User login
Crusted Plaque in the Umbilicus
The Diagnosis: Sister Mary Joseph Nodule
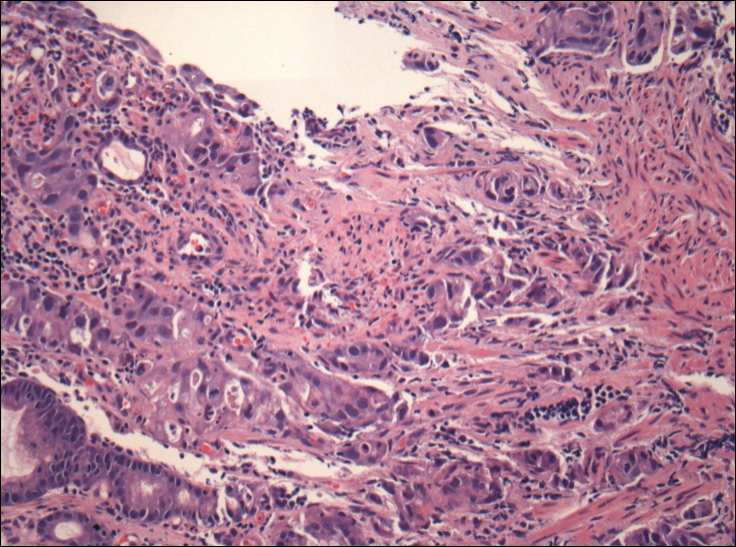
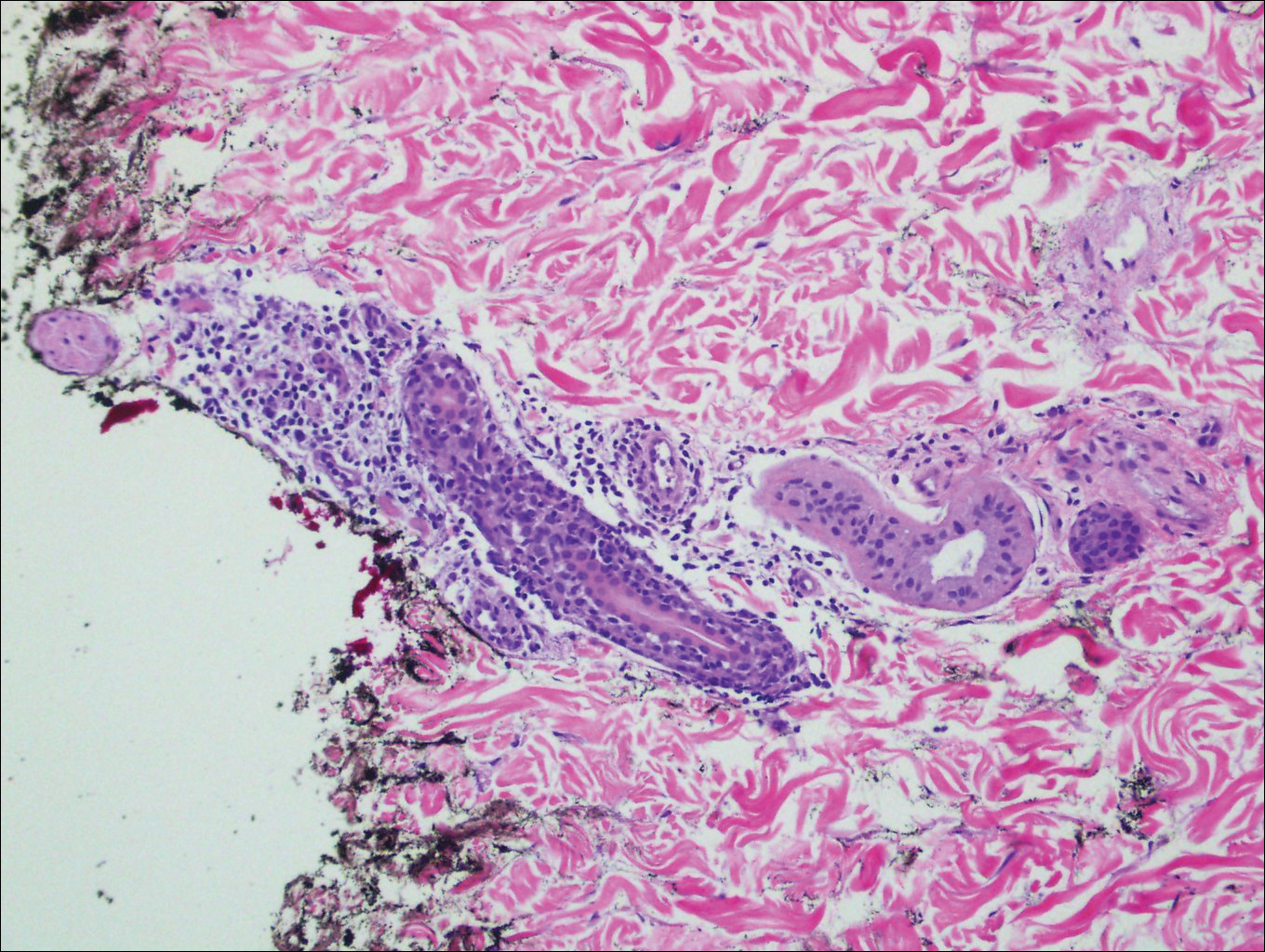
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
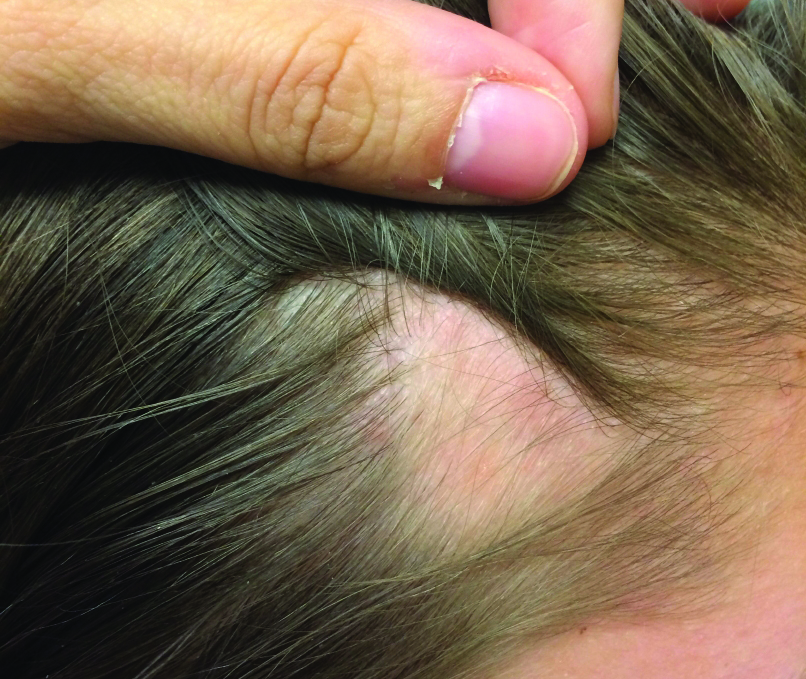
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
The Diagnosis: Sister Mary Joseph Nodule
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
The Diagnosis: Sister Mary Joseph Nodule
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
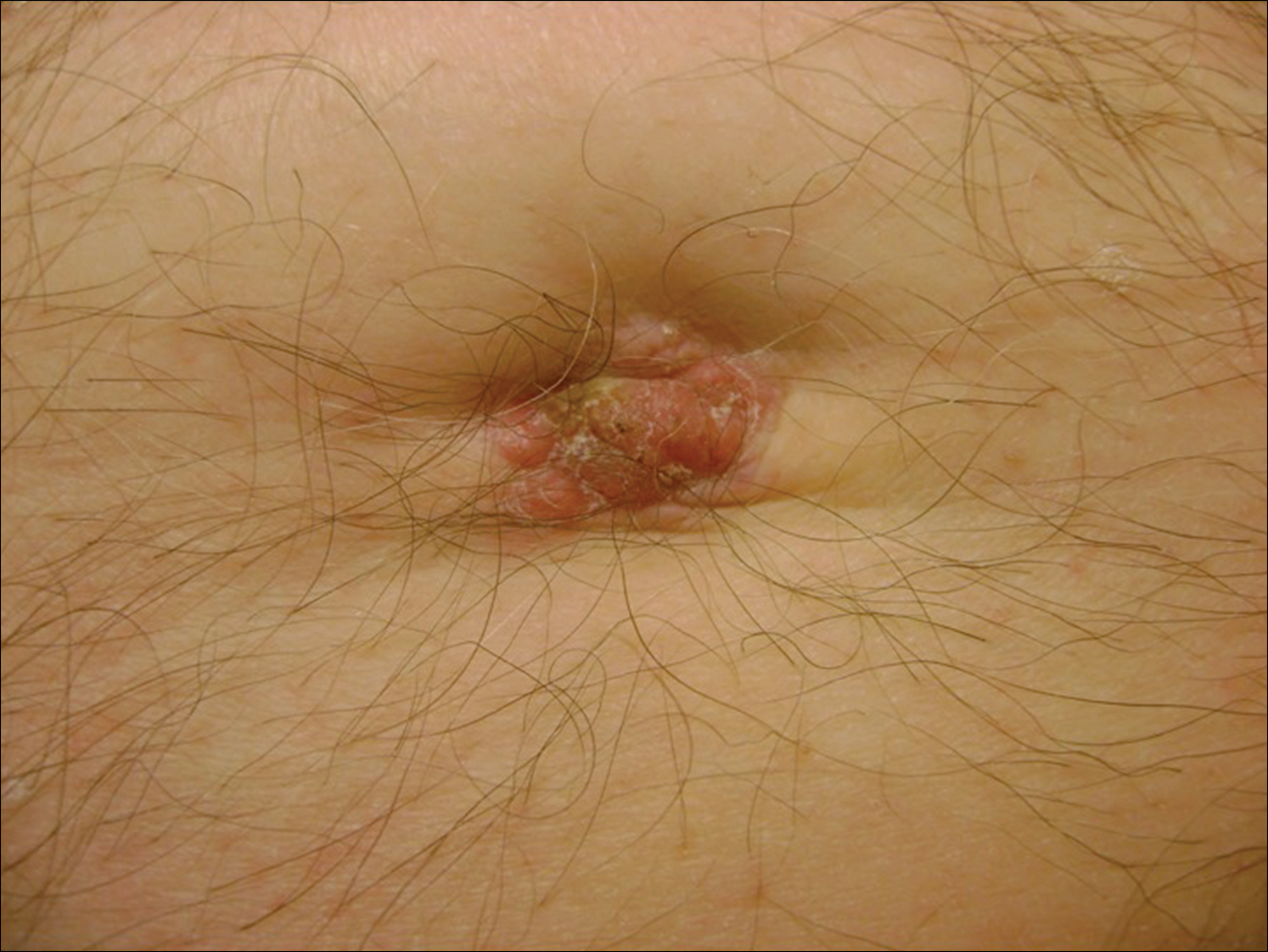
A 74-year-old man presented to our outpatient dermatology clinic with an asymptomatic umbilical lesion of unknown duration. The patient believed the lesion was a scar resulting from a prior laparoscopic repair of an umbilical hernia. However, the patient reported epigastric abdominal pain and diarrhea of 1 month's duration that he believed was due to the stomach flu. The patient denied fever, chills, loss of appetite, or weight loss. History was remarkable for hypertension, hyperlipidemia, coronary artery disease, chronic kidney disease, and emphysema. The patient had a surgical history of percutaneous transluminal coronary angioplasty in addition to the laparoscopic umbilical hernia repair. The patient's medications included pantoprazole, ondansetron, diphenoxylate-atropine as needed, amlodipine, lisinopril-hydrochlorothiazide, simvastatin, and aspirin. Physical examination revealed a 1×2-cm pink, nodular, firm plaque with crust at the umbilicus that was tender on palpation. A shave biopsy of the umbilicus was performed and sent for both pathological and immunohistochemical analysis.
