User login
Acute-Onset Alopecia
The Diagnosis: Thallium-Induced Alopecia
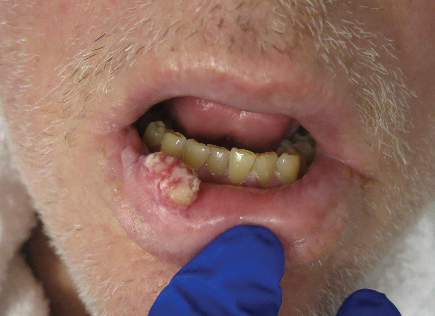
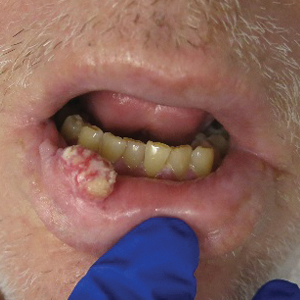
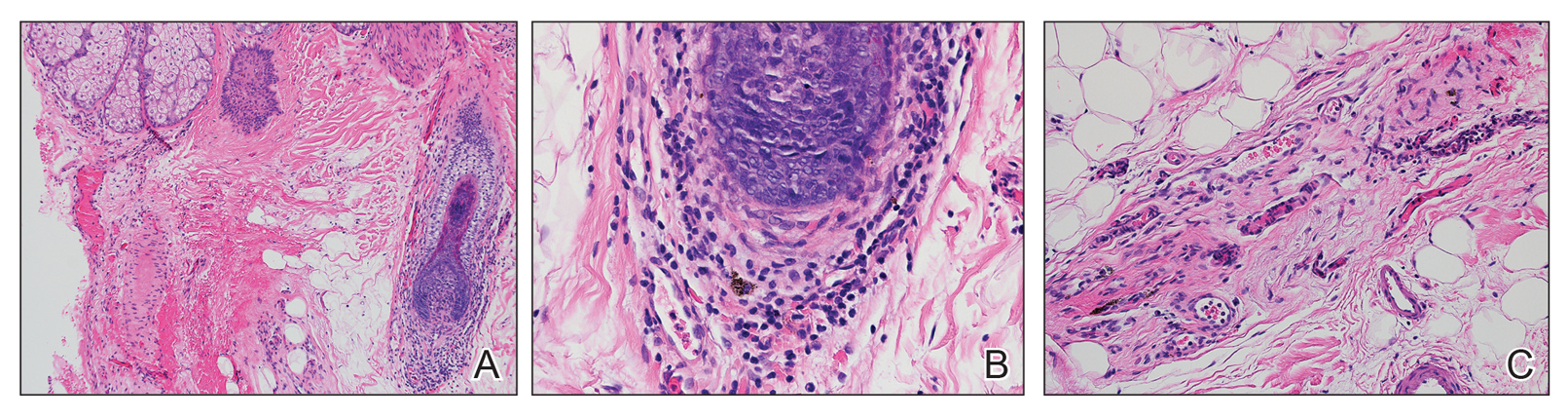
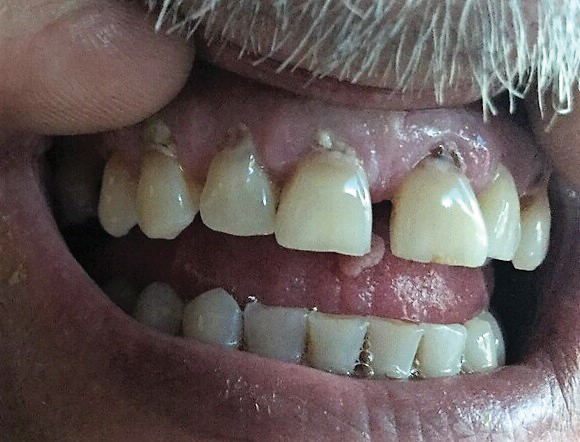
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.
The Diagnosis: Thallium-Induced Alopecia
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
The Diagnosis: Thallium-Induced Alopecia
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.

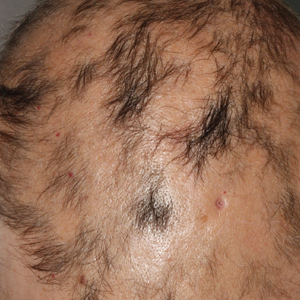
A previously healthy 45-year-old man presented to the dermatology department with abrupt onset of patchy, progressively worsening alopecia of the scalp as well as nausea with emesis and blurry vision of a few weeks' duration. All symptoms were temporally associated with a new demolition job the patient had started at an industrial site. He reported 10 other contractors were similarly affected. The patient denied paresthesia or other skin changes. On physical examination, large patches of smooth alopecia without erythema, scale, scarring, tenderness, or edema that coalesced to involve the majority of the scalp, eyebrows, and eyelashes (inset) were noted.
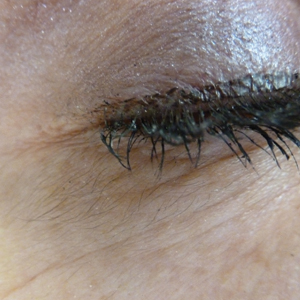
Acquired Hypertrichosis of the Periorbital Area and Malar Cheek
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
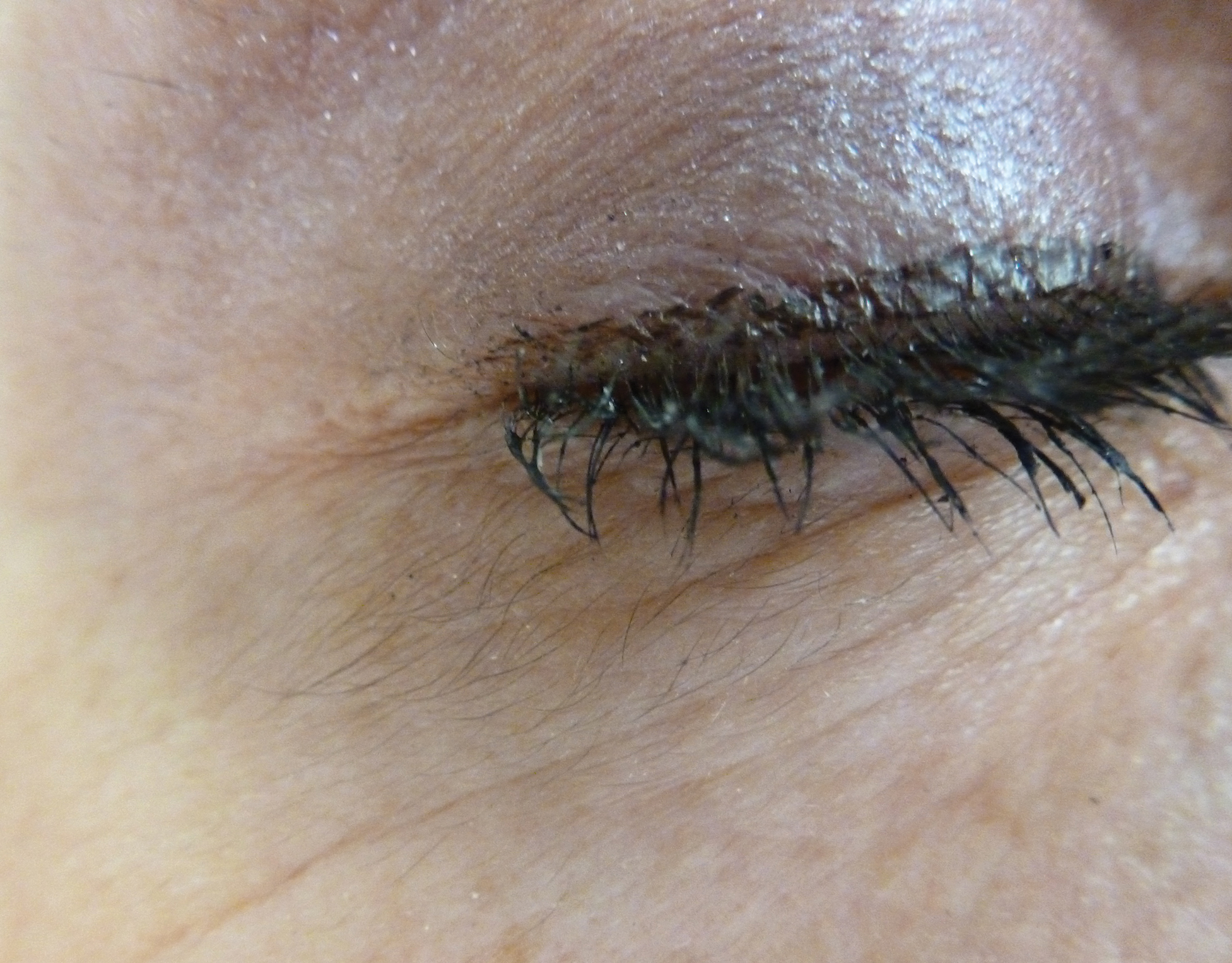
An otherwise healthy woman in her late 50s with Fitzpatrick skin type II presented to the dermatology department for a scheduled cosmetic botulinum toxin injection. Her medical history was notable only for periodic nonsurgical cosmetic procedures including botulinum toxin and dermal fillers, and she was not taking any daily systemic medications. During the preoperative assessment, subtle bilateral and symmetric hypertrichosis with darker terminal hair formation was noted on the periorbital skin and zygomatic cheek. Upon inquiry, the patient admitted to purchasing a “special eye drop” from Mexico and using it regularly. After instillation of 2 to 3 drops per eye, she would laterally wipe the resulting excess drops away from the eyes with her hands and then wash her hands. She denied a change in eye color from their natural brown but did report using blue color contact lenses. She denied an increase in hair growth elsewhere including the upper lip, chin, upper chest, forearms, and hands. She denied deepening of her voice, acne, or hair thinning.
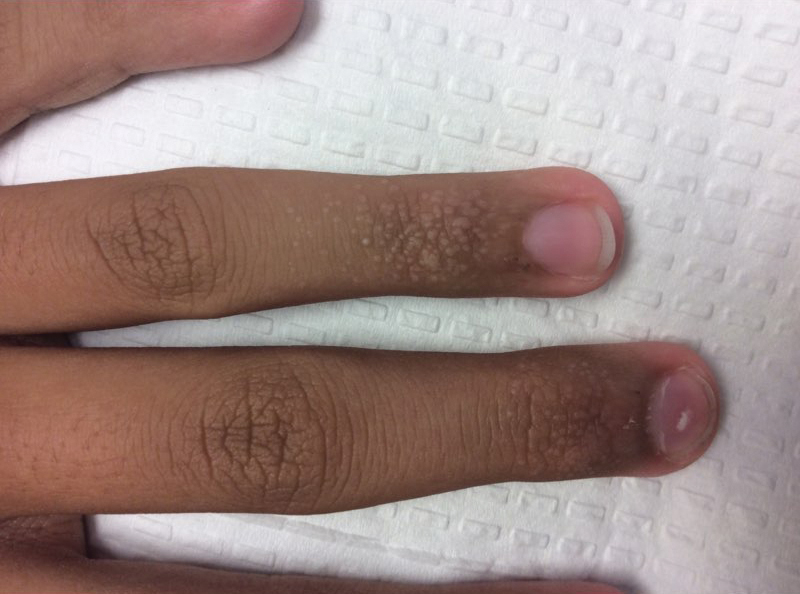
Acral Flesh-Colored Papules on the Fingers
The Diagnosis: Lichen Nitidus
Our patient represents a case of lichen nitidus (LN) that was diagnosed through clinicopathologic correlation, with the pathology results showing a lymphohistiocytic infiltrate in the papillary dermis enclosed by acanthotic rete ridges on either side. Lichen nitidus was first described by Pinkus in 1901 as a variant of lichen planus.1 It is a rare chronic inflammatory disease that is most prevalent in children and adolescents.2 Clinically, the lesions appear as 1- to 2-mm, shiny, flesh-colored papules with central umbilication.3 Typically, lesions are localized and discrete; however, vesicular, hemorrhagic, perforating, spinous follicular, linear, generalized, and actinic variants all have been reported in the literature. Lichen nitidus has a predilection for the lower abdomen, medial thighs, penis, forearms, ventral wrists, and hands.4 Cases of LN have been reported on the palms, soles, nails, and mucosa, presenting a diagnostic challenge.5 The pathogenesis of LN is unknown, and all races and sexes are affected equally.6
Histopathologically, LN has distinct findings including a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis embraced by elongated and acanthotic rete ridges.2 These histopathologic characteristics were seen in our patient's biopsy specimen (Figure) and have been described as the ball-and-claw configuration. Lichen nitidus may be pruritic but typically is asymptomatic.7 It often spontaneously regresses within months to years without any treatment7; however, successful outcomes have been seen with topical steroids, UVA/UVB phototherapy, and retinoids.2 Our patient was treated with topical steroids.

