User login
Metformin use may curb BCC risk
in Iceland.
“In addition to general anticarcinogenic effects, metformin has also been shown to directly inhibit the sonic hedgehog pathway, a key pathway in basal cell carcinoma (BCC) pathogenesis,” Jonas A. Adalsteinsson, MD, of the University of Iceland, Reykjavik, and colleagues wrote. “The relationship between metformin and keratinocyte carcinoma has not been well-characterized but is of importance considering that metformin is a commonly prescribed medication.”
They added that the hedgehog pathway inhibitors vismodegib (Erivedge) and sonidegib (Odomzo), approved for treating BCC, “are highly effective for BCC prevention, but their broad use for BCC prophylaxis is limited due to numerous side effects.”
In the study, published in the Journal of the American Academy of Dermatology, the researchers identified 6,880 first-time cancer patients with BCC, squamous cell carcinoma in situ (SCCis), or invasive SCC, and 69,620 population controls using data from the Icelandic Cancer Registry and the Icelandic Prescription Medicine Register between 2003 and 2017. Metformin exposure was defined as having filled at least one prescription of metformin more than 2 years prior to cancer diagnosis. They used grams and daily dose units of metformin in their analysis; one DDU of metformin, “or its average daily maintenance dose when used for its primary indication, is 2 grams,” they noted.
Overall, metformin use was associated with a significantly lower risk of developing BCC, compared with nonuse (adjusted odds ratio, 0.71; 95% confidence interval, 0.61-0.83).
The reduced risk occurred similarly across age and gender subgroups, with the exception of individuals younger than 60 years, the researchers said. “This might signify that metformin has less of a protective effect in younger individuals, but we might also have lacked power in this category.” The association with reduced BCC risk remained significant at all three cumulative dose levels measured: 1-500 DDUs, 501-1,500 DDUs, and more than 1,500 DDUs.
Metformin use was not significantly associated with reduced risk of invasive SCC (aOR, 1.01) and in most cases of SCCis. However, the 501-1,500 DDU dose category was associated with a slight increase in risk of SCCis (aOR, 1.40; 95% CI, 1.00-1.96), “showing a possible increased risk of SCCis,” the authors wrote.
The decrease in BCC risk was seen across all metformin dosing levels, but the reason for this remains unclear, and might be related to a confounding factor that was not considered in this study, the researchers said. “It could also be that metformin’s BCC risk-lowering effect is immediate, with only a low dose being needed to see a clinical benefit.”
The study findings were limited by several factors, including the retrospective design and the inability to adjust for factors including ultraviolet exposure, Fitzpatrick skin type, and comorbidities. The frequent use of metformin by people with type 2 diabetes suggests diabetes itself or other diabetes medications could be possible confounding factors, the researchers wrote.
However, the results were strengthened by the large study population, and the data suggest an association between reduced risk of first-time BCC and metformin use, they added.
“Randomized, prospective trials are required to fully understand the effect metformin has on BCC and SCC risk,” the researchers concluded.

“There is a dire need to reduce incidence of skin cancers in general, and consequently a need for new non-surgical treatment options for keratinocytic nonmelanoma skin cancers,” Amor Khachemoune, MD, a dermatologist at the State University of New York, Brooklyn, and the department of dermatology of the Veteran Affairs NY Harbor Healthcare System, also in Brooklyn, said in an interview.
Dr. Khachemoune, who was not involved with the study, said that he was not surprised by the findings. “Like other well-studied sonic hedgehog inhibitors, vismodegib and sonidegib, metformin has a demonstrated effect on this pathway. The medical community outside of dermatology has extensive experience with the use of metformin for a host of other indications, including its role as anticarcinogenic, so it seemed natural that one would consider widening its use to quell the ever-expanding cases of basal cell carcinomas.”
However, complications from long-term use, though likely rare, could be a limitation in using metformin as a chemoprotective agent, Dr. Khachemoune said. Metformin-associated lactic acidosis is one example of a rare, but potentially life-threatening adverse event.
“Finding the right dosage and having an algorithm for follow up monitoring of side effects would certainly need to be put in place in a standardized way,” he emphasized. “As stated by the authors of this study, more inclusive research involving other groups with nonkeratinocytic malignancies in larger cohorts is needed.”
The study received no outside funding. The researchers and Dr. Khachemoune had no financial conflicts to disclose.
in Iceland.
“In addition to general anticarcinogenic effects, metformin has also been shown to directly inhibit the sonic hedgehog pathway, a key pathway in basal cell carcinoma (BCC) pathogenesis,” Jonas A. Adalsteinsson, MD, of the University of Iceland, Reykjavik, and colleagues wrote. “The relationship between metformin and keratinocyte carcinoma has not been well-characterized but is of importance considering that metformin is a commonly prescribed medication.”
They added that the hedgehog pathway inhibitors vismodegib (Erivedge) and sonidegib (Odomzo), approved for treating BCC, “are highly effective for BCC prevention, but their broad use for BCC prophylaxis is limited due to numerous side effects.”
In the study, published in the Journal of the American Academy of Dermatology, the researchers identified 6,880 first-time cancer patients with BCC, squamous cell carcinoma in situ (SCCis), or invasive SCC, and 69,620 population controls using data from the Icelandic Cancer Registry and the Icelandic Prescription Medicine Register between 2003 and 2017. Metformin exposure was defined as having filled at least one prescription of metformin more than 2 years prior to cancer diagnosis. They used grams and daily dose units of metformin in their analysis; one DDU of metformin, “or its average daily maintenance dose when used for its primary indication, is 2 grams,” they noted.
Overall, metformin use was associated with a significantly lower risk of developing BCC, compared with nonuse (adjusted odds ratio, 0.71; 95% confidence interval, 0.61-0.83).
The reduced risk occurred similarly across age and gender subgroups, with the exception of individuals younger than 60 years, the researchers said. “This might signify that metformin has less of a protective effect in younger individuals, but we might also have lacked power in this category.” The association with reduced BCC risk remained significant at all three cumulative dose levels measured: 1-500 DDUs, 501-1,500 DDUs, and more than 1,500 DDUs.
Metformin use was not significantly associated with reduced risk of invasive SCC (aOR, 1.01) and in most cases of SCCis. However, the 501-1,500 DDU dose category was associated with a slight increase in risk of SCCis (aOR, 1.40; 95% CI, 1.00-1.96), “showing a possible increased risk of SCCis,” the authors wrote.
The decrease in BCC risk was seen across all metformin dosing levels, but the reason for this remains unclear, and might be related to a confounding factor that was not considered in this study, the researchers said. “It could also be that metformin’s BCC risk-lowering effect is immediate, with only a low dose being needed to see a clinical benefit.”
The study findings were limited by several factors, including the retrospective design and the inability to adjust for factors including ultraviolet exposure, Fitzpatrick skin type, and comorbidities. The frequent use of metformin by people with type 2 diabetes suggests diabetes itself or other diabetes medications could be possible confounding factors, the researchers wrote.
However, the results were strengthened by the large study population, and the data suggest an association between reduced risk of first-time BCC and metformin use, they added.
“Randomized, prospective trials are required to fully understand the effect metformin has on BCC and SCC risk,” the researchers concluded.
“There is a dire need to reduce incidence of skin cancers in general, and consequently a need for new non-surgical treatment options for keratinocytic nonmelanoma skin cancers,” Amor Khachemoune, MD, a dermatologist at the State University of New York, Brooklyn, and the department of dermatology of the Veteran Affairs NY Harbor Healthcare System, also in Brooklyn, said in an interview.
Dr. Khachemoune, who was not involved with the study, said that he was not surprised by the findings. “Like other well-studied sonic hedgehog inhibitors, vismodegib and sonidegib, metformin has a demonstrated effect on this pathway. The medical community outside of dermatology has extensive experience with the use of metformin for a host of other indications, including its role as anticarcinogenic, so it seemed natural that one would consider widening its use to quell the ever-expanding cases of basal cell carcinomas.”
However, complications from long-term use, though likely rare, could be a limitation in using metformin as a chemoprotective agent, Dr. Khachemoune said. Metformin-associated lactic acidosis is one example of a rare, but potentially life-threatening adverse event.
“Finding the right dosage and having an algorithm for follow up monitoring of side effects would certainly need to be put in place in a standardized way,” he emphasized. “As stated by the authors of this study, more inclusive research involving other groups with nonkeratinocytic malignancies in larger cohorts is needed.”
The study received no outside funding. The researchers and Dr. Khachemoune had no financial conflicts to disclose.
in Iceland.
“In addition to general anticarcinogenic effects, metformin has also been shown to directly inhibit the sonic hedgehog pathway, a key pathway in basal cell carcinoma (BCC) pathogenesis,” Jonas A. Adalsteinsson, MD, of the University of Iceland, Reykjavik, and colleagues wrote. “The relationship between metformin and keratinocyte carcinoma has not been well-characterized but is of importance considering that metformin is a commonly prescribed medication.”
They added that the hedgehog pathway inhibitors vismodegib (Erivedge) and sonidegib (Odomzo), approved for treating BCC, “are highly effective for BCC prevention, but their broad use for BCC prophylaxis is limited due to numerous side effects.”
In the study, published in the Journal of the American Academy of Dermatology, the researchers identified 6,880 first-time cancer patients with BCC, squamous cell carcinoma in situ (SCCis), or invasive SCC, and 69,620 population controls using data from the Icelandic Cancer Registry and the Icelandic Prescription Medicine Register between 2003 and 2017. Metformin exposure was defined as having filled at least one prescription of metformin more than 2 years prior to cancer diagnosis. They used grams and daily dose units of metformin in their analysis; one DDU of metformin, “or its average daily maintenance dose when used for its primary indication, is 2 grams,” they noted.
Overall, metformin use was associated with a significantly lower risk of developing BCC, compared with nonuse (adjusted odds ratio, 0.71; 95% confidence interval, 0.61-0.83).
The reduced risk occurred similarly across age and gender subgroups, with the exception of individuals younger than 60 years, the researchers said. “This might signify that metformin has less of a protective effect in younger individuals, but we might also have lacked power in this category.” The association with reduced BCC risk remained significant at all three cumulative dose levels measured: 1-500 DDUs, 501-1,500 DDUs, and more than 1,500 DDUs.
Metformin use was not significantly associated with reduced risk of invasive SCC (aOR, 1.01) and in most cases of SCCis. However, the 501-1,500 DDU dose category was associated with a slight increase in risk of SCCis (aOR, 1.40; 95% CI, 1.00-1.96), “showing a possible increased risk of SCCis,” the authors wrote.
The decrease in BCC risk was seen across all metformin dosing levels, but the reason for this remains unclear, and might be related to a confounding factor that was not considered in this study, the researchers said. “It could also be that metformin’s BCC risk-lowering effect is immediate, with only a low dose being needed to see a clinical benefit.”
The study findings were limited by several factors, including the retrospective design and the inability to adjust for factors including ultraviolet exposure, Fitzpatrick skin type, and comorbidities. The frequent use of metformin by people with type 2 diabetes suggests diabetes itself or other diabetes medications could be possible confounding factors, the researchers wrote.
However, the results were strengthened by the large study population, and the data suggest an association between reduced risk of first-time BCC and metformin use, they added.
“Randomized, prospective trials are required to fully understand the effect metformin has on BCC and SCC risk,” the researchers concluded.
“There is a dire need to reduce incidence of skin cancers in general, and consequently a need for new non-surgical treatment options for keratinocytic nonmelanoma skin cancers,” Amor Khachemoune, MD, a dermatologist at the State University of New York, Brooklyn, and the department of dermatology of the Veteran Affairs NY Harbor Healthcare System, also in Brooklyn, said in an interview.
Dr. Khachemoune, who was not involved with the study, said that he was not surprised by the findings. “Like other well-studied sonic hedgehog inhibitors, vismodegib and sonidegib, metformin has a demonstrated effect on this pathway. The medical community outside of dermatology has extensive experience with the use of metformin for a host of other indications, including its role as anticarcinogenic, so it seemed natural that one would consider widening its use to quell the ever-expanding cases of basal cell carcinomas.”
However, complications from long-term use, though likely rare, could be a limitation in using metformin as a chemoprotective agent, Dr. Khachemoune said. Metformin-associated lactic acidosis is one example of a rare, but potentially life-threatening adverse event.
“Finding the right dosage and having an algorithm for follow up monitoring of side effects would certainly need to be put in place in a standardized way,” he emphasized. “As stated by the authors of this study, more inclusive research involving other groups with nonkeratinocytic malignancies in larger cohorts is needed.”
The study received no outside funding. The researchers and Dr. Khachemoune had no financial conflicts to disclose.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Do patients with cancer need a third shot of COVID vaccine?
Patients with cancer have shown varying responses to COVID-19 vaccination, with good responses in patients with solid tumors (even while on systemic therapy) and poor responses in patients with blood cancers, particularly those on immunosuppressive therapies.
The data are evolving to show factors associated with a poor response but are not strong enough yet to recommend booster shots, say researchers.
The work is defining who will likely need a COVID vaccine booster when they become available. “It’s definitely not all cancer patients,” said Dimpy Shah, MD, PhD, a cancer epidemiologist at the Mays Cancer Center, University of Texas, San Antonio.
Public anxiously awaiting boosters
Boosters aren’t recommended in the United States at the moment, in large part because the Emergency Use Authorization under which the vaccines are being administered allows for only two shots of the Pfizer and Moderna vaccines and one shot of the Johnson & Johnson vaccine.
Even so, regulators and policymakers are “keenly aware that physicians and patients alike are anxious to get going and start doing boosters,” Dr. Shah said. There’s concern that antibody response might wane over time, perhaps even more quickly in patients with cancer.
Pfizer is already in talks with the U.S. Food and Drug Administration to authorize a third dose of its vaccine in the United States. Guidelines could very well change in coming months, said Ghady Haidar, MD, a specialist in infectious diseases and cancer at the University of Pittsburgh.
However, it’s still early in the game, and it’s not clear yet if boosters are necessary in cancer, Dr. Haidar said in an interview.
For one thing, it’s unknown if poor antibody response really means that patients aren’t protected, he explained. The vaccines elicit T-cell responses that could protect patients regardless of antibody levels. It’s also unclear if antibody titer levels are clinically relevant, and there hasn’t been much indication yet that less-than-robust vaccine responses translate to worse COVID outcomes in patients with cancer.
Those and other questions are areas of active investigation by Dr. Shah, Dr. Haidar, and others. Dozens of clinical trials are investigating vaccine response in patients with cancer, including the use of boosters.
Meanwhile, some cancer patients aren’t waiting around for more study results. “I get many, many emails a day” about booster shots, Dr. Haidar said. “We recommend against” them for now but some people bend the rules and get an extra shot anyway. “I get it. People are apprehensive.”
Three COVID deaths despite full vaccination
The vaccine clinical trials had fewer patients with cancer, so researchers are moving fast to backfill the data. Although there is some variation in what’s being reported, an overall picture is slowly emerging.
Dr. Shah and her team reported on responses to the mRNA COVID vaccines from Pfizer and Moderna and found a 94% seroconversion rate in 131 patients with cancer 3-4 weeks after their second dose of vaccine. They also found good responses among patients on cytotoxic chemotherapy within 6 months of their first vaccine dose, although their antibody titer levels were significantly lower than seen in other patients with cancer.
Investigators from Montefiore Medical Center in New York City also recently reported a 94% seroconversion rate among 200 patients with cancer, including 98% seroconversion in patients with solid tumors. Rates were lower in patients with blood cancers but were still 85% overall, with 70% conversion among patients on anti-CD20 therapies and 73% among stem cell transplant patients.
Dr. Haidar’s group reported a seroconversion rate of 82.4% among patients with solid tumors but only 54.7% among those with blood cancer. Risk factors for poor response included treatment with antimetabolites and anti-CD20 therapies, and, in the solid tumor group, radiation therapy, likely because of its overall toxicity and impact on lymphocyte function.
Israeli investigators reported in May a 90% seroconversion rate after two doses of the Pfizer vaccine among 102 patients with solid tumors on active treatment, which compared favorably to the 100% conversion rate in healthy controls, but they noted that antibody titers were considerably lower in patients with cancer.
The only variable associated with lower titer levels was combined use of chemotherapy and immunotherapy, they noted. There were also three women on dose-dense chemotherapy for breast cancer who did not produce any antibodies.
In a study limited to patients with blood cancers, a Lithuanian team recently reported that among 885 patients, those on Bruton tyrosine kinase inhibitors, ruxolitinib (Jakafi), venetoclax (Venclexta), or anti-CD20 therapies mounted almost no antibody response to the Pfizer vaccine.
The Lithuanian group also reported nine breakthrough COVID infections among their fully vaccinated blood cancer patients, including three deaths.
A team from the Icahn School of Medicine at Mount Sinai, New York reported that more than 15% of 260 patients with multiple myeloma also had no response to the Pfizer or Moderna vaccine; they were on BCMA-targeted therapy or anti-CD38 monoclonal antibody therapy at the time of vaccination, but a few had undergone CAR-T cell therapy more than 3 months beforehand.
Heated debate about antibody testing
Despite these reports of some patients with cancer having poorer responses, there’s some uncertainty over the benefit of giving a third (booster) shot.
There’s the question about the clinical relevance of antibody titer levels, and very little work has been done to date on cellular T-cell immunity from the vaccines.
“Right now, we are using titer levels like they actually mean something when they might not,” said Ravi Parikh, MD, a genitourinary and thoracic oncologist at the University of Pennsylvania, Philadelphia, who co-wrote an editorial that accompanies the Israeli report.
That’s one of the reasons why the FDA and others do not currently recommend antibody tests for COVID vaccine decisions outside of a clinical trial, but not everyone agrees with that position.
There’s been “a lot of heated debate in the medical community” over the issue, Dr. Haidar said.
The Icahn team, for instance, said that their results “underscore the need for routine serological monitoring of [multiple myeloma] patients following COVID-19 vaccination” to see if they might still need to mask-up and socially distance.
There is precedence, too, for vaccine boosters in cancer. As Dr. Parikh noted in his editorial, guidelines recommend revaccination after stem cell transplant for meningococcus, tetanus, and varicella, and other infections.
In France, COVID booster shots are already standard care for patients on dialysis and those on anti-CD20 agents, as well as for solid organ transplant recipients, for whom the literature supporting the benefit of COVID boosters is much more evolved than in cancer.
Israel has also authorized vaccine boosters for immunocompromised patients, including those with cancer, according to news reports.
It is also almost certain that the FDA will grant a formal approval for the COVID vaccines, at which point doctors will be free to administer boosters as they see fit.
“People are going to have to think really hard about what to do with them” if guidance hasn’t changed by then, Dr. Haidar said.
As the story unfolds, Dr. Haidar and others said in an interview that the take-home message for oncologists remains largely what it has been – namely to get patients vaccinated but also to consider masks and social distancing afterward for those at risk of a poor response.
Dr. Shah, Dr. Haidar, and Dr. Parikh have disclosed no relevant financial relationships. Dr. Parikh is a regular contributor to Medscape Oncology.
A version of this article first appeared on Medscape.com.
Patients with cancer have shown varying responses to COVID-19 vaccination, with good responses in patients with solid tumors (even while on systemic therapy) and poor responses in patients with blood cancers, particularly those on immunosuppressive therapies.
The data are evolving to show factors associated with a poor response but are not strong enough yet to recommend booster shots, say researchers.
The work is defining who will likely need a COVID vaccine booster when they become available. “It’s definitely not all cancer patients,” said Dimpy Shah, MD, PhD, a cancer epidemiologist at the Mays Cancer Center, University of Texas, San Antonio.
Public anxiously awaiting boosters
Boosters aren’t recommended in the United States at the moment, in large part because the Emergency Use Authorization under which the vaccines are being administered allows for only two shots of the Pfizer and Moderna vaccines and one shot of the Johnson & Johnson vaccine.
Even so, regulators and policymakers are “keenly aware that physicians and patients alike are anxious to get going and start doing boosters,” Dr. Shah said. There’s concern that antibody response might wane over time, perhaps even more quickly in patients with cancer.
Pfizer is already in talks with the U.S. Food and Drug Administration to authorize a third dose of its vaccine in the United States. Guidelines could very well change in coming months, said Ghady Haidar, MD, a specialist in infectious diseases and cancer at the University of Pittsburgh.
However, it’s still early in the game, and it’s not clear yet if boosters are necessary in cancer, Dr. Haidar said in an interview.
For one thing, it’s unknown if poor antibody response really means that patients aren’t protected, he explained. The vaccines elicit T-cell responses that could protect patients regardless of antibody levels. It’s also unclear if antibody titer levels are clinically relevant, and there hasn’t been much indication yet that less-than-robust vaccine responses translate to worse COVID outcomes in patients with cancer.
Those and other questions are areas of active investigation by Dr. Shah, Dr. Haidar, and others. Dozens of clinical trials are investigating vaccine response in patients with cancer, including the use of boosters.
Meanwhile, some cancer patients aren’t waiting around for more study results. “I get many, many emails a day” about booster shots, Dr. Haidar said. “We recommend against” them for now but some people bend the rules and get an extra shot anyway. “I get it. People are apprehensive.”
Three COVID deaths despite full vaccination
The vaccine clinical trials had fewer patients with cancer, so researchers are moving fast to backfill the data. Although there is some variation in what’s being reported, an overall picture is slowly emerging.