Blaschkoid Unilateral Patch on the Chest
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
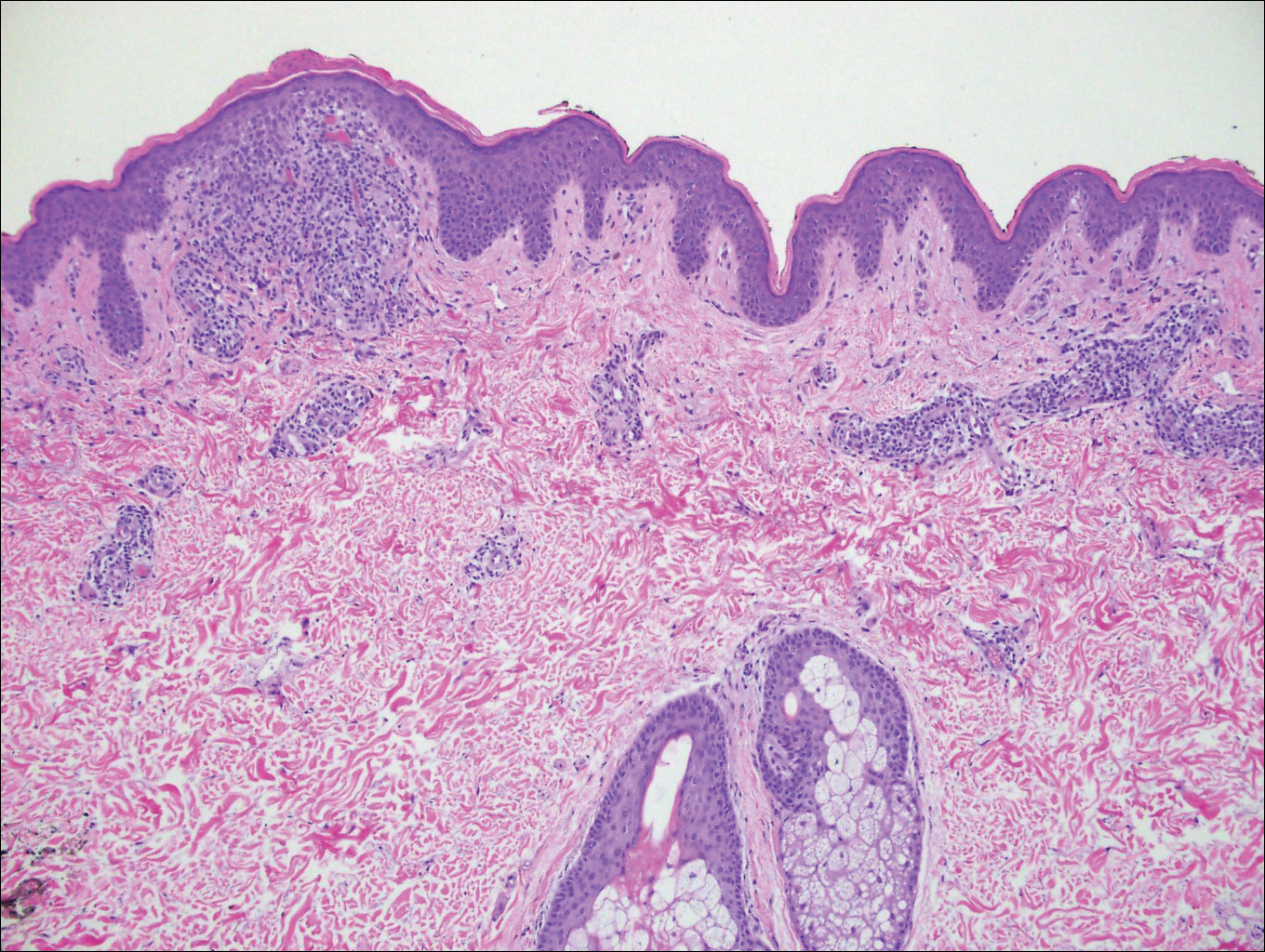
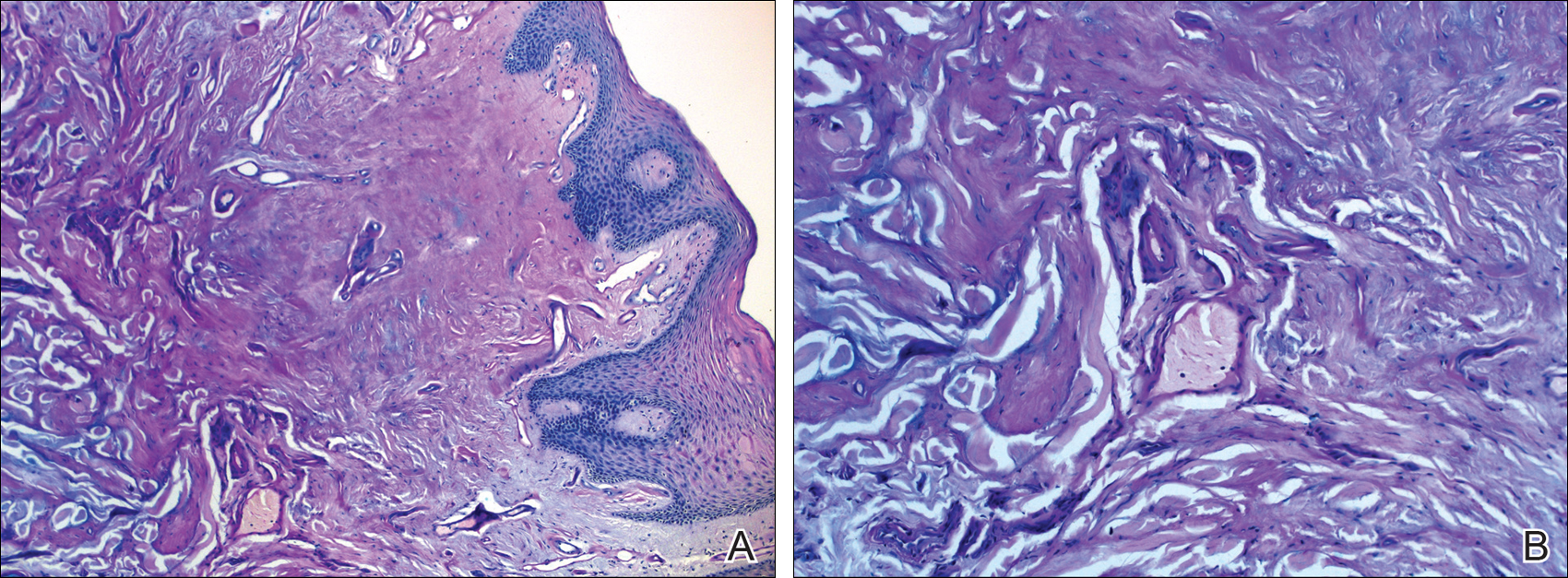
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
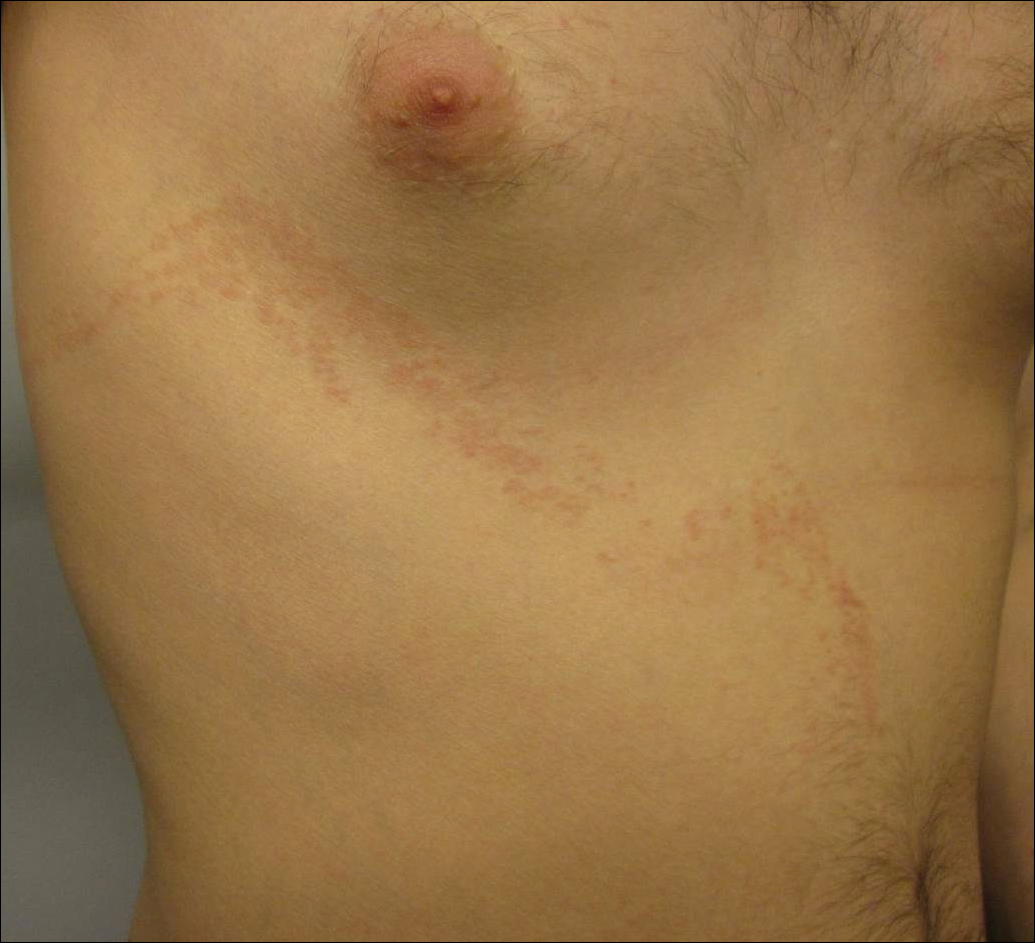
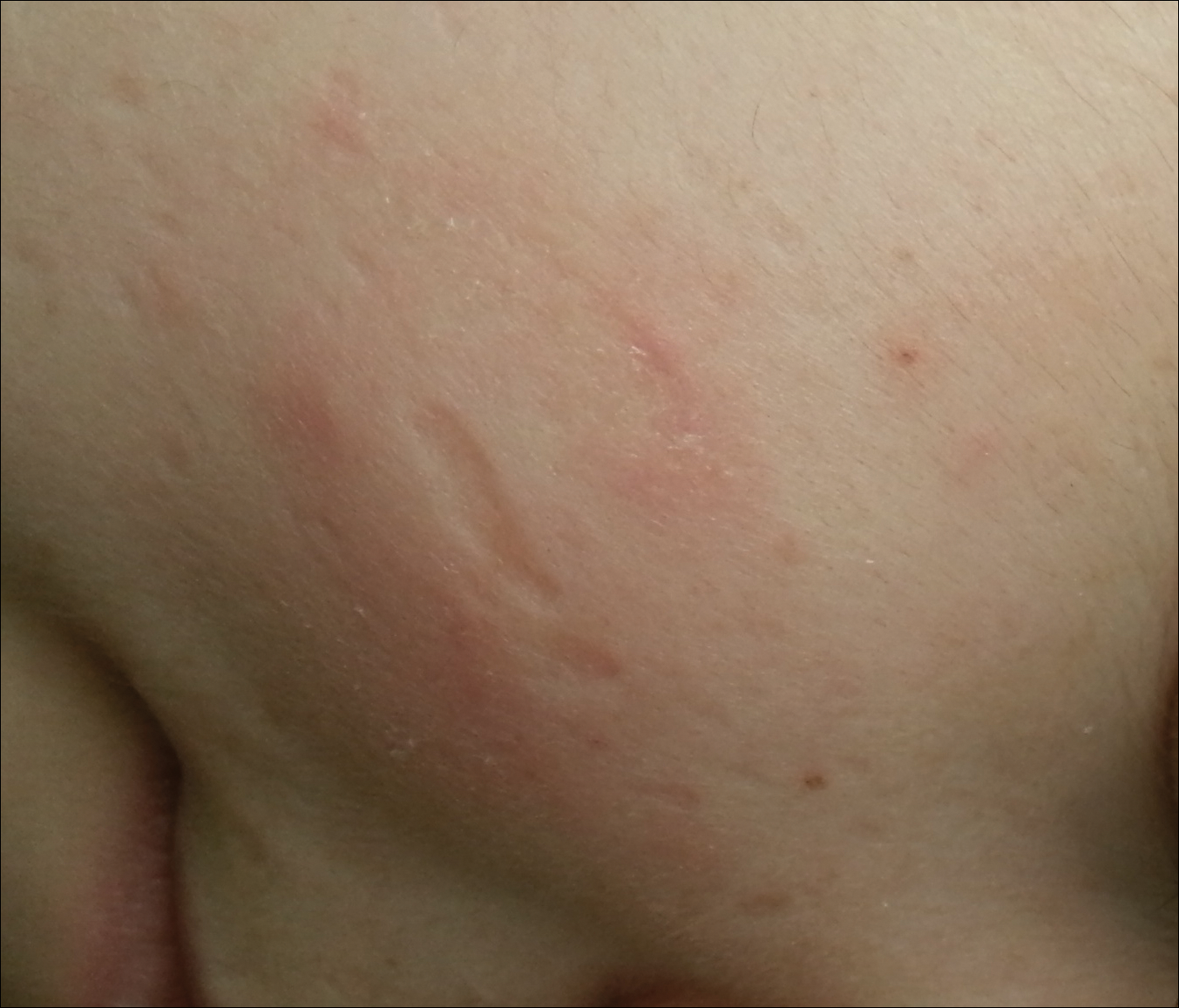
A 26-year-old man presented with erythematous, scaly, grouped papules along the right side of the chest of 3 weeks' duration, extending to the flank following a blaschkoid distribution on the right side of the chest and not crossing the midline. He reported occasional irritation but otherwise was asymptomatic. His medical history was unremarkable and he was not taking any medications. He also denied trauma to the area.