The differential diagnosis for LN includes verruca plana, dyshidrotic eczema, acral persistent papular mucinosis (APPM), and molluscum contagiosum. Verruca plana can occur as 1- to 5-mm, grouped, flesh-colored papules on the face, neck, dorsal hands, wrists, or knees.8 Most commonly, verruca plana occurs due to human papillomavirus type 3 and less commonly human papillomavirus types 10, 27, and 41. Verruca plana is easily differentiated from LN on pathology with findings of epidermal hyperkeratosis, irregular acanthosis, and koilocytic changes.8
Dyshidrotic eczema is a pruritic vesicular rash that is classically distributed symmetrically on the palmar aspects of the hands and lateral fingers.9 Histopathology of the lesions reveals spongiosis with an epidermal lymphocytic infiltrate. Exacerbating factors include exposure to allergens, stress, fungal infections, and genetic predisposition.9
Acral persistent papular mucinosis can present as multiple, 2- to 5-mm, flesh-colored papules on the dorsal aspects of the hands.10 However, the demographic is different from LN, as APPM most commonly affects middle-aged females versus adolescents. Lesions of APPM may multiply or spontaneously remit over time. Acral persistent papular mucinosis generally is asymptomatic but can be treated with cryotherapy, topical corticosteroids, electrodesiccation, or CO2 lasers for cosmetic purposes. Acral persistent papular mucinosis can be easily distinguished from LN on histology, as it will show areas of focal, well-circumscribed mucin in the papillary dermis and a spared Grenz zone.10
Molluscum contagiosum is a common viral skin infection caused by the poxvirus that affects children and adults.11 The skin lesions appear as 2- to 4-mm, dome-shaped, flesh-colored papules with central umbilication on the limbs, trunk, or face. Clinicians may choose to monitor lesions of molluscum contagiosum, as it is a self-limited condition, or it may be treated with cryotherapy, salicylic acid, imiquimod, curettage, laser, or cimetidine.11 On histology, epidermal budlike proliferations can be appreciated in the dermis, and characteristic large, eosinophilic, intracytoplasmic inclusion or molluscum bodies are found in the epidermis.12
- Barber HW. Case of lichen nitidus (Pinkus) or tuberculide lichéniforme et nitida (Chatellier). Proc R Soc Med. 1924;17:39.
- Frey MN, Luzzatto L, Seidel GB, et al. Case for diagnosis. An Bras Dermatol. 2010;85:561-563.
- Pielop JA, Hsu S. Tiny, skin-colored papules on the arms and hands. Am Fam Physician. 2005;72:343-344.
- Cho EB, Kim HY, Park EJ, et al. Three cases of lichen nitidus associated with various cutaneous diseases. Ann Dermatol. 2014;26:505-509.
- Podder I, Mohanty S, Chandra S, et al. Isolated palmar lichen nitidus--a diagnostic challenge: first case from Eastern India. Indian J Dermatol. 2015;60:308-309.
- Chen W, Schramm M, Zouboulis C. Generalized lichen nitidus. J Am Acad Dermatol. 1997;36:630-631.
- Rallis E, Verros C, Moussatou V, et al. Generalized purpuric lichen nitidus: a case report and review of the literature. Dermatol Online J. 2007;13:5.
- Pavithra S, Mallya H, Pai GS. Extensive presentation of verruca plana in a healthy individual. Indian J Dermatol. 2011;56:324-325.
- Paulsen L, Geller D, Guggenbiller M. Symmetrical vesicular eruption on the palms. Am Fam Physician. 2012;15:811-812.
- Alvarez-Garrido H, Najera L, Garrido-Rios A, et al. Acral persistent papular mucinosis: is it an under-diagnosed disease? Dermatol Online J. 2014;20:10
- Diaconu R, Oprea B, Vasilescu M, et al. Inflamed molluscum contagiosum in a 6-year-old boy: a case report. Rom J Morphol Embryol. 2015;56:843-845.
- Krishnamurthy J, Nagappa D. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74.
The Diagnosis: Lichen Nitidus
Our patient represents a case of lichen nitidus (LN) that was diagnosed through clinicopathologic correlation, with the pathology results showing a lymphohistiocytic infiltrate in the papillary dermis enclosed by acanthotic rete ridges on either side. Lichen nitidus was first described by Pinkus in 1901 as a variant of lichen planus.1 It is a rare chronic inflammatory disease that is most prevalent in children and adolescents.2 Clinically, the lesions appear as 1- to 2-mm, shiny, flesh-colored papules with central umbilication.3 Typically, lesions are localized and discrete; however, vesicular, hemorrhagic, perforating, spinous follicular, linear, generalized, and actinic variants all have been reported in the literature. Lichen nitidus has a predilection for the lower abdomen, medial thighs, penis, forearms, ventral wrists, and hands.4 Cases of LN have been reported on the palms, soles, nails, and mucosa, presenting a diagnostic challenge.5 The pathogenesis of LN is unknown, and all races and sexes are affected equally.6
Histopathologically, LN has distinct findings including a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis embraced by elongated and acanthotic rete ridges.2 These histopathologic characteristics were seen in our patient's biopsy specimen (Figure) and have been described as the ball-and-claw configuration. Lichen nitidus may be pruritic but typically is asymptomatic.7 It often spontaneously regresses within months to years without any treatment7; however, successful outcomes have been seen with topical steroids, UVA/UVB phototherapy, and retinoids.2 Our patient was treated with topical steroids.
The differential diagnosis for LN includes verruca plana, dyshidrotic eczema, acral persistent papular mucinosis (APPM), and molluscum contagiosum. Verruca plana can occur as 1- to 5-mm, grouped, flesh-colored papules on the face, neck, dorsal hands, wrists, or knees.8 Most commonly, verruca plana occurs due to human papillomavirus type 3 and less commonly human papillomavirus types 10, 27, and 41. Verruca plana is easily differentiated from LN on pathology with findings of epidermal hyperkeratosis, irregular acanthosis, and koilocytic changes.8
Dyshidrotic eczema is a pruritic vesicular rash that is classically distributed symmetrically on the palmar aspects of the hands and lateral fingers.9 Histopathology of the lesions reveals spongiosis with an epidermal lymphocytic infiltrate. Exacerbating factors include exposure to allergens, stress, fungal infections, and genetic predisposition.9
Acral persistent papular mucinosis can present as multiple, 2- to 5-mm, flesh-colored papules on the dorsal aspects of the hands.10 However, the demographic is different from LN, as APPM most commonly affects middle-aged females versus adolescents. Lesions of APPM may multiply or spontaneously remit over time. Acral persistent papular mucinosis generally is asymptomatic but can be treated with cryotherapy, topical corticosteroids, electrodesiccation, or CO2 lasers for cosmetic purposes. Acral persistent papular mucinosis can be easily distinguished from LN on histology, as it will show areas of focal, well-circumscribed mucin in the papillary dermis and a spared Grenz zone.10
Molluscum contagiosum is a common viral skin infection caused by the poxvirus that affects children and adults.11 The skin lesions appear as 2- to 4-mm, dome-shaped, flesh-colored papules with central umbilication on the limbs, trunk, or face. Clinicians may choose to monitor lesions of molluscum contagiosum, as it is a self-limited condition, or it may be treated with cryotherapy, salicylic acid, imiquimod, curettage, laser, or cimetidine.11 On histology, epidermal budlike proliferations can be appreciated in the dermis, and characteristic large, eosinophilic, intracytoplasmic inclusion or molluscum bodies are found in the epidermis.12
The Diagnosis: Lichen Nitidus
Our patient represents a case of lichen nitidus (LN) that was diagnosed through clinicopathologic correlation, with the pathology results showing a lymphohistiocytic infiltrate in the papillary dermis enclosed by acanthotic rete ridges on either side. Lichen nitidus was first described by Pinkus in 1901 as a variant of lichen planus.1 It is a rare chronic inflammatory disease that is most prevalent in children and adolescents.2 Clinically, the lesions appear as 1- to 2-mm, shiny, flesh-colored papules with central umbilication.3 Typically, lesions are localized and discrete; however, vesicular, hemorrhagic, perforating, spinous follicular, linear, generalized, and actinic variants all have been reported in the literature. Lichen nitidus has a predilection for the lower abdomen, medial thighs, penis, forearms, ventral wrists, and hands.4 Cases of LN have been reported on the palms, soles, nails, and mucosa, presenting a diagnostic challenge.5 The pathogenesis of LN is unknown, and all races and sexes are affected equally.6
Histopathologically, LN has distinct findings including a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis embraced by elongated and acanthotic rete ridges.2 These histopathologic characteristics were seen in our patient's biopsy specimen (Figure) and have been described as the ball-and-claw configuration. Lichen nitidus may be pruritic but typically is asymptomatic.7 It often spontaneously regresses within months to years without any treatment7; however, successful outcomes have been seen with topical steroids, UVA/UVB phototherapy, and retinoids.2 Our patient was treated with topical steroids.
The differential diagnosis for LN includes verruca plana, dyshidrotic eczema, acral persistent papular mucinosis (APPM), and molluscum contagiosum. Verruca plana can occur as 1- to 5-mm, grouped, flesh-colored papules on the face, neck, dorsal hands, wrists, or knees.8 Most commonly, verruca plana occurs due to human papillomavirus type 3 and less commonly human papillomavirus types 10, 27, and 41. Verruca plana is easily differentiated from LN on pathology with findings of epidermal hyperkeratosis, irregular acanthosis, and koilocytic changes.8
Dyshidrotic eczema is a pruritic vesicular rash that is classically distributed symmetrically on the palmar aspects of the hands and lateral fingers.9 Histopathology of the lesions reveals spongiosis with an epidermal lymphocytic infiltrate. Exacerbating factors include exposure to allergens, stress, fungal infections, and genetic predisposition.9
Acral persistent papular mucinosis can present as multiple, 2- to 5-mm, flesh-colored papules on the dorsal aspects of the hands.10 However, the demographic is different from LN, as APPM most commonly affects middle-aged females versus adolescents. Lesions of APPM may multiply or spontaneously remit over time. Acral persistent papular mucinosis generally is asymptomatic but can be treated with cryotherapy, topical corticosteroids, electrodesiccation, or CO2 lasers for cosmetic purposes. Acral persistent papular mucinosis can be easily distinguished from LN on histology, as it will show areas of focal, well-circumscribed mucin in the papillary dermis and a spared Grenz zone.10
Molluscum contagiosum is a common viral skin infection caused by the poxvirus that affects children and adults.11 The skin lesions appear as 2- to 4-mm, dome-shaped, flesh-colored papules with central umbilication on the limbs, trunk, or face. Clinicians may choose to monitor lesions of molluscum contagiosum, as it is a self-limited condition, or it may be treated with cryotherapy, salicylic acid, imiquimod, curettage, laser, or cimetidine.11 On histology, epidermal budlike proliferations can be appreciated in the dermis, and characteristic large, eosinophilic, intracytoplasmic inclusion or molluscum bodies are found in the epidermis.12
- Barber HW. Case of lichen nitidus (Pinkus) or tuberculide lichéniforme et nitida (Chatellier). Proc R Soc Med. 1924;17:39.
- Frey MN, Luzzatto L, Seidel GB, et al. Case for diagnosis. An Bras Dermatol. 2010;85:561-563.
- Pielop JA, Hsu S. Tiny, skin-colored papules on the arms and hands. Am Fam Physician. 2005;72:343-344.
- Cho EB, Kim HY, Park EJ, et al. Three cases of lichen nitidus associated with various cutaneous diseases. Ann Dermatol. 2014;26:505-509.
- Podder I, Mohanty S, Chandra S, et al. Isolated palmar lichen nitidus--a diagnostic challenge: first case from Eastern India. Indian J Dermatol. 2015;60:308-309.
- Chen W, Schramm M, Zouboulis C. Generalized lichen nitidus. J Am Acad Dermatol. 1997;36:630-631.
- Rallis E, Verros C, Moussatou V, et al. Generalized purpuric lichen nitidus: a case report and review of the literature. Dermatol Online J. 2007;13:5.
