outcomes analysts were masked to the treatment assignment.
The primary endpoint was the proportion of patients with an improvement in liver fibrosis of one stage or more without a worsening of NASH after 48 weeks, which was measured through biopsy in the intention-to-treat population. Safety was also assessed in all patients who received at least one dose of semaglutide.
Among the 71 patients, 47 were randomly assigned to the semaglutide group and 24 to the placebo group. About 90% completed treatment, and 63 had evaluable paired biopsies for primary endpoint assessment.
Between the groups, 49 participants (69%) were women and 22 were men. The average age was 59.5 years, and the average BMI was 34.9. About 75% of patients had diabetes at baseline, with an average hemoglobin A1c of 7.1%.
After 48 weeks, researchers found no statistically significant difference between the groups in the proportion of patients with an improvement in liver fibrosis of one stage or more without worsening of NASH. In the semaglutide group, five patients (11%) had an improvement, compared with seven patients (29%) in the placebo group (odds ratio, 0.28; 95% confidence interval, 0.06-1.24, P = .087).
There also wasn’t a significant difference between groups in the proportion of patients who achieved NASH resolution. In the semaglutide group, 16 patients (34%) had resolution, compared with 5 patients (21%) in the placebo group (OR, 1.97; 95% CI, 0.56-7.91; P = .29).
In addition, a lower proportion of patients achieved both NASH resolution and improvement in liver fibrosis with semaglutide versus placebo, although the difference wasn’t significant. In the semaglutide group, three patients (6%) achieved both, compared with three patients (13%) in the placebo group (OR, 0.48; 95% CI, 0.06-3.91; P = .4). A lower proportion of patients had an improvement in liver fibrosis stage with semaglutide versus placebo.
Some improvements seen
However, the semaglutide group had significantly greater improvements in liver steatosis (but not stiffness), liver fat volume, procollagen 3 peptide, and liver enzymes such as ALT, AST, and gamma-glutamyltransferase.
Body weight decreased by 8.83% in the semaglutide group, compared with 0.09% in the placebo group, which was a significant difference. BMI, waist circumference, triglycerides, and VLDL cholesterol were also significantly lower in the semaglutide group, but total cholesterol and blood pressure measurements weren’t significantly different. Among those with type 2 diabetes, A1c also decreased in the semaglutide group but did not in the placebo group.
Similar proportions of patients in each group reported adverse events. In the semaglutide group, 42 patients (89%) had an adverse event, compared with 19 patients (79%) in the placebo group. In addition, six patients (13%) in the semaglutide group and two patients (8%) in the placebo group reported serious adverse events.
The most common adverse events in the semaglutide and placebo groups were nausea (45% and 17%), diarrhea (19% and 8%), and vomiting (17% and none), which mainly occurred during treatment initiation or dose escalation. No patients withdrew from the trial because of adverse events, although five had a dose reduction. Hepatic and renal function remained stable after semaglutide treatment, and there were no decompensating events or deaths.
“GLP-1 analogue exposure – among patients with compensated cirrhosis who suffer from morbid obesity and type 2 diabetes – for the treatment of diabetes appears to be well-tolerated and may be safe,” Dr. Loomba said. “Further studies are needed in this study population.”
Considering next steps
Dr. Loomba and colleagues are continuing research around risk factors linked to advanced fibrosis, such as type 2 diabetes, a family history of cirrhosis, and the presence of key genetic risk alleles. Gut dysbiosis also appears to increase the risk for advanced fatty liver disease, he said.
Future clinical trials could focus on therapeutic options for patients with advanced fibrosis, particularly those with cirrhosis who face increased risks for liver-related complications and mortality.
“As these patients are oftentimes excluded from initial randomized controlled trials, we have significantly less information on how to address obesity, type 2 diabetes, and NASH in these patients,” Fernando Bril, MD, a physician-scientist focused on NASH-related research at the University of Alabama at Birmingham, said in an interview.
Dr. Bril, who wasn’t involved with this study, wrote an accompanying editorial in The Lancet Gastroenterology & Hepatology.
Patients with NASH-related cirrhosis may have progressed to a point of the disease where fibrosis regression may be more difficult to achieve, he said.
“This emphasizes that early diagnosis of patients with NASH is crucial,” he said.
“Therefore, primary care providers, endocrinologists, and diabetologists need to have a low threshold to suspect liver disease in patients with overweight, obesity, and/or type 2 diabetes. Only this will allow for early initiation of therapy, which may delay the progression of liver disease.”
In further research, investigators may want to consider the lack of NASH resolution, a result that could be caused by this study being underpowered, Dr. Bril noted. The trend in resolution in this study appeared similar to improvements seen in NASH patients without cirrhosis in other studies, he said. The weight reduction and improved diabetes control in this group also shows promise.
“While a purist may be adamant that this was a negative study for histological outcomes, it is essential to take note of the positive results in many secondary outcomes,” he said. “Improving cardiometabolic risk in these patients is essential because many still die of cardiovascular disease and not liver-related complications.”
At the same time, it’s important to note that NASH can’t be oversimplified as “a matter of weight,” Dr. Bril said. Significant weight loss in the study didn’t result in histologic improvement, which means other strategies are needed to treat the disease.
“Negative results from this study emphasize that monotherapy may not be enough to improve NASH and liver fibrosis,” he said. “In a similar way we treat type 2 diabetes and hypertension with combination therapy, we need to consider a similar approach for patients with NASH.”
The study was sponsored by Novo Nordisk, which manufactures semaglutide. The authors declared grant funding, speaker fees, and consultant roles with numerous pharmaceutical companies. Dr. Bril had no relevant disclosures.
A version of this article first appeared on Medscape.com.
according to a phase 2 trial.
However, the glucagonlike peptide–1 (GLP-1) receptor agonist led to improvements in liver enzymes, liver steatosis, weight, triglycerides, and very low-density lipoprotein (VLDL) cholesterol. Similar proportions of patients in each group reported adverse events, such as nausea, diarrhea, and vomiting.
“Previous studies in patients with NASH and stage 2 or 3 fibrosis have shown that semaglutide can improve NASH resolution over 72 weeks. However, there are limited data on whether any therapy is effective in patients with NASH cirrhosis,” lead author Rohit Loomba, MD, founding director of the NAFLD Research Center at the University of California, San Diego, said in an interview.
“Although semaglutide did not succeed in improving histological fibrosis, it had success in improving other clinically important parameters, such as cardiometabolic risk factors, liver enzymes, liver fat, and noninvasive biomarkers of fibrosis,” he said.
The study was published online in The Lancet Gastroenterology & Hepatology.
Analyzing safety and efficacy
Dr. Loomba and colleagues conducted a double-blind, placebo-controlled phase 2 trial that enrolled 71 patients at 38 centers in the United States and Europe between June 2019 and April 2021. Adults with biopsy-confirmed NASH-related cirrhosis and a body mass index (BMI) of at least 27 kg/m2 were randomly assigned 2:1 to receive either once-weekly subcutaneous semaglutide at 2.4 mg or a visually matching placebo.
Patients were randomly allocated through an interactive web system, which stratified participants on the basis of the presence or absence of type 2 diabetes. Patients, investigators, and outcomes analysts were masked to the treatment assignment.
The primary endpoint was the proportion of patients with an improvement in liver fibrosis of one stage or more without a worsening of NASH after 48 weeks, which was measured through biopsy in the intention-to-treat population. Safety was also assessed in all patients who received at least one dose of semaglutide.
Among the 71 patients, 47 were randomly assigned to the semaglutide group and 24 to the placebo group. About 90% completed treatment, and 63 had evaluable paired biopsies for primary endpoint assessment.
Between the groups, 49 participants (69%) were women and 22 were men. The average age was 59.5 years, and the average BMI was 34.9. About 75% of patients had diabetes at baseline, with an average hemoglobin A1c of 7.1%.
After 48 weeks, researchers found no statistically significant difference between the groups in the proportion of patients with an improvement in liver fibrosis of one stage or more without worsening of NASH. In the semaglutide group, five patients (11%) had an improvement, compared with seven patients (29%) in the placebo group (odds ratio, 0.28; 95% confidence interval, 0.06-1.24, P = .087).
There also wasn’t a significant difference between groups in the proportion of patients who achieved NASH resolution. In the semaglutide group, 16 patients (34%) had resolution, compared with 5 patients (21%) in the placebo group (OR, 1.97; 95% CI, 0.56-7.91; P = .29).
In addition, a lower proportion of patients achieved both NASH resolution and improvement in liver fibrosis with semaglutide versus placebo, although the difference wasn’t significant. In the semaglutide group, three patients (6%) achieved both, compared with three patients (13%) in the placebo group (OR, 0.48; 95% CI, 0.06-3.91; P = .4). A lower proportion of patients had an improvement in liver fibrosis stage with semaglutide versus placebo.
Some improvements seen
However, the semaglutide group had significantly greater improvements in liver steatosis (but not stiffness), liver fat volume, procollagen 3 peptide, and liver enzymes such as ALT, AST, and gamma-glutamyltransferase.
Body weight decreased by 8.83% in the semaglutide group, compared with 0.09% in the placebo group, which was a significant difference. BMI, waist circumference, triglycerides, and VLDL cholesterol were also significantly lower in the semaglutide group, but total cholesterol and blood pressure measurements weren’t significantly different. Among those with type 2 diabetes, A1c also decreased in the semaglutide group but did not in the placebo group.
Similar proportions of patients in each group reported adverse events. In the semaglutide group, 42 patients (89%) had an adverse event, compared with 19 patients (79%) in the placebo group. In addition, six patients (13%) in the semaglutide group and two patients (8%) in the placebo group reported serious adverse events.
The most common adverse events in the semaglutide and placebo groups were nausea (45% and 17%), diarrhea (19% and 8%), and vomiting (17% and none), which mainly occurred during treatment initiation or dose escalation. No patients withdrew from the trial because of adverse events, although five had a dose reduction. Hepatic and renal function remained stable after semaglutide treatment, and there were no decompensating events or deaths.
“GLP-1 analogue exposure – among patients with compensated cirrhosis who suffer from morbid obesity and type 2 diabetes – for the treatment of diabetes appears to be well-tolerated and may be safe,” Dr. Loomba said. “Further studies are needed in this study population.”
Considering next steps
Dr. Loomba and colleagues are continuing research around risk factors linked to advanced fibrosis, such as type 2 diabetes, a family history of cirrhosis, and the presence of key genetic risk alleles. Gut dysbiosis also appears to increase the risk for advanced fatty liver disease, he said.
Future clinical trials could focus on therapeutic options for patients with advanced fibrosis, particularly those with cirrhosis who face increased risks for liver-related complications and mortality.
“As these patients are oftentimes excluded from initial randomized controlled trials, we have significantly less information on how to address obesity, type 2 diabetes, and NASH in these patients,” Fernando Bril, MD, a physician-scientist focused on NASH-related research at the University of Alabama at Birmingham, said in an interview.
Dr. Bril, who wasn’t involved with this study, wrote an accompanying editorial in The Lancet Gastroenterology & Hepatology.
Patients with NASH-related cirrhosis may have progressed to a point of the disease where fibrosis regression may be more difficult to achieve, he said.
“This emphasizes that early diagnosis of patients with NASH is crucial,” he said.
“Therefore, primary care providers, endocrinologists, and diabetologists need to have a low threshold to suspect liver disease in patients with overweight, obesity, and/or type 2 diabetes. Only this will allow for early initiation of therapy, which may delay the progression of liver disease.”
In further research, investigators may want to consider the lack of NASH resolution, a result that could be caused by this study being underpowered, Dr. Bril noted. The trend in resolution in this study appeared similar to improvements seen in NASH patients without cirrhosis in other studies, he said. The weight reduction and improved diabetes control in this group also shows promise.
“While a purist may be adamant that this was a negative study for histological outcomes, it is essential to take note of the positive results in many secondary outcomes,” he said. “Improving cardiometabolic risk in these patients is essential because many still die of cardiovascular disease and not liver-related complications.”
At the same time, it’s important to note that NASH can’t be oversimplified as “a matter of weight,” Dr. Bril said. Significant weight loss in the study didn’t result in histologic improvement, which means other strategies are needed to treat the disease.
“Negative results from this study emphasize that monotherapy may not be enough to improve NASH and liver fibrosis,” he said. “In a similar way we treat type 2 diabetes and hypertension with combination therapy, we need to consider a similar approach for patients with NASH.”
The study was sponsored by Novo Nordisk, which manufactures semaglutide. The authors declared grant funding, speaker fees, and consultant roles with numerous pharmaceutical companies. Dr. Bril had no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM THE LANCET GASTROENTEROLOGY & HEPATOLOGY
Clozapine may curb schizophrenia’s ‘most dreaded outcome’
Investigators reviewed over 53,000 autopsy records, including over 600 from individuals whose autopsies revealed the presence of the antipsychotics clozapine or olanzapine, and found that those who took clozapine were significantly less likely to have died by suicide, compared with their counterparts who were taking olanzapine.
“Clozapine is an important and effective antisuicide medicine and should be strongly considered for treatment-resistant psychotic disorders, especially when the patient may be at risk for suicide,” study investigator Paul Nestadt, MD, associate professor, department of psychiatry and behavioral sciences, Johns Hopkins School of Medicine, Baltimore, told this news organization.
The study was published online in The Journal of Clinical Psychiatry.