Dr. Shah and her team reported on responses to the mRNA COVID vaccines from Pfizer and Moderna and found a 94% seroconversion rate in 131 patients with cancer 3-4 weeks after their second dose of vaccine. They also found good responses among patients on cytotoxic chemotherapy within 6 months of their first vaccine dose, although their antibody titer levels were significantly lower than seen in other patients with cancer.
Investigators from Montefiore Medical Center in New York City also recently reported a 94% seroconversion rate among 200 patients with cancer, including 98% seroconversion in patients with solid tumors. Rates were lower in patients with blood cancers but were still 85% overall, with 70% conversion among patients on anti-CD20 therapies and 73% among stem cell transplant patients.
Dr. Haidar’s group reported a seroconversion rate of 82.4% among patients with solid tumors but only 54.7% among those with blood cancer. Risk factors for poor response included treatment with antimetabolites and anti-CD20 therapies, and, in the solid tumor group, radiation therapy, likely because of its overall toxicity and impact on lymphocyte function.
Israeli investigators reported in May a 90% seroconversion rate after two doses of the Pfizer vaccine among 102 patients with solid tumors on active treatment, which compared favorably to the 100% conversion rate in healthy controls, but they noted that antibody titers were considerably lower in patients with cancer.
The only variable associated with lower titer levels was combined use of chemotherapy and immunotherapy, they noted. There were also three women on dose-dense chemotherapy for breast cancer who did not produce any antibodies.
In a study limited to patients with blood cancers, a Lithuanian team recently reported that among 885 patients, those on Bruton tyrosine kinase inhibitors, ruxolitinib (Jakafi), venetoclax (Venclexta), or anti-CD20 therapies mounted almost no antibody response to the Pfizer vaccine.
The Lithuanian group also reported nine breakthrough COVID infections among their fully vaccinated blood cancer patients, including three deaths.
A team from the Icahn School of Medicine at Mount Sinai, New York reported that more than 15% of 260 patients with multiple myeloma also had no response to the Pfizer or Moderna vaccine; they were on BCMA-targeted therapy or anti-CD38 monoclonal antibody therapy at the time of vaccination, but a few had undergone CAR-T cell therapy more than 3 months beforehand.
Heated debate about antibody testing
Despite these reports of some patients with cancer having poorer responses, there’s some uncertainty over the benefit of giving a third (booster) shot.
There’s the question about the clinical relevance of antibody titer levels, and very little work has been done to date on cellular T-cell immunity from the vaccines.
“Right now, we are using titer levels like they actually mean something when they might not,” said Ravi Parikh, MD, a genitourinary and thoracic oncologist at the University of Pennsylvania, Philadelphia, who co-wrote an editorial that accompanies the Israeli report.
That’s one of the reasons why the FDA and others do not currently recommend antibody tests for COVID vaccine decisions outside of a clinical trial, but not everyone agrees with that position.
There’s been “a lot of heated debate in the medical community” over the issue, Dr. Haidar said.
The Icahn team, for instance, said that their results “underscore the need for routine serological monitoring of [multiple myeloma] patients following COVID-19 vaccination” to see if they might still need to mask-up and socially distance.
There is precedence, too, for vaccine boosters in cancer. As Dr. Parikh noted in his editorial, guidelines recommend revaccination after stem cell transplant for meningococcus, tetanus, and varicella, and other infections.
In France, COVID booster shots are already standard care for patients on dialysis and those on anti-CD20 agents, as well as for solid organ transplant recipients, for whom the literature supporting the benefit of COVID boosters is much more evolved than in cancer.
Israel has also authorized vaccine boosters for immunocompromised patients, including those with cancer, according to news reports.
It is also almost certain that the FDA will grant a formal approval for the COVID vaccines, at which point doctors will be free to administer boosters as they see fit.
“People are going to have to think really hard about what to do with them” if guidance hasn’t changed by then, Dr. Haidar said.
As the story unfolds, Dr. Haidar and others said in an interview that the take-home message for oncologists remains largely what it has been – namely to get patients vaccinated but also to consider masks and social distancing afterward for those at risk of a poor response.
Dr. Shah, Dr. Haidar, and Dr. Parikh have disclosed no relevant financial relationships. Dr. Parikh is a regular contributor to Medscape Oncology.
A version of this article first appeared on Medscape.com.
Patients with cancer have shown varying responses to COVID-19 vaccination, with good responses in patients with solid tumors (even while on systemic therapy) and poor responses in patients with blood cancers, particularly those on immunosuppressive therapies.
The data are evolving to show factors associated with a poor response but are not strong enough yet to recommend booster shots, say researchers.
The work is defining who will likely need a COVID vaccine booster when they become available. “It’s definitely not all cancer patients,” said Dimpy Shah, MD, PhD, a cancer epidemiologist at the Mays Cancer Center, University of Texas, San Antonio.
Public anxiously awaiting boosters
Boosters aren’t recommended in the United States at the moment, in large part because the Emergency Use Authorization under which the vaccines are being administered allows for only two shots of the Pfizer and Moderna vaccines and one shot of the Johnson & Johnson vaccine.
Even so, regulators and policymakers are “keenly aware that physicians and patients alike are anxious to get going and start doing boosters,” Dr. Shah said. There’s concern that antibody response might wane over time, perhaps even more quickly in patients with cancer.
Pfizer is already in talks with the U.S. Food and Drug Administration to authorize a third dose of its vaccine in the United States. Guidelines could very well change in coming months, said Ghady Haidar, MD, a specialist in infectious diseases and cancer at the University of Pittsburgh.
However, it’s still early in the game, and it’s not clear yet if boosters are necessary in cancer, Dr. Haidar said in an interview.
For one thing, it’s unknown if poor antibody response really means that patients aren’t protected, he explained. The vaccines elicit T-cell responses that could protect patients regardless of antibody levels. It’s also unclear if antibody titer levels are clinically relevant, and there hasn’t been much indication yet that less-than-robust vaccine responses translate to worse COVID outcomes in patients with cancer.
Those and other questions are areas of active investigation by Dr. Shah, Dr. Haidar, and others. Dozens of clinical trials are investigating vaccine response in patients with cancer, including the use of boosters.
Meanwhile, some cancer patients aren’t waiting around for more study results. “I get many, many emails a day” about booster shots, Dr. Haidar said. “We recommend against” them for now but some people bend the rules and get an extra shot anyway. “I get it. People are apprehensive.”
Three COVID deaths despite full vaccination
The vaccine clinical trials had fewer patients with cancer, so researchers are moving fast to backfill the data. Although there is some variation in what’s being reported, an overall picture is slowly emerging.
Dr. Shah and her team reported on responses to the mRNA COVID vaccines from Pfizer and Moderna and found a 94% seroconversion rate in 131 patients with cancer 3-4 weeks after their second dose of vaccine. They also found good responses among patients on cytotoxic chemotherapy within 6 months of their first vaccine dose, although their antibody titer levels were significantly lower than seen in other patients with cancer.
Investigators from Montefiore Medical Center in New York City also recently reported a 94% seroconversion rate among 200 patients with cancer, including 98% seroconversion in patients with solid tumors. Rates were lower in patients with blood cancers but were still 85% overall, with 70% conversion among patients on anti-CD20 therapies and 73% among stem cell transplant patients.
Dr. Haidar’s group reported a seroconversion rate of 82.4% among patients with solid tumors but only 54.7% among those with blood cancer. Risk factors for poor response included treatment with antimetabolites and anti-CD20 therapies, and, in the solid tumor group, radiation therapy, likely because of its overall toxicity and impact on lymphocyte function.
Israeli investigators reported in May a 90% seroconversion rate after two doses of the Pfizer vaccine among 102 patients with solid tumors on active treatment, which compared favorably to the 100% conversion rate in healthy controls, but they noted that antibody titers were considerably lower in patients with cancer.
The only variable associated with lower titer levels was combined use of chemotherapy and immunotherapy, they noted. There were also three women on dose-dense chemotherapy for breast cancer who did not produce any antibodies.
In a study limited to patients with blood cancers, a Lithuanian team recently reported that among 885 patients, those on Bruton tyrosine kinase inhibitors, ruxolitinib (Jakafi), venetoclax (Venclexta), or anti-CD20 therapies mounted almost no antibody response to the Pfizer vaccine.
The Lithuanian group also reported nine breakthrough COVID infections among their fully vaccinated blood cancer patients, including three deaths.
A team from the Icahn School of Medicine at Mount Sinai, New York reported that more than 15% of 260 patients with multiple myeloma also had no response to the Pfizer or Moderna vaccine; they were on BCMA-targeted therapy or anti-CD38 monoclonal antibody therapy at the time of vaccination, but a few had undergone CAR-T cell therapy more than 3 months beforehand.
Heated debate about antibody testing
Despite these reports of some patients with cancer having poorer responses, there’s some uncertainty over the benefit of giving a third (booster) shot.
There’s the question about the clinical relevance of antibody titer levels, and very little work has been done to date on cellular T-cell immunity from the vaccines.
“Right now, we are using titer levels like they actually mean something when they might not,” said Ravi Parikh, MD, a genitourinary and thoracic oncologist at the University of Pennsylvania, Philadelphia, who co-wrote an editorial that accompanies the Israeli report.
That’s one of the reasons why the FDA and others do not currently recommend antibody tests for COVID vaccine decisions outside of a clinical trial, but not everyone agrees with that position.
There’s been “a lot of heated debate in the medical community” over the issue, Dr. Haidar said.
The Icahn team, for instance, said that their results “underscore the need for routine serological monitoring of [multiple myeloma] patients following COVID-19 vaccination” to see if they might still need to mask-up and socially distance.
There is precedence, too, for vaccine boosters in cancer. As Dr. Parikh noted in his editorial, guidelines recommend revaccination after stem cell transplant for meningococcus, tetanus, and varicella, and other infections.
In France, COVID booster shots are already standard care for patients on dialysis and those on anti-CD20 agents, as well as for solid organ transplant recipients, for whom the literature supporting the benefit of COVID boosters is much more evolved than in cancer.
Israel has also authorized vaccine boosters for immunocompromised patients, including those with cancer, according to news reports.
It is also almost certain that the FDA will grant a formal approval for the COVID vaccines, at which point doctors will be free to administer boosters as they see fit.
“People are going to have to think really hard about what to do with them” if guidance hasn’t changed by then, Dr. Haidar said.
As the story unfolds, Dr. Haidar and others said in an interview that the take-home message for oncologists remains largely what it has been – namely to get patients vaccinated but also to consider masks and social distancing afterward for those at risk of a poor response.
Dr. Shah, Dr. Haidar, and Dr. Parikh have disclosed no relevant financial relationships. Dr. Parikh is a regular contributor to Medscape Oncology.
A version of this article first appeared on Medscape.com.
Testosterone replacement shows CV benefit in hypogonadal men
Data from a long-term study suggest that testosterone replacement therapy (TRT) for men with hypogonadism may reduce the risk for major adverse cardiovascular events. Previous studies have yielded conflicting results on whether there is a benefit.
The latest results come from a study of 805 men with hypogonadism from Germany and Qatar who were followed for nearly a decade. For those who received parenteral testosterone 1,000 mg every 12 weeks, there were improvements in classical cardiovascular risk factors, such as obesity, lipid level, and inflammatory markers, whereas among those who chose not to take testosterone (control patients), all of these factors worsened.
In addition, there were only 16 deaths among patients in the TRT group, and none of the deaths were from myocardial infarction or stroke. In contrast, there were 74 deaths among the control patients, as well as 70 cases of MI and 59 strokes.
The men in the study were all at relatively high risk for cardiovascular adverse events. In the TRT group, the mean Framingham Risk score was 15.5; in the control group, it was 15.8. This translates into mean 10-year risks of 22.7% and 23.5%, respectively.
“Given that all these men would normally have been expected to suffer a heart attack or stroke in the next 5-10 years with no other intervention, it was a real surprise to see no cardiovascular events at all in the group on testosterone therapy. It’s clear that this treatment can significantly reduce the risks in this particular group,” commented lead investigator Omar Aboumarzouk, MD, from Hamad Medical in Doha, Qatar.
He presented the new data at the 2021 annual congress of the European Association of Urology.
Dr. Aboumarzouk emphasized, however, that, “while men need testosterone for certain psychological and biological functions, only those with low levels who display other symptoms are likely to benefit from testosterone therapy.”
Maarten Albersen, MD, a urologist at the University of Leuven (Belgium), who was not involved in the study, noted that, although the study showed a reduction in major adverse cardiovascular events and mortality among the men who received TRT, the risk scores were in the intermediate range, and the men in the TRT group were slightly younger and were at slightly lower risk at baseline.
“The study was long enough to see differences in the rate of cardiovascular events. However, the numbers involved and the fact that the trial was not randomized mean it’s still difficult to draw any hard conclusions,” he said.
Registry study
The data came from a cumulative registry study begun in 2004 to assess the long-term efficacy and safety of TRT every 3 months in men with hypogonadism. The study, conducted in Bremen, Dresden, and Muenster in Germany, as well as in Doha, Qatar, is ongoing.
At total of 805 men were enrolled; 412 received TRT, and 393 declined testosterone replacement and served as control patients.
The investigators reported 10-year data. Statistical models controlled for age, body mass index, smoking, alcohol, total and HDL cholesterol level, systolic blood pressure, and type 2 diabetes.
The median age at baseline was lower among those in the TRT arm, at 57.7 years versus 63.7 years for control patients (P < .001).
All classical cardiovascular risk factors, including obesity, glycemic control, lipid pattern, and C-reactive protein, improved in the TRT group and worsened in the control group.
Dr. Albersen noted that “a new trial is now underway, aiming to recruit 6,000 participants, and this should provide definitive answers on the cardiovascular risks or even benefits of hormone therapy in men with low testosterone.”
No funding source for the study was reported. Dr. Aboumarzouk and Dr. Albersen have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Data from a long-term study suggest that testosterone replacement therapy (TRT) for men with hypogonadism may reduce the risk for major adverse cardiovascular events. Previous studies have yielded conflicting results on whether there is a benefit.
The latest results come from a study of 805 men with hypogonadism from Germany and Qatar who were followed for nearly a decade. For those who received parenteral testosterone 1,000 mg every 12 weeks, there were improvements in classical cardiovascular risk factors, such as obesity, lipid level, and inflammatory markers, whereas among those who chose not to take testosterone (control patients), all of these factors worsened.
In addition, there were only 16 deaths among patients in the TRT group, and none of the deaths were from myocardial infarction or stroke. In contrast, there were 74 deaths among the control patients, as well as 70 cases of MI and 59 strokes.
The men in the study were all at relatively high risk for cardiovascular adverse events. In the TRT group, the mean Framingham Risk score was 15.5; in the control group, it was 15.8. This translates into mean 10-year risks of 22.7% and 23.5%, respectively.
“Given that all these men would normally have been expected to suffer a heart attack or stroke in the next 5-10 years with no other intervention, it was a real surprise to see no cardiovascular events at all in the group on testosterone therapy. It’s clear that this treatment can significantly reduce the risks in this particular group,” commented lead investigator Omar Aboumarzouk, MD, from Hamad Medical in Doha, Qatar.
He presented the new data at the 2021 annual congress of the European Association of Urology.
Dr. Aboumarzouk emphasized, however, that, “while men need testosterone for certain psychological and biological functions, only those with low levels who display other symptoms are likely to benefit from testosterone therapy.”
Maarten Albersen, MD, a urologist at the University of Leuven (Belgium), who was not involved in the study, noted that, although the study showed a reduction in major adverse cardiovascular events and mortality among the men who received TRT, the risk scores were in the intermediate range, and the men in the TRT group were slightly younger and were at slightly lower risk at baseline.
“The study was long enough to see differences in the rate of cardiovascular events. However, the numbers involved and the fact that the trial was not randomized mean it’s still difficult to draw any hard conclusions,” he said.
Registry study
The data came from a cumulative registry study begun in 2004 to assess the long-term efficacy and safety of TRT every 3 months in men with hypogonadism. The study, conducted in Bremen, Dresden, and Muenster in Germany, as well as in Doha, Qatar, is ongoing.
At total of 805 men were enrolled; 412 received TRT, and 393 declined testosterone replacement and served as control patients.
The investigators reported 10-year data. Statistical models controlled for age, body mass index, smoking, alcohol, total and HDL cholesterol level, systolic blood pressure, and type 2 diabetes.
The median age at baseline was lower among those in the TRT arm, at 57.7 years versus 63.7 years for control patients (P < .001).
All classical cardiovascular risk factors, including obesity, glycemic control, lipid pattern, and C-reactive protein, improved in the TRT group and worsened in the control group.
Dr. Albersen noted that “a new trial is now underway, aiming to recruit 6,000 participants, and this should provide definitive answers on the cardiovascular risks or even benefits of hormone therapy in men with low testosterone.”
No funding source for the study was reported. Dr. Aboumarzouk and Dr. Albersen have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Data from a long-term study suggest that testosterone replacement therapy (TRT) for men with hypogonadism may reduce the risk for major adverse cardiovascular events. Previous studies have yielded conflicting results on whether there is a benefit.
The latest results come from a study of 805 men with hypogonadism from Germany and Qatar who were followed for nearly a decade. For those who received parenteral testosterone 1,000 mg every 12 weeks, there were improvements in classical cardiovascular risk factors, such as obesity, lipid level, and inflammatory markers, whereas among those who chose not to take testosterone (control patients), all of these factors worsened.
In addition, there were only 16 deaths among patients in the TRT group, and none of the deaths were from myocardial infarction or stroke. In contrast, there were 74 deaths among the control patients, as well as 70 cases of MI and 59 strokes.
The men in the study were all at relatively high risk for cardiovascular adverse events. In the TRT group, the mean Framingham Risk score was 15.5; in the control group, it was 15.8. This translates into mean 10-year risks of 22.7% and 23.5%, respectively.
“Given that all these men would normally have been expected to suffer a heart attack or stroke in the next 5-10 years with no other intervention, it was a real surprise to see no cardiovascular events at all in the group on testosterone therapy. It’s clear that this treatment can significantly reduce the risks in this particular group,” commented lead investigator Omar Aboumarzouk, MD, from Hamad Medical in Doha, Qatar.
He presented the new data at the 2021 annual congress of the European Association of Urology.
Dr. Aboumarzouk emphasized, however, that, “while men need testosterone for certain psychological and biological functions, only those with low levels who display other symptoms are likely to benefit from testosterone therapy.”
Maarten Albersen, MD, a urologist at the University of Leuven (Belgium), who was not involved in the study, noted that, although the study showed a reduction in major adverse cardiovascular events and mortality among the men who received TRT, the risk scores were in the intermediate range, and the men in the TRT group were slightly younger and were at slightly lower risk at baseline.
“The study was long enough to see differences in the rate of cardiovascular events. However, the numbers involved and the fact that the trial was not randomized mean it’s still difficult to draw any hard conclusions,” he said.
Registry study
The data came from a cumulative registry study begun in 2004 to assess the long-term efficacy and safety of TRT every 3 months in men with hypogonadism. The study, conducted in Bremen, Dresden, and Muenster in Germany, as well as in Doha, Qatar, is ongoing.
At total of 805 men were enrolled; 412 received TRT, and 393 declined testosterone replacement and served as control patients.
The investigators reported 10-year data. Statistical models controlled for age, body mass index, smoking, alcohol, total and HDL cholesterol level, systolic blood pressure, and type 2 diabetes.
The median age at baseline was lower among those in the TRT arm, at 57.7 years versus 63.7 years for control patients (P < .001).
All classical cardiovascular risk factors, including obesity, glycemic control, lipid pattern, and C-reactive protein, improved in the TRT group and worsened in the control group.
Dr. Albersen noted that “a new trial is now underway, aiming to recruit 6,000 participants, and this should provide definitive answers on the cardiovascular risks or even benefits of hormone therapy in men with low testosterone.”
No funding source for the study was reported. Dr. Aboumarzouk and Dr. Albersen have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Assessment of a Medication Deprescribing Tool on Polypharmacy and Cost Avoidance
According to the Centers for Disease Control and Prevention National Center for Health Statistics (NCHS), the use of prescription drugs has increased in the past half century. Although prescription drugs have played an important role in preventing, controlling, and delaying onset or progression of disease, their growth in use also has posed many risks.1 One ramification of this growth is the occurrence of polypharmacy, which does not have a universal, clear definition. In general, it can be described as the concurrent use of multiple medications by a single patient to treat one or more medical ailments. Five or more medications taken simultaneously is the most common definition to date, but this is just one of many acceptable definitions and that varies from one health care facility to another.1,2
Regardless of the cutoffs established to indicate polypharmacy, its incidence can result in poor and potentially harmful health outcomes. Polypharmacy increases the risk of experiencing adverse drug events (ADEs), drug-drug interactions (DDIs), geriatric-related syndromes, falls, hospitalization, and mortality. Issues with adherence may begin to unfold secondary to increased pill burden. Both the patient and the health care system may encounter financial strain, as polypharmacy can lead to unnecessary and essentially preventable costs of care. When evaluating the likelihood of polypharmacy based on age group, NCHS found that 47.5% of patients taking ≥ 5 medications were aged ≥ 65 years.1-5 This indicates that polypharmacy is of great concern in the geriatric population, which also represents a large proportion of individuals accessing Veterans Health Administration (VHA) care.