Beaded Papules Along the Eyelid Margins
The Diagnosis: Lipoid Proteinosis
Lipoid proteinosis (LP), also known as hyalinosis cutis et mucosae or Urbach-Wiethe disease, is a rare autosomal-recessive disorder. It is characterized by deposition of hyalinelike material in multiple organs including the skin, oral mucosa, larynx, and brain. The underlying defect is mutations in the extracellular matrix protein 1 gene, ECM1, which binds to various proteins (eg, perlecan, fibulins, matrix metalloproteinase 9) and plays a role in angiogenesis and epidermal differentiation.1-4
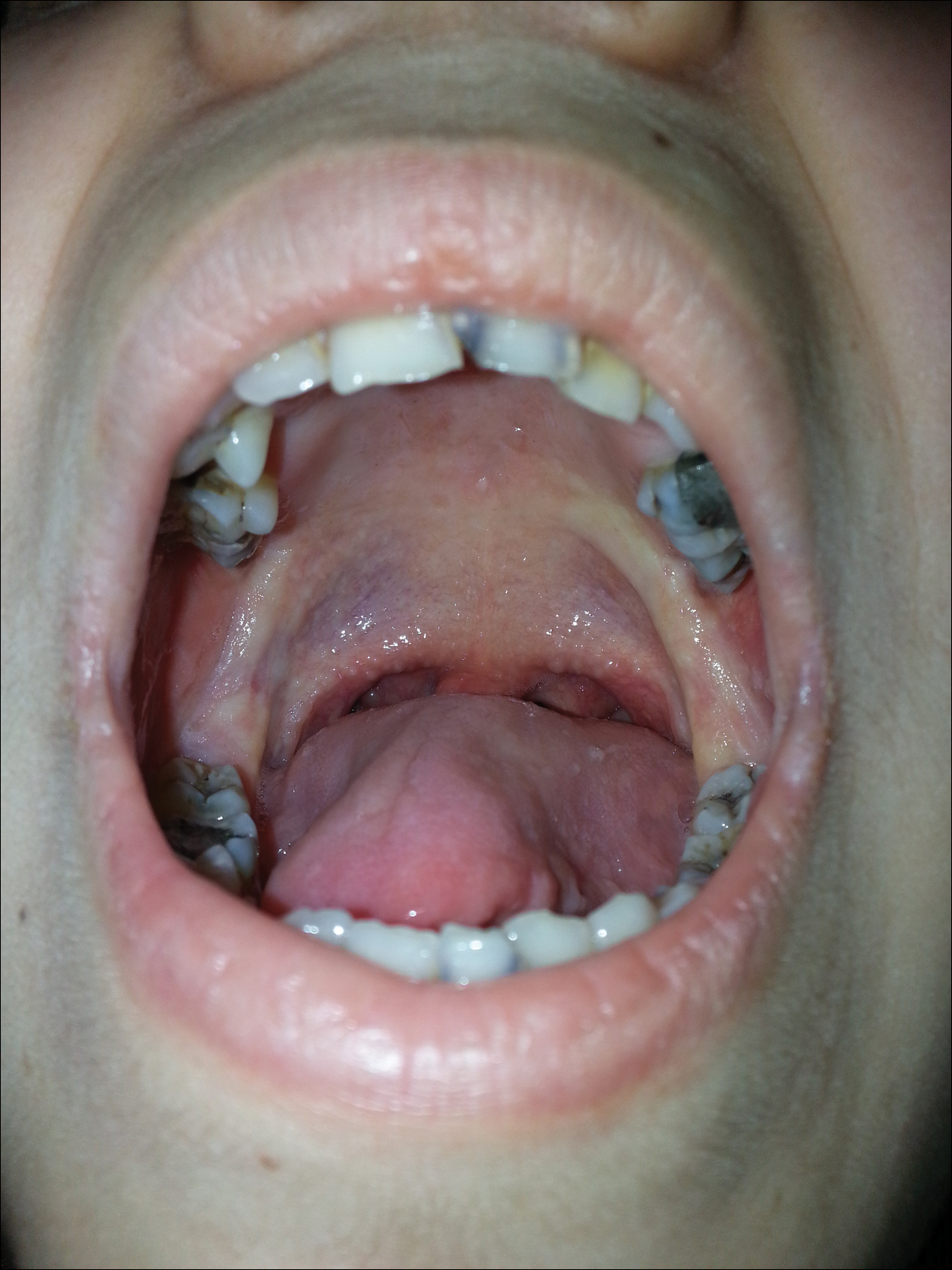
The clinical spectrum of LP is primarily related to respiratory, skin, and neurologic manifestations, but any organ involvement may be seen. A childhood-onset weak cry or hoarseness usually is the first clinical sign of LP due to infiltration of the laryngeal mucosa.3-6 A thickened frenulum, which manifests as restricted tongue movements, is another reliable clinical sign of LP.7 In addition, yellow-white submucous infiltrates on other mucosal surfaces (eg, pharynx, tongue, soft palate, esophagus)(Figure 1), occlusion of the salivary ducts (recurrent parotitis), dental anomalies, and dental caries (Figure 2) also may be seen.5,7

Related to cutaneous manifestations of LP, lesions that present in early childhood are characterized by vesicles, erosions, and hemorrhagic crusts that result in pocklike (Figure 3), linear, or cribriform scarring on the face and extremities, either following trauma or spontaneously.6,7 Second-stage skin lesions are beaded papules (moniliform blepharosis) along the eyelid margins; generalized cutaneous thickening with yellowish discoloration; and waxy papules, hyperkeratosis, or verrucous plaques/nodules on the hands, forehead, axillae, scrotum, elbows, or knees.1,5
Neurological manifestations usually occur as epilepsy and psychiatric problems, which are likely due to intracranial calcification within the amygdala or the temporal lobe. Bean-shaped calcification in the temporal lobe is seen as a pathognomonic radiographic finding.7 Other manifestations including drusenlike fundal lesions, corneal deposits with diminution of vision, and visceral involvement may be seen.7,8
Histologically, deposition of eosinophilic homogeneous material is seen around the blood vessels and sweat glands as well as in the dermis and dermoepidermal junction (Figure 4).1,5 Although most patients with LP have a slowly progressive benign course that stabilizes in early adult life, some morbidities and complications may occur (eg, rarely upper respiratory tract involvement can progress and require tracheostomy). There presently is no cure for LP, but some drugs (eg, oral dimethyl sulfoxide, etretinate, acitretin, penicillamine) and laser ablation/dermabrasion of papules are helpful in some cases.1,7
- Sarkany RPE, Breathnach S, Morris AAM, et al. Metabolic and nutritional disorders. In: Burns T, Breathnach S, Cox N, et al, eds. Rook's Textbook of Dermatology. 8th ed. Vol 2. Singapore: Wiley-Blackwell; 2010:59.41-59.42.
- Hamada T, McLean WH, Ramsay M, et al. Lipoid proteinosis maps to 1q21and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Hum Mol Genet. 2002;11:833-840.
- Bakry OA, Samaka RM, Houla NS, et al. Two Egyptian cases of lipoid proteinosis successfully treated with acitretin. J Dermatol Case Rep. 2014;8:29-34.
- Dogramaci AC, Celik MM, Celik E, et al. Lipoid proteinosis in the eastern Mediterranean region of Turkey. Indian J Dermatol Venereol Leprol. 2012;78:318-322.
- Franke I, Gollnick H. Deposition diseases. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008:633-640.
- Parmar NV, Krishna CV, De D, et al. Papules, pock-like scars, and hoarseness of voice. lipoid proteinosis. Indian J Dermatol Venereol Leprol. 2013;79:136.
- Dyer JA. Lipoid proteinosis. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick's Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:1288-1292.
- Gutte R, Sanghvi S, Tamhankar P, et al. Lipoid proteinosis: histopathological characterization of early papulovesicular lesions. Indian Dermatol Online J. 2012;3:148-149.
The Diagnosis: Lipoid Proteinosis
Lipoid proteinosis (LP), also known as hyalinosis cutis et mucosae or Urbach-Wiethe disease, is a rare autosomal-recessive disorder. It is characterized by deposition of hyalinelike material in multiple organs including the skin, oral mucosa, larynx, and brain. The underlying defect is mutations in the extracellular matrix protein 1 gene, ECM1, which binds to various proteins (eg, perlecan, fibulins, matrix metalloproteinase 9) and plays a role in angiogenesis and epidermal differentiation.1-4
The clinical spectrum of LP is primarily related to respiratory, skin, and neurologic manifestations, but any organ involvement may be seen. A childhood-onset weak cry or hoarseness usually is the first clinical sign of LP due to infiltration of the laryngeal mucosa.3-6 A thickened frenulum, which manifests as restricted tongue movements, is another reliable clinical sign of LP.7 In addition, yellow-white submucous infiltrates on other mucosal surfaces (eg, pharynx, tongue, soft palate, esophagus)(Figure 1), occlusion of the salivary ducts (recurrent parotitis), dental anomalies, and dental caries (Figure 2) also may be seen.5,7
Related to cutaneous manifestations of LP, lesions that present in early childhood are characterized by vesicles, erosions, and hemorrhagic crusts that result in pocklike (Figure 3), linear, or cribriform scarring on the face and extremities, either following trauma or spontaneously.6,7 Second-stage skin lesions are beaded papules (moniliform blepharosis) along the eyelid margins; generalized cutaneous thickening with yellowish discoloration; and waxy papules, hyperkeratosis, or verrucous plaques/nodules on the hands, forehead, axillae, scrotum, elbows, or knees.1,5
Neurological manifestations usually occur as epilepsy and psychiatric problems, which are likely due to intracranial calcification within the amygdala or the temporal lobe. Bean-shaped calcification in the temporal lobe is seen as a pathognomonic radiographic finding.7 Other manifestations including drusenlike fundal lesions, corneal deposits with diminution of vision, and visceral involvement may be seen.7,8
Histologically, deposition of eosinophilic homogeneous material is seen around the blood vessels and sweat glands as well as in the dermis and dermoepidermal junction (Figure 4).1,5 Although most patients with LP have a slowly progressive benign course that stabilizes in early adult life, some morbidities and complications may occur (eg, rarely upper respiratory tract involvement can progress and require tracheostomy). There presently is no cure for LP, but some drugs (eg, oral dimethyl sulfoxide, etretinate, acitretin, penicillamine) and laser ablation/dermabrasion of papules are helpful in some cases.1,7
The Diagnosis: Lipoid Proteinosis
Lipoid proteinosis (LP), also known as hyalinosis cutis et mucosae or Urbach-Wiethe disease, is a rare autosomal-recessive disorder. It is characterized by deposition of hyalinelike material in multiple organs including the skin, oral mucosa, larynx, and brain. The underlying defect is mutations in the extracellular matrix protein 1 gene, ECM1, which binds to various proteins (eg, perlecan, fibulins, matrix metalloproteinase 9) and plays a role in angiogenesis and epidermal differentiation.1-4
The clinical spectrum of LP is primarily related to respiratory, skin, and neurologic manifestations, but any organ involvement may be seen. A childhood-onset weak cry or hoarseness usually is the first clinical sign of LP due to infiltration of the laryngeal mucosa.3-6 A thickened frenulum, which manifests as restricted tongue movements, is another reliable clinical sign of LP.7 In addition, yellow-white submucous infiltrates on other mucosal surfaces (eg, pharynx, tongue, soft palate, esophagus)(Figure 1), occlusion of the salivary ducts (recurrent parotitis), dental anomalies, and dental caries (Figure 2) also may be seen.5,7
Related to cutaneous manifestations of LP, lesions that present in early childhood are characterized by vesicles, erosions, and hemorrhagic crusts that result in pocklike (Figure 3), linear, or cribriform scarring on the face and extremities, either following trauma or spontaneously.6,7 Second-stage skin lesions are beaded papules (moniliform blepharosis) along the eyelid margins; generalized cutaneous thickening with yellowish discoloration; and waxy papules, hyperkeratosis, or verrucous plaques/nodules on the hands, forehead, axillae, scrotum, elbows, or knees.1,5
Neurological manifestations usually occur as epilepsy and psychiatric problems, which are likely due to intracranial calcification within the amygdala or the temporal lobe. Bean-shaped calcification in the temporal lobe is seen as a pathognomonic radiographic finding.7 Other manifestations including drusenlike fundal lesions, corneal deposits with diminution of vision, and visceral involvement may be seen.7,8
Histologically, deposition of eosinophilic homogeneous material is seen around the blood vessels and sweat glands as well as in the dermis and dermoepidermal junction (Figure 4).1,5 Although most patients with LP have a slowly progressive benign course that stabilizes in early adult life, some morbidities and complications may occur (eg, rarely upper respiratory tract involvement can progress and require tracheostomy). There presently is no cure for LP, but some drugs (eg, oral dimethyl sulfoxide, etretinate, acitretin, penicillamine) and laser ablation/dermabrasion of papules are helpful in some cases.1,7
- Sarkany RPE, Breathnach S, Morris AAM, et al. Metabolic and nutritional disorders. In: Burns T, Breathnach S, Cox N, et al, eds. Rook's Textbook of Dermatology. 8th ed. Vol 2. Singapore: Wiley-Blackwell; 2010:59.41-59.42.
- Hamada T, McLean WH, Ramsay M, et al. Lipoid proteinosis maps to 1q21and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Hum Mol Genet. 2002;11:833-840.
- Bakry OA, Samaka RM, Houla NS, et al. Two Egyptian cases of lipoid proteinosis successfully treated with acitretin. J Dermatol Case Rep. 2014;8:29-34.
- Dogramaci AC, Celik MM, Celik E, et al. Lipoid proteinosis in the eastern Mediterranean region of Turkey. Indian J Dermatol Venereol Leprol. 2012;78:318-322.
- Franke I, Gollnick H. Deposition diseases. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008:633-640.
- Parmar NV, Krishna CV, De D, et al. Papules, pock-like scars, and hoarseness of voice. lipoid proteinosis. Indian J Dermatol Venereol Leprol. 2013;79:136.
- Dyer JA. Lipoid proteinosis. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick's Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:1288-1292.
- Gutte R, Sanghvi S, Tamhankar P, et al. Lipoid proteinosis: histopathological characterization of early papulovesicular lesions. Indian Dermatol Online J. 2012;3:148-149.
- Sarkany RPE, Breathnach S, Morris AAM, et al. Metabolic and nutritional disorders. In: Burns T, Breathnach S, Cox N, et al, eds. Rook's Textbook of Dermatology. 8th ed. Vol 2. Singapore: Wiley-Blackwell; 2010:59.41-59.42.
- Hamada T, McLean WH, Ramsay M, et al. Lipoid proteinosis maps to 1q21and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Hum Mol Genet. 2002;11:833-840.
- Bakry OA, Samaka RM, Houla NS, et al. Two Egyptian cases of lipoid proteinosis successfully treated with acitretin. J Dermatol Case Rep. 2014;8:29-34.
- Dogramaci AC, Celik MM, Celik E, et al. Lipoid proteinosis in the eastern Mediterranean region of Turkey. Indian J Dermatol Venereol Leprol. 2012;78:318-322.
- Franke I, Gollnick H. Deposition diseases. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008:633-640.
- Parmar NV, Krishna CV, De D, et al. Papules, pock-like scars, and hoarseness of voice. lipoid proteinosis. Indian J Dermatol Venereol Leprol. 2013;79:136.
- Dyer JA. Lipoid proteinosis. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick's Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:1288-1292.
- Gutte R, Sanghvi S, Tamhankar P, et al. Lipoid proteinosis: histopathological characterization of early papulovesicular lesions. Indian Dermatol Online J. 2012;3:148-149.
