User login
ASTRO Pushes Return to Direct Supervision in RT: Needed or ‘Babysitting’?
Although serious errors during virtual supervision are rare, ASTRO said radiation treatments (RT) should be done with a radiation oncologist on site to ensure high-quality care. But some radiation oncologists do not agree with the proposal to move back to direct in-person supervision only.
Changes to Direct Supervision
Most radiation oncology treatments are delivered in an outpatient setting under a physician’s direction and control.
During the COVID-19 pandemic when social distancing mandates were in place, CMS temporarily changed the definition of “direct supervision” to include telehealth, specifying that a physician must be immediately available to assist and direct a procedure virtually using real-time audio and video. In other words, a physician did not need to be physically present in the room when the treatment was being performed.
CMS has extended this rule until the end of 2024 and is considering making it a permanent change. In the Calendar Year (CY) 2024 Medicare Physician Fee Schedule (PFS) Final Rule, CMS asked for comments on whether to extend the rule.
“We received input from interested parties on potential patient safety or quality concerns when direct supervision occurs virtually, which we will consider for future rulemaking,” a CMS spokesperson told this news organization. “CMS is currently considering the best approach that will protect patient access and safety as well as quality of care and program integrity concerns following CY 2024.”
CMS also noted its concerns that an abrupt transition back to requiring a physician’s physical presence could interrupt care from practitioners who have established new patterns of practice with telehealth.
What Are ASTRO’s Concerns?
Late last month, ASTRO sent CMS a letter, asking the agency to change the rules back to direct in-person supervision for all radiation services, citing that virtual supervision jeopardizes patient safety and quality of care.
Jeff Michalski, MD, MBA, chair of the ASTRO Board of Directors, said in an interview that radiation oncologists should be physically present to supervise the treatments.
“ASTRO is concerned that blanket policies of general or virtual supervision could lead to patients not having direct, in-person access to their doctors’ care,” he said. “While serious errors are rare, real-world experiences of radiation oncologists across practice settings demonstrate how an in-person radiation oncology physician is best suited to ensure high-quality care.”
What Do Radiation Oncologists Think?
According to ASTRO, most radiation oncologists would agree that in-person supervision is best for patients.
But that might not be the case.
Radiation oncologists took to X (formerly Twitter) to voice their opinions about ASTRO’s letter.
Jason Beckta, MD, PhD, of Rutland Regional’s Foley Cancer Center, Vermont, said “the February 26th ASTRO letter reads like an Onion article.”
“I’m struggling to understand the Luddite-level myopia around this topic,” he said in another tweet. “Virtual direct/outpatient general supervision has done nothing but boost my productivity and in particular, face-to-face patient contact.”
Join Y. Luh, MD, with the Providence Medical Network in Eureka, California, said he understands the challenges faced by clinicians working in more isolated rural settings. “For them, it’s either having virtual supervision or closing the center,” Dr. Luh said.
“Virtual care is definitely at my clinic and is not only an option but is critical to my patients who are 2+ snowy, mountainous hours away,” Dr. Luh wrote. “But I’m still in the clinic directly supervising treatments.”
Sidney Roberts, MD, with the CHI St. Luke’s Health-Memorial, Texas, tweeted that supervision does require some face-to-face care but contended that “babysitting trained therapists for every routine treatment is a farce.”
Another issue Dr. Luh brought up is reimbursement for virtual supervision, noting that “the elephant in the room is whether that level of service should be reimbursed at the same rate. Reimbursement has not changed — but will it stay that way?”
ASTRO has acknowledged that radiation oncologists will have varying opinions and says it is working to balance these challenges.
CMS has not reached a decision on whether the change will be implemented permanently. The organization will assess concern, patient safety, and quality of care at the end of the year.
A version of this article first appeared on Medscape.com
Although serious errors during virtual supervision are rare, ASTRO said radiation treatments (RT) should be done with a radiation oncologist on site to ensure high-quality care. But some radiation oncologists do not agree with the proposal to move back to direct in-person supervision only.
Changes to Direct Supervision
Most radiation oncology treatments are delivered in an outpatient setting under a physician’s direction and control.
During the COVID-19 pandemic when social distancing mandates were in place, CMS temporarily changed the definition of “direct supervision” to include telehealth, specifying that a physician must be immediately available to assist and direct a procedure virtually using real-time audio and video. In other words, a physician did not need to be physically present in the room when the treatment was being performed.
CMS has extended this rule until the end of 2024 and is considering making it a permanent change. In the Calendar Year (CY) 2024 Medicare Physician Fee Schedule (PFS) Final Rule, CMS asked for comments on whether to extend the rule.
“We received input from interested parties on potential patient safety or quality concerns when direct supervision occurs virtually, which we will consider for future rulemaking,” a CMS spokesperson told this news organization. “CMS is currently considering the best approach that will protect patient access and safety as well as quality of care and program integrity concerns following CY 2024.”
CMS also noted its concerns that an abrupt transition back to requiring a physician’s physical presence could interrupt care from practitioners who have established new patterns of practice with telehealth.
What Are ASTRO’s Concerns?
Late last month, ASTRO sent CMS a letter, asking the agency to change the rules back to direct in-person supervision for all radiation services, citing that virtual supervision jeopardizes patient safety and quality of care.
Jeff Michalski, MD, MBA, chair of the ASTRO Board of Directors, said in an interview that radiation oncologists should be physically present to supervise the treatments.
“ASTRO is concerned that blanket policies of general or virtual supervision could lead to patients not having direct, in-person access to their doctors’ care,” he said. “While serious errors are rare, real-world experiences of radiation oncologists across practice settings demonstrate how an in-person radiation oncology physician is best suited to ensure high-quality care.”
What Do Radiation Oncologists Think?
According to ASTRO, most radiation oncologists would agree that in-person supervision is best for patients.
But that might not be the case.
Radiation oncologists took to X (formerly Twitter) to voice their opinions about ASTRO’s letter.
Jason Beckta, MD, PhD, of Rutland Regional’s Foley Cancer Center, Vermont, said “the February 26th ASTRO letter reads like an Onion article.”
“I’m struggling to understand the Luddite-level myopia around this topic,” he said in another tweet. “Virtual direct/outpatient general supervision has done nothing but boost my productivity and in particular, face-to-face patient contact.”
Join Y. Luh, MD, with the Providence Medical Network in Eureka, California, said he understands the challenges faced by clinicians working in more isolated rural settings. “For them, it’s either having virtual supervision or closing the center,” Dr. Luh said.
“Virtual care is definitely at my clinic and is not only an option but is critical to my patients who are 2+ snowy, mountainous hours away,” Dr. Luh wrote. “But I’m still in the clinic directly supervising treatments.”
Sidney Roberts, MD, with the CHI St. Luke’s Health-Memorial, Texas, tweeted that supervision does require some face-to-face care but contended that “babysitting trained therapists for every routine treatment is a farce.”
Another issue Dr. Luh brought up is reimbursement for virtual supervision, noting that “the elephant in the room is whether that level of service should be reimbursed at the same rate. Reimbursement has not changed — but will it stay that way?”
ASTRO has acknowledged that radiation oncologists will have varying opinions and says it is working to balance these challenges.
CMS has not reached a decision on whether the change will be implemented permanently. The organization will assess concern, patient safety, and quality of care at the end of the year.
A version of this article first appeared on Medscape.com
Although serious errors during virtual supervision are rare, ASTRO said radiation treatments (RT) should be done with a radiation oncologist on site to ensure high-quality care. But some radiation oncologists do not agree with the proposal to move back to direct in-person supervision only.
Changes to Direct Supervision
Most radiation oncology treatments are delivered in an outpatient setting under a physician’s direction and control.
During the COVID-19 pandemic when social distancing mandates were in place, CMS temporarily changed the definition of “direct supervision” to include telehealth, specifying that a physician must be immediately available to assist and direct a procedure virtually using real-time audio and video. In other words, a physician did not need to be physically present in the room when the treatment was being performed.
CMS has extended this rule until the end of 2024 and is considering making it a permanent change. In the Calendar Year (CY) 2024 Medicare Physician Fee Schedule (PFS) Final Rule, CMS asked for comments on whether to extend the rule.
“We received input from interested parties on potential patient safety or quality concerns when direct supervision occurs virtually, which we will consider for future rulemaking,” a CMS spokesperson told this news organization. “CMS is currently considering the best approach that will protect patient access and safety as well as quality of care and program integrity concerns following CY 2024.”
CMS also noted its concerns that an abrupt transition back to requiring a physician’s physical presence could interrupt care from practitioners who have established new patterns of practice with telehealth.
What Are ASTRO’s Concerns?
Late last month, ASTRO sent CMS a letter, asking the agency to change the rules back to direct in-person supervision for all radiation services, citing that virtual supervision jeopardizes patient safety and quality of care.
Jeff Michalski, MD, MBA, chair of the ASTRO Board of Directors, said in an interview that radiation oncologists should be physically present to supervise the treatments.
“ASTRO is concerned that blanket policies of general or virtual supervision could lead to patients not having direct, in-person access to their doctors’ care,” he said. “While serious errors are rare, real-world experiences of radiation oncologists across practice settings demonstrate how an in-person radiation oncology physician is best suited to ensure high-quality care.”
What Do Radiation Oncologists Think?
According to ASTRO, most radiation oncologists would agree that in-person supervision is best for patients.
But that might not be the case.
Radiation oncologists took to X (formerly Twitter) to voice their opinions about ASTRO’s letter.
Jason Beckta, MD, PhD, of Rutland Regional’s Foley Cancer Center, Vermont, said “the February 26th ASTRO letter reads like an Onion article.”
“I’m struggling to understand the Luddite-level myopia around this topic,” he said in another tweet. “Virtual direct/outpatient general supervision has done nothing but boost my productivity and in particular, face-to-face patient contact.”
Join Y. Luh, MD, with the Providence Medical Network in Eureka, California, said he understands the challenges faced by clinicians working in more isolated rural settings. “For them, it’s either having virtual supervision or closing the center,” Dr. Luh said.
“Virtual care is definitely at my clinic and is not only an option but is critical to my patients who are 2+ snowy, mountainous hours away,” Dr. Luh wrote. “But I’m still in the clinic directly supervising treatments.”
Sidney Roberts, MD, with the CHI St. Luke’s Health-Memorial, Texas, tweeted that supervision does require some face-to-face care but contended that “babysitting trained therapists for every routine treatment is a farce.”
Another issue Dr. Luh brought up is reimbursement for virtual supervision, noting that “the elephant in the room is whether that level of service should be reimbursed at the same rate. Reimbursement has not changed — but will it stay that way?”
ASTRO has acknowledged that radiation oncologists will have varying opinions and says it is working to balance these challenges.
CMS has not reached a decision on whether the change will be implemented permanently. The organization will assess concern, patient safety, and quality of care at the end of the year.
A version of this article first appeared on Medscape.com
FDA Removes Harmful Chemicals From Food Packaging
Issued on February 28, 2024, “this means the major source of dietary exposure to PFAS from food packaging like fast-food wrappers, microwave popcorn bags, take-out paperboard containers, and pet food bags is being eliminated,” the FDA said in a statement.
In 2020, the FDA had secured commitments from manufacturers to stop selling products containing PFAS used in the food packaging for grease-proofing. “Today’s announcement marks the fulfillment of these voluntary commitments,” according to the agency.
PFAS, a class of thousands of chemicals also called “forever chemicals” are widely used in consumer and industrial products. People may be exposed via contaminated food packaging (although perhaps no longer in the United States) or occupationally. Studies have found that some PFAS disrupt hormones including estrogen and testosterone, whereas others may impair thyroid function.