Deprescibing
Deprescribing is the act of withdrawing or discontinuing potentially inappropriate medications (PIM), or medications used by older patients harboring ADEs that generally outweigh the clinical benefits of the drug. Deprescribing is an effective tool for managing or reducing polypharmacy. A variety of tools have been created whose sole purpose is to simplify deprescribing. Some tools explicitly identify PIM and are widely familiar in medical practice. Examples are the Beers Criteria developed in 1991 or Screening Tool to Alert Right Treatment/Screening Tool of Older Persons Prescriptions (START/STOPP) criteria created in 2003. Other tools that are less commonplace but equally as resourceful are MedStopper and Deprescribing.org. The former was launched in 2015 and is a Canadian online system that provides risk assessments for medications with guidance for tapering or stopping medications if continuation of the drug presents higher risk than benefit.5-7 The latter is a full-fledged website developed by a physician, a pharmacist, and their research teams that serves as an exchange hub for deprescribing information.
In 2016, the VIONE (Vital, Important, Optional, Not indicated/treatment complete, and Every medication has an indication) deprescribing tool was developed by Saraswathy Battar, MD, at Central Arkansas Veterans Healthcare System (CAVHS) in Little Rock, as a system that could go beyond medication reconciliation (Table 1). Health care providers (HCPs) and pharmacists evaluate each medication that a patient has been prescribed and places each medication in a VIONE category. Prescribers may then take the opportunity to deprescribe or discontinue medications if deemed appropriate based on their clinical assessments and shared decision making.8 Traditionally, medication reconciliation involves the process of obtaining a complete and accurate list of medications as reported by a patient or caregiver to a HCP. VIONE encourages HCPs and pharmacists not only to ensure medication lists are accurate, but also that each medication reported is appropriate for continued use. In other words, VIONE is meant to help implement deprescribing at opportune times. More than 14,000 medications have been deprescribed using the VIONE method, resulting in more than $2,000,000 of annualized cost avoidance after just 1 year of implementation at CAVHS.9
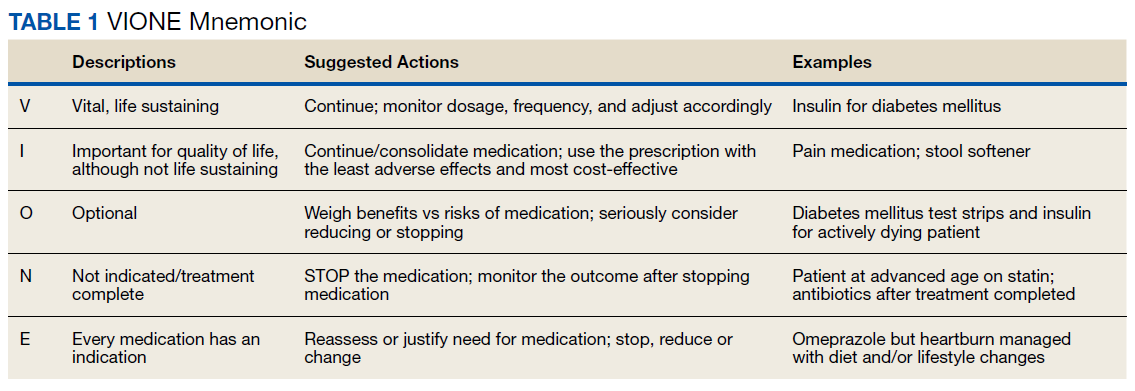
VIONE consists of 2 major components in the Computerized Patient Record System (CPRS): a template and a dropdown discontinuation menu. The template captured patient allergies, pertinent laboratory data, the patient’s active problem list and applicable diagnoses, and active medication list. Patient aligned care team (PACT) pharmacists used the information captured in the template to conduct medication reconciliations and polypharmacy reviews. Each medication is categorized in VIONE using data collected during reviews. A menu delineates reasons for discontinuation: optional, dose decrease, no diagnosis, not indicated/treatment complete, discontinue alternate medication prescribed, and patient reported no longer taking. The discontinuation menu allowed PACT pharmacists and physicians to choose 1 VIONE option per medication to clarify the reason for discontinuation. VIONE-based discontinuations are recorded in CPRS and identified as deprescribed.
At the time of this project, > 30 US Department of Veterans Affairs (VA) facilities had adopted VIONE. Use of VIONE at VA Southern Nevada Healthcare System (VASNHS) in North Las Vegas has been incorporated in the everyday practices of home-based primary care pharmacists and physicians but has yet to be implemented in other areas of the facility. The purpose of this project was to determine the impact of the VIONE tool on polypharmacy and cost avoidance at VASNHS when used by primary care physicians (PCPs) and PACT primary care clinics.
Methods
Veterans receiving care at VASNHS aged ≥ 65 years with ≥ 10 active medications noted in CPRS were included in this project. PACT pharmacists and physicians were educated on the proper use of the VIONE tool prior to its implementation. Education included a 15-minute slide presentation followed by dissemination of a 1-page VIONE tool handout during a PACT all-staff clinic meeting.
Data were collected for 3 months before and after the intervention. Data were made available for assessment by the Automated Data Processing Application Coordinator (ADPAC) at VASNHS. The ADPAC created and generated an Excel spreadsheet report, which listed all medications deprescribed using the VIONE method. The primary endpoint was the total number of medications discontinued using the VIONE template and/or discontinuation menu. For the purpose of this project, appropriate discontinuation was considered any prescription deprescribed, excluding medical supplies, by pharmacists and PCPs who received VIONE education.
![]()
The secondary endpoint was the estimated annualized cost avoidance for the facility (Figure). The calculation does not include medications discontinued due to the prescription of an alternative medication or dose decreases since these VIONE selections imply that a new prescription or order was placed and the original prescription was not deprescribed. Annualized cost avoidance was determined with use of the VIONE dashboard, a database that retrospectively gathers information regarding patients at risk of polypharmacy, polypharmacy-related ADEs, and cost. Manual adjustments were made to various parameters on the Veterans Integrated Service Network 15 VIONE dashboard by the author in order to obtain data specific to this project. These parameters allowed selection of service sections, specific staff members or the option to include or exclude chronic or nonchronic medications. The annualized cost avoidance figure was then compared to raw data pulled by a VIONE dashboard correspondent to ensure the manual calculation was accurate. Finally, the 5 most common classes of medications deprescribed were identified for information purposes and to provide a better postulation on the types of medications being discontinued using the VIONE method.
Results
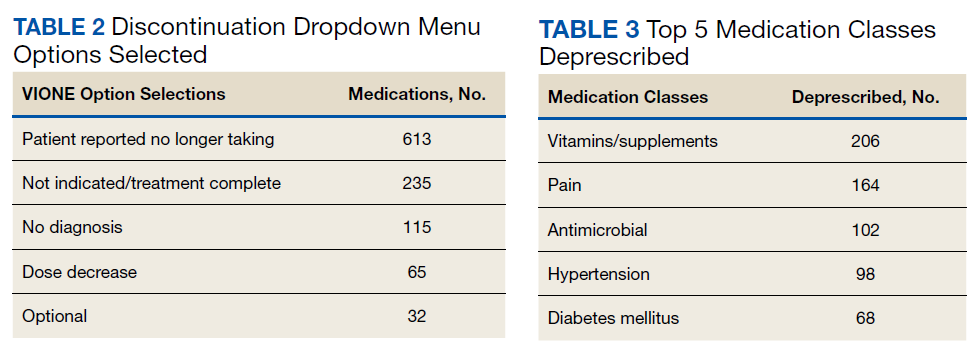
A total of 2,442 veterans met inclusion criteria, and the VIONE method was applied to 598 between late October 2018 and January 2019. The 13 PACT pharmacists contacted at least 10 veterans each, thus at least 130 were randomly selected for telephone calls to perform polypharmacy reviews using the VIONE note template. The discontinuation menu was used if a medication qualified to be deprescribed. After 3 months, 1986 prescriptions were deprescribed using VIONE; however, 1060 prescriptions were considered appropriately deprescribed (Table 2). The 13 PACT pharmacists deprescribed 361 medications, and the 29 PACT physicians deprescribed 699 medications. These prescriptions were then separated into medication categories to determine the most common discontinued classes. Vitamins and supplements were the medication class most frequently deprescribed (19.4%), followed by pain medications (15.5%), antimicrobial agents (9.6%), antihypertensive medications (9.2%), and diabetes medications (6.4%) (Table 3). The top 5 medication categories accounted for 60% of all medications appropriately deprescribed.
The estimated annualized cost avoidance for all medications deprescribed in the 3-month project period was $84,030.46. To provide the most appropriate and accurate calculation, medication classes excluded from this figure were acute or short-term prescriptions and antimicrobial agents. Medications prescribed short-term typically are not suitable to continue for an extended period, and antimicrobial agents were excluded since they are normally associated with higher costs, and may overestimate the cost avoidance calculation for the facility.
Discussion
The outcomes for the primary and secondary endpoints of this project illustrate that using VIONE in PACT primary care clinics had a notable impact on polypharmacy and cost avoidance over a short period. This outcome can be attributed to 2 significant effects of using the deprescribing tool. VIONE’s simplicity in application allowed clinicians to incorporate daily use of the tool with minimal effort. Education was all that was required to fully enable clinicians to work together successfully and exercise collaborative practice to promote deprescribing. VIONE also elicited a cascade of favorable effects that improve patient safety and health outcomes. The tool aided in identification of PIM, which helped reduce polypharmacy and medication burden. The risk for DDIs and ADEs may decrease; therefore, the incidence of falls, need for emergency department visits or inpatient care related to polypharmacy may decline. Less complex medication regimens may alleviate issues with adherence and avoid the various consequences of polypharmacy in theory. Simplified regimens can potentially improve disease management and quality of life for patients. Further studies are needed to substantiate deprescribing and its true effect on patient adherence and better health outcomes at this time.10
Reducing polypharmacy can lead to cost savings. Based on the results of this 3-month study, we expect that VASNHS would save more than $84,000 by reducing polypharmacy among its patients. Those savings can be funneled back into the health care system, and allotted to necessary patient care, prescriptions, and health care facility needs.
Limitations
There are some important limitations to this study. Definitions of polypharmacy may vary from one health care facility to another. The cutoffs for polypharmacy may differ, causing the prevalence of polypharmacy and potential costs savings to vary. Use of VIONE may be inconsistent among users if not previously educated or properly trained. For instance, VIONE selections are listed in the same menu as the standard CPRS discontinuation options, which may lead to discontinuation of medical supplies or laboratory orders instead of prescriptions.
The method of data analysis and project design used in this study may have been subject to error. For example, the list of PCPs may have been inaccurate or outdated, which would result in an over- or underrepresentation of those who contributed to data collection. Furthermore, there is some volatility in calculating the total cost avoidance. For example, medications for chronic conditions that were only taken on an as needed basis may have overestimated savings. Either under- or overestimations could occur when parameters are adjusted on the VIONE discontinuation dashboard without appropriate guidance. With the ability to manually adjust the dashboard parameters, dissimilarities in calculations may follow.
Conclusions
The VIONE tool may be useful in improving patient safety through deprescribing and discontinuing PIM. Decreasing the number of medications being taken concomitantly by a patient and continuing only those that are imperative in their medical treatment is the first step to reducing the incidence of polypharmacy. Consequently, chances of ADEs or DDIs are lessened, especially among older individuals who are considered high risk for experiencing the detrimental effects that may ensue. These effects include geriatric-related syndromes, increased risk of fall, hospital visits or admissions, or death. Use of VIONE easily promotes collaboration among clinicians to evaluate medications eligible for discontinuation more regularly. If this deprescribing tool is continuously used, costs avoided can likely be maximized within VA health care systems.
The results of this project should serve as an incentive to push for better prescribing practices and increase deprescribing efforts. It should provoke the need for change in regimens and the subsequent discontinuation of prescriptions that are not considered vital to continue. Finally, the result of this project should substantiate the positive impact a deprescribing tool can possess to avert the issues commonly associated with polypharmacy.
1. Centers for Disease Control and Prevention, National Center for Health Statistics. Health, United States, 2013: with special feature on prescription drugs. Published May 2014. Accessed May 13, 2021. https://www.cdc.gov/nchs/data/hus/hus13.pdf
2. Masnoon N, Shakib S, Kalisch-Ellett L, Caughey GE. What is polypharmacy? A systematic review of definitions. BMC Geriatr. 2017;17(1):230. Published 2017 Oct 10. doi:10.1186/s12877-017-0621-2
3. Parulekar MS, Rogers CK. Polypharmacy and mobility. In: Cifu DX, Lew HL, Oh-Park M., eds Geriatric Rehabilitation. Elsevier; 2018. doi:10.1016/B978-0-323-54454-2.12001-1
4. Rieckert A, Trampisch US, Klaaßen-Mielke R, et al. Polypharmacy in older patients with chronic diseases: a cross-sectional analysis of factors associated with excessive polypharmacy. BMC Fam Pract. 2018;19(1):113. Published 2018 Jul 18. doi:10.1186/s12875-018-0795-5
5. Thompson CA. New medication review method cuts veterans’ Rx load, saves millions. Am J Health Syst Pharm. 2018;75(8):502-503. doi:10.2146/news180023
6. Reeve E. Deprescribing tools: a review of the types of tools available to aid deprescribing in clinical practice. J Pharm Pract Res. 2020;50(1):98-107. doi:10.1002/jppr.1626
7. Fried TR, Niehoff KM, Street RL, et al. Effect of the Tool to Reduce Inappropriate Medications on Medication Communication and Deprescribing. J Am Geriatr Soc. 2017;65(10):2265-2271. doi:10.1111/jgs.15042
8. Battar S, Dickerson KR, Sedgwick C, et al. Understanding principles of high reliability organizations through the eyes of VIONE, a clinical program to improve patient safety by deprescribing potentially inappropriate medications and reducing polypharmacy. Fed Pract. 2019;36(12):564-568.
9. Battar S, Cmelik T, Dickerson K, Scott, M. Experience better health with VIONE a safe medication deprescribing tool [Nonpublic source, not verified]
10. Ulley J, Harrop D, Ali A, et al. Desprescribing interventions and their impact on medication adherence in community-dwelling older adults with polypharmacy: a systematic review. BMC Geriatr. 2019;19(15):1-13.
According to the Centers for Disease Control and Prevention National Center for Health Statistics (NCHS), the use of prescription drugs has increased in the past half century. Although prescription drugs have played an important role in preventing, controlling, and delaying onset or progression of disease, their growth in use also has posed many risks.1 One ramification of this growth is the occurrence of polypharmacy, which does not have a universal, clear definition. In general, it can be described as the concurrent use of multiple medications by a single patient to treat one or more medical ailments. Five or more medications taken simultaneously is the most common definition to date, but this is just one of many acceptable definitions and that varies from one health care facility to another.1,2
Regardless of the cutoffs established to indicate polypharmacy, its incidence can result in poor and potentially harmful health outcomes. Polypharmacy increases the risk of experiencing adverse drug events (ADEs), drug-drug interactions (DDIs), geriatric-related syndromes, falls, hospitalization, and mortality. Issues with adherence may begin to unfold secondary to increased pill burden. Both the patient and the health care system may encounter financial strain, as polypharmacy can lead to unnecessary and essentially preventable costs of care. When evaluating the likelihood of polypharmacy based on age group, NCHS found that 47.5% of patients taking ≥ 5 medications were aged ≥ 65 years.1-5 This indicates that polypharmacy is of great concern in the geriatric population, which also represents a large proportion of individuals accessing Veterans Health Administration (VHA) care.
Deprescibing
Deprescribing is the act of withdrawing or discontinuing potentially inappropriate medications (PIM), or medications used by older patients harboring ADEs that generally outweigh the clinical benefits of the drug. Deprescribing is an effective tool for managing or reducing polypharmacy. A variety of tools have been created whose sole purpose is to simplify deprescribing. Some tools explicitly identify PIM and are widely familiar in medical practice. Examples are the Beers Criteria developed in 1991 or Screening Tool to Alert Right Treatment/Screening Tool of Older Persons Prescriptions (START/STOPP) criteria created in 2003. Other tools that are less commonplace but equally as resourceful are MedStopper and Deprescribing.org. The former was launched in 2015 and is a Canadian online system that provides risk assessments for medications with guidance for tapering or stopping medications if continuation of the drug presents higher risk than benefit.5-7 The latter is a full-fledged website developed by a physician, a pharmacist, and their research teams that serves as an exchange hub for deprescribing information.
In 2016, the VIONE (Vital, Important, Optional, Not indicated/treatment complete, and Every medication has an indication) deprescribing tool was developed by Saraswathy Battar, MD, at Central Arkansas Veterans Healthcare System (CAVHS) in Little Rock, as a system that could go beyond medication reconciliation (Table 1). Health care providers (HCPs) and pharmacists evaluate each medication that a patient has been prescribed and places each medication in a VIONE category. Prescribers may then take the opportunity to deprescribe or discontinue medications if deemed appropriate based on their clinical assessments and shared decision making.8 Traditionally, medication reconciliation involves the process of obtaining a complete and accurate list of medications as reported by a patient or caregiver to a HCP. VIONE encourages HCPs and pharmacists not only to ensure medication lists are accurate, but also that each medication reported is appropriate for continued use. In other words, VIONE is meant to help implement deprescribing at opportune times. More than 14,000 medications have been deprescribed using the VIONE method, resulting in more than $2,000,000 of annualized cost avoidance after just 1 year of implementation at CAVHS.9
VIONE consists of 2 major components in the Computerized Patient Record System (CPRS): a template and a dropdown discontinuation menu. The template captured patient allergies, pertinent laboratory data, the patient’s active problem list and applicable diagnoses, and active medication list. Patient aligned care team (PACT) pharmacists used the information captured in the template to conduct medication reconciliations and polypharmacy reviews. Each medication is categorized in VIONE using data collected during reviews. A menu delineates reasons for discontinuation: optional, dose decrease, no diagnosis, not indicated/treatment complete, discontinue alternate medication prescribed, and patient reported no longer taking. The discontinuation menu allowed PACT pharmacists and physicians to choose 1 VIONE option per medication to clarify the reason for discontinuation. VIONE-based discontinuations are recorded in CPRS and identified as deprescribed.
At the time of this project, > 30 US Department of Veterans Affairs (VA) facilities had adopted VIONE. Use of VIONE at VA Southern Nevada Healthcare System (VASNHS) in North Las Vegas has been incorporated in the everyday practices of home-based primary care pharmacists and physicians but has yet to be implemented in other areas of the facility. The purpose of this project was to determine the impact of the VIONE tool on polypharmacy and cost avoidance at VASNHS when used by primary care physicians (PCPs) and PACT primary care clinics.
Methods
Veterans receiving care at VASNHS aged ≥ 65 years with ≥ 10 active medications noted in CPRS were included in this project. PACT pharmacists and physicians were educated on the proper use of the VIONE tool prior to its implementation. Education included a 15-minute slide presentation followed by dissemination of a 1-page VIONE tool handout during a PACT all-staff clinic meeting.
Data were collected for 3 months before and after the intervention. Data were made available for assessment by the Automated Data Processing Application Coordinator (ADPAC) at VASNHS. The ADPAC created and generated an Excel spreadsheet report, which listed all medications deprescribed using the VIONE method. The primary endpoint was the total number of medications discontinued using the VIONE template and/or discontinuation menu. For the purpose of this project, appropriate discontinuation was considered any prescription deprescribed, excluding medical supplies, by pharmacists and PCPs who received VIONE education.
![]()
The secondary endpoint was the estimated annualized cost avoidance for the facility (Figure). The calculation does not include medications discontinued due to the prescription of an alternative medication or dose decreases since these VIONE selections imply that a new prescription or order was placed and the original prescription was not deprescribed. Annualized cost avoidance was determined with use of the VIONE dashboard, a database that retrospectively gathers information regarding patients at risk of polypharmacy, polypharmacy-related ADEs, and cost. Manual adjustments were made to various parameters on the Veterans Integrated Service Network 15 VIONE dashboard by the author in order to obtain data specific to this project. These parameters allowed selection of service sections, specific staff members or the option to include or exclude chronic or nonchronic medications. The annualized cost avoidance figure was then compared to raw data pulled by a VIONE dashboard correspondent to ensure the manual calculation was accurate. Finally, the 5 most common classes of medications deprescribed were identified for information purposes and to provide a better postulation on the types of medications being discontinued using the VIONE method.
Results
A total of 2,442 veterans met inclusion criteria, and the VIONE method was applied to 598 between late October 2018 and January 2019. The 13 PACT pharmacists contacted at least 10 veterans each, thus at least 130 were randomly selected for telephone calls to perform polypharmacy reviews using the VIONE note template. The discontinuation menu was used if a medication qualified to be deprescribed. After 3 months, 1986 prescriptions were deprescribed using VIONE; however, 1060 prescriptions were considered appropriately deprescribed (Table 2). The 13 PACT pharmacists deprescribed 361 medications, and the 29 PACT physicians deprescribed 699 medications. These prescriptions were then separated into medication categories to determine the most common discontinued classes. Vitamins and supplements were the medication class most frequently deprescribed (19.4%), followed by pain medications (15.5%), antimicrobial agents (9.6%), antihypertensive medications (9.2%), and diabetes medications (6.4%) (Table 3). The top 5 medication categories accounted for 60% of all medications appropriately deprescribed.
The estimated annualized cost avoidance for all medications deprescribed in the 3-month project period was $84,030.46. To provide the most appropriate and accurate calculation, medication classes excluded from this figure were acute or short-term prescriptions and antimicrobial agents. Medications prescribed short-term typically are not suitable to continue for an extended period, and antimicrobial agents were excluded since they are normally associated with higher costs, and may overestimate the cost avoidance calculation for the facility.
