User login
PD-L1 cutoff for pembrolizumab in mTNBC confirmed
recently presented at the San Antonio Breast Cancer Symposium.
Patients enrolled in KEYNOTE-355 – which is a phase 3, placebo-controlled trial of 847 patients – were stratified by CPS scores of at least 1 and at least 10, with the latter group in which adding pembrolizumab to chemotherapy was shown to significantly improve both overall survival and progression-free survival.
As it was unclear whether taking a more fine-grained approach would reveal specific CPS scores at which pembrolizumab would be beneficial, Javier Cortes, MD, PhD, International Breast Cancer Center, Barcelona, and colleagues divided the patients into four CPS levels: less than 1, 1-9, 10-19, and at least 20.
Patients with a CPS 10-19 and at least 20 given pembrolizumab alongside chemotherapy had an overall survival benefit of 29% and 28%, respectively, while the PFS improvement was 30% and 38%. In the CPS of less than 1 and 1-9 groups, there were no discernible benefits from adding the checkpoint inhibitor.
“Given the similar outcomes in the CPS 10-19 and the CPS ≥20 subgroups, a CPS of 10 or more is a reasonable cutoff to define the population of patients with metastatic TNBC that might have benefit from the addition of pembrolizumab to chemotherapy,” Dr. Cortes said. “In my opinion, these results provide further support for pembrolizumab in combination with chemotherapy as a good option, maybe a standard of care for some patients ... with local recurrent unresectable or metastatic TNBC whose tumors express PD-1 CPS ≥10.”
Invited discussant Hope S. Rugo, MD, said the study demonstrates that PD-L1 CPS of at least 10 is “clearly the optimal cutoff for differentiating benefit from pembrolizumab” and confirms the combination with chemotherapy as a “standard of care in this population”.
However, there are a number of outstanding questions in the metastatic setting, she said, including the test used to determine PD-L1 expression.
“Clearly the test that you order should be matched to the planned checkpoint inhibitor, and we look forward to additional data” on the relative overlap of the assays used in both the current study and in KEYNOTE-522.
However, IMpassion130 showed there is “incomplete overlap in terms of the two antibodies and tests that have been used to define PD-L1 positivity in breast cancer,” said Dr. Rugo, professor of medicine in hematology and oncology at the University of California, San Francisco.
“For excellent responders, can chemotherapy and eventually immunotherapy be discontinued, and when is it optimal? How long should we be continuing the combination and how long should we continue the checkpoint inhibitor alone?” she asked.
“Certainly in my own clinical practice,” Dr. Rugo explained, “in those excellent responders, it’s difficult to know when to stop the checkpoint inhibitor, but sometimes toxicity tells us the answer to that question. At some point, we need to stop therapy and understand what happens to those patients.”
She said that only 38% of patients in the current study benefited from pembrolizumab. “How can we amplify the immune response in those patients who do not have PD-L1–positive disease to further extend this benefit, and can we extend the efficacy to other subtypes? There are ongoing studies evaluating this question,” Dr. Rugo said.
Dr. Cortes said that KEYNOTE-355 showed the addition of pembrolizumab to chemotherapy led to clinically meaningful improvements in both PFS and overall survival versus chemotherapy alone in the first-line treatment of mTNBC.
However, that benefit was seen only in patients with a PD-L1 CPS of at least 10, while there was no statistically significant improvement in either PFS or overall survival in those with a CPS of at least 1.
He explained that 847 patients with previously untreated locally recurrent or metastatic TNBC, or those who had been treated at least 6 months prior to disease recurrence, were randomized 2:1 to pembrolizumab or placebo plus chemotherapy.
For the current analysis, they substratified patients by PD-L1 CPS into less than 1, which accounted for 24.9% of patients; 1-9, seen in 36.2%-38.4%; 10-19, accounting for 13.9%-14.1%; and at least 20, seen in 22.8%-24.7% of patients.
Dr. Cortes said the overall survival rate among patients with CPS of at least 10 was 70.5% for patients treated with pembrolizumab plus chemotherapy versus 81.6% for those assigned to placebo, at a significant hazard ratio of 0.73 (P = .0093).
Among patients with CPS of at least 1, the overall survival rate was 79.1% with pembrolizumab plus chemotherapy and 83.9% in those given placebo, at a nonsignificant hazard ratio of 0.86. This translated into an HR of 0.89 in the intention-to-treat analysis.
Turning to the novel subgroups, Dr. Cortes showed that the HR for overall survival for pembrolizumab versus placebo was nonsignificant in patients with CPS of at least 1, at 0.97, and in those with CPS 1-9, at 1.09.
However, the HRs were markedly improved in patients with CPD 10-19, at 0.71, and in those with CPS of at least 20, at 0.72, showing that the “relative benefit of adding pembrolizumab to chemotherapy was pretty much the same ... suggesting that CPS ≥10 could be a reasonable cutoff.”
In both of these groups, there was a sustained separation in the overall survival curves starting at around 10 months.
Turning to the PFS results, Dr Cortes said the event-free rate was 65.5% with the addition of pembrolizumab to chemotherapy in patients with PD-L1 CPS of at least 10, while those given placebo had a rate of 78.6%, at an HR of 0.66.
In patients with PD-L1 CPS of at least 1, the HR was 0.75, or 0.82 in the intention-to-treat analysis.
“As with overall survival,” he said, there was a “trend toward improved efficacy with PD-L1 enrichment with the addition of pembrolizumab to chemotherapy, although the PFS benefit in the pembro arm was slightly greater in the CPS ≥20 subgroup, compared to the CPS 10-19 subgroup.”
However, they highlighted that the difference was “small and the confidence intervals clearly overlapped.”
Why does PD-L1 expression play a role in response to pembrolizumab in mTNBC, but not in the early disease setting as seen in KEYNOTE-522?
“This is a question we have raised many, many times and have had many debates on,” Dr. Cortes said. “They are two completely different populations with the early breast cancer setting completely different to that in metastatic disease. Maybe the microenvironment plays a different role there, maybe we have to explore more in detail other biomarkers. I also think that different drugs were used in the neoadjuvant setting. We still have many unanswered questions.”
Dr. Rugo suggested that previous studies have given some clues to these questions with reductions in PD-L1 expression and tumor-infiltrating leukocytes observed between primary and metastatic disease.
The immune differences between primary and metastatic disease lead to immune escape, she said, adding: “This is clearly complicated by mutational complexity under the pressure of treatment.”
The study was funded by Merck Sharp and Dohme. Dr. Cortes and Dr. Rugo reported relationships with numerous pharmaceutical companies.
recently presented at the San Antonio Breast Cancer Symposium.
Patients enrolled in KEYNOTE-355 – which is a phase 3, placebo-controlled trial of 847 patients – were stratified by CPS scores of at least 1 and at least 10, with the latter group in which adding pembrolizumab to chemotherapy was shown to significantly improve both overall survival and progression-free survival.
As it was unclear whether taking a more fine-grained approach would reveal specific CPS scores at which pembrolizumab would be beneficial, Javier Cortes, MD, PhD, International Breast Cancer Center, Barcelona, and colleagues divided the patients into four CPS levels: less than 1, 1-9, 10-19, and at least 20.
Patients with a CPS 10-19 and at least 20 given pembrolizumab alongside chemotherapy had an overall survival benefit of 29% and 28%, respectively, while the PFS improvement was 30% and 38%. In the CPS of less than 1 and 1-9 groups, there were no discernible benefits from adding the checkpoint inhibitor.
“Given the similar outcomes in the CPS 10-19 and the CPS ≥20 subgroups, a CPS of 10 or more is a reasonable cutoff to define the population of patients with metastatic TNBC that might have benefit from the addition of pembrolizumab to chemotherapy,” Dr. Cortes said. “In my opinion, these results provide further support for pembrolizumab in combination with chemotherapy as a good option, maybe a standard of care for some patients ... with local recurrent unresectable or metastatic TNBC whose tumors express PD-1 CPS ≥10.”
Invited discussant Hope S. Rugo, MD, said the study demonstrates that PD-L1 CPS of at least 10 is “clearly the optimal cutoff for differentiating benefit from pembrolizumab” and confirms the combination with chemotherapy as a “standard of care in this population”.
However, there are a number of outstanding questions in the metastatic setting, she said, including the test used to determine PD-L1 expression.
“Clearly the test that you order should be matched to the planned checkpoint inhibitor, and we look forward to additional data” on the relative overlap of the assays used in both the current study and in KEYNOTE-522.
However, IMpassion130 showed there is “incomplete overlap in terms of the two antibodies and tests that have been used to define PD-L1 positivity in breast cancer,” said Dr. Rugo, professor of medicine in hematology and oncology at the University of California, San Francisco.
“For excellent responders, can chemotherapy and eventually immunotherapy be discontinued, and when is it optimal? How long should we be continuing the combination and how long should we continue the checkpoint inhibitor alone?” she asked.
“Certainly in my own clinical practice,” Dr. Rugo explained, “in those excellent responders, it’s difficult to know when to stop the checkpoint inhibitor, but sometimes toxicity tells us the answer to that question. At some point, we need to stop therapy and understand what happens to those patients.”
She said that only 38% of patients in the current study benefited from pembrolizumab. “How can we amplify the immune response in those patients who do not have PD-L1–positive disease to further extend this benefit, and can we extend the efficacy to other subtypes? There are ongoing studies evaluating this question,” Dr. Rugo said.
Dr. Cortes said that KEYNOTE-355 showed the addition of pembrolizumab to chemotherapy led to clinically meaningful improvements in both PFS and overall survival versus chemotherapy alone in the first-line treatment of mTNBC.
However, that benefit was seen only in patients with a PD-L1 CPS of at least 10, while there was no statistically significant improvement in either PFS or overall survival in those with a CPS of at least 1.
He explained that 847 patients with previously untreated locally recurrent or metastatic TNBC, or those who had been treated at least 6 months prior to disease recurrence, were randomized 2:1 to pembrolizumab or placebo plus chemotherapy.
For the current analysis, they substratified patients by PD-L1 CPS into less than 1, which accounted for 24.9% of patients; 1-9, seen in 36.2%-38.4%; 10-19, accounting for 13.9%-14.1%; and at least 20, seen in 22.8%-24.7% of patients.
Dr. Cortes said the overall survival rate among patients with CPS of at least 10 was 70.5% for patients treated with pembrolizumab plus chemotherapy versus 81.6% for those assigned to placebo, at a significant hazard ratio of 0.73 (P = .0093).
Among patients with CPS of at least 1, the overall survival rate was 79.1% with pembrolizumab plus chemotherapy and 83.9% in those given placebo, at a nonsignificant hazard ratio of 0.86. This translated into an HR of 0.89 in the intention-to-treat analysis.
Turning to the novel subgroups, Dr. Cortes showed that the HR for overall survival for pembrolizumab versus placebo was nonsignificant in patients with CPS of at least 1, at 0.97, and in those with CPS 1-9, at 1.09.
However, the HRs were markedly improved in patients with CPD 10-19, at 0.71, and in those with CPS of at least 20, at 0.72, showing that the “relative benefit of adding pembrolizumab to chemotherapy was pretty much the same ... suggesting that CPS ≥10 could be a reasonable cutoff.”
In both of these groups, there was a sustained separation in the overall survival curves starting at around 10 months.
Turning to the PFS results, Dr Cortes said the event-free rate was 65.5% with the addition of pembrolizumab to chemotherapy in patients with PD-L1 CPS of at least 10, while those given placebo had a rate of 78.6%, at an HR of 0.66.
In patients with PD-L1 CPS of at least 1, the HR was 0.75, or 0.82 in the intention-to-treat analysis.
“As with overall survival,” he said, there was a “trend toward improved efficacy with PD-L1 enrichment with the addition of pembrolizumab to chemotherapy, although the PFS benefit in the pembro arm was slightly greater in the CPS ≥20 subgroup, compared to the CPS 10-19 subgroup.”
However, they highlighted that the difference was “small and the confidence intervals clearly overlapped.”
Why does PD-L1 expression play a role in response to pembrolizumab in mTNBC, but not in the early disease setting as seen in KEYNOTE-522?
“This is a question we have raised many, many times and have had many debates on,” Dr. Cortes said. “They are two completely different populations with the early breast cancer setting completely different to that in metastatic disease. Maybe the microenvironment plays a different role there, maybe we have to explore more in detail other biomarkers. I also think that different drugs were used in the neoadjuvant setting. We still have many unanswered questions.”
Dr. Rugo suggested that previous studies have given some clues to these questions with reductions in PD-L1 expression and tumor-infiltrating leukocytes observed between primary and metastatic disease.
The immune differences between primary and metastatic disease lead to immune escape, she said, adding: “This is clearly complicated by mutational complexity under the pressure of treatment.”
The study was funded by Merck Sharp and Dohme. Dr. Cortes and Dr. Rugo reported relationships with numerous pharmaceutical companies.
recently presented at the San Antonio Breast Cancer Symposium.
Patients enrolled in KEYNOTE-355 – which is a phase 3, placebo-controlled trial of 847 patients – were stratified by CPS scores of at least 1 and at least 10, with the latter group in which adding pembrolizumab to chemotherapy was shown to significantly improve both overall survival and progression-free survival.
As it was unclear whether taking a more fine-grained approach would reveal specific CPS scores at which pembrolizumab would be beneficial, Javier Cortes, MD, PhD, International Breast Cancer Center, Barcelona, and colleagues divided the patients into four CPS levels: less than 1, 1-9, 10-19, and at least 20.
Patients with a CPS 10-19 and at least 20 given pembrolizumab alongside chemotherapy had an overall survival benefit of 29% and 28%, respectively, while the PFS improvement was 30% and 38%. In the CPS of less than 1 and 1-9 groups, there were no discernible benefits from adding the checkpoint inhibitor.
“Given the similar outcomes in the CPS 10-19 and the CPS ≥20 subgroups, a CPS of 10 or more is a reasonable cutoff to define the population of patients with metastatic TNBC that might have benefit from the addition of pembrolizumab to chemotherapy,” Dr. Cortes said. “In my opinion, these results provide further support for pembrolizumab in combination with chemotherapy as a good option, maybe a standard of care for some patients ... with local recurrent unresectable or metastatic TNBC whose tumors express PD-1 CPS ≥10.”
Invited discussant Hope S. Rugo, MD, said the study demonstrates that PD-L1 CPS of at least 10 is “clearly the optimal cutoff for differentiating benefit from pembrolizumab” and confirms the combination with chemotherapy as a “standard of care in this population”.
However, there are a number of outstanding questions in the metastatic setting, she said, including the test used to determine PD-L1 expression.
“Clearly the test that you order should be matched to the planned checkpoint inhibitor, and we look forward to additional data” on the relative overlap of the assays used in both the current study and in KEYNOTE-522.
However, IMpassion130 showed there is “incomplete overlap in terms of the two antibodies and tests that have been used to define PD-L1 positivity in breast cancer,” said Dr. Rugo, professor of medicine in hematology and oncology at the University of California, San Francisco.
“For excellent responders, can chemotherapy and eventually immunotherapy be discontinued, and when is it optimal? How long should we be continuing the combination and how long should we continue the checkpoint inhibitor alone?” she asked.
“Certainly in my own clinical practice,” Dr. Rugo explained, “in those excellent responders, it’s difficult to know when to stop the checkpoint inhibitor, but sometimes toxicity tells us the answer to that question. At some point, we need to stop therapy and understand what happens to those patients.”
She said that only 38% of patients in the current study benefited from pembrolizumab. “How can we amplify the immune response in those patients who do not have PD-L1–positive disease to further extend this benefit, and can we extend the efficacy to other subtypes? There are ongoing studies evaluating this question,” Dr. Rugo said.
Dr. Cortes said that KEYNOTE-355 showed the addition of pembrolizumab to chemotherapy led to clinically meaningful improvements in both PFS and overall survival versus chemotherapy alone in the first-line treatment of mTNBC.
However, that benefit was seen only in patients with a PD-L1 CPS of at least 10, while there was no statistically significant improvement in either PFS or overall survival in those with a CPS of at least 1.
He explained that 847 patients with previously untreated locally recurrent or metastatic TNBC, or those who had been treated at least 6 months prior to disease recurrence, were randomized 2:1 to pembrolizumab or placebo plus chemotherapy.
For the current analysis, they substratified patients by PD-L1 CPS into less than 1, which accounted for 24.9% of patients; 1-9, seen in 36.2%-38.4%; 10-19, accounting for 13.9%-14.1%; and at least 20, seen in 22.8%-24.7% of patients.
Dr. Cortes said the overall survival rate among patients with CPS of at least 10 was 70.5% for patients treated with pembrolizumab plus chemotherapy versus 81.6% for those assigned to placebo, at a significant hazard ratio of 0.73 (P = .0093).
Among patients with CPS of at least 1, the overall survival rate was 79.1% with pembrolizumab plus chemotherapy and 83.9% in those given placebo, at a nonsignificant hazard ratio of 0.86. This translated into an HR of 0.89 in the intention-to-treat analysis.
Turning to the novel subgroups, Dr. Cortes showed that the HR for overall survival for pembrolizumab versus placebo was nonsignificant in patients with CPS of at least 1, at 0.97, and in those with CPS 1-9, at 1.09.
However, the HRs were markedly improved in patients with CPD 10-19, at 0.71, and in those with CPS of at least 20, at 0.72, showing that the “relative benefit of adding pembrolizumab to chemotherapy was pretty much the same ... suggesting that CPS ≥10 could be a reasonable cutoff.”
In both of these groups, there was a sustained separation in the overall survival curves starting at around 10 months.
Turning to the PFS results, Dr Cortes said the event-free rate was 65.5% with the addition of pembrolizumab to chemotherapy in patients with PD-L1 CPS of at least 10, while those given placebo had a rate of 78.6%, at an HR of 0.66.
In patients with PD-L1 CPS of at least 1, the HR was 0.75, or 0.82 in the intention-to-treat analysis.
“As with overall survival,” he said, there was a “trend toward improved efficacy with PD-L1 enrichment with the addition of pembrolizumab to chemotherapy, although the PFS benefit in the pembro arm was slightly greater in the CPS ≥20 subgroup, compared to the CPS 10-19 subgroup.”
However, they highlighted that the difference was “small and the confidence intervals clearly overlapped.”
Why does PD-L1 expression play a role in response to pembrolizumab in mTNBC, but not in the early disease setting as seen in KEYNOTE-522?
“This is a question we have raised many, many times and have had many debates on,” Dr. Cortes said. “They are two completely different populations with the early breast cancer setting completely different to that in metastatic disease. Maybe the microenvironment plays a different role there, maybe we have to explore more in detail other biomarkers. I also think that different drugs were used in the neoadjuvant setting. We still have many unanswered questions.”
Dr. Rugo suggested that previous studies have given some clues to these questions with reductions in PD-L1 expression and tumor-infiltrating leukocytes observed between primary and metastatic disease.
The immune differences between primary and metastatic disease lead to immune escape, she said, adding: “This is clearly complicated by mutational complexity under the pressure of treatment.”