Endocrine Society Report Sounds the Alarm About PFAS and Others
The FDA’s announcement came just 2 days after the Endocrine Society issued a new alarm about the human health dangers from environmental EDCs including PFAS in a report covering the latest science.
“Endocrine disrupting chemicals” are individual substances or mixtures that can interfere with natural hormonal function, leading to disease or even death. Many are ubiquitous in the modern environment and contribute to a wide range of human diseases.
The new report Endocrine Disrupting Chemicals: Threats to Human Health was issued jointly with the International Pollutants Elimination Network (IPEN), a global advocacy organization. It’s an update to the Endocrine Society’s 2015 report, providing new data on the endocrine-disrupting substances previously covered and adding four EDCs not discussed in that document: Pesticides, plastics, PFAS, and children’s products containing arsenic.
At a briefing held during the United Nations Environment Assembly meeting in Nairobi, Kenya, last week, the new report’s lead author Andrea C. Gore, PhD, of the University of Texas at Austin, noted, “A well-established body of scientific research indicates that endocrine-disrupting chemicals that are part of our daily lives are making us more susceptible to reproductive disorders, cancer, diabetes, obesity, heart disease, and other serious health conditions.”
Added Dr. Gore, who is also a member of the Endocrine Society’s Board of Directors, “These chemicals pose particularly serious risks to pregnant women and children. Now is the time for the UN Environment Assembly and other global policymakers to take action to address this threat to public health.”
While the science has been emerging rapidly, global and national chemical control policies haven’t kept up, the authors said. Of particular concern is that EDCs behave differently from other chemicals in many ways, including that even very low-dose exposures can pose health threats, but policies thus far haven’t dealt with that aspect.
Moreover, “the effects of low doses cannot be predicted by the effects observed at high doses. This means there may be no safe dose for exposure to EDCs,” according to the report.
Exposures can come from household products, including furniture, toys, and food packages, as well as electronics building materials and cosmetics. These chemicals are also in the outdoor environment, via pesticides, air pollution, and industrial waste.
“IPEN and the Endocrine Society call for chemical regulations based on the most modern scientific understanding of how hormones act and how EDCs can perturb these actions. We work to educate policy makers in global, regional, and national government assemblies and help ensure that regulations correlate with current scientific understanding,” they said in the report.
New Data on Four Classes of EDCs
Chapters of the report summarized the latest information about the science of EDCs and their links to endocrine disease and real-world exposure. It included a special section about “EDCs throughout the plastics life cycle” and a summary of the links between EDCs and climate change.
The report reviewed three pesticides, including the world’s most heavily applied herbicide, glycophosphate. Exposures can occur directly from the air, water, dust, and food residues. Recent data linked glycophosphate to adverse reproductive health outcomes.
Two toxic plastic chemicals, phthalates and bisphenols, are present in personal care products, among others. Emerging evidence links them with impaired neurodevelopment, leading to impaired cognitive function, learning, attention, and impulsivity.
Arsenic has long been linked to human health conditions including cancer, but more recent evidence finds it can disrupt multiple endocrine systems and lead to metabolic conditions including diabetes, reproductive dysfunction, and cardiovascular and neurocognitive conditions.
The special section about plastics noted that they are made from fossil fuels and chemicals, including many toxic substances that are known or suspected EDCs. People who live near plastic production facilities or waste dumps may be at greatest risk, but anyone can be exposed using any plastic product. Plastic waste disposal is increasingly problematic and often foisted on lower- and middle-income countries.
‘Additional Education and Awareness-Raising Among Stakeholders Remain Necessary’
Policies aimed at reducing human health risks from EDCs have included the 2022 Plastics Treaty, a resolution adopted by 175 countries at the United Nations Environmental Assembly that “may be a significant step toward global control of plastics and elimination of threats from exposures to EDCs in plastics,” the report said.
The authors added, “While significant progress has been made in recent years connecting scientific advances on EDCs with health-protective policies, additional education and awareness-raising among stakeholders remain necessary to achieve a safer and more sustainable environment that minimizes exposure to these harmful chemicals.”
The document was produced with financial contributions from the Government of Sweden, the Tides Foundation, Passport Foundation, and other donors.
A version of this article appeared on Medscape.com.
Issued on February 28, 2024, “this means the major source of dietary exposure to PFAS from food packaging like fast-food wrappers, microwave popcorn bags, take-out paperboard containers, and pet food bags is being eliminated,” the FDA said in a statement.
In 2020, the FDA had secured commitments from manufacturers to stop selling products containing PFAS used in the food packaging for grease-proofing. “Today’s announcement marks the fulfillment of these voluntary commitments,” according to the agency.
PFAS, a class of thousands of chemicals also called “forever chemicals” are widely used in consumer and industrial products. People may be exposed via contaminated food packaging (although perhaps no longer in the United States) or occupationally. Studies have found that some PFAS disrupt hormones including estrogen and testosterone, whereas others may impair thyroid function.
Endocrine Society Report Sounds the Alarm About PFAS and Others
The FDA’s announcement came just 2 days after the Endocrine Society issued a new alarm about the human health dangers from environmental EDCs including PFAS in a report covering the latest science.
“Endocrine disrupting chemicals” are individual substances or mixtures that can interfere with natural hormonal function, leading to disease or even death. Many are ubiquitous in the modern environment and contribute to a wide range of human diseases.
The new report Endocrine Disrupting Chemicals: Threats to Human Health was issued jointly with the International Pollutants Elimination Network (IPEN), a global advocacy organization. It’s an update to the Endocrine Society’s 2015 report, providing new data on the endocrine-disrupting substances previously covered and adding four EDCs not discussed in that document: Pesticides, plastics, PFAS, and children’s products containing arsenic.
At a briefing held during the United Nations Environment Assembly meeting in Nairobi, Kenya, last week, the new report’s lead author Andrea C. Gore, PhD, of the University of Texas at Austin, noted, “A well-established body of scientific research indicates that endocrine-disrupting chemicals that are part of our daily lives are making us more susceptible to reproductive disorders, cancer, diabetes, obesity, heart disease, and other serious health conditions.”
Added Dr. Gore, who is also a member of the Endocrine Society’s Board of Directors, “These chemicals pose particularly serious risks to pregnant women and children. Now is the time for the UN Environment Assembly and other global policymakers to take action to address this threat to public health.”
While the science has been emerging rapidly, global and national chemical control policies haven’t kept up, the authors said. Of particular concern is that EDCs behave differently from other chemicals in many ways, including that even very low-dose exposures can pose health threats, but policies thus far haven’t dealt with that aspect.
Moreover, “the effects of low doses cannot be predicted by the effects observed at high doses. This means there may be no safe dose for exposure to EDCs,” according to the report.
Exposures can come from household products, including furniture, toys, and food packages, as well as electronics building materials and cosmetics. These chemicals are also in the outdoor environment, via pesticides, air pollution, and industrial waste.
“IPEN and the Endocrine Society call for chemical regulations based on the most modern scientific understanding of how hormones act and how EDCs can perturb these actions. We work to educate policy makers in global, regional, and national government assemblies and help ensure that regulations correlate with current scientific understanding,” they said in the report.
New Data on Four Classes of EDCs
Chapters of the report summarized the latest information about the science of EDCs and their links to endocrine disease and real-world exposure. It included a special section about “EDCs throughout the plastics life cycle” and a summary of the links between EDCs and climate change.
The report reviewed three pesticides, including the world’s most heavily applied herbicide, glycophosphate. Exposures can occur directly from the air, water, dust, and food residues. Recent data linked glycophosphate to adverse reproductive health outcomes.
Two toxic plastic chemicals, phthalates and bisphenols, are present in personal care products, among others. Emerging evidence links them with impaired neurodevelopment, leading to impaired cognitive function, learning, attention, and impulsivity.
Arsenic has long been linked to human health conditions including cancer, but more recent evidence finds it can disrupt multiple endocrine systems and lead to metabolic conditions including diabetes, reproductive dysfunction, and cardiovascular and neurocognitive conditions.
The special section about plastics noted that they are made from fossil fuels and chemicals, including many toxic substances that are known or suspected EDCs. People who live near plastic production facilities or waste dumps may be at greatest risk, but anyone can be exposed using any plastic product. Plastic waste disposal is increasingly problematic and often foisted on lower- and middle-income countries.
‘Additional Education and Awareness-Raising Among Stakeholders Remain Necessary’
Policies aimed at reducing human health risks from EDCs have included the 2022 Plastics Treaty, a resolution adopted by 175 countries at the United Nations Environmental Assembly that “may be a significant step toward global control of plastics and elimination of threats from exposures to EDCs in plastics,” the report said.
The authors added, “While significant progress has been made in recent years connecting scientific advances on EDCs with health-protective policies, additional education and awareness-raising among stakeholders remain necessary to achieve a safer and more sustainable environment that minimizes exposure to these harmful chemicals.”
The document was produced with financial contributions from the Government of Sweden, the Tides Foundation, Passport Foundation, and other donors.
A version of this article appeared on Medscape.com.
Issued on February 28, 2024, “this means the major source of dietary exposure to PFAS from food packaging like fast-food wrappers, microwave popcorn bags, take-out paperboard containers, and pet food bags is being eliminated,” the FDA said in a statement.
In 2020, the FDA had secured commitments from manufacturers to stop selling products containing PFAS used in the food packaging for grease-proofing. “Today’s announcement marks the fulfillment of these voluntary commitments,” according to the agency.
PFAS, a class of thousands of chemicals also called “forever chemicals” are widely used in consumer and industrial products. People may be exposed via contaminated food packaging (although perhaps no longer in the United States) or occupationally. Studies have found that some PFAS disrupt hormones including estrogen and testosterone, whereas others may impair thyroid function.
Endocrine Society Report Sounds the Alarm About PFAS and Others
The FDA’s announcement came just 2 days after the Endocrine Society issued a new alarm about the human health dangers from environmental EDCs including PFAS in a report covering the latest science.
“Endocrine disrupting chemicals” are individual substances or mixtures that can interfere with natural hormonal function, leading to disease or even death. Many are ubiquitous in the modern environment and contribute to a wide range of human diseases.
The new report Endocrine Disrupting Chemicals: Threats to Human Health was issued jointly with the International Pollutants Elimination Network (IPEN), a global advocacy organization. It’s an update to the Endocrine Society’s 2015 report, providing new data on the endocrine-disrupting substances previously covered and adding four EDCs not discussed in that document: Pesticides, plastics, PFAS, and children’s products containing arsenic.
At a briefing held during the United Nations Environment Assembly meeting in Nairobi, Kenya, last week, the new report’s lead author Andrea C. Gore, PhD, of the University of Texas at Austin, noted, “A well-established body of scientific research indicates that endocrine-disrupting chemicals that are part of our daily lives are making us more susceptible to reproductive disorders, cancer, diabetes, obesity, heart disease, and other serious health conditions.”
Added Dr. Gore, who is also a member of the Endocrine Society’s Board of Directors, “These chemicals pose particularly serious risks to pregnant women and children. Now is the time for the UN Environment Assembly and other global policymakers to take action to address this threat to public health.”
While the science has been emerging rapidly, global and national chemical control policies haven’t kept up, the authors said. Of particular concern is that EDCs behave differently from other chemicals in many ways, including that even very low-dose exposures can pose health threats, but policies thus far haven’t dealt with that aspect.
Moreover, “the effects of low doses cannot be predicted by the effects observed at high doses. This means there may be no safe dose for exposure to EDCs,” according to the report.
Exposures can come from household products, including furniture, toys, and food packages, as well as electronics building materials and cosmetics. These chemicals are also in the outdoor environment, via pesticides, air pollution, and industrial waste.