Discussion
The outcomes for the primary and secondary endpoints of this project illustrate that using VIONE in PACT primary care clinics had a notable impact on polypharmacy and cost avoidance over a short period. This outcome can be attributed to 2 significant effects of using the deprescribing tool. VIONE’s simplicity in application allowed clinicians to incorporate daily use of the tool with minimal effort. Education was all that was required to fully enable clinicians to work together successfully and exercise collaborative practice to promote deprescribing. VIONE also elicited a cascade of favorable effects that improve patient safety and health outcomes. The tool aided in identification of PIM, which helped reduce polypharmacy and medication burden. The risk for DDIs and ADEs may decrease; therefore, the incidence of falls, need for emergency department visits or inpatient care related to polypharmacy may decline. Less complex medication regimens may alleviate issues with adherence and avoid the various consequences of polypharmacy in theory. Simplified regimens can potentially improve disease management and quality of life for patients. Further studies are needed to substantiate deprescribing and its true effect on patient adherence and better health outcomes at this time.10
Reducing polypharmacy can lead to cost savings. Based on the results of this 3-month study, we expect that VASNHS would save more than $84,000 by reducing polypharmacy among its patients. Those savings can be funneled back into the health care system, and allotted to necessary patient care, prescriptions, and health care facility needs.
Limitations
There are some important limitations to this study. Definitions of polypharmacy may vary from one health care facility to another. The cutoffs for polypharmacy may differ, causing the prevalence of polypharmacy and potential costs savings to vary. Use of VIONE may be inconsistent among users if not previously educated or properly trained. For instance, VIONE selections are listed in the same menu as the standard CPRS discontinuation options, which may lead to discontinuation of medical supplies or laboratory orders instead of prescriptions.
The method of data analysis and project design used in this study may have been subject to error. For example, the list of PCPs may have been inaccurate or outdated, which would result in an over- or underrepresentation of those who contributed to data collection. Furthermore, there is some volatility in calculating the total cost avoidance. For example, medications for chronic conditions that were only taken on an as needed basis may have overestimated savings. Either under- or overestimations could occur when parameters are adjusted on the VIONE discontinuation dashboard without appropriate guidance. With the ability to manually adjust the dashboard parameters, dissimilarities in calculations may follow.
Conclusions
The VIONE tool may be useful in improving patient safety through deprescribing and discontinuing PIM. Decreasing the number of medications being taken concomitantly by a patient and continuing only those that are imperative in their medical treatment is the first step to reducing the incidence of polypharmacy. Consequently, chances of ADEs or DDIs are lessened, especially among older individuals who are considered high risk for experiencing the detrimental effects that may ensue. These effects include geriatric-related syndromes, increased risk of fall, hospital visits or admissions, or death. Use of VIONE easily promotes collaboration among clinicians to evaluate medications eligible for discontinuation more regularly. If this deprescribing tool is continuously used, costs avoided can likely be maximized within VA health care systems.
The results of this project should serve as an incentive to push for better prescribing practices and increase deprescribing efforts. It should provoke the need for change in regimens and the subsequent discontinuation of prescriptions that are not considered vital to continue. Finally, the result of this project should substantiate the positive impact a deprescribing tool can possess to avert the issues commonly associated with polypharmacy.
According to the Centers for Disease Control and Prevention National Center for Health Statistics (NCHS), the use of prescription drugs has increased in the past half century. Although prescription drugs have played an important role in preventing, controlling, and delaying onset or progression of disease, their growth in use also has posed many risks.1 One ramification of this growth is the occurrence of polypharmacy, which does not have a universal, clear definition. In general, it can be described as the concurrent use of multiple medications by a single patient to treat one or more medical ailments. Five or more medications taken simultaneously is the most common definition to date, but this is just one of many acceptable definitions and that varies from one health care facility to another.1,2
Regardless of the cutoffs established to indicate polypharmacy, its incidence can result in poor and potentially harmful health outcomes. Polypharmacy increases the risk of experiencing adverse drug events (ADEs), drug-drug interactions (DDIs), geriatric-related syndromes, falls, hospitalization, and mortality. Issues with adherence may begin to unfold secondary to increased pill burden. Both the patient and the health care system may encounter financial strain, as polypharmacy can lead to unnecessary and essentially preventable costs of care. When evaluating the likelihood of polypharmacy based on age group, NCHS found that 47.5% of patients taking ≥ 5 medications were aged ≥ 65 years.1-5 This indicates that polypharmacy is of great concern in the geriatric population, which also represents a large proportion of individuals accessing Veterans Health Administration (VHA) care.
Deprescibing
Deprescribing is the act of withdrawing or discontinuing potentially inappropriate medications (PIM), or medications used by older patients harboring ADEs that generally outweigh the clinical benefits of the drug. Deprescribing is an effective tool for managing or reducing polypharmacy. A variety of tools have been created whose sole purpose is to simplify deprescribing. Some tools explicitly identify PIM and are widely familiar in medical practice. Examples are the Beers Criteria developed in 1991 or Screening Tool to Alert Right Treatment/Screening Tool of Older Persons Prescriptions (START/STOPP) criteria created in 2003. Other tools that are less commonplace but equally as resourceful are MedStopper and Deprescribing.org. The former was launched in 2015 and is a Canadian online system that provides risk assessments for medications with guidance for tapering or stopping medications if continuation of the drug presents higher risk than benefit.5-7 The latter is a full-fledged website developed by a physician, a pharmacist, and their research teams that serves as an exchange hub for deprescribing information.
In 2016, the VIONE (Vital, Important, Optional, Not indicated/treatment complete, and Every medication has an indication) deprescribing tool was developed by Saraswathy Battar, MD, at Central Arkansas Veterans Healthcare System (CAVHS) in Little Rock, as a system that could go beyond medication reconciliation (Table 1). Health care providers (HCPs) and pharmacists evaluate each medication that a patient has been prescribed and places each medication in a VIONE category. Prescribers may then take the opportunity to deprescribe or discontinue medications if deemed appropriate based on their clinical assessments and shared decision making.8 Traditionally, medication reconciliation involves the process of obtaining a complete and accurate list of medications as reported by a patient or caregiver to a HCP. VIONE encourages HCPs and pharmacists not only to ensure medication lists are accurate, but also that each medication reported is appropriate for continued use. In other words, VIONE is meant to help implement deprescribing at opportune times. More than 14,000 medications have been deprescribed using the VIONE method, resulting in more than $2,000,000 of annualized cost avoidance after just 1 year of implementation at CAVHS.9
VIONE consists of 2 major components in the Computerized Patient Record System (CPRS): a template and a dropdown discontinuation menu. The template captured patient allergies, pertinent laboratory data, the patient’s active problem list and applicable diagnoses, and active medication list. Patient aligned care team (PACT) pharmacists used the information captured in the template to conduct medication reconciliations and polypharmacy reviews. Each medication is categorized in VIONE using data collected during reviews. A menu delineates reasons for discontinuation: optional, dose decrease, no diagnosis, not indicated/treatment complete, discontinue alternate medication prescribed, and patient reported no longer taking. The discontinuation menu allowed PACT pharmacists and physicians to choose 1 VIONE option per medication to clarify the reason for discontinuation. VIONE-based discontinuations are recorded in CPRS and identified as deprescribed.
At the time of this project, > 30 US Department of Veterans Affairs (VA) facilities had adopted VIONE. Use of VIONE at VA Southern Nevada Healthcare System (VASNHS) in North Las Vegas has been incorporated in the everyday practices of home-based primary care pharmacists and physicians but has yet to be implemented in other areas of the facility. The purpose of this project was to determine the impact of the VIONE tool on polypharmacy and cost avoidance at VASNHS when used by primary care physicians (PCPs) and PACT primary care clinics.
Methods
Veterans receiving care at VASNHS aged ≥ 65 years with ≥ 10 active medications noted in CPRS were included in this project. PACT pharmacists and physicians were educated on the proper use of the VIONE tool prior to its implementation. Education included a 15-minute slide presentation followed by dissemination of a 1-page VIONE tool handout during a PACT all-staff clinic meeting.
Data were collected for 3 months before and after the intervention. Data were made available for assessment by the Automated Data Processing Application Coordinator (ADPAC) at VASNHS. The ADPAC created and generated an Excel spreadsheet report, which listed all medications deprescribed using the VIONE method. The primary endpoint was the total number of medications discontinued using the VIONE template and/or discontinuation menu. For the purpose of this project, appropriate discontinuation was considered any prescription deprescribed, excluding medical supplies, by pharmacists and PCPs who received VIONE education.
![]()
The secondary endpoint was the estimated annualized cost avoidance for the facility (Figure). The calculation does not include medications discontinued due to the prescription of an alternative medication or dose decreases since these VIONE selections imply that a new prescription or order was placed and the original prescription was not deprescribed. Annualized cost avoidance was determined with use of the VIONE dashboard, a database that retrospectively gathers information regarding patients at risk of polypharmacy, polypharmacy-related ADEs, and cost. Manual adjustments were made to various parameters on the Veterans Integrated Service Network 15 VIONE dashboard by the author in order to obtain data specific to this project. These parameters allowed selection of service sections, specific staff members or the option to include or exclude chronic or nonchronic medications. The annualized cost avoidance figure was then compared to raw data pulled by a VIONE dashboard correspondent to ensure the manual calculation was accurate. Finally, the 5 most common classes of medications deprescribed were identified for information purposes and to provide a better postulation on the types of medications being discontinued using the VIONE method.
Results
A total of 2,442 veterans met inclusion criteria, and the VIONE method was applied to 598 between late October 2018 and January 2019. The 13 PACT pharmacists contacted at least 10 veterans each, thus at least 130 were randomly selected for telephone calls to perform polypharmacy reviews using the VIONE note template. The discontinuation menu was used if a medication qualified to be deprescribed. After 3 months, 1986 prescriptions were deprescribed using VIONE; however, 1060 prescriptions were considered appropriately deprescribed (Table 2). The 13 PACT pharmacists deprescribed 361 medications, and the 29 PACT physicians deprescribed 699 medications. These prescriptions were then separated into medication categories to determine the most common discontinued classes. Vitamins and supplements were the medication class most frequently deprescribed (19.4%), followed by pain medications (15.5%), antimicrobial agents (9.6%), antihypertensive medications (9.2%), and diabetes medications (6.4%) (Table 3). The top 5 medication categories accounted for 60% of all medications appropriately deprescribed.
The estimated annualized cost avoidance for all medications deprescribed in the 3-month project period was $84,030.46. To provide the most appropriate and accurate calculation, medication classes excluded from this figure were acute or short-term prescriptions and antimicrobial agents. Medications prescribed short-term typically are not suitable to continue for an extended period, and antimicrobial agents were excluded since they are normally associated with higher costs, and may overestimate the cost avoidance calculation for the facility.
Discussion
The outcomes for the primary and secondary endpoints of this project illustrate that using VIONE in PACT primary care clinics had a notable impact on polypharmacy and cost avoidance over a short period. This outcome can be attributed to 2 significant effects of using the deprescribing tool. VIONE’s simplicity in application allowed clinicians to incorporate daily use of the tool with minimal effort. Education was all that was required to fully enable clinicians to work together successfully and exercise collaborative practice to promote deprescribing. VIONE also elicited a cascade of favorable effects that improve patient safety and health outcomes. The tool aided in identification of PIM, which helped reduce polypharmacy and medication burden. The risk for DDIs and ADEs may decrease; therefore, the incidence of falls, need for emergency department visits or inpatient care related to polypharmacy may decline. Less complex medication regimens may alleviate issues with adherence and avoid the various consequences of polypharmacy in theory. Simplified regimens can potentially improve disease management and quality of life for patients. Further studies are needed to substantiate deprescribing and its true effect on patient adherence and better health outcomes at this time.10
Reducing polypharmacy can lead to cost savings. Based on the results of this 3-month study, we expect that VASNHS would save more than $84,000 by reducing polypharmacy among its patients. Those savings can be funneled back into the health care system, and allotted to necessary patient care, prescriptions, and health care facility needs.
Limitations
There are some important limitations to this study. Definitions of polypharmacy may vary from one health care facility to another. The cutoffs for polypharmacy may differ, causing the prevalence of polypharmacy and potential costs savings to vary. Use of VIONE may be inconsistent among users if not previously educated or properly trained. For instance, VIONE selections are listed in the same menu as the standard CPRS discontinuation options, which may lead to discontinuation of medical supplies or laboratory orders instead of prescriptions.
The method of data analysis and project design used in this study may have been subject to error. For example, the list of PCPs may have been inaccurate or outdated, which would result in an over- or underrepresentation of those who contributed to data collection. Furthermore, there is some volatility in calculating the total cost avoidance. For example, medications for chronic conditions that were only taken on an as needed basis may have overestimated savings. Either under- or overestimations could occur when parameters are adjusted on the VIONE discontinuation dashboard without appropriate guidance. With the ability to manually adjust the dashboard parameters, dissimilarities in calculations may follow.
Conclusions
The VIONE tool may be useful in improving patient safety through deprescribing and discontinuing PIM. Decreasing the number of medications being taken concomitantly by a patient and continuing only those that are imperative in their medical treatment is the first step to reducing the incidence of polypharmacy. Consequently, chances of ADEs or DDIs are lessened, especially among older individuals who are considered high risk for experiencing the detrimental effects that may ensue. These effects include geriatric-related syndromes, increased risk of fall, hospital visits or admissions, or death. Use of VIONE easily promotes collaboration among clinicians to evaluate medications eligible for discontinuation more regularly. If this deprescribing tool is continuously used, costs avoided can likely be maximized within VA health care systems.
The results of this project should serve as an incentive to push for better prescribing practices and increase deprescribing efforts. It should provoke the need for change in regimens and the subsequent discontinuation of prescriptions that are not considered vital to continue. Finally, the result of this project should substantiate the positive impact a deprescribing tool can possess to avert the issues commonly associated with polypharmacy.
1. Centers for Disease Control and Prevention, National Center for Health Statistics. Health, United States, 2013: with special feature on prescription drugs. Published May 2014. Accessed May 13, 2021. https://www.cdc.gov/nchs/data/hus/hus13.pdf
2. Masnoon N, Shakib S, Kalisch-Ellett L, Caughey GE. What is polypharmacy? A systematic review of definitions. BMC Geriatr. 2017;17(1):230. Published 2017 Oct 10. doi:10.1186/s12877-017-0621-2
3. Parulekar MS, Rogers CK. Polypharmacy and mobility. In: Cifu DX, Lew HL, Oh-Park M., eds Geriatric Rehabilitation. Elsevier; 2018. doi:10.1016/B978-0-323-54454-2.12001-1
4. Rieckert A, Trampisch US, Klaaßen-Mielke R, et al. Polypharmacy in older patients with chronic diseases: a cross-sectional analysis of factors associated with excessive polypharmacy. BMC Fam Pract. 2018;19(1):113. Published 2018 Jul 18. doi:10.1186/s12875-018-0795-5
5. Thompson CA. New medication review method cuts veterans’ Rx load, saves millions. Am J Health Syst Pharm. 2018;75(8):502-503. doi:10.2146/news180023
6. Reeve E. Deprescribing tools: a review of the types of tools available to aid deprescribing in clinical practice. J Pharm Pract Res. 2020;50(1):98-107. doi:10.1002/jppr.1626
7. Fried TR, Niehoff KM, Street RL, et al. Effect of the Tool to Reduce Inappropriate Medications on Medication Communication and Deprescribing. J Am Geriatr Soc. 2017;65(10):2265-2271. doi:10.1111/jgs.15042
8. Battar S, Dickerson KR, Sedgwick C, et al. Understanding principles of high reliability organizations through the eyes of VIONE, a clinical program to improve patient safety by deprescribing potentially inappropriate medications and reducing polypharmacy. Fed Pract. 2019;36(12):564-568.
9. Battar S, Cmelik T, Dickerson K, Scott, M. Experience better health with VIONE a safe medication deprescribing tool [Nonpublic source, not verified]
10. Ulley J, Harrop D, Ali A, et al. Desprescribing interventions and their impact on medication adherence in community-dwelling older adults with polypharmacy: a systematic review. BMC Geriatr. 2019;19(15):1-13.
1. Centers for Disease Control and Prevention, National Center for Health Statistics. Health, United States, 2013: with special feature on prescription drugs. Published May 2014. Accessed May 13, 2021. https://www.cdc.gov/nchs/data/hus/hus13.pdf
2. Masnoon N, Shakib S, Kalisch-Ellett L, Caughey GE. What is polypharmacy? A systematic review of definitions. BMC Geriatr. 2017;17(1):230. Published 2017 Oct 10. doi:10.1186/s12877-017-0621-2
3. Parulekar MS, Rogers CK. Polypharmacy and mobility. In: Cifu DX, Lew HL, Oh-Park M., eds Geriatric Rehabilitation. Elsevier; 2018. doi:10.1016/B978-0-323-54454-2.12001-1
4. Rieckert A, Trampisch US, Klaaßen-Mielke R, et al. Polypharmacy in older patients with chronic diseases: a cross-sectional analysis of factors associated with excessive polypharmacy. BMC Fam Pract. 2018;19(1):113. Published 2018 Jul 18. doi:10.1186/s12875-018-0795-5
5. Thompson CA. New medication review method cuts veterans’ Rx load, saves millions. Am J Health Syst Pharm. 2018;75(8):502-503. doi:10.2146/news180023
6. Reeve E. Deprescribing tools: a review of the types of tools available to aid deprescribing in clinical practice. J Pharm Pract Res. 2020;50(1):98-107. doi:10.1002/jppr.1626
7. Fried TR, Niehoff KM, Street RL, et al. Effect of the Tool to Reduce Inappropriate Medications on Medication Communication and Deprescribing. J Am Geriatr Soc. 2017;65(10):2265-2271. doi:10.1111/jgs.15042
8. Battar S, Dickerson KR, Sedgwick C, et al. Understanding principles of high reliability organizations through the eyes of VIONE, a clinical program to improve patient safety by deprescribing potentially inappropriate medications and reducing polypharmacy. Fed Pract. 2019;36(12):564-568.
9. Battar S, Cmelik T, Dickerson K, Scott, M. Experience better health with VIONE a safe medication deprescribing tool [Nonpublic source, not verified]
10. Ulley J, Harrop D, Ali A, et al. Desprescribing interventions and their impact on medication adherence in community-dwelling older adults with polypharmacy: a systematic review. BMC Geriatr. 2019;19(15):1-13.
FDA updates label for controversial Alzheimer’s drug aducanumab (Aduhelm)
– the group studied in the clinical trials.
The FDA approved aducanumab in early June amid significant controversy and disregarding the recommendation by its own advisory panel not to approve the drug. The original prescribing information implied that the drug – which is administered intravenously and costs around $56,000 a year – could be used for treatment of any patient with Alzheimer’s disease.
The updated label now states that aducanumab should be initiated only in patients with mild cognitive impairment (MCI) or mild dementia stage of disease – the population in which treatment was initiated in the clinical trials leading to approval of the anti-amyloid drug.
The FDA granted accelerated approval of the drug based on data from clinical trials showing a reduction in amyloid beta plaques observed in patients with MCI or mild dementia stage of disease.
“Continued approval for the indication may be contingent upon verification of clinical benefit in confirmatory trial(s),” the label states. It emphasizes that there are no safety or effectiveness data on starting aducanumab treatment at earlier or later stages of the disease than were studied.
“Based on our ongoing conversations with prescribing physicians, FDA, and patient advocates, we submitted this label update with the goal to further clarify the patient population that was studied across the three Aduhelm clinical trials that supported approval,” Alfred Sandrock Jr., MD, PhD, Biogen’s head of research and development, said in a statement announcing the label update.
“We are committed to continue to listen to the community’s needs as clinical practice adapts to this important, first-in-class treatment option,” said Dr. Sandrock.
A version of this article first appeared on Medscape.com.
– the group studied in the clinical trials.
The FDA approved aducanumab in early June amid significant controversy and disregarding the recommendation by its own advisory panel not to approve the drug. The original prescribing information implied that the drug – which is administered intravenously and costs around $56,000 a year – could be used for treatment of any patient with Alzheimer’s disease.
The updated label now states that aducanumab should be initiated only in patients with mild cognitive impairment (MCI) or mild dementia stage of disease – the population in which treatment was initiated in the clinical trials leading to approval of the anti-amyloid drug.
The FDA granted accelerated approval of the drug based on data from clinical trials showing a reduction in amyloid beta plaques observed in patients with MCI or mild dementia stage of disease.
“Continued approval for the indication may be contingent upon verification of clinical benefit in confirmatory trial(s),” the label states. It emphasizes that there are no safety or effectiveness data on starting aducanumab treatment at earlier or later stages of the disease than were studied.
“Based on our ongoing conversations with prescribing physicians, FDA, and patient advocates, we submitted this label update with the goal to further clarify the patient population that was studied across the three Aduhelm clinical trials that supported approval,” Alfred Sandrock Jr., MD, PhD, Biogen’s head of research and development, said in a statement announcing the label update.
“We are committed to continue to listen to the community’s needs as clinical practice adapts to this important, first-in-class treatment option,” said Dr. Sandrock.
A version of this article first appeared on Medscape.com.
– the group studied in the clinical trials.
The FDA approved aducanumab in early June amid significant controversy and disregarding the recommendation by its own advisory panel not to approve the drug. The original prescribing information implied that the drug – which is administered intravenously and costs around $56,000 a year – could be used for treatment of any patient with Alzheimer’s disease.