The study was funded by Merck Sharp and Dohme. Dr. Cortes and Dr. Rugo reported relationships with numerous pharmaceutical companies.
FROM SABCS 2021
Isatuximab added to RVd boosts response in new myeloma
ATLANTA -
The drug is isatuximab (Sarclisa, Sanofi), an anti-CD38 antibody that was approved last year for use in patients with advanced disease.
Now it has shown benefit in patients who have been newly diagnosed with the disease. When isatuximab was added onto a usual triplet therapy for myeloma, it increased the likelihood that patients would be negative for minimal residual disease (MRD) at the end of the induction phase of treatment, thereby increasing their chances for a successful autologous stem cell transplant (ASCT).
The new results come from the GMMG-HD7 trial, in which all patients were treated with the triplet combination of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVd).
Some patients, after randomization, also received isatuximab, and in this group, the MRD-negativity rate was 50.1% at the end of induction therapy compared with 35.6% for patients treated with RVd alone.
Patients who are MRD-negative at the time of ASCT have significantly better outcomes than patients who remain MRD-positive.
“Isa-RVd is the first regimen to demonstrate significant MRD-negativity benefit at the end of induction versus RVd in a phase 3 trial,” reported Hartmut Goldschmidt, MD, from University Hospital Heidelberg, Germany.
“The benefits of the addition of Isa to RVd versus RVd regarding MRD negativity after induction therapy was consistent in all subgroups,” he added.
Dr. Goldschmidt spoke at a press briefing prior to his presentation of the data here at the annual meeting of the American Society of Hematology (ASH).
“I think that these data are encouraging, but they are preliminary, and we need mature data to be absolutely certain about whether this presents a major advance in treatment,” commented Ravi Vij, MD, from the Siteman Cancer Center and Washington University School of Medicine in St. Louis. Dr. Vij was not involved in the study.
“We know that for transplant-eligible patients, for whom this trial was conducted, the field is moving toward giving four drugs for induction,” he said in an interview with this news organization.
He noted that the combination of RVd with the other currently available anti-CD38 antibody, daratumumab (Darzalex), was approved for this indication in the United States in Jan. 2021.
Dr. Vij said that isatuximab has been slow to catch on in the United States both because it was approved after clinicians had already become familiar with daratumumab and because it is given intravenously, compared with subcutaneous administration of the latest formulation of daratumumab.
“Whereas isatuximab can take an hour-and-a-half with each infusion, daratumumab takes 5 minutes for an injection and the patient is out of there, so it is convenient both for the patient and the treating institution,” he said.
MRD vs. CR?
Dr. Goldschmidt was asked during the briefing about whether MRD-negativity or complete response rates are better predictors of progression-free survival (PFS). He replied that with current standardized sequencing techniques and sensitivity down to 10-6, “it’s a big benefit to analyze MRD negativity, and there is ongoing discussion between colleagues from the myeloma group with the Food and Drug Administration about how we can merge the data and predict PFS and overall survival.”
Laurie Sehn, MD, MPH, from the BC Cancer Centre for Lymphoid Cancer, Vancouver, who moderated the briefing, commented that “we’re desperately looking for surrogate markers to speed up answers to clinical trials, and I think MRD in myeloma is quickly becoming a very important surrogate marker.”
GMMG-7 results
For their trial, Dr. Goldschmidt and colleagues enrolled 662 patients with newly diagnosed multiple myeloma who were candidates for high-dose therapy and ASCT and after stratification by revised International Staging System (r-ISS) criteria, randomly assigned them six three-week cycles of induction therapy with Isa-RVd or RVd alone.
Following ASCT, patients were again randomized to maintenance with either isatuximab plus lenalidomide or lenalidomide alone.
As noted before, MRD rates at the end of induction were 50.1% with Isa-RVd versus 35.6% with RVd alone, translating to a hazard ratio favoring the four-drug combination of 1.83 (P < .001).
Treatment with Isa-RVd was the only significant predictor for the likelihood of MRD negativity in a multivariate analysis controlling for treatment group, r-ISS status, performance status, renal impairment, age, and sex.
Although the rate of complete responses at the end of induction was similar between the treatment groups, the rate of very good partial response or better was higher with the isatuximab-containing combination (77.3% vs. 60.5%; P < .001).
The respective rates of disease progression at the end of induction in the Isa-RVd and RVd groups were 1.5% versus 4.0%.
The rates of adverse events were generally similar between the groups, except a higher proportion of patients had leukocytopenia or neutropenia in the Isa-RVd than the RVdgroup (26.4% vs. 9.1%). There were four deaths in the Isa-RVd group and eight in the RVd group. Most of the deaths were attributable to disease progression or COVID-19, said Dr. Goldschmidt.
The study was funded by Sanofi. Dr. Goldschmidt has disclosed honoraria and research grants from Sanofi and others. Dr. Vij has disclosed honoraria or advisory board activities from various companies, including Sanofi. Dr. Sehn is a consultant for and has received honoraria from various companies, not including Sanofi.
A version of this article first appeared on Medscape.com.
ATLANTA -
The drug is isatuximab (Sarclisa, Sanofi), an anti-CD38 antibody that was approved last year for use in patients with advanced disease.
Now it has shown benefit in patients who have been newly diagnosed with the disease. When isatuximab was added onto a usual triplet therapy for myeloma, it increased the likelihood that patients would be negative for minimal residual disease (MRD) at the end of the induction phase of treatment, thereby increasing their chances for a successful autologous stem cell transplant (ASCT).
The new results come from the GMMG-HD7 trial, in which all patients were treated with the triplet combination of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVd).
Some patients, after randomization, also received isatuximab, and in this group, the MRD-negativity rate was 50.1% at the end of induction therapy compared with 35.6% for patients treated with RVd alone.
Patients who are MRD-negative at the time of ASCT have significantly better outcomes than patients who remain MRD-positive.
“Isa-RVd is the first regimen to demonstrate significant MRD-negativity benefit at the end of induction versus RVd in a phase 3 trial,” reported Hartmut Goldschmidt, MD, from University Hospital Heidelberg, Germany.
“The benefits of the addition of Isa to RVd versus RVd regarding MRD negativity after induction therapy was consistent in all subgroups,” he added.
Dr. Goldschmidt spoke at a press briefing prior to his presentation of the data here at the annual meeting of the American Society of Hematology (ASH).
“I think that these data are encouraging, but they are preliminary, and we need mature data to be absolutely certain about whether this presents a major advance in treatment,” commented Ravi Vij, MD, from the Siteman Cancer Center and Washington University School of Medicine in St. Louis. Dr. Vij was not involved in the study.
“We know that for transplant-eligible patients, for whom this trial was conducted, the field is moving toward giving four drugs for induction,” he said in an interview with this news organization.
He noted that the combination of RVd with the other currently available anti-CD38 antibody, daratumumab (Darzalex), was approved for this indication in the United States in Jan. 2021.
Dr. Vij said that isatuximab has been slow to catch on in the United States both because it was approved after clinicians had already become familiar with daratumumab and because it is given intravenously, compared with subcutaneous administration of the latest formulation of daratumumab.
“Whereas isatuximab can take an hour-and-a-half with each infusion, daratumumab takes 5 minutes for an injection and the patient is out of there, so it is convenient both for the patient and the treating institution,” he said.
MRD vs. CR?
Dr. Goldschmidt was asked during the briefing about whether MRD-negativity or complete response rates are better predictors of progression-free survival (PFS). He replied that with current standardized sequencing techniques and sensitivity down to 10-6, “it’s a big benefit to analyze MRD negativity, and there is ongoing discussion between colleagues from the myeloma group with the Food and Drug Administration about how we can merge the data and predict PFS and overall survival.”
Laurie Sehn, MD, MPH, from the BC Cancer Centre for Lymphoid Cancer, Vancouver, who moderated the briefing, commented that “we’re desperately looking for surrogate markers to speed up answers to clinical trials, and I think MRD in myeloma is quickly becoming a very important surrogate marker.”
GMMG-7 results
For their trial, Dr. Goldschmidt and colleagues enrolled 662 patients with newly diagnosed multiple myeloma who were candidates for high-dose therapy and ASCT and after stratification by revised International Staging System (r-ISS) criteria, randomly assigned them six three-week cycles of induction therapy with Isa-RVd or RVd alone.
Following ASCT, patients were again randomized to maintenance with either isatuximab plus lenalidomide or lenalidomide alone.
As noted before, MRD rates at the end of induction were 50.1% with Isa-RVd versus 35.6% with RVd alone, translating to a hazard ratio favoring the four-drug combination of 1.83 (P < .001).
Treatment with Isa-RVd was the only significant predictor for the likelihood of MRD negativity in a multivariate analysis controlling for treatment group, r-ISS status, performance status, renal impairment, age, and sex.
Although the rate of complete responses at the end of induction was similar between the treatment groups, the rate of very good partial response or better was higher with the isatuximab-containing combination (77.3% vs. 60.5%; P < .001).
The respective rates of disease progression at the end of induction in the Isa-RVd and RVd groups were 1.5% versus 4.0%.
The rates of adverse events were generally similar between the groups, except a higher proportion of patients had leukocytopenia or neutropenia in the Isa-RVd than the RVdgroup (26.4% vs. 9.1%). There were four deaths in the Isa-RVd group and eight in the RVd group. Most of the deaths were attributable to disease progression or COVID-19, said Dr. Goldschmidt.
The study was funded by Sanofi. Dr. Goldschmidt has disclosed honoraria and research grants from Sanofi and others. Dr. Vij has disclosed honoraria or advisory board activities from various companies, including Sanofi. Dr. Sehn is a consultant for and has received honoraria from various companies, not including Sanofi.
A version of this article first appeared on Medscape.com.
ATLANTA -
The drug is isatuximab (Sarclisa, Sanofi), an anti-CD38 antibody that was approved last year for use in patients with advanced disease.
Now it has shown benefit in patients who have been newly diagnosed with the disease. When isatuximab was added onto a usual triplet therapy for myeloma, it increased the likelihood that patients would be negative for minimal residual disease (MRD) at the end of the induction phase of treatment, thereby increasing their chances for a successful autologous stem cell transplant (ASCT).
The new results come from the GMMG-HD7 trial, in which all patients were treated with the triplet combination of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVd).
Some patients, after randomization, also received isatuximab, and in this group, the MRD-negativity rate was 50.1% at the end of induction therapy compared with 35.6% for patients treated with RVd alone.
Patients who are MRD-negative at the time of ASCT have significantly better outcomes than patients who remain MRD-positive.
“Isa-RVd is the first regimen to demonstrate significant MRD-negativity benefit at the end of induction versus RVd in a phase 3 trial,” reported Hartmut Goldschmidt, MD, from University Hospital Heidelberg, Germany.
“The benefits of the addition of Isa to RVd versus RVd regarding MRD negativity after induction therapy was consistent in all subgroups,” he added.
Dr. Goldschmidt spoke at a press briefing prior to his presentation of the data here at the annual meeting of the American Society of Hematology (ASH).
“I think that these data are encouraging, but they are preliminary, and we need mature data to be absolutely certain about whether this presents a major advance in treatment,” commented Ravi Vij, MD, from the Siteman Cancer Center and Washington University School of Medicine in St. Louis. Dr. Vij was not involved in the study.
“We know that for transplant-eligible patients, for whom this trial was conducted, the field is moving toward giving four drugs for induction,” he said in an interview with this news organization.
He noted that the combination of RVd with the other currently available anti-CD38 antibody, daratumumab (Darzalex), was approved for this indication in the United States in Jan. 2021.
Dr. Vij said that isatuximab has been slow to catch on in the United States both because it was approved after clinicians had already become familiar with daratumumab and because it is given intravenously, compared with subcutaneous administration of the latest formulation of daratumumab.
“Whereas isatuximab can take an hour-and-a-half with each infusion, daratumumab takes 5 minutes for an injection and the patient is out of there, so it is convenient both for the patient and the treating institution,” he said.
MRD vs. CR?
Dr. Goldschmidt was asked during the briefing about whether MRD-negativity or complete response rates are better predictors of progression-free survival (PFS). He replied that with current standardized sequencing techniques and sensitivity down to 10-6, “it’s a big benefit to analyze MRD negativity, and there is ongoing discussion between colleagues from the myeloma group with the Food and Drug Administration about how we can merge the data and predict PFS and overall survival.”
Laurie Sehn, MD, MPH, from the BC Cancer Centre for Lymphoid Cancer, Vancouver, who moderated the briefing, commented that “we’re desperately looking for surrogate markers to speed up answers to clinical trials, and I think MRD in myeloma is quickly becoming a very important surrogate marker.”
GMMG-7 results
For their trial, Dr. Goldschmidt and colleagues enrolled 662 patients with newly diagnosed multiple myeloma who were candidates for high-dose therapy and ASCT and after stratification by revised International Staging System (r-ISS) criteria, randomly assigned them six three-week cycles of induction therapy with Isa-RVd or RVd alone.
Following ASCT, patients were again randomized to maintenance with either isatuximab plus lenalidomide or lenalidomide alone.
As noted before, MRD rates at the end of induction were 50.1% with Isa-RVd versus 35.6% with RVd alone, translating to a hazard ratio favoring the four-drug combination of 1.83 (P < .001).
Treatment with Isa-RVd was the only significant predictor for the likelihood of MRD negativity in a multivariate analysis controlling for treatment group, r-ISS status, performance status, renal impairment, age, and sex.
Although the rate of complete responses at the end of induction was similar between the treatment groups, the rate of very good partial response or better was higher with the isatuximab-containing combination (77.3% vs. 60.5%; P < .001).
The respective rates of disease progression at the end of induction in the Isa-RVd and RVd groups were 1.5% versus 4.0%.
The rates of adverse events were generally similar between the groups, except a higher proportion of patients had leukocytopenia or neutropenia in the Isa-RVd than the RVdgroup (26.4% vs. 9.1%). There were four deaths in the Isa-RVd group and eight in the RVd group. Most of the deaths were attributable to disease progression or COVID-19, said Dr. Goldschmidt.
The study was funded by Sanofi. Dr. Goldschmidt has disclosed honoraria and research grants from Sanofi and others. Dr. Vij has disclosed honoraria or advisory board activities from various companies, including Sanofi. Dr. Sehn is a consultant for and has received honoraria from various companies, not including Sanofi.
A version of this article first appeared on Medscape.com.
AT ASH 2021
Antibiotic use associated with triple-negative breast cancer mortality
SAN ANTONIO –
The study was recently presented at the San Antonio Breast Cancer Symposium by Julia D. Ransohoff, MD, of Stanford (Calif.) University.
Gut-associated lymphoid tissues are the largest component of the immune system. They influence both local and systemic immune responses, but the use of antimicrobials can decrease circulating and tumor-infiltrating lymphocytes that effect the immune repertoire and in turn, the survival of women with triple-negative breast cancer.
Dr. Ransohoff and colleagues hypothesized that increasing antimicrobial exposure in the presence of time-varying absolute lymphocyte counts may lead to higher overall and breast cancer–specific mortality. Their analysis is based on data from the population-based Surveillance, Epidemiology, and End Results registry and electronic medical records from Stanford University and Sutter Health. It included 772 women who were treated for triple-negative breast cancer between 2000 and 2014. The women were followed for an average of 104 months.
In an earlier analysis of this same group, Dr. Ransohoff found that higher minimum absolute lymphocyte counts were associated with lower overall mortality (hazard ratio, 0.23; 95% confidence interval, 0.16-0.35) and breast cancer mortality (HR, 0.19; 95% CI, 0.11-0.34) The association between higher peripheral lymphocyte counts and tumor-infiltrating lymphocytes was significant.
In the analysis of relationships between antibiotic use and mortality, 85% of women (n = 654) were prescribed antibiotics after having been diagnosed with triple-negative breast cancer. The death rate among patients who were prescribed antibiotics was 23% (153/654), compared with 20% (24/118) among the patients who were not treated with antibiotics (which accounts for 15% of the entire group).
For total antibiotic exposure, the HR for overall mortality was 1.06 (95% CI, 1.03-1.09; P < .001) and 1.07 for breast cancer–specific mortality (95% CI, 1.04-1.10; P < .001). For unique antibiotic exposure (not counting repeat prescriptions of the same antibiotic), the HR for overall mortality was 1.17 (95% CI, 1.12-1.22; P < .001) and 1.18 for breast cancer–specific mortality (95% CI, 1.12-1.24; P < .001).
“These were all statistically significant associations derived from a statistical model that takes into account baseline patient characteristics, so the reported hazard ratios, to the best of our ability, represent the risk of death associated with antibiotic use adjusted for other baseline covariates. We’ve attempted to account for differences at baseline that may indicate patients are sicker, and so the reported risk represents mortality related with antibiotic exposure,” Dr. Ransohoff said.
Elucidating the role of the microbiome in mediating absolute lymphocyte counts and immune response may inform interventions to reduce triple-negative mortality, she said.
SAN ANTONIO –
The study was recently presented at the San Antonio Breast Cancer Symposium by Julia D. Ransohoff, MD, of Stanford (Calif.) University.
Gut-associated lymphoid tissues are the largest component of the immune system. They influence both local and systemic immune responses, but the use of antimicrobials can decrease circulating and tumor-infiltrating lymphocytes that effect the immune repertoire and in turn, the survival of women with triple-negative breast cancer.
Dr. Ransohoff and colleagues hypothesized that increasing antimicrobial exposure in the presence of time-varying absolute lymphocyte counts may lead to higher overall and breast cancer–specific mortality. Their analysis is based on data from the population-based Surveillance, Epidemiology, and End Results registry and electronic medical records from Stanford University and Sutter Health. It included 772 women who were treated for triple-negative breast cancer between 2000 and 2014. The women were followed for an average of 104 months.
In an earlier analysis of this same group, Dr. Ransohoff found that higher minimum absolute lymphocyte counts were associated with lower overall mortality (hazard ratio, 0.23; 95% confidence interval, 0.16-0.35) and breast cancer mortality (HR, 0.19; 95% CI, 0.11-0.34) The association between higher peripheral lymphocyte counts and tumor-infiltrating lymphocytes was significant.
In the analysis of relationships between antibiotic use and mortality, 85% of women (n = 654) were prescribed antibiotics after having been diagnosed with triple-negative breast cancer. The death rate among patients who were prescribed antibiotics was 23% (153/654), compared with 20% (24/118) among the patients who were not treated with antibiotics (which accounts for 15% of the entire group).
For total antibiotic exposure, the HR for overall mortality was 1.06 (95% CI, 1.03-1.09; P < .001) and 1.07 for breast cancer–specific mortality (95% CI, 1.04-1.10; P < .001). For unique antibiotic exposure (not counting repeat prescriptions of the same antibiotic), the HR for overall mortality was 1.17 (95% CI, 1.12-1.22; P < .001) and 1.18 for breast cancer–specific mortality (95% CI, 1.12-1.24; P < .001).