Underutilized medication
Clozapine is the only medication indicated for treatment-resistant schizophrenia and is considered “the most efficacious antipsychotic,” the investigators note. Unfortunately, it has “long been underutilized” for several reasons, including prescriber hesitancy and concerns about side effects.
The authors note that its mechanism of action and the basis for superior efficacy are “still poorly understood” but “may extend beyond neurotransmitter receptor binding.”
Importantly, it may have a beneficial impact on domains other than positive symptoms of schizophrenia, including suicidality. Several studies have shown that it’s beneficial in this regard, but it is “unclear whether the unique antisuicidal properties of clozapine are related to better symptom control ... or to the closer monitoring and follow-up mandated for clozapine use,” they note.
A previous trial, the International Suicide Prevention Trial (InterSePT), demonstrated that clozapine is associated with a greater reduction in suicidality, and the findings “led to an FDA indication for clozapine in reducing the risk of recurrent suicidal behavior.”
However, the authors note, “in the severely ill populations in these studies, it is difficult to be certain about patients’ adherence to prescribed clozapine.”
“Other studies, such as InterSePT, have shown some evidence of clozapine working to reduce suicide-related outcomes, such as attempts or suicidal ideation, but few have been sufficiently powered to measure an effect on actual suicide deaths,” said Dr. Nestadt.

“As a suicidologist, I feel it is very important that we understand what treatments and interventions can actually prevent suicide deaths, as most suicides are not associated with past attempts or ideation, with suicide decedents usually looking very different from characteristic nonfatal attempters, from a clinical or epidemiological standpoint,” he added.
“If we could show that clozapine actually decreases the likelihood of suicide deaths in our patients, it gives us more reason to choose it over less effective neuroleptics in our clinics – especially for patients at high risk of suicide,” he said.
For the study, the researchers reviewed 19-year state-wide autopsy records of Maryland’s Office of the Chief Medical Examiner, which “performs uniquely comprehensive death investigations.” Data included in these investigations are full toxicologic panels with postmortem blood levels of antipsychotics.
The researchers compared decedents who tested positive for clozapine and decedents who tested positive for olanzapine. They evaluated demographics, clinical features, and manner-of-death outcomes.
‘Untapped resource’
Of 53,133 decedents, olanzapine or clozapine was detected in the blood of 621 persons (n = 571 and n = 50, respectively).
There were no significant differences in age, sex, race, or urban residence between the decedents who were treated with olanzapine and those who received clozapine.
The odds of a death by suicide in those treated with clozapine were less than half of the odds among decedents who had been treated with olanzapine (odds ratio, 0.47; 95% confidence interval, 0.26-0.84; P = .011).
In sensitivity analyses, the investigators reanalyzed the data to compare clozapine with other antipsychotics, including chlorpromazine, thioridazine, quetiapine, and olanzapine, and the results were similar. The odds of suicide (compared with accident) in those taking clozapine were much lower than in those taking any other tested antipsychotics individually or in combination (OR, 0.42; 95% CI, 0.24-0.73; P = .002).
Dr. Nestadt outlined several hypotheses regarding the mechanism of clozapine’s antisuicidal properties.
“Most theories stem from the differences in its receptor affinity, compared [with] the other neuroleptics,” he said. “In addition to the more typical dopaminergic blockade seen in neuroleptics, clozapine enhances serotonin release and greatly increases peripheral norepinephrine.”
This has been shown to “grant clozapine a greater antidepressant effect than other neuroleptics while also potentially decreasing aggression and impulsivity, which are both strongly associated with suicide risk,” he said.
Clozapine may also “work to reduce the inflammation-triggered activation of the kynurenine pathway, which otherwise contributes to serotonin depletion,” he added.
He noted that some studies have shown that as many as 1 in 10 patients with schizophrenia die by suicide, “so addressing this risk is paramount,” and that clozapine can play an important role in this.
The authors note that the findings “also highlight the utility of state-wide autopsy records, an untapped resource for investigating the potential protective effect of psychiatric medications on suicide at a population level.
“Importantly, we can be certain that this was not an issue of nonadherence to treatment in either group, which is a common issue in the use of these drugs because, instead of prescription records or self-report, we used actual measurements of drug presence in decedents’ blood at death,” said Dr. Nestadt.
‘Strongly suggestive’ data
Commenting on the study, Maria Oquendo, MD, PhD, Ruth Meltzer Professor and chair of psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, said most work on antisuicidal psychopharmacologic approaches “focuses on suicidal ideation or suicide attempts, due to the rarity of suicide death, even in high-risk populations.”
“Showing that clozapine may decrease risk for the most dreaded outcome of schizophrenia – suicide – is critically important,” said Dr. Oquendo, past president of the American Psychiatric Association.
Nevertheless, some questions remain, said Dr. Oquendo, who was not involved with the study. “Comparison of suicides to only accidental deaths has limitations. Many individuals who die due to accidents, like many suicides, are not similar to the general population,” she added.
However, she acknowledged, the data are strongly suggestive that clozapine protects against suicide.
“While not definitive, ideally these findings will stimulate changes in prescribing practices which may be lifesaving both literally – in terms of preventing suicides – and figuratively, given the drug’s effect on symptoms that impact quality of life and functioning,” said Dr. Oquendo.
The study received no funding or support. Dr. Nestadt is supported by the American Foundation for Suicide prevention and the National Institute on Drug Abuse. The other authors’ disclosures are listed in the original article. Dr. Oquendo receives royalties from the Research Foundation for Mental Hygiene for the commercial use of the Columbia Suicide Severity Rating Scale. She serves as an advisor to Alkermes, Mind Medicine, Sage Therapeutics, St. George’s University, and Fundacion Jimenez Diaz. Her family owns stock in Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
Investigators reviewed over 53,000 autopsy records, including over 600 from individuals whose autopsies revealed the presence of the antipsychotics clozapine or olanzapine, and found that those who took clozapine were significantly less likely to have died by suicide, compared with their counterparts who were taking olanzapine.
“Clozapine is an important and effective antisuicide medicine and should be strongly considered for treatment-resistant psychotic disorders, especially when the patient may be at risk for suicide,” study investigator Paul Nestadt, MD, associate professor, department of psychiatry and behavioral sciences, Johns Hopkins School of Medicine, Baltimore, told this news organization.
The study was published online in The Journal of Clinical Psychiatry.
Underutilized medication
Clozapine is the only medication indicated for treatment-resistant schizophrenia and is considered “the most efficacious antipsychotic,” the investigators note. Unfortunately, it has “long been underutilized” for several reasons, including prescriber hesitancy and concerns about side effects.
The authors note that its mechanism of action and the basis for superior efficacy are “still poorly understood” but “may extend beyond neurotransmitter receptor binding.”
Importantly, it may have a beneficial impact on domains other than positive symptoms of schizophrenia, including suicidality. Several studies have shown that it’s beneficial in this regard, but it is “unclear whether the unique antisuicidal properties of clozapine are related to better symptom control ... or to the closer monitoring and follow-up mandated for clozapine use,” they note.
A previous trial, the International Suicide Prevention Trial (InterSePT), demonstrated that clozapine is associated with a greater reduction in suicidality, and the findings “led to an FDA indication for clozapine in reducing the risk of recurrent suicidal behavior.”
However, the authors note, “in the severely ill populations in these studies, it is difficult to be certain about patients’ adherence to prescribed clozapine.”
“Other studies, such as InterSePT, have shown some evidence of clozapine working to reduce suicide-related outcomes, such as attempts or suicidal ideation, but few have been sufficiently powered to measure an effect on actual suicide deaths,” said Dr. Nestadt.
“As a suicidologist, I feel it is very important that we understand what treatments and interventions can actually prevent suicide deaths, as most suicides are not associated with past attempts or ideation, with suicide decedents usually looking very different from characteristic nonfatal attempters, from a clinical or epidemiological standpoint,” he added.
“If we could show that clozapine actually decreases the likelihood of suicide deaths in our patients, it gives us more reason to choose it over less effective neuroleptics in our clinics – especially for patients at high risk of suicide,” he said.
For the study, the researchers reviewed 19-year state-wide autopsy records of Maryland’s Office of the Chief Medical Examiner, which “performs uniquely comprehensive death investigations.” Data included in these investigations are full toxicologic panels with postmortem blood levels of antipsychotics.
The researchers compared decedents who tested positive for clozapine and decedents who tested positive for olanzapine. They evaluated demographics, clinical features, and manner-of-death outcomes.
‘Untapped resource’
Of 53,133 decedents, olanzapine or clozapine was detected in the blood of 621 persons (n = 571 and n = 50, respectively).
There were no significant differences in age, sex, race, or urban residence between the decedents who were treated with olanzapine and those who received clozapine.
The odds of a death by suicide in those treated with clozapine were less than half of the odds among decedents who had been treated with olanzapine (odds ratio, 0.47; 95% confidence interval, 0.26-0.84; P = .011).
In sensitivity analyses, the investigators reanalyzed the data to compare clozapine with other antipsychotics, including chlorpromazine, thioridazine, quetiapine, and olanzapine, and the results were similar. The odds of suicide (compared with accident) in those taking clozapine were much lower than in those taking any other tested antipsychotics individually or in combination (OR, 0.42; 95% CI, 0.24-0.73; P = .002).
Dr. Nestadt outlined several hypotheses regarding the mechanism of clozapine’s antisuicidal properties.
“Most theories stem from the differences in its receptor affinity, compared [with] the other neuroleptics,” he said. “In addition to the more typical dopaminergic blockade seen in neuroleptics, clozapine enhances serotonin release and greatly increases peripheral norepinephrine.”
This has been shown to “grant clozapine a greater antidepressant effect than other neuroleptics while also potentially decreasing aggression and impulsivity, which are both strongly associated with suicide risk,” he said.
Clozapine may also “work to reduce the inflammation-triggered activation of the kynurenine pathway, which otherwise contributes to serotonin depletion,” he added.
He noted that some studies have shown that as many as 1 in 10 patients with schizophrenia die by suicide, “so addressing this risk is paramount,” and that clozapine can play an important role in this.
The authors note that the findings “also highlight the utility of state-wide autopsy records, an untapped resource for investigating the potential protective effect of psychiatric medications on suicide at a population level.
“Importantly, we can be certain that this was not an issue of nonadherence to treatment in either group, which is a common issue in the use of these drugs because, instead of prescription records or self-report, we used actual measurements of drug presence in decedents’ blood at death,” said Dr. Nestadt.
‘Strongly suggestive’ data
Commenting on the study, Maria Oquendo, MD, PhD, Ruth Meltzer Professor and chair of psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, said most work on antisuicidal psychopharmacologic approaches “focuses on suicidal ideation or suicide attempts, due to the rarity of suicide death, even in high-risk populations.”
“Showing that clozapine may decrease risk for the most dreaded outcome of schizophrenia – suicide – is critically important,” said Dr. Oquendo, past president of the American Psychiatric Association.
Nevertheless, some questions remain, said Dr. Oquendo, who was not involved with the study. “Comparison of suicides to only accidental deaths has limitations. Many individuals who die due to accidents, like many suicides, are not similar to the general population,” she added.
However, she acknowledged, the data are strongly suggestive that clozapine protects against suicide.
“While not definitive, ideally these findings will stimulate changes in prescribing practices which may be lifesaving both literally – in terms of preventing suicides – and figuratively, given the drug’s effect on symptoms that impact quality of life and functioning,” said Dr. Oquendo.
The study received no funding or support. Dr. Nestadt is supported by the American Foundation for Suicide prevention and the National Institute on Drug Abuse. The other authors’ disclosures are listed in the original article. Dr. Oquendo receives royalties from the Research Foundation for Mental Hygiene for the commercial use of the Columbia Suicide Severity Rating Scale. She serves as an advisor to Alkermes, Mind Medicine, Sage Therapeutics, St. George’s University, and Fundacion Jimenez Diaz. Her family owns stock in Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
Investigators reviewed over 53,000 autopsy records, including over 600 from individuals whose autopsies revealed the presence of the antipsychotics clozapine or olanzapine, and found that those who took clozapine were significantly less likely to have died by suicide, compared with their counterparts who were taking olanzapine.
“Clozapine is an important and effective antisuicide medicine and should be strongly considered for treatment-resistant psychotic disorders, especially when the patient may be at risk for suicide,” study investigator Paul Nestadt, MD, associate professor, department of psychiatry and behavioral sciences, Johns Hopkins School of Medicine, Baltimore, told this news organization.
The study was published online in The Journal of Clinical Psychiatry.
Underutilized medication
Clozapine is the only medication indicated for treatment-resistant schizophrenia and is considered “the most efficacious antipsychotic,” the investigators note. Unfortunately, it has “long been underutilized” for several reasons, including prescriber hesitancy and concerns about side effects.
The authors note that its mechanism of action and the basis for superior efficacy are “still poorly understood” but “may extend beyond neurotransmitter receptor binding.”
Importantly, it may have a beneficial impact on domains other than positive symptoms of schizophrenia, including suicidality. Several studies have shown that it’s beneficial in this regard, but it is “unclear whether the unique antisuicidal properties of clozapine are related to better symptom control ... or to the closer monitoring and follow-up mandated for clozapine use,” they note.
A previous trial, the International Suicide Prevention Trial (InterSePT), demonstrated that clozapine is associated with a greater reduction in suicidality, and the findings “led to an FDA indication for clozapine in reducing the risk of recurrent suicidal behavior.”