“IPEN and the Endocrine Society call for chemical regulations based on the most modern scientific understanding of how hormones act and how EDCs can perturb these actions. We work to educate policy makers in global, regional, and national government assemblies and help ensure that regulations correlate with current scientific understanding,” they said in the report.
New Data on Four Classes of EDCs
Chapters of the report summarized the latest information about the science of EDCs and their links to endocrine disease and real-world exposure. It included a special section about “EDCs throughout the plastics life cycle” and a summary of the links between EDCs and climate change.
The report reviewed three pesticides, including the world’s most heavily applied herbicide, glycophosphate. Exposures can occur directly from the air, water, dust, and food residues. Recent data linked glycophosphate to adverse reproductive health outcomes.
Two toxic plastic chemicals, phthalates and bisphenols, are present in personal care products, among others. Emerging evidence links them with impaired neurodevelopment, leading to impaired cognitive function, learning, attention, and impulsivity.
Arsenic has long been linked to human health conditions including cancer, but more recent evidence finds it can disrupt multiple endocrine systems and lead to metabolic conditions including diabetes, reproductive dysfunction, and cardiovascular and neurocognitive conditions.
The special section about plastics noted that they are made from fossil fuels and chemicals, including many toxic substances that are known or suspected EDCs. People who live near plastic production facilities or waste dumps may be at greatest risk, but anyone can be exposed using any plastic product. Plastic waste disposal is increasingly problematic and often foisted on lower- and middle-income countries.
‘Additional Education and Awareness-Raising Among Stakeholders Remain Necessary’
Policies aimed at reducing human health risks from EDCs have included the 2022 Plastics Treaty, a resolution adopted by 175 countries at the United Nations Environmental Assembly that “may be a significant step toward global control of plastics and elimination of threats from exposures to EDCs in plastics,” the report said.
The authors added, “While significant progress has been made in recent years connecting scientific advances on EDCs with health-protective policies, additional education and awareness-raising among stakeholders remain necessary to achieve a safer and more sustainable environment that minimizes exposure to these harmful chemicals.”
The document was produced with financial contributions from the Government of Sweden, the Tides Foundation, Passport Foundation, and other donors.
A version of this article appeared on Medscape.com.
AML: Genetic Testing Unlocks Hope
For adult patients, “we’ve seen a series of remarkable and well-overdue advances in a space that had not changed much over the prior decades,” hematologist/oncologist Thomas William LeBlanc, MD, associate professor of medicine at Duke University School of Medicine, Durham, North Carolina, said in an interview.
According to the National Cancer Institute, AML will be newly diagnosed in 20,800 patients in 2024, at a median age of 69, and will cause 11,220 deaths. As many as 70% of adult patients will reach complete remission, and 45% of those will live for more than 3 years and potentially be cured. As for children, the Leukemia & Lymphoma Society says the 5-year survival rate from 2012-2018 was 69% for those under 15 years old.
As the American Cancer Society notes, the goal of AML treatment “is to put the leukemia into complete remission (the bone marrow and blood cell counts return to normal), preferably a complete molecular remission (no signs of leukemia in the bone marrow, even using sensitive lab tests), and to keep it that way.”
Chemotherapy Strategies Shift Over Time
In terms of the treatment of adults with AML, “targeted therapies, in addition to the expanding role of venetoclax, has really altered our approach to AML from diagnosis, including after relapse, and later in the disease,” hematologist/oncologist Andrew M. Brunner, MD, of Harvard Medical School and Massachusetts General Hospital, Boston, said in an interview. “The ability to explore these options as monotherapy and in novel combinations has dramatically expanded our treatment options.”
Much depends on the underlying genetic profile of the disease, he said. “There certainly have been gains in patient survival in AML, but those improvements remain fairly heterogeneous and dependent on the underlying genetic profile of the disease. For instance, advances in FLT3- and IDH1/2-mutated AML are a direct result of the improvements in targeted therapies directed at these mutations. Similarly, some molecular and cytogenetic subtypes of AML are particularly responsive to venetoclax-based regimens, and these regimens have been expanded to previously undertreated populations, particularly those over age 60.”
Specifically, Dr. LeBlanc said, the Food and Drug Administration has approved “3 different FLT3 inhibitors, 2 IDH1 inhibitors, 1 IDH2 inhibitor, a BCL-2 inhibitor, a smoothened/hedgehog pathway inhibitor, an oral maintenance chemotherapy/hypomethylating agent (CC-486/oral azacitidine), a CD33-targeting antibody-drug conjugate, and even a novel formulation of two older chemotherapies that improves efficacy in a poor prognosis subgroup (CPX-351/liposomal daunorubicin and cytarabine).”
There’s also been a shift in treatment protocols for patients who were not fit for intensive chemotherapy. In the past, he said, it was standard “to give single-agent hypomethylating chemotherapy with azacitidine or decitabine, or in some contexts, low-dose chemotherapy with cytarabine. Today, many patients who are older and/or more frail are receiving novel therapies either alone or in combination, with greater efficacy and longer duration of response than previously seen with chemotherapy alone.”
Outcomes Improve but Remain Grim in High-Risk Cases
As a result, Dr. LeBlanc said, “we’re definitely seeing much better outcomes in AML overall. It takes some time to prove this via outcomes data assessments in a large population, but I expect that registries will show significant improvements in overall survival in the coming years, owing to the many new FDA approvals in AML”
Dr. LeBlanc highlighted national data from 2013-2019 showing that the 5-year relative survival rate from AML is 31.7%. That’s up from 26% just a few years ago, and the numbers “always lag several years behind the current year of practice,” he said. However, “the major area where we still have relatively poor outcomes and significant unmet needs remains the ‘adverse risk’ group of patients, particularly those who are older and/or not candidates for hematopoietic stem cell transplantation, which generally is the only potentially curative option for adverse-risk AML.”
He went on to say that “this risk grouping includes those with TP53 mutations, most of which confer a particularly poor prognosis. Exciting therapies that many of us were hoping would prove effective in this subgroup have unfortunately failed in recent clinical trials. We still have a lot of work to do in adverse-risk AML particularly, and also for those whose leukemia has relapsed.”
Mikkael Sekeres, MD, MS, chief of the Division of Hematology at the University of Miami Miller School of Medicine/Sylvester Comprehensive Cancer Center, agreed that more progress is needed, since survival rates are low even as lifespans improve. One key will be “better identifying subtypes of acute myeloid leukemia, and identifying the therapies that will benefit those people most,” he said in an interview. On the other side, it’s important to identify “when aggressive therapies aren’t going to work in somebody and maybe turn toward less-aggressive approaches so we can maximize that person’s quality of life.”
What advice do AML experts have for their colleagues? Dr. LeBlanc said “older patients are not often enough considered for allogeneic stem cell transplantation, which could potentially cure their AML when given as a consolidation treatment for those in remission. I have several patients who are healthy and in their 70s who have enormously benefited from transplants and are now being several years out from transplant with adverse risk AML and without relapse. They’ve had no significant impairments of their quality of life, including no significant graft vs. host disease.”
Dr. Sekeres highlighted the American Society of Hematology’s guidelines for treating older adults with AML, which are currently being updated. It’s crucial to order genetic testing “up front,” he said. “I’m often pleasantly surprised when genetic testing returns and reveals that I have other treatment options.”
However, it’s crucial to understand a patient’s priorities. “I’ve had patients who are 75 who say to me, ‘Do everything under the sun to get rid of my leukemia, I want to live as long as possible.’ And I’ve had patients who say, ‘I want to see as little of doctors and nurses as I can. I want you to maximize my quality of life and keep me out of the hospital.’ ”
Dr. Sekeres also noted that insurers may not cover some pill-based AML treatments such as venetoclax. “We work with our patients and assistance programs. For the most part, we’re pretty successful at getting these drugs for our patients,” he said.
In Pediatrics, Clinical Trials Are Crucial
AML in children is less well-known than in adults, since the number of cases is so small. The disease is diagnosed in about 500 children a year in the United States, according to St. Jude Children’s Research Hospital, adding, however, that AML is “the most common second cancer among children treated for other cancers.”
AML in children gained attention earlier this year when the 2-year-old daughter of a Boston Herald NFL reporter died of the disease following a bone marrow transplant and chemotherapy. Despite the agonies of her treatment, reporter Doug Kyed told a reporter that his daughter Hallie “was still able to find joy every day.”
In an interview, hematologist/oncologist Sarah K. Tasian, MD, of Children’s Hospital of Philadelphia, said researchers are discovering that pediatric AML is significantly different on from a biological perspective from adult AML. “We’ve come to understand a lot more about who these patients are, what makes these leukemias tick, and what their Achilles’ heels are. Then we can align that with the clinical trials outcome data that we have.”
About 80%-90% of pediatric patients with AML nationwide are enrolled in clinical trials, Dr. Tasian said, and an international consortium called the Children’s Oncology Group gathers data about genetics. About 60%-70% of patients will be cured, she added.
However, “we’ve kind of been stuck for about the last 20 years,” she said. “A lot of improving the survival of patients has not been because we’ve been better at chemotherapy or using new chemo, but because we’ve gotten better at supportive care, at treating infections that can be fatal.”
There haven’t been major conflicts with insurers over coverage, she said, although drug shortages are a problem, especially in relapsed AML.
As for advice to colleagues, Dr. Tasian counseled them to understand the importance of genetic testing and the expanding role of stem cell transplants. “We are now transplanting somewhere between 30% and 50% of children with AML, which is a higher rate than we used to do,” she said. The number is up thanks to genetic testing that reveals which patients are most likely to benefit.
Also, she noted, “the chemotherapy that we get to these patients is really strong, and patients have a lot of complications. Really pay attention to supportive care.”
Dr. LeBlanc reported ties with AbbVie, Agios/Servier, Astellas, BMS/Celgene, Genentech, Pfizer, Incyte, Rige, Deverra, GSK, Jazz, and Seattle Genetics. Dr. Sekeres discloses relationships with BMS and Kurome. Dr. Tasian serves as the Leukemia & Lymphoma Society Pediatric Acute Leukemia consortium clinical trials leader and works with pharmaceutical companies on clinical trials under confidentiality agreements. Dr. Brunner has no disclosures.
For adult patients, “we’ve seen a series of remarkable and well-overdue advances in a space that had not changed much over the prior decades,” hematologist/oncologist Thomas William LeBlanc, MD, associate professor of medicine at Duke University School of Medicine, Durham, North Carolina, said in an interview.
According to the National Cancer Institute, AML will be newly diagnosed in 20,800 patients in 2024, at a median age of 69, and will cause 11,220 deaths. As many as 70% of adult patients will reach complete remission, and 45% of those will live for more than 3 years and potentially be cured. As for children, the Leukemia & Lymphoma Society says the 5-year survival rate from 2012-2018 was 69% for those under 15 years old.
As the American Cancer Society notes, the goal of AML treatment “is to put the leukemia into complete remission (the bone marrow and blood cell counts return to normal), preferably a complete molecular remission (no signs of leukemia in the bone marrow, even using sensitive lab tests), and to keep it that way.”
Chemotherapy Strategies Shift Over Time
In terms of the treatment of adults with AML, “targeted therapies, in addition to the expanding role of venetoclax, has really altered our approach to AML from diagnosis, including after relapse, and later in the disease,” hematologist/oncologist Andrew M. Brunner, MD, of Harvard Medical School and Massachusetts General Hospital, Boston, said in an interview. “The ability to explore these options as monotherapy and in novel combinations has dramatically expanded our treatment options.”