- Pavithra S, Mallya H, Pai GS. Extensive presentation of verruca plana in a healthy individual. Indian J Dermatol. 2011;56:324-325.
- Paulsen L, Geller D, Guggenbiller M. Symmetrical vesicular eruption on the palms. Am Fam Physician. 2012;15:811-812.
- Alvarez-Garrido H, Najera L, Garrido-Rios A, et al. Acral persistent papular mucinosis: is it an under-diagnosed disease? Dermatol Online J. 2014;20:10
- Diaconu R, Oprea B, Vasilescu M, et al. Inflamed molluscum contagiosum in a 6-year-old boy: a case report. Rom J Morphol Embryol. 2015;56:843-845.
- Krishnamurthy J, Nagappa D. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74.
- Barber HW. Case of lichen nitidus (Pinkus) or tuberculide lichéniforme et nitida (Chatellier). Proc R Soc Med. 1924;17:39.
- Frey MN, Luzzatto L, Seidel GB, et al. Case for diagnosis. An Bras Dermatol. 2010;85:561-563.
- Pielop JA, Hsu S. Tiny, skin-colored papules on the arms and hands. Am Fam Physician. 2005;72:343-344.
- Cho EB, Kim HY, Park EJ, et al. Three cases of lichen nitidus associated with various cutaneous diseases. Ann Dermatol. 2014;26:505-509.
- Podder I, Mohanty S, Chandra S, et al. Isolated palmar lichen nitidus--a diagnostic challenge: first case from Eastern India. Indian J Dermatol. 2015;60:308-309.
- Chen W, Schramm M, Zouboulis C. Generalized lichen nitidus. J Am Acad Dermatol. 1997;36:630-631.
- Rallis E, Verros C, Moussatou V, et al. Generalized purpuric lichen nitidus: a case report and review of the literature. Dermatol Online J. 2007;13:5.
- Pavithra S, Mallya H, Pai GS. Extensive presentation of verruca plana in a healthy individual. Indian J Dermatol. 2011;56:324-325.
- Paulsen L, Geller D, Guggenbiller M. Symmetrical vesicular eruption on the palms. Am Fam Physician. 2012;15:811-812.
- Alvarez-Garrido H, Najera L, Garrido-Rios A, et al. Acral persistent papular mucinosis: is it an under-diagnosed disease? Dermatol Online J. 2014;20:10
- Diaconu R, Oprea B, Vasilescu M, et al. Inflamed molluscum contagiosum in a 6-year-old boy: a case report. Rom J Morphol Embryol. 2015;56:843-845.
- Krishnamurthy J, Nagappa D. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74.
A 13-year-old otherwise healthy adolescent boy presented to the dermatology clinic for a rash on the bilateral dorsal hands of approximately 1 year’s duration. The rash was asymptomatic with no pain or pruritus reported. Physical examination revealed a well-nourished adolescent boy in no acute distress with 1- to 2-mm flesh-colored papules clustered on the bilateral dorsal fingers.
Papules and Telangiectases on the Distal Fingers of a Child
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
The Diagnosis: Juvenile Dermatomyositis
Juvenile dermatomyositis (JDM) is a rare idiopathic inflammatory myopathy of childhood that is autoimmune in nature with an annual incidence ranging from 2.5 to 4.1 cases per million children. Its peak incidence is between 5 and 10 years of age, and it affects girls more than boys at a 2-fold to 5-fold greater rate.1 Juvenile dermatomyositis is characterized by skeletal muscle weakness in the presence of distinctive rashes, including Gottron papules and heliotrope erythema. Muscle weakness typically is proximal and symmetrical, and eventually patients may have trouble rising from a seated position or lifting objects overhead. Other skin manifestations include nail fold capillary changes, calcinosis cutis, and less commonly ulcerations signifying vasculopathy of the skin.2 A subset of patients will present with juvenile amyopathic dermatomyositis. These children have the characteristic skin changes without the muscle weakness or elevated muscle enzymes for more than 6 months; however, one-quarter may go on to develop mysositis.3
Diagnosis of JDM traditionally was based on the following 5 diagnostic criteria: characteristic skin rash, proximal muscle weakness, elevated muscle enzymes, myopathic changes on electromyogram, and typical muscle biopsy.1 Current practice shows a broadening of diagnostic criteria using new techniques in the diagnosis of JDM. To make the diagnosis, the patient must have the characteristic skin manifestations with a minimum of 3 other criteria.4 A 2006 international consensus survey expanded the list of criteria to include typical findings on magnetic resonance imaging (MRI), nail fold capillaroscopy abnormalities, calcinosis, and
dysphonia.5
To assess muscle disease, MRI is utilized because it is a reliable noninvasive tool to assess muscle inflammation. Muscle biopsy is only recommended if the diagnosis is unclear.5 The results of the MRI in our patient displayed symmetric mild fatty atrophy of the gluteus maximus muscle, as well as edema in the right rectus femoris and left vastus lateralis muscles, suggesting early findings of myositis. Muscle enzymes may not be diagnostic because they are not always elevated at diagnosis. Our patient had a normal creatinine kinase level (92 U/L [reference range, <190 U/L]), and both aldolase and lactate dehydrogenase also were within reference range. Conversely, antinuclear antibodies frequently are positive in patients with JDM, such as in our patient at a 1:320 dilution, but are nonspecific and nondiagnostic. It is recommended to include nail fold capillaroscopy to evaluate periungual capillary changes because nailfold capillary density is a sensitive measure of both skin and muscle disease.5 Using dermoscopy, nail fold capillary dilation was observed in our patient.
Other differential diagnoses can have somewhat similar clinical features to JDM. Infantile papular acrodermatitis, commonly referred to as Gianotti-Crosti syndrome, is a viral exanthem that affects children (median age, 2 years).6 The rash appears as monomorphous, flat-topped, pink to brown papules affecting the face, buttocks, and arms; it typically spontaneously resolves in 10 days.6
Juvenile-onset lupus is a chronic autoimmune disorder that can involve any organ system and typically affects children aged 11 to 12 years with a female preponderance. Skin manifestations are similar to adult-onset lupus and include malar rash, discoid rash, oral ulcerations, petechiae, palpable purpura, and digital telangiectasia and ulcers. 7
Juvenile scleroderma is rare connective-tissue disorder that also has multiple organ involvement. Cutaneous involvement can range from isolated morphealike plaques to diffuse sclerotic lesions with growth disturbances, contractures, and facial atrophy.8
Verrucae planae, commonly referred to as flat warts, are papules caused primarily by human papillomavirus types 3, 10, 28, and 41. Children and young adults commonly are affected, and warts can appear on the hands, as in our patient.6
Treatment of JDM depends on disease severity at initial presentation and requires a multidisciplinary approach. The mainstay of treatment is high-dose oral prednisone in combination with disease-modifying drugs such as methotrexate and cyclosporin A. Patients with more severe presentations (eg, ulcerative skin disease) or life-threatening organ involvement are treated with cyclophosphamide, usually in combination with high-dose glucocorticoids.9
Early detection with aggressive treatment is vital to reduce morbidity and mortality from organ damage and disease complications. Mortality rates have dropped to 3%10 in recent decades with the use of systemic glucocorticoids. Delayed treatment is associated with a prolonged disease course and poorer outcomes. Disease complications in children with JDM include osteoporosis, calcinosis, and intestinal perforation; however, with early treatment, children with JDM can expect full recovery and to live a normal life as compared to adults with dermatomyositis.10
Prior to our patient's diagnosis, the family was assigned to move to an overseas location through the US Military with no direct access to advanced medical care. Early detection and diagnosis of JDM through an astute clinical examination allowed the patient and her family to remain in the continental United States to continue receiving specialty care.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
- Mendez EP, Lipton R, Ramsey-Goldman R, et al. US incidence of juvenile dermatomyositis,1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49:300-305.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92:25-41.
- Gerami P, Walling HW, Lewis J, et al. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;57:637-644.
- Enders FB, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329-340.
- Brown VE, Pilkington CA, Feldman BM, et al. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford). 2006;45:990-993.
- William JD, Berger TG, Elston DM. Viral diseases. In: William JD, Berger TG, Elston DM. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:360-413.
- Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. 2012;59:345-364.
- Li SC, Torok KS, Pope E, et al; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Localized Scleroderma Workgroup. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175-1185.
- Stringer E, Ota S, Bohnsack J, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM treatment study. J Rhematol. 2010;37:S1953-S1961.
- Huber AM, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. 2005;7:441-446.
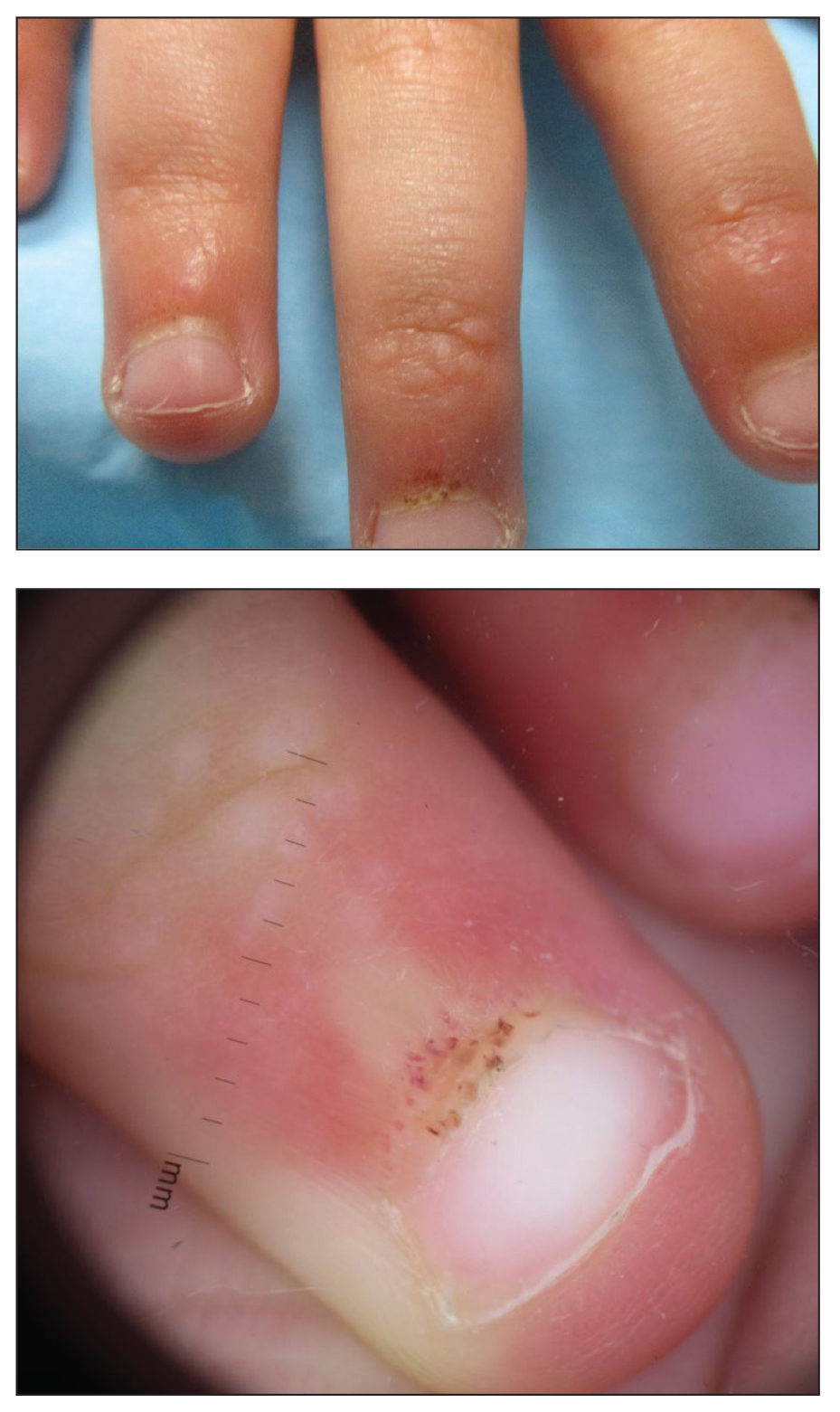
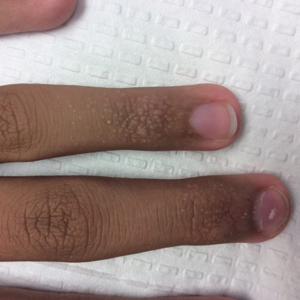
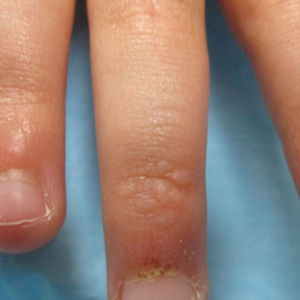
A 4-year-old girl presented to our dermatology clinic with asymptomatic flesh-colored bumps on the fingers of 2 to 3 months’ duration. Prior to presentation the patient was otherwise healthy with normal growth and development. She was referred to dermatology for recommended treatment options for suspected flat warts. On physical examination, grouped 1- to 3-mm, smooth, flat-topped papules were found on the dorsal aspects of the distal interphalangeal joints of all fingers (top). The papules were nonpruritic. Additionally, there were nail findings of ragged cuticles and dilated capillary loops in the proximal nail folds (bottom). The patient did not bite her nails, per the mother’s report, and no other rashes were noted. There were no systemic symptoms or reports of muscle fatigue. She was positive for antinuclear antibodies at 1:320 dilution. Magnetic resonance imaging of the thighs and pelvis was ordered.