However, the authors note, “in the severely ill populations in these studies, it is difficult to be certain about patients’ adherence to prescribed clozapine.”
“Other studies, such as InterSePT, have shown some evidence of clozapine working to reduce suicide-related outcomes, such as attempts or suicidal ideation, but few have been sufficiently powered to measure an effect on actual suicide deaths,” said Dr. Nestadt.
“As a suicidologist, I feel it is very important that we understand what treatments and interventions can actually prevent suicide deaths, as most suicides are not associated with past attempts or ideation, with suicide decedents usually looking very different from characteristic nonfatal attempters, from a clinical or epidemiological standpoint,” he added.
“If we could show that clozapine actually decreases the likelihood of suicide deaths in our patients, it gives us more reason to choose it over less effective neuroleptics in our clinics – especially for patients at high risk of suicide,” he said.
For the study, the researchers reviewed 19-year state-wide autopsy records of Maryland’s Office of the Chief Medical Examiner, which “performs uniquely comprehensive death investigations.” Data included in these investigations are full toxicologic panels with postmortem blood levels of antipsychotics.
The researchers compared decedents who tested positive for clozapine and decedents who tested positive for olanzapine. They evaluated demographics, clinical features, and manner-of-death outcomes.
‘Untapped resource’
Of 53,133 decedents, olanzapine or clozapine was detected in the blood of 621 persons (n = 571 and n = 50, respectively).
There were no significant differences in age, sex, race, or urban residence between the decedents who were treated with olanzapine and those who received clozapine.
The odds of a death by suicide in those treated with clozapine were less than half of the odds among decedents who had been treated with olanzapine (odds ratio, 0.47; 95% confidence interval, 0.26-0.84; P = .011).
In sensitivity analyses, the investigators reanalyzed the data to compare clozapine with other antipsychotics, including chlorpromazine, thioridazine, quetiapine, and olanzapine, and the results were similar. The odds of suicide (compared with accident) in those taking clozapine were much lower than in those taking any other tested antipsychotics individually or in combination (OR, 0.42; 95% CI, 0.24-0.73; P = .002).
Dr. Nestadt outlined several hypotheses regarding the mechanism of clozapine’s antisuicidal properties.
“Most theories stem from the differences in its receptor affinity, compared [with] the other neuroleptics,” he said. “In addition to the more typical dopaminergic blockade seen in neuroleptics, clozapine enhances serotonin release and greatly increases peripheral norepinephrine.”
This has been shown to “grant clozapine a greater antidepressant effect than other neuroleptics while also potentially decreasing aggression and impulsivity, which are both strongly associated with suicide risk,” he said.
Clozapine may also “work to reduce the inflammation-triggered activation of the kynurenine pathway, which otherwise contributes to serotonin depletion,” he added.
He noted that some studies have shown that as many as 1 in 10 patients with schizophrenia die by suicide, “so addressing this risk is paramount,” and that clozapine can play an important role in this.
The authors note that the findings “also highlight the utility of state-wide autopsy records, an untapped resource for investigating the potential protective effect of psychiatric medications on suicide at a population level.
“Importantly, we can be certain that this was not an issue of nonadherence to treatment in either group, which is a common issue in the use of these drugs because, instead of prescription records or self-report, we used actual measurements of drug presence in decedents’ blood at death,” said Dr. Nestadt.
‘Strongly suggestive’ data
Commenting on the study, Maria Oquendo, MD, PhD, Ruth Meltzer Professor and chair of psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, said most work on antisuicidal psychopharmacologic approaches “focuses on suicidal ideation or suicide attempts, due to the rarity of suicide death, even in high-risk populations.”
“Showing that clozapine may decrease risk for the most dreaded outcome of schizophrenia – suicide – is critically important,” said Dr. Oquendo, past president of the American Psychiatric Association.
Nevertheless, some questions remain, said Dr. Oquendo, who was not involved with the study. “Comparison of suicides to only accidental deaths has limitations. Many individuals who die due to accidents, like many suicides, are not similar to the general population,” she added.
However, she acknowledged, the data are strongly suggestive that clozapine protects against suicide.
“While not definitive, ideally these findings will stimulate changes in prescribing practices which may be lifesaving both literally – in terms of preventing suicides – and figuratively, given the drug’s effect on symptoms that impact quality of life and functioning,” said Dr. Oquendo.
The study received no funding or support. Dr. Nestadt is supported by the American Foundation for Suicide prevention and the National Institute on Drug Abuse. The other authors’ disclosures are listed in the original article. Dr. Oquendo receives royalties from the Research Foundation for Mental Hygiene for the commercial use of the Columbia Suicide Severity Rating Scale. She serves as an advisor to Alkermes, Mind Medicine, Sage Therapeutics, St. George’s University, and Fundacion Jimenez Diaz. Her family owns stock in Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF CLINICAL PSYCHIATRY
Low-dose olanzapine improves appetite in chemotherapy patients
Anorexia is a problem in approximately 50% of newly-diagnosed cancer patients, and can compromise survival, wrote study author Lakshmi Sandhya, MD, of Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), Puducherry, India, and colleagues. In particular, patients with lung and gastrointestinal tract cancers are prone to anorexia during chemotherapy, they said. Olanzapine is a demonstrated appetite stimulant and has been used in cancer patients as a short-term antiemetic, but its use for long-term appetite stimulation has not been well-studied, they said.
In the study, published in the Journal of Clinical Oncology, the researchers randomized 124 adults aged 18 years and older to a 2.5 grams of olanzapine or placebo for 12 weeks. The participants had untreated, locally advanced, or metastatic gastric, hepatopancreaticobiliary (HPB), or lung cancers.
The median age of the participants was 55 years. The primary outcome was a weight gain greater than 5% and improved appetite based on the visual analog scale (VAS) and questionnaires. A change in nutritional status, quality of life (QOL), and chemotherapy toxicity, were secondary endpoints.
After 12 weeks, complete data were available for 58 patients in the olanzapine group and 54 in the placebo group. Of these, 60% of the olanzapine group and 9% of the placebo group met the primary endpoint of a weight gain greater than 5%. The proportion of patients with improved appetite based on VAS scores and questionnaire scores was significantly higher in olanzapine patients vs. placebo patients (43% vs. 13% and 22% vs. 4%, respectively).
In addition, 52% of the olanzapine group vs. 18% of the placebo group achieved more than 75% intake of recommended daily calories.
Most of the reported toxicities were not hematological and similar between the groups (85% for olanzapine vs. 88% for placebo). The proportion of patients with toxicities of grade 3 or higher was lower in the olanzapine group vs. the placebo group (12% vs. 37%, P = .002). Patients in the olanzapine group also reported significantly improved quality of life from baseline compared to the placebo patients.
The findings were limited by several factors including the heterogeneous cancers and treatment regimens, the lack of data on weight beyond 12 weeks, the relatively small study population, and the subjective nature of anorexia measurements, the researchers noted.
However, the results suggest that low-dose olanzapine is an effective and well-tolerated add-on intervention for the subset of patients at risk for anorexia at the start of chemotherapy, they said.
“Future studies could look at various cancers in a multicentric setting and long-term endpoints such as patient survival,” they concluded.
The study drug and placebo were funded by an intramural grant from Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER). The researchers had no financial conflicts to disclose.
Anorexia is a problem in approximately 50% of newly-diagnosed cancer patients, and can compromise survival, wrote study author Lakshmi Sandhya, MD, of Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), Puducherry, India, and colleagues. In particular, patients with lung and gastrointestinal tract cancers are prone to anorexia during chemotherapy, they said. Olanzapine is a demonstrated appetite stimulant and has been used in cancer patients as a short-term antiemetic, but its use for long-term appetite stimulation has not been well-studied, they said.
In the study, published in the Journal of Clinical Oncology, the researchers randomized 124 adults aged 18 years and older to a 2.5 grams of olanzapine or placebo for 12 weeks. The participants had untreated, locally advanced, or metastatic gastric, hepatopancreaticobiliary (HPB), or lung cancers.
The median age of the participants was 55 years. The primary outcome was a weight gain greater than 5% and improved appetite based on the visual analog scale (VAS) and questionnaires. A change in nutritional status, quality of life (QOL), and chemotherapy toxicity, were secondary endpoints.
After 12 weeks, complete data were available for 58 patients in the olanzapine group and 54 in the placebo group. Of these, 60% of the olanzapine group and 9% of the placebo group met the primary endpoint of a weight gain greater than 5%. The proportion of patients with improved appetite based on VAS scores and questionnaire scores was significantly higher in olanzapine patients vs. placebo patients (43% vs. 13% and 22% vs. 4%, respectively).
In addition, 52% of the olanzapine group vs. 18% of the placebo group achieved more than 75% intake of recommended daily calories.
Most of the reported toxicities were not hematological and similar between the groups (85% for olanzapine vs. 88% for placebo). The proportion of patients with toxicities of grade 3 or higher was lower in the olanzapine group vs. the placebo group (12% vs. 37%, P = .002). Patients in the olanzapine group also reported significantly improved quality of life from baseline compared to the placebo patients.
The findings were limited by several factors including the heterogeneous cancers and treatment regimens, the lack of data on weight beyond 12 weeks, the relatively small study population, and the subjective nature of anorexia measurements, the researchers noted.
However, the results suggest that low-dose olanzapine is an effective and well-tolerated add-on intervention for the subset of patients at risk for anorexia at the start of chemotherapy, they said.
“Future studies could look at various cancers in a multicentric setting and long-term endpoints such as patient survival,” they concluded.
The study drug and placebo were funded by an intramural grant from Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER). The researchers had no financial conflicts to disclose.
Anorexia is a problem in approximately 50% of newly-diagnosed cancer patients, and can compromise survival, wrote study author Lakshmi Sandhya, MD, of Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), Puducherry, India, and colleagues. In particular, patients with lung and gastrointestinal tract cancers are prone to anorexia during chemotherapy, they said. Olanzapine is a demonstrated appetite stimulant and has been used in cancer patients as a short-term antiemetic, but its use for long-term appetite stimulation has not been well-studied, they said.
In the study, published in the Journal of Clinical Oncology, the researchers randomized 124 adults aged 18 years and older to a 2.5 grams of olanzapine or placebo for 12 weeks. The participants had untreated, locally advanced, or metastatic gastric, hepatopancreaticobiliary (HPB), or lung cancers.
The median age of the participants was 55 years. The primary outcome was a weight gain greater than 5% and improved appetite based on the visual analog scale (VAS) and questionnaires. A change in nutritional status, quality of life (QOL), and chemotherapy toxicity, were secondary endpoints.
After 12 weeks, complete data were available for 58 patients in the olanzapine group and 54 in the placebo group. Of these, 60% of the olanzapine group and 9% of the placebo group met the primary endpoint of a weight gain greater than 5%. The proportion of patients with improved appetite based on VAS scores and questionnaire scores was significantly higher in olanzapine patients vs. placebo patients (43% vs. 13% and 22% vs. 4%, respectively).
In addition, 52% of the olanzapine group vs. 18% of the placebo group achieved more than 75% intake of recommended daily calories.
Most of the reported toxicities were not hematological and similar between the groups (85% for olanzapine vs. 88% for placebo). The proportion of patients with toxicities of grade 3 or higher was lower in the olanzapine group vs. the placebo group (12% vs. 37%, P = .002). Patients in the olanzapine group also reported significantly improved quality of life from baseline compared to the placebo patients.
The findings were limited by several factors including the heterogeneous cancers and treatment regimens, the lack of data on weight beyond 12 weeks, the relatively small study population, and the subjective nature of anorexia measurements, the researchers noted.
However, the results suggest that low-dose olanzapine is an effective and well-tolerated add-on intervention for the subset of patients at risk for anorexia at the start of chemotherapy, they said.
“Future studies could look at various cancers in a multicentric setting and long-term endpoints such as patient survival,” they concluded.
The study drug and placebo were funded by an intramural grant from Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER). The researchers had no financial conflicts to disclose.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
SGLT2 inhibitors: Real-world data show benefits outweigh risks
Starting therapy with an SGLT2 inhibitor versus a glucagon-like peptide-1 (GLP-1) receptor agonist was associated with more lower limb amputations, nonvertebral fractures, and genital infections, but these risks need to be balanced against cardiovascular and renoprotective benefits, according to the researchers.
The analysis showed that there would be 2.1 more lower limb amputations, 2.5 more nonvertebral fractures, and 41 more genital infections per 1,000 patients per year among those receiving SGLT2 inhibitors versus an equal number of patients receiving GLP-1 agonists, lead author Edouard Fu, PhD, explained to this news organization in an email.
“On the other hand, we know from the evidence from randomized controlled trials that taking an SGLT2 inhibitor compared with placebo lowers the risk of developing kidney failure,” said Dr. Fu, who is a research fellow in the division of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital, Boston.
“For instance,” he continued, “in the DAPA-CKD clinical trial, dapagliflozin versus placebo led to 29 fewer events per 1,000 patients per year of the composite outcome (50% decline in estimated glomerular filtration rate [eGFR], kidney failure, cardiovascular or kidney death).”
In the CREDENCE trial, canagliflozin versus placebo led to 18 fewer events per 1,000 person-years for the composite outcome of doubling of serum creatinine, kidney failure, and cardiovascular or kidney death.
And in the EMPA-KIDNEY study, empagliflozin versus placebo led to 21 fewer events per 1,000 person-years for the composite outcome of progression of kidney disease or cardiovascular death.