Much depends on the underlying genetic profile of the disease, he said. “There certainly have been gains in patient survival in AML, but those improvements remain fairly heterogeneous and dependent on the underlying genetic profile of the disease. For instance, advances in FLT3- and IDH1/2-mutated AML are a direct result of the improvements in targeted therapies directed at these mutations. Similarly, some molecular and cytogenetic subtypes of AML are particularly responsive to venetoclax-based regimens, and these regimens have been expanded to previously undertreated populations, particularly those over age 60.”
Specifically, Dr. LeBlanc said, the Food and Drug Administration has approved “3 different FLT3 inhibitors, 2 IDH1 inhibitors, 1 IDH2 inhibitor, a BCL-2 inhibitor, a smoothened/hedgehog pathway inhibitor, an oral maintenance chemotherapy/hypomethylating agent (CC-486/oral azacitidine), a CD33-targeting antibody-drug conjugate, and even a novel formulation of two older chemotherapies that improves efficacy in a poor prognosis subgroup (CPX-351/liposomal daunorubicin and cytarabine).”
There’s also been a shift in treatment protocols for patients who were not fit for intensive chemotherapy. In the past, he said, it was standard “to give single-agent hypomethylating chemotherapy with azacitidine or decitabine, or in some contexts, low-dose chemotherapy with cytarabine. Today, many patients who are older and/or more frail are receiving novel therapies either alone or in combination, with greater efficacy and longer duration of response than previously seen with chemotherapy alone.”
Outcomes Improve but Remain Grim in High-Risk Cases
As a result, Dr. LeBlanc said, “we’re definitely seeing much better outcomes in AML overall. It takes some time to prove this via outcomes data assessments in a large population, but I expect that registries will show significant improvements in overall survival in the coming years, owing to the many new FDA approvals in AML”
Dr. LeBlanc highlighted national data from 2013-2019 showing that the 5-year relative survival rate from AML is 31.7%. That’s up from 26% just a few years ago, and the numbers “always lag several years behind the current year of practice,” he said. However, “the major area where we still have relatively poor outcomes and significant unmet needs remains the ‘adverse risk’ group of patients, particularly those who are older and/or not candidates for hematopoietic stem cell transplantation, which generally is the only potentially curative option for adverse-risk AML.”
He went on to say that “this risk grouping includes those with TP53 mutations, most of which confer a particularly poor prognosis. Exciting therapies that many of us were hoping would prove effective in this subgroup have unfortunately failed in recent clinical trials. We still have a lot of work to do in adverse-risk AML particularly, and also for those whose leukemia has relapsed.”
Mikkael Sekeres, MD, MS, chief of the Division of Hematology at the University of Miami Miller School of Medicine/Sylvester Comprehensive Cancer Center, agreed that more progress is needed, since survival rates are low even as lifespans improve. One key will be “better identifying subtypes of acute myeloid leukemia, and identifying the therapies that will benefit those people most,” he said in an interview. On the other side, it’s important to identify “when aggressive therapies aren’t going to work in somebody and maybe turn toward less-aggressive approaches so we can maximize that person’s quality of life.”
What advice do AML experts have for their colleagues? Dr. LeBlanc said “older patients are not often enough considered for allogeneic stem cell transplantation, which could potentially cure their AML when given as a consolidation treatment for those in remission. I have several patients who are healthy and in their 70s who have enormously benefited from transplants and are now being several years out from transplant with adverse risk AML and without relapse. They’ve had no significant impairments of their quality of life, including no significant graft vs. host disease.”
Dr. Sekeres highlighted the American Society of Hematology’s guidelines for treating older adults with AML, which are currently being updated. It’s crucial to order genetic testing “up front,” he said. “I’m often pleasantly surprised when genetic testing returns and reveals that I have other treatment options.”
However, it’s crucial to understand a patient’s priorities. “I’ve had patients who are 75 who say to me, ‘Do everything under the sun to get rid of my leukemia, I want to live as long as possible.’ And I’ve had patients who say, ‘I want to see as little of doctors and nurses as I can. I want you to maximize my quality of life and keep me out of the hospital.’ ”
Dr. Sekeres also noted that insurers may not cover some pill-based AML treatments such as venetoclax. “We work with our patients and assistance programs. For the most part, we’re pretty successful at getting these drugs for our patients,” he said.
In Pediatrics, Clinical Trials Are Crucial
AML in children is less well-known than in adults, since the number of cases is so small. The disease is diagnosed in about 500 children a year in the United States, according to St. Jude Children’s Research Hospital, adding, however, that AML is “the most common second cancer among children treated for other cancers.”
AML in children gained attention earlier this year when the 2-year-old daughter of a Boston Herald NFL reporter died of the disease following a bone marrow transplant and chemotherapy. Despite the agonies of her treatment, reporter Doug Kyed told a reporter that his daughter Hallie “was still able to find joy every day.”
In an interview, hematologist/oncologist Sarah K. Tasian, MD, of Children’s Hospital of Philadelphia, said researchers are discovering that pediatric AML is significantly different on from a biological perspective from adult AML. “We’ve come to understand a lot more about who these patients are, what makes these leukemias tick, and what their Achilles’ heels are. Then we can align that with the clinical trials outcome data that we have.”
About 80%-90% of pediatric patients with AML nationwide are enrolled in clinical trials, Dr. Tasian said, and an international consortium called the Children’s Oncology Group gathers data about genetics. About 60%-70% of patients will be cured, she added.
However, “we’ve kind of been stuck for about the last 20 years,” she said. “A lot of improving the survival of patients has not been because we’ve been better at chemotherapy or using new chemo, but because we’ve gotten better at supportive care, at treating infections that can be fatal.”
There haven’t been major conflicts with insurers over coverage, she said, although drug shortages are a problem, especially in relapsed AML.
As for advice to colleagues, Dr. Tasian counseled them to understand the importance of genetic testing and the expanding role of stem cell transplants. “We are now transplanting somewhere between 30% and 50% of children with AML, which is a higher rate than we used to do,” she said. The number is up thanks to genetic testing that reveals which patients are most likely to benefit.
Also, she noted, “the chemotherapy that we get to these patients is really strong, and patients have a lot of complications. Really pay attention to supportive care.”
Dr. LeBlanc reported ties with AbbVie, Agios/Servier, Astellas, BMS/Celgene, Genentech, Pfizer, Incyte, Rige, Deverra, GSK, Jazz, and Seattle Genetics. Dr. Sekeres discloses relationships with BMS and Kurome. Dr. Tasian serves as the Leukemia & Lymphoma Society Pediatric Acute Leukemia consortium clinical trials leader and works with pharmaceutical companies on clinical trials under confidentiality agreements. Dr. Brunner has no disclosures.
For adult patients, “we’ve seen a series of remarkable and well-overdue advances in a space that had not changed much over the prior decades,” hematologist/oncologist Thomas William LeBlanc, MD, associate professor of medicine at Duke University School of Medicine, Durham, North Carolina, said in an interview.
According to the National Cancer Institute, AML will be newly diagnosed in 20,800 patients in 2024, at a median age of 69, and will cause 11,220 deaths. As many as 70% of adult patients will reach complete remission, and 45% of those will live for more than 3 years and potentially be cured. As for children, the Leukemia & Lymphoma Society says the 5-year survival rate from 2012-2018 was 69% for those under 15 years old.
As the American Cancer Society notes, the goal of AML treatment “is to put the leukemia into complete remission (the bone marrow and blood cell counts return to normal), preferably a complete molecular remission (no signs of leukemia in the bone marrow, even using sensitive lab tests), and to keep it that way.”
Chemotherapy Strategies Shift Over Time
In terms of the treatment of adults with AML, “targeted therapies, in addition to the expanding role of venetoclax, has really altered our approach to AML from diagnosis, including after relapse, and later in the disease,” hematologist/oncologist Andrew M. Brunner, MD, of Harvard Medical School and Massachusetts General Hospital, Boston, said in an interview. “The ability to explore these options as monotherapy and in novel combinations has dramatically expanded our treatment options.”
Much depends on the underlying genetic profile of the disease, he said. “There certainly have been gains in patient survival in AML, but those improvements remain fairly heterogeneous and dependent on the underlying genetic profile of the disease. For instance, advances in FLT3- and IDH1/2-mutated AML are a direct result of the improvements in targeted therapies directed at these mutations. Similarly, some molecular and cytogenetic subtypes of AML are particularly responsive to venetoclax-based regimens, and these regimens have been expanded to previously undertreated populations, particularly those over age 60.”
Specifically, Dr. LeBlanc said, the Food and Drug Administration has approved “3 different FLT3 inhibitors, 2 IDH1 inhibitors, 1 IDH2 inhibitor, a BCL-2 inhibitor, a smoothened/hedgehog pathway inhibitor, an oral maintenance chemotherapy/hypomethylating agent (CC-486/oral azacitidine), a CD33-targeting antibody-drug conjugate, and even a novel formulation of two older chemotherapies that improves efficacy in a poor prognosis subgroup (CPX-351/liposomal daunorubicin and cytarabine).”
There’s also been a shift in treatment protocols for patients who were not fit for intensive chemotherapy. In the past, he said, it was standard “to give single-agent hypomethylating chemotherapy with azacitidine or decitabine, or in some contexts, low-dose chemotherapy with cytarabine. Today, many patients who are older and/or more frail are receiving novel therapies either alone or in combination, with greater efficacy and longer duration of response than previously seen with chemotherapy alone.”
Outcomes Improve but Remain Grim in High-Risk Cases
As a result, Dr. LeBlanc said, “we’re definitely seeing much better outcomes in AML overall. It takes some time to prove this via outcomes data assessments in a large population, but I expect that registries will show significant improvements in overall survival in the coming years, owing to the many new FDA approvals in AML”
Dr. LeBlanc highlighted national data from 2013-2019 showing that the 5-year relative survival rate from AML is 31.7%. That’s up from 26% just a few years ago, and the numbers “always lag several years behind the current year of practice,” he said. However, “the major area where we still have relatively poor outcomes and significant unmet needs remains the ‘adverse risk’ group of patients, particularly those who are older and/or not candidates for hematopoietic stem cell transplantation, which generally is the only potentially curative option for adverse-risk AML.”
He went on to say that “this risk grouping includes those with TP53 mutations, most of which confer a particularly poor prognosis. Exciting therapies that many of us were hoping would prove effective in this subgroup have unfortunately failed in recent clinical trials. We still have a lot of work to do in adverse-risk AML particularly, and also for those whose leukemia has relapsed.”
Mikkael Sekeres, MD, MS, chief of the Division of Hematology at the University of Miami Miller School of Medicine/Sylvester Comprehensive Cancer Center, agreed that more progress is needed, since survival rates are low even as lifespans improve. One key will be “better identifying subtypes of acute myeloid leukemia, and identifying the therapies that will benefit those people most,” he said in an interview. On the other side, it’s important to identify “when aggressive therapies aren’t going to work in somebody and maybe turn toward less-aggressive approaches so we can maximize that person’s quality of life.”
What advice do AML experts have for their colleagues? Dr. LeBlanc said “older patients are not often enough considered for allogeneic stem cell transplantation, which could potentially cure their AML when given as a consolidation treatment for those in remission. I have several patients who are healthy and in their 70s who have enormously benefited from transplants and are now being several years out from transplant with adverse risk AML and without relapse. They’ve had no significant impairments of their quality of life, including no significant graft vs. host disease.”
Dr. Sekeres highlighted the American Society of Hematology’s guidelines for treating older adults with AML, which are currently being updated. It’s crucial to order genetic testing “up front,” he said. “I’m often pleasantly surprised when genetic testing returns and reveals that I have other treatment options.”