Asymptomatic Nodule on the Back
The Diagnosis: Primary Cutaneous Perivascular Epithelioid Cell Tumor
Perivascular epithelioid cell tumors (PEComas) were first described in 1996.1 They comprise a family of rare mesenchymal neoplasms that have a unique characteristic of staining positive for melanocytic and smooth muscle markers on immunohistochemistry.2 These neoplasms have been described in many areas of the body including the uterus, bladder, heart, pancreas, and prostate. The majority of PEComas are extracutaneous, with only 8% of reported cases originating on the skin.3 A case of primary cutaneous PEComa (pcPEComa) was described in 2003.4 The primary cutaneous form is extremely rare.3,5-7
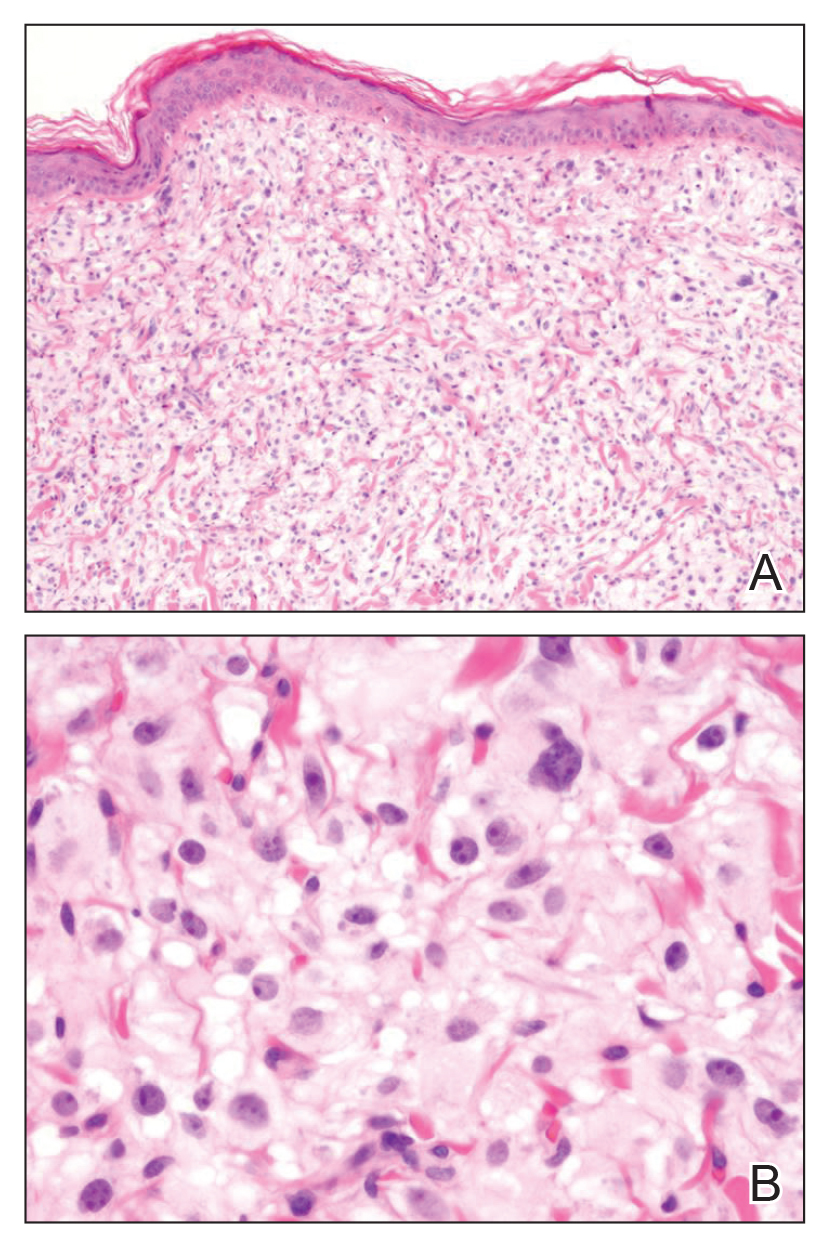
A broad deep shave biopsy was performed in our patient in an attempt to sample the entire lesion. Histopathologic examination of the nodule demonstrated a dermal neoplasm comprised of a diffuse proliferation of large polygonal cells with abundant clear cytoplasm, fine chromatin, and prominent nucleoli (Figure 1A). Higher-power magnification showed moderate nuclear pleomorphism and only rare mitotic figures (Figure 1B).
Immunohistochemical staining revealed positivity for myomelanocytic markers with positivity for human melanoma black 45 (HMB-45)(Figure 2) and desmin (not shown). Additionally, the tumor was positive for CD163 and negative for smooth muscle actin, cytokeratin, and S-100 protein.
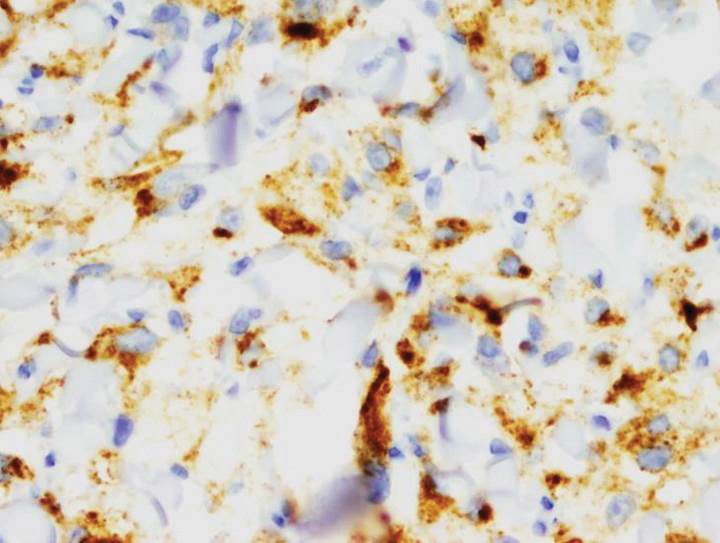
Perivascular epithelioid cell tumors are characterized histologically as mesenchymal neoplasms containing large epithelioid to spindled cells with a slightly granular, vacuolated cytoplasm. These cells often are found in close proximity to vascular structures.3,5,8 The hallmark of PEComas is the expression of both melanocytic and muscle markers.3,8 A review of staining patterns of pcPEComas emphasized that immunophenotypes between visceral and primary cutaneous forms may vary considerably.3,5,8 The most consistent and sensitive melanocytic marker is HMB-45 (88%-92% positive).3,8 Positive Melan-A staining varies in the literature from 0% to 50% of cases.3 Our patient's neoplasm expressed the characteristic myomelanocytic immunophenotype with both HMB-45 and desmin positivity.
Given the histologic characteristics, these lesions can be mistaken for melanocytic and other nonmelanocytic tumors with a clear cell morphology such as balloon cell nevus, hypomelanotic blue nevus, and melanoma.2,3 A pigmented case of pcPEComa was reported in 2015 and was originally diagnosed as metastatic melanoma.6 Unlike pcPEComa, melanoma usually stains positive with S-100 protein in up to 99% of cases8 and is negative for muscle markers; however, a case series reported S-100 protein positivity in 38% of pcPEComas.3 Nonmelanocytic neoplasms in the histologic differential diagnosis include clear cell sarcoma and clear cell renal cell carcinoma, both of which show immunoreactivity for cytokeratin.9
Histologic criteria exist for establishing malignancy potential for visceral PEComas but not for pcPEComas, though it has been suggested that the same malignancy criteria should be applied to pcPEComas.3,9 Features associated with malignancy include size greater than 8 cm, mitotic activity greater than 1 mitosis per 50 high-power fields, infiltrative growth pattern, high nuclear grade, necrosis, and vascular invasion. Based on these criteria, fulfilling 2 or more features technically classifies the lesion as malignant, 1 feature classifies it as uncertain malignant potential, and a lack of these features renders the lesion benign.9
The overwhelming majority of pcPEComas are considered benign. One case of pcPEComa was considered malignant with a high mitotic rate (5 mitoses per 10 high-power fields) and nuclear atypia.10 Further workup with thoracic computed tomography and positron emission tomography-computed tomography was negative for metastasis. Treatment with wide excision and radiotherapy was performed with no sign of recurrence at 24-month follow-up.10
Although pcPEComas arising from the dermis seem to be benign overall, PEComas originating from the subcutaneous tissue may have greater malignancy potential. Two cases of subcutaneous PEComas presenting as nodules resulted in metastasis; one case had local nodal metastasis and another developed metastasis to the lungs months later.10,11
- Zamboni G, Pea M, Martignoni G, et al. Clear cell “sugar” tumorof the pancreas. a novel member of the family of lesions characterizedby the presence of perivascular epithelioid cells. Am J Surg Pathol.1996;20:722-730.
- Folpe AK, Wiatkowski D. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1-15.
- Charli-Joseph Y, Saggini A, Vemula S, et al. Primary cutaneous perivascularepithelioid cell tumor: a clinicopathological and molecular reappraisal. J Am Acad Dermatol. 2014;71:1127-1136.
- Crowson AN, Taylor JR, Magro CM. Cutaneous clear cell myomelanocytictumor-perivascular epithelioid cell tumor: first reported case. Mod Pathol. 2003;16:90A.
- Chaplin A, Conrad D, Tatlidil C, et al. Primary cutaneous PEComa. Am J Dermatopathol. 2010;32:310-312.
- Navale P, Asgari M, Chen S. Pigmented perivascular epithelioid cell tumor of the skin. Am J Dermatopathol. 2015;37:866-869.
- Ieremia E, Robson A. Cutaneous PEComa. Am J Dermatopathol. 2014;36:E198-E201.
- Calder K, Schlauder S, Morgan M. Malignant perivascularepithelioid cell tumor (‘PEComa’): a case report and literature review of cutaneous/subcutaneous presentations. J Cutan Pathol. 2008;35:499-503.
- Folpe A, Mentzel T, Lehr H, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Dermatopathol. 2005; 29:1558-1575.
- Greveling K, Winnepenninckx V, Nagtzaam I, et al. Malignant perivascular epithelioid cell tumor: a case report of a cutaneous tumor on the cheek of a male patient. J Am Acad Dermatol. 2013;69:E262-E264.
- Shon W, Kim J, Sukov W, et al. Malignant TFE3-rearranged perivascular epithelioid cell neoplasm (PEComa) presenting as a subcutaneous mass. Br J Dermatol. 2015;174:617-620.
The Diagnosis: Primary Cutaneous Perivascular Epithelioid Cell Tumor
Perivascular epithelioid cell tumors (PEComas) were first described in 1996.1 They comprise a family of rare mesenchymal neoplasms that have a unique characteristic of staining positive for melanocytic and smooth muscle markers on immunohistochemistry.2 These neoplasms have been described in many areas of the body including the uterus, bladder, heart, pancreas, and prostate. The majority of PEComas are extracutaneous, with only 8% of reported cases originating on the skin.3 A case of primary cutaneous PEComa (pcPEComa) was described in 2003.4 The primary cutaneous form is extremely rare.3,5-7
A broad deep shave biopsy was performed in our patient in an attempt to sample the entire lesion. Histopathologic examination of the nodule demonstrated a dermal neoplasm comprised of a diffuse proliferation of large polygonal cells with abundant clear cytoplasm, fine chromatin, and prominent nucleoli (Figure 1A). Higher-power magnification showed moderate nuclear pleomorphism and only rare mitotic figures (Figure 1B).