“Thus, benefits would still outweigh the risks,” Dr. Fu emphasized.
‘Quantifies absolute rate of events among routine care patients’
“The importance of our paper,” he summarized, “is that it quantifies the absolute rate of events among routine care patients and may be used to inform shared decision-making.”
The analysis also found that the risks of diabetic ketoacidosis (DKA), hypovolemia, hypoglycemia, and severe urinary tract infection (UTI) were similar with SGLT2 inhibitors versus GLP-1 agonists, but the risk of developing acute kidney injury (AKI) was lower with an SGLT2 inhibitor.
“Our study can help inform patient-physician decision-making regarding risks and benefits before prescribing SGLT2 inhibitors in this population” of patients with CKD and diabetes treated in clinical practice, the researchers conclude, “but needs to be interpreted in light of its limitations, including residual confounding, short follow-up time, and the use of diagnosis codes to identify patients with CKD.”
The study was recently published in the Clinical Journal of the American Society of Nephrology.
Slow uptake, safety concerns
SGLT2 inhibitors are recommended as first-line therapy in patients with type 2 diabetes and CKD who have an eGFR equal to or greater than 20 mL/min per 1.73 m2, and thus are at high risk for cardiovascular disease and kidney disease progression, Dr. Fu and colleagues write.
However, studies report that as few as 6% of patients with CKD and type 2 diabetes are currently prescribed SGLT2 inhibitors in the United States.
This slow uptake of SGLT2 inhibitors among patients with CKD may be partly due to concerns about DKA, fractures, amputations, and urogenital infections observed in clinical trials.
However, such trials are generally underpowered to assess rare adverse events, use monitoring protocols to lower the risk of adverse events, and include a highly selected patient population, and so safety in routine clinical practice is often unclear.
To examine this, the researchers identified health insurance claims data from 96,128 individuals (from Optum, IBM MarketScan, and Medicare databases) who were 18 years or older (65 years or older for Medicare) and had type 2 diabetes and at least one inpatient or two outpatient diagnostic codes for stage 3 or 4 CKD.
Of these patients, 32,192 had a newly filled prescription for an SGLT2 inhibitor (empagliflozin, dapagliflozin, canagliflozin, or ertugliflozin) and 63,936 had a newly filled prescription for a GLP-1 agonist (liraglutide, dulaglutide, semaglutide, exenatide, albiglutide, or lixisenatide) between April 2013, when the first SGLT2 inhibitor was available in the United States, and 2021.
The researchers matched 28,847 individuals who were initiated on an SGLT2 inhibitor with an equal number who were initiated on a GLP-1 agonist, based on propensity scores, adjusting for more than 120 baseline characteristics.
Safety outcomes were based on previously identified potential safety signals.
Patients who were initiated on an SGLT2 inhibitor had 1.30-fold, 2.13-fold, and 3.08-fold higher risks of having a nonvertebral fracture, a lower limb amputation, and a genital infection, respectively, compared with patients who were initiated on a GLP-1 agonist, after a mean on-treatment time of 7.5 months,
Risks of DKA, hypovolemia, hypoglycemia, and severe UTI were similar in both groups.
Patients initiated on an SGLT2 inhibitor versus a GLP-1 agonist had a lower risk of AKI (hazard ratio, 0.93) equivalent to 6.75 fewer cases of AKI per 1,000 patients per year.
Patients had higher risks for lower limb amputation, genital infections, and nonvertebral fractures with SGLT2 inhibitors versus GLP-1 agonists across most of the prespecified subgroups by age, sex, cardiovascular disease, heart failure, and use of metformin, insulin, or sulfonylurea, but with wider confidence intervals.
Dr. Fu was supported by a Rubicon grant from the Dutch Research Council and has reported no relevant financial relationships. Disclosures for the other authors are listed with the article.
A version of this article originally appeared on Medscape.com.
Starting therapy with an SGLT2 inhibitor versus a glucagon-like peptide-1 (GLP-1) receptor agonist was associated with more lower limb amputations, nonvertebral fractures, and genital infections, but these risks need to be balanced against cardiovascular and renoprotective benefits, according to the researchers.
The analysis showed that there would be 2.1 more lower limb amputations, 2.5 more nonvertebral fractures, and 41 more genital infections per 1,000 patients per year among those receiving SGLT2 inhibitors versus an equal number of patients receiving GLP-1 agonists, lead author Edouard Fu, PhD, explained to this news organization in an email.
“On the other hand, we know from the evidence from randomized controlled trials that taking an SGLT2 inhibitor compared with placebo lowers the risk of developing kidney failure,” said Dr. Fu, who is a research fellow in the division of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital, Boston.
“For instance,” he continued, “in the DAPA-CKD clinical trial, dapagliflozin versus placebo led to 29 fewer events per 1,000 patients per year of the composite outcome (50% decline in estimated glomerular filtration rate [eGFR], kidney failure, cardiovascular or kidney death).”
In the CREDENCE trial, canagliflozin versus placebo led to 18 fewer events per 1,000 person-years for the composite outcome of doubling of serum creatinine, kidney failure, and cardiovascular or kidney death.
And in the EMPA-KIDNEY study, empagliflozin versus placebo led to 21 fewer events per 1,000 person-years for the composite outcome of progression of kidney disease or cardiovascular death.
“Thus, benefits would still outweigh the risks,” Dr. Fu emphasized.
‘Quantifies absolute rate of events among routine care patients’
“The importance of our paper,” he summarized, “is that it quantifies the absolute rate of events among routine care patients and may be used to inform shared decision-making.”
The analysis also found that the risks of diabetic ketoacidosis (DKA), hypovolemia, hypoglycemia, and severe urinary tract infection (UTI) were similar with SGLT2 inhibitors versus GLP-1 agonists, but the risk of developing acute kidney injury (AKI) was lower with an SGLT2 inhibitor.
“Our study can help inform patient-physician decision-making regarding risks and benefits before prescribing SGLT2 inhibitors in this population” of patients with CKD and diabetes treated in clinical practice, the researchers conclude, “but needs to be interpreted in light of its limitations, including residual confounding, short follow-up time, and the use of diagnosis codes to identify patients with CKD.”
The study was recently published in the Clinical Journal of the American Society of Nephrology.
Slow uptake, safety concerns
SGLT2 inhibitors are recommended as first-line therapy in patients with type 2 diabetes and CKD who have an eGFR equal to or greater than 20 mL/min per 1.73 m2, and thus are at high risk for cardiovascular disease and kidney disease progression, Dr. Fu and colleagues write.
However, studies report that as few as 6% of patients with CKD and type 2 diabetes are currently prescribed SGLT2 inhibitors in the United States.
This slow uptake of SGLT2 inhibitors among patients with CKD may be partly due to concerns about DKA, fractures, amputations, and urogenital infections observed in clinical trials.
However, such trials are generally underpowered to assess rare adverse events, use monitoring protocols to lower the risk of adverse events, and include a highly selected patient population, and so safety in routine clinical practice is often unclear.
To examine this, the researchers identified health insurance claims data from 96,128 individuals (from Optum, IBM MarketScan, and Medicare databases) who were 18 years or older (65 years or older for Medicare) and had type 2 diabetes and at least one inpatient or two outpatient diagnostic codes for stage 3 or 4 CKD.
Of these patients, 32,192 had a newly filled prescription for an SGLT2 inhibitor (empagliflozin, dapagliflozin, canagliflozin, or ertugliflozin) and 63,936 had a newly filled prescription for a GLP-1 agonist (liraglutide, dulaglutide, semaglutide, exenatide, albiglutide, or lixisenatide) between April 2013, when the first SGLT2 inhibitor was available in the United States, and 2021.
The researchers matched 28,847 individuals who were initiated on an SGLT2 inhibitor with an equal number who were initiated on a GLP-1 agonist, based on propensity scores, adjusting for more than 120 baseline characteristics.
Safety outcomes were based on previously identified potential safety signals.
Patients who were initiated on an SGLT2 inhibitor had 1.30-fold, 2.13-fold, and 3.08-fold higher risks of having a nonvertebral fracture, a lower limb amputation, and a genital infection, respectively, compared with patients who were initiated on a GLP-1 agonist, after a mean on-treatment time of 7.5 months,
Risks of DKA, hypovolemia, hypoglycemia, and severe UTI were similar in both groups.
Patients initiated on an SGLT2 inhibitor versus a GLP-1 agonist had a lower risk of AKI (hazard ratio, 0.93) equivalent to 6.75 fewer cases of AKI per 1,000 patients per year.
Patients had higher risks for lower limb amputation, genital infections, and nonvertebral fractures with SGLT2 inhibitors versus GLP-1 agonists across most of the prespecified subgroups by age, sex, cardiovascular disease, heart failure, and use of metformin, insulin, or sulfonylurea, but with wider confidence intervals.
Dr. Fu was supported by a Rubicon grant from the Dutch Research Council and has reported no relevant financial relationships. Disclosures for the other authors are listed with the article.
A version of this article originally appeared on Medscape.com.
Starting therapy with an SGLT2 inhibitor versus a glucagon-like peptide-1 (GLP-1) receptor agonist was associated with more lower limb amputations, nonvertebral fractures, and genital infections, but these risks need to be balanced against cardiovascular and renoprotective benefits, according to the researchers.
The analysis showed that there would be 2.1 more lower limb amputations, 2.5 more nonvertebral fractures, and 41 more genital infections per 1,000 patients per year among those receiving SGLT2 inhibitors versus an equal number of patients receiving GLP-1 agonists, lead author Edouard Fu, PhD, explained to this news organization in an email.
“On the other hand, we know from the evidence from randomized controlled trials that taking an SGLT2 inhibitor compared with placebo lowers the risk of developing kidney failure,” said Dr. Fu, who is a research fellow in the division of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital, Boston.
“For instance,” he continued, “in the DAPA-CKD clinical trial, dapagliflozin versus placebo led to 29 fewer events per 1,000 patients per year of the composite outcome (50% decline in estimated glomerular filtration rate [eGFR], kidney failure, cardiovascular or kidney death).”
In the CREDENCE trial, canagliflozin versus placebo led to 18 fewer events per 1,000 person-years for the composite outcome of doubling of serum creatinine, kidney failure, and cardiovascular or kidney death.
And in the EMPA-KIDNEY study, empagliflozin versus placebo led to 21 fewer events per 1,000 person-years for the composite outcome of progression of kidney disease or cardiovascular death.
“Thus, benefits would still outweigh the risks,” Dr. Fu emphasized.
‘Quantifies absolute rate of events among routine care patients’
“The importance of our paper,” he summarized, “is that it quantifies the absolute rate of events among routine care patients and may be used to inform shared decision-making.”
The analysis also found that the risks of diabetic ketoacidosis (DKA), hypovolemia, hypoglycemia, and severe urinary tract infection (UTI) were similar with SGLT2 inhibitors versus GLP-1 agonists, but the risk of developing acute kidney injury (AKI) was lower with an SGLT2 inhibitor.
“Our study can help inform patient-physician decision-making regarding risks and benefits before prescribing SGLT2 inhibitors in this population” of patients with CKD and diabetes treated in clinical practice, the researchers conclude, “but needs to be interpreted in light of its limitations, including residual confounding, short follow-up time, and the use of diagnosis codes to identify patients with CKD.”
The study was recently published in the Clinical Journal of the American Society of Nephrology.
Slow uptake, safety concerns
SGLT2 inhibitors are recommended as first-line therapy in patients with type 2 diabetes and CKD who have an eGFR equal to or greater than 20 mL/min per 1.73 m2, and thus are at high risk for cardiovascular disease and kidney disease progression, Dr. Fu and colleagues write.
However, studies report that as few as 6% of patients with CKD and type 2 diabetes are currently prescribed SGLT2 inhibitors in the United States.
This slow uptake of SGLT2 inhibitors among patients with CKD may be partly due to concerns about DKA, fractures, amputations, and urogenital infections observed in clinical trials.
However, such trials are generally underpowered to assess rare adverse events, use monitoring protocols to lower the risk of adverse events, and include a highly selected patient population, and so safety in routine clinical practice is often unclear.
To examine this, the researchers identified health insurance claims data from 96,128 individuals (from Optum, IBM MarketScan, and Medicare databases) who were 18 years or older (65 years or older for Medicare) and had type 2 diabetes and at least one inpatient or two outpatient diagnostic codes for stage 3 or 4 CKD.
Of these patients, 32,192 had a newly filled prescription for an SGLT2 inhibitor (empagliflozin, dapagliflozin, canagliflozin, or ertugliflozin) and 63,936 had a newly filled prescription for a GLP-1 agonist (liraglutide, dulaglutide, semaglutide, exenatide, albiglutide, or lixisenatide) between April 2013, when the first SGLT2 inhibitor was available in the United States, and 2021.
The researchers matched 28,847 individuals who were initiated on an SGLT2 inhibitor with an equal number who were initiated on a GLP-1 agonist, based on propensity scores, adjusting for more than 120 baseline characteristics.
Safety outcomes were based on previously identified potential safety signals.
Patients who were initiated on an SGLT2 inhibitor had 1.30-fold, 2.13-fold, and 3.08-fold higher risks of having a nonvertebral fracture, a lower limb amputation, and a genital infection, respectively, compared with patients who were initiated on a GLP-1 agonist, after a mean on-treatment time of 7.5 months,
Risks of DKA, hypovolemia, hypoglycemia, and severe UTI were similar in both groups.