“These were all statistically significant associations derived from a statistical model that takes into account baseline patient characteristics, so the reported hazard ratios, to the best of our ability, represent the risk of death associated with antibiotic use adjusted for other baseline covariates. We’ve attempted to account for differences at baseline that may indicate patients are sicker, and so the reported risk represents mortality related with antibiotic exposure,” Dr. Ransohoff said.
Elucidating the role of the microbiome in mediating absolute lymphocyte counts and immune response may inform interventions to reduce triple-negative mortality, she said.
SAN ANTONIO –
The study was recently presented at the San Antonio Breast Cancer Symposium by Julia D. Ransohoff, MD, of Stanford (Calif.) University.
Gut-associated lymphoid tissues are the largest component of the immune system. They influence both local and systemic immune responses, but the use of antimicrobials can decrease circulating and tumor-infiltrating lymphocytes that effect the immune repertoire and in turn, the survival of women with triple-negative breast cancer.
Dr. Ransohoff and colleagues hypothesized that increasing antimicrobial exposure in the presence of time-varying absolute lymphocyte counts may lead to higher overall and breast cancer–specific mortality. Their analysis is based on data from the population-based Surveillance, Epidemiology, and End Results registry and electronic medical records from Stanford University and Sutter Health. It included 772 women who were treated for triple-negative breast cancer between 2000 and 2014. The women were followed for an average of 104 months.
In an earlier analysis of this same group, Dr. Ransohoff found that higher minimum absolute lymphocyte counts were associated with lower overall mortality (hazard ratio, 0.23; 95% confidence interval, 0.16-0.35) and breast cancer mortality (HR, 0.19; 95% CI, 0.11-0.34) The association between higher peripheral lymphocyte counts and tumor-infiltrating lymphocytes was significant.
In the analysis of relationships between antibiotic use and mortality, 85% of women (n = 654) were prescribed antibiotics after having been diagnosed with triple-negative breast cancer. The death rate among patients who were prescribed antibiotics was 23% (153/654), compared with 20% (24/118) among the patients who were not treated with antibiotics (which accounts for 15% of the entire group).
For total antibiotic exposure, the HR for overall mortality was 1.06 (95% CI, 1.03-1.09; P < .001) and 1.07 for breast cancer–specific mortality (95% CI, 1.04-1.10; P < .001). For unique antibiotic exposure (not counting repeat prescriptions of the same antibiotic), the HR for overall mortality was 1.17 (95% CI, 1.12-1.22; P < .001) and 1.18 for breast cancer–specific mortality (95% CI, 1.12-1.24; P < .001).
“These were all statistically significant associations derived from a statistical model that takes into account baseline patient characteristics, so the reported hazard ratios, to the best of our ability, represent the risk of death associated with antibiotic use adjusted for other baseline covariates. We’ve attempted to account for differences at baseline that may indicate patients are sicker, and so the reported risk represents mortality related with antibiotic exposure,” Dr. Ransohoff said.
Elucidating the role of the microbiome in mediating absolute lymphocyte counts and immune response may inform interventions to reduce triple-negative mortality, she said.
AT SABCS 2021
Women struggle with benzodiazepine addiction post chemotherapy treatment
SAN ANTONIO – shows a new study.
While benzodiazepines and nonbenzodiazepine sedative-hypnotics are effective for these indications, misuse and increased health care utilization can ensue from their prolonged use, said Jacob C. Cogan, MD, a fellow in oncology/hematology at the Herbert Irving Comprehensive Cancer Center, Columbia University, New York. Dr. Cogan recently presented the results of the study at the San Antonio Breast Cancer Symposium.
The study included patients with breast cancer who received adjuvant chemotherapy between 2008 and 2017. Prescriptions for sedatives were divided into three periods: 365 days prior to chemotherapy to the start of chemotherapy (period one); start of chemotherapy to 90 days after the end of chemotherapy (period two); and 90-365 days after chemotherapy (period three). Patients who filled at least one benzodiazepine prescription in period two and patients who filled at least two benzodiazepine in period three were classified as new persistent benzodiazepine users. The same definitions were then used for nonbenzodiazepine sedative-hypnotics.
Among 17,532 benzodiazepine-naive patients (mean age, 57 years) and 21,863 nonbenzodiazepine sedative-hypnotic drug–naive patients (mean age, 56 years) who received adjuvant chemotherapy for breast cancer, lumpectomies were performed for a small majority (56.6% benzodiazepine naive, 55.1% nonbenzodiazepine sedative-hypnotics naive) versus mastectomy, and about half of patients received less than 4 months of chemotherapy (48.0% benzodiazepine naive, 48.6% nonbenzodiazepine sedative-hypnotics naive). Among benzodiazepine-naive patients, 4,447 (25.4%) filled at least one benzodiazepine prescription during chemotherapy, and 2,160 (9.9%) filled at least one nonbenzodiazepine sedative-hypnotic prescription during chemotherapy. The rate of new persistent benzodiazepine use after initial exposure during chemotherapy was 26.8% (n = 1,192). Similarly, 33.8% (n = 730) of nonbenzodiazepine sedative-hypnotics users became new persistent users. In addition, 115 patients became new persistent users of both types of sedative-hypnotics.
New persistent benzodiazepine use was associated with several characteristics: age 50-65 (odds ratio, 1.23; P = .01) and age greater than 65 (OR, 1.38, P = .005) relative to age less than 49; as well as Medicaid insurance, relative to commercial and Medicare insurance (OR, 1.68; P < .0001). Both new persistent benzodiazepine and nonbenzodiazepine sedative-hypnotics use was associated with chemotherapy duration of less than 4 months relative to 4 or more months of chemotherapy (OR, 1.17; P = .03 for benzodiazepines; OR, 1.58; P < .0001 for nonbenzodiazepine sedative-hypnotics).
It is not clear why shorter chemotherapy duration is associated with more new persistent use, Dr. Cogan said. “It may be that, paradoxically, a shorter duration of treatment could lead to more anxiety about recurrence. These patients may need closer monitoring of mental health symptoms and earlier referral for psychological services.”
Dr. Cogan said that providers should take steps to ensure that benzodiazepines and nonbenzodiazepine sedatives are used appropriately, which includes tapering dosages and, when appropriate, encouraging nonpharmacologic strategies.
There were no funding or other conflicts of interest associated with this study.
SAN ANTONIO – shows a new study.
While benzodiazepines and nonbenzodiazepine sedative-hypnotics are effective for these indications, misuse and increased health care utilization can ensue from their prolonged use, said Jacob C. Cogan, MD, a fellow in oncology/hematology at the Herbert Irving Comprehensive Cancer Center, Columbia University, New York. Dr. Cogan recently presented the results of the study at the San Antonio Breast Cancer Symposium.
The study included patients with breast cancer who received adjuvant chemotherapy between 2008 and 2017. Prescriptions for sedatives were divided into three periods: 365 days prior to chemotherapy to the start of chemotherapy (period one); start of chemotherapy to 90 days after the end of chemotherapy (period two); and 90-365 days after chemotherapy (period three). Patients who filled at least one benzodiazepine prescription in period two and patients who filled at least two benzodiazepine in period three were classified as new persistent benzodiazepine users. The same definitions were then used for nonbenzodiazepine sedative-hypnotics.
Among 17,532 benzodiazepine-naive patients (mean age, 57 years) and 21,863 nonbenzodiazepine sedative-hypnotic drug–naive patients (mean age, 56 years) who received adjuvant chemotherapy for breast cancer, lumpectomies were performed for a small majority (56.6% benzodiazepine naive, 55.1% nonbenzodiazepine sedative-hypnotics naive) versus mastectomy, and about half of patients received less than 4 months of chemotherapy (48.0% benzodiazepine naive, 48.6% nonbenzodiazepine sedative-hypnotics naive). Among benzodiazepine-naive patients, 4,447 (25.4%) filled at least one benzodiazepine prescription during chemotherapy, and 2,160 (9.9%) filled at least one nonbenzodiazepine sedative-hypnotic prescription during chemotherapy. The rate of new persistent benzodiazepine use after initial exposure during chemotherapy was 26.8% (n = 1,192). Similarly, 33.8% (n = 730) of nonbenzodiazepine sedative-hypnotics users became new persistent users. In addition, 115 patients became new persistent users of both types of sedative-hypnotics.
New persistent benzodiazepine use was associated with several characteristics: age 50-65 (odds ratio, 1.23; P = .01) and age greater than 65 (OR, 1.38, P = .005) relative to age less than 49; as well as Medicaid insurance, relative to commercial and Medicare insurance (OR, 1.68; P < .0001). Both new persistent benzodiazepine and nonbenzodiazepine sedative-hypnotics use was associated with chemotherapy duration of less than 4 months relative to 4 or more months of chemotherapy (OR, 1.17; P = .03 for benzodiazepines; OR, 1.58; P < .0001 for nonbenzodiazepine sedative-hypnotics).
It is not clear why shorter chemotherapy duration is associated with more new persistent use, Dr. Cogan said. “It may be that, paradoxically, a shorter duration of treatment could lead to more anxiety about recurrence. These patients may need closer monitoring of mental health symptoms and earlier referral for psychological services.”
Dr. Cogan said that providers should take steps to ensure that benzodiazepines and nonbenzodiazepine sedatives are used appropriately, which includes tapering dosages and, when appropriate, encouraging nonpharmacologic strategies.
There were no funding or other conflicts of interest associated with this study.
SAN ANTONIO – shows a new study.
While benzodiazepines and nonbenzodiazepine sedative-hypnotics are effective for these indications, misuse and increased health care utilization can ensue from their prolonged use, said Jacob C. Cogan, MD, a fellow in oncology/hematology at the Herbert Irving Comprehensive Cancer Center, Columbia University, New York. Dr. Cogan recently presented the results of the study at the San Antonio Breast Cancer Symposium.
The study included patients with breast cancer who received adjuvant chemotherapy between 2008 and 2017. Prescriptions for sedatives were divided into three periods: 365 days prior to chemotherapy to the start of chemotherapy (period one); start of chemotherapy to 90 days after the end of chemotherapy (period two); and 90-365 days after chemotherapy (period three). Patients who filled at least one benzodiazepine prescription in period two and patients who filled at least two benzodiazepine in period three were classified as new persistent benzodiazepine users. The same definitions were then used for nonbenzodiazepine sedative-hypnotics.
Among 17,532 benzodiazepine-naive patients (mean age, 57 years) and 21,863 nonbenzodiazepine sedative-hypnotic drug–naive patients (mean age, 56 years) who received adjuvant chemotherapy for breast cancer, lumpectomies were performed for a small majority (56.6% benzodiazepine naive, 55.1% nonbenzodiazepine sedative-hypnotics naive) versus mastectomy, and about half of patients received less than 4 months of chemotherapy (48.0% benzodiazepine naive, 48.6% nonbenzodiazepine sedative-hypnotics naive). Among benzodiazepine-naive patients, 4,447 (25.4%) filled at least one benzodiazepine prescription during chemotherapy, and 2,160 (9.9%) filled at least one nonbenzodiazepine sedative-hypnotic prescription during chemotherapy. The rate of new persistent benzodiazepine use after initial exposure during chemotherapy was 26.8% (n = 1,192). Similarly, 33.8% (n = 730) of nonbenzodiazepine sedative-hypnotics users became new persistent users. In addition, 115 patients became new persistent users of both types of sedative-hypnotics.
New persistent benzodiazepine use was associated with several characteristics: age 50-65 (odds ratio, 1.23; P = .01) and age greater than 65 (OR, 1.38, P = .005) relative to age less than 49; as well as Medicaid insurance, relative to commercial and Medicare insurance (OR, 1.68; P < .0001). Both new persistent benzodiazepine and nonbenzodiazepine sedative-hypnotics use was associated with chemotherapy duration of less than 4 months relative to 4 or more months of chemotherapy (OR, 1.17; P = .03 for benzodiazepines; OR, 1.58; P < .0001 for nonbenzodiazepine sedative-hypnotics).
It is not clear why shorter chemotherapy duration is associated with more new persistent use, Dr. Cogan said. “It may be that, paradoxically, a shorter duration of treatment could lead to more anxiety about recurrence. These patients may need closer monitoring of mental health symptoms and earlier referral for psychological services.”
Dr. Cogan said that providers should take steps to ensure that benzodiazepines and nonbenzodiazepine sedatives are used appropriately, which includes tapering dosages and, when appropriate, encouraging nonpharmacologic strategies.
There were no funding or other conflicts of interest associated with this study.
AT SABCS 2021
Assessing Outcomes Between Risperidone Microspheres and Paliperidone Palmitate Long-Acting Injectable Antipsychotics Among Veterans
Medication nonadherence is common with oral antipsychotic formulations, resulting in relapse, increased morbidity, and more frequent psychiatric hospitalization.1-7 Psychiatric hospitalization and illness decompensation is costly to health care systems and leads to reduced quality of life for veterans and families.6,7 Long-acting injectable antipsychotics (LAIAs) were developed to enhance antipsychotic adherence and improve patient outcomes, including reduced psychiatric hospitalization.8-12
Little outcomes data exist comparing LAIAs, including biweekly risperidone microspheres and monthly paliperidone palmitate.10-13 Risperidone microspheres require a 3-week oral crossover and are administered every 2 weeks, whereas paliperidone palmitate does not require an oral crossover and is administered every 4 weeks. The paliperidone palmitate loading regimen replaces an oral crossover.
The primary objective of this study was to compare the number of psychiatric hospitalizations between veterans administered risperidone microspheres and those on paliperidone palmitate pre- and post-LAIA initiation. Secondary objectives were to assess rehospitalization rates between patients taking risperidone microspheres and paliperidone palmitate, reduction in pre- and posthospitalization rates with LAIAs, and medication adherence.
Methods
This observational study with a retrospective cohort design was conducted at the Veterans Affairs Loma Linda Healthcare System (VALLHS) in California. We examined veterans who were initiated on LAIAs risperidone microspheres or paliperidone palmitate from January 01, 2016 through December 31, 2018. Veterans who were aged ≥ 18 years and received ≥ 2 injections of either risperidone microspheres or paliperidone palmitate during the study period were included. Veterans were excluded if they had received < 2 doses of either LAIA, received the LAIA outside of the review period, were nonadherent to risperidone crossover if they received risperidone microspheres, or transferred their care to another facility. At VALLHS, LAIA injections are administered by a nurse, and veterans must travel to the facility to receive the injections.
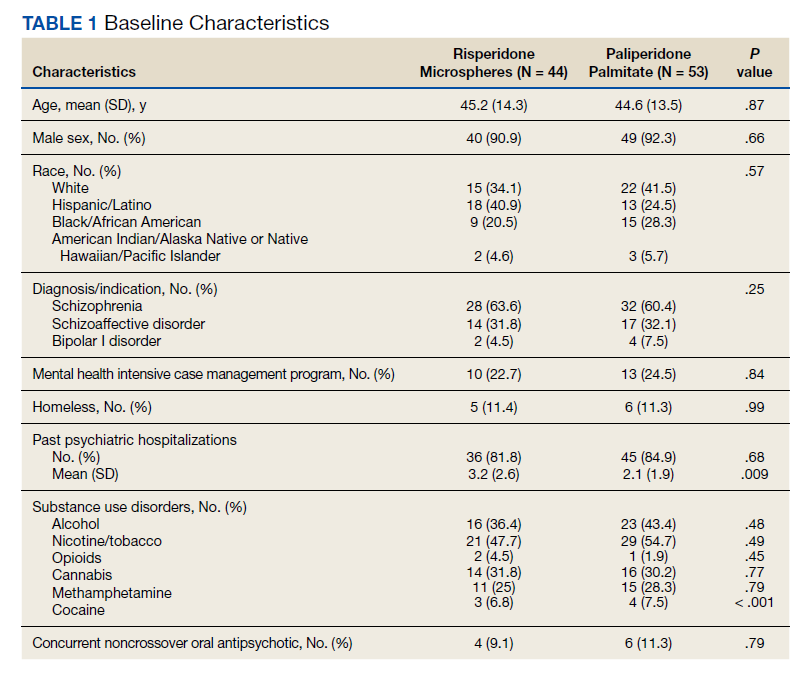
Extracted patient chart elements included participant demographics; diagnoses; comorbid alcohol, nicotine, opioid, or other substance use; duration on LAIA; psychiatric hospitalizations pre- and postinitiation of the LAIA; medication adherence; and medication discontinuation based on clinician documentation and clinic orders (Table 1).
Nonadherence to LAIA was defined as missing an injection by > 3 days for risperidone microspheres and > 7 days for paliperidone palmitate. This time frame was based on pharmacokinetic information listed in the products’ package inserts.14,15 Nonadherence to oral risperidone crossover with risperidone microspheres was defined as ≤ 80% of days covered.
Data Analysis
Patient demographics were analyzed using descriptive statistics and experimental comparisons between the risperidone microspheres and paliperidone palmitate groups to assess baseline differences between groups. Psychiatric hospitalizations pre- and post-LAIA were analyzed with parallel group (between veterans–independent groups) and pre-post (within veterans–dependent groups) designs. Index hospitalizations were examined for a period equivalent to the length of time veterans were on the LAIA. Psychiatric rehospitalization rates were analyzed for patients who had index hospitalizations and were rehospitalized for any period when they were receiving the LAIA. Incidences of pre- and post-LAIA hospitalizations were calculated in 100 person-years.
Parallel-group analysis was analyzed using the χ2 and Mann-Whitney U tests. Pre-post analyses were analyzed using the Wilcoxon rank sum test. P was set at < .05 for statistical significance.
Results
We screened 111 veterans, and 97 were included in this study (risperidone microspheres, 44; paliperidone palmitate, 53). Mean (SD) age was 46 (13.8) years, 92% were male, 38% were White, 94% were diagnosed with schizophrenia or schizoaffective disorder, and 11% were homeless. Substance use was documented as 52% for nicotine products, 40% for alcohol, 31% for cannabis, 27% for methamphetamine, 7% for cocaine, and 3% for opioids. Cannabis, methamphetamine, cocaine, and opioid use were based on clinician documentation and listed as active diagnoses at the time of LAIA initiation. Statistical significance was found in index hospitalizations P = .009) and history of cocaine use disorder (6.8% vs 7.5%, P < .001).