Immunohistochemical staining revealed positivity for myomelanocytic markers with positivity for human melanoma black 45 (HMB-45)(Figure 2) and desmin (not shown). Additionally, the tumor was positive for CD163 and negative for smooth muscle actin, cytokeratin, and S-100 protein.
Perivascular epithelioid cell tumors are characterized histologically as mesenchymal neoplasms containing large epithelioid to spindled cells with a slightly granular, vacuolated cytoplasm. These cells often are found in close proximity to vascular structures.3,5,8 The hallmark of PEComas is the expression of both melanocytic and muscle markers.3,8 A review of staining patterns of pcPEComas emphasized that immunophenotypes between visceral and primary cutaneous forms may vary considerably.3,5,8 The most consistent and sensitive melanocytic marker is HMB-45 (88%-92% positive).3,8 Positive Melan-A staining varies in the literature from 0% to 50% of cases.3 Our patient's neoplasm expressed the characteristic myomelanocytic immunophenotype with both HMB-45 and desmin positivity.
Given the histologic characteristics, these lesions can be mistaken for melanocytic and other nonmelanocytic tumors with a clear cell morphology such as balloon cell nevus, hypomelanotic blue nevus, and melanoma.2,3 A pigmented case of pcPEComa was reported in 2015 and was originally diagnosed as metastatic melanoma.6 Unlike pcPEComa, melanoma usually stains positive with S-100 protein in up to 99% of cases8 and is negative for muscle markers; however, a case series reported S-100 protein positivity in 38% of pcPEComas.3 Nonmelanocytic neoplasms in the histologic differential diagnosis include clear cell sarcoma and clear cell renal cell carcinoma, both of which show immunoreactivity for cytokeratin.9
Histologic criteria exist for establishing malignancy potential for visceral PEComas but not for pcPEComas, though it has been suggested that the same malignancy criteria should be applied to pcPEComas.3,9 Features associated with malignancy include size greater than 8 cm, mitotic activity greater than 1 mitosis per 50 high-power fields, infiltrative growth pattern, high nuclear grade, necrosis, and vascular invasion. Based on these criteria, fulfilling 2 or more features technically classifies the lesion as malignant, 1 feature classifies it as uncertain malignant potential, and a lack of these features renders the lesion benign.9
The overwhelming majority of pcPEComas are considered benign. One case of pcPEComa was considered malignant with a high mitotic rate (5 mitoses per 10 high-power fields) and nuclear atypia.10 Further workup with thoracic computed tomography and positron emission tomography-computed tomography was negative for metastasis. Treatment with wide excision and radiotherapy was performed with no sign of recurrence at 24-month follow-up.10
Although pcPEComas arising from the dermis seem to be benign overall, PEComas originating from the subcutaneous tissue may have greater malignancy potential. Two cases of subcutaneous PEComas presenting as nodules resulted in metastasis; one case had local nodal metastasis and another developed metastasis to the lungs months later.10,11
The Diagnosis: Primary Cutaneous Perivascular Epithelioid Cell Tumor
Perivascular epithelioid cell tumors (PEComas) were first described in 1996.1 They comprise a family of rare mesenchymal neoplasms that have a unique characteristic of staining positive for melanocytic and smooth muscle markers on immunohistochemistry.2 These neoplasms have been described in many areas of the body including the uterus, bladder, heart, pancreas, and prostate. The majority of PEComas are extracutaneous, with only 8% of reported cases originating on the skin.3 A case of primary cutaneous PEComa (pcPEComa) was described in 2003.4 The primary cutaneous form is extremely rare.3,5-7
A broad deep shave biopsy was performed in our patient in an attempt to sample the entire lesion. Histopathologic examination of the nodule demonstrated a dermal neoplasm comprised of a diffuse proliferation of large polygonal cells with abundant clear cytoplasm, fine chromatin, and prominent nucleoli (Figure 1A). Higher-power magnification showed moderate nuclear pleomorphism and only rare mitotic figures (Figure 1B).
Immunohistochemical staining revealed positivity for myomelanocytic markers with positivity for human melanoma black 45 (HMB-45)(Figure 2) and desmin (not shown). Additionally, the tumor was positive for CD163 and negative for smooth muscle actin, cytokeratin, and S-100 protein.
Perivascular epithelioid cell tumors are characterized histologically as mesenchymal neoplasms containing large epithelioid to spindled cells with a slightly granular, vacuolated cytoplasm. These cells often are found in close proximity to vascular structures.3,5,8 The hallmark of PEComas is the expression of both melanocytic and muscle markers.3,8 A review of staining patterns of pcPEComas emphasized that immunophenotypes between visceral and primary cutaneous forms may vary considerably.3,5,8 The most consistent and sensitive melanocytic marker is HMB-45 (88%-92% positive).3,8 Positive Melan-A staining varies in the literature from 0% to 50% of cases.3 Our patient's neoplasm expressed the characteristic myomelanocytic immunophenotype with both HMB-45 and desmin positivity.
Given the histologic characteristics, these lesions can be mistaken for melanocytic and other nonmelanocytic tumors with a clear cell morphology such as balloon cell nevus, hypomelanotic blue nevus, and melanoma.2,3 A pigmented case of pcPEComa was reported in 2015 and was originally diagnosed as metastatic melanoma.6 Unlike pcPEComa, melanoma usually stains positive with S-100 protein in up to 99% of cases8 and is negative for muscle markers; however, a case series reported S-100 protein positivity in 38% of pcPEComas.3 Nonmelanocytic neoplasms in the histologic differential diagnosis include clear cell sarcoma and clear cell renal cell carcinoma, both of which show immunoreactivity for cytokeratin.9
Histologic criteria exist for establishing malignancy potential for visceral PEComas but not for pcPEComas, though it has been suggested that the same malignancy criteria should be applied to pcPEComas.3,9 Features associated with malignancy include size greater than 8 cm, mitotic activity greater than 1 mitosis per 50 high-power fields, infiltrative growth pattern, high nuclear grade, necrosis, and vascular invasion. Based on these criteria, fulfilling 2 or more features technically classifies the lesion as malignant, 1 feature classifies it as uncertain malignant potential, and a lack of these features renders the lesion benign.9
The overwhelming majority of pcPEComas are considered benign. One case of pcPEComa was considered malignant with a high mitotic rate (5 mitoses per 10 high-power fields) and nuclear atypia.10 Further workup with thoracic computed tomography and positron emission tomography-computed tomography was negative for metastasis. Treatment with wide excision and radiotherapy was performed with no sign of recurrence at 24-month follow-up.10
Although pcPEComas arising from the dermis seem to be benign overall, PEComas originating from the subcutaneous tissue may have greater malignancy potential. Two cases of subcutaneous PEComas presenting as nodules resulted in metastasis; one case had local nodal metastasis and another developed metastasis to the lungs months later.10,11
- Zamboni G, Pea M, Martignoni G, et al. Clear cell “sugar” tumorof the pancreas. a novel member of the family of lesions characterizedby the presence of perivascular epithelioid cells. Am J Surg Pathol.1996;20:722-730.
- Folpe AK, Wiatkowski D. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1-15.
- Charli-Joseph Y, Saggini A, Vemula S, et al. Primary cutaneous perivascularepithelioid cell tumor: a clinicopathological and molecular reappraisal. J Am Acad Dermatol. 2014;71:1127-1136.
- Crowson AN, Taylor JR, Magro CM. Cutaneous clear cell myomelanocytictumor-perivascular epithelioid cell tumor: first reported case. Mod Pathol. 2003;16:90A.
- Chaplin A, Conrad D, Tatlidil C, et al. Primary cutaneous PEComa. Am J Dermatopathol. 2010;32:310-312.
- Navale P, Asgari M, Chen S. Pigmented perivascular epithelioid cell tumor of the skin. Am J Dermatopathol. 2015;37:866-869.
- Ieremia E, Robson A. Cutaneous PEComa. Am J Dermatopathol. 2014;36:E198-E201.
- Calder K, Schlauder S, Morgan M. Malignant perivascularepithelioid cell tumor (‘PEComa’): a case report and literature review of cutaneous/subcutaneous presentations. J Cutan Pathol. 2008;35:499-503.
- Folpe A, Mentzel T, Lehr H, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Dermatopathol. 2005; 29:1558-1575.
- Greveling K, Winnepenninckx V, Nagtzaam I, et al. Malignant perivascular epithelioid cell tumor: a case report of a cutaneous tumor on the cheek of a male patient. J Am Acad Dermatol. 2013;69:E262-E264.
- Shon W, Kim J, Sukov W, et al. Malignant TFE3-rearranged perivascular epithelioid cell neoplasm (PEComa) presenting as a subcutaneous mass. Br J Dermatol. 2015;174:617-620.
- Zamboni G, Pea M, Martignoni G, et al. Clear cell “sugar” tumorof the pancreas. a novel member of the family of lesions characterizedby the presence of perivascular epithelioid cells. Am J Surg Pathol.1996;20:722-730.
- Folpe AK, Wiatkowski D. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1-15.
- Charli-Joseph Y, Saggini A, Vemula S, et al. Primary cutaneous perivascularepithelioid cell tumor: a clinicopathological and molecular reappraisal. J Am Acad Dermatol. 2014;71:1127-1136.
- Crowson AN, Taylor JR, Magro CM. Cutaneous clear cell myomelanocytictumor-perivascular epithelioid cell tumor: first reported case. Mod Pathol. 2003;16:90A.
- Chaplin A, Conrad D, Tatlidil C, et al. Primary cutaneous PEComa. Am J Dermatopathol. 2010;32:310-312.
- Navale P, Asgari M, Chen S. Pigmented perivascular epithelioid cell tumor of the skin. Am J Dermatopathol. 2015;37:866-869.
- Ieremia E, Robson A. Cutaneous PEComa. Am J Dermatopathol. 2014;36:E198-E201.
- Calder K, Schlauder S, Morgan M. Malignant perivascularepithelioid cell tumor (‘PEComa’): a case report and literature review of cutaneous/subcutaneous presentations. J Cutan Pathol. 2008;35:499-503.
- Folpe A, Mentzel T, Lehr H, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Dermatopathol. 2005; 29:1558-1575.
- Greveling K, Winnepenninckx V, Nagtzaam I, et al. Malignant perivascular epithelioid cell tumor: a case report of a cutaneous tumor on the cheek of a male patient. J Am Acad Dermatol. 2013;69:E262-E264.
- Shon W, Kim J, Sukov W, et al. Malignant TFE3-rearranged perivascular epithelioid cell neoplasm (PEComa) presenting as a subcutaneous mass. Br J Dermatol. 2015;174:617-620.
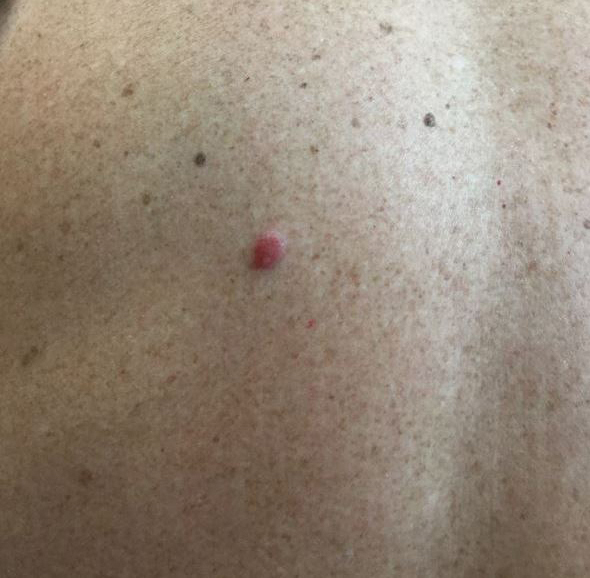
A 54-year-old man presented with an asymptomatic nodule on the left side of the mid back that had been slowly growing in size over the last 12 months. The patient had 2 other lesions on the nasal supratip and left upper arm that were concerning for basal cell carcinoma. The patient’s medical history was notable for stage IV mantle cell lymphoma diagnosed 8 years prior by lymph node biopsy. He completed multiple rounds of methotrexate and CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy over 2 years and later received a stem cell transplant; he had been in clinical remission for the last 6 years. On review of symptoms he denied any fevers, chills, fatigue, night sweats, or constitutional symptoms. The remainder of the review of symptoms was negative. Physical examination showed a 1.5×1.0-cm pink, firm, nontender nodule on the left side of the mid back.