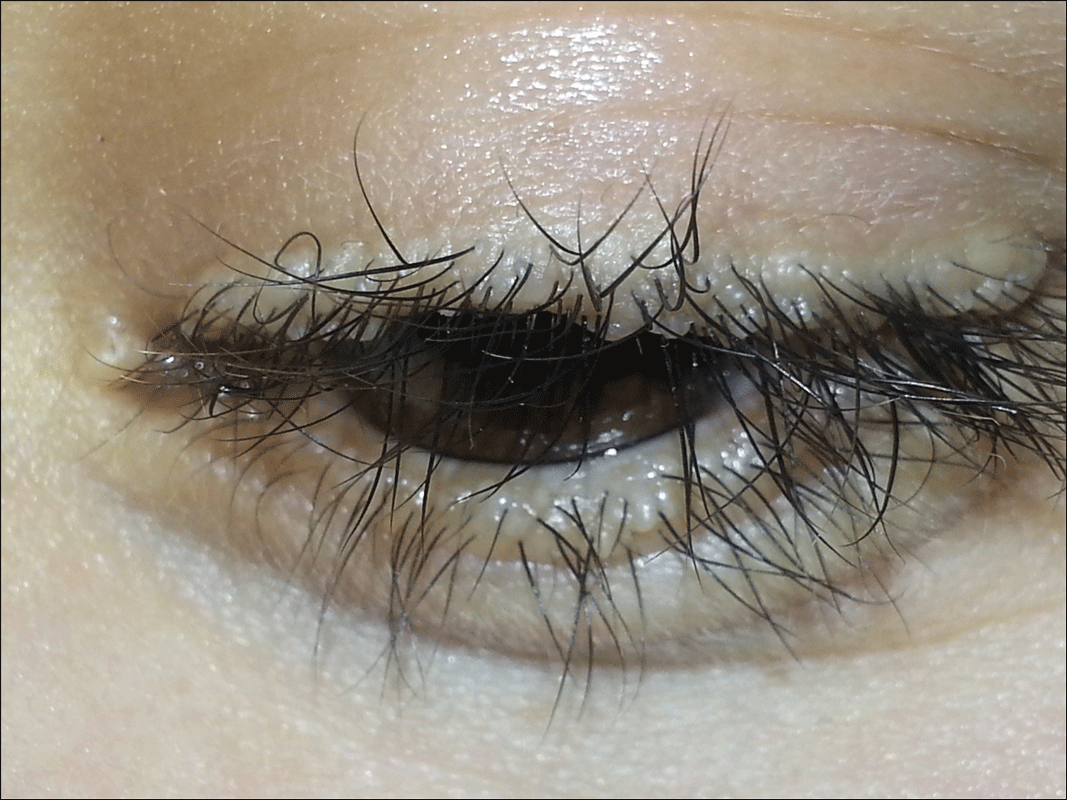
A 21-year-old woman (born of consanguineous parents) presented with asymptomatic, waxy, white, beaded papules along the eyelid margins of 6 years' duration. Physical examination revealed moniliform blepharosis over the eyelid margins, multiple linear and pocklike scars on the face and arm, pebbling on the lower lip and oropharynx, and hoarseness that was present since early infancy. Medical history was unremarkable for systemic disorders and routine laboratory tests were within reference range. Pathological examination of a papule on the lower lip mucosae revealed perivascular deposition of eosinophilic homogeneous material.
Reticular Hyperpigmentation on the Lower Legs
The Diagnosis: Erythema Ab Igne
Given the patient's reticulated hyperpigmented lesions in the setting of recent space heater use with heater closer to the more affected leg, erythema ab igne was diagnosed. Patient education was provided and moving the heater away from the lower extremities was advised.
Erythema ab igne first was described by German dermatologist Abraham Buschke as hitze melanose, meaning melanosis induced by heat. The classic skin findings were first observed on the lower legs of patients who worked in front of open fires or coal stoves.1 Over the years, new causes of erythema ab igne secondary to prolonged thermal radiation exposure have been reported.1 In the elderly, hospitalized, and chronic pain patients, erythema ab igne has been observed in areas treated with heating pads and blankets.2 Other triggers such as frequent hot bathing, furniture, steam radiators, space heaters, and laptops also have been reported.3-6 Laptop-induced erythema ab igne is a diagnosis that has been reported in the last decade and its incidence likely will increase in the future.6
The clinical manifestations of erythema ab igne correlate with the frequency and duration of heat exposure. Acutely, a mild and transient erythema develops in the affected area. With chronic heat exposure, these areas subsequently develop a permanent reticulated hyperpigmented pattern and may eventually become atrophic.2,6 All body surfaces are at risk, but erythema ab igne classically involves the legs, lower back, and/or abdomen. Lesions typically are asymptomatic; however, burning and pruritus can be present.2,6 Bullous erythema ab igne, though rare, has been reported,7 suggesting a potential transition from erythema ab igne to burns.6
Biopsy is not recommended for diagnosis; however, the histopathologic changes of erythema ab igne include hyperkeratosis, interface dermatitis, epidermal atrophy with apoptotic keratinocytes, and melanin incontinence. Although this condition typically is benign, histologic findings could resemble actinic keratosis, suggesting that chronic changes induced by infrared thermal radiation may lead to squamous cell carcinoma or rarely Merkel cell carcinoma. The latency for developing carcinoma appears to extend 30 years, with a 30% tendency for recurrence or metastasis. Given the possibility of an increase in erythema ab igne in the pediatric population in the upcoming years, as displayed by our patient, and increasing laptop and electronic use in children and adolescents, it is important to be aware of this skin condition and the potential complications of it going undiagnosed.2,6
No specific therapy for erythema ab igne exists. Treatment is centered on eliminating exposure to the heat source. With appropriate removal, the reticulated hyperpigmented lesions will resolve, sometimes taking several months.
Differential diagnosis includes livedo reticularis, livedoid vasculopathy, and cutis marmorata. The reticulated purpuric lesions of livedo reticularis involving the extremities often mimic erythema ab igne's cutaneous morphology; however, livedo reticularis frequently is associated with conditions such as drug reactions, infections, thrombosis, and vasculitides,2 as opposed to erythema ab igne, which frequently is associated with conditions causing pain or decreased body temperature, thus necessitating use of heating devices, as seen in our patient. Livedoid vasculopathy is characterized by purpuric macules involving the lower legs and feet that progress to recurrent leg ulcers. Our patient's asymptomatic lesions and absence of ulcers excluded this diagnosis.8 Lastly, cutis marmorata, a congenital condition, is characterized by blue-violet vascular networks that often display ulceration and atrophy of the involved skin as well as hypertrophy or atrophy of the involved limb9; these clinical findings were not present in our patient and this diagnosis would not explain the relationship between the cutaneous lesions and heat exposure.
- Nilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
- Riahi RR, Cohen PR, Robinson FW, et al. Erythema ab igne mimicking livedo reticularis. Int J Dermatol. 2010;49:1314-1317.
- Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol. 2002;82:478-479.
- Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol. 1996;34:516-517.
- Kligman LH, Kligman AM. Reflections on heat. Br J Dermatol. 1984;110:369-375.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:e1227-e1230.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Khenifer S, Thomas L, Balme B, et al. Livedoid vasculopathy: thrombotic or inflammatory disease? Clin Exp Dermatol. 2009;35:693-698.
- Pernet C, Guillot B, Bigorre M, et al. Focal and atrophic cutis marmorata telangiectatica congenital. J Am Acad Dermatol. 2013;69:e268-e269.
The Diagnosis: Erythema Ab Igne
Given the patient's reticulated hyperpigmented lesions in the setting of recent space heater use with heater closer to the more affected leg, erythema ab igne was diagnosed. Patient education was provided and moving the heater away from the lower extremities was advised.
Erythema ab igne first was described by German dermatologist Abraham Buschke as hitze melanose, meaning melanosis induced by heat. The classic skin findings were first observed on the lower legs of patients who worked in front of open fires or coal stoves.1 Over the years, new causes of erythema ab igne secondary to prolonged thermal radiation exposure have been reported.1 In the elderly, hospitalized, and chronic pain patients, erythema ab igne has been observed in areas treated with heating pads and blankets.2 Other triggers such as frequent hot bathing, furniture, steam radiators, space heaters, and laptops also have been reported.3-6 Laptop-induced erythema ab igne is a diagnosis that has been reported in the last decade and its incidence likely will increase in the future.6
The clinical manifestations of erythema ab igne correlate with the frequency and duration of heat exposure. Acutely, a mild and transient erythema develops in the affected area. With chronic heat exposure, these areas subsequently develop a permanent reticulated hyperpigmented pattern and may eventually become atrophic.2,6 All body surfaces are at risk, but erythema ab igne classically involves the legs, lower back, and/or abdomen. Lesions typically are asymptomatic; however, burning and pruritus can be present.2,6 Bullous erythema ab igne, though rare, has been reported,7 suggesting a potential transition from erythema ab igne to burns.6
Biopsy is not recommended for diagnosis; however, the histopathologic changes of erythema ab igne include hyperkeratosis, interface dermatitis, epidermal atrophy with apoptotic keratinocytes, and melanin incontinence. Although this condition typically is benign, histologic findings could resemble actinic keratosis, suggesting that chronic changes induced by infrared thermal radiation may lead to squamous cell carcinoma or rarely Merkel cell carcinoma. The latency for developing carcinoma appears to extend 30 years, with a 30% tendency for recurrence or metastasis. Given the possibility of an increase in erythema ab igne in the pediatric population in the upcoming years, as displayed by our patient, and increasing laptop and electronic use in children and adolescents, it is important to be aware of this skin condition and the potential complications of it going undiagnosed.2,6
No specific therapy for erythema ab igne exists. Treatment is centered on eliminating exposure to the heat source. With appropriate removal, the reticulated hyperpigmented lesions will resolve, sometimes taking several months.
Differential diagnosis includes livedo reticularis, livedoid vasculopathy, and cutis marmorata. The reticulated purpuric lesions of livedo reticularis involving the extremities often mimic erythema ab igne's cutaneous morphology; however, livedo reticularis frequently is associated with conditions such as drug reactions, infections, thrombosis, and vasculitides,2 as opposed to erythema ab igne, which frequently is associated with conditions causing pain or decreased body temperature, thus necessitating use of heating devices, as seen in our patient. Livedoid vasculopathy is characterized by purpuric macules involving the lower legs and feet that progress to recurrent leg ulcers. Our patient's asymptomatic lesions and absence of ulcers excluded this diagnosis.8 Lastly, cutis marmorata, a congenital condition, is characterized by blue-violet vascular networks that often display ulceration and atrophy of the involved skin as well as hypertrophy or atrophy of the involved limb9; these clinical findings were not present in our patient and this diagnosis would not explain the relationship between the cutaneous lesions and heat exposure.
The Diagnosis: Erythema Ab Igne
Given the patient's reticulated hyperpigmented lesions in the setting of recent space heater use with heater closer to the more affected leg, erythema ab igne was diagnosed. Patient education was provided and moving the heater away from the lower extremities was advised.
Erythema ab igne first was described by German dermatologist Abraham Buschke as hitze melanose, meaning melanosis induced by heat. The classic skin findings were first observed on the lower legs of patients who worked in front of open fires or coal stoves.1 Over the years, new causes of erythema ab igne secondary to prolonged thermal radiation exposure have been reported.1 In the elderly, hospitalized, and chronic pain patients, erythema ab igne has been observed in areas treated with heating pads and blankets.2 Other triggers such as frequent hot bathing, furniture, steam radiators, space heaters, and laptops also have been reported.3-6 Laptop-induced erythema ab igne is a diagnosis that has been reported in the last decade and its incidence likely will increase in the future.6
The clinical manifestations of erythema ab igne correlate with the frequency and duration of heat exposure. Acutely, a mild and transient erythema develops in the affected area. With chronic heat exposure, these areas subsequently develop a permanent reticulated hyperpigmented pattern and may eventually become atrophic.2,6 All body surfaces are at risk, but erythema ab igne classically involves the legs, lower back, and/or abdomen. Lesions typically are asymptomatic; however, burning and pruritus can be present.2,6 Bullous erythema ab igne, though rare, has been reported,7 suggesting a potential transition from erythema ab igne to burns.6
Biopsy is not recommended for diagnosis; however, the histopathologic changes of erythema ab igne include hyperkeratosis, interface dermatitis, epidermal atrophy with apoptotic keratinocytes, and melanin incontinence. Although this condition typically is benign, histologic findings could resemble actinic keratosis, suggesting that chronic changes induced by infrared thermal radiation may lead to squamous cell carcinoma or rarely Merkel cell carcinoma. The latency for developing carcinoma appears to extend 30 years, with a 30% tendency for recurrence or metastasis. Given the possibility of an increase in erythema ab igne in the pediatric population in the upcoming years, as displayed by our patient, and increasing laptop and electronic use in children and adolescents, it is important to be aware of this skin condition and the potential complications of it going undiagnosed.2,6
No specific therapy for erythema ab igne exists. Treatment is centered on eliminating exposure to the heat source. With appropriate removal, the reticulated hyperpigmented lesions will resolve, sometimes taking several months.
Differential diagnosis includes livedo reticularis, livedoid vasculopathy, and cutis marmorata. The reticulated purpuric lesions of livedo reticularis involving the extremities often mimic erythema ab igne's cutaneous morphology; however, livedo reticularis frequently is associated with conditions such as drug reactions, infections, thrombosis, and vasculitides,2 as opposed to erythema ab igne, which frequently is associated with conditions causing pain or decreased body temperature, thus necessitating use of heating devices, as seen in our patient. Livedoid vasculopathy is characterized by purpuric macules involving the lower legs and feet that progress to recurrent leg ulcers. Our patient's asymptomatic lesions and absence of ulcers excluded this diagnosis.8 Lastly, cutis marmorata, a congenital condition, is characterized by blue-violet vascular networks that often display ulceration and atrophy of the involved skin as well as hypertrophy or atrophy of the involved limb9; these clinical findings were not present in our patient and this diagnosis would not explain the relationship between the cutaneous lesions and heat exposure.
- Nilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
- Riahi RR, Cohen PR, Robinson FW, et al. Erythema ab igne mimicking livedo reticularis. Int J Dermatol. 2010;49:1314-1317.
- Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol. 2002;82:478-479.
- Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol. 1996;34:516-517.
- Kligman LH, Kligman AM. Reflections on heat. Br J Dermatol. 1984;110:369-375.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:e1227-e1230.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Khenifer S, Thomas L, Balme B, et al. Livedoid vasculopathy: thrombotic or inflammatory disease? Clin Exp Dermatol. 2009;35:693-698.
- Pernet C, Guillot B, Bigorre M, et al. Focal and atrophic cutis marmorata telangiectatica congenital. J Am Acad Dermatol. 2013;69:e268-e269.
- Nilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
- Riahi RR, Cohen PR, Robinson FW, et al. Erythema ab igne mimicking livedo reticularis. Int J Dermatol. 2010;49:1314-1317.
- Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol. 2002;82:478-479.
- Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol. 1996;34:516-517.
- Kligman LH, Kligman AM. Reflections on heat. Br J Dermatol. 1984;110:369-375.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:e1227-e1230.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Khenifer S, Thomas L, Balme B, et al. Livedoid vasculopathy: thrombotic or inflammatory disease? Clin Exp Dermatol. 2009;35:693-698.
- Pernet C, Guillot B, Bigorre M, et al. Focal and atrophic cutis marmorata telangiectatica congenital. J Am Acad Dermatol. 2013;69:e268-e269.
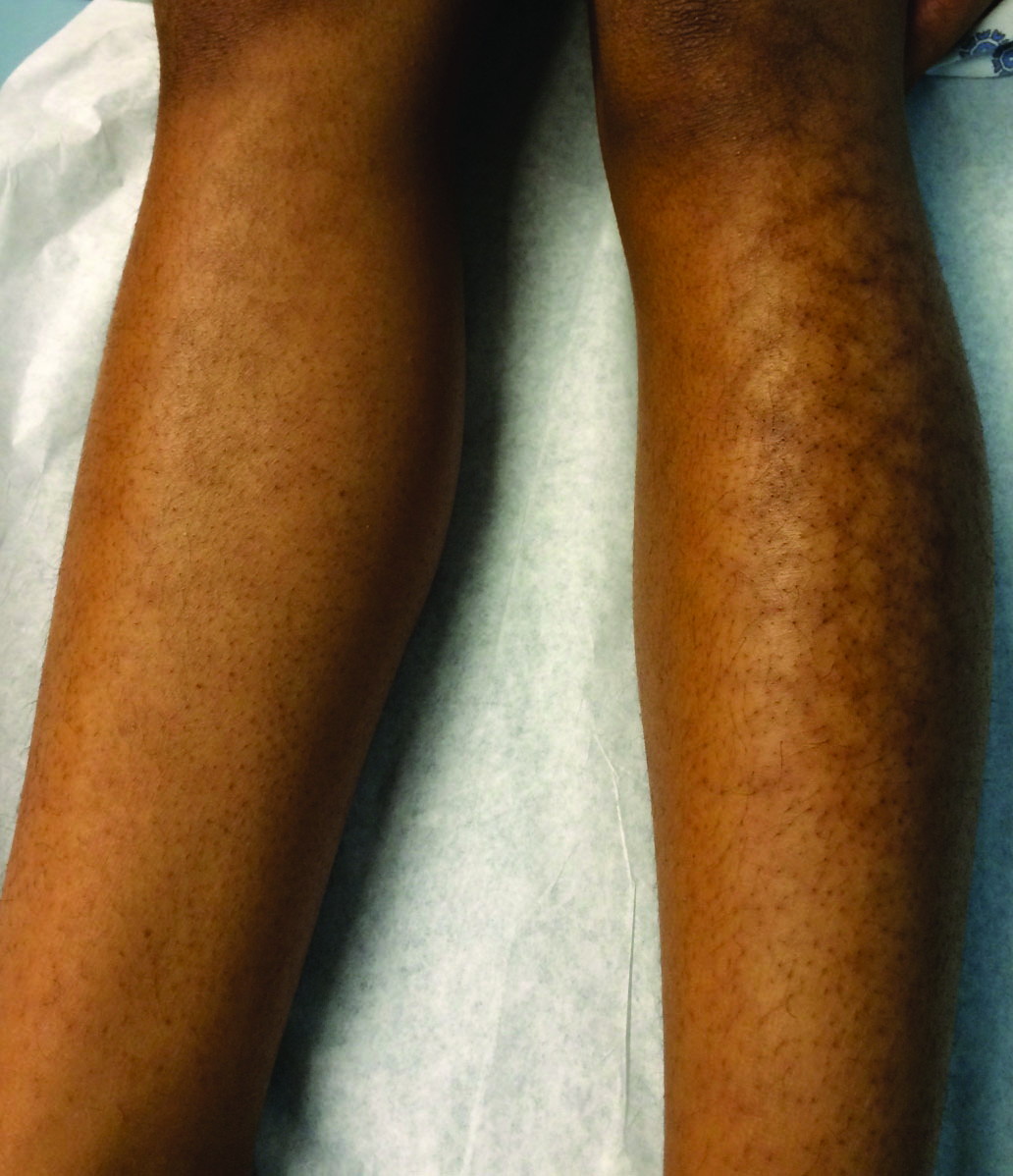
A 13-year-old otherwise healthy adolescent girl presented to the pediatric dermatology clinic for evaluation of a rash on the legs. The patient noticed the rash 1 month prior to presentation. The rash initially involved the left shin and gradually spread to involve the shins bilaterally. The rash was asymptomatic with no pain, pruritus, or muscular asymmetry of the legs. She denied recent fevers, chills, or travel. The patient reported using a space heater daily that was directed at the legs, approximately 0.5 m away. Physical examination revealed a well-nourished adolescent girl in no acute distress with reticular hyperpigmentation of the lower extremities located on the left anterior shin and knee, with mild involvement of the right shin. The reticulated hyperpigmented areas were arranged in a rectangular distribution. Lower extremity musculoskeletal examination was symmetric.
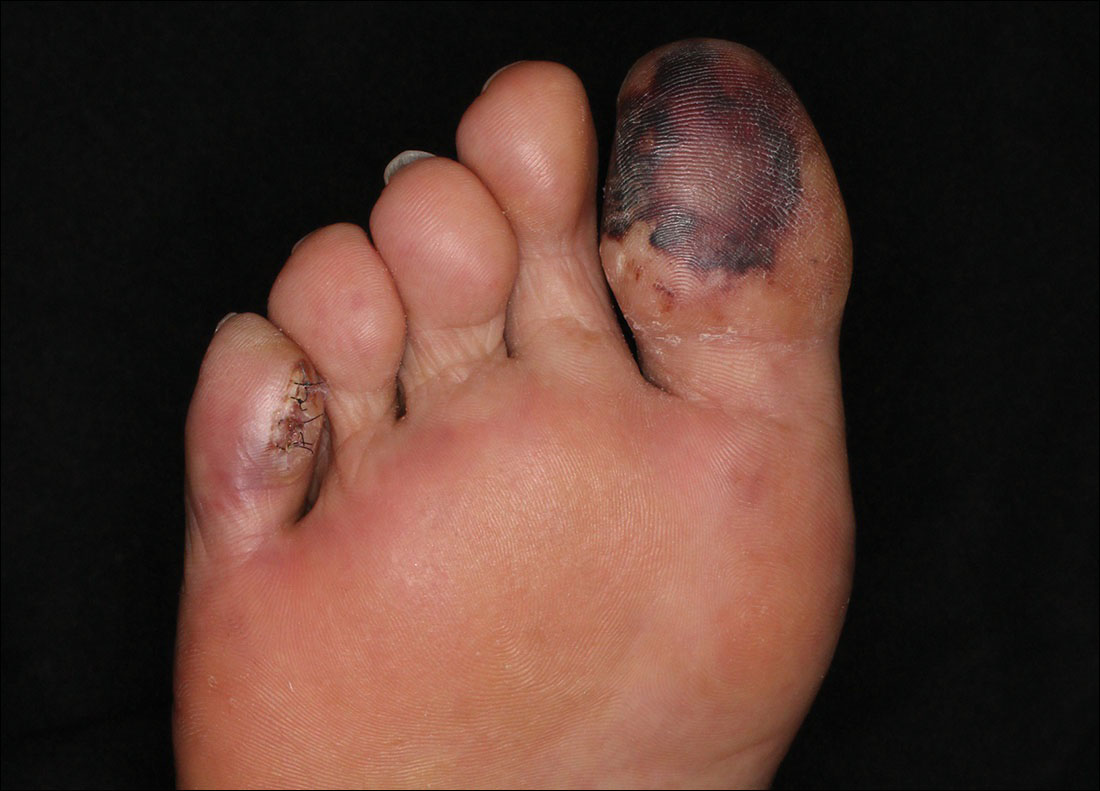
Painful Purple Toes
The Diagnosis: Blue Toe Syndrome
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
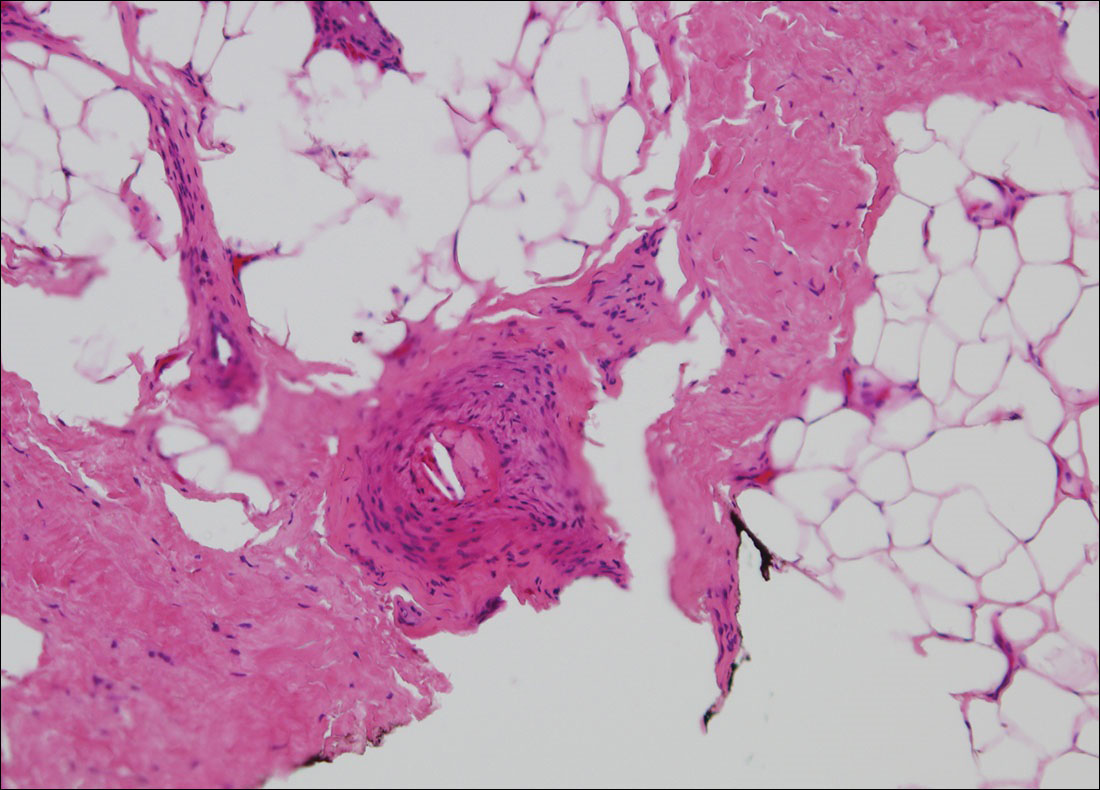
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
The Diagnosis: Blue Toe Syndrome
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
The Diagnosis: Blue Toe Syndrome
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
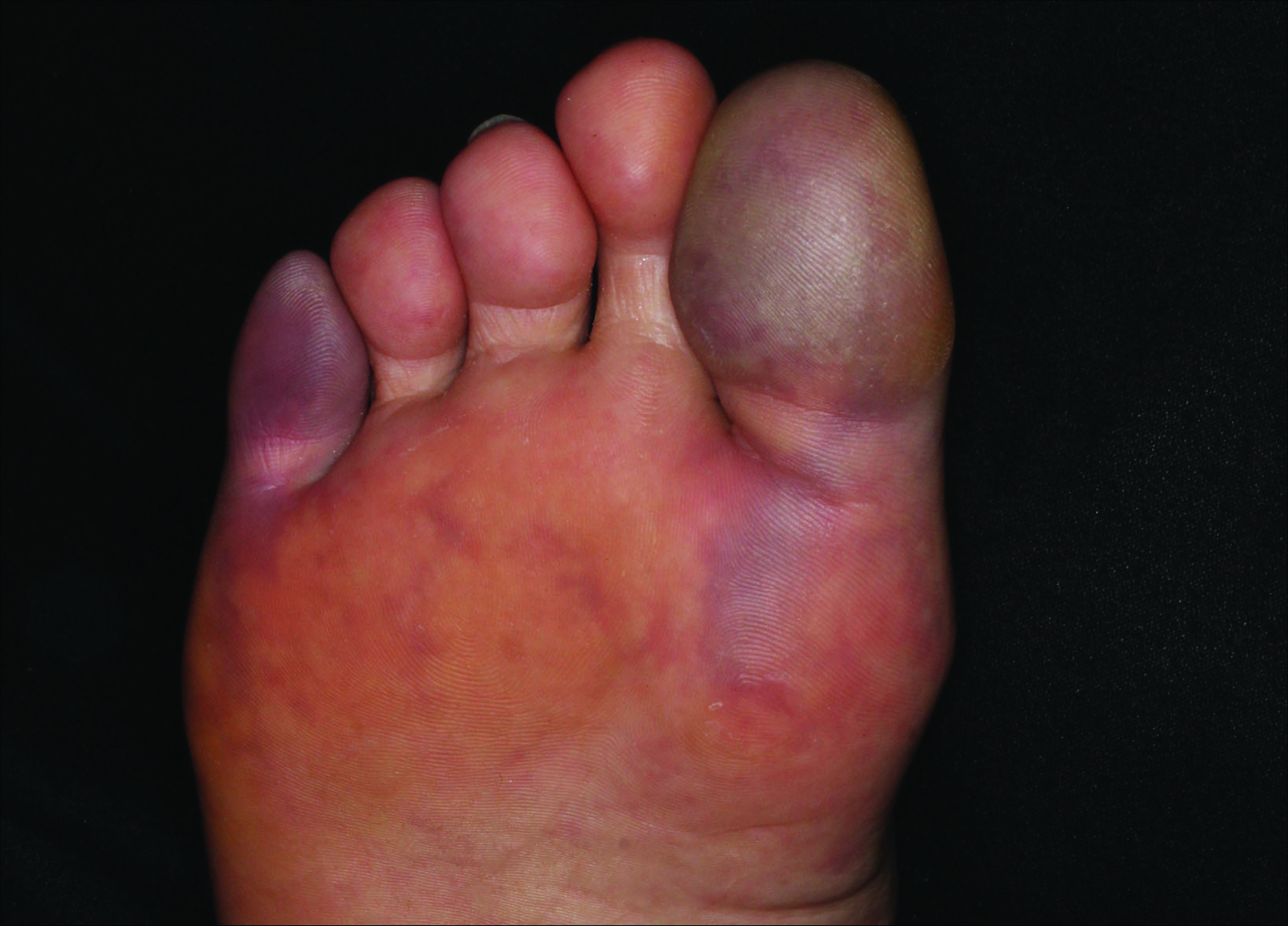
A 63-year-old man presented with sudden onset of severe pain in the right hallux and fifth toe of 3 days' duration. The patient had hypertension and hyperlipidemia with a 45-year history of smoking and had not undergone any vascular procedures. Physical examination revealed relatively well-defined cyanotic change with remarkable coldness on the affected toes as well as livedo reticularis on the underside of the toes. All peripheral pulses were present. Laboratory investigation revealed no remarkable changes with eosinophil counts within reference range and normal renal function. A biopsy taken from the fifth toe revealed thrombotic arterioles with cholesterol clefts.