However, it’s crucial to understand a patient’s priorities. “I’ve had patients who are 75 who say to me, ‘Do everything under the sun to get rid of my leukemia, I want to live as long as possible.’ And I’ve had patients who say, ‘I want to see as little of doctors and nurses as I can. I want you to maximize my quality of life and keep me out of the hospital.’ ”
Dr. Sekeres also noted that insurers may not cover some pill-based AML treatments such as venetoclax. “We work with our patients and assistance programs. For the most part, we’re pretty successful at getting these drugs for our patients,” he said.
In Pediatrics, Clinical Trials Are Crucial
AML in children is less well-known than in adults, since the number of cases is so small. The disease is diagnosed in about 500 children a year in the United States, according to St. Jude Children’s Research Hospital, adding, however, that AML is “the most common second cancer among children treated for other cancers.”
AML in children gained attention earlier this year when the 2-year-old daughter of a Boston Herald NFL reporter died of the disease following a bone marrow transplant and chemotherapy. Despite the agonies of her treatment, reporter Doug Kyed told a reporter that his daughter Hallie “was still able to find joy every day.”
In an interview, hematologist/oncologist Sarah K. Tasian, MD, of Children’s Hospital of Philadelphia, said researchers are discovering that pediatric AML is significantly different on from a biological perspective from adult AML. “We’ve come to understand a lot more about who these patients are, what makes these leukemias tick, and what their Achilles’ heels are. Then we can align that with the clinical trials outcome data that we have.”
About 80%-90% of pediatric patients with AML nationwide are enrolled in clinical trials, Dr. Tasian said, and an international consortium called the Children’s Oncology Group gathers data about genetics. About 60%-70% of patients will be cured, she added.
However, “we’ve kind of been stuck for about the last 20 years,” she said. “A lot of improving the survival of patients has not been because we’ve been better at chemotherapy or using new chemo, but because we’ve gotten better at supportive care, at treating infections that can be fatal.”
There haven’t been major conflicts with insurers over coverage, she said, although drug shortages are a problem, especially in relapsed AML.
As for advice to colleagues, Dr. Tasian counseled them to understand the importance of genetic testing and the expanding role of stem cell transplants. “We are now transplanting somewhere between 30% and 50% of children with AML, which is a higher rate than we used to do,” she said. The number is up thanks to genetic testing that reveals which patients are most likely to benefit.
Also, she noted, “the chemotherapy that we get to these patients is really strong, and patients have a lot of complications. Really pay attention to supportive care.”
Dr. LeBlanc reported ties with AbbVie, Agios/Servier, Astellas, BMS/Celgene, Genentech, Pfizer, Incyte, Rige, Deverra, GSK, Jazz, and Seattle Genetics. Dr. Sekeres discloses relationships with BMS and Kurome. Dr. Tasian serves as the Leukemia & Lymphoma Society Pediatric Acute Leukemia consortium clinical trials leader and works with pharmaceutical companies on clinical trials under confidentiality agreements. Dr. Brunner has no disclosures.
Are Food Emulsifiers Associated With Increased Cancer Risk?
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Democratic Lawmakers Press Pfizer on Chemotherapy Drug Shortages
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
Unleashing Our Immune Response to Quash Cancer
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
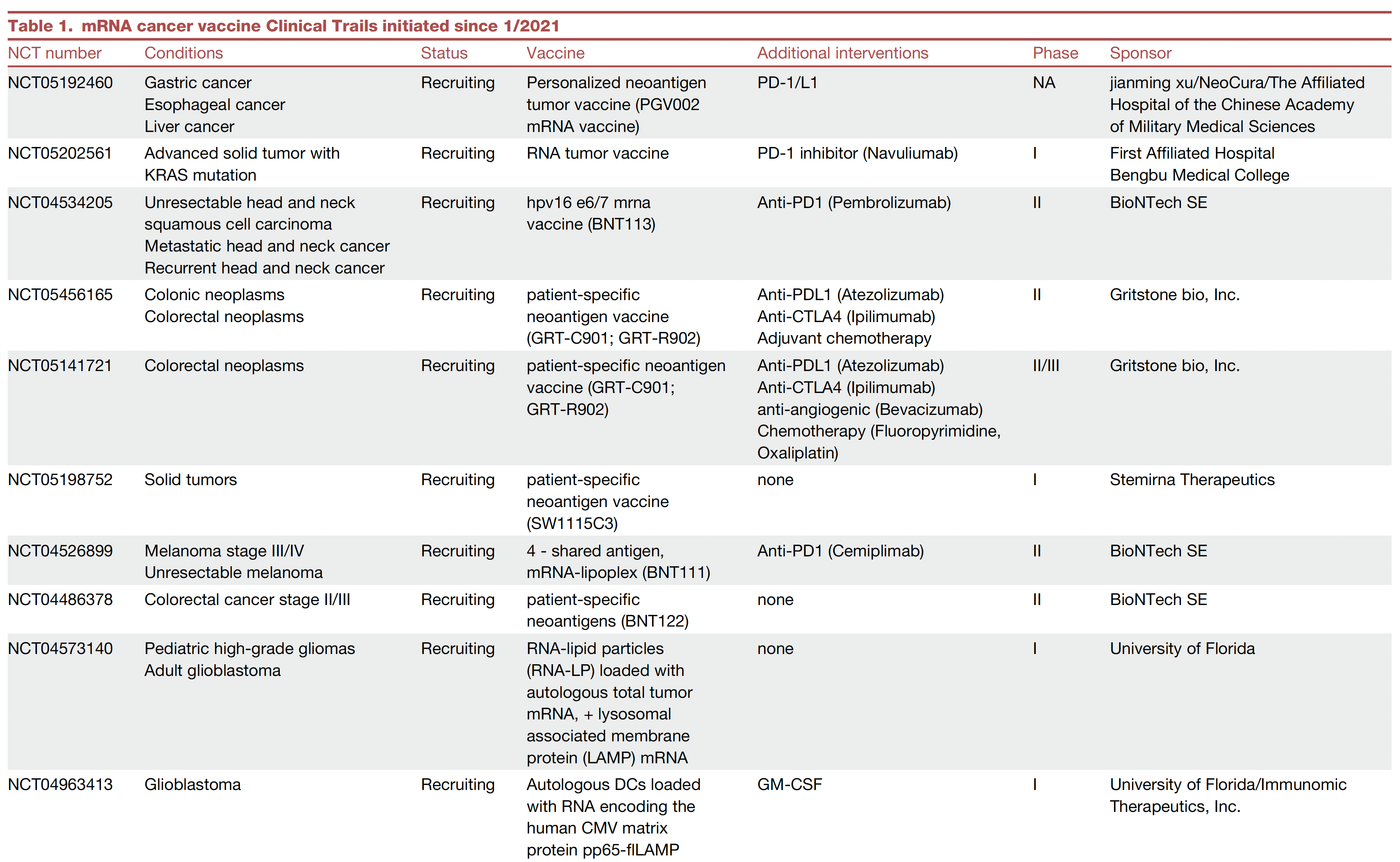{kind=link}
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
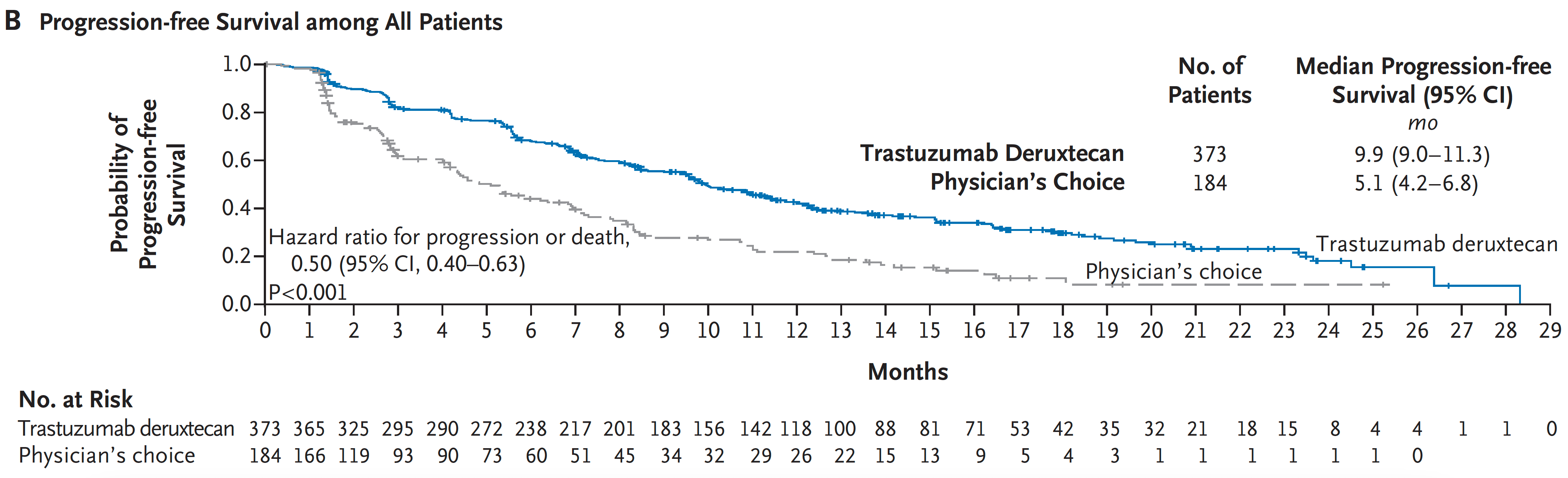{kind=link}
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
MRD status predicts transplant benefit in NPM1-mutated AML
.
This survival benefit did not extend to patients who were MRD-negative after their second induction therapy, Jad Othman, MBBS, reported at the American Society of Hematology annual meeting.
The findings confirm the value of assessing MRD after induction chemotherapy to help identify patients with NPM1-mutated AML in first complete remission who are more likely to benefit from allogeneic transplant, said Dr. Othman, of King’s College London and Guy’s and St Thomas’ NHS Foundation Trust, London, and the University of Sydney, Australia.
Recently, updated European LeukemiaNet recommendations, which stratify patients with AML by favorable, intermediate, and adverse prognoses, now include a revised genetic-risk classification. This classification generally considers NPM1-mutated AML favorable risk. However, having a co-mutation with FLT3-ITD raises the risk to intermediate.
Despite this increased granularity in risk stratification, “it’s still not really clear who should have transplant in first remission with NPM1-mutated AML,” Dr. Othman said. “And there is still significant variation in practice, not just worldwide but even center to center.”
Although accumulating evidence suggests that MRD-negative patients with intermediate-risk AML are unlikely to benefit from allogeneic transplant in first complete remission, the presence of a FLT3-ITD mutation is often considered an indication for transplant, Othman explained. However, most studies supporting this view occurred before the development of sensitive molecular MRD measurement techniques.
The latest findings, from two sequential prospective randomized trials of intensive chemotherapy in adults aged 18-60 years with newly diagnosed AML may help clarify who will probably benefit from transplant and who won’t based on MRD status and relevant molecular features.
The first study (AML17), conducted from 2009 to 2014, selected patients for transplant in first complete remission using a validated risk score that incorporated features including age, sex, and response after therapy. The other (AML19), conducted from 2015 to 2020, selected patients with NPM1-mutated AML for transplant only if they tested positive for MRD in peripheral blood after their second course of treatment, regardless of FLT3-ITD status or other baseline risk factors.
Overall, the current analysis included the 737 patients with NPM1-mutated AML, 348 from AML17 and 389 from AML19, who were in complete remission after two courses of treatment and had an MRD sample at that point.
In AML17, 27% of MRD-positive patients (16 of 60) and 18% of MRD-negative patients (52 of 288) underwent transplant in first complete remission compared with 60% (50 of 83) and 16% (49 of 306), respectively, in AML19.