The updated label now states that aducanumab should be initiated only in patients with mild cognitive impairment (MCI) or mild dementia stage of disease – the population in which treatment was initiated in the clinical trials leading to approval of the anti-amyloid drug.
The FDA granted accelerated approval of the drug based on data from clinical trials showing a reduction in amyloid beta plaques observed in patients with MCI or mild dementia stage of disease.
“Continued approval for the indication may be contingent upon verification of clinical benefit in confirmatory trial(s),” the label states. It emphasizes that there are no safety or effectiveness data on starting aducanumab treatment at earlier or later stages of the disease than were studied.
“Based on our ongoing conversations with prescribing physicians, FDA, and patient advocates, we submitted this label update with the goal to further clarify the patient population that was studied across the three Aduhelm clinical trials that supported approval,” Alfred Sandrock Jr., MD, PhD, Biogen’s head of research and development, said in a statement announcing the label update.
“We are committed to continue to listen to the community’s needs as clinical practice adapts to this important, first-in-class treatment option,” said Dr. Sandrock.
A version of this article first appeared on Medscape.com.
Extra COVID-19 vaccine could help immunocompromised people
People whose immune systems are compromised by therapy or disease may benefit from additional doses of vaccines against SARS-CoV-2, researchers say.
In a study involving 101 people with solid-organ transplants, there was a significant boost in antibodies after the patients received third doses of the Pfizer vaccine, said Nassim Kamar, MD, PhD, professor of nephrology at Toulouse University Hospital, France.
None of the transplant patients had antibodies against the virus before their first dose of the vaccine, and only 4% produced antibodies after the first dose. That proportion rose to 40% after the second dose and to 68% after the third dose.
The effect is so strong that Dr. Kamar and colleagues at Toulouse University Hospital routinely administer three doses of mRNA vaccines to patients with solid-organ transplant without testing them for antibodies.
“When we observed that the second dose was not sufficient to have an immune response, the Francophone Society of Transplantation asked the National Health Authority to allow the third dose,” he told this news organization.
That agency on April 11 approved third doses of mRNA vaccines not only for people with solid-organ transplants but also for those with recent bone marrow transplants, those undergoing dialysis, and those with autoimmune diseases who were receiving strong immunosuppressive treatment, such as anti-CD20 or antimetabolites. Contrary to their procedure for people with solid-organ transplants, clinicians at Toulouse University Hospital test these patients for antibodies and administer third doses of vaccine only to those who test negative or have very low titers.
The researchers’ findings, published on June 23 as a letter to the editor of The New England Journal of Medicine, come as other researchers document more and more categories of patients whose responses to the vaccines typically fall short.
A study at the University of Pittsburgh that was published as a preprint on MedRxiv compared people with various health conditions to healthy health care workers. People with HIV who were taking antivirals against that virus responded almost as well as did the health care workers, said John Mellors, MD, chief of infectious diseases at the university. But people whose immune systems were compromised for other reasons fared less well.
“The areas of concern are hematological malignancy and solid-organ transplants, with the most nonresponsive groups being those who have had lung transplantation,” he said in an interview.
For patients with liver disease, mixed news came from the International Liver Congress (ILC) 2021 annual meeting.
In a study involving patients with liver disease who had received the Pfizer vaccine at Hadassah University Medical Center in Jerusalem, antibody titers were lower in patients who had received liver transplants or who had advanced liver fibrosis, as reported by this news organization.
A multicenter study in China that was presented at the ILC meeting and that was also published in the Journal of Hepatology, provided a more optimistic picture. Among patients with nonalcoholic fatty liver disease who were immunized against SARS-CoV-2 with the Sinopharm vaccine, 95.5% had neutralizing antibodies; the median neutralizing antibody titer was 32.
In the Toulouse University Hospital study, for the 40 patients who were seropositive after the second dose, antibody titers increased from 36 before the third dose to 2,676 a month after the third dose, a statistically significant result (P < .001).
For patients whose immune systems are compromised for reasons other than having received a transplant, clinicians at Toulouse University Hospital use a titer of 14 as the threshold below which they administer a third dose of mRNA vaccines. But Dr. Kamar acknowledged that the threshold is arbitrary and that the assays for antibodies with different vaccines in different populations can’t be compared head to head.
“We can’t tell you simply on the basis of the amount of antibody in your laboratory report how protected you are,” agreed William Schaffner, MD, professor of infectious diseases at Vanderbilt University, Nashville, Tenn., who is a spokesperson for the Infectious Diseases Society of America.
Not enough research has been done to establish that relationship, and results vary from one laboratory to another, he said.
That doesn’t mean that antibody tests don’t help, Dr. Schaffner said. On the basis of views of other experts he has consulted, Dr. Schaffner recommends that people who are immunocompromised undergo an antibody test. If the test is positive – meaning they have some antibodies to SARS-CoV-2, however low the titers – patients can take fewer precautions than before they were vaccinated.
But they should still be more cautious than people with healthy immune systems, he said. “Would I be going to large indoor gatherings without a mask? No. Would I be outside without a mask? Yes. Would I gather with three other people who are vaccinated to play a game of bridge? Yes. You have to titrate things a little and use some common sense,” he added.
If the results are negative, such patients may still be protected. Much research remains to be done on T-cell immunity, a second line of defense against the virus. And the current assays often produce false negative results. But to be on the safe side, people with this result should assume that their vaccine is not protecting them, Dr. Schaffner said.
That suggestion contradicts the Food and Drug Administration, which issued a recommendation on May 19 against using antibody tests to check the effectiveness of SARS-CoV-2 vaccination.
The studies so far suggest that vaccines are safe for people whose immune systems are compromised, Dr. Schaffner and Dr. Kamar agreed. Dr. Kamar is aware of only two case reports of transplant patients rejecting their transplants after vaccination. One of these was his own patient, and the rejection occurred after her second dose. She has not needed dialysis, although her kidney function was impaired.
But the FDA has not approved additional doses of SARS-CoV-2 vaccine to treat patients who are immunocompromised, and Dr. Kamar has not heard of any other national regulatory agencies that have.
In the United States, it may be difficult for anyone to obtain a third dose of vaccine outside of a clinical trial, Dr. Schaffner said, because vaccinators are likely to check databases and deny vaccination to anyone who has already received the recommended number.
Dr. Kamar, Dr. Mellors, and Dr. Schaffner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
People whose immune systems are compromised by therapy or disease may benefit from additional doses of vaccines against SARS-CoV-2, researchers say.
In a study involving 101 people with solid-organ transplants, there was a significant boost in antibodies after the patients received third doses of the Pfizer vaccine, said Nassim Kamar, MD, PhD, professor of nephrology at Toulouse University Hospital, France.
None of the transplant patients had antibodies against the virus before their first dose of the vaccine, and only 4% produced antibodies after the first dose. That proportion rose to 40% after the second dose and to 68% after the third dose.
The effect is so strong that Dr. Kamar and colleagues at Toulouse University Hospital routinely administer three doses of mRNA vaccines to patients with solid-organ transplant without testing them for antibodies.
“When we observed that the second dose was not sufficient to have an immune response, the Francophone Society of Transplantation asked the National Health Authority to allow the third dose,” he told this news organization.
That agency on April 11 approved third doses of mRNA vaccines not only for people with solid-organ transplants but also for those with recent bone marrow transplants, those undergoing dialysis, and those with autoimmune diseases who were receiving strong immunosuppressive treatment, such as anti-CD20 or antimetabolites. Contrary to their procedure for people with solid-organ transplants, clinicians at Toulouse University Hospital test these patients for antibodies and administer third doses of vaccine only to those who test negative or have very low titers.
The researchers’ findings, published on June 23 as a letter to the editor of The New England Journal of Medicine, come as other researchers document more and more categories of patients whose responses to the vaccines typically fall short.
A study at the University of Pittsburgh that was published as a preprint on MedRxiv compared people with various health conditions to healthy health care workers. People with HIV who were taking antivirals against that virus responded almost as well as did the health care workers, said John Mellors, MD, chief of infectious diseases at the university. But people whose immune systems were compromised for other reasons fared less well.
“The areas of concern are hematological malignancy and solid-organ transplants, with the most nonresponsive groups being those who have had lung transplantation,” he said in an interview.
For patients with liver disease, mixed news came from the International Liver Congress (ILC) 2021 annual meeting.
In a study involving patients with liver disease who had received the Pfizer vaccine at Hadassah University Medical Center in Jerusalem, antibody titers were lower in patients who had received liver transplants or who had advanced liver fibrosis, as reported by this news organization.
A multicenter study in China that was presented at the ILC meeting and that was also published in the Journal of Hepatology, provided a more optimistic picture. Among patients with nonalcoholic fatty liver disease who were immunized against SARS-CoV-2 with the Sinopharm vaccine, 95.5% had neutralizing antibodies; the median neutralizing antibody titer was 32.
In the Toulouse University Hospital study, for the 40 patients who were seropositive after the second dose, antibody titers increased from 36 before the third dose to 2,676 a month after the third dose, a statistically significant result (P < .001).
For patients whose immune systems are compromised for reasons other than having received a transplant, clinicians at Toulouse University Hospital use a titer of 14 as the threshold below which they administer a third dose of mRNA vaccines. But Dr. Kamar acknowledged that the threshold is arbitrary and that the assays for antibodies with different vaccines in different populations can’t be compared head to head.
“We can’t tell you simply on the basis of the amount of antibody in your laboratory report how protected you are,” agreed William Schaffner, MD, professor of infectious diseases at Vanderbilt University, Nashville, Tenn., who is a spokesperson for the Infectious Diseases Society of America.
Not enough research has been done to establish that relationship, and results vary from one laboratory to another, he said.
That doesn’t mean that antibody tests don’t help, Dr. Schaffner said. On the basis of views of other experts he has consulted, Dr. Schaffner recommends that people who are immunocompromised undergo an antibody test. If the test is positive – meaning they have some antibodies to SARS-CoV-2, however low the titers – patients can take fewer precautions than before they were vaccinated.
But they should still be more cautious than people with healthy immune systems, he said. “Would I be going to large indoor gatherings without a mask? No. Would I be outside without a mask? Yes. Would I gather with three other people who are vaccinated to play a game of bridge? Yes. You have to titrate things a little and use some common sense,” he added.
If the results are negative, such patients may still be protected. Much research remains to be done on T-cell immunity, a second line of defense against the virus. And the current assays often produce false negative results. But to be on the safe side, people with this result should assume that their vaccine is not protecting them, Dr. Schaffner said.
That suggestion contradicts the Food and Drug Administration, which issued a recommendation on May 19 against using antibody tests to check the effectiveness of SARS-CoV-2 vaccination.
The studies so far suggest that vaccines are safe for people whose immune systems are compromised, Dr. Schaffner and Dr. Kamar agreed. Dr. Kamar is aware of only two case reports of transplant patients rejecting their transplants after vaccination. One of these was his own patient, and the rejection occurred after her second dose. She has not needed dialysis, although her kidney function was impaired.
But the FDA has not approved additional doses of SARS-CoV-2 vaccine to treat patients who are immunocompromised, and Dr. Kamar has not heard of any other national regulatory agencies that have.
In the United States, it may be difficult for anyone to obtain a third dose of vaccine outside of a clinical trial, Dr. Schaffner said, because vaccinators are likely to check databases and deny vaccination to anyone who has already received the recommended number.
Dr. Kamar, Dr. Mellors, and Dr. Schaffner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
People whose immune systems are compromised by therapy or disease may benefit from additional doses of vaccines against SARS-CoV-2, researchers say.
In a study involving 101 people with solid-organ transplants, there was a significant boost in antibodies after the patients received third doses of the Pfizer vaccine, said Nassim Kamar, MD, PhD, professor of nephrology at Toulouse University Hospital, France.
None of the transplant patients had antibodies against the virus before their first dose of the vaccine, and only 4% produced antibodies after the first dose. That proportion rose to 40% after the second dose and to 68% after the third dose.
The effect is so strong that Dr. Kamar and colleagues at Toulouse University Hospital routinely administer three doses of mRNA vaccines to patients with solid-organ transplant without testing them for antibodies.
“When we observed that the second dose was not sufficient to have an immune response, the Francophone Society of Transplantation asked the National Health Authority to allow the third dose,” he told this news organization.
That agency on April 11 approved third doses of mRNA vaccines not only for people with solid-organ transplants but also for those with recent bone marrow transplants, those undergoing dialysis, and those with autoimmune diseases who were receiving strong immunosuppressive treatment, such as anti-CD20 or antimetabolites. Contrary to their procedure for people with solid-organ transplants, clinicians at Toulouse University Hospital test these patients for antibodies and administer third doses of vaccine only to those who test negative or have very low titers.
The researchers’ findings, published on June 23 as a letter to the editor of The New England Journal of Medicine, come as other researchers document more and more categories of patients whose responses to the vaccines typically fall short.
A study at the University of Pittsburgh that was published as a preprint on MedRxiv compared people with various health conditions to healthy health care workers. People with HIV who were taking antivirals against that virus responded almost as well as did the health care workers, said John Mellors, MD, chief of infectious diseases at the university. But people whose immune systems were compromised for other reasons fared less well.
“The areas of concern are hematological malignancy and solid-organ transplants, with the most nonresponsive groups being those who have had lung transplantation,” he said in an interview.
For patients with liver disease, mixed news came from the International Liver Congress (ILC) 2021 annual meeting.
In a study involving patients with liver disease who had received the Pfizer vaccine at Hadassah University Medical Center in Jerusalem, antibody titers were lower in patients who had received liver transplants or who had advanced liver fibrosis, as reported by this news organization.
A multicenter study in China that was presented at the ILC meeting and that was also published in the Journal of Hepatology, provided a more optimistic picture. Among patients with nonalcoholic fatty liver disease who were immunized against SARS-CoV-2 with the Sinopharm vaccine, 95.5% had neutralizing antibodies; the median neutralizing antibody titer was 32.
In the Toulouse University Hospital study, for the 40 patients who were seropositive after the second dose, antibody titers increased from 36 before the third dose to 2,676 a month after the third dose, a statistically significant result (P < .001).
For patients whose immune systems are compromised for reasons other than having received a transplant, clinicians at Toulouse University Hospital use a titer of 14 as the threshold below which they administer a third dose of mRNA vaccines. But Dr. Kamar acknowledged that the threshold is arbitrary and that the assays for antibodies with different vaccines in different populations can’t be compared head to head.
“We can’t tell you simply on the basis of the amount of antibody in your laboratory report how protected you are,” agreed William Schaffner, MD, professor of infectious diseases at Vanderbilt University, Nashville, Tenn., who is a spokesperson for the Infectious Diseases Society of America.
Not enough research has been done to establish that relationship, and results vary from one laboratory to another, he said.
That doesn’t mean that antibody tests don’t help, Dr. Schaffner said. On the basis of views of other experts he has consulted, Dr. Schaffner recommends that people who are immunocompromised undergo an antibody test. If the test is positive – meaning they have some antibodies to SARS-CoV-2, however low the titers – patients can take fewer precautions than before they were vaccinated.
But they should still be more cautious than people with healthy immune systems, he said. “Would I be going to large indoor gatherings without a mask? No. Would I be outside without a mask? Yes. Would I gather with three other people who are vaccinated to play a game of bridge? Yes. You have to titrate things a little and use some common sense,” he added.
If the results are negative, such patients may still be protected. Much research remains to be done on T-cell immunity, a second line of defense against the virus. And the current assays often produce false negative results. But to be on the safe side, people with this result should assume that their vaccine is not protecting them, Dr. Schaffner said.
That suggestion contradicts the Food and Drug Administration, which issued a recommendation on May 19 against using antibody tests to check the effectiveness of SARS-CoV-2 vaccination.
The studies so far suggest that vaccines are safe for people whose immune systems are compromised, Dr. Schaffner and Dr. Kamar agreed. Dr. Kamar is aware of only two case reports of transplant patients rejecting their transplants after vaccination. One of these was his own patient, and the rejection occurred after her second dose. She has not needed dialysis, although her kidney function was impaired.
But the FDA has not approved additional doses of SARS-CoV-2 vaccine to treat patients who are immunocompromised, and Dr. Kamar has not heard of any other national regulatory agencies that have.
In the United States, it may be difficult for anyone to obtain a third dose of vaccine outside of a clinical trial, Dr. Schaffner said, because vaccinators are likely to check databases and deny vaccination to anyone who has already received the recommended number.
Dr. Kamar, Dr. Mellors, and Dr. Schaffner have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Heart failure med undertreatment because of older age common, flouts evidence
, suggests a large cohort study.
About 80% of patients aged 80 years or older were prescribed renin-angiotensin-system inhibitors (RASi) in a multivariate-adjusted analysis of more than 27,000 patients in the Swedish Heart Failure Registry (SwedeHF). In contrast, such drugs – which included angiotensin receptor-neprilysin inhibitors (ARNi), angiotensin receptor blockers, and ACE inhibitors – were prescribed to 95% of patients younger than 70 years.
Similarly, fewer of the oldest patients were offered meds from the two other drug classes core to HF management at the time: Beta blockers and mineralocorticoid receptor antagonists (MRA).
And among those in the 80-and-older age group who were prescribed RASi or beta blockers, their uptitration more often fell short of even half the target dosage, compared with the youngest patients in the analysis.
Physicians may hold back on full guideline-directed medical therapy in their very elderly patients with HFrEF for many reasons, including a perceived likelihood of drug intolerance due to frailty or multiple comorbidities, including renal dysfunction, Davide Stolfo, MD, Karolinska Institutet, Stockholm, and University of Trieste, Italy, told this news organization.
But the current analysis was adjusted for about 80 variables “that in our interpretation may be main reasons for not introducing drugs and using them in the older patients,” he said. They included care setting (that is, inpatient or outpatient), HF severity by several measures, a range of comorbidities, renal dysfunction, and history of serious illness such as cancer.
Even then, age emerged as a significant, independent predictor of medical therapy underuse in the oldest patients. Some physicians apparently see advanced age, by itself, as an “intrinsic reason” not to abide by HFrEF medical therapy recommendations, said Dr. Stolfo, who presented the analysis at HFA 2021, the annual meeting of the Heart Failure Association of the European Society of Cardiology (ESC-HFA), conducted both virtually and live in Florence, Italy.
Most major HF-drug trials have excluded or admitted few patients aged 80 years or older, but “the guidelines recommend treatment regardless of age, and in the trials there has been no influence from age on the effectiveness of drugs,” Dr. Stolfo observed.
Moreover, in a prior SwedeHF analysis with propensity matching, patients with HFrEF aged 80 or older showed steeper reductions in risk for death or HF hospitalization from treatment with RASi than those in younger age groups.
One of the few randomized trials to focus on the very elderly, called SENIORS, enrolled patients aged 70 years and older – the average age was 76 – and saw a significantly reduced risk of death or cardiovascular hospitalization for those assigned to the beta blocker nebivolol. The benefits in the trial, which was conducted 15 years ago, were independent of left ventricular function.
So in the oldest patients, “we could question the need to achieve full dose of an evidence-based drug, but we shouldn’t question the use of these drugs.”
The findings are consistent with a need to individualize medical therapy in senior patients with HFrEF, especially those of more advanced age, some of whom may be robust enough to be managed similarly to younger patients while others who may be less suitable for full guideline-directed medical therapy, Dr. Stolfo said.
Even for those who are more frail or have major comorbidities, drug therapy of HFrEF continues to be important for symptom control even if competing causes of death make it harder to prolong survival, Dr. Stolfo said.
“We should provide to all patients the best strategy they can tolerate,” he said. “If we cannot greatly impact on the long-term survival for these patients, treatment can be aimed to improve the quality of life and keep the patient out of the hospital.”
The analysis was supported by Boehringer Ingelheim. Dr. Stolfo disclosed personal fees from Novartis, Merck, GlaxoSmithKline, and Acceleron.
A version of this article first appeared on Medscape.com.
, suggests a large cohort study.
About 80% of patients aged 80 years or older were prescribed renin-angiotensin-system inhibitors (RASi) in a multivariate-adjusted analysis of more than 27,000 patients in the Swedish Heart Failure Registry (SwedeHF). In contrast, such drugs – which included angiotensin receptor-neprilysin inhibitors (ARNi), angiotensin receptor blockers, and ACE inhibitors – were prescribed to 95% of patients younger than 70 years.
Similarly, fewer of the oldest patients were offered meds from the two other drug classes core to HF management at the time: Beta blockers and mineralocorticoid receptor antagonists (MRA).
And among those in the 80-and-older age group who were prescribed RASi or beta blockers, their uptitration more often fell short of even half the target dosage, compared with the youngest patients in the analysis.
Physicians may hold back on full guideline-directed medical therapy in their very elderly patients with HFrEF for many reasons, including a perceived likelihood of drug intolerance due to frailty or multiple comorbidities, including renal dysfunction, Davide Stolfo, MD, Karolinska Institutet, Stockholm, and University of Trieste, Italy, told this news organization.
But the current analysis was adjusted for about 80 variables “that in our interpretation may be main reasons for not introducing drugs and using them in the older patients,” he said. They included care setting (that is, inpatient or outpatient), HF severity by several measures, a range of comorbidities, renal dysfunction, and history of serious illness such as cancer.