Erythematous and Necrotic Papules in an Immunosuppressed Woman
The Diagnosis: Disseminated Fusariosis
Histologic evaluation of the punch biopsy demonstrated thrombosed vessels in the deep dermis and along fibrous septae of subcutaneous tissue, as well as delicate, thin-walled, branching hyphae with vesicular swellings (Figure). The hyphae were present within the vascular thrombi and extended into surrounding tissue. The fungal tissue culture eventually grew scant Fusarium. At the time of biopsy, there was a high index of suspicion for fungal infection, which supported the decision to empirically treat with anidulafungin and voriconazole.
Differentiating the diagnosis in this case was done primarily with histopathology. Although Aspergillus also has slender hyphae, it lacks the vesicular swellings characteristic of fusariosis. Disseminated candidiasis would demonstrate budding yeast and pseudohyphae in the dermis. Ecthyma gangrenosum histologically presents as necrotizing hemorrhagic vasculitis with gram-negative rods in the walls of deeper vessels, characteristically sparing the intima. Leukemia cutis histologically varies but would display a neoplastic infiltrate of atypical monocytoid cells with nuclear pleomorphism.
Our patient had been treated with palliative chemotherapy as a salvage regimen with idarubicin and cytarabine. She had persistent pancytopenia despite granulocyte-macrophage colony-stimulating factor therapy. The mortality rate for disseminated Fusarium infection approaches 100% when risk factors such as angiotropism and prolonged neutropenia are present.1,2 Additionally, our patient's susceptibility profile subsequently demonstrated an elevated minimum inhibitory concentration to amphotericin B, itraconazole, voriconazole, and posaconazole. The neutropenia and Fusarium infection were not responsive to treatment. She was discharged on palliative voriconazole with home hospice care.
Fusarium species are soil-dwelling saprophytes and important plant pathogens that have increasingly emerged as rare but notable causes of morbidity and mortality in immunocompromised patients.1-3 More specifically, Fusarium infection is most commonly observed in patients with hematologic malignancy complicated by persistent neutropenia. The 3 most frequently encountered Fusarium species in human disease are Fusarium solani, Fusarium oxysporum, and Fusarium moniliforme, with F solani being the most virulent.1,2 Infection with Fusarium may manifest as a broad range of presentations depending on the route of entry, such as endophthalmitis, sinusitis, pneumonia, and cutaneous lesions.1 Disseminated infection is marked by skin lesions or positive blood cultures for Fusarium.3 This fungus is notorious for its limited susceptibility profile.1 It requires systemic antifungal medications such as triazoles and amphotericin B. Fusarium is most susceptible in vitro to amphotericin B but often requires toxic dosages to be effective in decreasing fungal load.2,3 The high mortality rate of disseminated fusariosis further emphasizes that prevention is an important component to protecting high-risk patients. Keeping patients in rooms with high-efficiency particulate arresting filters and limiting exposure to unsanitized tap water faucets can help decrease exposure; however, reducing immunosuppression and improving neutropenia are the most effective ways to prevent fusariosis.1 Although skin breakdown can facilitate the spread of infection, it has been observed that immunosuppressed individuals do not necessarily have this finding.4
This case emphasizes the importance of considering disseminated fusariosis in patients with hematologic malignancy or other immunosuppressed conditions. The most important factors that should raise clinical suspicion are persistent neutropenia and recent corticosteroid therapy.1 A clinical picture that suggests fungal infection should warrant consideration of prophylactic treatment as well as tissue and blood cultures to determine species and susceptibility.
- Nucci M, Anaissie E. Fusarium infections in immunocompromised patients. Clin Microbiol Rev. 2007;20:695-704.
- Jossi M, Ambrosioni J, Macedo-Vinas M, et al. Invasive fusariosis with prolonged fungemia in a patient with acute lymphoblastic leukemia: case report and review of the literature. Int J Infect Dis. 2010;14:E354-E356.
- Tan R, Ng KP, Gan GG, et al. Fusarium sp. infection in a patient with Acute Lymphoblastic Leukaemia. Med J Malaysia. 2013;68:479-480.
- Nucci M, Anaissie E. Cutaneous infection by Fusarium species in healthy and immunocompromised hosts: implications for diagnosis and management. Clin Infect Dis. 2002;35:909-920.
The Diagnosis: Disseminated Fusariosis
Histologic evaluation of the punch biopsy demonstrated thrombosed vessels in the deep dermis and along fibrous septae of subcutaneous tissue, as well as delicate, thin-walled, branching hyphae with vesicular swellings (Figure). The hyphae were present within the vascular thrombi and extended into surrounding tissue. The fungal tissue culture eventually grew scant Fusarium. At the time of biopsy, there was a high index of suspicion for fungal infection, which supported the decision to empirically treat with anidulafungin and voriconazole.
Differentiating the diagnosis in this case was done primarily with histopathology. Although Aspergillus also has slender hyphae, it lacks the vesicular swellings characteristic of fusariosis. Disseminated candidiasis would demonstrate budding yeast and pseudohyphae in the dermis. Ecthyma gangrenosum histologically presents as necrotizing hemorrhagic vasculitis with gram-negative rods in the walls of deeper vessels, characteristically sparing the intima. Leukemia cutis histologically varies but would display a neoplastic infiltrate of atypical monocytoid cells with nuclear pleomorphism.
Our patient had been treated with palliative chemotherapy as a salvage regimen with idarubicin and cytarabine. She had persistent pancytopenia despite granulocyte-macrophage colony-stimulating factor therapy. The mortality rate for disseminated Fusarium infection approaches 100% when risk factors such as angiotropism and prolonged neutropenia are present.1,2 Additionally, our patient's susceptibility profile subsequently demonstrated an elevated minimum inhibitory concentration to amphotericin B, itraconazole, voriconazole, and posaconazole. The neutropenia and Fusarium infection were not responsive to treatment. She was discharged on palliative voriconazole with home hospice care.
Fusarium species are soil-dwelling saprophytes and important plant pathogens that have increasingly emerged as rare but notable causes of morbidity and mortality in immunocompromised patients.1-3 More specifically, Fusarium infection is most commonly observed in patients with hematologic malignancy complicated by persistent neutropenia. The 3 most frequently encountered Fusarium species in human disease are Fusarium solani, Fusarium oxysporum, and Fusarium moniliforme, with F solani being the most virulent.1,2 Infection with Fusarium may manifest as a broad range of presentations depending on the route of entry, such as endophthalmitis, sinusitis, pneumonia, and cutaneous lesions.1 Disseminated infection is marked by skin lesions or positive blood cultures for Fusarium.3 This fungus is notorious for its limited susceptibility profile.1 It requires systemic antifungal medications such as triazoles and amphotericin B. Fusarium is most susceptible in vitro to amphotericin B but often requires toxic dosages to be effective in decreasing fungal load.2,3 The high mortality rate of disseminated fusariosis further emphasizes that prevention is an important component to protecting high-risk patients. Keeping patients in rooms with high-efficiency particulate arresting filters and limiting exposure to unsanitized tap water faucets can help decrease exposure; however, reducing immunosuppression and improving neutropenia are the most effective ways to prevent fusariosis.1 Although skin breakdown can facilitate the spread of infection, it has been observed that immunosuppressed individuals do not necessarily have this finding.4
This case emphasizes the importance of considering disseminated fusariosis in patients with hematologic malignancy or other immunosuppressed conditions. The most important factors that should raise clinical suspicion are persistent neutropenia and recent corticosteroid therapy.1 A clinical picture that suggests fungal infection should warrant consideration of prophylactic treatment as well as tissue and blood cultures to determine species and susceptibility.
The Diagnosis: Disseminated Fusariosis
Histologic evaluation of the punch biopsy demonstrated thrombosed vessels in the deep dermis and along fibrous septae of subcutaneous tissue, as well as delicate, thin-walled, branching hyphae with vesicular swellings (Figure). The hyphae were present within the vascular thrombi and extended into surrounding tissue. The fungal tissue culture eventually grew scant Fusarium. At the time of biopsy, there was a high index of suspicion for fungal infection, which supported the decision to empirically treat with anidulafungin and voriconazole.
Differentiating the diagnosis in this case was done primarily with histopathology. Although Aspergillus also has slender hyphae, it lacks the vesicular swellings characteristic of fusariosis. Disseminated candidiasis would demonstrate budding yeast and pseudohyphae in the dermis. Ecthyma gangrenosum histologically presents as necrotizing hemorrhagic vasculitis with gram-negative rods in the walls of deeper vessels, characteristically sparing the intima. Leukemia cutis histologically varies but would display a neoplastic infiltrate of atypical monocytoid cells with nuclear pleomorphism.
Our patient had been treated with palliative chemotherapy as a salvage regimen with idarubicin and cytarabine. She had persistent pancytopenia despite granulocyte-macrophage colony-stimulating factor therapy. The mortality rate for disseminated Fusarium infection approaches 100% when risk factors such as angiotropism and prolonged neutropenia are present.1,2 Additionally, our patient's susceptibility profile subsequently demonstrated an elevated minimum inhibitory concentration to amphotericin B, itraconazole, voriconazole, and posaconazole. The neutropenia and Fusarium infection were not responsive to treatment. She was discharged on palliative voriconazole with home hospice care.
Fusarium species are soil-dwelling saprophytes and important plant pathogens that have increasingly emerged as rare but notable causes of morbidity and mortality in immunocompromised patients.1-3 More specifically, Fusarium infection is most commonly observed in patients with hematologic malignancy complicated by persistent neutropenia. The 3 most frequently encountered Fusarium species in human disease are Fusarium solani, Fusarium oxysporum, and Fusarium moniliforme, with F solani being the most virulent.1,2 Infection with Fusarium may manifest as a broad range of presentations depending on the route of entry, such as endophthalmitis, sinusitis, pneumonia, and cutaneous lesions.1 Disseminated infection is marked by skin lesions or positive blood cultures for Fusarium.3 This fungus is notorious for its limited susceptibility profile.1 It requires systemic antifungal medications such as triazoles and amphotericin B. Fusarium is most susceptible in vitro to amphotericin B but often requires toxic dosages to be effective in decreasing fungal load.2,3 The high mortality rate of disseminated fusariosis further emphasizes that prevention is an important component to protecting high-risk patients. Keeping patients in rooms with high-efficiency particulate arresting filters and limiting exposure to unsanitized tap water faucets can help decrease exposure; however, reducing immunosuppression and improving neutropenia are the most effective ways to prevent fusariosis.1 Although skin breakdown can facilitate the spread of infection, it has been observed that immunosuppressed individuals do not necessarily have this finding.4
This case emphasizes the importance of considering disseminated fusariosis in patients with hematologic malignancy or other immunosuppressed conditions. The most important factors that should raise clinical suspicion are persistent neutropenia and recent corticosteroid therapy.1 A clinical picture that suggests fungal infection should warrant consideration of prophylactic treatment as well as tissue and blood cultures to determine species and susceptibility.
- Nucci M, Anaissie E. Fusarium infections in immunocompromised patients. Clin Microbiol Rev. 2007;20:695-704.
- Jossi M, Ambrosioni J, Macedo-Vinas M, et al. Invasive fusariosis with prolonged fungemia in a patient with acute lymphoblastic leukemia: case report and review of the literature. Int J Infect Dis. 2010;14:E354-E356.
- Tan R, Ng KP, Gan GG, et al. Fusarium sp. infection in a patient with Acute Lymphoblastic Leukaemia. Med J Malaysia. 2013;68:479-480.
- Nucci M, Anaissie E. Cutaneous infection by Fusarium species in healthy and immunocompromised hosts: implications for diagnosis and management. Clin Infect Dis. 2002;35:909-920.
- Nucci M, Anaissie E. Fusarium infections in immunocompromised patients. Clin Microbiol Rev. 2007;20:695-704.
- Jossi M, Ambrosioni J, Macedo-Vinas M, et al. Invasive fusariosis with prolonged fungemia in a patient with acute lymphoblastic leukemia: case report and review of the literature. Int J Infect Dis. 2010;14:E354-E356.
- Tan R, Ng KP, Gan GG, et al. Fusarium sp. infection in a patient with Acute Lymphoblastic Leukaemia. Med J Malaysia. 2013;68:479-480.