Among all 737 patients, Dr. Othman and colleagues did not observe an overall survival benefit among those who underwent transplant vs those who did not (hazard ratio [HR], 1.01) or among patients who were MRD-negative (HR, 0.82).
However, patients who were MRD-positive did have a significant survival advantage after transplant (HR, 0.39). In these patients, 3-year overall survival was 61% among those who underwent transplant vs 24% among those who did not.
In MRD-negative patients, transplant in first complete remission did not improve overall survival despite improved relapse-free survival (HR, 0.50). This outcome, Othman explained, probably occurred because most patients who did not undergo transplant and who relapsed were salvaged, with about two thirds undergoing a transplant during their second complete response.
Results in patients with NPM1 FLT3-ITD co-mutation mirrored those in the overall population: MRD-positive patients in first complete remission who underwent transplant demonstrated improved overall survival compared with those without transplant (HR, 0.52), but the overall survival benefit did not extend to MRD-negative patients (HR, 0.80).
The findings show that molecular MRD after induction chemotherapy can identify patients with NPM1-mutated AML who are more likely to benefit from transplant in first remission, Dr. Othman concluded. However, he noted, because only 16% of patients overall were older than 60 years, the results may not be generalizable to older patients.
A version of this article appeared on Medscape.com.
.
This survival benefit did not extend to patients who were MRD-negative after their second induction therapy, Jad Othman, MBBS, reported at the American Society of Hematology annual meeting.
The findings confirm the value of assessing MRD after induction chemotherapy to help identify patients with NPM1-mutated AML in first complete remission who are more likely to benefit from allogeneic transplant, said Dr. Othman, of King’s College London and Guy’s and St Thomas’ NHS Foundation Trust, London, and the University of Sydney, Australia.
Recently, updated European LeukemiaNet recommendations, which stratify patients with AML by favorable, intermediate, and adverse prognoses, now include a revised genetic-risk classification. This classification generally considers NPM1-mutated AML favorable risk. However, having a co-mutation with FLT3-ITD raises the risk to intermediate.
Despite this increased granularity in risk stratification, “it’s still not really clear who should have transplant in first remission with NPM1-mutated AML,” Dr. Othman said. “And there is still significant variation in practice, not just worldwide but even center to center.”
Although accumulating evidence suggests that MRD-negative patients with intermediate-risk AML are unlikely to benefit from allogeneic transplant in first complete remission, the presence of a FLT3-ITD mutation is often considered an indication for transplant, Othman explained. However, most studies supporting this view occurred before the development of sensitive molecular MRD measurement techniques.
The latest findings, from two sequential prospective randomized trials of intensive chemotherapy in adults aged 18-60 years with newly diagnosed AML may help clarify who will probably benefit from transplant and who won’t based on MRD status and relevant molecular features.
The first study (AML17), conducted from 2009 to 2014, selected patients for transplant in first complete remission using a validated risk score that incorporated features including age, sex, and response after therapy. The other (AML19), conducted from 2015 to 2020, selected patients with NPM1-mutated AML for transplant only if they tested positive for MRD in peripheral blood after their second course of treatment, regardless of FLT3-ITD status or other baseline risk factors.
Overall, the current analysis included the 737 patients with NPM1-mutated AML, 348 from AML17 and 389 from AML19, who were in complete remission after two courses of treatment and had an MRD sample at that point.
In AML17, 27% of MRD-positive patients (16 of 60) and 18% of MRD-negative patients (52 of 288) underwent transplant in first complete remission compared with 60% (50 of 83) and 16% (49 of 306), respectively, in AML19.
Among all 737 patients, Dr. Othman and colleagues did not observe an overall survival benefit among those who underwent transplant vs those who did not (hazard ratio [HR], 1.01) or among patients who were MRD-negative (HR, 0.82).
However, patients who were MRD-positive did have a significant survival advantage after transplant (HR, 0.39). In these patients, 3-year overall survival was 61% among those who underwent transplant vs 24% among those who did not.
In MRD-negative patients, transplant in first complete remission did not improve overall survival despite improved relapse-free survival (HR, 0.50). This outcome, Othman explained, probably occurred because most patients who did not undergo transplant and who relapsed were salvaged, with about two thirds undergoing a transplant during their second complete response.
Results in patients with NPM1 FLT3-ITD co-mutation mirrored those in the overall population: MRD-positive patients in first complete remission who underwent transplant demonstrated improved overall survival compared with those without transplant (HR, 0.52), but the overall survival benefit did not extend to MRD-negative patients (HR, 0.80).
The findings show that molecular MRD after induction chemotherapy can identify patients with NPM1-mutated AML who are more likely to benefit from transplant in first remission, Dr. Othman concluded. However, he noted, because only 16% of patients overall were older than 60 years, the results may not be generalizable to older patients.
A version of this article appeared on Medscape.com.
.
This survival benefit did not extend to patients who were MRD-negative after their second induction therapy, Jad Othman, MBBS, reported at the American Society of Hematology annual meeting.
The findings confirm the value of assessing MRD after induction chemotherapy to help identify patients with NPM1-mutated AML in first complete remission who are more likely to benefit from allogeneic transplant, said Dr. Othman, of King’s College London and Guy’s and St Thomas’ NHS Foundation Trust, London, and the University of Sydney, Australia.
Recently, updated European LeukemiaNet recommendations, which stratify patients with AML by favorable, intermediate, and adverse prognoses, now include a revised genetic-risk classification. This classification generally considers NPM1-mutated AML favorable risk. However, having a co-mutation with FLT3-ITD raises the risk to intermediate.
Despite this increased granularity in risk stratification, “it’s still not really clear who should have transplant in first remission with NPM1-mutated AML,” Dr. Othman said. “And there is still significant variation in practice, not just worldwide but even center to center.”
Although accumulating evidence suggests that MRD-negative patients with intermediate-risk AML are unlikely to benefit from allogeneic transplant in first complete remission, the presence of a FLT3-ITD mutation is often considered an indication for transplant, Othman explained. However, most studies supporting this view occurred before the development of sensitive molecular MRD measurement techniques.
The latest findings, from two sequential prospective randomized trials of intensive chemotherapy in adults aged 18-60 years with newly diagnosed AML may help clarify who will probably benefit from transplant and who won’t based on MRD status and relevant molecular features.
The first study (AML17), conducted from 2009 to 2014, selected patients for transplant in first complete remission using a validated risk score that incorporated features including age, sex, and response after therapy. The other (AML19), conducted from 2015 to 2020, selected patients with NPM1-mutated AML for transplant only if they tested positive for MRD in peripheral blood after their second course of treatment, regardless of FLT3-ITD status or other baseline risk factors.
Overall, the current analysis included the 737 patients with NPM1-mutated AML, 348 from AML17 and 389 from AML19, who were in complete remission after two courses of treatment and had an MRD sample at that point.
In AML17, 27% of MRD-positive patients (16 of 60) and 18% of MRD-negative patients (52 of 288) underwent transplant in first complete remission compared with 60% (50 of 83) and 16% (49 of 306), respectively, in AML19.
Among all 737 patients, Dr. Othman and colleagues did not observe an overall survival benefit among those who underwent transplant vs those who did not (hazard ratio [HR], 1.01) or among patients who were MRD-negative (HR, 0.82).
However, patients who were MRD-positive did have a significant survival advantage after transplant (HR, 0.39). In these patients, 3-year overall survival was 61% among those who underwent transplant vs 24% among those who did not.
In MRD-negative patients, transplant in first complete remission did not improve overall survival despite improved relapse-free survival (HR, 0.50). This outcome, Othman explained, probably occurred because most patients who did not undergo transplant and who relapsed were salvaged, with about two thirds undergoing a transplant during their second complete response.
Results in patients with NPM1 FLT3-ITD co-mutation mirrored those in the overall population: MRD-positive patients in first complete remission who underwent transplant demonstrated improved overall survival compared with those without transplant (HR, 0.52), but the overall survival benefit did not extend to MRD-negative patients (HR, 0.80).
The findings show that molecular MRD after induction chemotherapy can identify patients with NPM1-mutated AML who are more likely to benefit from transplant in first remission, Dr. Othman concluded. However, he noted, because only 16% of patients overall were older than 60 years, the results may not be generalizable to older patients.
A version of this article appeared on Medscape.com.
FROM ASH 2023
This test may guide AML therapy for Black pediatric patients
.
The score, dubbed ACS10 and initially highlighted in a 2022 report, predicts how well patients will respond to cytarabine based on their genetic make-up, and has the potential to personalize treatment for Black pediatric patients, a group that often has worse outcomes than White patients.
In the current study, presented at the annual meeting of the American Society of Hematology (ASH) , Black patients with low ACS10 scores had significantly worse outcomes compared with those with high scores when initially treated with low-dose cytarabine, daunorubicin, and etoposide.
The difference in outcomes disappeared, however, for patients who received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine.
The genetic traits revealed by the test likely help explain why Black patients with AML typically fare worse on certain regimens, Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute, commented in an ASH press preview briefing.
This study also suggests that clinicians should perform testing for genetic variants and biomarkers that impact outcomes “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity,” said Dr. Dunbar, also secretary of ASH.
The ACS10 test, derived from a combination of 10 single nucleotide polymorphisms, is not yet available, but one could be developed to help guide treatment decisions for clinicians, especially those in developing countries where AML treatment can be very expensive, said study lead author Jatinder Lamba, PhD, MSc, of the University of Florida College of Pharmacy, Gainesville, at an ASH press briefing on Thursday.
Prior research shows that Black pediatric patients with AML often have worse outcomes than White patients. A recent study , for instance, found Black patients with AML, especially those aged 18 to 29 years, had a higher early death rate compared with White patients (16% vs 3%) and significantly lower 5-year overall survival rates (22% vs 51%). The authors of this study suggested that genetic differences between young Black and White patients could help explain the disparity.
In the new analysis, Dr. Lamba and colleagues explored how outcomes by race and cytarabine pharmacogenomics varied in pediatric patients with AML.
The study included 86 Black patients and 359 White patients with newly diagnosed AML treated on two multi-institutional clinical trials. The patients received one of three initial treatments that included cytarabine: high-dose or low-dose cytarabine, daunorubicin, and etoposide, or clofarabine and cytarabine.
Most Black patients in the analysis (73%) had low ACS10 scores compared with 30% of White patients.
Unlike other recent reports, this study found that Black and White patients had similar complete remission rates following two courses of induction therapy (92.6% vs 95%) as well as similar rates of minimal residual disease negativity after one course (55.8% vs 55.4%).
Event-free survival (EFS) and overall survival rates were also similar, with 5-year EFS estimates at 58.3% for Black patients and 58.2% for White patients and overall survival rates at 63.8% vs 69.4%, respectively (P = .24).
However, when separating outcomes by ACS10 scores, Black patients with low scores had significantly worse EFS following low-dose cytarabine, daunorubicin, and etoposide compared with those with high ACS10 scores. And when these patients received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine induction therapy instead, the differences went away.
Overall, Black patients demonstrated significantly better EFS following treatment with clofarabine and cytarabine compared with the low-dose cytarabine triple therapy (hazard ratio, 0.17; P = .01). After adjusting for cofounders, clofarabine and cytarabine induction was the best treatment for Black patients with low ACS10 scores (HR for EFS, 0.2).
“Our results suggest that pharmacogenomics differences between Black and White patients should be considered when tailoring induction regimens to improve outcomes of Black patients and bridge the racial disparity gap in AML treatment,” the researchers concluded.
In developing countries, especially in Africa, starting patients on high-dose cytarabine, daunorubicin, and etoposide can lead to better results “without increasing much of the economic burden” since this treatment is the cheapest, Dr. Lamba said. “At the same time, if the patients have high ACS10 score, you can reduce their economic burden by giving them standard dose” cytarabine, daunorubicin, and etoposide and achieve similar results.