Even then, age emerged as a significant, independent predictor of medical therapy underuse in the oldest patients. Some physicians apparently see advanced age, by itself, as an “intrinsic reason” not to abide by HFrEF medical therapy recommendations, said Dr. Stolfo, who presented the analysis at HFA 2021, the annual meeting of the Heart Failure Association of the European Society of Cardiology (ESC-HFA), conducted both virtually and live in Florence, Italy.
Most major HF-drug trials have excluded or admitted few patients aged 80 years or older, but “the guidelines recommend treatment regardless of age, and in the trials there has been no influence from age on the effectiveness of drugs,” Dr. Stolfo observed.
Moreover, in a prior SwedeHF analysis with propensity matching, patients with HFrEF aged 80 or older showed steeper reductions in risk for death or HF hospitalization from treatment with RASi than those in younger age groups.
One of the few randomized trials to focus on the very elderly, called SENIORS, enrolled patients aged 70 years and older – the average age was 76 – and saw a significantly reduced risk of death or cardiovascular hospitalization for those assigned to the beta blocker nebivolol. The benefits in the trial, which was conducted 15 years ago, were independent of left ventricular function.
So in the oldest patients, “we could question the need to achieve full dose of an evidence-based drug, but we shouldn’t question the use of these drugs.”
The findings are consistent with a need to individualize medical therapy in senior patients with HFrEF, especially those of more advanced age, some of whom may be robust enough to be managed similarly to younger patients while others who may be less suitable for full guideline-directed medical therapy, Dr. Stolfo said.
Even for those who are more frail or have major comorbidities, drug therapy of HFrEF continues to be important for symptom control even if competing causes of death make it harder to prolong survival, Dr. Stolfo said.
“We should provide to all patients the best strategy they can tolerate,” he said. “If we cannot greatly impact on the long-term survival for these patients, treatment can be aimed to improve the quality of life and keep the patient out of the hospital.”
The analysis was supported by Boehringer Ingelheim. Dr. Stolfo disclosed personal fees from Novartis, Merck, GlaxoSmithKline, and Acceleron.
A version of this article first appeared on Medscape.com.
, suggests a large cohort study.
About 80% of patients aged 80 years or older were prescribed renin-angiotensin-system inhibitors (RASi) in a multivariate-adjusted analysis of more than 27,000 patients in the Swedish Heart Failure Registry (SwedeHF). In contrast, such drugs – which included angiotensin receptor-neprilysin inhibitors (ARNi), angiotensin receptor blockers, and ACE inhibitors – were prescribed to 95% of patients younger than 70 years.
Similarly, fewer of the oldest patients were offered meds from the two other drug classes core to HF management at the time: Beta blockers and mineralocorticoid receptor antagonists (MRA).
And among those in the 80-and-older age group who were prescribed RASi or beta blockers, their uptitration more often fell short of even half the target dosage, compared with the youngest patients in the analysis.
Physicians may hold back on full guideline-directed medical therapy in their very elderly patients with HFrEF for many reasons, including a perceived likelihood of drug intolerance due to frailty or multiple comorbidities, including renal dysfunction, Davide Stolfo, MD, Karolinska Institutet, Stockholm, and University of Trieste, Italy, told this news organization.
But the current analysis was adjusted for about 80 variables “that in our interpretation may be main reasons for not introducing drugs and using them in the older patients,” he said. They included care setting (that is, inpatient or outpatient), HF severity by several measures, a range of comorbidities, renal dysfunction, and history of serious illness such as cancer.
Even then, age emerged as a significant, independent predictor of medical therapy underuse in the oldest patients. Some physicians apparently see advanced age, by itself, as an “intrinsic reason” not to abide by HFrEF medical therapy recommendations, said Dr. Stolfo, who presented the analysis at HFA 2021, the annual meeting of the Heart Failure Association of the European Society of Cardiology (ESC-HFA), conducted both virtually and live in Florence, Italy.
Most major HF-drug trials have excluded or admitted few patients aged 80 years or older, but “the guidelines recommend treatment regardless of age, and in the trials there has been no influence from age on the effectiveness of drugs,” Dr. Stolfo observed.
Moreover, in a prior SwedeHF analysis with propensity matching, patients with HFrEF aged 80 or older showed steeper reductions in risk for death or HF hospitalization from treatment with RASi than those in younger age groups.
One of the few randomized trials to focus on the very elderly, called SENIORS, enrolled patients aged 70 years and older – the average age was 76 – and saw a significantly reduced risk of death or cardiovascular hospitalization for those assigned to the beta blocker nebivolol. The benefits in the trial, which was conducted 15 years ago, were independent of left ventricular function.
So in the oldest patients, “we could question the need to achieve full dose of an evidence-based drug, but we shouldn’t question the use of these drugs.”
The findings are consistent with a need to individualize medical therapy in senior patients with HFrEF, especially those of more advanced age, some of whom may be robust enough to be managed similarly to younger patients while others who may be less suitable for full guideline-directed medical therapy, Dr. Stolfo said.
Even for those who are more frail or have major comorbidities, drug therapy of HFrEF continues to be important for symptom control even if competing causes of death make it harder to prolong survival, Dr. Stolfo said.
“We should provide to all patients the best strategy they can tolerate,” he said. “If we cannot greatly impact on the long-term survival for these patients, treatment can be aimed to improve the quality of life and keep the patient out of the hospital.”
The analysis was supported by Boehringer Ingelheim. Dr. Stolfo disclosed personal fees from Novartis, Merck, GlaxoSmithKline, and Acceleron.
A version of this article first appeared on Medscape.com.
Huge trial casts doubt on bisphosphonates for breast cancer
say researchers reporting new results from a phase 3 trial with almost 3,000 women.
Current guidelines call for 3-5 years of bisphosphonate therapy on the theory that these drugs might reduce breast cancer recurrence as well as treatment-related bone problems.
However, the new results show no difference in disease-free survival, distant disease-free survival, and overall survival – regardless of menopausal status – between the 1,540 women who received intravenous zoledronate over a 5-year period and 1,447 women who received such therapy over a 2-year period.
What they did find was a substantially higher risk for adverse events with prolonged bisphosphonate treatment, including risks for grade 3/4 events, bone pain, bone fractures, arthralgia, and jaw necrosis, a rare but well- recognized possibility with bisphosphonates.
Lead investigator Thomas Friedl, PhD, a statistician at University Hospital Ulm (Germany), and colleagues concluded that the current duration of treatment can be reduced and that, short of good reason to use bisphosphonates longer, such as decreased bone density, “treatment with zoledronate for 5 years should not be considered in patients with early breast cancer.”
The study was published online on June 24 in JAMA Oncology.
An accompanying editorial went even further, stating not only that “shorter duration of treatment is sufficient” but also that the whole idea of bisphosphonates for breast cancer is in doubt.
With “the modest outcomes of bisphosphonates, compared with no bone-targeted therapy, in historical trials” and the low rates of recurrence with modern treatment – less than 10% in the trial – “what, if any, is the benefit from adjuvant bisphosphonates? It’s time to reevaluate the guidelines,” said the editorialists, led by Alexandra Desnoyers, MD, a breast cancer fellow at the University of Toronto.
“We suggest that zoledronate or other amino-bisphosphonates should not be given as standard adjuvant therapy for unselected women with breast cancer,” they wrote.
Risk for necrosis with 5 years of zoledronate
The women in the trial had primary invasive breast cancer and were at high risk for recurrence. They had either positive nodes or high-risk features, including age (median, 53 years). They were treated at 250 centers in Germany.
The first part of the trial was to see whether use of gemcitabine improved outcomes when added to docetaxel after standard fluorouracil, epirubicin, and cyclophosphamide adjuvant therapy following surgery. It did not, and the authors reported in 2020 that adjuvant gemcitabine should not be used in the treatment of high-risk early breast cancer.
The next phase of the trial involved zoledronate. Women were randomly assigned to receive zoledronate for 2 or 5 years after surgery and after undergoing chemotherapy. Dosing was 4 mg IV every 3 months for 2 years. The women in the 5-year group went on to receive 4 mg IV every 6 months for another 3 years.
At a mean of 5 years’ follow-up after the first zoledronate dose, there was no difference in any of the survival measures between the two dosage groups.
There was also no difference in rates of bone recurrence or in circulating tumor cells, which the bisphosphonates theory would have predicted. For instance, 10.5% of women in the 5-year group had one or more circulating tumor cells on follow-up versus 7.2% in the 2-year group.
Almost half of the women in the 5-year treatment group experienced adverse events with zoledronate – including 7.6% with grade 3/4 events – versus just over a quarter in the 2-year arm and only 5.1% with grade 3/4 events.
In the 5-year group, 8.3% of patients experienced bone pain and 5.1% experienced arthralgia versus 3.7% and 3.1%, respectively, in the 2-year arm.
Atypical fractures, such as femoral spiral fractures, are another concern with bisphosphonates. Although this trial did not report on fracture type, fractures were reported in 14 women in the 5-year group but in only 3 in the 2-year arm.
Jaw necrosis, another known adverse effect of bisphosphonates, was reported in 11 women in the 5-year group and in 5 in the 2-year group.
The study was funded by several pharmaceutical companies, including Novartis, the maker of zoledronate. The investigators have numerous industry ties. Dr. Friedl has received payments from Novartis.
A version of this article first appeared on Medscape.com.
say researchers reporting new results from a phase 3 trial with almost 3,000 women.
Current guidelines call for 3-5 years of bisphosphonate therapy on the theory that these drugs might reduce breast cancer recurrence as well as treatment-related bone problems.
However, the new results show no difference in disease-free survival, distant disease-free survival, and overall survival – regardless of menopausal status – between the 1,540 women who received intravenous zoledronate over a 5-year period and 1,447 women who received such therapy over a 2-year period.
What they did find was a substantially higher risk for adverse events with prolonged bisphosphonate treatment, including risks for grade 3/4 events, bone pain, bone fractures, arthralgia, and jaw necrosis, a rare but well- recognized possibility with bisphosphonates.
Lead investigator Thomas Friedl, PhD, a statistician at University Hospital Ulm (Germany), and colleagues concluded that the current duration of treatment can be reduced and that, short of good reason to use bisphosphonates longer, such as decreased bone density, “treatment with zoledronate for 5 years should not be considered in patients with early breast cancer.”
The study was published online on June 24 in JAMA Oncology.
An accompanying editorial went even further, stating not only that “shorter duration of treatment is sufficient” but also that the whole idea of bisphosphonates for breast cancer is in doubt.
With “the modest outcomes of bisphosphonates, compared with no bone-targeted therapy, in historical trials” and the low rates of recurrence with modern treatment – less than 10% in the trial – “what, if any, is the benefit from adjuvant bisphosphonates? It’s time to reevaluate the guidelines,” said the editorialists, led by Alexandra Desnoyers, MD, a breast cancer fellow at the University of Toronto.
“We suggest that zoledronate or other amino-bisphosphonates should not be given as standard adjuvant therapy for unselected women with breast cancer,” they wrote.
Risk for necrosis with 5 years of zoledronate
The women in the trial had primary invasive breast cancer and were at high risk for recurrence. They had either positive nodes or high-risk features, including age (median, 53 years). They were treated at 250 centers in Germany.
The first part of the trial was to see whether use of gemcitabine improved outcomes when added to docetaxel after standard fluorouracil, epirubicin, and cyclophosphamide adjuvant therapy following surgery. It did not, and the authors reported in 2020 that adjuvant gemcitabine should not be used in the treatment of high-risk early breast cancer.
The next phase of the trial involved zoledronate. Women were randomly assigned to receive zoledronate for 2 or 5 years after surgery and after undergoing chemotherapy. Dosing was 4 mg IV every 3 months for 2 years. The women in the 5-year group went on to receive 4 mg IV every 6 months for another 3 years.
At a mean of 5 years’ follow-up after the first zoledronate dose, there was no difference in any of the survival measures between the two dosage groups.
There was also no difference in rates of bone recurrence or in circulating tumor cells, which the bisphosphonates theory would have predicted. For instance, 10.5% of women in the 5-year group had one or more circulating tumor cells on follow-up versus 7.2% in the 2-year group.
Almost half of the women in the 5-year treatment group experienced adverse events with zoledronate – including 7.6% with grade 3/4 events – versus just over a quarter in the 2-year arm and only 5.1% with grade 3/4 events.
In the 5-year group, 8.3% of patients experienced bone pain and 5.1% experienced arthralgia versus 3.7% and 3.1%, respectively, in the 2-year arm.
Atypical fractures, such as femoral spiral fractures, are another concern with bisphosphonates. Although this trial did not report on fracture type, fractures were reported in 14 women in the 5-year group but in only 3 in the 2-year arm.
Jaw necrosis, another known adverse effect of bisphosphonates, was reported in 11 women in the 5-year group and in 5 in the 2-year group.
The study was funded by several pharmaceutical companies, including Novartis, the maker of zoledronate. The investigators have numerous industry ties. Dr. Friedl has received payments from Novartis.
A version of this article first appeared on Medscape.com.
say researchers reporting new results from a phase 3 trial with almost 3,000 women.
Current guidelines call for 3-5 years of bisphosphonate therapy on the theory that these drugs might reduce breast cancer recurrence as well as treatment-related bone problems.
However, the new results show no difference in disease-free survival, distant disease-free survival, and overall survival – regardless of menopausal status – between the 1,540 women who received intravenous zoledronate over a 5-year period and 1,447 women who received such therapy over a 2-year period.
What they did find was a substantially higher risk for adverse events with prolonged bisphosphonate treatment, including risks for grade 3/4 events, bone pain, bone fractures, arthralgia, and jaw necrosis, a rare but well- recognized possibility with bisphosphonates.
Lead investigator Thomas Friedl, PhD, a statistician at University Hospital Ulm (Germany), and colleagues concluded that the current duration of treatment can be reduced and that, short of good reason to use bisphosphonates longer, such as decreased bone density, “treatment with zoledronate for 5 years should not be considered in patients with early breast cancer.”
The study was published online on June 24 in JAMA Oncology.
An accompanying editorial went even further, stating not only that “shorter duration of treatment is sufficient” but also that the whole idea of bisphosphonates for breast cancer is in doubt.
With “the modest outcomes of bisphosphonates, compared with no bone-targeted therapy, in historical trials” and the low rates of recurrence with modern treatment – less than 10% in the trial – “what, if any, is the benefit from adjuvant bisphosphonates? It’s time to reevaluate the guidelines,” said the editorialists, led by Alexandra Desnoyers, MD, a breast cancer fellow at the University of Toronto.
“We suggest that zoledronate or other amino-bisphosphonates should not be given as standard adjuvant therapy for unselected women with breast cancer,” they wrote.
Risk for necrosis with 5 years of zoledronate
The women in the trial had primary invasive breast cancer and were at high risk for recurrence. They had either positive nodes or high-risk features, including age (median, 53 years). They were treated at 250 centers in Germany.
The first part of the trial was to see whether use of gemcitabine improved outcomes when added to docetaxel after standard fluorouracil, epirubicin, and cyclophosphamide adjuvant therapy following surgery. It did not, and the authors reported in 2020 that adjuvant gemcitabine should not be used in the treatment of high-risk early breast cancer.
The next phase of the trial involved zoledronate. Women were randomly assigned to receive zoledronate for 2 or 5 years after surgery and after undergoing chemotherapy. Dosing was 4 mg IV every 3 months for 2 years. The women in the 5-year group went on to receive 4 mg IV every 6 months for another 3 years.
At a mean of 5 years’ follow-up after the first zoledronate dose, there was no difference in any of the survival measures between the two dosage groups.
There was also no difference in rates of bone recurrence or in circulating tumor cells, which the bisphosphonates theory would have predicted. For instance, 10.5% of women in the 5-year group had one or more circulating tumor cells on follow-up versus 7.2% in the 2-year group.
Almost half of the women in the 5-year treatment group experienced adverse events with zoledronate – including 7.6% with grade 3/4 events – versus just over a quarter in the 2-year arm and only 5.1% with grade 3/4 events.
In the 5-year group, 8.3% of patients experienced bone pain and 5.1% experienced arthralgia versus 3.7% and 3.1%, respectively, in the 2-year arm.
Atypical fractures, such as femoral spiral fractures, are another concern with bisphosphonates. Although this trial did not report on fracture type, fractures were reported in 14 women in the 5-year group but in only 3 in the 2-year arm.
Jaw necrosis, another known adverse effect of bisphosphonates, was reported in 11 women in the 5-year group and in 5 in the 2-year group.
The study was funded by several pharmaceutical companies, including Novartis, the maker of zoledronate. The investigators have numerous industry ties. Dr. Friedl has received payments from Novartis.
A version of this article first appeared on Medscape.com.
What’s best for diabetes after metformin? GRADE outdated at outset
Liraglutide and insulin glargine outperformed glimepiride and sitagliptin as single add-on agents to metformin for treating patients with type 2 diabetes in a multicenter U.S. trial that randomized just over 5,000 patients.
. Results were reported at the virtual American Diabetes Association (ADA) 81st Scientific Sessions.
The comparison included two oral medications – the sulfonylurea glimepiride and dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin – and two injectable medications – insulin glargine and glucagon-like peptide 1 (GLP-1) receptor agonist liraglutide.
The primary endpoint was change in A1c level and overall glycemic control. Secondary endpoints include changes in weight, as well as cardiovascular, renal, gastrointestinal, and other complications.
For the primary endpoint – keeping A1c levels below 7% – liraglutide and the basal insulin glargine both did this best and were almost equivalent.
During the average 5-year follow-up, the rates of patients progressing to a confirmed A1c of 7% or higher were 67% among patients randomized to insulin glargine, 68% maintained on liraglutide, 72% taking the sulfonylurea glimepiride, and 77% taking sitagliptin, reported John M. Lachin, ScD, a biostatistician at George Washington University, Washington.
Too soon for take-aways, or are the data already obsolete?
“The ultimate goal of GRADE is to help clinicians select the therapies that will work best for individual patients, as diabetes care is not a one-size-fits all approach,” noted David M. Nathan, MD, chair of the study and director of the Diabetes Center at Massachusetts General Hospital, in an ADA press release.
Dr. Nathan, as well as several other members of the GRADE trial steering committee who presented results, repeatedly cautioned that the findings were preliminary because they represent 90% of outcomes, with the remaining 10% still to be adjudicated.
“We undertook this study to fill a gap in the guidelines,” said investigator Deborah J. Wexler, MD, clinical director of the Diabetes Center at Massachusetts General Hospital in Boston. “I would like to have all the results in ... before I comment on how the guidelines should change.”
“The metabolic data are solid, but the cardiovascular disease data are preliminary,” warned Dr. Nathan.
But that didn’t stop some from drawing their own conclusions, with Julio Rosenstock, MD, who comoderated the session but was not involved with the study, giving his own opinion.
“A pleasant surprise was the performance of basal insulin,” he said, calling the findings “a vindication” for basal insulin as a treatment for the types of patients with type 2 diabetes that enrolled in the study.
Steven E. Kahn, MB, ChB, another GRADE co-investigator agreed. “Based on the results, guidelines should say that you add insulin early on,” he observed.
A generic basal insulin and a generic sulfonylurea are both reasonable options, after metformin, for patients with limited resources, added Dr. Kahn, an endocrinologist and professor at the University of Washington, Seattle.
Dr. Rosenstock, director of the Dallas Diabetes Research Center, also saw the results as an indictment of agents in the DDP-4 inhibitor class, such as sitagliptin.
The DPP-4 inhibitors generate $9 billion a year, he said, wondering whether it “is justifiable to put them on the same level as other agents?”
Meanwhile the assigned discussant, David R. Matthews, DPhil, a professor of diabetes medicine at the University of Oxford, England – while congratulating the investigators on certain aspects of the study – said it ultimately fell short because it didn’t include an arm with an SGLT2 inhibitor.
“We should kick the authors for missing out on SGLT2 inhibitors,” Dr. Matthews said. “The omission means that the GRADE data are already obsolescent.”
In reply, Dr. Nathan admitted “we feel bad we did not include” an SGLT2 inhibitor, but he vigorously defended the dilemma faced by the trial’s organizers.
Oral SGLT2 inhibitors were not “well-established drugs” for type 2 diabetes when enrollment launched in 2013, and the researchers were wary of including what could turn out to be a problematic agent soon after controversy over the safety of agents in the thiazolidinedione drug class (such as rosiglitazone), he explained.
They also realized that adding a fifth drug to the study would necessitate doubling enrollment size, which would have undercut the funding plans already in place.
Dr. Matthews also derided GRADE as being underpowered to adequately address the impact of the tested agents on major adverse cardiovascular events (MACE) and hospitalizations for heart failure and too U.S.-centric to be generalizable elsewhere.
A study with lots of data
The roughly 5,000 patients enrolled in GRADE were an average age of 57 years old, 64% were men, 66% were White, and 20% were Black. They had had type 2 diabetes, on average, for 4.2 years. Mean body mass index at entry was about 34 kg/m2, average A1c was 7.5%, and average estimated glomerular filtration rate was 95 mL/min/1.73m2. The trial included a 6-12 week run-in period during which background metformin treatment was optimized and led to average A1c levels less than 7%.
Patients were then randomized to one of the four agents as add-on treatment.
Both liraglutide and insulin glargine performed well on many of the numerous metrics in the data-rich trial, largely funded by two branches of the National Institutes of Health, with commercial involvement limited to free supplies of the study drugs.
The secondary metabolic outcome, of disease progressing to a confirmed A1c of 7.5%, was reached by 39% of patients taking insulin glargine, significantly lower than the rate of 46% among patients taking liraglutide, and that rate, in turn, was significantly below the 50% rate among patients taking glimepiride and the 55% rate of those taking sitagliptin.