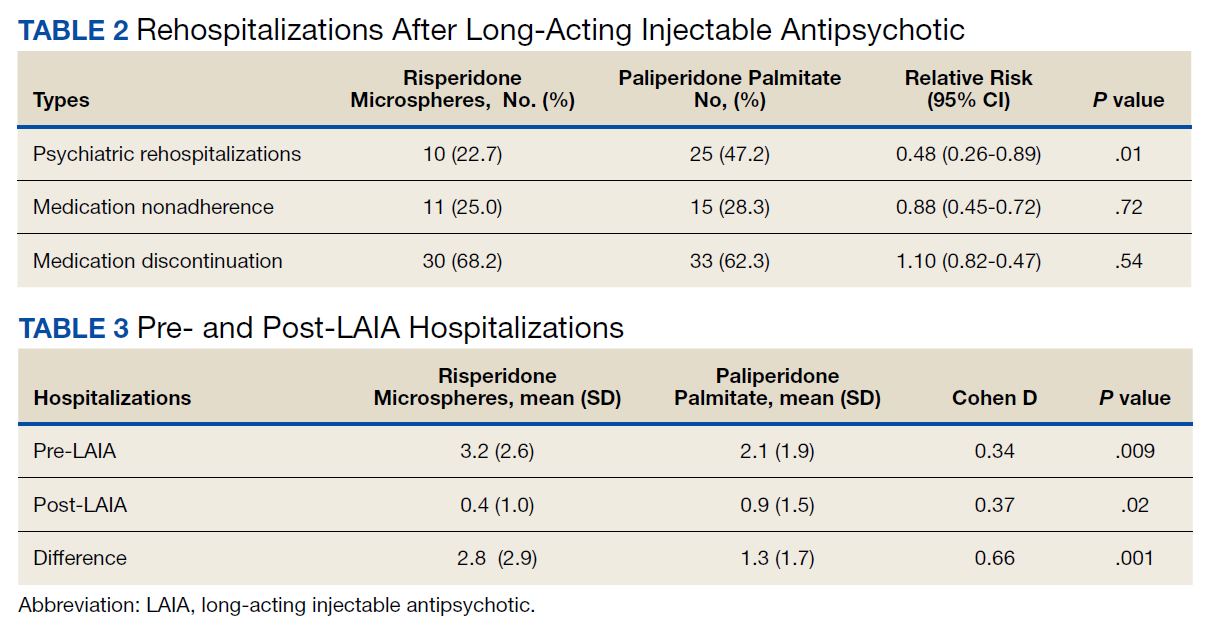
Veterans administered risperidone microspheres had fewer mean (SD) post-LAIA hospitalizations (0.4 [1.0] vs 0.9 [1.5]; P = .02) and were less likely to be rehospitalized (22.7% vs 47.2%, P = .01) compared with paliperidone palmitate. However, veterans taking risperidone microspheres had a shorter mean (SD) treatment duration (41.6 [40.2] vs 58.2 [45.7] weeks, P = .04) compared with paliperidone palmitate, mainly because patients switched to a different LAIA or oral antipsychotic. No differences were detected in nonadherence and discontinuation between risperidone microspheres and paliperidone palmitate. All veterans in the risperidone microspheres group adhered to oral risperidone crossover with an average 87.8% days covered (Table 2).
The average maintenance dose of risperidone microspheres was 42 mg every 2 weeks and 153 mg every 4 weeks for paliperidone palmitate.
Across the sample, 84% of veterans had a previous psychiatric hospitalization, although veterans initiated on risperidone microspheres had significantly higher mean (SD) index hospitalizations than those started on paliperidone palmitate (3.2 [2.6] risperidone microspheres vs 2.1 [1.9] paliperidone palmitate, P = .009). Both groups had significant decreases in mean (SD) hospitalizations (3.2 [2.6] to 0.4 [1.0], risperidone microspheres vs 2.1 [1.9] to 0.9 [1.5] paliperidone palmitate). The risperidone microspheres group had a larger decrease in mean (SD) hospitalizations post-LAIA (2.8 [2.9] risperidone microspheres vs 1.3 [1.7] paliperidone palmitate, P = .001) (Table 3).
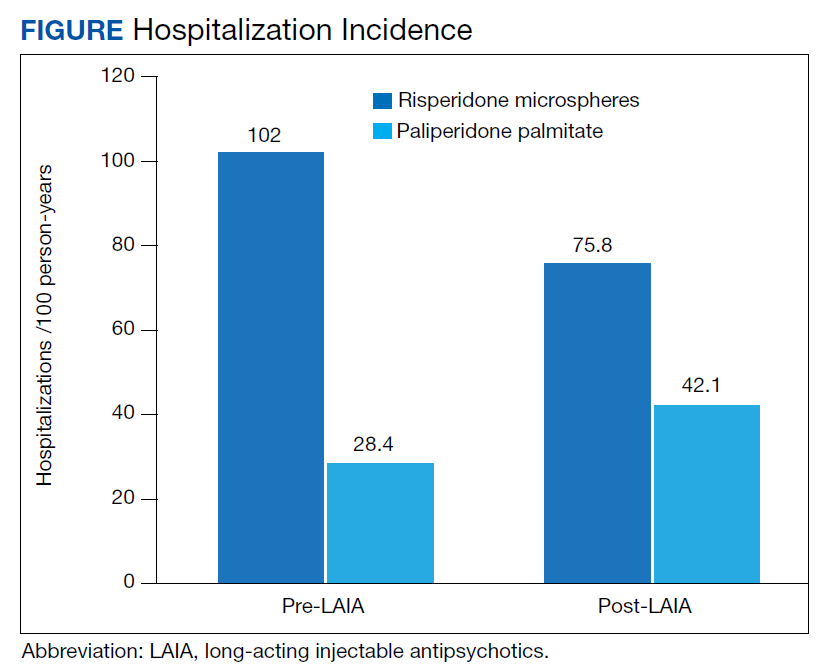
Differences in incidence per 100 person-years between pre- and post-LAIA hospitalizations were larger in risperidone microspheres users than in paliperidone palmitate (73.8 vs 33.7, P = .01) (Figure). No differences between risperidone microspheres and paliperidone palmitate were detected when looking at incidence pre-LAIA (102.2 vs 75.8, P = .22) and post-LAIA (28.4 vs 42.1, P = .38) separately.
Thirty veterans in the risperidone microspheres group discontinued LAIA: 11 were nonadherent, 5 experienced adverse effects (AEs), and 14 discontinued due to inconvenience. Among 33 veterans in the paliperidone palmitate group who discontinued the LAIA, 15 were nonadherent, 11 experienced AEs, 4 stopped due to of inconvenience, and 3 switched to a less frequently administered LAIA. The most common AEs reported were injection site reactions, cholinergic AEs (salivation, lacrimation, urination), orthostasis, and weight gain.
Discussion
The main finding of this study was that initiation of LAIAs significantly reduced hospitalizations. Veterans taking risperidone microspheres had higher index hospitalizations and lower posttreatment hospitalizations compared with paliperidone palmitate. We found that patients initiated on risperidone microspheres had more hospitalizations before use of a LAIA than those initiated on paliperidone palmitate. Risperidone microspheres reduced the number of hospitalization post-LAIA significantly more than paliperidone palmitate. We also found that veterans taking risperidone microspheres were on the medication for less mean (SD) time than those on paliperidone palmitate (41.6 [40.2] vs 58.2 [45.7] weeks; P = .04).
To our knowledge, this is one of the few studies that compared outcomes of psychiatric hospitalizations, medication adherence, and treatment discontinuation between risperidone microspheres and paliperidone palmitate, specifically in a veteran population.16-19 Limosin and colleagues aimed to compare length of stay during the initial hospitalization, rehospitalization risk, and treatment duration between risperidone microspheres and paliperidone palmitate in patients with schizophrenia.16 These researchers detected no differences in initial hospitalization duration and time to rehospitalization between risperidone microspheres and paliperidone palmitate.16 The study revealed a more favorable trend in time to discontinuation for paliperidone palmitate, but switching between LAIAs might have confounded the data.16 The authors note that their study lacked power, and patients on paliperidone palmitate had significantly more nonpsychiatric comorbidities.16 Joshi and colleagues looked at adherence, medication discontinuation, hospitalization rates, emergency department visits, and hospitalization costs between risperidone microspheres and paliperidone palmitate in patients identified in Truven MarketScan Commercial, Medicare Supplemental, and Medicaid Multi-State insurance databases.17 The authors found paliperidone palmitate to be superior in all objectives with better adherence, lower discontinuation rates, less likelihood of hospitalization, fewer emergency department visits, and lower hospitalization costs compared with risperidone microspheres.17 Korell and colleagues aimed to establish reference ranges for plasma concentrations of risperidone and paliperidone among adherent patients.18
The researchers established reference ranges for risperidone and paliperidone plasma concentrations that represented expected variability within a population and were derived from population pharmacokinetic models.18 Gopal and colleagues conducted a post hoc comparison between paliperidone palmitate and oral risperidone during initiation of long-acting injectable risperidone in patients with acute schizophrenia.19 The researchers found that during the first month after initiating long-acting injectable risperidone, paliperidone palmitate without oral supplementation had similar efficacy and safety to oral risperidone among these patients.19
LAIAs can create a steadier drug plasma concentration compared with oral antipsychotics and do not need to be taken daily. These agents improve adherence by reducing the frequency of medication administrations.20-24 Assessing nonadherence is easier with LAIAs by counting missed injections compared with oral antipsychotics that require calculation of percentage of days covered.25
The results in our study are somewhat unexpected in part because of the close relationship between risperidone and paliperidone. Risperidone is converted to paliperidone (9-OH-risperidone) via hepatic cytochrome P450 2D6. Although the molecules do not have identical pharmacologic profiles, it is accepted that they are similar enough that risperidone can establish oral tolerability when transitioning therapy to paliperidone palmitate and vice versa.24 Although the active moiety in risperidone microspheres and paliperidone palmitate is similar, the dosing interval for risperidone microspheres is 2 weeks compared with 4 weeks with paliperidone palmitate. One potential explanation as to why veterans started on risperidone microspheres experienced better outcomes is because they had twice as many office visits with the health care team. Facility procedures dictate veterans receive the LAIA at an on-site clinic. During the visits, a licensed vocational nurse administers the injection and monitors the patient for 15 to 30 minutes afterward.
Despite new LAIAs coming to market, high-quality data examining potential differences in treatment outcomes among agents are limited. This is problematic for clinicians who want to optimize care by understanding how administration schedules or other aspects of LAIA use could modify treatment outcomes. Our results suggest that an advantage might exist in selecting an agent with a more frequent administration schedule, at least initially. This could allow for close monitoring and regular therapeutic contact, which could improve short-term outcomes. This conclusion is supported by meta-analyses, randomized controlled trials, and conceptual articles conducted by Wehring and colleagues, Berwaerts and colleagues, and Parellada and colleagues, respectively, who examined patients on different LAIAs and contact with health care professionals as part of their research.26-28 These researchers concluded that patients who had regular contact with a health care professional had better outcomes when initiated on a LAIA.26-28
Limitations
There are several limitations in this study. Retrospective and observational methods introduce risks of bias and confounding variables. Sample size might have limited statistical power to detect differences. Veterans might have had undocumented pre- or posthospitalizations at other institutions, which was not accounted for and lack of rehospitalization is not conclusive of a positive outcome. Institutions could improve on our study and help to fill gaps in comparative data by conducting larger analyses over longer periods and including more LAIA agents.
Conclusions
Although veterans that were administered risperidone microspheres had a shorter treatment duration, they were less likely to be rehospitalized, had a fewer mean number of post-LAIA hospitalizations, and had a larger difference in incidence in 100 person-years compared with veterans on paliperidone palmitate. Nonadherence and discontinuation rates were comparable between risperidone microspheres and paliperidone palmitate. Future studies could aim to further clarify differences in outcomes among agents or administration schedules.
1. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
2. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223. doi:10.1056/NEJMoa051688
3. Swartz MS, Stroup TS, McEvoy JP, et al. What CATIE found: results from the schizophrenia trial. Psychiatr Serv. 2008;59(5):500-506. doi:10.1176/ps.2008.59.5.500
4. Haywood TW, Kravitz HM, Grossman LS, Cavanaugh JL Jr, Davis JM, Lewis DA. Predicting the “revolving door” phenomenon among patients with schizophrenic, schizoaffective, and affective disorders. Am J Psychiatry. 1995;152(6):856-561. doi:10.1176/ajp.152.6.856
5. Morken G, Widen JH, Grawe RW. Non-adherence to antipsychotic medication, relapse and rehospitalisation in recent-onset schizophrenia. BMC Psychiatry. 2008;8:32. doi:10.1186/1471-244X-8-32
6. Weiden PJ, Kozma C, Grogg A, Locklear J. Partial compliance and risk of rehospitalization among California Medicaid patients with schizophrenia. Psychiatr Serv. 2004;55(8):886-891. doi:10.1176/appi.ps.55.8.886
7. Gilmer TP, Dolder CR, Lacro JP, et al. Adherence to treatment with antipsychotic medication and health care costs among Medicaid beneficiaries with schizophrenia. Am J Psychiatry. 2004;161(4):692-699. doi:10.1176/appi.ajp.161.4.692
8. Lafeuille MH, Dean J, Carter V, et al. Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia. Curr Med Res Opin. 2014;30(8):1643-1655. doi:10.1185/03007995.2014.915211
9. Yu W, Wagner TH, Chen S, Barnett PG. Average cost of VA rehabilitation, mental health, and long-term hospital stays. Med Care Res Rev. 2003;60(3 suppl):40S-53S. doi:10.1177/1077558703256724
10. Duncan EJ, Woolson SL, Hamer RM. Treatment compliance in veterans administration schizophrenia spectrum patients treated with risperidone long-acting injectable. Int Clin Psychopharmacol. 2012;27(5):283-290. doi:10.1097/YIC.0b013e328354b534
11. Romstadt N, Wonson E. Outcomes comparison of long-acting injectable antipsychotic initiation in treatment-naïve veterans in the inpatient versus outpatient setting. Ment Health Clin. 2018;8(1):24-27. doi:10.9740/mhc.2018.01.024
12. Dimitropoulos E, Drogemuller L, Wong K. Evaluation of concurrent oral and long-acting injectable antipsychotic prescribing at the Minneapolis Veterans Affairs Health Care System. J Clin Psychopharmacol. 2017;37(5):605-608. doi:10.1097/JCP.0000000000000755
13. Marcus SC, Zummo J, Pettit AR, Stoddard J, Doshi JA. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768. doi:10.18553/jmcp.2015.21.9.754
14. Risperdal Consta. Package insert. Janssen Pharmaceutical; 2007.
15. Invega Sustenna. Package insert. Janssen Pharmaceutical; 2009.
16. Limosin F, Belhadi D, Comet D, et al. Comparison of paliperidone palmitate and risperidone long-acting injection in schizophrenic patients: results from a multicenter retrospective cohort study in France. J Clin Psychopharmacol. 2018;38(1):19-26. doi:10.1097/JCP.0000000000000827
17. Joshi K, Pan X, Wang R, Yang E, Benson C. Healthcare resource utilization of second-generation long-acting injectable antipsychotics in schizophrenia: risperidone versus paliperidone palmitate. Curr Med Res Opin. 2016;32(11):1873-1881. doi: 10.1080/03007995.2016.1219706
18. Korell J, Green B, Remmerie B, Vermeulen A. Determination of plasma concentration reference ranges for risperidone and paliperidone. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):589-595. doi:10.1002/psp4.12217
19. Gopal S, Pandina G, Lane R, et al. A post-hoc comparison of paliperidone palmitate to oral risperidone during initiation of long-acting risperidone injection in patients with acute schizophrenia. Innov Clin Neurosci. 2011;8(8):26-33.
20. Marcus SC, Zummo J, Pettit AR, Stoddard J, Doshi JA. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768. doi:10.18553/jmcp.2015.21.9.754
21. Romstadt N, Wonson E. Outcomes comparison of long-acting injectable antipsychotic initiation in treatment-naïve veterans in the inpatient versus outpatient setting. Ment Health Clin. 2018;8(1):24-27. doi:10.9740/mhc.2018.01.024
22. Green AI, Brunette MF, Dawson R, et al. Long-acting injectable vs oral risperidone for schizophrenia and co-occurring alcohol use disorder: a randomized trial. J Clin Psychiatry. 2015;76(10):1359-1365. doi:10.4088/JCP.13m08838
23. Rezansoff SN, Moniruzzaman A, Fazel S, Procyshyn R, Somers JM. Adherence to antipsychotic medication among homeless adults in Vancouver, Canada: a 15-year retrospective cohort study. Soc Psychiatry Psychiatr Epidemiol. 2016;51(12):1623-1632. doi:10.1007/s00127-016-1259-7
24. Castillo EG, Stroup TS. Effectiveness of long-acting injectable antipsychotics: a clinical perspective. Evid Based Ment Health. 2015;18(2):36-39. doi:10.1136/eb-2015-102086
25. Marder SR. Overview of partial compliance. J Clin Psychiatry. 2003;64 (suppl 16):3-9.
26. Wehring HJ, Thedford S, Koola M, Kelly DL. Patient and health care provider perspectives on long acting injectable antipsychotics in schizophrenia and the introduction of olanzapine long-acting injection. J Cent Nerv Syst Dis. 2011;2011(3):107-123. doi:10.4137/JCNSD.S4091
27. Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2015;72(8):830-839. doi:10.1001/jamapsychiatry.2015.0241
28. Parellada E, Bioque M. Barriers to the use of long-acting injectable antipsychotics in the management of schizophrenia. CNS Drugs. 2016;30(8):689-701. doi:10.1007/s40263-016-0350-7
Medication nonadherence is common with oral antipsychotic formulations, resulting in relapse, increased morbidity, and more frequent psychiatric hospitalization.1-7 Psychiatric hospitalization and illness decompensation is costly to health care systems and leads to reduced quality of life for veterans and families.6,7 Long-acting injectable antipsychotics (LAIAs) were developed to enhance antipsychotic adherence and improve patient outcomes, including reduced psychiatric hospitalization.8-12
Little outcomes data exist comparing LAIAs, including biweekly risperidone microspheres and monthly paliperidone palmitate.10-13 Risperidone microspheres require a 3-week oral crossover and are administered every 2 weeks, whereas paliperidone palmitate does not require an oral crossover and is administered every 4 weeks. The paliperidone palmitate loading regimen replaces an oral crossover.
The primary objective of this study was to compare the number of psychiatric hospitalizations between veterans administered risperidone microspheres and those on paliperidone palmitate pre- and post-LAIA initiation. Secondary objectives were to assess rehospitalization rates between patients taking risperidone microspheres and paliperidone palmitate, reduction in pre- and posthospitalization rates with LAIAs, and medication adherence.
Methods
This observational study with a retrospective cohort design was conducted at the Veterans Affairs Loma Linda Healthcare System (VALLHS) in California. We examined veterans who were initiated on LAIAs risperidone microspheres or paliperidone palmitate from January 01, 2016 through December 31, 2018. Veterans who were aged ≥ 18 years and received ≥ 2 injections of either risperidone microspheres or paliperidone palmitate during the study period were included. Veterans were excluded if they had received < 2 doses of either LAIA, received the LAIA outside of the review period, were nonadherent to risperidone crossover if they received risperidone microspheres, or transferred their care to another facility. At VALLHS, LAIA injections are administered by a nurse, and veterans must travel to the facility to receive the injections.
Extracted patient chart elements included participant demographics; diagnoses; comorbid alcohol, nicotine, opioid, or other substance use; duration on LAIA; psychiatric hospitalizations pre- and postinitiation of the LAIA; medication adherence; and medication discontinuation based on clinician documentation and clinic orders (Table 1).