No study funding was reported. Dr. Lamba reported no relevant financial relationships, and three other authors reported various disclosures. Disclosures for Dr. Dunbar were unavailable..
A version of this article appeared on Medscape.com.
.
The score, dubbed ACS10 and initially highlighted in a 2022 report, predicts how well patients will respond to cytarabine based on their genetic make-up, and has the potential to personalize treatment for Black pediatric patients, a group that often has worse outcomes than White patients.
In the current study, presented at the annual meeting of the American Society of Hematology (ASH) , Black patients with low ACS10 scores had significantly worse outcomes compared with those with high scores when initially treated with low-dose cytarabine, daunorubicin, and etoposide.
The difference in outcomes disappeared, however, for patients who received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine.
The genetic traits revealed by the test likely help explain why Black patients with AML typically fare worse on certain regimens, Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute, commented in an ASH press preview briefing.
This study also suggests that clinicians should perform testing for genetic variants and biomarkers that impact outcomes “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity,” said Dr. Dunbar, also secretary of ASH.
The ACS10 test, derived from a combination of 10 single nucleotide polymorphisms, is not yet available, but one could be developed to help guide treatment decisions for clinicians, especially those in developing countries where AML treatment can be very expensive, said study lead author Jatinder Lamba, PhD, MSc, of the University of Florida College of Pharmacy, Gainesville, at an ASH press briefing on Thursday.
Prior research shows that Black pediatric patients with AML often have worse outcomes than White patients. A recent study , for instance, found Black patients with AML, especially those aged 18 to 29 years, had a higher early death rate compared with White patients (16% vs 3%) and significantly lower 5-year overall survival rates (22% vs 51%). The authors of this study suggested that genetic differences between young Black and White patients could help explain the disparity.
In the new analysis, Dr. Lamba and colleagues explored how outcomes by race and cytarabine pharmacogenomics varied in pediatric patients with AML.
The study included 86 Black patients and 359 White patients with newly diagnosed AML treated on two multi-institutional clinical trials. The patients received one of three initial treatments that included cytarabine: high-dose or low-dose cytarabine, daunorubicin, and etoposide, or clofarabine and cytarabine.
Most Black patients in the analysis (73%) had low ACS10 scores compared with 30% of White patients.
Unlike other recent reports, this study found that Black and White patients had similar complete remission rates following two courses of induction therapy (92.6% vs 95%) as well as similar rates of minimal residual disease negativity after one course (55.8% vs 55.4%).
Event-free survival (EFS) and overall survival rates were also similar, with 5-year EFS estimates at 58.3% for Black patients and 58.2% for White patients and overall survival rates at 63.8% vs 69.4%, respectively (P = .24).
However, when separating outcomes by ACS10 scores, Black patients with low scores had significantly worse EFS following low-dose cytarabine, daunorubicin, and etoposide compared with those with high ACS10 scores. And when these patients received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine induction therapy instead, the differences went away.
Overall, Black patients demonstrated significantly better EFS following treatment with clofarabine and cytarabine compared with the low-dose cytarabine triple therapy (hazard ratio, 0.17; P = .01). After adjusting for cofounders, clofarabine and cytarabine induction was the best treatment for Black patients with low ACS10 scores (HR for EFS, 0.2).
“Our results suggest that pharmacogenomics differences between Black and White patients should be considered when tailoring induction regimens to improve outcomes of Black patients and bridge the racial disparity gap in AML treatment,” the researchers concluded.
In developing countries, especially in Africa, starting patients on high-dose cytarabine, daunorubicin, and etoposide can lead to better results “without increasing much of the economic burden” since this treatment is the cheapest, Dr. Lamba said. “At the same time, if the patients have high ACS10 score, you can reduce their economic burden by giving them standard dose” cytarabine, daunorubicin, and etoposide and achieve similar results.
No study funding was reported. Dr. Lamba reported no relevant financial relationships, and three other authors reported various disclosures. Disclosures for Dr. Dunbar were unavailable..
A version of this article appeared on Medscape.com.
.
The score, dubbed ACS10 and initially highlighted in a 2022 report, predicts how well patients will respond to cytarabine based on their genetic make-up, and has the potential to personalize treatment for Black pediatric patients, a group that often has worse outcomes than White patients.
In the current study, presented at the annual meeting of the American Society of Hematology (ASH) , Black patients with low ACS10 scores had significantly worse outcomes compared with those with high scores when initially treated with low-dose cytarabine, daunorubicin, and etoposide.
The difference in outcomes disappeared, however, for patients who received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine.
The genetic traits revealed by the test likely help explain why Black patients with AML typically fare worse on certain regimens, Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute, commented in an ASH press preview briefing.
This study also suggests that clinicians should perform testing for genetic variants and biomarkers that impact outcomes “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity,” said Dr. Dunbar, also secretary of ASH.
The ACS10 test, derived from a combination of 10 single nucleotide polymorphisms, is not yet available, but one could be developed to help guide treatment decisions for clinicians, especially those in developing countries where AML treatment can be very expensive, said study lead author Jatinder Lamba, PhD, MSc, of the University of Florida College of Pharmacy, Gainesville, at an ASH press briefing on Thursday.
Prior research shows that Black pediatric patients with AML often have worse outcomes than White patients. A recent study , for instance, found Black patients with AML, especially those aged 18 to 29 years, had a higher early death rate compared with White patients (16% vs 3%) and significantly lower 5-year overall survival rates (22% vs 51%). The authors of this study suggested that genetic differences between young Black and White patients could help explain the disparity.
In the new analysis, Dr. Lamba and colleagues explored how outcomes by race and cytarabine pharmacogenomics varied in pediatric patients with AML.
The study included 86 Black patients and 359 White patients with newly diagnosed AML treated on two multi-institutional clinical trials. The patients received one of three initial treatments that included cytarabine: high-dose or low-dose cytarabine, daunorubicin, and etoposide, or clofarabine and cytarabine.
Most Black patients in the analysis (73%) had low ACS10 scores compared with 30% of White patients.
Unlike other recent reports, this study found that Black and White patients had similar complete remission rates following two courses of induction therapy (92.6% vs 95%) as well as similar rates of minimal residual disease negativity after one course (55.8% vs 55.4%).
Event-free survival (EFS) and overall survival rates were also similar, with 5-year EFS estimates at 58.3% for Black patients and 58.2% for White patients and overall survival rates at 63.8% vs 69.4%, respectively (P = .24).
However, when separating outcomes by ACS10 scores, Black patients with low scores had significantly worse EFS following low-dose cytarabine, daunorubicin, and etoposide compared with those with high ACS10 scores. And when these patients received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine induction therapy instead, the differences went away.
Overall, Black patients demonstrated significantly better EFS following treatment with clofarabine and cytarabine compared with the low-dose cytarabine triple therapy (hazard ratio, 0.17; P = .01). After adjusting for cofounders, clofarabine and cytarabine induction was the best treatment for Black patients with low ACS10 scores (HR for EFS, 0.2).
“Our results suggest that pharmacogenomics differences between Black and White patients should be considered when tailoring induction regimens to improve outcomes of Black patients and bridge the racial disparity gap in AML treatment,” the researchers concluded.
In developing countries, especially in Africa, starting patients on high-dose cytarabine, daunorubicin, and etoposide can lead to better results “without increasing much of the economic burden” since this treatment is the cheapest, Dr. Lamba said. “At the same time, if the patients have high ACS10 score, you can reduce their economic burden by giving them standard dose” cytarabine, daunorubicin, and etoposide and achieve similar results.
No study funding was reported. Dr. Lamba reported no relevant financial relationships, and three other authors reported various disclosures. Disclosures for Dr. Dunbar were unavailable..
A version of this article appeared on Medscape.com.
FROM ASH 2023
ASH 2023: Equity, Sickle Cell, and Real-Life Outcomes
Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute and secretary of ASH, added that insight into actual patient experiences also will be a major theme at ASH 2023.
“There is a huge growth in research on outcomes and focusing on using real-world data and how important that is,” Dr. Dunbar said. “Academic research and hematology is really focusing on patient-reported outcomes and how care is delivered in a real-world setting – actually looking at what matters to patients. Are they alive in a certain number of years? And how are they feeling?”
As an example, Dr. Dunbar pointed to an abstract that examined clinical databases in Canada and found that real-world outcomes in multiple myeloma treatments were much worse than those in the original clinical trials for the therapies. Patients reached relapse 44% faster and their overall survival was 75% worse.
In the media briefing, ASH chair of communications Mikkael A. Sekeres, MD, MS, of the Sylvester Comprehensive Cancer Center at the University of Miami, noted that patients in these types of clinical trials “are just these pristine specimens of human beings except for the cancer that’s being treated.”
Dr. Dunbar agreed, noting that “patients who are able to enroll in clinical trials are more likely to be able to show up at the treatment center at the right time and for every dose, have transportation, and afford drugs to prevent side effects. They might stay on the drug for longer, or they have nurses who are always encouraging them of how to make it through a toxicity.”
Hematologists and patients should consider randomized controlled trials to be “the best possible outcome, and perhaps adjust their thinking if an individual patient is older, sicker, or less able to follow a regimen exactly,” she said.
Another highlighted study linked worse outcomes in African-Americans with pediatric acute myeloid leukemia to genetic traits that are more common in that population. The traits “likely explain at least in part the worst outcomes in Black patients in prior studies and on some regimens,” Dr. Dunbar said.
She added that the findings emphasize how testing for genetic variants and biomarkers that impact outcomes should be performed “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity.”
ASH President Robert A. Brodsky, MD, of Johns Hopkins University School of Medicine, Baltimore, highlighted an abstract that reported on the use of AI as a clinical decision support tool to differentiate two easily confused conditions — prefibrotic primary myelofibrosis and essential thrombocythemia.
AI “is a tool that’s going to help pathologists make more accurate and faster diagnoses,” he said. He also spotlighted an abstract about the use of “social media listening” to understand the experiences of patients with SCD and their caregivers. “There can be a lot of misuse and waste of time with social media, but they used this in a way to try and gain insight as to what’s really important to the patients and the caregiver.”
Also, in regard to SCD, Dr. Dunbar pointed to a study that reports on outcomes in patients who received lovotibeglogene autotemcel (lovo-cel) gene therapy for up to 60 months. Both this treatment and a CRISPR-based therapy called exa-cel “appear to result in comparable very impressive efficacy in terms of pain crises and organ dysfunction,” she said. “The hurdle is going to be figuring out how to deliver what will be very expensive and complicated therapies — but likely curative — therapies to patients.”
Another study to be presented at ASH — coauthored by Dr. Brodsky — shows promising results from reduced-intensity haploidentical bone marrow transplantation in adults with severe SCD. Results were similar to those seen with bone marrow from matched siblings, Dr. Sekeres said.
He added that more clarity is needed about new treatment options for SCD, perhaps through a “randomized trial where patients upfront get a haploidentical bone marrow transplant or fully matched bone marrow transplant. Then other patients are randomized to some of these other, newer technology therapies, and we follow them over time. We’re looking not only for overall survival but complications of the therapy itself and how many patients relapse from the treatment.”
Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute and secretary of ASH, added that insight into actual patient experiences also will be a major theme at ASH 2023.
“There is a huge growth in research on outcomes and focusing on using real-world data and how important that is,” Dr. Dunbar said. “Academic research and hematology is really focusing on patient-reported outcomes and how care is delivered in a real-world setting – actually looking at what matters to patients. Are they alive in a certain number of years? And how are they feeling?”