- Nucci M, Anaissie E. Cutaneous infection by Fusarium species in healthy and immunocompromised hosts: implications for diagnosis and management. Clin Infect Dis. 2002;35:909-920.
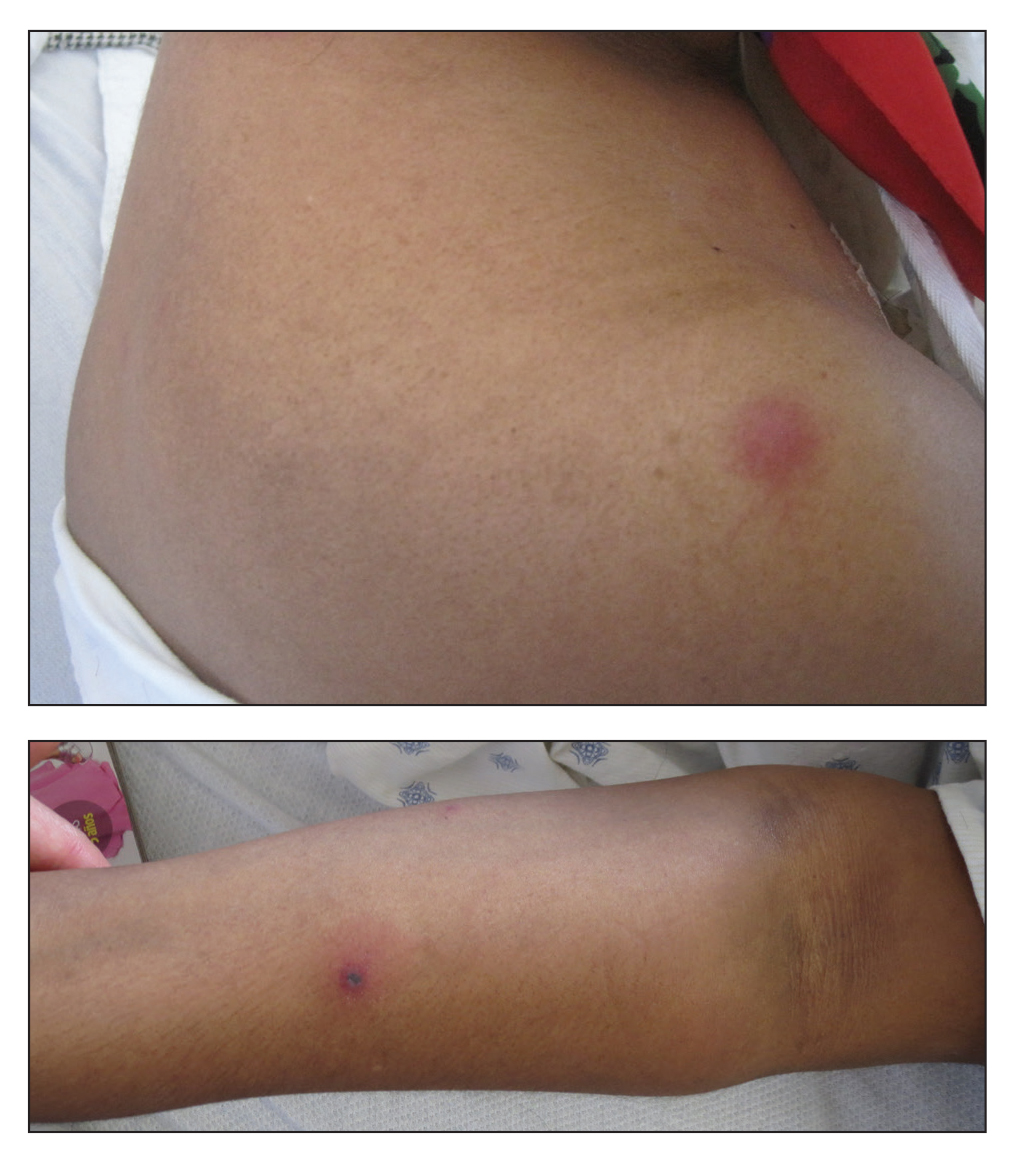

Pruritic Nodules on the Breast
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
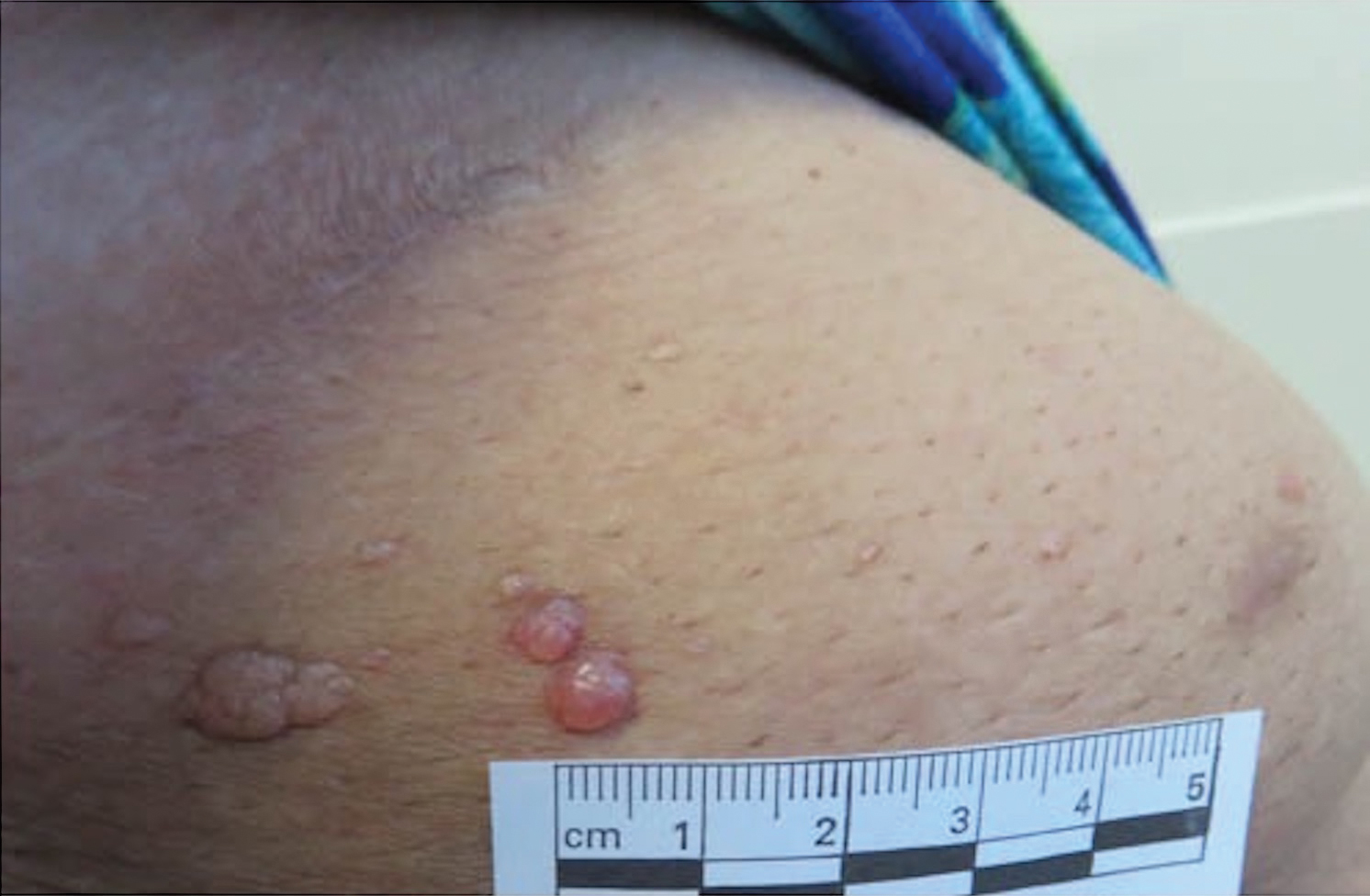
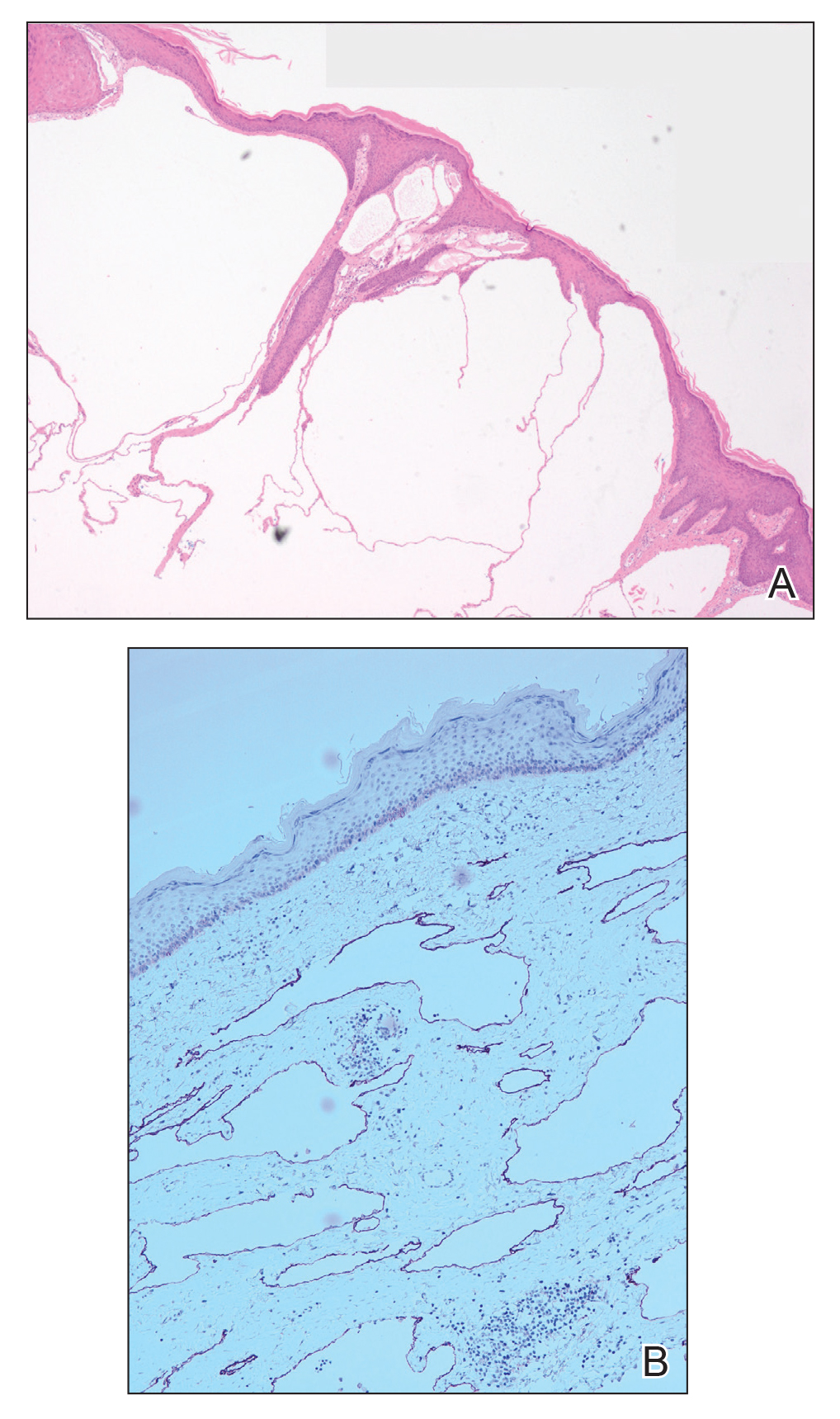
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
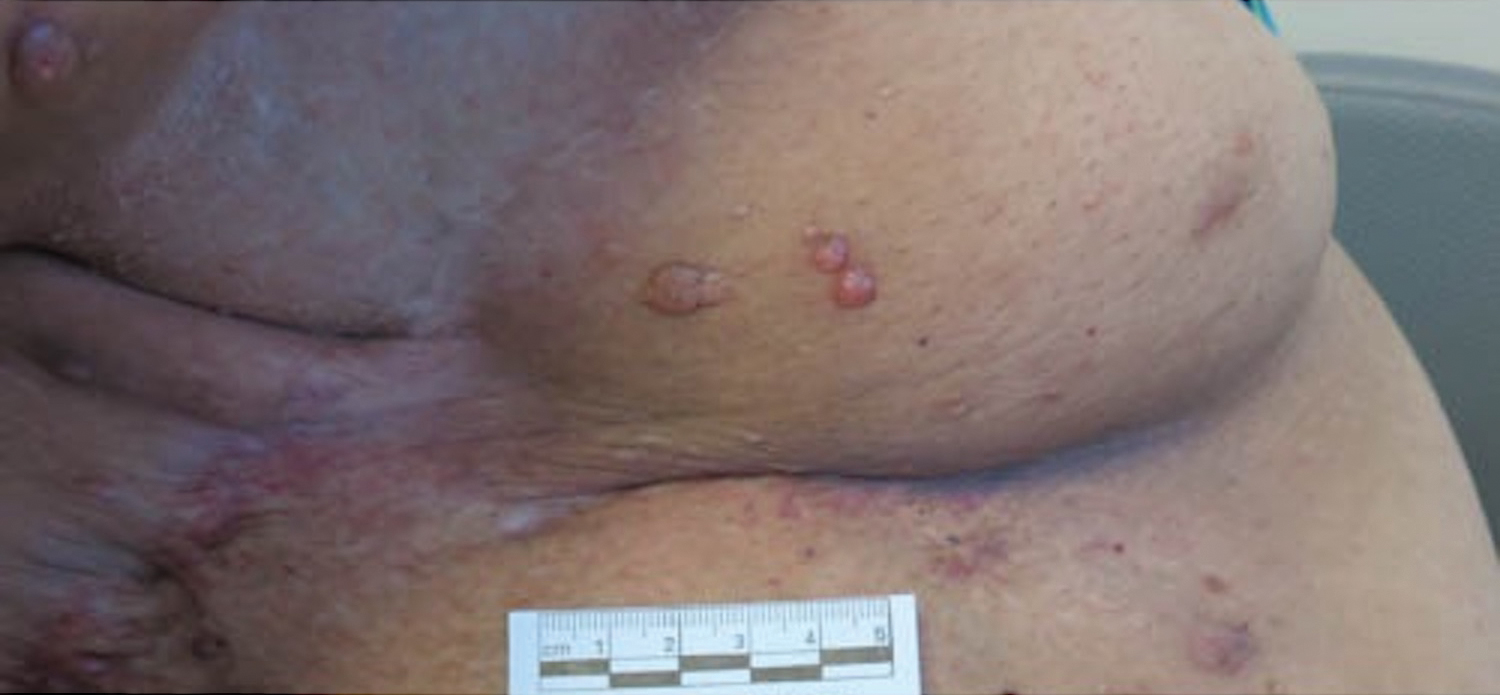
A 51-year-old woman with a history of bilateral breast cancer presented for evaluation of lesions on the underside of the right breast. She was first diagnosed with stage II cancer of the right breast that was subsequently treated with a mastectomy and adjuvant chemotherapy 7 years prior to presentation. One year later, she developed stage IIIC adenocarcinoma of the left breast and was treated with a modified radical mastectomy, adjuvant chemotherapy, and radiation. She had been followed closely by her oncologist with regular surveillance imaging (last at 7 months prior to presentation) that had all been negative for recurrent breast cancer. She presented to our dermatology clinic for evaluation of lesions on the underside of the right breast that were pruritic and occasionally painful with a burning quality. These lesions had recently begun to bleed when scratched but were not otherwise growing or spreading. On physical examination she was afebrile with stable vital signs. Skin examination was notable for numerous violaceous and translucent papules and nodules underneath the right breast and axilla overlying a well-healed mastectomy scar. No lymphadenopathy was present. Shave biopsies were performed and showed well-circumscribed nodular lesions with ectatic vascular channels separated by thin fibrous walls and filled with eosinophilic proteinaceous material and scattered red blood cells. Immunohistochemical staining also showed positivity for D2-40.