Thick Scaly Plaques on the Wrists, Knees, and Feet
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
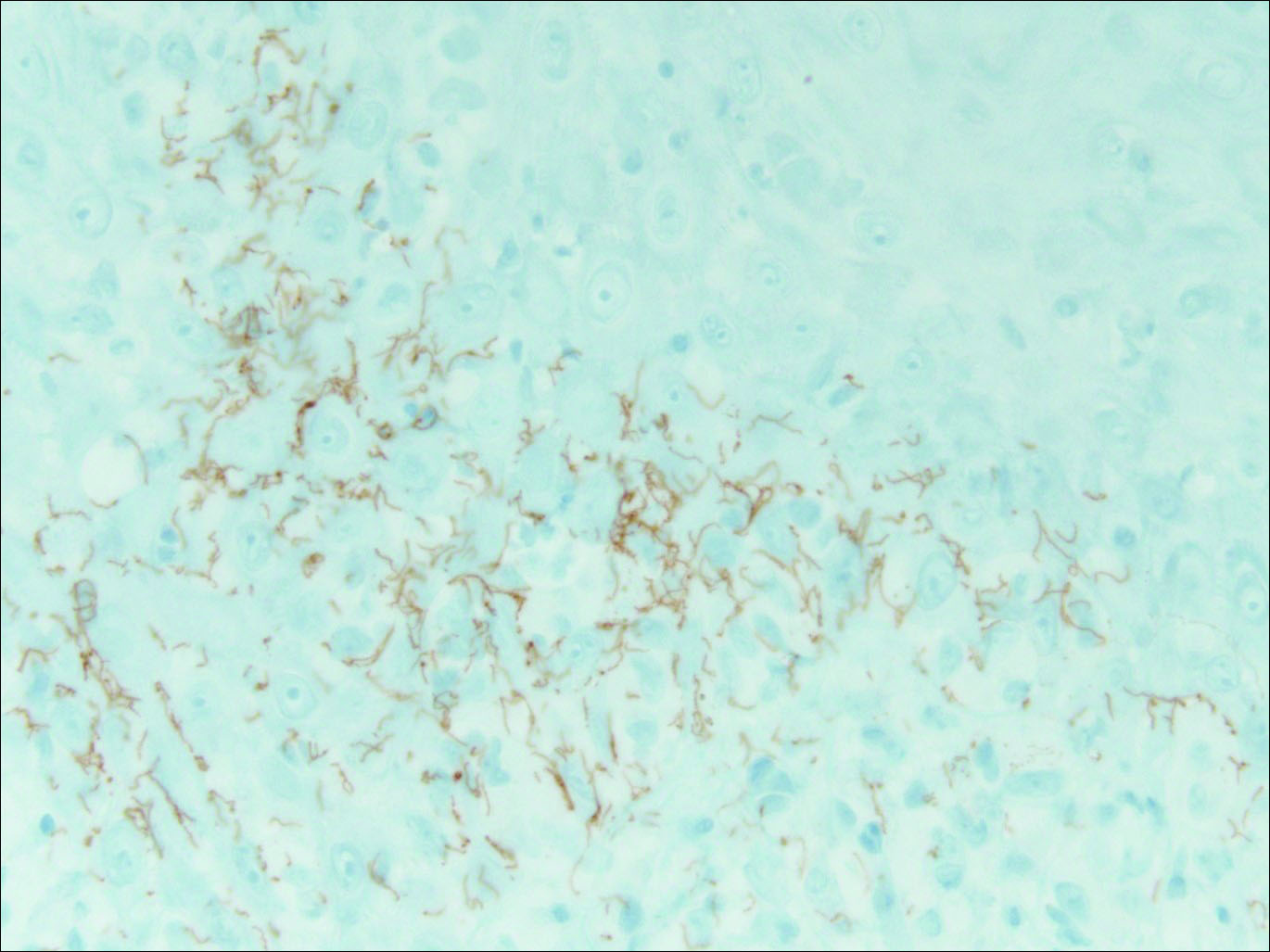
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
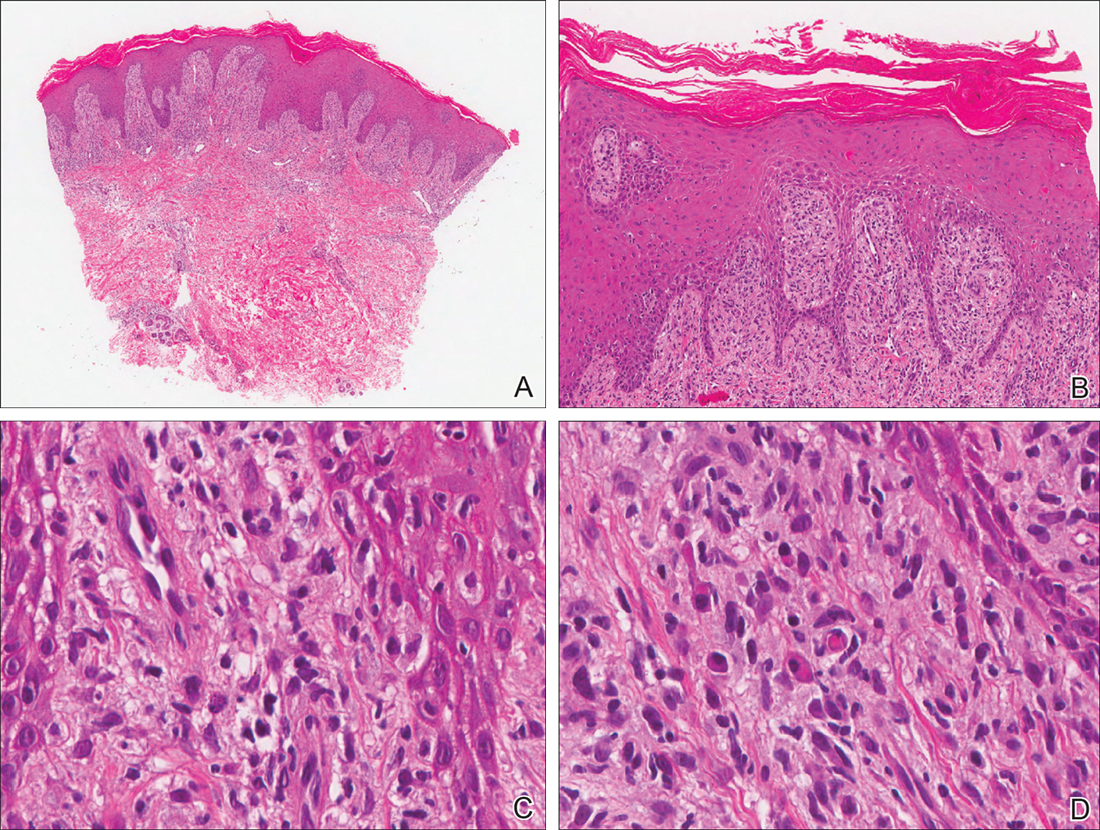
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
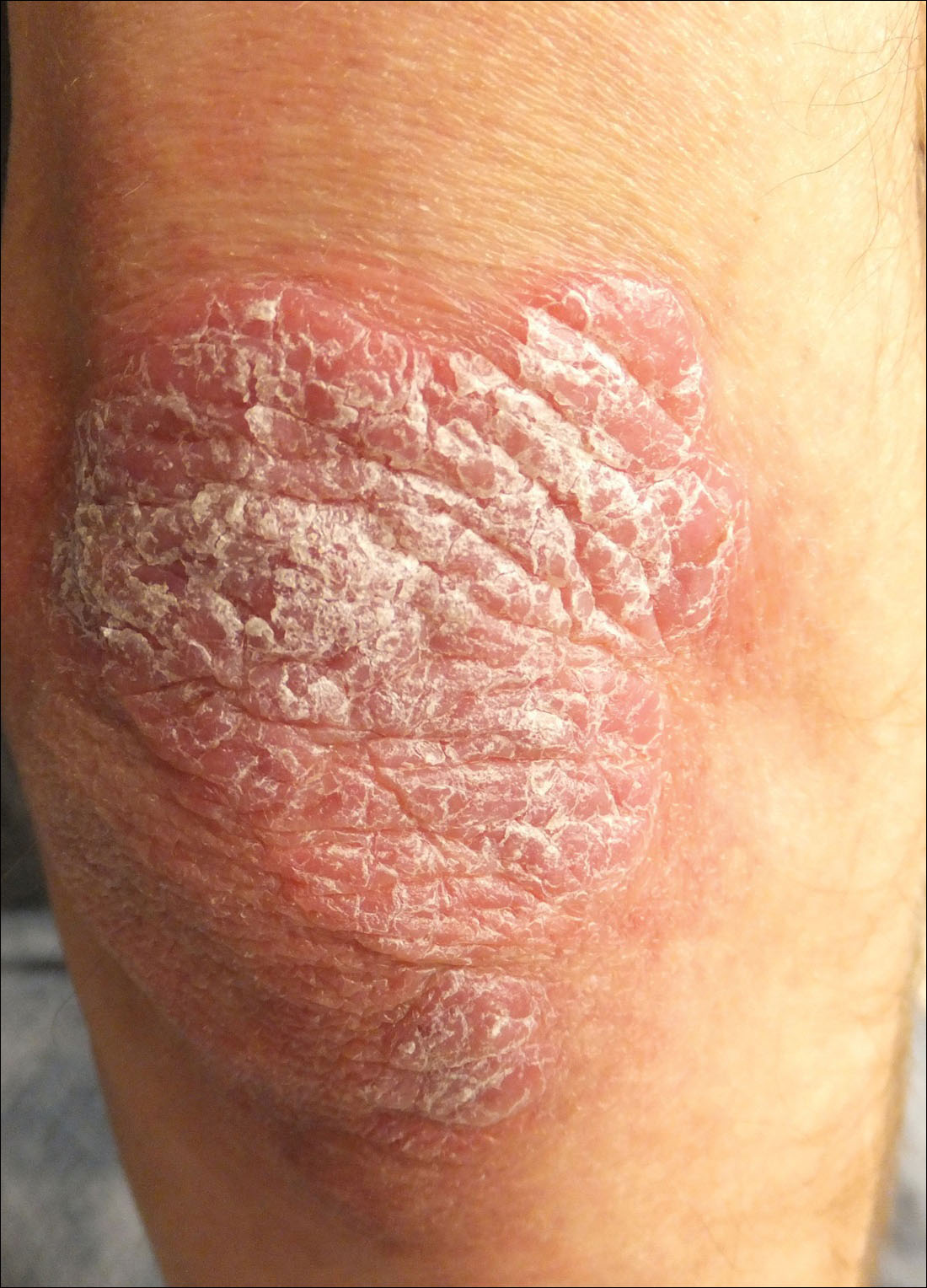
A 34-year-old man presented with thick scaly plaques on the wrists, knees, and feet of 2 years' duration. He had seen several dermatologists, and despite the use of topical steroids, he had no improvement.