Patients initiated on an SGLT2 inhibitor versus a GLP-1 agonist had a lower risk of AKI (hazard ratio, 0.93) equivalent to 6.75 fewer cases of AKI per 1,000 patients per year.
Patients had higher risks for lower limb amputation, genital infections, and nonvertebral fractures with SGLT2 inhibitors versus GLP-1 agonists across most of the prespecified subgroups by age, sex, cardiovascular disease, heart failure, and use of metformin, insulin, or sulfonylurea, but with wider confidence intervals.
Dr. Fu was supported by a Rubicon grant from the Dutch Research Council and has reported no relevant financial relationships. Disclosures for the other authors are listed with the article.
A version of this article originally appeared on Medscape.com.
Nasal COVID treatment shows early promise against multiple variants
if used within 4 hours after infection inside the nose, new research reveals.
Known as TriSb92 (brand name Covidin, from drugmaker Pandemblock Oy in Finland), the viral inhibitor also appears effective against all coronavirus variants of concern, neutralizing even the Omicron variants BA.5, XBB, and BQ.1.1 in laboratory and mice studies.
Unlike a COVID vaccine that boosts a person’s immune system as protection, the antiviral nasal spray works more directly by blocking the virus, acting as a “biological mask in the nasal cavity,” according to the biotechnology company set up to develop the treatment.
The product targets a stable site on the spike protein of the virus that is not known to mutate. This same site is shared among many variants of the COVID virus, so it could be effective against future variants as well, researchers note.
“In animal models, by directly inactivating the virus, TriSb92 offers immediate and robust protection” against coronavirus infection and severe COVID, said Anna R. Mäkelä, PhD, lead author of the study and a senior scientist in the department of virology at the University of Helsinki.
The study was published online in Nature Communications.
A potential first line of defense
Even in cases where the antiviral does not prevent coronavirus infection, the treatment could slow infection. This could happen by limiting how much virus could replicate early in the skin inside the nose and nasopharynx (the upper part of the throat), said Dr. Mäkelä, who is also CEO of Pandemblock Oy, the company set up to develop the product.
“TriSb92 could effectively tip the balance in favor of the [the person] and thereby help to reduce the risk of severe COVID-19 disease,” she said.
The antiviral also could offer an alternative to people who cannot or do not respond to a vaccine.
“Many elderly people as well as individuals who are immunodeficient for various reasons do not respond to vaccines and are in the need of other protective measures,” said Kalle Saksela, MD, PhD, senior author of the study and a virologist at the University of Helsinki.
Multiple doses needed?
TriSb92 is “one of multiple nasal spray approaches but unlikely to be as durable as effective nasal vaccines,” said Eric Topol, MD, a professor of molecular medicine and executive vice president of Scripps Research in La Jolla, Calif. Dr. Topol is also editor-in-chief of Medscape, WebMD’s sister site for medical professionals.
“The sprays generally require multiple doses per day, whereas a single dose of a nasal vaccine may protect for months,” he said.
“Both have the allure of being variant-proof,” Dr. Topol added.
Thinking small
Many laboratories are shifting from treatments using monoclonal antibodies to treatments using smaller antibody fragments called “nanobodies” because they are more cost-effective and are able to last longer in storage, Dr. Mäkelä and colleagues noted.
Several of these nanobodies have shown promise against viruses in cell culture or animal models, including as an intranasal preventive treatment for SARS-CoV-2.
One of these smaller antibodies is being developed from llamas for example; another comes from experiments with yeast to develop synthetic nanobodies; and in a third case, researchers isolated nanobodies from llamas and from mice and showed they could neutralize the SARS-CoV-2 virus.
These nanobodies and TriSb92 target a specific part of the coronavirus spike protein called the receptor-binding domain (RBD). The RBD is where the coronavirus attaches to cells in the body. These agents essentially trick the virus by changing the structure of the outside of cells, so they look like a virus has already fused to them. This way, the virus moves on.
Key findings
The researchers compared mice treated with TriSb92 before and after exposure to SARS-CoV-2. When given in advance, none of the treated mice had SARS-CoV-2 RNA in their lungs, while untreated mice in the comparison group had “abundant” levels.
Other evidence of viral infection showed similar differences between treated and untreated mice in the protective lining of cells called the epithelium inside the nose, nasal mucosa, and airways.
Similarly, when given 2 or 4 hours after SARS-CoV-2 had already infected the epithelium, TriSb92 was linked to a complete lack of the virus’s RNA in the lungs.
It was more effective against the virus, though, when given before infection rather than after, “perhaps due to the initial establishment of the infection,” the researchers note.
The company led by Dr. Mäkelä is now working to secure funding for clinical trials of TriSb92 in humans.
A version of this article first appeared on WebMD.com.
if used within 4 hours after infection inside the nose, new research reveals.
Known as TriSb92 (brand name Covidin, from drugmaker Pandemblock Oy in Finland), the viral inhibitor also appears effective against all coronavirus variants of concern, neutralizing even the Omicron variants BA.5, XBB, and BQ.1.1 in laboratory and mice studies.
Unlike a COVID vaccine that boosts a person’s immune system as protection, the antiviral nasal spray works more directly by blocking the virus, acting as a “biological mask in the nasal cavity,” according to the biotechnology company set up to develop the treatment.
The product targets a stable site on the spike protein of the virus that is not known to mutate. This same site is shared among many variants of the COVID virus, so it could be effective against future variants as well, researchers note.
“In animal models, by directly inactivating the virus, TriSb92 offers immediate and robust protection” against coronavirus infection and severe COVID, said Anna R. Mäkelä, PhD, lead author of the study and a senior scientist in the department of virology at the University of Helsinki.
The study was published online in Nature Communications.
A potential first line of defense
Even in cases where the antiviral does not prevent coronavirus infection, the treatment could slow infection. This could happen by limiting how much virus could replicate early in the skin inside the nose and nasopharynx (the upper part of the throat), said Dr. Mäkelä, who is also CEO of Pandemblock Oy, the company set up to develop the product.
“TriSb92 could effectively tip the balance in favor of the [the person] and thereby help to reduce the risk of severe COVID-19 disease,” she said.
The antiviral also could offer an alternative to people who cannot or do not respond to a vaccine.
“Many elderly people as well as individuals who are immunodeficient for various reasons do not respond to vaccines and are in the need of other protective measures,” said Kalle Saksela, MD, PhD, senior author of the study and a virologist at the University of Helsinki.
Multiple doses needed?
TriSb92 is “one of multiple nasal spray approaches but unlikely to be as durable as effective nasal vaccines,” said Eric Topol, MD, a professor of molecular medicine and executive vice president of Scripps Research in La Jolla, Calif. Dr. Topol is also editor-in-chief of Medscape, WebMD’s sister site for medical professionals.
“The sprays generally require multiple doses per day, whereas a single dose of a nasal vaccine may protect for months,” he said.
“Both have the allure of being variant-proof,” Dr. Topol added.
Thinking small
Many laboratories are shifting from treatments using monoclonal antibodies to treatments using smaller antibody fragments called “nanobodies” because they are more cost-effective and are able to last longer in storage, Dr. Mäkelä and colleagues noted.
Several of these nanobodies have shown promise against viruses in cell culture or animal models, including as an intranasal preventive treatment for SARS-CoV-2.
One of these smaller antibodies is being developed from llamas for example; another comes from experiments with yeast to develop synthetic nanobodies; and in a third case, researchers isolated nanobodies from llamas and from mice and showed they could neutralize the SARS-CoV-2 virus.
These nanobodies and TriSb92 target a specific part of the coronavirus spike protein called the receptor-binding domain (RBD). The RBD is where the coronavirus attaches to cells in the body. These agents essentially trick the virus by changing the structure of the outside of cells, so they look like a virus has already fused to them. This way, the virus moves on.
Key findings
The researchers compared mice treated with TriSb92 before and after exposure to SARS-CoV-2. When given in advance, none of the treated mice had SARS-CoV-2 RNA in their lungs, while untreated mice in the comparison group had “abundant” levels.
Other evidence of viral infection showed similar differences between treated and untreated mice in the protective lining of cells called the epithelium inside the nose, nasal mucosa, and airways.
Similarly, when given 2 or 4 hours after SARS-CoV-2 had already infected the epithelium, TriSb92 was linked to a complete lack of the virus’s RNA in the lungs.
It was more effective against the virus, though, when given before infection rather than after, “perhaps due to the initial establishment of the infection,” the researchers note.
The company led by Dr. Mäkelä is now working to secure funding for clinical trials of TriSb92 in humans.
A version of this article first appeared on WebMD.com.
if used within 4 hours after infection inside the nose, new research reveals.
Known as TriSb92 (brand name Covidin, from drugmaker Pandemblock Oy in Finland), the viral inhibitor also appears effective against all coronavirus variants of concern, neutralizing even the Omicron variants BA.5, XBB, and BQ.1.1 in laboratory and mice studies.
Unlike a COVID vaccine that boosts a person’s immune system as protection, the antiviral nasal spray works more directly by blocking the virus, acting as a “biological mask in the nasal cavity,” according to the biotechnology company set up to develop the treatment.
The product targets a stable site on the spike protein of the virus that is not known to mutate. This same site is shared among many variants of the COVID virus, so it could be effective against future variants as well, researchers note.
“In animal models, by directly inactivating the virus, TriSb92 offers immediate and robust protection” against coronavirus infection and severe COVID, said Anna R. Mäkelä, PhD, lead author of the study and a senior scientist in the department of virology at the University of Helsinki.
The study was published online in Nature Communications.
A potential first line of defense
Even in cases where the antiviral does not prevent coronavirus infection, the treatment could slow infection. This could happen by limiting how much virus could replicate early in the skin inside the nose and nasopharynx (the upper part of the throat), said Dr. Mäkelä, who is also CEO of Pandemblock Oy, the company set up to develop the product.
“TriSb92 could effectively tip the balance in favor of the [the person] and thereby help to reduce the risk of severe COVID-19 disease,” she said.
The antiviral also could offer an alternative to people who cannot or do not respond to a vaccine.
“Many elderly people as well as individuals who are immunodeficient for various reasons do not respond to vaccines and are in the need of other protective measures,” said Kalle Saksela, MD, PhD, senior author of the study and a virologist at the University of Helsinki.
Multiple doses needed?
TriSb92 is “one of multiple nasal spray approaches but unlikely to be as durable as effective nasal vaccines,” said Eric Topol, MD, a professor of molecular medicine and executive vice president of Scripps Research in La Jolla, Calif. Dr. Topol is also editor-in-chief of Medscape, WebMD’s sister site for medical professionals.
“The sprays generally require multiple doses per day, whereas a single dose of a nasal vaccine may protect for months,” he said.
“Both have the allure of being variant-proof,” Dr. Topol added.
Thinking small
Many laboratories are shifting from treatments using monoclonal antibodies to treatments using smaller antibody fragments called “nanobodies” because they are more cost-effective and are able to last longer in storage, Dr. Mäkelä and colleagues noted.
Several of these nanobodies have shown promise against viruses in cell culture or animal models, including as an intranasal preventive treatment for SARS-CoV-2.
One of these smaller antibodies is being developed from llamas for example; another comes from experiments with yeast to develop synthetic nanobodies; and in a third case, researchers isolated nanobodies from llamas and from mice and showed they could neutralize the SARS-CoV-2 virus.
These nanobodies and TriSb92 target a specific part of the coronavirus spike protein called the receptor-binding domain (RBD). The RBD is where the coronavirus attaches to cells in the body. These agents essentially trick the virus by changing the structure of the outside of cells, so they look like a virus has already fused to them. This way, the virus moves on.
Key findings
The researchers compared mice treated with TriSb92 before and after exposure to SARS-CoV-2. When given in advance, none of the treated mice had SARS-CoV-2 RNA in their lungs, while untreated mice in the comparison group had “abundant” levels.
Other evidence of viral infection showed similar differences between treated and untreated mice in the protective lining of cells called the epithelium inside the nose, nasal mucosa, and airways.
Similarly, when given 2 or 4 hours after SARS-CoV-2 had already infected the epithelium, TriSb92 was linked to a complete lack of the virus’s RNA in the lungs.
It was more effective against the virus, though, when given before infection rather than after, “perhaps due to the initial establishment of the infection,” the researchers note.
The company led by Dr. Mäkelä is now working to secure funding for clinical trials of TriSb92 in humans.
A version of this article first appeared on WebMD.com.
FROM NATURE COMMUNICATIONS
High-dose prophylactic anticoagulation benefits patients with COVID-19 pneumonia
High-dose prophylactic anticoagulation or therapeutic anticoagulation reduced de novo thrombosis in patients with hypoxemic COVID-19 pneumonia, based on data from 334 adults.
Patients with hypoxemic COVID-19 pneumonia are at increased risk of thrombosis and anticoagulation-related bleeding, therefore data to identify the lowest effective anticoagulant dose are needed, wrote Vincent Labbé, MD, of Sorbonne University, Paris, and colleagues.
Previous studies of different anticoagulation strategies for noncritically ill and critically ill patients with COVID-19 pneumonia have shown contrasting results, but some institutions recommend a high-dose regimen in the wake of data showing macrovascular thrombosis in patients with COVID-19 who were treated with standard anticoagulation, the authors wrote.
However, no previously published studies have compared the effectiveness of the three anticoagulation strategies: high-dose prophylactic anticoagulation (HD-PA), standard dose prophylactic anticoagulation (SD-PA), and therapeutic anticoagulation (TA), they said.