Nonadherence to LAIA was defined as missing an injection by > 3 days for risperidone microspheres and > 7 days for paliperidone palmitate. This time frame was based on pharmacokinetic information listed in the products’ package inserts.14,15 Nonadherence to oral risperidone crossover with risperidone microspheres was defined as ≤ 80% of days covered.
Data Analysis
Patient demographics were analyzed using descriptive statistics and experimental comparisons between the risperidone microspheres and paliperidone palmitate groups to assess baseline differences between groups. Psychiatric hospitalizations pre- and post-LAIA were analyzed with parallel group (between veterans–independent groups) and pre-post (within veterans–dependent groups) designs. Index hospitalizations were examined for a period equivalent to the length of time veterans were on the LAIA. Psychiatric rehospitalization rates were analyzed for patients who had index hospitalizations and were rehospitalized for any period when they were receiving the LAIA. Incidences of pre- and post-LAIA hospitalizations were calculated in 100 person-years.
Parallel-group analysis was analyzed using the χ2 and Mann-Whitney U tests. Pre-post analyses were analyzed using the Wilcoxon rank sum test. P was set at < .05 for statistical significance.
Results
We screened 111 veterans, and 97 were included in this study (risperidone microspheres, 44; paliperidone palmitate, 53). Mean (SD) age was 46 (13.8) years, 92% were male, 38% were White, 94% were diagnosed with schizophrenia or schizoaffective disorder, and 11% were homeless. Substance use was documented as 52% for nicotine products, 40% for alcohol, 31% for cannabis, 27% for methamphetamine, 7% for cocaine, and 3% for opioids. Cannabis, methamphetamine, cocaine, and opioid use were based on clinician documentation and listed as active diagnoses at the time of LAIA initiation. Statistical significance was found in index hospitalizations P = .009) and history of cocaine use disorder (6.8% vs 7.5%, P < .001).
Veterans administered risperidone microspheres had fewer mean (SD) post-LAIA hospitalizations (0.4 [1.0] vs 0.9 [1.5]; P = .02) and were less likely to be rehospitalized (22.7% vs 47.2%, P = .01) compared with paliperidone palmitate. However, veterans taking risperidone microspheres had a shorter mean (SD) treatment duration (41.6 [40.2] vs 58.2 [45.7] weeks, P = .04) compared with paliperidone palmitate, mainly because patients switched to a different LAIA or oral antipsychotic. No differences were detected in nonadherence and discontinuation between risperidone microspheres and paliperidone palmitate. All veterans in the risperidone microspheres group adhered to oral risperidone crossover with an average 87.8% days covered (Table 2).
The average maintenance dose of risperidone microspheres was 42 mg every 2 weeks and 153 mg every 4 weeks for paliperidone palmitate.
Across the sample, 84% of veterans had a previous psychiatric hospitalization, although veterans initiated on risperidone microspheres had significantly higher mean (SD) index hospitalizations than those started on paliperidone palmitate (3.2 [2.6] risperidone microspheres vs 2.1 [1.9] paliperidone palmitate, P = .009). Both groups had significant decreases in mean (SD) hospitalizations (3.2 [2.6] to 0.4 [1.0], risperidone microspheres vs 2.1 [1.9] to 0.9 [1.5] paliperidone palmitate). The risperidone microspheres group had a larger decrease in mean (SD) hospitalizations post-LAIA (2.8 [2.9] risperidone microspheres vs 1.3 [1.7] paliperidone palmitate, P = .001) (Table 3).
Differences in incidence per 100 person-years between pre- and post-LAIA hospitalizations were larger in risperidone microspheres users than in paliperidone palmitate (73.8 vs 33.7, P = .01) (Figure). No differences between risperidone microspheres and paliperidone palmitate were detected when looking at incidence pre-LAIA (102.2 vs 75.8, P = .22) and post-LAIA (28.4 vs 42.1, P = .38) separately.
Thirty veterans in the risperidone microspheres group discontinued LAIA: 11 were nonadherent, 5 experienced adverse effects (AEs), and 14 discontinued due to inconvenience. Among 33 veterans in the paliperidone palmitate group who discontinued the LAIA, 15 were nonadherent, 11 experienced AEs, 4 stopped due to of inconvenience, and 3 switched to a less frequently administered LAIA. The most common AEs reported were injection site reactions, cholinergic AEs (salivation, lacrimation, urination), orthostasis, and weight gain.
Discussion
The main finding of this study was that initiation of LAIAs significantly reduced hospitalizations. Veterans taking risperidone microspheres had higher index hospitalizations and lower posttreatment hospitalizations compared with paliperidone palmitate. We found that patients initiated on risperidone microspheres had more hospitalizations before use of a LAIA than those initiated on paliperidone palmitate. Risperidone microspheres reduced the number of hospitalization post-LAIA significantly more than paliperidone palmitate. We also found that veterans taking risperidone microspheres were on the medication for less mean (SD) time than those on paliperidone palmitate (41.6 [40.2] vs 58.2 [45.7] weeks; P = .04).
To our knowledge, this is one of the few studies that compared outcomes of psychiatric hospitalizations, medication adherence, and treatment discontinuation between risperidone microspheres and paliperidone palmitate, specifically in a veteran population.16-19 Limosin and colleagues aimed to compare length of stay during the initial hospitalization, rehospitalization risk, and treatment duration between risperidone microspheres and paliperidone palmitate in patients with schizophrenia.16 These researchers detected no differences in initial hospitalization duration and time to rehospitalization between risperidone microspheres and paliperidone palmitate.16 The study revealed a more favorable trend in time to discontinuation for paliperidone palmitate, but switching between LAIAs might have confounded the data.16 The authors note that their study lacked power, and patients on paliperidone palmitate had significantly more nonpsychiatric comorbidities.16 Joshi and colleagues looked at adherence, medication discontinuation, hospitalization rates, emergency department visits, and hospitalization costs between risperidone microspheres and paliperidone palmitate in patients identified in Truven MarketScan Commercial, Medicare Supplemental, and Medicaid Multi-State insurance databases.17 The authors found paliperidone palmitate to be superior in all objectives with better adherence, lower discontinuation rates, less likelihood of hospitalization, fewer emergency department visits, and lower hospitalization costs compared with risperidone microspheres.17 Korell and colleagues aimed to establish reference ranges for plasma concentrations of risperidone and paliperidone among adherent patients.18
The researchers established reference ranges for risperidone and paliperidone plasma concentrations that represented expected variability within a population and were derived from population pharmacokinetic models.18 Gopal and colleagues conducted a post hoc comparison between paliperidone palmitate and oral risperidone during initiation of long-acting injectable risperidone in patients with acute schizophrenia.19 The researchers found that during the first month after initiating long-acting injectable risperidone, paliperidone palmitate without oral supplementation had similar efficacy and safety to oral risperidone among these patients.19
LAIAs can create a steadier drug plasma concentration compared with oral antipsychotics and do not need to be taken daily. These agents improve adherence by reducing the frequency of medication administrations.20-24 Assessing nonadherence is easier with LAIAs by counting missed injections compared with oral antipsychotics that require calculation of percentage of days covered.25
The results in our study are somewhat unexpected in part because of the close relationship between risperidone and paliperidone. Risperidone is converted to paliperidone (9-OH-risperidone) via hepatic cytochrome P450 2D6. Although the molecules do not have identical pharmacologic profiles, it is accepted that they are similar enough that risperidone can establish oral tolerability when transitioning therapy to paliperidone palmitate and vice versa.24 Although the active moiety in risperidone microspheres and paliperidone palmitate is similar, the dosing interval for risperidone microspheres is 2 weeks compared with 4 weeks with paliperidone palmitate. One potential explanation as to why veterans started on risperidone microspheres experienced better outcomes is because they had twice as many office visits with the health care team. Facility procedures dictate veterans receive the LAIA at an on-site clinic. During the visits, a licensed vocational nurse administers the injection and monitors the patient for 15 to 30 minutes afterward.
Despite new LAIAs coming to market, high-quality data examining potential differences in treatment outcomes among agents are limited. This is problematic for clinicians who want to optimize care by understanding how administration schedules or other aspects of LAIA use could modify treatment outcomes. Our results suggest that an advantage might exist in selecting an agent with a more frequent administration schedule, at least initially. This could allow for close monitoring and regular therapeutic contact, which could improve short-term outcomes. This conclusion is supported by meta-analyses, randomized controlled trials, and conceptual articles conducted by Wehring and colleagues, Berwaerts and colleagues, and Parellada and colleagues, respectively, who examined patients on different LAIAs and contact with health care professionals as part of their research.26-28 These researchers concluded that patients who had regular contact with a health care professional had better outcomes when initiated on a LAIA.26-28
Limitations
There are several limitations in this study. Retrospective and observational methods introduce risks of bias and confounding variables. Sample size might have limited statistical power to detect differences. Veterans might have had undocumented pre- or posthospitalizations at other institutions, which was not accounted for and lack of rehospitalization is not conclusive of a positive outcome. Institutions could improve on our study and help to fill gaps in comparative data by conducting larger analyses over longer periods and including more LAIA agents.
Conclusions
Although veterans that were administered risperidone microspheres had a shorter treatment duration, they were less likely to be rehospitalized, had a fewer mean number of post-LAIA hospitalizations, and had a larger difference in incidence in 100 person-years compared with veterans on paliperidone palmitate. Nonadherence and discontinuation rates were comparable between risperidone microspheres and paliperidone palmitate. Future studies could aim to further clarify differences in outcomes among agents or administration schedules.
Medication nonadherence is common with oral antipsychotic formulations, resulting in relapse, increased morbidity, and more frequent psychiatric hospitalization.1-7 Psychiatric hospitalization and illness decompensation is costly to health care systems and leads to reduced quality of life for veterans and families.6,7 Long-acting injectable antipsychotics (LAIAs) were developed to enhance antipsychotic adherence and improve patient outcomes, including reduced psychiatric hospitalization.8-12
Little outcomes data exist comparing LAIAs, including biweekly risperidone microspheres and monthly paliperidone palmitate.10-13 Risperidone microspheres require a 3-week oral crossover and are administered every 2 weeks, whereas paliperidone palmitate does not require an oral crossover and is administered every 4 weeks. The paliperidone palmitate loading regimen replaces an oral crossover.
The primary objective of this study was to compare the number of psychiatric hospitalizations between veterans administered risperidone microspheres and those on paliperidone palmitate pre- and post-LAIA initiation. Secondary objectives were to assess rehospitalization rates between patients taking risperidone microspheres and paliperidone palmitate, reduction in pre- and posthospitalization rates with LAIAs, and medication adherence.
Methods
This observational study with a retrospective cohort design was conducted at the Veterans Affairs Loma Linda Healthcare System (VALLHS) in California. We examined veterans who were initiated on LAIAs risperidone microspheres or paliperidone palmitate from January 01, 2016 through December 31, 2018. Veterans who were aged ≥ 18 years and received ≥ 2 injections of either risperidone microspheres or paliperidone palmitate during the study period were included. Veterans were excluded if they had received < 2 doses of either LAIA, received the LAIA outside of the review period, were nonadherent to risperidone crossover if they received risperidone microspheres, or transferred their care to another facility. At VALLHS, LAIA injections are administered by a nurse, and veterans must travel to the facility to receive the injections.
Extracted patient chart elements included participant demographics; diagnoses; comorbid alcohol, nicotine, opioid, or other substance use; duration on LAIA; psychiatric hospitalizations pre- and postinitiation of the LAIA; medication adherence; and medication discontinuation based on clinician documentation and clinic orders (Table 1).
Nonadherence to LAIA was defined as missing an injection by > 3 days for risperidone microspheres and > 7 days for paliperidone palmitate. This time frame was based on pharmacokinetic information listed in the products’ package inserts.14,15 Nonadherence to oral risperidone crossover with risperidone microspheres was defined as ≤ 80% of days covered.
Data Analysis
Patient demographics were analyzed using descriptive statistics and experimental comparisons between the risperidone microspheres and paliperidone palmitate groups to assess baseline differences between groups. Psychiatric hospitalizations pre- and post-LAIA were analyzed with parallel group (between veterans–independent groups) and pre-post (within veterans–dependent groups) designs. Index hospitalizations were examined for a period equivalent to the length of time veterans were on the LAIA. Psychiatric rehospitalization rates were analyzed for patients who had index hospitalizations and were rehospitalized for any period when they were receiving the LAIA. Incidences of pre- and post-LAIA hospitalizations were calculated in 100 person-years.
Parallel-group analysis was analyzed using the χ2 and Mann-Whitney U tests. Pre-post analyses were analyzed using the Wilcoxon rank sum test. P was set at < .05 for statistical significance.
Results
We screened 111 veterans, and 97 were included in this study (risperidone microspheres, 44; paliperidone palmitate, 53). Mean (SD) age was 46 (13.8) years, 92% were male, 38% were White, 94% were diagnosed with schizophrenia or schizoaffective disorder, and 11% were homeless. Substance use was documented as 52% for nicotine products, 40% for alcohol, 31% for cannabis, 27% for methamphetamine, 7% for cocaine, and 3% for opioids. Cannabis, methamphetamine, cocaine, and opioid use were based on clinician documentation and listed as active diagnoses at the time of LAIA initiation. Statistical significance was found in index hospitalizations P = .009) and history of cocaine use disorder (6.8% vs 7.5%, P < .001).
Veterans administered risperidone microspheres had fewer mean (SD) post-LAIA hospitalizations (0.4 [1.0] vs 0.9 [1.5]; P = .02) and were less likely to be rehospitalized (22.7% vs 47.2%, P = .01) compared with paliperidone palmitate. However, veterans taking risperidone microspheres had a shorter mean (SD) treatment duration (41.6 [40.2] vs 58.2 [45.7] weeks, P = .04) compared with paliperidone palmitate, mainly because patients switched to a different LAIA or oral antipsychotic. No differences were detected in nonadherence and discontinuation between risperidone microspheres and paliperidone palmitate. All veterans in the risperidone microspheres group adhered to oral risperidone crossover with an average 87.8% days covered (Table 2).
The average maintenance dose of risperidone microspheres was 42 mg every 2 weeks and 153 mg every 4 weeks for paliperidone palmitate.
Across the sample, 84% of veterans had a previous psychiatric hospitalization, although veterans initiated on risperidone microspheres had significantly higher mean (SD) index hospitalizations than those started on paliperidone palmitate (3.2 [2.6] risperidone microspheres vs 2.1 [1.9] paliperidone palmitate, P = .009). Both groups had significant decreases in mean (SD) hospitalizations (3.2 [2.6] to 0.4 [1.0], risperidone microspheres vs 2.1 [1.9] to 0.9 [1.5] paliperidone palmitate). The risperidone microspheres group had a larger decrease in mean (SD) hospitalizations post-LAIA (2.8 [2.9] risperidone microspheres vs 1.3 [1.7] paliperidone palmitate, P = .001) (Table 3).
Differences in incidence per 100 person-years between pre- and post-LAIA hospitalizations were larger in risperidone microspheres users than in paliperidone palmitate (73.8 vs 33.7, P = .01) (Figure). No differences between risperidone microspheres and paliperidone palmitate were detected when looking at incidence pre-LAIA (102.2 vs 75.8, P = .22) and post-LAIA (28.4 vs 42.1, P = .38) separately.
Thirty veterans in the risperidone microspheres group discontinued LAIA: 11 were nonadherent, 5 experienced adverse effects (AEs), and 14 discontinued due to inconvenience. Among 33 veterans in the paliperidone palmitate group who discontinued the LAIA, 15 were nonadherent, 11 experienced AEs, 4 stopped due to of inconvenience, and 3 switched to a less frequently administered LAIA. The most common AEs reported were injection site reactions, cholinergic AEs (salivation, lacrimation, urination), orthostasis, and weight gain.
Discussion
The main finding of this study was that initiation of LAIAs significantly reduced hospitalizations. Veterans taking risperidone microspheres had higher index hospitalizations and lower posttreatment hospitalizations compared with paliperidone palmitate. We found that patients initiated on risperidone microspheres had more hospitalizations before use of a LAIA than those initiated on paliperidone palmitate. Risperidone microspheres reduced the number of hospitalization post-LAIA significantly more than paliperidone palmitate. We also found that veterans taking risperidone microspheres were on the medication for less mean (SD) time than those on paliperidone palmitate (41.6 [40.2] vs 58.2 [45.7] weeks; P = .04).
To our knowledge, this is one of the few studies that compared outcomes of psychiatric hospitalizations, medication adherence, and treatment discontinuation between risperidone microspheres and paliperidone palmitate, specifically in a veteran population.16-19 Limosin and colleagues aimed to compare length of stay during the initial hospitalization, rehospitalization risk, and treatment duration between risperidone microspheres and paliperidone palmitate in patients with schizophrenia.16 These researchers detected no differences in initial hospitalization duration and time to rehospitalization between risperidone microspheres and paliperidone palmitate.16 The study revealed a more favorable trend in time to discontinuation for paliperidone palmitate, but switching between LAIAs might have confounded the data.16 The authors note that their study lacked power, and patients on paliperidone palmitate had significantly more nonpsychiatric comorbidities.16 Joshi and colleagues looked at adherence, medication discontinuation, hospitalization rates, emergency department visits, and hospitalization costs between risperidone microspheres and paliperidone palmitate in patients identified in Truven MarketScan Commercial, Medicare Supplemental, and Medicaid Multi-State insurance databases.17 The authors found paliperidone palmitate to be superior in all objectives with better adherence, lower discontinuation rates, less likelihood of hospitalization, fewer emergency department visits, and lower hospitalization costs compared with risperidone microspheres.17 Korell and colleagues aimed to establish reference ranges for plasma concentrations of risperidone and paliperidone among adherent patients.18
The researchers established reference ranges for risperidone and paliperidone plasma concentrations that represented expected variability within a population and were derived from population pharmacokinetic models.18 Gopal and colleagues conducted a post hoc comparison between paliperidone palmitate and oral risperidone during initiation of long-acting injectable risperidone in patients with acute schizophrenia.19 The researchers found that during the first month after initiating long-acting injectable risperidone, paliperidone palmitate without oral supplementation had similar efficacy and safety to oral risperidone among these patients.19
LAIAs can create a steadier drug plasma concentration compared with oral antipsychotics and do not need to be taken daily. These agents improve adherence by reducing the frequency of medication administrations.20-24 Assessing nonadherence is easier with LAIAs by counting missed injections compared with oral antipsychotics that require calculation of percentage of days covered.25
The results in our study are somewhat unexpected in part because of the close relationship between risperidone and paliperidone. Risperidone is converted to paliperidone (9-OH-risperidone) via hepatic cytochrome P450 2D6. Although the molecules do not have identical pharmacologic profiles, it is accepted that they are similar enough that risperidone can establish oral tolerability when transitioning therapy to paliperidone palmitate and vice versa.24 Although the active moiety in risperidone microspheres and paliperidone palmitate is similar, the dosing interval for risperidone microspheres is 2 weeks compared with 4 weeks with paliperidone palmitate. One potential explanation as to why veterans started on risperidone microspheres experienced better outcomes is because they had twice as many office visits with the health care team. Facility procedures dictate veterans receive the LAIA at an on-site clinic. During the visits, a licensed vocational nurse administers the injection and monitors the patient for 15 to 30 minutes afterward.