As an example, Dr. Dunbar pointed to an abstract that examined clinical databases in Canada and found that real-world outcomes in multiple myeloma treatments were much worse than those in the original clinical trials for the therapies. Patients reached relapse 44% faster and their overall survival was 75% worse.
In the media briefing, ASH chair of communications Mikkael A. Sekeres, MD, MS, of the Sylvester Comprehensive Cancer Center at the University of Miami, noted that patients in these types of clinical trials “are just these pristine specimens of human beings except for the cancer that’s being treated.”
Dr. Dunbar agreed, noting that “patients who are able to enroll in clinical trials are more likely to be able to show up at the treatment center at the right time and for every dose, have transportation, and afford drugs to prevent side effects. They might stay on the drug for longer, or they have nurses who are always encouraging them of how to make it through a toxicity.”
Hematologists and patients should consider randomized controlled trials to be “the best possible outcome, and perhaps adjust their thinking if an individual patient is older, sicker, or less able to follow a regimen exactly,” she said.
Another highlighted study linked worse outcomes in African-Americans with pediatric acute myeloid leukemia to genetic traits that are more common in that population. The traits “likely explain at least in part the worst outcomes in Black patients in prior studies and on some regimens,” Dr. Dunbar said.
She added that the findings emphasize how testing for genetic variants and biomarkers that impact outcomes should be performed “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity.”
ASH President Robert A. Brodsky, MD, of Johns Hopkins University School of Medicine, Baltimore, highlighted an abstract that reported on the use of AI as a clinical decision support tool to differentiate two easily confused conditions — prefibrotic primary myelofibrosis and essential thrombocythemia.
AI “is a tool that’s going to help pathologists make more accurate and faster diagnoses,” he said. He also spotlighted an abstract about the use of “social media listening” to understand the experiences of patients with SCD and their caregivers. “There can be a lot of misuse and waste of time with social media, but they used this in a way to try and gain insight as to what’s really important to the patients and the caregiver.”
Also, in regard to SCD, Dr. Dunbar pointed to a study that reports on outcomes in patients who received lovotibeglogene autotemcel (lovo-cel) gene therapy for up to 60 months. Both this treatment and a CRISPR-based therapy called exa-cel “appear to result in comparable very impressive efficacy in terms of pain crises and organ dysfunction,” she said. “The hurdle is going to be figuring out how to deliver what will be very expensive and complicated therapies — but likely curative — therapies to patients.”
Another study to be presented at ASH — coauthored by Dr. Brodsky — shows promising results from reduced-intensity haploidentical bone marrow transplantation in adults with severe SCD. Results were similar to those seen with bone marrow from matched siblings, Dr. Sekeres said.
He added that more clarity is needed about new treatment options for SCD, perhaps through a “randomized trial where patients upfront get a haploidentical bone marrow transplant or fully matched bone marrow transplant. Then other patients are randomized to some of these other, newer technology therapies, and we follow them over time. We’re looking not only for overall survival but complications of the therapy itself and how many patients relapse from the treatment.”
Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute and secretary of ASH, added that insight into actual patient experiences also will be a major theme at ASH 2023.
“There is a huge growth in research on outcomes and focusing on using real-world data and how important that is,” Dr. Dunbar said. “Academic research and hematology is really focusing on patient-reported outcomes and how care is delivered in a real-world setting – actually looking at what matters to patients. Are they alive in a certain number of years? And how are they feeling?”
As an example, Dr. Dunbar pointed to an abstract that examined clinical databases in Canada and found that real-world outcomes in multiple myeloma treatments were much worse than those in the original clinical trials for the therapies. Patients reached relapse 44% faster and their overall survival was 75% worse.
In the media briefing, ASH chair of communications Mikkael A. Sekeres, MD, MS, of the Sylvester Comprehensive Cancer Center at the University of Miami, noted that patients in these types of clinical trials “are just these pristine specimens of human beings except for the cancer that’s being treated.”
Dr. Dunbar agreed, noting that “patients who are able to enroll in clinical trials are more likely to be able to show up at the treatment center at the right time and for every dose, have transportation, and afford drugs to prevent side effects. They might stay on the drug for longer, or they have nurses who are always encouraging them of how to make it through a toxicity.”
Hematologists and patients should consider randomized controlled trials to be “the best possible outcome, and perhaps adjust their thinking if an individual patient is older, sicker, or less able to follow a regimen exactly,” she said.
Another highlighted study linked worse outcomes in African-Americans with pediatric acute myeloid leukemia to genetic traits that are more common in that population. The traits “likely explain at least in part the worst outcomes in Black patients in prior studies and on some regimens,” Dr. Dunbar said.
She added that the findings emphasize how testing for genetic variants and biomarkers that impact outcomes should be performed “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity.”
ASH President Robert A. Brodsky, MD, of Johns Hopkins University School of Medicine, Baltimore, highlighted an abstract that reported on the use of AI as a clinical decision support tool to differentiate two easily confused conditions — prefibrotic primary myelofibrosis and essential thrombocythemia.
AI “is a tool that’s going to help pathologists make more accurate and faster diagnoses,” he said. He also spotlighted an abstract about the use of “social media listening” to understand the experiences of patients with SCD and their caregivers. “There can be a lot of misuse and waste of time with social media, but they used this in a way to try and gain insight as to what’s really important to the patients and the caregiver.”
Also, in regard to SCD, Dr. Dunbar pointed to a study that reports on outcomes in patients who received lovotibeglogene autotemcel (lovo-cel) gene therapy for up to 60 months. Both this treatment and a CRISPR-based therapy called exa-cel “appear to result in comparable very impressive efficacy in terms of pain crises and organ dysfunction,” she said. “The hurdle is going to be figuring out how to deliver what will be very expensive and complicated therapies — but likely curative — therapies to patients.”
Another study to be presented at ASH — coauthored by Dr. Brodsky — shows promising results from reduced-intensity haploidentical bone marrow transplantation in adults with severe SCD. Results were similar to those seen with bone marrow from matched siblings, Dr. Sekeres said.
He added that more clarity is needed about new treatment options for SCD, perhaps through a “randomized trial where patients upfront get a haploidentical bone marrow transplant or fully matched bone marrow transplant. Then other patients are randomized to some of these other, newer technology therapies, and we follow them over time. We’re looking not only for overall survival but complications of the therapy itself and how many patients relapse from the treatment.”
AT ASH 2023
FDA panel voices concerns over 2 lymphoma accelerated approvals
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”
At a Nov. 16 meeting, the Oncologic Drugs Advisory Committee of the Food and Drug Administration reviewed the reasons for delays in confirmatory trials for pralatrexate (Folotyn) and belinostat (Beleodaq), both now owned by East Windsor, N.J.–based Acrotech. The FDA granted accelerated approval for pralatrexate in 2009 and belinostat in 2014.
“The consensus of the advisory committee is that we have significant concerns about the very prolonged delay and getting these confirmatory studies underway,” said Andy Chen, MD, PhD, of Oregon Health & Science University, Portland, who served as acting ODAC chair for the meeting.
Corporate ownership changes were among the reasons Acrotech cited for the long delays in producing the confirmatory research on pralatrexate and belinostat. Allos Therapeutics won the FDA approval of pralatrexate in 2009. In 2012, Spectrum Pharmaceuticals acquired Acrotech. Spectrum won approval of belinostat in 2014. Acrotech acquired Spectrum in 2019.
The FDA didn’t ask ODAC to take votes on any questions at the meeting. Instead, the FDA sought its expert feedback about how to address the prolonged delays with pralatrexate and belinostat research and, in general, how to promote more timely completion of confirmatory trials for drugs cleared by accelerated approval.
Pralatrexate and belinostat are both used to treat relapsed or refractory peripheral T-cell lymphoma, a rare and aggressive disease affecting about 10,000-15,000 people annually in the United States.
Through the accelerated approval process, the FDA seeks to speed medicines to people with fatal and serious conditions based on promising signs in clinical testing.
The initial pralatrexate and belinostat were based on phase 2, single-arm, monotherapy studies, with about 109 evaluable patients in the key pralatrexate study and 120 evaluable patients in the belinostat study. As is common, these phase 2 tests used measurements of cancer progression, known as the overall response rate.
The FDA then expects companies to show through more extensive testing that medicines cleared with accelerated approvals can deliver significant benefits, such as extending lives. When there are delays in confirmatory trials, patients can be exposed to medicines, often with significant side effects, that are unlikely to benefit them.
For example, the FDA granted an accelerated approval in 2011 for romidepsin for this use for peripheral T-cell lymphoma, the same condition for which pralatrexate and belinostat are used. But in 2021, Bristol-Myers Squibb withdrew the approval for that use of romidepsin when a confirmatory trial failed to meet the primary efficacy endpoint of progression free survival.
At the meeting, Richard Pazdur, MD, who leads oncology medicine at the FDA, urged Acrotech to shorten the time needed to determine whether its medicines deliver significant benefits to patients and thus merit full approval, or whether they too may fall short.
“We’re really in a situation where patients are caught in the middle here,” Dr. Pazdur said. “I feel very bad for that situation and very bad for the patients that they don’t have this information.”
‘Dangerous precedent’
The FDA in recent years has stepped up its efforts to get companies to complete their required studies on drugs cleared by accelerated approvals. The FDA has granted a total of 187 accelerated approvals for cancer drugs. Many of these cover new uses of established drugs and others serve to allow the introduction of new medicines.
For more than half of these cases, 96 of 187, the FDA already has learned that it made the right call in allowing early access to medicines. Companies have presented study results that confirmed the benefit of drugs and thus been able to convert accelerated approvals to traditional approvals.
But 27 of the 187 oncology accelerated approvals have been withdrawn. In these cases, subsequent research failed to establish the expected benefits of these cancer drugs.
And in 95 cases, the FDA and companies are still waiting for the results of studies to confirm the expected benefit of drugs granted accelerated approvals. The FDA classifies these as ongoing accelerated approvals. About 85% of these ongoing approvals were granted in the past 5 years, in contrast to 14 years for pralatrexate and 9 for belinostat.
“It sets a dangerous precedent for the other sponsors and drug companies to have such outliers from the same company,” said ODAC member Toni K. Choueiri, MD, of Harvard Medical School and the Dana-Farber Cancer Institute, both in Boston.
The current agreement between the FDA and Acrotech focuses on a phase 3 trial, SPI-BEL-301 as the confirmatory study. Acrotech’s plan is to start with dose optimization studies in part 1 of the trial, with part 2 meant to see if its medicines provide a significant benefit as measured by progression-free survival.
The plan is to compare treatments. One group of patients would get belinostat plus a common cancer regimen known as CHOP, another group would get pralatrexate plus the COP cancer regimen, which is CHOP without doxorubicin, and a third group would get CHOP.
Acrotech’s current time line is for part 1, which began in October, to finish by December 2025. Then the part 2 timeline would run from 2026 to 2030, with interim progression-free survival possible by 2028.
ODAC member Ashley Rosko, MD, a hematologist from Ohio State University, Columbus, asked Acrotech what steps it will take to try to speed recruitment for the study.
“We are going to implement many strategies,” including what’s called digital amplification, replied Ashish Anvekar, president of Acrotech. This will help identify patients and channel them toward participating clinical sites.
Alexander A. Vinks, PhD, PharmD, who served as a temporary member of ODAC for the Nov. 16 meeting, said many clinicians will not be excited about enrolling patients in this kind of large, traditionally designed study.
Dr. Vinks, who is professor emeritus at Cincinnati Children’s Hospital Medical Center and University of Cincinnati, now works with consultant group NDA, a firm that advises companies on developing drugs.
Dr. Vinks advised Acrotech should try “to pin down what is most likely a smaller study that could be simpler, but still give robust, informative data.”