In the open-label Anticoagulation COVID-19 (ANTICOVID) trial, published in JAMA Internal Medicine, the researchers identified consecutively hospitalized adults aged 18 years and older being treated for hypoxemic COVID-19 pneumonia in 23 centers in France between April 2021 and December 2021.
The patients were randomly assigned to SD-PA (116 patients), HD-PA (111 patients), and TA (112 patients) using low-molecular-weight heparin for 14 days, or until either hospital discharge or weaning from supplemental oxygen for 48 consecutive hours, whichever outcome occurred first. The HD-PA patients received two times the SD-PA dose. The mean age of the patients was 58.3 years, and approximately two-thirds were men; race and ethnicity data were not collected. Participants had no macrovascular thrombosis at the start of the study.
The primary outcomes were all-cause mortality and time to clinical improvement (defined as the time from randomization to a 2-point improvement on a 7-category respiratory function scale).
The secondary outcome was a combination of safety and efficacy at day 28 that included a composite of thrombosis (ischemic stroke, noncerebrovascular arterial thrombosis, deep venous thrombosis, pulmonary artery thrombosis, and central venous catheter–related deep venous thrombosis), major bleeding, or all-cause death.
For the primary outcome, results were similar among the groups; HD-PA had no significant benefit over SD-PA or TA. All-cause death rates for SD-PA, HD-PA, and TA patients were 14%, 12%, and 13%, respectively. The time to clinical improvement for the three groups was approximately 8 days, 9 days, and 8 days, respectively. Results for the primary outcome were consistent across all prespecified subgroups.
However, HD-PA was associated with a significant fourfold reduced risk of de novo thrombosis compared with SD-PA (5.5% vs. 20.2%) with no observed increase in major bleeding. TA was not associated with any significant improvement in primary or secondary outcomes compared with HD-PA or SD-PA.
The current study findings of no improvement in survival or disease resolution in patients with a higher anticoagulant dose reflects data from previous studies, the researchers wrote in their discussion. “Our study results together with those of previous RCTs support the premise that the role of microvascular thrombosis in worsening organ dysfunction may be narrower than estimated,” they said.
The findings were limited by several factors including the open-label design and the relatively small sample size, the lack of data on microvascular (vs. macrovascular) thrombosis at baseline, and the predominance of the Delta variant of COVID-19 among the study participants, which may have contributed to a lower mortality rate, the researchers noted.
However, given the significant reduction in de novo thrombosis, the results support the routine use of HD-PA in patients with severe hypoxemic COVID-19 pneumonia, they concluded.
Results inform current clinical practice
Over the course of the COVID-19 pandemic, “Patients hospitalized with COVID-19 manifested the highest risk for thromboembolic complications, especially patients in the intensive care setting,” and early reports suggested that standard prophylactic doses of anticoagulant therapy might be insufficient to prevent thrombotic events, Richard C. Becker, MD, of the University of Cincinnati, and Thomas L. Ortel, MD, of Duke University, Durham, N.C., wrote in an accompanying editorial.
“Although there have been several studies that have investigated the role of anticoagulant therapy in hospitalized patients with COVID-19, this is the first study that specifically compared a standard, prophylactic dose of low-molecular-weight heparin to a ‘high-dose’ prophylactic regimen and to a full therapeutic dose regimen,” Dr. Ortel said in an interview.
“Given the concerns about an increased thrombotic risk with prophylactic dose anticoagulation, and the potential bleeding risk associated with a full therapeutic dose of anticoagulation, this approach enabled the investigators to explore the efficacy and safety of an intermediate dose between these two extremes,” he said.
In the current study, , a finding that was not observed in other studies investigating anticoagulant therapy in hospitalized patients with severe COVID-19,” Dr. Ortel told this news organization. “Much initial concern about progression of disease in patients hospitalized with severe COVID-19 focused on the role of microvascular thrombosis, which appears to be less important in this process, or, alternatively, less responsive to anticoagulant therapy.”
The clinical takeaway from the study, Dr. Ortel said, is the decreased risk for venous thromboembolism with a high-dose prophylactic anticoagulation strategy compared with a standard-dose prophylactic regimen for patients hospitalized with hypoxemic COVID-19 pneumonia, “leading to an improved net clinical outcome.”
Looking ahead, “Additional research is needed to determine whether a higher dose of prophylactic anticoagulation would be beneficial for patients hospitalized with COVID-19 pneumonia who are not in an intensive care unit setting,” Dr. Ortel said. Studies are needed to determine whether therapeutic interventions are equally beneficial in patients with different coronavirus variants, since most patients in the current study were infected with the Delta variant, he added.
The study was supported by LEO Pharma. Dr. Labbé disclosed grants from LEO Pharma during the study and fees from AOP Health unrelated to the current study.
Dr. Becker disclosed personal fees from Novartis Data Safety Monitoring Board, Ionis Data Safety Monitoring Board, and Basking Biosciences Scientific Advisory Board unrelated to the current study. Dr. Ortel disclosed grants from the National Institutes of Health, Instrumentation Laboratory, Stago, and Siemens; contract fees from the Centers for Disease Control and Prevention; and honoraria from UpToDate unrelated to the current study.
A version of this article originally appeared on Medscape.com.
High-dose prophylactic anticoagulation or therapeutic anticoagulation reduced de novo thrombosis in patients with hypoxemic COVID-19 pneumonia, based on data from 334 adults.
Patients with hypoxemic COVID-19 pneumonia are at increased risk of thrombosis and anticoagulation-related bleeding, therefore data to identify the lowest effective anticoagulant dose are needed, wrote Vincent Labbé, MD, of Sorbonne University, Paris, and colleagues.
Previous studies of different anticoagulation strategies for noncritically ill and critically ill patients with COVID-19 pneumonia have shown contrasting results, but some institutions recommend a high-dose regimen in the wake of data showing macrovascular thrombosis in patients with COVID-19 who were treated with standard anticoagulation, the authors wrote.
However, no previously published studies have compared the effectiveness of the three anticoagulation strategies: high-dose prophylactic anticoagulation (HD-PA), standard dose prophylactic anticoagulation (SD-PA), and therapeutic anticoagulation (TA), they said.
In the open-label Anticoagulation COVID-19 (ANTICOVID) trial, published in JAMA Internal Medicine, the researchers identified consecutively hospitalized adults aged 18 years and older being treated for hypoxemic COVID-19 pneumonia in 23 centers in France between April 2021 and December 2021.
The patients were randomly assigned to SD-PA (116 patients), HD-PA (111 patients), and TA (112 patients) using low-molecular-weight heparin for 14 days, or until either hospital discharge or weaning from supplemental oxygen for 48 consecutive hours, whichever outcome occurred first. The HD-PA patients received two times the SD-PA dose. The mean age of the patients was 58.3 years, and approximately two-thirds were men; race and ethnicity data were not collected. Participants had no macrovascular thrombosis at the start of the study.
The primary outcomes were all-cause mortality and time to clinical improvement (defined as the time from randomization to a 2-point improvement on a 7-category respiratory function scale).
The secondary outcome was a combination of safety and efficacy at day 28 that included a composite of thrombosis (ischemic stroke, noncerebrovascular arterial thrombosis, deep venous thrombosis, pulmonary artery thrombosis, and central venous catheter–related deep venous thrombosis), major bleeding, or all-cause death.
For the primary outcome, results were similar among the groups; HD-PA had no significant benefit over SD-PA or TA. All-cause death rates for SD-PA, HD-PA, and TA patients were 14%, 12%, and 13%, respectively. The time to clinical improvement for the three groups was approximately 8 days, 9 days, and 8 days, respectively. Results for the primary outcome were consistent across all prespecified subgroups.
However, HD-PA was associated with a significant fourfold reduced risk of de novo thrombosis compared with SD-PA (5.5% vs. 20.2%) with no observed increase in major bleeding. TA was not associated with any significant improvement in primary or secondary outcomes compared with HD-PA or SD-PA.
The current study findings of no improvement in survival or disease resolution in patients with a higher anticoagulant dose reflects data from previous studies, the researchers wrote in their discussion. “Our study results together with those of previous RCTs support the premise that the role of microvascular thrombosis in worsening organ dysfunction may be narrower than estimated,” they said.
The findings were limited by several factors including the open-label design and the relatively small sample size, the lack of data on microvascular (vs. macrovascular) thrombosis at baseline, and the predominance of the Delta variant of COVID-19 among the study participants, which may have contributed to a lower mortality rate, the researchers noted.
However, given the significant reduction in de novo thrombosis, the results support the routine use of HD-PA in patients with severe hypoxemic COVID-19 pneumonia, they concluded.
Results inform current clinical practice
Over the course of the COVID-19 pandemic, “Patients hospitalized with COVID-19 manifested the highest risk for thromboembolic complications, especially patients in the intensive care setting,” and early reports suggested that standard prophylactic doses of anticoagulant therapy might be insufficient to prevent thrombotic events, Richard C. Becker, MD, of the University of Cincinnati, and Thomas L. Ortel, MD, of Duke University, Durham, N.C., wrote in an accompanying editorial.
“Although there have been several studies that have investigated the role of anticoagulant therapy in hospitalized patients with COVID-19, this is the first study that specifically compared a standard, prophylactic dose of low-molecular-weight heparin to a ‘high-dose’ prophylactic regimen and to a full therapeutic dose regimen,” Dr. Ortel said in an interview.
“Given the concerns about an increased thrombotic risk with prophylactic dose anticoagulation, and the potential bleeding risk associated with a full therapeutic dose of anticoagulation, this approach enabled the investigators to explore the efficacy and safety of an intermediate dose between these two extremes,” he said.
In the current study, , a finding that was not observed in other studies investigating anticoagulant therapy in hospitalized patients with severe COVID-19,” Dr. Ortel told this news organization. “Much initial concern about progression of disease in patients hospitalized with severe COVID-19 focused on the role of microvascular thrombosis, which appears to be less important in this process, or, alternatively, less responsive to anticoagulant therapy.”
The clinical takeaway from the study, Dr. Ortel said, is the decreased risk for venous thromboembolism with a high-dose prophylactic anticoagulation strategy compared with a standard-dose prophylactic regimen for patients hospitalized with hypoxemic COVID-19 pneumonia, “leading to an improved net clinical outcome.”
Looking ahead, “Additional research is needed to determine whether a higher dose of prophylactic anticoagulation would be beneficial for patients hospitalized with COVID-19 pneumonia who are not in an intensive care unit setting,” Dr. Ortel said. Studies are needed to determine whether therapeutic interventions are equally beneficial in patients with different coronavirus variants, since most patients in the current study were infected with the Delta variant, he added.
The study was supported by LEO Pharma. Dr. Labbé disclosed grants from LEO Pharma during the study and fees from AOP Health unrelated to the current study.
Dr. Becker disclosed personal fees from Novartis Data Safety Monitoring Board, Ionis Data Safety Monitoring Board, and Basking Biosciences Scientific Advisory Board unrelated to the current study. Dr. Ortel disclosed grants from the National Institutes of Health, Instrumentation Laboratory, Stago, and Siemens; contract fees from the Centers for Disease Control and Prevention; and honoraria from UpToDate unrelated to the current study.
A version of this article originally appeared on Medscape.com.
High-dose prophylactic anticoagulation or therapeutic anticoagulation reduced de novo thrombosis in patients with hypoxemic COVID-19 pneumonia, based on data from 334 adults.
Patients with hypoxemic COVID-19 pneumonia are at increased risk of thrombosis and anticoagulation-related bleeding, therefore data to identify the lowest effective anticoagulant dose are needed, wrote Vincent Labbé, MD, of Sorbonne University, Paris, and colleagues.
Previous studies of different anticoagulation strategies for noncritically ill and critically ill patients with COVID-19 pneumonia have shown contrasting results, but some institutions recommend a high-dose regimen in the wake of data showing macrovascular thrombosis in patients with COVID-19 who were treated with standard anticoagulation, the authors wrote.
However, no previously published studies have compared the effectiveness of the three anticoagulation strategies: high-dose prophylactic anticoagulation (HD-PA), standard dose prophylactic anticoagulation (SD-PA), and therapeutic anticoagulation (TA), they said.
In the open-label Anticoagulation COVID-19 (ANTICOVID) trial, published in JAMA Internal Medicine, the researchers identified consecutively hospitalized adults aged 18 years and older being treated for hypoxemic COVID-19 pneumonia in 23 centers in France between April 2021 and December 2021.
The patients were randomly assigned to SD-PA (116 patients), HD-PA (111 patients), and TA (112 patients) using low-molecular-weight heparin for 14 days, or until either hospital discharge or weaning from supplemental oxygen for 48 consecutive hours, whichever outcome occurred first. The HD-PA patients received two times the SD-PA dose. The mean age of the patients was 58.3 years, and approximately two-thirds were men; race and ethnicity data were not collected. Participants had no macrovascular thrombosis at the start of the study.
The primary outcomes were all-cause mortality and time to clinical improvement (defined as the time from randomization to a 2-point improvement on a 7-category respiratory function scale).
The secondary outcome was a combination of safety and efficacy at day 28 that included a composite of thrombosis (ischemic stroke, noncerebrovascular arterial thrombosis, deep venous thrombosis, pulmonary artery thrombosis, and central venous catheter–related deep venous thrombosis), major bleeding, or all-cause death.