Despite new LAIAs coming to market, high-quality data examining potential differences in treatment outcomes among agents are limited. This is problematic for clinicians who want to optimize care by understanding how administration schedules or other aspects of LAIA use could modify treatment outcomes. Our results suggest that an advantage might exist in selecting an agent with a more frequent administration schedule, at least initially. This could allow for close monitoring and regular therapeutic contact, which could improve short-term outcomes. This conclusion is supported by meta-analyses, randomized controlled trials, and conceptual articles conducted by Wehring and colleagues, Berwaerts and colleagues, and Parellada and colleagues, respectively, who examined patients on different LAIAs and contact with health care professionals as part of their research.26-28 These researchers concluded that patients who had regular contact with a health care professional had better outcomes when initiated on a LAIA.26-28
Limitations
There are several limitations in this study. Retrospective and observational methods introduce risks of bias and confounding variables. Sample size might have limited statistical power to detect differences. Veterans might have had undocumented pre- or posthospitalizations at other institutions, which was not accounted for and lack of rehospitalization is not conclusive of a positive outcome. Institutions could improve on our study and help to fill gaps in comparative data by conducting larger analyses over longer periods and including more LAIA agents.
Conclusions
Although veterans that were administered risperidone microspheres had a shorter treatment duration, they were less likely to be rehospitalized, had a fewer mean number of post-LAIA hospitalizations, and had a larger difference in incidence in 100 person-years compared with veterans on paliperidone palmitate. Nonadherence and discontinuation rates were comparable between risperidone microspheres and paliperidone palmitate. Future studies could aim to further clarify differences in outcomes among agents or administration schedules.
1. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
2. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223. doi:10.1056/NEJMoa051688
3. Swartz MS, Stroup TS, McEvoy JP, et al. What CATIE found: results from the schizophrenia trial. Psychiatr Serv. 2008;59(5):500-506. doi:10.1176/ps.2008.59.5.500
4. Haywood TW, Kravitz HM, Grossman LS, Cavanaugh JL Jr, Davis JM, Lewis DA. Predicting the “revolving door” phenomenon among patients with schizophrenic, schizoaffective, and affective disorders. Am J Psychiatry. 1995;152(6):856-561. doi:10.1176/ajp.152.6.856
5. Morken G, Widen JH, Grawe RW. Non-adherence to antipsychotic medication, relapse and rehospitalisation in recent-onset schizophrenia. BMC Psychiatry. 2008;8:32. doi:10.1186/1471-244X-8-32
6. Weiden PJ, Kozma C, Grogg A, Locklear J. Partial compliance and risk of rehospitalization among California Medicaid patients with schizophrenia. Psychiatr Serv. 2004;55(8):886-891. doi:10.1176/appi.ps.55.8.886
7. Gilmer TP, Dolder CR, Lacro JP, et al. Adherence to treatment with antipsychotic medication and health care costs among Medicaid beneficiaries with schizophrenia. Am J Psychiatry. 2004;161(4):692-699. doi:10.1176/appi.ajp.161.4.692
8. Lafeuille MH, Dean J, Carter V, et al. Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia. Curr Med Res Opin. 2014;30(8):1643-1655. doi:10.1185/03007995.2014.915211
9. Yu W, Wagner TH, Chen S, Barnett PG. Average cost of VA rehabilitation, mental health, and long-term hospital stays. Med Care Res Rev. 2003;60(3 suppl):40S-53S. doi:10.1177/1077558703256724
10. Duncan EJ, Woolson SL, Hamer RM. Treatment compliance in veterans administration schizophrenia spectrum patients treated with risperidone long-acting injectable. Int Clin Psychopharmacol. 2012;27(5):283-290. doi:10.1097/YIC.0b013e328354b534
11. Romstadt N, Wonson E. Outcomes comparison of long-acting injectable antipsychotic initiation in treatment-naïve veterans in the inpatient versus outpatient setting. Ment Health Clin. 2018;8(1):24-27. doi:10.9740/mhc.2018.01.024
12. Dimitropoulos E, Drogemuller L, Wong K. Evaluation of concurrent oral and long-acting injectable antipsychotic prescribing at the Minneapolis Veterans Affairs Health Care System. J Clin Psychopharmacol. 2017;37(5):605-608. doi:10.1097/JCP.0000000000000755
13. Marcus SC, Zummo J, Pettit AR, Stoddard J, Doshi JA. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768. doi:10.18553/jmcp.2015.21.9.754
14. Risperdal Consta. Package insert. Janssen Pharmaceutical; 2007.
15. Invega Sustenna. Package insert. Janssen Pharmaceutical; 2009.
16. Limosin F, Belhadi D, Comet D, et al. Comparison of paliperidone palmitate and risperidone long-acting injection in schizophrenic patients: results from a multicenter retrospective cohort study in France. J Clin Psychopharmacol. 2018;38(1):19-26. doi:10.1097/JCP.0000000000000827
17. Joshi K, Pan X, Wang R, Yang E, Benson C. Healthcare resource utilization of second-generation long-acting injectable antipsychotics in schizophrenia: risperidone versus paliperidone palmitate. Curr Med Res Opin. 2016;32(11):1873-1881. doi: 10.1080/03007995.2016.1219706
18. Korell J, Green B, Remmerie B, Vermeulen A. Determination of plasma concentration reference ranges for risperidone and paliperidone. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):589-595. doi:10.1002/psp4.12217
19. Gopal S, Pandina G, Lane R, et al. A post-hoc comparison of paliperidone palmitate to oral risperidone during initiation of long-acting risperidone injection in patients with acute schizophrenia. Innov Clin Neurosci. 2011;8(8):26-33.
20. Marcus SC, Zummo J, Pettit AR, Stoddard J, Doshi JA. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768. doi:10.18553/jmcp.2015.21.9.754
21. Romstadt N, Wonson E. Outcomes comparison of long-acting injectable antipsychotic initiation in treatment-naïve veterans in the inpatient versus outpatient setting. Ment Health Clin. 2018;8(1):24-27. doi:10.9740/mhc.2018.01.024
22. Green AI, Brunette MF, Dawson R, et al. Long-acting injectable vs oral risperidone for schizophrenia and co-occurring alcohol use disorder: a randomized trial. J Clin Psychiatry. 2015;76(10):1359-1365. doi:10.4088/JCP.13m08838
23. Rezansoff SN, Moniruzzaman A, Fazel S, Procyshyn R, Somers JM. Adherence to antipsychotic medication among homeless adults in Vancouver, Canada: a 15-year retrospective cohort study. Soc Psychiatry Psychiatr Epidemiol. 2016;51(12):1623-1632. doi:10.1007/s00127-016-1259-7
24. Castillo EG, Stroup TS. Effectiveness of long-acting injectable antipsychotics: a clinical perspective. Evid Based Ment Health. 2015;18(2):36-39. doi:10.1136/eb-2015-102086
25. Marder SR. Overview of partial compliance. J Clin Psychiatry. 2003;64 (suppl 16):3-9.
26. Wehring HJ, Thedford S, Koola M, Kelly DL. Patient and health care provider perspectives on long acting injectable antipsychotics in schizophrenia and the introduction of olanzapine long-acting injection. J Cent Nerv Syst Dis. 2011;2011(3):107-123. doi:10.4137/JCNSD.S4091
27. Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2015;72(8):830-839. doi:10.1001/jamapsychiatry.2015.0241
28. Parellada E, Bioque M. Barriers to the use of long-acting injectable antipsychotics in the management of schizophrenia. CNS Drugs. 2016;30(8):689-701. doi:10.1007/s40263-016-0350-7
1. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
2. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223. doi:10.1056/NEJMoa051688
3. Swartz MS, Stroup TS, McEvoy JP, et al. What CATIE found: results from the schizophrenia trial. Psychiatr Serv. 2008;59(5):500-506. doi:10.1176/ps.2008.59.5.500
4. Haywood TW, Kravitz HM, Grossman LS, Cavanaugh JL Jr, Davis JM, Lewis DA. Predicting the “revolving door” phenomenon among patients with schizophrenic, schizoaffective, and affective disorders. Am J Psychiatry. 1995;152(6):856-561. doi:10.1176/ajp.152.6.856
5. Morken G, Widen JH, Grawe RW. Non-adherence to antipsychotic medication, relapse and rehospitalisation in recent-onset schizophrenia. BMC Psychiatry. 2008;8:32. doi:10.1186/1471-244X-8-32
6. Weiden PJ, Kozma C, Grogg A, Locklear J. Partial compliance and risk of rehospitalization among California Medicaid patients with schizophrenia. Psychiatr Serv. 2004;55(8):886-891. doi:10.1176/appi.ps.55.8.886
7. Gilmer TP, Dolder CR, Lacro JP, et al. Adherence to treatment with antipsychotic medication and health care costs among Medicaid beneficiaries with schizophrenia. Am J Psychiatry. 2004;161(4):692-699. doi:10.1176/appi.ajp.161.4.692
8. Lafeuille MH, Dean J, Carter V, et al. Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia. Curr Med Res Opin. 2014;30(8):1643-1655. doi:10.1185/03007995.2014.915211
9. Yu W, Wagner TH, Chen S, Barnett PG. Average cost of VA rehabilitation, mental health, and long-term hospital stays. Med Care Res Rev. 2003;60(3 suppl):40S-53S. doi:10.1177/1077558703256724
10. Duncan EJ, Woolson SL, Hamer RM. Treatment compliance in veterans administration schizophrenia spectrum patients treated with risperidone long-acting injectable. Int Clin Psychopharmacol. 2012;27(5):283-290. doi:10.1097/YIC.0b013e328354b534
11. Romstadt N, Wonson E. Outcomes comparison of long-acting injectable antipsychotic initiation in treatment-naïve veterans in the inpatient versus outpatient setting. Ment Health Clin. 2018;8(1):24-27. doi:10.9740/mhc.2018.01.024
12. Dimitropoulos E, Drogemuller L, Wong K. Evaluation of concurrent oral and long-acting injectable antipsychotic prescribing at the Minneapolis Veterans Affairs Health Care System. J Clin Psychopharmacol. 2017;37(5):605-608. doi:10.1097/JCP.0000000000000755
13. Marcus SC, Zummo J, Pettit AR, Stoddard J, Doshi JA. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768. doi:10.18553/jmcp.2015.21.9.754
14. Risperdal Consta. Package insert. Janssen Pharmaceutical; 2007.
15. Invega Sustenna. Package insert. Janssen Pharmaceutical; 2009.
16. Limosin F, Belhadi D, Comet D, et al. Comparison of paliperidone palmitate and risperidone long-acting injection in schizophrenic patients: results from a multicenter retrospective cohort study in France. J Clin Psychopharmacol. 2018;38(1):19-26. doi:10.1097/JCP.0000000000000827
17. Joshi K, Pan X, Wang R, Yang E, Benson C. Healthcare resource utilization of second-generation long-acting injectable antipsychotics in schizophrenia: risperidone versus paliperidone palmitate. Curr Med Res Opin. 2016;32(11):1873-1881. doi: 10.1080/03007995.2016.1219706
18. Korell J, Green B, Remmerie B, Vermeulen A. Determination of plasma concentration reference ranges for risperidone and paliperidone. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):589-595. doi:10.1002/psp4.12217
19. Gopal S, Pandina G, Lane R, et al. A post-hoc comparison of paliperidone palmitate to oral risperidone during initiation of long-acting risperidone injection in patients with acute schizophrenia. Innov Clin Neurosci. 2011;8(8):26-33.
20. Marcus SC, Zummo J, Pettit AR, Stoddard J, Doshi JA. Antipsychotic adherence and rehospitalization in schizophrenia patients receiving oral versus long-acting injectable antipsychotics following hospital discharge. J Manag Care Spec Pharm. 2015;21(9):754-768. doi:10.18553/jmcp.2015.21.9.754
21. Romstadt N, Wonson E. Outcomes comparison of long-acting injectable antipsychotic initiation in treatment-naïve veterans in the inpatient versus outpatient setting. Ment Health Clin. 2018;8(1):24-27. doi:10.9740/mhc.2018.01.024
22. Green AI, Brunette MF, Dawson R, et al. Long-acting injectable vs oral risperidone for schizophrenia and co-occurring alcohol use disorder: a randomized trial. J Clin Psychiatry. 2015;76(10):1359-1365. doi:10.4088/JCP.13m08838
23. Rezansoff SN, Moniruzzaman A, Fazel S, Procyshyn R, Somers JM. Adherence to antipsychotic medication among homeless adults in Vancouver, Canada: a 15-year retrospective cohort study. Soc Psychiatry Psychiatr Epidemiol. 2016;51(12):1623-1632. doi:10.1007/s00127-016-1259-7
24. Castillo EG, Stroup TS. Effectiveness of long-acting injectable antipsychotics: a clinical perspective. Evid Based Ment Health. 2015;18(2):36-39. doi:10.1136/eb-2015-102086
25. Marder SR. Overview of partial compliance. J Clin Psychiatry. 2003;64 (suppl 16):3-9.
26. Wehring HJ, Thedford S, Koola M, Kelly DL. Patient and health care provider perspectives on long acting injectable antipsychotics in schizophrenia and the introduction of olanzapine long-acting injection. J Cent Nerv Syst Dis. 2011;2011(3):107-123. doi:10.4137/JCNSD.S4091
27. Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2015;72(8):830-839. doi:10.1001/jamapsychiatry.2015.0241
28. Parellada E, Bioque M. Barriers to the use of long-acting injectable antipsychotics in the management of schizophrenia. CNS Drugs. 2016;30(8):689-701. doi:10.1007/s40263-016-0350-7
New data on rare myocarditis after COVID-19 vaccination
Adolescents and adults younger than age 21 who develop myocarditis after mRNA COVID-19 vaccination frequently have abnormal findings on cardiac MRI (cMRI) but most have a mild clinical course with rapid resolution of symptoms, a new study concludes.

“This study supports what we’ve been seeing. People identified and treated early and appropriately for the rare complication of COVID-19 vaccine-related myocarditis typically experienced only mild cases and short recovery times,” American Heart Association President Donald M. Lloyd-Jones, MD, said in a podcast.
“Overwhelmingly, the data continue to indicate [that] the benefits of COVID-19 vaccine far outweigh any very rare risks of adverse events from the vaccine, including myocarditis,” Dr. Lloyd-Jones added.
The study was published online Dec. 6 in Circulation.
Using data from 26 pediatric medical centers across the United States and Canada, the researchers reviewed the medical records of 139 patients younger than 21 with suspected myocarditis within 1 month of receiving a COVID-19 vaccination.
They made the following key observations:
- Most patients were male (90.6%), White (66.2%) and with a median age of 15.8 years.
- Suspected myocarditis occurred in 136 patients (97.8%) following mRNA vaccine, with 131 (94.2%) following the Pfizer-BioNTech vaccine; 128 cases (91.4%) occurred after the second dose.
- Symptoms started a median of 2 days (range 0 to 22 days) following vaccination administration.
- Chest pain was the most common symptom (99.3%), with fever present in 30.9% of patients and shortness of breath in 27.3%.
- Patients were treated with nonsteroidal anti-inflammatory drugs (81.3%), intravenous immunoglobulin (21.6%), glucocorticoids (21.6%), colchicine (7.9%) or no anti-inflammatory therapies (8.6%).
- Twenty-six patients (18.7%) were admitted to the intensive care unit; 2 received inotropic/vasoactive support; none required extracorporeal membrane oxygenation or died.
- Median time spent in the hospital was 2 days.
- A total of 111 patients had elevated troponin I (8.12 ng/mL) and 28 had elevated troponin T (0.61 ng/mL).
- More than two-thirds (69.8%) had abnormal electrocardiograms and/or arrhythmias (7 with nonsustained ventricular tachycardia).
- Twenty-six patients (18.7%) had left ventricular ejection fraction (LVEF) less than 55% on echocardiogram; LVEF had returned to normal in the 25 who returned for follow-up.
- 75 of 97 patients (77.3%) who underwent cMRI at a median of 5 days from symptom onset had abnormal findings; 74 (76.3%) had late gadolinium enhancement, 54 (55.7%) had myocardial edema, and 49 (50.5%) met Lake Louise criteria for myocarditis.
“These data suggest that most cases of suspected COVID-19 vaccine–related myocarditis in people younger than 21 are mild and resolve quickly,” corresponding author Dongngan Truong, MD, Division of Pediatric Cardiology, University of Utah and Primary Children’s Hospital, Salt Lake City, said in a statement.
“We were very happy to see that type of recovery. However, we are awaiting further studies to better understand the long-term outcomes of patients who have had COVID-19 vaccination-related myocarditis. We also need to study the risk factors and mechanisms for this rare complication,” Dr. Truong added.
Dr. Lloyd-Jones said these findings support the AHA’s position that COVID-19 vaccines are “safe, highly effective, and fundamental to saving lives, protecting our families and communities against COVID-19, and ending the pandemic.”
The study received no funding. Dr. Truong consults for Pfizer on vaccine-associated myocarditis. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.
Adolescents and adults younger than age 21 who develop myocarditis after mRNA COVID-19 vaccination frequently have abnormal findings on cardiac MRI (cMRI) but most have a mild clinical course with rapid resolution of symptoms, a new study concludes.
“This study supports what we’ve been seeing. People identified and treated early and appropriately for the rare complication of COVID-19 vaccine-related myocarditis typically experienced only mild cases and short recovery times,” American Heart Association President Donald M. Lloyd-Jones, MD, said in a podcast.
“Overwhelmingly, the data continue to indicate [that] the benefits of COVID-19 vaccine far outweigh any very rare risks of adverse events from the vaccine, including myocarditis,” Dr. Lloyd-Jones added.
The study was published online Dec. 6 in Circulation.
Using data from 26 pediatric medical centers across the United States and Canada, the researchers reviewed the medical records of 139 patients younger than 21 with suspected myocarditis within 1 month of receiving a COVID-19 vaccination.
They made the following key observations:
- Most patients were male (90.6%), White (66.2%) and with a median age of 15.8 years.
- Suspected myocarditis occurred in 136 patients (97.8%) following mRNA vaccine, with 131 (94.2%) following the Pfizer-BioNTech vaccine; 128 cases (91.4%) occurred after the second dose.
- Symptoms started a median of 2 days (range 0 to 22 days) following vaccination administration.
- Chest pain was the most common symptom (99.3%), with fever present in 30.9% of patients and shortness of breath in 27.3%.
- Patients were treated with nonsteroidal anti-inflammatory drugs (81.3%), intravenous immunoglobulin (21.6%), glucocorticoids (21.6%), colchicine (7.9%) or no anti-inflammatory therapies (8.6%).