Painful Ulcerations Above the Malleoli
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
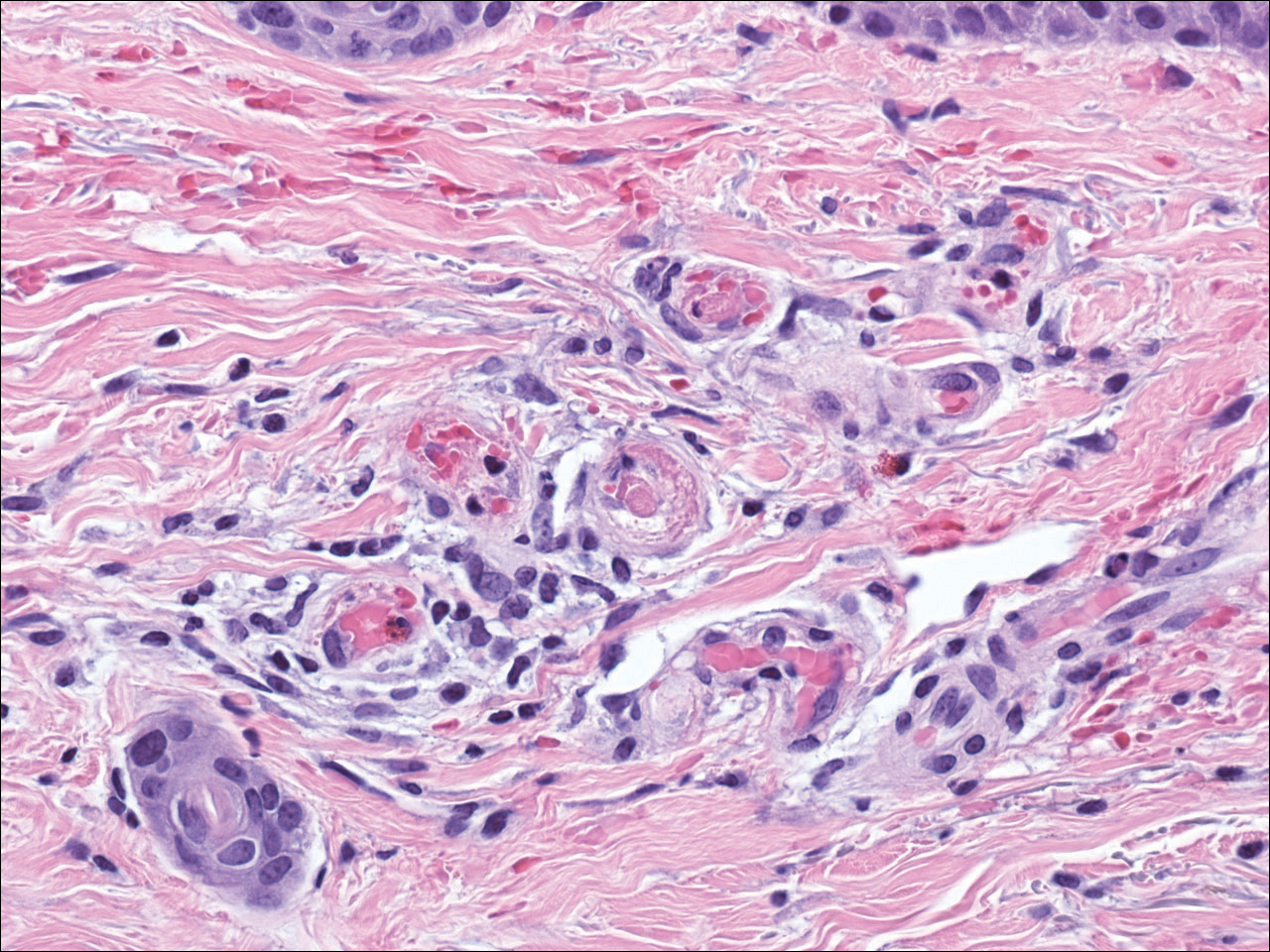
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
A 58-year-old woman presented in the summertime with skin discoloration of the bilateral lower legs and painful ulcerations above the medial and lateral malleoli of 15 years’ duration. She denied any recent trauma to the area or change in skin lesion appearance with cold exposure. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities was negative. A punch biopsy specimen obtained from the left anterior lower leg revealed vascular thrombi with extravasated erythrocytes and a sparse perivascular inflammatory cell infiltrate.
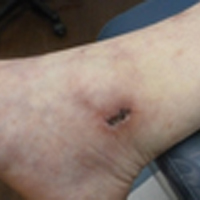
Enlarging Breast Lesion
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
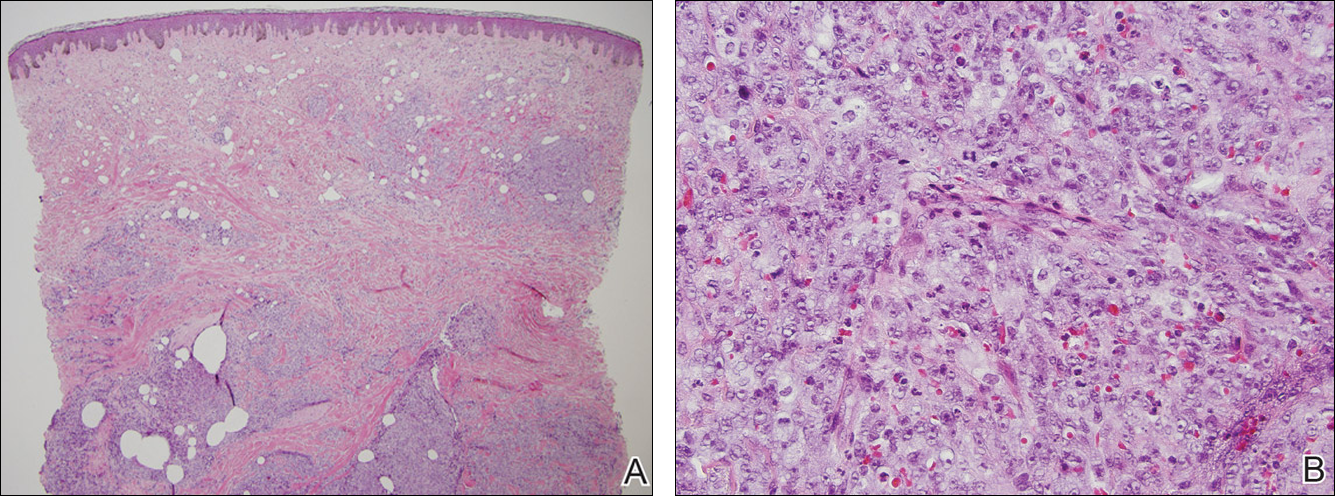
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.

A 75-year-old woman with a history of stage II invasive ductal carcinoma of the right breast presented to the dermatology clinic with an enlarging, indurated, ecchymotic plaque on the inferior aspect of the right breast of 2 months’ duration. The patient underwent a lumpectomy, radiation, and adjuvant chemotherapy 13 years prior to presentation. Review of systems was otherwise noncontributory.
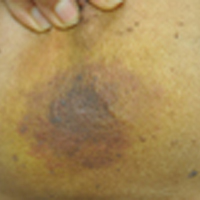
Sporotrichoid Fluctuant Nodules
The Diagnosis: Atypical Mycobacterial Infection
Punch biopsy specimens demonstrated necrotizing granulomatous inflammation in the dermis and subcutis (Figure). Special staining for microorganisms was negative. Tissue culture grew Mycobacterium avium-intracellulare (MAI). The patient began treatment with azithromycin, ethambutol, and rifabutin. Tissue susceptibilities later showed resistance to rifabutin and sensitivity to clarithromycin, moxifloxacin, and clofazimine. She subsequently was switched to azithromycin, clofazimine, and moxifloxacin with good response.
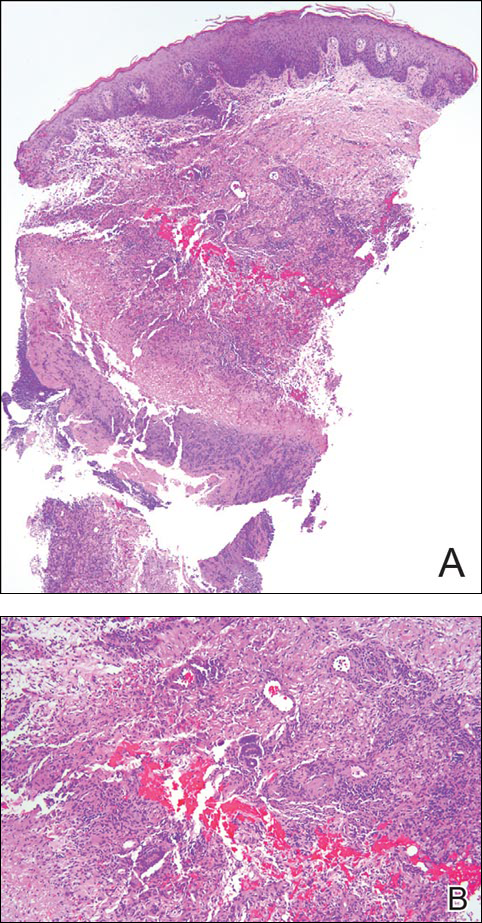
Mycobacterium avium-intracellulare is a slow-growing, nonchromogenic, atypical mycobacteria. Although ubiquitous, it tends to only cause serious infection in the setting of immunosuppression. Transmission usually is through the respiratory or gastrointestinal tract.1 Skin infections with MAI are uncommon and usually are secondary to seeding from disseminated infection or from direct inoculation.2
The clinical presentations of primary cutaneous MAI are myriad, including an isolated red nodule, multiple ulcers, abscesses, draining sinuses, facial nodules, granulomatous plaques, and panniculitis.2,3 Of 3 reported cases of primary cutaneous MAI in the form of sporotrichoid lesions, 2 involved patients with AIDS2 and 1 involved a cardiac transplant recipient.4
Cutaneous MAI is typically diagnosed with skin biopsy and tissue culture. Tissue culture is critical for determining the specific mycobacterial species and antibiotic susceptibilities. Polymerase chain reaction has been utilized to rapidly diagnose cutaneous MAI infection from an acid-fast bacilli–positive tissue sample in which the tissue culture was negative.5
Recommended treatment protocols for MAI involve multidrug regimens because of the intrinsic resistance of MAI and the concern for development of resistance with monotherapy.2 No definitive guidelines exist for treatment of primary cutaneous MAI infections. However, regimens for the treatment of pulmonary infection that also have been successfully utilized for cutaneous infection include a macrolide, ethambutol, and a rifamycin.6 Clinicians should be aware of MAI as a cause of primary cutaneous infections presenting as lymphocutaneous suppurative nodules and ulcerations.
- Hautmann G, Lotti T. Atypical mycobacterial infections of the skin. Dermatol Clin. 1994;12:657-668.
- Kayal JD, McCall CO. Sporotrichoid cutaneous Mycobacterium avium complex infection. J Am Acad Dermatol. 2002;47(5 suppl):S249-S250.
- Kullavanijaya P, Sirimachan S, Surarak S. Primary cutaneous infection with Mycobacterium avium-intracellulare complex resembling lupus vulgaris. Br J Dermatol. 1997;136:264-266.
- Wood C, Nickoloff BJ, Todes-Taylor NR. Pseudotumor resulting from atypical mycobacterial infection: a “histoid” variety of Mycobacterium avium-intracellulare complex infection. Am J Clin Pathol. 1985;83:524-527.
- Carlos CA, Tang YW, Adler DJ, et al. Mycobacterial infection identified with broad-range PCR amplification and suspension array identification. J Cutan Pathol. 2012;39:795-797.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
The Diagnosis: Atypical Mycobacterial Infection
Punch biopsy specimens demonstrated necrotizing granulomatous inflammation in the dermis and subcutis (Figure). Special staining for microorganisms was negative. Tissue culture grew Mycobacterium avium-intracellulare (MAI). The patient began treatment with azithromycin, ethambutol, and rifabutin. Tissue susceptibilities later showed resistance to rifabutin and sensitivity to clarithromycin, moxifloxacin, and clofazimine. She subsequently was switched to azithromycin, clofazimine, and moxifloxacin with good response.
Mycobacterium avium-intracellulare is a slow-growing, nonchromogenic, atypical mycobacteria. Although ubiquitous, it tends to only cause serious infection in the setting of immunosuppression. Transmission usually is through the respiratory or gastrointestinal tract.1 Skin infections with MAI are uncommon and usually are secondary to seeding from disseminated infection or from direct inoculation.2
The clinical presentations of primary cutaneous MAI are myriad, including an isolated red nodule, multiple ulcers, abscesses, draining sinuses, facial nodules, granulomatous plaques, and panniculitis.2,3 Of 3 reported cases of primary cutaneous MAI in the form of sporotrichoid lesions, 2 involved patients with AIDS2 and 1 involved a cardiac transplant recipient.4
Cutaneous MAI is typically diagnosed with skin biopsy and tissue culture. Tissue culture is critical for determining the specific mycobacterial species and antibiotic susceptibilities. Polymerase chain reaction has been utilized to rapidly diagnose cutaneous MAI infection from an acid-fast bacilli–positive tissue sample in which the tissue culture was negative.5
Recommended treatment protocols for MAI involve multidrug regimens because of the intrinsic resistance of MAI and the concern for development of resistance with monotherapy.2 No definitive guidelines exist for treatment of primary cutaneous MAI infections. However, regimens for the treatment of pulmonary infection that also have been successfully utilized for cutaneous infection include a macrolide, ethambutol, and a rifamycin.6 Clinicians should be aware of MAI as a cause of primary cutaneous infections presenting as lymphocutaneous suppurative nodules and ulcerations.
The Diagnosis: Atypical Mycobacterial Infection
Punch biopsy specimens demonstrated necrotizing granulomatous inflammation in the dermis and subcutis (Figure). Special staining for microorganisms was negative. Tissue culture grew Mycobacterium avium-intracellulare (MAI). The patient began treatment with azithromycin, ethambutol, and rifabutin. Tissue susceptibilities later showed resistance to rifabutin and sensitivity to clarithromycin, moxifloxacin, and clofazimine. She subsequently was switched to azithromycin, clofazimine, and moxifloxacin with good response.