For the primary outcome, results were similar among the groups; HD-PA had no significant benefit over SD-PA or TA. All-cause death rates for SD-PA, HD-PA, and TA patients were 14%, 12%, and 13%, respectively. The time to clinical improvement for the three groups was approximately 8 days, 9 days, and 8 days, respectively. Results for the primary outcome were consistent across all prespecified subgroups.
However, HD-PA was associated with a significant fourfold reduced risk of de novo thrombosis compared with SD-PA (5.5% vs. 20.2%) with no observed increase in major bleeding. TA was not associated with any significant improvement in primary or secondary outcomes compared with HD-PA or SD-PA.
The current study findings of no improvement in survival or disease resolution in patients with a higher anticoagulant dose reflects data from previous studies, the researchers wrote in their discussion. “Our study results together with those of previous RCTs support the premise that the role of microvascular thrombosis in worsening organ dysfunction may be narrower than estimated,” they said.
The findings were limited by several factors including the open-label design and the relatively small sample size, the lack of data on microvascular (vs. macrovascular) thrombosis at baseline, and the predominance of the Delta variant of COVID-19 among the study participants, which may have contributed to a lower mortality rate, the researchers noted.
However, given the significant reduction in de novo thrombosis, the results support the routine use of HD-PA in patients with severe hypoxemic COVID-19 pneumonia, they concluded.
Results inform current clinical practice
Over the course of the COVID-19 pandemic, “Patients hospitalized with COVID-19 manifested the highest risk for thromboembolic complications, especially patients in the intensive care setting,” and early reports suggested that standard prophylactic doses of anticoagulant therapy might be insufficient to prevent thrombotic events, Richard C. Becker, MD, of the University of Cincinnati, and Thomas L. Ortel, MD, of Duke University, Durham, N.C., wrote in an accompanying editorial.
“Although there have been several studies that have investigated the role of anticoagulant therapy in hospitalized patients with COVID-19, this is the first study that specifically compared a standard, prophylactic dose of low-molecular-weight heparin to a ‘high-dose’ prophylactic regimen and to a full therapeutic dose regimen,” Dr. Ortel said in an interview.
“Given the concerns about an increased thrombotic risk with prophylactic dose anticoagulation, and the potential bleeding risk associated with a full therapeutic dose of anticoagulation, this approach enabled the investigators to explore the efficacy and safety of an intermediate dose between these two extremes,” he said.
In the current study, , a finding that was not observed in other studies investigating anticoagulant therapy in hospitalized patients with severe COVID-19,” Dr. Ortel told this news organization. “Much initial concern about progression of disease in patients hospitalized with severe COVID-19 focused on the role of microvascular thrombosis, which appears to be less important in this process, or, alternatively, less responsive to anticoagulant therapy.”
The clinical takeaway from the study, Dr. Ortel said, is the decreased risk for venous thromboembolism with a high-dose prophylactic anticoagulation strategy compared with a standard-dose prophylactic regimen for patients hospitalized with hypoxemic COVID-19 pneumonia, “leading to an improved net clinical outcome.”
Looking ahead, “Additional research is needed to determine whether a higher dose of prophylactic anticoagulation would be beneficial for patients hospitalized with COVID-19 pneumonia who are not in an intensive care unit setting,” Dr. Ortel said. Studies are needed to determine whether therapeutic interventions are equally beneficial in patients with different coronavirus variants, since most patients in the current study were infected with the Delta variant, he added.
The study was supported by LEO Pharma. Dr. Labbé disclosed grants from LEO Pharma during the study and fees from AOP Health unrelated to the current study.
Dr. Becker disclosed personal fees from Novartis Data Safety Monitoring Board, Ionis Data Safety Monitoring Board, and Basking Biosciences Scientific Advisory Board unrelated to the current study. Dr. Ortel disclosed grants from the National Institutes of Health, Instrumentation Laboratory, Stago, and Siemens; contract fees from the Centers for Disease Control and Prevention; and honoraria from UpToDate unrelated to the current study.
A version of this article originally appeared on Medscape.com.
Brain stimulation can improve prognosis following a stroke and other neurological diseases
HAMBURG, GERMANY – Around 86 billion nerve cells in our brain work together in complex dynamic networks to control almost every sensorimotor and cognitive process. However, the way in which the information is processed in the different regions of the brain is still unclear. There are already some promising approaches to specifically influence the dynamics of neuronal networks to treat neurological and psychiatric diseases.
One of the main topics at the Congress for Clinical Neuroscience of the German Society for Clinical Neurophysiology and Functional Neuroimaging (DGKN), recently held in Hamburg, Germany, was the dynamics of cerebral networks in sensorimotor and cognitive processes, as well as disruptions to network dynamics in neurological and psychiatric diseases.
“We will be unable to develop innovative therapies for widespread neurological and psychiatric diseases until we understand neuronal functions on every level of complexity,” Andreas K. Engel, PhD, director of the Institute for Neurophysiology and Pathophysiology at the University Hospital of Hamburg-Eppendorf, president of the DGKN, and congress president, said during an online press conference.
Characterizing states of consciousness
For more than 30 years, it has been known that neuronal signals in the brain are dynamically coupled. Despite intensive research, the functional significance of this coupling on information processing is still largely unknown.
Neuroimaging methods such as electroencephalography (EEG), magnetoencephalography (MEG), structural and functional magnetic resonance imaging (MRI), and electrophysiological examinations were used. Model calculations of the data suggest that dynamic couplings of signals in the cortex play a crucial role in memory performance, thinking processes, and developing perception, among other things.
It has already been shown that the network dynamics of neuronal signals could possibly characterize states of consciousness. Neuronal signals and coupling patterns differ significantly between healthy individuals in a waking state and those who are asleep, under general anesthetic, or in a vegetative state. In Dr. Engel’s view, it may be possible in the future for machine learning algorithms to be used to classify states of consciousness.
Changes in brain activity as a biomarker?
The differences in the dynamics of neuronal signals between healthy individuals and patients with psychiatric diseases such as schizophrenia appear much more important for clinical practice. “The characteristic changes in brain activity in the primary auditory cortex could be considered a potential biomarker and used to predict the clinical course of psychiatric diseases, such as psychoses,” reported Dr. Engel.
The gamma-band activity in the auditory cortex could be a potential marker for schizophrenia. According to MEG examinations, the values are decreased both in people at increased risk of psychosis and experiencing first symptoms compared with controls.
Activation or inhibition of cerebral networks as new therapeutic approaches
New therapeutic approaches based on the activation or inhibition of cerebral networks are currently areas of intensive research. Close interdisciplinary collaboration between basic science researchers and clinicians is necessary, stressed Dr. Engel. The use of noninvasive brain stimulation is already within reach for the neurorehabilitation of stroke patients. “I am optimistic that in a few years brain stimulation will be established as an integral element of stroke therapy,” said Christian Grefkes-Hermann, MD, PhD, director of the department of neurology at University Hospital of Frankfurt and first vice president of the DGKN.
Despite great advances in acute stroke therapy, many patients must endure permanent deficits in their everyday life, he said. According to Dr. Grefkes-Hermann, rehabilitation procedures often have a dissatisfactory effect, and results greatly vary. He hopes that in the future it may be possible to personalize therapy by using network patterns, thereby improving results.
“The most important factor for functional recovery after a stroke is neuronal reorganization,” said Dr. Grefkes-Hermann. With the new methods of neurorehabilitation, network-connectivity disruptions, which are associated with motor function deficits, are first visualized using functional MRI (fMRI).
The imaging or the EEG makes visible the area of the brain that may benefit most from neurostimulation. Subsequently, nerve cells in this region may be precisely stimulated with TMS. Because the healthy hemisphere of the brain is usually overactive after a stroke, there are simultaneous attempts to inhibit the contralesional motor cortex.
Initial results are hopeful. In the initial period after a stroke, TMS can be used in some patients to correct pathological connectivities and thereby improve motor deficits, reported Dr. Grefkes-Hermann. The fMRI pattern can also be used to predict recovery and intervention effects on an individual basis. A phase 3 trial is currently underway of 150 patients who have had a stroke and aims to study the efficacy of the new procedure.
Combined TMS and EEG
With the combination of TMS and the simultaneous measurement of EEG activity, a further development of fMRI connectivity analyses is currently being tested. Dr. Grefkes-Hermann believes that this procedure, which is more cost-effective, has higher temporal resolution, can be used directly at the bedside, and has more potential for personalized therapy planning in clinical practice.
The TMS-EEG procedure also makes it possible to predict the risk of post-stroke delirium, which affects around 30% of stroke patients and greatly worsens the outcome, underlined Ulf Ziemann, MD, medical director of the department of neurology at Tübingen (Germany) University Hospital. In a study of 33 patients with acute stroke, the onset of post-stroke delirium could be predicted with a high degree of accuracy by using the TMS-EEG procedure no later than 48 hours after the event.
Other promising, noninvasive methods for neuron activation mentioned by Dr. Ziemann include transcranial focused ultrasound stimulation (tFUS) with low intensity, which is being studied for chronic pain, dementia, epilepsy, traumatic brain injury, and depression, as well as transcranial pulse stimulation (TPS), which is also based on ultrasound. In a pilot study of 35 patients with Alzheimer’s disease, use of TPS within 3 months had positive effects on cognition. However, the study was not controlled and therefore further assessments are needed.
Custom deep brain stimulation
For deep brain stimulation (DBS), an established therapy for Parkinson’s disease and other movement disorders, the aim is individualized, symptom-related network stimulation, reported Andrea Kühn, MD, head of the movement disorders and neuromodulation section in the department of neurology at Charité University Hospital Berlin.
At the panregional collaborative research center ReTune, which has been supported for 4 years now by €10 million from the German Research Foundation (DFG), imaging and computer-assisted programming algorithms are being developed for DBS. They will greatly simplify the time-consuming standard procedure for the best possible setting of the stimulation parameters, which requires a hospital stay of several days.
A randomized crossover study of 35 patients with Parkinson’s disease proved the equivalence of the fast, algorithm-assisted DBS for the control of motor symptoms compared with standard procedures.
The new methods have the potential to considerably improve the outcome of patients with neurological and psychiatric diseases, according to scientists. However, the positive data must still be validated in further studies.
This article was translated from Medscape’s German edition. A version of this article appeared on Medscape.com.
HAMBURG, GERMANY – Around 86 billion nerve cells in our brain work together in complex dynamic networks to control almost every sensorimotor and cognitive process. However, the way in which the information is processed in the different regions of the brain is still unclear. There are already some promising approaches to specifically influence the dynamics of neuronal networks to treat neurological and psychiatric diseases.
One of the main topics at the Congress for Clinical Neuroscience of the German Society for Clinical Neurophysiology and Functional Neuroimaging (DGKN), recently held in Hamburg, Germany, was the dynamics of cerebral networks in sensorimotor and cognitive processes, as well as disruptions to network dynamics in neurological and psychiatric diseases.
“We will be unable to develop innovative therapies for widespread neurological and psychiatric diseases until we understand neuronal functions on every level of complexity,” Andreas K. Engel, PhD, director of the Institute for Neurophysiology and Pathophysiology at the University Hospital of Hamburg-Eppendorf, president of the DGKN, and congress president, said during an online press conference.
Characterizing states of consciousness
For more than 30 years, it has been known that neuronal signals in the brain are dynamically coupled. Despite intensive research, the functional significance of this coupling on information processing is still largely unknown.
Neuroimaging methods such as electroencephalography (EEG), magnetoencephalography (MEG), structural and functional magnetic resonance imaging (MRI), and electrophysiological examinations were used. Model calculations of the data suggest that dynamic couplings of signals in the cortex play a crucial role in memory performance, thinking processes, and developing perception, among other things.
It has already been shown that the network dynamics of neuronal signals could possibly characterize states of consciousness. Neuronal signals and coupling patterns differ significantly between healthy individuals in a waking state and those who are asleep, under general anesthetic, or in a vegetative state. In Dr. Engel’s view, it may be possible in the future for machine learning algorithms to be used to classify states of consciousness.
Changes in brain activity as a biomarker?
The differences in the dynamics of neuronal signals between healthy individuals and patients with psychiatric diseases such as schizophrenia appear much more important for clinical practice. “The characteristic changes in brain activity in the primary auditory cortex could be considered a potential biomarker and used to predict the clinical course of psychiatric diseases, such as psychoses,” reported Dr. Engel.
The gamma-band activity in the auditory cortex could be a potential marker for schizophrenia. According to MEG examinations, the values are decreased both in people at increased risk of psychosis and experiencing first symptoms compared with controls.
Activation or inhibition of cerebral networks as new therapeutic approaches
New therapeutic approaches based on the activation or inhibition of cerebral networks are currently areas of intensive research. Close interdisciplinary collaboration between basic science researchers and clinicians is necessary, stressed Dr. Engel. The use of noninvasive brain stimulation is already within reach for the neurorehabilitation of stroke patients. “I am optimistic that in a few years brain stimulation will be established as an integral element of stroke therapy,” said Christian Grefkes-Hermann, MD, PhD, director of the department of neurology at University Hospital of Frankfurt and first vice president of the DGKN.
Despite great advances in acute stroke therapy, many patients must endure permanent deficits in their everyday life, he said. According to Dr. Grefkes-Hermann, rehabilitation procedures often have a dissatisfactory effect, and results greatly vary. He hopes that in the future it may be possible to personalize therapy by using network patterns, thereby improving results.