- Twenty-six patients (18.7%) were admitted to the intensive care unit; 2 received inotropic/vasoactive support; none required extracorporeal membrane oxygenation or died.
- Median time spent in the hospital was 2 days.
- A total of 111 patients had elevated troponin I (8.12 ng/mL) and 28 had elevated troponin T (0.61 ng/mL).
- More than two-thirds (69.8%) had abnormal electrocardiograms and/or arrhythmias (7 with nonsustained ventricular tachycardia).
- Twenty-six patients (18.7%) had left ventricular ejection fraction (LVEF) less than 55% on echocardiogram; LVEF had returned to normal in the 25 who returned for follow-up.
- 75 of 97 patients (77.3%) who underwent cMRI at a median of 5 days from symptom onset had abnormal findings; 74 (76.3%) had late gadolinium enhancement, 54 (55.7%) had myocardial edema, and 49 (50.5%) met Lake Louise criteria for myocarditis.
“These data suggest that most cases of suspected COVID-19 vaccine–related myocarditis in people younger than 21 are mild and resolve quickly,” corresponding author Dongngan Truong, MD, Division of Pediatric Cardiology, University of Utah and Primary Children’s Hospital, Salt Lake City, said in a statement.
“We were very happy to see that type of recovery. However, we are awaiting further studies to better understand the long-term outcomes of patients who have had COVID-19 vaccination-related myocarditis. We also need to study the risk factors and mechanisms for this rare complication,” Dr. Truong added.
Dr. Lloyd-Jones said these findings support the AHA’s position that COVID-19 vaccines are “safe, highly effective, and fundamental to saving lives, protecting our families and communities against COVID-19, and ending the pandemic.”
The study received no funding. Dr. Truong consults for Pfizer on vaccine-associated myocarditis. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.
Adolescents and adults younger than age 21 who develop myocarditis after mRNA COVID-19 vaccination frequently have abnormal findings on cardiac MRI (cMRI) but most have a mild clinical course with rapid resolution of symptoms, a new study concludes.
“This study supports what we’ve been seeing. People identified and treated early and appropriately for the rare complication of COVID-19 vaccine-related myocarditis typically experienced only mild cases and short recovery times,” American Heart Association President Donald M. Lloyd-Jones, MD, said in a podcast.
“Overwhelmingly, the data continue to indicate [that] the benefits of COVID-19 vaccine far outweigh any very rare risks of adverse events from the vaccine, including myocarditis,” Dr. Lloyd-Jones added.
The study was published online Dec. 6 in Circulation.
Using data from 26 pediatric medical centers across the United States and Canada, the researchers reviewed the medical records of 139 patients younger than 21 with suspected myocarditis within 1 month of receiving a COVID-19 vaccination.
They made the following key observations:
- Most patients were male (90.6%), White (66.2%) and with a median age of 15.8 years.
- Suspected myocarditis occurred in 136 patients (97.8%) following mRNA vaccine, with 131 (94.2%) following the Pfizer-BioNTech vaccine; 128 cases (91.4%) occurred after the second dose.
- Symptoms started a median of 2 days (range 0 to 22 days) following vaccination administration.
- Chest pain was the most common symptom (99.3%), with fever present in 30.9% of patients and shortness of breath in 27.3%.
- Patients were treated with nonsteroidal anti-inflammatory drugs (81.3%), intravenous immunoglobulin (21.6%), glucocorticoids (21.6%), colchicine (7.9%) or no anti-inflammatory therapies (8.6%).
- Twenty-six patients (18.7%) were admitted to the intensive care unit; 2 received inotropic/vasoactive support; none required extracorporeal membrane oxygenation or died.
- Median time spent in the hospital was 2 days.
- A total of 111 patients had elevated troponin I (8.12 ng/mL) and 28 had elevated troponin T (0.61 ng/mL).
- More than two-thirds (69.8%) had abnormal electrocardiograms and/or arrhythmias (7 with nonsustained ventricular tachycardia).
- Twenty-six patients (18.7%) had left ventricular ejection fraction (LVEF) less than 55% on echocardiogram; LVEF had returned to normal in the 25 who returned for follow-up.
- 75 of 97 patients (77.3%) who underwent cMRI at a median of 5 days from symptom onset had abnormal findings; 74 (76.3%) had late gadolinium enhancement, 54 (55.7%) had myocardial edema, and 49 (50.5%) met Lake Louise criteria for myocarditis.
“These data suggest that most cases of suspected COVID-19 vaccine–related myocarditis in people younger than 21 are mild and resolve quickly,” corresponding author Dongngan Truong, MD, Division of Pediatric Cardiology, University of Utah and Primary Children’s Hospital, Salt Lake City, said in a statement.
“We were very happy to see that type of recovery. However, we are awaiting further studies to better understand the long-term outcomes of patients who have had COVID-19 vaccination-related myocarditis. We also need to study the risk factors and mechanisms for this rare complication,” Dr. Truong added.
Dr. Lloyd-Jones said these findings support the AHA’s position that COVID-19 vaccines are “safe, highly effective, and fundamental to saving lives, protecting our families and communities against COVID-19, and ending the pandemic.”
The study received no funding. Dr. Truong consults for Pfizer on vaccine-associated myocarditis. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.
FDA expands pembrolizumab approval for advanced melanoma
The over age 12 years. The FDA also extended the approval to those with stage III disease.
The FDA approval on Dec. 3 was based on first interim findings from the randomized, placebo-controlled KEYNOTE-716 trial, which evaluated patients with stage IIB and IIC disease.
Since the anti-PD-1 therapy was approved in metastatic melanoma 7 years ago, “we have built on this foundation in melanoma and have expanded the use of KEYTRUDA into earlier stages of this disease,” said Scot Ebbinghaus, MD, vice president, clinical research, Merck Research Laboratories, in a press release. “With today’s approval, we can now offer health care providers and patients 12 years and older the opportunity to help prevent melanoma recurrence with Keytruda across resected stage IIB, stage IIC, and stage III melanoma.”
In KEYNOTE-716, patients with completely resected stage IIB or IIC melanoma were randomly assigned to receive 200 mg of intravenous pembrolizumab, the pediatric dose 2 mg/kg (up to a maximum of 200 mg) every 3 weeks, or placebo for up to 1 year until disease recurrence or unacceptable toxicity.
After a median follow-up of 14.4 months, investigators reported a statistically significant 35% improvement in recurrence-free survival (RFS) in those treated with pembrolizumab, compared with those who received placebo (hazard ratio, 0.65).
The most common adverse reactions reported in patients receiving pembrolizumab in KEYNOTE-716 were fatigue, diarrhea, pruritus, and arthralgia, each occurring in at least 20% of patients.
“Early identification and management of immune-mediated adverse reactions are essential to ensure safe use of Keytruda,” according to Merck.
A version of this article first appeared on Medscape.com.
The over age 12 years. The FDA also extended the approval to those with stage III disease.
The FDA approval on Dec. 3 was based on first interim findings from the randomized, placebo-controlled KEYNOTE-716 trial, which evaluated patients with stage IIB and IIC disease.
Since the anti-PD-1 therapy was approved in metastatic melanoma 7 years ago, “we have built on this foundation in melanoma and have expanded the use of KEYTRUDA into earlier stages of this disease,” said Scot Ebbinghaus, MD, vice president, clinical research, Merck Research Laboratories, in a press release. “With today’s approval, we can now offer health care providers and patients 12 years and older the opportunity to help prevent melanoma recurrence with Keytruda across resected stage IIB, stage IIC, and stage III melanoma.”
In KEYNOTE-716, patients with completely resected stage IIB or IIC melanoma were randomly assigned to receive 200 mg of intravenous pembrolizumab, the pediatric dose 2 mg/kg (up to a maximum of 200 mg) every 3 weeks, or placebo for up to 1 year until disease recurrence or unacceptable toxicity.
After a median follow-up of 14.4 months, investigators reported a statistically significant 35% improvement in recurrence-free survival (RFS) in those treated with pembrolizumab, compared with those who received placebo (hazard ratio, 0.65).
The most common adverse reactions reported in patients receiving pembrolizumab in KEYNOTE-716 were fatigue, diarrhea, pruritus, and arthralgia, each occurring in at least 20% of patients.
“Early identification and management of immune-mediated adverse reactions are essential to ensure safe use of Keytruda,” according to Merck.
A version of this article first appeared on Medscape.com.
The over age 12 years. The FDA also extended the approval to those with stage III disease.
The FDA approval on Dec. 3 was based on first interim findings from the randomized, placebo-controlled KEYNOTE-716 trial, which evaluated patients with stage IIB and IIC disease.
Since the anti-PD-1 therapy was approved in metastatic melanoma 7 years ago, “we have built on this foundation in melanoma and have expanded the use of KEYTRUDA into earlier stages of this disease,” said Scot Ebbinghaus, MD, vice president, clinical research, Merck Research Laboratories, in a press release. “With today’s approval, we can now offer health care providers and patients 12 years and older the opportunity to help prevent melanoma recurrence with Keytruda across resected stage IIB, stage IIC, and stage III melanoma.”
In KEYNOTE-716, patients with completely resected stage IIB or IIC melanoma were randomly assigned to receive 200 mg of intravenous pembrolizumab, the pediatric dose 2 mg/kg (up to a maximum of 200 mg) every 3 weeks, or placebo for up to 1 year until disease recurrence or unacceptable toxicity.
After a median follow-up of 14.4 months, investigators reported a statistically significant 35% improvement in recurrence-free survival (RFS) in those treated with pembrolizumab, compared with those who received placebo (hazard ratio, 0.65).
The most common adverse reactions reported in patients receiving pembrolizumab in KEYNOTE-716 were fatigue, diarrhea, pruritus, and arthralgia, each occurring in at least 20% of patients.
“Early identification and management of immune-mediated adverse reactions are essential to ensure safe use of Keytruda,” according to Merck.
A version of this article first appeared on Medscape.com.
Could Viagra help prevent Alzheimer’s?
published in the journal Nature Aging.
Patients who used the drug sildenafil, the generic name for Viagra, were 69% less likely to develop the disease than were nonusers.
“Sildenafil, which has been shown to significantly improve cognition and memory in preclinical models, presented as the best drug candidate,” Feixiong Cheng, PhD, the lead study author in the Cleveland Clinic’s Genomic Medicine Institute, said in a statement.
“Notably, we found that sildenafil use reduced the likelihood of Alzheimer’s in individuals with coronary artery disease, hypertension, and type 2 diabetes, all of which are comorbidities significantly associated with risk of the disease, as well as in those without,” he said.
Alzheimer’s, which is the most common form of age-related dementia, affects hundreds of millions of people worldwide. The disease is expected to affect nearly 14 million Americans by 2050. There is no approved treatment for it.
Dr. Cheng and colleagues at the Cleveland Clinic used a large gene-mapping network to analyze whether more than 1,600 Food and Drug Administration–approved drugs could work against Alzheimer’s. They gave higher scores to drugs that target both amyloid and tau proteins in the brain, which are two hallmarks of the disease. Sildenafil appeared at the top of the list.
Then the researchers used a database of health insurance claims for more than 7 million people in the U.S. to understand the relationship between sildenafil and Alzheimer’s disease outcomes. They compared sildenafil users to nonusers and found that those who used the drug were 69% less likely to have the neurodegenerative disease, even after 6 years of follow-up.
After that, the research team came up with a lab model that showed the sildenafil increased brain cell growth and targeted tau proteins. The lab model could indicate how the drug influences disease-related brain changes.
But Dr. Cheng cautioned against drawing strong conclusions. The study doesn’t demonstrate a causal relationship between sildenafil and Alzheimer’s disease. Researchers will need to conduct clinical trials with a placebo control to see how well the drug works.
Other researchers said the findings offer a new avenue for research but don’t yet provide solid answers.
“Being able to repurpose a drug already licensed for health conditions could help speed up the drug discovery process and bring about life-changing dementia treatments sooner,” Susan Kohlhaas, PhD, director of research at Alzheimer’s Research UK, told the Science Media Centre.
“Importantly, this research doesn’t prove that sildenafil is responsible for reducing dementia risk, or that it slows or stops the disease,” she continued. “If you want to discuss any treatments you are receiving, the first port of call is to speak to your doctor.”
And doctors won’t likely recommend it as a treatment just yet either.
“While these data are interesting scientifically, based on this study, I would not rush out to start taking sildenafil as a prevention for Alzheimer’s disease,” Tara Spires-Jones, PhD, deputy director of the Centre for Discovery Brain Sciences at the University of Edinburgh, told the Science Media Centre.
A version of this article first appeared on WebMD.com.
published in the journal Nature Aging.
Patients who used the drug sildenafil, the generic name for Viagra, were 69% less likely to develop the disease than were nonusers.
“Sildenafil, which has been shown to significantly improve cognition and memory in preclinical models, presented as the best drug candidate,” Feixiong Cheng, PhD, the lead study author in the Cleveland Clinic’s Genomic Medicine Institute, said in a statement.
“Notably, we found that sildenafil use reduced the likelihood of Alzheimer’s in individuals with coronary artery disease, hypertension, and type 2 diabetes, all of which are comorbidities significantly associated with risk of the disease, as well as in those without,” he said.
Alzheimer’s, which is the most common form of age-related dementia, affects hundreds of millions of people worldwide. The disease is expected to affect nearly 14 million Americans by 2050. There is no approved treatment for it.
Dr. Cheng and colleagues at the Cleveland Clinic used a large gene-mapping network to analyze whether more than 1,600 Food and Drug Administration–approved drugs could work against Alzheimer’s. They gave higher scores to drugs that target both amyloid and tau proteins in the brain, which are two hallmarks of the disease. Sildenafil appeared at the top of the list.
Then the researchers used a database of health insurance claims for more than 7 million people in the U.S. to understand the relationship between sildenafil and Alzheimer’s disease outcomes. They compared sildenafil users to nonusers and found that those who used the drug were 69% less likely to have the neurodegenerative disease, even after 6 years of follow-up.
After that, the research team came up with a lab model that showed the sildenafil increased brain cell growth and targeted tau proteins. The lab model could indicate how the drug influences disease-related brain changes.
But Dr. Cheng cautioned against drawing strong conclusions. The study doesn’t demonstrate a causal relationship between sildenafil and Alzheimer’s disease. Researchers will need to conduct clinical trials with a placebo control to see how well the drug works.
Other researchers said the findings offer a new avenue for research but don’t yet provide solid answers.
“Being able to repurpose a drug already licensed for health conditions could help speed up the drug discovery process and bring about life-changing dementia treatments sooner,” Susan Kohlhaas, PhD, director of research at Alzheimer’s Research UK, told the Science Media Centre.
“Importantly, this research doesn’t prove that sildenafil is responsible for reducing dementia risk, or that it slows or stops the disease,” she continued. “If you want to discuss any treatments you are receiving, the first port of call is to speak to your doctor.”
And doctors won’t likely recommend it as a treatment just yet either.
“While these data are interesting scientifically, based on this study, I would not rush out to start taking sildenafil as a prevention for Alzheimer’s disease,” Tara Spires-Jones, PhD, deputy director of the Centre for Discovery Brain Sciences at the University of Edinburgh, told the Science Media Centre.
A version of this article first appeared on WebMD.com.
published in the journal Nature Aging.
Patients who used the drug sildenafil, the generic name for Viagra, were 69% less likely to develop the disease than were nonusers.
“Sildenafil, which has been shown to significantly improve cognition and memory in preclinical models, presented as the best drug candidate,” Feixiong Cheng, PhD, the lead study author in the Cleveland Clinic’s Genomic Medicine Institute, said in a statement.
“Notably, we found that sildenafil use reduced the likelihood of Alzheimer’s in individuals with coronary artery disease, hypertension, and type 2 diabetes, all of which are comorbidities significantly associated with risk of the disease, as well as in those without,” he said.
Alzheimer’s, which is the most common form of age-related dementia, affects hundreds of millions of people worldwide. The disease is expected to affect nearly 14 million Americans by 2050. There is no approved treatment for it.
Dr. Cheng and colleagues at the Cleveland Clinic used a large gene-mapping network to analyze whether more than 1,600 Food and Drug Administration–approved drugs could work against Alzheimer’s. They gave higher scores to drugs that target both amyloid and tau proteins in the brain, which are two hallmarks of the disease. Sildenafil appeared at the top of the list.
Then the researchers used a database of health insurance claims for more than 7 million people in the U.S. to understand the relationship between sildenafil and Alzheimer’s disease outcomes. They compared sildenafil users to nonusers and found that those who used the drug were 69% less likely to have the neurodegenerative disease, even after 6 years of follow-up.
After that, the research team came up with a lab model that showed the sildenafil increased brain cell growth and targeted tau proteins. The lab model could indicate how the drug influences disease-related brain changes.
But Dr. Cheng cautioned against drawing strong conclusions. The study doesn’t demonstrate a causal relationship between sildenafil and Alzheimer’s disease. Researchers will need to conduct clinical trials with a placebo control to see how well the drug works.
Other researchers said the findings offer a new avenue for research but don’t yet provide solid answers.
“Being able to repurpose a drug already licensed for health conditions could help speed up the drug discovery process and bring about life-changing dementia treatments sooner,” Susan Kohlhaas, PhD, director of research at Alzheimer’s Research UK, told the Science Media Centre.
“Importantly, this research doesn’t prove that sildenafil is responsible for reducing dementia risk, or that it slows or stops the disease,” she continued. “If you want to discuss any treatments you are receiving, the first port of call is to speak to your doctor.”
And doctors won’t likely recommend it as a treatment just yet either.
“While these data are interesting scientifically, based on this study, I would not rush out to start taking sildenafil as a prevention for Alzheimer’s disease,” Tara Spires-Jones, PhD, deputy director of the Centre for Discovery Brain Sciences at the University of Edinburgh, told the Science Media Centre.
A version of this article first appeared on WebMD.com.
FROM NATURE AGING
Apixaban outmatches rivaroxaban for VTE in study
Recurrent venous thromboembolism (VTE) – a composite of pulmonary embolism and deep vein thrombosis – was the primary effectiveness outcome in the retrospective analysis of new-user data from almost 40,000 patients, which was published in Annals of Internal Medicine. Safety was evaluated through a composite of intracranial and gastrointestinal bleeding.
After a median follow-up of 102 days in the apixaban group and 105 days in the rivaroxaban group, apixaban demonstrated superiority for both primary outcomes.
These real-world findings may guide selection of initial anticoagulant therapy, reported lead author Ghadeer K. Dawwas, PhD, MSc, MBA, of the University of Pennsylvania, Philadelphia, and colleagues.
“Randomized clinical trials comparing apixaban with rivaroxaban in patients with VTE are under way (for example, COBRRA (NCT03266783),” the investigators wrote. “Until the results from these trials become available (The estimated completion date for COBRRA is December 2023.), observational studies that use existing data can provide evidence on the effectiveness and safety of these alternatives to inform clinical practice.”