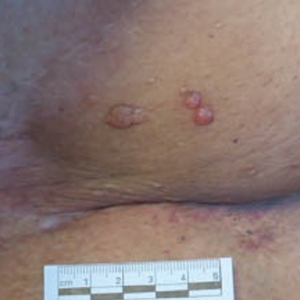
Annular Atrophic Plaques on the Forearm
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
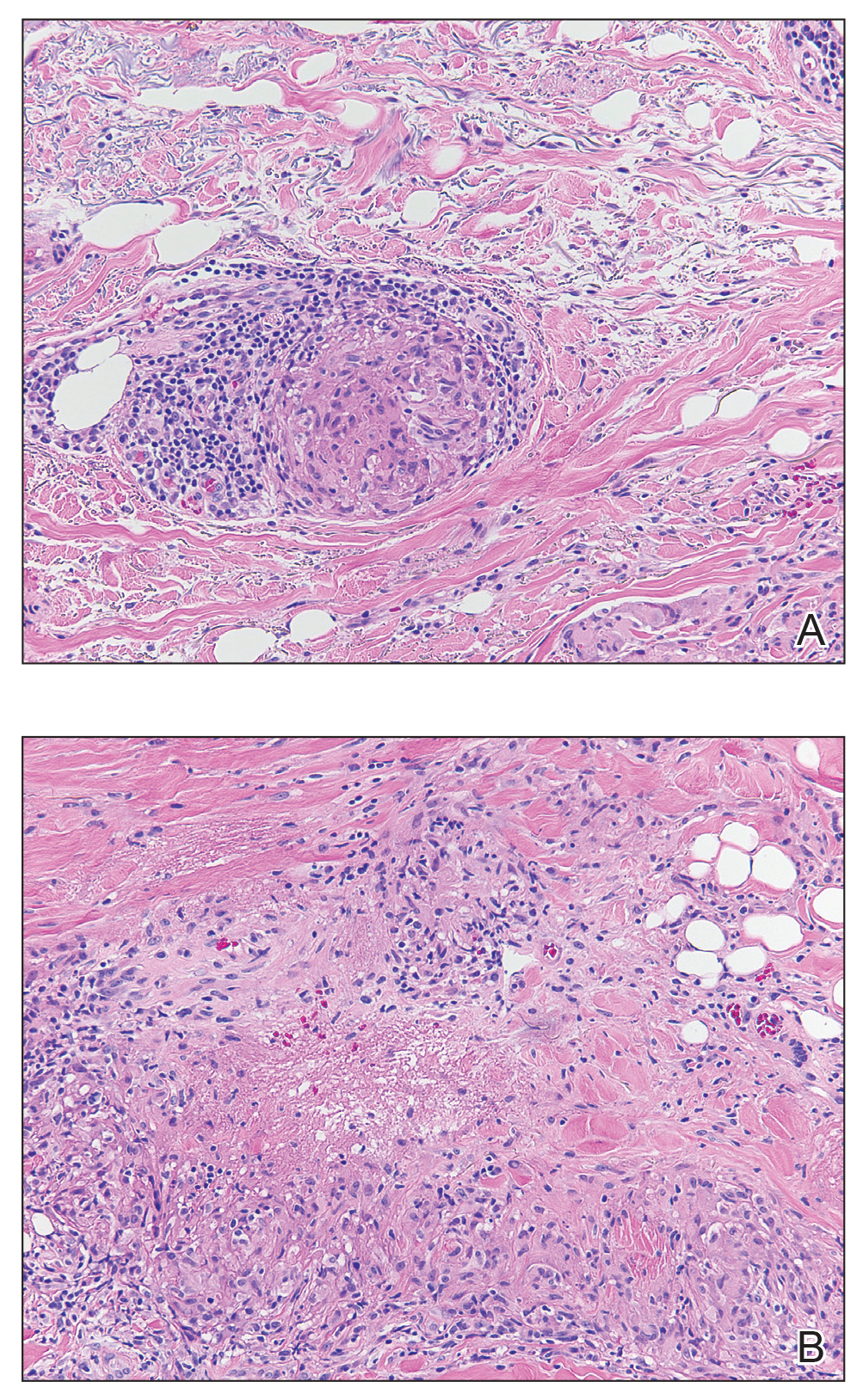
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
A 57-year-old woman presented with several lesions on the left extensor forearm of 10 years’ duration. A single annular indurated lesion with central atrophy initially developed near a prior surgical site. The lesions were pruritic with no associated pain or bleeding. Over 5 years, similar lesions had developed extending up the arm. No benefit was seen with low-potency topical steroid application. Biopsy for histopathologic examination was performed to confirm the diagnosis.
Erythematous Edematous Plaques on the Dorsal Aspects of the Hands
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
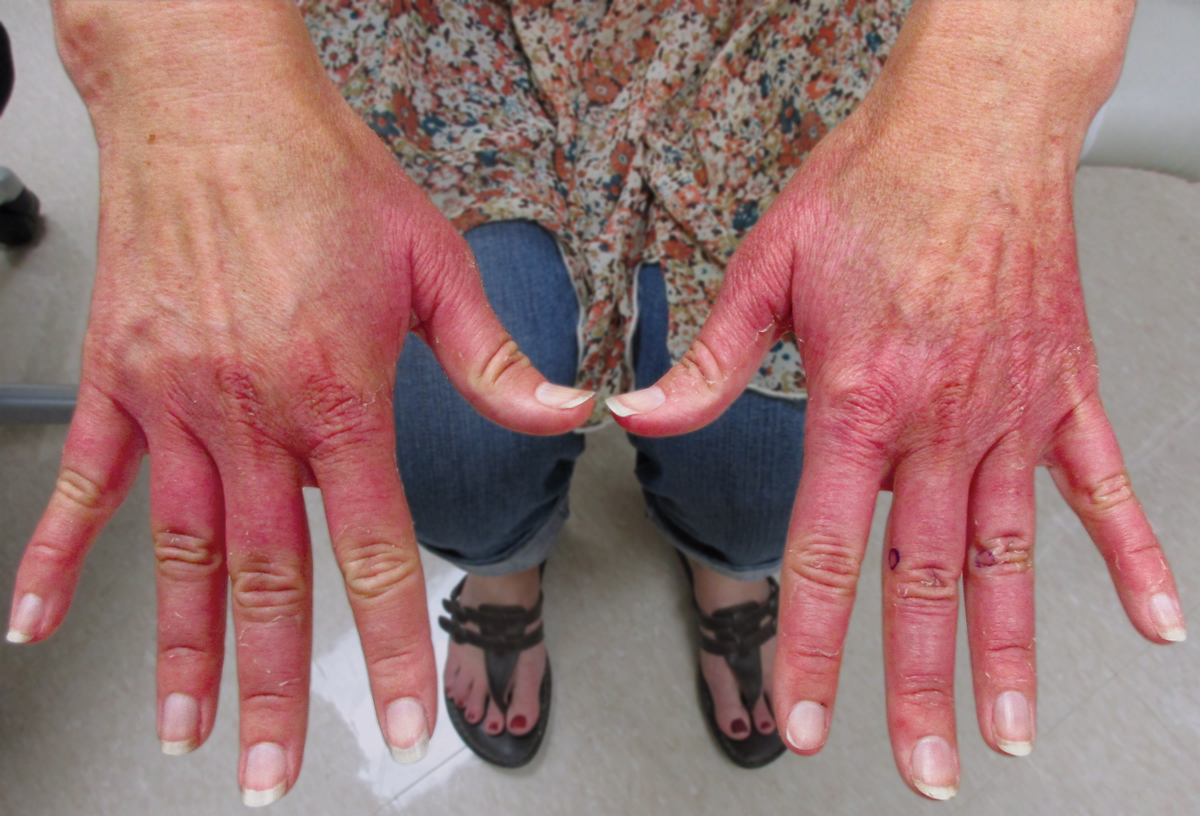
A 48-year-old woman presented with erythematous swelling of the dorsal aspects of the bilateral hands followed by desquamation and pruritus of 2 weeks’ duration. She denied any recent contact with plants, chemicals, or topical products or use of over-the-counter medications. A 6-day course of prednisone provided by her primary care physician relieved the swelling and pruritus; however, the erythema persisted. Physical examination revealed clearly demarcated, erythematous to violaceous, edematous plaques with peripheral scaling that involved all digits. There was notable sparing of the proximal interphalangeal joints and volar aspects of the hands extending proximally to the metacarpophalangeal joints.
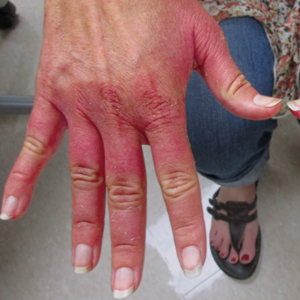
Growing Painful Nodule on the Lower Lip
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.