Mycobacterium avium-intracellulare is a slow-growing, nonchromogenic, atypical mycobacteria. Although ubiquitous, it tends to only cause serious infection in the setting of immunosuppression. Transmission usually is through the respiratory or gastrointestinal tract.1 Skin infections with MAI are uncommon and usually are secondary to seeding from disseminated infection or from direct inoculation.2
The clinical presentations of primary cutaneous MAI are myriad, including an isolated red nodule, multiple ulcers, abscesses, draining sinuses, facial nodules, granulomatous plaques, and panniculitis.2,3 Of 3 reported cases of primary cutaneous MAI in the form of sporotrichoid lesions, 2 involved patients with AIDS2 and 1 involved a cardiac transplant recipient.4
Cutaneous MAI is typically diagnosed with skin biopsy and tissue culture. Tissue culture is critical for determining the specific mycobacterial species and antibiotic susceptibilities. Polymerase chain reaction has been utilized to rapidly diagnose cutaneous MAI infection from an acid-fast bacilli–positive tissue sample in which the tissue culture was negative.5
Recommended treatment protocols for MAI involve multidrug regimens because of the intrinsic resistance of MAI and the concern for development of resistance with monotherapy.2 No definitive guidelines exist for treatment of primary cutaneous MAI infections. However, regimens for the treatment of pulmonary infection that also have been successfully utilized for cutaneous infection include a macrolide, ethambutol, and a rifamycin.6 Clinicians should be aware of MAI as a cause of primary cutaneous infections presenting as lymphocutaneous suppurative nodules and ulcerations.
- Hautmann G, Lotti T. Atypical mycobacterial infections of the skin. Dermatol Clin. 1994;12:657-668.
- Kayal JD, McCall CO. Sporotrichoid cutaneous Mycobacterium avium complex infection. J Am Acad Dermatol. 2002;47(5 suppl):S249-S250.
- Kullavanijaya P, Sirimachan S, Surarak S. Primary cutaneous infection with Mycobacterium avium-intracellulare complex resembling lupus vulgaris. Br J Dermatol. 1997;136:264-266.
- Wood C, Nickoloff BJ, Todes-Taylor NR. Pseudotumor resulting from atypical mycobacterial infection: a “histoid” variety of Mycobacterium avium-intracellulare complex infection. Am J Clin Pathol. 1985;83:524-527.
- Carlos CA, Tang YW, Adler DJ, et al. Mycobacterial infection identified with broad-range PCR amplification and suspension array identification. J Cutan Pathol. 2012;39:795-797.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Hautmann G, Lotti T. Atypical mycobacterial infections of the skin. Dermatol Clin. 1994;12:657-668.
- Kayal JD, McCall CO. Sporotrichoid cutaneous Mycobacterium avium complex infection. J Am Acad Dermatol. 2002;47(5 suppl):S249-S250.
- Kullavanijaya P, Sirimachan S, Surarak S. Primary cutaneous infection with Mycobacterium avium-intracellulare complex resembling lupus vulgaris. Br J Dermatol. 1997;136:264-266.
- Wood C, Nickoloff BJ, Todes-Taylor NR. Pseudotumor resulting from atypical mycobacterial infection: a “histoid” variety of Mycobacterium avium-intracellulare complex infection. Am J Clin Pathol. 1985;83:524-527.
- Carlos CA, Tang YW, Adler DJ, et al. Mycobacterial infection identified with broad-range PCR amplification and suspension array identification. J Cutan Pathol. 2012;39:795-797.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
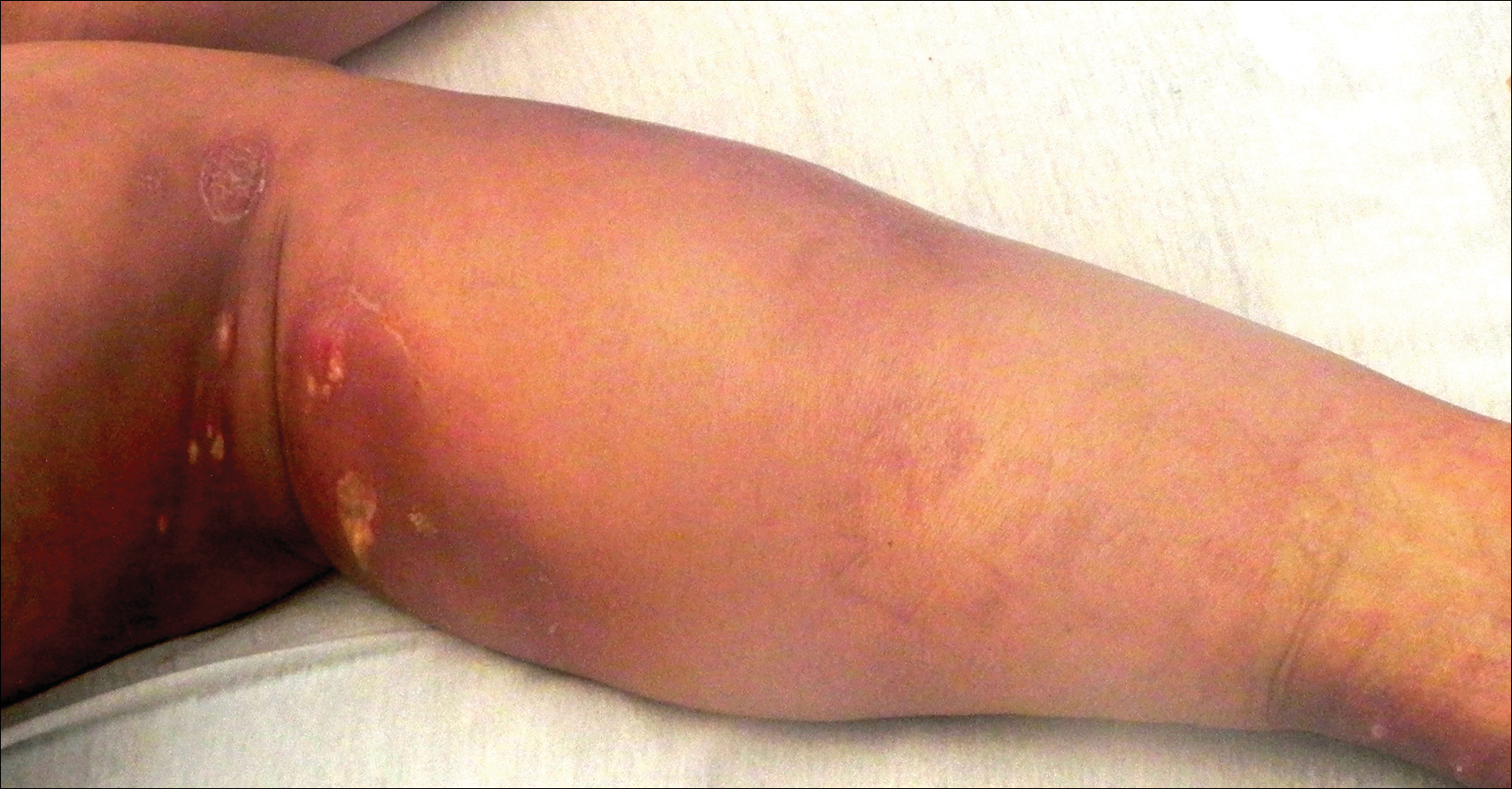
A woman in her 50s presented with low-grade subjective intermittent fevers and painful draining ulcerations on the legs of 7 months’ duration. Her medical history was remarkable for polymyositis and interstitial lung disease managed with prednisone and mycophenolate mofetil. While living in Taiwan, she developed lower extremity abscesses and persistent fevers. The patient denied any skin injuries or exposure to animals or brackish water. Mycophenolate mofetil was discontinued, and she was treated with multiple antibiotics alone and in combination without improvement, including amoxicillin–clavulanic acid, levofloxacin, azithromycin, moxifloxacin, rifampin, rifabutin, and ethambutol. She returned to the United States for evaluation. Physical examination revealed ulcerations with purulent drainage and interconnected sinus tracts with rare fluctuant nodules along the right leg. A single similar lesion was present on the right chest wall. There was no clinical evidence of disseminated disease.
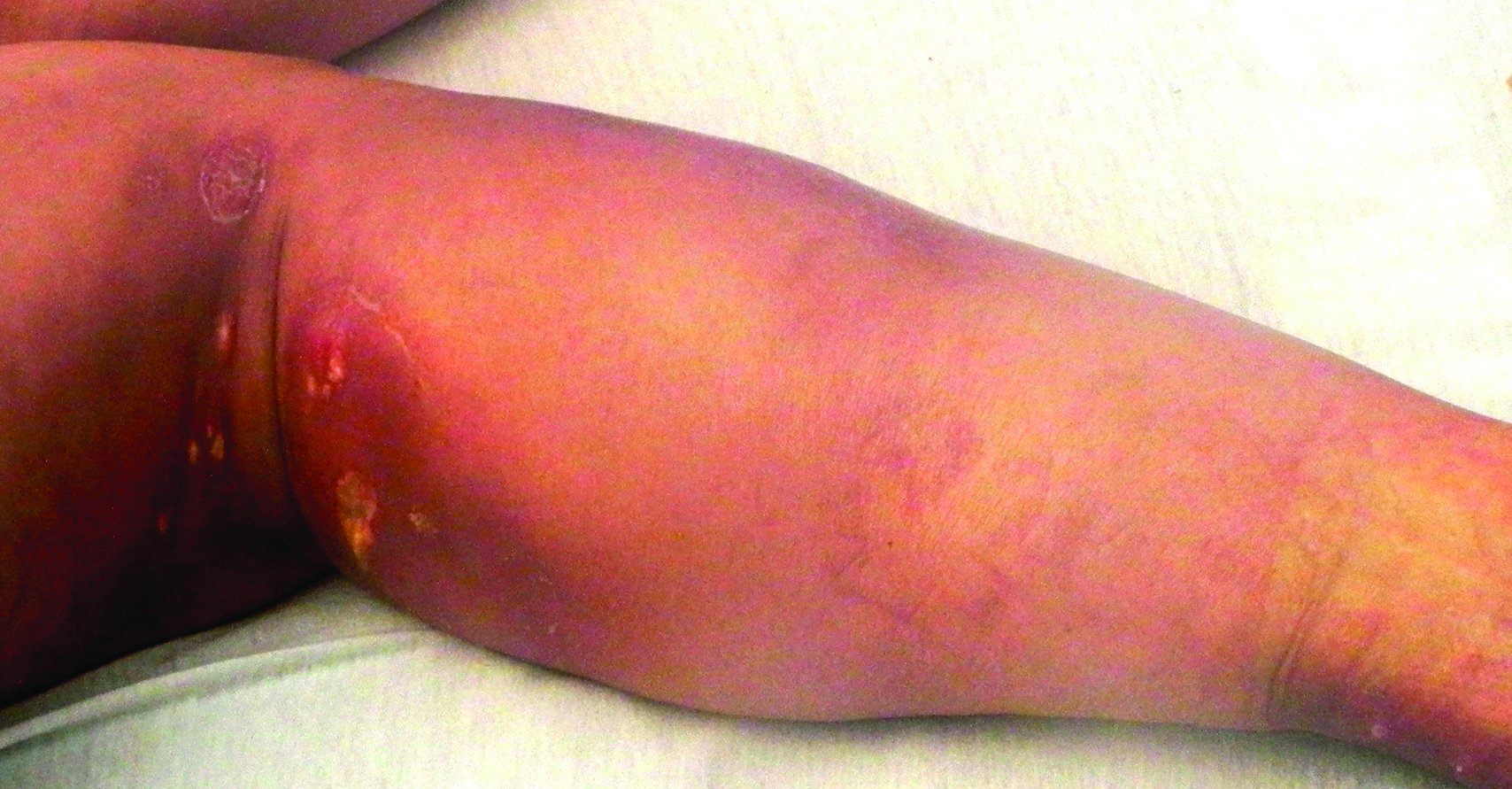
Patch of Hair Loss on the Scalp
The Diagnosis: Temporal Triangular Alopecia
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
The Diagnosis: Temporal Triangular Alopecia
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
The Diagnosis: Temporal Triangular Alopecia
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
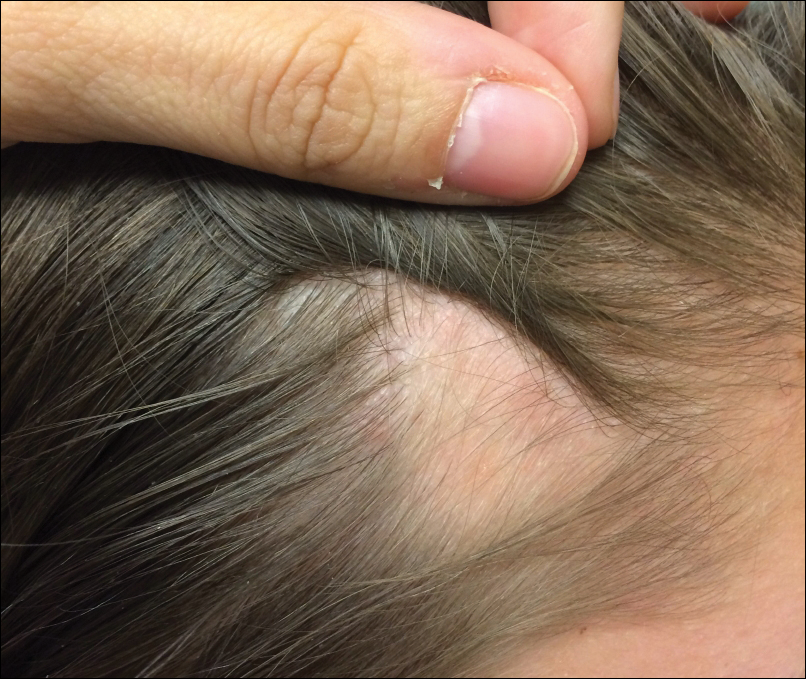
An 11-year-old girl presented for evaluation of a patch of hair loss on the right parietal scalp that had been present and stable for 2.5 years. Physical examination revealed a unilateral area of hair loss that was triangular in shape on the right parietal/temporal region, measuring 2.1×2.2 cm. Dermatoscope examination showed vellus hairs throughout. A hair-pull test was negative and the patient confirmed that the area had never been completely smooth. There were no associated symptoms and no family history of autoimmune disease or hair loss. Prior to presentation, the patient underwent a trial of intralesional steroids and topical steroids to the area without effect.