Mean doses of the second-line agents after 4 years of treatment were 38.3 units/day for glargine, 3.5 mg/day for glimepiride, 1.3 mg/day for subcutaneous liraglutide, and 82.9 mg/day for sitagliptin.
A trio of cardiovascular outcomes showed one significant benefit of liraglutide over the other three drugs for the endpoint of any cardiovascular event, which included not only major adverse cardiovascular events (MACE; cardiovascular death, myocardial infarction, or stroke), but also several other event types, including heart failure requiring hospitalization, unstable angina requiring hospitalization, revascularization or any arterial repairs, stent thrombosis, or transient ischemic attack.
For the endpoint of any cardiovascular event, the rate was 5.8% for patients taking liraglutide, significantly less than the rate of 7.6% of those taking insulin glargine, 8.0% for glimepiride, and 8.6% for sitagliptin, reported John B. Buse, MD, PhD, professor, chief of endocrinology, and director of the Diabetes Center at the University of North Carolina at Chapel Hill.
For each of the other two main cardiovascular endpoints – MACE and hospitalization for heart failure – liraglutide had a numeric advantage over the other three drugs but failed to reach significance.
Patients taking liraglutide also had a smaller but not significantly different point estimate for all-cause death, at 2.1%, compared with 3.1%-3.4% in the other three groups.
And, Dr. Nathan emphasized, the cardiovascular disease data are still considered preliminary.
Liraglutide scored a pair of additional outcome victories. Its use resulted in a significantly lower rate of patients who progressed during follow-up to either needing antihypertensive medications or having their blood pressure rise above 140/90 mm Hg compared with the other three drugs. (At baseline, average blood pressure for all patients was 128/77 mm Hg.)
And after 4 years, patients taking liraglutide lost an average of about 4 kg (8.8 lb) from their baseline weight (which averaged about 100 kg [220 lb]), roughly the same as patients taking sitagliptin but significantly better than with glimepiride or insulin glargine. Patients taking glargine gained a small amount of weight on average during their first couple of years of treatment, roughly 1 kg, but returned to around their baseline weight by the end of 4 years.
Four drugs performed equally well for some outcomes
Finally, the four drugs had similar results for some outcomes. This included their effects on renal function, distal sensory polyneuropathy, and low-density lipoprotein (LDL) cholesterol.
The four agents also had roughly similar safety profiles, with rates of serious adverse events all falling within the tight range of 33%-37%.
But the rate of severe hypoglycemic episodes that required assistance to treat showed significant separation, ranging from 2.3% for glimepiride, 1.4% for glargine, 0.9% for liraglutide, and 0.7% for sitagliptin. Gastrointestinal symptoms occurred in about 50% of patients in three of the treatment groups but were significantly higher in those taking liraglutide, affecting 60%.
GRADE received no commercial funding. Dr. Wexler has reported serving on data monitoring committees for Novo Nordisk. Dr. Buse has reported being a consultant for and holding stock in numerous companies. Dr. Rosenstock has reported being an advisor or consultant to Applied Therapeutics, Boehringer Ingelheim, Hanmi Pharmaceutical, Intarcia Therapeutics, Lilly, Novo Nordisk, Oramed, and Sanofi and has received research support from numerous companies. Dr. Kahn has reported being an advisor to or speaker on behalf of Bayer, Boehringer Ingelheim, Casma Therapeutics, Intarcia Therapeutics, Lilly, Merck, Novo Nordisk, Pfizer, and Third Rock Ventures. Dr. Matthews has reported receiving lecture and advisor fees from Merck, Novartis, Novo Nordisk, Sanofi Aventis, and Servier. Dr. Lachin and Dr. Nathan have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Liraglutide and insulin glargine outperformed glimepiride and sitagliptin as single add-on agents to metformin for treating patients with type 2 diabetes in a multicenter U.S. trial that randomized just over 5,000 patients.
. Results were reported at the virtual American Diabetes Association (ADA) 81st Scientific Sessions.
The comparison included two oral medications – the sulfonylurea glimepiride and dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin – and two injectable medications – insulin glargine and glucagon-like peptide 1 (GLP-1) receptor agonist liraglutide.
The primary endpoint was change in A1c level and overall glycemic control. Secondary endpoints include changes in weight, as well as cardiovascular, renal, gastrointestinal, and other complications.
For the primary endpoint – keeping A1c levels below 7% – liraglutide and the basal insulin glargine both did this best and were almost equivalent.
During the average 5-year follow-up, the rates of patients progressing to a confirmed A1c of 7% or higher were 67% among patients randomized to insulin glargine, 68% maintained on liraglutide, 72% taking the sulfonylurea glimepiride, and 77% taking sitagliptin, reported John M. Lachin, ScD, a biostatistician at George Washington University, Washington.
Too soon for take-aways, or are the data already obsolete?
“The ultimate goal of GRADE is to help clinicians select the therapies that will work best for individual patients, as diabetes care is not a one-size-fits all approach,” noted David M. Nathan, MD, chair of the study and director of the Diabetes Center at Massachusetts General Hospital, in an ADA press release.
Dr. Nathan, as well as several other members of the GRADE trial steering committee who presented results, repeatedly cautioned that the findings were preliminary because they represent 90% of outcomes, with the remaining 10% still to be adjudicated.
“We undertook this study to fill a gap in the guidelines,” said investigator Deborah J. Wexler, MD, clinical director of the Diabetes Center at Massachusetts General Hospital in Boston. “I would like to have all the results in ... before I comment on how the guidelines should change.”
“The metabolic data are solid, but the cardiovascular disease data are preliminary,” warned Dr. Nathan.
But that didn’t stop some from drawing their own conclusions, with Julio Rosenstock, MD, who comoderated the session but was not involved with the study, giving his own opinion.
“A pleasant surprise was the performance of basal insulin,” he said, calling the findings “a vindication” for basal insulin as a treatment for the types of patients with type 2 diabetes that enrolled in the study.
Steven E. Kahn, MB, ChB, another GRADE co-investigator agreed. “Based on the results, guidelines should say that you add insulin early on,” he observed.
A generic basal insulin and a generic sulfonylurea are both reasonable options, after metformin, for patients with limited resources, added Dr. Kahn, an endocrinologist and professor at the University of Washington, Seattle.
Dr. Rosenstock, director of the Dallas Diabetes Research Center, also saw the results as an indictment of agents in the DDP-4 inhibitor class, such as sitagliptin.
The DPP-4 inhibitors generate $9 billion a year, he said, wondering whether it “is justifiable to put them on the same level as other agents?”
Meanwhile the assigned discussant, David R. Matthews, DPhil, a professor of diabetes medicine at the University of Oxford, England – while congratulating the investigators on certain aspects of the study – said it ultimately fell short because it didn’t include an arm with an SGLT2 inhibitor.
“We should kick the authors for missing out on SGLT2 inhibitors,” Dr. Matthews said. “The omission means that the GRADE data are already obsolescent.”
In reply, Dr. Nathan admitted “we feel bad we did not include” an SGLT2 inhibitor, but he vigorously defended the dilemma faced by the trial’s organizers.
Oral SGLT2 inhibitors were not “well-established drugs” for type 2 diabetes when enrollment launched in 2013, and the researchers were wary of including what could turn out to be a problematic agent soon after controversy over the safety of agents in the thiazolidinedione drug class (such as rosiglitazone), he explained.
They also realized that adding a fifth drug to the study would necessitate doubling enrollment size, which would have undercut the funding plans already in place.
Dr. Matthews also derided GRADE as being underpowered to adequately address the impact of the tested agents on major adverse cardiovascular events (MACE) and hospitalizations for heart failure and too U.S.-centric to be generalizable elsewhere.
A study with lots of data
The roughly 5,000 patients enrolled in GRADE were an average age of 57 years old, 64% were men, 66% were White, and 20% were Black. They had had type 2 diabetes, on average, for 4.2 years. Mean body mass index at entry was about 34 kg/m2, average A1c was 7.5%, and average estimated glomerular filtration rate was 95 mL/min/1.73m2. The trial included a 6-12 week run-in period during which background metformin treatment was optimized and led to average A1c levels less than 7%.
Patients were then randomized to one of the four agents as add-on treatment.
Both liraglutide and insulin glargine performed well on many of the numerous metrics in the data-rich trial, largely funded by two branches of the National Institutes of Health, with commercial involvement limited to free supplies of the study drugs.
The secondary metabolic outcome, of disease progressing to a confirmed A1c of 7.5%, was reached by 39% of patients taking insulin glargine, significantly lower than the rate of 46% among patients taking liraglutide, and that rate, in turn, was significantly below the 50% rate among patients taking glimepiride and the 55% rate of those taking sitagliptin.
Mean doses of the second-line agents after 4 years of treatment were 38.3 units/day for glargine, 3.5 mg/day for glimepiride, 1.3 mg/day for subcutaneous liraglutide, and 82.9 mg/day for sitagliptin.
A trio of cardiovascular outcomes showed one significant benefit of liraglutide over the other three drugs for the endpoint of any cardiovascular event, which included not only major adverse cardiovascular events (MACE; cardiovascular death, myocardial infarction, or stroke), but also several other event types, including heart failure requiring hospitalization, unstable angina requiring hospitalization, revascularization or any arterial repairs, stent thrombosis, or transient ischemic attack.
For the endpoint of any cardiovascular event, the rate was 5.8% for patients taking liraglutide, significantly less than the rate of 7.6% of those taking insulin glargine, 8.0% for glimepiride, and 8.6% for sitagliptin, reported John B. Buse, MD, PhD, professor, chief of endocrinology, and director of the Diabetes Center at the University of North Carolina at Chapel Hill.
For each of the other two main cardiovascular endpoints – MACE and hospitalization for heart failure – liraglutide had a numeric advantage over the other three drugs but failed to reach significance.
Patients taking liraglutide also had a smaller but not significantly different point estimate for all-cause death, at 2.1%, compared with 3.1%-3.4% in the other three groups.
And, Dr. Nathan emphasized, the cardiovascular disease data are still considered preliminary.
Liraglutide scored a pair of additional outcome victories. Its use resulted in a significantly lower rate of patients who progressed during follow-up to either needing antihypertensive medications or having their blood pressure rise above 140/90 mm Hg compared with the other three drugs. (At baseline, average blood pressure for all patients was 128/77 mm Hg.)
And after 4 years, patients taking liraglutide lost an average of about 4 kg (8.8 lb) from their baseline weight (which averaged about 100 kg [220 lb]), roughly the same as patients taking sitagliptin but significantly better than with glimepiride or insulin glargine. Patients taking glargine gained a small amount of weight on average during their first couple of years of treatment, roughly 1 kg, but returned to around their baseline weight by the end of 4 years.
Four drugs performed equally well for some outcomes
Finally, the four drugs had similar results for some outcomes. This included their effects on renal function, distal sensory polyneuropathy, and low-density lipoprotein (LDL) cholesterol.
The four agents also had roughly similar safety profiles, with rates of serious adverse events all falling within the tight range of 33%-37%.
But the rate of severe hypoglycemic episodes that required assistance to treat showed significant separation, ranging from 2.3% for glimepiride, 1.4% for glargine, 0.9% for liraglutide, and 0.7% for sitagliptin. Gastrointestinal symptoms occurred in about 50% of patients in three of the treatment groups but were significantly higher in those taking liraglutide, affecting 60%.
GRADE received no commercial funding. Dr. Wexler has reported serving on data monitoring committees for Novo Nordisk. Dr. Buse has reported being a consultant for and holding stock in numerous companies. Dr. Rosenstock has reported being an advisor or consultant to Applied Therapeutics, Boehringer Ingelheim, Hanmi Pharmaceutical, Intarcia Therapeutics, Lilly, Novo Nordisk, Oramed, and Sanofi and has received research support from numerous companies. Dr. Kahn has reported being an advisor to or speaker on behalf of Bayer, Boehringer Ingelheim, Casma Therapeutics, Intarcia Therapeutics, Lilly, Merck, Novo Nordisk, Pfizer, and Third Rock Ventures. Dr. Matthews has reported receiving lecture and advisor fees from Merck, Novartis, Novo Nordisk, Sanofi Aventis, and Servier. Dr. Lachin and Dr. Nathan have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Liraglutide and insulin glargine outperformed glimepiride and sitagliptin as single add-on agents to metformin for treating patients with type 2 diabetes in a multicenter U.S. trial that randomized just over 5,000 patients.
. Results were reported at the virtual American Diabetes Association (ADA) 81st Scientific Sessions.
The comparison included two oral medications – the sulfonylurea glimepiride and dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin – and two injectable medications – insulin glargine and glucagon-like peptide 1 (GLP-1) receptor agonist liraglutide.
The primary endpoint was change in A1c level and overall glycemic control. Secondary endpoints include changes in weight, as well as cardiovascular, renal, gastrointestinal, and other complications.
For the primary endpoint – keeping A1c levels below 7% – liraglutide and the basal insulin glargine both did this best and were almost equivalent.
During the average 5-year follow-up, the rates of patients progressing to a confirmed A1c of 7% or higher were 67% among patients randomized to insulin glargine, 68% maintained on liraglutide, 72% taking the sulfonylurea glimepiride, and 77% taking sitagliptin, reported John M. Lachin, ScD, a biostatistician at George Washington University, Washington.
Too soon for take-aways, or are the data already obsolete?
“The ultimate goal of GRADE is to help clinicians select the therapies that will work best for individual patients, as diabetes care is not a one-size-fits all approach,” noted David M. Nathan, MD, chair of the study and director of the Diabetes Center at Massachusetts General Hospital, in an ADA press release.
Dr. Nathan, as well as several other members of the GRADE trial steering committee who presented results, repeatedly cautioned that the findings were preliminary because they represent 90% of outcomes, with the remaining 10% still to be adjudicated.
“We undertook this study to fill a gap in the guidelines,” said investigator Deborah J. Wexler, MD, clinical director of the Diabetes Center at Massachusetts General Hospital in Boston. “I would like to have all the results in ... before I comment on how the guidelines should change.”
“The metabolic data are solid, but the cardiovascular disease data are preliminary,” warned Dr. Nathan.
But that didn’t stop some from drawing their own conclusions, with Julio Rosenstock, MD, who comoderated the session but was not involved with the study, giving his own opinion.
“A pleasant surprise was the performance of basal insulin,” he said, calling the findings “a vindication” for basal insulin as a treatment for the types of patients with type 2 diabetes that enrolled in the study.
Steven E. Kahn, MB, ChB, another GRADE co-investigator agreed. “Based on the results, guidelines should say that you add insulin early on,” he observed.
A generic basal insulin and a generic sulfonylurea are both reasonable options, after metformin, for patients with limited resources, added Dr. Kahn, an endocrinologist and professor at the University of Washington, Seattle.
Dr. Rosenstock, director of the Dallas Diabetes Research Center, also saw the results as an indictment of agents in the DDP-4 inhibitor class, such as sitagliptin.
The DPP-4 inhibitors generate $9 billion a year, he said, wondering whether it “is justifiable to put them on the same level as other agents?”
Meanwhile the assigned discussant, David R. Matthews, DPhil, a professor of diabetes medicine at the University of Oxford, England – while congratulating the investigators on certain aspects of the study – said it ultimately fell short because it didn’t include an arm with an SGLT2 inhibitor.
“We should kick the authors for missing out on SGLT2 inhibitors,” Dr. Matthews said. “The omission means that the GRADE data are already obsolescent.”
In reply, Dr. Nathan admitted “we feel bad we did not include” an SGLT2 inhibitor, but he vigorously defended the dilemma faced by the trial’s organizers.
Oral SGLT2 inhibitors were not “well-established drugs” for type 2 diabetes when enrollment launched in 2013, and the researchers were wary of including what could turn out to be a problematic agent soon after controversy over the safety of agents in the thiazolidinedione drug class (such as rosiglitazone), he explained.
They also realized that adding a fifth drug to the study would necessitate doubling enrollment size, which would have undercut the funding plans already in place.
Dr. Matthews also derided GRADE as being underpowered to adequately address the impact of the tested agents on major adverse cardiovascular events (MACE) and hospitalizations for heart failure and too U.S.-centric to be generalizable elsewhere.
A study with lots of data
The roughly 5,000 patients enrolled in GRADE were an average age of 57 years old, 64% were men, 66% were White, and 20% were Black. They had had type 2 diabetes, on average, for 4.2 years. Mean body mass index at entry was about 34 kg/m2, average A1c was 7.5%, and average estimated glomerular filtration rate was 95 mL/min/1.73m2. The trial included a 6-12 week run-in period during which background metformin treatment was optimized and led to average A1c levels less than 7%.
Patients were then randomized to one of the four agents as add-on treatment.
Both liraglutide and insulin glargine performed well on many of the numerous metrics in the data-rich trial, largely funded by two branches of the National Institutes of Health, with commercial involvement limited to free supplies of the study drugs.
The secondary metabolic outcome, of disease progressing to a confirmed A1c of 7.5%, was reached by 39% of patients taking insulin glargine, significantly lower than the rate of 46% among patients taking liraglutide, and that rate, in turn, was significantly below the 50% rate among patients taking glimepiride and the 55% rate of those taking sitagliptin.
Mean doses of the second-line agents after 4 years of treatment were 38.3 units/day for glargine, 3.5 mg/day for glimepiride, 1.3 mg/day for subcutaneous liraglutide, and 82.9 mg/day for sitagliptin.
A trio of cardiovascular outcomes showed one significant benefit of liraglutide over the other three drugs for the endpoint of any cardiovascular event, which included not only major adverse cardiovascular events (MACE; cardiovascular death, myocardial infarction, or stroke), but also several other event types, including heart failure requiring hospitalization, unstable angina requiring hospitalization, revascularization or any arterial repairs, stent thrombosis, or transient ischemic attack.
For the endpoint of any cardiovascular event, the rate was 5.8% for patients taking liraglutide, significantly less than the rate of 7.6% of those taking insulin glargine, 8.0% for glimepiride, and 8.6% for sitagliptin, reported John B. Buse, MD, PhD, professor, chief of endocrinology, and director of the Diabetes Center at the University of North Carolina at Chapel Hill.
For each of the other two main cardiovascular endpoints – MACE and hospitalization for heart failure – liraglutide had a numeric advantage over the other three drugs but failed to reach significance.
Patients taking liraglutide also had a smaller but not significantly different point estimate for all-cause death, at 2.1%, compared with 3.1%-3.4% in the other three groups.
And, Dr. Nathan emphasized, the cardiovascular disease data are still considered preliminary.
Liraglutide scored a pair of additional outcome victories. Its use resulted in a significantly lower rate of patients who progressed during follow-up to either needing antihypertensive medications or having their blood pressure rise above 140/90 mm Hg compared with the other three drugs. (At baseline, average blood pressure for all patients was 128/77 mm Hg.)
And after 4 years, patients taking liraglutide lost an average of about 4 kg (8.8 lb) from their baseline weight (which averaged about 100 kg [220 lb]), roughly the same as patients taking sitagliptin but significantly better than with glimepiride or insulin glargine. Patients taking glargine gained a small amount of weight on average during their first couple of years of treatment, roughly 1 kg, but returned to around their baseline weight by the end of 4 years.
Four drugs performed equally well for some outcomes
Finally, the four drugs had similar results for some outcomes. This included their effects on renal function, distal sensory polyneuropathy, and low-density lipoprotein (LDL) cholesterol.
The four agents also had roughly similar safety profiles, with rates of serious adverse events all falling within the tight range of 33%-37%.
But the rate of severe hypoglycemic episodes that required assistance to treat showed significant separation, ranging from 2.3% for glimepiride, 1.4% for glargine, 0.9% for liraglutide, and 0.7% for sitagliptin. Gastrointestinal symptoms occurred in about 50% of patients in three of the treatment groups but were significantly higher in those taking liraglutide, affecting 60%.
GRADE received no commercial funding. Dr. Wexler has reported serving on data monitoring committees for Novo Nordisk. Dr. Buse has reported being a consultant for and holding stock in numerous companies. Dr. Rosenstock has reported being an advisor or consultant to Applied Therapeutics, Boehringer Ingelheim, Hanmi Pharmaceutical, Intarcia Therapeutics, Lilly, Novo Nordisk, Oramed, and Sanofi and has received research support from numerous companies. Dr. Kahn has reported being an advisor to or speaker on behalf of Bayer, Boehringer Ingelheim, Casma Therapeutics, Intarcia Therapeutics, Lilly, Merck, Novo Nordisk, Pfizer, and Third Rock Ventures. Dr. Matthews has reported receiving lecture and advisor fees from Merck, Novartis, Novo Nordisk, Sanofi Aventis, and Servier. Dr. Lachin and Dr. Nathan have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Nocturnal hypoglycemia halved with insulin degludec vs. glargine
Patients with type 1 diabetes who used insulin degludec as their basal insulin had fewer than half the number of nocturnal hypoglycemia events, compared with patients who used insulin glargine U100, in a head-to-head crossover study with 51 patients who had a history of nighttime hypoglycemia episodes.
Patients with type 1 diabetes who are “struggling with nocturnal hypoglycemia would benefit from insulin degludec treatment,” said Julie M. Brøsen, MD, at the annual scientific sessions of the American Diabetes Association.