“The most important factor for functional recovery after a stroke is neuronal reorganization,” said Dr. Grefkes-Hermann. With the new methods of neurorehabilitation, network-connectivity disruptions, which are associated with motor function deficits, are first visualized using functional MRI (fMRI).
The imaging or the EEG makes visible the area of the brain that may benefit most from neurostimulation. Subsequently, nerve cells in this region may be precisely stimulated with TMS. Because the healthy hemisphere of the brain is usually overactive after a stroke, there are simultaneous attempts to inhibit the contralesional motor cortex.
Initial results are hopeful. In the initial period after a stroke, TMS can be used in some patients to correct pathological connectivities and thereby improve motor deficits, reported Dr. Grefkes-Hermann. The fMRI pattern can also be used to predict recovery and intervention effects on an individual basis. A phase 3 trial is currently underway of 150 patients who have had a stroke and aims to study the efficacy of the new procedure.
Combined TMS and EEG
With the combination of TMS and the simultaneous measurement of EEG activity, a further development of fMRI connectivity analyses is currently being tested. Dr. Grefkes-Hermann believes that this procedure, which is more cost-effective, has higher temporal resolution, can be used directly at the bedside, and has more potential for personalized therapy planning in clinical practice.
The TMS-EEG procedure also makes it possible to predict the risk of post-stroke delirium, which affects around 30% of stroke patients and greatly worsens the outcome, underlined Ulf Ziemann, MD, medical director of the department of neurology at Tübingen (Germany) University Hospital. In a study of 33 patients with acute stroke, the onset of post-stroke delirium could be predicted with a high degree of accuracy by using the TMS-EEG procedure no later than 48 hours after the event.
Other promising, noninvasive methods for neuron activation mentioned by Dr. Ziemann include transcranial focused ultrasound stimulation (tFUS) with low intensity, which is being studied for chronic pain, dementia, epilepsy, traumatic brain injury, and depression, as well as transcranial pulse stimulation (TPS), which is also based on ultrasound. In a pilot study of 35 patients with Alzheimer’s disease, use of TPS within 3 months had positive effects on cognition. However, the study was not controlled and therefore further assessments are needed.
Custom deep brain stimulation
For deep brain stimulation (DBS), an established therapy for Parkinson’s disease and other movement disorders, the aim is individualized, symptom-related network stimulation, reported Andrea Kühn, MD, head of the movement disorders and neuromodulation section in the department of neurology at Charité University Hospital Berlin.
At the panregional collaborative research center ReTune, which has been supported for 4 years now by €10 million from the German Research Foundation (DFG), imaging and computer-assisted programming algorithms are being developed for DBS. They will greatly simplify the time-consuming standard procedure for the best possible setting of the stimulation parameters, which requires a hospital stay of several days.
A randomized crossover study of 35 patients with Parkinson’s disease proved the equivalence of the fast, algorithm-assisted DBS for the control of motor symptoms compared with standard procedures.
The new methods have the potential to considerably improve the outcome of patients with neurological and psychiatric diseases, according to scientists. However, the positive data must still be validated in further studies.
This article was translated from Medscape’s German edition. A version of this article appeared on Medscape.com.
HAMBURG, GERMANY – Around 86 billion nerve cells in our brain work together in complex dynamic networks to control almost every sensorimotor and cognitive process. However, the way in which the information is processed in the different regions of the brain is still unclear. There are already some promising approaches to specifically influence the dynamics of neuronal networks to treat neurological and psychiatric diseases.
One of the main topics at the Congress for Clinical Neuroscience of the German Society for Clinical Neurophysiology and Functional Neuroimaging (DGKN), recently held in Hamburg, Germany, was the dynamics of cerebral networks in sensorimotor and cognitive processes, as well as disruptions to network dynamics in neurological and psychiatric diseases.
“We will be unable to develop innovative therapies for widespread neurological and psychiatric diseases until we understand neuronal functions on every level of complexity,” Andreas K. Engel, PhD, director of the Institute for Neurophysiology and Pathophysiology at the University Hospital of Hamburg-Eppendorf, president of the DGKN, and congress president, said during an online press conference.
Characterizing states of consciousness
For more than 30 years, it has been known that neuronal signals in the brain are dynamically coupled. Despite intensive research, the functional significance of this coupling on information processing is still largely unknown.
Neuroimaging methods such as electroencephalography (EEG), magnetoencephalography (MEG), structural and functional magnetic resonance imaging (MRI), and electrophysiological examinations were used. Model calculations of the data suggest that dynamic couplings of signals in the cortex play a crucial role in memory performance, thinking processes, and developing perception, among other things.
It has already been shown that the network dynamics of neuronal signals could possibly characterize states of consciousness. Neuronal signals and coupling patterns differ significantly between healthy individuals in a waking state and those who are asleep, under general anesthetic, or in a vegetative state. In Dr. Engel’s view, it may be possible in the future for machine learning algorithms to be used to classify states of consciousness.
Changes in brain activity as a biomarker?
The differences in the dynamics of neuronal signals between healthy individuals and patients with psychiatric diseases such as schizophrenia appear much more important for clinical practice. “The characteristic changes in brain activity in the primary auditory cortex could be considered a potential biomarker and used to predict the clinical course of psychiatric diseases, such as psychoses,” reported Dr. Engel.
The gamma-band activity in the auditory cortex could be a potential marker for schizophrenia. According to MEG examinations, the values are decreased both in people at increased risk of psychosis and experiencing first symptoms compared with controls.
Activation or inhibition of cerebral networks as new therapeutic approaches
New therapeutic approaches based on the activation or inhibition of cerebral networks are currently areas of intensive research. Close interdisciplinary collaboration between basic science researchers and clinicians is necessary, stressed Dr. Engel. The use of noninvasive brain stimulation is already within reach for the neurorehabilitation of stroke patients. “I am optimistic that in a few years brain stimulation will be established as an integral element of stroke therapy,” said Christian Grefkes-Hermann, MD, PhD, director of the department of neurology at University Hospital of Frankfurt and first vice president of the DGKN.
Despite great advances in acute stroke therapy, many patients must endure permanent deficits in their everyday life, he said. According to Dr. Grefkes-Hermann, rehabilitation procedures often have a dissatisfactory effect, and results greatly vary. He hopes that in the future it may be possible to personalize therapy by using network patterns, thereby improving results.
“The most important factor for functional recovery after a stroke is neuronal reorganization,” said Dr. Grefkes-Hermann. With the new methods of neurorehabilitation, network-connectivity disruptions, which are associated with motor function deficits, are first visualized using functional MRI (fMRI).
The imaging or the EEG makes visible the area of the brain that may benefit most from neurostimulation. Subsequently, nerve cells in this region may be precisely stimulated with TMS. Because the healthy hemisphere of the brain is usually overactive after a stroke, there are simultaneous attempts to inhibit the contralesional motor cortex.
Initial results are hopeful. In the initial period after a stroke, TMS can be used in some patients to correct pathological connectivities and thereby improve motor deficits, reported Dr. Grefkes-Hermann. The fMRI pattern can also be used to predict recovery and intervention effects on an individual basis. A phase 3 trial is currently underway of 150 patients who have had a stroke and aims to study the efficacy of the new procedure.
Combined TMS and EEG
With the combination of TMS and the simultaneous measurement of EEG activity, a further development of fMRI connectivity analyses is currently being tested. Dr. Grefkes-Hermann believes that this procedure, which is more cost-effective, has higher temporal resolution, can be used directly at the bedside, and has more potential for personalized therapy planning in clinical practice.
The TMS-EEG procedure also makes it possible to predict the risk of post-stroke delirium, which affects around 30% of stroke patients and greatly worsens the outcome, underlined Ulf Ziemann, MD, medical director of the department of neurology at Tübingen (Germany) University Hospital. In a study of 33 patients with acute stroke, the onset of post-stroke delirium could be predicted with a high degree of accuracy by using the TMS-EEG procedure no later than 48 hours after the event.
Other promising, noninvasive methods for neuron activation mentioned by Dr. Ziemann include transcranial focused ultrasound stimulation (tFUS) with low intensity, which is being studied for chronic pain, dementia, epilepsy, traumatic brain injury, and depression, as well as transcranial pulse stimulation (TPS), which is also based on ultrasound. In a pilot study of 35 patients with Alzheimer’s disease, use of TPS within 3 months had positive effects on cognition. However, the study was not controlled and therefore further assessments are needed.
Custom deep brain stimulation
For deep brain stimulation (DBS), an established therapy for Parkinson’s disease and other movement disorders, the aim is individualized, symptom-related network stimulation, reported Andrea Kühn, MD, head of the movement disorders and neuromodulation section in the department of neurology at Charité University Hospital Berlin.
At the panregional collaborative research center ReTune, which has been supported for 4 years now by €10 million from the German Research Foundation (DFG), imaging and computer-assisted programming algorithms are being developed for DBS. They will greatly simplify the time-consuming standard procedure for the best possible setting of the stimulation parameters, which requires a hospital stay of several days.
A randomized crossover study of 35 patients with Parkinson’s disease proved the equivalence of the fast, algorithm-assisted DBS for the control of motor symptoms compared with standard procedures.
The new methods have the potential to considerably improve the outcome of patients with neurological and psychiatric diseases, according to scientists. However, the positive data must still be validated in further studies.
This article was translated from Medscape’s German edition. A version of this article appeared on Medscape.com.
Antidepressants benefit some patients with osteoarthritis pain
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
FROM OARSI 2023
JAK inhibitor ivarmacitinib shows efficacy for atopic dermatitis in a pivotal trial
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.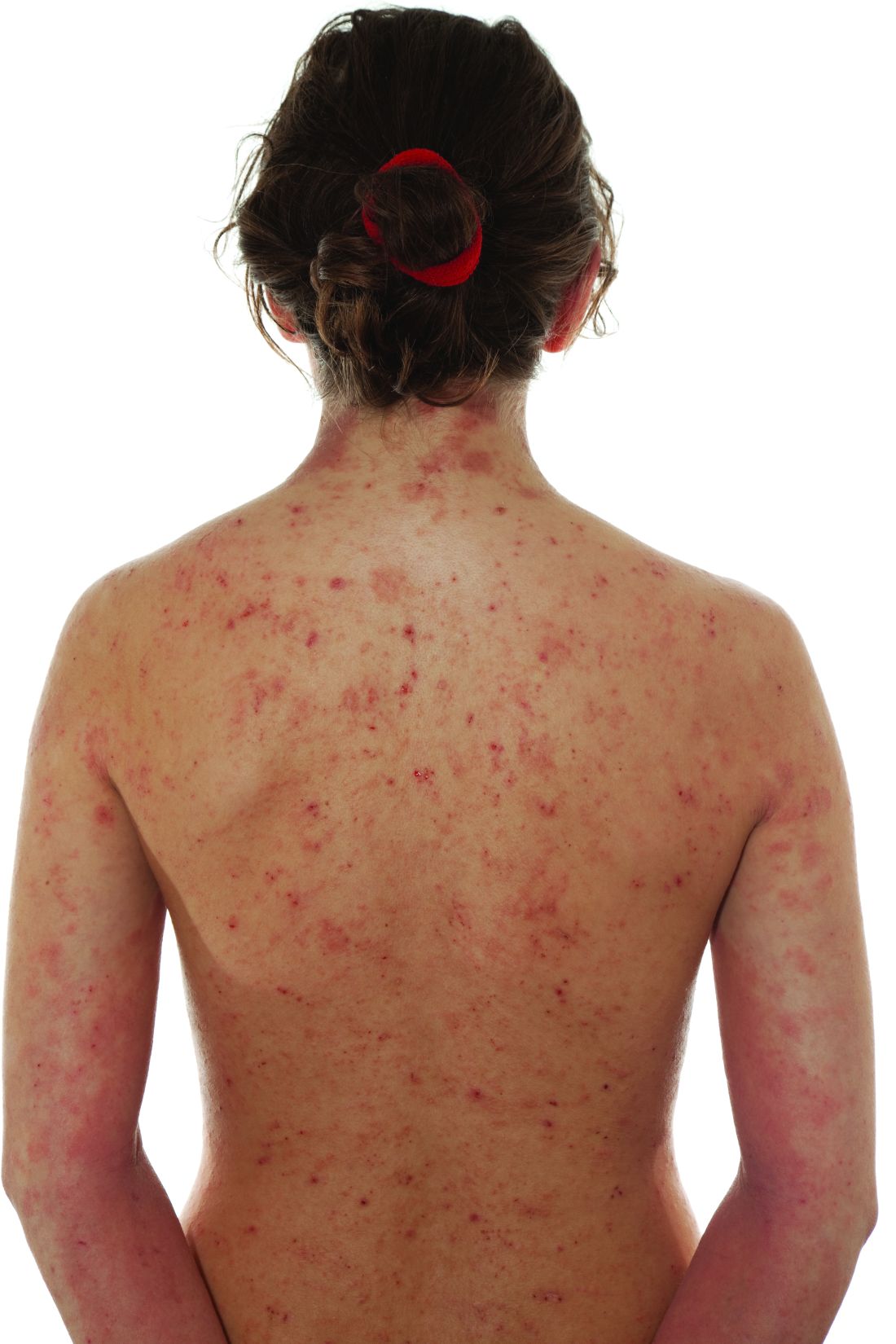
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
AT AAD 2023
Melatonin: A new way to reduce self-harm?
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF CHILD PSYCHOLOGY AND PSYCHIATRY