In the new research, apixaban was associated with a 23% lower rate of recurrent VTE (hazard ratio, 0.77; 95% confidence interval, 0.69-0.87), including a 15% lower rate of deep vein thrombosis and a 41% lower rate of pulmonary embolism. Apixaban was associated with 40% fewer bleeding events (HR, 0.60; 95% CI, 0.53-0.69]), including a 40% lower rate of GI bleeding and a 46% lower rate of intracranial bleeding.
The study involved 37,236 patients with VTE, all of whom were diagnosed in at least one inpatient encounter and initiated direct oral anticoagulant (DOAC) therapy within 30 days, according to Optum’s deidentified Clinformatics Data Mart Database. Patients were evenly split into apixaban and rivaroxaban groups, with 18,618 individuals in each. Propensity score matching was used to minimize differences in baseline characteristics.
Apixaban was associated with an absolute reduction in recurrent VTE of 0.6% and 1.1% over 2 and 6 months, respectively, as well as reductions in bleeding of 1.1% and 1.5% over the same respective time periods.
The investigators noted that these findings were maintained in various sensitivity and subgroup analyses, including a model in which patients with VTE who had transient risk factors were compared with VTE patients exhibiting chronic risk factors.
“These findings suggest that apixaban has superior effectiveness and safety, compared with rivaroxaban and may provide guidance to clinicians and patients regarding selection of an anticoagulant for treatment of VTE,” Dr. Dawwas and colleagues concluded.
Study may have missed some nuance in possible outcomes, according to vascular surgeon
Thomas Wakefield, MD, a vascular surgeon and a professor of surgery at the University of Michigan Health Frankel Cardiovascular Center, Ann Arbor, generally agreed with the investigators’ conclusion, although he noted that DOAC selection may also be influenced by other considerations.

“The results of this study suggest that, when choosing an agent for an individual patient, apixaban does appear to have an advantage over rivaroxaban related to recurrent VTE and bleeding,” Dr. Wakefield said in an interview. “One must keep in mind that these are not the only factors that are considered when choosing an agent and these are not the only two DOACs available. For example, rivaroxaban is given once per day while apixaban is given twice per day, and rivaroxaban has been shown to be successful in the treatment of other thrombotic disorders.”
Dr. Wakefield also pointed out that the study may have missed some nuance in possible outcomes.
“The current study looked at severe outcomes that resulted in inpatient hospitalization, so the generalization to strictly outpatient treatment and less severe outcomes cannot be inferred,” he said.
Damon E. Houghton, MD, of the department of medicine and a consultant in the department of vascular medicine and hematology at Mayo Clinic, Rochester, Minn., called the study a “very nice analysis,” highlighting the large sample size.
“The results are not a reason to abandon rivaroxaban altogether, but do suggest that, when otherwise appropriate for a patient, apixaban should be the first choice,” Dr. Houghton said in a written comment. “Hopefully this analysis will encourage more payers to create financial incentives that facilitate the use of apixaban in more patients.”
Randomized trial needed, says hematologist
Colleen Edwards, MD, of the departments of medicine, hematology, and medical oncology, at the Icahn School of Medicine at Mount Sinai, New York, had a more guarded view of the findings than Dr. Wakefield and Dr. Houghton.

“[The investigators] certainly seem to be doing a lot of statistical gymnastics in this paper,” Dr. Edwards said in an interview. “They used all kinds of surrogates in place of real data that you would get from a randomized trial.”
For example, Dr. Edwards noted the use of prescription refills as a surrogate for medication adherence, and emphasized that inpatient observational data may not reflect outpatient therapy.
“Inpatients are constantly missing their medicines all the time,” she said. “They’re holding it for procedures, they’re NPO, they’re off the floor, so they missed their medicine. So it’s just a very different patient population than the outpatient population, which is where venous thromboembolism is treated now, by and large.”
Although Dr. Edwards suggested that the findings might guide treatment selection “a little bit,” she noted that insurance constraints and costs play a greater role, and ultimately concluded that a randomized trial is needed to materially alter clinical decision-making.
“I think we really have to wait for randomized trial before we abandon our other choices,” she said.
The investigators disclosed relationships with Merck, Celgene, UCB, and others. Dr. Wakefield reported awaiting disclosures. Dr. Houghton and Dr. Edwards reported no relevant conflicts of interest.
Recurrent venous thromboembolism (VTE) – a composite of pulmonary embolism and deep vein thrombosis – was the primary effectiveness outcome in the retrospective analysis of new-user data from almost 40,000 patients, which was published in Annals of Internal Medicine. Safety was evaluated through a composite of intracranial and gastrointestinal bleeding.
After a median follow-up of 102 days in the apixaban group and 105 days in the rivaroxaban group, apixaban demonstrated superiority for both primary outcomes.
These real-world findings may guide selection of initial anticoagulant therapy, reported lead author Ghadeer K. Dawwas, PhD, MSc, MBA, of the University of Pennsylvania, Philadelphia, and colleagues.
“Randomized clinical trials comparing apixaban with rivaroxaban in patients with VTE are under way (for example, COBRRA (NCT03266783),” the investigators wrote. “Until the results from these trials become available (The estimated completion date for COBRRA is December 2023.), observational studies that use existing data can provide evidence on the effectiveness and safety of these alternatives to inform clinical practice.”
In the new research, apixaban was associated with a 23% lower rate of recurrent VTE (hazard ratio, 0.77; 95% confidence interval, 0.69-0.87), including a 15% lower rate of deep vein thrombosis and a 41% lower rate of pulmonary embolism. Apixaban was associated with 40% fewer bleeding events (HR, 0.60; 95% CI, 0.53-0.69]), including a 40% lower rate of GI bleeding and a 46% lower rate of intracranial bleeding.
The study involved 37,236 patients with VTE, all of whom were diagnosed in at least one inpatient encounter and initiated direct oral anticoagulant (DOAC) therapy within 30 days, according to Optum’s deidentified Clinformatics Data Mart Database. Patients were evenly split into apixaban and rivaroxaban groups, with 18,618 individuals in each. Propensity score matching was used to minimize differences in baseline characteristics.
Apixaban was associated with an absolute reduction in recurrent VTE of 0.6% and 1.1% over 2 and 6 months, respectively, as well as reductions in bleeding of 1.1% and 1.5% over the same respective time periods.
The investigators noted that these findings were maintained in various sensitivity and subgroup analyses, including a model in which patients with VTE who had transient risk factors were compared with VTE patients exhibiting chronic risk factors.
“These findings suggest that apixaban has superior effectiveness and safety, compared with rivaroxaban and may provide guidance to clinicians and patients regarding selection of an anticoagulant for treatment of VTE,” Dr. Dawwas and colleagues concluded.
Study may have missed some nuance in possible outcomes, according to vascular surgeon
Thomas Wakefield, MD, a vascular surgeon and a professor of surgery at the University of Michigan Health Frankel Cardiovascular Center, Ann Arbor, generally agreed with the investigators’ conclusion, although he noted that DOAC selection may also be influenced by other considerations.
“The results of this study suggest that, when choosing an agent for an individual patient, apixaban does appear to have an advantage over rivaroxaban related to recurrent VTE and bleeding,” Dr. Wakefield said in an interview. “One must keep in mind that these are not the only factors that are considered when choosing an agent and these are not the only two DOACs available. For example, rivaroxaban is given once per day while apixaban is given twice per day, and rivaroxaban has been shown to be successful in the treatment of other thrombotic disorders.”
Dr. Wakefield also pointed out that the study may have missed some nuance in possible outcomes.
“The current study looked at severe outcomes that resulted in inpatient hospitalization, so the generalization to strictly outpatient treatment and less severe outcomes cannot be inferred,” he said.
Damon E. Houghton, MD, of the department of medicine and a consultant in the department of vascular medicine and hematology at Mayo Clinic, Rochester, Minn., called the study a “very nice analysis,” highlighting the large sample size.
“The results are not a reason to abandon rivaroxaban altogether, but do suggest that, when otherwise appropriate for a patient, apixaban should be the first choice,” Dr. Houghton said in a written comment. “Hopefully this analysis will encourage more payers to create financial incentives that facilitate the use of apixaban in more patients.”
Randomized trial needed, says hematologist
Colleen Edwards, MD, of the departments of medicine, hematology, and medical oncology, at the Icahn School of Medicine at Mount Sinai, New York, had a more guarded view of the findings than Dr. Wakefield and Dr. Houghton.
“[The investigators] certainly seem to be doing a lot of statistical gymnastics in this paper,” Dr. Edwards said in an interview. “They used all kinds of surrogates in place of real data that you would get from a randomized trial.”
For example, Dr. Edwards noted the use of prescription refills as a surrogate for medication adherence, and emphasized that inpatient observational data may not reflect outpatient therapy.
“Inpatients are constantly missing their medicines all the time,” she said. “They’re holding it for procedures, they’re NPO, they’re off the floor, so they missed their medicine. So it’s just a very different patient population than the outpatient population, which is where venous thromboembolism is treated now, by and large.”
Although Dr. Edwards suggested that the findings might guide treatment selection “a little bit,” she noted that insurance constraints and costs play a greater role, and ultimately concluded that a randomized trial is needed to materially alter clinical decision-making.
“I think we really have to wait for randomized trial before we abandon our other choices,” she said.
The investigators disclosed relationships with Merck, Celgene, UCB, and others. Dr. Wakefield reported awaiting disclosures. Dr. Houghton and Dr. Edwards reported no relevant conflicts of interest.
Recurrent venous thromboembolism (VTE) – a composite of pulmonary embolism and deep vein thrombosis – was the primary effectiveness outcome in the retrospective analysis of new-user data from almost 40,000 patients, which was published in Annals of Internal Medicine. Safety was evaluated through a composite of intracranial and gastrointestinal bleeding.
After a median follow-up of 102 days in the apixaban group and 105 days in the rivaroxaban group, apixaban demonstrated superiority for both primary outcomes.
These real-world findings may guide selection of initial anticoagulant therapy, reported lead author Ghadeer K. Dawwas, PhD, MSc, MBA, of the University of Pennsylvania, Philadelphia, and colleagues.
“Randomized clinical trials comparing apixaban with rivaroxaban in patients with VTE are under way (for example, COBRRA (NCT03266783),” the investigators wrote. “Until the results from these trials become available (The estimated completion date for COBRRA is December 2023.), observational studies that use existing data can provide evidence on the effectiveness and safety of these alternatives to inform clinical practice.”
In the new research, apixaban was associated with a 23% lower rate of recurrent VTE (hazard ratio, 0.77; 95% confidence interval, 0.69-0.87), including a 15% lower rate of deep vein thrombosis and a 41% lower rate of pulmonary embolism. Apixaban was associated with 40% fewer bleeding events (HR, 0.60; 95% CI, 0.53-0.69]), including a 40% lower rate of GI bleeding and a 46% lower rate of intracranial bleeding.
The study involved 37,236 patients with VTE, all of whom were diagnosed in at least one inpatient encounter and initiated direct oral anticoagulant (DOAC) therapy within 30 days, according to Optum’s deidentified Clinformatics Data Mart Database. Patients were evenly split into apixaban and rivaroxaban groups, with 18,618 individuals in each. Propensity score matching was used to minimize differences in baseline characteristics.
Apixaban was associated with an absolute reduction in recurrent VTE of 0.6% and 1.1% over 2 and 6 months, respectively, as well as reductions in bleeding of 1.1% and 1.5% over the same respective time periods.
The investigators noted that these findings were maintained in various sensitivity and subgroup analyses, including a model in which patients with VTE who had transient risk factors were compared with VTE patients exhibiting chronic risk factors.
“These findings suggest that apixaban has superior effectiveness and safety, compared with rivaroxaban and may provide guidance to clinicians and patients regarding selection of an anticoagulant for treatment of VTE,” Dr. Dawwas and colleagues concluded.
Study may have missed some nuance in possible outcomes, according to vascular surgeon
Thomas Wakefield, MD, a vascular surgeon and a professor of surgery at the University of Michigan Health Frankel Cardiovascular Center, Ann Arbor, generally agreed with the investigators’ conclusion, although he noted that DOAC selection may also be influenced by other considerations.
“The results of this study suggest that, when choosing an agent for an individual patient, apixaban does appear to have an advantage over rivaroxaban related to recurrent VTE and bleeding,” Dr. Wakefield said in an interview. “One must keep in mind that these are not the only factors that are considered when choosing an agent and these are not the only two DOACs available. For example, rivaroxaban is given once per day while apixaban is given twice per day, and rivaroxaban has been shown to be successful in the treatment of other thrombotic disorders.”
Dr. Wakefield also pointed out that the study may have missed some nuance in possible outcomes.
“The current study looked at severe outcomes that resulted in inpatient hospitalization, so the generalization to strictly outpatient treatment and less severe outcomes cannot be inferred,” he said.
Damon E. Houghton, MD, of the department of medicine and a consultant in the department of vascular medicine and hematology at Mayo Clinic, Rochester, Minn., called the study a “very nice analysis,” highlighting the large sample size.
“The results are not a reason to abandon rivaroxaban altogether, but do suggest that, when otherwise appropriate for a patient, apixaban should be the first choice,” Dr. Houghton said in a written comment. “Hopefully this analysis will encourage more payers to create financial incentives that facilitate the use of apixaban in more patients.”
Randomized trial needed, says hematologist
Colleen Edwards, MD, of the departments of medicine, hematology, and medical oncology, at the Icahn School of Medicine at Mount Sinai, New York, had a more guarded view of the findings than Dr. Wakefield and Dr. Houghton.
“[The investigators] certainly seem to be doing a lot of statistical gymnastics in this paper,” Dr. Edwards said in an interview. “They used all kinds of surrogates in place of real data that you would get from a randomized trial.”
For example, Dr. Edwards noted the use of prescription refills as a surrogate for medication adherence, and emphasized that inpatient observational data may not reflect outpatient therapy.
“Inpatients are constantly missing their medicines all the time,” she said. “They’re holding it for procedures, they’re NPO, they’re off the floor, so they missed their medicine. So it’s just a very different patient population than the outpatient population, which is where venous thromboembolism is treated now, by and large.”
Although Dr. Edwards suggested that the findings might guide treatment selection “a little bit,” she noted that insurance constraints and costs play a greater role, and ultimately concluded that a randomized trial is needed to materially alter clinical decision-making.
“I think we really have to wait for randomized trial before we abandon our other choices,” she said.
The investigators disclosed relationships with Merck, Celgene, UCB, and others. Dr. Wakefield reported awaiting disclosures. Dr. Houghton and Dr. Edwards reported no relevant conflicts of interest.
FROM ANNALS OF INTERNAL MEDICINE
FDA approves time-saving combo for r/r multiple myeloma
The U.S. Food and Drug Administration who have had one to three prior lines of therapy.

Using the newly approved combination in this setting is a time-saver for patients and clinics, observed an investigator.
“The approval of subcutaneous daratumumab in combination with Kd will help clinicians address unmet patient needs by reducing the administration time from hours to just minutes and reducing the frequency of infusion-related reactions, as compared to the intravenous daratumumab formulation in combination with Kd,” said Ajai Chari, MD, of Mount Sinai Cancer Clinical Trials Office in New York City in a Janssen press statement.
Efficacy data for the new approval come from a single-arm cohort of PLEIADES, a multicohort, open-label trial. The cohort included 66 patients with relapsed or refractory multiple myeloma who had received one or more prior lines of therapy. Patients received daratumumab + hyaluronidase-fihj subcutaneously in combination with carfilzomib and dexamethasone.
The main efficacy outcome measure was overall response rate, which was 84.8%. At a median follow-up of 9.2 months, the median duration of response had not been reached.
The response rate with the new combination, which features a subcutaneous injection, was akin to those with the older combination, which features the more time-consuming IV administration and was FDA approved, according to the company press release.
The most common adverse reactions (≥20%) occurring in patients treated with Darzalex Faspro, Kyprolis, and dexamethasone were upper respiratory tract infections, fatigue, insomnia, hypertension, diarrhea, cough, dyspnea, headache, pyrexia, nausea, and edema peripheral.
A version of this article first appeared on Medscape.com .
The U.S. Food and Drug Administration who have had one to three prior lines of therapy.
Using the newly approved combination in this setting is a time-saver for patients and clinics, observed an investigator.
“The approval of subcutaneous daratumumab in combination with Kd will help clinicians address unmet patient needs by reducing the administration time from hours to just minutes and reducing the frequency of infusion-related reactions, as compared to the intravenous daratumumab formulation in combination with Kd,” said Ajai Chari, MD, of Mount Sinai Cancer Clinical Trials Office in New York City in a Janssen press statement.
Efficacy data for the new approval come from a single-arm cohort of PLEIADES, a multicohort, open-label trial. The cohort included 66 patients with relapsed or refractory multiple myeloma who had received one or more prior lines of therapy. Patients received daratumumab + hyaluronidase-fihj subcutaneously in combination with carfilzomib and dexamethasone.
The main efficacy outcome measure was overall response rate, which was 84.8%. At a median follow-up of 9.2 months, the median duration of response had not been reached.
The response rate with the new combination, which features a subcutaneous injection, was akin to those with the older combination, which features the more time-consuming IV administration and was FDA approved, according to the company press release.
The most common adverse reactions (≥20%) occurring in patients treated with Darzalex Faspro, Kyprolis, and dexamethasone were upper respiratory tract infections, fatigue, insomnia, hypertension, diarrhea, cough, dyspnea, headache, pyrexia, nausea, and edema peripheral.
A version of this article first appeared on Medscape.com .
The U.S. Food and Drug Administration who have had one to three prior lines of therapy.
Using the newly approved combination in this setting is a time-saver for patients and clinics, observed an investigator.
“The approval of subcutaneous daratumumab in combination with Kd will help clinicians address unmet patient needs by reducing the administration time from hours to just minutes and reducing the frequency of infusion-related reactions, as compared to the intravenous daratumumab formulation in combination with Kd,” said Ajai Chari, MD, of Mount Sinai Cancer Clinical Trials Office in New York City in a Janssen press statement.
Efficacy data for the new approval come from a single-arm cohort of PLEIADES, a multicohort, open-label trial. The cohort included 66 patients with relapsed or refractory multiple myeloma who had received one or more prior lines of therapy. Patients received daratumumab + hyaluronidase-fihj subcutaneously in combination with carfilzomib and dexamethasone.
The main efficacy outcome measure was overall response rate, which was 84.8%. At a median follow-up of 9.2 months, the median duration of response had not been reached.
The response rate with the new combination, which features a subcutaneous injection, was akin to those with the older combination, which features the more time-consuming IV administration and was FDA approved, according to the company press release.
The most common adverse reactions (≥20%) occurring in patients treated with Darzalex Faspro, Kyprolis, and dexamethasone were upper respiratory tract infections, fatigue, insomnia, hypertension, diarrhea, cough, dyspnea, headache, pyrexia, nausea, and edema peripheral.
A version of this article first appeared on Medscape.com .