Accumulating evidence for less hypoglycemia with insulin degludec
Results from several studies comparing insulin degludec (Tresiba), a second-generation, longer-acting insulin with more stable steady-state performance, with the first-generation basal insulin analogue glargine (Lantus), have built the case that degludec produces fewer hypoglycemia events.
The landmark SWITCH 1 crossover study published in 2017 showed in about 500 patients with type 1 diabetes and a risk factor for hypoglycemia that treatment with insulin degludec led to significantly few total hypoglycemia episodes and significantly fewer nocturnal episodes, compared with insulin glargine.
Next came similar findings from ReFLeCT, a multicenter observational study that followed 556 unselected patients with type 1 diabetes in routine practice settings who switched to insulin degludec following treatment with a different basal insulin. The results again showed a significant drop-off in total, nonsevere, severe, and nocturnal hypoglycemia events.
Homing in on higher-risk patients
The current study, HypoDeg (Insulin Degludec and Symptomatic Nocturnal Hypoglycaemia), ran at 10 Danish centers and enrolled 149 adults with type 1 diabetes who had at least one episode of severe nocturnal hypoglycemia within the prior 2 years, focusing on patients most at risk for future nocturnal hypoglycemia events. In an unusual study design, researchers identified nocturnal hypoglycemic episodes with hourly venous blood samples drawn from a subcutaneous line.
They randomized the patients to basal insulin treatment with either insulin degludec or to insulin glargine U100, allowed their treatment to stabilize for 3 months, and then tallied nocturnal hypoglycemia events for 9 months. They then crossed patients to the alternative basal insulin and repeated the process.
Results from the full study have not yet appeared in published form but were in a pair of reports at the 2020 scientific sessions of the ADA.
One report included findings based on 136 episodes of severe hypoglycemia identified clinically and showed these events occurred 35% less often during treatment with insulin degludec, a significant difference. The overall finding was primarily driven by 48% fewer episodes of severe nocturnal hypoglycemia, but this difference was not significant.
The second report identified hypoglycemia events with continuous glucose monitoring in 74 of the study participants, which identified 193 episodes of nonsevere nocturnal hypoglycemia and found that treatment with insulin degludec cut the rate by 47%, primarily by reducing asymptomatic episodes.
Hourly blood draws track overnight hypoglycemia
The current study included 51 of the 149 HypoDeg patients who agreed to undergo overnight blood sampling and had this done at least once while treated with each of the two study insulins. (The study design called for two blood sampling nights for each willing patient during each of the two treatment periods.) The 51 patients had type 1 diabetes for an average of 28 years and an average age of 58 years. Two-thirds were men, their baseline A1c was 7.8%, and on average had 2.6 episodes of severe nocturnal hypoglycemia during the prior 2 years.
The researchers drew hourly blood specimens on a total of 196 nights from the 51 participating patients and identified 57 nights when blood glucose levels reached hypoglycemia thresholds in 33 patients. One-third of the events occurred when patients were on insulin degludec treatment, and two-thirds when they were on insulin glargine, reported Dr. Brøsen.
She presented three separate analyses of the data. One analysis focused on level 1 hypoglycemia events, when blood glucose dips to 70 mg/dL or less, which occurred 54% less often when patients were on insulin degludec. A second analysis looked at level 2 events, when blood glucose falls below 54 mg/dL, and treatment with insulin degludec cut this by 64% compared with insulin glargine. The third analysis focused on symptomatic events when blood glucose was 70 mg/dL or less, and treatment with insulin degludec linked with a 62% cut in this metric. All three between-group differences were significant.
Evidence supports already-changed practice
This new evidence “supports recommending” insulin degludec over insulin glargine, commented Bastiaan E. de Galan, MD, PhD, an endocrinologist and professor at Maastrict (the Netherlands) University Medical Center. The new results “extend those from previous trials in populations with type 1 diabetes that were unselected for the risk of hypoglycemia. In clinical practice, insulin degludec is already considered for patients who reported nocturnal hypoglycemia while on insulin glargine U100, but it’s great this study provides the scientific evidence,” said Dr. de Galan in an interview.

“The lower rate of nocturnal hypoglycemia with degludec, compared with glargine U100 is well established. Inpatient assessment of hypoglycemia with measurement of hourly plasma glucose allowed HypoDeg to provide stronger evidence than prior studies. The benefit of delgudec versus glargine U100 was significant and clinically meaningful, in hypo-prone patients who would benefit the most” by using insulin degludec, commented Gian Paolo Fadini, MD, an endocrinologist at the University of Padova (Italy), and a lead investigator on the ReFLeCT study.
But insulin degludec is not a completely silver bullet. Its prolonged duration of action and stability that may in part explain why it limits hypoglycemia events can also be a drawback: “It probably offers fewer options for flexibility. Any change in dose takes at least a day or 2 to settle, which may be unfavorable in certain circumstances,” noted Dr. de Galan.
“I wouldn’t recommend insulin degludec for all patients with type 1 diabetes. It’s an individual evaluation in each patient,” said Dr. Brøsen. “We will be looking into whether some patients are better off on insulin glargine.”
Cost makes a difference
Another, potentially more consequential flaw is insulin degludec’s relative expense.
“To date, use of degludec in routine practice has been limited by its cost, compared with older basal insulins,” observed Dr. Fadini in an interview. “In several countries, including the United States, degludec is substantially more expensive than glargine.”
The ADA’s Standards of Medical Care in Diabetes–2021 includes table 9.3 that lists the costs of various insulins and shows the median average wholesale price of insulin glargine U100 follow-on products as $190/vial, compared with a $407 price for a similar vial of insulin degludec.

Insulin degludec “is clearly superior from a hypoglycemia standpoint. Patients with type 1 diabetes like the reduction because hypoglycemia is scary, and dangerous. The main issue is cost, and the extent to which it may be covered by insurance,” commented Lisa Chow, MD, an endocrinologist at the University of Minnesota, Minneapolis. “We generally won’t prescribe degludec unless it is at a price affordable to the patient. We try to use patient assistance programs sponsored by the company [that markets insulin degludec: Novo Nordisk] to try to make it more affordable.”
Dr. Chow also highlighted that a new wrinkle has been introduction of a more concentrated formulation of insulin glargine, U300, which appears to cause less hypoglycemia than insulin glargine U100. Recent study results indicated that no significant difference exists in the incidence of hypoglycemia among patients treated with insulin glargine U300 and those treated with insulin degludec, such as findings from the BRIGHT trial, which included just over 900 patients, and in the CONCLUDE trial, which randomized more than 1,600 patients.
The HypoDeg study was sponsored by Novo Nordisk, the company that markets insulin degludec. Dr. Brøsen had no personal disclosures, but several of her coauthors were either Novo Nordisk employees or had financial relationships with the company. Dr. de Galan has received research funding from Novo Nordisk. Dr. Fadini has received lecture fees and research funding from Novo Nordisk, from Sanofi, the company that markets insulin glargine, and from several other companies. Dr. Chow has received research funding from Dexcom.
Patients with type 1 diabetes who used insulin degludec as their basal insulin had fewer than half the number of nocturnal hypoglycemia events, compared with patients who used insulin glargine U100, in a head-to-head crossover study with 51 patients who had a history of nighttime hypoglycemia episodes.
Patients with type 1 diabetes who are “struggling with nocturnal hypoglycemia would benefit from insulin degludec treatment,” said Julie M. Brøsen, MD, at the annual scientific sessions of the American Diabetes Association.
Accumulating evidence for less hypoglycemia with insulin degludec
Results from several studies comparing insulin degludec (Tresiba), a second-generation, longer-acting insulin with more stable steady-state performance, with the first-generation basal insulin analogue glargine (Lantus), have built the case that degludec produces fewer hypoglycemia events.
The landmark SWITCH 1 crossover study published in 2017 showed in about 500 patients with type 1 diabetes and a risk factor for hypoglycemia that treatment with insulin degludec led to significantly few total hypoglycemia episodes and significantly fewer nocturnal episodes, compared with insulin glargine.
Next came similar findings from ReFLeCT, a multicenter observational study that followed 556 unselected patients with type 1 diabetes in routine practice settings who switched to insulin degludec following treatment with a different basal insulin. The results again showed a significant drop-off in total, nonsevere, severe, and nocturnal hypoglycemia events.
Homing in on higher-risk patients
The current study, HypoDeg (Insulin Degludec and Symptomatic Nocturnal Hypoglycaemia), ran at 10 Danish centers and enrolled 149 adults with type 1 diabetes who had at least one episode of severe nocturnal hypoglycemia within the prior 2 years, focusing on patients most at risk for future nocturnal hypoglycemia events. In an unusual study design, researchers identified nocturnal hypoglycemic episodes with hourly venous blood samples drawn from a subcutaneous line.
They randomized the patients to basal insulin treatment with either insulin degludec or to insulin glargine U100, allowed their treatment to stabilize for 3 months, and then tallied nocturnal hypoglycemia events for 9 months. They then crossed patients to the alternative basal insulin and repeated the process.
Results from the full study have not yet appeared in published form but were in a pair of reports at the 2020 scientific sessions of the ADA.
One report included findings based on 136 episodes of severe hypoglycemia identified clinically and showed these events occurred 35% less often during treatment with insulin degludec, a significant difference. The overall finding was primarily driven by 48% fewer episodes of severe nocturnal hypoglycemia, but this difference was not significant.
The second report identified hypoglycemia events with continuous glucose monitoring in 74 of the study participants, which identified 193 episodes of nonsevere nocturnal hypoglycemia and found that treatment with insulin degludec cut the rate by 47%, primarily by reducing asymptomatic episodes.
Hourly blood draws track overnight hypoglycemia
The current study included 51 of the 149 HypoDeg patients who agreed to undergo overnight blood sampling and had this done at least once while treated with each of the two study insulins. (The study design called for two blood sampling nights for each willing patient during each of the two treatment periods.) The 51 patients had type 1 diabetes for an average of 28 years and an average age of 58 years. Two-thirds were men, their baseline A1c was 7.8%, and on average had 2.6 episodes of severe nocturnal hypoglycemia during the prior 2 years.
The researchers drew hourly blood specimens on a total of 196 nights from the 51 participating patients and identified 57 nights when blood glucose levels reached hypoglycemia thresholds in 33 patients. One-third of the events occurred when patients were on insulin degludec treatment, and two-thirds when they were on insulin glargine, reported Dr. Brøsen.
She presented three separate analyses of the data. One analysis focused on level 1 hypoglycemia events, when blood glucose dips to 70 mg/dL or less, which occurred 54% less often when patients were on insulin degludec. A second analysis looked at level 2 events, when blood glucose falls below 54 mg/dL, and treatment with insulin degludec cut this by 64% compared with insulin glargine. The third analysis focused on symptomatic events when blood glucose was 70 mg/dL or less, and treatment with insulin degludec linked with a 62% cut in this metric. All three between-group differences were significant.
Evidence supports already-changed practice
This new evidence “supports recommending” insulin degludec over insulin glargine, commented Bastiaan E. de Galan, MD, PhD, an endocrinologist and professor at Maastrict (the Netherlands) University Medical Center. The new results “extend those from previous trials in populations with type 1 diabetes that were unselected for the risk of hypoglycemia. In clinical practice, insulin degludec is already considered for patients who reported nocturnal hypoglycemia while on insulin glargine U100, but it’s great this study provides the scientific evidence,” said Dr. de Galan in an interview.
“The lower rate of nocturnal hypoglycemia with degludec, compared with glargine U100 is well established. Inpatient assessment of hypoglycemia with measurement of hourly plasma glucose allowed HypoDeg to provide stronger evidence than prior studies. The benefit of delgudec versus glargine U100 was significant and clinically meaningful, in hypo-prone patients who would benefit the most” by using insulin degludec, commented Gian Paolo Fadini, MD, an endocrinologist at the University of Padova (Italy), and a lead investigator on the ReFLeCT study.
But insulin degludec is not a completely silver bullet. Its prolonged duration of action and stability that may in part explain why it limits hypoglycemia events can also be a drawback: “It probably offers fewer options for flexibility. Any change in dose takes at least a day or 2 to settle, which may be unfavorable in certain circumstances,” noted Dr. de Galan.
“I wouldn’t recommend insulin degludec for all patients with type 1 diabetes. It’s an individual evaluation in each patient,” said Dr. Brøsen. “We will be looking into whether some patients are better off on insulin glargine.”
Cost makes a difference
Another, potentially more consequential flaw is insulin degludec’s relative expense.
“To date, use of degludec in routine practice has been limited by its cost, compared with older basal insulins,” observed Dr. Fadini in an interview. “In several countries, including the United States, degludec is substantially more expensive than glargine.”
The ADA’s Standards of Medical Care in Diabetes–2021 includes table 9.3 that lists the costs of various insulins and shows the median average wholesale price of insulin glargine U100 follow-on products as $190/vial, compared with a $407 price for a similar vial of insulin degludec.
Insulin degludec “is clearly superior from a hypoglycemia standpoint. Patients with type 1 diabetes like the reduction because hypoglycemia is scary, and dangerous. The main issue is cost, and the extent to which it may be covered by insurance,” commented Lisa Chow, MD, an endocrinologist at the University of Minnesota, Minneapolis. “We generally won’t prescribe degludec unless it is at a price affordable to the patient. We try to use patient assistance programs sponsored by the company [that markets insulin degludec: Novo Nordisk] to try to make it more affordable.”
Dr. Chow also highlighted that a new wrinkle has been introduction of a more concentrated formulation of insulin glargine, U300, which appears to cause less hypoglycemia than insulin glargine U100. Recent study results indicated that no significant difference exists in the incidence of hypoglycemia among patients treated with insulin glargine U300 and those treated with insulin degludec, such as findings from the BRIGHT trial, which included just over 900 patients, and in the CONCLUDE trial, which randomized more than 1,600 patients.
The HypoDeg study was sponsored by Novo Nordisk, the company that markets insulin degludec. Dr. Brøsen had no personal disclosures, but several of her coauthors were either Novo Nordisk employees or had financial relationships with the company. Dr. de Galan has received research funding from Novo Nordisk. Dr. Fadini has received lecture fees and research funding from Novo Nordisk, from Sanofi, the company that markets insulin glargine, and from several other companies. Dr. Chow has received research funding from Dexcom.
Patients with type 1 diabetes who used insulin degludec as their basal insulin had fewer than half the number of nocturnal hypoglycemia events, compared with patients who used insulin glargine U100, in a head-to-head crossover study with 51 patients who had a history of nighttime hypoglycemia episodes.
Patients with type 1 diabetes who are “struggling with nocturnal hypoglycemia would benefit from insulin degludec treatment,” said Julie M. Brøsen, MD, at the annual scientific sessions of the American Diabetes Association.
Accumulating evidence for less hypoglycemia with insulin degludec
Results from several studies comparing insulin degludec (Tresiba), a second-generation, longer-acting insulin with more stable steady-state performance, with the first-generation basal insulin analogue glargine (Lantus), have built the case that degludec produces fewer hypoglycemia events.
The landmark SWITCH 1 crossover study published in 2017 showed in about 500 patients with type 1 diabetes and a risk factor for hypoglycemia that treatment with insulin degludec led to significantly few total hypoglycemia episodes and significantly fewer nocturnal episodes, compared with insulin glargine.
Next came similar findings from ReFLeCT, a multicenter observational study that followed 556 unselected patients with type 1 diabetes in routine practice settings who switched to insulin degludec following treatment with a different basal insulin. The results again showed a significant drop-off in total, nonsevere, severe, and nocturnal hypoglycemia events.
Homing in on higher-risk patients
The current study, HypoDeg (Insulin Degludec and Symptomatic Nocturnal Hypoglycaemia), ran at 10 Danish centers and enrolled 149 adults with type 1 diabetes who had at least one episode of severe nocturnal hypoglycemia within the prior 2 years, focusing on patients most at risk for future nocturnal hypoglycemia events. In an unusual study design, researchers identified nocturnal hypoglycemic episodes with hourly venous blood samples drawn from a subcutaneous line.
They randomized the patients to basal insulin treatment with either insulin degludec or to insulin glargine U100, allowed their treatment to stabilize for 3 months, and then tallied nocturnal hypoglycemia events for 9 months. They then crossed patients to the alternative basal insulin and repeated the process.
Results from the full study have not yet appeared in published form but were in a pair of reports at the 2020 scientific sessions of the ADA.
One report included findings based on 136 episodes of severe hypoglycemia identified clinically and showed these events occurred 35% less often during treatment with insulin degludec, a significant difference. The overall finding was primarily driven by 48% fewer episodes of severe nocturnal hypoglycemia, but this difference was not significant.
The second report identified hypoglycemia events with continuous glucose monitoring in 74 of the study participants, which identified 193 episodes of nonsevere nocturnal hypoglycemia and found that treatment with insulin degludec cut the rate by 47%, primarily by reducing asymptomatic episodes.
Hourly blood draws track overnight hypoglycemia
The current study included 51 of the 149 HypoDeg patients who agreed to undergo overnight blood sampling and had this done at least once while treated with each of the two study insulins. (The study design called for two blood sampling nights for each willing patient during each of the two treatment periods.) The 51 patients had type 1 diabetes for an average of 28 years and an average age of 58 years. Two-thirds were men, their baseline A1c was 7.8%, and on average had 2.6 episodes of severe nocturnal hypoglycemia during the prior 2 years.
The researchers drew hourly blood specimens on a total of 196 nights from the 51 participating patients and identified 57 nights when blood glucose levels reached hypoglycemia thresholds in 33 patients. One-third of the events occurred when patients were on insulin degludec treatment, and two-thirds when they were on insulin glargine, reported Dr. Brøsen.
She presented three separate analyses of the data. One analysis focused on level 1 hypoglycemia events, when blood glucose dips to 70 mg/dL or less, which occurred 54% less often when patients were on insulin degludec. A second analysis looked at level 2 events, when blood glucose falls below 54 mg/dL, and treatment with insulin degludec cut this by 64% compared with insulin glargine. The third analysis focused on symptomatic events when blood glucose was 70 mg/dL or less, and treatment with insulin degludec linked with a 62% cut in this metric. All three between-group differences were significant.
Evidence supports already-changed practice
This new evidence “supports recommending” insulin degludec over insulin glargine, commented Bastiaan E. de Galan, MD, PhD, an endocrinologist and professor at Maastrict (the Netherlands) University Medical Center. The new results “extend those from previous trials in populations with type 1 diabetes that were unselected for the risk of hypoglycemia. In clinical practice, insulin degludec is already considered for patients who reported nocturnal hypoglycemia while on insulin glargine U100, but it’s great this study provides the scientific evidence,” said Dr. de Galan in an interview.
“The lower rate of nocturnal hypoglycemia with degludec, compared with glargine U100 is well established. Inpatient assessment of hypoglycemia with measurement of hourly plasma glucose allowed HypoDeg to provide stronger evidence than prior studies. The benefit of delgudec versus glargine U100 was significant and clinically meaningful, in hypo-prone patients who would benefit the most” by using insulin degludec, commented Gian Paolo Fadini, MD, an endocrinologist at the University of Padova (Italy), and a lead investigator on the ReFLeCT study.
But insulin degludec is not a completely silver bullet. Its prolonged duration of action and stability that may in part explain why it limits hypoglycemia events can also be a drawback: “It probably offers fewer options for flexibility. Any change in dose takes at least a day or 2 to settle, which may be unfavorable in certain circumstances,” noted Dr. de Galan.
“I wouldn’t recommend insulin degludec for all patients with type 1 diabetes. It’s an individual evaluation in each patient,” said Dr. Brøsen. “We will be looking into whether some patients are better off on insulin glargine.”
Cost makes a difference
Another, potentially more consequential flaw is insulin degludec’s relative expense.
“To date, use of degludec in routine practice has been limited by its cost, compared with older basal insulins,” observed Dr. Fadini in an interview. “In several countries, including the United States, degludec is substantially more expensive than glargine.”
The ADA’s Standards of Medical Care in Diabetes–2021 includes table 9.3 that lists the costs of various insulins and shows the median average wholesale price of insulin glargine U100 follow-on products as $190/vial, compared with a $407 price for a similar vial of insulin degludec.
Insulin degludec “is clearly superior from a hypoglycemia standpoint. Patients with type 1 diabetes like the reduction because hypoglycemia is scary, and dangerous. The main issue is cost, and the extent to which it may be covered by insurance,” commented Lisa Chow, MD, an endocrinologist at the University of Minnesota, Minneapolis. “We generally won’t prescribe degludec unless it is at a price affordable to the patient. We try to use patient assistance programs sponsored by the company [that markets insulin degludec: Novo Nordisk] to try to make it more affordable.”
Dr. Chow also highlighted that a new wrinkle has been introduction of a more concentrated formulation of insulin glargine, U300, which appears to cause less hypoglycemia than insulin glargine U100. Recent study results indicated that no significant difference exists in the incidence of hypoglycemia among patients treated with insulin glargine U300 and those treated with insulin degludec, such as findings from the BRIGHT trial, which included just over 900 patients, and in the CONCLUDE trial, which randomized more than 1,600 patients.
The HypoDeg study was sponsored by Novo Nordisk, the company that markets insulin degludec. Dr. Brøsen had no personal disclosures, but several of her coauthors were either Novo Nordisk employees or had financial relationships with the company. Dr. de Galan has received research funding from Novo Nordisk. Dr. Fadini has received lecture fees and research funding from Novo Nordisk, from Sanofi, the company that markets insulin glargine, and from several other companies. Dr. Chow has received research funding from Dexcom.
FROM ADA 2021